User login
Incidentally Discovered Ochronosis Explaining Decades of Chronic Pain
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”

At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
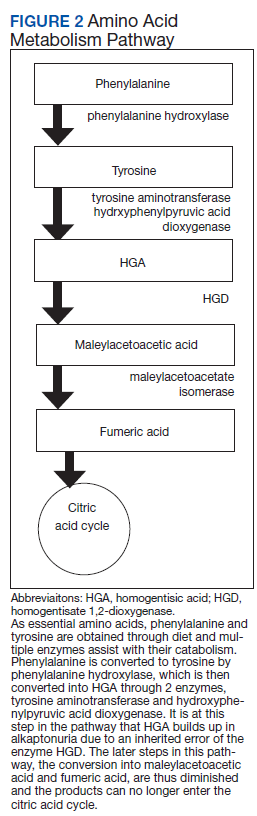
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
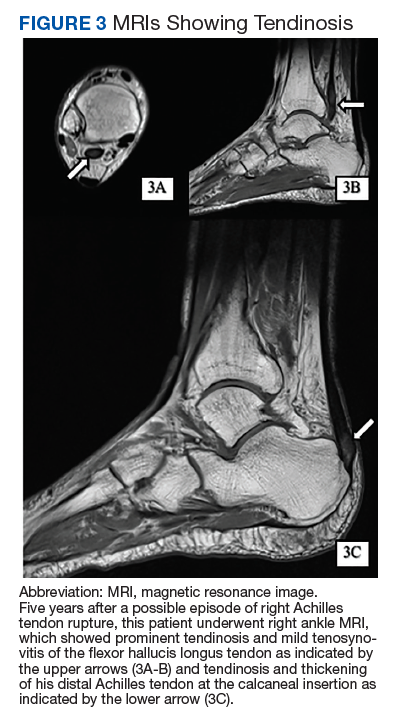
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
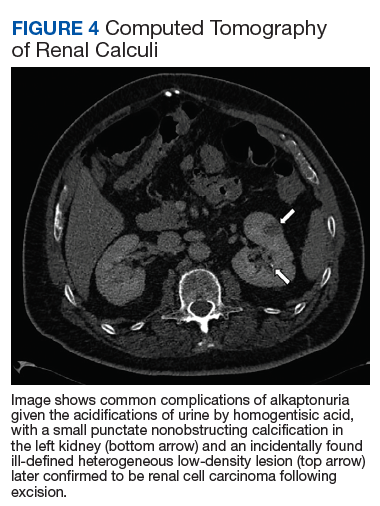
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”
At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”
At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
Annual Skin Check: Examining the Dermatology Headlines of 2019
From chemical sunscreen to the measles outbreak and drug approvals to product recalls, dermatology experienced its share of firsts and controversies in 2019.
Chemical Sunscreen Controversies
Controversial concerns about the effects of chemical sunscreen on coral reefs took an unprecedented turn in the United States this last year. On February 5, 2019, an ordinance was passed in Key West, Florida, prohibiting the sale of sunscreen containing the organic UV filters oxybenzone and/or octinoxate within city limits.1 On June 25, 2019, a similar law that also included octocrylene was passed in the US Virgin Islands.2 In so doing, these areas joined Hawaii, the Republic of Palau, and parts of Mexico in restricting chemical sunscreen sales.1 Although the Key West ordinance is set to take effect in January 2021, opponents, including dermatologists who believe it will discourage sunscreen use, currently are trying to overturn the ban.3 In the US Virgin Islands, part of the ban went into effect in September 2019, with the rest of the ban set to start in March 2020.2 Companies have started to follow suit. On August 1, 2019, CVS Pharmacy announced that, by the end of 2020, it will remove oxybenzone and octinoxate from some of its store-brand chemical sunscreens.4
On February 26, 2019, the US Food and Drug Administration (FDA) proposed that there are insufficient data to determine if 12 organic UV filters—including the aforementioned oxybenzone, octinoxate, and octocrylene—are generally recognized as safe and effective (GRASE).5 Although these ingredients were listed as GRASE by the FDA in 2011, the rise in sunscreen use since then, as well as changes in sunscreen formulations, prompted the FDA to ask manufacturers to perform additional studies on safety parameters such as systemic absorption.5,6 One study conducted by the FDA itself was published in May 2019 and showed that maximal use of 4 sunscreens resulted in systemic absorption of 4 organic UV filters above 0.5 ng/mL, the FDA’s threshold for requiring nonclinical toxicology assessment. The study authors concluded that “further studies [are needed] to determine the clinical significance of these findings. [But] These results do not indicate that individuals should refrain from the use of
End of the New York City Measles Outbreak
On September 3, 2019, New York City’s largest measles outbreak in nearly 30 years was declared over. This announcement reflected the fact that 2 incubation periods for measles—42 days—had passed since the last measles patient was considered contagious. In total, there were 654 cases of measles and 52 associated hospitalizations, including 16 admissions to the intensive care unit. Most patients were younger than 18 years and unvaccinated.8
The outbreak began in October 2018 after Orthodox Jewish children from Brooklyn became infected while visiting Israel and imported the measles virus upon their return home.8,9 All 5 boroughs in New York City were ultimately affected, although 4 zip codes in Williamsburg, a neighborhood in Brooklyn with an undervaccinated Orthodox Jewish community, accounted for 72% of cases.8,10 As part of a $6 million effort to stop the outbreak, an emergency order was placed on these 4 zip codes, posing potential fines on people living or working there if they were unvaccinated.8 In addition, a bill was passed and signed into law in New York State that eliminated religious exemptions for immunizations.11 In collaboration with Jewish leaders, these efforts increased the administration of measles-mumps-rubella vaccines by 41% compared with the year before in Williamsburg and Borough Park, another heavily Orthodox Jewish neighborhood in Brooklyn.8
Drug Approvals for Pediatric Dermatology
On March 11, 2019, the IL-4/IL-13 inhibitor dupilumab became the third biologic with a pediatric dermatology indication when the FDA extended its approval to adolescents for the treatment of atopic dermatitis.12 The FDA approval was based on a randomized, double-blind, placebo-controlled trial in which 42% (34/82) of adolescents treated with dupilumab monotherapy every other week achieved 75% or more improvement in the Eczema Area and Severity Index at week 16 compared with 8% (7/85) in the placebo group (P<.001).13
In October 2019, trifarotene cream and minocycline foam were approved by the FDA for the treatment of acne in patients 9 years and older.14,15 As such, both became the first acne therapies to include patients as young as 9 years in their studies and indication—a milestone, considering the fact that children have historically been excluded from clinical trials.16 The 2 topical treatments also are noteworthy for being first in class: trifarotene cream is the only topical retinoid to selectively target the retinoic acid receptor γ and to have been studied specifically for both facial and truncal acne,14,17 and minocycline foam is the first topical tetracycline.15
Drug Approvals for Rare Dermatologic Diseases
On July 19, 2019, apremilast, a phosphodiesterase 4 inhibitor, became the first medication approved by the FDA for the treatment of adults with oral ulcers due to Behçet disease, a rare multisystem inflammatory disease.18 The FDA approval was based on a double-blind, randomized, placebo-controlled trial in which 53% (55/104) of patients receiving apremilast monotherapy were ulcer free at week 12 compared to 22% (23/103) receiving placebo (P<.0001)(ClinicalTrials.gov Identifier NCT02307513).19
On October 8, 2019, afamelanotide was approved by the FDA to increase pain-free light exposure in adults with erythropoietic protoporphyria, a rare metabolic disorder associated with photosensitivity.20 A melanocortin receptor agonist, afamelanotide is believed to confer photoprotection by increasing the production of eumelanin in the epidermis. The FDA approval was based on 2 randomized, double-blind, placebo-controlled trials, both of which found that patients given afamelanotide spent significantly more time in direct sunlight without pain compared to patients in the placebo group (P=.005 and P=.04).21
Recalls of Popular Skin Products
On July 5, 2019, Neutrogena recalled its cult-favorite Light Therapy Acne Mask. The recall was driven by rare reports of transient visual side effects due to insufficient eye protection from the mask’s light-emitting diodes.22,23 Reported in association with 0.02% of masks sold at the time of the recall, these side effects included eye pain, irritation, tearing, blurry vision, seeing spots, and changes in color vision.24 In addition, a risk for potentially irreversible eye injury from the mask was cited in people taking photosensitizing medications, such as doxycycline, and people with certain underlying eye conditions, such as retinitis pigmentosa and ocular albinism.22,24,25
Following decades of asbestos-related controversy, 1 lot of the iconic Johnson’s Baby Powder was recalled for the first time on October 18, 2019, after the FDA found subtrace levels of asbestos in 1 of the lot’s bottles.26 After the recall, Johnson & Johnson reported that 2 third-party laboratories did not ultimately find asbestos when they tested the bottle of interest as well as other bottles from the recalled lot. Three of 5 samples prepared in 1 room by the third-party laboratories initially did test positive for asbestos, but this result was attributed to the room’s air conditioner, which was found to be contaminated with asbestos. When the same samples were prepared in another room, no asbestos was detected.27 The FDA maintained there was “no indication of cross-contamination” when they originally tested the implicated bottle.28
- Zraick K. Key West bans sunscreen containing chemicals believed to harm coral reefs. New York Times. February 7, 2019. https://www.nytimes.com/2019/02/07/us/sunscreen-coral-reef-key-west.html. Accessed December 23, 2019.
- Gies H. The U.S. Virigin Islands becomes the first American jurisdiction to ban common chemical sunscreens. Pacific Standard. July 18, 2019. https://psmag.com/environment/sunscreen-is-corals-biggest-anemone. Accessed December 23, 2019.
- Luscombe R. Republicans seek to overturn Key West ban on coral-damaging sunscreens. The Guardian. November 9, 2019. https://www.theguardian.com/us-news/2019/nov/09/key-west-sunscreen-coral-reef-backlash-skin-cancer. Accessed December 23, 2019.
- Salazar D. CVS to remove 2 chemicals from 60 store-brand sunscreens. Drug Store News. August 2, 2019. https://drugstorenews.com/retail-news/cvs-to-remove-2-chemicals-from-60-store-brand-sunscreens. Accessed December 23, 2019.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 2019;84(38):6204-6275. To be codified at 21 CFR §201, 310, 347, and 352.
- DeLeo VA. Sunscreen regulations and advice for your patients. Cutis. 2019;103:251-253.
- Matta MK, Zusterzeel R, Pilli NR, et al. Effect of sunscreen application under maximal use conditions on plasma concentration of sunscreen active ingredients: a randomized clinical trial. JAMA. 2019;321:2082-2091.
- Mayor de Blasio, health officials declare end of measles outbreak in New York City [news release]. New York, NY: City of New York; September 3, 2019. https://www1.nyc.gov/office-of-the-mayor/news/409-19/mayor-de-blasio-health-officials-declare-end-measles-outbreak-new-york-city. Accessed December 23, 2019.
- Health department reports eleven new cases of measles in Brooklyn’s Orthodox Jewish community, urges on time vaccination for all children, especially before traveling to Israel and other countries experiencing measles outbreaks [news release]. New York, NY: City of New York; November 2, 2018. https://www1.nyc.gov/site/doh/about/press/pr2018/pr091-18.page. Accessed December 23, 2019.
- Centers for Disease Control and Prevention. Measles elimination. https://www.cdc.gov/measles/elimination.html. Updated October 4, 2019. Accessed December 23, 2019.
- McKinley J. Measles outbreak: N.Y. eliminates religious exemptions for vaccinations. New York Times. June 13, 2019. https://www.nytimes.com/2019/06/13/nyregion/measles-vaccinations-new-york.html. Accessed December 23, 2019.
- FDA approves Dupixent® (dupilumab) for moderate-to-severe atopic dermatitis in adolescents [news release]. Cambridge, MA: Sanofi; March 11, 2019. http://www.news.sanofi.us/2019-03-11-FDA-approves-Dupixent-R-dupilumab-for-moderate-to-severe-atopic-dermatitis-in-adolescents. Accessed December 23, 2019.
- Simpson EL, Paller AS, Siegfried EC, et al. Efficacy and safety of dupilumab in adolescents with uncontrolled moderate to severe atopic dermatitis: a phase 3 randomized clinical trial [published online ahead of print November 6, 2019]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.3336.
- Galderma receives FDA approval for AKLIEF® (trifarotene) cream, 0.005%, the first new retinoid molecule for the treatment of acne in over 20 years [news release]. Fort Worth, TX: Galderma Laboratories, LP; October 4, 2019. https://www.multivu.com/players/English/8613051-galderma-aklief-retinoid-molecule-acne-treatment/. Accessed December 23, 2019.
- Update—Foamix receives FDA approval of AMZEEQ™ topical minocycline treatment for millions of moderate to severe acne sufferers [news release]. Bridgewater, NJ: Foamix Pharmaceuticals Ltd; October 18, 2019. http://www.foamix.com/news-releases/news-release-details/update-foamix-receives-fda-approval-amzeeqtm-topical-minocycline. Accessed December 23, 2019.
- Redfearn S. Clinical trial patient inclusion and exclusion criteria need an overhaul, say experts. CenterWatch website. April 23, 2018. https://www.centerwatch.com/cwweekly/2018/04/23/clinical-trial-patient-inclusion-and-exclusion-criteria-need-an-overhaul-say-experts. Accessed December 23, 2019.
- Tan J, Thiboutot D, Popp G, et al. Randomized phase 3 evaluation of trifarotene 50 mug/g cream treatment of moderate facial and truncal acne. J Am Acad Dermatol. 2019;80:1691-1699.
- FDA approves OTEZLA® (apremilast) for the treatment of oral ulcers associated with Behçet’s disease [news release]. Summit, NJ: Celgene; July 19, 2019. https://ir.celgene.com/press-releases/press-release-details/2019/FDA-Approves-OTEZLA-apremilast-for-the-Treatment-of-Oral-Ulcers-Associated-with-Behets-Disease/default.aspx. Accessed December 23, 2019.
- Apremilast [package insert]. Summit, NJ: Celgene Corporation; 2019.
- FDA approves first treatment to increase pain-free light exposure in patients with a rare disorder [news release]. Silver Spring, MD: US Food and Drug Administration; October 8, 2019. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-increase-pain-free-light-exposure-patients-rare-disorder. Accessed December 23, 2019.
- Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for erythropoietic protoporphyria. N Engl J Med. 2015;373:48-59.
- Light Therapy Mask recall statement. Neutrogena website. https://www.neutrogena.com/light-therapy-statement.html. Accessed December 23, 2019.
- Bromwich JE. Neutrogena recalls Light Therapy Masks, citing risk of eye injury. New York Times. July 18, 2019. https://www.nytimes.com/2019/07/18/style/neutrogena-light-therapy-mask-recall.html. Accessed December 23, 2019, 2019.
- Nguyen T. Neutrogena recalls acne mask over concerns about blue light. Chemical & Engineering News. August 6, 2019. https://cen.acs.org/safety/lab-safety/Neutrogena-recalls-acne-mask-over-concerns-about-blue-light/97/web/2019/08. Accessed November 16, 2019.
- Australian Government Department of Health, Therapeutic Goods Administration. Neutrogena Visibly Clear Light Therapy Acne Mask and Activator: Recall - potential for eye damage. https://www.tga.gov.au/alert/neutrogena-visibly-clear-light-therapy-acne-mask-and-activator. Published July 17, 2019. Accessed December 23, 2019.
- Johnson & Johnson Consumer Inc. to voluntarily recall a single lot of Johnson’s Baby Powder in the United States [press release]. New Brunswick, NJ: Johnson & Johnson Consumer Inc; October 18, 2019. https://www.factsabouttalc.com/_document/15-new-tests-from-the-same-bottle-of-johnsons-baby-powder-previously-tested-by-fda-find-no-asbestos?id=0000016e-1915-dc68-af7e-df3f147c0000. Accessed December 23, 2019.
- 15 new tests from the same bottle of Johnson’s Baby Powder previously tested by FDA find no asbestos [press release]. New Brunswick, NJ: Johnson & Johnson Consumer Inc; October 29, 2019. https://www.factsabouttalc.com/_document/johnson-johnson-consumer-inc-to-voluntarily-recall-a-single-lot-of-johnsons-baby-powder-in-the-united-states?id=0000016d-debf-d71d-a77d-dfbfebeb0000. Accessed December 23, 2019.
- Hsu T. Johnson & Johnson says recalled baby powder doesn’t have asbestos. New York Times. October 29, 2019. https://www.nytimes.com/2019/10/29/business/johnson-baby-powder-asbestos.html. Accessed December 23, 2019.
From chemical sunscreen to the measles outbreak and drug approvals to product recalls, dermatology experienced its share of firsts and controversies in 2019.
