User login
A Practical Overview of Pediatric Atopic Dermatitis, Part 2: Triggers and Grading
Atopic dermatitis (AD) may be triggered by viral infections, food allergens, weather, and other causes, and it may trigger an inflammatory progression known as the atopic march. This article reviews research on triggers of pediatric AD so that dermatologists may discuss trigger avoidance with patients and guardians. Other factors affecting AD development include genetics and hygiene. Grading of AD also is discussed.
The Atopic March
The persistence of AD in untreated skin can trigger an inflammatory progression called the atopic march in which food and environmental allergies as well as asthma may occur progressively due to ongoing inflammatory triggering.1 In a study of asthma and food allergy reporting and management in public schools in Chicago, Illinois, food allergies were seen in 9.3% of asthmatic students (n=18,000), and 40.1% of food allergic students (n=4000) had asthma.2 An observational study by Flohr et al3 in London, England, included 619 exclusively breastfed infants who were recruited at 3 months of age. The investigators determined that food sensitization was unrelated to the presence of filaggrin mutations, type of eczema (flexural vs nonflexural), and transepidermal water loss but was associated with AD severity as determined by SCORAD (SCORing Atopic Dermatitis), a composite score of AD that includes pruritus as a factor in severity. Other AD associations included 3 leading food allergens: eggs, milk, and peanuts. No association with cod, wheat, or sesame allergy was noted. The investigators concluded that AD and AD severity were the leading skin-related risk factors for food allergies and therefore food allergy development in breastfed infants was probably mediated by cutaneous antigen-presenting cells.3
The skin has been documented to react to contact with known food allergens4 and is known to be a route of allergic sensitization to allergens such as fragrance in patients with AD.5,6 Two phenotypes of eczema that have been associated with asthma development are severe AD disease and multiple environmental allergies, supporting the theory of the atopic march.7 There also is evidence that release of danger-associated proteins from an impaired barrier also may trigger asthma.8 An analysis of the 2007 National Survey of Children’s Health, a population-based study of91,642 children aged 0 to 17 years, showed that children with AD had a higher prevalence of comorbid asthma (25.1% vs 12.3%), hay fever (34.4% vs 14.3%), and food allergies (15.1% vs 3.6%) compared to children without AD.9 A recent article provided detailed information on how food and diet interplay with AD.10
Triggers of Disease Flares
Triggers are the leading source of AD flare initiation, and avoidance of triggers is an important mechanism by which patients can control disease activity. Despite the best skin care and trigger avoidance, disease flares occur, sometimes due to ongoing inflammation and other times due to inability to prevent flares such as heat and humidity. A survey of patients with AD in Spain identified the following triggers: cosmetic products, clothing, mites, detergents/soaps, and temperature changes.11 In childhood, wool also is a known trigger of AD.12 Viral infections including respiratory syncytial virus may trigger the first onset of AD.13 Patients with AD may become allergic to fragrance and metals causing disease exacerbation on exposure.14,15 Food allergens contribute to approximately 40% of cases of AD in infancy but are not the cause of AD. The best evidence for improvement of AD with food allergen avoidance exists for egg white allergy.16 Food avoidance programs should be developed in conjunction with an allergist, as it is no longer advised in many cases to completely withdraw foods; therefore, an allergist has to assess the level of allergic severity and the risk-benefit ratio of food avoidance or introduction.17 Emotional stressors, heat, and humidity, as well as indoor heating in the winter months, can cause AD flares.18
A study by Silverberg et al19 provided evidence of climate influences on the US prevalence of childhood eczema using a merged analysis of the 2007 National Survey of Children’s Health and the 2006-2007 National Climate Data Center and Weather Service. Results showed that eczema prevalence was significantly lower when associated with higher annual relative humidity (P=.01), UV index (P<.0001), and highest-quartile air temperature (P=.002).19 The Pediatric Eczema Elective Registry also showed that warm, humid, and high-sun-exposure climates are associated with poorly controlled eczema in affected patients.20 The association of eczema with latitude as well as its negative association with mean annual outdoor temperature has been described by Weiland et al21 in the ISAAC (International Study of Asthma and Allergies in Childhood) study. Long airplane flights in low humidity can trigger eczema in adults. Climate has been postulated to affect eczema through alterations in filaggrin and skin barrier function.22 Indoor temperature and humidity regulation may be used adjunctively for daily flare prevention.
Genetics and AD
Of 762 infants in a birth cohort with a parent with atopy in Cincinnati, Ohio, 39% developed eczema by the age of 3 years. Single nucleotide polymorphisms of IL-4Rα 175 V and CD14-159 C/T were linked to greater eczema risk at 2 to 3 years of age.23 Monozygotic twins have a concordance rate of 0.72 to 0.86 versus 0.21 to 0.23 in dizygotic twins, demonstrating a strong genetic component in the development of AD.24 Linkage to AD has been positively made to the epidermal differentiation complex on human chromosome 1q21, which contains the genes for filaggrin and other proteins such as loricrin. Other genes linked to AD include the serine protease inhibitor SPINK5 (serine peptidase inhibitor, Kazal type 5) implicated in Netherton syndrome (triad of ichthyosis linearis circumflexa, bamboo hair, and atopic disorders); RANTES (regulated on activation, normal T-expressed, and secreted), which has been associated with severity of AD; IL-4; and IL-13.5,25,26
The Hygiene Hypothesis
Atopic dermatitis is more common in wealthy developed countries, leading some to believe that hygiene and relative reduction in illness via vaccination have contributed to the rise of AD prevalence in developed nations.13,27 There currently is evidence demonstrating that wild-type varicella infection confers long-standing protection against AD and mediates reduced total IgE and peripheral blood lymphocytes.27
Grading of AD
Grading of AD is a subject of controversy, as there currently are no uniform grading scales.28 A recent outcomes group attempted to determine the best scale for disease monitoring. Schmitt et al29 presented the Harmonizing Outcome Measures for Eczema (HOME) roadmap, which was intended to determine a core outcome set for eczema; however, because these outcome measurements have not yet been standardized, only the eczema assessment and severity index (EASI) scoring system meets criteria for standardization. In clinical practice, physicians often assign mild, moderate, or severe labeling based on their general sense of the disease extent using an investigator global assessment score.28
The EASI score is a well-validated composite score of AD severity based on 4 body regions: (1) head and neck, (2) trunk (including genital area), (3) upper limbs, and (4) lower limbs (including buttocks). The total area of involvement in each region is graded on a scale of 0 to 6, and AD severity is graded as a composite of 4 parameters (ranked on a scale of 0–3), including redness (erythema, inflammation), thickness (induration, papulation, swelling [acute eczema]), scratching (excoriation), and lichenification (prurigo nodules [chronic eczema]). The surface area of each region relative to body size is used as a multiplying factor, resulting in the following severity strata: 0=clear; 0.1–1.0=almost clear; 1.1–7.0=mild; 7.1–21.0=moderate; 21.1–50.0=severe; 50.1–72.0=very severe (κ=0.75).30-32 The six area, six sign AD (SASSAD) score32,33 is a similar score without adjustment for body surface area by region.34
An older, now less frequently used eczema score is the SCORAD, which addressed surface area by rule of nines and severity of 6 features—redness, swelling, oozing/crusting, scratch marks, skin thickening (lichenification), dryness (assessed in an area with no inflammation)—by region on a scale of 0 to 3. A subjective symptom parameter for itching and sleeplessness helped highlight that these comorbidities are important in gauging disease activity and impact on a child’s life.35
Natural History of AD
The clinical dogma has been that AD would improve with age, with reduction at grade school entry and perhaps full disappearance in adulthood; however, 3 recent surveys have suggested otherwise. The ISAAC group has found prevalence of AD in wealthy developed countries among children aged 6 to 7 years to be at a consistent increase.36 A US-based survey from the National Health Interview Survey showed a 1-year prevalence of 10.2% of active AD in adults and 9.8% when occupational dermatitis was excluded.37 Halvorsen et al38 demonstrated that eczema prevalence is 9.7% in individuals aged 18 to 19 years.
A prospective trial of eighth graders followed from 1995 to 2010 demonstrated that AD persisted in 50% at school age. Persistent eczema into adulthood was associated with early-onset childhood allergic rhinitis and hand eczema.39 In a cohort of hand eczema patients (N=368), 28% had AD and 39% had an atopic illness.40 An association with allergic contact dermatitis and increased IgE to Malassezia furfur was further associated.41
Conclusion
The role of triggers and allergens in disease activity in AD is an important consideration in children with AD and requires ongoing consideration with age and varied exposures. Understanding the grading of AD is important in evaluating clinical trial data. The natural history of AD has changed, which is important for the practitioner to note when counseling patients and guardians.
- Li M. Current evidence of epidermal barrier dysfunction and thymic stromal lymphopoietin in the atopic march. Eur Respir Rev. 2014;23:292-298.
- Gupta RS, Rivkina V, DeSantiago-Cardenas L, et al. Asthma and food allergy management in Chicago public schools. Pediatrics. 2014;134:729-736.
- Flohr C, Perkin M, Logan K, et al. Atopic dermatitis and disease severity are the main risk factors for food sensitization in exclusively breastfed infants. J Invest Dermatol. 2014;134:345-350.
- Silverberg NB. Food, glorious food. Cutis. 2011;87:267-268.
- De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol. 2012;132:949-963.
- Thyssen JP, McFadden JP, Kimber I. The multiple factors affecting the association between atopic dermatitis and contact sensitization. Allergy. 2014;69:28-36.
- Amat F, Saint-Pierre P, Bourrat E, et al. Early-onset atopic dermatitis in children: which are the phenotypes at risk of asthma? results from the ORCA Cohort. PLoS One. 2015;10:e0131369.
- Demehri S, Morimoto M, Holtzman MJ, et al. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. 2009;7:e1000067.
- Silverberg JI, Simpson EL. Association between severe eczema in children and multiple comorbid conditions and increased healthcare utilization. Pediatr Allergy Immunol. 2013;24:476-486.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Ortiz de Frutos FJ, Torrelo A, de Lucas R, et al. Patient perspectives on triggers, adherence to medical recommendations, and disease control in atopic dermatitis: the DATOP study. Actas Dermosifiliogr. 2014;105:487-496.
- Ricci G, Patrizi A, Bellini F, et al. Use of textiles in atopic dermatitis: care of atopic dermatitis. Curr Probl Dermatol. 2006;33:127-143.
- Welliver RC, Wong DT, Sun M, et al. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N Engl J Med. 1981;305:841-846.
- Aquino M, Fonacier L. The role of contact dermatitis in patients with atopic dermatitis. J Allergy Clin Immunol Pract. 2014;2:382-387.
- Brod BA, Treat JR, Rothe MJ, et al. Allergic contact dermatitis: kids are not just little people. Clin Dermatol. 2015;33:605-612.
- Martorell A, Alonso E, Boné J, et al. Position document: IgE-mediated allergy to egg protein. Allergol Immunopathol (Madr). 2013;41:320-336.
- Sicherer SH. Early introduction of peanut to infants at high allergic risk can reduce peanut allergy at age 5 years [published online September 17, 2015]. Evid Based Med. 2015;20:204.
- Kiken DA, Silverberg NB. Atopic dermatitis in children, part 1: epidemiology, clinical features, and complications. Cutis. 2006;78:241-247.
- Silverberg JI, Hanifin J, Simpson EL. Climatic factors are associated with childhood eczema prevalence in the United States. J Invest Dermatol. 2013;133:1752-1759.
- Sargen MR, Hoffstad O, Margolis DJ. Warm, humid, and high sun exposure climates are associated with poorly controlled eczema: PEER (Pediatric Eczema Elective Registry) cohort, 2004-2012. J Invest Dermatol. 2014;134:51-57.
- Weiland SK, Hüsing A, Strachan DP, et al. Climate and the prevalence of symptoms of asthma, allergic rhinitis, and atopic eczema in children. Occup Environ Med. 2004;61:609-615.
- Langan SM, Irvine AD. Childhood eczema and the importance of the physical environment. J Invest Dermatol. 2013;133:1706-1709.
- Biagini Myers JM, Wang N, LeMasters GK, et al. Genetic and environmental risk factors for childhood eczema development and allergic sensitization in the CCAAPS cohort. J Invest Dermatol. 2010;130:430-437.
- Brown SJ, McLean WH. Eczema genetics: current state of knowledge and future goals. J Invest Dermatol. 2009;129:543-552.
- Hanifin JM. Evolving concepts of pathogenesis in atopic dermatitis and other eczemas. J Invest Dermatol. 2009;129:320-322.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Silverberg JI, Norowitz KB, Kleiman E, et al. Association between varicella zoster virus infection and atopic dermatitis in early and late childhood: a case-control study. J Allergy Clin Immunol. 2010;126:300-305.
- Futamura M, Leshem YA, Thomas KS, et al. A systematic review of Investigator Global Assessment (IGA) in atopic dermatitis (AD) trials: many options, no standards. J Am Acad Dermatol. 2016;74:288-294.
- Schmitt J, Apfelbacher C, Spuls PI, et al. The Harmonizing Outcome Measures for Eczema (HOME) roadmap: a methodological framework to develop core sets of outcome measurements in dermatology. J Invest Dermatol. 2015;135:24-30.
- Hanifin JM, Thurston M, Omoto M, et al. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10:11-18.
- Leshem YA, Hajar T, Hanifin JM, et al. What the Eczema Area and Severity Index score tells us about the severity of atopic dermatitis: an interpretability study. Br J Dermatol. 2015;172:1353-1357.
- Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150:96-102.
- Berth-Jones J. Six area, six sign atopic dermatitis (SASSAD) severity score: a simple system for monitoring disease activity in atopic dermatitis. Br J Dermatol. 1996;135(suppl 48):25-30.
- Zhao CY, Tran AQ, Lazo-Dizon JP, et al. A pilot comparison study of four clinician-rated atopic dermatitis severity scales. Br J Dermatol. 2015;173:488-497.
- Kunz B, Oranje AP, Labrèze L, et al. Clinical validation and guidelines for the SCORAD index: consensus report of the European Task Force on Atopic Dermatitis. Dermatology. 1997;195:10-19.
- Williams H, Stewart A, von Mutius E, et al. Is eczema really on the increase worldwide? J Allergy Clin Immunol. 2008;121:947-954.
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: a US population-based study. J Allergy Clin Immunol. 2013;132:1132-1138.
- Halvorsen JA, Lien L, Dalgard F, et al. Suicidal ideation, mental health problems, and social function in adolescents with eczema: a population-based study. J Invest Dermatol. 2014;134:1847-1854.
- Mortz CG, Andersen KE, Dellgren C, et al. Atopic dermatitis from adolescence to adulthood in the TOACS cohort: prevalence, persistence, and comorbidities. Allergy. 2015;70:836-845.
- Rystedt I. Atopic background in patients with occupational hand eczema. Contact Dermatitis. 1985;12:247-254.
- Mortz CG, Andersen KE, Dellgren C, et al. Atopic dermatitis from adolescence to adulthood in the TOACS cohort: prevalence, persistence and comorbidities. Allergy. 2015;70:836-845.
Atopic dermatitis (AD) may be triggered by viral infections, food allergens, weather, and other causes, and it may trigger an inflammatory progression known as the atopic march. This article reviews research on triggers of pediatric AD so that dermatologists may discuss trigger avoidance with patients and guardians. Other factors affecting AD development include genetics and hygiene. Grading of AD also is discussed.
The Atopic March
The persistence of AD in untreated skin can trigger an inflammatory progression called the atopic march in which food and environmental allergies as well as asthma may occur progressively due to ongoing inflammatory triggering.1 In a study of asthma and food allergy reporting and management in public schools in Chicago, Illinois, food allergies were seen in 9.3% of asthmatic students (n=18,000), and 40.1% of food allergic students (n=4000) had asthma.2 An observational study by Flohr et al3 in London, England, included 619 exclusively breastfed infants who were recruited at 3 months of age. The investigators determined that food sensitization was unrelated to the presence of filaggrin mutations, type of eczema (flexural vs nonflexural), and transepidermal water loss but was associated with AD severity as determined by SCORAD (SCORing Atopic Dermatitis), a composite score of AD that includes pruritus as a factor in severity. Other AD associations included 3 leading food allergens: eggs, milk, and peanuts. No association with cod, wheat, or sesame allergy was noted. The investigators concluded that AD and AD severity were the leading skin-related risk factors for food allergies and therefore food allergy development in breastfed infants was probably mediated by cutaneous antigen-presenting cells.3
The skin has been documented to react to contact with known food allergens4 and is known to be a route of allergic sensitization to allergens such as fragrance in patients with AD.5,6 Two phenotypes of eczema that have been associated with asthma development are severe AD disease and multiple environmental allergies, supporting the theory of the atopic march.7 There also is evidence that release of danger-associated proteins from an impaired barrier also may trigger asthma.8 An analysis of the 2007 National Survey of Children’s Health, a population-based study of91,642 children aged 0 to 17 years, showed that children with AD had a higher prevalence of comorbid asthma (25.1% vs 12.3%), hay fever (34.4% vs 14.3%), and food allergies (15.1% vs 3.6%) compared to children without AD.9 A recent article provided detailed information on how food and diet interplay with AD.10
Triggers of Disease Flares
Triggers are the leading source of AD flare initiation, and avoidance of triggers is an important mechanism by which patients can control disease activity. Despite the best skin care and trigger avoidance, disease flares occur, sometimes due to ongoing inflammation and other times due to inability to prevent flares such as heat and humidity. A survey of patients with AD in Spain identified the following triggers: cosmetic products, clothing, mites, detergents/soaps, and temperature changes.11 In childhood, wool also is a known trigger of AD.12 Viral infections including respiratory syncytial virus may trigger the first onset of AD.13 Patients with AD may become allergic to fragrance and metals causing disease exacerbation on exposure.14,15 Food allergens contribute to approximately 40% of cases of AD in infancy but are not the cause of AD. The best evidence for improvement of AD with food allergen avoidance exists for egg white allergy.16 Food avoidance programs should be developed in conjunction with an allergist, as it is no longer advised in many cases to completely withdraw foods; therefore, an allergist has to assess the level of allergic severity and the risk-benefit ratio of food avoidance or introduction.17 Emotional stressors, heat, and humidity, as well as indoor heating in the winter months, can cause AD flares.18
A study by Silverberg et al19 provided evidence of climate influences on the US prevalence of childhood eczema using a merged analysis of the 2007 National Survey of Children’s Health and the 2006-2007 National Climate Data Center and Weather Service. Results showed that eczema prevalence was significantly lower when associated with higher annual relative humidity (P=.01), UV index (P<.0001), and highest-quartile air temperature (P=.002).19 The Pediatric Eczema Elective Registry also showed that warm, humid, and high-sun-exposure climates are associated with poorly controlled eczema in affected patients.20 The association of eczema with latitude as well as its negative association with mean annual outdoor temperature has been described by Weiland et al21 in the ISAAC (International Study of Asthma and Allergies in Childhood) study. Long airplane flights in low humidity can trigger eczema in adults. Climate has been postulated to affect eczema through alterations in filaggrin and skin barrier function.22 Indoor temperature and humidity regulation may be used adjunctively for daily flare prevention.
Genetics and AD
Of 762 infants in a birth cohort with a parent with atopy in Cincinnati, Ohio, 39% developed eczema by the age of 3 years. Single nucleotide polymorphisms of IL-4Rα 175 V and CD14-159 C/T were linked to greater eczema risk at 2 to 3 years of age.23 Monozygotic twins have a concordance rate of 0.72 to 0.86 versus 0.21 to 0.23 in dizygotic twins, demonstrating a strong genetic component in the development of AD.24 Linkage to AD has been positively made to the epidermal differentiation complex on human chromosome 1q21, which contains the genes for filaggrin and other proteins such as loricrin. Other genes linked to AD include the serine protease inhibitor SPINK5 (serine peptidase inhibitor, Kazal type 5) implicated in Netherton syndrome (triad of ichthyosis linearis circumflexa, bamboo hair, and atopic disorders); RANTES (regulated on activation, normal T-expressed, and secreted), which has been associated with severity of AD; IL-4; and IL-13.5,25,26
The Hygiene Hypothesis
Atopic dermatitis is more common in wealthy developed countries, leading some to believe that hygiene and relative reduction in illness via vaccination have contributed to the rise of AD prevalence in developed nations.13,27 There currently is evidence demonstrating that wild-type varicella infection confers long-standing protection against AD and mediates reduced total IgE and peripheral blood lymphocytes.27
Grading of AD
Grading of AD is a subject of controversy, as there currently are no uniform grading scales.28 A recent outcomes group attempted to determine the best scale for disease monitoring. Schmitt et al29 presented the Harmonizing Outcome Measures for Eczema (HOME) roadmap, which was intended to determine a core outcome set for eczema; however, because these outcome measurements have not yet been standardized, only the eczema assessment and severity index (EASI) scoring system meets criteria for standardization. In clinical practice, physicians often assign mild, moderate, or severe labeling based on their general sense of the disease extent using an investigator global assessment score.28
The EASI score is a well-validated composite score of AD severity based on 4 body regions: (1) head and neck, (2) trunk (including genital area), (3) upper limbs, and (4) lower limbs (including buttocks). The total area of involvement in each region is graded on a scale of 0 to 6, and AD severity is graded as a composite of 4 parameters (ranked on a scale of 0–3), including redness (erythema, inflammation), thickness (induration, papulation, swelling [acute eczema]), scratching (excoriation), and lichenification (prurigo nodules [chronic eczema]). The surface area of each region relative to body size is used as a multiplying factor, resulting in the following severity strata: 0=clear; 0.1–1.0=almost clear; 1.1–7.0=mild; 7.1–21.0=moderate; 21.1–50.0=severe; 50.1–72.0=very severe (κ=0.75).30-32 The six area, six sign AD (SASSAD) score32,33 is a similar score without adjustment for body surface area by region.34
An older, now less frequently used eczema score is the SCORAD, which addressed surface area by rule of nines and severity of 6 features—redness, swelling, oozing/crusting, scratch marks, skin thickening (lichenification), dryness (assessed in an area with no inflammation)—by region on a scale of 0 to 3. A subjective symptom parameter for itching and sleeplessness helped highlight that these comorbidities are important in gauging disease activity and impact on a child’s life.35
Natural History of AD
The clinical dogma has been that AD would improve with age, with reduction at grade school entry and perhaps full disappearance in adulthood; however, 3 recent surveys have suggested otherwise. The ISAAC group has found prevalence of AD in wealthy developed countries among children aged 6 to 7 years to be at a consistent increase.36 A US-based survey from the National Health Interview Survey showed a 1-year prevalence of 10.2% of active AD in adults and 9.8% when occupational dermatitis was excluded.37 Halvorsen et al38 demonstrated that eczema prevalence is 9.7% in individuals aged 18 to 19 years.
