User login
Topical tacrolimus and corticosteroids show similar efficacy and impact on airways in childhood atopic dermatitis
Key clinical point: Long-term topical corticosteroid (TCS) and tacrolimus (TAC) treatments had similar efficacy and impact on airway hyperresponsiveness or inflammation in children with moderate-to-severe atopic dermatitis (AD).
Major finding: At month 36, children treated with TCS and TAC had no significant difference in the mean body surface area (P = .12), mean Eczema Area and Severity Index score (P = .2), mean Investigator’s Global Assessment Score (P = .12), mean transepidermal water loss at eczema site (P = .96) and control site (P = .19), median exhaled nitric oxide level (P = .71), or median bronchial hyperresponsiveness to methacholine (P = .7).
Study details: Findings are from a single-center 3-year follow-up study including 152 children aged 1-3 years with moderate-to-severe AD who were randomly assigned to receive TCS (n = 75) or TAC (n = 77).
Disclosures: This study was supported by the Foundation for Paediatric Research, Finland, Orion Research Foundation, Finland, Orion Pharma, Finland, Astellas Pharma, Japan, and others. Orion and Astellas are commercial manufacturers of the TCS used in this study.
Source: Perälä M et al. Topical tacrolimus versus corticosteroids in childhood moderate-to-severe atopic dermatitis with impact on airways: A long-term randomized open-label study. Clin Exp Dermatol. 2023 (Mar 14). Doi: 10.1093/ced/llad098
Key clinical point: Long-term topical corticosteroid (TCS) and tacrolimus (TAC) treatments had similar efficacy and impact on airway hyperresponsiveness or inflammation in children with moderate-to-severe atopic dermatitis (AD).
Major finding: At month 36, children treated with TCS and TAC had no significant difference in the mean body surface area (P = .12), mean Eczema Area and Severity Index score (P = .2), mean Investigator’s Global Assessment Score (P = .12), mean transepidermal water loss at eczema site (P = .96) and control site (P = .19), median exhaled nitric oxide level (P = .71), or median bronchial hyperresponsiveness to methacholine (P = .7).
Study details: Findings are from a single-center 3-year follow-up study including 152 children aged 1-3 years with moderate-to-severe AD who were randomly assigned to receive TCS (n = 75) or TAC (n = 77).
Disclosures: This study was supported by the Foundation for Paediatric Research, Finland, Orion Research Foundation, Finland, Orion Pharma, Finland, Astellas Pharma, Japan, and others. Orion and Astellas are commercial manufacturers of the TCS used in this study.
Source: Perälä M et al. Topical tacrolimus versus corticosteroids in childhood moderate-to-severe atopic dermatitis with impact on airways: A long-term randomized open-label study. Clin Exp Dermatol. 2023 (Mar 14). Doi: 10.1093/ced/llad098
Key clinical point: Long-term topical corticosteroid (TCS) and tacrolimus (TAC) treatments had similar efficacy and impact on airway hyperresponsiveness or inflammation in children with moderate-to-severe atopic dermatitis (AD).
Major finding: At month 36, children treated with TCS and TAC had no significant difference in the mean body surface area (P = .12), mean Eczema Area and Severity Index score (P = .2), mean Investigator’s Global Assessment Score (P = .12), mean transepidermal water loss at eczema site (P = .96) and control site (P = .19), median exhaled nitric oxide level (P = .71), or median bronchial hyperresponsiveness to methacholine (P = .7).
Study details: Findings are from a single-center 3-year follow-up study including 152 children aged 1-3 years with moderate-to-severe AD who were randomly assigned to receive TCS (n = 75) or TAC (n = 77).
Disclosures: This study was supported by the Foundation for Paediatric Research, Finland, Orion Research Foundation, Finland, Orion Pharma, Finland, Astellas Pharma, Japan, and others. Orion and Astellas are commercial manufacturers of the TCS used in this study.
Source: Perälä M et al. Topical tacrolimus versus corticosteroids in childhood moderate-to-severe atopic dermatitis with impact on airways: A long-term randomized open-label study. Clin Exp Dermatol. 2023 (Mar 14). Doi: 10.1093/ced/llad098
Baricitinib allows flexibility in dosing regimens up to 104 weeks in moderate-to-severe atopic dermatitis
Key clinical point: Baricitinib offers the flexibility of dose down-titration and improvement in atopic dermatitis (AD) signs and symptoms up to treatment week 104 in patients with moderate-to-severe AD.
Major finding: Among patients receiving 4 mg baricitinib, 51.2% and 47.6% achieved or maintained a validated Investigator’s Global Assessment for AD score of 0 or 1 and 82.1%, and 73.8% maintained an Eczema Area and Severity Index 75 response at weeks 52 and 104, respectively. The clinical response was maintained after down-titration to 2 mg baricitinib.
Study details: This sub-study of the BREEZE-AD3 trial included 168 patients with moderate-to-severe AD who were responders or partial responders to 4 mg baricitinib in originating studies and were re-assigned (1:1) to receive 4 or 2 mg baricitinib at week 52.
Disclosures: This study was funded by Eli Lilly and Company under license from Incyte Corporation. Some authors reported ties with various organizations, including Eli Lilly. Four authors declared being employees and stockholders of Eli Lilly.
Source: Thyssen JP et al. Maintained improvement in physician- and patient-reported outcomes with baricitinib in adults with moderate-to-severe atopic dermatitis who were treated for up to 104 weeks in a randomized trial. J Dermatolog Treat. 2023;34(1):2190430 (Apr 5). Doi: 10.1080/09546634.2023.2190430
Key clinical point: Baricitinib offers the flexibility of dose down-titration and improvement in atopic dermatitis (AD) signs and symptoms up to treatment week 104 in patients with moderate-to-severe AD.
Major finding: Among patients receiving 4 mg baricitinib, 51.2% and 47.6% achieved or maintained a validated Investigator’s Global Assessment for AD score of 0 or 1 and 82.1%, and 73.8% maintained an Eczema Area and Severity Index 75 response at weeks 52 and 104, respectively. The clinical response was maintained after down-titration to 2 mg baricitinib.
Study details: This sub-study of the BREEZE-AD3 trial included 168 patients with moderate-to-severe AD who were responders or partial responders to 4 mg baricitinib in originating studies and were re-assigned (1:1) to receive 4 or 2 mg baricitinib at week 52.
Disclosures: This study was funded by Eli Lilly and Company under license from Incyte Corporation. Some authors reported ties with various organizations, including Eli Lilly. Four authors declared being employees and stockholders of Eli Lilly.
Source: Thyssen JP et al. Maintained improvement in physician- and patient-reported outcomes with baricitinib in adults with moderate-to-severe atopic dermatitis who were treated for up to 104 weeks in a randomized trial. J Dermatolog Treat. 2023;34(1):2190430 (Apr 5). Doi: 10.1080/09546634.2023.2190430
Key clinical point: Baricitinib offers the flexibility of dose down-titration and improvement in atopic dermatitis (AD) signs and symptoms up to treatment week 104 in patients with moderate-to-severe AD.
Major finding: Among patients receiving 4 mg baricitinib, 51.2% and 47.6% achieved or maintained a validated Investigator’s Global Assessment for AD score of 0 or 1 and 82.1%, and 73.8% maintained an Eczema Area and Severity Index 75 response at weeks 52 and 104, respectively. The clinical response was maintained after down-titration to 2 mg baricitinib.
Study details: This sub-study of the BREEZE-AD3 trial included 168 patients with moderate-to-severe AD who were responders or partial responders to 4 mg baricitinib in originating studies and were re-assigned (1:1) to receive 4 or 2 mg baricitinib at week 52.
Disclosures: This study was funded by Eli Lilly and Company under license from Incyte Corporation. Some authors reported ties with various organizations, including Eli Lilly. Four authors declared being employees and stockholders of Eli Lilly.
Source: Thyssen JP et al. Maintained improvement in physician- and patient-reported outcomes with baricitinib in adults with moderate-to-severe atopic dermatitis who were treated for up to 104 weeks in a randomized trial. J Dermatolog Treat. 2023;34(1):2190430 (Apr 5). Doi: 10.1080/09546634.2023.2190430
Baricitinib shows promise in pediatric patients with moderate-to-severe atopic dermatitis
Key clinical point: A dose of 4 mg baricitinib plus topical corticosteroids (TCS) demonstrated superior efficacy compared to placebo+TCS in pediatric patients with moderate-to-severe atopic dermatitis (AD), with the safety profile being consistent with previous studies involving adults with moderate-to-severe AD.
Major finding: At week 16, a significantly higher proportion of patients receiving 4 mg baricitinib+TCS vs placebo+TCS achieved a validated Investigator’s Global Assessment for AD score of 0 or 1 with a ≥2-point improvement (41.7% vs 16.4%; P < .0001); no significant difference was observed between the 2 mg/1 mg baricitinib and placebo groups. No new safety signals were reported.