Chemical Sunscreen Controversies
Controversial concerns about the effects of chemical sunscreen on coral reefs took an unprecedented turn in the United States this last year. On February 5, 2019, an ordinance was passed in Key West, Florida, prohibiting the sale of sunscreen containing the organic UV filters oxybenzone and/or octinoxate within city limits.1 On June 25, 2019, a similar law that also included octocrylene was passed in the US Virgin Islands.2 In so doing, these areas joined Hawaii, the Republic of Palau, and parts of Mexico in restricting chemical sunscreen sales.1 Although the Key West ordinance is set to take effect in January 2021, opponents, including dermatologists who believe it will discourage sunscreen use, currently are trying to overturn the ban.3 In the US Virgin Islands, part of the ban went into effect in September 2019, with the rest of the ban set to start in March 2020.2 Companies have started to follow suit. On August 1, 2019, CVS Pharmacy announced that, by the end of 2020, it will remove oxybenzone and octinoxate from some of its store-brand chemical sunscreens.4
On February 26, 2019, the US Food and Drug Administration (FDA) proposed that there are insufficient data to determine if 12 organic UV filters—including the aforementioned oxybenzone, octinoxate, and octocrylene—are generally recognized as safe and effective (GRASE).5 Although these ingredients were listed as GRASE by the FDA in 2011, the rise in sunscreen use since then, as well as changes in sunscreen formulations, prompted the FDA to ask manufacturers to perform additional studies on safety parameters such as systemic absorption.5,6 One study conducted by the FDA itself was published in May 2019 and showed that maximal use of 4 sunscreens resulted in systemic absorption of 4 organic UV filters above 0.5 ng/mL, the FDA’s threshold for requiring nonclinical toxicology assessment. The study authors concluded that “further studies [are needed] to determine the clinical significance of these findings. [But] These results do not indicate that individuals should refrain from the use of
End of the New York City Measles Outbreak
On September 3, 2019, New York City’s largest measles outbreak in nearly 30 years was declared over. This announcement reflected the fact that 2 incubation periods for measles—42 days—had passed since the last measles patient was considered contagious. In total, there were 654 cases of measles and 52 associated hospitalizations, including 16 admissions to the intensive care unit. Most patients were younger than 18 years and unvaccinated.8
The outbreak began in October 2018 after Orthodox Jewish children from Brooklyn became infected while visiting Israel and imported the measles virus upon their return home.8,9 All 5 boroughs in New York City were ultimately affected, although 4 zip codes in Williamsburg, a neighborhood in Brooklyn with an undervaccinated Orthodox Jewish community, accounted for 72% of cases.8,10 As part of a $6 million effort to stop the outbreak, an emergency order was placed on these 4 zip codes, posing potential fines on people living or working there if they were unvaccinated.8 In addition, a bill was passed and signed into law in New York State that eliminated religious exemptions for immunizations.11 In collaboration with Jewish leaders, these efforts increased the administration of measles-mumps-rubella vaccines by 41% compared with the year before in Williamsburg and Borough Park, another heavily Orthodox Jewish neighborhood in Brooklyn.8
Drug Approvals for Pediatric Dermatology
On March 11, 2019, the IL-4/IL-13 inhibitor dupilumab became the third biologic with a pediatric dermatology indication when the FDA extended its approval to adolescents for the treatment of atopic dermatitis.12 The FDA approval was based on a randomized, double-blind, placebo-controlled trial in which 42% (34/82) of adolescents treated with dupilumab monotherapy every other week achieved 75% or more improvement in the Eczema Area and Severity Index at week 16 compared with 8% (7/85) in the placebo group (P<.001).13
In October 2019, trifarotene cream and minocycline foam were approved by the FDA for the treatment of acne in patients 9 years and older.14,15 As such, both became the first acne therapies to include patients as young as 9 years in their studies and indication—a milestone, considering the fact that children have historically been excluded from clinical trials.16 The 2 topical treatments also are noteworthy for being first in class: trifarotene cream is the only topical retinoid to selectively target the retinoic acid receptor γ and to have been studied specifically for both facial and truncal acne,14,17 and minocycline foam is the first topical tetracycline.15
Drug Approvals for Rare Dermatologic Diseases
On July 19, 2019, apremilast, a phosphodiesterase 4 inhibitor, became the first medication approved by the FDA for the treatment of adults with oral ulcers due to Behçet disease, a rare multisystem inflammatory disease.18 The FDA approval was based on a double-blind, randomized, placebo-controlled trial in which 53% (55/104) of patients receiving apremilast monotherapy were ulcer free at week 12 compared to 22% (23/103) receiving placebo (P<.0001)(ClinicalTrials.gov Identifier NCT02307513).19
On October 8, 2019, afamelanotide was approved by the FDA to increase pain-free light exposure in adults with erythropoietic protoporphyria, a rare metabolic disorder associated with photosensitivity.20 A melanocortin receptor agonist, afamelanotide is believed to confer photoprotection by increasing the production of eumelanin in the epidermis. The FDA approval was based on 2 randomized, double-blind, placebo-controlled trials, both of which found that patients given afamelanotide spent significantly more time in direct sunlight without pain compared to patients in the placebo group (P=.005 and P=.04).21
Recalls of Popular Skin Products
On July 5, 2019, Neutrogena recalled its cult-favorite Light Therapy Acne Mask. The recall was driven by rare reports of transient visual side effects due to insufficient eye protection from the mask’s light-emitting diodes.22,23 Reported in association with 0.02% of masks sold at the time of the recall, these side effects included eye pain, irritation, tearing, blurry vision, seeing spots, and changes in color vision.24 In addition, a risk for potentially irreversible eye injury from the mask was cited in people taking photosensitizing medications, such as doxycycline, and people with certain underlying eye conditions, such as retinitis pigmentosa and ocular albinism.22,24,25
Following decades of asbestos-related controversy, 1 lot of the iconic Johnson’s Baby Powder was recalled for the first time on October 18, 2019, after the FDA found subtrace levels of asbestos in 1 of the lot’s bottles.26 After the recall, Johnson & Johnson reported that 2 third-party laboratories did not ultimately find asbestos when they tested the bottle of interest as well as other bottles from the recalled lot. Three of 5 samples prepared in 1 room by the third-party laboratories initially did test positive for asbestos, but this result was attributed to the room’s air conditioner, which was found to be contaminated with asbestos. When the same samples were prepared in another room, no asbestos was detected.27 The FDA maintained there was “no indication of cross-contamination” when they originally tested the implicated bottle.28
From chemical sunscreen to the measles outbreak and drug approvals to product recalls, dermatology experienced its share of firsts and controversies in 2019.
Chemical Sunscreen Controversies
Controversial concerns about the effects of chemical sunscreen on coral reefs took an unprecedented turn in the United States this last year. On February 5, 2019, an ordinance was passed in Key West, Florida, prohibiting the sale of sunscreen containing the organic UV filters oxybenzone and/or octinoxate within city limits.1 On June 25, 2019, a similar law that also included octocrylene was passed in the US Virgin Islands.2 In so doing, these areas joined Hawaii, the Republic of Palau, and parts of Mexico in restricting chemical sunscreen sales.1 Although the Key West ordinance is set to take effect in January 2021, opponents, including dermatologists who believe it will discourage sunscreen use, currently are trying to overturn the ban.3 In the US Virgin Islands, part of the ban went into effect in September 2019, with the rest of the ban set to start in March 2020.2 Companies have started to follow suit. On August 1, 2019, CVS Pharmacy announced that, by the end of 2020, it will remove oxybenzone and octinoxate from some of its store-brand chemical sunscreens.4
On February 26, 2019, the US Food and Drug Administration (FDA) proposed that there are insufficient data to determine if 12 organic UV filters—including the aforementioned oxybenzone, octinoxate, and octocrylene—are generally recognized as safe and effective (GRASE).5 Although these ingredients were listed as GRASE by the FDA in 2011, the rise in sunscreen use since then, as well as changes in sunscreen formulations, prompted the FDA to ask manufacturers to perform additional studies on safety parameters such as systemic absorption.5,6 One study conducted by the FDA itself was published in May 2019 and showed that maximal use of 4 sunscreens resulted in systemic absorption of 4 organic UV filters above 0.5 ng/mL, the FDA’s threshold for requiring nonclinical toxicology assessment. The study authors concluded that “further studies [are needed] to determine the clinical significance of these findings. [But] These results do not indicate that individuals should refrain from the use of
End of the New York City Measles Outbreak
On September 3, 2019, New York City’s largest measles outbreak in nearly 30 years was declared over. This announcement reflected the fact that 2 incubation periods for measles—42 days—had passed since the last measles patient was considered contagious. In total, there were 654 cases of measles and 52 associated hospitalizations, including 16 admissions to the intensive care unit. Most patients were younger than 18 years and unvaccinated.8
The outbreak began in October 2018 after Orthodox Jewish children from Brooklyn became infected while visiting Israel and imported the measles virus upon their return home.8,9 All 5 boroughs in New York City were ultimately affected, although 4 zip codes in Williamsburg, a neighborhood in Brooklyn with an undervaccinated Orthodox Jewish community, accounted for 72% of cases.8,10 As part of a $6 million effort to stop the outbreak, an emergency order was placed on these 4 zip codes, posing potential fines on people living or working there if they were unvaccinated.8 In addition, a bill was passed and signed into law in New York State that eliminated religious exemptions for immunizations.11 In collaboration with Jewish leaders, these efforts increased the administration of measles-mumps-rubella vaccines by 41% compared with the year before in Williamsburg and Borough Park, another heavily Orthodox Jewish neighborhood in Brooklyn.8
Drug Approvals for Pediatric Dermatology
On March 11, 2019, the IL-4/IL-13 inhibitor dupilumab became the third biologic with a pediatric dermatology indication when the FDA extended its approval to adolescents for the treatment of atopic dermatitis.12 The FDA approval was based on a randomized, double-blind, placebo-controlled trial in which 42% (34/82) of adolescents treated with dupilumab monotherapy every other week achieved 75% or more improvement in the Eczema Area and Severity Index at week 16 compared with 8% (7/85) in the placebo group (P<.001).13
In October 2019, trifarotene cream and minocycline foam were approved by the FDA for the treatment of acne in patients 9 years and older.14,15 As such, both became the first acne therapies to include patients as young as 9 years in their studies and indication—a milestone, considering the fact that children have historically been excluded from clinical trials.16 The 2 topical treatments also are noteworthy for being first in class: trifarotene cream is the only topical retinoid to selectively target the retinoic acid receptor γ and to have been studied specifically for both facial and truncal acne,14,17 and minocycline foam is the first topical tetracycline.15
Drug Approvals for Rare Dermatologic Diseases
On July 19, 2019, apremilast, a phosphodiesterase 4 inhibitor, became the first medication approved by the FDA for the treatment of adults with oral ulcers due to Behçet disease, a rare multisystem inflammatory disease.18 The FDA approval was based on a double-blind, randomized, placebo-controlled trial in which 53% (55/104) of patients receiving apremilast monotherapy were ulcer free at week 12 compared to 22% (23/103) receiving placebo (P<.0001)(ClinicalTrials.gov Identifier NCT02307513).19
On October 8, 2019, afamelanotide was approved by the FDA to increase pain-free light exposure in adults with erythropoietic protoporphyria, a rare metabolic disorder associated with photosensitivity.20 A melanocortin receptor agonist, afamelanotide is believed to confer photoprotection by increasing the production of eumelanin in the epidermis. The FDA approval was based on 2 randomized, double-blind, placebo-controlled trials, both of which found that patients given afamelanotide spent significantly more time in direct sunlight without pain compared to patients in the placebo group (P=.005 and P=.04).21
Recalls of Popular Skin Products
On July 5, 2019, Neutrogena recalled its cult-favorite Light Therapy Acne Mask. The recall was driven by rare reports of transient visual side effects due to insufficient eye protection from the mask’s light-emitting diodes.22,23 Reported in association with 0.02% of masks sold at the time of the recall, these side effects included eye pain, irritation, tearing, blurry vision, seeing spots, and changes in color vision.24 In addition, a risk for potentially irreversible eye injury from the mask was cited in people taking photosensitizing medications, such as doxycycline, and people with certain underlying eye conditions, such as retinitis pigmentosa and ocular albinism.22,24,25
Following decades of asbestos-related controversy, 1 lot of the iconic Johnson’s Baby Powder was recalled for the first time on October 18, 2019, after the FDA found subtrace levels of asbestos in 1 of the lot’s bottles.26 After the recall, Johnson & Johnson reported that 2 third-party laboratories did not ultimately find asbestos when they tested the bottle of interest as well as other bottles from the recalled lot. Three of 5 samples prepared in 1 room by the third-party laboratories initially did test positive for asbestos, but this result was attributed to the room’s air conditioner, which was found to be contaminated with asbestos. When the same samples were prepared in another room, no asbestos was detected.27 The FDA maintained there was “no indication of cross-contamination” when they originally tested the implicated bottle.28
- Zraick K. Key West bans sunscreen containing chemicals believed to harm coral reefs. New York Times. February 7, 2019. https://www.nytimes.com/2019/02/07/us/sunscreen-coral-reef-key-west.html. Accessed December 23, 2019.
- Gies H. The U.S. Virigin Islands becomes the first American jurisdiction to ban common chemical sunscreens. Pacific Standard. July 18, 2019. https://psmag.com/environment/sunscreen-is-corals-biggest-anemone. Accessed December 23, 2019.
- Luscombe R. Republicans seek to overturn Key West ban on coral-damaging sunscreens. The Guardian. November 9, 2019. https://www.theguardian.com/us-news/2019/nov/09/key-west-sunscreen-coral-reef-backlash-skin-cancer. Accessed December 23, 2019.
- Salazar D. CVS to remove 2 chemicals from 60 store-brand sunscreens. Drug Store News. August 2, 2019. https://drugstorenews.com/retail-news/cvs-to-remove-2-chemicals-from-60-store-brand-sunscreens. Accessed December 23, 2019.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 2019;84(38):6204-6275. To be codified at 21 CFR §201, 310, 347, and 352.
- DeLeo VA. Sunscreen regulations and advice for your patients. Cutis. 2019;103:251-253.
- Matta MK, Zusterzeel R, Pilli NR, et al. Effect of sunscreen application under maximal use conditions on plasma concentration of sunscreen active ingredients: a randomized clinical trial. JAMA. 2019;321:2082-2091.
- Mayor de Blasio, health officials declare end of measles outbreak in New York City [news release]. New York, NY: City of New York; September 3, 2019. https://www1.nyc.gov/office-of-the-mayor/news/409-19/mayor-de-blasio-health-officials-declare-end-measles-outbreak-new-york-city. Accessed December 23, 2019.
- Health department reports eleven new cases of measles in Brooklyn’s Orthodox Jewish community, urges on time vaccination for all children, especially before traveling to Israel and other countries experiencing measles outbreaks [news release]. New York, NY: City of New York; November 2, 2018. https://www1.nyc.gov/site/doh/about/press/pr2018/pr091-18.page. Accessed December 23, 2019.
- Centers for Disease Control and Prevention. Measles elimination. https://www.cdc.gov/measles/elimination.html. Updated October 4, 2019. Accessed December 23, 2019.
- McKinley J. Measles outbreak: N.Y. eliminates religious exemptions for vaccinations. New York Times. June 13, 2019. https://www.nytimes.com/2019/06/13/nyregion/measles-vaccinations-new-york.html. Accessed December 23, 2019.
- FDA approves Dupixent® (dupilumab) for moderate-to-severe atopic dermatitis in adolescents [news release]. Cambridge, MA: Sanofi; March 11, 2019. http://www.news.sanofi.us/2019-03-11-FDA-approves-Dupixent-R-dupilumab-for-moderate-to-severe-atopic-dermatitis-in-adolescents. Accessed December 23, 2019.
- Simpson EL, Paller AS, Siegfried EC, et al. Efficacy and safety of dupilumab in adolescents with uncontrolled moderate to severe atopic dermatitis: a phase 3 randomized clinical trial [published online ahead of print November 6, 2019]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.3336.
- Galderma receives FDA approval for AKLIEF® (trifarotene) cream, 0.005%, the first new retinoid molecule for the treatment of acne in over 20 years [news release]. Fort Worth, TX: Galderma Laboratories, LP; October 4, 2019. https://www.multivu.com/players/English/8613051-galderma-aklief-retinoid-molecule-acne-treatment/. Accessed December 23, 2019.
- Update—Foamix receives FDA approval of AMZEEQ™ topical minocycline treatment for millions of moderate to severe acne sufferers [news release]. Bridgewater, NJ: Foamix Pharmaceuticals Ltd; October 18, 2019. http://www.foamix.com/news-releases/news-release-details/update-foamix-receives-fda-approval-amzeeqtm-topical-minocycline. Accessed December 23, 2019.
- Redfearn S. Clinical trial patient inclusion and exclusion criteria need an overhaul, say experts. CenterWatch website. April 23, 2018. https://www.centerwatch.com/cwweekly/2018/04/23/clinical-trial-patient-inclusion-and-exclusion-criteria-need-an-overhaul-say-experts. Accessed December 23, 2019.
- Tan J, Thiboutot D, Popp G, et al. Randomized phase 3 evaluation of trifarotene 50 mug/g cream treatment of moderate facial and truncal acne. J Am Acad Dermatol. 2019;80:1691-1699.
- FDA approves OTEZLA® (apremilast) for the treatment of oral ulcers associated with Behçet’s disease [news release]. Summit, NJ: Celgene; July 19, 2019. https://ir.celgene.com/press-releases/press-release-details/2019/FDA-Approves-OTEZLA-apremilast-for-the-Treatment-of-Oral-Ulcers-Associated-with-Behets-Disease/default.aspx. Accessed December 23, 2019.
- Apremilast [package insert]. Summit, NJ: Celgene Corporation; 2019.
- FDA approves first treatment to increase pain-free light exposure in patients with a rare disorder [news release]. Silver Spring, MD: US Food and Drug Administration; October 8, 2019. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-increase-pain-free-light-exposure-patients-rare-disorder. Accessed December 23, 2019.
- Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for erythropoietic protoporphyria. N Engl J Med. 2015;373:48-59.
