User login
Changing Public Perception of Vitiligo


Bilateral Brown Plaques Behind the Ears
The Diagnosis: Terra Firma-Forme Dermatosis
Terra firma-forme dermatosis (TFFD), also known as Duncan dirty dermatosis, is an idiopathic benign cutaneous condition that is easily misdiagnosed or mismanaged. In 1987, Duncan et al1 first described the condition in children who had mothers that lamented over dirty skin spots that could not be washed off. The term terra firma translates in Latin to solid ground, which describes the characteristic dirtlike appearance of these lesions.
Terra firma-forme dermatosis most commonly affects children and young adults, though it can present in patients of any age without any known predisposing risk factors.1-4 The lesions have a predilection for the face, neck, shoulders, trunk, and ankles. Terra firma-forme dermatosis has no association with bathing and hygiene habits, and most patients describe unsuccessful removal of the lesions, even after vigorous scrubbing with soaps and detergents at home. The lesions are asymptomatic, and many patients present to dermatology for cosmetic concerns.1-8
The etiology of TFFD is not well understood and is considered a retention hyperkeratosis. Duncan et al1 postulated that TFFD is the result of partial or improper maturation of keratinocytes leading to keratinocyte and melanin retention. Hematoxylin and eosin stains demonstrate lamellar hyperkeratosis of the stratum corneum without parakeratosis as well as keratin pearls scattered throughout. Mild acanthosis and papillomatosis also have been reported.1,5-7 Fontana-Masson stain shows excess melanin in these lesions, extending from the basal layer to the stratum corneum. Fungal and bacterial stains as well as cultures often have no notable findings.1,7 Similarly, histopathologic examination of our patient's biopsy with hematoxylin and eosin stain revealed hyperorthokeratosis with scattered naked vellus hair shafts and incidental yeast forms (Figure 1).
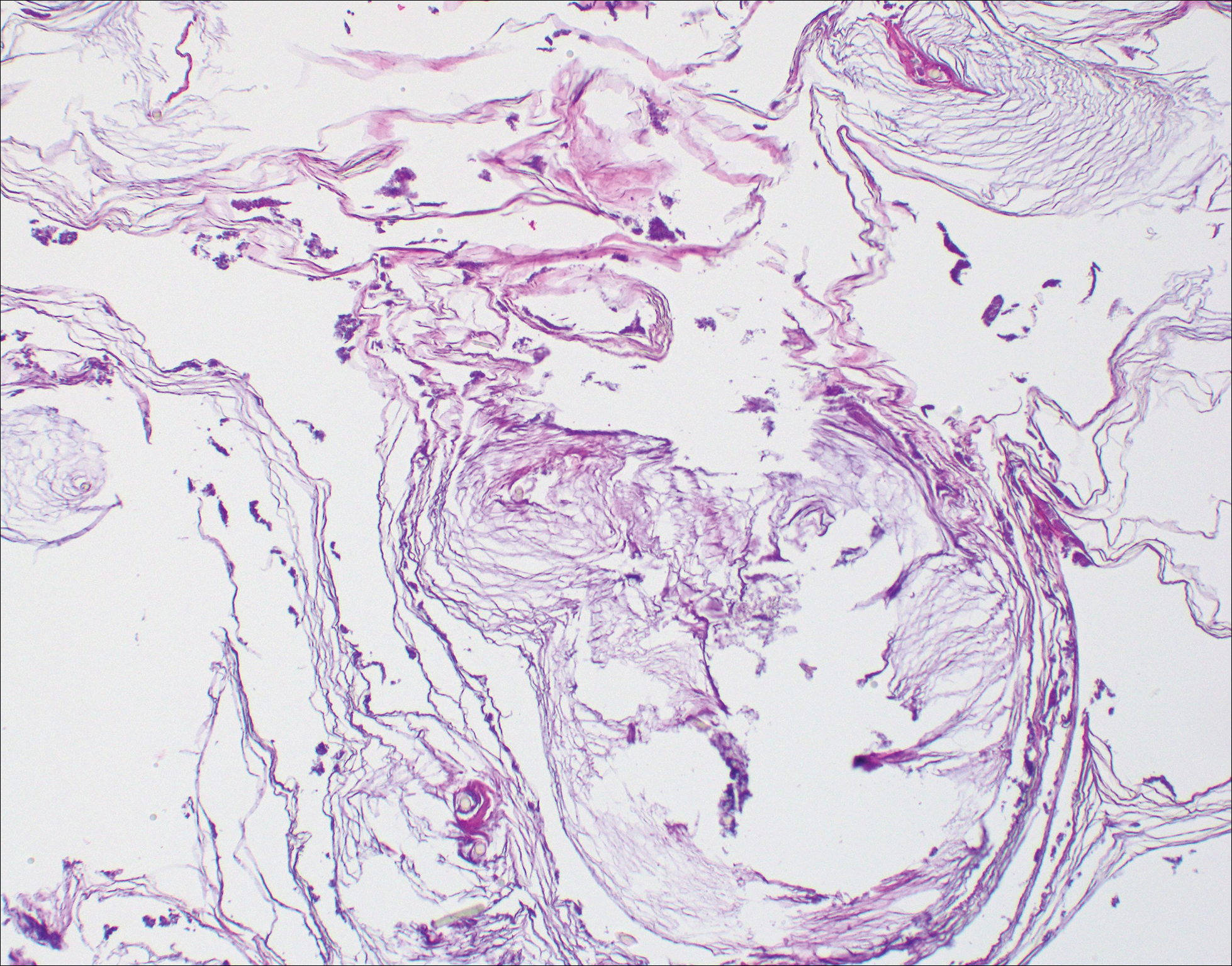
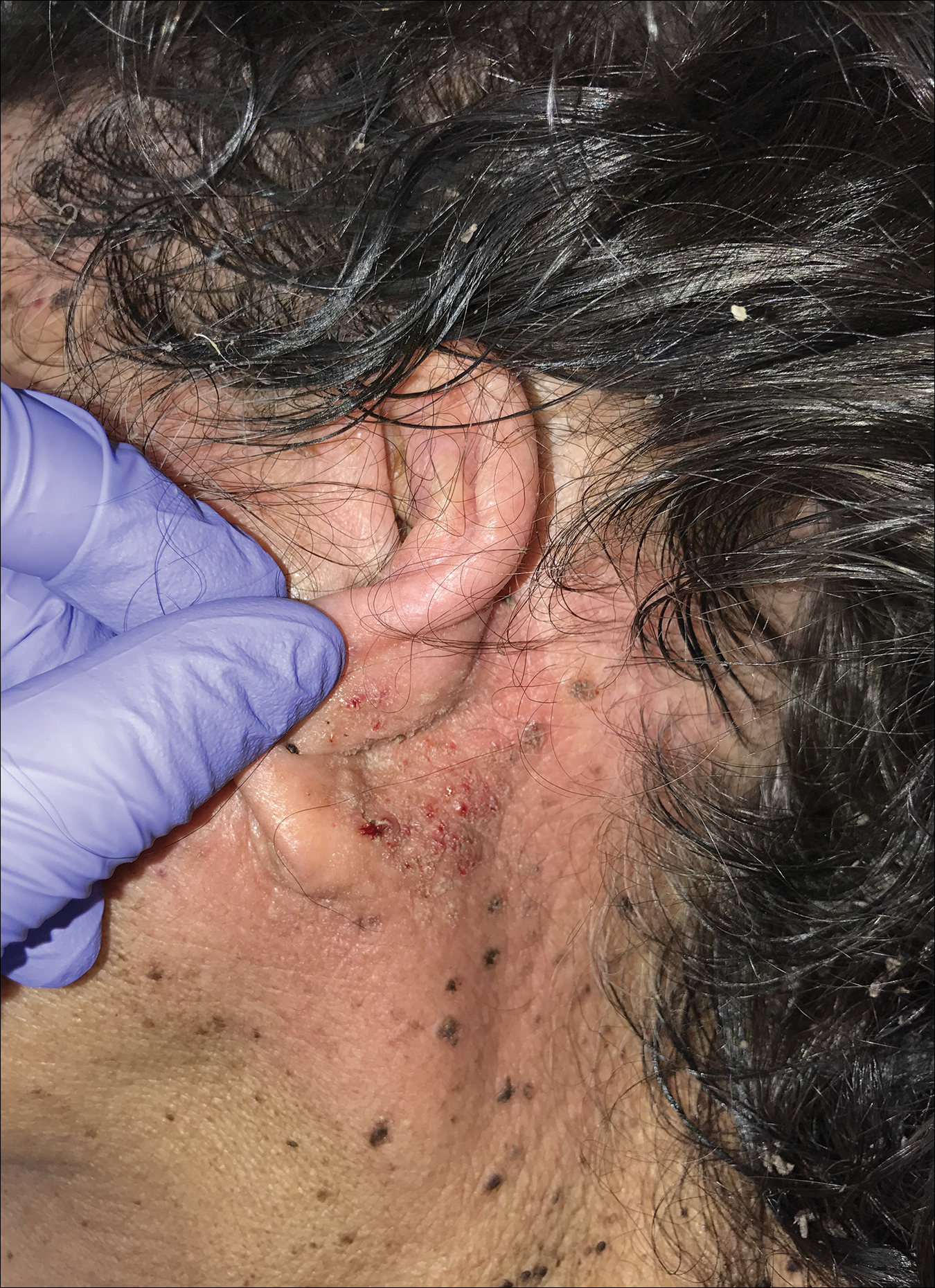
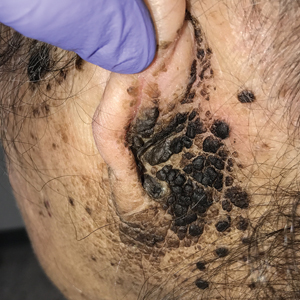
The differential diagnosis for TFFD may include pityriasis versicolor, confluent and reticulated papillomatosis, acanthosis nigricans, ichthyosis, malignant melanoma, and seborrheic keratosis. All of these diagnoses can be ruled out by the easy removal of the lesions with isopropyl alcohol 70%, which was performed on our patient by scrubbing the lesions with soaked gauze (Figure 2). Indeed, removal with isopropyl alcohol 70% is both the therapeutic and diagnostic procedure for TFFD.1-8 Of note, dermatitis neglecta is histologically and clinically identical to TFFD, albeit with a history of uncleanly habits or exposure to dirty environments.
The diagnosis of TFFD often is discovered incidentally as physicians wipe the area with alcohol to prepare for biopsy.1 Occasionally, vigorous scrubbing is needed to completely remove the lesions, and without this effort the lesions may be easily mistaken for another cutaneous process.3 Failure to consider TFFD as a diagnosis has led to unnecessary endocrine workups and invasive biopsies.4 Therefore, physicians should have early clinical suspicion of TFFD and be aware of the bedside diagnostic procedure using isopropyl alcohol.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan's dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Moon J, Kim MW, Yoon HS, et al. A case of terra firma-forme dermatosis: differentiation from other dirty-appearing diseases. Ann Dermatol. 2016;28:413-415.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Chun SW, Lee SY, Kim JB, et al. A case of terra firma-forme dermatosis treated with salicylic acid alcohol peeling. Ann Dermatol. 2017;29:83-85.
- Aslan NC, Guler S, Demirci K, et al. Features of terra firma-forme dermatosis. Ann Fam Med. 2018;16:52-54.
The Diagnosis: Terra Firma-Forme Dermatosis
Terra firma-forme dermatosis (TFFD), also known as Duncan dirty dermatosis, is an idiopathic benign cutaneous condition that is easily misdiagnosed or mismanaged. In 1987, Duncan et al1 first described the condition in children who had mothers that lamented over dirty skin spots that could not be washed off. The term terra firma translates in Latin to solid ground, which describes the characteristic dirtlike appearance of these lesions.