Study details: This multicenter phase 3 study, BREEZE-AD-PEDS, included 483 pediatric patients aged 2 to <18 years with moderate-to-severe AD and inadequate response to TCS or systemic treatments who were randomly assigned to receive 4 mg baricitinib+TCS, 2 mg baricitinib+TCS, 1 mg baricitinib+TCS, or placebo+TCS.
Disclosures: Baricitinib was developed by Eli Lilly and Company, under license from Incyte Corporation. Some authors reported various ties, including employment and stock ownership, with Eli Lilly or others.
Source: Torrelo A et al. Efficacy and safety of baricitinib in combination with topical corticosteroids in pediatric patients with moderate-to-severe atopic dermatitis with inadequate response to topical corticosteroids: Results from a phase 3, randomized, double-blind, placebo-controlled study (BREEZE-AD PEDS). Br J Dermatol. 2023 (Mar 31). Doi: 10.1093/bjd/ljad096
Key clinical point: A dose of 4 mg baricitinib plus topical corticosteroids (TCS) demonstrated superior efficacy compared to placebo+TCS in pediatric patients with moderate-to-severe atopic dermatitis (AD), with the safety profile being consistent with previous studies involving adults with moderate-to-severe AD.
Major finding: At week 16, a significantly higher proportion of patients receiving 4 mg baricitinib+TCS vs placebo+TCS achieved a validated Investigator’s Global Assessment for AD score of 0 or 1 with a ≥2-point improvement (41.7% vs 16.4%; P < .0001); no significant difference was observed between the 2 mg/1 mg baricitinib and placebo groups. No new safety signals were reported.
Study details: This multicenter phase 3 study, BREEZE-AD-PEDS, included 483 pediatric patients aged 2 to <18 years with moderate-to-severe AD and inadequate response to TCS or systemic treatments who were randomly assigned to receive 4 mg baricitinib+TCS, 2 mg baricitinib+TCS, 1 mg baricitinib+TCS, or placebo+TCS.
Disclosures: Baricitinib was developed by Eli Lilly and Company, under license from Incyte Corporation. Some authors reported various ties, including employment and stock ownership, with Eli Lilly or others.
Source: Torrelo A et al. Efficacy and safety of baricitinib in combination with topical corticosteroids in pediatric patients with moderate-to-severe atopic dermatitis with inadequate response to topical corticosteroids: Results from a phase 3, randomized, double-blind, placebo-controlled study (BREEZE-AD PEDS). Br J Dermatol. 2023 (Mar 31). Doi: 10.1093/bjd/ljad096
Key clinical point: A dose of 4 mg baricitinib plus topical corticosteroids (TCS) demonstrated superior efficacy compared to placebo+TCS in pediatric patients with moderate-to-severe atopic dermatitis (AD), with the safety profile being consistent with previous studies involving adults with moderate-to-severe AD.
Major finding: At week 16, a significantly higher proportion of patients receiving 4 mg baricitinib+TCS vs placebo+TCS achieved a validated Investigator’s Global Assessment for AD score of 0 or 1 with a ≥2-point improvement (41.7% vs 16.4%; P < .0001); no significant difference was observed between the 2 mg/1 mg baricitinib and placebo groups. No new safety signals were reported.
Study details: This multicenter phase 3 study, BREEZE-AD-PEDS, included 483 pediatric patients aged 2 to <18 years with moderate-to-severe AD and inadequate response to TCS or systemic treatments who were randomly assigned to receive 4 mg baricitinib+TCS, 2 mg baricitinib+TCS, 1 mg baricitinib+TCS, or placebo+TCS.
Disclosures: Baricitinib was developed by Eli Lilly and Company, under license from Incyte Corporation. Some authors reported various ties, including employment and stock ownership, with Eli Lilly or others.
Source: Torrelo A et al. Efficacy and safety of baricitinib in combination with topical corticosteroids in pediatric patients with moderate-to-severe atopic dermatitis with inadequate response to topical corticosteroids: Results from a phase 3, randomized, double-blind, placebo-controlled study (BREEZE-AD PEDS). Br J Dermatol. 2023 (Mar 31). Doi: 10.1093/bjd/ljad096
Phase 3 trials support lebrikizumab for moderate-to-severe atopic dermatitis
Key clinical point: Lebrikizumab significantly improved the signs and symptoms of moderate-to-severe atopic dermatitis (AD) in adults and adolescents.
Major finding: At week 16, in the ADvocate1 and ADvocate2 trials, a significantly higher proportion of patients receiving lebrikizumab vs placebo achieved an Investigator’s Global Assessment score of 0 or 1 (43.1% vs 12.7% and 33.2% vs 10.8%, respectively) and a ≥75% improvement in the Eczema Area and Severity Index (58.8% vs 16.2% and 52.1% vs 18.1%, respectively; all P < .001). Most treatment-emergent adverse events were of mild-to-moderate severity.
Study details: Findings are from two identical phase 3 studies, ADvocate1 (n = 424) and ADvocate2 (n = 427), including adults (≥18 years) and adolescents (12 to <18 years) with moderate-to-severe AD who were randomly assigned to receive lebrikizumab or placebo over 16-week induction and 36-week maintenance periods.
Disclosures: This study was sponsored by Dermira, a wholly owned subsidiary of Eli Lilly and Company. Some authors reported various ties, including employment, with Eli Lilly or others.
Source: Silverberg JI et al for the ADvocate1 and ADvocate2 Investigators. Two phase 3 trials of lebrikizumab for moderate-to-severe atopic dermatitis. N Engl J Med. 2023;388(12):1080-1091 (Mar 15). Doi: 10.1056/NEJMoa2206714
Key clinical point: Lebrikizumab significantly improved the signs and symptoms of moderate-to-severe atopic dermatitis (AD) in adults and adolescents.
Major finding: At week 16, in the ADvocate1 and ADvocate2 trials, a significantly higher proportion of patients receiving lebrikizumab vs placebo achieved an Investigator’s Global Assessment score of 0 or 1 (43.1% vs 12.7% and 33.2% vs 10.8%, respectively) and a ≥75% improvement in the Eczema Area and Severity Index (58.8% vs 16.2% and 52.1% vs 18.1%, respectively; all P < .001). Most treatment-emergent adverse events were of mild-to-moderate severity.
Study details: Findings are from two identical phase 3 studies, ADvocate1 (n = 424) and ADvocate2 (n = 427), including adults (≥18 years) and adolescents (12 to <18 years) with moderate-to-severe AD who were randomly assigned to receive lebrikizumab or placebo over 16-week induction and 36-week maintenance periods.
Disclosures: This study was sponsored by Dermira, a wholly owned subsidiary of Eli Lilly and Company. Some authors reported various ties, including employment, with Eli Lilly or others.
Source: Silverberg JI et al for the ADvocate1 and ADvocate2 Investigators. Two phase 3 trials of lebrikizumab for moderate-to-severe atopic dermatitis. N Engl J Med. 2023;388(12):1080-1091 (Mar 15). Doi: 10.1056/NEJMoa2206714
Key clinical point: Lebrikizumab significantly improved the signs and symptoms of moderate-to-severe atopic dermatitis (AD) in adults and adolescents.
Major finding: At week 16, in the ADvocate1 and ADvocate2 trials, a significantly higher proportion of patients receiving lebrikizumab vs placebo achieved an Investigator’s Global Assessment score of 0 or 1 (43.1% vs 12.7% and 33.2% vs 10.8%, respectively) and a ≥75% improvement in the Eczema Area and Severity Index (58.8% vs 16.2% and 52.1% vs 18.1%, respectively; all P < .001). Most treatment-emergent adverse events were of mild-to-moderate severity.
Study details: Findings are from two identical phase 3 studies, ADvocate1 (n = 424) and ADvocate2 (n = 427), including adults (≥18 years) and adolescents (12 to <18 years) with moderate-to-severe AD who were randomly assigned to receive lebrikizumab or placebo over 16-week induction and 36-week maintenance periods.
Disclosures: This study was sponsored by Dermira, a wholly owned subsidiary of Eli Lilly and Company. Some authors reported various ties, including employment, with Eli Lilly or others.
Source: Silverberg JI et al for the ADvocate1 and ADvocate2 Investigators. Two phase 3 trials of lebrikizumab for moderate-to-severe atopic dermatitis. N Engl J Med. 2023;388(12):1080-1091 (Mar 15). Doi: 10.1056/NEJMoa2206714
What are the clinical implications of recent skin dysbiosis discoveries?
NEW ORLEANS – .
“There’s still a lot for us to learn,” Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, said at the annual meeting of the American Academy of Dermatology. “Multiple factors contribute to the variability in the skin microbiota, including age, sex, environment, immune system, host genotype, lifestyle, and pathobiology. The question becomes, when do these factors or impacts on the microbiota become clinically significant?”
According to Dr. Friedman, there are 10 times more bacteria cells than human cells in the human body, “but it’s not a fight to the finish; it’s not us versus them,” he said. “Together, we are a super organism.” There are also more than 500 species of bacteria on human skin excluding viruses and fungi, and each person carries up to 5 pounds of bacteria, which is akin to finding a new organ in the body.
