User login
To have not and then to have: A challenging immune paradox

The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.
The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.

Drug reaction or metastatic lung cancer?
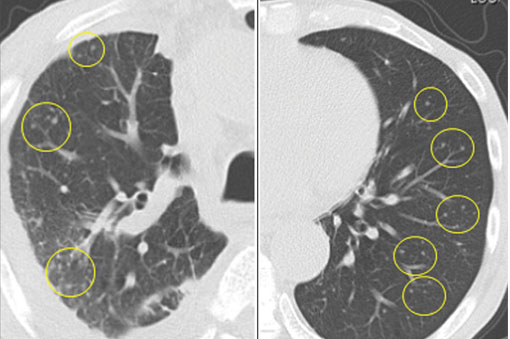
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
. The left lung appeared normal (B).")

Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
Anaphylaxis Controversy and Consensus

Abdominal pain and bloody diarrhea in a 32-year-old woman
A 32-year-old woman presented to our emergency department with chest pain and painful ulcerations on her arms, abdomen, back, groin, axillae, and in her mouth. She first noticed the ulcers 7 days earlier.
She also reported bloody diarrhea, which had started 2 years earlier, with 10 or more bowel movements daily. She described her stools as semiformed and associated with urgency and painful abdominal cramps.
Medical history
Her medical history included obstructive sleep apnea and morbid obesity. She had first presented 2 years earlier to another hospital with diarrhea, abdominal pain, and rectal bleeding. At that time, results of esophagogastroduodenoscopy and colonoscopy were reported as normal. Later, she became pregnant, and her symptoms went away. She had a normal pregnancy and delivery.
About 1 year postpartum, her abdominal pain and bloody diarrhea recurred. Colonoscopy showed severe sigmoid inflammation with small, shallow ulcerations and friable mucosa interrupted by areas of normal mucosa. Histopathologic study of the colonic mucosa indicated mild to moderate chronic active colitis consisting of focal areas of cryptitis with occasional crypt abscess formation. She was diagnosed with Crohn colitis based on the endoscopic appearance, histopathology, and clinical presentation. The endoscope, however, could not be advanced beyond the sigmoid colon, which suggested stenosis. She was started on 5-aminosalicylic acid (5-ASA) but developed visual hallucinations, and the medication was stopped.
Her symptoms continued, and she developed worsening rectal bleeding and anemia that required hospitalization and blood transfusions. Another colonoscopy performed 1 month before this emergency department visit had shown multiple mucosal ulcerations, but again, the colonoscope could not be advanced beyond the sigmoid colon. She was started on oral corticosteroids, which provided only minimal clinical improvement.
Her current medications included atenolol (for sinus tachycardia), prednisone (initial dose 60 mg/day tapered to 20 mg/day at presentation), and ciprofloxacin.
Her family history was unknown because she had been adopted.
About 1 week before presentation, she had noticed ulcers developing on her arms, abdomen, back, groin, oral mucosa, and axillae. The ulcers were large and painful, with occasional spontaneous bleeding. She also reported pustules and ulcerations at sites of previous skin punctures, consistent with pathergy.
Findings on presentation
- Temperature 99.5°F (37.5°C)
- Heart rate 124 beats per minute
- Respiratory rate 22 breaths per minute
- Oxygen saturation 100% on room air
- Blood pressure 128/81 mm Hg
- Body mass index 67 kg/m2 (morbidly obese).
She had multiple greyish-white patches and erosions over the soft palate, tongue, and upper and lower lip mucosa, erythematous pustules in the axillae bilaterally, and large erythematous, sharply demarcated ulcerations with a fibrinous base bilaterally covering her arms, thighs, groin, and abdomen.

Blood testing showed multiple abnormal results (Table 1). Urinalysis revealed a urine protein concentration of 100 mg/dL (reference range 0), more than 25 white blood cells per high-power field (reference range < 5), 6 to 10 red blood cells per high-power field (0–3), and more than 10 casts per low-power field (0), which suggested a urinary tract infection with hematuria.
Computed tomography (CT) of the abdomen and pelvis with intravenous and oral contrast showed diffuse fatty infiltration of the liver and wall thickening of the rectum and sigmoid colon.
She was admitted to the medical intensive care unit for potential septic shock. Intravenous vancomycin and ciprofloxacin were started (the latter owing to penicillin allergy).
CAUSES OF DIARRHEA AND SKIN CHANGES
1. What is the most likely diagnosis in our patient?
- Ulcerative colitis
- Crohn disease
- Behçet disease
- Intestinal tuberculosis
- Herpes simplex virus infection
- Cytomegalovirus infection
All of the above can cause diarrhea in combination with mucocutaneous lesions and other manifestations.
Ulcerative colitis and Crohn disease: Mucocutaneous findings
Extraintestinal manifestations of inflammatory bowel diseases (Crohn disease, ulcerative colitis, and Behçet disease) include arthritis, ocular involvement, mucocutaneous manifestations, and liver involvement in the form of primary sclerosing cholangitis. Less common extraintestinal manifestations include vascular, renal, pulmonary, cardiac, and neurologic involvement.
Mucocutaneous findings are observed in 5% to 10% of patients with ulcerative colitis and 20% to 75% of patients with Crohn disease.1–3 The most common are erythema nodosum and pyoderma gangrenosum.4
Yüksel et al5 reported that of 352 patients with inflammatory bowel disease, 7.4% had erythema nodosum and 2.3% had pyoderma gangrenosum. Erythema nodosum was significantly more common in patients with Crohn disease than in those with ulcerative colitis, and its severity was linked with higher disease activity. Lesions frequently resolved when bowel disease subsided.
Lebwohl and Lebwohl6 reported that pyoderma gangrenosum occurred in up to 20% of patients with Crohn disease and up to 10% of those with ulcerative colitis. It is not known whether pyoderma gangrenosum correlates with intestinal disease severity.
Other mucocutaneous manifestations of inflammatory bowel disease include oral aphthous ulcers, acute febrile neutrophilic dermatosis (Sweet syndrome), and metastatic Crohn disease. Aphthous ulcers in the oral cavity, often observed in both Crohn disease and ulcerative colitis, cannot be differentiated on clinical examination from herpes simplex virus (HSV) type 1-induced or idiopathic mucous membrane ulcers. The most common ulcer locations are the lips and buccal mucosa. If biopsied (seldom required), noncaseating granulomas can be identified that are comparable with intestinal mucosal granulomas found in Crohn disease.7
Behçet disease has similar signs
Oral aphthous ulcers are also the most frequent symptom in Behçet disease, occurring in 97% to 100% of cases.8 They most commonly affect the tongue, lips, buccal mucosa, and gingiva.
Cutaneous manifestations include erythema nodosum-like lesions, which present as erythematous painful nodules over pretibial surfaces of the lower limbs but can also affect the arms and thighs; they can also present as papulopustular rosacea eruptions composed of papules, pustules, and noninflammatory comedones, most commonly on the chest, back, and shoulders.8,9
Pathergy, ie, skin hyperresponse to minor trauma such as a bump or bruise, is a typical trait of Behçet disease. A positive pathergy test (ie, skin hyperreactivity to a needlestick or intracutaneous injection) has a specificity of 98.4% in patients with Behçet disease.10
Interestingly, there appears to be a regional difference in the susceptibility to pathergy. While a pathergy response in patients with Behçet disease is rare in the United States and the United Kingdom, it is very common in Japan, Turkey, and Israel.11
Patient demographics also distinguish Behçet disease from Crohn disease. The prevalence of Behçet disease is highest along the Silk Road from the Mediterranean Basin to East Asia and lowest in North America and Northern Europe.12 The mean age at onset is around the third and fourth decades. In males, the prevalence is highest in Mediterranean, Middle Eastern, and Asian countries. In females, the prevalence is highest in the United States, Northern Europe, and East Asia.10
Tuberculosis
Tubercular skin lesions can present in different forms.13 Lupus vulgaris, the most common, occurs after primary infection and presents as translucent brown nodules, mainly over the face and neck. So-called scrofuloderma is common at the site of a lymph node. It appears as a gradually enlarging subcutaneous nodule followed by skin breaks and ulcerations. Tuberculosis verrucosa cutis, also known as warty tuberculosis, is common in developing countries and presents as warty plaque over the hands, knees, and buttocks.14 Tuberculids are skin reactions to systemic tuberculosis infection.
Herpes simplex virus
Mucocutaneous manifestations of herpes simplex virus affect the oral cavity (gingivostomatitis, pharyngitis, and lip border lesions), the entire integumentary system, the eyes (HSV-1), and the genital region (HSV-2). The classic presentation is systemic symptoms (fever and malaise) associated with multiple vesicles on an erythematous base in a distinct region of skin. The virus can remain latent with reactivation occurring because of illness, immunosuppression, or stress. Pruritus and pain precede the appearance of these lesions.
Cytomegalovirus
Primary cytomegalovirus infection is subclinical in almost all cases unless the patient is immunocompromised, and it presents similarly to mononucleosis induced by Epstein-Barr virus. The skin manifestations are nonspecific and can include macular, maculopapular, morbilliform, and urticarial rashes, but usually not ulcerations.15
OUR PATIENT: BEHÇET DISEASE OR CROHN DISEASE?
In our patient, oral mucosal aphthous ulcers and the location of pustular skin lesions, in addition to pathergy, were highly suggestive of Behçet disease. However, Crohn disease with mucocutaneous manifestations remained in the differential diagnosis.
Because there is significant overlap between these diseases, it is important to know the key distinguishing features. Oral aphthous ulcers, pathergy, uveitis, skin and genital lesions, and neurologic involvement are much more common in Behçet disease than in Crohn disease.16,17 Demographic information was not helpful in this case, given that the patient was adopted.
FURTHER WORKUP
2. What should be the next step in the work-up?
- CT enterography
- Skin biopsy
- Colonoscopy with biopsy
- C-reactive protein, erythrocyte sedimentation rate, and fecal calprotecting testing
The endoscopic appearance and histopathology of the affected tissues are crucial for the diagnosis. Differentiating between Crohn disease and Behçet disease can be particularly challenging because of significant overlap between the intestinal and extraintestinal manifestations of the two diseases, especially the oral lesions and arthralgias. Thus, both colonoscopy with biopsy of the intestinal lesions and biopsy of a cutaneous ulceration should be pursued.

No single test or feature is pathognomonic for Behçet disease. Although many diagnostic criteria have been established, those of the International Study Group (Table 2) are the most widely used.18 Their sensitivity for Behçet disease has been found to be 92%, and their specificity 97%.19
Both CT enterography and inflammatory markers would depict inflammation, but since this is present in both Crohn disease and Behçet disease, these tests would not be helpful in this situation.
Endoscopic appearance of Crohn disease and Behçet disease
Intestinal Behçet disease, like Crohn disease, is an inflammatory bowel disease occurring throughout the gastrointestinal tract (small and large bowel). Both are chronic diseases with a waxing and waning course and have similar extraintestinal manifestations. Typical endoscopic lesions are deep, sharply demarcated (“punched-out”), round ulcers. The intestinal Behçet disease and Crohn disease ulcer phenotype and distribution can look the same, and in both entities, rectal sparing and “skip lesions” have been described.20–22
Nevertheless, findings on endoscopy have been analyzed to try to differentiate between Crohn disease and Behçet disease.
In 2009, Lee et al23 published a simple and accurate strategy for distinguishing the two diseases endoscopically. The authors reviewed 250 patients (115 with Behçet disease, 135 with Crohn disease) with ulcers on colonoscopy and identified 5 endoscopic findings indicative of intestinal Behçet disease:
- Round ulcers
- Focal single or focal multiple distribution of ulcers
- Fewer than 6 ulcers
- Absence of a “cobblestone” appearance
- Absence of aphthous lesions.
The two most accurate factors were absence of a cobblestone appearance (sensitivity 100%) and round ulcer shape (specificity 97.5 %). When more than one factor was present, specificity increased but sensitivity decreased.

Using a classification and regression tree analysis, the investigators created an algorithm that endoscopically differentiates between Crohn disease and Behçet disease (Figure 1) with an accuracy of 92 %.23
Histopathologic analysis of both colonic and skin lesions can provide additional clues to the correct diagnosis. Vasculitis suggests Behçet disease, whereas granulomas suggest Crohn disease.
CASE CONTINUED: SKIN BIOPSY AND COLONOSCOPY
Punch biopsy of the skin was performed on the right anterior thigh. Histopathologic analysis revealed acanthotic epidermis, a discrete full-thickness necrotic ulcer with a neutrophilic base, granulation tissue, and vasculitic changes. There were no vasculitic changes or granulomas outside the ulcer base. Cytomegalovirus staining was negative. An interferon-gamma release assay for tuberculosis was negative. Eye examination results were normal.

