User login
Epidermolysis bullosa classification criteria refined and ready
LONDON – have come in understanding this debilitating group of genetic skin diseases, but also how far there is still to go towards improving the management of those affected.

Previous criteria issued in 2014 represented “important progress” and “built on the achievements of several generations of physicians and researchers who described the phenotypes, the level of skin cleavage, developed and characterized antibodies, and discovered EB-associated genes,” Cristina Has, MD, said at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA).
Dr. Has, a senior dermatologist and professor of experimental dermatology at the University of Freiburg (Germany), observed that prior criteria had “introduced genetic and molecular data in a so-called onion-skin classification of EB, and removed most of the eponyms,” which had been maintained in the latest update.
“What is new, and probably the most important change, is making the distinction between classical EB and other disorders with skin fragility,” she said, noting that the revised classification criteria for EB included minor changes to the nomenclature of EB. Six new EB subtypes and genes have also been added, and there are new sections on genotype/phenotype correlations, disease modifying factors, and the natural history of EB. Furthermore, supporting information included a concise description of clinical and genetic features of all EB types and subtypes.
The updated criteria are the result of an expert meeting held in April 2019 and have been accepted for publication. The expert panel that developed the criteria think that the revised classification criteria will be “useful and, we hope, inspiring and motivating for the young generation of dermatologists, pediatricians, and for the researchers who work in this field,” Dr. Has said.
“The term EB has been used in the last years for many new disorders, and this is the reason why we thought we have to somehow control this, and to make the distinction between classical epidermolysis bullosa due to defects at the dermal junction and other disorders with skin fragility where the anomalies occur within other layers of the epidermis or in the dermis,” Dr. Has explained.
There are still 4 main types of classical EB: EB simplex (EBS), dystrophic EB (DEB), junctional EB, and Kindler EB, but there are now 34 subtypes, slightly fewer than before. The updated criteria distinguish between the types and subtypes according to the level of skin cleavage, the inheritance pattern, the mutated gene, and the targeted protein, Dr. Has said.
As for peeling disorders, these have been classified as being erosive or hyperkeratotic, or as affecting the connective tissue with skin blistering. Similar to classical EB, these disorders are associated with fragility of the skin and mucosa and share some pathogenetic mechanisms. Moreover, as “the suffering of the patient is similar,” Dr. Has said, “we’d like to consider them under the umbrella of EB.” Most of the disorders she listed were inherited via an autosomal recessive mechanism, with intraepidermal disorders inherited via an autosomal dominant mechanism. New genes are being identified the time, she added, so these groupings will no doubt be subject to future revisions.
Minor changes to nomenclature were made to avoid confusion among clinicians and those living with the condition. As such, Kindler EB replaces Kindler syndrome, names of some subtypes were simplified, and a new “self-improving” type of DEB was introduced to replace the term “transient dermolysis of the newborn.” Altogether, there are now 11 subtypes of DEB. A distinction was also made between syndromic and nonsyndromic EB. “We all know that EB can be a systemic disorder with secondary manifestations within different organs,” Dr. Has told conference attendees. Anemia and failure to thrive can be associated, but it still remains a nonsyndromic disorder, she said. By contrast, “syndromic EB is due to genetic defects, which are also expressed in other organs than the skin or mucosal membranes, and lead to primary extracutaneous manifestations, such as cardiomyopathy, nephropathy, and so on.”
There are fewer subtypes of EBS and “we think they are better defined,” Dr. Has stated. “EB simplex is the most heterogenous EB type, clinically and genetically, and includes several syndromic disorders,” and the new classification criteria should be useful in helping categorize individuals with EBS and thus help target their management.
One of the six new subtypes of EB included in the revised classification criteria is “syndromic EBS with cardiomyopathy” caused by the KLH24 mutation. This gene was discovered in 2016 and more than 40 cases have so far been identified, 50% of which have been sporadic de novo mutations.
Other new EB subtypes are:
- “EBS with localized nephropathy” caused by a mutation in the CD151 gene.
- An autosomal recessive EBS linked to the KRT5 gene.
- A new phenotype that manifests with oral mucosal blisters linked to the DSG3 gene. (Although only a single case has been reported to date, it was felt worthy of inclusion.)
- Another linked to DSG3 that leads to skin fragility and hypertrichosis.
- A new dystrophic EB subtype linked to mutations in the PLOD3 gene.
In an interview, Dr. Has reiterated the importance of keeping classification criteria updated in line with current research findings. She emphasized that there were many types of EB and how important it was to refine how these were classified based on the underlying genetics.
“We brought much more genetic data into the paper, because we are in the era of personalized medicine,” she said. “There are specific therapies for mutations and for different subtypes and that’s why we think that, step by step, we have to bring in more and more data into the classification.”
There are many people with EBS, she observed, and while these individuals may not have such a dramatic clinical presentation as those with recessive DEB, for example, the effect of the condition on their daily lives is no less. “These people are active, they have jobs, they have to work, and they have pain, they have blister,” Dr. Has said.
While the criteria are intended only for classification of EB, they might help in practice. Dr. Has gave an anecdotal example of a woman that has been misdiagnosed as having a type of DEB with a high risk of squamous cell carcinoma but in fact had a different form of EB with no risk of developing SCC. “That’s why criteria are important,” she said.
Dr. Has had no conflicts of interest to disclose.
LONDON – have come in understanding this debilitating group of genetic skin diseases, but also how far there is still to go towards improving the management of those affected.
Previous criteria issued in 2014 represented “important progress” and “built on the achievements of several generations of physicians and researchers who described the phenotypes, the level of skin cleavage, developed and characterized antibodies, and discovered EB-associated genes,” Cristina Has, MD, said at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA).
Dr. Has, a senior dermatologist and professor of experimental dermatology at the University of Freiburg (Germany), observed that prior criteria had “introduced genetic and molecular data in a so-called onion-skin classification of EB, and removed most of the eponyms,” which had been maintained in the latest update.
“What is new, and probably the most important change, is making the distinction between classical EB and other disorders with skin fragility,” she said, noting that the revised classification criteria for EB included minor changes to the nomenclature of EB. Six new EB subtypes and genes have also been added, and there are new sections on genotype/phenotype correlations, disease modifying factors, and the natural history of EB. Furthermore, supporting information included a concise description of clinical and genetic features of all EB types and subtypes.
The updated criteria are the result of an expert meeting held in April 2019 and have been accepted for publication. The expert panel that developed the criteria think that the revised classification criteria will be “useful and, we hope, inspiring and motivating for the young generation of dermatologists, pediatricians, and for the researchers who work in this field,” Dr. Has said.
“The term EB has been used in the last years for many new disorders, and this is the reason why we thought we have to somehow control this, and to make the distinction between classical epidermolysis bullosa due to defects at the dermal junction and other disorders with skin fragility where the anomalies occur within other layers of the epidermis or in the dermis,” Dr. Has explained.
There are still 4 main types of classical EB: EB simplex (EBS), dystrophic EB (DEB), junctional EB, and Kindler EB, but there are now 34 subtypes, slightly fewer than before. The updated criteria distinguish between the types and subtypes according to the level of skin cleavage, the inheritance pattern, the mutated gene, and the targeted protein, Dr. Has said.
As for peeling disorders, these have been classified as being erosive or hyperkeratotic, or as affecting the connective tissue with skin blistering. Similar to classical EB, these disorders are associated with fragility of the skin and mucosa and share some pathogenetic mechanisms. Moreover, as “the suffering of the patient is similar,” Dr. Has said, “we’d like to consider them under the umbrella of EB.” Most of the disorders she listed were inherited via an autosomal recessive mechanism, with intraepidermal disorders inherited via an autosomal dominant mechanism. New genes are being identified the time, she added, so these groupings will no doubt be subject to future revisions.
Minor changes to nomenclature were made to avoid confusion among clinicians and those living with the condition. As such, Kindler EB replaces Kindler syndrome, names of some subtypes were simplified, and a new “self-improving” type of DEB was introduced to replace the term “transient dermolysis of the newborn.” Altogether, there are now 11 subtypes of DEB. A distinction was also made between syndromic and nonsyndromic EB. “We all know that EB can be a systemic disorder with secondary manifestations within different organs,” Dr. Has told conference attendees. Anemia and failure to thrive can be associated, but it still remains a nonsyndromic disorder, she said. By contrast, “syndromic EB is due to genetic defects, which are also expressed in other organs than the skin or mucosal membranes, and lead to primary extracutaneous manifestations, such as cardiomyopathy, nephropathy, and so on.”
There are fewer subtypes of EBS and “we think they are better defined,” Dr. Has stated. “EB simplex is the most heterogenous EB type, clinically and genetically, and includes several syndromic disorders,” and the new classification criteria should be useful in helping categorize individuals with EBS and thus help target their management.
One of the six new subtypes of EB included in the revised classification criteria is “syndromic EBS with cardiomyopathy” caused by the KLH24 mutation. This gene was discovered in 2016 and more than 40 cases have so far been identified, 50% of which have been sporadic de novo mutations.
Other new EB subtypes are:
- “EBS with localized nephropathy” caused by a mutation in the CD151 gene.
- An autosomal recessive EBS linked to the KRT5 gene.
- A new phenotype that manifests with oral mucosal blisters linked to the DSG3 gene. (Although only a single case has been reported to date, it was felt worthy of inclusion.)
- Another linked to DSG3 that leads to skin fragility and hypertrichosis.
- A new dystrophic EB subtype linked to mutations in the PLOD3 gene.
In an interview, Dr. Has reiterated the importance of keeping classification criteria updated in line with current research findings. She emphasized that there were many types of EB and how important it was to refine how these were classified based on the underlying genetics.
“We brought much more genetic data into the paper, because we are in the era of personalized medicine,” she said. “There are specific therapies for mutations and for different subtypes and that’s why we think that, step by step, we have to bring in more and more data into the classification.”
There are many people with EBS, she observed, and while these individuals may not have such a dramatic clinical presentation as those with recessive DEB, for example, the effect of the condition on their daily lives is no less. “These people are active, they have jobs, they have to work, and they have pain, they have blister,” Dr. Has said.
While the criteria are intended only for classification of EB, they might help in practice. Dr. Has gave an anecdotal example of a woman that has been misdiagnosed as having a type of DEB with a high risk of squamous cell carcinoma but in fact had a different form of EB with no risk of developing SCC. “That’s why criteria are important,” she said.
Dr. Has had no conflicts of interest to disclose.
LONDON – have come in understanding this debilitating group of genetic skin diseases, but also how far there is still to go towards improving the management of those affected.
Previous criteria issued in 2014 represented “important progress” and “built on the achievements of several generations of physicians and researchers who described the phenotypes, the level of skin cleavage, developed and characterized antibodies, and discovered EB-associated genes,” Cristina Has, MD, said at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA).
Dr. Has, a senior dermatologist and professor of experimental dermatology at the University of Freiburg (Germany), observed that prior criteria had “introduced genetic and molecular data in a so-called onion-skin classification of EB, and removed most of the eponyms,” which had been maintained in the latest update.
“What is new, and probably the most important change, is making the distinction between classical EB and other disorders with skin fragility,” she said, noting that the revised classification criteria for EB included minor changes to the nomenclature of EB. Six new EB subtypes and genes have also been added, and there are new sections on genotype/phenotype correlations, disease modifying factors, and the natural history of EB. Furthermore, supporting information included a concise description of clinical and genetic features of all EB types and subtypes.
The updated criteria are the result of an expert meeting held in April 2019 and have been accepted for publication. The expert panel that developed the criteria think that the revised classification criteria will be “useful and, we hope, inspiring and motivating for the young generation of dermatologists, pediatricians, and for the researchers who work in this field,” Dr. Has said.
“The term EB has been used in the last years for many new disorders, and this is the reason why we thought we have to somehow control this, and to make the distinction between classical epidermolysis bullosa due to defects at the dermal junction and other disorders with skin fragility where the anomalies occur within other layers of the epidermis or in the dermis,” Dr. Has explained.
There are still 4 main types of classical EB: EB simplex (EBS), dystrophic EB (DEB), junctional EB, and Kindler EB, but there are now 34 subtypes, slightly fewer than before. The updated criteria distinguish between the types and subtypes according to the level of skin cleavage, the inheritance pattern, the mutated gene, and the targeted protein, Dr. Has said.
As for peeling disorders, these have been classified as being erosive or hyperkeratotic, or as affecting the connective tissue with skin blistering. Similar to classical EB, these disorders are associated with fragility of the skin and mucosa and share some pathogenetic mechanisms. Moreover, as “the suffering of the patient is similar,” Dr. Has said, “we’d like to consider them under the umbrella of EB.” Most of the disorders she listed were inherited via an autosomal recessive mechanism, with intraepidermal disorders inherited via an autosomal dominant mechanism. New genes are being identified the time, she added, so these groupings will no doubt be subject to future revisions.
