User login
Skip ultrasound in acute UTI in small children
MALMO, SWEDEN – Ultrasound of the kidneys and urinary tract in the acute phase of a first urinary tract infection in young children has an unacceptably high false-positive rate, Magdalena Okarska-Napierala, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.

“Sonography performed 2 weeks after treatment initiation seems to be more reliable,” said Dr. Okarska-Napierala, a pediatrician at the Medical University of Warsaw Children’s Hospital.
Broad agreement exists that imaging is warranted in all children with a first urinary tract infection (UTI), because this infection can be the first signal of a structural abnormality of the kidneys or urinary tract. Abdominal ultrasound is the first-choice imaging modality in this setting because it is noninvasive, widely available, and inexpensive. But there remains controversy – and guidelines differ – regarding when to perform the ultrasound in children with UTI who respond well to therapy. This was the impetus for Dr. Okarska-Napierala and her coinvestigators to launch a prospective, single-center study examining the issue.
“The theory beneath it is the possibility that diffuse inflammation affects the ultrasound picture of the kidneys and urinary tract and may give us false-positive results, so we shouldn’t base our decisions on those results,” she explained.
This theory has been provisionally confirmed by the preliminary results of the study, which is continuing to enroll patients.
To date, the study includes 48 children, mean age 10.4 months, hospitalized for their first UTI. Participation was restricted to patients with no known congenital abnormalities of the kidneys or urinary tract and who were not on antibiotics at enrollment. Of the 48 children, 44 had an Escherichia coli infection. The predominant treatment was a second-generation cephalosporin for a median of 10 days.
On day 1 of treatment all patients underwent an ultrasound exam evaluating kidney size, anterior-posterior renal pelvis diameter, and the urinary tract based upon a grading system for urinary tract dilation developed by multidisciplinary consensus (J Pediatr Urol. 2014 Dec;10[6]:982-98). The ultrasound exam was repeated 2 weeks later, and again 2 weeks after that.
The most striking findings were a significantly increased kidney size and more prevalent urinary tract dilation on the day 1 ultrasound exam than on repeat ultrasound 2 weeks later. The average length of the left and right kidneys was 67.0 and 64.5 mm, respectively, on day 1, dropping off to 64.3 and 62.0 mm at 2 weeks, with a smaller and statistically nonsignificant further drop-off to 61.9 and 60.0 mm on the week 4 ultrasound.
“We saw a strong correlation between initial kidney size and CRP [C-reactive protein] value: The higher the CRP you have initially, the bigger the kidneys. It’s an interesting finding, but not so very practical. The only practical conclusion is that if we perform ultrasound at this stage and the child has big kidneys, it doesn’t mean anything. We have to check it again later,” she said.
Also, the number of renal units with urinary tract dilation went from 29 on day 1 ultrasound to 20 at 2 weeks and 19 at 4 weeks. Of the 48 children, 28 had urinary tract dilation on day 1, compared with 18 at 2 weeks and 16 at 4 weeks.
“If we look at this practically, if we base our decision on the day 1 ultrasound we would qualify half of all children for voiding cystourethrography, which is harmful, but if we wait 2 weeks to do the ultrasound we would reduce this number by six children. So I think we can call this a clinically significant difference,” she continued.
Of the 48 children, 11 have undergone voiding cystourethrography, revealing 2 mild cases of vesicoureteral reflux, which is the most common congenital abnormality of the urinary tract.
“I would like to emphasize that there is no real benefit in performing an ultrasound exam in children in this acute phase of infection. And there is harm in that we have to repeat the exam later, the parents are worried, the doctor is worried,” Dr. Okarska-Napierala concluded.
She reported having no relevant financial conflicts, and the study was conducted free of commercial support.
MALMO, SWEDEN – Ultrasound of the kidneys and urinary tract in the acute phase of a first urinary tract infection in young children has an unacceptably high false-positive rate, Magdalena Okarska-Napierala, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“Sonography performed 2 weeks after treatment initiation seems to be more reliable,” said Dr. Okarska-Napierala, a pediatrician at the Medical University of Warsaw Children’s Hospital.
Broad agreement exists that imaging is warranted in all children with a first urinary tract infection (UTI), because this infection can be the first signal of a structural abnormality of the kidneys or urinary tract. Abdominal ultrasound is the first-choice imaging modality in this setting because it is noninvasive, widely available, and inexpensive. But there remains controversy – and guidelines differ – regarding when to perform the ultrasound in children with UTI who respond well to therapy. This was the impetus for Dr. Okarska-Napierala and her coinvestigators to launch a prospective, single-center study examining the issue.
“The theory beneath it is the possibility that diffuse inflammation affects the ultrasound picture of the kidneys and urinary tract and may give us false-positive results, so we shouldn’t base our decisions on those results,” she explained.
This theory has been provisionally confirmed by the preliminary results of the study, which is continuing to enroll patients.
To date, the study includes 48 children, mean age 10.4 months, hospitalized for their first UTI. Participation was restricted to patients with no known congenital abnormalities of the kidneys or urinary tract and who were not on antibiotics at enrollment. Of the 48 children, 44 had an Escherichia coli infection. The predominant treatment was a second-generation cephalosporin for a median of 10 days.
On day 1 of treatment all patients underwent an ultrasound exam evaluating kidney size, anterior-posterior renal pelvis diameter, and the urinary tract based upon a grading system for urinary tract dilation developed by multidisciplinary consensus (J Pediatr Urol. 2014 Dec;10[6]:982-98). The ultrasound exam was repeated 2 weeks later, and again 2 weeks after that.
The most striking findings were a significantly increased kidney size and more prevalent urinary tract dilation on the day 1 ultrasound exam than on repeat ultrasound 2 weeks later. The average length of the left and right kidneys was 67.0 and 64.5 mm, respectively, on day 1, dropping off to 64.3 and 62.0 mm at 2 weeks, with a smaller and statistically nonsignificant further drop-off to 61.9 and 60.0 mm on the week 4 ultrasound.
“We saw a strong correlation between initial kidney size and CRP [C-reactive protein] value: The higher the CRP you have initially, the bigger the kidneys. It’s an interesting finding, but not so very practical. The only practical conclusion is that if we perform ultrasound at this stage and the child has big kidneys, it doesn’t mean anything. We have to check it again later,” she said.
Also, the number of renal units with urinary tract dilation went from 29 on day 1 ultrasound to 20 at 2 weeks and 19 at 4 weeks. Of the 48 children, 28 had urinary tract dilation on day 1, compared with 18 at 2 weeks and 16 at 4 weeks.
“If we look at this practically, if we base our decision on the day 1 ultrasound we would qualify half of all children for voiding cystourethrography, which is harmful, but if we wait 2 weeks to do the ultrasound we would reduce this number by six children. So I think we can call this a clinically significant difference,” she continued.
Of the 48 children, 11 have undergone voiding cystourethrography, revealing 2 mild cases of vesicoureteral reflux, which is the most common congenital abnormality of the urinary tract.
“I would like to emphasize that there is no real benefit in performing an ultrasound exam in children in this acute phase of infection. And there is harm in that we have to repeat the exam later, the parents are worried, the doctor is worried,” Dr. Okarska-Napierala concluded.
She reported having no relevant financial conflicts, and the study was conducted free of commercial support.
MALMO, SWEDEN – Ultrasound of the kidneys and urinary tract in the acute phase of a first urinary tract infection in young children has an unacceptably high false-positive rate, Magdalena Okarska-Napierala, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“Sonography performed 2 weeks after treatment initiation seems to be more reliable,” said Dr. Okarska-Napierala, a pediatrician at the Medical University of Warsaw Children’s Hospital.
Broad agreement exists that imaging is warranted in all children with a first urinary tract infection (UTI), because this infection can be the first signal of a structural abnormality of the kidneys or urinary tract. Abdominal ultrasound is the first-choice imaging modality in this setting because it is noninvasive, widely available, and inexpensive. But there remains controversy – and guidelines differ – regarding when to perform the ultrasound in children with UTI who respond well to therapy. This was the impetus for Dr. Okarska-Napierala and her coinvestigators to launch a prospective, single-center study examining the issue.
“The theory beneath it is the possibility that diffuse inflammation affects the ultrasound picture of the kidneys and urinary tract and may give us false-positive results, so we shouldn’t base our decisions on those results,” she explained.
This theory has been provisionally confirmed by the preliminary results of the study, which is continuing to enroll patients.
To date, the study includes 48 children, mean age 10.4 months, hospitalized for their first UTI. Participation was restricted to patients with no known congenital abnormalities of the kidneys or urinary tract and who were not on antibiotics at enrollment. Of the 48 children, 44 had an Escherichia coli infection. The predominant treatment was a second-generation cephalosporin for a median of 10 days.
On day 1 of treatment all patients underwent an ultrasound exam evaluating kidney size, anterior-posterior renal pelvis diameter, and the urinary tract based upon a grading system for urinary tract dilation developed by multidisciplinary consensus (J Pediatr Urol. 2014 Dec;10[6]:982-98). The ultrasound exam was repeated 2 weeks later, and again 2 weeks after that.
The most striking findings were a significantly increased kidney size and more prevalent urinary tract dilation on the day 1 ultrasound exam than on repeat ultrasound 2 weeks later. The average length of the left and right kidneys was 67.0 and 64.5 mm, respectively, on day 1, dropping off to 64.3 and 62.0 mm at 2 weeks, with a smaller and statistically nonsignificant further drop-off to 61.9 and 60.0 mm on the week 4 ultrasound.
“We saw a strong correlation between initial kidney size and CRP [C-reactive protein] value: The higher the CRP you have initially, the bigger the kidneys. It’s an interesting finding, but not so very practical. The only practical conclusion is that if we perform ultrasound at this stage and the child has big kidneys, it doesn’t mean anything. We have to check it again later,” she said.
Also, the number of renal units with urinary tract dilation went from 29 on day 1 ultrasound to 20 at 2 weeks and 19 at 4 weeks. Of the 48 children, 28 had urinary tract dilation on day 1, compared with 18 at 2 weeks and 16 at 4 weeks.
“If we look at this practically, if we base our decision on the day 1 ultrasound we would qualify half of all children for voiding cystourethrography, which is harmful, but if we wait 2 weeks to do the ultrasound we would reduce this number by six children. So I think we can call this a clinically significant difference,” she continued.
Of the 48 children, 11 have undergone voiding cystourethrography, revealing 2 mild cases of vesicoureteral reflux, which is the most common congenital abnormality of the urinary tract.
“I would like to emphasize that there is no real benefit in performing an ultrasound exam in children in this acute phase of infection. And there is harm in that we have to repeat the exam later, the parents are worried, the doctor is worried,” Dr. Okarska-Napierala concluded.
She reported having no relevant financial conflicts, and the study was conducted free of commercial support.
REPORTING FROM ESPID 2018
Key clinical point:
Major finding: Average left kidney length dropped from 67.0 mm on treatment day 1 to 64.3 mm 2 weeks later.
Study details: This interim report from an ongoing, prospective, single-center study included 48 children up to age 3 years who were hospitalized for their first urinary tract infection.
Disclosures: The presenter reported no relevant financial conflicts.
Recommendations aim to reduce pediatric nephrology testing
Evidence-based recommendations for appropriate nephrology testing in children are the latest installment of the American Board of Internal Medicine Foundation’s “Choosing Wisely” campaign.

The list includes recommendations on when not to order screening urine analyses and urine cultures, initiate hypertension workups, and place central lines. “Sometimes parents or physicians want to ensure all available testing is done, but unnecessary testing can create more fear, cost, and risk for children. Good communication and discussion of options can help reduce the likelihood of unnecessary testing,” said Doug Silverstein, MD, chairperson of the AAP section on nephrology.
Evidence-based recommendations for appropriate nephrology testing in children are the latest installment of the American Board of Internal Medicine Foundation’s “Choosing Wisely” campaign.
The list includes recommendations on when not to order screening urine analyses and urine cultures, initiate hypertension workups, and place central lines. “Sometimes parents or physicians want to ensure all available testing is done, but unnecessary testing can create more fear, cost, and risk for children. Good communication and discussion of options can help reduce the likelihood of unnecessary testing,” said Doug Silverstein, MD, chairperson of the AAP section on nephrology.
Evidence-based recommendations for appropriate nephrology testing in children are the latest installment of the American Board of Internal Medicine Foundation’s “Choosing Wisely” campaign.
The list includes recommendations on when not to order screening urine analyses and urine cultures, initiate hypertension workups, and place central lines. “Sometimes parents or physicians want to ensure all available testing is done, but unnecessary testing can create more fear, cost, and risk for children. Good communication and discussion of options can help reduce the likelihood of unnecessary testing,” said Doug Silverstein, MD, chairperson of the AAP section on nephrology.
CREDENCE canagliflozin trial halted because of efficacy
The CREDENCE trial, which was investigating whether the antidiabetes drug canagliflozin (Invokana) plus standard of care could safely help prevent or slow chronic kidney disease (CKD) in patients with type 2 diabetes, has been ended early because it has already achieved prespecified efficacy criteria, Janssen announced in a press release. These criteria included risk reduction in the composite endpoint of time to dialysis or kidney transplant, doubling of serum creatinine, and renal or cardiovascular death.
In CANVAS, the cardiovascular outcomes trial for canagliflozin, treatment was linked to reductions in progression of albuminuria and the composite outcome of a sustained 40% reduction in the estimated glomerular filtration rate, the need for renal replacement therapy, or death from renal causes, compared with placebo, but those didn’t reach statistical significance.
CREDENCE (Evaluation of the Effects of Canagliflozin on Renal and Cardiovascular Outcomes in Participants With Diabetic Nephropathy) is a randomized, double-blind, placebo-controlled, parallel-group, multicenter trial that enrolled roughly 4,400 patients with type 2 diabetes and established kidney disease who had been receiving ACE inhibitors or angiotensin II receptor blockers for at least 4 weeks prior to randomization.
The decision to halt CREDENCE came about after a review of data by the study’s independent data monitoring committee during a planned interim analysis. The resulting recommendation was based on the efficacy findings, the exact data for which have not yet been released.
Canagliflozin, a sodium-glucose transporter 2 (SGLT2) inhibitor, in conjunction with diet and exercise, can help improve glycemic control. In the context of kidney disease and type 2 diabetes, canagliflozin has been associated with increased risk of dehydration, vaginal or penile yeast infections, and amputations of all or part of the foot or leg. It has also been associated with ketoacidosis, kidney problems, hyperkalemia, hypoglycemia, and urinary tract infections.
More information can be found in the press release. Full prescribing information can be found on the Food and Drug Administration website.
The CREDENCE trial, which was investigating whether the antidiabetes drug canagliflozin (Invokana) plus standard of care could safely help prevent or slow chronic kidney disease (CKD) in patients with type 2 diabetes, has been ended early because it has already achieved prespecified efficacy criteria, Janssen announced in a press release. These criteria included risk reduction in the composite endpoint of time to dialysis or kidney transplant, doubling of serum creatinine, and renal or cardiovascular death.
In CANVAS, the cardiovascular outcomes trial for canagliflozin, treatment was linked to reductions in progression of albuminuria and the composite outcome of a sustained 40% reduction in the estimated glomerular filtration rate, the need for renal replacement therapy, or death from renal causes, compared with placebo, but those didn’t reach statistical significance.
CREDENCE (Evaluation of the Effects of Canagliflozin on Renal and Cardiovascular Outcomes in Participants With Diabetic Nephropathy) is a randomized, double-blind, placebo-controlled, parallel-group, multicenter trial that enrolled roughly 4,400 patients with type 2 diabetes and established kidney disease who had been receiving ACE inhibitors or angiotensin II receptor blockers for at least 4 weeks prior to randomization.
The decision to halt CREDENCE came about after a review of data by the study’s independent data monitoring committee during a planned interim analysis. The resulting recommendation was based on the efficacy findings, the exact data for which have not yet been released.
Canagliflozin, a sodium-glucose transporter 2 (SGLT2) inhibitor, in conjunction with diet and exercise, can help improve glycemic control. In the context of kidney disease and type 2 diabetes, canagliflozin has been associated with increased risk of dehydration, vaginal or penile yeast infections, and amputations of all or part of the foot or leg. It has also been associated with ketoacidosis, kidney problems, hyperkalemia, hypoglycemia, and urinary tract infections.
More information can be found in the press release. Full prescribing information can be found on the Food and Drug Administration website.
The CREDENCE trial, which was investigating whether the antidiabetes drug canagliflozin (Invokana) plus standard of care could safely help prevent or slow chronic kidney disease (CKD) in patients with type 2 diabetes, has been ended early because it has already achieved prespecified efficacy criteria, Janssen announced in a press release. These criteria included risk reduction in the composite endpoint of time to dialysis or kidney transplant, doubling of serum creatinine, and renal or cardiovascular death.
In CANVAS, the cardiovascular outcomes trial for canagliflozin, treatment was linked to reductions in progression of albuminuria and the composite outcome of a sustained 40% reduction in the estimated glomerular filtration rate, the need for renal replacement therapy, or death from renal causes, compared with placebo, but those didn’t reach statistical significance.
CREDENCE (Evaluation of the Effects of Canagliflozin on Renal and Cardiovascular Outcomes in Participants With Diabetic Nephropathy) is a randomized, double-blind, placebo-controlled, parallel-group, multicenter trial that enrolled roughly 4,400 patients with type 2 diabetes and established kidney disease who had been receiving ACE inhibitors or angiotensin II receptor blockers for at least 4 weeks prior to randomization.
The decision to halt CREDENCE came about after a review of data by the study’s independent data monitoring committee during a planned interim analysis. The resulting recommendation was based on the efficacy findings, the exact data for which have not yet been released.
Canagliflozin, a sodium-glucose transporter 2 (SGLT2) inhibitor, in conjunction with diet and exercise, can help improve glycemic control. In the context of kidney disease and type 2 diabetes, canagliflozin has been associated with increased risk of dehydration, vaginal or penile yeast infections, and amputations of all or part of the foot or leg. It has also been associated with ketoacidosis, kidney problems, hyperkalemia, hypoglycemia, and urinary tract infections.
More information can be found in the press release. Full prescribing information can be found on the Food and Drug Administration website.
Abatacept loses ALLURE in lupus nephritis
AMSTERDAM – Abatacept used on top of the standard of care did not improve the primary endpoint of a complete renal response versus placebo in the ALLURE phase 3 study.
Criteria for a complete renal response (CRR) at 1 year was met by 35.1% of abatacept-treated and 33.5% of placebo-treated patients (P = .73). CRR criteria included having a urine protein to creatinine ratio (UPCR) of less than 0.5, a normal estimated glomerular filtration rate (eGFR) or an eGFR of 85% or more of baseline values, no cellular casts, and a daily corticosteroid dose of 10 mg or less.

