User login
Diffuse Dermal Angiomatosis
Diffuse dermal angiomatosis (DDA) is a rare acquired, cutaneous, reactive, vascular disorder that was originally thought to be a variant of cutaneous reactive angiomatosis (CREA) but is now considered to be on the spectrum of CREA. This article will focus on DDA and review the literature of prior case reports with brief descriptions of the differential diagnosis.
Case Report
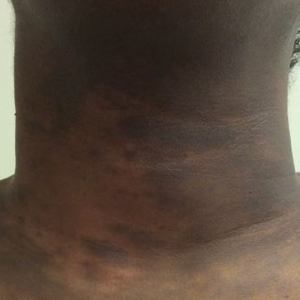
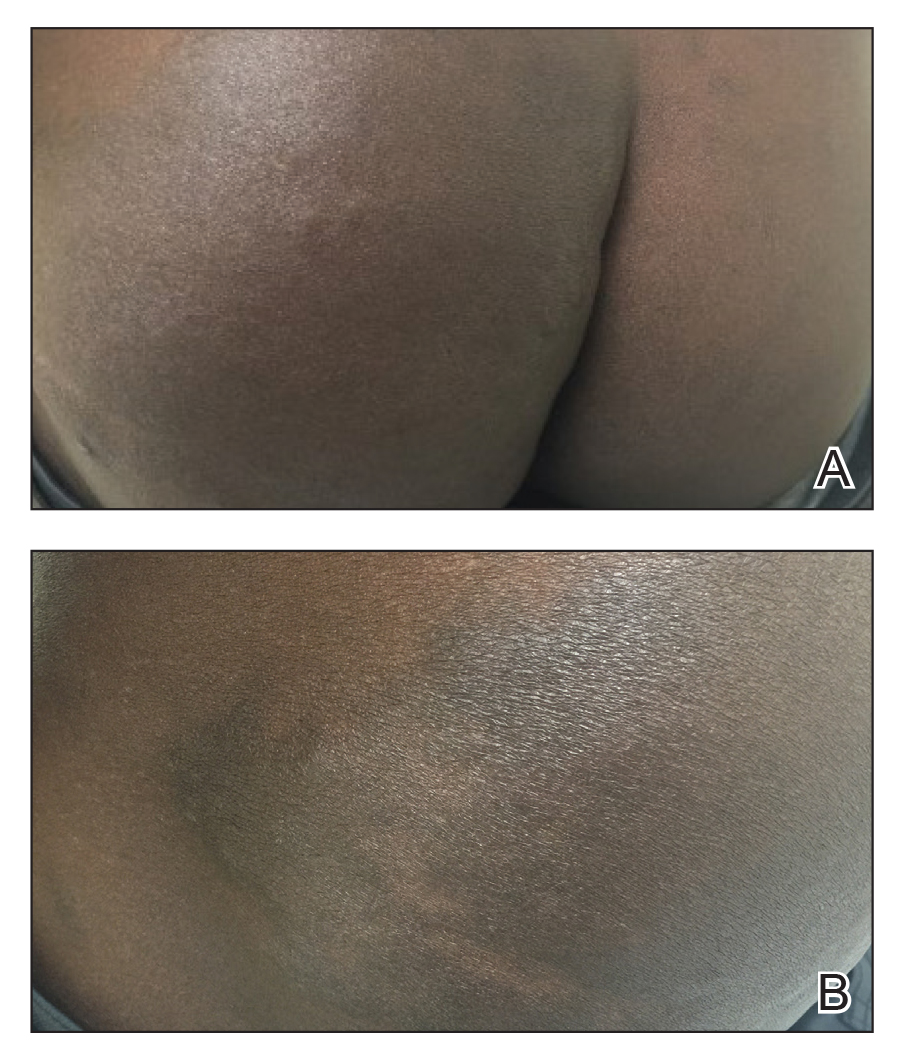
A 43-year-old Haitian man presented to the clinic with a lesion on the left buttock that had developed over the last 6 years. The patient stated the lesion had been enlarging over the last several months. Upon examination, there was a large (15-cm diameter), indurated, hyperpigmented plaque covering the left buttock (Figure 1). The patient reported no medical or contributory family history. Upon review of systems, he described a burning sensation sometimes in the area of the lesion that would develop randomly throughout the year.
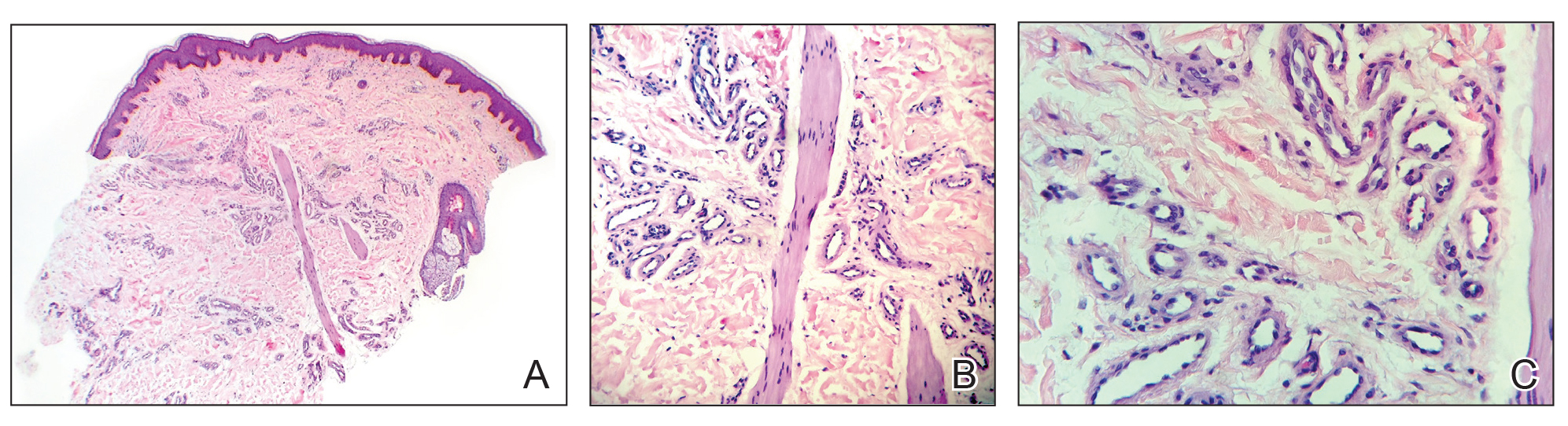
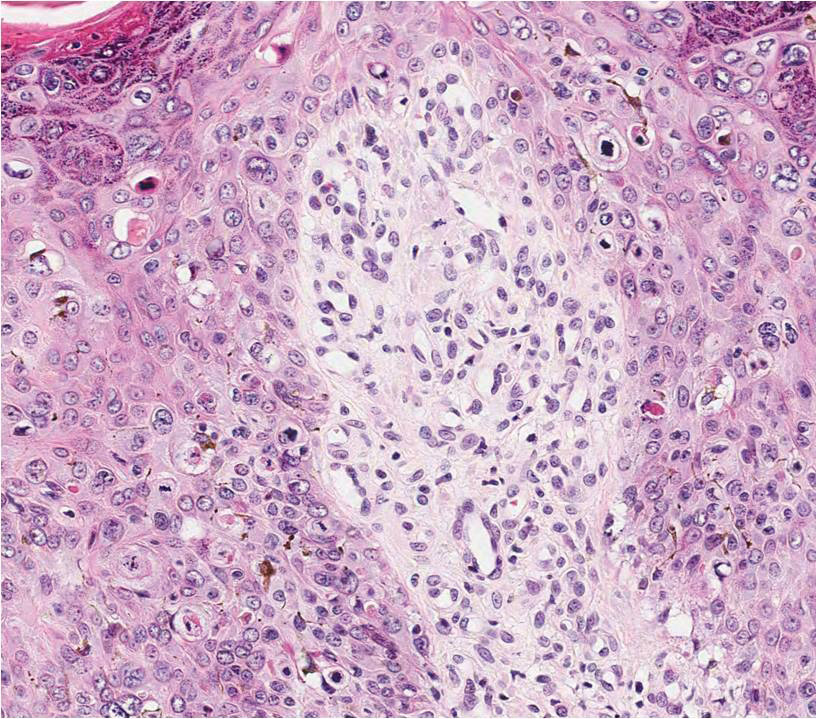
Three biopsies were performed, which revealed a collection of slightly dilated blood vessels with normal-appearing endothelial cells occupying the mid dermis and deep dermis (Figure 2). Immunohistochemical stains with antibodies were directed against human herpesvirus 8 (HHV-8), CD31, CD34, the cell surface glycoprotein podoplanin, Ki-67, and smooth muscle actin antigens, with appropriate controls. The vessel walls were positive for CD31, CD34, and smooth muscle actin, and negative for HHV-8 and podoplanin; Ki-67 was not increased. These histologic findings were consistent with a diagnosis of DDA. A detailed history was taken. The cause of DDA in our patient was uncertain.
Comment
Classification and Epidemiology
Diffuse dermal angiomatosis is a rare acquired, cutaneous, reactive, vascular disorder first described by Krell et al1 in 1994. Diffuse dermal angiomatosis is benign and is classified in the group of cutaneous reactive angiomatoses,2 which are benign vascular disorders marked by intravascular and extravascular hyperplasia of endothelial cells that may or may not include pericytes.2 Diffuse dermal angiomatosis was originally described as a variant of CREA, which is characterized by hyperplasia of endothelial dermal cells and intravascular proliferation.3 However, DDA has more recently been identified as a distinct disorder on the spectrum of CREA rather than as a variant of CREA.2 Given the recent reclassification, not all physicians make this distinction. However, as more case reports of DDA are published, physicians continue to support this change.4 Nevertheless, DDA has been an established disorder since 1994.1
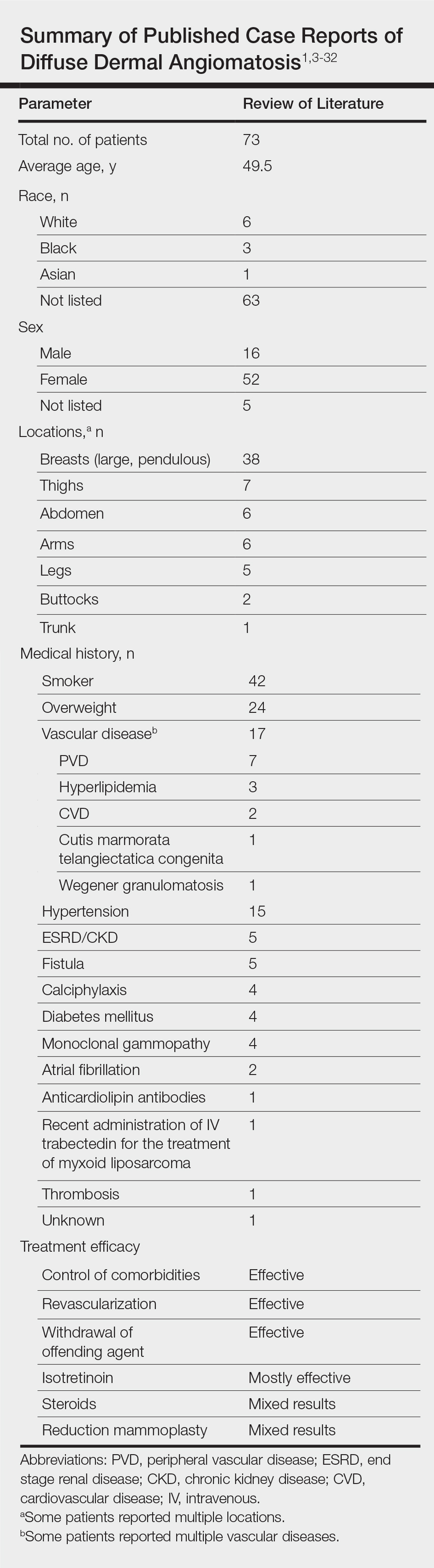
Vascular proliferation in DDA is hypothesized to stem from ischemia or inflammation.5 Peripheral vascular atherosclerosis has been associated with DDA.6 The epidemiology of DDA is not well known because of the rarity of the disease. We performed a more specific review of the literature by limiting the PubMed search of articles indexed for MEDLINE to the term diffuse dermal angiomatosis rather than a broader search including all reactive angioendotheliomatoses. Only 31 case reports have been published1,3-32; of them, only adults were affected. Most reported cases were in middle-aged females. A summary of the demographics of DDA is provided in the Table.1,3-32
Pathophysiology
The pathophysiology of DDA remains unclear. It has been hypothesized that ischemia or inflammation creates local hypoxia, leading to an increase in vascular endothelial growth factor with subsequent endothelial proliferation and neovascularization.5 Rongioletti and Robora2 supported this hypothesis, proposing that occlusion or inflammation of the vasculature creates microthrombi and thus hypoxia. Afterward, histiocytes are recruited to reabsorb the microthrombi while hyperplasia of endothelial cells and pericytes ensues.7 Complete resolution of skin lesions following revascularization provides support for this theory.8
Etiology
Diffuse dermal angiomatosis is a rare complication of ischemia that may be secondary to atherosclerosis, arteriovenous fistula, or macromastia.9-11 In DDA of the breasts, ulcerations of fatty tissue occur due to trauma in these patients who have large pendulous breasts, causing angiogenesis resembling DDA histologically.2 One case of DDA was reported secondary to relative ischemia from cutis marmorata telangiectatica congenita,12 whereas another case highlighted Wegener granulomatosis as the cause of ischemia.7 There also have been reported cases associated with calciphylaxis and anticardiolipin antibiodies.13 In general, any medical condition that can lead to ischemia can cause DDA. Comorbid conditions for DDA include cardiovascular disease, hypertension, diabetes mellitus, and most often severe peripheral vascular disease. Many patients also have a history of smoking.14 Diffuse dermal angiomatosis rarely presents without underlying comorbidity, with only 1 case report of unknown cause (Table).
Presentation, Histopathology, and Differential Diagnosis
Cutaneous reactive angiomatosis disorders present the same clinically, with multiple erythematous to violaceous purpuric patches and plaques that can progress to necrosis and ulceration. Lesions are widely distributed but are predisposed to the upper and lower extremities.2 The differential diagnosis of DDA includes CREA, acroangiodermatitis (pseudo–Kaposi sarcoma), or vascular malignancies such as Kaposi sarcoma and low-grade angiosarcoma.7
In DDA, lesions may be painful and sometimes have a central ulceration.15 They often are associated with notable peripheral vascular atherosclerotic disease and are mainly found on the lower extremities.12,16 Histologically, DDA presents as a diffuse proliferation of endothelial cells between collagen bundles. The endothelial cells are distributed throughout the papillary and reticular dermis and develop into vascular lumina.17 Furthermore, the proliferating endothelial cells are spindle shaped and contain vacuolated cytoplasm.14
Acroangiodermatitis, or pseudo–Kaposi sarcoma, presents as slow-growing, erythematous to violaceous, brown, or dusky macules, papules, or plaques of the legs.14 Histologically, acroangiodermatitis presents with relatively less proliferation of endothelial cells found intravascularly rather than extravascularly, as in DDA, forming new thick-walled vessels in a lobular pattern in the papillary dermis.14
Vascular malignancies, such as Kaposi sarcoma and angiosarcoma, may present similarly to DDA. Kaposi sarcoma, for example, presents as erythematous to violaceous patches, plaques, or nodules found mostly on the extremities.7 Histologically, spindle cells and vascular structures also are found but in a clefting pattern representative of Kaposi sarcoma (so-called vascular slits).7 Diffuse dermal angiomatosis and vascular malignancies can further be distinguished based on atypia of the proliferations and staining for HHV-8.7,14 Lastly, DDA differs from vascular tumors in that vascular tumors are reactive to locations of occluded vessels, with vascular proliferation ceasing once the underlying cause of hypoxia is removed.2
Treatment
There is no standard treatment of DDA.7 Treatment of the underlying cause of ischemia is the primary goal, which will cause the DDA to resolve in most cases. Stenting, removal of an arteriovenous fistula, or other forms of revascularization may be warranted.1,5,6,10,17,29,30
Reported medical therapies for DDA include systemic or topical corticosteroids used for their antiangiogenic properties with varying results.7 Isotretinoin also has been used, which has been found to be effective in several cases of DDA of the breast, though 1 study reported a subsequent elevated lipid profile, requiring a decrease in dosage.14,15,27,31
Most interestingly, a study by Sanz-Motilva et al16 demonstrated that control of comorbidities, especially smoking cessation, led to improvement, which highlights the importance of incorporating nonpharmacotherapy rather than initiating treatment solely with medication. The Table summarizes treatments used and their efficacy.
Conclusion
Diffuse dermal angiomatosis is associated with medical conditions that predispose an individual to ischemia. Although rare, DDA can present as painful and visibly disturbing lesions that can affect the daily life of afflicted patients. By reporting the few cases that do arise and reviewing prior cases and their treatments, physicians can consider DDA within the differential diagnosis and identify which treatment is most efficient for a given patient. For all DDA patients, strict control of comorbidities, especially smoking cessation, should be incorporated into the treatment plan. When DDA affects the breasts, isotretinoin appears to provide the best relief. Otherwise, treatment of the underlying cause, revascularization, withdrawal of the offending agent, or steroids seem to be the best treatment options.
- Krell JM, Sanchez RL, Solomon AR. Diffuse dermal angiomatosis: a variant of reactive cutaneous angioendotheliomatosis. J Cutan Pathol. 1994;21:363-370.
- Rongioletti F, Robora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Crickx E, Saussine A, Vignon-Pennamen MD, et al. Diffuse dermal angiomatosis associated with severe atherosclerosis: two cases and review of the literature. Clin Exp Dermatol. 2015;40:521-524.
- Reusche R, Winocour S, Degnim A, et al. Diffuse dermal angiomatosis of the breast: a series of 22 cases from a single institution. Gland Surg. 2015;4:554-560.
- Sriphojanart T, Vachiramon V. Diffuse dermal angiomatosis: a clue to the diagnosis of atherosclerotic vascular disease. Case Rep Dermatol. 2015;7:100-106.
- Kimyai-Asadi A, Nousari HC, Ketabchi N, et al. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with atherosclerosis. J Am Acad Dermatol. 1999;40:257-259.
- Bassi A, Arunachalam M, Maio V, et al. Diffuse dermal angiomatosis in a patient with an iatrogenic arterio-venous fistula and Wegener’s granulomatosis. Acta Derm Venereol. 2013;93:93-94.
- Ormerod E, Miller K, Kennedy C. Diffuse dermal angiomatosis: a contributory factor to ulceration in a patient with renal transplant. Clin Exp Dermatol. 2015;40:48-51.
- Kim S, Elenitsas R, James WD. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with peripheral vascular atherosclerosis. Arch Dermatol. 2002;138:456-458.
- Requena L, Fariña MC, Renedo G, et al. Intravascular and diffuse dermal reactive angioendotheliomatosis secondary to iatrogenic arteriovenous fistulas. J Cutan Pathol. 1999;26:159-164.
- Villa MT, White LE, Petronic-Rosic V, et al. The treatment of diffuse dermal angiomatosis of the breast with reduction mammoplasty. Arch Dermatol. 2008;144:693-694.
- Halbesleben JJ, Cleveland MG, Stone MS. Diffuse dermal angiomatosis arising in cutis marmorata telangiectatica congenita. Arch Dermatol. 2010;146:1311-1313.
- Ferreli C, Atzori L, Pinna AL, et al. Diffuse dermal angiomatosis: a clinical mimicker of vasculitis associated with calciphylaxis and monoclonal gammopathy. G Ital Dermatol Venereol. 2015;150:115-121.
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347.
- Steele KT, Sullivan BJ, Wanat KA, et al. Diffuse dermal angiomatosis associated with calciphylaxis in a patient with end-stage renal disease.J Cutan Pathol. 2013;40:829-832.
- Sanz-Motilva V, Martorell-Calatayud A, Rongioletti F, et al. Diffuse dermal angiomatosis of the breast: clinical and histopathological features. Int J Dermatol. 2014;53:445-449.
