User login
TDF preferred in PrEP for Blacks and women, studies indicate
Although the efficacy of two pre-exposure prophylaxis (PrEP) regimens containing differing prodrug formulations of tenofovir are virtually identical, the balance between benefit and risk tips in favor of the combination using the older formulation, tenofovir disoproxil fumarate (TDF), a pharmacology researcher said.
An analysis of the pharmacologic profiles of TDF plus emtricitabine (FTC; Truvada and generic) with tenofovir alafenamide (TAF) plus FTC (Descovy) shows that the risk of decreased bone mineral density and renal toxicity with TDF are significantly lower than those of weight gain and related metabolic and cardiovascular problems associated with the newer tenofovir formulation TAF, according to pharmacology research fellow Andrew Hill, MD, PhD, from the University of Liverpool (England).
“I think when we’re comparing these two drugs overall, we have a clear benefit/risk, and we need to take both of these potential toxicities seriously, “ he said in an online presentation during IDWeek 2020, an annual scientific meeting on infectious diseases held virtually this year.
“But in my view, treating women – Black women – with TAF/FTC is a bad thing,” he continued. “I think it’s going lead to more harm, more myocardial infarctions, more cases of diabetes, and potentially more adverse birth outcomes, and I think that is a risk that is not worth taking, given that the apparent benefit in terms of bone mineral density and renal markers is a hypothesis at best, and is not translated into hard clinical endpoints.”
Adverse event profiles
Dr. Hill compared the side effect profiles of the two agents when used both in antiretroviral therapy (ART) in combination the integrase inhibitor dolutegravir (DTG; Tivicay), and in PrEP.
World Health Organization guidelines for first-line ART recommend the use of TDF/FTC/DTG, reserving TAF plus lamivudine (3TC) and DTG for use in special circumstances only, Dr. Hill noted.
He pointed to a pooled analysis of data from eight randomized, controlled trials of treatment-naive people living with HIV who started on ART from 2003 to 2015. The authors found that demographic factors associated with weight gain included lower CD4 cell counter, higher levels of HIV type 1 RNA, no injection drug use, female sex, and Black race.
They also found that, among nucleoside/nucleotide reverse transcriptase inhibitors, TAF was associated with more weight gain than TDF, abacavir, or zidovudine.
“This pattern is seen consistently across studies both of pre-exposure prophylaxis or treatment comparing tenofovir with either TAF or other nucleoside analogs,” he said.
The greater weight gain with TAF versus TDF was seen in both treatment trials and in the DISCOVER PrEP trial.
In addition, in a crossover trial conducted in Germany, patients who switched from TDF to TAF had an approximately 2 kg increase in body weight.
TAF has also been associated with higher grade 3 or 4 glucose and LDL cholesterol than TDF in clinical trials for the treatment of hepatitis B infections, and with higher LDL cholesterol and total cholesterol levels as well as diabetes in patients treated with the drugs in combination in the EMERALD HIV trial.
Clinical trials also tend to underestimate the real-world population of persons at highest risk for adverse events from TAF, Dr. Hill said, noting that the percentage of Black women in phase 3 trials for dolutegravir was 9%, compared with 42% among persons infected with HIV worldwide. The respective percentages for Black men are 16% versus 30%. These differences are similar across clinical trial programs for other ART agents.
“Generally, it’s women and Black people who seem to be at greatest risk for safety issues,” he said.
In the ADVANCE trial comparing TAF/FTC/DTG with TDF/FTC/DTG and a control arm of TDF, FTC and efavirenz, the mean change in weight among men after 3 years on the TAF-based regimen was a gain of 7.2 kg (15.9 lbs), compared with 5.5 kg (12 lbs) with TDF, and 2.6 kg (5.7 lbs) with the efavirenz-containing regimen.
In women enrolled in the same trial, the respective mean weight gains were 12.3 kg (27 lbs), 7.4 kg (16.3 lbs), and 5.5 kg (12 lbs).
“All of our analyses so far have shown that the weight continues to go up. We’re actually seeing people doubling in their body weight. We’ve seen some women come into clinic and their doctors don’t even recognize them because they’ve put on so much weight,” he said.
In women, most of the gain in weight occurs as limb or trunk fat, with a predominance of visceral fat.
People taking TAF in the trial were also at significantly greater risk for developing the metabolic syndrome, and at week 96, 27% of women on TAF/FTC/DTG had treatment-emergent obesity, compared with 17% for those on TDF/FTC/DTG and 11% for those on TDF/FTC/EFV. In men, the respective 96-week rates of treatment-emergent obesity were 7%, 3%, and 2%.
Clinical obesity itself is a risk factor for obstetric complications and birth outcomes, Alzheimer’s disease, type 2 diabetes, cardiovascular disease, hypertension and cancer, and an average 4-year reduction in life expectancy, Dr. Hill said. “I think it’s actually very unlikely that the [World Health Organization] guidelines will now change and allow the widespread use of TAF/FTC in combination with integrase inhibitors worldwide given these potential implications.”
Modern times
The bad rap that TDF gets for its alleged effects on bone mineral density and kidneys comes from studies where the drug was given in a boosted regimen that can amplify tenofovir toxicities, Dr. Hill said.
He noted that data from Gilead Sciences shows through 7 years of therapy in previously ART-naive patients, the combination of TDF/3TC/EFV showed sustained durable efficacy, no discontinuations to renal adverse effects, and no evidence of clinically relevant bone effects.
“I think we need to be very careful when we look at tenofovir and TAF. We need to look at the more modern way that these drugs are used, which is not with pharmacokinetic boosters anymore, and in that situation the toxicity profile of tenofovir/3TC – the original TDF – is very favorable,” he said.
Robert Goldstein, MD, PhD, an infectious disease specialist and medical director of the transgender health program at Massachusetts General Hospital in Boston, who comoderated the session where Dr. Hill presented his data, said that his clinical experience mirrors the pharmacokinetic findings.
“I certainly have strong feelings about the use of TDF in pre-exposure prophylaxis,” he said in an interview. “TDF is an effective and safe formulation of tenofovir to be used in pre-exposure prophylaxis, and one that we have more experience with. It’s the formulation of tenofovir that I use for all of my patients who are on pre-exposure prophylaxis, and I think it is the most cost-effective.’
No funding source was reported. Andrew Hill consults for Tibotec on clinical trial programs for darunavir, etravirine, and rilpivirine. Dr. Goldstein reported having no relevant disclosures.
Although the efficacy of two pre-exposure prophylaxis (PrEP) regimens containing differing prodrug formulations of tenofovir are virtually identical, the balance between benefit and risk tips in favor of the combination using the older formulation, tenofovir disoproxil fumarate (TDF), a pharmacology researcher said.
An analysis of the pharmacologic profiles of TDF plus emtricitabine (FTC; Truvada and generic) with tenofovir alafenamide (TAF) plus FTC (Descovy) shows that the risk of decreased bone mineral density and renal toxicity with TDF are significantly lower than those of weight gain and related metabolic and cardiovascular problems associated with the newer tenofovir formulation TAF, according to pharmacology research fellow Andrew Hill, MD, PhD, from the University of Liverpool (England).
“I think when we’re comparing these two drugs overall, we have a clear benefit/risk, and we need to take both of these potential toxicities seriously, “ he said in an online presentation during IDWeek 2020, an annual scientific meeting on infectious diseases held virtually this year.
“But in my view, treating women – Black women – with TAF/FTC is a bad thing,” he continued. “I think it’s going lead to more harm, more myocardial infarctions, more cases of diabetes, and potentially more adverse birth outcomes, and I think that is a risk that is not worth taking, given that the apparent benefit in terms of bone mineral density and renal markers is a hypothesis at best, and is not translated into hard clinical endpoints.”
Adverse event profiles
Dr. Hill compared the side effect profiles of the two agents when used both in antiretroviral therapy (ART) in combination the integrase inhibitor dolutegravir (DTG; Tivicay), and in PrEP.
World Health Organization guidelines for first-line ART recommend the use of TDF/FTC/DTG, reserving TAF plus lamivudine (3TC) and DTG for use in special circumstances only, Dr. Hill noted.
He pointed to a pooled analysis of data from eight randomized, controlled trials of treatment-naive people living with HIV who started on ART from 2003 to 2015. The authors found that demographic factors associated with weight gain included lower CD4 cell counter, higher levels of HIV type 1 RNA, no injection drug use, female sex, and Black race.
They also found that, among nucleoside/nucleotide reverse transcriptase inhibitors, TAF was associated with more weight gain than TDF, abacavir, or zidovudine.
“This pattern is seen consistently across studies both of pre-exposure prophylaxis or treatment comparing tenofovir with either TAF or other nucleoside analogs,” he said.
The greater weight gain with TAF versus TDF was seen in both treatment trials and in the DISCOVER PrEP trial.
In addition, in a crossover trial conducted in Germany, patients who switched from TDF to TAF had an approximately 2 kg increase in body weight.
TAF has also been associated with higher grade 3 or 4 glucose and LDL cholesterol than TDF in clinical trials for the treatment of hepatitis B infections, and with higher LDL cholesterol and total cholesterol levels as well as diabetes in patients treated with the drugs in combination in the EMERALD HIV trial.
Clinical trials also tend to underestimate the real-world population of persons at highest risk for adverse events from TAF, Dr. Hill said, noting that the percentage of Black women in phase 3 trials for dolutegravir was 9%, compared with 42% among persons infected with HIV worldwide. The respective percentages for Black men are 16% versus 30%. These differences are similar across clinical trial programs for other ART agents.
“Generally, it’s women and Black people who seem to be at greatest risk for safety issues,” he said.
In the ADVANCE trial comparing TAF/FTC/DTG with TDF/FTC/DTG and a control arm of TDF, FTC and efavirenz, the mean change in weight among men after 3 years on the TAF-based regimen was a gain of 7.2 kg (15.9 lbs), compared with 5.5 kg (12 lbs) with TDF, and 2.6 kg (5.7 lbs) with the efavirenz-containing regimen.
In women enrolled in the same trial, the respective mean weight gains were 12.3 kg (27 lbs), 7.4 kg (16.3 lbs), and 5.5 kg (12 lbs).
“All of our analyses so far have shown that the weight continues to go up. We’re actually seeing people doubling in their body weight. We’ve seen some women come into clinic and their doctors don’t even recognize them because they’ve put on so much weight,” he said.
In women, most of the gain in weight occurs as limb or trunk fat, with a predominance of visceral fat.
People taking TAF in the trial were also at significantly greater risk for developing the metabolic syndrome, and at week 96, 27% of women on TAF/FTC/DTG had treatment-emergent obesity, compared with 17% for those on TDF/FTC/DTG and 11% for those on TDF/FTC/EFV. In men, the respective 96-week rates of treatment-emergent obesity were 7%, 3%, and 2%.
Clinical obesity itself is a risk factor for obstetric complications and birth outcomes, Alzheimer’s disease, type 2 diabetes, cardiovascular disease, hypertension and cancer, and an average 4-year reduction in life expectancy, Dr. Hill said. “I think it’s actually very unlikely that the [World Health Organization] guidelines will now change and allow the widespread use of TAF/FTC in combination with integrase inhibitors worldwide given these potential implications.”
Modern times
The bad rap that TDF gets for its alleged effects on bone mineral density and kidneys comes from studies where the drug was given in a boosted regimen that can amplify tenofovir toxicities, Dr. Hill said.
He noted that data from Gilead Sciences shows through 7 years of therapy in previously ART-naive patients, the combination of TDF/3TC/EFV showed sustained durable efficacy, no discontinuations to renal adverse effects, and no evidence of clinically relevant bone effects.
“I think we need to be very careful when we look at tenofovir and TAF. We need to look at the more modern way that these drugs are used, which is not with pharmacokinetic boosters anymore, and in that situation the toxicity profile of tenofovir/3TC – the original TDF – is very favorable,” he said.
Robert Goldstein, MD, PhD, an infectious disease specialist and medical director of the transgender health program at Massachusetts General Hospital in Boston, who comoderated the session where Dr. Hill presented his data, said that his clinical experience mirrors the pharmacokinetic findings.
“I certainly have strong feelings about the use of TDF in pre-exposure prophylaxis,” he said in an interview. “TDF is an effective and safe formulation of tenofovir to be used in pre-exposure prophylaxis, and one that we have more experience with. It’s the formulation of tenofovir that I use for all of my patients who are on pre-exposure prophylaxis, and I think it is the most cost-effective.’
No funding source was reported. Andrew Hill consults for Tibotec on clinical trial programs for darunavir, etravirine, and rilpivirine. Dr. Goldstein reported having no relevant disclosures.
Although the efficacy of two pre-exposure prophylaxis (PrEP) regimens containing differing prodrug formulations of tenofovir are virtually identical, the balance between benefit and risk tips in favor of the combination using the older formulation, tenofovir disoproxil fumarate (TDF), a pharmacology researcher said.
An analysis of the pharmacologic profiles of TDF plus emtricitabine (FTC; Truvada and generic) with tenofovir alafenamide (TAF) plus FTC (Descovy) shows that the risk of decreased bone mineral density and renal toxicity with TDF are significantly lower than those of weight gain and related metabolic and cardiovascular problems associated with the newer tenofovir formulation TAF, according to pharmacology research fellow Andrew Hill, MD, PhD, from the University of Liverpool (England).
“I think when we’re comparing these two drugs overall, we have a clear benefit/risk, and we need to take both of these potential toxicities seriously, “ he said in an online presentation during IDWeek 2020, an annual scientific meeting on infectious diseases held virtually this year.
“But in my view, treating women – Black women – with TAF/FTC is a bad thing,” he continued. “I think it’s going lead to more harm, more myocardial infarctions, more cases of diabetes, and potentially more adverse birth outcomes, and I think that is a risk that is not worth taking, given that the apparent benefit in terms of bone mineral density and renal markers is a hypothesis at best, and is not translated into hard clinical endpoints.”
Adverse event profiles
Dr. Hill compared the side effect profiles of the two agents when used both in antiretroviral therapy (ART) in combination the integrase inhibitor dolutegravir (DTG; Tivicay), and in PrEP.
World Health Organization guidelines for first-line ART recommend the use of TDF/FTC/DTG, reserving TAF plus lamivudine (3TC) and DTG for use in special circumstances only, Dr. Hill noted.
He pointed to a pooled analysis of data from eight randomized, controlled trials of treatment-naive people living with HIV who started on ART from 2003 to 2015. The authors found that demographic factors associated with weight gain included lower CD4 cell counter, higher levels of HIV type 1 RNA, no injection drug use, female sex, and Black race.
They also found that, among nucleoside/nucleotide reverse transcriptase inhibitors, TAF was associated with more weight gain than TDF, abacavir, or zidovudine.
“This pattern is seen consistently across studies both of pre-exposure prophylaxis or treatment comparing tenofovir with either TAF or other nucleoside analogs,” he said.
The greater weight gain with TAF versus TDF was seen in both treatment trials and in the DISCOVER PrEP trial.
In addition, in a crossover trial conducted in Germany, patients who switched from TDF to TAF had an approximately 2 kg increase in body weight.
TAF has also been associated with higher grade 3 or 4 glucose and LDL cholesterol than TDF in clinical trials for the treatment of hepatitis B infections, and with higher LDL cholesterol and total cholesterol levels as well as diabetes in patients treated with the drugs in combination in the EMERALD HIV trial.
Clinical trials also tend to underestimate the real-world population of persons at highest risk for adverse events from TAF, Dr. Hill said, noting that the percentage of Black women in phase 3 trials for dolutegravir was 9%, compared with 42% among persons infected with HIV worldwide. The respective percentages for Black men are 16% versus 30%. These differences are similar across clinical trial programs for other ART agents.
“Generally, it’s women and Black people who seem to be at greatest risk for safety issues,” he said.
In the ADVANCE trial comparing TAF/FTC/DTG with TDF/FTC/DTG and a control arm of TDF, FTC and efavirenz, the mean change in weight among men after 3 years on the TAF-based regimen was a gain of 7.2 kg (15.9 lbs), compared with 5.5 kg (12 lbs) with TDF, and 2.6 kg (5.7 lbs) with the efavirenz-containing regimen.
In women enrolled in the same trial, the respective mean weight gains were 12.3 kg (27 lbs), 7.4 kg (16.3 lbs), and 5.5 kg (12 lbs).
“All of our analyses so far have shown that the weight continues to go up. We’re actually seeing people doubling in their body weight. We’ve seen some women come into clinic and their doctors don’t even recognize them because they’ve put on so much weight,” he said.
In women, most of the gain in weight occurs as limb or trunk fat, with a predominance of visceral fat.
People taking TAF in the trial were also at significantly greater risk for developing the metabolic syndrome, and at week 96, 27% of women on TAF/FTC/DTG had treatment-emergent obesity, compared with 17% for those on TDF/FTC/DTG and 11% for those on TDF/FTC/EFV. In men, the respective 96-week rates of treatment-emergent obesity were 7%, 3%, and 2%.
Clinical obesity itself is a risk factor for obstetric complications and birth outcomes, Alzheimer’s disease, type 2 diabetes, cardiovascular disease, hypertension and cancer, and an average 4-year reduction in life expectancy, Dr. Hill said. “I think it’s actually very unlikely that the [World Health Organization] guidelines will now change and allow the widespread use of TAF/FTC in combination with integrase inhibitors worldwide given these potential implications.”
Modern times
The bad rap that TDF gets for its alleged effects on bone mineral density and kidneys comes from studies where the drug was given in a boosted regimen that can amplify tenofovir toxicities, Dr. Hill said.
He noted that data from Gilead Sciences shows through 7 years of therapy in previously ART-naive patients, the combination of TDF/3TC/EFV showed sustained durable efficacy, no discontinuations to renal adverse effects, and no evidence of clinically relevant bone effects.
“I think we need to be very careful when we look at tenofovir and TAF. We need to look at the more modern way that these drugs are used, which is not with pharmacokinetic boosters anymore, and in that situation the toxicity profile of tenofovir/3TC – the original TDF – is very favorable,” he said.
Robert Goldstein, MD, PhD, an infectious disease specialist and medical director of the transgender health program at Massachusetts General Hospital in Boston, who comoderated the session where Dr. Hill presented his data, said that his clinical experience mirrors the pharmacokinetic findings.
“I certainly have strong feelings about the use of TDF in pre-exposure prophylaxis,” he said in an interview. “TDF is an effective and safe formulation of tenofovir to be used in pre-exposure prophylaxis, and one that we have more experience with. It’s the formulation of tenofovir that I use for all of my patients who are on pre-exposure prophylaxis, and I think it is the most cost-effective.’
No funding source was reported. Andrew Hill consults for Tibotec on clinical trial programs for darunavir, etravirine, and rilpivirine. Dr. Goldstein reported having no relevant disclosures.
FROM IDWEEK 2020
Cutaneous Filariasis in an American Traveler
To the Editor:
Cutaneous filariasis is a group of infectious diseases caused by more than 60 different nematode species and endemic to approximately 83 countries.1,2 These infections are transmitted to humans by vectors such as mosquitoes, blackflies, biting midges, or Tabanid flies.1 The blood meal taken by vectors allow microfilariae to enter the skin and develop into adult worms.1-3 It is postulated that filarial infections were first described in 2000
Filarial parasites have been categorized into groups based on the site of adult worm stage habitat. The cutaneous group includes Loa loa, Onchocerca volvulus, Mansonella perstans, and Dipetalonema streptocerca. The lymphatic group includes Wuchereria bancrofti, Brugia malayi, and Brugia timori. Lastly, the body cavity group includes Mansonella ozzardi. Because humidity is required for survival of the infective larval stage, individuals from tropical countries in Africa, Central America, and South America most commonly are affected.1 These diseases also are related to poor housing quality and inadequate sanitation.1,3,4 Travel for business, medical missions, pleasure, or emigration has caused increased filarial infections globally. In fact, dermatologic disorders with infectious etiologies cause approximately 17% of travelers to seek medical attention.3
Dermatologic manifestations indicative of a potential cutaneous filarial infection include papules, nodules, excoriations with secondary xerosis, lichenification, skin pigment changes, and/or severe pruritus. However, individuals from filarial endemic regions may not demonstrate any clinical signs or symptoms, despite having microfilariae in their blood,1 which enables the disease to propagate and poses a major public health concern. In fact, patients with chronic filarial infections are at increased risk for developing lymphedema, elephantiasis, or blindness, which are hypothesized to be the second largest cause of permanent disability worldwide.1 Although rarely seen in US citizens, cutaneous filariasis should always be a diagnostic consideration in patients who present with a pruritic eruption and have a travel history to tropical countries. We report a case of cutaneous onchocerciasis in a US citizen who developed a pruritic eczematous eruption on the right upper arm following an arthropod assault while working in an Onchocerca endemic region approximately 1.5 years prior.
