User login
White Atrophic Plaques on the Thighs
White Atrophic Plaques on the Thighs
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
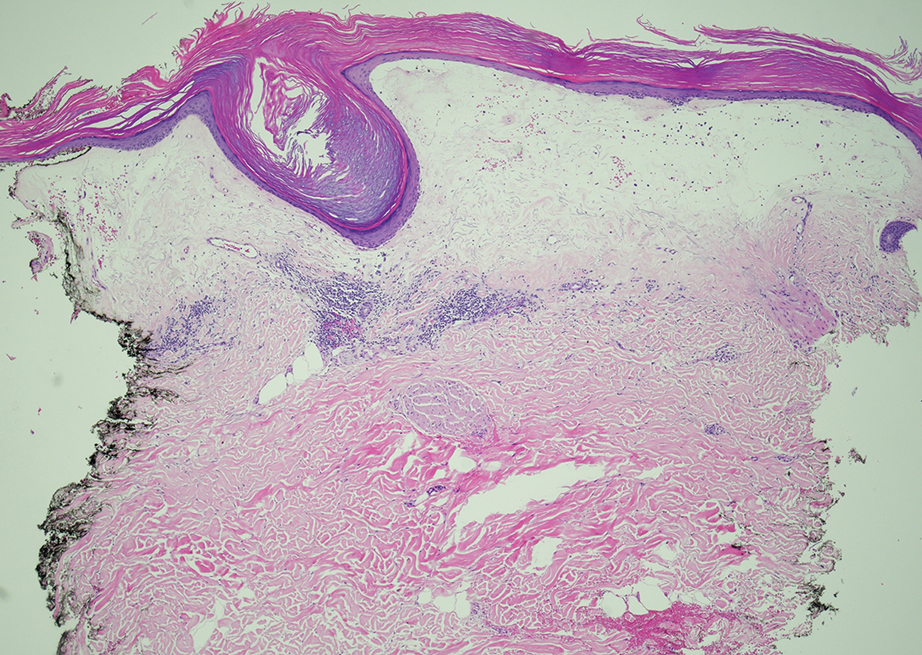
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
THE DIAGNOSIS: Lichen Sclerosus
Given the clinical appearance of white atrophic plaques with characteristic wrinkling of the skin, a diagnosis of lichen sclerosus was strongly suspected. At the initial office visit, the patient was prescribed clobetasol 0.05% ointment twice daily for 6 weeks. Histopathology revealed hyperkeratosis, follicular plugging, papillary dermal pallor, and adjacent lymphocytic inflammation, confirming the clinical diagnosis of lichen sclerosus (Figure). The patient then was lost to follow-up.
Lichen sclerosus is a chronic benign dermatologic condition of unknown etiology that is characterized by epidermal atrophy and inflammation and is common in postmenopausal women. It features pale, ivory-colored lesions with partially atrophic skin and a wrinkled cigarette paper appearance.1 The differential for lichen sclerosus is broad, and definitive diagnosis is made via biopsy to rule out potential malignancy and other inflammatory skin diseases.1 Lichen sclerosus is an immune-mediated disorder driven by type 1 T helper cells and regulated by miR-155. There has been an association with extracellular matrix protein 1, a glycoprotein that is found in the dermal-epidermal basement membrane zone, which provides structural integrity to the skin. Autoantibodies against extracellular matrix protein 1 and other antigens in the basement membrane generally are found in anogenital lichen sclerosus; however, their precise roles in the pathogenesis of lichen sclerosus remains unclear.1
The differential diagnoses for lichen sclerosus include psoriasis, tinea corporis, lichen simplex chronicus, and atopic dermatitis. Psoriasis typically manifests as pink plaques with silver scales on the elbows, knees, and scalp in adult patients.2 Our patient’s white plaques may have suggested psoriasis, but the partially atrophic skin with a wrinkled cigarette paper appearance was not compatible with that diagnosis.
Tinea corporis, a superficial fungal infection of the skin, manifests as circular or ovoid lesions with raised erythematous scaly borders, often with central clearing resembling a ring, that can occur anywhere on the body other than the feet, groin, face, scalp, or beard area.3 The fact that our patient previously had tried topical antifungal medications with no relief and that the skin lesions were atrophic rather than ring shaped made the diagnosis of tinea corporis unlikely.
Lichen simplex chronicus is a chronic condition caused by friction or scratching that is characterized by dry, patchy, scaly, and thickened areas of the skin. Typically affecting the head, arms, neck, scalp, and genital region, lichen simplex chronicus manifests with violaceous or hyperpigmented lesions.4 The nonpruritic atrophic plaques on the inner thighs and the presence of white patches on the vaginal area were not indicative of lichen simplex chronicus in our patient.
Atopic dermatitis manifests as pruritic erythematous scaly papules and plaques with secondary excoriation and possible lichenification. In adults, atopic dermatitis commonly appears on flexural surfaces.2 Atopic dermatitis does not manifest with atrophy and skin wrinkling as seen in our patient.
In the management of lichen sclerosus, the standard treatment is potent topical corticosteroids. Alternatively, topical calcineurin inhibitors can be employed; however, due to the unknown nature of the condition’s underlying cause, targeted treatment is challenging. Our case underscores how lichen sclerosus can be misdiagnosed, highlighting the need for more frequent reporting in the literature to enhance early recognition and reduce delays in patient treatment.
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389 /fmed.2023.1106318
- Chovatiya R, Silverberg JI. Pathophysiology of atopic dermatitis and psoriasis: implications for management in children. Children (Basel). 2019;6:108. doi:10.3390/children6100108
- Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291.
- Ju T, Vander Does A, Mohsin N, et al. Lichen simplex chronicus itch: an update. Acta Derm Venereol. 2022;102:adv00796. doi:10.2340 /actadv.v102.4367
White Atrophic Plaques on the Thighs
White Atrophic Plaques on the Thighs
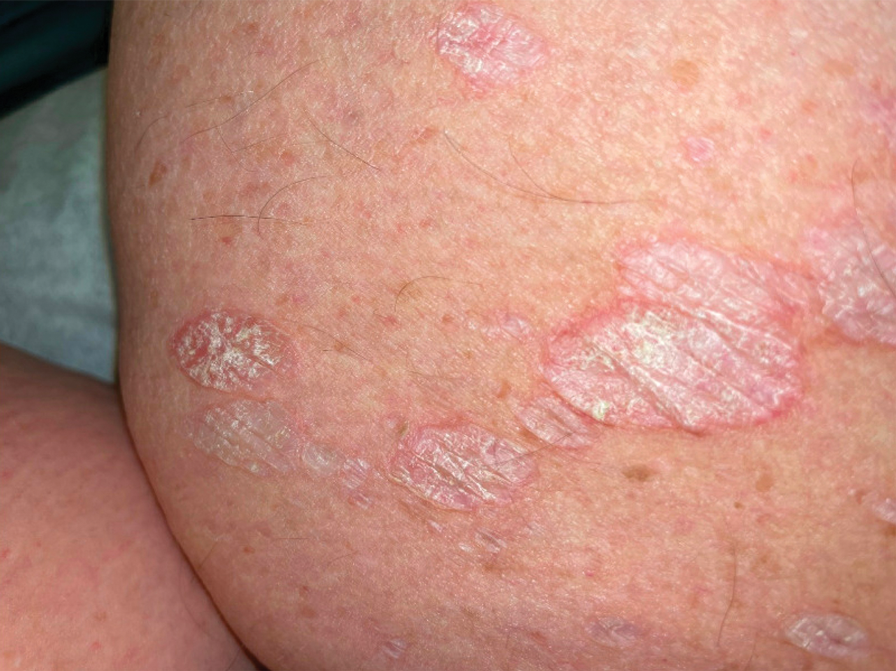
A 71-year-old woman presented to the dermatology clinic for evaluation of intense pruritus of the vaginal region and a nonpruritic rash on the inner thighs of 7 months’ duration. Physical examination revealed white atrophic plaques with scaling and a wrinkled appearance on the inner thighs. White atrophic patches also were noted on the vulva. The patient reported that she had tried over-the-counter antifungals with no improvement. A punch biopsy was performed.

Bilateral Brownish-Red Indurated Facial Plaques in an Adult Man
Bilateral Brownish-Red Indurated Facial Plaques in an Adult Man
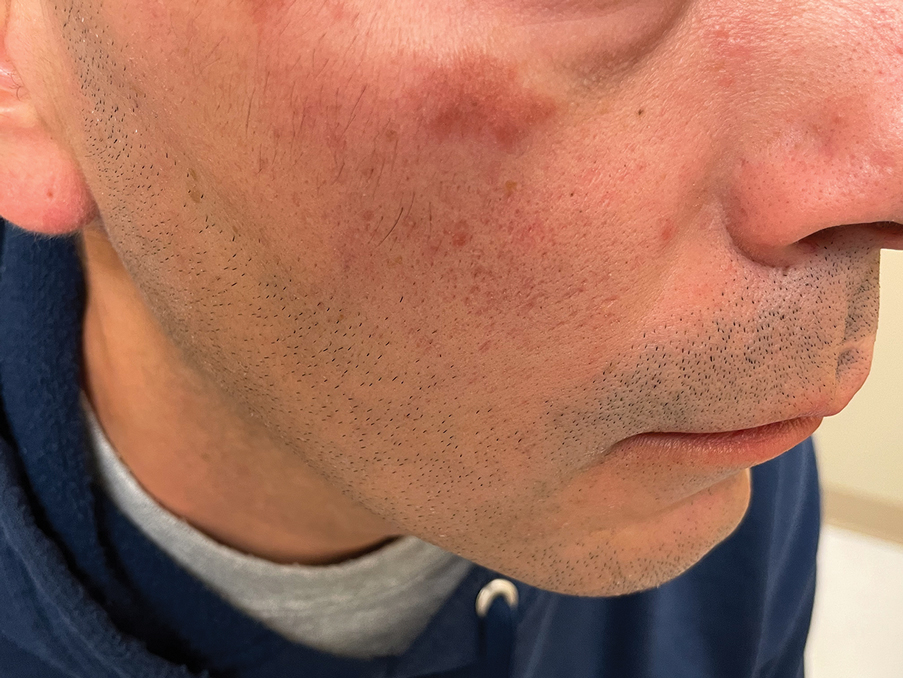
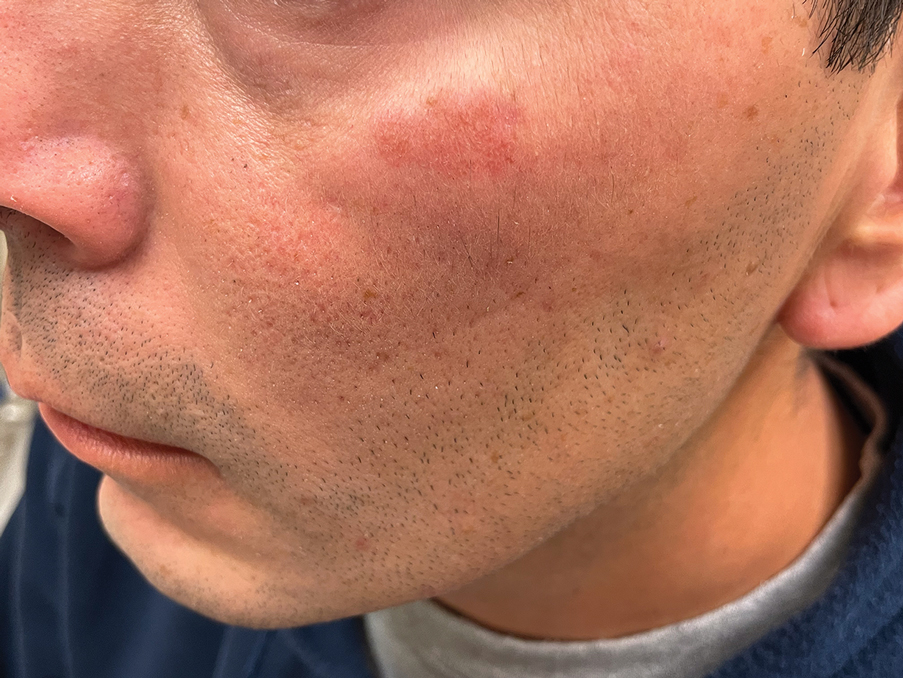
THE DIAGNOSIS: Granuloma Faciale
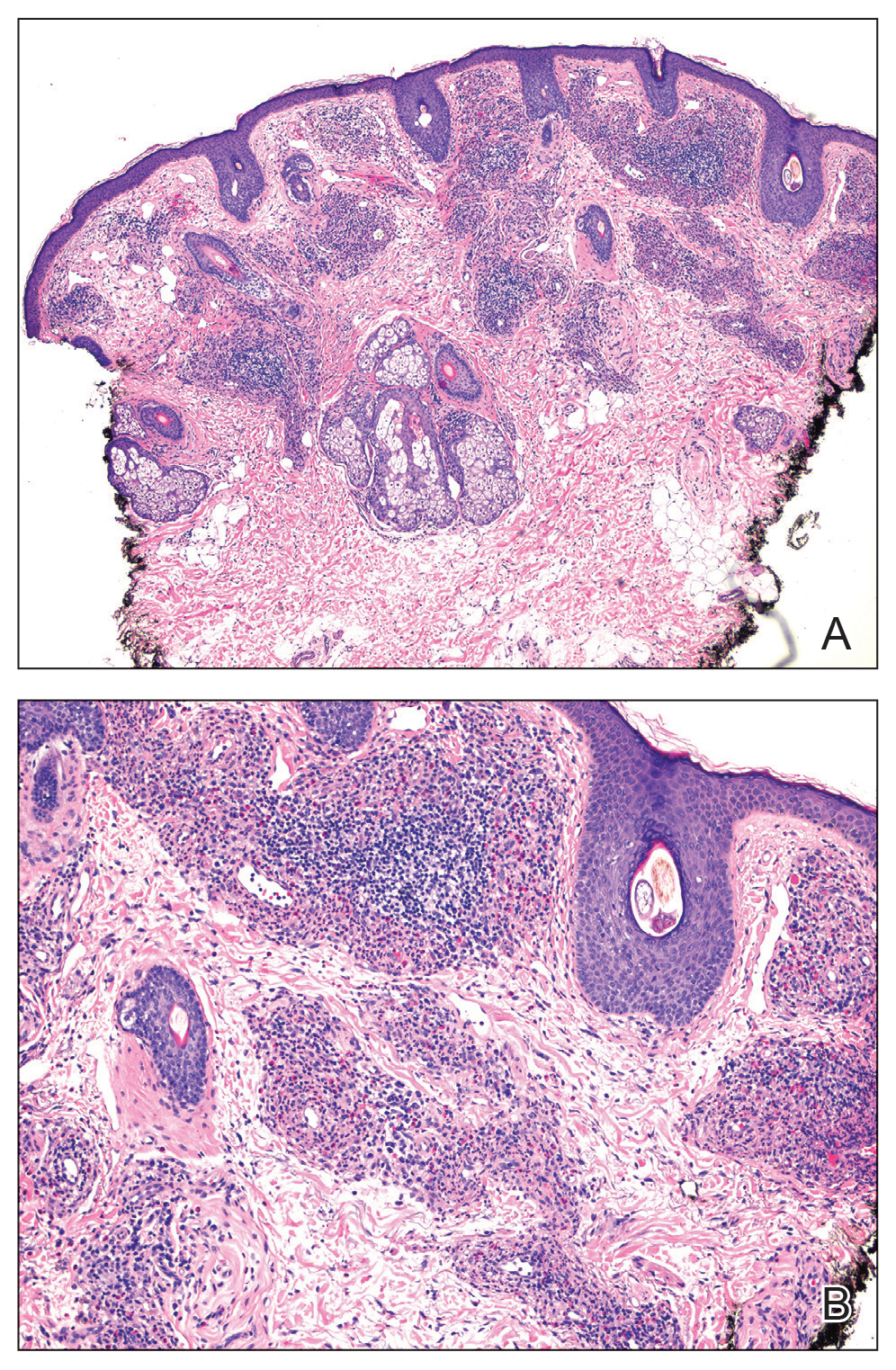
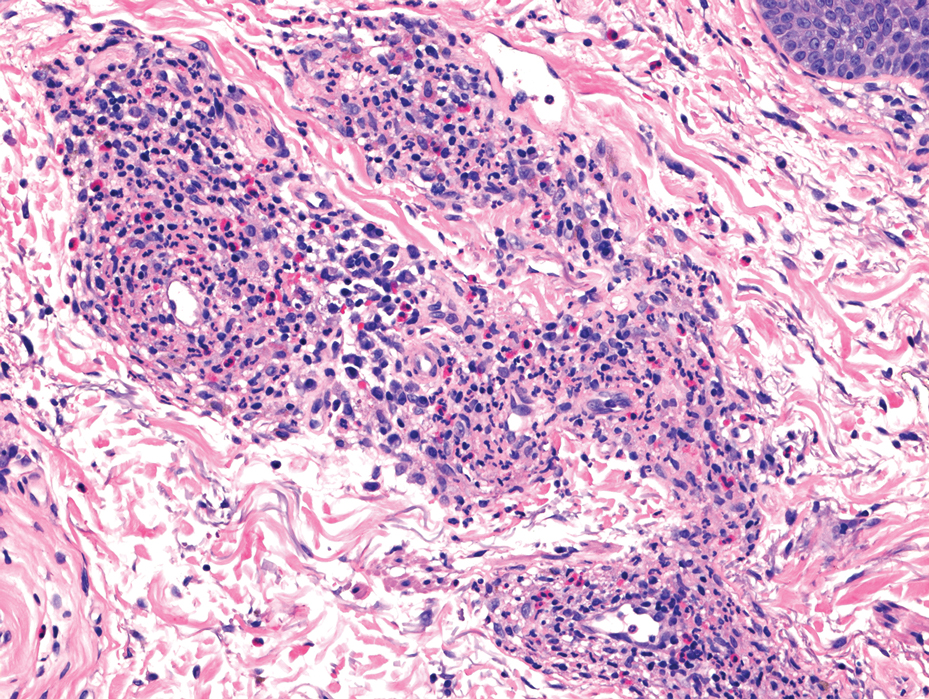
Histology revealed a dense mixed inflammatory cell infiltrate with conspicuous neutrophils and eosinophils in the upper to mid dermis with a narrow uninvolved grenz zone beneath the epidermis (Figures 1 and 2). These findings along with the clinical presentation (Figure 3) were consistent with a diagnosis of granuloma faciale (GF). Most often seen in middle-aged White men, GF is an uncommon localized inflammatory skin condition that often manifests as a single, well-defined, red-to-brown papule, nodule, or plaque on the face or other sun-exposed areas of the skin. Since numerous other skin diseases manifest similarly to GF, biopsy is necessary for definitive diagnosis.1 Histopathology of GF classically shows a mixed inflammatory infiltrate with a narrow band of uninvolved dermis separating it from the epidermis (grenz zone). Dilated follicular plugs and vascular changes frequently are appreciated. Despite its name, GF does not include granulomas and is thought to be similar to leukocytoclastic vasculitis.1 Reports of GF in the literature have shown immunohistochemical staining with the presence of CD4+ lymphocytes that secrete IL-5, a chemotactic agent responsible for attracting eosinophils that contributes to the eosinophilic infiltrate on histology.2
Topical corticosteroids and topical tacrolimus are the first-line treatments for GF. Intralesional corticosteroids also are a treatment option and can be used in combination with cryotherapy.1,3 Additionally, both topical and oral dapsone have been shown to be effective for GF.1 Oral dapsone is given at a dose of 50 mg to 150 mg once daily.1 Clofazimine, typically used as an antileprosy treatment, also has been efficacious in treating GF. Clofazimine has anti-inflammatory and antiproliferative effects on lymphocytes that may attenuate the inflammation underlying GF. It is prescribed at a dose of 300 mg once daily for 3 to 5 months.1
The differential diagnosis for GF is broad and includes tumid lupus erythematosus, Jessner lymphocytic infiltrate (JLI), cutaneous sarcoidosis, and mycosis fungoides. Tumid lupus erythematosus is a subtype of cutaneous lupus erythematosus that rarely is associated with systemic lupus manifestations. Tumid lupus erythematosus manifests as annular, indurated, erythematous plaques, whereas JLI manifests with erythematous papular to nodular lesions without scale on the upper back or face.4 Jessner lymphocytic infiltrate and tumid lupus erythematosus are histopathologically identical, with abundant dermal mucin deposition and a superficial and deep perivascular and periadnexal lymphocytic infiltrate. It is debatable whether JLI is a separate entity or a variant of tumid lupus erythematosus. Sarcoidosis is a granulomatous disease that manifests with a myriad of clinical features. The skin is the second most commonly involved organ.5 The most common morphology is numerous small, firm, nonscaly papules, typically on the face. Histology in cutaneous sarcoidosis will show lymphocyte-poor, noncaseating epithelioid cell granulomas with positive reticulin staining, which were not seen in our patient.6 Lastly, mycosis fungoides is the most common type of cutaneous T-cell lymphoma. It can manifest as patches, plaques, or tumors. The plaque stage may mimic GF as lesions are infiltrative, annular, and raised, with well-defined margins. Histopathology will show intraepidermal lymphocytes out of proportion with spongiosis.7
- Al Dhafiri M, Kaliyadan F. Granuloma faciale. StatPearls Publishing. Updated July 4, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539832/
- Chen A, Harview CL, Rand SE, et al. Refractory granuloma faciale successfully treated with adjunct topical JAK inhibitor. JAAD Case Rep. 2023;33:91-94. doi:10.1016/j.jdcr.2023.01.016
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551. doi:10.1046 /j.1365-4362.1997.00161.x
- Koritala T, Grubbs H, Crane J. Tumid lupus erythematosus. StatPearls Publishing. Updated June 28, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482515/
- Caplan A, Rosenbach M, Imadojemu S. Cutaneous sarcoidosis. Semin Respir Crit Care Med. 2020;41:689-699. doi:10.1055/s-0040-1713130
- Singh P, Jain E, Dhingra H, et al. Clinico-pathological spectrum of cutaneous sarcoidosis: an experience from a government institute in North India. Med Pharm Rep. 2020;93:241-245. doi:10.15386 /mpr-1384
- Vaidya T, Badri T. Mycosis fungoides. StatPearls Publishing. Updated July 31, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519572/
THE DIAGNOSIS: Granuloma Faciale