- Light Therapy Mask recall statement. Neutrogena website. https://www.neutrogena.com/light-therapy-statement.html. Accessed December 23, 2019.
- Bromwich JE. Neutrogena recalls Light Therapy Masks, citing risk of eye injury. New York Times. July 18, 2019. https://www.nytimes.com/2019/07/18/style/neutrogena-light-therapy-mask-recall.html. Accessed December 23, 2019, 2019.
- Nguyen T. Neutrogena recalls acne mask over concerns about blue light. Chemical & Engineering News. August 6, 2019. https://cen.acs.org/safety/lab-safety/Neutrogena-recalls-acne-mask-over-concerns-about-blue-light/97/web/2019/08. Accessed November 16, 2019.
- Australian Government Department of Health, Therapeutic Goods Administration. Neutrogena Visibly Clear Light Therapy Acne Mask and Activator: Recall - potential for eye damage. https://www.tga.gov.au/alert/neutrogena-visibly-clear-light-therapy-acne-mask-and-activator. Published July 17, 2019. Accessed December 23, 2019.
- Johnson & Johnson Consumer Inc. to voluntarily recall a single lot of Johnson’s Baby Powder in the United States [press release]. New Brunswick, NJ: Johnson & Johnson Consumer Inc; October 18, 2019. https://www.factsabouttalc.com/_document/15-new-tests-from-the-same-bottle-of-johnsons-baby-powder-previously-tested-by-fda-find-no-asbestos?id=0000016e-1915-dc68-af7e-df3f147c0000. Accessed December 23, 2019.
- 15 new tests from the same bottle of Johnson’s Baby Powder previously tested by FDA find no asbestos [press release]. New Brunswick, NJ: Johnson & Johnson Consumer Inc; October 29, 2019. https://www.factsabouttalc.com/_document/johnson-johnson-consumer-inc-to-voluntarily-recall-a-single-lot-of-johnsons-baby-powder-in-the-united-states?id=0000016d-debf-d71d-a77d-dfbfebeb0000. Accessed December 23, 2019.
- Hsu T. Johnson & Johnson says recalled baby powder doesn’t have asbestos. New York Times. October 29, 2019. https://www.nytimes.com/2019/10/29/business/johnson-baby-powder-asbestos.html. Accessed December 23, 2019.
- Zraick K. Key West bans sunscreen containing chemicals believed to harm coral reefs. New York Times. February 7, 2019. https://www.nytimes.com/2019/02/07/us/sunscreen-coral-reef-key-west.html. Accessed December 23, 2019.
- Gies H. The U.S. Virigin Islands becomes the first American jurisdiction to ban common chemical sunscreens. Pacific Standard. July 18, 2019. https://psmag.com/environment/sunscreen-is-corals-biggest-anemone. Accessed December 23, 2019.
- Luscombe R. Republicans seek to overturn Key West ban on coral-damaging sunscreens. The Guardian. November 9, 2019. https://www.theguardian.com/us-news/2019/nov/09/key-west-sunscreen-coral-reef-backlash-skin-cancer. Accessed December 23, 2019.
- Salazar D. CVS to remove 2 chemicals from 60 store-brand sunscreens. Drug Store News. August 2, 2019. https://drugstorenews.com/retail-news/cvs-to-remove-2-chemicals-from-60-store-brand-sunscreens. Accessed December 23, 2019.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 2019;84(38):6204-6275. To be codified at 21 CFR §201, 310, 347, and 352.
- DeLeo VA. Sunscreen regulations and advice for your patients. Cutis. 2019;103:251-253.
- Matta MK, Zusterzeel R, Pilli NR, et al. Effect of sunscreen application under maximal use conditions on plasma concentration of sunscreen active ingredients: a randomized clinical trial. JAMA. 2019;321:2082-2091.
- Mayor de Blasio, health officials declare end of measles outbreak in New York City [news release]. New York, NY: City of New York; September 3, 2019. https://www1.nyc.gov/office-of-the-mayor/news/409-19/mayor-de-blasio-health-officials-declare-end-measles-outbreak-new-york-city. Accessed December 23, 2019.
- Health department reports eleven new cases of measles in Brooklyn’s Orthodox Jewish community, urges on time vaccination for all children, especially before traveling to Israel and other countries experiencing measles outbreaks [news release]. New York, NY: City of New York; November 2, 2018. https://www1.nyc.gov/site/doh/about/press/pr2018/pr091-18.page. Accessed December 23, 2019.
- Centers for Disease Control and Prevention. Measles elimination. https://www.cdc.gov/measles/elimination.html. Updated October 4, 2019. Accessed December 23, 2019.
- McKinley J. Measles outbreak: N.Y. eliminates religious exemptions for vaccinations. New York Times. June 13, 2019. https://www.nytimes.com/2019/06/13/nyregion/measles-vaccinations-new-york.html. Accessed December 23, 2019.
- FDA approves Dupixent® (dupilumab) for moderate-to-severe atopic dermatitis in adolescents [news release]. Cambridge, MA: Sanofi; March 11, 2019. http://www.news.sanofi.us/2019-03-11-FDA-approves-Dupixent-R-dupilumab-for-moderate-to-severe-atopic-dermatitis-in-adolescents. Accessed December 23, 2019.
- Simpson EL, Paller AS, Siegfried EC, et al. Efficacy and safety of dupilumab in adolescents with uncontrolled moderate to severe atopic dermatitis: a phase 3 randomized clinical trial [published online ahead of print November 6, 2019]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.3336.
- Galderma receives FDA approval for AKLIEF® (trifarotene) cream, 0.005%, the first new retinoid molecule for the treatment of acne in over 20 years [news release]. Fort Worth, TX: Galderma Laboratories, LP; October 4, 2019. https://www.multivu.com/players/English/8613051-galderma-aklief-retinoid-molecule-acne-treatment/. Accessed December 23, 2019.
- Update—Foamix receives FDA approval of AMZEEQ™ topical minocycline treatment for millions of moderate to severe acne sufferers [news release]. Bridgewater, NJ: Foamix Pharmaceuticals Ltd; October 18, 2019. http://www.foamix.com/news-releases/news-release-details/update-foamix-receives-fda-approval-amzeeqtm-topical-minocycline. Accessed December 23, 2019.
- Redfearn S. Clinical trial patient inclusion and exclusion criteria need an overhaul, say experts. CenterWatch website. April 23, 2018. https://www.centerwatch.com/cwweekly/2018/04/23/clinical-trial-patient-inclusion-and-exclusion-criteria-need-an-overhaul-say-experts. Accessed December 23, 2019.
- Tan J, Thiboutot D, Popp G, et al. Randomized phase 3 evaluation of trifarotene 50 mug/g cream treatment of moderate facial and truncal acne. J Am Acad Dermatol. 2019;80:1691-1699.
- FDA approves OTEZLA® (apremilast) for the treatment of oral ulcers associated with Behçet’s disease [news release]. Summit, NJ: Celgene; July 19, 2019. https://ir.celgene.com/press-releases/press-release-details/2019/FDA-Approves-OTEZLA-apremilast-for-the-Treatment-of-Oral-Ulcers-Associated-with-Behets-Disease/default.aspx. Accessed December 23, 2019.
- Apremilast [package insert]. Summit, NJ: Celgene Corporation; 2019.
- FDA approves first treatment to increase pain-free light exposure in patients with a rare disorder [news release]. Silver Spring, MD: US Food and Drug Administration; October 8, 2019. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-increase-pain-free-light-exposure-patients-rare-disorder. Accessed December 23, 2019.
- Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for erythropoietic protoporphyria. N Engl J Med. 2015;373:48-59.
- Light Therapy Mask recall statement. Neutrogena website. https://www.neutrogena.com/light-therapy-statement.html. Accessed December 23, 2019.
- Bromwich JE. Neutrogena recalls Light Therapy Masks, citing risk of eye injury. New York Times. July 18, 2019. https://www.nytimes.com/2019/07/18/style/neutrogena-light-therapy-mask-recall.html. Accessed December 23, 2019, 2019.
- Nguyen T. Neutrogena recalls acne mask over concerns about blue light. Chemical & Engineering News. August 6, 2019. https://cen.acs.org/safety/lab-safety/Neutrogena-recalls-acne-mask-over-concerns-about-blue-light/97/web/2019/08. Accessed November 16, 2019.
- Australian Government Department of Health, Therapeutic Goods Administration. Neutrogena Visibly Clear Light Therapy Acne Mask and Activator: Recall - potential for eye damage. https://www.tga.gov.au/alert/neutrogena-visibly-clear-light-therapy-acne-mask-and-activator. Published July 17, 2019. Accessed December 23, 2019.
- Johnson & Johnson Consumer Inc. to voluntarily recall a single lot of Johnson’s Baby Powder in the United States [press release]. New Brunswick, NJ: Johnson & Johnson Consumer Inc; October 18, 2019. https://www.factsabouttalc.com/_document/15-new-tests-from-the-same-bottle-of-johnsons-baby-powder-previously-tested-by-fda-find-no-asbestos?id=0000016e-1915-dc68-af7e-df3f147c0000. Accessed December 23, 2019.
- 15 new tests from the same bottle of Johnson’s Baby Powder previously tested by FDA find no asbestos [press release]. New Brunswick, NJ: Johnson & Johnson Consumer Inc; October 29, 2019. https://www.factsabouttalc.com/_document/johnson-johnson-consumer-inc-to-voluntarily-recall-a-single-lot-of-johnsons-baby-powder-in-the-united-states?id=0000016d-debf-d71d-a77d-dfbfebeb0000. Accessed December 23, 2019.
- Hsu T. Johnson & Johnson says recalled baby powder doesn’t have asbestos. New York Times. October 29, 2019. https://www.nytimes.com/2019/10/29/business/johnson-baby-powder-asbestos.html. Accessed December 23, 2019.
Resident Pearls
- Chemical sunscreen made headlines in 2019 due to concerns over coral reef toxicity and systemic absorption in humans.
- With a total of 654 cases, New York City’s largest measles outbreak in nearly 30 years ended in September 2019.
- From dupilumab for adolescent atopic dermatitis to apremilast for Behçet disease, the US Food and Drug Administration approved several therapies for pediatric dermatology and rare dermatologic conditions in 2019.
- Two popular skin care products—the Neutrogena Light Therapy Acne Mask and Johnson’s Baby Powder—were involved in recalls in 2019.

North American Blastomycosis in an Immunocompromised Patient
Blastomycosis is a systemic fungal infection that is endemic in the South Central, Midwest, and southeastern regions of the United States, as well as in provinces of Canada bordering the Great Lakes. After inhalation of Blastomyces dermatitidis spores, which are taken up by bronchopulmonary macrophages, there is an approximate 30- to 45-day incubation period. The initial response at the infected site is suppurative, which progresses to granuloma formation. Blastomyces dermatitidis most commonly infects the lungs, followed by the skin, bones, prostate, and central nervous system (CNS). Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent.
We present the case of a 38-year-old man with a medical history of human immunodeficiency virus (HIV) infection and AIDS who reported a 3- to 4-week history of respiratory and cutaneous symptoms. Initial clinical impression favored secondary syphilis; however, after laboratory evaluation and lack of response to treatment for syphilis, further investigation revealed a diagnosis of widespread cutaneous North American blastomycosis.
Case Report
A 38-year-old man with a medical history of HIV infection and AIDS presented to the emergency department at a medical center in Minneapolis, Minnesota, with a cough; chest discomfort; and concomitant nonpainful, mildly pruritic papules and plaques of 3 to 4 weeks’ duration that initially appeared on the face and ears and spread to the trunk, arms, palms, legs, and feet. He had a nonpainful ulcer on the glans penis. Symptoms began while he was living in Atlanta, Georgia, before relocating to Minneapolis. A chest radiograph was negative.
The initial clinical impression favored secondary syphilis. Intramuscular penicillin G benzathine (2.4 million U) weekly for 3 weeks was initiated by the primary care team based on clinical suspicion alone without laboratory evidence of a positive rapid plasma reagin or VDRL test. Because laboratory evaluation and lack of response to treatment did not support syphilis, dermatology consultation was requested.
The patient had a history of crack cocaine abuse. He reported sexual activity with a single female partner while living in a halfway house in the Minneapolis–St. Paul area. Physical examination showed an age-appropriate man in no acute distress who was alert and oriented. He had well-demarcated papules and plaques on the forehead, ears, nose, cutaneous and mucosal lips, chest, back, arms, legs, palms, and soles. Many of the facial papules were pink, nonscaly, and concentrated around the nose and mouth; some were umbilicated (Figure 1). Trunk and extensor papules and plaques were well demarcated, oval, and scaly; some had erosions centrally and were excoriated. Palmar papules were round and had peripheral brown hyperpigmentation and central scale (Figure 2). A 1-cm, shallow, nontender, oval ulceration withraised borders was located on the glans penis under the foreskin (Figure 3).
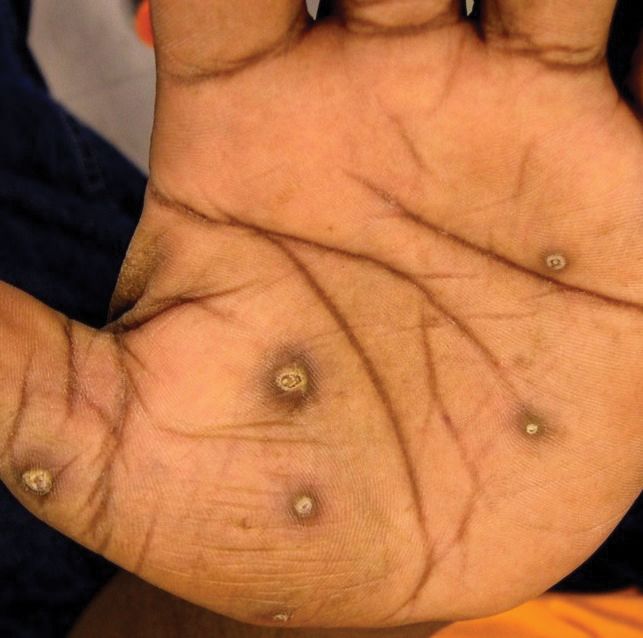
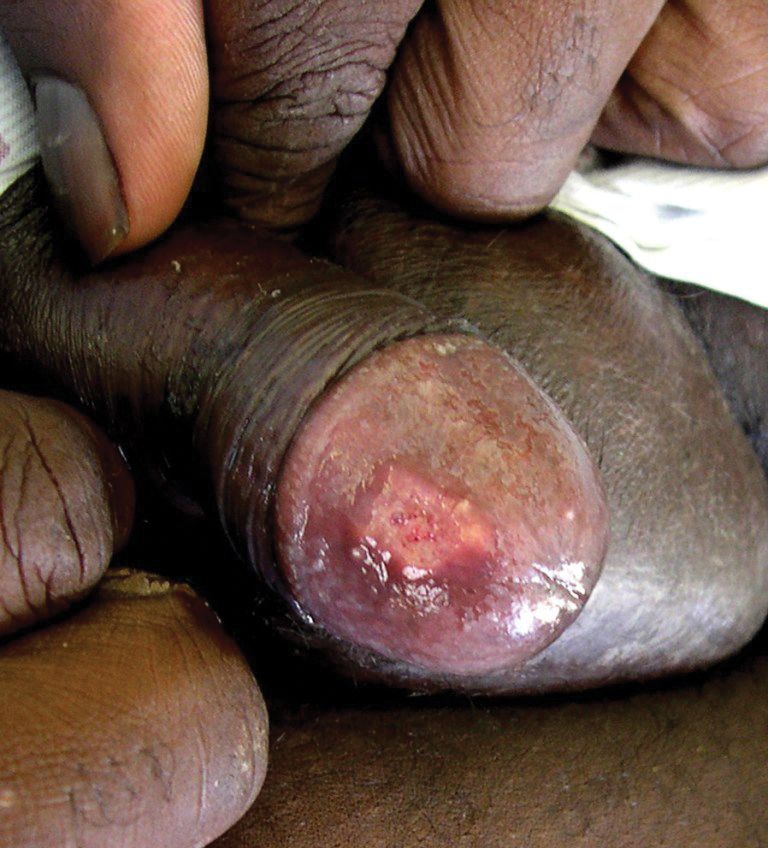
A rapid plasma reagin test was nonreactive; a fluorescent treponemal antibody absorption test was negative. Chest radiograph, magnetic resonance imaging, and electroencephalogram were normal. In addition, spinal fluid drawn from a tap was negative on India ink and Gram stain preparations and was negative for cryptococcal antigen. In addition, spinal fluid was negative for fungal and bacterial growth, as were blood cultures.
Abnormal tests included a positive enzyme-linked immunosorbent assay and Western blot test for HIV, with an absolute CD4 count of 6 cells/mL and a viral load more than 100,000 copies/mL. Urine histoplasmosis antigen was markedly elevated. A potassium hydroxide preparation was performed on the skin of the right forearm, revealing broad-based budding yeast, later confirmed on skin and sputum cultures to be B dermatitidis.
Punch biopsy from the upper back revealed a mixed acute and granulomatous infiltrate with numerous yeast forms (Figure 4A) that were highlighted by Grocott-Gomori methenamine-silver (Figure 4B) and periodic acid–Schiff (Figure 4C) stains.
