User login
Arthritis joint pain, inactivity vary greatly across U.S.
Almost 31% of the estimated 54 million adults in the United States with arthritis have severe joint pain, according to the Centers for Disease Control and Prevention.

Nationally, the prevalence of severe joint pain was 30.8% in adults with arthritis in 2017, but state-specific, age-standardized prevalences varied from a low of 20.8% in Colorado to 45.2% in Mississippi. Regionally, prevalences of both severe joint pain and physical inactivity in arthritis patients were highest in the Southeast, noted Dana Guglielmo, MPH, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and associates (MMWR 2019 May 3;68(17):381-7).
The prevalence of arthritis itself was lowest in the District of Columbia at 15.7% and highest in West Virginia at 34.6%. Alabama, at 30.4%, was the only other state above 30%. Colorado had the lowest physical inactivity rate (23.2%), while Kentucky had the highest (44.4%), the investigators said.
The differences among arthritis patients were demographic as well as geographic in 2017. The prevalence of severe joint pain was 33.0% among those aged 18-44 years and 35.6% in those 45-64 but only 25.1% in those aged 65 and older. Whites had a 27.4% prevalence of severe joint pain, compared with 42.0% for Hispanics and 50.9% for blacks. For arthritis patients with a college degree, the age-standardized prevalence of severe joint pain was 15.1%, compared with 35.5% for high school graduates and 54.1% for those with less than a high school degree, based on data from the Behavioral Risk Factor Surveillance System.
“Although persons with arthritis report that pain, or fear of causing or worsening it, is a substantial barrier to exercising, physical activity is an inexpensive intervention that can reduce pain, prevent or delay disability and limitations, and improve mental health, physical functioning, and quality of life with few adverse effects,” wrote Ms. Guglielmo and associates. Adults with severe joint pain “should engage in regular physical activity according to their abilities and avoid physical inactivity [since] even small amounts of physical activity can improve physical functioning in adults with joint conditions.”
SOURCE: Guglielmo D et al. MMWR 2019 May 3;68(17):381-7.
Almost 31% of the estimated 54 million adults in the United States with arthritis have severe joint pain, according to the Centers for Disease Control and Prevention.
Nationally, the prevalence of severe joint pain was 30.8% in adults with arthritis in 2017, but state-specific, age-standardized prevalences varied from a low of 20.8% in Colorado to 45.2% in Mississippi. Regionally, prevalences of both severe joint pain and physical inactivity in arthritis patients were highest in the Southeast, noted Dana Guglielmo, MPH, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and associates (MMWR 2019 May 3;68(17):381-7).
The prevalence of arthritis itself was lowest in the District of Columbia at 15.7% and highest in West Virginia at 34.6%. Alabama, at 30.4%, was the only other state above 30%. Colorado had the lowest physical inactivity rate (23.2%), while Kentucky had the highest (44.4%), the investigators said.
The differences among arthritis patients were demographic as well as geographic in 2017. The prevalence of severe joint pain was 33.0% among those aged 18-44 years and 35.6% in those 45-64 but only 25.1% in those aged 65 and older. Whites had a 27.4% prevalence of severe joint pain, compared with 42.0% for Hispanics and 50.9% for blacks. For arthritis patients with a college degree, the age-standardized prevalence of severe joint pain was 15.1%, compared with 35.5% for high school graduates and 54.1% for those with less than a high school degree, based on data from the Behavioral Risk Factor Surveillance System.
“Although persons with arthritis report that pain, or fear of causing or worsening it, is a substantial barrier to exercising, physical activity is an inexpensive intervention that can reduce pain, prevent or delay disability and limitations, and improve mental health, physical functioning, and quality of life with few adverse effects,” wrote Ms. Guglielmo and associates. Adults with severe joint pain “should engage in regular physical activity according to their abilities and avoid physical inactivity [since] even small amounts of physical activity can improve physical functioning in adults with joint conditions.”
SOURCE: Guglielmo D et al. MMWR 2019 May 3;68(17):381-7.
Almost 31% of the estimated 54 million adults in the United States with arthritis have severe joint pain, according to the Centers for Disease Control and Prevention.
Nationally, the prevalence of severe joint pain was 30.8% in adults with arthritis in 2017, but state-specific, age-standardized prevalences varied from a low of 20.8% in Colorado to 45.2% in Mississippi. Regionally, prevalences of both severe joint pain and physical inactivity in arthritis patients were highest in the Southeast, noted Dana Guglielmo, MPH, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and associates (MMWR 2019 May 3;68(17):381-7).
The prevalence of arthritis itself was lowest in the District of Columbia at 15.7% and highest in West Virginia at 34.6%. Alabama, at 30.4%, was the only other state above 30%. Colorado had the lowest physical inactivity rate (23.2%), while Kentucky had the highest (44.4%), the investigators said.
The differences among arthritis patients were demographic as well as geographic in 2017. The prevalence of severe joint pain was 33.0% among those aged 18-44 years and 35.6% in those 45-64 but only 25.1% in those aged 65 and older. Whites had a 27.4% prevalence of severe joint pain, compared with 42.0% for Hispanics and 50.9% for blacks. For arthritis patients with a college degree, the age-standardized prevalence of severe joint pain was 15.1%, compared with 35.5% for high school graduates and 54.1% for those with less than a high school degree, based on data from the Behavioral Risk Factor Surveillance System.
“Although persons with arthritis report that pain, or fear of causing or worsening it, is a substantial barrier to exercising, physical activity is an inexpensive intervention that can reduce pain, prevent or delay disability and limitations, and improve mental health, physical functioning, and quality of life with few adverse effects,” wrote Ms. Guglielmo and associates. Adults with severe joint pain “should engage in regular physical activity according to their abilities and avoid physical inactivity [since] even small amounts of physical activity can improve physical functioning in adults with joint conditions.”
SOURCE: Guglielmo D et al. MMWR 2019 May 3;68(17):381-7.
FROM MMWR
Benlysta approved for children with SLE

The B-lymphocyte stimulator–inhibitor called Benlysta already is approved for use in adults alongside standard therapy for SLE, and this approval marks the first such treatment available for children. Although there are regulatory submissions for use of this drug among children elsewhere, the United States is the first to approve its use among this age group, according to a press release from GSK. According to an FDA press announcement, the agency expedited the review and approval because belimumab could fulfill an unmet need.
The approval is based on a 1-year postapproval commitment study, which assessed efficacy, safety, and pharmacokinetics of 10 mg/kg belimumab plus standard therapy versus placebo plus standard therapy among children with SLE aged 5-11 years (n = 13) and those aged 12-17 years (n = 80). Although the study was not fully powered because of the rarity of the disease in this age group, it did find numerically higher SLE responder index response rates over 1 year among children treated with belimumab plus standard therapy (53%) than in those treated with placebo and standard therapy (44%).
Adverse reactions seen among this age group were consistent with those seen in adults, including nausea, diarrhea, pyrexia, nasopharyngitis, and bronchitis. The most common serious adverse reactions were serious infections. Belimumab has not been studied in combination with certain other drugs, such as other biologics or cyclophosphamide; therefore, combining it with such treatments is not recommended. Acute hypersensitivity reactions – including anaphylaxis and death – have been observed, even among patients who had previously tolerated belimumab.
Infusion reactions were common, so pretreat patients with an antihistamine. Also, do not administer the drug with live vaccines, the FDA noted.
More information can be found in the press announcement on the FDA website.
The B-lymphocyte stimulator–inhibitor called Benlysta already is approved for use in adults alongside standard therapy for SLE, and this approval marks the first such treatment available for children. Although there are regulatory submissions for use of this drug among children elsewhere, the United States is the first to approve its use among this age group, according to a press release from GSK. According to an FDA press announcement, the agency expedited the review and approval because belimumab could fulfill an unmet need.
The approval is based on a 1-year postapproval commitment study, which assessed efficacy, safety, and pharmacokinetics of 10 mg/kg belimumab plus standard therapy versus placebo plus standard therapy among children with SLE aged 5-11 years (n = 13) and those aged 12-17 years (n = 80). Although the study was not fully powered because of the rarity of the disease in this age group, it did find numerically higher SLE responder index response rates over 1 year among children treated with belimumab plus standard therapy (53%) than in those treated with placebo and standard therapy (44%).
Adverse reactions seen among this age group were consistent with those seen in adults, including nausea, diarrhea, pyrexia, nasopharyngitis, and bronchitis. The most common serious adverse reactions were serious infections. Belimumab has not been studied in combination with certain other drugs, such as other biologics or cyclophosphamide; therefore, combining it with such treatments is not recommended. Acute hypersensitivity reactions – including anaphylaxis and death – have been observed, even among patients who had previously tolerated belimumab.
Infusion reactions were common, so pretreat patients with an antihistamine. Also, do not administer the drug with live vaccines, the FDA noted.
More information can be found in the press announcement on the FDA website.
The B-lymphocyte stimulator–inhibitor called Benlysta already is approved for use in adults alongside standard therapy for SLE, and this approval marks the first such treatment available for children. Although there are regulatory submissions for use of this drug among children elsewhere, the United States is the first to approve its use among this age group, according to a press release from GSK. According to an FDA press announcement, the agency expedited the review and approval because belimumab could fulfill an unmet need.
The approval is based on a 1-year postapproval commitment study, which assessed efficacy, safety, and pharmacokinetics of 10 mg/kg belimumab plus standard therapy versus placebo plus standard therapy among children with SLE aged 5-11 years (n = 13) and those aged 12-17 years (n = 80). Although the study was not fully powered because of the rarity of the disease in this age group, it did find numerically higher SLE responder index response rates over 1 year among children treated with belimumab plus standard therapy (53%) than in those treated with placebo and standard therapy (44%).
Adverse reactions seen among this age group were consistent with those seen in adults, including nausea, diarrhea, pyrexia, nasopharyngitis, and bronchitis. The most common serious adverse reactions were serious infections. Belimumab has not been studied in combination with certain other drugs, such as other biologics or cyclophosphamide; therefore, combining it with such treatments is not recommended. Acute hypersensitivity reactions – including anaphylaxis and death – have been observed, even among patients who had previously tolerated belimumab.
Infusion reactions were common, so pretreat patients with an antihistamine. Also, do not administer the drug with live vaccines, the FDA noted.
More information can be found in the press announcement on the FDA website.
Twitter chat recap: Take-homes from LUPUS 2019
Despite negative trial findings, belimumab (Benlysta) remains a valid option for black lupus patients, so long as they have high disease activity and positive serology.

That was just one of the many useful messages from a robust question-and-answer session on Twitter April 23, about important findings from the recent LUPUS 2019 Congress in San Francisco. The Twitter chat was hosted by MDedge Rheumatology and led by Jinoos Yazdany, MD, and Gabriela Schmajuk, MD, both associate professors of rheumatology at the University of California, San Francisco (UCSF). The chat included scores of posts from over a dozen participants, most of them rheumatologists, and it’s worth a recap.
The EMBRACE trial

The belimumab EMBRACE trial was the first topic up to bat. The Food and Drug Administration ordered GlaxoSmithKline to conduct the trial as a condition of approval for lupus in 2011; phase 3 trials found no benefit among a small number of black subjects and even a suggestion of harm.
Although lupus is highly prevalent among black people, and outcomes are generally worse, EMBRACE was the first lupus trial to enroll an entirely black population; 345 patients were treated for a year at 10 mg/kg IV every 4 weeks. Inclusion criteria included disease activity scores of at least 8.
Overall, 49% of belimumab patients, and 42% on placebo, had a positive response, which meant a drop of 4 or more disease activity points, among other things. The difference was not statistically significant (P = .11).
However, GSK’s data showed a statistically significant benefit in favor of belimumab among people who entered with a disease activity score of at least 10 (53% vs. 41% for placebo), as well as for those with low complement levels (47% vs. 25%) and both anti–double stranded DNA antibodies and low complement (45% vs. 24%). Response rates were also significantly higher among the 57% of subjects who lived outside of the United States and Canada.
So what to make of the results?

“The EMBRACE efficacy data seems to reinforce what we already know: belimumab doesn’t work in a lot of patients, and it [has] the best chance of working in patients with higher baseline disease activity,” tweeted Dr. Yazdany.