Histology revealed a dense mixed inflammatory cell infiltrate with conspicuous neutrophils and eosinophils in the upper to mid dermis with a narrow uninvolved grenz zone beneath the epidermis (Figures 1 and 2). These findings along with the clinical presentation (Figure 3) were consistent with a diagnosis of granuloma faciale (GF). Most often seen in middle-aged White men, GF is an uncommon localized inflammatory skin condition that often manifests as a single, well-defined, red-to-brown papule, nodule, or plaque on the face or other sun-exposed areas of the skin. Since numerous other skin diseases manifest similarly to GF, biopsy is necessary for definitive diagnosis.1 Histopathology of GF classically shows a mixed inflammatory infiltrate with a narrow band of uninvolved dermis separating it from the epidermis (grenz zone). Dilated follicular plugs and vascular changes frequently are appreciated. Despite its name, GF does not include granulomas and is thought to be similar to leukocytoclastic vasculitis.1 Reports of GF in the literature have shown immunohistochemical staining with the presence of CD4+ lymphocytes that secrete IL-5, a chemotactic agent responsible for attracting eosinophils that contributes to the eosinophilic infiltrate on histology.2
Topical corticosteroids and topical tacrolimus are the first-line treatments for GF. Intralesional corticosteroids also are a treatment option and can be used in combination with cryotherapy.1,3 Additionally, both topical and oral dapsone have been shown to be effective for GF.1 Oral dapsone is given at a dose of 50 mg to 150 mg once daily.1 Clofazimine, typically used as an antileprosy treatment, also has been efficacious in treating GF. Clofazimine has anti-inflammatory and antiproliferative effects on lymphocytes that may attenuate the inflammation underlying GF. It is prescribed at a dose of 300 mg once daily for 3 to 5 months.1
The differential diagnosis for GF is broad and includes tumid lupus erythematosus, Jessner lymphocytic infiltrate (JLI), cutaneous sarcoidosis, and mycosis fungoides. Tumid lupus erythematosus is a subtype of cutaneous lupus erythematosus that rarely is associated with systemic lupus manifestations. Tumid lupus erythematosus manifests as annular, indurated, erythematous plaques, whereas JLI manifests with erythematous papular to nodular lesions without scale on the upper back or face.4 Jessner lymphocytic infiltrate and tumid lupus erythematosus are histopathologically identical, with abundant dermal mucin deposition and a superficial and deep perivascular and periadnexal lymphocytic infiltrate. It is debatable whether JLI is a separate entity or a variant of tumid lupus erythematosus. Sarcoidosis is a granulomatous disease that manifests with a myriad of clinical features. The skin is the second most commonly involved organ.5 The most common morphology is numerous small, firm, nonscaly papules, typically on the face. Histology in cutaneous sarcoidosis will show lymphocyte-poor, noncaseating epithelioid cell granulomas with positive reticulin staining, which were not seen in our patient.6 Lastly, mycosis fungoides is the most common type of cutaneous T-cell lymphoma. It can manifest as patches, plaques, or tumors. The plaque stage may mimic GF as lesions are infiltrative, annular, and raised, with well-defined margins. Histopathology will show intraepidermal lymphocytes out of proportion with spongiosis.7
THE DIAGNOSIS: Granuloma Faciale
Histology revealed a dense mixed inflammatory cell infiltrate with conspicuous neutrophils and eosinophils in the upper to mid dermis with a narrow uninvolved grenz zone beneath the epidermis (Figures 1 and 2). These findings along with the clinical presentation (Figure 3) were consistent with a diagnosis of granuloma faciale (GF). Most often seen in middle-aged White men, GF is an uncommon localized inflammatory skin condition that often manifests as a single, well-defined, red-to-brown papule, nodule, or plaque on the face or other sun-exposed areas of the skin. Since numerous other skin diseases manifest similarly to GF, biopsy is necessary for definitive diagnosis.1 Histopathology of GF classically shows a mixed inflammatory infiltrate with a narrow band of uninvolved dermis separating it from the epidermis (grenz zone). Dilated follicular plugs and vascular changes frequently are appreciated. Despite its name, GF does not include granulomas and is thought to be similar to leukocytoclastic vasculitis.1 Reports of GF in the literature have shown immunohistochemical staining with the presence of CD4+ lymphocytes that secrete IL-5, a chemotactic agent responsible for attracting eosinophils that contributes to the eosinophilic infiltrate on histology.2
Topical corticosteroids and topical tacrolimus are the first-line treatments for GF. Intralesional corticosteroids also are a treatment option and can be used in combination with cryotherapy.1,3 Additionally, both topical and oral dapsone have been shown to be effective for GF.1 Oral dapsone is given at a dose of 50 mg to 150 mg once daily.1 Clofazimine, typically used as an antileprosy treatment, also has been efficacious in treating GF. Clofazimine has anti-inflammatory and antiproliferative effects on lymphocytes that may attenuate the inflammation underlying GF. It is prescribed at a dose of 300 mg once daily for 3 to 5 months.1
The differential diagnosis for GF is broad and includes tumid lupus erythematosus, Jessner lymphocytic infiltrate (JLI), cutaneous sarcoidosis, and mycosis fungoides. Tumid lupus erythematosus is a subtype of cutaneous lupus erythematosus that rarely is associated with systemic lupus manifestations. Tumid lupus erythematosus manifests as annular, indurated, erythematous plaques, whereas JLI manifests with erythematous papular to nodular lesions without scale on the upper back or face.4 Jessner lymphocytic infiltrate and tumid lupus erythematosus are histopathologically identical, with abundant dermal mucin deposition and a superficial and deep perivascular and periadnexal lymphocytic infiltrate. It is debatable whether JLI is a separate entity or a variant of tumid lupus erythematosus. Sarcoidosis is a granulomatous disease that manifests with a myriad of clinical features. The skin is the second most commonly involved organ.5 The most common morphology is numerous small, firm, nonscaly papules, typically on the face. Histology in cutaneous sarcoidosis will show lymphocyte-poor, noncaseating epithelioid cell granulomas with positive reticulin staining, which were not seen in our patient.6 Lastly, mycosis fungoides is the most common type of cutaneous T-cell lymphoma. It can manifest as patches, plaques, or tumors. The plaque stage may mimic GF as lesions are infiltrative, annular, and raised, with well-defined margins. Histopathology will show intraepidermal lymphocytes out of proportion with spongiosis.7
- Al Dhafiri M, Kaliyadan F. Granuloma faciale. StatPearls Publishing. Updated July 4, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539832/
- Chen A, Harview CL, Rand SE, et al. Refractory granuloma faciale successfully treated with adjunct topical JAK inhibitor. JAAD Case Rep. 2023;33:91-94. doi:10.1016/j.jdcr.2023.01.016
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551. doi:10.1046 /j.1365-4362.1997.00161.x
- Koritala T, Grubbs H, Crane J. Tumid lupus erythematosus. StatPearls Publishing. Updated June 28, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482515/
- Caplan A, Rosenbach M, Imadojemu S. Cutaneous sarcoidosis. Semin Respir Crit Care Med. 2020;41:689-699. doi:10.1055/s-0040-1713130
- Singh P, Jain E, Dhingra H, et al. Clinico-pathological spectrum of cutaneous sarcoidosis: an experience from a government institute in North India. Med Pharm Rep. 2020;93:241-245. doi:10.15386 /mpr-1384
- Vaidya T, Badri T. Mycosis fungoides. StatPearls Publishing. Updated July 31, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519572/
- Al Dhafiri M, Kaliyadan F. Granuloma faciale. StatPearls Publishing. Updated July 4, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539832/
- Chen A, Harview CL, Rand SE, et al. Refractory granuloma faciale successfully treated with adjunct topical JAK inhibitor. JAAD Case Rep. 2023;33:91-94. doi:10.1016/j.jdcr.2023.01.016
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551. doi:10.1046 /j.1365-4362.1997.00161.x
- Koritala T, Grubbs H, Crane J. Tumid lupus erythematosus. StatPearls Publishing. Updated June 28, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482515/
- Caplan A, Rosenbach M, Imadojemu S. Cutaneous sarcoidosis. Semin Respir Crit Care Med. 2020;41:689-699. doi:10.1055/s-0040-1713130
- Singh P, Jain E, Dhingra H, et al. Clinico-pathological spectrum of cutaneous sarcoidosis: an experience from a government institute in North India. Med Pharm Rep. 2020;93:241-245. doi:10.15386 /mpr-1384
- Vaidya T, Badri T. Mycosis fungoides. StatPearls Publishing. Updated July 31, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519572/
Bilateral Brownish-Red Indurated Facial Plaques in an Adult Man
Bilateral Brownish-Red Indurated Facial Plaques in an Adult Man
A 44-year-old man presented to the dermatology clinic with a facial rash of 2 years’ duration. The patient reported associated pruritus but no systemic symptoms. His medical history was relevant for childhood eczema. He had tried various over-the-counter treatments for the facial rash, including topical hydrocortisone, neomycin/bacitracin/polymyxin antibiotic ointment, moisturizers, and antihistamines, with no success. Physical examination demonstrated symmetric, well-circumscribed, circinate, brownish-red, indurated plaques without scaling on the cheeks. A 4-mm punch biopsy was obtained from a plaque on the left cheek.