Colonoscopy showed multiple deep, round, and confluent ulcers with a punched-out appearance and fissures with normal intervening mucosa in the entire examined colon (Figure 2). The terminal ileal mucosa was normal. Colonic biopsies were consistent with cryptitis and rare crypt abscesses. Vasculitis was not identified.
Although the histologic changes were nonspecific, at this point we considered Behçet disease to be more likely than Crohn disease, given the typical endoscopic appearance and skin changes.
TREATING INTESTINAL BEHÇET DISEASE
3. Which is not considered a standard treatment for intestinal Behçet disease?
- Mesalamine (5-ASA)
- Corticosteroids
- Immunosuppressants
- Mycophenolate mofetil
- Surgery
Overall, data on the management of intestinal Behçet disease are limited. The data that do exist have shown that 5-ASA, corticosteroids, immunosuppressants, and surgery are options, but not mycophenolate mofetil.
Consensus recommendations from the Japanese IBD Research Group,24 published in 2007, included 5-ASA, corticosteroids, immunosuppressants, enteral and total parenteral nutrition, and surgical resection. In 2014, the group published a second consensus statement, adding anti-tumor necrosis factor (TNF) agents as standard therapy for this disease.22
Mycophenolate mofetil has not been shown to be effective in the treatment of mucocutaneous Behçet disease,25 although it may be effective in the treatment of its neurologic manifestations.26 Data regarding its efficacy in intestinal Behçet disease are sparse.
Differences in treatment for Crohn and Behçet disease
Although the treatment options are comparable for Behçet disease and Crohn disease, certain features differ.
Doses of 5-ASA and immunnosuppressive agents are typically higher in Crohn disease. For example, the optimal dose of 5-ASA is up to 3 g/day for Behçet disease but up to 4.8 g/day for Crohn disease.
Standard dosing for azathioprine is 50 to 100 mg/day for Behçet disease but 2 to 2.5 mg/kg/day (eg, 168 to 210 mg/day for a 185-lb patient) for Crohn disease.
In addition, evidence supporting the use of biologic agents such as anti-TNF agents or vedolizumab is more abundant in Crohn disease.
Finally, data on monitoring drug levels of immunomodulators or biologics are available only for patients with Crohn disease, not Behçet disease. Thus, an accurate diagnosis is important.
CASE CONTINUED: EMERGENCY LAPAROTOMY
Our patient continued to experience abdominal pain and bloody diarrhea despite receiving corticosteroids intravenously in high doses. We were also considering anti-TNF therapy.
At this point, CT of her abdomen and pelvis was repeated and showed free intraperitoneal air consistent with a perforation of the transverse colon.
She underwent emergency exploratory laparotomy. Intraoperative findings included pneumoperitoneum but no gross peritoneal contamination, extensive colitis with a contained splenic flexure perforation, and normal small-bowel features without evidence of enteritis. Subtotal colectomy, implantation of the rectal stump into the subcutaneous tissue, and end-ileostomy were performed.
After 23 days of recovery in the hospital, she was discharged on oral antibiotics and 4 weeks of steroid taper.
PROGNOSIS OF INTESTINAL BEHÇET DISEASE
4. What can the patient expect from her intestinal Behçet disease in the future?
- The disease is cured after resection of the diseased segments
- Behçet disease is a progressive lifelong disorder that can recur after surgery
Like Crohn disease, Behçet disease should be considered a lifelong progressive disorder, even after surgical resection of diseased segments.
It is unclear which patients will have a complicated disease course and need treatment with stronger immunosuppression. In patients with intestinal Behçet disease whose disease is in remission on thiopurine therapy, the 1-year relapse rate has been reported as 5.8%, and the 5-year relapse rate 51.7%.27,28 After surgical resection, the 5-year recurrence rate was 47.2%, and 30.6% of patients needed repeat surgery.29 Predictors of poor prognosis were younger age, higher erythrocyte sedimentation rate, higher C-reactive protein level, low albumin level at diagnosis, and a high disease-activity index for intestinal Behçet disease.30
The Korean IBD Study Group has developed and validated a disease activity index for intestinal Behçet disease.28 The index has a list of weighted scores for 8 symptoms, which provides for a more objective assessment of disease activity for determining the best treatment approach.
CASE CONTINUED
The patient has continued with her follow-up care and appointments in gastroenterology, rheumatology, and dermatology clinics. She still complains of intermittent abdominal pain, occasional bleeding at the rectal stump, intermittent skin lesions mainly in the form of pustular lesions, and intermittent joint pain. If symptoms persist, anti-TNF therapy is an option.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol 1981; 5:689–695.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol 2008; 22:1033–1043.
- Timani S, Mutasim DF. Skin manifestations of inflammatory bowel disease. Clin Dermatol 2008; 26:265–273.
- Tavarela Veloso F. Skin complications associated with inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20(suppl 4):50–53.
- Yüksel I, Basar O, Ataseven H, et al. Mucocutaneous manifestations in inflammatory bowel disease. Inflamm Bowel Dis 2009; 15:546–550.
- Lebwohl M, Lebwohl O. Cutaneous manifestations of inflammatory bowel disease. Inflamm Bowel Dis 1998; 4:142–148.
- Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (NY) 2011; 7:235–241.
- Mat C, Yurdakul S, Sevim A, Özyazgan Y, Tüzün Y. Behçet’s syndrome: facts and controversies. Clin Dermatol 2013; 31:352–361.
- Lee ES, Bangz D, Lee S. Dermatologic manifestation of Behçet’s disease. Yonsei Med J 1997; 38:380–389.
- Davatchi F, Chams-Davatchi C, Ghodsi Z, et al. Diagnostic value of pathergy test in Behçet’s disease according to the change of incidence over the time. Clin Rheumatol 2011; 30:1151–1155.
- Friedman-Birnbaum R, Bergman R, Aizen E. Sensitivity and specificity of pathergy test results in Israeli patients with Behçet’s disease. Cutis 1990; 45:261–264.
- Mahr A, Maldini C. Epidemiology of Behçet’s disease. Rev Med Interne 2014; 35:81–89. French.
- Barbagallo J, Tager P, Ingleton R, Hirsch RJ, Weinberg JM. Cutaneous tuberculosis. Am J Clin Dermatol 2002; 3:319–328.
- Padmavathy L, Lakshmana Rao L, Ethirajan N, Ramakrishna Rao M, Subrahmanyan EN, Manohar U. Tuberculosis verrucosa cutis (TBVC)—foot with miliary tuberculosis. Indian J Tuberc 2007; 54:145–148.
- Drago F, Aragone MG, Lugani C, Rebora A. Cytomegalovirus infection in normal and immunocompromised humans. A review. Dermatology 2000; 200:189–195.
- Yazısız V. Similarities and differences between Behçet’s disease and Crohn’s disease. World J Gastrointest Pathophysiol 2014; 5:228–238.
- Chin AB, Kumar AS. Behçet colitis. Clin Colon Rectal Surg 2015; 28:99–102.
- International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990; 335:1078–1080.
- Davatchi F. Diagnosis/classification criteria for Behcet’s disease. Patholog Res Int 2012; 2012:607921.
- Chang DK, Kim JJ, Choi H, et al. Double balloon endoscopy in small intestinal Crohn’s disease and other inflammatory diseases such as cryptogenic multifocal ulcerous stenosing enteritis (CMUSE). Gastrointest Endosc 2007; 66(suppl):S96–S98.
- Hamdulay SS, Cheent K, Ghosh C, Stocks J, Ghosh S, Haskard DO. Wireless capsule endoscopy in the investigation of intestinal Behçet’s syndrome. Rheumatology (Oxford) 2008; 47:1231–1234.
- Hisamatsu T, Ueno F, Matsumoto T, et al. The 2nd edition of consensus statements for the diagnosis and management of intestinal Behçet’s disease: indication of anti-TNFa monoclonal antibodies. J Gastroenterol 2014; 49:156–162.
- Lee SK, Kim BK, Kim TI, Kim WH. Differential diagnosis of intestinal Behçet’s disease and Crohn’s disease by colonoscopic findings. Endoscopy 2009; 41:9–16.
- Kobayashi K, Ueno F, Bito S, et al. Development of consensus statements for the diagnosis and management of intestinal Behçet’s disease using a modified Delphi approach. J Gastroenterol 2007; 42:737–745.
- Adler YD, Mansmann U, Zouboulis CC. Mycophenolate mofetil is ineffective in the treatment of mucocutaneous Adamantiades-Behçet’s disease. Dermatology 2001; 203:322–324.
- Shugaiv E, Tüzün E, Mutlu M, Kiyat-Atamer A, Kurtuncu M, Akman-Demir G. Mycophenolate mofetil as a novel immunosuppressant in the treatment of neuro-Behçet’s disease with parenchymal involvement: presentation of four cases. Clin Exp Rheumatol 2011; 29(suppl 67):S64–S67.
- Jung YS, Cheon JH, Hong SP, Kim TI, Kim WH. Clinical outcomes and prognostic factors for thiopurine maintenance therapy in patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2012; 18:750–757.
- Cheon JH, Han DS, Park JY, et al; Korean IBD Study Group. Development, validation, and responsiveness of a novel disease activity index for intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:605–613.
- Jung YS, Yoon JY, Lee JH, et al. Prognostic factors and long-term clinical outcomes for surgical patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:1594–1602.
- Jung YS, Cheon JH, Park SJ, Hong SP, Kim TI, Kim WH. Clinical course of intestinal Behçet’s disease during the first five years. Dig Dis Sci 2013; 58:496–503.
A 32-year-old woman presented to our emergency department with chest pain and painful ulcerations on her arms, abdomen, back, groin, axillae, and in her mouth. She first noticed the ulcers 7 days earlier.
She also reported bloody diarrhea, which had started 2 years earlier, with 10 or more bowel movements daily. She described her stools as semiformed and associated with urgency and painful abdominal cramps.
Medical history
Her medical history included obstructive sleep apnea and morbid obesity. She had first presented 2 years earlier to another hospital with diarrhea, abdominal pain, and rectal bleeding. At that time, results of esophagogastroduodenoscopy and colonoscopy were reported as normal. Later, she became pregnant, and her symptoms went away. She had a normal pregnancy and delivery.
About 1 year postpartum, her abdominal pain and bloody diarrhea recurred. Colonoscopy showed severe sigmoid inflammation with small, shallow ulcerations and friable mucosa interrupted by areas of normal mucosa. Histopathologic study of the colonic mucosa indicated mild to moderate chronic active colitis consisting of focal areas of cryptitis with occasional crypt abscess formation. She was diagnosed with Crohn colitis based on the endoscopic appearance, histopathology, and clinical presentation. The endoscope, however, could not be advanced beyond the sigmoid colon, which suggested stenosis. She was started on 5-aminosalicylic acid (5-ASA) but developed visual hallucinations, and the medication was stopped.
Her symptoms continued, and she developed worsening rectal bleeding and anemia that required hospitalization and blood transfusions. Another colonoscopy performed 1 month before this emergency department visit had shown multiple mucosal ulcerations, but again, the colonoscope could not be advanced beyond the sigmoid colon. She was started on oral corticosteroids, which provided only minimal clinical improvement.
Her current medications included atenolol (for sinus tachycardia), prednisone (initial dose 60 mg/day tapered to 20 mg/day at presentation), and ciprofloxacin.
Her family history was unknown because she had been adopted.
About 1 week before presentation, she had noticed ulcers developing on her arms, abdomen, back, groin, oral mucosa, and axillae. The ulcers were large and painful, with occasional spontaneous bleeding. She also reported pustules and ulcerations at sites of previous skin punctures, consistent with pathergy.
Findings on presentation
- Temperature 99.5°F (37.5°C)
- Heart rate 124 beats per minute
- Respiratory rate 22 breaths per minute
- Oxygen saturation 100% on room air
- Blood pressure 128/81 mm Hg
- Body mass index 67 kg/m2 (morbidly obese).
She had multiple greyish-white patches and erosions over the soft palate, tongue, and upper and lower lip mucosa, erythematous pustules in the axillae bilaterally, and large erythematous, sharply demarcated ulcerations with a fibrinous base bilaterally covering her arms, thighs, groin, and abdomen.
Blood testing showed multiple abnormal results (Table 1). Urinalysis revealed a urine protein concentration of 100 mg/dL (reference range 0), more than 25 white blood cells per high-power field (reference range < 5), 6 to 10 red blood cells per high-power field (0–3), and more than 10 casts per low-power field (0), which suggested a urinary tract infection with hematuria.
Computed tomography (CT) of the abdomen and pelvis with intravenous and oral contrast showed diffuse fatty infiltration of the liver and wall thickening of the rectum and sigmoid colon.
She was admitted to the medical intensive care unit for potential septic shock. Intravenous vancomycin and ciprofloxacin were started (the latter owing to penicillin allergy).
CAUSES OF DIARRHEA AND SKIN CHANGES
1. What is the most likely diagnosis in our patient?
- Ulcerative colitis
- Crohn disease
- Behçet disease
- Intestinal tuberculosis
- Herpes simplex virus infection
- Cytomegalovirus infection
All of the above can cause diarrhea in combination with mucocutaneous lesions and other manifestations.
Ulcerative colitis and Crohn disease: Mucocutaneous findings
Extraintestinal manifestations of inflammatory bowel diseases (Crohn disease, ulcerative colitis, and Behçet disease) include arthritis, ocular involvement, mucocutaneous manifestations, and liver involvement in the form of primary sclerosing cholangitis. Less common extraintestinal manifestations include vascular, renal, pulmonary, cardiac, and neurologic involvement.
Mucocutaneous findings are observed in 5% to 10% of patients with ulcerative colitis and 20% to 75% of patients with Crohn disease.1–3 The most common are erythema nodosum and pyoderma gangrenosum.4
Yüksel et al5 reported that of 352 patients with inflammatory bowel disease, 7.4% had erythema nodosum and 2.3% had pyoderma gangrenosum. Erythema nodosum was significantly more common in patients with Crohn disease than in those with ulcerative colitis, and its severity was linked with higher disease activity. Lesions frequently resolved when bowel disease subsided.
Lebwohl and Lebwohl6 reported that pyoderma gangrenosum occurred in up to 20% of patients with Crohn disease and up to 10% of those with ulcerative colitis. It is not known whether pyoderma gangrenosum correlates with intestinal disease severity.
Other mucocutaneous manifestations of inflammatory bowel disease include oral aphthous ulcers, acute febrile neutrophilic dermatosis (Sweet syndrome), and metastatic Crohn disease. Aphthous ulcers in the oral cavity, often observed in both Crohn disease and ulcerative colitis, cannot be differentiated on clinical examination from herpes simplex virus (HSV) type 1-induced or idiopathic mucous membrane ulcers. The most common ulcer locations are the lips and buccal mucosa. If biopsied (seldom required), noncaseating granulomas can be identified that are comparable with intestinal mucosal granulomas found in Crohn disease.7
Behçet disease has similar signs
Oral aphthous ulcers are also the most frequent symptom in Behçet disease, occurring in 97% to 100% of cases.8 They most commonly affect the tongue, lips, buccal mucosa, and gingiva.
Cutaneous manifestations include erythema nodosum-like lesions, which present as erythematous painful nodules over pretibial surfaces of the lower limbs but can also affect the arms and thighs; they can also present as papulopustular rosacea eruptions composed of papules, pustules, and noninflammatory comedones, most commonly on the chest, back, and shoulders.8,9
Pathergy, ie, skin hyperresponse to minor trauma such as a bump or bruise, is a typical trait of Behçet disease. A positive pathergy test (ie, skin hyperreactivity to a needlestick or intracutaneous injection) has a specificity of 98.4% in patients with Behçet disease.10
Interestingly, there appears to be a regional difference in the susceptibility to pathergy. While a pathergy response in patients with Behçet disease is rare in the United States and the United Kingdom, it is very common in Japan, Turkey, and Israel.11
Patient demographics also distinguish Behçet disease from Crohn disease. The prevalence of Behçet disease is highest along the Silk Road from the Mediterranean Basin to East Asia and lowest in North America and Northern Europe.12 The mean age at onset is around the third and fourth decades. In males, the prevalence is highest in Mediterranean, Middle Eastern, and Asian countries. In females, the prevalence is highest in the United States, Northern Europe, and East Asia.10
Tuberculosis
Tubercular skin lesions can present in different forms.13 Lupus vulgaris, the most common, occurs after primary infection and presents as translucent brown nodules, mainly over the face and neck. So-called scrofuloderma is common at the site of a lymph node. It appears as a gradually enlarging subcutaneous nodule followed by skin breaks and ulcerations. Tuberculosis verrucosa cutis, also known as warty tuberculosis, is common in developing countries and presents as warty plaque over the hands, knees, and buttocks.14 Tuberculids are skin reactions to systemic tuberculosis infection.
Herpes simplex virus
Mucocutaneous manifestations of herpes simplex virus affect the oral cavity (gingivostomatitis, pharyngitis, and lip border lesions), the entire integumentary system, the eyes (HSV-1), and the genital region (HSV-2). The classic presentation is systemic symptoms (fever and malaise) associated with multiple vesicles on an erythematous base in a distinct region of skin. The virus can remain latent with reactivation occurring because of illness, immunosuppression, or stress. Pruritus and pain precede the appearance of these lesions.
Cytomegalovirus
Primary cytomegalovirus infection is subclinical in almost all cases unless the patient is immunocompromised, and it presents similarly to mononucleosis induced by Epstein-Barr virus. The skin manifestations are nonspecific and can include macular, maculopapular, morbilliform, and urticarial rashes, but usually not ulcerations.15
OUR PATIENT: BEHÇET DISEASE OR CROHN DISEASE?
In our patient, oral mucosal aphthous ulcers and the location of pustular skin lesions, in addition to pathergy, were highly suggestive of Behçet disease. However, Crohn disease with mucocutaneous manifestations remained in the differential diagnosis.
Because there is significant overlap between these diseases, it is important to know the key distinguishing features. Oral aphthous ulcers, pathergy, uveitis, skin and genital lesions, and neurologic involvement are much more common in Behçet disease than in Crohn disease.16,17 Demographic information was not helpful in this case, given that the patient was adopted.
FURTHER WORKUP
2. What should be the next step in the work-up?
- CT enterography
- Skin biopsy
- Colonoscopy with biopsy
- C-reactive protein, erythrocyte sedimentation rate, and fecal calprotecting testing
The endoscopic appearance and histopathology of the affected tissues are crucial for the diagnosis. Differentiating between Crohn disease and Behçet disease can be particularly challenging because of significant overlap between the intestinal and extraintestinal manifestations of the two diseases, especially the oral lesions and arthralgias. Thus, both colonoscopy with biopsy of the intestinal lesions and biopsy of a cutaneous ulceration should be pursued.
No single test or feature is pathognomonic for Behçet disease. Although many diagnostic criteria have been established, those of the International Study Group (Table 2) are the most widely used.18 Their sensitivity for Behçet disease has been found to be 92%, and their specificity 97%.19
Both CT enterography and inflammatory markers would depict inflammation, but since this is present in both Crohn disease and Behçet disease, these tests would not be helpful in this situation.
Endoscopic appearance of Crohn disease and Behçet disease
Intestinal Behçet disease, like Crohn disease, is an inflammatory bowel disease occurring throughout the gastrointestinal tract (small and large bowel). Both are chronic diseases with a waxing and waning course and have similar extraintestinal manifestations. Typical endoscopic lesions are deep, sharply demarcated (“punched-out”), round ulcers. The intestinal Behçet disease and Crohn disease ulcer phenotype and distribution can look the same, and in both entities, rectal sparing and “skip lesions” have been described.20–22
Nevertheless, findings on endoscopy have been analyzed to try to differentiate between Crohn disease and Behçet disease.
In 2009, Lee et al23 published a simple and accurate strategy for distinguishing the two diseases endoscopically. The authors reviewed 250 patients (115 with Behçet disease, 135 with Crohn disease) with ulcers on colonoscopy and identified 5 endoscopic findings indicative of intestinal Behçet disease:
- Round ulcers
- Focal single or focal multiple distribution of ulcers
- Fewer than 6 ulcers
- Absence of a “cobblestone” appearance
- Absence of aphthous lesions.
The two most accurate factors were absence of a cobblestone appearance (sensitivity 100%) and round ulcer shape (specificity 97.5 %). When more than one factor was present, specificity increased but sensitivity decreased.
Using a classification and regression tree analysis, the investigators created an algorithm that endoscopically differentiates between Crohn disease and Behçet disease (Figure 1) with an accuracy of 92 %.23
Histopathologic analysis of both colonic and skin lesions can provide additional clues to the correct diagnosis. Vasculitis suggests Behçet disease, whereas granulomas suggest Crohn disease.
CASE CONTINUED: SKIN BIOPSY AND COLONOSCOPY
Punch biopsy of the skin was performed on the right anterior thigh. Histopathologic analysis revealed acanthotic epidermis, a discrete full-thickness necrotic ulcer with a neutrophilic base, granulation tissue, and vasculitic changes. There were no vasculitic changes or granulomas outside the ulcer base. Cytomegalovirus staining was negative. An interferon-gamma release assay for tuberculosis was negative. Eye examination results were normal.
Colonoscopy showed multiple deep, round, and confluent ulcers with a punched-out appearance and fissures with normal intervening mucosa in the entire examined colon (Figure 2). The terminal ileal mucosa was normal. Colonic biopsies were consistent with cryptitis and rare crypt abscesses. Vasculitis was not identified.
Although the histologic changes were nonspecific, at this point we considered Behçet disease to be more likely than Crohn disease, given the typical endoscopic appearance and skin changes.
TREATING INTESTINAL BEHÇET DISEASE
3. Which is not considered a standard treatment for intestinal Behçet disease?
- Mesalamine (5-ASA)
- Corticosteroids
- Immunosuppressants
- Mycophenolate mofetil
- Surgery
Overall, data on the management of intestinal Behçet disease are limited. The data that do exist have shown that 5-ASA, corticosteroids, immunosuppressants, and surgery are options, but not mycophenolate mofetil.
Consensus recommendations from the Japanese IBD Research Group,24 published in 2007, included 5-ASA, corticosteroids, immunosuppressants, enteral and total parenteral nutrition, and surgical resection. In 2014, the group published a second consensus statement, adding anti-tumor necrosis factor (TNF) agents as standard therapy for this disease.22
Mycophenolate mofetil has not been shown to be effective in the treatment of mucocutaneous Behçet disease,25 although it may be effective in the treatment of its neurologic manifestations.26 Data regarding its efficacy in intestinal Behçet disease are sparse.
Differences in treatment for Crohn and Behçet disease
Although the treatment options are comparable for Behçet disease and Crohn disease, certain features differ.
Doses of 5-ASA and immunnosuppressive agents are typically higher in Crohn disease. For example, the optimal dose of 5-ASA is up to 3 g/day for Behçet disease but up to 4.8 g/day for Crohn disease.
Standard dosing for azathioprine is 50 to 100 mg/day for Behçet disease but 2 to 2.5 mg/kg/day (eg, 168 to 210 mg/day for a 185-lb patient) for Crohn disease.
In addition, evidence supporting the use of biologic agents such as anti-TNF agents or vedolizumab is more abundant in Crohn disease.
Finally, data on monitoring drug levels of immunomodulators or biologics are available only for patients with Crohn disease, not Behçet disease. Thus, an accurate diagnosis is important.
CASE CONTINUED: EMERGENCY LAPAROTOMY
Our patient continued to experience abdominal pain and bloody diarrhea despite receiving corticosteroids intravenously in high doses. We were also considering anti-TNF therapy.
At this point, CT of her abdomen and pelvis was repeated and showed free intraperitoneal air consistent with a perforation of the transverse colon.
She underwent emergency exploratory laparotomy. Intraoperative findings included pneumoperitoneum but no gross peritoneal contamination, extensive colitis with a contained splenic flexure perforation, and normal small-bowel features without evidence of enteritis. Subtotal colectomy, implantation of the rectal stump into the subcutaneous tissue, and end-ileostomy were performed.
After 23 days of recovery in the hospital, she was discharged on oral antibiotics and 4 weeks of steroid taper.
PROGNOSIS OF INTESTINAL BEHÇET DISEASE
4. What can the patient expect from her intestinal Behçet disease in the future?
- The disease is cured after resection of the diseased segments
- Behçet disease is a progressive lifelong disorder that can recur after surgery
Like Crohn disease, Behçet disease should be considered a lifelong progressive disorder, even after surgical resection of diseased segments.
It is unclear which patients will have a complicated disease course and need treatment with stronger immunosuppression. In patients with intestinal Behçet disease whose disease is in remission on thiopurine therapy, the 1-year relapse rate has been reported as 5.8%, and the 5-year relapse rate 51.7%.27,28 After surgical resection, the 5-year recurrence rate was 47.2%, and 30.6% of patients needed repeat surgery.29 Predictors of poor prognosis were younger age, higher erythrocyte sedimentation rate, higher C-reactive protein level, low albumin level at diagnosis, and a high disease-activity index for intestinal Behçet disease.30
The Korean IBD Study Group has developed and validated a disease activity index for intestinal Behçet disease.28 The index has a list of weighted scores for 8 symptoms, which provides for a more objective assessment of disease activity for determining the best treatment approach.
CASE CONTINUED
The patient has continued with her follow-up care and appointments in gastroenterology, rheumatology, and dermatology clinics. She still complains of intermittent abdominal pain, occasional bleeding at the rectal stump, intermittent skin lesions mainly in the form of pustular lesions, and intermittent joint pain. If symptoms persist, anti-TNF therapy is an option.
