User login
Adding a blood test to standard screening may improve early cancer detection
A minimally invasive multicancer blood test used with standard-of-care screening is safe, effective, and feasible for use in routine clinical care, according to interim findings from a large, prospective study.
The DETECT-A blood test, an early version of the CancerSEEK test currently in development, effectively guided patient management in real time, in some cases leading to diagnosis of early cancer and potentially curative surgery in asymptomatic women with no history of cancer.
Nickolas Papadopoulos, PhD, of Johns Hopkins Medicine in Baltimore, reported these findings at the AACR virtual meeting I. The findings were simultaneously published in Science.
The study enrolled 10,006 women, aged 65-75 years, with no prior cancer diagnosis. After exclusion and loss to follow-up, 9,911 women remained.
There were 26 patients who had cancer detected by the DETECT-A blood test, 15 of whom underwent follow-up PET-CT imaging and 9 of whom underwent surgical excision. An additional 24 cancers were detected by standard screening, and 46 were detected by other means.
The positive predictive value of the blood test was 19%. When the blood test was combined with imaging, the positive predictive value was 41%.
Improving upon standard screening
“Standard-of-care screening [was used] for three different organs: breast, lung, and colon. It was more sensitive for breast cancer,” Dr. Papadopoulos noted. “Blood testing, though, identified cancer in 10 different organs.”
In fact, the DETECT-A blood test detected 14 of 45 cancers in 7 organs for which no standard screening test is available.
In addition, 12 cancers in 3 organs (breast, lung, and colon) were first detected by DETECT-A rather than by standard screening. This increased the sensitivity of cancer detection from 47% with standard screening alone to 71% with standard screening plus blood testing.
“More important, 65% [of the cancers detected by blood test] were localized or regional, which have higher chance of successful treatment with intent to cure,” Dr. Papadopoulos said.
DETECT-A covers regions of 16 commonly mutated genes and 9 proteins known to be associated with cancer. In this study, 57% of cancers were detected by mutations.
Safety and additional screening
DETECT-A also proved safe, “without incurring a large number of futile invasive follow-up tests,” Dr. Papadopoulos said.
In fact, only 1% of patients without cancer underwent PET-CT imaging, and only 0.22% underwent a “futile” invasive follow-up procedure.
Three surgeries occurred in patients who were counted as false-positives, but the surgeries were determined to be indicated, Dr. Papadopoulos said. He explained that one was for large colonic polyps with high-grade dysplasia that could not be removed endoscopically, one was for an in situ carcinoma of the appendix, and one was for a 10-cm ovarian lesion that was found to be a mucinous cystadenoma.
The investigators also analyzed whether the availability of a “liquid biopsy” test like DETECT-A would inadvertently reduce patients’ use of standard screening and found that it did not. Mammography screening habits after receiving the baseline DETECT-A blood test did not differ significantly from those prior to study enrollment.
These findings are important because early detection is a key factor in reducing cancer-specific morbidity and mortality, and although minimally invasive screening tests, including liquid biopsies like DETECT-A, hold great promise, prospective clinical studies of these new methods are needed to ensure that the anticipated benefits outweigh the potential risks, Dr. Papadopoulos explained.
“The problem is that most cancers are detected at advanced stages when they are difficult to treat,” he said. “The earlier cancer is detected, the greater the chance of successful treatment.”
Unanswered questions and future studies
This study demonstrates that it is feasible for a minimally invasive blood test to safely detect multiple cancer types in patients without a history of cancer and to enable treatment with curative intent, at least in a subset of individuals, Dr. Papadopoulos said. He added that the findings also inform the design of future randomized trials “to establish clinical utility, cost-effectiveness, and benefit-to-risk ratio of future tests.”
Further studies will also be required to determine the clinical validity and utility of the strategy of using liquid biopsy as a complement to standard-of-care screening, Dr. Papadopoulos said.
Invited discussant David G. Huntsman, MD, of the University of British Columbia in Vancouver, applauded the investigators, saying this study serves to “move the field forward.” However, it still isn’t clear how sensitivity and negative predictive value will be determined and what the optimal testing schedule is.
“This is a prospective study that will provide the data on how this assay will be used [and] whether it should be used going forward,” Dr. Huntsman said, noting that the “much bigger and more important question” is whether it improves survival.
Cost-effectiveness will also be critical, he said.
This research was supported by The Marcus Foundation, Lustgarten Foundation for Pancreatic Cancer Research, The Virginia and D.K. Ludwig Fund for Cancer Research, The Sol Goldman Center for Pancreatic Cancer Research, Susan Wojcicki and Dennis Troper, the Rolfe Foundation, The Conrad R. Hilton Foundation, The John Templeton Foundation, Burroughs Wellcome Career Award For Medical Scientists, and grants/contracts from the National Institutes of Health.
Dr. Papadopoulos disclosed relationships with Thrive Earlier Detection Inc., PGDx Inc., NeoPhore, Cage Pharma, and other companies. Dr. Huntsman is a founder, shareholder, and chief medical officer for Contextual Genomics.
SOURCE: Papadopoulos N et al. AACR 2020, Abstract CT022; Lennon AM et al. Science. 2020 Apr 28. pii: eabb9601. doi: 10.1126/science.abb9601.
A minimally invasive multicancer blood test used with standard-of-care screening is safe, effective, and feasible for use in routine clinical care, according to interim findings from a large, prospective study.
The DETECT-A blood test, an early version of the CancerSEEK test currently in development, effectively guided patient management in real time, in some cases leading to diagnosis of early cancer and potentially curative surgery in asymptomatic women with no history of cancer.
Nickolas Papadopoulos, PhD, of Johns Hopkins Medicine in Baltimore, reported these findings at the AACR virtual meeting I. The findings were simultaneously published in Science.
The study enrolled 10,006 women, aged 65-75 years, with no prior cancer diagnosis. After exclusion and loss to follow-up, 9,911 women remained.
There were 26 patients who had cancer detected by the DETECT-A blood test, 15 of whom underwent follow-up PET-CT imaging and 9 of whom underwent surgical excision. An additional 24 cancers were detected by standard screening, and 46 were detected by other means.
The positive predictive value of the blood test was 19%. When the blood test was combined with imaging, the positive predictive value was 41%.
Improving upon standard screening
“Standard-of-care screening [was used] for three different organs: breast, lung, and colon. It was more sensitive for breast cancer,” Dr. Papadopoulos noted. “Blood testing, though, identified cancer in 10 different organs.”
In fact, the DETECT-A blood test detected 14 of 45 cancers in 7 organs for which no standard screening test is available.
In addition, 12 cancers in 3 organs (breast, lung, and colon) were first detected by DETECT-A rather than by standard screening. This increased the sensitivity of cancer detection from 47% with standard screening alone to 71% with standard screening plus blood testing.
“More important, 65% [of the cancers detected by blood test] were localized or regional, which have higher chance of successful treatment with intent to cure,” Dr. Papadopoulos said.
DETECT-A covers regions of 16 commonly mutated genes and 9 proteins known to be associated with cancer. In this study, 57% of cancers were detected by mutations.
Safety and additional screening
DETECT-A also proved safe, “without incurring a large number of futile invasive follow-up tests,” Dr. Papadopoulos said.
In fact, only 1% of patients without cancer underwent PET-CT imaging, and only 0.22% underwent a “futile” invasive follow-up procedure.
Three surgeries occurred in patients who were counted as false-positives, but the surgeries were determined to be indicated, Dr. Papadopoulos said. He explained that one was for large colonic polyps with high-grade dysplasia that could not be removed endoscopically, one was for an in situ carcinoma of the appendix, and one was for a 10-cm ovarian lesion that was found to be a mucinous cystadenoma.
The investigators also analyzed whether the availability of a “liquid biopsy” test like DETECT-A would inadvertently reduce patients’ use of standard screening and found that it did not. Mammography screening habits after receiving the baseline DETECT-A blood test did not differ significantly from those prior to study enrollment.
These findings are important because early detection is a key factor in reducing cancer-specific morbidity and mortality, and although minimally invasive screening tests, including liquid biopsies like DETECT-A, hold great promise, prospective clinical studies of these new methods are needed to ensure that the anticipated benefits outweigh the potential risks, Dr. Papadopoulos explained.
“The problem is that most cancers are detected at advanced stages when they are difficult to treat,” he said. “The earlier cancer is detected, the greater the chance of successful treatment.”
Unanswered questions and future studies
This study demonstrates that it is feasible for a minimally invasive blood test to safely detect multiple cancer types in patients without a history of cancer and to enable treatment with curative intent, at least in a subset of individuals, Dr. Papadopoulos said. He added that the findings also inform the design of future randomized trials “to establish clinical utility, cost-effectiveness, and benefit-to-risk ratio of future tests.”
Further studies will also be required to determine the clinical validity and utility of the strategy of using liquid biopsy as a complement to standard-of-care screening, Dr. Papadopoulos said.
Invited discussant David G. Huntsman, MD, of the University of British Columbia in Vancouver, applauded the investigators, saying this study serves to “move the field forward.” However, it still isn’t clear how sensitivity and negative predictive value will be determined and what the optimal testing schedule is.
“This is a prospective study that will provide the data on how this assay will be used [and] whether it should be used going forward,” Dr. Huntsman said, noting that the “much bigger and more important question” is whether it improves survival.
Cost-effectiveness will also be critical, he said.
This research was supported by The Marcus Foundation, Lustgarten Foundation for Pancreatic Cancer Research, The Virginia and D.K. Ludwig Fund for Cancer Research, The Sol Goldman Center for Pancreatic Cancer Research, Susan Wojcicki and Dennis Troper, the Rolfe Foundation, The Conrad R. Hilton Foundation, The John Templeton Foundation, Burroughs Wellcome Career Award For Medical Scientists, and grants/contracts from the National Institutes of Health.
Dr. Papadopoulos disclosed relationships with Thrive Earlier Detection Inc., PGDx Inc., NeoPhore, Cage Pharma, and other companies. Dr. Huntsman is a founder, shareholder, and chief medical officer for Contextual Genomics.
SOURCE: Papadopoulos N et al. AACR 2020, Abstract CT022; Lennon AM et al. Science. 2020 Apr 28. pii: eabb9601. doi: 10.1126/science.abb9601.
A minimally invasive multicancer blood test used with standard-of-care screening is safe, effective, and feasible for use in routine clinical care, according to interim findings from a large, prospective study.
The DETECT-A blood test, an early version of the CancerSEEK test currently in development, effectively guided patient management in real time, in some cases leading to diagnosis of early cancer and potentially curative surgery in asymptomatic women with no history of cancer.
Nickolas Papadopoulos, PhD, of Johns Hopkins Medicine in Baltimore, reported these findings at the AACR virtual meeting I. The findings were simultaneously published in Science.
The study enrolled 10,006 women, aged 65-75 years, with no prior cancer diagnosis. After exclusion and loss to follow-up, 9,911 women remained.
There were 26 patients who had cancer detected by the DETECT-A blood test, 15 of whom underwent follow-up PET-CT imaging and 9 of whom underwent surgical excision. An additional 24 cancers were detected by standard screening, and 46 were detected by other means.
The positive predictive value of the blood test was 19%. When the blood test was combined with imaging, the positive predictive value was 41%.
Improving upon standard screening
“Standard-of-care screening [was used] for three different organs: breast, lung, and colon. It was more sensitive for breast cancer,” Dr. Papadopoulos noted. “Blood testing, though, identified cancer in 10 different organs.”
In fact, the DETECT-A blood test detected 14 of 45 cancers in 7 organs for which no standard screening test is available.
In addition, 12 cancers in 3 organs (breast, lung, and colon) were first detected by DETECT-A rather than by standard screening. This increased the sensitivity of cancer detection from 47% with standard screening alone to 71% with standard screening plus blood testing.
“More important, 65% [of the cancers detected by blood test] were localized or regional, which have higher chance of successful treatment with intent to cure,” Dr. Papadopoulos said.
DETECT-A covers regions of 16 commonly mutated genes and 9 proteins known to be associated with cancer. In this study, 57% of cancers were detected by mutations.
Safety and additional screening
DETECT-A also proved safe, “without incurring a large number of futile invasive follow-up tests,” Dr. Papadopoulos said.
In fact, only 1% of patients without cancer underwent PET-CT imaging, and only 0.22% underwent a “futile” invasive follow-up procedure.
Three surgeries occurred in patients who were counted as false-positives, but the surgeries were determined to be indicated, Dr. Papadopoulos said. He explained that one was for large colonic polyps with high-grade dysplasia that could not be removed endoscopically, one was for an in situ carcinoma of the appendix, and one was for a 10-cm ovarian lesion that was found to be a mucinous cystadenoma.
The investigators also analyzed whether the availability of a “liquid biopsy” test like DETECT-A would inadvertently reduce patients’ use of standard screening and found that it did not. Mammography screening habits after receiving the baseline DETECT-A blood test did not differ significantly from those prior to study enrollment.
These findings are important because early detection is a key factor in reducing cancer-specific morbidity and mortality, and although minimally invasive screening tests, including liquid biopsies like DETECT-A, hold great promise, prospective clinical studies of these new methods are needed to ensure that the anticipated benefits outweigh the potential risks, Dr. Papadopoulos explained.
“The problem is that most cancers are detected at advanced stages when they are difficult to treat,” he said. “The earlier cancer is detected, the greater the chance of successful treatment.”
Unanswered questions and future studies
This study demonstrates that it is feasible for a minimally invasive blood test to safely detect multiple cancer types in patients without a history of cancer and to enable treatment with curative intent, at least in a subset of individuals, Dr. Papadopoulos said. He added that the findings also inform the design of future randomized trials “to establish clinical utility, cost-effectiveness, and benefit-to-risk ratio of future tests.”
Further studies will also be required to determine the clinical validity and utility of the strategy of using liquid biopsy as a complement to standard-of-care screening, Dr. Papadopoulos said.
Invited discussant David G. Huntsman, MD, of the University of British Columbia in Vancouver, applauded the investigators, saying this study serves to “move the field forward.” However, it still isn’t clear how sensitivity and negative predictive value will be determined and what the optimal testing schedule is.
“This is a prospective study that will provide the data on how this assay will be used [and] whether it should be used going forward,” Dr. Huntsman said, noting that the “much bigger and more important question” is whether it improves survival.
Cost-effectiveness will also be critical, he said.
This research was supported by The Marcus Foundation, Lustgarten Foundation for Pancreatic Cancer Research, The Virginia and D.K. Ludwig Fund for Cancer Research, The Sol Goldman Center for Pancreatic Cancer Research, Susan Wojcicki and Dennis Troper, the Rolfe Foundation, The Conrad R. Hilton Foundation, The John Templeton Foundation, Burroughs Wellcome Career Award For Medical Scientists, and grants/contracts from the National Institutes of Health.
Dr. Papadopoulos disclosed relationships with Thrive Earlier Detection Inc., PGDx Inc., NeoPhore, Cage Pharma, and other companies. Dr. Huntsman is a founder, shareholder, and chief medical officer for Contextual Genomics.
SOURCE: Papadopoulos N et al. AACR 2020, Abstract CT022; Lennon AM et al. Science. 2020 Apr 28. pii: eabb9601. doi: 10.1126/science.abb9601.
FROM AACR 2020
Progress report: Elimination of neonatal tetanus
Worldwide cases of neonatal tetanus fell by 90% from 2000 to 2018, deaths dropped by 85%, and 45 countries achieved elimination of maternal and neonatal tetanus (MNT), according to the Centers for Disease Control and Prevention.
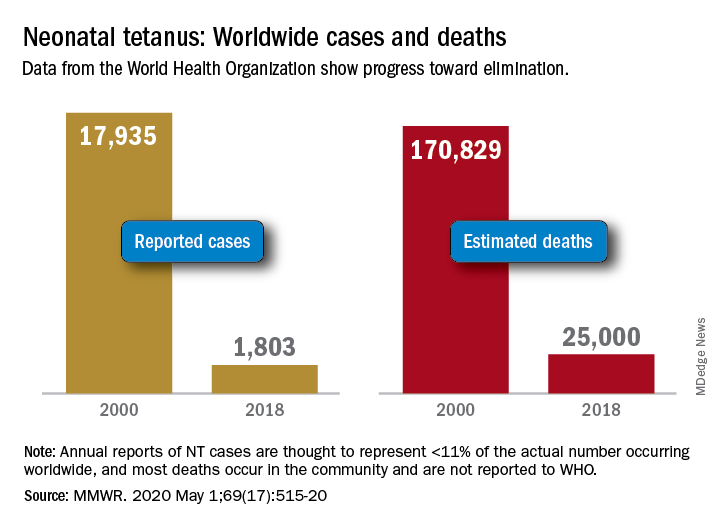
“Despite this progress, some countries that achieved elimination are still struggling to sustain performance indicators; war and insecurity pose challenges in countries that have not achieved MNT elimination,” Henry N. Njuguna, MD, of the CDC’s global immunization division, and associates wrote in the Morbidity and Mortality Weekly Report.
Other worldwide measures also improved from 2000 to 2018: and the percentage of deliveries attended by a skilled birth attendant increased from 62% during 2000-2005 to 81% in 2013-2018, they reported.
The MNT elimination initiative, which began in 1999 and targeted 59 priority countries, immunized approximately 154 million women of reproductive age with at least two doses of tetanus toxoid–containing vaccine from 2000 to 2018, the investigators wrote, based on data from the World Health Organization and the United Nations Children’s Fund.
With 14 of the priority countries – including Nigeria, Pakistan, and Yemen – still dealing with MNT, however, numerous challenges remain, they noted. About 47 million women and their babies are still unprotected, and 49 million women have not received tetanus toxoid–containing vaccine.
This lack of coverage “can be attributed to weak health systems, including conflict and security issues that limit access to vaccination services, competing priorities that limit the implementation of planned MNT elimination activities, and withdrawal of donor funding,” Dr. Njuguna and associates wrote.
SOURCE: Njuguna HN et al. MMWR. 2020 May 1;69(17):515-20.
Worldwide cases of neonatal tetanus fell by 90% from 2000 to 2018, deaths dropped by 85%, and 45 countries achieved elimination of maternal and neonatal tetanus (MNT), according to the Centers for Disease Control and Prevention.
“Despite this progress, some countries that achieved elimination are still struggling to sustain performance indicators; war and insecurity pose challenges in countries that have not achieved MNT elimination,” Henry N. Njuguna, MD, of the CDC’s global immunization division, and associates wrote in the Morbidity and Mortality Weekly Report.
Other worldwide measures also improved from 2000 to 2018: and the percentage of deliveries attended by a skilled birth attendant increased from 62% during 2000-2005 to 81% in 2013-2018, they reported.
The MNT elimination initiative, which began in 1999 and targeted 59 priority countries, immunized approximately 154 million women of reproductive age with at least two doses of tetanus toxoid–containing vaccine from 2000 to 2018, the investigators wrote, based on data from the World Health Organization and the United Nations Children’s Fund.
With 14 of the priority countries – including Nigeria, Pakistan, and Yemen – still dealing with MNT, however, numerous challenges remain, they noted. About 47 million women and their babies are still unprotected, and 49 million women have not received tetanus toxoid–containing vaccine.
This lack of coverage “can be attributed to weak health systems, including conflict and security issues that limit access to vaccination services, competing priorities that limit the implementation of planned MNT elimination activities, and withdrawal of donor funding,” Dr. Njuguna and associates wrote.
SOURCE: Njuguna HN et al. MMWR. 2020 May 1;69(17):515-20.
Worldwide cases of neonatal tetanus fell by 90% from 2000 to 2018, deaths dropped by 85%, and 45 countries achieved elimination of maternal and neonatal tetanus (MNT), according to the Centers for Disease Control and Prevention.
“Despite this progress, some countries that achieved elimination are still struggling to sustain performance indicators; war and insecurity pose challenges in countries that have not achieved MNT elimination,” Henry N. Njuguna, MD, of the CDC’s global immunization division, and associates wrote in the Morbidity and Mortality Weekly Report.
