User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
nav[contains(@class, 'nav-ce-stack nav-ce-stack__large-screen')]
header[@id='header']
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
‘Myriad’ dermatologic reactions after COVID-19 vaccination
GLASGOW – Individuals given COVID-19 vaccination may experience a wide range of dermatologic reactions, some of which may be life-threatening, reveals a prospective Indian study that suggests histopathological assessment is key to understanding the cause.
Studying more than 130 patients who presented with vaccine-related dermatologic reactions, the researchers found that the most common acute adverse events were acute urticaria, generalized pruritus, and maculopapular rash.
Dermal hypersensitivity reactions occurred within 3 days of vaccination, which suggests the culprit is an immediate type 1 hypersensitivity reaction, said study presenter Alpana Mohta, MD, department of dermatology, Sardar Patel Medical College, Bikaner, Rajasthan, India. Most of the patients had received the AstraZeneca vaccine, she said.
, which occurred within 3-4 weeks of vaccination and could be a result of delayed hypersensitivity or a T cell–mediated skin reaction caused by “molecular mimicry with a viral epitope,” Dr. Mohta said.
The research was presented at the British Association of Dermatologists (BAD) 2022 Annual Meeting on July 5.
Dr. Mohta said that, given the “surge” in the number of people who have been vaccinated, it is “imperative as dermatologists” to maintain a “very high index of suspicion to differentiate reactions caused by vaccination” from other causes, and a proper assessment should be performed in “every patient” who presents with a possible reaction.
She also emphasized that “since so many clinical [COVID-19] variants are being encountered,” histopathological assessment could “help in better understanding the underlying pathophysiology” of every reaction.
Dr. Mohta began her presentation by explaining that India is running one of the “world’s largest vaccination drives” for COVID-19, with almost 90% of adults fully vaccinated.
She added that studies have indicated that the incidence of cutaneous adverse reactions following COVID-19 vaccination ranges from 1.0% to 1.9% and that dermatologists have encountered a “plethora” of related reactions.
Dr. Mohta emphasized that the “myriad presentations” of these reactions means that correlating clinical and pathological findings is “key” to understanding the underlying pathophysiology.
She and her colleagues therefore conducted a prospective, hospital-based study of patients who self-reported mucocutaneous adverse reactions from April to December 2021, within 4 weeks of receiving a COVID-19 vaccine.
They gathered information on the patients’ signs and symptoms, as well as the date of vaccine administration and the type of vaccine given, alongside a detailed medical history, including previous allergies, prior COVID-19 infection, and any comorbidities.
The patients also underwent a clinical examination and laboratory investigations, and their cases were assessed by two senior dermatologists to determine whether the association between the adverse event and COVID-19 vaccination was likely causal.
Dr. Mohta said that 132 adult patients, with an average age of 38.2 years, were identified as having vaccine-related reactions.
This included 84 (63.6%) patients with a mild reaction, defined as resolving with symptomatic treatment; 43 (32.6%) patients with a moderate reaction, defined as extensive and lasting for more than 4 weeks; and five (3.8%) patients with severe reactions, defined as systemic and potentially life-threatening.
The mild group included 21 patients with acute urticaria, with a mean onset of 1.2 days following vaccination, as well as 20 cases of maculopapular rash, with a mean onset of 2.4 days; 18 cases of pityriasis rosea, with a mean onset of 17.4 days; and nine cases of eruptive pseudoangioma, with a mean onset of 3.5 days.
There were 16 cases of lichen planus in the moderate group, with a mean onset of 22.7 days after COVID-19 vaccination; nine cases of herpes zoster, with a mean onset of 15.3 days; and one case of pityriasis lichenoides et varioliformis acuta (PLEVA), among others.
The severe group included two cases of erythroderma, with a mean onset of 9 days after vaccination; one case of drug rash with eosinophilia and systemic symptoms (DRESS), with a mean onset of 20 days; and one case each of subacute cutaneous lupus erythematosus (SCLE) and bullous pemphigoid, with mean onsets of 15 days and 14 days, respectively.
Turning to the histopathological results, Dr. Mohta explained that only 57 patients from their cohort agreed to have a skin biopsy.
Results of those skin biopsies showed that 21 (36.8%) patients had vaccine-related eruption of papules and plaques, predominantly spongiotic dermatitis. This correlated with the clinical diagnoses of pityriasis rosea, maculopapular and papulosquamous rash, and DRESS.
Lichenoid and interface dermatitis were seen in 13 (22.8%) patients, which correlated with the clinical diagnoses of lichen planus, PLEVA, and SCLE. Eleven (19.3%) patients had a dermal hypersensitivity reaction, equated to the clinical diagnoses of urticaria, and eruptive pseudoangioma.
Dr. Mohta acknowledged that the study was limited by the inability to calculate the “true prevalence of vaccine-associated reactions,” and because immunohistochemistry was not performed.
Session chair Saleem Taibjee, MD, department of dermatology, Dorset County Hospital NHS Foundation Trust, Dorchester, United Kingdom, congratulated Dr. Mohta on her “very interesting” presentation, highlighting their “extensive experience in such a large cohort of patients.”
He asked what type of COVID-19 vaccines the patients had received, and whether Dr. Mohta could provide any “insights into which patients you can safely give the vaccine again to, and those [to whom] you may avoid giving further doses.”
Dr. Mohta said that the majority of the patients in the study received the AstraZeneca COVID-19 vaccine, as that was the one most commonly used in India at the time, with around 30 patients receiving the Indian Covishield version of the AstraZeneca vaccine. (The two-dose AstraZeneca vaccine, which is cheaper to manufacture and easier to store at typical refrigerated temperatures than mRNA-based vaccines, has been authorized by the World Health Organization, the European Medicines Agency, and over 50 countries but has not been authorized in the United States.)
She added that none of the patients in the study with mild-to-moderate skin reactions were advised against receiving further doses” but that those with severe reactions “were advised not to take any further doses.”
Consequently, in the case of mild reactions, “further doses are not a contraindication,” Dr. Mohta said, but patients with more severe reactions should be considered on a “case by case basis.”
A version of this article first appeared on Medscape.com.
GLASGOW – Individuals given COVID-19 vaccination may experience a wide range of dermatologic reactions, some of which may be life-threatening, reveals a prospective Indian study that suggests histopathological assessment is key to understanding the cause.
Studying more than 130 patients who presented with vaccine-related dermatologic reactions, the researchers found that the most common acute adverse events were acute urticaria, generalized pruritus, and maculopapular rash.
Dermal hypersensitivity reactions occurred within 3 days of vaccination, which suggests the culprit is an immediate type 1 hypersensitivity reaction, said study presenter Alpana Mohta, MD, department of dermatology, Sardar Patel Medical College, Bikaner, Rajasthan, India. Most of the patients had received the AstraZeneca vaccine, she said.
, which occurred within 3-4 weeks of vaccination and could be a result of delayed hypersensitivity or a T cell–mediated skin reaction caused by “molecular mimicry with a viral epitope,” Dr. Mohta said.
The research was presented at the British Association of Dermatologists (BAD) 2022 Annual Meeting on July 5.
Dr. Mohta said that, given the “surge” in the number of people who have been vaccinated, it is “imperative as dermatologists” to maintain a “very high index of suspicion to differentiate reactions caused by vaccination” from other causes, and a proper assessment should be performed in “every patient” who presents with a possible reaction.
She also emphasized that “since so many clinical [COVID-19] variants are being encountered,” histopathological assessment could “help in better understanding the underlying pathophysiology” of every reaction.
Dr. Mohta began her presentation by explaining that India is running one of the “world’s largest vaccination drives” for COVID-19, with almost 90% of adults fully vaccinated.
She added that studies have indicated that the incidence of cutaneous adverse reactions following COVID-19 vaccination ranges from 1.0% to 1.9% and that dermatologists have encountered a “plethora” of related reactions.
Dr. Mohta emphasized that the “myriad presentations” of these reactions means that correlating clinical and pathological findings is “key” to understanding the underlying pathophysiology.
She and her colleagues therefore conducted a prospective, hospital-based study of patients who self-reported mucocutaneous adverse reactions from April to December 2021, within 4 weeks of receiving a COVID-19 vaccine.
They gathered information on the patients’ signs and symptoms, as well as the date of vaccine administration and the type of vaccine given, alongside a detailed medical history, including previous allergies, prior COVID-19 infection, and any comorbidities.
The patients also underwent a clinical examination and laboratory investigations, and their cases were assessed by two senior dermatologists to determine whether the association between the adverse event and COVID-19 vaccination was likely causal.
Dr. Mohta said that 132 adult patients, with an average age of 38.2 years, were identified as having vaccine-related reactions.
This included 84 (63.6%) patients with a mild reaction, defined as resolving with symptomatic treatment; 43 (32.6%) patients with a moderate reaction, defined as extensive and lasting for more than 4 weeks; and five (3.8%) patients with severe reactions, defined as systemic and potentially life-threatening.
The mild group included 21 patients with acute urticaria, with a mean onset of 1.2 days following vaccination, as well as 20 cases of maculopapular rash, with a mean onset of 2.4 days; 18 cases of pityriasis rosea, with a mean onset of 17.4 days; and nine cases of eruptive pseudoangioma, with a mean onset of 3.5 days.
There were 16 cases of lichen planus in the moderate group, with a mean onset of 22.7 days after COVID-19 vaccination; nine cases of herpes zoster, with a mean onset of 15.3 days; and one case of pityriasis lichenoides et varioliformis acuta (PLEVA), among others.
The severe group included two cases of erythroderma, with a mean onset of 9 days after vaccination; one case of drug rash with eosinophilia and systemic symptoms (DRESS), with a mean onset of 20 days; and one case each of subacute cutaneous lupus erythematosus (SCLE) and bullous pemphigoid, with mean onsets of 15 days and 14 days, respectively.
Turning to the histopathological results, Dr. Mohta explained that only 57 patients from their cohort agreed to have a skin biopsy.
Results of those skin biopsies showed that 21 (36.8%) patients had vaccine-related eruption of papules and plaques, predominantly spongiotic dermatitis. This correlated with the clinical diagnoses of pityriasis rosea, maculopapular and papulosquamous rash, and DRESS.
Lichenoid and interface dermatitis were seen in 13 (22.8%) patients, which correlated with the clinical diagnoses of lichen planus, PLEVA, and SCLE. Eleven (19.3%) patients had a dermal hypersensitivity reaction, equated to the clinical diagnoses of urticaria, and eruptive pseudoangioma.
Dr. Mohta acknowledged that the study was limited by the inability to calculate the “true prevalence of vaccine-associated reactions,” and because immunohistochemistry was not performed.
Session chair Saleem Taibjee, MD, department of dermatology, Dorset County Hospital NHS Foundation Trust, Dorchester, United Kingdom, congratulated Dr. Mohta on her “very interesting” presentation, highlighting their “extensive experience in such a large cohort of patients.”
He asked what type of COVID-19 vaccines the patients had received, and whether Dr. Mohta could provide any “insights into which patients you can safely give the vaccine again to, and those [to whom] you may avoid giving further doses.”
Dr. Mohta said that the majority of the patients in the study received the AstraZeneca COVID-19 vaccine, as that was the one most commonly used in India at the time, with around 30 patients receiving the Indian Covishield version of the AstraZeneca vaccine. (The two-dose AstraZeneca vaccine, which is cheaper to manufacture and easier to store at typical refrigerated temperatures than mRNA-based vaccines, has been authorized by the World Health Organization, the European Medicines Agency, and over 50 countries but has not been authorized in the United States.)
She added that none of the patients in the study with mild-to-moderate skin reactions were advised against receiving further doses” but that those with severe reactions “were advised not to take any further doses.”
Consequently, in the case of mild reactions, “further doses are not a contraindication,” Dr. Mohta said, but patients with more severe reactions should be considered on a “case by case basis.”
A version of this article first appeared on Medscape.com.
GLASGOW – Individuals given COVID-19 vaccination may experience a wide range of dermatologic reactions, some of which may be life-threatening, reveals a prospective Indian study that suggests histopathological assessment is key to understanding the cause.
Studying more than 130 patients who presented with vaccine-related dermatologic reactions, the researchers found that the most common acute adverse events were acute urticaria, generalized pruritus, and maculopapular rash.
Dermal hypersensitivity reactions occurred within 3 days of vaccination, which suggests the culprit is an immediate type 1 hypersensitivity reaction, said study presenter Alpana Mohta, MD, department of dermatology, Sardar Patel Medical College, Bikaner, Rajasthan, India. Most of the patients had received the AstraZeneca vaccine, she said.
, which occurred within 3-4 weeks of vaccination and could be a result of delayed hypersensitivity or a T cell–mediated skin reaction caused by “molecular mimicry with a viral epitope,” Dr. Mohta said.
The research was presented at the British Association of Dermatologists (BAD) 2022 Annual Meeting on July 5.
Dr. Mohta said that, given the “surge” in the number of people who have been vaccinated, it is “imperative as dermatologists” to maintain a “very high index of suspicion to differentiate reactions caused by vaccination” from other causes, and a proper assessment should be performed in “every patient” who presents with a possible reaction.
She also emphasized that “since so many clinical [COVID-19] variants are being encountered,” histopathological assessment could “help in better understanding the underlying pathophysiology” of every reaction.
Dr. Mohta began her presentation by explaining that India is running one of the “world’s largest vaccination drives” for COVID-19, with almost 90% of adults fully vaccinated.
She added that studies have indicated that the incidence of cutaneous adverse reactions following COVID-19 vaccination ranges from 1.0% to 1.9% and that dermatologists have encountered a “plethora” of related reactions.
Dr. Mohta emphasized that the “myriad presentations” of these reactions means that correlating clinical and pathological findings is “key” to understanding the underlying pathophysiology.
She and her colleagues therefore conducted a prospective, hospital-based study of patients who self-reported mucocutaneous adverse reactions from April to December 2021, within 4 weeks of receiving a COVID-19 vaccine.
They gathered information on the patients’ signs and symptoms, as well as the date of vaccine administration and the type of vaccine given, alongside a detailed medical history, including previous allergies, prior COVID-19 infection, and any comorbidities.
The patients also underwent a clinical examination and laboratory investigations, and their cases were assessed by two senior dermatologists to determine whether the association between the adverse event and COVID-19 vaccination was likely causal.
Dr. Mohta said that 132 adult patients, with an average age of 38.2 years, were identified as having vaccine-related reactions.
This included 84 (63.6%) patients with a mild reaction, defined as resolving with symptomatic treatment; 43 (32.6%) patients with a moderate reaction, defined as extensive and lasting for more than 4 weeks; and five (3.8%) patients with severe reactions, defined as systemic and potentially life-threatening.
The mild group included 21 patients with acute urticaria, with a mean onset of 1.2 days following vaccination, as well as 20 cases of maculopapular rash, with a mean onset of 2.4 days; 18 cases of pityriasis rosea, with a mean onset of 17.4 days; and nine cases of eruptive pseudoangioma, with a mean onset of 3.5 days.
There were 16 cases of lichen planus in the moderate group, with a mean onset of 22.7 days after COVID-19 vaccination; nine cases of herpes zoster, with a mean onset of 15.3 days; and one case of pityriasis lichenoides et varioliformis acuta (PLEVA), among others.
The severe group included two cases of erythroderma, with a mean onset of 9 days after vaccination; one case of drug rash with eosinophilia and systemic symptoms (DRESS), with a mean onset of 20 days; and one case each of subacute cutaneous lupus erythematosus (SCLE) and bullous pemphigoid, with mean onsets of 15 days and 14 days, respectively.
Turning to the histopathological results, Dr. Mohta explained that only 57 patients from their cohort agreed to have a skin biopsy.
Results of those skin biopsies showed that 21 (36.8%) patients had vaccine-related eruption of papules and plaques, predominantly spongiotic dermatitis. This correlated with the clinical diagnoses of pityriasis rosea, maculopapular and papulosquamous rash, and DRESS.
Lichenoid and interface dermatitis were seen in 13 (22.8%) patients, which correlated with the clinical diagnoses of lichen planus, PLEVA, and SCLE. Eleven (19.3%) patients had a dermal hypersensitivity reaction, equated to the clinical diagnoses of urticaria, and eruptive pseudoangioma.
Dr. Mohta acknowledged that the study was limited by the inability to calculate the “true prevalence of vaccine-associated reactions,” and because immunohistochemistry was not performed.
Session chair Saleem Taibjee, MD, department of dermatology, Dorset County Hospital NHS Foundation Trust, Dorchester, United Kingdom, congratulated Dr. Mohta on her “very interesting” presentation, highlighting their “extensive experience in such a large cohort of patients.”
