User login
Emotions, worse attention linked to pain-related health care use in SCD
The cognitive and emotional status of children with sickle cell disease (SCD) appears to have a significant effect on how they cope with pain and use health care resources, investigators have found.
Results of a retrospective study of 112 children and adolescents with SCD, the majority of whom had sickle cell anemia, showed that ED visits and hospitalizations were significantly lower among children with SCD who performed better on an attention task, as well as those who were better able to cope emotionally with having SCD and pain, reported Zaria Williams, a second-year medical student at Howard University, Washington, and colleagues.
“Since I started learning more about sickle cell disease, I’ve been very concerned about the great disease burden that this condition can place on pediatric patients, particularly those who suffer from pain,” Ms. Williams said in an oral abstract presented at the annual meeting of the American Society of Hematology.
Although many children and adolescents with SCD can have their pain effectively managed at home with opioids and other medications, some require ED visits and potentially hospitalizations for pain management.
“There is great variability in health care utilization among patients with sickle cell disease, with some having to come to the ED and be admit to the hospital more than others. In searching for reasons why this might be the case, we thought about cognitive function and emotional differences between children with sickle cell disease as potentially affecting disease management,” she said.
Anxiety and catastrophizing
Children with SCD are known to be susceptible to affective comorbidities such as anxiety and catastrophizing, and to conditions that have the potential for deleterious effects on executive function, attention, and working memory. To determine whether cognitive and emotional factors affect the disease self-management in children and adolescents with SCD, Ms. Williams and coinvestigators looked at a cohort of 112 SCD patients aged 7-16 years treated at Children’s National Hospital in Washington, D.C.
The patients had participated in a previous pilot study of computerized working memory training. The authors reviewed charts for data on health care utilization, focusing on ED visits and hospitalization for pain 1 and 3 years after enrollment in the study.
They collected data on SCD genotype, disease-related variables, psychosocial information, and measures of cognition and emotion from the dataset. The information included socioeconomic status, parent education level, household income, and number of adults in the household.
Cognitive measures included the Weschler Intelligence Scale for Children full scale IQ, and the Cogstate computerized cognitive assessment system, which measures attention, executive function, and working memory.
Emotional measures were captured from the Pediatric Quality of Life Inventory Sickle Cell Disease module, including questions about worrying and emotions such as anger regarding SCD and pain.
The mean age of participants was 10.61 years. Of the 112 children/adolescents in the study, 65 (58%) were female, and 83 (74%) had sickle cell anemia (either HbSS or HbSβ0 thalassemia).
The participants had a median number of ED visits for pain of one within a year of enrollment, and a median of three within 3 years of enrollment,
The median number of hospital admissions for pain was zero and one, respectively.
Attention, emotions linked to higher use
Factors significantly associated with ED visits for pain within the first year were higher (worse) scores for attention (P = .001) and self-reported emotion (P = .049). ED visits within 3 years of enrollment were associated with attention (P = .003) and working memory (P = .039).
Similarly, hospitalizations for pain within the first year were significantly associated with worse attention scores (P = .009) and child-reported emotion (P = .013). Hospitalizations for pain within 3 years of enrollment were also significantly associated with attention deficits (P = .006) and with worse emotional function as reported by a parent (P = .020).
There was no significant effect of SCD genotype or socioeconomic status on either pain-related ED visits or hospitalizations, however.
The investigators theorized that poor attention may make it difficult to distract children from focusing on their pain, and could also hamper disease self-management strategies such as medication adherence and avoiding pain triggers.
Age-related differences?
In the question-and-answer session following her presentation, comoderator Susanna A Curtis, MD, from Yale New Haven (Conn.) Hospital, commented that “some previous work has shown that adolescents and young adults with sickle cell disease have higher utilization as compared to their younger counterparts,” and asked whether the investigators found differences between cognition and utilization among different age groups within the cohort.
“We didn’t find a significant association with age, but I’m also very interested in that as well, especially considering that maybe there is more or less parent involvement, considering how old the child is,” Ms. Williams said.
Dr. Curtis noted that many of the comorbidities of sickle cell disease such as stroke or degree of anemia can affect cognitive function, but can also have an effect on health care utilization as well, asked whether the investigators were able to look at the potential confounding effects of comorbidities.
Ms. Williams said that, although they have not looked at potential confounders as yet, they hope to do so in future research.
Asked by another audience member whether the authors had considered using the Pain Catastrophizing Scale for children and/or their parents, in addition to other markers, Ms. Williams replied that “I definitely have considered it. Under recommendations from my mentors, we just focused on the quality-of-life scale first, but catastrophizing is something I’m very interested in. Especially, I would love to have the parent factors as well, so along the journey I hope to include that.”
The study was sponsored in part by a grant from the Doris Duke Charitable Foundation. Ms Williams is the recipient of an ASH Minority Medical Student Award. Dr. Curtis and Ms. Williams both reported no relevant conflicts of interest to disclose.
SOURCE: Williams Z et al. ASH 2020, Abstract 366
The cognitive and emotional status of children with sickle cell disease (SCD) appears to have a significant effect on how they cope with pain and use health care resources, investigators have found.
Results of a retrospective study of 112 children and adolescents with SCD, the majority of whom had sickle cell anemia, showed that ED visits and hospitalizations were significantly lower among children with SCD who performed better on an attention task, as well as those who were better able to cope emotionally with having SCD and pain, reported Zaria Williams, a second-year medical student at Howard University, Washington, and colleagues.
“Since I started learning more about sickle cell disease, I’ve been very concerned about the great disease burden that this condition can place on pediatric patients, particularly those who suffer from pain,” Ms. Williams said in an oral abstract presented at the annual meeting of the American Society of Hematology.
Although many children and adolescents with SCD can have their pain effectively managed at home with opioids and other medications, some require ED visits and potentially hospitalizations for pain management.
“There is great variability in health care utilization among patients with sickle cell disease, with some having to come to the ED and be admit to the hospital more than others. In searching for reasons why this might be the case, we thought about cognitive function and emotional differences between children with sickle cell disease as potentially affecting disease management,” she said.
Anxiety and catastrophizing
Children with SCD are known to be susceptible to affective comorbidities such as anxiety and catastrophizing, and to conditions that have the potential for deleterious effects on executive function, attention, and working memory. To determine whether cognitive and emotional factors affect the disease self-management in children and adolescents with SCD, Ms. Williams and coinvestigators looked at a cohort of 112 SCD patients aged 7-16 years treated at Children’s National Hospital in Washington, D.C.
The patients had participated in a previous pilot study of computerized working memory training. The authors reviewed charts for data on health care utilization, focusing on ED visits and hospitalization for pain 1 and 3 years after enrollment in the study.
They collected data on SCD genotype, disease-related variables, psychosocial information, and measures of cognition and emotion from the dataset. The information included socioeconomic status, parent education level, household income, and number of adults in the household.
Cognitive measures included the Weschler Intelligence Scale for Children full scale IQ, and the Cogstate computerized cognitive assessment system, which measures attention, executive function, and working memory.
Emotional measures were captured from the Pediatric Quality of Life Inventory Sickle Cell Disease module, including questions about worrying and emotions such as anger regarding SCD and pain.
The mean age of participants was 10.61 years. Of the 112 children/adolescents in the study, 65 (58%) were female, and 83 (74%) had sickle cell anemia (either HbSS or HbSβ0 thalassemia).
The participants had a median number of ED visits for pain of one within a year of enrollment, and a median of three within 3 years of enrollment,
The median number of hospital admissions for pain was zero and one, respectively.
Attention, emotions linked to higher use
Factors significantly associated with ED visits for pain within the first year were higher (worse) scores for attention (P = .001) and self-reported emotion (P = .049). ED visits within 3 years of enrollment were associated with attention (P = .003) and working memory (P = .039).
Similarly, hospitalizations for pain within the first year were significantly associated with worse attention scores (P = .009) and child-reported emotion (P = .013). Hospitalizations for pain within 3 years of enrollment were also significantly associated with attention deficits (P = .006) and with worse emotional function as reported by a parent (P = .020).
There was no significant effect of SCD genotype or socioeconomic status on either pain-related ED visits or hospitalizations, however.
The investigators theorized that poor attention may make it difficult to distract children from focusing on their pain, and could also hamper disease self-management strategies such as medication adherence and avoiding pain triggers.
Age-related differences?
In the question-and-answer session following her presentation, comoderator Susanna A Curtis, MD, from Yale New Haven (Conn.) Hospital, commented that “some previous work has shown that adolescents and young adults with sickle cell disease have higher utilization as compared to their younger counterparts,” and asked whether the investigators found differences between cognition and utilization among different age groups within the cohort.
“We didn’t find a significant association with age, but I’m also very interested in that as well, especially considering that maybe there is more or less parent involvement, considering how old the child is,” Ms. Williams said.
Dr. Curtis noted that many of the comorbidities of sickle cell disease such as stroke or degree of anemia can affect cognitive function, but can also have an effect on health care utilization as well, asked whether the investigators were able to look at the potential confounding effects of comorbidities.
Ms. Williams said that, although they have not looked at potential confounders as yet, they hope to do so in future research.
Asked by another audience member whether the authors had considered using the Pain Catastrophizing Scale for children and/or their parents, in addition to other markers, Ms. Williams replied that “I definitely have considered it. Under recommendations from my mentors, we just focused on the quality-of-life scale first, but catastrophizing is something I’m very interested in. Especially, I would love to have the parent factors as well, so along the journey I hope to include that.”
The study was sponsored in part by a grant from the Doris Duke Charitable Foundation. Ms Williams is the recipient of an ASH Minority Medical Student Award. Dr. Curtis and Ms. Williams both reported no relevant conflicts of interest to disclose.
SOURCE: Williams Z et al. ASH 2020, Abstract 366
The cognitive and emotional status of children with sickle cell disease (SCD) appears to have a significant effect on how they cope with pain and use health care resources, investigators have found.
Results of a retrospective study of 112 children and adolescents with SCD, the majority of whom had sickle cell anemia, showed that ED visits and hospitalizations were significantly lower among children with SCD who performed better on an attention task, as well as those who were better able to cope emotionally with having SCD and pain, reported Zaria Williams, a second-year medical student at Howard University, Washington, and colleagues.
“Since I started learning more about sickle cell disease, I’ve been very concerned about the great disease burden that this condition can place on pediatric patients, particularly those who suffer from pain,” Ms. Williams said in an oral abstract presented at the annual meeting of the American Society of Hematology.
Although many children and adolescents with SCD can have their pain effectively managed at home with opioids and other medications, some require ED visits and potentially hospitalizations for pain management.
“There is great variability in health care utilization among patients with sickle cell disease, with some having to come to the ED and be admit to the hospital more than others. In searching for reasons why this might be the case, we thought about cognitive function and emotional differences between children with sickle cell disease as potentially affecting disease management,” she said.
Anxiety and catastrophizing
Children with SCD are known to be susceptible to affective comorbidities such as anxiety and catastrophizing, and to conditions that have the potential for deleterious effects on executive function, attention, and working memory. To determine whether cognitive and emotional factors affect the disease self-management in children and adolescents with SCD, Ms. Williams and coinvestigators looked at a cohort of 112 SCD patients aged 7-16 years treated at Children’s National Hospital in Washington, D.C.
The patients had participated in a previous pilot study of computerized working memory training. The authors reviewed charts for data on health care utilization, focusing on ED visits and hospitalization for pain 1 and 3 years after enrollment in the study.
They collected data on SCD genotype, disease-related variables, psychosocial information, and measures of cognition and emotion from the dataset. The information included socioeconomic status, parent education level, household income, and number of adults in the household.
Cognitive measures included the Weschler Intelligence Scale for Children full scale IQ, and the Cogstate computerized cognitive assessment system, which measures attention, executive function, and working memory.
Emotional measures were captured from the Pediatric Quality of Life Inventory Sickle Cell Disease module, including questions about worrying and emotions such as anger regarding SCD and pain.
The mean age of participants was 10.61 years. Of the 112 children/adolescents in the study, 65 (58%) were female, and 83 (74%) had sickle cell anemia (either HbSS or HbSβ0 thalassemia).
The participants had a median number of ED visits for pain of one within a year of enrollment, and a median of three within 3 years of enrollment,
The median number of hospital admissions for pain was zero and one, respectively.
Attention, emotions linked to higher use
Factors significantly associated with ED visits for pain within the first year were higher (worse) scores for attention (P = .001) and self-reported emotion (P = .049). ED visits within 3 years of enrollment were associated with attention (P = .003) and working memory (P = .039).
Similarly, hospitalizations for pain within the first year were significantly associated with worse attention scores (P = .009) and child-reported emotion (P = .013). Hospitalizations for pain within 3 years of enrollment were also significantly associated with attention deficits (P = .006) and with worse emotional function as reported by a parent (P = .020).
There was no significant effect of SCD genotype or socioeconomic status on either pain-related ED visits or hospitalizations, however.
The investigators theorized that poor attention may make it difficult to distract children from focusing on their pain, and could also hamper disease self-management strategies such as medication adherence and avoiding pain triggers.
Age-related differences?
In the question-and-answer session following her presentation, comoderator Susanna A Curtis, MD, from Yale New Haven (Conn.) Hospital, commented that “some previous work has shown that adolescents and young adults with sickle cell disease have higher utilization as compared to their younger counterparts,” and asked whether the investigators found differences between cognition and utilization among different age groups within the cohort.
“We didn’t find a significant association with age, but I’m also very interested in that as well, especially considering that maybe there is more or less parent involvement, considering how old the child is,” Ms. Williams said.
Dr. Curtis noted that many of the comorbidities of sickle cell disease such as stroke or degree of anemia can affect cognitive function, but can also have an effect on health care utilization as well, asked whether the investigators were able to look at the potential confounding effects of comorbidities.
Ms. Williams said that, although they have not looked at potential confounders as yet, they hope to do so in future research.
Asked by another audience member whether the authors had considered using the Pain Catastrophizing Scale for children and/or their parents, in addition to other markers, Ms. Williams replied that “I definitely have considered it. Under recommendations from my mentors, we just focused on the quality-of-life scale first, but catastrophizing is something I’m very interested in. Especially, I would love to have the parent factors as well, so along the journey I hope to include that.”
The study was sponsored in part by a grant from the Doris Duke Charitable Foundation. Ms Williams is the recipient of an ASH Minority Medical Student Award. Dr. Curtis and Ms. Williams both reported no relevant conflicts of interest to disclose.