Painful Plaque on the Forearm
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
A 30-year-old woman presented to the dermatology clinic with lesions on the right forearm of 2 years’ duration. Her medical history was unremarkable. She reported working as a chef and caring for multiple pets in her home, including 3 cats, 6 fish tanks, 3 dogs, and 3 lizards. Physical examination revealed a painful, indurated, red-violaceous plaque on the right forearm with satellite pink nodules that had been slowly migrating proximally up the forearm. An outside excisional biopsy performed 1 year prior had shown suppurative granulomatous dermatitis with negative stains for infectious organisms and negative tissue cultures. At that time, the patient was diagnosed with ruptured folliculitis; however, a subsequent lack of clinical improvement prompted her to seek a second opinion at our clinic.
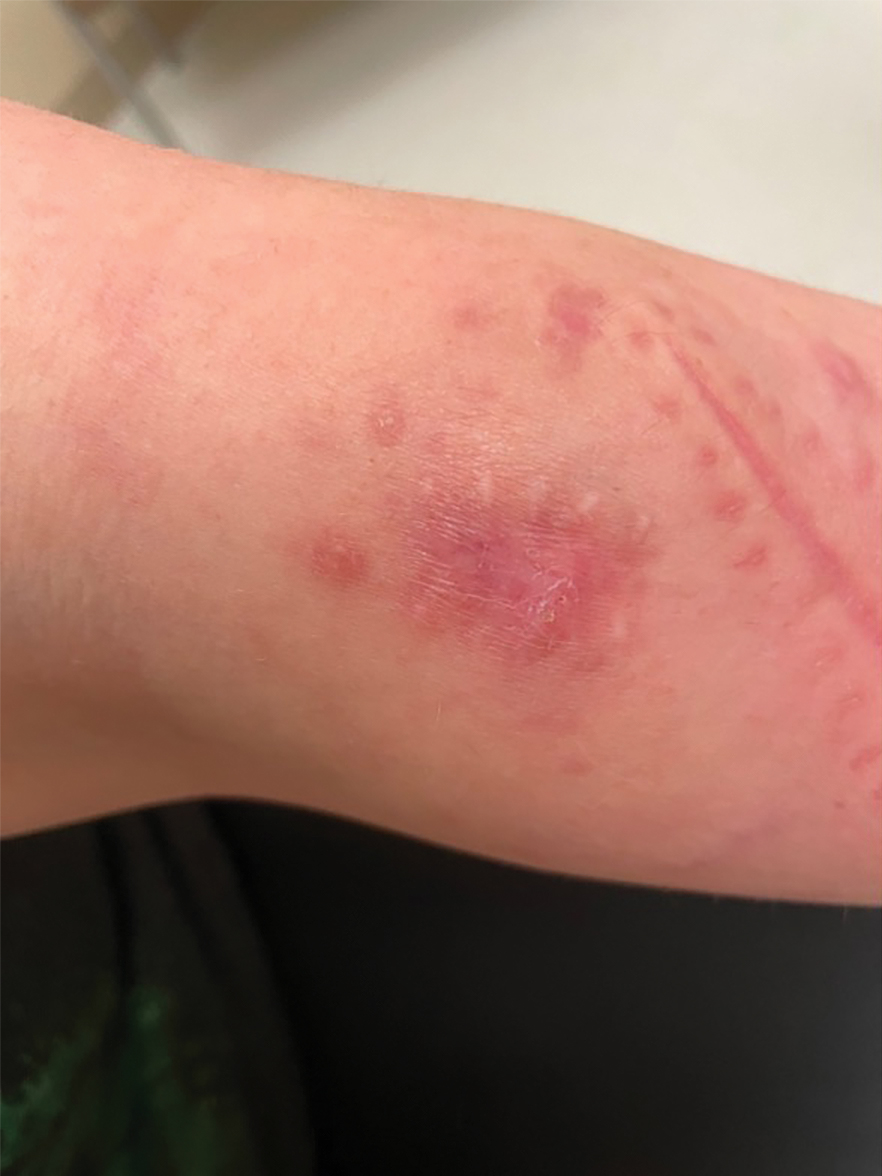
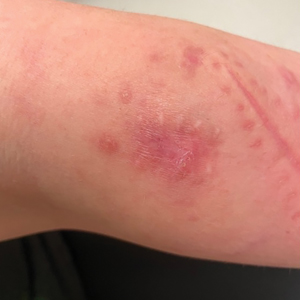
Erythematous Plaques on the Dorsal Aspect of the Hand
The Diagnosis: Majocchi Granuloma
Histopathology showed rare follicular-based organisms highlighted by periodic acid–Schiff staining. This finding along with her use of clobetasol ointment on the hands led to a diagnosis of Majocchi granuloma in our patient. Clobetasol and crisaborole ointments were discontinued, and she was started on oral terbinafine 250 mg daily for 4 weeks, which resulted in resolution of the rash.
Majocchi granuloma (also known as nodular granulomatous perifolliculitis) is a perifollicular granulomatous process caused by a dermatophyte infection of the hair follicles. Trichophyton rubrum is the most commonly implicated organism, followed by Trichophyton mentagrophytes and Epidermophyton floccosum, which also cause tinea corporis and tinea pedis.1 This condition most commonly occurs in women aged 20 to 35 years. Risk factors include trauma, occlusion of the hair follicles, immunosuppression, and use of potent topical corticosteroids in patients with tinea.2,3 Immunocompetent patients present with perifollicular papules or pustules with erythematous scaly plaques on the extremities, while immunocompromised patients may have subcutaneous nodules or abscesses on any hair-bearing parts of the body.3
Majocchi granuloma is considered a dermal fungal infection in which the disruption of hair follicles from occlusion or trauma allows fungal organisms and keratinaceous material substrates to be introduced into the dermis. The differential diagnosis is based on the types of presenting lesions. The papules of Majocchi granuloma can resemble folliculitis, acne, or insect bites, while nodules can resemble erythema nodosum or furunculosis.4 Plaques, such as those seen in our patient, can mimic cellulitis and allergic or irritant contact dermatitis.4 Additionally, the plaques may appear annular or figurate, which may resemble erythema gyratum repens or erythema annulare centrifugum.
The diagnosis of Majocchi granuloma often requires fungal culture and biopsy because a potassium hydroxide preparation is unable to distinguish between superficial and invasive dermatophytes.3 Histopathology will show perifollicular granulomatous inflammation. Fungal elements can be detected with periodic acid–Schiff or Grocott-Gomori methenamine-silver staining of the hairs and hair follicles as well as dermal infiltrates.4
Topical corticosteroids should be discontinued. Systemic antifungals are the treatment of choice for Majocchi granuloma, as topical antifungals are not effective against deep fungal infections. Although there are no standard guidelines on duration or dosage, recommended regimens in immunocompetent patients include terbinafine 250 mg/d for 4 weeks; itraconazole pulse therapy consisting of 200 mg twice daily for 1 week with 2 weeks off therapy, then repeat the cycle for a total of 2 to 3 pulses; and griseofulvin 500 mg twice daily for 8 to 12 weeks (Table).3 For immunocompromised patients, combination therapy with more than one antifungal may be necessary.

- James WD, Berger T, Elston DM. Diseases resulting from fungi and yeasts. In: James WD, Berger T, Elston D, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Saunders Elsevier; 2016:285-318.
- Li FQ, Lv S, Xia JX. Majocchi’s granuloma after topical corticosteroids therapy. Case Rep Dermatol Med. 2014;2014:507176.
- Boral H, Durdu M, Ilkit M. Majocchi’s granuloma: current perspectives. Infect Drug Resist. 2018;11:751-760.
- I˙lkit M, Durdu M, Karakas¸ M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
The Diagnosis: Majocchi Granuloma
Histopathology showed rare follicular-based organisms highlighted by periodic acid–Schiff staining. This finding along with her use of clobetasol ointment on the hands led to a diagnosis of Majocchi granuloma in our patient. Clobetasol and crisaborole ointments were discontinued, and she was started on oral terbinafine 250 mg daily for 4 weeks, which resulted in resolution of the rash.
Majocchi granuloma (also known as nodular granulomatous perifolliculitis) is a perifollicular granulomatous process caused by a dermatophyte infection of the hair follicles. Trichophyton rubrum is the most commonly implicated organism, followed by Trichophyton mentagrophytes and Epidermophyton floccosum, which also cause tinea corporis and tinea pedis.1 This condition most commonly occurs in women aged 20 to 35 years. Risk factors include trauma, occlusion of the hair follicles, immunosuppression, and use of potent topical corticosteroids in patients with tinea.2,3 Immunocompetent patients present with perifollicular papules or pustules with erythematous scaly plaques on the extremities, while immunocompromised patients may have subcutaneous nodules or abscesses on any hair-bearing parts of the body.3
Majocchi granuloma is considered a dermal fungal infection in which the disruption of hair follicles from occlusion or trauma allows fungal organisms and keratinaceous material substrates to be introduced into the dermis. The differential diagnosis is based on the types of presenting lesions. The papules of Majocchi granuloma can resemble folliculitis, acne, or insect bites, while nodules can resemble erythema nodosum or furunculosis.4 Plaques, such as those seen in our patient, can mimic cellulitis and allergic or irritant contact dermatitis.4 Additionally, the plaques may appear annular or figurate, which may resemble erythema gyratum repens or erythema annulare centrifugum.
The diagnosis of Majocchi granuloma often requires fungal culture and biopsy because a potassium hydroxide preparation is unable to distinguish between superficial and invasive dermatophytes.3 Histopathology will show perifollicular granulomatous inflammation. Fungal elements can be detected with periodic acid–Schiff or Grocott-Gomori methenamine-silver staining of the hairs and hair follicles as well as dermal infiltrates.4
Topical corticosteroids should be discontinued. Systemic antifungals are the treatment of choice for Majocchi granuloma, as topical antifungals are not effective against deep fungal infections. Although there are no standard guidelines on duration or dosage, recommended regimens in immunocompetent patients include terbinafine 250 mg/d for 4 weeks; itraconazole pulse therapy consisting of 200 mg twice daily for 1 week with 2 weeks off therapy, then repeat the cycle for a total of 2 to 3 pulses; and griseofulvin 500 mg twice daily for 8 to 12 weeks (Table).3 For immunocompromised patients, combination therapy with more than one antifungal may be necessary.
The Diagnosis: Majocchi Granuloma
Histopathology showed rare follicular-based organisms highlighted by periodic acid–Schiff staining. This finding along with her use of clobetasol ointment on the hands led to a diagnosis of Majocchi granuloma in our patient. Clobetasol and crisaborole ointments were discontinued, and she was started on oral terbinafine 250 mg daily for 4 weeks, which resulted in resolution of the rash.
Majocchi granuloma (also known as nodular granulomatous perifolliculitis) is a perifollicular granulomatous process caused by a dermatophyte infection of the hair follicles. Trichophyton rubrum is the most commonly implicated organism, followed by Trichophyton mentagrophytes and Epidermophyton floccosum, which also cause tinea corporis and tinea pedis.1 This condition most commonly occurs in women aged 20 to 35 years. Risk factors include trauma, occlusion of the hair follicles, immunosuppression, and use of potent topical corticosteroids in patients with tinea.2,3 Immunocompetent patients present with perifollicular papules or pustules with erythematous scaly plaques on the extremities, while immunocompromised patients may have subcutaneous nodules or abscesses on any hair-bearing parts of the body.3
Majocchi granuloma is considered a dermal fungal infection in which the disruption of hair follicles from occlusion or trauma allows fungal organisms and keratinaceous material substrates to be introduced into the dermis. The differential diagnosis is based on the types of presenting lesions. The papules of Majocchi granuloma can resemble folliculitis, acne, or insect bites, while nodules can resemble erythema nodosum or furunculosis.4 Plaques, such as those seen in our patient, can mimic cellulitis and allergic or irritant contact dermatitis.4 Additionally, the plaques may appear annular or figurate, which may resemble erythema gyratum repens or erythema annulare centrifugum.
The diagnosis of Majocchi granuloma often requires fungal culture and biopsy because a potassium hydroxide preparation is unable to distinguish between superficial and invasive dermatophytes.3 Histopathology will show perifollicular granulomatous inflammation. Fungal elements can be detected with periodic acid–Schiff or Grocott-Gomori methenamine-silver staining of the hairs and hair follicles as well as dermal infiltrates.4
Topical corticosteroids should be discontinued. Systemic antifungals are the treatment of choice for Majocchi granuloma, as topical antifungals are not effective against deep fungal infections. Although there are no standard guidelines on duration or dosage, recommended regimens in immunocompetent patients include terbinafine 250 mg/d for 4 weeks; itraconazole pulse therapy consisting of 200 mg twice daily for 1 week with 2 weeks off therapy, then repeat the cycle for a total of 2 to 3 pulses; and griseofulvin 500 mg twice daily for 8 to 12 weeks (Table).3 For immunocompromised patients, combination therapy with more than one antifungal may be necessary.
- James WD, Berger T, Elston DM. Diseases resulting from fungi and yeasts. In: James WD, Berger T, Elston D, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Saunders Elsevier; 2016:285-318.
- Li FQ, Lv S, Xia JX. Majocchi’s granuloma after topical corticosteroids therapy. Case Rep Dermatol Med. 2014;2014:507176.
- Boral H, Durdu M, Ilkit M. Majocchi’s granuloma: current perspectives. Infect Drug Resist. 2018;11:751-760.
- I˙lkit M, Durdu M, Karakas¸ M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- James WD, Berger T, Elston DM. Diseases resulting from fungi and yeasts. In: James WD, Berger T, Elston D, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Saunders Elsevier; 2016:285-318.
- Li FQ, Lv S, Xia JX. Majocchi’s granuloma after topical corticosteroids therapy. Case Rep Dermatol Med. 2014;2014:507176.
- Boral H, Durdu M, Ilkit M. Majocchi’s granuloma: current perspectives. Infect Drug Resist. 2018;11:751-760.