Other worldwide measures also improved from 2000 to 2018: and the percentage of deliveries attended by a skilled birth attendant increased from 62% during 2000-2005 to 81% in 2013-2018, they reported.
The MNT elimination initiative, which began in 1999 and targeted 59 priority countries, immunized approximately 154 million women of reproductive age with at least two doses of tetanus toxoid–containing vaccine from 2000 to 2018, the investigators wrote, based on data from the World Health Organization and the United Nations Children’s Fund.
With 14 of the priority countries – including Nigeria, Pakistan, and Yemen – still dealing with MNT, however, numerous challenges remain, they noted. About 47 million women and their babies are still unprotected, and 49 million women have not received tetanus toxoid–containing vaccine.
This lack of coverage “can be attributed to weak health systems, including conflict and security issues that limit access to vaccination services, competing priorities that limit the implementation of planned MNT elimination activities, and withdrawal of donor funding,” Dr. Njuguna and associates wrote.
SOURCE: Njuguna HN et al. MMWR. 2020 May 1;69(17):515-20.
FROM MMWR
Ob.gyns., peds, other PCPs seeking COVID-19 financial relief from feds
A handful of specialties – including family medicine, obstetrics/gynecology, pediatrics, and other primary care specialties – are calling for targeted and urgent relief payments from the federal government, saying that they have been left out of distributions aimed at alleviating the financial fallout associated with the novel coronavirus.
The federal government has already distributed about $150 billion – through direct payments and advances on reimbursement – to clinicians, but, to date, the money has only been given to providers who bill Medicare, and not even all of those individuals have received payments.
“It is critical that frontline physicians who may not participate in Medicare fee-for-service, in whole or in part, including obstetrician/gynecologists, pediatricians, and family physicians, have the resources they need to continue providing essential health care to patients amid the pandemic and in the months to come,” said the American Academy of Family Physicians, the American Academy of Pediatrics, and the American College of Obstetricians and Gynecologists in a letter to Health & Human Services (Secretary Alex Azar.
In particular, the organizations are concerned that no money has been distributed or earmarked for clinicians who serve Medicaid recipients.
“The organizations that signed that letter are the primary providers of care to the Medicaid population,” Shawn Martin, senior VP for the AAFP, said in an interview. That’s true even for family physicians.
“Typically, in an average family medicine practice, their Medicaid panel size is equal to if not greater than the Medicare panel size,” he said.
On April 23, Mr. Azar said HHS was working on a distribution plan for providers who only take Medicaid, as well as for dentists and skilled nursing facilities. An HHS spokesperson confirmed that the agency still intends to provide money to those groups of providers and that the agency is committed to distributing funds quickly and with transparency.
Mr. Azar had also announced that the government would soon start distributing $20 billion in payments to Medicare providers, on top of the $30 billion that had already been handed out to clinicians on April 10 and 17.
That $50 billion came from the COVID-19–related $100 billion Provider Relief Fund, which was part of the Coronavirus Aid, Relief, and Economic Security Act, signed into law on March 27.
Additionally, the Centers for Medicare & Medicaid Services had distributed some $100 billion to providers who participated in Medicare Part A or B through the Medicare Advance Payment program, which is a deferred loan. The agency brought that program to a halt on April 27.
An additional $75 billion will now be available through the Public Health and Social Services Emergency Fund (PHSSEF) as part of the third congressional COVID relief package, signed into law on April 24.
Mr. Martin said that the AAFP and other physician organizations have been talking with HHS about how to distribute money from that new pool of funds. “There’s been a lot of progress, but there hasn’t been any action,” he said, adding that the purpose of the joint letter to HHS “is to say it’s time for action.”
COVID-19 damage
AAFP, AAP, and ACOG noted in the letter the damage that’s being inflicted by COVID-19. They cited data that show a 50% decline in measles, mumps, and rubella shots, a 42% drop in diphtheria and whooping cough vaccinations, and a 73% decline in human papillomavirus shots. The groups also noted a rise in child abuse injuries that are being seen in EDs and the potential for a worsening of the maternal mortality crisis in the United States.
Primary care physicians are also the go-to doctors for upper respiratory infections, noted the groups in the letter.
“Put simply, our physician members need to be able to keep their doors open and continue treating patients,” said the groups.
A study by Harvard University and Phreesia, a health care technology company, found that ambulatory practice visits had declined by at least half since early February, with a 71% drop in visits by 7- to 17-year-olds and a 59% decline in visits by neonates, infants, and toddlers (up to age 6). Overall, pediatric practices experienced a 62% drop-off in visits.
Research conducted by the Physicians Foundation and Merritt Hawkins shows that 21% of 842 physicians who responded to an early April survey said they’d been furloughed or been given a pay cut. That number rose to 30% among doctors who are not treating COVID-19 patients.
Although the majority in the survey (66%) said they planned to keep practicing in the same manner during the pandemic, 32% said they planned to change practices, opt out of patient care roles, close their practices temporarily, or retire. The survey has a margin of error of ±3.5%.
Internists seek consideration, too
The American College of Physicians also has urged HHS to give special consideration to its members. The group wrote to Mr. Azar on April 28, recommending that payments from the new $75 billion PHSSEF be prioritized for primary care, as well as for smaller practices, those that provide care in underserved areas, and internal medicine subspecialty practices.
“Internal medicine specialists and other primary care physicians have an essential role in delivering primary, preventive, and comprehensive care not only to patients with symptoms or diagnoses of COVID-19, but also to patients with other underlying medical conditions, including conditions like heart disease and diabetes that put them at greater risk of mortality from COVID-19,” wrote ACP President Jacqueline Fincher, MD, MACP.
ACP said the government could pay physicians on the basis of the amount of additional expenses incurred that were related to COVID-19, such as extra staffing or temporary relocation of their place of residence to prevent exposing family members to the virus. Pay should also be based on the percentage of revenue losses from all payers, including Medicare, Medicaid, and commercial insurers, Dr. Fincher said in the letter.
AAFP, AAP, and ACOG also had a suggestion for distributing payments to non-Medicare providers. “Given that most women’s health, pediatric, and family practices have received less financial relief to date, we recommend that HHS provide these practices with a larger proportion of funds relative to their reported revenue than is provided on average across specialties,” they wrote.
A version of this article originally appeared on Medscape.com.
A handful of specialties – including family medicine, obstetrics/gynecology, pediatrics, and other primary care specialties – are calling for targeted and urgent relief payments from the federal government, saying that they have been left out of distributions aimed at alleviating the financial fallout associated with the novel coronavirus.
The federal government has already distributed about $150 billion – through direct payments and advances on reimbursement – to clinicians, but, to date, the money has only been given to providers who bill Medicare, and not even all of those individuals have received payments.
“It is critical that frontline physicians who may not participate in Medicare fee-for-service, in whole or in part, including obstetrician/gynecologists, pediatricians, and family physicians, have the resources they need to continue providing essential health care to patients amid the pandemic and in the months to come,” said the American Academy of Family Physicians, the American Academy of Pediatrics, and the American College of Obstetricians and Gynecologists in a letter to Health & Human Services (Secretary Alex Azar.
In particular, the organizations are concerned that no money has been distributed or earmarked for clinicians who serve Medicaid recipients.
“The organizations that signed that letter are the primary providers of care to the Medicaid population,” Shawn Martin, senior VP for the AAFP, said in an interview. That’s true even for family physicians.
“Typically, in an average family medicine practice, their Medicaid panel size is equal to if not greater than the Medicare panel size,” he said.
On April 23, Mr. Azar said HHS was working on a distribution plan for providers who only take Medicaid, as well as for dentists and skilled nursing facilities. An HHS spokesperson confirmed that the agency still intends to provide money to those groups of providers and that the agency is committed to distributing funds quickly and with transparency.
Mr. Azar had also announced that the government would soon start distributing $20 billion in payments to Medicare providers, on top of the $30 billion that had already been handed out to clinicians on April 10 and 17.
That $50 billion came from the COVID-19–related $100 billion Provider Relief Fund, which was part of the Coronavirus Aid, Relief, and Economic Security Act, signed into law on March 27.
Additionally, the Centers for Medicare & Medicaid Services had distributed some $100 billion to providers who participated in Medicare Part A or B through the Medicare Advance Payment program, which is a deferred loan. The agency brought that program to a halt on April 27.
An additional $75 billion will now be available through the Public Health and Social Services Emergency Fund (PHSSEF) as part of the third congressional COVID relief package, signed into law on April 24.
Mr. Martin said that the AAFP and other physician organizations have been talking with HHS about how to distribute money from that new pool of funds. “There’s been a lot of progress, but there hasn’t been any action,” he said, adding that the purpose of the joint letter to HHS “is to say it’s time for action.”
COVID-19 damage
AAFP, AAP, and ACOG noted in the letter the damage that’s being inflicted by COVID-19. They cited data that show a 50% decline in measles, mumps, and rubella shots, a 42% drop in diphtheria and whooping cough vaccinations, and a 73% decline in human papillomavirus shots. The groups also noted a rise in child abuse injuries that are being seen in EDs and the potential for a worsening of the maternal mortality crisis in the United States.
Primary care physicians are also the go-to doctors for upper respiratory infections, noted the groups in the letter.
“Put simply, our physician members need to be able to keep their doors open and continue treating patients,” said the groups.
A study by Harvard University and Phreesia, a health care technology company, found that ambulatory practice visits had declined by at least half since early February, with a 71% drop in visits by 7- to 17-year-olds and a 59% decline in visits by neonates, infants, and toddlers (up to age 6). Overall, pediatric practices experienced a 62% drop-off in visits.
Research conducted by the Physicians Foundation and Merritt Hawkins shows that 21% of 842 physicians who responded to an early April survey said they’d been furloughed or been given a pay cut. That number rose to 30% among doctors who are not treating COVID-19 patients.
Although the majority in the survey (66%) said they planned to keep practicing in the same manner during the pandemic, 32% said they planned to change practices, opt out of patient care roles, close their practices temporarily, or retire. The survey has a margin of error of ±3.5%.
Internists seek consideration, too
The American College of Physicians also has urged HHS to give special consideration to its members. The group wrote to Mr. Azar on April 28, recommending that payments from the new $75 billion PHSSEF be prioritized for primary care, as well as for smaller practices, those that provide care in underserved areas, and internal medicine subspecialty practices.
“Internal medicine specialists and other primary care physicians have an essential role in delivering primary, preventive, and comprehensive care not only to patients with symptoms or diagnoses of COVID-19, but also to patients with other underlying medical conditions, including conditions like heart disease and diabetes that put them at greater risk of mortality from COVID-19,” wrote ACP President Jacqueline Fincher, MD, MACP.
ACP said the government could pay physicians on the basis of the amount of additional expenses incurred that were related to COVID-19, such as extra staffing or temporary relocation of their place of residence to prevent exposing family members to the virus. Pay should also be based on the percentage of revenue losses from all payers, including Medicare, Medicaid, and commercial insurers, Dr. Fincher said in the letter.
AAFP, AAP, and ACOG also had a suggestion for distributing payments to non-Medicare providers. “Given that most women’s health, pediatric, and family practices have received less financial relief to date, we recommend that HHS provide these practices with a larger proportion of funds relative to their reported revenue than is provided on average across specialties,” they wrote.
A version of this article originally appeared on Medscape.com.
A handful of specialties – including family medicine, obstetrics/gynecology, pediatrics, and other primary care specialties – are calling for targeted and urgent relief payments from the federal government, saying that they have been left out of distributions aimed at alleviating the financial fallout associated with the novel coronavirus.
The federal government has already distributed about $150 billion – through direct payments and advances on reimbursement – to clinicians, but, to date, the money has only been given to providers who bill Medicare, and not even all of those individuals have received payments.
“It is critical that frontline physicians who may not participate in Medicare fee-for-service, in whole or in part, including obstetrician/gynecologists, pediatricians, and family physicians, have the resources they need to continue providing essential health care to patients amid the pandemic and in the months to come,” said the American Academy of Family Physicians, the American Academy of Pediatrics, and the American College of Obstetricians and Gynecologists in a letter to Health & Human Services (Secretary Alex Azar.
In particular, the organizations are concerned that no money has been distributed or earmarked for clinicians who serve Medicaid recipients.
“The organizations that signed that letter are the primary providers of care to the Medicaid population,” Shawn Martin, senior VP for the AAFP, said in an interview. That’s true even for family physicians.
“Typically, in an average family medicine practice, their Medicaid panel size is equal to if not greater than the Medicare panel size,” he said.
On April 23, Mr. Azar said HHS was working on a distribution plan for providers who only take Medicaid, as well as for dentists and skilled nursing facilities. An HHS spokesperson confirmed that the agency still intends to provide money to those groups of providers and that the agency is committed to distributing funds quickly and with transparency.
Mr. Azar had also announced that the government would soon start distributing $20 billion in payments to Medicare providers, on top of the $30 billion that had already been handed out to clinicians on April 10 and 17.
That $50 billion came from the COVID-19–related $100 billion Provider Relief Fund, which was part of the Coronavirus Aid, Relief, and Economic Security Act, signed into law on March 27.
Additionally, the Centers for Medicare & Medicaid Services had distributed some $100 billion to providers who participated in Medicare Part A or B through the Medicare Advance Payment program, which is a deferred loan. The agency brought that program to a halt on April 27.
An additional $75 billion will now be available through the Public Health and Social Services Emergency Fund (PHSSEF) as part of the third congressional COVID relief package, signed into law on April 24.
Mr. Martin said that the AAFP and other physician organizations have been talking with HHS about how to distribute money from that new pool of funds. “There’s been a lot of progress, but there hasn’t been any action,” he said, adding that the purpose of the joint letter to HHS “is to say it’s time for action.”
COVID-19 damage
AAFP, AAP, and ACOG noted in the letter the damage that’s being inflicted by COVID-19. They cited data that show a 50% decline in measles, mumps, and rubella shots, a 42% drop in diphtheria and whooping cough vaccinations, and a 73% decline in human papillomavirus shots. The groups also noted a rise in child abuse injuries that are being seen in EDs and the potential for a worsening of the maternal mortality crisis in the United States.
Primary care physicians are also the go-to doctors for upper respiratory infections, noted the groups in the letter.
“Put simply, our physician members need to be able to keep their doors open and continue treating patients,” said the groups.
A study by Harvard University and Phreesia, a health care technology company, found that ambulatory practice visits had declined by at least half since early February, with a 71% drop in visits by 7- to 17-year-olds and a 59% decline in visits by neonates, infants, and toddlers (up to age 6). Overall, pediatric practices experienced a 62% drop-off in visits.
Research conducted by the Physicians Foundation and Merritt Hawkins shows that 21% of 842 physicians who responded to an early April survey said they’d been furloughed or been given a pay cut. That number rose to 30% among doctors who are not treating COVID-19 patients.
Although the majority in the survey (66%) said they planned to keep practicing in the same manner during the pandemic, 32% said they planned to change practices, opt out of patient care roles, close their practices temporarily, or retire. The survey has a margin of error of ±3.5%.
Internists seek consideration, too
The American College of Physicians also has urged HHS to give special consideration to its members. The group wrote to Mr. Azar on April 28, recommending that payments from the new $75 billion PHSSEF be prioritized for primary care, as well as for smaller practices, those that provide care in underserved areas, and internal medicine subspecialty practices.
“Internal medicine specialists and other primary care physicians have an essential role in delivering primary, preventive, and comprehensive care not only to patients with symptoms or diagnoses of COVID-19, but also to patients with other underlying medical conditions, including conditions like heart disease and diabetes that put them at greater risk of mortality from COVID-19,” wrote ACP President Jacqueline Fincher, MD, MACP.
ACP said the government could pay physicians on the basis of the amount of additional expenses incurred that were related to COVID-19, such as extra staffing or temporary relocation of their place of residence to prevent exposing family members to the virus. Pay should also be based on the percentage of revenue losses from all payers, including Medicare, Medicaid, and commercial insurers, Dr. Fincher said in the letter.
AAFP, AAP, and ACOG also had a suggestion for distributing payments to non-Medicare providers. “Given that most women’s health, pediatric, and family practices have received less financial relief to date, we recommend that HHS provide these practices with a larger proportion of funds relative to their reported revenue than is provided on average across specialties,” they wrote.
A version of this article originally appeared on Medscape.com.
Novel combo boosts response in HER2-negative breast cancer
A novel combination has boosted responses in women with high-risk, HER2-negative breast cancer.
The new combo comprises the immune checkpoint inhibitor durvalumab (Imfinzi, AstraZeneca) and the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib (Lynparza, AstraZeneca).
When this combo was added to standard neoadjuvant chemotherapy, it yielded a significantly higher pathologic complete response (pCR) rate at the time of surgery than was seen with chemotherapy alone.
The superior pCR rate was seen across all HER2-negative breast cancer subtypes, including HER2-negative, estrogen receptor–positive tumors (Mammaprint high risk), and in women with triple-negative breast cancer (TNBC), reported lead investigator Lajos Pusztai, MD, DPhil, from Yale Cancer Center in New Haven, Connecticut.
“These results provide further evidence for the clinical value of immunotherapy in early-stage breast cancer and suggest new avenues for how to exploit these drugs in hormone receptor [HR]–positive breast cancers,” said Pusztai.
He presented the results at the American Association for Cancer Research (AACR) virtual annual meeting, which took place online, owing to the COVID-19 pandemic.
Toxicities, including financial
“The benefits from immunotherapy are clearly emerging in early and metastatic triple-negative breast cancer, likely with PD-L1 expression. However, there’s much more uncertainty in patients with hormone-sensitive tumors whether there is benefit and who will benefit from immunotherapy,” commented Pamela N. Munster, MD, from the University of California, San Francisco, who was the invited discussant.
“The signal of a better pCR rate among patients in the ultra-high Mammaprint group may allow selection of patients with HR-positive disease who may benefit from immunotherapeutic agents and/or PARP inhibitors,” she said.
Munster noted that the approximately 10% higher rate of immune-related adverse events of grade 3 or greater that was seen with the combination appears similar to that seen in other studies, but anemia and fatigue appeared to be less frequent with durvalumab/olaparaib and paclitaxel in comparison with paclitaxel alone.
“In the absence of a clear delineation of the contribution of olaparib, some weight should be given to the financial burden of adding both durvalumab and olaparib to a preoperative regimen,” she said.
The additional cost of durvalumab is approximately $34,000, and adding olaparib boosts that by about $22,000 more, Munster said.
Ongoing platform trial
The new results come from one arm of the ongoing Investigation of Serial Studies to Predict Your Therapeutic Response Through Imaging and Molecular Analysis 2 (I-SPY-2) trial. This is an ongoing platform trial that is exploring the use of new drugs in combination with a standard neoadjuvant therapy backbone for the treatment of high-risk cancers.
In this trial, women with stage II or III breast cancer with tumors 2.5 cm or larger are assessed for one of eight biomarker subtypes according to HER2 status, HR status, and genetic risk factors, as determined on the basis of a 70-gene assay. The patients within each biomarker subtype are randomly assigned to receive standard therapy either with or without an investigational agent.
For each subtrial, a primary endpoint is an improvement in pCR in comparison with the standard of care.
Changes in tumor volume on MRI are used to predict whether patients will achieve a pCR. Those who are considered to have a high Bayesian predictive probability of success are eligible for moving on to phase 3 trials.
The I-SPY-2 trial was described in detail by principal investigator Laura J. Esserman, MD, MBA, from the Helen Diller Comprehensive Cancer Center at the University of California, San Francisco, in a 2017 interview from the annual meeting of the American Society of Clinical Oncology.
As previously reported, the first drugs to “graduate” from the trial were the HER2/HER4 inhibitor neratinib (Nerlynx, Puma Biotechnology) and the investigational PARP inhibitor veliparib (AbbVie).
Study details
The I-SPY-2 results that Pusztai reported at the AACR meeting were based on an analysis of 73 HER2-negative patients, including 21 patients with TNBC and 52 with HR-positive tumors with Mammaprint high-risk features. These patients underwent treatment with durvalumab 1500 mg every 4 weeks for three cycles, olaparib 100 mg twice daily for weeks 1 through 11, and paclitaxel 80 mg/m2 weekly for 12 weeks, followed by AC chemotherapy (doxorubicin and cyclophosphamide) for four cycles.