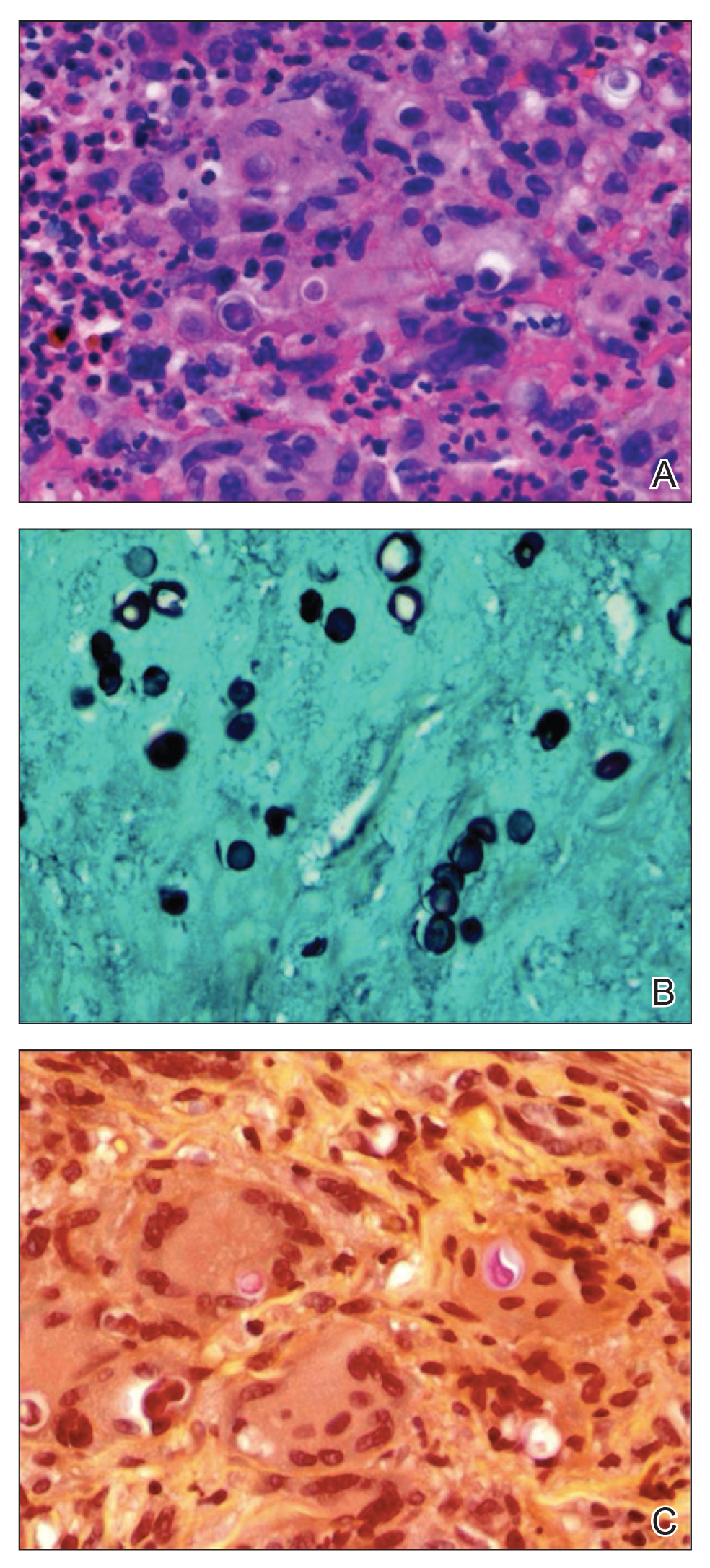
The patient was treated with intravenous amphotericin with improvement in skin lesions. A healing ointment and occlusive dressing were used on eroded skin lesions. The patient was discharged on oral itraconazole 200 mg twice daily for 6 months (for blastomycosis); oral sulfamethoxazole-trimethoprim 15 mg/kg/d every 8 hours for 21 days (for Pneumocystis carinii pneumonia prophylaxis); oral azithromycin 500 mg daily (for Mycobacterium avium-intracellulare prophylaxis); oral levetiracetam 500 mg every 12 hours (as an antiseizure agent); albuterol 90 µg per actuation; and healing ointment. He continues his chemical dependency program and is being followed by the neurology seizure clinic as well as the outpatient HIV infectious disease clinic for planned reinitiation of highly active antiretroviral therapy.
Comment
Diagnosis
Our patient had an interesting and dramatic presentation of widespread cutaneous North American blastomycosis that was initially considered to be secondary syphilis because of involvement of the palms and soles and the presence of the painless penile ulcer. In addition, the initial skin biopsy finding was considered morphologically consistent with Cryptococcus neoformans based on positive Grocott-Gomori methenamine-silver and periodic acid–Schiff stains and an equivocal mucicarmine stain. However, the potassium hydroxide preparation of skin and positive urine histoplasmosis antigen strongly suggested blastomycosis, which was confirmed by culture of B dermatitidis. The urine histoplasmosis antigen can cross-react with B dermatitidis and other mycoses (eg, Paracoccidioides brasiliensis and Penicillium marneffei); however, because the treatment of either of these mycoses is similar, the value of the test remains high.1
Skin tests and serologic markers are useful epidemiologic tools but are of inadequate sensitivity and specificity to be diagnostic for B dermatitidis. Diagnosis depends on direct examination of tissue or isolation of the fungus in culture.2
Source of Infection
The probable occult source of cutaneous infection was the lungs, given the natural history of disseminated blastomycosis; the history of cough and chest discomfort; the widespread nature of skin lesions; and the ultimate growth of rare yeast forms in sputum. Cutaneous infection generally is from disseminated disease and rarely from direct inoculation.
Unlike many other systemic dimorphic mycoses, blastomycosis usually occurs in healthy hosts and is frequently associated with point-source outbreak. Immunosuppressed patients typically develop infection following exposure to the organism, but reactivation also can occur. Blastomycosis is uncommon among HIV-infected individuals and is not recognized as an AIDS-defining illness.
In a review from Canada of 133 patients with blastomycosis, nearly half had an underlying medical condition but not one typically associated with marked immunosuppression.3 Only 2 of 133 patients had HIV infection. Overall mortality was 6.3%, and the average duration of symptoms before diagnosis was less in those who died vs those who survived the disease.3 In the setting of AIDS or other marked immunosuppression, disease usually is more severe, with multiple-system involvement, including the CNS, and can progress rapidly to death.2
Treatment
Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent. There are no randomized, blinded trials comparing antifungal agents, and data on the treatment of blastomycosis in patients infected with HIV are limited. Amphotericin B 3 mg/kg every 24 hours is recommended in life-threatening systemic disease and CNS disease as well as in patients with immune suppression, including AIDS.4 In a retrospective study of 326 patients with blastomycosis, those receiving amphotericin B had a cure rate of 86.5% with a relapse rate of 3.9%; patients receiving ketoconazole had a cure rate of 81.7% with a relapse rate of 14%.4 Although data are limited, chronic suppressive therapy generally is recommended in patients with HIV who have been treated for blastomycosis. Fluconazole, itraconazole, and ketoconazole are all used as chronic suppressive therapy; however, given the higher relapse rate observed with ketoconazole, itraconazole is preferred. Because neither ketoconazole nor itraconazole penetrates the blood-brain barrier, these drugs are not recommended in cases of CNS involvement. Patients with CNS disease or intolerance to itraconazole should be treated with fluconazole for chronic suppression.3
- Wheat J, Wheat H, Connolly P, et al. Cross-reactivity in Histoplasma capsulatum variety capsulatum antigen assays of urine samples from patients with endemic mycoses. Clin Infect Dis. 1997;24:1169-1171.
- Pappas PG, Pottage JC, Powderly WG, et al. Blastomycosis in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1992;116:847-853.
- Crampton TL, Light RB, Berg GM, et al. Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals. Clin Infect Dis. 2002;34:1310-1316. Cited by: Aberg JA. Blastomycosis and HIV. HIV In Site Knowledge Base Chapter. http://hivinsite.ucsf.edu/InSite?page=kb-05-02-09#SIX. Published April 2003. Updated January 2006. Accessed December 16, 2019.
- Chapman SW, Bradsher RW Jr, Campbell GD Jr, et al. Practice guidelines for the management of patients with blastomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:679-683.
Blastomycosis is a systemic fungal infection that is endemic in the South Central, Midwest, and southeastern regions of the United States, as well as in provinces of Canada bordering the Great Lakes. After inhalation of Blastomyces dermatitidis spores, which are taken up by bronchopulmonary macrophages, there is an approximate 30- to 45-day incubation period. The initial response at the infected site is suppurative, which progresses to granuloma formation. Blastomyces dermatitidis most commonly infects the lungs, followed by the skin, bones, prostate, and central nervous system (CNS). Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent.
We present the case of a 38-year-old man with a medical history of human immunodeficiency virus (HIV) infection and AIDS who reported a 3- to 4-week history of respiratory and cutaneous symptoms. Initial clinical impression favored secondary syphilis; however, after laboratory evaluation and lack of response to treatment for syphilis, further investigation revealed a diagnosis of widespread cutaneous North American blastomycosis.
Case Report
A 38-year-old man with a medical history of HIV infection and AIDS presented to the emergency department at a medical center in Minneapolis, Minnesota, with a cough; chest discomfort; and concomitant nonpainful, mildly pruritic papules and plaques of 3 to 4 weeks’ duration that initially appeared on the face and ears and spread to the trunk, arms, palms, legs, and feet. He had a nonpainful ulcer on the glans penis. Symptoms began while he was living in Atlanta, Georgia, before relocating to Minneapolis. A chest radiograph was negative.
The initial clinical impression favored secondary syphilis. Intramuscular penicillin G benzathine (2.4 million U) weekly for 3 weeks was initiated by the primary care team based on clinical suspicion alone without laboratory evidence of a positive rapid plasma reagin or VDRL test. Because laboratory evaluation and lack of response to treatment did not support syphilis, dermatology consultation was requested.
The patient had a history of crack cocaine abuse. He reported sexual activity with a single female partner while living in a halfway house in the Minneapolis–St. Paul area. Physical examination showed an age-appropriate man in no acute distress who was alert and oriented. He had well-demarcated papules and plaques on the forehead, ears, nose, cutaneous and mucosal lips, chest, back, arms, legs, palms, and soles. Many of the facial papules were pink, nonscaly, and concentrated around the nose and mouth; some were umbilicated (Figure 1). Trunk and extensor papules and plaques were well demarcated, oval, and scaly; some had erosions centrally and were excoriated. Palmar papules were round and had peripheral brown hyperpigmentation and central scale (Figure 2). A 1-cm, shallow, nontender, oval ulceration withraised borders was located on the glans penis under the foreskin (Figure 3).
A rapid plasma reagin test was nonreactive; a fluorescent treponemal antibody absorption test was negative. Chest radiograph, magnetic resonance imaging, and electroencephalogram were normal. In addition, spinal fluid drawn from a tap was negative on India ink and Gram stain preparations and was negative for cryptococcal antigen. In addition, spinal fluid was negative for fungal and bacterial growth, as were blood cultures.
Abnormal tests included a positive enzyme-linked immunosorbent assay and Western blot test for HIV, with an absolute CD4 count of 6 cells/mL and a viral load more than 100,000 copies/mL. Urine histoplasmosis antigen was markedly elevated. A potassium hydroxide preparation was performed on the skin of the right forearm, revealing broad-based budding yeast, later confirmed on skin and sputum cultures to be B dermatitidis.
Punch biopsy from the upper back revealed a mixed acute and granulomatous infiltrate with numerous yeast forms (Figure 4A) that were highlighted by Grocott-Gomori methenamine-silver (Figure 4B) and periodic acid–Schiff (Figure 4C) stains.
The patient was treated with intravenous amphotericin with improvement in skin lesions. A healing ointment and occlusive dressing were used on eroded skin lesions. The patient was discharged on oral itraconazole 200 mg twice daily for 6 months (for blastomycosis); oral sulfamethoxazole-trimethoprim 15 mg/kg/d every 8 hours for 21 days (for Pneumocystis carinii pneumonia prophylaxis); oral azithromycin 500 mg daily (for Mycobacterium avium-intracellulare prophylaxis); oral levetiracetam 500 mg every 12 hours (as an antiseizure agent); albuterol 90 µg per actuation; and healing ointment. He continues his chemical dependency program and is being followed by the neurology seizure clinic as well as the outpatient HIV infectious disease clinic for planned reinitiation of highly active antiretroviral therapy.
Comment
Diagnosis
Our patient had an interesting and dramatic presentation of widespread cutaneous North American blastomycosis that was initially considered to be secondary syphilis because of involvement of the palms and soles and the presence of the painless penile ulcer. In addition, the initial skin biopsy finding was considered morphologically consistent with Cryptococcus neoformans based on positive Grocott-Gomori methenamine-silver and periodic acid–Schiff stains and an equivocal mucicarmine stain. However, the potassium hydroxide preparation of skin and positive urine histoplasmosis antigen strongly suggested blastomycosis, which was confirmed by culture of B dermatitidis. The urine histoplasmosis antigen can cross-react with B dermatitidis and other mycoses (eg, Paracoccidioides brasiliensis and Penicillium marneffei); however, because the treatment of either of these mycoses is similar, the value of the test remains high.1
Skin tests and serologic markers are useful epidemiologic tools but are of inadequate sensitivity and specificity to be diagnostic for B dermatitidis. Diagnosis depends on direct examination of tissue or isolation of the fungus in culture.2
Source of Infection
The probable occult source of cutaneous infection was the lungs, given the natural history of disseminated blastomycosis; the history of cough and chest discomfort; the widespread nature of skin lesions; and the ultimate growth of rare yeast forms in sputum. Cutaneous infection generally is from disseminated disease and rarely from direct inoculation.
Unlike many other systemic dimorphic mycoses, blastomycosis usually occurs in healthy hosts and is frequently associated with point-source outbreak. Immunosuppressed patients typically develop infection following exposure to the organism, but reactivation also can occur. Blastomycosis is uncommon among HIV-infected individuals and is not recognized as an AIDS-defining illness.
In a review from Canada of 133 patients with blastomycosis, nearly half had an underlying medical condition but not one typically associated with marked immunosuppression.3 Only 2 of 133 patients had HIV infection. Overall mortality was 6.3%, and the average duration of symptoms before diagnosis was less in those who died vs those who survived the disease.3 In the setting of AIDS or other marked immunosuppression, disease usually is more severe, with multiple-system involvement, including the CNS, and can progress rapidly to death.2
Treatment
Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent. There are no randomized, blinded trials comparing antifungal agents, and data on the treatment of blastomycosis in patients infected with HIV are limited. Amphotericin B 3 mg/kg every 24 hours is recommended in life-threatening systemic disease and CNS disease as well as in patients with immune suppression, including AIDS.4 In a retrospective study of 326 patients with blastomycosis, those receiving amphotericin B had a cure rate of 86.5% with a relapse rate of 3.9%; patients receiving ketoconazole had a cure rate of 81.7% with a relapse rate of 14%.4 Although data are limited, chronic suppressive therapy generally is recommended in patients with HIV who have been treated for blastomycosis. Fluconazole, itraconazole, and ketoconazole are all used as chronic suppressive therapy; however, given the higher relapse rate observed with ketoconazole, itraconazole is preferred. Because neither ketoconazole nor itraconazole penetrates the blood-brain barrier, these drugs are not recommended in cases of CNS involvement. Patients with CNS disease or intolerance to itraconazole should be treated with fluconazole for chronic suppression.3
Blastomycosis is a systemic fungal infection that is endemic in the South Central, Midwest, and southeastern regions of the United States, as well as in provinces of Canada bordering the Great Lakes. After inhalation of Blastomyces dermatitidis spores, which are taken up by bronchopulmonary macrophages, there is an approximate 30- to 45-day incubation period. The initial response at the infected site is suppurative, which progresses to granuloma formation. Blastomyces dermatitidis most commonly infects the lungs, followed by the skin, bones, prostate, and central nervous system (CNS). Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent.
We present the case of a 38-year-old man with a medical history of human immunodeficiency virus (HIV) infection and AIDS who reported a 3- to 4-week history of respiratory and cutaneous symptoms. Initial clinical impression favored secondary syphilis; however, after laboratory evaluation and lack of response to treatment for syphilis, further investigation revealed a diagnosis of widespread cutaneous North American blastomycosis.
Case Report
A 38-year-old man with a medical history of HIV infection and AIDS presented to the emergency department at a medical center in Minneapolis, Minnesota, with a cough; chest discomfort; and concomitant nonpainful, mildly pruritic papules and plaques of 3 to 4 weeks’ duration that initially appeared on the face and ears and spread to the trunk, arms, palms, legs, and feet. He had a nonpainful ulcer on the glans penis. Symptoms began while he was living in Atlanta, Georgia, before relocating to Minneapolis. A chest radiograph was negative.
The initial clinical impression favored secondary syphilis. Intramuscular penicillin G benzathine (2.4 million U) weekly for 3 weeks was initiated by the primary care team based on clinical suspicion alone without laboratory evidence of a positive rapid plasma reagin or VDRL test. Because laboratory evaluation and lack of response to treatment did not support syphilis, dermatology consultation was requested.
The patient had a history of crack cocaine abuse. He reported sexual activity with a single female partner while living in a halfway house in the Minneapolis–St. Paul area. Physical examination showed an age-appropriate man in no acute distress who was alert and oriented. He had well-demarcated papules and plaques on the forehead, ears, nose, cutaneous and mucosal lips, chest, back, arms, legs, palms, and soles. Many of the facial papules were pink, nonscaly, and concentrated around the nose and mouth; some were umbilicated (Figure 1). Trunk and extensor papules and plaques were well demarcated, oval, and scaly; some had erosions centrally and were excoriated. Palmar papules were round and had peripheral brown hyperpigmentation and central scale (Figure 2). A 1-cm, shallow, nontender, oval ulceration withraised borders was located on the glans penis under the foreskin (Figure 3).