- I˙lkit M, Durdu M, Karakas¸ M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
A 33-year-old woman presented with an asymptomatic rash on the left hand that was suspected by her primary care physician to be a flare of hand dermatitis. The patient had a history of irritant hand dermatitis diagnosed 2 years prior that was suspected to be secondary to frequent handwashing and was well controlled with clobetasol and crisaborole ointments for 1 year. Four months prior to the current presentation, she developed a flare that was refractory to these topical therapies; treatment with biweekly dupilumab 300 mg was initiated by dermatology, but the rash continued to evolve. A punch biopsy was performed to confirm the diagnosis.


Diffuse Papular Eruption With Erosions and Ulcerations
The Diagnosis: Immunotherapy-Related Lichenoid Drug Eruption
Direct immunofluorescence was negative, and histopathology revealed a lichenoid interface dermatitis, minimal parakeratosis, and saw-toothed rete ridges (Figure 1). He was diagnosed with an immunotherapyrelated lichenoid drug eruption based on the morphology of the skin lesions and clinicopathologic correlation. Bullous pemphigoid and lichen planus pemphigoides were ruled out given the negative direct immunofluorescence findings. Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) was not consistent with the clinical presentation, especially given the lack of mucosal findings. The histology also was not consistent, as the biopsy specimen lacked apoptotic and necrotic keratinocytes to the degree seen in SJS/TEN and also had a greater degree of inflammatory infiltrate. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome was ruled out given the lack of systemic findings, including facial swelling and lymphadenopathy and the clinical appearance of the rash. No morbilliform features were present, which is the most common presentation of DRESS syndrome.
.")
Checkpoint inhibitor (CPI) therapy has become the cornerstone in management of certain advanced malignancies.1 Checkpoint inhibitors block cytotoxic T lymphocyte–associated protein 4, programmed cell death-1, and/or programmed cell death ligand-1, allowing activated T cells to infiltrate the tumor microenvironment and destroy malignant cells. Checkpoint inhibitors are approved for the treatment of melanoma, cutaneous squamous cell carcinoma, and Merkel cell carcinoma and are being investigated in various other cutaneous and soft tissue malignancies.1-3
Although CPIs have shown substantial efficacy in the management of advanced malignancies, immune-related adverse events (AEs) are common due to nonspecific immune activation.2 Immune-related cutaneous AEs are the most common immune-related AEs, occurring in 30% to 50% of patients who undergo treatment.2-5 Common immune-related cutaneous AEs include maculopapular, psoriasiform, and lichenoid dermatitis, as well as pruritus without dermatitis.2,3,6 Other reactions include but are not limited to bullous pemphigoid, vitiligolike depigmentation, and alopecia.2,3 Immune-related cutaneous AEs usually are self-limited; however, severe life-threatening reactions such as the spectrum of SJS/TEN and DRESS syndrome also can occur.2-4 Immune-related cutaneous AEs are graded based on the Common Terminology Criteria for Adverse Events: grade 1 reactions are asymptomatic and cover less than 10% of the patient’s body surface area (BSA), grade 2 reactions have mild symptoms and cover 10% to 30% of the patient’s BSA, grade 3 reactions have moderate to severe symptoms and cover greater than 30% of the patient’s BSA, and grade 4 reactions are life-threatening.2,3 With prompt recognition and adequate treatment, mild to moderate immune-related cutaneous AEs—grades 1 and 2—largely are reversible, and less than 5% require discontinuation of therapy.2,3,6 It has been suggested that immune-related cutaneous AEs may be a positive prognostic factor in the treatment of underlying malignancy, indicating adequate immune activation targeting the malignant cells.6
Although our patient had some typical violaceous, flat-topped papules and plaques with Wickham striae, he also had atypical findings for a lichenoid reaction. Given the endorsement of blisters, it is possible that some of these lesions initially were bullous and subsequently ruptured, leaving behind erosions. However, in other areas, there also were eroded papules and ulcerations without a reported history of excoriation, scratching, picking, or prior bullae, including difficult-to-reach areas such as the back. It is favored that these lesions represented a robust lichenoid dermatitis leading to erosive and ulcerated lesions, similar to the formation of bullous lichen planus. Lichenoid eruptions secondary to immunotherapy are well-known phenomena, but a PubMed search of articles indexed for MEDLINE using the terms ulcer, lichenoid, and immunotherapy revealed only 2 cases of ulcerative lichenoid eruptions: a localized digital erosive lichenoid dermatitis and a widespread ulcerative lichenoid drug eruption without true erosions.7,8 However, widespread erosive and ulcerated lichenoid reactions are rare.
Lichenoid eruptions most strongly are associated with anti–programmed cell death-1/ programmed cell death ligand-1 therapy, occurring in 20% of patients undergoing treatment.3 Lichenoid eruptions present as discrete, pruritic, erythematous, violaceous papules and plaques on the chest and back and rarely may involve the limbs, palmoplantar surfaces, and oral mucosa.2,3,6 Histopathologic features include a dense bandlike lymphocytic infiltrate in the dermis with scattered apoptotic keratinocytes in the basal layer of the epidermis.2,4,6 Grades 1 to 2 lesions can be managed with high-potency topical corticosteroids without CPI dose interruption, with more extensive grade 2 lesions requiring systemic corticosteroids.2,6,9 Lichenoid eruptions grade 3 or higher also require systemic corticosteroid therapy CPI therapy cessation until the eruption has receded to grade 0 to 1.2 Alternative treatment options for high-grade toxicity include phototherapy and acitretin.2,4,9
Our patient was treated with cessation of immunotherapy and initiation of a systemic corticosteroid taper, acitretin, and narrowband UVB therapy. After 6 weeks of treatment, the pain and pruritus improved and the rash had resolved in some areas while it had taken on a more classic lichenoid appearance with violaceous scaly papules and plaques (Figure 2) in areas of prior ulcers and erosions. He no longer had any bullae, erosions, or ulcers.

- Barrios DM, Do MH, Phillips GS, et al. Immune checkpoint inhibitors to treat cutaneous malignancies. J Am Acad Dermatol. 2020;83:1239-1253. doi:10.1016/j.jaad.2020.03.131
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Tattersall IW, Leventhal JS. Cutaneous toxicities of immune checkpoint inhibitors: the role of the dermatologist. Yale J Biol Med. 2020;93:123-132.
- Si X, He C, Zhang L, et al. Management of immune checkpoint inhibitor-related dermatologic adverse events. Thorac Cancer. 2020;11:488-492. doi:10.1111/1759-7714.13275
- Eggermont AMM, Kicinski M, Blank CU, et al. Association between immune-related adverse events and recurrence-free survival among patients with stage III melanoma randomized to receive pembrolizumab or placebo: a secondary analysis of a randomized clinical trial. JAMA Oncol. 2020;6:519-527. doi:10.1001 /jamaoncol.2019.5570
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263. doi:10.1097/CCO.0000000000000290
- Martínez-Doménech Á, García-Legaz Martínez M, Magdaleno-Tapial J, et al. Digital ulcerative lichenoid dermatitis in a patient receiving anti-PD-1 therapy. Dermatol Online J. 2019;25:13030/qt8sm0j7t7.
- Davis MJ, Wilken R, Fung MA, et al. Debilitating erosive lichenoid interface dermatitis from checkpoint inhibitor therapy. Dermatol Online J. 2018;24:13030/qt3vq6b04v.
- Apalla Z, Papageorgiou C, Lallas A, et al. Cutaneous adverse events of immune checkpoint inhibitors: a literature review [published online January 29, 2021]. Dermatol Pract Concept. 2021;11:E2021155. doi:10.5826/dpc.1101a155
The Diagnosis: Immunotherapy-Related Lichenoid Drug Eruption
Direct immunofluorescence was negative, and histopathology revealed a lichenoid interface dermatitis, minimal parakeratosis, and saw-toothed rete ridges (Figure 1). He was diagnosed with an immunotherapyrelated lichenoid drug eruption based on the morphology of the skin lesions and clinicopathologic correlation. Bullous pemphigoid and lichen planus pemphigoides were ruled out given the negative direct immunofluorescence findings. Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) was not consistent with the clinical presentation, especially given the lack of mucosal findings. The histology also was not consistent, as the biopsy specimen lacked apoptotic and necrotic keratinocytes to the degree seen in SJS/TEN and also had a greater degree of inflammatory infiltrate. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome was ruled out given the lack of systemic findings, including facial swelling and lymphadenopathy and the clinical appearance of the rash. No morbilliform features were present, which is the most common presentation of DRESS syndrome.
Checkpoint inhibitor (CPI) therapy has become the cornerstone in management of certain advanced malignancies.1 Checkpoint inhibitors block cytotoxic T lymphocyte–associated protein 4, programmed cell death-1, and/or programmed cell death ligand-1, allowing activated T cells to infiltrate the tumor microenvironment and destroy malignant cells. Checkpoint inhibitors are approved for the treatment of melanoma, cutaneous squamous cell carcinoma, and Merkel cell carcinoma and are being investigated in various other cutaneous and soft tissue malignancies.1-3
Although CPIs have shown substantial efficacy in the management of advanced malignancies, immune-related adverse events (AEs) are common due to nonspecific immune activation.2 Immune-related cutaneous AEs are the most common immune-related AEs, occurring in 30% to 50% of patients who undergo treatment.2-5 Common immune-related cutaneous AEs include maculopapular, psoriasiform, and lichenoid dermatitis, as well as pruritus without dermatitis.2,3,6 Other reactions include but are not limited to bullous pemphigoid, vitiligolike depigmentation, and alopecia.2,3 Immune-related cutaneous AEs usually are self-limited; however, severe life-threatening reactions such as the spectrum of SJS/TEN and DRESS syndrome also can occur.2-4 Immune-related cutaneous AEs are graded based on the Common Terminology Criteria for Adverse Events: grade 1 reactions are asymptomatic and cover less than 10% of the patient’s body surface area (BSA), grade 2 reactions have mild symptoms and cover 10% to 30% of the patient’s BSA, grade 3 reactions have moderate to severe symptoms and cover greater than 30% of the patient’s BSA, and grade 4 reactions are life-threatening.2,3 With prompt recognition and adequate treatment, mild to moderate immune-related cutaneous AEs—grades 1 and 2—largely are reversible, and less than 5% require discontinuation of therapy.2,3,6 It has been suggested that immune-related cutaneous AEs may be a positive prognostic factor in the treatment of underlying malignancy, indicating adequate immune activation targeting the malignant cells.6
Although our patient had some typical violaceous, flat-topped papules and plaques with Wickham striae, he also had atypical findings for a lichenoid reaction. Given the endorsement of blisters, it is possible that some of these lesions initially were bullous and subsequently ruptured, leaving behind erosions. However, in other areas, there also were eroded papules and ulcerations without a reported history of excoriation, scratching, picking, or prior bullae, including difficult-to-reach areas such as the back. It is favored that these lesions represented a robust lichenoid dermatitis leading to erosive and ulcerated lesions, similar to the formation of bullous lichen planus. Lichenoid eruptions secondary to immunotherapy are well-known phenomena, but a PubMed search of articles indexed for MEDLINE using the terms ulcer, lichenoid, and immunotherapy revealed only 2 cases of ulcerative lichenoid eruptions: a localized digital erosive lichenoid dermatitis and a widespread ulcerative lichenoid drug eruption without true erosions.7,8 However, widespread erosive and ulcerated lichenoid reactions are rare.
Lichenoid eruptions most strongly are associated with anti–programmed cell death-1/ programmed cell death ligand-1 therapy, occurring in 20% of patients undergoing treatment.3 Lichenoid eruptions present as discrete, pruritic, erythematous, violaceous papules and plaques on the chest and back and rarely may involve the limbs, palmoplantar surfaces, and oral mucosa.2,3,6 Histopathologic features include a dense bandlike lymphocytic infiltrate in the dermis with scattered apoptotic keratinocytes in the basal layer of the epidermis.2,4,6 Grades 1 to 2 lesions can be managed with high-potency topical corticosteroids without CPI dose interruption, with more extensive grade 2 lesions requiring systemic corticosteroids.2,6,9 Lichenoid eruptions grade 3 or higher also require systemic corticosteroid therapy CPI therapy cessation until the eruption has receded to grade 0 to 1.2 Alternative treatment options for high-grade toxicity include phototherapy and acitretin.2,4,9
Our patient was treated with cessation of immunotherapy and initiation of a systemic corticosteroid taper, acitretin, and narrowband UVB therapy. After 6 weeks of treatment, the pain and pruritus improved and the rash had resolved in some areas while it had taken on a more classic lichenoid appearance with violaceous scaly papules and plaques (Figure 2) in areas of prior ulcers and erosions. He no longer had any bullae, erosions, or ulcers.
The Diagnosis: Immunotherapy-Related Lichenoid Drug Eruption
Direct immunofluorescence was negative, and histopathology revealed a lichenoid interface dermatitis, minimal parakeratosis, and saw-toothed rete ridges (Figure 1). He was diagnosed with an immunotherapyrelated lichenoid drug eruption based on the morphology of the skin lesions and clinicopathologic correlation. Bullous pemphigoid and lichen planus pemphigoides were ruled out given the negative direct immunofluorescence findings. Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) was not consistent with the clinical presentation, especially given the lack of mucosal findings. The histology also was not consistent, as the biopsy specimen lacked apoptotic and necrotic keratinocytes to the degree seen in SJS/TEN and also had a greater degree of inflammatory infiltrate. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome was ruled out given the lack of systemic findings, including facial swelling and lymphadenopathy and the clinical appearance of the rash. No morbilliform features were present, which is the most common presentation of DRESS syndrome.
Checkpoint inhibitor (CPI) therapy has become the cornerstone in management of certain advanced malignancies.1 Checkpoint inhibitors block cytotoxic T lymphocyte–associated protein 4, programmed cell death-1, and/or programmed cell death ligand-1, allowing activated T cells to infiltrate the tumor microenvironment and destroy malignant cells. Checkpoint inhibitors are approved for the treatment of melanoma, cutaneous squamous cell carcinoma, and Merkel cell carcinoma and are being investigated in various other cutaneous and soft tissue malignancies.1-3
Although CPIs have shown substantial efficacy in the management of advanced malignancies, immune-related adverse events (AEs) are common due to nonspecific immune activation.2 Immune-related cutaneous AEs are the most common immune-related AEs, occurring in 30% to 50% of patients who undergo treatment.2-5 Common immune-related cutaneous AEs include maculopapular, psoriasiform, and lichenoid dermatitis, as well as pruritus without dermatitis.2,3,6 Other reactions include but are not limited to bullous pemphigoid, vitiligolike depigmentation, and alopecia.2,3 Immune-related cutaneous AEs usually are self-limited; however, severe life-threatening reactions such as the spectrum of SJS/TEN and DRESS syndrome also can occur.2-4 Immune-related cutaneous AEs are graded based on the Common Terminology Criteria for Adverse Events: grade 1 reactions are asymptomatic and cover less than 10% of the patient’s body surface area (BSA), grade 2 reactions have mild symptoms and cover 10% to 30% of the patient’s BSA, grade 3 reactions have moderate to severe symptoms and cover greater than 30% of the patient’s BSA, and grade 4 reactions are life-threatening.2,3 With prompt recognition and adequate treatment, mild to moderate immune-related cutaneous AEs—grades 1 and 2—largely are reversible, and less than 5% require discontinuation of therapy.2,3,6 It has been suggested that immune-related cutaneous AEs may be a positive prognostic factor in the treatment of underlying malignancy, indicating adequate immune activation targeting the malignant cells.6
Although our patient had some typical violaceous, flat-topped papules and plaques with Wickham striae, he also had atypical findings for a lichenoid reaction. Given the endorsement of blisters, it is possible that some of these lesions initially were bullous and subsequently ruptured, leaving behind erosions. However, in other areas, there also were eroded papules and ulcerations without a reported history of excoriation, scratching, picking, or prior bullae, including difficult-to-reach areas such as the back. It is favored that these lesions represented a robust lichenoid dermatitis leading to erosive and ulcerated lesions, similar to the formation of bullous lichen planus. Lichenoid eruptions secondary to immunotherapy are well-known phenomena, but a PubMed search of articles indexed for MEDLINE using the terms ulcer, lichenoid, and immunotherapy revealed only 2 cases of ulcerative lichenoid eruptions: a localized digital erosive lichenoid dermatitis and a widespread ulcerative lichenoid drug eruption without true erosions.7,8 However, widespread erosive and ulcerated lichenoid reactions are rare.