He asked what type of COVID-19 vaccines the patients had received, and whether Dr. Mohta could provide any “insights into which patients you can safely give the vaccine again to, and those [to whom] you may avoid giving further doses.”
Dr. Mohta said that the majority of the patients in the study received the AstraZeneca COVID-19 vaccine, as that was the one most commonly used in India at the time, with around 30 patients receiving the Indian Covishield version of the AstraZeneca vaccine. (The two-dose AstraZeneca vaccine, which is cheaper to manufacture and easier to store at typical refrigerated temperatures than mRNA-based vaccines, has been authorized by the World Health Organization, the European Medicines Agency, and over 50 countries but has not been authorized in the United States.)
She added that none of the patients in the study with mild-to-moderate skin reactions were advised against receiving further doses” but that those with severe reactions “were advised not to take any further doses.”
Consequently, in the case of mild reactions, “further doses are not a contraindication,” Dr. Mohta said, but patients with more severe reactions should be considered on a “case by case basis.”
A version of this article first appeared on Medscape.com.
U.S. allows pharmacists to prescribe Paxlovid directly
The Food and Drug Administration revised the drug’s emergency use authorization on July 6, letting state-licensed pharmacists screen patients and determine if they are eligible for Paxlovid, according to The Associated Press.
Previously, only doctors could prescribe the antiviral drug, the AP reported. With some limits, pharmacists can now prescribe the medication for patients who face high risks for severe COVID-19.
“The FDA recognizes the important role pharmacists have played and continue to play in combating this pandemic,” Patrizia Cavazzoni, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement.
“Since Paxlovid must be taken within 5 days after symptoms begin, authorizing state-licensed pharmacists to prescribe Paxlovid could expand access to timely treatment for some patients who are eligible to receive this drug for the treatment of COVID-19,” she said.
Tom Kraus, the vice president of government relations at the American Society of Health-System Pharmacists, said in a statement that the organization was “pleased to see the FDA remove this barrier to patients’ access to this critical treatment.”
“Pharmacists have played a vital role in our pandemic response efforts and are well-positioned to help patients, particularly those in rural and underserved communities, benefit from this medication,” he said.
But some doctor’s groups questioned the FDA’s move. Jack Resneck Jr., MD, the president of the American Medical Association, said in a statement that prescribing Paxlovid “requires knowledge of a patient’s medical history, as well as clinical monitoring for side effects and follow-up care to determine whether a patient is improving” – requirements that are “far beyond a pharmacist’s scope and training.”
“In the fight against a virus that has killed more than a million people in the United States and is still extremely present and transmissible, patients will get the best, most comprehensive care from physician-led teams – teams that include pharmacists. But, whenever possible, prescribing decisions should be made by a physician with knowledge of a patient’s medical history and the ability to follow up. To ensure the best possible care for COVID-19 patients, we urge people who test positive to discuss treatment options with their physician, if they have one,” he said.
After testing positive for COVID-19, patients should first consider seeking care from their regular health care provider or locating a Test-to-Treat site in their area, the FDA said. Although the latest update allows pharmacists to prescribe Paxlovid, community pharmacies that don’t yet take part in the Test-to-Treat program can decide if they will offer the prescription service to patients.
Paxlovid is authorized to treat mild to moderate COVID-19 in adults and in kids ages 12 and older who weigh at least 88 pounds. Patients who report a positive at-home test are eligible for Paxlovid under the FDA authorization.
If patients want to seek a prescription directly from a pharmacist, they should bring electronic or printed health records from the past year, including their most recent reports of blood work, so the pharmacist can review for kidney or liver problems. Pharmacists can also get this information from the patient’s health care provider.
In addition, patients should bring a list of all medications they are taking, including over-the-counter medications, so the pharmacist can screen for drugs that can have serious interactions with Paxlovid.
Under the limits in the updated FDA authorization, pharmacists should refer patients for more screening if Paxlovid isn’t a good option or if there’s not enough information to find out how well their kidneys or liver works, as well as potential drug interactions.
Paxlovid is intended for people with COVID-19 who face the highest risks for serious disease, the AP reported, including older adults and those with health conditions such as heart disease, obesity, cancer, or diabetes. It isn’t recommended for people with severe kidney or liver problems. A course of treatment requires three pills twice a day for 5 days.
A version of this article first appeared on WebMD.com.
The Food and Drug Administration revised the drug’s emergency use authorization on July 6, letting state-licensed pharmacists screen patients and determine if they are eligible for Paxlovid, according to The Associated Press.
Previously, only doctors could prescribe the antiviral drug, the AP reported. With some limits, pharmacists can now prescribe the medication for patients who face high risks for severe COVID-19.
“The FDA recognizes the important role pharmacists have played and continue to play in combating this pandemic,” Patrizia Cavazzoni, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement.
“Since Paxlovid must be taken within 5 days after symptoms begin, authorizing state-licensed pharmacists to prescribe Paxlovid could expand access to timely treatment for some patients who are eligible to receive this drug for the treatment of COVID-19,” she said.
Tom Kraus, the vice president of government relations at the American Society of Health-System Pharmacists, said in a statement that the organization was “pleased to see the FDA remove this barrier to patients’ access to this critical treatment.”
“Pharmacists have played a vital role in our pandemic response efforts and are well-positioned to help patients, particularly those in rural and underserved communities, benefit from this medication,” he said.
But some doctor’s groups questioned the FDA’s move. Jack Resneck Jr., MD, the president of the American Medical Association, said in a statement that prescribing Paxlovid “requires knowledge of a patient’s medical history, as well as clinical monitoring for side effects and follow-up care to determine whether a patient is improving” – requirements that are “far beyond a pharmacist’s scope and training.”
“In the fight against a virus that has killed more than a million people in the United States and is still extremely present and transmissible, patients will get the best, most comprehensive care from physician-led teams – teams that include pharmacists. But, whenever possible, prescribing decisions should be made by a physician with knowledge of a patient’s medical history and the ability to follow up. To ensure the best possible care for COVID-19 patients, we urge people who test positive to discuss treatment options with their physician, if they have one,” he said.
After testing positive for COVID-19, patients should first consider seeking care from their regular health care provider or locating a Test-to-Treat site in their area, the FDA said. Although the latest update allows pharmacists to prescribe Paxlovid, community pharmacies that don’t yet take part in the Test-to-Treat program can decide if they will offer the prescription service to patients.
Paxlovid is authorized to treat mild to moderate COVID-19 in adults and in kids ages 12 and older who weigh at least 88 pounds. Patients who report a positive at-home test are eligible for Paxlovid under the FDA authorization.
If patients want to seek a prescription directly from a pharmacist, they should bring electronic or printed health records from the past year, including their most recent reports of blood work, so the pharmacist can review for kidney or liver problems. Pharmacists can also get this information from the patient’s health care provider.
In addition, patients should bring a list of all medications they are taking, including over-the-counter medications, so the pharmacist can screen for drugs that can have serious interactions with Paxlovid.
Under the limits in the updated FDA authorization, pharmacists should refer patients for more screening if Paxlovid isn’t a good option or if there’s not enough information to find out how well their kidneys or liver works, as well as potential drug interactions.
Paxlovid is intended for people with COVID-19 who face the highest risks for serious disease, the AP reported, including older adults and those with health conditions such as heart disease, obesity, cancer, or diabetes. It isn’t recommended for people with severe kidney or liver problems. A course of treatment requires three pills twice a day for 5 days.
A version of this article first appeared on WebMD.com.
The Food and Drug Administration revised the drug’s emergency use authorization on July 6, letting state-licensed pharmacists screen patients and determine if they are eligible for Paxlovid, according to The Associated Press.
Previously, only doctors could prescribe the antiviral drug, the AP reported. With some limits, pharmacists can now prescribe the medication for patients who face high risks for severe COVID-19.
“The FDA recognizes the important role pharmacists have played and continue to play in combating this pandemic,” Patrizia Cavazzoni, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement.
“Since Paxlovid must be taken within 5 days after symptoms begin, authorizing state-licensed pharmacists to prescribe Paxlovid could expand access to timely treatment for some patients who are eligible to receive this drug for the treatment of COVID-19,” she said.
Tom Kraus, the vice president of government relations at the American Society of Health-System Pharmacists, said in a statement that the organization was “pleased to see the FDA remove this barrier to patients’ access to this critical treatment.”
“Pharmacists have played a vital role in our pandemic response efforts and are well-positioned to help patients, particularly those in rural and underserved communities, benefit from this medication,” he said.
But some doctor’s groups questioned the FDA’s move. Jack Resneck Jr., MD, the president of the American Medical Association, said in a statement that prescribing Paxlovid “requires knowledge of a patient’s medical history, as well as clinical monitoring for side effects and follow-up care to determine whether a patient is improving” – requirements that are “far beyond a pharmacist’s scope and training.”
“In the fight against a virus that has killed more than a million people in the United States and is still extremely present and transmissible, patients will get the best, most comprehensive care from physician-led teams – teams that include pharmacists. But, whenever possible, prescribing decisions should be made by a physician with knowledge of a patient’s medical history and the ability to follow up. To ensure the best possible care for COVID-19 patients, we urge people who test positive to discuss treatment options with their physician, if they have one,” he said.
After testing positive for COVID-19, patients should first consider seeking care from their regular health care provider or locating a Test-to-Treat site in their area, the FDA said. Although the latest update allows pharmacists to prescribe Paxlovid, community pharmacies that don’t yet take part in the Test-to-Treat program can decide if they will offer the prescription service to patients.
Paxlovid is authorized to treat mild to moderate COVID-19 in adults and in kids ages 12 and older who weigh at least 88 pounds. Patients who report a positive at-home test are eligible for Paxlovid under the FDA authorization.
If patients want to seek a prescription directly from a pharmacist, they should bring electronic or printed health records from the past year, including their most recent reports of blood work, so the pharmacist can review for kidney or liver problems. Pharmacists can also get this information from the patient’s health care provider.
In addition, patients should bring a list of all medications they are taking, including over-the-counter medications, so the pharmacist can screen for drugs that can have serious interactions with Paxlovid.
Under the limits in the updated FDA authorization, pharmacists should refer patients for more screening if Paxlovid isn’t a good option or if there’s not enough information to find out how well their kidneys or liver works, as well as potential drug interactions.
Paxlovid is intended for people with COVID-19 who face the highest risks for serious disease, the AP reported, including older adults and those with health conditions such as heart disease, obesity, cancer, or diabetes. It isn’t recommended for people with severe kidney or liver problems. A course of treatment requires three pills twice a day for 5 days.
A version of this article first appeared on WebMD.com.
Eczema severity, time spent on management strongly associated with overall disease burden
. However, AD severity and spending 11 hours or more per week managing the condition did correlate with higher overall disease burden.
“Research has documented the disease burden of AD, including its visible nature and the effect on itch and sleep, but knowledge gaps remain,” Aaron M. Drucker, MD, of the division of dermatology at the University of Toronto, and colleagues wrote in the study published online in JAMA Dermatology. “Gaps include a poor understanding of symptoms other than itch, patients’ treatment experience, and how different elements of burden of disease interact.”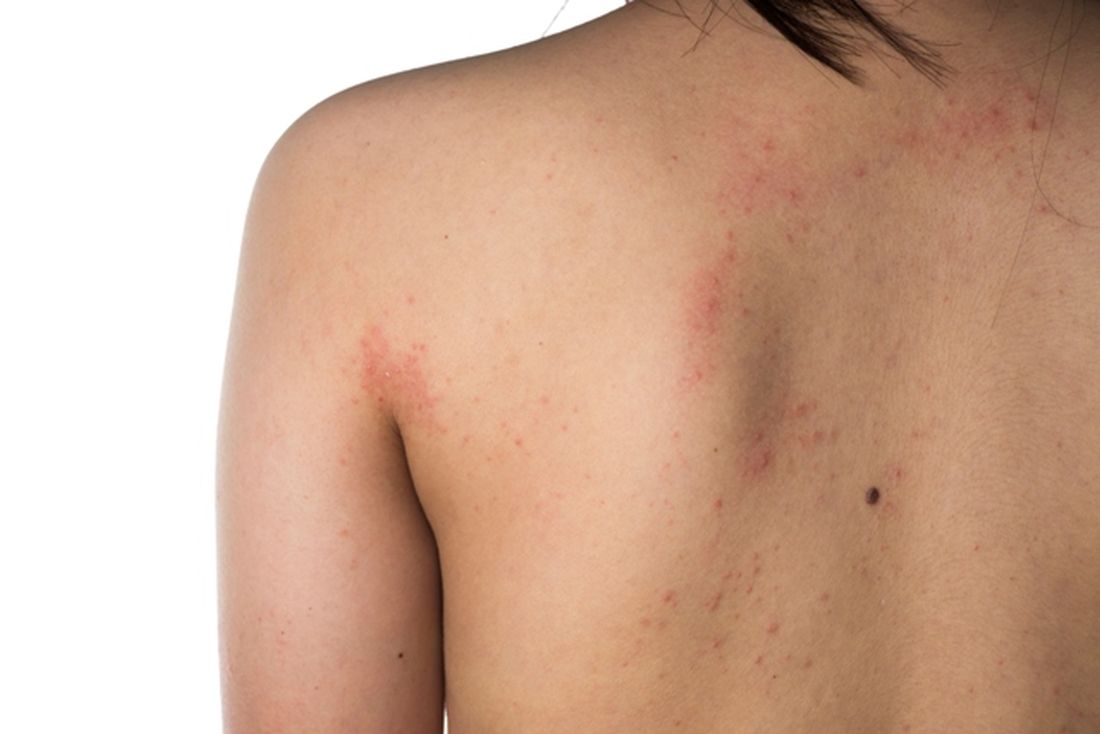
Dr. Drucker and colleagues collected data from an externally led patient-focused drug development survey on AD, a 32-item questionnaire that was administered electronically between Aug. 1, 2019, and Oct. 11, 2019. Respondents were asked to rate the overall impact of their AD in the past months and the specific elements of disease burden on a 1-5 scale, with 1 meaning no impact, and 5 meaning a significant impact. They were also asked to rate current mood changes and mood changes at the worst point of AD on a 4-point scale that ranged from “not present” to “severe.” The researchers used multivariable ordinal regression to examine associations between demographic and clinical variables and patient-reported overall AD impact scores.
Survey results
Of the 1,065 respondents, 33% were aged 18-34 years, 50% were aged 35-50 years, 17% were aged 65 years or older, and 83% were female. Nearly half (45%) reported having moderate AD, while 28% had severe AD. When asked about the overall disease burden of AD symptoms in the past month, 30% reported a significant impact on life, 28% reported a moderate impact score, 21% reported a high impact score, 18% reported a low impact score, and 3% of respondents reported no impact.
In the multivariable proportional odds analysis, moderate AD (odds ratio [OR], 4.13) and severe AD (OR, 13.63) were both associated with greater disease burden compared with mild AD. Also, spending 11 or more hours per week managing AD symptoms was associated with greater disease burden compared with 0 to 4 hours (an OR of 2.67 for 11-20 hours per week spent managing AD and OR of 5.34 for 21 or more hours per week spent managing AD).
Correlations between specific impact domains such as sleep, cognitive thinking, and physical activity and overall AD impact scores ranged from weak to moderate, and no individual aspect of disease burden correlated strongly with overall impact scores. The researchers observed similar results after they stratified the analysis by age, current severity, and time spent managing AD.
In other findings, 40% of study participants reported mild changes in mood related to their AD, 30% reported moderate changes, 9% reported severe changes, while the remainder reported no changes in mood. The variable most strongly associated with current mood changes was having severe AD at the time of the survey (OR 5.29).
Understanding of disease burden ‘limited’
“Atopic dermatitis is associated with an immense clinical burden,” said Raj Chovatiya, MD, PhD, assistant professor in the department of dermatology at Northwestern University, Chicago, who was asked to comment on the study. “However, our understanding of disease burden from the patient perspective is limited,” he added.

“Interestingly, no single specific element of disease burden was strongly correlated with overall burden, further supporting the complex, multidimensional nature” of the impact of AD, he said, noting that the study “highlights the need for clinicians to look beyond the skin when it comes to AD and underscores the need for additional research to better understand the patient and caregiver perspective.”
Zelma Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was also asked to comment on the study, noted that aside from the well discussed impact and burden of itch and its impact on sleep loss, much remains to be learned about the full impact of AD, particularly among adults.