Minor changes to nomenclature were made to avoid confusion among clinicians and those living with the condition. As such, Kindler EB replaces Kindler syndrome, names of some subtypes were simplified, and a new “self-improving” type of DEB was introduced to replace the term “transient dermolysis of the newborn.” Altogether, there are now 11 subtypes of DEB. A distinction was also made between syndromic and nonsyndromic EB. “We all know that EB can be a systemic disorder with secondary manifestations within different organs,” Dr. Has told conference attendees. Anemia and failure to thrive can be associated, but it still remains a nonsyndromic disorder, she said. By contrast, “syndromic EB is due to genetic defects, which are also expressed in other organs than the skin or mucosal membranes, and lead to primary extracutaneous manifestations, such as cardiomyopathy, nephropathy, and so on.”
There are fewer subtypes of EBS and “we think they are better defined,” Dr. Has stated. “EB simplex is the most heterogenous EB type, clinically and genetically, and includes several syndromic disorders,” and the new classification criteria should be useful in helping categorize individuals with EBS and thus help target their management.
One of the six new subtypes of EB included in the revised classification criteria is “syndromic EBS with cardiomyopathy” caused by the KLH24 mutation. This gene was discovered in 2016 and more than 40 cases have so far been identified, 50% of which have been sporadic de novo mutations.
Other new EB subtypes are:
- “EBS with localized nephropathy” caused by a mutation in the CD151 gene.
- An autosomal recessive EBS linked to the KRT5 gene.
- A new phenotype that manifests with oral mucosal blisters linked to the DSG3 gene. (Although only a single case has been reported to date, it was felt worthy of inclusion.)
- Another linked to DSG3 that leads to skin fragility and hypertrichosis.
- A new dystrophic EB subtype linked to mutations in the PLOD3 gene.
In an interview, Dr. Has reiterated the importance of keeping classification criteria updated in line with current research findings. She emphasized that there were many types of EB and how important it was to refine how these were classified based on the underlying genetics.
“We brought much more genetic data into the paper, because we are in the era of personalized medicine,” she said. “There are specific therapies for mutations and for different subtypes and that’s why we think that, step by step, we have to bring in more and more data into the classification.”
There are many people with EBS, she observed, and while these individuals may not have such a dramatic clinical presentation as those with recessive DEB, for example, the effect of the condition on their daily lives is no less. “These people are active, they have jobs, they have to work, and they have pain, they have blister,” Dr. Has said.
While the criteria are intended only for classification of EB, they might help in practice. Dr. Has gave an anecdotal example of a woman that has been misdiagnosed as having a type of DEB with a high risk of squamous cell carcinoma but in fact had a different form of EB with no risk of developing SCC. “That’s why criteria are important,” she said.
Dr. Has had no conflicts of interest to disclose.
REPORTING FROM EB 2020
Psoriasis ointment helped with itch, healing in phase 2 EB study
LONDON – , in a small, placebo-controlled, phase 2 study.

More importantly, use of the ointment promoted wound healing in those with the severe skin-blistering condition. Indeed, compared with placebo, a greater reduction in wound size was observed after 2 weeks when the ointment was applied (a mean reduction of 65.5% vs. 88.4%; P less than .006). However, at 1 month, no significant differences were seen in the size of the wounds between the two treatment arms.
“Calcipotriol is a vitamin D analog and it is well known that vitamin D is a very critical factor for skin homeostasis and proper wound healing,” Christina Guttmann-Gruber, PhD, said at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA). Dr. Guttmann-Gruber, a group lead researcher for EB House Austria, which is based at the Salzburg (Austria) University Clinic for Dermatology, noted that vitamin D also helps with tissue repair and immune modulation, and enhances local antimicrobial activity.
During an oral poster presentation at the meeting, Dr. Guttmann-Gruber explained that in previous in vitro studies, it was found that low concentrations (100 nmol) of calcipotriol inhibited proliferation of RDEB tumor cells (Sci Rep. 2018 Sep 7;8:13430). Calcipotriol (also known as calcipotriene) also was found to improve the expression of antimicrobial peptides and promote wound closure. “Therefore, we thought that applying calcipotriol at the site of injury, on chronic wounds prone to superinfection where it is needed, might be beneficial for our patients.”
She and her associates designed a two-arm, randomized, double-blind crossover study to assess the effects of an existing calcipotriol-containing ointment on wound healing in patients with RDEB. The ointment used in the study is approved for treating psoriasis but was adapted by the in-house pharmacy team to reduce the concentration of calcipotriol to about 0.05 mcg/g, or around 121 nmol. The reason for the reduction was that, at higher doses, keratinocyte proliferation was reduced, which would be detrimental in RDEB patients.
Nine patients were included in the study and were randomized to either apply 1 g of the active or placebo ointment to each of two designated wounds, of at least 6 cm2 in size, every day for 4 weeks. A 2-month washout period then followed before the groups switched to use the other ointment for 1 month. Six out of the nine patients completed both treatment phases. The reasons for the patients not completing both intervention phases were not related to the drug.
Calcipotriol treatment resulted in a significant and steady reduction in itch over the entire course of treatment, which was not seen among those on placebo, Dr. Guttmann-Gruber observed. The reduction in itch was “striking,” but only while the treatment was being used, she said. Results for pain were less clear, with a significant reduction in pain after 2 weeks seen only in the placebo group, while both treatments reduced pain to the same degree by 1 month.
No serious adverse events were observed at any time point and topical use of the low-dose calcipotriol did not significantly change serum levels of calcium or vitamin D in the two patients in which this was studied, Dr. Guttmann-Gruber said.
“This is an approved drug; it’s used in psoriasis, but at a very high concentration. We were able to use it off label and make a diluted version,” she observed. “Any pharmacy can do it.” Although it was applied topically, it could be done by applying it to the dressing rather directly onto the wounded skin, she said.
Data on the skin microbiome response to treatment were also collected but were not available to analyze in time for presentation, but it appeared that there was improvement with the low-dose calcipotriol treatment, Dr. Guttmann-Gruber said. “When the wounds are healing, the microbial flora is improving.”
The next step will probably be to plan a multicenter trial of this treatment, Dr. Guttmann-Gruber said in an interview. The questions is whether such a trial would get the financial backing it needed, but if an orphan drug designation could be obtained for calcipotriol for EB, then it would be possible to conduct such a trial.
The study was funded by DEBRA Austria. The presenting author, Dr. Guttmann-Gruber, had no conflicts of interest to disclose.
SOURCE: Guttmann-Gruber C et al. EB World Congress 2020. Poster 34.
LONDON – , in a small, placebo-controlled, phase 2 study.
More importantly, use of the ointment promoted wound healing in those with the severe skin-blistering condition. Indeed, compared with placebo, a greater reduction in wound size was observed after 2 weeks when the ointment was applied (a mean reduction of 65.5% vs. 88.4%; P less than .006). However, at 1 month, no significant differences were seen in the size of the wounds between the two treatment arms.
“Calcipotriol is a vitamin D analog and it is well known that vitamin D is a very critical factor for skin homeostasis and proper wound healing,” Christina Guttmann-Gruber, PhD, said at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA). Dr. Guttmann-Gruber, a group lead researcher for EB House Austria, which is based at the Salzburg (Austria) University Clinic for Dermatology, noted that vitamin D also helps with tissue repair and immune modulation, and enhances local antimicrobial activity.
During an oral poster presentation at the meeting, Dr. Guttmann-Gruber explained that in previous in vitro studies, it was found that low concentrations (100 nmol) of calcipotriol inhibited proliferation of RDEB tumor cells (Sci Rep. 2018 Sep 7;8:13430). Calcipotriol (also known as calcipotriene) also was found to improve the expression of antimicrobial peptides and promote wound closure. “Therefore, we thought that applying calcipotriol at the site of injury, on chronic wounds prone to superinfection where it is needed, might be beneficial for our patients.”
She and her associates designed a two-arm, randomized, double-blind crossover study to assess the effects of an existing calcipotriol-containing ointment on wound healing in patients with RDEB. The ointment used in the study is approved for treating psoriasis but was adapted by the in-house pharmacy team to reduce the concentration of calcipotriol to about 0.05 mcg/g, or around 121 nmol. The reason for the reduction was that, at higher doses, keratinocyte proliferation was reduced, which would be detrimental in RDEB patients.
Nine patients were included in the study and were randomized to either apply 1 g of the active or placebo ointment to each of two designated wounds, of at least 6 cm2 in size, every day for 4 weeks. A 2-month washout period then followed before the groups switched to use the other ointment for 1 month. Six out of the nine patients completed both treatment phases. The reasons for the patients not completing both intervention phases were not related to the drug.
Calcipotriol treatment resulted in a significant and steady reduction in itch over the entire course of treatment, which was not seen among those on placebo, Dr. Guttmann-Gruber observed. The reduction in itch was “striking,” but only while the treatment was being used, she said. Results for pain were less clear, with a significant reduction in pain after 2 weeks seen only in the placebo group, while both treatments reduced pain to the same degree by 1 month.
No serious adverse events were observed at any time point and topical use of the low-dose calcipotriol did not significantly change serum levels of calcium or vitamin D in the two patients in which this was studied, Dr. Guttmann-Gruber said.
“This is an approved drug; it’s used in psoriasis, but at a very high concentration. We were able to use it off label and make a diluted version,” she observed. “Any pharmacy can do it.” Although it was applied topically, it could be done by applying it to the dressing rather directly onto the wounded skin, she said.
Data on the skin microbiome response to treatment were also collected but were not available to analyze in time for presentation, but it appeared that there was improvement with the low-dose calcipotriol treatment, Dr. Guttmann-Gruber said. “When the wounds are healing, the microbial flora is improving.”
The next step will probably be to plan a multicenter trial of this treatment, Dr. Guttmann-Gruber said in an interview. The questions is whether such a trial would get the financial backing it needed, but if an orphan drug designation could be obtained for calcipotriol for EB, then it would be possible to conduct such a trial.
The study was funded by DEBRA Austria. The presenting author, Dr. Guttmann-Gruber, had no conflicts of interest to disclose.
SOURCE: Guttmann-Gruber C et al. EB World Congress 2020. Poster 34.
LONDON – , in a small, placebo-controlled, phase 2 study.
More importantly, use of the ointment promoted wound healing in those with the severe skin-blistering condition. Indeed, compared with placebo, a greater reduction in wound size was observed after 2 weeks when the ointment was applied (a mean reduction of 65.5% vs. 88.4%; P less than .006). However, at 1 month, no significant differences were seen in the size of the wounds between the two treatment arms.
“Calcipotriol is a vitamin D analog and it is well known that vitamin D is a very critical factor for skin homeostasis and proper wound healing,” Christina Guttmann-Gruber, PhD, said at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA). Dr. Guttmann-Gruber, a group lead researcher for EB House Austria, which is based at the Salzburg (Austria) University Clinic for Dermatology, noted that vitamin D also helps with tissue repair and immune modulation, and enhances local antimicrobial activity.
During an oral poster presentation at the meeting, Dr. Guttmann-Gruber explained that in previous in vitro studies, it was found that low concentrations (100 nmol) of calcipotriol inhibited proliferation of RDEB tumor cells (Sci Rep. 2018 Sep 7;8:13430). Calcipotriol (also known as calcipotriene) also was found to improve the expression of antimicrobial peptides and promote wound closure. “Therefore, we thought that applying calcipotriol at the site of injury, on chronic wounds prone to superinfection where it is needed, might be beneficial for our patients.”
She and her associates designed a two-arm, randomized, double-blind crossover study to assess the effects of an existing calcipotriol-containing ointment on wound healing in patients with RDEB. The ointment used in the study is approved for treating psoriasis but was adapted by the in-house pharmacy team to reduce the concentration of calcipotriol to about 0.05 mcg/g, or around 121 nmol. The reason for the reduction was that, at higher doses, keratinocyte proliferation was reduced, which would be detrimental in RDEB patients.
Nine patients were included in the study and were randomized to either apply 1 g of the active or placebo ointment to each of two designated wounds, of at least 6 cm2 in size, every day for 4 weeks. A 2-month washout period then followed before the groups switched to use the other ointment for 1 month. Six out of the nine patients completed both treatment phases. The reasons for the patients not completing both intervention phases were not related to the drug.
Calcipotriol treatment resulted in a significant and steady reduction in itch over the entire course of treatment, which was not seen among those on placebo, Dr. Guttmann-Gruber observed. The reduction in itch was “striking,” but only while the treatment was being used, she said. Results for pain were less clear, with a significant reduction in pain after 2 weeks seen only in the placebo group, while both treatments reduced pain to the same degree by 1 month.
No serious adverse events were observed at any time point and topical use of the low-dose calcipotriol did not significantly change serum levels of calcium or vitamin D in the two patients in which this was studied, Dr. Guttmann-Gruber said.
“This is an approved drug; it’s used in psoriasis, but at a very high concentration. We were able to use it off label and make a diluted version,” she observed. “Any pharmacy can do it.” Although it was applied topically, it could be done by applying it to the dressing rather directly onto the wounded skin, she said.
Data on the skin microbiome response to treatment were also collected but were not available to analyze in time for presentation, but it appeared that there was improvement with the low-dose calcipotriol treatment, Dr. Guttmann-Gruber said. “When the wounds are healing, the microbial flora is improving.”