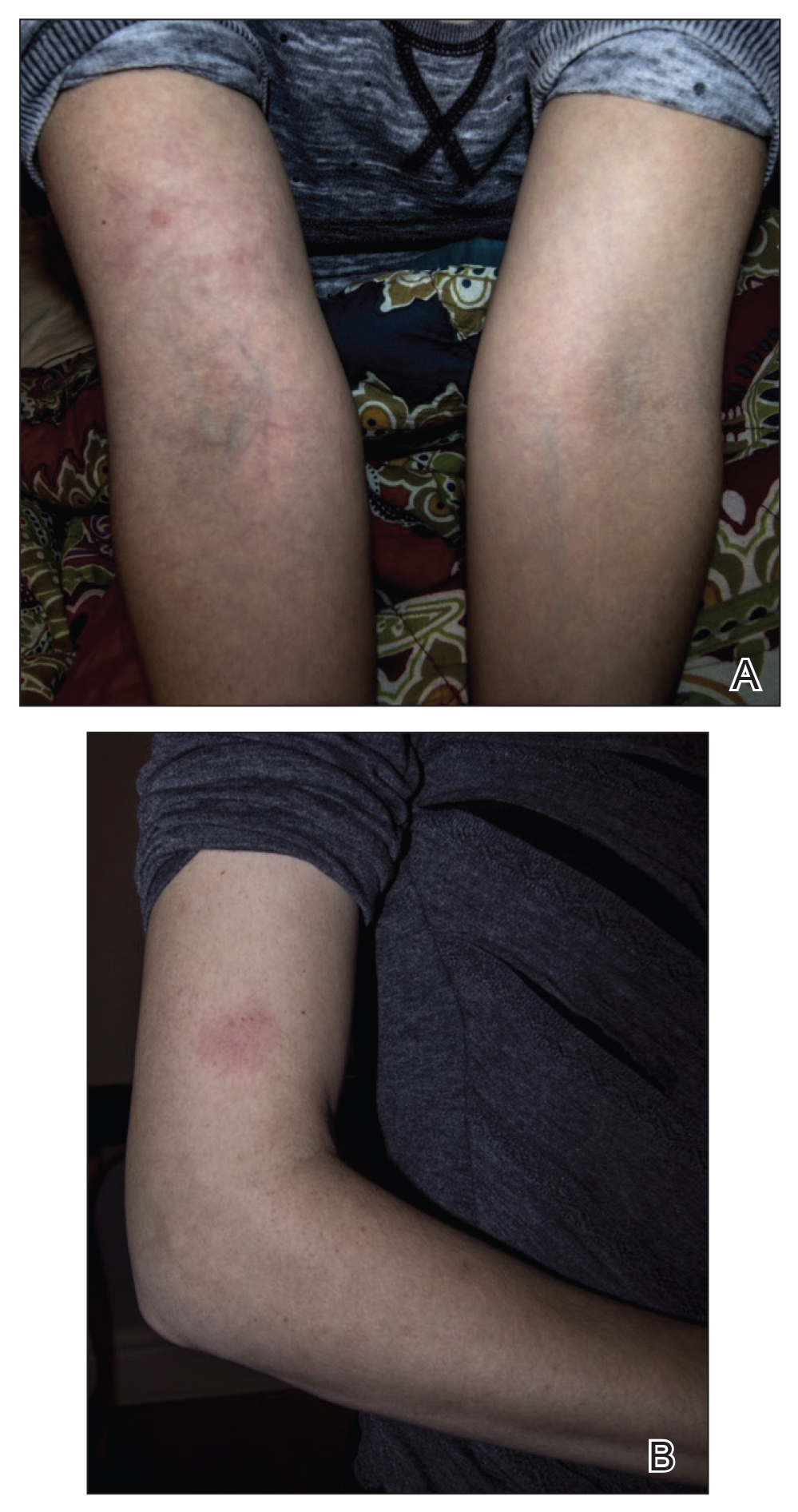
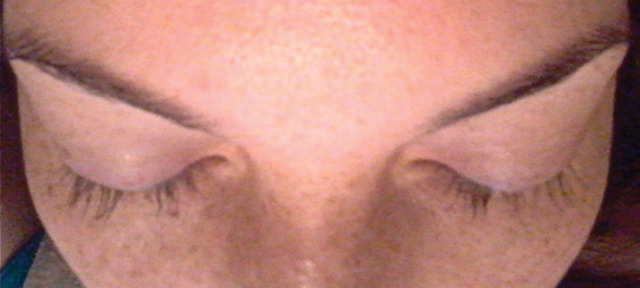
A 33-year-old woman presented to our outpatient dermatology clinic with the chief concern of a pruritic rash on the right arm (Figure 1). She revealed a history of travel to Peru, associated symptoms, prior diagnoses, and attempted treatment regimens. Over a 1.5-year period, the patient traveled for work-related reasons to Madre de Dios, Peru. During that time, the patient experienced 2 bouts of severe abdominal pain, nausea, and vomiting that she attributed to poor food and water quality. She did not immediately seek medical attention. She also developed skin manifestations that began as a pruritic and irritating pinpoint-sized red papule on the right eyelid, possibly a site of an arthropod assault. She then experienced episodic eyelash loss (Figure 2) and subsequent vision changes, including blurry vision, halos around lights, and dimming around the periphery. When the patient developed circular, pink, pruritic plaques on the right upper extremity, she sought medical attention in Lima, Peru. She was diagnosed with tinea corporis and prescribed an oral antibiotic and topical antifungal.
This treatment regimen did not result in improvement. In the following months, the patient noticed worsening of the ocular symptoms, and she developed a dry cough with occasional dyspnea. She again sought medical attention in Peru and was referred to an ophthalmologist who suspected Demodex mite or fungal infection; however, workup for those pathologies was negative. The respiratory symptoms continued to worsen, particularly at night, and she began to experience palpitations.
Upon returning to the United States, the patient was evaluated by her primary care physician who ordered the following laboratory tests: a complete blood cell count with differential, comprehensive metabolic panel, vitamin D level, blood culture, lipid panel, ferritin level, and thyroid function. An electrocardiogram, throat culture, and stool culture for ova and parasites also were obtained. The electrocardiogram showed sinus tachycardia, and the stool culture revealed blastocystis, for which she was prescribed oral metronidazole and tinidazole. The other results were within reference range, and the throat culture showed normal oropharyngeal microbes.
A few days after the treatment for the blastocystis, the gastrointestinal tract symptoms improved, but dermatologic, ocular, pulmonary, and cardiac symptoms persisted. She also began experiencing night sweats and hot flashes. In between visits to the primary care physician, she sought medical attention at an urgent care clinic for worsening pulmonary and cardiac symptoms. At that time, results of a chest radiograph were normal.
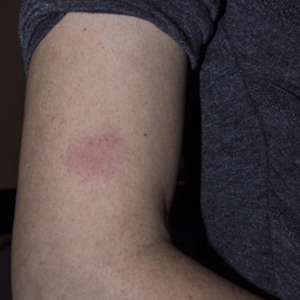
The primary care physician referred her to infectious disease and dermatology for further evaluation within a few weeks of returning to the United States. Upon presentation to dermatology, the patient described a waxing and waning nature to the rash on the arm with associated intermittent pain. Physical examination revealed several erythematous patches on the triceps and a 1-cm subcutaneous nodule on the right elbow with overlying skin discoloration. The patient stated that she first noticed the inflamed painful nodule when she returned to the United States. Since its discovery, the nodule increased in size and became firm. The patient also described occasional limited range of motion of the right arm due to the discomfort of the rash and inflamed nodule. Interestingly, the patient’s partner who accompanied her to Peru also developed a similar pruritic rash on the chest. He was evaluated by dermatology and infectious disease; however, biopsies of his rash performed at a different practice revealed nonspecific results. Due to her partner’s inconclusive results, our patient initially refused a biopsy; however, she returned to the office after 1 week with worsening symptoms, and a 4-mm punch biopsy of the right arm was obtained in addition to rapid plasma reagin and QuantiFERON-TB Gold test.
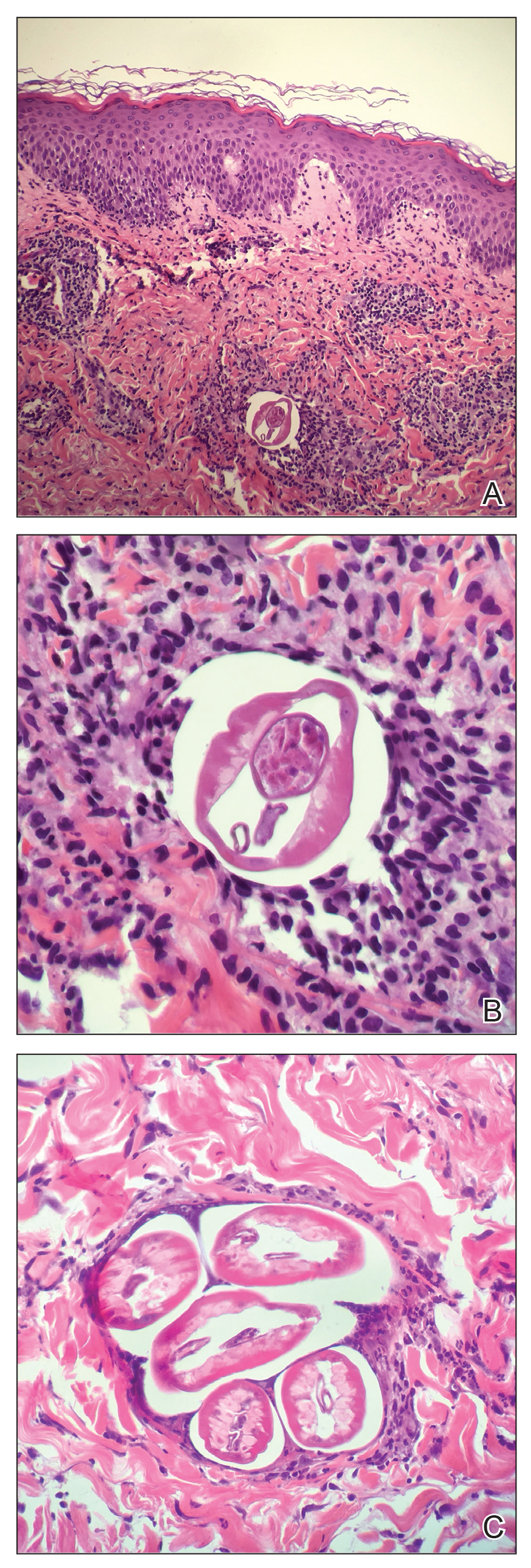
Based on the patient’s travel history, clinical course, and physical examination, the clinical differential diagnosis included nummular dermatitis, panniculitis/nodular vasculitis, tinea corporis, secondary syphilis, pinta, tuberculosis, or cutaneous filariasis. Rapid plasma reagin and QuantiFERON-TB Gold test results were negative, and a periodic acid–Schiff stain of the biopsy was negative for fungal elements. Dermatopathology revealed intradermal filarial nematodes of 120 to 150 µm in diameter and 1-to 2-mm cuticles eliciting a predominantly superficial and deep lymphohistiocytic reaction (Figure 3). The histopathologic differential diagnosis based on the diameter of the nematode included M perstans, W bancrofti, and O volvulus. Clinical correlation was highly recommended to obtain a final diagnosis and management plan. A portion of the biopsy was sent to the Centers for Disease Control and Prevention, which confirmed the presence of a filarial nematode consistent with a zoonotic Brugia but also with zoonotic Onchocerca as a possible differential. Therefore, considering the history, clinical course, and histopathologic analysis, the final diagnosis of cutaneous onchocerciasis was made.
The patient was referred back to infectious disease and started on combination therapy of ivermectin (25.5 mg) plus albendazole (800 mg) taken at once followed by doxycycline (200 mg) for 6 weeks. The symptoms initially worsened, then she experienced near-complete resolution of dermatologic, pulmonary, and ocular symptoms after approximately 2 weeks of treatment. She continued to report fatigue and palpitations, for she continued to see a cardiologist. We highly encouraged her to continue following up with infectious disease to ensure complete eradication of the infection.
Onchocerciasis is common in individuals who live or work in equatorial Africa, Central America, and South America. The disease process has dermatologic, ocular, and other systemic manifestations if the infection continues long-term without treatment.2,5 Although rare, this infection has been observed in US citizens who have traveled to filarial endemic regions for a period of time ranging from 2 weeks to 39 years.2,6,7
Blackflies (genus Simulium), the intermediate host of O volvulus, breed in areas close to freely flowing water. The blackfly bites a human host, and within 7 days the microfilariae undergo 2 molts to reach the infective stage. The larvae remain in the dermis and subcutaneous tissue for 2 additional molts until they develop into adult worms. Then, adult worms may become encapsulated, eliciting and forming subcutaneous nodules, also known as onchocercomas.2,3
A fertilized female in the subcutaneous tissue releases microfilariae that may remain in the skin or travel to the eyes of the host.3 The host may demonstrate signs of infection within 3 to 15 months.2 Most commonly, patients report localized or generalized pruritus, and one extremity develops swelling, papules, lymphadenopathy, and alterations of skin pigment (hypopigmentation or hyperpigmentation).5-8 Our patient developed a pruritic eczematous eruption that only affected her right arm with an inflamed firm nodule overlying the elbow, a suspected onchocercoma. Onchocercomas are the direct result of adult worms coiled in the dermis, subcutaneous tissue, and deep fascia. These commonly are found in close proximity to bony prominences with the specific location pending the geographic area of infection.8 For example, onchocercomas more commonly are found over bony prominences in the head and upper body region in patients who acquired the disease from Central or South America, whereas individuals infected in Africa more commonly develop onchocercomas near the femoral, coccyx, or sacral regions.2
The immune response mounted by the infected host is responsible for the ocular signs and symptoms.2 Specifically, the inflammatory reaction may impact the patient’s cornea, anterior uveal tract, chorioretinal zone, or optic nerve. In fact, onchocerciasis, or river blindness, is the leading cause of blindness in the world.2 The host immune response also is responsible for the dermatologic manifestations of this disease. In addition to the dermatologic manifestations already mentioned, others include epidermal atrophy, ulcerations, femoral or inguinal lymphadenitis, lymphedema, and/or general wasting in more severe long-standing infections. Additionally, patients may experience systemic signs resulting from an underlying O volvulus infection.8 Our patient demonstrated a subjectively remarkable systemic response manifested by shortness of breath, dyspnea, cough, and palpitations, likely the result of her existing filarial infection.
Diagnosis of onchocerciasis is made by identification of microfilariae or adult worms in skin snips or punch biopsies. Histopathologic analysis of onchocerciasis has been described as several adult worms in the subcutaneous tissue surrounded by a granulomatous, fibrotic, calcified, or sometimes ossified inflammatory reaction.8 Microfilariae may migrate to the upper dermis and typically are surrounded by lymphocytes, histiocytes, plasma cells, and eosinophils with an absence of neutrophils,5,8 which directly causes the overlying epidermis to undergo reactive changes. Measurement of filarial nematodes often helps differentiate species. For example, O volvulus typically measures 230 to 500 mm in length for females and 16 to 42 mm in length for males.8 The thickness of cuticles typically measures 4 to 8 µm for females and 3 to 5 µm for males. Microfilariae of O volvulus also have been identified in patient’s sputum, urine, blood, and spinal fluid.8 An alternative diagnostic method described by Stingl et al9 is a diethylcarbamazine (DEC) patch test that was 92% accurate in onchocerciasis cases diagnosed by skin snip analysis.9 Therefore, this test may be a practical alternative when skin specimens are inconclusive.
Management of onchocerciasis should include excision of subcutaneous nodules to remove adult worms. In the past, DEC with suramin was prescribed and kills both microfilariae and adult worms.8 However, DEC may induce pruritus, chorioretinal damage, and optic neuritis and suramin is nephrotoxic.2 Therefore, oral ivermectin (0.15–0.20 mg/kg one-time dose, may be repeated in 3–12 months) is supported to be a less harmful option. Patients treated with ivermectin have a decreased risk for transmitting infection to the blackfly vector for up to 6 months after treatment, and it is a more effective microfilaricidal agent than DEC.2,3 Oral doxycycline (100 mg/d for 6 weeks) commonly is added as adjuvant therapy because it kills Wolbachia species, a bacterium that is needed for O volvulus reproduction.3
Although rare in the United States, cutaneous filariasis has become a prevalent public health concern, especially in tropical countries. Individuals with chronic cutaneous filarial infections are at increased risk for debilitating complications that negatively affect their quality of life and productivity. Although the World Health Organization has attempted to eradicate cutaneous filarial infections by mass drug administration, transmission of these diseases remains a challenge. Further research on treatment and methods to prevent transmission by controlling arthropod vectors is required to avoid short-term and long-term health consequences caused by cutaneous filariasis.
Parasitic infections should always be a diagnostic consideration in individuals who present with a pruritic eruption and a history of travel to foreign countries located in Africa, Central America, and South America. Dermatologists in the United States should increase familiarity with these infections because of increased travel, economic globalization, and the impact of global climate change on the geographic distribution of vector arthropods. To control these infections, research efforts should focus on improved sanitation; drug treatment; transmission prevention; and improved education of their clinical manifestations, especially mucocutaneous signs.
- Mendoza N, Li A, Gill A, et al. Filariasis: diagnosis and treatment. Dermatol Ther. 2009;22:475-490.
- Maso MJ, Kapila R, Schwartz RA, et al. Cutaneous onchocerciasis. Int J Dermatol. 1987;26:593-596.
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- Bolivar-Meija A, Alarcon-Olave C, Rodriguez-Morales AJ. Skin manifestations of arthropod-borne infection in Latin America. Curr Opin. 2014;27:288-294.
- Okulicz JF, Stibich AS, Elston DM, et al. Cutaneous onchocercoma. Int J Dermatol. 2004;43:170-172.
- Nguyen JC, Murphy ME, Nutman TB, et al. Cutaneous onchocerciasis in an American traveler. Int Soc Dermatol. 2005;44:125-128.
- Toovey S, Moerman F, van Gompel A. Special infectious disease risks of expatriates and long-term travelers in tropical countries part II: infections other than malaria. J Travel Med. 2007;14:50-60.
- Meyers WM, Neafie RC, Connor DH. Bancroftian and malayan filariasis. In Binford CH, ed. Pathology of Tropical and Extraordinary Diseases. Vol 2. Fort Sam Houston, TX: Armed Forces Institute of Pathology; 1976:340-355.
- Stingl P, Ross M, Gibson DW, et al. A diagnostic “patch-test” for onchocerciasis using topical diethylcarbamazine. Trans R Soc Trop Med Hyg.
To the Editor:
Cutaneous filariasis is a group of infectious diseases caused by more than 60 different nematode species and endemic to approximately 83 countries.1,2 These infections are transmitted to humans by vectors such as mosquitoes, blackflies, biting midges, or Tabanid flies.1 The blood meal taken by vectors allow microfilariae to enter the skin and develop into adult worms.1-3 It is postulated that filarial infections were first described in 2000
Filarial parasites have been categorized into groups based on the site of adult worm stage habitat. The cutaneous group includes Loa loa, Onchocerca volvulus, Mansonella perstans, and Dipetalonema streptocerca. The lymphatic group includes Wuchereria bancrofti, Brugia malayi, and Brugia timori. Lastly, the body cavity group includes Mansonella ozzardi. Because humidity is required for survival of the infective larval stage, individuals from tropical countries in Africa, Central America, and South America most commonly are affected.1 These diseases also are related to poor housing quality and inadequate sanitation.1,3,4 Travel for business, medical missions, pleasure, or emigration has caused increased filarial infections globally. In fact, dermatologic disorders with infectious etiologies cause approximately 17% of travelers to seek medical attention.3
Dermatologic manifestations indicative of a potential cutaneous filarial infection include papules, nodules, excoriations with secondary xerosis, lichenification, skin pigment changes, and/or severe pruritus. However, individuals from filarial endemic regions may not demonstrate any clinical signs or symptoms, despite having microfilariae in their blood,1 which enables the disease to propagate and poses a major public health concern. In fact, patients with chronic filarial infections are at increased risk for developing lymphedema, elephantiasis, or blindness, which are hypothesized to be the second largest cause of permanent disability worldwide.1 Although rarely seen in US citizens, cutaneous filariasis should always be a diagnostic consideration in patients who present with a pruritic eruption and have a travel history to tropical countries. We report a case of cutaneous onchocerciasis in a US citizen who developed a pruritic eczematous eruption on the right upper arm following an arthropod assault while working in an Onchocerca endemic region approximately 1.5 years prior.
A 33-year-old woman presented to our outpatient dermatology clinic with the chief concern of a pruritic rash on the right arm (Figure 1). She revealed a history of travel to Peru, associated symptoms, prior diagnoses, and attempted treatment regimens. Over a 1.5-year period, the patient traveled for work-related reasons to Madre de Dios, Peru. During that time, the patient experienced 2 bouts of severe abdominal pain, nausea, and vomiting that she attributed to poor food and water quality. She did not immediately seek medical attention. She also developed skin manifestations that began as a pruritic and irritating pinpoint-sized red papule on the right eyelid, possibly a site of an arthropod assault. She then experienced episodic eyelash loss (Figure 2) and subsequent vision changes, including blurry vision, halos around lights, and dimming around the periphery. When the patient developed circular, pink, pruritic plaques on the right upper extremity, she sought medical attention in Lima, Peru. She was diagnosed with tinea corporis and prescribed an oral antibiotic and topical antifungal.