“We also saw a more rapid decline in proteinuria in those people treated with abatacept, and that seemed to be sustained over the course of the study,” said Dr. Furie, professor of medicine at Hofstra University, Hempstead, N.Y., chief of the division of rheumatology at Northwell Health in Great Neck, N.Y., and a professor at the Center for Autoimmune, Musculoskeletal, and Hematopoietic Diseases in the Feinstein Institute for Medical Research in Manhasset, N.Y. After about 12 weeks, the adjusted mean change in UPCR from baseline was –2.5 for abatacept and –2.0 for placebo; the values at 1 year were a respective –2.95 vs. –2.68 and at 2 years were –3.13 vs. –2.72.
Renal function was not negatively impacted by treatment with abatacept, with about a 5%-8% increase in eGFR seen in both groups.
Furthermore, improvements in lupus-related biomarkers were more pronounced in patients treated with abatacept than placebo, Dr. Furie said. This included a greater decrease in anti–double-stranded DNA autoantibody titers and an increase in complement C3 and C4 levels.
Eric Morand, MD, who was not involved in the ALLURE study, commented during discussion that the main result of the study was “very sad.”
Dr. Morand of Monash University in Melbourne observed that the duration of renal disease at study entry was about 14 months and that around 38% had been previously treated with mycophenolate mofetil (MMF). So, could this have influenced the findings?
Dr. Furie was unable to answer the question but confirmed that MMF was one of two background medications given in the trial, at an oral dose of 1.5 g/day, alongside of oral prednisone up to 60-mg daily.
ALLURE was a 2-year randomized, double-blind study with an open-ended, blinded, long-term extension in 405 patients with active class III or IV lupus nephritis. The aim of the trial was to determine the efficacy and safety of abatacept versus placebo in the treatment of active proliferative lupus nephritis.
Abatacept was given intravenously, first at a dose of 30 mg/kg on days 1, 15, 29, and 57, and then at a dose of 10 mg/kg every 4 weeks.
In terms of safety, 14 deaths occurred during the course of the study and its long-term extension. Seven abatacept patients died in year 1, two of whom died more than 56 days after discontinuing the study drug. Five patients in the placebo group died in year 1, one in year 2, and one in the long-term extension. Rates of any or serious adverse events were similar among the groups, decreasing over time.
“The safety signals were really no different to what we already know about abatacept,” Dr. Furie said. As for the future, more analyses from the trial can be expected, he added.
The study was sponsored by Bristol-Myers Squibb. Dr. Furie disclosed receiving grant or research support from, and acting as a consultant to, the company. All but 3 of the study’s 12 authors had financial ties to many pharmaceutical companies, some of which included Bristol-Myers Squibb. Two authors are employees of Bristol-Myers Squibb. Dr. Monash was not involved in the ALLURE study but has received research support from Bristol-Myers Squibb, among other pharmaceutical companies.
SOURCE: Furie RA et al. EULAR 2018. Abstract OP0253.
AMSTERDAM – Abatacept used on top of the standard of care did not improve the primary endpoint of a complete renal response versus placebo in the ALLURE phase 3 study.
Criteria for a complete renal response (CRR) at 1 year was met by 35.1% of abatacept-treated and 33.5% of placebo-treated patients (P = .73). CRR criteria included having a urine protein to creatinine ratio (UPCR) of less than 0.5, a normal estimated glomerular filtration rate (eGFR) or an eGFR of 85% or more of baseline values, no cellular casts, and a daily corticosteroid dose of 10 mg or less.
“We also saw a more rapid decline in proteinuria in those people treated with abatacept, and that seemed to be sustained over the course of the study,” said Dr. Furie, professor of medicine at Hofstra University, Hempstead, N.Y., chief of the division of rheumatology at Northwell Health in Great Neck, N.Y., and a professor at the Center for Autoimmune, Musculoskeletal, and Hematopoietic Diseases in the Feinstein Institute for Medical Research in Manhasset, N.Y. After about 12 weeks, the adjusted mean change in UPCR from baseline was –2.5 for abatacept and –2.0 for placebo; the values at 1 year were a respective –2.95 vs. –2.68 and at 2 years were –3.13 vs. –2.72.
Renal function was not negatively impacted by treatment with abatacept, with about a 5%-8% increase in eGFR seen in both groups.
Furthermore, improvements in lupus-related biomarkers were more pronounced in patients treated with abatacept than placebo, Dr. Furie said. This included a greater decrease in anti–double-stranded DNA autoantibody titers and an increase in complement C3 and C4 levels.
Eric Morand, MD, who was not involved in the ALLURE study, commented during discussion that the main result of the study was “very sad.”
Dr. Morand of Monash University in Melbourne observed that the duration of renal disease at study entry was about 14 months and that around 38% had been previously treated with mycophenolate mofetil (MMF). So, could this have influenced the findings?
Dr. Furie was unable to answer the question but confirmed that MMF was one of two background medications given in the trial, at an oral dose of 1.5 g/day, alongside of oral prednisone up to 60-mg daily.
ALLURE was a 2-year randomized, double-blind study with an open-ended, blinded, long-term extension in 405 patients with active class III or IV lupus nephritis. The aim of the trial was to determine the efficacy and safety of abatacept versus placebo in the treatment of active proliferative lupus nephritis.
Abatacept was given intravenously, first at a dose of 30 mg/kg on days 1, 15, 29, and 57, and then at a dose of 10 mg/kg every 4 weeks.
In terms of safety, 14 deaths occurred during the course of the study and its long-term extension. Seven abatacept patients died in year 1, two of whom died more than 56 days after discontinuing the study drug. Five patients in the placebo group died in year 1, one in year 2, and one in the long-term extension. Rates of any or serious adverse events were similar among the groups, decreasing over time.
“The safety signals were really no different to what we already know about abatacept,” Dr. Furie said. As for the future, more analyses from the trial can be expected, he added.
The study was sponsored by Bristol-Myers Squibb. Dr. Furie disclosed receiving grant or research support from, and acting as a consultant to, the company. All but 3 of the study’s 12 authors had financial ties to many pharmaceutical companies, some of which included Bristol-Myers Squibb. Two authors are employees of Bristol-Myers Squibb. Dr. Monash was not involved in the ALLURE study but has received research support from Bristol-Myers Squibb, among other pharmaceutical companies.
SOURCE: Furie RA et al. EULAR 2018. Abstract OP0253.
AMSTERDAM – Abatacept used on top of the standard of care did not improve the primary endpoint of a complete renal response versus placebo in the ALLURE phase 3 study.
Criteria for a complete renal response (CRR) at 1 year was met by 35.1% of abatacept-treated and 33.5% of placebo-treated patients (P = .73). CRR criteria included having a urine protein to creatinine ratio (UPCR) of less than 0.5, a normal estimated glomerular filtration rate (eGFR) or an eGFR of 85% or more of baseline values, no cellular casts, and a daily corticosteroid dose of 10 mg or less.
“We also saw a more rapid decline in proteinuria in those people treated with abatacept, and that seemed to be sustained over the course of the study,” said Dr. Furie, professor of medicine at Hofstra University, Hempstead, N.Y., chief of the division of rheumatology at Northwell Health in Great Neck, N.Y., and a professor at the Center for Autoimmune, Musculoskeletal, and Hematopoietic Diseases in the Feinstein Institute for Medical Research in Manhasset, N.Y. After about 12 weeks, the adjusted mean change in UPCR from baseline was –2.5 for abatacept and –2.0 for placebo; the values at 1 year were a respective –2.95 vs. –2.68 and at 2 years were –3.13 vs. –2.72.
Renal function was not negatively impacted by treatment with abatacept, with about a 5%-8% increase in eGFR seen in both groups.
Furthermore, improvements in lupus-related biomarkers were more pronounced in patients treated with abatacept than placebo, Dr. Furie said. This included a greater decrease in anti–double-stranded DNA autoantibody titers and an increase in complement C3 and C4 levels.
Eric Morand, MD, who was not involved in the ALLURE study, commented during discussion that the main result of the study was “very sad.”
Dr. Morand of Monash University in Melbourne observed that the duration of renal disease at study entry was about 14 months and that around 38% had been previously treated with mycophenolate mofetil (MMF). So, could this have influenced the findings?
Dr. Furie was unable to answer the question but confirmed that MMF was one of two background medications given in the trial, at an oral dose of 1.5 g/day, alongside of oral prednisone up to 60-mg daily.
ALLURE was a 2-year randomized, double-blind study with an open-ended, blinded, long-term extension in 405 patients with active class III or IV lupus nephritis. The aim of the trial was to determine the efficacy and safety of abatacept versus placebo in the treatment of active proliferative lupus nephritis.
Abatacept was given intravenously, first at a dose of 30 mg/kg on days 1, 15, 29, and 57, and then at a dose of 10 mg/kg every 4 weeks.
In terms of safety, 14 deaths occurred during the course of the study and its long-term extension. Seven abatacept patients died in year 1, two of whom died more than 56 days after discontinuing the study drug. Five patients in the placebo group died in year 1, one in year 2, and one in the long-term extension. Rates of any or serious adverse events were similar among the groups, decreasing over time.
“The safety signals were really no different to what we already know about abatacept,” Dr. Furie said. As for the future, more analyses from the trial can be expected, he added.
The study was sponsored by Bristol-Myers Squibb. Dr. Furie disclosed receiving grant or research support from, and acting as a consultant to, the company. All but 3 of the study’s 12 authors had financial ties to many pharmaceutical companies, some of which included Bristol-Myers Squibb. Two authors are employees of Bristol-Myers Squibb. Dr. Monash was not involved in the ALLURE study but has received research support from Bristol-Myers Squibb, among other pharmaceutical companies.
SOURCE: Furie RA et al. EULAR 2018. Abstract OP0253.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point: Abatacept treatment did not improve the complete renal response rate versus placebo.
Major finding: A complete renal response rate at 1 year was seen in 35.1% of abatacept-treated and 33.5% of placebo-treated patients (P = .73).
Study details: The phase 3 ALLURE study, a 2-year, randomized, double-blind study with an open-ended, blinded, long-term extension in 405 patients with active class III or IV lupus nephritis.
Disclosures: The study was sponsored by Bristol-Myers Squibb. Dr. Furie disclosed receiving grant or research support from, and acting as a consultant to, the company. All but 3 of the study’s 12 authors had financial ties to many pharmaceutical companies, some of which included Bristol-Myers Squibb. Two authors are employees of Bristol-Myers Squibb. Dr. Monash was not involved in the ALLURE study but has received research support from Bristol-Myers Squibb, among other pharmaceutical companies.
Source: Furie RA et al. EULAR 2018. Abstract OP0253.
Early BCC seen in teen kidney transplant patient
A 17-year-old girl seen in a Portuguese dermatology clinic was found to have a nodular basal cell carcinoma on the parietal region of her scalp. The nodule appeared 6 years after she had received a kidney transplant, according to João Borges-Costa, MD, PhD, who submitted the case report.
Since the transplant, the girl had been maintained on immunosuppressive medication of tacrolimus 1 mg twice daily, mycophenolate sodium 360 mg twice daily, and prednisolone 10 mg every other day. The 1-cm nodule was pigmented; dermatoscopy did not yield clarity about whether the lesion was melanocytic. An excisional biopsy with 0.5-cm margins was performed, and histology confirmed that the lesion was a nodular pigmented basal cell carcinoma that had been excised completely.
The case, said Dr. Borges-Costa, shows that skin cancers can develop earlier than the typical 12-18 years after pediatric transplantation. Most reported cases have been squamous cell cancers and melanomas, and often are associated with lack of appropriate sun protection behavior.
The patient, a Caucasian, was a sailor who used sunscreen but did not typically wear a hat while sailing, reported Dr. Borges-Costa, a dermatologist at the University of Lisbon. Her family history was significant for a grandparent with melanoma.
Dr. Borges noted that the parents and patient were given advice regarding the importance of the lifelong use of sun-protective clothing and headgear. “Education of pediatric organ recipients and their parents about sun protection is important because, as occurred with our patient, protective clothing and hats are frequently forgotten.”
Because of the ongoing potential for skin malignancies, early referral “after transplantation to specialized dermatology outpatient clinics, similar to what is now advocated for transplanted adults, could help in surveillance and improve adherence to sun-protective measures,” he added.
SOURCE: Borges-Costa J et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13537..
A 17-year-old girl seen in a Portuguese dermatology clinic was found to have a nodular basal cell carcinoma on the parietal region of her scalp. The nodule appeared 6 years after she had received a kidney transplant, according to João Borges-Costa, MD, PhD, who submitted the case report.
Since the transplant, the girl had been maintained on immunosuppressive medication of tacrolimus 1 mg twice daily, mycophenolate sodium 360 mg twice daily, and prednisolone 10 mg every other day. The 1-cm nodule was pigmented; dermatoscopy did not yield clarity about whether the lesion was melanocytic. An excisional biopsy with 0.5-cm margins was performed, and histology confirmed that the lesion was a nodular pigmented basal cell carcinoma that had been excised completely.
The case, said Dr. Borges-Costa, shows that skin cancers can develop earlier than the typical 12-18 years after pediatric transplantation. Most reported cases have been squamous cell cancers and melanomas, and often are associated with lack of appropriate sun protection behavior.
The patient, a Caucasian, was a sailor who used sunscreen but did not typically wear a hat while sailing, reported Dr. Borges-Costa, a dermatologist at the University of Lisbon. Her family history was significant for a grandparent with melanoma.
Dr. Borges noted that the parents and patient were given advice regarding the importance of the lifelong use of sun-protective clothing and headgear. “Education of pediatric organ recipients and their parents about sun protection is important because, as occurred with our patient, protective clothing and hats are frequently forgotten.”
Because of the ongoing potential for skin malignancies, early referral “after transplantation to specialized dermatology outpatient clinics, similar to what is now advocated for transplanted adults, could help in surveillance and improve adherence to sun-protective measures,” he added.
SOURCE: Borges-Costa J et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13537..
A 17-year-old girl seen in a Portuguese dermatology clinic was found to have a nodular basal cell carcinoma on the parietal region of her scalp. The nodule appeared 6 years after she had received a kidney transplant, according to João Borges-Costa, MD, PhD, who submitted the case report.
Since the transplant, the girl had been maintained on immunosuppressive medication of tacrolimus 1 mg twice daily, mycophenolate sodium 360 mg twice daily, and prednisolone 10 mg every other day. The 1-cm nodule was pigmented; dermatoscopy did not yield clarity about whether the lesion was melanocytic. An excisional biopsy with 0.5-cm margins was performed, and histology confirmed that the lesion was a nodular pigmented basal cell carcinoma that had been excised completely.
The case, said Dr. Borges-Costa, shows that skin cancers can develop earlier than the typical 12-18 years after pediatric transplantation. Most reported cases have been squamous cell cancers and melanomas, and often are associated with lack of appropriate sun protection behavior.
The patient, a Caucasian, was a sailor who used sunscreen but did not typically wear a hat while sailing, reported Dr. Borges-Costa, a dermatologist at the University of Lisbon. Her family history was significant for a grandparent with melanoma.
Dr. Borges noted that the parents and patient were given advice regarding the importance of the lifelong use of sun-protective clothing and headgear. “Education of pediatric organ recipients and their parents about sun protection is important because, as occurred with our patient, protective clothing and hats are frequently forgotten.”
Because of the ongoing potential for skin malignancies, early referral “after transplantation to specialized dermatology outpatient clinics, similar to what is now advocated for transplanted adults, could help in surveillance and improve adherence to sun-protective measures,” he added.
SOURCE: Borges-Costa J et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13537..
FROM PEDIATRIC DERMATOLOGY
Uric acid tied to pediatric diabetic kidney disease
ORLANDO – , according to a 7-year investigation of 539 children.
Every 1-mg/dL climb in baseline serum uric acid increased the risk of subsequent elevated urine albumin excretion 1.23 fold, after adjustment for potential confounders (P = .02).
The finding adds to growing evidence that serum uric acid (SUA) isn’t just a marker of diabetic kidney disease, but a contributor to it. “There is definitely” cross-talk between gout and diabetes, said lead investigator Petter Bjornstad, MD, assistant professor of pediatric endocrinology at the University of Colorado, Aurora.
Elevated SUA is common in both conditions and a risk factor for kidney disease. Newer studies have linked higher levels to nephron number decline and other pathologies, perhaps through renal inflammation. Allopurinol, the traditional uric acid lowering agent in gout, is already under investigation to prevent kidney decline in adults with type 1 diabetes mellitus. There’s also evidence that the potent uric acid lowering agent, febuxostat (Uloric), attenuates hypofiltration in early diabetic kidney disease.

The 539 children, all part of the Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) trial, were assessed annually over a mean of 5.7 years. At baseline, they were 13.9 years old and had T2DM for 7.9 months, on average. The mean body mass index was 34.6 kg/m2, mean hemoglobin A1c was 6%.
Almost 20% of the children were hypertensive at baseline (130/80 mm Hg or higher); 26% were hyperuricemic (6.8 mg/dL or higher); and 6.1% had elevated urine albumin excretion (urine albumin creatinine ratio of at least 30 mg/g), a marker of renal pathology. At the end of follow-up, 18% had elevated albumin excretion and 37.4% were hypertensive.
“Hyperuricemia was common in youth with type 2 diabetes,” just as it’s been shown in adults with the disease. “Higher baseline SUA independently increase[s] risk for onset of hypertension and elevated urine albumin excretion,” Dr. Bjornstad said.
However, the association between SUA and elevated albumin excretion was statistically significant only in boys – 36% of the study population – and non-Hispanic whites, 20% of the subjects, after adjustment for BMI, hemoglobin A1c, estimated glomerular filtration rate, and use of ACE inhibitors and angiotensin II receptor blockers.
The National Institutes of Health funded the work. Dr. Bjornstad is a consultant for Boehringer Ingelheim.
SOURCE: Bjornstad P et al. ADA 2018, abstract 339-OR.
ORLANDO – , according to a 7-year investigation of 539 children.
Every 1-mg/dL climb in baseline serum uric acid increased the risk of subsequent elevated urine albumin excretion 1.23 fold, after adjustment for potential confounders (P = .02).
The finding adds to growing evidence that serum uric acid (SUA) isn’t just a marker of diabetic kidney disease, but a contributor to it. “There is definitely” cross-talk between gout and diabetes, said lead investigator Petter Bjornstad, MD, assistant professor of pediatric endocrinology at the University of Colorado, Aurora.
Elevated SUA is common in both conditions and a risk factor for kidney disease. Newer studies have linked higher levels to nephron number decline and other pathologies, perhaps through renal inflammation. Allopurinol, the traditional uric acid lowering agent in gout, is already under investigation to prevent kidney decline in adults with type 1 diabetes mellitus. There’s also evidence that the potent uric acid lowering agent, febuxostat (Uloric), attenuates hypofiltration in early diabetic kidney disease.
The 539 children, all part of the Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) trial, were assessed annually over a mean of 5.7 years. At baseline, they were 13.9 years old and had T2DM for 7.9 months, on average. The mean body mass index was 34.6 kg/m2, mean hemoglobin A1c was 6%.
Almost 20% of the children were hypertensive at baseline (130/80 mm Hg or higher); 26% were hyperuricemic (6.8 mg/dL or higher); and 6.1% had elevated urine albumin excretion (urine albumin creatinine ratio of at least 30 mg/g), a marker of renal pathology. At the end of follow-up, 18% had elevated albumin excretion and 37.4% were hypertensive.
“Hyperuricemia was common in youth with type 2 diabetes,” just as it’s been shown in adults with the disease. “Higher baseline SUA independently increase[s] risk for onset of hypertension and elevated urine albumin excretion,” Dr. Bjornstad said.
However, the association between SUA and elevated albumin excretion was statistically significant only in boys – 36% of the study population – and non-Hispanic whites, 20% of the subjects, after adjustment for BMI, hemoglobin A1c, estimated glomerular filtration rate, and use of ACE inhibitors and angiotensin II receptor blockers.
The National Institutes of Health funded the work. Dr. Bjornstad is a consultant for Boehringer Ingelheim.
SOURCE: Bjornstad P et al. ADA 2018, abstract 339-OR.
ORLANDO – , according to a 7-year investigation of 539 children.
Every 1-mg/dL climb in baseline serum uric acid increased the risk of subsequent elevated urine albumin excretion 1.23 fold, after adjustment for potential confounders (P = .02).
The finding adds to growing evidence that serum uric acid (SUA) isn’t just a marker of diabetic kidney disease, but a contributor to it. “There is definitely” cross-talk between gout and diabetes, said lead investigator Petter Bjornstad, MD, assistant professor of pediatric endocrinology at the University of Colorado, Aurora.
Elevated SUA is common in both conditions and a risk factor for kidney disease. Newer studies have linked higher levels to nephron number decline and other pathologies, perhaps through renal inflammation. Allopurinol, the traditional uric acid lowering agent in gout, is already under investigation to prevent kidney decline in adults with type 1 diabetes mellitus. There’s also evidence that the potent uric acid lowering agent, febuxostat (Uloric), attenuates hypofiltration in early diabetic kidney disease.
The 539 children, all part of the Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) trial, were assessed annually over a mean of 5.7 years. At baseline, they were 13.9 years old and had T2DM for 7.9 months, on average. The mean body mass index was 34.6 kg/m2, mean hemoglobin A1c was 6%.
Almost 20% of the children were hypertensive at baseline (130/80 mm Hg or higher); 26% were hyperuricemic (6.8 mg/dL or higher); and 6.1% had elevated urine albumin excretion (urine albumin creatinine ratio of at least 30 mg/g), a marker of renal pathology. At the end of follow-up, 18% had elevated albumin excretion and 37.4% were hypertensive.
“Hyperuricemia was common in youth with type 2 diabetes,” just as it’s been shown in adults with the disease. “Higher baseline SUA independently increase[s] risk for onset of hypertension and elevated urine albumin excretion,” Dr. Bjornstad said.
However, the association between SUA and elevated albumin excretion was statistically significant only in boys – 36% of the study population – and non-Hispanic whites, 20% of the subjects, after adjustment for BMI, hemoglobin A1c, estimated glomerular filtration rate, and use of ACE inhibitors and angiotensin II receptor blockers.
The National Institutes of Health funded the work. Dr. Bjornstad is a consultant for Boehringer Ingelheim.
SOURCE: Bjornstad P et al. ADA 2018, abstract 339-OR.
REPORTING FROM ADA 2018
Key clinical point: Serum uric acid lowering might help prevent kidney disease in children with T2DM.
Major finding: Every1-mg/dL climb in baseline serum uric acid increased the risk of subsequent elevated urine albumin excretion 1.23 fold, after adjustment for potential confounders (P = .02)
Study details: Seven-year investigation of 539 children with new-onset T2DM.
Disclosures: The National Institutes of Health funded the work. The study lead is a consultant for Boehringer Ingelheim.
Source: Bjornstad P et al. ADA 2018 Abstract 339-OR.
Study spotlights risk factors for albuminuria in youth with T2DM
TORONTO – When Brandy Wicklow, MD, began her pediatric endocrinology fellowship at McGill University in 2006, about 12 per 100,000 children in Manitoba, Canada, were diagnosed with type 2 diabetes mellitus each year. By 2016 that rate had more than doubled, to 26 per 100,000 children.
“If you look just at indigenous youth in our province, it’s probably one of the highest rates ever reported, with 95 per 100,000 Manitoba First Nation children diagnosed with type 2 diabetes,” said Dr. Wicklow, a pediatric endocrinologist at the University of Manitoba and the Children’s Hospital Research Institute of Manitoba.
Many indigenous populations also face an increased risk for primary renal disease. One study reviewed the charts 90 of Canadian First Nation children and adolescents with T2DM (Diabetes Care. 2009;32[5]:786-90). Of 10 who had renal biopsies performed, nine had immune complex disease/glomerulosclerosis, two had mild diabetes-related lesions, and seven had focal segmental glomerulosclerosis (FSGS); yet none had classic nephropathy. An analysis of Chinese youth that included 216 renal biopsies yielded similar findings (Intl Urol Nephrol. 2012;45[1]:173-9).
It’s also known that early-onset T2DM is associated with substantially increased incidence of end-stage renal disease (ESRD) and mortality in middle age. For example, one study of Pima Indians found that those who were diagnosed with T2DM earlier than 20 years of age had a one in five chance of developing ESRD, while those who were diagnosed at age 20 years or older had a one in two chance of ESRD (JAMA. 2006;296[4]:421-6). In a separate analysis, researchers estimated the remaining lifetime risks for ESRD among Aboriginal people in Australia with and without diabetes (Diabetes Res Clin Pract. 2014;103[3]:e24-6). The value for young adults with diabetes was high, about one in two at the age of 30 years, while it decreased with age to one in seven at 60 years.
“One of the first biomarkers we see in terms of renal disease in kids with T2DM is albuminuria,” Dr. Wicklow said at the Pediatric Academic Societies meeting. “The question is, why do kids with type 2 get more renal disease than kids with type 1 diabetes?” The SEARCH for Diabetes in Youth (SEARCH) study from 2006 found that hypertension, increased body mass index, increased weight circumference, and increased lipids were factors, while the SEARCH study from 2015 found that ethnicity, increased weight to height ratio, and mean arterial pressure were factors.
“Insulin resistance is significantly associated with albuminuria,” Dr. Wicklow continued. “It’s also been shown to be associated with hyperfiltration. Some of the markers of insulin resistance are important but they make up about 19% of the variance between type 1 and type 2, which means there are other variables that we’re not measuring.”
Enter ICARE (Improving Renal Complications in Adolescents with Type 2 Diabetes through Research), an ongoing prospective cohort study that Dr. Wicklow and her associates launched in 2014 at eight centers in Canada. It aims to examine the biopsychosocial risk factors for albuminuria in youth with T2DM and the mechanisms for renal injury. “Our theoretical framework was that biological exposures that we are aware of, such as glycemic control, hypertension, and lipids, would all be important in the development of albuminuria and renal disease in kids,” said Dr. Wicklow, who is the study’s coprimary investigator along with Allison Dart, MD. “But what we thought was novel was that psychological exposures either as socioeconomic status or as mental health factors would also directly impinge on renal health with respect to chronic inflammation in the body, inflammation in the kidneys, and long-term kidney damage.”

The first phase of ICARE involved a detailed phenotypic assessment of youth, including anthropometrics, biochemistry, 24-hour ambulatory blood pressure monitoring, overnight urine collections for albumin excretion, renal ultrasound, and iohexol-derived glomerular filtration rate (GFR). Phase 2 included an evaluation of psychological factors, including hair-derived cortisol; validated questionnaires for perceived stress, distress, and resiliency; and a detailed evaluation of systemic and urine inflammatory biomarkers. Annual follow-up is carried out to assess temporal associations between clinical risk factors and renal outcomes, including progression of albuminuria.
At the meeting, Dr. Wicklow reported on 187 youth enrolled to date. Of these, 96% were of indigenous ethnicity, 57 had albuminuria and 130 did not, and the mean ages of the groups were 16 years and 15 years, respectively. At baseline, a higher proportion of those in the albuminuria group were female (74% vs. 64% of those in the no albuminuria group, respectively), had a higher mean hemoglobin A1c (11% vs. 9%), and had hypertension (94% vs. 72%). She noted that upon presentation to the clinic, only 23% of participants had HbA1c levels less than 7%, only 26% had ranges between 7% and 9%, and about 40% did not have any hypertension. Of those who did, 27% had nighttime-only hypertension, and only 2% had daytime-only hypertension.
“The other risk factor these kids have for developing ESRD is that the majority were exposed to diabetes in pregnancy,” Dr. Wicklow said. “Murine models of maternal diabetes exposure have demonstrated that offspring have small kidneys, less ureteric bud branching, and a lower number of nephrons. Most of the human clinical cohort studies look at associations between development of diabetes and parental hypertension, maternal smoking, and maternal education. There is likely an impact at birth that sets these kids up for development of type 2 diabetes.”
In addition, results from clinical cohort studies have found that depression, mental stress, and distress are high in youth with T2DM. “Preliminary data suggest that if you have positive mental health, or coping strategies, or someone has worked through this with you and you are resilient, you might benefit in terms of overall glycemic control,” she said. For example, ICARE investigators have found that the higher the score on the Kessler Psychological Distress Scale (K6), the greater the risk of renal inflammation as measured by monocyte chemotactic protein-1 (MCP-1; P = .02). “Mental health seems to be something that can directly impact your health from a biological standpoint, and we might be able to find biomarkers of that risk,” Dr. Wicklow said. “Where does the stress come from? Most of my patients are indigenous, so it’s not surprising that the history in Canada of colonization of residential schools has left a lasting impression on these families and communities in terms of loss of language, loss of culture, and loss of land. There’s a community-based stress and a family-based stress that these children feel.”
Social factors also play a big role. She presented baseline findings from 196 youth with T2DM and 456 with T1DM, including measures such as the Socioeconomic Factor Index-Version 2 (SEFI-2), a way to assess socioeconomic characteristics based on Canadian Census data that reflects nonmedical social determinants of health. “It looks at factors like number of rooms in the house, single-parent households, maternal education attainment, and family income,” Dr. Wicklow explained. “The higher the SEFI-2 score, the lower your socioeconomic status is for the area you live in. Kids with T2DM generally live in areas of lower SES and lower socioeconomic index. They often live far away from health care providers. Many do not attend school and many are not with their biologic families, so we’ve had a lot of issues addressing child and family services, in particular in the phase of a chronic illness where our expectation is one thing and the family’s and community’s expectations of what’s realistic in terms of treatment and goals is another. We also have a lot of adolescent pregnancies.”