Terra firma-forme dermatosis most commonly affects children and young adults, though it can present in patients of any age without any known predisposing risk factors.1-4 The lesions have a predilection for the face, neck, shoulders, trunk, and ankles. Terra firma-forme dermatosis has no association with bathing and hygiene habits, and most patients describe unsuccessful removal of the lesions, even after vigorous scrubbing with soaps and detergents at home. The lesions are asymptomatic, and many patients present to dermatology for cosmetic concerns.1-8
The etiology of TFFD is not well understood and is considered a retention hyperkeratosis. Duncan et al1 postulated that TFFD is the result of partial or improper maturation of keratinocytes leading to keratinocyte and melanin retention. Hematoxylin and eosin stains demonstrate lamellar hyperkeratosis of the stratum corneum without parakeratosis as well as keratin pearls scattered throughout. Mild acanthosis and papillomatosis also have been reported.1,5-7 Fontana-Masson stain shows excess melanin in these lesions, extending from the basal layer to the stratum corneum. Fungal and bacterial stains as well as cultures often have no notable findings.1,7 Similarly, histopathologic examination of our patient's biopsy with hematoxylin and eosin stain revealed hyperorthokeratosis with scattered naked vellus hair shafts and incidental yeast forms (Figure 1).
The differential diagnosis for TFFD may include pityriasis versicolor, confluent and reticulated papillomatosis, acanthosis nigricans, ichthyosis, malignant melanoma, and seborrheic keratosis. All of these diagnoses can be ruled out by the easy removal of the lesions with isopropyl alcohol 70%, which was performed on our patient by scrubbing the lesions with soaked gauze (Figure 2). Indeed, removal with isopropyl alcohol 70% is both the therapeutic and diagnostic procedure for TFFD.1-8 Of note, dermatitis neglecta is histologically and clinically identical to TFFD, albeit with a history of uncleanly habits or exposure to dirty environments.
The diagnosis of TFFD often is discovered incidentally as physicians wipe the area with alcohol to prepare for biopsy.1 Occasionally, vigorous scrubbing is needed to completely remove the lesions, and without this effort the lesions may be easily mistaken for another cutaneous process.3 Failure to consider TFFD as a diagnosis has led to unnecessary endocrine workups and invasive biopsies.4 Therefore, physicians should have early clinical suspicion of TFFD and be aware of the bedside diagnostic procedure using isopropyl alcohol.
The Diagnosis: Terra Firma-Forme Dermatosis
Terra firma-forme dermatosis (TFFD), also known as Duncan dirty dermatosis, is an idiopathic benign cutaneous condition that is easily misdiagnosed or mismanaged. In 1987, Duncan et al1 first described the condition in children who had mothers that lamented over dirty skin spots that could not be washed off. The term terra firma translates in Latin to solid ground, which describes the characteristic dirtlike appearance of these lesions.
Terra firma-forme dermatosis most commonly affects children and young adults, though it can present in patients of any age without any known predisposing risk factors.1-4 The lesions have a predilection for the face, neck, shoulders, trunk, and ankles. Terra firma-forme dermatosis has no association with bathing and hygiene habits, and most patients describe unsuccessful removal of the lesions, even after vigorous scrubbing with soaps and detergents at home. The lesions are asymptomatic, and many patients present to dermatology for cosmetic concerns.1-8
The etiology of TFFD is not well understood and is considered a retention hyperkeratosis. Duncan et al1 postulated that TFFD is the result of partial or improper maturation of keratinocytes leading to keratinocyte and melanin retention. Hematoxylin and eosin stains demonstrate lamellar hyperkeratosis of the stratum corneum without parakeratosis as well as keratin pearls scattered throughout. Mild acanthosis and papillomatosis also have been reported.1,5-7 Fontana-Masson stain shows excess melanin in these lesions, extending from the basal layer to the stratum corneum. Fungal and bacterial stains as well as cultures often have no notable findings.1,7 Similarly, histopathologic examination of our patient's biopsy with hematoxylin and eosin stain revealed hyperorthokeratosis with scattered naked vellus hair shafts and incidental yeast forms (Figure 1).
The differential diagnosis for TFFD may include pityriasis versicolor, confluent and reticulated papillomatosis, acanthosis nigricans, ichthyosis, malignant melanoma, and seborrheic keratosis. All of these diagnoses can be ruled out by the easy removal of the lesions with isopropyl alcohol 70%, which was performed on our patient by scrubbing the lesions with soaked gauze (Figure 2). Indeed, removal with isopropyl alcohol 70% is both the therapeutic and diagnostic procedure for TFFD.1-8 Of note, dermatitis neglecta is histologically and clinically identical to TFFD, albeit with a history of uncleanly habits or exposure to dirty environments.
The diagnosis of TFFD often is discovered incidentally as physicians wipe the area with alcohol to prepare for biopsy.1 Occasionally, vigorous scrubbing is needed to completely remove the lesions, and without this effort the lesions may be easily mistaken for another cutaneous process.3 Failure to consider TFFD as a diagnosis has led to unnecessary endocrine workups and invasive biopsies.4 Therefore, physicians should have early clinical suspicion of TFFD and be aware of the bedside diagnostic procedure using isopropyl alcohol.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan's dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Moon J, Kim MW, Yoon HS, et al. A case of terra firma-forme dermatosis: differentiation from other dirty-appearing diseases. Ann Dermatol. 2016;28:413-415.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Chun SW, Lee SY, Kim JB, et al. A case of terra firma-forme dermatosis treated with salicylic acid alcohol peeling. Ann Dermatol. 2017;29:83-85.
- Aslan NC, Guler S, Demirci K, et al. Features of terra firma-forme dermatosis. Ann Fam Med. 2018;16:52-54.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan's dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Moon J, Kim MW, Yoon HS, et al. A case of terra firma-forme dermatosis: differentiation from other dirty-appearing diseases. Ann Dermatol. 2016;28:413-415.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Chun SW, Lee SY, Kim JB, et al. A case of terra firma-forme dermatosis treated with salicylic acid alcohol peeling. Ann Dermatol. 2017;29:83-85.
- Aslan NC, Guler S, Demirci K, et al. Features of terra firma-forme dermatosis. Ann Fam Med. 2018;16:52-54.
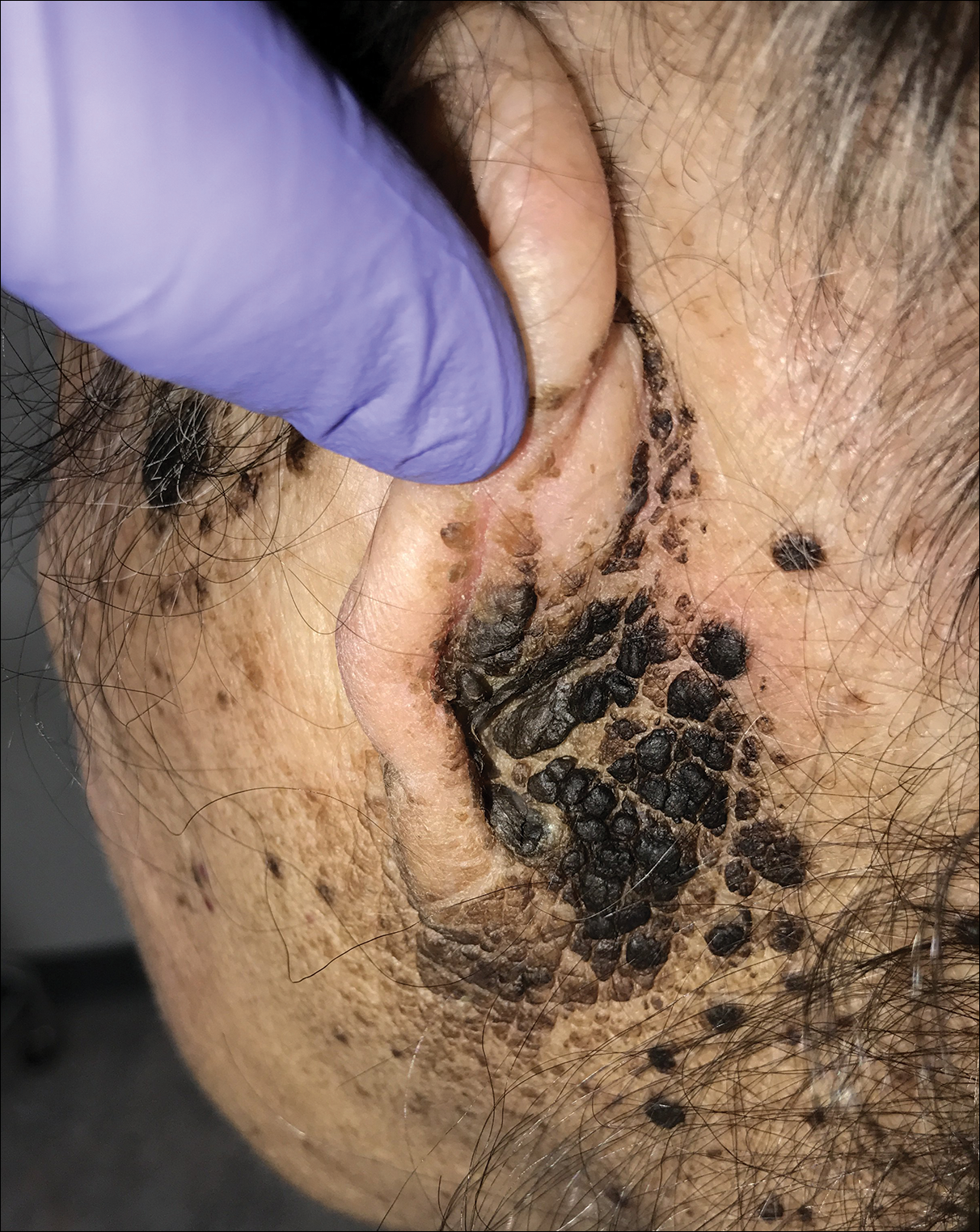
A 94-year-old woman was referred to the dermatology department for biopsy of pigmented tumors behind the ears of unknown duration. The growths were asymptomatic. Her medical history included the early stages of Alzheimer disease. On physical examination dark brown, smooth, coalescing papules and plaques were noted extending from the posterior neck to the conchal bowls and ear folds bilaterally. The nodules were removed by scrubbing with isopropyl alcohol 70%. A nodule was submitted for histopathologic review.
Is Vitiligo in Vogue? The Changing Face of Vitiligo
Vitiligo is a disfiguring skin condition that is thought to result from autoimmune destruction of melanocytes in the skin, leading to patchy depigmentation. The prevalence of vitiligo is estimated at 1% worldwide.1 Once seen as merely a cosmetic disorder, it is increasingly recognized for its devastating psychological effects. As skin quality, texture, and color are a few of the first things people notice about others, skin plays a major role in our daily interactions with the world. Vitiligo often affects the face and other visible areas of the body; thus, it is associated with impaired quality of life, and affected individuals often experience psychosocial impairment including anxiety, depression, stigmatization, and self-harm ideation.2 Indeed, vitiligo is a condition with not only a visible skin component but a deeper psychological component that also is important to recognize and address. However, due in large part to recent exposure to vitiligo through mainstream media, general understanding about and attitudes toward this condition are changing. As a result, vitiligo has seen a surge in outreach by those affected by the disease.
