User login
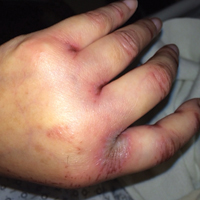
Purpuric Macule of the Right Axilla
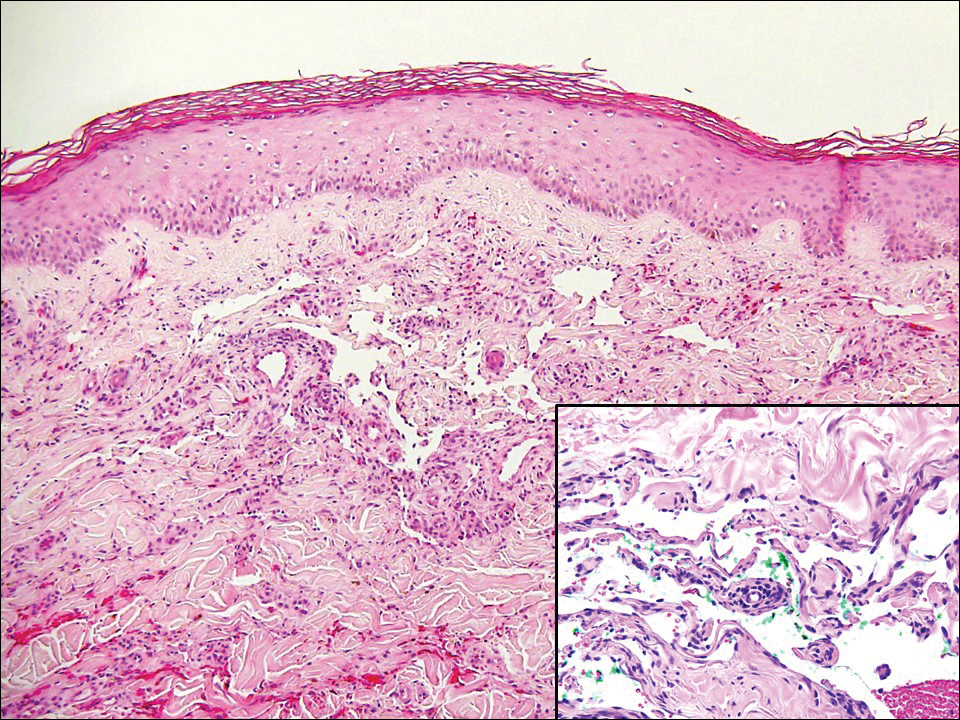
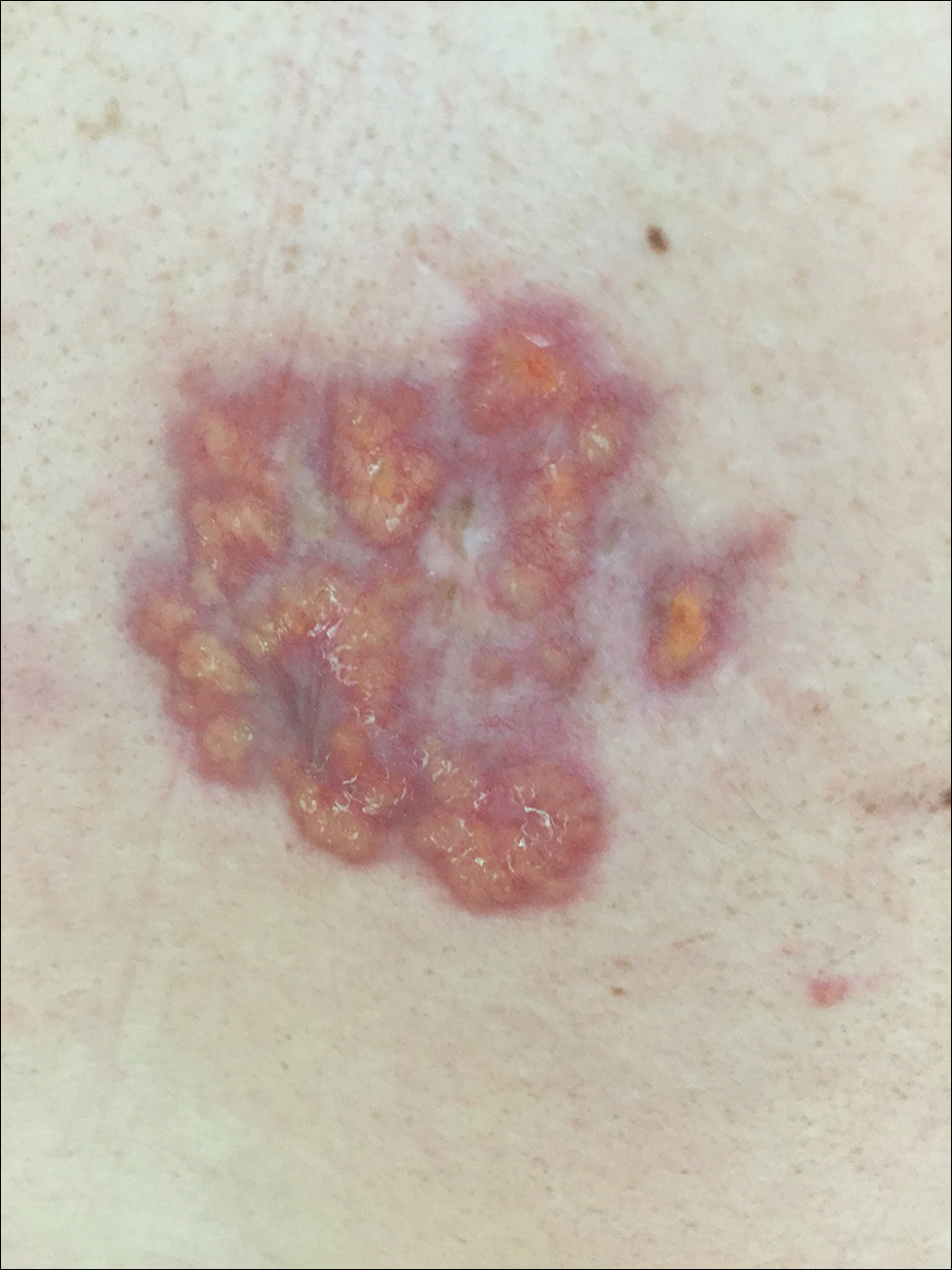
The Diagnosis: Atypical Vascular Lesion
Atypical vascular lesion (AVL)(quiz image), named by Fineberg and Rosen,1 is a vascular lesion that arises on mammary skin with a history of radiation exposure. Clinically, AVL can present as a papule or erythematous patch that manifests 3 to 7 years after radiation therapy.2,3 There are 2 histologic subtypes of AVL: lymphatic and vascular.2,4 Lymphatic-type AVL is comprised of a symmetric distribution of thin, dilated, and anastomosing vessels usually found in the superficial and mid dermis. The vessels are lined by flat or hobnail protuberant endothelial cells that lack nuclear irregularity or pleomorphism; however, hyperchromatism of endothelial cell nuclei is a common finding. Vascular-type AVL is morphologically similar to a capillary hemangioma, and histologic features include irregular growth of capillary-sized vessels that extend to the dermis and subcutis.2,4 Atypical vascular lesions are benign lesions but may be a precursor to angiosarcoma. Along with vascular markers, D2-40 typically is positive. Surgical excision with clear margins is recommended when the lesion is small.4,5 Observation is more appropriate for extensive lesions.
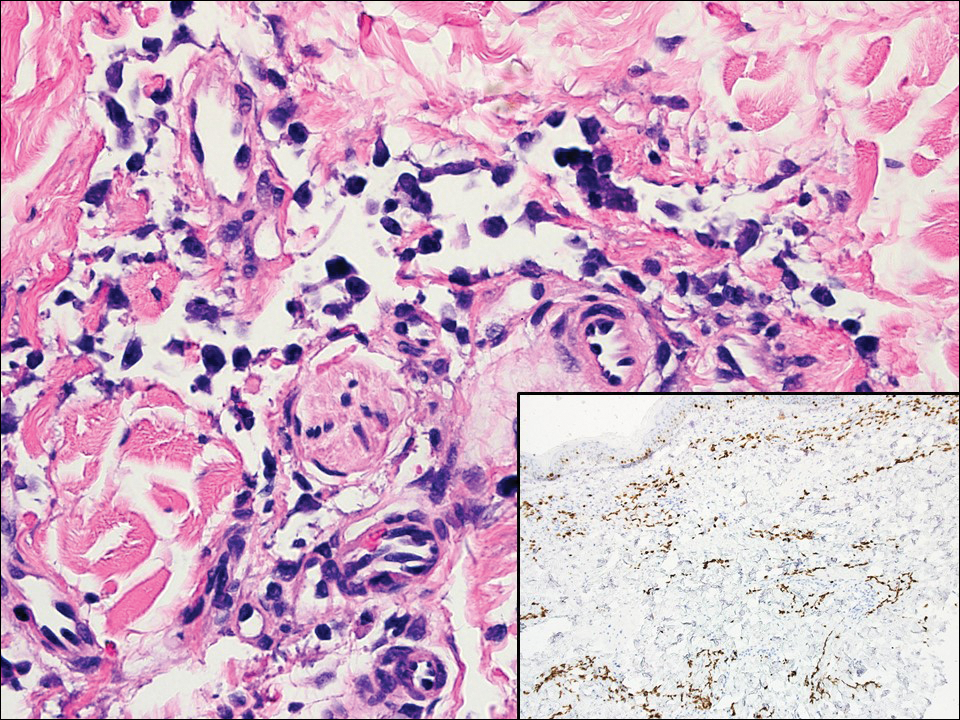
Angiosarcoma can arise spontaneously or in association with radiation or chronic lymphedema. Given the shared risk factors and presentation with AVL, it is essential to differentiate angiosarcoma from AVL. Primary cutaneous angiosarcoma usually presents on the head of elderly patients as an ecchymotic patch or plaque with ulceration.4 Secondary angiosarcoma may arise following radiation or chronic lymphedema (Stewart-Treves syndrome); however, some authors now prefer to consider lymphangiosarcoma arising in chronic lymphedematous limbs a distinct entity.6 Surgical excision with wide margins is the mainstay of therapy, but angiosarcoma has high recurrence rates, and the 5-year survival rate has been reported to be as low as 35%.7 Histologic overlap with AVL includes dissecting anastomosing vessels lined by hyperchromatic nuclei; however, angiosarcoma is distinguished by endothelial cell layering, nuclear pleomorphism, and prominent nucleoli (Figure 1).4,8 Increased positivity for Ki-67 immunostain, which indicates cell proliferation, may be used to distinguish angiosarcoma from an AVL (Figure 1 [inset]).9 Further, in contrast to AVL, radiation-induced angiosarcoma is characterized by amplification of C-MYC, a regulator gene, and FLT4 (FMS-related tyrosine kinase 4), a gene encoding vascular endothelial growth factor receptor 3. Gene amplification may be detected through immunohistochemistry or fluorescence in situ hybridization.10 Ki-67 labeling showed less than 10% staining in endothelial cells in our case (quiz image [inset]), and fluorescence in situ hybridization was negative for C-MYC amplification, supporting the diagnosis of AVL.
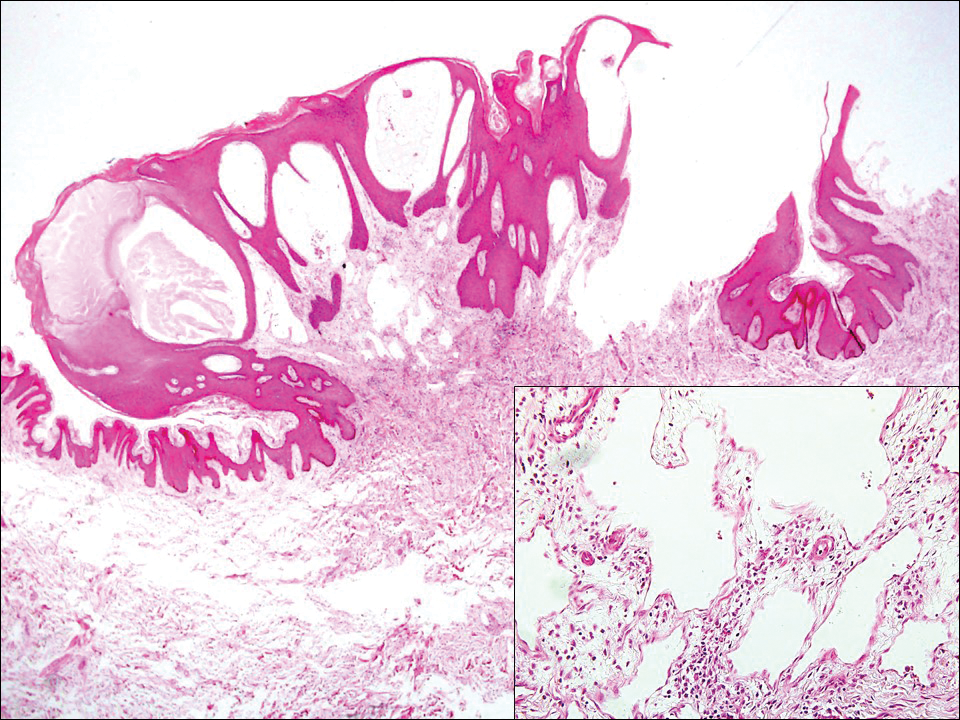
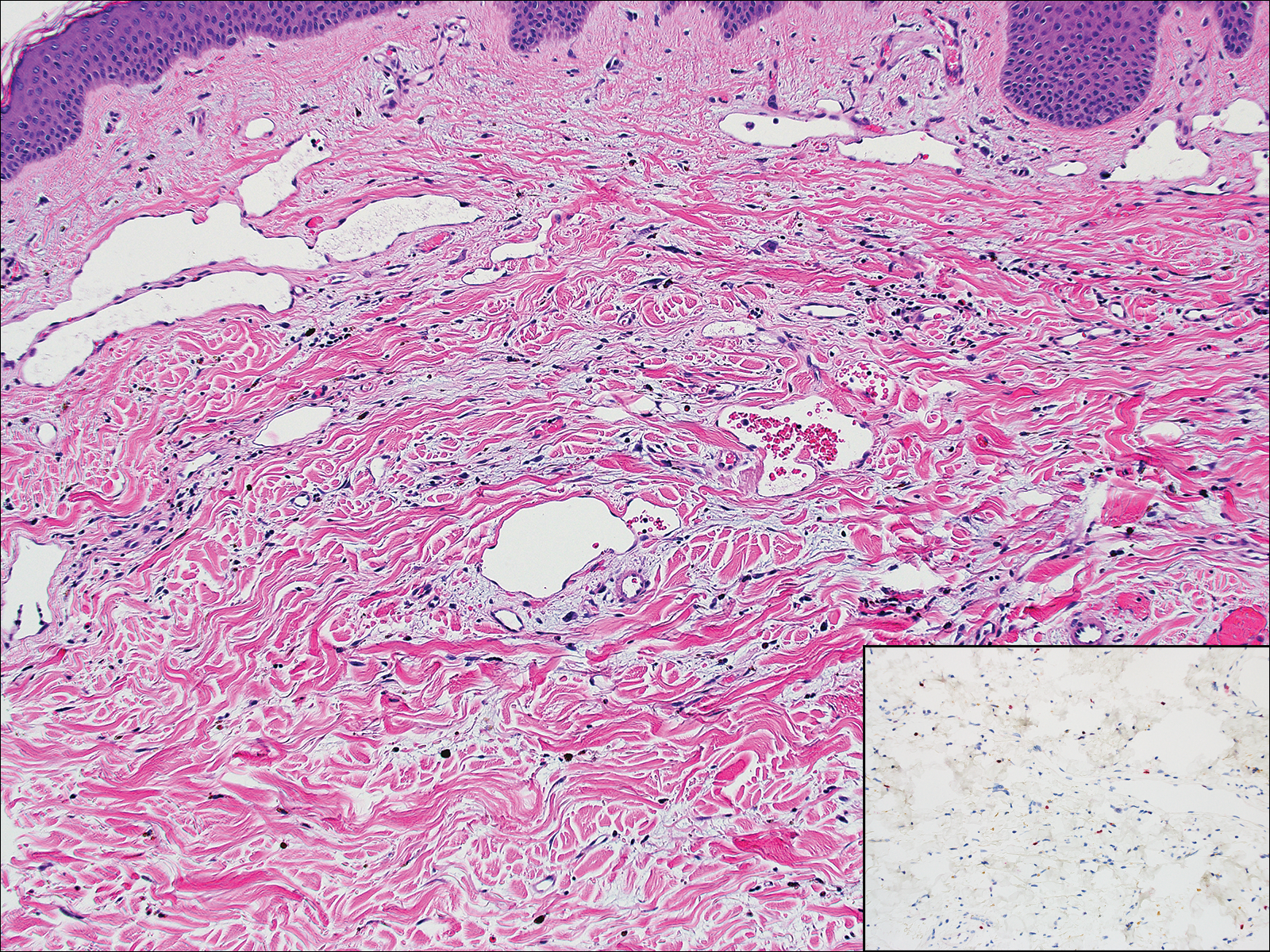
Lymphangioma circumscriptum, the most common superficial lymphangioma, is a hamartomatous malformation that usually occurs at the axillary folds, neck, and trunk. It clinically presents as small agminated vesicles with a characteristic frog spawn appearance.11 Dermoscopic features include yellow lacunae that may alternate with a dark red color secondary to extravasation of erythrocytes.12 These clinical features often lead to a differential diagnosis of verrucae, angiokeratoma, and angiosarcoma. Lymphangioma circumscriptum histologically is characterized by an overgrowth of dilated lymphatic vessels that fill the papillary dermis. The vessels are composed of flat endothelial cells typically filled with acellular proteinaceous debris and occasional erythrocytes (Figure 2). As the lesion traverses deeper into the dermis, the caliber of the lymphatic channel becomes narrower. The presence of deep lymphatic cisterns with surrounding smooth muscle is helpful to differentiate lymphangioma circumscriptum from other lymphatic malformations such as acquired lymphangiectasia. Treatment options include surgical excision, sclerosing agents, and destructive modalities such as cryotherapy.
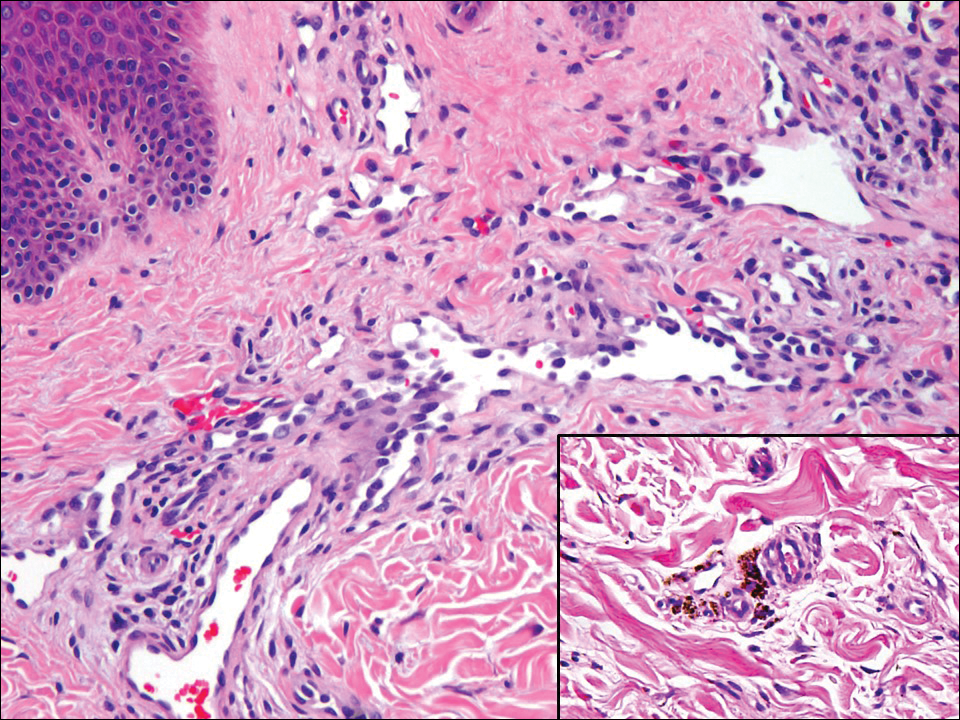
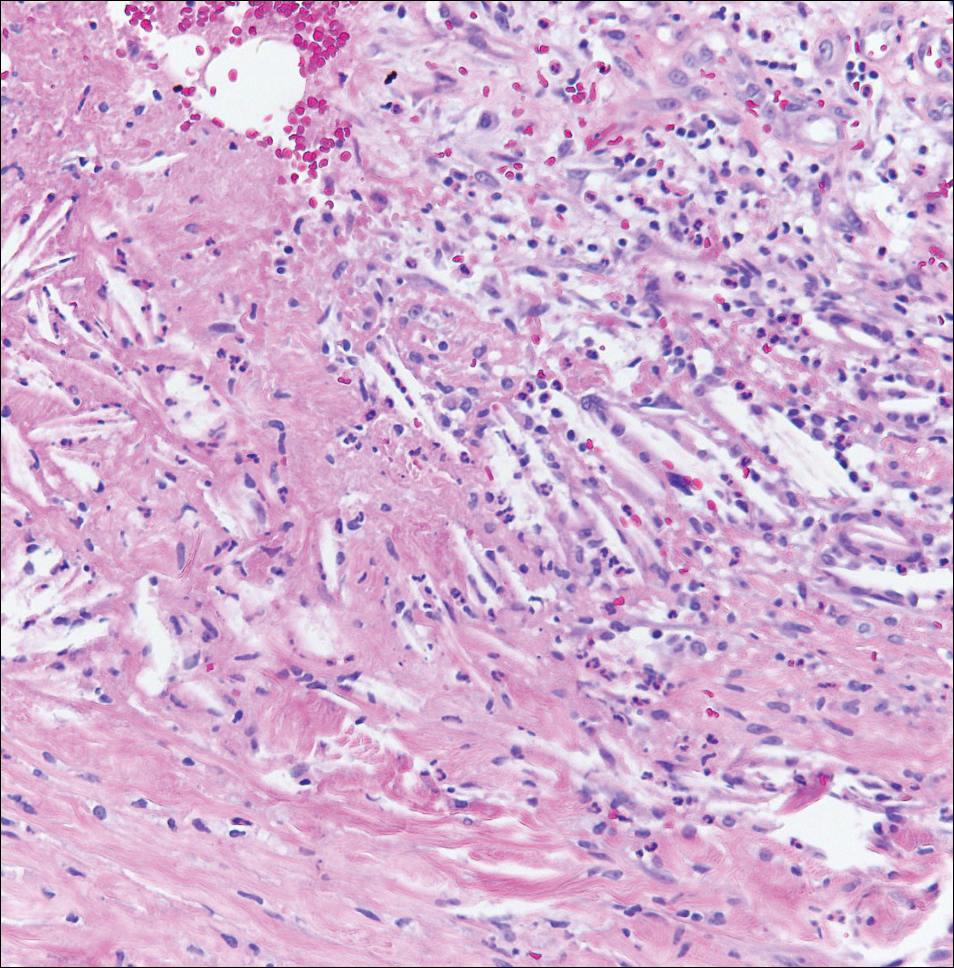
Hobnail hemangioma, originally termed targetoid hemosiderotic hemangioma by Santa Cruz and Aronberg,13 presents as a violaceous papule or nodule surrounded by a characteristic brown halo on the leg. Trauma has been proposed as the inciting factor for the clinical appearance of hobnail hemangioma.14 Microscopically, the lesion shows vessels in a wedge shape. The superficial component has telangiectatic vessels with focal areas of papillary projections lined by endothelial cells. Although the endothelial nuclei typically project into the lumen, the nuclei are small, bland, and without mitotic activity.15 Deeper components show slit-shaped vasculature with dermal collagen dissection. Hemosiderin, extravasated red blood cells, and inflammation are found adjacent to the vessels (Figure 3). Given the benign nature, hobnail hemangiomas may be monitored.
Kaposi sarcoma (KS) is a low-grade vascular neoplasm associated with human herpesvirus 8 that arises in multiple clinical settings, especially in immunosuppression secondary to human immunodeficiency virus. There are 3 distinct clinical stages: patch, plaque, and tumor. The patch stage appears as red macules that blend into larger plaques; the tumor stage is defined as larger nodules developing from plaques. Histologic features differ by stage. Similar to angiosarcoma, KS is comprised of anastomosing vessels that dissect collagen bundles; endothelial cell atypia is minimal. A useful feature of KS is its propensity to involve adnexa and display the promontory sign, which involves the tumor growing into normal vasculature (Figure 4).16 Positive immunohistochemistry for human herpesvirus 8 aids in confirmation of the diagnosis. Treatment options for KS are numerous but include destructive modalities, chemotherapeutic agents such as doxorubicin, or highly active antiretroviral therapy for AIDS-related KS.17
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Billings SD, McKenney JK, Folpe AL, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation: an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Udager AM, Ishikawa MK, Lucas DR, et al. MYC immunohistochemistry in angiosarcoma and atypical vascular lesions: practical considerations based on a single institutional experience. Pathology. 2016;48:697-704.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Elsevier; 2016:1069-1115.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fraga-Guedes C, Gobbi H, Mastropasqua MG, et al. Clinicopathological and immunohistochemical study of 30 cases of post-radiation atypical vascular lesion of the breast. Breast Cancer Res Treat. 2014;146:347-354.
- Shin SJ, Lesser M, Rosen PP. Hemangiomas and angiosarcomas of the breast: diagnostic utility of cell cycle markers with emphasis on Ki-67. Arch Pathol Lab Med. 2007;131:538-544.
- Cornejo KM, Deng A, Wu H, et al. The utility of MYC and FLT4 in the diagnosis and treatment of postradiation atypical vascular lesion and angiosarcoma of the breast. Hum Pathol. 2015;46:868-875.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
- Massa AF, Menezes N, Baptista A, et al. Cutaneous lymphangioma circumscriptum--dermoscopic features. An Bras Dermatol. 2015;90:262-264.
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Christenson LJ, Stone MS. Trauma-induced simulator of targetoid hemosiderotic hemangioma. Am J Dermatopathol. 2001;23:221-223.
- Trindade F, Kutzner H, Tellechea O, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer. 2008;112:1147-1152.
The Diagnosis: Atypical Vascular Lesion
Atypical vascular lesion (AVL)(quiz image), named by Fineberg and Rosen,1 is a vascular lesion that arises on mammary skin with a history of radiation exposure. Clinically, AVL can present as a papule or erythematous patch that manifests 3 to 7 years after radiation therapy.2,3 There are 2 histologic subtypes of AVL: lymphatic and vascular.2,4 Lymphatic-type AVL is comprised of a symmetric distribution of thin, dilated, and anastomosing vessels usually found in the superficial and mid dermis. The vessels are lined by flat or hobnail protuberant endothelial cells that lack nuclear irregularity or pleomorphism; however, hyperchromatism of endothelial cell nuclei is a common finding. Vascular-type AVL is morphologically similar to a capillary hemangioma, and histologic features include irregular growth of capillary-sized vessels that extend to the dermis and subcutis.2,4 Atypical vascular lesions are benign lesions but may be a precursor to angiosarcoma. Along with vascular markers, D2-40 typically is positive. Surgical excision with clear margins is recommended when the lesion is small.4,5 Observation is more appropriate for extensive lesions.
Angiosarcoma can arise spontaneously or in association with radiation or chronic lymphedema. Given the shared risk factors and presentation with AVL, it is essential to differentiate angiosarcoma from AVL. Primary cutaneous angiosarcoma usually presents on the head of elderly patients as an ecchymotic patch or plaque with ulceration.4 Secondary angiosarcoma may arise following radiation or chronic lymphedema (Stewart-Treves syndrome); however, some authors now prefer to consider lymphangiosarcoma arising in chronic lymphedematous limbs a distinct entity.6 Surgical excision with wide margins is the mainstay of therapy, but angiosarcoma has high recurrence rates, and the 5-year survival rate has been reported to be as low as 35%.7 Histologic overlap with AVL includes dissecting anastomosing vessels lined by hyperchromatic nuclei; however, angiosarcoma is distinguished by endothelial cell layering, nuclear pleomorphism, and prominent nucleoli (Figure 1).4,8 Increased positivity for Ki-67 immunostain, which indicates cell proliferation, may be used to distinguish angiosarcoma from an AVL (Figure 1 [inset]).9 Further, in contrast to AVL, radiation-induced angiosarcoma is characterized by amplification of C-MYC, a regulator gene, and FLT4 (FMS-related tyrosine kinase 4), a gene encoding vascular endothelial growth factor receptor 3. Gene amplification may be detected through immunohistochemistry or fluorescence in situ hybridization.10 Ki-67 labeling showed less than 10% staining in endothelial cells in our case (quiz image [inset]), and fluorescence in situ hybridization was negative for C-MYC amplification, supporting the diagnosis of AVL.
Lymphangioma circumscriptum, the most common superficial lymphangioma, is a hamartomatous malformation that usually occurs at the axillary folds, neck, and trunk. It clinically presents as small agminated vesicles with a characteristic frog spawn appearance.11 Dermoscopic features include yellow lacunae that may alternate with a dark red color secondary to extravasation of erythrocytes.12 These clinical features often lead to a differential diagnosis of verrucae, angiokeratoma, and angiosarcoma. Lymphangioma circumscriptum histologically is characterized by an overgrowth of dilated lymphatic vessels that fill the papillary dermis. The vessels are composed of flat endothelial cells typically filled with acellular proteinaceous debris and occasional erythrocytes (Figure 2). As the lesion traverses deeper into the dermis, the caliber of the lymphatic channel becomes narrower. The presence of deep lymphatic cisterns with surrounding smooth muscle is helpful to differentiate lymphangioma circumscriptum from other lymphatic malformations such as acquired lymphangiectasia. Treatment options include surgical excision, sclerosing agents, and destructive modalities such as cryotherapy.
