User login
Does Vitamin D Supplementation Improve Lower Extremity Power and Function in Community-Dwelling Older Adults?
Study Overview
Objective. To test the effect of 12 months of vitamin D supplementation on lower-extremity power and function in older community-dwelling adults screened for low serum 25-hydroxyvitamin D (25(OH)D).
Design. A single-center, double-blind, randomized placebo-controlled study in which participants were assigned to 800 IU of vitamin D3 supplementation or placebo daily and were followed over a total period of 12 months.
Setting and participants. A total of 100 community-dwelling men and women aged ≥ 60 years with serum 25(OH)D ≤ 20 ng/mL at screening participated. Participants were prescreened by phone, and were excluded if they met any of the following exclusion criteria: vitamin D supplement use > 600 IU/day (for age 60-70 years) or > 800 IU/day (for age ≥ 71 years); vitamin D injection within the previous 3 months; > 2 falls or 1 fall with injury in past year; use of cane, walker, or other indoor walking aid; history of kidney stones within past 3 years; hypercalcemia (serum calcium > 10.8 mg/dL); renal dysfunction (glomerular filtration rate, < 30 mL/min); history of liver disease, sarcoidosis, lymphoma, dysphagia, or other gastrointestinal disorder; neuromuscular disorder affecting lower-extremity function; hip replacement within the past year; cancer treatment in the past 3 years; treatment with thiazide diuretics > 37.5 mg, teriparatide, denosumab, or bisphosphonates within the past 2 years; oral steroids (for > 3 weeks in the past 6 months); and use of fat malabsorption products or anticonvulsive therapy.
Main outcome measures. The primary outcome was leg extensor power assessed using a computer-interfaced bilateral Keiser pneumatic leg press. Secondary outcomes to measure physical function included: (1) backward tandem walk test (which is an indicator of balance and postural control during movement1); (2) Short Physical Performance Battery (SPPB) testing, which includes a balance assessment (ability to stand with feet positioned normally, semi-tandem, and tandem for 10s), a timed 4-m walk, and a chair stand test (time to complete 5 repeated chair stands); (3) stair climbing (ie, time to climb 10 steps, as a measure of knee extensor strength and functional capacity); and (4) handgrip strength (using a dynamometer). Lean tissue mass was assessed by dual X-ray absorptiometry (DEXA scan). Finally, other measures included serum total 25(OH)D levels measured at baseline, 4, 8, and 12 months, as well as 24-hour urine collection for urea-nitrogen and creatinine measurements.
Main results. Of the 2289 individuals screened for the study, 100 met eligibility criteria and underwent randomization to receive either 800 IU vitamin D supplementation daily (n = 49) or placebo (n = 51). Three patients (2 in vitamin D group and 1 in placebo group) were lost to follow up. The mean age of all participants was 69.6 ± 6.9 years. In the vitamin D group versus the control group, respectively, the percent male: female ratio was 66:34 versus 63:37, and percent Caucasian was 75% versus 82%. Mean body mass index was 28.2 ± 7.0 and mean serum 25(OH)D was 20.2 ± 6.7 ng/mL. At the end of the study (12 months), 70% of participants given vitamin D supplementation had 25(OH)D levels ≥ 30 ng/mL and all participants had levels ≥ 20 ng/mL. In the placebo group, the serum 25(OH)D level was ≥ 20 ng/mL in 54% and ≥ 30 ng/mL in 6%. The mean serum 25(OH)D level increased to 32.5 ± 5.1 ng/mL in the vitamin D–supplemented group, but no significant change was found in the placebo group (treatment × time, P < 0.001). Overall, the serum 1,25 (OH)2D3 levels did not differ between the 2 groups over the intervention period (time, P = 0.49; treatment × time, P = 0.27). Dietary intake of vitamin D, calcium, nitrogen, and protein did not differ or change over time between the 2 groups. The change in leg press power, function, and strength did not differ between the groups over 12 months (all treatment × time, P values ≥ 0.60). A total of 27 falls were reported (14 in vitamin D versus 9 in control group), of which 9 were associated with injuries. There was no significant change in lean body mass at the end of the study period in either group (treatment × time, P = 0.98).
Conclusion. In community-dwelling older adults with vitamin D deficiency (≤ 20 ng/mL), 12-month daily supplementation with 800 IU of vitamin D3 resulted in sufficient increases in serum 25(OH)D levels, but did not improve lower-extremity power, strength, or lean mass.
Commentary
Vitamin D deficiency is common in older adults (prevalence of about 41% in US adults ≥ 65 years old, according to Forrest et al2) and is likely due to dietary deficiency, reduced sun exposure (lifestyle), and decreased intestinal calcium absorption. As such, vitamin D deficiency has historically been a topic of debate and of interest in geriatric medicine, as it relates to muscle weakness, which in turn leads to increased susceptibility to falls.3 Interestingly, vitamin D receptors are expressed in human skeletal muscle,4 and in one study, 3-month supplementation of vitamin D led to an increase in type II skeletal muscle fibers in older women.5 Similarly, results from a meta-analysis of 5 randomized controlled trials (RCTs)6 showed that vitamin D supplementation may reduce fall risk in older adults by 22% (corrected odds ratio, 0.78; 95% confidence interval, 0.64-0.92). Thus, in keeping with this general theme of vitamin D supplementation yielding beneficial effects in clinical outcomes, clinicians have long accepted and practiced routine vitamin D supplementation in caring for older adults.
In more recent years, the role of vitamin D supplementation in primary care has become controversial,7 as observed in a recent paradigm shift of moving away from routine supplementation for fall and fracture prevention in clinical practice.8 In a recent meta-analysis of 33 RCTs in older community-dwelling adults, supplementation with vitamin D with or without calcium did not result in a reduction of hip fracture or total number of fractures.9 Moreover, the United States Preventive Services Task Force (USPSTF) recently published updated recommendations on the use of vitamin D supplementation for primary prevention of fractures10 and prevention of falls11 in community-dwelling adults. In these updated recommendations, the USPSTF indicated that insufficient evidence exists to re
Vitamin D supplementation is no longer routinely recommended for fall and fracture prevention. However, if we believe that poor lower extremity muscle strength is a risk factor for falls,12 then the question of whether vitamin D has a beneficial role in improving lower extremity strength in older adults needs to be addressed. Results regarding the effect of vitamin D supplementation on muscle function have so far been mixed. For example, in a randomized, double-blinded, placebo-controlled trial of 160 postmenopausal women with low vitamin D level (< 20 ng/mL), vitamin D3 supplementation at 1000 IU/day for 9 months showed a significant increase in lower extremity muscle strength.13 However, in another randomized double-blinded, placebo-controlled trial of 130 men aged 65 to 90 years with low vitamin D level (< 30 ng/mL) and an SPPB score of ≤ 9 (mild-moderate limitation in mobility), daily supplementation with 4000 IU of vitamin D3 for 9 months did not result in improved SPPB score or gait speed.14 In the study reported by Shea et al, the authors showed that 800 IU of daily vitamin D supplementation (consistent with the Institute of Medicine [IOM] recommendations for older adults15) in community-dwelling older adults with vitamin D deficiency (< 20 ng/mL) did not improve lower extremity muscle strength. This finding is significant in that it adds further evidence to support the rationale against using vitamin D supplementation for the sole purpose of improving lower extremity muscle function in older adults with vitamin D deficiency.
Valuable strengths of this study include its randomized, double-blinded, placebo-controlled trial design testing the IOM recommended dose of daily vitamin D supplementation for older adults. In addition, compared to some of the prior studies mentioned above, the study population included both males and females, although the final study population resulted in some gender bias (with male predominance). Moreover, participants were followed for a sufficient amount of time (1 year), with an excellent adherence rate (only 3 were lost to follow-up) and with corresponding improvement in vitamin D levels. Finally, the use of SPPB as a readout for primary outcome should also be commended, as this assessment is a well-validated method for measuring lower extremity function with scaled scores that predict poor outcomes.16 However, some limitations include the aforementioned predominance of male participants and Caucasian race in both intervention and control groups, as well as discrepancies between the measurement methods for serum vitamin D levels (ie, finger-stick cards versus clinical lab measurement) that may have underestimated the actual serum 25(OH)D levels.
Applications for Clinical Practice
While the null findings from the Shea and colleagues study are applicable to healthier community-dwelling older adults, they may not be generalizable to the care of more frail older patients due to their increased risks for falls and high vulnerability to adverse outcomes. Thus, further studies that account for baseline sarcopenia, frailty, and other fall-risk factors (eg, polypharmacy) are needed to better evaluate the value of vitamin D supplementation in this most vulnerable population.
— Caroline Park, MD, PhD, and Fred Ko, MD
Icahn School of Medicine at Mount Sinai, New York, NY
1. Husu P, Suni J, Pasanen M, Miilunpalo S. Health-related fitness tests as predictors of difficulties in long-distance walking among high-functioning older adults. Aging Clin Exp Res. 2007;19:444-450.
2. Forrest KYZ, Stuhldreher WL. Prevalence and correlates of vitamin D deficiency in US adults. Nutr Res. 2011;31:48-54.
3. Bischoff-Ferrari HA, Giovannucci E, Willett WC, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:1253.
4. Simpson RU, Thomas GA, Arnold AJ. Identification of 1,25-dihydroxyvitamin-D3 receptors and activities in muscle. J Biol Chem. 1985;260:8882-8891.
5. Sorensen OH, Lund BI, Saltin B, et al. Myopathy in bone loss ofaging - improvement by treatment with 1alpha-hydroxycholecalciferol and calcium. Clinical Science. 1979;56:157-161.
6. Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls - A meta-analysis. JAMA. 2004;291:1999-2006.
7. Lewis JR SM, Daly RM. The vitamin D and calcium controversy: an update. Curr Opin Rheumatol. 2019;31:91-97.
8. Schwenk T. No value for routine vitamin D supplementation. NEJM Journal Watch. December 26, 2018.
9. Zhao JG, Zeng XT, Wang J, Liu L. Association between calcium or vitamin D supplementation and fracture incidence in community-dwelling older adults: a systematic review and meta-analysis. JAMA. 2017;318:2466-2482.
10. Grossman DC, Curry SJ, Owens DK, et al. Vitamin D, calcium, or combined supplementation for the primary prevention of fractures in community-dwelling adults US Preventive Services Task Force Recommendation Statement. JAMA. 2018;319:1592-1599.
11. Grossman DC, Curry SJ, Owens DK, et al. Interventions to prevent falls in community-dwelling older adults US Preventive Services Task Force Recommendation Statement. JAMA. 2018;319:1696-1704.
12. Tinetti ME, Speechley M, Ginter SF. Risk-factors for falls among elderly persons living in the community. N Engl J Med. 1988;319:1701-1707.
13. Cangussu LM, Nahas-Neto J, Orsatti CL, et al. Effect of vitamin D supplementation alone on muscle function in postmenopausal women: a randomized, double-blind, placebo-controlled clinical trial. Osteoporos Int. 2015;26:2413-2421.
14. Levis S, Gomez-Marin O. Vitamin D and physical function in sedentary older men. J Am Geriatr Soc. 2017;65:323-331.
15. Ross CA TC, Yaktine AL, Del Valle HB. Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium. National Academies Press. 2011.
16. Guralnik JM, Ferrucci L, Simonsick EM, et al. Lower-extremity function in persons over the age of 70 years as a predictor of subsequent disability. N Engl J Med. 1995;332:556-561
Study Overview
Objective. To test the effect of 12 months of vitamin D supplementation on lower-extremity power and function in older community-dwelling adults screened for low serum 25-hydroxyvitamin D (25(OH)D).
Design. A single-center, double-blind, randomized placebo-controlled study in which participants were assigned to 800 IU of vitamin D3 supplementation or placebo daily and were followed over a total period of 12 months.
Setting and participants. A total of 100 community-dwelling men and women aged ≥ 60 years with serum 25(OH)D ≤ 20 ng/mL at screening participated. Participants were prescreened by phone, and were excluded if they met any of the following exclusion criteria: vitamin D supplement use > 600 IU/day (for age 60-70 years) or > 800 IU/day (for age ≥ 71 years); vitamin D injection within the previous 3 months; > 2 falls or 1 fall with injury in past year; use of cane, walker, or other indoor walking aid; history of kidney stones within past 3 years; hypercalcemia (serum calcium > 10.8 mg/dL); renal dysfunction (glomerular filtration rate, < 30 mL/min); history of liver disease, sarcoidosis, lymphoma, dysphagia, or other gastrointestinal disorder; neuromuscular disorder affecting lower-extremity function; hip replacement within the past year; cancer treatment in the past 3 years; treatment with thiazide diuretics > 37.5 mg, teriparatide, denosumab, or bisphosphonates within the past 2 years; oral steroids (for > 3 weeks in the past 6 months); and use of fat malabsorption products or anticonvulsive therapy.
Main outcome measures. The primary outcome was leg extensor power assessed using a computer-interfaced bilateral Keiser pneumatic leg press. Secondary outcomes to measure physical function included: (1) backward tandem walk test (which is an indicator of balance and postural control during movement1); (2) Short Physical Performance Battery (SPPB) testing, which includes a balance assessment (ability to stand with feet positioned normally, semi-tandem, and tandem for 10s), a timed 4-m walk, and a chair stand test (time to complete 5 repeated chair stands); (3) stair climbing (ie, time to climb 10 steps, as a measure of knee extensor strength and functional capacity); and (4) handgrip strength (using a dynamometer). Lean tissue mass was assessed by dual X-ray absorptiometry (DEXA scan). Finally, other measures included serum total 25(OH)D levels measured at baseline, 4, 8, and 12 months, as well as 24-hour urine collection for urea-nitrogen and creatinine measurements.
Main results. Of the 2289 individuals screened for the study, 100 met eligibility criteria and underwent randomization to receive either 800 IU vitamin D supplementation daily (n = 49) or placebo (n = 51). Three patients (2 in vitamin D group and 1 in placebo group) were lost to follow up. The mean age of all participants was 69.6 ± 6.9 years. In the vitamin D group versus the control group, respectively, the percent male: female ratio was 66:34 versus 63:37, and percent Caucasian was 75% versus 82%. Mean body mass index was 28.2 ± 7.0 and mean serum 25(OH)D was 20.2 ± 6.7 ng/mL. At the end of the study (12 months), 70% of participants given vitamin D supplementation had 25(OH)D levels ≥ 30 ng/mL and all participants had levels ≥ 20 ng/mL. In the placebo group, the serum 25(OH)D level was ≥ 20 ng/mL in 54% and ≥ 30 ng/mL in 6%. The mean serum 25(OH)D level increased to 32.5 ± 5.1 ng/mL in the vitamin D–supplemented group, but no significant change was found in the placebo group (treatment × time, P < 0.001). Overall, the serum 1,25 (OH)2D3 levels did not differ between the 2 groups over the intervention period (time, P = 0.49; treatment × time, P = 0.27). Dietary intake of vitamin D, calcium, nitrogen, and protein did not differ or change over time between the 2 groups. The change in leg press power, function, and strength did not differ between the groups over 12 months (all treatment × time, P values ≥ 0.60). A total of 27 falls were reported (14 in vitamin D versus 9 in control group), of which 9 were associated with injuries. There was no significant change in lean body mass at the end of the study period in either group (treatment × time, P = 0.98).
Conclusion. In community-dwelling older adults with vitamin D deficiency (≤ 20 ng/mL), 12-month daily supplementation with 800 IU of vitamin D3 resulted in sufficient increases in serum 25(OH)D levels, but did not improve lower-extremity power, strength, or lean mass.
Commentary
Vitamin D deficiency is common in older adults (prevalence of about 41% in US adults ≥ 65 years old, according to Forrest et al2) and is likely due to dietary deficiency, reduced sun exposure (lifestyle), and decreased intestinal calcium absorption. As such, vitamin D deficiency has historically been a topic of debate and of interest in geriatric medicine, as it relates to muscle weakness, which in turn leads to increased susceptibility to falls.3 Interestingly, vitamin D receptors are expressed in human skeletal muscle,4 and in one study, 3-month supplementation of vitamin D led to an increase in type II skeletal muscle fibers in older women.5 Similarly, results from a meta-analysis of 5 randomized controlled trials (RCTs)6 showed that vitamin D supplementation may reduce fall risk in older adults by 22% (corrected odds ratio, 0.78; 95% confidence interval, 0.64-0.92). Thus, in keeping with this general theme of vitamin D supplementation yielding beneficial effects in clinical outcomes, clinicians have long accepted and practiced routine vitamin D supplementation in caring for older adults.
In more recent years, the role of vitamin D supplementation in primary care has become controversial,7 as observed in a recent paradigm shift of moving away from routine supplementation for fall and fracture prevention in clinical practice.8 In a recent meta-analysis of 33 RCTs in older community-dwelling adults, supplementation with vitamin D with or without calcium did not result in a reduction of hip fracture or total number of fractures.9 Moreover, the United States Preventive Services Task Force (USPSTF) recently published updated recommendations on the use of vitamin D supplementation for primary prevention of fractures10 and prevention of falls11 in community-dwelling adults. In these updated recommendations, the USPSTF indicated that insufficient evidence exists to re
Vitamin D supplementation is no longer routinely recommended for fall and fracture prevention. However, if we believe that poor lower extremity muscle strength is a risk factor for falls,12 then the question of whether vitamin D has a beneficial role in improving lower extremity strength in older adults needs to be addressed. Results regarding the effect of vitamin D supplementation on muscle function have so far been mixed. For example, in a randomized, double-blinded, placebo-controlled trial of 160 postmenopausal women with low vitamin D level (< 20 ng/mL), vitamin D3 supplementation at 1000 IU/day for 9 months showed a significant increase in lower extremity muscle strength.13 However, in another randomized double-blinded, placebo-controlled trial of 130 men aged 65 to 90 years with low vitamin D level (< 30 ng/mL) and an SPPB score of ≤ 9 (mild-moderate limitation in mobility), daily supplementation with 4000 IU of vitamin D3 for 9 months did not result in improved SPPB score or gait speed.14 In the study reported by Shea et al, the authors showed that 800 IU of daily vitamin D supplementation (consistent with the Institute of Medicine [IOM] recommendations for older adults15) in community-dwelling older adults with vitamin D deficiency (< 20 ng/mL) did not improve lower extremity muscle strength. This finding is significant in that it adds further evidence to support the rationale against using vitamin D supplementation for the sole purpose of improving lower extremity muscle function in older adults with vitamin D deficiency.