This treatment regimen did not result in improvement. In the following months, the patient noticed worsening of the ocular symptoms, and she developed a dry cough with occasional dyspnea. She again sought medical attention in Peru and was referred to an ophthalmologist who suspected Demodex mite or fungal infection; however, workup for those pathologies was negative. The respiratory symptoms continued to worsen, particularly at night, and she began to experience palpitations.
Upon returning to the United States, the patient was evaluated by her primary care physician who ordered the following laboratory tests: a complete blood cell count with differential, comprehensive metabolic panel, vitamin D level, blood culture, lipid panel, ferritin level, and thyroid function. An electrocardiogram, throat culture, and stool culture for ova and parasites also were obtained. The electrocardiogram showed sinus tachycardia, and the stool culture revealed blastocystis, for which she was prescribed oral metronidazole and tinidazole. The other results were within reference range, and the throat culture showed normal oropharyngeal microbes.
A few days after the treatment for the blastocystis, the gastrointestinal tract symptoms improved, but dermatologic, ocular, pulmonary, and cardiac symptoms persisted. She also began experiencing night sweats and hot flashes. In between visits to the primary care physician, she sought medical attention at an urgent care clinic for worsening pulmonary and cardiac symptoms. At that time, results of a chest radiograph were normal.
The primary care physician referred her to infectious disease and dermatology for further evaluation within a few weeks of returning to the United States. Upon presentation to dermatology, the patient described a waxing and waning nature to the rash on the arm with associated intermittent pain. Physical examination revealed several erythematous patches on the triceps and a 1-cm subcutaneous nodule on the right elbow with overlying skin discoloration. The patient stated that she first noticed the inflamed painful nodule when she returned to the United States. Since its discovery, the nodule increased in size and became firm. The patient also described occasional limited range of motion of the right arm due to the discomfort of the rash and inflamed nodule. Interestingly, the patient’s partner who accompanied her to Peru also developed a similar pruritic rash on the chest. He was evaluated by dermatology and infectious disease; however, biopsies of his rash performed at a different practice revealed nonspecific results. Due to her partner’s inconclusive results, our patient initially refused a biopsy; however, she returned to the office after 1 week with worsening symptoms, and a 4-mm punch biopsy of the right arm was obtained in addition to rapid plasma reagin and QuantiFERON-TB Gold test.
Based on the patient’s travel history, clinical course, and physical examination, the clinical differential diagnosis included nummular dermatitis, panniculitis/nodular vasculitis, tinea corporis, secondary syphilis, pinta, tuberculosis, or cutaneous filariasis. Rapid plasma reagin and QuantiFERON-TB Gold test results were negative, and a periodic acid–Schiff stain of the biopsy was negative for fungal elements. Dermatopathology revealed intradermal filarial nematodes of 120 to 150 µm in diameter and 1-to 2-mm cuticles eliciting a predominantly superficial and deep lymphohistiocytic reaction (Figure 3). The histopathologic differential diagnosis based on the diameter of the nematode included M perstans, W bancrofti, and O volvulus. Clinical correlation was highly recommended to obtain a final diagnosis and management plan. A portion of the biopsy was sent to the Centers for Disease Control and Prevention, which confirmed the presence of a filarial nematode consistent with a zoonotic Brugia but also with zoonotic Onchocerca as a possible differential. Therefore, considering the history, clinical course, and histopathologic analysis, the final diagnosis of cutaneous onchocerciasis was made.
The patient was referred back to infectious disease and started on combination therapy of ivermectin (25.5 mg) plus albendazole (800 mg) taken at once followed by doxycycline (200 mg) for 6 weeks. The symptoms initially worsened, then she experienced near-complete resolution of dermatologic, pulmonary, and ocular symptoms after approximately 2 weeks of treatment. She continued to report fatigue and palpitations, for she continued to see a cardiologist. We highly encouraged her to continue following up with infectious disease to ensure complete eradication of the infection.
Onchocerciasis is common in individuals who live or work in equatorial Africa, Central America, and South America. The disease process has dermatologic, ocular, and other systemic manifestations if the infection continues long-term without treatment.2,5 Although rare, this infection has been observed in US citizens who have traveled to filarial endemic regions for a period of time ranging from 2 weeks to 39 years.2,6,7
Blackflies (genus Simulium), the intermediate host of O volvulus, breed in areas close to freely flowing water. The blackfly bites a human host, and within 7 days the microfilariae undergo 2 molts to reach the infective stage. The larvae remain in the dermis and subcutaneous tissue for 2 additional molts until they develop into adult worms. Then, adult worms may become encapsulated, eliciting and forming subcutaneous nodules, also known as onchocercomas.2,3
A fertilized female in the subcutaneous tissue releases microfilariae that may remain in the skin or travel to the eyes of the host.3 The host may demonstrate signs of infection within 3 to 15 months.2 Most commonly, patients report localized or generalized pruritus, and one extremity develops swelling, papules, lymphadenopathy, and alterations of skin pigment (hypopigmentation or hyperpigmentation).5-8 Our patient developed a pruritic eczematous eruption that only affected her right arm with an inflamed firm nodule overlying the elbow, a suspected onchocercoma. Onchocercomas are the direct result of adult worms coiled in the dermis, subcutaneous tissue, and deep fascia. These commonly are found in close proximity to bony prominences with the specific location pending the geographic area of infection.8 For example, onchocercomas more commonly are found over bony prominences in the head and upper body region in patients who acquired the disease from Central or South America, whereas individuals infected in Africa more commonly develop onchocercomas near the femoral, coccyx, or sacral regions.2
The immune response mounted by the infected host is responsible for the ocular signs and symptoms.2 Specifically, the inflammatory reaction may impact the patient’s cornea, anterior uveal tract, chorioretinal zone, or optic nerve. In fact, onchocerciasis, or river blindness, is the leading cause of blindness in the world.2 The host immune response also is responsible for the dermatologic manifestations of this disease. In addition to the dermatologic manifestations already mentioned, others include epidermal atrophy, ulcerations, femoral or inguinal lymphadenitis, lymphedema, and/or general wasting in more severe long-standing infections. Additionally, patients may experience systemic signs resulting from an underlying O volvulus infection.8 Our patient demonstrated a subjectively remarkable systemic response manifested by shortness of breath, dyspnea, cough, and palpitations, likely the result of her existing filarial infection.
Diagnosis of onchocerciasis is made by identification of microfilariae or adult worms in skin snips or punch biopsies. Histopathologic analysis of onchocerciasis has been described as several adult worms in the subcutaneous tissue surrounded by a granulomatous, fibrotic, calcified, or sometimes ossified inflammatory reaction.8 Microfilariae may migrate to the upper dermis and typically are surrounded by lymphocytes, histiocytes, plasma cells, and eosinophils with an absence of neutrophils,5,8 which directly causes the overlying epidermis to undergo reactive changes. Measurement of filarial nematodes often helps differentiate species. For example, O volvulus typically measures 230 to 500 mm in length for females and 16 to 42 mm in length for males.8 The thickness of cuticles typically measures 4 to 8 µm for females and 3 to 5 µm for males. Microfilariae of O volvulus also have been identified in patient’s sputum, urine, blood, and spinal fluid.8 An alternative diagnostic method described by Stingl et al9 is a diethylcarbamazine (DEC) patch test that was 92% accurate in onchocerciasis cases diagnosed by skin snip analysis.9 Therefore, this test may be a practical alternative when skin specimens are inconclusive.
Management of onchocerciasis should include excision of subcutaneous nodules to remove adult worms. In the past, DEC with suramin was prescribed and kills both microfilariae and adult worms.8 However, DEC may induce pruritus, chorioretinal damage, and optic neuritis and suramin is nephrotoxic.2 Therefore, oral ivermectin (0.15–0.20 mg/kg one-time dose, may be repeated in 3–12 months) is supported to be a less harmful option. Patients treated with ivermectin have a decreased risk for transmitting infection to the blackfly vector for up to 6 months after treatment, and it is a more effective microfilaricidal agent than DEC.2,3 Oral doxycycline (100 mg/d for 6 weeks) commonly is added as adjuvant therapy because it kills Wolbachia species, a bacterium that is needed for O volvulus reproduction.3
Although rare in the United States, cutaneous filariasis has become a prevalent public health concern, especially in tropical countries. Individuals with chronic cutaneous filarial infections are at increased risk for debilitating complications that negatively affect their quality of life and productivity. Although the World Health Organization has attempted to eradicate cutaneous filarial infections by mass drug administration, transmission of these diseases remains a challenge. Further research on treatment and methods to prevent transmission by controlling arthropod vectors is required to avoid short-term and long-term health consequences caused by cutaneous filariasis.
Parasitic infections should always be a diagnostic consideration in individuals who present with a pruritic eruption and a history of travel to foreign countries located in Africa, Central America, and South America. Dermatologists in the United States should increase familiarity with these infections because of increased travel, economic globalization, and the impact of global climate change on the geographic distribution of vector arthropods. To control these infections, research efforts should focus on improved sanitation; drug treatment; transmission prevention; and improved education of their clinical manifestations, especially mucocutaneous signs.
To the Editor:
Cutaneous filariasis is a group of infectious diseases caused by more than 60 different nematode species and endemic to approximately 83 countries.1,2 These infections are transmitted to humans by vectors such as mosquitoes, blackflies, biting midges, or Tabanid flies.1 The blood meal taken by vectors allow microfilariae to enter the skin and develop into adult worms.1-3 It is postulated that filarial infections were first described in 2000
Filarial parasites have been categorized into groups based on the site of adult worm stage habitat. The cutaneous group includes Loa loa, Onchocerca volvulus, Mansonella perstans, and Dipetalonema streptocerca. The lymphatic group includes Wuchereria bancrofti, Brugia malayi, and Brugia timori. Lastly, the body cavity group includes Mansonella ozzardi. Because humidity is required for survival of the infective larval stage, individuals from tropical countries in Africa, Central America, and South America most commonly are affected.1 These diseases also are related to poor housing quality and inadequate sanitation.1,3,4 Travel for business, medical missions, pleasure, or emigration has caused increased filarial infections globally. In fact, dermatologic disorders with infectious etiologies cause approximately 17% of travelers to seek medical attention.3
Dermatologic manifestations indicative of a potential cutaneous filarial infection include papules, nodules, excoriations with secondary xerosis, lichenification, skin pigment changes, and/or severe pruritus. However, individuals from filarial endemic regions may not demonstrate any clinical signs or symptoms, despite having microfilariae in their blood,1 which enables the disease to propagate and poses a major public health concern. In fact, patients with chronic filarial infections are at increased risk for developing lymphedema, elephantiasis, or blindness, which are hypothesized to be the second largest cause of permanent disability worldwide.1 Although rarely seen in US citizens, cutaneous filariasis should always be a diagnostic consideration in patients who present with a pruritic eruption and have a travel history to tropical countries. We report a case of cutaneous onchocerciasis in a US citizen who developed a pruritic eczematous eruption on the right upper arm following an arthropod assault while working in an Onchocerca endemic region approximately 1.5 years prior.
A 33-year-old woman presented to our outpatient dermatology clinic with the chief concern of a pruritic rash on the right arm (Figure 1). She revealed a history of travel to Peru, associated symptoms, prior diagnoses, and attempted treatment regimens. Over a 1.5-year period, the patient traveled for work-related reasons to Madre de Dios, Peru. During that time, the patient experienced 2 bouts of severe abdominal pain, nausea, and vomiting that she attributed to poor food and water quality. She did not immediately seek medical attention. She also developed skin manifestations that began as a pruritic and irritating pinpoint-sized red papule on the right eyelid, possibly a site of an arthropod assault. She then experienced episodic eyelash loss (Figure 2) and subsequent vision changes, including blurry vision, halos around lights, and dimming around the periphery. When the patient developed circular, pink, pruritic plaques on the right upper extremity, she sought medical attention in Lima, Peru. She was diagnosed with tinea corporis and prescribed an oral antibiotic and topical antifungal.
This treatment regimen did not result in improvement. In the following months, the patient noticed worsening of the ocular symptoms, and she developed a dry cough with occasional dyspnea. She again sought medical attention in Peru and was referred to an ophthalmologist who suspected Demodex mite or fungal infection; however, workup for those pathologies was negative. The respiratory symptoms continued to worsen, particularly at night, and she began to experience palpitations.
Upon returning to the United States, the patient was evaluated by her primary care physician who ordered the following laboratory tests: a complete blood cell count with differential, comprehensive metabolic panel, vitamin D level, blood culture, lipid panel, ferritin level, and thyroid function. An electrocardiogram, throat culture, and stool culture for ova and parasites also were obtained. The electrocardiogram showed sinus tachycardia, and the stool culture revealed blastocystis, for which she was prescribed oral metronidazole and tinidazole. The other results were within reference range, and the throat culture showed normal oropharyngeal microbes.
A few days after the treatment for the blastocystis, the gastrointestinal tract symptoms improved, but dermatologic, ocular, pulmonary, and cardiac symptoms persisted. She also began experiencing night sweats and hot flashes. In between visits to the primary care physician, she sought medical attention at an urgent care clinic for worsening pulmonary and cardiac symptoms. At that time, results of a chest radiograph were normal.
The primary care physician referred her to infectious disease and dermatology for further evaluation within a few weeks of returning to the United States. Upon presentation to dermatology, the patient described a waxing and waning nature to the rash on the arm with associated intermittent pain. Physical examination revealed several erythematous patches on the triceps and a 1-cm subcutaneous nodule on the right elbow with overlying skin discoloration. The patient stated that she first noticed the inflamed painful nodule when she returned to the United States. Since its discovery, the nodule increased in size and became firm. The patient also described occasional limited range of motion of the right arm due to the discomfort of the rash and inflamed nodule. Interestingly, the patient’s partner who accompanied her to Peru also developed a similar pruritic rash on the chest. He was evaluated by dermatology and infectious disease; however, biopsies of his rash performed at a different practice revealed nonspecific results. Due to her partner’s inconclusive results, our patient initially refused a biopsy; however, she returned to the office after 1 week with worsening symptoms, and a 4-mm punch biopsy of the right arm was obtained in addition to rapid plasma reagin and QuantiFERON-TB Gold test.
Based on the patient’s travel history, clinical course, and physical examination, the clinical differential diagnosis included nummular dermatitis, panniculitis/nodular vasculitis, tinea corporis, secondary syphilis, pinta, tuberculosis, or cutaneous filariasis. Rapid plasma reagin and QuantiFERON-TB Gold test results were negative, and a periodic acid–Schiff stain of the biopsy was negative for fungal elements. Dermatopathology revealed intradermal filarial nematodes of 120 to 150 µm in diameter and 1-to 2-mm cuticles eliciting a predominantly superficial and deep lymphohistiocytic reaction (Figure 3). The histopathologic differential diagnosis based on the diameter of the nematode included M perstans, W bancrofti, and O volvulus. Clinical correlation was highly recommended to obtain a final diagnosis and management plan. A portion of the biopsy was sent to the Centers for Disease Control and Prevention, which confirmed the presence of a filarial nematode consistent with a zoonotic Brugia but also with zoonotic Onchocerca as a possible differential. Therefore, considering the history, clinical course, and histopathologic analysis, the final diagnosis of cutaneous onchocerciasis was made.
The patient was referred back to infectious disease and started on combination therapy of ivermectin (25.5 mg) plus albendazole (800 mg) taken at once followed by doxycycline (200 mg) for 6 weeks. The symptoms initially worsened, then she experienced near-complete resolution of dermatologic, pulmonary, and ocular symptoms after approximately 2 weeks of treatment. She continued to report fatigue and palpitations, for she continued to see a cardiologist. We highly encouraged her to continue following up with infectious disease to ensure complete eradication of the infection.
Onchocerciasis is common in individuals who live or work in equatorial Africa, Central America, and South America. The disease process has dermatologic, ocular, and other systemic manifestations if the infection continues long-term without treatment.2,5 Although rare, this infection has been observed in US citizens who have traveled to filarial endemic regions for a period of time ranging from 2 weeks to 39 years.2,6,7
Blackflies (genus Simulium), the intermediate host of O volvulus, breed in areas close to freely flowing water. The blackfly bites a human host, and within 7 days the microfilariae undergo 2 molts to reach the infective stage. The larvae remain in the dermis and subcutaneous tissue for 2 additional molts until they develop into adult worms. Then, adult worms may become encapsulated, eliciting and forming subcutaneous nodules, also known as onchocercomas.2,3
A fertilized female in the subcutaneous tissue releases microfilariae that may remain in the skin or travel to the eyes of the host.3 The host may demonstrate signs of infection within 3 to 15 months.2 Most commonly, patients report localized or generalized pruritus, and one extremity develops swelling, papules, lymphadenopathy, and alterations of skin pigment (hypopigmentation or hyperpigmentation).5-8 Our patient developed a pruritic eczematous eruption that only affected her right arm with an inflamed firm nodule overlying the elbow, a suspected onchocercoma. Onchocercomas are the direct result of adult worms coiled in the dermis, subcutaneous tissue, and deep fascia. These commonly are found in close proximity to bony prominences with the specific location pending the geographic area of infection.8 For example, onchocercomas more commonly are found over bony prominences in the head and upper body region in patients who acquired the disease from Central or South America, whereas individuals infected in Africa more commonly develop onchocercomas near the femoral, coccyx, or sacral regions.2
The immune response mounted by the infected host is responsible for the ocular signs and symptoms.2 Specifically, the inflammatory reaction may impact the patient’s cornea, anterior uveal tract, chorioretinal zone, or optic nerve. In fact, onchocerciasis, or river blindness, is the leading cause of blindness in the world.2 The host immune response also is responsible for the dermatologic manifestations of this disease. In addition to the dermatologic manifestations already mentioned, others include epidermal atrophy, ulcerations, femoral or inguinal lymphadenitis, lymphedema, and/or general wasting in more severe long-standing infections. Additionally, patients may experience systemic signs resulting from an underlying O volvulus infection.8 Our patient demonstrated a subjectively remarkable systemic response manifested by shortness of breath, dyspnea, cough, and palpitations, likely the result of her existing filarial infection.
Diagnosis of onchocerciasis is made by identification of microfilariae or adult worms in skin snips or punch biopsies. Histopathologic analysis of onchocerciasis has been described as several adult worms in the subcutaneous tissue surrounded by a granulomatous, fibrotic, calcified, or sometimes ossified inflammatory reaction.8 Microfilariae may migrate to the upper dermis and typically are surrounded by lymphocytes, histiocytes, plasma cells, and eosinophils with an absence of neutrophils,5,8 which directly causes the overlying epidermis to undergo reactive changes. Measurement of filarial nematodes often helps differentiate species. For example, O volvulus typically measures 230 to 500 mm in length for females and 16 to 42 mm in length for males.8 The thickness of cuticles typically measures 4 to 8 µm for females and 3 to 5 µm for males. Microfilariae of O volvulus also have been identified in patient’s sputum, urine, blood, and spinal fluid.8 An alternative diagnostic method described by Stingl et al9 is a diethylcarbamazine (DEC) patch test that was 92% accurate in onchocerciasis cases diagnosed by skin snip analysis.9 Therefore, this test may be a practical alternative when skin specimens are inconclusive.
Management of onchocerciasis should include excision of subcutaneous nodules to remove adult worms. In the past, DEC with suramin was prescribed and kills both microfilariae and adult worms.8 However, DEC may induce pruritus, chorioretinal damage, and optic neuritis and suramin is nephrotoxic.2 Therefore, oral ivermectin (0.15–0.20 mg/kg one-time dose, may be repeated in 3–12 months) is supported to be a less harmful option. Patients treated with ivermectin have a decreased risk for transmitting infection to the blackfly vector for up to 6 months after treatment, and it is a more effective microfilaricidal agent than DEC.2,3 Oral doxycycline (100 mg/d for 6 weeks) commonly is added as adjuvant therapy because it kills Wolbachia species, a bacterium that is needed for O volvulus reproduction.3
Although rare in the United States, cutaneous filariasis has become a prevalent public health concern, especially in tropical countries. Individuals with chronic cutaneous filarial infections are at increased risk for debilitating complications that negatively affect their quality of life and productivity. Although the World Health Organization has attempted to eradicate cutaneous filarial infections by mass drug administration, transmission of these diseases remains a challenge. Further research on treatment and methods to prevent transmission by controlling arthropod vectors is required to avoid short-term and long-term health consequences caused by cutaneous filariasis.
Parasitic infections should always be a diagnostic consideration in individuals who present with a pruritic eruption and a history of travel to foreign countries located in Africa, Central America, and South America. Dermatologists in the United States should increase familiarity with these infections because of increased travel, economic globalization, and the impact of global climate change on the geographic distribution of vector arthropods. To control these infections, research efforts should focus on improved sanitation; drug treatment; transmission prevention; and improved education of their clinical manifestations, especially mucocutaneous signs.