- Kirkland CR, Hawayek LH, Mutasim DF. Atherosclerosis-induced diffuse dermal angiomatosis with fatal outcome. Arch Dermatol. 2010;146:684-685.
- Sommer S, Merchant WJ, Wilson CL. Diffuse dermal angiomatosis due to an iatrogenic arteriovenous fistula. Acta Derm Venereol. 2004;84:251-252.
- Corti MA, Rongioletti F, Borradori L, et al. Cutaneous reactive angiomatosis with combined histological pattern mimicking a cellulitis. Dermatology. 2013;227:226-230.
- Tollefson MM, McEvoy MT, Torgerson RR, et al. Diffuse dermal angiomatosis of the breast: clinicopathologic study of 5 patients. J Am Acad Dermatol. 2014;71:1212-1217.
- Walton K, Liggett J. Diffuse dermal angiomatosis: a case report. J Am Acad Dermatol. 2012;66(suppl 1):AB49.
- Mayor-Ibarguren A, Gómez-Fernández C, Beato-Merino MJ, et al. Diffuse reactive angioendotheliomatosis secondary to the administration of trabectedin and pegfilgrastim. Am J Dermatopathol. 2015;37:581-584.
- Lora V, Cota C, Cerroni L. Diffuse dermal angiomatosis of the abdomen. Eur J Dermatol. 2015;25:350-352.
- Pichardo RO, Lu D, Sangueza OP, et al. What is your diagnosis? diffuse dermal angiomatosis secondary to anticardiolipin antibodies. Am J Dermatopathol. 2002;24:502.
- Kutzner H, Requena L, Mentzel T, et al. Diffuse dermal angiomatosis. Hautarzt. 2002;53:808-812.
- McLaughlin ER, Morris R, Weiss SW, et al. Diffuse dermal angiomatosis of the breast: response to isotretinoin. J Am Acad Dermatol. 2001;45:462-465.
- Prinz Vavricka BM, Barry C, Victor T, et al. Diffuse dermal angiomatosis associated with calciphylaxis. Am J Dermatopathol. 2009;31:653-657.
- Müller CS, Wagner A, Pföhler C, et al. Cup-shaped painful ulcer of abdominal wall. Hautarzt. 2008;59:656-658.
- Draper BK, Boyd AS. Diffuse dermal angiomatosis. J Cutan Pathol. 2006;33:646-648.
- Adams BJ, Goldberg S, Massey HD, et al. A cause of unbearably painful breast, diffuse dermal angiomatosis. Gland Surg. 2012;1. doi:10.3978/j.issn.2227-684X.2012.07.02.
- Quatresooz P, Fumal I, Willemaers V, et al. Diffuse dermal angiomatosis: a previously undescribed pattern of immunoglobulin and complement deposits in two cases. Am J Dermatopathol. 2006;28:150-154.
- Morimoto K, Ioka H, Asada H, et al. Diffuse dermal angiomatosis. Eur J Vasc Endovasc Surg. 2011;42:381-383.
Diffuse dermal angiomatosis (DDA) is a rare acquired, cutaneous, reactive, vascular disorder that was originally thought to be a variant of cutaneous reactive angiomatosis (CREA) but is now considered to be on the spectrum of CREA. This article will focus on DDA and review the literature of prior case reports with brief descriptions of the differential diagnosis.
Case Report
A 43-year-old Haitian man presented to the clinic with a lesion on the left buttock that had developed over the last 6 years. The patient stated the lesion had been enlarging over the last several months. Upon examination, there was a large (15-cm diameter), indurated, hyperpigmented plaque covering the left buttock (Figure 1). The patient reported no medical or contributory family history. Upon review of systems, he described a burning sensation sometimes in the area of the lesion that would develop randomly throughout the year.
Three biopsies were performed, which revealed a collection of slightly dilated blood vessels with normal-appearing endothelial cells occupying the mid dermis and deep dermis (Figure 2). Immunohistochemical stains with antibodies were directed against human herpesvirus 8 (HHV-8), CD31, CD34, the cell surface glycoprotein podoplanin, Ki-67, and smooth muscle actin antigens, with appropriate controls. The vessel walls were positive for CD31, CD34, and smooth muscle actin, and negative for HHV-8 and podoplanin; Ki-67 was not increased. These histologic findings were consistent with a diagnosis of DDA. A detailed history was taken. The cause of DDA in our patient was uncertain.
Comment
Classification and Epidemiology
Diffuse dermal angiomatosis is a rare acquired, cutaneous, reactive, vascular disorder first described by Krell et al1 in 1994. Diffuse dermal angiomatosis is benign and is classified in the group of cutaneous reactive angiomatoses,2 which are benign vascular disorders marked by intravascular and extravascular hyperplasia of endothelial cells that may or may not include pericytes.2 Diffuse dermal angiomatosis was originally described as a variant of CREA, which is characterized by hyperplasia of endothelial dermal cells and intravascular proliferation.3 However, DDA has more recently been identified as a distinct disorder on the spectrum of CREA rather than as a variant of CREA.2 Given the recent reclassification, not all physicians make this distinction. However, as more case reports of DDA are published, physicians continue to support this change.4 Nevertheless, DDA has been an established disorder since 1994.1
Vascular proliferation in DDA is hypothesized to stem from ischemia or inflammation.5 Peripheral vascular atherosclerosis has been associated with DDA.6 The epidemiology of DDA is not well known because of the rarity of the disease. We performed a more specific review of the literature by limiting the PubMed search of articles indexed for MEDLINE to the term diffuse dermal angiomatosis rather than a broader search including all reactive angioendotheliomatoses. Only 31 case reports have been published1,3-32; of them, only adults were affected. Most reported cases were in middle-aged females. A summary of the demographics of DDA is provided in the Table.1,3-32
Pathophysiology
The pathophysiology of DDA remains unclear. It has been hypothesized that ischemia or inflammation creates local hypoxia, leading to an increase in vascular endothelial growth factor with subsequent endothelial proliferation and neovascularization.5 Rongioletti and Robora2 supported this hypothesis, proposing that occlusion or inflammation of the vasculature creates microthrombi and thus hypoxia. Afterward, histiocytes are recruited to reabsorb the microthrombi while hyperplasia of endothelial cells and pericytes ensues.7 Complete resolution of skin lesions following revascularization provides support for this theory.8
Etiology
Diffuse dermal angiomatosis is a rare complication of ischemia that may be secondary to atherosclerosis, arteriovenous fistula, or macromastia.9-11 In DDA of the breasts, ulcerations of fatty tissue occur due to trauma in these patients who have large pendulous breasts, causing angiogenesis resembling DDA histologically.2 One case of DDA was reported secondary to relative ischemia from cutis marmorata telangiectatica congenita,12 whereas another case highlighted Wegener granulomatosis as the cause of ischemia.7 There also have been reported cases associated with calciphylaxis and anticardiolipin antibiodies.13 In general, any medical condition that can lead to ischemia can cause DDA. Comorbid conditions for DDA include cardiovascular disease, hypertension, diabetes mellitus, and most often severe peripheral vascular disease. Many patients also have a history of smoking.14 Diffuse dermal angiomatosis rarely presents without underlying comorbidity, with only 1 case report of unknown cause (Table).
Presentation, Histopathology, and Differential Diagnosis
Cutaneous reactive angiomatosis disorders present the same clinically, with multiple erythematous to violaceous purpuric patches and plaques that can progress to necrosis and ulceration. Lesions are widely distributed but are predisposed to the upper and lower extremities.2 The differential diagnosis of DDA includes CREA, acroangiodermatitis (pseudo–Kaposi sarcoma), or vascular malignancies such as Kaposi sarcoma and low-grade angiosarcoma.7
In DDA, lesions may be painful and sometimes have a central ulceration.15 They often are associated with notable peripheral vascular atherosclerotic disease and are mainly found on the lower extremities.12,16 Histologically, DDA presents as a diffuse proliferation of endothelial cells between collagen bundles. The endothelial cells are distributed throughout the papillary and reticular dermis and develop into vascular lumina.17 Furthermore, the proliferating endothelial cells are spindle shaped and contain vacuolated cytoplasm.14
Acroangiodermatitis, or pseudo–Kaposi sarcoma, presents as slow-growing, erythematous to violaceous, brown, or dusky macules, papules, or plaques of the legs.14 Histologically, acroangiodermatitis presents with relatively less proliferation of endothelial cells found intravascularly rather than extravascularly, as in DDA, forming new thick-walled vessels in a lobular pattern in the papillary dermis.14
Vascular malignancies, such as Kaposi sarcoma and angiosarcoma, may present similarly to DDA. Kaposi sarcoma, for example, presents as erythematous to violaceous patches, plaques, or nodules found mostly on the extremities.7 Histologically, spindle cells and vascular structures also are found but in a clefting pattern representative of Kaposi sarcoma (so-called vascular slits).7 Diffuse dermal angiomatosis and vascular malignancies can further be distinguished based on atypia of the proliferations and staining for HHV-8.7,14 Lastly, DDA differs from vascular tumors in that vascular tumors are reactive to locations of occluded vessels, with vascular proliferation ceasing once the underlying cause of hypoxia is removed.2
Treatment
There is no standard treatment of DDA.7 Treatment of the underlying cause of ischemia is the primary goal, which will cause the DDA to resolve in most cases. Stenting, removal of an arteriovenous fistula, or other forms of revascularization may be warranted.1,5,6,10,17,29,30
Reported medical therapies for DDA include systemic or topical corticosteroids used for their antiangiogenic properties with varying results.7 Isotretinoin also has been used, which has been found to be effective in several cases of DDA of the breast, though 1 study reported a subsequent elevated lipid profile, requiring a decrease in dosage.14,15,27,31
Most interestingly, a study by Sanz-Motilva et al16 demonstrated that control of comorbidities, especially smoking cessation, led to improvement, which highlights the importance of incorporating nonpharmacotherapy rather than initiating treatment solely with medication. The Table summarizes treatments used and their efficacy.
Conclusion
Diffuse dermal angiomatosis is associated with medical conditions that predispose an individual to ischemia. Although rare, DDA can present as painful and visibly disturbing lesions that can affect the daily life of afflicted patients. By reporting the few cases that do arise and reviewing prior cases and their treatments, physicians can consider DDA within the differential diagnosis and identify which treatment is most efficient for a given patient. For all DDA patients, strict control of comorbidities, especially smoking cessation, should be incorporated into the treatment plan. When DDA affects the breasts, isotretinoin appears to provide the best relief. Otherwise, treatment of the underlying cause, revascularization, withdrawal of the offending agent, or steroids seem to be the best treatment options.
Diffuse dermal angiomatosis (DDA) is a rare acquired, cutaneous, reactive, vascular disorder that was originally thought to be a variant of cutaneous reactive angiomatosis (CREA) but is now considered to be on the spectrum of CREA. This article will focus on DDA and review the literature of prior case reports with brief descriptions of the differential diagnosis.
Case Report
A 43-year-old Haitian man presented to the clinic with a lesion on the left buttock that had developed over the last 6 years. The patient stated the lesion had been enlarging over the last several months. Upon examination, there was a large (15-cm diameter), indurated, hyperpigmented plaque covering the left buttock (Figure 1). The patient reported no medical or contributory family history. Upon review of systems, he described a burning sensation sometimes in the area of the lesion that would develop randomly throughout the year.
Three biopsies were performed, which revealed a collection of slightly dilated blood vessels with normal-appearing endothelial cells occupying the mid dermis and deep dermis (Figure 2). Immunohistochemical stains with antibodies were directed against human herpesvirus 8 (HHV-8), CD31, CD34, the cell surface glycoprotein podoplanin, Ki-67, and smooth muscle actin antigens, with appropriate controls. The vessel walls were positive for CD31, CD34, and smooth muscle actin, and negative for HHV-8 and podoplanin; Ki-67 was not increased. These histologic findings were consistent with a diagnosis of DDA. A detailed history was taken. The cause of DDA in our patient was uncertain.
Comment
Classification and Epidemiology
Diffuse dermal angiomatosis is a rare acquired, cutaneous, reactive, vascular disorder first described by Krell et al1 in 1994. Diffuse dermal angiomatosis is benign and is classified in the group of cutaneous reactive angiomatoses,2 which are benign vascular disorders marked by intravascular and extravascular hyperplasia of endothelial cells that may or may not include pericytes.2 Diffuse dermal angiomatosis was originally described as a variant of CREA, which is characterized by hyperplasia of endothelial dermal cells and intravascular proliferation.3 However, DDA has more recently been identified as a distinct disorder on the spectrum of CREA rather than as a variant of CREA.2 Given the recent reclassification, not all physicians make this distinction. However, as more case reports of DDA are published, physicians continue to support this change.4 Nevertheless, DDA has been an established disorder since 1994.1
Vascular proliferation in DDA is hypothesized to stem from ischemia or inflammation.5 Peripheral vascular atherosclerosis has been associated with DDA.6 The epidemiology of DDA is not well known because of the rarity of the disease. We performed a more specific review of the literature by limiting the PubMed search of articles indexed for MEDLINE to the term diffuse dermal angiomatosis rather than a broader search including all reactive angioendotheliomatoses. Only 31 case reports have been published1,3-32; of them, only adults were affected. Most reported cases were in middle-aged females. A summary of the demographics of DDA is provided in the Table.1,3-32
Pathophysiology
The pathophysiology of DDA remains unclear. It has been hypothesized that ischemia or inflammation creates local hypoxia, leading to an increase in vascular endothelial growth factor with subsequent endothelial proliferation and neovascularization.5 Rongioletti and Robora2 supported this hypothesis, proposing that occlusion or inflammation of the vasculature creates microthrombi and thus hypoxia. Afterward, histiocytes are recruited to reabsorb the microthrombi while hyperplasia of endothelial cells and pericytes ensues.7 Complete resolution of skin lesions following revascularization provides support for this theory.8
Etiology
Diffuse dermal angiomatosis is a rare complication of ischemia that may be secondary to atherosclerosis, arteriovenous fistula, or macromastia.9-11 In DDA of the breasts, ulcerations of fatty tissue occur due to trauma in these patients who have large pendulous breasts, causing angiogenesis resembling DDA histologically.2 One case of DDA was reported secondary to relative ischemia from cutis marmorata telangiectatica congenita,12 whereas another case highlighted Wegener granulomatosis as the cause of ischemia.7 There also have been reported cases associated with calciphylaxis and anticardiolipin antibiodies.13 In general, any medical condition that can lead to ischemia can cause DDA. Comorbid conditions for DDA include cardiovascular disease, hypertension, diabetes mellitus, and most often severe peripheral vascular disease. Many patients also have a history of smoking.14 Diffuse dermal angiomatosis rarely presents without underlying comorbidity, with only 1 case report of unknown cause (Table).
Presentation, Histopathology, and Differential Diagnosis
Cutaneous reactive angiomatosis disorders present the same clinically, with multiple erythematous to violaceous purpuric patches and plaques that can progress to necrosis and ulceration. Lesions are widely distributed but are predisposed to the upper and lower extremities.2 The differential diagnosis of DDA includes CREA, acroangiodermatitis (pseudo–Kaposi sarcoma), or vascular malignancies such as Kaposi sarcoma and low-grade angiosarcoma.7
In DDA, lesions may be painful and sometimes have a central ulceration.15 They often are associated with notable peripheral vascular atherosclerotic disease and are mainly found on the lower extremities.12,16 Histologically, DDA presents as a diffuse proliferation of endothelial cells between collagen bundles. The endothelial cells are distributed throughout the papillary and reticular dermis and develop into vascular lumina.17 Furthermore, the proliferating endothelial cells are spindle shaped and contain vacuolated cytoplasm.14
Acroangiodermatitis, or pseudo–Kaposi sarcoma, presents as slow-growing, erythematous to violaceous, brown, or dusky macules, papules, or plaques of the legs.14 Histologically, acroangiodermatitis presents with relatively less proliferation of endothelial cells found intravascularly rather than extravascularly, as in DDA, forming new thick-walled vessels in a lobular pattern in the papillary dermis.14
Vascular malignancies, such as Kaposi sarcoma and angiosarcoma, may present similarly to DDA. Kaposi sarcoma, for example, presents as erythematous to violaceous patches, plaques, or nodules found mostly on the extremities.7 Histologically, spindle cells and vascular structures also are found but in a clefting pattern representative of Kaposi sarcoma (so-called vascular slits).7 Diffuse dermal angiomatosis and vascular malignancies can further be distinguished based on atypia of the proliferations and staining for HHV-8.7,14 Lastly, DDA differs from vascular tumors in that vascular tumors are reactive to locations of occluded vessels, with vascular proliferation ceasing once the underlying cause of hypoxia is removed.2
Treatment
There is no standard treatment of DDA.7 Treatment of the underlying cause of ischemia is the primary goal, which will cause the DDA to resolve in most cases. Stenting, removal of an arteriovenous fistula, or other forms of revascularization may be warranted.1,5,6,10,17,29,30
Reported medical therapies for DDA include systemic or topical corticosteroids used for their antiangiogenic properties with varying results.7 Isotretinoin also has been used, which has been found to be effective in several cases of DDA of the breast, though 1 study reported a subsequent elevated lipid profile, requiring a decrease in dosage.14,15,27,31
Most interestingly, a study by Sanz-Motilva et al16 demonstrated that control of comorbidities, especially smoking cessation, led to improvement, which highlights the importance of incorporating nonpharmacotherapy rather than initiating treatment solely with medication. The Table summarizes treatments used and their efficacy.
Conclusion
Diffuse dermal angiomatosis is associated with medical conditions that predispose an individual to ischemia. Although rare, DDA can present as painful and visibly disturbing lesions that can affect the daily life of afflicted patients. By reporting the few cases that do arise and reviewing prior cases and their treatments, physicians can consider DDA within the differential diagnosis and identify which treatment is most efficient for a given patient. For all DDA patients, strict control of comorbidities, especially smoking cessation, should be incorporated into the treatment plan. When DDA affects the breasts, isotretinoin appears to provide the best relief. Otherwise, treatment of the underlying cause, revascularization, withdrawal of the offending agent, or steroids seem to be the best treatment options.
- Krell JM, Sanchez RL, Solomon AR. Diffuse dermal angiomatosis: a variant of reactive cutaneous angioendotheliomatosis. J Cutan Pathol. 1994;21:363-370.
- Rongioletti F, Robora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Crickx E, Saussine A, Vignon-Pennamen MD, et al. Diffuse dermal angiomatosis associated with severe atherosclerosis: two cases and review of the literature. Clin Exp Dermatol. 2015;40:521-524.
- Reusche R, Winocour S, Degnim A, et al. Diffuse dermal angiomatosis of the breast: a series of 22 cases from a single institution. Gland Surg. 2015;4:554-560.
- Sriphojanart T, Vachiramon V. Diffuse dermal angiomatosis: a clue to the diagnosis of atherosclerotic vascular disease. Case Rep Dermatol. 2015;7:100-106.
- Kimyai-Asadi A, Nousari HC, Ketabchi N, et al. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with atherosclerosis. J Am Acad Dermatol. 1999;40:257-259.
- Bassi A, Arunachalam M, Maio V, et al. Diffuse dermal angiomatosis in a patient with an iatrogenic arterio-venous fistula and Wegener’s granulomatosis. Acta Derm Venereol. 2013;93:93-94.
- Ormerod E, Miller K, Kennedy C. Diffuse dermal angiomatosis: a contributory factor to ulceration in a patient with renal transplant. Clin Exp Dermatol. 2015;40:48-51.
- Kim S, Elenitsas R, James WD. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with peripheral vascular atherosclerosis. Arch Dermatol. 2002;138:456-458.
- Requena L, Fariña MC, Renedo G, et al. Intravascular and diffuse dermal reactive angioendotheliomatosis secondary to iatrogenic arteriovenous fistulas. J Cutan Pathol. 1999;26:159-164.
- Villa MT, White LE, Petronic-Rosic V, et al. The treatment of diffuse dermal angiomatosis of the breast with reduction mammoplasty. Arch Dermatol. 2008;144:693-694.