The next step will probably be to plan a multicenter trial of this treatment, Dr. Guttmann-Gruber said in an interview. The questions is whether such a trial would get the financial backing it needed, but if an orphan drug designation could be obtained for calcipotriol for EB, then it would be possible to conduct such a trial.
The study was funded by DEBRA Austria. The presenting author, Dr. Guttmann-Gruber, had no conflicts of interest to disclose.
SOURCE: Guttmann-Gruber C et al. EB World Congress 2020. Poster 34.
REPORTING FROM EB 2020
Losartan showing promise in pediatric epidermolysis bullosa trial
LONDON – Treatment with the in an early clinical study.
In the ongoing phase 1/2 REFLECT (Recessive dystrophic EB: Mechanisms of fibrosis and its prevention with Losartan in vivo) trial, involving 29 children, no severe complications have been noted so far, according to one of the study investigators, Dimitra Kiritsi, MD, of the University of Freiburg, Germany. At the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA), she presented interim data on 18 patients in the trial, emphasizing that the primary aim of the trial was to evaluate the safety of this treatment approach.
Over the 2 years the trial has been underway, 65 adverse events have been reported, of which 4 have been severe. Two of these were bacterial infections that required hospital treatment and the other two were a reduction in the general health condition of the child.
Losartan is an angiotensin-II receptor blocker (ARB) that has been in clinical use for more than 25 years in adults and 15 years in children over the age of 6 years.
The drug may be used for treating recessive dystrophic EB (RDEB) in the future, Dr. Kiritsi said, because it attenuates tumor necrosis factor–beta (TGF-beta) signaling, which is thought to be involved in the fibrotic process. So while it may not target the genetic defect, it could help ameliorate the effects of the disease.
The precursor to REFLECT was a study performed in a mouse disease model of EB (EMBO Mol Med. 2015;7:1211-28) where a reduction in fibrotic scarring was seen with losartan with “remarkable effects” on “mitten” deformity, Dr. Kiritsi said. The results of that study suggested that the earlier treatment with losartan was started in the course of the disease, the better the effect, she added. (Mitten deformity is the result of fused skin between the fingers or toes, and the subsequent buildup of fibrotic tissue causes the hand or foot to contract.)
REFLECT is an investigator-initiated trial that started in 2017 and is being funded by DEBRA International. It is a dual-center, nonrandomized, single-arm study in which children aged 3-16 years with RDEB are treated with losartan for 10 months, with follow-up at 3 months.
Various secondary endpoints were included to look for the first signs of any efficacy: the Physician’s Global Assessment (PGA), the Birmingham Epidermolysis Bullosa Severity Score (BEBS), the Epidermolysis Bullosa Disease Activity and Scarring Index (EBDASI), the Itch Assessment Scale for the Pediatric Burn Patients, and two quality of life indices: the Quality of Life in EB (QOLEB) questionnaire and the Children’s Dermatology Life Quality Index (CDLQI).
Dr. Kiritsi highlighted a few of the secondary endpoint findings, saying that reduced BEBS scores showed there was “amelioration of the patients’ phenotype” and that EBDASI scores also decreased, with “nearly 60% of the patients having significant improvement of their skin disease.” Importantly, itch improved in most of the patients, she said. Reductions in CDLQI were observed, “meaning that quality of life was significantly better at the end of the trial.” There were also decreases in inflammatory markers, such as C-reactive protein, interleukin-6, and TNF-alpha.
Although there is no validated tool available to assess hand function, Dr. Kiritsi and her team used their own morphometric scoring instrument to measure how far the hand could stretch; their evaluations suggested that this measure improved – or at least did not worsen – with losartan treatment, she noted.
A larger, randomized trial is needed to confirm if there is any benefit of losartan, but first, a new, easy-to-swallow losartan formulation needs to be developed specifically for EB in the pediatric population, Dr. Kiritsi said. Although a pediatric suspension of losartan was previously available, it is no longer on the market, so the next step is to develop a formulation that could be used in a pivotal clinical trial, she noted.
“Losartan faces fewer technical hurdles compared to other novel treatments as it is an established medicine,” Dr. Kiritsi and associates observed in a poster presentation. There are still economic hurdles, however, since “with losartan patents expired, companies cannot expect to recoup an investment into clinical studies” and alternative funding sources are needed.
In 2019, losartan was granted an orphan drug designation for the treatment of EB from both the Food and Drug Administration and the European Medicines Agency, but its use remains off label in children. “We decided to treat children,” Dr. Kiritsi said, “because we wanted to start as early as possible. If you already have mitten deformities, these cannot be reversed.”
DEBRA International funded the study. Dr. Kiritsi received research support from Rheacell GmbH and honoraria or consultation fees from Amryt Pharma and Rheacell GmbH. She has received other support from DEBRA International, EB Research Partnership, Fritz Thyssen Stiftung, German Research Foundation (funding of research projects), and 3R Pharma Consulting and Midas Pharma GmbH (consultation for losartan new drug formulation).
SOURCE: Kiritsi D et al. EB 2020. Poster 47.
LONDON – Treatment with the in an early clinical study.
In the ongoing phase 1/2 REFLECT (Recessive dystrophic EB: Mechanisms of fibrosis and its prevention with Losartan in vivo) trial, involving 29 children, no severe complications have been noted so far, according to one of the study investigators, Dimitra Kiritsi, MD, of the University of Freiburg, Germany. At the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA), she presented interim data on 18 patients in the trial, emphasizing that the primary aim of the trial was to evaluate the safety of this treatment approach.
Over the 2 years the trial has been underway, 65 adverse events have been reported, of which 4 have been severe. Two of these were bacterial infections that required hospital treatment and the other two were a reduction in the general health condition of the child.
Losartan is an angiotensin-II receptor blocker (ARB) that has been in clinical use for more than 25 years in adults and 15 years in children over the age of 6 years.
The drug may be used for treating recessive dystrophic EB (RDEB) in the future, Dr. Kiritsi said, because it attenuates tumor necrosis factor–beta (TGF-beta) signaling, which is thought to be involved in the fibrotic process. So while it may not target the genetic defect, it could help ameliorate the effects of the disease.
The precursor to REFLECT was a study performed in a mouse disease model of EB (EMBO Mol Med. 2015;7:1211-28) where a reduction in fibrotic scarring was seen with losartan with “remarkable effects” on “mitten” deformity, Dr. Kiritsi said. The results of that study suggested that the earlier treatment with losartan was started in the course of the disease, the better the effect, she added. (Mitten deformity is the result of fused skin between the fingers or toes, and the subsequent buildup of fibrotic tissue causes the hand or foot to contract.)
REFLECT is an investigator-initiated trial that started in 2017 and is being funded by DEBRA International. It is a dual-center, nonrandomized, single-arm study in which children aged 3-16 years with RDEB are treated with losartan for 10 months, with follow-up at 3 months.
Various secondary endpoints were included to look for the first signs of any efficacy: the Physician’s Global Assessment (PGA), the Birmingham Epidermolysis Bullosa Severity Score (BEBS), the Epidermolysis Bullosa Disease Activity and Scarring Index (EBDASI), the Itch Assessment Scale for the Pediatric Burn Patients, and two quality of life indices: the Quality of Life in EB (QOLEB) questionnaire and the Children’s Dermatology Life Quality Index (CDLQI).
Dr. Kiritsi highlighted a few of the secondary endpoint findings, saying that reduced BEBS scores showed there was “amelioration of the patients’ phenotype” and that EBDASI scores also decreased, with “nearly 60% of the patients having significant improvement of their skin disease.” Importantly, itch improved in most of the patients, she said. Reductions in CDLQI were observed, “meaning that quality of life was significantly better at the end of the trial.” There were also decreases in inflammatory markers, such as C-reactive protein, interleukin-6, and TNF-alpha.
Although there is no validated tool available to assess hand function, Dr. Kiritsi and her team used their own morphometric scoring instrument to measure how far the hand could stretch; their evaluations suggested that this measure improved – or at least did not worsen – with losartan treatment, she noted.
A larger, randomized trial is needed to confirm if there is any benefit of losartan, but first, a new, easy-to-swallow losartan formulation needs to be developed specifically for EB in the pediatric population, Dr. Kiritsi said. Although a pediatric suspension of losartan was previously available, it is no longer on the market, so the next step is to develop a formulation that could be used in a pivotal clinical trial, she noted.
“Losartan faces fewer technical hurdles compared to other novel treatments as it is an established medicine,” Dr. Kiritsi and associates observed in a poster presentation. There are still economic hurdles, however, since “with losartan patents expired, companies cannot expect to recoup an investment into clinical studies” and alternative funding sources are needed.
In 2019, losartan was granted an orphan drug designation for the treatment of EB from both the Food and Drug Administration and the European Medicines Agency, but its use remains off label in children. “We decided to treat children,” Dr. Kiritsi said, “because we wanted to start as early as possible. If you already have mitten deformities, these cannot be reversed.”
DEBRA International funded the study. Dr. Kiritsi received research support from Rheacell GmbH and honoraria or consultation fees from Amryt Pharma and Rheacell GmbH. She has received other support from DEBRA International, EB Research Partnership, Fritz Thyssen Stiftung, German Research Foundation (funding of research projects), and 3R Pharma Consulting and Midas Pharma GmbH (consultation for losartan new drug formulation).
SOURCE: Kiritsi D et al. EB 2020. Poster 47.
LONDON – Treatment with the in an early clinical study.
In the ongoing phase 1/2 REFLECT (Recessive dystrophic EB: Mechanisms of fibrosis and its prevention with Losartan in vivo) trial, involving 29 children, no severe complications have been noted so far, according to one of the study investigators, Dimitra Kiritsi, MD, of the University of Freiburg, Germany. At the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA), she presented interim data on 18 patients in the trial, emphasizing that the primary aim of the trial was to evaluate the safety of this treatment approach.
Over the 2 years the trial has been underway, 65 adverse events have been reported, of which 4 have been severe. Two of these were bacterial infections that required hospital treatment and the other two were a reduction in the general health condition of the child.
Losartan is an angiotensin-II receptor blocker (ARB) that has been in clinical use for more than 25 years in adults and 15 years in children over the age of 6 years.
The drug may be used for treating recessive dystrophic EB (RDEB) in the future, Dr. Kiritsi said, because it attenuates tumor necrosis factor–beta (TGF-beta) signaling, which is thought to be involved in the fibrotic process. So while it may not target the genetic defect, it could help ameliorate the effects of the disease.
The precursor to REFLECT was a study performed in a mouse disease model of EB (EMBO Mol Med. 2015;7:1211-28) where a reduction in fibrotic scarring was seen with losartan with “remarkable effects” on “mitten” deformity, Dr. Kiritsi said. The results of that study suggested that the earlier treatment with losartan was started in the course of the disease, the better the effect, she added. (Mitten deformity is the result of fused skin between the fingers or toes, and the subsequent buildup of fibrotic tissue causes the hand or foot to contract.)
REFLECT is an investigator-initiated trial that started in 2017 and is being funded by DEBRA International. It is a dual-center, nonrandomized, single-arm study in which children aged 3-16 years with RDEB are treated with losartan for 10 months, with follow-up at 3 months.
Various secondary endpoints were included to look for the first signs of any efficacy: the Physician’s Global Assessment (PGA), the Birmingham Epidermolysis Bullosa Severity Score (BEBS), the Epidermolysis Bullosa Disease Activity and Scarring Index (EBDASI), the Itch Assessment Scale for the Pediatric Burn Patients, and two quality of life indices: the Quality of Life in EB (QOLEB) questionnaire and the Children’s Dermatology Life Quality Index (CDLQI).
Dr. Kiritsi highlighted a few of the secondary endpoint findings, saying that reduced BEBS scores showed there was “amelioration of the patients’ phenotype” and that EBDASI scores also decreased, with “nearly 60% of the patients having significant improvement of their skin disease.” Importantly, itch improved in most of the patients, she said. Reductions in CDLQI were observed, “meaning that quality of life was significantly better at the end of the trial.” There were also decreases in inflammatory markers, such as C-reactive protein, interleukin-6, and TNF-alpha.
Although there is no validated tool available to assess hand function, Dr. Kiritsi and her team used their own morphometric scoring instrument to measure how far the hand could stretch; their evaluations suggested that this measure improved – or at least did not worsen – with losartan treatment, she noted.
A larger, randomized trial is needed to confirm if there is any benefit of losartan, but first, a new, easy-to-swallow losartan formulation needs to be developed specifically for EB in the pediatric population, Dr. Kiritsi said. Although a pediatric suspension of losartan was previously available, it is no longer on the market, so the next step is to develop a formulation that could be used in a pivotal clinical trial, she noted.
“Losartan faces fewer technical hurdles compared to other novel treatments as it is an established medicine,” Dr. Kiritsi and associates observed in a poster presentation. There are still economic hurdles, however, since “with losartan patents expired, companies cannot expect to recoup an investment into clinical studies” and alternative funding sources are needed.
In 2019, losartan was granted an orphan drug designation for the treatment of EB from both the Food and Drug Administration and the European Medicines Agency, but its use remains off label in children. “We decided to treat children,” Dr. Kiritsi said, “because we wanted to start as early as possible. If you already have mitten deformities, these cannot be reversed.”