A 32-year-old woman presented to our emergency department with chest pain and painful ulcerations on her arms, abdomen, back, groin, axillae, and in her mouth. She first noticed the ulcers 7 days earlier.
She also reported bloody diarrhea, which had started 2 years earlier, with 10 or more bowel movements daily. She described her stools as semiformed and associated with urgency and painful abdominal cramps.
Medical history
Her medical history included obstructive sleep apnea and morbid obesity. She had first presented 2 years earlier to another hospital with diarrhea, abdominal pain, and rectal bleeding. At that time, results of esophagogastroduodenoscopy and colonoscopy were reported as normal. Later, she became pregnant, and her symptoms went away. She had a normal pregnancy and delivery.
About 1 year postpartum, her abdominal pain and bloody diarrhea recurred. Colonoscopy showed severe sigmoid inflammation with small, shallow ulcerations and friable mucosa interrupted by areas of normal mucosa. Histopathologic study of the colonic mucosa indicated mild to moderate chronic active colitis consisting of focal areas of cryptitis with occasional crypt abscess formation. She was diagnosed with Crohn colitis based on the endoscopic appearance, histopathology, and clinical presentation. The endoscope, however, could not be advanced beyond the sigmoid colon, which suggested stenosis. She was started on 5-aminosalicylic acid (5-ASA) but developed visual hallucinations, and the medication was stopped.
Her symptoms continued, and she developed worsening rectal bleeding and anemia that required hospitalization and blood transfusions. Another colonoscopy performed 1 month before this emergency department visit had shown multiple mucosal ulcerations, but again, the colonoscope could not be advanced beyond the sigmoid colon. She was started on oral corticosteroids, which provided only minimal clinical improvement.
Her current medications included atenolol (for sinus tachycardia), prednisone (initial dose 60 mg/day tapered to 20 mg/day at presentation), and ciprofloxacin.
Her family history was unknown because she had been adopted.
About 1 week before presentation, she had noticed ulcers developing on her arms, abdomen, back, groin, oral mucosa, and axillae. The ulcers were large and painful, with occasional spontaneous bleeding. She also reported pustules and ulcerations at sites of previous skin punctures, consistent with pathergy.
Findings on presentation
- Temperature 99.5°F (37.5°C)
- Heart rate 124 beats per minute
- Respiratory rate 22 breaths per minute
- Oxygen saturation 100% on room air
- Blood pressure 128/81 mm Hg
- Body mass index 67 kg/m2 (morbidly obese).
She had multiple greyish-white patches and erosions over the soft palate, tongue, and upper and lower lip mucosa, erythematous pustules in the axillae bilaterally, and large erythematous, sharply demarcated ulcerations with a fibrinous base bilaterally covering her arms, thighs, groin, and abdomen.
Blood testing showed multiple abnormal results (Table 1). Urinalysis revealed a urine protein concentration of 100 mg/dL (reference range 0), more than 25 white blood cells per high-power field (reference range < 5), 6 to 10 red blood cells per high-power field (0–3), and more than 10 casts per low-power field (0), which suggested a urinary tract infection with hematuria.
Computed tomography (CT) of the abdomen and pelvis with intravenous and oral contrast showed diffuse fatty infiltration of the liver and wall thickening of the rectum and sigmoid colon.
She was admitted to the medical intensive care unit for potential septic shock. Intravenous vancomycin and ciprofloxacin were started (the latter owing to penicillin allergy).
CAUSES OF DIARRHEA AND SKIN CHANGES
1. What is the most likely diagnosis in our patient?
- Ulcerative colitis
- Crohn disease
- Behçet disease
- Intestinal tuberculosis
- Herpes simplex virus infection
- Cytomegalovirus infection
All of the above can cause diarrhea in combination with mucocutaneous lesions and other manifestations.
Ulcerative colitis and Crohn disease: Mucocutaneous findings
Extraintestinal manifestations of inflammatory bowel diseases (Crohn disease, ulcerative colitis, and Behçet disease) include arthritis, ocular involvement, mucocutaneous manifestations, and liver involvement in the form of primary sclerosing cholangitis. Less common extraintestinal manifestations include vascular, renal, pulmonary, cardiac, and neurologic involvement.
Mucocutaneous findings are observed in 5% to 10% of patients with ulcerative colitis and 20% to 75% of patients with Crohn disease.1–3 The most common are erythema nodosum and pyoderma gangrenosum.4
Yüksel et al5 reported that of 352 patients with inflammatory bowel disease, 7.4% had erythema nodosum and 2.3% had pyoderma gangrenosum. Erythema nodosum was significantly more common in patients with Crohn disease than in those with ulcerative colitis, and its severity was linked with higher disease activity. Lesions frequently resolved when bowel disease subsided.
Lebwohl and Lebwohl6 reported that pyoderma gangrenosum occurred in up to 20% of patients with Crohn disease and up to 10% of those with ulcerative colitis. It is not known whether pyoderma gangrenosum correlates with intestinal disease severity.
Other mucocutaneous manifestations of inflammatory bowel disease include oral aphthous ulcers, acute febrile neutrophilic dermatosis (Sweet syndrome), and metastatic Crohn disease. Aphthous ulcers in the oral cavity, often observed in both Crohn disease and ulcerative colitis, cannot be differentiated on clinical examination from herpes simplex virus (HSV) type 1-induced or idiopathic mucous membrane ulcers. The most common ulcer locations are the lips and buccal mucosa. If biopsied (seldom required), noncaseating granulomas can be identified that are comparable with intestinal mucosal granulomas found in Crohn disease.7
Behçet disease has similar signs
Oral aphthous ulcers are also the most frequent symptom in Behçet disease, occurring in 97% to 100% of cases.8 They most commonly affect the tongue, lips, buccal mucosa, and gingiva.
Cutaneous manifestations include erythema nodosum-like lesions, which present as erythematous painful nodules over pretibial surfaces of the lower limbs but can also affect the arms and thighs; they can also present as papulopustular rosacea eruptions composed of papules, pustules, and noninflammatory comedones, most commonly on the chest, back, and shoulders.8,9
Pathergy, ie, skin hyperresponse to minor trauma such as a bump or bruise, is a typical trait of Behçet disease. A positive pathergy test (ie, skin hyperreactivity to a needlestick or intracutaneous injection) has a specificity of 98.4% in patients with Behçet disease.10
Interestingly, there appears to be a regional difference in the susceptibility to pathergy. While a pathergy response in patients with Behçet disease is rare in the United States and the United Kingdom, it is very common in Japan, Turkey, and Israel.11
Patient demographics also distinguish Behçet disease from Crohn disease. The prevalence of Behçet disease is highest along the Silk Road from the Mediterranean Basin to East Asia and lowest in North America and Northern Europe.12 The mean age at onset is around the third and fourth decades. In males, the prevalence is highest in Mediterranean, Middle Eastern, and Asian countries. In females, the prevalence is highest in the United States, Northern Europe, and East Asia.10
Tuberculosis
Tubercular skin lesions can present in different forms.13 Lupus vulgaris, the most common, occurs after primary infection and presents as translucent brown nodules, mainly over the face and neck. So-called scrofuloderma is common at the site of a lymph node. It appears as a gradually enlarging subcutaneous nodule followed by skin breaks and ulcerations. Tuberculosis verrucosa cutis, also known as warty tuberculosis, is common in developing countries and presents as warty plaque over the hands, knees, and buttocks.14 Tuberculids are skin reactions to systemic tuberculosis infection.
Herpes simplex virus
Mucocutaneous manifestations of herpes simplex virus affect the oral cavity (gingivostomatitis, pharyngitis, and lip border lesions), the entire integumentary system, the eyes (HSV-1), and the genital region (HSV-2). The classic presentation is systemic symptoms (fever and malaise) associated with multiple vesicles on an erythematous base in a distinct region of skin. The virus can remain latent with reactivation occurring because of illness, immunosuppression, or stress. Pruritus and pain precede the appearance of these lesions.
Cytomegalovirus
Primary cytomegalovirus infection is subclinical in almost all cases unless the patient is immunocompromised, and it presents similarly to mononucleosis induced by Epstein-Barr virus. The skin manifestations are nonspecific and can include macular, maculopapular, morbilliform, and urticarial rashes, but usually not ulcerations.15
OUR PATIENT: BEHÇET DISEASE OR CROHN DISEASE?
In our patient, oral mucosal aphthous ulcers and the location of pustular skin lesions, in addition to pathergy, were highly suggestive of Behçet disease. However, Crohn disease with mucocutaneous manifestations remained in the differential diagnosis.
Because there is significant overlap between these diseases, it is important to know the key distinguishing features. Oral aphthous ulcers, pathergy, uveitis, skin and genital lesions, and neurologic involvement are much more common in Behçet disease than in Crohn disease.16,17 Demographic information was not helpful in this case, given that the patient was adopted.
FURTHER WORKUP
2. What should be the next step in the work-up?
- CT enterography
- Skin biopsy
- Colonoscopy with biopsy
- C-reactive protein, erythrocyte sedimentation rate, and fecal calprotecting testing
The endoscopic appearance and histopathology of the affected tissues are crucial for the diagnosis. Differentiating between Crohn disease and Behçet disease can be particularly challenging because of significant overlap between the intestinal and extraintestinal manifestations of the two diseases, especially the oral lesions and arthralgias. Thus, both colonoscopy with biopsy of the intestinal lesions and biopsy of a cutaneous ulceration should be pursued.
No single test or feature is pathognomonic for Behçet disease. Although many diagnostic criteria have been established, those of the International Study Group (Table 2) are the most widely used.18 Their sensitivity for Behçet disease has been found to be 92%, and their specificity 97%.19
Both CT enterography and inflammatory markers would depict inflammation, but since this is present in both Crohn disease and Behçet disease, these tests would not be helpful in this situation.
Endoscopic appearance of Crohn disease and Behçet disease
Intestinal Behçet disease, like Crohn disease, is an inflammatory bowel disease occurring throughout the gastrointestinal tract (small and large bowel). Both are chronic diseases with a waxing and waning course and have similar extraintestinal manifestations. Typical endoscopic lesions are deep, sharply demarcated (“punched-out”), round ulcers. The intestinal Behçet disease and Crohn disease ulcer phenotype and distribution can look the same, and in both entities, rectal sparing and “skip lesions” have been described.20–22
Nevertheless, findings on endoscopy have been analyzed to try to differentiate between Crohn disease and Behçet disease.
In 2009, Lee et al23 published a simple and accurate strategy for distinguishing the two diseases endoscopically. The authors reviewed 250 patients (115 with Behçet disease, 135 with Crohn disease) with ulcers on colonoscopy and identified 5 endoscopic findings indicative of intestinal Behçet disease:
- Round ulcers
- Focal single or focal multiple distribution of ulcers
- Fewer than 6 ulcers
- Absence of a “cobblestone” appearance
- Absence of aphthous lesions.
The two most accurate factors were absence of a cobblestone appearance (sensitivity 100%) and round ulcer shape (specificity 97.5 %). When more than one factor was present, specificity increased but sensitivity decreased.
Using a classification and regression tree analysis, the investigators created an algorithm that endoscopically differentiates between Crohn disease and Behçet disease (Figure 1) with an accuracy of 92 %.23
Histopathologic analysis of both colonic and skin lesions can provide additional clues to the correct diagnosis. Vasculitis suggests Behçet disease, whereas granulomas suggest Crohn disease.
CASE CONTINUED: SKIN BIOPSY AND COLONOSCOPY
Punch biopsy of the skin was performed on the right anterior thigh. Histopathologic analysis revealed acanthotic epidermis, a discrete full-thickness necrotic ulcer with a neutrophilic base, granulation tissue, and vasculitic changes. There were no vasculitic changes or granulomas outside the ulcer base. Cytomegalovirus staining was negative. An interferon-gamma release assay for tuberculosis was negative. Eye examination results were normal.
Colonoscopy showed multiple deep, round, and confluent ulcers with a punched-out appearance and fissures with normal intervening mucosa in the entire examined colon (Figure 2). The terminal ileal mucosa was normal. Colonic biopsies were consistent with cryptitis and rare crypt abscesses. Vasculitis was not identified.
Although the histologic changes were nonspecific, at this point we considered Behçet disease to be more likely than Crohn disease, given the typical endoscopic appearance and skin changes.
TREATING INTESTINAL BEHÇET DISEASE
3. Which is not considered a standard treatment for intestinal Behçet disease?
- Mesalamine (5-ASA)
- Corticosteroids
- Immunosuppressants
- Mycophenolate mofetil
- Surgery
Overall, data on the management of intestinal Behçet disease are limited. The data that do exist have shown that 5-ASA, corticosteroids, immunosuppressants, and surgery are options, but not mycophenolate mofetil.
Consensus recommendations from the Japanese IBD Research Group,24 published in 2007, included 5-ASA, corticosteroids, immunosuppressants, enteral and total parenteral nutrition, and surgical resection. In 2014, the group published a second consensus statement, adding anti-tumor necrosis factor (TNF) agents as standard therapy for this disease.22
Mycophenolate mofetil has not been shown to be effective in the treatment of mucocutaneous Behçet disease,25 although it may be effective in the treatment of its neurologic manifestations.26 Data regarding its efficacy in intestinal Behçet disease are sparse.
Differences in treatment for Crohn and Behçet disease
Although the treatment options are comparable for Behçet disease and Crohn disease, certain features differ.
Doses of 5-ASA and immunnosuppressive agents are typically higher in Crohn disease. For example, the optimal dose of 5-ASA is up to 3 g/day for Behçet disease but up to 4.8 g/day for Crohn disease.
Standard dosing for azathioprine is 50 to 100 mg/day for Behçet disease but 2 to 2.5 mg/kg/day (eg, 168 to 210 mg/day for a 185-lb patient) for Crohn disease.
In addition, evidence supporting the use of biologic agents such as anti-TNF agents or vedolizumab is more abundant in Crohn disease.
Finally, data on monitoring drug levels of immunomodulators or biologics are available only for patients with Crohn disease, not Behçet disease. Thus, an accurate diagnosis is important.
CASE CONTINUED: EMERGENCY LAPAROTOMY
Our patient continued to experience abdominal pain and bloody diarrhea despite receiving corticosteroids intravenously in high doses. We were also considering anti-TNF therapy.
At this point, CT of her abdomen and pelvis was repeated and showed free intraperitoneal air consistent with a perforation of the transverse colon.
She underwent emergency exploratory laparotomy. Intraoperative findings included pneumoperitoneum but no gross peritoneal contamination, extensive colitis with a contained splenic flexure perforation, and normal small-bowel features without evidence of enteritis. Subtotal colectomy, implantation of the rectal stump into the subcutaneous tissue, and end-ileostomy were performed.
After 23 days of recovery in the hospital, she was discharged on oral antibiotics and 4 weeks of steroid taper.
PROGNOSIS OF INTESTINAL BEHÇET DISEASE
4. What can the patient expect from her intestinal Behçet disease in the future?
- The disease is cured after resection of the diseased segments
- Behçet disease is a progressive lifelong disorder that can recur after surgery
Like Crohn disease, Behçet disease should be considered a lifelong progressive disorder, even after surgical resection of diseased segments.
It is unclear which patients will have a complicated disease course and need treatment with stronger immunosuppression. In patients with intestinal Behçet disease whose disease is in remission on thiopurine therapy, the 1-year relapse rate has been reported as 5.8%, and the 5-year relapse rate 51.7%.27,28 After surgical resection, the 5-year recurrence rate was 47.2%, and 30.6% of patients needed repeat surgery.29 Predictors of poor prognosis were younger age, higher erythrocyte sedimentation rate, higher C-reactive protein level, low albumin level at diagnosis, and a high disease-activity index for intestinal Behçet disease.30
The Korean IBD Study Group has developed and validated a disease activity index for intestinal Behçet disease.28 The index has a list of weighted scores for 8 symptoms, which provides for a more objective assessment of disease activity for determining the best treatment approach.
CASE CONTINUED
The patient has continued with her follow-up care and appointments in gastroenterology, rheumatology, and dermatology clinics. She still complains of intermittent abdominal pain, occasional bleeding at the rectal stump, intermittent skin lesions mainly in the form of pustular lesions, and intermittent joint pain. If symptoms persist, anti-TNF therapy is an option.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol 1981; 5:689–695.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol 2008; 22:1033–1043.
- Timani S, Mutasim DF. Skin manifestations of inflammatory bowel disease. Clin Dermatol 2008; 26:265–273.
- Tavarela Veloso F. Skin complications associated with inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20(suppl 4):50–53.
- Yüksel I, Basar O, Ataseven H, et al. Mucocutaneous manifestations in inflammatory bowel disease. Inflamm Bowel Dis 2009; 15:546–550.
- Lebwohl M, Lebwohl O. Cutaneous manifestations of inflammatory bowel disease. Inflamm Bowel Dis 1998; 4:142–148.
- Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (NY) 2011; 7:235–241.
- Mat C, Yurdakul S, Sevim A, Özyazgan Y, Tüzün Y. Behçet’s syndrome: facts and controversies. Clin Dermatol 2013; 31:352–361.
- Lee ES, Bangz D, Lee S. Dermatologic manifestation of Behçet’s disease. Yonsei Med J 1997; 38:380–389.
- Davatchi F, Chams-Davatchi C, Ghodsi Z, et al. Diagnostic value of pathergy test in Behçet’s disease according to the change of incidence over the time. Clin Rheumatol 2011; 30:1151–1155.
- Friedman-Birnbaum R, Bergman R, Aizen E. Sensitivity and specificity of pathergy test results in Israeli patients with Behçet’s disease. Cutis 1990; 45:261–264.
- Mahr A, Maldini C. Epidemiology of Behçet’s disease. Rev Med Interne 2014; 35:81–89. French.
- Barbagallo J, Tager P, Ingleton R, Hirsch RJ, Weinberg JM. Cutaneous tuberculosis. Am J Clin Dermatol 2002; 3:319–328.
- Padmavathy L, Lakshmana Rao L, Ethirajan N, Ramakrishna Rao M, Subrahmanyan EN, Manohar U. Tuberculosis verrucosa cutis (TBVC)—foot with miliary tuberculosis. Indian J Tuberc 2007; 54:145–148.
- Drago F, Aragone MG, Lugani C, Rebora A. Cytomegalovirus infection in normal and immunocompromised humans. A review. Dermatology 2000; 200:189–195.
- Yazısız V. Similarities and differences between Behçet’s disease and Crohn’s disease. World J Gastrointest Pathophysiol 2014; 5:228–238.
- Chin AB, Kumar AS. Behçet colitis. Clin Colon Rectal Surg 2015; 28:99–102.
- International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990; 335:1078–1080.
- Davatchi F. Diagnosis/classification criteria for Behcet’s disease. Patholog Res Int 2012; 2012:607921.
- Chang DK, Kim JJ, Choi H, et al. Double balloon endoscopy in small intestinal Crohn’s disease and other inflammatory diseases such as cryptogenic multifocal ulcerous stenosing enteritis (CMUSE). Gastrointest Endosc 2007; 66(suppl):S96–S98.
- Hamdulay SS, Cheent K, Ghosh C, Stocks J, Ghosh S, Haskard DO. Wireless capsule endoscopy in the investigation of intestinal Behçet’s syndrome. Rheumatology (Oxford) 2008; 47:1231–1234.
- Hisamatsu T, Ueno F, Matsumoto T, et al. The 2nd edition of consensus statements for the diagnosis and management of intestinal Behçet’s disease: indication of anti-TNFa monoclonal antibodies. J Gastroenterol 2014; 49:156–162.
- Lee SK, Kim BK, Kim TI, Kim WH. Differential diagnosis of intestinal Behçet’s disease and Crohn’s disease by colonoscopic findings. Endoscopy 2009; 41:9–16.
- Kobayashi K, Ueno F, Bito S, et al. Development of consensus statements for the diagnosis and management of intestinal Behçet’s disease using a modified Delphi approach. J Gastroenterol 2007; 42:737–745.
- Adler YD, Mansmann U, Zouboulis CC. Mycophenolate mofetil is ineffective in the treatment of mucocutaneous Adamantiades-Behçet’s disease. Dermatology 2001; 203:322–324.
- Shugaiv E, Tüzün E, Mutlu M, Kiyat-Atamer A, Kurtuncu M, Akman-Demir G. Mycophenolate mofetil as a novel immunosuppressant in the treatment of neuro-Behçet’s disease with parenchymal involvement: presentation of four cases. Clin Exp Rheumatol 2011; 29(suppl 67):S64–S67.
- Jung YS, Cheon JH, Hong SP, Kim TI, Kim WH. Clinical outcomes and prognostic factors for thiopurine maintenance therapy in patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2012; 18:750–757.
- Cheon JH, Han DS, Park JY, et al; Korean IBD Study Group. Development, validation, and responsiveness of a novel disease activity index for intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:605–613.
- Jung YS, Yoon JY, Lee JH, et al. Prognostic factors and long-term clinical outcomes for surgical patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:1594–1602.
- Jung YS, Cheon JH, Park SJ, Hong SP, Kim TI, Kim WH. Clinical course of intestinal Behçet’s disease during the first five years. Dig Dis Sci 2013; 58:496–503.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol 1981; 5:689–695.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol 2008; 22:1033–1043.
- Timani S, Mutasim DF. Skin manifestations of inflammatory bowel disease. Clin Dermatol 2008; 26:265–273.
- Tavarela Veloso F. Skin complications associated with inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20(suppl 4):50–53.
- Yüksel I, Basar O, Ataseven H, et al. Mucocutaneous manifestations in inflammatory bowel disease. Inflamm Bowel Dis 2009; 15:546–550.
- Lebwohl M, Lebwohl O. Cutaneous manifestations of inflammatory bowel disease. Inflamm Bowel Dis 1998; 4:142–148.
- Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (NY) 2011; 7:235–241.
- Mat C, Yurdakul S, Sevim A, Özyazgan Y, Tüzün Y. Behçet’s syndrome: facts and controversies. Clin Dermatol 2013; 31:352–361.
- Lee ES, Bangz D, Lee S. Dermatologic manifestation of Behçet’s disease. Yonsei Med J 1997; 38:380–389.
- Davatchi F, Chams-Davatchi C, Ghodsi Z, et al. Diagnostic value of pathergy test in Behçet’s disease according to the change of incidence over the time. Clin Rheumatol 2011; 30:1151–1155.
- Friedman-Birnbaum R, Bergman R, Aizen E. Sensitivity and specificity of pathergy test results in Israeli patients with Behçet’s disease. Cutis 1990; 45:261–264.
- Mahr A, Maldini C. Epidemiology of Behçet’s disease. Rev Med Interne 2014; 35:81–89. French.
- Barbagallo J, Tager P, Ingleton R, Hirsch RJ, Weinberg JM. Cutaneous tuberculosis. Am J Clin Dermatol 2002; 3:319–328.
- Padmavathy L, Lakshmana Rao L, Ethirajan N, Ramakrishna Rao M, Subrahmanyan EN, Manohar U. Tuberculosis verrucosa cutis (TBVC)—foot with miliary tuberculosis. Indian J Tuberc 2007; 54:145–148.
- Drago F, Aragone MG, Lugani C, Rebora A. Cytomegalovirus infection in normal and immunocompromised humans. A review. Dermatology 2000; 200:189–195.
- Yazısız V. Similarities and differences between Behçet’s disease and Crohn’s disease. World J Gastrointest Pathophysiol 2014; 5:228–238.
- Chin AB, Kumar AS. Behçet colitis. Clin Colon Rectal Surg 2015; 28:99–102.
- International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990; 335:1078–1080.
- Davatchi F. Diagnosis/classification criteria for Behcet’s disease. Patholog Res Int 2012; 2012:607921.
- Chang DK, Kim JJ, Choi H, et al. Double balloon endoscopy in small intestinal Crohn’s disease and other inflammatory diseases such as cryptogenic multifocal ulcerous stenosing enteritis (CMUSE). Gastrointest Endosc 2007; 66(suppl):S96–S98.
- Hamdulay SS, Cheent K, Ghosh C, Stocks J, Ghosh S, Haskard DO. Wireless capsule endoscopy in the investigation of intestinal Behçet’s syndrome. Rheumatology (Oxford) 2008; 47:1231–1234.
- Hisamatsu T, Ueno F, Matsumoto T, et al. The 2nd edition of consensus statements for the diagnosis and management of intestinal Behçet’s disease: indication of anti-TNFa monoclonal antibodies. J Gastroenterol 2014; 49:156–162.
- Lee SK, Kim BK, Kim TI, Kim WH. Differential diagnosis of intestinal Behçet’s disease and Crohn’s disease by colonoscopic findings. Endoscopy 2009; 41:9–16.
- Kobayashi K, Ueno F, Bito S, et al. Development of consensus statements for the diagnosis and management of intestinal Behçet’s disease using a modified Delphi approach. J Gastroenterol 2007; 42:737–745.
- Adler YD, Mansmann U, Zouboulis CC. Mycophenolate mofetil is ineffective in the treatment of mucocutaneous Adamantiades-Behçet’s disease. Dermatology 2001; 203:322–324.
- Shugaiv E, Tüzün E, Mutlu M, Kiyat-Atamer A, Kurtuncu M, Akman-Demir G. Mycophenolate mofetil as a novel immunosuppressant in the treatment of neuro-Behçet’s disease with parenchymal involvement: presentation of four cases. Clin Exp Rheumatol 2011; 29(suppl 67):S64–S67.
- Jung YS, Cheon JH, Hong SP, Kim TI, Kim WH. Clinical outcomes and prognostic factors for thiopurine maintenance therapy in patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2012; 18:750–757.
- Cheon JH, Han DS, Park JY, et al; Korean IBD Study Group. Development, validation, and responsiveness of a novel disease activity index for intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:605–613.
- Jung YS, Yoon JY, Lee JH, et al. Prognostic factors and long-term clinical outcomes for surgical patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:1594–1602.
- Jung YS, Cheon JH, Park SJ, Hong SP, Kim TI, Kim WH. Clinical course of intestinal Behçet’s disease during the first five years. Dig Dis Sci 2013; 58:496–503.
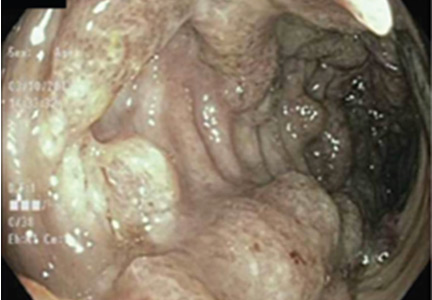
Tdap during pregnancy, or before, offers infants pertussis protection
during the early months of life, according to Tami H. Skoff of the Centers for Disease Control and Prevention, Atlanta, and her associates.
In an analysis of 240 infants younger than 2 months with pertussis cough onset between 2011 and 2015 and 535 control infants, 57% of case mothers and 67% of control mothers had at least one valid Tdap dose; 13% of vaccinated case mothers and 14% of vaccinated control mothers had more than one valid dose of Tdap reported.