Perhaps the most well-known current face of vitiligo is Chantelle Brown-Young, a black fashion model, activist, and vitiligo spokesperson known professionally as Winnie Harlow. Diagnosed with vitiligo in childhood, she revealed she was teased and bullied and at one point contemplated suicide. “The continuous harassment and the despair that [vitiligo] brought on my life was so unbearably dehumanizing that I wanted to kill myself,” she disclosed.3 After competing on America’s Next Top Model in 2014, Winnie Harlow became a household name for redefining global standards of beauty and, in her own words, accepting the differences that make us unique and authentic.4 She went on to speak at the Dove Self-Esteem Project panel at the 2015 Women in the World London Summit and was presented with the Role Model award at the Portuguese GQ Men of the Year event that same year.5
More recently, Amy Deanna, a model with vitiligo, was featured in videos for CoverGirl’s 2018 “I Am What I Make Up” campaign in which she is shown enhancing her various skin tones rather than hiding them by applying both light and dark shades of makeup on her face. In a press release she stated, “Vitiligo awareness is something that is very important to me. Being given a platform to [raise awareness] means so much.”6
Additionally, Brock Elbank, a London-based photographer, recently launched a photograph series of men and women with vitiligo on the digital platform Instagram.7 In a recent interview he stated, “I see beauty in what many see as different. Unique individuals who stand out from the crowd are what inspire me to do what I do.”7
Lee Thomas, a television broadcaster and author of the book Turning White: A Memoir of Change is yet another example of a vitiligo patient who recently stopped hiding his condition. He admitted he has had people refuse to shake his hand due to his condition but has used the experience to educate others. He stated, “Because I’m in this position, I think this is where my next thing is supposed to be. It’s supposed to be about sharing and helping, and hopefully leaving the planet a little better for everybody else who comes along with vitiligo.”8 Thomas is dedicated to inspiring others with the condition and started the Clarity Lee Thomas Foundation to provide emotional and mental support to those with vitiligo.
Critics may say this vitiligo movement is merely another example of exploitation of what is unique or different by mainstream media and the fashion industry, similar to prior movements for plus-sized models, natural hairstyles in black women, and transgender identification. Even if partially true, the ultimate effect has been an increase in attention and representation of individuals with vitiligo in mainstream media. At the time this article was being published (September 2018), an Instagram search for #vitiligo yielded approximately 226,000 posts. For comparison with other much more common dermatologic conditions, #eczema returned approximately 958,000 results, #moles returned approximately 65,000 results, and #skincancer returned approximately 104,000 results. Additionally, the Vitiligo Research Foundation currently has more than 5000 followers on Instagram, which is as many as the Melanoma Research Foundation and almost twice as many as the Skin Cancer Foundation, supporting the idea that mainstream representation of individuals with vitiligo is contributing to raising awareness and backing of organizations aimed at making advancements in this area of dermatology.
As more individuals gain an understanding and curiosity about this disease, perhaps more research and investigation will be done to improve treatment options and outcomes for patients with vitiligo. With this movement, perhaps vitiligo patients will feel more comfortable and confident in their skin.
- Ezzedine K, Eleftheriadou V, Whitton M, et al. Vitiligo. Lancet. 2015;386:74-84.
- Tomas‐Aragones L, Marron SE. Body image and body dysmorphic concerns. Acta Derm Venereol. 2016;96:47-50.
- Rodney D. From suicide thoughts to finalist in America’s Next Top Model. The Gleaner. February 25, 2014. http://jamaica-gleaner.com/gleaner/20140225/news/news1.html. Accessed September 7, 2018.
- Keyes-Bevan B. Winnie Harlow: her emotional story with vitiligo. Personal Health News website. http://www.personalhealthnews.ca/prevention-and-treatment/her-emotional-story-with-vitiligo. Accessed September 7, 2018.
- Giles K, Davidson R. ‘I think I’m beautiful’: model Winnie Harlow, who suffers from rare vitiligo skin condition, gives empowering talk at Women in the World event. Daily Mail. October 9, 2015. http://www.dailymail.co.uk/tvshowbiz/article-3266579/I-think-m-beautiful-Model-Winnie-Harlow-suffers-rare-Vitiligo-skin-condition-gives-empowering-talk-Women-World-event.html. Updated October 13, 2015. Accessed September 7, 2018.
- Ruffo J. CoverGirl’s first model with vitiligo stars in new campaign: ‘w
e have to be more inclusive.’ People. February 20, 2018. https://people.com/style/covergirl-first-model-with-vitiligo-interview/. Accessed September 25, 2018. - Blair O. This vitiligo photo series is absolutely breathtaking. Cosmopolitan. March 23, 2018. https://www.cosmopolitan.com/uk/beauty-hair/a19494259/vitiligo-photo-series-instagram/. Accessed September 7, 2018.
- Broadcaster opens up about living with vitiligo. People. February 20, 2018. http://people.com/health/lee-thomas-tv-reporter-on-his-vitiligo/. Accessed April 1, 2018.
Vitiligo is a disfiguring skin condition that is thought to result from autoimmune destruction of melanocytes in the skin, leading to patchy depigmentation. The prevalence of vitiligo is estimated at 1% worldwide.1 Once seen as merely a cosmetic disorder, it is increasingly recognized for its devastating psychological effects. As skin quality, texture, and color are a few of the first things people notice about others, skin plays a major role in our daily interactions with the world. Vitiligo often affects the face and other visible areas of the body; thus, it is associated with impaired quality of life, and affected individuals often experience psychosocial impairment including anxiety, depression, stigmatization, and self-harm ideation.2 Indeed, vitiligo is a condition with not only a visible skin component but a deeper psychological component that also is important to recognize and address. However, due in large part to recent exposure to vitiligo through mainstream media, general understanding about and attitudes toward this condition are changing. As a result, vitiligo has seen a surge in outreach by those affected by the disease.
Perhaps the most well-known current face of vitiligo is Chantelle Brown-Young, a black fashion model, activist, and vitiligo spokesperson known professionally as Winnie Harlow. Diagnosed with vitiligo in childhood, she revealed she was teased and bullied and at one point contemplated suicide. “The continuous harassment and the despair that [vitiligo] brought on my life was so unbearably dehumanizing that I wanted to kill myself,” she disclosed.3 After competing on America’s Next Top Model in 2014, Winnie Harlow became a household name for redefining global standards of beauty and, in her own words, accepting the differences that make us unique and authentic.4 She went on to speak at the Dove Self-Esteem Project panel at the 2015 Women in the World London Summit and was presented with the Role Model award at the Portuguese GQ Men of the Year event that same year.5
More recently, Amy Deanna, a model with vitiligo, was featured in videos for CoverGirl’s 2018 “I Am What I Make Up” campaign in which she is shown enhancing her various skin tones rather than hiding them by applying both light and dark shades of makeup on her face. In a press release she stated, “Vitiligo awareness is something that is very important to me. Being given a platform to [raise awareness] means so much.”6
Additionally, Brock Elbank, a London-based photographer, recently launched a photograph series of men and women with vitiligo on the digital platform Instagram.7 In a recent interview he stated, “I see beauty in what many see as different. Unique individuals who stand out from the crowd are what inspire me to do what I do.”7
Lee Thomas, a television broadcaster and author of the book Turning White: A Memoir of Change is yet another example of a vitiligo patient who recently stopped hiding his condition. He admitted he has had people refuse to shake his hand due to his condition but has used the experience to educate others. He stated, “Because I’m in this position, I think this is where my next thing is supposed to be. It’s supposed to be about sharing and helping, and hopefully leaving the planet a little better for everybody else who comes along with vitiligo.”8 Thomas is dedicated to inspiring others with the condition and started the Clarity Lee Thomas Foundation to provide emotional and mental support to those with vitiligo.
Critics may say this vitiligo movement is merely another example of exploitation of what is unique or different by mainstream media and the fashion industry, similar to prior movements for plus-sized models, natural hairstyles in black women, and transgender identification. Even if partially true, the ultimate effect has been an increase in attention and representation of individuals with vitiligo in mainstream media. At the time this article was being published (September 2018), an Instagram search for #vitiligo yielded approximately 226,000 posts. For comparison with other much more common dermatologic conditions, #eczema returned approximately 958,000 results, #moles returned approximately 65,000 results, and #skincancer returned approximately 104,000 results. Additionally, the Vitiligo Research Foundation currently has more than 5000 followers on Instagram, which is as many as the Melanoma Research Foundation and almost twice as many as the Skin Cancer Foundation, supporting the idea that mainstream representation of individuals with vitiligo is contributing to raising awareness and backing of organizations aimed at making advancements in this area of dermatology.
As more individuals gain an understanding and curiosity about this disease, perhaps more research and investigation will be done to improve treatment options and outcomes for patients with vitiligo. With this movement, perhaps vitiligo patients will feel more comfortable and confident in their skin.
Vitiligo is a disfiguring skin condition that is thought to result from autoimmune destruction of melanocytes in the skin, leading to patchy depigmentation. The prevalence of vitiligo is estimated at 1% worldwide.1 Once seen as merely a cosmetic disorder, it is increasingly recognized for its devastating psychological effects. As skin quality, texture, and color are a few of the first things people notice about others, skin plays a major role in our daily interactions with the world. Vitiligo often affects the face and other visible areas of the body; thus, it is associated with impaired quality of life, and affected individuals often experience psychosocial impairment including anxiety, depression, stigmatization, and self-harm ideation.2 Indeed, vitiligo is a condition with not only a visible skin component but a deeper psychological component that also is important to recognize and address. However, due in large part to recent exposure to vitiligo through mainstream media, general understanding about and attitudes toward this condition are changing. As a result, vitiligo has seen a surge in outreach by those affected by the disease.
Perhaps the most well-known current face of vitiligo is Chantelle Brown-Young, a black fashion model, activist, and vitiligo spokesperson known professionally as Winnie Harlow. Diagnosed with vitiligo in childhood, she revealed she was teased and bullied and at one point contemplated suicide. “The continuous harassment and the despair that [vitiligo] brought on my life was so unbearably dehumanizing that I wanted to kill myself,” she disclosed.3 After competing on America’s Next Top Model in 2014, Winnie Harlow became a household name for redefining global standards of beauty and, in her own words, accepting the differences that make us unique and authentic.4 She went on to speak at the Dove Self-Esteem Project panel at the 2015 Women in the World London Summit and was presented with the Role Model award at the Portuguese GQ Men of the Year event that same year.5
More recently, Amy Deanna, a model with vitiligo, was featured in videos for CoverGirl’s 2018 “I Am What I Make Up” campaign in which she is shown enhancing her various skin tones rather than hiding them by applying both light and dark shades of makeup on her face. In a press release she stated, “Vitiligo awareness is something that is very important to me. Being given a platform to [raise awareness] means so much.”6
Additionally, Brock Elbank, a London-based photographer, recently launched a photograph series of men and women with vitiligo on the digital platform Instagram.7 In a recent interview he stated, “I see beauty in what many see as different. Unique individuals who stand out from the crowd are what inspire me to do what I do.”7
Lee Thomas, a television broadcaster and author of the book Turning White: A Memoir of Change is yet another example of a vitiligo patient who recently stopped hiding his condition. He admitted he has had people refuse to shake his hand due to his condition but has used the experience to educate others. He stated, “Because I’m in this position, I think this is where my next thing is supposed to be. It’s supposed to be about sharing and helping, and hopefully leaving the planet a little better for everybody else who comes along with vitiligo.”8 Thomas is dedicated to inspiring others with the condition and started the Clarity Lee Thomas Foundation to provide emotional and mental support to those with vitiligo.
Critics may say this vitiligo movement is merely another example of exploitation of what is unique or different by mainstream media and the fashion industry, similar to prior movements for plus-sized models, natural hairstyles in black women, and transgender identification. Even if partially true, the ultimate effect has been an increase in attention and representation of individuals with vitiligo in mainstream media. At the time this article was being published (September 2018), an Instagram search for #vitiligo yielded approximately 226,000 posts. For comparison with other much more common dermatologic conditions, #eczema returned approximately 958,000 results, #moles returned approximately 65,000 results, and #skincancer returned approximately 104,000 results. Additionally, the Vitiligo Research Foundation currently has more than 5000 followers on Instagram, which is as many as the Melanoma Research Foundation and almost twice as many as the Skin Cancer Foundation, supporting the idea that mainstream representation of individuals with vitiligo is contributing to raising awareness and backing of organizations aimed at making advancements in this area of dermatology.