“For example, it is commonly accepted and expected that patients with more severe AD likely experience higher disease burden, but are there other factors that can influence this risk?” she asked. “Can we explain the high impact of AD disease aside from the level of disease severity, particularly among adults with AD?”
The study, she added, “is important because it provides additional insights into those possible factors, including ‘time spent managing their disease’ and ‘associated depression.’ In particular, understanding the association between ‘time spent managing their disease’ and higher disease burden is critical because, in my opinion, it emphasizes the need to develop better strategies for improving the care of patients with AD including the development of more efficacious and safer treatment strategies.”
Dr. Drucker and colleagues acknowledged certain limitations of the analysis, including its cross-sectional design, the potential for selection bias, and the fact that it did not use the patient-oriented outcome measure or the dermatology life quality index. “Further work to address the complex burden of AD, including strategies to reduce time spent managing AD, and understanding the fullness of the patient experience is needed,” they concluded.
The work was supported in part by a grant from the National Eczema Association (NEA). Dr. Drucker reported that he receives compensation from the British Journal of Dermatology (as reviewer and section editor), American Academy of Dermatology (guidelines writer), and NEA (grant reviewer). Coauthors representing the NEA and other patient organizations including the Allergy & Asthma Network, Asthma and Allergy Foundation of America, Global Parents for Eczema Research, and International Topical Steroid Awareness Network received organizational grants (Pfizer) and sponsorship funding for these analyses from AbbVie, Eli Lilly, Incyte, LEO Pharma, Regeneron Pharmaceuticals, and Sanofi Genzyme.
Dr. Chovatiya disclosed that he has served as an advisory board member, consultant, speaker, and/or investigator for AbbVie, Arcutis, Arena, Beiersdorf, Bristol Myers Squibb, Dermavant, Eli Lilly and Company, EPI Health, Incyte, L’Oréal, the NEA, Pfizer, Regeneron, Sanofi, and UCB.
Dr. Chiesa Fuxench disclosed that she has received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, and Vanda for work related to AD She has served as consultant for the Asthma and Allergy Foundation of America, NEA, AbbVie, Incyte Corporation, and Pfizer; and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer.
. However, AD severity and spending 11 hours or more per week managing the condition did correlate with higher overall disease burden.
“Research has documented the disease burden of AD, including its visible nature and the effect on itch and sleep, but knowledge gaps remain,” Aaron M. Drucker, MD, of the division of dermatology at the University of Toronto, and colleagues wrote in the study published online in JAMA Dermatology. “Gaps include a poor understanding of symptoms other than itch, patients’ treatment experience, and how different elements of burden of disease interact.”
Dr. Drucker and colleagues collected data from an externally led patient-focused drug development survey on AD, a 32-item questionnaire that was administered electronically between Aug. 1, 2019, and Oct. 11, 2019. Respondents were asked to rate the overall impact of their AD in the past months and the specific elements of disease burden on a 1-5 scale, with 1 meaning no impact, and 5 meaning a significant impact. They were also asked to rate current mood changes and mood changes at the worst point of AD on a 4-point scale that ranged from “not present” to “severe.” The researchers used multivariable ordinal regression to examine associations between demographic and clinical variables and patient-reported overall AD impact scores.
Survey results
Of the 1,065 respondents, 33% were aged 18-34 years, 50% were aged 35-50 years, 17% were aged 65 years or older, and 83% were female. Nearly half (45%) reported having moderate AD, while 28% had severe AD. When asked about the overall disease burden of AD symptoms in the past month, 30% reported a significant impact on life, 28% reported a moderate impact score, 21% reported a high impact score, 18% reported a low impact score, and 3% of respondents reported no impact.
In the multivariable proportional odds analysis, moderate AD (odds ratio [OR], 4.13) and severe AD (OR, 13.63) were both associated with greater disease burden compared with mild AD. Also, spending 11 or more hours per week managing AD symptoms was associated with greater disease burden compared with 0 to 4 hours (an OR of 2.67 for 11-20 hours per week spent managing AD and OR of 5.34 for 21 or more hours per week spent managing AD).
Correlations between specific impact domains such as sleep, cognitive thinking, and physical activity and overall AD impact scores ranged from weak to moderate, and no individual aspect of disease burden correlated strongly with overall impact scores. The researchers observed similar results after they stratified the analysis by age, current severity, and time spent managing AD.
In other findings, 40% of study participants reported mild changes in mood related to their AD, 30% reported moderate changes, 9% reported severe changes, while the remainder reported no changes in mood. The variable most strongly associated with current mood changes was having severe AD at the time of the survey (OR 5.29).
Understanding of disease burden ‘limited’
“Atopic dermatitis is associated with an immense clinical burden,” said Raj Chovatiya, MD, PhD, assistant professor in the department of dermatology at Northwestern University, Chicago, who was asked to comment on the study. “However, our understanding of disease burden from the patient perspective is limited,” he added.
“Interestingly, no single specific element of disease burden was strongly correlated with overall burden, further supporting the complex, multidimensional nature” of the impact of AD, he said, noting that the study “highlights the need for clinicians to look beyond the skin when it comes to AD and underscores the need for additional research to better understand the patient and caregiver perspective.”
Zelma Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was also asked to comment on the study, noted that aside from the well discussed impact and burden of itch and its impact on sleep loss, much remains to be learned about the full impact of AD, particularly among adults.
“For example, it is commonly accepted and expected that patients with more severe AD likely experience higher disease burden, but are there other factors that can influence this risk?” she asked. “Can we explain the high impact of AD disease aside from the level of disease severity, particularly among adults with AD?”
The study, she added, “is important because it provides additional insights into those possible factors, including ‘time spent managing their disease’ and ‘associated depression.’ In particular, understanding the association between ‘time spent managing their disease’ and higher disease burden is critical because, in my opinion, it emphasizes the need to develop better strategies for improving the care of patients with AD including the development of more efficacious and safer treatment strategies.”
Dr. Drucker and colleagues acknowledged certain limitations of the analysis, including its cross-sectional design, the potential for selection bias, and the fact that it did not use the patient-oriented outcome measure or the dermatology life quality index. “Further work to address the complex burden of AD, including strategies to reduce time spent managing AD, and understanding the fullness of the patient experience is needed,” they concluded.
The work was supported in part by a grant from the National Eczema Association (NEA). Dr. Drucker reported that he receives compensation from the British Journal of Dermatology (as reviewer and section editor), American Academy of Dermatology (guidelines writer), and NEA (grant reviewer). Coauthors representing the NEA and other patient organizations including the Allergy & Asthma Network, Asthma and Allergy Foundation of America, Global Parents for Eczema Research, and International Topical Steroid Awareness Network received organizational grants (Pfizer) and sponsorship funding for these analyses from AbbVie, Eli Lilly, Incyte, LEO Pharma, Regeneron Pharmaceuticals, and Sanofi Genzyme.
Dr. Chovatiya disclosed that he has served as an advisory board member, consultant, speaker, and/or investigator for AbbVie, Arcutis, Arena, Beiersdorf, Bristol Myers Squibb, Dermavant, Eli Lilly and Company, EPI Health, Incyte, L’Oréal, the NEA, Pfizer, Regeneron, Sanofi, and UCB.
Dr. Chiesa Fuxench disclosed that she has received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, and Vanda for work related to AD She has served as consultant for the Asthma and Allergy Foundation of America, NEA, AbbVie, Incyte Corporation, and Pfizer; and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer.
. However, AD severity and spending 11 hours or more per week managing the condition did correlate with higher overall disease burden.
“Research has documented the disease burden of AD, including its visible nature and the effect on itch and sleep, but knowledge gaps remain,” Aaron M. Drucker, MD, of the division of dermatology at the University of Toronto, and colleagues wrote in the study published online in JAMA Dermatology. “Gaps include a poor understanding of symptoms other than itch, patients’ treatment experience, and how different elements of burden of disease interact.”
Dr. Drucker and colleagues collected data from an externally led patient-focused drug development survey on AD, a 32-item questionnaire that was administered electronically between Aug. 1, 2019, and Oct. 11, 2019. Respondents were asked to rate the overall impact of their AD in the past months and the specific elements of disease burden on a 1-5 scale, with 1 meaning no impact, and 5 meaning a significant impact. They were also asked to rate current mood changes and mood changes at the worst point of AD on a 4-point scale that ranged from “not present” to “severe.” The researchers used multivariable ordinal regression to examine associations between demographic and clinical variables and patient-reported overall AD impact scores.
Survey results
Of the 1,065 respondents, 33% were aged 18-34 years, 50% were aged 35-50 years, 17% were aged 65 years or older, and 83% were female. Nearly half (45%) reported having moderate AD, while 28% had severe AD. When asked about the overall disease burden of AD symptoms in the past month, 30% reported a significant impact on life, 28% reported a moderate impact score, 21% reported a high impact score, 18% reported a low impact score, and 3% of respondents reported no impact.
In the multivariable proportional odds analysis, moderate AD (odds ratio [OR], 4.13) and severe AD (OR, 13.63) were both associated with greater disease burden compared with mild AD. Also, spending 11 or more hours per week managing AD symptoms was associated with greater disease burden compared with 0 to 4 hours (an OR of 2.67 for 11-20 hours per week spent managing AD and OR of 5.34 for 21 or more hours per week spent managing AD).
Correlations between specific impact domains such as sleep, cognitive thinking, and physical activity and overall AD impact scores ranged from weak to moderate, and no individual aspect of disease burden correlated strongly with overall impact scores. The researchers observed similar results after they stratified the analysis by age, current severity, and time spent managing AD.
In other findings, 40% of study participants reported mild changes in mood related to their AD, 30% reported moderate changes, 9% reported severe changes, while the remainder reported no changes in mood. The variable most strongly associated with current mood changes was having severe AD at the time of the survey (OR 5.29).
Understanding of disease burden ‘limited’
“Atopic dermatitis is associated with an immense clinical burden,” said Raj Chovatiya, MD, PhD, assistant professor in the department of dermatology at Northwestern University, Chicago, who was asked to comment on the study. “However, our understanding of disease burden from the patient perspective is limited,” he added.
“Interestingly, no single specific element of disease burden was strongly correlated with overall burden, further supporting the complex, multidimensional nature” of the impact of AD, he said, noting that the study “highlights the need for clinicians to look beyond the skin when it comes to AD and underscores the need for additional research to better understand the patient and caregiver perspective.”
Zelma Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, who was also asked to comment on the study, noted that aside from the well discussed impact and burden of itch and its impact on sleep loss, much remains to be learned about the full impact of AD, particularly among adults.
“For example, it is commonly accepted and expected that patients with more severe AD likely experience higher disease burden, but are there other factors that can influence this risk?” she asked. “Can we explain the high impact of AD disease aside from the level of disease severity, particularly among adults with AD?”
The study, she added, “is important because it provides additional insights into those possible factors, including ‘time spent managing their disease’ and ‘associated depression.’ In particular, understanding the association between ‘time spent managing their disease’ and higher disease burden is critical because, in my opinion, it emphasizes the need to develop better strategies for improving the care of patients with AD including the development of more efficacious and safer treatment strategies.”
Dr. Drucker and colleagues acknowledged certain limitations of the analysis, including its cross-sectional design, the potential for selection bias, and the fact that it did not use the patient-oriented outcome measure or the dermatology life quality index. “Further work to address the complex burden of AD, including strategies to reduce time spent managing AD, and understanding the fullness of the patient experience is needed,” they concluded.
The work was supported in part by a grant from the National Eczema Association (NEA). Dr. Drucker reported that he receives compensation from the British Journal of Dermatology (as reviewer and section editor), American Academy of Dermatology (guidelines writer), and NEA (grant reviewer). Coauthors representing the NEA and other patient organizations including the Allergy & Asthma Network, Asthma and Allergy Foundation of America, Global Parents for Eczema Research, and International Topical Steroid Awareness Network received organizational grants (Pfizer) and sponsorship funding for these analyses from AbbVie, Eli Lilly, Incyte, LEO Pharma, Regeneron Pharmaceuticals, and Sanofi Genzyme.
Dr. Chovatiya disclosed that he has served as an advisory board member, consultant, speaker, and/or investigator for AbbVie, Arcutis, Arena, Beiersdorf, Bristol Myers Squibb, Dermavant, Eli Lilly and Company, EPI Health, Incyte, L’Oréal, the NEA, Pfizer, Regeneron, Sanofi, and UCB.
Dr. Chiesa Fuxench disclosed that she has received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, and Vanda for work related to AD She has served as consultant for the Asthma and Allergy Foundation of America, NEA, AbbVie, Incyte Corporation, and Pfizer; and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer.
FROM JAMA DERMATOLOGY
Study explores gender differences in pediatric melanoma
INDIANAPOLIS – .
In addition, male gender was independently associated with increased mortality, but age was not.
Those are key findings from a retrospective cohort analysis of nearly 5,000 records from the National Cancer Database.

“There are multiple studies from primarily adult populations showing females with melanoma have a different presentation and better outcomes than males,” co-first author Rebecca M. Thiede, MD, a dermatologist at the University of Arizona, Tucson, said in an interview with this news organization in advance of the annual meeting of the Society for Pediatric Dermatology, where the abstract was presented during a poster session. “However, because melanoma is so rare in younger patients, little is known about gender differences in presentation and survival in pediatric and adolescent patients. To our knowledge, this is one of the largest studies to date in this population, and the first to explore gender differences in detail in pediatric and adolescent patients with melanoma.”
Working with co-first author Sabrina Dahak, a fourth-year medical student at the University of Arizona, Phoenix, Dr. Thiede and colleagues retrospectively analyzed the National Cancer Database to identify biopsy-confirmed invasive primary cutaneous melanoma cases diagnosed in patients 0-21 years of age between 2004 and 2018. The search yielded 4,645 cases, and the researchers used American Academy of Pediatrics definitions to categorize the patients by age, from infancy (birth to 2 years), to childhood (3-10 years), early adolescence (11-14 years), middle adolescence (15-17 years), and late adolescence (18-21 years). They used the Kaplan Meier analysis to determine overall survival and multivariate Cox regression to determine independent survival predictors.
Of the 4,645 pediatric melanoma cases, 63.4% were in females and 36.6% were in males, a difference that was significant (P < .001). Dr. Thiede and colleagues also observed a significant relationship between primary site and gender (P < .001). Primary sites included the trunk (34.3% of females vs. 32.9% of males, respectively), head and neck (16.4% vs. 30.9%), upper extremities (19.5% vs. 16%), lower extremities (27.9% vs. 16.5%), and “unspecified” (1.9% vs. 3.7%).
Females had higher rates of superficial spreading melanoma while males were affected by nodular melanoma more often. For example, the median Breslow depth was higher for males (1.05 mm; interquartile range [IQR] 0.50-2.31) than for females (0.80 mm; IQR, 0.40-1.67; P < .001).
Although females accounted for a higher percentage of cases than males overall, from birth to 17 years, a higher percentage of males than females were found to have later stage of melanoma at time of diagnosis: Females were more likely to be diagnosed with stage I disease (67.8%) than were males (53.6%), and males were more likely than were females to be diagnosed with stages II (15.9% vs. 12.3%), III (27.1% vs. 18.3%), and IV disease (3.3% vs. 1.6%; P < .001 for all).
In other findings, the 5- and 10-year overall survival rates were higher for females (95.9% and 93.9%, respectively) than for males (92.0% vs. 86.7%, respectively; P < .001). However, by age group, overall survival rates were similar between females and males among infants, children, and those in early adolescence – but not for those in middle adolescence (96.7% vs. 91.9%; P < .001) or late adolescence (95.7% vs. 90.4%; P < .001).
When the researchers adjusted for confounding variables, male gender was independently associated with an increased risk of death (adjusted hazard ratio 1.37; P < .001), but age was not.
“It was particularly surprising to see that even at such a young age, there is a significant difference in overall survival between males and females, where females have better outcomes than males,” Dr. Thiede said. “When examining pediatric and adolescent patients, it is essential to maintain cutaneous melanoma on the differential,” she advised. “It is important for clinicians to perform a thorough exam at annual visits particularly for those at high risk for melanoma to catch this rare but potentially devastating diagnosis.”
She acknowledged certain limitations of the study, including its reliance on one database, “as comparing multiple databases would strengthen the conclusions,” she said. “There was some missing data present in our dataset, and a large percentage of the histologic subtypes were unspecified, both of which are common issues with cancer registries. An additional limitation is related to the low death rates in adolescent and pediatric patients, which may impact the analysis related to survival and independent predictors of survival.”