Hobnail hemangioma, originally termed targetoid hemosiderotic hemangioma by Santa Cruz and Aronberg,13 presents as a violaceous papule or nodule surrounded by a characteristic brown halo on the leg. Trauma has been proposed as the inciting factor for the clinical appearance of hobnail hemangioma.14 Microscopically, the lesion shows vessels in a wedge shape. The superficial component has telangiectatic vessels with focal areas of papillary projections lined by endothelial cells. Although the endothelial nuclei typically project into the lumen, the nuclei are small, bland, and without mitotic activity.15 Deeper components show slit-shaped vasculature with dermal collagen dissection. Hemosiderin, extravasated red blood cells, and inflammation are found adjacent to the vessels (Figure 3). Given the benign nature, hobnail hemangiomas may be monitored.
Kaposi sarcoma (KS) is a low-grade vascular neoplasm associated with human herpesvirus 8 that arises in multiple clinical settings, especially in immunosuppression secondary to human immunodeficiency virus. There are 3 distinct clinical stages: patch, plaque, and tumor. The patch stage appears as red macules that blend into larger plaques; the tumor stage is defined as larger nodules developing from plaques. Histologic features differ by stage. Similar to angiosarcoma, KS is comprised of anastomosing vessels that dissect collagen bundles; endothelial cell atypia is minimal. A useful feature of KS is its propensity to involve adnexa and display the promontory sign, which involves the tumor growing into normal vasculature (Figure 4).16 Positive immunohistochemistry for human herpesvirus 8 aids in confirmation of the diagnosis. Treatment options for KS are numerous but include destructive modalities, chemotherapeutic agents such as doxorubicin, or highly active antiretroviral therapy for AIDS-related KS.17
The Diagnosis: Atypical Vascular Lesion
Atypical vascular lesion (AVL)(quiz image), named by Fineberg and Rosen,1 is a vascular lesion that arises on mammary skin with a history of radiation exposure. Clinically, AVL can present as a papule or erythematous patch that manifests 3 to 7 years after radiation therapy.2,3 There are 2 histologic subtypes of AVL: lymphatic and vascular.2,4 Lymphatic-type AVL is comprised of a symmetric distribution of thin, dilated, and anastomosing vessels usually found in the superficial and mid dermis. The vessels are lined by flat or hobnail protuberant endothelial cells that lack nuclear irregularity or pleomorphism; however, hyperchromatism of endothelial cell nuclei is a common finding. Vascular-type AVL is morphologically similar to a capillary hemangioma, and histologic features include irregular growth of capillary-sized vessels that extend to the dermis and subcutis.2,4 Atypical vascular lesions are benign lesions but may be a precursor to angiosarcoma. Along with vascular markers, D2-40 typically is positive. Surgical excision with clear margins is recommended when the lesion is small.4,5 Observation is more appropriate for extensive lesions.
Angiosarcoma can arise spontaneously or in association with radiation or chronic lymphedema. Given the shared risk factors and presentation with AVL, it is essential to differentiate angiosarcoma from AVL. Primary cutaneous angiosarcoma usually presents on the head of elderly patients as an ecchymotic patch or plaque with ulceration.4 Secondary angiosarcoma may arise following radiation or chronic lymphedema (Stewart-Treves syndrome); however, some authors now prefer to consider lymphangiosarcoma arising in chronic lymphedematous limbs a distinct entity.6 Surgical excision with wide margins is the mainstay of therapy, but angiosarcoma has high recurrence rates, and the 5-year survival rate has been reported to be as low as 35%.7 Histologic overlap with AVL includes dissecting anastomosing vessels lined by hyperchromatic nuclei; however, angiosarcoma is distinguished by endothelial cell layering, nuclear pleomorphism, and prominent nucleoli (Figure 1).4,8 Increased positivity for Ki-67 immunostain, which indicates cell proliferation, may be used to distinguish angiosarcoma from an AVL (Figure 1 [inset]).9 Further, in contrast to AVL, radiation-induced angiosarcoma is characterized by amplification of C-MYC, a regulator gene, and FLT4 (FMS-related tyrosine kinase 4), a gene encoding vascular endothelial growth factor receptor 3. Gene amplification may be detected through immunohistochemistry or fluorescence in situ hybridization.10 Ki-67 labeling showed less than 10% staining in endothelial cells in our case (quiz image [inset]), and fluorescence in situ hybridization was negative for C-MYC amplification, supporting the diagnosis of AVL.
Lymphangioma circumscriptum, the most common superficial lymphangioma, is a hamartomatous malformation that usually occurs at the axillary folds, neck, and trunk. It clinically presents as small agminated vesicles with a characteristic frog spawn appearance.11 Dermoscopic features include yellow lacunae that may alternate with a dark red color secondary to extravasation of erythrocytes.12 These clinical features often lead to a differential diagnosis of verrucae, angiokeratoma, and angiosarcoma. Lymphangioma circumscriptum histologically is characterized by an overgrowth of dilated lymphatic vessels that fill the papillary dermis. The vessels are composed of flat endothelial cells typically filled with acellular proteinaceous debris and occasional erythrocytes (Figure 2). As the lesion traverses deeper into the dermis, the caliber of the lymphatic channel becomes narrower. The presence of deep lymphatic cisterns with surrounding smooth muscle is helpful to differentiate lymphangioma circumscriptum from other lymphatic malformations such as acquired lymphangiectasia. Treatment options include surgical excision, sclerosing agents, and destructive modalities such as cryotherapy.
Hobnail hemangioma, originally termed targetoid hemosiderotic hemangioma by Santa Cruz and Aronberg,13 presents as a violaceous papule or nodule surrounded by a characteristic brown halo on the leg. Trauma has been proposed as the inciting factor for the clinical appearance of hobnail hemangioma.14 Microscopically, the lesion shows vessels in a wedge shape. The superficial component has telangiectatic vessels with focal areas of papillary projections lined by endothelial cells. Although the endothelial nuclei typically project into the lumen, the nuclei are small, bland, and without mitotic activity.15 Deeper components show slit-shaped vasculature with dermal collagen dissection. Hemosiderin, extravasated red blood cells, and inflammation are found adjacent to the vessels (Figure 3). Given the benign nature, hobnail hemangiomas may be monitored.
Kaposi sarcoma (KS) is a low-grade vascular neoplasm associated with human herpesvirus 8 that arises in multiple clinical settings, especially in immunosuppression secondary to human immunodeficiency virus. There are 3 distinct clinical stages: patch, plaque, and tumor. The patch stage appears as red macules that blend into larger plaques; the tumor stage is defined as larger nodules developing from plaques. Histologic features differ by stage. Similar to angiosarcoma, KS is comprised of anastomosing vessels that dissect collagen bundles; endothelial cell atypia is minimal. A useful feature of KS is its propensity to involve adnexa and display the promontory sign, which involves the tumor growing into normal vasculature (Figure 4).16 Positive immunohistochemistry for human herpesvirus 8 aids in confirmation of the diagnosis. Treatment options for KS are numerous but include destructive modalities, chemotherapeutic agents such as doxorubicin, or highly active antiretroviral therapy for AIDS-related KS.17
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Billings SD, McKenney JK, Folpe AL, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation: an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Udager AM, Ishikawa MK, Lucas DR, et al. MYC immunohistochemistry in angiosarcoma and atypical vascular lesions: practical considerations based on a single institutional experience. Pathology. 2016;48:697-704.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Elsevier; 2016:1069-1115.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fraga-Guedes C, Gobbi H, Mastropasqua MG, et al. Clinicopathological and immunohistochemical study of 30 cases of post-radiation atypical vascular lesion of the breast. Breast Cancer Res Treat. 2014;146:347-354.
- Shin SJ, Lesser M, Rosen PP. Hemangiomas and angiosarcomas of the breast: diagnostic utility of cell cycle markers with emphasis on Ki-67. Arch Pathol Lab Med. 2007;131:538-544.
- Cornejo KM, Deng A, Wu H, et al. The utility of MYC and FLT4 in the diagnosis and treatment of postradiation atypical vascular lesion and angiosarcoma of the breast. Hum Pathol. 2015;46:868-875.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
- Massa AF, Menezes N, Baptista A, et al. Cutaneous lymphangioma circumscriptum--dermoscopic features. An Bras Dermatol. 2015;90:262-264.
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Christenson LJ, Stone MS. Trauma-induced simulator of targetoid hemosiderotic hemangioma. Am J Dermatopathol. 2001;23:221-223.
- Trindade F, Kutzner H, Tellechea O, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer. 2008;112:1147-1152.
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102:757-763.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Billings SD, McKenney JK, Folpe AL, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation: an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Udager AM, Ishikawa MK, Lucas DR, et al. MYC immunohistochemistry in angiosarcoma and atypical vascular lesions: practical considerations based on a single institutional experience. Pathology. 2016;48:697-704.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Elsevier; 2016:1069-1115.
- Shin JY, Roh SG, Lee NH, et al. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face: systematic review and meta-analysis. Head Neck. 2017;39:380-386.
- Fraga-Guedes C, Gobbi H, Mastropasqua MG, et al. Clinicopathological and immunohistochemical study of 30 cases of post-radiation atypical vascular lesion of the breast. Breast Cancer Res Treat. 2014;146:347-354.
- Shin SJ, Lesser M, Rosen PP. Hemangiomas and angiosarcomas of the breast: diagnostic utility of cell cycle markers with emphasis on Ki-67. Arch Pathol Lab Med. 2007;131:538-544.
- Cornejo KM, Deng A, Wu H, et al. The utility of MYC and FLT4 in the diagnosis and treatment of postradiation atypical vascular lesion and angiosarcoma of the breast. Hum Pathol. 2015;46:868-875.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
- Massa AF, Menezes N, Baptista A, et al. Cutaneous lymphangioma circumscriptum--dermoscopic features. An Bras Dermatol. 2015;90:262-264.
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Christenson LJ, Stone MS. Trauma-induced simulator of targetoid hemosiderotic hemangioma. Am J Dermatopathol. 2001;23:221-223.
- Trindade F, Kutzner H, Tellechea O, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Di Lorenzo G, Di Trolio R, Montesarchio V, et al. Pegylated liposomal doxorubicin as second-line therapy in the treatment of patients with advanced classic Kaposi sarcoma: a retrospective study. Cancer. 2008;112:1147-1152.
A 67-year-old woman presented with a lesion on the medial aspect of the right axilla of 2 weeks' duration. The patient had a history of cancer of the right breast treated with a mastectomy and adjuvant radiation. She denied pain, bleeding, pruritus, or rapid growth, as well as any changes in medication or recent trauma. Physical examination revealed a 5-mm purpuric macule of the right axilla. A punch biopsy was performed. Amplification for the C-MYC gene was negative by fluorescence in situ hybridization.
Irregular Yellow-Brown Plaques on the Trunk and Thighs
The Diagnosis: Necrobiotic Xanthogranuloma
A 4-mm punch biopsy was performed for routine stain with hematoxylin and eosin. The differential diagnosis included sarcoidosis, necrobiosis lipoidica, xanthoma disseminatum, and multicentric reticulohistiocytosis. Histopathologic examination demonstrated a dermal infiltrate of foamy histiocytes and neutrophils (Figure). There were surrounding areas of degenerated collagen containing numerous cholesterol clefts. After clinical pathologic correlation, a diagnosis of necrobiotic xanthogranuloma (NXG) was elucidated.
The patient was referred to general surgery for elective excision of 1 or more of the lesions. Excision of an abdominal lesion was performed without complication. After several months, a new lesion reformed within the excisional scar that also was consistent with NXG. At further dermatologic visits, a trial of intralesional corticosteroids was attempted to the largest lesions with modest improvement. In addition, follow-up with hematology and oncology was recommended for routine surveillance of the known blood dyscrasia.
Necrobiotic xanthogranuloma is a multisystem non-Langerhans cell histiocytic disease. Clinically, NXG is characterized by infiltrative plaques and ulcerative nodules. Lesions may appear red, brown, or yellow with associated atrophy and telangiectasia.1 Koch et al2 described a predilection for granuloma formation within preexisting scars. Periorbital location is the most common cutaneous site of involvement of NXG, seen in 80% of cases, but the trunk and extremities also may be involved.1,3 Approximately half of those with periocular involvement experience ocular symptoms including prop- tosis, blepharoptosis, and restricted eye movements.4 The onset of NXG most commonly is seen in middle age.
Characteristic systemic associations have been reported in the setting of NXG. More than 20% of patients may exhibit hepatomegaly. Hematologic abnormalities, hyperlipidemia, and cryoglobulinemia also may be seen.1 In addition, a monoclonal gammopathy of uncertain significance is found in more than 80% of NXG cases. The IgG κ light chain is most commonly identified.2 A foreign body reaction is incited by the immunoglobulin-lipid complex, which is thought to contribute to the formation of cutaneous lesions. There may be associated plasma cell dyscrasia such as multiple myeloma or B-cell lymphoma in approximately 13% of cases.2 Evaluation for underlying plasma cell dyscrasia or lymphoproliferative disorder should be performed regularly with serum protein electrophoresis or immunofixation electrophoresis, and in some cases full-body imaging with computed tomography or magnetic resonance imaging may be warranted.1
Treatment of NXG often is unsuccessful. Surgical excision, systemic immunosuppressive agents, electron beam radiation, and destructive therapies such as cryotherapy may be trialed, often with little success.1 Cutaneous regression has been reported with combination treatment of high-dose dexamethasone and high-dose lenalidomide.5
- Efebera Y, Blanchard E, Allam C, et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk. 2011;11:298-302.
- Koch PS, Goerdt S, Géraud C. Erythematous papules, plaques, and nodular lesions on the trunk and within preexisting scars. JAMA Dermatol. 2013;149:1103-1104.
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Dholaria BR, Cappel M, Roy V. Necrobiotic xanthogranuloma associated with monoclonal gammopathy: successful treatment with lenalidomide and dexamethasone [published online Jan 27, 2016]. Ann Hematol. 2016;95:671-672.
The Diagnosis: Necrobiotic Xanthogranuloma
A 4-mm punch biopsy was performed for routine stain with hematoxylin and eosin. The differential diagnosis included sarcoidosis, necrobiosis lipoidica, xanthoma disseminatum, and multicentric reticulohistiocytosis. Histopathologic examination demonstrated a dermal infiltrate of foamy histiocytes and neutrophils (Figure). There were surrounding areas of degenerated collagen containing numerous cholesterol clefts. After clinical pathologic correlation, a diagnosis of necrobiotic xanthogranuloma (NXG) was elucidated.
The patient was referred to general surgery for elective excision of 1 or more of the lesions. Excision of an abdominal lesion was performed without complication. After several months, a new lesion reformed within the excisional scar that also was consistent with NXG. At further dermatologic visits, a trial of intralesional corticosteroids was attempted to the largest lesions with modest improvement. In addition, follow-up with hematology and oncology was recommended for routine surveillance of the known blood dyscrasia.
Necrobiotic xanthogranuloma is a multisystem non-Langerhans cell histiocytic disease. Clinically, NXG is characterized by infiltrative plaques and ulcerative nodules. Lesions may appear red, brown, or yellow with associated atrophy and telangiectasia.1 Koch et al2 described a predilection for granuloma formation within preexisting scars. Periorbital location is the most common cutaneous site of involvement of NXG, seen in 80% of cases, but the trunk and extremities also may be involved.1,3 Approximately half of those with periocular involvement experience ocular symptoms including prop- tosis, blepharoptosis, and restricted eye movements.4 The onset of NXG most commonly is seen in middle age.
Characteristic systemic associations have been reported in the setting of NXG. More than 20% of patients may exhibit hepatomegaly. Hematologic abnormalities, hyperlipidemia, and cryoglobulinemia also may be seen.1 In addition, a monoclonal gammopathy of uncertain significance is found in more than 80% of NXG cases. The IgG κ light chain is most commonly identified.2 A foreign body reaction is incited by the immunoglobulin-lipid complex, which is thought to contribute to the formation of cutaneous lesions. There may be associated plasma cell dyscrasia such as multiple myeloma or B-cell lymphoma in approximately 13% of cases.2 Evaluation for underlying plasma cell dyscrasia or lymphoproliferative disorder should be performed regularly with serum protein electrophoresis or immunofixation electrophoresis, and in some cases full-body imaging with computed tomography or magnetic resonance imaging may be warranted.1
Treatment of NXG often is unsuccessful. Surgical excision, systemic immunosuppressive agents, electron beam radiation, and destructive therapies such as cryotherapy may be trialed, often with little success.1 Cutaneous regression has been reported with combination treatment of high-dose dexamethasone and high-dose lenalidomide.5
The Diagnosis: Necrobiotic Xanthogranuloma
A 4-mm punch biopsy was performed for routine stain with hematoxylin and eosin. The differential diagnosis included sarcoidosis, necrobiosis lipoidica, xanthoma disseminatum, and multicentric reticulohistiocytosis. Histopathologic examination demonstrated a dermal infiltrate of foamy histiocytes and neutrophils (Figure). There were surrounding areas of degenerated collagen containing numerous cholesterol clefts. After clinical pathologic correlation, a diagnosis of necrobiotic xanthogranuloma (NXG) was elucidated.
The patient was referred to general surgery for elective excision of 1 or more of the lesions. Excision of an abdominal lesion was performed without complication. After several months, a new lesion reformed within the excisional scar that also was consistent with NXG. At further dermatologic visits, a trial of intralesional corticosteroids was attempted to the largest lesions with modest improvement. In addition, follow-up with hematology and oncology was recommended for routine surveillance of the known blood dyscrasia.
Necrobiotic xanthogranuloma is a multisystem non-Langerhans cell histiocytic disease. Clinically, NXG is characterized by infiltrative plaques and ulcerative nodules. Lesions may appear red, brown, or yellow with associated atrophy and telangiectasia.1 Koch et al2 described a predilection for granuloma formation within preexisting scars. Periorbital location is the most common cutaneous site of involvement of NXG, seen in 80% of cases, but the trunk and extremities also may be involved.1,3 Approximately half of those with periocular involvement experience ocular symptoms including prop- tosis, blepharoptosis, and restricted eye movements.4 The onset of NXG most commonly is seen in middle age.
Characteristic systemic associations have been reported in the setting of NXG. More than 20% of patients may exhibit hepatomegaly. Hematologic abnormalities, hyperlipidemia, and cryoglobulinemia also may be seen.1 In addition, a monoclonal gammopathy of uncertain significance is found in more than 80% of NXG cases. The IgG κ light chain is most commonly identified.2 A foreign body reaction is incited by the immunoglobulin-lipid complex, which is thought to contribute to the formation of cutaneous lesions. There may be associated plasma cell dyscrasia such as multiple myeloma or B-cell lymphoma in approximately 13% of cases.2 Evaluation for underlying plasma cell dyscrasia or lymphoproliferative disorder should be performed regularly with serum protein electrophoresis or immunofixation electrophoresis, and in some cases full-body imaging with computed tomography or magnetic resonance imaging may be warranted.1
Treatment of NXG often is unsuccessful. Surgical excision, systemic immunosuppressive agents, electron beam radiation, and destructive therapies such as cryotherapy may be trialed, often with little success.1 Cutaneous regression has been reported with combination treatment of high-dose dexamethasone and high-dose lenalidomide.5
- Efebera Y, Blanchard E, Allam C, et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk. 2011;11:298-302.
- Koch PS, Goerdt S, Géraud C. Erythematous papules, plaques, and nodular lesions on the trunk and within preexisting scars. JAMA Dermatol. 2013;149:1103-1104.
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Dholaria BR, Cappel M, Roy V. Necrobiotic xanthogranuloma associated with monoclonal gammopathy: successful treatment with lenalidomide and dexamethasone [published online Jan 27, 2016]. Ann Hematol. 2016;95:671-672.
- Efebera Y, Blanchard E, Allam C, et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk. 2011;11:298-302.
- Koch PS, Goerdt S, Géraud C. Erythematous papules, plaques, and nodular lesions on the trunk and within preexisting scars. JAMA Dermatol. 2013;149:1103-1104.
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Dholaria BR, Cappel M, Roy V. Necrobiotic xanthogranuloma associated with monoclonal gammopathy: successful treatment with lenalidomide and dexamethasone [published online Jan 27, 2016]. Ann Hematol. 2016;95:671-672.
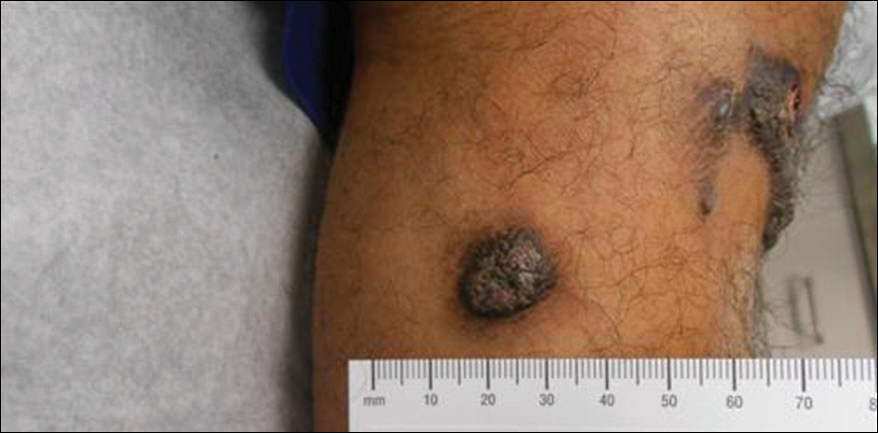
A 40-year-old man presented with tender lesions on the back, abdomen, and thighs of 10 years' duration. His medical history was remarkable for follicular lymphoma treated with chemotherapy and a monoclonal gammopathy of uncertain significance diagnosed 5 years after the onset of skin symptoms. Physical examination revealed numerous irregularly shaped, yellow plaques on the back, abdomen, and thighs with overlying telangiectasia. A single lesion was noted to extend from a scar.
Pseudomyogenic Hemangioendothelioma
Pseudomyogenic hemangioendothelioma (PMHE), also referred to as epithelioid sarcoma–like hemangioendothelioma,1 is a rare soft tissue tumor that was described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. It predominantly affects males between the second and fifth decades of life and most commonly presents as multiple nodules that may involve either the superficial or deep soft tissues of the legs and less often the arms. It also can arise on the trunk. We present a case of PMHE occurring in a young man and briefly review the literature on clinical presentation and histologic differentiation of this unique tumor, comparing these findings to its mimickers.
Case Report
A 20-year-old man presented with skin lesions on the left leg that had been present for 1 year. The patient described the lesions as tender pimples that would drain yellow discharge on occasion but had now transformed into large brown plaques. Physical examination showed 4 verrucous plaques ranging in size from 1 to 3 cm with hyperpigmentation and a central crust (Figure 1). Initially, the patient thought the lesions appeared due to shaving his legs for sports. He presented to the emergency department multiple times over the past year; pain control was provided and local skin care was recommended. Culture of the discharge had been performed 6 months prior to biopsy with negative results. No biopsy was performed on initial presentation and the lesions were diagnosed in the emergency department clinically as boils.