- Halbesleben JJ, Cleveland MG, Stone MS. Diffuse dermal angiomatosis arising in cutis marmorata telangiectatica congenita. Arch Dermatol. 2010;146:1311-1313.
- Ferreli C, Atzori L, Pinna AL, et al. Diffuse dermal angiomatosis: a clinical mimicker of vasculitis associated with calciphylaxis and monoclonal gammopathy. G Ital Dermatol Venereol. 2015;150:115-121.
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347.
- Steele KT, Sullivan BJ, Wanat KA, et al. Diffuse dermal angiomatosis associated with calciphylaxis in a patient with end-stage renal disease.J Cutan Pathol. 2013;40:829-832.
- Sanz-Motilva V, Martorell-Calatayud A, Rongioletti F, et al. Diffuse dermal angiomatosis of the breast: clinical and histopathological features. Int J Dermatol. 2014;53:445-449.
- Kirkland CR, Hawayek LH, Mutasim DF. Atherosclerosis-induced diffuse dermal angiomatosis with fatal outcome. Arch Dermatol. 2010;146:684-685.
- Sommer S, Merchant WJ, Wilson CL. Diffuse dermal angiomatosis due to an iatrogenic arteriovenous fistula. Acta Derm Venereol. 2004;84:251-252.
- Corti MA, Rongioletti F, Borradori L, et al. Cutaneous reactive angiomatosis with combined histological pattern mimicking a cellulitis. Dermatology. 2013;227:226-230.
- Tollefson MM, McEvoy MT, Torgerson RR, et al. Diffuse dermal angiomatosis of the breast: clinicopathologic study of 5 patients. J Am Acad Dermatol. 2014;71:1212-1217.
- Walton K, Liggett J. Diffuse dermal angiomatosis: a case report. J Am Acad Dermatol. 2012;66(suppl 1):AB49.
- Mayor-Ibarguren A, Gómez-Fernández C, Beato-Merino MJ, et al. Diffuse reactive angioendotheliomatosis secondary to the administration of trabectedin and pegfilgrastim. Am J Dermatopathol. 2015;37:581-584.
- Lora V, Cota C, Cerroni L. Diffuse dermal angiomatosis of the abdomen. Eur J Dermatol. 2015;25:350-352.
- Pichardo RO, Lu D, Sangueza OP, et al. What is your diagnosis? diffuse dermal angiomatosis secondary to anticardiolipin antibodies. Am J Dermatopathol. 2002;24:502.
- Kutzner H, Requena L, Mentzel T, et al. Diffuse dermal angiomatosis. Hautarzt. 2002;53:808-812.
- McLaughlin ER, Morris R, Weiss SW, et al. Diffuse dermal angiomatosis of the breast: response to isotretinoin. J Am Acad Dermatol. 2001;45:462-465.
- Prinz Vavricka BM, Barry C, Victor T, et al. Diffuse dermal angiomatosis associated with calciphylaxis. Am J Dermatopathol. 2009;31:653-657.
- Müller CS, Wagner A, Pföhler C, et al. Cup-shaped painful ulcer of abdominal wall. Hautarzt. 2008;59:656-658.
- Draper BK, Boyd AS. Diffuse dermal angiomatosis. J Cutan Pathol. 2006;33:646-648.
- Adams BJ, Goldberg S, Massey HD, et al. A cause of unbearably painful breast, diffuse dermal angiomatosis. Gland Surg. 2012;1. doi:10.3978/j.issn.2227-684X.2012.07.02.
- Quatresooz P, Fumal I, Willemaers V, et al. Diffuse dermal angiomatosis: a previously undescribed pattern of immunoglobulin and complement deposits in two cases. Am J Dermatopathol. 2006;28:150-154.
- Morimoto K, Ioka H, Asada H, et al. Diffuse dermal angiomatosis. Eur J Vasc Endovasc Surg. 2011;42:381-383.
- Krell JM, Sanchez RL, Solomon AR. Diffuse dermal angiomatosis: a variant of reactive cutaneous angioendotheliomatosis. J Cutan Pathol. 1994;21:363-370.
- Rongioletti F, Robora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Crickx E, Saussine A, Vignon-Pennamen MD, et al. Diffuse dermal angiomatosis associated with severe atherosclerosis: two cases and review of the literature. Clin Exp Dermatol. 2015;40:521-524.
- Reusche R, Winocour S, Degnim A, et al. Diffuse dermal angiomatosis of the breast: a series of 22 cases from a single institution. Gland Surg. 2015;4:554-560.
- Sriphojanart T, Vachiramon V. Diffuse dermal angiomatosis: a clue to the diagnosis of atherosclerotic vascular disease. Case Rep Dermatol. 2015;7:100-106.
- Kimyai-Asadi A, Nousari HC, Ketabchi N, et al. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with atherosclerosis. J Am Acad Dermatol. 1999;40:257-259.
- Bassi A, Arunachalam M, Maio V, et al. Diffuse dermal angiomatosis in a patient with an iatrogenic arterio-venous fistula and Wegener’s granulomatosis. Acta Derm Venereol. 2013;93:93-94.
- Ormerod E, Miller K, Kennedy C. Diffuse dermal angiomatosis: a contributory factor to ulceration in a patient with renal transplant. Clin Exp Dermatol. 2015;40:48-51.
- Kim S, Elenitsas R, James WD. Diffuse dermal angiomatosis: a variant of reactive angioendotheliomatosis associated with peripheral vascular atherosclerosis. Arch Dermatol. 2002;138:456-458.
- Requena L, Fariña MC, Renedo G, et al. Intravascular and diffuse dermal reactive angioendotheliomatosis secondary to iatrogenic arteriovenous fistulas. J Cutan Pathol. 1999;26:159-164.
- Villa MT, White LE, Petronic-Rosic V, et al. The treatment of diffuse dermal angiomatosis of the breast with reduction mammoplasty. Arch Dermatol. 2008;144:693-694.
- Halbesleben JJ, Cleveland MG, Stone MS. Diffuse dermal angiomatosis arising in cutis marmorata telangiectatica congenita. Arch Dermatol. 2010;146:1311-1313.
- Ferreli C, Atzori L, Pinna AL, et al. Diffuse dermal angiomatosis: a clinical mimicker of vasculitis associated with calciphylaxis and monoclonal gammopathy. G Ital Dermatol Venereol. 2015;150:115-121.
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347.
- Steele KT, Sullivan BJ, Wanat KA, et al. Diffuse dermal angiomatosis associated with calciphylaxis in a patient with end-stage renal disease.J Cutan Pathol. 2013;40:829-832.
- Sanz-Motilva V, Martorell-Calatayud A, Rongioletti F, et al. Diffuse dermal angiomatosis of the breast: clinical and histopathological features. Int J Dermatol. 2014;53:445-449.
- Kirkland CR, Hawayek LH, Mutasim DF. Atherosclerosis-induced diffuse dermal angiomatosis with fatal outcome. Arch Dermatol. 2010;146:684-685.
- Sommer S, Merchant WJ, Wilson CL. Diffuse dermal angiomatosis due to an iatrogenic arteriovenous fistula. Acta Derm Venereol. 2004;84:251-252.
- Corti MA, Rongioletti F, Borradori L, et al. Cutaneous reactive angiomatosis with combined histological pattern mimicking a cellulitis. Dermatology. 2013;227:226-230.
- Tollefson MM, McEvoy MT, Torgerson RR, et al. Diffuse dermal angiomatosis of the breast: clinicopathologic study of 5 patients. J Am Acad Dermatol. 2014;71:1212-1217.
- Walton K, Liggett J. Diffuse dermal angiomatosis: a case report. J Am Acad Dermatol. 2012;66(suppl 1):AB49.
- Mayor-Ibarguren A, Gómez-Fernández C, Beato-Merino MJ, et al. Diffuse reactive angioendotheliomatosis secondary to the administration of trabectedin and pegfilgrastim. Am J Dermatopathol. 2015;37:581-584.
- Lora V, Cota C, Cerroni L. Diffuse dermal angiomatosis of the abdomen. Eur J Dermatol. 2015;25:350-352.
- Pichardo RO, Lu D, Sangueza OP, et al. What is your diagnosis? diffuse dermal angiomatosis secondary to anticardiolipin antibodies. Am J Dermatopathol. 2002;24:502.
- Kutzner H, Requena L, Mentzel T, et al. Diffuse dermal angiomatosis. Hautarzt. 2002;53:808-812.
- McLaughlin ER, Morris R, Weiss SW, et al. Diffuse dermal angiomatosis of the breast: response to isotretinoin. J Am Acad Dermatol. 2001;45:462-465.
- Prinz Vavricka BM, Barry C, Victor T, et al. Diffuse dermal angiomatosis associated with calciphylaxis. Am J Dermatopathol. 2009;31:653-657.
- Müller CS, Wagner A, Pföhler C, et al. Cup-shaped painful ulcer of abdominal wall. Hautarzt. 2008;59:656-658.
- Draper BK, Boyd AS. Diffuse dermal angiomatosis. J Cutan Pathol. 2006;33:646-648.
- Adams BJ, Goldberg S, Massey HD, et al. A cause of unbearably painful breast, diffuse dermal angiomatosis. Gland Surg. 2012;1. doi:10.3978/j.issn.2227-684X.2012.07.02.
- Quatresooz P, Fumal I, Willemaers V, et al. Diffuse dermal angiomatosis: a previously undescribed pattern of immunoglobulin and complement deposits in two cases. Am J Dermatopathol. 2006;28:150-154.
- Morimoto K, Ioka H, Asada H, et al. Diffuse dermal angiomatosis. Eur J Vasc Endovasc Surg. 2011;42:381-383.
Practice Points
- Diffuse dermal angiomatosis is commonly reported in patients with hypoxic comorbidities such as smoking or vascular disease as well as in women with large pendulous breasts.
- Effective treatments include control of comorbidities, revascularization, withdrawal of the offending agent, steroids, and isotretinoin.
Depigmentation therapy may be appropriate for patients with vitiligo
WASHINGTON – Seemal Desai, MD, said at the annual meeting of the American Academy of Dermatology.

Depigmentation therapy is “an underutilized resource for those patients who have recalcitrant disease or whose disease is so bad that you have not been able to improve their clinical, visible outcome,” said Dr. Desai, clinical assistant professor of dermatology at the University of Texas, Dallas. “Some of these patients simply desire to be one uniform color.”
Therapy is administered through medications that destroy residual melanocytes with the goal of achieving a uniform appearance of the skin. Monobenzyl ether of hydroquinone (MBEH) is the most common drug used in depigmentation therapy, according to Dr. Desai. It is available in concentrations of 20% and 40% and can be compounded in other concentrations. At first, patients should apply MBEH to a nickel-sized area of the skin (such as the forearm, the back of the hand, or the thigh) for 3 or 4 days. Treatment is administered in the morning and evening, but not at bedtime. A common side effect is irritant contact dermatitis.
When the patient is able to tolerate the medication in the first area, he or she can apply it to larger areas of the skin. Parts of the body are treated one at a time, and successful treatment takes time, he noted. Hair may become depigmented, but patients can be assured that the eyes will not.
Repigmentation has been reported after sun exposure. For all patients undergoing depigmentation therapy, dermatologists should provide extensive counseling about the need for lifelong photoprotection, said Dr. Desai. Protective measures can include wide-brimmed hats and broad-spectrum sunscreen. Paradoxical repigmentation can be treated with a stronger concentration of MBEH, liquid nitrogen therapy, microdermabrasion, and peels.
For patients with vitiligo, which results from the immune-mediated destruction of melanocytes, dermatologists should assess the patient’s psychological status and discuss “the impact that the disease has on not only the patient, but also the family,” Dr. Desai said. “The psychological trauma from this disease, especially for our patients who have rapidly progressing vitiligo or who have failed multiple therapies, is something that we cannot discount.”
Patients with vitiligo often have comorbid depression, and evening of the skin tone that depigmentation provides can benefit the patient’s mental state, he observed. Patients with severe depression related to vitiligo should be referred to a psychologist or psychiatrist. Finally, counseling and appropriate patient selection for depigmentation is of paramount importance.
During the presentation, he noted that hair dyes, resin products and adhesives, detergents, and leather preservatives have been associated with vitiligo. Dermatologic drugs such as imiquimod, chemotherapeutic agents, and interferon also may cause the condition.
He referred to the thousands of reported cases of vitiligo related to rhododenol, a phenolic compound that has been used in cosmetics and topical products. Most cases resolved when those affected stopped using the product, but some developed vitiligo vulgaris. Cosmetics containing rhododenol have been recalled in Japan since 2013, but over-the-counter products in Asia and Africa have been found to contain similar compounds, so dermatologists should ask patients about their travel history and about what products they are using for their skin, Dr. Desai advised.
He reported receiving grants and research funding from AbbVie, Dermira, Dr. Reddy’s Laboratories, and Menlo Therapeutics and serving as a consultant for several pharmaceutical companies.
WASHINGTON – Seemal Desai, MD, said at the annual meeting of the American Academy of Dermatology.
Depigmentation therapy is “an underutilized resource for those patients who have recalcitrant disease or whose disease is so bad that you have not been able to improve their clinical, visible outcome,” said Dr. Desai, clinical assistant professor of dermatology at the University of Texas, Dallas. “Some of these patients simply desire to be one uniform color.”
Therapy is administered through medications that destroy residual melanocytes with the goal of achieving a uniform appearance of the skin. Monobenzyl ether of hydroquinone (MBEH) is the most common drug used in depigmentation therapy, according to Dr. Desai. It is available in concentrations of 20% and 40% and can be compounded in other concentrations. At first, patients should apply MBEH to a nickel-sized area of the skin (such as the forearm, the back of the hand, or the thigh) for 3 or 4 days. Treatment is administered in the morning and evening, but not at bedtime. A common side effect is irritant contact dermatitis.
When the patient is able to tolerate the medication in the first area, he or she can apply it to larger areas of the skin. Parts of the body are treated one at a time, and successful treatment takes time, he noted. Hair may become depigmented, but patients can be assured that the eyes will not.
Repigmentation has been reported after sun exposure. For all patients undergoing depigmentation therapy, dermatologists should provide extensive counseling about the need for lifelong photoprotection, said Dr. Desai. Protective measures can include wide-brimmed hats and broad-spectrum sunscreen. Paradoxical repigmentation can be treated with a stronger concentration of MBEH, liquid nitrogen therapy, microdermabrasion, and peels.
For patients with vitiligo, which results from the immune-mediated destruction of melanocytes, dermatologists should assess the patient’s psychological status and discuss “the impact that the disease has on not only the patient, but also the family,” Dr. Desai said. “The psychological trauma from this disease, especially for our patients who have rapidly progressing vitiligo or who have failed multiple therapies, is something that we cannot discount.”
Patients with vitiligo often have comorbid depression, and evening of the skin tone that depigmentation provides can benefit the patient’s mental state, he observed. Patients with severe depression related to vitiligo should be referred to a psychologist or psychiatrist. Finally, counseling and appropriate patient selection for depigmentation is of paramount importance.
During the presentation, he noted that hair dyes, resin products and adhesives, detergents, and leather preservatives have been associated with vitiligo. Dermatologic drugs such as imiquimod, chemotherapeutic agents, and interferon also may cause the condition.
He referred to the thousands of reported cases of vitiligo related to rhododenol, a phenolic compound that has been used in cosmetics and topical products. Most cases resolved when those affected stopped using the product, but some developed vitiligo vulgaris. Cosmetics containing rhododenol have been recalled in Japan since 2013, but over-the-counter products in Asia and Africa have been found to contain similar compounds, so dermatologists should ask patients about their travel history and about what products they are using for their skin, Dr. Desai advised.
He reported receiving grants and research funding from AbbVie, Dermira, Dr. Reddy’s Laboratories, and Menlo Therapeutics and serving as a consultant for several pharmaceutical companies.
WASHINGTON – Seemal Desai, MD, said at the annual meeting of the American Academy of Dermatology.
Depigmentation therapy is “an underutilized resource for those patients who have recalcitrant disease or whose disease is so bad that you have not been able to improve their clinical, visible outcome,” said Dr. Desai, clinical assistant professor of dermatology at the University of Texas, Dallas. “Some of these patients simply desire to be one uniform color.”
Therapy is administered through medications that destroy residual melanocytes with the goal of achieving a uniform appearance of the skin. Monobenzyl ether of hydroquinone (MBEH) is the most common drug used in depigmentation therapy, according to Dr. Desai. It is available in concentrations of 20% and 40% and can be compounded in other concentrations. At first, patients should apply MBEH to a nickel-sized area of the skin (such as the forearm, the back of the hand, or the thigh) for 3 or 4 days. Treatment is administered in the morning and evening, but not at bedtime. A common side effect is irritant contact dermatitis.
When the patient is able to tolerate the medication in the first area, he or she can apply it to larger areas of the skin. Parts of the body are treated one at a time, and successful treatment takes time, he noted. Hair may become depigmented, but patients can be assured that the eyes will not.
Repigmentation has been reported after sun exposure. For all patients undergoing depigmentation therapy, dermatologists should provide extensive counseling about the need for lifelong photoprotection, said Dr. Desai. Protective measures can include wide-brimmed hats and broad-spectrum sunscreen. Paradoxical repigmentation can be treated with a stronger concentration of MBEH, liquid nitrogen therapy, microdermabrasion, and peels.
For patients with vitiligo, which results from the immune-mediated destruction of melanocytes, dermatologists should assess the patient’s psychological status and discuss “the impact that the disease has on not only the patient, but also the family,” Dr. Desai said. “The psychological trauma from this disease, especially for our patients who have rapidly progressing vitiligo or who have failed multiple therapies, is something that we cannot discount.”
Patients with vitiligo often have comorbid depression, and evening of the skin tone that depigmentation provides can benefit the patient’s mental state, he observed. Patients with severe depression related to vitiligo should be referred to a psychologist or psychiatrist. Finally, counseling and appropriate patient selection for depigmentation is of paramount importance.
During the presentation, he noted that hair dyes, resin products and adhesives, detergents, and leather preservatives have been associated with vitiligo. Dermatologic drugs such as imiquimod, chemotherapeutic agents, and interferon also may cause the condition.
He referred to the thousands of reported cases of vitiligo related to rhododenol, a phenolic compound that has been used in cosmetics and topical products. Most cases resolved when those affected stopped using the product, but some developed vitiligo vulgaris. Cosmetics containing rhododenol have been recalled in Japan since 2013, but over-the-counter products in Asia and Africa have been found to contain similar compounds, so dermatologists should ask patients about their travel history and about what products they are using for their skin, Dr. Desai advised.
He reported receiving grants and research funding from AbbVie, Dermira, Dr. Reddy’s Laboratories, and Menlo Therapeutics and serving as a consultant for several pharmaceutical companies.
EXPERT ANALYSIS FROM AAD 2019
Trametinib effectively treats case of giant congenital melanocytic nevus
according to a case report presented in Pediatrics.
Her nevus covered most of her back and much of her torso and had thickened significantly over the years since initial presentation to the point of disfigurement, even invading the fascia and musculature of the trunk and pelvis, reported Adnan Mir, MD, PhD, of the University of Texas Southwestern Medical Center, Dallas, and his coauthors. Furthermore, she presented with intractable pruritus and pain that interfered with sleep and responded minimally to treatments. Although initial immunohistochemical staining and gene sequencing did not reveal any mutations, such as BRAF V600E, further testing uncovered an AKAP9-BRAF fusion.
There are few if any effective ways of treating GCMNs. With that knowledge, as well as general theories of the mechanism GCMNs in mind, the patient’s health care team decided to try a 0.5-mg daily dose of trametinib when she was 7 years old. Her pruritus and pain resolved completely, and after 6 months of treatment with trametinib, repeat MRI “revealed decreased thickening of the dermis and near resolutions of muscular invasion.” According to the patient’s family, her quality of life improved dramatically.
SOURCE: Mir A et al. Pediatrics. 2019;143(3):e20182469.
according to a case report presented in Pediatrics.