Asked to comment on the study results, Carrie C. Coughlin, MD, who directs the section of pediatric dermatology Washington University/St. Louis Children’s Hospital, said that the finding that males were more likely to present with stage II or higher disease compared with females “could be related to their finding that females had more superficial spreading melanomas, whereas males had more nodular melanoma.” Those differences “could influence how providers evaluate melanocytic lesions in children,” she added.

Dr. Coughlin, who directs the pediatric dermatology fellowship at Washington University/St. Louis Children’s Hospital, said it was “interesting” that the authors found no association between older age and an increased risk of death. “It would be helpful to have more data about melanoma subtype, including information about Spitz or Spitzoid melanomas,” she said. “Also, knowing the distribution of melanoma across the age categories could provide more insight into their data.”
Ms. Dahak received an award from the National Cancer Institute to fund travel for presentation of this study at the SPD meeting. No other financial conflicts were reported by the researchers. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance (PeDRA) and the International Immunosuppression and Transplant Skin Cancer Collaborative.
INDIANAPOLIS – .
In addition, male gender was independently associated with increased mortality, but age was not.
Those are key findings from a retrospective cohort analysis of nearly 5,000 records from the National Cancer Database.
“There are multiple studies from primarily adult populations showing females with melanoma have a different presentation and better outcomes than males,” co-first author Rebecca M. Thiede, MD, a dermatologist at the University of Arizona, Tucson, said in an interview with this news organization in advance of the annual meeting of the Society for Pediatric Dermatology, where the abstract was presented during a poster session. “However, because melanoma is so rare in younger patients, little is known about gender differences in presentation and survival in pediatric and adolescent patients. To our knowledge, this is one of the largest studies to date in this population, and the first to explore gender differences in detail in pediatric and adolescent patients with melanoma.”
Working with co-first author Sabrina Dahak, a fourth-year medical student at the University of Arizona, Phoenix, Dr. Thiede and colleagues retrospectively analyzed the National Cancer Database to identify biopsy-confirmed invasive primary cutaneous melanoma cases diagnosed in patients 0-21 years of age between 2004 and 2018. The search yielded 4,645 cases, and the researchers used American Academy of Pediatrics definitions to categorize the patients by age, from infancy (birth to 2 years), to childhood (3-10 years), early adolescence (11-14 years), middle adolescence (15-17 years), and late adolescence (18-21 years). They used the Kaplan Meier analysis to determine overall survival and multivariate Cox regression to determine independent survival predictors.
Of the 4,645 pediatric melanoma cases, 63.4% were in females and 36.6% were in males, a difference that was significant (P < .001). Dr. Thiede and colleagues also observed a significant relationship between primary site and gender (P < .001). Primary sites included the trunk (34.3% of females vs. 32.9% of males, respectively), head and neck (16.4% vs. 30.9%), upper extremities (19.5% vs. 16%), lower extremities (27.9% vs. 16.5%), and “unspecified” (1.9% vs. 3.7%).
Females had higher rates of superficial spreading melanoma while males were affected by nodular melanoma more often. For example, the median Breslow depth was higher for males (1.05 mm; interquartile range [IQR] 0.50-2.31) than for females (0.80 mm; IQR, 0.40-1.67; P < .001).
Although females accounted for a higher percentage of cases than males overall, from birth to 17 years, a higher percentage of males than females were found to have later stage of melanoma at time of diagnosis: Females were more likely to be diagnosed with stage I disease (67.8%) than were males (53.6%), and males were more likely than were females to be diagnosed with stages II (15.9% vs. 12.3%), III (27.1% vs. 18.3%), and IV disease (3.3% vs. 1.6%; P < .001 for all).
In other findings, the 5- and 10-year overall survival rates were higher for females (95.9% and 93.9%, respectively) than for males (92.0% vs. 86.7%, respectively; P < .001). However, by age group, overall survival rates were similar between females and males among infants, children, and those in early adolescence – but not for those in middle adolescence (96.7% vs. 91.9%; P < .001) or late adolescence (95.7% vs. 90.4%; P < .001).
When the researchers adjusted for confounding variables, male gender was independently associated with an increased risk of death (adjusted hazard ratio 1.37; P < .001), but age was not.
“It was particularly surprising to see that even at such a young age, there is a significant difference in overall survival between males and females, where females have better outcomes than males,” Dr. Thiede said. “When examining pediatric and adolescent patients, it is essential to maintain cutaneous melanoma on the differential,” she advised. “It is important for clinicians to perform a thorough exam at annual visits particularly for those at high risk for melanoma to catch this rare but potentially devastating diagnosis.”
She acknowledged certain limitations of the study, including its reliance on one database, “as comparing multiple databases would strengthen the conclusions,” she said. “There was some missing data present in our dataset, and a large percentage of the histologic subtypes were unspecified, both of which are common issues with cancer registries. An additional limitation is related to the low death rates in adolescent and pediatric patients, which may impact the analysis related to survival and independent predictors of survival.”
Asked to comment on the study results, Carrie C. Coughlin, MD, who directs the section of pediatric dermatology Washington University/St. Louis Children’s Hospital, said that the finding that males were more likely to present with stage II or higher disease compared with females “could be related to their finding that females had more superficial spreading melanomas, whereas males had more nodular melanoma.” Those differences “could influence how providers evaluate melanocytic lesions in children,” she added.
Dr. Coughlin, who directs the pediatric dermatology fellowship at Washington University/St. Louis Children’s Hospital, said it was “interesting” that the authors found no association between older age and an increased risk of death. “It would be helpful to have more data about melanoma subtype, including information about Spitz or Spitzoid melanomas,” she said. “Also, knowing the distribution of melanoma across the age categories could provide more insight into their data.”
Ms. Dahak received an award from the National Cancer Institute to fund travel for presentation of this study at the SPD meeting. No other financial conflicts were reported by the researchers. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance (PeDRA) and the International Immunosuppression and Transplant Skin Cancer Collaborative.
INDIANAPOLIS – .
In addition, male gender was independently associated with increased mortality, but age was not.
Those are key findings from a retrospective cohort analysis of nearly 5,000 records from the National Cancer Database.
“There are multiple studies from primarily adult populations showing females with melanoma have a different presentation and better outcomes than males,” co-first author Rebecca M. Thiede, MD, a dermatologist at the University of Arizona, Tucson, said in an interview with this news organization in advance of the annual meeting of the Society for Pediatric Dermatology, where the abstract was presented during a poster session. “However, because melanoma is so rare in younger patients, little is known about gender differences in presentation and survival in pediatric and adolescent patients. To our knowledge, this is one of the largest studies to date in this population, and the first to explore gender differences in detail in pediatric and adolescent patients with melanoma.”
Working with co-first author Sabrina Dahak, a fourth-year medical student at the University of Arizona, Phoenix, Dr. Thiede and colleagues retrospectively analyzed the National Cancer Database to identify biopsy-confirmed invasive primary cutaneous melanoma cases diagnosed in patients 0-21 years of age between 2004 and 2018. The search yielded 4,645 cases, and the researchers used American Academy of Pediatrics definitions to categorize the patients by age, from infancy (birth to 2 years), to childhood (3-10 years), early adolescence (11-14 years), middle adolescence (15-17 years), and late adolescence (18-21 years). They used the Kaplan Meier analysis to determine overall survival and multivariate Cox regression to determine independent survival predictors.
Of the 4,645 pediatric melanoma cases, 63.4% were in females and 36.6% were in males, a difference that was significant (P < .001). Dr. Thiede and colleagues also observed a significant relationship between primary site and gender (P < .001). Primary sites included the trunk (34.3% of females vs. 32.9% of males, respectively), head and neck (16.4% vs. 30.9%), upper extremities (19.5% vs. 16%), lower extremities (27.9% vs. 16.5%), and “unspecified” (1.9% vs. 3.7%).
Females had higher rates of superficial spreading melanoma while males were affected by nodular melanoma more often. For example, the median Breslow depth was higher for males (1.05 mm; interquartile range [IQR] 0.50-2.31) than for females (0.80 mm; IQR, 0.40-1.67; P < .001).
Although females accounted for a higher percentage of cases than males overall, from birth to 17 years, a higher percentage of males than females were found to have later stage of melanoma at time of diagnosis: Females were more likely to be diagnosed with stage I disease (67.8%) than were males (53.6%), and males were more likely than were females to be diagnosed with stages II (15.9% vs. 12.3%), III (27.1% vs. 18.3%), and IV disease (3.3% vs. 1.6%; P < .001 for all).
In other findings, the 5- and 10-year overall survival rates were higher for females (95.9% and 93.9%, respectively) than for males (92.0% vs. 86.7%, respectively; P < .001). However, by age group, overall survival rates were similar between females and males among infants, children, and those in early adolescence – but not for those in middle adolescence (96.7% vs. 91.9%; P < .001) or late adolescence (95.7% vs. 90.4%; P < .001).
When the researchers adjusted for confounding variables, male gender was independently associated with an increased risk of death (adjusted hazard ratio 1.37; P < .001), but age was not.
“It was particularly surprising to see that even at such a young age, there is a significant difference in overall survival between males and females, where females have better outcomes than males,” Dr. Thiede said. “When examining pediatric and adolescent patients, it is essential to maintain cutaneous melanoma on the differential,” she advised. “It is important for clinicians to perform a thorough exam at annual visits particularly for those at high risk for melanoma to catch this rare but potentially devastating diagnosis.”
She acknowledged certain limitations of the study, including its reliance on one database, “as comparing multiple databases would strengthen the conclusions,” she said. “There was some missing data present in our dataset, and a large percentage of the histologic subtypes were unspecified, both of which are common issues with cancer registries. An additional limitation is related to the low death rates in adolescent and pediatric patients, which may impact the analysis related to survival and independent predictors of survival.”
Asked to comment on the study results, Carrie C. Coughlin, MD, who directs the section of pediatric dermatology Washington University/St. Louis Children’s Hospital, said that the finding that males were more likely to present with stage II or higher disease compared with females “could be related to their finding that females had more superficial spreading melanomas, whereas males had more nodular melanoma.” Those differences “could influence how providers evaluate melanocytic lesions in children,” she added.
Dr. Coughlin, who directs the pediatric dermatology fellowship at Washington University/St. Louis Children’s Hospital, said it was “interesting” that the authors found no association between older age and an increased risk of death. “It would be helpful to have more data about melanoma subtype, including information about Spitz or Spitzoid melanomas,” she said. “Also, knowing the distribution of melanoma across the age categories could provide more insight into their data.”
Ms. Dahak received an award from the National Cancer Institute to fund travel for presentation of this study at the SPD meeting. No other financial conflicts were reported by the researchers. Dr. Coughlin is on the board of the Pediatric Dermatology Research Alliance (PeDRA) and the International Immunosuppression and Transplant Skin Cancer Collaborative.
AT SPD 2022
To vaccinate 6-month- to 5-year-olds against SARS-CoV-2 or not to vaccinate
A family’s decision to vaccinate their child is best made jointly with a trusted medical provider who knows the child and family. The American Academy of Pediatrics created a toolkit with resources for answering questions about the recently authorized SARS-CoV-2 mRNA vaccines (Pfizer and Moderna) for 6-month- to 5-year-olds with science-backed vaccine facts, including links to other useful AAP information websites, talking points, graphics, and videos.1

SARS-CoV-2 seasonality
SARS-CoV-2 is now endemic, not a once-a-year seasonal virus. Seasons (aka surges) will occur whenever a new variant arises (twice yearly since 2020, Omicron BA.4/BA.5 currently), or when enough vaccine holdouts, newborns, and/or those with waning of prior immunity (vaccine or infection induced) accrue.
Emergency use authorization submission data for mRNA vaccine responses in young children2,3
Moderna in 6-month- through 5-year-olds. Two 25-mcg doses given 4-8 weeks apart produced 37.8% (95% confidence interval, 20.9%-51.1%) protection against symptomatic Omicron SARS-CoV-2 infections through 3 months of follow-up. Immunobridging analysis of antibody responses compared to 18- to 25-year-olds (100-mcg doses) showed the children’s responses were noninferior. Thus, the committee inferred that vaccine effectiveness in children should be similar to that in 18- to 25-year-olds. Fever, irritability, or local reaction/pain occurred in two-thirds after the second dose. Grade 3 reactions were noted in less than 5%.
Pfizer in 6-month- through 4-year-olds. Three 3-mcg doses, two doses 3-8 weeks apart and the third dose at least 8 weeks later (median 16 weeks), produced 80.3% (95% CI, 13.9%-96.7%) protection against symptomatic COVID-19 during the 6 weeks after the third dose. Local and systemic reactions occurred in 63.8%; less than 5% had grade 3 reactions (fever in about 3%, irritability in 1.3%, fatigue in 0.8%) mostly after second dose.
Neither duration of follow-up is very long. The Moderna data tell me that a third primary dose would have been better but restarting the trial to evaluate third doses would have delayed Moderna’s EUA another 4-6 months. The three-dose Pfizer data look better but may not have been as good with another 6 weeks of follow-up.
Additional post-EUA data will be collected. Boosters will be needed when immunity from both vaccines wanes (one estimate is about 6 months after the primary series). The Advisory Committee on Immunization Practices noted in their deliberations that vaccine-induced antibody responses are higher and cross-neutralize variants (even Omicron) better than infection-induced immunity.4
Are there downsides to the vaccines? Naysayers question vaccinating children less than 5 years old with reasons containing enough “truth” that they catch people’s attention, for example, “young children don’t get very sick with COVID-19,” “most have been infected already,” “RNA for the spike protein stays in the body for months,” or “myocarditis.” Naysayers can quote references in reputable journals but seem to spin selected data out of context or quote unconfirmed data from the Vaccine Adverse Event Reporting System.
Reasons to vaccinate
- While children have milder disease than adults, mid-June 2022 surveillance indicated 50 hospitalizations and 1 pediatric death each day from SARS-CoV-2.5
- Vaccinating young children endows a foundation of vaccine-induced SARS-CoV-2 immunity that is superior to infection-induced immunity.4
- Long-term effects of large numbers of SARS-CoV-2 particles that enter every organ of a developing child have not been determined.
- Viral loads are lowered by prior vaccine; fewer viral replications lessen chances for newer variants to arise.
- Transmission is less in breakthrough infections than infections in the unvaccinated.
- Thirty percent of 5- to 11-year-olds hospitalized for SARS-CoV-2 had no underlying conditions;6 hospitalization rates in newborn to 4-year-olds have been the highest in the Omicron surge.7
- No myocarditis or pericarditis episodes have been detected in 6-month- to 11-year-old trials.
- The AAP and ACIP recommend the mRNA vaccines.
My thoughts are that SARS-CoV-2 vaccine is just another “routine” childhood vaccine that prepares children for healthier futures, pandemic or not, and the vaccines are as safe as other routine vaccines.
And like other pediatric vaccines, it should be no surprise that boosters will be needed, even if no newer variants than Omicron BA.4/BA.5 arise. But we know newer variants will arise and, similar to influenza vaccine, new formulations, perhaps with multiple SARS-CoV-2 strain antigens, will be needed every year or so. Everyone will get SARS-CoV-2 multiple times in their lives no matter how careful they are. So isn’t it good medical practice to establish early the best available foundation for maintaining lifelong SARS-CoV-2 immunity?
To me it is like pertussis. Most pertussis-infected children are sick enough to be hospitalized; very few die. They are miserable with illnesses that take weeks to months to subside. The worst disease usually occurs in unvaccinated young children or those with underlying conditions. Reactogenicity was reduced with acellular vaccine but resulted in less immunogenicity, so we give boosters at intervals that best match waning immunity. Circulating strains can be different than the vaccine strain, so protection against infection is 80%. Finally, even the safest vaccine may very rarely have sequelae. That is why The National Vaccine Injury Compensation Program was created. Yet the benefit-to-harm ratio for children and society favors universal pertussis vaccine use. And we vaccinate even those who have had pertussis because even infection-based immunity is incomplete and protection wanes. If arguments similar to those by SARS-CoV-2 vaccine naysayers were applied to acellular pertussis vaccine, it seems they would argue against pertussis vaccine for young children.
Another major issue has been “safety concerns” about the vaccines’ small amount of mRNA for the spike protein encased in microscopic lipid bubbles injected in the arm or leg. This mRNA is picked up by human cells, and in the cytoplasm (not the nucleus where our DNA resides) produces a limited supply of spike protein that is then picked up by antigen-presenting cells for short-lived distribution (days to 2 weeks at most) to regional lymph nodes where immune-memory processes are jump-started. Contrast that to even asymptomatic SARS-CoV-2 infection where multibillions of virus particles are produced for up to 14 days with access to every bodily organ that contains ACE-2 receptors (they all do). Each virus particle hijacks a human cell producing thousands of mRNA for spike protein (and multiple other SARS-CoV-2 proteins), eventually releasing multibillions of lipid fragments from the ruptured cell. Comparing the amount of these components in the mRNA vaccines to those from infection is like comparing a campfire to the many-thousand-acre wildfire. So, if one is worried about the effects of spike protein and lipid fragments, the limited localized amounts in mRNA vaccines should make one much less concerned than the enormous amounts circulating throughout the body as a result of a SARS-CoV-2 infection.