“What’s so unique is that we each have our own bacterial fingerprint,” he said. “Whoever is sitting next to you? Their microbiota makeup is different than yours.”
Beyond genetics and environment, activities that can contribute to alterations in skin flora or skin dysbiosis include topical application of steroids, antibiotics, retinoids, harsh soaps, chemical and physical exfoliants, and resurfacing techniques. “With anything we apply or do to the skin, we are literally changing the home of many microorganisms, for good or bad,” he said.
In the realm of atopic dermatitis (AD), Staphylococcus aureus has been implicated as an offender in the pathophysiology of the disease. “It’s not about one single species of Staphylococcus, though,” said Dr. Friedman, who also is director of translational research at George Washington University. “We’re finding out that, depending on the severity of disease, Staph. epidermis may be part of the problem as opposed to it just being about Staph. aureus. Furthermore, and more importantly, these changes in the microbiota, specifically a decrease in microbial diversity, has been shown to precede a disease flare, highlighting the central role of maintaining microbial diversity and by definition, supporting the living barrier in our management of AD.”
With this in mind, researchers in one study used high-throughput sequencing to evaluate the microbial communities associated with affected and unaffected skin of 49 patients with AD before and after emollient treatment. Following 84 days of emollient application, clinical symptoms of AD improved in 72% of the study population and Stenotrophomonas species were significantly more abundant among responders.
Prebiotics, probiotics
“Our treatments certainly can positively impact the microbiota, as we have seen even recently with some of our new targeted therapies, but we can also directly provide support,” he continued. Prebiotics, which he defined as supplements or foods that contain a nondigestible ingredient that selectively stimulates the growth and/or activity of indigenous bacteria, can be found in many over-the-counter moisturizers.

For example, colloidal oatmeal has been found to support the growth of S. epidermidis and enhance the production of lactic acid. “We really don’t know much about what these induced changes mean from a clinical perspective; that has yet to be elucidated,” Dr. Friedman said.
In light of the recent attention to the early application of moisturizers in infants at high risk of developing AD in an effort to prevent or limit AD, “maybe part of this has to do with applying something that’s nurturing an evolving microbiota,” Dr. Friedman noted. “It’s something to think about.”
Yet another area of study involves the use of probiotics, which Dr. Friedman defined as supplements or foods that contain viable microorganisms that alter the microflora of the host. In a first-of-its-kind trial, researchers evaluated the safety and efficacy of self-administered topical Roseomonas mucosa in 10 adults and 5 children with AD. No adverse events or treatment complications were observed, and the topical R. mucosa was associated with significant decreases in measures of disease severity, topical steroid requirement, and S. aureus burden
In a more recent randomized trial of 11 patients with AD, Richard L. Gallo, MD, PhD, chair of dermatology, University of California, San Diego, and colleagues found that application of a personalized topical cream formulated from coagulase-negative Staphylococcus with antimicrobial activity against S. aureus reduced colonization of S. aureus and improved disease severity.
And in another randomized, controlled trial, Italian researchers enrolled 80 adults with mild to severe AD to receive a placebo or a supplement that was a mixture of lactobacilli for 56 days. They found that adults in the treatment arm showed an improvement in skin smoothness, skin moisturization, self-perception, and a decrease in the SCORing Atopic Dermatitis (SCORAD) index as well as in levels of inflammatory markers associated with AD.
Dr. Friedman also discussed postbiotics, nonviable bacterial products or metabolic byproducts from probiotic microorganisms that have biologic activity in the host. In one trial, French researchers enrolled 75 people with AD who ranged in age from 6 to 70 years to receive a cream containing a 5% lysate of the nonpathogenic bacteria Vitreoscilla filiformis, or a vehicle cream for 30 days. They found that compared with the vehicle, V. filiformis lysate significantly decreased SCORAD levels and pruritus; active cream was shown to significantly decrease loss of sleep from day 0 to day 29.
Dr. Friedman characterized these novel approaches to AD as “an exciting area, one we need to pay attention to. But what I really want to know is, aside from these purposefully made and marketed products that have pre- and postprobiotics, is there a difference with some of the products we use already? My assumption is that there is, but we need to see that data.”
Dr. Friedman disclosed that he is a consultant and/or advisory board member for Medscape/SanovaWorks, Oakstone Institute, L’Oréal, La Roche Posay, Galderma, Aveeno, Ortho Dermatologic, Microcures, Pfizer, Novartis, Lilly, Hoth Therapeutics, Zylo Therapeutics, BMS, Vial, Janssen, Novocure, Dermavant, Regeneron/Sanofi, and Incyte. He has also received grants from Pfizer, the Dermatology Foundation, Lilly, Janssen, Incyte, and Galderma.
NEW ORLEANS – .
“There’s still a lot for us to learn,” Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, said at the annual meeting of the American Academy of Dermatology. “Multiple factors contribute to the variability in the skin microbiota, including age, sex, environment, immune system, host genotype, lifestyle, and pathobiology. The question becomes, when do these factors or impacts on the microbiota become clinically significant?”
According to Dr. Friedman, there are 10 times more bacteria cells than human cells in the human body, “but it’s not a fight to the finish; it’s not us versus them,” he said. “Together, we are a super organism.” There are also more than 500 species of bacteria on human skin excluding viruses and fungi, and each person carries up to 5 pounds of bacteria, which is akin to finding a new organ in the body.
“What’s so unique is that we each have our own bacterial fingerprint,” he said. “Whoever is sitting next to you? Their microbiota makeup is different than yours.”
Beyond genetics and environment, activities that can contribute to alterations in skin flora or skin dysbiosis include topical application of steroids, antibiotics, retinoids, harsh soaps, chemical and physical exfoliants, and resurfacing techniques. “With anything we apply or do to the skin, we are literally changing the home of many microorganisms, for good or bad,” he said.
In the realm of atopic dermatitis (AD), Staphylococcus aureus has been implicated as an offender in the pathophysiology of the disease. “It’s not about one single species of Staphylococcus, though,” said Dr. Friedman, who also is director of translational research at George Washington University. “We’re finding out that, depending on the severity of disease, Staph. epidermis may be part of the problem as opposed to it just being about Staph. aureus. Furthermore, and more importantly, these changes in the microbiota, specifically a decrease in microbial diversity, has been shown to precede a disease flare, highlighting the central role of maintaining microbial diversity and by definition, supporting the living barrier in our management of AD.”
With this in mind, researchers in one study used high-throughput sequencing to evaluate the microbial communities associated with affected and unaffected skin of 49 patients with AD before and after emollient treatment. Following 84 days of emollient application, clinical symptoms of AD improved in 72% of the study population and Stenotrophomonas species were significantly more abundant among responders.
Prebiotics, probiotics
“Our treatments certainly can positively impact the microbiota, as we have seen even recently with some of our new targeted therapies, but we can also directly provide support,” he continued. Prebiotics, which he defined as supplements or foods that contain a nondigestible ingredient that selectively stimulates the growth and/or activity of indigenous bacteria, can be found in many over-the-counter moisturizers.
For example, colloidal oatmeal has been found to support the growth of S. epidermidis and enhance the production of lactic acid. “We really don’t know much about what these induced changes mean from a clinical perspective; that has yet to be elucidated,” Dr. Friedman said.
In light of the recent attention to the early application of moisturizers in infants at high risk of developing AD in an effort to prevent or limit AD, “maybe part of this has to do with applying something that’s nurturing an evolving microbiota,” Dr. Friedman noted. “It’s something to think about.”
Yet another area of study involves the use of probiotics, which Dr. Friedman defined as supplements or foods that contain viable microorganisms that alter the microflora of the host. In a first-of-its-kind trial, researchers evaluated the safety and efficacy of self-administered topical Roseomonas mucosa in 10 adults and 5 children with AD. No adverse events or treatment complications were observed, and the topical R. mucosa was associated with significant decreases in measures of disease severity, topical steroid requirement, and S. aureus burden
In a more recent randomized trial of 11 patients with AD, Richard L. Gallo, MD, PhD, chair of dermatology, University of California, San Diego, and colleagues found that application of a personalized topical cream formulated from coagulase-negative Staphylococcus with antimicrobial activity against S. aureus reduced colonization of S. aureus and improved disease severity.
And in another randomized, controlled trial, Italian researchers enrolled 80 adults with mild to severe AD to receive a placebo or a supplement that was a mixture of lactobacilli for 56 days. They found that adults in the treatment arm showed an improvement in skin smoothness, skin moisturization, self-perception, and a decrease in the SCORing Atopic Dermatitis (SCORAD) index as well as in levels of inflammatory markers associated with AD.