- Mendoza N, Li A, Gill A, et al. Filariasis: diagnosis and treatment. Dermatol Ther. 2009;22:475-490.
- Maso MJ, Kapila R, Schwartz RA, et al. Cutaneous onchocerciasis. Int J Dermatol. 1987;26:593-596.
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- Bolivar-Meija A, Alarcon-Olave C, Rodriguez-Morales AJ. Skin manifestations of arthropod-borne infection in Latin America. Curr Opin. 2014;27:288-294.
- Okulicz JF, Stibich AS, Elston DM, et al. Cutaneous onchocercoma. Int J Dermatol. 2004;43:170-172.
- Nguyen JC, Murphy ME, Nutman TB, et al. Cutaneous onchocerciasis in an American traveler. Int Soc Dermatol. 2005;44:125-128.
- Toovey S, Moerman F, van Gompel A. Special infectious disease risks of expatriates and long-term travelers in tropical countries part II: infections other than malaria. J Travel Med. 2007;14:50-60.
- Meyers WM, Neafie RC, Connor DH. Bancroftian and malayan filariasis. In Binford CH, ed. Pathology of Tropical and Extraordinary Diseases. Vol 2. Fort Sam Houston, TX: Armed Forces Institute of Pathology; 1976:340-355.
- Stingl P, Ross M, Gibson DW, et al. A diagnostic “patch-test” for onchocerciasis using topical diethylcarbamazine. Trans R Soc Trop Med Hyg.
- Mendoza N, Li A, Gill A, et al. Filariasis: diagnosis and treatment. Dermatol Ther. 2009;22:475-490.
- Maso MJ, Kapila R, Schwartz RA, et al. Cutaneous onchocerciasis. Int J Dermatol. 1987;26:593-596.
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- Bolivar-Meija A, Alarcon-Olave C, Rodriguez-Morales AJ. Skin manifestations of arthropod-borne infection in Latin America. Curr Opin. 2014;27:288-294.
- Okulicz JF, Stibich AS, Elston DM, et al. Cutaneous onchocercoma. Int J Dermatol. 2004;43:170-172.
- Nguyen JC, Murphy ME, Nutman TB, et al. Cutaneous onchocerciasis in an American traveler. Int Soc Dermatol. 2005;44:125-128.
- Toovey S, Moerman F, van Gompel A. Special infectious disease risks of expatriates and long-term travelers in tropical countries part II: infections other than malaria. J Travel Med. 2007;14:50-60.
- Meyers WM, Neafie RC, Connor DH. Bancroftian and malayan filariasis. In Binford CH, ed. Pathology of Tropical and Extraordinary Diseases. Vol 2. Fort Sam Houston, TX: Armed Forces Institute of Pathology; 1976:340-355.
- Stingl P, Ross M, Gibson DW, et al. A diagnostic “patch-test” for onchocerciasis using topical diethylcarbamazine. Trans R Soc Trop Med Hyg.
Practice Points
- Parasitic infections should always be a diagnostic consideration in individuals who present with a pruritic eruption and a history of travel.
- To control parasitic infections, research efforts should focus on improved sanitation, drug treatment, transmission prevention, and improved education of clinical manifestations to increase early detection.
The Dermatologist Nose Best: Correlation of Nose-Picking Habits and <i>Staphylococcus aureus</i>–Related Dermatologic Disease
Primitive human habits have withstood the test of time but can pose health risks. Exploring a nasal cavity with a finger might have first occurred shortly after whichever species first developed a nasal opening and a digit able to reach it. Humans have been keen on continuing the long-standing yet taboo habit of nose-picking (rhinotillexis).
Even though nose-picking is stigmatized, anonymous surveys show that almost all adolescents and adults do it.1 People are typically unaware of the risks of regular rhinotillexis. Studies exploring the intranasal human microbiome have elicited asymptomatic yet potential disease-causing microbes, including the notorious bacterium Staphylococcus aureus. As many as 30% of humans are asymptomatically permanently colonized with S aureus in their anterior nares.2 These natural reservoirs can be the source of opportunistic infection that increases morbidity, mortality, and overall health care costs.
With the rise of antimicrobial resistance, especially methicillin-resistant S aureus (MRSA), a more direct approach might be necessary to curb nasally sourced cutaneous infection. Since dermatology patients deal with a wide array of skin barrier defects that put them at risk for S aureus–related infection, a medical provider’s understanding about the role of nasal colonization and transmission is important. Addressing the awkward question of “Do you pick your nose?” and providing education on the topic might be uncomfortable, but it might be necessary for dermatology patients at risk for S aureus–related cutaneous disease.
Staphylococcus aureus colonizes the anterior nares of 20% to 80% of humans; nasal colonization can begin during the first days of life.2 The anterior nares are noted as the main reservoir of chronic carriage of S aureus, but carriage can occur at various body sites, including the rectum, vagina, gastrointestinal tract, and axilla, as well as other cutaneous sites. Hands are noted as the main vector of S aureus transmission from source to nose; a positive correlation between nose-picking habits and nasal carriage of S aureus has been noted.2
The percentage of S aureus–colonized humans who harbor MRSA is unknown, but it is a topic of concern with the rise of MRSA-related infection. Multisite MRSA carriage increases the risk for nasal MRSA colonization, and nasal MRSA has been noted to be more difficult to decolonize than nonresistant strains. Health care workers carrying S aureus can trigger a potential hospital outbreak of MRSA. Studies have shown that bacterial transmission is increased 40-fold when the nasal host is co-infected by rhinovirus.2 Health care workers can be a source of MRSA during outbreaks, but they have not been shown to be more likely to carry MRSA than the general population.2 Understanding which patients might be at risk for S aureus–associated disease in dermatology can lead clinicians to consider decolonization strategies.
Nasal colonization has been noted more frequently in patients with predisposing risk factors, including human immunodeficiency virus infection, obesity, diabetes mellitus, granulomatosis with polyangiitis, HLA-DR3 phenotype, skin and soft-tissue infections, atopic dermatitis, impetigo, and recurrent furunculosis.2Staphylococcus aureus is the most frequently noted pathogen in diabetic foot infection. A study found that 36% of sampled diabetic foot-infection patients also had S aureus isolated from both nares and the foot wound, with 65% of isolated strains being identical.2 Although there are clear data on decolonization of patients prior to heart and orthopedic surgery, more data are needed to determine the benefit of screening and treating nasal carriers in populations with diabetic foot ulcers.
Staphylococcus aureus nasal colonization also has been shown in approximately 60% of patients with recurrent furunculosis and impetigo.2 Although it is clear that there is a correlation between S aureus–related skin infection and nasal colonization, it is unclear what role nose-picking might have in perpetuating these complications.
There are multiple approaches to S aureus decolonization, including intranasal mupirocin, chlorhexidine body wipes, bleach baths, and even oral antibiotics (eg, trimethoprim-sulfamethoxazole, clindamycin). The Infectious Diseases Society of America has published guidelines for treating recurrent MRSA infection, including 5 to 10 days of intranasal mupirocin plus either body decolonization with a daily chlorhexidine wash for 5 to 14 days or a 15-minute dilute bleach bath twice weekly for 3 months.3,4
There are ample meta-analyses and systematic reviews regarding S aureus decolonization and management in patients undergoing dialysis or surgery but limited data when it comes to this topic in dermatology. Those limited studies do show a benefit to decolonization in several diseases, including atopic dermatitis, hand dermatitis, recurrent skin and soft-tissue infections, cutaneous T-cell lymphoma, and surgical infection following Mohs micrographic surgery.4 Typically, it also is necessary to treat those who might come in contact with the patient or caregiver; in theory, treating contacts helps reduce the chance that the patient will become recolonized shortly afterward, but the data are limited regarding long-term colonization status following treatment. Contact surfaces, especially cell phones, are noted to be a contributing factor to nares colonization; therefore, it also may be necessary to educate patients on surface-cleaning techniques.5 Because there are multiple sources of S aureus that patients can come in contact with after decolonization attempts, a nose-picking habit might play a vital role in recolonization.
Due to rising bacterial resistance to mupirocin and chlorhexidine decolonization strategies, there is a growing need for more effective, long-term decolonization strategies.4 These strategies must address patients’ environmental exposure and nasal-touching habits. Overcoming the habit of nose-picking might aid S aureus decolonization strategies and thus aid in preventing future antimicrobial resistance.
But are at-risk patients receiving sufficient screening and education on the dangers of a nose-picking habit? Effective strategies to assess these practices and recommend the discontinuation of the habit could have positive effects in maintaining long-term decolonization. Potential euphemistic ways to approach this somewhat taboo topic include questions that elicit information on whether the patient ever touches the inside of his/her nose, washes his/her hands before and after touching the inside of the nose, knows about transfer of bacteria from hand to nose, or understands what decolonization is doing for them. The patient might be inclined to deny such activity, but education on nasal hygiene should be provided regardless, especially in pediatric patients.
Staphylococcus aureus might be a normal human nasal inhabitant, but it can cause a range of problems for dermatologic disease. Although pharmacotherapeutic decolonization strategies can have a positive effect on dermatologic disease, growing antibiotic resistance calls for health care providers to assess patients’ nose picking-habits and educate them on effective ways to prevent finger-to-nose practices.
- Andrade C, Srihari BS. A preliminary survey of rhinotillexomania in an adolescent sample. J Clin Psychiatry. 2001;62:426-431.
- Sakr A, Brégeon F, Mège J-L, et al. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections. Front Microbiol. 2018;9:2419.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52:285-292.
- Kuraitis D, Williams L. Decolonization of Staphylococcus aureus in healthcare: a dermatology perspective. J Healthc Eng. 2018;2018:2382050.
- Creech CB, Al-Zubeidi DN, Fritz SA. Prevention of recurrent staphylococcal kin infections. Infect Dis Clin North Am. 2015;29:429-464.
Primitive human habits have withstood the test of time but can pose health risks. Exploring a nasal cavity with a finger might have first occurred shortly after whichever species first developed a nasal opening and a digit able to reach it. Humans have been keen on continuing the long-standing yet taboo habit of nose-picking (rhinotillexis).
Even though nose-picking is stigmatized, anonymous surveys show that almost all adolescents and adults do it.1 People are typically unaware of the risks of regular rhinotillexis. Studies exploring the intranasal human microbiome have elicited asymptomatic yet potential disease-causing microbes, including the notorious bacterium Staphylococcus aureus. As many as 30% of humans are asymptomatically permanently colonized with S aureus in their anterior nares.2 These natural reservoirs can be the source of opportunistic infection that increases morbidity, mortality, and overall health care costs.
With the rise of antimicrobial resistance, especially methicillin-resistant S aureus (MRSA), a more direct approach might be necessary to curb nasally sourced cutaneous infection. Since dermatology patients deal with a wide array of skin barrier defects that put them at risk for S aureus–related infection, a medical provider’s understanding about the role of nasal colonization and transmission is important. Addressing the awkward question of “Do you pick your nose?” and providing education on the topic might be uncomfortable, but it might be necessary for dermatology patients at risk for S aureus–related cutaneous disease.
Staphylococcus aureus colonizes the anterior nares of 20% to 80% of humans; nasal colonization can begin during the first days of life.2 The anterior nares are noted as the main reservoir of chronic carriage of S aureus, but carriage can occur at various body sites, including the rectum, vagina, gastrointestinal tract, and axilla, as well as other cutaneous sites. Hands are noted as the main vector of S aureus transmission from source to nose; a positive correlation between nose-picking habits and nasal carriage of S aureus has been noted.2
The percentage of S aureus–colonized humans who harbor MRSA is unknown, but it is a topic of concern with the rise of MRSA-related infection. Multisite MRSA carriage increases the risk for nasal MRSA colonization, and nasal MRSA has been noted to be more difficult to decolonize than nonresistant strains. Health care workers carrying S aureus can trigger a potential hospital outbreak of MRSA. Studies have shown that bacterial transmission is increased 40-fold when the nasal host is co-infected by rhinovirus.2 Health care workers can be a source of MRSA during outbreaks, but they have not been shown to be more likely to carry MRSA than the general population.2 Understanding which patients might be at risk for S aureus–associated disease in dermatology can lead clinicians to consider decolonization strategies.
Nasal colonization has been noted more frequently in patients with predisposing risk factors, including human immunodeficiency virus infection, obesity, diabetes mellitus, granulomatosis with polyangiitis, HLA-DR3 phenotype, skin and soft-tissue infections, atopic dermatitis, impetigo, and recurrent furunculosis.2Staphylococcus aureus is the most frequently noted pathogen in diabetic foot infection. A study found that 36% of sampled diabetic foot-infection patients also had S aureus isolated from both nares and the foot wound, with 65% of isolated strains being identical.2 Although there are clear data on decolonization of patients prior to heart and orthopedic surgery, more data are needed to determine the benefit of screening and treating nasal carriers in populations with diabetic foot ulcers.
Staphylococcus aureus nasal colonization also has been shown in approximately 60% of patients with recurrent furunculosis and impetigo.2 Although it is clear that there is a correlation between S aureus–related skin infection and nasal colonization, it is unclear what role nose-picking might have in perpetuating these complications.
There are multiple approaches to S aureus decolonization, including intranasal mupirocin, chlorhexidine body wipes, bleach baths, and even oral antibiotics (eg, trimethoprim-sulfamethoxazole, clindamycin). The Infectious Diseases Society of America has published guidelines for treating recurrent MRSA infection, including 5 to 10 days of intranasal mupirocin plus either body decolonization with a daily chlorhexidine wash for 5 to 14 days or a 15-minute dilute bleach bath twice weekly for 3 months.3,4
There are ample meta-analyses and systematic reviews regarding S aureus decolonization and management in patients undergoing dialysis or surgery but limited data when it comes to this topic in dermatology. Those limited studies do show a benefit to decolonization in several diseases, including atopic dermatitis, hand dermatitis, recurrent skin and soft-tissue infections, cutaneous T-cell lymphoma, and surgical infection following Mohs micrographic surgery.4 Typically, it also is necessary to treat those who might come in contact with the patient or caregiver; in theory, treating contacts helps reduce the chance that the patient will become recolonized shortly afterward, but the data are limited regarding long-term colonization status following treatment. Contact surfaces, especially cell phones, are noted to be a contributing factor to nares colonization; therefore, it also may be necessary to educate patients on surface-cleaning techniques.5 Because there are multiple sources of S aureus that patients can come in contact with after decolonization attempts, a nose-picking habit might play a vital role in recolonization.
Due to rising bacterial resistance to mupirocin and chlorhexidine decolonization strategies, there is a growing need for more effective, long-term decolonization strategies.4 These strategies must address patients’ environmental exposure and nasal-touching habits. Overcoming the habit of nose-picking might aid S aureus decolonization strategies and thus aid in preventing future antimicrobial resistance.
But are at-risk patients receiving sufficient screening and education on the dangers of a nose-picking habit? Effective strategies to assess these practices and recommend the discontinuation of the habit could have positive effects in maintaining long-term decolonization. Potential euphemistic ways to approach this somewhat taboo topic include questions that elicit information on whether the patient ever touches the inside of his/her nose, washes his/her hands before and after touching the inside of the nose, knows about transfer of bacteria from hand to nose, or understands what decolonization is doing for them. The patient might be inclined to deny such activity, but education on nasal hygiene should be provided regardless, especially in pediatric patients.
Staphylococcus aureus might be a normal human nasal inhabitant, but it can cause a range of problems for dermatologic disease. Although pharmacotherapeutic decolonization strategies can have a positive effect on dermatologic disease, growing antibiotic resistance calls for health care providers to assess patients’ nose picking-habits and educate them on effective ways to prevent finger-to-nose practices.
Primitive human habits have withstood the test of time but can pose health risks. Exploring a nasal cavity with a finger might have first occurred shortly after whichever species first developed a nasal opening and a digit able to reach it. Humans have been keen on continuing the long-standing yet taboo habit of nose-picking (rhinotillexis).
Even though nose-picking is stigmatized, anonymous surveys show that almost all adolescents and adults do it.1 People are typically unaware of the risks of regular rhinotillexis. Studies exploring the intranasal human microbiome have elicited asymptomatic yet potential disease-causing microbes, including the notorious bacterium Staphylococcus aureus. As many as 30% of humans are asymptomatically permanently colonized with S aureus in their anterior nares.2 These natural reservoirs can be the source of opportunistic infection that increases morbidity, mortality, and overall health care costs.
With the rise of antimicrobial resistance, especially methicillin-resistant S aureus (MRSA), a more direct approach might be necessary to curb nasally sourced cutaneous infection. Since dermatology patients deal with a wide array of skin barrier defects that put them at risk for S aureus–related infection, a medical provider’s understanding about the role of nasal colonization and transmission is important. Addressing the awkward question of “Do you pick your nose?” and providing education on the topic might be uncomfortable, but it might be necessary for dermatology patients at risk for S aureus–related cutaneous disease.
Staphylococcus aureus colonizes the anterior nares of 20% to 80% of humans; nasal colonization can begin during the first days of life.2 The anterior nares are noted as the main reservoir of chronic carriage of S aureus, but carriage can occur at various body sites, including the rectum, vagina, gastrointestinal tract, and axilla, as well as other cutaneous sites. Hands are noted as the main vector of S aureus transmission from source to nose; a positive correlation between nose-picking habits and nasal carriage of S aureus has been noted.2
The percentage of S aureus–colonized humans who harbor MRSA is unknown, but it is a topic of concern with the rise of MRSA-related infection. Multisite MRSA carriage increases the risk for nasal MRSA colonization, and nasal MRSA has been noted to be more difficult to decolonize than nonresistant strains. Health care workers carrying S aureus can trigger a potential hospital outbreak of MRSA. Studies have shown that bacterial transmission is increased 40-fold when the nasal host is co-infected by rhinovirus.2 Health care workers can be a source of MRSA during outbreaks, but they have not been shown to be more likely to carry MRSA than the general population.2 Understanding which patients might be at risk for S aureus–associated disease in dermatology can lead clinicians to consider decolonization strategies.
Nasal colonization has been noted more frequently in patients with predisposing risk factors, including human immunodeficiency virus infection, obesity, diabetes mellitus, granulomatosis with polyangiitis, HLA-DR3 phenotype, skin and soft-tissue infections, atopic dermatitis, impetigo, and recurrent furunculosis.2Staphylococcus aureus is the most frequently noted pathogen in diabetic foot infection. A study found that 36% of sampled diabetic foot-infection patients also had S aureus isolated from both nares and the foot wound, with 65% of isolated strains being identical.2 Although there are clear data on decolonization of patients prior to heart and orthopedic surgery, more data are needed to determine the benefit of screening and treating nasal carriers in populations with diabetic foot ulcers.
Staphylococcus aureus nasal colonization also has been shown in approximately 60% of patients with recurrent furunculosis and impetigo.2 Although it is clear that there is a correlation between S aureus–related skin infection and nasal colonization, it is unclear what role nose-picking might have in perpetuating these complications.
There are multiple approaches to S aureus decolonization, including intranasal mupirocin, chlorhexidine body wipes, bleach baths, and even oral antibiotics (eg, trimethoprim-sulfamethoxazole, clindamycin). The Infectious Diseases Society of America has published guidelines for treating recurrent MRSA infection, including 5 to 10 days of intranasal mupirocin plus either body decolonization with a daily chlorhexidine wash for 5 to 14 days or a 15-minute dilute bleach bath twice weekly for 3 months.3,4
There are ample meta-analyses and systematic reviews regarding S aureus decolonization and management in patients undergoing dialysis or surgery but limited data when it comes to this topic in dermatology. Those limited studies do show a benefit to decolonization in several diseases, including atopic dermatitis, hand dermatitis, recurrent skin and soft-tissue infections, cutaneous T-cell lymphoma, and surgical infection following Mohs micrographic surgery.4 Typically, it also is necessary to treat those who might come in contact with the patient or caregiver; in theory, treating contacts helps reduce the chance that the patient will become recolonized shortly afterward, but the data are limited regarding long-term colonization status following treatment. Contact surfaces, especially cell phones, are noted to be a contributing factor to nares colonization; therefore, it also may be necessary to educate patients on surface-cleaning techniques.5 Because there are multiple sources of S aureus that patients can come in contact with after decolonization attempts, a nose-picking habit might play a vital role in recolonization.
Due to rising bacterial resistance to mupirocin and chlorhexidine decolonization strategies, there is a growing need for more effective, long-term decolonization strategies.4 These strategies must address patients’ environmental exposure and nasal-touching habits. Overcoming the habit of nose-picking might aid S aureus decolonization strategies and thus aid in preventing future antimicrobial resistance.
But are at-risk patients receiving sufficient screening and education on the dangers of a nose-picking habit? Effective strategies to assess these practices and recommend the discontinuation of the habit could have positive effects in maintaining long-term decolonization. Potential euphemistic ways to approach this somewhat taboo topic include questions that elicit information on whether the patient ever touches the inside of his/her nose, washes his/her hands before and after touching the inside of the nose, knows about transfer of bacteria from hand to nose, or understands what decolonization is doing for them. The patient might be inclined to deny such activity, but education on nasal hygiene should be provided regardless, especially in pediatric patients.