My take is that children 6-months to 5-years-old deserve SARS-CoV-2–induced vaccine protection and we can and should strongly recommend it as medical providers and child advocates.
*Dr. Harrison is professor, University of Missouri Kansas City School of Medicine, department of medicine, infectious diseases section, Kansas City. Email him at [email protected].
References
1. AAP. 2022 Jun 21. As COVID-19 vaccines become available for children ages 6 months to 4 years, AAP urges families to reach out to pediatricians to ask questions and access vaccine. www.aap.org.
2. CDC. Grading of recommendations, assessment, development, and evaluation (GRADE): Moderna COVID-19 vaccine for children aged 6 months–5 years. www.cdc.gov.
3. CDC. ACIP evidence to recommendations for use of Moderna COVID-19 vaccine in children ages 6 months–5 years and Pfizer-BioNTech COVID-19 vaccine in children ages 6 months–4 years under an emergency use authorization. www.cdc.gov.
4. Tang J et al. Nat Commun. 2022;13:2979.
5. Children and COVID-19: State Data Report. 2022 Jun 30. www.aap.org.
6. Shi DS et al. MMWR Morb Mortal Wkly Rep. 2022;71:574-81.
7. Marks KJ et al. MMWR Morb Mortal Wkly Rep. 2022;71:429-36.
Other good resources for families are https://getvaccineanswers.org/ or www.mayoclinic.org/diseases-conditions/coronavirus/in-depth/coronavirus-in-babies-and-children/art-20484405.
*This story was updated on July 19, 2022.
A family’s decision to vaccinate their child is best made jointly with a trusted medical provider who knows the child and family. The American Academy of Pediatrics created a toolkit with resources for answering questions about the recently authorized SARS-CoV-2 mRNA vaccines (Pfizer and Moderna) for 6-month- to 5-year-olds with science-backed vaccine facts, including links to other useful AAP information websites, talking points, graphics, and videos.1
SARS-CoV-2 seasonality
SARS-CoV-2 is now endemic, not a once-a-year seasonal virus. Seasons (aka surges) will occur whenever a new variant arises (twice yearly since 2020, Omicron BA.4/BA.5 currently), or when enough vaccine holdouts, newborns, and/or those with waning of prior immunity (vaccine or infection induced) accrue.
Emergency use authorization submission data for mRNA vaccine responses in young children2,3
Moderna in 6-month- through 5-year-olds. Two 25-mcg doses given 4-8 weeks apart produced 37.8% (95% confidence interval, 20.9%-51.1%) protection against symptomatic Omicron SARS-CoV-2 infections through 3 months of follow-up. Immunobridging analysis of antibody responses compared to 18- to 25-year-olds (100-mcg doses) showed the children’s responses were noninferior. Thus, the committee inferred that vaccine effectiveness in children should be similar to that in 18- to 25-year-olds. Fever, irritability, or local reaction/pain occurred in two-thirds after the second dose. Grade 3 reactions were noted in less than 5%.
Pfizer in 6-month- through 4-year-olds. Three 3-mcg doses, two doses 3-8 weeks apart and the third dose at least 8 weeks later (median 16 weeks), produced 80.3% (95% CI, 13.9%-96.7%) protection against symptomatic COVID-19 during the 6 weeks after the third dose. Local and systemic reactions occurred in 63.8%; less than 5% had grade 3 reactions (fever in about 3%, irritability in 1.3%, fatigue in 0.8%) mostly after second dose.
Neither duration of follow-up is very long. The Moderna data tell me that a third primary dose would have been better but restarting the trial to evaluate third doses would have delayed Moderna’s EUA another 4-6 months. The three-dose Pfizer data look better but may not have been as good with another 6 weeks of follow-up.
Additional post-EUA data will be collected. Boosters will be needed when immunity from both vaccines wanes (one estimate is about 6 months after the primary series). The Advisory Committee on Immunization Practices noted in their deliberations that vaccine-induced antibody responses are higher and cross-neutralize variants (even Omicron) better than infection-induced immunity.4
Are there downsides to the vaccines? Naysayers question vaccinating children less than 5 years old with reasons containing enough “truth” that they catch people’s attention, for example, “young children don’t get very sick with COVID-19,” “most have been infected already,” “RNA for the spike protein stays in the body for months,” or “myocarditis.” Naysayers can quote references in reputable journals but seem to spin selected data out of context or quote unconfirmed data from the Vaccine Adverse Event Reporting System.
Reasons to vaccinate
- While children have milder disease than adults, mid-June 2022 surveillance indicated 50 hospitalizations and 1 pediatric death each day from SARS-CoV-2.5
- Vaccinating young children endows a foundation of vaccine-induced SARS-CoV-2 immunity that is superior to infection-induced immunity.4
- Long-term effects of large numbers of SARS-CoV-2 particles that enter every organ of a developing child have not been determined.
- Viral loads are lowered by prior vaccine; fewer viral replications lessen chances for newer variants to arise.
- Transmission is less in breakthrough infections than infections in the unvaccinated.
- Thirty percent of 5- to 11-year-olds hospitalized for SARS-CoV-2 had no underlying conditions;6 hospitalization rates in newborn to 4-year-olds have been the highest in the Omicron surge.7
- No myocarditis or pericarditis episodes have been detected in 6-month- to 11-year-old trials.
- The AAP and ACIP recommend the mRNA vaccines.
My thoughts are that SARS-CoV-2 vaccine is just another “routine” childhood vaccine that prepares children for healthier futures, pandemic or not, and the vaccines are as safe as other routine vaccines.
And like other pediatric vaccines, it should be no surprise that boosters will be needed, even if no newer variants than Omicron BA.4/BA.5 arise. But we know newer variants will arise and, similar to influenza vaccine, new formulations, perhaps with multiple SARS-CoV-2 strain antigens, will be needed every year or so. Everyone will get SARS-CoV-2 multiple times in their lives no matter how careful they are. So isn’t it good medical practice to establish early the best available foundation for maintaining lifelong SARS-CoV-2 immunity?
To me it is like pertussis. Most pertussis-infected children are sick enough to be hospitalized; very few die. They are miserable with illnesses that take weeks to months to subside. The worst disease usually occurs in unvaccinated young children or those with underlying conditions. Reactogenicity was reduced with acellular vaccine but resulted in less immunogenicity, so we give boosters at intervals that best match waning immunity. Circulating strains can be different than the vaccine strain, so protection against infection is 80%. Finally, even the safest vaccine may very rarely have sequelae. That is why The National Vaccine Injury Compensation Program was created. Yet the benefit-to-harm ratio for children and society favors universal pertussis vaccine use. And we vaccinate even those who have had pertussis because even infection-based immunity is incomplete and protection wanes. If arguments similar to those by SARS-CoV-2 vaccine naysayers were applied to acellular pertussis vaccine, it seems they would argue against pertussis vaccine for young children.
Another major issue has been “safety concerns” about the vaccines’ small amount of mRNA for the spike protein encased in microscopic lipid bubbles injected in the arm or leg. This mRNA is picked up by human cells, and in the cytoplasm (not the nucleus where our DNA resides) produces a limited supply of spike protein that is then picked up by antigen-presenting cells for short-lived distribution (days to 2 weeks at most) to regional lymph nodes where immune-memory processes are jump-started. Contrast that to even asymptomatic SARS-CoV-2 infection where multibillions of virus particles are produced for up to 14 days with access to every bodily organ that contains ACE-2 receptors (they all do). Each virus particle hijacks a human cell producing thousands of mRNA for spike protein (and multiple other SARS-CoV-2 proteins), eventually releasing multibillions of lipid fragments from the ruptured cell. Comparing the amount of these components in the mRNA vaccines to those from infection is like comparing a campfire to the many-thousand-acre wildfire. So, if one is worried about the effects of spike protein and lipid fragments, the limited localized amounts in mRNA vaccines should make one much less concerned than the enormous amounts circulating throughout the body as a result of a SARS-CoV-2 infection.
My take is that children 6-months to 5-years-old deserve SARS-CoV-2–induced vaccine protection and we can and should strongly recommend it as medical providers and child advocates.
*Dr. Harrison is professor, University of Missouri Kansas City School of Medicine, department of medicine, infectious diseases section, Kansas City. Email him at [email protected].
References
1. AAP. 2022 Jun 21. As COVID-19 vaccines become available for children ages 6 months to 4 years, AAP urges families to reach out to pediatricians to ask questions and access vaccine. www.aap.org.
2. CDC. Grading of recommendations, assessment, development, and evaluation (GRADE): Moderna COVID-19 vaccine for children aged 6 months–5 years. www.cdc.gov.
3. CDC. ACIP evidence to recommendations for use of Moderna COVID-19 vaccine in children ages 6 months–5 years and Pfizer-BioNTech COVID-19 vaccine in children ages 6 months–4 years under an emergency use authorization. www.cdc.gov.
4. Tang J et al. Nat Commun. 2022;13:2979.
5. Children and COVID-19: State Data Report. 2022 Jun 30. www.aap.org.
6. Shi DS et al. MMWR Morb Mortal Wkly Rep. 2022;71:574-81.
7. Marks KJ et al. MMWR Morb Mortal Wkly Rep. 2022;71:429-36.
Other good resources for families are https://getvaccineanswers.org/ or www.mayoclinic.org/diseases-conditions/coronavirus/in-depth/coronavirus-in-babies-and-children/art-20484405.
*This story was updated on July 19, 2022.
A family’s decision to vaccinate their child is best made jointly with a trusted medical provider who knows the child and family. The American Academy of Pediatrics created a toolkit with resources for answering questions about the recently authorized SARS-CoV-2 mRNA vaccines (Pfizer and Moderna) for 6-month- to 5-year-olds with science-backed vaccine facts, including links to other useful AAP information websites, talking points, graphics, and videos.1
SARS-CoV-2 seasonality
SARS-CoV-2 is now endemic, not a once-a-year seasonal virus. Seasons (aka surges) will occur whenever a new variant arises (twice yearly since 2020, Omicron BA.4/BA.5 currently), or when enough vaccine holdouts, newborns, and/or those with waning of prior immunity (vaccine or infection induced) accrue.
Emergency use authorization submission data for mRNA vaccine responses in young children2,3
Moderna in 6-month- through 5-year-olds. Two 25-mcg doses given 4-8 weeks apart produced 37.8% (95% confidence interval, 20.9%-51.1%) protection against symptomatic Omicron SARS-CoV-2 infections through 3 months of follow-up. Immunobridging analysis of antibody responses compared to 18- to 25-year-olds (100-mcg doses) showed the children’s responses were noninferior. Thus, the committee inferred that vaccine effectiveness in children should be similar to that in 18- to 25-year-olds. Fever, irritability, or local reaction/pain occurred in two-thirds after the second dose. Grade 3 reactions were noted in less than 5%.
Pfizer in 6-month- through 4-year-olds. Three 3-mcg doses, two doses 3-8 weeks apart and the third dose at least 8 weeks later (median 16 weeks), produced 80.3% (95% CI, 13.9%-96.7%) protection against symptomatic COVID-19 during the 6 weeks after the third dose. Local and systemic reactions occurred in 63.8%; less than 5% had grade 3 reactions (fever in about 3%, irritability in 1.3%, fatigue in 0.8%) mostly after second dose.
Neither duration of follow-up is very long. The Moderna data tell me that a third primary dose would have been better but restarting the trial to evaluate third doses would have delayed Moderna’s EUA another 4-6 months. The three-dose Pfizer data look better but may not have been as good with another 6 weeks of follow-up.
Additional post-EUA data will be collected. Boosters will be needed when immunity from both vaccines wanes (one estimate is about 6 months after the primary series). The Advisory Committee on Immunization Practices noted in their deliberations that vaccine-induced antibody responses are higher and cross-neutralize variants (even Omicron) better than infection-induced immunity.4
Are there downsides to the vaccines? Naysayers question vaccinating children less than 5 years old with reasons containing enough “truth” that they catch people’s attention, for example, “young children don’t get very sick with COVID-19,” “most have been infected already,” “RNA for the spike protein stays in the body for months,” or “myocarditis.” Naysayers can quote references in reputable journals but seem to spin selected data out of context or quote unconfirmed data from the Vaccine Adverse Event Reporting System.
Reasons to vaccinate
- While children have milder disease than adults, mid-June 2022 surveillance indicated 50 hospitalizations and 1 pediatric death each day from SARS-CoV-2.5
- Vaccinating young children endows a foundation of vaccine-induced SARS-CoV-2 immunity that is superior to infection-induced immunity.4
- Long-term effects of large numbers of SARS-CoV-2 particles that enter every organ of a developing child have not been determined.
- Viral loads are lowered by prior vaccine; fewer viral replications lessen chances for newer variants to arise.
- Transmission is less in breakthrough infections than infections in the unvaccinated.
- Thirty percent of 5- to 11-year-olds hospitalized for SARS-CoV-2 had no underlying conditions;6 hospitalization rates in newborn to 4-year-olds have been the highest in the Omicron surge.7
- No myocarditis or pericarditis episodes have been detected in 6-month- to 11-year-old trials.
- The AAP and ACIP recommend the mRNA vaccines.
My thoughts are that SARS-CoV-2 vaccine is just another “routine” childhood vaccine that prepares children for healthier futures, pandemic or not, and the vaccines are as safe as other routine vaccines.
And like other pediatric vaccines, it should be no surprise that boosters will be needed, even if no newer variants than Omicron BA.4/BA.5 arise. But we know newer variants will arise and, similar to influenza vaccine, new formulations, perhaps with multiple SARS-CoV-2 strain antigens, will be needed every year or so. Everyone will get SARS-CoV-2 multiple times in their lives no matter how careful they are. So isn’t it good medical practice to establish early the best available foundation for maintaining lifelong SARS-CoV-2 immunity?
To me it is like pertussis. Most pertussis-infected children are sick enough to be hospitalized; very few die. They are miserable with illnesses that take weeks to months to subside. The worst disease usually occurs in unvaccinated young children or those with underlying conditions. Reactogenicity was reduced with acellular vaccine but resulted in less immunogenicity, so we give boosters at intervals that best match waning immunity. Circulating strains can be different than the vaccine strain, so protection against infection is 80%. Finally, even the safest vaccine may very rarely have sequelae. That is why The National Vaccine Injury Compensation Program was created. Yet the benefit-to-harm ratio for children and society favors universal pertussis vaccine use. And we vaccinate even those who have had pertussis because even infection-based immunity is incomplete and protection wanes. If arguments similar to those by SARS-CoV-2 vaccine naysayers were applied to acellular pertussis vaccine, it seems they would argue against pertussis vaccine for young children.
Another major issue has been “safety concerns” about the vaccines’ small amount of mRNA for the spike protein encased in microscopic lipid bubbles injected in the arm or leg. This mRNA is picked up by human cells, and in the cytoplasm (not the nucleus where our DNA resides) produces a limited supply of spike protein that is then picked up by antigen-presenting cells for short-lived distribution (days to 2 weeks at most) to regional lymph nodes where immune-memory processes are jump-started. Contrast that to even asymptomatic SARS-CoV-2 infection where multibillions of virus particles are produced for up to 14 days with access to every bodily organ that contains ACE-2 receptors (they all do). Each virus particle hijacks a human cell producing thousands of mRNA for spike protein (and multiple other SARS-CoV-2 proteins), eventually releasing multibillions of lipid fragments from the ruptured cell. Comparing the amount of these components in the mRNA vaccines to those from infection is like comparing a campfire to the many-thousand-acre wildfire. So, if one is worried about the effects of spike protein and lipid fragments, the limited localized amounts in mRNA vaccines should make one much less concerned than the enormous amounts circulating throughout the body as a result of a SARS-CoV-2 infection.
My take is that children 6-months to 5-years-old deserve SARS-CoV-2–induced vaccine protection and we can and should strongly recommend it as medical providers and child advocates.
*Dr. Harrison is professor, University of Missouri Kansas City School of Medicine, department of medicine, infectious diseases section, Kansas City. Email him at [email protected].