Of Tdap doses received during pregnancy in 22 cases and 117 controls, 77% were received during the third trimester, most during the Advisory Committee on Immunization Practices’ recommended 27-36 weeks of gestation. Of the Tdap doses received before pregnancy in mothers of 24 cases and 67 controls, 25% of the case mothers and 67% of the control mothers received Tdap 2 or fewer years before pregnancy.
The effectiveness of Tdap vaccination during the third trimester of pregnancy was 78%, and effectiveness during the first or second trimester was 64%. Effectiveness of Tdap given 2 or fewer years before pregnancy was 83%. This study was not powered to determine a difference if the vaccine was administered in the ACIP-recommended time period during the third trimester.
A reported 49% of U.S. pregnant women received Tdap during the 2015-2016 flu season, an increase of 22% from the 2013-2014 season, according to a CDC Internet panel survey.
“While maternal immunization during pregnancy will help bridge the gap until next-generation pertussis vaccines are licensed and available for use, this highly effective strategy will likely remain an integral component of pertussis prevention and control, even in the setting of new vaccines,” the investigators said.
Read more in Clinical Infectious Diseases (2017 Sep 28. doi: 10.1093/cid/cix724).
during the early months of life, according to Tami H. Skoff of the Centers for Disease Control and Prevention, Atlanta, and her associates.
In an analysis of 240 infants younger than 2 months with pertussis cough onset between 2011 and 2015 and 535 control infants, 57% of case mothers and 67% of control mothers had at least one valid Tdap dose; 13% of vaccinated case mothers and 14% of vaccinated control mothers had more than one valid dose of Tdap reported.
Of Tdap doses received during pregnancy in 22 cases and 117 controls, 77% were received during the third trimester, most during the Advisory Committee on Immunization Practices’ recommended 27-36 weeks of gestation. Of the Tdap doses received before pregnancy in mothers of 24 cases and 67 controls, 25% of the case mothers and 67% of the control mothers received Tdap 2 or fewer years before pregnancy.
The effectiveness of Tdap vaccination during the third trimester of pregnancy was 78%, and effectiveness during the first or second trimester was 64%. Effectiveness of Tdap given 2 or fewer years before pregnancy was 83%. This study was not powered to determine a difference if the vaccine was administered in the ACIP-recommended time period during the third trimester.
A reported 49% of U.S. pregnant women received Tdap during the 2015-2016 flu season, an increase of 22% from the 2013-2014 season, according to a CDC Internet panel survey.
“While maternal immunization during pregnancy will help bridge the gap until next-generation pertussis vaccines are licensed and available for use, this highly effective strategy will likely remain an integral component of pertussis prevention and control, even in the setting of new vaccines,” the investigators said.
Read more in Clinical Infectious Diseases (2017 Sep 28. doi: 10.1093/cid/cix724).
during the early months of life, according to Tami H. Skoff of the Centers for Disease Control and Prevention, Atlanta, and her associates.
In an analysis of 240 infants younger than 2 months with pertussis cough onset between 2011 and 2015 and 535 control infants, 57% of case mothers and 67% of control mothers had at least one valid Tdap dose; 13% of vaccinated case mothers and 14% of vaccinated control mothers had more than one valid dose of Tdap reported.
Of Tdap doses received during pregnancy in 22 cases and 117 controls, 77% were received during the third trimester, most during the Advisory Committee on Immunization Practices’ recommended 27-36 weeks of gestation. Of the Tdap doses received before pregnancy in mothers of 24 cases and 67 controls, 25% of the case mothers and 67% of the control mothers received Tdap 2 or fewer years before pregnancy.
The effectiveness of Tdap vaccination during the third trimester of pregnancy was 78%, and effectiveness during the first or second trimester was 64%. Effectiveness of Tdap given 2 or fewer years before pregnancy was 83%. This study was not powered to determine a difference if the vaccine was administered in the ACIP-recommended time period during the third trimester.
A reported 49% of U.S. pregnant women received Tdap during the 2015-2016 flu season, an increase of 22% from the 2013-2014 season, according to a CDC Internet panel survey.
“While maternal immunization during pregnancy will help bridge the gap until next-generation pertussis vaccines are licensed and available for use, this highly effective strategy will likely remain an integral component of pertussis prevention and control, even in the setting of new vaccines,” the investigators said.
Read more in Clinical Infectious Diseases (2017 Sep 28. doi: 10.1093/cid/cix724).
FROM CLINICAL INFECTIOUS DISEASES
Acute monocular vision loss: Don’t lose sight of the differential
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.

Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history

A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.

An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations

A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).

For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history
A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.
An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations
A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).
For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history
A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.
An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations
A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).
For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
Immunologic testing is key to diagnosing autoimmune blistering diseases
SAN FRANCISCO –
“You have to have some kind of immunological test,” according to Peter Marinkovich, MD. “Pathologists will try to give you as much information as they can on the routine histology, but don’t use that as a diagnostic.”
If not properly identified, autoimmune blistering diseases can lead to chronic overexposure to steroids and resultant side effects without addressing the underlying problem, said Dr. Marinkovich of the department of dermatology at Stanford (Calif.) University.
Dr. Marinkovich gave one example of a patient who had been diagnosed with bullous pemphigoid several years before, and who was becoming Cushingoid as a result of steroids. But the diagnosis was made on the basis of histopathology and clinical appearance alone.
“Nobody had done the immunofluorescence test,” he explained at the annual meeting of the Pacific Dermatologic Association. “I did it, and it turned out she had linear IgA disease. The patient went through 2 years of toxicity just because nobody had done the immunofluorescence test.” Instead, the patient improved when placed on dapsone, which is much less toxic than prednisone.
Direct/indirect immunofluorescence is the highest-yield test for patients with blistering disease. “It’s the best way, I believe, to make the diagnosis,” Dr. Marinkovich said. If that test isn’t available, serum taken during an active phase can also be used. But serum samples can turn up false negatives, so dermatologists should consider collecting and testing serum samples several times.
Another useful tool is salt-split skin analysis, which will demarcate antigens to the roof or floor of the blister. Specifically, it helps distinguish bullous pemphigoid and epidermolysis bullosa acquisita.
In the future, Dr. Marinkovich said, ELISA (enzyme-linked immunosorbent assay) testing will have greater importance for diagnosis and disease monitoring, not just for pemphigus but for subepidermal bullous disorders as well.
Autoimmune blistering diseases do respond to prednisone treatment, although not as well as some other conditions. However, symptom improvement can mask the true cause of the disease.
“It’s easy for physicians to give steroids, and the patients will be happy for the time being; but that doesn’t solve the problem in the long term,” Dr. Marinkovich cautioned. “These are chronic conditions, and the patient will continue to require prednisone, and they’ll get more and more side effects, which could have been avoided if someone had done a more thorough investigation.”
Topical agents such as tetracycline, niacinamide, and topical steroids are more useful in pemphigoid than for pemphigus, because pemphigoid involves local immune factors that the agents can target, while pemphigus can be driven by antibodies alone, which are not as responsive to these treatments.
When systemic therapies are necessary, prednisone is a useful tool, but aim for the lowest possible dose, he said. Reducing prednisone dose is challenging in and of itself. Dropping the dose too quickly can lead to more long-term exposure, because a steep drop can lead to a rebound in the disease, which leads to a higher dose.
“The patient is on this roller coaster ride, up and down, up and down, and that alone can ramp up disease activity,” said Dr. Marinkovich. “Lowering steroid more steadily is a better way to go. This calms the disease down by itself.”
When steroids can’t be completely tapered, which is almost always the case in pemphigus and common in pemphigoid, add steroid-sparing agents such as mycophenolate and azathioprine.
If the steroid-sparing agents don’t get patients down to 10 mg/day prednisone or below, then consider using rituximab and intravenous IgG.
In Europe, physicians are using rituximab earlier in the course of disease, a strategy that appeared effective in a study published in the Lancet (2017 May 20;389[10083]:2031-40). “The evidence suggests to me that earlier use of rituximab tends to reduce the total amount of steroids that the patients are using and has the potential to reduce the duration of the disease,” Dr. Marinkovich said. “That’s a trend that will be going on in the next couple of years here in the United States.”
Dr. Marinkovich is an investigator on a clinical trial funded by Syntimmune.
SAN FRANCISCO –
“You have to have some kind of immunological test,” according to Peter Marinkovich, MD. “Pathologists will try to give you as much information as they can on the routine histology, but don’t use that as a diagnostic.”
If not properly identified, autoimmune blistering diseases can lead to chronic overexposure to steroids and resultant side effects without addressing the underlying problem, said Dr. Marinkovich of the department of dermatology at Stanford (Calif.) University.
Dr. Marinkovich gave one example of a patient who had been diagnosed with bullous pemphigoid several years before, and who was becoming Cushingoid as a result of steroids. But the diagnosis was made on the basis of histopathology and clinical appearance alone.
“Nobody had done the immunofluorescence test,” he explained at the annual meeting of the Pacific Dermatologic Association. “I did it, and it turned out she had linear IgA disease. The patient went through 2 years of toxicity just because nobody had done the immunofluorescence test.” Instead, the patient improved when placed on dapsone, which is much less toxic than prednisone.
Direct/indirect immunofluorescence is the highest-yield test for patients with blistering disease. “It’s the best way, I believe, to make the diagnosis,” Dr. Marinkovich said. If that test isn’t available, serum taken during an active phase can also be used. But serum samples can turn up false negatives, so dermatologists should consider collecting and testing serum samples several times.
Another useful tool is salt-split skin analysis, which will demarcate antigens to the roof or floor of the blister. Specifically, it helps distinguish bullous pemphigoid and epidermolysis bullosa acquisita.
In the future, Dr. Marinkovich said, ELISA (enzyme-linked immunosorbent assay) testing will have greater importance for diagnosis and disease monitoring, not just for pemphigus but for subepidermal bullous disorders as well.
Autoimmune blistering diseases do respond to prednisone treatment, although not as well as some other conditions. However, symptom improvement can mask the true cause of the disease.
“It’s easy for physicians to give steroids, and the patients will be happy for the time being; but that doesn’t solve the problem in the long term,” Dr. Marinkovich cautioned. “These are chronic conditions, and the patient will continue to require prednisone, and they’ll get more and more side effects, which could have been avoided if someone had done a more thorough investigation.”
Topical agents such as tetracycline, niacinamide, and topical steroids are more useful in pemphigoid than for pemphigus, because pemphigoid involves local immune factors that the agents can target, while pemphigus can be driven by antibodies alone, which are not as responsive to these treatments.
When systemic therapies are necessary, prednisone is a useful tool, but aim for the lowest possible dose, he said. Reducing prednisone dose is challenging in and of itself. Dropping the dose too quickly can lead to more long-term exposure, because a steep drop can lead to a rebound in the disease, which leads to a higher dose.
“The patient is on this roller coaster ride, up and down, up and down, and that alone can ramp up disease activity,” said Dr. Marinkovich. “Lowering steroid more steadily is a better way to go. This calms the disease down by itself.”
When steroids can’t be completely tapered, which is almost always the case in pemphigus and common in pemphigoid, add steroid-sparing agents such as mycophenolate and azathioprine.
If the steroid-sparing agents don’t get patients down to 10 mg/day prednisone or below, then consider using rituximab and intravenous IgG.
In Europe, physicians are using rituximab earlier in the course of disease, a strategy that appeared effective in a study published in the Lancet (2017 May 20;389[10083]:2031-40). “The evidence suggests to me that earlier use of rituximab tends to reduce the total amount of steroids that the patients are using and has the potential to reduce the duration of the disease,” Dr. Marinkovich said. “That’s a trend that will be going on in the next couple of years here in the United States.”
Dr. Marinkovich is an investigator on a clinical trial funded by Syntimmune.
SAN FRANCISCO –
“You have to have some kind of immunological test,” according to Peter Marinkovich, MD. “Pathologists will try to give you as much information as they can on the routine histology, but don’t use that as a diagnostic.”
If not properly identified, autoimmune blistering diseases can lead to chronic overexposure to steroids and resultant side effects without addressing the underlying problem, said Dr. Marinkovich of the department of dermatology at Stanford (Calif.) University.
Dr. Marinkovich gave one example of a patient who had been diagnosed with bullous pemphigoid several years before, and who was becoming Cushingoid as a result of steroids. But the diagnosis was made on the basis of histopathology and clinical appearance alone.
“Nobody had done the immunofluorescence test,” he explained at the annual meeting of the Pacific Dermatologic Association. “I did it, and it turned out she had linear IgA disease. The patient went through 2 years of toxicity just because nobody had done the immunofluorescence test.” Instead, the patient improved when placed on dapsone, which is much less toxic than prednisone.
Direct/indirect immunofluorescence is the highest-yield test for patients with blistering disease. “It’s the best way, I believe, to make the diagnosis,” Dr. Marinkovich said. If that test isn’t available, serum taken during an active phase can also be used. But serum samples can turn up false negatives, so dermatologists should consider collecting and testing serum samples several times.
Another useful tool is salt-split skin analysis, which will demarcate antigens to the roof or floor of the blister. Specifically, it helps distinguish bullous pemphigoid and epidermolysis bullosa acquisita.
In the future, Dr. Marinkovich said, ELISA (enzyme-linked immunosorbent assay) testing will have greater importance for diagnosis and disease monitoring, not just for pemphigus but for subepidermal bullous disorders as well.
Autoimmune blistering diseases do respond to prednisone treatment, although not as well as some other conditions. However, symptom improvement can mask the true cause of the disease.
“It’s easy for physicians to give steroids, and the patients will be happy for the time being; but that doesn’t solve the problem in the long term,” Dr. Marinkovich cautioned. “These are chronic conditions, and the patient will continue to require prednisone, and they’ll get more and more side effects, which could have been avoided if someone had done a more thorough investigation.”
Topical agents such as tetracycline, niacinamide, and topical steroids are more useful in pemphigoid than for pemphigus, because pemphigoid involves local immune factors that the agents can target, while pemphigus can be driven by antibodies alone, which are not as responsive to these treatments.
When systemic therapies are necessary, prednisone is a useful tool, but aim for the lowest possible dose, he said. Reducing prednisone dose is challenging in and of itself. Dropping the dose too quickly can lead to more long-term exposure, because a steep drop can lead to a rebound in the disease, which leads to a higher dose.
“The patient is on this roller coaster ride, up and down, up and down, and that alone can ramp up disease activity,” said Dr. Marinkovich. “Lowering steroid more steadily is a better way to go. This calms the disease down by itself.”
When steroids can’t be completely tapered, which is almost always the case in pemphigus and common in pemphigoid, add steroid-sparing agents such as mycophenolate and azathioprine.
If the steroid-sparing agents don’t get patients down to 10 mg/day prednisone or below, then consider using rituximab and intravenous IgG.
In Europe, physicians are using rituximab earlier in the course of disease, a strategy that appeared effective in a study published in the Lancet (2017 May 20;389[10083]:2031-40). “The evidence suggests to me that earlier use of rituximab tends to reduce the total amount of steroids that the patients are using and has the potential to reduce the duration of the disease,” Dr. Marinkovich said. “That’s a trend that will be going on in the next couple of years here in the United States.”
Dr. Marinkovich is an investigator on a clinical trial funded by Syntimmune.
AT PDA 2017
The Benefits of Exercise for Patients With Multiple Sclerosis
Q) Should I recommend exercise to my patients living with MS?
Multiple sclerosis (MS) causes varied symptoms and functional impairment, depending on what part of the central nervous system is involved. Currently, many patients living with MS have sedentary lifestyles, which increases the risk for comorbidities such as cardiovascular disease, type 2 diabetes, and osteoporosis.1-3
Some MS symptoms—ambulatory difficulty, balance impairment, heat intolerance, muscle weakness, spasticity, visual impairment, and fatigue—act as obstacles to routine physical exercise; they also typically worsen over the course of the disease.2-5 In addition, psychosocial factors such as lower levels of education, single status, smoking, and depression or anxiety have been shown to increase the likelihood that a patient will not meet the World Health Organization’s recommendations on physical activity for health.1
For many years, MS patients were advised against physical activity out of concern that it would exacerbate symptoms.6 It is likely still true that patients who fear worsened symptoms or have higher levels of disability avoid physical activity.2-5 Unfortunately, for persons living with MS, this cycle of fear and reduced activity perpetuates itself, resulting in increased disability and decreased quality of life. Thankfully, many of the physical and social factors that prevent patients from exercising are modifiable.1,4
Many types of exercise have been studied in patients living with MS; those shown to be beneficial include regimens focused on cardiovascular fitness, resistance training, balance, and flexibility. Evidence supports the benefits of exercise training for improving overall fitness, muscle strength, ambulation, cognition, spasticity, fatigue, and anxiety and depression in patients with MS.2-4,6-9 Exercise with aerobic, anaerobic, or resistance training has been considered an important nonpharmacologic treatment for MS patients to improve quality of life without worsening disease symptoms.9 There is increasing evidence that engaging in more physical activity and improving physical fitness is an important modality to improve disease course and slow progression over time.
Any increase in symptoms related to exercise is transient, and there is no evidence of lasting harmful effects on overall day-to-day functioning or association with disease progression.6,10 Patient reports of the perceived benefits of exercise include maintenance of physical function, increased social involvement, and feelings of self-management and control.5 Thus, if patients can comply with an exercise regimen, much of the initial disability that limited their activity may be reduced.
More research is needed to fully elucidate what type of exercise is most beneficial for an individual patient.4,5,8,9 However, the benefits of exercise are clear: It can significantly improve quality of life by enhancing psychologic and physical functioning.1,3,5,6,8 Given this information, patients living with MS have incentives to exercise. Health care providers should endorse the benefits of exercise and work to help patients reduce barriers to physical activity.1-5 —RR
Rebecca Rahn, MPA-C, MSCS
Augusta MS Center
Neurology Department, Augusta University, Georgia
1. Reider N, Salter AR, Cutter GR, et al. Potentially modifiable factors associated with physical activity in people living with multiple sclerosis. Res Nurs Health. 2017;40(2):143-152.
2. Sebastiao E, Learmonth YC, Motl RM. Lower physical activity in persons with multiple sclerosis at increased fall risk: a cross sectional study. Am J Phys Med Rehabil. 2017;96:357-361.
3. Vister E, Tijsma ME, Hoang PD, Lord SR. Fatigue, physical activity, quality of life, and fall risk in people with multiple sclerosis. Int J MS Care. 2017;19:91-98.
4. Edwards T, Pilutti LA. The effect of exercise training in adults with multiple sclerosis with severe disability: a systematic review and future research directions. Mult Scler Relat Disord. 2017;16:31-39.
5. Learmonth YC, Motl RW. Physical activity and exercise training in multiple sclerosis: a review and content analysis of qualitative research identifying perceived determinants and consequences. Disabil Rehabil. 2016;38(13):1227-1242.
6. Paul L, Coote S, Crosbie J, et al. Core outcome measures for exercise studies in people with multiple sclerosis: recommendations from a multidisciplinary consensus meeting. Mult Scler. 2014;20(12):1641-1650.
7. Sandroff BM, Motl RW, Scuddler MR, Deluca J. Systematic, evidence-based review of exercise, physical activity, and physical fitness effects on cognition in persons with multiple sclerosis. Neuropsychol Rev. 2016;26(3):271-294.
8. Hugos CL, Bourdette D, Chen YCZ, Cameron M. A group-delivered self-management program reduces spasticity in people with multiple sclerosis: a randomized, controlled pilot trial. Mult Scler J Exp Transl Clin. 2017;3(1):1-11.
9. Alvarenga-Filho H, Sacramento PM, Ferreira TB, et al. Combined exercise training reduces fatigue and modulates the cytokine profile of T-cells from multiple sclerosis patients in response to neuromediators. J Neuroimmunol. 2016;293:91-99.
10. Smith RM, Adeney-Steel M, Fulcher G, Longley WA. Symptom change with exercise is a temporary phenomenon for people with multiple sclerosis. Arch Phys Med Rehabil. 2006;87(5):723-727.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Stacey Panasci, MSPAS, PA-C, who practices at Springfield Neurology Associates, LLC, in Massachusetts, and Rebecca Rahn, MPA-C, MSCS, who is Associate Director of the Augusta MS Center in the Neurology Department of Augusta University in Georgia.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Stacey Panasci, MSPAS, PA-C, who practices at Springfield Neurology Associates, LLC, in Massachusetts, and Rebecca Rahn, MPA-C, MSCS, who is Associate Director of the Augusta MS Center in the Neurology Department of Augusta University in Georgia.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Stacey Panasci, MSPAS, PA-C, who practices at Springfield Neurology Associates, LLC, in Massachusetts, and Rebecca Rahn, MPA-C, MSCS, who is Associate Director of the Augusta MS Center in the Neurology Department of Augusta University in Georgia.
Q) Should I recommend exercise to my patients living with MS?
Multiple sclerosis (MS) causes varied symptoms and functional impairment, depending on what part of the central nervous system is involved. Currently, many patients living with MS have sedentary lifestyles, which increases the risk for comorbidities such as cardiovascular disease, type 2 diabetes, and osteoporosis.1-3
Some MS symptoms—ambulatory difficulty, balance impairment, heat intolerance, muscle weakness, spasticity, visual impairment, and fatigue—act as obstacles to routine physical exercise; they also typically worsen over the course of the disease.2-5 In addition, psychosocial factors such as lower levels of education, single status, smoking, and depression or anxiety have been shown to increase the likelihood that a patient will not meet the World Health Organization’s recommendations on physical activity for health.1
For many years, MS patients were advised against physical activity out of concern that it would exacerbate symptoms.6 It is likely still true that patients who fear worsened symptoms or have higher levels of disability avoid physical activity.2-5 Unfortunately, for persons living with MS, this cycle of fear and reduced activity perpetuates itself, resulting in increased disability and decreased quality of life. Thankfully, many of the physical and social factors that prevent patients from exercising are modifiable.1,4
Many types of exercise have been studied in patients living with MS; those shown to be beneficial include regimens focused on cardiovascular fitness, resistance training, balance, and flexibility. Evidence supports the benefits of exercise training for improving overall fitness, muscle strength, ambulation, cognition, spasticity, fatigue, and anxiety and depression in patients with MS.2-4,6-9 Exercise with aerobic, anaerobic, or resistance training has been considered an important nonpharmacologic treatment for MS patients to improve quality of life without worsening disease symptoms.9 There is increasing evidence that engaging in more physical activity and improving physical fitness is an important modality to improve disease course and slow progression over time.
Any increase in symptoms related to exercise is transient, and there is no evidence of lasting harmful effects on overall day-to-day functioning or association with disease progression.6,10 Patient reports of the perceived benefits of exercise include maintenance of physical function, increased social involvement, and feelings of self-management and control.5 Thus, if patients can comply with an exercise regimen, much of the initial disability that limited their activity may be reduced.
More research is needed to fully elucidate what type of exercise is most beneficial for an individual patient.4,5,8,9 However, the benefits of exercise are clear: It can significantly improve quality of life by enhancing psychologic and physical functioning.1,3,5,6,8 Given this information, patients living with MS have incentives to exercise. Health care providers should endorse the benefits of exercise and work to help patients reduce barriers to physical activity.1-5 —RR
Rebecca Rahn, MPA-C, MSCS
Augusta MS Center
Neurology Department, Augusta University, Georgia
Q) Should I recommend exercise to my patients living with MS?
Multiple sclerosis (MS) causes varied symptoms and functional impairment, depending on what part of the central nervous system is involved. Currently, many patients living with MS have sedentary lifestyles, which increases the risk for comorbidities such as cardiovascular disease, type 2 diabetes, and osteoporosis.1-3
Some MS symptoms—ambulatory difficulty, balance impairment, heat intolerance, muscle weakness, spasticity, visual impairment, and fatigue—act as obstacles to routine physical exercise; they also typically worsen over the course of the disease.2-5 In addition, psychosocial factors such as lower levels of education, single status, smoking, and depression or anxiety have been shown to increase the likelihood that a patient will not meet the World Health Organization’s recommendations on physical activity for health.1
For many years, MS patients were advised against physical activity out of concern that it would exacerbate symptoms.6 It is likely still true that patients who fear worsened symptoms or have higher levels of disability avoid physical activity.2-5 Unfortunately, for persons living with MS, this cycle of fear and reduced activity perpetuates itself, resulting in increased disability and decreased quality of life. Thankfully, many of the physical and social factors that prevent patients from exercising are modifiable.1,4
Many types of exercise have been studied in patients living with MS; those shown to be beneficial include regimens focused on cardiovascular fitness, resistance training, balance, and flexibility. Evidence supports the benefits of exercise training for improving overall fitness, muscle strength, ambulation, cognition, spasticity, fatigue, and anxiety and depression in patients with MS.2-4,6-9 Exercise with aerobic, anaerobic, or resistance training has been considered an important nonpharmacologic treatment for MS patients to improve quality of life without worsening disease symptoms.9 There is increasing evidence that engaging in more physical activity and improving physical fitness is an important modality to improve disease course and slow progression over time.
Any increase in symptoms related to exercise is transient, and there is no evidence of lasting harmful effects on overall day-to-day functioning or association with disease progression.6,10 Patient reports of the perceived benefits of exercise include maintenance of physical function, increased social involvement, and feelings of self-management and control.5 Thus, if patients can comply with an exercise regimen, much of the initial disability that limited their activity may be reduced.
More research is needed to fully elucidate what type of exercise is most beneficial for an individual patient.4,5,8,9 However, the benefits of exercise are clear: It can significantly improve quality of life by enhancing psychologic and physical functioning.1,3,5,6,8 Given this information, patients living with MS have incentives to exercise. Health care providers should endorse the benefits of exercise and work to help patients reduce barriers to physical activity.1-5 —RR
Rebecca Rahn, MPA-C, MSCS
Augusta MS Center
Neurology Department, Augusta University, Georgia
1. Reider N, Salter AR, Cutter GR, et al. Potentially modifiable factors associated with physical activity in people living with multiple sclerosis. Res Nurs Health. 2017;40(2):143-152.
2. Sebastiao E, Learmonth YC, Motl RM. Lower physical activity in persons with multiple sclerosis at increased fall risk: a cross sectional study. Am J Phys Med Rehabil. 2017;96:357-361.
3. Vister E, Tijsma ME, Hoang PD, Lord SR. Fatigue, physical activity, quality of life, and fall risk in people with multiple sclerosis. Int J MS Care. 2017;19:91-98.
4. Edwards T, Pilutti LA. The effect of exercise training in adults with multiple sclerosis with severe disability: a systematic review and future research directions. Mult Scler Relat Disord. 2017;16:31-39.
5. Learmonth YC, Motl RW. Physical activity and exercise training in multiple sclerosis: a review and content analysis of qualitative research identifying perceived determinants and consequences. Disabil Rehabil. 2016;38(13):1227-1242.
6. Paul L, Coote S, Crosbie J, et al. Core outcome measures for exercise studies in people with multiple sclerosis: recommendations from a multidisciplinary consensus meeting. Mult Scler. 2014;20(12):1641-1650.
7. Sandroff BM, Motl RW, Scuddler MR, Deluca J. Systematic, evidence-based review of exercise, physical activity, and physical fitness effects on cognition in persons with multiple sclerosis. Neuropsychol Rev. 2016;26(3):271-294.
8. Hugos CL, Bourdette D, Chen YCZ, Cameron M. A group-delivered self-management program reduces spasticity in people with multiple sclerosis: a randomized, controlled pilot trial. Mult Scler J Exp Transl Clin. 2017;3(1):1-11.
9. Alvarenga-Filho H, Sacramento PM, Ferreira TB, et al. Combined exercise training reduces fatigue and modulates the cytokine profile of T-cells from multiple sclerosis patients in response to neuromediators. J Neuroimmunol. 2016;293:91-99.
10. Smith RM, Adeney-Steel M, Fulcher G, Longley WA. Symptom change with exercise is a temporary phenomenon for people with multiple sclerosis. Arch Phys Med Rehabil. 2006;87(5):723-727.
1. Reider N, Salter AR, Cutter GR, et al. Potentially modifiable factors associated with physical activity in people living with multiple sclerosis. Res Nurs Health. 2017;40(2):143-152.
2. Sebastiao E, Learmonth YC, Motl RM. Lower physical activity in persons with multiple sclerosis at increased fall risk: a cross sectional study. Am J Phys Med Rehabil. 2017;96:357-361.
3. Vister E, Tijsma ME, Hoang PD, Lord SR. Fatigue, physical activity, quality of life, and fall risk in people with multiple sclerosis. Int J MS Care. 2017;19:91-98.
4. Edwards T, Pilutti LA. The effect of exercise training in adults with multiple sclerosis with severe disability: a systematic review and future research directions. Mult Scler Relat Disord. 2017;16:31-39.
5. Learmonth YC, Motl RW. Physical activity and exercise training in multiple sclerosis: a review and content analysis of qualitative research identifying perceived determinants and consequences. Disabil Rehabil. 2016;38(13):1227-1242.
6. Paul L, Coote S, Crosbie J, et al. Core outcome measures for exercise studies in people with multiple sclerosis: recommendations from a multidisciplinary consensus meeting. Mult Scler. 2014;20(12):1641-1650.
7. Sandroff BM, Motl RW, Scuddler MR, Deluca J. Systematic, evidence-based review of exercise, physical activity, and physical fitness effects on cognition in persons with multiple sclerosis. Neuropsychol Rev. 2016;26(3):271-294.
8. Hugos CL, Bourdette D, Chen YCZ, Cameron M. A group-delivered self-management program reduces spasticity in people with multiple sclerosis: a randomized, controlled pilot trial. Mult Scler J Exp Transl Clin. 2017;3(1):1-11.
9. Alvarenga-Filho H, Sacramento PM, Ferreira TB, et al. Combined exercise training reduces fatigue and modulates the cytokine profile of T-cells from multiple sclerosis patients in response to neuromediators. J Neuroimmunol. 2016;293:91-99.
10. Smith RM, Adeney-Steel M, Fulcher G, Longley WA. Symptom change with exercise is a temporary phenomenon for people with multiple sclerosis. Arch Phys Med Rehabil. 2006;87(5):723-727.