As for prescribing, Sarah Patterson, MD, a postdoctoral fellow in the UCSF Division of Rheumatology, tweeted that the results “support the use of belimumab for black lupus patients with high disease activity” and positive serology.
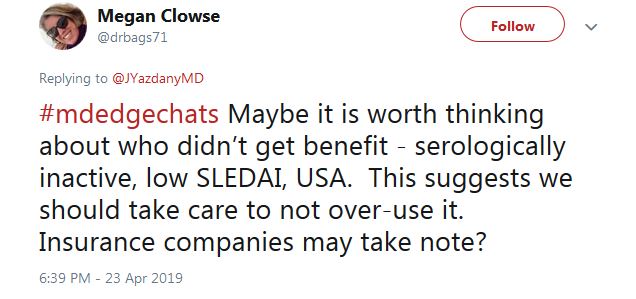
For those who don’t fit the treatment profile, “we should take care to not over-use it,” said Megan Clowse, MD, an associate professor of rheumatology at Duke University, Durham, N.C., in a tweet.
The HCQ adherence fail
Poor hydroxychloroquine (HCQ) adherence came up next on Twitter. The chat participants agreed it’s a huge problem, but no one knows why. Perhaps it’s because patients don’t feel a therapeutic effect or perhaps because GI problems and other side effects are worse than doctors think. Maybe there’s simply not enough social support to encourage people to stay on the drug, even though it’s the single most important medication in lupus.
A nine-study meta-analysis presented at LUPUS 2019 suggested a solution: blood levels. The odds of nonadherence were three times higher in patients below threshold HCQ levels, and although not statistically significant, the mean lupus disease activity index score was more than 3 points higher.
A rheumatologist on the chat said that he’s already checking them.
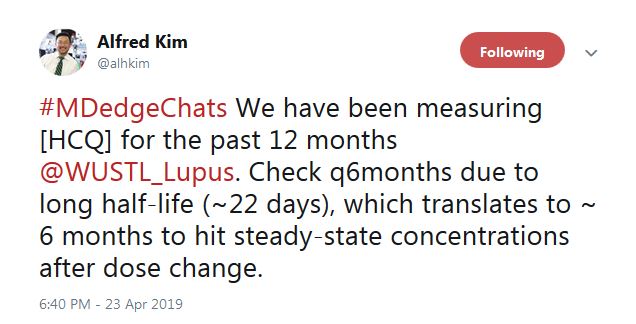
“We have been measuring HCQ for the past 12 months” at Washington University, St. Louis, according to Alfred Kim, MD, PhD, an assistant professor of rheumatology at the school. The data are just now coming in, he said, but it seems to be catching people.
That raised another question on the chat, however: What do you do with people who aren’t down with the program? They’ll be out the door and gone with the wrong words.
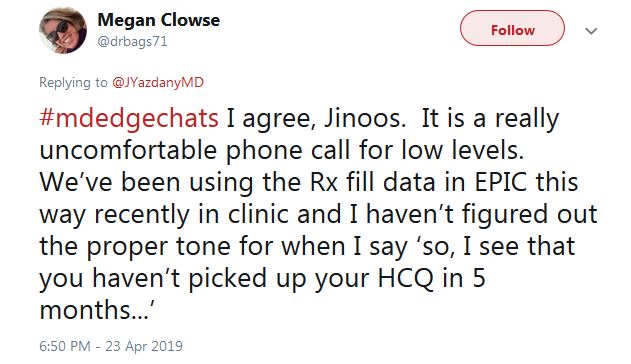
“I haven’t figured out the proper tone for when I say ‘So, I see that you haven’t picked up your HCQ in 5 months,’ ” tweeted Dr. Clowse. “It is a really uncomfortable phone call.”
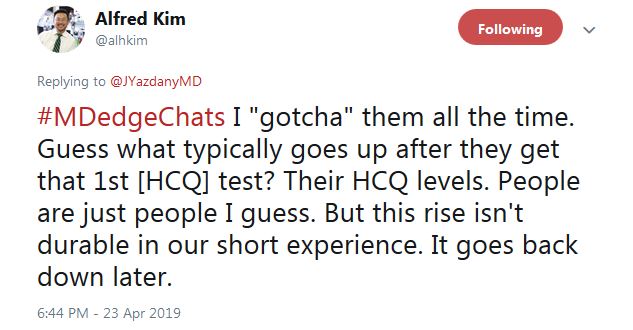
Dr. Kim said that “I ‘gotcha’ them all the time. Guess what typically goes up after they get that 1st HCQ test? Their HCQ levels ... But this rise isn’t durable in our short experience. It goes back down later.”
He tweeted that overall “this is an opportunity to educate patients on the benefits of HCQ ... Most actually do not [know] why they are taking this medication, in our experience.” In another tweet, Dr. Kim said “I tell them I care,” and that checking HCQ levels “is one way of demonstrating how I want to improve their outcomes.”
Tiffany from #LupusChat thought that it’s time for doctors to sit down with patient advocates and hash it out. She tweeted that “I truly feel this would be a positive 1st step ... a two-way conversation that’s non-judgmental and focused on improving” outcomes and quality of life.
Baricitinib for lupus?
The final topic was baricitinib (Olumiant).
There were modest indications of benefit at 4 mg/day oral after 6 months in a phase 2 trial with 314 people. There were also serious infections in 6% versus 2% on 2 mg/day and 1% on placebo. One patient in the 4-mg/day arm (1%) developed deep vein thrombosis (DVT), but they were positive for antiphospholipid antibodies, which raise the clot risk.
Baricitinib is FDA approved as second line at 2 mg/day for adult rheumatoid arthritis; there’s a black box warning of malignancies, thrombosis, and serious infections.

“I’m not sure” the risk-benefit is in the right direction for baricitinib in lupus. “There were a significant number of serious infections ... for not a lot of effect on” disease activity, tweeted the Twitter chat coleader, Dr. Schmajuk. In a separate tweet, she said that, even in nonlupus patients, “we are avoiding [Janus kinase inhibitors] in patients with DVT risk factors. If I were a patient, [I’m] not sure I would want to take the risk.”

“Future studies with larger sample size and longer follow up ... are needed to address some of the concerns related to DVT,” said Zahi Touma, MD, an assistant professor of rheumatology at the University of Toronto.
Despite negative trial findings, belimumab (Benlysta) remains a valid option for black lupus patients, so long as they have high disease activity and positive serology.
That was just one of the many useful messages from a robust question-and-answer session on Twitter April 23, about important findings from the recent LUPUS 2019 Congress in San Francisco. The Twitter chat was hosted by MDedge Rheumatology and led by Jinoos Yazdany, MD, and Gabriela Schmajuk, MD, both associate professors of rheumatology at the University of California, San Francisco (UCSF). The chat included scores of posts from over a dozen participants, most of them rheumatologists, and it’s worth a recap.
The EMBRACE trial
The belimumab EMBRACE trial was the first topic up to bat. The Food and Drug Administration ordered GlaxoSmithKline to conduct the trial as a condition of approval for lupus in 2011; phase 3 trials found no benefit among a small number of black subjects and even a suggestion of harm.
Although lupus is highly prevalent among black people, and outcomes are generally worse, EMBRACE was the first lupus trial to enroll an entirely black population; 345 patients were treated for a year at 10 mg/kg IV every 4 weeks. Inclusion criteria included disease activity scores of at least 8.
Overall, 49% of belimumab patients, and 42% on placebo, had a positive response, which meant a drop of 4 or more disease activity points, among other things. The difference was not statistically significant (P = .11).
However, GSK’s data showed a statistically significant benefit in favor of belimumab among people who entered with a disease activity score of at least 10 (53% vs. 41% for placebo), as well as for those with low complement levels (47% vs. 25%) and both anti–double stranded DNA antibodies and low complement (45% vs. 24%). Response rates were also significantly higher among the 57% of subjects who lived outside of the United States and Canada.
So what to make of the results?
“The EMBRACE efficacy data seems to reinforce what we already know: belimumab doesn’t work in a lot of patients, and it [has] the best chance of working in patients with higher baseline disease activity,” tweeted Dr. Yazdany.
As for prescribing, Sarah Patterson, MD, a postdoctoral fellow in the UCSF Division of Rheumatology, tweeted that the results “support the use of belimumab for black lupus patients with high disease activity” and positive serology.
For those who don’t fit the treatment profile, “we should take care to not over-use it,” said Megan Clowse, MD, an associate professor of rheumatology at Duke University, Durham, N.C., in a tweet.
The HCQ adherence fail
Poor hydroxychloroquine (HCQ) adherence came up next on Twitter. The chat participants agreed it’s a huge problem, but no one knows why. Perhaps it’s because patients don’t feel a therapeutic effect or perhaps because GI problems and other side effects are worse than doctors think. Maybe there’s simply not enough social support to encourage people to stay on the drug, even though it’s the single most important medication in lupus.
A nine-study meta-analysis presented at LUPUS 2019 suggested a solution: blood levels. The odds of nonadherence were three times higher in patients below threshold HCQ levels, and although not statistically significant, the mean lupus disease activity index score was more than 3 points higher.
A rheumatologist on the chat said that he’s already checking them.
“We have been measuring HCQ for the past 12 months” at Washington University, St. Louis, according to Alfred Kim, MD, PhD, an assistant professor of rheumatology at the school. The data are just now coming in, he said, but it seems to be catching people.
That raised another question on the chat, however: What do you do with people who aren’t down with the program? They’ll be out the door and gone with the wrong words.
“I haven’t figured out the proper tone for when I say ‘So, I see that you haven’t picked up your HCQ in 5 months,’ ” tweeted Dr. Clowse. “It is a really uncomfortable phone call.”
Dr. Kim said that “I ‘gotcha’ them all the time. Guess what typically goes up after they get that 1st HCQ test? Their HCQ levels ... But this rise isn’t durable in our short experience. It goes back down later.”
He tweeted that overall “this is an opportunity to educate patients on the benefits of HCQ ... Most actually do not [know] why they are taking this medication, in our experience.” In another tweet, Dr. Kim said “I tell them I care,” and that checking HCQ levels “is one way of demonstrating how I want to improve their outcomes.”
Tiffany from #LupusChat thought that it’s time for doctors to sit down with patient advocates and hash it out. She tweeted that “I truly feel this would be a positive 1st step ... a two-way conversation that’s non-judgmental and focused on improving” outcomes and quality of life.
Baricitinib for lupus?
The final topic was baricitinib (Olumiant).
There were modest indications of benefit at 4 mg/day oral after 6 months in a phase 2 trial with 314 people. There were also serious infections in 6% versus 2% on 2 mg/day and 1% on placebo. One patient in the 4-mg/day arm (1%) developed deep vein thrombosis (DVT), but they were positive for antiphospholipid antibodies, which raise the clot risk.
Baricitinib is FDA approved as second line at 2 mg/day for adult rheumatoid arthritis; there’s a black box warning of malignancies, thrombosis, and serious infections.
“I’m not sure” the risk-benefit is in the right direction for baricitinib in lupus. “There were a significant number of serious infections ... for not a lot of effect on” disease activity, tweeted the Twitter chat coleader, Dr. Schmajuk. In a separate tweet, she said that, even in nonlupus patients, “we are avoiding [Janus kinase inhibitors] in patients with DVT risk factors. If I were a patient, [I’m] not sure I would want to take the risk.”
“Future studies with larger sample size and longer follow up ... are needed to address some of the concerns related to DVT,” said Zahi Touma, MD, an assistant professor of rheumatology at the University of Toronto.
Despite negative trial findings, belimumab (Benlysta) remains a valid option for black lupus patients, so long as they have high disease activity and positive serology.
That was just one of the many useful messages from a robust question-and-answer session on Twitter April 23, about important findings from the recent LUPUS 2019 Congress in San Francisco. The Twitter chat was hosted by MDedge Rheumatology and led by Jinoos Yazdany, MD, and Gabriela Schmajuk, MD, both associate professors of rheumatology at the University of California, San Francisco (UCSF). The chat included scores of posts from over a dozen participants, most of them rheumatologists, and it’s worth a recap.
The EMBRACE trial
The belimumab EMBRACE trial was the first topic up to bat. The Food and Drug Administration ordered GlaxoSmithKline to conduct the trial as a condition of approval for lupus in 2011; phase 3 trials found no benefit among a small number of black subjects and even a suggestion of harm.
Although lupus is highly prevalent among black people, and outcomes are generally worse, EMBRACE was the first lupus trial to enroll an entirely black population; 345 patients were treated for a year at 10 mg/kg IV every 4 weeks. Inclusion criteria included disease activity scores of at least 8.
Overall, 49% of belimumab patients, and 42% on placebo, had a positive response, which meant a drop of 4 or more disease activity points, among other things. The difference was not statistically significant (P = .11).
However, GSK’s data showed a statistically significant benefit in favor of belimumab among people who entered with a disease activity score of at least 10 (53% vs. 41% for placebo), as well as for those with low complement levels (47% vs. 25%) and both anti–double stranded DNA antibodies and low complement (45% vs. 24%). Response rates were also significantly higher among the 57% of subjects who lived outside of the United States and Canada.
So what to make of the results?
“The EMBRACE efficacy data seems to reinforce what we already know: belimumab doesn’t work in a lot of patients, and it [has] the best chance of working in patients with higher baseline disease activity,” tweeted Dr. Yazdany.
As for prescribing, Sarah Patterson, MD, a postdoctoral fellow in the UCSF Division of Rheumatology, tweeted that the results “support the use of belimumab for black lupus patients with high disease activity” and positive serology.
For those who don’t fit the treatment profile, “we should take care to not over-use it,” said Megan Clowse, MD, an associate professor of rheumatology at Duke University, Durham, N.C., in a tweet.
The HCQ adherence fail
Poor hydroxychloroquine (HCQ) adherence came up next on Twitter. The chat participants agreed it’s a huge problem, but no one knows why. Perhaps it’s because patients don’t feel a therapeutic effect or perhaps because GI problems and other side effects are worse than doctors think. Maybe there’s simply not enough social support to encourage people to stay on the drug, even though it’s the single most important medication in lupus.
A nine-study meta-analysis presented at LUPUS 2019 suggested a solution: blood levels. The odds of nonadherence were three times higher in patients below threshold HCQ levels, and although not statistically significant, the mean lupus disease activity index score was more than 3 points higher.
A rheumatologist on the chat said that he’s already checking them.
“We have been measuring HCQ for the past 12 months” at Washington University, St. Louis, according to Alfred Kim, MD, PhD, an assistant professor of rheumatology at the school. The data are just now coming in, he said, but it seems to be catching people.
That raised another question on the chat, however: What do you do with people who aren’t down with the program? They’ll be out the door and gone with the wrong words.
“I haven’t figured out the proper tone for when I say ‘So, I see that you haven’t picked up your HCQ in 5 months,’ ” tweeted Dr. Clowse. “It is a really uncomfortable phone call.”
Dr. Kim said that “I ‘gotcha’ them all the time. Guess what typically goes up after they get that 1st HCQ test? Their HCQ levels ... But this rise isn’t durable in our short experience. It goes back down later.”
He tweeted that overall “this is an opportunity to educate patients on the benefits of HCQ ... Most actually do not [know] why they are taking this medication, in our experience.” In another tweet, Dr. Kim said “I tell them I care,” and that checking HCQ levels “is one way of demonstrating how I want to improve their outcomes.”
Tiffany from #LupusChat thought that it’s time for doctors to sit down with patient advocates and hash it out. She tweeted that “I truly feel this would be a positive 1st step ... a two-way conversation that’s non-judgmental and focused on improving” outcomes and quality of life.
Baricitinib for lupus?
The final topic was baricitinib (Olumiant).
There were modest indications of benefit at 4 mg/day oral after 6 months in a phase 2 trial with 314 people. There were also serious infections in 6% versus 2% on 2 mg/day and 1% on placebo. One patient in the 4-mg/day arm (1%) developed deep vein thrombosis (DVT), but they were positive for antiphospholipid antibodies, which raise the clot risk.
Baricitinib is FDA approved as second line at 2 mg/day for adult rheumatoid arthritis; there’s a black box warning of malignancies, thrombosis, and serious infections.
“I’m not sure” the risk-benefit is in the right direction for baricitinib in lupus. “There were a significant number of serious infections ... for not a lot of effect on” disease activity, tweeted the Twitter chat coleader, Dr. Schmajuk. In a separate tweet, she said that, even in nonlupus patients, “we are avoiding [Janus kinase inhibitors] in patients with DVT risk factors. If I were a patient, [I’m] not sure I would want to take the risk.”
“Future studies with larger sample size and longer follow up ... are needed to address some of the concerns related to DVT,” said Zahi Touma, MD, an assistant professor of rheumatology at the University of Toronto.
FROM LUPUS 2019
Measuring hydroxychloroquine blood levels could inform safe, optimal dosing
SAN FRANCISCO – , according to an investigation of the Hopkins Lupus Cohort, an ongoing longitudinal study of lupus patients in the Baltimore area.