References
1. AAP. 2022 Jun 21. As COVID-19 vaccines become available for children ages 6 months to 4 years, AAP urges families to reach out to pediatricians to ask questions and access vaccine. www.aap.org.
2. CDC. Grading of recommendations, assessment, development, and evaluation (GRADE): Moderna COVID-19 vaccine for children aged 6 months–5 years. www.cdc.gov.
3. CDC. ACIP evidence to recommendations for use of Moderna COVID-19 vaccine in children ages 6 months–5 years and Pfizer-BioNTech COVID-19 vaccine in children ages 6 months–4 years under an emergency use authorization. www.cdc.gov.
4. Tang J et al. Nat Commun. 2022;13:2979.
5. Children and COVID-19: State Data Report. 2022 Jun 30. www.aap.org.
6. Shi DS et al. MMWR Morb Mortal Wkly Rep. 2022;71:574-81.
7. Marks KJ et al. MMWR Morb Mortal Wkly Rep. 2022;71:429-36.
Other good resources for families are https://getvaccineanswers.org/ or www.mayoclinic.org/diseases-conditions/coronavirus/in-depth/coronavirus-in-babies-and-children/art-20484405.
*This story was updated on July 19, 2022.
Experimental cancer drug promising for hospitalized COVID patients
, a new study shows.
The medication, called sabizabulin and given as a pill, reduced by half the risk of death among participants. It could be more effective than other drugs for those severely sick with COVID-19, The New York Times reports.
The manufacturer, Veru, is seeking emergency use authorization from the Food and Drug Administration. Hospitalized COVID-19 patients currently have only a few pharmaceutical options.
Sabizabulin blocks cells from building molecular cables that carry material from one part of a cell to another. It was created to fight cancer, because tumor cells need those cables (called microtubules) to grow quickly.
Researchers tried it against COVID-19 2 years ago, because viral replication also requires microtubules to bring pieces of new viruses together.
To participate in the small trial, patients had to be receiving oxygen or on a ventilator and at a high risk of dying from COVID-19, “with risk factors such as hypertension, advanced age or obesity,” the Times reported.
A total of 134 patients received the medicine; 70 got a placebo. Among those receiving sabizabulin, 20.2% died within 2 months; 45.1% of those who took the placebo died.
One infectious disease expert told the Times that the high mortality rate of those on the placebo could mean the study was too small to offer conclusive results.
“The 45% mortality rate in the control group jumps out at me as rather high,” said David Boulware, MD, of the University of Minnesota.
A version of this article first appeared on WebMD.com.
, a new study shows.
The medication, called sabizabulin and given as a pill, reduced by half the risk of death among participants. It could be more effective than other drugs for those severely sick with COVID-19, The New York Times reports.
The manufacturer, Veru, is seeking emergency use authorization from the Food and Drug Administration. Hospitalized COVID-19 patients currently have only a few pharmaceutical options.
Sabizabulin blocks cells from building molecular cables that carry material from one part of a cell to another. It was created to fight cancer, because tumor cells need those cables (called microtubules) to grow quickly.
Researchers tried it against COVID-19 2 years ago, because viral replication also requires microtubules to bring pieces of new viruses together.
To participate in the small trial, patients had to be receiving oxygen or on a ventilator and at a high risk of dying from COVID-19, “with risk factors such as hypertension, advanced age or obesity,” the Times reported.
A total of 134 patients received the medicine; 70 got a placebo. Among those receiving sabizabulin, 20.2% died within 2 months; 45.1% of those who took the placebo died.
One infectious disease expert told the Times that the high mortality rate of those on the placebo could mean the study was too small to offer conclusive results.
“The 45% mortality rate in the control group jumps out at me as rather high,” said David Boulware, MD, of the University of Minnesota.
A version of this article first appeared on WebMD.com.
, a new study shows.
The medication, called sabizabulin and given as a pill, reduced by half the risk of death among participants. It could be more effective than other drugs for those severely sick with COVID-19, The New York Times reports.
The manufacturer, Veru, is seeking emergency use authorization from the Food and Drug Administration. Hospitalized COVID-19 patients currently have only a few pharmaceutical options.
Sabizabulin blocks cells from building molecular cables that carry material from one part of a cell to another. It was created to fight cancer, because tumor cells need those cables (called microtubules) to grow quickly.
Researchers tried it against COVID-19 2 years ago, because viral replication also requires microtubules to bring pieces of new viruses together.
To participate in the small trial, patients had to be receiving oxygen or on a ventilator and at a high risk of dying from COVID-19, “with risk factors such as hypertension, advanced age or obesity,” the Times reported.
A total of 134 patients received the medicine; 70 got a placebo. Among those receiving sabizabulin, 20.2% died within 2 months; 45.1% of those who took the placebo died.
One infectious disease expert told the Times that the high mortality rate of those on the placebo could mean the study was too small to offer conclusive results.
“The 45% mortality rate in the control group jumps out at me as rather high,” said David Boulware, MD, of the University of Minnesota.
A version of this article first appeared on WebMD.com.
CDC recommends high-dose flu vaccines for seniors
In an online statement Fluzone High-Dose Quadrivalent, Flublok Quadrivalent, and Fluad Quadrivalent flu vaccines are among those specified in the release.
The organization says that these higher-dose vaccines may be more effective for the aging population, who often have difficulty mounting a strong enough immune response to protect themselves against the flu virus. People older than 65 years struggle the most during flu season and have the highest proportion of hospitalizations and deaths from flu, according to the release.
But the CDC believes that higher-dose vaccines have the potential to better protect against that danger. One study, from The New England Journal of Medicine, reported that high-dose/adjuvanted vaccines prevented flu in older patients 24% better than did lower-dose/nonadjuvanted vaccines.
These types of vaccines work by creating a larger immune response than a standard vaccine dose. In particular, adjuvanted vaccines contain an extra ingredient within them that helps the immune system produce a stronger reaction to the vaccine. These may be things like aluminum salts, which signal the body to respond faster. Higher-dose vaccines similarly promote a stronger immune response by having more particles of the target virus in their mixture. In theory, this means the body will create an enhanced response to the vaccine. For example, a higher-dose vaccine may quadruple the amount of antigens, compared with the standard dose.
The hope is that this recommendation may increase vaccine use across the board, says José Romero, MD, the director of the CDC’s National Center for Immunization and Respiratory Diseases. As quoted in the CDC announcement, Dr. Romero said that this may help reduce racial inequities in access to flu vaccines. A 2019 meta-analysis concluded that Black and Hispanic people are around 30%-40% less likely to get the flu vaccine. So increasing the access to this medication “could help reduce health disparities by making these vaccines more available to racial and ethnic minority groups,” said Dr. Romero.
The decision, spearheaded by CDC Director Rochelle Walensky, MD, follows recommendations from the Advisory Committee on Immunization Practices, which presented on this topic during a June 22 meeting. It is now part of official CDC policy and will continue to be developed as the 2022-2023 flu season approaches.
In addition, the organization says they’ll reveal more details for their plan later this summer, in their Morbidity and Mortality Weekly Report (MMWR). For now, seniors should know that they should try to get the recommended high-dose vaccines, but if they can’t, then a standard dose of whatever their provider has on hand will do.
At this point, there is still no specific vaccine recommendation for people aged under 65 years. The CDC historically avoids specifying one type of vaccine over another and says each should still be effective in younger patients.
A version of this article first appeared on Medscape.com.
In an online statement Fluzone High-Dose Quadrivalent, Flublok Quadrivalent, and Fluad Quadrivalent flu vaccines are among those specified in the release.
The organization says that these higher-dose vaccines may be more effective for the aging population, who often have difficulty mounting a strong enough immune response to protect themselves against the flu virus. People older than 65 years struggle the most during flu season and have the highest proportion of hospitalizations and deaths from flu, according to the release.
But the CDC believes that higher-dose vaccines have the potential to better protect against that danger. One study, from The New England Journal of Medicine, reported that high-dose/adjuvanted vaccines prevented flu in older patients 24% better than did lower-dose/nonadjuvanted vaccines.
These types of vaccines work by creating a larger immune response than a standard vaccine dose. In particular, adjuvanted vaccines contain an extra ingredient within them that helps the immune system produce a stronger reaction to the vaccine. These may be things like aluminum salts, which signal the body to respond faster. Higher-dose vaccines similarly promote a stronger immune response by having more particles of the target virus in their mixture. In theory, this means the body will create an enhanced response to the vaccine. For example, a higher-dose vaccine may quadruple the amount of antigens, compared with the standard dose.
The hope is that this recommendation may increase vaccine use across the board, says José Romero, MD, the director of the CDC’s National Center for Immunization and Respiratory Diseases. As quoted in the CDC announcement, Dr. Romero said that this may help reduce racial inequities in access to flu vaccines. A 2019 meta-analysis concluded that Black and Hispanic people are around 30%-40% less likely to get the flu vaccine. So increasing the access to this medication “could help reduce health disparities by making these vaccines more available to racial and ethnic minority groups,” said Dr. Romero.
The decision, spearheaded by CDC Director Rochelle Walensky, MD, follows recommendations from the Advisory Committee on Immunization Practices, which presented on this topic during a June 22 meeting. It is now part of official CDC policy and will continue to be developed as the 2022-2023 flu season approaches.
In addition, the organization says they’ll reveal more details for their plan later this summer, in their Morbidity and Mortality Weekly Report (MMWR). For now, seniors should know that they should try to get the recommended high-dose vaccines, but if they can’t, then a standard dose of whatever their provider has on hand will do.
At this point, there is still no specific vaccine recommendation for people aged under 65 years. The CDC historically avoids specifying one type of vaccine over another and says each should still be effective in younger patients.
A version of this article first appeared on Medscape.com.
In an online statement Fluzone High-Dose Quadrivalent, Flublok Quadrivalent, and Fluad Quadrivalent flu vaccines are among those specified in the release.
The organization says that these higher-dose vaccines may be more effective for the aging population, who often have difficulty mounting a strong enough immune response to protect themselves against the flu virus. People older than 65 years struggle the most during flu season and have the highest proportion of hospitalizations and deaths from flu, according to the release.
But the CDC believes that higher-dose vaccines have the potential to better protect against that danger. One study, from The New England Journal of Medicine, reported that high-dose/adjuvanted vaccines prevented flu in older patients 24% better than did lower-dose/nonadjuvanted vaccines.
These types of vaccines work by creating a larger immune response than a standard vaccine dose. In particular, adjuvanted vaccines contain an extra ingredient within them that helps the immune system produce a stronger reaction to the vaccine. These may be things like aluminum salts, which signal the body to respond faster. Higher-dose vaccines similarly promote a stronger immune response by having more particles of the target virus in their mixture. In theory, this means the body will create an enhanced response to the vaccine. For example, a higher-dose vaccine may quadruple the amount of antigens, compared with the standard dose.
The hope is that this recommendation may increase vaccine use across the board, says José Romero, MD, the director of the CDC’s National Center for Immunization and Respiratory Diseases. As quoted in the CDC announcement, Dr. Romero said that this may help reduce racial inequities in access to flu vaccines. A 2019 meta-analysis concluded that Black and Hispanic people are around 30%-40% less likely to get the flu vaccine. So increasing the access to this medication “could help reduce health disparities by making these vaccines more available to racial and ethnic minority groups,” said Dr. Romero.
The decision, spearheaded by CDC Director Rochelle Walensky, MD, follows recommendations from the Advisory Committee on Immunization Practices, which presented on this topic during a June 22 meeting. It is now part of official CDC policy and will continue to be developed as the 2022-2023 flu season approaches.
In addition, the organization says they’ll reveal more details for their plan later this summer, in their Morbidity and Mortality Weekly Report (MMWR). For now, seniors should know that they should try to get the recommended high-dose vaccines, but if they can’t, then a standard dose of whatever their provider has on hand will do.
At this point, there is still no specific vaccine recommendation for people aged under 65 years. The CDC historically avoids specifying one type of vaccine over another and says each should still be effective in younger patients.
A version of this article first appeared on Medscape.com.
Baricitinib’s approval for alopecia areata: Considerations for starting patients on treatment
On June 13, the FDA approved baricitinib, a Janus kinase inhibitor (Olumiant, Lilly), for severe AA, and two other options may not be far behind. Pfizer and Concert Pharmaceuticals have JAK inhibitors in late-stage development for AA. JAK inhibitors, including baricitinib, are already on the market for treating rheumatoid arthritis and other autoimmune diseases.
Meanwhile, dermatologists have been fielding calls from hopeful patients and sorting out who should get the treatment, how to advise patients on risks and benefits, and what tests should be used before and after starting treatment.

Uptake for new systemic drugs, such as biologics, can be slow in dermatology, noted Adam Friedman, MD, professor and chair of dermatology, George Washington University, Washington, as some doctors like to stick with what they know.
He told this news organization that he hopes that uptake for baricitinib is quicker, as it is the only approved oral systemic treatment for patients with severe alopecia areata, which affects about 300,000 people a year in the United States. Other treatments, including steroid injections in the scalp, have lacked efficacy and convenience.
Beyond the physical effects, the mental toll of patchy hair clumps and missing brows and lashes can be devastating for patients with alopecia areata.
Fielding patient inquiries
Word of the FDA approval spread fast, and calls and emails are coming into dermatologists’ offices and clinics from interested patients.
Physicians should be ready for patients with any kind of hair loss, not just severe alopecia areata, to ask about the drug, Dr. Friedman said. Some patients contacting him don’t fit the indication, which “highlights how disabling hair loss” is for people, considering that, in general, “people see this and think it is for them.”
Baricitinib is not a new drug, but a drug with a new indication. It had already been approved for treating moderate to severe RA in patients who have had an inadequate response to one or more tumor necrosis factor blockers, and for treating COVID-19 in certain hospitalized adults.
Boxed warning
Patients may ask about the boxed warning in the baricitinib label about the increased risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Natasha A. Mesinkovska, MD, PhD, an investigator in the clinical trials that led to FDA approval of baricitinib and the chief scientific officer at the National Alopecia Areata Foundation, told this news organization that several aspects of the label are important to point out.
One is that the warning is for all the JAK inhibitors used to treat RA and other inflammatory conditions, not just baricitinib. Also, the warning is based mostly on data on patients with RA who, she noted, have substantial comorbidities and have been taking toxic immunosuppressive medications. The RA population is also typically many years older than the alopecia areata population.
“Whether the warnings apply to the alopecia areata patients is as yet unclear,” said Dr. Mesinkovska, who is also an associate professor of dermatology at the University of California, Irvine.
Patients are also asking about how well it works.
In one of the two trials that led up to the FDA approval, which enrolled patients with at least 50% scalp hair loss for over 6 months, 22% of the patients who received 2 mg of baricitinib and 35% of those who received 4 mg saw adequate hair coverage (at least 80%) at week 36, compared with 5% on placebo. In the second trial, 17% of those who received 2 mg and 32% who received 4 mg saw adequate hair coverage, compared with 3% on placebo.
Common side effects associated with baricitinib, according to the FDA, are lower respiratory tract infections, headache, acne, high cholesterol, increased creatinine phosphokinase, urinary tract infection, liver enzyme elevations, folliculitis, fatigue, nausea, genital yeast infections, anemia, neutropenia, abdominal pain, herpes zoster (shingles), and weight gain.
The risk-benefit discussions with patients should also include potential benefits beyond hair regrowth on the scalp. Loss of hair in the ears and nose can affect hearing and allergies, Dr. Mesinkovska said.
“About 30%-50% with alopecia areata, depending on age group or part of the world, will have allergies,” she said.
Patients should also know that baricitinib will need to be taken “for a very long time,” Dr. Mesinkovska noted. It’s possible that could be forever and that stopping the medication at any point may result in hair falling out again, she says, but duration will vary from case to case.
The good news is that it has been well tolerated. “We give a lot of medications for acne like doxycycline and other antibiotics and people have more stomach problems and angst with those than with [baricitinib],” she said.
Regrowth takes time
Benjamin Ungar, MD, a dermatologist at the Alopecia Center of Excellence at Mount Sinai, New York, told this news organization that an important message for patients is that hair regrowth takes time. For some other skin conditions, patients start treatment and see almost instant improvement.

“That is not the case for alopecia areata,” he said. “The expectation is that it will take months for regrowth in general.”
He said he hasn’t started prescribing baricitinib yet, but plans to do so soon.
“Obviously, I’ll have conversations with patients about it, but it’s a medication I’m going to be using, definitely. I have no reservations,” Dr. Ungar said.
After initial testing, physicians may find that some patients might not be ideal candidates, he added. People with liver disease, a history of blood clots, abnormal blood counts, or low neutrophils are among those who may not be the best candidates for baricitinib.