Lichenoid eruptions most strongly are associated with anti–programmed cell death-1/ programmed cell death ligand-1 therapy, occurring in 20% of patients undergoing treatment.3 Lichenoid eruptions present as discrete, pruritic, erythematous, violaceous papules and plaques on the chest and back and rarely may involve the limbs, palmoplantar surfaces, and oral mucosa.2,3,6 Histopathologic features include a dense bandlike lymphocytic infiltrate in the dermis with scattered apoptotic keratinocytes in the basal layer of the epidermis.2,4,6 Grades 1 to 2 lesions can be managed with high-potency topical corticosteroids without CPI dose interruption, with more extensive grade 2 lesions requiring systemic corticosteroids.2,6,9 Lichenoid eruptions grade 3 or higher also require systemic corticosteroid therapy CPI therapy cessation until the eruption has receded to grade 0 to 1.2 Alternative treatment options for high-grade toxicity include phototherapy and acitretin.2,4,9
Our patient was treated with cessation of immunotherapy and initiation of a systemic corticosteroid taper, acitretin, and narrowband UVB therapy. After 6 weeks of treatment, the pain and pruritus improved and the rash had resolved in some areas while it had taken on a more classic lichenoid appearance with violaceous scaly papules and plaques (Figure 2) in areas of prior ulcers and erosions. He no longer had any bullae, erosions, or ulcers.
- Barrios DM, Do MH, Phillips GS, et al. Immune checkpoint inhibitors to treat cutaneous malignancies. J Am Acad Dermatol. 2020;83:1239-1253. doi:10.1016/j.jaad.2020.03.131
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Tattersall IW, Leventhal JS. Cutaneous toxicities of immune checkpoint inhibitors: the role of the dermatologist. Yale J Biol Med. 2020;93:123-132.
- Si X, He C, Zhang L, et al. Management of immune checkpoint inhibitor-related dermatologic adverse events. Thorac Cancer. 2020;11:488-492. doi:10.1111/1759-7714.13275
- Eggermont AMM, Kicinski M, Blank CU, et al. Association between immune-related adverse events and recurrence-free survival among patients with stage III melanoma randomized to receive pembrolizumab or placebo: a secondary analysis of a randomized clinical trial. JAMA Oncol. 2020;6:519-527. doi:10.1001 /jamaoncol.2019.5570
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263. doi:10.1097/CCO.0000000000000290
- Martínez-Doménech Á, García-Legaz Martínez M, Magdaleno-Tapial J, et al. Digital ulcerative lichenoid dermatitis in a patient receiving anti-PD-1 therapy. Dermatol Online J. 2019;25:13030/qt8sm0j7t7.
- Davis MJ, Wilken R, Fung MA, et al. Debilitating erosive lichenoid interface dermatitis from checkpoint inhibitor therapy. Dermatol Online J. 2018;24:13030/qt3vq6b04v.
- Apalla Z, Papageorgiou C, Lallas A, et al. Cutaneous adverse events of immune checkpoint inhibitors: a literature review [published online January 29, 2021]. Dermatol Pract Concept. 2021;11:E2021155. doi:10.5826/dpc.1101a155
- Barrios DM, Do MH, Phillips GS, et al. Immune checkpoint inhibitors to treat cutaneous malignancies. J Am Acad Dermatol. 2020;83:1239-1253. doi:10.1016/j.jaad.2020.03.131
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Tattersall IW, Leventhal JS. Cutaneous toxicities of immune checkpoint inhibitors: the role of the dermatologist. Yale J Biol Med. 2020;93:123-132.
- Si X, He C, Zhang L, et al. Management of immune checkpoint inhibitor-related dermatologic adverse events. Thorac Cancer. 2020;11:488-492. doi:10.1111/1759-7714.13275
- Eggermont AMM, Kicinski M, Blank CU, et al. Association between immune-related adverse events and recurrence-free survival among patients with stage III melanoma randomized to receive pembrolizumab or placebo: a secondary analysis of a randomized clinical trial. JAMA Oncol. 2020;6:519-527. doi:10.1001 /jamaoncol.2019.5570
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263. doi:10.1097/CCO.0000000000000290
- Martínez-Doménech Á, García-Legaz Martínez M, Magdaleno-Tapial J, et al. Digital ulcerative lichenoid dermatitis in a patient receiving anti-PD-1 therapy. Dermatol Online J. 2019;25:13030/qt8sm0j7t7.
- Davis MJ, Wilken R, Fung MA, et al. Debilitating erosive lichenoid interface dermatitis from checkpoint inhibitor therapy. Dermatol Online J. 2018;24:13030/qt3vq6b04v.
- Apalla Z, Papageorgiou C, Lallas A, et al. Cutaneous adverse events of immune checkpoint inhibitors: a literature review [published online January 29, 2021]. Dermatol Pract Concept. 2021;11:E2021155. doi:10.5826/dpc.1101a155
A 70-year-old man presented with a painful, pruritic, diffuse eruption on the trunk, legs, and arms of 2 months’ duration. He had a history of stage IV pleomorphic cell sarcoma of the retroperitoneum and was started on pembrolizumab therapy 6 weeks prior to the eruption. Physical examination revealed violaceous papules and plaques with shiny reticulated scaling as well as multiple scattered eroded papules and shallow ulcerations. The oral mucosa and genitals were spared. The patient endorsed blisters followed by open sores that were both itchy and painful. He denied self-infliction. Both the patient and his wife denied scratching. Two biopsies for direct immunofluorescence and histopathology were performed.


Progressive Axillary Hyperpigmentation
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.

Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.
Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.
Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.

A 50-year-old Hispanic woman presented with asymptomatic, progressive, brown hyperpigmentation involving the axillae, neck, upper back, and inframammary areas of 5 years’ duration. She had no other notable medical history; family history was unremarkable. She had been treated with topical hydroquinone and tretinoin by an outside physician without improvement. Physical examination revealed reticulated hyperpigmented macules and patches involving the inverse regions of the neck, axillae, and inframammary regions. Additionally, acneform pitted scars involving the perioral region were seen. A 4.0-mm punch biopsy of the right axilla was performed.
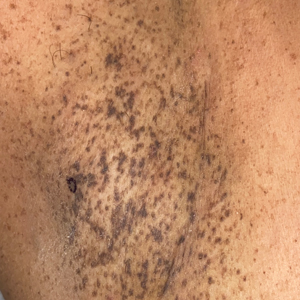
Hyperpigmentation on the Head and Neck
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
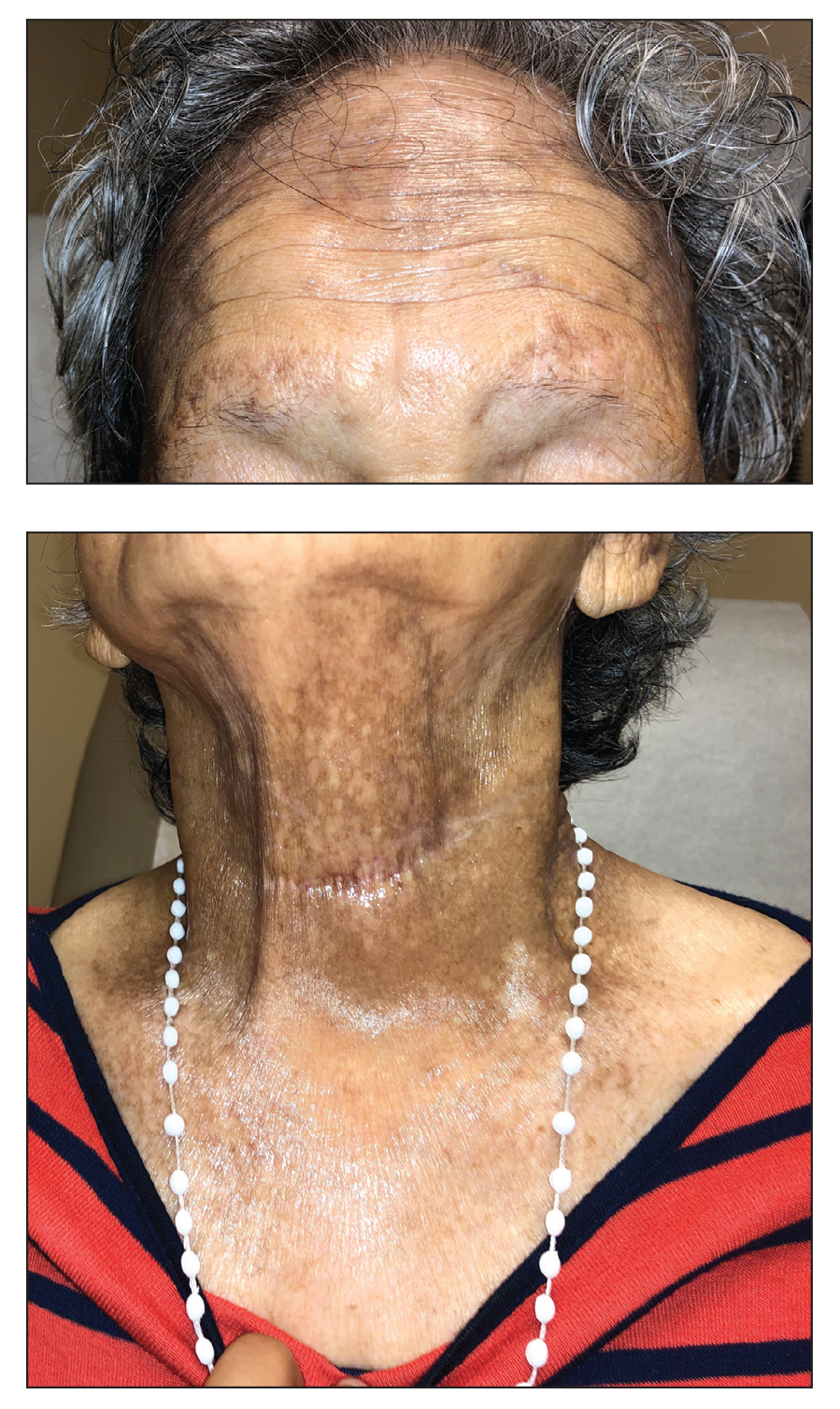
A 78-year-old Asian woman presented to the dermatology clinic with progressively worsening dark spots on the forehead and neck of 3 months’ duration. She noted mild pruritis and hair loss involving the eyebrows and anterior scalp. Her medical history was notable for type 2 diabetes mellitus. She denied any new medical conditions or medications and had no prior history of similar symptoms. Physical examination showed hyperpigmented brown macules and patches on the forehead (top) and anterior neck (bottom) with sparing of the posterior neck and lower face. Alopecia with areas of perifollicular erythema and hyperpigmentation with reduced follicular openings were present on the eyebrows and anterior forehead. Two punch biopsies of head and neck lesions were performed.
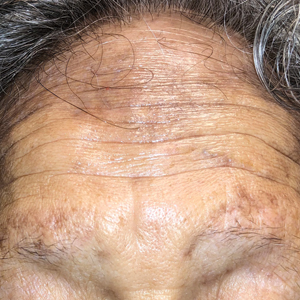
High-Potency Topical Steroid Treatment of Multiple Keratoacanthomas Associated With Prurigo Nodularis
Practice Gap
Multiple keratoacanthomas (KAs) of the legs often are a challenge to treat, especially when these lesions appear within a field of prurigo nodules. Multiple KAs associated with prurigo nodularis is a rarer finding; more often, the condition is reported on the lower limbs of elderly women with actinically damaged skin.1,2 At times, it can be difficult to distinguish between KA and prurigo nodularis in these patients, who often report notable pruritus and might have associated eczematous dermatitis.2
Keratoacanthomas often are treated with aggressive modalities, such as Mohs micrographic surgery, excision, and electrodesiccation and curettage. Some patients are hesitant to undergo surgical treatment, however, preferring a less invasive approach. Trauma from these aggressive modalities also can be associated with recurrence of existing lesions or development of new KAs, possibly related to stimulation of a local inflammatory response and upregulation of helper T cells.2-4
Acitretin and other systemic retinoids often are considered first-line therapy for multiple KAs. Cyclosporine has been added as adjunctive treatment in cases associated with prurigo nodularis or eczematous dermatitis1,2; however, these treatments have a high rate of discontinuation because of adverse effects, including transaminitis, xerostomia, alopecia (acitretin), and renal toxicity (cyclosporine).2
Another treatment option for patients with coexisting KA and prurigo nodularis is intralesional corticosteroids, often administered in combination with systemic retinoids.3 Topical 5-fluorouracil (5-FU) has been used successfully for KA, but topical treatment options are limited if 5-FU fails. Topical imiquimod and cryotherapy are thought to be of little benefit, and the appearance of new KA within imiquimod and cryotherapy treatment fields has been reported.1,2 Topical corticosteroids have been used as an adjuvant therapy for multiple KAs associated with prurigo nodularis; however, a PubMed search of articles indexed for MEDLINE using the terms keratoacanthoma and steroid and keratoacanthoma and prurigo nodularis yielded no published reports of successful use of topical corticosteroids as monotherapy.2
The Technique
For patients who want to continue topical treatment of coexisting KA and prurigo nodularis after topical 5-FU fails, we have found success applying a high-potency topical corticosteroid to affected areas under occlusion nightly for 6 to 8 weeks. This treatment not only leads to resolution of KA but also simultaneously treats prurigo nodules that might be clinically difficult to distinguish from KA in some presentations. This regimen has been implemented in our practice with remarkable reduction of KA burden and relief of pruritus.
In a 68-year-old woman who was treated with this technique, multiple biopsies had shown KA (or well-differentiated squamous cell carcinoma that appeared clinically as KA) on the shin (Figure, A) arising amid many lesions consistent with prurigo nodules. Topical 5-FU had failed, but the patient did not want to be treated with a more invasive modality, such as excision or injection.