A rapid plasma reagin test was nonreactive; a fluorescent treponemal antibody absorption test was negative. Chest radiograph, magnetic resonance imaging, and electroencephalogram were normal. In addition, spinal fluid drawn from a tap was negative on India ink and Gram stain preparations and was negative for cryptococcal antigen. In addition, spinal fluid was negative for fungal and bacterial growth, as were blood cultures.
Abnormal tests included a positive enzyme-linked immunosorbent assay and Western blot test for HIV, with an absolute CD4 count of 6 cells/mL and a viral load more than 100,000 copies/mL. Urine histoplasmosis antigen was markedly elevated. A potassium hydroxide preparation was performed on the skin of the right forearm, revealing broad-based budding yeast, later confirmed on skin and sputum cultures to be B dermatitidis.
Punch biopsy from the upper back revealed a mixed acute and granulomatous infiltrate with numerous yeast forms (Figure 4A) that were highlighted by Grocott-Gomori methenamine-silver (Figure 4B) and periodic acid–Schiff (Figure 4C) stains.
The patient was treated with intravenous amphotericin with improvement in skin lesions. A healing ointment and occlusive dressing were used on eroded skin lesions. The patient was discharged on oral itraconazole 200 mg twice daily for 6 months (for blastomycosis); oral sulfamethoxazole-trimethoprim 15 mg/kg/d every 8 hours for 21 days (for Pneumocystis carinii pneumonia prophylaxis); oral azithromycin 500 mg daily (for Mycobacterium avium-intracellulare prophylaxis); oral levetiracetam 500 mg every 12 hours (as an antiseizure agent); albuterol 90 µg per actuation; and healing ointment. He continues his chemical dependency program and is being followed by the neurology seizure clinic as well as the outpatient HIV infectious disease clinic for planned reinitiation of highly active antiretroviral therapy.
Comment
Diagnosis
Our patient had an interesting and dramatic presentation of widespread cutaneous North American blastomycosis that was initially considered to be secondary syphilis because of involvement of the palms and soles and the presence of the painless penile ulcer. In addition, the initial skin biopsy finding was considered morphologically consistent with Cryptococcus neoformans based on positive Grocott-Gomori methenamine-silver and periodic acid–Schiff stains and an equivocal mucicarmine stain. However, the potassium hydroxide preparation of skin and positive urine histoplasmosis antigen strongly suggested blastomycosis, which was confirmed by culture of B dermatitidis. The urine histoplasmosis antigen can cross-react with B dermatitidis and other mycoses (eg, Paracoccidioides brasiliensis and Penicillium marneffei); however, because the treatment of either of these mycoses is similar, the value of the test remains high.1
Skin tests and serologic markers are useful epidemiologic tools but are of inadequate sensitivity and specificity to be diagnostic for B dermatitidis. Diagnosis depends on direct examination of tissue or isolation of the fungus in culture.2
Source of Infection
The probable occult source of cutaneous infection was the lungs, given the natural history of disseminated blastomycosis; the history of cough and chest discomfort; the widespread nature of skin lesions; and the ultimate growth of rare yeast forms in sputum. Cutaneous infection generally is from disseminated disease and rarely from direct inoculation.
Unlike many other systemic dimorphic mycoses, blastomycosis usually occurs in healthy hosts and is frequently associated with point-source outbreak. Immunosuppressed patients typically develop infection following exposure to the organism, but reactivation also can occur. Blastomycosis is uncommon among HIV-infected individuals and is not recognized as an AIDS-defining illness.
In a review from Canada of 133 patients with blastomycosis, nearly half had an underlying medical condition but not one typically associated with marked immunosuppression.3 Only 2 of 133 patients had HIV infection. Overall mortality was 6.3%, and the average duration of symptoms before diagnosis was less in those who died vs those who survived the disease.3 In the setting of AIDS or other marked immunosuppression, disease usually is more severe, with multiple-system involvement, including the CNS, and can progress rapidly to death.2
Treatment
Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent. There are no randomized, blinded trials comparing antifungal agents, and data on the treatment of blastomycosis in patients infected with HIV are limited. Amphotericin B 3 mg/kg every 24 hours is recommended in life-threatening systemic disease and CNS disease as well as in patients with immune suppression, including AIDS.4 In a retrospective study of 326 patients with blastomycosis, those receiving amphotericin B had a cure rate of 86.5% with a relapse rate of 3.9%; patients receiving ketoconazole had a cure rate of 81.7% with a relapse rate of 14%.4 Although data are limited, chronic suppressive therapy generally is recommended in patients with HIV who have been treated for blastomycosis. Fluconazole, itraconazole, and ketoconazole are all used as chronic suppressive therapy; however, given the higher relapse rate observed with ketoconazole, itraconazole is preferred. Because neither ketoconazole nor itraconazole penetrates the blood-brain barrier, these drugs are not recommended in cases of CNS involvement. Patients with CNS disease or intolerance to itraconazole should be treated with fluconazole for chronic suppression.3
- Wheat J, Wheat H, Connolly P, et al. Cross-reactivity in Histoplasma capsulatum variety capsulatum antigen assays of urine samples from patients with endemic mycoses. Clin Infect Dis. 1997;24:1169-1171.
- Pappas PG, Pottage JC, Powderly WG, et al. Blastomycosis in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1992;116:847-853.
- Crampton TL, Light RB, Berg GM, et al. Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals. Clin Infect Dis. 2002;34:1310-1316. Cited by: Aberg JA. Blastomycosis and HIV. HIV In Site Knowledge Base Chapter. http://hivinsite.ucsf.edu/InSite?page=kb-05-02-09#SIX. Published April 2003. Updated January 2006. Accessed December 16, 2019.
- Chapman SW, Bradsher RW Jr, Campbell GD Jr, et al. Practice guidelines for the management of patients with blastomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:679-683.
- Wheat J, Wheat H, Connolly P, et al. Cross-reactivity in Histoplasma capsulatum variety capsulatum antigen assays of urine samples from patients with endemic mycoses. Clin Infect Dis. 1997;24:1169-1171.
- Pappas PG, Pottage JC, Powderly WG, et al. Blastomycosis in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1992;116:847-853.
- Crampton TL, Light RB, Berg GM, et al. Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals. Clin Infect Dis. 2002;34:1310-1316. Cited by: Aberg JA. Blastomycosis and HIV. HIV In Site Knowledge Base Chapter. http://hivinsite.ucsf.edu/InSite?page=kb-05-02-09#SIX. Published April 2003. Updated January 2006. Accessed December 16, 2019.
- Chapman SW, Bradsher RW Jr, Campbell GD Jr, et al. Practice guidelines for the management of patients with blastomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:679-683.
Practice Points
- Blastomycosis generally produces a pulmonary form of the disease and, to a lesser extent, extrapulmonary forms, such as cutaneous, osteoarticular, and genitourinary.
- Blastomycosis can be diagnosed by culture, direct visualization of the yeast in affected tissue, antigen testing, or a combination of these methods.
- After inhalation of Blastomyces dermatitidis spores, which are taken up by bronchopulmonary macrophages, there is an approximate 30- to 45-day incubation period.
Spotlight on SMA: The urgent need for early diagnosis in spinal muscular atrophy

The diagnosis of spinal muscular atrophy (SMA), especially Type 1, is a medical emergency, as SMA is a leading genetic cause of death in infants. In infants with SMA Type 1, the onset of irreversible denervation occurs within the first 3 months with loss of 90% of motor units occurring within 6 months of age.
This supplement examines the clinical implications of delayed diagnosis of SMA, as well as assessment tools, treatment methods, and resources that are available for physicians, patients, and caregivers to better manage this rare disease.
The diagnosis of spinal muscular atrophy (SMA), especially Type 1, is a medical emergency, as SMA is a leading genetic cause of death in infants. In infants with SMA Type 1, the onset of irreversible denervation occurs within the first 3 months with loss of 90% of motor units occurring within 6 months of age.
This supplement examines the clinical implications of delayed diagnosis of SMA, as well as assessment tools, treatment methods, and resources that are available for physicians, patients, and caregivers to better manage this rare disease.
The diagnosis of spinal muscular atrophy (SMA), especially Type 1, is a medical emergency, as SMA is a leading genetic cause of death in infants. In infants with SMA Type 1, the onset of irreversible denervation occurs within the first 3 months with loss of 90% of motor units occurring within 6 months of age.
This supplement examines the clinical implications of delayed diagnosis of SMA, as well as assessment tools, treatment methods, and resources that are available for physicians, patients, and caregivers to better manage this rare disease.
Phase 2 studies show potential of FcRn blockade in primary ITP
ORLANDO – Treatments targeted to the neonatal Fc receptor are showing promise in phase 2 studies in primary immune thrombocytopenia, investigators reported at the annual meeting of the American Society of Hematology.
Encouraging outcomes support the continued phase 3 development of these agents, which are designed to block the neonatal Fc receptor (FcRn) in patients with this IgG-mediated disease.
Blocking FcRN is intended to prevent recycling of IgG, resulting in IgG degradation, according to the authors of studies evaluating rozanolixizumab, a subcutaneously administered monoclonal antibody, and efgartigimod, an intravenously administered antibody fragment, in primary immune thrombocytopenia (ITP).
Rozanolixizumab
Results of the phase 2 study of rozanolixizumab demonstrated that this agent reduced IgG levels and improved platelet counts at all doses tested, according to the investigators, led by Tadeusz Robak, MD, of the department of hematology at the Medical University of Lodz (Poland).
Efficacy endpoints were seen more quickly – by day 8 of treatment – with single subcutaneous infusions at higher doses, according to the researchers.
Headaches of mild to moderate severity were noted at higher doses, and no patients left the study because of adverse events, they reported.
“These safety, tolerability, and efficacy data support phase 3 development of rozanolixizumab in patients with primary ITP,” wrote Dr. Robak and coauthors in the abstract for their study.
A total of 66 adult patients with primary ITP were enrolled and treated with single or multiple subcutaneous doses of rozanolixizumab administered at 1-week intervals.
Baseline characteristics suggested a “difficult-to-treat” patient cohort that had a median ITP duration of nearly 6 years and a median of four prior therapies, including thrombopoietin receptor agonists in about one-third of patients, according to the investigators.
Platelet counts of at least 50 x 109/L were achieved by day 8 in more than half of patients who received single doses of rozanolixizumab at higher dose levels of 15 mg/kg (58.3%) and 20 mg/kg (54.5%), Dr. Robak and colleagues reported.
Mild to moderate headaches were seen in about 40% of patients over an 8-week observation period. There were no serious infections and, of four serious adverse events occurring during the study, none were deemed to be treatment related, according to the investigators.
“People who have primary ITP may experience low platelet count that can put them at risk for severe bleeding, and there are limited options that provide a rapid increase in platelet count to reduce this risk,” Dr. Robak said in an interview. “These data build on the growing body of evidence that suggest targeting the FcRn pathway could have the potential to transform the treatment experience for people with rare IgG autoantibody–mediated diseases such as primary ITP.”
Efgartigimod
Substantial reductions in IgG levels and clinically relevant increases in platelet counts were seen following a 3-week treatment cycle with efgartigimod in patients with treatment-refractory ITP, according to investigator Adrian C. Newland, MB, BCh, of the Royal London Hospital and coinvestigators.
The human IgG1 antibody Fc-fragment, a natural ligand of FcRN, is engineered to have increased affinity to FcRn, while preserving its pH‐dependent binding, according to the investigators.
Efgartigimod treatment was well tolerated and reduced the proportion of patients with bleeding in the phase 2 study presented at ASH 2019.
“This suggests that targeted IgG reduction with efgartigimod is a potential new treatment modality in primary ITP, and warrants evaluation of longer-term treatment in a larger phase 3 study,” the investigators reported in the abstract for their study.
The report described 38 patients randomized to four weekly intravenous infusions of placebo or efgartigimod at one of two dosing levels. Patients had long-standing ITP, with a median 4.8 years disease duration, and all had either failed splenectomy or had inadequate response to prior treatment.
Efgartigimod treatment rapidly reduced total IgG in all patients who received it, with a mean change from baseline of up to 63.7%, according to investigators.
Platelet counts favored the investigational treatment over placebo by several measures. Platelet counts of at least 50 x 109/L on two or more occasions were seen in 46% of efgartigimod-treated patients and 25% of the placebo group; that platelet count was achieved for 10 or more days in 38% and 0% of the efgartigimod and placebo groups, respectively.
Treatment was well tolerated, according to the investigators, who said there were “no dose-related safety observations.” Full results of the phase 2 investigation were published in the American Journal of Hematology, concurrent with the meeting (2019 Dec 10. doi.org/10.1002/ajh.25680).
The study of rozanolixizumab was supported by UCB; Dr. Robak reported disclosures related to UCB (honoraria, research funding), as well as Takeda, Janssen, Amgen, Roche, AbbVie, Gilead, BeiGene, Acerta, and MorphoSys. The study of efgartigimod was supported by argenx; Dr. Newland reported disclosures related to argenx, Novartis, Angle, Amgen, Ono Pharmaceutical, Shionogi, Rigel, and Dova Pharmaceuticals.
SOURCEs: Robak T et al. ASH 2019, Abstract 897; Newland AC et al. ASH 2019, Abstract 895.
ORLANDO – Treatments targeted to the neonatal Fc receptor are showing promise in phase 2 studies in primary immune thrombocytopenia, investigators reported at the annual meeting of the American Society of Hematology.
Encouraging outcomes support the continued phase 3 development of these agents, which are designed to block the neonatal Fc receptor (FcRn) in patients with this IgG-mediated disease.
Blocking FcRN is intended to prevent recycling of IgG, resulting in IgG degradation, according to the authors of studies evaluating rozanolixizumab, a subcutaneously administered monoclonal antibody, and efgartigimod, an intravenously administered antibody fragment, in primary immune thrombocytopenia (ITP).
Rozanolixizumab
Results of the phase 2 study of rozanolixizumab demonstrated that this agent reduced IgG levels and improved platelet counts at all doses tested, according to the investigators, led by Tadeusz Robak, MD, of the department of hematology at the Medical University of Lodz (Poland).
Efficacy endpoints were seen more quickly – by day 8 of treatment – with single subcutaneous infusions at higher doses, according to the researchers.
Headaches of mild to moderate severity were noted at higher doses, and no patients left the study because of adverse events, they reported.
“These safety, tolerability, and efficacy data support phase 3 development of rozanolixizumab in patients with primary ITP,” wrote Dr. Robak and coauthors in the abstract for their study.
A total of 66 adult patients with primary ITP were enrolled and treated with single or multiple subcutaneous doses of rozanolixizumab administered at 1-week intervals.
Baseline characteristics suggested a “difficult-to-treat” patient cohort that had a median ITP duration of nearly 6 years and a median of four prior therapies, including thrombopoietin receptor agonists in about one-third of patients, according to the investigators.
Platelet counts of at least 50 x 109/L were achieved by day 8 in more than half of patients who received single doses of rozanolixizumab at higher dose levels of 15 mg/kg (58.3%) and 20 mg/kg (54.5%), Dr. Robak and colleagues reported.
Mild to moderate headaches were seen in about 40% of patients over an 8-week observation period. There were no serious infections and, of four serious adverse events occurring during the study, none were deemed to be treatment related, according to the investigators.
“People who have primary ITP may experience low platelet count that can put them at risk for severe bleeding, and there are limited options that provide a rapid increase in platelet count to reduce this risk,” Dr. Robak said in an interview. “These data build on the growing body of evidence that suggest targeting the FcRn pathway could have the potential to transform the treatment experience for people with rare IgG autoantibody–mediated diseases such as primary ITP.”
Efgartigimod
Substantial reductions in IgG levels and clinically relevant increases in platelet counts were seen following a 3-week treatment cycle with efgartigimod in patients with treatment-refractory ITP, according to investigator Adrian C. Newland, MB, BCh, of the Royal London Hospital and coinvestigators.
The human IgG1 antibody Fc-fragment, a natural ligand of FcRN, is engineered to have increased affinity to FcRn, while preserving its pH‐dependent binding, according to the investigators.
Efgartigimod treatment was well tolerated and reduced the proportion of patients with bleeding in the phase 2 study presented at ASH 2019.
“This suggests that targeted IgG reduction with efgartigimod is a potential new treatment modality in primary ITP, and warrants evaluation of longer-term treatment in a larger phase 3 study,” the investigators reported in the abstract for their study.
The report described 38 patients randomized to four weekly intravenous infusions of placebo or efgartigimod at one of two dosing levels. Patients had long-standing ITP, with a median 4.8 years disease duration, and all had either failed splenectomy or had inadequate response to prior treatment.
Efgartigimod treatment rapidly reduced total IgG in all patients who received it, with a mean change from baseline of up to 63.7%, according to investigators.
Platelet counts favored the investigational treatment over placebo by several measures. Platelet counts of at least 50 x 109/L on two or more occasions were seen in 46% of efgartigimod-treated patients and 25% of the placebo group; that platelet count was achieved for 10 or more days in 38% and 0% of the efgartigimod and placebo groups, respectively.