DEBRA International funded the study. Dr. Kiritsi received research support from Rheacell GmbH and honoraria or consultation fees from Amryt Pharma and Rheacell GmbH. She has received other support from DEBRA International, EB Research Partnership, Fritz Thyssen Stiftung, German Research Foundation (funding of research projects), and 3R Pharma Consulting and Midas Pharma GmbH (consultation for losartan new drug formulation).
SOURCE: Kiritsi D et al. EB 2020. Poster 47.
REPORTING FROM EB 2020
High cost of wound dressings for epidermolysis bullosa highlighted
LONDON – More than £2.8 million (RDEB), according to a report at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA).

Results from the Prospective Epidermolysis Bullosa Longitudinal Evaluation Study (PEBLES), which is looking at the natural history of RDEB, showed that wound dressing and bandage costs were highest for study participants with the generalized severe (RDEB-GS) subtype, at just over £85,156 (about $112,450) per patient annually. Respective yearly costs for the generalized intermediate (RDEB-GI) and inversa (RDEB-INV) subtypes were £10,112 (about $13,350) and £1,699 (about $2,240) per patient.
Looking at the costs associated with EB is important, said one of the lead investigators for PEBLES, Jemima Mellerio, MD, FRCP, consultant dermatologist at St John’s Institute of Dermatology, at Guy’s & St. Thomas’ NHS Foundation Trust, London.
“If we are going to justify the kind of expenditure [associated with new treatments], we need to know that what we are treating is already a significant burden on our health care systems,” Dr. Mellerio said.
PEBLES is an ongoing London-based registry study that is enrolling patients with all subtypes of RDEB. Data are collected via a tablet device and include demographic data, information on clinical features, results of skin biopsies and genetic tests, and laboratory findings, as well as objective disease severity and subjective patient-orientated outcome scores.
So far, 60 patients – 49 adults and 11 children – have been enrolled in PEBLES since November 2014: 26 with RDEB-GS, 23 with RDEB-GI, 9 with RDEB-INV, and 2 with the pruriginosa RDEB subtype (RDEB-PR).
Most of the participants (71%) changed all their wound dressings at one time, patching up when required. Fourteen of 49 participants had paid people to help them change their dressings and when the total cost of combined wound dressings and paid care was taken into consideration, the mean annual cost per patient was around £2,500 (about $3,300) for RDEB-INV, £10,375 (about $13,700) per patient for RDEB-GS, and a staggering £98,000 (about $129,000) per patient for RDEB-GS. The total annual cost of dressings and associated care was an estimated £3,184,229 (about $4.2 million).
In addition to data on the cost of wound dressings, data on itch and pain and quality of life were presented at the EB World Congress and discussed by Dr. Mellerio.
A total of 42 participants older than 8 years of age had itch measured via the Leuven Itch Scale, she reported, noting that itch was a consistent symptom across all subtypes of RDEB. Itch is important as it not only causes problems with skin lesions and healing, but also significantly affects sleep and has a negative impact on patients’ mood, she emphasized.

Despite experiencing itch, more than half (58%) of participants were not using any kind of treatment for itch. This “likely reflects the lack of effectiveness of current medication for this debilitating symptom,” Dr. Mellerio and associates noted in one of their poster presentations of PEBLES data.
When treatment was used for itch, it consisted mainly of antihistamines (19% of patients), emollients (19%), or a combination of both (4%). However, treatment was generally “not very good,” with a satisfaction score of just 5 on a scale of 10, Dr. Mellerio pointed out. Participants “reported frustration with the lack of effective treatment for itch,” she said.
Itch was associated with disturbed sleep 1-3 nights per week in 20%-40% of participants, and every night in 20%-30%.
Pain was found to be a significant problem, with a median level of background pain scored as 4 on a 10-cm visual analog scale and a higher level (6) when associated with dressing changes.
Data on how RDEB affected quality of life were reported for 39 adults completing the 17-item Quality of Life in EB Questionnaire (QOLEB) and eight children who were able to complete the Pediatric Quality of Life Inventory (PedsQL) with the aid of their parents.
Dr. Mellerio reported that adults with RDEB-GS had an overall QOLEB score of 24 out of 50, an indication that their condition had a severe impact on their quality of life. The effect on quality of life was greater in terms of their physical functioning than emotional well-being, with respective scores of 19 out of 36, and 5 out of a possible 15. Less impact on quality of life was reported by participants with other RDEB subtypes.
PedsQL scores for the children indicated there might be a lesser effect of physical functioning on quality of life but a greater effect of emotional well-being on quality of life, but the numbers were small. “Interestingly, parents tended to rate their children’s impact on quality of life much higher than the children themselves,” Dr. Mellerio said.
The point of PEBLES is to start to understand the natural history of RDEB and to identify endpoints that might help in clinical trials of potential new treatments. Discussing the next steps for PEBLES, Dr. Mellerio said the aim was to recruit more pediatric patients and look at other data sets, such as bone health. The PEBLES team also hopes to extend recruitment to include other United Kingdom, and ultimately international, EB centers and, perhaps eventually to start to include other types of EB, such as EB simplex.
PEBLES is funded by DEBRA UK. Dr. Mellerio is a PEBLES investigator but had no conflicts of interest to disclose.
SOURCE: Mellerio JE et al. EB 2020. Pillay EI et al. Poster 77; Jeffs E et al. Poster 74; Jeffs et al. Poster 75. https://ebworldcongress.org/.
LONDON – More than £2.8 million (RDEB), according to a report at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA).
Results from the Prospective Epidermolysis Bullosa Longitudinal Evaluation Study (PEBLES), which is looking at the natural history of RDEB, showed that wound dressing and bandage costs were highest for study participants with the generalized severe (RDEB-GS) subtype, at just over £85,156 (about $112,450) per patient annually. Respective yearly costs for the generalized intermediate (RDEB-GI) and inversa (RDEB-INV) subtypes were £10,112 (about $13,350) and £1,699 (about $2,240) per patient.
Looking at the costs associated with EB is important, said one of the lead investigators for PEBLES, Jemima Mellerio, MD, FRCP, consultant dermatologist at St John’s Institute of Dermatology, at Guy’s & St. Thomas’ NHS Foundation Trust, London.
“If we are going to justify the kind of expenditure [associated with new treatments], we need to know that what we are treating is already a significant burden on our health care systems,” Dr. Mellerio said.
PEBLES is an ongoing London-based registry study that is enrolling patients with all subtypes of RDEB. Data are collected via a tablet device and include demographic data, information on clinical features, results of skin biopsies and genetic tests, and laboratory findings, as well as objective disease severity and subjective patient-orientated outcome scores.
So far, 60 patients – 49 adults and 11 children – have been enrolled in PEBLES since November 2014: 26 with RDEB-GS, 23 with RDEB-GI, 9 with RDEB-INV, and 2 with the pruriginosa RDEB subtype (RDEB-PR).
Most of the participants (71%) changed all their wound dressings at one time, patching up when required. Fourteen of 49 participants had paid people to help them change their dressings and when the total cost of combined wound dressings and paid care was taken into consideration, the mean annual cost per patient was around £2,500 (about $3,300) for RDEB-INV, £10,375 (about $13,700) per patient for RDEB-GS, and a staggering £98,000 (about $129,000) per patient for RDEB-GS. The total annual cost of dressings and associated care was an estimated £3,184,229 (about $4.2 million).
In addition to data on the cost of wound dressings, data on itch and pain and quality of life were presented at the EB World Congress and discussed by Dr. Mellerio.
A total of 42 participants older than 8 years of age had itch measured via the Leuven Itch Scale, she reported, noting that itch was a consistent symptom across all subtypes of RDEB. Itch is important as it not only causes problems with skin lesions and healing, but also significantly affects sleep and has a negative impact on patients’ mood, she emphasized.
Despite experiencing itch, more than half (58%) of participants were not using any kind of treatment for itch. This “likely reflects the lack of effectiveness of current medication for this debilitating symptom,” Dr. Mellerio and associates noted in one of their poster presentations of PEBLES data.
When treatment was used for itch, it consisted mainly of antihistamines (19% of patients), emollients (19%), or a combination of both (4%). However, treatment was generally “not very good,” with a satisfaction score of just 5 on a scale of 10, Dr. Mellerio pointed out. Participants “reported frustration with the lack of effective treatment for itch,” she said.
Itch was associated with disturbed sleep 1-3 nights per week in 20%-40% of participants, and every night in 20%-30%.
Pain was found to be a significant problem, with a median level of background pain scored as 4 on a 10-cm visual analog scale and a higher level (6) when associated with dressing changes.
Data on how RDEB affected quality of life were reported for 39 adults completing the 17-item Quality of Life in EB Questionnaire (QOLEB) and eight children who were able to complete the Pediatric Quality of Life Inventory (PedsQL) with the aid of their parents.
Dr. Mellerio reported that adults with RDEB-GS had an overall QOLEB score of 24 out of 50, an indication that their condition had a severe impact on their quality of life. The effect on quality of life was greater in terms of their physical functioning than emotional well-being, with respective scores of 19 out of 36, and 5 out of a possible 15. Less impact on quality of life was reported by participants with other RDEB subtypes.
PedsQL scores for the children indicated there might be a lesser effect of physical functioning on quality of life but a greater effect of emotional well-being on quality of life, but the numbers were small. “Interestingly, parents tended to rate their children’s impact on quality of life much higher than the children themselves,” Dr. Mellerio said.
The point of PEBLES is to start to understand the natural history of RDEB and to identify endpoints that might help in clinical trials of potential new treatments. Discussing the next steps for PEBLES, Dr. Mellerio said the aim was to recruit more pediatric patients and look at other data sets, such as bone health. The PEBLES team also hopes to extend recruitment to include other United Kingdom, and ultimately international, EB centers and, perhaps eventually to start to include other types of EB, such as EB simplex.
PEBLES is funded by DEBRA UK. Dr. Mellerio is a PEBLES investigator but had no conflicts of interest to disclose.
SOURCE: Mellerio JE et al. EB 2020. Pillay EI et al. Poster 77; Jeffs E et al. Poster 74; Jeffs et al. Poster 75. https://ebworldcongress.org/.
LONDON – More than £2.8 million (RDEB), according to a report at the EB World Congress, organized by the Dystrophic Epidermolysis Bullosa Association (DEBRA).
Results from the Prospective Epidermolysis Bullosa Longitudinal Evaluation Study (PEBLES), which is looking at the natural history of RDEB, showed that wound dressing and bandage costs were highest for study participants with the generalized severe (RDEB-GS) subtype, at just over £85,156 (about $112,450) per patient annually. Respective yearly costs for the generalized intermediate (RDEB-GI) and inversa (RDEB-INV) subtypes were £10,112 (about $13,350) and £1,699 (about $2,240) per patient.
Looking at the costs associated with EB is important, said one of the lead investigators for PEBLES, Jemima Mellerio, MD, FRCP, consultant dermatologist at St John’s Institute of Dermatology, at Guy’s & St. Thomas’ NHS Foundation Trust, London.
“If we are going to justify the kind of expenditure [associated with new treatments], we need to know that what we are treating is already a significant burden on our health care systems,” Dr. Mellerio said.
PEBLES is an ongoing London-based registry study that is enrolling patients with all subtypes of RDEB. Data are collected via a tablet device and include demographic data, information on clinical features, results of skin biopsies and genetic tests, and laboratory findings, as well as objective disease severity and subjective patient-orientated outcome scores.
So far, 60 patients – 49 adults and 11 children – have been enrolled in PEBLES since November 2014: 26 with RDEB-GS, 23 with RDEB-GI, 9 with RDEB-INV, and 2 with the pruriginosa RDEB subtype (RDEB-PR).
Most of the participants (71%) changed all their wound dressings at one time, patching up when required. Fourteen of 49 participants had paid people to help them change their dressings and when the total cost of combined wound dressings and paid care was taken into consideration, the mean annual cost per patient was around £2,500 (about $3,300) for RDEB-INV, £10,375 (about $13,700) per patient for RDEB-GS, and a staggering £98,000 (about $129,000) per patient for RDEB-GS. The total annual cost of dressings and associated care was an estimated £3,184,229 (about $4.2 million).
In addition to data on the cost of wound dressings, data on itch and pain and quality of life were presented at the EB World Congress and discussed by Dr. Mellerio.
A total of 42 participants older than 8 years of age had itch measured via the Leuven Itch Scale, she reported, noting that itch was a consistent symptom across all subtypes of RDEB. Itch is important as it not only causes problems with skin lesions and healing, but also significantly affects sleep and has a negative impact on patients’ mood, she emphasized.
Despite experiencing itch, more than half (58%) of participants were not using any kind of treatment for itch. This “likely reflects the lack of effectiveness of current medication for this debilitating symptom,” Dr. Mellerio and associates noted in one of their poster presentations of PEBLES data.
When treatment was used for itch, it consisted mainly of antihistamines (19% of patients), emollients (19%), or a combination of both (4%). However, treatment was generally “not very good,” with a satisfaction score of just 5 on a scale of 10, Dr. Mellerio pointed out. Participants “reported frustration with the lack of effective treatment for itch,” she said.