Valuable strengths of this study include its randomized, double-blinded, placebo-controlled trial design testing the IOM recommended dose of daily vitamin D supplementation for older adults. In addition, compared to some of the prior studies mentioned above, the study population included both males and females, although the final study population resulted in some gender bias (with male predominance). Moreover, participants were followed for a sufficient amount of time (1 year), with an excellent adherence rate (only 3 were lost to follow-up) and with corresponding improvement in vitamin D levels. Finally, the use of SPPB as a readout for primary outcome should also be commended, as this assessment is a well-validated method for measuring lower extremity function with scaled scores that predict poor outcomes.16 However, some limitations include the aforementioned predominance of male participants and Caucasian race in both intervention and control groups, as well as discrepancies between the measurement methods for serum vitamin D levels (ie, finger-stick cards versus clinical lab measurement) that may have underestimated the actual serum 25(OH)D levels.
Applications for Clinical Practice
While the null findings from the Shea and colleagues study are applicable to healthier community-dwelling older adults, they may not be generalizable to the care of more frail older patients due to their increased risks for falls and high vulnerability to adverse outcomes. Thus, further studies that account for baseline sarcopenia, frailty, and other fall-risk factors (eg, polypharmacy) are needed to better evaluate the value of vitamin D supplementation in this most vulnerable population.
— Caroline Park, MD, PhD, and Fred Ko, MD
Icahn School of Medicine at Mount Sinai, New York, NY
Study Overview
Objective. To test the effect of 12 months of vitamin D supplementation on lower-extremity power and function in older community-dwelling adults screened for low serum 25-hydroxyvitamin D (25(OH)D).
Design. A single-center, double-blind, randomized placebo-controlled study in which participants were assigned to 800 IU of vitamin D3 supplementation or placebo daily and were followed over a total period of 12 months.
Setting and participants. A total of 100 community-dwelling men and women aged ≥ 60 years with serum 25(OH)D ≤ 20 ng/mL at screening participated. Participants were prescreened by phone, and were excluded if they met any of the following exclusion criteria: vitamin D supplement use > 600 IU/day (for age 60-70 years) or > 800 IU/day (for age ≥ 71 years); vitamin D injection within the previous 3 months; > 2 falls or 1 fall with injury in past year; use of cane, walker, or other indoor walking aid; history of kidney stones within past 3 years; hypercalcemia (serum calcium > 10.8 mg/dL); renal dysfunction (glomerular filtration rate, < 30 mL/min); history of liver disease, sarcoidosis, lymphoma, dysphagia, or other gastrointestinal disorder; neuromuscular disorder affecting lower-extremity function; hip replacement within the past year; cancer treatment in the past 3 years; treatment with thiazide diuretics > 37.5 mg, teriparatide, denosumab, or bisphosphonates within the past 2 years; oral steroids (for > 3 weeks in the past 6 months); and use of fat malabsorption products or anticonvulsive therapy.
Main outcome measures. The primary outcome was leg extensor power assessed using a computer-interfaced bilateral Keiser pneumatic leg press. Secondary outcomes to measure physical function included: (1) backward tandem walk test (which is an indicator of balance and postural control during movement1); (2) Short Physical Performance Battery (SPPB) testing, which includes a balance assessment (ability to stand with feet positioned normally, semi-tandem, and tandem for 10s), a timed 4-m walk, and a chair stand test (time to complete 5 repeated chair stands); (3) stair climbing (ie, time to climb 10 steps, as a measure of knee extensor strength and functional capacity); and (4) handgrip strength (using a dynamometer). Lean tissue mass was assessed by dual X-ray absorptiometry (DEXA scan). Finally, other measures included serum total 25(OH)D levels measured at baseline, 4, 8, and 12 months, as well as 24-hour urine collection for urea-nitrogen and creatinine measurements.
Main results. Of the 2289 individuals screened for the study, 100 met eligibility criteria and underwent randomization to receive either 800 IU vitamin D supplementation daily (n = 49) or placebo (n = 51). Three patients (2 in vitamin D group and 1 in placebo group) were lost to follow up. The mean age of all participants was 69.6 ± 6.9 years. In the vitamin D group versus the control group, respectively, the percent male: female ratio was 66:34 versus 63:37, and percent Caucasian was 75% versus 82%. Mean body mass index was 28.2 ± 7.0 and mean serum 25(OH)D was 20.2 ± 6.7 ng/mL. At the end of the study (12 months), 70% of participants given vitamin D supplementation had 25(OH)D levels ≥ 30 ng/mL and all participants had levels ≥ 20 ng/mL. In the placebo group, the serum 25(OH)D level was ≥ 20 ng/mL in 54% and ≥ 30 ng/mL in 6%. The mean serum 25(OH)D level increased to 32.5 ± 5.1 ng/mL in the vitamin D–supplemented group, but no significant change was found in the placebo group (treatment × time, P < 0.001). Overall, the serum 1,25 (OH)2D3 levels did not differ between the 2 groups over the intervention period (time, P = 0.49; treatment × time, P = 0.27). Dietary intake of vitamin D, calcium, nitrogen, and protein did not differ or change over time between the 2 groups. The change in leg press power, function, and strength did not differ between the groups over 12 months (all treatment × time, P values ≥ 0.60). A total of 27 falls were reported (14 in vitamin D versus 9 in control group), of which 9 were associated with injuries. There was no significant change in lean body mass at the end of the study period in either group (treatment × time, P = 0.98).
Conclusion. In community-dwelling older adults with vitamin D deficiency (≤ 20 ng/mL), 12-month daily supplementation with 800 IU of vitamin D3 resulted in sufficient increases in serum 25(OH)D levels, but did not improve lower-extremity power, strength, or lean mass.
Commentary
Vitamin D deficiency is common in older adults (prevalence of about 41% in US adults ≥ 65 years old, according to Forrest et al2) and is likely due to dietary deficiency, reduced sun exposure (lifestyle), and decreased intestinal calcium absorption. As such, vitamin D deficiency has historically been a topic of debate and of interest in geriatric medicine, as it relates to muscle weakness, which in turn leads to increased susceptibility to falls.3 Interestingly, vitamin D receptors are expressed in human skeletal muscle,4 and in one study, 3-month supplementation of vitamin D led to an increase in type II skeletal muscle fibers in older women.5 Similarly, results from a meta-analysis of 5 randomized controlled trials (RCTs)6 showed that vitamin D supplementation may reduce fall risk in older adults by 22% (corrected odds ratio, 0.78; 95% confidence interval, 0.64-0.92). Thus, in keeping with this general theme of vitamin D supplementation yielding beneficial effects in clinical outcomes, clinicians have long accepted and practiced routine vitamin D supplementation in caring for older adults.
In more recent years, the role of vitamin D supplementation in primary care has become controversial,7 as observed in a recent paradigm shift of moving away from routine supplementation for fall and fracture prevention in clinical practice.8 In a recent meta-analysis of 33 RCTs in older community-dwelling adults, supplementation with vitamin D with or without calcium did not result in a reduction of hip fracture or total number of fractures.9 Moreover, the United States Preventive Services Task Force (USPSTF) recently published updated recommendations on the use of vitamin D supplementation for primary prevention of fractures10 and prevention of falls11 in community-dwelling adults. In these updated recommendations, the USPSTF indicated that insufficient evidence exists to re
Vitamin D supplementation is no longer routinely recommended for fall and fracture prevention. However, if we believe that poor lower extremity muscle strength is a risk factor for falls,12 then the question of whether vitamin D has a beneficial role in improving lower extremity strength in older adults needs to be addressed. Results regarding the effect of vitamin D supplementation on muscle function have so far been mixed. For example, in a randomized, double-blinded, placebo-controlled trial of 160 postmenopausal women with low vitamin D level (< 20 ng/mL), vitamin D3 supplementation at 1000 IU/day for 9 months showed a significant increase in lower extremity muscle strength.13 However, in another randomized double-blinded, placebo-controlled trial of 130 men aged 65 to 90 years with low vitamin D level (< 30 ng/mL) and an SPPB score of ≤ 9 (mild-moderate limitation in mobility), daily supplementation with 4000 IU of vitamin D3 for 9 months did not result in improved SPPB score or gait speed.14 In the study reported by Shea et al, the authors showed that 800 IU of daily vitamin D supplementation (consistent with the Institute of Medicine [IOM] recommendations for older adults15) in community-dwelling older adults with vitamin D deficiency (< 20 ng/mL) did not improve lower extremity muscle strength. This finding is significant in that it adds further evidence to support the rationale against using vitamin D supplementation for the sole purpose of improving lower extremity muscle function in older adults with vitamin D deficiency.
Valuable strengths of this study include its randomized, double-blinded, placebo-controlled trial design testing the IOM recommended dose of daily vitamin D supplementation for older adults. In addition, compared to some of the prior studies mentioned above, the study population included both males and females, although the final study population resulted in some gender bias (with male predominance). Moreover, participants were followed for a sufficient amount of time (1 year), with an excellent adherence rate (only 3 were lost to follow-up) and with corresponding improvement in vitamin D levels. Finally, the use of SPPB as a readout for primary outcome should also be commended, as this assessment is a well-validated method for measuring lower extremity function with scaled scores that predict poor outcomes.16 However, some limitations include the aforementioned predominance of male participants and Caucasian race in both intervention and control groups, as well as discrepancies between the measurement methods for serum vitamin D levels (ie, finger-stick cards versus clinical lab measurement) that may have underestimated the actual serum 25(OH)D levels.
Applications for Clinical Practice
While the null findings from the Shea and colleagues study are applicable to healthier community-dwelling older adults, they may not be generalizable to the care of more frail older patients due to their increased risks for falls and high vulnerability to adverse outcomes. Thus, further studies that account for baseline sarcopenia, frailty, and other fall-risk factors (eg, polypharmacy) are needed to better evaluate the value of vitamin D supplementation in this most vulnerable population.
— Caroline Park, MD, PhD, and Fred Ko, MD
Icahn School of Medicine at Mount Sinai, New York, NY
1. Husu P, Suni J, Pasanen M, Miilunpalo S. Health-related fitness tests as predictors of difficulties in long-distance walking among high-functioning older adults. Aging Clin Exp Res. 2007;19:444-450.
2. Forrest KYZ, Stuhldreher WL. Prevalence and correlates of vitamin D deficiency in US adults. Nutr Res. 2011;31:48-54.
3. Bischoff-Ferrari HA, Giovannucci E, Willett WC, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:1253.
4. Simpson RU, Thomas GA, Arnold AJ. Identification of 1,25-dihydroxyvitamin-D3 receptors and activities in muscle. J Biol Chem. 1985;260:8882-8891.
5. Sorensen OH, Lund BI, Saltin B, et al. Myopathy in bone loss ofaging - improvement by treatment with 1alpha-hydroxycholecalciferol and calcium. Clinical Science. 1979;56:157-161.
6. Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls - A meta-analysis. JAMA. 2004;291:1999-2006.
7. Lewis JR SM, Daly RM. The vitamin D and calcium controversy: an update. Curr Opin Rheumatol. 2019;31:91-97.
8. Schwenk T. No value for routine vitamin D supplementation. NEJM Journal Watch. December 26, 2018.
9. Zhao JG, Zeng XT, Wang J, Liu L. Association between calcium or vitamin D supplementation and fracture incidence in community-dwelling older adults: a systematic review and meta-analysis. JAMA. 2017;318:2466-2482.
10. Grossman DC, Curry SJ, Owens DK, et al. Vitamin D, calcium, or combined supplementation for the primary prevention of fractures in community-dwelling adults US Preventive Services Task Force Recommendation Statement. JAMA. 2018;319:1592-1599.
11. Grossman DC, Curry SJ, Owens DK, et al. Interventions to prevent falls in community-dwelling older adults US Preventive Services Task Force Recommendation Statement. JAMA. 2018;319:1696-1704.
12. Tinetti ME, Speechley M, Ginter SF. Risk-factors for falls among elderly persons living in the community. N Engl J Med. 1988;319:1701-1707.
13. Cangussu LM, Nahas-Neto J, Orsatti CL, et al. Effect of vitamin D supplementation alone on muscle function in postmenopausal women: a randomized, double-blind, placebo-controlled clinical trial. Osteoporos Int. 2015;26:2413-2421.
14. Levis S, Gomez-Marin O. Vitamin D and physical function in sedentary older men. J Am Geriatr Soc. 2017;65:323-331.
15. Ross CA TC, Yaktine AL, Del Valle HB. Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium. National Academies Press. 2011.
16. Guralnik JM, Ferrucci L, Simonsick EM, et al. Lower-extremity function in persons over the age of 70 years as a predictor of subsequent disability. N Engl J Med. 1995;332:556-561
1. Husu P, Suni J, Pasanen M, Miilunpalo S. Health-related fitness tests as predictors of difficulties in long-distance walking among high-functioning older adults. Aging Clin Exp Res. 2007;19:444-450.
2. Forrest KYZ, Stuhldreher WL. Prevalence and correlates of vitamin D deficiency in US adults. Nutr Res. 2011;31:48-54.
3. Bischoff-Ferrari HA, Giovannucci E, Willett WC, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:1253.
4. Simpson RU, Thomas GA, Arnold AJ. Identification of 1,25-dihydroxyvitamin-D3 receptors and activities in muscle. J Biol Chem. 1985;260:8882-8891.
5. Sorensen OH, Lund BI, Saltin B, et al. Myopathy in bone loss ofaging - improvement by treatment with 1alpha-hydroxycholecalciferol and calcium. Clinical Science. 1979;56:157-161.
6. Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls - A meta-analysis. JAMA. 2004;291:1999-2006.
7. Lewis JR SM, Daly RM. The vitamin D and calcium controversy: an update. Curr Opin Rheumatol. 2019;31:91-97.
8. Schwenk T. No value for routine vitamin D supplementation. NEJM Journal Watch. December 26, 2018.
9. Zhao JG, Zeng XT, Wang J, Liu L. Association between calcium or vitamin D supplementation and fracture incidence in community-dwelling older adults: a systematic review and meta-analysis. JAMA. 2017;318:2466-2482.
10. Grossman DC, Curry SJ, Owens DK, et al. Vitamin D, calcium, or combined supplementation for the primary prevention of fractures in community-dwelling adults US Preventive Services Task Force Recommendation Statement. JAMA. 2018;319:1592-1599.
11. Grossman DC, Curry SJ, Owens DK, et al. Interventions to prevent falls in community-dwelling older adults US Preventive Services Task Force Recommendation Statement. JAMA. 2018;319:1696-1704.
12. Tinetti ME, Speechley M, Ginter SF. Risk-factors for falls among elderly persons living in the community. N Engl J Med. 1988;319:1701-1707.
13. Cangussu LM, Nahas-Neto J, Orsatti CL, et al. Effect of vitamin D supplementation alone on muscle function in postmenopausal women: a randomized, double-blind, placebo-controlled clinical trial. Osteoporos Int. 2015;26:2413-2421.
14. Levis S, Gomez-Marin O. Vitamin D and physical function in sedentary older men. J Am Geriatr Soc. 2017;65:323-331.
15. Ross CA TC, Yaktine AL, Del Valle HB. Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium. National Academies Press. 2011.
16. Guralnik JM, Ferrucci L, Simonsick EM, et al. Lower-extremity function in persons over the age of 70 years as a predictor of subsequent disability. N Engl J Med. 1995;332:556-561
Meta-analysis finds no link between PPI use and risk of dementia

The finding runs counter to recent studies, including a large pharmacoepidemiological claims data analysis from Germany, that propose an association between proton pump inhibitor (PPI) use and the development of dementia (JAMA Neurol. 2016;73[4]:410-6). “The issue with these studies is that they’re based on retrospective claims data and pharmacoepidemiological studies and insurance databases that don’t really give you a good causality basis,” lead study author Saad Alrajhi, MD, said in an interview at the annual Digestive Disease Week.
In an effort to better characterize the association between PPI exposure and dementia, Dr. Alrajhi, a gastroenterology fellow at McGill University, Montreal, and colleagues conducted a meta-analysis of all fully published randomized clinical trials or observational studies comparing use of PPIs and occurrence of dementia. The researchers queried Embase, MEDLINE, and ISI Web of Knowledge for relevant studies that were published from 1995 through September 2018. Next, they assessed the quality of the studies by using the Cochrane risk assessment tool for RCTs or the Newcastle-Ottawa Scale for observational studies.
As the primary outcome, the researchers compared dementia incidence after PPI exposure (experimental group) versus no PPI exposure (control group). Development of Alzheimer’s dementia was a secondary outcome. Sensitivity analyses consisted of excluding one study at a time, and assessing results among studies of highest qualities. Subgroup analyses included stratifying patients by age. To report odds ratios, Dr. Alrajhi and colleagues used fixed or random effects models based on the absence or presence of heterogeneity.
Of 549 studies assessed, 5 met the criteria for inclusion in the final analysis: 3 case-control studies and 2 cohort studies, with a total of 472,933 patients. All of the studies scored 8 or 9 on the Newcastle-Ottawa scale, indicating high quality. Significant heterogeneity was noted for all analyses. The researchers found that the incidence of dementia was not significantly increased among patients in the PPI-exposed group (odd ratio, 1.08 (95% confidence interval, 0.97-1.20; P = .18). Sensitivity analyses confirmed the robustness of the results. Subgroup analysis showed no between-group differences among studies that included a minimum age above 65 years (three studies) or less than age 65 (two studies). PPI exposure was not associated with the development of Alzheimer’s dementia (two studies) (OR, 1.32 (95% CI, 0.80-2.17; P = .27).
“In the absence of randomized trial evidence, a PPI prescribing approach based on appropriate utilization of guideline-based prescription should be done without the extra fear of the association of dementia,” Dr. Alrajhi said.
The researchers reported having no financial disclosures.
The finding runs counter to recent studies, including a large pharmacoepidemiological claims data analysis from Germany, that propose an association between proton pump inhibitor (PPI) use and the development of dementia (JAMA Neurol. 2016;73[4]:410-6). “The issue with these studies is that they’re based on retrospective claims data and pharmacoepidemiological studies and insurance databases that don’t really give you a good causality basis,” lead study author Saad Alrajhi, MD, said in an interview at the annual Digestive Disease Week.