To date, about 80% of youth with T1D have seen a health care provider within the first year after transition from the pediatric diabetes clinic, compared with just over 50% of kids with T2D. “We transition youth with T1DM to internists, while our youth with T2DM go to itinerant physicians often back in their communities and/or rural family physicians,” she said. Between baseline and year 2, the rate of hospital admissions remained similar among T1DM at 11.6 and 11.8 admissions per 100 patient-years, respectively, but the number of hospital admissions for T2DM patients jumped from 20.1 to 25.5 admissions per 100 patient-years. “Kids with type 2 are showing up in the hospital a lot more than those with type 1 diabetes, but not for diabetes-related diagnoses,” Dr. Wicklow said. “We’re starting to look through the data now, and most of our kids are showing up with mental health complaints and issues. That’s why they’re getting hospitalized.”
Among ICARE study participants who have completed 3 years of follow-up, about 52% had albuminuria at their baseline visit and 48% sustained albuminuria throughout the study. About 26% progressed from normal levels of albuminuria to microalbuminuria, from microalbuminuria to macroalbuminuria, or from normal levels of albuminuria to macroalbuminuria. In addition, 16% persisted in the category that they were in, and 10% regressed. “The good news is, some of our kids get better over time,” Dr. Wicklow said. “The bad news is that the majority do not.”

Going forward, Dr. Wicklow and her associates work with an ICARE advisory group composed of children and families “who sit with us and talk about what mental health needs might be important, and how we should organize our study in a follow-up of the kids, to try and answer some of the questions that are important,” she said. “Working with the concept of the study’s theoretical framework, they acknowledged that the biological exposures are important, but they were also concerned about food security, finding strength/resilience within the community, and finding coping factors in terms of keeping themselves healthy with their diabetes. For some communities, they are concerned with basic needs. We’re working with them to help them progress, and to figure out how to best study children with type 2 diabetes.”

ICARE has received support from Diabetes Canada, Research Manitoba, the Canadian Institutes of Health Research, the Children’s Hospital Research Institute of Manitoba (specifically the Diabetes Research Envisioned and Accomplished in Manitoba (DREAM) theme), and the University of Manitoba. Dr. Wicklow reported having no financial disclosures.
TORONTO – When Brandy Wicklow, MD, began her pediatric endocrinology fellowship at McGill University in 2006, about 12 per 100,000 children in Manitoba, Canada, were diagnosed with type 2 diabetes mellitus each year. By 2016 that rate had more than doubled, to 26 per 100,000 children.
“If you look just at indigenous youth in our province, it’s probably one of the highest rates ever reported, with 95 per 100,000 Manitoba First Nation children diagnosed with type 2 diabetes,” said Dr. Wicklow, a pediatric endocrinologist at the University of Manitoba and the Children’s Hospital Research Institute of Manitoba.
Many indigenous populations also face an increased risk for primary renal disease. One study reviewed the charts 90 of Canadian First Nation children and adolescents with T2DM (Diabetes Care. 2009;32[5]:786-90). Of 10 who had renal biopsies performed, nine had immune complex disease/glomerulosclerosis, two had mild diabetes-related lesions, and seven had focal segmental glomerulosclerosis (FSGS); yet none had classic nephropathy. An analysis of Chinese youth that included 216 renal biopsies yielded similar findings (Intl Urol Nephrol. 2012;45[1]:173-9).
It’s also known that early-onset T2DM is associated with substantially increased incidence of end-stage renal disease (ESRD) and mortality in middle age. For example, one study of Pima Indians found that those who were diagnosed with T2DM earlier than 20 years of age had a one in five chance of developing ESRD, while those who were diagnosed at age 20 years or older had a one in two chance of ESRD (JAMA. 2006;296[4]:421-6). In a separate analysis, researchers estimated the remaining lifetime risks for ESRD among Aboriginal people in Australia with and without diabetes (Diabetes Res Clin Pract. 2014;103[3]:e24-6). The value for young adults with diabetes was high, about one in two at the age of 30 years, while it decreased with age to one in seven at 60 years.
“One of the first biomarkers we see in terms of renal disease in kids with T2DM is albuminuria,” Dr. Wicklow said at the Pediatric Academic Societies meeting. “The question is, why do kids with type 2 get more renal disease than kids with type 1 diabetes?” The SEARCH for Diabetes in Youth (SEARCH) study from 2006 found that hypertension, increased body mass index, increased weight circumference, and increased lipids were factors, while the SEARCH study from 2015 found that ethnicity, increased weight to height ratio, and mean arterial pressure were factors.
“Insulin resistance is significantly associated with albuminuria,” Dr. Wicklow continued. “It’s also been shown to be associated with hyperfiltration. Some of the markers of insulin resistance are important but they make up about 19% of the variance between type 1 and type 2, which means there are other variables that we’re not measuring.”
Enter ICARE (Improving Renal Complications in Adolescents with Type 2 Diabetes through Research), an ongoing prospective cohort study that Dr. Wicklow and her associates launched in 2014 at eight centers in Canada. It aims to examine the biopsychosocial risk factors for albuminuria in youth with T2DM and the mechanisms for renal injury. “Our theoretical framework was that biological exposures that we are aware of, such as glycemic control, hypertension, and lipids, would all be important in the development of albuminuria and renal disease in kids,” said Dr. Wicklow, who is the study’s coprimary investigator along with Allison Dart, MD. “But what we thought was novel was that psychological exposures either as socioeconomic status or as mental health factors would also directly impinge on renal health with respect to chronic inflammation in the body, inflammation in the kidneys, and long-term kidney damage.”
The first phase of ICARE involved a detailed phenotypic assessment of youth, including anthropometrics, biochemistry, 24-hour ambulatory blood pressure monitoring, overnight urine collections for albumin excretion, renal ultrasound, and iohexol-derived glomerular filtration rate (GFR). Phase 2 included an evaluation of psychological factors, including hair-derived cortisol; validated questionnaires for perceived stress, distress, and resiliency; and a detailed evaluation of systemic and urine inflammatory biomarkers. Annual follow-up is carried out to assess temporal associations between clinical risk factors and renal outcomes, including progression of albuminuria.
At the meeting, Dr. Wicklow reported on 187 youth enrolled to date. Of these, 96% were of indigenous ethnicity, 57 had albuminuria and 130 did not, and the mean ages of the groups were 16 years and 15 years, respectively. At baseline, a higher proportion of those in the albuminuria group were female (74% vs. 64% of those in the no albuminuria group, respectively), had a higher mean hemoglobin A1c (11% vs. 9%), and had hypertension (94% vs. 72%). She noted that upon presentation to the clinic, only 23% of participants had HbA1c levels less than 7%, only 26% had ranges between 7% and 9%, and about 40% did not have any hypertension. Of those who did, 27% had nighttime-only hypertension, and only 2% had daytime-only hypertension.
“The other risk factor these kids have for developing ESRD is that the majority were exposed to diabetes in pregnancy,” Dr. Wicklow said. “Murine models of maternal diabetes exposure have demonstrated that offspring have small kidneys, less ureteric bud branching, and a lower number of nephrons. Most of the human clinical cohort studies look at associations between development of diabetes and parental hypertension, maternal smoking, and maternal education. There is likely an impact at birth that sets these kids up for development of type 2 diabetes.”
In addition, results from clinical cohort studies have found that depression, mental stress, and distress are high in youth with T2DM. “Preliminary data suggest that if you have positive mental health, or coping strategies, or someone has worked through this with you and you are resilient, you might benefit in terms of overall glycemic control,” she said. For example, ICARE investigators have found that the higher the score on the Kessler Psychological Distress Scale (K6), the greater the risk of renal inflammation as measured by monocyte chemotactic protein-1 (MCP-1; P = .02). “Mental health seems to be something that can directly impact your health from a biological standpoint, and we might be able to find biomarkers of that risk,” Dr. Wicklow said. “Where does the stress come from? Most of my patients are indigenous, so it’s not surprising that the history in Canada of colonization of residential schools has left a lasting impression on these families and communities in terms of loss of language, loss of culture, and loss of land. There’s a community-based stress and a family-based stress that these children feel.”
Social factors also play a big role. She presented baseline findings from 196 youth with T2DM and 456 with T1DM, including measures such as the Socioeconomic Factor Index-Version 2 (SEFI-2), a way to assess socioeconomic characteristics based on Canadian Census data that reflects nonmedical social determinants of health. “It looks at factors like number of rooms in the house, single-parent households, maternal education attainment, and family income,” Dr. Wicklow explained. “The higher the SEFI-2 score, the lower your socioeconomic status is for the area you live in. Kids with T2DM generally live in areas of lower SES and lower socioeconomic index. They often live far away from health care providers. Many do not attend school and many are not with their biologic families, so we’ve had a lot of issues addressing child and family services, in particular in the phase of a chronic illness where our expectation is one thing and the family’s and community’s expectations of what’s realistic in terms of treatment and goals is another. We also have a lot of adolescent pregnancies.”
To date, about 80% of youth with T1D have seen a health care provider within the first year after transition from the pediatric diabetes clinic, compared with just over 50% of kids with T2D. “We transition youth with T1DM to internists, while our youth with T2DM go to itinerant physicians often back in their communities and/or rural family physicians,” she said. Between baseline and year 2, the rate of hospital admissions remained similar among T1DM at 11.6 and 11.8 admissions per 100 patient-years, respectively, but the number of hospital admissions for T2DM patients jumped from 20.1 to 25.5 admissions per 100 patient-years. “Kids with type 2 are showing up in the hospital a lot more than those with type 1 diabetes, but not for diabetes-related diagnoses,” Dr. Wicklow said. “We’re starting to look through the data now, and most of our kids are showing up with mental health complaints and issues. That’s why they’re getting hospitalized.”
Among ICARE study participants who have completed 3 years of follow-up, about 52% had albuminuria at their baseline visit and 48% sustained albuminuria throughout the study. About 26% progressed from normal levels of albuminuria to microalbuminuria, from microalbuminuria to macroalbuminuria, or from normal levels of albuminuria to macroalbuminuria. In addition, 16% persisted in the category that they were in, and 10% regressed. “The good news is, some of our kids get better over time,” Dr. Wicklow said. “The bad news is that the majority do not.”
Going forward, Dr. Wicklow and her associates work with an ICARE advisory group composed of children and families “who sit with us and talk about what mental health needs might be important, and how we should organize our study in a follow-up of the kids, to try and answer some of the questions that are important,” she said. “Working with the concept of the study’s theoretical framework, they acknowledged that the biological exposures are important, but they were also concerned about food security, finding strength/resilience within the community, and finding coping factors in terms of keeping themselves healthy with their diabetes. For some communities, they are concerned with basic needs. We’re working with them to help them progress, and to figure out how to best study children with type 2 diabetes.”
ICARE has received support from Diabetes Canada, Research Manitoba, the Canadian Institutes of Health Research, the Children’s Hospital Research Institute of Manitoba (specifically the Diabetes Research Envisioned and Accomplished in Manitoba (DREAM) theme), and the University of Manitoba. Dr. Wicklow reported having no financial disclosures.
TORONTO – When Brandy Wicklow, MD, began her pediatric endocrinology fellowship at McGill University in 2006, about 12 per 100,000 children in Manitoba, Canada, were diagnosed with type 2 diabetes mellitus each year. By 2016 that rate had more than doubled, to 26 per 100,000 children.
“If you look just at indigenous youth in our province, it’s probably one of the highest rates ever reported, with 95 per 100,000 Manitoba First Nation children diagnosed with type 2 diabetes,” said Dr. Wicklow, a pediatric endocrinologist at the University of Manitoba and the Children’s Hospital Research Institute of Manitoba.
Many indigenous populations also face an increased risk for primary renal disease. One study reviewed the charts 90 of Canadian First Nation children and adolescents with T2DM (Diabetes Care. 2009;32[5]:786-90). Of 10 who had renal biopsies performed, nine had immune complex disease/glomerulosclerosis, two had mild diabetes-related lesions, and seven had focal segmental glomerulosclerosis (FSGS); yet none had classic nephropathy. An analysis of Chinese youth that included 216 renal biopsies yielded similar findings (Intl Urol Nephrol. 2012;45[1]:173-9).
It’s also known that early-onset T2DM is associated with substantially increased incidence of end-stage renal disease (ESRD) and mortality in middle age. For example, one study of Pima Indians found that those who were diagnosed with T2DM earlier than 20 years of age had a one in five chance of developing ESRD, while those who were diagnosed at age 20 years or older had a one in two chance of ESRD (JAMA. 2006;296[4]:421-6). In a separate analysis, researchers estimated the remaining lifetime risks for ESRD among Aboriginal people in Australia with and without diabetes (Diabetes Res Clin Pract. 2014;103[3]:e24-6). The value for young adults with diabetes was high, about one in two at the age of 30 years, while it decreased with age to one in seven at 60 years.
“One of the first biomarkers we see in terms of renal disease in kids with T2DM is albuminuria,” Dr. Wicklow said at the Pediatric Academic Societies meeting. “The question is, why do kids with type 2 get more renal disease than kids with type 1 diabetes?” The SEARCH for Diabetes in Youth (SEARCH) study from 2006 found that hypertension, increased body mass index, increased weight circumference, and increased lipids were factors, while the SEARCH study from 2015 found that ethnicity, increased weight to height ratio, and mean arterial pressure were factors.
“Insulin resistance is significantly associated with albuminuria,” Dr. Wicklow continued. “It’s also been shown to be associated with hyperfiltration. Some of the markers of insulin resistance are important but they make up about 19% of the variance between type 1 and type 2, which means there are other variables that we’re not measuring.”
Enter ICARE (Improving Renal Complications in Adolescents with Type 2 Diabetes through Research), an ongoing prospective cohort study that Dr. Wicklow and her associates launched in 2014 at eight centers in Canada. It aims to examine the biopsychosocial risk factors for albuminuria in youth with T2DM and the mechanisms for renal injury. “Our theoretical framework was that biological exposures that we are aware of, such as glycemic control, hypertension, and lipids, would all be important in the development of albuminuria and renal disease in kids,” said Dr. Wicklow, who is the study’s coprimary investigator along with Allison Dart, MD. “But what we thought was novel was that psychological exposures either as socioeconomic status or as mental health factors would also directly impinge on renal health with respect to chronic inflammation in the body, inflammation in the kidneys, and long-term kidney damage.”
The first phase of ICARE involved a detailed phenotypic assessment of youth, including anthropometrics, biochemistry, 24-hour ambulatory blood pressure monitoring, overnight urine collections for albumin excretion, renal ultrasound, and iohexol-derived glomerular filtration rate (GFR). Phase 2 included an evaluation of psychological factors, including hair-derived cortisol; validated questionnaires for perceived stress, distress, and resiliency; and a detailed evaluation of systemic and urine inflammatory biomarkers. Annual follow-up is carried out to assess temporal associations between clinical risk factors and renal outcomes, including progression of albuminuria.
At the meeting, Dr. Wicklow reported on 187 youth enrolled to date. Of these, 96% were of indigenous ethnicity, 57 had albuminuria and 130 did not, and the mean ages of the groups were 16 years and 15 years, respectively. At baseline, a higher proportion of those in the albuminuria group were female (74% vs. 64% of those in the no albuminuria group, respectively), had a higher mean hemoglobin A1c (11% vs. 9%), and had hypertension (94% vs. 72%). She noted that upon presentation to the clinic, only 23% of participants had HbA1c levels less than 7%, only 26% had ranges between 7% and 9%, and about 40% did not have any hypertension. Of those who did, 27% had nighttime-only hypertension, and only 2% had daytime-only hypertension.
“The other risk factor these kids have for developing ESRD is that the majority were exposed to diabetes in pregnancy,” Dr. Wicklow said. “Murine models of maternal diabetes exposure have demonstrated that offspring have small kidneys, less ureteric bud branching, and a lower number of nephrons. Most of the human clinical cohort studies look at associations between development of diabetes and parental hypertension, maternal smoking, and maternal education. There is likely an impact at birth that sets these kids up for development of type 2 diabetes.”
In addition, results from clinical cohort studies have found that depression, mental stress, and distress are high in youth with T2DM. “Preliminary data suggest that if you have positive mental health, or coping strategies, or someone has worked through this with you and you are resilient, you might benefit in terms of overall glycemic control,” she said. For example, ICARE investigators have found that the higher the score on the Kessler Psychological Distress Scale (K6), the greater the risk of renal inflammation as measured by monocyte chemotactic protein-1 (MCP-1; P = .02). “Mental health seems to be something that can directly impact your health from a biological standpoint, and we might be able to find biomarkers of that risk,” Dr. Wicklow said. “Where does the stress come from? Most of my patients are indigenous, so it’s not surprising that the history in Canada of colonization of residential schools has left a lasting impression on these families and communities in terms of loss of language, loss of culture, and loss of land. There’s a community-based stress and a family-based stress that these children feel.”
Social factors also play a big role. She presented baseline findings from 196 youth with T2DM and 456 with T1DM, including measures such as the Socioeconomic Factor Index-Version 2 (SEFI-2), a way to assess socioeconomic characteristics based on Canadian Census data that reflects nonmedical social determinants of health. “It looks at factors like number of rooms in the house, single-parent households, maternal education attainment, and family income,” Dr. Wicklow explained. “The higher the SEFI-2 score, the lower your socioeconomic status is for the area you live in. Kids with T2DM generally live in areas of lower SES and lower socioeconomic index. They often live far away from health care providers. Many do not attend school and many are not with their biologic families, so we’ve had a lot of issues addressing child and family services, in particular in the phase of a chronic illness where our expectation is one thing and the family’s and community’s expectations of what’s realistic in terms of treatment and goals is another. We also have a lot of adolescent pregnancies.”
To date, about 80% of youth with T1D have seen a health care provider within the first year after transition from the pediatric diabetes clinic, compared with just over 50% of kids with T2D. “We transition youth with T1DM to internists, while our youth with T2DM go to itinerant physicians often back in their communities and/or rural family physicians,” she said. Between baseline and year 2, the rate of hospital admissions remained similar among T1DM at 11.6 and 11.8 admissions per 100 patient-years, respectively, but the number of hospital admissions for T2DM patients jumped from 20.1 to 25.5 admissions per 100 patient-years. “Kids with type 2 are showing up in the hospital a lot more than those with type 1 diabetes, but not for diabetes-related diagnoses,” Dr. Wicklow said. “We’re starting to look through the data now, and most of our kids are showing up with mental health complaints and issues. That’s why they’re getting hospitalized.”
Among ICARE study participants who have completed 3 years of follow-up, about 52% had albuminuria at their baseline visit and 48% sustained albuminuria throughout the study. About 26% progressed from normal levels of albuminuria to microalbuminuria, from microalbuminuria to macroalbuminuria, or from normal levels of albuminuria to macroalbuminuria. In addition, 16% persisted in the category that they were in, and 10% regressed. “The good news is, some of our kids get better over time,” Dr. Wicklow said. “The bad news is that the majority do not.”
Going forward, Dr. Wicklow and her associates work with an ICARE advisory group composed of children and families “who sit with us and talk about what mental health needs might be important, and how we should organize our study in a follow-up of the kids, to try and answer some of the questions that are important,” she said. “Working with the concept of the study’s theoretical framework, they acknowledged that the biological exposures are important, but they were also concerned about food security, finding strength/resilience within the community, and finding coping factors in terms of keeping themselves healthy with their diabetes. For some communities, they are concerned with basic needs. We’re working with them to help them progress, and to figure out how to best study children with type 2 diabetes.”
ICARE has received support from Diabetes Canada, Research Manitoba, the Canadian Institutes of Health Research, the Children’s Hospital Research Institute of Manitoba (specifically the Diabetes Research Envisioned and Accomplished in Manitoba (DREAM) theme), and the University of Manitoba. Dr. Wicklow reported having no financial disclosures.
AT PAS 18
Key clinical point: Mental health may indirectly increase inflammation, which contributes to kidney health.
Major finding: The higher the score on the Kessler Psychological Distress Scale (K6), the greater the risk of renal inflammation as measured by MCP-1 (P = .02).
Study details: Preliminary results from ICARE (Improving Renal Complications in Adolescents with Type 2 Diabetes through Research), an ongoing prospective cohort study.
Disclosures: ICARE has received support from Diabetes Canada, Research Manitoba, the Canadian Institutes of Health Research, the Children’s Hospital Research Institute of Manitoba (specifically the Diabetes Research Envisioned and Accomplished in Manitoba [DREAM] theme), and the University of Manitoba. Dr. Wicklow reported having no financial disclosures.
How the IHS Reduced Kidney Disease in the Highest-risk Population
Alaska is a vast state—larger than Texas, Montana, and California combined. It is also home to the highest percentage of American Indian (AI) and Alaska Native (AN) persons in the United States. These two populations—collectively referred to as Native Americans—have been served by the Indian Health Services (IHS) since it was established through the Snyder Act of 1921, in response to the dismal health conditions of the indigenous tribes in this country.1 Across the US (not only in Alaska), the IHS has partnered with AI/AN peoples to decrease health disparities in a culturally acceptable manner that honors and protects their traditions and values.
The IHS—which in 2016 comprised 2,500 nurses, 750 physicians, 700 pharmacists, 200 PAs and NPs, and 280 dentists, as well as nutritionists, diabetes educators, administrators, and other professionals—has made huge advances in decreasing health disparities in their populations. Among them: decreased rates of tuberculosis and of maternal and infant deaths.
However, life expectancy among Native Americans remains four years shorter than that of the rest of the US population. This disparity can be traced to three recalcitrant factors: unintentional injuries, liver disease, and diabetes.
The IHS practitioners decided to tackle diabetes with a multipronged approach. And what they achieved is astonishing.
WHAT THEY DID
Worldwide, diabetes is the most common cause of kidney failure; identifying patients with diabetes and early-stage chronic kidney disease allows for aggressive treatment that can slow progression to kidney failure and dialysis.