As innocuous as the assertions seem, they are anything but. They directly contradict a 2014 investigation from Kaiser Permanente that put the retinopathy risk after 20 years at almost 40%; that finding led directly to an American Academy of Ophthalmology recommendation to reduce the maximum hydroxychloroquine dose from 6.5 mg/kg per day ideal weight to 5 mg/kg real weight, where it remains to this day.
Meanwhile, very few rheumatologists have access to hydroxychloroquine (HCQ) blood levels because most commercial labs don’t offer them. Plasma testing is widely available, but it’s nowhere near as good, according to Michelle Petri, MD, a rheumatology professor at Johns Hopkins University, Baltimore; director of the Hopkins Lupus Cohort; and a respected authority on lupus management.
“The Kaiser Permanente study was very worrisome,” she said. “I remember that I thought it didn’t fit my practice at all; I don’t see 40%. It made me even more concerned when the ophthalmologists” reduced the dose, “because hydroxychloroquine is the most important medicine I have for my lupus patients; it is the only one that improves survival. We don’t want to scare our patients into thinking that 40% of them are going to have retinopathy.”
Dueling studies
Dr. Petri’s concerns led her and her team to launch their own investigation; they followed 537 Baltimore cohort members on HCQ as they went through eye exams by Hopkins retinopathy specialists, often with optical coherence tomography (OCT). With a sensitivity of 93% and specificity of 84%, OCT is the best screening method available.
“We found that the risk of retinopathy is not nearly as high as Kaiser Permanente found,” just 11.46% (11/96) with 16-20 years of use, and 8% (6/75) with 21 or more years. On average, “the risk is probably about 10% after 16 or more years, not 40%,” Dr. Petri said at an international congress on systemic lupus erythematosus.
Patients with “possible” retinopathy were not included in the analysis.
When asked for comment, Ronald Melles, MD, a Kaiser ophthalmologist in Redwood City, Calif.; one of the two authors on the Kaiser study; and an author on the subsequent AAO recommendations, stood by his work.
“A rate of 12% retinopathy after 16 years of use ... seems right in line with what we found.” However, “the fact that the rate went down to 8%” after 20 years does not make sense; “the longer you are on the medicine, the more likely you would be to develop the toxicity,” he said.
Maybe the fluctuation had to do with the fact that there were only 75 patients in the Hopkins study on HCQ past 20 years, whereas “we looked at 2,361 patients, and 238 were on the medication for” 20 years or more. Patients over 5.0 mg/kg per day had a 5.67-fold higher risk”of retinopathy, he said (univariate analysis, P less than .001).
Dr. Petri wasn’t buying it. The across the board recommendation was made “without any recognition that if you reduce the dose, you reduce the benefit,” she said.
A new referee: blood levels
Dr. Petri and her team also found that HCQ blood levels correlated with retinopathy, and it was a direct relationship. Patients in the highest maximum tertile (1,753-6,281 ng/mL) had a retinopathy rate of 6.7%, a good deal higher than patients in lower tertiles. It was the same story with the highest mean tertile (1,117-3,513 ng/mL). Retinopathy in that group occurred in 7.9% versus 3.7% or less in lower tertiles. The findings were statistically significant.
Patients in the third tertile “are at the greatest risk, so I reduce their dose,” but “I do not want to reduce the dose across the board” to 5 mg/kg per day; that’s overreach. The tertile approach, if it pans out, might be a better way, she said.
The problem with plasma levels is that HCQ binds to red blood cells, so plasma levels are artificially low and do not indicate the true HCQ load. For now, just one company in the United States offers HCQ blood levels: Exagen.
“We have to get the big companies to start offering” this, and “I want rheumatologists to adopt it. I am lucky at Hopkins [because] we have our own homegrown blood level assay,” she said.
Dr. Melles agreed that tracking blood level makes sense, “but the literature I am aware of has not been able to closely correlate either lupus disease activity or retinal toxicity with blood levels. Also, we have seen some patients at lower doses develop toxicity and other patients on higher doses without any detectable changes.”
Still, “we would like to see [this] studied more, perhaps with newer analytic methods,” said his coauthor on the Kaiser study, and also the lead author on the AAO guidelines, Michael Marmor, MD, professor emeritus of ophthalmology at Stanford (Calif.) University.
In the end, on the same team
Dr. Petri said there is interest among some of her fellow members of the American College of Rheumatology to work with AAO to revise the guidelines. “Until then,” she said, “I want the ophthalmologists to withdraw” them.
She’s worried about undertreatment and believes that the previous AAO guideline, up to 6.5 mg/kg per day ideal weight, was fine, “with some understanding that there are high-risk groups, such as the elderly and people with renal impairment, where the dose should be reduced.”
“No matter how obese a patient is, I cap it at 400 mg/day,” she said, and, with the luxury of HCQ blood level testing, “no matter the weight, if the person is in the upper tertile, I reduce the dose.”

Dr. Marmor agreed that “if rheumatologists prescribe 5 mg/kg real weight and do not stress compliance, some patients may indeed be underdosed.”
“However, that is a fault of the doctor and patient relationship,” he said, “not the guideline; we do not feel it ethical to prescribe higher doses which could increase toxicity in reliable patients ... just because some patients might be unreliable.”
Overall, “I have not heard complaints from rheumatologists in our area, who try hard to follow the current recommendations. ... any doctor can use the dose he or she feels is necessary for a patient. Several recent reports [also] suggest the incidence of toxicity is falling now with usage of AAO guidelines, [and] I am not aware of any data” showing that management has become less effective, he said.
In the meantime, “I assure you that AAO wants ... to serve both specialties, and will change the guidelines when there is new, defensible data,” he added.
The Hopkins team found that the risk of HCQ retinopathy was highest in men and white patients, as well as older people. Body mass index and hypertension also predicted retina issues.
“As screening tests are frequently abnormal due to causes other than HCQ ... stopping [it] based on an abnormal test without confirmation from a retinopathy expert could needlessly deprive an SLE patient of an important medication,” they said.
The Hopkins Lupus Cohort is funded by the National Institutes of Health. The physicians didn’t have any relevant disclosures.
SOURCES: Petri M et al. Lupus Sci Med. 2019;6(suppl 1). Abstracts 15 and 16.
SAN FRANCISCO – , according to an investigation of the Hopkins Lupus Cohort, an ongoing longitudinal study of lupus patients in the Baltimore area.
As innocuous as the assertions seem, they are anything but. They directly contradict a 2014 investigation from Kaiser Permanente that put the retinopathy risk after 20 years at almost 40%; that finding led directly to an American Academy of Ophthalmology recommendation to reduce the maximum hydroxychloroquine dose from 6.5 mg/kg per day ideal weight to 5 mg/kg real weight, where it remains to this day.
Meanwhile, very few rheumatologists have access to hydroxychloroquine (HCQ) blood levels because most commercial labs don’t offer them. Plasma testing is widely available, but it’s nowhere near as good, according to Michelle Petri, MD, a rheumatology professor at Johns Hopkins University, Baltimore; director of the Hopkins Lupus Cohort; and a respected authority on lupus management.
“The Kaiser Permanente study was very worrisome,” she said. “I remember that I thought it didn’t fit my practice at all; I don’t see 40%. It made me even more concerned when the ophthalmologists” reduced the dose, “because hydroxychloroquine is the most important medicine I have for my lupus patients; it is the only one that improves survival. We don’t want to scare our patients into thinking that 40% of them are going to have retinopathy.”
Dueling studies
Dr. Petri’s concerns led her and her team to launch their own investigation; they followed 537 Baltimore cohort members on HCQ as they went through eye exams by Hopkins retinopathy specialists, often with optical coherence tomography (OCT). With a sensitivity of 93% and specificity of 84%, OCT is the best screening method available.
“We found that the risk of retinopathy is not nearly as high as Kaiser Permanente found,” just 11.46% (11/96) with 16-20 years of use, and 8% (6/75) with 21 or more years. On average, “the risk is probably about 10% after 16 or more years, not 40%,” Dr. Petri said at an international congress on systemic lupus erythematosus.
Patients with “possible” retinopathy were not included in the analysis.
When asked for comment, Ronald Melles, MD, a Kaiser ophthalmologist in Redwood City, Calif.; one of the two authors on the Kaiser study; and an author on the subsequent AAO recommendations, stood by his work.
“A rate of 12% retinopathy after 16 years of use ... seems right in line with what we found.” However, “the fact that the rate went down to 8%” after 20 years does not make sense; “the longer you are on the medicine, the more likely you would be to develop the toxicity,” he said.
Maybe the fluctuation had to do with the fact that there were only 75 patients in the Hopkins study on HCQ past 20 years, whereas “we looked at 2,361 patients, and 238 were on the medication for” 20 years or more. Patients over 5.0 mg/kg per day had a 5.67-fold higher risk”of retinopathy, he said (univariate analysis, P less than .001).
Dr. Petri wasn’t buying it. The across the board recommendation was made “without any recognition that if you reduce the dose, you reduce the benefit,” she said.
A new referee: blood levels
Dr. Petri and her team also found that HCQ blood levels correlated with retinopathy, and it was a direct relationship. Patients in the highest maximum tertile (1,753-6,281 ng/mL) had a retinopathy rate of 6.7%, a good deal higher than patients in lower tertiles. It was the same story with the highest mean tertile (1,117-3,513 ng/mL). Retinopathy in that group occurred in 7.9% versus 3.7% or less in lower tertiles. The findings were statistically significant.
Patients in the third tertile “are at the greatest risk, so I reduce their dose,” but “I do not want to reduce the dose across the board” to 5 mg/kg per day; that’s overreach. The tertile approach, if it pans out, might be a better way, she said.
The problem with plasma levels is that HCQ binds to red blood cells, so plasma levels are artificially low and do not indicate the true HCQ load. For now, just one company in the United States offers HCQ blood levels: Exagen.
“We have to get the big companies to start offering” this, and “I want rheumatologists to adopt it. I am lucky at Hopkins [because] we have our own homegrown blood level assay,” she said.
Dr. Melles agreed that tracking blood level makes sense, “but the literature I am aware of has not been able to closely correlate either lupus disease activity or retinal toxicity with blood levels. Also, we have seen some patients at lower doses develop toxicity and other patients on higher doses without any detectable changes.”
Still, “we would like to see [this] studied more, perhaps with newer analytic methods,” said his coauthor on the Kaiser study, and also the lead author on the AAO guidelines, Michael Marmor, MD, professor emeritus of ophthalmology at Stanford (Calif.) University.
In the end, on the same team
Dr. Petri said there is interest among some of her fellow members of the American College of Rheumatology to work with AAO to revise the guidelines. “Until then,” she said, “I want the ophthalmologists to withdraw” them.
She’s worried about undertreatment and believes that the previous AAO guideline, up to 6.5 mg/kg per day ideal weight, was fine, “with some understanding that there are high-risk groups, such as the elderly and people with renal impairment, where the dose should be reduced.”
“No matter how obese a patient is, I cap it at 400 mg/day,” she said, and, with the luxury of HCQ blood level testing, “no matter the weight, if the person is in the upper tertile, I reduce the dose.”
Dr. Marmor agreed that “if rheumatologists prescribe 5 mg/kg real weight and do not stress compliance, some patients may indeed be underdosed.”
“However, that is a fault of the doctor and patient relationship,” he said, “not the guideline; we do not feel it ethical to prescribe higher doses which could increase toxicity in reliable patients ... just because some patients might be unreliable.”
Overall, “I have not heard complaints from rheumatologists in our area, who try hard to follow the current recommendations. ... any doctor can use the dose he or she feels is necessary for a patient. Several recent reports [also] suggest the incidence of toxicity is falling now with usage of AAO guidelines, [and] I am not aware of any data” showing that management has become less effective, he said.
In the meantime, “I assure you that AAO wants ... to serve both specialties, and will change the guidelines when there is new, defensible data,” he added.
The Hopkins team found that the risk of HCQ retinopathy was highest in men and white patients, as well as older people. Body mass index and hypertension also predicted retina issues.
“As screening tests are frequently abnormal due to causes other than HCQ ... stopping [it] based on an abnormal test without confirmation from a retinopathy expert could needlessly deprive an SLE patient of an important medication,” they said.
The Hopkins Lupus Cohort is funded by the National Institutes of Health. The physicians didn’t have any relevant disclosures.
SOURCES: Petri M et al. Lupus Sci Med. 2019;6(suppl 1). Abstracts 15 and 16.
SAN FRANCISCO – , according to an investigation of the Hopkins Lupus Cohort, an ongoing longitudinal study of lupus patients in the Baltimore area.
As innocuous as the assertions seem, they are anything but. They directly contradict a 2014 investigation from Kaiser Permanente that put the retinopathy risk after 20 years at almost 40%; that finding led directly to an American Academy of Ophthalmology recommendation to reduce the maximum hydroxychloroquine dose from 6.5 mg/kg per day ideal weight to 5 mg/kg real weight, where it remains to this day.
Meanwhile, very few rheumatologists have access to hydroxychloroquine (HCQ) blood levels because most commercial labs don’t offer them. Plasma testing is widely available, but it’s nowhere near as good, according to Michelle Petri, MD, a rheumatology professor at Johns Hopkins University, Baltimore; director of the Hopkins Lupus Cohort; and a respected authority on lupus management.
“The Kaiser Permanente study was very worrisome,” she said. “I remember that I thought it didn’t fit my practice at all; I don’t see 40%. It made me even more concerned when the ophthalmologists” reduced the dose, “because hydroxychloroquine is the most important medicine I have for my lupus patients; it is the only one that improves survival. We don’t want to scare our patients into thinking that 40% of them are going to have retinopathy.”
Dueling studies
Dr. Petri’s concerns led her and her team to launch their own investigation; they followed 537 Baltimore cohort members on HCQ as they went through eye exams by Hopkins retinopathy specialists, often with optical coherence tomography (OCT). With a sensitivity of 93% and specificity of 84%, OCT is the best screening method available.
“We found that the risk of retinopathy is not nearly as high as Kaiser Permanente found,” just 11.46% (11/96) with 16-20 years of use, and 8% (6/75) with 21 or more years. On average, “the risk is probably about 10% after 16 or more years, not 40%,” Dr. Petri said at an international congress on systemic lupus erythematosus.
Patients with “possible” retinopathy were not included in the analysis.
When asked for comment, Ronald Melles, MD, a Kaiser ophthalmologist in Redwood City, Calif.; one of the two authors on the Kaiser study; and an author on the subsequent AAO recommendations, stood by his work.
“A rate of 12% retinopathy after 16 years of use ... seems right in line with what we found.” However, “the fact that the rate went down to 8%” after 20 years does not make sense; “the longer you are on the medicine, the more likely you would be to develop the toxicity,” he said.
Maybe the fluctuation had to do with the fact that there were only 75 patients in the Hopkins study on HCQ past 20 years, whereas “we looked at 2,361 patients, and 238 were on the medication for” 20 years or more. Patients over 5.0 mg/kg per day had a 5.67-fold higher risk”of retinopathy, he said (univariate analysis, P less than .001).
Dr. Petri wasn’t buying it. The across the board recommendation was made “without any recognition that if you reduce the dose, you reduce the benefit,” she said.
A new referee: blood levels
Dr. Petri and her team also found that HCQ blood levels correlated with retinopathy, and it was a direct relationship. Patients in the highest maximum tertile (1,753-6,281 ng/mL) had a retinopathy rate of 6.7%, a good deal higher than patients in lower tertiles. It was the same story with the highest mean tertile (1,117-3,513 ng/mL). Retinopathy in that group occurred in 7.9% versus 3.7% or less in lower tertiles. The findings were statistically significant.
Patients in the third tertile “are at the greatest risk, so I reduce their dose,” but “I do not want to reduce the dose across the board” to 5 mg/kg per day; that’s overreach. The tertile approach, if it pans out, might be a better way, she said.
The problem with plasma levels is that HCQ binds to red blood cells, so plasma levels are artificially low and do not indicate the true HCQ load. For now, just one company in the United States offers HCQ blood levels: Exagen.
“We have to get the big companies to start offering” this, and “I want rheumatologists to adopt it. I am lucky at Hopkins [because] we have our own homegrown blood level assay,” she said.
Dr. Melles agreed that tracking blood level makes sense, “but the literature I am aware of has not been able to closely correlate either lupus disease activity or retinal toxicity with blood levels. Also, we have seen some patients at lower doses develop toxicity and other patients on higher doses without any detectable changes.”
Still, “we would like to see [this] studied more, perhaps with newer analytic methods,” said his coauthor on the Kaiser study, and also the lead author on the AAO guidelines, Michael Marmor, MD, professor emeritus of ophthalmology at Stanford (Calif.) University.
In the end, on the same team
Dr. Petri said there is interest among some of her fellow members of the American College of Rheumatology to work with AAO to revise the guidelines. “Until then,” she said, “I want the ophthalmologists to withdraw” them.
She’s worried about undertreatment and believes that the previous AAO guideline, up to 6.5 mg/kg per day ideal weight, was fine, “with some understanding that there are high-risk groups, such as the elderly and people with renal impairment, where the dose should be reduced.”
“No matter how obese a patient is, I cap it at 400 mg/day,” she said, and, with the luxury of HCQ blood level testing, “no matter the weight, if the person is in the upper tertile, I reduce the dose.”
Dr. Marmor agreed that “if rheumatologists prescribe 5 mg/kg real weight and do not stress compliance, some patients may indeed be underdosed.”
“However, that is a fault of the doctor and patient relationship,” he said, “not the guideline; we do not feel it ethical to prescribe higher doses which could increase toxicity in reliable patients ... just because some patients might be unreliable.”
Overall, “I have not heard complaints from rheumatologists in our area, who try hard to follow the current recommendations. ... any doctor can use the dose he or she feels is necessary for a patient. Several recent reports [also] suggest the incidence of toxicity is falling now with usage of AAO guidelines, [and] I am not aware of any data” showing that management has become less effective, he said.
In the meantime, “I assure you that AAO wants ... to serve both specialties, and will change the guidelines when there is new, defensible data,” he added.
The Hopkins team found that the risk of HCQ retinopathy was highest in men and white patients, as well as older people. Body mass index and hypertension also predicted retina issues.
“As screening tests are frequently abnormal due to causes other than HCQ ... stopping [it] based on an abnormal test without confirmation from a retinopathy expert could needlessly deprive an SLE patient of an important medication,” they said.
The Hopkins Lupus Cohort is funded by the National Institutes of Health. The physicians didn’t have any relevant disclosures.
SOURCES: Petri M et al. Lupus Sci Med. 2019;6(suppl 1). Abstracts 15 and 16.
REPORTING FROM LUPUS 2019
Studies begin to pinpoint ways to diagnose SLE earlier
SAN FRANCISCO – A host of novel clinical and serologic findings that physicians can put to good use right now in helping to distinguish early SLE from its many mimickers have been identified in a large study conducted on four continents, Marta Mosca, MD, PhD, observed at an international congress on systemic lupus erythematosus.

