User login
AVAHO
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
VA Cancer Clinical Trials as a Strategy for Increasing Accrual of Racial and Ethnic Underrepresented Groups
Background
Cancer clinical trials (CCTs) are central to improving cancer care. However, generalizability of findings from CCTs is difficult due to the lack of diversity in most United States CCTs. Clinical trial accrual of underrepresented groups, is low throughout the United States and is approximately 4-5% in most CCTs. Reasons for low accrual in this population are multifactorial. Despite numerous factors related to accruing racial and ethnic underrepresented groups, many institutions have sought to address these barriers. We conducted a scoping review to identify evidence-based approaches to increase participation in cancer treatment clinical trials.
Methods
We reviewed the Salisbury VA Medical Center Oncology clinical trial database from October 2019 to June 2024. The participants in these clinical trials required consent. These clinical trials included treatment interventional as well as non-treatment interventional. Fifteen studies were included and over 260 Veterans participated.
Results
Key themes emerged that included a focus on patient education, cultural competency, and building capacity in the clinics to care for the Veteran population at three separate sites in the Salisbury VA system. The Black Veteran accrual rate of 29% was achieved. This accrual rate is representative of our VA catchment population of 33% for Black Veterans, and is five times the national average.
Conclusions
The research team’s success in enrolling Black Veterans in clinical trials is attributed to several factors. The demographic composition of Veterans served by the Salisbury, Charlotte, and Kernersville VA provided a diverse population that included a 33% Black group. The type of clinical trials focused on patients who were most impacted by the disease. The VA did afford less barriers to access to health care.
Background
Cancer clinical trials (CCTs) are central to improving cancer care. However, generalizability of findings from CCTs is difficult due to the lack of diversity in most United States CCTs. Clinical trial accrual of underrepresented groups, is low throughout the United States and is approximately 4-5% in most CCTs. Reasons for low accrual in this population are multifactorial. Despite numerous factors related to accruing racial and ethnic underrepresented groups, many institutions have sought to address these barriers. We conducted a scoping review to identify evidence-based approaches to increase participation in cancer treatment clinical trials.
Methods
We reviewed the Salisbury VA Medical Center Oncology clinical trial database from October 2019 to June 2024. The participants in these clinical trials required consent. These clinical trials included treatment interventional as well as non-treatment interventional. Fifteen studies were included and over 260 Veterans participated.
Results
Key themes emerged that included a focus on patient education, cultural competency, and building capacity in the clinics to care for the Veteran population at three separate sites in the Salisbury VA system. The Black Veteran accrual rate of 29% was achieved. This accrual rate is representative of our VA catchment population of 33% for Black Veterans, and is five times the national average.
Conclusions
The research team’s success in enrolling Black Veterans in clinical trials is attributed to several factors. The demographic composition of Veterans served by the Salisbury, Charlotte, and Kernersville VA provided a diverse population that included a 33% Black group. The type of clinical trials focused on patients who were most impacted by the disease. The VA did afford less barriers to access to health care.
Background
Cancer clinical trials (CCTs) are central to improving cancer care. However, generalizability of findings from CCTs is difficult due to the lack of diversity in most United States CCTs. Clinical trial accrual of underrepresented groups, is low throughout the United States and is approximately 4-5% in most CCTs. Reasons for low accrual in this population are multifactorial. Despite numerous factors related to accruing racial and ethnic underrepresented groups, many institutions have sought to address these barriers. We conducted a scoping review to identify evidence-based approaches to increase participation in cancer treatment clinical trials.
Methods
We reviewed the Salisbury VA Medical Center Oncology clinical trial database from October 2019 to June 2024. The participants in these clinical trials required consent. These clinical trials included treatment interventional as well as non-treatment interventional. Fifteen studies were included and over 260 Veterans participated.
Results
Key themes emerged that included a focus on patient education, cultural competency, and building capacity in the clinics to care for the Veteran population at three separate sites in the Salisbury VA system. The Black Veteran accrual rate of 29% was achieved. This accrual rate is representative of our VA catchment population of 33% for Black Veterans, and is five times the national average.
Conclusions
The research team’s success in enrolling Black Veterans in clinical trials is attributed to several factors. The demographic composition of Veterans served by the Salisbury, Charlotte, and Kernersville VA provided a diverse population that included a 33% Black group. The type of clinical trials focused on patients who were most impacted by the disease. The VA did afford less barriers to access to health care.

Patient Navigators for Serious Illnesses Can Now Bill Under New Medicare Codes
In a move that acknowledges the gauntlet the US health system poses for people facing serious and fatal illnesses, Medicare will pay for a new class of workers to help patients manage treatments for conditions like cancer and heart failure.
The 2024 Medicare physician fee schedule includes new billing codes, including G0023, to pay for 60 minutes a month of care coordination by certified or trained auxiliary personnel working under the direction of a clinician.
A diagnosis of cancer or another serious illness takes a toll beyond the physical effects of the disease. Patients often scramble to make adjustments in family and work schedules to manage treatment, said Samyukta Mullangi, MD, MBA, medical director of oncology at Thyme Care, a Nashville, Tennessee–based firm that provides navigation and coordination services to oncology practices and insurers.

“It just really does create a bit of a pressure cooker for patients,” Dr. Mullangi told this news organization.
Medicare has for many years paid for medical professionals to help patients cope with the complexities of disease, such as chronic care management (CCM) provided by physicians, nurses, and physician assistants.
The new principal illness navigation (PIN) payments are intended to pay for work that to date typically has been done by people without medical degrees, including those involved in peer support networks and community health programs. The US Centers for Medicare and Medicaid Services(CMS) expects these navigators will undergo training and work under the supervision of clinicians.
The new navigators may coordinate care transitions between medical settings, follow up with patients after emergency department (ED) visits, or communicate with skilled nursing facilities regarding the psychosocial needs and functional deficits of a patient, among other functions.
CMS expects the new navigators may:
- Conduct assessments to understand a patient’s life story, strengths, needs, goals, preferences, and desired outcomes, including understanding cultural and linguistic factors.
- Provide support to accomplish the clinician’s treatment plan.
- Coordinate the receipt of needed services from healthcare facilities, home- and community-based service providers, and caregivers.
Peers as Navigators
The new navigators can be former patients who have undergone similar treatments for serious diseases, CMS said. This approach sets the new program apart from other care management services Medicare already covers, program officials wrote in the 2024 physician fee schedule.
“For some conditions, patients are best able to engage with the healthcare system and access care if they have assistance from a single, dedicated individual who has ‘lived experience,’ ” according to the rule.
The agency has taken a broad initial approach in defining what kinds of illnesses a patient may have to qualify for services. Patients must have a serious condition that is expected to last at least 3 months, such as cancer, heart failure, or substance use disorder.
But those without a definitive diagnosis may also qualify to receive navigator services.
In the rule, CMS cited a case in which a CT scan identified a suspicious mass in a patient’s colon. A clinician might decide this person would benefit from navigation services due to the potential risks for an undiagnosed illness.
“Regardless of the definitive diagnosis of the mass, presence of a colonic mass for that patient may be a serious high-risk condition that could, for example, cause obstruction and lead the patient to present to the emergency department, as well as be potentially indicative of an underlying life-threatening illness such as colon cancer,” CMS wrote in the rule.
Navigators often start their work when cancer patients are screened and guide them through initial diagnosis, potential surgery, radiation, or chemotherapy, said Sharon Gentry, MSN, RN, a former nurse navigator who is now the editor in chief of the Journal of the Academy of Oncology Nurse & Patient Navigators.
The navigators are meant to be a trusted and continual presence for patients, who otherwise might be left to start anew in finding help at each phase of care.
The navigators “see the whole picture. They see the whole journey the patient takes, from pre-diagnosis all the way through diagnosis care out through survival,” Ms. Gentry said.

Gaining a special Medicare payment for these kinds of services will elevate this work, she said.
Many newer drugs can target specific mechanisms and proteins of cancer. Often, oncology treatment involves testing to find out if mutations are allowing the cancer cells to evade a patient’s immune system.
Checking these biomarkers takes time, however. Patients sometimes become frustrated because they are anxious to begin treatment. Patients may receive inaccurate information from friends or family who went through treatment previously. Navigators can provide knowledge on the current state of care for a patient’s disease, helping them better manage anxieties.
“You have to explain to them that things have changed since the guy you drink coffee with was diagnosed with cancer, and there may be a drug that could target that,” Ms. Gentry said.
Potential Challenges
Initial uptake of the new PIN codes may be slow going, however, as clinicians and health systems may already use well-established codes. These include CCM and principal care management services, which may pay higher rates, Mullangi said.
“There might be sensitivity around not wanting to cannibalize existing programs with a new program,” Dr. Mullangi said.
In addition, many patients will have a copay for the services of principal illness navigators, Dr. Mullangi said.
While many patients have additional insurance that would cover the service, not all do. People with traditional Medicare coverage can sometimes pay 20% of the cost of some medical services.
“I think that may give patients pause, particularly if they’re already feeling the financial burden of a cancer treatment journey,” Dr. Mullangi said.
Pay rates for PIN services involve calculations of regional price differences, which are posted publicly by CMS, and potential added fees for services provided by hospital-affiliated organizations.
Consider payments for code G0023, covering 60 minutes of principal navigation services provided in a single month.
A set reimbursement for patients cared for in independent medical practices exists, with variation for local costs. Medicare’s non-facility price for G0023 would be $102.41 in some parts of Silicon Valley in California, including San Jose. In Arkansas, where costs are lower, reimbursement would be $73.14 for this same service.
Patients who get services covered by code G0023 in independent medical practices would have monthly copays of about $15-$20, depending on where they live.
The tab for patients tends to be higher for these same services if delivered through a medical practice owned by a hospital, as this would trigger the addition of facility fees to the payments made to cover the services. Facility fees are difficult for the public to ascertain before getting a treatment or service.
Dr. Mullangi and Ms. Gentry reported no relevant financial disclosures outside of their employers.
A version of this article first appeared on Medscape.com.
In a move that acknowledges the gauntlet the US health system poses for people facing serious and fatal illnesses, Medicare will pay for a new class of workers to help patients manage treatments for conditions like cancer and heart failure.
The 2024 Medicare physician fee schedule includes new billing codes, including G0023, to pay for 60 minutes a month of care coordination by certified or trained auxiliary personnel working under the direction of a clinician.
A diagnosis of cancer or another serious illness takes a toll beyond the physical effects of the disease. Patients often scramble to make adjustments in family and work schedules to manage treatment, said Samyukta Mullangi, MD, MBA, medical director of oncology at Thyme Care, a Nashville, Tennessee–based firm that provides navigation and coordination services to oncology practices and insurers.
“It just really does create a bit of a pressure cooker for patients,” Dr. Mullangi told this news organization.
Medicare has for many years paid for medical professionals to help patients cope with the complexities of disease, such as chronic care management (CCM) provided by physicians, nurses, and physician assistants.
The new principal illness navigation (PIN) payments are intended to pay for work that to date typically has been done by people without medical degrees, including those involved in peer support networks and community health programs. The US Centers for Medicare and Medicaid Services(CMS) expects these navigators will undergo training and work under the supervision of clinicians.
The new navigators may coordinate care transitions between medical settings, follow up with patients after emergency department (ED) visits, or communicate with skilled nursing facilities regarding the psychosocial needs and functional deficits of a patient, among other functions.
CMS expects the new navigators may:
- Conduct assessments to understand a patient’s life story, strengths, needs, goals, preferences, and desired outcomes, including understanding cultural and linguistic factors.
- Provide support to accomplish the clinician’s treatment plan.
- Coordinate the receipt of needed services from healthcare facilities, home- and community-based service providers, and caregivers.
Peers as Navigators
The new navigators can be former patients who have undergone similar treatments for serious diseases, CMS said. This approach sets the new program apart from other care management services Medicare already covers, program officials wrote in the 2024 physician fee schedule.
“For some conditions, patients are best able to engage with the healthcare system and access care if they have assistance from a single, dedicated individual who has ‘lived experience,’ ” according to the rule.
The agency has taken a broad initial approach in defining what kinds of illnesses a patient may have to qualify for services. Patients must have a serious condition that is expected to last at least 3 months, such as cancer, heart failure, or substance use disorder.
But those without a definitive diagnosis may also qualify to receive navigator services.
In the rule, CMS cited a case in which a CT scan identified a suspicious mass in a patient’s colon. A clinician might decide this person would benefit from navigation services due to the potential risks for an undiagnosed illness.
“Regardless of the definitive diagnosis of the mass, presence of a colonic mass for that patient may be a serious high-risk condition that could, for example, cause obstruction and lead the patient to present to the emergency department, as well as be potentially indicative of an underlying life-threatening illness such as colon cancer,” CMS wrote in the rule.
Navigators often start their work when cancer patients are screened and guide them through initial diagnosis, potential surgery, radiation, or chemotherapy, said Sharon Gentry, MSN, RN, a former nurse navigator who is now the editor in chief of the Journal of the Academy of Oncology Nurse & Patient Navigators.
The navigators are meant to be a trusted and continual presence for patients, who otherwise might be left to start anew in finding help at each phase of care.
The navigators “see the whole picture. They see the whole journey the patient takes, from pre-diagnosis all the way through diagnosis care out through survival,” Ms. Gentry said.
Gaining a special Medicare payment for these kinds of services will elevate this work, she said.
Many newer drugs can target specific mechanisms and proteins of cancer. Often, oncology treatment involves testing to find out if mutations are allowing the cancer cells to evade a patient’s immune system.
Checking these biomarkers takes time, however. Patients sometimes become frustrated because they are anxious to begin treatment. Patients may receive inaccurate information from friends or family who went through treatment previously. Navigators can provide knowledge on the current state of care for a patient’s disease, helping them better manage anxieties.
“You have to explain to them that things have changed since the guy you drink coffee with was diagnosed with cancer, and there may be a drug that could target that,” Ms. Gentry said.
Potential Challenges
Initial uptake of the new PIN codes may be slow going, however, as clinicians and health systems may already use well-established codes. These include CCM and principal care management services, which may pay higher rates, Mullangi said.
“There might be sensitivity around not wanting to cannibalize existing programs with a new program,” Dr. Mullangi said.
In addition, many patients will have a copay for the services of principal illness navigators, Dr. Mullangi said.
While many patients have additional insurance that would cover the service, not all do. People with traditional Medicare coverage can sometimes pay 20% of the cost of some medical services.
“I think that may give patients pause, particularly if they’re already feeling the financial burden of a cancer treatment journey,” Dr. Mullangi said.
Pay rates for PIN services involve calculations of regional price differences, which are posted publicly by CMS, and potential added fees for services provided by hospital-affiliated organizations.
Consider payments for code G0023, covering 60 minutes of principal navigation services provided in a single month.
A set reimbursement for patients cared for in independent medical practices exists, with variation for local costs. Medicare’s non-facility price for G0023 would be $102.41 in some parts of Silicon Valley in California, including San Jose. In Arkansas, where costs are lower, reimbursement would be $73.14 for this same service.
Patients who get services covered by code G0023 in independent medical practices would have monthly copays of about $15-$20, depending on where they live.