SOURCE: Williams Z et al. ASH 2020, Abstract 366
FROM ASH 2020
Landmark sickle cell report targets massive failures, calls for action
The National Academies of Science, Engineering, and Medicine have just released a 522-page report, but it’s not the usual compilation of guidelines for treatment of a disease. Instead, the authors of “Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action” argue in stark terms that the American society has colossally failed individuals living with sickle cell disease (SCD), who are mostly Black or Brown. A dramatic overhaul of the country’s medical and societal priorities is needed to turn things around to improve health and longevity among this rare disease population.
The findings from the NASEM report are explicit: “There has been substantial success in increasing the survival of children with SCD, but this success had not been translated to similar success as they become adults.” One factor posited to contribute to the slow progress in the improvement of quality and quantity of life for adults living with this disease is the fact that “SCD is largely a disease of African Americans, and as such exists in a context of racial discrimination, health and other societal disparities, mistrust of the health care system, and the effects of poverty.” The report also cites the substantial evidence that those with SCD may receive poorer quality of care.
The report’s 14 authors were made up of an ad hoc committee formed at the request of the Department of Health & Human Services’ Office of Minority Health. The office asked NASEM to convene the committee to develop a strategic plan and blueprint for the United States and others regarding SCD.
The NASEM SCD committee members “realized that we can’t address the medical components of SCD if we don’t explore societal issues and why it’s been so hard to get good care for people with sickle cell disease,” hematologist and report coauthor Ifeyinwa (Ify) Osunkwo, MD, professor of medicine and pediatrics at Atrium Health and director of the Sickle Cell Disease Enterprise, Levine Cancer Institute, Charlotte, N.C., said in an interview. Dr. Osunkwo is also the medical editor of Hematology News.
“After almost a year of meetings and digging into the background and history of SCD care, we came out with very comprehensive summary of where we were and where we want to be,” she said. “The report provides short-, intermediate- and long-term recommendations and identifies which entity and organization should be responsible for implementing them.”
The report authors, led by pediatrician and committee chair Marie Clare McCormick, MD, of the Harvard School of Public Health, Boston, stated that about 100,000 people in the United States and millions worldwide live with SCD. The disease kills more than 700 people per year in the United States, and treatment costs an estimated $2 billion a year.
When judged by disability-adjusted life-years lost – a measurement of expected healthy years of life without an illness – the impact of SCD on individuals is estimated to be greater than a long list of other diseases such as Alzheimer’s disease, breast cancer, type 1 diabetes, and AIDS/HIV, the report noted.
“The health care needs of individuals living with SCD have been neglected by the U.S. and global health care systems, causing them and their families to suffer,” the report said. “Many of the complications that afflict individuals with SCD, particularly pain, are invisible. Pain is only diagnosed by self-reports, and in SCD there are few to no external indicators of the pain experience. Nevertheless, the pain can be excruciatingly severe and requires treatment with strong analgesics.”
There’s even more misery to the story of SCD, the report said, and Dr. Osunkwo agreed. “It’s not just about pain. These individuals suffer from multiple organ-system complications that are physical but also psychological and societal. They experience a lot of disparities in every aspect of their lives. You’re sick, so then you can’t get a job or health insurance, you can’t get Social Security benefits. You can’t get the type of health care you need nor can you access the other forms of support you need and often you are judged as a drug seeker for complaining of pain or repeatedly seeking acute care for unresolved pain.”
Multiple factors exacerbate the experience of people living with SCD in America, the report said. “Because of systemic racism, unconscious bias, and the stigma associated with the diagnosis, the disease brings with it a much broader burden.”

Dr. Osunkwo put it this way: “SCD is a disease that mostly affects Brown and Black people, and that gets layered into the whole discrimination issues that Black and Brown people face compounding the health burden from their disease.”
The report added that “the SCD community has developed a significant lack of trust in the health care system due to the nearly universal stigma and lack of belief in their reports of pain, a lack of trust that has been further reinforced by historical events, such as the Tuskegee experiment.”
The report highlighted research that finds that Blacks “are more likely to receive a lower quality of pain management than white patients and may be perceived as having drug-seeking behavior.”
The report also identified gaps in treatment, noting that “many SCD complications are not restricted to any one organ system, and the impact of the disease on [quality of life] can be profound but hard to define and compartmentalize.”
Dr. Osunkwo said medical professionals often fail to understand the full breadth of the disease. “There’s no particular look to SCD. When you have cancer, you come in, and you look like you’re sick because you’re bald. Everyone clues into that cancer look and knows it’s lethal, that you’re may likely die early. We don’t have that “look that generates empathy” for SCD, and people don’t understand the burden on those affected. They don’t understand or appreciate that SCD shortens your lifespan as well ... that people living with SCD die 3 decades earlier than their ethnically matched peers. Also, SCD is associated with a lot of pain, and pain and the treatment of pain with opioids makes people [health care providers] uncomfortable unless it’s cancer pain.”
She added: “People also assume that, if it’s not pain, it’s not SCD even though SCD can cause leg ulcers and blood clots and even affect the tonsils, or lead to a stroke. When a disease complexity is too difficult for providers to understand, they either avoid it or don’t do anything for the patient.”
Screening and surveillance for SCD and sickle cell trait is insufficient, the report said, and the potential cost of missed childhood cases is large. Detecting the condition at birth allows the implementation of appropriate comprehensive care and treatment to prevent early death from infections and strokes. As the authors noted, “tremendous strides have been made in the past few decades in the care of children with SCD, which have led to almost all children in high-income settings surviving to adulthood.” However, there remains gaps in care coordination and follow-up of babies screened at birth and even bigger gaps in translating these life span gains to adults particularly around the period of transition from pediatrics to adult care when there appears to be a spike in morbidity and mortality.
The report summarized current treatments for SCD and noted “an influx of pipeline products” after years of little progress and identifies “a need for targeted SCD therapies that address the underlying cause of the disease.”
While treatment recommendations exist, Dr. Osunkwo said, “the evidence for them is very poor and many SCD complications have no evidence-based guidelines for providers to follow. We need more research to provide high quality evidence to make guidelines for SCD treatment stronger and more robust.”
In its final section, the report offers a “strategic plan and blueprint for sickle cell disease action.” It offers several strategies to achieve the vision of “long healthy productive lives for those living with sickle cell disease and sickle cell trait”:
- Establish and fund a research agenda to inform effective programs and policies across the life span.
- Implement efforts to advance understanding of the full impact of sickle cell trait on individuals and society.
- Address barriers to accessing current and pipeline therapies for SCD.
- Improve SCD awareness and strengthen advocacy efforts.
- Increase the number of qualified health professionals providing SCD care.
- Strengthen the evidence base for interventions and disease management and implement widespread efforts to monitor the quality of SCD care.
- Establish organized systems of care assuring both clinical and nonclinical supportive services to all persons living with SCD.
- Establish a national system to collect and link data to characterize the burden of disease, outcomes, and the needs of those with SCD across the life span.
“Right now, the average lifespan for SCD is in the mid-40s to mid-50s,” Dr. Osunkwo said. “That’s a horrible statistic. Even if we just take up half of these recommendations, people will live longer with SCD, and they’ll be more productive and contribute more to society. If we value a cancer life the same as a sickle cell life, we’ll be halfway across the finish line. But the stigma of SCD being a Black and Brown problem is going to be the hardest to confront as it requires a systemic change in our culture as a country and a health care system.”
Still, she said, the commissioning of the report “shows that there is a desire to understand the issue in better detail and try to mitigate it.”
Dr. Osunkwo and Dr. McCormick had no relevant disclosures.
SOURCE: National Academies of Sciences, Engineering, and Medicine. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. Washington, D.C.: National Academies Press, 2020.
The National Academies of Science, Engineering, and Medicine have just released a 522-page report, but it’s not the usual compilation of guidelines for treatment of a disease. Instead, the authors of “Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action” argue in stark terms that the American society has colossally failed individuals living with sickle cell disease (SCD), who are mostly Black or Brown. A dramatic overhaul of the country’s medical and societal priorities is needed to turn things around to improve health and longevity among this rare disease population.
The findings from the NASEM report are explicit: “There has been substantial success in increasing the survival of children with SCD, but this success had not been translated to similar success as they become adults.” One factor posited to contribute to the slow progress in the improvement of quality and quantity of life for adults living with this disease is the fact that “SCD is largely a disease of African Americans, and as such exists in a context of racial discrimination, health and other societal disparities, mistrust of the health care system, and the effects of poverty.” The report also cites the substantial evidence that those with SCD may receive poorer quality of care.
The report’s 14 authors were made up of an ad hoc committee formed at the request of the Department of Health & Human Services’ Office of Minority Health. The office asked NASEM to convene the committee to develop a strategic plan and blueprint for the United States and others regarding SCD.
The NASEM SCD committee members “realized that we can’t address the medical components of SCD if we don’t explore societal issues and why it’s been so hard to get good care for people with sickle cell disease,” hematologist and report coauthor Ifeyinwa (Ify) Osunkwo, MD, professor of medicine and pediatrics at Atrium Health and director of the Sickle Cell Disease Enterprise, Levine Cancer Institute, Charlotte, N.C., said in an interview. Dr. Osunkwo is also the medical editor of Hematology News.
“After almost a year of meetings and digging into the background and history of SCD care, we came out with very comprehensive summary of where we were and where we want to be,” she said. “The report provides short-, intermediate- and long-term recommendations and identifies which entity and organization should be responsible for implementing them.”
The report authors, led by pediatrician and committee chair Marie Clare McCormick, MD, of the Harvard School of Public Health, Boston, stated that about 100,000 people in the United States and millions worldwide live with SCD. The disease kills more than 700 people per year in the United States, and treatment costs an estimated $2 billion a year.
When judged by disability-adjusted life-years lost – a measurement of expected healthy years of life without an illness – the impact of SCD on individuals is estimated to be greater than a long list of other diseases such as Alzheimer’s disease, breast cancer, type 1 diabetes, and AIDS/HIV, the report noted.
“The health care needs of individuals living with SCD have been neglected by the U.S. and global health care systems, causing them and their families to suffer,” the report said. “Many of the complications that afflict individuals with SCD, particularly pain, are invisible. Pain is only diagnosed by self-reports, and in SCD there are few to no external indicators of the pain experience. Nevertheless, the pain can be excruciatingly severe and requires treatment with strong analgesics.”
There’s even more misery to the story of SCD, the report said, and Dr. Osunkwo agreed. “It’s not just about pain. These individuals suffer from multiple organ-system complications that are physical but also psychological and societal. They experience a lot of disparities in every aspect of their lives. You’re sick, so then you can’t get a job or health insurance, you can’t get Social Security benefits. You can’t get the type of health care you need nor can you access the other forms of support you need and often you are judged as a drug seeker for complaining of pain or repeatedly seeking acute care for unresolved pain.”
Multiple factors exacerbate the experience of people living with SCD in America, the report said. “Because of systemic racism, unconscious bias, and the stigma associated with the diagnosis, the disease brings with it a much broader burden.”
Dr. Osunkwo put it this way: “SCD is a disease that mostly affects Brown and Black people, and that gets layered into the whole discrimination issues that Black and Brown people face compounding the health burden from their disease.”
The report added that “the SCD community has developed a significant lack of trust in the health care system due to the nearly universal stigma and lack of belief in their reports of pain, a lack of trust that has been further reinforced by historical events, such as the Tuskegee experiment.”
The report highlighted research that finds that Blacks “are more likely to receive a lower quality of pain management than white patients and may be perceived as having drug-seeking behavior.”
The report also identified gaps in treatment, noting that “many SCD complications are not restricted to any one organ system, and the impact of the disease on [quality of life] can be profound but hard to define and compartmentalize.”
Dr. Osunkwo said medical professionals often fail to understand the full breadth of the disease. “There’s no particular look to SCD. When you have cancer, you come in, and you look like you’re sick because you’re bald. Everyone clues into that cancer look and knows it’s lethal, that you’re may likely die early. We don’t have that “look that generates empathy” for SCD, and people don’t understand the burden on those affected. They don’t understand or appreciate that SCD shortens your lifespan as well ... that people living with SCD die 3 decades earlier than their ethnically matched peers. Also, SCD is associated with a lot of pain, and pain and the treatment of pain with opioids makes people [health care providers] uncomfortable unless it’s cancer pain.”
She added: “People also assume that, if it’s not pain, it’s not SCD even though SCD can cause leg ulcers and blood clots and even affect the tonsils, or lead to a stroke. When a disease complexity is too difficult for providers to understand, they either avoid it or don’t do anything for the patient.”
Screening and surveillance for SCD and sickle cell trait is insufficient, the report said, and the potential cost of missed childhood cases is large. Detecting the condition at birth allows the implementation of appropriate comprehensive care and treatment to prevent early death from infections and strokes. As the authors noted, “tremendous strides have been made in the past few decades in the care of children with SCD, which have led to almost all children in high-income settings surviving to adulthood.” However, there remains gaps in care coordination and follow-up of babies screened at birth and even bigger gaps in translating these life span gains to adults particularly around the period of transition from pediatrics to adult care when there appears to be a spike in morbidity and mortality.
The report summarized current treatments for SCD and noted “an influx of pipeline products” after years of little progress and identifies “a need for targeted SCD therapies that address the underlying cause of the disease.”
While treatment recommendations exist, Dr. Osunkwo said, “the evidence for them is very poor and many SCD complications have no evidence-based guidelines for providers to follow. We need more research to provide high quality evidence to make guidelines for SCD treatment stronger and more robust.”
In its final section, the report offers a “strategic plan and blueprint for sickle cell disease action.” It offers several strategies to achieve the vision of “long healthy productive lives for those living with sickle cell disease and sickle cell trait”:
- Establish and fund a research agenda to inform effective programs and policies across the life span.
- Implement efforts to advance understanding of the full impact of sickle cell trait on individuals and society.
- Address barriers to accessing current and pipeline therapies for SCD.
- Improve SCD awareness and strengthen advocacy efforts.
- Increase the number of qualified health professionals providing SCD care.
- Strengthen the evidence base for interventions and disease management and implement widespread efforts to monitor the quality of SCD care.
- Establish organized systems of care assuring both clinical and nonclinical supportive services to all persons living with SCD.
- Establish a national system to collect and link data to characterize the burden of disease, outcomes, and the needs of those with SCD across the life span.