The 299 patients in the control arm received paclitaxel and chemotherapy only.
In all three biomarker subsets studied, durvalumab and olaparib increased pCR rates compared with controls, as shown in the table.
The probability that the combination was superior to control in each subgroup approached 100%, Pusztai noted.
Adverse events with the combination were consistent with known side effects of the drugs, he commented. Immune-related grade 3 adverse events occurred in 19% of patients in the combination therapy arm, compared with 1.6% in the control arm.
Higher pCR rates were seen in the subset of immune-rich tumors among all cancer subtypes and in both study arms.
“Exploratory analysis suggests several potential predictive markers of durvalumab/olaparib benefit over chemotherapy alone,” Pusztai reported.
These markers included Mammaprint MP2 (ultra high) versus MP1 in HR+/HER2– tumors, and low CD3/CD8 gene signature ratio, high macrophage/Tc-class 2 gene signature ratio, and high proliferation signature, all of which were associated with higher pCR rates in the experimental arm among patients with TNBC.
The trial was supported by the William K. Bowes Jr Foundation, Foundation for the NIH, Give Breast Cancer the Boot, UCSF, the Biomarkers Consortium, IQVIA, the Breast Cancer Research Foundation, Safeway, California Breast Cancer Research Program, Breast Cancer Research–Atwater Trust, and Stand Up to Cancer. Pusztai has received honoraria and consulting fees from AstraZeneca and other companies. Munster has received research and travel support from and has served on the scientific advisory boards of AstraZeneca and other companies.
This article first appeared on Medscape.com.
A novel combination has boosted responses in women with high-risk, HER2-negative breast cancer.
The new combo comprises the immune checkpoint inhibitor durvalumab (Imfinzi, AstraZeneca) and the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib (Lynparza, AstraZeneca).
When this combo was added to standard neoadjuvant chemotherapy, it yielded a significantly higher pathologic complete response (pCR) rate at the time of surgery than was seen with chemotherapy alone.
The superior pCR rate was seen across all HER2-negative breast cancer subtypes, including HER2-negative, estrogen receptor–positive tumors (Mammaprint high risk), and in women with triple-negative breast cancer (TNBC), reported lead investigator Lajos Pusztai, MD, DPhil, from Yale Cancer Center in New Haven, Connecticut.
“These results provide further evidence for the clinical value of immunotherapy in early-stage breast cancer and suggest new avenues for how to exploit these drugs in hormone receptor [HR]–positive breast cancers,” said Pusztai.
He presented the results at the American Association for Cancer Research (AACR) virtual annual meeting, which took place online, owing to the COVID-19 pandemic.
Toxicities, including financial
“The benefits from immunotherapy are clearly emerging in early and metastatic triple-negative breast cancer, likely with PD-L1 expression. However, there’s much more uncertainty in patients with hormone-sensitive tumors whether there is benefit and who will benefit from immunotherapy,” commented Pamela N. Munster, MD, from the University of California, San Francisco, who was the invited discussant.
“The signal of a better pCR rate among patients in the ultra-high Mammaprint group may allow selection of patients with HR-positive disease who may benefit from immunotherapeutic agents and/or PARP inhibitors,” she said.
Munster noted that the approximately 10% higher rate of immune-related adverse events of grade 3 or greater that was seen with the combination appears similar to that seen in other studies, but anemia and fatigue appeared to be less frequent with durvalumab/olaparaib and paclitaxel in comparison with paclitaxel alone.
“In the absence of a clear delineation of the contribution of olaparib, some weight should be given to the financial burden of adding both durvalumab and olaparib to a preoperative regimen,” she said.
The additional cost of durvalumab is approximately $34,000, and adding olaparib boosts that by about $22,000 more, Munster said.
Ongoing platform trial
The new results come from one arm of the ongoing Investigation of Serial Studies to Predict Your Therapeutic Response Through Imaging and Molecular Analysis 2 (I-SPY-2) trial. This is an ongoing platform trial that is exploring the use of new drugs in combination with a standard neoadjuvant therapy backbone for the treatment of high-risk cancers.
In this trial, women with stage II or III breast cancer with tumors 2.5 cm or larger are assessed for one of eight biomarker subtypes according to HER2 status, HR status, and genetic risk factors, as determined on the basis of a 70-gene assay. The patients within each biomarker subtype are randomly assigned to receive standard therapy either with or without an investigational agent.
For each subtrial, a primary endpoint is an improvement in pCR in comparison with the standard of care.
Changes in tumor volume on MRI are used to predict whether patients will achieve a pCR. Those who are considered to have a high Bayesian predictive probability of success are eligible for moving on to phase 3 trials.
The I-SPY-2 trial was described in detail by principal investigator Laura J. Esserman, MD, MBA, from the Helen Diller Comprehensive Cancer Center at the University of California, San Francisco, in a 2017 interview from the annual meeting of the American Society of Clinical Oncology.
As previously reported, the first drugs to “graduate” from the trial were the HER2/HER4 inhibitor neratinib (Nerlynx, Puma Biotechnology) and the investigational PARP inhibitor veliparib (AbbVie).
Study details
The I-SPY-2 results that Pusztai reported at the AACR meeting were based on an analysis of 73 HER2-negative patients, including 21 patients with TNBC and 52 with HR-positive tumors with Mammaprint high-risk features. These patients underwent treatment with durvalumab 1500 mg every 4 weeks for three cycles, olaparib 100 mg twice daily for weeks 1 through 11, and paclitaxel 80 mg/m2 weekly for 12 weeks, followed by AC chemotherapy (doxorubicin and cyclophosphamide) for four cycles.
The 299 patients in the control arm received paclitaxel and chemotherapy only.
In all three biomarker subsets studied, durvalumab and olaparib increased pCR rates compared with controls, as shown in the table.
The probability that the combination was superior to control in each subgroup approached 100%, Pusztai noted.
Adverse events with the combination were consistent with known side effects of the drugs, he commented. Immune-related grade 3 adverse events occurred in 19% of patients in the combination therapy arm, compared with 1.6% in the control arm.
Higher pCR rates were seen in the subset of immune-rich tumors among all cancer subtypes and in both study arms.
“Exploratory analysis suggests several potential predictive markers of durvalumab/olaparib benefit over chemotherapy alone,” Pusztai reported.
These markers included Mammaprint MP2 (ultra high) versus MP1 in HR+/HER2– tumors, and low CD3/CD8 gene signature ratio, high macrophage/Tc-class 2 gene signature ratio, and high proliferation signature, all of which were associated with higher pCR rates in the experimental arm among patients with TNBC.
The trial was supported by the William K. Bowes Jr Foundation, Foundation for the NIH, Give Breast Cancer the Boot, UCSF, the Biomarkers Consortium, IQVIA, the Breast Cancer Research Foundation, Safeway, California Breast Cancer Research Program, Breast Cancer Research–Atwater Trust, and Stand Up to Cancer. Pusztai has received honoraria and consulting fees from AstraZeneca and other companies. Munster has received research and travel support from and has served on the scientific advisory boards of AstraZeneca and other companies.
This article first appeared on Medscape.com.
A novel combination has boosted responses in women with high-risk, HER2-negative breast cancer.
The new combo comprises the immune checkpoint inhibitor durvalumab (Imfinzi, AstraZeneca) and the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib (Lynparza, AstraZeneca).
When this combo was added to standard neoadjuvant chemotherapy, it yielded a significantly higher pathologic complete response (pCR) rate at the time of surgery than was seen with chemotherapy alone.
The superior pCR rate was seen across all HER2-negative breast cancer subtypes, including HER2-negative, estrogen receptor–positive tumors (Mammaprint high risk), and in women with triple-negative breast cancer (TNBC), reported lead investigator Lajos Pusztai, MD, DPhil, from Yale Cancer Center in New Haven, Connecticut.
“These results provide further evidence for the clinical value of immunotherapy in early-stage breast cancer and suggest new avenues for how to exploit these drugs in hormone receptor [HR]–positive breast cancers,” said Pusztai.
He presented the results at the American Association for Cancer Research (AACR) virtual annual meeting, which took place online, owing to the COVID-19 pandemic.
Toxicities, including financial
“The benefits from immunotherapy are clearly emerging in early and metastatic triple-negative breast cancer, likely with PD-L1 expression. However, there’s much more uncertainty in patients with hormone-sensitive tumors whether there is benefit and who will benefit from immunotherapy,” commented Pamela N. Munster, MD, from the University of California, San Francisco, who was the invited discussant.
“The signal of a better pCR rate among patients in the ultra-high Mammaprint group may allow selection of patients with HR-positive disease who may benefit from immunotherapeutic agents and/or PARP inhibitors,” she said.
Munster noted that the approximately 10% higher rate of immune-related adverse events of grade 3 or greater that was seen with the combination appears similar to that seen in other studies, but anemia and fatigue appeared to be less frequent with durvalumab/olaparaib and paclitaxel in comparison with paclitaxel alone.
“In the absence of a clear delineation of the contribution of olaparib, some weight should be given to the financial burden of adding both durvalumab and olaparib to a preoperative regimen,” she said.
The additional cost of durvalumab is approximately $34,000, and adding olaparib boosts that by about $22,000 more, Munster said.
Ongoing platform trial
The new results come from one arm of the ongoing Investigation of Serial Studies to Predict Your Therapeutic Response Through Imaging and Molecular Analysis 2 (I-SPY-2) trial. This is an ongoing platform trial that is exploring the use of new drugs in combination with a standard neoadjuvant therapy backbone for the treatment of high-risk cancers.
In this trial, women with stage II or III breast cancer with tumors 2.5 cm or larger are assessed for one of eight biomarker subtypes according to HER2 status, HR status, and genetic risk factors, as determined on the basis of a 70-gene assay. The patients within each biomarker subtype are randomly assigned to receive standard therapy either with or without an investigational agent.
For each subtrial, a primary endpoint is an improvement in pCR in comparison with the standard of care.
Changes in tumor volume on MRI are used to predict whether patients will achieve a pCR. Those who are considered to have a high Bayesian predictive probability of success are eligible for moving on to phase 3 trials.
The I-SPY-2 trial was described in detail by principal investigator Laura J. Esserman, MD, MBA, from the Helen Diller Comprehensive Cancer Center at the University of California, San Francisco, in a 2017 interview from the annual meeting of the American Society of Clinical Oncology.
As previously reported, the first drugs to “graduate” from the trial were the HER2/HER4 inhibitor neratinib (Nerlynx, Puma Biotechnology) and the investigational PARP inhibitor veliparib (AbbVie).
Study details
The I-SPY-2 results that Pusztai reported at the AACR meeting were based on an analysis of 73 HER2-negative patients, including 21 patients with TNBC and 52 with HR-positive tumors with Mammaprint high-risk features. These patients underwent treatment with durvalumab 1500 mg every 4 weeks for three cycles, olaparib 100 mg twice daily for weeks 1 through 11, and paclitaxel 80 mg/m2 weekly for 12 weeks, followed by AC chemotherapy (doxorubicin and cyclophosphamide) for four cycles.
The 299 patients in the control arm received paclitaxel and chemotherapy only.
In all three biomarker subsets studied, durvalumab and olaparib increased pCR rates compared with controls, as shown in the table.
The probability that the combination was superior to control in each subgroup approached 100%, Pusztai noted.
Adverse events with the combination were consistent with known side effects of the drugs, he commented. Immune-related grade 3 adverse events occurred in 19% of patients in the combination therapy arm, compared with 1.6% in the control arm.
Higher pCR rates were seen in the subset of immune-rich tumors among all cancer subtypes and in both study arms.
“Exploratory analysis suggests several potential predictive markers of durvalumab/olaparib benefit over chemotherapy alone,” Pusztai reported.
These markers included Mammaprint MP2 (ultra high) versus MP1 in HR+/HER2– tumors, and low CD3/CD8 gene signature ratio, high macrophage/Tc-class 2 gene signature ratio, and high proliferation signature, all of which were associated with higher pCR rates in the experimental arm among patients with TNBC.
The trial was supported by the William K. Bowes Jr Foundation, Foundation for the NIH, Give Breast Cancer the Boot, UCSF, the Biomarkers Consortium, IQVIA, the Breast Cancer Research Foundation, Safeway, California Breast Cancer Research Program, Breast Cancer Research–Atwater Trust, and Stand Up to Cancer. Pusztai has received honoraria and consulting fees from AstraZeneca and other companies. Munster has received research and travel support from and has served on the scientific advisory boards of AstraZeneca and other companies.
This article first appeared on Medscape.com.
FROM AACR 2020
Reproductive psychiatry during the COVID-19 pandemic
When last I wrote this column, I was preparing for travel to professional meetings in the spring, planning a presentation for an upcoming grand rounds, and readying to host a scientific advisory board meeting as part of a large scientific project we conduct in Center for Women’s Mental Health. We were also awaiting the relocation of several junior faculty and research staff to Boston this spring and summer as we build our team.

It is now obvious that the COVID-19 pandemic is not a passing squall, but rather a persistent gale that has placed our collective sails in the water. It has not capsized the boat, however, thanks in part to the actions of courageous frontline caregivers and first responders who have mobilized in the wake of this recent public health crisis. From doctors, nurses, and hospital staff to grocery store clerks, home health aides, and neighbors checking in on the elderly – to name just a few – a whole crew of members across society have helped buoy our collective ship. Resilience also is required by all of us who are managing the array of feelings brought about by the day-in, day-out challenges of living life with restricted movement and freedom to engage in pre-COVID-19 activities we took for granted. What seemed like a temporary workaround is now becoming the “new normal” for an unknown amount of time looking forward.
For over 3 decades, my colleagues and I have worked with women who suffer from serious psychiatric disorders and whose treatment has required psychiatric medications such as antidepressants, mood stabilizers, and anxiolytics. The challenge of our work with women who are pregnant or planning pregnancy has been the configuration of the safest ways to navigate treatment on an individual basis for these women across pregnancy and post partum, with continual assessments of how to minimize the risk to fetus from in utero exposure to medications that have been instrumental in the treatment of psychiatric disorders on one hand versus the risks of untreated psychiatric disorder on the other. This work has been the essence of the clinical mission and the cornerstone of the research conducted at the Center for Women’s Mental Health since its inception.
While I have worked shoulder to shoulder with obstetricians for years, my respect for these colleagues during these past weeks has only grown as they have instituted the swiftest protocols to mitigate risk associated with COVID-19 for our pregnant patients, some of whom have tested positive for COVID-19, all in an effort to keep both mother, fetus, and newborn as safe as possible.
For those of us providing mental health services to pregnant women during this time, certain clinical situations have arisen in the context of the COVID-19 pandemic which require particular attention and discussion.
Planned pregnancy and contraception during the COVID-19 pandemic
Half of the pregnancies in this country are unplanned. Now more than ever, it is critical that decisions about moving forward with a plan to conceive be deliberate. These considerations range from the existential to the most concrete. For example, during these last weeks, we have consulted on cases where couples on the cusp of attempts to conceive face concerns about COVID-19, hence making more complicated their timeline with respect to actual plans to get pregnant. These are complicated decisions, particularly for women who may be slightly older and at the reproductive age where delaying pregnancy may have an adverse effect on fertility.
A concrete example of how the pandemic has affected fertility is evident as we encounter situations where women may defer starting a prescription oral contraceptive or lapse in its use because they have had difficulty coordinating visits with health care providers and may fear picking up prescriptions from pharmacies. We also have seen that procedures such as IUD placements have been deferred or canceled, or that some patients decline trips to the hospital or clinic to receive this type of service. These new barriers to access of contraception may require more planning at this time so that decisions about family planning are by design and not default during a time as complicated as the current public health crisis.
Telemedicine: telepsychiatry and obstetrics virtual visits
While wide-scale use of telemedicine platforms was not the standard day-to-day practice in either obstetrics or psychiatry prior to the pandemic, telepsychiatry has come up to speed within a short number of weeks. At our institution, 85% of outpatient visits are being conducted remotely, with in-person visits being reserved for only urgent or emergent visits. Our inpatient psychiatry service remains a setting where psychiatric patients, regardless of their COVID-19 status, can receive necessary care.

The use of telemedicine and specifically telepsychiatry is critical to mitigate the likelihood of exposure to SARS-CoV-2. On our reproductive psychiatry service, it has actually been an opportunity to engage with patients for comprehensive initial consults about reproductive safety of psychiatric medications currently being taken, or for ongoing consultation and direct patient care during follow-up visits during pregnancy to see that patients are sustaining emotional well-being or have changes for treatment implemented if they are not well. An increased frequency of visits allows us more opportunity to capture any signs of early clinical worsening of symptoms that might have been missed previously using the more traditional in-person setting.
Telepsychiatry and “virtual visits” have allowed us to do real-time, nimble modifications of treatment regimens with both pharmacologic and nonpharmacologic interventions to keep women well and to keep them out of the hospital for psychiatric care as often as possible. It also has facilitated a closer collaboration with our colleagues in obstetrics. In a way, the team of providers, including psychiatrists, obstetrical providers, social workers, and therapists can more easily communicate virtually than has sometimes been the case previously, when day-to-day use of telemedicine and virtual team meetings was less common.
Recognition and treatment of anxiety in perinatal patients
Even pregnant women without preexisting anxiety disorders may have heightened anxiety during usual times, and women and their partners cope with this typically in numerous ways including participation in peer-support opportunities, wellness and self-care activities, leveraging support from care providers, and engaging with family. But the previously “typical pregnancy experience” has shifted in the context of COVID-19. Specifically, added concerns of pregnant women about becoming infected, of potential separation from family if they do become ill, or of separation from partners or support systems during labor and delivery (an issue that has been largely resolved in many hospitals), as well as the possibility that a neonate might become ill with exposure to the coronavirus are obviously understandable and real. Such contingencies are unsettling, even for the most settled of our patients. Labor and delivery plans, and plans for outside help from family or others with the baby and older children in the postpartum period, have been upended for many patients.
These are anxious times. The number of nonpharmacologic virtual interventions available to mitigate anxiety are filling email inboxes daily. Curating these options can be a challenge, although several resources are worth noting, such as our department’s page on mental health resources.
During these past weeks, we have seen growing numbers of women for whom the normative anxiety of pregnancy is increasing to the point of causing distress to the level of functional impairment. Many patients for the first time meet criteria for frank anxiety disorders. These patients deserve prompt evaluation by mental health professionals and treatment with evidence-based therapies for anxiety disorders whether nonpharmacologic or pharmacologic so as to mitigate the risk of untreated anxiety on maternal and fetal well-being and also to limit risk for postpartum depression and postpartum anxiety disorders.
Miscarriage and infertility
A 36-year-old patient came to see me in clinic in late January following a miscarriage. She had a history of a previous miscarriage a year before and had an episode of major depression to follow for which she received treatment with an antidepressant and cognitive-behavioral therapy; she also attended a perinatal loss support group. She saw me in early March, anxious to try to conceive but extremely concerned about the risks associated with becoming pregnant at this point in time. Following a lengthy discussion with me and her obstetrician, the patient decided to wait until “the curve flattened” in Boston in terms of new cases of COVID-19, and then start trying to conceive. The case of another patient with a very similar history was presented at our rounds a few weeks ago; she also elected to defer attempts to conceive until life is more settled.
Perhaps one of the most dramatic examples of the impact of COVID-19 on fertility has been for those women with plans to pursue treatment with one of the assisted reproductive technologies. They have been told that professional societies have made recommendations regarding use of assisted reproductive technologies that are not entirely consistent across the country, but where in many places such interventions have been suspended during the COVID-19 pandemic. For many women near the end of their reproductive years, delays in trying to conceive either with or without the aid of fertility treatments may indelibly shape their plans to have children.