The tab for patients tends to be higher for these same services if delivered through a medical practice owned by a hospital, as this would trigger the addition of facility fees to the payments made to cover the services. Facility fees are difficult for the public to ascertain before getting a treatment or service.
Dr. Mullangi and Ms. Gentry reported no relevant financial disclosures outside of their employers.
A version of this article first appeared on Medscape.com.
In a move that acknowledges the gauntlet the US health system poses for people facing serious and fatal illnesses, Medicare will pay for a new class of workers to help patients manage treatments for conditions like cancer and heart failure.
The 2024 Medicare physician fee schedule includes new billing codes, including G0023, to pay for 60 minutes a month of care coordination by certified or trained auxiliary personnel working under the direction of a clinician.
A diagnosis of cancer or another serious illness takes a toll beyond the physical effects of the disease. Patients often scramble to make adjustments in family and work schedules to manage treatment, said Samyukta Mullangi, MD, MBA, medical director of oncology at Thyme Care, a Nashville, Tennessee–based firm that provides navigation and coordination services to oncology practices and insurers.
“It just really does create a bit of a pressure cooker for patients,” Dr. Mullangi told this news organization.
Medicare has for many years paid for medical professionals to help patients cope with the complexities of disease, such as chronic care management (CCM) provided by physicians, nurses, and physician assistants.
The new principal illness navigation (PIN) payments are intended to pay for work that to date typically has been done by people without medical degrees, including those involved in peer support networks and community health programs. The US Centers for Medicare and Medicaid Services(CMS) expects these navigators will undergo training and work under the supervision of clinicians.
The new navigators may coordinate care transitions between medical settings, follow up with patients after emergency department (ED) visits, or communicate with skilled nursing facilities regarding the psychosocial needs and functional deficits of a patient, among other functions.
CMS expects the new navigators may:
- Conduct assessments to understand a patient’s life story, strengths, needs, goals, preferences, and desired outcomes, including understanding cultural and linguistic factors.
- Provide support to accomplish the clinician’s treatment plan.
- Coordinate the receipt of needed services from healthcare facilities, home- and community-based service providers, and caregivers.
Peers as Navigators
The new navigators can be former patients who have undergone similar treatments for serious diseases, CMS said. This approach sets the new program apart from other care management services Medicare already covers, program officials wrote in the 2024 physician fee schedule.
“For some conditions, patients are best able to engage with the healthcare system and access care if they have assistance from a single, dedicated individual who has ‘lived experience,’ ” according to the rule.
The agency has taken a broad initial approach in defining what kinds of illnesses a patient may have to qualify for services. Patients must have a serious condition that is expected to last at least 3 months, such as cancer, heart failure, or substance use disorder.
But those without a definitive diagnosis may also qualify to receive navigator services.
In the rule, CMS cited a case in which a CT scan identified a suspicious mass in a patient’s colon. A clinician might decide this person would benefit from navigation services due to the potential risks for an undiagnosed illness.
“Regardless of the definitive diagnosis of the mass, presence of a colonic mass for that patient may be a serious high-risk condition that could, for example, cause obstruction and lead the patient to present to the emergency department, as well as be potentially indicative of an underlying life-threatening illness such as colon cancer,” CMS wrote in the rule.
Navigators often start their work when cancer patients are screened and guide them through initial diagnosis, potential surgery, radiation, or chemotherapy, said Sharon Gentry, MSN, RN, a former nurse navigator who is now the editor in chief of the Journal of the Academy of Oncology Nurse & Patient Navigators.
The navigators are meant to be a trusted and continual presence for patients, who otherwise might be left to start anew in finding help at each phase of care.
The navigators “see the whole picture. They see the whole journey the patient takes, from pre-diagnosis all the way through diagnosis care out through survival,” Ms. Gentry said.
Gaining a special Medicare payment for these kinds of services will elevate this work, she said.
Many newer drugs can target specific mechanisms and proteins of cancer. Often, oncology treatment involves testing to find out if mutations are allowing the cancer cells to evade a patient’s immune system.
Checking these biomarkers takes time, however. Patients sometimes become frustrated because they are anxious to begin treatment. Patients may receive inaccurate information from friends or family who went through treatment previously. Navigators can provide knowledge on the current state of care for a patient’s disease, helping them better manage anxieties.
“You have to explain to them that things have changed since the guy you drink coffee with was diagnosed with cancer, and there may be a drug that could target that,” Ms. Gentry said.
Potential Challenges
Initial uptake of the new PIN codes may be slow going, however, as clinicians and health systems may already use well-established codes. These include CCM and principal care management services, which may pay higher rates, Mullangi said.
“There might be sensitivity around not wanting to cannibalize existing programs with a new program,” Dr. Mullangi said.
In addition, many patients will have a copay for the services of principal illness navigators, Dr. Mullangi said.
While many patients have additional insurance that would cover the service, not all do. People with traditional Medicare coverage can sometimes pay 20% of the cost of some medical services.
“I think that may give patients pause, particularly if they’re already feeling the financial burden of a cancer treatment journey,” Dr. Mullangi said.
Pay rates for PIN services involve calculations of regional price differences, which are posted publicly by CMS, and potential added fees for services provided by hospital-affiliated organizations.
Consider payments for code G0023, covering 60 minutes of principal navigation services provided in a single month.
A set reimbursement for patients cared for in independent medical practices exists, with variation for local costs. Medicare’s non-facility price for G0023 would be $102.41 in some parts of Silicon Valley in California, including San Jose. In Arkansas, where costs are lower, reimbursement would be $73.14 for this same service.
Patients who get services covered by code G0023 in independent medical practices would have monthly copays of about $15-$20, depending on where they live.
The tab for patients tends to be higher for these same services if delivered through a medical practice owned by a hospital, as this would trigger the addition of facility fees to the payments made to cover the services. Facility fees are difficult for the public to ascertain before getting a treatment or service.
Dr. Mullangi and Ms. Gentry reported no relevant financial disclosures outside of their employers.
A version of this article first appeared on Medscape.com.

Improving Colorectal Cancer Screening via Mailed Fecal Immunochemical Testing in a Veterans Affairs Health System
Colorectal cancer (CRC) is among the most common cancers and causes of cancer-related deaths in the United States.1 Reflective of a nationwide trend, CRC screening rates at the Veterans Affairs Connecticut Healthcare System (VACHS) decreased during the COVID-19 pandemic.2-5 Contributing factors to this decrease included cancellations of elective colonoscopies during the initial phase of the pandemic and concurrent turnover of endoscopists. In 2021, the US Preventive Services Task Force lowered the recommended initial CRC screening age from 50 years to 45 years, further increasing the backlog of unscreened patients.6
Fecal immunochemical testing (FIT) is a noninvasive screening method in which antibodies are used to detect hemoglobin in the stool. The sensitivity and specificity of 1-time FIT are 79% to 80% and 94%, respectively, for the detection of CRC, with sensitivity improving with successive testing.7,8 Annual FIT is recognized as a tier 1 preferred screening method by the US Multi-Society Task Force on Colorectal Cancer.7,9 Programs that mail FIT kits to eligible patients outside of physician visits have been successfully implemented in health care systems.10,11
The VACHS designed and implemented a mailed FIT program using existing infrastructure and staffing.
Program Description
A team of local stakeholders comprised of VACHS leadership, primary care, nursing, and gastroenterology staff, as well as representatives from laboratory, informatics, mail services, and group practice management, was established to execute the project. The team met monthly to plan the project.
The team developed a dataset consisting of patients aged 45 to 75 years who were at average risk for CRC and due for CRC screening. Patients were defined as due for CRC screening if they had not had a colonoscopy in the previous 9 years or a FIT or fecal occult blood test in the previous 11 months. Average risk for CRC was defined by excluding patients with associated diagnosis codes for CRC, colectomy, inflammatory bowel disease, and anemia. The program also excluded patients with diagnosis codes associated with dementia, deferring discussions about cancer screening to their primary care practitioners (PCPs). Patients with invalid mailing addresses were also excluded, as well as those whose PCPs had indicated in the electronic health record that the patient received CRC screening outside the US Department of Veterans Affairs (VA) system.
Letter Templates
Two patient letter electronic health record templates were developed. The first was a primer letter, which was mailed to patients 2 to 3 weeks before the mailed FIT kit as an introduction to the program.12 The purpose of the primer letter was to give advance notice to patients that they could expect a FIT kit to arrive in the mail. The goal was to prepare patients to complete FIT when the kit arrived and prompt them to call the VA to opt out of the mailed FIT program if they were up to date with CRC screening or if they had a condition which made them at high risk for CRC.
The second FIT letter arrived with the FIT kit, introduced FIT and described the importance of CRC screening. The letter detailed instructions for completing FIT and automatically created a FIT order. It also included a list of common conditions that may exclude patients, with a recommendation for patients to contact their medical team if they felt they were not candidates for FIT.
Staff Education
A previous VACHS pilot project demonstrated the success of a mailed FIT program to increase FIT use. Implemented as part of the pilot program, staff education consisted of a session for clinicians about the role of FIT in CRC screening and an all-staff education session. An additional education session about CRC and FIT for all staff was repeated with the program launch.
Program Launch
The mailed FIT program was introduced during a VACHS primary care all-staff meeting. After the meeting, each patient aligned care team (PACT) received an encrypted email that included a list of the patients on their team who were candidates for the program, a patient-facing FIT instruction sheet, detailed instructions on how to send the FIT primer letter, and a FIT package consisting of the labeled FIT kit, FIT letter, and patient instruction sheet. A reminder letter was sent to each patient 3 weeks after the FIT package was mailed. The patient lists were populated into a shared, encrypted Microsoft Teams folder that was edited in real time by PACT teams and viewed by VACHS leadership to track progress.
Program Metrics
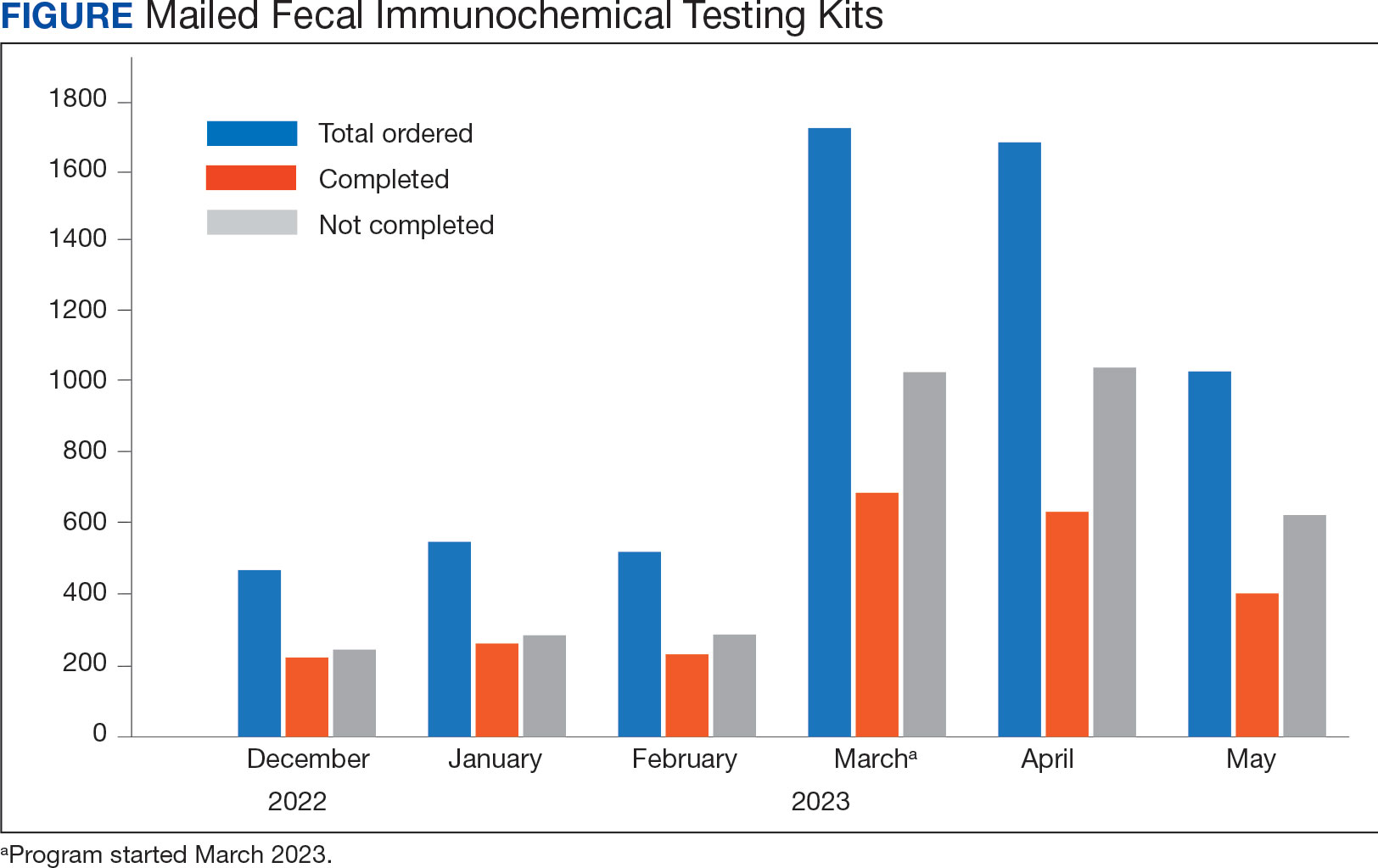
At program launch, the VACHS had 4642 patients due for CRC screening who were eligible for the mailed FIT program. On March 7, 2023, the data consisting of FIT tests ordered between December 2022 and May 2023—3 months before and after the launch of the program—were reviewed and categorized. In the 3 months before program launch, 1528 FIT were ordered and 714 were returned (46.7%). In the 3 months after the launch of the program, 4383 FIT were ordered and 1712 were returned (39.1%) (Figure). Test orders increased 287% from the preintervention to the postintervention period. The mean (SD) number of monthly FIT tests prelaunch was 509 (32.7), which increased to 1461 (331.6) postlaunch.
At the VACHS, 61.4% of patients aged 45 to 75 years were up to date with CRC screening before the program launch. In the 3 months after program launch, the rate increased to 63.8% among patients aged 45 to 75 years, the highest rate in our Veterans Integrated Services Network and exceeding the VA national average CRC screening rate, according to unpublished VA Monthly Management Report data.
In the 3 months following the program launch, 139 FIT kits tested positive for potential CRC. Of these, 79 (56.8%) patients had completed a diagnostic colonoscopy. PACT PCPs and nurses received reports on patients with positive FIT tests and those with no colonoscopy scheduled or completed and were asked to follow up.
Discussion
Through a proactive, population-based CRC screening program centered on mailed FIT kits outside of the traditional patient visit, the VACHS increased the use of FIT and rates of CRC screening. The numbers of FIT kits ordered and completed substantially increased in the 3 months after program launch.