“Right now, the average lifespan for SCD is in the mid-40s to mid-50s,” Dr. Osunkwo said. “That’s a horrible statistic. Even if we just take up half of these recommendations, people will live longer with SCD, and they’ll be more productive and contribute more to society. If we value a cancer life the same as a sickle cell life, we’ll be halfway across the finish line. But the stigma of SCD being a Black and Brown problem is going to be the hardest to confront as it requires a systemic change in our culture as a country and a health care system.”
Still, she said, the commissioning of the report “shows that there is a desire to understand the issue in better detail and try to mitigate it.”
Dr. Osunkwo and Dr. McCormick had no relevant disclosures.
SOURCE: National Academies of Sciences, Engineering, and Medicine. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. Washington, D.C.: National Academies Press, 2020.
The National Academies of Science, Engineering, and Medicine have just released a 522-page report, but it’s not the usual compilation of guidelines for treatment of a disease. Instead, the authors of “Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action” argue in stark terms that the American society has colossally failed individuals living with sickle cell disease (SCD), who are mostly Black or Brown. A dramatic overhaul of the country’s medical and societal priorities is needed to turn things around to improve health and longevity among this rare disease population.
The findings from the NASEM report are explicit: “There has been substantial success in increasing the survival of children with SCD, but this success had not been translated to similar success as they become adults.” One factor posited to contribute to the slow progress in the improvement of quality and quantity of life for adults living with this disease is the fact that “SCD is largely a disease of African Americans, and as such exists in a context of racial discrimination, health and other societal disparities, mistrust of the health care system, and the effects of poverty.” The report also cites the substantial evidence that those with SCD may receive poorer quality of care.
The report’s 14 authors were made up of an ad hoc committee formed at the request of the Department of Health & Human Services’ Office of Minority Health. The office asked NASEM to convene the committee to develop a strategic plan and blueprint for the United States and others regarding SCD.
The NASEM SCD committee members “realized that we can’t address the medical components of SCD if we don’t explore societal issues and why it’s been so hard to get good care for people with sickle cell disease,” hematologist and report coauthor Ifeyinwa (Ify) Osunkwo, MD, professor of medicine and pediatrics at Atrium Health and director of the Sickle Cell Disease Enterprise, Levine Cancer Institute, Charlotte, N.C., said in an interview. Dr. Osunkwo is also the medical editor of Hematology News.
“After almost a year of meetings and digging into the background and history of SCD care, we came out with very comprehensive summary of where we were and where we want to be,” she said. “The report provides short-, intermediate- and long-term recommendations and identifies which entity and organization should be responsible for implementing them.”
The report authors, led by pediatrician and committee chair Marie Clare McCormick, MD, of the Harvard School of Public Health, Boston, stated that about 100,000 people in the United States and millions worldwide live with SCD. The disease kills more than 700 people per year in the United States, and treatment costs an estimated $2 billion a year.
When judged by disability-adjusted life-years lost – a measurement of expected healthy years of life without an illness – the impact of SCD on individuals is estimated to be greater than a long list of other diseases such as Alzheimer’s disease, breast cancer, type 1 diabetes, and AIDS/HIV, the report noted.
“The health care needs of individuals living with SCD have been neglected by the U.S. and global health care systems, causing them and their families to suffer,” the report said. “Many of the complications that afflict individuals with SCD, particularly pain, are invisible. Pain is only diagnosed by self-reports, and in SCD there are few to no external indicators of the pain experience. Nevertheless, the pain can be excruciatingly severe and requires treatment with strong analgesics.”
There’s even more misery to the story of SCD, the report said, and Dr. Osunkwo agreed. “It’s not just about pain. These individuals suffer from multiple organ-system complications that are physical but also psychological and societal. They experience a lot of disparities in every aspect of their lives. You’re sick, so then you can’t get a job or health insurance, you can’t get Social Security benefits. You can’t get the type of health care you need nor can you access the other forms of support you need and often you are judged as a drug seeker for complaining of pain or repeatedly seeking acute care for unresolved pain.”
Multiple factors exacerbate the experience of people living with SCD in America, the report said. “Because of systemic racism, unconscious bias, and the stigma associated with the diagnosis, the disease brings with it a much broader burden.”
Dr. Osunkwo put it this way: “SCD is a disease that mostly affects Brown and Black people, and that gets layered into the whole discrimination issues that Black and Brown people face compounding the health burden from their disease.”
The report added that “the SCD community has developed a significant lack of trust in the health care system due to the nearly universal stigma and lack of belief in their reports of pain, a lack of trust that has been further reinforced by historical events, such as the Tuskegee experiment.”
The report highlighted research that finds that Blacks “are more likely to receive a lower quality of pain management than white patients and may be perceived as having drug-seeking behavior.”
The report also identified gaps in treatment, noting that “many SCD complications are not restricted to any one organ system, and the impact of the disease on [quality of life] can be profound but hard to define and compartmentalize.”
Dr. Osunkwo said medical professionals often fail to understand the full breadth of the disease. “There’s no particular look to SCD. When you have cancer, you come in, and you look like you’re sick because you’re bald. Everyone clues into that cancer look and knows it’s lethal, that you’re may likely die early. We don’t have that “look that generates empathy” for SCD, and people don’t understand the burden on those affected. They don’t understand or appreciate that SCD shortens your lifespan as well ... that people living with SCD die 3 decades earlier than their ethnically matched peers. Also, SCD is associated with a lot of pain, and pain and the treatment of pain with opioids makes people [health care providers] uncomfortable unless it’s cancer pain.”
She added: “People also assume that, if it’s not pain, it’s not SCD even though SCD can cause leg ulcers and blood clots and even affect the tonsils, or lead to a stroke. When a disease complexity is too difficult for providers to understand, they either avoid it or don’t do anything for the patient.”
Screening and surveillance for SCD and sickle cell trait is insufficient, the report said, and the potential cost of missed childhood cases is large. Detecting the condition at birth allows the implementation of appropriate comprehensive care and treatment to prevent early death from infections and strokes. As the authors noted, “tremendous strides have been made in the past few decades in the care of children with SCD, which have led to almost all children in high-income settings surviving to adulthood.” However, there remains gaps in care coordination and follow-up of babies screened at birth and even bigger gaps in translating these life span gains to adults particularly around the period of transition from pediatrics to adult care when there appears to be a spike in morbidity and mortality.
The report summarized current treatments for SCD and noted “an influx of pipeline products” after years of little progress and identifies “a need for targeted SCD therapies that address the underlying cause of the disease.”
While treatment recommendations exist, Dr. Osunkwo said, “the evidence for them is very poor and many SCD complications have no evidence-based guidelines for providers to follow. We need more research to provide high quality evidence to make guidelines for SCD treatment stronger and more robust.”
In its final section, the report offers a “strategic plan and blueprint for sickle cell disease action.” It offers several strategies to achieve the vision of “long healthy productive lives for those living with sickle cell disease and sickle cell trait”:
- Establish and fund a research agenda to inform effective programs and policies across the life span.
- Implement efforts to advance understanding of the full impact of sickle cell trait on individuals and society.
- Address barriers to accessing current and pipeline therapies for SCD.
- Improve SCD awareness and strengthen advocacy efforts.
- Increase the number of qualified health professionals providing SCD care.
- Strengthen the evidence base for interventions and disease management and implement widespread efforts to monitor the quality of SCD care.
- Establish organized systems of care assuring both clinical and nonclinical supportive services to all persons living with SCD.
- Establish a national system to collect and link data to characterize the burden of disease, outcomes, and the needs of those with SCD across the life span.
“Right now, the average lifespan for SCD is in the mid-40s to mid-50s,” Dr. Osunkwo said. “That’s a horrible statistic. Even if we just take up half of these recommendations, people will live longer with SCD, and they’ll be more productive and contribute more to society. If we value a cancer life the same as a sickle cell life, we’ll be halfway across the finish line. But the stigma of SCD being a Black and Brown problem is going to be the hardest to confront as it requires a systemic change in our culture as a country and a health care system.”
Still, she said, the commissioning of the report “shows that there is a desire to understand the issue in better detail and try to mitigate it.”
Dr. Osunkwo and Dr. McCormick had no relevant disclosures.
SOURCE: National Academies of Sciences, Engineering, and Medicine. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. Washington, D.C.: National Academies Press, 2020.
Survey quantifies COVID-19’s impact on oncology
An international survey provides new insights into how COVID-19 has affected, and may continue to affect, the field of oncology.
The survey showed that “COVID-19 has had a major impact on the organization of patient care, on the well-being of caregivers, on continued medical education, and on clinical trial activities in oncology,” stated Guy Jerusalem, MD, PhD, of Centre Hospitalier Universitaire de Liège (Belgium).
Dr. Jerusalem presented these findings at the European Society for Medical Oncology Virtual Congress 2020.
The survey was distributed by 20 oncologists from 10 of the countries most affected by COVID-19. Responses were obtained from 109 oncologists representing centers in 18 countries. The responses were recorded between June 17 and July 14, 2020.
The survey consisted of 95 items intended to evaluate the impact of COVID-19 on the organization of oncologic care. Questions encompassed the capacity and service offered at each center, the magnitude of COVID-19–based care interruptions and the reasons for them, the ensuing challenges faced, interventions implemented, and the estimated harms to patients during the pandemic.
The 109 oncologists surveyed had a median of 20 years of oncology experience. A majority of respondents were men (61.5%), and the median age was 48.5 years.
The respondents had worked predominantly (62.4%) at academic hospitals, with 29.6% at community hospitals. Most respondents worked at general hospitals with an oncology unit (66.1%) rather than a specialized separate cancer center (32.1%).
The most common specialty was breast cancer (60.6%), followed by gastrointestinal cancer (10.1%), urogenital cancer (9.2%), and lung cancer (8.3%).
Impact on treatment
The treatment modalities affected by the pandemic – through cancellations or delays in more than 10% of patients – included surgery (in 34% of centers), chemotherapy (22%), radiotherapy (13.7%), checkpoint inhibitor therapy (9.1%), monoclonal antibodies (9%), and oral targeted therapy (3.7%).
Among oncologists treating breast cancer, cancellations/delays in more than 10% of patients were reported for everolimus (18%), CDK4/6 inhibitors (8.9%), and endocrine therapy (2.2%).
Overall, 34.8% of respondents reported increased use of granulocyte colony–stimulating factor, and 6.4% reported increased use of erythropoietin.
On the other hand, 11.1% of respondents reported a decrease in the use of double immunotherapy, and 21.9% reported decreased use of corticosteroids.
Not only can the immunosuppressive effects of steroid use increase infection risks, Dr. Jerusalem noted, fever suppression can lead to a delayed diagnosis of COVID-19.
“To circumvent potential higher infection risks or greater disease severity, we use lower doses of steroids, but this is not based on studies,” he said.
“Previous exposure to steroids or being on steroids at the time of COVID-19 infection is a detrimental factor for complications and mortality,” commented ESMO President Solange Peters, MD, PhD, of Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland.

Dr. Peters noted that the observation was based on lung cancer registry findings. Furthermore, because data from smaller outbreaks of other coronavirus infections suggested worse prognosis and increased mortality, steroid use was already feared in the very early days of the COVID-19 pandemic.
Lastly, earlier cessation of palliative treatment was observed in 32.1% of centers, and 64.2% of respondents agreed that undertreatment because of COVID-19 is a major concern.
Dr. Jerusalem noted that the survey data do not explain the early cessation of palliative treatment. “I suspect that many patients died at home rather than alone in institutions because it was the only way they could die with their families around them.”
Telehealth, meetings, and trials
The survey also revealed rationales for the use of teleconsultation, including follow-up (94.5%), oral therapy (92.7%), immunotherapy (57.8%), and chemotherapy (55%).
Most respondents reported more frequent use of virtual meetings for continuing medical education (94%), oncologic team meetings (92%), and tumor boards (82%).
While about 82% of respondents said they were likely to continue the use of telemedicine, 45% said virtual conferences are not an acceptable alternative to live international conferences such as ESMO, Dr. Jerusalem said.
Finally, nearly three-quarters of respondents (72.5%) said all clinical trial activities are or will soon be activated, or never stopped, at their centers. On the other hand, 27.5% of respondents reported that their centers had major protocol violations or deviations, and 37% of respondents said they expect significant reductions in clinical trial activities this year.
Dr. Jerusalem concluded that COVID-19 is having a major, long-term impact on the organization of patient care, caregivers, continued medical education, and clinical trial activities in oncology.
He cautioned that “the risk of a delayed diagnosis of new cancers and economic consequences of COVID-19 on access to health care and cancer treatments have to be carefully evaluated.”
This research was funded by Fondation Léon Fredericq. Dr. Jerusalem disclosed relationships with Novartis, Roche, Lilly, Pfizer, Amgen, Bristol-Myers Squibb, AstraZeneca, Daiichi Sankyo, AbbVie, MedImmune, and Merck. Dr. Peters disclosed relationships with AbbVie, Amgen, AstraZeneca, and many other companies.
SOURCE: Jerusalem G et al. ESMO 2020, Abstract LBA76.
An international survey provides new insights into how COVID-19 has affected, and may continue to affect, the field of oncology.
The survey showed that “COVID-19 has had a major impact on the organization of patient care, on the well-being of caregivers, on continued medical education, and on clinical trial activities in oncology,” stated Guy Jerusalem, MD, PhD, of Centre Hospitalier Universitaire de Liège (Belgium).
Dr. Jerusalem presented these findings at the European Society for Medical Oncology Virtual Congress 2020.
The survey was distributed by 20 oncologists from 10 of the countries most affected by COVID-19. Responses were obtained from 109 oncologists representing centers in 18 countries. The responses were recorded between June 17 and July 14, 2020.
The survey consisted of 95 items intended to evaluate the impact of COVID-19 on the organization of oncologic care. Questions encompassed the capacity and service offered at each center, the magnitude of COVID-19–based care interruptions and the reasons for them, the ensuing challenges faced, interventions implemented, and the estimated harms to patients during the pandemic.
The 109 oncologists surveyed had a median of 20 years of oncology experience. A majority of respondents were men (61.5%), and the median age was 48.5 years.
The respondents had worked predominantly (62.4%) at academic hospitals, with 29.6% at community hospitals. Most respondents worked at general hospitals with an oncology unit (66.1%) rather than a specialized separate cancer center (32.1%).
The most common specialty was breast cancer (60.6%), followed by gastrointestinal cancer (10.1%), urogenital cancer (9.2%), and lung cancer (8.3%).