Her nevus covered most of her back and much of her torso and had thickened significantly over the years since initial presentation to the point of disfigurement, even invading the fascia and musculature of the trunk and pelvis, reported Adnan Mir, MD, PhD, of the University of Texas Southwestern Medical Center, Dallas, and his coauthors. Furthermore, she presented with intractable pruritus and pain that interfered with sleep and responded minimally to treatments. Although initial immunohistochemical staining and gene sequencing did not reveal any mutations, such as BRAF V600E, further testing uncovered an AKAP9-BRAF fusion.
There are few if any effective ways of treating GCMNs. With that knowledge, as well as general theories of the mechanism GCMNs in mind, the patient’s health care team decided to try a 0.5-mg daily dose of trametinib when she was 7 years old. Her pruritus and pain resolved completely, and after 6 months of treatment with trametinib, repeat MRI “revealed decreased thickening of the dermis and near resolutions of muscular invasion.” According to the patient’s family, her quality of life improved dramatically.
SOURCE: Mir A et al. Pediatrics. 2019;143(3):e20182469.
according to a case report presented in Pediatrics.
Her nevus covered most of her back and much of her torso and had thickened significantly over the years since initial presentation to the point of disfigurement, even invading the fascia and musculature of the trunk and pelvis, reported Adnan Mir, MD, PhD, of the University of Texas Southwestern Medical Center, Dallas, and his coauthors. Furthermore, she presented with intractable pruritus and pain that interfered with sleep and responded minimally to treatments. Although initial immunohistochemical staining and gene sequencing did not reveal any mutations, such as BRAF V600E, further testing uncovered an AKAP9-BRAF fusion.
There are few if any effective ways of treating GCMNs. With that knowledge, as well as general theories of the mechanism GCMNs in mind, the patient’s health care team decided to try a 0.5-mg daily dose of trametinib when she was 7 years old. Her pruritus and pain resolved completely, and after 6 months of treatment with trametinib, repeat MRI “revealed decreased thickening of the dermis and near resolutions of muscular invasion.” According to the patient’s family, her quality of life improved dramatically.
SOURCE: Mir A et al. Pediatrics. 2019;143(3):e20182469.
FROM PEDIATRICS
Irregularly Hyperpigmented Plaque on the Right Heel
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
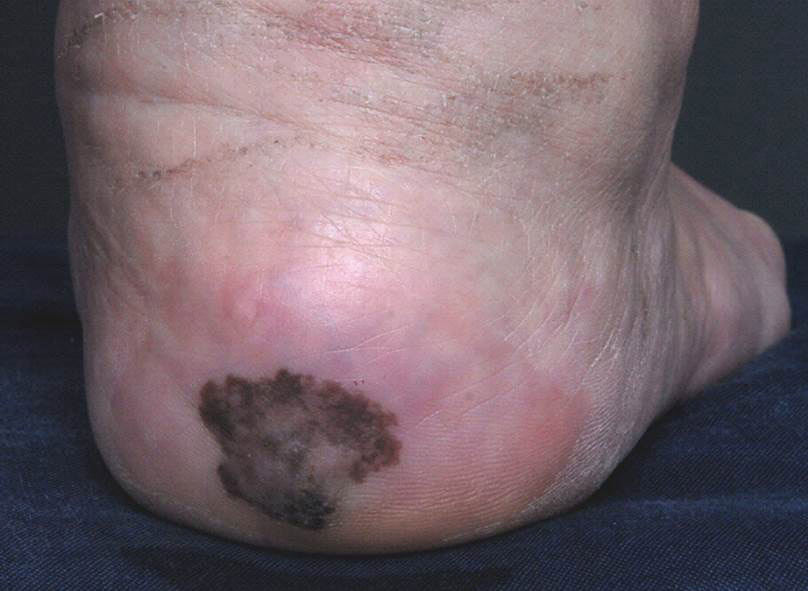
A 56-year-old woman presented with an asymptomatic plaque on the right heel that had grown
steadily over the last year. Pigmented lesions were not appreciated on other sites, and lymph nodes were not enlarged. Her medical history was otherwise normal, except for bilateral hearing loss due to encephalitis at the age of 5 years. None of her family members had similar symptoms. Physical examination revealed a well-defined, irregularly hyperpigmented plaque on the right heel.
Neurofibromatosis Type 1 in the Setting of Systemic Lupus Erythematosus
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
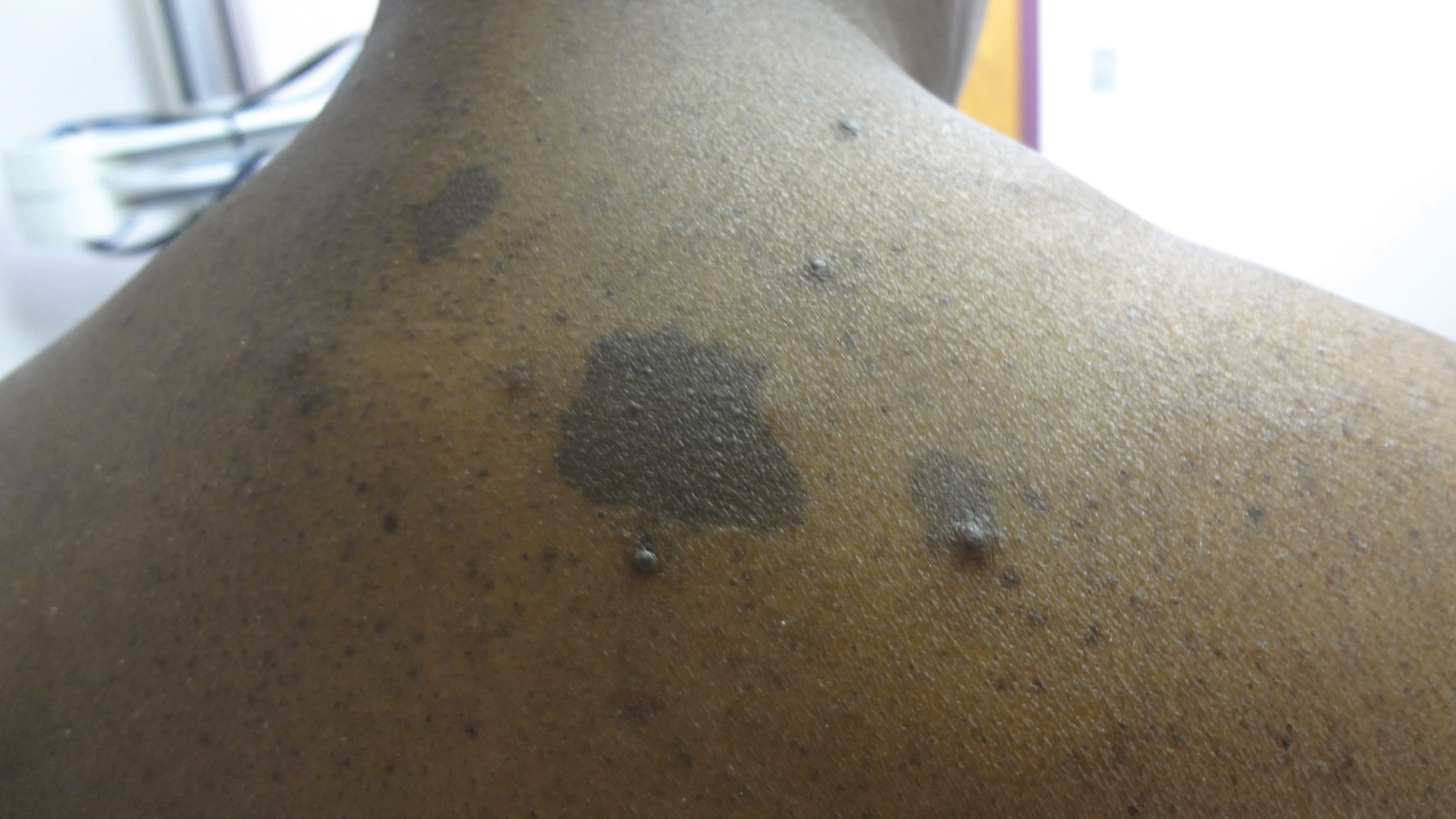
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
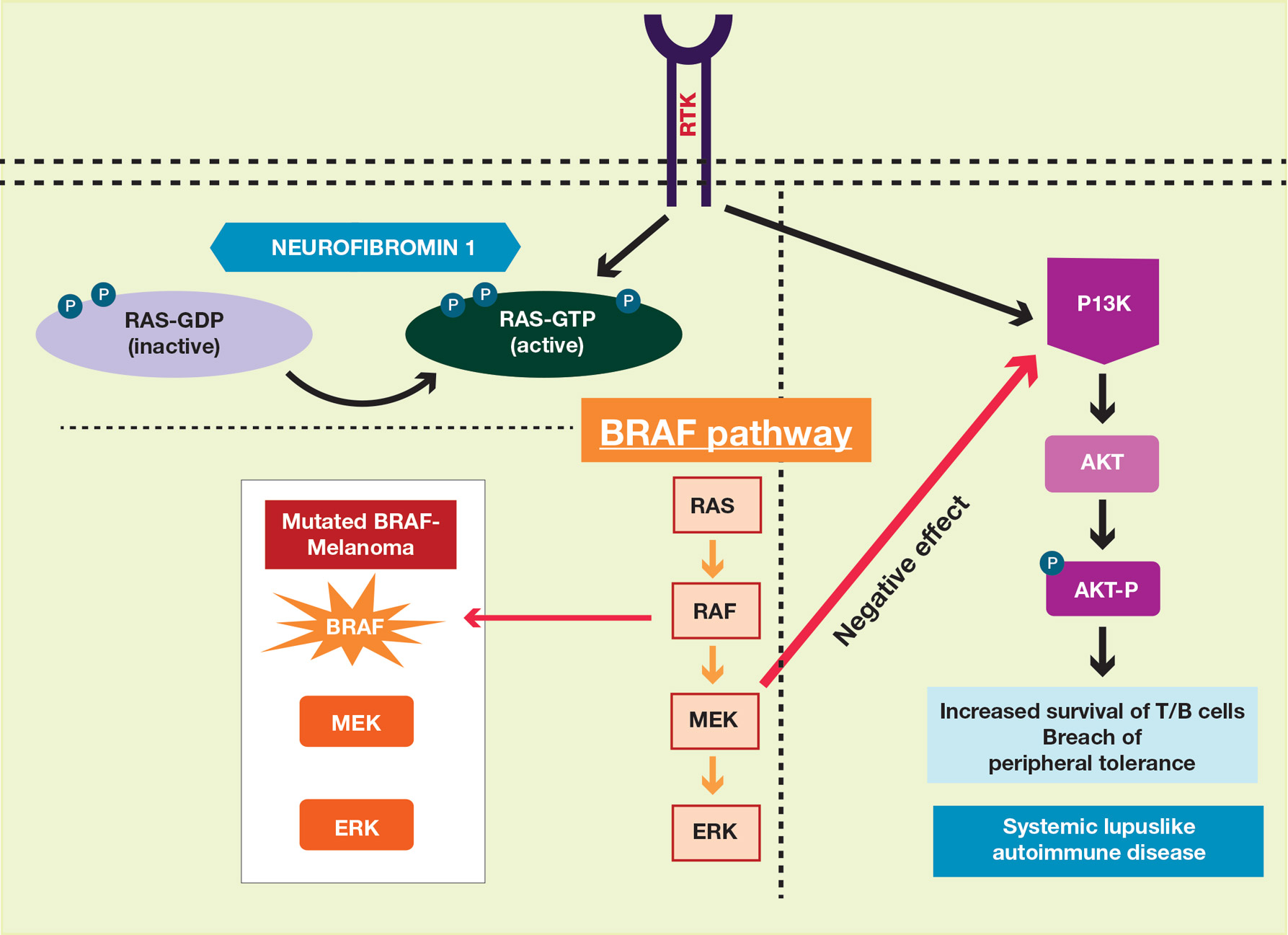
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.

We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.
We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.
We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
Practice Points
- Patients with neurofibromatosis type 1 (NF-1) benefit from early diagnosis and long-term follow-up.
- Patients with systemic lupus erythematosus (SLE) may develop different malignancies given the immune dysregulation. We recommend that patients with SLE undergo detailed skin examinations to check for subtle clues for NF-1.
- Similarly, patients with NF-1 can develop SLE later in life.
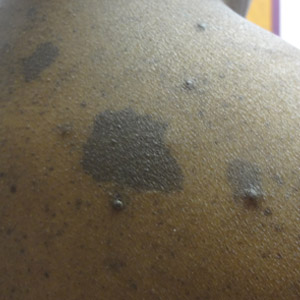
Managing Postinflammatory Hyperpigmentation in Pediatric Patients With Skin of Color
Postnflammatory hyperpigmentation (PIH) is an acquired hypermelanosis that can occur in children and adults following an inflammatory cutaneous disease or trauma. Postinflammatory hyperpigmentation may last for months to even years. Although PIH may occur in all skin types, it is more common and presents with greater severity and intensity in individuals with skin of color. By the year 2050, 1 in 3 US residents is projected to be Hispanic.1 It is projected that by 2044, non-Hispanic white individuals (all ages) will make up less than 50% of the US population.2 Currently, the majority of the US residents younger than 18 years are minorities. The majority minority population in the United States already exists in those younger than 18 years and is predicted to occur in the adult population by 2044.2
Effective treatment options and management strategies for PIH in adults with skin of color have been described in the literature.3 Due to a paucity of research, the approach to management of PIH in children with skin of color has been based on clinical experience and lessons learned from adult patients. This article focuses on management of PIH in pediatric patients with skin of color, which includes black/African American, African-Caribbean, Hispanic, Asian, Pacific Islander, and American Indian individuals.
Underlying Inflammatory Dermatoses Resulting in PIH
There are numerous conditions that may result in PIH, including but not limited to atopic dermatitis (AD), acne, arthropod bites, and injuries to the skin. Postinflammatory hyperpigmentation may have more of a psychological impact than the inciting disease or injury itself. The most important step in the approach to managing PIH is treating the underlying inflammatory condition that caused the pigmentation.
Parents/guardians may report a chief concern of dark spots, manchas (stains), blemishes, or stains on the skin, often with no mention of a coexisting inflammatory dermatosis. Parents/guardians of children with skin of color often have personally experienced PIH and may be determined to shield their children from similar angst associated with the condition. Although physicians may see just another pediatric patient with PIH, the child’s parents/guardians may see a condition that will be readily perceptible during major life events, such as the child’s prom or even his/her wedding day. Promptly diagnosing and instituting early treatment of inflammatory conditions associated with PIH may accelerate resolution and prevent worsening of the pigmentation.3
Select inflammatory dermatoses that are common in children with skin of color and may lead to PIH are highlighted below. Although this list is not comprehensive, the approach and management strategies should prompt creation of plans that keep PIH in mind when treating primary inflammatory skin diseases.
Atopic Dermatitis
Atopic dermatitis may induce PIH or hypopigmentation of the skin in children with skin of color. Developing a plan for AD flare prevention, as well as management of mild, moderate, and severe AD flares, is imperative in pediatric patients. Prevention plans should include gentle skin care, twice-daily application of emollients to the full body, and reduction of Staphylococcus aureus loads on the skin. The treatment action plan for mild to moderate flares may include topical corticosteroids, immunomodulators, and nonsteroidal agents. Treatment options for severe AD or patients who were unsuccessfully treated with other therapies may include phototherapy, biologics, and methotrexate, among others.4 Creating action plans for AD flares is a vital step in the prevention of PIH in patients with skin of color. Additionally, PIH should not be considered a sign of AD treatment failure.
Acne
Acne is a common skin disorder seen in patients with skin of color.5 A prospective observational study found that 34.3% of 683 children aged 9 to 14 years in a pediatric ambulatory clinic had acne.6 The number of preadolescents with acne is growing. Most cases are not associated with underlying endocrinopathy.7 With the growing population of children with skin of color in the United States along with the increasing childhood acne rate and subsequent inherent risk for hyperpigmentation, early acne interventions should be considered in pediatric acne patients with skin of color to reduce the impact of PIH in those at risk.
In a survey study of 313 adult acne patients with skin of color, 37.2% reported the presence of dark marks lasting 4 months or longer.5 Regardless of the severity of the acne, treatment should be initiated as tolerated in those with PIH. Adolescent acne patients with skin of color may develop PIH that is more severe and longer lasting than the acne itself.
The foundation for treatment of acne in adolescent skin of color patients is the same as those without skin of color, including topical retinoids, topical antibiotics, oral antibiotics, and isotretinoin when needed. Topical tretinoin, adapalene, azelaic acid, and tazarotene not only treat acne but also are a valuable part of the treatment armamentarium for PIH. Several studies in adults with skin of color have demonstrated improvement of PIH from the use of topical retinoids alone.8-10 Despite wanting to treat the acne aggressively, special guidance should be given to prevent retinoid dermatitis, which may lead to PIH.10 Demonstrating the application of the topical acne medications, discussing how to avoid potential side effects, and giving permission to skip applications, if needed, may empower families to make adjustments between visits to limit irritation that might prompt further PIH. Incorporating α-hydroxy acid–based cleansers, α-hydroxy acid–based chemical peels, or salicylic acid chemical peels may be warranted in the setting of intense PIH. Selecting treatments that not only help the inflammatory disease leading to the PIH but also can help improve the pigmentation are preferred; however, the risks and benefits have to be weighed because many treatments that work well for PIH also may cause irritation, leading to new or worsening PIH.
Arthropod Bites
Arthropod bites cause inflamed pruritic papules and nodules, and the resulting PIH in those with darker skin types may be quite dramatic. Parents/guardians should be instructed to have a low-potency topical corticosteroid on hand to use on bites for a few days when they appear, which will not only help with the inflammation associated with the bite but also will help decrease pruritus and subsequently skin injury from scratching. In homes with pets, checking animals routinely for fleas and other infestations is helpful. In the setting of repeated arthropod bites in the spring and summer, applying bug repellant with 10% to 30% DEET (N,N-diethyl-meta-toluamide) on the child’s clothing and exposed body areas before playing outside or in the morning before school or camp may prevent some bites. There are DEET alternatives, such as picaridin, that may be used. Product instructions should be followed when using insect repellants in the pediatric population.11
PIH Management Strategies
Gentle Skin Care Routine
There are misconceptions that areas of hyperpigmentation on the skin are caused by dirt and that scrubbing the skin harder may help to lighten the affected areas. Parents/guardians may report that the child’s skin looks dirty or, in the setting of acne, view dirt as the cause of the skin condition, which may prompt the patient to scrub the skin and the friction further worsens the PIH. Use of daily gentle cleansers and moisturizers is advised to keep the skin moisturized and free of further potential irritation and dryness that may prompt scratching or flares of the underlying condition.
Photoprotection
During the treatment course for PIH, using sun protection is helpful to prevent further darkening of the PIH areas. Sun protection may be in the form of broad-spectrum sunscreen, hats, or sun-protective clothing. Patients should be encouraged to apply sunscreen daily and to reapply every 2 hours and after water-based activities.12 For pediatric and adolescent populations, practicing sun-protective behaviors before school or outdoor activities also should be advised, as many families only think about sun protection in the setting of sunny vacation activities. Research has demonstrated that individuals with skin of color may not realize that they can be affected by skin cancer,13 thus they may not have any experience selecting, applying, or regularly using sunscreens. Products that do not leave a white hue on the skin are suggested for adolescents who may be sensitive about their appearance following sunscreen application.
Skin Lightening Treatments
Although the most important therapy for PIH is to treat the underlying inflammatory conditions, some parents/guardians may desire additional options due to the extent of involvement of the PIH, its psychological impact on the child, or adverse effect on the child’s quality of life.14 In adolescents, incorporating an α-hydroxy acid–based cleanser, glycolic acid chemical peels, salicylic acid chemical peels, and topical cosmeceuticals may be warranted in the setting of intense PIH and acne. However, irritation may lead to further dyspigmentation.
Topical ammonium lactate 12% is lactic acid neutralized with ammonium hydroxide that is formulated as a lotion or a cream. It is used to hydrate dry skin and may decrease corneocyte cohesion.15 Topical ammonium lactate also has been used anecdotally for PIH on the body during periods of watchful waiting.