As more individuals gain an understanding and curiosity about this disease, perhaps more research and investigation will be done to improve treatment options and outcomes for patients with vitiligo. With this movement, perhaps vitiligo patients will feel more comfortable and confident in their skin.
- Ezzedine K, Eleftheriadou V, Whitton M, et al. Vitiligo. Lancet. 2015;386:74-84.
- Tomas‐Aragones L, Marron SE. Body image and body dysmorphic concerns. Acta Derm Venereol. 2016;96:47-50.
- Rodney D. From suicide thoughts to finalist in America’s Next Top Model. The Gleaner. February 25, 2014. http://jamaica-gleaner.com/gleaner/20140225/news/news1.html. Accessed September 7, 2018.
- Keyes-Bevan B. Winnie Harlow: her emotional story with vitiligo. Personal Health News website. http://www.personalhealthnews.ca/prevention-and-treatment/her-emotional-story-with-vitiligo. Accessed September 7, 2018.
- Giles K, Davidson R. ‘I think I’m beautiful’: model Winnie Harlow, who suffers from rare vitiligo skin condition, gives empowering talk at Women in the World event. Daily Mail. October 9, 2015. http://www.dailymail.co.uk/tvshowbiz/article-3266579/I-think-m-beautiful-Model-Winnie-Harlow-suffers-rare-Vitiligo-skin-condition-gives-empowering-talk-Women-World-event.html. Updated October 13, 2015. Accessed September 7, 2018.
- Ruffo J. CoverGirl’s first model with vitiligo stars in new campaign: ‘w
e have to be more inclusive.’ People. February 20, 2018. https://people.com/style/covergirl-first-model-with-vitiligo-interview/. Accessed September 25, 2018. - Blair O. This vitiligo photo series is absolutely breathtaking. Cosmopolitan. March 23, 2018. https://www.cosmopolitan.com/uk/beauty-hair/a19494259/vitiligo-photo-series-instagram/. Accessed September 7, 2018.
- Broadcaster opens up about living with vitiligo. People. February 20, 2018. http://people.com/health/lee-thomas-tv-reporter-on-his-vitiligo/. Accessed April 1, 2018.
- Ezzedine K, Eleftheriadou V, Whitton M, et al. Vitiligo. Lancet. 2015;386:74-84.
- Tomas‐Aragones L, Marron SE. Body image and body dysmorphic concerns. Acta Derm Venereol. 2016;96:47-50.
- Rodney D. From suicide thoughts to finalist in America’s Next Top Model. The Gleaner. February 25, 2014. http://jamaica-gleaner.com/gleaner/20140225/news/news1.html. Accessed September 7, 2018.
- Keyes-Bevan B. Winnie Harlow: her emotional story with vitiligo. Personal Health News website. http://www.personalhealthnews.ca/prevention-and-treatment/her-emotional-story-with-vitiligo. Accessed September 7, 2018.
- Giles K, Davidson R. ‘I think I’m beautiful’: model Winnie Harlow, who suffers from rare vitiligo skin condition, gives empowering talk at Women in the World event. Daily Mail. October 9, 2015. http://www.dailymail.co.uk/tvshowbiz/article-3266579/I-think-m-beautiful-Model-Winnie-Harlow-suffers-rare-Vitiligo-skin-condition-gives-empowering-talk-Women-World-event.html. Updated October 13, 2015. Accessed September 7, 2018.
- Ruffo J. CoverGirl’s first model with vitiligo stars in new campaign: ‘w
e have to be more inclusive.’ People. February 20, 2018. https://people.com/style/covergirl-first-model-with-vitiligo-interview/. Accessed September 25, 2018. - Blair O. This vitiligo photo series is absolutely breathtaking. Cosmopolitan. March 23, 2018. https://www.cosmopolitan.com/uk/beauty-hair/a19494259/vitiligo-photo-series-instagram/. Accessed September 7, 2018.
- Broadcaster opens up about living with vitiligo. People. February 20, 2018. http://people.com/health/lee-thomas-tv-reporter-on-his-vitiligo/. Accessed April 1, 2018.

Nevus of Ota Associated With a Primary Uveal Melanoma and Intracranial Melanoma Metastasis
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
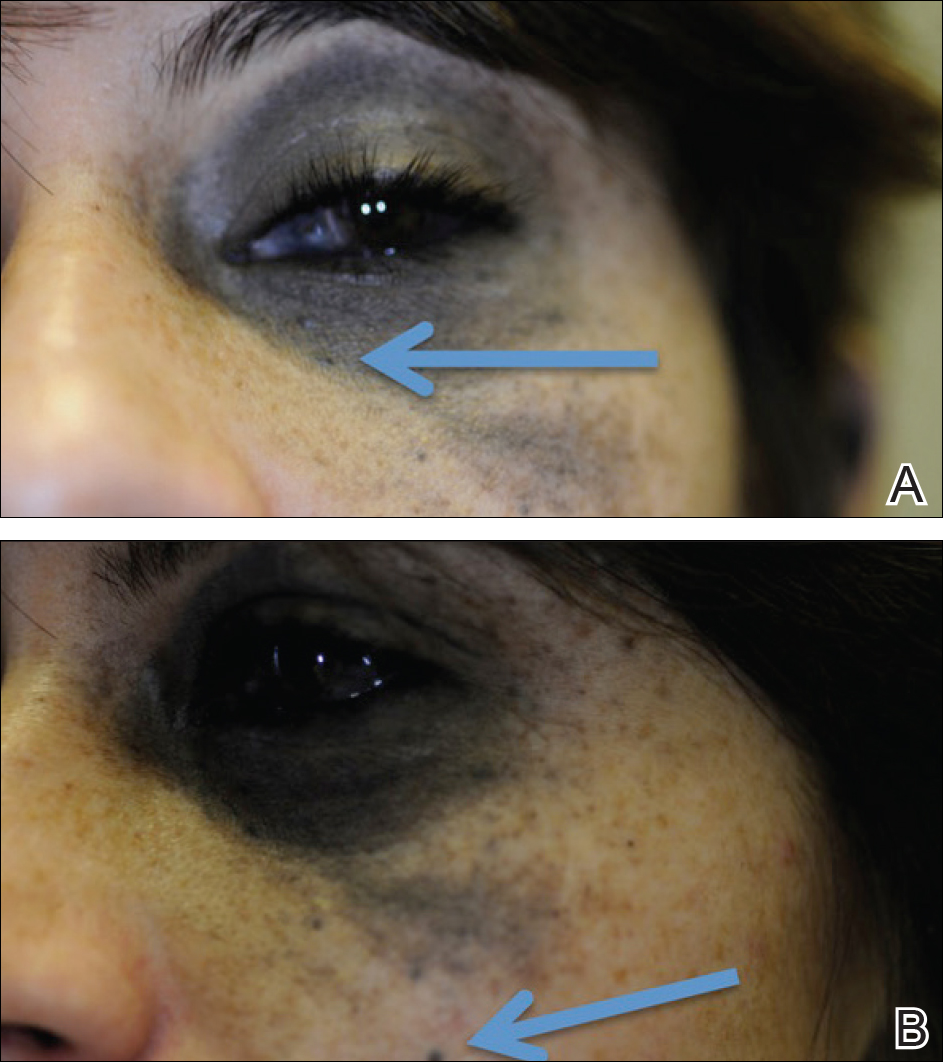
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
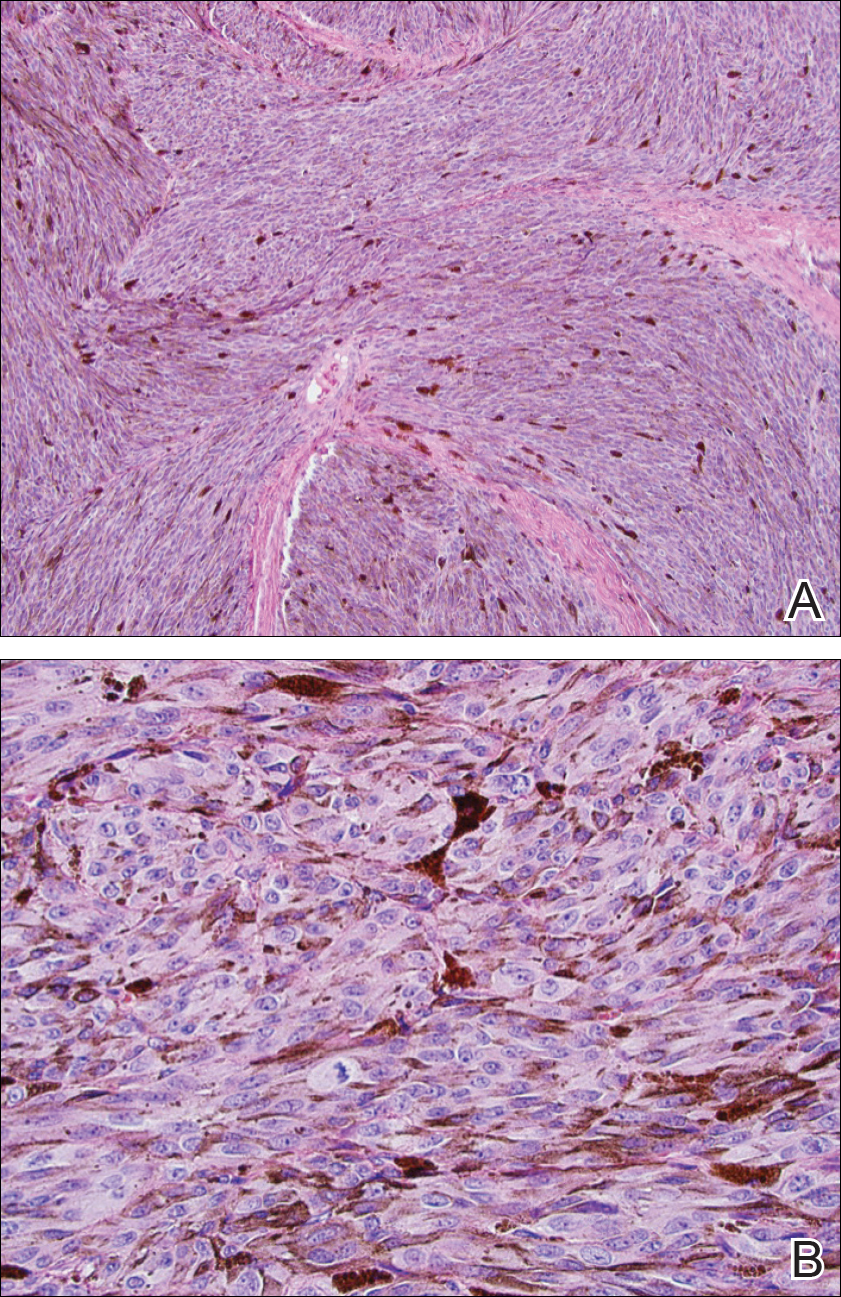
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.

The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.
The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.
The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
Practice Points
- Nevus of Ota is a hyperpigmented dermatosis that typically is distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.
- GNAQ and BAP1 mutations in patients with nevus of Ota confer a greater risk for malignant melanoma and metastatic progression.
- Ongoing ophthalmologic screening is paramount in patients with nevus of Ota and may prevent devastating sequelae.