Compared to mailed FIT programs described in the literature that rely on centralized processes in that a separate team operates the mailed FIT program for the entire organization, this program used existing PACT infrastructure and staff.10,11 This strategy allowed VACHS to design and implement the program in several months. Not needing to hire new staff or create a central team for the sole purpose of implementing the program allowed us to save on any organizational funding and efforts that would have accompanied the additional staff. The program described in this article may be more attainable for primary care practices or smaller health systems that do not have the capacity for the creation of a centralized process.
Limitations
Although the total number of FIT completions substantially increased during the program, the rate of FIT completion during the mailed FIT program was lower than the rate of completion prior to program launch. This decreased rate of FIT kit completion may be related to separation from a patient visit and potential loss of real-time education with a clinician. The program’s decentralized design increased the existing workload for primary care staff, and as a result, consideration must be given to local staffing levels. Additionally, the report of eligible patients depended on diagnosis codes and may have captured patients with higher-than-average risk of CRC, such as patients with prior history of adenomatous polyps, family history of CRC, or other medical or genetic conditions. We attempted to mitigate this by including a list of conditions that would exclude patients from FIT eligibility in the FIT letter and giving them the option to opt out.
Conclusions
CRC screening rates improved following implementation of a primary care team-centered quality improvement process to proactively identify patients appropriate for FIT and mail them FIT kits. This project highlights that population-health interventions around CRC screening via use of FIT can be successful within a primary care patient-centered medical home model, considering the increases in both CRC screening rates and increase in FIT tests ordered.
1. American Cancer Society. Key statistics for colorectal cancer. Revised January 29, 2024. Accessed June 11, 2024. https://www.cancer.org/cancer/types/colon-rectal-cancer/about/key-statistics.html
2. Chen RC, Haynes K, Du S, Barron J, Katz AJ. Association of cancer screening deficit in the United States with the COVID-19 pandemic. JAMA Oncol. 2021;7(6):878-884. doi:10.1001/jamaoncol.2021.0884
3. Mazidimoradi A, Tiznobaik A, Salehiniya H. Impact of the COVID-19 pandemic on colorectal cancer screening: a systematic review. J Gastrointest Cancer. 2022;53(3):730-744. doi:10.1007/s12029-021-00679-x
4. Adams MA, Kurlander JE, Gao Y, Yankey N, Saini SD. Impact of coronavirus disease 2019 on screening colonoscopy utilization in a large integrated health system. Gastroenterology. 2022;162(7):2098-2100.e2. doi:10.1053/j.gastro.2022.02.034
5. Sundaram S, Olson S, Sharma P, Rajendra S. A review of the impact of the COVID-19 pandemic on colorectal cancer screening: implications and solutions. Pathogens. 2021;10(11):558. doi:10.3390/pathogens10111508
6. US Preventive Services Task Force. Screening for colorectal cancer: US Preventive Services Task Force recommendation statement. JAMA. 2021;325(19):1965-1977. doi:10.1001/jama.2021.6238
7. Robertson DJ, Lee JK, Boland CR, et al. Recommendations on fecal immunochemical testing to screen for colorectal neoplasia: a consensus statement by the US Multi-Society Task Force on Colorectal Cancer. Gastrointest Endosc. 2017;85(1):2-21.e3. doi:10.1016/j.gie.2016.09.025
8. Lee JK, Liles EG, Bent S, Levin TR, Corley DA. Accuracy of fecal immunochemical tests for colorectal cancer: systematic review and meta-analysis. Ann Intern Med. 2014;160(3):171. doi:10.7326/M13-1484
9. Rex DK, Boland CR, Dominitz JA, et al. Colorectal cancer screening: recommendations for physicians and patients from the U.S. Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2017;153(1):307-323. doi:10.1053/j.gastro.2017.05.013
10. Deeds SA, Moore CB, Gunnink EJ, et al. Implementation of a mailed faecal immunochemical test programme for colorectal cancer screening among veterans. BMJ Open Qual. 2022;11(4):e001927. doi:10.1136/bmjoq-2022-001927
11. Selby K, Jensen CD, Levin TR, et al. Program components and results from an organized colorectal cancer screening program using annual fecal immunochemical testing. Clin Gastroenterol Hepatol. 2022;20(1):145-152. doi:10.1016/j.cgh.2020.09.042
12. Deeds S, Liu T, Schuttner L, et al. A postcard primer prior to mailed fecal immunochemical test among veterans: a randomized controlled trial. J Gen Intern Med. 2023:38(14):3235-3241. doi:10.1007/s11606-023-08248-7
Colorectal cancer (CRC) is among the most common cancers and causes of cancer-related deaths in the United States.1 Reflective of a nationwide trend, CRC screening rates at the Veterans Affairs Connecticut Healthcare System (VACHS) decreased during the COVID-19 pandemic.2-5 Contributing factors to this decrease included cancellations of elective colonoscopies during the initial phase of the pandemic and concurrent turnover of endoscopists. In 2021, the US Preventive Services Task Force lowered the recommended initial CRC screening age from 50 years to 45 years, further increasing the backlog of unscreened patients.6
Fecal immunochemical testing (FIT) is a noninvasive screening method in which antibodies are used to detect hemoglobin in the stool. The sensitivity and specificity of 1-time FIT are 79% to 80% and 94%, respectively, for the detection of CRC, with sensitivity improving with successive testing.7,8 Annual FIT is recognized as a tier 1 preferred screening method by the US Multi-Society Task Force on Colorectal Cancer.7,9 Programs that mail FIT kits to eligible patients outside of physician visits have been successfully implemented in health care systems.10,11
The VACHS designed and implemented a mailed FIT program using existing infrastructure and staffing.
Program Description
A team of local stakeholders comprised of VACHS leadership, primary care, nursing, and gastroenterology staff, as well as representatives from laboratory, informatics, mail services, and group practice management, was established to execute the project. The team met monthly to plan the project.
The team developed a dataset consisting of patients aged 45 to 75 years who were at average risk for CRC and due for CRC screening. Patients were defined as due for CRC screening if they had not had a colonoscopy in the previous 9 years or a FIT or fecal occult blood test in the previous 11 months. Average risk for CRC was defined by excluding patients with associated diagnosis codes for CRC, colectomy, inflammatory bowel disease, and anemia. The program also excluded patients with diagnosis codes associated with dementia, deferring discussions about cancer screening to their primary care practitioners (PCPs). Patients with invalid mailing addresses were also excluded, as well as those whose PCPs had indicated in the electronic health record that the patient received CRC screening outside the US Department of Veterans Affairs (VA) system.
Letter Templates
Two patient letter electronic health record templates were developed. The first was a primer letter, which was mailed to patients 2 to 3 weeks before the mailed FIT kit as an introduction to the program.12 The purpose of the primer letter was to give advance notice to patients that they could expect a FIT kit to arrive in the mail. The goal was to prepare patients to complete FIT when the kit arrived and prompt them to call the VA to opt out of the mailed FIT program if they were up to date with CRC screening or if they had a condition which made them at high risk for CRC.
The second FIT letter arrived with the FIT kit, introduced FIT and described the importance of CRC screening. The letter detailed instructions for completing FIT and automatically created a FIT order. It also included a list of common conditions that may exclude patients, with a recommendation for patients to contact their medical team if they felt they were not candidates for FIT.
Staff Education
A previous VACHS pilot project demonstrated the success of a mailed FIT program to increase FIT use. Implemented as part of the pilot program, staff education consisted of a session for clinicians about the role of FIT in CRC screening and an all-staff education session. An additional education session about CRC and FIT for all staff was repeated with the program launch.
Program Launch
The mailed FIT program was introduced during a VACHS primary care all-staff meeting. After the meeting, each patient aligned care team (PACT) received an encrypted email that included a list of the patients on their team who were candidates for the program, a patient-facing FIT instruction sheet, detailed instructions on how to send the FIT primer letter, and a FIT package consisting of the labeled FIT kit, FIT letter, and patient instruction sheet. A reminder letter was sent to each patient 3 weeks after the FIT package was mailed. The patient lists were populated into a shared, encrypted Microsoft Teams folder that was edited in real time by PACT teams and viewed by VACHS leadership to track progress.
Program Metrics
At program launch, the VACHS had 4642 patients due for CRC screening who were eligible for the mailed FIT program. On March 7, 2023, the data consisting of FIT tests ordered between December 2022 and May 2023—3 months before and after the launch of the program—were reviewed and categorized. In the 3 months before program launch, 1528 FIT were ordered and 714 were returned (46.7%). In the 3 months after the launch of the program, 4383 FIT were ordered and 1712 were returned (39.1%) (Figure). Test orders increased 287% from the preintervention to the postintervention period. The mean (SD) number of monthly FIT tests prelaunch was 509 (32.7), which increased to 1461 (331.6) postlaunch.
At the VACHS, 61.4% of patients aged 45 to 75 years were up to date with CRC screening before the program launch. In the 3 months after program launch, the rate increased to 63.8% among patients aged 45 to 75 years, the highest rate in our Veterans Integrated Services Network and exceeding the VA national average CRC screening rate, according to unpublished VA Monthly Management Report data.
In the 3 months following the program launch, 139 FIT kits tested positive for potential CRC. Of these, 79 (56.8%) patients had completed a diagnostic colonoscopy. PACT PCPs and nurses received reports on patients with positive FIT tests and those with no colonoscopy scheduled or completed and were asked to follow up.
Discussion
Through a proactive, population-based CRC screening program centered on mailed FIT kits outside of the traditional patient visit, the VACHS increased the use of FIT and rates of CRC screening. The numbers of FIT kits ordered and completed substantially increased in the 3 months after program launch.
Compared to mailed FIT programs described in the literature that rely on centralized processes in that a separate team operates the mailed FIT program for the entire organization, this program used existing PACT infrastructure and staff.10,11 This strategy allowed VACHS to design and implement the program in several months. Not needing to hire new staff or create a central team for the sole purpose of implementing the program allowed us to save on any organizational funding and efforts that would have accompanied the additional staff. The program described in this article may be more attainable for primary care practices or smaller health systems that do not have the capacity for the creation of a centralized process.
Limitations
Although the total number of FIT completions substantially increased during the program, the rate of FIT completion during the mailed FIT program was lower than the rate of completion prior to program launch. This decreased rate of FIT kit completion may be related to separation from a patient visit and potential loss of real-time education with a clinician. The program’s decentralized design increased the existing workload for primary care staff, and as a result, consideration must be given to local staffing levels. Additionally, the report of eligible patients depended on diagnosis codes and may have captured patients with higher-than-average risk of CRC, such as patients with prior history of adenomatous polyps, family history of CRC, or other medical or genetic conditions. We attempted to mitigate this by including a list of conditions that would exclude patients from FIT eligibility in the FIT letter and giving them the option to opt out.
Conclusions
CRC screening rates improved following implementation of a primary care team-centered quality improvement process to proactively identify patients appropriate for FIT and mail them FIT kits. This project highlights that population-health interventions around CRC screening via use of FIT can be successful within a primary care patient-centered medical home model, considering the increases in both CRC screening rates and increase in FIT tests ordered.
Colorectal cancer (CRC) is among the most common cancers and causes of cancer-related deaths in the United States.1 Reflective of a nationwide trend, CRC screening rates at the Veterans Affairs Connecticut Healthcare System (VACHS) decreased during the COVID-19 pandemic.2-5 Contributing factors to this decrease included cancellations of elective colonoscopies during the initial phase of the pandemic and concurrent turnover of endoscopists. In 2021, the US Preventive Services Task Force lowered the recommended initial CRC screening age from 50 years to 45 years, further increasing the backlog of unscreened patients.6
Fecal immunochemical testing (FIT) is a noninvasive screening method in which antibodies are used to detect hemoglobin in the stool. The sensitivity and specificity of 1-time FIT are 79% to 80% and 94%, respectively, for the detection of CRC, with sensitivity improving with successive testing.7,8 Annual FIT is recognized as a tier 1 preferred screening method by the US Multi-Society Task Force on Colorectal Cancer.7,9 Programs that mail FIT kits to eligible patients outside of physician visits have been successfully implemented in health care systems.10,11
The VACHS designed and implemented a mailed FIT program using existing infrastructure and staffing.
Program Description
A team of local stakeholders comprised of VACHS leadership, primary care, nursing, and gastroenterology staff, as well as representatives from laboratory, informatics, mail services, and group practice management, was established to execute the project. The team met monthly to plan the project.
The team developed a dataset consisting of patients aged 45 to 75 years who were at average risk for CRC and due for CRC screening. Patients were defined as due for CRC screening if they had not had a colonoscopy in the previous 9 years or a FIT or fecal occult blood test in the previous 11 months. Average risk for CRC was defined by excluding patients with associated diagnosis codes for CRC, colectomy, inflammatory bowel disease, and anemia. The program also excluded patients with diagnosis codes associated with dementia, deferring discussions about cancer screening to their primary care practitioners (PCPs). Patients with invalid mailing addresses were also excluded, as well as those whose PCPs had indicated in the electronic health record that the patient received CRC screening outside the US Department of Veterans Affairs (VA) system.
Letter Templates
Two patient letter electronic health record templates were developed. The first was a primer letter, which was mailed to patients 2 to 3 weeks before the mailed FIT kit as an introduction to the program.12 The purpose of the primer letter was to give advance notice to patients that they could expect a FIT kit to arrive in the mail. The goal was to prepare patients to complete FIT when the kit arrived and prompt them to call the VA to opt out of the mailed FIT program if they were up to date with CRC screening or if they had a condition which made them at high risk for CRC.
The second FIT letter arrived with the FIT kit, introduced FIT and described the importance of CRC screening. The letter detailed instructions for completing FIT and automatically created a FIT order. It also included a list of common conditions that may exclude patients, with a recommendation for patients to contact their medical team if they felt they were not candidates for FIT.
Staff Education
A previous VACHS pilot project demonstrated the success of a mailed FIT program to increase FIT use. Implemented as part of the pilot program, staff education consisted of a session for clinicians about the role of FIT in CRC screening and an all-staff education session. An additional education session about CRC and FIT for all staff was repeated with the program launch.
Program Launch
The mailed FIT program was introduced during a VACHS primary care all-staff meeting. After the meeting, each patient aligned care team (PACT) received an encrypted email that included a list of the patients on their team who were candidates for the program, a patient-facing FIT instruction sheet, detailed instructions on how to send the FIT primer letter, and a FIT package consisting of the labeled FIT kit, FIT letter, and patient instruction sheet. A reminder letter was sent to each patient 3 weeks after the FIT package was mailed. The patient lists were populated into a shared, encrypted Microsoft Teams folder that was edited in real time by PACT teams and viewed by VACHS leadership to track progress.
Program Metrics
At program launch, the VACHS had 4642 patients due for CRC screening who were eligible for the mailed FIT program. On March 7, 2023, the data consisting of FIT tests ordered between December 2022 and May 2023—3 months before and after the launch of the program—were reviewed and categorized. In the 3 months before program launch, 1528 FIT were ordered and 714 were returned (46.7%). In the 3 months after the launch of the program, 4383 FIT were ordered and 1712 were returned (39.1%) (Figure). Test orders increased 287% from the preintervention to the postintervention period. The mean (SD) number of monthly FIT tests prelaunch was 509 (32.7), which increased to 1461 (331.6) postlaunch.