Staphylococcus aureus might be a normal human nasal inhabitant, but it can cause a range of problems for dermatologic disease. Although pharmacotherapeutic decolonization strategies can have a positive effect on dermatologic disease, growing antibiotic resistance calls for health care providers to assess patients’ nose picking-habits and educate them on effective ways to prevent finger-to-nose practices.
- Andrade C, Srihari BS. A preliminary survey of rhinotillexomania in an adolescent sample. J Clin Psychiatry. 2001;62:426-431.
- Sakr A, Brégeon F, Mège J-L, et al. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections. Front Microbiol. 2018;9:2419.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52:285-292.
- Kuraitis D, Williams L. Decolonization of Staphylococcus aureus in healthcare: a dermatology perspective. J Healthc Eng. 2018;2018:2382050.
- Creech CB, Al-Zubeidi DN, Fritz SA. Prevention of recurrent staphylococcal kin infections. Infect Dis Clin North Am. 2015;29:429-464.
- Andrade C, Srihari BS. A preliminary survey of rhinotillexomania in an adolescent sample. J Clin Psychiatry. 2001;62:426-431.
- Sakr A, Brégeon F, Mège J-L, et al. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections. Front Microbiol. 2018;9:2419.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52:285-292.
- Kuraitis D, Williams L. Decolonization of Staphylococcus aureus in healthcare: a dermatology perspective. J Healthc Eng. 2018;2018:2382050.
- Creech CB, Al-Zubeidi DN, Fritz SA. Prevention of recurrent staphylococcal kin infections. Infect Dis Clin North Am. 2015;29:429-464.
Practice Points
- Staphylococcus aureus colonizes the anterior nares of approximately 20% to 80% of humans and can play a large factor in dermatologic disease.
- Staphylococcus aureus decolonization practices for at-risk dermatology patients may overlook the role that nose-picking plays.
Shared decision-making aids choice of PrEP
A patient-centered approach can help guide persons at risk for HIV exposure to decide on the best choice of pre-exposure prophylaxis (PrEP) regimens for them, stifling the noise generated by direct-to-consumer advertising, an infectious disease specialist recommends.
The decision for patients whether to start or remain on the PrEP combination of tenofovir disoproxil fumarate (TDF) plus emtricitabine (FTC; Truvada and generic) or on tenofovir alafenamide (TAF) plus FTC (Descovy) is made more fraught by confusion regarding the use of the newer and allegedly safer TAF prodrug of tenofovir in HIV treatment regimens, said Oni Blackstock, MD, founder and executive director of Health Justice and an attending physician in the division of infectious diseases at Harlem Hospital, New York.
“There have been commercials on TV as well as on social media around class-action lawsuits against [Truvada maker] Gilead,” she said in an online presentation during IDWeek 2020, an annual scientific meeting on infectious diseases, held virtually this year.
“These lawsuits focus on TDF for HIV treatment, but they have sown a great deal of confusion about TDF versus TAF for PrEP among potential and actual PrEP users,” she added.
Dr. Blackstock described her approach to shared decision-making regarding TDF/FTC versus TAF/FTC, and to helping patients understand the relative benefits and risks of each formulation.
In January of 2020, Dr. Blackstock, who was then assistant commissioner of the HIV bureau of the New York City Department of Health and Mental Hygiene, issued with other Bureau members a “Dear colleague” letter stating why they believed that TDF/FTC should remain the first-line regimen for PrEP.
That opinion, she said, was bolstered by an editorial published in February 2020 by Douglas E. Krakower, MD, from Beth Israel Deaconess Medical Center in Boston, and colleagues, which questioned the rush to shift from TDF to TAF in HIV treatment, and cautioned against the same approach to PrEP.
“Despite evidence that TAF/FTC would not be cost-effective, compared with generic TDF/FTC , the newer regimen quickly and irrevocably displaced TDF/FTC for HIV treatment in the U.S. A similar shift for PrEP – especially for populations in which TAF/FTC is untested – would be premature, costly, and counterproductive for population impact,” Krakower et al. wrote.
Shared decision-making
Clinicians can help patients who may be a candidate for either PrEP regimen by engaging them in shared decision-making.
“The clinician provides information in this case about a prevention strategy, options, benefits and risks, alternatives, and the patient provides their preferences and values, and together the clinician and patient make a decision,” Dr. Blackstock said.
The process differs from the model of informed decision-making, where the clinician gives the patient the information and the patient comes to a decision, or the old, “paternalistic” model in which the clinician gives information and makes recommendations to the patient.
“Shared decision-making has been studied extensively and has been shown to improve patient satisfaction, patient communication, and also potentially reducing health inequities that we see,” she said.
The model for shared decision-making for clinical practice includes three distinct portions: a choice talk, option talk, and decision talk.
Choice
To begin the discussion, the physician informs the patients of the availability of choices and justifies them, saying, for example, “there is good information about how these two PrEP options differ that I’d like to discuss with you,” and “the two PrEP options have different side effects … some will matter more to you than other people.”
At this stage the clinician should defer closure by offering a more detailed discussion of the choices.
Option
Here the clinician solicits information about what the patient has heard or read about PrEP, describes each option in practical terms, and points out where the two regimens differ, being specific about the pros and cons of each (for example, potential bone mineral density loss or renal complications with TDF, and potential weight gain with subsequent metabolic and cardiovascular consequences with TAF).
The TDF versus TAF-based PrEP discussion could also focus on what’s known about the comparative effectiveness of each regimen.
For example, TDF/FTC has been shown to be about 99% effective at preventing infection in men who have sex with men and in transgender women, also about 99% effective in heterosexual women and men, and 74%-84% effective in persons who inject drugs.
In contrast, TAF/FTC has been shown to be about 99% effective in men who have sex with men and transgender women, but it’s efficacy in the other two categories is unknown, Dr. Blackstock said.
The option discussion should include a comparison of the evidence base for each regimen, including the real-world experience with TDF/FTC since 2012, and much more limited experience with TAF/FTC.
Discussing relative costs, although the wholesale costs of the regimens are similar, there is now a generic version of TDF/FTC made by Teva Pharmaceuticals that sells for about $400 less per month than the brand name, which might make the option more acceptable to health insurers.
Decision
The decision talk is about considering the patients preferences and deciding with them what is best.
The clinician could say, for example: “What, from your point of view, matters most to you?”
The clinician should also be willing to allow the patient to defer a decision or to guide them depending on their stated wish, asking something like: “Are you ready to decide, or do you want more time? Do you have more questions? Are there more things we should discuss?
Offering the patient a chance to review the decision can also be a good way to arrive at closure, Dr. Blackstock said.
Robert Goldstein, MD, PhD, an infectious disease specialist and medical director of the transgender health program at Massachusetts General Hospital in Boston, who comoderated the session where Dr. Blackstock presented her talk, said that he also uses a similar approach to the PrEP discussion.
“I use a very patient-centered approach of providing information and talking through the data that are available,” he said.
“I will say that, as patients come to me talking about the transition from TDF to TAF for pre-exposure prophylaxis, I am very clear with them about the limited benefit or no benefit that I see with TAF for pre-exposure prophylaxis, and all of my patients have remained on TDF for pre-exposure prophylaxis,” he added.
No funding source for the presentation was reported. Dr. Blackstock and Dr. Goldstein reported having no conflicts of interest to disclose.
A patient-centered approach can help guide persons at risk for HIV exposure to decide on the best choice of pre-exposure prophylaxis (PrEP) regimens for them, stifling the noise generated by direct-to-consumer advertising, an infectious disease specialist recommends.
The decision for patients whether to start or remain on the PrEP combination of tenofovir disoproxil fumarate (TDF) plus emtricitabine (FTC; Truvada and generic) or on tenofovir alafenamide (TAF) plus FTC (Descovy) is made more fraught by confusion regarding the use of the newer and allegedly safer TAF prodrug of tenofovir in HIV treatment regimens, said Oni Blackstock, MD, founder and executive director of Health Justice and an attending physician in the division of infectious diseases at Harlem Hospital, New York.
“There have been commercials on TV as well as on social media around class-action lawsuits against [Truvada maker] Gilead,” she said in an online presentation during IDWeek 2020, an annual scientific meeting on infectious diseases, held virtually this year.
“These lawsuits focus on TDF for HIV treatment, but they have sown a great deal of confusion about TDF versus TAF for PrEP among potential and actual PrEP users,” she added.
Dr. Blackstock described her approach to shared decision-making regarding TDF/FTC versus TAF/FTC, and to helping patients understand the relative benefits and risks of each formulation.
In January of 2020, Dr. Blackstock, who was then assistant commissioner of the HIV bureau of the New York City Department of Health and Mental Hygiene, issued with other Bureau members a “Dear colleague” letter stating why they believed that TDF/FTC should remain the first-line regimen for PrEP.
That opinion, she said, was bolstered by an editorial published in February 2020 by Douglas E. Krakower, MD, from Beth Israel Deaconess Medical Center in Boston, and colleagues, which questioned the rush to shift from TDF to TAF in HIV treatment, and cautioned against the same approach to PrEP.
“Despite evidence that TAF/FTC would not be cost-effective, compared with generic TDF/FTC , the newer regimen quickly and irrevocably displaced TDF/FTC for HIV treatment in the U.S. A similar shift for PrEP – especially for populations in which TAF/FTC is untested – would be premature, costly, and counterproductive for population impact,” Krakower et al. wrote.
Shared decision-making
Clinicians can help patients who may be a candidate for either PrEP regimen by engaging them in shared decision-making.
“The clinician provides information in this case about a prevention strategy, options, benefits and risks, alternatives, and the patient provides their preferences and values, and together the clinician and patient make a decision,” Dr. Blackstock said.
The process differs from the model of informed decision-making, where the clinician gives the patient the information and the patient comes to a decision, or the old, “paternalistic” model in which the clinician gives information and makes recommendations to the patient.
“Shared decision-making has been studied extensively and has been shown to improve patient satisfaction, patient communication, and also potentially reducing health inequities that we see,” she said.
The model for shared decision-making for clinical practice includes three distinct portions: a choice talk, option talk, and decision talk.
Choice
To begin the discussion, the physician informs the patients of the availability of choices and justifies them, saying, for example, “there is good information about how these two PrEP options differ that I’d like to discuss with you,” and “the two PrEP options have different side effects … some will matter more to you than other people.”
At this stage the clinician should defer closure by offering a more detailed discussion of the choices.
Option
Here the clinician solicits information about what the patient has heard or read about PrEP, describes each option in practical terms, and points out where the two regimens differ, being specific about the pros and cons of each (for example, potential bone mineral density loss or renal complications with TDF, and potential weight gain with subsequent metabolic and cardiovascular consequences with TAF).
The TDF versus TAF-based PrEP discussion could also focus on what’s known about the comparative effectiveness of each regimen.
For example, TDF/FTC has been shown to be about 99% effective at preventing infection in men who have sex with men and in transgender women, also about 99% effective in heterosexual women and men, and 74%-84% effective in persons who inject drugs.
In contrast, TAF/FTC has been shown to be about 99% effective in men who have sex with men and transgender women, but it’s efficacy in the other two categories is unknown, Dr. Blackstock said.
The option discussion should include a comparison of the evidence base for each regimen, including the real-world experience with TDF/FTC since 2012, and much more limited experience with TAF/FTC.
Discussing relative costs, although the wholesale costs of the regimens are similar, there is now a generic version of TDF/FTC made by Teva Pharmaceuticals that sells for about $400 less per month than the brand name, which might make the option more acceptable to health insurers.
Decision
The decision talk is about considering the patients preferences and deciding with them what is best.
The clinician could say, for example: “What, from your point of view, matters most to you?”
The clinician should also be willing to allow the patient to defer a decision or to guide them depending on their stated wish, asking something like: “Are you ready to decide, or do you want more time? Do you have more questions? Are there more things we should discuss?
Offering the patient a chance to review the decision can also be a good way to arrive at closure, Dr. Blackstock said.
Robert Goldstein, MD, PhD, an infectious disease specialist and medical director of the transgender health program at Massachusetts General Hospital in Boston, who comoderated the session where Dr. Blackstock presented her talk, said that he also uses a similar approach to the PrEP discussion.
“I use a very patient-centered approach of providing information and talking through the data that are available,” he said.
“I will say that, as patients come to me talking about the transition from TDF to TAF for pre-exposure prophylaxis, I am very clear with them about the limited benefit or no benefit that I see with TAF for pre-exposure prophylaxis, and all of my patients have remained on TDF for pre-exposure prophylaxis,” he added.
No funding source for the presentation was reported. Dr. Blackstock and Dr. Goldstein reported having no conflicts of interest to disclose.
A patient-centered approach can help guide persons at risk for HIV exposure to decide on the best choice of pre-exposure prophylaxis (PrEP) regimens for them, stifling the noise generated by direct-to-consumer advertising, an infectious disease specialist recommends.
The decision for patients whether to start or remain on the PrEP combination of tenofovir disoproxil fumarate (TDF) plus emtricitabine (FTC; Truvada and generic) or on tenofovir alafenamide (TAF) plus FTC (Descovy) is made more fraught by confusion regarding the use of the newer and allegedly safer TAF prodrug of tenofovir in HIV treatment regimens, said Oni Blackstock, MD, founder and executive director of Health Justice and an attending physician in the division of infectious diseases at Harlem Hospital, New York.
“There have been commercials on TV as well as on social media around class-action lawsuits against [Truvada maker] Gilead,” she said in an online presentation during IDWeek 2020, an annual scientific meeting on infectious diseases, held virtually this year.
“These lawsuits focus on TDF for HIV treatment, but they have sown a great deal of confusion about TDF versus TAF for PrEP among potential and actual PrEP users,” she added.
Dr. Blackstock described her approach to shared decision-making regarding TDF/FTC versus TAF/FTC, and to helping patients understand the relative benefits and risks of each formulation.
In January of 2020, Dr. Blackstock, who was then assistant commissioner of the HIV bureau of the New York City Department of Health and Mental Hygiene, issued with other Bureau members a “Dear colleague” letter stating why they believed that TDF/FTC should remain the first-line regimen for PrEP.
That opinion, she said, was bolstered by an editorial published in February 2020 by Douglas E. Krakower, MD, from Beth Israel Deaconess Medical Center in Boston, and colleagues, which questioned the rush to shift from TDF to TAF in HIV treatment, and cautioned against the same approach to PrEP.
“Despite evidence that TAF/FTC would not be cost-effective, compared with generic TDF/FTC , the newer regimen quickly and irrevocably displaced TDF/FTC for HIV treatment in the U.S. A similar shift for PrEP – especially for populations in which TAF/FTC is untested – would be premature, costly, and counterproductive for population impact,” Krakower et al. wrote.
Shared decision-making
Clinicians can help patients who may be a candidate for either PrEP regimen by engaging them in shared decision-making.
“The clinician provides information in this case about a prevention strategy, options, benefits and risks, alternatives, and the patient provides their preferences and values, and together the clinician and patient make a decision,” Dr. Blackstock said.
The process differs from the model of informed decision-making, where the clinician gives the patient the information and the patient comes to a decision, or the old, “paternalistic” model in which the clinician gives information and makes recommendations to the patient.
“Shared decision-making has been studied extensively and has been shown to improve patient satisfaction, patient communication, and also potentially reducing health inequities that we see,” she said.
The model for shared decision-making for clinical practice includes three distinct portions: a choice talk, option talk, and decision talk.
Choice
To begin the discussion, the physician informs the patients of the availability of choices and justifies them, saying, for example, “there is good information about how these two PrEP options differ that I’d like to discuss with you,” and “the two PrEP options have different side effects … some will matter more to you than other people.”
At this stage the clinician should defer closure by offering a more detailed discussion of the choices.
Option
Here the clinician solicits information about what the patient has heard or read about PrEP, describes each option in practical terms, and points out where the two regimens differ, being specific about the pros and cons of each (for example, potential bone mineral density loss or renal complications with TDF, and potential weight gain with subsequent metabolic and cardiovascular consequences with TAF).
The TDF versus TAF-based PrEP discussion could also focus on what’s known about the comparative effectiveness of each regimen.
For example, TDF/FTC has been shown to be about 99% effective at preventing infection in men who have sex with men and in transgender women, also about 99% effective in heterosexual women and men, and 74%-84% effective in persons who inject drugs.
In contrast, TAF/FTC has been shown to be about 99% effective in men who have sex with men and transgender women, but it’s efficacy in the other two categories is unknown, Dr. Blackstock said.
The option discussion should include a comparison of the evidence base for each regimen, including the real-world experience with TDF/FTC since 2012, and much more limited experience with TAF/FTC.
Discussing relative costs, although the wholesale costs of the regimens are similar, there is now a generic version of TDF/FTC made by Teva Pharmaceuticals that sells for about $400 less per month than the brand name, which might make the option more acceptable to health insurers.
Decision
The decision talk is about considering the patients preferences and deciding with them what is best.
The clinician could say, for example: “What, from your point of view, matters most to you?”
The clinician should also be willing to allow the patient to defer a decision or to guide them depending on their stated wish, asking something like: “Are you ready to decide, or do you want more time? Do you have more questions? Are there more things we should discuss?
Offering the patient a chance to review the decision can also be a good way to arrive at closure, Dr. Blackstock said.
Robert Goldstein, MD, PhD, an infectious disease specialist and medical director of the transgender health program at Massachusetts General Hospital in Boston, who comoderated the session where Dr. Blackstock presented her talk, said that he also uses a similar approach to the PrEP discussion.
“I use a very patient-centered approach of providing information and talking through the data that are available,” he said.
“I will say that, as patients come to me talking about the transition from TDF to TAF for pre-exposure prophylaxis, I am very clear with them about the limited benefit or no benefit that I see with TAF for pre-exposure prophylaxis, and all of my patients have remained on TDF for pre-exposure prophylaxis,” he added.
No funding source for the presentation was reported. Dr. Blackstock and Dr. Goldstein reported having no conflicts of interest to disclose.
FROM IDWEEK 2020
COVID-19 vaccine standards questioned at FDA advisory meeting

The FDA’s Vaccines and Related Biological Products Advisory Committee met for a wide-ranging discussion beginning around 10 am. The FDA did not ask the panel to weigh in on any particular vaccine. Instead, the FDA asked for the panel’s feedback on a series of questions, including considerations for continuing phase 3 trials if a product were to get an interim clearance known as an emergency use authorization (EUA).
Speakers at the hearing made a variety of requests, including asking for data showing COVID-19 vaccines can prevent serious illness and urging transparency about the agency’s deliberations for each product to be considered.
FDA staff are closely tracking the crop of experimental vaccines that have made it into advanced stages of testing, including products from Pfizer Inc, AstraZeneca, Johnson & Johnson, and Moderna.
‘Time for a reset’
Among the speakers at the public hearing was Peter Lurie, MD, who served as an FDA associate commissioner from 2014 to 2017. Now the president of the Center for Science in the Public Interest, Lurie was among the speakers who asked the agency to make its independence clear.
President Donald Trump has for months been making predictions about COVID-19 vaccine approvals that have been overly optimistic. In one example, the president, who is seeking re-election on November 3, last month spoke about being able to begin distributing a vaccine in October.
“Until now the process of developing candidate vaccines has been inappropriately politicized with an eye on the election calendar, rather than the deliberate timeframe science requires,” Lurie told the FDA advisory panel. “Now is the time for a reset. This committee has a unique opportunity to set a new tone for vaccine deliberations going forward.”
Lurie asked the panel to press the FDA to commit to hold an advisory committee meeting on requests by drugmakers for EUAs. He also asked the panel to demand that informed consent forms and minutes from institutional review board (IRB) discussions of COVID-19 vaccines trials be made public.
Also among the speakers at the public hearing was Peter Doshi, PhD, an associate professor at the University of Maryland School of Pharmacy, who argued that the current trials won’t answer the right questions about the COVID-19 vaccines.
“We could end up with approved vaccines that reduce the risk of mild infection, but do not decrease the risk of hospitalization, ICU use, or death — either at all or by a clinically relevant amount,” Doshi told the panel.
In his presentation, he reiterated points he had made previously, including in an October 21 article in the BMJ, for which he is an associate editor. Doshi also raised these concerns in a September opinion article in The New York Times, co-authored with Eric Topol, MD, director of the Scripps Research Translational Institute and editor-in-chief of Medscape.
Risks of a ‘rushed vaccine’
Other complaints about the FDA’s approach included criticism of a 2-month follow-up time after vaccination, which was seen as too short. ECRI, a nonprofit organization that seeks to improve the safety, quality, and cost-effectiveness of medicines, has argued that approving a weak COVID-19 vaccine might worsen the pandemic.
In an October 21 statement, ECRI noted the risk of a partially effective vaccine, which could be welcomed as a means of slowing transmission of the virus. But public response and attitudes over the past 9 months in the United States suggest that people would relax their precautions as soon as a vaccine is available.
“Resulting infections may offset the vaccine’s impact and end up increasing the mortality and morbidity burden,” ECRI said in the brief.