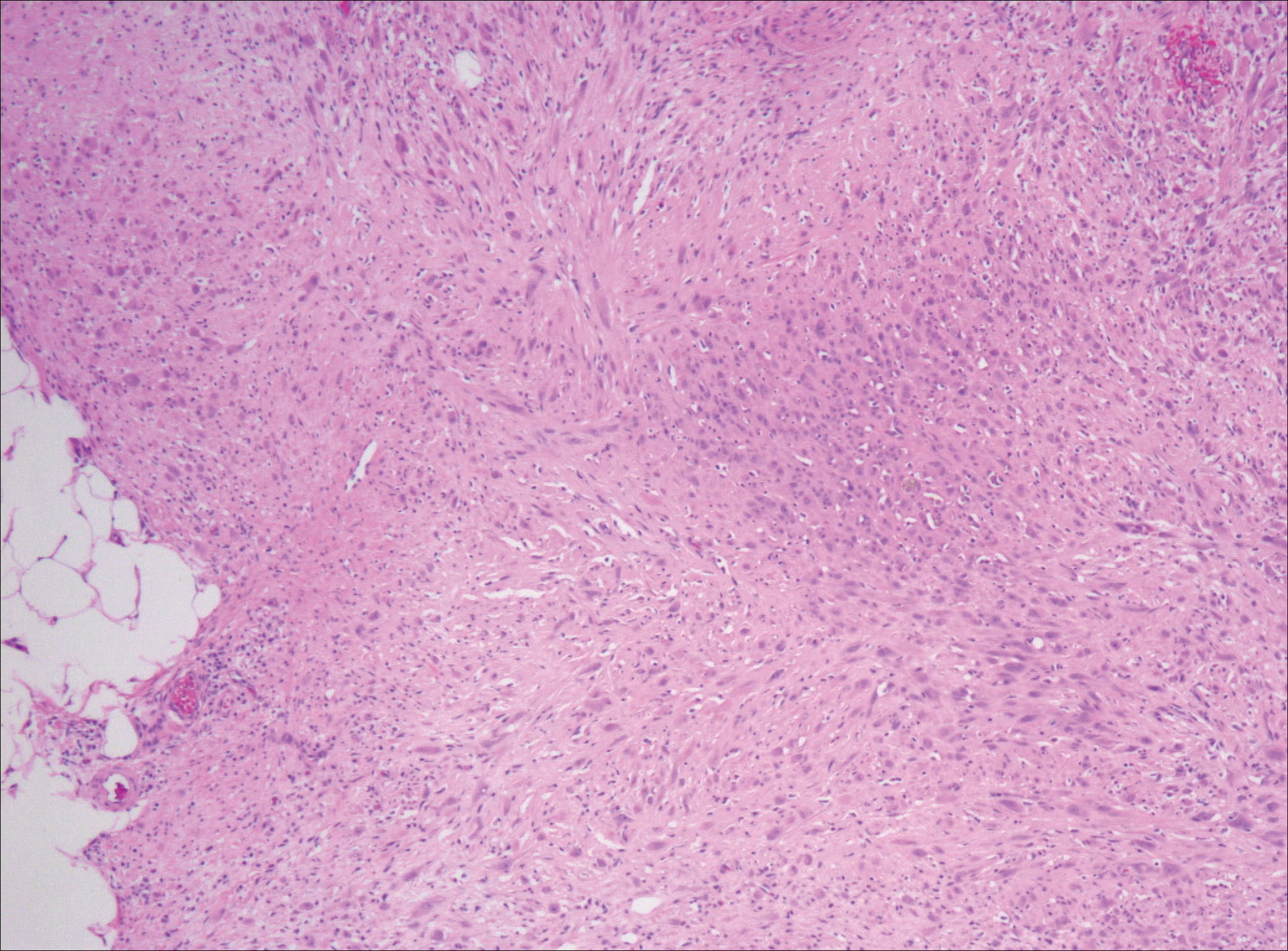
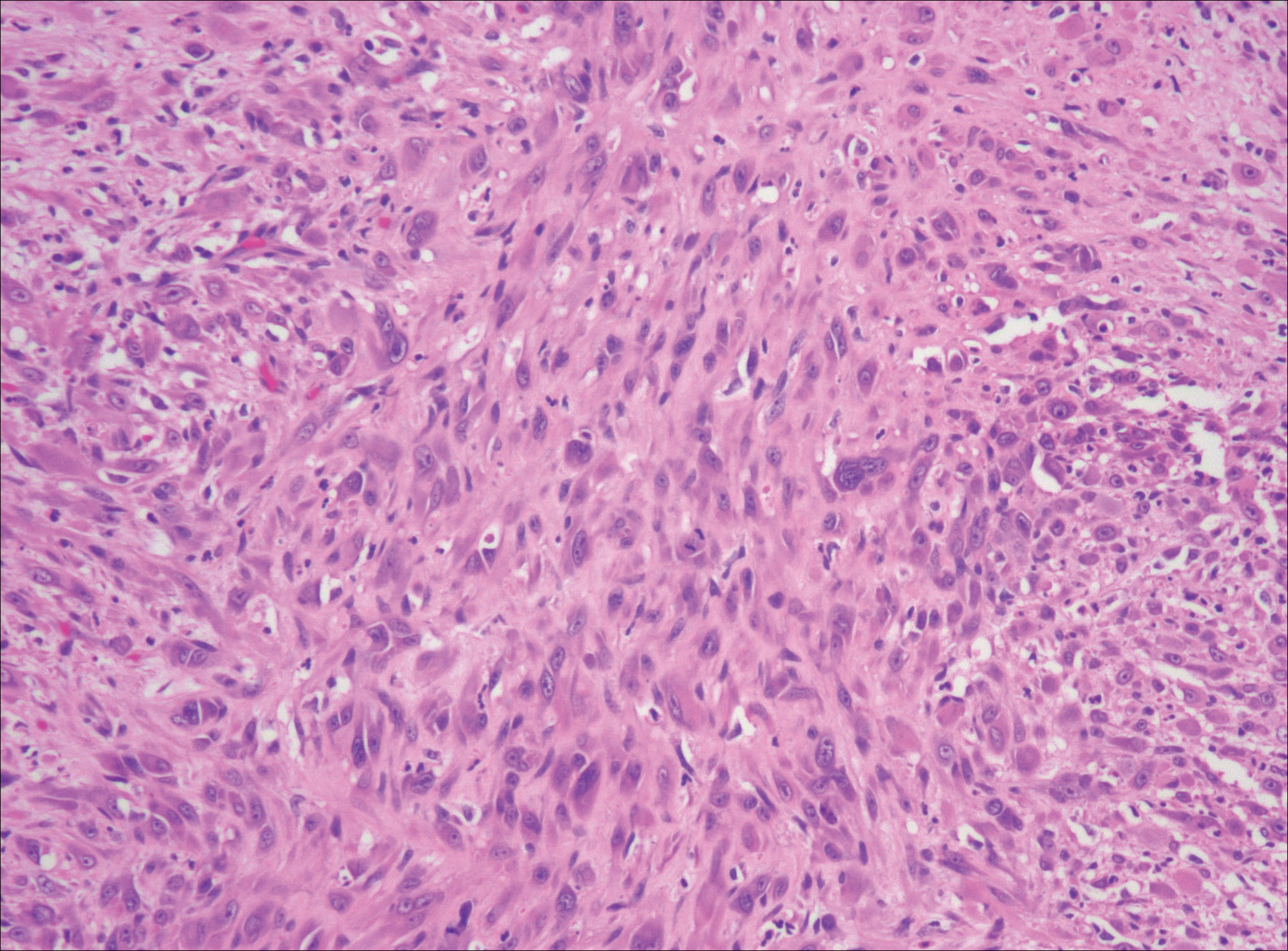
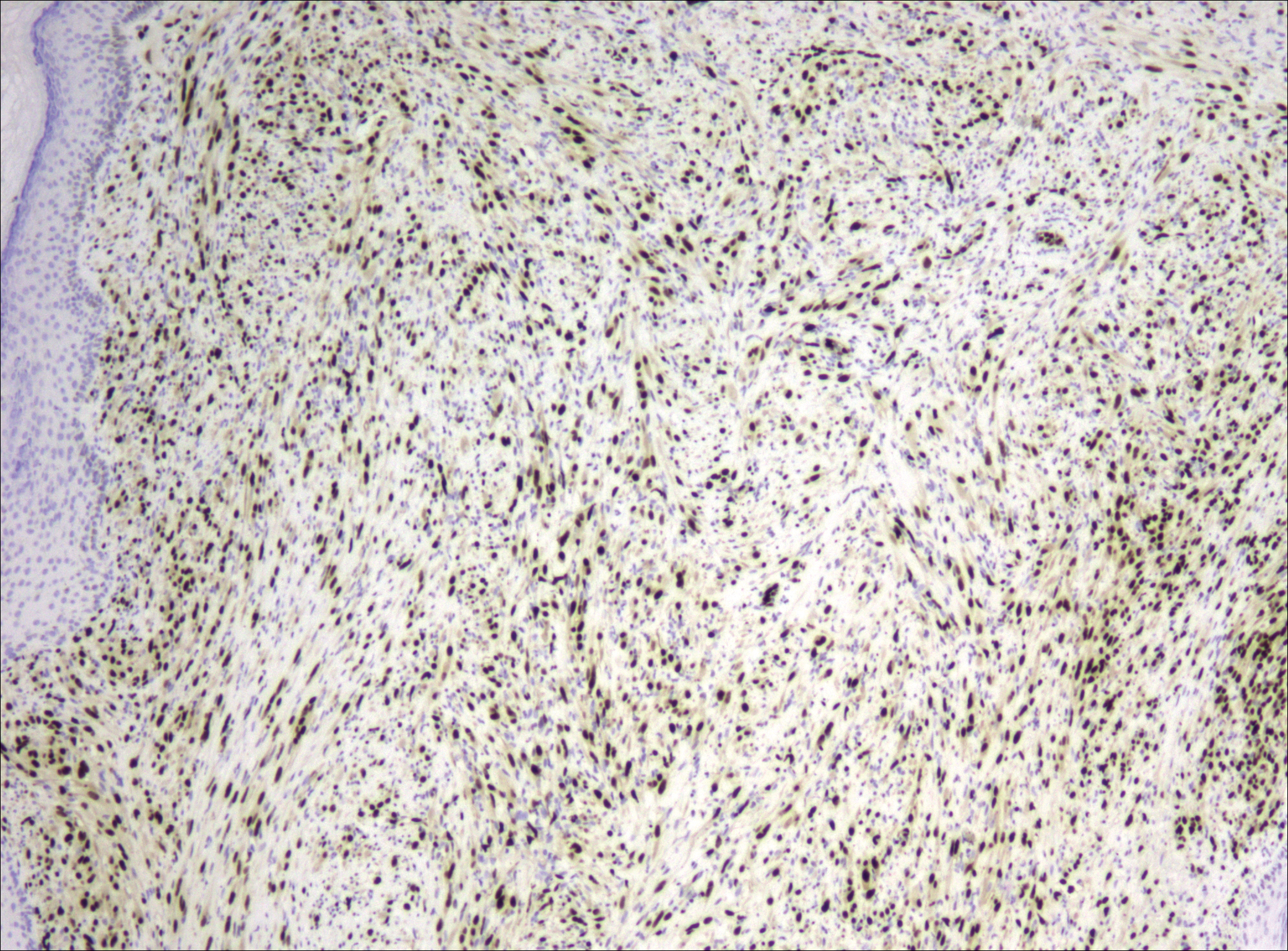
After failing to improve, the patient was seen by an outside dermatologist and the clinical differential diagnosis included deep fungal infection, atypical mycobacterial infection, and keloids. A 4-mm punch biopsy was taken from the periphery of one of the lesions and demonstrated hyperkeratosis, papillomatosis, and acanthosis (Figure 2). Within the superficial and deep dermis and focally extending into the subcutaneous tissue, there were sheets of spindled to epithelioid-appearing cells with moderate cytologic atypia (Figure 3). The tumor showed infiltrative margins. There was moderate cellularity. The individual cells had a rhabdoid appearance with large eccentric vesicular nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm (Figure 4). No definitive evidence of glandular, squamous, or vascular differentiation was present. There was an associated moderate inflammatory host response composed of neutrophils and lymphocytes. Occasional extravasated red blood cells were present. Immunohistochemistry staining was performed and the atypical cells demonstrated diffuse positive staining for friend leukemia integration 1 transcription factor (FLI-1), erythroblastosis virus E26 transforming sequence-related gene (ERG)(Figure 5), CD31, and CD68. There was patchy positive staining for cytokeratin AE1/AE3, CD10, and factor VIII. There was no remarkable staining for human herpesvirus 8, epithelial membrane antigen, S-100, CD34, cytokeratin 903, and desmin. Overall, the histologic features in conjunction with the immunohistochemistry staining were consistent with a diagnosis of PMHE.

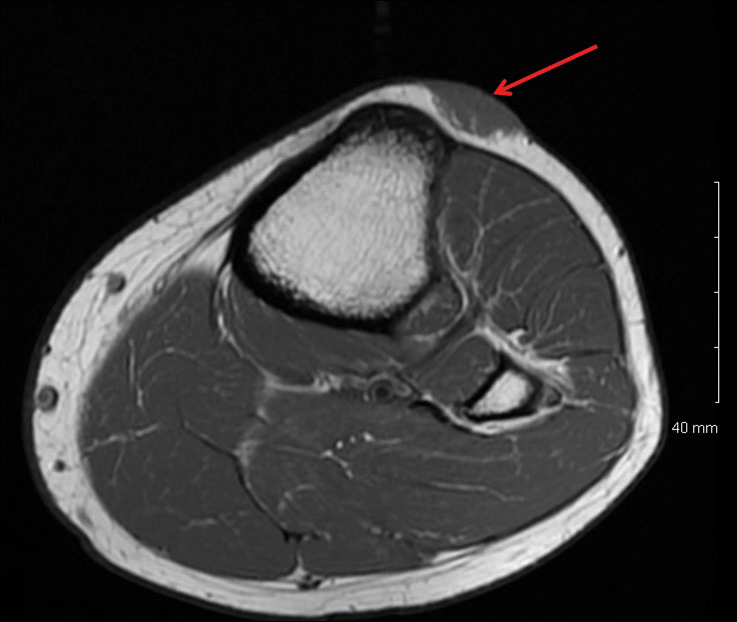
Magnetic resonance imaging was then performed to evaluate the depth and extent of the lesions for surgical excision planning (Figure 6), which showed 5 nodular lesions within the dermis and subcutis adjacent to the proximal aspect of the left tibia and medial aspect of the left knee. An additional lesion was noted between the sartorius and semimembranosus muscles, which was thought to represent either a lymph node or an additional neoplastic lesion. Chest computed tomography also displayed indeterminate lesions in the lungs.
Excision of the superficial lesions was performed. All of the lesions demonstrated similar histologic changes to the previously described biopsy specimen. The tumor was limited to the dermis and subcutaneous tissue. The patient was lost to follow-up and the etiology of the lung lesions was unknown.
Comment
Nomenclature
Pseudomyogenic hemangioendothelioma is a relatively new type of vascular tumor that has been included in the updated 2013 edition of the World Health Organization classification as an intermediate malignant tumor that rarely metastasizes.3 It typically involves multiple tissue planes, most notably the dermis and subcutaneous layers but also muscle and bone.4 The term pseudomyogenic refers to the histologic resemblance of some of the cells to rhabdomyoblasts; however, these tumors are negative for all immunohistochemical muscle markers, most notably myogenin, desmin, and α-smooth muscle actin.5
Clinical Presentation
Gross features of PMHE typically include multiple firm nodules with ill-defined margins. The tumor was initially described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. In 2003, a series of 7 cases of PMHE was reported by Billings et al6 under the term epithelioid sarcomalike hemangioendothelioma. Other than the predominance of an epithelioid morphology, the cases reported as epithelioid sarcomalike hemangioendothelioma had similar clinical features and immunophenotype to what has been reported as PMHE.
Based on a PubMed search of articles indexed for MEDLINE using the term pseudomyogenic hemangioendothelioma, the 2 largest case series were reported by Pradhan et al7 (N=8) in 2017 and Hornick and Fletcher4 (N=50) in 2011. Hornick and Fletcher4 reported a male (41/50 [82.0%]) to female (9/50 [18.0%]) ratio of 4.6 to 1, and an average age at presentation of 31 years with 82% (41/50) of patients 40 years or younger. Pradhan et al7 also reported a male predominance (7/8 [87.5%]) with a similar average age at presentation of 29 years (age range, 9–62 years). The size of individual tumors ranged from 0.3 to 5.5 cm (mean size, 1.9 cm) in the series by Hornick and Fletcher4 and 0.3 to 6.0 com in the series by Pradhan et al.7 Hornick and Fletcher4 reported the most common site of involvement was the leg (27/50 [54.0%]), followed by the arm (12/50 [24.0%]), trunk (9/50 [18.0%]), and head and neck (2/50 [4.0%]). The leg (6/8 [75.0%]) also was the most common site of involvement in the series by Pradhan et al,7 with 2 cases occurring on the arm. In the series by Hornick and Fletcher,4 the tumors typically involved the dermis and subcutaneous tissue (26/50 [52%]) with a smaller number involving skeletal muscle (17/50 [34%]) and bone (7/50 [14%]). They reported 66% of their patients (33/50) had multifocal disease at presentation.4 Pradhan et al7 also reported 2 (25.0%) cases being limited to the superficial soft tissue, 2 (25.0%) being limited to the deep soft tissue, and 4 (50.0%) involving the bone; 5 (62.5%) patients had multifocal disease at presentation. The presentation of our patient in regards to gender, age, and tumor characteristics is consistent with other published cases.5-10
Histopathology
Microscopic features of PMHE include sheets of spindled to epithelioid-appearing cells with mild to moderate nuclear atypia and eosinophilic cytoplasm. The tumor has an infiltrative growth pattern. Some of the cells may resemble rhabdomyoblastlike cells, hence the moniker pseudomyogenic. There is no recapitulation of vascular structures or remarkable cytoplasmic vacuolization. Mitotic rate is low and there is no tumor necrosis.4 The tumor cells do not appear to arise from a vessel or display an angiocentric growth pattern. Many cases report the presence of an inflammatory infiltrate containing neutrophils interspersed within the tumor.4,5,7 The overlying epidermis will commonly show hyperkeratosis, epidermal hyperplasia, and acanthosis.4,11
Differential Diagnosis
The histopathologic differential diagnosis would include epithelioid sarcoma, epithelioid hemangioendothelioma, and to a lesser extent dermatofibrosarcoma protuberans (DFSP) and rhabdomyosarcoma. Dermatofibrosarcoma protuberans is the most commonly encountered of these tumors. Histologically, DFSP is characterized by a cellular proliferation of small spindle cells with plump nuclei arranged in a storiform or cartwheel pattern. Dermatofibrosarcoma protuberans tends to be limited to the dermis and subcutaneous tissue and only rarely involves underlying skeletal muscle. The presence of the storiform growth pattern in conjunction with the lack of rhabdoid changes would favor a diagnosis of DFSP. Another characteristic histologic finding typically only associated with DFSP is the interdigitating growth pattern of the spindle cells within the lobules of the subcutaneous tissue, creating a lacelike or honeycomb appearance.
Immunohistochemistry staining is necessary to help differentiate PMHE from other tumors in the differential diagnosis. Pseudomyogenic hemangioendothelioma stains positive for cytokeratin AE1/AE3; integrase interactor 1; and vascular markers FLI-1, CD31, and ERG, and negative for CD34.4,6,12-15 In contrast to epithelioid hemangioendothelioma, DFSP, and to a lesser extent epithelioid sarcoma, all of which are positive for CD34, epithelioid sarcoma is negative for both CD31 and integrase interactor 1. Dermatofibrosarcoma protuberans is negative for cytokeratin AE1/AE3. Rhabdomyosarcomas are positive for myogenic markers such as MyoD1 and myogenin, unlike any of the other tumors mentioned. Histologically, epithelioid sarcomas will tend to have a granulomalike growth pattern with central necrosis, unlike PMHE.12 Epithelioid hemangioendothelioma often will have a cordlike growth pattern in a myxochondroid background. Unlike PMHE, these tumors often will appear to be arising from vessels, and intracytoplasmic vacuoles are common. Three cases of PMHE have been reported to have a t(7;19)(q22;q13) chromosomal anomaly, which is not consistent with every case.16
Treatment Options
Standard treatment typically includes wide excision of the lesions, as was done in our case. Because of the substantial risk of local recurrence, which was up to 58% in the series by Hornick and Fletcher,4 adjuvant therapy may be considered if positive margins are found on excision. Metastasis to lymph nodes and the lungs has been reported but is rare.2,4 Most cases have been shown to have a favorable prognosis; however, local recurrence seems to be common. Rarely, amputation of the limb may be required.5 In contrast, epithelioid sarcomas have been found to spread to lymph nodes and the lungs in up to 50% of cases with a 5-year survival rate of 10% to 30%.13
Conclusion
In summary, we describe a case of PMHE involving the lower leg in a 20-year-old man. These tumors often are multinodular and multiplanar, with the dermis and subcutaneous tissues being the most common areas affected. It has a high rate of local recurrence but rarely has distant metastasis. Pseudomyogenic hemangioendothelioma, similar to other soft tissue tumors, can be difficult to diagnose on shave biopsy or superficial punch biopsy not extending into subcutaneous tissue. Deep incisional or punch biopsies are required to more definitively diagnose these types of tumors. The diagnosis of PMHE versus other soft tissue tumors requires correlation of histology and immunohistochemistry staining with clinical information and radiographic findings.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma (pseudomyogenic hemangioendothelioma). Am J Surg Pathol. 2011;35:1088; author reply 1088-1089.
- Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
- Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014;46:95-104.
- Hornick JL, Fletcher CD. Pseudomyogenic hemangioendothelioma: a distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol. 2011;35:190-201.
- Sheng W, Pan Y, Wang J. Pseudomyogenic hemangioendothelioma: report of an additional case with aggressive clinical course. Am J Dermatopathol. 2013;35:597-600.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol. 2003;27:48-57.
- Pradhan D, Schoedel K, McGough RL, et al. Pseudomyogenic hemangioendothelioma of skin, bone and soft tissue—a clinicopathological, immunohistochemical and fluorescence in situ hybridization study [published online November 2, 2017]. Hum Pathol. 2017. doi:0.1016/j.humpath.2017.10.023.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- McGinity M, Bartanusz V, Dengler B, et al. Pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma, fibroma-like variant of epithelioid sarcoma) of the thoracic spine. Eur Spine J. 2013;22(suppl 3):S506-S511.
- Stuart LN, Gardner JM, Lauer SR, et al. Epithelioid sarcoma-like (pseudomyogenic) hemangioendothelioma, clinically mimicking dermatofibroma, diagnosed by skin biopsy in a 30-year-old man. J Cutan Pathol. 2013;40:909-913.
- Amary MF, O’Donnell P, Berisha F, et al. Pseudomyogenic (epithelioid sarcoma-like) hemangioendothelioma: characterization of five cases. Skeletal Radiol. 2013;42:947-957.
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542-550.
- Chbani L, Guillou L, Terrier P, et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French Sarcoma Group. Am J Clin Pathol. 2009;131:222-227.
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- Trombetta D, Magnusson L, von Steyern FV, et al. Translocation t(7;19)(q22;q13)—a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet. 2011;204:211-215.
Pseudomyogenic hemangioendothelioma (PMHE), also referred to as epithelioid sarcoma–like hemangioendothelioma,1 is a rare soft tissue tumor that was described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. It predominantly affects males between the second and fifth decades of life and most commonly presents as multiple nodules that may involve either the superficial or deep soft tissues of the legs and less often the arms. It also can arise on the trunk. We present a case of PMHE occurring in a young man and briefly review the literature on clinical presentation and histologic differentiation of this unique tumor, comparing these findings to its mimickers.
Case Report
A 20-year-old man presented with skin lesions on the left leg that had been present for 1 year. The patient described the lesions as tender pimples that would drain yellow discharge on occasion but had now transformed into large brown plaques. Physical examination showed 4 verrucous plaques ranging in size from 1 to 3 cm with hyperpigmentation and a central crust (Figure 1). Initially, the patient thought the lesions appeared due to shaving his legs for sports. He presented to the emergency department multiple times over the past year; pain control was provided and local skin care was recommended. Culture of the discharge had been performed 6 months prior to biopsy with negative results. No biopsy was performed on initial presentation and the lesions were diagnosed in the emergency department clinically as boils.
After failing to improve, the patient was seen by an outside dermatologist and the clinical differential diagnosis included deep fungal infection, atypical mycobacterial infection, and keloids. A 4-mm punch biopsy was taken from the periphery of one of the lesions and demonstrated hyperkeratosis, papillomatosis, and acanthosis (Figure 2). Within the superficial and deep dermis and focally extending into the subcutaneous tissue, there were sheets of spindled to epithelioid-appearing cells with moderate cytologic atypia (Figure 3). The tumor showed infiltrative margins. There was moderate cellularity. The individual cells had a rhabdoid appearance with large eccentric vesicular nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm (Figure 4). No definitive evidence of glandular, squamous, or vascular differentiation was present. There was an associated moderate inflammatory host response composed of neutrophils and lymphocytes. Occasional extravasated red blood cells were present. Immunohistochemistry staining was performed and the atypical cells demonstrated diffuse positive staining for friend leukemia integration 1 transcription factor (FLI-1), erythroblastosis virus E26 transforming sequence-related gene (ERG)(Figure 5), CD31, and CD68. There was patchy positive staining for cytokeratin AE1/AE3, CD10, and factor VIII. There was no remarkable staining for human herpesvirus 8, epithelial membrane antigen, S-100, CD34, cytokeratin 903, and desmin. Overall, the histologic features in conjunction with the immunohistochemistry staining were consistent with a diagnosis of PMHE.
Magnetic resonance imaging was then performed to evaluate the depth and extent of the lesions for surgical excision planning (Figure 6), which showed 5 nodular lesions within the dermis and subcutis adjacent to the proximal aspect of the left tibia and medial aspect of the left knee. An additional lesion was noted between the sartorius and semimembranosus muscles, which was thought to represent either a lymph node or an additional neoplastic lesion. Chest computed tomography also displayed indeterminate lesions in the lungs.
Excision of the superficial lesions was performed. All of the lesions demonstrated similar histologic changes to the previously described biopsy specimen. The tumor was limited to the dermis and subcutaneous tissue. The patient was lost to follow-up and the etiology of the lung lesions was unknown.
Comment
Nomenclature
Pseudomyogenic hemangioendothelioma is a relatively new type of vascular tumor that has been included in the updated 2013 edition of the World Health Organization classification as an intermediate malignant tumor that rarely metastasizes.3 It typically involves multiple tissue planes, most notably the dermis and subcutaneous layers but also muscle and bone.4 The term pseudomyogenic refers to the histologic resemblance of some of the cells to rhabdomyoblasts; however, these tumors are negative for all immunohistochemical muscle markers, most notably myogenin, desmin, and α-smooth muscle actin.5
Clinical Presentation
Gross features of PMHE typically include multiple firm nodules with ill-defined margins. The tumor was initially described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. In 2003, a series of 7 cases of PMHE was reported by Billings et al6 under the term epithelioid sarcomalike hemangioendothelioma. Other than the predominance of an epithelioid morphology, the cases reported as epithelioid sarcomalike hemangioendothelioma had similar clinical features and immunophenotype to what has been reported as PMHE.
Based on a PubMed search of articles indexed for MEDLINE using the term pseudomyogenic hemangioendothelioma, the 2 largest case series were reported by Pradhan et al7 (N=8) in 2017 and Hornick and Fletcher4 (N=50) in 2011. Hornick and Fletcher4 reported a male (41/50 [82.0%]) to female (9/50 [18.0%]) ratio of 4.6 to 1, and an average age at presentation of 31 years with 82% (41/50) of patients 40 years or younger. Pradhan et al7 also reported a male predominance (7/8 [87.5%]) with a similar average age at presentation of 29 years (age range, 9–62 years). The size of individual tumors ranged from 0.3 to 5.5 cm (mean size, 1.9 cm) in the series by Hornick and Fletcher4 and 0.3 to 6.0 com in the series by Pradhan et al.7 Hornick and Fletcher4 reported the most common site of involvement was the leg (27/50 [54.0%]), followed by the arm (12/50 [24.0%]), trunk (9/50 [18.0%]), and head and neck (2/50 [4.0%]). The leg (6/8 [75.0%]) also was the most common site of involvement in the series by Pradhan et al,7 with 2 cases occurring on the arm. In the series by Hornick and Fletcher,4 the tumors typically involved the dermis and subcutaneous tissue (26/50 [52%]) with a smaller number involving skeletal muscle (17/50 [34%]) and bone (7/50 [14%]). They reported 66% of their patients (33/50) had multifocal disease at presentation.4 Pradhan et al7 also reported 2 (25.0%) cases being limited to the superficial soft tissue, 2 (25.0%) being limited to the deep soft tissue, and 4 (50.0%) involving the bone; 5 (62.5%) patients had multifocal disease at presentation. The presentation of our patient in regards to gender, age, and tumor characteristics is consistent with other published cases.5-10
Histopathology
Microscopic features of PMHE include sheets of spindled to epithelioid-appearing cells with mild to moderate nuclear atypia and eosinophilic cytoplasm. The tumor has an infiltrative growth pattern. Some of the cells may resemble rhabdomyoblastlike cells, hence the moniker pseudomyogenic. There is no recapitulation of vascular structures or remarkable cytoplasmic vacuolization. Mitotic rate is low and there is no tumor necrosis.4 The tumor cells do not appear to arise from a vessel or display an angiocentric growth pattern. Many cases report the presence of an inflammatory infiltrate containing neutrophils interspersed within the tumor.4,5,7 The overlying epidermis will commonly show hyperkeratosis, epidermal hyperplasia, and acanthosis.4,11
Differential Diagnosis
The histopathologic differential diagnosis would include epithelioid sarcoma, epithelioid hemangioendothelioma, and to a lesser extent dermatofibrosarcoma protuberans (DFSP) and rhabdomyosarcoma. Dermatofibrosarcoma protuberans is the most commonly encountered of these tumors. Histologically, DFSP is characterized by a cellular proliferation of small spindle cells with plump nuclei arranged in a storiform or cartwheel pattern. Dermatofibrosarcoma protuberans tends to be limited to the dermis and subcutaneous tissue and only rarely involves underlying skeletal muscle. The presence of the storiform growth pattern in conjunction with the lack of rhabdoid changes would favor a diagnosis of DFSP. Another characteristic histologic finding typically only associated with DFSP is the interdigitating growth pattern of the spindle cells within the lobules of the subcutaneous tissue, creating a lacelike or honeycomb appearance.
Immunohistochemistry staining is necessary to help differentiate PMHE from other tumors in the differential diagnosis. Pseudomyogenic hemangioendothelioma stains positive for cytokeratin AE1/AE3; integrase interactor 1; and vascular markers FLI-1, CD31, and ERG, and negative for CD34.4,6,12-15 In contrast to epithelioid hemangioendothelioma, DFSP, and to a lesser extent epithelioid sarcoma, all of which are positive for CD34, epithelioid sarcoma is negative for both CD31 and integrase interactor 1. Dermatofibrosarcoma protuberans is negative for cytokeratin AE1/AE3. Rhabdomyosarcomas are positive for myogenic markers such as MyoD1 and myogenin, unlike any of the other tumors mentioned. Histologically, epithelioid sarcomas will tend to have a granulomalike growth pattern with central necrosis, unlike PMHE.12 Epithelioid hemangioendothelioma often will have a cordlike growth pattern in a myxochondroid background. Unlike PMHE, these tumors often will appear to be arising from vessels, and intracytoplasmic vacuoles are common. Three cases of PMHE have been reported to have a t(7;19)(q22;q13) chromosomal anomaly, which is not consistent with every case.16
Treatment Options
Standard treatment typically includes wide excision of the lesions, as was done in our case. Because of the substantial risk of local recurrence, which was up to 58% in the series by Hornick and Fletcher,4 adjuvant therapy may be considered if positive margins are found on excision. Metastasis to lymph nodes and the lungs has been reported but is rare.2,4 Most cases have been shown to have a favorable prognosis; however, local recurrence seems to be common. Rarely, amputation of the limb may be required.5 In contrast, epithelioid sarcomas have been found to spread to lymph nodes and the lungs in up to 50% of cases with a 5-year survival rate of 10% to 30%.13
Conclusion
In summary, we describe a case of PMHE involving the lower leg in a 20-year-old man. These tumors often are multinodular and multiplanar, with the dermis and subcutaneous tissues being the most common areas affected. It has a high rate of local recurrence but rarely has distant metastasis. Pseudomyogenic hemangioendothelioma, similar to other soft tissue tumors, can be difficult to diagnose on shave biopsy or superficial punch biopsy not extending into subcutaneous tissue. Deep incisional or punch biopsies are required to more definitively diagnose these types of tumors. The diagnosis of PMHE versus other soft tissue tumors requires correlation of histology and immunohistochemistry staining with clinical information and radiographic findings.
Pseudomyogenic hemangioendothelioma (PMHE), also referred to as epithelioid sarcoma–like hemangioendothelioma,1 is a rare soft tissue tumor that was described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. It predominantly affects males between the second and fifth decades of life and most commonly presents as multiple nodules that may involve either the superficial or deep soft tissues of the legs and less often the arms. It also can arise on the trunk. We present a case of PMHE occurring in a young man and briefly review the literature on clinical presentation and histologic differentiation of this unique tumor, comparing these findings to its mimickers.
Case Report
A 20-year-old man presented with skin lesions on the left leg that had been present for 1 year. The patient described the lesions as tender pimples that would drain yellow discharge on occasion but had now transformed into large brown plaques. Physical examination showed 4 verrucous plaques ranging in size from 1 to 3 cm with hyperpigmentation and a central crust (Figure 1). Initially, the patient thought the lesions appeared due to shaving his legs for sports. He presented to the emergency department multiple times over the past year; pain control was provided and local skin care was recommended. Culture of the discharge had been performed 6 months prior to biopsy with negative results. No biopsy was performed on initial presentation and the lesions were diagnosed in the emergency department clinically as boils.
After failing to improve, the patient was seen by an outside dermatologist and the clinical differential diagnosis included deep fungal infection, atypical mycobacterial infection, and keloids. A 4-mm punch biopsy was taken from the periphery of one of the lesions and demonstrated hyperkeratosis, papillomatosis, and acanthosis (Figure 2). Within the superficial and deep dermis and focally extending into the subcutaneous tissue, there were sheets of spindled to epithelioid-appearing cells with moderate cytologic atypia (Figure 3). The tumor showed infiltrative margins. There was moderate cellularity. The individual cells had a rhabdoid appearance with large eccentric vesicular nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm (Figure 4). No definitive evidence of glandular, squamous, or vascular differentiation was present. There was an associated moderate inflammatory host response composed of neutrophils and lymphocytes. Occasional extravasated red blood cells were present. Immunohistochemistry staining was performed and the atypical cells demonstrated diffuse positive staining for friend leukemia integration 1 transcription factor (FLI-1), erythroblastosis virus E26 transforming sequence-related gene (ERG)(Figure 5), CD31, and CD68. There was patchy positive staining for cytokeratin AE1/AE3, CD10, and factor VIII. There was no remarkable staining for human herpesvirus 8, epithelial membrane antigen, S-100, CD34, cytokeratin 903, and desmin. Overall, the histologic features in conjunction with the immunohistochemistry staining were consistent with a diagnosis of PMHE.