Dr. Friedman also discussed postbiotics, nonviable bacterial products or metabolic byproducts from probiotic microorganisms that have biologic activity in the host. In one trial, French researchers enrolled 75 people with AD who ranged in age from 6 to 70 years to receive a cream containing a 5% lysate of the nonpathogenic bacteria Vitreoscilla filiformis, or a vehicle cream for 30 days. They found that compared with the vehicle, V. filiformis lysate significantly decreased SCORAD levels and pruritus; active cream was shown to significantly decrease loss of sleep from day 0 to day 29.
Dr. Friedman characterized these novel approaches to AD as “an exciting area, one we need to pay attention to. But what I really want to know is, aside from these purposefully made and marketed products that have pre- and postprobiotics, is there a difference with some of the products we use already? My assumption is that there is, but we need to see that data.”
Dr. Friedman disclosed that he is a consultant and/or advisory board member for Medscape/SanovaWorks, Oakstone Institute, L’Oréal, La Roche Posay, Galderma, Aveeno, Ortho Dermatologic, Microcures, Pfizer, Novartis, Lilly, Hoth Therapeutics, Zylo Therapeutics, BMS, Vial, Janssen, Novocure, Dermavant, Regeneron/Sanofi, and Incyte. He has also received grants from Pfizer, the Dermatology Foundation, Lilly, Janssen, Incyte, and Galderma.
NEW ORLEANS – .
“There’s still a lot for us to learn,” Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, said at the annual meeting of the American Academy of Dermatology. “Multiple factors contribute to the variability in the skin microbiota, including age, sex, environment, immune system, host genotype, lifestyle, and pathobiology. The question becomes, when do these factors or impacts on the microbiota become clinically significant?”
According to Dr. Friedman, there are 10 times more bacteria cells than human cells in the human body, “but it’s not a fight to the finish; it’s not us versus them,” he said. “Together, we are a super organism.” There are also more than 500 species of bacteria on human skin excluding viruses and fungi, and each person carries up to 5 pounds of bacteria, which is akin to finding a new organ in the body.
“What’s so unique is that we each have our own bacterial fingerprint,” he said. “Whoever is sitting next to you? Their microbiota makeup is different than yours.”
Beyond genetics and environment, activities that can contribute to alterations in skin flora or skin dysbiosis include topical application of steroids, antibiotics, retinoids, harsh soaps, chemical and physical exfoliants, and resurfacing techniques. “With anything we apply or do to the skin, we are literally changing the home of many microorganisms, for good or bad,” he said.
In the realm of atopic dermatitis (AD), Staphylococcus aureus has been implicated as an offender in the pathophysiology of the disease. “It’s not about one single species of Staphylococcus, though,” said Dr. Friedman, who also is director of translational research at George Washington University. “We’re finding out that, depending on the severity of disease, Staph. epidermis may be part of the problem as opposed to it just being about Staph. aureus. Furthermore, and more importantly, these changes in the microbiota, specifically a decrease in microbial diversity, has been shown to precede a disease flare, highlighting the central role of maintaining microbial diversity and by definition, supporting the living barrier in our management of AD.”
With this in mind, researchers in one study used high-throughput sequencing to evaluate the microbial communities associated with affected and unaffected skin of 49 patients with AD before and after emollient treatment. Following 84 days of emollient application, clinical symptoms of AD improved in 72% of the study population and Stenotrophomonas species were significantly more abundant among responders.
Prebiotics, probiotics
“Our treatments certainly can positively impact the microbiota, as we have seen even recently with some of our new targeted therapies, but we can also directly provide support,” he continued. Prebiotics, which he defined as supplements or foods that contain a nondigestible ingredient that selectively stimulates the growth and/or activity of indigenous bacteria, can be found in many over-the-counter moisturizers.
For example, colloidal oatmeal has been found to support the growth of S. epidermidis and enhance the production of lactic acid. “We really don’t know much about what these induced changes mean from a clinical perspective; that has yet to be elucidated,” Dr. Friedman said.
In light of the recent attention to the early application of moisturizers in infants at high risk of developing AD in an effort to prevent or limit AD, “maybe part of this has to do with applying something that’s nurturing an evolving microbiota,” Dr. Friedman noted. “It’s something to think about.”
Yet another area of study involves the use of probiotics, which Dr. Friedman defined as supplements or foods that contain viable microorganisms that alter the microflora of the host. In a first-of-its-kind trial, researchers evaluated the safety and efficacy of self-administered topical Roseomonas mucosa in 10 adults and 5 children with AD. No adverse events or treatment complications were observed, and the topical R. mucosa was associated with significant decreases in measures of disease severity, topical steroid requirement, and S. aureus burden
In a more recent randomized trial of 11 patients with AD, Richard L. Gallo, MD, PhD, chair of dermatology, University of California, San Diego, and colleagues found that application of a personalized topical cream formulated from coagulase-negative Staphylococcus with antimicrobial activity against S. aureus reduced colonization of S. aureus and improved disease severity.
And in another randomized, controlled trial, Italian researchers enrolled 80 adults with mild to severe AD to receive a placebo or a supplement that was a mixture of lactobacilli for 56 days. They found that adults in the treatment arm showed an improvement in skin smoothness, skin moisturization, self-perception, and a decrease in the SCORing Atopic Dermatitis (SCORAD) index as well as in levels of inflammatory markers associated with AD.
Dr. Friedman also discussed postbiotics, nonviable bacterial products or metabolic byproducts from probiotic microorganisms that have biologic activity in the host. In one trial, French researchers enrolled 75 people with AD who ranged in age from 6 to 70 years to receive a cream containing a 5% lysate of the nonpathogenic bacteria Vitreoscilla filiformis, or a vehicle cream for 30 days. They found that compared with the vehicle, V. filiformis lysate significantly decreased SCORAD levels and pruritus; active cream was shown to significantly decrease loss of sleep from day 0 to day 29.
Dr. Friedman characterized these novel approaches to AD as “an exciting area, one we need to pay attention to. But what I really want to know is, aside from these purposefully made and marketed products that have pre- and postprobiotics, is there a difference with some of the products we use already? My assumption is that there is, but we need to see that data.”
Dr. Friedman disclosed that he is a consultant and/or advisory board member for Medscape/SanovaWorks, Oakstone Institute, L’Oréal, La Roche Posay, Galderma, Aveeno, Ortho Dermatologic, Microcures, Pfizer, Novartis, Lilly, Hoth Therapeutics, Zylo Therapeutics, BMS, Vial, Janssen, Novocure, Dermavant, Regeneron/Sanofi, and Incyte. He has also received grants from Pfizer, the Dermatology Foundation, Lilly, Janssen, Incyte, and Galderma.
AT AAD 2023

Commentary: IL-31 inhibitor, e-cigarettes, and upadacitinib in AD, April 2023

Good news! There's not a lot to say about this. Dupilumab is so easy. No blood work, no immunosuppression. Dupilumab is highly effective and very safe. It's safe enough for children as young as 6 months! It's so effective that if it is not working, I question my diagnosis (Could it be contact dermatitis or mycosis fungoides instead?) and whether the patient is taking the medication properly.
Boesjes and colleagues describe in Acta Dermato-Venereologica the Dutch experience with upadacitinib in patients who have not been successfully treated with dupilumab or baricitinib. Presumably, such patients, because treatment with dupilumab or baricitinib or both was unsuccessful, have very resistant atopic dermatitis (either due to strong genetic propensity or perhaps because they don't take their medications). Despite having such refractory disease, most patients did well on the treatment with rapid disease improvement. Upadacitinib didn't work for everyone, though. About 30% of the patients discontinued upadacitinib treatment due to ineffectiveness, adverse events, or both (8.5%, 14.9%, and 6.4%, respectively).
How much of that ineffectiveness was due to poor adherence to taking the treatment was not assessed. Upadacitinib is extraordinarily effective for atopic dermatitis. I didn't think I would ever see a drug more effective than dupilumab for atopic dermatitis, but a low dose of upadacitinib (15 mg/day) seems about twice as effective as dupilumab for complete clearing of atopic dermatitis. The higher dose of 30 mg may be 3.5 times as effective as dupiliumab at getting atopic dermatitis completely clear.1
I dislike the word significant. Significant is ambiguous. It could mean that an observed association would not be likely to occur by chance, or it could mean that an observed association is clinically meaningful. Smith and colleagues in "Association between electronic cigarette use and atopic dermatitis among United States adults" reported finding a "significant" association between e-cigarette use and atopic dermatitis. A total of 23% of 2119 e-cigarette users had atopic dermatitis vs 17.1% of 26,444 nonusers. Clearly, the observed association was statistically significant (the 6% difference was not likely to occur due to chance alone). Is the finding clinically meaningful? I don't think it would affect our practice in any way.
The authors made the point that the study doesn't tell us whether e-cigarette use causes atopic dermatitis or if atopic dermatitis causes people to smoke. I wonder if just being younger (or some other factor) might make people more likely to use e-cigarettes and more likely to have atopic dermatitis (assuming atopic dermatitis gradually subsides over time, a dogma that may not be true).