In an effort to better characterize the association between PPI exposure and dementia, Dr. Alrajhi, a gastroenterology fellow at McGill University, Montreal, and colleagues conducted a meta-analysis of all fully published randomized clinical trials or observational studies comparing use of PPIs and occurrence of dementia. The researchers queried Embase, MEDLINE, and ISI Web of Knowledge for relevant studies that were published from 1995 through September 2018. Next, they assessed the quality of the studies by using the Cochrane risk assessment tool for RCTs or the Newcastle-Ottawa Scale for observational studies.
As the primary outcome, the researchers compared dementia incidence after PPI exposure (experimental group) versus no PPI exposure (control group). Development of Alzheimer’s dementia was a secondary outcome. Sensitivity analyses consisted of excluding one study at a time, and assessing results among studies of highest qualities. Subgroup analyses included stratifying patients by age. To report odds ratios, Dr. Alrajhi and colleagues used fixed or random effects models based on the absence or presence of heterogeneity.
Of 549 studies assessed, 5 met the criteria for inclusion in the final analysis: 3 case-control studies and 2 cohort studies, with a total of 472,933 patients. All of the studies scored 8 or 9 on the Newcastle-Ottawa scale, indicating high quality. Significant heterogeneity was noted for all analyses. The researchers found that the incidence of dementia was not significantly increased among patients in the PPI-exposed group (odd ratio, 1.08 (95% confidence interval, 0.97-1.20; P = .18). Sensitivity analyses confirmed the robustness of the results. Subgroup analysis showed no between-group differences among studies that included a minimum age above 65 years (three studies) or less than age 65 (two studies). PPI exposure was not associated with the development of Alzheimer’s dementia (two studies) (OR, 1.32 (95% CI, 0.80-2.17; P = .27).
“In the absence of randomized trial evidence, a PPI prescribing approach based on appropriate utilization of guideline-based prescription should be done without the extra fear of the association of dementia,” Dr. Alrajhi said.
The researchers reported having no financial disclosures.
The finding runs counter to recent studies, including a large pharmacoepidemiological claims data analysis from Germany, that propose an association between proton pump inhibitor (PPI) use and the development of dementia (JAMA Neurol. 2016;73[4]:410-6). “The issue with these studies is that they’re based on retrospective claims data and pharmacoepidemiological studies and insurance databases that don’t really give you a good causality basis,” lead study author Saad Alrajhi, MD, said in an interview at the annual Digestive Disease Week.
In an effort to better characterize the association between PPI exposure and dementia, Dr. Alrajhi, a gastroenterology fellow at McGill University, Montreal, and colleagues conducted a meta-analysis of all fully published randomized clinical trials or observational studies comparing use of PPIs and occurrence of dementia. The researchers queried Embase, MEDLINE, and ISI Web of Knowledge for relevant studies that were published from 1995 through September 2018. Next, they assessed the quality of the studies by using the Cochrane risk assessment tool for RCTs or the Newcastle-Ottawa Scale for observational studies.
As the primary outcome, the researchers compared dementia incidence after PPI exposure (experimental group) versus no PPI exposure (control group). Development of Alzheimer’s dementia was a secondary outcome. Sensitivity analyses consisted of excluding one study at a time, and assessing results among studies of highest qualities. Subgroup analyses included stratifying patients by age. To report odds ratios, Dr. Alrajhi and colleagues used fixed or random effects models based on the absence or presence of heterogeneity.
Of 549 studies assessed, 5 met the criteria for inclusion in the final analysis: 3 case-control studies and 2 cohort studies, with a total of 472,933 patients. All of the studies scored 8 or 9 on the Newcastle-Ottawa scale, indicating high quality. Significant heterogeneity was noted for all analyses. The researchers found that the incidence of dementia was not significantly increased among patients in the PPI-exposed group (odd ratio, 1.08 (95% confidence interval, 0.97-1.20; P = .18). Sensitivity analyses confirmed the robustness of the results. Subgroup analysis showed no between-group differences among studies that included a minimum age above 65 years (three studies) or less than age 65 (two studies). PPI exposure was not associated with the development of Alzheimer’s dementia (two studies) (OR, 1.32 (95% CI, 0.80-2.17; P = .27).
“In the absence of randomized trial evidence, a PPI prescribing approach based on appropriate utilization of guideline-based prescription should be done without the extra fear of the association of dementia,” Dr. Alrajhi said.
The researchers reported having no financial disclosures.
REPORTING FROM DDW 2019
A sleeping beast: Obstructive sleep apnea and stroke
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.

Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.

A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.
Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.
A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.
Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.
A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
KEY POINTS
- A low threshold for evaluating for OSA after a stroke is warranted: the prevalence is high in this population, and risk factors for OSA and its typical clinical picture may not be present.
- Questionnaires can help screen for the likelihood of OSA and the need for more definitive assessment with polysomnography or home sleep apnea testing, tests that pose additional challenges after stroke.
- Positive airway pressure (PAP) therapy remains the first-line treatment for OSA after stroke; it may improve recovery and reduce long-term sequelae of untreated OSA.
- Acceptance of and adherence to PAP therapy can be especially problematic in this population, and alternatives should be considered if needed.
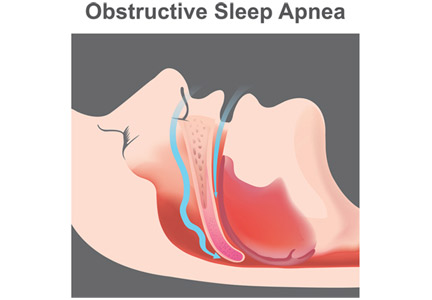
Correction: Subclinical hypothyroidism
In Azim S, Nasr C, “Subclinical hypothyroidism: When to treat,” Cleve Clin J Med 2019; 86(2):101–110, on page 103, in the section “Subclinical hypothyroidism can resolve or progress,” the sentence “The rate of progression to overt hypothyroidism is estimated to be 33% to 35% over 10 to 20 years of follow-up” contained an error. The correct rate of progression is 33% to 55%. This error has been corrected online.
In Azim S, Nasr C, “Subclinical hypothyroidism: When to treat,” Cleve Clin J Med 2019; 86(2):101–110, on page 103, in the section “Subclinical hypothyroidism can resolve or progress,” the sentence “The rate of progression to overt hypothyroidism is estimated to be 33% to 35% over 10 to 20 years of follow-up” contained an error. The correct rate of progression is 33% to 55%. This error has been corrected online.
In Azim S, Nasr C, “Subclinical hypothyroidism: When to treat,” Cleve Clin J Med 2019; 86(2):101–110, on page 103, in the section “Subclinical hypothyroidism can resolve or progress,” the sentence “The rate of progression to overt hypothyroidism is estimated to be 33% to 35% over 10 to 20 years of follow-up” contained an error. The correct rate of progression is 33% to 55%. This error has been corrected online.
From sweet to belligerent in the blink of an eye
CASE Combative and agitated
Ms. P, age 87, presents to the emergency department (ED) with her caregiver, who says Ms. P has new-onset altered mental status, agitation, and combativeness.
Ms. P resides at a long-term care (LTC) facility, where according to the nurses she normally is pleasant, well-oriented, and cooperative. Ms. P’s medical history includes major depressive disorder, generalized anxiety disorder, hypertension, chronic kidney disease (CKD) stage III, peptic ulcer disease, gastroesophageal reflux disease, coronary artery disease with 2 past myocardial infarctions requiring stents, chronic obstructive pulmonary disease, hyperlipidemia, bradycardia requiring a pacemaker, paroxysmal atrial fibrillation, asthma, aortic stenosis, peripheral vascular disease, esophageal stricture requiring dilation, deep vein thrombosis, and migraines.
Mr. P’s medication list includes acetaminophen, 650 mg every 6 hours; ipratropium/albuterol nebulized solution, 3 mL 4 times a day; aspirin, 81 mg/d; atorvastatin, 40 mg/d; calcitonin, 1 spray nasally at bedtime; clopidogrel, 75 mg/d; ezetimibe, 10 mg/d; fluoxetine, 20 mg/d; furosemide, 20 mg/d; isosorbide dinitrate, 120 mg/d; lisinopril, 15 mg/d; risperidone, 0.5 mg/d; magnesium oxide, 800 mg/d; pantoprazole, 40 mg/d; polyethylene glycol, 17 g/d; sotalol, 160 mg/d; olanzapine, 5 mg IM every 6 hours as needed for agitation; and tramadol, 50 mg every 8 hours as needed for headache.
Seven days before coming to the ED, Ms. P was started on ceftriaxone, 1 g/d, for suspected community-acquired pneumonia. At that time, the nursing staff noticed behavioral changes. Soon after, Ms. P began refusing all her medications. Two days before presenting to the ED, Ms. P was started on nitrofurantoin, 200 mg/d, for a suspected urinary tract infection, but it was discontinued because of an allergy.
Her caregiver reports that while at the LTC facility, Ms. P’s behavioral changes worsened. Ms. P claimed to be Jesus Christ and said she was talking to the devil; she chased other residents around the facility and slapped medications away from the nursing staff. According to caregivers, this behavior was out of character.
Shortly after arriving in the ED, Ms. P is admitted to the psychiatric unit.
[polldaddy:10332748]

The authors’ observations
Delirium is a complex, acute alteration in a patient’s mental status compared with his/her baseline functioning1 (Table 12). The onset of delirium is quick, happening within hours to days, with fluctuations in mental function. Patients might present with hyperactive, hypoactive, or mixed delirium.3 Patients with hyperactive delirium often have delusions and hallucinations; these patients might be agitated and could become violent with family and caregivers.3 Patients with hypoactive delirium are less likely to experience hallucinations and more likely to show symptoms of sedation.3 Patients with hypoactive delirium can be difficult to diagnose because it is challenging to interview them and understand what might be the cause of their sedated state. Patients also can exhibit a mixed delirium in which they fluctuate between periods of hyperactivity and hypoactivity.3
Continue to: Suspected delirium...
Suspected delirium should be considered a medical emergency because the outcome could be fatal.1 It is important to uncover and treat the underlying cause(s) of delirium rather than solely administering antipsychotics, which might mask the presenting symptoms. In an older study, Francis and Kapoor4 reported that 56% of geriatric patients with delirium had a single definite or probable etiology, while the other 44% had about 2.8 etiologies per patient on average. Delirium risk factors, causes, and factors to consider during patient evaluation are listed in Table 21,3,5-7 and Table 3.1,3,5-7

A synergistic relationship between comorbidities, environment, and medications can induce delirium.5 Identifying irreversible and reversible causes is the key to treating delirium. After the cause has been identified, it can be addressed and the patient could return to his/her previous level of functioning. If the delirium is the result of multiple irreversible causes, it could become chronic.

[polldaddy:10332749]
EVALUATION Cardiac dysfunction
Ms. P undergoes laboratory testing. The results include: white blood cell count, 5.9/µL; hemoglobin, 13.6 g/dL; hematocrit, 42.6%; platelets, 304 × 103/µL; sodium,143 mEq/L; potassium, 3.2 mEq/L; chloride, 96 mEq/L; carbon dioxide, 23 mEq/L; blood glucose, 87 mg/dL; creatinine, 1.2 mg/dL; estimated creatinine clearance (eCrCl) level of 33 mL/min/1.73 m2; calcium, 9.5 mg/dL; albumin, 3.6 g/dL; liver enzymes within normal limits; thyroid-stimulating hormone, 0.78 mIU/L; vitamin B12, 995 pg/mL; folic acid, 16.6 ng/mL; vitamin D, 31 pg/mL; and rapid plasma reagin: nonreactive. Urinalysis is unremarkable, and no culture is performed. Urine drug screening/toxicology is positive for the benzodiazepines that she received in the ED (oral alprazolam 0.25 mg given once and oral lorazepam 0.5 mg given once).
Electrocardiogram (ECG) shows atrial flutter/tachycardia with rapid ventricular response, marked left axis deviation, nonspecific ST- and T-wave abnormality, QT/QTC of 301/387 ms, and ventricular rate 151 beats per minute. A CT scan of the head and brain without contrast shows mild atrophy and chronic white matter changes and no acute intracranial abnormality. A two-view chest radiography shows no acute cardiopulmonary findings. Her temperature is 98.4°F; heart rate is 122 beats per minute; respiratory rate is 20 breaths per minute; blood pressure is 161/98 mm Hg; and oxygen saturation is 86% on room air.
Based on this data, Ms. P’s cardiac condition seems to be worsening, which is thought to be caused by her refusal of furosemide, lisinopril, isosorbide, sotalol, clopidogrel, and aspirin. The treatment team plans to work on compliance to resolve these cardiac issues and places Ms. P on 1:1 observation with a sitter and music in attempt to calm her.
Continue to: The authors' observations
The authors’ observations
Many factors can contribute to behavioral or cognitive changes in geriatric patients. Often, a major change noted in an older patient can be attributed to new-onset dementia, dementia with behavioral disturbances, delirium, depression, or acute psychosis. These potential causes should be considered and ruled out in a step-by-step progression. Because patients are unreliable historians during acute distress, a complete history from family or caregivers and exhaustive workup is paramount.
TREATMENT Medication adjustments
In an attempt to resolve Ms. P’s disruptive behaviors, her risperidone dosage is changed to 0.5 mg twice daily. Ms. P is encouraged to use the provided oxygen to raise her saturation level.
On hospital Day 3, a loose stool prompts a Clostridium difficile test as a possible source of delirium; however, the results are negative.
On hospital Day 4, Ms. P is confused and irritable overnight, yelling profanities at staff, refusing care, inappropriately disrobing, and having difficulty falling asleep and staying asleep. Risperidone is discontinued because it appears to have had little or no effect on Ms. P’s disruptive behaviors. Olanzapine, 10 mg/d, is initiated with mirtazapine, 7.5 mg/d, to help with mood, appetite, and sleep. Fluoxetine is also discontinued because of a possible interaction with clopidogrel.
On hospital Days 6 to 8, Ms. P remains upset and unable to follow instructions. Melatonin is initiated to improve her sleep cycle. On Day 9, she continues to decline and is cursing at hospital staff; haloperidol is initiated at 5 mg every morning, 10 mg at bedtime, and 5 mg IM as needed for agitation. Her sleep improves with melatonin and mirtazapine. IV hydration also is initiated. Ms. P has a slight improvement in medication compliance. On Day 11, haloperidol is increased to 5 mg in the morning, 5 mg in the afternoon, and 10 mg at bedtime. On Day 12, haloperidol is changed to 7.5 mg twice daily; a slight improvement in Ms. P’s behavior is noted.
Continue to: On hospital Day 13...
On hospital Day 13, Ms. P’s behavior declines again. She screams profanities at staff and does not recognize the clinicians who have been providing care to her. The physician initiates valproic acid, 125 mg, 3 times a day, to target Ms. P’s behavioral disturbances. A pharmacist notes that the patient’s sotalol could be contributing to Ms. P’s psychiatric presentation, and that based on her eCrCl level of 33 mL/min/1.73 m2, a dosage adjustment or medication change might be warranted.
On Day 14, Ms. P displays erratic behavior and intermittent tachycardia. A cardiac consultation is ordered. A repeat ECG reveals atrial fibrillation with rapid rate and a QT/QTc of 409/432 ms. Ms. P is transferred to the telemetry unit, where the cardiologist discontinues sotalol because the dosage is not properly renally adjusted. Sotalol hydrochloride has been associated with life-threatening ventricular tachycardia.8 Diltiazem, 30 mg every 6 hours is initiated to replace sotalol.
By Day 16, the treatment team notes improved cognition and behavior. On Day 17, the cardiologist reports that Ms. P’s atrial fibrillation is controlled. An ECG reveals mild left ventricular hypertrophy, an ejection fraction of 50% to 55%, no stenosis in the mitral or tricuspid valves, no valvular pulmonic stenosis, and moderate aortic sclerosis. Cardiac markers also are evaluated (creatinine phosphokinase: 105 U/L; creatinine kinase–MB fraction: 2.6 ng/mL; troponin: 0.01 ng/mL; pro-B-type natriuretic peptide: 2,073 pg/mL); and myocardial infarction is ruled out.
On Day 19, Ms. P’s diltiazem is consolidated to a controlled-delivery formulation, 180 mg/d, along with the addition of metoprolol, 12.5 mg twice daily. Ms. P is transferred back to the psychiatric unit.
OUTCOME Gradual improvement
On Days 20 to 23, Ms. P shows remarkable progress, and her mental status, cognition, and behavior slowly return to baseline. Haloperidol and valproic acid are tapered and discontinued. Ms. P is observed to be healthy and oriented to person, place, and time.
Continue to: On Day 25...
On Day 25, she is discharged from the hospital, and returns to the LTC facility.
The authors’ observations
Ms. P’s delirium was a combination of her older age, non-renally adjusted sotalol, and CKD. At admission, the hospital treatment team first thought that pneumonia or antibiotic use could have caused delirium. However, Ms. P’s condition did not improve after antibiotics were stopped. In addition, several chest radiographs found no evidence of pneumonia. It is important to check for any source of infection because infection is a common source of delirium in older patients.1 Urine samples revealed no pathogens, a C. difficile test was negative, and the patient’s white blood cell counts remained within normal limits. Physicians began looking elsewhere for potential causes of Ms. P’s delirium.
Ms. P’s vital signs ruled out a temperature irregularity or hypertension as the cause of her delirium. She has a slightly low oxygen saturation when she first presented, but this quickly returned to normal with administration of oxygen, which ruled out hypoxemia. Laboratory results concluded that Ms. P’s glucose levels were within a normal range and she had no electrolyte imbalances. A head CT scan showed slight atrophy of white matter that is consistent with Ms. P’s age. The head CT scan also showed that Ms. P had no acute condition or head trauma.
In terms of organ function, Ms. P was in relatively healthy condition other than paroxysmal atrial fibrillation and CKD. Chronic kidney disease can interrupt the normal pharmacokinetics of medications. Reviewing Ms. P’s medication list, several agents could have induced delirium, including antidepressants, antipsychotics, cardiovascular medications (beta blocker/antiarrhythmic [sotalol]), and opioid analgesics such as tramadol.5 Ms. P’s condition did not improve after discontinuing fluoxetine, risperidone, or olanzapine, although haloperidol was started in their place. Ms. P scored an 8 on the Naranjo Adverse Drug Reaction Probability Scale, indicating this event was a probable adverse drug reaction.9
Identifying a cause
This was a unique case where sotalol was identified as the culprit for inducing Ms. P’s delirium, because her age and CKD are irreversible. It is important to note that antiarrhythmics can induce arrhythmias when present in high concentrations or administered without appropriate renal dose adjustments. Although Ms. P’s serum levels of sotalol were not evaluated, because of her renal impairment, it is possible that toxic levels of sotalol accumulated and lead to arrhythmias and delirium. Of note, a cardiologist was consulted to safely change Ms. P to a calcium channel blocker so she could undergo cardiac monitoring. With the addition of diltiazem and metoprolol, the patient’s delirium subsided and her arrhythmia was controlled. Once the source of Ms. P’s delirium had been identified, antipsychotics were no longer needed.