Treatment was well tolerated, according to the investigators, who said there were “no dose-related safety observations.” Full results of the phase 2 investigation were published in the American Journal of Hematology, concurrent with the meeting (2019 Dec 10. doi.org/10.1002/ajh.25680).
The study of rozanolixizumab was supported by UCB; Dr. Robak reported disclosures related to UCB (honoraria, research funding), as well as Takeda, Janssen, Amgen, Roche, AbbVie, Gilead, BeiGene, Acerta, and MorphoSys. The study of efgartigimod was supported by argenx; Dr. Newland reported disclosures related to argenx, Novartis, Angle, Amgen, Ono Pharmaceutical, Shionogi, Rigel, and Dova Pharmaceuticals.
SOURCEs: Robak T et al. ASH 2019, Abstract 897; Newland AC et al. ASH 2019, Abstract 895.
ORLANDO – Treatments targeted to the neonatal Fc receptor are showing promise in phase 2 studies in primary immune thrombocytopenia, investigators reported at the annual meeting of the American Society of Hematology.
Encouraging outcomes support the continued phase 3 development of these agents, which are designed to block the neonatal Fc receptor (FcRn) in patients with this IgG-mediated disease.
Blocking FcRN is intended to prevent recycling of IgG, resulting in IgG degradation, according to the authors of studies evaluating rozanolixizumab, a subcutaneously administered monoclonal antibody, and efgartigimod, an intravenously administered antibody fragment, in primary immune thrombocytopenia (ITP).
Rozanolixizumab
Results of the phase 2 study of rozanolixizumab demonstrated that this agent reduced IgG levels and improved platelet counts at all doses tested, according to the investigators, led by Tadeusz Robak, MD, of the department of hematology at the Medical University of Lodz (Poland).
Efficacy endpoints were seen more quickly – by day 8 of treatment – with single subcutaneous infusions at higher doses, according to the researchers.
Headaches of mild to moderate severity were noted at higher doses, and no patients left the study because of adverse events, they reported.
“These safety, tolerability, and efficacy data support phase 3 development of rozanolixizumab in patients with primary ITP,” wrote Dr. Robak and coauthors in the abstract for their study.
A total of 66 adult patients with primary ITP were enrolled and treated with single or multiple subcutaneous doses of rozanolixizumab administered at 1-week intervals.
Baseline characteristics suggested a “difficult-to-treat” patient cohort that had a median ITP duration of nearly 6 years and a median of four prior therapies, including thrombopoietin receptor agonists in about one-third of patients, according to the investigators.
Platelet counts of at least 50 x 109/L were achieved by day 8 in more than half of patients who received single doses of rozanolixizumab at higher dose levels of 15 mg/kg (58.3%) and 20 mg/kg (54.5%), Dr. Robak and colleagues reported.
Mild to moderate headaches were seen in about 40% of patients over an 8-week observation period. There were no serious infections and, of four serious adverse events occurring during the study, none were deemed to be treatment related, according to the investigators.
“People who have primary ITP may experience low platelet count that can put them at risk for severe bleeding, and there are limited options that provide a rapid increase in platelet count to reduce this risk,” Dr. Robak said in an interview. “These data build on the growing body of evidence that suggest targeting the FcRn pathway could have the potential to transform the treatment experience for people with rare IgG autoantibody–mediated diseases such as primary ITP.”
Efgartigimod
Substantial reductions in IgG levels and clinically relevant increases in platelet counts were seen following a 3-week treatment cycle with efgartigimod in patients with treatment-refractory ITP, according to investigator Adrian C. Newland, MB, BCh, of the Royal London Hospital and coinvestigators.
The human IgG1 antibody Fc-fragment, a natural ligand of FcRN, is engineered to have increased affinity to FcRn, while preserving its pH‐dependent binding, according to the investigators.
Efgartigimod treatment was well tolerated and reduced the proportion of patients with bleeding in the phase 2 study presented at ASH 2019.
“This suggests that targeted IgG reduction with efgartigimod is a potential new treatment modality in primary ITP, and warrants evaluation of longer-term treatment in a larger phase 3 study,” the investigators reported in the abstract for their study.
The report described 38 patients randomized to four weekly intravenous infusions of placebo or efgartigimod at one of two dosing levels. Patients had long-standing ITP, with a median 4.8 years disease duration, and all had either failed splenectomy or had inadequate response to prior treatment.
Efgartigimod treatment rapidly reduced total IgG in all patients who received it, with a mean change from baseline of up to 63.7%, according to investigators.
Platelet counts favored the investigational treatment over placebo by several measures. Platelet counts of at least 50 x 109/L on two or more occasions were seen in 46% of efgartigimod-treated patients and 25% of the placebo group; that platelet count was achieved for 10 or more days in 38% and 0% of the efgartigimod and placebo groups, respectively.
Treatment was well tolerated, according to the investigators, who said there were “no dose-related safety observations.” Full results of the phase 2 investigation were published in the American Journal of Hematology, concurrent with the meeting (2019 Dec 10. doi.org/10.1002/ajh.25680).
The study of rozanolixizumab was supported by UCB; Dr. Robak reported disclosures related to UCB (honoraria, research funding), as well as Takeda, Janssen, Amgen, Roche, AbbVie, Gilead, BeiGene, Acerta, and MorphoSys. The study of efgartigimod was supported by argenx; Dr. Newland reported disclosures related to argenx, Novartis, Angle, Amgen, Ono Pharmaceutical, Shionogi, Rigel, and Dova Pharmaceuticals.
SOURCEs: Robak T et al. ASH 2019, Abstract 897; Newland AC et al. ASH 2019, Abstract 895.
REPORTING FROM ASH 2019
Sutimlimab boosts hemoglobin, quality of life in cold agglutinin disease
ORLANDO – An investigational selective inhibitor of the complement pathway, sutimlimab, induced rapid and sustained benefits in patients with cold agglutinin disease, a rare autoimmune hemolytic anemia with no currently approved effective therapies.

Among 24 patients with cold agglutinin disease who received at least one dose of sutimlimab in a phase 3 trial, 20 had a mean increase in hemoglobin of at least 1 g/dL, and 17 remained transfusion free from weeks 5 to 26 following sutimlimab infusion.
“Sutimlimab has the potential to change treatment practices for patients with this disease,” said lead author Alexander Röth, MD, from the University of Duisburg-Essen (Germany), at a late-breaking abstract session at the annual meeting of the American Society of Hematology.
Mean total bilirubin rapidly normalized within 1-3 weeks of infusion of sutimlimab, and patients had a mean improvement of 11 points on the Functional Assessment of Chronic Illness Therapy–Fatigue scale (FACIT-F), indicating a substantial improvement in their quality of life, Dr. Röth said.
Cold agglutinin disease is an acquired hemolytic anemia with an underlying lymphoproliferative disorder. The estimated prevalence of the disease is 16 per 1 million persons. The disease is characterized by hemolysis driven by activation of the complement pathway, leading to opsonization of erythrocytes (coating of erythrocytes with particles that facilitate phagocytosis and other immune reactions), extravascular hemolysis (primarily in the liver), intravascular hemolysis, and anemia.
Patients experience severe fatigue and poor quality of life, as well as increased risk for thrombosis and mortality, compared with matched cohorts.
Sutimlimab is a humanized monoclonal antibody that blocks the C1s component of the classical complement pathway, thereby stopping pathway activation while leaving alternative lectin pathways intact.
Dr. Röth presented results of the phase 3, open-label Cardinal study. Patients with cold agglutinin disease with baseline hemoglobin of 10 g/dL or less, active hemolysis signaled by total bilirubin levels above normal, and at least one blood transfusion within the past 6 months were eligible for the study. Patients with secondary cold agglutinin syndrome or rituximab therapy within the last 3 months or combination therapies within the last 6 months were excluded.
Sutimlimab was delivered intravenously at a dose of 6.5 g for patients under 75 kg in weight and 7.5 g for those 75 kg and over at day 0 and 7, then every 2 weeks thereafter.
A total of 24 patients with a mean age of 71 years were enrolled. Of the 24 patients, 15 (62.5%) were women.
The patients had received a mean of 3.2 transfusions (range 1-19) in the previous 6 months, and 15 had received one or more prior targeted therapies for the disease within the last 5 years. The mean baseline hemoglobin level was 8.6 (range 4.9-11.1) g/dL.
Hemoglobin levels increased rapidly after the first infusion, with a mean increase of 1.2 g/dL at the end of week 1, and 2.3 g/dL after week 3.
The estimated mean increase at treatment assessment (an average of weeks 23, 25, and 26) – the primary endpoint – was 2.6 g/dL, exceeding the prespecified increase of at least 2 g/dL. Normalization of hemoglobin to 12 g/dL or greater was an alternative primary endpoint. The trial met the primary endpoint, with 13 of 24 patients (54.2%) achieving either of the two prespecified events.
The mean overall hemoglobin level was maintained above 11 g/dL after week 3. Of the 24 patients, 20 had hemoglobin increases of 1 g/dL or greater.
Mean total bilirubin, a marker of hemolysis, dropped markedly within hours of infusion and was normalized by week 3.
As noted before, patient quality of life, as measured by the FACIT-F scale, improved by a mean of 11 points from a mean baseline of 32 out of 52 points.
All but two patients had one or more treatment-emergent adverse events, and seven of these patients had a serious treatment-related event, although none of the serious events were thought to be related to sutimlimab. One patient with liver cancer died from causes deemed unrelated to the drug. There were no meningococcal infections.
All 22 patients who completed the 26 weeks of therapy continued on an extended safety phase of the study.
The study results demonstrate that targeting the complement pathways is an novel and effective approach to managing cold agglutinin disease, Dr. Röth concluded.

In a press briefing the day before the presentation, moderator Robert Brodsky, MD, professor of medicine and director of the division of hematology at Johns Hopkins School, Baltimore, who treats patients with cold agglutinin disease, said that the results “are very exciting.”
“These patients are very difficult to treat and there really is no approved drug,” he said. “Right now, we usually use [rituximab] first line, but only half of those patients respond, and usually it only lasts for 6 months or so, so this is a welcome addition.”
Sutimlimab was granted Breakthrough Therapy designation by the Food and Drug Administration, and Orphan Drug status by the FDA, European Medicines Agency, and the Pharmaceuticals and Medical Devices Agency in Japan.
The study was supported by Sanofi. Dr. Röth reported financial relationships with Sanofi and other companies.
SOURCE: Röth A et al. ASH 2019, Abstract LBA-2.
ORLANDO – An investigational selective inhibitor of the complement pathway, sutimlimab, induced rapid and sustained benefits in patients with cold agglutinin disease, a rare autoimmune hemolytic anemia with no currently approved effective therapies.
Among 24 patients with cold agglutinin disease who received at least one dose of sutimlimab in a phase 3 trial, 20 had a mean increase in hemoglobin of at least 1 g/dL, and 17 remained transfusion free from weeks 5 to 26 following sutimlimab infusion.
“Sutimlimab has the potential to change treatment practices for patients with this disease,” said lead author Alexander Röth, MD, from the University of Duisburg-Essen (Germany), at a late-breaking abstract session at the annual meeting of the American Society of Hematology.
Mean total bilirubin rapidly normalized within 1-3 weeks of infusion of sutimlimab, and patients had a mean improvement of 11 points on the Functional Assessment of Chronic Illness Therapy–Fatigue scale (FACIT-F), indicating a substantial improvement in their quality of life, Dr. Röth said.
Cold agglutinin disease is an acquired hemolytic anemia with an underlying lymphoproliferative disorder. The estimated prevalence of the disease is 16 per 1 million persons. The disease is characterized by hemolysis driven by activation of the complement pathway, leading to opsonization of erythrocytes (coating of erythrocytes with particles that facilitate phagocytosis and other immune reactions), extravascular hemolysis (primarily in the liver), intravascular hemolysis, and anemia.
Patients experience severe fatigue and poor quality of life, as well as increased risk for thrombosis and mortality, compared with matched cohorts.
Sutimlimab is a humanized monoclonal antibody that blocks the C1s component of the classical complement pathway, thereby stopping pathway activation while leaving alternative lectin pathways intact.
Dr. Röth presented results of the phase 3, open-label Cardinal study. Patients with cold agglutinin disease with baseline hemoglobin of 10 g/dL or less, active hemolysis signaled by total bilirubin levels above normal, and at least one blood transfusion within the past 6 months were eligible for the study. Patients with secondary cold agglutinin syndrome or rituximab therapy within the last 3 months or combination therapies within the last 6 months were excluded.
Sutimlimab was delivered intravenously at a dose of 6.5 g for patients under 75 kg in weight and 7.5 g for those 75 kg and over at day 0 and 7, then every 2 weeks thereafter.
A total of 24 patients with a mean age of 71 years were enrolled. Of the 24 patients, 15 (62.5%) were women.
The patients had received a mean of 3.2 transfusions (range 1-19) in the previous 6 months, and 15 had received one or more prior targeted therapies for the disease within the last 5 years. The mean baseline hemoglobin level was 8.6 (range 4.9-11.1) g/dL.
Hemoglobin levels increased rapidly after the first infusion, with a mean increase of 1.2 g/dL at the end of week 1, and 2.3 g/dL after week 3.
The estimated mean increase at treatment assessment (an average of weeks 23, 25, and 26) – the primary endpoint – was 2.6 g/dL, exceeding the prespecified increase of at least 2 g/dL. Normalization of hemoglobin to 12 g/dL or greater was an alternative primary endpoint. The trial met the primary endpoint, with 13 of 24 patients (54.2%) achieving either of the two prespecified events.
The mean overall hemoglobin level was maintained above 11 g/dL after week 3. Of the 24 patients, 20 had hemoglobin increases of 1 g/dL or greater.
Mean total bilirubin, a marker of hemolysis, dropped markedly within hours of infusion and was normalized by week 3.
As noted before, patient quality of life, as measured by the FACIT-F scale, improved by a mean of 11 points from a mean baseline of 32 out of 52 points.
All but two patients had one or more treatment-emergent adverse events, and seven of these patients had a serious treatment-related event, although none of the serious events were thought to be related to sutimlimab. One patient with liver cancer died from causes deemed unrelated to the drug. There were no meningococcal infections.
All 22 patients who completed the 26 weeks of therapy continued on an extended safety phase of the study.
The study results demonstrate that targeting the complement pathways is an novel and effective approach to managing cold agglutinin disease, Dr. Röth concluded.
In a press briefing the day before the presentation, moderator Robert Brodsky, MD, professor of medicine and director of the division of hematology at Johns Hopkins School, Baltimore, who treats patients with cold agglutinin disease, said that the results “are very exciting.”
“These patients are very difficult to treat and there really is no approved drug,” he said. “Right now, we usually use [rituximab] first line, but only half of those patients respond, and usually it only lasts for 6 months or so, so this is a welcome addition.”
Sutimlimab was granted Breakthrough Therapy designation by the Food and Drug Administration, and Orphan Drug status by the FDA, European Medicines Agency, and the Pharmaceuticals and Medical Devices Agency in Japan.
The study was supported by Sanofi. Dr. Röth reported financial relationships with Sanofi and other companies.
SOURCE: Röth A et al. ASH 2019, Abstract LBA-2.
ORLANDO – An investigational selective inhibitor of the complement pathway, sutimlimab, induced rapid and sustained benefits in patients with cold agglutinin disease, a rare autoimmune hemolytic anemia with no currently approved effective therapies.
Among 24 patients with cold agglutinin disease who received at least one dose of sutimlimab in a phase 3 trial, 20 had a mean increase in hemoglobin of at least 1 g/dL, and 17 remained transfusion free from weeks 5 to 26 following sutimlimab infusion.
“Sutimlimab has the potential to change treatment practices for patients with this disease,” said lead author Alexander Röth, MD, from the University of Duisburg-Essen (Germany), at a late-breaking abstract session at the annual meeting of the American Society of Hematology.
Mean total bilirubin rapidly normalized within 1-3 weeks of infusion of sutimlimab, and patients had a mean improvement of 11 points on the Functional Assessment of Chronic Illness Therapy–Fatigue scale (FACIT-F), indicating a substantial improvement in their quality of life, Dr. Röth said.
Cold agglutinin disease is an acquired hemolytic anemia with an underlying lymphoproliferative disorder. The estimated prevalence of the disease is 16 per 1 million persons. The disease is characterized by hemolysis driven by activation of the complement pathway, leading to opsonization of erythrocytes (coating of erythrocytes with particles that facilitate phagocytosis and other immune reactions), extravascular hemolysis (primarily in the liver), intravascular hemolysis, and anemia.
Patients experience severe fatigue and poor quality of life, as well as increased risk for thrombosis and mortality, compared with matched cohorts.
Sutimlimab is a humanized monoclonal antibody that blocks the C1s component of the classical complement pathway, thereby stopping pathway activation while leaving alternative lectin pathways intact.
Dr. Röth presented results of the phase 3, open-label Cardinal study. Patients with cold agglutinin disease with baseline hemoglobin of 10 g/dL or less, active hemolysis signaled by total bilirubin levels above normal, and at least one blood transfusion within the past 6 months were eligible for the study. Patients with secondary cold agglutinin syndrome or rituximab therapy within the last 3 months or combination therapies within the last 6 months were excluded.
Sutimlimab was delivered intravenously at a dose of 6.5 g for patients under 75 kg in weight and 7.5 g for those 75 kg and over at day 0 and 7, then every 2 weeks thereafter.
A total of 24 patients with a mean age of 71 years were enrolled. Of the 24 patients, 15 (62.5%) were women.