“The risks and consequences of a rushed vaccine could be very severe if the review is anything shy of thorough,” ECRI Chief Executive Officer Marcus Schabacker, MD, PhD, said in a statement prepared for the hearing.
This article first appeared on Medscape.com.
The FDA’s Vaccines and Related Biological Products Advisory Committee met for a wide-ranging discussion beginning around 10 am. The FDA did not ask the panel to weigh in on any particular vaccine. Instead, the FDA asked for the panel’s feedback on a series of questions, including considerations for continuing phase 3 trials if a product were to get an interim clearance known as an emergency use authorization (EUA).
Speakers at the hearing made a variety of requests, including asking for data showing COVID-19 vaccines can prevent serious illness and urging transparency about the agency’s deliberations for each product to be considered.
FDA staff are closely tracking the crop of experimental vaccines that have made it into advanced stages of testing, including products from Pfizer Inc, AstraZeneca, Johnson & Johnson, and Moderna.
‘Time for a reset’
Among the speakers at the public hearing was Peter Lurie, MD, who served as an FDA associate commissioner from 2014 to 2017. Now the president of the Center for Science in the Public Interest, Lurie was among the speakers who asked the agency to make its independence clear.
President Donald Trump has for months been making predictions about COVID-19 vaccine approvals that have been overly optimistic. In one example, the president, who is seeking re-election on November 3, last month spoke about being able to begin distributing a vaccine in October.
“Until now the process of developing candidate vaccines has been inappropriately politicized with an eye on the election calendar, rather than the deliberate timeframe science requires,” Lurie told the FDA advisory panel. “Now is the time for a reset. This committee has a unique opportunity to set a new tone for vaccine deliberations going forward.”
Lurie asked the panel to press the FDA to commit to hold an advisory committee meeting on requests by drugmakers for EUAs. He also asked the panel to demand that informed consent forms and minutes from institutional review board (IRB) discussions of COVID-19 vaccines trials be made public.
Also among the speakers at the public hearing was Peter Doshi, PhD, an associate professor at the University of Maryland School of Pharmacy, who argued that the current trials won’t answer the right questions about the COVID-19 vaccines.
“We could end up with approved vaccines that reduce the risk of mild infection, but do not decrease the risk of hospitalization, ICU use, or death — either at all or by a clinically relevant amount,” Doshi told the panel.
In his presentation, he reiterated points he had made previously, including in an October 21 article in the BMJ, for which he is an associate editor. Doshi also raised these concerns in a September opinion article in The New York Times, co-authored with Eric Topol, MD, director of the Scripps Research Translational Institute and editor-in-chief of Medscape.
Risks of a ‘rushed vaccine’
Other complaints about the FDA’s approach included criticism of a 2-month follow-up time after vaccination, which was seen as too short. ECRI, a nonprofit organization that seeks to improve the safety, quality, and cost-effectiveness of medicines, has argued that approving a weak COVID-19 vaccine might worsen the pandemic.
In an October 21 statement, ECRI noted the risk of a partially effective vaccine, which could be welcomed as a means of slowing transmission of the virus. But public response and attitudes over the past 9 months in the United States suggest that people would relax their precautions as soon as a vaccine is available.
“Resulting infections may offset the vaccine’s impact and end up increasing the mortality and morbidity burden,” ECRI said in the brief.
“The risks and consequences of a rushed vaccine could be very severe if the review is anything shy of thorough,” ECRI Chief Executive Officer Marcus Schabacker, MD, PhD, said in a statement prepared for the hearing.
This article first appeared on Medscape.com.
The FDA’s Vaccines and Related Biological Products Advisory Committee met for a wide-ranging discussion beginning around 10 am. The FDA did not ask the panel to weigh in on any particular vaccine. Instead, the FDA asked for the panel’s feedback on a series of questions, including considerations for continuing phase 3 trials if a product were to get an interim clearance known as an emergency use authorization (EUA).
Speakers at the hearing made a variety of requests, including asking for data showing COVID-19 vaccines can prevent serious illness and urging transparency about the agency’s deliberations for each product to be considered.
FDA staff are closely tracking the crop of experimental vaccines that have made it into advanced stages of testing, including products from Pfizer Inc, AstraZeneca, Johnson & Johnson, and Moderna.
‘Time for a reset’
Among the speakers at the public hearing was Peter Lurie, MD, who served as an FDA associate commissioner from 2014 to 2017. Now the president of the Center for Science in the Public Interest, Lurie was among the speakers who asked the agency to make its independence clear.
President Donald Trump has for months been making predictions about COVID-19 vaccine approvals that have been overly optimistic. In one example, the president, who is seeking re-election on November 3, last month spoke about being able to begin distributing a vaccine in October.
“Until now the process of developing candidate vaccines has been inappropriately politicized with an eye on the election calendar, rather than the deliberate timeframe science requires,” Lurie told the FDA advisory panel. “Now is the time for a reset. This committee has a unique opportunity to set a new tone for vaccine deliberations going forward.”
Lurie asked the panel to press the FDA to commit to hold an advisory committee meeting on requests by drugmakers for EUAs. He also asked the panel to demand that informed consent forms and minutes from institutional review board (IRB) discussions of COVID-19 vaccines trials be made public.
Also among the speakers at the public hearing was Peter Doshi, PhD, an associate professor at the University of Maryland School of Pharmacy, who argued that the current trials won’t answer the right questions about the COVID-19 vaccines.
“We could end up with approved vaccines that reduce the risk of mild infection, but do not decrease the risk of hospitalization, ICU use, or death — either at all or by a clinically relevant amount,” Doshi told the panel.
In his presentation, he reiterated points he had made previously, including in an October 21 article in the BMJ, for which he is an associate editor. Doshi also raised these concerns in a September opinion article in The New York Times, co-authored with Eric Topol, MD, director of the Scripps Research Translational Institute and editor-in-chief of Medscape.
Risks of a ‘rushed vaccine’
Other complaints about the FDA’s approach included criticism of a 2-month follow-up time after vaccination, which was seen as too short. ECRI, a nonprofit organization that seeks to improve the safety, quality, and cost-effectiveness of medicines, has argued that approving a weak COVID-19 vaccine might worsen the pandemic.
In an October 21 statement, ECRI noted the risk of a partially effective vaccine, which could be welcomed as a means of slowing transmission of the virus. But public response and attitudes over the past 9 months in the United States suggest that people would relax their precautions as soon as a vaccine is available.
“Resulting infections may offset the vaccine’s impact and end up increasing the mortality and morbidity burden,” ECRI said in the brief.
“The risks and consequences of a rushed vaccine could be very severe if the review is anything shy of thorough,” ECRI Chief Executive Officer Marcus Schabacker, MD, PhD, said in a statement prepared for the hearing.
This article first appeared on Medscape.com.
Direct-acting agents cure hepatitis C in children
Between 23,000 and 46,000 U.S. children live with chronic hepatitis C virus with a prevalence of 0.17% anti–hepatitis C virus (HCV) antibody positivity in those aged 6-11 years and 0.39% among children aged 12-19 years. In the United States, genotype 1 is most frequent, followed by genotypes 2 and 3. About 99% of cases result from vertical transmission; transfusion-related cases have not been observed in recent decades.Only viremic mothers are at risk of transmission as those who have spontaneously cleared HCV viremia or have been treated successfully do not risk transmission. Maternal HCV viral load appears to be a risk factor for HCV transmission, however transmission is reported at all levels of viremia.

In conjunction with the opioid epidemics, the prevalence of HCV infection has increased over the last decade. The Centers for Disease Control and Prevention reported that, between 2009 and 2014, the prevalence of HCV infection increased from 1.8 to 3.4 per 1,000 live births. They identified substantial state-to-state variation with the highest rate in West Virginia (22.6 per 1,000 live births), and the lowest in Hawaii (0.7 per 1,000 live births). The implications are clear that increasing numbers of newborns are exposed to HCV and, if transmission rates are between 1% and 5%, 80-400 U.S. infants each year acquire HCV infection.
HCV in children
HCV in children is almost always associated with persistent transaminitis. Chronic infection is defined as the persistence of HCV RNA for at least 6 months, and clearance of HCV infection is determined by the persistent disappearance of HCV RNA. Regardless of infection status, an infant may have detectable maternal anti-HCV antibody in serum until 18 months of age, resulting from passive transfer. In addition, prolonged infection can lead to cirrhosis, hepatocellular carcinoma, or decompensated liver disease. Potential extrahepatic manifestations including reduced physical and psychosocial health also are linked to chronic HCV. Autoimmune disease also has been reported in children with HCV. As well, the stigma of HCV elicits fear in school and child care settings that is a result of public misunderstanding regarding routes of hepatitis C transmission. No restriction of regular childhood activities is required in the daily life of HCV-infected children.
Taken together, increasing rates of HCV infection in pregnant women, increasing numbers of exposed and infected infants annually, potential for both short- and long-term morbidity, and curative nontoxic treatment,
Screening for HCV
There is considerable discussion about which strategy for screening of at-risk infants is more appropriate. Some groups advocate for HCV-RNA testing within the first year of life. Proponents argue the use of a highly sensitive RNA assay early in life has potential to increase detection of infected infants while a negative result allows the conclusion the infant is not infected. Advocates hypothesize that early identification has potential to improve continued follow-up.
Opponents argue that early testing does not change the need for repeat testing after 18 months to confirm diagnosis. They also argue that HCV RNA is more expensive than an antibody-based testing; and treatment will not begin prior to age 3 as there is still opportunity for viremia to spontaneously clear.
Direct acting agents licensed
Ledipasvir/sofosbuvir (Harvoni) was initially demonstrated as curative for genotype 1, 4, 5, or 6 infection in a phase 2, multicenter, open-label study of 100 adolescents with genotype 1 treated for 12 weeks. Sustained virologic response (SVR) was documented in 98% of participants.The regimen was safe and well tolerated in this population, and the adult dosage formulation resulted in pharmacokinetic characteristics similar to those observed in adults. Two clinical trials supported the efficacy of ledipasvir/sofosbuvir in the pediatric population aged 3-11 years. This regimen also is recommended for interferon-experienced (± ribavirin, with or without an HCV protease inhibitor) children and adolescents aged 3 years or older with genotype 1 or 4. A 12-week course is recommended for patients without cirrhosis; 24 weeks is recommended for those with compensated cirrhosis. The combination of ledipasvir/sofosbuvir is the only treatment option for children aged 3-6 years with genotype 1, 4, 5, or 6 infection.

The efficacy of sofosbuvir/velpatasvir (Epclusa) once daily for 12 weeks was first evaluated in an open-label trial among children aged 6 years and older with genotype 1, 2, 3, 4, or 6 infection, without cirrhosis or with compensated cirrhosis. Subsequently, the “cocktail” was evaluated in children aged 6-12 years, with 76% genotype 1, 3% genotype 2, 15% genotype 3, and 6% genotype 4. SVR12 rates were 93% (50/54) in children with genotype 1, 91% (10/11) in those with genotype 3, and 100% in participants with genotype 2 (2/2) or genotype 4 (4/4). Sofosbuvir/velpatasvir was approved in March 2020 by the Food and Drug Administration for pediatric patients aged 6 years and older. Given its pangenotypic activity, safety, and efficacy, sofosbuvir/velpatasvir is currently recommended as a first choice for HCV treatment in children and adolescents aged at least 6 years.
The daily fixed-dose combination of glecaprevir/pibrentasvir (Mavyret) was approved in April 2019 for adolescents aged 12-17 years, and weighing at least 45 kg.Treatment is for 8 weeks, and includes treatment-naive patients without cirrhosis or those with compensated cirrhosis. SVR12 rates for Mavyret have ranged from 91% to 100 % across clinic trials. FDA approval and HCV guideline treatment recommendations for direct-acting antiviral (DAA)–experienced adolescents are based on clinical trial data from adults. Given its pangenotypic activity, safety, and efficacy record in adult patients, glecaprevir/pibrentasvir is recommended as a first choice for adolescent HCV treatment. Glecaprevir/pibrentasvir once approved for children less than 3 years of age will be safe and efficacious as a pangenotypic treatment option in children with chronic HCV infection.
Current recommendations
Tools for identifying HCV infected infants as early as a few months of age are available, yet studies demonstrate that a minority of at-risk children are tested for HCV using either an HCV polymerase chain reaction strategy early in life or an anti-HCV antibody strategy after 18 months of age.
Therapy with direct-acting agents is now licensed to those aged 3 years and offers the potential for cure, eliminating concern for possible progression after prolonged infection. Such therapy offers the potential to eliminate the stigma faced by many children as well as the hepatic and extrahepatic manifestations observed in children. Medication formulation and the child’s abilities to take the medication needs to be considered when prescribing DAAs. It is important to assess if the child can successfully swallow pills. Currently, Harvoni is the only medication that comes in both pellet and pill formulations. The dose is based on weight. The pellets need to be given in a small amount of nonacidic food; they cannot be chewed.
All children with chronic HCV infection are candidates for treatment. When significant fibrosis and/or cirrhosis is present treatment should not be delayed once the child is age 3 years; when only transaminitis is present, treatment can be delayed. In our experience, parents are eager to complete treatment before starting kindergarten.
Liver biopsy for obtaining liver tissue for histopathologic examination is not routinely indicated in children with chronic HCV infection but should be evaluated case by case. Noninvasive tests of hepatic fibrosis have been used in children, these include serologic markers (i.e., FibroSure) and radiologic tests such as ultrasound-based transient elastography (i.e., Fibroscan). Validation for pediatric patients is variable for the different serologic tests. Studies have shown that Fibroscan using the M probe is feasible for a wide range of ages, but poor patient cooperation may make measurement difficult.
Further details regarding dosing and choice of formulation is available at https://www.hcvguidelines.org/unique-populations/children.
Dr. Sabharwal is assistant professor of pediatrics at Boston University and attending physician in pediatric infectious diseases at Boston Medical Center. Ms. Moloney is an instructor in pediatrics at Boston University and a pediatric nurse practitioner in pediatric infectious diseases at Boston Medicine Center. Dr. Pelton is professor of pediatrics and epidemiology at Boston University and public health and senior attending physician at Boston Medical Center. Boston Medical Center received funding from AbbVie for study of Harvoni in Children 3 years of age and older. Email them at [email protected].
References
MMWR Morb Mortal Wkly Rep. 2017 May 12;66(18):470-3. Hepatol Commun. 2017 March 23. doi: 10.1002/hep4.1028. Hepatology. 2020 Feb;71(2):422-30. Lancet Gastroenterol Hepatol. 2019 Apr 11. doi: 10.1016/S2468-1253(19)30046-9. Arch Dis Child. 2006 Sep;91(9):781-5. J Pediatr Gastroenterol Nutr. 2010 Feb;50(2):123-31.
Between 23,000 and 46,000 U.S. children live with chronic hepatitis C virus with a prevalence of 0.17% anti–hepatitis C virus (HCV) antibody positivity in those aged 6-11 years and 0.39% among children aged 12-19 years. In the United States, genotype 1 is most frequent, followed by genotypes 2 and 3. About 99% of cases result from vertical transmission; transfusion-related cases have not been observed in recent decades.Only viremic mothers are at risk of transmission as those who have spontaneously cleared HCV viremia or have been treated successfully do not risk transmission. Maternal HCV viral load appears to be a risk factor for HCV transmission, however transmission is reported at all levels of viremia.
In conjunction with the opioid epidemics, the prevalence of HCV infection has increased over the last decade. The Centers for Disease Control and Prevention reported that, between 2009 and 2014, the prevalence of HCV infection increased from 1.8 to 3.4 per 1,000 live births. They identified substantial state-to-state variation with the highest rate in West Virginia (22.6 per 1,000 live births), and the lowest in Hawaii (0.7 per 1,000 live births). The implications are clear that increasing numbers of newborns are exposed to HCV and, if transmission rates are between 1% and 5%, 80-400 U.S. infants each year acquire HCV infection.
HCV in children
HCV in children is almost always associated with persistent transaminitis. Chronic infection is defined as the persistence of HCV RNA for at least 6 months, and clearance of HCV infection is determined by the persistent disappearance of HCV RNA. Regardless of infection status, an infant may have detectable maternal anti-HCV antibody in serum until 18 months of age, resulting from passive transfer. In addition, prolonged infection can lead to cirrhosis, hepatocellular carcinoma, or decompensated liver disease. Potential extrahepatic manifestations including reduced physical and psychosocial health also are linked to chronic HCV. Autoimmune disease also has been reported in children with HCV. As well, the stigma of HCV elicits fear in school and child care settings that is a result of public misunderstanding regarding routes of hepatitis C transmission. No restriction of regular childhood activities is required in the daily life of HCV-infected children.
Taken together, increasing rates of HCV infection in pregnant women, increasing numbers of exposed and infected infants annually, potential for both short- and long-term morbidity, and curative nontoxic treatment,
Screening for HCV
There is considerable discussion about which strategy for screening of at-risk infants is more appropriate. Some groups advocate for HCV-RNA testing within the first year of life. Proponents argue the use of a highly sensitive RNA assay early in life has potential to increase detection of infected infants while a negative result allows the conclusion the infant is not infected. Advocates hypothesize that early identification has potential to improve continued follow-up.
Opponents argue that early testing does not change the need for repeat testing after 18 months to confirm diagnosis. They also argue that HCV RNA is more expensive than an antibody-based testing; and treatment will not begin prior to age 3 as there is still opportunity for viremia to spontaneously clear.
Direct acting agents licensed
Ledipasvir/sofosbuvir (Harvoni) was initially demonstrated as curative for genotype 1, 4, 5, or 6 infection in a phase 2, multicenter, open-label study of 100 adolescents with genotype 1 treated for 12 weeks. Sustained virologic response (SVR) was documented in 98% of participants.The regimen was safe and well tolerated in this population, and the adult dosage formulation resulted in pharmacokinetic characteristics similar to those observed in adults. Two clinical trials supported the efficacy of ledipasvir/sofosbuvir in the pediatric population aged 3-11 years. This regimen also is recommended for interferon-experienced (± ribavirin, with or without an HCV protease inhibitor) children and adolescents aged 3 years or older with genotype 1 or 4. A 12-week course is recommended for patients without cirrhosis; 24 weeks is recommended for those with compensated cirrhosis. The combination of ledipasvir/sofosbuvir is the only treatment option for children aged 3-6 years with genotype 1, 4, 5, or 6 infection.
The efficacy of sofosbuvir/velpatasvir (Epclusa) once daily for 12 weeks was first evaluated in an open-label trial among children aged 6 years and older with genotype 1, 2, 3, 4, or 6 infection, without cirrhosis or with compensated cirrhosis. Subsequently, the “cocktail” was evaluated in children aged 6-12 years, with 76% genotype 1, 3% genotype 2, 15% genotype 3, and 6% genotype 4. SVR12 rates were 93% (50/54) in children with genotype 1, 91% (10/11) in those with genotype 3, and 100% in participants with genotype 2 (2/2) or genotype 4 (4/4). Sofosbuvir/velpatasvir was approved in March 2020 by the Food and Drug Administration for pediatric patients aged 6 years and older. Given its pangenotypic activity, safety, and efficacy, sofosbuvir/velpatasvir is currently recommended as a first choice for HCV treatment in children and adolescents aged at least 6 years.
The daily fixed-dose combination of glecaprevir/pibrentasvir (Mavyret) was approved in April 2019 for adolescents aged 12-17 years, and weighing at least 45 kg.Treatment is for 8 weeks, and includes treatment-naive patients without cirrhosis or those with compensated cirrhosis. SVR12 rates for Mavyret have ranged from 91% to 100 % across clinic trials. FDA approval and HCV guideline treatment recommendations for direct-acting antiviral (DAA)–experienced adolescents are based on clinical trial data from adults. Given its pangenotypic activity, safety, and efficacy record in adult patients, glecaprevir/pibrentasvir is recommended as a first choice for adolescent HCV treatment. Glecaprevir/pibrentasvir once approved for children less than 3 years of age will be safe and efficacious as a pangenotypic treatment option in children with chronic HCV infection.
Current recommendations
Tools for identifying HCV infected infants as early as a few months of age are available, yet studies demonstrate that a minority of at-risk children are tested for HCV using either an HCV polymerase chain reaction strategy early in life or an anti-HCV antibody strategy after 18 months of age.
Therapy with direct-acting agents is now licensed to those aged 3 years and offers the potential for cure, eliminating concern for possible progression after prolonged infection. Such therapy offers the potential to eliminate the stigma faced by many children as well as the hepatic and extrahepatic manifestations observed in children. Medication formulation and the child’s abilities to take the medication needs to be considered when prescribing DAAs. It is important to assess if the child can successfully swallow pills. Currently, Harvoni is the only medication that comes in both pellet and pill formulations. The dose is based on weight. The pellets need to be given in a small amount of nonacidic food; they cannot be chewed.
All children with chronic HCV infection are candidates for treatment. When significant fibrosis and/or cirrhosis is present treatment should not be delayed once the child is age 3 years; when only transaminitis is present, treatment can be delayed. In our experience, parents are eager to complete treatment before starting kindergarten.