At the VACHS, 61.4% of patients aged 45 to 75 years were up to date with CRC screening before the program launch. In the 3 months after program launch, the rate increased to 63.8% among patients aged 45 to 75 years, the highest rate in our Veterans Integrated Services Network and exceeding the VA national average CRC screening rate, according to unpublished VA Monthly Management Report data.
In the 3 months following the program launch, 139 FIT kits tested positive for potential CRC. Of these, 79 (56.8%) patients had completed a diagnostic colonoscopy. PACT PCPs and nurses received reports on patients with positive FIT tests and those with no colonoscopy scheduled or completed and were asked to follow up.
Discussion
Through a proactive, population-based CRC screening program centered on mailed FIT kits outside of the traditional patient visit, the VACHS increased the use of FIT and rates of CRC screening. The numbers of FIT kits ordered and completed substantially increased in the 3 months after program launch.
Compared to mailed FIT programs described in the literature that rely on centralized processes in that a separate team operates the mailed FIT program for the entire organization, this program used existing PACT infrastructure and staff.10,11 This strategy allowed VACHS to design and implement the program in several months. Not needing to hire new staff or create a central team for the sole purpose of implementing the program allowed us to save on any organizational funding and efforts that would have accompanied the additional staff. The program described in this article may be more attainable for primary care practices or smaller health systems that do not have the capacity for the creation of a centralized process.
Limitations
Although the total number of FIT completions substantially increased during the program, the rate of FIT completion during the mailed FIT program was lower than the rate of completion prior to program launch. This decreased rate of FIT kit completion may be related to separation from a patient visit and potential loss of real-time education with a clinician. The program’s decentralized design increased the existing workload for primary care staff, and as a result, consideration must be given to local staffing levels. Additionally, the report of eligible patients depended on diagnosis codes and may have captured patients with higher-than-average risk of CRC, such as patients with prior history of adenomatous polyps, family history of CRC, or other medical or genetic conditions. We attempted to mitigate this by including a list of conditions that would exclude patients from FIT eligibility in the FIT letter and giving them the option to opt out.
Conclusions
CRC screening rates improved following implementation of a primary care team-centered quality improvement process to proactively identify patients appropriate for FIT and mail them FIT kits. This project highlights that population-health interventions around CRC screening via use of FIT can be successful within a primary care patient-centered medical home model, considering the increases in both CRC screening rates and increase in FIT tests ordered.
1. American Cancer Society. Key statistics for colorectal cancer. Revised January 29, 2024. Accessed June 11, 2024. https://www.cancer.org/cancer/types/colon-rectal-cancer/about/key-statistics.html
2. Chen RC, Haynes K, Du S, Barron J, Katz AJ. Association of cancer screening deficit in the United States with the COVID-19 pandemic. JAMA Oncol. 2021;7(6):878-884. doi:10.1001/jamaoncol.2021.0884
3. Mazidimoradi A, Tiznobaik A, Salehiniya H. Impact of the COVID-19 pandemic on colorectal cancer screening: a systematic review. J Gastrointest Cancer. 2022;53(3):730-744. doi:10.1007/s12029-021-00679-x
4. Adams MA, Kurlander JE, Gao Y, Yankey N, Saini SD. Impact of coronavirus disease 2019 on screening colonoscopy utilization in a large integrated health system. Gastroenterology. 2022;162(7):2098-2100.e2. doi:10.1053/j.gastro.2022.02.034
5. Sundaram S, Olson S, Sharma P, Rajendra S. A review of the impact of the COVID-19 pandemic on colorectal cancer screening: implications and solutions. Pathogens. 2021;10(11):558. doi:10.3390/pathogens10111508
6. US Preventive Services Task Force. Screening for colorectal cancer: US Preventive Services Task Force recommendation statement. JAMA. 2021;325(19):1965-1977. doi:10.1001/jama.2021.6238
7. Robertson DJ, Lee JK, Boland CR, et al. Recommendations on fecal immunochemical testing to screen for colorectal neoplasia: a consensus statement by the US Multi-Society Task Force on Colorectal Cancer. Gastrointest Endosc. 2017;85(1):2-21.e3. doi:10.1016/j.gie.2016.09.025
8. Lee JK, Liles EG, Bent S, Levin TR, Corley DA. Accuracy of fecal immunochemical tests for colorectal cancer: systematic review and meta-analysis. Ann Intern Med. 2014;160(3):171. doi:10.7326/M13-1484
9. Rex DK, Boland CR, Dominitz JA, et al. Colorectal cancer screening: recommendations for physicians and patients from the U.S. Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2017;153(1):307-323. doi:10.1053/j.gastro.2017.05.013
10. Deeds SA, Moore CB, Gunnink EJ, et al. Implementation of a mailed faecal immunochemical test programme for colorectal cancer screening among veterans. BMJ Open Qual. 2022;11(4):e001927. doi:10.1136/bmjoq-2022-001927
11. Selby K, Jensen CD, Levin TR, et al. Program components and results from an organized colorectal cancer screening program using annual fecal immunochemical testing. Clin Gastroenterol Hepatol. 2022;20(1):145-152. doi:10.1016/j.cgh.2020.09.042
12. Deeds S, Liu T, Schuttner L, et al. A postcard primer prior to mailed fecal immunochemical test among veterans: a randomized controlled trial. J Gen Intern Med. 2023:38(14):3235-3241. doi:10.1007/s11606-023-08248-7
1. American Cancer Society. Key statistics for colorectal cancer. Revised January 29, 2024. Accessed June 11, 2024. https://www.cancer.org/cancer/types/colon-rectal-cancer/about/key-statistics.html
2. Chen RC, Haynes K, Du S, Barron J, Katz AJ. Association of cancer screening deficit in the United States with the COVID-19 pandemic. JAMA Oncol. 2021;7(6):878-884. doi:10.1001/jamaoncol.2021.0884
3. Mazidimoradi A, Tiznobaik A, Salehiniya H. Impact of the COVID-19 pandemic on colorectal cancer screening: a systematic review. J Gastrointest Cancer. 2022;53(3):730-744. doi:10.1007/s12029-021-00679-x
4. Adams MA, Kurlander JE, Gao Y, Yankey N, Saini SD. Impact of coronavirus disease 2019 on screening colonoscopy utilization in a large integrated health system. Gastroenterology. 2022;162(7):2098-2100.e2. doi:10.1053/j.gastro.2022.02.034
5. Sundaram S, Olson S, Sharma P, Rajendra S. A review of the impact of the COVID-19 pandemic on colorectal cancer screening: implications and solutions. Pathogens. 2021;10(11):558. doi:10.3390/pathogens10111508
6. US Preventive Services Task Force. Screening for colorectal cancer: US Preventive Services Task Force recommendation statement. JAMA. 2021;325(19):1965-1977. doi:10.1001/jama.2021.6238
7. Robertson DJ, Lee JK, Boland CR, et al. Recommendations on fecal immunochemical testing to screen for colorectal neoplasia: a consensus statement by the US Multi-Society Task Force on Colorectal Cancer. Gastrointest Endosc. 2017;85(1):2-21.e3. doi:10.1016/j.gie.2016.09.025
8. Lee JK, Liles EG, Bent S, Levin TR, Corley DA. Accuracy of fecal immunochemical tests for colorectal cancer: systematic review and meta-analysis. Ann Intern Med. 2014;160(3):171. doi:10.7326/M13-1484
9. Rex DK, Boland CR, Dominitz JA, et al. Colorectal cancer screening: recommendations for physicians and patients from the U.S. Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2017;153(1):307-323. doi:10.1053/j.gastro.2017.05.013
10. Deeds SA, Moore CB, Gunnink EJ, et al. Implementation of a mailed faecal immunochemical test programme for colorectal cancer screening among veterans. BMJ Open Qual. 2022;11(4):e001927. doi:10.1136/bmjoq-2022-001927
11. Selby K, Jensen CD, Levin TR, et al. Program components and results from an organized colorectal cancer screening program using annual fecal immunochemical testing. Clin Gastroenterol Hepatol. 2022;20(1):145-152. doi:10.1016/j.cgh.2020.09.042
12. Deeds S, Liu T, Schuttner L, et al. A postcard primer prior to mailed fecal immunochemical test among veterans: a randomized controlled trial. J Gen Intern Med. 2023:38(14):3235-3241. doi:10.1007/s11606-023-08248-7
Why Does the Heart Rarely Develop Cancer?
Why Does the Heart Rarely Develop Cancer?
The heart is one of the organs least likely to develop cancer, a long-standing biologic puzzle that may now have an explanation. A study published in Science found that the mechanical load generated by the beating heart suppresses tumor cell proliferation through a molecular pathway that alters gene expression, raising the possibility of new therapeutic targets.
Mechanical Protection
Tumors that originate directly in the myocardium are exceptionally rare, occurring in < 1% of autopsies. Even cardiac metastases, which have been reported in up to 18% of autopsies, are often small, asymptomatic, and incidentally discovered. Although this phenomenon has long been recognized, its biologic basis remains unclear.
The heart is notable for its limited capacity for regeneration. After birth, cardiomyocytes stop dividing and subsequently renew at a rate of about 1% per year. However, when the mechanical load is reduced, such as in patients supported by left ventricular assist devices, cardiomyocytes once again show signs of proliferation.
This observation prompted researchers to investigate whether the same mechanical load that restrains normal cardiac cells might also suppress cancer growth.
More Load, Less Growth
To investigate this question, researchers introduced two genetic alterations commonly found in human cancers, activation of the KRAS oncogene and loss of the TP53, into the liver, skeletal muscle, and hearts of mice. Tumors developed in multiple organs, but not in the heart.
The researchers then used a heterotopic heart transplant model in which a donor mouse’s heart is surgically connected to the neck (cervical) or abdominal vessels of a recipient mouse. The transplanted heart remained perfused but lost its normal mechanical loading (constant beating).
When researchers injected lung adenocarcinoma cells into 2 different hearts of the same animal, they observed entirely different outcomes. The cancer cells did not grow in the native mechanically loaded heart. However, the same cells grew rapidly and extensively in the mechanically unloaded transplanted heart.
Tumor cells had replaced nearly all normal tissue in the unloaded heart, whereas they occupied only approximately 20% of the ventricle in the native heart in 14 days. This difference could not be explained by differences in the initial tumor engraftment or cell death. Instead, the findings pointed to substantial differences in tumor cell proliferation.
Similar results were observed in bioengineered cardiac tissues exposed to varying degrees of mechanical stress. Tumor cells proliferated under conditions of low mechanical load but ceased proliferating as the mechanical load increased. Tumor growth was lowest in regions exposed to the greatest mechanical stimulation of cardiomyocytes in vitro.
However, the possibility of metabolic competition between cardiac and tumor cells for nutrition was ruled out.
From Mechanics to Genes
Next, we examined the influence of mechanical forces on tumor cell behavior.
Gene expression analyses of both human cardiac metastases and mouse tumor cells showed that mechanical stimulation altered chromatin accessibility through the activation of genes involved in chromatin remodeling. These changes promoted the expression of genes that suppress cell division.
The study also identified Nesprin-2, a part of the linker of the nucleoskeleton and cytoskeleton complex, which acts as a physical bridge. It is a multitasking protein that connects the cell’s outer structural network (cytoskeleton) to its inner genetic storage (nucleus) and appears to play a significant role in converting mechanical signals into changes in gene expression.
When Nesprin-2 was inactivated, cancer cells resumed proliferation despite exposure to a mechanical load, both in engineered tissues and animal models.
“Collectively, these results shed light on the role of mechanical forces in protecting the heart from cancer and may pave the way to cancer therapies based on mechanical stimulation,” concluded the authors.
An Actively Protected Organ
Speaking with Univadis Italy, part of the Medscape Professional Network, Giorgio Scita, PhD, director of the Mechanisms of Tumor Cell Migration research unit at AIRC Institute of Molecular Oncology and professor of general pathology at the University of Milan in Milan, Italy, said, “The study addressed a simple but fundamental question: Why is the heart largely resistant to cancer despite being highly vascularized and continuously exposed to circulating tumor cells?
These findings suggest that the heartbeat itself creates a mechanical environment that is hostile to tumor growth. The compressive forces generated by rhythmic myocardial contraction are sensed by cancer cells and translated into biochemical signals that limit their proliferation.
In this view, the heart is not simply an organ that is unfavorable for cancer growth but a tissue actively protected by its own mechanical forces.”
Speaking with Univadis Italy, Serena Zacchigna, PhD, study coauthor and head of the Cardiovascular Biology Laboratory at the International Centre for Genetic Engineering and Biotechnology, Trieste, Italy, said, “Until now, however, attention had focused primarily on signals from the extracellular matrix, such as tissue stiffness. Our study adds a new element: even forces generated directly by the movement of an organ — in this case, cardiac contraction — can influence the growth of cancer cells.”
Beyond the Heart
Scita said the findings have implications that extend well beyond the heart.
“The most significant aspect is that this work identifies tissue mechanics as an active regulator of tumor behavior,” he said. Stiffness, compression, tension, and confinement are not merely consequences of tumor growth, but factors capable of influencing proliferation, invasion, and dormancy.
The concept may apply to many solid tumors. Scita noted that cancer cells growing in confined environments, such as ductal carcinoma in situ of the breast, are exposed to substantial mechanical constraints. Understanding why some tumor cells remain susceptible to these forces whereas others evade them and become invasive remains a major unanswered question in cancer biology.
Research on these mechanisms is expanding internationally and in Italy as well. One example is the AIRC “5 per mille” (5 per thousand) research programs on metastatic disease, which includes projects designed to clarify how the mechanical properties of tumor tissue influence cancer initiation, metastatic spread, and disease progression.
Therapeutic Potential
According to Zacchigna, these findings open 2 principal avenues for future research.
“The first focuses on mechanical stimulation itself. In collaboration with engineers at the University of Siena, including a group led by Domenico Prattichizzo, researchers are developing wearable robotic devices designed to mimic the heartbeat and deliver mechanical stimulation to superficial solid tumors such as certain skin cancers.
The second approach is pharmacology. Researchers are investigating whether epigenetic therapies capable of modifying chromatin remodeling can reproduce the effects of cardiac contraction and suppress tumor cell proliferation.
However, Zacchigna cautioned that this work remains at an early experimental phase.”
However, before therapeutic applications can be pursued, important mechanistic questions remain unanswered.
Zacchigna noted that although the linker of nucleoskeleton and cytoskeleton (LINC) complex and Nesprin-2 are involved in signal transduction leading to chromatin reorganization and activation of cell cycle inhibitory loci, the molecular intermediates involved have yet to be fully defined.
Researchers also need to determine which genes are most critical, whether the mechanism operates across different tumor types, and whether it can be safely manipulated for therapeutic purposes.
In an accompanying commentary published in Science, Wyatt G. Paltzer, PhD, and James F. Martin, MD, from the Department of Integrative Physiology at the Baylor College of Medicine in Houston, noted that the findings suggest enhancing LINC complex activity could potentially suppress tumor growth.