The patients had received a mean of 3.2 transfusions (range 1-19) in the previous 6 months, and 15 had received one or more prior targeted therapies for the disease within the last 5 years. The mean baseline hemoglobin level was 8.6 (range 4.9-11.1) g/dL.
Hemoglobin levels increased rapidly after the first infusion, with a mean increase of 1.2 g/dL at the end of week 1, and 2.3 g/dL after week 3.
The estimated mean increase at treatment assessment (an average of weeks 23, 25, and 26) – the primary endpoint – was 2.6 g/dL, exceeding the prespecified increase of at least 2 g/dL. Normalization of hemoglobin to 12 g/dL or greater was an alternative primary endpoint. The trial met the primary endpoint, with 13 of 24 patients (54.2%) achieving either of the two prespecified events.
The mean overall hemoglobin level was maintained above 11 g/dL after week 3. Of the 24 patients, 20 had hemoglobin increases of 1 g/dL or greater.
Mean total bilirubin, a marker of hemolysis, dropped markedly within hours of infusion and was normalized by week 3.
As noted before, patient quality of life, as measured by the FACIT-F scale, improved by a mean of 11 points from a mean baseline of 32 out of 52 points.
All but two patients had one or more treatment-emergent adverse events, and seven of these patients had a serious treatment-related event, although none of the serious events were thought to be related to sutimlimab. One patient with liver cancer died from causes deemed unrelated to the drug. There were no meningococcal infections.
All 22 patients who completed the 26 weeks of therapy continued on an extended safety phase of the study.
The study results demonstrate that targeting the complement pathways is an novel and effective approach to managing cold agglutinin disease, Dr. Röth concluded.
In a press briefing the day before the presentation, moderator Robert Brodsky, MD, professor of medicine and director of the division of hematology at Johns Hopkins School, Baltimore, who treats patients with cold agglutinin disease, said that the results “are very exciting.”
“These patients are very difficult to treat and there really is no approved drug,” he said. “Right now, we usually use [rituximab] first line, but only half of those patients respond, and usually it only lasts for 6 months or so, so this is a welcome addition.”
Sutimlimab was granted Breakthrough Therapy designation by the Food and Drug Administration, and Orphan Drug status by the FDA, European Medicines Agency, and the Pharmaceuticals and Medical Devices Agency in Japan.
The study was supported by Sanofi. Dr. Röth reported financial relationships with Sanofi and other companies.
SOURCE: Röth A et al. ASH 2019, Abstract LBA-2.
REPORTING FROM ASH 2019
VEDOSS study describes predictors of progression to systemic sclerosis
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.

“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.
“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
ATLANTA – , according to recent results from the Very Early Diagnosis Of Systemic Sclerosis (VEDOSS) study presented at the annual meeting of the American College of Rheumatology.
“Our data show that thanks [to a] combination of the signs that characterize the various phases of the disease, patients can be diagnosed [with systemic sclerosis] in the very early stages,” first author Silvia Bellando-Randone, MD, PhD, assistant professor in the division of rheumatology at the University of Florence (Italy), said in her presentation.
Dr. Bellando-Randone and colleagues performed a longitudinal, observational study of 742 patients (mean 45.7 years old) at 42 centers in a cohort of mostly women (90%), nearly all of whom had had Raynaud’s phenomenon for longer than 36 months (97.5%). Patients were excluded if they had systemic sclerosis based on ACR 1980 classification criteria and/or ACR–European League Against Rheumatism 2013 criteria, had systemic sclerosis together with other connective-tissue diseases, or were unlikely to be present for three consecutive annual exams. Data collection began in March 2012 with follow-up of 5 years.
The researchers determined the positive predictive values (PPV) and negative predictive values (NPV) of clinical features, systemic sclerosis–specific antibodies, and nailfold video capillaroscopy (NVC) abnormalities on progression from Raynaud’s phenomenon to systemic sclerosis. Laboratory data collected at baseline included presence of antinuclear antibodies (ANA), anticentromere antibodies (ACA), anti-DNA topoisomerase I antibodies (anti-Scl-70), anti-U1RNP antibodies, anti-RNA polymerase III antibodies (ARA), N-terminal pro b-type natriuretic peptides (NT-proBNP), and C-reactive protein/erythrocyte sedimentation rate. Dr. Bellando-Randone and colleagues also collected clinical, pulmonary function, lung high-resolution CT, echocardiographic, and ECG data at baseline.
Predictions were based on these factors alone and in combination. Overall, 65% of patients had positive ANA. Other baseline characteristics present in patients that predicted systemic sclerosis included positive ACA/anti-Scl-70/ARA (32%), NVC abnormalities such as giant capillaries (25%), and puffy fingers (17%).
Using Kaplan-Meier analysis, the researchers found 7.4% of 401 patients who were ANA positive progressed to meet ACR-EULAR 2013 criteria, and the percentage of these patients increased to 29.3% at 3 years and 44.1% at 5 years. When the researchers considered disease-specific antibodies alone, 10.6% of 90 patients progressed from Raynaud’s phenomenon to systemic sclerosis within 1 year, 39.6% within 3 years, and 50.3% within 5 years. When the researchers analyzed disease-specific antibodies and NVC abnormalities together, 16% of 72 patients progressed to systemic sclerosis within 1 year, 61.7% within 3 years, and 77.4% within 5 years.
Puffy fingers also were a predictor of progression, and 14.4% of 69 patients with puffy fingers alone progressed from Raynaud’s phenomenon to systemic sclerosis at 1 year, 47.7% at 3 years, and 67.9% at 5 years. Considering puffy fingers and disease-specific antibodies together, 20% of 27 patients progressed at 1 year, 56.3% at 3 years, and 91.3% at 5 years. No patients with puffy fingers together and NVC abnormalities progressed to systemic sclerosis at 1 year, but 60.4% of 22 patients progressed at 3 years before plateauing at 5 years. For patients with NVC abnormalities alone, 7.1% progressed to systemic sclerosis from Raynaud’s phenomenon at 1 year, 39.4% at 3 years, and 52.7% at 5 years.
“Regarding capillaroscopy, we have to say that not all centers that participated were equally screened in capillaroscopy, and so we cannot assume the accuracy of this data,” she said.
Dr. Bellando-Randone noted that, apart from puffy fingers, disease-specific antibodies, and NVC abnormalities, patients were more likely to have a history of esophageal symptoms if they progressed to systemic sclerosis (37.3%), compared with patients who did not progress (23.6%; P = .003).
Puffy fingers alone were an independent predictor of systemic sclerosis (PPV, 78.9%; NPV, 45.1%) as well as in combination with disease-specific antibodies (PPV, 94.1%; NPV, 43.9%). The combination of disease-specific antibodies plus NVC abnormalities also independently predicted progression to systemic sclerosis (PPV, 82.2%; NPV, 50.4%). In a Cox multivariate analysis, disease-specific antibodies (relative risk, 5.4; 95% confidence interval, 3.7-7.9) and puffy fingers (RR, 3.0; 95% CI, 2.0-4.4) together were strongly predictive of progression from Raynaud’s phenomenon to systemic sclerosis (RR, 4.3; 95% CI, 2.6-7.3).
“This is really important for the risk stratification of patients [in] the very early stages of the disease, even if these data should be corroborated by larger data in larger studies in the future,” said Dr. Bellando-Randone.
The investigators reported having no conflicts of interest.
SOURCE: Bellando-Randone S et al. Arthritis Rheumatol. 2019;71(suppl 10). Abstract 2914.
REPORTING FROM ACR 2019
ADA2 is a potent new biomarker for macrophage activation syndrome
ATLANTA – Adenosine deaminase 2 above the upper limit of normal is 86% sensitive and 94% specific for distinguishing macrophage activation syndrome from active systemic juvenile idiopathic arthritis, making it perhaps the most potent blood marker yet identified to differentiate the two, according to a report presented at the annual meeting of the American College of Rheumatology.

The upper limit of normal was 27.8 U/L, two standard deviations above the median of 13 U/L (interquartile range, 10.6-16.1) in 174 healthy children. The work was published simultaneously in Annals of the Rheumatic Diseases.
In children with active systemic juvenile idiopathic arthritis (JIA), adenosine deaminase 2 (ADA2) “beyond the upper limit of normal is strong evidence for concomitant” macrophage activation syndrome (MAS). “Our work represents a new method to diagnose this condition,” said lead investigator Pui Y. Lee, MD, PhD, a pediatric rheumatologist at Boston Children’s Hospital.
The hope, he said, is that the finding will lead to quicker recognition and treatment of MAS, a devastating complication of systemic JIA in which rampant inflammation begets further inflammation in a downward spiral that ultimately proves fatal in about 20% of cases. The problem is that the clinical features of MAS overlap with those of active systemic JIA, which makes early diagnosis difficult.
Ferritin and other common markers are not very specific unless “the cutoff is raised significantly to distinguish MAS from general inflammation. Most labs will not tell you ‘this is an active systemic JIA range; this is an MAS-like range.’ It’s hard for them to define that for you. ADA2 is more black and white; if you go above the upper limit, you most likely have MAS,” Dr. Lee explained at the meeting.
Potentially, “we can combine this test with other tests to define a single MAS panel,” he said.
ADA2 is measured by a simple, inexpensive enzyme assay that’s been around for 20 years, but it hasn’t caught on because the protein’s function is unknown and the clinical relevance of ADA2 levels has been uncertain. With the new findings, “it is our hope that ADA2 testing will become more available,” Dr. Lee said.
The protein appears to be a product of monocytes and macrophages, and a genetic deficiency has recently been linked to congenital vasculitis, which made Dr. Lee and colleagues curious about ADA2 in other rheumatic diseases. The first step was to define normal limits in healthy controls; the 13 U/L median in children proved to be a bit higher than in 150 healthy adults.
The team then found that levels were completely normal in 25 children with active Kawasaki disease, and only mildly elevated in 13 children with systemic lupus and 13 with juvenile dermatomyositis. The Kawasaki children, in particular “were highly inflamed, so this protein is not just simply a marker of inflammation,” Dr. Lee said.
They next turned to 120 children with JIA, with a mix of systemic and nonsystemic cases. “The ones with very high levels, far beyond the upper limit of normal, were” almost exclusively the 23 children with systemic JIA and clinically diagnosed MAS. “As long as [JIA children] didn’t have MAS, their levels were pretty much close to normal,” he said.
In eight MAS children with repeat testing, levels fell below the upper limit of normal with treatment and remission, but children prone to repeat MAS seemed to hover closer to the limit even when they were well.
Blood sample testing showed that interleukin-18 and interferon-gamma were the main drivers of ADA2 expression in the periphery, “which makes sense because these two cytokines are very involved in the process of MAS,” Dr. Lee said.
The work was funded by the National Institutes of Health, among others. Dr. Lee didn’t have any disclosures.
SOURCE: Lee PY et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 920.
ATLANTA – Adenosine deaminase 2 above the upper limit of normal is 86% sensitive and 94% specific for distinguishing macrophage activation syndrome from active systemic juvenile idiopathic arthritis, making it perhaps the most potent blood marker yet identified to differentiate the two, according to a report presented at the annual meeting of the American College of Rheumatology.
The upper limit of normal was 27.8 U/L, two standard deviations above the median of 13 U/L (interquartile range, 10.6-16.1) in 174 healthy children. The work was published simultaneously in Annals of the Rheumatic Diseases.
In children with active systemic juvenile idiopathic arthritis (JIA), adenosine deaminase 2 (ADA2) “beyond the upper limit of normal is strong evidence for concomitant” macrophage activation syndrome (MAS). “Our work represents a new method to diagnose this condition,” said lead investigator Pui Y. Lee, MD, PhD, a pediatric rheumatologist at Boston Children’s Hospital.
The hope, he said, is that the finding will lead to quicker recognition and treatment of MAS, a devastating complication of systemic JIA in which rampant inflammation begets further inflammation in a downward spiral that ultimately proves fatal in about 20% of cases. The problem is that the clinical features of MAS overlap with those of active systemic JIA, which makes early diagnosis difficult.
Ferritin and other common markers are not very specific unless “the cutoff is raised significantly to distinguish MAS from general inflammation. Most labs will not tell you ‘this is an active systemic JIA range; this is an MAS-like range.’ It’s hard for them to define that for you. ADA2 is more black and white; if you go above the upper limit, you most likely have MAS,” Dr. Lee explained at the meeting.
Potentially, “we can combine this test with other tests to define a single MAS panel,” he said.
ADA2 is measured by a simple, inexpensive enzyme assay that’s been around for 20 years, but it hasn’t caught on because the protein’s function is unknown and the clinical relevance of ADA2 levels has been uncertain. With the new findings, “it is our hope that ADA2 testing will become more available,” Dr. Lee said.
The protein appears to be a product of monocytes and macrophages, and a genetic deficiency has recently been linked to congenital vasculitis, which made Dr. Lee and colleagues curious about ADA2 in other rheumatic diseases. The first step was to define normal limits in healthy controls; the 13 U/L median in children proved to be a bit higher than in 150 healthy adults.
The team then found that levels were completely normal in 25 children with active Kawasaki disease, and only mildly elevated in 13 children with systemic lupus and 13 with juvenile dermatomyositis. The Kawasaki children, in particular “were highly inflamed, so this protein is not just simply a marker of inflammation,” Dr. Lee said.
They next turned to 120 children with JIA, with a mix of systemic and nonsystemic cases. “The ones with very high levels, far beyond the upper limit of normal, were” almost exclusively the 23 children with systemic JIA and clinically diagnosed MAS. “As long as [JIA children] didn’t have MAS, their levels were pretty much close to normal,” he said.
In eight MAS children with repeat testing, levels fell below the upper limit of normal with treatment and remission, but children prone to repeat MAS seemed to hover closer to the limit even when they were well.
Blood sample testing showed that interleukin-18 and interferon-gamma were the main drivers of ADA2 expression in the periphery, “which makes sense because these two cytokines are very involved in the process of MAS,” Dr. Lee said.
The work was funded by the National Institutes of Health, among others. Dr. Lee didn’t have any disclosures.
SOURCE: Lee PY et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 920.
ATLANTA – Adenosine deaminase 2 above the upper limit of normal is 86% sensitive and 94% specific for distinguishing macrophage activation syndrome from active systemic juvenile idiopathic arthritis, making it perhaps the most potent blood marker yet identified to differentiate the two, according to a report presented at the annual meeting of the American College of Rheumatology.
The upper limit of normal was 27.8 U/L, two standard deviations above the median of 13 U/L (interquartile range, 10.6-16.1) in 174 healthy children. The work was published simultaneously in Annals of the Rheumatic Diseases.
In children with active systemic juvenile idiopathic arthritis (JIA), adenosine deaminase 2 (ADA2) “beyond the upper limit of normal is strong evidence for concomitant” macrophage activation syndrome (MAS). “Our work represents a new method to diagnose this condition,” said lead investigator Pui Y. Lee, MD, PhD, a pediatric rheumatologist at Boston Children’s Hospital.
The hope, he said, is that the finding will lead to quicker recognition and treatment of MAS, a devastating complication of systemic JIA in which rampant inflammation begets further inflammation in a downward spiral that ultimately proves fatal in about 20% of cases. The problem is that the clinical features of MAS overlap with those of active systemic JIA, which makes early diagnosis difficult.
Ferritin and other common markers are not very specific unless “the cutoff is raised significantly to distinguish MAS from general inflammation. Most labs will not tell you ‘this is an active systemic JIA range; this is an MAS-like range.’ It’s hard for them to define that for you. ADA2 is more black and white; if you go above the upper limit, you most likely have MAS,” Dr. Lee explained at the meeting.
Potentially, “we can combine this test with other tests to define a single MAS panel,” he said.
ADA2 is measured by a simple, inexpensive enzyme assay that’s been around for 20 years, but it hasn’t caught on because the protein’s function is unknown and the clinical relevance of ADA2 levels has been uncertain. With the new findings, “it is our hope that ADA2 testing will become more available,” Dr. Lee said.
The protein appears to be a product of monocytes and macrophages, and a genetic deficiency has recently been linked to congenital vasculitis, which made Dr. Lee and colleagues curious about ADA2 in other rheumatic diseases. The first step was to define normal limits in healthy controls; the 13 U/L median in children proved to be a bit higher than in 150 healthy adults.
The team then found that levels were completely normal in 25 children with active Kawasaki disease, and only mildly elevated in 13 children with systemic lupus and 13 with juvenile dermatomyositis. The Kawasaki children, in particular “were highly inflamed, so this protein is not just simply a marker of inflammation,” Dr. Lee said.
They next turned to 120 children with JIA, with a mix of systemic and nonsystemic cases. “The ones with very high levels, far beyond the upper limit of normal, were” almost exclusively the 23 children with systemic JIA and clinically diagnosed MAS. “As long as [JIA children] didn’t have MAS, their levels were pretty much close to normal,” he said.
In eight MAS children with repeat testing, levels fell below the upper limit of normal with treatment and remission, but children prone to repeat MAS seemed to hover closer to the limit even when they were well.
Blood sample testing showed that interleukin-18 and interferon-gamma were the main drivers of ADA2 expression in the periphery, “which makes sense because these two cytokines are very involved in the process of MAS,” Dr. Lee said.
The work was funded by the National Institutes of Health, among others. Dr. Lee didn’t have any disclosures.
SOURCE: Lee PY et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract 920.