Impact on treatment
The treatment modalities affected by the pandemic – through cancellations or delays in more than 10% of patients – included surgery (in 34% of centers), chemotherapy (22%), radiotherapy (13.7%), checkpoint inhibitor therapy (9.1%), monoclonal antibodies (9%), and oral targeted therapy (3.7%).
Among oncologists treating breast cancer, cancellations/delays in more than 10% of patients were reported for everolimus (18%), CDK4/6 inhibitors (8.9%), and endocrine therapy (2.2%).
Overall, 34.8% of respondents reported increased use of granulocyte colony–stimulating factor, and 6.4% reported increased use of erythropoietin.
On the other hand, 11.1% of respondents reported a decrease in the use of double immunotherapy, and 21.9% reported decreased use of corticosteroids.
Not only can the immunosuppressive effects of steroid use increase infection risks, Dr. Jerusalem noted, fever suppression can lead to a delayed diagnosis of COVID-19.
“To circumvent potential higher infection risks or greater disease severity, we use lower doses of steroids, but this is not based on studies,” he said.
“Previous exposure to steroids or being on steroids at the time of COVID-19 infection is a detrimental factor for complications and mortality,” commented ESMO President Solange Peters, MD, PhD, of Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland.
Dr. Peters noted that the observation was based on lung cancer registry findings. Furthermore, because data from smaller outbreaks of other coronavirus infections suggested worse prognosis and increased mortality, steroid use was already feared in the very early days of the COVID-19 pandemic.
Lastly, earlier cessation of palliative treatment was observed in 32.1% of centers, and 64.2% of respondents agreed that undertreatment because of COVID-19 is a major concern.
Dr. Jerusalem noted that the survey data do not explain the early cessation of palliative treatment. “I suspect that many patients died at home rather than alone in institutions because it was the only way they could die with their families around them.”
Telehealth, meetings, and trials
The survey also revealed rationales for the use of teleconsultation, including follow-up (94.5%), oral therapy (92.7%), immunotherapy (57.8%), and chemotherapy (55%).
Most respondents reported more frequent use of virtual meetings for continuing medical education (94%), oncologic team meetings (92%), and tumor boards (82%).
While about 82% of respondents said they were likely to continue the use of telemedicine, 45% said virtual conferences are not an acceptable alternative to live international conferences such as ESMO, Dr. Jerusalem said.
Finally, nearly three-quarters of respondents (72.5%) said all clinical trial activities are or will soon be activated, or never stopped, at their centers. On the other hand, 27.5% of respondents reported that their centers had major protocol violations or deviations, and 37% of respondents said they expect significant reductions in clinical trial activities this year.
Dr. Jerusalem concluded that COVID-19 is having a major, long-term impact on the organization of patient care, caregivers, continued medical education, and clinical trial activities in oncology.
He cautioned that “the risk of a delayed diagnosis of new cancers and economic consequences of COVID-19 on access to health care and cancer treatments have to be carefully evaluated.”
This research was funded by Fondation Léon Fredericq. Dr. Jerusalem disclosed relationships with Novartis, Roche, Lilly, Pfizer, Amgen, Bristol-Myers Squibb, AstraZeneca, Daiichi Sankyo, AbbVie, MedImmune, and Merck. Dr. Peters disclosed relationships with AbbVie, Amgen, AstraZeneca, and many other companies.
SOURCE: Jerusalem G et al. ESMO 2020, Abstract LBA76.
An international survey provides new insights into how COVID-19 has affected, and may continue to affect, the field of oncology.
The survey showed that “COVID-19 has had a major impact on the organization of patient care, on the well-being of caregivers, on continued medical education, and on clinical trial activities in oncology,” stated Guy Jerusalem, MD, PhD, of Centre Hospitalier Universitaire de Liège (Belgium).
Dr. Jerusalem presented these findings at the European Society for Medical Oncology Virtual Congress 2020.
The survey was distributed by 20 oncologists from 10 of the countries most affected by COVID-19. Responses were obtained from 109 oncologists representing centers in 18 countries. The responses were recorded between June 17 and July 14, 2020.
The survey consisted of 95 items intended to evaluate the impact of COVID-19 on the organization of oncologic care. Questions encompassed the capacity and service offered at each center, the magnitude of COVID-19–based care interruptions and the reasons for them, the ensuing challenges faced, interventions implemented, and the estimated harms to patients during the pandemic.
The 109 oncologists surveyed had a median of 20 years of oncology experience. A majority of respondents were men (61.5%), and the median age was 48.5 years.
The respondents had worked predominantly (62.4%) at academic hospitals, with 29.6% at community hospitals. Most respondents worked at general hospitals with an oncology unit (66.1%) rather than a specialized separate cancer center (32.1%).
The most common specialty was breast cancer (60.6%), followed by gastrointestinal cancer (10.1%), urogenital cancer (9.2%), and lung cancer (8.3%).
Impact on treatment
The treatment modalities affected by the pandemic – through cancellations or delays in more than 10% of patients – included surgery (in 34% of centers), chemotherapy (22%), radiotherapy (13.7%), checkpoint inhibitor therapy (9.1%), monoclonal antibodies (9%), and oral targeted therapy (3.7%).
Among oncologists treating breast cancer, cancellations/delays in more than 10% of patients were reported for everolimus (18%), CDK4/6 inhibitors (8.9%), and endocrine therapy (2.2%).
Overall, 34.8% of respondents reported increased use of granulocyte colony–stimulating factor, and 6.4% reported increased use of erythropoietin.
On the other hand, 11.1% of respondents reported a decrease in the use of double immunotherapy, and 21.9% reported decreased use of corticosteroids.
Not only can the immunosuppressive effects of steroid use increase infection risks, Dr. Jerusalem noted, fever suppression can lead to a delayed diagnosis of COVID-19.
“To circumvent potential higher infection risks or greater disease severity, we use lower doses of steroids, but this is not based on studies,” he said.
“Previous exposure to steroids or being on steroids at the time of COVID-19 infection is a detrimental factor for complications and mortality,” commented ESMO President Solange Peters, MD, PhD, of Centre Hospitalier Universitaire Vaudois in Lausanne, Switzerland.
Dr. Peters noted that the observation was based on lung cancer registry findings. Furthermore, because data from smaller outbreaks of other coronavirus infections suggested worse prognosis and increased mortality, steroid use was already feared in the very early days of the COVID-19 pandemic.
Lastly, earlier cessation of palliative treatment was observed in 32.1% of centers, and 64.2% of respondents agreed that undertreatment because of COVID-19 is a major concern.
Dr. Jerusalem noted that the survey data do not explain the early cessation of palliative treatment. “I suspect that many patients died at home rather than alone in institutions because it was the only way they could die with their families around them.”
Telehealth, meetings, and trials
The survey also revealed rationales for the use of teleconsultation, including follow-up (94.5%), oral therapy (92.7%), immunotherapy (57.8%), and chemotherapy (55%).
Most respondents reported more frequent use of virtual meetings for continuing medical education (94%), oncologic team meetings (92%), and tumor boards (82%).
While about 82% of respondents said they were likely to continue the use of telemedicine, 45% said virtual conferences are not an acceptable alternative to live international conferences such as ESMO, Dr. Jerusalem said.
Finally, nearly three-quarters of respondents (72.5%) said all clinical trial activities are or will soon be activated, or never stopped, at their centers. On the other hand, 27.5% of respondents reported that their centers had major protocol violations or deviations, and 37% of respondents said they expect significant reductions in clinical trial activities this year.
Dr. Jerusalem concluded that COVID-19 is having a major, long-term impact on the organization of patient care, caregivers, continued medical education, and clinical trial activities in oncology.
He cautioned that “the risk of a delayed diagnosis of new cancers and economic consequences of COVID-19 on access to health care and cancer treatments have to be carefully evaluated.”
This research was funded by Fondation Léon Fredericq. Dr. Jerusalem disclosed relationships with Novartis, Roche, Lilly, Pfizer, Amgen, Bristol-Myers Squibb, AstraZeneca, Daiichi Sankyo, AbbVie, MedImmune, and Merck. Dr. Peters disclosed relationships with AbbVie, Amgen, AstraZeneca, and many other companies.
SOURCE: Jerusalem G et al. ESMO 2020, Abstract LBA76.
FROM ESMO 2020
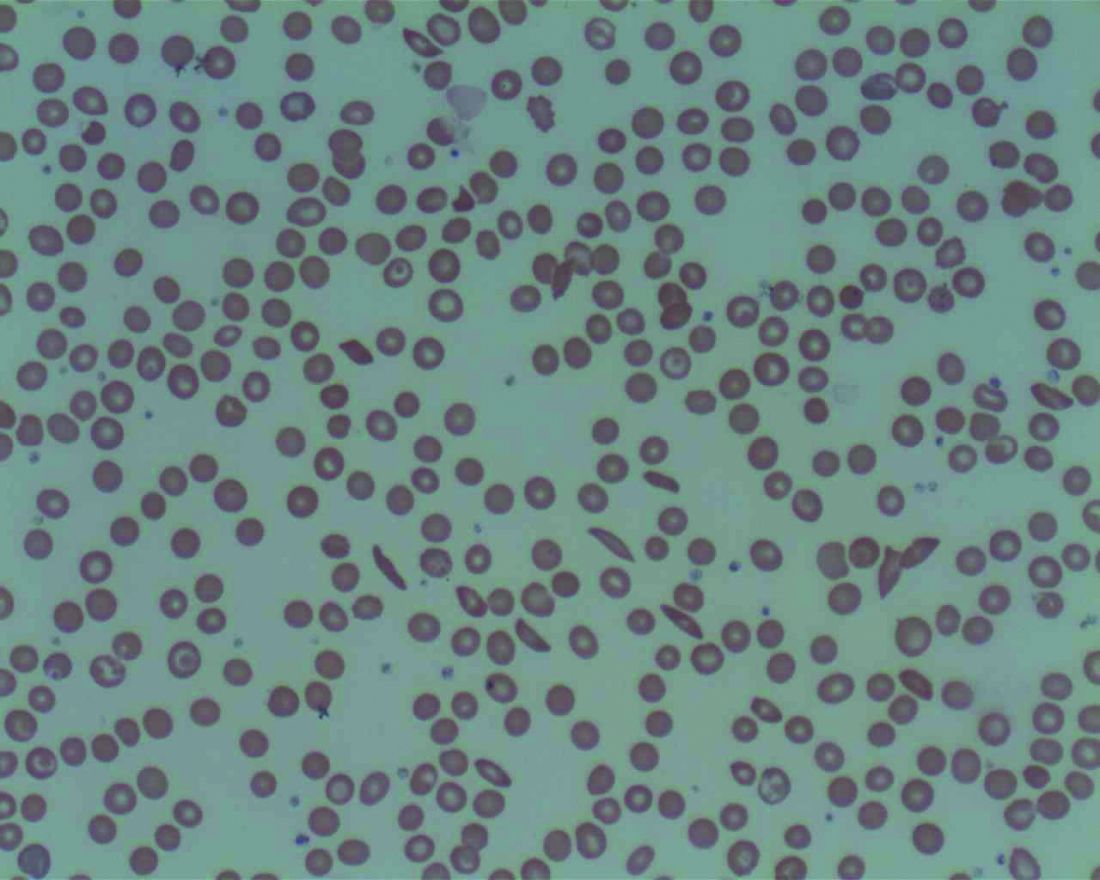
Oxidatative stress–related genetic variant tied to stroke risk in sickle cell patients
Oxidative stress-related genetic variants, in particular, the according to Igor F. Domingos of the Genetics Postgraduate Program, Federal University of Pernambuco, Recife, Brazil, and colleagues.
The researchers genotyped 499 unrelated adult patients with sickle cell anemia (SCA) for a variety of polymorphisms, along with alpha-thalassemia status and beta-globin gene haplotypes.
They found that SOD2 Val16Ala polymorphism was independently associated with risk of stroke (odds ratio, 1.98; 95% confidence interval, 1.18-3.32; P = .009) and with the long-term cumulative incidence of stroke (hazard ratio, 2.24; 95% CI, 1.3-3.9; P = .004).
A crucial limitation identified by the authors was the inability to replicate their results in a validation cohort. They suggested that there could have been genetic differences between the Brazilian population and the validation cohort of 231 patients followed at King’s College London for whom biological samples was available. They also suggested that patient treatment history between the two countries may be a factor.
“We believe that our study represents an alternative for understudied SCA populations with no access to TCD [transcranial Doppler ultrasound screening] and imaging exams, in which genetic modifiers may be a useful tool for predicting stroke in SCA,” the authors concluded.
The authors reported that they had no competing financial interests.
[email protected]
SOURCE: Domingos IF et al. J Neurol Sci. 2020 Apr 16; doi: 10.1016/j.jns.2020.116839.
Oxidative stress-related genetic variants, in particular, the according to Igor F. Domingos of the Genetics Postgraduate Program, Federal University of Pernambuco, Recife, Brazil, and colleagues.
The researchers genotyped 499 unrelated adult patients with sickle cell anemia (SCA) for a variety of polymorphisms, along with alpha-thalassemia status and beta-globin gene haplotypes.
They found that SOD2 Val16Ala polymorphism was independently associated with risk of stroke (odds ratio, 1.98; 95% confidence interval, 1.18-3.32; P = .009) and with the long-term cumulative incidence of stroke (hazard ratio, 2.24; 95% CI, 1.3-3.9; P = .004).
A crucial limitation identified by the authors was the inability to replicate their results in a validation cohort. They suggested that there could have been genetic differences between the Brazilian population and the validation cohort of 231 patients followed at King’s College London for whom biological samples was available. They also suggested that patient treatment history between the two countries may be a factor.
“We believe that our study represents an alternative for understudied SCA populations with no access to TCD [transcranial Doppler ultrasound screening] and imaging exams, in which genetic modifiers may be a useful tool for predicting stroke in SCA,” the authors concluded.
The authors reported that they had no competing financial interests.
[email protected]
SOURCE: Domingos IF et al. J Neurol Sci. 2020 Apr 16; doi: 10.1016/j.jns.2020.116839.
Oxidative stress-related genetic variants, in particular, the according to Igor F. Domingos of the Genetics Postgraduate Program, Federal University of Pernambuco, Recife, Brazil, and colleagues.
The researchers genotyped 499 unrelated adult patients with sickle cell anemia (SCA) for a variety of polymorphisms, along with alpha-thalassemia status and beta-globin gene haplotypes.