These useful findings aren’t incorporated into the current American College of Rheumatology (ACR) or Systemic Lupus International Collaborating Clinics (SLICC) lupus classification criteria, which have come under criticism for limited sensitivity in identifying early SLE. Some of the novel findings provide support for increased suspicion of early SLE, while others suggest a need to veer in another direction and assess a patient for a disease other than lupus. The study has served to provide input for the proposed new ACR/EULAR weighted SLE classification criteria, although that scheme is meant to be used only for research and not in clinical practice, explained Dr. Mosca of the University of Pisa (Italy).
She was lead author of the four-continent study, which included 616 patients referred to experienced academic lupus centers for possible SLE with a symptom duration of less than 1 year. During up to 3 years of follow-up, 389 patients were diagnosed as having SLE by experienced rheumatologists based upon their clinical judgment, without any requirement to meet the full ACR or SLICC classification criteria. The other 227 patients were determined to be SLE mimickers with conditions including lymphoma, Sjögren’s syndrome, systemic sclerosis, interstitial lung disease, fibromyalgia, antinuclear antibody–positive thyroiditis, and undifferentiated connective tissue disease.
Dr. Mosca also highlighted key recent work by other investigators aimed at speeding the diagnosis of SLE and shortening the duration of what she called “the gray zone” of diagnostic uncertainty, when autoantibodies and insidious symptoms are present but not yet sufficient to make the diagnosis of SLE by conventional criteria. It’s well established that 60%-70% of patients with mild undifferentiated connective tissue disease will remain stable without evolving into SLE during long years of follow-up.
The ultimate objective of all this work is to try to change the natural history of the disease through targeted early aggressive therapy aimed at minimizing the extent of active disease and preventing severe organ involvement.
Among the key takeaways from the four-continent study led by Dr. Mosca: Fever not related to infection was far more prevalent in early SLE than in mimicking conditions, by a margin of 34.5% versus 13.7%. On the other hand, Raynaud’s phenomenon was more than twice as prevalent among patients with mimicking conditions: 22.1% in early SLE, compared with 48.5% in SLE mimickers. Sicca symptoms were present in just 4.4% of early SLE patients versus 34.4% of SLE mimickers. Only 0.3% of early SLE patients complained of dysphagia; the rate was 20-fold higher in the SLE mimickers. Rashes atypical for lupus were twice as frequent in the SLE mimicking conditions (Arthritis Rheumatol. 2019 Jan;71[1]:91-8).
Turning to key differentiating serologic findings, Dr. Mosca noted that anti-double stranded DNA (anti-dsDNA) and anti-Sm antibodies were present in 71.7% and 30.2% of early SLE patients, respectively, compared with 6.9% and 2.6% of SLE mimickers. Anticardiolipin IgM, a positive Coombs test, anti-beta2 glycoprotein-I antibodies, leukopenia, autoimmune hemolytic anemia, and hypocomplementemia were all significantly more common in the early SLE cohort.
In contrast, antibodies to Ro (SS-A) and La (SS-B) were of no value in separating early SLE from its mimickers, according to Dr. Mosca.
Other tipoffs to early SLE
Two separate teams of British researchers have advanced the field in a highly practical way. One group showed in a study of 1,739 newly diagnosed SLE patients and 6,956 controls that in the 5 years prior to diagnosis, the SLE group averaged 9.2 primary care visits per year, compared with 3.8 for controls. The visits clustered around nonspecific complaints of arthritis and arthralgias, alopecia, and rash (Arthritis Care Res. 2017 Jun;69[6]:833-41).
“An accumulating number of primary care office visits and referrals over time should raise suspicion,” Dr. Mosca said.
Other investigators, working with 1,426 cases of newly diagnosed SLE in the U.K. Clinical Practice Research Database, observed that the proportion of patients with disease manifestations in three or more British Isles Lupus Activity Group (BILAG) symptom domains rose from 18.7% at 3 years prior to diagnosis to 39.7% in the year before diagnosis (Lupus Sci Med. 2017 Feb 10;4[1]:e000172. doi: 10.1136/lupus-2016-000172).
“These patients accrue clinical manifestations. It’s not just one symptom, it’s more of a state of being unwell. This is a suspicious factor for the development of lupus,” she continued.
And just as patients who will eventually be diagnosed with SLE accrue a growing number of signs and symptoms during the run up to diagnosis, they also accrue multiple autoantibodies. Moreover, as demonstrated by a multicenter group of U.S. investigators, patients also develop elevated levels of multiple soluble inflammatory markers more than 3.5 years prior to diagnosis of SLE. These include interleukins-5 and -6 and interferon-gamma. And less than 10 months prior to being classified as having SLE, patients develop significantly higher levels of B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand known as APRIL. The investigators developed a predictive model incorporating IL-5, -6, and interferon-gamma levels with antinuclear antibody status that identified future SLE patients with 84% accuracy more than 3.5 years before diagnosis. This could prove useful in selecting high-risk patients for clinical prevention trials (J Autoimmun. 2016 Nov;74:182-93).
Researchers at the University of Leeds (England) have also zeroed in on interferon activity as playing a key role in progression from asymptomatic antinuclear antigen positivity, which is present in up to 25% of the general population, to symptomatic autoimmune connective tissue disease, which affects less than 1%. A multivariate logistic regression analysis identified two independent predictors of development of autoimmune connective tissue disease within the next 12 months: a family history of autoimmune rheumatic disease, which was associated with an 8.2-fold increased risk; and positivity for a pattern of interferon-stimulated gene activity they call IFN-Score-B (Ann Rheum Dis. 2018 Oct;77[10]:1432-9).
All of this work has led up to what Dr. Mosca called “a glance into the future”: the National Institutes of Health–supported Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE), which is now recruiting patients. This randomized, double-blind, placebo-controlled multicenter U.S. trial involves patients who are antinuclear antibody positive at a titer of 1:80 or more plus one or two additional criteria from the SLICC classification scheme. Participants are being randomized to 96 weeks of hydroxychloroquine or placebo. The goal is to learn whether hydroxychloroquine can slow disease progression, with the primary endpoint being the number of SLICC criteria met at the study’s end. The trial will also scrutinize potential biomarkers that could be used to guide treatment decisions (Trials. 2018 Dec 20;19[1]:694. doi: 10.1186/s13063-018-3076-7).
Dr. Mosca reported serving as an adviser to UCB and Lilly.
SAN FRANCISCO – A host of novel clinical and serologic findings that physicians can put to good use right now in helping to distinguish early SLE from its many mimickers have been identified in a large study conducted on four continents, Marta Mosca, MD, PhD, observed at an international congress on systemic lupus erythematosus.
These useful findings aren’t incorporated into the current American College of Rheumatology (ACR) or Systemic Lupus International Collaborating Clinics (SLICC) lupus classification criteria, which have come under criticism for limited sensitivity in identifying early SLE. Some of the novel findings provide support for increased suspicion of early SLE, while others suggest a need to veer in another direction and assess a patient for a disease other than lupus. The study has served to provide input for the proposed new ACR/EULAR weighted SLE classification criteria, although that scheme is meant to be used only for research and not in clinical practice, explained Dr. Mosca of the University of Pisa (Italy).
She was lead author of the four-continent study, which included 616 patients referred to experienced academic lupus centers for possible SLE with a symptom duration of less than 1 year. During up to 3 years of follow-up, 389 patients were diagnosed as having SLE by experienced rheumatologists based upon their clinical judgment, without any requirement to meet the full ACR or SLICC classification criteria. The other 227 patients were determined to be SLE mimickers with conditions including lymphoma, Sjögren’s syndrome, systemic sclerosis, interstitial lung disease, fibromyalgia, antinuclear antibody–positive thyroiditis, and undifferentiated connective tissue disease.
Dr. Mosca also highlighted key recent work by other investigators aimed at speeding the diagnosis of SLE and shortening the duration of what she called “the gray zone” of diagnostic uncertainty, when autoantibodies and insidious symptoms are present but not yet sufficient to make the diagnosis of SLE by conventional criteria. It’s well established that 60%-70% of patients with mild undifferentiated connective tissue disease will remain stable without evolving into SLE during long years of follow-up.
The ultimate objective of all this work is to try to change the natural history of the disease through targeted early aggressive therapy aimed at minimizing the extent of active disease and preventing severe organ involvement.
Among the key takeaways from the four-continent study led by Dr. Mosca: Fever not related to infection was far more prevalent in early SLE than in mimicking conditions, by a margin of 34.5% versus 13.7%. On the other hand, Raynaud’s phenomenon was more than twice as prevalent among patients with mimicking conditions: 22.1% in early SLE, compared with 48.5% in SLE mimickers. Sicca symptoms were present in just 4.4% of early SLE patients versus 34.4% of SLE mimickers. Only 0.3% of early SLE patients complained of dysphagia; the rate was 20-fold higher in the SLE mimickers. Rashes atypical for lupus were twice as frequent in the SLE mimicking conditions (Arthritis Rheumatol. 2019 Jan;71[1]:91-8).
Turning to key differentiating serologic findings, Dr. Mosca noted that anti-double stranded DNA (anti-dsDNA) and anti-Sm antibodies were present in 71.7% and 30.2% of early SLE patients, respectively, compared with 6.9% and 2.6% of SLE mimickers. Anticardiolipin IgM, a positive Coombs test, anti-beta2 glycoprotein-I antibodies, leukopenia, autoimmune hemolytic anemia, and hypocomplementemia were all significantly more common in the early SLE cohort.
In contrast, antibodies to Ro (SS-A) and La (SS-B) were of no value in separating early SLE from its mimickers, according to Dr. Mosca.
Other tipoffs to early SLE
Two separate teams of British researchers have advanced the field in a highly practical way. One group showed in a study of 1,739 newly diagnosed SLE patients and 6,956 controls that in the 5 years prior to diagnosis, the SLE group averaged 9.2 primary care visits per year, compared with 3.8 for controls. The visits clustered around nonspecific complaints of arthritis and arthralgias, alopecia, and rash (Arthritis Care Res. 2017 Jun;69[6]:833-41).
“An accumulating number of primary care office visits and referrals over time should raise suspicion,” Dr. Mosca said.
Other investigators, working with 1,426 cases of newly diagnosed SLE in the U.K. Clinical Practice Research Database, observed that the proportion of patients with disease manifestations in three or more British Isles Lupus Activity Group (BILAG) symptom domains rose from 18.7% at 3 years prior to diagnosis to 39.7% in the year before diagnosis (Lupus Sci Med. 2017 Feb 10;4[1]:e000172. doi: 10.1136/lupus-2016-000172).
“These patients accrue clinical manifestations. It’s not just one symptom, it’s more of a state of being unwell. This is a suspicious factor for the development of lupus,” she continued.
And just as patients who will eventually be diagnosed with SLE accrue a growing number of signs and symptoms during the run up to diagnosis, they also accrue multiple autoantibodies. Moreover, as demonstrated by a multicenter group of U.S. investigators, patients also develop elevated levels of multiple soluble inflammatory markers more than 3.5 years prior to diagnosis of SLE. These include interleukins-5 and -6 and interferon-gamma. And less than 10 months prior to being classified as having SLE, patients develop significantly higher levels of B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand known as APRIL. The investigators developed a predictive model incorporating IL-5, -6, and interferon-gamma levels with antinuclear antibody status that identified future SLE patients with 84% accuracy more than 3.5 years before diagnosis. This could prove useful in selecting high-risk patients for clinical prevention trials (J Autoimmun. 2016 Nov;74:182-93).