Kabashima and colleagues report on the efficacy of the interleukin (IL)–31 antagonist nemolizumab. IL-31 mediates itch and having a new drug to block IL-31 may be a great treatment for our itchy patients. In this study, patients who had greater itch reduction had greater improvement in eczema and in quality of life. I'm quite sure that reducing itch improves patients' quality of life. But when it comes to the itch and the inflammation, I'm not sure which comes first. Does controlling the itch make the inflammation better? Maybe. Does controlling inflammation make itch better? Certainly.
For atopic patients with inflammation, controlling that inflammation seems to me to be the best approach, and we don't need more new treatments to accomplish that. For those patients who have a lot of itch and little inflammation, an IL-31 antagonist may be a revolutionary addition to our treatment options.
Additional References
1. Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157:1047-1055. doi: 10.1001/jamadermatol.2021.3023. Erratum in: JAMA Dermatol. 2022;158:219. doi: 10.1001/jamadermatol.2021.5451
Good news! There's not a lot to say about this. Dupilumab is so easy. No blood work, no immunosuppression. Dupilumab is highly effective and very safe. It's safe enough for children as young as 6 months! It's so effective that if it is not working, I question my diagnosis (Could it be contact dermatitis or mycosis fungoides instead?) and whether the patient is taking the medication properly.
Boesjes and colleagues describe in Acta Dermato-Venereologica the Dutch experience with upadacitinib in patients who have not been successfully treated with dupilumab or baricitinib. Presumably, such patients, because treatment with dupilumab or baricitinib or both was unsuccessful, have very resistant atopic dermatitis (either due to strong genetic propensity or perhaps because they don't take their medications). Despite having such refractory disease, most patients did well on the treatment with rapid disease improvement. Upadacitinib didn't work for everyone, though. About 30% of the patients discontinued upadacitinib treatment due to ineffectiveness, adverse events, or both (8.5%, 14.9%, and 6.4%, respectively).
How much of that ineffectiveness was due to poor adherence to taking the treatment was not assessed. Upadacitinib is extraordinarily effective for atopic dermatitis. I didn't think I would ever see a drug more effective than dupilumab for atopic dermatitis, but a low dose of upadacitinib (15 mg/day) seems about twice as effective as dupilumab for complete clearing of atopic dermatitis. The higher dose of 30 mg may be 3.5 times as effective as dupiliumab at getting atopic dermatitis completely clear.1
I dislike the word significant. Significant is ambiguous. It could mean that an observed association would not be likely to occur by chance, or it could mean that an observed association is clinically meaningful. Smith and colleagues in "Association between electronic cigarette use and atopic dermatitis among United States adults" reported finding a "significant" association between e-cigarette use and atopic dermatitis. A total of 23% of 2119 e-cigarette users had atopic dermatitis vs 17.1% of 26,444 nonusers. Clearly, the observed association was statistically significant (the 6% difference was not likely to occur due to chance alone). Is the finding clinically meaningful? I don't think it would affect our practice in any way.
The authors made the point that the study doesn't tell us whether e-cigarette use causes atopic dermatitis or if atopic dermatitis causes people to smoke. I wonder if just being younger (or some other factor) might make people more likely to use e-cigarettes and more likely to have atopic dermatitis (assuming atopic dermatitis gradually subsides over time, a dogma that may not be true).
Kabashima and colleagues report on the efficacy of the interleukin (IL)–31 antagonist nemolizumab. IL-31 mediates itch and having a new drug to block IL-31 may be a great treatment for our itchy patients. In this study, patients who had greater itch reduction had greater improvement in eczema and in quality of life. I'm quite sure that reducing itch improves patients' quality of life. But when it comes to the itch and the inflammation, I'm not sure which comes first. Does controlling the itch make the inflammation better? Maybe. Does controlling inflammation make itch better? Certainly.
For atopic patients with inflammation, controlling that inflammation seems to me to be the best approach, and we don't need more new treatments to accomplish that. For those patients who have a lot of itch and little inflammation, an IL-31 antagonist may be a revolutionary addition to our treatment options.
Additional References
1. Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157:1047-1055. doi: 10.1001/jamadermatol.2021.3023. Erratum in: JAMA Dermatol. 2022;158:219. doi: 10.1001/jamadermatol.2021.5451
Good news! There's not a lot to say about this. Dupilumab is so easy. No blood work, no immunosuppression. Dupilumab is highly effective and very safe. It's safe enough for children as young as 6 months! It's so effective that if it is not working, I question my diagnosis (Could it be contact dermatitis or mycosis fungoides instead?) and whether the patient is taking the medication properly.
Boesjes and colleagues describe in Acta Dermato-Venereologica the Dutch experience with upadacitinib in patients who have not been successfully treated with dupilumab or baricitinib. Presumably, such patients, because treatment with dupilumab or baricitinib or both was unsuccessful, have very resistant atopic dermatitis (either due to strong genetic propensity or perhaps because they don't take their medications). Despite having such refractory disease, most patients did well on the treatment with rapid disease improvement. Upadacitinib didn't work for everyone, though. About 30% of the patients discontinued upadacitinib treatment due to ineffectiveness, adverse events, or both (8.5%, 14.9%, and 6.4%, respectively).
How much of that ineffectiveness was due to poor adherence to taking the treatment was not assessed. Upadacitinib is extraordinarily effective for atopic dermatitis. I didn't think I would ever see a drug more effective than dupilumab for atopic dermatitis, but a low dose of upadacitinib (15 mg/day) seems about twice as effective as dupilumab for complete clearing of atopic dermatitis. The higher dose of 30 mg may be 3.5 times as effective as dupiliumab at getting atopic dermatitis completely clear.1
I dislike the word significant. Significant is ambiguous. It could mean that an observed association would not be likely to occur by chance, or it could mean that an observed association is clinically meaningful. Smith and colleagues in "Association between electronic cigarette use and atopic dermatitis among United States adults" reported finding a "significant" association between e-cigarette use and atopic dermatitis. A total of 23% of 2119 e-cigarette users had atopic dermatitis vs 17.1% of 26,444 nonusers. Clearly, the observed association was statistically significant (the 6% difference was not likely to occur due to chance alone). Is the finding clinically meaningful? I don't think it would affect our practice in any way.
The authors made the point that the study doesn't tell us whether e-cigarette use causes atopic dermatitis or if atopic dermatitis causes people to smoke. I wonder if just being younger (or some other factor) might make people more likely to use e-cigarettes and more likely to have atopic dermatitis (assuming atopic dermatitis gradually subsides over time, a dogma that may not be true).
Kabashima and colleagues report on the efficacy of the interleukin (IL)–31 antagonist nemolizumab. IL-31 mediates itch and having a new drug to block IL-31 may be a great treatment for our itchy patients. In this study, patients who had greater itch reduction had greater improvement in eczema and in quality of life. I'm quite sure that reducing itch improves patients' quality of life. But when it comes to the itch and the inflammation, I'm not sure which comes first. Does controlling the itch make the inflammation better? Maybe. Does controlling inflammation make itch better? Certainly.
For atopic patients with inflammation, controlling that inflammation seems to me to be the best approach, and we don't need more new treatments to accomplish that. For those patients who have a lot of itch and little inflammation, an IL-31 antagonist may be a revolutionary addition to our treatment options.