Topical hydroquinone, the gold standard for treating hyperpigmentation,3,16 is not approved in children, but some parents/guardians elect to utilize hydroquinone off label to accelerate the clearing of distressing PIH in adolescents. Careful consideration including a discussion of potential risks and alternatives (eg, watchful waiting) should be highlighted.
In the setting of chronic inflammatory conditions that recur and remit, potentially irritating topical treatments should be used only during periods when symptoms of inflammation such as itching or erythema are absent.
Conclusion
Despite the best management efforts, PIH in some patients with skin of color may be present for months to years. In the pediatric skin of color population, treatment of the underlying inflammatory condition, gentle skin care, use of photoprotection, and time may be all that is needed for PIH resolution. With their parent/guardians’ consent, adolescents distressed by PIH may decide to pursue more aggressive, potentially irritating treatments. Above all, the most important management in the setting of PIH is to treat the underlying inflammatory condition causing the PIH and set reasonable expectations. For challenging cases, pediatric dermatologists with special expertise in treating pediatric and adolescent patients with skin of color may be consulted.
- Broughton A. Minorities expected to be majority in 2050. CNN. August 13, 2008. Accessed January 2, 2019.
- Colby SL, Ortman JM. Projections of the Size and Composition of the US Population: 2014 to 2060. Washington, DC: US Census Bureau; 2014. Current Population Reports, P25-1143. Published March 2015. Accessed January 23, 2019.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Eichenfield LF, Ahluwalia J, Waldman A, et al. Current guidelines for the evaluation and management of atopic dermatitis: a comparison of the Joint Task Force Practice Parameter and American Academy of Dermatology guidelines. J Allergy Clin Immunol. 2017;139(4S):S49-S57.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Napolitano M, Ruggiero G, Monfrecola G, et al. Acne prevalence in 9 to 14-year-old patients attending pediatric ambulatory clinics in Italy. Int J Dermatol. 2018;57:1320-1323.
- Mancini AJ, Baldwin HE, Eichenfield LF. Acne life cycle: the spectrum of pediatric disease. Semin Cutan Med Surg 2011;30:2-5.
- Lowe NJ, Rizk D, Grimes P, et al. Azelaic acid 20% cream in the treatment of facial hyperpigmentation in darker-skinned patients. Clin Ther. 1998;20:945-959.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Bulengo-Ransby SM, Griffiths CE, Kimbrough-Green CK, et al. Topical tretinoin (retinoid acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- American Academy of Pediatrics. Choosing an insect repellent for your child. Healthy Children website. Updated July 18, 2018. Accessed January 8, 2019.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:312-317.
- Buster KJ, You Z, Fouad M, et al. Skin cancer risk perceptions: a comparison across ethnicity, age, education, gender, and income. J Am Acad Dermatol. 2012;66:771-779.
- Downie J. Help prevent and reverse post-inflammatory hyperpigmentation. Pract Dermatol Pediatr. May/June 2011:12-14. Accessed January 18, 2019.
- Ammonium lactate lotion 12% [package insert]. Bronx, New York: Perrigo New York, Inc; 2006.
- Grimes PE. Management of hyperpigmentation in darker racial ethnic groups. Semin Cutan Med Surg. 2009;28:77-85.
Postnflammatory hyperpigmentation (PIH) is an acquired hypermelanosis that can occur in children and adults following an inflammatory cutaneous disease or trauma. Postinflammatory hyperpigmentation may last for months to even years. Although PIH may occur in all skin types, it is more common and presents with greater severity and intensity in individuals with skin of color. By the year 2050, 1 in 3 US residents is projected to be Hispanic.1 It is projected that by 2044, non-Hispanic white individuals (all ages) will make up less than 50% of the US population.2 Currently, the majority of the US residents younger than 18 years are minorities. The majority minority population in the United States already exists in those younger than 18 years and is predicted to occur in the adult population by 2044.2
Effective treatment options and management strategies for PIH in adults with skin of color have been described in the literature.3 Due to a paucity of research, the approach to management of PIH in children with skin of color has been based on clinical experience and lessons learned from adult patients. This article focuses on management of PIH in pediatric patients with skin of color, which includes black/African American, African-Caribbean, Hispanic, Asian, Pacific Islander, and American Indian individuals.
Underlying Inflammatory Dermatoses Resulting in PIH
There are numerous conditions that may result in PIH, including but not limited to atopic dermatitis (AD), acne, arthropod bites, and injuries to the skin. Postinflammatory hyperpigmentation may have more of a psychological impact than the inciting disease or injury itself. The most important step in the approach to managing PIH is treating the underlying inflammatory condition that caused the pigmentation.
Parents/guardians may report a chief concern of dark spots, manchas (stains), blemishes, or stains on the skin, often with no mention of a coexisting inflammatory dermatosis. Parents/guardians of children with skin of color often have personally experienced PIH and may be determined to shield their children from similar angst associated with the condition. Although physicians may see just another pediatric patient with PIH, the child’s parents/guardians may see a condition that will be readily perceptible during major life events, such as the child’s prom or even his/her wedding day. Promptly diagnosing and instituting early treatment of inflammatory conditions associated with PIH may accelerate resolution and prevent worsening of the pigmentation.3
Select inflammatory dermatoses that are common in children with skin of color and may lead to PIH are highlighted below. Although this list is not comprehensive, the approach and management strategies should prompt creation of plans that keep PIH in mind when treating primary inflammatory skin diseases.
Atopic Dermatitis
Atopic dermatitis may induce PIH or hypopigmentation of the skin in children with skin of color. Developing a plan for AD flare prevention, as well as management of mild, moderate, and severe AD flares, is imperative in pediatric patients. Prevention plans should include gentle skin care, twice-daily application of emollients to the full body, and reduction of Staphylococcus aureus loads on the skin. The treatment action plan for mild to moderate flares may include topical corticosteroids, immunomodulators, and nonsteroidal agents. Treatment options for severe AD or patients who were unsuccessfully treated with other therapies may include phototherapy, biologics, and methotrexate, among others.4 Creating action plans for AD flares is a vital step in the prevention of PIH in patients with skin of color. Additionally, PIH should not be considered a sign of AD treatment failure.
Acne
Acne is a common skin disorder seen in patients with skin of color.5 A prospective observational study found that 34.3% of 683 children aged 9 to 14 years in a pediatric ambulatory clinic had acne.6 The number of preadolescents with acne is growing. Most cases are not associated with underlying endocrinopathy.7 With the growing population of children with skin of color in the United States along with the increasing childhood acne rate and subsequent inherent risk for hyperpigmentation, early acne interventions should be considered in pediatric acne patients with skin of color to reduce the impact of PIH in those at risk.
In a survey study of 313 adult acne patients with skin of color, 37.2% reported the presence of dark marks lasting 4 months or longer.5 Regardless of the severity of the acne, treatment should be initiated as tolerated in those with PIH. Adolescent acne patients with skin of color may develop PIH that is more severe and longer lasting than the acne itself.
The foundation for treatment of acne in adolescent skin of color patients is the same as those without skin of color, including topical retinoids, topical antibiotics, oral antibiotics, and isotretinoin when needed. Topical tretinoin, adapalene, azelaic acid, and tazarotene not only treat acne but also are a valuable part of the treatment armamentarium for PIH. Several studies in adults with skin of color have demonstrated improvement of PIH from the use of topical retinoids alone.8-10 Despite wanting to treat the acne aggressively, special guidance should be given to prevent retinoid dermatitis, which may lead to PIH.10 Demonstrating the application of the topical acne medications, discussing how to avoid potential side effects, and giving permission to skip applications, if needed, may empower families to make adjustments between visits to limit irritation that might prompt further PIH. Incorporating α-hydroxy acid–based cleansers, α-hydroxy acid–based chemical peels, or salicylic acid chemical peels may be warranted in the setting of intense PIH. Selecting treatments that not only help the inflammatory disease leading to the PIH but also can help improve the pigmentation are preferred; however, the risks and benefits have to be weighed because many treatments that work well for PIH also may cause irritation, leading to new or worsening PIH.
Arthropod Bites
Arthropod bites cause inflamed pruritic papules and nodules, and the resulting PIH in those with darker skin types may be quite dramatic. Parents/guardians should be instructed to have a low-potency topical corticosteroid on hand to use on bites for a few days when they appear, which will not only help with the inflammation associated with the bite but also will help decrease pruritus and subsequently skin injury from scratching. In homes with pets, checking animals routinely for fleas and other infestations is helpful. In the setting of repeated arthropod bites in the spring and summer, applying bug repellant with 10% to 30% DEET (N,N-diethyl-meta-toluamide) on the child’s clothing and exposed body areas before playing outside or in the morning before school or camp may prevent some bites. There are DEET alternatives, such as picaridin, that may be used. Product instructions should be followed when using insect repellants in the pediatric population.11
PIH Management Strategies
Gentle Skin Care Routine
There are misconceptions that areas of hyperpigmentation on the skin are caused by dirt and that scrubbing the skin harder may help to lighten the affected areas. Parents/guardians may report that the child’s skin looks dirty or, in the setting of acne, view dirt as the cause of the skin condition, which may prompt the patient to scrub the skin and the friction further worsens the PIH. Use of daily gentle cleansers and moisturizers is advised to keep the skin moisturized and free of further potential irritation and dryness that may prompt scratching or flares of the underlying condition.
Photoprotection
During the treatment course for PIH, using sun protection is helpful to prevent further darkening of the PIH areas. Sun protection may be in the form of broad-spectrum sunscreen, hats, or sun-protective clothing. Patients should be encouraged to apply sunscreen daily and to reapply every 2 hours and after water-based activities.12 For pediatric and adolescent populations, practicing sun-protective behaviors before school or outdoor activities also should be advised, as many families only think about sun protection in the setting of sunny vacation activities. Research has demonstrated that individuals with skin of color may not realize that they can be affected by skin cancer,13 thus they may not have any experience selecting, applying, or regularly using sunscreens. Products that do not leave a white hue on the skin are suggested for adolescents who may be sensitive about their appearance following sunscreen application.
Skin Lightening Treatments
Although the most important therapy for PIH is to treat the underlying inflammatory conditions, some parents/guardians may desire additional options due to the extent of involvement of the PIH, its psychological impact on the child, or adverse effect on the child’s quality of life.14 In adolescents, incorporating an α-hydroxy acid–based cleanser, glycolic acid chemical peels, salicylic acid chemical peels, and topical cosmeceuticals may be warranted in the setting of intense PIH and acne. However, irritation may lead to further dyspigmentation.
Topical ammonium lactate 12% is lactic acid neutralized with ammonium hydroxide that is formulated as a lotion or a cream. It is used to hydrate dry skin and may decrease corneocyte cohesion.15 Topical ammonium lactate also has been used anecdotally for PIH on the body during periods of watchful waiting.
Topical hydroquinone, the gold standard for treating hyperpigmentation,3,16 is not approved in children, but some parents/guardians elect to utilize hydroquinone off label to accelerate the clearing of distressing PIH in adolescents. Careful consideration including a discussion of potential risks and alternatives (eg, watchful waiting) should be highlighted.
In the setting of chronic inflammatory conditions that recur and remit, potentially irritating topical treatments should be used only during periods when symptoms of inflammation such as itching or erythema are absent.
Conclusion
Despite the best management efforts, PIH in some patients with skin of color may be present for months to years. In the pediatric skin of color population, treatment of the underlying inflammatory condition, gentle skin care, use of photoprotection, and time may be all that is needed for PIH resolution. With their parent/guardians’ consent, adolescents distressed by PIH may decide to pursue more aggressive, potentially irritating treatments. Above all, the most important management in the setting of PIH is to treat the underlying inflammatory condition causing the PIH and set reasonable expectations. For challenging cases, pediatric dermatologists with special expertise in treating pediatric and adolescent patients with skin of color may be consulted.
Postnflammatory hyperpigmentation (PIH) is an acquired hypermelanosis that can occur in children and adults following an inflammatory cutaneous disease or trauma. Postinflammatory hyperpigmentation may last for months to even years. Although PIH may occur in all skin types, it is more common and presents with greater severity and intensity in individuals with skin of color. By the year 2050, 1 in 3 US residents is projected to be Hispanic.1 It is projected that by 2044, non-Hispanic white individuals (all ages) will make up less than 50% of the US population.2 Currently, the majority of the US residents younger than 18 years are minorities. The majority minority population in the United States already exists in those younger than 18 years and is predicted to occur in the adult population by 2044.2
Effective treatment options and management strategies for PIH in adults with skin of color have been described in the literature.3 Due to a paucity of research, the approach to management of PIH in children with skin of color has been based on clinical experience and lessons learned from adult patients. This article focuses on management of PIH in pediatric patients with skin of color, which includes black/African American, African-Caribbean, Hispanic, Asian, Pacific Islander, and American Indian individuals.
Underlying Inflammatory Dermatoses Resulting in PIH
There are numerous conditions that may result in PIH, including but not limited to atopic dermatitis (AD), acne, arthropod bites, and injuries to the skin. Postinflammatory hyperpigmentation may have more of a psychological impact than the inciting disease or injury itself. The most important step in the approach to managing PIH is treating the underlying inflammatory condition that caused the pigmentation.
Parents/guardians may report a chief concern of dark spots, manchas (stains), blemishes, or stains on the skin, often with no mention of a coexisting inflammatory dermatosis. Parents/guardians of children with skin of color often have personally experienced PIH and may be determined to shield their children from similar angst associated with the condition. Although physicians may see just another pediatric patient with PIH, the child’s parents/guardians may see a condition that will be readily perceptible during major life events, such as the child’s prom or even his/her wedding day. Promptly diagnosing and instituting early treatment of inflammatory conditions associated with PIH may accelerate resolution and prevent worsening of the pigmentation.3
Select inflammatory dermatoses that are common in children with skin of color and may lead to PIH are highlighted below. Although this list is not comprehensive, the approach and management strategies should prompt creation of plans that keep PIH in mind when treating primary inflammatory skin diseases.
Atopic Dermatitis
Atopic dermatitis may induce PIH or hypopigmentation of the skin in children with skin of color. Developing a plan for AD flare prevention, as well as management of mild, moderate, and severe AD flares, is imperative in pediatric patients. Prevention plans should include gentle skin care, twice-daily application of emollients to the full body, and reduction of Staphylococcus aureus loads on the skin. The treatment action plan for mild to moderate flares may include topical corticosteroids, immunomodulators, and nonsteroidal agents. Treatment options for severe AD or patients who were unsuccessfully treated with other therapies may include phototherapy, biologics, and methotrexate, among others.4 Creating action plans for AD flares is a vital step in the prevention of PIH in patients with skin of color. Additionally, PIH should not be considered a sign of AD treatment failure.
Acne
Acne is a common skin disorder seen in patients with skin of color.5 A prospective observational study found that 34.3% of 683 children aged 9 to 14 years in a pediatric ambulatory clinic had acne.6 The number of preadolescents with acne is growing. Most cases are not associated with underlying endocrinopathy.7 With the growing population of children with skin of color in the United States along with the increasing childhood acne rate and subsequent inherent risk for hyperpigmentation, early acne interventions should be considered in pediatric acne patients with skin of color to reduce the impact of PIH in those at risk.
In a survey study of 313 adult acne patients with skin of color, 37.2% reported the presence of dark marks lasting 4 months or longer.5 Regardless of the severity of the acne, treatment should be initiated as tolerated in those with PIH. Adolescent acne patients with skin of color may develop PIH that is more severe and longer lasting than the acne itself.
The foundation for treatment of acne in adolescent skin of color patients is the same as those without skin of color, including topical retinoids, topical antibiotics, oral antibiotics, and isotretinoin when needed. Topical tretinoin, adapalene, azelaic acid, and tazarotene not only treat acne but also are a valuable part of the treatment armamentarium for PIH. Several studies in adults with skin of color have demonstrated improvement of PIH from the use of topical retinoids alone.8-10 Despite wanting to treat the acne aggressively, special guidance should be given to prevent retinoid dermatitis, which may lead to PIH.10 Demonstrating the application of the topical acne medications, discussing how to avoid potential side effects, and giving permission to skip applications, if needed, may empower families to make adjustments between visits to limit irritation that might prompt further PIH. Incorporating α-hydroxy acid–based cleansers, α-hydroxy acid–based chemical peels, or salicylic acid chemical peels may be warranted in the setting of intense PIH. Selecting treatments that not only help the inflammatory disease leading to the PIH but also can help improve the pigmentation are preferred; however, the risks and benefits have to be weighed because many treatments that work well for PIH also may cause irritation, leading to new or worsening PIH.
Arthropod Bites
Arthropod bites cause inflamed pruritic papules and nodules, and the resulting PIH in those with darker skin types may be quite dramatic. Parents/guardians should be instructed to have a low-potency topical corticosteroid on hand to use on bites for a few days when they appear, which will not only help with the inflammation associated with the bite but also will help decrease pruritus and subsequently skin injury from scratching. In homes with pets, checking animals routinely for fleas and other infestations is helpful. In the setting of repeated arthropod bites in the spring and summer, applying bug repellant with 10% to 30% DEET (N,N-diethyl-meta-toluamide) on the child’s clothing and exposed body areas before playing outside or in the morning before school or camp may prevent some bites. There are DEET alternatives, such as picaridin, that may be used. Product instructions should be followed when using insect repellants in the pediatric population.11
PIH Management Strategies
Gentle Skin Care Routine
There are misconceptions that areas of hyperpigmentation on the skin are caused by dirt and that scrubbing the skin harder may help to lighten the affected areas. Parents/guardians may report that the child’s skin looks dirty or, in the setting of acne, view dirt as the cause of the skin condition, which may prompt the patient to scrub the skin and the friction further worsens the PIH. Use of daily gentle cleansers and moisturizers is advised to keep the skin moisturized and free of further potential irritation and dryness that may prompt scratching or flares of the underlying condition.
Photoprotection
During the treatment course for PIH, using sun protection is helpful to prevent further darkening of the PIH areas. Sun protection may be in the form of broad-spectrum sunscreen, hats, or sun-protective clothing. Patients should be encouraged to apply sunscreen daily and to reapply every 2 hours and after water-based activities.12 For pediatric and adolescent populations, practicing sun-protective behaviors before school or outdoor activities also should be advised, as many families only think about sun protection in the setting of sunny vacation activities. Research has demonstrated that individuals with skin of color may not realize that they can be affected by skin cancer,13 thus they may not have any experience selecting, applying, or regularly using sunscreens. Products that do not leave a white hue on the skin are suggested for adolescents who may be sensitive about their appearance following sunscreen application.
Skin Lightening Treatments
Although the most important therapy for PIH is to treat the underlying inflammatory conditions, some parents/guardians may desire additional options due to the extent of involvement of the PIH, its psychological impact on the child, or adverse effect on the child’s quality of life.14 In adolescents, incorporating an α-hydroxy acid–based cleanser, glycolic acid chemical peels, salicylic acid chemical peels, and topical cosmeceuticals may be warranted in the setting of intense PIH and acne. However, irritation may lead to further dyspigmentation.
Topical ammonium lactate 12% is lactic acid neutralized with ammonium hydroxide that is formulated as a lotion or a cream. It is used to hydrate dry skin and may decrease corneocyte cohesion.15 Topical ammonium lactate also has been used anecdotally for PIH on the body during periods of watchful waiting.
Topical hydroquinone, the gold standard for treating hyperpigmentation,3,16 is not approved in children, but some parents/guardians elect to utilize hydroquinone off label to accelerate the clearing of distressing PIH in adolescents. Careful consideration including a discussion of potential risks and alternatives (eg, watchful waiting) should be highlighted.
In the setting of chronic inflammatory conditions that recur and remit, potentially irritating topical treatments should be used only during periods when symptoms of inflammation such as itching or erythema are absent.
Conclusion
Despite the best management efforts, PIH in some patients with skin of color may be present for months to years. In the pediatric skin of color population, treatment of the underlying inflammatory condition, gentle skin care, use of photoprotection, and time may be all that is needed for PIH resolution. With their parent/guardians’ consent, adolescents distressed by PIH may decide to pursue more aggressive, potentially irritating treatments. Above all, the most important management in the setting of PIH is to treat the underlying inflammatory condition causing the PIH and set reasonable expectations. For challenging cases, pediatric dermatologists with special expertise in treating pediatric and adolescent patients with skin of color may be consulted.