A prospective trial of eighth graders followed from 1995 to 2010 demonstrated that AD persisted in 50% at school age. Persistent eczema into adulthood was associated with early-onset childhood allergic rhinitis and hand eczema.39 In a cohort of hand eczema patients (N=368), 28% had AD and 39% had an atopic illness.40 An association with allergic contact dermatitis and increased IgE to Malassezia furfur was further associated.41
Conclusion
The role of triggers and allergens in disease activity in AD is an important consideration in children with AD and requires ongoing consideration with age and varied exposures. Understanding the grading of AD is important in evaluating clinical trial data. The natural history of AD has changed, which is important for the practitioner to note when counseling patients and guardians.
Atopic dermatitis (AD) may be triggered by viral infections, food allergens, weather, and other causes, and it may trigger an inflammatory progression known as the atopic march. This article reviews research on triggers of pediatric AD so that dermatologists may discuss trigger avoidance with patients and guardians. Other factors affecting AD development include genetics and hygiene. Grading of AD also is discussed.
The Atopic March
The persistence of AD in untreated skin can trigger an inflammatory progression called the atopic march in which food and environmental allergies as well as asthma may occur progressively due to ongoing inflammatory triggering.1 In a study of asthma and food allergy reporting and management in public schools in Chicago, Illinois, food allergies were seen in 9.3% of asthmatic students (n=18,000), and 40.1% of food allergic students (n=4000) had asthma.2 An observational study by Flohr et al3 in London, England, included 619 exclusively breastfed infants who were recruited at 3 months of age. The investigators determined that food sensitization was unrelated to the presence of filaggrin mutations, type of eczema (flexural vs nonflexural), and transepidermal water loss but was associated with AD severity as determined by SCORAD (SCORing Atopic Dermatitis), a composite score of AD that includes pruritus as a factor in severity. Other AD associations included 3 leading food allergens: eggs, milk, and peanuts. No association with cod, wheat, or sesame allergy was noted. The investigators concluded that AD and AD severity were the leading skin-related risk factors for food allergies and therefore food allergy development in breastfed infants was probably mediated by cutaneous antigen-presenting cells.3
The skin has been documented to react to contact with known food allergens4 and is known to be a route of allergic sensitization to allergens such as fragrance in patients with AD.5,6 Two phenotypes of eczema that have been associated with asthma development are severe AD disease and multiple environmental allergies, supporting the theory of the atopic march.7 There also is evidence that release of danger-associated proteins from an impaired barrier also may trigger asthma.8 An analysis of the 2007 National Survey of Children’s Health, a population-based study of91,642 children aged 0 to 17 years, showed that children with AD had a higher prevalence of comorbid asthma (25.1% vs 12.3%), hay fever (34.4% vs 14.3%), and food allergies (15.1% vs 3.6%) compared to children without AD.9 A recent article provided detailed information on how food and diet interplay with AD.10
Triggers of Disease Flares
Triggers are the leading source of AD flare initiation, and avoidance of triggers is an important mechanism by which patients can control disease activity. Despite the best skin care and trigger avoidance, disease flares occur, sometimes due to ongoing inflammation and other times due to inability to prevent flares such as heat and humidity. A survey of patients with AD in Spain identified the following triggers: cosmetic products, clothing, mites, detergents/soaps, and temperature changes.11 In childhood, wool also is a known trigger of AD.12 Viral infections including respiratory syncytial virus may trigger the first onset of AD.13 Patients with AD may become allergic to fragrance and metals causing disease exacerbation on exposure.14,15 Food allergens contribute to approximately 40% of cases of AD in infancy but are not the cause of AD. The best evidence for improvement of AD with food allergen avoidance exists for egg white allergy.16 Food avoidance programs should be developed in conjunction with an allergist, as it is no longer advised in many cases to completely withdraw foods; therefore, an allergist has to assess the level of allergic severity and the risk-benefit ratio of food avoidance or introduction.17 Emotional stressors, heat, and humidity, as well as indoor heating in the winter months, can cause AD flares.18
A study by Silverberg et al19 provided evidence of climate influences on the US prevalence of childhood eczema using a merged analysis of the 2007 National Survey of Children’s Health and the 2006-2007 National Climate Data Center and Weather Service. Results showed that eczema prevalence was significantly lower when associated with higher annual relative humidity (P=.01), UV index (P<.0001), and highest-quartile air temperature (P=.002).19 The Pediatric Eczema Elective Registry also showed that warm, humid, and high-sun-exposure climates are associated with poorly controlled eczema in affected patients.20 The association of eczema with latitude as well as its negative association with mean annual outdoor temperature has been described by Weiland et al21 in the ISAAC (International Study of Asthma and Allergies in Childhood) study. Long airplane flights in low humidity can trigger eczema in adults. Climate has been postulated to affect eczema through alterations in filaggrin and skin barrier function.22 Indoor temperature and humidity regulation may be used adjunctively for daily flare prevention.
Genetics and AD
Of 762 infants in a birth cohort with a parent with atopy in Cincinnati, Ohio, 39% developed eczema by the age of 3 years. Single nucleotide polymorphisms of IL-4Rα 175 V and CD14-159 C/T were linked to greater eczema risk at 2 to 3 years of age.23 Monozygotic twins have a concordance rate of 0.72 to 0.86 versus 0.21 to 0.23 in dizygotic twins, demonstrating a strong genetic component in the development of AD.24 Linkage to AD has been positively made to the epidermal differentiation complex on human chromosome 1q21, which contains the genes for filaggrin and other proteins such as loricrin. Other genes linked to AD include the serine protease inhibitor SPINK5 (serine peptidase inhibitor, Kazal type 5) implicated in Netherton syndrome (triad of ichthyosis linearis circumflexa, bamboo hair, and atopic disorders); RANTES (regulated on activation, normal T-expressed, and secreted), which has been associated with severity of AD; IL-4; and IL-13.5,25,26
The Hygiene Hypothesis
Atopic dermatitis is more common in wealthy developed countries, leading some to believe that hygiene and relative reduction in illness via vaccination have contributed to the rise of AD prevalence in developed nations.13,27 There currently is evidence demonstrating that wild-type varicella infection confers long-standing protection against AD and mediates reduced total IgE and peripheral blood lymphocytes.27
Grading of AD
Grading of AD is a subject of controversy, as there currently are no uniform grading scales.28 A recent outcomes group attempted to determine the best scale for disease monitoring. Schmitt et al29 presented the Harmonizing Outcome Measures for Eczema (HOME) roadmap, which was intended to determine a core outcome set for eczema; however, because these outcome measurements have not yet been standardized, only the eczema assessment and severity index (EASI) scoring system meets criteria for standardization. In clinical practice, physicians often assign mild, moderate, or severe labeling based on their general sense of the disease extent using an investigator global assessment score.28
The EASI score is a well-validated composite score of AD severity based on 4 body regions: (1) head and neck, (2) trunk (including genital area), (3) upper limbs, and (4) lower limbs (including buttocks). The total area of involvement in each region is graded on a scale of 0 to 6, and AD severity is graded as a composite of 4 parameters (ranked on a scale of 0–3), including redness (erythema, inflammation), thickness (induration, papulation, swelling [acute eczema]), scratching (excoriation), and lichenification (prurigo nodules [chronic eczema]). The surface area of each region relative to body size is used as a multiplying factor, resulting in the following severity strata: 0=clear; 0.1–1.0=almost clear; 1.1–7.0=mild; 7.1–21.0=moderate; 21.1–50.0=severe; 50.1–72.0=very severe (κ=0.75).30-32 The six area, six sign AD (SASSAD) score32,33 is a similar score without adjustment for body surface area by region.34
An older, now less frequently used eczema score is the SCORAD, which addressed surface area by rule of nines and severity of 6 features—redness, swelling, oozing/crusting, scratch marks, skin thickening (lichenification), dryness (assessed in an area with no inflammation)—by region on a scale of 0 to 3. A subjective symptom parameter for itching and sleeplessness helped highlight that these comorbidities are important in gauging disease activity and impact on a child’s life.35
Natural History of AD
The clinical dogma has been that AD would improve with age, with reduction at grade school entry and perhaps full disappearance in adulthood; however, 3 recent surveys have suggested otherwise. The ISAAC group has found prevalence of AD in wealthy developed countries among children aged 6 to 7 years to be at a consistent increase.36 A US-based survey from the National Health Interview Survey showed a 1-year prevalence of 10.2% of active AD in adults and 9.8% when occupational dermatitis was excluded.37 Halvorsen et al38 demonstrated that eczema prevalence is 9.7% in individuals aged 18 to 19 years.
A prospective trial of eighth graders followed from 1995 to 2010 demonstrated that AD persisted in 50% at school age. Persistent eczema into adulthood was associated with early-onset childhood allergic rhinitis and hand eczema.39 In a cohort of hand eczema patients (N=368), 28% had AD and 39% had an atopic illness.40 An association with allergic contact dermatitis and increased IgE to Malassezia furfur was further associated.41
Conclusion
The role of triggers and allergens in disease activity in AD is an important consideration in children with AD and requires ongoing consideration with age and varied exposures. Understanding the grading of AD is important in evaluating clinical trial data. The natural history of AD has changed, which is important for the practitioner to note when counseling patients and guardians.
- Li M. Current evidence of epidermal barrier dysfunction and thymic stromal lymphopoietin in the atopic march. Eur Respir Rev. 2014;23:292-298.
- Gupta RS, Rivkina V, DeSantiago-Cardenas L, et al. Asthma and food allergy management in Chicago public schools. Pediatrics. 2014;134:729-736.
- Flohr C, Perkin M, Logan K, et al. Atopic dermatitis and disease severity are the main risk factors for food sensitization in exclusively breastfed infants. J Invest Dermatol. 2014;134:345-350.
- Silverberg NB. Food, glorious food. Cutis. 2011;87:267-268.
- De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol. 2012;132:949-963.
- Thyssen JP, McFadden JP, Kimber I. The multiple factors affecting the association between atopic dermatitis and contact sensitization. Allergy. 2014;69:28-36.
- Amat F, Saint-Pierre P, Bourrat E, et al. Early-onset atopic dermatitis in children: which are the phenotypes at risk of asthma? results from the ORCA Cohort. PLoS One. 2015;10:e0131369.
- Demehri S, Morimoto M, Holtzman MJ, et al. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. 2009;7:e1000067.
- Silverberg JI, Simpson EL. Association between severe eczema in children and multiple comorbid conditions and increased healthcare utilization. Pediatr Allergy Immunol. 2013;24:476-486.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Ortiz de Frutos FJ, Torrelo A, de Lucas R, et al. Patient perspectives on triggers, adherence to medical recommendations, and disease control in atopic dermatitis: the DATOP study. Actas Dermosifiliogr. 2014;105:487-496.
- Ricci G, Patrizi A, Bellini F, et al. Use of textiles in atopic dermatitis: care of atopic dermatitis. Curr Probl Dermatol. 2006;33:127-143.
- Welliver RC, Wong DT, Sun M, et al. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N Engl J Med. 1981;305:841-846.
- Aquino M, Fonacier L. The role of contact dermatitis in patients with atopic dermatitis. J Allergy Clin Immunol Pract. 2014;2:382-387.
- Brod BA, Treat JR, Rothe MJ, et al. Allergic contact dermatitis: kids are not just little people. Clin Dermatol. 2015;33:605-612.
- Martorell A, Alonso E, Boné J, et al. Position document: IgE-mediated allergy to egg protein. Allergol Immunopathol (Madr). 2013;41:320-336.
- Sicherer SH. Early introduction of peanut to infants at high allergic risk can reduce peanut allergy at age 5 years [published online September 17, 2015]. Evid Based Med. 2015;20:204.
- Kiken DA, Silverberg NB. Atopic dermatitis in children, part 1: epidemiology, clinical features, and complications. Cutis. 2006;78:241-247.
- Silverberg JI, Hanifin J, Simpson EL. Climatic factors are associated with childhood eczema prevalence in the United States. J Invest Dermatol. 2013;133:1752-1759.
- Sargen MR, Hoffstad O, Margolis DJ. Warm, humid, and high sun exposure climates are associated with poorly controlled eczema: PEER (Pediatric Eczema Elective Registry) cohort, 2004-2012. J Invest Dermatol. 2014;134:51-57.
- Weiland SK, Hüsing A, Strachan DP, et al. Climate and the prevalence of symptoms of asthma, allergic rhinitis, and atopic eczema in children. Occup Environ Med. 2004;61:609-615.
- Langan SM, Irvine AD. Childhood eczema and the importance of the physical environment. J Invest Dermatol. 2013;133:1706-1709.
- Biagini Myers JM, Wang N, LeMasters GK, et al. Genetic and environmental risk factors for childhood eczema development and allergic sensitization in the CCAAPS cohort. J Invest Dermatol. 2010;130:430-437.
- Brown SJ, McLean WH. Eczema genetics: current state of knowledge and future goals. J Invest Dermatol. 2009;129:543-552.
- Hanifin JM. Evolving concepts of pathogenesis in atopic dermatitis and other eczemas. J Invest Dermatol. 2009;129:320-322.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Silverberg JI, Norowitz KB, Kleiman E, et al. Association between varicella zoster virus infection and atopic dermatitis in early and late childhood: a case-control study. J Allergy Clin Immunol. 2010;126:300-305.
- Futamura M, Leshem YA, Thomas KS, et al. A systematic review of Investigator Global Assessment (IGA) in atopic dermatitis (AD) trials: many options, no standards. J Am Acad Dermatol. 2016;74:288-294.
- Schmitt J, Apfelbacher C, Spuls PI, et al. The Harmonizing Outcome Measures for Eczema (HOME) roadmap: a methodological framework to develop core sets of outcome measurements in dermatology. J Invest Dermatol. 2015;135:24-30.
- Hanifin JM, Thurston M, Omoto M, et al. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10:11-18.
- Leshem YA, Hajar T, Hanifin JM, et al. What the Eczema Area and Severity Index score tells us about the severity of atopic dermatitis: an interpretability study. Br J Dermatol. 2015;172:1353-1357.
- Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150:96-102.
- Berth-Jones J. Six area, six sign atopic dermatitis (SASSAD) severity score: a simple system for monitoring disease activity in atopic dermatitis. Br J Dermatol. 1996;135(suppl 48):25-30.
- Zhao CY, Tran AQ, Lazo-Dizon JP, et al. A pilot comparison study of four clinician-rated atopic dermatitis severity scales. Br J Dermatol. 2015;173:488-497.
- Kunz B, Oranje AP, Labrèze L, et al. Clinical validation and guidelines for the SCORAD index: consensus report of the European Task Force on Atopic Dermatitis. Dermatology. 1997;195:10-19.
- Williams H, Stewart A, von Mutius E, et al. Is eczema really on the increase worldwide? J Allergy Clin Immunol. 2008;121:947-954.
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: a US population-based study. J Allergy Clin Immunol. 2013;132:1132-1138.
- Halvorsen JA, Lien L, Dalgard F, et al. Suicidal ideation, mental health problems, and social function in adolescents with eczema: a population-based study. J Invest Dermatol. 2014;134:1847-1854.
- Mortz CG, Andersen KE, Dellgren C, et al. Atopic dermatitis from adolescence to adulthood in the TOACS cohort: prevalence, persistence, and comorbidities. Allergy. 2015;70:836-845.
- Rystedt I. Atopic background in patients with occupational hand eczema. Contact Dermatitis. 1985;12:247-254.
- Mortz CG, Andersen KE, Dellgren C, et al. Atopic dermatitis from adolescence to adulthood in the TOACS cohort: prevalence, persistence and comorbidities. Allergy. 2015;70:836-845.
- Li M. Current evidence of epidermal barrier dysfunction and thymic stromal lymphopoietin in the atopic march. Eur Respir Rev. 2014;23:292-298.
- Gupta RS, Rivkina V, DeSantiago-Cardenas L, et al. Asthma and food allergy management in Chicago public schools. Pediatrics. 2014;134:729-736.
- Flohr C, Perkin M, Logan K, et al. Atopic dermatitis and disease severity are the main risk factors for food sensitization in exclusively breastfed infants. J Invest Dermatol. 2014;134:345-350.
- Silverberg NB. Food, glorious food. Cutis. 2011;87:267-268.
- De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol. 2012;132:949-963.
- Thyssen JP, McFadden JP, Kimber I. The multiple factors affecting the association between atopic dermatitis and contact sensitization. Allergy. 2014;69:28-36.
- Amat F, Saint-Pierre P, Bourrat E, et al. Early-onset atopic dermatitis in children: which are the phenotypes at risk of asthma? results from the ORCA Cohort. PLoS One. 2015;10:e0131369.
- Demehri S, Morimoto M, Holtzman MJ, et al. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. 2009;7:e1000067.
- Silverberg JI, Simpson EL. Association between severe eczema in children and multiple comorbid conditions and increased healthcare utilization. Pediatr Allergy Immunol. 2013;24:476-486.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Ortiz de Frutos FJ, Torrelo A, de Lucas R, et al. Patient perspectives on triggers, adherence to medical recommendations, and disease control in atopic dermatitis: the DATOP study. Actas Dermosifiliogr. 2014;105:487-496.
- Ricci G, Patrizi A, Bellini F, et al. Use of textiles in atopic dermatitis: care of atopic dermatitis. Curr Probl Dermatol. 2006;33:127-143.
- Welliver RC, Wong DT, Sun M, et al. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N Engl J Med. 1981;305:841-846.
- Aquino M, Fonacier L. The role of contact dermatitis in patients with atopic dermatitis. J Allergy Clin Immunol Pract. 2014;2:382-387.
- Brod BA, Treat JR, Rothe MJ, et al. Allergic contact dermatitis: kids are not just little people. Clin Dermatol. 2015;33:605-612.
- Martorell A, Alonso E, Boné J, et al. Position document: IgE-mediated allergy to egg protein. Allergol Immunopathol (Madr). 2013;41:320-336.
- Sicherer SH. Early introduction of peanut to infants at high allergic risk can reduce peanut allergy at age 5 years [published online September 17, 2015]. Evid Based Med. 2015;20:204.
- Kiken DA, Silverberg NB. Atopic dermatitis in children, part 1: epidemiology, clinical features, and complications. Cutis. 2006;78:241-247.
- Silverberg JI, Hanifin J, Simpson EL. Climatic factors are associated with childhood eczema prevalence in the United States. J Invest Dermatol. 2013;133:1752-1759.
- Sargen MR, Hoffstad O, Margolis DJ. Warm, humid, and high sun exposure climates are associated with poorly controlled eczema: PEER (Pediatric Eczema Elective Registry) cohort, 2004-2012. J Invest Dermatol. 2014;134:51-57.
- Weiland SK, Hüsing A, Strachan DP, et al. Climate and the prevalence of symptoms of asthma, allergic rhinitis, and atopic eczema in children. Occup Environ Med. 2004;61:609-615.
- Langan SM, Irvine AD. Childhood eczema and the importance of the physical environment. J Invest Dermatol. 2013;133:1706-1709.
- Biagini Myers JM, Wang N, LeMasters GK, et al. Genetic and environmental risk factors for childhood eczema development and allergic sensitization in the CCAAPS cohort. J Invest Dermatol. 2010;130:430-437.
- Brown SJ, McLean WH. Eczema genetics: current state of knowledge and future goals. J Invest Dermatol. 2009;129:543-552.
- Hanifin JM. Evolving concepts of pathogenesis in atopic dermatitis and other eczemas. J Invest Dermatol. 2009;129:320-322.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Silverberg JI, Norowitz KB, Kleiman E, et al. Association between varicella zoster virus infection and atopic dermatitis in early and late childhood: a case-control study. J Allergy Clin Immunol. 2010;126:300-305.
- Futamura M, Leshem YA, Thomas KS, et al. A systematic review of Investigator Global Assessment (IGA) in atopic dermatitis (AD) trials: many options, no standards. J Am Acad Dermatol. 2016;74:288-294.
- Schmitt J, Apfelbacher C, Spuls PI, et al. The Harmonizing Outcome Measures for Eczema (HOME) roadmap: a methodological framework to develop core sets of outcome measurements in dermatology. J Invest Dermatol. 2015;135:24-30.