However, because the complex has broad biologic functions, it may prove difficult to target therapeutically. The authors suggested that future studies should focus on identifying proteins that interact with Nesprin-2 or other components of the LINC complex and play a more specific role in inhibiting cancer cell proliferation.
Looking Ahead
Despite these challenges, Scita said that the study’s conceptual significance is already clear.
“Even if therapeutic applications remain years away, the findings suggest that cancer may one day be targeted by altering how tumor cells perceive and interpret physical forces.”
Scita and Zacchigna reported having no relevant conflicts of interest.
This story was translated from Univadis Italy.
A version of this article first appeared on Medscape.com.
The heart is one of the organs least likely to develop cancer, a long-standing biologic puzzle that may now have an explanation. A study published in Science found that the mechanical load generated by the beating heart suppresses tumor cell proliferation through a molecular pathway that alters gene expression, raising the possibility of new therapeutic targets.
Mechanical Protection
Tumors that originate directly in the myocardium are exceptionally rare, occurring in < 1% of autopsies. Even cardiac metastases, which have been reported in up to 18% of autopsies, are often small, asymptomatic, and incidentally discovered. Although this phenomenon has long been recognized, its biologic basis remains unclear.
The heart is notable for its limited capacity for regeneration. After birth, cardiomyocytes stop dividing and subsequently renew at a rate of about 1% per year. However, when the mechanical load is reduced, such as in patients supported by left ventricular assist devices, cardiomyocytes once again show signs of proliferation.
This observation prompted researchers to investigate whether the same mechanical load that restrains normal cardiac cells might also suppress cancer growth.
More Load, Less Growth
To investigate this question, researchers introduced two genetic alterations commonly found in human cancers, activation of the KRAS oncogene and loss of the TP53, into the liver, skeletal muscle, and hearts of mice. Tumors developed in multiple organs, but not in the heart.
The researchers then used a heterotopic heart transplant model in which a donor mouse’s heart is surgically connected to the neck (cervical) or abdominal vessels of a recipient mouse. The transplanted heart remained perfused but lost its normal mechanical loading (constant beating).
When researchers injected lung adenocarcinoma cells into 2 different hearts of the same animal, they observed entirely different outcomes. The cancer cells did not grow in the native mechanically loaded heart. However, the same cells grew rapidly and extensively in the mechanically unloaded transplanted heart.
Tumor cells had replaced nearly all normal tissue in the unloaded heart, whereas they occupied only approximately 20% of the ventricle in the native heart in 14 days. This difference could not be explained by differences in the initial tumor engraftment or cell death. Instead, the findings pointed to substantial differences in tumor cell proliferation.
Similar results were observed in bioengineered cardiac tissues exposed to varying degrees of mechanical stress. Tumor cells proliferated under conditions of low mechanical load but ceased proliferating as the mechanical load increased. Tumor growth was lowest in regions exposed to the greatest mechanical stimulation of cardiomyocytes in vitro.
However, the possibility of metabolic competition between cardiac and tumor cells for nutrition was ruled out.
From Mechanics to Genes
Next, we examined the influence of mechanical forces on tumor cell behavior.
Gene expression analyses of both human cardiac metastases and mouse tumor cells showed that mechanical stimulation altered chromatin accessibility through the activation of genes involved in chromatin remodeling. These changes promoted the expression of genes that suppress cell division.
The study also identified Nesprin-2, a part of the linker of the nucleoskeleton and cytoskeleton complex, which acts as a physical bridge. It is a multitasking protein that connects the cell’s outer structural network (cytoskeleton) to its inner genetic storage (nucleus) and appears to play a significant role in converting mechanical signals into changes in gene expression.
When Nesprin-2 was inactivated, cancer cells resumed proliferation despite exposure to a mechanical load, both in engineered tissues and animal models.
“Collectively, these results shed light on the role of mechanical forces in protecting the heart from cancer and may pave the way to cancer therapies based on mechanical stimulation,” concluded the authors.
An Actively Protected Organ
Speaking with Univadis Italy, part of the Medscape Professional Network, Giorgio Scita, PhD, director of the Mechanisms of Tumor Cell Migration research unit at AIRC Institute of Molecular Oncology and professor of general pathology at the University of Milan in Milan, Italy, said, “The study addressed a simple but fundamental question: Why is the heart largely resistant to cancer despite being highly vascularized and continuously exposed to circulating tumor cells?
These findings suggest that the heartbeat itself creates a mechanical environment that is hostile to tumor growth. The compressive forces generated by rhythmic myocardial contraction are sensed by cancer cells and translated into biochemical signals that limit their proliferation.
In this view, the heart is not simply an organ that is unfavorable for cancer growth but a tissue actively protected by its own mechanical forces.”
Speaking with Univadis Italy, Serena Zacchigna, PhD, study coauthor and head of the Cardiovascular Biology Laboratory at the International Centre for Genetic Engineering and Biotechnology, Trieste, Italy, said, “Until now, however, attention had focused primarily on signals from the extracellular matrix, such as tissue stiffness. Our study adds a new element: even forces generated directly by the movement of an organ — in this case, cardiac contraction — can influence the growth of cancer cells.”
Beyond the Heart
Scita said the findings have implications that extend well beyond the heart.
“The most significant aspect is that this work identifies tissue mechanics as an active regulator of tumor behavior,” he said. Stiffness, compression, tension, and confinement are not merely consequences of tumor growth, but factors capable of influencing proliferation, invasion, and dormancy.
The concept may apply to many solid tumors. Scita noted that cancer cells growing in confined environments, such as ductal carcinoma in situ of the breast, are exposed to substantial mechanical constraints. Understanding why some tumor cells remain susceptible to these forces whereas others evade them and become invasive remains a major unanswered question in cancer biology.
Research on these mechanisms is expanding internationally and in Italy as well. One example is the AIRC “5 per mille” (5 per thousand) research programs on metastatic disease, which includes projects designed to clarify how the mechanical properties of tumor tissue influence cancer initiation, metastatic spread, and disease progression.
Therapeutic Potential
According to Zacchigna, these findings open 2 principal avenues for future research.
“The first focuses on mechanical stimulation itself. In collaboration with engineers at the University of Siena, including a group led by Domenico Prattichizzo, researchers are developing wearable robotic devices designed to mimic the heartbeat and deliver mechanical stimulation to superficial solid tumors such as certain skin cancers.
The second approach is pharmacology. Researchers are investigating whether epigenetic therapies capable of modifying chromatin remodeling can reproduce the effects of cardiac contraction and suppress tumor cell proliferation.
However, Zacchigna cautioned that this work remains at an early experimental phase.”
However, before therapeutic applications can be pursued, important mechanistic questions remain unanswered.
Zacchigna noted that although the linker of nucleoskeleton and cytoskeleton (LINC) complex and Nesprin-2 are involved in signal transduction leading to chromatin reorganization and activation of cell cycle inhibitory loci, the molecular intermediates involved have yet to be fully defined.
Researchers also need to determine which genes are most critical, whether the mechanism operates across different tumor types, and whether it can be safely manipulated for therapeutic purposes.
In an accompanying commentary published in Science, Wyatt G. Paltzer, PhD, and James F. Martin, MD, from the Department of Integrative Physiology at the Baylor College of Medicine in Houston, noted that the findings suggest enhancing LINC complex activity could potentially suppress tumor growth.
However, because the complex has broad biologic functions, it may prove difficult to target therapeutically. The authors suggested that future studies should focus on identifying proteins that interact with Nesprin-2 or other components of the LINC complex and play a more specific role in inhibiting cancer cell proliferation.
Looking Ahead
Despite these challenges, Scita said that the study’s conceptual significance is already clear.
“Even if therapeutic applications remain years away, the findings suggest that cancer may one day be targeted by altering how tumor cells perceive and interpret physical forces.”
Scita and Zacchigna reported having no relevant conflicts of interest.
This story was translated from Univadis Italy.
A version of this article first appeared on Medscape.com.
The heart is one of the organs least likely to develop cancer, a long-standing biologic puzzle that may now have an explanation. A study published in Science found that the mechanical load generated by the beating heart suppresses tumor cell proliferation through a molecular pathway that alters gene expression, raising the possibility of new therapeutic targets.
Mechanical Protection
Tumors that originate directly in the myocardium are exceptionally rare, occurring in < 1% of autopsies. Even cardiac metastases, which have been reported in up to 18% of autopsies, are often small, asymptomatic, and incidentally discovered. Although this phenomenon has long been recognized, its biologic basis remains unclear.
The heart is notable for its limited capacity for regeneration. After birth, cardiomyocytes stop dividing and subsequently renew at a rate of about 1% per year. However, when the mechanical load is reduced, such as in patients supported by left ventricular assist devices, cardiomyocytes once again show signs of proliferation.
This observation prompted researchers to investigate whether the same mechanical load that restrains normal cardiac cells might also suppress cancer growth.
More Load, Less Growth
To investigate this question, researchers introduced two genetic alterations commonly found in human cancers, activation of the KRAS oncogene and loss of the TP53, into the liver, skeletal muscle, and hearts of mice. Tumors developed in multiple organs, but not in the heart.
The researchers then used a heterotopic heart transplant model in which a donor mouse’s heart is surgically connected to the neck (cervical) or abdominal vessels of a recipient mouse. The transplanted heart remained perfused but lost its normal mechanical loading (constant beating).
When researchers injected lung adenocarcinoma cells into 2 different hearts of the same animal, they observed entirely different outcomes. The cancer cells did not grow in the native mechanically loaded heart. However, the same cells grew rapidly and extensively in the mechanically unloaded transplanted heart.
Tumor cells had replaced nearly all normal tissue in the unloaded heart, whereas they occupied only approximately 20% of the ventricle in the native heart in 14 days. This difference could not be explained by differences in the initial tumor engraftment or cell death. Instead, the findings pointed to substantial differences in tumor cell proliferation.
Similar results were observed in bioengineered cardiac tissues exposed to varying degrees of mechanical stress. Tumor cells proliferated under conditions of low mechanical load but ceased proliferating as the mechanical load increased. Tumor growth was lowest in regions exposed to the greatest mechanical stimulation of cardiomyocytes in vitro.
However, the possibility of metabolic competition between cardiac and tumor cells for nutrition was ruled out.
From Mechanics to Genes
Next, we examined the influence of mechanical forces on tumor cell behavior.
Gene expression analyses of both human cardiac metastases and mouse tumor cells showed that mechanical stimulation altered chromatin accessibility through the activation of genes involved in chromatin remodeling. These changes promoted the expression of genes that suppress cell division.
The study also identified Nesprin-2, a part of the linker of the nucleoskeleton and cytoskeleton complex, which acts as a physical bridge. It is a multitasking protein that connects the cell’s outer structural network (cytoskeleton) to its inner genetic storage (nucleus) and appears to play a significant role in converting mechanical signals into changes in gene expression.
When Nesprin-2 was inactivated, cancer cells resumed proliferation despite exposure to a mechanical load, both in engineered tissues and animal models.
“Collectively, these results shed light on the role of mechanical forces in protecting the heart from cancer and may pave the way to cancer therapies based on mechanical stimulation,” concluded the authors.
An Actively Protected Organ
Speaking with Univadis Italy, part of the Medscape Professional Network, Giorgio Scita, PhD, director of the Mechanisms of Tumor Cell Migration research unit at AIRC Institute of Molecular Oncology and professor of general pathology at the University of Milan in Milan, Italy, said, “The study addressed a simple but fundamental question: Why is the heart largely resistant to cancer despite being highly vascularized and continuously exposed to circulating tumor cells?
These findings suggest that the heartbeat itself creates a mechanical environment that is hostile to tumor growth. The compressive forces generated by rhythmic myocardial contraction are sensed by cancer cells and translated into biochemical signals that limit their proliferation.
In this view, the heart is not simply an organ that is unfavorable for cancer growth but a tissue actively protected by its own mechanical forces.”
Speaking with Univadis Italy, Serena Zacchigna, PhD, study coauthor and head of the Cardiovascular Biology Laboratory at the International Centre for Genetic Engineering and Biotechnology, Trieste, Italy, said, “Until now, however, attention had focused primarily on signals from the extracellular matrix, such as tissue stiffness. Our study adds a new element: even forces generated directly by the movement of an organ — in this case, cardiac contraction — can influence the growth of cancer cells.”
Beyond the Heart
Scita said the findings have implications that extend well beyond the heart.
“The most significant aspect is that this work identifies tissue mechanics as an active regulator of tumor behavior,” he said. Stiffness, compression, tension, and confinement are not merely consequences of tumor growth, but factors capable of influencing proliferation, invasion, and dormancy.
The concept may apply to many solid tumors. Scita noted that cancer cells growing in confined environments, such as ductal carcinoma in situ of the breast, are exposed to substantial mechanical constraints. Understanding why some tumor cells remain susceptible to these forces whereas others evade them and become invasive remains a major unanswered question in cancer biology.
Research on these mechanisms is expanding internationally and in Italy as well. One example is the AIRC “5 per mille” (5 per thousand) research programs on metastatic disease, which includes projects designed to clarify how the mechanical properties of tumor tissue influence cancer initiation, metastatic spread, and disease progression.
Therapeutic Potential
According to Zacchigna, these findings open 2 principal avenues for future research.
“The first focuses on mechanical stimulation itself. In collaboration with engineers at the University of Siena, including a group led by Domenico Prattichizzo, researchers are developing wearable robotic devices designed to mimic the heartbeat and deliver mechanical stimulation to superficial solid tumors such as certain skin cancers.
The second approach is pharmacology. Researchers are investigating whether epigenetic therapies capable of modifying chromatin remodeling can reproduce the effects of cardiac contraction and suppress tumor cell proliferation.
However, Zacchigna cautioned that this work remains at an early experimental phase.”
However, before therapeutic applications can be pursued, important mechanistic questions remain unanswered.
Zacchigna noted that although the linker of nucleoskeleton and cytoskeleton (LINC) complex and Nesprin-2 are involved in signal transduction leading to chromatin reorganization and activation of cell cycle inhibitory loci, the molecular intermediates involved have yet to be fully defined.
Researchers also need to determine which genes are most critical, whether the mechanism operates across different tumor types, and whether it can be safely manipulated for therapeutic purposes.
In an accompanying commentary published in Science, Wyatt G. Paltzer, PhD, and James F. Martin, MD, from the Department of Integrative Physiology at the Baylor College of Medicine in Houston, noted that the findings suggest enhancing LINC complex activity could potentially suppress tumor growth.
However, because the complex has broad biologic functions, it may prove difficult to target therapeutically. The authors suggested that future studies should focus on identifying proteins that interact with Nesprin-2 or other components of the LINC complex and play a more specific role in inhibiting cancer cell proliferation.
Looking Ahead
Despite these challenges, Scita said that the study’s conceptual significance is already clear.
“Even if therapeutic applications remain years away, the findings suggest that cancer may one day be targeted by altering how tumor cells perceive and interpret physical forces.”
Scita and Zacchigna reported having no relevant conflicts of interest.
This story was translated from Univadis Italy.
A version of this article first appeared on Medscape.com.
Why Does the Heart Rarely Develop Cancer?
Why Does the Heart Rarely Develop Cancer?