Liver biopsy for obtaining liver tissue for histopathologic examination is not routinely indicated in children with chronic HCV infection but should be evaluated case by case. Noninvasive tests of hepatic fibrosis have been used in children, these include serologic markers (i.e., FibroSure) and radiologic tests such as ultrasound-based transient elastography (i.e., Fibroscan). Validation for pediatric patients is variable for the different serologic tests. Studies have shown that Fibroscan using the M probe is feasible for a wide range of ages, but poor patient cooperation may make measurement difficult.
Further details regarding dosing and choice of formulation is available at https://www.hcvguidelines.org/unique-populations/children.
Dr. Sabharwal is assistant professor of pediatrics at Boston University and attending physician in pediatric infectious diseases at Boston Medical Center. Ms. Moloney is an instructor in pediatrics at Boston University and a pediatric nurse practitioner in pediatric infectious diseases at Boston Medicine Center. Dr. Pelton is professor of pediatrics and epidemiology at Boston University and public health and senior attending physician at Boston Medical Center. Boston Medical Center received funding from AbbVie for study of Harvoni in Children 3 years of age and older. Email them at [email protected].
References
MMWR Morb Mortal Wkly Rep. 2017 May 12;66(18):470-3. Hepatol Commun. 2017 March 23. doi: 10.1002/hep4.1028. Hepatology. 2020 Feb;71(2):422-30. Lancet Gastroenterol Hepatol. 2019 Apr 11. doi: 10.1016/S2468-1253(19)30046-9. Arch Dis Child. 2006 Sep;91(9):781-5. J Pediatr Gastroenterol Nutr. 2010 Feb;50(2):123-31.
Between 23,000 and 46,000 U.S. children live with chronic hepatitis C virus with a prevalence of 0.17% anti–hepatitis C virus (HCV) antibody positivity in those aged 6-11 years and 0.39% among children aged 12-19 years. In the United States, genotype 1 is most frequent, followed by genotypes 2 and 3. About 99% of cases result from vertical transmission; transfusion-related cases have not been observed in recent decades.Only viremic mothers are at risk of transmission as those who have spontaneously cleared HCV viremia or have been treated successfully do not risk transmission. Maternal HCV viral load appears to be a risk factor for HCV transmission, however transmission is reported at all levels of viremia.
In conjunction with the opioid epidemics, the prevalence of HCV infection has increased over the last decade. The Centers for Disease Control and Prevention reported that, between 2009 and 2014, the prevalence of HCV infection increased from 1.8 to 3.4 per 1,000 live births. They identified substantial state-to-state variation with the highest rate in West Virginia (22.6 per 1,000 live births), and the lowest in Hawaii (0.7 per 1,000 live births). The implications are clear that increasing numbers of newborns are exposed to HCV and, if transmission rates are between 1% and 5%, 80-400 U.S. infants each year acquire HCV infection.
HCV in children
HCV in children is almost always associated with persistent transaminitis. Chronic infection is defined as the persistence of HCV RNA for at least 6 months, and clearance of HCV infection is determined by the persistent disappearance of HCV RNA. Regardless of infection status, an infant may have detectable maternal anti-HCV antibody in serum until 18 months of age, resulting from passive transfer. In addition, prolonged infection can lead to cirrhosis, hepatocellular carcinoma, or decompensated liver disease. Potential extrahepatic manifestations including reduced physical and psychosocial health also are linked to chronic HCV. Autoimmune disease also has been reported in children with HCV. As well, the stigma of HCV elicits fear in school and child care settings that is a result of public misunderstanding regarding routes of hepatitis C transmission. No restriction of regular childhood activities is required in the daily life of HCV-infected children.
Taken together, increasing rates of HCV infection in pregnant women, increasing numbers of exposed and infected infants annually, potential for both short- and long-term morbidity, and curative nontoxic treatment,
Screening for HCV
There is considerable discussion about which strategy for screening of at-risk infants is more appropriate. Some groups advocate for HCV-RNA testing within the first year of life. Proponents argue the use of a highly sensitive RNA assay early in life has potential to increase detection of infected infants while a negative result allows the conclusion the infant is not infected. Advocates hypothesize that early identification has potential to improve continued follow-up.
Opponents argue that early testing does not change the need for repeat testing after 18 months to confirm diagnosis. They also argue that HCV RNA is more expensive than an antibody-based testing; and treatment will not begin prior to age 3 as there is still opportunity for viremia to spontaneously clear.
Direct acting agents licensed
Ledipasvir/sofosbuvir (Harvoni) was initially demonstrated as curative for genotype 1, 4, 5, or 6 infection in a phase 2, multicenter, open-label study of 100 adolescents with genotype 1 treated for 12 weeks. Sustained virologic response (SVR) was documented in 98% of participants.The regimen was safe and well tolerated in this population, and the adult dosage formulation resulted in pharmacokinetic characteristics similar to those observed in adults. Two clinical trials supported the efficacy of ledipasvir/sofosbuvir in the pediatric population aged 3-11 years. This regimen also is recommended for interferon-experienced (± ribavirin, with or without an HCV protease inhibitor) children and adolescents aged 3 years or older with genotype 1 or 4. A 12-week course is recommended for patients without cirrhosis; 24 weeks is recommended for those with compensated cirrhosis. The combination of ledipasvir/sofosbuvir is the only treatment option for children aged 3-6 years with genotype 1, 4, 5, or 6 infection.
The efficacy of sofosbuvir/velpatasvir (Epclusa) once daily for 12 weeks was first evaluated in an open-label trial among children aged 6 years and older with genotype 1, 2, 3, 4, or 6 infection, without cirrhosis or with compensated cirrhosis. Subsequently, the “cocktail” was evaluated in children aged 6-12 years, with 76% genotype 1, 3% genotype 2, 15% genotype 3, and 6% genotype 4. SVR12 rates were 93% (50/54) in children with genotype 1, 91% (10/11) in those with genotype 3, and 100% in participants with genotype 2 (2/2) or genotype 4 (4/4). Sofosbuvir/velpatasvir was approved in March 2020 by the Food and Drug Administration for pediatric patients aged 6 years and older. Given its pangenotypic activity, safety, and efficacy, sofosbuvir/velpatasvir is currently recommended as a first choice for HCV treatment in children and adolescents aged at least 6 years.
The daily fixed-dose combination of glecaprevir/pibrentasvir (Mavyret) was approved in April 2019 for adolescents aged 12-17 years, and weighing at least 45 kg.Treatment is for 8 weeks, and includes treatment-naive patients without cirrhosis or those with compensated cirrhosis. SVR12 rates for Mavyret have ranged from 91% to 100 % across clinic trials. FDA approval and HCV guideline treatment recommendations for direct-acting antiviral (DAA)–experienced adolescents are based on clinical trial data from adults. Given its pangenotypic activity, safety, and efficacy record in adult patients, glecaprevir/pibrentasvir is recommended as a first choice for adolescent HCV treatment. Glecaprevir/pibrentasvir once approved for children less than 3 years of age will be safe and efficacious as a pangenotypic treatment option in children with chronic HCV infection.
Current recommendations
Tools for identifying HCV infected infants as early as a few months of age are available, yet studies demonstrate that a minority of at-risk children are tested for HCV using either an HCV polymerase chain reaction strategy early in life or an anti-HCV antibody strategy after 18 months of age.
Therapy with direct-acting agents is now licensed to those aged 3 years and offers the potential for cure, eliminating concern for possible progression after prolonged infection. Such therapy offers the potential to eliminate the stigma faced by many children as well as the hepatic and extrahepatic manifestations observed in children. Medication formulation and the child’s abilities to take the medication needs to be considered when prescribing DAAs. It is important to assess if the child can successfully swallow pills. Currently, Harvoni is the only medication that comes in both pellet and pill formulations. The dose is based on weight. The pellets need to be given in a small amount of nonacidic food; they cannot be chewed.
All children with chronic HCV infection are candidates for treatment. When significant fibrosis and/or cirrhosis is present treatment should not be delayed once the child is age 3 years; when only transaminitis is present, treatment can be delayed. In our experience, parents are eager to complete treatment before starting kindergarten.
Liver biopsy for obtaining liver tissue for histopathologic examination is not routinely indicated in children with chronic HCV infection but should be evaluated case by case. Noninvasive tests of hepatic fibrosis have been used in children, these include serologic markers (i.e., FibroSure) and radiologic tests such as ultrasound-based transient elastography (i.e., Fibroscan). Validation for pediatric patients is variable for the different serologic tests. Studies have shown that Fibroscan using the M probe is feasible for a wide range of ages, but poor patient cooperation may make measurement difficult.
Further details regarding dosing and choice of formulation is available at https://www.hcvguidelines.org/unique-populations/children.
Dr. Sabharwal is assistant professor of pediatrics at Boston University and attending physician in pediatric infectious diseases at Boston Medical Center. Ms. Moloney is an instructor in pediatrics at Boston University and a pediatric nurse practitioner in pediatric infectious diseases at Boston Medicine Center. Dr. Pelton is professor of pediatrics and epidemiology at Boston University and public health and senior attending physician at Boston Medical Center. Boston Medical Center received funding from AbbVie for study of Harvoni in Children 3 years of age and older. Email them at [email protected].
References
MMWR Morb Mortal Wkly Rep. 2017 May 12;66(18):470-3. Hepatol Commun. 2017 March 23. doi: 10.1002/hep4.1028. Hepatology. 2020 Feb;71(2):422-30. Lancet Gastroenterol Hepatol. 2019 Apr 11. doi: 10.1016/S2468-1253(19)30046-9. Arch Dis Child. 2006 Sep;91(9):781-5. J Pediatr Gastroenterol Nutr. 2010 Feb;50(2):123-31.
CDC expands definition of COVID-19 exposure from ‘close contact’
![]()
New data suggest each close encounter – coming within 6 feet of an infected person – can increase the risk for transmission, CDC director Robert Redfield, MD, said during a media briefing.
“As we get more data and understand the science of COVID, we’re going to continue to incorporate that in our recommendations,” Dr. Redfield said in response to a reporter’s question about a recent study.
Previously, the CDC cautioned against spending 15 minutes or longer in close proximity to an infected person, particularly in enclosed indoor spaces.
In a new report published online Oct. 21 in Morbidity and Mortality Weekly Report, however, investigators “determined that an individual who had a series of shorter contacts that over time added up to more than 15 minutes became infected.”
Beware of brief encounters?
On July 28, a 20-year-old male correctional officer in Vermont had multiple brief encounters with six transferred incarcerated or detained people while their SARS-CoV-2 test results were pending. The six were asymptomatic at the time and were housed in a quarantine unit, reported CDC researcher Julia Pringle, PhD, and colleagues.
The following day, all six inmates tested polymerase chain reaction (PCR) positive for COVID-19. The correctional officer did not spend 15 minutes or more within 6 feet of any of the inmates, according to video surveillance footage, and he continued to work.
On Aug. 4, however, he developed symptoms that included loss of smell and taste, myalgia, runny nose, cough, shortness of breath, headache, loss of appetite, and gastrointestinal symptoms. He stayed home starting the next day and tested PCR positive for COVID-19 on Aug. 11.
Further review of the surveillance video showed that the officer had numerous brief encounters of approximately 1 minute each that cumulatively exceeded 15 minutes over a 24-hour period, the researchers reported.
During all the interactions with inmates, the correctional officer wore a cloth mask, gown, and eye protection. The inmates wore masks while in their cells but did not have them on during brief cell doorway interactions or in the recreation room, according to the report.
No interaction is 100% safe
“We know that every activity that involves interacting with others has some degree of risk right now,” said Jay Butler, MD, CDC deputy director for infectious diseases.
“Unfortunately, we’re seeing a distressing trend here in the United States with COVID-19 cases increasing in nearly 75% of the country,” he said. “We’ve confirmed 8.1 million cases and, sadly, over 220,000 deaths since January.
“I know these are numbers, but these are also people,” Dr. Butler added.
“The pandemic is not over,” Dr. Redfield said. “Earlier this week, COVID virus cases reached over 40 million globally. Here in the United States we are approaching a critical phase.”
Four factors associated with higher risk for transmission are the proximity of each encounter, its duration, whether an interaction takes place indoors or outdoors, and the number of people encountered, Dr. Butler said.
Dr. Butler acknowledged widespread fatigue with adherence to personal protection measures, but added that social distancing, mask-wearing, and other measures are more important now than ever. He noted that more Americans will be spending time indoors with the onset of cooler weather and the upcoming holidays.
A note of optimism
Dr. Redfield remains optimistic about the limited availability of a vaccine or vaccines by year’s end but added that “it’s important for all of us to remain diligent in our efforts to defeat this virus.”
“There is hope on the way, in the form of safe and effective vaccines in a matter of weeks or months. To bridge to that next phase, we have to take steps to keep ourselves, our families, and our communities safe,” said Alex Azar, secretary of the Department of Health & Human Services.
“I know it’s been a difficult year for Americans, but we are going to come through this on the other side,” Dr. Redfield said.
![]()
New data suggest each close encounter – coming within 6 feet of an infected person – can increase the risk for transmission, CDC director Robert Redfield, MD, said during a media briefing.
“As we get more data and understand the science of COVID, we’re going to continue to incorporate that in our recommendations,” Dr. Redfield said in response to a reporter’s question about a recent study.
Previously, the CDC cautioned against spending 15 minutes or longer in close proximity to an infected person, particularly in enclosed indoor spaces.
In a new report published online Oct. 21 in Morbidity and Mortality Weekly Report, however, investigators “determined that an individual who had a series of shorter contacts that over time added up to more than 15 minutes became infected.”
Beware of brief encounters?
On July 28, a 20-year-old male correctional officer in Vermont had multiple brief encounters with six transferred incarcerated or detained people while their SARS-CoV-2 test results were pending. The six were asymptomatic at the time and were housed in a quarantine unit, reported CDC researcher Julia Pringle, PhD, and colleagues.
The following day, all six inmates tested polymerase chain reaction (PCR) positive for COVID-19. The correctional officer did not spend 15 minutes or more within 6 feet of any of the inmates, according to video surveillance footage, and he continued to work.
On Aug. 4, however, he developed symptoms that included loss of smell and taste, myalgia, runny nose, cough, shortness of breath, headache, loss of appetite, and gastrointestinal symptoms. He stayed home starting the next day and tested PCR positive for COVID-19 on Aug. 11.
Further review of the surveillance video showed that the officer had numerous brief encounters of approximately 1 minute each that cumulatively exceeded 15 minutes over a 24-hour period, the researchers reported.
During all the interactions with inmates, the correctional officer wore a cloth mask, gown, and eye protection. The inmates wore masks while in their cells but did not have them on during brief cell doorway interactions or in the recreation room, according to the report.
No interaction is 100% safe
“We know that every activity that involves interacting with others has some degree of risk right now,” said Jay Butler, MD, CDC deputy director for infectious diseases.
“Unfortunately, we’re seeing a distressing trend here in the United States with COVID-19 cases increasing in nearly 75% of the country,” he said. “We’ve confirmed 8.1 million cases and, sadly, over 220,000 deaths since January.
“I know these are numbers, but these are also people,” Dr. Butler added.
“The pandemic is not over,” Dr. Redfield said. “Earlier this week, COVID virus cases reached over 40 million globally. Here in the United States we are approaching a critical phase.”
Four factors associated with higher risk for transmission are the proximity of each encounter, its duration, whether an interaction takes place indoors or outdoors, and the number of people encountered, Dr. Butler said.
Dr. Butler acknowledged widespread fatigue with adherence to personal protection measures, but added that social distancing, mask-wearing, and other measures are more important now than ever. He noted that more Americans will be spending time indoors with the onset of cooler weather and the upcoming holidays.
A note of optimism
Dr. Redfield remains optimistic about the limited availability of a vaccine or vaccines by year’s end but added that “it’s important for all of us to remain diligent in our efforts to defeat this virus.”
“There is hope on the way, in the form of safe and effective vaccines in a matter of weeks or months. To bridge to that next phase, we have to take steps to keep ourselves, our families, and our communities safe,” said Alex Azar, secretary of the Department of Health & Human Services.
“I know it’s been a difficult year for Americans, but we are going to come through this on the other side,” Dr. Redfield said.
![]()
New data suggest each close encounter – coming within 6 feet of an infected person – can increase the risk for transmission, CDC director Robert Redfield, MD, said during a media briefing.
“As we get more data and understand the science of COVID, we’re going to continue to incorporate that in our recommendations,” Dr. Redfield said in response to a reporter’s question about a recent study.
Previously, the CDC cautioned against spending 15 minutes or longer in close proximity to an infected person, particularly in enclosed indoor spaces.
In a new report published online Oct. 21 in Morbidity and Mortality Weekly Report, however, investigators “determined that an individual who had a series of shorter contacts that over time added up to more than 15 minutes became infected.”
Beware of brief encounters?
On July 28, a 20-year-old male correctional officer in Vermont had multiple brief encounters with six transferred incarcerated or detained people while their SARS-CoV-2 test results were pending. The six were asymptomatic at the time and were housed in a quarantine unit, reported CDC researcher Julia Pringle, PhD, and colleagues.
The following day, all six inmates tested polymerase chain reaction (PCR) positive for COVID-19. The correctional officer did not spend 15 minutes or more within 6 feet of any of the inmates, according to video surveillance footage, and he continued to work.
On Aug. 4, however, he developed symptoms that included loss of smell and taste, myalgia, runny nose, cough, shortness of breath, headache, loss of appetite, and gastrointestinal symptoms. He stayed home starting the next day and tested PCR positive for COVID-19 on Aug. 11.
Further review of the surveillance video showed that the officer had numerous brief encounters of approximately 1 minute each that cumulatively exceeded 15 minutes over a 24-hour period, the researchers reported.
During all the interactions with inmates, the correctional officer wore a cloth mask, gown, and eye protection. The inmates wore masks while in their cells but did not have them on during brief cell doorway interactions or in the recreation room, according to the report.
No interaction is 100% safe
“We know that every activity that involves interacting with others has some degree of risk right now,” said Jay Butler, MD, CDC deputy director for infectious diseases.
“Unfortunately, we’re seeing a distressing trend here in the United States with COVID-19 cases increasing in nearly 75% of the country,” he said. “We’ve confirmed 8.1 million cases and, sadly, over 220,000 deaths since January.
“I know these are numbers, but these are also people,” Dr. Butler added.
“The pandemic is not over,” Dr. Redfield said. “Earlier this week, COVID virus cases reached over 40 million globally. Here in the United States we are approaching a critical phase.”
Four factors associated with higher risk for transmission are the proximity of each encounter, its duration, whether an interaction takes place indoors or outdoors, and the number of people encountered, Dr. Butler said.
Dr. Butler acknowledged widespread fatigue with adherence to personal protection measures, but added that social distancing, mask-wearing, and other measures are more important now than ever. He noted that more Americans will be spending time indoors with the onset of cooler weather and the upcoming holidays.
A note of optimism
Dr. Redfield remains optimistic about the limited availability of a vaccine or vaccines by year’s end but added that “it’s important for all of us to remain diligent in our efforts to defeat this virus.”
“There is hope on the way, in the form of safe and effective vaccines in a matter of weeks or months. To bridge to that next phase, we have to take steps to keep ourselves, our families, and our communities safe,” said Alex Azar, secretary of the Department of Health & Human Services.
“I know it’s been a difficult year for Americans, but we are going to come through this on the other side,” Dr. Redfield said.
FDA approves remdesivir, first treatment for COVID-19
making it the first and only approved treatment for COVID-19, according to a release from drug manufacturer Gilead Sciences.
The FDA’s initial Emergency Use Authorization (EUA) of the antiviral, issued in May, allowed the drug to be used only for patients with severe COVID-19, specifically, COVID-19 patients with low blood oxygen levels or who needed oxygen therapy or mechanical ventilation.
An August EUA expanded treatment to include all adult and pediatric hospitalized COVID-19 patients, regardless of the severity of their disease. The FDA also issued a new EUA for remdesivir Oct. 22 allowing treatment of hospitalized pediatric patients younger than 12 weighing at least 3.5 kg.
Today’s approval is based on three randomized controlled trials, according to Gilead.
Final trial results from one of them, the National Institute of Allergy and Infectious Disease–funded ACTT-1 trial, published earlier in October, showed that hospitalized patients with COVID-19 who received remdesivir had a shorter median recovery time than those who received a placebo – 10 days versus 15 days.
This difference and some related secondary endpoints were statistically significant in the randomized trial, but there was not a statistically significant difference in mortality between the treatment and placebo groups.
The other two trials used for the approval, the SIMPLE trials, were open-label phase 3 trials conducted in countries with a high prevalence of COVID-19 infections, according to Gilead.
The SIMPLE-Severe trial was a randomized, multicenter study that evaluated the efficacy and safety of 5-day and 10-day dosing plus standard of care in 397 hospitalized adult patients with severe COVID-19. The primary endpoint was clinical status on day 14 assessed on a 7-point ordinal scale, according to Gilead.
The trial found that a 5-day or a 10-day treatment course of Veklury achieved similar clinical outcomes to the ACTT-1 trial (odds ratio, 0.75; 95% confidence interval, 0.51-1.12).