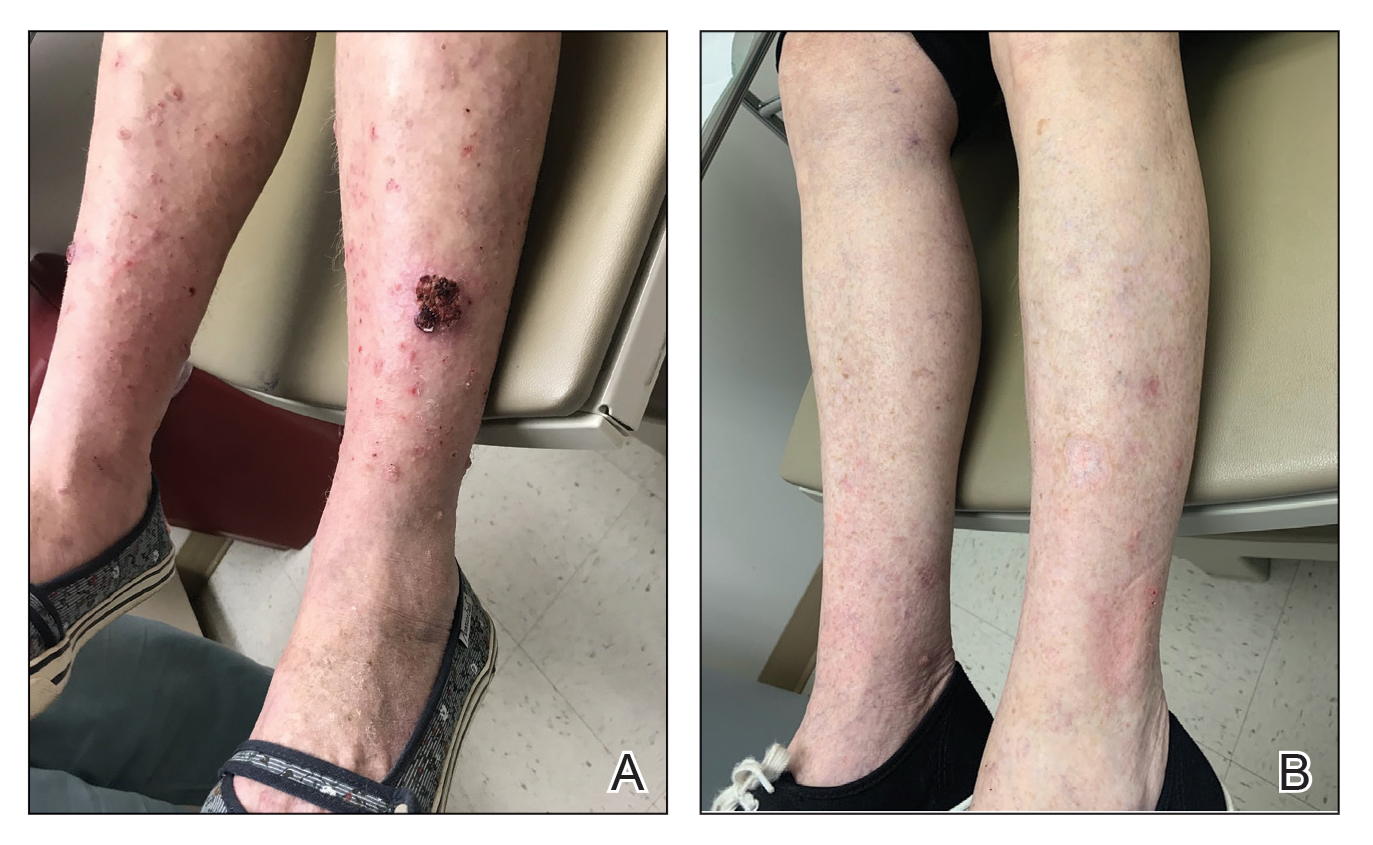
Instead, we treated the patient with clobetasol propionate ointment 0.05% under occlusion nightly for 6 weeks. This strategy produced resolution of both KA and prurigo nodules (Figure, B). When lesions recurred after a few months, they were successfully re-treated with topical clobetasol under occlusion in a second 6-week course.
Practical Implications
Treatment of multiple KAs associated with prurigo nodularis can present a distinct challenge. For the subset of patients who want to pursue topical treatment, options reported in the literature are limited. We have found success treating multiple KAs and associated prurigo nodules with a high-potency topical corticosteroid under occlusion, with minimal or no adverse effects. We believe that a topical corticosteroid can be implemented easily in clinical practice before a more invasive surgical or intralesional modality is considered.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233. doi:10.1016/j.jaad.2015.11.033
- Wu TP, Miller K, Cohen DE, et al. Keratoacanthomas arising in association with prurigo nodules in pruritic, actinically damaged skin. J Am Acad Dermatol. 2013;69:426-430. doi:10.1016/J.JAAD.2013.03.035
- Sanders S, Busam KJ, Halpern AC, et al. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958. doi:10.1046/j.1524-4725.2002.02069.x
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:E117-E119. doi:10.1111/ajd.12501
Practice Gap
Multiple keratoacanthomas (KAs) of the legs often are a challenge to treat, especially when these lesions appear within a field of prurigo nodules. Multiple KAs associated with prurigo nodularis is a rarer finding; more often, the condition is reported on the lower limbs of elderly women with actinically damaged skin.1,2 At times, it can be difficult to distinguish between KA and prurigo nodularis in these patients, who often report notable pruritus and might have associated eczematous dermatitis.2
Keratoacanthomas often are treated with aggressive modalities, such as Mohs micrographic surgery, excision, and electrodesiccation and curettage. Some patients are hesitant to undergo surgical treatment, however, preferring a less invasive approach. Trauma from these aggressive modalities also can be associated with recurrence of existing lesions or development of new KAs, possibly related to stimulation of a local inflammatory response and upregulation of helper T cells.2-4
Acitretin and other systemic retinoids often are considered first-line therapy for multiple KAs. Cyclosporine has been added as adjunctive treatment in cases associated with prurigo nodularis or eczematous dermatitis1,2; however, these treatments have a high rate of discontinuation because of adverse effects, including transaminitis, xerostomia, alopecia (acitretin), and renal toxicity (cyclosporine).2
Another treatment option for patients with coexisting KA and prurigo nodularis is intralesional corticosteroids, often administered in combination with systemic retinoids.3 Topical 5-fluorouracil (5-FU) has been used successfully for KA, but topical treatment options are limited if 5-FU fails. Topical imiquimod and cryotherapy are thought to be of little benefit, and the appearance of new KA within imiquimod and cryotherapy treatment fields has been reported.1,2 Topical corticosteroids have been used as an adjuvant therapy for multiple KAs associated with prurigo nodularis; however, a PubMed search of articles indexed for MEDLINE using the terms keratoacanthoma and steroid and keratoacanthoma and prurigo nodularis yielded no published reports of successful use of topical corticosteroids as monotherapy.2
The Technique
For patients who want to continue topical treatment of coexisting KA and prurigo nodularis after topical 5-FU fails, we have found success applying a high-potency topical corticosteroid to affected areas under occlusion nightly for 6 to 8 weeks. This treatment not only leads to resolution of KA but also simultaneously treats prurigo nodules that might be clinically difficult to distinguish from KA in some presentations. This regimen has been implemented in our practice with remarkable reduction of KA burden and relief of pruritus.
In a 68-year-old woman who was treated with this technique, multiple biopsies had shown KA (or well-differentiated squamous cell carcinoma that appeared clinically as KA) on the shin (Figure, A) arising amid many lesions consistent with prurigo nodules. Topical 5-FU had failed, but the patient did not want to be treated with a more invasive modality, such as excision or injection.
Instead, we treated the patient with clobetasol propionate ointment 0.05% under occlusion nightly for 6 weeks. This strategy produced resolution of both KA and prurigo nodules (Figure, B). When lesions recurred after a few months, they were successfully re-treated with topical clobetasol under occlusion in a second 6-week course.
Practical Implications
Treatment of multiple KAs associated with prurigo nodularis can present a distinct challenge. For the subset of patients who want to pursue topical treatment, options reported in the literature are limited. We have found success treating multiple KAs and associated prurigo nodules with a high-potency topical corticosteroid under occlusion, with minimal or no adverse effects. We believe that a topical corticosteroid can be implemented easily in clinical practice before a more invasive surgical or intralesional modality is considered.
Practice Gap
Multiple keratoacanthomas (KAs) of the legs often are a challenge to treat, especially when these lesions appear within a field of prurigo nodules. Multiple KAs associated with prurigo nodularis is a rarer finding; more often, the condition is reported on the lower limbs of elderly women with actinically damaged skin.1,2 At times, it can be difficult to distinguish between KA and prurigo nodularis in these patients, who often report notable pruritus and might have associated eczematous dermatitis.2
Keratoacanthomas often are treated with aggressive modalities, such as Mohs micrographic surgery, excision, and electrodesiccation and curettage. Some patients are hesitant to undergo surgical treatment, however, preferring a less invasive approach. Trauma from these aggressive modalities also can be associated with recurrence of existing lesions or development of new KAs, possibly related to stimulation of a local inflammatory response and upregulation of helper T cells.2-4
Acitretin and other systemic retinoids often are considered first-line therapy for multiple KAs. Cyclosporine has been added as adjunctive treatment in cases associated with prurigo nodularis or eczematous dermatitis1,2; however, these treatments have a high rate of discontinuation because of adverse effects, including transaminitis, xerostomia, alopecia (acitretin), and renal toxicity (cyclosporine).2
Another treatment option for patients with coexisting KA and prurigo nodularis is intralesional corticosteroids, often administered in combination with systemic retinoids.3 Topical 5-fluorouracil (5-FU) has been used successfully for KA, but topical treatment options are limited if 5-FU fails. Topical imiquimod and cryotherapy are thought to be of little benefit, and the appearance of new KA within imiquimod and cryotherapy treatment fields has been reported.1,2 Topical corticosteroids have been used as an adjuvant therapy for multiple KAs associated with prurigo nodularis; however, a PubMed search of articles indexed for MEDLINE using the terms keratoacanthoma and steroid and keratoacanthoma and prurigo nodularis yielded no published reports of successful use of topical corticosteroids as monotherapy.2
The Technique
For patients who want to continue topical treatment of coexisting KA and prurigo nodularis after topical 5-FU fails, we have found success applying a high-potency topical corticosteroid to affected areas under occlusion nightly for 6 to 8 weeks. This treatment not only leads to resolution of KA but also simultaneously treats prurigo nodules that might be clinically difficult to distinguish from KA in some presentations. This regimen has been implemented in our practice with remarkable reduction of KA burden and relief of pruritus.
In a 68-year-old woman who was treated with this technique, multiple biopsies had shown KA (or well-differentiated squamous cell carcinoma that appeared clinically as KA) on the shin (Figure, A) arising amid many lesions consistent with prurigo nodules. Topical 5-FU had failed, but the patient did not want to be treated with a more invasive modality, such as excision or injection.
Instead, we treated the patient with clobetasol propionate ointment 0.05% under occlusion nightly for 6 weeks. This strategy produced resolution of both KA and prurigo nodules (Figure, B). When lesions recurred after a few months, they were successfully re-treated with topical clobetasol under occlusion in a second 6-week course.