Oral Nicotinamide: Cost-Effective for Reducing Keratinocyte Carcinoma Risk
Oral Nicotinamide: Cost-Effective for Reducing Keratinocyte Carcinoma Risk
Oral nicotinamide was cost-effective for reducing keratinocyte carcinoma (KC) risk in US veterans with a history of the disease, according to an economic analysis of Veterans Health Administration (VHA) data.
The findings, published online June 10 in JAMA Dermatology, “support strong consideration of nicotinamide for KC prevention in high-risk populations like veterans, particularly given its safety and tolerability,” wrote senior author Rebecca I. Hartman, MD, chief of the Dermatology Section at VA Boston and assistant professor of dermatology at Brigham and Women’s Hospital and Harvard Medical School, Boston, and co-authors.
Nicotinamide supplementation is “not only a cost-effective and patient-centric strategy for KC prevention, but it also remains economically favorable under a range of assumptions and may become even more cost-effective under higher procedure costs and frequency,” noted the authors.
The analysis included 33,822 individuals from the VHA database, all with a history of one or more KCs, including those with nicotinamide exposure for 30 or more days (n = 12,287) and those without that exposure (n = 21,535).
The mean ages in the unexposed and exposed cohorts were 76.9 and 77.2 years, respectively, and 98% were men. Procedural US VHA costs for KC treatment were estimated from previous research and adjusted for inflation. Nicotinamide pricing was obtained from the VHA.
KC incidence among nicotinamide-exposed and unexposed individuals was 0.204 and 0.255 events per person-year, respectively, reflecting an absolute risk reduction of 0.051 and 624 KCs prevented annually with nicotinamide supplementation.
With an estimated cost of $843 per KC, the yearly KC treatment expense was estimated at approximately $2.64 million, and the annual nicotinamide cost was estimated at $161,451, resulting in net savings of $364,581 — a 19.9% reduction in cohort-specific costs.
Assuming a quality-adjusted life-year (QALY) decrement of -0.01 per KC, nicotinamide use yielded an annual gain of 6.24 QALYs across the cohort and a savings of $58,426 per QALY gained.
A calculation of non-VHA cost-effectiveness, estimated with civilian prices and distributions, showed savings of $14,407 per QALY gained.
The authors concluded that oral nicotinamide was “a cost-effective and patient-centric preventive approach for KC, particularly in individuals with KC history at high risk of multiple primary KC.”
In an accompanying editorial, Ivo Abraham, PhD, JAMA Dermatology’s associate editor for quantitative methods and chief scientist at Matrix45, a health economics research and consulting group in Tucson, Arizona, and co-authors noted that although nicotinamide “is inexpensive, widely available, and mechanistically plausible for chemoprevention of actinic keratoses and KCs…stronger evidence remains required to support clinical recommendations.”
“Broader nicotinamide implementation might impart substantial population health benefits and cost savings to the VHA,” they wrote, while also asking, “do we truly know whether nicotinamide is effective for KC chemoprevention in broader populations?” They suggested that only an adequately powered randomized clinical trial in representative nonimmunosuppressed populations would provide the answer.
“Additional randomized controlled trials in non-VHA populations would provide further insight into generalizability beyond the VA healthcare system,” Hartman told Medscape Medical News.
“We are aiming to conduct a large [randomized controlled trial] in the VA to provide a more definitive answer,” added Lee Wheless, MD, one of Hartman’s coauthors, from Vanderbilt University Medical Center and the Tennessee Valley Healthcare System VA Medical Center, both in Nashville, Tennessee. “Doing so would also give a much better estimate of any potential side effects, though we and others have found no increased rate, and sometimes even a decreased rate, of major adverse cardiovascular events.”
Sarah Arron, MD, dermatologic surgeon with Palo Alto Foundation Medical Group in Palo Alto, California, and Premier Aesthetic Dermatology in San Carlos, California, who was not involved in the research, said, “It is gratifying to see that in the veteran population, nicotinamide affords protection against nonmelanoma skin cancer and is a cost-effective intervention. For a healthcare system such as the VHA, providing this over-the-counter vitamin through pharmacy benefits is an excellent method for reducing the overall cost of skin cancer treatment.”
Although Arron agreed that a randomized trial would offer a higher level of evidence for this intervention, she said the real-world obstacle is that nicotinamide is such an easily available, low-cost vitamin with a high safety profile. “Patients are not likely to sign up for a possible placebo when they can purchase nicotinamide online or at the drugstore,” she said. “This was reflected in Australia; once the positive data from the ONTRAC trial was publicized, investigators on the ONTRANS trial had difficulty enrolling patients because they were already taking the vitamin. The second study closed without meeting its enrollment goals and thus did not have power to show statistical significance.”
Hartman is supported by the US Department of Defense and the US Department of Veterans Affairs. Wheless is also supported by the US Department of Veterans Affairs. Arron is a speaker for Regeneron and Castle Biosciences; a consultant for Regeneron, Replimune, Castle, Lumenis, and Enspectra Health; an unpaid ambassador for HarkenDerm, which makes sunscreen as well as a sun and eye health supplement that includes nicotinamide as one of the ingredients.
The study authors reported having no conflicts of interest. Of the editorial authors, Abraham disclosed owning stock in Matrix45, which has received contract funding from companies outside this work, one author had disclosures not related to the work, and the third author had no disclosures.
Kate Johnson is a Montreal-based freelance medical journalist who has been writing for > 30 years about all areas of medicine.
A version of this article first appeared on Medscape.com.
Oral nicotinamide was cost-effective for reducing keratinocyte carcinoma (KC) risk in US veterans with a history of the disease, according to an economic analysis of Veterans Health Administration (VHA) data.
The findings, published online June 10 in JAMA Dermatology, “support strong consideration of nicotinamide for KC prevention in high-risk populations like veterans, particularly given its safety and tolerability,” wrote senior author Rebecca I. Hartman, MD, chief of the Dermatology Section at VA Boston and assistant professor of dermatology at Brigham and Women’s Hospital and Harvard Medical School, Boston, and co-authors.
Nicotinamide supplementation is “not only a cost-effective and patient-centric strategy for KC prevention, but it also remains economically favorable under a range of assumptions and may become even more cost-effective under higher procedure costs and frequency,” noted the authors.
The analysis included 33,822 individuals from the VHA database, all with a history of one or more KCs, including those with nicotinamide exposure for 30 or more days (n = 12,287) and those without that exposure (n = 21,535).
The mean ages in the unexposed and exposed cohorts were 76.9 and 77.2 years, respectively, and 98% were men. Procedural US VHA costs for KC treatment were estimated from previous research and adjusted for inflation. Nicotinamide pricing was obtained from the VHA.
KC incidence among nicotinamide-exposed and unexposed individuals was 0.204 and 0.255 events per person-year, respectively, reflecting an absolute risk reduction of 0.051 and 624 KCs prevented annually with nicotinamide supplementation.
With an estimated cost of $843 per KC, the yearly KC treatment expense was estimated at approximately $2.64 million, and the annual nicotinamide cost was estimated at $161,451, resulting in net savings of $364,581 — a 19.9% reduction in cohort-specific costs.
Assuming a quality-adjusted life-year (QALY) decrement of -0.01 per KC, nicotinamide use yielded an annual gain of 6.24 QALYs across the cohort and a savings of $58,426 per QALY gained.
A calculation of non-VHA cost-effectiveness, estimated with civilian prices and distributions, showed savings of $14,407 per QALY gained.
The authors concluded that oral nicotinamide was “a cost-effective and patient-centric preventive approach for KC, particularly in individuals with KC history at high risk of multiple primary KC.”
In an accompanying editorial, Ivo Abraham, PhD, JAMA Dermatology’s associate editor for quantitative methods and chief scientist at Matrix45, a health economics research and consulting group in Tucson, Arizona, and co-authors noted that although nicotinamide “is inexpensive, widely available, and mechanistically plausible for chemoprevention of actinic keratoses and KCs…stronger evidence remains required to support clinical recommendations.”
“Broader nicotinamide implementation might impart substantial population health benefits and cost savings to the VHA,” they wrote, while also asking, “do we truly know whether nicotinamide is effective for KC chemoprevention in broader populations?” They suggested that only an adequately powered randomized clinical trial in representative nonimmunosuppressed populations would provide the answer.
“Additional randomized controlled trials in non-VHA populations would provide further insight into generalizability beyond the VA healthcare system,” Hartman told Medscape Medical News.
“We are aiming to conduct a large [randomized controlled trial] in the VA to provide a more definitive answer,” added Lee Wheless, MD, one of Hartman’s coauthors, from Vanderbilt University Medical Center and the Tennessee Valley Healthcare System VA Medical Center, both in Nashville, Tennessee. “Doing so would also give a much better estimate of any potential side effects, though we and others have found no increased rate, and sometimes even a decreased rate, of major adverse cardiovascular events.”
Sarah Arron, MD, dermatologic surgeon with Palo Alto Foundation Medical Group in Palo Alto, California, and Premier Aesthetic Dermatology in San Carlos, California, who was not involved in the research, said, “It is gratifying to see that in the veteran population, nicotinamide affords protection against nonmelanoma skin cancer and is a cost-effective intervention. For a healthcare system such as the VHA, providing this over-the-counter vitamin through pharmacy benefits is an excellent method for reducing the overall cost of skin cancer treatment.”
Although Arron agreed that a randomized trial would offer a higher level of evidence for this intervention, she said the real-world obstacle is that nicotinamide is such an easily available, low-cost vitamin with a high safety profile. “Patients are not likely to sign up for a possible placebo when they can purchase nicotinamide online or at the drugstore,” she said. “This was reflected in Australia; once the positive data from the ONTRAC trial was publicized, investigators on the ONTRANS trial had difficulty enrolling patients because they were already taking the vitamin. The second study closed without meeting its enrollment goals and thus did not have power to show statistical significance.”
Hartman is supported by the US Department of Defense and the US Department of Veterans Affairs. Wheless is also supported by the US Department of Veterans Affairs. Arron is a speaker for Regeneron and Castle Biosciences; a consultant for Regeneron, Replimune, Castle, Lumenis, and Enspectra Health; an unpaid ambassador for HarkenDerm, which makes sunscreen as well as a sun and eye health supplement that includes nicotinamide as one of the ingredients.
The study authors reported having no conflicts of interest. Of the editorial authors, Abraham disclosed owning stock in Matrix45, which has received contract funding from companies outside this work, one author had disclosures not related to the work, and the third author had no disclosures.
Kate Johnson is a Montreal-based freelance medical journalist who has been writing for > 30 years about all areas of medicine.
A version of this article first appeared on Medscape.com.
Oral nicotinamide was cost-effective for reducing keratinocyte carcinoma (KC) risk in US veterans with a history of the disease, according to an economic analysis of Veterans Health Administration (VHA) data.
The findings, published online June 10 in JAMA Dermatology, “support strong consideration of nicotinamide for KC prevention in high-risk populations like veterans, particularly given its safety and tolerability,” wrote senior author Rebecca I. Hartman, MD, chief of the Dermatology Section at VA Boston and assistant professor of dermatology at Brigham and Women’s Hospital and Harvard Medical School, Boston, and co-authors.
Nicotinamide supplementation is “not only a cost-effective and patient-centric strategy for KC prevention, but it also remains economically favorable under a range of assumptions and may become even more cost-effective under higher procedure costs and frequency,” noted the authors.
The analysis included 33,822 individuals from the VHA database, all with a history of one or more KCs, including those with nicotinamide exposure for 30 or more days (n = 12,287) and those without that exposure (n = 21,535).
The mean ages in the unexposed and exposed cohorts were 76.9 and 77.2 years, respectively, and 98% were men. Procedural US VHA costs for KC treatment were estimated from previous research and adjusted for inflation. Nicotinamide pricing was obtained from the VHA.
KC incidence among nicotinamide-exposed and unexposed individuals was 0.204 and 0.255 events per person-year, respectively, reflecting an absolute risk reduction of 0.051 and 624 KCs prevented annually with nicotinamide supplementation.
With an estimated cost of $843 per KC, the yearly KC treatment expense was estimated at approximately $2.64 million, and the annual nicotinamide cost was estimated at $161,451, resulting in net savings of $364,581 — a 19.9% reduction in cohort-specific costs.
Assuming a quality-adjusted life-year (QALY) decrement of -0.01 per KC, nicotinamide use yielded an annual gain of 6.24 QALYs across the cohort and a savings of $58,426 per QALY gained.
A calculation of non-VHA cost-effectiveness, estimated with civilian prices and distributions, showed savings of $14,407 per QALY gained.
The authors concluded that oral nicotinamide was “a cost-effective and patient-centric preventive approach for KC, particularly in individuals with KC history at high risk of multiple primary KC.”
In an accompanying editorial, Ivo Abraham, PhD, JAMA Dermatology’s associate editor for quantitative methods and chief scientist at Matrix45, a health economics research and consulting group in Tucson, Arizona, and co-authors noted that although nicotinamide “is inexpensive, widely available, and mechanistically plausible for chemoprevention of actinic keratoses and KCs…stronger evidence remains required to support clinical recommendations.”
“Broader nicotinamide implementation might impart substantial population health benefits and cost savings to the VHA,” they wrote, while also asking, “do we truly know whether nicotinamide is effective for KC chemoprevention in broader populations?” They suggested that only an adequately powered randomized clinical trial in representative nonimmunosuppressed populations would provide the answer.
“Additional randomized controlled trials in non-VHA populations would provide further insight into generalizability beyond the VA healthcare system,” Hartman told Medscape Medical News.
“We are aiming to conduct a large [randomized controlled trial] in the VA to provide a more definitive answer,” added Lee Wheless, MD, one of Hartman’s coauthors, from Vanderbilt University Medical Center and the Tennessee Valley Healthcare System VA Medical Center, both in Nashville, Tennessee. “Doing so would also give a much better estimate of any potential side effects, though we and others have found no increased rate, and sometimes even a decreased rate, of major adverse cardiovascular events.”
Sarah Arron, MD, dermatologic surgeon with Palo Alto Foundation Medical Group in Palo Alto, California, and Premier Aesthetic Dermatology in San Carlos, California, who was not involved in the research, said, “It is gratifying to see that in the veteran population, nicotinamide affords protection against nonmelanoma skin cancer and is a cost-effective intervention. For a healthcare system such as the VHA, providing this over-the-counter vitamin through pharmacy benefits is an excellent method for reducing the overall cost of skin cancer treatment.”
Although Arron agreed that a randomized trial would offer a higher level of evidence for this intervention, she said the real-world obstacle is that nicotinamide is such an easily available, low-cost vitamin with a high safety profile. “Patients are not likely to sign up for a possible placebo when they can purchase nicotinamide online or at the drugstore,” she said. “This was reflected in Australia; once the positive data from the ONTRAC trial was publicized, investigators on the ONTRANS trial had difficulty enrolling patients because they were already taking the vitamin. The second study closed without meeting its enrollment goals and thus did not have power to show statistical significance.”