Continue to: Bottom Line
Bottom Line
Delirium is a complex disorder that often has multiple causes, both reversible and irreversible. A “process of elimination” approach should be used to accurately identify and manage delirium. If a patient with delirium has little to no response to antipsychotic medications, the underlying cause or causes likely has not yet been addressed, and the evaluation should continue.
Related Resources
- Marcantonio ER. Delirium in hospitalized older adults. N Engl J Med. 2017;377:1456-1466.
- Inouye SK, Westendorp RGJ, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
Drug Brand Names
Acyclovir • Zovirax
Alprazolam • Niravam, Xanax
Amantadine • Symmetrel
Amphotericin B • Abelcet
Atorvastatin • Lipitor
Atropine • Atropen
Baclofen • EnovaRX-Baclofen
Benztropine • Cogentin
Bromocriptine • Cycloset
Calcitonin • Miacalcin
Carbamazepine • Tegretol
Carbidopa-levodopa • Duopa
Ceftriaxone • Rocephin
Chlorpromazine • Thorazine
Clonidine • Catapres
Clopidogrel • Plavix
Cyclobenzaprine • Amrix
Digoxin • Lanoxin
Diltiazem • Cardizem
Disulfiram • Antabuse
Ezetimibe • Zetia
Fluoxetine • Prozac
Fluphenazine • Prolixin
Furosemide • Lasix
Haloperidol • Haldol
Ipratropium/albuterol nebulized solution • Combivent Respimat
Isoniazid • Isotamine
Isosorbide nitrate • Dilatrate
Levetiracetam • Keppra
Levodopa • Stalevo
Linezolid • Zyvox
Lisinopril • Zestril
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Magnesium Oxide • Mag-200
Meperidine • Demerol
Methyldopa • Aldomet
Metoprolol • Lopressor
Metronidazole • Flagyl
Mirtazapine • Remeron
Nitrofurantoin • Macrobid
Olanzapine • Zyprexa
Pantoprazole • Protonix
Phenytoin • Dilantin
Pramipexole • Mirapex
Rifampin • Rifadin
Risperidone • Risperdal
Ropinirole • Requip
Sotalol hydrochloride • Betapace AF
Tramadol • Ultram
Trihexyphenidyl • Trihexane
Valproic acid • Depakote
1. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention, and treatment. Nat Rev Neurol. 2009;5(4):210-220.
2. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
3. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
4. Francis J, Kapoor WN. Delirium in hospitalized elderly. J Gen Intern Med. 1990;5(1):65-79.
5. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
6. Cook IA. Guideline watch: practice guideline for the treatment of patients with delirium. Arlington, VA: American Psychiatric Publishing; 2004.
7. Bourgeois J, Ategan A, Losier B. Delirium in the hospital: emphasis on the management of geriatric patients. Current Psychiatry. 2014;13(8):29,36-42.
8. Betapace AF [package insert]. Zug, Switzerland: Covis Pharma; 2016.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
CASE Combative and agitated
Ms. P, age 87, presents to the emergency department (ED) with her caregiver, who says Ms. P has new-onset altered mental status, agitation, and combativeness.
Ms. P resides at a long-term care (LTC) facility, where according to the nurses she normally is pleasant, well-oriented, and cooperative. Ms. P’s medical history includes major depressive disorder, generalized anxiety disorder, hypertension, chronic kidney disease (CKD) stage III, peptic ulcer disease, gastroesophageal reflux disease, coronary artery disease with 2 past myocardial infarctions requiring stents, chronic obstructive pulmonary disease, hyperlipidemia, bradycardia requiring a pacemaker, paroxysmal atrial fibrillation, asthma, aortic stenosis, peripheral vascular disease, esophageal stricture requiring dilation, deep vein thrombosis, and migraines.
Mr. P’s medication list includes acetaminophen, 650 mg every 6 hours; ipratropium/albuterol nebulized solution, 3 mL 4 times a day; aspirin, 81 mg/d; atorvastatin, 40 mg/d; calcitonin, 1 spray nasally at bedtime; clopidogrel, 75 mg/d; ezetimibe, 10 mg/d; fluoxetine, 20 mg/d; furosemide, 20 mg/d; isosorbide dinitrate, 120 mg/d; lisinopril, 15 mg/d; risperidone, 0.5 mg/d; magnesium oxide, 800 mg/d; pantoprazole, 40 mg/d; polyethylene glycol, 17 g/d; sotalol, 160 mg/d; olanzapine, 5 mg IM every 6 hours as needed for agitation; and tramadol, 50 mg every 8 hours as needed for headache.
Seven days before coming to the ED, Ms. P was started on ceftriaxone, 1 g/d, for suspected community-acquired pneumonia. At that time, the nursing staff noticed behavioral changes. Soon after, Ms. P began refusing all her medications. Two days before presenting to the ED, Ms. P was started on nitrofurantoin, 200 mg/d, for a suspected urinary tract infection, but it was discontinued because of an allergy.
Her caregiver reports that while at the LTC facility, Ms. P’s behavioral changes worsened. Ms. P claimed to be Jesus Christ and said she was talking to the devil; she chased other residents around the facility and slapped medications away from the nursing staff. According to caregivers, this behavior was out of character.
Shortly after arriving in the ED, Ms. P is admitted to the psychiatric unit.
[polldaddy:10332748]
The authors’ observations
Delirium is a complex, acute alteration in a patient’s mental status compared with his/her baseline functioning1 (Table 12). The onset of delirium is quick, happening within hours to days, with fluctuations in mental function. Patients might present with hyperactive, hypoactive, or mixed delirium.3 Patients with hyperactive delirium often have delusions and hallucinations; these patients might be agitated and could become violent with family and caregivers.3 Patients with hypoactive delirium are less likely to experience hallucinations and more likely to show symptoms of sedation.3 Patients with hypoactive delirium can be difficult to diagnose because it is challenging to interview them and understand what might be the cause of their sedated state. Patients also can exhibit a mixed delirium in which they fluctuate between periods of hyperactivity and hypoactivity.3
Continue to: Suspected delirium...
Suspected delirium should be considered a medical emergency because the outcome could be fatal.1 It is important to uncover and treat the underlying cause(s) of delirium rather than solely administering antipsychotics, which might mask the presenting symptoms. In an older study, Francis and Kapoor4 reported that 56% of geriatric patients with delirium had a single definite or probable etiology, while the other 44% had about 2.8 etiologies per patient on average. Delirium risk factors, causes, and factors to consider during patient evaluation are listed in Table 21,3,5-7 and Table 3.1,3,5-7
A synergistic relationship between comorbidities, environment, and medications can induce delirium.5 Identifying irreversible and reversible causes is the key to treating delirium. After the cause has been identified, it can be addressed and the patient could return to his/her previous level of functioning. If the delirium is the result of multiple irreversible causes, it could become chronic.
[polldaddy:10332749]
EVALUATION Cardiac dysfunction
Ms. P undergoes laboratory testing. The results include: white blood cell count, 5.9/µL; hemoglobin, 13.6 g/dL; hematocrit, 42.6%; platelets, 304 × 103/µL; sodium,143 mEq/L; potassium, 3.2 mEq/L; chloride, 96 mEq/L; carbon dioxide, 23 mEq/L; blood glucose, 87 mg/dL; creatinine, 1.2 mg/dL; estimated creatinine clearance (eCrCl) level of 33 mL/min/1.73 m2; calcium, 9.5 mg/dL; albumin, 3.6 g/dL; liver enzymes within normal limits; thyroid-stimulating hormone, 0.78 mIU/L; vitamin B12, 995 pg/mL; folic acid, 16.6 ng/mL; vitamin D, 31 pg/mL; and rapid plasma reagin: nonreactive. Urinalysis is unremarkable, and no culture is performed. Urine drug screening/toxicology is positive for the benzodiazepines that she received in the ED (oral alprazolam 0.25 mg given once and oral lorazepam 0.5 mg given once).
Electrocardiogram (ECG) shows atrial flutter/tachycardia with rapid ventricular response, marked left axis deviation, nonspecific ST- and T-wave abnormality, QT/QTC of 301/387 ms, and ventricular rate 151 beats per minute. A CT scan of the head and brain without contrast shows mild atrophy and chronic white matter changes and no acute intracranial abnormality. A two-view chest radiography shows no acute cardiopulmonary findings. Her temperature is 98.4°F; heart rate is 122 beats per minute; respiratory rate is 20 breaths per minute; blood pressure is 161/98 mm Hg; and oxygen saturation is 86% on room air.
Based on this data, Ms. P’s cardiac condition seems to be worsening, which is thought to be caused by her refusal of furosemide, lisinopril, isosorbide, sotalol, clopidogrel, and aspirin. The treatment team plans to work on compliance to resolve these cardiac issues and places Ms. P on 1:1 observation with a sitter and music in attempt to calm her.
Continue to: The authors' observations
The authors’ observations
Many factors can contribute to behavioral or cognitive changes in geriatric patients. Often, a major change noted in an older patient can be attributed to new-onset dementia, dementia with behavioral disturbances, delirium, depression, or acute psychosis. These potential causes should be considered and ruled out in a step-by-step progression. Because patients are unreliable historians during acute distress, a complete history from family or caregivers and exhaustive workup is paramount.
TREATMENT Medication adjustments
In an attempt to resolve Ms. P’s disruptive behaviors, her risperidone dosage is changed to 0.5 mg twice daily. Ms. P is encouraged to use the provided oxygen to raise her saturation level.
On hospital Day 3, a loose stool prompts a Clostridium difficile test as a possible source of delirium; however, the results are negative.
On hospital Day 4, Ms. P is confused and irritable overnight, yelling profanities at staff, refusing care, inappropriately disrobing, and having difficulty falling asleep and staying asleep. Risperidone is discontinued because it appears to have had little or no effect on Ms. P’s disruptive behaviors. Olanzapine, 10 mg/d, is initiated with mirtazapine, 7.5 mg/d, to help with mood, appetite, and sleep. Fluoxetine is also discontinued because of a possible interaction with clopidogrel.
On hospital Days 6 to 8, Ms. P remains upset and unable to follow instructions. Melatonin is initiated to improve her sleep cycle. On Day 9, she continues to decline and is cursing at hospital staff; haloperidol is initiated at 5 mg every morning, 10 mg at bedtime, and 5 mg IM as needed for agitation. Her sleep improves with melatonin and mirtazapine. IV hydration also is initiated. Ms. P has a slight improvement in medication compliance. On Day 11, haloperidol is increased to 5 mg in the morning, 5 mg in the afternoon, and 10 mg at bedtime. On Day 12, haloperidol is changed to 7.5 mg twice daily; a slight improvement in Ms. P’s behavior is noted.
Continue to: On hospital Day 13...
On hospital Day 13, Ms. P’s behavior declines again. She screams profanities at staff and does not recognize the clinicians who have been providing care to her. The physician initiates valproic acid, 125 mg, 3 times a day, to target Ms. P’s behavioral disturbances. A pharmacist notes that the patient’s sotalol could be contributing to Ms. P’s psychiatric presentation, and that based on her eCrCl level of 33 mL/min/1.73 m2, a dosage adjustment or medication change might be warranted.
On Day 14, Ms. P displays erratic behavior and intermittent tachycardia. A cardiac consultation is ordered. A repeat ECG reveals atrial fibrillation with rapid rate and a QT/QTc of 409/432 ms. Ms. P is transferred to the telemetry unit, where the cardiologist discontinues sotalol because the dosage is not properly renally adjusted. Sotalol hydrochloride has been associated with life-threatening ventricular tachycardia.8 Diltiazem, 30 mg every 6 hours is initiated to replace sotalol.
By Day 16, the treatment team notes improved cognition and behavior. On Day 17, the cardiologist reports that Ms. P’s atrial fibrillation is controlled. An ECG reveals mild left ventricular hypertrophy, an ejection fraction of 50% to 55%, no stenosis in the mitral or tricuspid valves, no valvular pulmonic stenosis, and moderate aortic sclerosis. Cardiac markers also are evaluated (creatinine phosphokinase: 105 U/L; creatinine kinase–MB fraction: 2.6 ng/mL; troponin: 0.01 ng/mL; pro-B-type natriuretic peptide: 2,073 pg/mL); and myocardial infarction is ruled out.
On Day 19, Ms. P’s diltiazem is consolidated to a controlled-delivery formulation, 180 mg/d, along with the addition of metoprolol, 12.5 mg twice daily. Ms. P is transferred back to the psychiatric unit.
OUTCOME Gradual improvement
On Days 20 to 23, Ms. P shows remarkable progress, and her mental status, cognition, and behavior slowly return to baseline. Haloperidol and valproic acid are tapered and discontinued. Ms. P is observed to be healthy and oriented to person, place, and time.
Continue to: On Day 25...
On Day 25, she is discharged from the hospital, and returns to the LTC facility.
The authors’ observations
Ms. P’s delirium was a combination of her older age, non-renally adjusted sotalol, and CKD. At admission, the hospital treatment team first thought that pneumonia or antibiotic use could have caused delirium. However, Ms. P’s condition did not improve after antibiotics were stopped. In addition, several chest radiographs found no evidence of pneumonia. It is important to check for any source of infection because infection is a common source of delirium in older patients.1 Urine samples revealed no pathogens, a C. difficile test was negative, and the patient’s white blood cell counts remained within normal limits. Physicians began looking elsewhere for potential causes of Ms. P’s delirium.
Ms. P’s vital signs ruled out a temperature irregularity or hypertension as the cause of her delirium. She has a slightly low oxygen saturation when she first presented, but this quickly returned to normal with administration of oxygen, which ruled out hypoxemia. Laboratory results concluded that Ms. P’s glucose levels were within a normal range and she had no electrolyte imbalances. A head CT scan showed slight atrophy of white matter that is consistent with Ms. P’s age. The head CT scan also showed that Ms. P had no acute condition or head trauma.
In terms of organ function, Ms. P was in relatively healthy condition other than paroxysmal atrial fibrillation and CKD. Chronic kidney disease can interrupt the normal pharmacokinetics of medications. Reviewing Ms. P’s medication list, several agents could have induced delirium, including antidepressants, antipsychotics, cardiovascular medications (beta blocker/antiarrhythmic [sotalol]), and opioid analgesics such as tramadol.5 Ms. P’s condition did not improve after discontinuing fluoxetine, risperidone, or olanzapine, although haloperidol was started in their place. Ms. P scored an 8 on the Naranjo Adverse Drug Reaction Probability Scale, indicating this event was a probable adverse drug reaction.9
Identifying a cause
This was a unique case where sotalol was identified as the culprit for inducing Ms. P’s delirium, because her age and CKD are irreversible. It is important to note that antiarrhythmics can induce arrhythmias when present in high concentrations or administered without appropriate renal dose adjustments. Although Ms. P’s serum levels of sotalol were not evaluated, because of her renal impairment, it is possible that toxic levels of sotalol accumulated and lead to arrhythmias and delirium. Of note, a cardiologist was consulted to safely change Ms. P to a calcium channel blocker so she could undergo cardiac monitoring. With the addition of diltiazem and metoprolol, the patient’s delirium subsided and her arrhythmia was controlled. Once the source of Ms. P’s delirium had been identified, antipsychotics were no longer needed.
Continue to: Bottom Line
Bottom Line
Delirium is a complex disorder that often has multiple causes, both reversible and irreversible. A “process of elimination” approach should be used to accurately identify and manage delirium. If a patient with delirium has little to no response to antipsychotic medications, the underlying cause or causes likely has not yet been addressed, and the evaluation should continue.
Related Resources
- Marcantonio ER. Delirium in hospitalized older adults. N Engl J Med. 2017;377:1456-1466.
- Inouye SK, Westendorp RGJ, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
Drug Brand Names
Acyclovir • Zovirax
Alprazolam • Niravam, Xanax
Amantadine • Symmetrel
Amphotericin B • Abelcet
Atorvastatin • Lipitor
Atropine • Atropen
Baclofen • EnovaRX-Baclofen
Benztropine • Cogentin
Bromocriptine • Cycloset
Calcitonin • Miacalcin
Carbamazepine • Tegretol
Carbidopa-levodopa • Duopa
Ceftriaxone • Rocephin
Chlorpromazine • Thorazine
Clonidine • Catapres
Clopidogrel • Plavix
Cyclobenzaprine • Amrix
Digoxin • Lanoxin
Diltiazem • Cardizem
Disulfiram • Antabuse
Ezetimibe • Zetia
Fluoxetine • Prozac
Fluphenazine • Prolixin
Furosemide • Lasix
Haloperidol • Haldol
Ipratropium/albuterol nebulized solution • Combivent Respimat
Isoniazid • Isotamine
Isosorbide nitrate • Dilatrate
Levetiracetam • Keppra
Levodopa • Stalevo
Linezolid • Zyvox
Lisinopril • Zestril
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Magnesium Oxide • Mag-200
Meperidine • Demerol
Methyldopa • Aldomet
Metoprolol • Lopressor
Metronidazole • Flagyl
Mirtazapine • Remeron
Nitrofurantoin • Macrobid
Olanzapine • Zyprexa
Pantoprazole • Protonix
Phenytoin • Dilantin
Pramipexole • Mirapex
Rifampin • Rifadin
Risperidone • Risperdal
Ropinirole • Requip
Sotalol hydrochloride • Betapace AF
Tramadol • Ultram
Trihexyphenidyl • Trihexane
Valproic acid • Depakote
CASE Combative and agitated
Ms. P, age 87, presents to the emergency department (ED) with her caregiver, who says Ms. P has new-onset altered mental status, agitation, and combativeness.
Ms. P resides at a long-term care (LTC) facility, where according to the nurses she normally is pleasant, well-oriented, and cooperative. Ms. P’s medical history includes major depressive disorder, generalized anxiety disorder, hypertension, chronic kidney disease (CKD) stage III, peptic ulcer disease, gastroesophageal reflux disease, coronary artery disease with 2 past myocardial infarctions requiring stents, chronic obstructive pulmonary disease, hyperlipidemia, bradycardia requiring a pacemaker, paroxysmal atrial fibrillation, asthma, aortic stenosis, peripheral vascular disease, esophageal stricture requiring dilation, deep vein thrombosis, and migraines.