Additional References
1. Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157:1047-1055. doi: 10.1001/jamadermatol.2021.3023. Erratum in: JAMA Dermatol. 2022;158:219. doi: 10.1001/jamadermatol.2021.5451
JAK inhibitor ivarmacitinib shows efficacy for atopic dermatitis in a pivotal trial
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.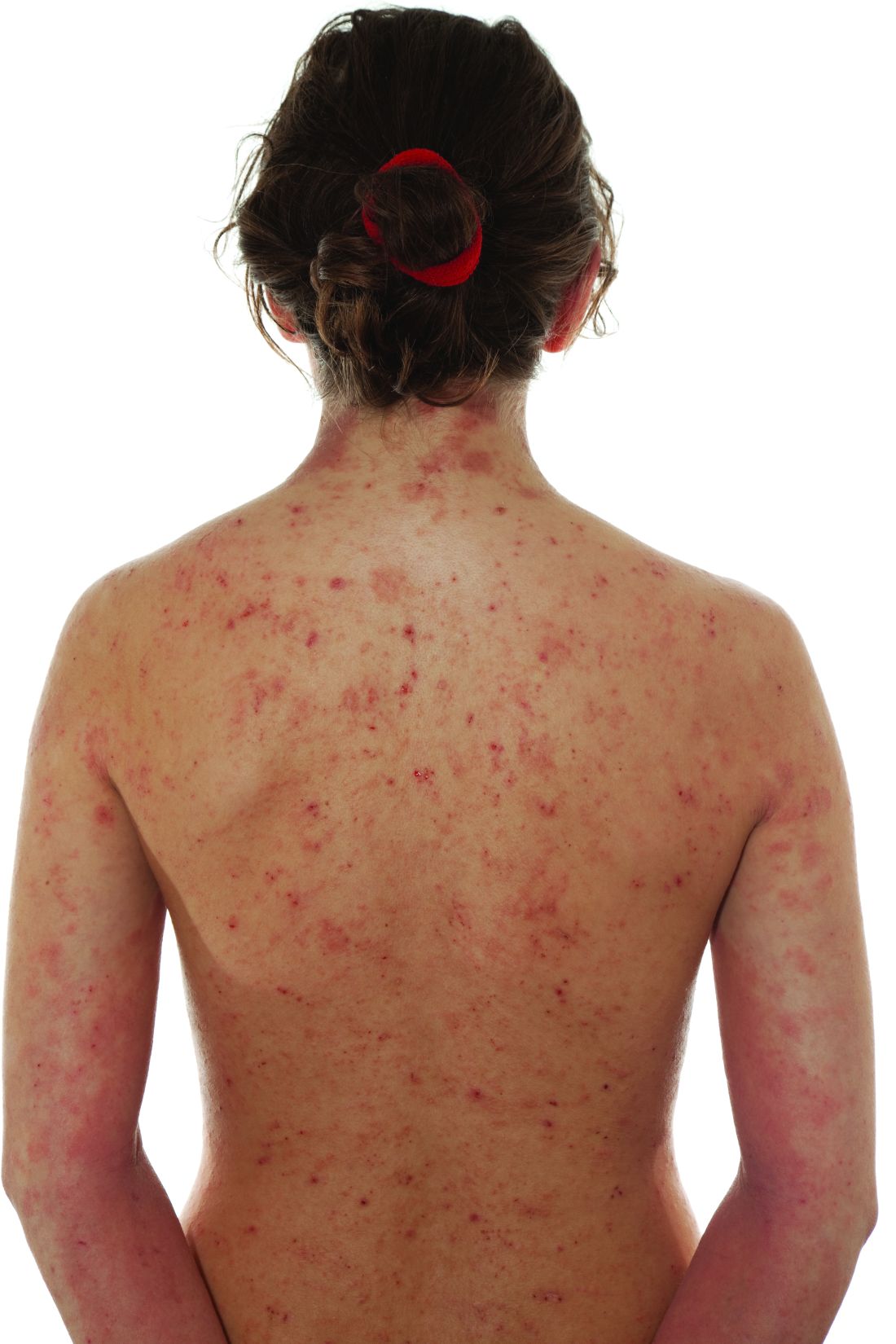
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
AT AAD 2023
Topical delgocitinib shows promise for chronic hand eczema, pivotal trial shows
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.

“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.
“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.
“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
AT AAD 2023
Air pollution may be causing eczema
The finding points scientists toward how to better treat the skin ailment. There are now more than three times as many eczema cases as there were in the 1970s, and it now affects as many as 20% of children and 10% of adults.
“I think these authors are spot-on in recognizing that the incidence of allergic conditions is increasing concurrently with how different pollutants are increasing in our environment,” said Denver-based pediatric allergist and immunologist Jessica Hui, MD, according to NBC News. “We’re finally understanding more about why people are getting eczema.”
Some people get eczema because of genetics, but the new research built on the previous understanding of how chemicals called diisocyanates can trigger the eczema symptoms of severe itching, skin redness, and oozing or painful rashes. An experiment on mice showed that exposure to a specific part of diisocyanates, called isocyanates, disrupted oil production that the skin needs to stay healthy.
Researchers at the National Institutes of Health “found that when bacteria that live on healthy skin are exposed to isocyanate, they must adapt to survive,” the agency summarized in a news release. “When they adapt, these bacteria shift their metabolism away from making the lipids, or oils, that skin needs to stay healthy. This finding suggests that eczema may be treatable by replacing the modified skin bacteria with healthy bacteria.”
The study was published in the journal Science Advances.
The chemicals also trigger a message to the brain that causes skin inflammation and itching, lead researcher Ian Myles, MD, told NBC News. Dr. Myles is also chief of the Epithelial Research Unit in the National Institute of Allergy and Infectious Diseases Laboratory of Clinical Immunology and Microbiology.
“So much of this is out of our control. I mean, you can’t shut the highways down,” he said of the environmental sources.
Previous research that explored attempting to restore healthy skin bacteria called Roseomonas mucosa to treat eczema symptoms had mixed results. The NIH says it has made the bacteria available “for commercial, nontherapeutic development ... as a potentially beneficial probiotic.”
A version of this article first appeared on WebMD.com.
The finding points scientists toward how to better treat the skin ailment. There are now more than three times as many eczema cases as there were in the 1970s, and it now affects as many as 20% of children and 10% of adults.
“I think these authors are spot-on in recognizing that the incidence of allergic conditions is increasing concurrently with how different pollutants are increasing in our environment,” said Denver-based pediatric allergist and immunologist Jessica Hui, MD, according to NBC News. “We’re finally understanding more about why people are getting eczema.”
Some people get eczema because of genetics, but the new research built on the previous understanding of how chemicals called diisocyanates can trigger the eczema symptoms of severe itching, skin redness, and oozing or painful rashes. An experiment on mice showed that exposure to a specific part of diisocyanates, called isocyanates, disrupted oil production that the skin needs to stay healthy.
Researchers at the National Institutes of Health “found that when bacteria that live on healthy skin are exposed to isocyanate, they must adapt to survive,” the agency summarized in a news release. “When they adapt, these bacteria shift their metabolism away from making the lipids, or oils, that skin needs to stay healthy. This finding suggests that eczema may be treatable by replacing the modified skin bacteria with healthy bacteria.”
The study was published in the journal Science Advances.
The chemicals also trigger a message to the brain that causes skin inflammation and itching, lead researcher Ian Myles, MD, told NBC News. Dr. Myles is also chief of the Epithelial Research Unit in the National Institute of Allergy and Infectious Diseases Laboratory of Clinical Immunology and Microbiology.
“So much of this is out of our control. I mean, you can’t shut the highways down,” he said of the environmental sources.
Previous research that explored attempting to restore healthy skin bacteria called Roseomonas mucosa to treat eczema symptoms had mixed results. The NIH says it has made the bacteria available “for commercial, nontherapeutic development ... as a potentially beneficial probiotic.”
A version of this article first appeared on WebMD.com.
The finding points scientists toward how to better treat the skin ailment. There are now more than three times as many eczema cases as there were in the 1970s, and it now affects as many as 20% of children and 10% of adults.
“I think these authors are spot-on in recognizing that the incidence of allergic conditions is increasing concurrently with how different pollutants are increasing in our environment,” said Denver-based pediatric allergist and immunologist Jessica Hui, MD, according to NBC News. “We’re finally understanding more about why people are getting eczema.”
Some people get eczema because of genetics, but the new research built on the previous understanding of how chemicals called diisocyanates can trigger the eczema symptoms of severe itching, skin redness, and oozing or painful rashes. An experiment on mice showed that exposure to a specific part of diisocyanates, called isocyanates, disrupted oil production that the skin needs to stay healthy.
Researchers at the National Institutes of Health “found that when bacteria that live on healthy skin are exposed to isocyanate, they must adapt to survive,” the agency summarized in a news release. “When they adapt, these bacteria shift their metabolism away from making the lipids, or oils, that skin needs to stay healthy. This finding suggests that eczema may be treatable by replacing the modified skin bacteria with healthy bacteria.”
The study was published in the journal Science Advances.
The chemicals also trigger a message to the brain that causes skin inflammation and itching, lead researcher Ian Myles, MD, told NBC News. Dr. Myles is also chief of the Epithelial Research Unit in the National Institute of Allergy and Infectious Diseases Laboratory of Clinical Immunology and Microbiology.
“So much of this is out of our control. I mean, you can’t shut the highways down,” he said of the environmental sources.
Previous research that explored attempting to restore healthy skin bacteria called Roseomonas mucosa to treat eczema symptoms had mixed results. The NIH says it has made the bacteria available “for commercial, nontherapeutic development ... as a potentially beneficial probiotic.”
A version of this article first appeared on WebMD.com.
FROM SCIENCE ADVANCES
New data forecast more oral PDE4 inhibitors for psoriasis
NEW ORLEANS – according to results of a phase 2 clinical trial presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
The phase 2b data, which are prompting a phase 3 trial, suggest that the drug, called orismilast, “is a potential new addition to the psoriasis armamentarium,” reported Lars E. French, MD, professor and chair, department of dermatology, Ludwig Maximilian University of Munich (Germany).

At the same session, findings from another study supported off-label use of oral roflumilast (Daliresp and generic), a PDE4 inhibitor approved for severe chronic obstructive pulmonary disease (COPD). The only PDE4 inhibitors with an indication for psoriasis are roflumilast, approved as a cream (Zoryve), and apremilast (Otezla), approved as an oral therapy.
Phase 2 study of orismilast
In the orismilast trial, Dr. French attributed the efficacy observed to the potency of orismilast on the B and D subtypes of PDE4 associated with inflammation. One clue is that these specific subtypes are overly expressed in the skin of patients with either psoriasis or atopic dermatitis.