- Hanifin JM, Thurston M, Omoto M, et al. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10:11-18.
- Leshem YA, Hajar T, Hanifin JM, et al. What the Eczema Area and Severity Index score tells us about the severity of atopic dermatitis: an interpretability study. Br J Dermatol. 2015;172:1353-1357.
- Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150:96-102.
- Berth-Jones J. Six area, six sign atopic dermatitis (SASSAD) severity score: a simple system for monitoring disease activity in atopic dermatitis. Br J Dermatol. 1996;135(suppl 48):25-30.
- Zhao CY, Tran AQ, Lazo-Dizon JP, et al. A pilot comparison study of four clinician-rated atopic dermatitis severity scales. Br J Dermatol. 2015;173:488-497.
- Kunz B, Oranje AP, Labrèze L, et al. Clinical validation and guidelines for the SCORAD index: consensus report of the European Task Force on Atopic Dermatitis. Dermatology. 1997;195:10-19.
- Williams H, Stewart A, von Mutius E, et al. Is eczema really on the increase worldwide? J Allergy Clin Immunol. 2008;121:947-954.
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: a US population-based study. J Allergy Clin Immunol. 2013;132:1132-1138.
- Halvorsen JA, Lien L, Dalgard F, et al. Suicidal ideation, mental health problems, and social function in adolescents with eczema: a population-based study. J Invest Dermatol. 2014;134:1847-1854.
- Mortz CG, Andersen KE, Dellgren C, et al. Atopic dermatitis from adolescence to adulthood in the TOACS cohort: prevalence, persistence, and comorbidities. Allergy. 2015;70:836-845.
- Rystedt I. Atopic background in patients with occupational hand eczema. Contact Dermatitis. 1985;12:247-254.
- Mortz CG, Andersen KE, Dellgren C, et al. Atopic dermatitis from adolescence to adulthood in the TOACS cohort: prevalence, persistence and comorbidities. Allergy. 2015;70:836-845.
Practice Points
- Atopic dermatitis (AD) can be triggered by viral infections, weather, and food allergens.
- The scoring of AD is largely used experimentally and includes the eczema assessment and severity index; the SCORAD (SCORing Atopic Dermatitis); and the six area, six sign AD (SASSAD) scores.
- There is a strong genetic contribution to the development of AD.
- Children with AD may have persistent disease into adulthood in half of cases.
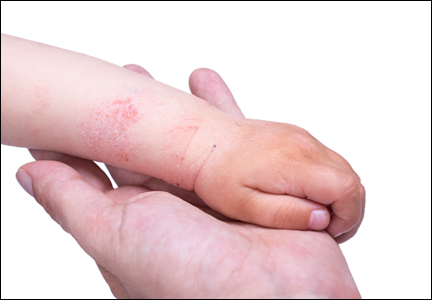
Patch Testing: Working With Patients to Find a Relevant Allergen

What do your patients need to know at the first visit?
Patients with chronic dermatitis are frequently referred for patch testing. An in-depth conversation reviewing the patch test procedure and the many potential causes of dermatitis (eg, endogenous, allergic, irritant, seborrheic) is needed. Patients should understand the patch test process. The testing extends over a week, requiring 3 days of visits. The patches are applied at day 1 and must be kept dry and in place for 48 hours, then they are removed and evaluated. A second follow-up visit at 96 hours to 1 week after the patches are applied is done to perform a final read, interpret, and explain the final results. The patient needs to know that we are looking for an allergen that might be causing the eruption through contact exposure with the skin. The difference between patch testing and prick testing often needs to be discussed, as patients are not always aware of the difference. Explaining the need to avoid topical steroids at the patch test site, sunburn, or systemic steroids during the patch test period is also important to obtain optimal testing conditions.
Querying all exposures including work, home, personal care products, and hobbies is important to help determine which allergen series should be tested to obtain the best results. Patients need to understand that even small intermittent exposures can cause an ongoing dermatitis. If a causative allergen(s) is identified, strict avoidance can lead to clearance and resolution.
Setting expectations is important, and therefore you should discuss the possibility that no allergen will be identified while letting the patient know that this information is also useful. Also, let patients know there are other things that can be done if patch testing is negative to try and gain control of the dermatitis including laboratory tests and biopsies, which may be needed to help direct future management.
What are your go-to treatments? What are the side effects?
The beauty of patch testing is that finding a relevant allergen and subsequent avoidance of that allergen often is sufficient to improve or clear the dermatitis. Detailed education regarding the allergen, where it is found, and how to avoid it are imperative in patient management. I provide the patient with information sheets or narratives found on the American Contact Dermatitis Society website (http://www.contactderm.org) as well as a list of safe products found on the Contact Allergen Management Program (CAMP) area of the site. These tools help in patient compliance.
Go-to treatments for relevant patch test dermatitis involve topical steroids to calm the acute dermatitis while educating and instituting a personal environment free of the identified allergens. Occasionally, systemic steroids are used to provide relief and calm down an extensive dermatitis while educating, identifying, and eliminating known allergens from the patient’s environment. Identifying and eliminating an allergen can mitigate the need for chronic steroids, and the resultant side effects of hypertension, osteoporosis, avascular necrosis, hyperglycemia, and gastrointestinal tract problems can be avoided. Likewise, avoidance of allergens can lead to the elimination of the need for chronic topical steroids and the resultant atrophy and striae.
Side effects of the patch test procedure itself include an allergic reaction to one of the chemicals tested (eg, gold), which is what you are looking for; persistent reactions; flaring of existing dermatitis; irritation; hyperpigmentation; and rarely anaphylaxis or infection at a patch test site. If no allergy is found, treatment of generalized dermatitis can include topical steroids. Topical calcineurin inhibitors can be useful as well as narrowband UV light. Several oral medications can be used for recalcitrant patch test–negative dermatitis and the selection of the right medication is based on the patient’s comorbidities and extent of dermatitis, including systemic steroids, though long-term use is not recommended. Mycophenolate mofetil, methotrexate, cyclosporine, and azathioprine all have side effects including liver and renal toxicity, immunosuppression, and risk for malignancy and therefore need to be considered on a case-by-case basis.
How do you keep the patient compliant with treatment?
Treating allergic contact dermatitis once an allergen(s) has been identified can be challenging. Education is key so that the patient understands where the allergen is found in his/her environment and how to avoid it. Teaching the patient to read labels also is important. Providing a list of safe products simplifies compliance. Reinforcing the need for ongoing vigilance in allergen avoidance is critical to resolution of the dermatitis. Reinforcing the need for continuous avoidance is imperative, as patients sometimes become less vigilant once the dermatitis resolves and the allergen can sneak back into their environment.
What do I do if a patient refuses treatment?
Sometimes patients are so attached to a product that they do not want to stop using it even though they know it is the cause of their dermatitis. If I can help them identify a comparable product, I introduce them to it, but ultimately they get to decide if they prefer to use a product that they know is the cause of their rash or if they want to avoid it and be clear of the dermatitis. For those who do not have an allergen identified through patch testing, alternative treatments can be used. If they do not want systemic medication, I try and optimize their skin care regimen with mild soaps, bland moisturizing creams, and short lukewarm showers, which often is not enough and eventually due to ongoing itch patients decide to discuss and pursue treatment options.
What do your patients need to know at the first visit?
Patients with chronic dermatitis are frequently referred for patch testing. An in-depth conversation reviewing the patch test procedure and the many potential causes of dermatitis (eg, endogenous, allergic, irritant, seborrheic) is needed. Patients should understand the patch test process. The testing extends over a week, requiring 3 days of visits. The patches are applied at day 1 and must be kept dry and in place for 48 hours, then they are removed and evaluated. A second follow-up visit at 96 hours to 1 week after the patches are applied is done to perform a final read, interpret, and explain the final results. The patient needs to know that we are looking for an allergen that might be causing the eruption through contact exposure with the skin. The difference between patch testing and prick testing often needs to be discussed, as patients are not always aware of the difference. Explaining the need to avoid topical steroids at the patch test site, sunburn, or systemic steroids during the patch test period is also important to obtain optimal testing conditions.
Querying all exposures including work, home, personal care products, and hobbies is important to help determine which allergen series should be tested to obtain the best results. Patients need to understand that even small intermittent exposures can cause an ongoing dermatitis. If a causative allergen(s) is identified, strict avoidance can lead to clearance and resolution.
Setting expectations is important, and therefore you should discuss the possibility that no allergen will be identified while letting the patient know that this information is also useful. Also, let patients know there are other things that can be done if patch testing is negative to try and gain control of the dermatitis including laboratory tests and biopsies, which may be needed to help direct future management.
What are your go-to treatments? What are the side effects?
The beauty of patch testing is that finding a relevant allergen and subsequent avoidance of that allergen often is sufficient to improve or clear the dermatitis. Detailed education regarding the allergen, where it is found, and how to avoid it are imperative in patient management. I provide the patient with information sheets or narratives found on the American Contact Dermatitis Society website (http://www.contactderm.org) as well as a list of safe products found on the Contact Allergen Management Program (CAMP) area of the site. These tools help in patient compliance.
Go-to treatments for relevant patch test dermatitis involve topical steroids to calm the acute dermatitis while educating and instituting a personal environment free of the identified allergens. Occasionally, systemic steroids are used to provide relief and calm down an extensive dermatitis while educating, identifying, and eliminating known allergens from the patient’s environment. Identifying and eliminating an allergen can mitigate the need for chronic steroids, and the resultant side effects of hypertension, osteoporosis, avascular necrosis, hyperglycemia, and gastrointestinal tract problems can be avoided. Likewise, avoidance of allergens can lead to the elimination of the need for chronic topical steroids and the resultant atrophy and striae.
Side effects of the patch test procedure itself include an allergic reaction to one of the chemicals tested (eg, gold), which is what you are looking for; persistent reactions; flaring of existing dermatitis; irritation; hyperpigmentation; and rarely anaphylaxis or infection at a patch test site. If no allergy is found, treatment of generalized dermatitis can include topical steroids. Topical calcineurin inhibitors can be useful as well as narrowband UV light. Several oral medications can be used for recalcitrant patch test–negative dermatitis and the selection of the right medication is based on the patient’s comorbidities and extent of dermatitis, including systemic steroids, though long-term use is not recommended. Mycophenolate mofetil, methotrexate, cyclosporine, and azathioprine all have side effects including liver and renal toxicity, immunosuppression, and risk for malignancy and therefore need to be considered on a case-by-case basis.
How do you keep the patient compliant with treatment?
Treating allergic contact dermatitis once an allergen(s) has been identified can be challenging. Education is key so that the patient understands where the allergen is found in his/her environment and how to avoid it. Teaching the patient to read labels also is important. Providing a list of safe products simplifies compliance. Reinforcing the need for ongoing vigilance in allergen avoidance is critical to resolution of the dermatitis. Reinforcing the need for continuous avoidance is imperative, as patients sometimes become less vigilant once the dermatitis resolves and the allergen can sneak back into their environment.
What do I do if a patient refuses treatment?
Sometimes patients are so attached to a product that they do not want to stop using it even though they know it is the cause of their dermatitis. If I can help them identify a comparable product, I introduce them to it, but ultimately they get to decide if they prefer to use a product that they know is the cause of their rash or if they want to avoid it and be clear of the dermatitis. For those who do not have an allergen identified through patch testing, alternative treatments can be used. If they do not want systemic medication, I try and optimize their skin care regimen with mild soaps, bland moisturizing creams, and short lukewarm showers, which often is not enough and eventually due to ongoing itch patients decide to discuss and pursue treatment options.
What do your patients need to know at the first visit?
Patients with chronic dermatitis are frequently referred for patch testing. An in-depth conversation reviewing the patch test procedure and the many potential causes of dermatitis (eg, endogenous, allergic, irritant, seborrheic) is needed. Patients should understand the patch test process. The testing extends over a week, requiring 3 days of visits. The patches are applied at day 1 and must be kept dry and in place for 48 hours, then they are removed and evaluated. A second follow-up visit at 96 hours to 1 week after the patches are applied is done to perform a final read, interpret, and explain the final results. The patient needs to know that we are looking for an allergen that might be causing the eruption through contact exposure with the skin. The difference between patch testing and prick testing often needs to be discussed, as patients are not always aware of the difference. Explaining the need to avoid topical steroids at the patch test site, sunburn, or systemic steroids during the patch test period is also important to obtain optimal testing conditions.
Querying all exposures including work, home, personal care products, and hobbies is important to help determine which allergen series should be tested to obtain the best results. Patients need to understand that even small intermittent exposures can cause an ongoing dermatitis. If a causative allergen(s) is identified, strict avoidance can lead to clearance and resolution.
Setting expectations is important, and therefore you should discuss the possibility that no allergen will be identified while letting the patient know that this information is also useful. Also, let patients know there are other things that can be done if patch testing is negative to try and gain control of the dermatitis including laboratory tests and biopsies, which may be needed to help direct future management.
What are your go-to treatments? What are the side effects?
The beauty of patch testing is that finding a relevant allergen and subsequent avoidance of that allergen often is sufficient to improve or clear the dermatitis. Detailed education regarding the allergen, where it is found, and how to avoid it are imperative in patient management. I provide the patient with information sheets or narratives found on the American Contact Dermatitis Society website (http://www.contactderm.org) as well as a list of safe products found on the Contact Allergen Management Program (CAMP) area of the site. These tools help in patient compliance.
Go-to treatments for relevant patch test dermatitis involve topical steroids to calm the acute dermatitis while educating and instituting a personal environment free of the identified allergens. Occasionally, systemic steroids are used to provide relief and calm down an extensive dermatitis while educating, identifying, and eliminating known allergens from the patient’s environment. Identifying and eliminating an allergen can mitigate the need for chronic steroids, and the resultant side effects of hypertension, osteoporosis, avascular necrosis, hyperglycemia, and gastrointestinal tract problems can be avoided. Likewise, avoidance of allergens can lead to the elimination of the need for chronic topical steroids and the resultant atrophy and striae.
Side effects of the patch test procedure itself include an allergic reaction to one of the chemicals tested (eg, gold), which is what you are looking for; persistent reactions; flaring of existing dermatitis; irritation; hyperpigmentation; and rarely anaphylaxis or infection at a patch test site. If no allergy is found, treatment of generalized dermatitis can include topical steroids. Topical calcineurin inhibitors can be useful as well as narrowband UV light. Several oral medications can be used for recalcitrant patch test–negative dermatitis and the selection of the right medication is based on the patient’s comorbidities and extent of dermatitis, including systemic steroids, though long-term use is not recommended. Mycophenolate mofetil, methotrexate, cyclosporine, and azathioprine all have side effects including liver and renal toxicity, immunosuppression, and risk for malignancy and therefore need to be considered on a case-by-case basis.
How do you keep the patient compliant with treatment?
Treating allergic contact dermatitis once an allergen(s) has been identified can be challenging. Education is key so that the patient understands where the allergen is found in his/her environment and how to avoid it. Teaching the patient to read labels also is important. Providing a list of safe products simplifies compliance. Reinforcing the need for ongoing vigilance in allergen avoidance is critical to resolution of the dermatitis. Reinforcing the need for continuous avoidance is imperative, as patients sometimes become less vigilant once the dermatitis resolves and the allergen can sneak back into their environment.
What do I do if a patient refuses treatment?
Sometimes patients are so attached to a product that they do not want to stop using it even though they know it is the cause of their dermatitis. If I can help them identify a comparable product, I introduce them to it, but ultimately they get to decide if they prefer to use a product that they know is the cause of their rash or if they want to avoid it and be clear of the dermatitis. For those who do not have an allergen identified through patch testing, alternative treatments can be used. If they do not want systemic medication, I try and optimize their skin care regimen with mild soaps, bland moisturizing creams, and short lukewarm showers, which often is not enough and eventually due to ongoing itch patients decide to discuss and pursue treatment options.

Atopic Dermatitis Treatments Moving Forward: Report From the AAD Meeting
Although psoriasis was once at the forefront of therapeutic advancements in dermatology, atopic dermatitis (AD) is now taking center stage with several new treatments in the pipeline. Dr. Emma Guttman-Yassky provides an overview of the future of AD treatment, which includes new topical and systemic agents that currently are moving forward in advanced clinical trials or are close to registration. She also discusses strategies for improving disease management in AD patients, noting that prevention and education of both patients and their caregivers are key to effective treatment.
Although psoriasis was once at the forefront of therapeutic advancements in dermatology, atopic dermatitis (AD) is now taking center stage with several new treatments in the pipeline. Dr. Emma Guttman-Yassky provides an overview of the future of AD treatment, which includes new topical and systemic agents that currently are moving forward in advanced clinical trials or are close to registration. She also discusses strategies for improving disease management in AD patients, noting that prevention and education of both patients and their caregivers are key to effective treatment.
Although psoriasis was once at the forefront of therapeutic advancements in dermatology, atopic dermatitis (AD) is now taking center stage with several new treatments in the pipeline. Dr. Emma Guttman-Yassky provides an overview of the future of AD treatment, which includes new topical and systemic agents that currently are moving forward in advanced clinical trials or are close to registration. She also discusses strategies for improving disease management in AD patients, noting that prevention and education of both patients and their caregivers are key to effective treatment.

Night of the Living Thrips: An Unusual Outbreak of Thysanoptera Dermatitis
Case Reports
A platoon of 24 US Marines participated in a 1-week outdoor training exercise (February 4–8) at the Marine Corps Training Area Bellows in Oahu, Hawaii. During the last 3 days of training, 15 (62.5%) marines presented to the same primary care provider with what appeared to be diffuse scattered lesions on the face, neck, and dorsal aspect of the hands. All 15 patients reported that they noticed the lesions upon waking up the morning after their second night at the training area. The patients were unable to recollect specific direct arthropod interactions, but they reported the presence of “bugs” in the training area and denied use of any insect repellents, insect nets, or sunscreen. Sleeping arrangements varied from covered vehicles and cots to sleeping bags on the ground, which were laundered independently by each marine and thereby were ruled out as a commonality. The patients denied working with any chemicals or cleansers while in the field. Further questioning of all 15 patients revealed a history of extended contact with live foliage as branches were broken off to build camouflaged sites.
The following week, a second platoon of 20 marines occupied a separate undisturbed portion of the same training area for a similar 1-week training evolution. Manifestation of similar symptoms among members of the second group, who had no contact with the initial 15 patients, supported the likely environmental etiology of the eruptions.
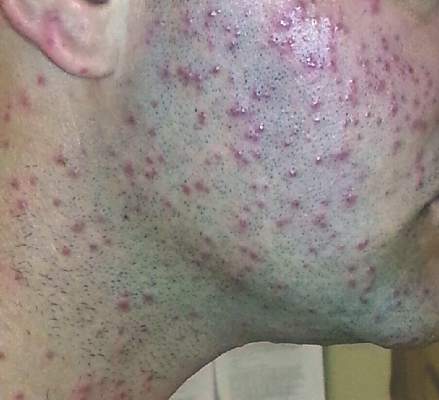
|
| Figure 1. Numerous well-circumscribed, discrete, pink-red papules diffusely scattered across the face. |

|
| Figure 2. Papules with classic anemic halos. |
Referral
Two patients from the first group were evaluated at the dermatology clinic at Tripler Army Medical Center (Honolulu, Hawaii) on day 10 of the initial outbreak. Cutaneous examination revealed numerous discrete, pink-red, well-circumscribed, 2- to 4-mm, dome-shaped papules exclusive to exposed areas on the face, neck, and dorsal aspect of the hands (Figures 1 and 2). Anemic halos surrounding the hand papules were noted (Figure 2). A punch biopsy in both patients revealed spongiotic dermatitis with superficial perivascular and interstitial lymphohistiocytic inflammation with eosinophils, suggestive of an arthropod bite (Figure 3). No retained arthropod parts wereidentified. Both patients were treated with triamcinolone ointment twice daily for 7 days with total resolution of the lesions.
Site Survey Results
Five days following the initial presentation of the first outbreak, a daytime site survey of the training area was conducted by a medical entomologist, an environmental health scientist, and a wildlife biologist. Records indicated that prior to the current utilization, the training area had not been used for 9 months. Approximately half of the training area was covered with mixed scrub vegetation and the remainder was clear pavement or sand (clear of vegetation). Feral hogs (Sus scrofa), cats (Felis domesticus), and mongooses (Herpestes javanicus) were observed at the site. Patient interviews and site survey ruled out a number of potential environmental irritants, including contact with fresh or salt water and chemical contaminants in the air or soil.
Because biting insects were suspected as the cause of the eruptions, an overnight entomological survey was conducted 3 weeks after the first outbreak under similar weather conditions and was centered in the area of an Australian pine (Casuarina equisetifolia) forest where most of the marines had slept during training. Mosquitoes (Aedes albopictus and Culex quinquefasciatus) were observed in the area, with an estimated biting rate of 1 to 2 bites per hour. Centipedes (Scolopendra subspinipes) were commonly observed after dark. There was no sign of heavy bird roosting or nesting, which would be a possible source of biting ectoparasites. Other than the Australian pine, notable vegetation present included Christmasberry (Schinus terebinthifolius), koa haole (Leucaena leucocephala), and Chinese banyan (Ficus microcarpa). A survey of the vegetation uncovered no notable insects, and no damage to the leaves of the Chinese banyans, which is typical of thrip infestation, was noted.