Mr. P’s medication list includes acetaminophen, 650 mg every 6 hours; ipratropium/albuterol nebulized solution, 3 mL 4 times a day; aspirin, 81 mg/d; atorvastatin, 40 mg/d; calcitonin, 1 spray nasally at bedtime; clopidogrel, 75 mg/d; ezetimibe, 10 mg/d; fluoxetine, 20 mg/d; furosemide, 20 mg/d; isosorbide dinitrate, 120 mg/d; lisinopril, 15 mg/d; risperidone, 0.5 mg/d; magnesium oxide, 800 mg/d; pantoprazole, 40 mg/d; polyethylene glycol, 17 g/d; sotalol, 160 mg/d; olanzapine, 5 mg IM every 6 hours as needed for agitation; and tramadol, 50 mg every 8 hours as needed for headache.
Seven days before coming to the ED, Ms. P was started on ceftriaxone, 1 g/d, for suspected community-acquired pneumonia. At that time, the nursing staff noticed behavioral changes. Soon after, Ms. P began refusing all her medications. Two days before presenting to the ED, Ms. P was started on nitrofurantoin, 200 mg/d, for a suspected urinary tract infection, but it was discontinued because of an allergy.
Her caregiver reports that while at the LTC facility, Ms. P’s behavioral changes worsened. Ms. P claimed to be Jesus Christ and said she was talking to the devil; she chased other residents around the facility and slapped medications away from the nursing staff. According to caregivers, this behavior was out of character.
Shortly after arriving in the ED, Ms. P is admitted to the psychiatric unit.
[polldaddy:10332748]
The authors’ observations
Delirium is a complex, acute alteration in a patient’s mental status compared with his/her baseline functioning1 (Table 12). The onset of delirium is quick, happening within hours to days, with fluctuations in mental function. Patients might present with hyperactive, hypoactive, or mixed delirium.3 Patients with hyperactive delirium often have delusions and hallucinations; these patients might be agitated and could become violent with family and caregivers.3 Patients with hypoactive delirium are less likely to experience hallucinations and more likely to show symptoms of sedation.3 Patients with hypoactive delirium can be difficult to diagnose because it is challenging to interview them and understand what might be the cause of their sedated state. Patients also can exhibit a mixed delirium in which they fluctuate between periods of hyperactivity and hypoactivity.3
Continue to: Suspected delirium...
Suspected delirium should be considered a medical emergency because the outcome could be fatal.1 It is important to uncover and treat the underlying cause(s) of delirium rather than solely administering antipsychotics, which might mask the presenting symptoms. In an older study, Francis and Kapoor4 reported that 56% of geriatric patients with delirium had a single definite or probable etiology, while the other 44% had about 2.8 etiologies per patient on average. Delirium risk factors, causes, and factors to consider during patient evaluation are listed in Table 21,3,5-7 and Table 3.1,3,5-7
A synergistic relationship between comorbidities, environment, and medications can induce delirium.5 Identifying irreversible and reversible causes is the key to treating delirium. After the cause has been identified, it can be addressed and the patient could return to his/her previous level of functioning. If the delirium is the result of multiple irreversible causes, it could become chronic.
[polldaddy:10332749]
EVALUATION Cardiac dysfunction
Ms. P undergoes laboratory testing. The results include: white blood cell count, 5.9/µL; hemoglobin, 13.6 g/dL; hematocrit, 42.6%; platelets, 304 × 103/µL; sodium,143 mEq/L; potassium, 3.2 mEq/L; chloride, 96 mEq/L; carbon dioxide, 23 mEq/L; blood glucose, 87 mg/dL; creatinine, 1.2 mg/dL; estimated creatinine clearance (eCrCl) level of 33 mL/min/1.73 m2; calcium, 9.5 mg/dL; albumin, 3.6 g/dL; liver enzymes within normal limits; thyroid-stimulating hormone, 0.78 mIU/L; vitamin B12, 995 pg/mL; folic acid, 16.6 ng/mL; vitamin D, 31 pg/mL; and rapid plasma reagin: nonreactive. Urinalysis is unremarkable, and no culture is performed. Urine drug screening/toxicology is positive for the benzodiazepines that she received in the ED (oral alprazolam 0.25 mg given once and oral lorazepam 0.5 mg given once).
Electrocardiogram (ECG) shows atrial flutter/tachycardia with rapid ventricular response, marked left axis deviation, nonspecific ST- and T-wave abnormality, QT/QTC of 301/387 ms, and ventricular rate 151 beats per minute. A CT scan of the head and brain without contrast shows mild atrophy and chronic white matter changes and no acute intracranial abnormality. A two-view chest radiography shows no acute cardiopulmonary findings. Her temperature is 98.4°F; heart rate is 122 beats per minute; respiratory rate is 20 breaths per minute; blood pressure is 161/98 mm Hg; and oxygen saturation is 86% on room air.
Based on this data, Ms. P’s cardiac condition seems to be worsening, which is thought to be caused by her refusal of furosemide, lisinopril, isosorbide, sotalol, clopidogrel, and aspirin. The treatment team plans to work on compliance to resolve these cardiac issues and places Ms. P on 1:1 observation with a sitter and music in attempt to calm her.
Continue to: The authors' observations
The authors’ observations
Many factors can contribute to behavioral or cognitive changes in geriatric patients. Often, a major change noted in an older patient can be attributed to new-onset dementia, dementia with behavioral disturbances, delirium, depression, or acute psychosis. These potential causes should be considered and ruled out in a step-by-step progression. Because patients are unreliable historians during acute distress, a complete history from family or caregivers and exhaustive workup is paramount.
TREATMENT Medication adjustments
In an attempt to resolve Ms. P’s disruptive behaviors, her risperidone dosage is changed to 0.5 mg twice daily. Ms. P is encouraged to use the provided oxygen to raise her saturation level.
On hospital Day 3, a loose stool prompts a Clostridium difficile test as a possible source of delirium; however, the results are negative.
On hospital Day 4, Ms. P is confused and irritable overnight, yelling profanities at staff, refusing care, inappropriately disrobing, and having difficulty falling asleep and staying asleep. Risperidone is discontinued because it appears to have had little or no effect on Ms. P’s disruptive behaviors. Olanzapine, 10 mg/d, is initiated with mirtazapine, 7.5 mg/d, to help with mood, appetite, and sleep. Fluoxetine is also discontinued because of a possible interaction with clopidogrel.
On hospital Days 6 to 8, Ms. P remains upset and unable to follow instructions. Melatonin is initiated to improve her sleep cycle. On Day 9, she continues to decline and is cursing at hospital staff; haloperidol is initiated at 5 mg every morning, 10 mg at bedtime, and 5 mg IM as needed for agitation. Her sleep improves with melatonin and mirtazapine. IV hydration also is initiated. Ms. P has a slight improvement in medication compliance. On Day 11, haloperidol is increased to 5 mg in the morning, 5 mg in the afternoon, and 10 mg at bedtime. On Day 12, haloperidol is changed to 7.5 mg twice daily; a slight improvement in Ms. P’s behavior is noted.
Continue to: On hospital Day 13...
On hospital Day 13, Ms. P’s behavior declines again. She screams profanities at staff and does not recognize the clinicians who have been providing care to her. The physician initiates valproic acid, 125 mg, 3 times a day, to target Ms. P’s behavioral disturbances. A pharmacist notes that the patient’s sotalol could be contributing to Ms. P’s psychiatric presentation, and that based on her eCrCl level of 33 mL/min/1.73 m2, a dosage adjustment or medication change might be warranted.
On Day 14, Ms. P displays erratic behavior and intermittent tachycardia. A cardiac consultation is ordered. A repeat ECG reveals atrial fibrillation with rapid rate and a QT/QTc of 409/432 ms. Ms. P is transferred to the telemetry unit, where the cardiologist discontinues sotalol because the dosage is not properly renally adjusted. Sotalol hydrochloride has been associated with life-threatening ventricular tachycardia.8 Diltiazem, 30 mg every 6 hours is initiated to replace sotalol.
By Day 16, the treatment team notes improved cognition and behavior. On Day 17, the cardiologist reports that Ms. P’s atrial fibrillation is controlled. An ECG reveals mild left ventricular hypertrophy, an ejection fraction of 50% to 55%, no stenosis in the mitral or tricuspid valves, no valvular pulmonic stenosis, and moderate aortic sclerosis. Cardiac markers also are evaluated (creatinine phosphokinase: 105 U/L; creatinine kinase–MB fraction: 2.6 ng/mL; troponin: 0.01 ng/mL; pro-B-type natriuretic peptide: 2,073 pg/mL); and myocardial infarction is ruled out.
On Day 19, Ms. P’s diltiazem is consolidated to a controlled-delivery formulation, 180 mg/d, along with the addition of metoprolol, 12.5 mg twice daily. Ms. P is transferred back to the psychiatric unit.
OUTCOME Gradual improvement
On Days 20 to 23, Ms. P shows remarkable progress, and her mental status, cognition, and behavior slowly return to baseline. Haloperidol and valproic acid are tapered and discontinued. Ms. P is observed to be healthy and oriented to person, place, and time.
Continue to: On Day 25...
On Day 25, she is discharged from the hospital, and returns to the LTC facility.
The authors’ observations
Ms. P’s delirium was a combination of her older age, non-renally adjusted sotalol, and CKD. At admission, the hospital treatment team first thought that pneumonia or antibiotic use could have caused delirium. However, Ms. P’s condition did not improve after antibiotics were stopped. In addition, several chest radiographs found no evidence of pneumonia. It is important to check for any source of infection because infection is a common source of delirium in older patients.1 Urine samples revealed no pathogens, a C. difficile test was negative, and the patient’s white blood cell counts remained within normal limits. Physicians began looking elsewhere for potential causes of Ms. P’s delirium.
Ms. P’s vital signs ruled out a temperature irregularity or hypertension as the cause of her delirium. She has a slightly low oxygen saturation when she first presented, but this quickly returned to normal with administration of oxygen, which ruled out hypoxemia. Laboratory results concluded that Ms. P’s glucose levels were within a normal range and she had no electrolyte imbalances. A head CT scan showed slight atrophy of white matter that is consistent with Ms. P’s age. The head CT scan also showed that Ms. P had no acute condition or head trauma.
In terms of organ function, Ms. P was in relatively healthy condition other than paroxysmal atrial fibrillation and CKD. Chronic kidney disease can interrupt the normal pharmacokinetics of medications. Reviewing Ms. P’s medication list, several agents could have induced delirium, including antidepressants, antipsychotics, cardiovascular medications (beta blocker/antiarrhythmic [sotalol]), and opioid analgesics such as tramadol.5 Ms. P’s condition did not improve after discontinuing fluoxetine, risperidone, or olanzapine, although haloperidol was started in their place. Ms. P scored an 8 on the Naranjo Adverse Drug Reaction Probability Scale, indicating this event was a probable adverse drug reaction.9
Identifying a cause
This was a unique case where sotalol was identified as the culprit for inducing Ms. P’s delirium, because her age and CKD are irreversible. It is important to note that antiarrhythmics can induce arrhythmias when present in high concentrations or administered without appropriate renal dose adjustments. Although Ms. P’s serum levels of sotalol were not evaluated, because of her renal impairment, it is possible that toxic levels of sotalol accumulated and lead to arrhythmias and delirium. Of note, a cardiologist was consulted to safely change Ms. P to a calcium channel blocker so she could undergo cardiac monitoring. With the addition of diltiazem and metoprolol, the patient’s delirium subsided and her arrhythmia was controlled. Once the source of Ms. P’s delirium had been identified, antipsychotics were no longer needed.
Continue to: Bottom Line
Bottom Line
Delirium is a complex disorder that often has multiple causes, both reversible and irreversible. A “process of elimination” approach should be used to accurately identify and manage delirium. If a patient with delirium has little to no response to antipsychotic medications, the underlying cause or causes likely has not yet been addressed, and the evaluation should continue.
Related Resources
- Marcantonio ER. Delirium in hospitalized older adults. N Engl J Med. 2017;377:1456-1466.
- Inouye SK, Westendorp RGJ, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
Drug Brand Names
Acyclovir • Zovirax
Alprazolam • Niravam, Xanax
Amantadine • Symmetrel
Amphotericin B • Abelcet
Atorvastatin • Lipitor
Atropine • Atropen
Baclofen • EnovaRX-Baclofen
Benztropine • Cogentin
Bromocriptine • Cycloset
Calcitonin • Miacalcin
Carbamazepine • Tegretol
Carbidopa-levodopa • Duopa
Ceftriaxone • Rocephin
Chlorpromazine • Thorazine
Clonidine • Catapres
Clopidogrel • Plavix
Cyclobenzaprine • Amrix
Digoxin • Lanoxin
Diltiazem • Cardizem
Disulfiram • Antabuse
Ezetimibe • Zetia
Fluoxetine • Prozac
Fluphenazine • Prolixin
Furosemide • Lasix
Haloperidol • Haldol
Ipratropium/albuterol nebulized solution • Combivent Respimat
Isoniazid • Isotamine
Isosorbide nitrate • Dilatrate
Levetiracetam • Keppra
Levodopa • Stalevo
Linezolid • Zyvox
Lisinopril • Zestril
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Magnesium Oxide • Mag-200
Meperidine • Demerol
Methyldopa • Aldomet
Metoprolol • Lopressor
Metronidazole • Flagyl
Mirtazapine • Remeron
Nitrofurantoin • Macrobid
Olanzapine • Zyprexa
Pantoprazole • Protonix
Phenytoin • Dilantin
Pramipexole • Mirapex
Rifampin • Rifadin
Risperidone • Risperdal
Ropinirole • Requip
Sotalol hydrochloride • Betapace AF
Tramadol • Ultram
Trihexyphenidyl • Trihexane
Valproic acid • Depakote
1. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention, and treatment. Nat Rev Neurol. 2009;5(4):210-220.
2. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
3. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
4. Francis J, Kapoor WN. Delirium in hospitalized elderly. J Gen Intern Med. 1990;5(1):65-79.
5. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
6. Cook IA. Guideline watch: practice guideline for the treatment of patients with delirium. Arlington, VA: American Psychiatric Publishing; 2004.
7. Bourgeois J, Ategan A, Losier B. Delirium in the hospital: emphasis on the management of geriatric patients. Current Psychiatry. 2014;13(8):29,36-42.
8. Betapace AF [package insert]. Zug, Switzerland: Covis Pharma; 2016.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
1. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention, and treatment. Nat Rev Neurol. 2009;5(4):210-220.
2. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
3. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
4. Francis J, Kapoor WN. Delirium in hospitalized elderly. J Gen Intern Med. 1990;5(1):65-79.
5. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
6. Cook IA. Guideline watch: practice guideline for the treatment of patients with delirium. Arlington, VA: American Psychiatric Publishing; 2004.
7. Bourgeois J, Ategan A, Losier B. Delirium in the hospital: emphasis on the management of geriatric patients. Current Psychiatry. 2014;13(8):29,36-42.
8. Betapace AF [package insert]. Zug, Switzerland: Covis Pharma; 2016.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.

Coding variants in apolipoprotein B may be associated with early-onset Alzheimer’s disease
Variants in the apolipoprotein B gene (APOB), which creates the main protein in low-density and very low-density cholesterol, may be associated with early-onset Alzheimer’s disease, Thomas Wingo, MD, and his colleagues have determined.

The finding may help fill out the genetic risk picture for early-onset Alzheimer’s disease (EOAD), said Dr. Wingo of the Atlanta Veterans Affairs Medical Center. The study found that the already-known genetic markers for EOAD – mutations of the presenilin (PSEN) 1 and 2 genes and amyloid precursor protein (APP) – account for just a small fraction of cases.
“To place the genetic association between APOB and EOAD in context, we note that only 3.4% of all EOAD cases in our combined data set showed a known pathogenic mutation, and we found a stronger association between EOAD and rare coding variants in APOB, compared with PSEN1 in our fully adjusted analysis,” the team wrote. However, “approximately 5.0% of patients with EOAD and 1.7% of controls were found to harbor a rare coding polymorphism in APOB that is likely to disrupt the structure, functions, or abundance of ApoB protein.”
The team conducted genetic analysis on plasma samples from 2,125 EOAD and control subjects included in several research cohorts. They first determined the association between cholesterol and EOAD, and then the frequency of variants in apolipoprotein E epsilon 4 (APOE e4), APP, PSEN1, PSEN2, and ApoB. Gene sequencing revealed that 3.4% of samples showed mutations in APP, PSEN1, or PSEN2.
“Given the strong associations between APOE e4 and EOAD and elevated circulating LDL cholesterol levels, we expected individuals with EOAD to have elevated LDL levels,” the team said. But an analysis of 267 of the samples for lipid levels found that, even after the researchers controlled for APOE e4, EOAD cases had higher total cholesterol, low-density cholesterol, and plasma ApoB, compared with controls. However, they found no association between EOAD and high-density lipoprotein or triglycerides.
“Because total cholesterol largely consists of LDL-C, and ApoB is the main lipoprotein of LDL-C, these findings are consistent with one another.
“From these data, we estimated that LDL-C explains 7.6% of the variance in liability to EOAD, independently of APOE e4 ... These results demonstrate that elevated levels of LDL-C [and ApoB] were significantly associated with increased EOAD risk, and this effect was only partially mediated by APOE e4 genotype.”
The results also raised a question: What was driving the association between LDL and EOAD? Because variants of the ApoB gene can either raise or lower LDL, the team examined variants associated with coding changes. These variants were significantly more common in EOAD cases than in controls (5.0% vs. 1.7%).
“Two affected individuals ... were compound heterozygotes, with the remainder being heterozygotes,” the researchers wrote. “Each compound heterozygote case was heterozygous for two different rare coding sites ... Of these four variants, only [one] has been previously described.”
“Our finding of a significant association between rare coding variants in APOB and EOAD independently of APOE is novel, important, and consistent with multiple genome-wide association studies that revealed strong associations between late-onset AD and common intron markers of genes involved in brain cholesterol metabolism [ABCA7, BIN1, CLU, and SORL1]. Furthermore, mice overexpressing ApoB show hyperlipidemia, neurodegeneration, increases in APP, accumulation of amyloid plaques, and cognitive impairment similar to mice overexpressing wild-type human APP. Collectively, these studies and our findings suggest an important role of cholesterol metabolism in AD pathogenesis.”