Magnetic resonance imaging was then performed to evaluate the depth and extent of the lesions for surgical excision planning (Figure 6), which showed 5 nodular lesions within the dermis and subcutis adjacent to the proximal aspect of the left tibia and medial aspect of the left knee. An additional lesion was noted between the sartorius and semimembranosus muscles, which was thought to represent either a lymph node or an additional neoplastic lesion. Chest computed tomography also displayed indeterminate lesions in the lungs.
Excision of the superficial lesions was performed. All of the lesions demonstrated similar histologic changes to the previously described biopsy specimen. The tumor was limited to the dermis and subcutaneous tissue. The patient was lost to follow-up and the etiology of the lung lesions was unknown.
Comment
Nomenclature
Pseudomyogenic hemangioendothelioma is a relatively new type of vascular tumor that has been included in the updated 2013 edition of the World Health Organization classification as an intermediate malignant tumor that rarely metastasizes.3 It typically involves multiple tissue planes, most notably the dermis and subcutaneous layers but also muscle and bone.4 The term pseudomyogenic refers to the histologic resemblance of some of the cells to rhabdomyoblasts; however, these tumors are negative for all immunohistochemical muscle markers, most notably myogenin, desmin, and α-smooth muscle actin.5
Clinical Presentation
Gross features of PMHE typically include multiple firm nodules with ill-defined margins. The tumor was initially described in 1992 by Mirra et al2 as a fibromalike variant of epithelioid sarcoma. In 2003, a series of 7 cases of PMHE was reported by Billings et al6 under the term epithelioid sarcomalike hemangioendothelioma. Other than the predominance of an epithelioid morphology, the cases reported as epithelioid sarcomalike hemangioendothelioma had similar clinical features and immunophenotype to what has been reported as PMHE.
Based on a PubMed search of articles indexed for MEDLINE using the term pseudomyogenic hemangioendothelioma, the 2 largest case series were reported by Pradhan et al7 (N=8) in 2017 and Hornick and Fletcher4 (N=50) in 2011. Hornick and Fletcher4 reported a male (41/50 [82.0%]) to female (9/50 [18.0%]) ratio of 4.6 to 1, and an average age at presentation of 31 years with 82% (41/50) of patients 40 years or younger. Pradhan et al7 also reported a male predominance (7/8 [87.5%]) with a similar average age at presentation of 29 years (age range, 9–62 years). The size of individual tumors ranged from 0.3 to 5.5 cm (mean size, 1.9 cm) in the series by Hornick and Fletcher4 and 0.3 to 6.0 com in the series by Pradhan et al.7 Hornick and Fletcher4 reported the most common site of involvement was the leg (27/50 [54.0%]), followed by the arm (12/50 [24.0%]), trunk (9/50 [18.0%]), and head and neck (2/50 [4.0%]). The leg (6/8 [75.0%]) also was the most common site of involvement in the series by Pradhan et al,7 with 2 cases occurring on the arm. In the series by Hornick and Fletcher,4 the tumors typically involved the dermis and subcutaneous tissue (26/50 [52%]) with a smaller number involving skeletal muscle (17/50 [34%]) and bone (7/50 [14%]). They reported 66% of their patients (33/50) had multifocal disease at presentation.4 Pradhan et al7 also reported 2 (25.0%) cases being limited to the superficial soft tissue, 2 (25.0%) being limited to the deep soft tissue, and 4 (50.0%) involving the bone; 5 (62.5%) patients had multifocal disease at presentation. The presentation of our patient in regards to gender, age, and tumor characteristics is consistent with other published cases.5-10
Histopathology
Microscopic features of PMHE include sheets of spindled to epithelioid-appearing cells with mild to moderate nuclear atypia and eosinophilic cytoplasm. The tumor has an infiltrative growth pattern. Some of the cells may resemble rhabdomyoblastlike cells, hence the moniker pseudomyogenic. There is no recapitulation of vascular structures or remarkable cytoplasmic vacuolization. Mitotic rate is low and there is no tumor necrosis.4 The tumor cells do not appear to arise from a vessel or display an angiocentric growth pattern. Many cases report the presence of an inflammatory infiltrate containing neutrophils interspersed within the tumor.4,5,7 The overlying epidermis will commonly show hyperkeratosis, epidermal hyperplasia, and acanthosis.4,11
Differential Diagnosis
The histopathologic differential diagnosis would include epithelioid sarcoma, epithelioid hemangioendothelioma, and to a lesser extent dermatofibrosarcoma protuberans (DFSP) and rhabdomyosarcoma. Dermatofibrosarcoma protuberans is the most commonly encountered of these tumors. Histologically, DFSP is characterized by a cellular proliferation of small spindle cells with plump nuclei arranged in a storiform or cartwheel pattern. Dermatofibrosarcoma protuberans tends to be limited to the dermis and subcutaneous tissue and only rarely involves underlying skeletal muscle. The presence of the storiform growth pattern in conjunction with the lack of rhabdoid changes would favor a diagnosis of DFSP. Another characteristic histologic finding typically only associated with DFSP is the interdigitating growth pattern of the spindle cells within the lobules of the subcutaneous tissue, creating a lacelike or honeycomb appearance.
Immunohistochemistry staining is necessary to help differentiate PMHE from other tumors in the differential diagnosis. Pseudomyogenic hemangioendothelioma stains positive for cytokeratin AE1/AE3; integrase interactor 1; and vascular markers FLI-1, CD31, and ERG, and negative for CD34.4,6,12-15 In contrast to epithelioid hemangioendothelioma, DFSP, and to a lesser extent epithelioid sarcoma, all of which are positive for CD34, epithelioid sarcoma is negative for both CD31 and integrase interactor 1. Dermatofibrosarcoma protuberans is negative for cytokeratin AE1/AE3. Rhabdomyosarcomas are positive for myogenic markers such as MyoD1 and myogenin, unlike any of the other tumors mentioned. Histologically, epithelioid sarcomas will tend to have a granulomalike growth pattern with central necrosis, unlike PMHE.12 Epithelioid hemangioendothelioma often will have a cordlike growth pattern in a myxochondroid background. Unlike PMHE, these tumors often will appear to be arising from vessels, and intracytoplasmic vacuoles are common. Three cases of PMHE have been reported to have a t(7;19)(q22;q13) chromosomal anomaly, which is not consistent with every case.16
Treatment Options
Standard treatment typically includes wide excision of the lesions, as was done in our case. Because of the substantial risk of local recurrence, which was up to 58% in the series by Hornick and Fletcher,4 adjuvant therapy may be considered if positive margins are found on excision. Metastasis to lymph nodes and the lungs has been reported but is rare.2,4 Most cases have been shown to have a favorable prognosis; however, local recurrence seems to be common. Rarely, amputation of the limb may be required.5 In contrast, epithelioid sarcomas have been found to spread to lymph nodes and the lungs in up to 50% of cases with a 5-year survival rate of 10% to 30%.13
Conclusion
In summary, we describe a case of PMHE involving the lower leg in a 20-year-old man. These tumors often are multinodular and multiplanar, with the dermis and subcutaneous tissues being the most common areas affected. It has a high rate of local recurrence but rarely has distant metastasis. Pseudomyogenic hemangioendothelioma, similar to other soft tissue tumors, can be difficult to diagnose on shave biopsy or superficial punch biopsy not extending into subcutaneous tissue. Deep incisional or punch biopsies are required to more definitively diagnose these types of tumors. The diagnosis of PMHE versus other soft tissue tumors requires correlation of histology and immunohistochemistry staining with clinical information and radiographic findings.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma (pseudomyogenic hemangioendothelioma). Am J Surg Pathol. 2011;35:1088; author reply 1088-1089.
- Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
- Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014;46:95-104.
- Hornick JL, Fletcher CD. Pseudomyogenic hemangioendothelioma: a distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol. 2011;35:190-201.
- Sheng W, Pan Y, Wang J. Pseudomyogenic hemangioendothelioma: report of an additional case with aggressive clinical course. Am J Dermatopathol. 2013;35:597-600.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol. 2003;27:48-57.
- Pradhan D, Schoedel K, McGough RL, et al. Pseudomyogenic hemangioendothelioma of skin, bone and soft tissue—a clinicopathological, immunohistochemical and fluorescence in situ hybridization study [published online November 2, 2017]. Hum Pathol. 2017. doi:0.1016/j.humpath.2017.10.023.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- McGinity M, Bartanusz V, Dengler B, et al. Pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma, fibroma-like variant of epithelioid sarcoma) of the thoracic spine. Eur Spine J. 2013;22(suppl 3):S506-S511.
- Stuart LN, Gardner JM, Lauer SR, et al. Epithelioid sarcoma-like (pseudomyogenic) hemangioendothelioma, clinically mimicking dermatofibroma, diagnosed by skin biopsy in a 30-year-old man. J Cutan Pathol. 2013;40:909-913.
- Amary MF, O’Donnell P, Berisha F, et al. Pseudomyogenic (epithelioid sarcoma-like) hemangioendothelioma: characterization of five cases. Skeletal Radiol. 2013;42:947-957.
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542-550.
- Chbani L, Guillou L, Terrier P, et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French Sarcoma Group. Am J Clin Pathol. 2009;131:222-227.
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- Trombetta D, Magnusson L, von Steyern FV, et al. Translocation t(7;19)(q22;q13)—a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet. 2011;204:211-215.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma (pseudomyogenic hemangioendothelioma). Am J Surg Pathol. 2011;35:1088; author reply 1088-1089.
- Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
- Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014;46:95-104.
- Hornick JL, Fletcher CD. Pseudomyogenic hemangioendothelioma: a distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol. 2011;35:190-201.
- Sheng W, Pan Y, Wang J. Pseudomyogenic hemangioendothelioma: report of an additional case with aggressive clinical course. Am J Dermatopathol. 2013;35:597-600.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol. 2003;27:48-57.
- Pradhan D, Schoedel K, McGough RL, et al. Pseudomyogenic hemangioendothelioma of skin, bone and soft tissue—a clinicopathological, immunohistochemical and fluorescence in situ hybridization study [published online November 2, 2017]. Hum Pathol. 2017. doi:0.1016/j.humpath.2017.10.023.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- McGinity M, Bartanusz V, Dengler B, et al. Pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma, fibroma-like variant of epithelioid sarcoma) of the thoracic spine. Eur Spine J. 2013;22(suppl 3):S506-S511.
- Stuart LN, Gardner JM, Lauer SR, et al. Epithelioid sarcoma-like (pseudomyogenic) hemangioendothelioma, clinically mimicking dermatofibroma, diagnosed by skin biopsy in a 30-year-old man. J Cutan Pathol. 2013;40:909-913.
- Amary MF, O’Donnell P, Berisha F, et al. Pseudomyogenic (epithelioid sarcoma-like) hemangioendothelioma: characterization of five cases. Skeletal Radiol. 2013;42:947-957.
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542-550.
- Chbani L, Guillou L, Terrier P, et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French Sarcoma Group. Am J Clin Pathol. 2009;131:222-227.
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol. 2013;149:459-465.
- Trombetta D, Magnusson L, von Steyern FV, et al. Translocation t(7;19)(q22;q13)—a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet. 2011;204:211-215.
Practice Points
- Pseudomyogenic hemangioendothelioma (PMHE) is an uncommon vascular tumor that most often presents as multiple distinct nodules on the legs in young men.
- Pseudomyogenic hemangioendothelioma has an unusual immunohistochemistry staining pattern, with positive staining for cytokeratin AE1/AE3, CD31, and ERG but negative for CD34.
- Although local reoccurrence is common, PMHE metastasis is very uncommon.
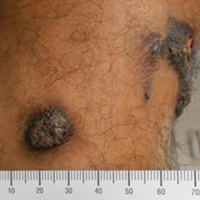
Lichen Planus Pemphigoides Treated With Ustekinumab
Case Report
A 71-year-old woman presented with pink to violaceous, flat-topped, polygonal papules consistent with lichen planus (LP) on the volar wrists, extensor elbows, and bilateral lower legs of 3 years’ duration. She also had erythematous, violaceous, infiltrated plaques with microvesiculation on the bilateral thighs of several months’ duration (Figure 1). She reported pruritus, burning, and discomfort. Her medical history included type 2 diabetes mellitus, hypertension, and asthma with no history of skin rashes. A complete physical examination was performed. Age-appropriate screening for malignancy was negative. Hepatitis B and C antibody serologies were negative. Her medications at the time included risedronate and atenolol, which she had been taking for several years.
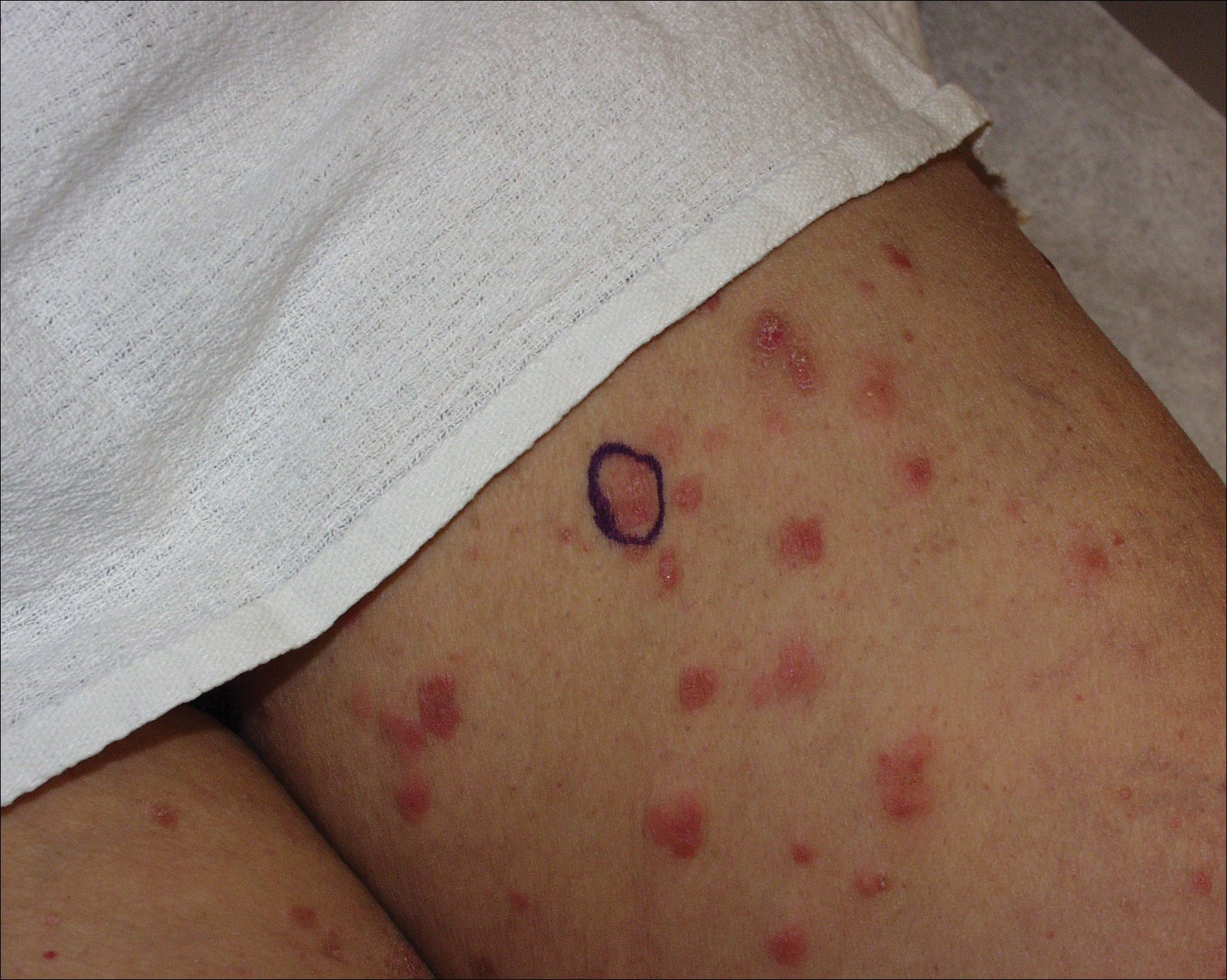
Punch biopsies from perilesional skin were submitted for hematoxylin and eosin staining and direct immunofluorescence (DIF). Histopathology showed a subepidermal blistering disease with tissue eosinophilia consistent with lichen planus pemphigoides (LPP)(Figure 2); direct immunofluorescence was positive for IgG, C3, and type IV collagen at the dermoepidermal junction. Serum BP180 was positive at 51 U/mL (reference range, <14 U/mL) and BP230 was negative. She was then started on tetracycline (500 mg twice daily), nicotinamide (500 mg twice daily), prednisone (5 mg daily), and dapsone (100 mg daily).
After 3 months without improvement, tetracycline and nicotinamide were discontinued, prednisone was increased to 10 mg daily, and dapsone was continued. A repeat biopsy was taken from a new area of involvement on the left lower leg, which revealed a psoriasiform dermatitis with interface changes. The DIF was positive for IgG and C3 along the basement membrane. A serum indirect immunofluorescence for BP180 also was positive.
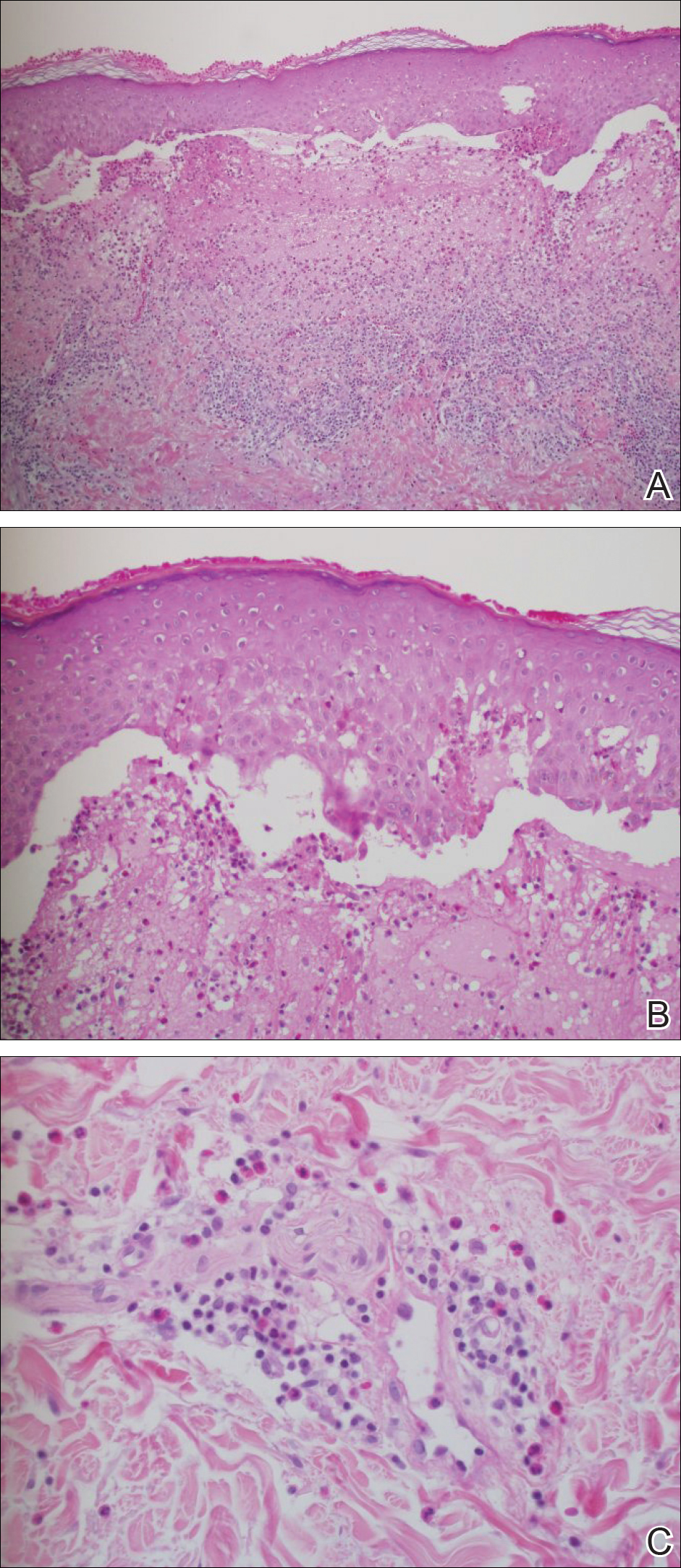
The patient developed mild hemolytic anemia on dapsone; the medication was eventually discontinued. Subsequent treatments included adequate trials of azathioprine, mycophenolate mofetil, and hydroxychloroquine. Azathioprine (150 mg daily) and hydroxychloroquine (400 mg daily) treatment failed. She initially improved on mycophenolate mofetil (500 mg in the morning and 1000 mg in the evening) with flattening of the papules on the arms and legs and decreased erythema. However, mycophenolate mofetil eventually lost its efficacy and was discontinued.
Because several medications failed (ie, tetracycline, nicotinamide, prednisone, dapsone, azathioprine, mycophenolate mofetil, hydroxychloroquine), she was started on ustekinumab (45 mg) initial loading dose by subcutaneous injection (patient’s weight, 63 kg). At 4 weeks, the patient was given the second subcutaneous injection of ustekinumab (45 mg). She experienced marked improvement with no new lesions. The prior lesions also had decreased in size and were only slightly pink. The prednisone dose was tapered to 5 mg daily.
She had near-complete resolution of the skin lesions 12 weeks after the second dose of ustekinumab. Since then, she has had some recrudescence of the papulosquamous lesions but no vesicles or bullae. With the exception of occasional scattered pink papules on the forearms, her condition greatly improved on ustekinumab. She is no longer taking any of the other medications with the exception of prednisone (down to 1 mg daily) with a plan to gradually taper completely off of it.
Comment
Clinical Presentation
Lichen planus pemphigoides is a rare autoimmune subepidermal blistering disease with few cases reported in the literature. It is considered a clinical variation of bullous pemphigoid (BP) or a coexistence of LP and BP.1,2 It is characterized by bullous lesions developing on LP papules as well as on clinically uninvolved areas of the skin. It has been reported that LPP is provoked by several medications including cinnarizine, captopril, ramipril, simvastatin, psoralen plus UVA, and antituberculous medications (eg, isoniazid, rifampin, ethambutol, pyrazinamide).1 Risedronate or atenolol have not been reported to cause LPP, LP, or BP; however, according to Litt,3 a lichenoid drug eruption has been associated with atenolol. Furthermore, some cases of LPP demonstrate overlapping characteristics with paraneoplastic pemphigus and have been associated with internal malignancy. Hamada et al4 described a case of LPP coupled with colon adenocarcinoma and numerous keratoacanthomas. The earliest depiction of the coexistence of a case of mainstream LP complicated by an extensive bullous eruption was by Kaposi5 in 1892. He coined the term lichen ruber pemphigoides.5
Compared to BP, LPP is believed to affect a younger age group and have a less serious clinical course. The mean age of onset of LPP is in the third to fourth decades of life, while BP typically presents in the sixth decade. When comparing the location of bullae in LPP versus BP, the lesions of LPP tend to occur on the limbs, while BP tends to occur on the trunk.6
Clinically, LPP is distinguished by the existence of bullous lesions developing atop of the lesions of LP as well as on normal skin, with the latter being more commonplace. A classic example of LPP is characterized by an initial episode of traditional LP lesions often having severe pruritus, with or without patches of erythema, with the sudden eruption of tense bullae. These bullae commonly appear on the extremities and can appear over the normal skin, erythematous patches, or preexisting papules.7 In the atypical clinical presentations of this dubious skin condition, the bullae may only be seen on the lesions of LP.8 There also could be a lichenoid erythrodermic manifestation of a bullous eruption.9
Oral lesions of LPP have been described but had not been studied immunopathologically until Allen et al10 portrayed a 59-year-old man with cutaneous and oral lesions of LPP. They performed biopsies on the oral lesions and examined them by routine light microscopy and immunofluorescent techniques. The fine keratotic striae on the anterior buccal mucosal lesions were clinically consistent with oral LP. Perilesional tissue in conjunction with ulceration of the posterior buccal mucosa demonstrated histologic and immunopathologic alterations consistent with BP.10
Histopathology
Histopathologically, the lesions of LP show a bandlike lymphohistiocytic infiltrate, colloid bodies in the dermis, irregular acanthosis with saw-toothed rete ridges, orthokeratosis, wedge-shaped hypergranulosis, and liquefaction degeneration of the basal layer. Direct immunofluorescence shows mainly IgM and C3 deposited on colloid bodies, fibrin, and fibrinogen.11 The histopathology of the bullous lesion of LPP depicts a subepidermal bulla with variable diffuse or sparse lymphohistiocytic infiltrate and frequent eosinophils with or without neutrophils in the upper dermis. The existence of C3 alone or with IgG along the dermoepidermal junction gives confirmation on DIF.7
Autoantibodies
The expression of IgG autoantibodies directed against the basement membrane zone distinguishes LPP from bullous LP.2 IgG autoantibodies to either one or both the 230-kDa and 180-kDa BP (type XVII collagen) antigens has been demonstrated with LPP.4,12-14 Hamada et al4 described a histologic pattern more consistent with paraneoplastic pemphigus. It has been suggested that injury to the basal cells in LP or damage due to other courses of therapy such as psoralen plus UVA unveil suppressed antigenic determinants or produce new antigens, leading to antibody development and production of BP.12,15
Zillikens et al2 performed a study to identify the target antigen of LPP autoantibodies. They used sera from patients with LPP (n=4) and stained the epidermal side of salt-split human skin in a configuration identical to BP sera. In BP, the autoimmune response is directed against BP180, a hemidesmosomal transmembrane collagenous glycoprotein. They demonstrated that sera from BP patients largely reacted with a set of 4 epitopes (MCW-0 through MCW-3) grouped within a 45 amino acid stretch of the major noncollagenous extracellular domain (NC16A) of BP180. By immunoblotting and enzyme-linked immunosorbent assay, LPP sera also were compellingly reactive with recombinant BP180 NC16A. Lichen planus pemphigoides epitopes were additionally mapped using a series of overlapping recombinant segments of the NC16A domain. The authors demonstrated that all LPP sera reacted with amino acids 46 through 59 of domain NC16A, a protein portion that was previously shown to be unreactive with BP sera. In addition, they showed that 2 LPP sera reacted with the immunodominant antigenic region related to BP. Furthermore, they identified a unique epitope within the BP180 NC16A domain—MCW-4—which was distinctively recognized by sera from patients with LPP.2
Pathogenesis
The pathogenesis of both LP and BP has been linked to multiple cytokines that induce apoptosis in basal keratinocytes. Implicated cytokines include IFN-γ, tumor necrosis factor α (TNF-α), IL-1, IL-6, and IL-8, as well as other apoptosis-related molecules, such as Fas/Apo-1 and Bcl-2 in LP.16-18 Soluble E-selectin, vascular endothelial growth factor, IL-1β, IL-8, IL-5, transforming growth factor β1, and TNF-α were found to be elevated in either blister fluid or sera of BP patients.15-17
Management
Lichen planus pemphigoides usually responds well to traditional therapies, with systemic steroids being the most efficacious treatment of extensive disease.12,13 Other options include tetracycline and nicotinamide, isotretinoin, dapsone, and immunosuppressive drugs such as systemic cortico-steroids.12 Demirçay et al12 described a patient with skin lesions that rapidly cleared after the administration of oral methylprednisolone (48 mg/d) and oral dapsone (100 mg/d). The methylprednisolone and dapsone were withdrawn after 12 and 16 weeks, respectively. There was no recurrence during the 1-year follow-up period.12 et al19 described a patient who was treated with pulsed intravenous corticosteroids and continued to develop new papular and vesicular skin lesions. However, when oral acitretin was added to the patient’s regimen, the skin lesions cleared.19 There are several case reports of the successful use of hydroxychloroquine in LP.20,21
Cutaneous, nail, and oral LP also can be treated with TNF-α inhibitors (eg, adalimumab, etanercept) with resolution of lesions.22-25 However, we have not been able to find any reports of treating LPP with biologic medications in a search of PubMed articles indexed for MEDLINE using the terms lichen planus pemphigoides and biologic treatments/therapies. Given the fact that TNF-α and other inflammatory cytokines are involved in the pathogenesis of BP and LP, it is feasible that they also may be involved in the pathogenesis of LPP.
In our patient with cutaneous LPP, we chose to use ustekinumab instead of a primary TNF-α inhibitor because ustekinumab indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22, which also could have played a role in the patient’s disease. Our goal was to use ustekinumab as a potential corticosteroid-sparing agent. Ustekinumab greatly improved her skin condition and allowed us to discontinue other medications.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Litt J. Litt’s Drug Eruptions and Reactions Manual. 18th Ed. London, England: Informa Healthcare; 2011.
- Hamada T, Fujimoto W, Okazaki F, et al. Lichen planus pemphigoides and multiple keratoacanthomas associated with colon adenocarcinoma. Br J Dermatol. 2004;151:252-254.
- Kaposi M. Lichen ruber pemphigoides. Arch Derm Syph. 1892;343-346.
- Swale VJ, Black MM, Bhogal BS. Lichen planus pemphigoides: two case reports. Clin Exp Dermatol. 1998;23:132-135.
- Okochi H, Nashiro K, Tsuchida T, et al. Lichen planus pemphigoides: case reports and results of immunofluorescence and immunoelectron microscopic study. J Am Acad Dermatol. 1990;22:626-631.
- Mendiratta V, Asati DP, Koranne RV. Lichen planus pemphigoides in an Indian female. Indian J Dermatol. 2005;50:224-226.