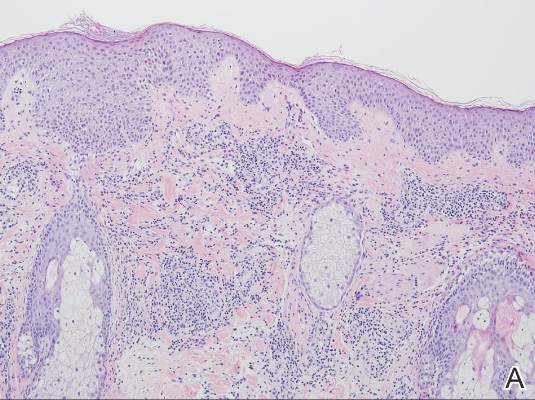
| 
|
| Figure 3. Superficial and deep perivascular and interstitial dermatitis (A)(H&E, original magnification ×10) with lymphocytic predominance (B)(H&E, original magnification ×40). | |
After completion of a resource-intensive investigation that included site survey, literature review, detailed patient history including thrips-associated skin manifestations, and thorough consultation with local dermatologists and entomologists, the findings seemingly pointed to thrips as the most likely etiology of the eruption seen in our patients and a diagnosis of Thysanoptera dermatitis was made.
Comment
Thrips are small winged insects in the order Thysanoptera, which comprises more than 5000 identified species ranging in size from 0.5 to 15 mm, though most are approximately 1 mm.1 The insects typically are phytophagous (feeding on plants) and are attracted to humidity and seemingly the sweat of animals and humans.2 Although largely a phytophagous organism, a few published cases of thrips exposure reported papular skin eruptions known as Thysanoptera dermatitis.3-8 Several species of thrips across the globe have been associated with incidental attacks on humans to include “Heliothrips indicus Bagnall, a cotton pest of the Sudan; Thrips imagines Bagnall, reported in Australia; Limothrips cerealium (Haliday), in Germany; Gynaitkothrips uzeli Zimmerman, in Algeria; and other species.”7 In Hawaii, Gynaikothrips ficorum (Cuban laurel thrips) is a common pest of the Chinese banyan tree (F microcarpa) tree.9
A case series reported by Goldstein and Skipworth5 in the late 1960s of military personnel stationed in Oahu described exposure to similar environmental conditions with resultant lesions that were nearly identical to those seen in our patients. The final conclusion of the investigation was that Cuban laurel thrips were the likely etiology, though mites also were considered.5 In a subsequent commentary in 1968, Waisman10 reported similar eruptions in hospitalized patients with further comment regarding the nocturnal occurrence of the bites. Additionally, the eruptions were reported to be short lasting and devoid of discomfort, similar to our patient population.10
Following suit, Aeling6 published a case series in 1974 depicting several service members who presented with symptoms that were nearly identical to the symptoms experienced by our patients as well as those of Goldstein and Skipworth.5 The investigator coined the term hypoanesthetic halos in Hawaii to describe the findings and further reported that Hawaiian dermatologists were familiar with the symptoms and clinical presentation of the disease. Patients in this outbreak had observed small flying insects, similar to the reports from our patients, and postulated that the symptoms occurred secondary to insect bites.6
Since the report by Goldstein and Skipworth5 in 1968, the majority of the literature regarding Thysanoptera dermatitis has largely been in case reports. In 1987, Fishman7 reported the case of a 43-year-old woman who presented with a palm-sized area of grouped red puncta on the lateral neck with the subsequent entrapment and identification of a flower thrips from the patient’s clothing. In 2005, Leigheb et al2 reported the case of a 30-year-old man with an erythematous papular cutaneous eruption on the anterior chest. In this case, the causative etiology was unequivocally confirmed upon identification of the presence of thrips on biopsy.2 In 2006, Guarneri et al1 reported the case of a 59-year-old farmer who had tentatively been diagnosed with delusional parasitosis until persistent presentation to a dermatologist for evaluation enabled the capture and identification of grain thrips. More recently, another case of likely Thysanoptera dermatitis was published in 2012 after a man presented with a slide-mounted thrip from his skin for evaluation as to a potential cause of a recurrent rash he had been experiencing.11 In all of these cases, it was fortunate that a specific organism could be identified for 2 reasons: (1) members of the order Thysanoptera have a biological cycle of only 11 to 36 days, and (2) thrips may go virtually unnoticed by humans, as they are often difficult to see due to their small size.2,12 Perhaps the most extensive report, however, comes from Childers et al8 in a descriptive case series published in 2005. In this report, the investigators provided a thorough detailing of multiple encounters dating back to 1883 through which patients were inadvertently exposed to various species of thrips and subsequently presented with arthropod bites.
Conclusion
The rapid and clustered manner of patient presentation in this case series makes it unique and highlights the need for further consideration of Thysanoptera dermatitis as a potential etiology for an outbreak of a papular eruption. Further reporting may help to better contextualize the true epidemiology of the condition and subsequently may trigger its greater inclusion in the differential diagnosis for a pruritic papular eruption.
Acknowledgments
We would like to extend our appreciation to Amy Spizuoco, DO (New York, New York), for her assistance with the initial diagnosis; Steve Montgomery, PhD (Honolulu, Hawaii), for his assistance with further entomological discussion of potential etiologies; and John R. Gilstad, MD (Honolulu, Hawaii), for contributing his thoughts on the differential diagnosis of the presenting symptoms.
1. Guarneri F, Guarneri C, Mento G, et al. Pseudo‐delusory syndrome caused by Limothrips cerealium. Int J Dermatol. 2006;45:197-199.
2. Leigheb G, Tiberio R, Filosa G, et al. Thysanoptera dermatitis. J Eur Acad Dermatol Venereol. 2005;19:722-724.
3. Williams CB. A blood sucking thrips. The Entomologist. 1921;54:164.
4. Bailey SF. Thrips attacking man. Can Entomol. 1936;68:95-98.
5. Goldstein N, Skipworth GB. Papular eruption secondary to thrips bites. JAMA. 1968;203:53-55.
6. Aeling JL. Hypoanesthetic halos in Hawaii. Cutis. 1974;14:541-544.
7. Fishman HC. Thrips. Arch Dermatol. 1987;123:993.
8. Childers CC, Beshear RJ, Frantz G, et al. A review of thrips species biting man including records in Florida and Georgia between 1986-1997. Florida Entomologist. 2005;88:447-451.
9. Funasaki GY. Studies on the life cycle and propagation technique of Montandoniola moraguesi (Puton)(Heteroptera: Anthocoridae). Proc Hawaii Entomol Soc. 1966;XIX.2:209-211.
10. Waisman M. Thrips bites dermatitis. JAMA. 1968;204:82.
11. Martin J, Richmond A, Davis BM, et al. Thysanoptera dermatitis presenting as folie à deux. Arch Dermatol. 2012;148:864-865.
12. Cooper RG. Dermatitis & conjunctivitis in workers on an ostrich farm following thrips infestation. Indian J Med Res. 2007;125:588-589.
Case Reports
A platoon of 24 US Marines participated in a 1-week outdoor training exercise (February 4–8) at the Marine Corps Training Area Bellows in Oahu, Hawaii. During the last 3 days of training, 15 (62.5%) marines presented to the same primary care provider with what appeared to be diffuse scattered lesions on the face, neck, and dorsal aspect of the hands. All 15 patients reported that they noticed the lesions upon waking up the morning after their second night at the training area. The patients were unable to recollect specific direct arthropod interactions, but they reported the presence of “bugs” in the training area and denied use of any insect repellents, insect nets, or sunscreen. Sleeping arrangements varied from covered vehicles and cots to sleeping bags on the ground, which were laundered independently by each marine and thereby were ruled out as a commonality. The patients denied working with any chemicals or cleansers while in the field. Further questioning of all 15 patients revealed a history of extended contact with live foliage as branches were broken off to build camouflaged sites.
The following week, a second platoon of 20 marines occupied a separate undisturbed portion of the same training area for a similar 1-week training evolution. Manifestation of similar symptoms among members of the second group, who had no contact with the initial 15 patients, supported the likely environmental etiology of the eruptions.
|
|
| Figure 1. Numerous well-circumscribed, discrete, pink-red papules diffusely scattered across the face. |
|
|
| Figure 2. Papules with classic anemic halos. |
Referral
Two patients from the first group were evaluated at the dermatology clinic at Tripler Army Medical Center (Honolulu, Hawaii) on day 10 of the initial outbreak. Cutaneous examination revealed numerous discrete, pink-red, well-circumscribed, 2- to 4-mm, dome-shaped papules exclusive to exposed areas on the face, neck, and dorsal aspect of the hands (Figures 1 and 2). Anemic halos surrounding the hand papules were noted (Figure 2). A punch biopsy in both patients revealed spongiotic dermatitis with superficial perivascular and interstitial lymphohistiocytic inflammation with eosinophils, suggestive of an arthropod bite (Figure 3). No retained arthropod parts wereidentified. Both patients were treated with triamcinolone ointment twice daily for 7 days with total resolution of the lesions.
Site Survey Results
Five days following the initial presentation of the first outbreak, a daytime site survey of the training area was conducted by a medical entomologist, an environmental health scientist, and a wildlife biologist. Records indicated that prior to the current utilization, the training area had not been used for 9 months. Approximately half of the training area was covered with mixed scrub vegetation and the remainder was clear pavement or sand (clear of vegetation). Feral hogs (Sus scrofa), cats (Felis domesticus), and mongooses (Herpestes javanicus) were observed at the site. Patient interviews and site survey ruled out a number of potential environmental irritants, including contact with fresh or salt water and chemical contaminants in the air or soil.
Because biting insects were suspected as the cause of the eruptions, an overnight entomological survey was conducted 3 weeks after the first outbreak under similar weather conditions and was centered in the area of an Australian pine (Casuarina equisetifolia) forest where most of the marines had slept during training. Mosquitoes (Aedes albopictus and Culex quinquefasciatus) were observed in the area, with an estimated biting rate of 1 to 2 bites per hour. Centipedes (Scolopendra subspinipes) were commonly observed after dark. There was no sign of heavy bird roosting or nesting, which would be a possible source of biting ectoparasites. Other than the Australian pine, notable vegetation present included Christmasberry (Schinus terebinthifolius), koa haole (Leucaena leucocephala), and Chinese banyan (Ficus microcarpa). A survey of the vegetation uncovered no notable insects, and no damage to the leaves of the Chinese banyans, which is typical of thrip infestation, was noted.
|
|
|
| Figure 3. Superficial and deep perivascular and interstitial dermatitis (A)(H&E, original magnification ×10) with lymphocytic predominance (B)(H&E, original magnification ×40). | |
After completion of a resource-intensive investigation that included site survey, literature review, detailed patient history including thrips-associated skin manifestations, and thorough consultation with local dermatologists and entomologists, the findings seemingly pointed to thrips as the most likely etiology of the eruption seen in our patients and a diagnosis of Thysanoptera dermatitis was made.
Comment
Thrips are small winged insects in the order Thysanoptera, which comprises more than 5000 identified species ranging in size from 0.5 to 15 mm, though most are approximately 1 mm.1 The insects typically are phytophagous (feeding on plants) and are attracted to humidity and seemingly the sweat of animals and humans.2 Although largely a phytophagous organism, a few published cases of thrips exposure reported papular skin eruptions known as Thysanoptera dermatitis.3-8 Several species of thrips across the globe have been associated with incidental attacks on humans to include “Heliothrips indicus Bagnall, a cotton pest of the Sudan; Thrips imagines Bagnall, reported in Australia; Limothrips cerealium (Haliday), in Germany; Gynaitkothrips uzeli Zimmerman, in Algeria; and other species.”7 In Hawaii, Gynaikothrips ficorum (Cuban laurel thrips) is a common pest of the Chinese banyan tree (F microcarpa) tree.9
A case series reported by Goldstein and Skipworth5 in the late 1960s of military personnel stationed in Oahu described exposure to similar environmental conditions with resultant lesions that were nearly identical to those seen in our patients. The final conclusion of the investigation was that Cuban laurel thrips were the likely etiology, though mites also were considered.5 In a subsequent commentary in 1968, Waisman10 reported similar eruptions in hospitalized patients with further comment regarding the nocturnal occurrence of the bites. Additionally, the eruptions were reported to be short lasting and devoid of discomfort, similar to our patient population.10
Following suit, Aeling6 published a case series in 1974 depicting several service members who presented with symptoms that were nearly identical to the symptoms experienced by our patients as well as those of Goldstein and Skipworth.5 The investigator coined the term hypoanesthetic halos in Hawaii to describe the findings and further reported that Hawaiian dermatologists were familiar with the symptoms and clinical presentation of the disease. Patients in this outbreak had observed small flying insects, similar to the reports from our patients, and postulated that the symptoms occurred secondary to insect bites.6
Since the report by Goldstein and Skipworth5 in 1968, the majority of the literature regarding Thysanoptera dermatitis has largely been in case reports. In 1987, Fishman7 reported the case of a 43-year-old woman who presented with a palm-sized area of grouped red puncta on the lateral neck with the subsequent entrapment and identification of a flower thrips from the patient’s clothing. In 2005, Leigheb et al2 reported the case of a 30-year-old man with an erythematous papular cutaneous eruption on the anterior chest. In this case, the causative etiology was unequivocally confirmed upon identification of the presence of thrips on biopsy.2 In 2006, Guarneri et al1 reported the case of a 59-year-old farmer who had tentatively been diagnosed with delusional parasitosis until persistent presentation to a dermatologist for evaluation enabled the capture and identification of grain thrips. More recently, another case of likely Thysanoptera dermatitis was published in 2012 after a man presented with a slide-mounted thrip from his skin for evaluation as to a potential cause of a recurrent rash he had been experiencing.11 In all of these cases, it was fortunate that a specific organism could be identified for 2 reasons: (1) members of the order Thysanoptera have a biological cycle of only 11 to 36 days, and (2) thrips may go virtually unnoticed by humans, as they are often difficult to see due to their small size.2,12 Perhaps the most extensive report, however, comes from Childers et al8 in a descriptive case series published in 2005. In this report, the investigators provided a thorough detailing of multiple encounters dating back to 1883 through which patients were inadvertently exposed to various species of thrips and subsequently presented with arthropod bites.
Conclusion
The rapid and clustered manner of patient presentation in this case series makes it unique and highlights the need for further consideration of Thysanoptera dermatitis as a potential etiology for an outbreak of a papular eruption. Further reporting may help to better contextualize the true epidemiology of the condition and subsequently may trigger its greater inclusion in the differential diagnosis for a pruritic papular eruption.
Acknowledgments
We would like to extend our appreciation to Amy Spizuoco, DO (New York, New York), for her assistance with the initial diagnosis; Steve Montgomery, PhD (Honolulu, Hawaii), for his assistance with further entomological discussion of potential etiologies; and John R. Gilstad, MD (Honolulu, Hawaii), for contributing his thoughts on the differential diagnosis of the presenting symptoms.
Case Reports
A platoon of 24 US Marines participated in a 1-week outdoor training exercise (February 4–8) at the Marine Corps Training Area Bellows in Oahu, Hawaii. During the last 3 days of training, 15 (62.5%) marines presented to the same primary care provider with what appeared to be diffuse scattered lesions on the face, neck, and dorsal aspect of the hands. All 15 patients reported that they noticed the lesions upon waking up the morning after their second night at the training area. The patients were unable to recollect specific direct arthropod interactions, but they reported the presence of “bugs” in the training area and denied use of any insect repellents, insect nets, or sunscreen. Sleeping arrangements varied from covered vehicles and cots to sleeping bags on the ground, which were laundered independently by each marine and thereby were ruled out as a commonality. The patients denied working with any chemicals or cleansers while in the field. Further questioning of all 15 patients revealed a history of extended contact with live foliage as branches were broken off to build camouflaged sites.
The following week, a second platoon of 20 marines occupied a separate undisturbed portion of the same training area for a similar 1-week training evolution. Manifestation of similar symptoms among members of the second group, who had no contact with the initial 15 patients, supported the likely environmental etiology of the eruptions.
|
|
| Figure 1. Numerous well-circumscribed, discrete, pink-red papules diffusely scattered across the face. |
|
|
| Figure 2. Papules with classic anemic halos. |
Referral
Two patients from the first group were evaluated at the dermatology clinic at Tripler Army Medical Center (Honolulu, Hawaii) on day 10 of the initial outbreak. Cutaneous examination revealed numerous discrete, pink-red, well-circumscribed, 2- to 4-mm, dome-shaped papules exclusive to exposed areas on the face, neck, and dorsal aspect of the hands (Figures 1 and 2). Anemic halos surrounding the hand papules were noted (Figure 2). A punch biopsy in both patients revealed spongiotic dermatitis with superficial perivascular and interstitial lymphohistiocytic inflammation with eosinophils, suggestive of an arthropod bite (Figure 3). No retained arthropod parts wereidentified. Both patients were treated with triamcinolone ointment twice daily for 7 days with total resolution of the lesions.
Site Survey Results
Five days following the initial presentation of the first outbreak, a daytime site survey of the training area was conducted by a medical entomologist, an environmental health scientist, and a wildlife biologist. Records indicated that prior to the current utilization, the training area had not been used for 9 months. Approximately half of the training area was covered with mixed scrub vegetation and the remainder was clear pavement or sand (clear of vegetation). Feral hogs (Sus scrofa), cats (Felis domesticus), and mongooses (Herpestes javanicus) were observed at the site. Patient interviews and site survey ruled out a number of potential environmental irritants, including contact with fresh or salt water and chemical contaminants in the air or soil.
Because biting insects were suspected as the cause of the eruptions, an overnight entomological survey was conducted 3 weeks after the first outbreak under similar weather conditions and was centered in the area of an Australian pine (Casuarina equisetifolia) forest where most of the marines had slept during training. Mosquitoes (Aedes albopictus and Culex quinquefasciatus) were observed in the area, with an estimated biting rate of 1 to 2 bites per hour. Centipedes (Scolopendra subspinipes) were commonly observed after dark. There was no sign of heavy bird roosting or nesting, which would be a possible source of biting ectoparasites. Other than the Australian pine, notable vegetation present included Christmasberry (Schinus terebinthifolius), koa haole (Leucaena leucocephala), and Chinese banyan (Ficus microcarpa). A survey of the vegetation uncovered no notable insects, and no damage to the leaves of the Chinese banyans, which is typical of thrip infestation, was noted.
|
|
|
| Figure 3. Superficial and deep perivascular and interstitial dermatitis (A)(H&E, original magnification ×10) with lymphocytic predominance (B)(H&E, original magnification ×40). | |
After completion of a resource-intensive investigation that included site survey, literature review, detailed patient history including thrips-associated skin manifestations, and thorough consultation with local dermatologists and entomologists, the findings seemingly pointed to thrips as the most likely etiology of the eruption seen in our patients and a diagnosis of Thysanoptera dermatitis was made.
Comment
Thrips are small winged insects in the order Thysanoptera, which comprises more than 5000 identified species ranging in size from 0.5 to 15 mm, though most are approximately 1 mm.1 The insects typically are phytophagous (feeding on plants) and are attracted to humidity and seemingly the sweat of animals and humans.2 Although largely a phytophagous organism, a few published cases of thrips exposure reported papular skin eruptions known as Thysanoptera dermatitis.3-8 Several species of thrips across the globe have been associated with incidental attacks on humans to include “Heliothrips indicus Bagnall, a cotton pest of the Sudan; Thrips imagines Bagnall, reported in Australia; Limothrips cerealium (Haliday), in Germany; Gynaitkothrips uzeli Zimmerman, in Algeria; and other species.”7 In Hawaii, Gynaikothrips ficorum (Cuban laurel thrips) is a common pest of the Chinese banyan tree (F microcarpa) tree.9
A case series reported by Goldstein and Skipworth5 in the late 1960s of military personnel stationed in Oahu described exposure to similar environmental conditions with resultant lesions that were nearly identical to those seen in our patients. The final conclusion of the investigation was that Cuban laurel thrips were the likely etiology, though mites also were considered.5 In a subsequent commentary in 1968, Waisman10 reported similar eruptions in hospitalized patients with further comment regarding the nocturnal occurrence of the bites. Additionally, the eruptions were reported to be short lasting and devoid of discomfort, similar to our patient population.10
Following suit, Aeling6 published a case series in 1974 depicting several service members who presented with symptoms that were nearly identical to the symptoms experienced by our patients as well as those of Goldstein and Skipworth.5 The investigator coined the term hypoanesthetic halos in Hawaii to describe the findings and further reported that Hawaiian dermatologists were familiar with the symptoms and clinical presentation of the disease. Patients in this outbreak had observed small flying insects, similar to the reports from our patients, and postulated that the symptoms occurred secondary to insect bites.6
Since the report by Goldstein and Skipworth5 in 1968, the majority of the literature regarding Thysanoptera dermatitis has largely been in case reports. In 1987, Fishman7 reported the case of a 43-year-old woman who presented with a palm-sized area of grouped red puncta on the lateral neck with the subsequent entrapment and identification of a flower thrips from the patient’s clothing. In 2005, Leigheb et al2 reported the case of a 30-year-old man with an erythematous papular cutaneous eruption on the anterior chest. In this case, the causative etiology was unequivocally confirmed upon identification of the presence of thrips on biopsy.2 In 2006, Guarneri et al1 reported the case of a 59-year-old farmer who had tentatively been diagnosed with delusional parasitosis until persistent presentation to a dermatologist for evaluation enabled the capture and identification of grain thrips. More recently, another case of likely Thysanoptera dermatitis was published in 2012 after a man presented with a slide-mounted thrip from his skin for evaluation as to a potential cause of a recurrent rash he had been experiencing.11 In all of these cases, it was fortunate that a specific organism could be identified for 2 reasons: (1) members of the order Thysanoptera have a biological cycle of only 11 to 36 days, and (2) thrips may go virtually unnoticed by humans, as they are often difficult to see due to their small size.2,12 Perhaps the most extensive report, however, comes from Childers et al8 in a descriptive case series published in 2005. In this report, the investigators provided a thorough detailing of multiple encounters dating back to 1883 through which patients were inadvertently exposed to various species of thrips and subsequently presented with arthropod bites.