This research was supported by grants from the Veterans Health Administration, the National Institutes of Health, the To Remember Foundation, the Douglas French Alzheimer’s Foundation, and a contract with the State of California Department of Health Services. Several authors reported financial ties to pharmaceutical companies outside of this work.
SOURCE: Wingo TS et al. JAMA Neurol. 2019 May 28. doi: 10.1001/jamaneurol.2019.0648.
This important study provides the first evidence that rare genetic coding variants of apolipoprotein B may contribute to the risk of early-onset Alzheimer’s disease, Makoto Ishii, MD, PhD, wrote in an accompanying editorial (JAMA Neurol. 2019 May 28. doi: 10.1001/jamaneurol.2019.0212).
But the study by Wingo et al. doesn’t tell the entire tale, he wrote.
The results from this study “found that there are likely to be additional contributing factors independent of APOB and APOE. These may include rare variants in other genes involved directly in LDL cholesterol metabolism, such as the LDL receptor and proprotein convertase subtilisin/kexin type 913 or factors known to modulate circulating LDL cholesterol levels, such as thyroid hormones.”
Although intriguing, “Clearly, additional studies looking at these factors are needed to fully elucidate the association between LDL cholesterol and EOAD. Furthermore, as the authors of this study note, it is not known if there are protective variants of APOB that would decrease the risk for developing EOAD. Identifying such a protective coding variant of APOB would greatly strengthen the link between APOB and AD pathogenesis.”
Prior studies of circulating APOB levels in humans have reached disparate conclusions. A large population-based study found no association between APOB levels and incident dementia or Alzheimer’s, he noted.
“Therefore, whether these findings can be verified in individuals with late-onset AD remains to be determined.”
Dr. Ishii is with the Feil Family Brain and Mind Research Institute in the department of neurology at Cornell University, New York. He has no relevant disclosures.
This important study provides the first evidence that rare genetic coding variants of apolipoprotein B may contribute to the risk of early-onset Alzheimer’s disease, Makoto Ishii, MD, PhD, wrote in an accompanying editorial (JAMA Neurol. 2019 May 28. doi: 10.1001/jamaneurol.2019.0212).
But the study by Wingo et al. doesn’t tell the entire tale, he wrote.
The results from this study “found that there are likely to be additional contributing factors independent of APOB and APOE. These may include rare variants in other genes involved directly in LDL cholesterol metabolism, such as the LDL receptor and proprotein convertase subtilisin/kexin type 913 or factors known to modulate circulating LDL cholesterol levels, such as thyroid hormones.”
Although intriguing, “Clearly, additional studies looking at these factors are needed to fully elucidate the association between LDL cholesterol and EOAD. Furthermore, as the authors of this study note, it is not known if there are protective variants of APOB that would decrease the risk for developing EOAD. Identifying such a protective coding variant of APOB would greatly strengthen the link between APOB and AD pathogenesis.”
Prior studies of circulating APOB levels in humans have reached disparate conclusions. A large population-based study found no association between APOB levels and incident dementia or Alzheimer’s, he noted.
“Therefore, whether these findings can be verified in individuals with late-onset AD remains to be determined.”
Dr. Ishii is with the Feil Family Brain and Mind Research Institute in the department of neurology at Cornell University, New York. He has no relevant disclosures.
This important study provides the first evidence that rare genetic coding variants of apolipoprotein B may contribute to the risk of early-onset Alzheimer’s disease, Makoto Ishii, MD, PhD, wrote in an accompanying editorial (JAMA Neurol. 2019 May 28. doi: 10.1001/jamaneurol.2019.0212).
But the study by Wingo et al. doesn’t tell the entire tale, he wrote.
The results from this study “found that there are likely to be additional contributing factors independent of APOB and APOE. These may include rare variants in other genes involved directly in LDL cholesterol metabolism, such as the LDL receptor and proprotein convertase subtilisin/kexin type 913 or factors known to modulate circulating LDL cholesterol levels, such as thyroid hormones.”
Although intriguing, “Clearly, additional studies looking at these factors are needed to fully elucidate the association between LDL cholesterol and EOAD. Furthermore, as the authors of this study note, it is not known if there are protective variants of APOB that would decrease the risk for developing EOAD. Identifying such a protective coding variant of APOB would greatly strengthen the link between APOB and AD pathogenesis.”
Prior studies of circulating APOB levels in humans have reached disparate conclusions. A large population-based study found no association between APOB levels and incident dementia or Alzheimer’s, he noted.
“Therefore, whether these findings can be verified in individuals with late-onset AD remains to be determined.”
Dr. Ishii is with the Feil Family Brain and Mind Research Institute in the department of neurology at Cornell University, New York. He has no relevant disclosures.
Variants in the apolipoprotein B gene (APOB), which creates the main protein in low-density and very low-density cholesterol, may be associated with early-onset Alzheimer’s disease, Thomas Wingo, MD, and his colleagues have determined.
The finding may help fill out the genetic risk picture for early-onset Alzheimer’s disease (EOAD), said Dr. Wingo of the Atlanta Veterans Affairs Medical Center. The study found that the already-known genetic markers for EOAD – mutations of the presenilin (PSEN) 1 and 2 genes and amyloid precursor protein (APP) – account for just a small fraction of cases.
“To place the genetic association between APOB and EOAD in context, we note that only 3.4% of all EOAD cases in our combined data set showed a known pathogenic mutation, and we found a stronger association between EOAD and rare coding variants in APOB, compared with PSEN1 in our fully adjusted analysis,” the team wrote. However, “approximately 5.0% of patients with EOAD and 1.7% of controls were found to harbor a rare coding polymorphism in APOB that is likely to disrupt the structure, functions, or abundance of ApoB protein.”
The team conducted genetic analysis on plasma samples from 2,125 EOAD and control subjects included in several research cohorts. They first determined the association between cholesterol and EOAD, and then the frequency of variants in apolipoprotein E epsilon 4 (APOE e4), APP, PSEN1, PSEN2, and ApoB. Gene sequencing revealed that 3.4% of samples showed mutations in APP, PSEN1, or PSEN2.
“Given the strong associations between APOE e4 and EOAD and elevated circulating LDL cholesterol levels, we expected individuals with EOAD to have elevated LDL levels,” the team said. But an analysis of 267 of the samples for lipid levels found that, even after the researchers controlled for APOE e4, EOAD cases had higher total cholesterol, low-density cholesterol, and plasma ApoB, compared with controls. However, they found no association between EOAD and high-density lipoprotein or triglycerides.
“Because total cholesterol largely consists of LDL-C, and ApoB is the main lipoprotein of LDL-C, these findings are consistent with one another.
“From these data, we estimated that LDL-C explains 7.6% of the variance in liability to EOAD, independently of APOE e4 ... These results demonstrate that elevated levels of LDL-C [and ApoB] were significantly associated with increased EOAD risk, and this effect was only partially mediated by APOE e4 genotype.”
The results also raised a question: What was driving the association between LDL and EOAD? Because variants of the ApoB gene can either raise or lower LDL, the team examined variants associated with coding changes. These variants were significantly more common in EOAD cases than in controls (5.0% vs. 1.7%).
“Two affected individuals ... were compound heterozygotes, with the remainder being heterozygotes,” the researchers wrote. “Each compound heterozygote case was heterozygous for two different rare coding sites ... Of these four variants, only [one] has been previously described.”
“Our finding of a significant association between rare coding variants in APOB and EOAD independently of APOE is novel, important, and consistent with multiple genome-wide association studies that revealed strong associations between late-onset AD and common intron markers of genes involved in brain cholesterol metabolism [ABCA7, BIN1, CLU, and SORL1]. Furthermore, mice overexpressing ApoB show hyperlipidemia, neurodegeneration, increases in APP, accumulation of amyloid plaques, and cognitive impairment similar to mice overexpressing wild-type human APP. Collectively, these studies and our findings suggest an important role of cholesterol metabolism in AD pathogenesis.”
This research was supported by grants from the Veterans Health Administration, the National Institutes of Health, the To Remember Foundation, the Douglas French Alzheimer’s Foundation, and a contract with the State of California Department of Health Services. Several authors reported financial ties to pharmaceutical companies outside of this work.
SOURCE: Wingo TS et al. JAMA Neurol. 2019 May 28. doi: 10.1001/jamaneurol.2019.0648.
Variants in the apolipoprotein B gene (APOB), which creates the main protein in low-density and very low-density cholesterol, may be associated with early-onset Alzheimer’s disease, Thomas Wingo, MD, and his colleagues have determined.
The finding may help fill out the genetic risk picture for early-onset Alzheimer’s disease (EOAD), said Dr. Wingo of the Atlanta Veterans Affairs Medical Center. The study found that the already-known genetic markers for EOAD – mutations of the presenilin (PSEN) 1 and 2 genes and amyloid precursor protein (APP) – account for just a small fraction of cases.
“To place the genetic association between APOB and EOAD in context, we note that only 3.4% of all EOAD cases in our combined data set showed a known pathogenic mutation, and we found a stronger association between EOAD and rare coding variants in APOB, compared with PSEN1 in our fully adjusted analysis,” the team wrote. However, “approximately 5.0% of patients with EOAD and 1.7% of controls were found to harbor a rare coding polymorphism in APOB that is likely to disrupt the structure, functions, or abundance of ApoB protein.”
The team conducted genetic analysis on plasma samples from 2,125 EOAD and control subjects included in several research cohorts. They first determined the association between cholesterol and EOAD, and then the frequency of variants in apolipoprotein E epsilon 4 (APOE e4), APP, PSEN1, PSEN2, and ApoB. Gene sequencing revealed that 3.4% of samples showed mutations in APP, PSEN1, or PSEN2.
“Given the strong associations between APOE e4 and EOAD and elevated circulating LDL cholesterol levels, we expected individuals with EOAD to have elevated LDL levels,” the team said. But an analysis of 267 of the samples for lipid levels found that, even after the researchers controlled for APOE e4, EOAD cases had higher total cholesterol, low-density cholesterol, and plasma ApoB, compared with controls. However, they found no association between EOAD and high-density lipoprotein or triglycerides.
“Because total cholesterol largely consists of LDL-C, and ApoB is the main lipoprotein of LDL-C, these findings are consistent with one another.
“From these data, we estimated that LDL-C explains 7.6% of the variance in liability to EOAD, independently of APOE e4 ... These results demonstrate that elevated levels of LDL-C [and ApoB] were significantly associated with increased EOAD risk, and this effect was only partially mediated by APOE e4 genotype.”
The results also raised a question: What was driving the association between LDL and EOAD? Because variants of the ApoB gene can either raise or lower LDL, the team examined variants associated with coding changes. These variants were significantly more common in EOAD cases than in controls (5.0% vs. 1.7%).
“Two affected individuals ... were compound heterozygotes, with the remainder being heterozygotes,” the researchers wrote. “Each compound heterozygote case was heterozygous for two different rare coding sites ... Of these four variants, only [one] has been previously described.”
“Our finding of a significant association between rare coding variants in APOB and EOAD independently of APOE is novel, important, and consistent with multiple genome-wide association studies that revealed strong associations between late-onset AD and common intron markers of genes involved in brain cholesterol metabolism [ABCA7, BIN1, CLU, and SORL1]. Furthermore, mice overexpressing ApoB show hyperlipidemia, neurodegeneration, increases in APP, accumulation of amyloid plaques, and cognitive impairment similar to mice overexpressing wild-type human APP. Collectively, these studies and our findings suggest an important role of cholesterol metabolism in AD pathogenesis.”
This research was supported by grants from the Veterans Health Administration, the National Institutes of Health, the To Remember Foundation, the Douglas French Alzheimer’s Foundation, and a contract with the State of California Department of Health Services. Several authors reported financial ties to pharmaceutical companies outside of this work.
SOURCE: Wingo TS et al. JAMA Neurol. 2019 May 28. doi: 10.1001/jamaneurol.2019.0648.
FROM JAMA NEUROLOGY
mTORC1 inhibitor protects elderly asthmatics from viral respiratory tract infections
DALLAS – A molecule that boosts innate viral immunity may protect elderly people with asthma from the root cause of most exacerbations – viral respiratory tract infections.

Dubbed RTB101, the oral medication is a selective, potent inhibitor of target of rapamycin complex 1 (TORC1). In phase 2b data presented at the American Thoracic Society’s international conference, RTB101 decreased by 52% the number of elderly subjects with severe, lab-confirmed respiratory tract infections (RTI) symptoms.
But the molecule was even more effective in patients with asthma aged 65 years and older, Joan Mannick, MD, said in an interview during the meeting. In this group, it reduced by 69% the percentage of subjects who developed RTIs and reduced the rate of infection by about 79%, compared with placebo.
“The core cause of asthma exacerbations in these patients is viral respiratory tract infection,” said Dr. Mannick, chief medical officer of resTORbio, the Boston company developing RTB101. “About 80% of the viruses detected in these infections are rhinoviruses, and there are 170 rhinovirus serotypes. We have never been able to develop a vaccine against rhinovirus, and we have no treatment other than to treat the inflammation caused by the infection.”
Centers for Disease Control and Prevention mortality records confirm the impact of viral respiratory infections on older people who experience asthma exacerbations: 6 of 10,000 will die, compared with less than 2 per 10,000 for all other age groups. Decreasing the number of these infections in older people with asthma would prevent morbidity and mortality and save considerable health care dollars.
“One of the reasons that asthmatics have such difficulty when they get respiratory infections is that they seem to have deficient antiviral immunity in the airways,” Dr. Mannick said. She pointed to a 2008 study of bronchial epithelial cells from both patients with asthma and healthy controls. When inoculated with rhinovirus, the cells from asthmatic airways were unable to mount a healthy immune response and were particularly deficient in producing interferon-beta.
By inhibiting mammalian TORC1 (mTORC1), RBT101 also inhibits sterol regulatory element binding transcription factor 2, a pathway that influences cholesterol synthesis. Cells perceive cholesterol synthesis attenuation as a threat, Dr. Mannick said, and react by up-regulating a number of immune response genes – including some specifically antiviral genes that up-regulate interferon-alpha and -beta production and immune cytokine signaling pathways.
RTB101 is not a particularly new molecule; Novartis originally investigated it as an anticancer agent. “It failed, because it was too selective for mTORC1,” Dr. Mannick said. After Novartis dropped the molecule, resTORbio, a Novartis spin-off, began to investigate it as an immunotherapy for RTIs, particularly in patients with asthma.
reSTORbio’s phase 2 studies on RTB101 comprised 264 healthy subjects aged 65 years and older, who received placebo or 10 mg RTB101 daily for 6 weeks, during cold and flu season. They were followed for a year, confirming the antiviral gene up-regulation. Treatment was also associated with a 42% reduction in the rate of respiratory tract infections.
Conversations with the Food and Drug Administration and payers collected, Dr. Mannick said. “They said that where this drug could really make a difference was if it could decrease these infections in high-risk elderly, who are expensive to treat. So, we targeted people 65 years and older with asthma, chronic obstructive pulmonary disease, and smokers, and people who are 85 years or older.”
The phase 2b trial comprised 652 of these elderly high-risk subjects randomized to the following treatment arms: RTB101 5 mg once daily (n = 61), RTB101 10 mg once daily (n = 176), RTB101 10 mg b.i.d. (n = 120), RTB101 10 mg plus everolimus 0.1 mg daily (n = 115), or matching placebo (n = 180) over 16 weeks, during the entire cold and flu season. The primary endpoint was laboratory-confirmed RTIs in all groups.
The RTB101 10-mg, once-daily group had the best results with a 30.6% reduction in the percentage of patients with lab-confirmed RTIs, compared with placebo, and a 52% reduction in the percentage with severe symptoms.
A subgroup analysis found even more benefit to those with asthma. Among these patients, RTB101 effected a 58.2% decrease in patients with RTIs, and a 66.4% decrease in the rate of infections, compared with placebo.
RTB101 was most effective against rhinoviruses, but it also prevented RTIs associated with influenza A and coronavirus OC43. It also decreased the incidence of RTIs caused by respiratory syncytial virus, parainfluenza 4, influenza B, metapneumovirus, or other coronavirus serotypes.
There were no safety signals, Dr. Mannick noted. Adverse events were similar in both placebo and active groups, and none were deemed related to the study drug. About 5% of each group discontinued the drug because an adverse event.
Plans for a phase 3 trial are underway. A phase 3, placebo-controlled study in the Southern Hemisphere is now ongoing, during the winter cold and flu season. The Northern Hemisphere phase 3 will commence fall and winter of 2019.
Whether RBT101 can help younger people with asthma is an open question. Elderly patients not only have the asthma-related immune deficiency, but also the general age-related immune issues. Younger patients, however, still express the same asthma-related impairment of bronchial immunity.
“We would like to investigate this in younger people and in children, but that will have to wait until our other phase 3 studies are complete,” Dr. Mannick said.
The trial was sponsored by resTORbio.
SOURCE: Mannick J et al. ATS 2019, Abstract A2623.
CORRECTION 5/24/2019 The article was corrected to state a decreased the incidence of RTIs caused by respiratory syncytial virus, parainfluenza 4, influenza B, metapneumovirus, or other coronavirus serotypes.
DALLAS – A molecule that boosts innate viral immunity may protect elderly people with asthma from the root cause of most exacerbations – viral respiratory tract infections.
Dubbed RTB101, the oral medication is a selective, potent inhibitor of target of rapamycin complex 1 (TORC1). In phase 2b data presented at the American Thoracic Society’s international conference, RTB101 decreased by 52% the number of elderly subjects with severe, lab-confirmed respiratory tract infections (RTI) symptoms.
But the molecule was even more effective in patients with asthma aged 65 years and older, Joan Mannick, MD, said in an interview during the meeting. In this group, it reduced by 69% the percentage of subjects who developed RTIs and reduced the rate of infection by about 79%, compared with placebo.
“The core cause of asthma exacerbations in these patients is viral respiratory tract infection,” said Dr. Mannick, chief medical officer of resTORbio, the Boston company developing RTB101. “About 80% of the viruses detected in these infections are rhinoviruses, and there are 170 rhinovirus serotypes. We have never been able to develop a vaccine against rhinovirus, and we have no treatment other than to treat the inflammation caused by the infection.”