The IHS providers knew when they decided to tackle the problem of diabetes in the AI/AN population that the incidence was 16%—and the rate of diabetes leading to kidney failure in this population was the highest for any ethnic group in the US.2,3 And yet …
From 1996 to 2013, the rate of diabetes-related kidney failure among Native Americans dropped by 54%.3 Yes—the group of patients with the highest percentage of diabetes diagnoses has had the greatest improvement in prevention of kidney failure.4
Continue to: Some of the clinical achievements that contributed to...
Some of the clinical achievements that contributed to this significant change include
- Increased use of ACE inhibitors or angiotensin receptor blockers (ARBs) (from 42% to 74% over a five-year period)
- Reduced average blood pressure among hypertensive patients (to 133/76 mm Hg)
- Improved blood glucose control (by 10%)
- Increased testing for kidney disease among older patients (50% higher than the rest of the Medicare diabetes population).3
HOW THEY DID IT
This is not rocket science. The IHS staff integrated both population- and team-based approaches to achieve a more impressive decrease than ever could have been expected. In retrospect, perhaps this success should not come as such a surprise—many religious beliefs held by Native Americans focus around society, communal harmony, kinship, and cooperation.
The population health approach focused on promoting the wellness of the entire community and connecting people to local resources, including healthy food, transportation, housing, and mental health care. In the team approach, IHS medical experts implemented strategies to improve patient education, community outreach, care coordination, health outcome tracking, and access to a wide variety of health care providers.3,5
In a place like Alaska—where the northernmost city, Barrow, is more than 700 miles (two hours by plane) from Anchorage, and the southeastern Annette Island is more than 1,000 miles (six hours by plane) from the capital—this can be an especially challenging prospect. To reduce travel burden for rural patients, the IHS sponsors a diabetes team that travels from village to village. Nephrology services are not included in these field visits, however, so the kidney team relies heavily on telehealth. This requires extensive clinic staff coordination, as well as equipment and knowledgeable information systems support teams.
Other challenges require educational and logistical solutions. As noted, the use of ACE inhibitors and ARBs increased through the IHS’s efforts—and contributed to the delayed progression of diabetic kidney disease—but those additional prescriptions necessitate patient education. Understanding of these medications can be limited; many rural patients trust that when the bottle is empty, their practitioner has treated and cured their disease—mistakenly believing that no refills are needed. And even when the need to continue the prescription is understood, rural clinics may have difficulty tracking appointments and prescriptions written by providers at specialty clinics in Anchorage, making ongoing refills an issue.
Continue to: The necessary dietary changes can also be...
The necessary dietary changes can also be difficult for AI/AN populations. For example, in rural Alaska, tap water may not be safe to drink, and soda costs less than bottled water. Fresh produce is expensive and has often begun to spoil by the time it reaches local stores. The Native villagers often prefer their usual diets of gathered berries, fish, and red meat from subsistence hunting, making implementation of dietary changes difficult.
However, as the success of the IHS initiative shows, challenges can be met and overcome by practitioners who see a need, formulate a solution individualized to the circumstance, and think outside the box. One of the keys is developing a trusting relationship with patients. Another is to recognize informational needs and utilize available resources to educate patients. For example, visual representations of kidney function tend to be helpful in explaining the nature and course of disease; the National Kidney Disease Education Program uses an illustration similar to a gas gauge to demonstrate glomerular filtration rate (which would otherwise seem abstract and hard to understand for some patients; see below).6 When you understand your patient population and their needs, it makes addressing the challenging aspects of health care and prevention easier.

CONCLUSION
The results that the IHS achieved should serve as an example for all Americans with diabetes and their health care providers. We must be open to delivery of care via different approaches and practitioners in order to successfully help patients of different backgrounds and circumstances. This is the individualization of care that we hear so much about.
In 2016, the costs of caring for the kidney failure population were greater than the entire budget of the NIH. By aggressively identifying and treating patients at risk for kidney failure, we can slow disease progression—saving society money, but more importantly allowing our patients many more years of life free from the constraints of dialysis. —MET, RB
Mandy E. Thompson, PA-C
Kidney Center of Denver Health
Robin Bassett, DNP
Nephrology and Hypertension Associates, Anchorage
Adjunct Professor, NP program, University of Alaska, Anchorage
1. Indian Health Service. Legislation. www.ihs.gov/aboutihs/legislation. Accessed June 13, 2018.
2. National Health Interview Survey and Indian Health Service, 2010-2012.
3. CDC. Native Americans with diabetes. www.cdc.gov/vitalsigns/aian-diabetes/. Accessed June 13, 2018.
4. United States Renal Data System. Figure 1.5: Trends in adjusted* ESRD incidence rate (per million/year), by race, in the U.S. population, 1996-2014. In: 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2016.
5. Indian Health Service. Special diabetes program for Indians. www.ihs.gov/newsroom/factsheets/diabetes. Accessed June 13, 2018.
6. National Kidney Disease Education Program. How well are your kidneys working? Explaining your kidney test results. www.niddk.nih.gov/health-information/professionals/clinical-tools-patient-education-outreach/explain-kidney-test-results. Accessed June 13, 2018.
Clinician Reviews in partnership with
The National Kidney Foundation Council of Advanced Practitioners' (NKF-CAP) mission is to serve as an advisory resource for the NKF, nurse practitioners, physician assistants, clinical nurse specialists, and the community in advancing the care, treatment, and education of patients with kidney disease and their families. CAP is an advocate for professional development, research, and health policies that impact the delivery of patient care and professional practice. For more information on NKF-CAP, visit www.kidney.org/CAP. Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF- CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi- retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's column was authored by Mandy E. Thompson, PA-C, who practices at the Kidney Center of Denver Health, and Robin Bassett, DNP, who practices with Nephrology and Hypertension Associates in Anchorage and is an Adjunct Professor in the NP program at the University of Alaska-Anchorage. Dr. Bassett was assigned to the IHS Alaska Native Medical Center in Anchorage for 13 years during her employment with the United States Public Health Service.
Clinician Reviews in partnership with
The National Kidney Foundation Council of Advanced Practitioners' (NKF-CAP) mission is to serve as an advisory resource for the NKF, nurse practitioners, physician assistants, clinical nurse specialists, and the community in advancing the care, treatment, and education of patients with kidney disease and their families. CAP is an advocate for professional development, research, and health policies that impact the delivery of patient care and professional practice. For more information on NKF-CAP, visit www.kidney.org/CAP. Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF- CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi- retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's column was authored by Mandy E. Thompson, PA-C, who practices at the Kidney Center of Denver Health, and Robin Bassett, DNP, who practices with Nephrology and Hypertension Associates in Anchorage and is an Adjunct Professor in the NP program at the University of Alaska-Anchorage. Dr. Bassett was assigned to the IHS Alaska Native Medical Center in Anchorage for 13 years during her employment with the United States Public Health Service.
Clinician Reviews in partnership with
The National Kidney Foundation Council of Advanced Practitioners' (NKF-CAP) mission is to serve as an advisory resource for the NKF, nurse practitioners, physician assistants, clinical nurse specialists, and the community in advancing the care, treatment, and education of patients with kidney disease and their families. CAP is an advocate for professional development, research, and health policies that impact the delivery of patient care and professional practice. For more information on NKF-CAP, visit www.kidney.org/CAP. Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF- CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi- retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month's column was authored by Mandy E. Thompson, PA-C, who practices at the Kidney Center of Denver Health, and Robin Bassett, DNP, who practices with Nephrology and Hypertension Associates in Anchorage and is an Adjunct Professor in the NP program at the University of Alaska-Anchorage. Dr. Bassett was assigned to the IHS Alaska Native Medical Center in Anchorage for 13 years during her employment with the United States Public Health Service.
Alaska is a vast state—larger than Texas, Montana, and California combined. It is also home to the highest percentage of American Indian (AI) and Alaska Native (AN) persons in the United States. These two populations—collectively referred to as Native Americans—have been served by the Indian Health Services (IHS) since it was established through the Snyder Act of 1921, in response to the dismal health conditions of the indigenous tribes in this country.1 Across the US (not only in Alaska), the IHS has partnered with AI/AN peoples to decrease health disparities in a culturally acceptable manner that honors and protects their traditions and values.
The IHS—which in 2016 comprised 2,500 nurses, 750 physicians, 700 pharmacists, 200 PAs and NPs, and 280 dentists, as well as nutritionists, diabetes educators, administrators, and other professionals—has made huge advances in decreasing health disparities in their populations. Among them: decreased rates of tuberculosis and of maternal and infant deaths.
However, life expectancy among Native Americans remains four years shorter than that of the rest of the US population. This disparity can be traced to three recalcitrant factors: unintentional injuries, liver disease, and diabetes.
The IHS practitioners decided to tackle diabetes with a multipronged approach. And what they achieved is astonishing.
WHAT THEY DID
Worldwide, diabetes is the most common cause of kidney failure; identifying patients with diabetes and early-stage chronic kidney disease allows for aggressive treatment that can slow progression to kidney failure and dialysis.
The IHS providers knew when they decided to tackle the problem of diabetes in the AI/AN population that the incidence was 16%—and the rate of diabetes leading to kidney failure in this population was the highest for any ethnic group in the US.2,3 And yet …
From 1996 to 2013, the rate of diabetes-related kidney failure among Native Americans dropped by 54%.3 Yes—the group of patients with the highest percentage of diabetes diagnoses has had the greatest improvement in prevention of kidney failure.4
Continue to: Some of the clinical achievements that contributed to...
Some of the clinical achievements that contributed to this significant change include
- Increased use of ACE inhibitors or angiotensin receptor blockers (ARBs) (from 42% to 74% over a five-year period)
- Reduced average blood pressure among hypertensive patients (to 133/76 mm Hg)
- Improved blood glucose control (by 10%)
- Increased testing for kidney disease among older patients (50% higher than the rest of the Medicare diabetes population).3
HOW THEY DID IT
This is not rocket science. The IHS staff integrated both population- and team-based approaches to achieve a more impressive decrease than ever could have been expected. In retrospect, perhaps this success should not come as such a surprise—many religious beliefs held by Native Americans focus around society, communal harmony, kinship, and cooperation.
The population health approach focused on promoting the wellness of the entire community and connecting people to local resources, including healthy food, transportation, housing, and mental health care. In the team approach, IHS medical experts implemented strategies to improve patient education, community outreach, care coordination, health outcome tracking, and access to a wide variety of health care providers.3,5
In a place like Alaska—where the northernmost city, Barrow, is more than 700 miles (two hours by plane) from Anchorage, and the southeastern Annette Island is more than 1,000 miles (six hours by plane) from the capital—this can be an especially challenging prospect. To reduce travel burden for rural patients, the IHS sponsors a diabetes team that travels from village to village. Nephrology services are not included in these field visits, however, so the kidney team relies heavily on telehealth. This requires extensive clinic staff coordination, as well as equipment and knowledgeable information systems support teams.
Other challenges require educational and logistical solutions. As noted, the use of ACE inhibitors and ARBs increased through the IHS’s efforts—and contributed to the delayed progression of diabetic kidney disease—but those additional prescriptions necessitate patient education. Understanding of these medications can be limited; many rural patients trust that when the bottle is empty, their practitioner has treated and cured their disease—mistakenly believing that no refills are needed. And even when the need to continue the prescription is understood, rural clinics may have difficulty tracking appointments and prescriptions written by providers at specialty clinics in Anchorage, making ongoing refills an issue.
Continue to: The necessary dietary changes can also be...
The necessary dietary changes can also be difficult for AI/AN populations. For example, in rural Alaska, tap water may not be safe to drink, and soda costs less than bottled water. Fresh produce is expensive and has often begun to spoil by the time it reaches local stores. The Native villagers often prefer their usual diets of gathered berries, fish, and red meat from subsistence hunting, making implementation of dietary changes difficult.
However, as the success of the IHS initiative shows, challenges can be met and overcome by practitioners who see a need, formulate a solution individualized to the circumstance, and think outside the box. One of the keys is developing a trusting relationship with patients. Another is to recognize informational needs and utilize available resources to educate patients. For example, visual representations of kidney function tend to be helpful in explaining the nature and course of disease; the National Kidney Disease Education Program uses an illustration similar to a gas gauge to demonstrate glomerular filtration rate (which would otherwise seem abstract and hard to understand for some patients; see below).6 When you understand your patient population and their needs, it makes addressing the challenging aspects of health care and prevention easier.
CONCLUSION
The results that the IHS achieved should serve as an example for all Americans with diabetes and their health care providers. We must be open to delivery of care via different approaches and practitioners in order to successfully help patients of different backgrounds and circumstances. This is the individualization of care that we hear so much about.
In 2016, the costs of caring for the kidney failure population were greater than the entire budget of the NIH. By aggressively identifying and treating patients at risk for kidney failure, we can slow disease progression—saving society money, but more importantly allowing our patients many more years of life free from the constraints of dialysis. —MET, RB
Mandy E. Thompson, PA-C
Kidney Center of Denver Health
Robin Bassett, DNP
Nephrology and Hypertension Associates, Anchorage
Adjunct Professor, NP program, University of Alaska, Anchorage
Alaska is a vast state—larger than Texas, Montana, and California combined. It is also home to the highest percentage of American Indian (AI) and Alaska Native (AN) persons in the United States. These two populations—collectively referred to as Native Americans—have been served by the Indian Health Services (IHS) since it was established through the Snyder Act of 1921, in response to the dismal health conditions of the indigenous tribes in this country.1 Across the US (not only in Alaska), the IHS has partnered with AI/AN peoples to decrease health disparities in a culturally acceptable manner that honors and protects their traditions and values.
The IHS—which in 2016 comprised 2,500 nurses, 750 physicians, 700 pharmacists, 200 PAs and NPs, and 280 dentists, as well as nutritionists, diabetes educators, administrators, and other professionals—has made huge advances in decreasing health disparities in their populations. Among them: decreased rates of tuberculosis and of maternal and infant deaths.
However, life expectancy among Native Americans remains four years shorter than that of the rest of the US population. This disparity can be traced to three recalcitrant factors: unintentional injuries, liver disease, and diabetes.
The IHS practitioners decided to tackle diabetes with a multipronged approach. And what they achieved is astonishing.
WHAT THEY DID
Worldwide, diabetes is the most common cause of kidney failure; identifying patients with diabetes and early-stage chronic kidney disease allows for aggressive treatment that can slow progression to kidney failure and dialysis.
The IHS providers knew when they decided to tackle the problem of diabetes in the AI/AN population that the incidence was 16%—and the rate of diabetes leading to kidney failure in this population was the highest for any ethnic group in the US.2,3 And yet …
From 1996 to 2013, the rate of diabetes-related kidney failure among Native Americans dropped by 54%.3 Yes—the group of patients with the highest percentage of diabetes diagnoses has had the greatest improvement in prevention of kidney failure.4
Continue to: Some of the clinical achievements that contributed to...
Some of the clinical achievements that contributed to this significant change include
- Increased use of ACE inhibitors or angiotensin receptor blockers (ARBs) (from 42% to 74% over a five-year period)
- Reduced average blood pressure among hypertensive patients (to 133/76 mm Hg)
- Improved blood glucose control (by 10%)
- Increased testing for kidney disease among older patients (50% higher than the rest of the Medicare diabetes population).3
HOW THEY DID IT
This is not rocket science. The IHS staff integrated both population- and team-based approaches to achieve a more impressive decrease than ever could have been expected. In retrospect, perhaps this success should not come as such a surprise—many religious beliefs held by Native Americans focus around society, communal harmony, kinship, and cooperation.
The population health approach focused on promoting the wellness of the entire community and connecting people to local resources, including healthy food, transportation, housing, and mental health care. In the team approach, IHS medical experts implemented strategies to improve patient education, community outreach, care coordination, health outcome tracking, and access to a wide variety of health care providers.3,5
In a place like Alaska—where the northernmost city, Barrow, is more than 700 miles (two hours by plane) from Anchorage, and the southeastern Annette Island is more than 1,000 miles (six hours by plane) from the capital—this can be an especially challenging prospect. To reduce travel burden for rural patients, the IHS sponsors a diabetes team that travels from village to village. Nephrology services are not included in these field visits, however, so the kidney team relies heavily on telehealth. This requires extensive clinic staff coordination, as well as equipment and knowledgeable information systems support teams.
Other challenges require educational and logistical solutions. As noted, the use of ACE inhibitors and ARBs increased through the IHS’s efforts—and contributed to the delayed progression of diabetic kidney disease—but those additional prescriptions necessitate patient education. Understanding of these medications can be limited; many rural patients trust that when the bottle is empty, their practitioner has treated and cured their disease—mistakenly believing that no refills are needed. And even when the need to continue the prescription is understood, rural clinics may have difficulty tracking appointments and prescriptions written by providers at specialty clinics in Anchorage, making ongoing refills an issue.
Continue to: The necessary dietary changes can also be...
The necessary dietary changes can also be difficult for AI/AN populations. For example, in rural Alaska, tap water may not be safe to drink, and soda costs less than bottled water. Fresh produce is expensive and has often begun to spoil by the time it reaches local stores. The Native villagers often prefer their usual diets of gathered berries, fish, and red meat from subsistence hunting, making implementation of dietary changes difficult.
However, as the success of the IHS initiative shows, challenges can be met and overcome by practitioners who see a need, formulate a solution individualized to the circumstance, and think outside the box. One of the keys is developing a trusting relationship with patients. Another is to recognize informational needs and utilize available resources to educate patients. For example, visual representations of kidney function tend to be helpful in explaining the nature and course of disease; the National Kidney Disease Education Program uses an illustration similar to a gas gauge to demonstrate glomerular filtration rate (which would otherwise seem abstract and hard to understand for some patients; see below).6 When you understand your patient population and their needs, it makes addressing the challenging aspects of health care and prevention easier.
CONCLUSION
The results that the IHS achieved should serve as an example for all Americans with diabetes and their health care providers. We must be open to delivery of care via different approaches and practitioners in order to successfully help patients of different backgrounds and circumstances. This is the individualization of care that we hear so much about.
In 2016, the costs of caring for the kidney failure population were greater than the entire budget of the NIH. By aggressively identifying and treating patients at risk for kidney failure, we can slow disease progression—saving society money, but more importantly allowing our patients many more years of life free from the constraints of dialysis. —MET, RB
Mandy E. Thompson, PA-C
Kidney Center of Denver Health
Robin Bassett, DNP
Nephrology and Hypertension Associates, Anchorage
Adjunct Professor, NP program, University of Alaska, Anchorage
1. Indian Health Service. Legislation. www.ihs.gov/aboutihs/legislation. Accessed June 13, 2018.
2. National Health Interview Survey and Indian Health Service, 2010-2012.
3. CDC. Native Americans with diabetes. www.cdc.gov/vitalsigns/aian-diabetes/. Accessed June 13, 2018.
4. United States Renal Data System. Figure 1.5: Trends in adjusted* ESRD incidence rate (per million/year), by race, in the U.S. population, 1996-2014. In: 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2016.
5. Indian Health Service. Special diabetes program for Indians. www.ihs.gov/newsroom/factsheets/diabetes. Accessed June 13, 2018.
6. National Kidney Disease Education Program. How well are your kidneys working? Explaining your kidney test results. www.niddk.nih.gov/health-information/professionals/clinical-tools-patient-education-outreach/explain-kidney-test-results. Accessed June 13, 2018.
1. Indian Health Service. Legislation. www.ihs.gov/aboutihs/legislation. Accessed June 13, 2018.
2. National Health Interview Survey and Indian Health Service, 2010-2012.
3. CDC. Native Americans with diabetes. www.cdc.gov/vitalsigns/aian-diabetes/. Accessed June 13, 2018.
4. United States Renal Data System. Figure 1.5: Trends in adjusted* ESRD incidence rate (per million/year), by race, in the U.S. population, 1996-2014. In: 2016 USRDS annual data report: Epidemiology of kidney disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2016.
5. Indian Health Service. Special diabetes program for Indians. www.ihs.gov/newsroom/factsheets/diabetes. Accessed June 13, 2018.
6. National Kidney Disease Education Program. How well are your kidneys working? Explaining your kidney test results. www.niddk.nih.gov/health-information/professionals/clinical-tools-patient-education-outreach/explain-kidney-test-results. Accessed June 13, 2018.

Renal disease and the surgical patient: Minimizing the impact
Chronic kidney disease (CKD) is estimated to affect 14% of Americans, but it is likely underdiagnosed because it is often asymptomatic.1,2 Its prevalence is even higher in patients who undergo surgery—up to 30% in cardiac surgery.3 Its impact on surgical outcomes is substantial.4 Importantly, patients with CKD are at higher risk of postoperative acute kidney injury (AKI), which is also associated with adverse outcomes. Thus, it is important to recognize, assess, and manage abnormal renal function in surgical patients.
WHAT IS THE IMPACT ON POSTOPERATIVE OUTCOMES?

Cardiac surgery outcomes

Moreover, in patients undergoing coronary artery bypass grafting (CABG), the worse the renal dysfunction, the higher the long-term mortality rate. Patients with moderate (stage 3) CKD had a 3.5 times higher odds of in-hospital mortality compared with patients with normal renal function, rising to 8.8 with severe (stage 4) and to 9.6 with dialysis-dependent (stage 5) CKD.11
The mechanisms linking CKD with negative cardiac outcomes are unclear, but many possibilities exist. CKD is an independent risk factor for coronary artery disease and shares underlying risk factors such as hypertension and diabetes. Cardiac surgery patients with CKD are also more likely to have diabetes, left ventricular dysfunction, and peripheral vascular disease.
Noncardiac surgery outcomes
CKD is also associated with adverse outcomes in noncardiac surgery patients, especially at higher levels of renal dysfunction.12–14 For example, in patients who underwent major noncardiac surgery, compared with patients in stage 1 (estimated GFR > 90 mL/min/1.73 m2), the odds ratios for all-cause mortality were as follows:
- 0.8 for patients with stage 2 CKD
- 2.2 in stage 3a
- 2.8 in stage 3b
- 11.3 in stage 4
- 5.8 in stage 5.14
The association between estimated GFR and all-cause mortality was not statistically significant (P = .071), but statistically significant associations were observed between estimated GFR and major adverse cardiovascular events (P < .001) and hospital length of stay (P < .001).
The association of CKD with major adverse outcomes and death in both cardiac and noncardiac surgical patients demonstrates the importance of understanding this risk, identifying patients with CKD preoperatively, and taking steps to lower the risk.
WHAT IS THE IMPACT OF ACUTE KIDNEY INJURY?
AKI is a common and serious complication of surgery, especially cardiac surgery. It has been associated with higher rates of morbidity, mortality, and cardiovascular events, longer hospital length of stay, and higher cost.
Several groups have proposed criteria for defining AKI and its severity; the KDIGO criteria are the most widely accepted.15 These define AKI as an increase in serum creatinine concentration of 0.3 mg/dL or more within 48 hours or at least 1.5 times the baseline value within 7 days, or urine volume less than 0.5 mL/kg/hour for more than 6 hours. There are 3 stages of severity:
- Stage 1—an increase in serum creatinine of 1.5 to 1.9 times baseline, an absolute increase of at least 0.3 mg/dL, or urine output less than 0.5 mL/kg/hour for 6 to 12 hours
- Stage 2—an increase in serum creatinine of 2.0 to 2.9 times baseline or urine output less than 0.5 mmL/kg/hour for 12 or more hours
- Stage 3—an increase in serum creatinine of 3 times baseline, an absolute increase of at least 4 mg/dL, initiation of renal replacement therapy, urine output less than 0.3 mL/kg/hour for 24 or more hours, or anuria for 12 or more hours.15
Multiple factors associated with surgery may contribute to AKI, including hemodynamic instability, volume shifts, blood loss, use of heart-lung bypass, new medications, activation of the inflammatory cascade, oxidative stress, and anemia.
AKI in cardiac surgery
The incidence of AKI is high in cardiac surgery. In a meta-analysis of 46 studies (N = 242,000), its incidence in cardiopulmonary bypass surgery was about 18%, with 2.1% of patients needing renal replacement therapy.16 However, the incidence varied considerably from study to study, ranging from 1% to 53%, and was influenced by the definition of AKI, the type of cardiac surgery, and the patient population.16
Cardiac surgery-associated AKI adversely affects outcomes. Several studies have shown that cardiac surgery patients who develop AKI have higher rates of death and stroke.16–21 More severe AKI confers higher mortality rates, with the highest mortality rate in patients who need renal replacement therapy, approximately 37%.17 Patients with cardiac surgery-associated AKI also have a longer hospital length of stay and significantly higher costs of care.17,18
Long-term outcomes are also negatively affected by AKI. In cardiac surgery patients with AKI who had completely recovered renal function by the time they left the hospital, the 2-year incidence rate of CKD was 6.8%, significantly higher than the 0.2% rate in patients who did not develop AKI.19 The 2-year survival rates also were significantly worse for patients who developed postoperative AKI (82.3% vs 93.7%). Similarly, in patients undergoing CABG who had normal renal function before surgery, those who developed AKI postoperatively had significantly shorter long-term survival rates.20 The effect does not require a large change in renal function. An increase in creatinine as small as 0.3 mg/dL has been associated with a higher rate of death and a long-term risk of end-stage renal disease that is 3 times higher.21
WHAT ARE THE RISK FACTORS FOR ACUTE KIDNEY INJURY?