Conclusion
The rapid and clustered manner of patient presentation in this case series makes it unique and highlights the need for further consideration of Thysanoptera dermatitis as a potential etiology for an outbreak of a papular eruption. Further reporting may help to better contextualize the true epidemiology of the condition and subsequently may trigger its greater inclusion in the differential diagnosis for a pruritic papular eruption.
Acknowledgments
We would like to extend our appreciation to Amy Spizuoco, DO (New York, New York), for her assistance with the initial diagnosis; Steve Montgomery, PhD (Honolulu, Hawaii), for his assistance with further entomological discussion of potential etiologies; and John R. Gilstad, MD (Honolulu, Hawaii), for contributing his thoughts on the differential diagnosis of the presenting symptoms.
1. Guarneri F, Guarneri C, Mento G, et al. Pseudo‐delusory syndrome caused by Limothrips cerealium. Int J Dermatol. 2006;45:197-199.
2. Leigheb G, Tiberio R, Filosa G, et al. Thysanoptera dermatitis. J Eur Acad Dermatol Venereol. 2005;19:722-724.
3. Williams CB. A blood sucking thrips. The Entomologist. 1921;54:164.
4. Bailey SF. Thrips attacking man. Can Entomol. 1936;68:95-98.
5. Goldstein N, Skipworth GB. Papular eruption secondary to thrips bites. JAMA. 1968;203:53-55.
6. Aeling JL. Hypoanesthetic halos in Hawaii. Cutis. 1974;14:541-544.
7. Fishman HC. Thrips. Arch Dermatol. 1987;123:993.
8. Childers CC, Beshear RJ, Frantz G, et al. A review of thrips species biting man including records in Florida and Georgia between 1986-1997. Florida Entomologist. 2005;88:447-451.
9. Funasaki GY. Studies on the life cycle and propagation technique of Montandoniola moraguesi (Puton)(Heteroptera: Anthocoridae). Proc Hawaii Entomol Soc. 1966;XIX.2:209-211.
10. Waisman M. Thrips bites dermatitis. JAMA. 1968;204:82.
11. Martin J, Richmond A, Davis BM, et al. Thysanoptera dermatitis presenting as folie à deux. Arch Dermatol. 2012;148:864-865.
12. Cooper RG. Dermatitis & conjunctivitis in workers on an ostrich farm following thrips infestation. Indian J Med Res. 2007;125:588-589.
1. Guarneri F, Guarneri C, Mento G, et al. Pseudo‐delusory syndrome caused by Limothrips cerealium. Int J Dermatol. 2006;45:197-199.
2. Leigheb G, Tiberio R, Filosa G, et al. Thysanoptera dermatitis. J Eur Acad Dermatol Venereol. 2005;19:722-724.
3. Williams CB. A blood sucking thrips. The Entomologist. 1921;54:164.
4. Bailey SF. Thrips attacking man. Can Entomol. 1936;68:95-98.
5. Goldstein N, Skipworth GB. Papular eruption secondary to thrips bites. JAMA. 1968;203:53-55.
6. Aeling JL. Hypoanesthetic halos in Hawaii. Cutis. 1974;14:541-544.
7. Fishman HC. Thrips. Arch Dermatol. 1987;123:993.
8. Childers CC, Beshear RJ, Frantz G, et al. A review of thrips species biting man including records in Florida and Georgia between 1986-1997. Florida Entomologist. 2005;88:447-451.
9. Funasaki GY. Studies on the life cycle and propagation technique of Montandoniola moraguesi (Puton)(Heteroptera: Anthocoridae). Proc Hawaii Entomol Soc. 1966;XIX.2:209-211.
10. Waisman M. Thrips bites dermatitis. JAMA. 1968;204:82.
11. Martin J, Richmond A, Davis BM, et al. Thysanoptera dermatitis presenting as folie à deux. Arch Dermatol. 2012;148:864-865.
12. Cooper RG. Dermatitis & conjunctivitis in workers on an ostrich farm following thrips infestation. Indian J Med Res. 2007;125:588-589.
Practice Points
- Thysanoptera dermatitis presents as a diffuse cutaneous eruption consisting of scattered pruritic papules to exposed skin surfaces.
- The importance of considering the environmental component of a cutaneous eruption via a thorough understanding of local flora and fauna cannot be underestimated.
- The role of a dermatologist in the rapid identification of a cutaneous eruption in the setting of an acute cluster outbreak is of utmost importance to assist with eliminating infectious and environmental public health threats from the differential diagnosis.
Prolonged Pustular Eruption From Hydroxychloroquine: An Unusual Case of Acute Generalized Exanthematous Pustulosis
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
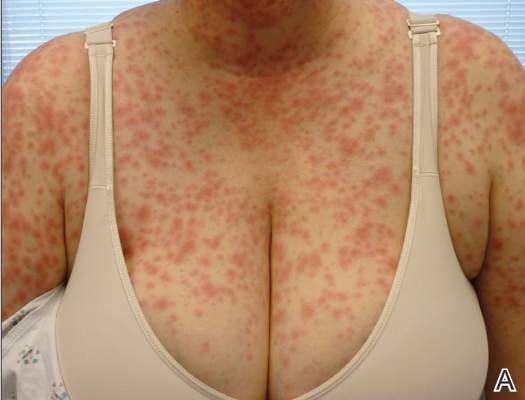

Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
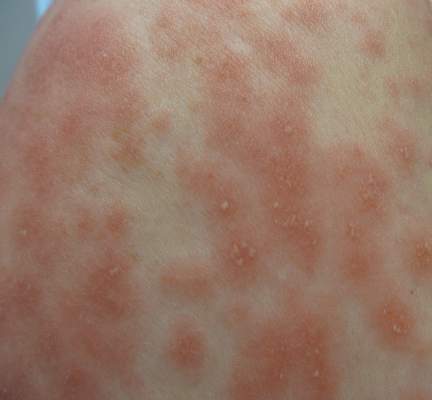
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
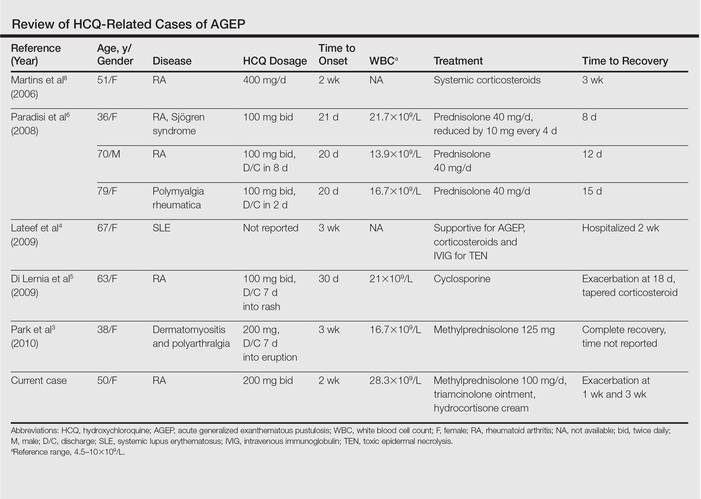
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
Practice Points
- Acute generalized exanthematous pustulosis (AGEP) is most commonly caused by antibiotics (eg, aminopenicillins, macrolides, cephalosporins) followed by calcium channel blockers.
- The main treatment of AGEP is discontinuation of the culprit medication, which typically results in resolution within 2 weeks. Treatment also can symptomatically include topical or systemic corticosteroids and antipyretics.
- Hydroxychloroquine (HCQ) can be a culprit of AGEP with a prolonged recovery course. It is important to inform patients with HCQ-associated AGEP that the clearance of their lesions may take longer than the typical 2 weeks.
Therapies to Improve the Cosmetic Symptoms of Atopic Dermatitis
Atopic dermatitis (AD), more commonly referred to as eczema, is a chronic pruritic inflammatory skin disease that frequently affects both children and adults. Atopic dermatitis is most common in urban and developed countries, with a prevalence of approximately 11% in the United States.1 The pathophysiology of AD is complex and not fully understood, despite the increasing incidence of the disease.2 A myriad of factors, including genetics, defects in the innate and adaptive immune response, and skin barrier abnormalities all contribute to the pathogenesis.3,4 As a result of these abnormalities, patients with AD are more prone to damage from environmental irritants and allergens.
The diagnosis of AD is made clinically based on patient history and visual assessment of the skin.5 Atopic dermatitis follows a chronic and relapsing course characterized by severe pruritus and visible skin changes including xerosis, redness, blistering, oozing, crusting, scaling, thickening, and color change.6,7 Due to the genetic predisposition to make IgE antibodies in response to common environmental and food antigens, patients also may develop allergic rhinitis, asthma, and food-induced anaphylaxis.8,9 Patients also are susceptible to cutaneous viral, fungal, and bacterial infections, the most common of which is an infection with Staphylococcus aureus.10
Atopic dermatitis can have a substantial impact on quality of life, which has been revealed in studies linking chronic skin conditions to depression, impairment of self-esteem, and financial hardship.11 Because skin appearance impacts how a person is initially perceived by others, patients often report feeling self-conscious about their disease and experience teasing or bullying.12 To improve their physical appearance, patients may incur considerable medical expenses. According to 2 population-based studies comprising more than 60,000 adults aged 18 to 85 years, individuals with AD face substantial financial burdens and utilize the health care system more than those without the disease. On average, patients with AD spend $371 to $489 per year on costly out-of-pocket medical expenses and report more absences from work.13
Although there currently is no cure for AD, treatment is aimed at relieving its symptoms and preventing acute exacerbations as well as improving cosmetic appearance to enhance quality of life. Treatment must follow a stepwise approach, which focuses on hydrating the skin, repairing the dysfunctional epithelial barrier, and controlling inflammation. Thus, the standard of care focuses on avoiding skin irritants and triggers along with the use of moisturizers and topical corticosteroids (TCs). In patients with recurring severe disease, topical calcineurin inhibitors, phototherapy, and systemic agents also may be utilized.14
Avoiding Irritants and Triggers
Atopic dermatitis is worsened by skin contact with physical and chemical irritants. Exacerbating factors in AD include exposure to food allergens, dust, emotional stress, detergents, fragranced soaps, textiles, and ingredients in cosmetic products. Patients should be advised to use mild detergents and fragrance-free soaps and to avoid harsh materials such as wool. However, avoidance of specific ingredients in cosmetic products is not as straightforward because manufacturers are not required to disclose certain ingredients. In general, fragrances such as balsam of Peru and cinnamaldehyde, as well as preservatives such as parabens, isothiazolinones, and formaldehyde, should be avoided when selecting cosmetic products. Patients with AD should purchase fragrance-free products that are specifically formulated for sensitive skin. Additionally, patients should not apply makeup if their skin is irritated or oozing, as the flare may worsen.15
Moisturizers
Due to the impaired skin barrier function in patients with AD, regular application of fragrance-free moisturizers is essential to maintain hydration and to reduce xerosis. Various classes of moisturizers may be prescribed (eg, lotions, creams, gels, ointments) based on disease severity and patient preference. Light preparations such as lotions, creams, and gels have a high water content and generally are more appealing from a cosmetic standpoint because they do not create any residue on the skin. However, these options may require more frequent application because they are absorbed quickly. Heavy preparations such as ointments have longer-lasting effects due to their high oil content but tend to be less cosmetically appealing because of their greasiness.16
Although the amount and frequency of application of moisturizers has not been defined, liberal application several times daily is generally advised to minimize xerosis.17 Most physicians recommend applying moisturizer to the skin immediately after bathing to seal in moisture. Some patients prefer to use lotions and creams during the day because these products make the skin feel smooth and reserve the greasier ointments for nighttime application.
Topical Corticosteroids
Prescribed in conjunction with moisturizers, TCs are the mainstay of anti-inflammatory therapy in AD. Topical corticosteroids are classified into 7 groups based on potency, ranging from superpotent (class 1) to least potent (class 7). For acute AD flares, TCs should be applied daily for up to several weeks. Once the inflammation has resolved, it is recommended to apply TCs once to twice weekly to reduce the rate of relapse.18 Despite their effectiveness in the treatment of acute AD flares, TCs have a considerable side-effect profile. Potential adverse effects include skin atrophy, striae, telangiectasia, hypopigmentation, increased hair growth, steroid acne, growth retardation, and Cushing syndrome. Skin atrophy, which is the most common complication associated with TCs, results in shiny transparent skin, allowing for visualization of veins.19,20 Although many of these side effects will resolve after discontinuing the TCs, they are aesthetically displeasing during treatment, making it crucial for physicians to educate their patients on the proper usage of TCs to prevent negative outcomes.
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors (TCIs) are a class of anti-inflammatories that are used to overcome the adverse effects of TCs. They are approved as alternatives to TCs in patients who have failed to respond to other topical treatments as well as those who have developed cutaneous atrophy from the use of TCs or have AD in sensitive areas such as the face, neck, and/or skin folds. Unlike TCs, TCIs do not cause atrophy, striae, or discoloration of the skin, which makes them more desirable from a cosmetic perspective. Their mechanism of action is distinct from TCs in that they inhibit calcineurin-dependent T-cell activation, thus preventing the transcription of inflammatory cytokines.21 Two TCIs are currently available: tacrolimus ointment 0.03% and 0.1% concentrations for moderate to severe AD and pimecrolimus cream 1% for mild to moderate AD.22 Twice-daily application of TCIs is recommended to decrease inflammation and pruritus associated with AD. Studies also have shown that intermittent use of TCIs 3 times weekly can aid in reducing relapses.23-25
The results from clinical trials demonstrate the rapid and continuous effects of both pimecrolimus and tacrolimus. In a controlled long-term study of adults, pimecrolimus provided significant relief of pruritus as soon as day 3 (P<.001).26,27 Pimecrolimus also provides long-term relief by preventing disease progression to flares, which was exemplified in a study (N=713) with no flares in 51% of pimecrolimus patients at 12 months versus 28% in the conventional treatment group (P<.001).28 Similarly, long-term studies of tacrolimus demonstrated an improvement of all symptoms of AD after 1 week of treatment. Maximal improvement was achieved with continued use of tacrolimus, and up to 1 year of tacrolimus use was found to be safe and effective.29,30 Thus, TCIs have been proven to be an effective choice in maintenance therapy for AD and have a good safety profile. The most common adverse effects of TCIs are local skin reactions, such as stinging and burning at the site of application. Rare cases of skin cancer and lymphoma have been reported; however, a causal relationship has yet to be established.31,32
Additional Therapies
Wet wrap therapy is effective for rapid control of flares and in controlling recalcitrant AD. Wet wraps function via several mechanisms; they provide a mechanical barrier against scratching, increase moisture and soften the skin, and enhance absorption of topical medications.33,34 The following method is employed when using wet wraps: an emollient or TC is applied to the area, a tubular bandage soaked in warm water is wrapped over the area, and dry bandages are used to form the outermost layer. Although wet wrap therapy is beneficial in treating AD, it is labor intensive and may require the expertise of a nurse. Thus, unlike other therapies, which patients can easily apply without interfering with their day, wet wraps must be applied at home or in a hospital setting.
Light therapy is another effective method of controlling AD. Although multiple forms of UV phototherapy are beneficial for symptom control in AD, there is no definitive recommendation regarding the specific type of light therapy due to a lack of comparative studies. Natural sunlight, narrowband UVB, broadband UVB, UVA, oral or topical psoralen plus UVA, as well as UVA and UVB can all be utilized in the treatment of AD. However, similar to natural sunlight, artificial light therapy can cause burning, blistering, hyperpigmentation, dark spots, and wrinkles. Because society places a large emphasis on maintaining a youthful appearance, patients may be hesitant to use a treatment that could potentially advance the skin’s aging process. Thus, it is important that this therapy is properly controlled to prevent further skin damage.35-37
When optimal topical regimens and phototherapy have failed to control AD, systemic immunomodulation therapies may be used. Currently, the most commonly used medications are cyclosporine 150 to 300 mg daily, methotrexate 7.5 to 25 mg weekly, mycophenolate mofetil 0.5 to 3 g daily, and azathioprine 1 to 3 mg/kg daily.38,39 Decisions regarding the specific class of drugs should be based on the patient’s AD status, comorbidities, and personal preference.
Conclusion
Atopic dermatitis is a common chronic condition that can occur at any age and cause substantial physical, psychological, social, and/or emotional stress for patients and their families. Although TCs have been the standard of treatment for many years, ongoing concerns regarding their safety have led to the use of TCIs, which overcome some of the drawbacks of steroid therapy. Phototherapy and systemic immunosuppressant therapy are reserved for patients who have not responded to optimal topical therapies. Although several therapeutic avenues exist for patients, there is a need for the development of more effective and safer drugs. Furthermore, cosmetic products created specifically for patients with AD would be beneficial, as patients often struggle to select products that do not cause more harm than good. Given the complexity of the pathogenesis of AD, further research must focus on defining the specific pathways involved in the disease and targeting these pathways with therapies.
1. Shaw TE, Currie GP, Koudelka CW, et al. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131:67-73.
2. Deckers IA, McLean S, Linssen S, et al. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: a systematic review of epidemiological studies. PLoS One. 2012;7:e39803.
3. Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233-246.
4. Peate I. Eczema: causes, symptoms and treatment in the community. Br J Community Nurs. 2011;16:324, 326-331.
5. Williams HC, Burney PG, Pembroke AC, et al. The U.K. Working Party’s diagnostic criteria for atopic dermatitis. III. independent hospital validation. Br J Dermatol. 1994;131:406-416.
6. Magin P, Adams J, Heading G, et al. Experiences of appearance-related teasing and bullying in skin diseases and their psychological sequelae: results of a qualitative study. Scand J Caring Sci. 2008;22:430-436.
7. Beattie P, Lewis-Jones M. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol. 2006;155:145-151.
8. Spergel JM. From atopic dermatitis to asthma: the atopic march [published online January 22, 2010]. Ann Allergy Asthma Immunol. 2010;105:99-106; quiz 107-109, 117.
9. Leung DY. New insights into atopic dermatitis: role of skin barrier and immune dysregulation. Allergol Int. 2013;62:151-161.
10. Balma-Mena A, Lara-Corrales I, Zeller J, et al. Colonization with community-acquired methicillin-resistant Staphylococcus aureus in children with atopic dermatitis: a cross-sectional study. Int J Dermatol. 2011;50:682-688.
11. Strawser MS, Storch EA, Roberti JW. The Teasing Questionnaire-Revised: measurement of childhood teasing in adults. J Anxiety Disord. 2005;19:780-792.
12. Magin P, Adams J, Heading G, et al. Experiences of appearance-related teasing and bullying in skin diseases and their psychological sequelae: results of a qualitative study. Scand J Caring Sci. 2008;22:430-436.
13. Silverberg J. Health care utilization, patient costs, and access to care in US adults with eczema: a population-based study. JAMA Dermatol. 2015;151:743-752.
14. Ellis C, Luger T, Abeck D, et al. International Consensus Conference on Atopic Dermatitis II (ICCAD II): clinical update and current treatment strategies. Br J Dermatol. 2003;148(suppl 63):3-10.
15. Kim K. Influences of environmental chemicals on atopic dermatitis. Toxicol Res. 2015;31:89-96.
16. Ridd M, Redmond N, Hollinghurst S, et al. Choice of Moisturiser for Eczema Treatment (COMET): study protocol for a randomized controlled trial. Trials. 2015;16:304.
17. Hon KL, Ching GK, Leung TF, et al. Estimating emollient usage in patients with eczema. Clin Exp Dermatol. 2010;35:22-26.
18. Hanifin J, Gupta AK, Rajagopalan R. Intermittent dosing of fluticasone propionate cream for reducing the risk of relapse in atopic dermatitis patients. Br J Dermatol. 2002;147:528-537.
19. Hill CJ, Rostenberg A Jr. Adverse effects from topical steroids. Cutis. 1978;21:624-628.
20. Ruiz-Maldonado R, Zapata G, Lourdes T, et al. Cushing’s syndrome after topical application of corticosteroids. Am J Dis Child. 1982;136:274-275.
21. Grassberger M, Baumruker T, Enz A, et al. A novel anti-inflammatory drug, SDZ ASM 981, for the treatment of skin diseases: in vitro pharmacology. Br J Dermatol. 1999;141:264-273.
22. Eichenfield L, Wynnis T, Berger T. Guidelines of care for the management of atopic dermatitis: management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
23. Reitamo S, Harper J, Bos JD, et al. 0.03% Tacrolimus ointment applied once or twice daily is more efficacious than 1% hydrocortisone acetate in children with moderate to severe atopic dermatitis: results of a randomized double-blind controlled trial. Br J Dermatol. 2004;150:554-562.
24. Ruer-Mulard M, Aberer W, Gunstone A, et al. Twice-daily versus once-daily applications of pimecrolimus cream 1% for the prevention of disease relapse in pediatric patients with atopic dermatitis. Pediatr Dermatol. 2009;26:551-558.