Researchers at the University of Leeds (England) have also zeroed in on interferon activity as playing a key role in progression from asymptomatic antinuclear antigen positivity, which is present in up to 25% of the general population, to symptomatic autoimmune connective tissue disease, which affects less than 1%. A multivariate logistic regression analysis identified two independent predictors of development of autoimmune connective tissue disease within the next 12 months: a family history of autoimmune rheumatic disease, which was associated with an 8.2-fold increased risk; and positivity for a pattern of interferon-stimulated gene activity they call IFN-Score-B (Ann Rheum Dis. 2018 Oct;77[10]:1432-9).
All of this work has led up to what Dr. Mosca called “a glance into the future”: the National Institutes of Health–supported Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE), which is now recruiting patients. This randomized, double-blind, placebo-controlled multicenter U.S. trial involves patients who are antinuclear antibody positive at a titer of 1:80 or more plus one or two additional criteria from the SLICC classification scheme. Participants are being randomized to 96 weeks of hydroxychloroquine or placebo. The goal is to learn whether hydroxychloroquine can slow disease progression, with the primary endpoint being the number of SLICC criteria met at the study’s end. The trial will also scrutinize potential biomarkers that could be used to guide treatment decisions (Trials. 2018 Dec 20;19[1]:694. doi: 10.1186/s13063-018-3076-7).
Dr. Mosca reported serving as an adviser to UCB and Lilly.
SAN FRANCISCO – A host of novel clinical and serologic findings that physicians can put to good use right now in helping to distinguish early SLE from its many mimickers have been identified in a large study conducted on four continents, Marta Mosca, MD, PhD, observed at an international congress on systemic lupus erythematosus.
These useful findings aren’t incorporated into the current American College of Rheumatology (ACR) or Systemic Lupus International Collaborating Clinics (SLICC) lupus classification criteria, which have come under criticism for limited sensitivity in identifying early SLE. Some of the novel findings provide support for increased suspicion of early SLE, while others suggest a need to veer in another direction and assess a patient for a disease other than lupus. The study has served to provide input for the proposed new ACR/EULAR weighted SLE classification criteria, although that scheme is meant to be used only for research and not in clinical practice, explained Dr. Mosca of the University of Pisa (Italy).
She was lead author of the four-continent study, which included 616 patients referred to experienced academic lupus centers for possible SLE with a symptom duration of less than 1 year. During up to 3 years of follow-up, 389 patients were diagnosed as having SLE by experienced rheumatologists based upon their clinical judgment, without any requirement to meet the full ACR or SLICC classification criteria. The other 227 patients were determined to be SLE mimickers with conditions including lymphoma, Sjögren’s syndrome, systemic sclerosis, interstitial lung disease, fibromyalgia, antinuclear antibody–positive thyroiditis, and undifferentiated connective tissue disease.
Dr. Mosca also highlighted key recent work by other investigators aimed at speeding the diagnosis of SLE and shortening the duration of what she called “the gray zone” of diagnostic uncertainty, when autoantibodies and insidious symptoms are present but not yet sufficient to make the diagnosis of SLE by conventional criteria. It’s well established that 60%-70% of patients with mild undifferentiated connective tissue disease will remain stable without evolving into SLE during long years of follow-up.
The ultimate objective of all this work is to try to change the natural history of the disease through targeted early aggressive therapy aimed at minimizing the extent of active disease and preventing severe organ involvement.
Among the key takeaways from the four-continent study led by Dr. Mosca: Fever not related to infection was far more prevalent in early SLE than in mimicking conditions, by a margin of 34.5% versus 13.7%. On the other hand, Raynaud’s phenomenon was more than twice as prevalent among patients with mimicking conditions: 22.1% in early SLE, compared with 48.5% in SLE mimickers. Sicca symptoms were present in just 4.4% of early SLE patients versus 34.4% of SLE mimickers. Only 0.3% of early SLE patients complained of dysphagia; the rate was 20-fold higher in the SLE mimickers. Rashes atypical for lupus were twice as frequent in the SLE mimicking conditions (Arthritis Rheumatol. 2019 Jan;71[1]:91-8).
Turning to key differentiating serologic findings, Dr. Mosca noted that anti-double stranded DNA (anti-dsDNA) and anti-Sm antibodies were present in 71.7% and 30.2% of early SLE patients, respectively, compared with 6.9% and 2.6% of SLE mimickers. Anticardiolipin IgM, a positive Coombs test, anti-beta2 glycoprotein-I antibodies, leukopenia, autoimmune hemolytic anemia, and hypocomplementemia were all significantly more common in the early SLE cohort.
In contrast, antibodies to Ro (SS-A) and La (SS-B) were of no value in separating early SLE from its mimickers, according to Dr. Mosca.
Other tipoffs to early SLE
Two separate teams of British researchers have advanced the field in a highly practical way. One group showed in a study of 1,739 newly diagnosed SLE patients and 6,956 controls that in the 5 years prior to diagnosis, the SLE group averaged 9.2 primary care visits per year, compared with 3.8 for controls. The visits clustered around nonspecific complaints of arthritis and arthralgias, alopecia, and rash (Arthritis Care Res. 2017 Jun;69[6]:833-41).
“An accumulating number of primary care office visits and referrals over time should raise suspicion,” Dr. Mosca said.
Other investigators, working with 1,426 cases of newly diagnosed SLE in the U.K. Clinical Practice Research Database, observed that the proportion of patients with disease manifestations in three or more British Isles Lupus Activity Group (BILAG) symptom domains rose from 18.7% at 3 years prior to diagnosis to 39.7% in the year before diagnosis (Lupus Sci Med. 2017 Feb 10;4[1]:e000172. doi: 10.1136/lupus-2016-000172).
“These patients accrue clinical manifestations. It’s not just one symptom, it’s more of a state of being unwell. This is a suspicious factor for the development of lupus,” she continued.
And just as patients who will eventually be diagnosed with SLE accrue a growing number of signs and symptoms during the run up to diagnosis, they also accrue multiple autoantibodies. Moreover, as demonstrated by a multicenter group of U.S. investigators, patients also develop elevated levels of multiple soluble inflammatory markers more than 3.5 years prior to diagnosis of SLE. These include interleukins-5 and -6 and interferon-gamma. And less than 10 months prior to being classified as having SLE, patients develop significantly higher levels of B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand known as APRIL. The investigators developed a predictive model incorporating IL-5, -6, and interferon-gamma levels with antinuclear antibody status that identified future SLE patients with 84% accuracy more than 3.5 years before diagnosis. This could prove useful in selecting high-risk patients for clinical prevention trials (J Autoimmun. 2016 Nov;74:182-93).
Researchers at the University of Leeds (England) have also zeroed in on interferon activity as playing a key role in progression from asymptomatic antinuclear antigen positivity, which is present in up to 25% of the general population, to symptomatic autoimmune connective tissue disease, which affects less than 1%. A multivariate logistic regression analysis identified two independent predictors of development of autoimmune connective tissue disease within the next 12 months: a family history of autoimmune rheumatic disease, which was associated with an 8.2-fold increased risk; and positivity for a pattern of interferon-stimulated gene activity they call IFN-Score-B (Ann Rheum Dis. 2018 Oct;77[10]:1432-9).
All of this work has led up to what Dr. Mosca called “a glance into the future”: the National Institutes of Health–supported Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE), which is now recruiting patients. This randomized, double-blind, placebo-controlled multicenter U.S. trial involves patients who are antinuclear antibody positive at a titer of 1:80 or more plus one or two additional criteria from the SLICC classification scheme. Participants are being randomized to 96 weeks of hydroxychloroquine or placebo. The goal is to learn whether hydroxychloroquine can slow disease progression, with the primary endpoint being the number of SLICC criteria met at the study’s end. The trial will also scrutinize potential biomarkers that could be used to guide treatment decisions (Trials. 2018 Dec 20;19[1]:694. doi: 10.1186/s13063-018-3076-7).
Dr. Mosca reported serving as an adviser to UCB and Lilly.
REPORTING FROM LUPUS 2019
Gut bacterium R. gnavus linked to lupus flares
SAN FRANCISCO –
Not only that, but those patients also had highly elevated antibodies to an endotoxin-like antigen released by one particular R. gnavus strain.
That antigen is “very proinflammatory, very immunogenic. We are wondering if this is actually [what drives] the immune activation that results in immune complexes in the glomeruli” of patients with lupus nephritis, said investigator Gregg Silverman, MD, a professor of medicine and pathology and head of the laboratory of B-cell immunobiology at New York University.
R. gnavus is an obligate anaerobe found in the guts of most people, but in lupus, it might be a problem.
“We are finding a very specific relationship with lupus patients and this bacteria – and this particular antibody,” Dr. Silverman explained in an interview at an international congress on systemic lupus erythematosus. “There’s an expansion of this particular bug, but also a contraction of others” as disease activity progresses.
“It speaks to an imbalance,” he added, and it suggests a role for probiotics or even fecal transplants to restore order.
“What if instead of killing the immune system” in lupus treatment, “we should be reducing or removing a single bacterium or a single molecule?” he asked.
Dr. Silverman is one of many researchers working to unravel the role of the human microbiome in both disease and health. His findings are preliminary, and, as he cautioned, correlation is not causation. But the implications are remarkable, Dr. Silverman noted.
SAN FRANCISCO –
Not only that, but those patients also had highly elevated antibodies to an endotoxin-like antigen released by one particular R. gnavus strain.
That antigen is “very proinflammatory, very immunogenic. We are wondering if this is actually [what drives] the immune activation that results in immune complexes in the glomeruli” of patients with lupus nephritis, said investigator Gregg Silverman, MD, a professor of medicine and pathology and head of the laboratory of B-cell immunobiology at New York University.
R. gnavus is an obligate anaerobe found in the guts of most people, but in lupus, it might be a problem.
“We are finding a very specific relationship with lupus patients and this bacteria – and this particular antibody,” Dr. Silverman explained in an interview at an international congress on systemic lupus erythematosus. “There’s an expansion of this particular bug, but also a contraction of others” as disease activity progresses.
“It speaks to an imbalance,” he added, and it suggests a role for probiotics or even fecal transplants to restore order.
“What if instead of killing the immune system” in lupus treatment, “we should be reducing or removing a single bacterium or a single molecule?” he asked.
Dr. Silverman is one of many researchers working to unravel the role of the human microbiome in both disease and health. His findings are preliminary, and, as he cautioned, correlation is not causation. But the implications are remarkable, Dr. Silverman noted.
SAN FRANCISCO –
Not only that, but those patients also had highly elevated antibodies to an endotoxin-like antigen released by one particular R. gnavus strain.
That antigen is “very proinflammatory, very immunogenic. We are wondering if this is actually [what drives] the immune activation that results in immune complexes in the glomeruli” of patients with lupus nephritis, said investigator Gregg Silverman, MD, a professor of medicine and pathology and head of the laboratory of B-cell immunobiology at New York University.
R. gnavus is an obligate anaerobe found in the guts of most people, but in lupus, it might be a problem.
“We are finding a very specific relationship with lupus patients and this bacteria – and this particular antibody,” Dr. Silverman explained in an interview at an international congress on systemic lupus erythematosus. “There’s an expansion of this particular bug, but also a contraction of others” as disease activity progresses.
“It speaks to an imbalance,” he added, and it suggests a role for probiotics or even fecal transplants to restore order.
“What if instead of killing the immune system” in lupus treatment, “we should be reducing or removing a single bacterium or a single molecule?” he asked.
Dr. Silverman is one of many researchers working to unravel the role of the human microbiome in both disease and health. His findings are preliminary, and, as he cautioned, correlation is not causation. But the implications are remarkable, Dr. Silverman noted.
REPORTING FROM LUPUS 2019