Cardiac surgery
CKD is a risk factor not only after cardiac surgery but also after percutaneous procedures. In a meta-analysis of 4,992 patients with CKD who underwent transcatheter aortic valve replacement, both moderate and severe CKD increased the odds of AKI, early stroke, the need for dialysis, and all-cause and cardiovascular mortality at 1 year.22,23 Increased rates of AKI also have been found in patients with CKD undergoing CABG surgery.24 These results point to a synergistic effect between AKI and CKD, with outcomes much worse in combination than alone.
In cardiac surgery, the most important patient risk factors associated with a higher incidence of postoperative AKI are age older than 75, CKD, preoperative heart failure, and prior myocardial infarction.19,25 Diabetes is an additional independent risk factor, with type 1 conferring higher risk than type 2.26 Preoperative use of angiotensin-converting enzyme (ACE) inhibitors may or may not be a risk factor for cardiac surgery-associated AKI, with some studies finding increased risk and others finding reduced rates.27,28
Anemia, which may be related to either patient or surgical risk factors (eg, intraoperative blood loss), also increases the risk of AKI in cardiac surgery.29,30 A retrospective study of CABG surgery patients found that intraoperative hemoglobin levels below 8 g/dL were associated with a 25% to 30% incidence of AKI, compared with 15% to 20% with hemoglobin levels above 9 g/dL.29 Additionally, having severe hypotension (mean arterial pressure < 50 mm Hg) significantly increased the AKI rates in the low-hemoglobin group.29 Similar results were reported in a later study.30
Among surgical factors, several randomized controlled trials have shown that off-pump CABG is associated with a significantly lower risk of postoperative AKI than on-pump CABG; however, this difference did not translate into any long-term difference in mortality rates.31,32 Longer cardiopulmonary bypass time is strongly associated with a higher incidence of AKI and postoperative death.33
Noncardiac surgery
AKI is less common after noncardiac surgery; however, outcomes are severe in patients in whom it occurs. In a study of 15,102 noncardiac surgery patients, only 0.8% developed AKI and 0.1% required renal replacement therapy.34
Risk factors after noncardiac surgery are similar to those after cardiac surgery (Table 3).34–36 Factors with the greatest impact are older age, peripheral vascular occlusive disease, chronic obstructive pulmonary disease necessitating chronic bronchodilator therapy, high-risk surgery, hepatic disease, emergent or urgent surgery, and high body mass index.
Surgical risk factors include total vasopressor dose administered, use of a vasopressor infusion, and diuretic administration.34 In addition, intraoperative hypotension is associated with a higher risk of AKI, major adverse cardiac events, and 30-day mortality.37
Noncardiac surgery patients with postoperative AKI have significantly higher rates of 30-day readmissions, 1-year progression to end-stage renal disease, and mortality than patients who do not develop AKI.35 Additionally, patients with AKI have significantly higher rates of cardiovascular complications (33.3% vs 11.3%) and death (6.1% vs 0.9%), as well as a significantly longer length of hospital stay.34,36
CAN WE DECREASE THE IMPACT OF RENAL DISEASE IN SURGERY?
Before surgery, practitioners need to identify patients at risk of AKI, implement possible risk-reduction measures, and, afterward, treat it early in its course if it occurs.
The preoperative visit is the ideal time to assess a patient’s risk of postoperative renal dysfunction. Laboratory tests can identify risks based on surgery type, age, hypertension, the presence of CKD, and medications that affect renal function. However, the basic chemistry panel is abnormal in only 8.2% of patients and affects management in just 2.6%, requiring the clinician to target testing to patients at high risk.38
Patients with a significant degree of renal dysfunction, particularly those previously undiagnosed, may benefit from additional preoperative testing and medication management. Perioperative management of medications that could adversely affect renal function should be carefully considered during the preoperative visit. In addition, the postoperative inpatient team needs to be informed about potentially nephrotoxic medications and medications that are renally cleared. Attention needs to be given to the renal impact of common perioperative medications such as nonsteroidal anti-inflammatory drugs, antibiotics, intravenous contrast, low-molecular-weight heparins, diuretics, ACE inhibitors, and angiotensin II receptor blockers. With the emphasis on opioid-sparing analgesics, it is particularly important to assess the risk of AKI if nonsteroidal anti-inflammatory drugs are part of the pain control plan.
Nephrology referral may help, especially for patients with a GFR less than 45 mL/min. This information enables more informed decision-making regarding the risks of adverse outcomes related to kidney disease.
WHAT TOOLS DO WE HAVE TO DIAGNOSE RENAL INJURY?
Several risk-prediction models have been developed to assess the postoperative risk of AKI in both cardiac and major noncardiac surgery patients. Although these models can identify risk factors, their clinical accuracy and utility have been questioned.
Biomarkers
Early diagnosis is the first step in managing AKI, allowing time to implement measures to minimize its impact.
Serum creatinine testing is widely used to measure renal function and diagnose AKI; however, it does not detect small reductions in renal function, and there is a time lag between renal insult and a rise in creatinine. The result is a delay to diagnosis of AKI.
Biomarkers other than creatinine have been studied for early detection of intraoperative and postoperative renal insult. These novel renal injury markers include the following:
Neutrophil gelatinase-associated lipocalin (NGAL). Two studies looked at plasma NGAL as an early marker of AKI in patients with CKD who were undergoing cardiac surgery.39,40 One study found that by using NGAL instead of creatinine, postoperative AKI could be diagnosed an average of 20 hours earlier.39 In addition, NGAL helped detect renal recovery earlier than creatinine.40 The diagnostic cut-off values of NGAL were different for patients with CKD than for those without CKD.39,40
Other novel markers include:
- Kidney injury marker 1
- N-acetyl-beta-D-glucosaminidase
- Cysteine C.
Although these biomarkers show some ability to detect renal injury, they provide only modest discrimination and are not widely available for clinical use.41 Current evidence does not support routine use of these markers in clinical settings.
CAN WE PROTECT RENAL FUNCTION?
Interventions to prevent or ameliorate the impact of CKD and AKI on surgical outcomes have been studied most extensively in cardiac surgery patients.
Aspirin. A retrospective study of 3,585 cardiac surgery patients with CKD found that preoperative aspirin use significantly lowered the incidence of postoperative AKI and 30-day mortality compared with patients not using aspirin.42 Aspirin use reduced 30-day mortality in CKD stages 1, 2, and 3 by 23.3%, 58%, and 70%, respectively. On the other hand, in the Perioperative Ischemic Evaluation (POISE) trial, in noncardiac surgery patients, neither aspirin nor clonidine started 2 to 4 hours preoperatively and continued up to 30 days after surgery altered the risk of AKI significantly more than placebo.43
Statins have been ineffective in reducing the incidence of AKI in cardiac surgery patients. In fact, a meta-analysis of 8 interventional trials found an increased incidence of AKI in patients in whom statins were started perioperatively.44 Erythropoietin was also found to be ineffective in the prevention of perioperative AKI in cardiac surgery patients in a separate study.45
The evidence regarding other therapies has also varied.
N-acetylcysteine in high doses reduced the incidence of AKI in patients with CKD stage 3 and 4 undergoing CABG.46 Another meta-analysis of 10 studies in cardiac surgery patients published recently did not show any benefit of N-acetylcysteine in reducing AKI.47
Human atrial natriuretic peptide, given preoperatively to patients with CKD, reduced the acute and long-term creatinine rise as well as the number of cardiac events after CABG; however, it did not reduce mortality rates.48
Renin-angiotensin system inhibitors, given preoperatively to patients with heart failure was associated with a decrease in the incidence of AKI in 1 study.49
Dexmedetomidine is a highly selective alpha 2 adrenoreceptor agonist. A recent meta-analysis of 10 clinical trials found it beneficial in reducing the risk of perioperative AKI in cardiac surgery patients.50 An earlier meta-analysis had similar results.51
Levosimendan is an inotropic vasodilator that improves cardiac output and renal perfusion in patients with systolic heart failure, and it has been hypothesized to decrease the risk of AKI after cardiac surgery. Previous data demonstrated that this drug reduced AKI and mortality; however, analysis was limited by small sample size and varying definitions of AKI.52 A recent meta-analysis showed that levosimendan was associated with a lower incidence of AKI but was also associated with an increased incidence of atrial fibrillation and no reduction in 30-day mortality.53
Remote ischemic preconditioning is a procedure that subjects the kidneys to brief episodes of ischemia before surgery, protecting them when they are later subjected to prolonged ischemia or reperfusion injury. It has shown initial promising results in preventing AKI. In a randomized controlled trial in 240 patients at high risk of AKI, those who received remote ischemic preconditioning had an AKI incidence of 37.5% compared with 52.5% for controls (P = .02); however, the mortality rate was the same.54 Similarly, remote ischemic preconditioning significantly lowered the incidence of AKI in nondiabetic patients undergoing CABG surgery compared with controls.55
Fluid management. Renal perfusion is intimately related to the development of AKI, and there is evidence that both hypovolemia and excessive fluid resuscitation can increase the risk of AKI in noncardiac surgery patients.56 Because of this, fluid management has also received attention in perioperative AKI. Goal-directed fluid management has been evaluated in noncardiac surgery patients, and it did not show any benefit in preventing AKI.57 However, in a more recent retrospective study, postoperative positive fluid balance was associated with increased incidence of AKI compared with zero fluid balance. Negative fluid balance did not appear to have a detrimental effect.58
RECOMMENDATIONS
No prophylactic therapy has yet been shown to definitively decrease the risk of postoperative AKI in all patients. Nevertheless, it is important to identify patients at risk during the preoperative visit, especially those with CKD. Many patients undergoing surgery have CKD, placing them at high risk of developing AKI in the perioperative period. The risk is particularly high with cardiac surgery.
Serum creatinine and urine output should be closely monitored perioperatively in at-risk patients. If AKI is diagnosed, practitioners need to identify and ameliorate the cause as early as possible.