Centers for Disease Control and Prevention mortality records confirm the impact of viral respiratory infections on older people who experience asthma exacerbations: 6 of 10,000 will die, compared with less than 2 per 10,000 for all other age groups. Decreasing the number of these infections in older people with asthma would prevent morbidity and mortality and save considerable health care dollars.
“One of the reasons that asthmatics have such difficulty when they get respiratory infections is that they seem to have deficient antiviral immunity in the airways,” Dr. Mannick said. She pointed to a 2008 study of bronchial epithelial cells from both patients with asthma and healthy controls. When inoculated with rhinovirus, the cells from asthmatic airways were unable to mount a healthy immune response and were particularly deficient in producing interferon-beta.
By inhibiting mammalian TORC1 (mTORC1), RBT101 also inhibits sterol regulatory element binding transcription factor 2, a pathway that influences cholesterol synthesis. Cells perceive cholesterol synthesis attenuation as a threat, Dr. Mannick said, and react by up-regulating a number of immune response genes – including some specifically antiviral genes that up-regulate interferon-alpha and -beta production and immune cytokine signaling pathways.
RTB101 is not a particularly new molecule; Novartis originally investigated it as an anticancer agent. “It failed, because it was too selective for mTORC1,” Dr. Mannick said. After Novartis dropped the molecule, resTORbio, a Novartis spin-off, began to investigate it as an immunotherapy for RTIs, particularly in patients with asthma.
reSTORbio’s phase 2 studies on RTB101 comprised 264 healthy subjects aged 65 years and older, who received placebo or 10 mg RTB101 daily for 6 weeks, during cold and flu season. They were followed for a year, confirming the antiviral gene up-regulation. Treatment was also associated with a 42% reduction in the rate of respiratory tract infections.
Conversations with the Food and Drug Administration and payers collected, Dr. Mannick said. “They said that where this drug could really make a difference was if it could decrease these infections in high-risk elderly, who are expensive to treat. So, we targeted people 65 years and older with asthma, chronic obstructive pulmonary disease, and smokers, and people who are 85 years or older.”
The phase 2b trial comprised 652 of these elderly high-risk subjects randomized to the following treatment arms: RTB101 5 mg once daily (n = 61), RTB101 10 mg once daily (n = 176), RTB101 10 mg b.i.d. (n = 120), RTB101 10 mg plus everolimus 0.1 mg daily (n = 115), or matching placebo (n = 180) over 16 weeks, during the entire cold and flu season. The primary endpoint was laboratory-confirmed RTIs in all groups.
The RTB101 10-mg, once-daily group had the best results with a 30.6% reduction in the percentage of patients with lab-confirmed RTIs, compared with placebo, and a 52% reduction in the percentage with severe symptoms.
A subgroup analysis found even more benefit to those with asthma. Among these patients, RTB101 effected a 58.2% decrease in patients with RTIs, and a 66.4% decrease in the rate of infections, compared with placebo.
RTB101 was most effective against rhinoviruses, but it also prevented RTIs associated with influenza A and coronavirus OC43. It also decreased the incidence of RTIs caused by respiratory syncytial virus, parainfluenza 4, influenza B, metapneumovirus, or other coronavirus serotypes.
There were no safety signals, Dr. Mannick noted. Adverse events were similar in both placebo and active groups, and none were deemed related to the study drug. About 5% of each group discontinued the drug because an adverse event.
Plans for a phase 3 trial are underway. A phase 3, placebo-controlled study in the Southern Hemisphere is now ongoing, during the winter cold and flu season. The Northern Hemisphere phase 3 will commence fall and winter of 2019.
Whether RBT101 can help younger people with asthma is an open question. Elderly patients not only have the asthma-related immune deficiency, but also the general age-related immune issues. Younger patients, however, still express the same asthma-related impairment of bronchial immunity.
“We would like to investigate this in younger people and in children, but that will have to wait until our other phase 3 studies are complete,” Dr. Mannick said.
The trial was sponsored by resTORbio.
SOURCE: Mannick J et al. ATS 2019, Abstract A2623.
CORRECTION 5/24/2019 The article was corrected to state a decreased the incidence of RTIs caused by respiratory syncytial virus, parainfluenza 4, influenza B, metapneumovirus, or other coronavirus serotypes.
DALLAS – A molecule that boosts innate viral immunity may protect elderly people with asthma from the root cause of most exacerbations – viral respiratory tract infections.
Dubbed RTB101, the oral medication is a selective, potent inhibitor of target of rapamycin complex 1 (TORC1). In phase 2b data presented at the American Thoracic Society’s international conference, RTB101 decreased by 52% the number of elderly subjects with severe, lab-confirmed respiratory tract infections (RTI) symptoms.
But the molecule was even more effective in patients with asthma aged 65 years and older, Joan Mannick, MD, said in an interview during the meeting. In this group, it reduced by 69% the percentage of subjects who developed RTIs and reduced the rate of infection by about 79%, compared with placebo.
“The core cause of asthma exacerbations in these patients is viral respiratory tract infection,” said Dr. Mannick, chief medical officer of resTORbio, the Boston company developing RTB101. “About 80% of the viruses detected in these infections are rhinoviruses, and there are 170 rhinovirus serotypes. We have never been able to develop a vaccine against rhinovirus, and we have no treatment other than to treat the inflammation caused by the infection.”
Centers for Disease Control and Prevention mortality records confirm the impact of viral respiratory infections on older people who experience asthma exacerbations: 6 of 10,000 will die, compared with less than 2 per 10,000 for all other age groups. Decreasing the number of these infections in older people with asthma would prevent morbidity and mortality and save considerable health care dollars.
“One of the reasons that asthmatics have such difficulty when they get respiratory infections is that they seem to have deficient antiviral immunity in the airways,” Dr. Mannick said. She pointed to a 2008 study of bronchial epithelial cells from both patients with asthma and healthy controls. When inoculated with rhinovirus, the cells from asthmatic airways were unable to mount a healthy immune response and were particularly deficient in producing interferon-beta.
By inhibiting mammalian TORC1 (mTORC1), RBT101 also inhibits sterol regulatory element binding transcription factor 2, a pathway that influences cholesterol synthesis. Cells perceive cholesterol synthesis attenuation as a threat, Dr. Mannick said, and react by up-regulating a number of immune response genes – including some specifically antiviral genes that up-regulate interferon-alpha and -beta production and immune cytokine signaling pathways.
RTB101 is not a particularly new molecule; Novartis originally investigated it as an anticancer agent. “It failed, because it was too selective for mTORC1,” Dr. Mannick said. After Novartis dropped the molecule, resTORbio, a Novartis spin-off, began to investigate it as an immunotherapy for RTIs, particularly in patients with asthma.
reSTORbio’s phase 2 studies on RTB101 comprised 264 healthy subjects aged 65 years and older, who received placebo or 10 mg RTB101 daily for 6 weeks, during cold and flu season. They were followed for a year, confirming the antiviral gene up-regulation. Treatment was also associated with a 42% reduction in the rate of respiratory tract infections.
Conversations with the Food and Drug Administration and payers collected, Dr. Mannick said. “They said that where this drug could really make a difference was if it could decrease these infections in high-risk elderly, who are expensive to treat. So, we targeted people 65 years and older with asthma, chronic obstructive pulmonary disease, and smokers, and people who are 85 years or older.”
The phase 2b trial comprised 652 of these elderly high-risk subjects randomized to the following treatment arms: RTB101 5 mg once daily (n = 61), RTB101 10 mg once daily (n = 176), RTB101 10 mg b.i.d. (n = 120), RTB101 10 mg plus everolimus 0.1 mg daily (n = 115), or matching placebo (n = 180) over 16 weeks, during the entire cold and flu season. The primary endpoint was laboratory-confirmed RTIs in all groups.
The RTB101 10-mg, once-daily group had the best results with a 30.6% reduction in the percentage of patients with lab-confirmed RTIs, compared with placebo, and a 52% reduction in the percentage with severe symptoms.
A subgroup analysis found even more benefit to those with asthma. Among these patients, RTB101 effected a 58.2% decrease in patients with RTIs, and a 66.4% decrease in the rate of infections, compared with placebo.
RTB101 was most effective against rhinoviruses, but it also prevented RTIs associated with influenza A and coronavirus OC43. It also decreased the incidence of RTIs caused by respiratory syncytial virus, parainfluenza 4, influenza B, metapneumovirus, or other coronavirus serotypes.
There were no safety signals, Dr. Mannick noted. Adverse events were similar in both placebo and active groups, and none were deemed related to the study drug. About 5% of each group discontinued the drug because an adverse event.
Plans for a phase 3 trial are underway. A phase 3, placebo-controlled study in the Southern Hemisphere is now ongoing, during the winter cold and flu season. The Northern Hemisphere phase 3 will commence fall and winter of 2019.
Whether RBT101 can help younger people with asthma is an open question. Elderly patients not only have the asthma-related immune deficiency, but also the general age-related immune issues. Younger patients, however, still express the same asthma-related impairment of bronchial immunity.
“We would like to investigate this in younger people and in children, but that will have to wait until our other phase 3 studies are complete,” Dr. Mannick said.
The trial was sponsored by resTORbio.
SOURCE: Mannick J et al. ATS 2019, Abstract A2623.
CORRECTION 5/24/2019 The article was corrected to state a decreased the incidence of RTIs caused by respiratory syncytial virus, parainfluenza 4, influenza B, metapneumovirus, or other coronavirus serotypes.
REPORTING FROM ATS 2019
Elderly concussion patients who used statins had lower dementia risk
, compared with similar adults not taking statins.
The findings come from a population-based double cohort study of 28,815 patients in the Ontario Health Insurance Plan. Study patients were enrolled over 20 years, and had a minimum follow-up of 3 years. The study excluded patients hospitalized caused by a severe concussion, those previously diagnosed with delirium or dementia, and those who died within 90 days of their concussions.
Concussions are a common injury in older adults and dementia may be a frequent outcome years afterward, Donald A. Redelmeier, MD, of the University of Toronto and colleagues wrote in a study published in JAMA Neurology. A concussion should not be interpreted as a reason to stop statins, and a potential neuroprotective benefit may encourage medication adherence among patients who are already prescribed a statin.
Of the 28,815 patients studied, 4,727 patients (1 case per 6 patients) developed dementia over the mean follow-up period of 3.9 years. The 7,058 patients who received a statin had a 13% reduced risk of developing dementia, compared with the 21,757 patients who did not (relative risk, 0.87; 95% confidence interval, 0.81-0.93; P less than .001).
Even though statin use was associated with a lower risk, the subsequent incidence of dementia was still twice the population norm in statin users who had concussions, the researchers wrote. The findings indicate concussions are a common injury in older adults and dementia may be a frequent outcome years after concussions.
Statin users who had concussions continued to have a reduced risk of developing dementia after adjustment for patient characteristics, use of other cardiovascular medications, dosage, and depression risk. The statin associated with the greatest risk reduction was rosuvastatin; simvastatin was associated with the least risk reduction. With the possible exception of angiotensin II receptor blockers, no other cardiovascular or noncardiovascular medications were associated with a decreased risk of dementia after a concussion, the researchers wrote.
They also examined data for elderly patients using statins after an ankle sprain and found the risk of dementia was similar for those who did and did not receive statins after the injury.
Factors such as smoking status, exercise, drug adherence, and other unknown aspects of patient health might have influenced the results of the study, the researchers acknowledged. Additionally, a secondary analysis was not statistically powered to distinguish the relative efficacy of statin use before a concussion.
This study was funded in part by a Canada Research Chair in Medical Decision Sciences, the Canadian Institutes of Health Research, the BrightFocus Foundation, and the Comprehensive Research Experience for Medical Students at the University of Toronto. The authors reported no relevant conflicts of interest.
SOURCE: Redelmeier DA et al. JAMA Neurol. 2019 May 20. doi: 10.1001/jamaneurol.2019.1148.
This appears to be the first large study to explore the relationship between statin use, concussions, and the development of dementia. Although statins have anti-inflammatory properties, no trials have linked statins to reduced cognitive impairment. Considering it can be difficult to mitigate against confounding by indication in pharmacologic studies, this observational study included a large group of diverse individuals who developed concussions over a period of 20 years.

Rachel A. Whitmer, PhD, is with the division of epidemiology and department of public health sciences at the University of California, Davis. She made her remarks in a related editorial published with the study, and reported no relevant conflicts of interest.
This appears to be the first large study to explore the relationship between statin use, concussions, and the development of dementia. Although statins have anti-inflammatory properties, no trials have linked statins to reduced cognitive impairment. Considering it can be difficult to mitigate against confounding by indication in pharmacologic studies, this observational study included a large group of diverse individuals who developed concussions over a period of 20 years.
Rachel A. Whitmer, PhD, is with the division of epidemiology and department of public health sciences at the University of California, Davis. She made her remarks in a related editorial published with the study, and reported no relevant conflicts of interest.
This appears to be the first large study to explore the relationship between statin use, concussions, and the development of dementia. Although statins have anti-inflammatory properties, no trials have linked statins to reduced cognitive impairment. Considering it can be difficult to mitigate against confounding by indication in pharmacologic studies, this observational study included a large group of diverse individuals who developed concussions over a period of 20 years.
Rachel A. Whitmer, PhD, is with the division of epidemiology and department of public health sciences at the University of California, Davis. She made her remarks in a related editorial published with the study, and reported no relevant conflicts of interest.
, compared with similar adults not taking statins.
The findings come from a population-based double cohort study of 28,815 patients in the Ontario Health Insurance Plan. Study patients were enrolled over 20 years, and had a minimum follow-up of 3 years. The study excluded patients hospitalized caused by a severe concussion, those previously diagnosed with delirium or dementia, and those who died within 90 days of their concussions.
Concussions are a common injury in older adults and dementia may be a frequent outcome years afterward, Donald A. Redelmeier, MD, of the University of Toronto and colleagues wrote in a study published in JAMA Neurology. A concussion should not be interpreted as a reason to stop statins, and a potential neuroprotective benefit may encourage medication adherence among patients who are already prescribed a statin.
Of the 28,815 patients studied, 4,727 patients (1 case per 6 patients) developed dementia over the mean follow-up period of 3.9 years. The 7,058 patients who received a statin had a 13% reduced risk of developing dementia, compared with the 21,757 patients who did not (relative risk, 0.87; 95% confidence interval, 0.81-0.93; P less than .001).
Even though statin use was associated with a lower risk, the subsequent incidence of dementia was still twice the population norm in statin users who had concussions, the researchers wrote. The findings indicate concussions are a common injury in older adults and dementia may be a frequent outcome years after concussions.
Statin users who had concussions continued to have a reduced risk of developing dementia after adjustment for patient characteristics, use of other cardiovascular medications, dosage, and depression risk. The statin associated with the greatest risk reduction was rosuvastatin; simvastatin was associated with the least risk reduction. With the possible exception of angiotensin II receptor blockers, no other cardiovascular or noncardiovascular medications were associated with a decreased risk of dementia after a concussion, the researchers wrote.
They also examined data for elderly patients using statins after an ankle sprain and found the risk of dementia was similar for those who did and did not receive statins after the injury.
Factors such as smoking status, exercise, drug adherence, and other unknown aspects of patient health might have influenced the results of the study, the researchers acknowledged. Additionally, a secondary analysis was not statistically powered to distinguish the relative efficacy of statin use before a concussion.
This study was funded in part by a Canada Research Chair in Medical Decision Sciences, the Canadian Institutes of Health Research, the BrightFocus Foundation, and the Comprehensive Research Experience for Medical Students at the University of Toronto. The authors reported no relevant conflicts of interest.
SOURCE: Redelmeier DA et al. JAMA Neurol. 2019 May 20. doi: 10.1001/jamaneurol.2019.1148.
, compared with similar adults not taking statins.
The findings come from a population-based double cohort study of 28,815 patients in the Ontario Health Insurance Plan. Study patients were enrolled over 20 years, and had a minimum follow-up of 3 years. The study excluded patients hospitalized caused by a severe concussion, those previously diagnosed with delirium or dementia, and those who died within 90 days of their concussions.
Concussions are a common injury in older adults and dementia may be a frequent outcome years afterward, Donald A. Redelmeier, MD, of the University of Toronto and colleagues wrote in a study published in JAMA Neurology. A concussion should not be interpreted as a reason to stop statins, and a potential neuroprotective benefit may encourage medication adherence among patients who are already prescribed a statin.
Of the 28,815 patients studied, 4,727 patients (1 case per 6 patients) developed dementia over the mean follow-up period of 3.9 years. The 7,058 patients who received a statin had a 13% reduced risk of developing dementia, compared with the 21,757 patients who did not (relative risk, 0.87; 95% confidence interval, 0.81-0.93; P less than .001).
Even though statin use was associated with a lower risk, the subsequent incidence of dementia was still twice the population norm in statin users who had concussions, the researchers wrote. The findings indicate concussions are a common injury in older adults and dementia may be a frequent outcome years after concussions.
Statin users who had concussions continued to have a reduced risk of developing dementia after adjustment for patient characteristics, use of other cardiovascular medications, dosage, and depression risk. The statin associated with the greatest risk reduction was rosuvastatin; simvastatin was associated with the least risk reduction. With the possible exception of angiotensin II receptor blockers, no other cardiovascular or noncardiovascular medications were associated with a decreased risk of dementia after a concussion, the researchers wrote.
They also examined data for elderly patients using statins after an ankle sprain and found the risk of dementia was similar for those who did and did not receive statins after the injury.
Factors such as smoking status, exercise, drug adherence, and other unknown aspects of patient health might have influenced the results of the study, the researchers acknowledged. Additionally, a secondary analysis was not statistically powered to distinguish the relative efficacy of statin use before a concussion.
This study was funded in part by a Canada Research Chair in Medical Decision Sciences, the Canadian Institutes of Health Research, the BrightFocus Foundation, and the Comprehensive Research Experience for Medical Students at the University of Toronto. The authors reported no relevant conflicts of interest.
SOURCE: Redelmeier DA et al. JAMA Neurol. 2019 May 20. doi: 10.1001/jamaneurol.2019.1148.
FROM JAMA NEUROLOGY
Key clinical point: Older adults taking a statin within 90 days after a concussion had a lower rate of dementia.
Major finding: Statin use within 90 days of a concussion in older adults was associated with a 13% reduced risk of dementia (relative risk, 0.87; 95% confidence interval, 0.81-0.93; P less than .001).
Study details: A population-based double cohort study of 28,815 elderly patients who had a concussion between April 1993 and April 2013.