Hartman is supported by the US Department of Defense and the US Department of Veterans Affairs. Wheless is also supported by the US Department of Veterans Affairs. Arron is a speaker for Regeneron and Castle Biosciences; a consultant for Regeneron, Replimune, Castle, Lumenis, and Enspectra Health; an unpaid ambassador for HarkenDerm, which makes sunscreen as well as a sun and eye health supplement that includes nicotinamide as one of the ingredients.
The study authors reported having no conflicts of interest. Of the editorial authors, Abraham disclosed owning stock in Matrix45, which has received contract funding from companies outside this work, one author had disclosures not related to the work, and the third author had no disclosures.
Kate Johnson is a Montreal-based freelance medical journalist who has been writing for > 30 years about all areas of medicine.
A version of this article first appeared on Medscape.com.
Oral Nicotinamide: Cost-Effective for Reducing Keratinocyte Carcinoma Risk
Oral Nicotinamide: Cost-Effective for Reducing Keratinocyte Carcinoma Risk
Simpler Screening Criteria Could Catch More Lung Cancers
Simpler Screening Criteria Could Catch More Lung Cancers
Offering lung cancer screening to everyone with a 20-year smoking history could expand access to screening, identify more cancers, and reduce disparities, new research suggests.
In an analysis of nearly 1 million US veterans, researchers estimated that a simplified approach to lung cancer screening — based on smoking duration rather than pack-years — would expand screening eligibility by nearly 30% and reduce potentially missed lung cancers by over 70%.
Those shifts would be especially pronounced among women and Black individuals — 2 groups that are underserved by current screening criteria.
The results, presented at the American Society of Clinical Oncology (ASCO) 2026, come at a time when some groups are revisiting their lung cancer screening guidelines.
And they support smoking duration as a “simpler, more sensitive, and more equitable metric for screening eligibility,” researcher Brendan T. Heiden, MD, MPHS, Washington University School of Medicine in St. Louis, St. Louis, told meeting attendees.
Toward a Better Metric
Current guidelines from the US Preventive Services Task Force (USPSTF) recommend annual lung cancer screening with low-dose CT for adults aged 50-80 years who have at least a 20 pack-year smoking history and either currently smoke or quit within the past 15 years.
The 20 pack-year metric is equivalent to smoking a pack of cigarettes per day for 20 years. Because it requires patients to remember their smoking intensity over decades, it can be challenging to calculate and translate into care, Heiden said.
As it stands, few Americans who are eligible under current USPSTF guidelines actually undergo lung cancer screening, at about 15%-20%, Heiden noted. Meanwhile, mounting evidence suggests that many lung cancers occur in individuals who never meet those eligibility criteria.
Boosting screening uptake, Heiden said, is not enough: There’s a need to revisit eligibility itself to reach more high-risk individuals.
Some groups are already taking steps in that direction. Recently updated guidelines from the National Comprehensive Cancer Network (NCCN) added a category 2B recommendation supporting screening for individuals with at least a 20-year smoking history, regardless of pack-years. (The guidelines also say former smokers are eligible no matter how long ago they quit.)
For their study, Heiden’s team sought to estimate the performance of that smoking-duration metric against current USPSTF pack-year criteria. They used Veterans Health Administration data on over 980,000 veterans whose smoking histories were prospectively collected; lung cancer diagnoses were identified through the Veterans Affairs Central Cancer Registry.
Most of the included veterans (67%) had a smoking history; their mean age was 64 years, and 21% were Black.
Overall, the researchers found that basing eligibility on 20-year smoking duration would substantially expand access to screening: Among veterans with a smoking history, 68% qualified for screening under current USPSTF criteria compared with 87% using the smoking-duration approach.
The gains were especially pronounced among women and Black individuals (who, based on prior research, typically smoke less intensely than White males). Under USPSTF criteria, only about 55% of female and Black veterans qualified for screening compared with 83% for both groups under the smoking-duration criterion.
Importantly, Heiden said, people meeting the smoking-duration threshold remained at substantially elevated risk for lung cancer, suggesting the broader screening criteria were not merely capturing low-risk smokers.
The 5-year lung cancer incidence among veterans eligible under the smoking-duration approach was 1.59% — 11 times the rate of 0.14% among never smokers.
Perhaps most striking, Heiden said, the proportion of potentially missed cancers dropped from 13% with the pack-year metric to just 4% using the smoking-duration metric — a relative reduction of more than 70%.
Again, women and Black individuals would see the largest gains: Among Black veterans, potentially missed cancers fell from 25% to 6%, whereas among female veterans they declined from 22% to 7%.
Optimal Approach Still Unclear
The analysis had limitations, including a predominantly male veteran population whose smoking exposure was far greater than that of the general US population, indicating high inherent lung cancer risk.
But the results support what the NCCN has already done, according to Mary Reid, PhD, MSPH, BSN, a member of the group’s lung cancer screening guideline panel and chief of cancer screening, survivorship and mentorship at Roswell Park Comprehensive Cancer Center in Buffalo, New York.
“Doing the calculation for pack-years can be difficult,” Reid told Medscape Medical News. “Smoking duration is easier to calculate and really the way to go.”
The USPSTF does not comment on individual studies outside of its recommendation development process.
At the meeting, study discussant Katharine A. Rendle, PhD, called the work “impressive,” citing the size of the cohort and strength of the data.
It’s particularly noteworthy that the simpler screening criteria improved sensitivity for all veterans, while largely eliminating disparities, according to Rendle, of the Abramson Cancer Center at the University of Pennsylvania in Philadelphia.
Still, she said, further research could better define the optimal screening strategy.
“Smoking duration is a promising approach, but in my opinion, guidelines likely need to account for the underlying risk in the population,” Rendle said, noting that current smoking prevalence in the US population is about 10%.
She suggested future studies consider other smoking-duration thresholds, such as 30 or 40 years, and look at other outcomes, including life-years gained.
“It’s critical that we prioritize strategies that maximize potential benefit from screening — not just identify those at lung cancer risk — given downstream costs and burden on populations and health care systems,” Rendle said.
The study had no commercial funding. Heiden, Rendle, and Reid had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Offering lung cancer screening to everyone with a 20-year smoking history could expand access to screening, identify more cancers, and reduce disparities, new research suggests.
In an analysis of nearly 1 million US veterans, researchers estimated that a simplified approach to lung cancer screening — based on smoking duration rather than pack-years — would expand screening eligibility by nearly 30% and reduce potentially missed lung cancers by over 70%.
Those shifts would be especially pronounced among women and Black individuals — 2 groups that are underserved by current screening criteria.
The results, presented at the American Society of Clinical Oncology (ASCO) 2026, come at a time when some groups are revisiting their lung cancer screening guidelines.
And they support smoking duration as a “simpler, more sensitive, and more equitable metric for screening eligibility,” researcher Brendan T. Heiden, MD, MPHS, Washington University School of Medicine in St. Louis, St. Louis, told meeting attendees.
Toward a Better Metric
Current guidelines from the US Preventive Services Task Force (USPSTF) recommend annual lung cancer screening with low-dose CT for adults aged 50-80 years who have at least a 20 pack-year smoking history and either currently smoke or quit within the past 15 years.
The 20 pack-year metric is equivalent to smoking a pack of cigarettes per day for 20 years. Because it requires patients to remember their smoking intensity over decades, it can be challenging to calculate and translate into care, Heiden said.
As it stands, few Americans who are eligible under current USPSTF guidelines actually undergo lung cancer screening, at about 15%-20%, Heiden noted. Meanwhile, mounting evidence suggests that many lung cancers occur in individuals who never meet those eligibility criteria.
Boosting screening uptake, Heiden said, is not enough: There’s a need to revisit eligibility itself to reach more high-risk individuals.
Some groups are already taking steps in that direction. Recently updated guidelines from the National Comprehensive Cancer Network (NCCN) added a category 2B recommendation supporting screening for individuals with at least a 20-year smoking history, regardless of pack-years. (The guidelines also say former smokers are eligible no matter how long ago they quit.)
For their study, Heiden’s team sought to estimate the performance of that smoking-duration metric against current USPSTF pack-year criteria. They used Veterans Health Administration data on over 980,000 veterans whose smoking histories were prospectively collected; lung cancer diagnoses were identified through the Veterans Affairs Central Cancer Registry.
Most of the included veterans (67%) had a smoking history; their mean age was 64 years, and 21% were Black.
Overall, the researchers found that basing eligibility on 20-year smoking duration would substantially expand access to screening: Among veterans with a smoking history, 68% qualified for screening under current USPSTF criteria compared with 87% using the smoking-duration approach.
The gains were especially pronounced among women and Black individuals (who, based on prior research, typically smoke less intensely than White males). Under USPSTF criteria, only about 55% of female and Black veterans qualified for screening compared with 83% for both groups under the smoking-duration criterion.
Importantly, Heiden said, people meeting the smoking-duration threshold remained at substantially elevated risk for lung cancer, suggesting the broader screening criteria were not merely capturing low-risk smokers.
The 5-year lung cancer incidence among veterans eligible under the smoking-duration approach was 1.59% — 11 times the rate of 0.14% among never smokers.
Perhaps most striking, Heiden said, the proportion of potentially missed cancers dropped from 13% with the pack-year metric to just 4% using the smoking-duration metric — a relative reduction of more than 70%.
Again, women and Black individuals would see the largest gains: Among Black veterans, potentially missed cancers fell from 25% to 6%, whereas among female veterans they declined from 22% to 7%.
Optimal Approach Still Unclear
The analysis had limitations, including a predominantly male veteran population whose smoking exposure was far greater than that of the general US population, indicating high inherent lung cancer risk.
But the results support what the NCCN has already done, according to Mary Reid, PhD, MSPH, BSN, a member of the group’s lung cancer screening guideline panel and chief of cancer screening, survivorship and mentorship at Roswell Park Comprehensive Cancer Center in Buffalo, New York.
“Doing the calculation for pack-years can be difficult,” Reid told Medscape Medical News. “Smoking duration is easier to calculate and really the way to go.”
The USPSTF does not comment on individual studies outside of its recommendation development process.
At the meeting, study discussant Katharine A. Rendle, PhD, called the work “impressive,” citing the size of the cohort and strength of the data.
It’s particularly noteworthy that the simpler screening criteria improved sensitivity for all veterans, while largely eliminating disparities, according to Rendle, of the Abramson Cancer Center at the University of Pennsylvania in Philadelphia.
Still, she said, further research could better define the optimal screening strategy.
“Smoking duration is a promising approach, but in my opinion, guidelines likely need to account for the underlying risk in the population,” Rendle said, noting that current smoking prevalence in the US population is about 10%.
She suggested future studies consider other smoking-duration thresholds, such as 30 or 40 years, and look at other outcomes, including life-years gained.
“It’s critical that we prioritize strategies that maximize potential benefit from screening — not just identify those at lung cancer risk — given downstream costs and burden on populations and health care systems,” Rendle said.
The study had no commercial funding. Heiden, Rendle, and Reid had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Offering lung cancer screening to everyone with a 20-year smoking history could expand access to screening, identify more cancers, and reduce disparities, new research suggests.
In an analysis of nearly 1 million US veterans, researchers estimated that a simplified approach to lung cancer screening — based on smoking duration rather than pack-years — would expand screening eligibility by nearly 30% and reduce potentially missed lung cancers by over 70%.
Those shifts would be especially pronounced among women and Black individuals — 2 groups that are underserved by current screening criteria.
The results, presented at the American Society of Clinical Oncology (ASCO) 2026, come at a time when some groups are revisiting their lung cancer screening guidelines.
And they support smoking duration as a “simpler, more sensitive, and more equitable metric for screening eligibility,” researcher Brendan T. Heiden, MD, MPHS, Washington University School of Medicine in St. Louis, St. Louis, told meeting attendees.
Toward a Better Metric
Current guidelines from the US Preventive Services Task Force (USPSTF) recommend annual lung cancer screening with low-dose CT for adults aged 50-80 years who have at least a 20 pack-year smoking history and either currently smoke or quit within the past 15 years.
The 20 pack-year metric is equivalent to smoking a pack of cigarettes per day for 20 years. Because it requires patients to remember their smoking intensity over decades, it can be challenging to calculate and translate into care, Heiden said.
As it stands, few Americans who are eligible under current USPSTF guidelines actually undergo lung cancer screening, at about 15%-20%, Heiden noted. Meanwhile, mounting evidence suggests that many lung cancers occur in individuals who never meet those eligibility criteria.
Boosting screening uptake, Heiden said, is not enough: There’s a need to revisit eligibility itself to reach more high-risk individuals.
Some groups are already taking steps in that direction. Recently updated guidelines from the National Comprehensive Cancer Network (NCCN) added a category 2B recommendation supporting screening for individuals with at least a 20-year smoking history, regardless of pack-years. (The guidelines also say former smokers are eligible no matter how long ago they quit.)
For their study, Heiden’s team sought to estimate the performance of that smoking-duration metric against current USPSTF pack-year criteria. They used Veterans Health Administration data on over 980,000 veterans whose smoking histories were prospectively collected; lung cancer diagnoses were identified through the Veterans Affairs Central Cancer Registry.
Most of the included veterans (67%) had a smoking history; their mean age was 64 years, and 21% were Black.
Overall, the researchers found that basing eligibility on 20-year smoking duration would substantially expand access to screening: Among veterans with a smoking history, 68% qualified for screening under current USPSTF criteria compared with 87% using the smoking-duration approach.
The gains were especially pronounced among women and Black individuals (who, based on prior research, typically smoke less intensely than White males). Under USPSTF criteria, only about 55% of female and Black veterans qualified for screening compared with 83% for both groups under the smoking-duration criterion.
Importantly, Heiden said, people meeting the smoking-duration threshold remained at substantially elevated risk for lung cancer, suggesting the broader screening criteria were not merely capturing low-risk smokers.
The 5-year lung cancer incidence among veterans eligible under the smoking-duration approach was 1.59% — 11 times the rate of 0.14% among never smokers.
Perhaps most striking, Heiden said, the proportion of potentially missed cancers dropped from 13% with the pack-year metric to just 4% using the smoking-duration metric — a relative reduction of more than 70%.
Again, women and Black individuals would see the largest gains: Among Black veterans, potentially missed cancers fell from 25% to 6%, whereas among female veterans they declined from 22% to 7%.
Optimal Approach Still Unclear
The analysis had limitations, including a predominantly male veteran population whose smoking exposure was far greater than that of the general US population, indicating high inherent lung cancer risk.
But the results support what the NCCN has already done, according to Mary Reid, PhD, MSPH, BSN, a member of the group’s lung cancer screening guideline panel and chief of cancer screening, survivorship and mentorship at Roswell Park Comprehensive Cancer Center in Buffalo, New York.
“Doing the calculation for pack-years can be difficult,” Reid told Medscape Medical News. “Smoking duration is easier to calculate and really the way to go.”
The USPSTF does not comment on individual studies outside of its recommendation development process.
At the meeting, study discussant Katharine A. Rendle, PhD, called the work “impressive,” citing the size of the cohort and strength of the data.
It’s particularly noteworthy that the simpler screening criteria improved sensitivity for all veterans, while largely eliminating disparities, according to Rendle, of the Abramson Cancer Center at the University of Pennsylvania in Philadelphia.