- Joly P, Tanasescu S, Wolkenstein P, et al. Lichenoid erythrodermic bullous pemphigoid of the African patient. J Am Acad Dermatol. 1998;39:691-697.
- Allen , , R. Lichen planus pemphigoides: report of a case with oral lesions. Oral Surg Oral Med Oral Pathol. 1987;63:184-188.
- Rapini RP. Practical Dermatopathology. Philadelphia, PA: Mosby Elsevier; 2005.
- Demirçay Z, Baykal C, Demirkesen C. Lichen planus pemphigoides: report of two cases. Int J Dermatol. 2001;40:757-759.
- Sakuma-Oyama Y, Powell AM, Albert S, et al. Lichen planus pemphigoides evolving into pemphigoid nodularis. Clin Exp Dermatol. 2004;28:613-616.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Ameglio F, D’Auria L, Cordiali-Fei P, et al. Bullous pemphigoid and pemphigus vulgaris: correlated behaviour of serum VEGF, sE-selectin and TNF-alpha levels. J Biol Regul Homeost Agents. 1997;11:148-153.
- Ameglio F, D’auria L, Bonifati C, et al. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. 1998;138:611-614.
- D’Auria L, Mussi A, Bonifati C, et al. Increased serum IL-6, TNF-alpha and IL-10 levels in patients with bullous pemphigoid: relationships with disease activity. J Eur Acad Dermatol Venereol. 1999;12:11-15.
- , ,, . Treatment of lichen planus pemphigoides with acitretin and pulsed corticosteroids. Hautarzt. 2003;54:268-273.
- Eisen D. Hydroxychloroquine sulfate (Plaquenil) improves oral lichen planus: an open trial. J Am Acad Dermatol. 1993;28:609-612.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 11th ed. Philadelphia, PA: Mosby Elsevier; 2011.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Yarom N. Etanercept for the management of oral lichen planus. Am J Clin Dermatol. 2007;8:121.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Irla N, Schneiter T, Haneke E, et al. Nail lichen planus: successful treatment with etanercept. Case Rep Dermatol. 2010;2:173-176.
Case Report
A 71-year-old woman presented with pink to violaceous, flat-topped, polygonal papules consistent with lichen planus (LP) on the volar wrists, extensor elbows, and bilateral lower legs of 3 years’ duration. She also had erythematous, violaceous, infiltrated plaques with microvesiculation on the bilateral thighs of several months’ duration (Figure 1). She reported pruritus, burning, and discomfort. Her medical history included type 2 diabetes mellitus, hypertension, and asthma with no history of skin rashes. A complete physical examination was performed. Age-appropriate screening for malignancy was negative. Hepatitis B and C antibody serologies were negative. Her medications at the time included risedronate and atenolol, which she had been taking for several years.
Punch biopsies from perilesional skin were submitted for hematoxylin and eosin staining and direct immunofluorescence (DIF). Histopathology showed a subepidermal blistering disease with tissue eosinophilia consistent with lichen planus pemphigoides (LPP)(Figure 2); direct immunofluorescence was positive for IgG, C3, and type IV collagen at the dermoepidermal junction. Serum BP180 was positive at 51 U/mL (reference range, <14 U/mL) and BP230 was negative. She was then started on tetracycline (500 mg twice daily), nicotinamide (500 mg twice daily), prednisone (5 mg daily), and dapsone (100 mg daily).
After 3 months without improvement, tetracycline and nicotinamide were discontinued, prednisone was increased to 10 mg daily, and dapsone was continued. A repeat biopsy was taken from a new area of involvement on the left lower leg, which revealed a psoriasiform dermatitis with interface changes. The DIF was positive for IgG and C3 along the basement membrane. A serum indirect immunofluorescence for BP180 also was positive.
The patient developed mild hemolytic anemia on dapsone; the medication was eventually discontinued. Subsequent treatments included adequate trials of azathioprine, mycophenolate mofetil, and hydroxychloroquine. Azathioprine (150 mg daily) and hydroxychloroquine (400 mg daily) treatment failed. She initially improved on mycophenolate mofetil (500 mg in the morning and 1000 mg in the evening) with flattening of the papules on the arms and legs and decreased erythema. However, mycophenolate mofetil eventually lost its efficacy and was discontinued.
Because several medications failed (ie, tetracycline, nicotinamide, prednisone, dapsone, azathioprine, mycophenolate mofetil, hydroxychloroquine), she was started on ustekinumab (45 mg) initial loading dose by subcutaneous injection (patient’s weight, 63 kg). At 4 weeks, the patient was given the second subcutaneous injection of ustekinumab (45 mg). She experienced marked improvement with no new lesions. The prior lesions also had decreased in size and were only slightly pink. The prednisone dose was tapered to 5 mg daily.
She had near-complete resolution of the skin lesions 12 weeks after the second dose of ustekinumab. Since then, she has had some recrudescence of the papulosquamous lesions but no vesicles or bullae. With the exception of occasional scattered pink papules on the forearms, her condition greatly improved on ustekinumab. She is no longer taking any of the other medications with the exception of prednisone (down to 1 mg daily) with a plan to gradually taper completely off of it.
Comment
Clinical Presentation
Lichen planus pemphigoides is a rare autoimmune subepidermal blistering disease with few cases reported in the literature. It is considered a clinical variation of bullous pemphigoid (BP) or a coexistence of LP and BP.1,2 It is characterized by bullous lesions developing on LP papules as well as on clinically uninvolved areas of the skin. It has been reported that LPP is provoked by several medications including cinnarizine, captopril, ramipril, simvastatin, psoralen plus UVA, and antituberculous medications (eg, isoniazid, rifampin, ethambutol, pyrazinamide).1 Risedronate or atenolol have not been reported to cause LPP, LP, or BP; however, according to Litt,3 a lichenoid drug eruption has been associated with atenolol. Furthermore, some cases of LPP demonstrate overlapping characteristics with paraneoplastic pemphigus and have been associated with internal malignancy. Hamada et al4 described a case of LPP coupled with colon adenocarcinoma and numerous keratoacanthomas. The earliest depiction of the coexistence of a case of mainstream LP complicated by an extensive bullous eruption was by Kaposi5 in 1892. He coined the term lichen ruber pemphigoides.5
Compared to BP, LPP is believed to affect a younger age group and have a less serious clinical course. The mean age of onset of LPP is in the third to fourth decades of life, while BP typically presents in the sixth decade. When comparing the location of bullae in LPP versus BP, the lesions of LPP tend to occur on the limbs, while BP tends to occur on the trunk.6
Clinically, LPP is distinguished by the existence of bullous lesions developing atop of the lesions of LP as well as on normal skin, with the latter being more commonplace. A classic example of LPP is characterized by an initial episode of traditional LP lesions often having severe pruritus, with or without patches of erythema, with the sudden eruption of tense bullae. These bullae commonly appear on the extremities and can appear over the normal skin, erythematous patches, or preexisting papules.7 In the atypical clinical presentations of this dubious skin condition, the bullae may only be seen on the lesions of LP.8 There also could be a lichenoid erythrodermic manifestation of a bullous eruption.9
Oral lesions of LPP have been described but had not been studied immunopathologically until Allen et al10 portrayed a 59-year-old man with cutaneous and oral lesions of LPP. They performed biopsies on the oral lesions and examined them by routine light microscopy and immunofluorescent techniques. The fine keratotic striae on the anterior buccal mucosal lesions were clinically consistent with oral LP. Perilesional tissue in conjunction with ulceration of the posterior buccal mucosa demonstrated histologic and immunopathologic alterations consistent with BP.10
Histopathology
Histopathologically, the lesions of LP show a bandlike lymphohistiocytic infiltrate, colloid bodies in the dermis, irregular acanthosis with saw-toothed rete ridges, orthokeratosis, wedge-shaped hypergranulosis, and liquefaction degeneration of the basal layer. Direct immunofluorescence shows mainly IgM and C3 deposited on colloid bodies, fibrin, and fibrinogen.11 The histopathology of the bullous lesion of LPP depicts a subepidermal bulla with variable diffuse or sparse lymphohistiocytic infiltrate and frequent eosinophils with or without neutrophils in the upper dermis. The existence of C3 alone or with IgG along the dermoepidermal junction gives confirmation on DIF.7
Autoantibodies
The expression of IgG autoantibodies directed against the basement membrane zone distinguishes LPP from bullous LP.2 IgG autoantibodies to either one or both the 230-kDa and 180-kDa BP (type XVII collagen) antigens has been demonstrated with LPP.4,12-14 Hamada et al4 described a histologic pattern more consistent with paraneoplastic pemphigus. It has been suggested that injury to the basal cells in LP or damage due to other courses of therapy such as psoralen plus UVA unveil suppressed antigenic determinants or produce new antigens, leading to antibody development and production of BP.12,15
Zillikens et al2 performed a study to identify the target antigen of LPP autoantibodies. They used sera from patients with LPP (n=4) and stained the epidermal side of salt-split human skin in a configuration identical to BP sera. In BP, the autoimmune response is directed against BP180, a hemidesmosomal transmembrane collagenous glycoprotein. They demonstrated that sera from BP patients largely reacted with a set of 4 epitopes (MCW-0 through MCW-3) grouped within a 45 amino acid stretch of the major noncollagenous extracellular domain (NC16A) of BP180. By immunoblotting and enzyme-linked immunosorbent assay, LPP sera also were compellingly reactive with recombinant BP180 NC16A. Lichen planus pemphigoides epitopes were additionally mapped using a series of overlapping recombinant segments of the NC16A domain. The authors demonstrated that all LPP sera reacted with amino acids 46 through 59 of domain NC16A, a protein portion that was previously shown to be unreactive with BP sera. In addition, they showed that 2 LPP sera reacted with the immunodominant antigenic region related to BP. Furthermore, they identified a unique epitope within the BP180 NC16A domain—MCW-4—which was distinctively recognized by sera from patients with LPP.2
Pathogenesis
The pathogenesis of both LP and BP has been linked to multiple cytokines that induce apoptosis in basal keratinocytes. Implicated cytokines include IFN-γ, tumor necrosis factor α (TNF-α), IL-1, IL-6, and IL-8, as well as other apoptosis-related molecules, such as Fas/Apo-1 and Bcl-2 in LP.16-18 Soluble E-selectin, vascular endothelial growth factor, IL-1β, IL-8, IL-5, transforming growth factor β1, and TNF-α were found to be elevated in either blister fluid or sera of BP patients.15-17
Management
Lichen planus pemphigoides usually responds well to traditional therapies, with systemic steroids being the most efficacious treatment of extensive disease.12,13 Other options include tetracycline and nicotinamide, isotretinoin, dapsone, and immunosuppressive drugs such as systemic cortico-steroids.12 Demirçay et al12 described a patient with skin lesions that rapidly cleared after the administration of oral methylprednisolone (48 mg/d) and oral dapsone (100 mg/d). The methylprednisolone and dapsone were withdrawn after 12 and 16 weeks, respectively. There was no recurrence during the 1-year follow-up period.12 et al19 described a patient who was treated with pulsed intravenous corticosteroids and continued to develop new papular and vesicular skin lesions. However, when oral acitretin was added to the patient’s regimen, the skin lesions cleared.19 There are several case reports of the successful use of hydroxychloroquine in LP.20,21
Cutaneous, nail, and oral LP also can be treated with TNF-α inhibitors (eg, adalimumab, etanercept) with resolution of lesions.22-25 However, we have not been able to find any reports of treating LPP with biologic medications in a search of PubMed articles indexed for MEDLINE using the terms lichen planus pemphigoides and biologic treatments/therapies. Given the fact that TNF-α and other inflammatory cytokines are involved in the pathogenesis of BP and LP, it is feasible that they also may be involved in the pathogenesis of LPP.
In our patient with cutaneous LPP, we chose to use ustekinumab instead of a primary TNF-α inhibitor because ustekinumab indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22, which also could have played a role in the patient’s disease. Our goal was to use ustekinumab as a potential corticosteroid-sparing agent. Ustekinumab greatly improved her skin condition and allowed us to discontinue other medications.
Case Report
A 71-year-old woman presented with pink to violaceous, flat-topped, polygonal papules consistent with lichen planus (LP) on the volar wrists, extensor elbows, and bilateral lower legs of 3 years’ duration. She also had erythematous, violaceous, infiltrated plaques with microvesiculation on the bilateral thighs of several months’ duration (Figure 1). She reported pruritus, burning, and discomfort. Her medical history included type 2 diabetes mellitus, hypertension, and asthma with no history of skin rashes. A complete physical examination was performed. Age-appropriate screening for malignancy was negative. Hepatitis B and C antibody serologies were negative. Her medications at the time included risedronate and atenolol, which she had been taking for several years.
Punch biopsies from perilesional skin were submitted for hematoxylin and eosin staining and direct immunofluorescence (DIF). Histopathology showed a subepidermal blistering disease with tissue eosinophilia consistent with lichen planus pemphigoides (LPP)(Figure 2); direct immunofluorescence was positive for IgG, C3, and type IV collagen at the dermoepidermal junction. Serum BP180 was positive at 51 U/mL (reference range, <14 U/mL) and BP230 was negative. She was then started on tetracycline (500 mg twice daily), nicotinamide (500 mg twice daily), prednisone (5 mg daily), and dapsone (100 mg daily).
After 3 months without improvement, tetracycline and nicotinamide were discontinued, prednisone was increased to 10 mg daily, and dapsone was continued. A repeat biopsy was taken from a new area of involvement on the left lower leg, which revealed a psoriasiform dermatitis with interface changes. The DIF was positive for IgG and C3 along the basement membrane. A serum indirect immunofluorescence for BP180 also was positive.
The patient developed mild hemolytic anemia on dapsone; the medication was eventually discontinued. Subsequent treatments included adequate trials of azathioprine, mycophenolate mofetil, and hydroxychloroquine. Azathioprine (150 mg daily) and hydroxychloroquine (400 mg daily) treatment failed. She initially improved on mycophenolate mofetil (500 mg in the morning and 1000 mg in the evening) with flattening of the papules on the arms and legs and decreased erythema. However, mycophenolate mofetil eventually lost its efficacy and was discontinued.
Because several medications failed (ie, tetracycline, nicotinamide, prednisone, dapsone, azathioprine, mycophenolate mofetil, hydroxychloroquine), she was started on ustekinumab (45 mg) initial loading dose by subcutaneous injection (patient’s weight, 63 kg). At 4 weeks, the patient was given the second subcutaneous injection of ustekinumab (45 mg). She experienced marked improvement with no new lesions. The prior lesions also had decreased in size and were only slightly pink. The prednisone dose was tapered to 5 mg daily.
She had near-complete resolution of the skin lesions 12 weeks after the second dose of ustekinumab. Since then, she has had some recrudescence of the papulosquamous lesions but no vesicles or bullae. With the exception of occasional scattered pink papules on the forearms, her condition greatly improved on ustekinumab. She is no longer taking any of the other medications with the exception of prednisone (down to 1 mg daily) with a plan to gradually taper completely off of it.
Comment
Clinical Presentation
Lichen planus pemphigoides is a rare autoimmune subepidermal blistering disease with few cases reported in the literature. It is considered a clinical variation of bullous pemphigoid (BP) or a coexistence of LP and BP.1,2 It is characterized by bullous lesions developing on LP papules as well as on clinically uninvolved areas of the skin. It has been reported that LPP is provoked by several medications including cinnarizine, captopril, ramipril, simvastatin, psoralen plus UVA, and antituberculous medications (eg, isoniazid, rifampin, ethambutol, pyrazinamide).1 Risedronate or atenolol have not been reported to cause LPP, LP, or BP; however, according to Litt,3 a lichenoid drug eruption has been associated with atenolol. Furthermore, some cases of LPP demonstrate overlapping characteristics with paraneoplastic pemphigus and have been associated with internal malignancy. Hamada et al4 described a case of LPP coupled with colon adenocarcinoma and numerous keratoacanthomas. The earliest depiction of the coexistence of a case of mainstream LP complicated by an extensive bullous eruption was by Kaposi5 in 1892. He coined the term lichen ruber pemphigoides.5
Compared to BP, LPP is believed to affect a younger age group and have a less serious clinical course. The mean age of onset of LPP is in the third to fourth decades of life, while BP typically presents in the sixth decade. When comparing the location of bullae in LPP versus BP, the lesions of LPP tend to occur on the limbs, while BP tends to occur on the trunk.6
Clinically, LPP is distinguished by the existence of bullous lesions developing atop of the lesions of LP as well as on normal skin, with the latter being more commonplace. A classic example of LPP is characterized by an initial episode of traditional LP lesions often having severe pruritus, with or without patches of erythema, with the sudden eruption of tense bullae. These bullae commonly appear on the extremities and can appear over the normal skin, erythematous patches, or preexisting papules.7 In the atypical clinical presentations of this dubious skin condition, the bullae may only be seen on the lesions of LP.8 There also could be a lichenoid erythrodermic manifestation of a bullous eruption.9
Oral lesions of LPP have been described but had not been studied immunopathologically until Allen et al10 portrayed a 59-year-old man with cutaneous and oral lesions of LPP. They performed biopsies on the oral lesions and examined them by routine light microscopy and immunofluorescent techniques. The fine keratotic striae on the anterior buccal mucosal lesions were clinically consistent with oral LP. Perilesional tissue in conjunction with ulceration of the posterior buccal mucosa demonstrated histologic and immunopathologic alterations consistent with BP.10
Histopathology
Histopathologically, the lesions of LP show a bandlike lymphohistiocytic infiltrate, colloid bodies in the dermis, irregular acanthosis with saw-toothed rete ridges, orthokeratosis, wedge-shaped hypergranulosis, and liquefaction degeneration of the basal layer. Direct immunofluorescence shows mainly IgM and C3 deposited on colloid bodies, fibrin, and fibrinogen.11 The histopathology of the bullous lesion of LPP depicts a subepidermal bulla with variable diffuse or sparse lymphohistiocytic infiltrate and frequent eosinophils with or without neutrophils in the upper dermis. The existence of C3 alone or with IgG along the dermoepidermal junction gives confirmation on DIF.7
Autoantibodies
The expression of IgG autoantibodies directed against the basement membrane zone distinguishes LPP from bullous LP.2 IgG autoantibodies to either one or both the 230-kDa and 180-kDa BP (type XVII collagen) antigens has been demonstrated with LPP.4,12-14 Hamada et al4 described a histologic pattern more consistent with paraneoplastic pemphigus. It has been suggested that injury to the basal cells in LP or damage due to other courses of therapy such as psoralen plus UVA unveil suppressed antigenic determinants or produce new antigens, leading to antibody development and production of BP.12,15
Zillikens et al2 performed a study to identify the target antigen of LPP autoantibodies. They used sera from patients with LPP (n=4) and stained the epidermal side of salt-split human skin in a configuration identical to BP sera. In BP, the autoimmune response is directed against BP180, a hemidesmosomal transmembrane collagenous glycoprotein. They demonstrated that sera from BP patients largely reacted with a set of 4 epitopes (MCW-0 through MCW-3) grouped within a 45 amino acid stretch of the major noncollagenous extracellular domain (NC16A) of BP180. By immunoblotting and enzyme-linked immunosorbent assay, LPP sera also were compellingly reactive with recombinant BP180 NC16A. Lichen planus pemphigoides epitopes were additionally mapped using a series of overlapping recombinant segments of the NC16A domain. The authors demonstrated that all LPP sera reacted with amino acids 46 through 59 of domain NC16A, a protein portion that was previously shown to be unreactive with BP sera. In addition, they showed that 2 LPP sera reacted with the immunodominant antigenic region related to BP. Furthermore, they identified a unique epitope within the BP180 NC16A domain—MCW-4—which was distinctively recognized by sera from patients with LPP.2
Pathogenesis
The pathogenesis of both LP and BP has been linked to multiple cytokines that induce apoptosis in basal keratinocytes. Implicated cytokines include IFN-γ, tumor necrosis factor α (TNF-α), IL-1, IL-6, and IL-8, as well as other apoptosis-related molecules, such as Fas/Apo-1 and Bcl-2 in LP.16-18 Soluble E-selectin, vascular endothelial growth factor, IL-1β, IL-8, IL-5, transforming growth factor β1, and TNF-α were found to be elevated in either blister fluid or sera of BP patients.15-17
Management
Lichen planus pemphigoides usually responds well to traditional therapies, with systemic steroids being the most efficacious treatment of extensive disease.12,13 Other options include tetracycline and nicotinamide, isotretinoin, dapsone, and immunosuppressive drugs such as systemic cortico-steroids.12 Demirçay et al12 described a patient with skin lesions that rapidly cleared after the administration of oral methylprednisolone (48 mg/d) and oral dapsone (100 mg/d). The methylprednisolone and dapsone were withdrawn after 12 and 16 weeks, respectively. There was no recurrence during the 1-year follow-up period.12 et al19 described a patient who was treated with pulsed intravenous corticosteroids and continued to develop new papular and vesicular skin lesions. However, when oral acitretin was added to the patient’s regimen, the skin lesions cleared.19 There are several case reports of the successful use of hydroxychloroquine in LP.20,21
Cutaneous, nail, and oral LP also can be treated with TNF-α inhibitors (eg, adalimumab, etanercept) with resolution of lesions.22-25 However, we have not been able to find any reports of treating LPP with biologic medications in a search of PubMed articles indexed for MEDLINE using the terms lichen planus pemphigoides and biologic treatments/therapies. Given the fact that TNF-α and other inflammatory cytokines are involved in the pathogenesis of BP and LP, it is feasible that they also may be involved in the pathogenesis of LPP.
In our patient with cutaneous LPP, we chose to use ustekinumab instead of a primary TNF-α inhibitor because ustekinumab indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22, which also could have played a role in the patient’s disease. Our goal was to use ustekinumab as a potential corticosteroid-sparing agent. Ustekinumab greatly improved her skin condition and allowed us to discontinue other medications.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Litt J. Litt’s Drug Eruptions and Reactions Manual. 18th Ed. London, England: Informa Healthcare; 2011.
- Hamada T, Fujimoto W, Okazaki F, et al. Lichen planus pemphigoides and multiple keratoacanthomas associated with colon adenocarcinoma. Br J Dermatol. 2004;151:252-254.
- Kaposi M. Lichen ruber pemphigoides. Arch Derm Syph. 1892;343-346.
- Swale VJ, Black MM, Bhogal BS. Lichen planus pemphigoides: two case reports. Clin Exp Dermatol. 1998;23:132-135.
- Okochi H, Nashiro K, Tsuchida T, et al. Lichen planus pemphigoides: case reports and results of immunofluorescence and immunoelectron microscopic study. J Am Acad Dermatol. 1990;22:626-631.
- Mendiratta V, Asati DP, Koranne RV. Lichen planus pemphigoides in an Indian female. Indian J Dermatol. 2005;50:224-226.
- Joly P, Tanasescu S, Wolkenstein P, et al. Lichenoid erythrodermic bullous pemphigoid of the African patient. J Am Acad Dermatol. 1998;39:691-697.
- Allen , , R. Lichen planus pemphigoides: report of a case with oral lesions. Oral Surg Oral Med Oral Pathol. 1987;63:184-188.
- Rapini RP. Practical Dermatopathology. Philadelphia, PA: Mosby Elsevier; 2005.
- Demirçay Z, Baykal C, Demirkesen C. Lichen planus pemphigoides: report of two cases. Int J Dermatol. 2001;40:757-759.
- Sakuma-Oyama Y, Powell AM, Albert S, et al. Lichen planus pemphigoides evolving into pemphigoid nodularis. Clin Exp Dermatol. 2004;28:613-616.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Ameglio F, D’Auria L, Cordiali-Fei P, et al. Bullous pemphigoid and pemphigus vulgaris: correlated behaviour of serum VEGF, sE-selectin and TNF-alpha levels. J Biol Regul Homeost Agents. 1997;11:148-153.
- Ameglio F, D’auria L, Bonifati C, et al. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. 1998;138:611-614.
- D’Auria L, Mussi A, Bonifati C, et al. Increased serum IL-6, TNF-alpha and IL-10 levels in patients with bullous pemphigoid: relationships with disease activity. J Eur Acad Dermatol Venereol. 1999;12:11-15.
- , ,, . Treatment of lichen planus pemphigoides with acitretin and pulsed corticosteroids. Hautarzt. 2003;54:268-273.
- Eisen D. Hydroxychloroquine sulfate (Plaquenil) improves oral lichen planus: an open trial. J Am Acad Dermatol. 1993;28:609-612.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 11th ed. Philadelphia, PA: Mosby Elsevier; 2011.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Yarom N. Etanercept for the management of oral lichen planus. Am J Clin Dermatol. 2007;8:121.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Irla N, Schneiter T, Haneke E, et al. Nail lichen planus: successful treatment with etanercept. Case Rep Dermatol. 2010;2:173-176.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Litt J. Litt’s Drug Eruptions and Reactions Manual. 18th Ed. London, England: Informa Healthcare; 2011.
- Hamada T, Fujimoto W, Okazaki F, et al. Lichen planus pemphigoides and multiple keratoacanthomas associated with colon adenocarcinoma. Br J Dermatol. 2004;151:252-254.
- Kaposi M. Lichen ruber pemphigoides. Arch Derm Syph. 1892;343-346.
- Swale VJ, Black MM, Bhogal BS. Lichen planus pemphigoides: two case reports. Clin Exp Dermatol. 1998;23:132-135.
- Okochi H, Nashiro K, Tsuchida T, et al. Lichen planus pemphigoides: case reports and results of immunofluorescence and immunoelectron microscopic study. J Am Acad Dermatol. 1990;22:626-631.
- Mendiratta V, Asati DP, Koranne RV. Lichen planus pemphigoides in an Indian female. Indian J Dermatol. 2005;50:224-226.
- Joly P, Tanasescu S, Wolkenstein P, et al. Lichenoid erythrodermic bullous pemphigoid of the African patient. J Am Acad Dermatol. 1998;39:691-697.
- Allen , , R. Lichen planus pemphigoides: report of a case with oral lesions. Oral Surg Oral Med Oral Pathol. 1987;63:184-188.
- Rapini RP. Practical Dermatopathology. Philadelphia, PA: Mosby Elsevier; 2005.
- Demirçay Z, Baykal C, Demirkesen C. Lichen planus pemphigoides: report of two cases. Int J Dermatol. 2001;40:757-759.
- Sakuma-Oyama Y, Powell AM, Albert S, et al. Lichen planus pemphigoides evolving into pemphigoid nodularis. Clin Exp Dermatol. 2004;28:613-616.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Ameglio F, D’Auria L, Cordiali-Fei P, et al. Bullous pemphigoid and pemphigus vulgaris: correlated behaviour of serum VEGF, sE-selectin and TNF-alpha levels. J Biol Regul Homeost Agents. 1997;11:148-153.
- Ameglio F, D’auria L, Bonifati C, et al. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. 1998;138:611-614.
- D’Auria L, Mussi A, Bonifati C, et al. Increased serum IL-6, TNF-alpha and IL-10 levels in patients with bullous pemphigoid: relationships with disease activity. J Eur Acad Dermatol Venereol. 1999;12:11-15.
- , ,, . Treatment of lichen planus pemphigoides with acitretin and pulsed corticosteroids. Hautarzt. 2003;54:268-273.
- Eisen D. Hydroxychloroquine sulfate (Plaquenil) improves oral lichen planus: an open trial. J Am Acad Dermatol. 1993;28:609-612.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 11th ed. Philadelphia, PA: Mosby Elsevier; 2011.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Yarom N. Etanercept for the management of oral lichen planus. Am J Clin Dermatol. 2007;8:121.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Irla N, Schneiter T, Haneke E, et al. Nail lichen planus: successful treatment with etanercept. Case Rep Dermatol. 2010;2:173-176.
- Lichen planus pemphigoides (LPP) is a rare autoimmune subepidermal blistering disease with few cases reported in the literature.
- Because tumor necrosis factor 11α (TNF-11α) and other inflammatory cytokines are involved in the pathogenesis of bullous pemphigoid and lichen planus, it is feasible that they also may be involved in the pathogenesis of LPP.
- Ustekinumab may be used to treat LPP as a potential corticosteroid-sparing agent because it indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22.

Multinucleate Cell Angiohistiocytoma
Multinucleate cell angiohistiocytoma (MCAH) is a rare benign, soft-tissue tumor first described in 1985 by Smith and Jones1 that presents clinically as erythematous to violaceous papules most commonly affecting females on the dorsal aspect of the hands and face.2 Multinucleate cell angiohistiocytoma is histologically characterized by vascular and histiocytic proliferations with dermal fibrosis. Few cases have been reported of lesions affecting the lower extremities. We report a case of MCAH affecting the legs.
Case Report
An 83-year-old white man with a history of basal cell carcinoma presented for evaluation of grouped, well-circumscribed, soft, red-violet, painless papules on the right anterior thigh that had been present for 8 months (Figure 1A). A review of symptoms was negative for immunologic, respiratory, and hematologic changes. The patient’s medical history also was remarkable for prostate cancer treated with radiation 18 years prior as well as right hip and left knee implants. The initial clinical impression was Kaposi sarcoma or a granulomatous disorder.
Histopathologic evaluation of a deep shave biopsy initially determined the lesion to be scar tissue without other pathologic findings. The patient returned to the clinic 12 months later for a complete skin examination given his history of skin cancer. Compared to clinical photographs taken a year prior, new violaceous papules were noted on the right thigh (Figure 1B) and left calf. Furthermore, there was no recurrence of the lesion at the prior biopsy site. Shave biopsies of the papules on the right thigh and left calf demonstrated similar histologic findings to each other. There was a mild increase in the number of small blood vessels in the superficial dermis (Figure 2A). A mild perivascular lymphocytic infiltrate surrounded some of these blood vessels. The endothelial cells had small nuclei with no evidence of nuclear pleomorphism. Careful examination of the interstitial dermis revealed scattered multinucleate cells with angulated cytoplasm (Figure 2B). Immunostaining for CD31 and human herpesvirus 8 were negative, excluding an infiltrative vascular tumor and Kaposi sarcoma, respectively. The diagnosis of MCAH was made based on the histopathologic findings.