For most with severe alopecia areata, though, baricitinib provides hope.
“Treatment options have been not readily available, often inaccessible, ineffective, often dangerous,” he said. “There’s a treatment now that can be accessed, generally is safe and is effective for many people.”
Be up front with patients about the unknown
Additionally, it’s important to tell patients what is not yet known, the experts interviewed say.
“Alopecia areata is a chronic disease. We don’t have long-term data on the patient population yet,” Dr. Friedman said.
Also unknown is how easy it will be for physicians to get insurance to reimburse for baricitinib, which, at the end of June, was priced at about $5,000 a month for the 4-mg dose. FDA approval was important in that regard. Previously, some claims had been rejected for drugs used off label for AA.
“We dermatologists know how much it affects patients,” Dr. Mesinkovska said. “As long as we stick by what we know and convey to insurers how much it affects people’s lives, they should cover it.”
Another unknown is what other drugs can be taken with baricitinib. In clinical trials, it was used alone, she said. Currently, concomitant use of other immune suppressants – such as methotrexate or prednisone – is not recommended. But it remains to be seen what other medications will be safe to use at the same time as more long-term data are available.

Lynne J. Goldberg, MD, professor of dermatology, pathology, and laboratory medicine, Boston University, and director of the Hair Clinic at Boston Medical Center, said that she received a slew of emails from patients asking about baricitinib, but most of them did not have alopecia areata and were not candidates for this treatment.
She said that nurses in her clinic have been instructed on what to tell patients about which patients the drug is meant to treat, side effects, and benefits.
Access won’t be immediate
Dr. Goldberg said the drug’s approval does not mean immediate access. The patient has to come in, discuss the treatment, and get lab tests first. “It’s not a casual drug. This is a potent immunosuppressant drug. You need lab tests and once you start it you need blood tests every 3 months to stay on it.”
Those tests may vary by physician, but people will generally need a standard blood count and a comprehensive metabolic panel and lipid panel. “There’s nothing esoteric,” she said.
She added that physicians will need to check for presence of infections including tuberculosis and hepatitis B and C before prescribing, just as they would before they start prescribing a biologic.
“You don’t want to reactivate something,” she noted.
But, Dr. Goldberg added, the benefits for all who have been either living with only patches of hair or no hair or who put on a wig or hat every day are “life changing.”
Dr. Mesinkovska is on the advisory boards and runs trials for Eli Lilly, Pfizer, and Concert Pharmaceuticals. Dr. Friedman, Dr. Goldberg, and Dr. Ungar reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
On June 13, the FDA approved baricitinib, a Janus kinase inhibitor (Olumiant, Lilly), for severe AA, and two other options may not be far behind. Pfizer and Concert Pharmaceuticals have JAK inhibitors in late-stage development for AA. JAK inhibitors, including baricitinib, are already on the market for treating rheumatoid arthritis and other autoimmune diseases.
Meanwhile, dermatologists have been fielding calls from hopeful patients and sorting out who should get the treatment, how to advise patients on risks and benefits, and what tests should be used before and after starting treatment.
Uptake for new systemic drugs, such as biologics, can be slow in dermatology, noted Adam Friedman, MD, professor and chair of dermatology, George Washington University, Washington, as some doctors like to stick with what they know.
He told this news organization that he hopes that uptake for baricitinib is quicker, as it is the only approved oral systemic treatment for patients with severe alopecia areata, which affects about 300,000 people a year in the United States. Other treatments, including steroid injections in the scalp, have lacked efficacy and convenience.
Beyond the physical effects, the mental toll of patchy hair clumps and missing brows and lashes can be devastating for patients with alopecia areata.
Fielding patient inquiries
Word of the FDA approval spread fast, and calls and emails are coming into dermatologists’ offices and clinics from interested patients.
Physicians should be ready for patients with any kind of hair loss, not just severe alopecia areata, to ask about the drug, Dr. Friedman said. Some patients contacting him don’t fit the indication, which “highlights how disabling hair loss” is for people, considering that, in general, “people see this and think it is for them.”
Baricitinib is not a new drug, but a drug with a new indication. It had already been approved for treating moderate to severe RA in patients who have had an inadequate response to one or more tumor necrosis factor blockers, and for treating COVID-19 in certain hospitalized adults.
Boxed warning
Patients may ask about the boxed warning in the baricitinib label about the increased risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Natasha A. Mesinkovska, MD, PhD, an investigator in the clinical trials that led to FDA approval of baricitinib and the chief scientific officer at the National Alopecia Areata Foundation, told this news organization that several aspects of the label are important to point out.
One is that the warning is for all the JAK inhibitors used to treat RA and other inflammatory conditions, not just baricitinib. Also, the warning is based mostly on data on patients with RA who, she noted, have substantial comorbidities and have been taking toxic immunosuppressive medications. The RA population is also typically many years older than the alopecia areata population.
“Whether the warnings apply to the alopecia areata patients is as yet unclear,” said Dr. Mesinkovska, who is also an associate professor of dermatology at the University of California, Irvine.
Patients are also asking about how well it works.
In one of the two trials that led up to the FDA approval, which enrolled patients with at least 50% scalp hair loss for over 6 months, 22% of the patients who received 2 mg of baricitinib and 35% of those who received 4 mg saw adequate hair coverage (at least 80%) at week 36, compared with 5% on placebo. In the second trial, 17% of those who received 2 mg and 32% who received 4 mg saw adequate hair coverage, compared with 3% on placebo.
Common side effects associated with baricitinib, according to the FDA, are lower respiratory tract infections, headache, acne, high cholesterol, increased creatinine phosphokinase, urinary tract infection, liver enzyme elevations, folliculitis, fatigue, nausea, genital yeast infections, anemia, neutropenia, abdominal pain, herpes zoster (shingles), and weight gain.
The risk-benefit discussions with patients should also include potential benefits beyond hair regrowth on the scalp. Loss of hair in the ears and nose can affect hearing and allergies, Dr. Mesinkovska said.
“About 30%-50% with alopecia areata, depending on age group or part of the world, will have allergies,” she said.
Patients should also know that baricitinib will need to be taken “for a very long time,” Dr. Mesinkovska noted. It’s possible that could be forever and that stopping the medication at any point may result in hair falling out again, she says, but duration will vary from case to case.
The good news is that it has been well tolerated. “We give a lot of medications for acne like doxycycline and other antibiotics and people have more stomach problems and angst with those than with [baricitinib],” she said.
Regrowth takes time
Benjamin Ungar, MD, a dermatologist at the Alopecia Center of Excellence at Mount Sinai, New York, told this news organization that an important message for patients is that hair regrowth takes time. For some other skin conditions, patients start treatment and see almost instant improvement.
“That is not the case for alopecia areata,” he said. “The expectation is that it will take months for regrowth in general.”
He said he hasn’t started prescribing baricitinib yet, but plans to do so soon.
“Obviously, I’ll have conversations with patients about it, but it’s a medication I’m going to be using, definitely. I have no reservations,” Dr. Ungar said.
After initial testing, physicians may find that some patients might not be ideal candidates, he added. People with liver disease, a history of blood clots, abnormal blood counts, or low neutrophils are among those who may not be the best candidates for baricitinib.
For most with severe alopecia areata, though, baricitinib provides hope.
“Treatment options have been not readily available, often inaccessible, ineffective, often dangerous,” he said. “There’s a treatment now that can be accessed, generally is safe and is effective for many people.”
Be up front with patients about the unknown
Additionally, it’s important to tell patients what is not yet known, the experts interviewed say.
“Alopecia areata is a chronic disease. We don’t have long-term data on the patient population yet,” Dr. Friedman said.
Also unknown is how easy it will be for physicians to get insurance to reimburse for baricitinib, which, at the end of June, was priced at about $5,000 a month for the 4-mg dose. FDA approval was important in that regard. Previously, some claims had been rejected for drugs used off label for AA.
“We dermatologists know how much it affects patients,” Dr. Mesinkovska said. “As long as we stick by what we know and convey to insurers how much it affects people’s lives, they should cover it.”
Another unknown is what other drugs can be taken with baricitinib. In clinical trials, it was used alone, she said. Currently, concomitant use of other immune suppressants – such as methotrexate or prednisone – is not recommended. But it remains to be seen what other medications will be safe to use at the same time as more long-term data are available.
Lynne J. Goldberg, MD, professor of dermatology, pathology, and laboratory medicine, Boston University, and director of the Hair Clinic at Boston Medical Center, said that she received a slew of emails from patients asking about baricitinib, but most of them did not have alopecia areata and were not candidates for this treatment.
She said that nurses in her clinic have been instructed on what to tell patients about which patients the drug is meant to treat, side effects, and benefits.
Access won’t be immediate
Dr. Goldberg said the drug’s approval does not mean immediate access. The patient has to come in, discuss the treatment, and get lab tests first. “It’s not a casual drug. This is a potent immunosuppressant drug. You need lab tests and once you start it you need blood tests every 3 months to stay on it.”
Those tests may vary by physician, but people will generally need a standard blood count and a comprehensive metabolic panel and lipid panel. “There’s nothing esoteric,” she said.
She added that physicians will need to check for presence of infections including tuberculosis and hepatitis B and C before prescribing, just as they would before they start prescribing a biologic.
“You don’t want to reactivate something,” she noted.
But, Dr. Goldberg added, the benefits for all who have been either living with only patches of hair or no hair or who put on a wig or hat every day are “life changing.”
Dr. Mesinkovska is on the advisory boards and runs trials for Eli Lilly, Pfizer, and Concert Pharmaceuticals. Dr. Friedman, Dr. Goldberg, and Dr. Ungar reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
On June 13, the FDA approved baricitinib, a Janus kinase inhibitor (Olumiant, Lilly), for severe AA, and two other options may not be far behind. Pfizer and Concert Pharmaceuticals have JAK inhibitors in late-stage development for AA. JAK inhibitors, including baricitinib, are already on the market for treating rheumatoid arthritis and other autoimmune diseases.
Meanwhile, dermatologists have been fielding calls from hopeful patients and sorting out who should get the treatment, how to advise patients on risks and benefits, and what tests should be used before and after starting treatment.
Uptake for new systemic drugs, such as biologics, can be slow in dermatology, noted Adam Friedman, MD, professor and chair of dermatology, George Washington University, Washington, as some doctors like to stick with what they know.
He told this news organization that he hopes that uptake for baricitinib is quicker, as it is the only approved oral systemic treatment for patients with severe alopecia areata, which affects about 300,000 people a year in the United States. Other treatments, including steroid injections in the scalp, have lacked efficacy and convenience.
Beyond the physical effects, the mental toll of patchy hair clumps and missing brows and lashes can be devastating for patients with alopecia areata.
Fielding patient inquiries
Word of the FDA approval spread fast, and calls and emails are coming into dermatologists’ offices and clinics from interested patients.
Physicians should be ready for patients with any kind of hair loss, not just severe alopecia areata, to ask about the drug, Dr. Friedman said. Some patients contacting him don’t fit the indication, which “highlights how disabling hair loss” is for people, considering that, in general, “people see this and think it is for them.”
Baricitinib is not a new drug, but a drug with a new indication. It had already been approved for treating moderate to severe RA in patients who have had an inadequate response to one or more tumor necrosis factor blockers, and for treating COVID-19 in certain hospitalized adults.
Boxed warning
Patients may ask about the boxed warning in the baricitinib label about the increased risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis.
Natasha A. Mesinkovska, MD, PhD, an investigator in the clinical trials that led to FDA approval of baricitinib and the chief scientific officer at the National Alopecia Areata Foundation, told this news organization that several aspects of the label are important to point out.
One is that the warning is for all the JAK inhibitors used to treat RA and other inflammatory conditions, not just baricitinib. Also, the warning is based mostly on data on patients with RA who, she noted, have substantial comorbidities and have been taking toxic immunosuppressive medications. The RA population is also typically many years older than the alopecia areata population.
“Whether the warnings apply to the alopecia areata patients is as yet unclear,” said Dr. Mesinkovska, who is also an associate professor of dermatology at the University of California, Irvine.
Patients are also asking about how well it works.
In one of the two trials that led up to the FDA approval, which enrolled patients with at least 50% scalp hair loss for over 6 months, 22% of the patients who received 2 mg of baricitinib and 35% of those who received 4 mg saw adequate hair coverage (at least 80%) at week 36, compared with 5% on placebo. In the second trial, 17% of those who received 2 mg and 32% who received 4 mg saw adequate hair coverage, compared with 3% on placebo.
Common side effects associated with baricitinib, according to the FDA, are lower respiratory tract infections, headache, acne, high cholesterol, increased creatinine phosphokinase, urinary tract infection, liver enzyme elevations, folliculitis, fatigue, nausea, genital yeast infections, anemia, neutropenia, abdominal pain, herpes zoster (shingles), and weight gain.
The risk-benefit discussions with patients should also include potential benefits beyond hair regrowth on the scalp. Loss of hair in the ears and nose can affect hearing and allergies, Dr. Mesinkovska said.
“About 30%-50% with alopecia areata, depending on age group or part of the world, will have allergies,” she said.
Patients should also know that baricitinib will need to be taken “for a very long time,” Dr. Mesinkovska noted. It’s possible that could be forever and that stopping the medication at any point may result in hair falling out again, she says, but duration will vary from case to case.
The good news is that it has been well tolerated. “We give a lot of medications for acne like doxycycline and other antibiotics and people have more stomach problems and angst with those than with [baricitinib],” she said.
Regrowth takes time
Benjamin Ungar, MD, a dermatologist at the Alopecia Center of Excellence at Mount Sinai, New York, told this news organization that an important message for patients is that hair regrowth takes time. For some other skin conditions, patients start treatment and see almost instant improvement.
“That is not the case for alopecia areata,” he said. “The expectation is that it will take months for regrowth in general.”
He said he hasn’t started prescribing baricitinib yet, but plans to do so soon.
“Obviously, I’ll have conversations with patients about it, but it’s a medication I’m going to be using, definitely. I have no reservations,” Dr. Ungar said.
After initial testing, physicians may find that some patients might not be ideal candidates, he added. People with liver disease, a history of blood clots, abnormal blood counts, or low neutrophils are among those who may not be the best candidates for baricitinib.
For most with severe alopecia areata, though, baricitinib provides hope.
“Treatment options have been not readily available, often inaccessible, ineffective, often dangerous,” he said. “There’s a treatment now that can be accessed, generally is safe and is effective for many people.”
Be up front with patients about the unknown
Additionally, it’s important to tell patients what is not yet known, the experts interviewed say.
“Alopecia areata is a chronic disease. We don’t have long-term data on the patient population yet,” Dr. Friedman said.
Also unknown is how easy it will be for physicians to get insurance to reimburse for baricitinib, which, at the end of June, was priced at about $5,000 a month for the 4-mg dose. FDA approval was important in that regard. Previously, some claims had been rejected for drugs used off label for AA.
“We dermatologists know how much it affects patients,” Dr. Mesinkovska said. “As long as we stick by what we know and convey to insurers how much it affects people’s lives, they should cover it.”
Another unknown is what other drugs can be taken with baricitinib. In clinical trials, it was used alone, she said. Currently, concomitant use of other immune suppressants – such as methotrexate or prednisone – is not recommended. But it remains to be seen what other medications will be safe to use at the same time as more long-term data are available.
Lynne J. Goldberg, MD, professor of dermatology, pathology, and laboratory medicine, Boston University, and director of the Hair Clinic at Boston Medical Center, said that she received a slew of emails from patients asking about baricitinib, but most of them did not have alopecia areata and were not candidates for this treatment.
She said that nurses in her clinic have been instructed on what to tell patients about which patients the drug is meant to treat, side effects, and benefits.
Access won’t be immediate
Dr. Goldberg said the drug’s approval does not mean immediate access. The patient has to come in, discuss the treatment, and get lab tests first. “It’s not a casual drug. This is a potent immunosuppressant drug. You need lab tests and once you start it you need blood tests every 3 months to stay on it.”
Those tests may vary by physician, but people will generally need a standard blood count and a comprehensive metabolic panel and lipid panel. “There’s nothing esoteric,” she said.
She added that physicians will need to check for presence of infections including tuberculosis and hepatitis B and C before prescribing, just as they would before they start prescribing a biologic.
“You don’t want to reactivate something,” she noted.
But, Dr. Goldberg added, the benefits for all who have been either living with only patches of hair or no hair or who put on a wig or hat every day are “life changing.”