Many devices optimal for treating vascular skin lesions
SAN DIEGO – According to J. Stuart Nelson, MD, PhD,

“You’re going to be using wavelengths of light generally in the green and yellow portion of the spectrum,” Dr. Nelson, professor of surgery and biomedical engineering at the Beckman Laser Institute and Medical Clinic at the University of California, Irvine, said at the annual Masters of Aesthetics Symposium. “Blue light is highly absorbed by hemoglobin but unfortunately, blue light is highly scattered by human skin, so it won’t penetrate deep enough into the dermis. So primarily, we’re targeting hemoglobin using green and yellow light sources.”
The second principle is to match the pulse width with the vessel size, while the third is to give sufficient energy to irreversibly injure vessels based on selective photothermolysis.
Next, he advised clinicians to ask themselves three questions: What is the vessel size? “The larger the vessel, the longer the thermal relaxation time,” he said. What is the vessel depth? Deeper vessels require longer wavelengths of light and larger spot sizes. What is the patient’s skin phototype? Darker skin contains more epidermal melanin and requires extra caution during treatment.
Dr. Nelson listed seven optimal devices for the treatment of vascular skin lesions: intense pulsed light (IPL) with wavelengths of 515-1,200 nm and pulse durations of 1-10 ms, pulsed green light with a wavelength of 532 nm and pulse durations of 1-50 ms, pulsed dye yellow light with wavelengths of 585-600 nm and pulse durations of 0.5-40 ms, pulsed dye plus Nd:YAG with wavelengths of 595 and 1,064 nm and pulse durations of 0.5-40 ms, alexandrite laser with a wavelength of 755 nm and pulse durations of 0.25-100 ms, diode laser with a wavelength of 940 nm and pulse durations of 5-100 ms, and the pulsed Nd:YAG laser with a wavelength of 1,064 nm and pulse durations of 0.25-100 ms.
“You can get good results with every one of these devices,” Dr. Nelson said. “What you need to do is pick one and become what R. Rox Anderson, MD, calls an ‘endpointologist,’ so you can understand the clinical endpoints. Do not use a cookbook approach by trying to memorize treatment settings.”
Pulsed dye lasers with a wavelength of 585-600 nm have been the standard of care for years, he said, and is the treatment of choice for port wine stains in infants and young children. Upsides include the ability to treat large areas quickly and the ability to use two to three separate passes. It also induces diminution in diffuse redness and telangiectasia. Drawbacks include its potential to cause purpura when short pulse durations are used, it requires several treatments, it can be painful, and it causes considerable edema and erythema.
Millisecond green lasers at a wavelength of 532 nm are also effective for treating vascular skin lesions. “The nice thing about these devices is that you can focus them down to very small spots, so you can literally trace out individual blood vessels,” Dr. Nelson said. Other upsides include the fact that it can be performed without producing purpura, only transient erythema if few areas are treated. Drawbacks are that it’s moderately painful and may cause considerable edema. It also causes significant melanin absorption so is not advised for use in tanned and darker-skinned individuals. For all patients, contact cooling must be assured.
IPL, meanwhile, “can be very useful for treating not only vascular lesions, but also concurrently pigmented lesions such as poikiloderma of Civatte,” Dr. Nelson said. Potential drawbacks to IPL therapy are that the spectrum of light emitted and the pulse duration characteristics vary between devices and multiple treatments are required.
Finally, in the millisecond domain, the pulsed alexandrite 755-nm and Nd:YAG 1,064-nm lasers “are very good when trying to target something very deep in the skin like a vein,” he said. “But when you’re using those devices, you’re coagulating a large volume of tissue, so you need to be very careful about the amount of heat that you’re generating deep in the skin.”
When consulting with patients who have rosacea or telangiectasia, Dr. Nelson tells them multiple treatments will be required. “These are chronic conditions, and they may need ongoing maintenance treatments. The nice thing about all these procedures you’re doing for rosacea and telangiectasia is that they can be combined with all of your FDA [Food and Drug Administration]-approved topical and oral treatment protocols. All of these drugs you have at your disposal to medically treat rosacea can be all used concurrently with your laser treatment. When you see a patient you need to emphasize to them: ‘I’m not treating your rosacea with the laser. I’m treating a symptom of your rosacea with the laser.’ ”
Dr. Nelson closed his presentation by offering basic principles for success, the first being do no harm. “That’s the single most important thing you want to remember. No one will get mad if the blood vessel’s still there, but they’ll get very mad if something bad happens. You also want to underpromise and overdeliver. I always tell patients it’s going to require two to four treatments. When in doubt, don’t treat or undertreat. You can always treat again.”
If you’re concerned, perform a test spot. “There’s nothing wrong with that, particularly in a patient where you’re not sure what the outcome will be,” he said. “Check for any unusual skin reaction and for potential success of the procedure. Finally, don’t treat patients who are tanned.”
Dr. Nelson reported having intellectual property rights with Syneron Candela.
SAN DIEGO – According to J. Stuart Nelson, MD, PhD,
“You’re going to be using wavelengths of light generally in the green and yellow portion of the spectrum,” Dr. Nelson, professor of surgery and biomedical engineering at the Beckman Laser Institute and Medical Clinic at the University of California, Irvine, said at the annual Masters of Aesthetics Symposium. “Blue light is highly absorbed by hemoglobin but unfortunately, blue light is highly scattered by human skin, so it won’t penetrate deep enough into the dermis. So primarily, we’re targeting hemoglobin using green and yellow light sources.”
The second principle is to match the pulse width with the vessel size, while the third is to give sufficient energy to irreversibly injure vessels based on selective photothermolysis.
Next, he advised clinicians to ask themselves three questions: What is the vessel size? “The larger the vessel, the longer the thermal relaxation time,” he said. What is the vessel depth? Deeper vessels require longer wavelengths of light and larger spot sizes. What is the patient’s skin phototype? Darker skin contains more epidermal melanin and requires extra caution during treatment.
Dr. Nelson listed seven optimal devices for the treatment of vascular skin lesions: intense pulsed light (IPL) with wavelengths of 515-1,200 nm and pulse durations of 1-10 ms, pulsed green light with a wavelength of 532 nm and pulse durations of 1-50 ms, pulsed dye yellow light with wavelengths of 585-600 nm and pulse durations of 0.5-40 ms, pulsed dye plus Nd:YAG with wavelengths of 595 and 1,064 nm and pulse durations of 0.5-40 ms, alexandrite laser with a wavelength of 755 nm and pulse durations of 0.25-100 ms, diode laser with a wavelength of 940 nm and pulse durations of 5-100 ms, and the pulsed Nd:YAG laser with a wavelength of 1,064 nm and pulse durations of 0.25-100 ms.
“You can get good results with every one of these devices,” Dr. Nelson said. “What you need to do is pick one and become what R. Rox Anderson, MD, calls an ‘endpointologist,’ so you can understand the clinical endpoints. Do not use a cookbook approach by trying to memorize treatment settings.”
Pulsed dye lasers with a wavelength of 585-600 nm have been the standard of care for years, he said, and is the treatment of choice for port wine stains in infants and young children. Upsides include the ability to treat large areas quickly and the ability to use two to three separate passes. It also induces diminution in diffuse redness and telangiectasia. Drawbacks include its potential to cause purpura when short pulse durations are used, it requires several treatments, it can be painful, and it causes considerable edema and erythema.
Millisecond green lasers at a wavelength of 532 nm are also effective for treating vascular skin lesions. “The nice thing about these devices is that you can focus them down to very small spots, so you can literally trace out individual blood vessels,” Dr. Nelson said. Other upsides include the fact that it can be performed without producing purpura, only transient erythema if few areas are treated. Drawbacks are that it’s moderately painful and may cause considerable edema. It also causes significant melanin absorption so is not advised for use in tanned and darker-skinned individuals. For all patients, contact cooling must be assured.
IPL, meanwhile, “can be very useful for treating not only vascular lesions, but also concurrently pigmented lesions such as poikiloderma of Civatte,” Dr. Nelson said. Potential drawbacks to IPL therapy are that the spectrum of light emitted and the pulse duration characteristics vary between devices and multiple treatments are required.
Finally, in the millisecond domain, the pulsed alexandrite 755-nm and Nd:YAG 1,064-nm lasers “are very good when trying to target something very deep in the skin like a vein,” he said. “But when you’re using those devices, you’re coagulating a large volume of tissue, so you need to be very careful about the amount of heat that you’re generating deep in the skin.”
When consulting with patients who have rosacea or telangiectasia, Dr. Nelson tells them multiple treatments will be required. “These are chronic conditions, and they may need ongoing maintenance treatments. The nice thing about all these procedures you’re doing for rosacea and telangiectasia is that they can be combined with all of your FDA [Food and Drug Administration]-approved topical and oral treatment protocols. All of these drugs you have at your disposal to medically treat rosacea can be all used concurrently with your laser treatment. When you see a patient you need to emphasize to them: ‘I’m not treating your rosacea with the laser. I’m treating a symptom of your rosacea with the laser.’ ”
Dr. Nelson closed his presentation by offering basic principles for success, the first being do no harm. “That’s the single most important thing you want to remember. No one will get mad if the blood vessel’s still there, but they’ll get very mad if something bad happens. You also want to underpromise and overdeliver. I always tell patients it’s going to require two to four treatments. When in doubt, don’t treat or undertreat. You can always treat again.”
If you’re concerned, perform a test spot. “There’s nothing wrong with that, particularly in a patient where you’re not sure what the outcome will be,” he said. “Check for any unusual skin reaction and for potential success of the procedure. Finally, don’t treat patients who are tanned.”
Dr. Nelson reported having intellectual property rights with Syneron Candela.
SAN DIEGO – According to J. Stuart Nelson, MD, PhD,
“You’re going to be using wavelengths of light generally in the green and yellow portion of the spectrum,” Dr. Nelson, professor of surgery and biomedical engineering at the Beckman Laser Institute and Medical Clinic at the University of California, Irvine, said at the annual Masters of Aesthetics Symposium. “Blue light is highly absorbed by hemoglobin but unfortunately, blue light is highly scattered by human skin, so it won’t penetrate deep enough into the dermis. So primarily, we’re targeting hemoglobin using green and yellow light sources.”
The second principle is to match the pulse width with the vessel size, while the third is to give sufficient energy to irreversibly injure vessels based on selective photothermolysis.
Next, he advised clinicians to ask themselves three questions: What is the vessel size? “The larger the vessel, the longer the thermal relaxation time,” he said. What is the vessel depth? Deeper vessels require longer wavelengths of light and larger spot sizes. What is the patient’s skin phototype? Darker skin contains more epidermal melanin and requires extra caution during treatment.
Dr. Nelson listed seven optimal devices for the treatment of vascular skin lesions: intense pulsed light (IPL) with wavelengths of 515-1,200 nm and pulse durations of 1-10 ms, pulsed green light with a wavelength of 532 nm and pulse durations of 1-50 ms, pulsed dye yellow light with wavelengths of 585-600 nm and pulse durations of 0.5-40 ms, pulsed dye plus Nd:YAG with wavelengths of 595 and 1,064 nm and pulse durations of 0.5-40 ms, alexandrite laser with a wavelength of 755 nm and pulse durations of 0.25-100 ms, diode laser with a wavelength of 940 nm and pulse durations of 5-100 ms, and the pulsed Nd:YAG laser with a wavelength of 1,064 nm and pulse durations of 0.25-100 ms.
“You can get good results with every one of these devices,” Dr. Nelson said. “What you need to do is pick one and become what R. Rox Anderson, MD, calls an ‘endpointologist,’ so you can understand the clinical endpoints. Do not use a cookbook approach by trying to memorize treatment settings.”