Itch was associated with disturbed sleep 1-3 nights per week in 20%-40% of participants, and every night in 20%-30%.
Pain was found to be a significant problem, with a median level of background pain scored as 4 on a 10-cm visual analog scale and a higher level (6) when associated with dressing changes.
Data on how RDEB affected quality of life were reported for 39 adults completing the 17-item Quality of Life in EB Questionnaire (QOLEB) and eight children who were able to complete the Pediatric Quality of Life Inventory (PedsQL) with the aid of their parents.
Dr. Mellerio reported that adults with RDEB-GS had an overall QOLEB score of 24 out of 50, an indication that their condition had a severe impact on their quality of life. The effect on quality of life was greater in terms of their physical functioning than emotional well-being, with respective scores of 19 out of 36, and 5 out of a possible 15. Less impact on quality of life was reported by participants with other RDEB subtypes.
PedsQL scores for the children indicated there might be a lesser effect of physical functioning on quality of life but a greater effect of emotional well-being on quality of life, but the numbers were small. “Interestingly, parents tended to rate their children’s impact on quality of life much higher than the children themselves,” Dr. Mellerio said.
The point of PEBLES is to start to understand the natural history of RDEB and to identify endpoints that might help in clinical trials of potential new treatments. Discussing the next steps for PEBLES, Dr. Mellerio said the aim was to recruit more pediatric patients and look at other data sets, such as bone health. The PEBLES team also hopes to extend recruitment to include other United Kingdom, and ultimately international, EB centers and, perhaps eventually to start to include other types of EB, such as EB simplex.
PEBLES is funded by DEBRA UK. Dr. Mellerio is a PEBLES investigator but had no conflicts of interest to disclose.
SOURCE: Mellerio JE et al. EB 2020. Pillay EI et al. Poster 77; Jeffs E et al. Poster 74; Jeffs et al. Poster 75. https://ebworldcongress.org/.
REPORTING FROM EB 2020
Presentation of a Rare Malignancy: Leiomyosarcoma of the Prostate (FULL)
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B). 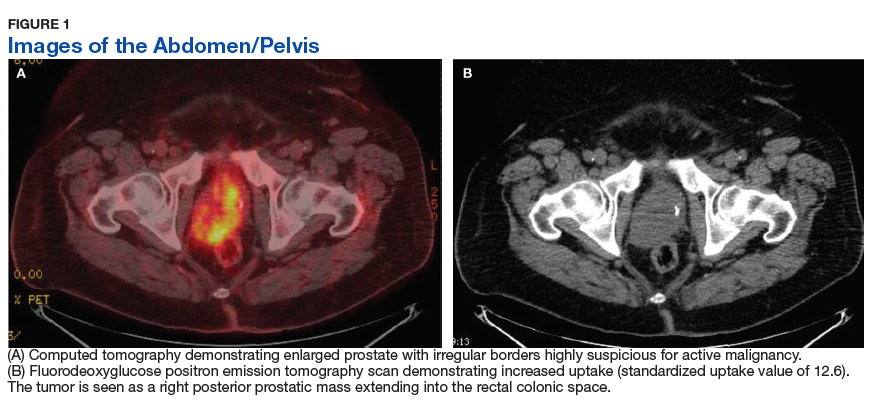
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues. 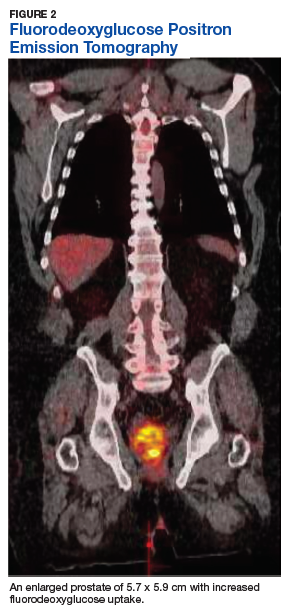
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is an aggressive malignancy with a high risk of metastasis and a poor prognosis that poses unique diagnostic and treatment challenges.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B).
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues.
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Prostatic leiomyosarcoma is a rare tumor.1 This neoplasm is composed of highly aggressive prostatic smooth muscle cells that present with nonspecific signs and symptoms mimicking other forms of prostatic pathology. Of the primary prostatic sarcomas, leiomyosarcoma represents the most common subtype in adults and is found in 38% to 52% of newly diagnosed prostate sarcoma.1,2 The prognosis is poor, and no clear guidelines exist regarding the optimal treatment approach. We report a case of prostate leiomyosarcoma and describe the disease characteristics, diagnostic modalities, and treatment approach regarding these rare malignancies.
Case Presentation
A 72-year-old male presented with 6 months of progressive severe lower urinary tract symptoms (LUTS) secondary to bladder outlet obstruction. The patient was refractory to medical management with combination α-blocker and 5-α-reductase inhibitor therapy and continued to require multiple emergent bladder catheterizations. Workup with urinalysis, blood biochemistry, and prostate specific antigen (PSA) levels were persistently normal. He reported no hematuria, weight loss, or perineal pain. The patient reported no history of tobacco use, exposure to hazardous chemicals, and had no family history of genitourinary cancers. On rectal exam, the prostate was firm and nodular, with induration noted along the right upper lobe of the prostate.
The patient was referred for a urology consultation and subsequently underwent transurethral resection of the prostate (TURP) for suspected severe benign prostatic hypertrophy (BPH). A histopathologic examination demonstrated atypical cytology consistent with high- grade leiomyosarcoma. Immunohistochemical analysis revealed positive staining for vimentin, smooth muscle actin, desmin (partial), cytokeratin, smooth muscle myosin, muscle specific actin, and Ki-67 (50%-60% expression).
Fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed a 5.7 x 5.9 cm tumor with a maximum standardized uptake value (SUVmax) of 12.6 in the right posterior prostate, without evidence of metastatic disease (Figures 1A and 1B).
Discussion
Originating from prostatic interstitial cells, prostatic leiomyosarcoma is a rare tumor that accounts for < 0.1% of all primary prostatic malignancies.1 Since its first description in 1950 by Riba and colleagues, < 200 cases have been reported worldwide.2 Among the sarcomas of the prostate, it is the most common tumor, accounting for around 38% to 52% of prostate sarcoma presentations.1,2
Patients typically present between the ages of 41 and 78 years (mean age 61 years).2,3 Signs and symptoms at presentation may vary; however, the most common symptoms are related to lower urinary tract obstruction (89.4% of patients). These symptoms include urinary frequency, urgency, nocturia, and may mimic the presentation of BPH.
Symptoms commonly associated with other malignancies, including constitutional symptoms such as weight loss, tend to occur less frequently or may be absent. Perineal or rectal pain may only be present in 25.6% of patients. Hematuria, burning on ejaculation, and constitutional symptoms are a less common presentation (< 10% of patients).3,4 PSA levels typically do not rise and are found to be within normal limits. The lack of PSA elevation is related to the tumors nonepithelial origin and may contribute to a delay in diagnosis.2,4,5
Diagnosis
Diagnosis may be further eluded as digital rectal exam (DRE) findings tend to reveal nonspecific enlargement of the prostate, resembling that of BPH. DRE may show a hard and firm prostate with nodular induration at the base or over the lobes of the prostate.6 At this stage a urology consultation is useful, as diagnosis is most commonly achieved using transrectal ultrasound (TRUS) with ultrasound-guided needle biopsy or after a TURP procedure.3
Prostate sarcoma is associated with markedly enlarged prostate volume, irregular margins with invasion, or heterogenous hypoechoic lesions on TRUS.7 Transperineal biopsy, computed tomography (CT)-guided biopsy, or suprapubic prostatectomy have been less frequently employed for diagnosis in previously reported cases.8 Specialized imaging modalities, such as CT scan or bone scan, do not show any specific findings with regards to these tumors; their role is limited to evaluation of the local and distant metastasis and for follow-up assessments.9 Transabdominal ultrasound may assess hydronephrosis or enlarged prostate and its relation to nearby structures, although it has not been shown to be helpful in establishing a specific diagnosis.6
Histologically, prostatic leiomyosarcoma is a distinct subtype of prostatic sarcoma. Other subtypes include stromal tumors such as rhabdomyosarcoma, fibrosarcoma, and spindle cell sarcoma.2 The majority of leiomyosarcomas are high-grade lesions demonstrating neoplastic spindle cells with nuclear atypia, multifocal necrosis, and cystic degeneration. Low-grade leiomyosarcomas are very rare.10 Immunohistochemistry is characteristically positive for vimentin, smooth muscle actin, and desmin expression. Cytokeratin may be positive in up to 25% of cases, whereas S-100, CD34, CD117, and PSA are negative.2,3 These histopathological findings help to differentiate leiomyosarcoma from other prostatic tumors.
Tumor size may vary greatly, and measurements have been reported to range from 3 cm to 21 cm, frequently presenting with invasion of local structures.11 Advanced stage disease is commonly found at initial diagnosis and is thought to be due to the lack of early specific symptoms. Metastatic disease at presentation may be found in up to one-third of patients, with the lungs being the most common site of metastasis followed by the liver. Local extent and distant spread of disease may be determined by CT or magnetic resonance imaging (MRI) scans, which provide clear delineation of neoplastic and nonneoplastic tissues.
Treatment
Treatment regimens may include a multimodal approach of combination surgery, radiation, and chemotherapy. However, there are currently no standardized guidelines for treatment and the optimal therapy remains unknown.2,3,6 Surgery remains the mainstay of treatment, and patients with surgically resectable tumors are treated with curative intent. Surgeries performed include radical retropubic prostatectomy, radical cystoprostatectomy, suprapubic prostatectomy, and pelvic exenteration.2,5,8,12 These operations may be preceded or followed by radiation therapy and/or chemotherapy depending on extent of disease.
It has been reported that neo-adjuvant chemotherapy and/or radiotherapy can aid in decreasing tumor burden to facilitate a complete resection.2,8,13,14 Patients who are determined to not be candidates for surgery or whom have widespread disease may be offered systemic chemotherapy. Chemotherapy regimens vary, but common regimens include anthracyclines (doxorubicin or epirubicin), alkylating agents (cyclophosphamide, ifosfamide, dacarbazine), and/or vinca alkaloids (vinblastine or vincristine). Patients who do not receive surgical intervention rarely achieve a sustained remission.3,5,8
The long-term prognosis of prostatic leiomyosarcoma is poor due to the aggressive nature of the neoplasm and the high chance of disease recurrence or metastasis. Median survival is estimated at 17 months, and from 50% to 75% of patients die within 2 to 5 years of diagnosis.2,3 Prognosis may be improved in patients with localized disease at diagnosis who are candidates for complete surgical resection with negative margins.13 Adverse prognostic factors include metastatic disease at presentation and the presence of positive surgical margins after surgery.
Overall survival is very poor, and it is estimated that the 1-, 3-, and 5-year survival rates are 68%, 34%, and 26%, respectively.3 However, some studies estimate the 5-year survival to be anywhere from 0 to 60%.8,9 Due to the substantially high risk of death, prostatic leiomyosarcoma may be one of the most aggressive and poorly prognostic malignancies involving the prostate.
Conclusion
Prostatic leiomyosarcoma poses a unique diagnostic challenge, as clinical presentation alone may not always be suggestive of underlying malignancy. This challenge is further exacerbated by its aggressive nature, high risk of metastasis, and difficulties with unclear treatment. Proper history and physical examination, differential diagnosis, and a multidisciplinary approach to patient care are the foundation for early detection and promoting improved survival.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29
1. Miedler JD, MacLennan GT. Leiomyosarcoma of the prostate. J Urol. 2007;178(2):668.
2. Zazzara M, Divenuto L, Scarcia M, Cardo G, Maselli FP, Ludovico GM. Leiomyosarcoma of prostate: case report and literature review. Urol Case Rep. 2018;17:4-6.
3. Vandoros GP, Manolidis T, Karamouzis MV, et al. Leiomyosarcoma of the prostate: case report and review of 54 previously published cases. Sarcoma. 2008;2008:458709.
4. Talapatra K, Nemade B, Bhutani R, et al. Recurrent episodes of hematuria: a rare presentation of leiomyosarcoma of prostate. J Cancer Res Ther. 2006;2(4):212-214.
5. Cheville JC, Dundore PA, Nascimento AG, et al. Leiomyosarcoma of the prostate. Report of 23 cases. Cancer. 1995;76(8):1422-1427.
6. Venyo AK. A review of the literature on primary leiomyosarcoma of the prostate gland. Adv Urol. 2015;2015:485786.
7. Stilgenbauer R, Benedict M, Bamshad R, Viduetsky A. Sarcoma of the prostate: sonographic findings and pathologic correlation. J Ultrasound Med. 2007;26(12):1789-1793.
8. Sexton WJ, Lance RE, Reyes AO, Pisters PW, Tu SM, Pisters LL. Adult prostate sarcoma: the M.D. Anderson Cancer Center experience. J Urol. 2001;166(2):521-525.