Dr. Mesinkovska is on the advisory boards and runs trials for Eli Lilly, Pfizer, and Concert Pharmaceuticals. Dr. Friedman, Dr. Goldberg, and Dr. Ungar reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Nail dystrophy and foot pain

These findings are consistent with a type of heritable keratoderma called pachyonychia congenita (also called twenty-nails dystrophy). It is easy to mistake this unusual cause of thickening nails with a more common cause: onychomycosis.
Pachyonychia congenita describes a set of disorders driven by heritable defects in 1 of 5 keratin genes. The disorder is often transmitted in an autosomal dominant fashion, although a third of patients are thought to have a spontaneous mutation.1 These gene changes can cause 1 or multiple dystrophic nails, thickened nail beds, natal teeth, thick plantar or palmar nodules or plaques, and hearing difficulties. Some patients may have symptoms at birth, while other patients do not develop symptoms until later in life.1
There is currently no cure for pachyonychia congenita. Patients with suspected heritable keratoderma benefit from referral to Medical Genetics and a dermatologist who is comfortable treating keratodermas. Patients can obtain free genetic testing, educational material, and additional resources through pachyonychia.org.
This patient was prescribed topical urea 40% cream that was to be applied to the feet nightly, until the nodules became less painful. He was also evaluated for pressure-offloading orthotics. Nails may be treated with topical urea lacquer nightly until patients are satisfied with the appearance, although this patient chose to forgo the lacquer.
Text courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. Photos courtesy of Jonathan Karnes, MD (copyright retained).
1. Smith FJD, Hansen CD, Hull PR, et al. Pachyonychia congenita. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews. Seattle (WA): University of Washington, Seattle; 2006. Updated November 30, 2017. Accessed June 27, 2022. https://www.ncbi.nlm.nih.gov/books/NBK1280/
These findings are consistent with a type of heritable keratoderma called pachyonychia congenita (also called twenty-nails dystrophy). It is easy to mistake this unusual cause of thickening nails with a more common cause: onychomycosis.
Pachyonychia congenita describes a set of disorders driven by heritable defects in 1 of 5 keratin genes. The disorder is often transmitted in an autosomal dominant fashion, although a third of patients are thought to have a spontaneous mutation.1 These gene changes can cause 1 or multiple dystrophic nails, thickened nail beds, natal teeth, thick plantar or palmar nodules or plaques, and hearing difficulties. Some patients may have symptoms at birth, while other patients do not develop symptoms until later in life.1
There is currently no cure for pachyonychia congenita. Patients with suspected heritable keratoderma benefit from referral to Medical Genetics and a dermatologist who is comfortable treating keratodermas. Patients can obtain free genetic testing, educational material, and additional resources through pachyonychia.org.
This patient was prescribed topical urea 40% cream that was to be applied to the feet nightly, until the nodules became less painful. He was also evaluated for pressure-offloading orthotics. Nails may be treated with topical urea lacquer nightly until patients are satisfied with the appearance, although this patient chose to forgo the lacquer.
Text courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. Photos courtesy of Jonathan Karnes, MD (copyright retained).
These findings are consistent with a type of heritable keratoderma called pachyonychia congenita (also called twenty-nails dystrophy). It is easy to mistake this unusual cause of thickening nails with a more common cause: onychomycosis.
Pachyonychia congenita describes a set of disorders driven by heritable defects in 1 of 5 keratin genes. The disorder is often transmitted in an autosomal dominant fashion, although a third of patients are thought to have a spontaneous mutation.1 These gene changes can cause 1 or multiple dystrophic nails, thickened nail beds, natal teeth, thick plantar or palmar nodules or plaques, and hearing difficulties. Some patients may have symptoms at birth, while other patients do not develop symptoms until later in life.1
There is currently no cure for pachyonychia congenita. Patients with suspected heritable keratoderma benefit from referral to Medical Genetics and a dermatologist who is comfortable treating keratodermas. Patients can obtain free genetic testing, educational material, and additional resources through pachyonychia.org.
This patient was prescribed topical urea 40% cream that was to be applied to the feet nightly, until the nodules became less painful. He was also evaluated for pressure-offloading orthotics. Nails may be treated with topical urea lacquer nightly until patients are satisfied with the appearance, although this patient chose to forgo the lacquer.
Text courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. Photos courtesy of Jonathan Karnes, MD (copyright retained).
1. Smith FJD, Hansen CD, Hull PR, et al. Pachyonychia congenita. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews. Seattle (WA): University of Washington, Seattle; 2006. Updated November 30, 2017. Accessed June 27, 2022. https://www.ncbi.nlm.nih.gov/books/NBK1280/
1. Smith FJD, Hansen CD, Hull PR, et al. Pachyonychia congenita. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews. Seattle (WA): University of Washington, Seattle; 2006. Updated November 30, 2017. Accessed June 27, 2022. https://www.ncbi.nlm.nih.gov/books/NBK1280/

‘Not their fault:’ Obesity warrants long-term management
This transcript has been edited for clarity.
It’s important to remember and to think about the first time when patients with obesity come to see us: What have they faced? What have been their struggles? What shame and blame and bias have they faced?
One of the first things that I do when a patient comes to see me is invite them to share their weight journey with me. I ask them to tell me about their struggles, about what’s worked and what hasn’t worked, what they would like, and what their health goals are.
As they share their stories, I look for the opportunity to share with them that obesity is not their fault, but that it’s biology driving their body to carry extra weight and their body is super smart. Neither their body nor their brain want them to starve.
Our bodies evolved during a time where there was food scarcity and the potential of famine. We have a complex system that was designed to make sure that we always held on to extra weight, specifically extra fat, because that’s how we store energy. In the current obesogenic environment, what happens is our bodies carry extra weight, or specifically, extra fat.
Again, I say to them, this is biology. Your body’s doing exactly what it was designed to do. Your body’s very smart, but now we have to figure out how to help your body want to carry less fat because it is impacting your health. This is not your fault. Having obesity is not your fault any more than having diabetes or hypertension is anyone’s fault. Now it’s time for all of us to use highly effective tools that target the pathophysiology of obesity.
When a patient comes to me for weight management or to help them treat their obesity, I listen to them, and I look for clues as to what might help that specific patient. Every patient deserves to have individualized treatment. One medicine may be right for one person, another medicine may be right for another, and surgery may be right for another patient. I really try to listen and hear what that patient is telling me.
What we as providers really need is tools – different options – to be able to provide for our patients and basically present them with different options, and then guide them toward the best therapy for them. Whether it’s semaglutide or tirzepatide potentially in the future, these types of medications are excellent options for our patients. They’re highly effective tools with safe profiles.
A question that I often get from providers or patients is, “Well, Doctor, I’ve lost the weight now. How long should I take this medicine? Can I stop it now?”
Then, we have a conversation, and we actually usually have this conversation even before we start the medicine. Basically, we talk about the fact that obesity is a chronic disease. There’s no cure for obesity. Because it’s a chronic disease, we need to treat it like we would treat any other chronic disease.
The example that I often use is, if you have a patient who has hypertension and you start them on an antihypertensive medication, what happens? Their blood pressure goes down. It improves. Now, if their blood pressure is improved with a specific antihypertensive, would you stop that medicine? What would happen if you stopped that antihypertensive? Well, their blood pressure would go up, and we wouldn’t be surprised.
In the same way, if you have a patient who has obesity and you start that patient on an antiobesity medication, and their weight decreases, and their body fat mass at that point decreases, what would happen if you stop that medicine? They lost the weight, but you stop the medicine. Well, their weight gain comes back. They regain the weight.
We should not be surprised that weight gain occurs when we stop the treatment. That really underscores the fact that treatment needs to be continued. If a patient is started on an antiobesity medication and they lose weight, that medication needs to be continued to maintain that weight loss.
Basically, we eat food and our body responds by releasing these hormones. The hormones are made in our gut and in our pancreas and these hormones inform our brain. Are we hungry? Are we full? Where are we with our homeostatic set point of fat mass? Based on that, our brain is like the sensor or the thermostat.
Obesity is a chronic, treatable disease. We should treat obesity as we treat any other chronic disease, with effective and safe approaches that target underlying disease mechanisms. These results in the SURMOUNT-1 trial underscore that tirzepatide may be doing just that. Remarkably, 9 in 10 individuals with obesity lost weight while taking tirzepatide. These results are impressive. They’re an important step forward in potentially expanding effective therapeutic options for people with obesity.
Dr. Jastreboff is an associate professor of medicine and pediatrics at Yale University, New Haven, Conn., and director of weight management and obesity prevention at Yale Stress Center. She reported conducting trials with Eli Lilly, Novo Nordisk, and Rhythm Pharmaceuticals; serving on scientific advisory boards for Ely Lilly, Intellihealth, Novo Nordisk, Pfizer, Rhythm Pharmaceuticals, and WW; and consulting for Boehringer Ingelheim and Scholar Rock.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
It’s important to remember and to think about the first time when patients with obesity come to see us: What have they faced? What have been their struggles? What shame and blame and bias have they faced?
One of the first things that I do when a patient comes to see me is invite them to share their weight journey with me. I ask them to tell me about their struggles, about what’s worked and what hasn’t worked, what they would like, and what their health goals are.
As they share their stories, I look for the opportunity to share with them that obesity is not their fault, but that it’s biology driving their body to carry extra weight and their body is super smart. Neither their body nor their brain want them to starve.
Our bodies evolved during a time where there was food scarcity and the potential of famine. We have a complex system that was designed to make sure that we always held on to extra weight, specifically extra fat, because that’s how we store energy. In the current obesogenic environment, what happens is our bodies carry extra weight, or specifically, extra fat.
Again, I say to them, this is biology. Your body’s doing exactly what it was designed to do. Your body’s very smart, but now we have to figure out how to help your body want to carry less fat because it is impacting your health. This is not your fault. Having obesity is not your fault any more than having diabetes or hypertension is anyone’s fault. Now it’s time for all of us to use highly effective tools that target the pathophysiology of obesity.
When a patient comes to me for weight management or to help them treat their obesity, I listen to them, and I look for clues as to what might help that specific patient. Every patient deserves to have individualized treatment. One medicine may be right for one person, another medicine may be right for another, and surgery may be right for another patient. I really try to listen and hear what that patient is telling me.
What we as providers really need is tools – different options – to be able to provide for our patients and basically present them with different options, and then guide them toward the best therapy for them. Whether it’s semaglutide or tirzepatide potentially in the future, these types of medications are excellent options for our patients. They’re highly effective tools with safe profiles.
A question that I often get from providers or patients is, “Well, Doctor, I’ve lost the weight now. How long should I take this medicine? Can I stop it now?”
Then, we have a conversation, and we actually usually have this conversation even before we start the medicine. Basically, we talk about the fact that obesity is a chronic disease. There’s no cure for obesity. Because it’s a chronic disease, we need to treat it like we would treat any other chronic disease.
The example that I often use is, if you have a patient who has hypertension and you start them on an antihypertensive medication, what happens? Their blood pressure goes down. It improves. Now, if their blood pressure is improved with a specific antihypertensive, would you stop that medicine? What would happen if you stopped that antihypertensive? Well, their blood pressure would go up, and we wouldn’t be surprised.
In the same way, if you have a patient who has obesity and you start that patient on an antiobesity medication, and their weight decreases, and their body fat mass at that point decreases, what would happen if you stop that medicine? They lost the weight, but you stop the medicine. Well, their weight gain comes back. They regain the weight.
We should not be surprised that weight gain occurs when we stop the treatment. That really underscores the fact that treatment needs to be continued. If a patient is started on an antiobesity medication and they lose weight, that medication needs to be continued to maintain that weight loss.
Basically, we eat food and our body responds by releasing these hormones. The hormones are made in our gut and in our pancreas and these hormones inform our brain. Are we hungry? Are we full? Where are we with our homeostatic set point of fat mass? Based on that, our brain is like the sensor or the thermostat.
Obesity is a chronic, treatable disease. We should treat obesity as we treat any other chronic disease, with effective and safe approaches that target underlying disease mechanisms. These results in the SURMOUNT-1 trial underscore that tirzepatide may be doing just that. Remarkably, 9 in 10 individuals with obesity lost weight while taking tirzepatide. These results are impressive. They’re an important step forward in potentially expanding effective therapeutic options for people with obesity.
Dr. Jastreboff is an associate professor of medicine and pediatrics at Yale University, New Haven, Conn., and director of weight management and obesity prevention at Yale Stress Center. She reported conducting trials with Eli Lilly, Novo Nordisk, and Rhythm Pharmaceuticals; serving on scientific advisory boards for Ely Lilly, Intellihealth, Novo Nordisk, Pfizer, Rhythm Pharmaceuticals, and WW; and consulting for Boehringer Ingelheim and Scholar Rock.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
It’s important to remember and to think about the first time when patients with obesity come to see us: What have they faced? What have been their struggles? What shame and blame and bias have they faced?
One of the first things that I do when a patient comes to see me is invite them to share their weight journey with me. I ask them to tell me about their struggles, about what’s worked and what hasn’t worked, what they would like, and what their health goals are.
As they share their stories, I look for the opportunity to share with them that obesity is not their fault, but that it’s biology driving their body to carry extra weight and their body is super smart. Neither their body nor their brain want them to starve.
Our bodies evolved during a time where there was food scarcity and the potential of famine. We have a complex system that was designed to make sure that we always held on to extra weight, specifically extra fat, because that’s how we store energy. In the current obesogenic environment, what happens is our bodies carry extra weight, or specifically, extra fat.
Again, I say to them, this is biology. Your body’s doing exactly what it was designed to do. Your body’s very smart, but now we have to figure out how to help your body want to carry less fat because it is impacting your health. This is not your fault. Having obesity is not your fault any more than having diabetes or hypertension is anyone’s fault. Now it’s time for all of us to use highly effective tools that target the pathophysiology of obesity.
When a patient comes to me for weight management or to help them treat their obesity, I listen to them, and I look for clues as to what might help that specific patient. Every patient deserves to have individualized treatment. One medicine may be right for one person, another medicine may be right for another, and surgery may be right for another patient. I really try to listen and hear what that patient is telling me.
What we as providers really need is tools – different options – to be able to provide for our patients and basically present them with different options, and then guide them toward the best therapy for them. Whether it’s semaglutide or tirzepatide potentially in the future, these types of medications are excellent options for our patients. They’re highly effective tools with safe profiles.
A question that I often get from providers or patients is, “Well, Doctor, I’ve lost the weight now. How long should I take this medicine? Can I stop it now?”
Then, we have a conversation, and we actually usually have this conversation even before we start the medicine. Basically, we talk about the fact that obesity is a chronic disease. There’s no cure for obesity. Because it’s a chronic disease, we need to treat it like we would treat any other chronic disease.
The example that I often use is, if you have a patient who has hypertension and you start them on an antihypertensive medication, what happens? Their blood pressure goes down. It improves. Now, if their blood pressure is improved with a specific antihypertensive, would you stop that medicine? What would happen if you stopped that antihypertensive? Well, their blood pressure would go up, and we wouldn’t be surprised.
In the same way, if you have a patient who has obesity and you start that patient on an antiobesity medication, and their weight decreases, and their body fat mass at that point decreases, what would happen if you stop that medicine? They lost the weight, but you stop the medicine. Well, their weight gain comes back. They regain the weight.
We should not be surprised that weight gain occurs when we stop the treatment. That really underscores the fact that treatment needs to be continued. If a patient is started on an antiobesity medication and they lose weight, that medication needs to be continued to maintain that weight loss.
Basically, we eat food and our body responds by releasing these hormones. The hormones are made in our gut and in our pancreas and these hormones inform our brain. Are we hungry? Are we full? Where are we with our homeostatic set point of fat mass? Based on that, our brain is like the sensor or the thermostat.
Obesity is a chronic, treatable disease. We should treat obesity as we treat any other chronic disease, with effective and safe approaches that target underlying disease mechanisms. These results in the SURMOUNT-1 trial underscore that tirzepatide may be doing just that. Remarkably, 9 in 10 individuals with obesity lost weight while taking tirzepatide. These results are impressive. They’re an important step forward in potentially expanding effective therapeutic options for people with obesity.
Dr. Jastreboff is an associate professor of medicine and pediatrics at Yale University, New Haven, Conn., and director of weight management and obesity prevention at Yale Stress Center. She reported conducting trials with Eli Lilly, Novo Nordisk, and Rhythm Pharmaceuticals; serving on scientific advisory boards for Ely Lilly, Intellihealth, Novo Nordisk, Pfizer, Rhythm Pharmaceuticals, and WW; and consulting for Boehringer Ingelheim and Scholar Rock.
A version of this article first appeared on Medscape.com.