“When compared to apremilast, orismilast is at least two to fivefold more potent on all PDE4 isoforms and up to 39 times more potent on some of the PDE4 B and D isoforms,” said Dr. French, referring to preclinical findings in human whole blood and blood cells and in a mouse model of chronic inflammation.
The efficacy of orismilast in an immediate-release oral formulation was previously demonstrated in a recently published phase 2a trial, but the newest study tested a modified-release formulation of orismilast to test its potential to improve tolerability.
In the study, 202 adult patients with moderate to severe psoriasis (Psoriasis Area Severity Index [PASI] score ≥ 12) were randomly assigned to one of three doses of orismilast or to placebo. Each of the three doses – 20 mg, 30 mg, or 40 mg – were administered twice daily. The primary endpoint was change in PASI score at 16 weeks. Secondary endpoints included PASI 75 responses (signifying 75% clearance) and safety.
Relative to placebo, which was associated with a PASI improvement of 17%, all three of the tested orismilast doses were superior in a dose-dependent manner. The rates of response were 53%, 61%, and 64% for the 20-mg, 30-mg, and 40-mg twice-daily doses, respectively.
The PASI improvements were rapid, Dr. French said. At 4 weeks, PASI scores climbed from baseline by nearly 40% for those on all orismilast doses, which was more than double the improvement in the placebo group.
In the intention-to-treat analysis with missing data counted as nonresponders, the proportion of patients reaching PASI-75 scores at 16 weeks were 39%, 49%, 45%, and 17%, in the 20-mg, 30-mg, 40-mg, and placebo groups, respectively. The proportion of patients experiencing complete or near-complete skin clearance defined by a PASI 90 were 24%, 22%, 28%, and 8%, respectively.
The side-effect profile was consistent with other PDE4 inhibitors. The most common adverse events included gastrointestinal complaints, such as diarrhea and nausea, as well as headache and dizziness. But the majority of these events were of low grade, and they were largely confined to the first 4 weeks of treatment, which is a pattern reported with other PDE4 inhibitors in psoriasis and other chronic inflammatory diseases, such as COPD, according to Dr. French.
“There were no discontinuations for a treatment-related adverse event in the arms receiving either the 20-mg or the 30-mg doses,” Dr. French reported. There were only two serious adverse events, and neither were considered by trial investigators to be related to orismilast.
Based on the limited therapeutic gain but greater risk for adverse events on the 40-mg twice-daily dose, “the question is now whether to move forward with the 20-mg or the 30-mg dose,” said Dr. French, who said planning of a phase 3 trial is underway.
Phase 2 study of roflumilast
However, this was not the only set of data on an oral PDE4 inhibitor presented as a late-breaker at the AAD meeting. For clinicians looking for a more immediate and less expensive alternative to apremilast, another study indicated that off-label use of oral roflumilast is an option.
In an investigator-initiated, multicenter, double-blind, placebo-controlled trial conducted in Denmark, the rate of response to oral roflumilast at 24 weeks, including the clear or almost clear response, was on the same general order of magnitude as that seen in the orismilast study, reported Alexander Egeberg, MD, PhD, professor of dermatology, University of Copenhagen.
“At 24 weeks, 21.7% had achieved a PASI 90, and 8.7% achieved a PASI 100,” Dr. Egeberg said.
Oral roflumilast has been available for the treatment of COPD for more than 10 years and is now available in a generic formulation. This study was conducted independent of any pharmaceutical company involvement, and the high rate of response and low risk of adverse events suggests that patients can benefit from a PDE4 inhibitor in a very low-cost form.
“Generic oral roflumilast is cheaper than a Starbucks coffee,” Dr. Egeberg said.
In this trial, 46 patients were randomly assigned to placebo or to the COPD-approved roflumilast dose of 500 mcg once daily. The primary endpoint was change in PASI scores from baseline to week 12, which Dr. Egeberg pointed out is a shorter time frame than the 16 weeks more typical of psoriasis treatment studies.
At week 12, the median improvement in PASI was 34.8% in the roflumilast group versus 0% in the placebo group. Patients were then followed for an additional 12 weeks, but those randomized to placebo were switched to the active treatment. By week 24, the switch patients had largely caught up to those initiated on roflumilast for median PASI improvement (39.1% vs. 43.5%).
Similar to orismilast, roflumilast “was generally well tolerated,” Dr. Egeberg said. The adverse events were consistent with those associated with PDE4 inhibitors in previous trials, whether in psoriasis or COPD. There was only one serious adverse event, and it was not considered treatment related. Discontinuations for adverse events “were very low.”
In a population with a relatively high rate of smoking, Dr. Egeberg further reported, lung function was improved, a remark initially interpreted as a joke by some attending the presentation. However, Dr. Egeberg confirmed that lung function was monitored, and objective improvements were recorded.
By Danish law, the investigators were required to inform the manufacturers of roflumilast. Despite the results of this study, he is not aware of any plans to seek an indication for roflumilast in psoriasis, but he noted that the drug is readily available at a low price.
For those willing to offer this therapy off label, “you can start using it tomorrow if you’d like,” he said.
Dr. French reports financial relationships with Almirall, Amgen, Biotest, Galderma, Janssen Cilag, Leo Pharma, Pincell, Regeneron, UCB, and UNION Therapeutics, which provided funding for this trial. Dr. Egeberg reports financial relationships with Eli Lilly, Galderma, Janssen-Cilag, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – according to results of a phase 2 clinical trial presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
The phase 2b data, which are prompting a phase 3 trial, suggest that the drug, called orismilast, “is a potential new addition to the psoriasis armamentarium,” reported Lars E. French, MD, professor and chair, department of dermatology, Ludwig Maximilian University of Munich (Germany).
At the same session, findings from another study supported off-label use of oral roflumilast (Daliresp and generic), a PDE4 inhibitor approved for severe chronic obstructive pulmonary disease (COPD). The only PDE4 inhibitors with an indication for psoriasis are roflumilast, approved as a cream (Zoryve), and apremilast (Otezla), approved as an oral therapy.
Phase 2 study of orismilast
In the orismilast trial, Dr. French attributed the efficacy observed to the potency of orismilast on the B and D subtypes of PDE4 associated with inflammation. One clue is that these specific subtypes are overly expressed in the skin of patients with either psoriasis or atopic dermatitis.
“When compared to apremilast, orismilast is at least two to fivefold more potent on all PDE4 isoforms and up to 39 times more potent on some of the PDE4 B and D isoforms,” said Dr. French, referring to preclinical findings in human whole blood and blood cells and in a mouse model of chronic inflammation.
The efficacy of orismilast in an immediate-release oral formulation was previously demonstrated in a recently published phase 2a trial, but the newest study tested a modified-release formulation of orismilast to test its potential to improve tolerability.
In the study, 202 adult patients with moderate to severe psoriasis (Psoriasis Area Severity Index [PASI] score ≥ 12) were randomly assigned to one of three doses of orismilast or to placebo. Each of the three doses – 20 mg, 30 mg, or 40 mg – were administered twice daily. The primary endpoint was change in PASI score at 16 weeks. Secondary endpoints included PASI 75 responses (signifying 75% clearance) and safety.
Relative to placebo, which was associated with a PASI improvement of 17%, all three of the tested orismilast doses were superior in a dose-dependent manner. The rates of response were 53%, 61%, and 64% for the 20-mg, 30-mg, and 40-mg twice-daily doses, respectively.
The PASI improvements were rapid, Dr. French said. At 4 weeks, PASI scores climbed from baseline by nearly 40% for those on all orismilast doses, which was more than double the improvement in the placebo group.
In the intention-to-treat analysis with missing data counted as nonresponders, the proportion of patients reaching PASI-75 scores at 16 weeks were 39%, 49%, 45%, and 17%, in the 20-mg, 30-mg, 40-mg, and placebo groups, respectively. The proportion of patients experiencing complete or near-complete skin clearance defined by a PASI 90 were 24%, 22%, 28%, and 8%, respectively.
The side-effect profile was consistent with other PDE4 inhibitors. The most common adverse events included gastrointestinal complaints, such as diarrhea and nausea, as well as headache and dizziness. But the majority of these events were of low grade, and they were largely confined to the first 4 weeks of treatment, which is a pattern reported with other PDE4 inhibitors in psoriasis and other chronic inflammatory diseases, such as COPD, according to Dr. French.
“There were no discontinuations for a treatment-related adverse event in the arms receiving either the 20-mg or the 30-mg doses,” Dr. French reported. There were only two serious adverse events, and neither were considered by trial investigators to be related to orismilast.
Based on the limited therapeutic gain but greater risk for adverse events on the 40-mg twice-daily dose, “the question is now whether to move forward with the 20-mg or the 30-mg dose,” said Dr. French, who said planning of a phase 3 trial is underway.
Phase 2 study of roflumilast
However, this was not the only set of data on an oral PDE4 inhibitor presented as a late-breaker at the AAD meeting. For clinicians looking for a more immediate and less expensive alternative to apremilast, another study indicated that off-label use of oral roflumilast is an option.