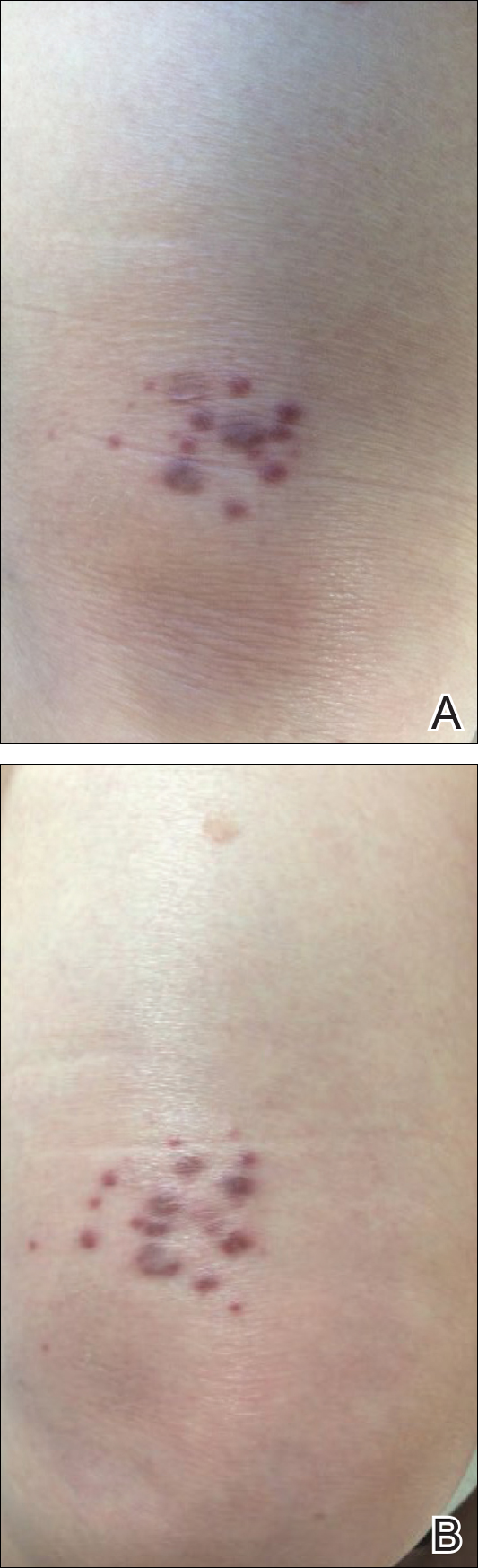
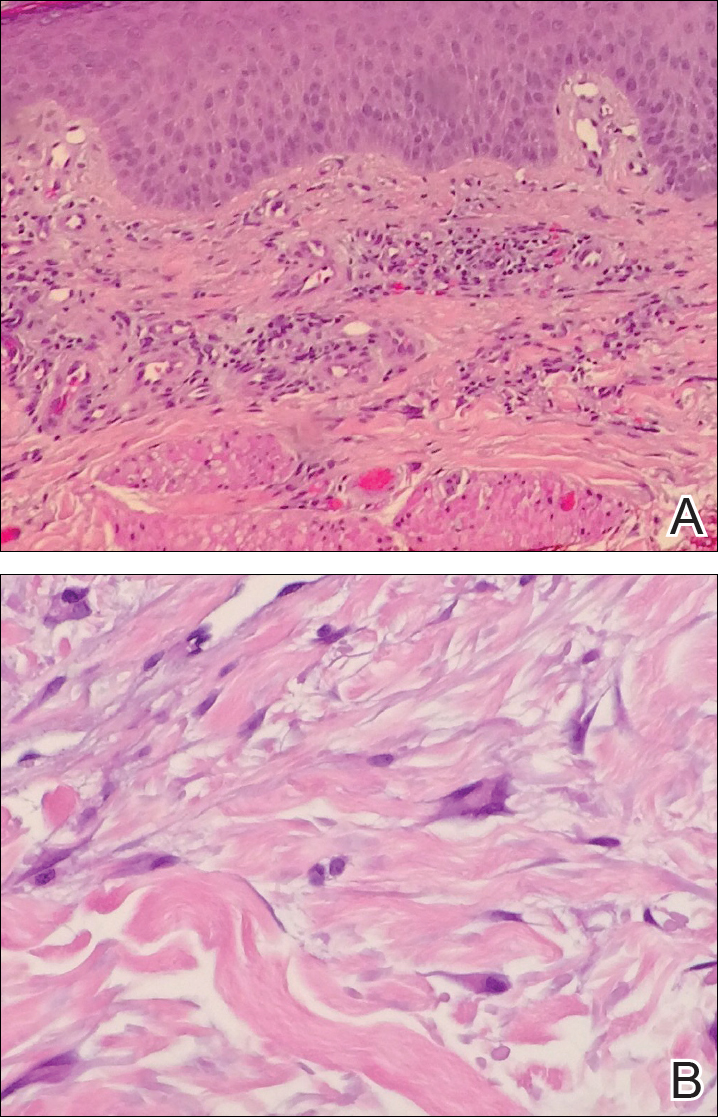
At 1-year follow-up, the condition was stable with no gross changes in the lesions based on prior photographs. Once again, there was no recurrence of the excised lesions at both biopsy sites.
Comment
Presentation
A systematic review of published reports determined that 79% of MCAH cases occur in females, with an average age of onset of 50.1 years.2 However, MCAH likely is underreported due to the overall lack of knowledge regarding this condition by physicians and pathologists. The hands and face are the most commonly affected areas, though other sites of involvement have been reported, including the lower extremities,3,4 oral mucosa and upper lip,5,6 and trunk,7,8 as well as generalized distribution.9-12 Additionally, 1 case presented as a single plaque on the trunk rather than having papular or nodular morphology.8 Multinucleate cell angiohistiocytoma lesions generally are asymptomatic, though pruritus may be present.13 The condition is regarded as benign, though a minority of cases have exhibited spontaneous resolution.14-16
Histopathology
Multinucleate cell angiohistiocytoma histology demonstrates full-thickness dermal microvessel proliferation and fibrosis with characteristic multinucleate giant cells.2,3 Vascular endothelial cells stained positive for CD68 in 60% of cases2 as well as the normal endothelial markers (ie, factor VIII, CD31, CD34). The multinucleate giant cells exhibit immunoreactivity for macrophage/histiocytic markers factor XIIIa and CD68.
Etiology
The pathogenesis of MCAH is not yet fully understood, but it is considered to be a benign vascular or fibrohistiocytic neoplasm.17 Calderaro et al18 described a series of 8 patients who developed MCAH either within a cutaneous neoplastic process or in conjunction with various cutaneous reactive conditions, including hidradenitis suppurativa and chronic radiation dermatitis, as well as overlying a bone prosthesis placed due to degenerative arthritis. These cases suggest that MCAH, or possibly a subset of the disease, is a reactive process.
Differential Diagnosis
The differential diagnosis for MCAH includes Kaposi sarcoma clinically and dermatofibroma and fibrous papules histologically. Sass et al21 determined the in vitro behavior of cultured MCAH cells to contrast markedly with Kaposi sarcoma–derived cells. Although Kaposi sarcoma–derived cells exhibited invasive behavior, cells isolated from MCAH lesions were less elongated and were unable to traverse basement membranes.
Treatment
Surgical excision or cryotherapy appear to be definitive treatments of MCAH; however, a number of cases have reported light and laser modalities as successful alternatives to excision. One case of MCAH affecting the face was treated with pulsed dye laser monotherapy.22 This modality allowed selective coagulation of the vascular structures in MCAH. At 8-month follow-up, the initial lesion was noted to be completely cleared, though similar lesions had recently appeared elsewhere on the face.22 Another case of MCAH affecting the leg was treated with pulsed dye laser and both topical and intralesional corticosteroid combination therapy. In this case, the lesion failed to respond to treatment, which may suggest that facial localization could influence response in pulsed dye laser treatment.3
Intense pulsed light also has been reported as a definitive treatment in 2 cases.2,13 Slight erythema and transient pruritus have been reported immediately following treatment. In this case, complete resolution with only residual hyperpigmentation was reported at 2-month follow-up, with no recurrence during 12 months of follow-up.13
Argon laser therapy has been used in 2 cases. After a single session, lesions were no longer palpable, with no scarring noted at 8 weeks follow-up.23 Lastly, 2 cases of MCAH have been successfully treated with the CO2 laser, with no relapse noted at 2.5- or 5-month follow-up, respectively.24
Conclusion
Multinucleate cell angiohistiocytoma is a rare and likely underdiagnosed dermatologic condition that is believed to be a reactive process. Characteristic histology of MCAH demonstrates microvascular proliferations of the dermis with multinucleate giant cells amidst a fibrous background. Although surgical excision is curative, there are reports in which laser and light therapies were used to effectively treat MCAH.
- Smith NP, Jones EW. Multinucleate cell angiohistiocytoma—a new entity. Br J Dermatol. 1985;113:15.
- Frew JW. Multinucleate cell angiohistiocytoma: clinicopathological correlation of 142 cases with insights into etiology and pathogenesis. Am J Dermatopathol. 2015;37:222-228.
- Applebaum DS, Shuja F, Hicks L, et al. Multinucleate cell angiohistiocytoma: a case report and review of the literature. Dermatol Online J. 2014;20:22610.
- Sagdeo A, Chu EY, Elenitsas R, et al. Multiple asymptomatic violaceous macules on the thigh. Multinucleate cell angiohistiocytoma (MCAH). JAMA Dermatol. 2013;149:357-363.
- Rawal YB, Anderson KM, Rawal SY. Multinucleate cell angiohistiocytoma: an uncommon mucosal tumour. Clin Exp Dermatol. 2009;34:333-336.
- Jones AC, Mullins D, Jimenez F. Multinucleate cell angiohistiocytoma of the upper lip. Oral Surg Oral Med Oral Pathol. 1994;78:743-747.
- Doshi-Chougule BN, Gust A, Mentzel T, et al. Multinucleate cell angiohistiocytoma with hypertrophic nerves. J Cutan Pathol. 2013;40:1048-1053.
- Issa AA, Lui H, Shapiro J, et al. Plaque-type multinucleate cell angiohistiocytoma. J Cutan Med Surg. 1998;3:112-114.
- Doane JA, Purdy K, Pasternak S. Generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2015;19:323-325.
- Marti N, Monteagudo C, Revert A, et al. Multiple papules on the trunk and extremities. generalized multinucleate cell angiohistiocytoma. Int J Dermatol. 2013;52:544-546.
- O’Blenes CA, Walsh NM, Green PJ, et al. Novel case of generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2010;14:178-180.
- Chang SN, Kim HS, Kim SC, et al. Generalized multinucleate cell angiohistiocytoma. J Am Acad Dermatol. 1996;35:320-322.
- Fernández-Jorge B, Del Pozo J, García-Silva J, et al. Multinucleate cell angiohistiocytoma: treatment using intense pulsed light. Dermatol Surg. 2009;35:1141-1143.
- Perez LP, Zulaica A, Rodriguez L, et al. Multinucleate cell angiohistiocytoma. report of five cases. J Cutan Pathol. 2006;33:349-352.
- Shapiro PE, Nova MP, Rosmarin LA, et al. Multinucleate cell angiohistiocytoma: a distinct entity diagnosable by clinical and histologic features. J Am Acad Dermatol. 1994;30:417-422.
- Jaconelli L, Kanitakis J, Ktiouet S, et al. Multinucleate cell angiohistiocytoma: report of three new cases and literature review. Dermatol Online J. 2009;15:4.
- Jones WE, Cerio R, Smith NP. Multinucleate cell angiohistiocytoma: an acquired vascular anomaly to be distinguished from Kaposi’s sarcoma. Br J Dermatol. 1990;122:651-663.
- Calderaro J, Rethers L, Ortonne N. Multinucleated cells angiohistiocytoma: a reactive lesion? Am J Dermatopathol. 2010;32:415-417.
- Cesinaro AM, Roncati L, Maiorana A. Estrogen receptor alpha overexpression in multinucleate cell angiohistiocytoma: new insights into the pathogenesis of a reactive process. Am J Dermatopathol. 2010;32:655-659.
- Losordo DW, Isner JM. Estrogen and angiogenesis: a review. Arterioscler Thromb Vasc Biol. 2001;21:6-12.
- Sass U, Noel JC, Andre J, et al. Multinucleate cell angiohistiocytoma: report of two cases with no evidence of human herpesvirus-8 infection. J Cutan Pathol. 2000;27:258-261.
- Richer V, Lui H. Facial multinucleate cell angiohistiocytoma: long-term remission with 585 nm pulsed dye laser. Clin Exp Dermatol. 2016;41:312-313.
- Kopera D, Smolle J, Kerl H. Multinucleate cell angiohistiocytoma: treatment with argon laser. Br J Dermatol. 1995;133:308-310.
- Väkevä L, Saksela O, Kariniemi AL. Multinucleate cell angiohistiocytoma: a report of four cases in Finland. Acta Derm Venereol. 2003;83:222-223.
Multinucleate cell angiohistiocytoma (MCAH) is a rare benign, soft-tissue tumor first described in 1985 by Smith and Jones1 that presents clinically as erythematous to violaceous papules most commonly affecting females on the dorsal aspect of the hands and face.2 Multinucleate cell angiohistiocytoma is histologically characterized by vascular and histiocytic proliferations with dermal fibrosis. Few cases have been reported of lesions affecting the lower extremities. We report a case of MCAH affecting the legs.
Case Report
An 83-year-old white man with a history of basal cell carcinoma presented for evaluation of grouped, well-circumscribed, soft, red-violet, painless papules on the right anterior thigh that had been present for 8 months (Figure 1A). A review of symptoms was negative for immunologic, respiratory, and hematologic changes. The patient’s medical history also was remarkable for prostate cancer treated with radiation 18 years prior as well as right hip and left knee implants. The initial clinical impression was Kaposi sarcoma or a granulomatous disorder.
Histopathologic evaluation of a deep shave biopsy initially determined the lesion to be scar tissue without other pathologic findings. The patient returned to the clinic 12 months later for a complete skin examination given his history of skin cancer. Compared to clinical photographs taken a year prior, new violaceous papules were noted on the right thigh (Figure 1B) and left calf. Furthermore, there was no recurrence of the lesion at the prior biopsy site. Shave biopsies of the papules on the right thigh and left calf demonstrated similar histologic findings to each other. There was a mild increase in the number of small blood vessels in the superficial dermis (Figure 2A). A mild perivascular lymphocytic infiltrate surrounded some of these blood vessels. The endothelial cells had small nuclei with no evidence of nuclear pleomorphism. Careful examination of the interstitial dermis revealed scattered multinucleate cells with angulated cytoplasm (Figure 2B). Immunostaining for CD31 and human herpesvirus 8 were negative, excluding an infiltrative vascular tumor and Kaposi sarcoma, respectively. The diagnosis of MCAH was made based on the histopathologic findings.
At 1-year follow-up, the condition was stable with no gross changes in the lesions based on prior photographs. Once again, there was no recurrence of the excised lesions at both biopsy sites.
Comment
Presentation
A systematic review of published reports determined that 79% of MCAH cases occur in females, with an average age of onset of 50.1 years.2 However, MCAH likely is underreported due to the overall lack of knowledge regarding this condition by physicians and pathologists. The hands and face are the most commonly affected areas, though other sites of involvement have been reported, including the lower extremities,3,4 oral mucosa and upper lip,5,6 and trunk,7,8 as well as generalized distribution.9-12 Additionally, 1 case presented as a single plaque on the trunk rather than having papular or nodular morphology.8 Multinucleate cell angiohistiocytoma lesions generally are asymptomatic, though pruritus may be present.13 The condition is regarded as benign, though a minority of cases have exhibited spontaneous resolution.14-16
Histopathology
Multinucleate cell angiohistiocytoma histology demonstrates full-thickness dermal microvessel proliferation and fibrosis with characteristic multinucleate giant cells.2,3 Vascular endothelial cells stained positive for CD68 in 60% of cases2 as well as the normal endothelial markers (ie, factor VIII, CD31, CD34). The multinucleate giant cells exhibit immunoreactivity for macrophage/histiocytic markers factor XIIIa and CD68.
Etiology
The pathogenesis of MCAH is not yet fully understood, but it is considered to be a benign vascular or fibrohistiocytic neoplasm.17 Calderaro et al18 described a series of 8 patients who developed MCAH either within a cutaneous neoplastic process or in conjunction with various cutaneous reactive conditions, including hidradenitis suppurativa and chronic radiation dermatitis, as well as overlying a bone prosthesis placed due to degenerative arthritis. These cases suggest that MCAH, or possibly a subset of the disease, is a reactive process.
Differential Diagnosis
The differential diagnosis for MCAH includes Kaposi sarcoma clinically and dermatofibroma and fibrous papules histologically. Sass et al21 determined the in vitro behavior of cultured MCAH cells to contrast markedly with Kaposi sarcoma–derived cells. Although Kaposi sarcoma–derived cells exhibited invasive behavior, cells isolated from MCAH lesions were less elongated and were unable to traverse basement membranes.
Treatment
Surgical excision or cryotherapy appear to be definitive treatments of MCAH; however, a number of cases have reported light and laser modalities as successful alternatives to excision. One case of MCAH affecting the face was treated with pulsed dye laser monotherapy.22 This modality allowed selective coagulation of the vascular structures in MCAH. At 8-month follow-up, the initial lesion was noted to be completely cleared, though similar lesions had recently appeared elsewhere on the face.22 Another case of MCAH affecting the leg was treated with pulsed dye laser and both topical and intralesional corticosteroid combination therapy. In this case, the lesion failed to respond to treatment, which may suggest that facial localization could influence response in pulsed dye laser treatment.3
Intense pulsed light also has been reported as a definitive treatment in 2 cases.2,13 Slight erythema and transient pruritus have been reported immediately following treatment. In this case, complete resolution with only residual hyperpigmentation was reported at 2-month follow-up, with no recurrence during 12 months of follow-up.13
Argon laser therapy has been used in 2 cases. After a single session, lesions were no longer palpable, with no scarring noted at 8 weeks follow-up.23 Lastly, 2 cases of MCAH have been successfully treated with the CO2 laser, with no relapse noted at 2.5- or 5-month follow-up, respectively.24
Conclusion
Multinucleate cell angiohistiocytoma is a rare and likely underdiagnosed dermatologic condition that is believed to be a reactive process. Characteristic histology of MCAH demonstrates microvascular proliferations of the dermis with multinucleate giant cells amidst a fibrous background. Although surgical excision is curative, there are reports in which laser and light therapies were used to effectively treat MCAH.
Multinucleate cell angiohistiocytoma (MCAH) is a rare benign, soft-tissue tumor first described in 1985 by Smith and Jones1 that presents clinically as erythematous to violaceous papules most commonly affecting females on the dorsal aspect of the hands and face.2 Multinucleate cell angiohistiocytoma is histologically characterized by vascular and histiocytic proliferations with dermal fibrosis. Few cases have been reported of lesions affecting the lower extremities. We report a case of MCAH affecting the legs.
Case Report
An 83-year-old white man with a history of basal cell carcinoma presented for evaluation of grouped, well-circumscribed, soft, red-violet, painless papules on the right anterior thigh that had been present for 8 months (Figure 1A). A review of symptoms was negative for immunologic, respiratory, and hematologic changes. The patient’s medical history also was remarkable for prostate cancer treated with radiation 18 years prior as well as right hip and left knee implants. The initial clinical impression was Kaposi sarcoma or a granulomatous disorder.
Histopathologic evaluation of a deep shave biopsy initially determined the lesion to be scar tissue without other pathologic findings. The patient returned to the clinic 12 months later for a complete skin examination given his history of skin cancer. Compared to clinical photographs taken a year prior, new violaceous papules were noted on the right thigh (Figure 1B) and left calf. Furthermore, there was no recurrence of the lesion at the prior biopsy site. Shave biopsies of the papules on the right thigh and left calf demonstrated similar histologic findings to each other. There was a mild increase in the number of small blood vessels in the superficial dermis (Figure 2A). A mild perivascular lymphocytic infiltrate surrounded some of these blood vessels. The endothelial cells had small nuclei with no evidence of nuclear pleomorphism. Careful examination of the interstitial dermis revealed scattered multinucleate cells with angulated cytoplasm (Figure 2B). Immunostaining for CD31 and human herpesvirus 8 were negative, excluding an infiltrative vascular tumor and Kaposi sarcoma, respectively. The diagnosis of MCAH was made based on the histopathologic findings.
At 1-year follow-up, the condition was stable with no gross changes in the lesions based on prior photographs. Once again, there was no recurrence of the excised lesions at both biopsy sites.
Comment
Presentation
A systematic review of published reports determined that 79% of MCAH cases occur in females, with an average age of onset of 50.1 years.2 However, MCAH likely is underreported due to the overall lack of knowledge regarding this condition by physicians and pathologists. The hands and face are the most commonly affected areas, though other sites of involvement have been reported, including the lower extremities,3,4 oral mucosa and upper lip,5,6 and trunk,7,8 as well as generalized distribution.9-12 Additionally, 1 case presented as a single plaque on the trunk rather than having papular or nodular morphology.8 Multinucleate cell angiohistiocytoma lesions generally are asymptomatic, though pruritus may be present.13 The condition is regarded as benign, though a minority of cases have exhibited spontaneous resolution.14-16
Histopathology
Multinucleate cell angiohistiocytoma histology demonstrates full-thickness dermal microvessel proliferation and fibrosis with characteristic multinucleate giant cells.2,3 Vascular endothelial cells stained positive for CD68 in 60% of cases2 as well as the normal endothelial markers (ie, factor VIII, CD31, CD34). The multinucleate giant cells exhibit immunoreactivity for macrophage/histiocytic markers factor XIIIa and CD68.
Etiology
The pathogenesis of MCAH is not yet fully understood, but it is considered to be a benign vascular or fibrohistiocytic neoplasm.17 Calderaro et al18 described a series of 8 patients who developed MCAH either within a cutaneous neoplastic process or in conjunction with various cutaneous reactive conditions, including hidradenitis suppurativa and chronic radiation dermatitis, as well as overlying a bone prosthesis placed due to degenerative arthritis. These cases suggest that MCAH, or possibly a subset of the disease, is a reactive process.
Differential Diagnosis
The differential diagnosis for MCAH includes Kaposi sarcoma clinically and dermatofibroma and fibrous papules histologically. Sass et al21 determined the in vitro behavior of cultured MCAH cells to contrast markedly with Kaposi sarcoma–derived cells. Although Kaposi sarcoma–derived cells exhibited invasive behavior, cells isolated from MCAH lesions were less elongated and were unable to traverse basement membranes.
Treatment
Surgical excision or cryotherapy appear to be definitive treatments of MCAH; however, a number of cases have reported light and laser modalities as successful alternatives to excision. One case of MCAH affecting the face was treated with pulsed dye laser monotherapy.22 This modality allowed selective coagulation of the vascular structures in MCAH. At 8-month follow-up, the initial lesion was noted to be completely cleared, though similar lesions had recently appeared elsewhere on the face.22 Another case of MCAH affecting the leg was treated with pulsed dye laser and both topical and intralesional corticosteroid combination therapy. In this case, the lesion failed to respond to treatment, which may suggest that facial localization could influence response in pulsed dye laser treatment.3
Intense pulsed light also has been reported as a definitive treatment in 2 cases.2,13 Slight erythema and transient pruritus have been reported immediately following treatment. In this case, complete resolution with only residual hyperpigmentation was reported at 2-month follow-up, with no recurrence during 12 months of follow-up.13
Argon laser therapy has been used in 2 cases. After a single session, lesions were no longer palpable, with no scarring noted at 8 weeks follow-up.23 Lastly, 2 cases of MCAH have been successfully treated with the CO2 laser, with no relapse noted at 2.5- or 5-month follow-up, respectively.24
Conclusion
Multinucleate cell angiohistiocytoma is a rare and likely underdiagnosed dermatologic condition that is believed to be a reactive process. Characteristic histology of MCAH demonstrates microvascular proliferations of the dermis with multinucleate giant cells amidst a fibrous background. Although surgical excision is curative, there are reports in which laser and light therapies were used to effectively treat MCAH.
- Smith NP, Jones EW. Multinucleate cell angiohistiocytoma—a new entity. Br J Dermatol. 1985;113:15.
- Frew JW. Multinucleate cell angiohistiocytoma: clinicopathological correlation of 142 cases with insights into etiology and pathogenesis. Am J Dermatopathol. 2015;37:222-228.
- Applebaum DS, Shuja F, Hicks L, et al. Multinucleate cell angiohistiocytoma: a case report and review of the literature. Dermatol Online J. 2014;20:22610.
- Sagdeo A, Chu EY, Elenitsas R, et al. Multiple asymptomatic violaceous macules on the thigh. Multinucleate cell angiohistiocytoma (MCAH). JAMA Dermatol. 2013;149:357-363.
- Rawal YB, Anderson KM, Rawal SY. Multinucleate cell angiohistiocytoma: an uncommon mucosal tumour. Clin Exp Dermatol. 2009;34:333-336.
- Jones AC, Mullins D, Jimenez F. Multinucleate cell angiohistiocytoma of the upper lip. Oral Surg Oral Med Oral Pathol. 1994;78:743-747.
- Doshi-Chougule BN, Gust A, Mentzel T, et al. Multinucleate cell angiohistiocytoma with hypertrophic nerves. J Cutan Pathol. 2013;40:1048-1053.
- Issa AA, Lui H, Shapiro J, et al. Plaque-type multinucleate cell angiohistiocytoma. J Cutan Med Surg. 1998;3:112-114.
- Doane JA, Purdy K, Pasternak S. Generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2015;19:323-325.
- Marti N, Monteagudo C, Revert A, et al. Multiple papules on the trunk and extremities. generalized multinucleate cell angiohistiocytoma. Int J Dermatol. 2013;52:544-546.
- O’Blenes CA, Walsh NM, Green PJ, et al. Novel case of generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2010;14:178-180.
- Chang SN, Kim HS, Kim SC, et al. Generalized multinucleate cell angiohistiocytoma. J Am Acad Dermatol. 1996;35:320-322.
- Fernández-Jorge B, Del Pozo J, García-Silva J, et al. Multinucleate cell angiohistiocytoma: treatment using intense pulsed light. Dermatol Surg. 2009;35:1141-1143.
- Perez LP, Zulaica A, Rodriguez L, et al. Multinucleate cell angiohistiocytoma. report of five cases. J Cutan Pathol. 2006;33:349-352.
- Shapiro PE, Nova MP, Rosmarin LA, et al. Multinucleate cell angiohistiocytoma: a distinct entity diagnosable by clinical and histologic features. J Am Acad Dermatol. 1994;30:417-422.
- Jaconelli L, Kanitakis J, Ktiouet S, et al. Multinucleate cell angiohistiocytoma: report of three new cases and literature review. Dermatol Online J. 2009;15:4.
- Jones WE, Cerio R, Smith NP. Multinucleate cell angiohistiocytoma: an acquired vascular anomaly to be distinguished from Kaposi’s sarcoma. Br J Dermatol. 1990;122:651-663.
- Calderaro J, Rethers L, Ortonne N. Multinucleated cells angiohistiocytoma: a reactive lesion? Am J Dermatopathol. 2010;32:415-417.
- Cesinaro AM, Roncati L, Maiorana A. Estrogen receptor alpha overexpression in multinucleate cell angiohistiocytoma: new insights into the pathogenesis of a reactive process. Am J Dermatopathol. 2010;32:655-659.
- Losordo DW, Isner JM. Estrogen and angiogenesis: a review. Arterioscler Thromb Vasc Biol. 2001;21:6-12.
- Sass U, Noel JC, Andre J, et al. Multinucleate cell angiohistiocytoma: report of two cases with no evidence of human herpesvirus-8 infection. J Cutan Pathol. 2000;27:258-261.
- Richer V, Lui H. Facial multinucleate cell angiohistiocytoma: long-term remission with 585 nm pulsed dye laser. Clin Exp Dermatol. 2016;41:312-313.
- Kopera D, Smolle J, Kerl H. Multinucleate cell angiohistiocytoma: treatment with argon laser. Br J Dermatol. 1995;133:308-310.
- Väkevä L, Saksela O, Kariniemi AL. Multinucleate cell angiohistiocytoma: a report of four cases in Finland. Acta Derm Venereol. 2003;83:222-223.
- Smith NP, Jones EW. Multinucleate cell angiohistiocytoma—a new entity. Br J Dermatol. 1985;113:15.
- Frew JW. Multinucleate cell angiohistiocytoma: clinicopathological correlation of 142 cases with insights into etiology and pathogenesis. Am J Dermatopathol. 2015;37:222-228.
- Applebaum DS, Shuja F, Hicks L, et al. Multinucleate cell angiohistiocytoma: a case report and review of the literature. Dermatol Online J. 2014;20:22610.
- Sagdeo A, Chu EY, Elenitsas R, et al. Multiple asymptomatic violaceous macules on the thigh. Multinucleate cell angiohistiocytoma (MCAH). JAMA Dermatol. 2013;149:357-363.