Practical Implications
Treatment of multiple KAs associated with prurigo nodularis can present a distinct challenge. For the subset of patients who want to pursue topical treatment, options reported in the literature are limited. We have found success treating multiple KAs and associated prurigo nodules with a high-potency topical corticosteroid under occlusion, with minimal or no adverse effects. We believe that a topical corticosteroid can be implemented easily in clinical practice before a more invasive surgical or intralesional modality is considered.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233. doi:10.1016/j.jaad.2015.11.033
- Wu TP, Miller K, Cohen DE, et al. Keratoacanthomas arising in association with prurigo nodules in pruritic, actinically damaged skin. J Am Acad Dermatol. 2013;69:426-430. doi:10.1016/J.JAAD.2013.03.035
- Sanders S, Busam KJ, Halpern AC, et al. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958. doi:10.1046/j.1524-4725.2002.02069.x
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:E117-E119. doi:10.1111/ajd.12501
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233. doi:10.1016/j.jaad.2015.11.033
- Wu TP, Miller K, Cohen DE, et al. Keratoacanthomas arising in association with prurigo nodules in pruritic, actinically damaged skin. J Am Acad Dermatol. 2013;69:426-430. doi:10.1016/J.JAAD.2013.03.035
- Sanders S, Busam KJ, Halpern AC, et al. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958. doi:10.1046/j.1524-4725.2002.02069.x
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:E117-E119. doi:10.1111/ajd.12501
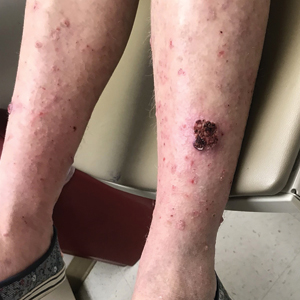
Asymptomatic Discolored Lesions on the Groin
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
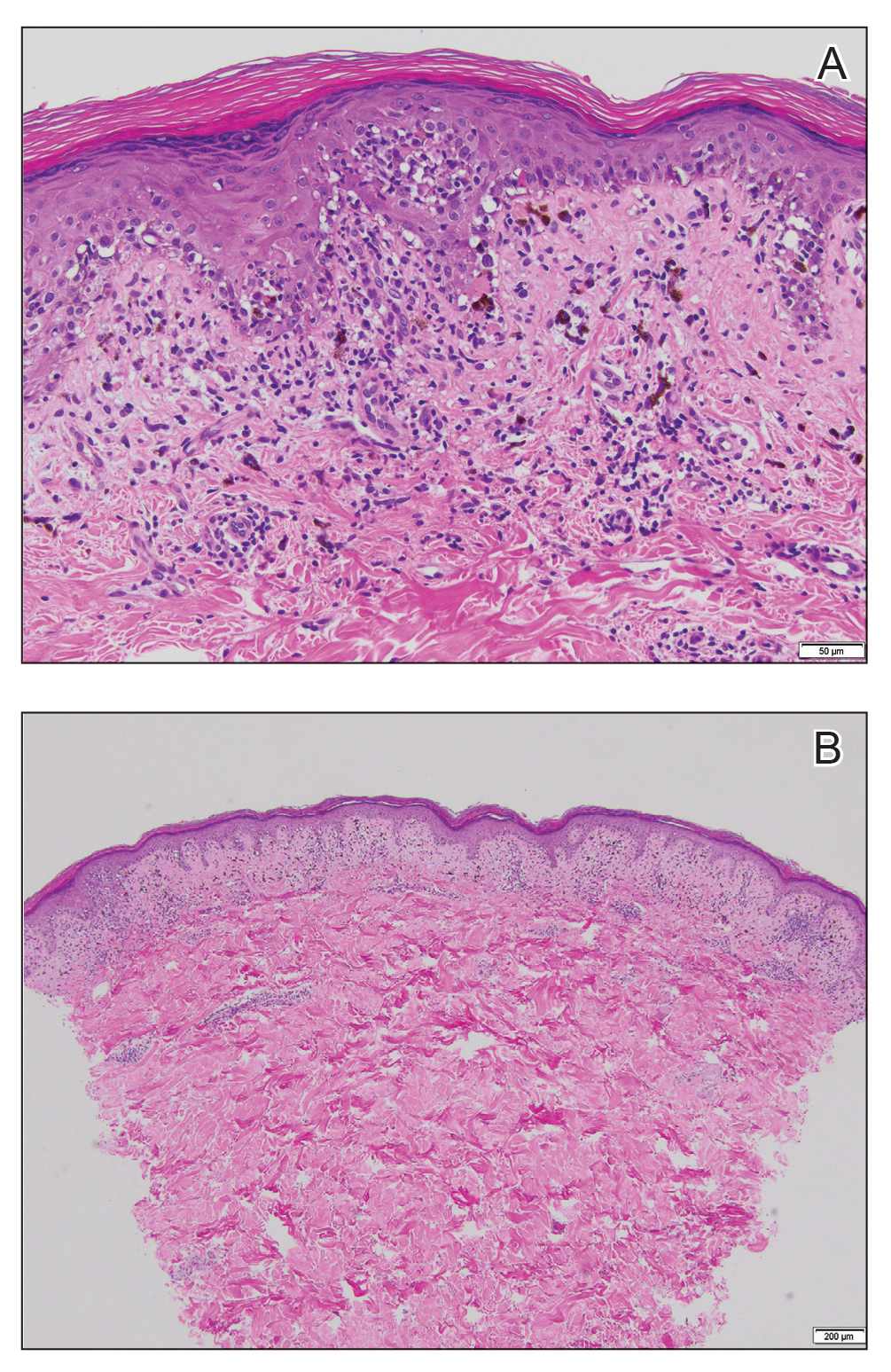
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
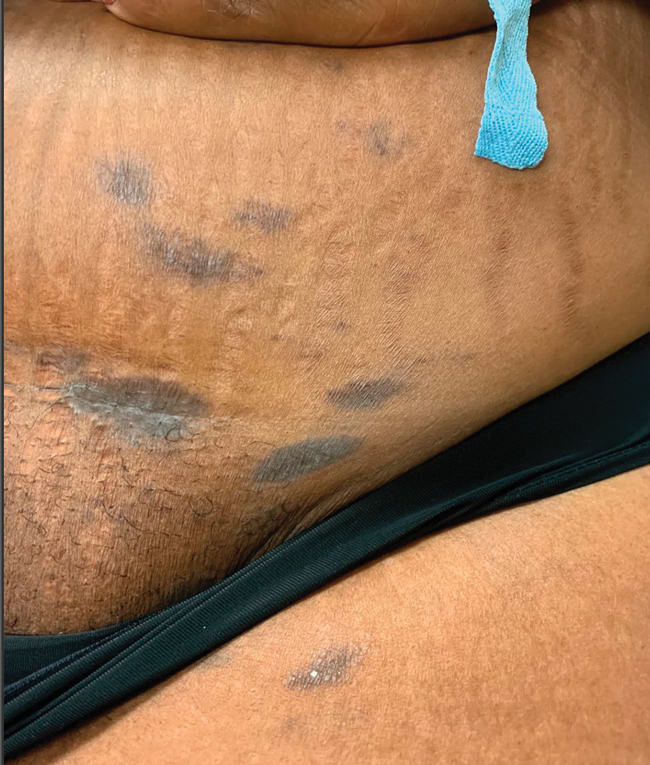
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
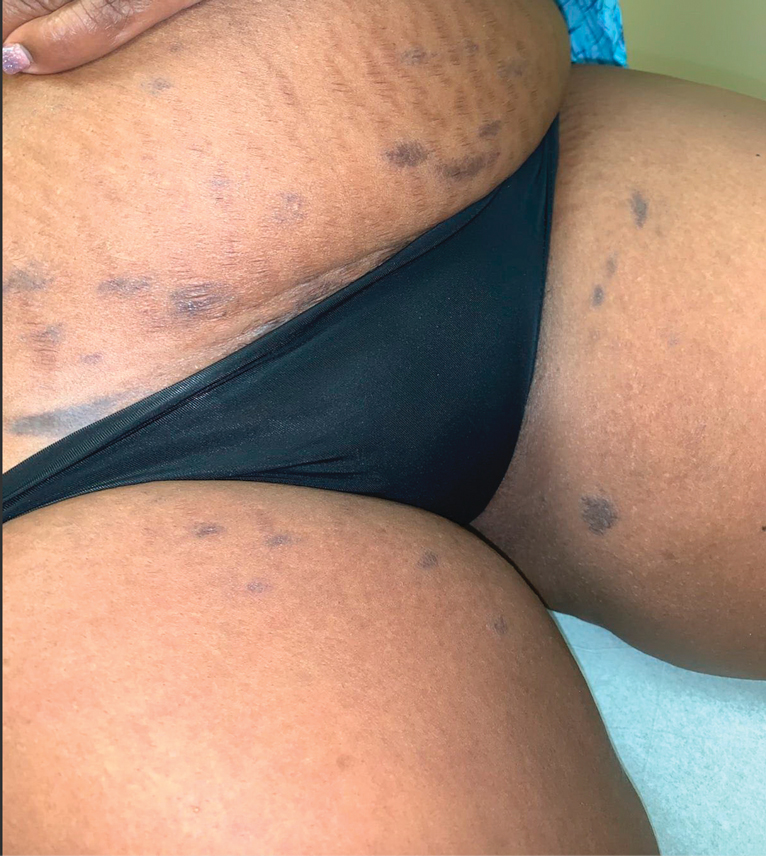
A 45-year-old African American woman presented with an asymptomatic rash that had worsened over the month prior to presentation. It initially began on the upper thighs and then spread to the abdomen, groin, and buttocks. The rash was mildly pruritic and had grown both in size and number of lesions. She had not tried any new over-the-counter medications. Her medical history was notable for late-stage breast cancer diagnosed 4 years prior that was treated with radiation and neoadjuvant NeoPACT—carboplatin, docetaxel, and pembrolizumab. One year prior to presentation, she underwent a lumpectomy that was complicated by gas gangrene of the finger. She has been in remission since the surgery. Physical examination at the current presentation was remarkable for multiple well-circumscribed, hyperpigmented macules on the medial thighs, lower abdomen, and buttocks. Syphilis antibody screening was negative.
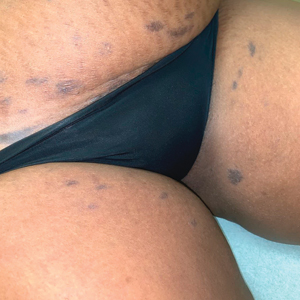
Scalp Wound Closures in Mohs Micrographic Surgery: A Survey of Staples vs Sutures
Limited data exist comparing staples and sutures for scalp closures during Mohs micrographic surgery (MMS). As a result, the closure method for these scalp wounds is based on surgeon preference without established consensus. The purpose of this study was to survey practicing Mohs surgeons on their scalp wound closure preferences as well as the clinical and economic variables that impact their decisions. Understanding practice habits can guide future trial design, with a goal of creating established criterion for MMS scalp wound closures.
Methods
An anonymous survey was distributed from April 2019 to June 2019 to fellowship-trained Mohs surgeons using an electronic mailing list from the American College of Mohs Surgery (ACMS). The 10-question survey was approved by the University of Kansas institutional review board and the executive committee of the ACMS. Surgeons were asked about their preferred method for scalp wound closure as well as clinical and economic variables that impacted those preferences. Respondents indicated their frequency of using deep sutures, epidermal sutures, and wound undermining on a sliding scale of 0% to 100%. Comparisons were made between practice habits, preferences, and surgeon demographics using t tests. Statistical significance was determined as P<.05.
Results
Sixty-eight ACMS fellowship-trained Mohs surgeons completed the survey. The average age of respondents was 45 years; 69.1% (n=47) of respondents were male, and 76.5% (n=52) practiced in a private setting (Table 1). Regardless of epidermal closure type, deep suture placement was used in an average (standard deviation [SD]) of 88.8% (19.5%) of cases overall, which did not statistically differ between years of Mohs experience or practice setting (Table 2). Wound undermining was performed in an average (SD) of 83.0% (24.3%) of cases overall and was more prevalent in private vs academic settings (87.6% [17.8%] vs 65.7% [35.0%]; P<.01). Epidermal sutures were used in an average (SD) of 27.1% (33.5%) of scalp wound cases overall. Surgeons with less experience (≤5 years) used them more frequently (average [SD], 42.7% [36.2%] of cases) than surgeons with more experience (≥16 years; average [SD], 18.8% [32.6%] of cases; P=.037). There was no significant difference between epidermal suture placement rates and practice setting (average [SD], 18.1% [28.1%] of cases for academic providers vs 30.0% [34.8%] of cases with private providers; P=.210).

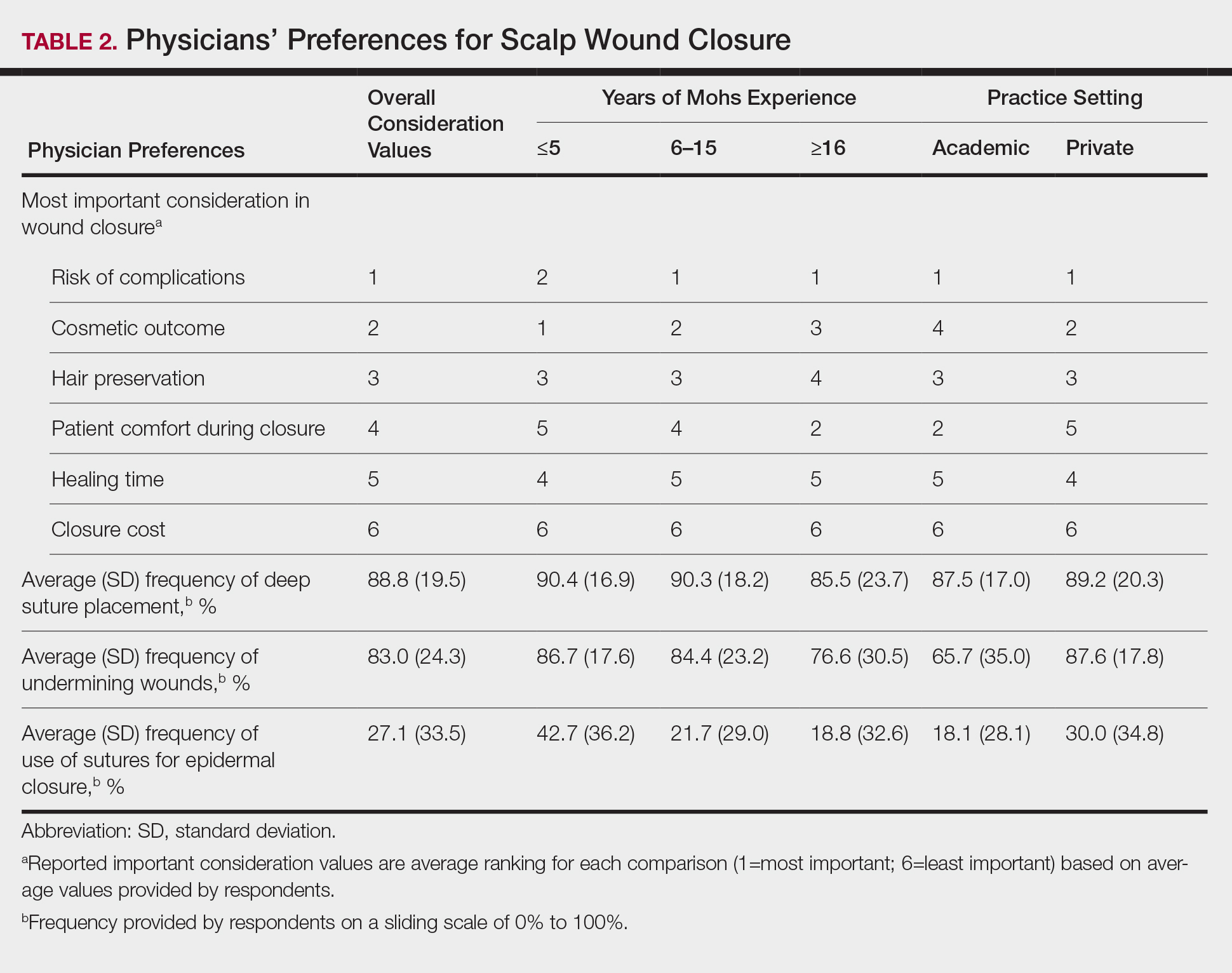
Clinical and economic factors that were most important during wound closure were ranked (beginning with most important) as the following: risk of complications, cosmetic outcome, hair preservation, patient comfort during closure, healing time, and closure cost. In all demographic cases, risk of complications was ranked 1 or 2 (1=most important; 6=least important) overall; cost was the least important factor overall (Table 2).
Surgeons perceived staples to be superior for speed of closure and for closing wounds in high-tension areas, whereas sutures were perceived as superior when considering cost of closure and ease of removal (Table 3). Successful healing rate, healing time, hair preservation, overall cosmetic outcome, and lower risk of complications were viewed as equivalent when comparing staples and sutures.
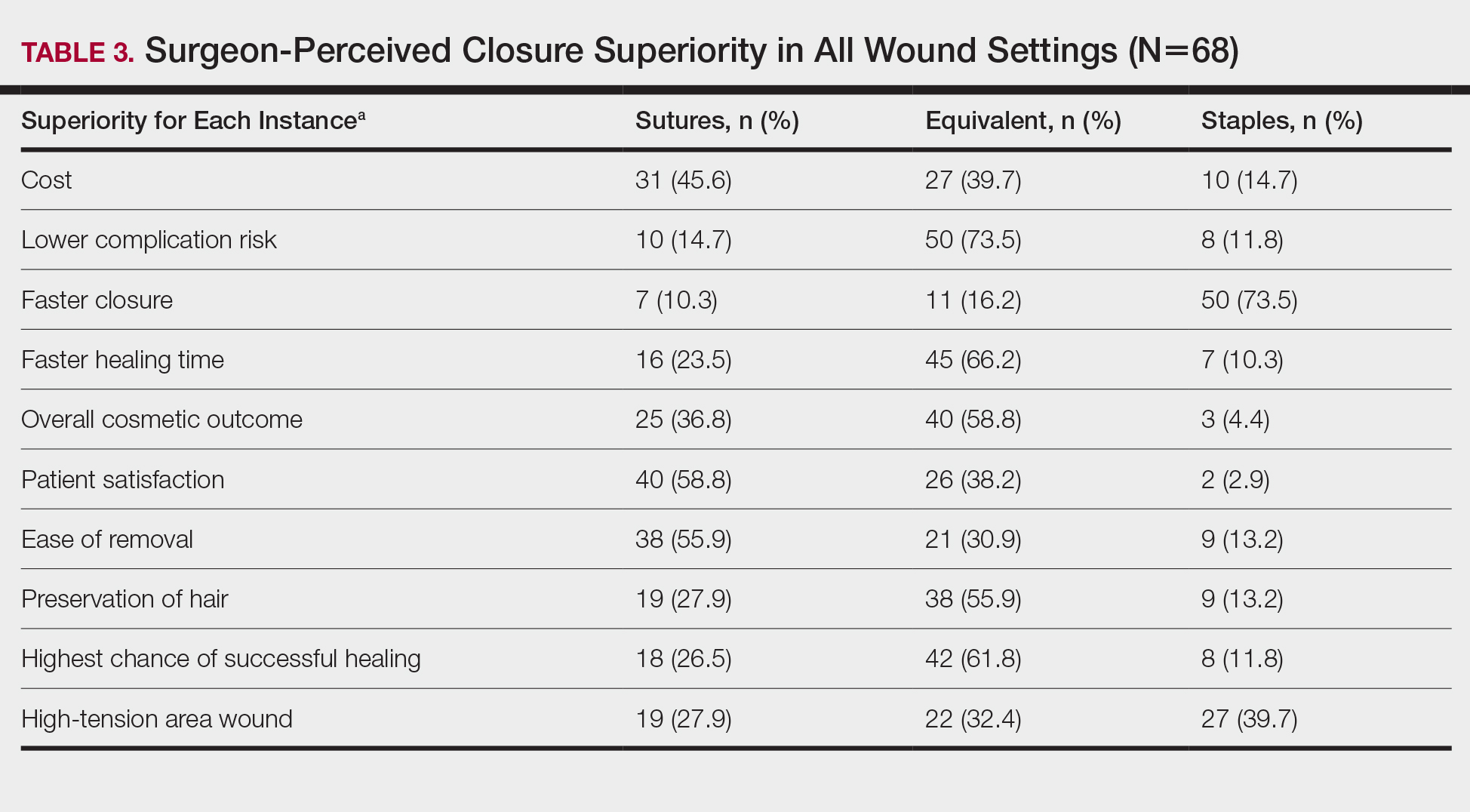
In cases in which surgeons did not use staples for closure, the most important factors for opting to not use them were patient discomfort (52.9% [n=36]), cost (25.0% [n=17]), and worse overall cosmetic outcome (23.5% [n=16])(Table 4). The most frequent locations outside of scalp wounds that physicians considered the use of staples for closure were the back (19.1% [n=13]), thigh (10.3% [n=7]), and shoulder (8.8% [n=6]).