Sustaining emotional well-being across pregnancy
Because most psychiatric disorders are chronic in course, it is often the situation where women are treated to wellness for serious psychiatric disorders, with the goal of maintaining wellness across pregnancy and the post partum. One of the most critical takeaway points from 30 years of working with psychiatrically ill pregnant women is the maxim that keeping women well during pregnancy is simply imperative. Maternal psychiatric well-being during pregnancy is a strong predictor of obstetrical and neonatal outcomes, postpartum mental health, and longer-term neurobehavioral outcomes in children. Critically, in the context of the pandemic, keeping women out of psychiatric crises mitigates the necessity of visits to urgent clinical settings such as EDs and psychiatric inpatient units, which can increase the likelihood of exposure to the coronavirus.
Preservation of sleep
Disruption in sleep (duration and quality) can be seen in well over half of women during pregnancy with and without psychiatric disorders, and our experience has been that this has been exacerbated for many women during the COVID-19 crisis. Yet there are very rich data showing that sleep deprivation or sleep dysregulation in women, for example, who suffer from bipolar disorder or major depression can be a strong trigger for psychiatric relapse of underlying illness during pregnancy and the postpartum period.
During a time when normal rhythms of day-to-day life have been shifted – if not frankly disrupted – by swift transitions to remote work, cancellation of school and associated school activities across the country, complaints of insomnia and non-restorative sleep have been exceedingly common. Relevant to all but particularly for pregnant women with histories of psychiatric disorder, attention to sleep hygiene, moderation of caffeine use (if any), and use of any number of biobehavioral interventions to enhance relaxation and modulate stress may be of great value.
Cognitive-behavioral therapy for insomnia (CBT-I) has been demonstrated to be effective in pregnant women. Fortunately, there are user-friendly options on digital platforms that can be used during the pandemic that may play an important role in sustaining emotional well-being for pregnant women who have frank symptoms of insomnia.
Maintenance of ongoing antidepressant treatment during pregnancy among women with histories of mood disorder
Over a decade ago, my colleagues and I wrote about the comparison of outcomes for women with histories of recurrent major depression, demonstrating the value of maintenance treatment with antidepressants, compared with discontinuation of these medications during pregnancy (JAMA. 2006 Feb 1;295[5]:499-507). Recently, I was asked if maintenance antidepressant use in women with histories of recurrent depression was still our clinical recommendation. Over the last decade, we have noted that nearly half of women treated with antidepressants, regardless of illness severity, will discontinue their use of these medications prior to or early on in pregnancy given concerns about potential unknown effects of fetal exposure to medications, even medications for which there are robust data supporting reproductive safety regarding risk of congenital malformations. Routine discontinuation of antidepressants prior to or during pregnancy continues, despite the fact that we showed nearly 70% of those women with past histories of depression on maintenance antidepressant treatment relapsed shortly after discontinuing medication.
While we do not dictate the decisions women make about antidepressant use before, during, or after pregnancy, women with the same severity of illness will frequently make different decisions (a good thing) but we are now having very frank discussions about the particular need during a pandemic to avoid the relapse of serious psychiatric disorders. We typically endorse maintenance medication use with all but a very few number of psychotropic medications for which benefit may not outweigh risk to the fetus. However, for women who have decided nonetheless to discontinue antidepressants or other psychotropics during pregnancy despite the known risk of relapse, we strongly advise that they initiate treatment with evidence-based nonpharmacologic intervention such as CBT or mindfulness-based cognitive therapy (MBCT).
As in other areas of medicine, the pandemic is prompting we professionals in psychiatry, and specifically in perinatal psychiatry, to use all of our tools to keep pregnant and postpartum women well. The availability of digital tools to deliver MBCT and CBT has made the use of such interventions particularly viable at a time of social distancing. That being said, for patients with highly recurrent affective disorder with histories of previous recurrence when they stop their antidepressants, we are more strongly recommending serious consideration of maintenance medication treatment.
Virtual rounds in reproductive psychiatry and women’s mental health
The use of virtual platforms to connect with both patients and colleagues also has provided new opportunities for interaction with the reproductive psychiatry community as a whole. Peer teaching and peer support has been a critical part of our mission, and we decided 1 month ago to establish Virtual Rounds at the Center for Women’s Mental Health. This is a free digital platform, held on a weekly basis with our colleagues from across the country, where we discuss cases that come up in our own clinical rounds and also questions that get put forth by our colleagues in the area of reproductive psychiatry as they manage patients during the pandemic.
Changes in the postpartum experience
The last decade has brought a growing appreciation of postpartum depression and the need to screen and treat postpartum psychiatric disorders, such as postpartum mood and anxiety disorders. Yet in the era of this pandemic, the postpartum experience is itself is changing. Changes in carefully configured plans for the postpartum period – from family coming and going to mobilizing extra support at home and to now having new moms having to manage families and their other children at home – has been an enormous stressor for many women. Plans to have more elderly parents visit during the acute postpartum period, and the increased concerns about people traveling to and from a home where there is a newborn and the need to quarantine, has made the transition to motherhood much more complicated for all postpartum women, let alone for those postpartum women who have histories of psychiatric disorder.
There is a risk of social isolation for postpartum women even under normal circumstances, and this is profoundly more likely during this pandemic. We are actively working with our postpartum patients and optimizing treatment, brainstorming options in terms of using both virtual and real-time support to the extent that it is safe in order to keep women healthy during such a stressful and critical time.
I am heartened by the efforts on the part of organizations such as Postpartum Support International to make available virtually their resources with respect to community-based support and education for women who feel increasingly isolated during the postpartum period, a time where connectedness is so critical.
Summarily, these have been challenging times, but also times of opportunity. The COVID-19 pandemic has prompted us to get even more creative as we configure ways to optimize the emotional well-being of our patients who are planning to get pregnant, who are pregnant, or who are post partum.
The current time, while challenging in so many ways and a time of great pain, loss, and grief for far too many, has also provided an opportunity to work even more collaboratively with our colleagues, coming up with new paradigms of treatments as we weather this historic challenge.
Dr. Cohen is the director of the Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital in Boston, which provides information resources and conducts clinical care and research in reproductive mental health. He has been a consultant to manufacturers of psychiatric medications. Email him at [email protected].
When last I wrote this column, I was preparing for travel to professional meetings in the spring, planning a presentation for an upcoming grand rounds, and readying to host a scientific advisory board meeting as part of a large scientific project we conduct in Center for Women’s Mental Health. We were also awaiting the relocation of several junior faculty and research staff to Boston this spring and summer as we build our team.
It is now obvious that the COVID-19 pandemic is not a passing squall, but rather a persistent gale that has placed our collective sails in the water. It has not capsized the boat, however, thanks in part to the actions of courageous frontline caregivers and first responders who have mobilized in the wake of this recent public health crisis. From doctors, nurses, and hospital staff to grocery store clerks, home health aides, and neighbors checking in on the elderly – to name just a few – a whole crew of members across society have helped buoy our collective ship. Resilience also is required by all of us who are managing the array of feelings brought about by the day-in, day-out challenges of living life with restricted movement and freedom to engage in pre-COVID-19 activities we took for granted. What seemed like a temporary workaround is now becoming the “new normal” for an unknown amount of time looking forward.
For over 3 decades, my colleagues and I have worked with women who suffer from serious psychiatric disorders and whose treatment has required psychiatric medications such as antidepressants, mood stabilizers, and anxiolytics. The challenge of our work with women who are pregnant or planning pregnancy has been the configuration of the safest ways to navigate treatment on an individual basis for these women across pregnancy and post partum, with continual assessments of how to minimize the risk to fetus from in utero exposure to medications that have been instrumental in the treatment of psychiatric disorders on one hand versus the risks of untreated psychiatric disorder on the other. This work has been the essence of the clinical mission and the cornerstone of the research conducted at the Center for Women’s Mental Health since its inception.
While I have worked shoulder to shoulder with obstetricians for years, my respect for these colleagues during these past weeks has only grown as they have instituted the swiftest protocols to mitigate risk associated with COVID-19 for our pregnant patients, some of whom have tested positive for COVID-19, all in an effort to keep both mother, fetus, and newborn as safe as possible.
For those of us providing mental health services to pregnant women during this time, certain clinical situations have arisen in the context of the COVID-19 pandemic which require particular attention and discussion.
Planned pregnancy and contraception during the COVID-19 pandemic
Half of the pregnancies in this country are unplanned. Now more than ever, it is critical that decisions about moving forward with a plan to conceive be deliberate. These considerations range from the existential to the most concrete. For example, during these last weeks, we have consulted on cases where couples on the cusp of attempts to conceive face concerns about COVID-19, hence making more complicated their timeline with respect to actual plans to get pregnant. These are complicated decisions, particularly for women who may be slightly older and at the reproductive age where delaying pregnancy may have an adverse effect on fertility.
A concrete example of how the pandemic has affected fertility is evident as we encounter situations where women may defer starting a prescription oral contraceptive or lapse in its use because they have had difficulty coordinating visits with health care providers and may fear picking up prescriptions from pharmacies. We also have seen that procedures such as IUD placements have been deferred or canceled, or that some patients decline trips to the hospital or clinic to receive this type of service. These new barriers to access of contraception may require more planning at this time so that decisions about family planning are by design and not default during a time as complicated as the current public health crisis.
Telemedicine: telepsychiatry and obstetrics virtual visits
While wide-scale use of telemedicine platforms was not the standard day-to-day practice in either obstetrics or psychiatry prior to the pandemic, telepsychiatry has come up to speed within a short number of weeks. At our institution, 85% of outpatient visits are being conducted remotely, with in-person visits being reserved for only urgent or emergent visits. Our inpatient psychiatry service remains a setting where psychiatric patients, regardless of their COVID-19 status, can receive necessary care.
The use of telemedicine and specifically telepsychiatry is critical to mitigate the likelihood of exposure to SARS-CoV-2. On our reproductive psychiatry service, it has actually been an opportunity to engage with patients for comprehensive initial consults about reproductive safety of psychiatric medications currently being taken, or for ongoing consultation and direct patient care during follow-up visits during pregnancy to see that patients are sustaining emotional well-being or have changes for treatment implemented if they are not well. An increased frequency of visits allows us more opportunity to capture any signs of early clinical worsening of symptoms that might have been missed previously using the more traditional in-person setting.
Telepsychiatry and “virtual visits” have allowed us to do real-time, nimble modifications of treatment regimens with both pharmacologic and nonpharmacologic interventions to keep women well and to keep them out of the hospital for psychiatric care as often as possible. It also has facilitated a closer collaboration with our colleagues in obstetrics. In a way, the team of providers, including psychiatrists, obstetrical providers, social workers, and therapists can more easily communicate virtually than has sometimes been the case previously, when day-to-day use of telemedicine and virtual team meetings was less common.
Recognition and treatment of anxiety in perinatal patients
Even pregnant women without preexisting anxiety disorders may have heightened anxiety during usual times, and women and their partners cope with this typically in numerous ways including participation in peer-support opportunities, wellness and self-care activities, leveraging support from care providers, and engaging with family. But the previously “typical pregnancy experience” has shifted in the context of COVID-19. Specifically, added concerns of pregnant women about becoming infected, of potential separation from family if they do become ill, or of separation from partners or support systems during labor and delivery (an issue that has been largely resolved in many hospitals), as well as the possibility that a neonate might become ill with exposure to the coronavirus are obviously understandable and real. Such contingencies are unsettling, even for the most settled of our patients. Labor and delivery plans, and plans for outside help from family or others with the baby and older children in the postpartum period, have been upended for many patients.
These are anxious times. The number of nonpharmacologic virtual interventions available to mitigate anxiety are filling email inboxes daily. Curating these options can be a challenge, although several resources are worth noting, such as our department’s page on mental health resources.
During these past weeks, we have seen growing numbers of women for whom the normative anxiety of pregnancy is increasing to the point of causing distress to the level of functional impairment. Many patients for the first time meet criteria for frank anxiety disorders. These patients deserve prompt evaluation by mental health professionals and treatment with evidence-based therapies for anxiety disorders whether nonpharmacologic or pharmacologic so as to mitigate the risk of untreated anxiety on maternal and fetal well-being and also to limit risk for postpartum depression and postpartum anxiety disorders.
Miscarriage and infertility
A 36-year-old patient came to see me in clinic in late January following a miscarriage. She had a history of a previous miscarriage a year before and had an episode of major depression to follow for which she received treatment with an antidepressant and cognitive-behavioral therapy; she also attended a perinatal loss support group. She saw me in early March, anxious to try to conceive but extremely concerned about the risks associated with becoming pregnant at this point in time. Following a lengthy discussion with me and her obstetrician, the patient decided to wait until “the curve flattened” in Boston in terms of new cases of COVID-19, and then start trying to conceive. The case of another patient with a very similar history was presented at our rounds a few weeks ago; she also elected to defer attempts to conceive until life is more settled.
Perhaps one of the most dramatic examples of the impact of COVID-19 on fertility has been for those women with plans to pursue treatment with one of the assisted reproductive technologies. They have been told that professional societies have made recommendations regarding use of assisted reproductive technologies that are not entirely consistent across the country, but where in many places such interventions have been suspended during the COVID-19 pandemic. For many women near the end of their reproductive years, delays in trying to conceive either with or without the aid of fertility treatments may indelibly shape their plans to have children.
Sustaining emotional well-being across pregnancy
Because most psychiatric disorders are chronic in course, it is often the situation where women are treated to wellness for serious psychiatric disorders, with the goal of maintaining wellness across pregnancy and the post partum. One of the most critical takeaway points from 30 years of working with psychiatrically ill pregnant women is the maxim that keeping women well during pregnancy is simply imperative. Maternal psychiatric well-being during pregnancy is a strong predictor of obstetrical and neonatal outcomes, postpartum mental health, and longer-term neurobehavioral outcomes in children. Critically, in the context of the pandemic, keeping women out of psychiatric crises mitigates the necessity of visits to urgent clinical settings such as EDs and psychiatric inpatient units, which can increase the likelihood of exposure to the coronavirus.
Preservation of sleep
Disruption in sleep (duration and quality) can be seen in well over half of women during pregnancy with and without psychiatric disorders, and our experience has been that this has been exacerbated for many women during the COVID-19 crisis. Yet there are very rich data showing that sleep deprivation or sleep dysregulation in women, for example, who suffer from bipolar disorder or major depression can be a strong trigger for psychiatric relapse of underlying illness during pregnancy and the postpartum period.
During a time when normal rhythms of day-to-day life have been shifted – if not frankly disrupted – by swift transitions to remote work, cancellation of school and associated school activities across the country, complaints of insomnia and non-restorative sleep have been exceedingly common. Relevant to all but particularly for pregnant women with histories of psychiatric disorder, attention to sleep hygiene, moderation of caffeine use (if any), and use of any number of biobehavioral interventions to enhance relaxation and modulate stress may be of great value.
Cognitive-behavioral therapy for insomnia (CBT-I) has been demonstrated to be effective in pregnant women. Fortunately, there are user-friendly options on digital platforms that can be used during the pandemic that may play an important role in sustaining emotional well-being for pregnant women who have frank symptoms of insomnia.
Maintenance of ongoing antidepressant treatment during pregnancy among women with histories of mood disorder
Over a decade ago, my colleagues and I wrote about the comparison of outcomes for women with histories of recurrent major depression, demonstrating the value of maintenance treatment with antidepressants, compared with discontinuation of these medications during pregnancy (JAMA. 2006 Feb 1;295[5]:499-507). Recently, I was asked if maintenance antidepressant use in women with histories of recurrent depression was still our clinical recommendation. Over the last decade, we have noted that nearly half of women treated with antidepressants, regardless of illness severity, will discontinue their use of these medications prior to or early on in pregnancy given concerns about potential unknown effects of fetal exposure to medications, even medications for which there are robust data supporting reproductive safety regarding risk of congenital malformations. Routine discontinuation of antidepressants prior to or during pregnancy continues, despite the fact that we showed nearly 70% of those women with past histories of depression on maintenance antidepressant treatment relapsed shortly after discontinuing medication.
While we do not dictate the decisions women make about antidepressant use before, during, or after pregnancy, women with the same severity of illness will frequently make different decisions (a good thing) but we are now having very frank discussions about the particular need during a pandemic to avoid the relapse of serious psychiatric disorders. We typically endorse maintenance medication use with all but a very few number of psychotropic medications for which benefit may not outweigh risk to the fetus. However, for women who have decided nonetheless to discontinue antidepressants or other psychotropics during pregnancy despite the known risk of relapse, we strongly advise that they initiate treatment with evidence-based nonpharmacologic intervention such as CBT or mindfulness-based cognitive therapy (MBCT).
As in other areas of medicine, the pandemic is prompting we professionals in psychiatry, and specifically in perinatal psychiatry, to use all of our tools to keep pregnant and postpartum women well. The availability of digital tools to deliver MBCT and CBT has made the use of such interventions particularly viable at a time of social distancing. That being said, for patients with highly recurrent affective disorder with histories of previous recurrence when they stop their antidepressants, we are more strongly recommending serious consideration of maintenance medication treatment.
Virtual rounds in reproductive psychiatry and women’s mental health
The use of virtual platforms to connect with both patients and colleagues also has provided new opportunities for interaction with the reproductive psychiatry community as a whole. Peer teaching and peer support has been a critical part of our mission, and we decided 1 month ago to establish Virtual Rounds at the Center for Women’s Mental Health. This is a free digital platform, held on a weekly basis with our colleagues from across the country, where we discuss cases that come up in our own clinical rounds and also questions that get put forth by our colleagues in the area of reproductive psychiatry as they manage patients during the pandemic.
Changes in the postpartum experience
The last decade has brought a growing appreciation of postpartum depression and the need to screen and treat postpartum psychiatric disorders, such as postpartum mood and anxiety disorders. Yet in the era of this pandemic, the postpartum experience is itself is changing. Changes in carefully configured plans for the postpartum period – from family coming and going to mobilizing extra support at home and to now having new moms having to manage families and their other children at home – has been an enormous stressor for many women. Plans to have more elderly parents visit during the acute postpartum period, and the increased concerns about people traveling to and from a home where there is a newborn and the need to quarantine, has made the transition to motherhood much more complicated for all postpartum women, let alone for those postpartum women who have histories of psychiatric disorder.
There is a risk of social isolation for postpartum women even under normal circumstances, and this is profoundly more likely during this pandemic. We are actively working with our postpartum patients and optimizing treatment, brainstorming options in terms of using both virtual and real-time support to the extent that it is safe in order to keep women healthy during such a stressful and critical time.
I am heartened by the efforts on the part of organizations such as Postpartum Support International to make available virtually their resources with respect to community-based support and education for women who feel increasingly isolated during the postpartum period, a time where connectedness is so critical.
Summarily, these have been challenging times, but also times of opportunity. The COVID-19 pandemic has prompted us to get even more creative as we configure ways to optimize the emotional well-being of our patients who are planning to get pregnant, who are pregnant, or who are post partum.
The current time, while challenging in so many ways and a time of great pain, loss, and grief for far too many, has also provided an opportunity to work even more collaboratively with our colleagues, coming up with new paradigms of treatments as we weather this historic challenge.
Dr. Cohen is the director of the Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital in Boston, which provides information resources and conducts clinical care and research in reproductive mental health. He has been a consultant to manufacturers of psychiatric medications. Email him at [email protected].
When last I wrote this column, I was preparing for travel to professional meetings in the spring, planning a presentation for an upcoming grand rounds, and readying to host a scientific advisory board meeting as part of a large scientific project we conduct in Center for Women’s Mental Health. We were also awaiting the relocation of several junior faculty and research staff to Boston this spring and summer as we build our team.
It is now obvious that the COVID-19 pandemic is not a passing squall, but rather a persistent gale that has placed our collective sails in the water. It has not capsized the boat, however, thanks in part to the actions of courageous frontline caregivers and first responders who have mobilized in the wake of this recent public health crisis. From doctors, nurses, and hospital staff to grocery store clerks, home health aides, and neighbors checking in on the elderly – to name just a few – a whole crew of members across society have helped buoy our collective ship. Resilience also is required by all of us who are managing the array of feelings brought about by the day-in, day-out challenges of living life with restricted movement and freedom to engage in pre-COVID-19 activities we took for granted. What seemed like a temporary workaround is now becoming the “new normal” for an unknown amount of time looking forward.