REPORTING FROM ACR 2019
Tofacitinib improves disease activity in patients with polyarticular-course JIA
ATLANTA – Treatment of polyarticular-course juvenile idiopathic arthritis with tofacitinib led to significantly fewer disease flares and greater improvement in disease activity than with placebo in a phase 3, multinational, randomized, double-blind, controlled withdrawal study presented at the annual meeting of the American College of Rheumatology.

Hermine I. Brunner, MD, director of the division of rheumatology at Cincinnati Children’s Hospital Medical Center, and colleagues conducted the study in 225 patients between 2 and less than 18 years old with polyarticular-course juvenile idiopathic arthritis (pJIA; n = 184), psoriatic arthritis (PsA; n = 20), or enthesitis-related arthritis (ERA; n = 21). Patients were included if they had an inadequate response or intolerance to a disease-modifying antirheumatic drug and active disease with five or more active joints in the case of pJIA and three or more active joints in PsA or ERA.
Dr. Brunner presented results only for pJIA patients; the results for PsA and ERA patients will be assessed and presented separately.
The researchers divided their study into two sections. In the open-label portion of the study, patients received twice-daily tofacitinib (Xeljanz) at a dose of 5 mg or a weight-based lower dose in patients under 40 kg for 18 weeks. A total of 173 patients met JIA ACR30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables, and then were randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. Dr. Brunner noted that most patients who discontinued treatment in parts 1 and 2 did so because of insufficient clinical response rather than from adverse events.
Disease flare occurrence between 18 and 44 weeks was measured as a primary endpoint, and key secondary endpoints included JIA ACR30/50/70 response and change in Childhood Health Assessment Questionnaire Disability Index (CHAQ-DI) scores from part 2 baseline. The researchers used a “gatekeeping approach” that sequenced outcome measures in their statistical analysis to control for false positives in primary and secondary outcomes, where statistical significance could be achieved only if the previous outcome in the sequence was statistically significant.
Patients had a median age of 13 years, and most were female, white (about 87%), and between one-third and one-half of patients were based in North America. JIA disease duration was a median of about 2.5 years, C-reactive protein was about 0.3 mg/dL, and median CHAQ-DI scores were about 0.9 across tofacitinib and placebo groups. Other baseline characteristics were balanced between the two groups, Dr. Brunner said.
“Patients with polyarticular-course JIA in the open-label study experienced a rapid improvement of their disease activity from very high to moderate within 18 weeks,” Dr. Brunner said in her presentation. “[T]ofacitinib demonstrated significantly greater efficacy versus placebo in patients with polyarticular-course JIA based on occurrence of fewer flares in part 2.”
Specifically, disease flare occurred in 29.2% of patients by 44 weeks in the tofacitinib group, compared with 52.9% of patients in the placebo group (P = .0031), for an overall 54% lower risk of flare among patients receiving tofacitinib (hazard ratio, 0.459; 95% confidence interval, 0.268-0.785; P = .0037). The response rate was higher for patients receiving tofacitinib at 44 weeks when measured by JIA ACR30 (70.8% vs. 47.1% with placebo; P = .0031) or by JIA ACR50 (66.7% vs. 47.1%; P = .0166) and JIA ACR70 criteria (54.2% vs. 37.1%; P = .0387). The change in CHAQ-DI score also improved at 44 weeks to a significantly greater extent in the tofacitinib group than with placebo (–0.09 vs. 0.03; P = .0292).
“The safety profile of tofacitinib in children with JIA was comparable to what you have seen or known in the [rheumatoid arthritis] population, and no new safety risks were identified in this pediatric population,” Dr. Brunner said.
The researchers reported ties with Pfizer, which funded the study, and more than two dozen other pharmaceutical companies.
SOURCE: Brunner HI et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract L22.
ATLANTA – Treatment of polyarticular-course juvenile idiopathic arthritis with tofacitinib led to significantly fewer disease flares and greater improvement in disease activity than with placebo in a phase 3, multinational, randomized, double-blind, controlled withdrawal study presented at the annual meeting of the American College of Rheumatology.
Hermine I. Brunner, MD, director of the division of rheumatology at Cincinnati Children’s Hospital Medical Center, and colleagues conducted the study in 225 patients between 2 and less than 18 years old with polyarticular-course juvenile idiopathic arthritis (pJIA; n = 184), psoriatic arthritis (PsA; n = 20), or enthesitis-related arthritis (ERA; n = 21). Patients were included if they had an inadequate response or intolerance to a disease-modifying antirheumatic drug and active disease with five or more active joints in the case of pJIA and three or more active joints in PsA or ERA.
Dr. Brunner presented results only for pJIA patients; the results for PsA and ERA patients will be assessed and presented separately.
The researchers divided their study into two sections. In the open-label portion of the study, patients received twice-daily tofacitinib (Xeljanz) at a dose of 5 mg or a weight-based lower dose in patients under 40 kg for 18 weeks. A total of 173 patients met JIA ACR30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables, and then were randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. Dr. Brunner noted that most patients who discontinued treatment in parts 1 and 2 did so because of insufficient clinical response rather than from adverse events.
Disease flare occurrence between 18 and 44 weeks was measured as a primary endpoint, and key secondary endpoints included JIA ACR30/50/70 response and change in Childhood Health Assessment Questionnaire Disability Index (CHAQ-DI) scores from part 2 baseline. The researchers used a “gatekeeping approach” that sequenced outcome measures in their statistical analysis to control for false positives in primary and secondary outcomes, where statistical significance could be achieved only if the previous outcome in the sequence was statistically significant.
Patients had a median age of 13 years, and most were female, white (about 87%), and between one-third and one-half of patients were based in North America. JIA disease duration was a median of about 2.5 years, C-reactive protein was about 0.3 mg/dL, and median CHAQ-DI scores were about 0.9 across tofacitinib and placebo groups. Other baseline characteristics were balanced between the two groups, Dr. Brunner said.
“Patients with polyarticular-course JIA in the open-label study experienced a rapid improvement of their disease activity from very high to moderate within 18 weeks,” Dr. Brunner said in her presentation. “[T]ofacitinib demonstrated significantly greater efficacy versus placebo in patients with polyarticular-course JIA based on occurrence of fewer flares in part 2.”
Specifically, disease flare occurred in 29.2% of patients by 44 weeks in the tofacitinib group, compared with 52.9% of patients in the placebo group (P = .0031), for an overall 54% lower risk of flare among patients receiving tofacitinib (hazard ratio, 0.459; 95% confidence interval, 0.268-0.785; P = .0037). The response rate was higher for patients receiving tofacitinib at 44 weeks when measured by JIA ACR30 (70.8% vs. 47.1% with placebo; P = .0031) or by JIA ACR50 (66.7% vs. 47.1%; P = .0166) and JIA ACR70 criteria (54.2% vs. 37.1%; P = .0387). The change in CHAQ-DI score also improved at 44 weeks to a significantly greater extent in the tofacitinib group than with placebo (–0.09 vs. 0.03; P = .0292).
“The safety profile of tofacitinib in children with JIA was comparable to what you have seen or known in the [rheumatoid arthritis] population, and no new safety risks were identified in this pediatric population,” Dr. Brunner said.
The researchers reported ties with Pfizer, which funded the study, and more than two dozen other pharmaceutical companies.
SOURCE: Brunner HI et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract L22.
ATLANTA – Treatment of polyarticular-course juvenile idiopathic arthritis with tofacitinib led to significantly fewer disease flares and greater improvement in disease activity than with placebo in a phase 3, multinational, randomized, double-blind, controlled withdrawal study presented at the annual meeting of the American College of Rheumatology.
Hermine I. Brunner, MD, director of the division of rheumatology at Cincinnati Children’s Hospital Medical Center, and colleagues conducted the study in 225 patients between 2 and less than 18 years old with polyarticular-course juvenile idiopathic arthritis (pJIA; n = 184), psoriatic arthritis (PsA; n = 20), or enthesitis-related arthritis (ERA; n = 21). Patients were included if they had an inadequate response or intolerance to a disease-modifying antirheumatic drug and active disease with five or more active joints in the case of pJIA and three or more active joints in PsA or ERA.
Dr. Brunner presented results only for pJIA patients; the results for PsA and ERA patients will be assessed and presented separately.
The researchers divided their study into two sections. In the open-label portion of the study, patients received twice-daily tofacitinib (Xeljanz) at a dose of 5 mg or a weight-based lower dose in patients under 40 kg for 18 weeks. A total of 173 patients met JIA ACR30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables, and then were randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. Dr. Brunner noted that most patients who discontinued treatment in parts 1 and 2 did so because of insufficient clinical response rather than from adverse events.
Disease flare occurrence between 18 and 44 weeks was measured as a primary endpoint, and key secondary endpoints included JIA ACR30/50/70 response and change in Childhood Health Assessment Questionnaire Disability Index (CHAQ-DI) scores from part 2 baseline. The researchers used a “gatekeeping approach” that sequenced outcome measures in their statistical analysis to control for false positives in primary and secondary outcomes, where statistical significance could be achieved only if the previous outcome in the sequence was statistically significant.
Patients had a median age of 13 years, and most were female, white (about 87%), and between one-third and one-half of patients were based in North America. JIA disease duration was a median of about 2.5 years, C-reactive protein was about 0.3 mg/dL, and median CHAQ-DI scores were about 0.9 across tofacitinib and placebo groups. Other baseline characteristics were balanced between the two groups, Dr. Brunner said.
“Patients with polyarticular-course JIA in the open-label study experienced a rapid improvement of their disease activity from very high to moderate within 18 weeks,” Dr. Brunner said in her presentation. “[T]ofacitinib demonstrated significantly greater efficacy versus placebo in patients with polyarticular-course JIA based on occurrence of fewer flares in part 2.”
Specifically, disease flare occurred in 29.2% of patients by 44 weeks in the tofacitinib group, compared with 52.9% of patients in the placebo group (P = .0031), for an overall 54% lower risk of flare among patients receiving tofacitinib (hazard ratio, 0.459; 95% confidence interval, 0.268-0.785; P = .0037). The response rate was higher for patients receiving tofacitinib at 44 weeks when measured by JIA ACR30 (70.8% vs. 47.1% with placebo; P = .0031) or by JIA ACR50 (66.7% vs. 47.1%; P = .0166) and JIA ACR70 criteria (54.2% vs. 37.1%; P = .0387). The change in CHAQ-DI score also improved at 44 weeks to a significantly greater extent in the tofacitinib group than with placebo (–0.09 vs. 0.03; P = .0292).
“The safety profile of tofacitinib in children with JIA was comparable to what you have seen or known in the [rheumatoid arthritis] population, and no new safety risks were identified in this pediatric population,” Dr. Brunner said.
The researchers reported ties with Pfizer, which funded the study, and more than two dozen other pharmaceutical companies.
SOURCE: Brunner HI et al. Arthritis Rheumatol. 2019;71(suppl 10), Abstract L22.
REPORTING FROM ACR 2019
Study delineates spectrum of Dravet syndrome phenotypes
BALTIMORE – , researchers said at the annual meeting of the American Epilepsy Society. About half of patients have an afebrile seizure as their first seizure, and it is common for patients to present with seizures before age 5 months. Patients also may have seizure onset after age 18 months, said Wenhui Li, a researcher affiliated with Children’s Hospital of Fudan University in Shanghai and University of Melbourne, and colleagues.
“Subtle differences in Dravet syndrome phenotypes lead to delayed diagnosis,” the researchers said. “Understanding key features within the phenotypic spectrum will assist clinicians in evaluating whether a child has Dravet syndrome, facilitating early diagnosis for precision therapies.”
Typically, Dravet syndrome is thought to begin with prolonged febrile hemiclonic or generalized tonic-clonic seizures at about age 6 months in normally developing infants. Multiple seizure types occur during subsequent years, including focal impaired awareness, bilateral tonic-clonic, absence, and myoclonic seizures.
Patients often do not receive a diagnosis of Dravet syndrome until they are older than 3 years, after “developmental plateau or regression occurs in the second year,” the investigators said. “Earlier diagnosis is critical for optimal management.”
To outline the range of phenotypes, researchers analyzed the clinical histories of 188 patients with Dravet syndrome and pathogenic SCN1A variants. They excluded from their analysis patients with SCN1A-positive genetic epilepsy with febrile seizures plus (GEFS+).
In all, 53% of the patients were female, and 2% had developmental delay prior to the onset of seizures. Age at seizure onset ranged from 1.5 months to 21 months (median, 5.75 months). Three patients had seizure onset after age 12 months, the authors noted.
In cases where the first seizure type could be classified, 52% had generalized tonic-clonic seizures at onset, 37% had hemiclonic seizures, 4% myoclonic seizures, 4% focal impaired awareness seizures, and 0.5% absence seizures. In addition, 1% had hemiclonic and myoclonic seizures, and 2% had tonic-clonic and myoclonic seizures.
Fifty-four percent of patients were febrile during their first seizure, and 46% were afebrile.
Status epilepticus as the first seizure occurred in about 44% of cases, while 35% of patients had a first seizure duration of 5 minutes or less.
The researchers had no disclosures.
SOURCE: Li W et al. AES 2019. Abstract 2.116.
BALTIMORE – , researchers said at the annual meeting of the American Epilepsy Society. About half of patients have an afebrile seizure as their first seizure, and it is common for patients to present with seizures before age 5 months. Patients also may have seizure onset after age 18 months, said Wenhui Li, a researcher affiliated with Children’s Hospital of Fudan University in Shanghai and University of Melbourne, and colleagues.
“Subtle differences in Dravet syndrome phenotypes lead to delayed diagnosis,” the researchers said. “Understanding key features within the phenotypic spectrum will assist clinicians in evaluating whether a child has Dravet syndrome, facilitating early diagnosis for precision therapies.”
Typically, Dravet syndrome is thought to begin with prolonged febrile hemiclonic or generalized tonic-clonic seizures at about age 6 months in normally developing infants. Multiple seizure types occur during subsequent years, including focal impaired awareness, bilateral tonic-clonic, absence, and myoclonic seizures.
Patients often do not receive a diagnosis of Dravet syndrome until they are older than 3 years, after “developmental plateau or regression occurs in the second year,” the investigators said. “Earlier diagnosis is critical for optimal management.”
To outline the range of phenotypes, researchers analyzed the clinical histories of 188 patients with Dravet syndrome and pathogenic SCN1A variants. They excluded from their analysis patients with SCN1A-positive genetic epilepsy with febrile seizures plus (GEFS+).
In all, 53% of the patients were female, and 2% had developmental delay prior to the onset of seizures. Age at seizure onset ranged from 1.5 months to 21 months (median, 5.75 months). Three patients had seizure onset after age 12 months, the authors noted.
In cases where the first seizure type could be classified, 52% had generalized tonic-clonic seizures at onset, 37% had hemiclonic seizures, 4% myoclonic seizures, 4% focal impaired awareness seizures, and 0.5% absence seizures. In addition, 1% had hemiclonic and myoclonic seizures, and 2% had tonic-clonic and myoclonic seizures.
Fifty-four percent of patients were febrile during their first seizure, and 46% were afebrile.
Status epilepticus as the first seizure occurred in about 44% of cases, while 35% of patients had a first seizure duration of 5 minutes or less.
The researchers had no disclosures.
SOURCE: Li W et al. AES 2019. Abstract 2.116.
BALTIMORE – , researchers said at the annual meeting of the American Epilepsy Society. About half of patients have an afebrile seizure as their first seizure, and it is common for patients to present with seizures before age 5 months. Patients also may have seizure onset after age 18 months, said Wenhui Li, a researcher affiliated with Children’s Hospital of Fudan University in Shanghai and University of Melbourne, and colleagues.
“Subtle differences in Dravet syndrome phenotypes lead to delayed diagnosis,” the researchers said. “Understanding key features within the phenotypic spectrum will assist clinicians in evaluating whether a child has Dravet syndrome, facilitating early diagnosis for precision therapies.”
Typically, Dravet syndrome is thought to begin with prolonged febrile hemiclonic or generalized tonic-clonic seizures at about age 6 months in normally developing infants. Multiple seizure types occur during subsequent years, including focal impaired awareness, bilateral tonic-clonic, absence, and myoclonic seizures.
Patients often do not receive a diagnosis of Dravet syndrome until they are older than 3 years, after “developmental plateau or regression occurs in the second year,” the investigators said. “Earlier diagnosis is critical for optimal management.”
To outline the range of phenotypes, researchers analyzed the clinical histories of 188 patients with Dravet syndrome and pathogenic SCN1A variants. They excluded from their analysis patients with SCN1A-positive genetic epilepsy with febrile seizures plus (GEFS+).
In all, 53% of the patients were female, and 2% had developmental delay prior to the onset of seizures. Age at seizure onset ranged from 1.5 months to 21 months (median, 5.75 months). Three patients had seizure onset after age 12 months, the authors noted.
In cases where the first seizure type could be classified, 52% had generalized tonic-clonic seizures at onset, 37% had hemiclonic seizures, 4% myoclonic seizures, 4% focal impaired awareness seizures, and 0.5% absence seizures. In addition, 1% had hemiclonic and myoclonic seizures, and 2% had tonic-clonic and myoclonic seizures.
Fifty-four percent of patients were febrile during their first seizure, and 46% were afebrile.
Status epilepticus as the first seizure occurred in about 44% of cases, while 35% of patients had a first seizure duration of 5 minutes or less.
The researchers had no disclosures.
SOURCE: Li W et al. AES 2019. Abstract 2.116.
REPORTING FROM AES 2019