25. Breneman D, Fleischer AB Jr, Abramovits W, et al. Intermittent therapy for flare prevention and long-term disease control in stabilized atopic dermatitis: a randomized comparison of 3-times-weekly applications of tacrolimus ointment versus vehicle. J Am Acad Dermatol. 2008;58:990-999.
26. Meurer M, Fölster-Holst R, Wozel G, et al. Pimecrolimus cream 1% (Elidel) provides significant and rapid relief of pruritus and improves disease control and quality of life in atopic dermatitis in adults. J Invest Dermatol. 2002;119:350.
27. Meurer M, Fölster-Holst R, Wozel G, et al. Pimecrolimus cream in the long-term management of atopic dermatitis in adults: a six-month study. Dermatology. 2002;205:271-277.
28. Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):e2.
29. Kang S, Lucky AW, Pariser D, et al. Long-term safety and efficacy of tacrolimus ointment for the treatment of atopic dermatitis in children. J Am Acad Dermatol. 2001;44(suppl 1):S58-S64.
30. Reitamo S, Wollenberg A, Schöpf E, et al. Safety and efficacy of 1 year of tacrolimus ointment monotherapy in adults with atopic dermatitis. the European Tacrolimus Ointment Study Group. Arch Dermatol. 2000;136:999-1006.
31. Frankel HC, Qureshi AA. Comparative effectiveness of topical calcineurin inhibitors in adult patients with atopic dermatitis. Am J Clin Dermatol. 2012;13:113-123.
32. Tennis P, Gelfand JM, Rothman KJ. Evaluation of cancer risk related to atopic dermatitis and use of topical calcineurin inhibitors. Br J Dermatol. 2011;165:465-473.
33. Dabade TS, Davis DM, Wetter DA, et al. Wet dressing therapy in conjunction with topical corticosteroids is effective for rapid control of severe pediatric atopic dermatitis: experience with 218 patients over 30 years at Mayo Clinic. J Am Acad Dermatol. 2012;67:100-106.
34. Devillers AC, Oranje AP. Efficacy and safety of ‘wet-wrap’ dressings as an intervention treatment in children with severe and/or refractory atopic dermatitis: a critical review of the literature. Br J Dermatol. 2006;154:579-585.
35. Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systematic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
36. Clayton TH, Clark SM, Turner D, et al. The treatment of severe atopic dermatitis in childhood with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2007;32:28-33.
37. Jekler J, Larko O. UVB phototherapy of atopic dermatitis. Br J Dermatol. 1988;119:697-705.
38. Roekevisch E, Spuls PI, Kuester D, et al. Efficacy and safety of systemic treatments for moderate-to-severe atopic dermatitis: a systematic review. J Allergy Clin Immunol. 2014;133:429-438.39. Hoare C, Li Wan Po A, Williams H. Systematic review of treatments for atopic eczema. Health Technol Assess. 2000;4:1-191.
39. Hoare C, Li Wan Po A, Williams H. Systematic review of treatments for atopic eczema. Health Technol Assess. 2000;4:1-191.
Atopic dermatitis (AD), more commonly referred to as eczema, is a chronic pruritic inflammatory skin disease that frequently affects both children and adults. Atopic dermatitis is most common in urban and developed countries, with a prevalence of approximately 11% in the United States.1 The pathophysiology of AD is complex and not fully understood, despite the increasing incidence of the disease.2 A myriad of factors, including genetics, defects in the innate and adaptive immune response, and skin barrier abnormalities all contribute to the pathogenesis.3,4 As a result of these abnormalities, patients with AD are more prone to damage from environmental irritants and allergens.
The diagnosis of AD is made clinically based on patient history and visual assessment of the skin.5 Atopic dermatitis follows a chronic and relapsing course characterized by severe pruritus and visible skin changes including xerosis, redness, blistering, oozing, crusting, scaling, thickening, and color change.6,7 Due to the genetic predisposition to make IgE antibodies in response to common environmental and food antigens, patients also may develop allergic rhinitis, asthma, and food-induced anaphylaxis.8,9 Patients also are susceptible to cutaneous viral, fungal, and bacterial infections, the most common of which is an infection with Staphylococcus aureus.10
Atopic dermatitis can have a substantial impact on quality of life, which has been revealed in studies linking chronic skin conditions to depression, impairment of self-esteem, and financial hardship.11 Because skin appearance impacts how a person is initially perceived by others, patients often report feeling self-conscious about their disease and experience teasing or bullying.12 To improve their physical appearance, patients may incur considerable medical expenses. According to 2 population-based studies comprising more than 60,000 adults aged 18 to 85 years, individuals with AD face substantial financial burdens and utilize the health care system more than those without the disease. On average, patients with AD spend $371 to $489 per year on costly out-of-pocket medical expenses and report more absences from work.13
Although there currently is no cure for AD, treatment is aimed at relieving its symptoms and preventing acute exacerbations as well as improving cosmetic appearance to enhance quality of life. Treatment must follow a stepwise approach, which focuses on hydrating the skin, repairing the dysfunctional epithelial barrier, and controlling inflammation. Thus, the standard of care focuses on avoiding skin irritants and triggers along with the use of moisturizers and topical corticosteroids (TCs). In patients with recurring severe disease, topical calcineurin inhibitors, phototherapy, and systemic agents also may be utilized.14
Avoiding Irritants and Triggers
Atopic dermatitis is worsened by skin contact with physical and chemical irritants. Exacerbating factors in AD include exposure to food allergens, dust, emotional stress, detergents, fragranced soaps, textiles, and ingredients in cosmetic products. Patients should be advised to use mild detergents and fragrance-free soaps and to avoid harsh materials such as wool. However, avoidance of specific ingredients in cosmetic products is not as straightforward because manufacturers are not required to disclose certain ingredients. In general, fragrances such as balsam of Peru and cinnamaldehyde, as well as preservatives such as parabens, isothiazolinones, and formaldehyde, should be avoided when selecting cosmetic products. Patients with AD should purchase fragrance-free products that are specifically formulated for sensitive skin. Additionally, patients should not apply makeup if their skin is irritated or oozing, as the flare may worsen.15
Moisturizers
Due to the impaired skin barrier function in patients with AD, regular application of fragrance-free moisturizers is essential to maintain hydration and to reduce xerosis. Various classes of moisturizers may be prescribed (eg, lotions, creams, gels, ointments) based on disease severity and patient preference. Light preparations such as lotions, creams, and gels have a high water content and generally are more appealing from a cosmetic standpoint because they do not create any residue on the skin. However, these options may require more frequent application because they are absorbed quickly. Heavy preparations such as ointments have longer-lasting effects due to their high oil content but tend to be less cosmetically appealing because of their greasiness.16
Although the amount and frequency of application of moisturizers has not been defined, liberal application several times daily is generally advised to minimize xerosis.17 Most physicians recommend applying moisturizer to the skin immediately after bathing to seal in moisture. Some patients prefer to use lotions and creams during the day because these products make the skin feel smooth and reserve the greasier ointments for nighttime application.
Topical Corticosteroids
Prescribed in conjunction with moisturizers, TCs are the mainstay of anti-inflammatory therapy in AD. Topical corticosteroids are classified into 7 groups based on potency, ranging from superpotent (class 1) to least potent (class 7). For acute AD flares, TCs should be applied daily for up to several weeks. Once the inflammation has resolved, it is recommended to apply TCs once to twice weekly to reduce the rate of relapse.18 Despite their effectiveness in the treatment of acute AD flares, TCs have a considerable side-effect profile. Potential adverse effects include skin atrophy, striae, telangiectasia, hypopigmentation, increased hair growth, steroid acne, growth retardation, and Cushing syndrome. Skin atrophy, which is the most common complication associated with TCs, results in shiny transparent skin, allowing for visualization of veins.19,20 Although many of these side effects will resolve after discontinuing the TCs, they are aesthetically displeasing during treatment, making it crucial for physicians to educate their patients on the proper usage of TCs to prevent negative outcomes.
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors (TCIs) are a class of anti-inflammatories that are used to overcome the adverse effects of TCs. They are approved as alternatives to TCs in patients who have failed to respond to other topical treatments as well as those who have developed cutaneous atrophy from the use of TCs or have AD in sensitive areas such as the face, neck, and/or skin folds. Unlike TCs, TCIs do not cause atrophy, striae, or discoloration of the skin, which makes them more desirable from a cosmetic perspective. Their mechanism of action is distinct from TCs in that they inhibit calcineurin-dependent T-cell activation, thus preventing the transcription of inflammatory cytokines.21 Two TCIs are currently available: tacrolimus ointment 0.03% and 0.1% concentrations for moderate to severe AD and pimecrolimus cream 1% for mild to moderate AD.22 Twice-daily application of TCIs is recommended to decrease inflammation and pruritus associated with AD. Studies also have shown that intermittent use of TCIs 3 times weekly can aid in reducing relapses.23-25
The results from clinical trials demonstrate the rapid and continuous effects of both pimecrolimus and tacrolimus. In a controlled long-term study of adults, pimecrolimus provided significant relief of pruritus as soon as day 3 (P<.001).26,27 Pimecrolimus also provides long-term relief by preventing disease progression to flares, which was exemplified in a study (N=713) with no flares in 51% of pimecrolimus patients at 12 months versus 28% in the conventional treatment group (P<.001).28 Similarly, long-term studies of tacrolimus demonstrated an improvement of all symptoms of AD after 1 week of treatment. Maximal improvement was achieved with continued use of tacrolimus, and up to 1 year of tacrolimus use was found to be safe and effective.29,30 Thus, TCIs have been proven to be an effective choice in maintenance therapy for AD and have a good safety profile. The most common adverse effects of TCIs are local skin reactions, such as stinging and burning at the site of application. Rare cases of skin cancer and lymphoma have been reported; however, a causal relationship has yet to be established.31,32
Additional Therapies
Wet wrap therapy is effective for rapid control of flares and in controlling recalcitrant AD. Wet wraps function via several mechanisms; they provide a mechanical barrier against scratching, increase moisture and soften the skin, and enhance absorption of topical medications.33,34 The following method is employed when using wet wraps: an emollient or TC is applied to the area, a tubular bandage soaked in warm water is wrapped over the area, and dry bandages are used to form the outermost layer. Although wet wrap therapy is beneficial in treating AD, it is labor intensive and may require the expertise of a nurse. Thus, unlike other therapies, which patients can easily apply without interfering with their day, wet wraps must be applied at home or in a hospital setting.
Light therapy is another effective method of controlling AD. Although multiple forms of UV phototherapy are beneficial for symptom control in AD, there is no definitive recommendation regarding the specific type of light therapy due to a lack of comparative studies. Natural sunlight, narrowband UVB, broadband UVB, UVA, oral or topical psoralen plus UVA, as well as UVA and UVB can all be utilized in the treatment of AD. However, similar to natural sunlight, artificial light therapy can cause burning, blistering, hyperpigmentation, dark spots, and wrinkles. Because society places a large emphasis on maintaining a youthful appearance, patients may be hesitant to use a treatment that could potentially advance the skin’s aging process. Thus, it is important that this therapy is properly controlled to prevent further skin damage.35-37
When optimal topical regimens and phototherapy have failed to control AD, systemic immunomodulation therapies may be used. Currently, the most commonly used medications are cyclosporine 150 to 300 mg daily, methotrexate 7.5 to 25 mg weekly, mycophenolate mofetil 0.5 to 3 g daily, and azathioprine 1 to 3 mg/kg daily.38,39 Decisions regarding the specific class of drugs should be based on the patient’s AD status, comorbidities, and personal preference.
Conclusion
Atopic dermatitis is a common chronic condition that can occur at any age and cause substantial physical, psychological, social, and/or emotional stress for patients and their families. Although TCs have been the standard of treatment for many years, ongoing concerns regarding their safety have led to the use of TCIs, which overcome some of the drawbacks of steroid therapy. Phototherapy and systemic immunosuppressant therapy are reserved for patients who have not responded to optimal topical therapies. Although several therapeutic avenues exist for patients, there is a need for the development of more effective and safer drugs. Furthermore, cosmetic products created specifically for patients with AD would be beneficial, as patients often struggle to select products that do not cause more harm than good. Given the complexity of the pathogenesis of AD, further research must focus on defining the specific pathways involved in the disease and targeting these pathways with therapies.
Atopic dermatitis (AD), more commonly referred to as eczema, is a chronic pruritic inflammatory skin disease that frequently affects both children and adults. Atopic dermatitis is most common in urban and developed countries, with a prevalence of approximately 11% in the United States.1 The pathophysiology of AD is complex and not fully understood, despite the increasing incidence of the disease.2 A myriad of factors, including genetics, defects in the innate and adaptive immune response, and skin barrier abnormalities all contribute to the pathogenesis.3,4 As a result of these abnormalities, patients with AD are more prone to damage from environmental irritants and allergens.
The diagnosis of AD is made clinically based on patient history and visual assessment of the skin.5 Atopic dermatitis follows a chronic and relapsing course characterized by severe pruritus and visible skin changes including xerosis, redness, blistering, oozing, crusting, scaling, thickening, and color change.6,7 Due to the genetic predisposition to make IgE antibodies in response to common environmental and food antigens, patients also may develop allergic rhinitis, asthma, and food-induced anaphylaxis.8,9 Patients also are susceptible to cutaneous viral, fungal, and bacterial infections, the most common of which is an infection with Staphylococcus aureus.10
Atopic dermatitis can have a substantial impact on quality of life, which has been revealed in studies linking chronic skin conditions to depression, impairment of self-esteem, and financial hardship.11 Because skin appearance impacts how a person is initially perceived by others, patients often report feeling self-conscious about their disease and experience teasing or bullying.12 To improve their physical appearance, patients may incur considerable medical expenses. According to 2 population-based studies comprising more than 60,000 adults aged 18 to 85 years, individuals with AD face substantial financial burdens and utilize the health care system more than those without the disease. On average, patients with AD spend $371 to $489 per year on costly out-of-pocket medical expenses and report more absences from work.13
Although there currently is no cure for AD, treatment is aimed at relieving its symptoms and preventing acute exacerbations as well as improving cosmetic appearance to enhance quality of life. Treatment must follow a stepwise approach, which focuses on hydrating the skin, repairing the dysfunctional epithelial barrier, and controlling inflammation. Thus, the standard of care focuses on avoiding skin irritants and triggers along with the use of moisturizers and topical corticosteroids (TCs). In patients with recurring severe disease, topical calcineurin inhibitors, phototherapy, and systemic agents also may be utilized.14
Avoiding Irritants and Triggers
Atopic dermatitis is worsened by skin contact with physical and chemical irritants. Exacerbating factors in AD include exposure to food allergens, dust, emotional stress, detergents, fragranced soaps, textiles, and ingredients in cosmetic products. Patients should be advised to use mild detergents and fragrance-free soaps and to avoid harsh materials such as wool. However, avoidance of specific ingredients in cosmetic products is not as straightforward because manufacturers are not required to disclose certain ingredients. In general, fragrances such as balsam of Peru and cinnamaldehyde, as well as preservatives such as parabens, isothiazolinones, and formaldehyde, should be avoided when selecting cosmetic products. Patients with AD should purchase fragrance-free products that are specifically formulated for sensitive skin. Additionally, patients should not apply makeup if their skin is irritated or oozing, as the flare may worsen.15
Moisturizers
Due to the impaired skin barrier function in patients with AD, regular application of fragrance-free moisturizers is essential to maintain hydration and to reduce xerosis. Various classes of moisturizers may be prescribed (eg, lotions, creams, gels, ointments) based on disease severity and patient preference. Light preparations such as lotions, creams, and gels have a high water content and generally are more appealing from a cosmetic standpoint because they do not create any residue on the skin. However, these options may require more frequent application because they are absorbed quickly. Heavy preparations such as ointments have longer-lasting effects due to their high oil content but tend to be less cosmetically appealing because of their greasiness.16
Although the amount and frequency of application of moisturizers has not been defined, liberal application several times daily is generally advised to minimize xerosis.17 Most physicians recommend applying moisturizer to the skin immediately after bathing to seal in moisture. Some patients prefer to use lotions and creams during the day because these products make the skin feel smooth and reserve the greasier ointments for nighttime application.
Topical Corticosteroids
Prescribed in conjunction with moisturizers, TCs are the mainstay of anti-inflammatory therapy in AD. Topical corticosteroids are classified into 7 groups based on potency, ranging from superpotent (class 1) to least potent (class 7). For acute AD flares, TCs should be applied daily for up to several weeks. Once the inflammation has resolved, it is recommended to apply TCs once to twice weekly to reduce the rate of relapse.18 Despite their effectiveness in the treatment of acute AD flares, TCs have a considerable side-effect profile. Potential adverse effects include skin atrophy, striae, telangiectasia, hypopigmentation, increased hair growth, steroid acne, growth retardation, and Cushing syndrome. Skin atrophy, which is the most common complication associated with TCs, results in shiny transparent skin, allowing for visualization of veins.19,20 Although many of these side effects will resolve after discontinuing the TCs, they are aesthetically displeasing during treatment, making it crucial for physicians to educate their patients on the proper usage of TCs to prevent negative outcomes.
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors (TCIs) are a class of anti-inflammatories that are used to overcome the adverse effects of TCs. They are approved as alternatives to TCs in patients who have failed to respond to other topical treatments as well as those who have developed cutaneous atrophy from the use of TCs or have AD in sensitive areas such as the face, neck, and/or skin folds. Unlike TCs, TCIs do not cause atrophy, striae, or discoloration of the skin, which makes them more desirable from a cosmetic perspective. Their mechanism of action is distinct from TCs in that they inhibit calcineurin-dependent T-cell activation, thus preventing the transcription of inflammatory cytokines.21 Two TCIs are currently available: tacrolimus ointment 0.03% and 0.1% concentrations for moderate to severe AD and pimecrolimus cream 1% for mild to moderate AD.22 Twice-daily application of TCIs is recommended to decrease inflammation and pruritus associated with AD. Studies also have shown that intermittent use of TCIs 3 times weekly can aid in reducing relapses.23-25
The results from clinical trials demonstrate the rapid and continuous effects of both pimecrolimus and tacrolimus. In a controlled long-term study of adults, pimecrolimus provided significant relief of pruritus as soon as day 3 (P<.001).26,27 Pimecrolimus also provides long-term relief by preventing disease progression to flares, which was exemplified in a study (N=713) with no flares in 51% of pimecrolimus patients at 12 months versus 28% in the conventional treatment group (P<.001).28 Similarly, long-term studies of tacrolimus demonstrated an improvement of all symptoms of AD after 1 week of treatment. Maximal improvement was achieved with continued use of tacrolimus, and up to 1 year of tacrolimus use was found to be safe and effective.29,30 Thus, TCIs have been proven to be an effective choice in maintenance therapy for AD and have a good safety profile. The most common adverse effects of TCIs are local skin reactions, such as stinging and burning at the site of application. Rare cases of skin cancer and lymphoma have been reported; however, a causal relationship has yet to be established.31,32
Additional Therapies
Wet wrap therapy is effective for rapid control of flares and in controlling recalcitrant AD. Wet wraps function via several mechanisms; they provide a mechanical barrier against scratching, increase moisture and soften the skin, and enhance absorption of topical medications.33,34 The following method is employed when using wet wraps: an emollient or TC is applied to the area, a tubular bandage soaked in warm water is wrapped over the area, and dry bandages are used to form the outermost layer. Although wet wrap therapy is beneficial in treating AD, it is labor intensive and may require the expertise of a nurse. Thus, unlike other therapies, which patients can easily apply without interfering with their day, wet wraps must be applied at home or in a hospital setting.
Light therapy is another effective method of controlling AD. Although multiple forms of UV phototherapy are beneficial for symptom control in AD, there is no definitive recommendation regarding the specific type of light therapy due to a lack of comparative studies. Natural sunlight, narrowband UVB, broadband UVB, UVA, oral or topical psoralen plus UVA, as well as UVA and UVB can all be utilized in the treatment of AD. However, similar to natural sunlight, artificial light therapy can cause burning, blistering, hyperpigmentation, dark spots, and wrinkles. Because society places a large emphasis on maintaining a youthful appearance, patients may be hesitant to use a treatment that could potentially advance the skin’s aging process. Thus, it is important that this therapy is properly controlled to prevent further skin damage.35-37
When optimal topical regimens and phototherapy have failed to control AD, systemic immunomodulation therapies may be used. Currently, the most commonly used medications are cyclosporine 150 to 300 mg daily, methotrexate 7.5 to 25 mg weekly, mycophenolate mofetil 0.5 to 3 g daily, and azathioprine 1 to 3 mg/kg daily.38,39 Decisions regarding the specific class of drugs should be based on the patient’s AD status, comorbidities, and personal preference.
Conclusion
Atopic dermatitis is a common chronic condition that can occur at any age and cause substantial physical, psychological, social, and/or emotional stress for patients and their families. Although TCs have been the standard of treatment for many years, ongoing concerns regarding their safety have led to the use of TCIs, which overcome some of the drawbacks of steroid therapy. Phototherapy and systemic immunosuppressant therapy are reserved for patients who have not responded to optimal topical therapies. Although several therapeutic avenues exist for patients, there is a need for the development of more effective and safer drugs. Furthermore, cosmetic products created specifically for patients with AD would be beneficial, as patients often struggle to select products that do not cause more harm than good. Given the complexity of the pathogenesis of AD, further research must focus on defining the specific pathways involved in the disease and targeting these pathways with therapies.
1. Shaw TE, Currie GP, Koudelka CW, et al. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131:67-73.
2. Deckers IA, McLean S, Linssen S, et al. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: a systematic review of epidemiological studies. PLoS One. 2012;7:e39803.
3. Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233-246.
4. Peate I. Eczema: causes, symptoms and treatment in the community. Br J Community Nurs. 2011;16:324, 326-331.
5. Williams HC, Burney PG, Pembroke AC, et al. The U.K. Working Party’s diagnostic criteria for atopic dermatitis. III. independent hospital validation. Br J Dermatol. 1994;131:406-416.
6. Magin P, Adams J, Heading G, et al. Experiences of appearance-related teasing and bullying in skin diseases and their psychological sequelae: results of a qualitative study. Scand J Caring Sci. 2008;22:430-436.
7. Beattie P, Lewis-Jones M. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol. 2006;155:145-151.
8. Spergel JM. From atopic dermatitis to asthma: the atopic march [published online January 22, 2010]. Ann Allergy Asthma Immunol. 2010;105:99-106; quiz 107-109, 117.
9. Leung DY. New insights into atopic dermatitis: role of skin barrier and immune dysregulation. Allergol Int. 2013;62:151-161.
10. Balma-Mena A, Lara-Corrales I, Zeller J, et al. Colonization with community-acquired methicillin-resistant Staphylococcus aureus in children with atopic dermatitis: a cross-sectional study. Int J Dermatol. 2011;50:682-688.
11. Strawser MS, Storch EA, Roberti JW. The Teasing Questionnaire-Revised: measurement of childhood teasing in adults. J Anxiety Disord. 2005;19:780-792.
12. Magin P, Adams J, Heading G, et al. Experiences of appearance-related teasing and bullying in skin diseases and their psychological sequelae: results of a qualitative study. Scand J Caring Sci. 2008;22:430-436.
13. Silverberg J. Health care utilization, patient costs, and access to care in US adults with eczema: a population-based study. JAMA Dermatol. 2015;151:743-752.
14. Ellis C, Luger T, Abeck D, et al. International Consensus Conference on Atopic Dermatitis II (ICCAD II): clinical update and current treatment strategies. Br J Dermatol. 2003;148(suppl 63):3-10.
15. Kim K. Influences of environmental chemicals on atopic dermatitis. Toxicol Res. 2015;31:89-96.
16. Ridd M, Redmond N, Hollinghurst S, et al. Choice of Moisturiser for Eczema Treatment (COMET): study protocol for a randomized controlled trial. Trials. 2015;16:304.
17. Hon KL, Ching GK, Leung TF, et al. Estimating emollient usage in patients with eczema. Clin Exp Dermatol. 2010;35:22-26.
18. Hanifin J, Gupta AK, Rajagopalan R. Intermittent dosing of fluticasone propionate cream for reducing the risk of relapse in atopic dermatitis patients. Br J Dermatol. 2002;147:528-537.
19. Hill CJ, Rostenberg A Jr. Adverse effects from topical steroids. Cutis. 1978;21:624-628.
20. Ruiz-Maldonado R, Zapata G, Lourdes T, et al. Cushing’s syndrome after topical application of corticosteroids. Am J Dis Child. 1982;136:274-275.
21. Grassberger M, Baumruker T, Enz A, et al. A novel anti-inflammatory drug, SDZ ASM 981, for the treatment of skin diseases: in vitro pharmacology. Br J Dermatol. 1999;141:264-273.
22. Eichenfield L, Wynnis T, Berger T. Guidelines of care for the management of atopic dermatitis: management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
23. Reitamo S, Harper J, Bos JD, et al. 0.03% Tacrolimus ointment applied once or twice daily is more efficacious than 1% hydrocortisone acetate in children with moderate to severe atopic dermatitis: results of a randomized double-blind controlled trial. Br J Dermatol. 2004;150:554-562.
24. Ruer-Mulard M, Aberer W, Gunstone A, et al. Twice-daily versus once-daily applications of pimecrolimus cream 1% for the prevention of disease relapse in pediatric patients with atopic dermatitis. Pediatr Dermatol. 2009;26:551-558.
25. Breneman D, Fleischer AB Jr, Abramovits W, et al. Intermittent therapy for flare prevention and long-term disease control in stabilized atopic dermatitis: a randomized comparison of 3-times-weekly applications of tacrolimus ointment versus vehicle. J Am Acad Dermatol. 2008;58:990-999.
26. Meurer M, Fölster-Holst R, Wozel G, et al. Pimecrolimus cream 1% (Elidel) provides significant and rapid relief of pruritus and improves disease control and quality of life in atopic dermatitis in adults. J Invest Dermatol. 2002;119:350.
27. Meurer M, Fölster-Holst R, Wozel G, et al. Pimecrolimus cream in the long-term management of atopic dermatitis in adults: a six-month study. Dermatology. 2002;205:271-277.
28. Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):e2.
29. Kang S, Lucky AW, Pariser D, et al. Long-term safety and efficacy of tacrolimus ointment for the treatment of atopic dermatitis in children. J Am Acad Dermatol. 2001;44(suppl 1):S58-S64.
30. Reitamo S, Wollenberg A, Schöpf E, et al. Safety and efficacy of 1 year of tacrolimus ointment monotherapy in adults with atopic dermatitis. the European Tacrolimus Ointment Study Group. Arch Dermatol. 2000;136:999-1006.
31. Frankel HC, Qureshi AA. Comparative effectiveness of topical calcineurin inhibitors in adult patients with atopic dermatitis. Am J Clin Dermatol. 2012;13:113-123.
32. Tennis P, Gelfand JM, Rothman KJ. Evaluation of cancer risk related to atopic dermatitis and use of topical calcineurin inhibitors. Br J Dermatol. 2011;165:465-473.
33. Dabade TS, Davis DM, Wetter DA, et al. Wet dressing therapy in conjunction with topical corticosteroids is effective for rapid control of severe pediatric atopic dermatitis: experience with 218 patients over 30 years at Mayo Clinic. J Am Acad Dermatol. 2012;67:100-106.
34. Devillers AC, Oranje AP. Efficacy and safety of ‘wet-wrap’ dressings as an intervention treatment in children with severe and/or refractory atopic dermatitis: a critical review of the literature. Br J Dermatol. 2006;154:579-585.
35. Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systematic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
36. Clayton TH, Clark SM, Turner D, et al. The treatment of severe atopic dermatitis in childhood with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2007;32:28-33.
37. Jekler J, Larko O. UVB phototherapy of atopic dermatitis. Br J Dermatol. 1988;119:697-705.
38. Roekevisch E, Spuls PI, Kuester D, et al. Efficacy and safety of systemic treatments for moderate-to-severe atopic dermatitis: a systematic review. J Allergy Clin Immunol. 2014;133:429-438.39. Hoare C, Li Wan Po A, Williams H. Systematic review of treatments for atopic eczema. Health Technol Assess. 2000;4:1-191.
39. Hoare C, Li Wan Po A, Williams H. Systematic review of treatments for atopic eczema. Health Technol Assess. 2000;4:1-191.
1. Shaw TE, Currie GP, Koudelka CW, et al. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131:67-73.
2. Deckers IA, McLean S, Linssen S, et al. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: a systematic review of epidemiological studies. PLoS One. 2012;7:e39803.
3. Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233-246.
4. Peate I. Eczema: causes, symptoms and treatment in the community. Br J Community Nurs. 2011;16:324, 326-331.
5. Williams HC, Burney PG, Pembroke AC, et al. The U.K. Working Party’s diagnostic criteria for atopic dermatitis. III. independent hospital validation. Br J Dermatol. 1994;131:406-416.
6. Magin P, Adams J, Heading G, et al. Experiences of appearance-related teasing and bullying in skin diseases and their psychological sequelae: results of a qualitative study. Scand J Caring Sci. 2008;22:430-436.
7. Beattie P, Lewis-Jones M. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol. 2006;155:145-151.
8. Spergel JM. From atopic dermatitis to asthma: the atopic march [published online January 22, 2010]. Ann Allergy Asthma Immunol. 2010;105:99-106; quiz 107-109, 117.
9. Leung DY. New insights into atopic dermatitis: role of skin barrier and immune dysregulation. Allergol Int. 2013;62:151-161.
10. Balma-Mena A, Lara-Corrales I, Zeller J, et al. Colonization with community-acquired methicillin-resistant Staphylococcus aureus in children with atopic dermatitis: a cross-sectional study. Int J Dermatol. 2011;50:682-688.
11. Strawser MS, Storch EA, Roberti JW. The Teasing Questionnaire-Revised: measurement of childhood teasing in adults. J Anxiety Disord. 2005;19:780-792.
12. Magin P, Adams J, Heading G, et al. Experiences of appearance-related teasing and bullying in skin diseases and their psychological sequelae: results of a qualitative study. Scand J Caring Sci. 2008;22:430-436.
13. Silverberg J. Health care utilization, patient costs, and access to care in US adults with eczema: a population-based study. JAMA Dermatol. 2015;151:743-752.
14. Ellis C, Luger T, Abeck D, et al. International Consensus Conference on Atopic Dermatitis II (ICCAD II): clinical update and current treatment strategies. Br J Dermatol. 2003;148(suppl 63):3-10.
15. Kim K. Influences of environmental chemicals on atopic dermatitis. Toxicol Res. 2015;31:89-96.
16. Ridd M, Redmond N, Hollinghurst S, et al. Choice of Moisturiser for Eczema Treatment (COMET): study protocol for a randomized controlled trial. Trials. 2015;16:304.
17. Hon KL, Ching GK, Leung TF, et al. Estimating emollient usage in patients with eczema. Clin Exp Dermatol. 2010;35:22-26.
18. Hanifin J, Gupta AK, Rajagopalan R. Intermittent dosing of fluticasone propionate cream for reducing the risk of relapse in atopic dermatitis patients. Br J Dermatol. 2002;147:528-537.
19. Hill CJ, Rostenberg A Jr. Adverse effects from topical steroids. Cutis. 1978;21:624-628.
20. Ruiz-Maldonado R, Zapata G, Lourdes T, et al. Cushing’s syndrome after topical application of corticosteroids. Am J Dis Child. 1982;136:274-275.
21. Grassberger M, Baumruker T, Enz A, et al. A novel anti-inflammatory drug, SDZ ASM 981, for the treatment of skin diseases: in vitro pharmacology. Br J Dermatol. 1999;141:264-273.
22. Eichenfield L, Wynnis T, Berger T. Guidelines of care for the management of atopic dermatitis: management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
23. Reitamo S, Harper J, Bos JD, et al. 0.03% Tacrolimus ointment applied once or twice daily is more efficacious than 1% hydrocortisone acetate in children with moderate to severe atopic dermatitis: results of a randomized double-blind controlled trial. Br J Dermatol. 2004;150:554-562.
24. Ruer-Mulard M, Aberer W, Gunstone A, et al. Twice-daily versus once-daily applications of pimecrolimus cream 1% for the prevention of disease relapse in pediatric patients with atopic dermatitis. Pediatr Dermatol. 2009;26:551-558.
25. Breneman D, Fleischer AB Jr, Abramovits W, et al. Intermittent therapy for flare prevention and long-term disease control in stabilized atopic dermatitis: a randomized comparison of 3-times-weekly applications of tacrolimus ointment versus vehicle. J Am Acad Dermatol. 2008;58:990-999.
26. Meurer M, Fölster-Holst R, Wozel G, et al. Pimecrolimus cream 1% (Elidel) provides significant and rapid relief of pruritus and improves disease control and quality of life in atopic dermatitis in adults. J Invest Dermatol. 2002;119:350.
27. Meurer M, Fölster-Holst R, Wozel G, et al. Pimecrolimus cream in the long-term management of atopic dermatitis in adults: a six-month study. Dermatology. 2002;205:271-277.
28. Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):e2.
29. Kang S, Lucky AW, Pariser D, et al. Long-term safety and efficacy of tacrolimus ointment for the treatment of atopic dermatitis in children. J Am Acad Dermatol. 2001;44(suppl 1):S58-S64.
30. Reitamo S, Wollenberg A, Schöpf E, et al. Safety and efficacy of 1 year of tacrolimus ointment monotherapy in adults with atopic dermatitis. the European Tacrolimus Ointment Study Group. Arch Dermatol. 2000;136:999-1006.
31. Frankel HC, Qureshi AA. Comparative effectiveness of topical calcineurin inhibitors in adult patients with atopic dermatitis. Am J Clin Dermatol. 2012;13:113-123.
32. Tennis P, Gelfand JM, Rothman KJ. Evaluation of cancer risk related to atopic dermatitis and use of topical calcineurin inhibitors. Br J Dermatol. 2011;165:465-473.
33. Dabade TS, Davis DM, Wetter DA, et al. Wet dressing therapy in conjunction with topical corticosteroids is effective for rapid control of severe pediatric atopic dermatitis: experience with 218 patients over 30 years at Mayo Clinic. J Am Acad Dermatol. 2012;67:100-106.
34. Devillers AC, Oranje AP. Efficacy and safety of ‘wet-wrap’ dressings as an intervention treatment in children with severe and/or refractory atopic dermatitis: a critical review of the literature. Br J Dermatol. 2006;154:579-585.
35. Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systematic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
36. Clayton TH, Clark SM, Turner D, et al. The treatment of severe atopic dermatitis in childhood with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2007;32:28-33.
37. Jekler J, Larko O. UVB phototherapy of atopic dermatitis. Br J Dermatol. 1988;119:697-705.
38. Roekevisch E, Spuls PI, Kuester D, et al. Efficacy and safety of systemic treatments for moderate-to-severe atopic dermatitis: a systematic review. J Allergy Clin Immunol. 2014;133:429-438.39. Hoare C, Li Wan Po A, Williams H. Systematic review of treatments for atopic eczema. Health Technol Assess. 2000;4:1-191.
39. Hoare C, Li Wan Po A, Williams H. Systematic review of treatments for atopic eczema. Health Technol Assess. 2000;4:1-191.
Practice Points
- Cosmetic symptoms of atopic dermatitis can have a serious impact on the patient’s quality of life.
- Avoidance of flares and prevention of triggers is an important aspect of care.
- Treatment options range from optimized skin care to topical prescription therapies to systemic medications.

An Eruption While on Total Parenteral Nutrition
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
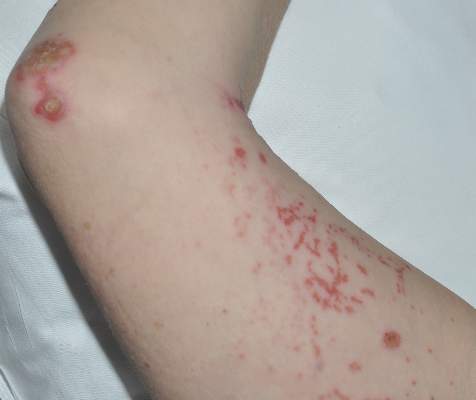
|
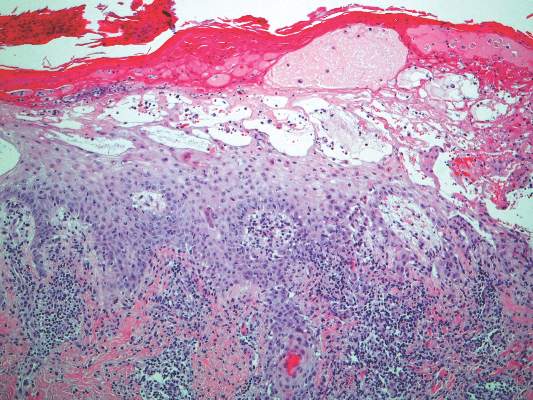
|
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.

Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
|
|
|
|
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.
Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
|
|
|
|
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.
Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
A 47-year-old woman with a history of bulimia and gastroparesis who had been on total parenteral nutrition for 8 weeks presented with a painful, perioral, perineal, and acral eruption of 7 weeks’ duration. Additionally, she had experienced diarrhea, vomiting, and a 13.5-kg weight loss in the last 4 months. Physical examination revealed perioral and perineal, well-demarcated, erythematous, scaly plaques with yellow crusting. She had edematous crusted erosions on the bilateral palms and soles and psoriasiform plaques along the right arm and flank. Punch biopsies (4 mm) from the right inguinal fold and right elbow were obtained.
Allergic Contact Dermatitis, Part 4

Practice Questions
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Question Answers
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Questions
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Question Answers
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Questions
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Question Answers
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix

What Is Your Diagnosis? Stinkbug Staining
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1

Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1
Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1
Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
A 56-year-old woman presented at the clinic for a total-body skin examination. A pigmented lesion was found on the medial aspect of the left first toe during the examination. The patient did not recognize this spot as a long-standing nevus. The area was scrubbed vigorously with an alcohol swab, which did not change the pigment. Clinically the lesion was concerning for an atypical nevus. Dermoscopic examination showed an unusual pattern with pigment deposition in ridges and on furrows.
Pruritic Dermatitis Caused by Bird Mite Infestation
To the Editor:
There are a wide variety of zoonotic diseases that can be transmitted from birds to humans. Pigeons, chickens, starlings, canaries, and parakeets are known reservoirs of one particular zoonotic infection caused by the parasitic arthropod Dermanyssus gallinae.1 Dermanyssus gallinae (chicken mite) and Ornithonyssus sylviarum (northern fowl mite) are collectively referred to as bird mites. When these mites are unable to take blood meals from birds, they search out alternative hosts2; in humans, this often leads to the development of pruritic dermatitis.3
A 30-year-old woman presented to our clinic for evaluation of severe generalized pruritus accompanied by a sensation of “bugs on the skin” of 2 weeks’ duration. She noted the pruritus worsened when she was sitting outside on her porch. A few days prior to presentation, she noticed a small, “pinpoint-sized bug” on her arm (<1 mm in size), which she brought in for identification (Figure).

The bug was identified as a bird mite (Dermanyssus gallinae) on light microscopy, which was later confirmed by a medical entomologist. After the diagnosis of bird mite dermatitis was made, the patient noted there was a nest of starlings above the light on her porch. When she later investigated the nest following the current presentation, she noted many small mites crawling around the nest. The nest was removed and her symptoms resolved completely within 2 weeks without treatment.
Bird mites belong to the Arachnida class, under the order Acari. In 1958, Williams4 noted D gallinae’s ability to feed on human blood. Bird mites have 5 stages of development: egg, larva, protonymph, deutonymph, and adult. Protonymphs, deutonymphs, and adults can bite humans for a blood meal.5 Bird mites range from 0.3 to 1 mm in length and have nonsegmented, egg-shaped bodies with 4 pairs of legs. Before taking a blood meal, bird mites generally are a translucent brown color, and appear red when engorged with blood.2 Their small size makes them barely visible to the unaided eye. Of note, D gallinae and O sylviarum can be distinguished from each other based on subtle differences in morphology; for instance, the posterior genitoventral shield of O sylviarum is narrowly rounded, whereas it is broadly rounded in D gallinae. The dorsal shield of O sylviarum abruptly narrows posteriorly but is more smoothly narrowed in D gallinae.6 Additionally, O sylviarum tends to cause more irritating dermatitis in humans than D gallinae.3
Although they can be found worldwide, D gallinae and O sylviarum undergo optimal development at 20°C to 25°C and 70% humidity.3,5,7 Bird mites generally develop over the course of 5 to 12 days; thus, the population of bird mites in a single nest may grow to the tens of thousands before young birds permanently leave. Dermanyssus gallinae can survive for months in abandoned nests without a blood meal, while O sylviarum can survive for several weeks.8 It is important to note that humans are not ideal hosts for bird mites, as they are unable to survive for extended periods of time or reproduce on human hosts.9
When bird mites are no longer able to obtain blood meals from nesting birds, they begin their nocturnal migration to find suitable hosts. Bird nests generally are abandoned in late spring; thus, most patients with bird mite dermatitis present to clinics with bird mite dermatitis in late spring and early summer.10 Mites often travel through cracks in doors, floors, walls, and ceilings but also can gain access to living areas through ventilation ducts and air conditioning units.1 The mite’s bite and crawling on the skin is sometimes noticed by the patient. In general, however, intense itching is not observed until about 1 to 3 days after the mite makes contact with the skin. Patients often report that pruritus is worst at night.9 Papules and vesicles (bite reactions) may accompany the pruritus, and physicians commonly find bloody crust and excoriations in particularly pruritic areas.5 Urticarial plaques and diffuse erythema occasionally also may be present.9 Bird mites sometimes can be scraped from the skin and observed under light microscopy.11 Blood eosinophilia is not found in bird mite dermatitis. On histologic examination, perivascular eosinophilic infiltration can be seen in the upper part of the dermis.12
The differential diagnosis in patients with pruritic dermatitis of unknown origin generally includes scabies, pediculosis, and dermatitis caused by other types of infestation. However, unlike scabies, bird mites do not cause burrows to form on the skin.9 The presence of a bird’s nest near the area where the patient lives places bird mite dermatitis higher in the differential.