9. Singh JP, Chakraborty D, Bera MK, Pal D. Leiomyosarcoma of prostate: a rare, aggressive tumor. J Cancer Res Ther. 2013;9(4):743-745.
10. Hansel DE, Herawi M, Montgomery E, Epstein JI. Spindle cell lesions of the adult prostate. Mod Pathol. 2007;20(1):148-158.
11. Punt SE, Eary JF, O'Sullivan J, Conrad EU. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: imaging characteristics. Nucl Med Commun. 2009;30(7):546-549.
12. Dotan ZA, Tal R, Golijanin D, et al. Adult genitourinary sarcoma: the 25-year Memorial Sloan-Kettering experience. J Urol. 2006;176(5):2033-2038.
13. Musser JE, Assel M, Mashni JW, Sjoberg DD, Russo P. Adult prostate sarcoma: the Memorial Sloan Kettering experience. Urology. 2014;84(3):624-628.
14. Janet NL, May AW, Akins RS. Sarcoma of the prostate: a single institutional review. Am J Clin Oncol. 2009;32:27-29
Deferiprone noninferior to deferoxamine for iron overload in SCD, rare anemias
ORLANDO – The oral iron chelator deferiprone showed noninferiority to deferoxamine for treating iron overload in patients with sickle cell disease and other rare anemias in a randomized open-label trial.
The least squares mean change from baseline in liver iron concentration (LIC) – the primary study endpoint – was –4.04 mg/g dry weight (dw) in 152 patients randomized to receive deferiprone, and –4.45 mg/g dw in 76 who received deferoxamine, Janet L. Kwiatkowski, MD, of the Children’s Hospital of Philadelphia reported at the annual meeting of the American Society of Hematology.
The upper limit of the stringent 96.01% confidence interval used for the evaluation of noninferiority in the study was 1.57, thus the findings demonstrated noninferiority of deferiprone, Dr. Kwiatkowski said.
Deferiprone also showed noninferiority for the secondary endpoints of change in cardiac iron (about –0.02 ms on T2* MRI, log-transformed for both groups) and serum ferritin levels (–415 vs. –750 mcg/L for deferiprone vs. deferoxamine) at 12 months. The difference between the groups was not statistically significant for either endpoint.
Study participants, who had a mean age of 16.9 years, were aged 2 years and older with LIC between 7 and 30 mg/g dw. They were recruited from 33 sites in nine countries and randomized 2:1 to receive deferiprone or deferoxamine for up to 12 months; in patients with lower transfusional iron input and/or less severe iron load, deferiprone was dosed at 75 mg/kg daily and deferoxamine was dosed at 20 mg/kg for children and 40 mg/kg for adults. In those with higher iron input and/or more severe iron load, the deferiprone dose was 99 mg/kg daily and the deferoxamine doses were up to 40 mg/kg in children and up to 50 mg/kg for adults.
“Over the course of the treatment period, the dosage could be adjusted downward if there were side effects, or upward if there was no improvement in iron burden,” Dr Kwiatkowski said, adding that after 12 months, patients had the option of continuing on to a 2-year extension trial in which everyone received deferiprone.
No significant demographic differences were noted between the groups; 84% in both groups had sickle cell disease, and the remaining patients had other, rarer forms of transfusion-dependent anemia. Baseline iron burden was similar in the groups.
The rates of acceptable compliance over the course of the study were also similar at 69% and 79% in the deferiprone and deferoxamine arms, respectively, she noted.
No statistically significant difference between the groups was seen in the overall rate of adverse events, treatment-related AEs, serious AEs, or withdrawals from the study due to AEs. Agranulocytosis occurred in one deferiprone patient and zero deferoxamine patients, and mild or moderate neutropenia occurred in four patients and one patient in the groups, respectively.
All episodes resolved, no difference was seen in the rates of any of the serious AEs, and no unexpected serious adverse events occurred, she said.
Patients with sickle cell disease or other rare anemias whose care includes chronic blood transfusions require iron chelation to prevent iron overload. Currently, only deferoxamine and deferasirox are approved chelators in these patient populations, she said, noting that in 2011 deferiprone received accelerated Food and Drug Administration approval for the treatment of thalassemia.
The current study was conducted because of an FDA requirement for postmarket assessment of deferiprone’s efficacy and safety in patients with sickle cell disease and other anemias who develop transfusional iron overload. It was initiated prior to the approval of deferasirox for the first-line treatment of SCD, therefore it was compared only with deferoxamine, she explained.
Dr. Kwiatkowski reported research funding from Apopharma, bluebird bio, Novartis, and Terumo, and consultancy for Agios, bluebird bio, Celgene, and Imara.
ORLANDO – The oral iron chelator deferiprone showed noninferiority to deferoxamine for treating iron overload in patients with sickle cell disease and other rare anemias in a randomized open-label trial.
The least squares mean change from baseline in liver iron concentration (LIC) – the primary study endpoint – was –4.04 mg/g dry weight (dw) in 152 patients randomized to receive deferiprone, and –4.45 mg/g dw in 76 who received deferoxamine, Janet L. Kwiatkowski, MD, of the Children’s Hospital of Philadelphia reported at the annual meeting of the American Society of Hematology.
The upper limit of the stringent 96.01% confidence interval used for the evaluation of noninferiority in the study was 1.57, thus the findings demonstrated noninferiority of deferiprone, Dr. Kwiatkowski said.
Deferiprone also showed noninferiority for the secondary endpoints of change in cardiac iron (about –0.02 ms on T2* MRI, log-transformed for both groups) and serum ferritin levels (–415 vs. –750 mcg/L for deferiprone vs. deferoxamine) at 12 months. The difference between the groups was not statistically significant for either endpoint.
Study participants, who had a mean age of 16.9 years, were aged 2 years and older with LIC between 7 and 30 mg/g dw. They were recruited from 33 sites in nine countries and randomized 2:1 to receive deferiprone or deferoxamine for up to 12 months; in patients with lower transfusional iron input and/or less severe iron load, deferiprone was dosed at 75 mg/kg daily and deferoxamine was dosed at 20 mg/kg for children and 40 mg/kg for adults. In those with higher iron input and/or more severe iron load, the deferiprone dose was 99 mg/kg daily and the deferoxamine doses were up to 40 mg/kg in children and up to 50 mg/kg for adults.
“Over the course of the treatment period, the dosage could be adjusted downward if there were side effects, or upward if there was no improvement in iron burden,” Dr Kwiatkowski said, adding that after 12 months, patients had the option of continuing on to a 2-year extension trial in which everyone received deferiprone.
No significant demographic differences were noted between the groups; 84% in both groups had sickle cell disease, and the remaining patients had other, rarer forms of transfusion-dependent anemia. Baseline iron burden was similar in the groups.
The rates of acceptable compliance over the course of the study were also similar at 69% and 79% in the deferiprone and deferoxamine arms, respectively, she noted.
No statistically significant difference between the groups was seen in the overall rate of adverse events, treatment-related AEs, serious AEs, or withdrawals from the study due to AEs. Agranulocytosis occurred in one deferiprone patient and zero deferoxamine patients, and mild or moderate neutropenia occurred in four patients and one patient in the groups, respectively.
All episodes resolved, no difference was seen in the rates of any of the serious AEs, and no unexpected serious adverse events occurred, she said.
Patients with sickle cell disease or other rare anemias whose care includes chronic blood transfusions require iron chelation to prevent iron overload. Currently, only deferoxamine and deferasirox are approved chelators in these patient populations, she said, noting that in 2011 deferiprone received accelerated Food and Drug Administration approval for the treatment of thalassemia.
The current study was conducted because of an FDA requirement for postmarket assessment of deferiprone’s efficacy and safety in patients with sickle cell disease and other anemias who develop transfusional iron overload. It was initiated prior to the approval of deferasirox for the first-line treatment of SCD, therefore it was compared only with deferoxamine, she explained.
Dr. Kwiatkowski reported research funding from Apopharma, bluebird bio, Novartis, and Terumo, and consultancy for Agios, bluebird bio, Celgene, and Imara.
ORLANDO – The oral iron chelator deferiprone showed noninferiority to deferoxamine for treating iron overload in patients with sickle cell disease and other rare anemias in a randomized open-label trial.
The least squares mean change from baseline in liver iron concentration (LIC) – the primary study endpoint – was –4.04 mg/g dry weight (dw) in 152 patients randomized to receive deferiprone, and –4.45 mg/g dw in 76 who received deferoxamine, Janet L. Kwiatkowski, MD, of the Children’s Hospital of Philadelphia reported at the annual meeting of the American Society of Hematology.
The upper limit of the stringent 96.01% confidence interval used for the evaluation of noninferiority in the study was 1.57, thus the findings demonstrated noninferiority of deferiprone, Dr. Kwiatkowski said.
Deferiprone also showed noninferiority for the secondary endpoints of change in cardiac iron (about –0.02 ms on T2* MRI, log-transformed for both groups) and serum ferritin levels (–415 vs. –750 mcg/L for deferiprone vs. deferoxamine) at 12 months. The difference between the groups was not statistically significant for either endpoint.
Study participants, who had a mean age of 16.9 years, were aged 2 years and older with LIC between 7 and 30 mg/g dw. They were recruited from 33 sites in nine countries and randomized 2:1 to receive deferiprone or deferoxamine for up to 12 months; in patients with lower transfusional iron input and/or less severe iron load, deferiprone was dosed at 75 mg/kg daily and deferoxamine was dosed at 20 mg/kg for children and 40 mg/kg for adults. In those with higher iron input and/or more severe iron load, the deferiprone dose was 99 mg/kg daily and the deferoxamine doses were up to 40 mg/kg in children and up to 50 mg/kg for adults.
“Over the course of the treatment period, the dosage could be adjusted downward if there were side effects, or upward if there was no improvement in iron burden,” Dr Kwiatkowski said, adding that after 12 months, patients had the option of continuing on to a 2-year extension trial in which everyone received deferiprone.
No significant demographic differences were noted between the groups; 84% in both groups had sickle cell disease, and the remaining patients had other, rarer forms of transfusion-dependent anemia. Baseline iron burden was similar in the groups.
The rates of acceptable compliance over the course of the study were also similar at 69% and 79% in the deferiprone and deferoxamine arms, respectively, she noted.
No statistically significant difference between the groups was seen in the overall rate of adverse events, treatment-related AEs, serious AEs, or withdrawals from the study due to AEs. Agranulocytosis occurred in one deferiprone patient and zero deferoxamine patients, and mild or moderate neutropenia occurred in four patients and one patient in the groups, respectively.
All episodes resolved, no difference was seen in the rates of any of the serious AEs, and no unexpected serious adverse events occurred, she said.
Patients with sickle cell disease or other rare anemias whose care includes chronic blood transfusions require iron chelation to prevent iron overload. Currently, only deferoxamine and deferasirox are approved chelators in these patient populations, she said, noting that in 2011 deferiprone received accelerated Food and Drug Administration approval for the treatment of thalassemia.
The current study was conducted because of an FDA requirement for postmarket assessment of deferiprone’s efficacy and safety in patients with sickle cell disease and other anemias who develop transfusional iron overload. It was initiated prior to the approval of deferasirox for the first-line treatment of SCD, therefore it was compared only with deferoxamine, she explained.
Dr. Kwiatkowski reported research funding from Apopharma, bluebird bio, Novartis, and Terumo, and consultancy for Agios, bluebird bio, Celgene, and Imara.
REPORTING FROM ASH 2019
Most epidermolysis bullosa patients turn to topical antimicrobials
Most patients with epidermolysis bullosa who use topical products choose antimicrobials, according to data from a survey of 202 children and adults.
Management of epidermolysis bullosa (EB) involves a combination of skin protection and infection management, but patient home care practices have not been well studied, wrote Leila Shayegan of Columbia University, New York, and colleagues.
In a study published in Pediatric Dermatology, the researchers surveyed 202 patients who were enrolled in the Epidermolysis Bullosa Clinical Characterization and Outcomes Database during 2017. The patients ranged in age from 1 month to 62 years with an average age of 11 years; 52% were female. The patients represented a range of EB subtypes, including 130 patients with dystrophic EB, 51 patients with EB simplex, 21 with junctional EB, and 3 patients each with Kindler syndrome and unspecified subtypes.
Overall, most of the patients reported cleaning their skin either every day (37%) or every other day (32%). Of the 188 patients who reported using topical products on their wounds, 131 (70%) said they used at least one antimicrobial product, while 125 patients (66%) reported using at least one emollient; 32 (17%) used emollients only, and 21(11%) reported no use of topical products.
The most popular topical antibiotics were mupirocin (31%) and bacitracin (31%). In addition, 14% of respondents used silver-containing products, and 16% used medical-grade honey. Roughly half (51%) of patients who reported use of at least one antimicrobial product used two or more different antimicrobial products.
A total of 38% of patients used only water for cleansing. Of the 131 patients who reported using additives in their cleansing water, 57% added salt, 54% added bleach, 27% added vinegar, and 26% reported “other” additive use, which could include Epsom salt, baking soda, oatmeal, or essential oils, the researchers said. The concentrations of these additives ranged from barely effective 0.002% sodium hypochlorite and 0.002% acetic acid solutions to potentially cytotoxic solutions of 0.09% sodium hypochlorite and 0.156% acetic acid.