Pulsed dye lasers with a wavelength of 585-600 nm have been the standard of care for years, he said, and is the treatment of choice for port wine stains in infants and young children. Upsides include the ability to treat large areas quickly and the ability to use two to three separate passes. It also induces diminution in diffuse redness and telangiectasia. Drawbacks include its potential to cause purpura when short pulse durations are used, it requires several treatments, it can be painful, and it causes considerable edema and erythema.
Millisecond green lasers at a wavelength of 532 nm are also effective for treating vascular skin lesions. “The nice thing about these devices is that you can focus them down to very small spots, so you can literally trace out individual blood vessels,” Dr. Nelson said. Other upsides include the fact that it can be performed without producing purpura, only transient erythema if few areas are treated. Drawbacks are that it’s moderately painful and may cause considerable edema. It also causes significant melanin absorption so is not advised for use in tanned and darker-skinned individuals. For all patients, contact cooling must be assured.
IPL, meanwhile, “can be very useful for treating not only vascular lesions, but also concurrently pigmented lesions such as poikiloderma of Civatte,” Dr. Nelson said. Potential drawbacks to IPL therapy are that the spectrum of light emitted and the pulse duration characteristics vary between devices and multiple treatments are required.
Finally, in the millisecond domain, the pulsed alexandrite 755-nm and Nd:YAG 1,064-nm lasers “are very good when trying to target something very deep in the skin like a vein,” he said. “But when you’re using those devices, you’re coagulating a large volume of tissue, so you need to be very careful about the amount of heat that you’re generating deep in the skin.”
When consulting with patients who have rosacea or telangiectasia, Dr. Nelson tells them multiple treatments will be required. “These are chronic conditions, and they may need ongoing maintenance treatments. The nice thing about all these procedures you’re doing for rosacea and telangiectasia is that they can be combined with all of your FDA [Food and Drug Administration]-approved topical and oral treatment protocols. All of these drugs you have at your disposal to medically treat rosacea can be all used concurrently with your laser treatment. When you see a patient you need to emphasize to them: ‘I’m not treating your rosacea with the laser. I’m treating a symptom of your rosacea with the laser.’ ”
Dr. Nelson closed his presentation by offering basic principles for success, the first being do no harm. “That’s the single most important thing you want to remember. No one will get mad if the blood vessel’s still there, but they’ll get very mad if something bad happens. You also want to underpromise and overdeliver. I always tell patients it’s going to require two to four treatments. When in doubt, don’t treat or undertreat. You can always treat again.”
If you’re concerned, perform a test spot. “There’s nothing wrong with that, particularly in a patient where you’re not sure what the outcome will be,” he said. “Check for any unusual skin reaction and for potential success of the procedure. Finally, don’t treat patients who are tanned.”
Dr. Nelson reported having intellectual property rights with Syneron Candela.
EXPERT ANALYSIS FROM MOAS 2018
Terra Firma-Forme Dermatosis Mimicking Livedo Racemosa
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
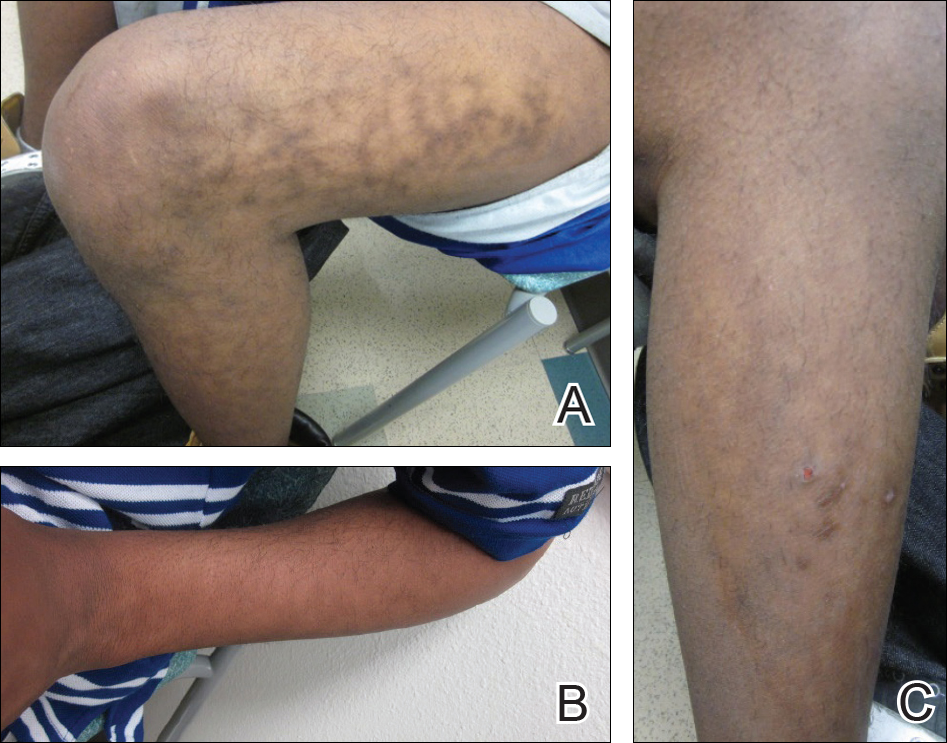
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
Practice Points
- Clinicians should include terra firma-forme dermatosis in the differential diagnosis of any hyperpigmented condition, regardless of pattern of presentation.
- Clean the skin with an alcohol wipe to rule out a diagnosis of terra firma-forme dermatosis.
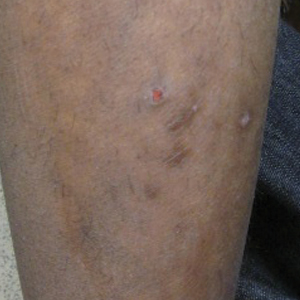
Extended propranolol use boosts success in high-risk infantile hemangioma
Extending oral propranolol treatment up to 12 months of age increased the success rate for high-risk infantile hemangioma, according to results published in Pediatrics.
Previous studies of oral propranol for infantile hemangiomas (IH) have revealed its efficacy, although there is no consensus on the optimal treatment duration. Nonetheless, treatment up to 12 months of age has been proposed if patients don’t respond after 6 months. Infants with high-risk hemangiomas, however, have been excluded from previous studies, authors of the current study explained.
In an open-label study of patients aged 35-150 days the success rate of oral propranolol was 47% after 6 months of treatment. The rate increased to 76% after the initial treatment period, reported Eulalia Baselga, MD, of the department of dermatology at Hospital de la Santa Creu i Sant Pau in Barcelona, and coauthors.
Investigators studied 45 patients from 10 hospitals in Spain and Poland between June 2015 and February 2017. The patients had high-risk IH in the proliferative phase. High-risk hemangiomas were defined as those that were life threatening, at risk for functional impact, disfiguring, or ulcerated nonresponsive to standard wound care measures.
Oral propranolol was administered twice daily at a dosage of 3 mg/kg per day. During the initial treatment period (ITP), patients received propranolol for a minimum of 6 months, and if treatment was not successful, it continued until success or up to 12 months of age.
Patients who achieved success in the initial phase were managed for 3 months with no treatment, and if there was rebound growth, treatment was restarted for up to 6 months at the provider’s discretion.
Treatment was considered a success if the target hemangioma resolved and there was no functional impact. The IH was considered resolved if it disappeared, even if there were minimal telangiectasias, erythema, skin thickening, soft tissue swelling, or the presence of sequelae.
Treatment success was achieved by 21 (47%) patients after 6 months and by 34 (76%) patients by the end of the ITP. Functional impact was determined using the Hemangioma Severity and Hemangioma Dynamic Complication scales. Adverse events, reported by 80% of patients, were resolved by the end of the study and included respiratory syncytial virus bronchiolitis, ulcerated hemangioma, pneumonia and respiratory failure, inguinal hernia, upper respiratory tract infection, dehydration, bronchitis, choking, and thermal burn. Although no patients experienced adverse events that resulted in discontinuation of treatment, 35 events led to temporary discontinuation, primarily due to respiratory events, the authors reported.
The results indicate that “oral propranolol is effective in treating high-risk IH with a favorable safety profile,” the authors concluded.
The study was funded by the Institut de Recherche Pierre Fabre Several authors were employed by or had other relationships with Pierre Fabre. The other authors had no conflicts of interest.
SOURCE: Baselga E et al. Pediatrics. 2018. doi: 10.1542/peds.2017-3866.
Extending oral propranolol treatment up to 12 months of age increased the success rate for high-risk infantile hemangioma, according to results published in Pediatrics.
Previous studies of oral propranol for infantile hemangiomas (IH) have revealed its efficacy, although there is no consensus on the optimal treatment duration. Nonetheless, treatment up to 12 months of age has been proposed if patients don’t respond after 6 months. Infants with high-risk hemangiomas, however, have been excluded from previous studies, authors of the current study explained.
In an open-label study of patients aged 35-150 days the success rate of oral propranolol was 47% after 6 months of treatment. The rate increased to 76% after the initial treatment period, reported Eulalia Baselga, MD, of the department of dermatology at Hospital de la Santa Creu i Sant Pau in Barcelona, and coauthors.
Investigators studied 45 patients from 10 hospitals in Spain and Poland between June 2015 and February 2017. The patients had high-risk IH in the proliferative phase. High-risk hemangiomas were defined as those that were life threatening, at risk for functional impact, disfiguring, or ulcerated nonresponsive to standard wound care measures.
Oral propranolol was administered twice daily at a dosage of 3 mg/kg per day. During the initial treatment period (ITP), patients received propranolol for a minimum of 6 months, and if treatment was not successful, it continued until success or up to 12 months of age.
Patients who achieved success in the initial phase were managed for 3 months with no treatment, and if there was rebound growth, treatment was restarted for up to 6 months at the provider’s discretion.
Treatment was considered a success if the target hemangioma resolved and there was no functional impact. The IH was considered resolved if it disappeared, even if there were minimal telangiectasias, erythema, skin thickening, soft tissue swelling, or the presence of sequelae.
Treatment success was achieved by 21 (47%) patients after 6 months and by 34 (76%) patients by the end of the ITP. Functional impact was determined using the Hemangioma Severity and Hemangioma Dynamic Complication scales. Adverse events, reported by 80% of patients, were resolved by the end of the study and included respiratory syncytial virus bronchiolitis, ulcerated hemangioma, pneumonia and respiratory failure, inguinal hernia, upper respiratory tract infection, dehydration, bronchitis, choking, and thermal burn. Although no patients experienced adverse events that resulted in discontinuation of treatment, 35 events led to temporary discontinuation, primarily due to respiratory events, the authors reported.
The results indicate that “oral propranolol is effective in treating high-risk IH with a favorable safety profile,” the authors concluded.
The study was funded by the Institut de Recherche Pierre Fabre Several authors were employed by or had other relationships with Pierre Fabre. The other authors had no conflicts of interest.
SOURCE: Baselga E et al. Pediatrics. 2018. doi: 10.1542/peds.2017-3866.
Extending oral propranolol treatment up to 12 months of age increased the success rate for high-risk infantile hemangioma, according to results published in Pediatrics.
Previous studies of oral propranol for infantile hemangiomas (IH) have revealed its efficacy, although there is no consensus on the optimal treatment duration. Nonetheless, treatment up to 12 months of age has been proposed if patients don’t respond after 6 months. Infants with high-risk hemangiomas, however, have been excluded from previous studies, authors of the current study explained.
In an open-label study of patients aged 35-150 days the success rate of oral propranolol was 47% after 6 months of treatment. The rate increased to 76% after the initial treatment period, reported Eulalia Baselga, MD, of the department of dermatology at Hospital de la Santa Creu i Sant Pau in Barcelona, and coauthors.