Dermanyssus gallinae is a known vector of bacteria (eg, Salmonella, Shigella, Staphylococcus, Spirochaete, Rickettsia, Pasteurella, Chlamydia psittaci, Erysipelothrix rhusiopathiae) as well as the viruses that cause Eastern and Western equine encephalitis and St. Louis encephalitis. Transmission of these bacteria and viruses is known in birds, but transmission to humans has not been reported.2,5,9,13
The management of bird mite dermatitis is straightforward. Usually mites can be successfully removed from the skin simply by bathing. Symptomatic treatment for bites with antihistamines and topical corticosteroids is sometimes but not always necessary.2 Unlike scabies or lice, there is no need for treatment with lindane.1 In terms of the prevention of additional bites, any bird nests located near living areas should be removed. Because bird mites often retreat back to nests between blood meals, insecticide sprays generally are unnecessary in interior spaces. Synthetic pyrethroids (eg, bifenthrin, cyfluthrin, cypermethrin, deltamethrin, cyhalothrin) can be used outside and in attics where nests may be located.2,14,15 However, the ability of bird mites to develop resistance to repeated chemical control could become a future concern.16
Research regarding the true incidence of bird mite dermatitis is lacking. Some researchers believe that the condition is underreported, possibly due to its uncommon environmental origin.3 Reports of bird mite dermatitis in the literature also are scarce. Our case demonstrates the importance of taking a thorough patient history to rule out exposure to bird mites. All patients with pruritic dermatitis of unknown origin should be questioned about possible contact or proximity to bird nests. These simple questions can lead to the correct diagnosis and a treatment plan that will quickly and effectively resolve the pruritic skin eruption.
- Regan AM, Metersky ML, Craven DE. Nosocomial dermatitis and pruritus caused by pigeon mite infestation. Arch Intern Med. 1987;147:2185-2187.
- Collgros H, Iglesias-Sancho M, Aldunce MJ, et al. Dermanyssus gallinae (chicken mite): an underdiagnosed environmental infestation. Clin Exp Dermatol. 2013;38:374-377.
- Bellanger AP, Boris C, Foulet F, et al. Nosocomial dermatitis caused by Dermanyssus gallinae. Infect Cont Hosp Ep. 2008;29:282-283.
- Williams RW. An infestation of a human habitation by Dermanyssus gallinae (de Geer, 1778) (Acarina: Dermanyssidae) in New York resulting in sanguisugent attacks upon the occupants. Am J Trop Med Hyg. 1958;7:627-629.
- Akdemir C, Gülcan E, Tanritanir P. Case report: Dermanyssus gallinae in a patient with pruritus and skin lesions. Turkiye Parazitol Derg. 2009;33:242-244.
- DiPalma A, Giangaspero A, Cafiero MA, et al. A gallery of the key characteristics to ease identification of Dermanyssus gallinae (Acari: Gamasida: Dermanyssidae) and allow differentiation from Ornithonyssus sylviarum (Acari: Gamasida: Macronyssidae). Parasites and Vectors. 2012;5:104.
- Maurer V, Baumgartner J. Temperature influence on life table statistics of the chicken mite Dermanyssus gallinae (Acari: Dermanyssidae). Exp Appl Acarol. 1992;15:27-40.
- Orton DI, Warren LJ, Wilkinson JD. Avian mite dermatitis. Clin Exper Dermatol. 2000;25:129-131.
- Auger P, Nantel J, Meunier N, et al. Skin acariasis caused by Dermanyssus gallinae (de Geer): an in-hospital outbreak. Can Med Assoc J. 1979;120:700-703.
- Kong TK, To WK. Bird mite infestation. N Engl J Med. 2006;354:1728.
- Koh WL, Liu TT, Tay YK. Formication due to true parasitic infection: bird mites. Arch Dermatol. 2011;147:508-509.
- Hidano A, Asanuma K. Letter: Acariasis caused by bird mites. Arch Dermatol. 1976;112:881-882.
- Valiente Moro C, Chauve C, Zenner L. Experimental infection of Salmonella Enteritidis by the poultry red mite, Dermanyssus gallinae. Vet Parasitol. 2007;146:329-336.
- Fletcher MG, Axtell RC. Susceptibilities of northern fowl mite, Ornithonyssus sylviarum (Acarina: Macronyssidae),and chicken mite, Dermanyssus gallinae (Acarina: Dermanyssidae), to selected acaricides. Exp Appl Acarol. 1991;13:137-142.
- Thind BB, Ford HL. Assessment of susceptibility of the poultry red mite Dermanyssus gallinae (Acari: Dermanyssidae) to some acaricides using an adapted filter paper based bioassay. Vet Parasitol. 2007;144:344-348.
- Chauve C. The poultry red mite Dermanyssus gallinae (De Geer, 1778): current situation and future prospects for control. Vet Parasitol. 1998;79:239-245.
To the Editor:
There are a wide variety of zoonotic diseases that can be transmitted from birds to humans. Pigeons, chickens, starlings, canaries, and parakeets are known reservoirs of one particular zoonotic infection caused by the parasitic arthropod Dermanyssus gallinae.1 Dermanyssus gallinae (chicken mite) and Ornithonyssus sylviarum (northern fowl mite) are collectively referred to as bird mites. When these mites are unable to take blood meals from birds, they search out alternative hosts2; in humans, this often leads to the development of pruritic dermatitis.3
A 30-year-old woman presented to our clinic for evaluation of severe generalized pruritus accompanied by a sensation of “bugs on the skin” of 2 weeks’ duration. She noted the pruritus worsened when she was sitting outside on her porch. A few days prior to presentation, she noticed a small, “pinpoint-sized bug” on her arm (<1 mm in size), which she brought in for identification (Figure).
The bug was identified as a bird mite (Dermanyssus gallinae) on light microscopy, which was later confirmed by a medical entomologist. After the diagnosis of bird mite dermatitis was made, the patient noted there was a nest of starlings above the light on her porch. When she later investigated the nest following the current presentation, she noted many small mites crawling around the nest. The nest was removed and her symptoms resolved completely within 2 weeks without treatment.
Bird mites belong to the Arachnida class, under the order Acari. In 1958, Williams4 noted D gallinae’s ability to feed on human blood. Bird mites have 5 stages of development: egg, larva, protonymph, deutonymph, and adult. Protonymphs, deutonymphs, and adults can bite humans for a blood meal.5 Bird mites range from 0.3 to 1 mm in length and have nonsegmented, egg-shaped bodies with 4 pairs of legs. Before taking a blood meal, bird mites generally are a translucent brown color, and appear red when engorged with blood.2 Their small size makes them barely visible to the unaided eye. Of note, D gallinae and O sylviarum can be distinguished from each other based on subtle differences in morphology; for instance, the posterior genitoventral shield of O sylviarum is narrowly rounded, whereas it is broadly rounded in D gallinae. The dorsal shield of O sylviarum abruptly narrows posteriorly but is more smoothly narrowed in D gallinae.6 Additionally, O sylviarum tends to cause more irritating dermatitis in humans than D gallinae.3
Although they can be found worldwide, D gallinae and O sylviarum undergo optimal development at 20°C to 25°C and 70% humidity.3,5,7 Bird mites generally develop over the course of 5 to 12 days; thus, the population of bird mites in a single nest may grow to the tens of thousands before young birds permanently leave. Dermanyssus gallinae can survive for months in abandoned nests without a blood meal, while O sylviarum can survive for several weeks.8 It is important to note that humans are not ideal hosts for bird mites, as they are unable to survive for extended periods of time or reproduce on human hosts.9
When bird mites are no longer able to obtain blood meals from nesting birds, they begin their nocturnal migration to find suitable hosts. Bird nests generally are abandoned in late spring; thus, most patients with bird mite dermatitis present to clinics with bird mite dermatitis in late spring and early summer.10 Mites often travel through cracks in doors, floors, walls, and ceilings but also can gain access to living areas through ventilation ducts and air conditioning units.1 The mite’s bite and crawling on the skin is sometimes noticed by the patient. In general, however, intense itching is not observed until about 1 to 3 days after the mite makes contact with the skin. Patients often report that pruritus is worst at night.9 Papules and vesicles (bite reactions) may accompany the pruritus, and physicians commonly find bloody crust and excoriations in particularly pruritic areas.5 Urticarial plaques and diffuse erythema occasionally also may be present.9 Bird mites sometimes can be scraped from the skin and observed under light microscopy.11 Blood eosinophilia is not found in bird mite dermatitis. On histologic examination, perivascular eosinophilic infiltration can be seen in the upper part of the dermis.12
The differential diagnosis in patients with pruritic dermatitis of unknown origin generally includes scabies, pediculosis, and dermatitis caused by other types of infestation. However, unlike scabies, bird mites do not cause burrows to form on the skin.9 The presence of a bird’s nest near the area where the patient lives places bird mite dermatitis higher in the differential.
Dermanyssus gallinae is a known vector of bacteria (eg, Salmonella, Shigella, Staphylococcus, Spirochaete, Rickettsia, Pasteurella, Chlamydia psittaci, Erysipelothrix rhusiopathiae) as well as the viruses that cause Eastern and Western equine encephalitis and St. Louis encephalitis. Transmission of these bacteria and viruses is known in birds, but transmission to humans has not been reported.2,5,9,13
The management of bird mite dermatitis is straightforward. Usually mites can be successfully removed from the skin simply by bathing. Symptomatic treatment for bites with antihistamines and topical corticosteroids is sometimes but not always necessary.2 Unlike scabies or lice, there is no need for treatment with lindane.1 In terms of the prevention of additional bites, any bird nests located near living areas should be removed. Because bird mites often retreat back to nests between blood meals, insecticide sprays generally are unnecessary in interior spaces. Synthetic pyrethroids (eg, bifenthrin, cyfluthrin, cypermethrin, deltamethrin, cyhalothrin) can be used outside and in attics where nests may be located.2,14,15 However, the ability of bird mites to develop resistance to repeated chemical control could become a future concern.16
Research regarding the true incidence of bird mite dermatitis is lacking. Some researchers believe that the condition is underreported, possibly due to its uncommon environmental origin.3 Reports of bird mite dermatitis in the literature also are scarce. Our case demonstrates the importance of taking a thorough patient history to rule out exposure to bird mites. All patients with pruritic dermatitis of unknown origin should be questioned about possible contact or proximity to bird nests. These simple questions can lead to the correct diagnosis and a treatment plan that will quickly and effectively resolve the pruritic skin eruption.
To the Editor:
There are a wide variety of zoonotic diseases that can be transmitted from birds to humans. Pigeons, chickens, starlings, canaries, and parakeets are known reservoirs of one particular zoonotic infection caused by the parasitic arthropod Dermanyssus gallinae.1 Dermanyssus gallinae (chicken mite) and Ornithonyssus sylviarum (northern fowl mite) are collectively referred to as bird mites. When these mites are unable to take blood meals from birds, they search out alternative hosts2; in humans, this often leads to the development of pruritic dermatitis.3
A 30-year-old woman presented to our clinic for evaluation of severe generalized pruritus accompanied by a sensation of “bugs on the skin” of 2 weeks’ duration. She noted the pruritus worsened when she was sitting outside on her porch. A few days prior to presentation, she noticed a small, “pinpoint-sized bug” on her arm (<1 mm in size), which she brought in for identification (Figure).
The bug was identified as a bird mite (Dermanyssus gallinae) on light microscopy, which was later confirmed by a medical entomologist. After the diagnosis of bird mite dermatitis was made, the patient noted there was a nest of starlings above the light on her porch. When she later investigated the nest following the current presentation, she noted many small mites crawling around the nest. The nest was removed and her symptoms resolved completely within 2 weeks without treatment.
Bird mites belong to the Arachnida class, under the order Acari. In 1958, Williams4 noted D gallinae’s ability to feed on human blood. Bird mites have 5 stages of development: egg, larva, protonymph, deutonymph, and adult. Protonymphs, deutonymphs, and adults can bite humans for a blood meal.5 Bird mites range from 0.3 to 1 mm in length and have nonsegmented, egg-shaped bodies with 4 pairs of legs. Before taking a blood meal, bird mites generally are a translucent brown color, and appear red when engorged with blood.2 Their small size makes them barely visible to the unaided eye. Of note, D gallinae and O sylviarum can be distinguished from each other based on subtle differences in morphology; for instance, the posterior genitoventral shield of O sylviarum is narrowly rounded, whereas it is broadly rounded in D gallinae. The dorsal shield of O sylviarum abruptly narrows posteriorly but is more smoothly narrowed in D gallinae.6 Additionally, O sylviarum tends to cause more irritating dermatitis in humans than D gallinae.3
Although they can be found worldwide, D gallinae and O sylviarum undergo optimal development at 20°C to 25°C and 70% humidity.3,5,7 Bird mites generally develop over the course of 5 to 12 days; thus, the population of bird mites in a single nest may grow to the tens of thousands before young birds permanently leave. Dermanyssus gallinae can survive for months in abandoned nests without a blood meal, while O sylviarum can survive for several weeks.8 It is important to note that humans are not ideal hosts for bird mites, as they are unable to survive for extended periods of time or reproduce on human hosts.9
When bird mites are no longer able to obtain blood meals from nesting birds, they begin their nocturnal migration to find suitable hosts. Bird nests generally are abandoned in late spring; thus, most patients with bird mite dermatitis present to clinics with bird mite dermatitis in late spring and early summer.10 Mites often travel through cracks in doors, floors, walls, and ceilings but also can gain access to living areas through ventilation ducts and air conditioning units.1 The mite’s bite and crawling on the skin is sometimes noticed by the patient. In general, however, intense itching is not observed until about 1 to 3 days after the mite makes contact with the skin. Patients often report that pruritus is worst at night.9 Papules and vesicles (bite reactions) may accompany the pruritus, and physicians commonly find bloody crust and excoriations in particularly pruritic areas.5 Urticarial plaques and diffuse erythema occasionally also may be present.9 Bird mites sometimes can be scraped from the skin and observed under light microscopy.11 Blood eosinophilia is not found in bird mite dermatitis. On histologic examination, perivascular eosinophilic infiltration can be seen in the upper part of the dermis.12
The differential diagnosis in patients with pruritic dermatitis of unknown origin generally includes scabies, pediculosis, and dermatitis caused by other types of infestation. However, unlike scabies, bird mites do not cause burrows to form on the skin.9 The presence of a bird’s nest near the area where the patient lives places bird mite dermatitis higher in the differential.
Dermanyssus gallinae is a known vector of bacteria (eg, Salmonella, Shigella, Staphylococcus, Spirochaete, Rickettsia, Pasteurella, Chlamydia psittaci, Erysipelothrix rhusiopathiae) as well as the viruses that cause Eastern and Western equine encephalitis and St. Louis encephalitis. Transmission of these bacteria and viruses is known in birds, but transmission to humans has not been reported.2,5,9,13
The management of bird mite dermatitis is straightforward. Usually mites can be successfully removed from the skin simply by bathing. Symptomatic treatment for bites with antihistamines and topical corticosteroids is sometimes but not always necessary.2 Unlike scabies or lice, there is no need for treatment with lindane.1 In terms of the prevention of additional bites, any bird nests located near living areas should be removed. Because bird mites often retreat back to nests between blood meals, insecticide sprays generally are unnecessary in interior spaces. Synthetic pyrethroids (eg, bifenthrin, cyfluthrin, cypermethrin, deltamethrin, cyhalothrin) can be used outside and in attics where nests may be located.2,14,15 However, the ability of bird mites to develop resistance to repeated chemical control could become a future concern.16
Research regarding the true incidence of bird mite dermatitis is lacking. Some researchers believe that the condition is underreported, possibly due to its uncommon environmental origin.3 Reports of bird mite dermatitis in the literature also are scarce. Our case demonstrates the importance of taking a thorough patient history to rule out exposure to bird mites. All patients with pruritic dermatitis of unknown origin should be questioned about possible contact or proximity to bird nests. These simple questions can lead to the correct diagnosis and a treatment plan that will quickly and effectively resolve the pruritic skin eruption.
- Regan AM, Metersky ML, Craven DE. Nosocomial dermatitis and pruritus caused by pigeon mite infestation. Arch Intern Med. 1987;147:2185-2187.
- Collgros H, Iglesias-Sancho M, Aldunce MJ, et al. Dermanyssus gallinae (chicken mite): an underdiagnosed environmental infestation. Clin Exp Dermatol. 2013;38:374-377.
- Bellanger AP, Boris C, Foulet F, et al. Nosocomial dermatitis caused by Dermanyssus gallinae. Infect Cont Hosp Ep. 2008;29:282-283.
- Williams RW. An infestation of a human habitation by Dermanyssus gallinae (de Geer, 1778) (Acarina: Dermanyssidae) in New York resulting in sanguisugent attacks upon the occupants. Am J Trop Med Hyg. 1958;7:627-629.
- Akdemir C, Gülcan E, Tanritanir P. Case report: Dermanyssus gallinae in a patient with pruritus and skin lesions. Turkiye Parazitol Derg. 2009;33:242-244.
- DiPalma A, Giangaspero A, Cafiero MA, et al. A gallery of the key characteristics to ease identification of Dermanyssus gallinae (Acari: Gamasida: Dermanyssidae) and allow differentiation from Ornithonyssus sylviarum (Acari: Gamasida: Macronyssidae). Parasites and Vectors. 2012;5:104.
- Maurer V, Baumgartner J. Temperature influence on life table statistics of the chicken mite Dermanyssus gallinae (Acari: Dermanyssidae). Exp Appl Acarol. 1992;15:27-40.
- Orton DI, Warren LJ, Wilkinson JD. Avian mite dermatitis. Clin Exper Dermatol. 2000;25:129-131.
- Auger P, Nantel J, Meunier N, et al. Skin acariasis caused by Dermanyssus gallinae (de Geer): an in-hospital outbreak. Can Med Assoc J. 1979;120:700-703.
- Kong TK, To WK. Bird mite infestation. N Engl J Med. 2006;354:1728.
- Koh WL, Liu TT, Tay YK. Formication due to true parasitic infection: bird mites. Arch Dermatol. 2011;147:508-509.
- Hidano A, Asanuma K. Letter: Acariasis caused by bird mites. Arch Dermatol. 1976;112:881-882.
- Valiente Moro C, Chauve C, Zenner L. Experimental infection of Salmonella Enteritidis by the poultry red mite, Dermanyssus gallinae. Vet Parasitol. 2007;146:329-336.
- Fletcher MG, Axtell RC. Susceptibilities of northern fowl mite, Ornithonyssus sylviarum (Acarina: Macronyssidae),and chicken mite, Dermanyssus gallinae (Acarina: Dermanyssidae), to selected acaricides. Exp Appl Acarol. 1991;13:137-142.
- Thind BB, Ford HL. Assessment of susceptibility of the poultry red mite Dermanyssus gallinae (Acari: Dermanyssidae) to some acaricides using an adapted filter paper based bioassay. Vet Parasitol. 2007;144:344-348.
- Chauve C. The poultry red mite Dermanyssus gallinae (De Geer, 1778): current situation and future prospects for control. Vet Parasitol. 1998;79:239-245.
- Regan AM, Metersky ML, Craven DE. Nosocomial dermatitis and pruritus caused by pigeon mite infestation. Arch Intern Med. 1987;147:2185-2187.
- Collgros H, Iglesias-Sancho M, Aldunce MJ, et al. Dermanyssus gallinae (chicken mite): an underdiagnosed environmental infestation. Clin Exp Dermatol. 2013;38:374-377.
- Bellanger AP, Boris C, Foulet F, et al. Nosocomial dermatitis caused by Dermanyssus gallinae. Infect Cont Hosp Ep. 2008;29:282-283.
- Williams RW. An infestation of a human habitation by Dermanyssus gallinae (de Geer, 1778) (Acarina: Dermanyssidae) in New York resulting in sanguisugent attacks upon the occupants. Am J Trop Med Hyg. 1958;7:627-629.
- Akdemir C, Gülcan E, Tanritanir P. Case report: Dermanyssus gallinae in a patient with pruritus and skin lesions. Turkiye Parazitol Derg. 2009;33:242-244.
- DiPalma A, Giangaspero A, Cafiero MA, et al. A gallery of the key characteristics to ease identification of Dermanyssus gallinae (Acari: Gamasida: Dermanyssidae) and allow differentiation from Ornithonyssus sylviarum (Acari: Gamasida: Macronyssidae). Parasites and Vectors. 2012;5:104.
- Maurer V, Baumgartner J. Temperature influence on life table statistics of the chicken mite Dermanyssus gallinae (Acari: Dermanyssidae). Exp Appl Acarol. 1992;15:27-40.
- Orton DI, Warren LJ, Wilkinson JD. Avian mite dermatitis. Clin Exper Dermatol. 2000;25:129-131.
- Auger P, Nantel J, Meunier N, et al. Skin acariasis caused by Dermanyssus gallinae (de Geer): an in-hospital outbreak. Can Med Assoc J. 1979;120:700-703.
- Kong TK, To WK. Bird mite infestation. N Engl J Med. 2006;354:1728.
- Koh WL, Liu TT, Tay YK. Formication due to true parasitic infection: bird mites. Arch Dermatol. 2011;147:508-509.
- Hidano A, Asanuma K. Letter: Acariasis caused by bird mites. Arch Dermatol. 1976;112:881-882.
- Valiente Moro C, Chauve C, Zenner L. Experimental infection of Salmonella Enteritidis by the poultry red mite, Dermanyssus gallinae. Vet Parasitol. 2007;146:329-336.
- Fletcher MG, Axtell RC. Susceptibilities of northern fowl mite, Ornithonyssus sylviarum (Acarina: Macronyssidae),and chicken mite, Dermanyssus gallinae (Acarina: Dermanyssidae), to selected acaricides. Exp Appl Acarol. 1991;13:137-142.
- Thind BB, Ford HL. Assessment of susceptibility of the poultry red mite Dermanyssus gallinae (Acari: Dermanyssidae) to some acaricides using an adapted filter paper based bioassay. Vet Parasitol. 2007;144:344-348.
- Chauve C. The poultry red mite Dermanyssus gallinae (De Geer, 1778): current situation and future prospects for control. Vet Parasitol. 1998;79:239-245.