- Rawal YB, Anderson KM, Rawal SY. Multinucleate cell angiohistiocytoma: an uncommon mucosal tumour. Clin Exp Dermatol. 2009;34:333-336.
- Jones AC, Mullins D, Jimenez F. Multinucleate cell angiohistiocytoma of the upper lip. Oral Surg Oral Med Oral Pathol. 1994;78:743-747.
- Doshi-Chougule BN, Gust A, Mentzel T, et al. Multinucleate cell angiohistiocytoma with hypertrophic nerves. J Cutan Pathol. 2013;40:1048-1053.
- Issa AA, Lui H, Shapiro J, et al. Plaque-type multinucleate cell angiohistiocytoma. J Cutan Med Surg. 1998;3:112-114.
- Doane JA, Purdy K, Pasternak S. Generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2015;19:323-325.
- Marti N, Monteagudo C, Revert A, et al. Multiple papules on the trunk and extremities. generalized multinucleate cell angiohistiocytoma. Int J Dermatol. 2013;52:544-546.
- O’Blenes CA, Walsh NM, Green PJ, et al. Novel case of generalized multinucleate cell angiohistiocytoma. J Cutan Med Surg. 2010;14:178-180.
- Chang SN, Kim HS, Kim SC, et al. Generalized multinucleate cell angiohistiocytoma. J Am Acad Dermatol. 1996;35:320-322.
- Fernández-Jorge B, Del Pozo J, García-Silva J, et al. Multinucleate cell angiohistiocytoma: treatment using intense pulsed light. Dermatol Surg. 2009;35:1141-1143.
- Perez LP, Zulaica A, Rodriguez L, et al. Multinucleate cell angiohistiocytoma. report of five cases. J Cutan Pathol. 2006;33:349-352.
- Shapiro PE, Nova MP, Rosmarin LA, et al. Multinucleate cell angiohistiocytoma: a distinct entity diagnosable by clinical and histologic features. J Am Acad Dermatol. 1994;30:417-422.
- Jaconelli L, Kanitakis J, Ktiouet S, et al. Multinucleate cell angiohistiocytoma: report of three new cases and literature review. Dermatol Online J. 2009;15:4.
- Jones WE, Cerio R, Smith NP. Multinucleate cell angiohistiocytoma: an acquired vascular anomaly to be distinguished from Kaposi’s sarcoma. Br J Dermatol. 1990;122:651-663.
- Calderaro J, Rethers L, Ortonne N. Multinucleated cells angiohistiocytoma: a reactive lesion? Am J Dermatopathol. 2010;32:415-417.
- Cesinaro AM, Roncati L, Maiorana A. Estrogen receptor alpha overexpression in multinucleate cell angiohistiocytoma: new insights into the pathogenesis of a reactive process. Am J Dermatopathol. 2010;32:655-659.
- Losordo DW, Isner JM. Estrogen and angiogenesis: a review. Arterioscler Thromb Vasc Biol. 2001;21:6-12.
- Sass U, Noel JC, Andre J, et al. Multinucleate cell angiohistiocytoma: report of two cases with no evidence of human herpesvirus-8 infection. J Cutan Pathol. 2000;27:258-261.
- Richer V, Lui H. Facial multinucleate cell angiohistiocytoma: long-term remission with 585 nm pulsed dye laser. Clin Exp Dermatol. 2016;41:312-313.
- Kopera D, Smolle J, Kerl H. Multinucleate cell angiohistiocytoma: treatment with argon laser. Br J Dermatol. 1995;133:308-310.
- Väkevä L, Saksela O, Kariniemi AL. Multinucleate cell angiohistiocytoma: a report of four cases in Finland. Acta Derm Venereol. 2003;83:222-223.
Practice Points
- Multinucleate cell angiohistiocytoma (MCAH) is a rare underrecognized cutaneous tumor presenting as erythematous to violaceous papules.
- Although it clinically mimics Kaposi sarcoma, MCAH may be distinguished histopathologically by negative immunostaining for human herpesvirus 8.
- Surgical excision and laser therapies are definitive treatments for MCAH, which is a benign lesion.
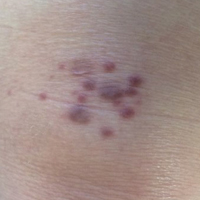
Cordlike Dermal Plaques and Nodules on the Neck and Hands
The Diagnosis: Fibroblastic Rheumatism
Routine histologic sections stained with hematoxylin and eosin demonstrated a noncircumscribed dermal proliferation of fibroblasts and myofibroblasts with thickened collagen bundles (Figure, A and B). Focally fragmented elastin fibers were noted with Verhoeff elastic tissue stain. Alcian blue stain did not show increased dermal mucin. With the clinical presentation and histologic findings described, we diagnosed the patient with fibroblastic rheumatism (FR). To date, the patient's condition has stabilized overall with skin lesions fading and minimal to no joint pain. Current therapies include adalimumab, mycophenolate mofetil 500 mg 3 times daily, and low-dose prednisone.
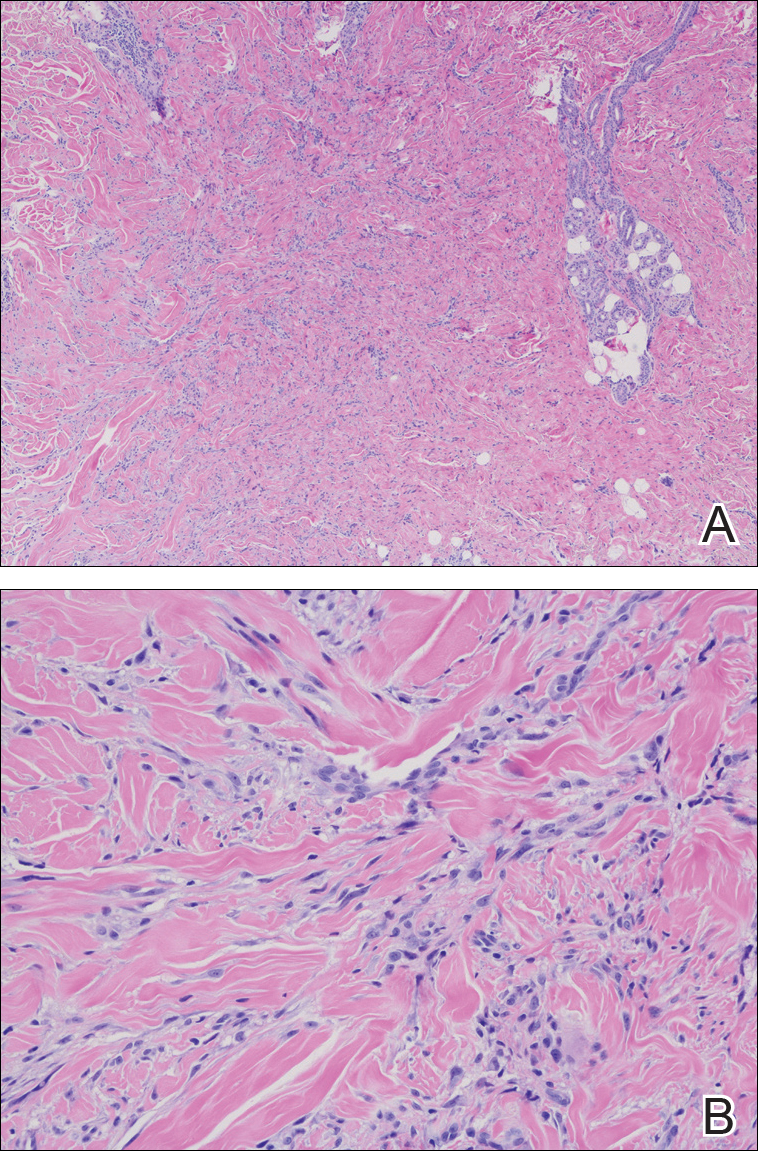
Fibroblastic rheumatism is a rare arthropathy with cutaneous findings initially described by Chaouat et al1 in 1980. Age of onset varies, and the condition also has been observed in pediatric patients.2 Fibroblastic rheumatism is characterized by sudden onset of firm, flesh-colored, subcutaneous nodules on periungual and periarticular surfaces.2 Neck lesions rarely are described,2-4 and cordlike plaques previously have not been reported in FR. Typically, patients develop diffusely swollen fingers, palmar thickening, sclerodactyly, and contractures. The eruption may be accompanied by Raynaud phenomenon as well as a progressive symmetric erosive arthropathy.2,5
The clinical course in FR is variable. The cutaneous findings spontaneously may regress in months to years.3,4 However, polyarthropathy often is destructive and progresses to disability.3 Response to therapy has been unpredictable, and the following treatments have been tried, generally with poor efficacy: aspirin, nonsteroidal anti-inflammatory drugs, hydroxychloroquine, colchicine, methotrexate, prednisone, infliximab, D-penicillamine, interferon alfa, and intensive physical therapy.2-4,6 Histologic characteristics may include thickened collagen bundles along with a fibroblastic and myofibroblastic proliferation. Elastic fibers may be decreased or absent.2,3,5
Clinical and histologic features in FR may mimic other entities; thus, clinical pathological correlation is essential in determining the correct diagnosis. Considerations in the differential diagnoses include multicentric reticulohistiocytosis (MRH), palisaded neutrophilic and granulomatous dermatitis, and scleroderma.
In MRH, a symmetric erosive arthritis of mainly distal interphalangeal joints typically precedes the cutaneous disease. Occurrence of arthritis mutilans is reported in approximately half of patients.4 Cutaneous manifestations typically include the presence of coral bead-like papules and nodules over the dorsal aspect of the hands, face, and neck. Unlike FR, MRH has a concomitant autoimmune disease in up to 20% of cases and an associated malignancy in up to 31% of cases, with breast and ovarian carcinomas most common. On histology, MRH is characterized by a nodular infiltrate of histiocytes and multinucleated giant cells with eosinophilic ground-glass cytoplasm.4 No notable collagen changes or fibroblastic proliferations typically are present.
Palisaded neutrophilic and granulomatous dermatitis, usually associated with rheumatoid arthritis or connective tissue disease, classically presents as annular plaques and indurated linear bands over the trunk and extremities. However, its clinical presentation is quite variable and may include pink to violaceous urticarialike; livedoid-appearing; or nonspecific papules, plaques, or nodules. Histology in palisaded neutrophilic and granulomatous dermatitis shows a dense dermal neutrophilic infiltrate associated with interstitial histiocytes having a palisading arrangement around degenerated collagen.7 No fibroblastic proliferation typically is present.
Scleroderma can be distinguished based on additional clinical and laboratory findings as well as histology showing thickened collagen bundles without fibroblastic proliferation.2 The histologic findings also may suggest inclusion of dermatofibroma or a scar in the differential diagnosis, though the clinical presentation of these entities would not support these diagnoses.
Acknowledgments
We thank the patient for granting permission to share this information. We also thank Sheng Chen, MD, PhD (Lake Success, New York), for his dermatopathological contributions to the case.
- Chaouat Y, Aron-Brunetiere R, Faures B, et al. Une nouvelle entité: le rhumatisme fibroblastique. a propos d'une observation [in French]. Rev Rhum Mal Osteoartic. 1980;47:34-35.
- Jurado SA, Alvin GG, Selim MA, et al. Fibroblastic rheumatism: a report of 4 cases with potential therapeutic implications. J Am Acad Dermatol. 2012;66:959-965.
- Colonna L, Barbieri C, Di Lella G, et al. Fibroblastic rheumatism: a case without rheumatological symptoms. Acta Derm Venereol. 2002;82:200-203.
- Trotta F, Colina M. Multicentric reticulohistiocytosis and fibroblastic rheumatism. Best Pract Res Clin Rheumatol. 2012;26:543-557.
- Lee JM, Sundel RP, Liang MG. Fibroblastic rheumatism: case report and review of the literature. Pediatr Dermatol. 2002;19:532-535.
- Kluger N, Dumas-Tesici A, Hamel D, et al. Fibroblastic rheumatism: fibromatosis rather than non-Langerhans cell histiocytosis. J Cutan Pathol. 2010;37:587-592.
- Stephenson SR, Campbell M, Dre GS, et al. Palisaded neutrophilic and granulomatous dermatitis presenting in a patient with rheumatoid arthritis on adalimumab. J Cutan Pathol. 2011;38:644-648.
The Diagnosis: Fibroblastic Rheumatism
Routine histologic sections stained with hematoxylin and eosin demonstrated a noncircumscribed dermal proliferation of fibroblasts and myofibroblasts with thickened collagen bundles (Figure, A and B). Focally fragmented elastin fibers were noted with Verhoeff elastic tissue stain. Alcian blue stain did not show increased dermal mucin. With the clinical presentation and histologic findings described, we diagnosed the patient with fibroblastic rheumatism (FR). To date, the patient's condition has stabilized overall with skin lesions fading and minimal to no joint pain. Current therapies include adalimumab, mycophenolate mofetil 500 mg 3 times daily, and low-dose prednisone.
Fibroblastic rheumatism is a rare arthropathy with cutaneous findings initially described by Chaouat et al1 in 1980. Age of onset varies, and the condition also has been observed in pediatric patients.2 Fibroblastic rheumatism is characterized by sudden onset of firm, flesh-colored, subcutaneous nodules on periungual and periarticular surfaces.2 Neck lesions rarely are described,2-4 and cordlike plaques previously have not been reported in FR. Typically, patients develop diffusely swollen fingers, palmar thickening, sclerodactyly, and contractures. The eruption may be accompanied by Raynaud phenomenon as well as a progressive symmetric erosive arthropathy.2,5
The clinical course in FR is variable. The cutaneous findings spontaneously may regress in months to years.3,4 However, polyarthropathy often is destructive and progresses to disability.3 Response to therapy has been unpredictable, and the following treatments have been tried, generally with poor efficacy: aspirin, nonsteroidal anti-inflammatory drugs, hydroxychloroquine, colchicine, methotrexate, prednisone, infliximab, D-penicillamine, interferon alfa, and intensive physical therapy.2-4,6 Histologic characteristics may include thickened collagen bundles along with a fibroblastic and myofibroblastic proliferation. Elastic fibers may be decreased or absent.2,3,5
Clinical and histologic features in FR may mimic other entities; thus, clinical pathological correlation is essential in determining the correct diagnosis. Considerations in the differential diagnoses include multicentric reticulohistiocytosis (MRH), palisaded neutrophilic and granulomatous dermatitis, and scleroderma.
In MRH, a symmetric erosive arthritis of mainly distal interphalangeal joints typically precedes the cutaneous disease. Occurrence of arthritis mutilans is reported in approximately half of patients.4 Cutaneous manifestations typically include the presence of coral bead-like papules and nodules over the dorsal aspect of the hands, face, and neck. Unlike FR, MRH has a concomitant autoimmune disease in up to 20% of cases and an associated malignancy in up to 31% of cases, with breast and ovarian carcinomas most common. On histology, MRH is characterized by a nodular infiltrate of histiocytes and multinucleated giant cells with eosinophilic ground-glass cytoplasm.4 No notable collagen changes or fibroblastic proliferations typically are present.
Palisaded neutrophilic and granulomatous dermatitis, usually associated with rheumatoid arthritis or connective tissue disease, classically presents as annular plaques and indurated linear bands over the trunk and extremities. However, its clinical presentation is quite variable and may include pink to violaceous urticarialike; livedoid-appearing; or nonspecific papules, plaques, or nodules. Histology in palisaded neutrophilic and granulomatous dermatitis shows a dense dermal neutrophilic infiltrate associated with interstitial histiocytes having a palisading arrangement around degenerated collagen.7 No fibroblastic proliferation typically is present.
Scleroderma can be distinguished based on additional clinical and laboratory findings as well as histology showing thickened collagen bundles without fibroblastic proliferation.2 The histologic findings also may suggest inclusion of dermatofibroma or a scar in the differential diagnosis, though the clinical presentation of these entities would not support these diagnoses.
Acknowledgments
We thank the patient for granting permission to share this information. We also thank Sheng Chen, MD, PhD (Lake Success, New York), for his dermatopathological contributions to the case.
The Diagnosis: Fibroblastic Rheumatism
Routine histologic sections stained with hematoxylin and eosin demonstrated a noncircumscribed dermal proliferation of fibroblasts and myofibroblasts with thickened collagen bundles (Figure, A and B). Focally fragmented elastin fibers were noted with Verhoeff elastic tissue stain. Alcian blue stain did not show increased dermal mucin. With the clinical presentation and histologic findings described, we diagnosed the patient with fibroblastic rheumatism (FR). To date, the patient's condition has stabilized overall with skin lesions fading and minimal to no joint pain. Current therapies include adalimumab, mycophenolate mofetil 500 mg 3 times daily, and low-dose prednisone.
Fibroblastic rheumatism is a rare arthropathy with cutaneous findings initially described by Chaouat et al1 in 1980. Age of onset varies, and the condition also has been observed in pediatric patients.2 Fibroblastic rheumatism is characterized by sudden onset of firm, flesh-colored, subcutaneous nodules on periungual and periarticular surfaces.2 Neck lesions rarely are described,2-4 and cordlike plaques previously have not been reported in FR. Typically, patients develop diffusely swollen fingers, palmar thickening, sclerodactyly, and contractures. The eruption may be accompanied by Raynaud phenomenon as well as a progressive symmetric erosive arthropathy.2,5
The clinical course in FR is variable. The cutaneous findings spontaneously may regress in months to years.3,4 However, polyarthropathy often is destructive and progresses to disability.3 Response to therapy has been unpredictable, and the following treatments have been tried, generally with poor efficacy: aspirin, nonsteroidal anti-inflammatory drugs, hydroxychloroquine, colchicine, methotrexate, prednisone, infliximab, D-penicillamine, interferon alfa, and intensive physical therapy.2-4,6 Histologic characteristics may include thickened collagen bundles along with a fibroblastic and myofibroblastic proliferation. Elastic fibers may be decreased or absent.2,3,5
Clinical and histologic features in FR may mimic other entities; thus, clinical pathological correlation is essential in determining the correct diagnosis. Considerations in the differential diagnoses include multicentric reticulohistiocytosis (MRH), palisaded neutrophilic and granulomatous dermatitis, and scleroderma.
In MRH, a symmetric erosive arthritis of mainly distal interphalangeal joints typically precedes the cutaneous disease. Occurrence of arthritis mutilans is reported in approximately half of patients.4 Cutaneous manifestations typically include the presence of coral bead-like papules and nodules over the dorsal aspect of the hands, face, and neck. Unlike FR, MRH has a concomitant autoimmune disease in up to 20% of cases and an associated malignancy in up to 31% of cases, with breast and ovarian carcinomas most common. On histology, MRH is characterized by a nodular infiltrate of histiocytes and multinucleated giant cells with eosinophilic ground-glass cytoplasm.4 No notable collagen changes or fibroblastic proliferations typically are present.
Palisaded neutrophilic and granulomatous dermatitis, usually associated with rheumatoid arthritis or connective tissue disease, classically presents as annular plaques and indurated linear bands over the trunk and extremities. However, its clinical presentation is quite variable and may include pink to violaceous urticarialike; livedoid-appearing; or nonspecific papules, plaques, or nodules. Histology in palisaded neutrophilic and granulomatous dermatitis shows a dense dermal neutrophilic infiltrate associated with interstitial histiocytes having a palisading arrangement around degenerated collagen.7 No fibroblastic proliferation typically is present.
Scleroderma can be distinguished based on additional clinical and laboratory findings as well as histology showing thickened collagen bundles without fibroblastic proliferation.2 The histologic findings also may suggest inclusion of dermatofibroma or a scar in the differential diagnosis, though the clinical presentation of these entities would not support these diagnoses.
Acknowledgments
We thank the patient for granting permission to share this information. We also thank Sheng Chen, MD, PhD (Lake Success, New York), for his dermatopathological contributions to the case.
- Chaouat Y, Aron-Brunetiere R, Faures B, et al. Une nouvelle entité: le rhumatisme fibroblastique. a propos d'une observation [in French]. Rev Rhum Mal Osteoartic. 1980;47:34-35.
- Jurado SA, Alvin GG, Selim MA, et al. Fibroblastic rheumatism: a report of 4 cases with potential therapeutic implications. J Am Acad Dermatol. 2012;66:959-965.
- Colonna L, Barbieri C, Di Lella G, et al. Fibroblastic rheumatism: a case without rheumatological symptoms. Acta Derm Venereol. 2002;82:200-203.
- Trotta F, Colina M. Multicentric reticulohistiocytosis and fibroblastic rheumatism. Best Pract Res Clin Rheumatol. 2012;26:543-557.
- Lee JM, Sundel RP, Liang MG. Fibroblastic rheumatism: case report and review of the literature. Pediatr Dermatol. 2002;19:532-535.
- Kluger N, Dumas-Tesici A, Hamel D, et al. Fibroblastic rheumatism: fibromatosis rather than non-Langerhans cell histiocytosis. J Cutan Pathol. 2010;37:587-592.
- Stephenson SR, Campbell M, Dre GS, et al. Palisaded neutrophilic and granulomatous dermatitis presenting in a patient with rheumatoid arthritis on adalimumab. J Cutan Pathol. 2011;38:644-648.
- Chaouat Y, Aron-Brunetiere R, Faures B, et al. Une nouvelle entité: le rhumatisme fibroblastique. a propos d'une observation [in French]. Rev Rhum Mal Osteoartic. 1980;47:34-35.
- Jurado SA, Alvin GG, Selim MA, et al. Fibroblastic rheumatism: a report of 4 cases with potential therapeutic implications. J Am Acad Dermatol. 2012;66:959-965.
- Colonna L, Barbieri C, Di Lella G, et al. Fibroblastic rheumatism: a case without rheumatological symptoms. Acta Derm Venereol. 2002;82:200-203.
- Trotta F, Colina M. Multicentric reticulohistiocytosis and fibroblastic rheumatism. Best Pract Res Clin Rheumatol. 2012;26:543-557.
- Lee JM, Sundel RP, Liang MG. Fibroblastic rheumatism: case report and review of the literature. Pediatr Dermatol. 2002;19:532-535.
- Kluger N, Dumas-Tesici A, Hamel D, et al. Fibroblastic rheumatism: fibromatosis rather than non-Langerhans cell histiocytosis. J Cutan Pathol. 2010;37:587-592.
- Stephenson SR, Campbell M, Dre GS, et al. Palisaded neutrophilic and granulomatous dermatitis presenting in a patient with rheumatoid arthritis on adalimumab. J Cutan Pathol. 2011;38:644-648.
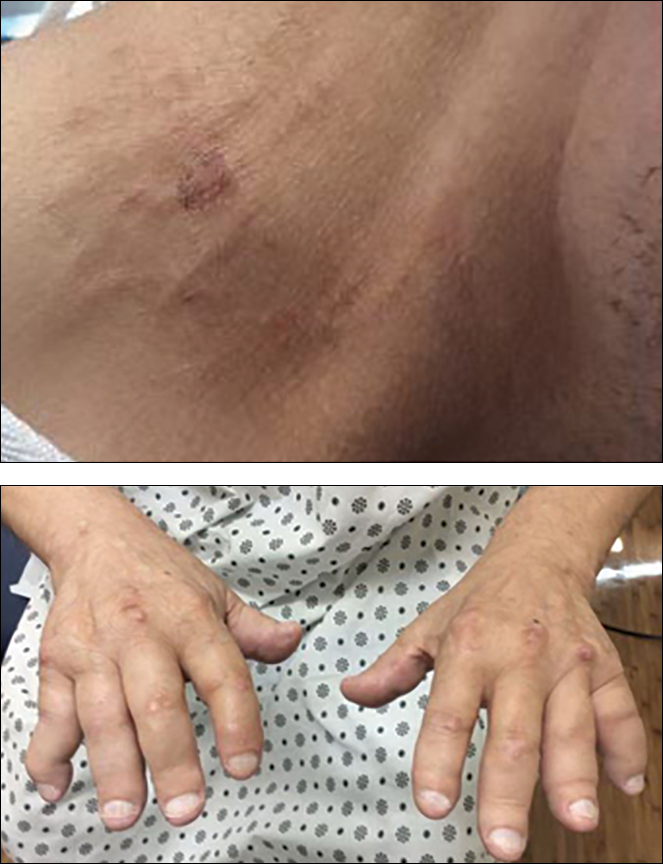
A 67-year-old man presented with asymptomatic plaques on the neck of 4 months' duration and nodules scattered over the hands, elbows, ears, and forehead of 3 years' duration. The eruption was associated with progressive thickening and contractures of the fingers, hand morning stiffness lasting less than 45 minutes, and Raynaud phenomenon. Physical examination revealed flesh-colored, firm, cordlike plaques on the neck bilaterally (top), with firm subcutaneous nodules on the helix and antihelix of the ears, forehead, elbows, and on the dorsal and ventral aspects of the hands (bottom). The largest nodules were approximately 5 cm. All fingers and first toes were thickened and firm with few contractile bands on the fingers. The patient had a persistently elevated erythrocyte sedimentation rate (80 mm/h)(reference range, 0-20 mm/h) and C-reactive protein level (3.27 mg/dL)(reference range, 0.00-0.40 mg/dL). Serologic workup was remarkable only for an antinuclear antibody titer of 1:80 (speckled). Plain radiographs confirmed an erosive arthropathy of the hands and feet. Erosions on the hands predominantly involved distal interphalangeal articulations, as well as, to a lesser extent, the proximal interphalangeal articulations, carpus, and the left distal radius. Erosive changes on the feet involved metatarsophalangeal, proximal interphalangeal, and distal interphalangeal articulations. Biopsies from the neck were performed for histopathologic correlation.
Indurated Plaque on the Eyebrow
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).

Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
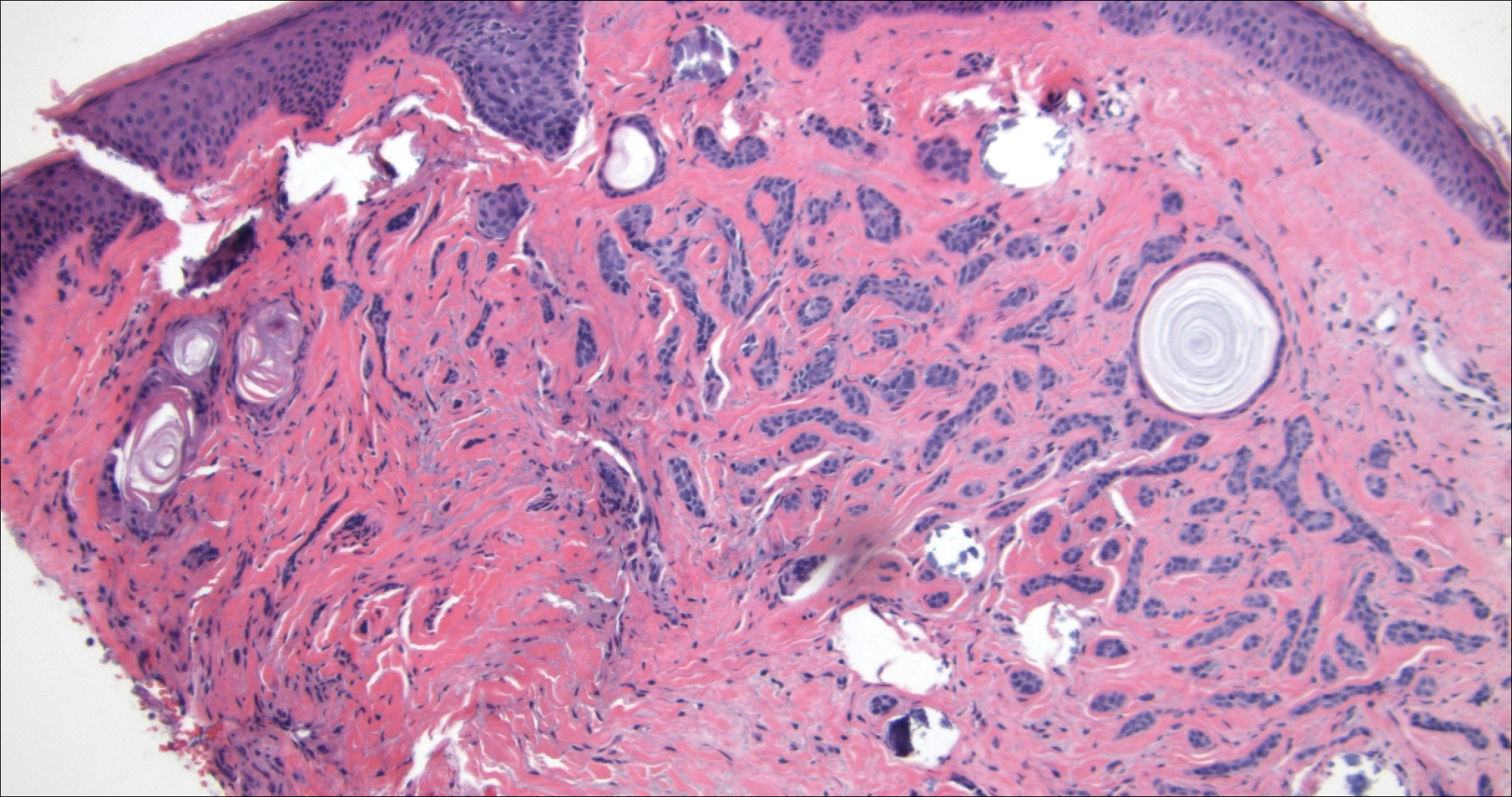
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
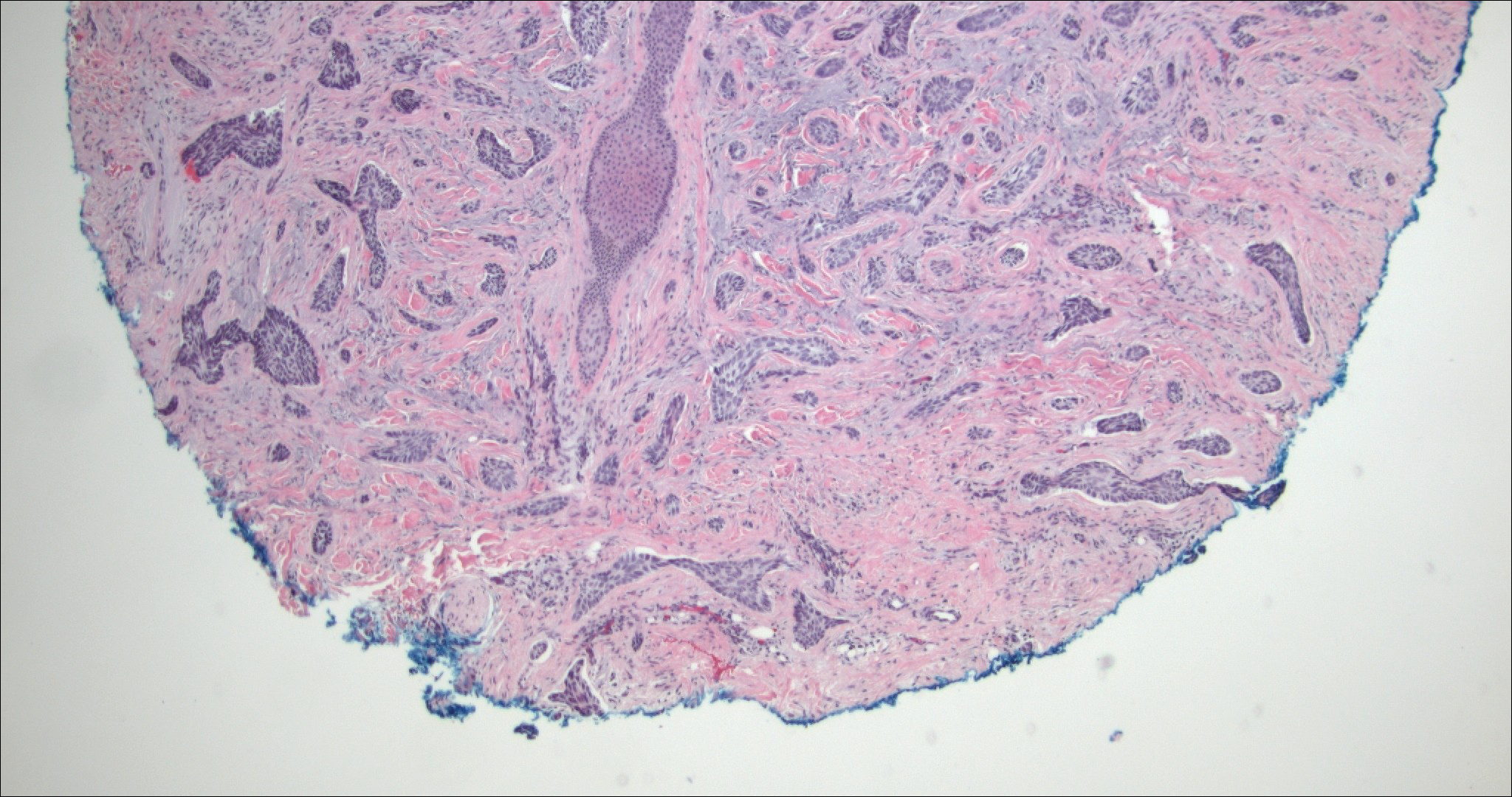
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
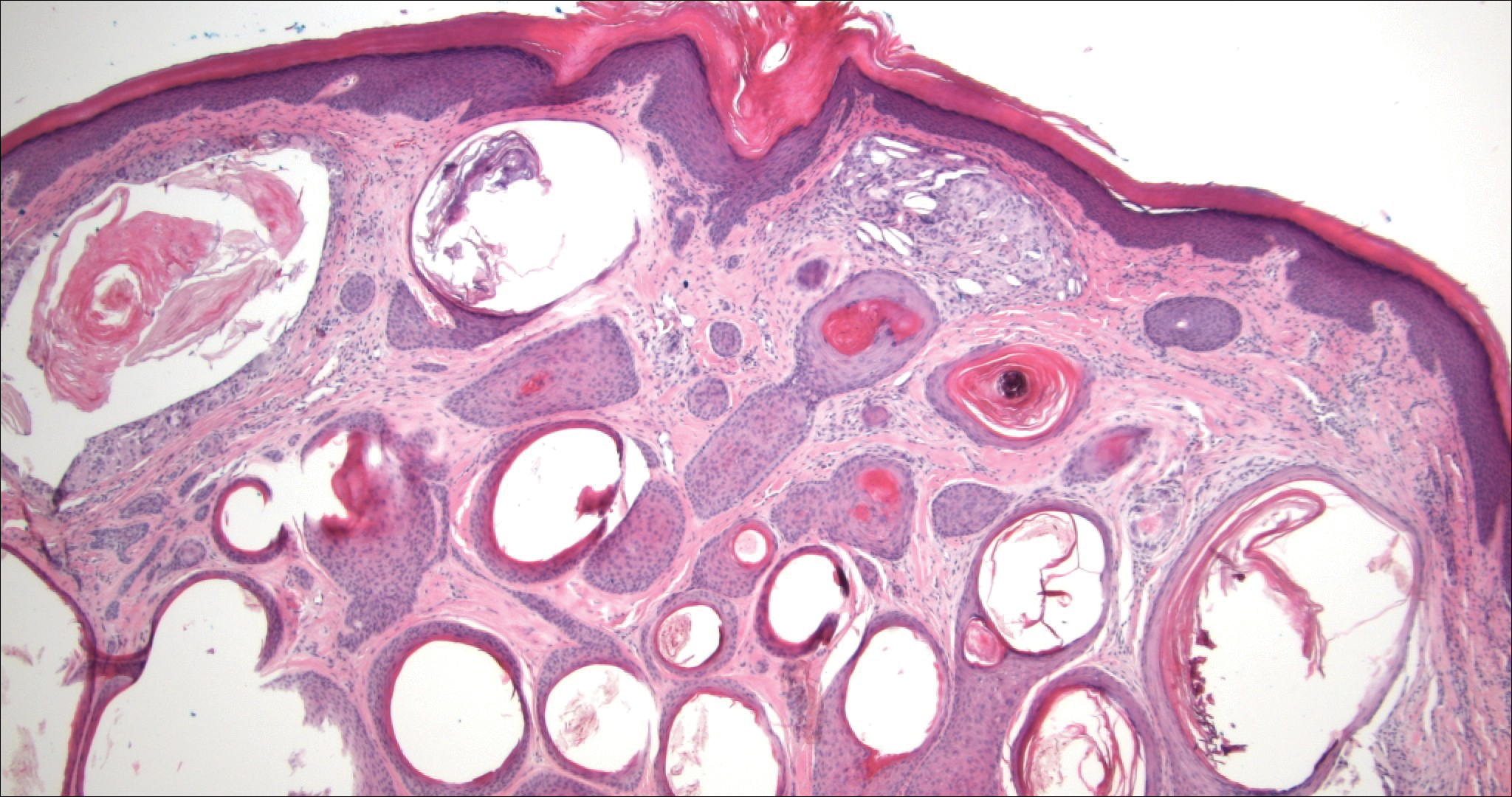
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).
Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).
Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
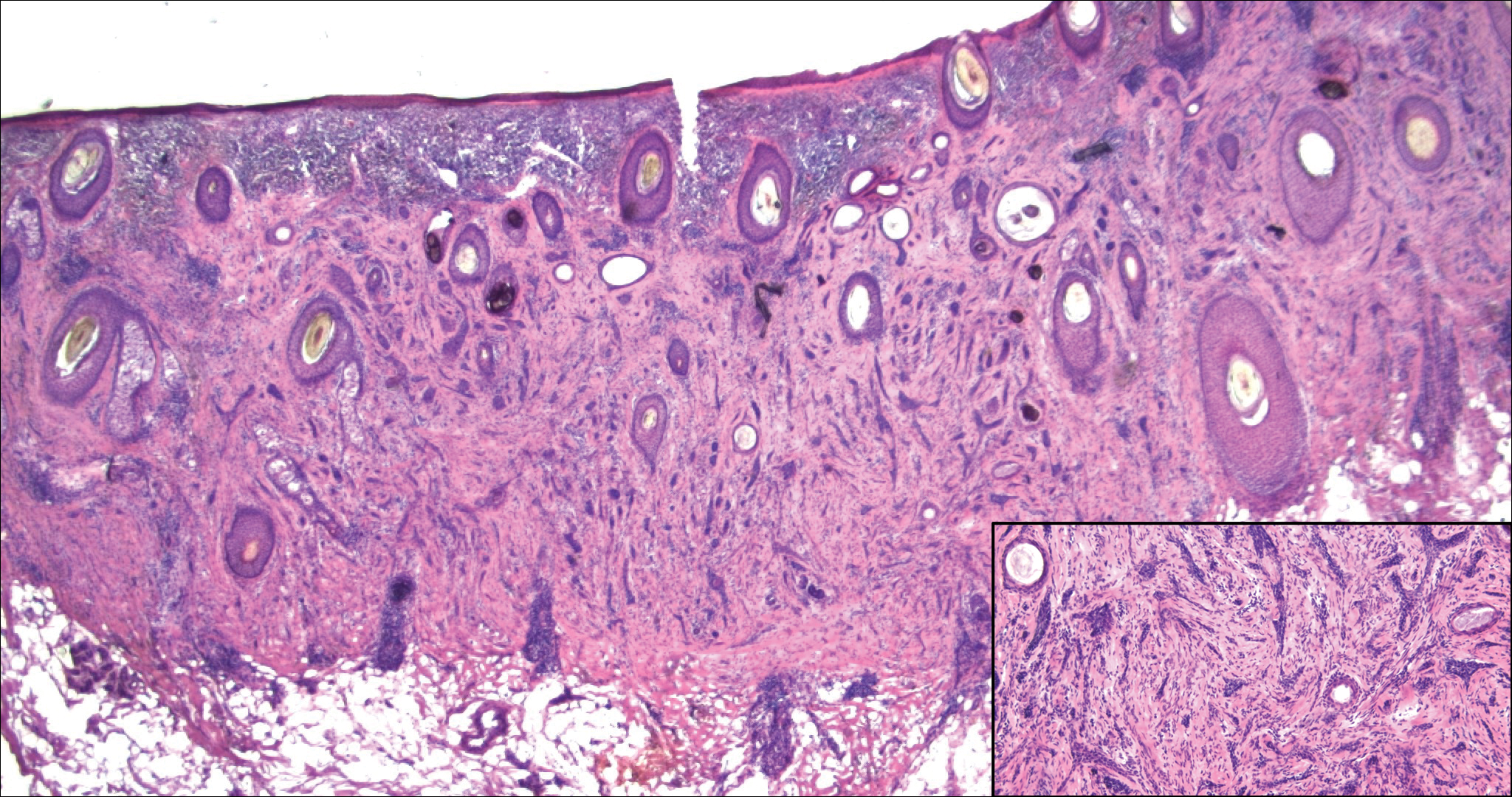
A 52-year-old woman presented with an indurated plaque on the right lateral eyebrow that had been slowly enlarging over the last 4 months.
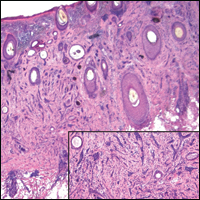
Asymptomatic Pink Plaque on the Scapula
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
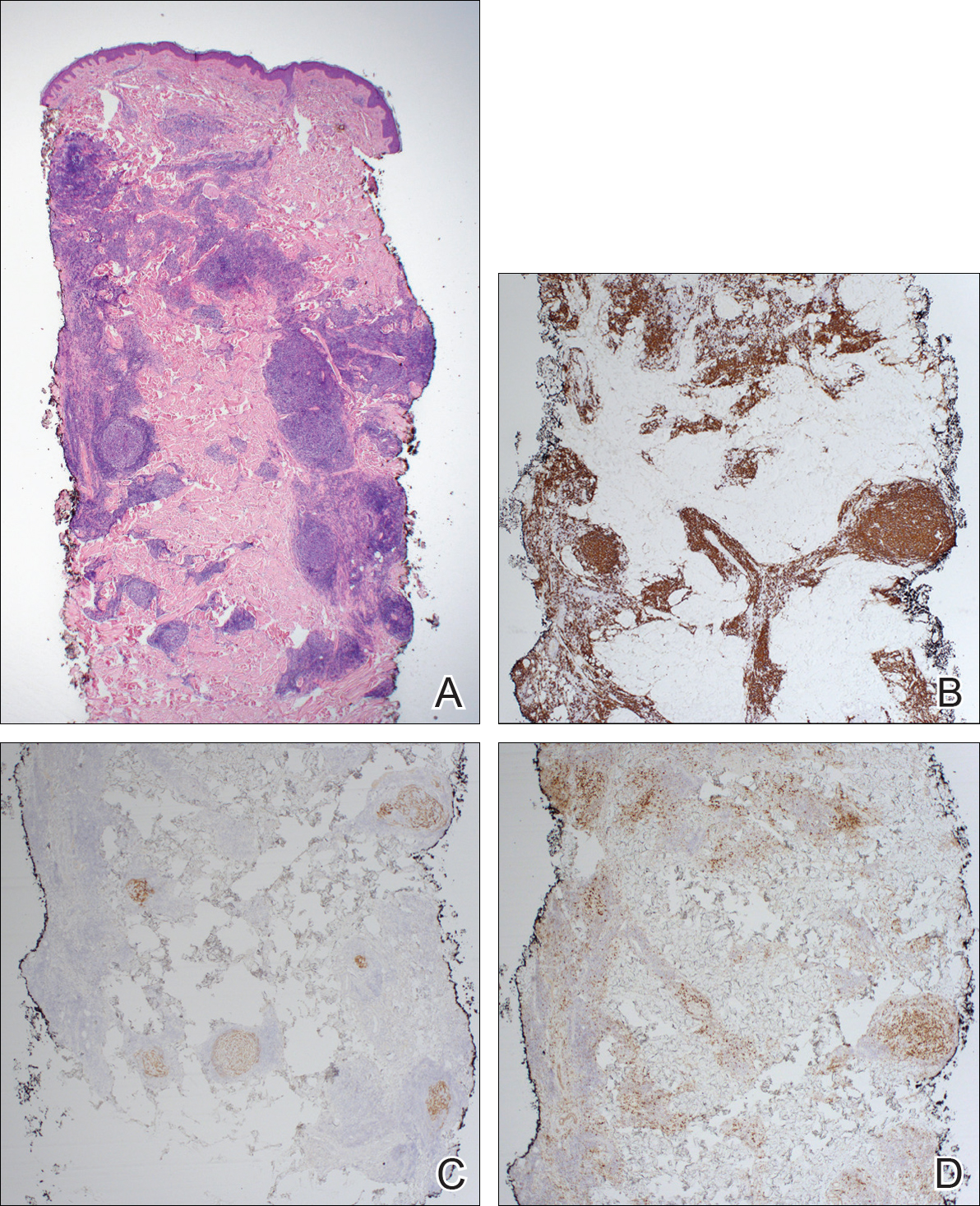
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
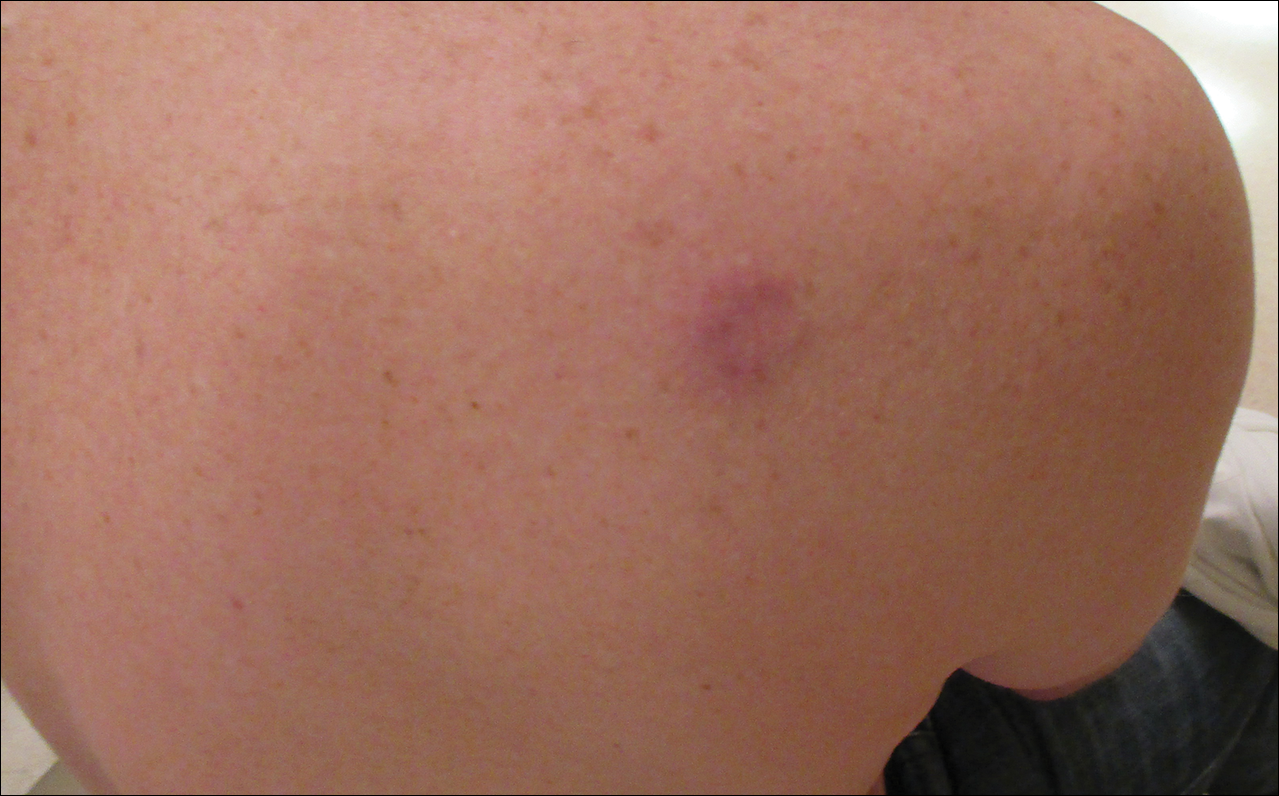
A 36-year-old man presented with a pink plaque on the right side of the scapula of 1 year's duration. The plaque had not grown and was completely asymptomatic. Physical examination revealed a violaceous, pink, 2-cm nodule with overlying telangiectasia. No other concerning lesions were identified on total-body skin examination. A punch biopsy was obtained.
Primary Mucinous Carcinoma of the Eyelid Treated With Mohs Micrographic Surgery
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
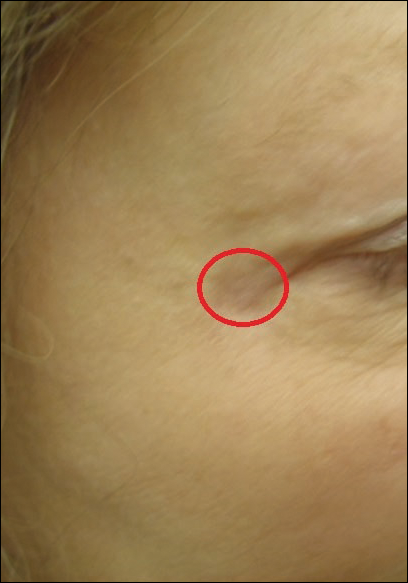
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
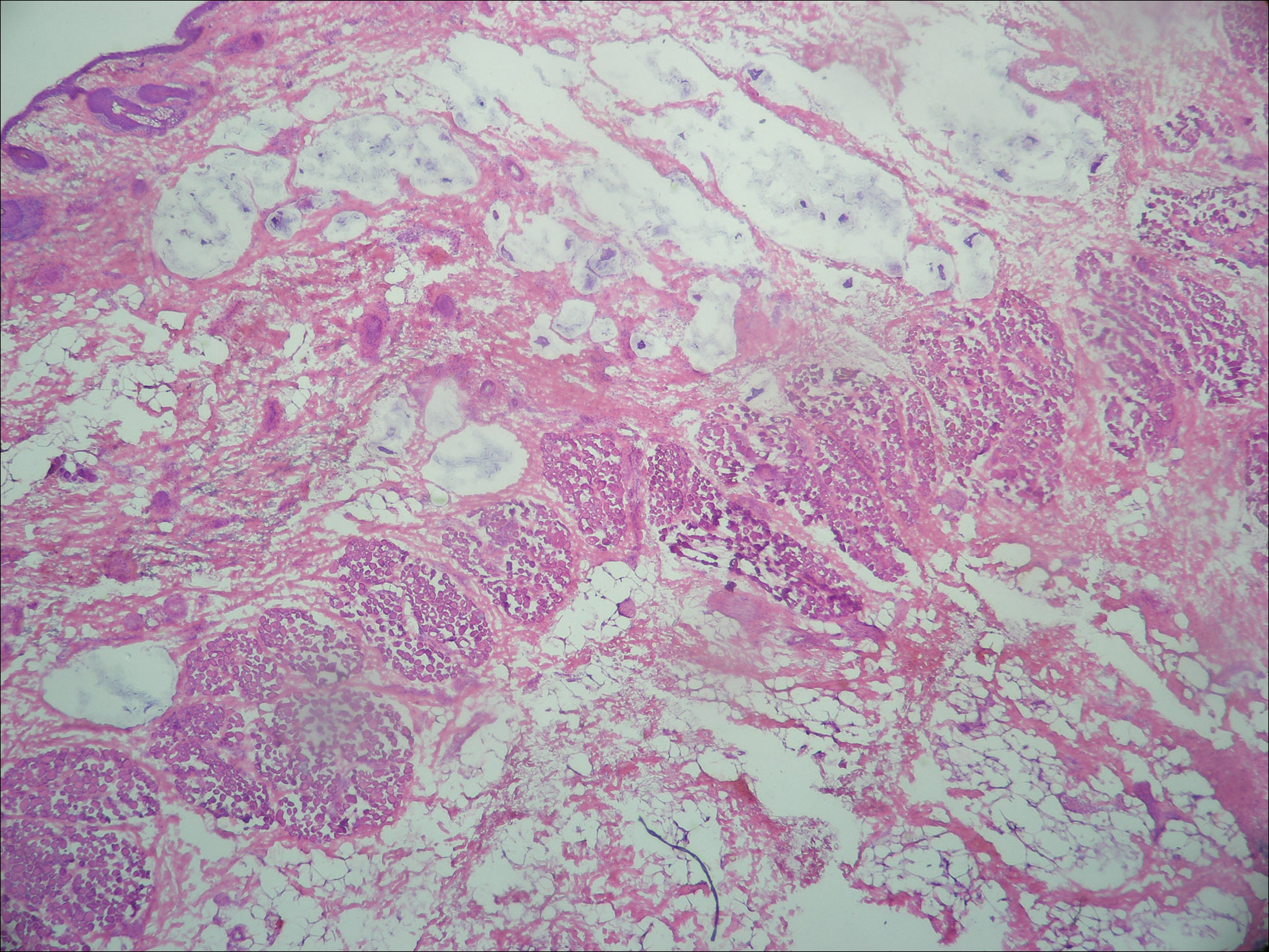
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
Practice Points
- Primary mucinous carcinoma (PMC) of the skin is a rare adnexal tumor.
- Prior to treatment, the diagnostic importance lies in distinguishing PMC from metastatic mucinous malignancies, which portend a poorer prognosis.
- Treatment primarily is surgical, with Mohs micrographic surgery offering improved tissue conservation and reduced recurrence rates.
Acrodermatitis Enteropathica in a Patient With Short Bowel Syndrome
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism

A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
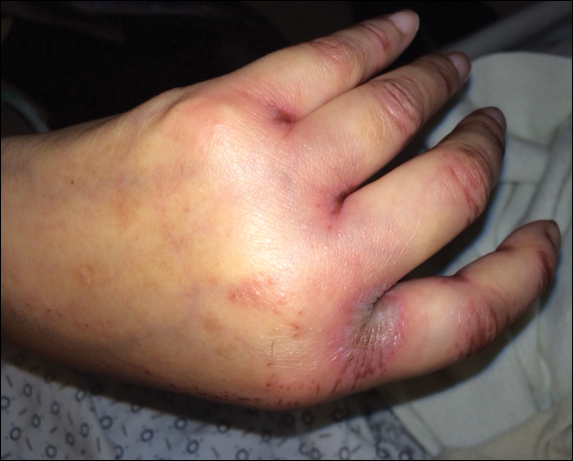
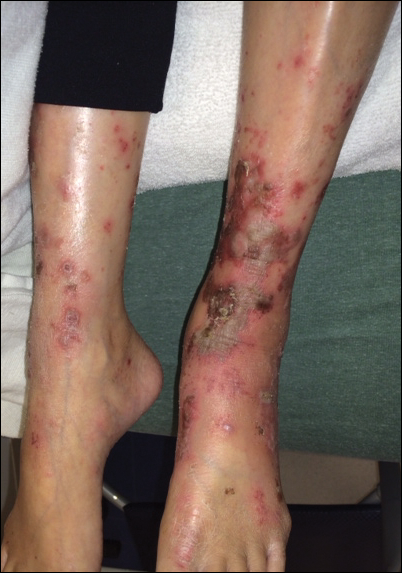
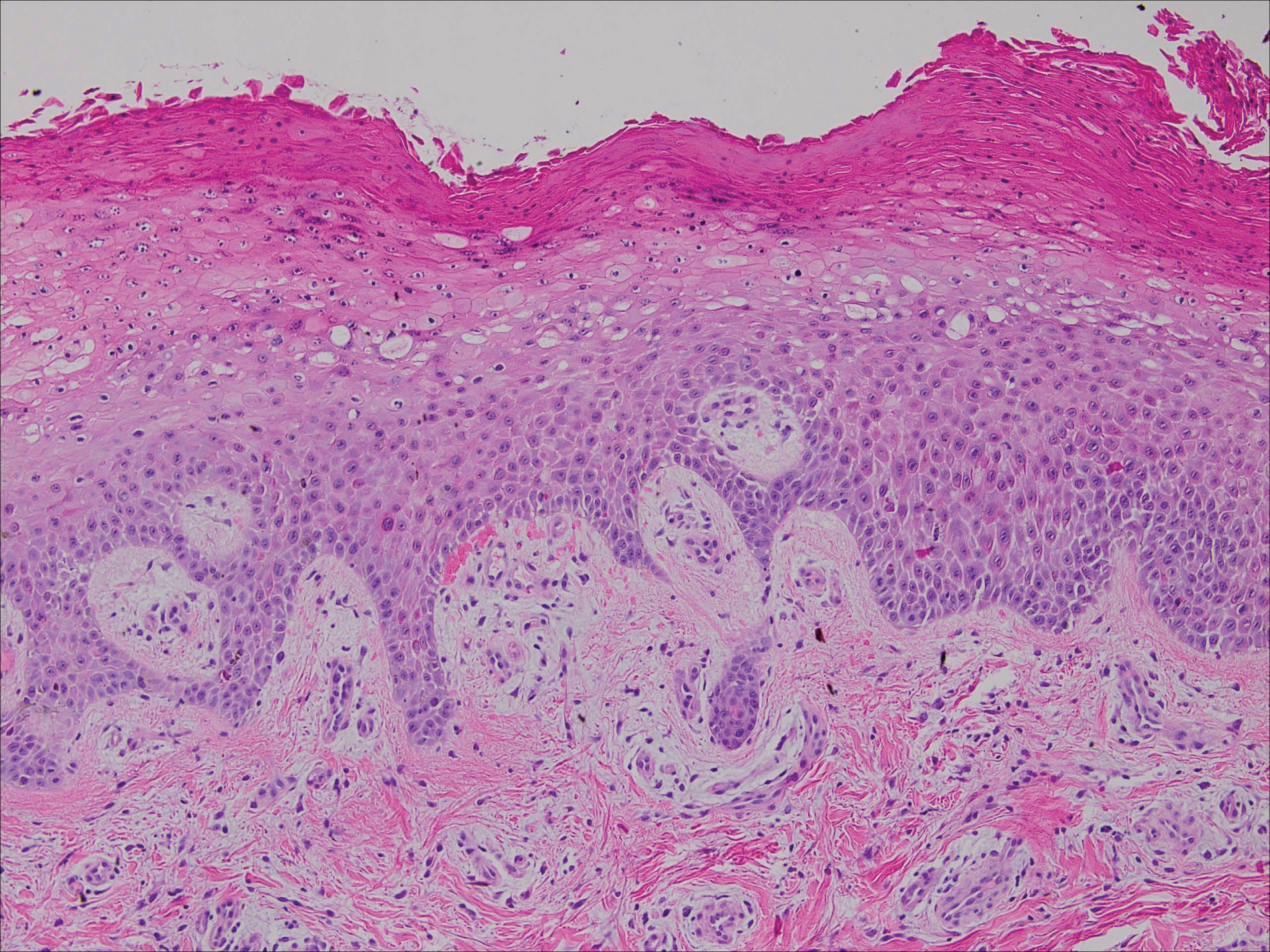
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
Practice Points
- Acrodermatitis enteropathica can be a manifestation of zinc deficiency.
- Acrodermatitis enteropathica should be considered in patients with poor intestinal absorption of nutrients.