The SIMPLE-Moderate trial was a randomized, controlled, multicenter study that evaluated the efficacy and safety of 5-day and 10-day dosing durations of Veklury plus standard of care, compared with standard of care alone in 600 hospitalized adult patients with moderate COVID-19, Gilead stated in its release.
The primary endpoint was clinical status on day 11 assessed on a 7-point ordinal scale.
The results showed statistically improved clinical outcomes with a 5-day treatment course of Veklury, compared with standard of care (OR, 1.65; 95% CI, 1.0-2.48; P = .017), according to Gilead.
This article first appeared on Medscape.com.
making it the first and only approved treatment for COVID-19, according to a release from drug manufacturer Gilead Sciences.
The FDA’s initial Emergency Use Authorization (EUA) of the antiviral, issued in May, allowed the drug to be used only for patients with severe COVID-19, specifically, COVID-19 patients with low blood oxygen levels or who needed oxygen therapy or mechanical ventilation.
An August EUA expanded treatment to include all adult and pediatric hospitalized COVID-19 patients, regardless of the severity of their disease. The FDA also issued a new EUA for remdesivir Oct. 22 allowing treatment of hospitalized pediatric patients younger than 12 weighing at least 3.5 kg.
Today’s approval is based on three randomized controlled trials, according to Gilead.
Final trial results from one of them, the National Institute of Allergy and Infectious Disease–funded ACTT-1 trial, published earlier in October, showed that hospitalized patients with COVID-19 who received remdesivir had a shorter median recovery time than those who received a placebo – 10 days versus 15 days.
This difference and some related secondary endpoints were statistically significant in the randomized trial, but there was not a statistically significant difference in mortality between the treatment and placebo groups.
The other two trials used for the approval, the SIMPLE trials, were open-label phase 3 trials conducted in countries with a high prevalence of COVID-19 infections, according to Gilead.
The SIMPLE-Severe trial was a randomized, multicenter study that evaluated the efficacy and safety of 5-day and 10-day dosing plus standard of care in 397 hospitalized adult patients with severe COVID-19. The primary endpoint was clinical status on day 14 assessed on a 7-point ordinal scale, according to Gilead.
The trial found that a 5-day or a 10-day treatment course of Veklury achieved similar clinical outcomes to the ACTT-1 trial (odds ratio, 0.75; 95% confidence interval, 0.51-1.12).
The SIMPLE-Moderate trial was a randomized, controlled, multicenter study that evaluated the efficacy and safety of 5-day and 10-day dosing durations of Veklury plus standard of care, compared with standard of care alone in 600 hospitalized adult patients with moderate COVID-19, Gilead stated in its release.
The primary endpoint was clinical status on day 11 assessed on a 7-point ordinal scale.
The results showed statistically improved clinical outcomes with a 5-day treatment course of Veklury, compared with standard of care (OR, 1.65; 95% CI, 1.0-2.48; P = .017), according to Gilead.
This article first appeared on Medscape.com.
making it the first and only approved treatment for COVID-19, according to a release from drug manufacturer Gilead Sciences.
The FDA’s initial Emergency Use Authorization (EUA) of the antiviral, issued in May, allowed the drug to be used only for patients with severe COVID-19, specifically, COVID-19 patients with low blood oxygen levels or who needed oxygen therapy or mechanical ventilation.
An August EUA expanded treatment to include all adult and pediatric hospitalized COVID-19 patients, regardless of the severity of their disease. The FDA also issued a new EUA for remdesivir Oct. 22 allowing treatment of hospitalized pediatric patients younger than 12 weighing at least 3.5 kg.
Today’s approval is based on three randomized controlled trials, according to Gilead.
Final trial results from one of them, the National Institute of Allergy and Infectious Disease–funded ACTT-1 trial, published earlier in October, showed that hospitalized patients with COVID-19 who received remdesivir had a shorter median recovery time than those who received a placebo – 10 days versus 15 days.
This difference and some related secondary endpoints were statistically significant in the randomized trial, but there was not a statistically significant difference in mortality between the treatment and placebo groups.
The other two trials used for the approval, the SIMPLE trials, were open-label phase 3 trials conducted in countries with a high prevalence of COVID-19 infections, according to Gilead.
The SIMPLE-Severe trial was a randomized, multicenter study that evaluated the efficacy and safety of 5-day and 10-day dosing plus standard of care in 397 hospitalized adult patients with severe COVID-19. The primary endpoint was clinical status on day 14 assessed on a 7-point ordinal scale, according to Gilead.
The trial found that a 5-day or a 10-day treatment course of Veklury achieved similar clinical outcomes to the ACTT-1 trial (odds ratio, 0.75; 95% confidence interval, 0.51-1.12).
The SIMPLE-Moderate trial was a randomized, controlled, multicenter study that evaluated the efficacy and safety of 5-day and 10-day dosing durations of Veklury plus standard of care, compared with standard of care alone in 600 hospitalized adult patients with moderate COVID-19, Gilead stated in its release.
The primary endpoint was clinical status on day 11 assessed on a 7-point ordinal scale.
The results showed statistically improved clinical outcomes with a 5-day treatment course of Veklury, compared with standard of care (OR, 1.65; 95% CI, 1.0-2.48; P = .017), according to Gilead.
This article first appeared on Medscape.com.
Rinse and repeat? Mouthwash might mitigate COVID-19 spread
Listerine Antiseptic led the list of most effective mouthwashes for inactivating the coronavirus. Interestingly, a 1% nasal rinse solution of Johnson’s Baby Shampoo also worked, eliminating up to 99.9% of the viral load in the in vitro experiments.
In contrast, use of a neti pot nasal solution yielded no decrease in virus levels.
The study was published in the Journal of Medical Virology.
Because the mouthwash and hydrogen peroxide oral rinses in the study are widely available and easy to use, “I would recommend the use of the rinses on top of wearing mask and social distancing. This could add a layer of protection for yourself and others,” lead study author Craig Meyers, PhD, professor of microbiology and immunology and obstetrics and gynecology, Penn State College of Medicine in Hershey, Pennsylvania, told Medscape Medical News.
Meyers and colleagues found that efficacy aligned with duration of time the cell cultures were exposed to each mouthwash or rinse product. Although it varied, the products required at least 30 seconds to kill most of the virus. Waiting 1 or 2 minutes tended to fortify results.
“This study adds to and further confirms the recently published evidence from virologists in Germany that mouthwashes can inactivate the virus that causes COVID-19 in a test tube,” Valerie O’Donnell, PhD, co-director of the Systems Immunity Research Institute of Cardiff University, Cardiff, Wales, said when asked to comment on the study.
“While this is great to see, what is still lacking is in vivo evidence, since we know the virus will be continually shed in the mouth,” O’Donnell said. “So, the question now becomes, by how much could mouthwashes reduce viral load in the oropharynx of infected people, and if so, then for how long?”
Meyers noted that studies of people positive for COVID-19 using each product would be informative. It remains unknown, for example, if swishing, gargling, and/or spitting out mouthwash would add or decrease the efficacy demonstrated in the lab.
The investigators used the human coronavirus HCoV‐229e as a surrogate for SARS-CoV-2. They noted HCoV-229e is analogous, and SARS-CoV-2 would have been more expensive, less available, and would have required biosafety level 3 laboratory conditions.
Listerine Antiseptic leads the way
“Surprisingly, we found that several of these common products had strong virucidal properties, inactivating from 2 log10 [or 99%] to greater than 4 log10 [or 99.99%] of infectious human coronavirus,” the researchers note.
The researchers added a small amount of organic material (extra protein) to each product to more closely mimic physiologic conditions in the nasopharynx.
Listerine Antiseptic “historically has claimed numerous antimicrobial properties,” the researchers note. Although the label currently only claims to kill germs that cause bad breath, “our tests show that it is highly effective at inactivating human coronavirus in solution. Even at the lowest contact time of 30 seconds, it inactivated greater than 99.99% of human coronavirus.”
Interestingly, the mouthwashes that contained the same active ingredients as Listerine Antiseptic — Listerine Ultra, Equate Antiseptic, and CVS Antiseptic Mouth Wash — were less efficacious. Meyers said the reason remains unclear, but he and colleagues found the same result when they repeated the comparisons.
Timing of the essence?
Meyers and colleagues also tested a nasal rinse solution of 1% baby shampoo because it is sometimes used to treat people with chronic rhinosinusitis. They found 30 seconds led to < 90% to < 99.99% effectiveness, but that, by 2 minutes, efficacy climbed to > 99.9% to > 99.99%.
“Thirty seconds for some products just was not enough time for the efficacy to be observed,” Meyers said. “Whereas, after a minute or two the active ingredient had enough time to work. Thirty seconds may be at the border to see full efficacy.” More research is needed to confirm the timing and determine which active ingredients are driving the findings.
A future trial could test the efficacy of mouthwash products to reduce the viral load in people with COVID-19. “If we are able to get funding to continue, I would like to see a small clinical trial as the next step,” Meyers said.
Meyers and O’Donnell disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Listerine Antiseptic led the list of most effective mouthwashes for inactivating the coronavirus. Interestingly, a 1% nasal rinse solution of Johnson’s Baby Shampoo also worked, eliminating up to 99.9% of the viral load in the in vitro experiments.
In contrast, use of a neti pot nasal solution yielded no decrease in virus levels.
The study was published in the Journal of Medical Virology.
Because the mouthwash and hydrogen peroxide oral rinses in the study are widely available and easy to use, “I would recommend the use of the rinses on top of wearing mask and social distancing. This could add a layer of protection for yourself and others,” lead study author Craig Meyers, PhD, professor of microbiology and immunology and obstetrics and gynecology, Penn State College of Medicine in Hershey, Pennsylvania, told Medscape Medical News.
Meyers and colleagues found that efficacy aligned with duration of time the cell cultures were exposed to each mouthwash or rinse product. Although it varied, the products required at least 30 seconds to kill most of the virus. Waiting 1 or 2 minutes tended to fortify results.
“This study adds to and further confirms the recently published evidence from virologists in Germany that mouthwashes can inactivate the virus that causes COVID-19 in a test tube,” Valerie O’Donnell, PhD, co-director of the Systems Immunity Research Institute of Cardiff University, Cardiff, Wales, said when asked to comment on the study.
“While this is great to see, what is still lacking is in vivo evidence, since we know the virus will be continually shed in the mouth,” O’Donnell said. “So, the question now becomes, by how much could mouthwashes reduce viral load in the oropharynx of infected people, and if so, then for how long?”
Meyers noted that studies of people positive for COVID-19 using each product would be informative. It remains unknown, for example, if swishing, gargling, and/or spitting out mouthwash would add or decrease the efficacy demonstrated in the lab.
The investigators used the human coronavirus HCoV‐229e as a surrogate for SARS-CoV-2. They noted HCoV-229e is analogous, and SARS-CoV-2 would have been more expensive, less available, and would have required biosafety level 3 laboratory conditions.
Listerine Antiseptic leads the way
“Surprisingly, we found that several of these common products had strong virucidal properties, inactivating from 2 log10 [or 99%] to greater than 4 log10 [or 99.99%] of infectious human coronavirus,” the researchers note.
The researchers added a small amount of organic material (extra protein) to each product to more closely mimic physiologic conditions in the nasopharynx.
Listerine Antiseptic “historically has claimed numerous antimicrobial properties,” the researchers note. Although the label currently only claims to kill germs that cause bad breath, “our tests show that it is highly effective at inactivating human coronavirus in solution. Even at the lowest contact time of 30 seconds, it inactivated greater than 99.99% of human coronavirus.”
Interestingly, the mouthwashes that contained the same active ingredients as Listerine Antiseptic — Listerine Ultra, Equate Antiseptic, and CVS Antiseptic Mouth Wash — were less efficacious. Meyers said the reason remains unclear, but he and colleagues found the same result when they repeated the comparisons.
Timing of the essence?
Meyers and colleagues also tested a nasal rinse solution of 1% baby shampoo because it is sometimes used to treat people with chronic rhinosinusitis. They found 30 seconds led to < 90% to < 99.99% effectiveness, but that, by 2 minutes, efficacy climbed to > 99.9% to > 99.99%.
“Thirty seconds for some products just was not enough time for the efficacy to be observed,” Meyers said. “Whereas, after a minute or two the active ingredient had enough time to work. Thirty seconds may be at the border to see full efficacy.” More research is needed to confirm the timing and determine which active ingredients are driving the findings.
A future trial could test the efficacy of mouthwash products to reduce the viral load in people with COVID-19. “If we are able to get funding to continue, I would like to see a small clinical trial as the next step,” Meyers said.
Meyers and O’Donnell disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Listerine Antiseptic led the list of most effective mouthwashes for inactivating the coronavirus. Interestingly, a 1% nasal rinse solution of Johnson’s Baby Shampoo also worked, eliminating up to 99.9% of the viral load in the in vitro experiments.
In contrast, use of a neti pot nasal solution yielded no decrease in virus levels.
The study was published in the Journal of Medical Virology.
Because the mouthwash and hydrogen peroxide oral rinses in the study are widely available and easy to use, “I would recommend the use of the rinses on top of wearing mask and social distancing. This could add a layer of protection for yourself and others,” lead study author Craig Meyers, PhD, professor of microbiology and immunology and obstetrics and gynecology, Penn State College of Medicine in Hershey, Pennsylvania, told Medscape Medical News.
Meyers and colleagues found that efficacy aligned with duration of time the cell cultures were exposed to each mouthwash or rinse product. Although it varied, the products required at least 30 seconds to kill most of the virus. Waiting 1 or 2 minutes tended to fortify results.
“This study adds to and further confirms the recently published evidence from virologists in Germany that mouthwashes can inactivate the virus that causes COVID-19 in a test tube,” Valerie O’Donnell, PhD, co-director of the Systems Immunity Research Institute of Cardiff University, Cardiff, Wales, said when asked to comment on the study.
“While this is great to see, what is still lacking is in vivo evidence, since we know the virus will be continually shed in the mouth,” O’Donnell said. “So, the question now becomes, by how much could mouthwashes reduce viral load in the oropharynx of infected people, and if so, then for how long?”
Meyers noted that studies of people positive for COVID-19 using each product would be informative. It remains unknown, for example, if swishing, gargling, and/or spitting out mouthwash would add or decrease the efficacy demonstrated in the lab.
The investigators used the human coronavirus HCoV‐229e as a surrogate for SARS-CoV-2. They noted HCoV-229e is analogous, and SARS-CoV-2 would have been more expensive, less available, and would have required biosafety level 3 laboratory conditions.
Listerine Antiseptic leads the way
“Surprisingly, we found that several of these common products had strong virucidal properties, inactivating from 2 log10 [or 99%] to greater than 4 log10 [or 99.99%] of infectious human coronavirus,” the researchers note.
The researchers added a small amount of organic material (extra protein) to each product to more closely mimic physiologic conditions in the nasopharynx.
Listerine Antiseptic “historically has claimed numerous antimicrobial properties,” the researchers note. Although the label currently only claims to kill germs that cause bad breath, “our tests show that it is highly effective at inactivating human coronavirus in solution. Even at the lowest contact time of 30 seconds, it inactivated greater than 99.99% of human coronavirus.”
Interestingly, the mouthwashes that contained the same active ingredients as Listerine Antiseptic — Listerine Ultra, Equate Antiseptic, and CVS Antiseptic Mouth Wash — were less efficacious. Meyers said the reason remains unclear, but he and colleagues found the same result when they repeated the comparisons.
Timing of the essence?
Meyers and colleagues also tested a nasal rinse solution of 1% baby shampoo because it is sometimes used to treat people with chronic rhinosinusitis. They found 30 seconds led to < 90% to < 99.99% effectiveness, but that, by 2 minutes, efficacy climbed to > 99.9% to > 99.99%.
“Thirty seconds for some products just was not enough time for the efficacy to be observed,” Meyers said. “Whereas, after a minute or two the active ingredient had enough time to work. Thirty seconds may be at the border to see full efficacy.” More research is needed to confirm the timing and determine which active ingredients are driving the findings.
A future trial could test the efficacy of mouthwash products to reduce the viral load in people with COVID-19. “If we are able to get funding to continue, I would like to see a small clinical trial as the next step,” Meyers said.
Meyers and O’Donnell disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Brazil confirms death of volunteer in COVID-19 vaccine trial
The Brazilian National Health Surveillance Agency (Anvisa) announced Oct. 21 that it is investigating data received on the death of a volunteer in a clinical trial of the COVID-19 vaccine developed by Oxford University and the pharmaceutical company AstraZeneca.
In an email sent to Medscape Medical News, the agency states that it was formally informed of the death on October 19. It has already received data regarding the investigation of the case, which is now being conducted by the Brazilian International Security Assessment Committee.
The identity of the volunteer and cause of death have not yet been confirmed by any official source linked to the study. In the email, Anvisa reiterated that “according to national and international regulations on good clinical practices, data on clinical research volunteers must be kept confidential, in accordance with the principles of confidentiality, human dignity, and protection of participants.”
A report in the Brazilian newspaper O Globo, however, states that the patient who died is a 28-year-old doctor, recently graduated, who worked on the front line of combating COVID-19 in three hospitals in Rio de Janeiro. . Due to the study design, it is impossible to know whether the volunteer received the vaccine or placebo.
It is imperative to wait for the results of the investigations, said Sergio Cimerman, MD, the scientific coordinator of the Brazilian Society of Infectious Diseases (SBI), because death is possible during any vaccine trial, even more so in cases in which the final goal is to immunize the population in record time.
“It is precisely the phase 3 study that assesses efficacy and safety so that the vaccine can be used for the entire population. We cannot let ourselves lose hope, and we must move forward, as safely as possible, in search of an ideal vaccine,” said Cimerman, who works at the Instituto de Infectologia Emílio Ribas and is also an advisor to the Portuguese edition of Medscape.
This article was translated and adapted from the Portuguese edition of Medscape.
The Brazilian National Health Surveillance Agency (Anvisa) announced Oct. 21 that it is investigating data received on the death of a volunteer in a clinical trial of the COVID-19 vaccine developed by Oxford University and the pharmaceutical company AstraZeneca.
In an email sent to Medscape Medical News, the agency states that it was formally informed of the death on October 19. It has already received data regarding the investigation of the case, which is now being conducted by the Brazilian International Security Assessment Committee.
The identity of the volunteer and cause of death have not yet been confirmed by any official source linked to the study. In the email, Anvisa reiterated that “according to national and international regulations on good clinical practices, data on clinical research volunteers must be kept confidential, in accordance with the principles of confidentiality, human dignity, and protection of participants.”
A report in the Brazilian newspaper O Globo, however, states that the patient who died is a 28-year-old doctor, recently graduated, who worked on the front line of combating COVID-19 in three hospitals in Rio de Janeiro. . Due to the study design, it is impossible to know whether the volunteer received the vaccine or placebo.
It is imperative to wait for the results of the investigations, said Sergio Cimerman, MD, the scientific coordinator of the Brazilian Society of Infectious Diseases (SBI), because death is possible during any vaccine trial, even more so in cases in which the final goal is to immunize the population in record time.
“It is precisely the phase 3 study that assesses efficacy and safety so that the vaccine can be used for the entire population. We cannot let ourselves lose hope, and we must move forward, as safely as possible, in search of an ideal vaccine,” said Cimerman, who works at the Instituto de Infectologia Emílio Ribas and is also an advisor to the Portuguese edition of Medscape.
This article was translated and adapted from the Portuguese edition of Medscape.
The Brazilian National Health Surveillance Agency (Anvisa) announced Oct. 21 that it is investigating data received on the death of a volunteer in a clinical trial of the COVID-19 vaccine developed by Oxford University and the pharmaceutical company AstraZeneca.
In an email sent to Medscape Medical News, the agency states that it was formally informed of the death on October 19. It has already received data regarding the investigation of the case, which is now being conducted by the Brazilian International Security Assessment Committee.
The identity of the volunteer and cause of death have not yet been confirmed by any official source linked to the study. In the email, Anvisa reiterated that “according to national and international regulations on good clinical practices, data on clinical research volunteers must be kept confidential, in accordance with the principles of confidentiality, human dignity, and protection of participants.”
A report in the Brazilian newspaper O Globo, however, states that the patient who died is a 28-year-old doctor, recently graduated, who worked on the front line of combating COVID-19 in three hospitals in Rio de Janeiro. . Due to the study design, it is impossible to know whether the volunteer received the vaccine or placebo.
It is imperative to wait for the results of the investigations, said Sergio Cimerman, MD, the scientific coordinator of the Brazilian Society of Infectious Diseases (SBI), because death is possible during any vaccine trial, even more so in cases in which the final goal is to immunize the population in record time.
“It is precisely the phase 3 study that assesses efficacy and safety so that the vaccine can be used for the entire population. We cannot let ourselves lose hope, and we must move forward, as safely as possible, in search of an ideal vaccine,” said Cimerman, who works at the Instituto de Infectologia Emílio Ribas and is also an advisor to the Portuguese edition of Medscape.
This article was translated and adapted from the Portuguese edition of Medscape.