For over 3 decades, my colleagues and I have worked with women who suffer from serious psychiatric disorders and whose treatment has required psychiatric medications such as antidepressants, mood stabilizers, and anxiolytics. The challenge of our work with women who are pregnant or planning pregnancy has been the configuration of the safest ways to navigate treatment on an individual basis for these women across pregnancy and post partum, with continual assessments of how to minimize the risk to fetus from in utero exposure to medications that have been instrumental in the treatment of psychiatric disorders on one hand versus the risks of untreated psychiatric disorder on the other. This work has been the essence of the clinical mission and the cornerstone of the research conducted at the Center for Women’s Mental Health since its inception.
While I have worked shoulder to shoulder with obstetricians for years, my respect for these colleagues during these past weeks has only grown as they have instituted the swiftest protocols to mitigate risk associated with COVID-19 for our pregnant patients, some of whom have tested positive for COVID-19, all in an effort to keep both mother, fetus, and newborn as safe as possible.
For those of us providing mental health services to pregnant women during this time, certain clinical situations have arisen in the context of the COVID-19 pandemic which require particular attention and discussion.
Planned pregnancy and contraception during the COVID-19 pandemic
Half of the pregnancies in this country are unplanned. Now more than ever, it is critical that decisions about moving forward with a plan to conceive be deliberate. These considerations range from the existential to the most concrete. For example, during these last weeks, we have consulted on cases where couples on the cusp of attempts to conceive face concerns about COVID-19, hence making more complicated their timeline with respect to actual plans to get pregnant. These are complicated decisions, particularly for women who may be slightly older and at the reproductive age where delaying pregnancy may have an adverse effect on fertility.
A concrete example of how the pandemic has affected fertility is evident as we encounter situations where women may defer starting a prescription oral contraceptive or lapse in its use because they have had difficulty coordinating visits with health care providers and may fear picking up prescriptions from pharmacies. We also have seen that procedures such as IUD placements have been deferred or canceled, or that some patients decline trips to the hospital or clinic to receive this type of service. These new barriers to access of contraception may require more planning at this time so that decisions about family planning are by design and not default during a time as complicated as the current public health crisis.
Telemedicine: telepsychiatry and obstetrics virtual visits
While wide-scale use of telemedicine platforms was not the standard day-to-day practice in either obstetrics or psychiatry prior to the pandemic, telepsychiatry has come up to speed within a short number of weeks. At our institution, 85% of outpatient visits are being conducted remotely, with in-person visits being reserved for only urgent or emergent visits. Our inpatient psychiatry service remains a setting where psychiatric patients, regardless of their COVID-19 status, can receive necessary care.
The use of telemedicine and specifically telepsychiatry is critical to mitigate the likelihood of exposure to SARS-CoV-2. On our reproductive psychiatry service, it has actually been an opportunity to engage with patients for comprehensive initial consults about reproductive safety of psychiatric medications currently being taken, or for ongoing consultation and direct patient care during follow-up visits during pregnancy to see that patients are sustaining emotional well-being or have changes for treatment implemented if they are not well. An increased frequency of visits allows us more opportunity to capture any signs of early clinical worsening of symptoms that might have been missed previously using the more traditional in-person setting.
Telepsychiatry and “virtual visits” have allowed us to do real-time, nimble modifications of treatment regimens with both pharmacologic and nonpharmacologic interventions to keep women well and to keep them out of the hospital for psychiatric care as often as possible. It also has facilitated a closer collaboration with our colleagues in obstetrics. In a way, the team of providers, including psychiatrists, obstetrical providers, social workers, and therapists can more easily communicate virtually than has sometimes been the case previously, when day-to-day use of telemedicine and virtual team meetings was less common.
Recognition and treatment of anxiety in perinatal patients
Even pregnant women without preexisting anxiety disorders may have heightened anxiety during usual times, and women and their partners cope with this typically in numerous ways including participation in peer-support opportunities, wellness and self-care activities, leveraging support from care providers, and engaging with family. But the previously “typical pregnancy experience” has shifted in the context of COVID-19. Specifically, added concerns of pregnant women about becoming infected, of potential separation from family if they do become ill, or of separation from partners or support systems during labor and delivery (an issue that has been largely resolved in many hospitals), as well as the possibility that a neonate might become ill with exposure to the coronavirus are obviously understandable and real. Such contingencies are unsettling, even for the most settled of our patients. Labor and delivery plans, and plans for outside help from family or others with the baby and older children in the postpartum period, have been upended for many patients.
These are anxious times. The number of nonpharmacologic virtual interventions available to mitigate anxiety are filling email inboxes daily. Curating these options can be a challenge, although several resources are worth noting, such as our department’s page on mental health resources.
During these past weeks, we have seen growing numbers of women for whom the normative anxiety of pregnancy is increasing to the point of causing distress to the level of functional impairment. Many patients for the first time meet criteria for frank anxiety disorders. These patients deserve prompt evaluation by mental health professionals and treatment with evidence-based therapies for anxiety disorders whether nonpharmacologic or pharmacologic so as to mitigate the risk of untreated anxiety on maternal and fetal well-being and also to limit risk for postpartum depression and postpartum anxiety disorders.
Miscarriage and infertility
A 36-year-old patient came to see me in clinic in late January following a miscarriage. She had a history of a previous miscarriage a year before and had an episode of major depression to follow for which she received treatment with an antidepressant and cognitive-behavioral therapy; she also attended a perinatal loss support group. She saw me in early March, anxious to try to conceive but extremely concerned about the risks associated with becoming pregnant at this point in time. Following a lengthy discussion with me and her obstetrician, the patient decided to wait until “the curve flattened” in Boston in terms of new cases of COVID-19, and then start trying to conceive. The case of another patient with a very similar history was presented at our rounds a few weeks ago; she also elected to defer attempts to conceive until life is more settled.
Perhaps one of the most dramatic examples of the impact of COVID-19 on fertility has been for those women with plans to pursue treatment with one of the assisted reproductive technologies. They have been told that professional societies have made recommendations regarding use of assisted reproductive technologies that are not entirely consistent across the country, but where in many places such interventions have been suspended during the COVID-19 pandemic. For many women near the end of their reproductive years, delays in trying to conceive either with or without the aid of fertility treatments may indelibly shape their plans to have children.
Sustaining emotional well-being across pregnancy
Because most psychiatric disorders are chronic in course, it is often the situation where women are treated to wellness for serious psychiatric disorders, with the goal of maintaining wellness across pregnancy and the post partum. One of the most critical takeaway points from 30 years of working with psychiatrically ill pregnant women is the maxim that keeping women well during pregnancy is simply imperative. Maternal psychiatric well-being during pregnancy is a strong predictor of obstetrical and neonatal outcomes, postpartum mental health, and longer-term neurobehavioral outcomes in children. Critically, in the context of the pandemic, keeping women out of psychiatric crises mitigates the necessity of visits to urgent clinical settings such as EDs and psychiatric inpatient units, which can increase the likelihood of exposure to the coronavirus.
Preservation of sleep
Disruption in sleep (duration and quality) can be seen in well over half of women during pregnancy with and without psychiatric disorders, and our experience has been that this has been exacerbated for many women during the COVID-19 crisis. Yet there are very rich data showing that sleep deprivation or sleep dysregulation in women, for example, who suffer from bipolar disorder or major depression can be a strong trigger for psychiatric relapse of underlying illness during pregnancy and the postpartum period.
During a time when normal rhythms of day-to-day life have been shifted – if not frankly disrupted – by swift transitions to remote work, cancellation of school and associated school activities across the country, complaints of insomnia and non-restorative sleep have been exceedingly common. Relevant to all but particularly for pregnant women with histories of psychiatric disorder, attention to sleep hygiene, moderation of caffeine use (if any), and use of any number of biobehavioral interventions to enhance relaxation and modulate stress may be of great value.
Cognitive-behavioral therapy for insomnia (CBT-I) has been demonstrated to be effective in pregnant women. Fortunately, there are user-friendly options on digital platforms that can be used during the pandemic that may play an important role in sustaining emotional well-being for pregnant women who have frank symptoms of insomnia.
Maintenance of ongoing antidepressant treatment during pregnancy among women with histories of mood disorder
Over a decade ago, my colleagues and I wrote about the comparison of outcomes for women with histories of recurrent major depression, demonstrating the value of maintenance treatment with antidepressants, compared with discontinuation of these medications during pregnancy (JAMA. 2006 Feb 1;295[5]:499-507). Recently, I was asked if maintenance antidepressant use in women with histories of recurrent depression was still our clinical recommendation. Over the last decade, we have noted that nearly half of women treated with antidepressants, regardless of illness severity, will discontinue their use of these medications prior to or early on in pregnancy given concerns about potential unknown effects of fetal exposure to medications, even medications for which there are robust data supporting reproductive safety regarding risk of congenital malformations. Routine discontinuation of antidepressants prior to or during pregnancy continues, despite the fact that we showed nearly 70% of those women with past histories of depression on maintenance antidepressant treatment relapsed shortly after discontinuing medication.
While we do not dictate the decisions women make about antidepressant use before, during, or after pregnancy, women with the same severity of illness will frequently make different decisions (a good thing) but we are now having very frank discussions about the particular need during a pandemic to avoid the relapse of serious psychiatric disorders. We typically endorse maintenance medication use with all but a very few number of psychotropic medications for which benefit may not outweigh risk to the fetus. However, for women who have decided nonetheless to discontinue antidepressants or other psychotropics during pregnancy despite the known risk of relapse, we strongly advise that they initiate treatment with evidence-based nonpharmacologic intervention such as CBT or mindfulness-based cognitive therapy (MBCT).
As in other areas of medicine, the pandemic is prompting we professionals in psychiatry, and specifically in perinatal psychiatry, to use all of our tools to keep pregnant and postpartum women well. The availability of digital tools to deliver MBCT and CBT has made the use of such interventions particularly viable at a time of social distancing. That being said, for patients with highly recurrent affective disorder with histories of previous recurrence when they stop their antidepressants, we are more strongly recommending serious consideration of maintenance medication treatment.
Virtual rounds in reproductive psychiatry and women’s mental health
The use of virtual platforms to connect with both patients and colleagues also has provided new opportunities for interaction with the reproductive psychiatry community as a whole. Peer teaching and peer support has been a critical part of our mission, and we decided 1 month ago to establish Virtual Rounds at the Center for Women’s Mental Health. This is a free digital platform, held on a weekly basis with our colleagues from across the country, where we discuss cases that come up in our own clinical rounds and also questions that get put forth by our colleagues in the area of reproductive psychiatry as they manage patients during the pandemic.
Changes in the postpartum experience
The last decade has brought a growing appreciation of postpartum depression and the need to screen and treat postpartum psychiatric disorders, such as postpartum mood and anxiety disorders. Yet in the era of this pandemic, the postpartum experience is itself is changing. Changes in carefully configured plans for the postpartum period – from family coming and going to mobilizing extra support at home and to now having new moms having to manage families and their other children at home – has been an enormous stressor for many women. Plans to have more elderly parents visit during the acute postpartum period, and the increased concerns about people traveling to and from a home where there is a newborn and the need to quarantine, has made the transition to motherhood much more complicated for all postpartum women, let alone for those postpartum women who have histories of psychiatric disorder.
There is a risk of social isolation for postpartum women even under normal circumstances, and this is profoundly more likely during this pandemic. We are actively working with our postpartum patients and optimizing treatment, brainstorming options in terms of using both virtual and real-time support to the extent that it is safe in order to keep women healthy during such a stressful and critical time.
I am heartened by the efforts on the part of organizations such as Postpartum Support International to make available virtually their resources with respect to community-based support and education for women who feel increasingly isolated during the postpartum period, a time where connectedness is so critical.
Summarily, these have been challenging times, but also times of opportunity. The COVID-19 pandemic has prompted us to get even more creative as we configure ways to optimize the emotional well-being of our patients who are planning to get pregnant, who are pregnant, or who are post partum.
The current time, while challenging in so many ways and a time of great pain, loss, and grief for far too many, has also provided an opportunity to work even more collaboratively with our colleagues, coming up with new paradigms of treatments as we weather this historic challenge.
Dr. Cohen is the director of the Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital in Boston, which provides information resources and conducts clinical care and research in reproductive mental health. He has been a consultant to manufacturers of psychiatric medications. Email him at [email protected].
REPLENISH: Oral estradiol/progesterone slowed bone turnover as it cut VMS
Menopausal women with an intact uterus treated for a year with a daily, oral, single-pill formulation of estradiol and progesterone for the primary goal of reducing moderate to severe vasomotor symptoms showed evidence of reduced bone turnover in a post hoc, subgroup analysis of 157 women enrolled in the REPLENISH trial.

These findings “provide support for a potential skeletal benefit” when menopausal women with an intact uterus regularly used the tested estradiol plus progesterone formulation to treat moderate to severe vasomotor symptoms (VMS), Risa Kagan, MD, and associates wrote in an abstract released ahead of the study’s scheduled presentation at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting, and released abstracts for press coverage.
The reductions reported for three different markers of bone turnover compared with placebo control after 6 and 12 months on treatment with a U.S.-marketed estradiol plus progesterone formulation were ”reassuring and in line with expectations” said Lubna Pal, MBBS, professor of obstetrics, gynecology, and reproductive sciences at Yale University, New Haven, Conn., who was not involved with the study. But the findings fell short of addressing the more clinically relevant issue of whether the tested regimen reduces the rate of bone fractures.

“Only longer-term data can tell us whether this magnitude of effect is sustainable,” which would need to happen to cut fracture incidence, said Dr. Pal, who directs the menopause program at her center. “The reduction in bone-turnover markers is adequate, but magnitude alone doesn’t entirely explain a reduction in fractures.”
The bone-marker findings came from a post hoc analysis of data collected in REPLENISH (Safety and Efficacy Study of the Combination Estradiol and Progesterone to Treat Vasomotor Symptoms in Postmenopausal Women With an Intact Uterus), a phase 3 randomized trial run at 119 U.S. sites that during 2013-2015 enrolled 1,845 menopausal women aged 65 years with an intact uterus and with moderate to severe VMS. The researchers randomized women to alternative daily treatment regimens with a single, oral pill that combined a bioidentical form of 17 beta-estradiol at dosages of 1.0 mg/day, 0.50 mg/day, or 0.25 mg/day, and dosages of bioidentical progesterone at dosages of 100 mg/day or 50 mg/day, or to a placebo control arm; not every possible dosage combination underwent testing.
The original study’s primary safety endpoint was the incidence of endometrial hyperplasia after 12 months on treatment, and the results showed no such events in any dosage arm. The primary efficacy endpoints assessed the frequency and severity of VMS after 4 and 12 weeks on treatment, and the results showed that all four tested formulations of estradiol plus progesterone had similar, statistically significant effects on decreasing VMS frequency, and the four formulations reduced severity in a dose-dependent way (Obstet Gynecol. 2018 Jul;132[1]:161-70). Based in part on results from this pivotal trial, the 1-mg estradiol/100 mg progesterone formulation (Bijuva) received Food and Drug Administration marketing approval in late 2018.
The new analysis of bone-turnover markers focused on a subgroup of women selected from two of the drug-treated arms and control patients who had at least 50 moderate to severe VMS events per week at baseline, were at least 5 years out from their last menstrual period, and had data recorded for three markers of bone turnover at baseline, and after 6 and 12 months on treatment, according to the abstract published in Obstetrics and Gynecology.
The analysis included 56 women treated with 1.0 mg estradiol and 100 mg progesterone daily, 56 who received 0.5 mg estradiol and 100 mg progesterone daily, and 45 women in the placebo arm. It assessed changes from baseline in the on-treatment compared with placebo groups using immunoassays for bone-specific alkaline phosphatase (BSAP), C-terminal telopeptide of type 1 collagen (CTX-1), and N-terminal propeptide of type 1 procollagen (PINP). For this analysis, the researchers combined the two active-treatment arms, and found that all three markers showed statistically significant drops from baseline with treatment, compared with placebo at both 6- and 12-month follow-up. For all three markers, the declines on active treatment grew larger with time, and after 12 months the mean drops from baseline compared with placebo were an 18% relative reduction in BSAP, a 41% relative reduction in CTX-1, and a 29% relative reduction in PINP, reported Dr. Kagan, a gynecologist based in Berkeley, Calif., who is affiliated with the University of California San Francisco.
In addition to not yet providing more clinically meaningful results on bone fracture rates, Dr. Pal cited additional issues that limit interpretation of both these findings and the broader REPLENISH results. First, the new analysis of bone-turnover markers as reported in the abstract does not allow assessment of a possible dose-dependent effect on bone turnover. More importantly, while the full REPLENISH trial provided reassuring data on endometrial protection, it has not yet reported data on the other major safety concern of hormone replacement: the effects of treatment on breast cancer rates. “They need to show no harm” in breast tissue, Dr. Pal said in an interview. In addition, the positive effect reported on markers of bone turnover is not unique to this formulation. Other formulations with estrogen at similar dosages would have similar effects, she said.
Other considerations when using an oral formulation include the reduced risk for prothrombotic effects from estradiol when delivered transdermally instead of orally, although some women have trouble getting insurance coverage for transdermal formulations, Dr. Pal said. Oral estrogen is appropriate only for women without an elevated clotting risk. Daily progesterone treatment also is more problematic than a cyclical dosage regimen, but noncontinuous progesterone results in monthly bleeding, something that some women prefer to avoid. “I’m not convinced that continuous progesterone is a physiologic approach,” said Dr. Pal, a member of the Ob.Gyn. News editorial advisory board. In addition, some women may find the formulation attractive because both the estradiol and progesterone are bioidentical and not synthetic molecules.
In short, the oral estradiol plus progesterone formulation tested in REPLENISH “is another tool” that’s an option for selected menopausal women with a uterus. “The bone findings are in line with expectations and are reassuring. Long-term data are keenly awaited to understand whether the bone marker changes lead to fewer fractures. The impact of the treatment on breast safety will be especially important.” For women who want a bioidentical, oral option and to not have bleeding, the tested formulation can be an effective option providing both symptom benefit and endometrial protection, Dr. Pal said.
REPLENISH was funded by TherapeuticsMD, the company that markets the tested estradiol plus progesterone formulation (Bijuva). Dr. Kagan has been a consultant and adviser to and speaker on behalf of Therapeutics MD, and she has also been a consultant or adviser to or has spoken on behalf of AMAG, Amgen, Astellas, Lupin, Radius, and Warner Chilcott/Allergan, and she has received research funding from Endoceutics. Dr. Pal has been a consultant to Flow Health.
SOURCE: Kagan R et al. Obstet Gynecol. 2020 May;135:62S. doi: 10.1097/01.AOG.0000665076.20231.a8.
Menopausal women with an intact uterus treated for a year with a daily, oral, single-pill formulation of estradiol and progesterone for the primary goal of reducing moderate to severe vasomotor symptoms showed evidence of reduced bone turnover in a post hoc, subgroup analysis of 157 women enrolled in the REPLENISH trial.
These findings “provide support for a potential skeletal benefit” when menopausal women with an intact uterus regularly used the tested estradiol plus progesterone formulation to treat moderate to severe vasomotor symptoms (VMS), Risa Kagan, MD, and associates wrote in an abstract released ahead of the study’s scheduled presentation at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting, and released abstracts for press coverage.
The reductions reported for three different markers of bone turnover compared with placebo control after 6 and 12 months on treatment with a U.S.-marketed estradiol plus progesterone formulation were ”reassuring and in line with expectations” said Lubna Pal, MBBS, professor of obstetrics, gynecology, and reproductive sciences at Yale University, New Haven, Conn., who was not involved with the study. But the findings fell short of addressing the more clinically relevant issue of whether the tested regimen reduces the rate of bone fractures.
“Only longer-term data can tell us whether this magnitude of effect is sustainable,” which would need to happen to cut fracture incidence, said Dr. Pal, who directs the menopause program at her center. “The reduction in bone-turnover markers is adequate, but magnitude alone doesn’t entirely explain a reduction in fractures.”