- Broughton A. Minorities expected to be majority in 2050. CNN. August 13, 2008. Accessed January 2, 2019.
- Colby SL, Ortman JM. Projections of the Size and Composition of the US Population: 2014 to 2060. Washington, DC: US Census Bureau; 2014. Current Population Reports, P25-1143. Published March 2015. Accessed January 23, 2019.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Eichenfield LF, Ahluwalia J, Waldman A, et al. Current guidelines for the evaluation and management of atopic dermatitis: a comparison of the Joint Task Force Practice Parameter and American Academy of Dermatology guidelines. J Allergy Clin Immunol. 2017;139(4S):S49-S57.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Napolitano M, Ruggiero G, Monfrecola G, et al. Acne prevalence in 9 to 14-year-old patients attending pediatric ambulatory clinics in Italy. Int J Dermatol. 2018;57:1320-1323.
- Mancini AJ, Baldwin HE, Eichenfield LF. Acne life cycle: the spectrum of pediatric disease. Semin Cutan Med Surg 2011;30:2-5.
- Lowe NJ, Rizk D, Grimes P, et al. Azelaic acid 20% cream in the treatment of facial hyperpigmentation in darker-skinned patients. Clin Ther. 1998;20:945-959.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Bulengo-Ransby SM, Griffiths CE, Kimbrough-Green CK, et al. Topical tretinoin (retinoid acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- American Academy of Pediatrics. Choosing an insect repellent for your child. Healthy Children website. Updated July 18, 2018. Accessed January 8, 2019.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:312-317.
- Buster KJ, You Z, Fouad M, et al. Skin cancer risk perceptions: a comparison across ethnicity, age, education, gender, and income. J Am Acad Dermatol. 2012;66:771-779.
- Downie J. Help prevent and reverse post-inflammatory hyperpigmentation. Pract Dermatol Pediatr. May/June 2011:12-14. Accessed January 18, 2019.
- Ammonium lactate lotion 12% [package insert]. Bronx, New York: Perrigo New York, Inc; 2006.
- Grimes PE. Management of hyperpigmentation in darker racial ethnic groups. Semin Cutan Med Surg. 2009;28:77-85.
- Broughton A. Minorities expected to be majority in 2050. CNN. August 13, 2008. Accessed January 2, 2019.
- Colby SL, Ortman JM. Projections of the Size and Composition of the US Population: 2014 to 2060. Washington, DC: US Census Bureau; 2014. Current Population Reports, P25-1143. Published March 2015. Accessed January 23, 2019.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Eichenfield LF, Ahluwalia J, Waldman A, et al. Current guidelines for the evaluation and management of atopic dermatitis: a comparison of the Joint Task Force Practice Parameter and American Academy of Dermatology guidelines. J Allergy Clin Immunol. 2017;139(4S):S49-S57.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Napolitano M, Ruggiero G, Monfrecola G, et al. Acne prevalence in 9 to 14-year-old patients attending pediatric ambulatory clinics in Italy. Int J Dermatol. 2018;57:1320-1323.
- Mancini AJ, Baldwin HE, Eichenfield LF. Acne life cycle: the spectrum of pediatric disease. Semin Cutan Med Surg 2011;30:2-5.
- Lowe NJ, Rizk D, Grimes P, et al. Azelaic acid 20% cream in the treatment of facial hyperpigmentation in darker-skinned patients. Clin Ther. 1998;20:945-959.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Bulengo-Ransby SM, Griffiths CE, Kimbrough-Green CK, et al. Topical tretinoin (retinoid acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- American Academy of Pediatrics. Choosing an insect repellent for your child. Healthy Children website. Updated July 18, 2018. Accessed January 8, 2019.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:312-317.
- Buster KJ, You Z, Fouad M, et al. Skin cancer risk perceptions: a comparison across ethnicity, age, education, gender, and income. J Am Acad Dermatol. 2012;66:771-779.
- Downie J. Help prevent and reverse post-inflammatory hyperpigmentation. Pract Dermatol Pediatr. May/June 2011:12-14. Accessed January 18, 2019.
- Ammonium lactate lotion 12% [package insert]. Bronx, New York: Perrigo New York, Inc; 2006.
- Grimes PE. Management of hyperpigmentation in darker racial ethnic groups. Semin Cutan Med Surg. 2009;28:77-85.
Practice Points
- The US population of children with skin of color is growing rapidly.
- Treating the underlying inflammatory dermatosis is the most important step in managing postinflammatory hyperpigmentation (PIH); however, many pediatric PIH patients and their parents/guardians presenting with a chief concern of pigmentary changes are unaware of the associated inflammatory condition.
- When appropriate, choose treatments for the underlying inflammatory condition that can simultaneously improve any existing PIH. Gentle skin care, avoidance of rubbing and scrubbing the skin, and photoprotection are essential to halt worsening of PIH.
- Patients’ parents/guardians may consent to more aggressive PIH treatment in select cases (eg, emotional distress in adolescents).
Bidirectional relationship found between depression, vitiligo
Vitiligo and major depressive disorder have a bidirectional relationship, according to a new study that examined data from a cohort of more than 6 million people.
“Ultimately, this suggests that mental health appears to play a large role in the pathogenesis of autoimmune diseases like vitiligo, which in turn can increase the risk of MDD, especially in younger patients,” wrote Isabelle Vallerand, PhD, and her colleagues. The report is in the Journal of the American Academy of Dermatology.
Dr. Vallerand and her colleagues found that patients with major depressive disorder (MDD, n = 405,397) had a 64% increased risk of vitiligo, compared with a referent cohort (n = 5,739,048; 95% confidence interval, 1.43-1.87; P less than .0001). Conversely, patients who had vitiligo also were at an increased risk of MDD. Patients who were younger than 30 years old at diagnosis (n = 7,104) had a hazard ratio of 1.31 for MDD (P less than .0001), compared with 1.22 for patients aged 30 years and older (P = .001).
Individuals who took antidepressants, whether or not they also had an MDD diagnosis, had a decreased risk for vitiligo.
Though it’s known that vitiligo increases the risk of MDD, less clarity has been in the literature about whether the converse also might be true. “The question of whether vitiligo onset can be precipitated by MDD has received less attention, despite the notion that patients often ask their dermatologists if stress or depression may have contributed to their disease,” wrote Dr. Vallerand, an epidemiologist and medical student at the University of Calgary, Alberta, and her colleagues.
There is a biologic plausibility for a bidirectional relationship, said Dr. Vallerand and her colleagues, since depression can boost systemic inflammation, and the risk for autoimmune disease such as vitiligo can be increased by proinflammatory states.
Access to a large dataset gave Dr. Vallerand and her collaborators the numbers to look at the relationship between vitiligo and MDD in the context of potential confounders, and to correct for those in their statistical analysis. Using medical records from The Health Improvement Network (THIN) database in the United Kingdom, the investigators conducted two independent population-based cohort studies. Each looked at risk in one direction of the MDD-vitiligo association.
The first analysis looked at MDD as a risk factor for vitiligo, following all patients with an incident diagnostic code for MDD. Patients without the MDD diagnosis code were the referent cohort. Patients in each cohort were followed until they reached the outcome of interest – a diagnosis of vitiligo – or were censored. Patients who had a vitiligo diagnosis before receiving an MDD diagnosis were not included.
The second analysis examined whether vitiligo was a risk factor for MDD, with a similar design that used nonvitiligo patients as the referent cohort. This analysis followed all patients until a diagnosis of MDD was recorded, or patients were censored. Again, patients with MDD diagnoses that came before the vitiligo diagnosis were excluded.
For the analysis of risk of vitiligo, the investigators looked at the effects of multiple covariates, including age, sex, alcohol use and smoking status, socioeconomic status, medical comorbidities, and whether patients were taking antidepressants. The covariates included in the analysis of risk of MDD were age, sex, medical comorbidities, and type of vitiligo treatment.
After the researchers determined unadjusted hazard ratios, each covariate was removed one at a time to see where there were substantial changes to the HR. Two additional models, one unadjusted and one that fully adjusted for all covariates, also were built.
The sensitivity analyses showed “an overall protective effect of antidepressants among both cohorts,” wrote Dr. Vallerand and her colleagues. The incidence rate of vitiligo among patients with MDD using antidepressants was 19.7 per 100,000 person-years, compared with 27.5 among MDD patients not using antidepressants (P = .0053).
“Similarly, those in the referent cohort who used antidepressants had about half the risk of vitiligo,” compared with the nonusers in the referent group, the investigators said. Serotonin also is present in the skin, and neurons and melanocytes share embryonic ectodermal origins, Dr. Vallerand and her colleagues said. Though the exact mechanisms are not known, in the THIN cohorts, they noted.
Though younger patients with vitiligo were at higher risk for MDD than were those aged 30 years and older, the overall cohort of individuals with vitiligo still had an unadjusted elevated risk for MDD, compared with the referent cohort (HR 1.27; 95% confidence interval, 1.16-1.40; P less than .0001).
“Unexpectedly, the magnitude of the reciprocal association was highest with MDD being a risk factor for vitiligo,” wrote Dr. Vallerand and her colleagues. “This highlights the notion that mental health may have a greater impact on the body, specifically with dermatologic manifestations, than previously thought.”
Some misclassification of both conditions is likely in such a large dataset, the investigators acknowledged. Also, subclinical depression was not evaluated, and there was no way to track the severity of either depression or vitiligo, they noted. Still, the big data approach “renders this one of the largest studies on psychodermatology to date,” said Dr. Vallerand and her colleagues, and the independent bidirectional analyses support causality.
Dr. Vallerand is a partner in a pharmaceutical consulting firm, GlacierRX, and was funded by Alberta Innovates. The authors reported having no conflicts of interest.
SOURCE: Vallerand IA et al. J Am Acad Dermatol. 2019. doi: 10.1016/j.jaad.2018.11.047.
Vitiligo and major depressive disorder have a bidirectional relationship, according to a new study that examined data from a cohort of more than 6 million people.
“Ultimately, this suggests that mental health appears to play a large role in the pathogenesis of autoimmune diseases like vitiligo, which in turn can increase the risk of MDD, especially in younger patients,” wrote Isabelle Vallerand, PhD, and her colleagues. The report is in the Journal of the American Academy of Dermatology.
Dr. Vallerand and her colleagues found that patients with major depressive disorder (MDD, n = 405,397) had a 64% increased risk of vitiligo, compared with a referent cohort (n = 5,739,048; 95% confidence interval, 1.43-1.87; P less than .0001). Conversely, patients who had vitiligo also were at an increased risk of MDD. Patients who were younger than 30 years old at diagnosis (n = 7,104) had a hazard ratio of 1.31 for MDD (P less than .0001), compared with 1.22 for patients aged 30 years and older (P = .001).
Individuals who took antidepressants, whether or not they also had an MDD diagnosis, had a decreased risk for vitiligo.
Though it’s known that vitiligo increases the risk of MDD, less clarity has been in the literature about whether the converse also might be true. “The question of whether vitiligo onset can be precipitated by MDD has received less attention, despite the notion that patients often ask their dermatologists if stress or depression may have contributed to their disease,” wrote Dr. Vallerand, an epidemiologist and medical student at the University of Calgary, Alberta, and her colleagues.
There is a biologic plausibility for a bidirectional relationship, said Dr. Vallerand and her colleagues, since depression can boost systemic inflammation, and the risk for autoimmune disease such as vitiligo can be increased by proinflammatory states.
Access to a large dataset gave Dr. Vallerand and her collaborators the numbers to look at the relationship between vitiligo and MDD in the context of potential confounders, and to correct for those in their statistical analysis. Using medical records from The Health Improvement Network (THIN) database in the United Kingdom, the investigators conducted two independent population-based cohort studies. Each looked at risk in one direction of the MDD-vitiligo association.
The first analysis looked at MDD as a risk factor for vitiligo, following all patients with an incident diagnostic code for MDD. Patients without the MDD diagnosis code were the referent cohort. Patients in each cohort were followed until they reached the outcome of interest – a diagnosis of vitiligo – or were censored. Patients who had a vitiligo diagnosis before receiving an MDD diagnosis were not included.
The second analysis examined whether vitiligo was a risk factor for MDD, with a similar design that used nonvitiligo patients as the referent cohort. This analysis followed all patients until a diagnosis of MDD was recorded, or patients were censored. Again, patients with MDD diagnoses that came before the vitiligo diagnosis were excluded.
For the analysis of risk of vitiligo, the investigators looked at the effects of multiple covariates, including age, sex, alcohol use and smoking status, socioeconomic status, medical comorbidities, and whether patients were taking antidepressants. The covariates included in the analysis of risk of MDD were age, sex, medical comorbidities, and type of vitiligo treatment.
After the researchers determined unadjusted hazard ratios, each covariate was removed one at a time to see where there were substantial changes to the HR. Two additional models, one unadjusted and one that fully adjusted for all covariates, also were built.
The sensitivity analyses showed “an overall protective effect of antidepressants among both cohorts,” wrote Dr. Vallerand and her colleagues. The incidence rate of vitiligo among patients with MDD using antidepressants was 19.7 per 100,000 person-years, compared with 27.5 among MDD patients not using antidepressants (P = .0053).
“Similarly, those in the referent cohort who used antidepressants had about half the risk of vitiligo,” compared with the nonusers in the referent group, the investigators said. Serotonin also is present in the skin, and neurons and melanocytes share embryonic ectodermal origins, Dr. Vallerand and her colleagues said. Though the exact mechanisms are not known, in the THIN cohorts, they noted.
Though younger patients with vitiligo were at higher risk for MDD than were those aged 30 years and older, the overall cohort of individuals with vitiligo still had an unadjusted elevated risk for MDD, compared with the referent cohort (HR 1.27; 95% confidence interval, 1.16-1.40; P less than .0001).
“Unexpectedly, the magnitude of the reciprocal association was highest with MDD being a risk factor for vitiligo,” wrote Dr. Vallerand and her colleagues. “This highlights the notion that mental health may have a greater impact on the body, specifically with dermatologic manifestations, than previously thought.”
Some misclassification of both conditions is likely in such a large dataset, the investigators acknowledged. Also, subclinical depression was not evaluated, and there was no way to track the severity of either depression or vitiligo, they noted. Still, the big data approach “renders this one of the largest studies on psychodermatology to date,” said Dr. Vallerand and her colleagues, and the independent bidirectional analyses support causality.
Dr. Vallerand is a partner in a pharmaceutical consulting firm, GlacierRX, and was funded by Alberta Innovates. The authors reported having no conflicts of interest.
SOURCE: Vallerand IA et al. J Am Acad Dermatol. 2019. doi: 10.1016/j.jaad.2018.11.047.
Vitiligo and major depressive disorder have a bidirectional relationship, according to a new study that examined data from a cohort of more than 6 million people.
“Ultimately, this suggests that mental health appears to play a large role in the pathogenesis of autoimmune diseases like vitiligo, which in turn can increase the risk of MDD, especially in younger patients,” wrote Isabelle Vallerand, PhD, and her colleagues. The report is in the Journal of the American Academy of Dermatology.
Dr. Vallerand and her colleagues found that patients with major depressive disorder (MDD, n = 405,397) had a 64% increased risk of vitiligo, compared with a referent cohort (n = 5,739,048; 95% confidence interval, 1.43-1.87; P less than .0001). Conversely, patients who had vitiligo also were at an increased risk of MDD. Patients who were younger than 30 years old at diagnosis (n = 7,104) had a hazard ratio of 1.31 for MDD (P less than .0001), compared with 1.22 for patients aged 30 years and older (P = .001).
Individuals who took antidepressants, whether or not they also had an MDD diagnosis, had a decreased risk for vitiligo.
Though it’s known that vitiligo increases the risk of MDD, less clarity has been in the literature about whether the converse also might be true. “The question of whether vitiligo onset can be precipitated by MDD has received less attention, despite the notion that patients often ask their dermatologists if stress or depression may have contributed to their disease,” wrote Dr. Vallerand, an epidemiologist and medical student at the University of Calgary, Alberta, and her colleagues.
There is a biologic plausibility for a bidirectional relationship, said Dr. Vallerand and her colleagues, since depression can boost systemic inflammation, and the risk for autoimmune disease such as vitiligo can be increased by proinflammatory states.
Access to a large dataset gave Dr. Vallerand and her collaborators the numbers to look at the relationship between vitiligo and MDD in the context of potential confounders, and to correct for those in their statistical analysis. Using medical records from The Health Improvement Network (THIN) database in the United Kingdom, the investigators conducted two independent population-based cohort studies. Each looked at risk in one direction of the MDD-vitiligo association.
The first analysis looked at MDD as a risk factor for vitiligo, following all patients with an incident diagnostic code for MDD. Patients without the MDD diagnosis code were the referent cohort. Patients in each cohort were followed until they reached the outcome of interest – a diagnosis of vitiligo – or were censored. Patients who had a vitiligo diagnosis before receiving an MDD diagnosis were not included.
The second analysis examined whether vitiligo was a risk factor for MDD, with a similar design that used nonvitiligo patients as the referent cohort. This analysis followed all patients until a diagnosis of MDD was recorded, or patients were censored. Again, patients with MDD diagnoses that came before the vitiligo diagnosis were excluded.
For the analysis of risk of vitiligo, the investigators looked at the effects of multiple covariates, including age, sex, alcohol use and smoking status, socioeconomic status, medical comorbidities, and whether patients were taking antidepressants. The covariates included in the analysis of risk of MDD were age, sex, medical comorbidities, and type of vitiligo treatment.
After the researchers determined unadjusted hazard ratios, each covariate was removed one at a time to see where there were substantial changes to the HR. Two additional models, one unadjusted and one that fully adjusted for all covariates, also were built.
The sensitivity analyses showed “an overall protective effect of antidepressants among both cohorts,” wrote Dr. Vallerand and her colleagues. The incidence rate of vitiligo among patients with MDD using antidepressants was 19.7 per 100,000 person-years, compared with 27.5 among MDD patients not using antidepressants (P = .0053).
“Similarly, those in the referent cohort who used antidepressants had about half the risk of vitiligo,” compared with the nonusers in the referent group, the investigators said. Serotonin also is present in the skin, and neurons and melanocytes share embryonic ectodermal origins, Dr. Vallerand and her colleagues said. Though the exact mechanisms are not known, in the THIN cohorts, they noted.
Though younger patients with vitiligo were at higher risk for MDD than were those aged 30 years and older, the overall cohort of individuals with vitiligo still had an unadjusted elevated risk for MDD, compared with the referent cohort (HR 1.27; 95% confidence interval, 1.16-1.40; P less than .0001).
“Unexpectedly, the magnitude of the reciprocal association was highest with MDD being a risk factor for vitiligo,” wrote Dr. Vallerand and her colleagues. “This highlights the notion that mental health may have a greater impact on the body, specifically with dermatologic manifestations, than previously thought.”
Some misclassification of both conditions is likely in such a large dataset, the investigators acknowledged. Also, subclinical depression was not evaluated, and there was no way to track the severity of either depression or vitiligo, they noted. Still, the big data approach “renders this one of the largest studies on psychodermatology to date,” said Dr. Vallerand and her colleagues, and the independent bidirectional analyses support causality.
Dr. Vallerand is a partner in a pharmaceutical consulting firm, GlacierRX, and was funded by Alberta Innovates. The authors reported having no conflicts of interest.
SOURCE: Vallerand IA et al. J Am Acad Dermatol. 2019. doi: 10.1016/j.jaad.2018.11.047.
FROM JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: The findings suggest that “mental health appears to play a large role in the pathogenesis of autoimmune diseases like vitiligo.”
Major finding: Patients with major depressive disorder had a 64% increased risk of vitiligo.
Study details: Retrospective records review of 405,397 patients with MDD and 5,738,048 patients in a referent cohort.
Disclosures: Dr. Vallerand is a partner in a pharmaceutical consulting firm, GlacierRx, and was funded by Alberta Innovates. The authors reported having no conflicts of interest.