“Although the survey was not designed to correlate skin care practices with wound culture results and resistance patterns, widespread use of topical antimicrobials described among EB patients highlights the need for increased emphasis on antibiotic stewardship,” the researchers noted. They added that health care providers should educate patients and families not only about mindful use of antibiotics, but also appropriate concentrations of cleansing additives.
“Optimizing EB patient home skin care routines, along with future longitudinal studies on the impact of EB skin care interventions on microbial resistance patterns, wound healing and [squamous cell carcinoma] risk are necessary to improve outcomes for patients with EB,” they emphasized.
The Epidermolysis Bullosa Clinical Characterization and Outcomes Database used in the study is funded by the Epidermolysis Bullosa Research Partnership and the Epidermolysis Bullosa Medical Research Foundation. Ms. Shayegan had no financial conflicts to disclose. Several coauthors disclosed relationships with multiple companies including Abeona Therapeutics, Castle Creek Pharmaceuticals, Fibrocell Science, ProQR, and Scioderm.
SOURCE: Shayegan L et al. Pediatr Dermatol. 2020. doi: 10.1111/pde.14102.
Most patients with epidermolysis bullosa who use topical products choose antimicrobials, according to data from a survey of 202 children and adults.
Management of epidermolysis bullosa (EB) involves a combination of skin protection and infection management, but patient home care practices have not been well studied, wrote Leila Shayegan of Columbia University, New York, and colleagues.
In a study published in Pediatric Dermatology, the researchers surveyed 202 patients who were enrolled in the Epidermolysis Bullosa Clinical Characterization and Outcomes Database during 2017. The patients ranged in age from 1 month to 62 years with an average age of 11 years; 52% were female. The patients represented a range of EB subtypes, including 130 patients with dystrophic EB, 51 patients with EB simplex, 21 with junctional EB, and 3 patients each with Kindler syndrome and unspecified subtypes.
Overall, most of the patients reported cleaning their skin either every day (37%) or every other day (32%). Of the 188 patients who reported using topical products on their wounds, 131 (70%) said they used at least one antimicrobial product, while 125 patients (66%) reported using at least one emollient; 32 (17%) used emollients only, and 21(11%) reported no use of topical products.
The most popular topical antibiotics were mupirocin (31%) and bacitracin (31%). In addition, 14% of respondents used silver-containing products, and 16% used medical-grade honey. Roughly half (51%) of patients who reported use of at least one antimicrobial product used two or more different antimicrobial products.
A total of 38% of patients used only water for cleansing. Of the 131 patients who reported using additives in their cleansing water, 57% added salt, 54% added bleach, 27% added vinegar, and 26% reported “other” additive use, which could include Epsom salt, baking soda, oatmeal, or essential oils, the researchers said. The concentrations of these additives ranged from barely effective 0.002% sodium hypochlorite and 0.002% acetic acid solutions to potentially cytotoxic solutions of 0.09% sodium hypochlorite and 0.156% acetic acid.
“Although the survey was not designed to correlate skin care practices with wound culture results and resistance patterns, widespread use of topical antimicrobials described among EB patients highlights the need for increased emphasis on antibiotic stewardship,” the researchers noted. They added that health care providers should educate patients and families not only about mindful use of antibiotics, but also appropriate concentrations of cleansing additives.
“Optimizing EB patient home skin care routines, along with future longitudinal studies on the impact of EB skin care interventions on microbial resistance patterns, wound healing and [squamous cell carcinoma] risk are necessary to improve outcomes for patients with EB,” they emphasized.
The Epidermolysis Bullosa Clinical Characterization and Outcomes Database used in the study is funded by the Epidermolysis Bullosa Research Partnership and the Epidermolysis Bullosa Medical Research Foundation. Ms. Shayegan had no financial conflicts to disclose. Several coauthors disclosed relationships with multiple companies including Abeona Therapeutics, Castle Creek Pharmaceuticals, Fibrocell Science, ProQR, and Scioderm.
SOURCE: Shayegan L et al. Pediatr Dermatol. 2020. doi: 10.1111/pde.14102.
Most patients with epidermolysis bullosa who use topical products choose antimicrobials, according to data from a survey of 202 children and adults.
Management of epidermolysis bullosa (EB) involves a combination of skin protection and infection management, but patient home care practices have not been well studied, wrote Leila Shayegan of Columbia University, New York, and colleagues.
In a study published in Pediatric Dermatology, the researchers surveyed 202 patients who were enrolled in the Epidermolysis Bullosa Clinical Characterization and Outcomes Database during 2017. The patients ranged in age from 1 month to 62 years with an average age of 11 years; 52% were female. The patients represented a range of EB subtypes, including 130 patients with dystrophic EB, 51 patients with EB simplex, 21 with junctional EB, and 3 patients each with Kindler syndrome and unspecified subtypes.
Overall, most of the patients reported cleaning their skin either every day (37%) or every other day (32%). Of the 188 patients who reported using topical products on their wounds, 131 (70%) said they used at least one antimicrobial product, while 125 patients (66%) reported using at least one emollient; 32 (17%) used emollients only, and 21(11%) reported no use of topical products.
The most popular topical antibiotics were mupirocin (31%) and bacitracin (31%). In addition, 14% of respondents used silver-containing products, and 16% used medical-grade honey. Roughly half (51%) of patients who reported use of at least one antimicrobial product used two or more different antimicrobial products.
A total of 38% of patients used only water for cleansing. Of the 131 patients who reported using additives in their cleansing water, 57% added salt, 54% added bleach, 27% added vinegar, and 26% reported “other” additive use, which could include Epsom salt, baking soda, oatmeal, or essential oils, the researchers said. The concentrations of these additives ranged from barely effective 0.002% sodium hypochlorite and 0.002% acetic acid solutions to potentially cytotoxic solutions of 0.09% sodium hypochlorite and 0.156% acetic acid.
“Although the survey was not designed to correlate skin care practices with wound culture results and resistance patterns, widespread use of topical antimicrobials described among EB patients highlights the need for increased emphasis on antibiotic stewardship,” the researchers noted. They added that health care providers should educate patients and families not only about mindful use of antibiotics, but also appropriate concentrations of cleansing additives.
“Optimizing EB patient home skin care routines, along with future longitudinal studies on the impact of EB skin care interventions on microbial resistance patterns, wound healing and [squamous cell carcinoma] risk are necessary to improve outcomes for patients with EB,” they emphasized.
The Epidermolysis Bullosa Clinical Characterization and Outcomes Database used in the study is funded by the Epidermolysis Bullosa Research Partnership and the Epidermolysis Bullosa Medical Research Foundation. Ms. Shayegan had no financial conflicts to disclose. Several coauthors disclosed relationships with multiple companies including Abeona Therapeutics, Castle Creek Pharmaceuticals, Fibrocell Science, ProQR, and Scioderm.
SOURCE: Shayegan L et al. Pediatr Dermatol. 2020. doi: 10.1111/pde.14102.
FROM PEDIATRIC DERMATOLOGY
Zika virus: Birth defects rose fourfold in U.S. hardest-hit areas
according to the Centers for Disease Control and Prevention.
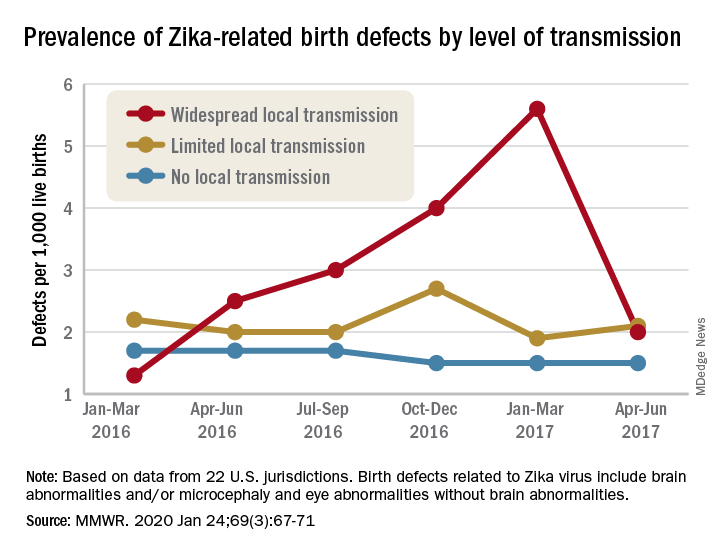
That spike in the prevalence of brain abnormalities and/or microcephaly or eye abnormalities without brain abnormalities came during January through March 2017, about 6 months after the Zika outbreak’s reported peak in the jurisdictions with widespread local transmission, Puerto Rico and the U.S. Virgin Islands, wrote Ashley N. Smoots, MPH, of the CDC’s National Center on Birth Defects and Developmental Disabilities and associates in the Morbidity and Mortality Weekly Report.
In those two territories, the prevalence of birth defects potentially related to Zika virus infection was 5.6 per 1,000 live births during January through March 2017, compared with 1.3 per 1,000 in January through March 2016, they reported.
In the southern areas of Florida and Texas, where there was limited local Zika transmission, the highest prevalence of birth defects, 2.7 per 1,000, occurred during October through December 2016, and was only slightly greater than the baseline rate of 2.2 per 1,000 in January through March 2016, the investigators reported.
Among the other 19 jurisdictions (including Illinois, Louisiana, New Jersey, South Carolina, and Virginia) involved in the analysis, the rate of Zika virus–related birth defects never reached any higher than the 1.7 per 1,000 recorded at the start of the study period in January through March 2016, they said.
“Population-based birth defects surveillance is critical for identifying infants and fetuses with birth defects potentially related to Zika virus regardless of whether Zika virus testing was conducted, especially given the high prevalence of asymptomatic disease. These data can be used to inform follow-up care and services as well as strengthen surveillance,” the investigators wrote.
SOURCE: Smoots AN et al. MMWR. 2020 Jan 24;69(3):67-71.
according to the Centers for Disease Control and Prevention.
That spike in the prevalence of brain abnormalities and/or microcephaly or eye abnormalities without brain abnormalities came during January through March 2017, about 6 months after the Zika outbreak’s reported peak in the jurisdictions with widespread local transmission, Puerto Rico and the U.S. Virgin Islands, wrote Ashley N. Smoots, MPH, of the CDC’s National Center on Birth Defects and Developmental Disabilities and associates in the Morbidity and Mortality Weekly Report.
In those two territories, the prevalence of birth defects potentially related to Zika virus infection was 5.6 per 1,000 live births during January through March 2017, compared with 1.3 per 1,000 in January through March 2016, they reported.
In the southern areas of Florida and Texas, where there was limited local Zika transmission, the highest prevalence of birth defects, 2.7 per 1,000, occurred during October through December 2016, and was only slightly greater than the baseline rate of 2.2 per 1,000 in January through March 2016, the investigators reported.
Among the other 19 jurisdictions (including Illinois, Louisiana, New Jersey, South Carolina, and Virginia) involved in the analysis, the rate of Zika virus–related birth defects never reached any higher than the 1.7 per 1,000 recorded at the start of the study period in January through March 2016, they said.
“Population-based birth defects surveillance is critical for identifying infants and fetuses with birth defects potentially related to Zika virus regardless of whether Zika virus testing was conducted, especially given the high prevalence of asymptomatic disease. These data can be used to inform follow-up care and services as well as strengthen surveillance,” the investigators wrote.
SOURCE: Smoots AN et al. MMWR. 2020 Jan 24;69(3):67-71.
according to the Centers for Disease Control and Prevention.
That spike in the prevalence of brain abnormalities and/or microcephaly or eye abnormalities without brain abnormalities came during January through March 2017, about 6 months after the Zika outbreak’s reported peak in the jurisdictions with widespread local transmission, Puerto Rico and the U.S. Virgin Islands, wrote Ashley N. Smoots, MPH, of the CDC’s National Center on Birth Defects and Developmental Disabilities and associates in the Morbidity and Mortality Weekly Report.
In those two territories, the prevalence of birth defects potentially related to Zika virus infection was 5.6 per 1,000 live births during January through March 2017, compared with 1.3 per 1,000 in January through March 2016, they reported.
In the southern areas of Florida and Texas, where there was limited local Zika transmission, the highest prevalence of birth defects, 2.7 per 1,000, occurred during October through December 2016, and was only slightly greater than the baseline rate of 2.2 per 1,000 in January through March 2016, the investigators reported.
Among the other 19 jurisdictions (including Illinois, Louisiana, New Jersey, South Carolina, and Virginia) involved in the analysis, the rate of Zika virus–related birth defects never reached any higher than the 1.7 per 1,000 recorded at the start of the study period in January through March 2016, they said.
“Population-based birth defects surveillance is critical for identifying infants and fetuses with birth defects potentially related to Zika virus regardless of whether Zika virus testing was conducted, especially given the high prevalence of asymptomatic disease. These data can be used to inform follow-up care and services as well as strengthen surveillance,” the investigators wrote.