Combo B-cell depletion advances in SLE
SAN FRANCISCO – The sequential combination of rituximab followed directly by maintenance belimumab shows considerable promise as a strategy to address the aberrant B-cell immunology present in systemic lupus erythematosus (SLE) – and thereby improve clinical outcomes, Y.K. Onno Teng, MD, PhD, reported at an international congress on systemic lupus erythematosus.

Dr. Teng, a nephrologist and clinical trialist at Leiden (the Netherlands) University, and his coworkers were pioneers of this one-two punch, in which a two-dose course of rituximab (Rituxan) is given to deplete CD20-positive B-cells, followed by long-term maintenance belimumab (Benlysta) to inhibit repopulation of specific problematic types of B-cells. The rationale for the use of belimumab here lies in the observation that the initial B-cell depletion induced by rituximab triggers a surge in B lymphocyte stimulator (BLyS), which signals the bone marrow to start making more B-cells. And belimumab famously inhibits BLyS, also known as B-cell activating factor, or BAFF.
Dr. Teng presented the 2-year extended results of Synergistic B-cell Immunomodulation in SLE (SYNBIoSe-1), a phase 2a, open-label, single-arm, proof-of-concept study whose 24-week immunologic results have previously been reported (J Autoimmun. 2018 Jul;91:45-54).
Based in part upon the encouraging SYNBIoSe-1 findings as well as the sound mechanistic rationale for this treatment strategy, the combination of rituximab and belimumab is picking up steam in the research world as a potentially important treatment advance in SLE. Currently underway in patients with nonrenal SLE is the phase 3, GlaxoSmithKline-sponsored, global BLISS-BELIEVE trial, as well as the phase 2 BEAT-LUPUS study, a University College London–based randomized trial of rituximab plus either placebo or belimumab. Also, Dr. Teng and his coworkers are now conducting SYNBIoSe-2, in which patients with lupus nephritis are being randomized to standard therapy with glucocorticoids and mycophenolate or to rituximab, belimumab, and mycophenolate.
SYNBIoSe-2 is a further exploration of the encouraging signal of efficacy for lupus nephritis noted in SYNBIoSe-1. Of the 12 participants in SYNBIoSe-1 who had baseline active lupus nephritis, 8 had a positive renal response to the rituximab/belimumab combo, including 6 patients who achieved a prolonged complete renal response through 104 weeks of follow-up.
SYNBIoSe-1 included 15 patients, all with severe refractory SLE as shown by a median 11-year disease duration and a baseline SLE Disease Activity Index score of 18. Two-thirds of patients achieved sustained low-level disease activity, interrupted in one case by a single major disease flare. Two patients stopped treatment because of a lack of response. Several others left the study because they were doing so well on treatment that they decided the time was right to become pregnant.
Immunologically, patients showed an 84% reduction in B-cell repopulation over the course of 2 years. Particularly striking was the long-term inhibition of double-negative B-cells and IgD-positive naive B-cells, which Dr. Teng described as “very trigger happy” in that they readily become transformed into activated antibody-producing cells.
Sustained specific reductions in anti-double-stranded DNA autoantibodies and other pathogenic antinuclear antibodies were also documented through 104 weeks.
SYNBIoSe-1 results at odds with CALIBRATE trial results
The favorable impact of the rituximab/belimumab combo on lupus nephritis seen in SYNBIoSe-1 is at odds with the results of CALIBRATE, a U.S. study in which 43 patients with active lupus nephritis despite conventional treatment were randomized open label to induction therapy with two doses of rituximab on top of standard background therapy, followed by either belimumab and prednisone or prednisone alone. In CALIBRATE, the anti-BLyS biologic didn’t improve clinical outcomes. Dr. Teng said he believes he knows why.
“There was an important difference in background immunosuppression in the two studies. We used mycophenolate in SYNBIoSe-1, while they used cyclophosphamide in CALIBRATE,” he noted. “Other investigators have shown that mycophenolate mostly depletes plasma cells, whereas cyclophosphamide is very much depleting proliferating cells, predominantly the B-cell population and to a lesser extent the plasma cell population. I think this phenomenon might explain why adding BLyS inhibition to patients treated with CellCept [mycophenolate] might be of more added value than adding it to cyclophosphamide therapy.”
Dr. Teng reported having no financial conflicts regarding the SYNBIoSe-1 study, which was funded by research grants from the Dutch Kidney Foundation and the Netherlands Organization for Health Research and Development.
SAN FRANCISCO – The sequential combination of rituximab followed directly by maintenance belimumab shows considerable promise as a strategy to address the aberrant B-cell immunology present in systemic lupus erythematosus (SLE) – and thereby improve clinical outcomes, Y.K. Onno Teng, MD, PhD, reported at an international congress on systemic lupus erythematosus.
Dr. Teng, a nephrologist and clinical trialist at Leiden (the Netherlands) University, and his coworkers were pioneers of this one-two punch, in which a two-dose course of rituximab (Rituxan) is given to deplete CD20-positive B-cells, followed by long-term maintenance belimumab (Benlysta) to inhibit repopulation of specific problematic types of B-cells. The rationale for the use of belimumab here lies in the observation that the initial B-cell depletion induced by rituximab triggers a surge in B lymphocyte stimulator (BLyS), which signals the bone marrow to start making more B-cells. And belimumab famously inhibits BLyS, also known as B-cell activating factor, or BAFF.
Dr. Teng presented the 2-year extended results of Synergistic B-cell Immunomodulation in SLE (SYNBIoSe-1), a phase 2a, open-label, single-arm, proof-of-concept study whose 24-week immunologic results have previously been reported (J Autoimmun. 2018 Jul;91:45-54).
Based in part upon the encouraging SYNBIoSe-1 findings as well as the sound mechanistic rationale for this treatment strategy, the combination of rituximab and belimumab is picking up steam in the research world as a potentially important treatment advance in SLE. Currently underway in patients with nonrenal SLE is the phase 3, GlaxoSmithKline-sponsored, global BLISS-BELIEVE trial, as well as the phase 2 BEAT-LUPUS study, a University College London–based randomized trial of rituximab plus either placebo or belimumab. Also, Dr. Teng and his coworkers are now conducting SYNBIoSe-2, in which patients with lupus nephritis are being randomized to standard therapy with glucocorticoids and mycophenolate or to rituximab, belimumab, and mycophenolate.
SYNBIoSe-2 is a further exploration of the encouraging signal of efficacy for lupus nephritis noted in SYNBIoSe-1. Of the 12 participants in SYNBIoSe-1 who had baseline active lupus nephritis, 8 had a positive renal response to the rituximab/belimumab combo, including 6 patients who achieved a prolonged complete renal response through 104 weeks of follow-up.
SYNBIoSe-1 included 15 patients, all with severe refractory SLE as shown by a median 11-year disease duration and a baseline SLE Disease Activity Index score of 18. Two-thirds of patients achieved sustained low-level disease activity, interrupted in one case by a single major disease flare. Two patients stopped treatment because of a lack of response. Several others left the study because they were doing so well on treatment that they decided the time was right to become pregnant.
Immunologically, patients showed an 84% reduction in B-cell repopulation over the course of 2 years. Particularly striking was the long-term inhibition of double-negative B-cells and IgD-positive naive B-cells, which Dr. Teng described as “very trigger happy” in that they readily become transformed into activated antibody-producing cells.
Sustained specific reductions in anti-double-stranded DNA autoantibodies and other pathogenic antinuclear antibodies were also documented through 104 weeks.
SYNBIoSe-1 results at odds with CALIBRATE trial results
The favorable impact of the rituximab/belimumab combo on lupus nephritis seen in SYNBIoSe-1 is at odds with the results of CALIBRATE, a U.S. study in which 43 patients with active lupus nephritis despite conventional treatment were randomized open label to induction therapy with two doses of rituximab on top of standard background therapy, followed by either belimumab and prednisone or prednisone alone. In CALIBRATE, the anti-BLyS biologic didn’t improve clinical outcomes. Dr. Teng said he believes he knows why.
“There was an important difference in background immunosuppression in the two studies. We used mycophenolate in SYNBIoSe-1, while they used cyclophosphamide in CALIBRATE,” he noted. “Other investigators have shown that mycophenolate mostly depletes plasma cells, whereas cyclophosphamide is very much depleting proliferating cells, predominantly the B-cell population and to a lesser extent the plasma cell population. I think this phenomenon might explain why adding BLyS inhibition to patients treated with CellCept [mycophenolate] might be of more added value than adding it to cyclophosphamide therapy.”
Dr. Teng reported having no financial conflicts regarding the SYNBIoSe-1 study, which was funded by research grants from the Dutch Kidney Foundation and the Netherlands Organization for Health Research and Development.
SAN FRANCISCO – The sequential combination of rituximab followed directly by maintenance belimumab shows considerable promise as a strategy to address the aberrant B-cell immunology present in systemic lupus erythematosus (SLE) – and thereby improve clinical outcomes, Y.K. Onno Teng, MD, PhD, reported at an international congress on systemic lupus erythematosus.
Dr. Teng, a nephrologist and clinical trialist at Leiden (the Netherlands) University, and his coworkers were pioneers of this one-two punch, in which a two-dose course of rituximab (Rituxan) is given to deplete CD20-positive B-cells, followed by long-term maintenance belimumab (Benlysta) to inhibit repopulation of specific problematic types of B-cells. The rationale for the use of belimumab here lies in the observation that the initial B-cell depletion induced by rituximab triggers a surge in B lymphocyte stimulator (BLyS), which signals the bone marrow to start making more B-cells. And belimumab famously inhibits BLyS, also known as B-cell activating factor, or BAFF.
Dr. Teng presented the 2-year extended results of Synergistic B-cell Immunomodulation in SLE (SYNBIoSe-1), a phase 2a, open-label, single-arm, proof-of-concept study whose 24-week immunologic results have previously been reported (J Autoimmun. 2018 Jul;91:45-54).
Based in part upon the encouraging SYNBIoSe-1 findings as well as the sound mechanistic rationale for this treatment strategy, the combination of rituximab and belimumab is picking up steam in the research world as a potentially important treatment advance in SLE. Currently underway in patients with nonrenal SLE is the phase 3, GlaxoSmithKline-sponsored, global BLISS-BELIEVE trial, as well as the phase 2 BEAT-LUPUS study, a University College London–based randomized trial of rituximab plus either placebo or belimumab. Also, Dr. Teng and his coworkers are now conducting SYNBIoSe-2, in which patients with lupus nephritis are being randomized to standard therapy with glucocorticoids and mycophenolate or to rituximab, belimumab, and mycophenolate.
SYNBIoSe-2 is a further exploration of the encouraging signal of efficacy for lupus nephritis noted in SYNBIoSe-1. Of the 12 participants in SYNBIoSe-1 who had baseline active lupus nephritis, 8 had a positive renal response to the rituximab/belimumab combo, including 6 patients who achieved a prolonged complete renal response through 104 weeks of follow-up.
SYNBIoSe-1 included 15 patients, all with severe refractory SLE as shown by a median 11-year disease duration and a baseline SLE Disease Activity Index score of 18. Two-thirds of patients achieved sustained low-level disease activity, interrupted in one case by a single major disease flare. Two patients stopped treatment because of a lack of response. Several others left the study because they were doing so well on treatment that they decided the time was right to become pregnant.
Immunologically, patients showed an 84% reduction in B-cell repopulation over the course of 2 years. Particularly striking was the long-term inhibition of double-negative B-cells and IgD-positive naive B-cells, which Dr. Teng described as “very trigger happy” in that they readily become transformed into activated antibody-producing cells.
Sustained specific reductions in anti-double-stranded DNA autoantibodies and other pathogenic antinuclear antibodies were also documented through 104 weeks.
SYNBIoSe-1 results at odds with CALIBRATE trial results
The favorable impact of the rituximab/belimumab combo on lupus nephritis seen in SYNBIoSe-1 is at odds with the results of CALIBRATE, a U.S. study in which 43 patients with active lupus nephritis despite conventional treatment were randomized open label to induction therapy with two doses of rituximab on top of standard background therapy, followed by either belimumab and prednisone or prednisone alone. In CALIBRATE, the anti-BLyS biologic didn’t improve clinical outcomes. Dr. Teng said he believes he knows why.
“There was an important difference in background immunosuppression in the two studies. We used mycophenolate in SYNBIoSe-1, while they used cyclophosphamide in CALIBRATE,” he noted. “Other investigators have shown that mycophenolate mostly depletes plasma cells, whereas cyclophosphamide is very much depleting proliferating cells, predominantly the B-cell population and to a lesser extent the plasma cell population. I think this phenomenon might explain why adding BLyS inhibition to patients treated with CellCept [mycophenolate] might be of more added value than adding it to cyclophosphamide therapy.”
Dr. Teng reported having no financial conflicts regarding the SYNBIoSe-1 study, which was funded by research grants from the Dutch Kidney Foundation and the Netherlands Organization for Health Research and Development.
REPORTING FROM LUPUS 2019
Hydroxychloroquine adherence in SLE: worse than you thought
SAN FRANCISCO – Routine office measurement of hydroxychloroquine blood levels in systemic lupus erythematosus (SLE) patients accomplishes two major objectives, Nathalie Costedoat-Chalumeau, MD, asserted at an international congress on systemic lupus erythematosus.