Disclosures: This study was funded in part by a Canada Research Chair in Medical Decision Sciences, the Canadian Institutes of Health Research, the BrightFocus Foundation, and the Comprehensive Research Experience for Medical Students at the University of Toronto. The authors reported no relevant conflicts of interest.
Source: Redelmeier DA et al. JAMA Neurol. 2019 May 20. doi: 10.1001/jamaneurol.2019.1148.
Report on newly recognized cause of dementia should be read widely
Alzheimer’s disease is recognized as the most common cause of dementia, and many in the laity use the two terms almost interchangeably. However, there is increasing recognition that dementia in old age is a complex disorder, with mixed neuropathologies being the norm rather than the exception (Ann Neurol. 2018 Jan;83[1]:74-83).

Alzheimer’s disease (AD) and cerebrovascular pathologies are the most common, but another pathology is receiving increasing attention in relation to cognitive disorders in very old individuals – that related to the transactive response DNA binding protein of 43 kDa (TDP-43). This protein is expressed in most human tissues, including the brain, is localized mostly in nuclei, and binds to RNA and DNA as well as numerous proteins, with the role of regulating gene expression.
It has been known for nearly 2 decades that TDP-43 can become abnormally phosphorylated and translocated to the cytoplasm to produce a proteinopathy that forms the basis of a significant proportion of frontotemporal dementia (FTD) and the majority of amyotrophic lateral sclerosis. More recently, it has also been reported to be common in the brains of older people (over age 80 years) and associated with a cognitive disorder characterized by an amnestic picture that mimics AD. Since the protein deposition is predominantly in the limbic regions (amygdala, hippocampus, insula), it has been termed “‘limbic-predominant, age-related TDP-43 encephalopathy”, or LATE.
A recently convened international working group has published consensus criteria for LATE and provided guidelines for its staging. Community-based autopsy studies suggest that 20%-50% of people aged over 80 years have the neuropathologic change associated with LATE. The clinical presentation resembles amnestic dementia syndrome, much like AD. Both LATE and AD pathologies often occur in the same individual, but the relative predominance of one or the other varies greatly between individuals. The genetic risks of LATE overlap with those for FTD and AD, and other risk factors may also be shared with AD, which remains an area for further investigation. There are at present no specific biomarkers of LATE. It is associated with hippocampal sclerosis in some cases, which may be visible on MRI, but hippocampal sclerosis itself is not specific to TDP-43 pathology.
of LATE and calls for systematic study of the causes of dementia – which may be nearly as common as AD in the very old. The report should be read widely and should remind us of the diverse pathologies that contribute to cognitive disorders, alone and in combination with one another.
Dr. Sachdev is Scientia Professor of Neuropsychiatry and codirector of the Center for Healthy Brain Aging at the University of New South Wales, Sydney; and clinical director of the Neuropsychiatric Institute at the Prince of Wales Hospital, also in Sydney. His major areas of research are drug-induced movement disorders, brain imaging, cognitive aging and dementia. Dr. Sachdev also served on the Neurocognitive Disorders Work Group of the DSM-5.
Alzheimer’s disease is recognized as the most common cause of dementia, and many in the laity use the two terms almost interchangeably. However, there is increasing recognition that dementia in old age is a complex disorder, with mixed neuropathologies being the norm rather than the exception (Ann Neurol. 2018 Jan;83[1]:74-83).
Alzheimer’s disease (AD) and cerebrovascular pathologies are the most common, but another pathology is receiving increasing attention in relation to cognitive disorders in very old individuals – that related to the transactive response DNA binding protein of 43 kDa (TDP-43). This protein is expressed in most human tissues, including the brain, is localized mostly in nuclei, and binds to RNA and DNA as well as numerous proteins, with the role of regulating gene expression.
It has been known for nearly 2 decades that TDP-43 can become abnormally phosphorylated and translocated to the cytoplasm to produce a proteinopathy that forms the basis of a significant proportion of frontotemporal dementia (FTD) and the majority of amyotrophic lateral sclerosis. More recently, it has also been reported to be common in the brains of older people (over age 80 years) and associated with a cognitive disorder characterized by an amnestic picture that mimics AD. Since the protein deposition is predominantly in the limbic regions (amygdala, hippocampus, insula), it has been termed “‘limbic-predominant, age-related TDP-43 encephalopathy”, or LATE.
A recently convened international working group has published consensus criteria for LATE and provided guidelines for its staging. Community-based autopsy studies suggest that 20%-50% of people aged over 80 years have the neuropathologic change associated with LATE. The clinical presentation resembles amnestic dementia syndrome, much like AD. Both LATE and AD pathologies often occur in the same individual, but the relative predominance of one or the other varies greatly between individuals. The genetic risks of LATE overlap with those for FTD and AD, and other risk factors may also be shared with AD, which remains an area for further investigation. There are at present no specific biomarkers of LATE. It is associated with hippocampal sclerosis in some cases, which may be visible on MRI, but hippocampal sclerosis itself is not specific to TDP-43 pathology.
of LATE and calls for systematic study of the causes of dementia – which may be nearly as common as AD in the very old. The report should be read widely and should remind us of the diverse pathologies that contribute to cognitive disorders, alone and in combination with one another.
Dr. Sachdev is Scientia Professor of Neuropsychiatry and codirector of the Center for Healthy Brain Aging at the University of New South Wales, Sydney; and clinical director of the Neuropsychiatric Institute at the Prince of Wales Hospital, also in Sydney. His major areas of research are drug-induced movement disorders, brain imaging, cognitive aging and dementia. Dr. Sachdev also served on the Neurocognitive Disorders Work Group of the DSM-5.
Alzheimer’s disease is recognized as the most common cause of dementia, and many in the laity use the two terms almost interchangeably. However, there is increasing recognition that dementia in old age is a complex disorder, with mixed neuropathologies being the norm rather than the exception (Ann Neurol. 2018 Jan;83[1]:74-83).
Alzheimer’s disease (AD) and cerebrovascular pathologies are the most common, but another pathology is receiving increasing attention in relation to cognitive disorders in very old individuals – that related to the transactive response DNA binding protein of 43 kDa (TDP-43). This protein is expressed in most human tissues, including the brain, is localized mostly in nuclei, and binds to RNA and DNA as well as numerous proteins, with the role of regulating gene expression.
It has been known for nearly 2 decades that TDP-43 can become abnormally phosphorylated and translocated to the cytoplasm to produce a proteinopathy that forms the basis of a significant proportion of frontotemporal dementia (FTD) and the majority of amyotrophic lateral sclerosis. More recently, it has also been reported to be common in the brains of older people (over age 80 years) and associated with a cognitive disorder characterized by an amnestic picture that mimics AD. Since the protein deposition is predominantly in the limbic regions (amygdala, hippocampus, insula), it has been termed “‘limbic-predominant, age-related TDP-43 encephalopathy”, or LATE.
A recently convened international working group has published consensus criteria for LATE and provided guidelines for its staging. Community-based autopsy studies suggest that 20%-50% of people aged over 80 years have the neuropathologic change associated with LATE. The clinical presentation resembles amnestic dementia syndrome, much like AD. Both LATE and AD pathologies often occur in the same individual, but the relative predominance of one or the other varies greatly between individuals. The genetic risks of LATE overlap with those for FTD and AD, and other risk factors may also be shared with AD, which remains an area for further investigation. There are at present no specific biomarkers of LATE. It is associated with hippocampal sclerosis in some cases, which may be visible on MRI, but hippocampal sclerosis itself is not specific to TDP-43 pathology.
of LATE and calls for systematic study of the causes of dementia – which may be nearly as common as AD in the very old. The report should be read widely and should remind us of the diverse pathologies that contribute to cognitive disorders, alone and in combination with one another.
Dr. Sachdev is Scientia Professor of Neuropsychiatry and codirector of the Center for Healthy Brain Aging at the University of New South Wales, Sydney; and clinical director of the Neuropsychiatric Institute at the Prince of Wales Hospital, also in Sydney. His major areas of research are drug-induced movement disorders, brain imaging, cognitive aging and dementia. Dr. Sachdev also served on the Neurocognitive Disorders Work Group of the DSM-5.
Rheumatoid arthritis treatment less aggressive, not less favorable in older adults
BIRMINGHAM, ENGLAND – Being diagnosed with rheumatoid arthritis at age 75 years or older made it less likely that patients would receive intensive therapy than their younger counterparts, but that did not mean that they were treated any less favorably overall, according to findings derived from the Early RA Network cohort.
“The claim that the elderly are treated less aggressively isn’t completely true throughout the whole treat-to-target strategy,” said Simone Howard of King’s College London at the British Society for Rheumatology annual conference. While older patients were less likely to receive intensive treatment up to 2 years after their diagnosis, there was a shorter delay between the onset of symptoms and the first outpatient visit to a rheumatology clinic.
When compared against patients who were younger than 65 years, those aged 65-74 years and 75 years and older were 11% (P = .02) and 15% (P = .02) more likely to have their first outpatient visit within 10 months.
Furthermore, no significant differences were seen between any age groups in the time to first initiation of a conventional synthetic disease-modifying antirheumatic drug (csDMARD), which averaged nearly 3 months after symptoms appeared.
Ms. Howard, who has previously worked at the European Medicines Agency, noted that, during her time at the EMA, “there was a real push to incorporate the elderly into the regulatory framework more. In parallel, there were also reports of the elderly being treated less aggressively. So the question was, where was that coming from?”
Similar therapeutic approaches are advocated for older and younger RA patients, and to look for any disparities, Ms. Howard and associates turned to the Early RA Network (ERAN) to “investigate potential treatment bias against the elderly.”
ERAN is a hospital-based inception cohort of 1,236 patients with early RA who were recruited across 23 centers in the United Kingdom and Ireland between 2002 and 2014.
Of 1,131 patients used in the analyses, 9.7% (n = 110) were 75 years or older, 21.5% (n = 243) were aged 65-74 years, and 68.8% (n = 778) were 65 years or younger. The majority (67.7%) of patients were female.
Patients aged 75 years and older were more likely to present with comorbidities than the youngest group, and they had higher health assessment questionnaire scores at baseline. However, they were no more likely to have high disease activity at the first visit, which was defined as a disease activity score in 28 joints of more than 5, and the older patients were 27% less likely to be seropositive (P = .004).
“It’s when we come to pharmacological aspects of care that we are seeing treatment biases,” Ms. Howard noted. Patients over 75 years were significantly more likely than the youngest age group to be treated with glucocorticoids or csDMARD monotherapy at 1 year, and 23% more likely to be on less aggressive therapy (P equal to or less than .0001). Aggressive therapy was defined as the use of a combination of csDMARDs or the use of biologic drugs.
At 2 years, the oldest patients were 46% more likely than those under 65 years to be on less-intensive therapies (P equal to or less than .0001), with those aged 65-74 years 19% more likely to be on glucocorticoid or csDMARD therapy (P = .005).
Factors such as patient choice and tolerance were not considered in the analyses and could be important, Ms. Howard conceded in response to a question after her presentation.
Another point raised was that perhaps the prescribing of aggressive therapy would rationally be different in someone diagnosed with RA at age 85 versus 65 because the duration of time that would be likely to be lived with accumulating joint damage would be shorter at the older age and that would be balanced against the other effects of the therapy. So, there may be important reasons in shared decision making that influenced the treatment choices other than the age of patients.
Ms. Howard agreed, noting that this demonstrated the need to be careful around the language used for defining what constituted aggressive or intensive therapy.
She and her coauthors reported no conflicts of interest.
SOURCE: Howard S et al. Rheumatology. 2019;58(suppl 3), Abstract 011.
BIRMINGHAM, ENGLAND – Being diagnosed with rheumatoid arthritis at age 75 years or older made it less likely that patients would receive intensive therapy than their younger counterparts, but that did not mean that they were treated any less favorably overall, according to findings derived from the Early RA Network cohort.
“The claim that the elderly are treated less aggressively isn’t completely true throughout the whole treat-to-target strategy,” said Simone Howard of King’s College London at the British Society for Rheumatology annual conference. While older patients were less likely to receive intensive treatment up to 2 years after their diagnosis, there was a shorter delay between the onset of symptoms and the first outpatient visit to a rheumatology clinic.
When compared against patients who were younger than 65 years, those aged 65-74 years and 75 years and older were 11% (P = .02) and 15% (P = .02) more likely to have their first outpatient visit within 10 months.
Furthermore, no significant differences were seen between any age groups in the time to first initiation of a conventional synthetic disease-modifying antirheumatic drug (csDMARD), which averaged nearly 3 months after symptoms appeared.
Ms. Howard, who has previously worked at the European Medicines Agency, noted that, during her time at the EMA, “there was a real push to incorporate the elderly into the regulatory framework more. In parallel, there were also reports of the elderly being treated less aggressively. So the question was, where was that coming from?”
Similar therapeutic approaches are advocated for older and younger RA patients, and to look for any disparities, Ms. Howard and associates turned to the Early RA Network (ERAN) to “investigate potential treatment bias against the elderly.”
ERAN is a hospital-based inception cohort of 1,236 patients with early RA who were recruited across 23 centers in the United Kingdom and Ireland between 2002 and 2014.
Of 1,131 patients used in the analyses, 9.7% (n = 110) were 75 years or older, 21.5% (n = 243) were aged 65-74 years, and 68.8% (n = 778) were 65 years or younger. The majority (67.7%) of patients were female.
Patients aged 75 years and older were more likely to present with comorbidities than the youngest group, and they had higher health assessment questionnaire scores at baseline. However, they were no more likely to have high disease activity at the first visit, which was defined as a disease activity score in 28 joints of more than 5, and the older patients were 27% less likely to be seropositive (P = .004).
“It’s when we come to pharmacological aspects of care that we are seeing treatment biases,” Ms. Howard noted. Patients over 75 years were significantly more likely than the youngest age group to be treated with glucocorticoids or csDMARD monotherapy at 1 year, and 23% more likely to be on less aggressive therapy (P equal to or less than .0001). Aggressive therapy was defined as the use of a combination of csDMARDs or the use of biologic drugs.
At 2 years, the oldest patients were 46% more likely than those under 65 years to be on less-intensive therapies (P equal to or less than .0001), with those aged 65-74 years 19% more likely to be on glucocorticoid or csDMARD therapy (P = .005).
Factors such as patient choice and tolerance were not considered in the analyses and could be important, Ms. Howard conceded in response to a question after her presentation.
Another point raised was that perhaps the prescribing of aggressive therapy would rationally be different in someone diagnosed with RA at age 85 versus 65 because the duration of time that would be likely to be lived with accumulating joint damage would be shorter at the older age and that would be balanced against the other effects of the therapy. So, there may be important reasons in shared decision making that influenced the treatment choices other than the age of patients.
Ms. Howard agreed, noting that this demonstrated the need to be careful around the language used for defining what constituted aggressive or intensive therapy.
She and her coauthors reported no conflicts of interest.
SOURCE: Howard S et al. Rheumatology. 2019;58(suppl 3), Abstract 011.
BIRMINGHAM, ENGLAND – Being diagnosed with rheumatoid arthritis at age 75 years or older made it less likely that patients would receive intensive therapy than their younger counterparts, but that did not mean that they were treated any less favorably overall, according to findings derived from the Early RA Network cohort.
“The claim that the elderly are treated less aggressively isn’t completely true throughout the whole treat-to-target strategy,” said Simone Howard of King’s College London at the British Society for Rheumatology annual conference. While older patients were less likely to receive intensive treatment up to 2 years after their diagnosis, there was a shorter delay between the onset of symptoms and the first outpatient visit to a rheumatology clinic.
When compared against patients who were younger than 65 years, those aged 65-74 years and 75 years and older were 11% (P = .02) and 15% (P = .02) more likely to have their first outpatient visit within 10 months.
Furthermore, no significant differences were seen between any age groups in the time to first initiation of a conventional synthetic disease-modifying antirheumatic drug (csDMARD), which averaged nearly 3 months after symptoms appeared.
Ms. Howard, who has previously worked at the European Medicines Agency, noted that, during her time at the EMA, “there was a real push to incorporate the elderly into the regulatory framework more. In parallel, there were also reports of the elderly being treated less aggressively. So the question was, where was that coming from?”
Similar therapeutic approaches are advocated for older and younger RA patients, and to look for any disparities, Ms. Howard and associates turned to the Early RA Network (ERAN) to “investigate potential treatment bias against the elderly.”
ERAN is a hospital-based inception cohort of 1,236 patients with early RA who were recruited across 23 centers in the United Kingdom and Ireland between 2002 and 2014.
Of 1,131 patients used in the analyses, 9.7% (n = 110) were 75 years or older, 21.5% (n = 243) were aged 65-74 years, and 68.8% (n = 778) were 65 years or younger. The majority (67.7%) of patients were female.
Patients aged 75 years and older were more likely to present with comorbidities than the youngest group, and they had higher health assessment questionnaire scores at baseline. However, they were no more likely to have high disease activity at the first visit, which was defined as a disease activity score in 28 joints of more than 5, and the older patients were 27% less likely to be seropositive (P = .004).
“It’s when we come to pharmacological aspects of care that we are seeing treatment biases,” Ms. Howard noted. Patients over 75 years were significantly more likely than the youngest age group to be treated with glucocorticoids or csDMARD monotherapy at 1 year, and 23% more likely to be on less aggressive therapy (P equal to or less than .0001). Aggressive therapy was defined as the use of a combination of csDMARDs or the use of biologic drugs.
At 2 years, the oldest patients were 46% more likely than those under 65 years to be on less-intensive therapies (P equal to or less than .0001), with those aged 65-74 years 19% more likely to be on glucocorticoid or csDMARD therapy (P = .005).
Factors such as patient choice and tolerance were not considered in the analyses and could be important, Ms. Howard conceded in response to a question after her presentation.
Another point raised was that perhaps the prescribing of aggressive therapy would rationally be different in someone diagnosed with RA at age 85 versus 65 because the duration of time that would be likely to be lived with accumulating joint damage would be shorter at the older age and that would be balanced against the other effects of the therapy. So, there may be important reasons in shared decision making that influenced the treatment choices other than the age of patients.
Ms. Howard agreed, noting that this demonstrated the need to be careful around the language used for defining what constituted aggressive or intensive therapy.
She and her coauthors reported no conflicts of interest.
SOURCE: Howard S et al. Rheumatology. 2019;58(suppl 3), Abstract 011.
REPORTING FROM BSR 2019