Does Diet Matter in Multiple Sclerosis?
Q) What is known about the impact of diet on multiple sclerosis? How can I advise my patients with MS?
Multiple sclerosis (MS) is a chronic inflammatory and degenerative central nervous system disease affecting more than 2.5 million people worldwide. Today, if a Google search is performed for “diet and MS,” more than 67 million results are obtained. Many tout specific protocols as beneficial for MS but have no substantial data to support these claims. This can be confusing for patients as well as providers. How should you advise those who ask for advice on dietary modifications to help control symptoms or disease course?
First, it’s important to remember that individuals with MS have a reduced median lifespan (by about seven years), compared to healthy controls. Furthermore, patients with MS commonly have comorbid conditions—such as diabetes, obesity, and ischemic heart disease—that increase mortality risk.1,2 Diet and nutrition are significant factors that impact the course of these diseases.
We must also bear in mind that patients with MS experience symptoms that may impede their efforts to prepare meals. In a 2008 study of 123 MS patients (more than 50% of whom were overweight or obese), fatigue was cited as a significant factor that limited cooking and food preparation. Cognitive impairment and depression also may affect dietary intake. Interestingly, the average recorded intake for all food groups was less than that recommended in the Dietary Guidelines for Americans.3
A web-based survey conducted by the German MS Society in 2011 revealed that 42% of the 337 respondents had modified their diet due to MS. These modifications included change in intake of fatty acids; decrease or elimination of meat, sugar, and additives; and introduction of a low-carb or Paleo diet.4
Among an international sample of 2,087 MS patients, a significant association was found between a healthy diet and improved quality of life (both physical and mental) and reduced disability. This “healthy consumption” of fruits, vegetables, and dietary fat was also associated with a marginally decreased risk for relapse. Patients who demonstrated increased disease activity were more likely to have poor consumption of fruits, vegetables, and fats and to consume more meat and dairy products.5
There has also been research on specific components of dietary intake. Antioxidant-containing foods, for example, may have an anti-inflammatory effect.6 Vitamin B12 deficiency plays a role in immunomodulatory effect, as well as formation of the myelin sheath, although its role (and the effect of biotin supplementation) in MS disease progression requires further study.7 Also ongoing is research into various calorie-restriction protocols, altering both timing and amount of caloric intake, since some data suggest this strategy reduces leptin, a satiety hormone that increases inflammation and has been shown to promote more aggressive MS in a mouse model.8
In the meantime, what can we conclude about diet and MS? A recent review determined that, although there is insufficient data to support one specific diet, there is sufficient evidence to recommend consumption of fish, foods lower in fat, whole grains, vitamin D, and supplemental omega fatty acids.5
It is important to discuss diet with our MS patients. In the German survey, 82% of patients felt that diet was important, yet only 10% had asked a provider for nutritional advice.4 In another study, patients indicated that food labels were their top source for nutrition information; only 20% sought advice from a nutritionist.3 We need to ask our MS patients if they are following a particular diet and be prepared to discuss potentially beneficial dietary choices with them—and offer referral to a nutritionist to those who require additional direction and support.—SP
Stacey Panasci, MSPAS, PA-C
Springfield Neurology Associates, LLC
Massachusetts
1. Marrie RA, Elliott L, Marriott J, et al. Effect of comorbidity on mortality in multiple sclerosis. Neurology. 2015;85(3):240-247.
2. Langer-Gould A, Brara SM, Beaber BE, Koebnick C. Childhood obesity and risk of pediatric multiple sclerosis and clinically isolated syndrome. Neurology. 2013;80(6):548-552.
3. Goodman S, Gulick EE. Dietary practices of people with multiple sclerosis. Int J MS Care. 2008;10:47-57.
4. Riemann- Lorenz K, Eilers M, von Geldern G, et al. Dietary interventions in multiple sclerosis: development and pilot testing of an evidence based patient education program. PLoS One. 2016;11(10):e0165246.
5. Hadgkiss EJ, Jekinek GA, Weiland TJ, et al. The association of diet with quality of life, disability, and relapse rate in an international sample of people with multiple sclerosis. Nutr Neurosci. 2015;18(3):125-136.
6. Khalili M, Azimi A, Izadi V, et al. Does lipoic acid consumption affect the cytokine profile in multiple sclerosis patients: a double-blind, placebo-controlled, randomized clinical trial. Neuroimmunomodulation. 2014;21(6):291-296.
7. Kocer B, Engur S, Ak F, Yilmaz M. Serum vitamin B12, folate, and homocysteine levels and their association with clinical and electrophysiological parameters in multiple sclerosis.
J Clin Neurosci. 2009;16:399-403.
8. Galgani M, Procaccini C, De Rosa V, et al. Leptin modulates the survival of autoreactive CD4+ T cells through the nutrient/energy-sensing mammalian target of rapamycin signaling pathway. J Immunol. 2010;185(12):7474-7479.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Stacey Panasci, MSPAS, PA-C, who practices at Springfield Neurology Associates, LLC, in Massachusetts, and Rebecca Rahn, MPA-C, MSCS, who is Associate Director of the Augusta MS Center in the Neurology Department of Augusta University in Georgia.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Stacey Panasci, MSPAS, PA-C, who practices at Springfield Neurology Associates, LLC, in Massachusetts, and Rebecca Rahn, MPA-C, MSCS, who is Associate Director of the Augusta MS Center in the Neurology Department of Augusta University in Georgia.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Stacey Panasci, MSPAS, PA-C, who practices at Springfield Neurology Associates, LLC, in Massachusetts, and Rebecca Rahn, MPA-C, MSCS, who is Associate Director of the Augusta MS Center in the Neurology Department of Augusta University in Georgia.
Q) What is known about the impact of diet on multiple sclerosis? How can I advise my patients with MS?
Multiple sclerosis (MS) is a chronic inflammatory and degenerative central nervous system disease affecting more than 2.5 million people worldwide. Today, if a Google search is performed for “diet and MS,” more than 67 million results are obtained. Many tout specific protocols as beneficial for MS but have no substantial data to support these claims. This can be confusing for patients as well as providers. How should you advise those who ask for advice on dietary modifications to help control symptoms or disease course?
First, it’s important to remember that individuals with MS have a reduced median lifespan (by about seven years), compared to healthy controls. Furthermore, patients with MS commonly have comorbid conditions—such as diabetes, obesity, and ischemic heart disease—that increase mortality risk.1,2 Diet and nutrition are significant factors that impact the course of these diseases.
We must also bear in mind that patients with MS experience symptoms that may impede their efforts to prepare meals. In a 2008 study of 123 MS patients (more than 50% of whom were overweight or obese), fatigue was cited as a significant factor that limited cooking and food preparation. Cognitive impairment and depression also may affect dietary intake. Interestingly, the average recorded intake for all food groups was less than that recommended in the Dietary Guidelines for Americans.3
A web-based survey conducted by the German MS Society in 2011 revealed that 42% of the 337 respondents had modified their diet due to MS. These modifications included change in intake of fatty acids; decrease or elimination of meat, sugar, and additives; and introduction of a low-carb or Paleo diet.4
Among an international sample of 2,087 MS patients, a significant association was found between a healthy diet and improved quality of life (both physical and mental) and reduced disability. This “healthy consumption” of fruits, vegetables, and dietary fat was also associated with a marginally decreased risk for relapse. Patients who demonstrated increased disease activity were more likely to have poor consumption of fruits, vegetables, and fats and to consume more meat and dairy products.5
There has also been research on specific components of dietary intake. Antioxidant-containing foods, for example, may have an anti-inflammatory effect.6 Vitamin B12 deficiency plays a role in immunomodulatory effect, as well as formation of the myelin sheath, although its role (and the effect of biotin supplementation) in MS disease progression requires further study.7 Also ongoing is research into various calorie-restriction protocols, altering both timing and amount of caloric intake, since some data suggest this strategy reduces leptin, a satiety hormone that increases inflammation and has been shown to promote more aggressive MS in a mouse model.8
In the meantime, what can we conclude about diet and MS? A recent review determined that, although there is insufficient data to support one specific diet, there is sufficient evidence to recommend consumption of fish, foods lower in fat, whole grains, vitamin D, and supplemental omega fatty acids.5
It is important to discuss diet with our MS patients. In the German survey, 82% of patients felt that diet was important, yet only 10% had asked a provider for nutritional advice.4 In another study, patients indicated that food labels were their top source for nutrition information; only 20% sought advice from a nutritionist.3 We need to ask our MS patients if they are following a particular diet and be prepared to discuss potentially beneficial dietary choices with them—and offer referral to a nutritionist to those who require additional direction and support.—SP
Stacey Panasci, MSPAS, PA-C
Springfield Neurology Associates, LLC
Massachusetts
Q) What is known about the impact of diet on multiple sclerosis? How can I advise my patients with MS?
Multiple sclerosis (MS) is a chronic inflammatory and degenerative central nervous system disease affecting more than 2.5 million people worldwide. Today, if a Google search is performed for “diet and MS,” more than 67 million results are obtained. Many tout specific protocols as beneficial for MS but have no substantial data to support these claims. This can be confusing for patients as well as providers. How should you advise those who ask for advice on dietary modifications to help control symptoms or disease course?
First, it’s important to remember that individuals with MS have a reduced median lifespan (by about seven years), compared to healthy controls. Furthermore, patients with MS commonly have comorbid conditions—such as diabetes, obesity, and ischemic heart disease—that increase mortality risk.1,2 Diet and nutrition are significant factors that impact the course of these diseases.
We must also bear in mind that patients with MS experience symptoms that may impede their efforts to prepare meals. In a 2008 study of 123 MS patients (more than 50% of whom were overweight or obese), fatigue was cited as a significant factor that limited cooking and food preparation. Cognitive impairment and depression also may affect dietary intake. Interestingly, the average recorded intake for all food groups was less than that recommended in the Dietary Guidelines for Americans.3
A web-based survey conducted by the German MS Society in 2011 revealed that 42% of the 337 respondents had modified their diet due to MS. These modifications included change in intake of fatty acids; decrease or elimination of meat, sugar, and additives; and introduction of a low-carb or Paleo diet.4
Among an international sample of 2,087 MS patients, a significant association was found between a healthy diet and improved quality of life (both physical and mental) and reduced disability. This “healthy consumption” of fruits, vegetables, and dietary fat was also associated with a marginally decreased risk for relapse. Patients who demonstrated increased disease activity were more likely to have poor consumption of fruits, vegetables, and fats and to consume more meat and dairy products.5
There has also been research on specific components of dietary intake. Antioxidant-containing foods, for example, may have an anti-inflammatory effect.6 Vitamin B12 deficiency plays a role in immunomodulatory effect, as well as formation of the myelin sheath, although its role (and the effect of biotin supplementation) in MS disease progression requires further study.7 Also ongoing is research into various calorie-restriction protocols, altering both timing and amount of caloric intake, since some data suggest this strategy reduces leptin, a satiety hormone that increases inflammation and has been shown to promote more aggressive MS in a mouse model.8
In the meantime, what can we conclude about diet and MS? A recent review determined that, although there is insufficient data to support one specific diet, there is sufficient evidence to recommend consumption of fish, foods lower in fat, whole grains, vitamin D, and supplemental omega fatty acids.5
It is important to discuss diet with our MS patients. In the German survey, 82% of patients felt that diet was important, yet only 10% had asked a provider for nutritional advice.4 In another study, patients indicated that food labels were their top source for nutrition information; only 20% sought advice from a nutritionist.3 We need to ask our MS patients if they are following a particular diet and be prepared to discuss potentially beneficial dietary choices with them—and offer referral to a nutritionist to those who require additional direction and support.—SP
Stacey Panasci, MSPAS, PA-C
Springfield Neurology Associates, LLC
Massachusetts
1. Marrie RA, Elliott L, Marriott J, et al. Effect of comorbidity on mortality in multiple sclerosis. Neurology. 2015;85(3):240-247.
2. Langer-Gould A, Brara SM, Beaber BE, Koebnick C. Childhood obesity and risk of pediatric multiple sclerosis and clinically isolated syndrome. Neurology. 2013;80(6):548-552.
3. Goodman S, Gulick EE. Dietary practices of people with multiple sclerosis. Int J MS Care. 2008;10:47-57.
4. Riemann- Lorenz K, Eilers M, von Geldern G, et al. Dietary interventions in multiple sclerosis: development and pilot testing of an evidence based patient education program. PLoS One. 2016;11(10):e0165246.
5. Hadgkiss EJ, Jekinek GA, Weiland TJ, et al. The association of diet with quality of life, disability, and relapse rate in an international sample of people with multiple sclerosis. Nutr Neurosci. 2015;18(3):125-136.
6. Khalili M, Azimi A, Izadi V, et al. Does lipoic acid consumption affect the cytokine profile in multiple sclerosis patients: a double-blind, placebo-controlled, randomized clinical trial. Neuroimmunomodulation. 2014;21(6):291-296.
7. Kocer B, Engur S, Ak F, Yilmaz M. Serum vitamin B12, folate, and homocysteine levels and their association with clinical and electrophysiological parameters in multiple sclerosis.
J Clin Neurosci. 2009;16:399-403.
8. Galgani M, Procaccini C, De Rosa V, et al. Leptin modulates the survival of autoreactive CD4+ T cells through the nutrient/energy-sensing mammalian target of rapamycin signaling pathway. J Immunol. 2010;185(12):7474-7479.
1. Marrie RA, Elliott L, Marriott J, et al. Effect of comorbidity on mortality in multiple sclerosis. Neurology. 2015;85(3):240-247.
2. Langer-Gould A, Brara SM, Beaber BE, Koebnick C. Childhood obesity and risk of pediatric multiple sclerosis and clinically isolated syndrome. Neurology. 2013;80(6):548-552.
3. Goodman S, Gulick EE. Dietary practices of people with multiple sclerosis. Int J MS Care. 2008;10:47-57.
4. Riemann- Lorenz K, Eilers M, von Geldern G, et al. Dietary interventions in multiple sclerosis: development and pilot testing of an evidence based patient education program. PLoS One. 2016;11(10):e0165246.
5. Hadgkiss EJ, Jekinek GA, Weiland TJ, et al. The association of diet with quality of life, disability, and relapse rate in an international sample of people with multiple sclerosis. Nutr Neurosci. 2015;18(3):125-136.
6. Khalili M, Azimi A, Izadi V, et al. Does lipoic acid consumption affect the cytokine profile in multiple sclerosis patients: a double-blind, placebo-controlled, randomized clinical trial. Neuroimmunomodulation. 2014;21(6):291-296.
7. Kocer B, Engur S, Ak F, Yilmaz M. Serum vitamin B12, folate, and homocysteine levels and their association with clinical and electrophysiological parameters in multiple sclerosis.
J Clin Neurosci. 2009;16:399-403.
8. Galgani M, Procaccini C, De Rosa V, et al. Leptin modulates the survival of autoreactive CD4+ T cells through the nutrient/energy-sensing mammalian target of rapamycin signaling pathway. J Immunol. 2010;185(12):7474-7479.
How to Increase HPV Vaccination Rates
CE/CME No: CR-1709
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand and identify the low- and high-risk human papillomavirus (HPV) types that can lead to benign and malignant manifestations.
• Know the recommended age range and dosing schedule for individuals who can and should receive the vaccination.
• Recognize important barriers to HPV vaccination in the health care setting.
• Understand how to promote HPV vaccination to parents/caregivers and patients.
• Find resources and educational material from national organizations that recommend and support HPV vaccination.
FACULTY
Tyler Cole practices at Coastal Community Health Services in Brunswick, Georgia, and is a clinical instructor in the DNP-APRN program at the Medical University of South Carolina (MUSC). Marie C. Thomas is a registered nurse on a surgical oncology unit at MUSC and will receive her DNP-FNP from MUSC in December 2017. Katlyn Straup practices at Roper St. Francis Healthcare and Southern Care Hospice in Charleston, South Carolina; she is also a clinical associate faculty member in the MUSC College of Nursing. Ashlyn Savage is an Associate Professor of Obstetrics and Gynecology at MUSC College of Nursing and is certified by the American Board of Obstetrics and Gynecology.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through August 31, 2018.
Article begins on next page >>
Although human papillomavirus (HPV) vaccine is a safe and effective means of preventing most HPV-related cancers, HPV vaccination rates lag well behind those of other vaccines recommended for children and adolescents. Understanding the barriers to HPV vaccine acceptance and effective strategies for overcoming them will improve vaccine uptake and completion in adolescents.
Human papillomavirus (HPV) infection is the most common sexually transmitted infection in the United States.1,2 HPV causes approximately 30,700 new cancer cases in the US annually.3 It is the primary cause of cervical cancer, which resulted in more than 4,000 deaths in the US in 2016.4 HPV is also associated with some vaginal, vulvar, penile, anal, and oropharyngeal cancers and causes anogenital warts.3
Although HPV vaccines are available to protect against infection with the HPV types that lead to these sequelae, HPV vaccination rates remain low compared with other routinely administered vaccines.5 Reasons for these lower rates include vaccine cost, lack of patient and provider education, providers’ failure to recommend, stigmas related to sexual behavior, and misconceptions about the vaccine, such as concerns about harm.5 This article discusses these barriers to better educate providers about the HPV vaccine and encourage them to assist in increasing vaccination rates.
EPIDEMIOLOGY
Approximately 79 million Americans are currently infected with HPV, and 14 million new cases are reported each year.2 In the US, the prevalence of HPV is highest among sexually active adolescents and young adults, especially those ages 20 to 24.2 Of the more than 150 types of HPV that have been identified, 40 infect the genital area. HPV genital infections are mainly spread through sexual intercourse but can also be spread through oral-to-genital contact.2
The genital HPV types are categorized as low-risk and high-risk based on their association with cervical cancer.2 High-risk types 16 and 18 are the most troublesome, accounting for 63% of all HPV-associated cancers, with HPV 16 posing the highest risk for cancer.3 High-risk types HPV 31, 33, 45, 52, and 58 account for another 10% of these cancers.3 Low-risk types, such as HPV 6 and 11, can cause low-grade cervical intraepithelial lesions, and HPV 6 and 11 account for more than 90% of genital warts.2
Most HPV infections, whether with high- or low-risk types, do not cause symptoms and resolve spontaneously in about two years.2 Persistent high-risk HPV infection is necessary for the development of cervical cancer precursor lesions—and therefore, once the infection has cleared, the risk for cancer declines.2
HPV VACCINES
Three HPV vaccines are licensed for use in the US: bivalent (Cervarix), quadrivalent (Gardasil), and 9-valent (Gardasil 9) vaccines (see Table 1).2,6,7 The bivalent, Cervarix, has recently been removed from the US market due to a decrease in product demand.6,8
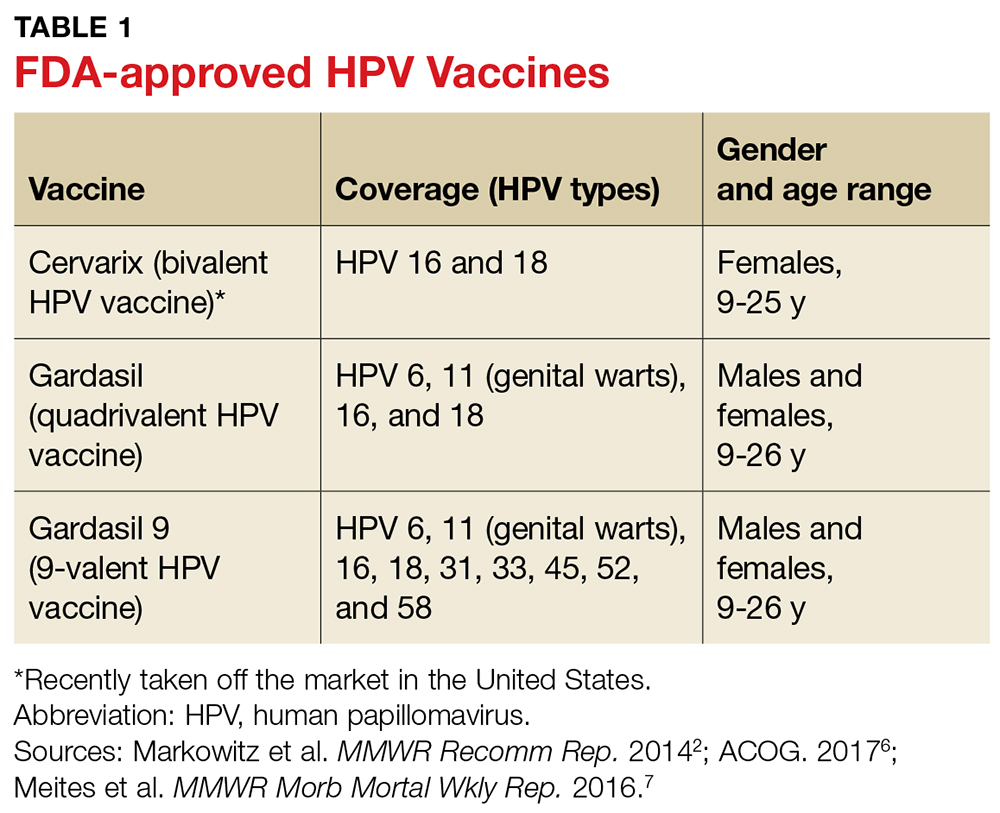
To ensure optimal protection, the vaccines must be administered in a series of scheduled doses over six to 12 months. The Advisory Committee on Immunization Practices (ACIP) recently updated their recommendations to include a two- or three-dose series based on age (see Table 2).7
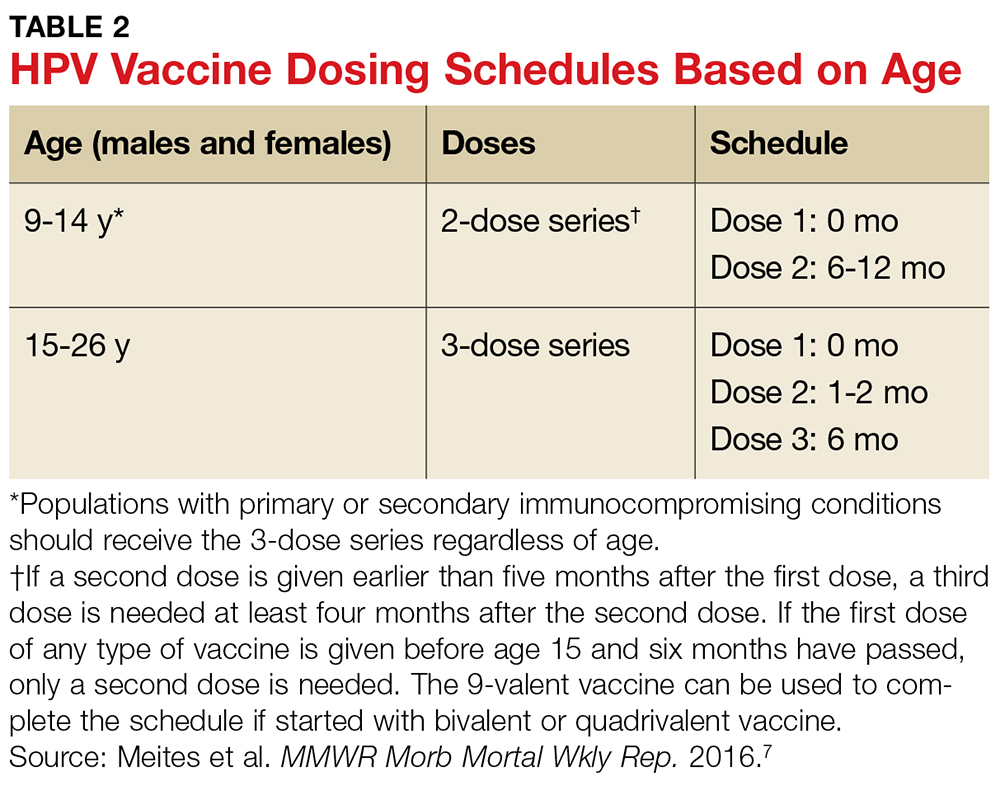
HPV vaccines are recommended for males and females between the ages of 9 and 26 years, but the ACIP and the American College of Obstetricians and Gynecologists (ACOG) strongly promote a targeted age range for vaccination between 11 and 12 years for both genders.6,7 Earlier vaccination is preferred because clinical data show a more rapid antibody response at a younger age, and because the vaccines are more effective if administered before an individual is exposed to or infected with HPV (ie, before the start of sexual activity).6,7
LOW VACCINATION RATES
HPV vaccination rates in the US are significantly lower than rates for other regularly administered vaccines; furthermore, they do not meet the Healthy People 2020 national goal of 80% for all vaccines.9 Immunization rates for most childhood vaccines range from 80% to 90%, but in 2015 only 28.1% of males and 41.9% of females ages 13 to 17 had completed the entire HPV vaccine series.9-11
The total HPV vaccination rates for male and female adolescents combined were 56.1% for one dose or more, 45.4% for two or more doses, and 34.9% for all three doses.9 In comparison, coverage rates for the meningococcal and Tdap (tetanus, diphtheria, and pertussis) immunizations, also recommended at the same age range as the HPV vaccine, were 81.3% and 86.4%, respectively.9
In addition to variation by gender and age, factors such as race, insurance coverage, and socioeconomic status influence vaccination rates.11 For the HPV vaccine specifically, Hispanic, non-Hispanic black, and American Indian/Alaska Native adolescents have higher rates of receiving each of the vaccine doses and higher rates of completing the vaccine series, compared to non-Hispanic white adolescents.9 Adolescents with Medicaid insurance and those living below the federal poverty level have better HPV vaccination coverage compared with adolescents with commercial insurance plans or those living at or above the poverty level.9,11
The HPV vaccine series completion rates in 2015 for males and females ages 13 to 17 living below the poverty level were 31.0% and 44.4%, respectively, compared to 27.4% and 41.3% for those living at or above the poverty level.9 One reason for increased rates among those living in lower-income households may be their eligibility for vaccinations at no cost through the Vaccines for Children (VFC) program, a federal program that provides vaccines to children who might otherwise forgo vaccination because of inability to pay.9
BARRIERS TO VACCINATION
Impediments that prevent adolescents and young adults from receiving the HPV vaccine exist throughout the vaccination process, with providers, parents, and the medical system itself contributing to low rates. Barriers to vaccination include fear and misconceptions, costs and socioeconomic status, lack of understanding and education, and logistic obstacles to completing the full series.5 Understanding these barriers, as well as discussing methods to overcome them, is key to increasing HPV vaccination rates and preventing the spread of this cancer-causing infection.
Health care provider barriers
Even though accredited national institutions and committees such as the CDC, ACIP, and ACOG strongly recommend vaccination based on current evidence, some health care providers still do not recommend the HPV vaccine to parents and patients.2,6,7,11 Lack of provider recommendation and the resulting lack of parental awareness of the vaccine account for many adolescents not receiving the vaccination.10,12
Providers do not recommend the vaccine for a number of reasons. Some have limited knowledge or conflicting ideas about the specific disease protection of the HPV vaccine, while others are hesitant to administer the vaccine before the onset of sexual activity, because they feel the suggested age for vaccination (11 to 12 years) is too young.10,11 Still other providers report difficulty approaching parents who they perceive as having concerns about the vaccine’s association with a sexually transmitted infection or believing that it might promote sexual activity.10
Some professionals simply claim that they forget to address the HPV vaccine at health visits, or that they propose it as optional and up to the parent’s discretion.5,10 Many providers do recommend and administer the initial dose of the vaccine, but have difficulties ensuring that patients complete the full multidose series.13 Evidence has shown that a strong provider recommendation is one of the most important incentives for parents and patients to accept vaccination.14
Parental and caregiver barriers
Lack of knowledge about the HPV vaccine and lack of recommendation from providers are two top reasons parents and caregivers cite for not vaccinating their children.5,10,14,15 In a national survey, almost all parents whose daughters completed the full vaccination series reported being counseled by their provider on the appropriate age for vaccination and the timeline of the series.14
Fears and apprehensions about side effects, especially with newer vaccines, can prevent some parents from having their children vaccinated.15 Although there is some stigma related to the vaccine’s association with the sexually transmitted HPV, this is a much less significant barrier than lack of provider recommendation or knowledge about the vaccine.5,11
Health care system barriers
Both providers and parents agree that system-level issues such as access, follow-up, and cost are barriers to initiating or completing the vaccination series.11,13 Many adolescents have few opportunities to receive the vaccine because they do not have a primary care provider.11 For those with access to primary care, visits are often problem-focused and frequently do not include a review of immunization history.13 Health care professionals also report challenges with scheduling follow-up visits for the second and third doses to complete the series within the recommended timeframe.13
Cost, insurance coverage, and reimbursement pose additional hurdles for both providers and patients, with some providers citing concerns about the cost of stocking the vaccine.16 Providers, both family practice providers and gynecologists, agree that reimbursement for administering the HPV vaccine in office poses a barrier when recommending the vaccine to patients.17 Lack of insurance coverage and type of insurance also pose barriers, with Medicaid patients more often completing the full series compared to those with private or no insurance, because Medicaid covers the cost of vaccination for men up to age 19.9,18 A national survey of males ages 9 to 17 found that the percentage of HPV vaccine initiation was double for those with public insurance compared to those with private insurance.19 Changes at the system-level, such as participation in the VFC program, in coordination with better provider recommendation should help increase HPV vaccination rates.9,11
STRATEGIES TO IMPROVE VACCINATION RATES
Many strategies for increasing HPV vaccination acceptance, decreasing barriers to access, and improving compliance with vaccine completion have been reported in the literature, with some strategies achieving more success than others. This section discusses interventions and strategies designed to help overcome provider-, parent-, and system-related barriers that have been shown to be effective (see Table 3).
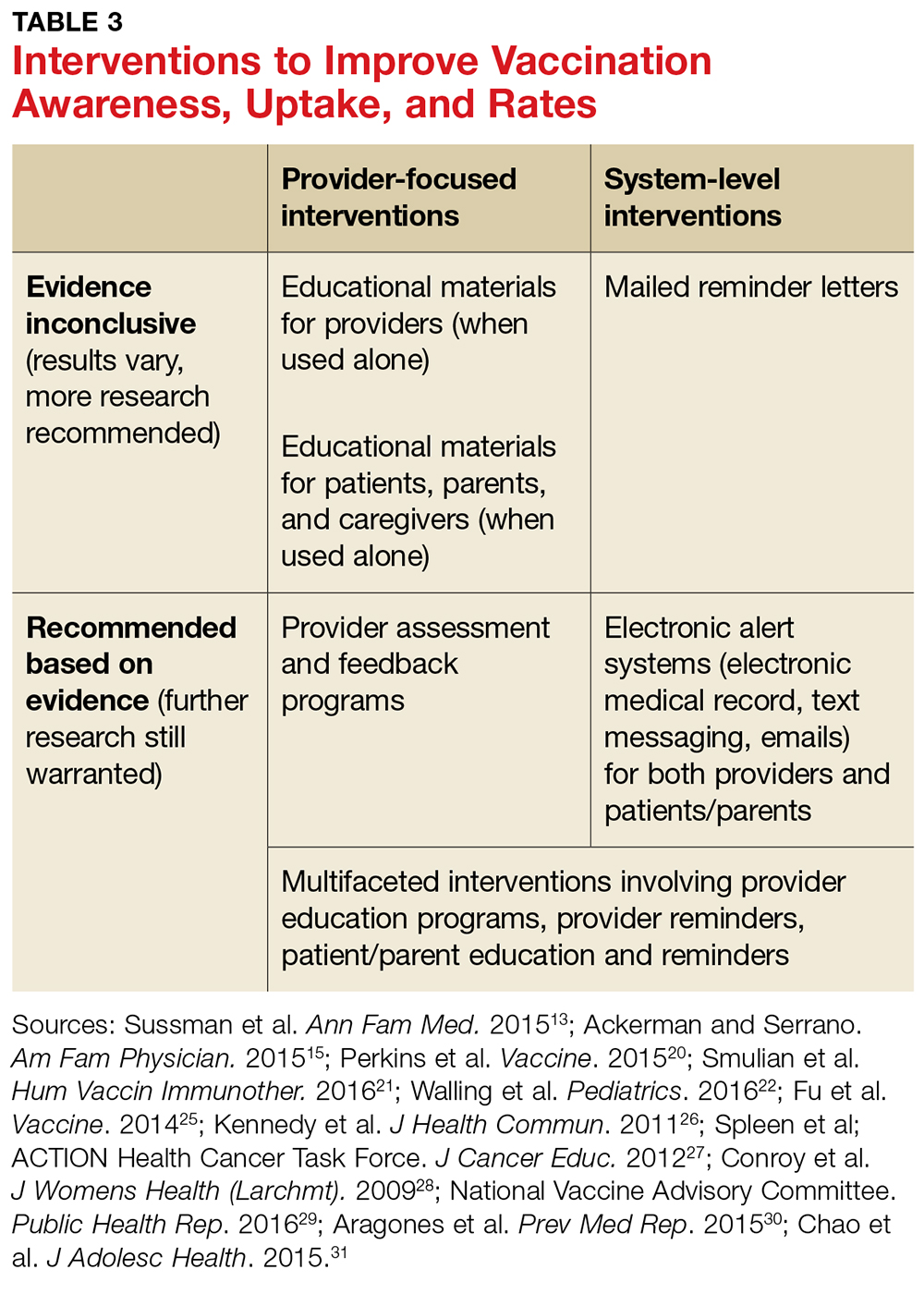
Health care provider interventions
Evidence supports a number of provider-level strategies to increase HPV vaccination rates (see Table 3). An improvement in vaccination acceptance was observed when providers promoted the vaccine as a safe, effective way to prevent cancer, rather than as a means to prevent a sexually transmitted infection.10,11,20