Comment
Epidermal closure with sutures was reportedly used in an average of only 27.1% of scalp wound cases, with clinical factors such as cosmetic outcome, risk of complications, and closure time seen as either equivalent or inferior to staples. Our data suggest that surgeon closure perceptions generally are in agreement with established head and neck literature within different medical specialties that favor staple closures, particularly in high-tension areas.1 Interestingly, the most common reasons given for not using staples included patient discomfort, cost, and worse cosmetic outcomes, which are unsubstantiated with head and neck comparative studies.2-4
Although cost was the least important variable for determining closure type in our surveyed cohort, it is likely that the overall cost of closure is frequently underestimated. A higher material cost is noted with staples; however, the largest determinant of overall cost remains the surgeon’s time, which is reduced by factors of 10 or more when closing with staples.2,3 This difference—coupled with the unchanged cosmetic outcome and complication rates—makes staples more advantageous for high-tension scalp wounds.4 Moreover, the stapling technique is more reproducible than suturing, which requires more surgical skill and experience.
Limitations of this study include a lack of directly comparable data for staple and suture scalp wound closures. In addition, the small cohort of respondents in this preliminary study can serve to guide future studies.
Conclusion
Scalp wounds during MMS were most frequently closed using staples vs sutures, with the perception that these methods are equivalent in complication risk, cosmetic outcome, and overall patient satisfaction. These results agree with comparative literature for head and neck surgery and assist with establishing an epidemiologic baseline for future studies comparing their use during MMS.
- Ritchie AJ, Rocke LG. Staples versus sutures in the closure of scalp wounds: a prospective, double-blind, randomized trial. Injury. 1989;20:217-218.
- Batra J, Bekal RK, Byadgi S, et al. Comparison of skin staples and standard sutures for closing incisions after head and neck cancer surgery: a double-blind, randomized and prospective study. J Maxillofac Oral Surg. 2016;15:243-250.
- Kanegaye JT, Vance CW, Chan L, et al. Comparison of skin stapling devices and standard sutures for pediatric scalp lacerations: a randomized study of cost and time benefits. J Pediatr. 1997;130:808-813.
- Khan ANGA, Dayan PS, Miller S, et al. Cosmetic outcome of scalp wound closure with staples in the pediatric emergency department: a prospective, randomized trial. Pediatr Emerg Care. 2002;18:171-173.
Limited data exist comparing staples and sutures for scalp closures during Mohs micrographic surgery (MMS). As a result, the closure method for these scalp wounds is based on surgeon preference without established consensus. The purpose of this study was to survey practicing Mohs surgeons on their scalp wound closure preferences as well as the clinical and economic variables that impact their decisions. Understanding practice habits can guide future trial design, with a goal of creating established criterion for MMS scalp wound closures.
Methods
An anonymous survey was distributed from April 2019 to June 2019 to fellowship-trained Mohs surgeons using an electronic mailing list from the American College of Mohs Surgery (ACMS). The 10-question survey was approved by the University of Kansas institutional review board and the executive committee of the ACMS. Surgeons were asked about their preferred method for scalp wound closure as well as clinical and economic variables that impacted those preferences. Respondents indicated their frequency of using deep sutures, epidermal sutures, and wound undermining on a sliding scale of 0% to 100%. Comparisons were made between practice habits, preferences, and surgeon demographics using t tests. Statistical significance was determined as P<.05.
Results
Sixty-eight ACMS fellowship-trained Mohs surgeons completed the survey. The average age of respondents was 45 years; 69.1% (n=47) of respondents were male, and 76.5% (n=52) practiced in a private setting (Table 1). Regardless of epidermal closure type, deep suture placement was used in an average (standard deviation [SD]) of 88.8% (19.5%) of cases overall, which did not statistically differ between years of Mohs experience or practice setting (Table 2). Wound undermining was performed in an average (SD) of 83.0% (24.3%) of cases overall and was more prevalent in private vs academic settings (87.6% [17.8%] vs 65.7% [35.0%]; P<.01). Epidermal sutures were used in an average (SD) of 27.1% (33.5%) of scalp wound cases overall. Surgeons with less experience (≤5 years) used them more frequently (average [SD], 42.7% [36.2%] of cases) than surgeons with more experience (≥16 years; average [SD], 18.8% [32.6%] of cases; P=.037). There was no significant difference between epidermal suture placement rates and practice setting (average [SD], 18.1% [28.1%] of cases for academic providers vs 30.0% [34.8%] of cases with private providers; P=.210).
Clinical and economic factors that were most important during wound closure were ranked (beginning with most important) as the following: risk of complications, cosmetic outcome, hair preservation, patient comfort during closure, healing time, and closure cost. In all demographic cases, risk of complications was ranked 1 or 2 (1=most important; 6=least important) overall; cost was the least important factor overall (Table 2).
Surgeons perceived staples to be superior for speed of closure and for closing wounds in high-tension areas, whereas sutures were perceived as superior when considering cost of closure and ease of removal (Table 3). Successful healing rate, healing time, hair preservation, overall cosmetic outcome, and lower risk of complications were viewed as equivalent when comparing staples and sutures.
In cases in which surgeons did not use staples for closure, the most important factors for opting to not use them were patient discomfort (52.9% [n=36]), cost (25.0% [n=17]), and worse overall cosmetic outcome (23.5% [n=16])(Table 4). The most frequent locations outside of scalp wounds that physicians considered the use of staples for closure were the back (19.1% [n=13]), thigh (10.3% [n=7]), and shoulder (8.8% [n=6]).
Comment
Epidermal closure with sutures was reportedly used in an average of only 27.1% of scalp wound cases, with clinical factors such as cosmetic outcome, risk of complications, and closure time seen as either equivalent or inferior to staples. Our data suggest that surgeon closure perceptions generally are in agreement with established head and neck literature within different medical specialties that favor staple closures, particularly in high-tension areas.1 Interestingly, the most common reasons given for not using staples included patient discomfort, cost, and worse cosmetic outcomes, which are unsubstantiated with head and neck comparative studies.2-4
Although cost was the least important variable for determining closure type in our surveyed cohort, it is likely that the overall cost of closure is frequently underestimated. A higher material cost is noted with staples; however, the largest determinant of overall cost remains the surgeon’s time, which is reduced by factors of 10 or more when closing with staples.2,3 This difference—coupled with the unchanged cosmetic outcome and complication rates—makes staples more advantageous for high-tension scalp wounds.4 Moreover, the stapling technique is more reproducible than suturing, which requires more surgical skill and experience.
Limitations of this study include a lack of directly comparable data for staple and suture scalp wound closures. In addition, the small cohort of respondents in this preliminary study can serve to guide future studies.
Conclusion
Scalp wounds during MMS were most frequently closed using staples vs sutures, with the perception that these methods are equivalent in complication risk, cosmetic outcome, and overall patient satisfaction. These results agree with comparative literature for head and neck surgery and assist with establishing an epidemiologic baseline for future studies comparing their use during MMS.
Limited data exist comparing staples and sutures for scalp closures during Mohs micrographic surgery (MMS). As a result, the closure method for these scalp wounds is based on surgeon preference without established consensus. The purpose of this study was to survey practicing Mohs surgeons on their scalp wound closure preferences as well as the clinical and economic variables that impact their decisions. Understanding practice habits can guide future trial design, with a goal of creating established criterion for MMS scalp wound closures.
Methods
An anonymous survey was distributed from April 2019 to June 2019 to fellowship-trained Mohs surgeons using an electronic mailing list from the American College of Mohs Surgery (ACMS). The 10-question survey was approved by the University of Kansas institutional review board and the executive committee of the ACMS. Surgeons were asked about their preferred method for scalp wound closure as well as clinical and economic variables that impacted those preferences. Respondents indicated their frequency of using deep sutures, epidermal sutures, and wound undermining on a sliding scale of 0% to 100%. Comparisons were made between practice habits, preferences, and surgeon demographics using t tests. Statistical significance was determined as P<.05.
Results
Sixty-eight ACMS fellowship-trained Mohs surgeons completed the survey. The average age of respondents was 45 years; 69.1% (n=47) of respondents were male, and 76.5% (n=52) practiced in a private setting (Table 1). Regardless of epidermal closure type, deep suture placement was used in an average (standard deviation [SD]) of 88.8% (19.5%) of cases overall, which did not statistically differ between years of Mohs experience or practice setting (Table 2). Wound undermining was performed in an average (SD) of 83.0% (24.3%) of cases overall and was more prevalent in private vs academic settings (87.6% [17.8%] vs 65.7% [35.0%]; P<.01). Epidermal sutures were used in an average (SD) of 27.1% (33.5%) of scalp wound cases overall. Surgeons with less experience (≤5 years) used them more frequently (average [SD], 42.7% [36.2%] of cases) than surgeons with more experience (≥16 years; average [SD], 18.8% [32.6%] of cases; P=.037). There was no significant difference between epidermal suture placement rates and practice setting (average [SD], 18.1% [28.1%] of cases for academic providers vs 30.0% [34.8%] of cases with private providers; P=.210).
Clinical and economic factors that were most important during wound closure were ranked (beginning with most important) as the following: risk of complications, cosmetic outcome, hair preservation, patient comfort during closure, healing time, and closure cost. In all demographic cases, risk of complications was ranked 1 or 2 (1=most important; 6=least important) overall; cost was the least important factor overall (Table 2).
Surgeons perceived staples to be superior for speed of closure and for closing wounds in high-tension areas, whereas sutures were perceived as superior when considering cost of closure and ease of removal (Table 3). Successful healing rate, healing time, hair preservation, overall cosmetic outcome, and lower risk of complications were viewed as equivalent when comparing staples and sutures.
In cases in which surgeons did not use staples for closure, the most important factors for opting to not use them were patient discomfort (52.9% [n=36]), cost (25.0% [n=17]), and worse overall cosmetic outcome (23.5% [n=16])(Table 4). The most frequent locations outside of scalp wounds that physicians considered the use of staples for closure were the back (19.1% [n=13]), thigh (10.3% [n=7]), and shoulder (8.8% [n=6]).
Comment
Epidermal closure with sutures was reportedly used in an average of only 27.1% of scalp wound cases, with clinical factors such as cosmetic outcome, risk of complications, and closure time seen as either equivalent or inferior to staples. Our data suggest that surgeon closure perceptions generally are in agreement with established head and neck literature within different medical specialties that favor staple closures, particularly in high-tension areas.1 Interestingly, the most common reasons given for not using staples included patient discomfort, cost, and worse cosmetic outcomes, which are unsubstantiated with head and neck comparative studies.2-4
Although cost was the least important variable for determining closure type in our surveyed cohort, it is likely that the overall cost of closure is frequently underestimated. A higher material cost is noted with staples; however, the largest determinant of overall cost remains the surgeon’s time, which is reduced by factors of 10 or more when closing with staples.2,3 This difference—coupled with the unchanged cosmetic outcome and complication rates—makes staples more advantageous for high-tension scalp wounds.4 Moreover, the stapling technique is more reproducible than suturing, which requires more surgical skill and experience.
Limitations of this study include a lack of directly comparable data for staple and suture scalp wound closures. In addition, the small cohort of respondents in this preliminary study can serve to guide future studies.
Conclusion
Scalp wounds during MMS were most frequently closed using staples vs sutures, with the perception that these methods are equivalent in complication risk, cosmetic outcome, and overall patient satisfaction. These results agree with comparative literature for head and neck surgery and assist with establishing an epidemiologic baseline for future studies comparing their use during MMS.
- Ritchie AJ, Rocke LG. Staples versus sutures in the closure of scalp wounds: a prospective, double-blind, randomized trial. Injury. 1989;20:217-218.
- Batra J, Bekal RK, Byadgi S, et al. Comparison of skin staples and standard sutures for closing incisions after head and neck cancer surgery: a double-blind, randomized and prospective study. J Maxillofac Oral Surg. 2016;15:243-250.
- Kanegaye JT, Vance CW, Chan L, et al. Comparison of skin stapling devices and standard sutures for pediatric scalp lacerations: a randomized study of cost and time benefits. J Pediatr. 1997;130:808-813.
- Khan ANGA, Dayan PS, Miller S, et al. Cosmetic outcome of scalp wound closure with staples in the pediatric emergency department: a prospective, randomized trial. Pediatr Emerg Care. 2002;18:171-173.
- Ritchie AJ, Rocke LG. Staples versus sutures in the closure of scalp wounds: a prospective, double-blind, randomized trial. Injury. 1989;20:217-218.
- Batra J, Bekal RK, Byadgi S, et al. Comparison of skin staples and standard sutures for closing incisions after head and neck cancer surgery: a double-blind, randomized and prospective study. J Maxillofac Oral Surg. 2016;15:243-250.
- Kanegaye JT, Vance CW, Chan L, et al. Comparison of skin stapling devices and standard sutures for pediatric scalp lacerations: a randomized study of cost and time benefits. J Pediatr. 1997;130:808-813.
- Khan ANGA, Dayan PS, Miller S, et al. Cosmetic outcome of scalp wound closure with staples in the pediatric emergency department: a prospective, randomized trial. Pediatr Emerg Care. 2002;18:171-173.
Practice Points
- Scalp wounds present a unique challenge for closure during Mohs micrographic surgery due to the scalp's tendency to bleed, limited elasticity, and hair-bearing nature.
- Among fellowship-trained Mohs surgeons, scalp wounds were closed with staples more often than with epidermal sutures.
- Staples and sutures for scalp wounds were perceived to be equivalent in risk of complications, cosmetic outcome, and overall patient satisfaction.
- Compared to epidermal sutures, staples were perceived as advantageous in high-tension areas and for speed of closure.