Recommendations from KDIGO for perioperative prevention and management of AKI are listed in Table 4.15 These include avoiding additional nephrotoxic medications and adjusting the doses of renally cleared medications. Also, some patients may benefit from preoperative counseling and specialist referral.
- Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA 2007; 298(17):2038–2047. doi:10.1001/jama.298.17.2038
- National Institute of Diabetes and Digestive and Kidney Diseases. Kidney Disease Statistics for the United States. www.niddk.nih.gov/health-information/health-statistics/kidney-disease. Accessed June 11, 2018.
- Rosner MH, Okusa MD. Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol 2006; 1(1):19–32. doi:10.2215/CJN.00240605
- Meersch M, Schmidt C, Zarbock A. Patient with chronic renal failure undergoing surgery. Curr Opin Anaesthesiol 2016; 29(3):413–420. doi:10.1097/ACO.0000000000000329
- Stevens PE, Levin A; Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: synopsis of the Kidney Disease: Improving Global Outcomes 2012 clinical practice guideline. Ann Intern Med 2013; 158(11):825–830. doi:10.7326/0003-4819-158-11-201306040-00007
- Levey AS, Eckardt KU, Tsukamoto Y, et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2005; 67(6):2089–2100. doi:10.1111/j.1523-1755.2005.00365.x
- Saitoh M, Takahashi T, Sakurada K, et al. Factors determining achievement of early postoperative cardiac rehabilitation goal in patients with or without preoperative kidney dysfunction undergoing isolated cardiac surgery. J Cardiol 2013; 61(4):299–303. doi:10.1016/j.jjcc.2012.12.014
- Minakata K, Bando K, Tanaka S, et al. Preoperative chronic kidney disease as a strong predictor of postoperative infection and mortality after coronary artery bypass grafting. Circ J 2014; 78(9):2225–2231. doi:10.1253/circj.CJ-14-0328
- Domoto S, Tagusari O, Nakamura Y, et al. Preoperative estimated glomerular filtration rate as a significant predictor of long-term outcomes after coronary artery bypass grafting in Japanese patients. Gen Thorac Cardiovasc Surg 2014; 62(2):95–102. doi:10.1007/s11748-013-0306-5
- Hedley AJ, Roberts MA, Hayward PA, et al. Impact of chronic kidney disease on patient outcome following cardiac surgery. Heart Lung Circ 2010; 19(8):453–459. doi:10.1016/j.hlc.2010.03.005
- Boulton BJ, Kilgo P, Guyton RA, et al. Impact of preoperative renal dysfunction in patients undergoing off-pump versus on-pump coronary artery bypass. Ann Thorac Surg 2011; 92(2):595–601. doi:10.1016/j.athoracsur.2011.04.023
- Prowle JR, Kam EP, Ahmad T, Smith NC, Protopapa K, Pearse RM. Preoperative renal dysfunction and mortality after non-cardiac surgery. Br J Surg 2016; 103(10):1316–1325. doi:10.1002/bjs.10186
- Gaber AO, Moore LW, Aloia TA, et al. Cross-sectional and case-control analyses of the association of kidney function staging with adverse postoperative outcomes in general and vascular surgery. Ann Surg 2013; 258(1):169–177. doi:10.1097/SLA.0b013e318288e18e
- Mases A, Sabaté S, Guilera N, et al. Preoperative estimated glomerular filtration rate and the risk of major adverse cardiovascular and cerebrovascular events in non-cardiac surgery. Br J Anaesth 2014; 113(4):644–651. doi:10.1093/bja/aeu134
- Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clinical Practice 2012; 120(4):c179–c184. doi:10.1159/000339789
- Pickering JW, James MT, Palmer SC. Acute kidney injury and prognosis after cardiopulmonary bypass: a meta-analysis of cohort studies. Am J Kidney Dis 2015; 65(2):283–293. doi:10.1053/j.ajkd.2014.09.008
- Dasta JF, Kane-Gill SL, Durtschi AJ, Pathak DS, Kellum JA. Costs and outcomes of acute kidney injury (AKI) following cardiac surgery. Nephrol Dial Transplant 2008; 23(6):1970-1974. doi:10.1093/ndt/gfm908
- Karkouti K, Wijeysundera DN, Yau TM, et al. Acute kidney injury after cardiac surgery focus on modifiable risk factors. Circulation 2009; 119(4):495–502. doi:10.1161/CIRCULATIONAHA.108.786913
- Xu JR, Zhu JM, Jiang J, et al. Risk factors for long-term mortality and progressive chronic kidney disease associated with acute kidney injury after cardiac surgery. Medicine (Baltimore) 2015; 94(45):e2025. doi:10.1097/MD.0000000000002025
- Chalmers J, Mediratta N, McShane J, Shaw M, Pullan M, Poullis M. The long-term effects of developing renal failure post-coronary artery bypass surgery, in patients with normal preoperative renal function. Eur J Cardiothorac Surg 2013; 43(3):555–559. doi:10.1093/ejcts/ezs329
- Ryden L, Sartipy U, Evans M, Holzmann MJ. Acute kidney injury after coronary artery bypass grafting and long-term risk of end-stage renal disease. Circulation 2014; 130(23):2005–2011. doi:10.1161/CIRCULATIONAHA.114.010622
- Gargiulo G, Capodanno D, Sannino A, et al. Impact of moderate preoperative chronic kidney disease on mortality after transcatheter aortic valve implantation. Int J Cardiol 2015; 189:77–78. doi:10.1016/j.ijcard.2015.04.077
- Gargiulo G, Capodanno D, Sannino A, et al. Moderate and severe preoperative chronic kidney disease worsen clinical outcomes after transcatheter aortic valve implantation meta-analysis of 4,992 patients. Circ Cardiovasc Interv 2015; 8(2):e002220. doi:10.1161/CIRCINTERVENTIONS.114.002220
- Han SS, Shin N, Baek SH, et al. Effects of acute kidney injury and chronic kidney disease on long-term mortality after coronary artery bypass grafting. Am Heart J 2015; 169(3):419–425. doi:10.1016/j.ahj.2014.12.019
- Aronson S, Fontes ML, Miao Y, Mangano DT; Investigators of the Multicenter Study of Perioperative Ischemia Research Group; Ischemia Research and Education Foundation. Risk index for perioperative renal dysfunction/failure: critical dependence on pulse pressure hypertension. Circulation 2007; 115(6):733–742. doi:10.1161/CIRCULATIONAHA.106.623538
- Hertzberg D, Sartipy U, Holzmann MJ. Type 1 and type 2 diabetes mellitus and risk of acute kidney injury after coronary artery bypass grafting. Am Heart J 2015; 170(5):895–902. doi:10.1016/j.ahj.2015.08.013
- Benedetto U, Sciarretta S, Roscitano A, et al. Preoperative angiotensin-converting enzyme inhibitors and acute kidney injury after coronary artery bypass grafting. Ann Thorac Surg 2008; 86(4):1160–1165. doi:10.1016/j.athoracsur.2008.06.018
- Arora P, Rajagopalam S, Ranjan R, et al. Preoperative use of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers is associated with increased risk for acute kidney injury after cardiovascular surgery. Clin J Am Soc Nephrol 2008; 3(5):1266–1273. doi:10.2215/CJN.05271107
- Haase M, Bellomo R, Story D, et al. Effect of mean arterial pressure, haemoglobin and blood transfusion during cardiopulmonary bypass on post-operative acute kidney injury. Nephrol Dial Transplant 2012; 27(1):153–160. doi:10.1093/ndt/gfr275
- Ono M, Arnaoutakis GJ, Fine DM, et al. Blood pressure excursions below the cerebral autoregulation threshold during cardiac surgery are associated with acute kidney injury. Crit Care Med 2013; 41(2):464-471. doi:10.1097/CCM.0b013e31826ab3a1
- Seabra VF, Alobaidi S, Balk EM, Poon AH, Jaber BL. Off-pump coronary artery bypass surgery and acute kidney injury: a meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol 2010; 5(10):1734–1744. doi:10.2215/CJN.02800310
- Garg AX, Devereaux PJ, Yusuf S, et al; CORONARY Investigators. Kidney function after off-pump or on-pump coronary artery bypass graft surgery: a randomized clinical trial. JAMA 2014; 311(21):2191–2198. doi:10.1001/jama.2014.4952
- Kumar AB, Suneja M, Bayman EO, Weide GD, Tarasi M. Association between postoperative acute kidney injury and duration of cardiopulmonary bypass: a meta-analysis. J Cardiothorac Vasc Anesth 2012; 26(1):64–69. doi:10.1053/j.jvca.2011.07.007
- Kheterpal S, Tremper KK, Englesbe MJ, et al. Predictors of postoperative acute renal failure after noncardiac surgery in patients with previously normal renal function. Anesthesiology 2007; 107(6):892–902. doi:10.1097/01.anes.0000290588.29668.38
- Grams ME, Sang Y, Coresh J, et al. Acute kidney injury after major surgery: a retrospective analysis of Veterans Health Administration data. Am J Kidney Dis 2016; 67(6):872–880. doi:10.1053/j.ajkd.2015.07.022
- Biteker M, Dayan A, Tekkesin AI, et al. Incidence, risk factors, and outcomes of perioperative acute kidney injury in noncardiac and nonvascular surgery. Am J Surg 2014: 207(1):53–59. doi:10.1016/j.amjsurg.2013.04.006
- Gu W-J, Hou B-L, Kwong JS, et al. Association between intraoperative hypotension and 30-day mortality, major adverse cardiac events, and acute kidney injury after non-cardiac surgery: a meta-analysis of cohort studies. Int J Cardiol 2018; 258:68–73. doi:10.1016/j.ijcard.2018.01.137
- Smetana GW, Macpherson DS. The case against routine preoperative laboratory testing. Med Clin North Am 2003; 87(1):7–40. pmid:12575882
- Perrotti A, Miltgen G, Chevet-Noel A, et al. Neutrophil gelatinase-associated lipocalin as early predictor of acute kidney injury after cardiac surgery in adults with chronic kidney failure. Ann Thorac Surg 2015; 99(3):864–869. doi:10.1016/j.athoracsur.2014.10.011
- Doi K, Urata M, Katagiri D, et al. Plasma neutrophil gelatinase-associated lipocalin in acute kidney injury superimposed on chronic kidney disease after cardiac surgery: a multicenter prospective study. Crit Care 2013; 17(6):R270. doi:10.1186/cc13104
- Ho J, Tangri N, Komenda P, et al. Urinary, plasma, and serum biomarkers’ utility for predicting acute kidney injury associated with cardiac surgery in adults: a meta-analysis. Am J Kidney Dis 2015; 66(6):993–1005. doi:10.1053/j.ajkd.2015.06.018
- Yao L, Young N, Liu H, et al. Evidence for preoperative aspirin improving major outcomes in patients with chronic kidney disease undergoing cardiac surgery: a cohort study. Ann Surg 2015; 261(1):207–212. doi:10.1097/SLA.0000000000000641
- Garg AX, Kurz A, Sessler DI, et al; POISE-2 Investigators. Aspirin and clonidine in non-cardiac surgery: acute kidney injury substudy protocol of the perioperative ischaemic evaluation (POISE) 2 randomised controlled trial. BMJ open 2014; 4(2):e004886. doi:10.1136/bmjopen-2014-004886
- He SJ, Liu Q, Li HQ, Tian F, Chen SY, Weng JX. Role of statins in preventing cardiac surgery-associated acute kidney injury: an updated meta-analysis of randomized controlled trials. Ther Clin Risk Manag 2018; 14:475–482. doi:10.2147/TCRM.S160298
- Tie HT, Luo MZ, Lin D, Zhang M, Wan JY, Wu QC. Erythropoietin administration for prevention of cardiac surgery-associated acute kidney injury: a meta-analysis of randomized controlled trials. Eur J Cardiothorac Surg 2015; 48(1):32–39. doi:10.1093/ejcts/ezu378
- Santana-Santos E, Gowdak LH, Gaiotto FA, et al. High dose of N-acetylcystein prevents acute kidney injury in chronic kidney disease patients undergoing myocardial revascularization. Ann Thorac Surg 2014; 97(5):1617–1623. doi:10.1016/j.athoracsur.2014.01.056
- Mei M, Zhao HW, Pan QG, Pu YM, Tang MZ, Shen BB. Efficacy of N-acetylcysteine in preventing acute kidney injury after cardiac surgery: a meta-analysis study. J Invest Surg 2018; 31(1):14–23. doi:10.1080/08941939.2016.1269853
- Sezai A, Hata M, Niino T, et al. Results of low-dose human atrial natriuretic peptide infusion in nondialysis patients with chronic kidney disease undergoing coronary artery bypass grafting: the NU-HIT (Nihon University working group study of low-dose HANP infusion therapy during cardiac surgery) trial for CKD. J Am Coll Cardiol 2011; 58(9):897–903. doi:10.1016/j.jacc.2011.03.056
- Xu N, Long Q, He T, et al. Association between preoperative renin-angiotensin system inhibitor use and postoperative acute kidney injury risk in patients with hypertension. Clin Nephrol 2018; 89(6):403–414. doi:10.5414/CN109319
- Liu Y, Sheng B, Wang S, Lu F, Zhen J, Chen W. Dexmedetomidine prevents acute kidney injury after adult cardiac surgery: a meta-analysis of randomized controlled trials. BMC Anesthesiol 2018; 18(1):7. doi:10.1186/s12871-018-0472-1
- Shi R, Tie H-T. Dexmedetomidine as a promising prevention strategy for cardiac surgery-associated acute kidney injury: a meta-analysis. Critical Care 2017; 21(1):198. doi:10.1186/s13054-017-1776-0
- Zhou C, Gong J, Chen D, Wang W, Liu M, Liu B. Levosimendan for prevention of acute kidney injury after cardiac surgery: a meta-analysis of randomized controlled trials. Am J Kidney Dis 2016; 67(3):408–416. doi:10.1053/j.ajkd.2015.09.015
- Elbadawi A, Elgendy IY, Saad M, et al. Meta-analysis of trials on prophylactic use of levosimendan in patients undergoing cardiac surgery. Ann Thorac Surg 2018; 105(5):1403–1410. doi:10.1016/j.athoracsur.2017.11.027
- Zarbock A, Schmidt C, Van Aken H, et al; RenalRIPC Investigators. Effect of remote ischemic preconditioning on kidney injury among high-risk patients undergoing cardiac surgery: a randomized clinical trial. JAMA 2015; 313(21):2133–2141. doi:10.1001/jama.2015.4189
- Venugopal V, Laing CM, Ludman A, Yellon DM, Hausenloy D. Effect of remote ischemic preconditioning on acute kidney injury in nondiabetic patients undergoing coronary artery bypass graft surgery: a secondary analysis of 2 small randomized trials. Am J Kidney Dis 2010; 56(6):1043–1049. doi:10.1053/j.ajkd.2010.07.014
- Futier E, Constantin JM, Petit A, et al. Conservative vs restrictive individualized goal-directed fluid replacement strategy in major abdominal surgery: a prospective randomized trial. Arch Surg 2010; 145(12):1193–1200. doi:10.1001/archsurg.2010.275
- Patel A, Prowle JR, Ackland GL. Postoperative goal-directed therapy and development of acute kidney injury following major elective noncardiac surgery: post-hoc analysis of POM-O randomized controlled trial. Clin Kidney J 2017; 10(3):348–356. doi:10.1093/ckj/sfw118
- Shen Y, Zhang W, Cheng X, Ying M. Association between postoperative fluid balance and acute kidney injury in patients after cardiac surgery: a retrospective cohort study. J Crit Care 2018; 44:273–277. doi:10.1016/j.jcrc.2017.11.041
Chronic kidney disease (CKD) is estimated to affect 14% of Americans, but it is likely underdiagnosed because it is often asymptomatic.1,2 Its prevalence is even higher in patients who undergo surgery—up to 30% in cardiac surgery.3 Its impact on surgical outcomes is substantial.4 Importantly, patients with CKD are at higher risk of postoperative acute kidney injury (AKI), which is also associated with adverse outcomes. Thus, it is important to recognize, assess, and manage abnormal renal function in surgical patients.
WHAT IS THE IMPACT ON POSTOPERATIVE OUTCOMES?
Cardiac surgery outcomes
Moreover, in patients undergoing coronary artery bypass grafting (CABG), the worse the renal dysfunction, the higher the long-term mortality rate. Patients with moderate (stage 3) CKD had a 3.5 times higher odds of in-hospital mortality compared with patients with normal renal function, rising to 8.8 with severe (stage 4) and to 9.6 with dialysis-dependent (stage 5) CKD.11
The mechanisms linking CKD with negative cardiac outcomes are unclear, but many possibilities exist. CKD is an independent risk factor for coronary artery disease and shares underlying risk factors such as hypertension and diabetes. Cardiac surgery patients with CKD are also more likely to have diabetes, left ventricular dysfunction, and peripheral vascular disease.
Noncardiac surgery outcomes
CKD is also associated with adverse outcomes in noncardiac surgery patients, especially at higher levels of renal dysfunction.12–14 For example, in patients who underwent major noncardiac surgery, compared with patients in stage 1 (estimated GFR > 90 mL/min/1.73 m2), the odds ratios for all-cause mortality were as follows:
- 0.8 for patients with stage 2 CKD
- 2.2 in stage 3a
- 2.8 in stage 3b
- 11.3 in stage 4
- 5.8 in stage 5.14
The association between estimated GFR and all-cause mortality was not statistically significant (P = .071), but statistically significant associations were observed between estimated GFR and major adverse cardiovascular events (P < .001) and hospital length of stay (P < .001).
The association of CKD with major adverse outcomes and death in both cardiac and noncardiac surgical patients demonstrates the importance of understanding this risk, identifying patients with CKD preoperatively, and taking steps to lower the risk.
WHAT IS THE IMPACT OF ACUTE KIDNEY INJURY?
AKI is a common and serious complication of surgery, especially cardiac surgery. It has been associated with higher rates of morbidity, mortality, and cardiovascular events, longer hospital length of stay, and higher cost.
Several groups have proposed criteria for defining AKI and its severity; the KDIGO criteria are the most widely accepted.15 These define AKI as an increase in serum creatinine concentration of 0.3 mg/dL or more within 48 hours or at least 1.5 times the baseline value within 7 days, or urine volume less than 0.5 mL/kg/hour for more than 6 hours. There are 3 stages of severity:
- Stage 1—an increase in serum creatinine of 1.5 to 1.9 times baseline, an absolute increase of at least 0.3 mg/dL, or urine output less than 0.5 mL/kg/hour for 6 to 12 hours
- Stage 2—an increase in serum creatinine of 2.0 to 2.9 times baseline or urine output less than 0.5 mmL/kg/hour for 12 or more hours
- Stage 3—an increase in serum creatinine of 3 times baseline, an absolute increase of at least 4 mg/dL, initiation of renal replacement therapy, urine output less than 0.3 mL/kg/hour for 24 or more hours, or anuria for 12 or more hours.15
Multiple factors associated with surgery may contribute to AKI, including hemodynamic instability, volume shifts, blood loss, use of heart-lung bypass, new medications, activation of the inflammatory cascade, oxidative stress, and anemia.
AKI in cardiac surgery
The incidence of AKI is high in cardiac surgery. In a meta-analysis of 46 studies (N = 242,000), its incidence in cardiopulmonary bypass surgery was about 18%, with 2.1% of patients needing renal replacement therapy.16 However, the incidence varied considerably from study to study, ranging from 1% to 53%, and was influenced by the definition of AKI, the type of cardiac surgery, and the patient population.16
Cardiac surgery-associated AKI adversely affects outcomes. Several studies have shown that cardiac surgery patients who develop AKI have higher rates of death and stroke.16–21 More severe AKI confers higher mortality rates, with the highest mortality rate in patients who need renal replacement therapy, approximately 37%.17 Patients with cardiac surgery-associated AKI also have a longer hospital length of stay and significantly higher costs of care.17,18
Long-term outcomes are also negatively affected by AKI. In cardiac surgery patients with AKI who had completely recovered renal function by the time they left the hospital, the 2-year incidence rate of CKD was 6.8%, significantly higher than the 0.2% rate in patients who did not develop AKI.19 The 2-year survival rates also were significantly worse for patients who developed postoperative AKI (82.3% vs 93.7%). Similarly, in patients undergoing CABG who had normal renal function before surgery, those who developed AKI postoperatively had significantly shorter long-term survival rates.20 The effect does not require a large change in renal function. An increase in creatinine as small as 0.3 mg/dL has been associated with a higher rate of death and a long-term risk of end-stage renal disease that is 3 times higher.21
WHAT ARE THE RISK FACTORS FOR ACUTE KIDNEY INJURY?
Cardiac surgery
CKD is a risk factor not only after cardiac surgery but also after percutaneous procedures. In a meta-analysis of 4,992 patients with CKD who underwent transcatheter aortic valve replacement, both moderate and severe CKD increased the odds of AKI, early stroke, the need for dialysis, and all-cause and cardiovascular mortality at 1 year.22,23 Increased rates of AKI also have been found in patients with CKD undergoing CABG surgery.24 These results point to a synergistic effect between AKI and CKD, with outcomes much worse in combination than alone.
In cardiac surgery, the most important patient risk factors associated with a higher incidence of postoperative AKI are age older than 75, CKD, preoperative heart failure, and prior myocardial infarction.19,25 Diabetes is an additional independent risk factor, with type 1 conferring higher risk than type 2.26 Preoperative use of angiotensin-converting enzyme (ACE) inhibitors may or may not be a risk factor for cardiac surgery-associated AKI, with some studies finding increased risk and others finding reduced rates.27,28
Anemia, which may be related to either patient or surgical risk factors (eg, intraoperative blood loss), also increases the risk of AKI in cardiac surgery.29,30 A retrospective study of CABG surgery patients found that intraoperative hemoglobin levels below 8 g/dL were associated with a 25% to 30% incidence of AKI, compared with 15% to 20% with hemoglobin levels above 9 g/dL.29 Additionally, having severe hypotension (mean arterial pressure < 50 mm Hg) significantly increased the AKI rates in the low-hemoglobin group.29 Similar results were reported in a later study.30
Among surgical factors, several randomized controlled trials have shown that off-pump CABG is associated with a significantly lower risk of postoperative AKI than on-pump CABG; however, this difference did not translate into any long-term difference in mortality rates.31,32 Longer cardiopulmonary bypass time is strongly associated with a higher incidence of AKI and postoperative death.33
Noncardiac surgery
AKI is less common after noncardiac surgery; however, outcomes are severe in patients in whom it occurs. In a study of 15,102 noncardiac surgery patients, only 0.8% developed AKI and 0.1% required renal replacement therapy.34
Risk factors after noncardiac surgery are similar to those after cardiac surgery (Table 3).34–36 Factors with the greatest impact are older age, peripheral vascular occlusive disease, chronic obstructive pulmonary disease necessitating chronic bronchodilator therapy, high-risk surgery, hepatic disease, emergent or urgent surgery, and high body mass index.
Surgical risk factors include total vasopressor dose administered, use of a vasopressor infusion, and diuretic administration.34 In addition, intraoperative hypotension is associated with a higher risk of AKI, major adverse cardiac events, and 30-day mortality.37
Noncardiac surgery patients with postoperative AKI have significantly higher rates of 30-day readmissions, 1-year progression to end-stage renal disease, and mortality than patients who do not develop AKI.35 Additionally, patients with AKI have significantly higher rates of cardiovascular complications (33.3% vs 11.3%) and death (6.1% vs 0.9%), as well as a significantly longer length of hospital stay.34,36
CAN WE DECREASE THE IMPACT OF RENAL DISEASE IN SURGERY?
Before surgery, practitioners need to identify patients at risk of AKI, implement possible risk-reduction measures, and, afterward, treat it early in its course if it occurs.
The preoperative visit is the ideal time to assess a patient’s risk of postoperative renal dysfunction. Laboratory tests can identify risks based on surgery type, age, hypertension, the presence of CKD, and medications that affect renal function. However, the basic chemistry panel is abnormal in only 8.2% of patients and affects management in just 2.6%, requiring the clinician to target testing to patients at high risk.38
Patients with a significant degree of renal dysfunction, particularly those previously undiagnosed, may benefit from additional preoperative testing and medication management. Perioperative management of medications that could adversely affect renal function should be carefully considered during the preoperative visit. In addition, the postoperative inpatient team needs to be informed about potentially nephrotoxic medications and medications that are renally cleared. Attention needs to be given to the renal impact of common perioperative medications such as nonsteroidal anti-inflammatory drugs, antibiotics, intravenous contrast, low-molecular-weight heparins, diuretics, ACE inhibitors, and angiotensin II receptor blockers. With the emphasis on opioid-sparing analgesics, it is particularly important to assess the risk of AKI if nonsteroidal anti-inflammatory drugs are part of the pain control plan.
Nephrology referral may help, especially for patients with a GFR less than 45 mL/min. This information enables more informed decision-making regarding the risks of adverse outcomes related to kidney disease.
WHAT TOOLS DO WE HAVE TO DIAGNOSE RENAL INJURY?
Several risk-prediction models have been developed to assess the postoperative risk of AKI in both cardiac and major noncardiac surgery patients. Although these models can identify risk factors, their clinical accuracy and utility have been questioned.
Biomarkers
Early diagnosis is the first step in managing AKI, allowing time to implement measures to minimize its impact.
Serum creatinine testing is widely used to measure renal function and diagnose AKI; however, it does not detect small reductions in renal function, and there is a time lag between renal insult and a rise in creatinine. The result is a delay to diagnosis of AKI.
Biomarkers other than creatinine have been studied for early detection of intraoperative and postoperative renal insult. These novel renal injury markers include the following:
Neutrophil gelatinase-associated lipocalin (NGAL). Two studies looked at plasma NGAL as an early marker of AKI in patients with CKD who were undergoing cardiac surgery.39,40 One study found that by using NGAL instead of creatinine, postoperative AKI could be diagnosed an average of 20 hours earlier.39 In addition, NGAL helped detect renal recovery earlier than creatinine.40 The diagnostic cut-off values of NGAL were different for patients with CKD than for those without CKD.39,40
Other novel markers include:
- Kidney injury marker 1
- N-acetyl-beta-D-glucosaminidase
- Cysteine C.
Although these biomarkers show some ability to detect renal injury, they provide only modest discrimination and are not widely available for clinical use.41 Current evidence does not support routine use of these markers in clinical settings.
CAN WE PROTECT RENAL FUNCTION?
Interventions to prevent or ameliorate the impact of CKD and AKI on surgical outcomes have been studied most extensively in cardiac surgery patients.
Aspirin. A retrospective study of 3,585 cardiac surgery patients with CKD found that preoperative aspirin use significantly lowered the incidence of postoperative AKI and 30-day mortality compared with patients not using aspirin.42 Aspirin use reduced 30-day mortality in CKD stages 1, 2, and 3 by 23.3%, 58%, and 70%, respectively. On the other hand, in the Perioperative Ischemic Evaluation (POISE) trial, in noncardiac surgery patients, neither aspirin nor clonidine started 2 to 4 hours preoperatively and continued up to 30 days after surgery altered the risk of AKI significantly more than placebo.43
Statins have been ineffective in reducing the incidence of AKI in cardiac surgery patients. In fact, a meta-analysis of 8 interventional trials found an increased incidence of AKI in patients in whom statins were started perioperatively.44 Erythropoietin was also found to be ineffective in the prevention of perioperative AKI in cardiac surgery patients in a separate study.45
The evidence regarding other therapies has also varied.
N-acetylcysteine in high doses reduced the incidence of AKI in patients with CKD stage 3 and 4 undergoing CABG.46 Another meta-analysis of 10 studies in cardiac surgery patients published recently did not show any benefit of N-acetylcysteine in reducing AKI.47
Human atrial natriuretic peptide, given preoperatively to patients with CKD, reduced the acute and long-term creatinine rise as well as the number of cardiac events after CABG; however, it did not reduce mortality rates.48
Renin-angiotensin system inhibitors, given preoperatively to patients with heart failure was associated with a decrease in the incidence of AKI in 1 study.49
Dexmedetomidine is a highly selective alpha 2 adrenoreceptor agonist. A recent meta-analysis of 10 clinical trials found it beneficial in reducing the risk of perioperative AKI in cardiac surgery patients.50 An earlier meta-analysis had similar results.51
Levosimendan is an inotropic vasodilator that improves cardiac output and renal perfusion in patients with systolic heart failure, and it has been hypothesized to decrease the risk of AKI after cardiac surgery. Previous data demonstrated that this drug reduced AKI and mortality; however, analysis was limited by small sample size and varying definitions of AKI.52 A recent meta-analysis showed that levosimendan was associated with a lower incidence of AKI but was also associated with an increased incidence of atrial fibrillation and no reduction in 30-day mortality.53
Remote ischemic preconditioning is a procedure that subjects the kidneys to brief episodes of ischemia before surgery, protecting them when they are later subjected to prolonged ischemia or reperfusion injury. It has shown initial promising results in preventing AKI. In a randomized controlled trial in 240 patients at high risk of AKI, those who received remote ischemic preconditioning had an AKI incidence of 37.5% compared with 52.5% for controls (P = .02); however, the mortality rate was the same.54 Similarly, remote ischemic preconditioning significantly lowered the incidence of AKI in nondiabetic patients undergoing CABG surgery compared with controls.55
Fluid management. Renal perfusion is intimately related to the development of AKI, and there is evidence that both hypovolemia and excessive fluid resuscitation can increase the risk of AKI in noncardiac surgery patients.56 Because of this, fluid management has also received attention in perioperative AKI. Goal-directed fluid management has been evaluated in noncardiac surgery patients, and it did not show any benefit in preventing AKI.57 However, in a more recent retrospective study, postoperative positive fluid balance was associated with increased incidence of AKI compared with zero fluid balance. Negative fluid balance did not appear to have a detrimental effect.58
RECOMMENDATIONS
No prophylactic therapy has yet been shown to definitively decrease the risk of postoperative AKI in all patients. Nevertheless, it is important to identify patients at risk during the preoperative visit, especially those with CKD. Many patients undergoing surgery have CKD, placing them at high risk of developing AKI in the perioperative period. The risk is particularly high with cardiac surgery.
Serum creatinine and urine output should be closely monitored perioperatively in at-risk patients. If AKI is diagnosed, practitioners need to identify and ameliorate the cause as early as possible.
Recommendations from KDIGO for perioperative prevention and management of AKI are listed in Table 4.15 These include avoiding additional nephrotoxic medications and adjusting the doses of renally cleared medications. Also, some patients may benefit from preoperative counseling and specialist referral.
Chronic kidney disease (CKD) is estimated to affect 14% of Americans, but it is likely underdiagnosed because it is often asymptomatic.1,2 Its prevalence is even higher in patients who undergo surgery—up to 30% in cardiac surgery.3 Its impact on surgical outcomes is substantial.4 Importantly, patients with CKD are at higher risk of postoperative acute kidney injury (AKI), which is also associated with adverse outcomes. Thus, it is important to recognize, assess, and manage abnormal renal function in surgical patients.
WHAT IS THE IMPACT ON POSTOPERATIVE OUTCOMES?
Cardiac surgery outcomes
Moreover, in patients undergoing coronary artery bypass grafting (CABG), the worse the renal dysfunction, the higher the long-term mortality rate. Patients with moderate (stage 3) CKD had a 3.5 times higher odds of in-hospital mortality compared with patients with normal renal function, rising to 8.8 with severe (stage 4) and to 9.6 with dialysis-dependent (stage 5) CKD.11
The mechanisms linking CKD with negative cardiac outcomes are unclear, but many possibilities exist. CKD is an independent risk factor for coronary artery disease and shares underlying risk factors such as hypertension and diabetes. Cardiac surgery patients with CKD are also more likely to have diabetes, left ventricular dysfunction, and peripheral vascular disease.
Noncardiac surgery outcomes
CKD is also associated with adverse outcomes in noncardiac surgery patients, especially at higher levels of renal dysfunction.12–14 For example, in patients who underwent major noncardiac surgery, compared with patients in stage 1 (estimated GFR > 90 mL/min/1.73 m2), the odds ratios for all-cause mortality were as follows:
- 0.8 for patients with stage 2 CKD
- 2.2 in stage 3a
- 2.8 in stage 3b
- 11.3 in stage 4
- 5.8 in stage 5.14
The association between estimated GFR and all-cause mortality was not statistically significant (P = .071), but statistically significant associations were observed between estimated GFR and major adverse cardiovascular events (P < .001) and hospital length of stay (P < .001).
The association of CKD with major adverse outcomes and death in both cardiac and noncardiac surgical patients demonstrates the importance of understanding this risk, identifying patients with CKD preoperatively, and taking steps to lower the risk.
WHAT IS THE IMPACT OF ACUTE KIDNEY INJURY?
AKI is a common and serious complication of surgery, especially cardiac surgery. It has been associated with higher rates of morbidity, mortality, and cardiovascular events, longer hospital length of stay, and higher cost.
Several groups have proposed criteria for defining AKI and its severity; the KDIGO criteria are the most widely accepted.15 These define AKI as an increase in serum creatinine concentration of 0.3 mg/dL or more within 48 hours or at least 1.5 times the baseline value within 7 days, or urine volume less than 0.5 mL/kg/hour for more than 6 hours. There are 3 stages of severity:
- Stage 1—an increase in serum creatinine of 1.5 to 1.9 times baseline, an absolute increase of at least 0.3 mg/dL, or urine output less than 0.5 mL/kg/hour for 6 to 12 hours
- Stage 2—an increase in serum creatinine of 2.0 to 2.9 times baseline or urine output less than 0.5 mmL/kg/hour for 12 or more hours
- Stage 3—an increase in serum creatinine of 3 times baseline, an absolute increase of at least 4 mg/dL, initiation of renal replacement therapy, urine output less than 0.3 mL/kg/hour for 24 or more hours, or anuria for 12 or more hours.15
Multiple factors associated with surgery may contribute to AKI, including hemodynamic instability, volume shifts, blood loss, use of heart-lung bypass, new medications, activation of the inflammatory cascade, oxidative stress, and anemia.
AKI in cardiac surgery
The incidence of AKI is high in cardiac surgery. In a meta-analysis of 46 studies (N = 242,000), its incidence in cardiopulmonary bypass surgery was about 18%, with 2.1% of patients needing renal replacement therapy.16 However, the incidence varied considerably from study to study, ranging from 1% to 53%, and was influenced by the definition of AKI, the type of cardiac surgery, and the patient population.16
Cardiac surgery-associated AKI adversely affects outcomes. Several studies have shown that cardiac surgery patients who develop AKI have higher rates of death and stroke.16–21 More severe AKI confers higher mortality rates, with the highest mortality rate in patients who need renal replacement therapy, approximately 37%.17 Patients with cardiac surgery-associated AKI also have a longer hospital length of stay and significantly higher costs of care.17,18
Long-term outcomes are also negatively affected by AKI. In cardiac surgery patients with AKI who had completely recovered renal function by the time they left the hospital, the 2-year incidence rate of CKD was 6.8%, significantly higher than the 0.2% rate in patients who did not develop AKI.19 The 2-year survival rates also were significantly worse for patients who developed postoperative AKI (82.3% vs 93.7%). Similarly, in patients undergoing CABG who had normal renal function before surgery, those who developed AKI postoperatively had significantly shorter long-term survival rates.20 The effect does not require a large change in renal function. An increase in creatinine as small as 0.3 mg/dL has been associated with a higher rate of death and a long-term risk of end-stage renal disease that is 3 times higher.21
WHAT ARE THE RISK FACTORS FOR ACUTE KIDNEY INJURY?
Cardiac surgery
CKD is a risk factor not only after cardiac surgery but also after percutaneous procedures. In a meta-analysis of 4,992 patients with CKD who underwent transcatheter aortic valve replacement, both moderate and severe CKD increased the odds of AKI, early stroke, the need for dialysis, and all-cause and cardiovascular mortality at 1 year.22,23 Increased rates of AKI also have been found in patients with CKD undergoing CABG surgery.24 These results point to a synergistic effect between AKI and CKD, with outcomes much worse in combination than alone.
In cardiac surgery, the most important patient risk factors associated with a higher incidence of postoperative AKI are age older than 75, CKD, preoperative heart failure, and prior myocardial infarction.19,25 Diabetes is an additional independent risk factor, with type 1 conferring higher risk than type 2.26 Preoperative use of angiotensin-converting enzyme (ACE) inhibitors may or may not be a risk factor for cardiac surgery-associated AKI, with some studies finding increased risk and others finding reduced rates.27,28
Anemia, which may be related to either patient or surgical risk factors (eg, intraoperative blood loss), also increases the risk of AKI in cardiac surgery.29,30 A retrospective study of CABG surgery patients found that intraoperative hemoglobin levels below 8 g/dL were associated with a 25% to 30% incidence of AKI, compared with 15% to 20% with hemoglobin levels above 9 g/dL.29 Additionally, having severe hypotension (mean arterial pressure < 50 mm Hg) significantly increased the AKI rates in the low-hemoglobin group.29 Similar results were reported in a later study.30
Among surgical factors, several randomized controlled trials have shown that off-pump CABG is associated with a significantly lower risk of postoperative AKI than on-pump CABG; however, this difference did not translate into any long-term difference in mortality rates.31,32 Longer cardiopulmonary bypass time is strongly associated with a higher incidence of AKI and postoperative death.33
Noncardiac surgery
AKI is less common after noncardiac surgery; however, outcomes are severe in patients in whom it occurs. In a study of 15,102 noncardiac surgery patients, only 0.8% developed AKI and 0.1% required renal replacement therapy.34
Risk factors after noncardiac surgery are similar to those after cardiac surgery (Table 3).34–36 Factors with the greatest impact are older age, peripheral vascular occlusive disease, chronic obstructive pulmonary disease necessitating chronic bronchodilator therapy, high-risk surgery, hepatic disease, emergent or urgent surgery, and high body mass index.
Surgical risk factors include total vasopressor dose administered, use of a vasopressor infusion, and diuretic administration.34 In addition, intraoperative hypotension is associated with a higher risk of AKI, major adverse cardiac events, and 30-day mortality.37
Noncardiac surgery patients with postoperative AKI have significantly higher rates of 30-day readmissions, 1-year progression to end-stage renal disease, and mortality than patients who do not develop AKI.35 Additionally, patients with AKI have significantly higher rates of cardiovascular complications (33.3% vs 11.3%) and death (6.1% vs 0.9%), as well as a significantly longer length of hospital stay.34,36
CAN WE DECREASE THE IMPACT OF RENAL DISEASE IN SURGERY?
Before surgery, practitioners need to identify patients at risk of AKI, implement possible risk-reduction measures, and, afterward, treat it early in its course if it occurs.
The preoperative visit is the ideal time to assess a patient’s risk of postoperative renal dysfunction. Laboratory tests can identify risks based on surgery type, age, hypertension, the presence of CKD, and medications that affect renal function. However, the basic chemistry panel is abnormal in only 8.2% of patients and affects management in just 2.6%, requiring the clinician to target testing to patients at high risk.38
Patients with a significant degree of renal dysfunction, particularly those previously undiagnosed, may benefit from additional preoperative testing and medication management. Perioperative management of medications that could adversely affect renal function should be carefully considered during the preoperative visit. In addition, the postoperative inpatient team needs to be informed about potentially nephrotoxic medications and medications that are renally cleared. Attention needs to be given to the renal impact of common perioperative medications such as nonsteroidal anti-inflammatory drugs, antibiotics, intravenous contrast, low-molecular-weight heparins, diuretics, ACE inhibitors, and angiotensin II receptor blockers. With the emphasis on opioid-sparing analgesics, it is particularly important to assess the risk of AKI if nonsteroidal anti-inflammatory drugs are part of the pain control plan.
Nephrology referral may help, especially for patients with a GFR less than 45 mL/min. This information enables more informed decision-making regarding the risks of adverse outcomes related to kidney disease.
WHAT TOOLS DO WE HAVE TO DIAGNOSE RENAL INJURY?
Several risk-prediction models have been developed to assess the postoperative risk of AKI in both cardiac and major noncardiac surgery patients. Although these models can identify risk factors, their clinical accuracy and utility have been questioned.
Biomarkers
Early diagnosis is the first step in managing AKI, allowing time to implement measures to minimize its impact.
Serum creatinine testing is widely used to measure renal function and diagnose AKI; however, it does not detect small reductions in renal function, and there is a time lag between renal insult and a rise in creatinine. The result is a delay to diagnosis of AKI.
Biomarkers other than creatinine have been studied for early detection of intraoperative and postoperative renal insult. These novel renal injury markers include the following:
Neutrophil gelatinase-associated lipocalin (NGAL). Two studies looked at plasma NGAL as an early marker of AKI in patients with CKD who were undergoing cardiac surgery.39,40 One study found that by using NGAL instead of creatinine, postoperative AKI could be diagnosed an average of 20 hours earlier.39 In addition, NGAL helped detect renal recovery earlier than creatinine.40 The diagnostic cut-off values of NGAL were different for patients with CKD than for those without CKD.39,40
Other novel markers include:
- Kidney injury marker 1
- N-acetyl-beta-D-glucosaminidase
- Cysteine C.
Although these biomarkers show some ability to detect renal injury, they provide only modest discrimination and are not widely available for clinical use.41 Current evidence does not support routine use of these markers in clinical settings.
CAN WE PROTECT RENAL FUNCTION?
Interventions to prevent or ameliorate the impact of CKD and AKI on surgical outcomes have been studied most extensively in cardiac surgery patients.
Aspirin. A retrospective study of 3,585 cardiac surgery patients with CKD found that preoperative aspirin use significantly lowered the incidence of postoperative AKI and 30-day mortality compared with patients not using aspirin.42 Aspirin use reduced 30-day mortality in CKD stages 1, 2, and 3 by 23.3%, 58%, and 70%, respectively. On the other hand, in the Perioperative Ischemic Evaluation (POISE) trial, in noncardiac surgery patients, neither aspirin nor clonidine started 2 to 4 hours preoperatively and continued up to 30 days after surgery altered the risk of AKI significantly more than placebo.43
Statins have been ineffective in reducing the incidence of AKI in cardiac surgery patients. In fact, a meta-analysis of 8 interventional trials found an increased incidence of AKI in patients in whom statins were started perioperatively.44 Erythropoietin was also found to be ineffective in the prevention of perioperative AKI in cardiac surgery patients in a separate study.45
The evidence regarding other therapies has also varied.
N-acetylcysteine in high doses reduced the incidence of AKI in patients with CKD stage 3 and 4 undergoing CABG.46 Another meta-analysis of 10 studies in cardiac surgery patients published recently did not show any benefit of N-acetylcysteine in reducing AKI.47
Human atrial natriuretic peptide, given preoperatively to patients with CKD, reduced the acute and long-term creatinine rise as well as the number of cardiac events after CABG; however, it did not reduce mortality rates.48
Renin-angiotensin system inhibitors, given preoperatively to patients with heart failure was associated with a decrease in the incidence of AKI in 1 study.49
Dexmedetomidine is a highly selective alpha 2 adrenoreceptor agonist. A recent meta-analysis of 10 clinical trials found it beneficial in reducing the risk of perioperative AKI in cardiac surgery patients.50 An earlier meta-analysis had similar results.51
Levosimendan is an inotropic vasodilator that improves cardiac output and renal perfusion in patients with systolic heart failure, and it has been hypothesized to decrease the risk of AKI after cardiac surgery. Previous data demonstrated that this drug reduced AKI and mortality; however, analysis was limited by small sample size and varying definitions of AKI.52 A recent meta-analysis showed that levosimendan was associated with a lower incidence of AKI but was also associated with an increased incidence of atrial fibrillation and no reduction in 30-day mortality.53
Remote ischemic preconditioning is a procedure that subjects the kidneys to brief episodes of ischemia before surgery, protecting them when they are later subjected to prolonged ischemia or reperfusion injury. It has shown initial promising results in preventing AKI. In a randomized controlled trial in 240 patients at high risk of AKI, those who received remote ischemic preconditioning had an AKI incidence of 37.5% compared with 52.5% for controls (P = .02); however, the mortality rate was the same.54 Similarly, remote ischemic preconditioning significantly lowered the incidence of AKI in nondiabetic patients undergoing CABG surgery compared with controls.55
Fluid management. Renal perfusion is intimately related to the development of AKI, and there is evidence that both hypovolemia and excessive fluid resuscitation can increase the risk of AKI in noncardiac surgery patients.56 Because of this, fluid management has also received attention in perioperative AKI. Goal-directed fluid management has been evaluated in noncardiac surgery patients, and it did not show any benefit in preventing AKI.57 However, in a more recent retrospective study, postoperative positive fluid balance was associated with increased incidence of AKI compared with zero fluid balance. Negative fluid balance did not appear to have a detrimental effect.58
RECOMMENDATIONS
No prophylactic therapy has yet been shown to definitively decrease the risk of postoperative AKI in all patients. Nevertheless, it is important to identify patients at risk during the preoperative visit, especially those with CKD. Many patients undergoing surgery have CKD, placing them at high risk of developing AKI in the perioperative period. The risk is particularly high with cardiac surgery.
Serum creatinine and urine output should be closely monitored perioperatively in at-risk patients. If AKI is diagnosed, practitioners need to identify and ameliorate the cause as early as possible.
Recommendations from KDIGO for perioperative prevention and management of AKI are listed in Table 4.15 These include avoiding additional nephrotoxic medications and adjusting the doses of renally cleared medications. Also, some patients may benefit from preoperative counseling and specialist referral.
- Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA 2007; 298(17):2038–2047. doi:10.1001/jama.298.17.2038
- National Institute of Diabetes and Digestive and Kidney Diseases. Kidney Disease Statistics for the United States. www.niddk.nih.gov/health-information/health-statistics/kidney-disease. Accessed June 11, 2018.
- Rosner MH, Okusa MD. Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol 2006; 1(1):19–32. doi:10.2215/CJN.00240605
- Meersch M, Schmidt C, Zarbock A. Patient with chronic renal failure undergoing surgery. Curr Opin Anaesthesiol 2016; 29(3):413–420. doi:10.1097/ACO.0000000000000329
- Stevens PE, Levin A; Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: synopsis of the Kidney Disease: Improving Global Outcomes 2012 clinical practice guideline. Ann Intern Med 2013; 158(11):825–830. doi:10.7326/0003-4819-158-11-201306040-00007
- Levey AS, Eckardt KU, Tsukamoto Y, et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2005; 67(6):2089–2100. doi:10.1111/j.1523-1755.2005.00365.x
- Saitoh M, Takahashi T, Sakurada K, et al. Factors determining achievement of early postoperative cardiac rehabilitation goal in patients with or without preoperative kidney dysfunction undergoing isolated cardiac surgery. J Cardiol 2013; 61(4):299–303. doi:10.1016/j.jjcc.2012.12.014
- Minakata K, Bando K, Tanaka S, et al. Preoperative chronic kidney disease as a strong predictor of postoperative infection and mortality after coronary artery bypass grafting. Circ J 2014; 78(9):2225–2231. doi:10.1253/circj.CJ-14-0328
- Domoto S, Tagusari O, Nakamura Y, et al. Preoperative estimated glomerular filtration rate as a significant predictor of long-term outcomes after coronary artery bypass grafting in Japanese patients. Gen Thorac Cardiovasc Surg 2014; 62(2):95–102. doi:10.1007/s11748-013-0306-5
- Hedley AJ, Roberts MA, Hayward PA, et al. Impact of chronic kidney disease on patient outcome following cardiac surgery. Heart Lung Circ 2010; 19(8):453–459. doi:10.1016/j.hlc.2010.03.005
- Boulton BJ, Kilgo P, Guyton RA, et al. Impact of preoperative renal dysfunction in patients undergoing off-pump versus on-pump coronary artery bypass. Ann Thorac Surg 2011; 92(2):595–601. doi:10.1016/j.athoracsur.2011.04.023
- Prowle JR, Kam EP, Ahmad T, Smith NC, Protopapa K, Pearse RM. Preoperative renal dysfunction and mortality after non-cardiac surgery. Br J Surg 2016; 103(10):1316–1325. doi:10.1002/bjs.10186
- Gaber AO, Moore LW, Aloia TA, et al. Cross-sectional and case-control analyses of the association of kidney function staging with adverse postoperative outcomes in general and vascular surgery. Ann Surg 2013; 258(1):169–177. doi:10.1097/SLA.0b013e318288e18e