The bone-marker findings came from a post hoc analysis of data collected in REPLENISH (Safety and Efficacy Study of the Combination Estradiol and Progesterone to Treat Vasomotor Symptoms in Postmenopausal Women With an Intact Uterus), a phase 3 randomized trial run at 119 U.S. sites that during 2013-2015 enrolled 1,845 menopausal women aged 65 years with an intact uterus and with moderate to severe VMS. The researchers randomized women to alternative daily treatment regimens with a single, oral pill that combined a bioidentical form of 17 beta-estradiol at dosages of 1.0 mg/day, 0.50 mg/day, or 0.25 mg/day, and dosages of bioidentical progesterone at dosages of 100 mg/day or 50 mg/day, or to a placebo control arm; not every possible dosage combination underwent testing.
The original study’s primary safety endpoint was the incidence of endometrial hyperplasia after 12 months on treatment, and the results showed no such events in any dosage arm. The primary efficacy endpoints assessed the frequency and severity of VMS after 4 and 12 weeks on treatment, and the results showed that all four tested formulations of estradiol plus progesterone had similar, statistically significant effects on decreasing VMS frequency, and the four formulations reduced severity in a dose-dependent way (Obstet Gynecol. 2018 Jul;132[1]:161-70). Based in part on results from this pivotal trial, the 1-mg estradiol/100 mg progesterone formulation (Bijuva) received Food and Drug Administration marketing approval in late 2018.
The new analysis of bone-turnover markers focused on a subgroup of women selected from two of the drug-treated arms and control patients who had at least 50 moderate to severe VMS events per week at baseline, were at least 5 years out from their last menstrual period, and had data recorded for three markers of bone turnover at baseline, and after 6 and 12 months on treatment, according to the abstract published in Obstetrics and Gynecology.
The analysis included 56 women treated with 1.0 mg estradiol and 100 mg progesterone daily, 56 who received 0.5 mg estradiol and 100 mg progesterone daily, and 45 women in the placebo arm. It assessed changes from baseline in the on-treatment compared with placebo groups using immunoassays for bone-specific alkaline phosphatase (BSAP), C-terminal telopeptide of type 1 collagen (CTX-1), and N-terminal propeptide of type 1 procollagen (PINP). For this analysis, the researchers combined the two active-treatment arms, and found that all three markers showed statistically significant drops from baseline with treatment, compared with placebo at both 6- and 12-month follow-up. For all three markers, the declines on active treatment grew larger with time, and after 12 months the mean drops from baseline compared with placebo were an 18% relative reduction in BSAP, a 41% relative reduction in CTX-1, and a 29% relative reduction in PINP, reported Dr. Kagan, a gynecologist based in Berkeley, Calif., who is affiliated with the University of California San Francisco.
In addition to not yet providing more clinically meaningful results on bone fracture rates, Dr. Pal cited additional issues that limit interpretation of both these findings and the broader REPLENISH results. First, the new analysis of bone-turnover markers as reported in the abstract does not allow assessment of a possible dose-dependent effect on bone turnover. More importantly, while the full REPLENISH trial provided reassuring data on endometrial protection, it has not yet reported data on the other major safety concern of hormone replacement: the effects of treatment on breast cancer rates. “They need to show no harm” in breast tissue, Dr. Pal said in an interview. In addition, the positive effect reported on markers of bone turnover is not unique to this formulation. Other formulations with estrogen at similar dosages would have similar effects, she said.
Other considerations when using an oral formulation include the reduced risk for prothrombotic effects from estradiol when delivered transdermally instead of orally, although some women have trouble getting insurance coverage for transdermal formulations, Dr. Pal said. Oral estrogen is appropriate only for women without an elevated clotting risk. Daily progesterone treatment also is more problematic than a cyclical dosage regimen, but noncontinuous progesterone results in monthly bleeding, something that some women prefer to avoid. “I’m not convinced that continuous progesterone is a physiologic approach,” said Dr. Pal, a member of the Ob.Gyn. News editorial advisory board. In addition, some women may find the formulation attractive because both the estradiol and progesterone are bioidentical and not synthetic molecules.
In short, the oral estradiol plus progesterone formulation tested in REPLENISH “is another tool” that’s an option for selected menopausal women with a uterus. “The bone findings are in line with expectations and are reassuring. Long-term data are keenly awaited to understand whether the bone marker changes lead to fewer fractures. The impact of the treatment on breast safety will be especially important.” For women who want a bioidentical, oral option and to not have bleeding, the tested formulation can be an effective option providing both symptom benefit and endometrial protection, Dr. Pal said.
REPLENISH was funded by TherapeuticsMD, the company that markets the tested estradiol plus progesterone formulation (Bijuva). Dr. Kagan has been a consultant and adviser to and speaker on behalf of Therapeutics MD, and she has also been a consultant or adviser to or has spoken on behalf of AMAG, Amgen, Astellas, Lupin, Radius, and Warner Chilcott/Allergan, and she has received research funding from Endoceutics. Dr. Pal has been a consultant to Flow Health.
SOURCE: Kagan R et al. Obstet Gynecol. 2020 May;135:62S. doi: 10.1097/01.AOG.0000665076.20231.a8.
Menopausal women with an intact uterus treated for a year with a daily, oral, single-pill formulation of estradiol and progesterone for the primary goal of reducing moderate to severe vasomotor symptoms showed evidence of reduced bone turnover in a post hoc, subgroup analysis of 157 women enrolled in the REPLENISH trial.
These findings “provide support for a potential skeletal benefit” when menopausal women with an intact uterus regularly used the tested estradiol plus progesterone formulation to treat moderate to severe vasomotor symptoms (VMS), Risa Kagan, MD, and associates wrote in an abstract released ahead of the study’s scheduled presentation at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting, and released abstracts for press coverage.
The reductions reported for three different markers of bone turnover compared with placebo control after 6 and 12 months on treatment with a U.S.-marketed estradiol plus progesterone formulation were ”reassuring and in line with expectations” said Lubna Pal, MBBS, professor of obstetrics, gynecology, and reproductive sciences at Yale University, New Haven, Conn., who was not involved with the study. But the findings fell short of addressing the more clinically relevant issue of whether the tested regimen reduces the rate of bone fractures.
“Only longer-term data can tell us whether this magnitude of effect is sustainable,” which would need to happen to cut fracture incidence, said Dr. Pal, who directs the menopause program at her center. “The reduction in bone-turnover markers is adequate, but magnitude alone doesn’t entirely explain a reduction in fractures.”
The bone-marker findings came from a post hoc analysis of data collected in REPLENISH (Safety and Efficacy Study of the Combination Estradiol and Progesterone to Treat Vasomotor Symptoms in Postmenopausal Women With an Intact Uterus), a phase 3 randomized trial run at 119 U.S. sites that during 2013-2015 enrolled 1,845 menopausal women aged 65 years with an intact uterus and with moderate to severe VMS. The researchers randomized women to alternative daily treatment regimens with a single, oral pill that combined a bioidentical form of 17 beta-estradiol at dosages of 1.0 mg/day, 0.50 mg/day, or 0.25 mg/day, and dosages of bioidentical progesterone at dosages of 100 mg/day or 50 mg/day, or to a placebo control arm; not every possible dosage combination underwent testing.
The original study’s primary safety endpoint was the incidence of endometrial hyperplasia after 12 months on treatment, and the results showed no such events in any dosage arm. The primary efficacy endpoints assessed the frequency and severity of VMS after 4 and 12 weeks on treatment, and the results showed that all four tested formulations of estradiol plus progesterone had similar, statistically significant effects on decreasing VMS frequency, and the four formulations reduced severity in a dose-dependent way (Obstet Gynecol. 2018 Jul;132[1]:161-70). Based in part on results from this pivotal trial, the 1-mg estradiol/100 mg progesterone formulation (Bijuva) received Food and Drug Administration marketing approval in late 2018.
The new analysis of bone-turnover markers focused on a subgroup of women selected from two of the drug-treated arms and control patients who had at least 50 moderate to severe VMS events per week at baseline, were at least 5 years out from their last menstrual period, and had data recorded for three markers of bone turnover at baseline, and after 6 and 12 months on treatment, according to the abstract published in Obstetrics and Gynecology.
The analysis included 56 women treated with 1.0 mg estradiol and 100 mg progesterone daily, 56 who received 0.5 mg estradiol and 100 mg progesterone daily, and 45 women in the placebo arm. It assessed changes from baseline in the on-treatment compared with placebo groups using immunoassays for bone-specific alkaline phosphatase (BSAP), C-terminal telopeptide of type 1 collagen (CTX-1), and N-terminal propeptide of type 1 procollagen (PINP). For this analysis, the researchers combined the two active-treatment arms, and found that all three markers showed statistically significant drops from baseline with treatment, compared with placebo at both 6- and 12-month follow-up. For all three markers, the declines on active treatment grew larger with time, and after 12 months the mean drops from baseline compared with placebo were an 18% relative reduction in BSAP, a 41% relative reduction in CTX-1, and a 29% relative reduction in PINP, reported Dr. Kagan, a gynecologist based in Berkeley, Calif., who is affiliated with the University of California San Francisco.
In addition to not yet providing more clinically meaningful results on bone fracture rates, Dr. Pal cited additional issues that limit interpretation of both these findings and the broader REPLENISH results. First, the new analysis of bone-turnover markers as reported in the abstract does not allow assessment of a possible dose-dependent effect on bone turnover. More importantly, while the full REPLENISH trial provided reassuring data on endometrial protection, it has not yet reported data on the other major safety concern of hormone replacement: the effects of treatment on breast cancer rates. “They need to show no harm” in breast tissue, Dr. Pal said in an interview. In addition, the positive effect reported on markers of bone turnover is not unique to this formulation. Other formulations with estrogen at similar dosages would have similar effects, she said.
Other considerations when using an oral formulation include the reduced risk for prothrombotic effects from estradiol when delivered transdermally instead of orally, although some women have trouble getting insurance coverage for transdermal formulations, Dr. Pal said. Oral estrogen is appropriate only for women without an elevated clotting risk. Daily progesterone treatment also is more problematic than a cyclical dosage regimen, but noncontinuous progesterone results in monthly bleeding, something that some women prefer to avoid. “I’m not convinced that continuous progesterone is a physiologic approach,” said Dr. Pal, a member of the Ob.Gyn. News editorial advisory board. In addition, some women may find the formulation attractive because both the estradiol and progesterone are bioidentical and not synthetic molecules.
In short, the oral estradiol plus progesterone formulation tested in REPLENISH “is another tool” that’s an option for selected menopausal women with a uterus. “The bone findings are in line with expectations and are reassuring. Long-term data are keenly awaited to understand whether the bone marker changes lead to fewer fractures. The impact of the treatment on breast safety will be especially important.” For women who want a bioidentical, oral option and to not have bleeding, the tested formulation can be an effective option providing both symptom benefit and endometrial protection, Dr. Pal said.
REPLENISH was funded by TherapeuticsMD, the company that markets the tested estradiol plus progesterone formulation (Bijuva). Dr. Kagan has been a consultant and adviser to and speaker on behalf of Therapeutics MD, and she has also been a consultant or adviser to or has spoken on behalf of AMAG, Amgen, Astellas, Lupin, Radius, and Warner Chilcott/Allergan, and she has received research funding from Endoceutics. Dr. Pal has been a consultant to Flow Health.
SOURCE: Kagan R et al. Obstet Gynecol. 2020 May;135:62S. doi: 10.1097/01.AOG.0000665076.20231.a8.
FROM ACOG 2020
ESMO gets creative with guidelines for breast cancer care in the COVID-19 era
Like other agencies, the European Society for Medical Oncology has developed guidelines for managing breast cancer patients during the COVID-19 pandemic, recommending when care should be prioritized, delayed, or modified.

ESMO’s breast cancer guidelines expand upon guidelines issued by other groups, addressing a broad spectrum of patient profiles and providing a creative array of treatment options in COVID-19–era clinical practice.
As with ESMO’s other disease-focused COVID-19 guidelines, the breast cancer guidelines are organized by priority levels – high, medium, and low – which are applied to several domains of diagnosis and treatment.
High-priority recommendations apply to patients whose condition is either clinically unstable or whose cancer burden is immediately life-threatening.
Medium-priority recommendations apply to patients for whom delaying care beyond 6 weeks would probably lower the likelihood of a significant benefit from the intervention.
Low-priority recommendations apply to patients for whom services can be delayed for the duration of the COVID-19 pandemic.
Personalized care and high-priority situations
ESMO’s guidelines suggest that multidisciplinary tumor boards should guide decisions about the urgency of care for individual patients, given the complexity of breast cancer biology, the multiplicity of evidence-based treatments, and the possibility of cure or durable high-quality remissions.
The guidelines deliver a clear message that prepandemic discussions about delivering personalized care are even more important now.
ESMO prioritizes investigating high-risk screening mammography results (i.e., BIRADS 5), lumps noted on breast self-examination, clinical evidence of local-regional recurrence, and breast cancer in pregnant women.
Making these scenarios “high priority” will facilitate the best long-term outcomes in time-sensitive scenarios and improve patient satisfaction with care.
Modifications to consider
ESMO provides explicit options for treatment of common breast cancer profiles in which short-term modifications of standard management strategies can safely be considered. Given the generally long natural history of most breast cancer subtypes, these temporary modifications are unlikely to compromise long-term outcomes.
For patients with a new diagnosis of localized breast cancer, the guidelines recommend neoadjuvant chemotherapy, targeted therapy, or hormonal therapy to achieve optimal breast cancer outcomes and safely delay surgery or radiotherapy.
In the metastatic setting, ESMO advises providers to consider:
- Symptom-oriented testing, recognizing the arguable benefit of frequent imaging or serum tumor marker measurement (J Clin Oncol. 2016 Aug 20;34[24]:2820-6).
- Drug holidays, de-escalated maintenance therapy, and protracted schedules of bone-modifying agents.
- Avoiding mTOR and PI3KCA inhibitors as an addition to standard hormonal therapy because of pneumonitis, hyperglycemia, and immunosuppression risks. The guidelines suggest careful thought about adding CDK4/6 inhibitors to standard hormonal therapy because of the added burden of remote safety monitoring with the biologic agents.
ESMO makes suggestions about trimming the duration of adjuvant trastuzumab to 6 months, as in the PERSEPHONE study (Lancet. 2019 Jun 29;393[10191]:2599-612), and modifying the schedule of luteinizing hormone–releasing hormone agonist administration, in an effort to reduce patient exposure to health care personnel (and vice versa).
The guidelines recommend continuing clinical trials if benefits to patients outweigh risks and trials can be modified to enhance patient safety while preserving study endpoint evaluations.
Lower-priority situations
ESMO pointedly assigns a low priority to follow-up of patients who are at high risk of relapse but lack signs or symptoms of relapse.
Like other groups, ESMO recommends that patients with equivocal (i.e., BIRADS 3) screening mammograms should have 6-month follow-up imaging in preference to immediate core needle biopsy of the area(s) of concern.
ESMO uses age to assign priority for postponing adjuvant breast radiation in patients with low- to moderate-risk lesions. However, the guidelines stop surprisingly short of recommending that adjuvant radiation be withheld for older patients with low-risk, stage I, hormonally sensitive, HER2-negative breast cancers who receive endocrine therapy.
Bottom line
The pragmatic adjustments ESMO suggests address the challenges of evaluating and treating breast cancer patients during the COVID-19 pandemic. The guidelines protect each patient’s right to care and safety as well as protecting the safety of caregivers.
The guidelines will likely heighten patients’ satisfaction with care and decrease concern about adequacy of timely evaluation and treatment.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
Like other agencies, the European Society for Medical Oncology has developed guidelines for managing breast cancer patients during the COVID-19 pandemic, recommending when care should be prioritized, delayed, or modified.
ESMO’s breast cancer guidelines expand upon guidelines issued by other groups, addressing a broad spectrum of patient profiles and providing a creative array of treatment options in COVID-19–era clinical practice.
As with ESMO’s other disease-focused COVID-19 guidelines, the breast cancer guidelines are organized by priority levels – high, medium, and low – which are applied to several domains of diagnosis and treatment.
High-priority recommendations apply to patients whose condition is either clinically unstable or whose cancer burden is immediately life-threatening.
Medium-priority recommendations apply to patients for whom delaying care beyond 6 weeks would probably lower the likelihood of a significant benefit from the intervention.
Low-priority recommendations apply to patients for whom services can be delayed for the duration of the COVID-19 pandemic.
Personalized care and high-priority situations
ESMO’s guidelines suggest that multidisciplinary tumor boards should guide decisions about the urgency of care for individual patients, given the complexity of breast cancer biology, the multiplicity of evidence-based treatments, and the possibility of cure or durable high-quality remissions.
The guidelines deliver a clear message that prepandemic discussions about delivering personalized care are even more important now.
ESMO prioritizes investigating high-risk screening mammography results (i.e., BIRADS 5), lumps noted on breast self-examination, clinical evidence of local-regional recurrence, and breast cancer in pregnant women.
Making these scenarios “high priority” will facilitate the best long-term outcomes in time-sensitive scenarios and improve patient satisfaction with care.
Modifications to consider
ESMO provides explicit options for treatment of common breast cancer profiles in which short-term modifications of standard management strategies can safely be considered. Given the generally long natural history of most breast cancer subtypes, these temporary modifications are unlikely to compromise long-term outcomes.
For patients with a new diagnosis of localized breast cancer, the guidelines recommend neoadjuvant chemotherapy, targeted therapy, or hormonal therapy to achieve optimal breast cancer outcomes and safely delay surgery or radiotherapy.
In the metastatic setting, ESMO advises providers to consider:
- Symptom-oriented testing, recognizing the arguable benefit of frequent imaging or serum tumor marker measurement (J Clin Oncol. 2016 Aug 20;34[24]:2820-6).
- Drug holidays, de-escalated maintenance therapy, and protracted schedules of bone-modifying agents.
- Avoiding mTOR and PI3KCA inhibitors as an addition to standard hormonal therapy because of pneumonitis, hyperglycemia, and immunosuppression risks. The guidelines suggest careful thought about adding CDK4/6 inhibitors to standard hormonal therapy because of the added burden of remote safety monitoring with the biologic agents.
ESMO makes suggestions about trimming the duration of adjuvant trastuzumab to 6 months, as in the PERSEPHONE study (Lancet. 2019 Jun 29;393[10191]:2599-612), and modifying the schedule of luteinizing hormone–releasing hormone agonist administration, in an effort to reduce patient exposure to health care personnel (and vice versa).
The guidelines recommend continuing clinical trials if benefits to patients outweigh risks and trials can be modified to enhance patient safety while preserving study endpoint evaluations.
Lower-priority situations
ESMO pointedly assigns a low priority to follow-up of patients who are at high risk of relapse but lack signs or symptoms of relapse.
Like other groups, ESMO recommends that patients with equivocal (i.e., BIRADS 3) screening mammograms should have 6-month follow-up imaging in preference to immediate core needle biopsy of the area(s) of concern.
ESMO uses age to assign priority for postponing adjuvant breast radiation in patients with low- to moderate-risk lesions. However, the guidelines stop surprisingly short of recommending that adjuvant radiation be withheld for older patients with low-risk, stage I, hormonally sensitive, HER2-negative breast cancers who receive endocrine therapy.
Bottom line
The pragmatic adjustments ESMO suggests address the challenges of evaluating and treating breast cancer patients during the COVID-19 pandemic. The guidelines protect each patient’s right to care and safety as well as protecting the safety of caregivers.
The guidelines will likely heighten patients’ satisfaction with care and decrease concern about adequacy of timely evaluation and treatment.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
Like other agencies, the European Society for Medical Oncology has developed guidelines for managing breast cancer patients during the COVID-19 pandemic, recommending when care should be prioritized, delayed, or modified.
ESMO’s breast cancer guidelines expand upon guidelines issued by other groups, addressing a broad spectrum of patient profiles and providing a creative array of treatment options in COVID-19–era clinical practice.
As with ESMO’s other disease-focused COVID-19 guidelines, the breast cancer guidelines are organized by priority levels – high, medium, and low – which are applied to several domains of diagnosis and treatment.