In an investigator-initiated, multicenter, double-blind, placebo-controlled trial conducted in Denmark, the rate of response to oral roflumilast at 24 weeks, including the clear or almost clear response, was on the same general order of magnitude as that seen in the orismilast study, reported Alexander Egeberg, MD, PhD, professor of dermatology, University of Copenhagen.
“At 24 weeks, 21.7% had achieved a PASI 90, and 8.7% achieved a PASI 100,” Dr. Egeberg said.
Oral roflumilast has been available for the treatment of COPD for more than 10 years and is now available in a generic formulation. This study was conducted independent of any pharmaceutical company involvement, and the high rate of response and low risk of adverse events suggests that patients can benefit from a PDE4 inhibitor in a very low-cost form.
“Generic oral roflumilast is cheaper than a Starbucks coffee,” Dr. Egeberg said.
In this trial, 46 patients were randomly assigned to placebo or to the COPD-approved roflumilast dose of 500 mcg once daily. The primary endpoint was change in PASI scores from baseline to week 12, which Dr. Egeberg pointed out is a shorter time frame than the 16 weeks more typical of psoriasis treatment studies.
At week 12, the median improvement in PASI was 34.8% in the roflumilast group versus 0% in the placebo group. Patients were then followed for an additional 12 weeks, but those randomized to placebo were switched to the active treatment. By week 24, the switch patients had largely caught up to those initiated on roflumilast for median PASI improvement (39.1% vs. 43.5%).
Similar to orismilast, roflumilast “was generally well tolerated,” Dr. Egeberg said. The adverse events were consistent with those associated with PDE4 inhibitors in previous trials, whether in psoriasis or COPD. There was only one serious adverse event, and it was not considered treatment related. Discontinuations for adverse events “were very low.”
In a population with a relatively high rate of smoking, Dr. Egeberg further reported, lung function was improved, a remark initially interpreted as a joke by some attending the presentation. However, Dr. Egeberg confirmed that lung function was monitored, and objective improvements were recorded.
By Danish law, the investigators were required to inform the manufacturers of roflumilast. Despite the results of this study, he is not aware of any plans to seek an indication for roflumilast in psoriasis, but he noted that the drug is readily available at a low price.
For those willing to offer this therapy off label, “you can start using it tomorrow if you’d like,” he said.
Dr. French reports financial relationships with Almirall, Amgen, Biotest, Galderma, Janssen Cilag, Leo Pharma, Pincell, Regeneron, UCB, and UNION Therapeutics, which provided funding for this trial. Dr. Egeberg reports financial relationships with Eli Lilly, Galderma, Janssen-Cilag, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – according to results of a phase 2 clinical trial presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
The phase 2b data, which are prompting a phase 3 trial, suggest that the drug, called orismilast, “is a potential new addition to the psoriasis armamentarium,” reported Lars E. French, MD, professor and chair, department of dermatology, Ludwig Maximilian University of Munich (Germany).
At the same session, findings from another study supported off-label use of oral roflumilast (Daliresp and generic), a PDE4 inhibitor approved for severe chronic obstructive pulmonary disease (COPD). The only PDE4 inhibitors with an indication for psoriasis are roflumilast, approved as a cream (Zoryve), and apremilast (Otezla), approved as an oral therapy.
Phase 2 study of orismilast
In the orismilast trial, Dr. French attributed the efficacy observed to the potency of orismilast on the B and D subtypes of PDE4 associated with inflammation. One clue is that these specific subtypes are overly expressed in the skin of patients with either psoriasis or atopic dermatitis.
“When compared to apremilast, orismilast is at least two to fivefold more potent on all PDE4 isoforms and up to 39 times more potent on some of the PDE4 B and D isoforms,” said Dr. French, referring to preclinical findings in human whole blood and blood cells and in a mouse model of chronic inflammation.
The efficacy of orismilast in an immediate-release oral formulation was previously demonstrated in a recently published phase 2a trial, but the newest study tested a modified-release formulation of orismilast to test its potential to improve tolerability.
In the study, 202 adult patients with moderate to severe psoriasis (Psoriasis Area Severity Index [PASI] score ≥ 12) were randomly assigned to one of three doses of orismilast or to placebo. Each of the three doses – 20 mg, 30 mg, or 40 mg – were administered twice daily. The primary endpoint was change in PASI score at 16 weeks. Secondary endpoints included PASI 75 responses (signifying 75% clearance) and safety.
Relative to placebo, which was associated with a PASI improvement of 17%, all three of the tested orismilast doses were superior in a dose-dependent manner. The rates of response were 53%, 61%, and 64% for the 20-mg, 30-mg, and 40-mg twice-daily doses, respectively.
The PASI improvements were rapid, Dr. French said. At 4 weeks, PASI scores climbed from baseline by nearly 40% for those on all orismilast doses, which was more than double the improvement in the placebo group.
In the intention-to-treat analysis with missing data counted as nonresponders, the proportion of patients reaching PASI-75 scores at 16 weeks were 39%, 49%, 45%, and 17%, in the 20-mg, 30-mg, 40-mg, and placebo groups, respectively. The proportion of patients experiencing complete or near-complete skin clearance defined by a PASI 90 were 24%, 22%, 28%, and 8%, respectively.
The side-effect profile was consistent with other PDE4 inhibitors. The most common adverse events included gastrointestinal complaints, such as diarrhea and nausea, as well as headache and dizziness. But the majority of these events were of low grade, and they were largely confined to the first 4 weeks of treatment, which is a pattern reported with other PDE4 inhibitors in psoriasis and other chronic inflammatory diseases, such as COPD, according to Dr. French.
“There were no discontinuations for a treatment-related adverse event in the arms receiving either the 20-mg or the 30-mg doses,” Dr. French reported. There were only two serious adverse events, and neither were considered by trial investigators to be related to orismilast.
Based on the limited therapeutic gain but greater risk for adverse events on the 40-mg twice-daily dose, “the question is now whether to move forward with the 20-mg or the 30-mg dose,” said Dr. French, who said planning of a phase 3 trial is underway.
Phase 2 study of roflumilast
However, this was not the only set of data on an oral PDE4 inhibitor presented as a late-breaker at the AAD meeting. For clinicians looking for a more immediate and less expensive alternative to apremilast, another study indicated that off-label use of oral roflumilast is an option.
In an investigator-initiated, multicenter, double-blind, placebo-controlled trial conducted in Denmark, the rate of response to oral roflumilast at 24 weeks, including the clear or almost clear response, was on the same general order of magnitude as that seen in the orismilast study, reported Alexander Egeberg, MD, PhD, professor of dermatology, University of Copenhagen.
“At 24 weeks, 21.7% had achieved a PASI 90, and 8.7% achieved a PASI 100,” Dr. Egeberg said.
Oral roflumilast has been available for the treatment of COPD for more than 10 years and is now available in a generic formulation. This study was conducted independent of any pharmaceutical company involvement, and the high rate of response and low risk of adverse events suggests that patients can benefit from a PDE4 inhibitor in a very low-cost form.
“Generic oral roflumilast is cheaper than a Starbucks coffee,” Dr. Egeberg said.
In this trial, 46 patients were randomly assigned to placebo or to the COPD-approved roflumilast dose of 500 mcg once daily. The primary endpoint was change in PASI scores from baseline to week 12, which Dr. Egeberg pointed out is a shorter time frame than the 16 weeks more typical of psoriasis treatment studies.
At week 12, the median improvement in PASI was 34.8% in the roflumilast group versus 0% in the placebo group. Patients were then followed for an additional 12 weeks, but those randomized to placebo were switched to the active treatment. By week 24, the switch patients had largely caught up to those initiated on roflumilast for median PASI improvement (39.1% vs. 43.5%).
Similar to orismilast, roflumilast “was generally well tolerated,” Dr. Egeberg said. The adverse events were consistent with those associated with PDE4 inhibitors in previous trials, whether in psoriasis or COPD. There was only one serious adverse event, and it was not considered treatment related. Discontinuations for adverse events “were very low.”
In a population with a relatively high rate of smoking, Dr. Egeberg further reported, lung function was improved, a remark initially interpreted as a joke by some attending the presentation. However, Dr. Egeberg confirmed that lung function was monitored, and objective improvements were recorded.
By Danish law, the investigators were required to inform the manufacturers of roflumilast. Despite the results of this study, he is not aware of any plans to seek an indication for roflumilast in psoriasis, but he noted that the drug is readily available at a low price.
For those willing to offer this therapy off label, “you can start using it tomorrow if you’d like,” he said.
Dr. French reports financial relationships with Almirall, Amgen, Biotest, Galderma, Janssen Cilag, Leo Pharma, Pincell, Regeneron, UCB, and UNION Therapeutics, which provided funding for this trial. Dr. Egeberg reports financial relationships with Eli Lilly, Galderma, Janssen-Cilag, Novartis, and Pfizer.
A version of this article first appeared on Medscape.com.
AT AAD 2023