Some primary care providers found that encouraging the HPV vaccine at the same time as the meningococcal and Tdap vaccinations, which are also recommended at age 11 to 12 years, increased vaccination rates as well.13,20 Another successful strategy is reviewing vaccination history at every visit, whether the visit is for an acute event or an annual well exam.10,13,20 These tactics are most useful when providers practice them consistently, which may require them to change or adapt their way of practice.
Provider-based trainings that educate and prepare them to consistently recommend the vaccine have demonstrated success in increasing HPV vaccination uptake.21,22 The CDC’s Assessment/Feedback/Incentive/eXchange (AFIX) quality improvement program to increase vaccination rates, which includes Web-based or in-person consults, has been shown to increase HPV vaccination rates.20-23 The Assessment phase of the AFIX program determines a practice’s current immunization practices and rates, while the Feedback portion provides strategies for increasing vaccination rates.23 A study by Perkins and colleagues utilized AFIX strategies, specifically for the HPV vaccine, such as focusing provider education on HPV-related cancers and vaccine efficacy, as well as preparing providers to discuss and answer questions through basic motivational interviewing tactics.20
The CDC also offers PowerPoint presentations, flyers, posters, videos, and other informational resources to guide and educate providers, parents, and patients about the HPV vaccine.24 Educational resources, such as pamphlets, flyers, or fact sheets given to parents and patients, have been shown to improve intent to vaccinate as well as awareness of the vaccine.25-27
Although Fu and colleagues in a systematic review concluded that there was insufficient data to support a specific educational intervention for widespread use, the authors did recommend utilizing educational pieces and adapting them to specific populations.25 These simple interventions help increase awareness and can be implemented with other interventions in health care offices by providers and other staff.
System-level interventions
The use of systems that track patients for necessary vaccines and remind providers, parents, and patients about vaccine appointments have increased vaccination rates.13,28 Facility-based interventions, such as electronic medical records (EMR) that track patients for scheduled vaccines and remind providers when patients are due for vaccinations, will help increase provider recommendations and completion of the entire vaccination series.13
The National Vaccine Advisory Committee (NVAC) suggests that provider offices implement reminder-recall systems and provide educational material for parents and patients to increase vaccination rates.29 One specific study using both educational material and text-message reminders for parents found that these interventions significantly increased vaccination rates.30 Health care facilities could also incorporate reminder letters mailed to patients and parents to promote vaccine initiation and completion.31 The evidence supports the use of reminder alerts and EMR tracking systems to increase rates, but more research is warranted to determine the most cost-effective approach.
National programs, committees, and organizations have provided recommendations for overcoming system-related barriers to HPV vaccination, such as access and cost.29,32 The NVAC recommends incorporating alternative venues for vaccination delivery, such as pharmacies, schools, and health clinics, to increase availability to the adolescent population, especially to those who do not have primary care providers.29 One study that addressed parental opinions of vaccination administration in schools found that the majority of parents were in favor of this type of program.33 Although these recommendations seem promising and are accepted by parents, logistical barriers such as reimbursement to the pharmacies, schools, and clinics and accurate documentation of the doses received need to be addressed.29 The NVAC recommends continued evaluation and efforts to develop these programs in the future.29
In addition to school-based interventions, providing home visits for vaccination and implementing standing orders are other suggestions to overcome access and cost barriers for vaccinations, including HPV.32 Standing orders allow for individuals to receive a vaccine by a health care professional in an approved institution, where allowed by state law.32 This provides easier access to vaccinations, especially for those who do not see a primary care provider.
Although some of the system-level interventions mentioned in this article are outside the realm of what providers can do in the office, understanding and advocating for these advancements will promote vaccine uptake.
CONCLUSION
Lack of provider recommendation, coupled with poor or no parental knowledge about the HPV vaccine, are significant factors affecting vaccination uptake. Evidence supports the use of multifaceted interventions that promote and support provider recommendation and parent/patient education. Studies of interventions that incorporated educational resources and alert systems for both providers and patients or their caregivers have shown these strategies to be effective in increasing vaccination uptake and completion.
In addition to recommending the HPV vaccine, providers must educate parents/caregivers and patients about it, particularly by presenting the vaccine as a means of cancer prevention. Primary care facilities should implement reminder plans and provide educational literature to promote vaccine uptake. Although the interventions highlighted here have increased HPV vaccination rates, further research is warranted to evaluate more effective strategies for overcoming barriers and to determine which strategies are most cost-effective.
1. Juckett G, Hartsman-Adams H. Human papillomavirus: clinical manifestations and prevention. Am Fam Physician. 2010;82(10):1209-1213.
2. Markowitz LE, Dunne EF, Saraiya M, et al. Human papillomavirus vaccination: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2014;63(RR-05):1-30.
3. Viens LJ, Henley SJ, Watson M, et al. Human papillomavirus-associated cancers—United States, 2008-2012. MMWR Morb Mortal Wkly Rep. 2016;65(26):661-666.
4. American Cancer Society. Cancer facts and figures 2016. Atlanta: American Cancer Society; 2016.
5. Holman DM, Benard V, Roland KB, et al. Barriers to the human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr. 2014;168(1):76-82.
6. American College of Obstetricians and Gynecologists. Human papillomavirus vaccination. Committee Opinion. Number 704. June 2017. www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Adolescent-Health-Care/Human-Papillomavirus-Vaccination. Accessed August 17, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendation of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65(49):1405-1408.
8. Mulcahy N. GSK’s HPV vaccine, Cervarix, no longer available in the US. Medscape Medical News. October 26, 2016. www.medscape.com/viewarticle/870853. Accessed June 11, 2017.
9. Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(29):784-792.
10. Muncie HL, Lebato AL. HPV vaccination: overcoming parental and physician impediments. Am Fam Physician. 2015;92(6):449-454.
11. Bratic JS, Seyferth ER, Bocchini JA Jr. Update on barriers to human papillomavirus vaccination and effective strategies to promote vaccine acceptance. Curr Opin Pediatr. 2016;28(3):407-412.
12. Rahman M, Laz TH, McGrath CJ, Berenson AB. Provider recommendation mediates the relationship between parental human papillomavirus (HPV) vaccine awareness and HPV vaccine initiation and completion among 13- to 17-year-old US adolescent children. Clin Pediatr. 2015;54(4):371-375.
13. Sussman AL, Helitzer D, Bennett A, et al. Catching up with the HPV vaccine: challenges and opportunities in primary care. Ann Fam Med. 2015;13(4):354-360.
14. Clark SJ, Cowan AE, Filipp SL, et al. Parent perception of provider interactions influences HPV vaccination status of adolescent females. Clin Pediatr. 2016;55(8):701-706.
15. Ackerman LK, Serrano JL. Update on routine childhood and adolescent immunizations. Am Fam Physician. 2015;92(6):460-468.
16. Tom A, Robinett H, Buenconsejo-Lum L, et al. Promoting and providing the HPV vaccination in Hawaii: barriers faced by health providers. J Community Health. 2016;41(5):1069-1077.
17. Young JL, Bernheim RG, Korte JE, et al. Human papillomavirus vaccination recommendation may be linked to reimbursement: A survey of Virginia family practitioners and gynecologists. J Pediat Adolesc Gynecol. 2011;24(6):380-385.
18. Thomas R, Higgins L, Ding L, et al. Factors associated with HPV vaccine initiation, vaccine completion, and accuracy of self-reported vaccination status among 13- to 26-year-old men. Am J Mens Health. 2016;1-9.
19. Laz TH, Rahman M, Berenson AB. Human papillomavirus vaccine uptake among 9-17 year old males in the United States: the National Health Interview Survey, 2010. Hum Vaccin Immunother. 2013;9(4):874-878.
20. Perkins RB, Zisblatt L, Legler A, et al. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine. 2015;33:1223-1229.
21. Smulian EA, Mitchell KR, Stokley S. Interventions to increase HPV vaccination coverage: a systematic review. Hum Vaccin Immunother. 2016;12:1566-1588.
22. Walling EB, Benzoni N, Dornfeld J, et al. Interventions to improve HPV vaccine uptake: a systematic review. Pediatrics. 2016;138(1).
23. CDC. Overview of AFIX. www.cdc.gov/vaccines/programs/afix/index.html. Accessed August 17, 2017.
24. CDC. Human papillomavirus (HPV). www.cdc.gov/hpv/. Accessed August 17, 2017.
25. Fu LY, Bonhomme LA, Cooper SC, et al. Educational interventions to increase HPV vaccination acceptance: a systematic review. Vaccine. 2014;32(17):1901-1920.
26. Kennedy A, Sapsis KF, Stokley S, et al. Parental attitudes toward human papillomavirus vaccination: evaluation of an educational intervention, 2008. J Health Commun. 2011;16(3):300-313.
27. Spleen AM, Kluhsman BC, Clark AD, et al; ACTION Health Cancer Task Force. An increase in HPV-related knowledge and vaccination intent among parental and non-parental caregivers of adolescent girls, ages 9-17 years, in Appalachian Pennsylvania. J Cancer Educ. 2012;27(2):312-319.
28. Conroy K, Rosenthal SL, Zimet GD, et al. Human papillomavirus vaccine uptake, predictors of vaccination, and self-reported barriers to vaccination. J Womens Health (Larchmt). 2009;18(10):1679-1686.
29. National Vaccine Advisory Committee. Overcoming barriers to low HPV vaccine uptake in the United States: recommendations from the National Vaccine Advisory Committee. Public Health Rep. 2016;131(1):17-25.
30. Aragones A, Bruno DM, Ehrenberg M, et al. Parental education and text messaging reminders as effective community based tools to increase HPV vaccination rates among Mexican American children. Prev Med Rep. 2015;2:554-558.
31. Chao C, Preciado M, Slezak J, Xu L. A randomized intervention of reminder letter for human papillomavirus vaccine series completion. J Adolesc Health. 2015;56(1):85-90.
32. Community Preventive Services Task Force. The community guide—guide to community preventive services: increasing appropriate vaccinations. Atlanta, GA: Community Preventive Services Task Force; 2016
33. Kelminson K, Saville A, Seewald L, et al. Parental views of school-located delivery of adolescent vaccines. J Adolesc Health. 2012;51(2):190-196.
CE/CME No: CR-1709
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand and identify the low- and high-risk human papillomavirus (HPV) types that can lead to benign and malignant manifestations.
• Know the recommended age range and dosing schedule for individuals who can and should receive the vaccination.
• Recognize important barriers to HPV vaccination in the health care setting.
• Understand how to promote HPV vaccination to parents/caregivers and patients.
• Find resources and educational material from national organizations that recommend and support HPV vaccination.
FACULTY
Tyler Cole practices at Coastal Community Health Services in Brunswick, Georgia, and is a clinical instructor in the DNP-APRN program at the Medical University of South Carolina (MUSC). Marie C. Thomas is a registered nurse on a surgical oncology unit at MUSC and will receive her DNP-FNP from MUSC in December 2017. Katlyn Straup practices at Roper St. Francis Healthcare and Southern Care Hospice in Charleston, South Carolina; she is also a clinical associate faculty member in the MUSC College of Nursing. Ashlyn Savage is an Associate Professor of Obstetrics and Gynecology at MUSC College of Nursing and is certified by the American Board of Obstetrics and Gynecology.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through August 31, 2018.
Article begins on next page >>
Although human papillomavirus (HPV) vaccine is a safe and effective means of preventing most HPV-related cancers, HPV vaccination rates lag well behind those of other vaccines recommended for children and adolescents. Understanding the barriers to HPV vaccine acceptance and effective strategies for overcoming them will improve vaccine uptake and completion in adolescents.
Human papillomavirus (HPV) infection is the most common sexually transmitted infection in the United States.1,2 HPV causes approximately 30,700 new cancer cases in the US annually.3 It is the primary cause of cervical cancer, which resulted in more than 4,000 deaths in the US in 2016.4 HPV is also associated with some vaginal, vulvar, penile, anal, and oropharyngeal cancers and causes anogenital warts.3
Although HPV vaccines are available to protect against infection with the HPV types that lead to these sequelae, HPV vaccination rates remain low compared with other routinely administered vaccines.5 Reasons for these lower rates include vaccine cost, lack of patient and provider education, providers’ failure to recommend, stigmas related to sexual behavior, and misconceptions about the vaccine, such as concerns about harm.5 This article discusses these barriers to better educate providers about the HPV vaccine and encourage them to assist in increasing vaccination rates.
EPIDEMIOLOGY
Approximately 79 million Americans are currently infected with HPV, and 14 million new cases are reported each year.2 In the US, the prevalence of HPV is highest among sexually active adolescents and young adults, especially those ages 20 to 24.2 Of the more than 150 types of HPV that have been identified, 40 infect the genital area. HPV genital infections are mainly spread through sexual intercourse but can also be spread through oral-to-genital contact.2
The genital HPV types are categorized as low-risk and high-risk based on their association with cervical cancer.2 High-risk types 16 and 18 are the most troublesome, accounting for 63% of all HPV-associated cancers, with HPV 16 posing the highest risk for cancer.3 High-risk types HPV 31, 33, 45, 52, and 58 account for another 10% of these cancers.3 Low-risk types, such as HPV 6 and 11, can cause low-grade cervical intraepithelial lesions, and HPV 6 and 11 account for more than 90% of genital warts.2
Most HPV infections, whether with high- or low-risk types, do not cause symptoms and resolve spontaneously in about two years.2 Persistent high-risk HPV infection is necessary for the development of cervical cancer precursor lesions—and therefore, once the infection has cleared, the risk for cancer declines.2
HPV VACCINES
Three HPV vaccines are licensed for use in the US: bivalent (Cervarix), quadrivalent (Gardasil), and 9-valent (Gardasil 9) vaccines (see Table 1).2,6,7 The bivalent, Cervarix, has recently been removed from the US market due to a decrease in product demand.6,8
To ensure optimal protection, the vaccines must be administered in a series of scheduled doses over six to 12 months. The Advisory Committee on Immunization Practices (ACIP) recently updated their recommendations to include a two- or three-dose series based on age (see Table 2).7
HPV vaccines are recommended for males and females between the ages of 9 and 26 years, but the ACIP and the American College of Obstetricians and Gynecologists (ACOG) strongly promote a targeted age range for vaccination between 11 and 12 years for both genders.6,7 Earlier vaccination is preferred because clinical data show a more rapid antibody response at a younger age, and because the vaccines are more effective if administered before an individual is exposed to or infected with HPV (ie, before the start of sexual activity).6,7
LOW VACCINATION RATES
HPV vaccination rates in the US are significantly lower than rates for other regularly administered vaccines; furthermore, they do not meet the Healthy People 2020 national goal of 80% for all vaccines.9 Immunization rates for most childhood vaccines range from 80% to 90%, but in 2015 only 28.1% of males and 41.9% of females ages 13 to 17 had completed the entire HPV vaccine series.9-11
The total HPV vaccination rates for male and female adolescents combined were 56.1% for one dose or more, 45.4% for two or more doses, and 34.9% for all three doses.9 In comparison, coverage rates for the meningococcal and Tdap (tetanus, diphtheria, and pertussis) immunizations, also recommended at the same age range as the HPV vaccine, were 81.3% and 86.4%, respectively.9
In addition to variation by gender and age, factors such as race, insurance coverage, and socioeconomic status influence vaccination rates.11 For the HPV vaccine specifically, Hispanic, non-Hispanic black, and American Indian/Alaska Native adolescents have higher rates of receiving each of the vaccine doses and higher rates of completing the vaccine series, compared to non-Hispanic white adolescents.9 Adolescents with Medicaid insurance and those living below the federal poverty level have better HPV vaccination coverage compared with adolescents with commercial insurance plans or those living at or above the poverty level.9,11
The HPV vaccine series completion rates in 2015 for males and females ages 13 to 17 living below the poverty level were 31.0% and 44.4%, respectively, compared to 27.4% and 41.3% for those living at or above the poverty level.9 One reason for increased rates among those living in lower-income households may be their eligibility for vaccinations at no cost through the Vaccines for Children (VFC) program, a federal program that provides vaccines to children who might otherwise forgo vaccination because of inability to pay.9
BARRIERS TO VACCINATION
Impediments that prevent adolescents and young adults from receiving the HPV vaccine exist throughout the vaccination process, with providers, parents, and the medical system itself contributing to low rates. Barriers to vaccination include fear and misconceptions, costs and socioeconomic status, lack of understanding and education, and logistic obstacles to completing the full series.5 Understanding these barriers, as well as discussing methods to overcome them, is key to increasing HPV vaccination rates and preventing the spread of this cancer-causing infection.
Health care provider barriers
Even though accredited national institutions and committees such as the CDC, ACIP, and ACOG strongly recommend vaccination based on current evidence, some health care providers still do not recommend the HPV vaccine to parents and patients.2,6,7,11 Lack of provider recommendation and the resulting lack of parental awareness of the vaccine account for many adolescents not receiving the vaccination.10,12
Providers do not recommend the vaccine for a number of reasons. Some have limited knowledge or conflicting ideas about the specific disease protection of the HPV vaccine, while others are hesitant to administer the vaccine before the onset of sexual activity, because they feel the suggested age for vaccination (11 to 12 years) is too young.10,11 Still other providers report difficulty approaching parents who they perceive as having concerns about the vaccine’s association with a sexually transmitted infection or believing that it might promote sexual activity.10
Some professionals simply claim that they forget to address the HPV vaccine at health visits, or that they propose it as optional and up to the parent’s discretion.5,10 Many providers do recommend and administer the initial dose of the vaccine, but have difficulties ensuring that patients complete the full multidose series.13 Evidence has shown that a strong provider recommendation is one of the most important incentives for parents and patients to accept vaccination.14
Parental and caregiver barriers
Lack of knowledge about the HPV vaccine and lack of recommendation from providers are two top reasons parents and caregivers cite for not vaccinating their children.5,10,14,15 In a national survey, almost all parents whose daughters completed the full vaccination series reported being counseled by their provider on the appropriate age for vaccination and the timeline of the series.14
Fears and apprehensions about side effects, especially with newer vaccines, can prevent some parents from having their children vaccinated.15 Although there is some stigma related to the vaccine’s association with the sexually transmitted HPV, this is a much less significant barrier than lack of provider recommendation or knowledge about the vaccine.5,11
Health care system barriers
Both providers and parents agree that system-level issues such as access, follow-up, and cost are barriers to initiating or completing the vaccination series.11,13 Many adolescents have few opportunities to receive the vaccine because they do not have a primary care provider.11 For those with access to primary care, visits are often problem-focused and frequently do not include a review of immunization history.13 Health care professionals also report challenges with scheduling follow-up visits for the second and third doses to complete the series within the recommended timeframe.13
Cost, insurance coverage, and reimbursement pose additional hurdles for both providers and patients, with some providers citing concerns about the cost of stocking the vaccine.16 Providers, both family practice providers and gynecologists, agree that reimbursement for administering the HPV vaccine in office poses a barrier when recommending the vaccine to patients.17 Lack of insurance coverage and type of insurance also pose barriers, with Medicaid patients more often completing the full series compared to those with private or no insurance, because Medicaid covers the cost of vaccination for men up to age 19.9,18 A national survey of males ages 9 to 17 found that the percentage of HPV vaccine initiation was double for those with public insurance compared to those with private insurance.19 Changes at the system-level, such as participation in the VFC program, in coordination with better provider recommendation should help increase HPV vaccination rates.9,11
STRATEGIES TO IMPROVE VACCINATION RATES
Many strategies for increasing HPV vaccination acceptance, decreasing barriers to access, and improving compliance with vaccine completion have been reported in the literature, with some strategies achieving more success than others. This section discusses interventions and strategies designed to help overcome provider-, parent-, and system-related barriers that have been shown to be effective (see Table 3).
Health care provider interventions
Evidence supports a number of provider-level strategies to increase HPV vaccination rates (see Table 3). An improvement in vaccination acceptance was observed when providers promoted the vaccine as a safe, effective way to prevent cancer, rather than as a means to prevent a sexually transmitted infection.10,11,20
Some primary care providers found that encouraging the HPV vaccine at the same time as the meningococcal and Tdap vaccinations, which are also recommended at age 11 to 12 years, increased vaccination rates as well.13,20 Another successful strategy is reviewing vaccination history at every visit, whether the visit is for an acute event or an annual well exam.10,13,20 These tactics are most useful when providers practice them consistently, which may require them to change or adapt their way of practice.
Provider-based trainings that educate and prepare them to consistently recommend the vaccine have demonstrated success in increasing HPV vaccination uptake.21,22 The CDC’s Assessment/Feedback/Incentive/eXchange (AFIX) quality improvement program to increase vaccination rates, which includes Web-based or in-person consults, has been shown to increase HPV vaccination rates.20-23 The Assessment phase of the AFIX program determines a practice’s current immunization practices and rates, while the Feedback portion provides strategies for increasing vaccination rates.23 A study by Perkins and colleagues utilized AFIX strategies, specifically for the HPV vaccine, such as focusing provider education on HPV-related cancers and vaccine efficacy, as well as preparing providers to discuss and answer questions through basic motivational interviewing tactics.20
The CDC also offers PowerPoint presentations, flyers, posters, videos, and other informational resources to guide and educate providers, parents, and patients about the HPV vaccine.24 Educational resources, such as pamphlets, flyers, or fact sheets given to parents and patients, have been shown to improve intent to vaccinate as well as awareness of the vaccine.25-27
Although Fu and colleagues in a systematic review concluded that there was insufficient data to support a specific educational intervention for widespread use, the authors did recommend utilizing educational pieces and adapting them to specific populations.25 These simple interventions help increase awareness and can be implemented with other interventions in health care offices by providers and other staff.
System-level interventions
The use of systems that track patients for necessary vaccines and remind providers, parents, and patients about vaccine appointments have increased vaccination rates.13,28 Facility-based interventions, such as electronic medical records (EMR) that track patients for scheduled vaccines and remind providers when patients are due for vaccinations, will help increase provider recommendations and completion of the entire vaccination series.13
The National Vaccine Advisory Committee (NVAC) suggests that provider offices implement reminder-recall systems and provide educational material for parents and patients to increase vaccination rates.29 One specific study using both educational material and text-message reminders for parents found that these interventions significantly increased vaccination rates.30 Health care facilities could also incorporate reminder letters mailed to patients and parents to promote vaccine initiation and completion.31 The evidence supports the use of reminder alerts and EMR tracking systems to increase rates, but more research is warranted to determine the most cost-effective approach.
National programs, committees, and organizations have provided recommendations for overcoming system-related barriers to HPV vaccination, such as access and cost.29,32 The NVAC recommends incorporating alternative venues for vaccination delivery, such as pharmacies, schools, and health clinics, to increase availability to the adolescent population, especially to those who do not have primary care providers.29 One study that addressed parental opinions of vaccination administration in schools found that the majority of parents were in favor of this type of program.33 Although these recommendations seem promising and are accepted by parents, logistical barriers such as reimbursement to the pharmacies, schools, and clinics and accurate documentation of the doses received need to be addressed.29 The NVAC recommends continued evaluation and efforts to develop these programs in the future.29
In addition to school-based interventions, providing home visits for vaccination and implementing standing orders are other suggestions to overcome access and cost barriers for vaccinations, including HPV.32 Standing orders allow for individuals to receive a vaccine by a health care professional in an approved institution, where allowed by state law.32 This provides easier access to vaccinations, especially for those who do not see a primary care provider.
Although some of the system-level interventions mentioned in this article are outside the realm of what providers can do in the office, understanding and advocating for these advancements will promote vaccine uptake.
CONCLUSION
Lack of provider recommendation, coupled with poor or no parental knowledge about the HPV vaccine, are significant factors affecting vaccination uptake. Evidence supports the use of multifaceted interventions that promote and support provider recommendation and parent/patient education. Studies of interventions that incorporated educational resources and alert systems for both providers and patients or their caregivers have shown these strategies to be effective in increasing vaccination uptake and completion.
In addition to recommending the HPV vaccine, providers must educate parents/caregivers and patients about it, particularly by presenting the vaccine as a means of cancer prevention. Primary care facilities should implement reminder plans and provide educational literature to promote vaccine uptake. Although the interventions highlighted here have increased HPV vaccination rates, further research is warranted to evaluate more effective strategies for overcoming barriers and to determine which strategies are most cost-effective.