They found that SOD2 Val16Ala polymorphism was independently associated with risk of stroke (odds ratio, 1.98; 95% confidence interval, 1.18-3.32; P = .009) and with the long-term cumulative incidence of stroke (hazard ratio, 2.24; 95% CI, 1.3-3.9; P = .004).
A crucial limitation identified by the authors was the inability to replicate their results in a validation cohort. They suggested that there could have been genetic differences between the Brazilian population and the validation cohort of 231 patients followed at King’s College London for whom biological samples was available. They also suggested that patient treatment history between the two countries may be a factor.
“We believe that our study represents an alternative for understudied SCA populations with no access to TCD [transcranial Doppler ultrasound screening] and imaging exams, in which genetic modifiers may be a useful tool for predicting stroke in SCA,” the authors concluded.
The authors reported that they had no competing financial interests.
[email protected]
SOURCE: Domingos IF et al. J Neurol Sci. 2020 Apr 16; doi: 10.1016/j.jns.2020.116839.
FROM THE JOURNAL OF NEUROLOGICAL SCIENCES
Key clinical point: SOD2 Val16Ala polymorphism may represent a simple and inexpensive alternative for identifying sickle cell patients at risk of stroke.
Major finding: SOD2 Val16Ala polymorphism was independently associated with an increased risk of stroke (odds ratio, 1.98; P = .009).
Study details: A total of 499 unrelated adult patients with sickle cell disease were genotyped.
Disclosures: The authors reported that they had no competing financial interests.
Source: Domingos IF et al. J Neurol Sci. 2020 Apr 16. doi: org/10.1016/j.jns.2020.116839.
Is anemia due to folate deficiency a myth?
A 46-year-old man who lives in Tacoma, Wash., is seen for fatigue. He has a no significant past medical history. He is not taking any medications. His physical exam is unremarkable. His hemoglobin is 12 gm/dL, hematocrit is 37 gm/dL, mean corpuscular volume (MCV) is 103 fL, and thyroid-stimulating hormone level is 1.2 mU/L.
What workup do you recommend?
A) B12, folate testing
B) Alcohol history, B12, folate testing
C) Alcohol history, B12 testing
I would choose doing a careful alcohol history and vitamin B12 testing.

Dr. Seppä and colleagues looked at all outpatients who had a blood count done over an 8-month period.1 A total of 9,527 blood counts were ordered, and 287 (3%) had macrocytosis.1 Further workup was done for 113 of the patients. The most common cause found for macrocytosis was alcohol abuse, in 74 (65%) of the patients (80% of the men and 36% of the women). In several studies, vitamin B12 deficiency was the cause of macrocytosis in 5%-7% of patients.2,3
In 1978, a study by Davidson and Hamilton looked at 200 consecutive patients with MCVs over 100, and were able to find a cause in 80%.4 Sixteen of these patients had a low B12 level and 10 had a low folate level.
In 1998, the Food and Drug Administration required folic acid fortification of enriched grain products in the United States to help decrease the risk of neural tube defects. Similar fortification efforts were undertaken in Canada. Since 1998, anemia due to folate deficiency has essentially disappeared in individuals who have access to fortified grain products.
Joelson and colleagues looked at data on folate testing from the year prior to fortification of the grain supply (1997) and after (2004).5 They found that, in 1997, 4.8% of tests had a folate level less than 160 ng/mL compared with only 0.6% of tests in 2004.
When a more stringent cutoff for deficiency was used (94 ng/mL) 0.98% of tests were below that level in 1997, and 0.09% in 2004. The mean RBC folate level in 1997 was 420 ng/mL and rose to 697 ng/mL in 2004. Of the patients who did have low folate levels, only a minority had elevated MCVs.
Shojania et al. looked at folate testing in Canada after widespread fortification had started.6 They found that 0.5% of 2,154 serum folate levels were low and 0.7% of 560 red blood cell folate levels were low. Folate deficiency was not the cause of anemia in any of the patients with low folate levels.
Theisen-Toupal and colleagues did a retrospective study looking at folate testing over an 11-year period after fortification.7 The researchers examined the results of 84,187 assessments of folate levels. Forty-seven (0.056%) of the tests found patients with folate deficiency, 166 (0.197%), found patients with low-normal folate levels, 57,411 (68.195%) of tests yielded normal results, and 26,563 (31.552%) of tests found high folate levels. The opinion of the authors was that folate testing should be severely reduced or eliminated. Furthermore, the American Society for Clinical Pathology, as part of the Choosing Wisely campaign, states: “Do not order red blood cell folate levels at all.”8
So what does this all mean? We have been taught to have a reflex response to the evaluation of macrocytosis to test for B12 and folate. Neither of these are particularly common causes of macrocytosis, and in countries where there is grain fortification, folate deficiency is exceedingly uncommon, and should not be tested for early in any diagnostic process.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. He is a member of the editorial advisory board of Internal Medicine News. Dr. Paauw has no conflicts to disclose. Contact him at [email protected].
References
1. Seppä K et al. Evaluation of macrocytosis by general practitioners. J Stud Alcohol. 1996 Jan;57(1):97-100.
2. Seppä K et al. Blood count and hematologic morphology in nonanemic macrocytosis: Differences between alcohol abuse and pernicious anemia. Alcohol. 1993 Sep-Oct;10(5):343-7.
3. Wymer A, Becker DM. Recognition and evaluation of red blood cell macrocytosis in the primary care setting. J Gen Intern Med. 1990 May-Jun;5(3):192-7.
4. Davidson RJ, Hamilton PJ. High mean red cell volume: Its incidence and significance in routine haematology. J Clin Pathol. 1978 May;31[5]:493-8.
5. Joelson DW, Fiebig EW. Diminished need for folate measurements among indigent populations in the post folic acid supplementation era. Arch Pathol Lab Med. 2007 Mar;131(3):477-80.
6. Shojania AM, von Kuster K. Ordering folate assays is no longer justified for investigation of anemias, in folic acid fortified countries. BMC Res Notes. 2010 Jan 25;3:22. doi: 10.1186/1756-0500-3-22.
7. Theisen-Toupal et al. Low yield of outpatient serum folate testing. JAMA Intern Med. 2014 Oct. doi: 10.1001/jamainternmed.2014.3593.
8. Choosing Wisely: American Society for Clinical Pathology, Oct. 19, 2017. Recommendation.
A 46-year-old man who lives in Tacoma, Wash., is seen for fatigue. He has a no significant past medical history. He is not taking any medications. His physical exam is unremarkable. His hemoglobin is 12 gm/dL, hematocrit is 37 gm/dL, mean corpuscular volume (MCV) is 103 fL, and thyroid-stimulating hormone level is 1.2 mU/L.
What workup do you recommend?
A) B12, folate testing
B) Alcohol history, B12, folate testing
C) Alcohol history, B12 testing
I would choose doing a careful alcohol history and vitamin B12 testing.
Dr. Seppä and colleagues looked at all outpatients who had a blood count done over an 8-month period.1 A total of 9,527 blood counts were ordered, and 287 (3%) had macrocytosis.1 Further workup was done for 113 of the patients. The most common cause found for macrocytosis was alcohol abuse, in 74 (65%) of the patients (80% of the men and 36% of the women). In several studies, vitamin B12 deficiency was the cause of macrocytosis in 5%-7% of patients.2,3
In 1978, a study by Davidson and Hamilton looked at 200 consecutive patients with MCVs over 100, and were able to find a cause in 80%.4 Sixteen of these patients had a low B12 level and 10 had a low folate level.
In 1998, the Food and Drug Administration required folic acid fortification of enriched grain products in the United States to help decrease the risk of neural tube defects. Similar fortification efforts were undertaken in Canada. Since 1998, anemia due to folate deficiency has essentially disappeared in individuals who have access to fortified grain products.
Joelson and colleagues looked at data on folate testing from the year prior to fortification of the grain supply (1997) and after (2004).5 They found that, in 1997, 4.8% of tests had a folate level less than 160 ng/mL compared with only 0.6% of tests in 2004.
When a more stringent cutoff for deficiency was used (94 ng/mL) 0.98% of tests were below that level in 1997, and 0.09% in 2004. The mean RBC folate level in 1997 was 420 ng/mL and rose to 697 ng/mL in 2004. Of the patients who did have low folate levels, only a minority had elevated MCVs.
Shojania et al. looked at folate testing in Canada after widespread fortification had started.6 They found that 0.5% of 2,154 serum folate levels were low and 0.7% of 560 red blood cell folate levels were low. Folate deficiency was not the cause of anemia in any of the patients with low folate levels.
Theisen-Toupal and colleagues did a retrospective study looking at folate testing over an 11-year period after fortification.7 The researchers examined the results of 84,187 assessments of folate levels. Forty-seven (0.056%) of the tests found patients with folate deficiency, 166 (0.197%), found patients with low-normal folate levels, 57,411 (68.195%) of tests yielded normal results, and 26,563 (31.552%) of tests found high folate levels. The opinion of the authors was that folate testing should be severely reduced or eliminated. Furthermore, the American Society for Clinical Pathology, as part of the Choosing Wisely campaign, states: “Do not order red blood cell folate levels at all.”8
So what does this all mean? We have been taught to have a reflex response to the evaluation of macrocytosis to test for B12 and folate. Neither of these are particularly common causes of macrocytosis, and in countries where there is grain fortification, folate deficiency is exceedingly uncommon, and should not be tested for early in any diagnostic process.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. He is a member of the editorial advisory board of Internal Medicine News. Dr. Paauw has no conflicts to disclose. Contact him at [email protected].
References
1. Seppä K et al. Evaluation of macrocytosis by general practitioners. J Stud Alcohol. 1996 Jan;57(1):97-100.
2. Seppä K et al. Blood count and hematologic morphology in nonanemic macrocytosis: Differences between alcohol abuse and pernicious anemia. Alcohol. 1993 Sep-Oct;10(5):343-7.
3. Wymer A, Becker DM. Recognition and evaluation of red blood cell macrocytosis in the primary care setting. J Gen Intern Med. 1990 May-Jun;5(3):192-7.
4. Davidson RJ, Hamilton PJ. High mean red cell volume: Its incidence and significance in routine haematology. J Clin Pathol. 1978 May;31[5]:493-8.
5. Joelson DW, Fiebig EW. Diminished need for folate measurements among indigent populations in the post folic acid supplementation era. Arch Pathol Lab Med. 2007 Mar;131(3):477-80.
6. Shojania AM, von Kuster K. Ordering folate assays is no longer justified for investigation of anemias, in folic acid fortified countries. BMC Res Notes. 2010 Jan 25;3:22. doi: 10.1186/1756-0500-3-22.
7. Theisen-Toupal et al. Low yield of outpatient serum folate testing. JAMA Intern Med. 2014 Oct. doi: 10.1001/jamainternmed.2014.3593.
8. Choosing Wisely: American Society for Clinical Pathology, Oct. 19, 2017. Recommendation.
A 46-year-old man who lives in Tacoma, Wash., is seen for fatigue. He has a no significant past medical history. He is not taking any medications. His physical exam is unremarkable. His hemoglobin is 12 gm/dL, hematocrit is 37 gm/dL, mean corpuscular volume (MCV) is 103 fL, and thyroid-stimulating hormone level is 1.2 mU/L.
What workup do you recommend?
A) B12, folate testing
B) Alcohol history, B12, folate testing
C) Alcohol history, B12 testing
I would choose doing a careful alcohol history and vitamin B12 testing.
Dr. Seppä and colleagues looked at all outpatients who had a blood count done over an 8-month period.1 A total of 9,527 blood counts were ordered, and 287 (3%) had macrocytosis.1 Further workup was done for 113 of the patients. The most common cause found for macrocytosis was alcohol abuse, in 74 (65%) of the patients (80% of the men and 36% of the women). In several studies, vitamin B12 deficiency was the cause of macrocytosis in 5%-7% of patients.2,3
In 1978, a study by Davidson and Hamilton looked at 200 consecutive patients with MCVs over 100, and were able to find a cause in 80%.4 Sixteen of these patients had a low B12 level and 10 had a low folate level.
In 1998, the Food and Drug Administration required folic acid fortification of enriched grain products in the United States to help decrease the risk of neural tube defects. Similar fortification efforts were undertaken in Canada. Since 1998, anemia due to folate deficiency has essentially disappeared in individuals who have access to fortified grain products.
Joelson and colleagues looked at data on folate testing from the year prior to fortification of the grain supply (1997) and after (2004).5 They found that, in 1997, 4.8% of tests had a folate level less than 160 ng/mL compared with only 0.6% of tests in 2004.
When a more stringent cutoff for deficiency was used (94 ng/mL) 0.98% of tests were below that level in 1997, and 0.09% in 2004. The mean RBC folate level in 1997 was 420 ng/mL and rose to 697 ng/mL in 2004. Of the patients who did have low folate levels, only a minority had elevated MCVs.
Shojania et al. looked at folate testing in Canada after widespread fortification had started.6 They found that 0.5% of 2,154 serum folate levels were low and 0.7% of 560 red blood cell folate levels were low. Folate deficiency was not the cause of anemia in any of the patients with low folate levels.
Theisen-Toupal and colleagues did a retrospective study looking at folate testing over an 11-year period after fortification.7 The researchers examined the results of 84,187 assessments of folate levels. Forty-seven (0.056%) of the tests found patients with folate deficiency, 166 (0.197%), found patients with low-normal folate levels, 57,411 (68.195%) of tests yielded normal results, and 26,563 (31.552%) of tests found high folate levels. The opinion of the authors was that folate testing should be severely reduced or eliminated. Furthermore, the American Society for Clinical Pathology, as part of the Choosing Wisely campaign, states: “Do not order red blood cell folate levels at all.”8
So what does this all mean? We have been taught to have a reflex response to the evaluation of macrocytosis to test for B12 and folate. Neither of these are particularly common causes of macrocytosis, and in countries where there is grain fortification, folate deficiency is exceedingly uncommon, and should not be tested for early in any diagnostic process.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. He is a member of the editorial advisory board of Internal Medicine News. Dr. Paauw has no conflicts to disclose. Contact him at [email protected].
References
1. Seppä K et al. Evaluation of macrocytosis by general practitioners. J Stud Alcohol. 1996 Jan;57(1):97-100.
2. Seppä K et al. Blood count and hematologic morphology in nonanemic macrocytosis: Differences between alcohol abuse and pernicious anemia. Alcohol. 1993 Sep-Oct;10(5):343-7.