First, measuring hydroxychloroquine levels identifies the surprisingly large number of individuals who are severely nonadherent to this cornerstone of lupus therapy despite its excellent benefit/risk ratio. Also, serial measurements coupled with brief counseling have been shown to boost poor adherence rates, noted Dr. Costedoat-Chalumeau, professor of rheumatology at Paris Descartes University.
Numerous studies have documented startlingly low adherence to hydroxychloroquine among SLE patients. Some of the same studies show prescribing physicians are often clueless as to their patients’ adherence or lack thereof.
Just how bad is the adherence problem? A recent study of 10,406 Medicaid patients with SLE who started on hydroxychloroquine showed that 85% of them were nonadherent as defined by pharmacy refill data, indicating insufficient medication on hand to cover a minimum of 80% of days during at least 1 year of follow-up.
In a novel finding, the investigators also broke down the Medicaid data month by month and identified four broad patterns of adherence/nonadherence. A total of 17% of patients were persistently adherent throughout the first year after the drug was dispensed. Another 36% were persistent nonadherers right from the get-go. A further 24% remained partially adherent, dropping down to a plateau of 30%-40% monthly adherence after the first couple of months and staying there. And 23% dropped steadily from roughly 50% adherence at month 3 to nearly total nonadherence from month 9 onward. Overall, adherence in the Medicaid cohort declined over the course of the first year (Semin Arthritis Rheum. 2018 Oct;48[2]:205-13).
Dr. Costedoat-Chalumeau was the lead investigator in a large French multicenter clinical trial known as the PLUS Study, which established that increasing the hydroxychloroquine daily dose to raise blood levels to a target of at least 1,000 ng/mL didn’t reduce the risk of flares (Ann Rheum Dis. 2013;72[11]:1786-92).
“So there is no reason to use blood drug measurements to adjust hydroxychloroquine daily dose or blood levels to prevent SLE flares. But drug levels teach us something regarding adherence,” she said.
Why routinely measuring hydroxychloroquine levels matters
Dr. Costedoat-Chalumeau and other investigators have shown that whole blood drug levels below 200 ng/mL indicate a patient is severely nonadherent. In various studies, that’s 7%-29% of SLE patients who are supposedly on hydroxychloroquine.
Also, investigators at Johns Hopkins University, Baltimore, have analyzed prospective data from the Hopkins Lupus Cohort and determined that at the first clinic visit after going on a maximum of 400 mg/day of hydroxychloroquine, only 44% of participants had a blood drug level above the 500-ng/mL threshold indicative of adherence.
Importantly, however, the Hopkins researchers also demonstrated that with repeated brief counseling of nonadherent patients as to why hydroxychloroquine is the most important medication they take for their SLE, adherence climbed in stepwise fashion with each visit in which the drug blood level was assessed: With no prior measurement, adherence was 56%; with one prior measurement, it jumped to 69%; with two, 77% of patients were adherent to hydroxychloroquine; and with three or more prior blood level checks, adherence rose to 80% (J Rheumatol. 2015 Nov;42[11]:2092-7).
It is well established that hydroxychloroquine prevents SLE flares, protects against thrombotic events, diabetes, dyslipidemia, and lupus-induced organ damage, and improves survival. Dr. Costedoat-Chalumeau’s final words on hydroxychloroquine adherence: ”Drugs don’t work in people who don’t take them.”
She reported having no financial conflicts regarding her presentation.
SAN FRANCISCO – Routine office measurement of hydroxychloroquine blood levels in systemic lupus erythematosus (SLE) patients accomplishes two major objectives, Nathalie Costedoat-Chalumeau, MD, asserted at an international congress on systemic lupus erythematosus.
First, measuring hydroxychloroquine levels identifies the surprisingly large number of individuals who are severely nonadherent to this cornerstone of lupus therapy despite its excellent benefit/risk ratio. Also, serial measurements coupled with brief counseling have been shown to boost poor adherence rates, noted Dr. Costedoat-Chalumeau, professor of rheumatology at Paris Descartes University.
Numerous studies have documented startlingly low adherence to hydroxychloroquine among SLE patients. Some of the same studies show prescribing physicians are often clueless as to their patients’ adherence or lack thereof.
Just how bad is the adherence problem? A recent study of 10,406 Medicaid patients with SLE who started on hydroxychloroquine showed that 85% of them were nonadherent as defined by pharmacy refill data, indicating insufficient medication on hand to cover a minimum of 80% of days during at least 1 year of follow-up.
In a novel finding, the investigators also broke down the Medicaid data month by month and identified four broad patterns of adherence/nonadherence. A total of 17% of patients were persistently adherent throughout the first year after the drug was dispensed. Another 36% were persistent nonadherers right from the get-go. A further 24% remained partially adherent, dropping down to a plateau of 30%-40% monthly adherence after the first couple of months and staying there. And 23% dropped steadily from roughly 50% adherence at month 3 to nearly total nonadherence from month 9 onward. Overall, adherence in the Medicaid cohort declined over the course of the first year (Semin Arthritis Rheum. 2018 Oct;48[2]:205-13).
Dr. Costedoat-Chalumeau was the lead investigator in a large French multicenter clinical trial known as the PLUS Study, which established that increasing the hydroxychloroquine daily dose to raise blood levels to a target of at least 1,000 ng/mL didn’t reduce the risk of flares (Ann Rheum Dis. 2013;72[11]:1786-92).
“So there is no reason to use blood drug measurements to adjust hydroxychloroquine daily dose or blood levels to prevent SLE flares. But drug levels teach us something regarding adherence,” she said.
Why routinely measuring hydroxychloroquine levels matters
Dr. Costedoat-Chalumeau and other investigators have shown that whole blood drug levels below 200 ng/mL indicate a patient is severely nonadherent. In various studies, that’s 7%-29% of SLE patients who are supposedly on hydroxychloroquine.
Also, investigators at Johns Hopkins University, Baltimore, have analyzed prospective data from the Hopkins Lupus Cohort and determined that at the first clinic visit after going on a maximum of 400 mg/day of hydroxychloroquine, only 44% of participants had a blood drug level above the 500-ng/mL threshold indicative of adherence.
Importantly, however, the Hopkins researchers also demonstrated that with repeated brief counseling of nonadherent patients as to why hydroxychloroquine is the most important medication they take for their SLE, adherence climbed in stepwise fashion with each visit in which the drug blood level was assessed: With no prior measurement, adherence was 56%; with one prior measurement, it jumped to 69%; with two, 77% of patients were adherent to hydroxychloroquine; and with three or more prior blood level checks, adherence rose to 80% (J Rheumatol. 2015 Nov;42[11]:2092-7).
It is well established that hydroxychloroquine prevents SLE flares, protects against thrombotic events, diabetes, dyslipidemia, and lupus-induced organ damage, and improves survival. Dr. Costedoat-Chalumeau’s final words on hydroxychloroquine adherence: ”Drugs don’t work in people who don’t take them.”
She reported having no financial conflicts regarding her presentation.
SAN FRANCISCO – Routine office measurement of hydroxychloroquine blood levels in systemic lupus erythematosus (SLE) patients accomplishes two major objectives, Nathalie Costedoat-Chalumeau, MD, asserted at an international congress on systemic lupus erythematosus.
First, measuring hydroxychloroquine levels identifies the surprisingly large number of individuals who are severely nonadherent to this cornerstone of lupus therapy despite its excellent benefit/risk ratio. Also, serial measurements coupled with brief counseling have been shown to boost poor adherence rates, noted Dr. Costedoat-Chalumeau, professor of rheumatology at Paris Descartes University.
Numerous studies have documented startlingly low adherence to hydroxychloroquine among SLE patients. Some of the same studies show prescribing physicians are often clueless as to their patients’ adherence or lack thereof.
Just how bad is the adherence problem? A recent study of 10,406 Medicaid patients with SLE who started on hydroxychloroquine showed that 85% of them were nonadherent as defined by pharmacy refill data, indicating insufficient medication on hand to cover a minimum of 80% of days during at least 1 year of follow-up.
In a novel finding, the investigators also broke down the Medicaid data month by month and identified four broad patterns of adherence/nonadherence. A total of 17% of patients were persistently adherent throughout the first year after the drug was dispensed. Another 36% were persistent nonadherers right from the get-go. A further 24% remained partially adherent, dropping down to a plateau of 30%-40% monthly adherence after the first couple of months and staying there. And 23% dropped steadily from roughly 50% adherence at month 3 to nearly total nonadherence from month 9 onward. Overall, adherence in the Medicaid cohort declined over the course of the first year (Semin Arthritis Rheum. 2018 Oct;48[2]:205-13).
Dr. Costedoat-Chalumeau was the lead investigator in a large French multicenter clinical trial known as the PLUS Study, which established that increasing the hydroxychloroquine daily dose to raise blood levels to a target of at least 1,000 ng/mL didn’t reduce the risk of flares (Ann Rheum Dis. 2013;72[11]:1786-92).
“So there is no reason to use blood drug measurements to adjust hydroxychloroquine daily dose or blood levels to prevent SLE flares. But drug levels teach us something regarding adherence,” she said.
Why routinely measuring hydroxychloroquine levels matters
Dr. Costedoat-Chalumeau and other investigators have shown that whole blood drug levels below 200 ng/mL indicate a patient is severely nonadherent. In various studies, that’s 7%-29% of SLE patients who are supposedly on hydroxychloroquine.
Also, investigators at Johns Hopkins University, Baltimore, have analyzed prospective data from the Hopkins Lupus Cohort and determined that at the first clinic visit after going on a maximum of 400 mg/day of hydroxychloroquine, only 44% of participants had a blood drug level above the 500-ng/mL threshold indicative of adherence.
Importantly, however, the Hopkins researchers also demonstrated that with repeated brief counseling of nonadherent patients as to why hydroxychloroquine is the most important medication they take for their SLE, adherence climbed in stepwise fashion with each visit in which the drug blood level was assessed: With no prior measurement, adherence was 56%; with one prior measurement, it jumped to 69%; with two, 77% of patients were adherent to hydroxychloroquine; and with three or more prior blood level checks, adherence rose to 80% (J Rheumatol. 2015 Nov;42[11]:2092-7).
It is well established that hydroxychloroquine prevents SLE flares, protects against thrombotic events, diabetes, dyslipidemia, and lupus-induced organ damage, and improves survival. Dr. Costedoat-Chalumeau’s final words on hydroxychloroquine adherence: ”Drugs don’t work in people who don’t take them.”
She reported having no financial conflicts regarding her presentation.
REPORTING FROM LUPUS 2019
‘Type II’ SLE assessment catches what matters to patients
SAN FRANCISCO – For almost a year, lupus patients at Duke University in Durham, N.C., have been getting two physician global assessments, the usual one for classic “type I” disease, and a new one for nonspecific “type II” symptoms: fatigue, widespread pain, depression, sleep disturbance, and cognitive dysfunction.