High-priority recommendations apply to patients whose condition is either clinically unstable or whose cancer burden is immediately life-threatening.
Medium-priority recommendations apply to patients for whom delaying care beyond 6 weeks would probably lower the likelihood of a significant benefit from the intervention.
Low-priority recommendations apply to patients for whom services can be delayed for the duration of the COVID-19 pandemic.
Personalized care and high-priority situations
ESMO’s guidelines suggest that multidisciplinary tumor boards should guide decisions about the urgency of care for individual patients, given the complexity of breast cancer biology, the multiplicity of evidence-based treatments, and the possibility of cure or durable high-quality remissions.
The guidelines deliver a clear message that prepandemic discussions about delivering personalized care are even more important now.
ESMO prioritizes investigating high-risk screening mammography results (i.e., BIRADS 5), lumps noted on breast self-examination, clinical evidence of local-regional recurrence, and breast cancer in pregnant women.
Making these scenarios “high priority” will facilitate the best long-term outcomes in time-sensitive scenarios and improve patient satisfaction with care.
Modifications to consider
ESMO provides explicit options for treatment of common breast cancer profiles in which short-term modifications of standard management strategies can safely be considered. Given the generally long natural history of most breast cancer subtypes, these temporary modifications are unlikely to compromise long-term outcomes.
For patients with a new diagnosis of localized breast cancer, the guidelines recommend neoadjuvant chemotherapy, targeted therapy, or hormonal therapy to achieve optimal breast cancer outcomes and safely delay surgery or radiotherapy.
In the metastatic setting, ESMO advises providers to consider:
- Symptom-oriented testing, recognizing the arguable benefit of frequent imaging or serum tumor marker measurement (J Clin Oncol. 2016 Aug 20;34[24]:2820-6).
- Drug holidays, de-escalated maintenance therapy, and protracted schedules of bone-modifying agents.
- Avoiding mTOR and PI3KCA inhibitors as an addition to standard hormonal therapy because of pneumonitis, hyperglycemia, and immunosuppression risks. The guidelines suggest careful thought about adding CDK4/6 inhibitors to standard hormonal therapy because of the added burden of remote safety monitoring with the biologic agents.
ESMO makes suggestions about trimming the duration of adjuvant trastuzumab to 6 months, as in the PERSEPHONE study (Lancet. 2019 Jun 29;393[10191]:2599-612), and modifying the schedule of luteinizing hormone–releasing hormone agonist administration, in an effort to reduce patient exposure to health care personnel (and vice versa).
The guidelines recommend continuing clinical trials if benefits to patients outweigh risks and trials can be modified to enhance patient safety while preserving study endpoint evaluations.
Lower-priority situations
ESMO pointedly assigns a low priority to follow-up of patients who are at high risk of relapse but lack signs or symptoms of relapse.
Like other groups, ESMO recommends that patients with equivocal (i.e., BIRADS 3) screening mammograms should have 6-month follow-up imaging in preference to immediate core needle biopsy of the area(s) of concern.
ESMO uses age to assign priority for postponing adjuvant breast radiation in patients with low- to moderate-risk lesions. However, the guidelines stop surprisingly short of recommending that adjuvant radiation be withheld for older patients with low-risk, stage I, hormonally sensitive, HER2-negative breast cancers who receive endocrine therapy.
Bottom line
The pragmatic adjustments ESMO suggests address the challenges of evaluating and treating breast cancer patients during the COVID-19 pandemic. The guidelines protect each patient’s right to care and safety as well as protecting the safety of caregivers.
The guidelines will likely heighten patients’ satisfaction with care and decrease concern about adequacy of timely evaluation and treatment.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
Decreased fetal movement: Time to educate patients and ourselves
We have all as providers experienced the tragic stillbirth of a term fetus for one of our patients. Too often no fetal movement was felt for days, but the patient never called. Or the patient did call, but the nonstress test (NST) was reactive or the ultrasound showed normal growth and fluid or the biophysical profile (BPP) was 8/8. Yet the patient still presented with a stillborn fetus a day later. Was the first patient simply so fearful of the likely deceased child within her that she did not call? Or did she simply not know to report it because she was not educated about what decreased fetal movement could mean? Could the second example have been prevented even though the testing was normal? I believe both scenarios could have been prevented with better education for both providers and patients.

The national stillbirth rate has remained relatively stagnant since 2000, despite many improvements in guidelines for the management of higher risk pregnancies.1 We follow the growth of these pregnancies, do NSTs, and often induce these patients prior to the due date. We do this in the hope of having a healthy mom and baby. However, an analysis of 614 stillbirth cases and 1,816 control deliveries found that 81% of patients presenting with a stillborn baby had no risks factors that required additional monitoring.2 Nearly 66% of 1,714 patients with a late stillbirth reported decreased fetal movement, no fetal movement, or a concerning increase in fetal movement in the days leading up to their baby’s death.3 Studies have suggested that persistent decreased fetal movement has an odds ratio for stillbirth of 4.51,4 which is higher than hypertensive disease and diabetes for this same outcome by nearly a factor of two. Yet there are no formal guidelines on education for patients or management of this chief complaint.
We assess fetal movement at every prenatal visit but patients who experienced stillbirth will say they didn’t know why. This is because as a culture and a profession we are afraid to talk about such a taboo subject as stillbirth. We are afraid we will scare our patients if we tell them that a decrease in fetal movement or no fetal movement may be because their baby is at risk for this dreaded complication. On one level this argument makes sense, but as soon as the baby is born we give parents plenty of education and advice to keep their children safe. Telling a parent to remove all bedding, put their baby on their back, and keep their baby from being too warm to prevent sudden infant death syndrome (SIDS) is very scary. However, this education is necessary. If moms simply know the reason why we ask about fetal movements, they may not wait 2 days before they call. We must have faith that pregnant women can handle this education about decreased fetal movement.
Next most important is our response to the complaint of decreased fetal movement. Often when the NST is reactive or the ultrasound is normal, we assume the baby is at no risk and we reassure the mother that everything is fine. We often tell moms the false myth that babies slow down at the end or advise kick counts after this complaint despite studies failing to show their utility. Because the education about kick count is frequency is what matters, a mother may not call if there is a change in pattern or strength – even if she is very worried about this. A baby may “pass” a kick count, but a mom still may be very worried, yet she will not call because the baby “passed.”
Protocols from the United Kingdom and Australia focus on the assumption that the complaint of decreased fetal movement may be the only warning sign of impending stillbirth. Harvey Kliman, MD, PhD, director of reproductive and placental research unit at Yale University, New Haven, Conn. said an analogy to this is a car driving 55 miles per hour despite only 10 miles of gas being left in the tank.* The car is running fine even when it is almost out of gas. That may be why we all have seen a fetus with recent reassuring tests in the last few days who presents stillborn. Perhaps the only warning sign is decreased fetal movement – not a nonreactive NST or low score BPP. Placental insufficiency is often the cause of initially unexplained stillbirth, far more common than “cord accidents.” If we liken the placenta to the “gas tank” for the pregnancy, then decreased fetal movement may be the “low gas” signal on the dashboard. After this patient has a reactive NST and/or reassuring ultrasound, we need to ask her if she is reassured. Data from a study of 380 women found that women who had a gut instinct that something was wrong were 23 times more likely to experience a stillbirth, according to the unadjusted odds ratio from the logistic regression model.5 We should follow up closely with moms who are not reassured and consider induction if they are over 39 weeks. We should tell every mom who presents with a concern about fetal movement that she did the right thing, and we want to hear from her again immediately if the movement is decreased again or persists. We cannot make women feel silly for calling. We should do an ultrasound for worried moms even if the NST is reactive to make sure we are not missing oligohydramnios or fetal growth restriction; the latter is the biggest known risk factor for stillbirth. We also should perform an ultrasound for moms with risk factors for stillbirth such as advanced maternal age or black race.

The education about and plan for management of decreased fetal movement are two components of the UK Saving Babies Lives Protocol; one study in the United Kingdom has shown a 20% decline in stillbirths from 2010 to 2017. The other two components are making sure to catch all fetal growth restricted babies and smoking cessation. We incorporated this protocol in my practice several months ago, and we have had very positive feedback from patients. We are not getting an increase in concerns/visits and have not had any patients call and say that they were upset about receiving this education. The Word Health Organization calls stillbirth a “neglected tragedy.” The United States has the lowest annual reduction of its stillbirth rate for all high-income nations in the Lancet 2015 series on stillbirth.6
Dr. Florescue is an ob.gyn. in private practice at Women Gynecology and Childbirth Associates in Rochester, N.Y. She delivers babies at Highland Hospital in Rochester. She has no relevant financial disclosures.
References
1. The Lancet. 2016, Jan 18;387(10018):587-603.
2. JAMA. 2011 Dec 14;306(22):2469-79.
3. BMC Pregnancy Childbirth. 2015 Aug 15;15:172.
4. BMJ Open. 2018 Jul 6;8(7):e020031.
5. Midwifery. 2018 Jul;62:171-6.
6. The Lancet. 2016, Jan 18;387(10019):691-702.
*This article was updated on 5/4/2020.
We have all as providers experienced the tragic stillbirth of a term fetus for one of our patients. Too often no fetal movement was felt for days, but the patient never called. Or the patient did call, but the nonstress test (NST) was reactive or the ultrasound showed normal growth and fluid or the biophysical profile (BPP) was 8/8. Yet the patient still presented with a stillborn fetus a day later. Was the first patient simply so fearful of the likely deceased child within her that she did not call? Or did she simply not know to report it because she was not educated about what decreased fetal movement could mean? Could the second example have been prevented even though the testing was normal? I believe both scenarios could have been prevented with better education for both providers and patients.
The national stillbirth rate has remained relatively stagnant since 2000, despite many improvements in guidelines for the management of higher risk pregnancies.1 We follow the growth of these pregnancies, do NSTs, and often induce these patients prior to the due date. We do this in the hope of having a healthy mom and baby. However, an analysis of 614 stillbirth cases and 1,816 control deliveries found that 81% of patients presenting with a stillborn baby had no risks factors that required additional monitoring.2 Nearly 66% of 1,714 patients with a late stillbirth reported decreased fetal movement, no fetal movement, or a concerning increase in fetal movement in the days leading up to their baby’s death.3 Studies have suggested that persistent decreased fetal movement has an odds ratio for stillbirth of 4.51,4 which is higher than hypertensive disease and diabetes for this same outcome by nearly a factor of two. Yet there are no formal guidelines on education for patients or management of this chief complaint.
We assess fetal movement at every prenatal visit but patients who experienced stillbirth will say they didn’t know why. This is because as a culture and a profession we are afraid to talk about such a taboo subject as stillbirth. We are afraid we will scare our patients if we tell them that a decrease in fetal movement or no fetal movement may be because their baby is at risk for this dreaded complication. On one level this argument makes sense, but as soon as the baby is born we give parents plenty of education and advice to keep their children safe. Telling a parent to remove all bedding, put their baby on their back, and keep their baby from being too warm to prevent sudden infant death syndrome (SIDS) is very scary. However, this education is necessary. If moms simply know the reason why we ask about fetal movements, they may not wait 2 days before they call. We must have faith that pregnant women can handle this education about decreased fetal movement.
Next most important is our response to the complaint of decreased fetal movement. Often when the NST is reactive or the ultrasound is normal, we assume the baby is at no risk and we reassure the mother that everything is fine. We often tell moms the false myth that babies slow down at the end or advise kick counts after this complaint despite studies failing to show their utility. Because the education about kick count is frequency is what matters, a mother may not call if there is a change in pattern or strength – even if she is very worried about this. A baby may “pass” a kick count, but a mom still may be very worried, yet she will not call because the baby “passed.”
Protocols from the United Kingdom and Australia focus on the assumption that the complaint of decreased fetal movement may be the only warning sign of impending stillbirth. Harvey Kliman, MD, PhD, director of reproductive and placental research unit at Yale University, New Haven, Conn. said an analogy to this is a car driving 55 miles per hour despite only 10 miles of gas being left in the tank.* The car is running fine even when it is almost out of gas. That may be why we all have seen a fetus with recent reassuring tests in the last few days who presents stillborn. Perhaps the only warning sign is decreased fetal movement – not a nonreactive NST or low score BPP. Placental insufficiency is often the cause of initially unexplained stillbirth, far more common than “cord accidents.” If we liken the placenta to the “gas tank” for the pregnancy, then decreased fetal movement may be the “low gas” signal on the dashboard. After this patient has a reactive NST and/or reassuring ultrasound, we need to ask her if she is reassured. Data from a study of 380 women found that women who had a gut instinct that something was wrong were 23 times more likely to experience a stillbirth, according to the unadjusted odds ratio from the logistic regression model.5 We should follow up closely with moms who are not reassured and consider induction if they are over 39 weeks. We should tell every mom who presents with a concern about fetal movement that she did the right thing, and we want to hear from her again immediately if the movement is decreased again or persists. We cannot make women feel silly for calling. We should do an ultrasound for worried moms even if the NST is reactive to make sure we are not missing oligohydramnios or fetal growth restriction; the latter is the biggest known risk factor for stillbirth. We also should perform an ultrasound for moms with risk factors for stillbirth such as advanced maternal age or black race.
The education about and plan for management of decreased fetal movement are two components of the UK Saving Babies Lives Protocol; one study in the United Kingdom has shown a 20% decline in stillbirths from 2010 to 2017. The other two components are making sure to catch all fetal growth restricted babies and smoking cessation. We incorporated this protocol in my practice several months ago, and we have had very positive feedback from patients. We are not getting an increase in concerns/visits and have not had any patients call and say that they were upset about receiving this education. The Word Health Organization calls stillbirth a “neglected tragedy.” The United States has the lowest annual reduction of its stillbirth rate for all high-income nations in the Lancet 2015 series on stillbirth.6
Dr. Florescue is an ob.gyn. in private practice at Women Gynecology and Childbirth Associates in Rochester, N.Y. She delivers babies at Highland Hospital in Rochester. She has no relevant financial disclosures.
References
1. The Lancet. 2016, Jan 18;387(10018):587-603.
2. JAMA. 2011 Dec 14;306(22):2469-79.
3. BMC Pregnancy Childbirth. 2015 Aug 15;15:172.
4. BMJ Open. 2018 Jul 6;8(7):e020031.
5. Midwifery. 2018 Jul;62:171-6.
6. The Lancet. 2016, Jan 18;387(10019):691-702.
*This article was updated on 5/4/2020.
We have all as providers experienced the tragic stillbirth of a term fetus for one of our patients. Too often no fetal movement was felt for days, but the patient never called. Or the patient did call, but the nonstress test (NST) was reactive or the ultrasound showed normal growth and fluid or the biophysical profile (BPP) was 8/8. Yet the patient still presented with a stillborn fetus a day later. Was the first patient simply so fearful of the likely deceased child within her that she did not call? Or did she simply not know to report it because she was not educated about what decreased fetal movement could mean? Could the second example have been prevented even though the testing was normal? I believe both scenarios could have been prevented with better education for both providers and patients.
The national stillbirth rate has remained relatively stagnant since 2000, despite many improvements in guidelines for the management of higher risk pregnancies.1 We follow the growth of these pregnancies, do NSTs, and often induce these patients prior to the due date. We do this in the hope of having a healthy mom and baby. However, an analysis of 614 stillbirth cases and 1,816 control deliveries found that 81% of patients presenting with a stillborn baby had no risks factors that required additional monitoring.2 Nearly 66% of 1,714 patients with a late stillbirth reported decreased fetal movement, no fetal movement, or a concerning increase in fetal movement in the days leading up to their baby’s death.3 Studies have suggested that persistent decreased fetal movement has an odds ratio for stillbirth of 4.51,4 which is higher than hypertensive disease and diabetes for this same outcome by nearly a factor of two. Yet there are no formal guidelines on education for patients or management of this chief complaint.
We assess fetal movement at every prenatal visit but patients who experienced stillbirth will say they didn’t know why. This is because as a culture and a profession we are afraid to talk about such a taboo subject as stillbirth. We are afraid we will scare our patients if we tell them that a decrease in fetal movement or no fetal movement may be because their baby is at risk for this dreaded complication. On one level this argument makes sense, but as soon as the baby is born we give parents plenty of education and advice to keep their children safe. Telling a parent to remove all bedding, put their baby on their back, and keep their baby from being too warm to prevent sudden infant death syndrome (SIDS) is very scary. However, this education is necessary. If moms simply know the reason why we ask about fetal movements, they may not wait 2 days before they call. We must have faith that pregnant women can handle this education about decreased fetal movement.
Next most important is our response to the complaint of decreased fetal movement. Often when the NST is reactive or the ultrasound is normal, we assume the baby is at no risk and we reassure the mother that everything is fine. We often tell moms the false myth that babies slow down at the end or advise kick counts after this complaint despite studies failing to show their utility. Because the education about kick count is frequency is what matters, a mother may not call if there is a change in pattern or strength – even if she is very worried about this. A baby may “pass” a kick count, but a mom still may be very worried, yet she will not call because the baby “passed.”
Protocols from the United Kingdom and Australia focus on the assumption that the complaint of decreased fetal movement may be the only warning sign of impending stillbirth. Harvey Kliman, MD, PhD, director of reproductive and placental research unit at Yale University, New Haven, Conn. said an analogy to this is a car driving 55 miles per hour despite only 10 miles of gas being left in the tank.* The car is running fine even when it is almost out of gas. That may be why we all have seen a fetus with recent reassuring tests in the last few days who presents stillborn. Perhaps the only warning sign is decreased fetal movement – not a nonreactive NST or low score BPP. Placental insufficiency is often the cause of initially unexplained stillbirth, far more common than “cord accidents.” If we liken the placenta to the “gas tank” for the pregnancy, then decreased fetal movement may be the “low gas” signal on the dashboard. After this patient has a reactive NST and/or reassuring ultrasound, we need to ask her if she is reassured. Data from a study of 380 women found that women who had a gut instinct that something was wrong were 23 times more likely to experience a stillbirth, according to the unadjusted odds ratio from the logistic regression model.5 We should follow up closely with moms who are not reassured and consider induction if they are over 39 weeks. We should tell every mom who presents with a concern about fetal movement that she did the right thing, and we want to hear from her again immediately if the movement is decreased again or persists. We cannot make women feel silly for calling. We should do an ultrasound for worried moms even if the NST is reactive to make sure we are not missing oligohydramnios or fetal growth restriction; the latter is the biggest known risk factor for stillbirth. We also should perform an ultrasound for moms with risk factors for stillbirth such as advanced maternal age or black race.
The education about and plan for management of decreased fetal movement are two components of the UK Saving Babies Lives Protocol; one study in the United Kingdom has shown a 20% decline in stillbirths from 2010 to 2017. The other two components are making sure to catch all fetal growth restricted babies and smoking cessation. We incorporated this protocol in my practice several months ago, and we have had very positive feedback from patients. We are not getting an increase in concerns/visits and have not had any patients call and say that they were upset about receiving this education. The Word Health Organization calls stillbirth a “neglected tragedy.” The United States has the lowest annual reduction of its stillbirth rate for all high-income nations in the Lancet 2015 series on stillbirth.6
Dr. Florescue is an ob.gyn. in private practice at Women Gynecology and Childbirth Associates in Rochester, N.Y. She delivers babies at Highland Hospital in Rochester. She has no relevant financial disclosures.
References
1. The Lancet. 2016, Jan 18;387(10018):587-603.
2. JAMA. 2011 Dec 14;306(22):2469-79.
3. BMC Pregnancy Childbirth. 2015 Aug 15;15:172.
4. BMJ Open. 2018 Jul 6;8(7):e020031.
5. Midwifery. 2018 Jul;62:171-6.
6. The Lancet. 2016, Jan 18;387(10019):691-702.
*This article was updated on 5/4/2020.
Postcesarean recovery protocols reduce opioid use
based on data from cohorts of women before and after the introduction of the protocols.

The findings were released ahead of the study’s scheduled presentation at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting and released abstracts for press coverage.