Investigators studied 45 patients from 10 hospitals in Spain and Poland between June 2015 and February 2017. The patients had high-risk IH in the proliferative phase. High-risk hemangiomas were defined as those that were life threatening, at risk for functional impact, disfiguring, or ulcerated nonresponsive to standard wound care measures.
Oral propranolol was administered twice daily at a dosage of 3 mg/kg per day. During the initial treatment period (ITP), patients received propranolol for a minimum of 6 months, and if treatment was not successful, it continued until success or up to 12 months of age.
Patients who achieved success in the initial phase were managed for 3 months with no treatment, and if there was rebound growth, treatment was restarted for up to 6 months at the provider’s discretion.
Treatment was considered a success if the target hemangioma resolved and there was no functional impact. The IH was considered resolved if it disappeared, even if there were minimal telangiectasias, erythema, skin thickening, soft tissue swelling, or the presence of sequelae.
Treatment success was achieved by 21 (47%) patients after 6 months and by 34 (76%) patients by the end of the ITP. Functional impact was determined using the Hemangioma Severity and Hemangioma Dynamic Complication scales. Adverse events, reported by 80% of patients, were resolved by the end of the study and included respiratory syncytial virus bronchiolitis, ulcerated hemangioma, pneumonia and respiratory failure, inguinal hernia, upper respiratory tract infection, dehydration, bronchitis, choking, and thermal burn. Although no patients experienced adverse events that resulted in discontinuation of treatment, 35 events led to temporary discontinuation, primarily due to respiratory events, the authors reported.
The results indicate that “oral propranolol is effective in treating high-risk IH with a favorable safety profile,” the authors concluded.
The study was funded by the Institut de Recherche Pierre Fabre Several authors were employed by or had other relationships with Pierre Fabre. The other authors had no conflicts of interest.
SOURCE: Baselga E et al. Pediatrics. 2018. doi: 10.1542/peds.2017-3866.
FROM PEDIATRICS
Key clinical point:
Major finding: After 6 months of treatment, the success rate was 47%, and it rose to 76% at the end of the treatment period.
Study details: A phase 3 study of 45 patients aged 35-150 days with high-risk IH.
Disclosures: The study was funded by the Institut de Recherche Pierre Fabre Several authors were employed by or had other relationships with Pierre Fabre. The other authors had no conflicts of interest.
Source: Baselga E et al. Pediatrics. 2018. doi: 10.1542/peds.2017-3866
Orodental issues often associated with facial port-wine stains
LAKE TAHOE, CALIF. – Several years ago, David H. Darrow, MD, DDS, began to notice a pattern in the conversation threads on websites dedicated to support for parents of children with facial port-wine stains (PWS).
, and that the alveolar ridge was lower on the side of the face that harbored the lesion. “Most importantly, parents were concerned that dentists were not touching their children because they were concerned about bleeding,” Dr. Darrow said at the annual meeting of the Society for Pediatric Dermatology. A search in the medical literature for port-wine stains and oral cavity changes, did not turn up much except for a few articles on bleeding. “One said that port-wine stains or capillary malformations rarely present major problems for the oral and maxillofacial surgeon. The other said that periodontal probing should not be done, as even gentle probing can result in uncontrolled bleeding,” he noted.
This prompted Dr. Darrow, who directs the Center for Hemangiomas and Vascular Birthmarks at the Eastern Virginia Medical School, Norfolk, and coinvestigators, Megan B. Dowling, MD, and Yueqin Zhao, PhD, to characterize manifestations of PWS in the oral cavity via an anonymous paired survey of volunteers with facial PWS and their dentists who were recruited from birthmarks.com and 10 collaborating physician practices (J Am Acad Dermatol 2012;67:687-93). Volunteers ranged in age from 1 to 62 years; mean age was 29 years. A total of 30 patients participated, and most (67%) were female.
The five most common oral manifestations reported by the patients were lip hyperplasia (53%), stained gingiva (47%), malocclusion (30%), bleeding gingiva (27%), and spacing between teeth (23%). Only 3% reported bleeding from dental procedures. When the researchers evaluated the orodental findings in the paired patient-physician responses, “most of the time there was good agreement between the patient and the dentist,” Dr. Darrow said. “The only one that fell out of agreement was lip hyperplasia. That’s probably because most dentists look right past the lips and into the oral cavity.”
When the researchers examined patients who had stained gingiva versus those who did not, they found that early-stage lesions demonstrated a reddish blush of the oral mucosa and gingiva, while late-stage lesions demonstrated increased blush of the oral tissues, as well as hyperplasia of the soft tissue or bone in the affected area. “Based on our review of the literature, bleeding of gums is rarely prolonged and dental procedures are safe,” Dr. Darrow said.
The findings are important, he continued, because capillary malformations of the oral cavity may result in periodontal disease associated with gingival overgrowth. The depth of the gingival pocket should be no more than 2-3 mm. “When you have areas of inflammation and deep-pocket formation, plaque and bacteria slowly erode the connection between the tooth and the soft tissue,” he explained. “At some point, that pocket becomes so deep that it reaches down into the bone in which the tooth is anchored. As that bone is eroded, the teeth loosen and begin to fall out. The goals of therapy are prevention of periodontal disease with meticulous oral hygiene.”
Soft tissue hyperplasia may be exacerbated by medications such as calcium channel blockers, cyclosporine, and phenytoin and phenobarbital, which are sometimes used by patients with Sturge-Weber syndrome, he said.
Dr. Darrow reported having no financial disclosures.
LAKE TAHOE, CALIF. – Several years ago, David H. Darrow, MD, DDS, began to notice a pattern in the conversation threads on websites dedicated to support for parents of children with facial port-wine stains (PWS).
, and that the alveolar ridge was lower on the side of the face that harbored the lesion. “Most importantly, parents were concerned that dentists were not touching their children because they were concerned about bleeding,” Dr. Darrow said at the annual meeting of the Society for Pediatric Dermatology. A search in the medical literature for port-wine stains and oral cavity changes, did not turn up much except for a few articles on bleeding. “One said that port-wine stains or capillary malformations rarely present major problems for the oral and maxillofacial surgeon. The other said that periodontal probing should not be done, as even gentle probing can result in uncontrolled bleeding,” he noted.
This prompted Dr. Darrow, who directs the Center for Hemangiomas and Vascular Birthmarks at the Eastern Virginia Medical School, Norfolk, and coinvestigators, Megan B. Dowling, MD, and Yueqin Zhao, PhD, to characterize manifestations of PWS in the oral cavity via an anonymous paired survey of volunteers with facial PWS and their dentists who were recruited from birthmarks.com and 10 collaborating physician practices (J Am Acad Dermatol 2012;67:687-93). Volunteers ranged in age from 1 to 62 years; mean age was 29 years. A total of 30 patients participated, and most (67%) were female.
The five most common oral manifestations reported by the patients were lip hyperplasia (53%), stained gingiva (47%), malocclusion (30%), bleeding gingiva (27%), and spacing between teeth (23%). Only 3% reported bleeding from dental procedures. When the researchers evaluated the orodental findings in the paired patient-physician responses, “most of the time there was good agreement between the patient and the dentist,” Dr. Darrow said. “The only one that fell out of agreement was lip hyperplasia. That’s probably because most dentists look right past the lips and into the oral cavity.”
When the researchers examined patients who had stained gingiva versus those who did not, they found that early-stage lesions demonstrated a reddish blush of the oral mucosa and gingiva, while late-stage lesions demonstrated increased blush of the oral tissues, as well as hyperplasia of the soft tissue or bone in the affected area. “Based on our review of the literature, bleeding of gums is rarely prolonged and dental procedures are safe,” Dr. Darrow said.
The findings are important, he continued, because capillary malformations of the oral cavity may result in periodontal disease associated with gingival overgrowth. The depth of the gingival pocket should be no more than 2-3 mm. “When you have areas of inflammation and deep-pocket formation, plaque and bacteria slowly erode the connection between the tooth and the soft tissue,” he explained. “At some point, that pocket becomes so deep that it reaches down into the bone in which the tooth is anchored. As that bone is eroded, the teeth loosen and begin to fall out. The goals of therapy are prevention of periodontal disease with meticulous oral hygiene.”
Soft tissue hyperplasia may be exacerbated by medications such as calcium channel blockers, cyclosporine, and phenytoin and phenobarbital, which are sometimes used by patients with Sturge-Weber syndrome, he said.
Dr. Darrow reported having no financial disclosures.
LAKE TAHOE, CALIF. – Several years ago, David H. Darrow, MD, DDS, began to notice a pattern in the conversation threads on websites dedicated to support for parents of children with facial port-wine stains (PWS).
, and that the alveolar ridge was lower on the side of the face that harbored the lesion. “Most importantly, parents were concerned that dentists were not touching their children because they were concerned about bleeding,” Dr. Darrow said at the annual meeting of the Society for Pediatric Dermatology. A search in the medical literature for port-wine stains and oral cavity changes, did not turn up much except for a few articles on bleeding. “One said that port-wine stains or capillary malformations rarely present major problems for the oral and maxillofacial surgeon. The other said that periodontal probing should not be done, as even gentle probing can result in uncontrolled bleeding,” he noted.
This prompted Dr. Darrow, who directs the Center for Hemangiomas and Vascular Birthmarks at the Eastern Virginia Medical School, Norfolk, and coinvestigators, Megan B. Dowling, MD, and Yueqin Zhao, PhD, to characterize manifestations of PWS in the oral cavity via an anonymous paired survey of volunteers with facial PWS and their dentists who were recruited from birthmarks.com and 10 collaborating physician practices (J Am Acad Dermatol 2012;67:687-93). Volunteers ranged in age from 1 to 62 years; mean age was 29 years. A total of 30 patients participated, and most (67%) were female.
The five most common oral manifestations reported by the patients were lip hyperplasia (53%), stained gingiva (47%), malocclusion (30%), bleeding gingiva (27%), and spacing between teeth (23%). Only 3% reported bleeding from dental procedures. When the researchers evaluated the orodental findings in the paired patient-physician responses, “most of the time there was good agreement between the patient and the dentist,” Dr. Darrow said. “The only one that fell out of agreement was lip hyperplasia. That’s probably because most dentists look right past the lips and into the oral cavity.”
When the researchers examined patients who had stained gingiva versus those who did not, they found that early-stage lesions demonstrated a reddish blush of the oral mucosa and gingiva, while late-stage lesions demonstrated increased blush of the oral tissues, as well as hyperplasia of the soft tissue or bone in the affected area. “Based on our review of the literature, bleeding of gums is rarely prolonged and dental procedures are safe,” Dr. Darrow said.
The findings are important, he continued, because capillary malformations of the oral cavity may result in periodontal disease associated with gingival overgrowth. The depth of the gingival pocket should be no more than 2-3 mm. “When you have areas of inflammation and deep-pocket formation, plaque and bacteria slowly erode the connection between the tooth and the soft tissue,” he explained. “At some point, that pocket becomes so deep that it reaches down into the bone in which the tooth is anchored. As that bone is eroded, the teeth loosen and begin to fall out. The goals of therapy are prevention of periodontal disease with meticulous oral hygiene.”
Soft tissue hyperplasia may be exacerbated by medications such as calcium channel blockers, cyclosporine, and phenytoin and phenobarbital, which are sometimes used by patients with Sturge-Weber syndrome, he said.
Dr. Darrow reported having no financial disclosures.
REPORTING FROM SPD 2018
Atrophodermalike Guttate Morphea
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
 as well as hypopigmented macules and a minimally depressed patch on the left thigh (B).")
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
(H&E, original magnification ×40)...")
(Verhoeff-van Gieson, original magnification ×40). Normal elastin network in the lower reticular dermis; note the normal size of elastin fibers (B)(Verhoeff-van Gieson, original magnification ×200).")