3. Wymer A, Becker DM. Recognition and evaluation of red blood cell macrocytosis in the primary care setting. J Gen Intern Med. 1990 May-Jun;5(3):192-7.
4. Davidson RJ, Hamilton PJ. High mean red cell volume: Its incidence and significance in routine haematology. J Clin Pathol. 1978 May;31[5]:493-8.
5. Joelson DW, Fiebig EW. Diminished need for folate measurements among indigent populations in the post folic acid supplementation era. Arch Pathol Lab Med. 2007 Mar;131(3):477-80.
6. Shojania AM, von Kuster K. Ordering folate assays is no longer justified for investigation of anemias, in folic acid fortified countries. BMC Res Notes. 2010 Jan 25;3:22. doi: 10.1186/1756-0500-3-22.
7. Theisen-Toupal et al. Low yield of outpatient serum folate testing. JAMA Intern Med. 2014 Oct. doi: 10.1001/jamainternmed.2014.3593.
8. Choosing Wisely: American Society for Clinical Pathology, Oct. 19, 2017. Recommendation.
FDA approves twice-daily formulation of key thalassemia drug
Chiesi Global Rare Diseases announced that the Food and Drug Administration has approved a new formulation of deferiprone (Ferriprox) for the treatment of patients with transfusional iron overload caused by thalassemia syndromes when current chelation therapy is inadequate. The new formulation of twice-a-day Ferriprox 1,000 mg oral tablets eliminates the midday dose, according to a company press release.

“A treatment option that reduces serum ferritin, cardiac iron, and liver iron with an established safety profile and now twice-a-day tablet dosing can represent a significant advantage for patients,” stated Thomas Coates, MD, in the press release. Dr. Coates is the section head of hematology at Children’s Hospital Los Angeles.
Deferiprone was originally approved by the FDA in 2011 for the treatment of transfusional iron overload caused by thalassemia syndromes. Ferriprox contains a label warning that it can cause agranulocytosis that can lead to serious infections and death. As neutropenia may precede the development of agranulocytosis, the warning advises measurement of the absolute neutrophil count before starting Ferriprox and monitoring the ANC weekly on therapy. In addition, Ferriprox should be interrupted if infection develops, and the ANC should be monitored more frequently.
As Ferriprox can cause fetal harm, women of reproductive potential should be advised to use an effective method of contraception during treatment and for at least 6 months after the last dose, according to the company release.
Chiesi Global Rare Diseases announced that the Food and Drug Administration has approved a new formulation of deferiprone (Ferriprox) for the treatment of patients with transfusional iron overload caused by thalassemia syndromes when current chelation therapy is inadequate. The new formulation of twice-a-day Ferriprox 1,000 mg oral tablets eliminates the midday dose, according to a company press release.
“A treatment option that reduces serum ferritin, cardiac iron, and liver iron with an established safety profile and now twice-a-day tablet dosing can represent a significant advantage for patients,” stated Thomas Coates, MD, in the press release. Dr. Coates is the section head of hematology at Children’s Hospital Los Angeles.
Deferiprone was originally approved by the FDA in 2011 for the treatment of transfusional iron overload caused by thalassemia syndromes. Ferriprox contains a label warning that it can cause agranulocytosis that can lead to serious infections and death. As neutropenia may precede the development of agranulocytosis, the warning advises measurement of the absolute neutrophil count before starting Ferriprox and monitoring the ANC weekly on therapy. In addition, Ferriprox should be interrupted if infection develops, and the ANC should be monitored more frequently.
As Ferriprox can cause fetal harm, women of reproductive potential should be advised to use an effective method of contraception during treatment and for at least 6 months after the last dose, according to the company release.
Chiesi Global Rare Diseases announced that the Food and Drug Administration has approved a new formulation of deferiprone (Ferriprox) for the treatment of patients with transfusional iron overload caused by thalassemia syndromes when current chelation therapy is inadequate. The new formulation of twice-a-day Ferriprox 1,000 mg oral tablets eliminates the midday dose, according to a company press release.
“A treatment option that reduces serum ferritin, cardiac iron, and liver iron with an established safety profile and now twice-a-day tablet dosing can represent a significant advantage for patients,” stated Thomas Coates, MD, in the press release. Dr. Coates is the section head of hematology at Children’s Hospital Los Angeles.
Deferiprone was originally approved by the FDA in 2011 for the treatment of transfusional iron overload caused by thalassemia syndromes. Ferriprox contains a label warning that it can cause agranulocytosis that can lead to serious infections and death. As neutropenia may precede the development of agranulocytosis, the warning advises measurement of the absolute neutrophil count before starting Ferriprox and monitoring the ANC weekly on therapy. In addition, Ferriprox should be interrupted if infection develops, and the ANC should be monitored more frequently.
As Ferriprox can cause fetal harm, women of reproductive potential should be advised to use an effective method of contraception during treatment and for at least 6 months after the last dose, according to the company release.
Protective levels of vitamin D achievable in SCD with oral supplementation
Sickle cell disease is associated with worse long-term bone health than that of the general population, and SCD patients are more likely to experience vitamin D [25(OH)D] deficiency. Oral vitamin D3 supplementation can achieve protective levels in children with sickle cell disease, and a daily dose was able to achieved optimal blood levels, according to a report published online in Bone.
The researchers performed a prospective, longitudinal, single-center study of 80 children with SCD. They collected demographic, clinical, and management data, as well as 25(OH)D levels. Bone densitometries (DXA) were also collected.
Among the 80 patients were included in the analysis, there were significant differences between the means of 25(OH)D levels based on whether the patient started prophylactic treatment as an infant or not (35.7 vs. 27.9 ng/mL, respectively [P = .014]), according to the researchers.
They also found that, in multivariate analysis, an oral 800 IU daily dose of vitamin D3 was shown to be a protective factor (P = .044) in reaching optimal 25(OH)D blood levels (≥ 30 ng/mL).
Kaplan-Meier analysis showed that those patients younger than 10 years of age reached optimal levels significantly earlier than older patients when on supplementation (P = .002), as did those patients who were not being treated with hydroxyurea (P = .039), the researchers wrote.
Significant differences were seen between the mean bone mineral density in both DXAs performed when comparing suboptimal vs. optimal blood levels of 25(OH)D (0.54 g/cm2 vs. 0.64 g/cm2, respectively, P = .001), for the initial DXA, and for the most recent DXA (0.59 g/cm2 vs. 0.77 g/cm2, respectively, P = .044). “VitD3 prophylaxis is a safe practice in SCD. It is important to start this prophylactic treatment when the child is an infant. The daily regimen with 800 IU could be more effective for reaching levels ≥ 30 ng/mL, and, especially in preadolescent and adolescent patients, we should raise awareness about the importance of good bone health,” the authors concluded.
The authors reported that they had no conflicts of interest.
SOURCE: Garrido C et al. Bone. 2020;133: doi.org/10.1016/j.bone.2020.115228.
Sickle cell disease is associated with worse long-term bone health than that of the general population, and SCD patients are more likely to experience vitamin D [25(OH)D] deficiency. Oral vitamin D3 supplementation can achieve protective levels in children with sickle cell disease, and a daily dose was able to achieved optimal blood levels, according to a report published online in Bone.
The researchers performed a prospective, longitudinal, single-center study of 80 children with SCD. They collected demographic, clinical, and management data, as well as 25(OH)D levels. Bone densitometries (DXA) were also collected.
Among the 80 patients were included in the analysis, there were significant differences between the means of 25(OH)D levels based on whether the patient started prophylactic treatment as an infant or not (35.7 vs. 27.9 ng/mL, respectively [P = .014]), according to the researchers.
They also found that, in multivariate analysis, an oral 800 IU daily dose of vitamin D3 was shown to be a protective factor (P = .044) in reaching optimal 25(OH)D blood levels (≥ 30 ng/mL).
Kaplan-Meier analysis showed that those patients younger than 10 years of age reached optimal levels significantly earlier than older patients when on supplementation (P = .002), as did those patients who were not being treated with hydroxyurea (P = .039), the researchers wrote.
Significant differences were seen between the mean bone mineral density in both DXAs performed when comparing suboptimal vs. optimal blood levels of 25(OH)D (0.54 g/cm2 vs. 0.64 g/cm2, respectively, P = .001), for the initial DXA, and for the most recent DXA (0.59 g/cm2 vs. 0.77 g/cm2, respectively, P = .044). “VitD3 prophylaxis is a safe practice in SCD. It is important to start this prophylactic treatment when the child is an infant. The daily regimen with 800 IU could be more effective for reaching levels ≥ 30 ng/mL, and, especially in preadolescent and adolescent patients, we should raise awareness about the importance of good bone health,” the authors concluded.
The authors reported that they had no conflicts of interest.
SOURCE: Garrido C et al. Bone. 2020;133: doi.org/10.1016/j.bone.2020.115228.
Sickle cell disease is associated with worse long-term bone health than that of the general population, and SCD patients are more likely to experience vitamin D [25(OH)D] deficiency. Oral vitamin D3 supplementation can achieve protective levels in children with sickle cell disease, and a daily dose was able to achieved optimal blood levels, according to a report published online in Bone.
The researchers performed a prospective, longitudinal, single-center study of 80 children with SCD. They collected demographic, clinical, and management data, as well as 25(OH)D levels. Bone densitometries (DXA) were also collected.
Among the 80 patients were included in the analysis, there were significant differences between the means of 25(OH)D levels based on whether the patient started prophylactic treatment as an infant or not (35.7 vs. 27.9 ng/mL, respectively [P = .014]), according to the researchers.
They also found that, in multivariate analysis, an oral 800 IU daily dose of vitamin D3 was shown to be a protective factor (P = .044) in reaching optimal 25(OH)D blood levels (≥ 30 ng/mL).
Kaplan-Meier analysis showed that those patients younger than 10 years of age reached optimal levels significantly earlier than older patients when on supplementation (P = .002), as did those patients who were not being treated with hydroxyurea (P = .039), the researchers wrote.
Significant differences were seen between the mean bone mineral density in both DXAs performed when comparing suboptimal vs. optimal blood levels of 25(OH)D (0.54 g/cm2 vs. 0.64 g/cm2, respectively, P = .001), for the initial DXA, and for the most recent DXA (0.59 g/cm2 vs. 0.77 g/cm2, respectively, P = .044). “VitD3 prophylaxis is a safe practice in SCD. It is important to start this prophylactic treatment when the child is an infant. The daily regimen with 800 IU could be more effective for reaching levels ≥ 30 ng/mL, and, especially in preadolescent and adolescent patients, we should raise awareness about the importance of good bone health,” the authors concluded.
The authors reported that they had no conflicts of interest.
SOURCE: Garrido C et al. Bone. 2020;133: doi.org/10.1016/j.bone.2020.115228.
FROM BONE
EHA and TIF explore how COVID-19 is affecting thalassemia and SCD patients
In a webinar designed to guide physicians in the care of hematology patients during the COVID-19 pandemic, three world experts on thalassemia and sickle cell disease (SCD) provided on-the-ground information from physicians who were dealing with the height of the crisis in their countries.
The webinar was organized by the European Hematology Association (EHA) and the Thalassemia International Federation (TIF).
Moderator Francesco Cerisoli, MD, head of research and mentoring at EHA, led the discussion with three guest speakers: Maria-Domenica Cappellini, MD, PhD, professor of hematology at the University of Milan; Androulla Eleftheriou, MD, executive director of TIF in Cyprus; and Raffaella Colombatti , MD, of the University of Padova in Italy, coordinator of the Red Cell Reserve Working Group of the Italian Association of Pediatric Hematology and Oncology.
Italian experience with thalassemia and COVID-19
Dr. Cappellini discussed the Italian experience with 11 thalassemia patients followed by a network survey who developed COVID-19 in the northern part of Italy, where the pandemic has been most widespread.
There are no published data focusing specifically on SARS-CoV-2 infection in patients with thalassemic syndromes, but patients with preexisting comorbidities are likely to be more severely affected by SARS-CoV-2, according to Dr. Cappellini.
Of particular concern is the fact that patients with thalassemia, especially older ones, are frequently splenectomized, which renders them more vulnerable to bacterial infections and can trigger life-threatening sepsis. However, splenectomy is not known to increase the risk of viral infection or severe viral illness. Of additional concern is the fact that many thalassemia patients need routine and frequent transfusions.
Overall, the 11 thalassemia patients who developed COVID-19 experienced only mild to moderate symptoms. This is despite the fact that 72% of the patients were splenectomized, which did not appear to affect the clinical course, and all of the patients had thalassemia-related comorbidities.
Around half of the patients were hospitalized, but none of them required transfer to the ICU. One patient who was treated with chemotherapy for diffuse large B-cell lymphoma in 2019 but is now in remission required more intense ventilation support with the use of continuous positive airway pressure.
Only three patients received specific treatment for COVID-19: one with hydroxychloroquine (HCQ) alone, one with HCQ plus anakinra, and one with HCQ plus ritonavir/darunavir.
Overall, “the number of infected thalassemia patients was lower than expected, likely due to earlier and more vigilant self-isolation compared to the general population,” Dr. Cappellini said. She pointed out that the first early response in February by thalassemia physicians was to warn their patients via email and phone calls about the need for self-isolation and precautions against the pandemic.
Physicians “rapidly reorganized activities, postponing nonessential ones” and managed to provide patients “a safe track at the hospital to receive their life-saving treatment in COVID-19–free areas with health care personnel wearing protective equipment” and assessment of all entering patients for COVID-19 infection, Dr. Cappellini said.
Results in additional thalassemia patients and SCD patients
Dr. Eleftheriou described 51 cases of thalassemia patients with SARS-CoV-2 infection reported to TIF as of April 16. Patients were from Cyprus, Italy, the United Kingdom, France, Turkey, Iran, Pakistan, and Indonesia.
Of the 51 patients, 46 presented with mild to moderate symptoms. Five patients had severe respiratory symptoms and required hospitalization, two were hospitalized and discharged, and three died between day 5 and day 15 post hospitalization.
Dr. Colombatti followed with a brief presentation of the intersection of COVID-19 with SCD patients. She presented anecdotal data involving 32 SCD patients who exhibited COVID-19 symptoms. Dr. Colombatti obtained the data via personal communication with Pablo Bartolucci, of Hôpitaux Universitaires Henri Mondor in Créteil, France.
All 32 SCD patients were screened and treated for COVID-19, and 17 of them continued treatment for 10 days. In all, 22 patients were hospitalized, 11 were transferred to the ICU, and 1 died.