ERAS protocols have been introduced in surgical specialties including colorectal, urologic, gynecologic, and hepatobiliary – with noted benefits to patients and the health care system, wrote Nnamdi I. Gwacham, DO, of Saint Barnabas Medical Center, Livingston, N.J., and colleagues.
The researchers explored the impact of ERAS on reduction in opioid use after cesarean sections at a community teaching hospital in a retrospective study also published in Obstetrics & Gynecology.
The study population included a historical cohort of 2,109 patients from 2018 before the establishment of the ERAS pathway and 1,463 patients since the ERAS pathway was established in 2019.*
Significantly fewer patients in the ERAS group required opioids, compared with the historical group (1,766 vs. 341). A total of 8,082 opioid units were used before the introduction of the ERAS pathway, compared with 803 units used since its introduction, Dr. Gwacham and associates reported. The study was a Donald F. Richardson Prize Paper.
The ERAS pathway consisted of received transversus abdominis plane blocks in the immediate postoperative period (given to 98% of the patients), and all patients were started on “a scheduled multimodal analgesia with a combination of ibuprofen, acetaminophen, and dextromethorphan until discharge,” the researchers wrote. Patients received opioids only if their pain was not well controlled with the ERAS protocol.
In addition, patients who received ERAS had significantly shorter hospital stays than the historical group (3.19 days vs. 2.63 days) and incurred a significantly lower average direct cost ($4,290 vs. $3,957).
The groups were not significantly different in age, race, or body mass index.

“Given the current opioid epidemic in America, researching ways to reduce their use is an urgent matter,” Angela Martin, MD, of the University of Kansas, Kansas City, said in an interview.
She thought the study findings were to be expected based on research in other areas. “Given the trends and ability to reduced opioid use with ERAS in other specialties, it does not surprise me that women recovering from cesarean sections are similar.”
The take-home message for clinicians: “Begin thinking outside of the box when it comes to pain control,” emphasized Dr. Martin, who was not a part of this study. “Opioids don’t have to be the first line medications for postoperative pain management.”
She added that additional directions for research could include the patient perspective on postoperative pain management after a cesarean delivery. “Alternative options to opioids would be even more enticing if the inpatient experience was also improved,” said Dr. Martin, who is a member of the Ob.Gyn News editorial advisory board.
The researchers had no financial conflicts to disclose. Dr. Martin had no relevant financial disclosures.
SOURCE: Gwacham NI et al. Obstet Gynecol. 2020 May;135:2S. doi: 10.1097/01.AOG.0000662880.08512.6b.
*This article was updated on 4/29/2020.
based on data from cohorts of women before and after the introduction of the protocols.
The findings were released ahead of the study’s scheduled presentation at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting and released abstracts for press coverage.
ERAS protocols have been introduced in surgical specialties including colorectal, urologic, gynecologic, and hepatobiliary – with noted benefits to patients and the health care system, wrote Nnamdi I. Gwacham, DO, of Saint Barnabas Medical Center, Livingston, N.J., and colleagues.
The researchers explored the impact of ERAS on reduction in opioid use after cesarean sections at a community teaching hospital in a retrospective study also published in Obstetrics & Gynecology.
The study population included a historical cohort of 2,109 patients from 2018 before the establishment of the ERAS pathway and 1,463 patients since the ERAS pathway was established in 2019.*
Significantly fewer patients in the ERAS group required opioids, compared with the historical group (1,766 vs. 341). A total of 8,082 opioid units were used before the introduction of the ERAS pathway, compared with 803 units used since its introduction, Dr. Gwacham and associates reported. The study was a Donald F. Richardson Prize Paper.
The ERAS pathway consisted of received transversus abdominis plane blocks in the immediate postoperative period (given to 98% of the patients), and all patients were started on “a scheduled multimodal analgesia with a combination of ibuprofen, acetaminophen, and dextromethorphan until discharge,” the researchers wrote. Patients received opioids only if their pain was not well controlled with the ERAS protocol.
In addition, patients who received ERAS had significantly shorter hospital stays than the historical group (3.19 days vs. 2.63 days) and incurred a significantly lower average direct cost ($4,290 vs. $3,957).
The groups were not significantly different in age, race, or body mass index.
“Given the current opioid epidemic in America, researching ways to reduce their use is an urgent matter,” Angela Martin, MD, of the University of Kansas, Kansas City, said in an interview.
She thought the study findings were to be expected based on research in other areas. “Given the trends and ability to reduced opioid use with ERAS in other specialties, it does not surprise me that women recovering from cesarean sections are similar.”
The take-home message for clinicians: “Begin thinking outside of the box when it comes to pain control,” emphasized Dr. Martin, who was not a part of this study. “Opioids don’t have to be the first line medications for postoperative pain management.”
She added that additional directions for research could include the patient perspective on postoperative pain management after a cesarean delivery. “Alternative options to opioids would be even more enticing if the inpatient experience was also improved,” said Dr. Martin, who is a member of the Ob.Gyn News editorial advisory board.
The researchers had no financial conflicts to disclose. Dr. Martin had no relevant financial disclosures.
SOURCE: Gwacham NI et al. Obstet Gynecol. 2020 May;135:2S. doi: 10.1097/01.AOG.0000662880.08512.6b.
*This article was updated on 4/29/2020.
based on data from cohorts of women before and after the introduction of the protocols.
The findings were released ahead of the study’s scheduled presentation at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting and released abstracts for press coverage.
ERAS protocols have been introduced in surgical specialties including colorectal, urologic, gynecologic, and hepatobiliary – with noted benefits to patients and the health care system, wrote Nnamdi I. Gwacham, DO, of Saint Barnabas Medical Center, Livingston, N.J., and colleagues.
The researchers explored the impact of ERAS on reduction in opioid use after cesarean sections at a community teaching hospital in a retrospective study also published in Obstetrics & Gynecology.
The study population included a historical cohort of 2,109 patients from 2018 before the establishment of the ERAS pathway and 1,463 patients since the ERAS pathway was established in 2019.*
Significantly fewer patients in the ERAS group required opioids, compared with the historical group (1,766 vs. 341). A total of 8,082 opioid units were used before the introduction of the ERAS pathway, compared with 803 units used since its introduction, Dr. Gwacham and associates reported. The study was a Donald F. Richardson Prize Paper.
The ERAS pathway consisted of received transversus abdominis plane blocks in the immediate postoperative period (given to 98% of the patients), and all patients were started on “a scheduled multimodal analgesia with a combination of ibuprofen, acetaminophen, and dextromethorphan until discharge,” the researchers wrote. Patients received opioids only if their pain was not well controlled with the ERAS protocol.
In addition, patients who received ERAS had significantly shorter hospital stays than the historical group (3.19 days vs. 2.63 days) and incurred a significantly lower average direct cost ($4,290 vs. $3,957).
The groups were not significantly different in age, race, or body mass index.
“Given the current opioid epidemic in America, researching ways to reduce their use is an urgent matter,” Angela Martin, MD, of the University of Kansas, Kansas City, said in an interview.
She thought the study findings were to be expected based on research in other areas. “Given the trends and ability to reduced opioid use with ERAS in other specialties, it does not surprise me that women recovering from cesarean sections are similar.”
The take-home message for clinicians: “Begin thinking outside of the box when it comes to pain control,” emphasized Dr. Martin, who was not a part of this study. “Opioids don’t have to be the first line medications for postoperative pain management.”
She added that additional directions for research could include the patient perspective on postoperative pain management after a cesarean delivery. “Alternative options to opioids would be even more enticing if the inpatient experience was also improved,” said Dr. Martin, who is a member of the Ob.Gyn News editorial advisory board.
The researchers had no financial conflicts to disclose. Dr. Martin had no relevant financial disclosures.
SOURCE: Gwacham NI et al. Obstet Gynecol. 2020 May;135:2S. doi: 10.1097/01.AOG.0000662880.08512.6b.
*This article was updated on 4/29/2020.
REPORTING FROM ACOG 2020
Vaginal cleansing at cesarean delivery works in practice
Vaginal cleansing before cesarean delivery was successfully implemented – and significantly decreased the rate of surgical site infections (SSI) – in a quality improvement study done at Thomas Jefferson University Hospital in Philadelphia.
“Our goal was not to prove that vaginal preparation [before cesarean section] works, because that’s already been shown in large randomized, controlled trials, but to show that we can implement it and that we can see the same results in real life,” lead investigator Johanna Quist-Nelson, MD, said in an interview.
Dr. Quist-Nelson, a third-year fellow at the hospital and the department of obstetrics and gynecology at Sidney Kimmel Medical College, Philadelphia, was scheduled to present the findings at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting and released abstracts for press coverage.
Resident and staff physicians as well as nursing and operating room staff were educated/reminded through a multipronged intervention about the benefits of vaginal cleansing with a sponge stick preparation of 10% povidone-iodine solution (Betadine) – and later about the potential benefits of intravenous azithromycin – immediately before cesarean delivery for women in labor and women with ruptured membranes.
Dr. Quist-Nelson and coinvestigators compared three periods of time: 12 months preintervention, 14 months with vaginal cleansing promoted for infection prophylaxis, and 16 months of instructions for both vaginal cleansing and intravenous azithromycin. The three periods captured 1,033 patients. The researchers used control charts – a tool “often used in implementation science,” she said – to analyze monthly data and assess trends for SSI rates and for compliance.
The rate of SSI – as defined by the Centers for Disease Control and Prevention – decreased by 33%, they found, from 23% to 15%. The drop occurred mainly 4 months into the vaginal cleansing portion of the study and was sustained during the following 26 months. The addition of intravenous azithromycin education did not result in any further change in the SSI rate, Dr. Quist-Nelson and associates reported in the study – the abstract for which was published in Obstetrics & Gynecology. It won a third-place prize among the papers on current clinical and basic investigation.
Compliance with the vaginal cleansing protocol increased from 60% at the start of the vaginal cleansing phase to 85% 1 year later. Azithromycin compliance rose to 75% over the third phase of the intervention.
Vaginal cleansing has received attention at Thomas Jefferson for several years. In 2017, researchers there collaborated with investigators in Italy on a systemic review and meta-analysis which concluded that women who received vaginal cleansing before cesarean delivery – most commonly with 10% povidine-iodine – had a significantly lower incidence of endometritis (Obstet Gynecol. 2017 Sep;130[3]:527-38).
A subgroup analysis showed that Dr. Quist-Nelson said.
Azithromycin similarly was found to reduce the risk of postoperative infection in women undergoing nonelective cesarean deliveries in a randomized trial published in 2016 (N Engl J Med. 2016 Sep 29;375[13]:1231-41). While the new quality improvement study did not suggest any additional benefit to intravenous azithromycin, “we continue to offer it [at our hospital] because it has been shown [in prior research] to be beneficial and because our study wasn’t [designed] to show benefit,” Dr. Quist-Nelson said.
The quality improvement intervention included hands-on training on vaginal cleansing for resident physicians and e-mail reminders for physician staff, and daily reviews for 1 week on intravenous azithromycin for resident physicians and EMR “best practice advisory” reminders for physician staff. “We also wrote a protocol available online, and put reminders in our OR notes, as well as trained the nursing staff and OR staff,” she said.

Catherine Cansino, MD, MPH, of the University of California, Davis, said in an interview that SSI rates are “problematic [in obstetrics], not only because of morbidity but also potential cost because of rehospitalization.” The study shows that vaginal cleansing “is absolutely a good target for quality improvement,” she said. “It’s promising, and very exciting to see something like this have such a dramatic positive result.” Dr. Cansino, who is a member of the Ob.Gyn News editorial advisory board, was not involved in this study.
Thomas Jefferson Hospital has had relatively high SSI rates, Dr. Quist-Nelson noted.
Dr. Quist-Nelson and coinvestigators did not report any potential conflicts of interest. Dr. Cansino also did not report any potential conflicts of interest.
SOURCE: Quist-Nelson J et al. Obstet. Gynecol. 2020 May;135:1S. doi: 10.1097/01.AOG.0000662876.23603.13.
Vaginal cleansing before cesarean delivery was successfully implemented – and significantly decreased the rate of surgical site infections (SSI) – in a quality improvement study done at Thomas Jefferson University Hospital in Philadelphia.
“Our goal was not to prove that vaginal preparation [before cesarean section] works, because that’s already been shown in large randomized, controlled trials, but to show that we can implement it and that we can see the same results in real life,” lead investigator Johanna Quist-Nelson, MD, said in an interview.
Dr. Quist-Nelson, a third-year fellow at the hospital and the department of obstetrics and gynecology at Sidney Kimmel Medical College, Philadelphia, was scheduled to present the findings at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting and released abstracts for press coverage.
Resident and staff physicians as well as nursing and operating room staff were educated/reminded through a multipronged intervention about the benefits of vaginal cleansing with a sponge stick preparation of 10% povidone-iodine solution (Betadine) – and later about the potential benefits of intravenous azithromycin – immediately before cesarean delivery for women in labor and women with ruptured membranes.
Dr. Quist-Nelson and coinvestigators compared three periods of time: 12 months preintervention, 14 months with vaginal cleansing promoted for infection prophylaxis, and 16 months of instructions for both vaginal cleansing and intravenous azithromycin. The three periods captured 1,033 patients. The researchers used control charts – a tool “often used in implementation science,” she said – to analyze monthly data and assess trends for SSI rates and for compliance.
The rate of SSI – as defined by the Centers for Disease Control and Prevention – decreased by 33%, they found, from 23% to 15%. The drop occurred mainly 4 months into the vaginal cleansing portion of the study and was sustained during the following 26 months. The addition of intravenous azithromycin education did not result in any further change in the SSI rate, Dr. Quist-Nelson and associates reported in the study – the abstract for which was published in Obstetrics & Gynecology. It won a third-place prize among the papers on current clinical and basic investigation.
Compliance with the vaginal cleansing protocol increased from 60% at the start of the vaginal cleansing phase to 85% 1 year later. Azithromycin compliance rose to 75% over the third phase of the intervention.
Vaginal cleansing has received attention at Thomas Jefferson for several years. In 2017, researchers there collaborated with investigators in Italy on a systemic review and meta-analysis which concluded that women who received vaginal cleansing before cesarean delivery – most commonly with 10% povidine-iodine – had a significantly lower incidence of endometritis (Obstet Gynecol. 2017 Sep;130[3]:527-38).
A subgroup analysis showed that Dr. Quist-Nelson said.
Azithromycin similarly was found to reduce the risk of postoperative infection in women undergoing nonelective cesarean deliveries in a randomized trial published in 2016 (N Engl J Med. 2016 Sep 29;375[13]:1231-41). While the new quality improvement study did not suggest any additional benefit to intravenous azithromycin, “we continue to offer it [at our hospital] because it has been shown [in prior research] to be beneficial and because our study wasn’t [designed] to show benefit,” Dr. Quist-Nelson said.
The quality improvement intervention included hands-on training on vaginal cleansing for resident physicians and e-mail reminders for physician staff, and daily reviews for 1 week on intravenous azithromycin for resident physicians and EMR “best practice advisory” reminders for physician staff. “We also wrote a protocol available online, and put reminders in our OR notes, as well as trained the nursing staff and OR staff,” she said.
Catherine Cansino, MD, MPH, of the University of California, Davis, said in an interview that SSI rates are “problematic [in obstetrics], not only because of morbidity but also potential cost because of rehospitalization.” The study shows that vaginal cleansing “is absolutely a good target for quality improvement,” she said. “It’s promising, and very exciting to see something like this have such a dramatic positive result.” Dr. Cansino, who is a member of the Ob.Gyn News editorial advisory board, was not involved in this study.
Thomas Jefferson Hospital has had relatively high SSI rates, Dr. Quist-Nelson noted.
Dr. Quist-Nelson and coinvestigators did not report any potential conflicts of interest. Dr. Cansino also did not report any potential conflicts of interest.
SOURCE: Quist-Nelson J et al. Obstet. Gynecol. 2020 May;135:1S. doi: 10.1097/01.AOG.0000662876.23603.13.
Vaginal cleansing before cesarean delivery was successfully implemented – and significantly decreased the rate of surgical site infections (SSI) – in a quality improvement study done at Thomas Jefferson University Hospital in Philadelphia.
“Our goal was not to prove that vaginal preparation [before cesarean section] works, because that’s already been shown in large randomized, controlled trials, but to show that we can implement it and that we can see the same results in real life,” lead investigator Johanna Quist-Nelson, MD, said in an interview.
Dr. Quist-Nelson, a third-year fellow at the hospital and the department of obstetrics and gynecology at Sidney Kimmel Medical College, Philadelphia, was scheduled to present the findings at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists. ACOG canceled the meeting and released abstracts for press coverage.
Resident and staff physicians as well as nursing and operating room staff were educated/reminded through a multipronged intervention about the benefits of vaginal cleansing with a sponge stick preparation of 10% povidone-iodine solution (Betadine) – and later about the potential benefits of intravenous azithromycin – immediately before cesarean delivery for women in labor and women with ruptured membranes.
Dr. Quist-Nelson and coinvestigators compared three periods of time: 12 months preintervention, 14 months with vaginal cleansing promoted for infection prophylaxis, and 16 months of instructions for both vaginal cleansing and intravenous azithromycin. The three periods captured 1,033 patients. The researchers used control charts – a tool “often used in implementation science,” she said – to analyze monthly data and assess trends for SSI rates and for compliance.
The rate of SSI – as defined by the Centers for Disease Control and Prevention – decreased by 33%, they found, from 23% to 15%. The drop occurred mainly 4 months into the vaginal cleansing portion of the study and was sustained during the following 26 months. The addition of intravenous azithromycin education did not result in any further change in the SSI rate, Dr. Quist-Nelson and associates reported in the study – the abstract for which was published in Obstetrics & Gynecology. It won a third-place prize among the papers on current clinical and basic investigation.
Compliance with the vaginal cleansing protocol increased from 60% at the start of the vaginal cleansing phase to 85% 1 year later. Azithromycin compliance rose to 75% over the third phase of the intervention.
Vaginal cleansing has received attention at Thomas Jefferson for several years. In 2017, researchers there collaborated with investigators in Italy on a systemic review and meta-analysis which concluded that women who received vaginal cleansing before cesarean delivery – most commonly with 10% povidine-iodine – had a significantly lower incidence of endometritis (Obstet Gynecol. 2017 Sep;130[3]:527-38).
A subgroup analysis showed that Dr. Quist-Nelson said.
Azithromycin similarly was found to reduce the risk of postoperative infection in women undergoing nonelective cesarean deliveries in a randomized trial published in 2016 (N Engl J Med. 2016 Sep 29;375[13]:1231-41). While the new quality improvement study did not suggest any additional benefit to intravenous azithromycin, “we continue to offer it [at our hospital] because it has been shown [in prior research] to be beneficial and because our study wasn’t [designed] to show benefit,” Dr. Quist-Nelson said.
The quality improvement intervention included hands-on training on vaginal cleansing for resident physicians and e-mail reminders for physician staff, and daily reviews for 1 week on intravenous azithromycin for resident physicians and EMR “best practice advisory” reminders for physician staff. “We also wrote a protocol available online, and put reminders in our OR notes, as well as trained the nursing staff and OR staff,” she said.
Catherine Cansino, MD, MPH, of the University of California, Davis, said in an interview that SSI rates are “problematic [in obstetrics], not only because of morbidity but also potential cost because of rehospitalization.” The study shows that vaginal cleansing “is absolutely a good target for quality improvement,” she said. “It’s promising, and very exciting to see something like this have such a dramatic positive result.” Dr. Cansino, who is a member of the Ob.Gyn News editorial advisory board, was not involved in this study.
Thomas Jefferson Hospital has had relatively high SSI rates, Dr. Quist-Nelson noted.
Dr. Quist-Nelson and coinvestigators did not report any potential conflicts of interest. Dr. Cansino also did not report any potential conflicts of interest.
SOURCE: Quist-Nelson J et al. Obstet. Gynecol. 2020 May;135:1S. doi: 10.1097/01.AOG.0000662876.23603.13.
REPORTING FROM ACOG 2020