Source: Vallerand IA et al. J Am Acad Dermatol. 2019. doi: 10.1016/j.jaad.2018.11.047.
Verrucous Coalescing Dry Papules and Plaques on the Hip and Flank
The Diagnosis: Blaschkitis
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
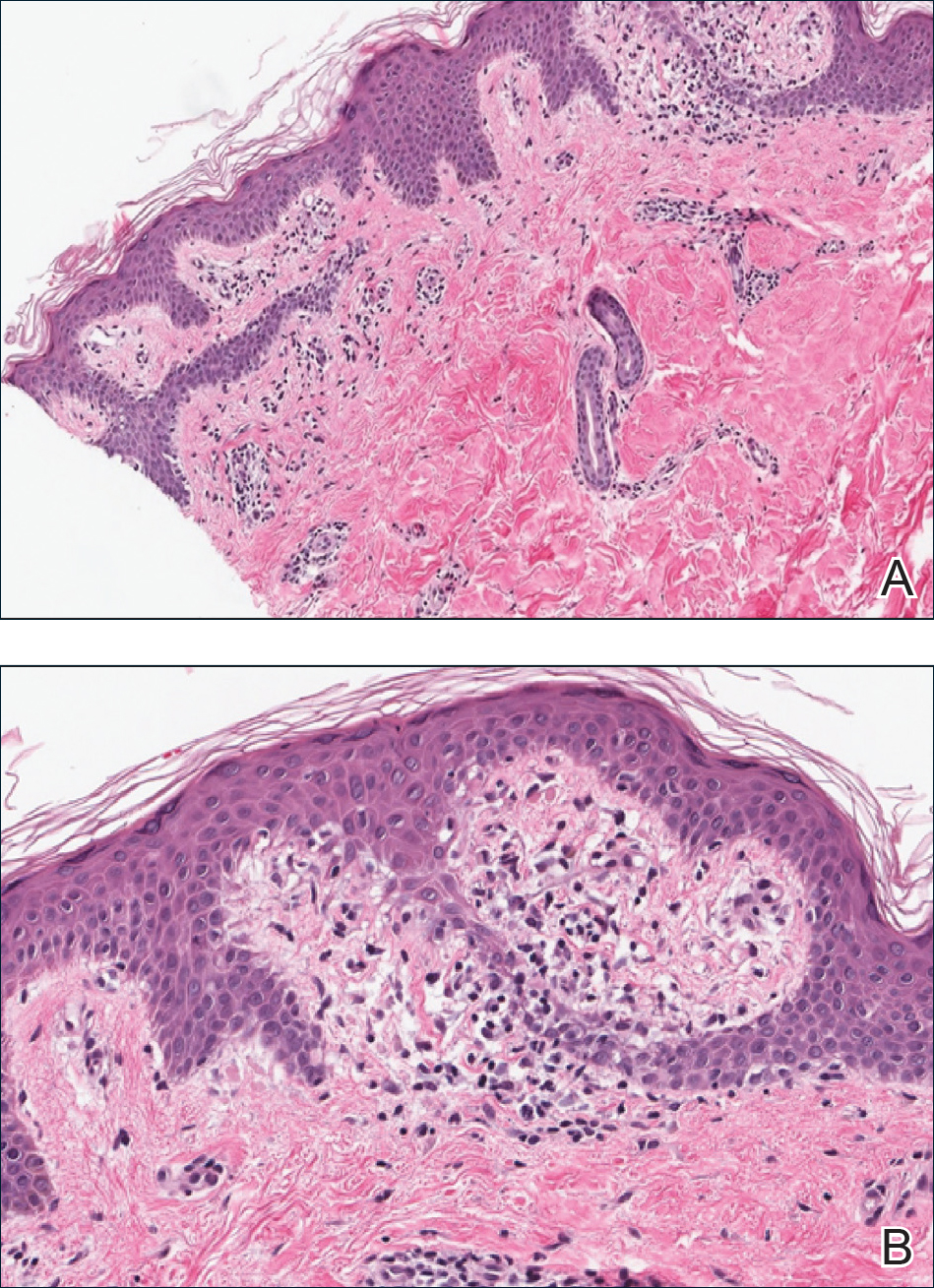
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
The Diagnosis: Blaschkitis
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
The Diagnosis: Blaschkitis
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
A 31-year-old man presented with a recurring pruritic rash on the right lateral flank and hip of 2 years’ duration. Physical examination revealed erythematous, verrucous, dry papules and plaques coalescing into larger plaques on the right flank and hip in dermatomal distributions involving the T10 and T11 dermatomes. A few papules were scattered in a linear eruption along the right flank and right upper thigh. Some postinflammatory changes were noted. No rash was noted over any other area of the body. Physical examination was otherwise unremarkable.

How to Identify Frontal Fibrosing Alopecia


Frontal Fibrosing Alopecia: Cutaneous Associations in Women With Skin of Color
Frontal fibrosing alopecia (FFA) has been reported in association with lichen planus pigmentosus (LPP) and facial papules.1-3 Lichen planus pigmentosus is a variant of lichen planus that causes hyperpigmentation of the face, neck, and/or intertriginous areas that may be useful as a clinical indicator in the development of FFA.1 Facial papules in association with FFA are secondary to fibrosed vellus hairs.2,3 Currently, reports of concomitant FFA, LPP, and facial papules in women with skin of color are limited in the literature. This case series includes 5 women of color (Hispanic and black) who presented to our clinic with FFA and various cutaneous associations. A review of the current literature on cutaneous associations of FFA also is provided.
Case Reports
Patient 1
A 50-year-old Hispanic woman who was previously presumed to have melasma by an outside physician presented with pruritus of the scalp and eyebrows of 1 month’s duration. Physical examination revealed decreased frontal scalp hair density with perifollicular erythema and scale with thinning of the lateral eyebrows. Hyperpigmented coalesced macules (Figure 1A) and erythematous perifollicular papules were noted along the temples and on the perioral skin. Depressed forehead and temporal veins also were noted (Figure 1B). A biopsy of the scalp demonstrated perifollicular and perivascular lymphocytic inflammation and fibrosed hair follicles, and a biopsy of the perioral skin demonstrated perivascular lymphocytic inflammation with melanophages in the papillary dermis. A diagnosis of FFA with LPP was established with these biopsies.
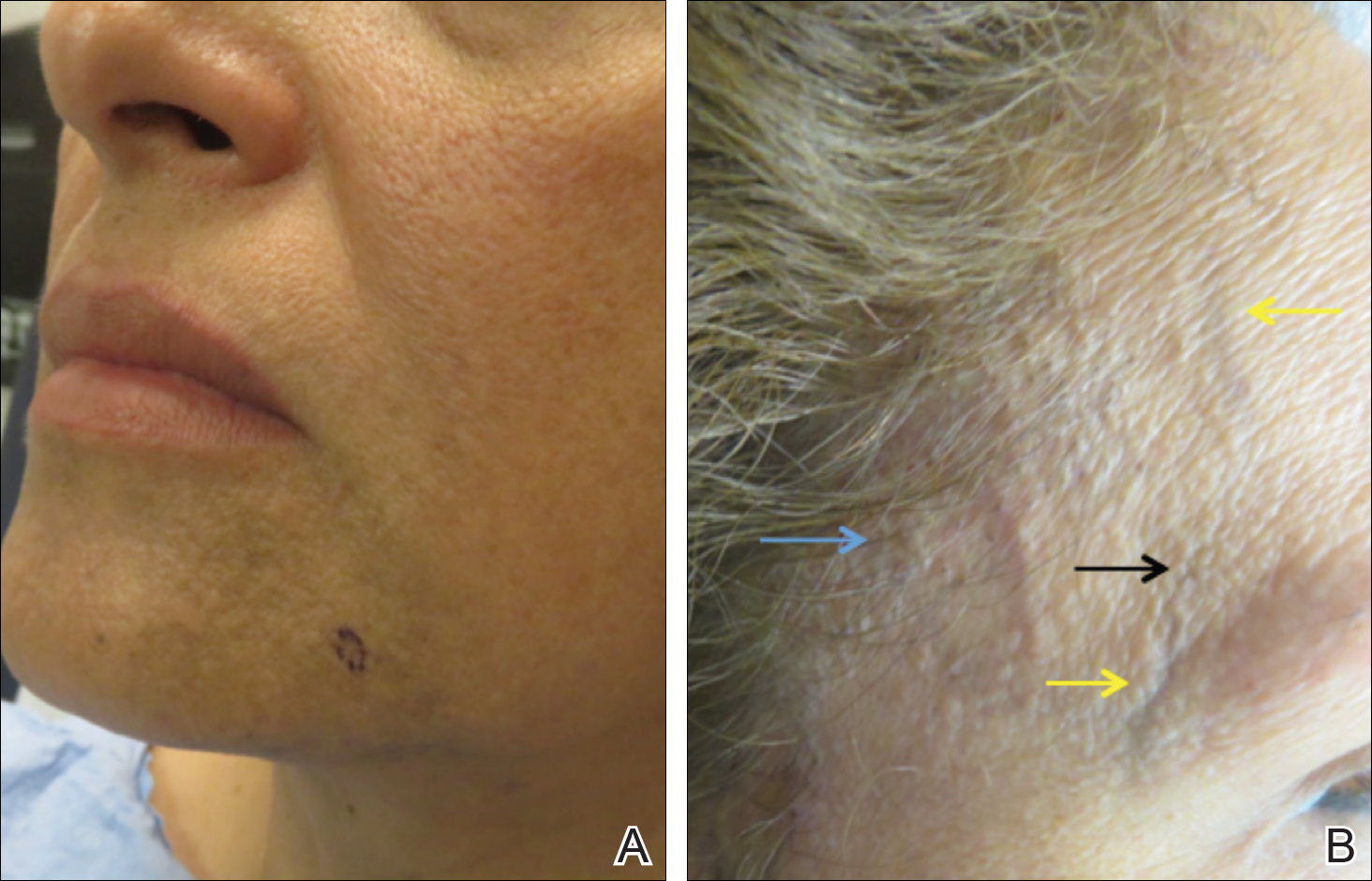
Patient 2
A 61-year-old black woman presented with asymptomatic hair loss along the frontal hairline for an unknown duration. On physical examination the frontal scalp and lateral eyebrows demonstrated decreased hair density with loss of follicular ostia. Fine, flesh-colored, monomorphic papules were scattered along the forehead and temples, and ill-defined brown pigmentation was present along the forehead, temples, and cheeks. Biopsy of the frontal scalp demonstrated patchy lichenoid inflammation with decreased number of follicles with replacement by follicular scars, confirming the diagnosis of FFA.
Patient 3
A 47-year-old Hispanic woman presented with hair loss of the frontal scalp and bilateral eyebrows with associated burning of 2 years’ duration. Physical examination demonstrated recession of the frontotemporal hairline with scattered lone hairs and thinning of the eyebrows. Innumerable flesh-colored papules were present on the forehead and temples (Figure 2A). Glabellar and eyebrow erythema was noted (Figure 2B). Biopsy of the frontal scalp demonstrated decreased terminal anagen hair follicles with perifollicular lymphoid infiltrate and fibrosis, consistent with a diagnosis of FFA. The patient was started on oral hydroxychloroquine 400 mg once daily, and 3 months later hyperpigmentation of the forehead and perioral skin was noted. The patient reported that she had facial hyperpigmentation prior to starting hydroxychloroquine and declined a biopsy.
Patient 4
A 40-year-old black woman presented with brown pruritic macles of the face, neck, arms, and forearms of 4 years’ duration. She also reported hair loss on the frontal and occipital scalp, eyebrows, and arms. On physical examination, ill-defined brown macules and patches were noted on the neck (Figure 3), face, arms, and forearms. Decreased hair density was noted on the frontal and occipital scalp with follicular dropout and perifollicular hyperpigmentation. Biopsy of the scalp demonstrated perivascular lymphocytic inflammation with sparse anagen follicles and fibrous tracts, and biopsy of the neck revealed superficial perivascular inflammation with numerous melanophages in the upper dermis; these histopathologic findings were consistent with FFA and LPP, respectively.
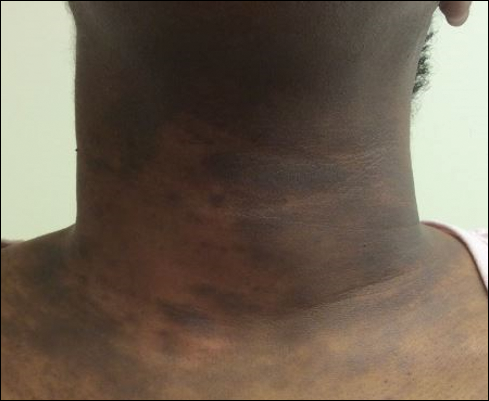
Patient 5
A 46-year-old black woman with history of hair loss presented with hyperpigmentation of the face and neck of 2 years’ duration. On physical examination decreased hair density of the frontal and vertex scalp and lateral eyebrows was noted. Flesh-colored papules were noted on the forehead and cheeks, and confluent dark brown patches were present on the temples and neck. Three punch biopsies were performed. Biopsy of the scalp revealed lymphocytic inflammation with surrounding fibroplasia with overlapping features of FFA and central centrifugal cicatricial alopecia (Figure 4). Biopsy of the neck revealed vacuolar interface dermatitis. Additionally, biopsy of a facial papule revealed lichenoid inflammation involving a vellus hair follicle. Clinical and histopathological correlation confirmed the diagnosis of FFA with LPP and facial papules.
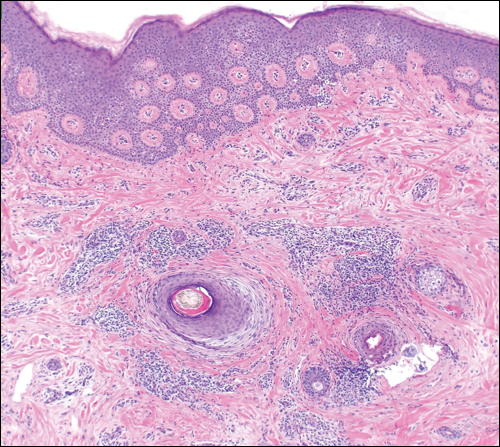
Comment
Current understanding of FFA as a progressive, lymphocytic, scarring alopecia has expanded in recent years. Clinical observation suggests that the incidence of FFA is increasing4; however more epidemiologic data are needed. Frontal fibrosing alopecia presents clinically with symmetrical frontotemporal hair loss with lone hairs. Trichoscopy reveals perifollicular hyperkeratosis, perifollicular erythema, and follicular plugging in 72%, 66%, and 44% of cases, respectively.5 In one study (N=242), patients were classified into 3 clinical patterns of FFA: pattern I (linear) showed bandlike loss of frontal hair with normal density directly behind the hairline; pattern II (diffuse) showed loss of density behind the frontal hairline; and pattern III (double line) showed a pseudo–“fringe sign” appearance. The majority of patients were classified as either pattern I or II, with pattern II predicting a poorer prognosis.6
rontal fibrosing alopecia is increasingly recognized in men, with prevalence as high as 5%.1 Facial hair involvement, particularly of the upper lip and sideburns, is an important consideration in men.7 Most studies suggest that 80% to 90% of affected women are postmenopausal,8 though a case series presented by Dlova1 identified 27% of affected women as postmenopausal. The coexistence of premature menopause and hysterectomy in FFA patients suggests a hormonal contribution, but this association is still poorly understood.8 Epidemiologic data on ethnicity in FFA are sparse but suggest that white individuals are more likely to be affected. Frontal fibrosing alopecia also may be misdiagnosed as traction alopecia in Hispanic and black patients.8
It is prudent for physicians to assess for and recognize clinical clues to severe forms of FFA. A 2014 multicenter review of 355 patients identified 3 clinical entities that predicted more severe forms of FFA: eyelash loss (madarosis), loss of body hair, and facial papules.8 Madarosis occurs due to perifollicular inflammation and fibrosis of eyelash hair follicles. Similarly, perifollicular inflammation of body hair was present in 24% of patients (N=86), most commonly of the axillary and pubic hair. Facial papules form due to facial vellus hair inflammation and fibrosis and were identified in 14% of patients (N=49).8 These clinical findings may allow providers to predict more extensive clinical involvement of FFA.
Frontal fibrosing alopecia and LPP occur concomitantly in up 54% of patients, more commonly in darker-skinned patients.1,9,10 Lichen planus pigmentosus frequently occurs on the face and neck, most commonly in a diffuse pattern, though reticulated and macular patterns also have been identified.11 In some patients, LPP precedes the development of FFA and may be useful as a herald sign1; therefore, it is important for dermatologists to evaluate for signs of FFA when evaluating those with LPP. Thorough evaluation in patients with skin of color also is important because FFA may be misdiagnosed as traction alopecia.
Additional cutaneous associations of FFA include eyebrow loss, glabellar red dots, and prominent frontal veins. Eyebrow loss occurs secondary to fibrosis of eyebrow hair follicles and has been found in 40% to 80% of patients with FFA; it is thought to be associated with milder forms of FFA.8 Glabellar red dots correlate with histopathologic lymphocytic inflammation of vellus hair follicles.12 Additionally, frontal vein prominence has been described in FFA and is thought to be secondary to atrophy in this scarring process, perhaps worsened by local steroid treatments.13 Mucocutaneous lichen planus, rosacea, thyroid disease, vitiligo, and other autoimmune disorders also have been reported in patients with FFA.14
Conclusion
Concomitant FFA, LPP, and facial papules have been rarely reported and exemplify the spectrum of cutaneous associations with FFA, particularly in individuals with skin of color. Clinical variants and associations of FFA are broad, including predictors of poorer prognosis such as eyelash loss and vellus hair involvement seen as facial papules. Lichen planus pigmentosus is well described in association with FFA and may serve as a herald sign that frontal hair loss should not be mistaken for traction alopecia in early stages. Eyebrow loss is thought to represent milder disease. It is important for dermatologists to be aware of these findings to understand the breadth of this disease and for appropriate evaluation and management of patients with FFA.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-432.
- Donati A, Molina L, Doche I, et al. Facial papules in frontal fibrosing alopecia: evidence of vellus follicle involvement. Arch Dermatol. 2011;147:1424-1427.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Rudnicka L, Rakowska A. The increasing incidence of frontal fibrosing alopecia. in search of triggering factors. J Eur Acad Dermatol Venereol. 2017;31:1579-1580.
- Toledo-Pastrana T, Hernández MJ, Camacho Martínez FM. Perifollicular erythema as a trichoscopy sign of progression in frontal fibrosing alopecia. Int J Trichology. 2013;5:151-153.
- Moreno-Arrones OM, Saceda-Corralo D, Fonda-Pascual P, et al. Frontal fibrosing alopecia: clinical and prognostic classification. J Eur Acad Dermatol Venereol. 2017;31:1739-1745.
- Tolkachjov SN, Chaudhry HM, Camilleri MJ, et al. Frontal fibrosing alopecia among men: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2017;77:683-690.e2.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Berliner JG, McCalmont TH, Price VH, et al. Frontal fibrosing alopecia and lichen planus pigmentosus. J Am Acad Dermatol. 2014;71:E26-E27.
- Rao R, Sarda A, Khanna R, et al. Coexistence of frontal fibrosing alopecia with lichen planus pigmentosus. Int J Dermatol. 2014;53:622-624.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Pirmez R, Donati A, Valente NS, et al. Glabellar red dots in frontal fibrosing alopecia: a further clinical sign of vellus follicle involvement. Br J Dermatol. 2014;170:745-746.
- Vañó-Galván S, Rodrigues-Barata AR, Urech M, et al. Depression of the frontal veins: a new clinical sign of frontal fibrosing alopecia. J Am Acad Dermatol. 2015;72:1087-1088.
- Pindado-Ortega C, Saceda-Corralo D, Buendía-Castaño D, et al. Frontal fibrosing alopecia and cutaneous comorbidities: a potential relationship with rosacea. J Am Acad Dermatol. 2018;78:596-597.e1.