Ensuring adequate blood supply
Dr. Eleftheriou also discussed the TIF response to the COVID-19 pandemic, which focused on the adequacy of blood supplies for these patients who so often need transfusions.
Dr. Eleftheriou stated that a shortage of blood was reported in 75% of the 62 member countries of the TIF, with 58% reporting severe shortages and 35% reporting moderate to severe shortages.
The shortages resulted in many countries returning to older family/friends donation practices, rare use of whole blood transfusions, and the use of older blood transfusions (older than 28 days).
In addition, physicians have modified their transfusion strategy. They have reduced the amount of blood given to thalassemia patients from two units to one unit during any transfusion, while making arrangements for more frequent transfusions; for example, one transfusion per week but with precautions made to “limit the time spent in the clinic and to control blood supplies while safeguarding that all [thalassemia] patients will be able to get their transfusion,” Dr. Eleftheriou said.
The information in the webinar was provided with the caveat that “no general evidence-based guidance can be derived from this discussion.” There were no other disclosures given.
In a webinar designed to guide physicians in the care of hematology patients during the COVID-19 pandemic, three world experts on thalassemia and sickle cell disease (SCD) provided on-the-ground information from physicians who were dealing with the height of the crisis in their countries.
The webinar was organized by the European Hematology Association (EHA) and the Thalassemia International Federation (TIF).
Moderator Francesco Cerisoli, MD, head of research and mentoring at EHA, led the discussion with three guest speakers: Maria-Domenica Cappellini, MD, PhD, professor of hematology at the University of Milan; Androulla Eleftheriou, MD, executive director of TIF in Cyprus; and Raffaella Colombatti , MD, of the University of Padova in Italy, coordinator of the Red Cell Reserve Working Group of the Italian Association of Pediatric Hematology and Oncology.
Italian experience with thalassemia and COVID-19
Dr. Cappellini discussed the Italian experience with 11 thalassemia patients followed by a network survey who developed COVID-19 in the northern part of Italy, where the pandemic has been most widespread.
There are no published data focusing specifically on SARS-CoV-2 infection in patients with thalassemic syndromes, but patients with preexisting comorbidities are likely to be more severely affected by SARS-CoV-2, according to Dr. Cappellini.
Of particular concern is the fact that patients with thalassemia, especially older ones, are frequently splenectomized, which renders them more vulnerable to bacterial infections and can trigger life-threatening sepsis. However, splenectomy is not known to increase the risk of viral infection or severe viral illness. Of additional concern is the fact that many thalassemia patients need routine and frequent transfusions.
Overall, the 11 thalassemia patients who developed COVID-19 experienced only mild to moderate symptoms. This is despite the fact that 72% of the patients were splenectomized, which did not appear to affect the clinical course, and all of the patients had thalassemia-related comorbidities.
Around half of the patients were hospitalized, but none of them required transfer to the ICU. One patient who was treated with chemotherapy for diffuse large B-cell lymphoma in 2019 but is now in remission required more intense ventilation support with the use of continuous positive airway pressure.
Only three patients received specific treatment for COVID-19: one with hydroxychloroquine (HCQ) alone, one with HCQ plus anakinra, and one with HCQ plus ritonavir/darunavir.
Overall, “the number of infected thalassemia patients was lower than expected, likely due to earlier and more vigilant self-isolation compared to the general population,” Dr. Cappellini said. She pointed out that the first early response in February by thalassemia physicians was to warn their patients via email and phone calls about the need for self-isolation and precautions against the pandemic.
Physicians “rapidly reorganized activities, postponing nonessential ones” and managed to provide patients “a safe track at the hospital to receive their life-saving treatment in COVID-19–free areas with health care personnel wearing protective equipment” and assessment of all entering patients for COVID-19 infection, Dr. Cappellini said.
Results in additional thalassemia patients and SCD patients
Dr. Eleftheriou described 51 cases of thalassemia patients with SARS-CoV-2 infection reported to TIF as of April 16. Patients were from Cyprus, Italy, the United Kingdom, France, Turkey, Iran, Pakistan, and Indonesia.
Of the 51 patients, 46 presented with mild to moderate symptoms. Five patients had severe respiratory symptoms and required hospitalization, two were hospitalized and discharged, and three died between day 5 and day 15 post hospitalization.
Dr. Colombatti followed with a brief presentation of the intersection of COVID-19 with SCD patients. She presented anecdotal data involving 32 SCD patients who exhibited COVID-19 symptoms. Dr. Colombatti obtained the data via personal communication with Pablo Bartolucci, of Hôpitaux Universitaires Henri Mondor in Créteil, France.
All 32 SCD patients were screened and treated for COVID-19, and 17 of them continued treatment for 10 days. In all, 22 patients were hospitalized, 11 were transferred to the ICU, and 1 died.
Ensuring adequate blood supply
Dr. Eleftheriou also discussed the TIF response to the COVID-19 pandemic, which focused on the adequacy of blood supplies for these patients who so often need transfusions.
Dr. Eleftheriou stated that a shortage of blood was reported in 75% of the 62 member countries of the TIF, with 58% reporting severe shortages and 35% reporting moderate to severe shortages.
The shortages resulted in many countries returning to older family/friends donation practices, rare use of whole blood transfusions, and the use of older blood transfusions (older than 28 days).
In addition, physicians have modified their transfusion strategy. They have reduced the amount of blood given to thalassemia patients from two units to one unit during any transfusion, while making arrangements for more frequent transfusions; for example, one transfusion per week but with precautions made to “limit the time spent in the clinic and to control blood supplies while safeguarding that all [thalassemia] patients will be able to get their transfusion,” Dr. Eleftheriou said.
The information in the webinar was provided with the caveat that “no general evidence-based guidance can be derived from this discussion.” There were no other disclosures given.
In a webinar designed to guide physicians in the care of hematology patients during the COVID-19 pandemic, three world experts on thalassemia and sickle cell disease (SCD) provided on-the-ground information from physicians who were dealing with the height of the crisis in their countries.
The webinar was organized by the European Hematology Association (EHA) and the Thalassemia International Federation (TIF).
Moderator Francesco Cerisoli, MD, head of research and mentoring at EHA, led the discussion with three guest speakers: Maria-Domenica Cappellini, MD, PhD, professor of hematology at the University of Milan; Androulla Eleftheriou, MD, executive director of TIF in Cyprus; and Raffaella Colombatti , MD, of the University of Padova in Italy, coordinator of the Red Cell Reserve Working Group of the Italian Association of Pediatric Hematology and Oncology.
Italian experience with thalassemia and COVID-19
Dr. Cappellini discussed the Italian experience with 11 thalassemia patients followed by a network survey who developed COVID-19 in the northern part of Italy, where the pandemic has been most widespread.
There are no published data focusing specifically on SARS-CoV-2 infection in patients with thalassemic syndromes, but patients with preexisting comorbidities are likely to be more severely affected by SARS-CoV-2, according to Dr. Cappellini.
Of particular concern is the fact that patients with thalassemia, especially older ones, are frequently splenectomized, which renders them more vulnerable to bacterial infections and can trigger life-threatening sepsis. However, splenectomy is not known to increase the risk of viral infection or severe viral illness. Of additional concern is the fact that many thalassemia patients need routine and frequent transfusions.
Overall, the 11 thalassemia patients who developed COVID-19 experienced only mild to moderate symptoms. This is despite the fact that 72% of the patients were splenectomized, which did not appear to affect the clinical course, and all of the patients had thalassemia-related comorbidities.
Around half of the patients were hospitalized, but none of them required transfer to the ICU. One patient who was treated with chemotherapy for diffuse large B-cell lymphoma in 2019 but is now in remission required more intense ventilation support with the use of continuous positive airway pressure.
Only three patients received specific treatment for COVID-19: one with hydroxychloroquine (HCQ) alone, one with HCQ plus anakinra, and one with HCQ plus ritonavir/darunavir.
Overall, “the number of infected thalassemia patients was lower than expected, likely due to earlier and more vigilant self-isolation compared to the general population,” Dr. Cappellini said. She pointed out that the first early response in February by thalassemia physicians was to warn their patients via email and phone calls about the need for self-isolation and precautions against the pandemic.
Physicians “rapidly reorganized activities, postponing nonessential ones” and managed to provide patients “a safe track at the hospital to receive their life-saving treatment in COVID-19–free areas with health care personnel wearing protective equipment” and assessment of all entering patients for COVID-19 infection, Dr. Cappellini said.
Results in additional thalassemia patients and SCD patients
Dr. Eleftheriou described 51 cases of thalassemia patients with SARS-CoV-2 infection reported to TIF as of April 16. Patients were from Cyprus, Italy, the United Kingdom, France, Turkey, Iran, Pakistan, and Indonesia.
Of the 51 patients, 46 presented with mild to moderate symptoms. Five patients had severe respiratory symptoms and required hospitalization, two were hospitalized and discharged, and three died between day 5 and day 15 post hospitalization.
Dr. Colombatti followed with a brief presentation of the intersection of COVID-19 with SCD patients. She presented anecdotal data involving 32 SCD patients who exhibited COVID-19 symptoms. Dr. Colombatti obtained the data via personal communication with Pablo Bartolucci, of Hôpitaux Universitaires Henri Mondor in Créteil, France.
All 32 SCD patients were screened and treated for COVID-19, and 17 of them continued treatment for 10 days. In all, 22 patients were hospitalized, 11 were transferred to the ICU, and 1 died.
Ensuring adequate blood supply
Dr. Eleftheriou also discussed the TIF response to the COVID-19 pandemic, which focused on the adequacy of blood supplies for these patients who so often need transfusions.
Dr. Eleftheriou stated that a shortage of blood was reported in 75% of the 62 member countries of the TIF, with 58% reporting severe shortages and 35% reporting moderate to severe shortages.
The shortages resulted in many countries returning to older family/friends donation practices, rare use of whole blood transfusions, and the use of older blood transfusions (older than 28 days).
In addition, physicians have modified their transfusion strategy. They have reduced the amount of blood given to thalassemia patients from two units to one unit during any transfusion, while making arrangements for more frequent transfusions; for example, one transfusion per week but with precautions made to “limit the time spent in the clinic and to control blood supplies while safeguarding that all [thalassemia] patients will be able to get their transfusion,” Dr. Eleftheriou said.
The information in the webinar was provided with the caveat that “no general evidence-based guidance can be derived from this discussion.” There were no other disclosures given.
Pandemic strains blood supply for COVID-19 and noninfected patients
The COVID-19 pandemic is putting a strain on the blood supply and could be putting people – including those who normally get transfusions, such as patients with sickle cell disease and cancer – at risk.
“Around the beginning of March, the hematology community got wind of what was going on because the blood banks were saying think about your patients and begin to restrict blood usage because we are expecting an increase in usage for COVID-positive ICU patients,” Ifeyinwa (Ify) Osunkwo, MD, a specialist in hematology and sickle cell disease at Levine Cancer Institute in Charlotte, N.C., said in an interview.
“I think that was the first call to arms around hematology ... you don’t want to shortchange somebody who is well and who is being sustained by life-giving transfusions and cut out their transfusion therapy because you are hoping to use the blood for people who are coming in with COVID-19,” she continued. “That is an ethical dilemma that no doctor wants to have to go through. But the reality is we have to do something to make it work for everybody.”
And the timing of the social restrictions due to the pandemic has added additional strain on the blood supply.
“Over the winter, traditionally, blood drives slow down because of the flu and different viruses,” she noted. “The spring and the summer are when we see the biggest recruitment and uptake of blood donation. COVID-19 hit [and] a lot of the blood drives that were traditionally scheduled to supply blood for the country have been canceled because of the new guidance for social distancing.”
Another big source of blood are health care professionals themselves and they may not be able to donate because of the extra hours being worked because of the pandemic.
In speaking about the needs for traditional patients such as those who are dealing with cancer or leukemia or sickle cell diseases as well as those who are being treated for COVID-19 in North Carolina, “we are not at the critical point, but I am a little bit nervous that we may get there because they are not going to up the usual blood drives anytime this summer. We project [sometime] in the fall, but maybe not even then. So there needs to be a significant call-out for people to make every effort to donate blood,” said Dr. Osunkwo. She added that in places such as New York City that are hot spots for the COVID-19 outbreak, the need is likely a lot greater.
She recalled a recent incident at a New York hospital that highlighted how those managing blood supplies are being restrictive and how this could be harming patients.
“A sickle cell patient came in with COVID-19 and the treatment recommendation was do a red blood cell exchange but the blood bank was nervous about getting enough blood to supply for that exchange transfusion,” she said, noting that the doctor still went to bat for that patient to get the needed treatment. “We gave her the supporting evidence that when you are on treatment for sickle cell disease, you tend to do better if you get COVID-19 or any other viral infection. The symptoms of COVID-19 in sickle cell disease is acute chest syndrome, for which the treatment is red blood cell exchange. Not doing that for [these patients] is really not giving them the optimal way of managing their disease, and managing their disease in the setting of COVID-19.”
To that end, Dr. Osunkwo stressed that doctors need to be doing all they can to get the word out that blood is needed and that the American Red Cross and other donation organizations are making it safe for people to donate. She has been using social media to highlight when her fellow doctors and others make donations as a way to motivate individuals.
“Everybody can do something during this pandemic,” she said. “Don’t feel like you are not working, that you are not a frontline worker, that you have nothing to contribute. You can donate blood. Your cousin can donate blood. You can tell your friends, your neighbors, your relatives, your enemies to go donate. We will take every kind of blood we can get because people are needing it more now. Even though we canceled elective surgeries, my patients when they get COVID-19, they need more blood ... than they usually do during their regular sickle cell admission. It is going to be the same for people who have other blood disorders like cancer and leukemia. We can’t stop life-saving treatments just because we have the COVID pandemic.”
Dr. Osunkwo also praised recent actions taken by the Food and Drug Administration to lessen some of the deferral periods for when an individual can donate.
The FDA on April 2 issued three sets of revised recommendations aimed at getting more people eligible to donate blood. All of the revised recommendations will remain in effect after the COVID-19 health emergency is declared over.
The first revised recommendation makes changes to December 2015 guidance.
For male blood donors who would have been deferred for having sex with another male partner, the deferral period has been reduced from 12 months to 3 months. That deferral period change also applies to female donors who had sex with a man who had sex with another man as well as for those with recent tattoos and piercings.
The second recommendation revises guidance from August 2013 and relates to the risk of transfusion-transmitted malaria.