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
Practice Points
- Atrophodermalike guttate morphea is a potentially underreported or undescribed entity consisting of a combination of clinicopathologic features.
- Widespread hypopigmented macules on the trunk and extremities marked by thinned collagen, fibroplasia, and altered fragmented elastin in the papillary dermis and upper reticular dermis are the key features.
- Atrophoderma, morphea, and lichen sclerosus et atrophicus should be ruled out during clinical workup.
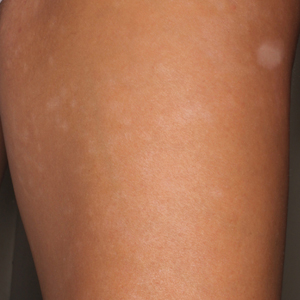
Melasma Treatment With Oral Tranexamic Acid and a Novel Adjuvant Topical Therapy
To the Editor:
I read with interest the informative article by Sheu1 published online in Cutis in February 2018, which succinctly described the pharmacologic characteristics of tranexamic acid, a synthetic lysine derivative, and its mechanism of action in the management of melasma by mitigating UV radiation-induced melanogenesis and neovascularization by inhibiting plasminogen activation. Additionally, the author summarized a study in which oral tranexamic acid was used to successfully treat melasma patients. After 4 months of treatment, 90% of 561 patients treated at a single center in Singapore demonstrated improvement in melasma severity.2 Sheu1 also discussed daily oral doses of tranexamic acid (500-1500 mg) that demonstrated improvement in melasma patients and reviewed potential adverse events (eg, abdominal pain and bloating, deep venous thrombosis, pulmonary embolism) for which patients should be screened and counseled prior to initiating treatment.
Recently, another study showed oral tranexamic acid to be an effective treatment in women with moderate to severe melasma. An important observation by the investigators was that once the initial phase of their study--250 mg of oral tranexamic acid twice daily and sunscreen applied to the face each morning and every 2 hours during daylight hours for 3 months--concluded and a second phase during which all participants only applied sunscreen for an additional 3 months, those with severe melasma lost most of their improvement.3 An adjuvant topical treatment, such as tranexamic acid or an inhibitor of tyrosinase (hydroquinone), might improve the results; however, initiating therapy with a topical agent whose mode of action is directed toward other melasma etiologic factors, such as the increased expression of estrogen receptors and vascular endothelial growth factor in affected skin, might be more beneficial.4,5
I recently proposed a novel approach for melasma management that would be appropriate as an adjuvant topical therapy for patients concurrently being treated with oral tranexamic acid.6 The therapeutic intervention utilizes active agents that specifically affect etiologic factors in the pathogenesis of melasma--estrogen and angiogenesis--that previously have not been targeted topically. Indeed, the topical agent contains an antiestrogen--either a selective estrogen receptor modulator (eg, tamoxifen, raloxifene), aromatase inhibitor (eg, anastrozole, letrozole, exemestane), or a selective estrogen receptor degrader (eg, fulvestrant)--and a vascular endothelial growth factor inhibitor (eg, bevacizumab).6
In conclusion, the therapeutic armamentarium for managing patients with melasma includes topical agents, oral therapies, and physical modalities. Optimizing the approach to treating melasma patients should incorporate therapies that are specifically directed toward various etiologic factors of the condition. The concurrent use of a topical agent that contains an antiestrogen and an inhibitor of vascular endothelial growth factor in women with melasma who are being treated with oral tranexamic acid warrants further investigation to assess not only for enhanced but also sustained reduction in facial skin pigmentation.
- Sheu SL. Treatment of melasma using tranexamic acid: what's known and what's next. Cutis. 2018;101:E7-E8.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma: a retrospective study. J Am Acad Dermatol. 2016;75:385-392.
- Del Rosaria E, Forez-Pollack S, Zapata L Jr, et al. Randomized, placebo-controlled, double-blind study of oral tranexamic acid in the treatment of moderate-to-severe melasma. J Am Acad Dermatol. 2018;78:363-369.
- Jang YH, Lee JY, Kang HY, et al. Oestrogen and progesterone receptor expression in melasma: an immunohistochemical analysis. J Eur Acad Dermatol Venereol. 2010;24:1312-1316.
- Kim EH, Kim YC, Lee ES, et al. The vascular characteristics of melasma. J Dermatol Sci. 2007;46:111-116.
- Cohen PR. Melasma treatment: a novel approach using a topical agent that contains an anti-estrogen and a vascular endothelial growth factor inhibitor [published online February 3, 2017]. Med Hypotheses. 2017;101:1-5.
To the Editor:
I read with interest the informative article by Sheu1 published online in Cutis in February 2018, which succinctly described the pharmacologic characteristics of tranexamic acid, a synthetic lysine derivative, and its mechanism of action in the management of melasma by mitigating UV radiation-induced melanogenesis and neovascularization by inhibiting plasminogen activation. Additionally, the author summarized a study in which oral tranexamic acid was used to successfully treat melasma patients. After 4 months of treatment, 90% of 561 patients treated at a single center in Singapore demonstrated improvement in melasma severity.2 Sheu1 also discussed daily oral doses of tranexamic acid (500-1500 mg) that demonstrated improvement in melasma patients and reviewed potential adverse events (eg, abdominal pain and bloating, deep venous thrombosis, pulmonary embolism) for which patients should be screened and counseled prior to initiating treatment.
Recently, another study showed oral tranexamic acid to be an effective treatment in women with moderate to severe melasma. An important observation by the investigators was that once the initial phase of their study--250 mg of oral tranexamic acid twice daily and sunscreen applied to the face each morning and every 2 hours during daylight hours for 3 months--concluded and a second phase during which all participants only applied sunscreen for an additional 3 months, those with severe melasma lost most of their improvement.3 An adjuvant topical treatment, such as tranexamic acid or an inhibitor of tyrosinase (hydroquinone), might improve the results; however, initiating therapy with a topical agent whose mode of action is directed toward other melasma etiologic factors, such as the increased expression of estrogen receptors and vascular endothelial growth factor in affected skin, might be more beneficial.4,5
I recently proposed a novel approach for melasma management that would be appropriate as an adjuvant topical therapy for patients concurrently being treated with oral tranexamic acid.6 The therapeutic intervention utilizes active agents that specifically affect etiologic factors in the pathogenesis of melasma--estrogen and angiogenesis--that previously have not been targeted topically. Indeed, the topical agent contains an antiestrogen--either a selective estrogen receptor modulator (eg, tamoxifen, raloxifene), aromatase inhibitor (eg, anastrozole, letrozole, exemestane), or a selective estrogen receptor degrader (eg, fulvestrant)--and a vascular endothelial growth factor inhibitor (eg, bevacizumab).6
In conclusion, the therapeutic armamentarium for managing patients with melasma includes topical agents, oral therapies, and physical modalities. Optimizing the approach to treating melasma patients should incorporate therapies that are specifically directed toward various etiologic factors of the condition. The concurrent use of a topical agent that contains an antiestrogen and an inhibitor of vascular endothelial growth factor in women with melasma who are being treated with oral tranexamic acid warrants further investigation to assess not only for enhanced but also sustained reduction in facial skin pigmentation.
To the Editor:
I read with interest the informative article by Sheu1 published online in Cutis in February 2018, which succinctly described the pharmacologic characteristics of tranexamic acid, a synthetic lysine derivative, and its mechanism of action in the management of melasma by mitigating UV radiation-induced melanogenesis and neovascularization by inhibiting plasminogen activation. Additionally, the author summarized a study in which oral tranexamic acid was used to successfully treat melasma patients. After 4 months of treatment, 90% of 561 patients treated at a single center in Singapore demonstrated improvement in melasma severity.2 Sheu1 also discussed daily oral doses of tranexamic acid (500-1500 mg) that demonstrated improvement in melasma patients and reviewed potential adverse events (eg, abdominal pain and bloating, deep venous thrombosis, pulmonary embolism) for which patients should be screened and counseled prior to initiating treatment.
Recently, another study showed oral tranexamic acid to be an effective treatment in women with moderate to severe melasma. An important observation by the investigators was that once the initial phase of their study--250 mg of oral tranexamic acid twice daily and sunscreen applied to the face each morning and every 2 hours during daylight hours for 3 months--concluded and a second phase during which all participants only applied sunscreen for an additional 3 months, those with severe melasma lost most of their improvement.3 An adjuvant topical treatment, such as tranexamic acid or an inhibitor of tyrosinase (hydroquinone), might improve the results; however, initiating therapy with a topical agent whose mode of action is directed toward other melasma etiologic factors, such as the increased expression of estrogen receptors and vascular endothelial growth factor in affected skin, might be more beneficial.4,5
I recently proposed a novel approach for melasma management that would be appropriate as an adjuvant topical therapy for patients concurrently being treated with oral tranexamic acid.6 The therapeutic intervention utilizes active agents that specifically affect etiologic factors in the pathogenesis of melasma--estrogen and angiogenesis--that previously have not been targeted topically. Indeed, the topical agent contains an antiestrogen--either a selective estrogen receptor modulator (eg, tamoxifen, raloxifene), aromatase inhibitor (eg, anastrozole, letrozole, exemestane), or a selective estrogen receptor degrader (eg, fulvestrant)--and a vascular endothelial growth factor inhibitor (eg, bevacizumab).6
In conclusion, the therapeutic armamentarium for managing patients with melasma includes topical agents, oral therapies, and physical modalities. Optimizing the approach to treating melasma patients should incorporate therapies that are specifically directed toward various etiologic factors of the condition. The concurrent use of a topical agent that contains an antiestrogen and an inhibitor of vascular endothelial growth factor in women with melasma who are being treated with oral tranexamic acid warrants further investigation to assess not only for enhanced but also sustained reduction in facial skin pigmentation.
- Sheu SL. Treatment of melasma using tranexamic acid: what's known and what's next. Cutis. 2018;101:E7-E8.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma: a retrospective study. J Am Acad Dermatol. 2016;75:385-392.
- Del Rosaria E, Forez-Pollack S, Zapata L Jr, et al. Randomized, placebo-controlled, double-blind study of oral tranexamic acid in the treatment of moderate-to-severe melasma. J Am Acad Dermatol. 2018;78:363-369.
- Jang YH, Lee JY, Kang HY, et al. Oestrogen and progesterone receptor expression in melasma: an immunohistochemical analysis. J Eur Acad Dermatol Venereol. 2010;24:1312-1316.
- Kim EH, Kim YC, Lee ES, et al. The vascular characteristics of melasma. J Dermatol Sci. 2007;46:111-116.
- Cohen PR. Melasma treatment: a novel approach using a topical agent that contains an anti-estrogen and a vascular endothelial growth factor inhibitor [published online February 3, 2017]. Med Hypotheses. 2017;101:1-5.
- Sheu SL. Treatment of melasma using tranexamic acid: what's known and what's next. Cutis. 2018;101:E7-E8.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma: a retrospective study. J Am Acad Dermatol. 2016;75:385-392.
- Del Rosaria E, Forez-Pollack S, Zapata L Jr, et al. Randomized, placebo-controlled, double-blind study of oral tranexamic acid in the treatment of moderate-to-severe melasma. J Am Acad Dermatol. 2018;78:363-369.
- Jang YH, Lee JY, Kang HY, et al. Oestrogen and progesterone receptor expression in melasma: an immunohistochemical analysis. J Eur Acad Dermatol Venereol. 2010;24:1312-1316.
- Kim EH, Kim YC, Lee ES, et al. The vascular characteristics of melasma. J Dermatol Sci. 2007;46:111-116.
- Cohen PR. Melasma treatment: a novel approach using a topical agent that contains an anti-estrogen and a vascular endothelial growth factor inhibitor [published online February 3, 2017]. Med Hypotheses. 2017;101:1-5.