CE/CME No: CR-1709
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand and identify the low- and high-risk human papillomavirus (HPV) types that can lead to benign and malignant manifestations.
• Know the recommended age range and dosing schedule for individuals who can and should receive the vaccination.
• Recognize important barriers to HPV vaccination in the health care setting.
• Understand how to promote HPV vaccination to parents/caregivers and patients.
• Find resources and educational material from national organizations that recommend and support HPV vaccination.
FACULTY
Tyler Cole practices at Coastal Community Health Services in Brunswick, Georgia, and is a clinical instructor in the DNP-APRN program at the Medical University of South Carolina (MUSC). Marie C. Thomas is a registered nurse on a surgical oncology unit at MUSC and will receive her DNP-FNP from MUSC in December 2017. Katlyn Straup practices at Roper St. Francis Healthcare and Southern Care Hospice in Charleston, South Carolina; she is also a clinical associate faculty member in the MUSC College of Nursing. Ashlyn Savage is an Associate Professor of Obstetrics and Gynecology at MUSC College of Nursing and is certified by the American Board of Obstetrics and Gynecology.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through August 31, 2018.
Article begins on next page >>
Although human papillomavirus (HPV) vaccine is a safe and effective means of preventing most HPV-related cancers, HPV vaccination rates lag well behind those of other vaccines recommended for children and adolescents. Understanding the barriers to HPV vaccine acceptance and effective strategies for overcoming them will improve vaccine uptake and completion in adolescents.
Human papillomavirus (HPV) infection is the most common sexually transmitted infection in the United States.1,2 HPV causes approximately 30,700 new cancer cases in the US annually.3 It is the primary cause of cervical cancer, which resulted in more than 4,000 deaths in the US in 2016.4 HPV is also associated with some vaginal, vulvar, penile, anal, and oropharyngeal cancers and causes anogenital warts.3
Although HPV vaccines are available to protect against infection with the HPV types that lead to these sequelae, HPV vaccination rates remain low compared with other routinely administered vaccines.5 Reasons for these lower rates include vaccine cost, lack of patient and provider education, providers’ failure to recommend, stigmas related to sexual behavior, and misconceptions about the vaccine, such as concerns about harm.5 This article discusses these barriers to better educate providers about the HPV vaccine and encourage them to assist in increasing vaccination rates.
EPIDEMIOLOGY
Approximately 79 million Americans are currently infected with HPV, and 14 million new cases are reported each year.2 In the US, the prevalence of HPV is highest among sexually active adolescents and young adults, especially those ages 20 to 24.2 Of the more than 150 types of HPV that have been identified, 40 infect the genital area. HPV genital infections are mainly spread through sexual intercourse but can also be spread through oral-to-genital contact.2
The genital HPV types are categorized as low-risk and high-risk based on their association with cervical cancer.2 High-risk types 16 and 18 are the most troublesome, accounting for 63% of all HPV-associated cancers, with HPV 16 posing the highest risk for cancer.3 High-risk types HPV 31, 33, 45, 52, and 58 account for another 10% of these cancers.3 Low-risk types, such as HPV 6 and 11, can cause low-grade cervical intraepithelial lesions, and HPV 6 and 11 account for more than 90% of genital warts.2
Most HPV infections, whether with high- or low-risk types, do not cause symptoms and resolve spontaneously in about two years.2 Persistent high-risk HPV infection is necessary for the development of cervical cancer precursor lesions—and therefore, once the infection has cleared, the risk for cancer declines.2
HPV VACCINES
Three HPV vaccines are licensed for use in the US: bivalent (Cervarix), quadrivalent (Gardasil), and 9-valent (Gardasil 9) vaccines (see Table 1).2,6,7 The bivalent, Cervarix, has recently been removed from the US market due to a decrease in product demand.6,8
To ensure optimal protection, the vaccines must be administered in a series of scheduled doses over six to 12 months. The Advisory Committee on Immunization Practices (ACIP) recently updated their recommendations to include a two- or three-dose series based on age (see Table 2).7
HPV vaccines are recommended for males and females between the ages of 9 and 26 years, but the ACIP and the American College of Obstetricians and Gynecologists (ACOG) strongly promote a targeted age range for vaccination between 11 and 12 years for both genders.6,7 Earlier vaccination is preferred because clinical data show a more rapid antibody response at a younger age, and because the vaccines are more effective if administered before an individual is exposed to or infected with HPV (ie, before the start of sexual activity).6,7
LOW VACCINATION RATES
HPV vaccination rates in the US are significantly lower than rates for other regularly administered vaccines; furthermore, they do not meet the Healthy People 2020 national goal of 80% for all vaccines.9 Immunization rates for most childhood vaccines range from 80% to 90%, but in 2015 only 28.1% of males and 41.9% of females ages 13 to 17 had completed the entire HPV vaccine series.9-11
The total HPV vaccination rates for male and female adolescents combined were 56.1% for one dose or more, 45.4% for two or more doses, and 34.9% for all three doses.9 In comparison, coverage rates for the meningococcal and Tdap (tetanus, diphtheria, and pertussis) immunizations, also recommended at the same age range as the HPV vaccine, were 81.3% and 86.4%, respectively.9
In addition to variation by gender and age, factors such as race, insurance coverage, and socioeconomic status influence vaccination rates.11 For the HPV vaccine specifically, Hispanic, non-Hispanic black, and American Indian/Alaska Native adolescents have higher rates of receiving each of the vaccine doses and higher rates of completing the vaccine series, compared to non-Hispanic white adolescents.9 Adolescents with Medicaid insurance and those living below the federal poverty level have better HPV vaccination coverage compared with adolescents with commercial insurance plans or those living at or above the poverty level.9,11
The HPV vaccine series completion rates in 2015 for males and females ages 13 to 17 living below the poverty level were 31.0% and 44.4%, respectively, compared to 27.4% and 41.3% for those living at or above the poverty level.9 One reason for increased rates among those living in lower-income households may be their eligibility for vaccinations at no cost through the Vaccines for Children (VFC) program, a federal program that provides vaccines to children who might otherwise forgo vaccination because of inability to pay.9
BARRIERS TO VACCINATION
Impediments that prevent adolescents and young adults from receiving the HPV vaccine exist throughout the vaccination process, with providers, parents, and the medical system itself contributing to low rates. Barriers to vaccination include fear and misconceptions, costs and socioeconomic status, lack of understanding and education, and logistic obstacles to completing the full series.5 Understanding these barriers, as well as discussing methods to overcome them, is key to increasing HPV vaccination rates and preventing the spread of this cancer-causing infection.
Health care provider barriers
Even though accredited national institutions and committees such as the CDC, ACIP, and ACOG strongly recommend vaccination based on current evidence, some health care providers still do not recommend the HPV vaccine to parents and patients.2,6,7,11 Lack of provider recommendation and the resulting lack of parental awareness of the vaccine account for many adolescents not receiving the vaccination.10,12
Providers do not recommend the vaccine for a number of reasons. Some have limited knowledge or conflicting ideas about the specific disease protection of the HPV vaccine, while others are hesitant to administer the vaccine before the onset of sexual activity, because they feel the suggested age for vaccination (11 to 12 years) is too young.10,11 Still other providers report difficulty approaching parents who they perceive as having concerns about the vaccine’s association with a sexually transmitted infection or believing that it might promote sexual activity.10
Some professionals simply claim that they forget to address the HPV vaccine at health visits, or that they propose it as optional and up to the parent’s discretion.5,10 Many providers do recommend and administer the initial dose of the vaccine, but have difficulties ensuring that patients complete the full multidose series.13 Evidence has shown that a strong provider recommendation is one of the most important incentives for parents and patients to accept vaccination.14
Parental and caregiver barriers
Lack of knowledge about the HPV vaccine and lack of recommendation from providers are two top reasons parents and caregivers cite for not vaccinating their children.5,10,14,15 In a national survey, almost all parents whose daughters completed the full vaccination series reported being counseled by their provider on the appropriate age for vaccination and the timeline of the series.14
Fears and apprehensions about side effects, especially with newer vaccines, can prevent some parents from having their children vaccinated.15 Although there is some stigma related to the vaccine’s association with the sexually transmitted HPV, this is a much less significant barrier than lack of provider recommendation or knowledge about the vaccine.5,11
Health care system barriers
Both providers and parents agree that system-level issues such as access, follow-up, and cost are barriers to initiating or completing the vaccination series.11,13 Many adolescents have few opportunities to receive the vaccine because they do not have a primary care provider.11 For those with access to primary care, visits are often problem-focused and frequently do not include a review of immunization history.13 Health care professionals also report challenges with scheduling follow-up visits for the second and third doses to complete the series within the recommended timeframe.13
Cost, insurance coverage, and reimbursement pose additional hurdles for both providers and patients, with some providers citing concerns about the cost of stocking the vaccine.16 Providers, both family practice providers and gynecologists, agree that reimbursement for administering the HPV vaccine in office poses a barrier when recommending the vaccine to patients.17 Lack of insurance coverage and type of insurance also pose barriers, with Medicaid patients more often completing the full series compared to those with private or no insurance, because Medicaid covers the cost of vaccination for men up to age 19.9,18 A national survey of males ages 9 to 17 found that the percentage of HPV vaccine initiation was double for those with public insurance compared to those with private insurance.19 Changes at the system-level, such as participation in the VFC program, in coordination with better provider recommendation should help increase HPV vaccination rates.9,11
STRATEGIES TO IMPROVE VACCINATION RATES
Many strategies for increasing HPV vaccination acceptance, decreasing barriers to access, and improving compliance with vaccine completion have been reported in the literature, with some strategies achieving more success than others. This section discusses interventions and strategies designed to help overcome provider-, parent-, and system-related barriers that have been shown to be effective (see Table 3).
Health care provider interventions
Evidence supports a number of provider-level strategies to increase HPV vaccination rates (see Table 3). An improvement in vaccination acceptance was observed when providers promoted the vaccine as a safe, effective way to prevent cancer, rather than as a means to prevent a sexually transmitted infection.10,11,20
Some primary care providers found that encouraging the HPV vaccine at the same time as the meningococcal and Tdap vaccinations, which are also recommended at age 11 to 12 years, increased vaccination rates as well.13,20 Another successful strategy is reviewing vaccination history at every visit, whether the visit is for an acute event or an annual well exam.10,13,20 These tactics are most useful when providers practice them consistently, which may require them to change or adapt their way of practice.
Provider-based trainings that educate and prepare them to consistently recommend the vaccine have demonstrated success in increasing HPV vaccination uptake.21,22 The CDC’s Assessment/Feedback/Incentive/eXchange (AFIX) quality improvement program to increase vaccination rates, which includes Web-based or in-person consults, has been shown to increase HPV vaccination rates.20-23 The Assessment phase of the AFIX program determines a practice’s current immunization practices and rates, while the Feedback portion provides strategies for increasing vaccination rates.23 A study by Perkins and colleagues utilized AFIX strategies, specifically for the HPV vaccine, such as focusing provider education on HPV-related cancers and vaccine efficacy, as well as preparing providers to discuss and answer questions through basic motivational interviewing tactics.20
The CDC also offers PowerPoint presentations, flyers, posters, videos, and other informational resources to guide and educate providers, parents, and patients about the HPV vaccine.24 Educational resources, such as pamphlets, flyers, or fact sheets given to parents and patients, have been shown to improve intent to vaccinate as well as awareness of the vaccine.25-27
Although Fu and colleagues in a systematic review concluded that there was insufficient data to support a specific educational intervention for widespread use, the authors did recommend utilizing educational pieces and adapting them to specific populations.25 These simple interventions help increase awareness and can be implemented with other interventions in health care offices by providers and other staff.
System-level interventions
The use of systems that track patients for necessary vaccines and remind providers, parents, and patients about vaccine appointments have increased vaccination rates.13,28 Facility-based interventions, such as electronic medical records (EMR) that track patients for scheduled vaccines and remind providers when patients are due for vaccinations, will help increase provider recommendations and completion of the entire vaccination series.13
The National Vaccine Advisory Committee (NVAC) suggests that provider offices implement reminder-recall systems and provide educational material for parents and patients to increase vaccination rates.29 One specific study using both educational material and text-message reminders for parents found that these interventions significantly increased vaccination rates.30 Health care facilities could also incorporate reminder letters mailed to patients and parents to promote vaccine initiation and completion.31 The evidence supports the use of reminder alerts and EMR tracking systems to increase rates, but more research is warranted to determine the most cost-effective approach.
National programs, committees, and organizations have provided recommendations for overcoming system-related barriers to HPV vaccination, such as access and cost.29,32 The NVAC recommends incorporating alternative venues for vaccination delivery, such as pharmacies, schools, and health clinics, to increase availability to the adolescent population, especially to those who do not have primary care providers.29 One study that addressed parental opinions of vaccination administration in schools found that the majority of parents were in favor of this type of program.33 Although these recommendations seem promising and are accepted by parents, logistical barriers such as reimbursement to the pharmacies, schools, and clinics and accurate documentation of the doses received need to be addressed.29 The NVAC recommends continued evaluation and efforts to develop these programs in the future.29
In addition to school-based interventions, providing home visits for vaccination and implementing standing orders are other suggestions to overcome access and cost barriers for vaccinations, including HPV.32 Standing orders allow for individuals to receive a vaccine by a health care professional in an approved institution, where allowed by state law.32 This provides easier access to vaccinations, especially for those who do not see a primary care provider.
Although some of the system-level interventions mentioned in this article are outside the realm of what providers can do in the office, understanding and advocating for these advancements will promote vaccine uptake.
CONCLUSION
Lack of provider recommendation, coupled with poor or no parental knowledge about the HPV vaccine, are significant factors affecting vaccination uptake. Evidence supports the use of multifaceted interventions that promote and support provider recommendation and parent/patient education. Studies of interventions that incorporated educational resources and alert systems for both providers and patients or their caregivers have shown these strategies to be effective in increasing vaccination uptake and completion.
In addition to recommending the HPV vaccine, providers must educate parents/caregivers and patients about it, particularly by presenting the vaccine as a means of cancer prevention. Primary care facilities should implement reminder plans and provide educational literature to promote vaccine uptake. Although the interventions highlighted here have increased HPV vaccination rates, further research is warranted to evaluate more effective strategies for overcoming barriers and to determine which strategies are most cost-effective.
1. Juckett G, Hartsman-Adams H. Human papillomavirus: clinical manifestations and prevention. Am Fam Physician. 2010;82(10):1209-1213.
2. Markowitz LE, Dunne EF, Saraiya M, et al. Human papillomavirus vaccination: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2014;63(RR-05):1-30.
3. Viens LJ, Henley SJ, Watson M, et al. Human papillomavirus-associated cancers—United States, 2008-2012. MMWR Morb Mortal Wkly Rep. 2016;65(26):661-666.
4. American Cancer Society. Cancer facts and figures 2016. Atlanta: American Cancer Society; 2016.
5. Holman DM, Benard V, Roland KB, et al. Barriers to the human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr. 2014;168(1):76-82.
6. American College of Obstetricians and Gynecologists. Human papillomavirus vaccination. Committee Opinion. Number 704. June 2017. www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Adolescent-Health-Care/Human-Papillomavirus-Vaccination. Accessed August 17, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendation of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65(49):1405-1408.
8. Mulcahy N. GSK’s HPV vaccine, Cervarix, no longer available in the US. Medscape Medical News. October 26, 2016. www.medscape.com/viewarticle/870853. Accessed June 11, 2017.
9. Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(29):784-792.
10. Muncie HL, Lebato AL. HPV vaccination: overcoming parental and physician impediments. Am Fam Physician. 2015;92(6):449-454.
11. Bratic JS, Seyferth ER, Bocchini JA Jr. Update on barriers to human papillomavirus vaccination and effective strategies to promote vaccine acceptance. Curr Opin Pediatr. 2016;28(3):407-412.
12. Rahman M, Laz TH, McGrath CJ, Berenson AB. Provider recommendation mediates the relationship between parental human papillomavirus (HPV) vaccine awareness and HPV vaccine initiation and completion among 13- to 17-year-old US adolescent children. Clin Pediatr. 2015;54(4):371-375.
13. Sussman AL, Helitzer D, Bennett A, et al. Catching up with the HPV vaccine: challenges and opportunities in primary care. Ann Fam Med. 2015;13(4):354-360.
14. Clark SJ, Cowan AE, Filipp SL, et al. Parent perception of provider interactions influences HPV vaccination status of adolescent females. Clin Pediatr. 2016;55(8):701-706.
15. Ackerman LK, Serrano JL. Update on routine childhood and adolescent immunizations. Am Fam Physician. 2015;92(6):460-468.
16. Tom A, Robinett H, Buenconsejo-Lum L, et al. Promoting and providing the HPV vaccination in Hawaii: barriers faced by health providers. J Community Health. 2016;41(5):1069-1077.
17. Young JL, Bernheim RG, Korte JE, et al. Human papillomavirus vaccination recommendation may be linked to reimbursement: A survey of Virginia family practitioners and gynecologists. J Pediat Adolesc Gynecol. 2011;24(6):380-385.
18. Thomas R, Higgins L, Ding L, et al. Factors associated with HPV vaccine initiation, vaccine completion, and accuracy of self-reported vaccination status among 13- to 26-year-old men. Am J Mens Health. 2016;1-9.
19. Laz TH, Rahman M, Berenson AB. Human papillomavirus vaccine uptake among 9-17 year old males in the United States: the National Health Interview Survey, 2010. Hum Vaccin Immunother. 2013;9(4):874-878.
20. Perkins RB, Zisblatt L, Legler A, et al. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine. 2015;33:1223-1229.
21. Smulian EA, Mitchell KR, Stokley S. Interventions to increase HPV vaccination coverage: a systematic review. Hum Vaccin Immunother. 2016;12:1566-1588.
22. Walling EB, Benzoni N, Dornfeld J, et al. Interventions to improve HPV vaccine uptake: a systematic review. Pediatrics. 2016;138(1).
23. CDC. Overview of AFIX. www.cdc.gov/vaccines/programs/afix/index.html. Accessed August 17, 2017.
24. CDC. Human papillomavirus (HPV). www.cdc.gov/hpv/. Accessed August 17, 2017.
25. Fu LY, Bonhomme LA, Cooper SC, et al. Educational interventions to increase HPV vaccination acceptance: a systematic review. Vaccine. 2014;32(17):1901-1920.
26. Kennedy A, Sapsis KF, Stokley S, et al. Parental attitudes toward human papillomavirus vaccination: evaluation of an educational intervention, 2008. J Health Commun. 2011;16(3):300-313.
27. Spleen AM, Kluhsman BC, Clark AD, et al; ACTION Health Cancer Task Force. An increase in HPV-related knowledge and vaccination intent among parental and non-parental caregivers of adolescent girls, ages 9-17 years, in Appalachian Pennsylvania. J Cancer Educ. 2012;27(2):312-319.
28. Conroy K, Rosenthal SL, Zimet GD, et al. Human papillomavirus vaccine uptake, predictors of vaccination, and self-reported barriers to vaccination. J Womens Health (Larchmt). 2009;18(10):1679-1686.
29. National Vaccine Advisory Committee. Overcoming barriers to low HPV vaccine uptake in the United States: recommendations from the National Vaccine Advisory Committee. Public Health Rep. 2016;131(1):17-25.
30. Aragones A, Bruno DM, Ehrenberg M, et al. Parental education and text messaging reminders as effective community based tools to increase HPV vaccination rates among Mexican American children. Prev Med Rep. 2015;2:554-558.
31. Chao C, Preciado M, Slezak J, Xu L. A randomized intervention of reminder letter for human papillomavirus vaccine series completion. J Adolesc Health. 2015;56(1):85-90.
32. Community Preventive Services Task Force. The community guide—guide to community preventive services: increasing appropriate vaccinations. Atlanta, GA: Community Preventive Services Task Force; 2016
33. Kelminson K, Saville A, Seewald L, et al. Parental views of school-located delivery of adolescent vaccines. J Adolesc Health. 2012;51(2):190-196.
1. Juckett G, Hartsman-Adams H. Human papillomavirus: clinical manifestations and prevention. Am Fam Physician. 2010;82(10):1209-1213.
2. Markowitz LE, Dunne EF, Saraiya M, et al. Human papillomavirus vaccination: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2014;63(RR-05):1-30.
3. Viens LJ, Henley SJ, Watson M, et al. Human papillomavirus-associated cancers—United States, 2008-2012. MMWR Morb Mortal Wkly Rep. 2016;65(26):661-666.
4. American Cancer Society. Cancer facts and figures 2016. Atlanta: American Cancer Society; 2016.
5. Holman DM, Benard V, Roland KB, et al. Barriers to the human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr. 2014;168(1):76-82.
6. American College of Obstetricians and Gynecologists. Human papillomavirus vaccination. Committee Opinion. Number 704. June 2017. www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Adolescent-Health-Care/Human-Papillomavirus-Vaccination. Accessed August 17, 2017.
7. Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendation of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep. 2016;65(49):1405-1408.
8. Mulcahy N. GSK’s HPV vaccine, Cervarix, no longer available in the US. Medscape Medical News. October 26, 2016. www.medscape.com/viewarticle/870853. Accessed June 11, 2017.
9. Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(29):784-792.
10. Muncie HL, Lebato AL. HPV vaccination: overcoming parental and physician impediments. Am Fam Physician. 2015;92(6):449-454.
11. Bratic JS, Seyferth ER, Bocchini JA Jr. Update on barriers to human papillomavirus vaccination and effective strategies to promote vaccine acceptance. Curr Opin Pediatr. 2016;28(3):407-412.
12. Rahman M, Laz TH, McGrath CJ, Berenson AB. Provider recommendation mediates the relationship between parental human papillomavirus (HPV) vaccine awareness and HPV vaccine initiation and completion among 13- to 17-year-old US adolescent children. Clin Pediatr. 2015;54(4):371-375.
13. Sussman AL, Helitzer D, Bennett A, et al. Catching up with the HPV vaccine: challenges and opportunities in primary care. Ann Fam Med. 2015;13(4):354-360.
14. Clark SJ, Cowan AE, Filipp SL, et al. Parent perception of provider interactions influences HPV vaccination status of adolescent females. Clin Pediatr. 2016;55(8):701-706.
15. Ackerman LK, Serrano JL. Update on routine childhood and adolescent immunizations. Am Fam Physician. 2015;92(6):460-468.
16. Tom A, Robinett H, Buenconsejo-Lum L, et al. Promoting and providing the HPV vaccination in Hawaii: barriers faced by health providers. J Community Health. 2016;41(5):1069-1077.
17. Young JL, Bernheim RG, Korte JE, et al. Human papillomavirus vaccination recommendation may be linked to reimbursement: A survey of Virginia family practitioners and gynecologists. J Pediat Adolesc Gynecol. 2011;24(6):380-385.
18. Thomas R, Higgins L, Ding L, et al. Factors associated with HPV vaccine initiation, vaccine completion, and accuracy of self-reported vaccination status among 13- to 26-year-old men. Am J Mens Health. 2016;1-9.
19. Laz TH, Rahman M, Berenson AB. Human papillomavirus vaccine uptake among 9-17 year old males in the United States: the National Health Interview Survey, 2010. Hum Vaccin Immunother. 2013;9(4):874-878.
20. Perkins RB, Zisblatt L, Legler A, et al. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine. 2015;33:1223-1229.
21. Smulian EA, Mitchell KR, Stokley S. Interventions to increase HPV vaccination coverage: a systematic review. Hum Vaccin Immunother. 2016;12:1566-1588.
22. Walling EB, Benzoni N, Dornfeld J, et al. Interventions to improve HPV vaccine uptake: a systematic review. Pediatrics. 2016;138(1).
23. CDC. Overview of AFIX. www.cdc.gov/vaccines/programs/afix/index.html. Accessed August 17, 2017.
24. CDC. Human papillomavirus (HPV). www.cdc.gov/hpv/. Accessed August 17, 2017.
25. Fu LY, Bonhomme LA, Cooper SC, et al. Educational interventions to increase HPV vaccination acceptance: a systematic review. Vaccine. 2014;32(17):1901-1920.
26. Kennedy A, Sapsis KF, Stokley S, et al. Parental attitudes toward human papillomavirus vaccination: evaluation of an educational intervention, 2008. J Health Commun. 2011;16(3):300-313.
27. Spleen AM, Kluhsman BC, Clark AD, et al; ACTION Health Cancer Task Force. An increase in HPV-related knowledge and vaccination intent among parental and non-parental caregivers of adolescent girls, ages 9-17 years, in Appalachian Pennsylvania. J Cancer Educ. 2012;27(2):312-319.
28. Conroy K, Rosenthal SL, Zimet GD, et al. Human papillomavirus vaccine uptake, predictors of vaccination, and self-reported barriers to vaccination. J Womens Health (Larchmt). 2009;18(10):1679-1686.
29. National Vaccine Advisory Committee. Overcoming barriers to low HPV vaccine uptake in the United States: recommendations from the National Vaccine Advisory Committee. Public Health Rep. 2016;131(1):17-25.
30. Aragones A, Bruno DM, Ehrenberg M, et al. Parental education and text messaging reminders as effective community based tools to increase HPV vaccination rates among Mexican American children. Prev Med Rep. 2015;2:554-558.
31. Chao C, Preciado M, Slezak J, Xu L. A randomized intervention of reminder letter for human papillomavirus vaccine series completion. J Adolesc Health. 2015;56(1):85-90.
32. Community Preventive Services Task Force. The community guide—guide to community preventive services: increasing appropriate vaccinations. Atlanta, GA: Community Preventive Services Task Force; 2016
33. Kelminson K, Saville A, Seewald L, et al. Parental views of school-located delivery of adolescent vaccines. J Adolesc Health. 2012;51(2):190-196.