Under the new recommendations, for those who traveled to malaria-endemic areas (and are residents of malaria non-endemic countries), the FDA is lowering the recommended deferral period from 12 months to 3 months, and also provides notices of an alternate procedure that permits donations without a deferral period provided the blood components are pathogen-reduced using an FDA-approved pathogen reduction device.
The third recommendation finalizes draft guidance from January that eliminates the referral period for donors who spent time in certain European countries or were on military bases in Europe and were previously considered to have been exposed to a potential risk of transmission of Creutzfeldt-Jakob Disease or Variant Creutzfeldt-Jakob Disease.
Dr. Osunkwo reports consultancy and being on the speakers bureau and participating in the advisory board for Novartis, and relationships with a variety of other pharmaceutical companies. She is the editor-in-chief for Hematology News.
The COVID-19 pandemic is putting a strain on the blood supply and could be putting people – including those who normally get transfusions, such as patients with sickle cell disease and cancer – at risk.
“Around the beginning of March, the hematology community got wind of what was going on because the blood banks were saying think about your patients and begin to restrict blood usage because we are expecting an increase in usage for COVID-positive ICU patients,” Ifeyinwa (Ify) Osunkwo, MD, a specialist in hematology and sickle cell disease at Levine Cancer Institute in Charlotte, N.C., said in an interview.
“I think that was the first call to arms around hematology ... you don’t want to shortchange somebody who is well and who is being sustained by life-giving transfusions and cut out their transfusion therapy because you are hoping to use the blood for people who are coming in with COVID-19,” she continued. “That is an ethical dilemma that no doctor wants to have to go through. But the reality is we have to do something to make it work for everybody.”
And the timing of the social restrictions due to the pandemic has added additional strain on the blood supply.
“Over the winter, traditionally, blood drives slow down because of the flu and different viruses,” she noted. “The spring and the summer are when we see the biggest recruitment and uptake of blood donation. COVID-19 hit [and] a lot of the blood drives that were traditionally scheduled to supply blood for the country have been canceled because of the new guidance for social distancing.”
Another big source of blood are health care professionals themselves and they may not be able to donate because of the extra hours being worked because of the pandemic.
In speaking about the needs for traditional patients such as those who are dealing with cancer or leukemia or sickle cell diseases as well as those who are being treated for COVID-19 in North Carolina, “we are not at the critical point, but I am a little bit nervous that we may get there because they are not going to up the usual blood drives anytime this summer. We project [sometime] in the fall, but maybe not even then. So there needs to be a significant call-out for people to make every effort to donate blood,” said Dr. Osunkwo. She added that in places such as New York City that are hot spots for the COVID-19 outbreak, the need is likely a lot greater.
She recalled a recent incident at a New York hospital that highlighted how those managing blood supplies are being restrictive and how this could be harming patients.
“A sickle cell patient came in with COVID-19 and the treatment recommendation was do a red blood cell exchange but the blood bank was nervous about getting enough blood to supply for that exchange transfusion,” she said, noting that the doctor still went to bat for that patient to get the needed treatment. “We gave her the supporting evidence that when you are on treatment for sickle cell disease, you tend to do better if you get COVID-19 or any other viral infection. The symptoms of COVID-19 in sickle cell disease is acute chest syndrome, for which the treatment is red blood cell exchange. Not doing that for [these patients] is really not giving them the optimal way of managing their disease, and managing their disease in the setting of COVID-19.”
To that end, Dr. Osunkwo stressed that doctors need to be doing all they can to get the word out that blood is needed and that the American Red Cross and other donation organizations are making it safe for people to donate. She has been using social media to highlight when her fellow doctors and others make donations as a way to motivate individuals.
“Everybody can do something during this pandemic,” she said. “Don’t feel like you are not working, that you are not a frontline worker, that you have nothing to contribute. You can donate blood. Your cousin can donate blood. You can tell your friends, your neighbors, your relatives, your enemies to go donate. We will take every kind of blood we can get because people are needing it more now. Even though we canceled elective surgeries, my patients when they get COVID-19, they need more blood ... than they usually do during their regular sickle cell admission. It is going to be the same for people who have other blood disorders like cancer and leukemia. We can’t stop life-saving treatments just because we have the COVID pandemic.”
Dr. Osunkwo also praised recent actions taken by the Food and Drug Administration to lessen some of the deferral periods for when an individual can donate.
The FDA on April 2 issued three sets of revised recommendations aimed at getting more people eligible to donate blood. All of the revised recommendations will remain in effect after the COVID-19 health emergency is declared over.
The first revised recommendation makes changes to December 2015 guidance.
For male blood donors who would have been deferred for having sex with another male partner, the deferral period has been reduced from 12 months to 3 months. That deferral period change also applies to female donors who had sex with a man who had sex with another man as well as for those with recent tattoos and piercings.
The second recommendation revises guidance from August 2013 and relates to the risk of transfusion-transmitted malaria.
Under the new recommendations, for those who traveled to malaria-endemic areas (and are residents of malaria non-endemic countries), the FDA is lowering the recommended deferral period from 12 months to 3 months, and also provides notices of an alternate procedure that permits donations without a deferral period provided the blood components are pathogen-reduced using an FDA-approved pathogen reduction device.
The third recommendation finalizes draft guidance from January that eliminates the referral period for donors who spent time in certain European countries or were on military bases in Europe and were previously considered to have been exposed to a potential risk of transmission of Creutzfeldt-Jakob Disease or Variant Creutzfeldt-Jakob Disease.
Dr. Osunkwo reports consultancy and being on the speakers bureau and participating in the advisory board for Novartis, and relationships with a variety of other pharmaceutical companies. She is the editor-in-chief for Hematology News.
The COVID-19 pandemic is putting a strain on the blood supply and could be putting people – including those who normally get transfusions, such as patients with sickle cell disease and cancer – at risk.
“Around the beginning of March, the hematology community got wind of what was going on because the blood banks were saying think about your patients and begin to restrict blood usage because we are expecting an increase in usage for COVID-positive ICU patients,” Ifeyinwa (Ify) Osunkwo, MD, a specialist in hematology and sickle cell disease at Levine Cancer Institute in Charlotte, N.C., said in an interview.
“I think that was the first call to arms around hematology ... you don’t want to shortchange somebody who is well and who is being sustained by life-giving transfusions and cut out their transfusion therapy because you are hoping to use the blood for people who are coming in with COVID-19,” she continued. “That is an ethical dilemma that no doctor wants to have to go through. But the reality is we have to do something to make it work for everybody.”
And the timing of the social restrictions due to the pandemic has added additional strain on the blood supply.
“Over the winter, traditionally, blood drives slow down because of the flu and different viruses,” she noted. “The spring and the summer are when we see the biggest recruitment and uptake of blood donation. COVID-19 hit [and] a lot of the blood drives that were traditionally scheduled to supply blood for the country have been canceled because of the new guidance for social distancing.”
Another big source of blood are health care professionals themselves and they may not be able to donate because of the extra hours being worked because of the pandemic.
In speaking about the needs for traditional patients such as those who are dealing with cancer or leukemia or sickle cell diseases as well as those who are being treated for COVID-19 in North Carolina, “we are not at the critical point, but I am a little bit nervous that we may get there because they are not going to up the usual blood drives anytime this summer. We project [sometime] in the fall, but maybe not even then. So there needs to be a significant call-out for people to make every effort to donate blood,” said Dr. Osunkwo. She added that in places such as New York City that are hot spots for the COVID-19 outbreak, the need is likely a lot greater.
She recalled a recent incident at a New York hospital that highlighted how those managing blood supplies are being restrictive and how this could be harming patients.
“A sickle cell patient came in with COVID-19 and the treatment recommendation was do a red blood cell exchange but the blood bank was nervous about getting enough blood to supply for that exchange transfusion,” she said, noting that the doctor still went to bat for that patient to get the needed treatment. “We gave her the supporting evidence that when you are on treatment for sickle cell disease, you tend to do better if you get COVID-19 or any other viral infection. The symptoms of COVID-19 in sickle cell disease is acute chest syndrome, for which the treatment is red blood cell exchange. Not doing that for [these patients] is really not giving them the optimal way of managing their disease, and managing their disease in the setting of COVID-19.”
To that end, Dr. Osunkwo stressed that doctors need to be doing all they can to get the word out that blood is needed and that the American Red Cross and other donation organizations are making it safe for people to donate. She has been using social media to highlight when her fellow doctors and others make donations as a way to motivate individuals.
“Everybody can do something during this pandemic,” she said. “Don’t feel like you are not working, that you are not a frontline worker, that you have nothing to contribute. You can donate blood. Your cousin can donate blood. You can tell your friends, your neighbors, your relatives, your enemies to go donate. We will take every kind of blood we can get because people are needing it more now. Even though we canceled elective surgeries, my patients when they get COVID-19, they need more blood ... than they usually do during their regular sickle cell admission. It is going to be the same for people who have other blood disorders like cancer and leukemia. We can’t stop life-saving treatments just because we have the COVID pandemic.”
Dr. Osunkwo also praised recent actions taken by the Food and Drug Administration to lessen some of the deferral periods for when an individual can donate.
The FDA on April 2 issued three sets of revised recommendations aimed at getting more people eligible to donate blood. All of the revised recommendations will remain in effect after the COVID-19 health emergency is declared over.
The first revised recommendation makes changes to December 2015 guidance.
For male blood donors who would have been deferred for having sex with another male partner, the deferral period has been reduced from 12 months to 3 months. That deferral period change also applies to female donors who had sex with a man who had sex with another man as well as for those with recent tattoos and piercings.
The second recommendation revises guidance from August 2013 and relates to the risk of transfusion-transmitted malaria.
Under the new recommendations, for those who traveled to malaria-endemic areas (and are residents of malaria non-endemic countries), the FDA is lowering the recommended deferral period from 12 months to 3 months, and also provides notices of an alternate procedure that permits donations without a deferral period provided the blood components are pathogen-reduced using an FDA-approved pathogen reduction device.
The third recommendation finalizes draft guidance from January that eliminates the referral period for donors who spent time in certain European countries or were on military bases in Europe and were previously considered to have been exposed to a potential risk of transmission of Creutzfeldt-Jakob Disease or Variant Creutzfeldt-Jakob Disease.
Dr. Osunkwo reports consultancy and being on the speakers bureau and participating in the advisory board for Novartis, and relationships with a variety of other pharmaceutical companies. She is the editor-in-chief for Hematology News.

Severe COVID-19 may lower hemoglobin levels
A meta-analysis of four applicable studies found that the hemoglobin value was significantly lower in COVID-19 patients with severe disease, compared with those with milder forms, according to a letter to the editor of Hematology Transfusion and Cell Therapy by Giuseppe Lippi, MD, of the University of Verona (Italy) and colleague.
The four studies comprised 1,210 COVID-19 patients (224 with severe disease; 18.5%). The primary endpoint was defined as a composite of admission to the ICU, need of mechanical ventilation or death. The heterogeneity among the studies was high.
Overall, the hemoglobin value was found to be significantly lower in COVID-19 patients with severe disease than in those with milder forms, yielding a weighted mean difference of −7.1 g/L, with a 95% confidence interval of −8.3 g/L to −5.9 g/L.
“Initial assessment and longitudinal monitoring of hemoglobin values seems advisable in patients with the SARS-CoV-2 infection, whereby a progressive decrease in the hemoglobin concentration may reflect a worse clinical progression,” the authors stated. They also suggested that studies should be “urgently planned to assess whether transfusion support (e.g., with administration of blood or packed red blood cells) may be helpful in this clinical setting to prevent evolution into severe disease and death.”
The authors declared the had no conflicts of interest.
SOURCE: Lippi G et al. Hematol Transfus Cell Ther. 2020 Apr 11; doi:10.1016/j.htct.2020.03.001.
A meta-analysis of four applicable studies found that the hemoglobin value was significantly lower in COVID-19 patients with severe disease, compared with those with milder forms, according to a letter to the editor of Hematology Transfusion and Cell Therapy by Giuseppe Lippi, MD, of the University of Verona (Italy) and colleague.
The four studies comprised 1,210 COVID-19 patients (224 with severe disease; 18.5%). The primary endpoint was defined as a composite of admission to the ICU, need of mechanical ventilation or death. The heterogeneity among the studies was high.
Overall, the hemoglobin value was found to be significantly lower in COVID-19 patients with severe disease than in those with milder forms, yielding a weighted mean difference of −7.1 g/L, with a 95% confidence interval of −8.3 g/L to −5.9 g/L.
“Initial assessment and longitudinal monitoring of hemoglobin values seems advisable in patients with the SARS-CoV-2 infection, whereby a progressive decrease in the hemoglobin concentration may reflect a worse clinical progression,” the authors stated. They also suggested that studies should be “urgently planned to assess whether transfusion support (e.g., with administration of blood or packed red blood cells) may be helpful in this clinical setting to prevent evolution into severe disease and death.”
The authors declared the had no conflicts of interest.
SOURCE: Lippi G et al. Hematol Transfus Cell Ther. 2020 Apr 11; doi:10.1016/j.htct.2020.03.001.
A meta-analysis of four applicable studies found that the hemoglobin value was significantly lower in COVID-19 patients with severe disease, compared with those with milder forms, according to a letter to the editor of Hematology Transfusion and Cell Therapy by Giuseppe Lippi, MD, of the University of Verona (Italy) and colleague.
The four studies comprised 1,210 COVID-19 patients (224 with severe disease; 18.5%). The primary endpoint was defined as a composite of admission to the ICU, need of mechanical ventilation or death. The heterogeneity among the studies was high.
Overall, the hemoglobin value was found to be significantly lower in COVID-19 patients with severe disease than in those with milder forms, yielding a weighted mean difference of −7.1 g/L, with a 95% confidence interval of −8.3 g/L to −5.9 g/L.
“Initial assessment and longitudinal monitoring of hemoglobin values seems advisable in patients with the SARS-CoV-2 infection, whereby a progressive decrease in the hemoglobin concentration may reflect a worse clinical progression,” the authors stated. They also suggested that studies should be “urgently planned to assess whether transfusion support (e.g., with administration of blood or packed red blood cells) may be helpful in this clinical setting to prevent evolution into severe disease and death.”
The authors declared the had no conflicts of interest.
SOURCE: Lippi G et al. Hematol Transfus Cell Ther. 2020 Apr 11; doi:10.1016/j.htct.2020.03.001.
FROM HEMATOLOGY, TRANSFUSION AND CELL THERAPY