It’s often the type II problems that affect patients the most, and what they are most concerned about; formally assessing them with the type II physician global assessment (PGA) – a 0- to 3-point visual analog scale – ensures they aren’t overlooked, said Jennifer Rogers, MD, assistant professor of rheumatology at Duke.
It “forces us to address these symptoms,” she said, and the approach seems to be working, according to a study Dr. Rogers presented at an international congress on systemic lupus erythematosus.
In the 5 months leading up to implementation of the PGA II in late spring 2018, type II problems had treatment recommendations in patients’ charts just 53% of the time; the number rose to 89% of the time during the PGA II’s first 5 months (P = .03). Type II PGA scores correlated strongly with patient-reported fibromyalgia and depression symptoms, but did not correlate with PGA scores for type I symptoms, such as nephritis and arthritis.
Type II problems are common in lupus. Patients’ joints might be fine, and their kidney disease in remission, but they can still feel miserable, and will often blame it on a lupus flare. Physicians who disagree end up at odds with their patients, Dr. Rogers explained.
“We decided to rethink how we address these patients, and came up with this new type I, type II categorization.” Now, when paints complain of brain fog, for example, “I say ‘yes, this is your lupus. I believe you,’ but we don’t need to give you more steroids or very expensive immunosuppressives for this. What you need to do is take your Cymbalta, work on your exercise, and maybe see your therapist,” she said.
It validates what people are going through, and builds trust. “Patients like it; they feel heard, and I walk out of the room, and I feel better,” she said.
During its first 5 months, 197 patients had PGAs for type II symptoms, along with type I PGAs. The average age of the patients was 46 years, and 92% were women.
Patients with predominately type II symptoms were more likely than were those with predominately type I disease to be depressed (84% versus 39%), and they reported higher lupus activity, greater symptom severity, and more severe fibromyalgia. The differences were statistically significant.
Type II treatments included medications in 60% of cases, exercise or physical therapy in almost 60% of cases, sleep studies or help with sleep hygiene in about 35%, and psychiatric or psychological referral in almost 20%. Less than 5% of patients were referred to a pain clinic.
There was no external funding for the study, and Dr. Rogers didn’t have any disclosures.
SOURCE: Rogers J et al. Lupus Sci Med. 2019;6[suppl 1]: Abstract 102. doi: 10.1136/lupus-2019-lsm.102
SAN FRANCISCO – For almost a year, lupus patients at Duke University in Durham, N.C., have been getting two physician global assessments, the usual one for classic “type I” disease, and a new one for nonspecific “type II” symptoms: fatigue, widespread pain, depression, sleep disturbance, and cognitive dysfunction.
It’s often the type II problems that affect patients the most, and what they are most concerned about; formally assessing them with the type II physician global assessment (PGA) – a 0- to 3-point visual analog scale – ensures they aren’t overlooked, said Jennifer Rogers, MD, assistant professor of rheumatology at Duke.
It “forces us to address these symptoms,” she said, and the approach seems to be working, according to a study Dr. Rogers presented at an international congress on systemic lupus erythematosus.
In the 5 months leading up to implementation of the PGA II in late spring 2018, type II problems had treatment recommendations in patients’ charts just 53% of the time; the number rose to 89% of the time during the PGA II’s first 5 months (P = .03). Type II PGA scores correlated strongly with patient-reported fibromyalgia and depression symptoms, but did not correlate with PGA scores for type I symptoms, such as nephritis and arthritis.
Type II problems are common in lupus. Patients’ joints might be fine, and their kidney disease in remission, but they can still feel miserable, and will often blame it on a lupus flare. Physicians who disagree end up at odds with their patients, Dr. Rogers explained.
“We decided to rethink how we address these patients, and came up with this new type I, type II categorization.” Now, when paints complain of brain fog, for example, “I say ‘yes, this is your lupus. I believe you,’ but we don’t need to give you more steroids or very expensive immunosuppressives for this. What you need to do is take your Cymbalta, work on your exercise, and maybe see your therapist,” she said.
It validates what people are going through, and builds trust. “Patients like it; they feel heard, and I walk out of the room, and I feel better,” she said.
During its first 5 months, 197 patients had PGAs for type II symptoms, along with type I PGAs. The average age of the patients was 46 years, and 92% were women.
Patients with predominately type II symptoms were more likely than were those with predominately type I disease to be depressed (84% versus 39%), and they reported higher lupus activity, greater symptom severity, and more severe fibromyalgia. The differences were statistically significant.
Type II treatments included medications in 60% of cases, exercise or physical therapy in almost 60% of cases, sleep studies or help with sleep hygiene in about 35%, and psychiatric or psychological referral in almost 20%. Less than 5% of patients were referred to a pain clinic.
There was no external funding for the study, and Dr. Rogers didn’t have any disclosures.
SOURCE: Rogers J et al. Lupus Sci Med. 2019;6[suppl 1]: Abstract 102. doi: 10.1136/lupus-2019-lsm.102
SAN FRANCISCO – For almost a year, lupus patients at Duke University in Durham, N.C., have been getting two physician global assessments, the usual one for classic “type I” disease, and a new one for nonspecific “type II” symptoms: fatigue, widespread pain, depression, sleep disturbance, and cognitive dysfunction.
It’s often the type II problems that affect patients the most, and what they are most concerned about; formally assessing them with the type II physician global assessment (PGA) – a 0- to 3-point visual analog scale – ensures they aren’t overlooked, said Jennifer Rogers, MD, assistant professor of rheumatology at Duke.
It “forces us to address these symptoms,” she said, and the approach seems to be working, according to a study Dr. Rogers presented at an international congress on systemic lupus erythematosus.
In the 5 months leading up to implementation of the PGA II in late spring 2018, type II problems had treatment recommendations in patients’ charts just 53% of the time; the number rose to 89% of the time during the PGA II’s first 5 months (P = .03). Type II PGA scores correlated strongly with patient-reported fibromyalgia and depression symptoms, but did not correlate with PGA scores for type I symptoms, such as nephritis and arthritis.
Type II problems are common in lupus. Patients’ joints might be fine, and their kidney disease in remission, but they can still feel miserable, and will often blame it on a lupus flare. Physicians who disagree end up at odds with their patients, Dr. Rogers explained.
“We decided to rethink how we address these patients, and came up with this new type I, type II categorization.” Now, when paints complain of brain fog, for example, “I say ‘yes, this is your lupus. I believe you,’ but we don’t need to give you more steroids or very expensive immunosuppressives for this. What you need to do is take your Cymbalta, work on your exercise, and maybe see your therapist,” she said.
It validates what people are going through, and builds trust. “Patients like it; they feel heard, and I walk out of the room, and I feel better,” she said.
During its first 5 months, 197 patients had PGAs for type II symptoms, along with type I PGAs. The average age of the patients was 46 years, and 92% were women.
Patients with predominately type II symptoms were more likely than were those with predominately type I disease to be depressed (84% versus 39%), and they reported higher lupus activity, greater symptom severity, and more severe fibromyalgia. The differences were statistically significant.
Type II treatments included medications in 60% of cases, exercise or physical therapy in almost 60% of cases, sleep studies or help with sleep hygiene in about 35%, and psychiatric or psychological referral in almost 20%. Less than 5% of patients were referred to a pain clinic.
There was no external funding for the study, and Dr. Rogers didn’t have any disclosures.
SOURCE: Rogers J et al. Lupus Sci Med. 2019;6[suppl 1]: Abstract 102. doi: 10.1136/lupus-2019-lsm.102
AT LUPUS 2019
Low-dose IL-2 found effective in SLE
SAN FRANCISCO – , according to the first randomized, double-blind, placebo-controlled clinical trial of the novel therapy.

Of note, more than half of the study participants with lupus nephritis experienced complete remission of their renal impairment, and another quarter had partial remission, Jing He, MD, PhD, reported at an international congress on systemic lupus erythematosus.
The mechanism of benefit appears to be the same as previously shown for low-dose interleukin-2 in patients with chronic graft versus host disease refractory to glucocorticoids (N Engl J Med. 2011 Dec 1;365[22]:2055-66): expansion of the deficient population of T regulatory cells, which is a hallmark of both inflammatory diseases.
“Low-dose IL-2 can reinstate the imbalance of T regulatory/T effector cells and improve immune homeostasis, which is critical in clinical remission of SLE,” said Dr. He of Peking University People’s Hospital in Beijing.
For nearly 20 years it has been known that SLE is characterized by very low levels of endogenous IL-2. Dr. He was lead author of the first proof-of-concept study, which showed low-dose subcutaneous IL-2 therapy resulted in markedly reduced SLE disease activity accompanied by expansion of the T regulatory cell population and suppression of follicular helper T cells and IL-17–producing helper T cells (Nat Med. 2016 Sep;22[9]:991-3). However, that was a small, single-center, uncontrolled study, so she and her coworkers have now carried out a 60-patient, double-blind, placebo-controlled randomized trial. In addition to hydroxychloroquine and other standard background medications, the patients in the active treatment arm received 1 million IU of IL-2 every other day for 2 weeks, followed by a 2-week hiatus, for a total of three courses.
At week 24 – 12 weeks after the last injection – the IL-2 recipients showed significantly greater improvement on numerous endpoints.
For example, the median SLE Disease Activity Index (SLEDAI) in the IL-2 group improved from 12 at baseline to 6 at week 12 and to 4 at week 24.
The marked improvement in renal impairment in the IL-2 recipients with lupus nephritis at baseline was accompanied by a significant increase in serum albumin and reduced 24-hour urinary protein, compared with controls.
The treatment was safe, with no increase in infections, severe or otherwise, and indeed with no serious adverse events of any kind, although nine patients in the IL-2 group experienced mild injection site reactions and three developed flu-like symptoms.
Dr. He reported having no financial conflicts regarding her study.
SAN FRANCISCO – , according to the first randomized, double-blind, placebo-controlled clinical trial of the novel therapy.
Of note, more than half of the study participants with lupus nephritis experienced complete remission of their renal impairment, and another quarter had partial remission, Jing He, MD, PhD, reported at an international congress on systemic lupus erythematosus.
The mechanism of benefit appears to be the same as previously shown for low-dose interleukin-2 in patients with chronic graft versus host disease refractory to glucocorticoids (N Engl J Med. 2011 Dec 1;365[22]:2055-66): expansion of the deficient population of T regulatory cells, which is a hallmark of both inflammatory diseases.
“Low-dose IL-2 can reinstate the imbalance of T regulatory/T effector cells and improve immune homeostasis, which is critical in clinical remission of SLE,” said Dr. He of Peking University People’s Hospital in Beijing.
For nearly 20 years it has been known that SLE is characterized by very low levels of endogenous IL-2. Dr. He was lead author of the first proof-of-concept study, which showed low-dose subcutaneous IL-2 therapy resulted in markedly reduced SLE disease activity accompanied by expansion of the T regulatory cell population and suppression of follicular helper T cells and IL-17–producing helper T cells (Nat Med. 2016 Sep;22[9]:991-3). However, that was a small, single-center, uncontrolled study, so she and her coworkers have now carried out a 60-patient, double-blind, placebo-controlled randomized trial. In addition to hydroxychloroquine and other standard background medications, the patients in the active treatment arm received 1 million IU of IL-2 every other day for 2 weeks, followed by a 2-week hiatus, for a total of three courses.
At week 24 – 12 weeks after the last injection – the IL-2 recipients showed significantly greater improvement on numerous endpoints.
For example, the median SLE Disease Activity Index (SLEDAI) in the IL-2 group improved from 12 at baseline to 6 at week 12 and to 4 at week 24.
The marked improvement in renal impairment in the IL-2 recipients with lupus nephritis at baseline was accompanied by a significant increase in serum albumin and reduced 24-hour urinary protein, compared with controls.
The treatment was safe, with no increase in infections, severe or otherwise, and indeed with no serious adverse events of any kind, although nine patients in the IL-2 group experienced mild injection site reactions and three developed flu-like symptoms.
Dr. He reported having no financial conflicts regarding her study.
SAN FRANCISCO – , according to the first randomized, double-blind, placebo-controlled clinical trial of the novel therapy.
Of note, more than half of the study participants with lupus nephritis experienced complete remission of their renal impairment, and another quarter had partial remission, Jing He, MD, PhD, reported at an international congress on systemic lupus erythematosus.
The mechanism of benefit appears to be the same as previously shown for low-dose interleukin-2 in patients with chronic graft versus host disease refractory to glucocorticoids (N Engl J Med. 2011 Dec 1;365[22]:2055-66): expansion of the deficient population of T regulatory cells, which is a hallmark of both inflammatory diseases.
“Low-dose IL-2 can reinstate the imbalance of T regulatory/T effector cells and improve immune homeostasis, which is critical in clinical remission of SLE,” said Dr. He of Peking University People’s Hospital in Beijing.
For nearly 20 years it has been known that SLE is characterized by very low levels of endogenous IL-2. Dr. He was lead author of the first proof-of-concept study, which showed low-dose subcutaneous IL-2 therapy resulted in markedly reduced SLE disease activity accompanied by expansion of the T regulatory cell population and suppression of follicular helper T cells and IL-17–producing helper T cells (Nat Med. 2016 Sep;22[9]:991-3). However, that was a small, single-center, uncontrolled study, so she and her coworkers have now carried out a 60-patient, double-blind, placebo-controlled randomized trial. In addition to hydroxychloroquine and other standard background medications, the patients in the active treatment arm received 1 million IU of IL-2 every other day for 2 weeks, followed by a 2-week hiatus, for a total of three courses.
At week 24 – 12 weeks after the last injection – the IL-2 recipients showed significantly greater improvement on numerous endpoints.
For example, the median SLE Disease Activity Index (SLEDAI) in the IL-2 group improved from 12 at baseline to 6 at week 12 and to 4 at week 24.
The marked improvement in renal impairment in the IL-2 recipients with lupus nephritis at baseline was accompanied by a significant increase in serum albumin and reduced 24-hour urinary protein, compared with controls.
The treatment was safe, with no increase in infections, severe or otherwise, and indeed with no serious adverse events of any kind, although nine patients in the IL-2 group experienced mild injection site reactions and three developed flu-like symptoms.
Dr. He reported having no financial conflicts regarding her study.
REPORTING FROM LUPUS 2019