SOURCE: Smoots AN et al. MMWR. 2020 Jan 24;69(3):67-71.
FROM MMWR
FDA approves first treatment for advanced epithelioid sarcoma
The Food and Drug Administration has granted accelerated approval to tazemetostat (Tazverik) for the treatment of adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection.

Approval was based on overall response rate in a trial enrolling 62 patients with metastatic or locally advanced epithelioid sarcoma. The overall response rate was 15%, with 1.6% of patients having a complete response and 13% having a partial response. Of the nine patients that had a response, six (67%) had a response lasting 6 months or longer, the FDA said in a press statement.
The most common side effects for patients taking tazemetostat were pain, fatigue, nausea, decreased appetite, vomiting, and constipation. Patients treated with tazemetostat are at increased risk of developing secondary malignancies, including T-cell lymphoblastic lymphoma, myelodysplastic syndrome, and acute myeloid leukemia.
“Epithelioid sarcoma accounts for less than 1% of all soft-tissue sarcomas,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research. “Until today, there were no treatment options specifically for patients with epithelioid sarcoma. The approval of Tazverik provides a treatment option that specifically targets this disease.”
Tazemetostat must be dispensed with a patient medication guide that describes important information about the drug’s uses and risks, the FDA said.
The Food and Drug Administration has granted accelerated approval to tazemetostat (Tazverik) for the treatment of adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection.
Approval was based on overall response rate in a trial enrolling 62 patients with metastatic or locally advanced epithelioid sarcoma. The overall response rate was 15%, with 1.6% of patients having a complete response and 13% having a partial response. Of the nine patients that had a response, six (67%) had a response lasting 6 months or longer, the FDA said in a press statement.
The most common side effects for patients taking tazemetostat were pain, fatigue, nausea, decreased appetite, vomiting, and constipation. Patients treated with tazemetostat are at increased risk of developing secondary malignancies, including T-cell lymphoblastic lymphoma, myelodysplastic syndrome, and acute myeloid leukemia.
“Epithelioid sarcoma accounts for less than 1% of all soft-tissue sarcomas,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research. “Until today, there were no treatment options specifically for patients with epithelioid sarcoma. The approval of Tazverik provides a treatment option that specifically targets this disease.”
Tazemetostat must be dispensed with a patient medication guide that describes important information about the drug’s uses and risks, the FDA said.
The Food and Drug Administration has granted accelerated approval to tazemetostat (Tazverik) for the treatment of adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection.
Approval was based on overall response rate in a trial enrolling 62 patients with metastatic or locally advanced epithelioid sarcoma. The overall response rate was 15%, with 1.6% of patients having a complete response and 13% having a partial response. Of the nine patients that had a response, six (67%) had a response lasting 6 months or longer, the FDA said in a press statement.
The most common side effects for patients taking tazemetostat were pain, fatigue, nausea, decreased appetite, vomiting, and constipation. Patients treated with tazemetostat are at increased risk of developing secondary malignancies, including T-cell lymphoblastic lymphoma, myelodysplastic syndrome, and acute myeloid leukemia.
“Epithelioid sarcoma accounts for less than 1% of all soft-tissue sarcomas,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the Center for Drug Evaluation and Research. “Until today, there were no treatment options specifically for patients with epithelioid sarcoma. The approval of Tazverik provides a treatment option that specifically targets this disease.”
Tazemetostat must be dispensed with a patient medication guide that describes important information about the drug’s uses and risks, the FDA said.
Expedited review programs not shortening drug development
Drug development times are not showing any signs of decreasing even though application review times at the Food and Drug Administration are improving, new research has shown.
“Despite the accumulation of expedited programs that have reduced FDA review times, overall clinical development times have not become shorter, although it is not possible to know what role these programs may have played in preventing an increase in development times,” Jonathan Darrow of Harvard Medical School, Boston, and colleagues wrote in a study published Jan. 14 in JAMA.
Researchers noted that FDA approval times declined from more than 3 years in 1983 to less than 1 year in 2017, although total time from the authorization of clinical testing to approval has remained steady across that period at about 8 years.
“The resistance of total development times to efforts to shorten them could be the result of more submissions of applications for rare disease drugs, which can sometimes pose trial recruitment challenges; a shifting emphasis to more challenging therapeutic categories, such as central nervous system disorder treatments; longer time horizons needed to establish efficacy when early intervention is important (e.g., cancer); or other factors,” wrote Mr. Darrow and colleagues.
The authors also note that the value of the expanded review toolbox the FDA now employs is “unclear” as “the number of new drug approvals has not increased substantially over the past three decades.” This observation does not count biologics or generic approvals.
Research revealed that the mean annual number of new drug approvals (including biologics) was 34 from 1990 to 1999, 25 from 2000 to 2009, and 41 from 2010 to 2018. In addition, while the number of new drug approvals has remained at or below 60 per year, the portion of drugs using at least one expedited review program increased over time, with more than 80% of the 59 new drugs approved in 2018 being subject to at least one expedited review program.
“Although the FDA has on average applied its expedited programs to drugs offering larger health gains, it is difficult to assess whether these programs have incentivized greater therapeutic innovation or merely allowed more appropriate agency prioritization,” noted Mr. Darrow and colleagues.
And while it may be hard to truly understand the impact of the many expedited review programs that have been introduced at the FDA, there is still room for improvement in the process.
In an editorial published in JAMA, Joshua Sharfstein, MD, of Johns Hopkins University, Baltimore, suggested four reforms the FDA could examine.
“First, Congress and the FDA should rationalize the various programs for expedited review,” he wrote, noting that companies are using the Orphan Drug Act to prevent competition. “Fixing the system requires, at a minimum, promoting meaningful competition for non-orphan uses.”
Second, he wrote, the FDA needs to strengthen oversight of postmarket safety though more diligence in the operation of its Risk Evaluation and Management Strategies (REMS) program.
“Third, Congress should recalibrate the programs that provide companies with special marketing protections,” added Dr. Sharfstein, former FDA principal deputy commissioner. “One place to look is pediatric exclusivity, which has been estimated to cost the health care system more than $6 for every $1 spent by a company on a pediatric trial.”
And finally, “Congress should use patent and pricing incentives to accelerate the generation of definitive evidence under accelerated approval,” citing proposals to limit pricing or market exclusivity until companies complete studies that assess clinically relevant endpoints.
“These changes would reflect an evolution, not a revolution, of the FDA’s approach to new drug approval,” Dr. Sharfstein said. “These reforms could also bring greater order and thoughtfulness to the regulation of important new therapies, while enhancing safety and creating greater capability to afford truly transformative medical products.”
SOURCE: Jonathan Darrow et al. JAMA 2020 Jan 14. doi: 10.1001/jama.2019.20288.
Drug development times are not showing any signs of decreasing even though application review times at the Food and Drug Administration are improving, new research has shown.
“Despite the accumulation of expedited programs that have reduced FDA review times, overall clinical development times have not become shorter, although it is not possible to know what role these programs may have played in preventing an increase in development times,” Jonathan Darrow of Harvard Medical School, Boston, and colleagues wrote in a study published Jan. 14 in JAMA.
Researchers noted that FDA approval times declined from more than 3 years in 1983 to less than 1 year in 2017, although total time from the authorization of clinical testing to approval has remained steady across that period at about 8 years.
“The resistance of total development times to efforts to shorten them could be the result of more submissions of applications for rare disease drugs, which can sometimes pose trial recruitment challenges; a shifting emphasis to more challenging therapeutic categories, such as central nervous system disorder treatments; longer time horizons needed to establish efficacy when early intervention is important (e.g., cancer); or other factors,” wrote Mr. Darrow and colleagues.
The authors also note that the value of the expanded review toolbox the FDA now employs is “unclear” as “the number of new drug approvals has not increased substantially over the past three decades.” This observation does not count biologics or generic approvals.
Research revealed that the mean annual number of new drug approvals (including biologics) was 34 from 1990 to 1999, 25 from 2000 to 2009, and 41 from 2010 to 2018. In addition, while the number of new drug approvals has remained at or below 60 per year, the portion of drugs using at least one expedited review program increased over time, with more than 80% of the 59 new drugs approved in 2018 being subject to at least one expedited review program.
“Although the FDA has on average applied its expedited programs to drugs offering larger health gains, it is difficult to assess whether these programs have incentivized greater therapeutic innovation or merely allowed more appropriate agency prioritization,” noted Mr. Darrow and colleagues.
And while it may be hard to truly understand the impact of the many expedited review programs that have been introduced at the FDA, there is still room for improvement in the process.
In an editorial published in JAMA, Joshua Sharfstein, MD, of Johns Hopkins University, Baltimore, suggested four reforms the FDA could examine.
“First, Congress and the FDA should rationalize the various programs for expedited review,” he wrote, noting that companies are using the Orphan Drug Act to prevent competition. “Fixing the system requires, at a minimum, promoting meaningful competition for non-orphan uses.”
Second, he wrote, the FDA needs to strengthen oversight of postmarket safety though more diligence in the operation of its Risk Evaluation and Management Strategies (REMS) program.
“Third, Congress should recalibrate the programs that provide companies with special marketing protections,” added Dr. Sharfstein, former FDA principal deputy commissioner. “One place to look is pediatric exclusivity, which has been estimated to cost the health care system more than $6 for every $1 spent by a company on a pediatric trial.”
And finally, “Congress should use patent and pricing incentives to accelerate the generation of definitive evidence under accelerated approval,” citing proposals to limit pricing or market exclusivity until companies complete studies that assess clinically relevant endpoints.
“These changes would reflect an evolution, not a revolution, of the FDA’s approach to new drug approval,” Dr. Sharfstein said. “These reforms could also bring greater order and thoughtfulness to the regulation of important new therapies, while enhancing safety and creating greater capability to afford truly transformative medical products.”
SOURCE: Jonathan Darrow et al. JAMA 2020 Jan 14. doi: 10.1001/jama.2019.20288.
Drug development times are not showing any signs of decreasing even though application review times at the Food and Drug Administration are improving, new research has shown.
“Despite the accumulation of expedited programs that have reduced FDA review times, overall clinical development times have not become shorter, although it is not possible to know what role these programs may have played in preventing an increase in development times,” Jonathan Darrow of Harvard Medical School, Boston, and colleagues wrote in a study published Jan. 14 in JAMA.
Researchers noted that FDA approval times declined from more than 3 years in 1983 to less than 1 year in 2017, although total time from the authorization of clinical testing to approval has remained steady across that period at about 8 years.
“The resistance of total development times to efforts to shorten them could be the result of more submissions of applications for rare disease drugs, which can sometimes pose trial recruitment challenges; a shifting emphasis to more challenging therapeutic categories, such as central nervous system disorder treatments; longer time horizons needed to establish efficacy when early intervention is important (e.g., cancer); or other factors,” wrote Mr. Darrow and colleagues.
The authors also note that the value of the expanded review toolbox the FDA now employs is “unclear” as “the number of new drug approvals has not increased substantially over the past three decades.” This observation does not count biologics or generic approvals.
Research revealed that the mean annual number of new drug approvals (including biologics) was 34 from 1990 to 1999, 25 from 2000 to 2009, and 41 from 2010 to 2018. In addition, while the number of new drug approvals has remained at or below 60 per year, the portion of drugs using at least one expedited review program increased over time, with more than 80% of the 59 new drugs approved in 2018 being subject to at least one expedited review program.
“Although the FDA has on average applied its expedited programs to drugs offering larger health gains, it is difficult to assess whether these programs have incentivized greater therapeutic innovation or merely allowed more appropriate agency prioritization,” noted Mr. Darrow and colleagues.
And while it may be hard to truly understand the impact of the many expedited review programs that have been introduced at the FDA, there is still room for improvement in the process.
In an editorial published in JAMA, Joshua Sharfstein, MD, of Johns Hopkins University, Baltimore, suggested four reforms the FDA could examine.
“First, Congress and the FDA should rationalize the various programs for expedited review,” he wrote, noting that companies are using the Orphan Drug Act to prevent competition. “Fixing the system requires, at a minimum, promoting meaningful competition for non-orphan uses.”
Second, he wrote, the FDA needs to strengthen oversight of postmarket safety though more diligence in the operation of its Risk Evaluation and Management Strategies (REMS) program.
“Third, Congress should recalibrate the programs that provide companies with special marketing protections,” added Dr. Sharfstein, former FDA principal deputy commissioner. “One place to look is pediatric exclusivity, which has been estimated to cost the health care system more than $6 for every $1 spent by a company on a pediatric trial.”
And finally, “Congress should use patent and pricing incentives to accelerate the generation of definitive evidence under accelerated approval,” citing proposals to limit pricing or market exclusivity until companies complete studies that assess clinically relevant endpoints.
“These changes would reflect an evolution, not a revolution, of the FDA’s approach to new drug approval,” Dr. Sharfstein said. “These reforms could also bring greater order and thoughtfulness to the regulation of important new therapies, while enhancing safety and creating greater capability to afford truly transformative medical products.”
SOURCE: Jonathan Darrow et al. JAMA 2020 Jan 14. doi: 10.1001/jama.2019.20288.
FROM JAMA