- Mases A, Sabaté S, Guilera N, et al. Preoperative estimated glomerular filtration rate and the risk of major adverse cardiovascular and cerebrovascular events in non-cardiac surgery. Br J Anaesth 2014; 113(4):644–651. doi:10.1093/bja/aeu134
- Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clinical Practice 2012; 120(4):c179–c184. doi:10.1159/000339789
- Pickering JW, James MT, Palmer SC. Acute kidney injury and prognosis after cardiopulmonary bypass: a meta-analysis of cohort studies. Am J Kidney Dis 2015; 65(2):283–293. doi:10.1053/j.ajkd.2014.09.008
- Dasta JF, Kane-Gill SL, Durtschi AJ, Pathak DS, Kellum JA. Costs and outcomes of acute kidney injury (AKI) following cardiac surgery. Nephrol Dial Transplant 2008; 23(6):1970-1974. doi:10.1093/ndt/gfm908
- Karkouti K, Wijeysundera DN, Yau TM, et al. Acute kidney injury after cardiac surgery focus on modifiable risk factors. Circulation 2009; 119(4):495–502. doi:10.1161/CIRCULATIONAHA.108.786913
- Xu JR, Zhu JM, Jiang J, et al. Risk factors for long-term mortality and progressive chronic kidney disease associated with acute kidney injury after cardiac surgery. Medicine (Baltimore) 2015; 94(45):e2025. doi:10.1097/MD.0000000000002025
- Chalmers J, Mediratta N, McShane J, Shaw M, Pullan M, Poullis M. The long-term effects of developing renal failure post-coronary artery bypass surgery, in patients with normal preoperative renal function. Eur J Cardiothorac Surg 2013; 43(3):555–559. doi:10.1093/ejcts/ezs329
- Ryden L, Sartipy U, Evans M, Holzmann MJ. Acute kidney injury after coronary artery bypass grafting and long-term risk of end-stage renal disease. Circulation 2014; 130(23):2005–2011. doi:10.1161/CIRCULATIONAHA.114.010622
- Gargiulo G, Capodanno D, Sannino A, et al. Impact of moderate preoperative chronic kidney disease on mortality after transcatheter aortic valve implantation. Int J Cardiol 2015; 189:77–78. doi:10.1016/j.ijcard.2015.04.077
- Gargiulo G, Capodanno D, Sannino A, et al. Moderate and severe preoperative chronic kidney disease worsen clinical outcomes after transcatheter aortic valve implantation meta-analysis of 4,992 patients. Circ Cardiovasc Interv 2015; 8(2):e002220. doi:10.1161/CIRCINTERVENTIONS.114.002220
- Han SS, Shin N, Baek SH, et al. Effects of acute kidney injury and chronic kidney disease on long-term mortality after coronary artery bypass grafting. Am Heart J 2015; 169(3):419–425. doi:10.1016/j.ahj.2014.12.019
- Aronson S, Fontes ML, Miao Y, Mangano DT; Investigators of the Multicenter Study of Perioperative Ischemia Research Group; Ischemia Research and Education Foundation. Risk index for perioperative renal dysfunction/failure: critical dependence on pulse pressure hypertension. Circulation 2007; 115(6):733–742. doi:10.1161/CIRCULATIONAHA.106.623538
- Hertzberg D, Sartipy U, Holzmann MJ. Type 1 and type 2 diabetes mellitus and risk of acute kidney injury after coronary artery bypass grafting. Am Heart J 2015; 170(5):895–902. doi:10.1016/j.ahj.2015.08.013
- Benedetto U, Sciarretta S, Roscitano A, et al. Preoperative angiotensin-converting enzyme inhibitors and acute kidney injury after coronary artery bypass grafting. Ann Thorac Surg 2008; 86(4):1160–1165. doi:10.1016/j.athoracsur.2008.06.018
- Arora P, Rajagopalam S, Ranjan R, et al. Preoperative use of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers is associated with increased risk for acute kidney injury after cardiovascular surgery. Clin J Am Soc Nephrol 2008; 3(5):1266–1273. doi:10.2215/CJN.05271107
- Haase M, Bellomo R, Story D, et al. Effect of mean arterial pressure, haemoglobin and blood transfusion during cardiopulmonary bypass on post-operative acute kidney injury. Nephrol Dial Transplant 2012; 27(1):153–160. doi:10.1093/ndt/gfr275
- Ono M, Arnaoutakis GJ, Fine DM, et al. Blood pressure excursions below the cerebral autoregulation threshold during cardiac surgery are associated with acute kidney injury. Crit Care Med 2013; 41(2):464-471. doi:10.1097/CCM.0b013e31826ab3a1
- Seabra VF, Alobaidi S, Balk EM, Poon AH, Jaber BL. Off-pump coronary artery bypass surgery and acute kidney injury: a meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol 2010; 5(10):1734–1744. doi:10.2215/CJN.02800310
- Garg AX, Devereaux PJ, Yusuf S, et al; CORONARY Investigators. Kidney function after off-pump or on-pump coronary artery bypass graft surgery: a randomized clinical trial. JAMA 2014; 311(21):2191–2198. doi:10.1001/jama.2014.4952
- Kumar AB, Suneja M, Bayman EO, Weide GD, Tarasi M. Association between postoperative acute kidney injury and duration of cardiopulmonary bypass: a meta-analysis. J Cardiothorac Vasc Anesth 2012; 26(1):64–69. doi:10.1053/j.jvca.2011.07.007
- Kheterpal S, Tremper KK, Englesbe MJ, et al. Predictors of postoperative acute renal failure after noncardiac surgery in patients with previously normal renal function. Anesthesiology 2007; 107(6):892–902. doi:10.1097/01.anes.0000290588.29668.38
- Grams ME, Sang Y, Coresh J, et al. Acute kidney injury after major surgery: a retrospective analysis of Veterans Health Administration data. Am J Kidney Dis 2016; 67(6):872–880. doi:10.1053/j.ajkd.2015.07.022
- Biteker M, Dayan A, Tekkesin AI, et al. Incidence, risk factors, and outcomes of perioperative acute kidney injury in noncardiac and nonvascular surgery. Am J Surg 2014: 207(1):53–59. doi:10.1016/j.amjsurg.2013.04.006
- Gu W-J, Hou B-L, Kwong JS, et al. Association between intraoperative hypotension and 30-day mortality, major adverse cardiac events, and acute kidney injury after non-cardiac surgery: a meta-analysis of cohort studies. Int J Cardiol 2018; 258:68–73. doi:10.1016/j.ijcard.2018.01.137
- Smetana GW, Macpherson DS. The case against routine preoperative laboratory testing. Med Clin North Am 2003; 87(1):7–40. pmid:12575882
- Perrotti A, Miltgen G, Chevet-Noel A, et al. Neutrophil gelatinase-associated lipocalin as early predictor of acute kidney injury after cardiac surgery in adults with chronic kidney failure. Ann Thorac Surg 2015; 99(3):864–869. doi:10.1016/j.athoracsur.2014.10.011
- Doi K, Urata M, Katagiri D, et al. Plasma neutrophil gelatinase-associated lipocalin in acute kidney injury superimposed on chronic kidney disease after cardiac surgery: a multicenter prospective study. Crit Care 2013; 17(6):R270. doi:10.1186/cc13104
- Ho J, Tangri N, Komenda P, et al. Urinary, plasma, and serum biomarkers’ utility for predicting acute kidney injury associated with cardiac surgery in adults: a meta-analysis. Am J Kidney Dis 2015; 66(6):993–1005. doi:10.1053/j.ajkd.2015.06.018
- Yao L, Young N, Liu H, et al. Evidence for preoperative aspirin improving major outcomes in patients with chronic kidney disease undergoing cardiac surgery: a cohort study. Ann Surg 2015; 261(1):207–212. doi:10.1097/SLA.0000000000000641
- Garg AX, Kurz A, Sessler DI, et al; POISE-2 Investigators. Aspirin and clonidine in non-cardiac surgery: acute kidney injury substudy protocol of the perioperative ischaemic evaluation (POISE) 2 randomised controlled trial. BMJ open 2014; 4(2):e004886. doi:10.1136/bmjopen-2014-004886
- He SJ, Liu Q, Li HQ, Tian F, Chen SY, Weng JX. Role of statins in preventing cardiac surgery-associated acute kidney injury: an updated meta-analysis of randomized controlled trials. Ther Clin Risk Manag 2018; 14:475–482. doi:10.2147/TCRM.S160298
- Tie HT, Luo MZ, Lin D, Zhang M, Wan JY, Wu QC. Erythropoietin administration for prevention of cardiac surgery-associated acute kidney injury: a meta-analysis of randomized controlled trials. Eur J Cardiothorac Surg 2015; 48(1):32–39. doi:10.1093/ejcts/ezu378
- Santana-Santos E, Gowdak LH, Gaiotto FA, et al. High dose of N-acetylcystein prevents acute kidney injury in chronic kidney disease patients undergoing myocardial revascularization. Ann Thorac Surg 2014; 97(5):1617–1623. doi:10.1016/j.athoracsur.2014.01.056
- Mei M, Zhao HW, Pan QG, Pu YM, Tang MZ, Shen BB. Efficacy of N-acetylcysteine in preventing acute kidney injury after cardiac surgery: a meta-analysis study. J Invest Surg 2018; 31(1):14–23. doi:10.1080/08941939.2016.1269853
- Sezai A, Hata M, Niino T, et al. Results of low-dose human atrial natriuretic peptide infusion in nondialysis patients with chronic kidney disease undergoing coronary artery bypass grafting: the NU-HIT (Nihon University working group study of low-dose HANP infusion therapy during cardiac surgery) trial for CKD. J Am Coll Cardiol 2011; 58(9):897–903. doi:10.1016/j.jacc.2011.03.056
- Xu N, Long Q, He T, et al. Association between preoperative renin-angiotensin system inhibitor use and postoperative acute kidney injury risk in patients with hypertension. Clin Nephrol 2018; 89(6):403–414. doi:10.5414/CN109319
- Liu Y, Sheng B, Wang S, Lu F, Zhen J, Chen W. Dexmedetomidine prevents acute kidney injury after adult cardiac surgery: a meta-analysis of randomized controlled trials. BMC Anesthesiol 2018; 18(1):7. doi:10.1186/s12871-018-0472-1
- Shi R, Tie H-T. Dexmedetomidine as a promising prevention strategy for cardiac surgery-associated acute kidney injury: a meta-analysis. Critical Care 2017; 21(1):198. doi:10.1186/s13054-017-1776-0
- Zhou C, Gong J, Chen D, Wang W, Liu M, Liu B. Levosimendan for prevention of acute kidney injury after cardiac surgery: a meta-analysis of randomized controlled trials. Am J Kidney Dis 2016; 67(3):408–416. doi:10.1053/j.ajkd.2015.09.015
- Elbadawi A, Elgendy IY, Saad M, et al. Meta-analysis of trials on prophylactic use of levosimendan in patients undergoing cardiac surgery. Ann Thorac Surg 2018; 105(5):1403–1410. doi:10.1016/j.athoracsur.2017.11.027
- Zarbock A, Schmidt C, Van Aken H, et al; RenalRIPC Investigators. Effect of remote ischemic preconditioning on kidney injury among high-risk patients undergoing cardiac surgery: a randomized clinical trial. JAMA 2015; 313(21):2133–2141. doi:10.1001/jama.2015.4189
- Venugopal V, Laing CM, Ludman A, Yellon DM, Hausenloy D. Effect of remote ischemic preconditioning on acute kidney injury in nondiabetic patients undergoing coronary artery bypass graft surgery: a secondary analysis of 2 small randomized trials. Am J Kidney Dis 2010; 56(6):1043–1049. doi:10.1053/j.ajkd.2010.07.014
- Futier E, Constantin JM, Petit A, et al. Conservative vs restrictive individualized goal-directed fluid replacement strategy in major abdominal surgery: a prospective randomized trial. Arch Surg 2010; 145(12):1193–1200. doi:10.1001/archsurg.2010.275
- Patel A, Prowle JR, Ackland GL. Postoperative goal-directed therapy and development of acute kidney injury following major elective noncardiac surgery: post-hoc analysis of POM-O randomized controlled trial. Clin Kidney J 2017; 10(3):348–356. doi:10.1093/ckj/sfw118
- Shen Y, Zhang W, Cheng X, Ying M. Association between postoperative fluid balance and acute kidney injury in patients after cardiac surgery: a retrospective cohort study. J Crit Care 2018; 44:273–277. doi:10.1016/j.jcrc.2017.11.041
- Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA 2007; 298(17):2038–2047. doi:10.1001/jama.298.17.2038
- National Institute of Diabetes and Digestive and Kidney Diseases. Kidney Disease Statistics for the United States. www.niddk.nih.gov/health-information/health-statistics/kidney-disease. Accessed June 11, 2018.
- Rosner MH, Okusa MD. Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol 2006; 1(1):19–32. doi:10.2215/CJN.00240605
- Meersch M, Schmidt C, Zarbock A. Patient with chronic renal failure undergoing surgery. Curr Opin Anaesthesiol 2016; 29(3):413–420. doi:10.1097/ACO.0000000000000329
- Stevens PE, Levin A; Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: synopsis of the Kidney Disease: Improving Global Outcomes 2012 clinical practice guideline. Ann Intern Med 2013; 158(11):825–830. doi:10.7326/0003-4819-158-11-201306040-00007
- Levey AS, Eckardt KU, Tsukamoto Y, et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2005; 67(6):2089–2100. doi:10.1111/j.1523-1755.2005.00365.x
- Saitoh M, Takahashi T, Sakurada K, et al. Factors determining achievement of early postoperative cardiac rehabilitation goal in patients with or without preoperative kidney dysfunction undergoing isolated cardiac surgery. J Cardiol 2013; 61(4):299–303. doi:10.1016/j.jjcc.2012.12.014
- Minakata K, Bando K, Tanaka S, et al. Preoperative chronic kidney disease as a strong predictor of postoperative infection and mortality after coronary artery bypass grafting. Circ J 2014; 78(9):2225–2231. doi:10.1253/circj.CJ-14-0328
- Domoto S, Tagusari O, Nakamura Y, et al. Preoperative estimated glomerular filtration rate as a significant predictor of long-term outcomes after coronary artery bypass grafting in Japanese patients. Gen Thorac Cardiovasc Surg 2014; 62(2):95–102. doi:10.1007/s11748-013-0306-5
- Hedley AJ, Roberts MA, Hayward PA, et al. Impact of chronic kidney disease on patient outcome following cardiac surgery. Heart Lung Circ 2010; 19(8):453–459. doi:10.1016/j.hlc.2010.03.005
- Boulton BJ, Kilgo P, Guyton RA, et al. Impact of preoperative renal dysfunction in patients undergoing off-pump versus on-pump coronary artery bypass. Ann Thorac Surg 2011; 92(2):595–601. doi:10.1016/j.athoracsur.2011.04.023
- Prowle JR, Kam EP, Ahmad T, Smith NC, Protopapa K, Pearse RM. Preoperative renal dysfunction and mortality after non-cardiac surgery. Br J Surg 2016; 103(10):1316–1325. doi:10.1002/bjs.10186
- Gaber AO, Moore LW, Aloia TA, et al. Cross-sectional and case-control analyses of the association of kidney function staging with adverse postoperative outcomes in general and vascular surgery. Ann Surg 2013; 258(1):169–177. doi:10.1097/SLA.0b013e318288e18e
- Mases A, Sabaté S, Guilera N, et al. Preoperative estimated glomerular filtration rate and the risk of major adverse cardiovascular and cerebrovascular events in non-cardiac surgery. Br J Anaesth 2014; 113(4):644–651. doi:10.1093/bja/aeu134
- Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clinical Practice 2012; 120(4):c179–c184. doi:10.1159/000339789
- Pickering JW, James MT, Palmer SC. Acute kidney injury and prognosis after cardiopulmonary bypass: a meta-analysis of cohort studies. Am J Kidney Dis 2015; 65(2):283–293. doi:10.1053/j.ajkd.2014.09.008
- Dasta JF, Kane-Gill SL, Durtschi AJ, Pathak DS, Kellum JA. Costs and outcomes of acute kidney injury (AKI) following cardiac surgery. Nephrol Dial Transplant 2008; 23(6):1970-1974. doi:10.1093/ndt/gfm908
- Karkouti K, Wijeysundera DN, Yau TM, et al. Acute kidney injury after cardiac surgery focus on modifiable risk factors. Circulation 2009; 119(4):495–502. doi:10.1161/CIRCULATIONAHA.108.786913
- Xu JR, Zhu JM, Jiang J, et al. Risk factors for long-term mortality and progressive chronic kidney disease associated with acute kidney injury after cardiac surgery. Medicine (Baltimore) 2015; 94(45):e2025. doi:10.1097/MD.0000000000002025
- Chalmers J, Mediratta N, McShane J, Shaw M, Pullan M, Poullis M. The long-term effects of developing renal failure post-coronary artery bypass surgery, in patients with normal preoperative renal function. Eur J Cardiothorac Surg 2013; 43(3):555–559. doi:10.1093/ejcts/ezs329
- Ryden L, Sartipy U, Evans M, Holzmann MJ. Acute kidney injury after coronary artery bypass grafting and long-term risk of end-stage renal disease. Circulation 2014; 130(23):2005–2011. doi:10.1161/CIRCULATIONAHA.114.010622
- Gargiulo G, Capodanno D, Sannino A, et al. Impact of moderate preoperative chronic kidney disease on mortality after transcatheter aortic valve implantation. Int J Cardiol 2015; 189:77–78. doi:10.1016/j.ijcard.2015.04.077
- Gargiulo G, Capodanno D, Sannino A, et al. Moderate and severe preoperative chronic kidney disease worsen clinical outcomes after transcatheter aortic valve implantation meta-analysis of 4,992 patients. Circ Cardiovasc Interv 2015; 8(2):e002220. doi:10.1161/CIRCINTERVENTIONS.114.002220
- Han SS, Shin N, Baek SH, et al. Effects of acute kidney injury and chronic kidney disease on long-term mortality after coronary artery bypass grafting. Am Heart J 2015; 169(3):419–425. doi:10.1016/j.ahj.2014.12.019
- Aronson S, Fontes ML, Miao Y, Mangano DT; Investigators of the Multicenter Study of Perioperative Ischemia Research Group; Ischemia Research and Education Foundation. Risk index for perioperative renal dysfunction/failure: critical dependence on pulse pressure hypertension. Circulation 2007; 115(6):733–742. doi:10.1161/CIRCULATIONAHA.106.623538
- Hertzberg D, Sartipy U, Holzmann MJ. Type 1 and type 2 diabetes mellitus and risk of acute kidney injury after coronary artery bypass grafting. Am Heart J 2015; 170(5):895–902. doi:10.1016/j.ahj.2015.08.013
- Benedetto U, Sciarretta S, Roscitano A, et al. Preoperative angiotensin-converting enzyme inhibitors and acute kidney injury after coronary artery bypass grafting. Ann Thorac Surg 2008; 86(4):1160–1165. doi:10.1016/j.athoracsur.2008.06.018
- Arora P, Rajagopalam S, Ranjan R, et al. Preoperative use of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers is associated with increased risk for acute kidney injury after cardiovascular surgery. Clin J Am Soc Nephrol 2008; 3(5):1266–1273. doi:10.2215/CJN.05271107
- Haase M, Bellomo R, Story D, et al. Effect of mean arterial pressure, haemoglobin and blood transfusion during cardiopulmonary bypass on post-operative acute kidney injury. Nephrol Dial Transplant 2012; 27(1):153–160. doi:10.1093/ndt/gfr275
- Ono M, Arnaoutakis GJ, Fine DM, et al. Blood pressure excursions below the cerebral autoregulation threshold during cardiac surgery are associated with acute kidney injury. Crit Care Med 2013; 41(2):464-471. doi:10.1097/CCM.0b013e31826ab3a1
- Seabra VF, Alobaidi S, Balk EM, Poon AH, Jaber BL. Off-pump coronary artery bypass surgery and acute kidney injury: a meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol 2010; 5(10):1734–1744. doi:10.2215/CJN.02800310
- Garg AX, Devereaux PJ, Yusuf S, et al; CORONARY Investigators. Kidney function after off-pump or on-pump coronary artery bypass graft surgery: a randomized clinical trial. JAMA 2014; 311(21):2191–2198. doi:10.1001/jama.2014.4952
- Kumar AB, Suneja M, Bayman EO, Weide GD, Tarasi M. Association between postoperative acute kidney injury and duration of cardiopulmonary bypass: a meta-analysis. J Cardiothorac Vasc Anesth 2012; 26(1):64–69. doi:10.1053/j.jvca.2011.07.007
- Kheterpal S, Tremper KK, Englesbe MJ, et al. Predictors of postoperative acute renal failure after noncardiac surgery in patients with previously normal renal function. Anesthesiology 2007; 107(6):892–902. doi:10.1097/01.anes.0000290588.29668.38
- Grams ME, Sang Y, Coresh J, et al. Acute kidney injury after major surgery: a retrospective analysis of Veterans Health Administration data. Am J Kidney Dis 2016; 67(6):872–880. doi:10.1053/j.ajkd.2015.07.022
- Biteker M, Dayan A, Tekkesin AI, et al. Incidence, risk factors, and outcomes of perioperative acute kidney injury in noncardiac and nonvascular surgery. Am J Surg 2014: 207(1):53–59. doi:10.1016/j.amjsurg.2013.04.006
- Gu W-J, Hou B-L, Kwong JS, et al. Association between intraoperative hypotension and 30-day mortality, major adverse cardiac events, and acute kidney injury after non-cardiac surgery: a meta-analysis of cohort studies. Int J Cardiol 2018; 258:68–73. doi:10.1016/j.ijcard.2018.01.137
- Smetana GW, Macpherson DS. The case against routine preoperative laboratory testing. Med Clin North Am 2003; 87(1):7–40. pmid:12575882
- Perrotti A, Miltgen G, Chevet-Noel A, et al. Neutrophil gelatinase-associated lipocalin as early predictor of acute kidney injury after cardiac surgery in adults with chronic kidney failure. Ann Thorac Surg 2015; 99(3):864–869. doi:10.1016/j.athoracsur.2014.10.011
- Doi K, Urata M, Katagiri D, et al. Plasma neutrophil gelatinase-associated lipocalin in acute kidney injury superimposed on chronic kidney disease after cardiac surgery: a multicenter prospective study. Crit Care 2013; 17(6):R270. doi:10.1186/cc13104
- Ho J, Tangri N, Komenda P, et al. Urinary, plasma, and serum biomarkers’ utility for predicting acute kidney injury associated with cardiac surgery in adults: a meta-analysis. Am J Kidney Dis 2015; 66(6):993–1005. doi:10.1053/j.ajkd.2015.06.018
- Yao L, Young N, Liu H, et al. Evidence for preoperative aspirin improving major outcomes in patients with chronic kidney disease undergoing cardiac surgery: a cohort study. Ann Surg 2015; 261(1):207–212. doi:10.1097/SLA.0000000000000641
- Garg AX, Kurz A, Sessler DI, et al; POISE-2 Investigators. Aspirin and clonidine in non-cardiac surgery: acute kidney injury substudy protocol of the perioperative ischaemic evaluation (POISE) 2 randomised controlled trial. BMJ open 2014; 4(2):e004886. doi:10.1136/bmjopen-2014-004886
- He SJ, Liu Q, Li HQ, Tian F, Chen SY, Weng JX. Role of statins in preventing cardiac surgery-associated acute kidney injury: an updated meta-analysis of randomized controlled trials. Ther Clin Risk Manag 2018; 14:475–482. doi:10.2147/TCRM.S160298
- Tie HT, Luo MZ, Lin D, Zhang M, Wan JY, Wu QC. Erythropoietin administration for prevention of cardiac surgery-associated acute kidney injury: a meta-analysis of randomized controlled trials. Eur J Cardiothorac Surg 2015; 48(1):32–39. doi:10.1093/ejcts/ezu378
- Santana-Santos E, Gowdak LH, Gaiotto FA, et al. High dose of N-acetylcystein prevents acute kidney injury in chronic kidney disease patients undergoing myocardial revascularization. Ann Thorac Surg 2014; 97(5):1617–1623. doi:10.1016/j.athoracsur.2014.01.056
- Mei M, Zhao HW, Pan QG, Pu YM, Tang MZ, Shen BB. Efficacy of N-acetylcysteine in preventing acute kidney injury after cardiac surgery: a meta-analysis study. J Invest Surg 2018; 31(1):14–23. doi:10.1080/08941939.2016.1269853
- Sezai A, Hata M, Niino T, et al. Results of low-dose human atrial natriuretic peptide infusion in nondialysis patients with chronic kidney disease undergoing coronary artery bypass grafting: the NU-HIT (Nihon University working group study of low-dose HANP infusion therapy during cardiac surgery) trial for CKD. J Am Coll Cardiol 2011; 58(9):897–903. doi:10.1016/j.jacc.2011.03.056
- Xu N, Long Q, He T, et al. Association between preoperative renin-angiotensin system inhibitor use and postoperative acute kidney injury risk in patients with hypertension. Clin Nephrol 2018; 89(6):403–414. doi:10.5414/CN109319
- Liu Y, Sheng B, Wang S, Lu F, Zhen J, Chen W. Dexmedetomidine prevents acute kidney injury after adult cardiac surgery: a meta-analysis of randomized controlled trials. BMC Anesthesiol 2018; 18(1):7. doi:10.1186/s12871-018-0472-1
- Shi R, Tie H-T. Dexmedetomidine as a promising prevention strategy for cardiac surgery-associated acute kidney injury: a meta-analysis. Critical Care 2017; 21(1):198. doi:10.1186/s13054-017-1776-0
- Zhou C, Gong J, Chen D, Wang W, Liu M, Liu B. Levosimendan for prevention of acute kidney injury after cardiac surgery: a meta-analysis of randomized controlled trials. Am J Kidney Dis 2016; 67(3):408–416. doi:10.1053/j.ajkd.2015.09.015
- Elbadawi A, Elgendy IY, Saad M, et al. Meta-analysis of trials on prophylactic use of levosimendan in patients undergoing cardiac surgery. Ann Thorac Surg 2018; 105(5):1403–1410. doi:10.1016/j.athoracsur.2017.11.027
- Zarbock A, Schmidt C, Van Aken H, et al; RenalRIPC Investigators. Effect of remote ischemic preconditioning on kidney injury among high-risk patients undergoing cardiac surgery: a randomized clinical trial. JAMA 2015; 313(21):2133–2141. doi:10.1001/jama.2015.4189
- Venugopal V, Laing CM, Ludman A, Yellon DM, Hausenloy D. Effect of remote ischemic preconditioning on acute kidney injury in nondiabetic patients undergoing coronary artery bypass graft surgery: a secondary analysis of 2 small randomized trials. Am J Kidney Dis 2010; 56(6):1043–1049. doi:10.1053/j.ajkd.2010.07.014
- Futier E, Constantin JM, Petit A, et al. Conservative vs restrictive individualized goal-directed fluid replacement strategy in major abdominal surgery: a prospective randomized trial. Arch Surg 2010; 145(12):1193–1200. doi:10.1001/archsurg.2010.275
- Patel A, Prowle JR, Ackland GL. Postoperative goal-directed therapy and development of acute kidney injury following major elective noncardiac surgery: post-hoc analysis of POM-O randomized controlled trial. Clin Kidney J 2017; 10(3):348–356. doi:10.1093/ckj/sfw118
- Shen Y, Zhang W, Cheng X, Ying M. Association between postoperative fluid balance and acute kidney injury in patients after cardiac surgery: a retrospective cohort study. J Crit Care 2018; 44:273–277. doi:10.1016/j.jcrc.2017.11.041
KEY POINTS
- Many patients undergoing surgery have CKD—up to 30% in some cardiac surgery populations.
- CKD is a risk factor for perioperative complications including acute kidney injury and death.
- Although challenging, early detection of renal injury is crucial to improving outcomes in this patient population. New biomarkers are being investigated.
- Preoperative assessment and perioperative management of renal dysfunction may reduce the risk of adverse postoperative outcomes.
Nephrogenic Systemic Fibrosis in a Patient With Multiple Inflammatory Disorders
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1) 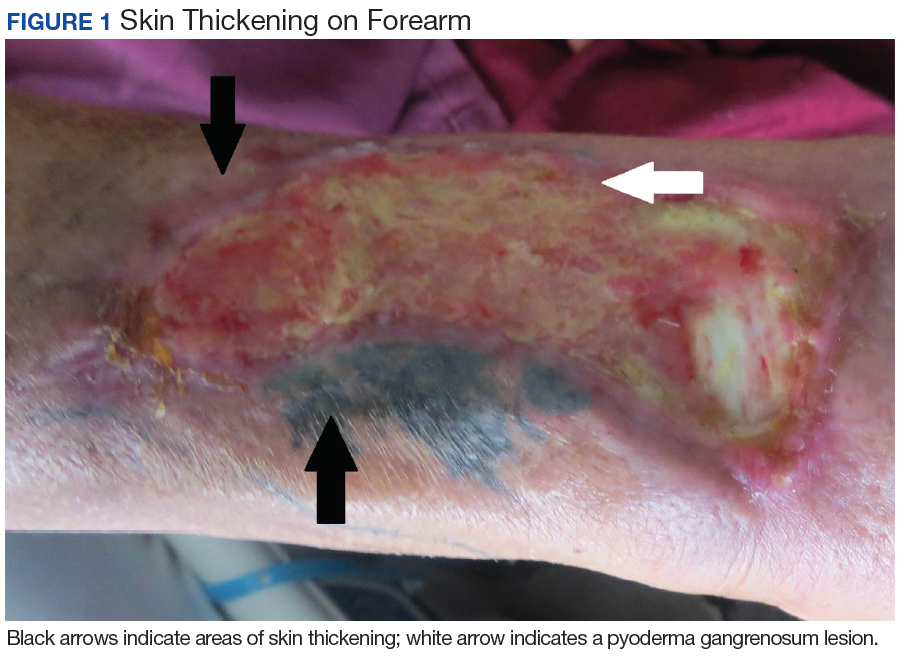
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available. 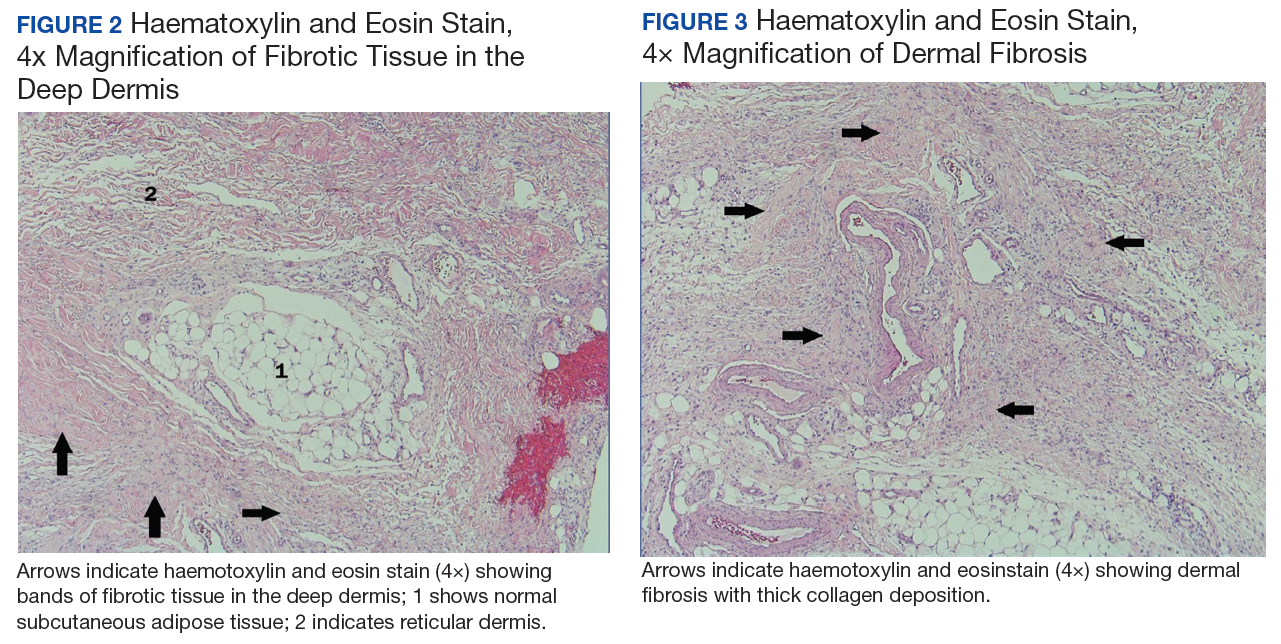
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1)
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available.
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1)
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available.
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.