Frontal fibrosing alopecia (FFA) has been reported in association with lichen planus pigmentosus (LPP) and facial papules.1-3 Lichen planus pigmentosus is a variant of lichen planus that causes hyperpigmentation of the face, neck, and/or intertriginous areas that may be useful as a clinical indicator in the development of FFA.1 Facial papules in association with FFA are secondary to fibrosed vellus hairs.2,3 Currently, reports of concomitant FFA, LPP, and facial papules in women with skin of color are limited in the literature. This case series includes 5 women of color (Hispanic and black) who presented to our clinic with FFA and various cutaneous associations. A review of the current literature on cutaneous associations of FFA also is provided.
Case Reports
Patient 1
A 50-year-old Hispanic woman who was previously presumed to have melasma by an outside physician presented with pruritus of the scalp and eyebrows of 1 month’s duration. Physical examination revealed decreased frontal scalp hair density with perifollicular erythema and scale with thinning of the lateral eyebrows. Hyperpigmented coalesced macules (Figure 1A) and erythematous perifollicular papules were noted along the temples and on the perioral skin. Depressed forehead and temporal veins also were noted (Figure 1B). A biopsy of the scalp demonstrated perifollicular and perivascular lymphocytic inflammation and fibrosed hair follicles, and a biopsy of the perioral skin demonstrated perivascular lymphocytic inflammation with melanophages in the papillary dermis. A diagnosis of FFA with LPP was established with these biopsies.
Patient 2
A 61-year-old black woman presented with asymptomatic hair loss along the frontal hairline for an unknown duration. On physical examination the frontal scalp and lateral eyebrows demonstrated decreased hair density with loss of follicular ostia. Fine, flesh-colored, monomorphic papules were scattered along the forehead and temples, and ill-defined brown pigmentation was present along the forehead, temples, and cheeks. Biopsy of the frontal scalp demonstrated patchy lichenoid inflammation with decreased number of follicles with replacement by follicular scars, confirming the diagnosis of FFA.
Patient 3
A 47-year-old Hispanic woman presented with hair loss of the frontal scalp and bilateral eyebrows with associated burning of 2 years’ duration. Physical examination demonstrated recession of the frontotemporal hairline with scattered lone hairs and thinning of the eyebrows. Innumerable flesh-colored papules were present on the forehead and temples (Figure 2A). Glabellar and eyebrow erythema was noted (Figure 2B). Biopsy of the frontal scalp demonstrated decreased terminal anagen hair follicles with perifollicular lymphoid infiltrate and fibrosis, consistent with a diagnosis of FFA. The patient was started on oral hydroxychloroquine 400 mg once daily, and 3 months later hyperpigmentation of the forehead and perioral skin was noted. The patient reported that she had facial hyperpigmentation prior to starting hydroxychloroquine and declined a biopsy.
Patient 4
A 40-year-old black woman presented with brown pruritic macles of the face, neck, arms, and forearms of 4 years’ duration. She also reported hair loss on the frontal and occipital scalp, eyebrows, and arms. On physical examination, ill-defined brown macules and patches were noted on the neck (Figure 3), face, arms, and forearms. Decreased hair density was noted on the frontal and occipital scalp with follicular dropout and perifollicular hyperpigmentation. Biopsy of the scalp demonstrated perivascular lymphocytic inflammation with sparse anagen follicles and fibrous tracts, and biopsy of the neck revealed superficial perivascular inflammation with numerous melanophages in the upper dermis; these histopathologic findings were consistent with FFA and LPP, respectively.
Patient 5
A 46-year-old black woman with history of hair loss presented with hyperpigmentation of the face and neck of 2 years’ duration. On physical examination decreased hair density of the frontal and vertex scalp and lateral eyebrows was noted. Flesh-colored papules were noted on the forehead and cheeks, and confluent dark brown patches were present on the temples and neck. Three punch biopsies were performed. Biopsy of the scalp revealed lymphocytic inflammation with surrounding fibroplasia with overlapping features of FFA and central centrifugal cicatricial alopecia (Figure 4). Biopsy of the neck revealed vacuolar interface dermatitis. Additionally, biopsy of a facial papule revealed lichenoid inflammation involving a vellus hair follicle. Clinical and histopathological correlation confirmed the diagnosis of FFA with LPP and facial papules.
Comment
Current understanding of FFA as a progressive, lymphocytic, scarring alopecia has expanded in recent years. Clinical observation suggests that the incidence of FFA is increasing4; however more epidemiologic data are needed. Frontal fibrosing alopecia presents clinically with symmetrical frontotemporal hair loss with lone hairs. Trichoscopy reveals perifollicular hyperkeratosis, perifollicular erythema, and follicular plugging in 72%, 66%, and 44% of cases, respectively.5 In one study (N=242), patients were classified into 3 clinical patterns of FFA: pattern I (linear) showed bandlike loss of frontal hair with normal density directly behind the hairline; pattern II (diffuse) showed loss of density behind the frontal hairline; and pattern III (double line) showed a pseudo–“fringe sign” appearance. The majority of patients were classified as either pattern I or II, with pattern II predicting a poorer prognosis.6
rontal fibrosing alopecia is increasingly recognized in men, with prevalence as high as 5%.1 Facial hair involvement, particularly of the upper lip and sideburns, is an important consideration in men.7 Most studies suggest that 80% to 90% of affected women are postmenopausal,8 though a case series presented by Dlova1 identified 27% of affected women as postmenopausal. The coexistence of premature menopause and hysterectomy in FFA patients suggests a hormonal contribution, but this association is still poorly understood.8 Epidemiologic data on ethnicity in FFA are sparse but suggest that white individuals are more likely to be affected. Frontal fibrosing alopecia also may be misdiagnosed as traction alopecia in Hispanic and black patients.8
It is prudent for physicians to assess for and recognize clinical clues to severe forms of FFA. A 2014 multicenter review of 355 patients identified 3 clinical entities that predicted more severe forms of FFA: eyelash loss (madarosis), loss of body hair, and facial papules.8 Madarosis occurs due to perifollicular inflammation and fibrosis of eyelash hair follicles. Similarly, perifollicular inflammation of body hair was present in 24% of patients (N=86), most commonly of the axillary and pubic hair. Facial papules form due to facial vellus hair inflammation and fibrosis and were identified in 14% of patients (N=49).8 These clinical findings may allow providers to predict more extensive clinical involvement of FFA.
Frontal fibrosing alopecia and LPP occur concomitantly in up 54% of patients, more commonly in darker-skinned patients.1,9,10 Lichen planus pigmentosus frequently occurs on the face and neck, most commonly in a diffuse pattern, though reticulated and macular patterns also have been identified.11 In some patients, LPP precedes the development of FFA and may be useful as a herald sign1; therefore, it is important for dermatologists to evaluate for signs of FFA when evaluating those with LPP. Thorough evaluation in patients with skin of color also is important because FFA may be misdiagnosed as traction alopecia.
Additional cutaneous associations of FFA include eyebrow loss, glabellar red dots, and prominent frontal veins. Eyebrow loss occurs secondary to fibrosis of eyebrow hair follicles and has been found in 40% to 80% of patients with FFA; it is thought to be associated with milder forms of FFA.8 Glabellar red dots correlate with histopathologic lymphocytic inflammation of vellus hair follicles.12 Additionally, frontal vein prominence has been described in FFA and is thought to be secondary to atrophy in this scarring process, perhaps worsened by local steroid treatments.13 Mucocutaneous lichen planus, rosacea, thyroid disease, vitiligo, and other autoimmune disorders also have been reported in patients with FFA.14
Conclusion
Concomitant FFA, LPP, and facial papules have been rarely reported and exemplify the spectrum of cutaneous associations with FFA, particularly in individuals with skin of color. Clinical variants and associations of FFA are broad, including predictors of poorer prognosis such as eyelash loss and vellus hair involvement seen as facial papules. Lichen planus pigmentosus is well described in association with FFA and may serve as a herald sign that frontal hair loss should not be mistaken for traction alopecia in early stages. Eyebrow loss is thought to represent milder disease. It is important for dermatologists to be aware of these findings to understand the breadth of this disease and for appropriate evaluation and management of patients with FFA.
Frontal fibrosing alopecia (FFA) has been reported in association with lichen planus pigmentosus (LPP) and facial papules.1-3 Lichen planus pigmentosus is a variant of lichen planus that causes hyperpigmentation of the face, neck, and/or intertriginous areas that may be useful as a clinical indicator in the development of FFA.1 Facial papules in association with FFA are secondary to fibrosed vellus hairs.2,3 Currently, reports of concomitant FFA, LPP, and facial papules in women with skin of color are limited in the literature. This case series includes 5 women of color (Hispanic and black) who presented to our clinic with FFA and various cutaneous associations. A review of the current literature on cutaneous associations of FFA also is provided.
Case Reports
Patient 1
A 50-year-old Hispanic woman who was previously presumed to have melasma by an outside physician presented with pruritus of the scalp and eyebrows of 1 month’s duration. Physical examination revealed decreased frontal scalp hair density with perifollicular erythema and scale with thinning of the lateral eyebrows. Hyperpigmented coalesced macules (Figure 1A) and erythematous perifollicular papules were noted along the temples and on the perioral skin. Depressed forehead and temporal veins also were noted (Figure 1B). A biopsy of the scalp demonstrated perifollicular and perivascular lymphocytic inflammation and fibrosed hair follicles, and a biopsy of the perioral skin demonstrated perivascular lymphocytic inflammation with melanophages in the papillary dermis. A diagnosis of FFA with LPP was established with these biopsies.
Patient 2
A 61-year-old black woman presented with asymptomatic hair loss along the frontal hairline for an unknown duration. On physical examination the frontal scalp and lateral eyebrows demonstrated decreased hair density with loss of follicular ostia. Fine, flesh-colored, monomorphic papules were scattered along the forehead and temples, and ill-defined brown pigmentation was present along the forehead, temples, and cheeks. Biopsy of the frontal scalp demonstrated patchy lichenoid inflammation with decreased number of follicles with replacement by follicular scars, confirming the diagnosis of FFA.
Patient 3
A 47-year-old Hispanic woman presented with hair loss of the frontal scalp and bilateral eyebrows with associated burning of 2 years’ duration. Physical examination demonstrated recession of the frontotemporal hairline with scattered lone hairs and thinning of the eyebrows. Innumerable flesh-colored papules were present on the forehead and temples (Figure 2A). Glabellar and eyebrow erythema was noted (Figure 2B). Biopsy of the frontal scalp demonstrated decreased terminal anagen hair follicles with perifollicular lymphoid infiltrate and fibrosis, consistent with a diagnosis of FFA. The patient was started on oral hydroxychloroquine 400 mg once daily, and 3 months later hyperpigmentation of the forehead and perioral skin was noted. The patient reported that she had facial hyperpigmentation prior to starting hydroxychloroquine and declined a biopsy.
Patient 4
A 40-year-old black woman presented with brown pruritic macles of the face, neck, arms, and forearms of 4 years’ duration. She also reported hair loss on the frontal and occipital scalp, eyebrows, and arms. On physical examination, ill-defined brown macules and patches were noted on the neck (Figure 3), face, arms, and forearms. Decreased hair density was noted on the frontal and occipital scalp with follicular dropout and perifollicular hyperpigmentation. Biopsy of the scalp demonstrated perivascular lymphocytic inflammation with sparse anagen follicles and fibrous tracts, and biopsy of the neck revealed superficial perivascular inflammation with numerous melanophages in the upper dermis; these histopathologic findings were consistent with FFA and LPP, respectively.
Patient 5
A 46-year-old black woman with history of hair loss presented with hyperpigmentation of the face and neck of 2 years’ duration. On physical examination decreased hair density of the frontal and vertex scalp and lateral eyebrows was noted. Flesh-colored papules were noted on the forehead and cheeks, and confluent dark brown patches were present on the temples and neck. Three punch biopsies were performed. Biopsy of the scalp revealed lymphocytic inflammation with surrounding fibroplasia with overlapping features of FFA and central centrifugal cicatricial alopecia (Figure 4). Biopsy of the neck revealed vacuolar interface dermatitis. Additionally, biopsy of a facial papule revealed lichenoid inflammation involving a vellus hair follicle. Clinical and histopathological correlation confirmed the diagnosis of FFA with LPP and facial papules.
Comment
Current understanding of FFA as a progressive, lymphocytic, scarring alopecia has expanded in recent years. Clinical observation suggests that the incidence of FFA is increasing4; however more epidemiologic data are needed. Frontal fibrosing alopecia presents clinically with symmetrical frontotemporal hair loss with lone hairs. Trichoscopy reveals perifollicular hyperkeratosis, perifollicular erythema, and follicular plugging in 72%, 66%, and 44% of cases, respectively.5 In one study (N=242), patients were classified into 3 clinical patterns of FFA: pattern I (linear) showed bandlike loss of frontal hair with normal density directly behind the hairline; pattern II (diffuse) showed loss of density behind the frontal hairline; and pattern III (double line) showed a pseudo–“fringe sign” appearance. The majority of patients were classified as either pattern I or II, with pattern II predicting a poorer prognosis.6
rontal fibrosing alopecia is increasingly recognized in men, with prevalence as high as 5%.1 Facial hair involvement, particularly of the upper lip and sideburns, is an important consideration in men.7 Most studies suggest that 80% to 90% of affected women are postmenopausal,8 though a case series presented by Dlova1 identified 27% of affected women as postmenopausal. The coexistence of premature menopause and hysterectomy in FFA patients suggests a hormonal contribution, but this association is still poorly understood.8 Epidemiologic data on ethnicity in FFA are sparse but suggest that white individuals are more likely to be affected. Frontal fibrosing alopecia also may be misdiagnosed as traction alopecia in Hispanic and black patients.8
It is prudent for physicians to assess for and recognize clinical clues to severe forms of FFA. A 2014 multicenter review of 355 patients identified 3 clinical entities that predicted more severe forms of FFA: eyelash loss (madarosis), loss of body hair, and facial papules.8 Madarosis occurs due to perifollicular inflammation and fibrosis of eyelash hair follicles. Similarly, perifollicular inflammation of body hair was present in 24% of patients (N=86), most commonly of the axillary and pubic hair. Facial papules form due to facial vellus hair inflammation and fibrosis and were identified in 14% of patients (N=49).8 These clinical findings may allow providers to predict more extensive clinical involvement of FFA.
Frontal fibrosing alopecia and LPP occur concomitantly in up 54% of patients, more commonly in darker-skinned patients.1,9,10 Lichen planus pigmentosus frequently occurs on the face and neck, most commonly in a diffuse pattern, though reticulated and macular patterns also have been identified.11 In some patients, LPP precedes the development of FFA and may be useful as a herald sign1; therefore, it is important for dermatologists to evaluate for signs of FFA when evaluating those with LPP. Thorough evaluation in patients with skin of color also is important because FFA may be misdiagnosed as traction alopecia.
Additional cutaneous associations of FFA include eyebrow loss, glabellar red dots, and prominent frontal veins. Eyebrow loss occurs secondary to fibrosis of eyebrow hair follicles and has been found in 40% to 80% of patients with FFA; it is thought to be associated with milder forms of FFA.8 Glabellar red dots correlate with histopathologic lymphocytic inflammation of vellus hair follicles.12 Additionally, frontal vein prominence has been described in FFA and is thought to be secondary to atrophy in this scarring process, perhaps worsened by local steroid treatments.13 Mucocutaneous lichen planus, rosacea, thyroid disease, vitiligo, and other autoimmune disorders also have been reported in patients with FFA.14
Conclusion
Concomitant FFA, LPP, and facial papules have been rarely reported and exemplify the spectrum of cutaneous associations with FFA, particularly in individuals with skin of color. Clinical variants and associations of FFA are broad, including predictors of poorer prognosis such as eyelash loss and vellus hair involvement seen as facial papules. Lichen planus pigmentosus is well described in association with FFA and may serve as a herald sign that frontal hair loss should not be mistaken for traction alopecia in early stages. Eyebrow loss is thought to represent milder disease. It is important for dermatologists to be aware of these findings to understand the breadth of this disease and for appropriate evaluation and management of patients with FFA.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-432.
- Donati A, Molina L, Doche I, et al. Facial papules in frontal fibrosing alopecia: evidence of vellus follicle involvement. Arch Dermatol. 2011;147:1424-1427.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Rudnicka L, Rakowska A. The increasing incidence of frontal fibrosing alopecia. in search of triggering factors. J Eur Acad Dermatol Venereol. 2017;31:1579-1580.
- Toledo-Pastrana T, Hernández MJ, Camacho Martínez FM. Perifollicular erythema as a trichoscopy sign of progression in frontal fibrosing alopecia. Int J Trichology. 2013;5:151-153.
- Moreno-Arrones OM, Saceda-Corralo D, Fonda-Pascual P, et al. Frontal fibrosing alopecia: clinical and prognostic classification. J Eur Acad Dermatol Venereol. 2017;31:1739-1745.
- Tolkachjov SN, Chaudhry HM, Camilleri MJ, et al. Frontal fibrosing alopecia among men: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2017;77:683-690.e2.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Berliner JG, McCalmont TH, Price VH, et al. Frontal fibrosing alopecia and lichen planus pigmentosus. J Am Acad Dermatol. 2014;71:E26-E27.
- Rao R, Sarda A, Khanna R, et al. Coexistence of frontal fibrosing alopecia with lichen planus pigmentosus. Int J Dermatol. 2014;53:622-624.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Pirmez R, Donati A, Valente NS, et al. Glabellar red dots in frontal fibrosing alopecia: a further clinical sign of vellus follicle involvement. Br J Dermatol. 2014;170:745-746.
- Vañó-Galván S, Rodrigues-Barata AR, Urech M, et al. Depression of the frontal veins: a new clinical sign of frontal fibrosing alopecia. J Am Acad Dermatol. 2015;72:1087-1088.
- Pindado-Ortega C, Saceda-Corralo D, Buendía-Castaño D, et al. Frontal fibrosing alopecia and cutaneous comorbidities: a potential relationship with rosacea. J Am Acad Dermatol. 2018;78:596-597.e1.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-432.
- Donati A, Molina L, Doche I, et al. Facial papules in frontal fibrosing alopecia: evidence of vellus follicle involvement. Arch Dermatol. 2011;147:1424-1427.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Rudnicka L, Rakowska A. The increasing incidence of frontal fibrosing alopecia. in search of triggering factors. J Eur Acad Dermatol Venereol. 2017;31:1579-1580.
- Toledo-Pastrana T, Hernández MJ, Camacho Martínez FM. Perifollicular erythema as a trichoscopy sign of progression in frontal fibrosing alopecia. Int J Trichology. 2013;5:151-153.
- Moreno-Arrones OM, Saceda-Corralo D, Fonda-Pascual P, et al. Frontal fibrosing alopecia: clinical and prognostic classification. J Eur Acad Dermatol Venereol. 2017;31:1739-1745.
- Tolkachjov SN, Chaudhry HM, Camilleri MJ, et al. Frontal fibrosing alopecia among men: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2017;77:683-690.e2.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Berliner JG, McCalmont TH, Price VH, et al. Frontal fibrosing alopecia and lichen planus pigmentosus. J Am Acad Dermatol. 2014;71:E26-E27.
- Rao R, Sarda A, Khanna R, et al. Coexistence of frontal fibrosing alopecia with lichen planus pigmentosus. Int J Dermatol. 2014;53:622-624.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Pirmez R, Donati A, Valente NS, et al. Glabellar red dots in frontal fibrosing alopecia: a further clinical sign of vellus follicle involvement. Br J Dermatol. 2014;170:745-746.
- Vañó-Galván S, Rodrigues-Barata AR, Urech M, et al. Depression of the frontal veins: a new clinical sign of frontal fibrosing alopecia. J Am Acad Dermatol. 2015;72:1087-1088.
- Pindado-Ortega C, Saceda-Corralo D, Buendía-Castaño D, et al. Frontal fibrosing alopecia and cutaneous comorbidities: a potential relationship with rosacea. J Am Acad Dermatol. 2018;78:596-597.e1.
Practice Points
- Frontal fibrosing alopecia (FFA) is associated with lichen planus pigmentosus, especially in patients with skin of color.
- Patients with FFA should be evaluated for additional cutaneous features including facial papules, glabellar red dots, and depressed frontal veins.