Still, she said, further research could better define the optimal screening strategy.
“Smoking duration is a promising approach, but in my opinion, guidelines likely need to account for the underlying risk in the population,” Rendle said, noting that current smoking prevalence in the US population is about 10%.
She suggested future studies consider other smoking-duration thresholds, such as 30 or 40 years, and look at other outcomes, including life-years gained.
“It’s critical that we prioritize strategies that maximize potential benefit from screening — not just identify those at lung cancer risk — given downstream costs and burden on populations and health care systems,” Rendle said.
The study had no commercial funding. Heiden, Rendle, and Reid had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Simpler Screening Criteria Could Catch More Lung Cancers
Simpler Screening Criteria Could Catch More Lung Cancers
Alcohol Intake Tied to Increased Colorectal Cancer Risk
Alcohol Intake Tied to Increased Colorectal Cancer Risk
Transcript generated from video captions.
Hello. I’m Dr Maurie Markman, from City of Hope. I’d like to discuss a very interesting paper that appeared in Cancer, entitled, “Association of alcohol intake over the lifetime with colorectal adenoma and colorectal cancer risk in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial.”
This is an important paper. It is very clear, certainly to those in the public health community and cancer doctors, that there is an association with alcohol intake and the risk of cancer. However, in population-based surveys, there is a very large percentage of individuals who do not appear to see the risk of alcohol intake, particularly excessive alcohol intake, related to cancer, or an even larger segment of population simply doesn’t know. This analysis is important to help address this question.
The investigators looked at adults in the United States who were enrolled in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial, which is a very important study that had been ongoing for many years. Individuals in this study reported at several timepoints during their participation what their current and past alcohol intake was.
What these investigators found is as follows. Among the 88,092 participants, there were a total of 1679 incident colorectal cancers that developed over 20 years of follow-up. Investigators demonstrated that current drinkers with an average lifetime alcohol intake of 14 or more drinks per week, or approximately 2 drinks per day, had a higher risk of colorectal cancer with a hazard ratio of 1.25, a 25% increase, compared to those individuals who had ≤ 1 drink per week.
Very importantly, individuals were characterized by their own information that they provided as consistent heavy drinking versus light drinking. This was associated with almost a doubling of the risk of the development of colorectal cancer over that 20-year period.
Clearly, heavy drinking and higher lifetime alcohol intake is associated with an increased risk of colorectal cancer. This is relevant information for public health officials, primary care doctors, and the public in general to understand that there is a risk if you drink often, particularly heavy drinking, with increased development of cancer in general — but in this case, we’re talking specifically about colorectal cancer.
I thank you for your attention.
A version of this article first appeared on Medscape.com.
Transcript generated from video captions.
Hello. I’m Dr Maurie Markman, from City of Hope. I’d like to discuss a very interesting paper that appeared in Cancer, entitled, “Association of alcohol intake over the lifetime with colorectal adenoma and colorectal cancer risk in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial.”
This is an important paper. It is very clear, certainly to those in the public health community and cancer doctors, that there is an association with alcohol intake and the risk of cancer. However, in population-based surveys, there is a very large percentage of individuals who do not appear to see the risk of alcohol intake, particularly excessive alcohol intake, related to cancer, or an even larger segment of population simply doesn’t know. This analysis is important to help address this question.
The investigators looked at adults in the United States who were enrolled in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial, which is a very important study that had been ongoing for many years. Individuals in this study reported at several timepoints during their participation what their current and past alcohol intake was.
What these investigators found is as follows. Among the 88,092 participants, there were a total of 1679 incident colorectal cancers that developed over 20 years of follow-up. Investigators demonstrated that current drinkers with an average lifetime alcohol intake of 14 or more drinks per week, or approximately 2 drinks per day, had a higher risk of colorectal cancer with a hazard ratio of 1.25, a 25% increase, compared to those individuals who had ≤ 1 drink per week.
Very importantly, individuals were characterized by their own information that they provided as consistent heavy drinking versus light drinking. This was associated with almost a doubling of the risk of the development of colorectal cancer over that 20-year period.
Clearly, heavy drinking and higher lifetime alcohol intake is associated with an increased risk of colorectal cancer. This is relevant information for public health officials, primary care doctors, and the public in general to understand that there is a risk if you drink often, particularly heavy drinking, with increased development of cancer in general — but in this case, we’re talking specifically about colorectal cancer.
I thank you for your attention.
A version of this article first appeared on Medscape.com.
Transcript generated from video captions.
Hello. I’m Dr Maurie Markman, from City of Hope. I’d like to discuss a very interesting paper that appeared in Cancer, entitled, “Association of alcohol intake over the lifetime with colorectal adenoma and colorectal cancer risk in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial.”
This is an important paper. It is very clear, certainly to those in the public health community and cancer doctors, that there is an association with alcohol intake and the risk of cancer. However, in population-based surveys, there is a very large percentage of individuals who do not appear to see the risk of alcohol intake, particularly excessive alcohol intake, related to cancer, or an even larger segment of population simply doesn’t know. This analysis is important to help address this question.
The investigators looked at adults in the United States who were enrolled in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial, which is a very important study that had been ongoing for many years. Individuals in this study reported at several timepoints during their participation what their current and past alcohol intake was.
What these investigators found is as follows. Among the 88,092 participants, there were a total of 1679 incident colorectal cancers that developed over 20 years of follow-up. Investigators demonstrated that current drinkers with an average lifetime alcohol intake of 14 or more drinks per week, or approximately 2 drinks per day, had a higher risk of colorectal cancer with a hazard ratio of 1.25, a 25% increase, compared to those individuals who had ≤ 1 drink per week.
Very importantly, individuals were characterized by their own information that they provided as consistent heavy drinking versus light drinking. This was associated with almost a doubling of the risk of the development of colorectal cancer over that 20-year period.
Clearly, heavy drinking and higher lifetime alcohol intake is associated with an increased risk of colorectal cancer. This is relevant information for public health officials, primary care doctors, and the public in general to understand that there is a risk if you drink often, particularly heavy drinking, with increased development of cancer in general — but in this case, we’re talking specifically about colorectal cancer.
I thank you for your attention.
A version of this article first appeared on Medscape.com.
Alcohol Intake Tied to Increased Colorectal Cancer Risk
Alcohol Intake Tied to Increased Colorectal Cancer Risk
GLP-1s Tied to Lower Cancer Risk in Patients With Obesity
GLP-1s Tied to Lower Cancer Risk in Patients With Obesity
Individuals with obesity without diabetes taking GLP-1 receptor agonists (RAs) may have a reduced risk for certain cancers, a new study suggests.
The target trial emulation of more than 160,000 patients found that individuals receiving the medications had a 41% lower risk for obesity-associated cancers (OACs), with an even more substantial 68% risk reduction among men.
“If confirmed in prospective studies, GLP-1 RAs may be associated with a broader clinical profile that extends beyond obesity management to include potential effects on cancer risk,” wrote lead author A.H.-C. Hsu, of Houston Methodist Neal Cancer Center, Houston, and colleagues in Annals of Oncology.
How Are GLP-1 RAs Linked to Reduced Cancer Risk?
According to the investigators, obesity is a recognized risk factor for 13 malignancies: breast, colorectal, endometrial, kidney, pancreatic, thyroid, ovarian, esophageal, gastric, liver, and gallbladder cancers, as well as multiple myeloma and meningioma. These conditions account for about 40% of cancers diagnosed in high-income countries, with incidence rising fastest among younger adults.
Preclinical work suggests GLP-1 receptor activation can suppress proliferation in cancer cells that express the receptor, although these mechanisms remain poorly understood. Observational clinical data have linked GLP-1 RAs to lower cancer risk, mainly in people with type 2 diabetes, but it has remained unclear whether the same association would hold among patients with obesity without diabetes.
How Was the New Study Designed?
The investigators first identified 229,467 adults with obesity without diabetes or a prior OAC in the TriNetX federated database. These patients were entered into a target trial emulation framework, which uses trial-like eligibility, treatment, and follow-up rules to approximate a randomized comparison from observational data.
The population was divided into two cohorts; the first comprised patients who filled at least two prescriptions for a GLP-1 RA and the second included patients who received only diet or exercise counseling. Groups were balanced using 1:1 propensity score matching, yielding 80,899 patients per group. The mean age was 47 years, and about 72% of the population were women.
The primary outcome was the cumulative incidence of OACs over 2 years. Further analyses characterized the same outcome based on sex, BMI, race, and the two most-used drugs, semaglutide and tirzepatide.
What Were the Key Findings?
GLP-1 RA use was associated with a 41% lower incidence of OACs overall (hazard ratio [HR], 0.59). The association held across most subgroups, with a more pronounced benefit among men (HR, 0.32) compared with women (HR, 0.65).
Taking tirzepatide (HR, 0.31) was also associated with a greater risk reduction than taking semaglutide (HR, 0.80); however, the investigators encouraged a cautious interpretation of these findings because the study was not designed for a head-to-head comparison between the agents.
In contrast with the above positive findings, no significant risk reduction was observed among Black patients (HR, 0.77; 95% CI, 0.58-1.03) vs a roughly 50% risk reduction among White patients (HR, 0.54; 95% CI, 0.47-0.62).
The reason for the lack of benefit among Black patients is unclear.
Among specific OACs, greatest risk reductions were reported for multiple myeloma (HR, 0.37), pancreatic cancer (HR, 0.40), endometrial cancer (HR, 0.42), and colorectal cancer (HR, 0.49).
No significant risk reductions were observed for breast cancer, ovarian cancer, or meningioma.
What Are the Clinical Implications?
The investigators emphasized that the study, being observational, is insufficient to confirm that GLP-1 RAs prevent cancer in this patient population. Still, the findings may be worth mentioning to patients considering GLP-1 RAs.
While using GLP-1 RAs specifically to lower cancer risk is “not ready for prime time yet,” the data may offer some reassurance for patients concerned about OACs who already have reason to be prescribed GLP-1 RAs, Michael D. Iglesia, MD, PhD, told Medscape Medical News.
Iglesia, who was not involved in the study, took a more cautious stance on the subgroup findings, first pointing out that women report higher rates of gastrointestinal side effects with GLP-1 RAs, so “the men in this study may have received more consistent GLP-1 RA exposure.”
Commenting on the negative findings among Black patients in the study, Iglesia, who is a medical oncologist at the UNC Lineberger Comprehensive Cancer Center pointed out that this subgroup comprised only 82 GLP-1 RA users and 106 people on diet or exercise.
“Issues related to structural inequality and access to care often affect this kind of study,” he said, adding that further work on social and structural factors is needed “before we posit any biological underpinnings for this observed difference.”
What Questions Remain?
Hsu and colleagues called for long-term prospective trials and postmarketing surveillance to track cancer outcomes among GLP-1 RA users, particularly younger patients.
Underlying mechanisms remain a key unknown, the investigators noted, as it remains unclear whether GLP-1 RAs lower cancer risk through weight loss, direct effects on cancer cells, or some combination thereof.
“Knowing the mechanism behind the association shown in this paper would be important for understanding which cancer types may be treated best with GLP-1 RAs,” Iglesia said.
The investigators and Iglesia reported having no conflicts of interest.
A version of this article first appeared on Medscape.com.
Individuals with obesity without diabetes taking GLP-1 receptor agonists (RAs) may have a reduced risk for certain cancers, a new study suggests.
The target trial emulation of more than 160,000 patients found that individuals receiving the medications had a 41% lower risk for obesity-associated cancers (OACs), with an even more substantial 68% risk reduction among men.
“If confirmed in prospective studies, GLP-1 RAs may be associated with a broader clinical profile that extends beyond obesity management to include potential effects on cancer risk,” wrote lead author A.H.-C. Hsu, of Houston Methodist Neal Cancer Center, Houston, and colleagues in Annals of Oncology.
How Are GLP-1 RAs Linked to Reduced Cancer Risk?
According to the investigators, obesity is a recognized risk factor for 13 malignancies: breast, colorectal, endometrial, kidney, pancreatic, thyroid, ovarian, esophageal, gastric, liver, and gallbladder cancers, as well as multiple myeloma and meningioma. These conditions account for about 40% of cancers diagnosed in high-income countries, with incidence rising fastest among younger adults.
Preclinical work suggests GLP-1 receptor activation can suppress proliferation in cancer cells that express the receptor, although these mechanisms remain poorly understood. Observational clinical data have linked GLP-1 RAs to lower cancer risk, mainly in people with type 2 diabetes, but it has remained unclear whether the same association would hold among patients with obesity without diabetes.
How Was the New Study Designed?
The investigators first identified 229,467 adults with obesity without diabetes or a prior OAC in the TriNetX federated database. These patients were entered into a target trial emulation framework, which uses trial-like eligibility, treatment, and follow-up rules to approximate a randomized comparison from observational data.
The population was divided into two cohorts; the first comprised patients who filled at least two prescriptions for a GLP-1 RA and the second included patients who received only diet or exercise counseling. Groups were balanced using 1:1 propensity score matching, yielding 80,899 patients per group. The mean age was 47 years, and about 72% of the population were women.
The primary outcome was the cumulative incidence of OACs over 2 years. Further analyses characterized the same outcome based on sex, BMI, race, and the two most-used drugs, semaglutide and tirzepatide.
What Were the Key Findings?
GLP-1 RA use was associated with a 41% lower incidence of OACs overall (hazard ratio [HR], 0.59). The association held across most subgroups, with a more pronounced benefit among men (HR, 0.32) compared with women (HR, 0.65).
Taking tirzepatide (HR, 0.31) was also associated with a greater risk reduction than taking semaglutide (HR, 0.80); however, the investigators encouraged a cautious interpretation of these findings because the study was not designed for a head-to-head comparison between the agents.
In contrast with the above positive findings, no significant risk reduction was observed among Black patients (HR, 0.77; 95% CI, 0.58-1.03) vs a roughly 50% risk reduction among White patients (HR, 0.54; 95% CI, 0.47-0.62).
The reason for the lack of benefit among Black patients is unclear.
Among specific OACs, greatest risk reductions were reported for multiple myeloma (HR, 0.37), pancreatic cancer (HR, 0.40), endometrial cancer (HR, 0.42), and colorectal cancer (HR, 0.49).
No significant risk reductions were observed for breast cancer, ovarian cancer, or meningioma.
What Are the Clinical Implications?
The investigators emphasized that the study, being observational, is insufficient to confirm that GLP-1 RAs prevent cancer in this patient population. Still, the findings may be worth mentioning to patients considering GLP-1 RAs.
While using GLP-1 RAs specifically to lower cancer risk is “not ready for prime time yet,” the data may offer some reassurance for patients concerned about OACs who already have reason to be prescribed GLP-1 RAs, Michael D. Iglesia, MD, PhD, told Medscape Medical News.
Iglesia, who was not involved in the study, took a more cautious stance on the subgroup findings, first pointing out that women report higher rates of gastrointestinal side effects with GLP-1 RAs, so “the men in this study may have received more consistent GLP-1 RA exposure.”
Commenting on the negative findings among Black patients in the study, Iglesia, who is a medical oncologist at the UNC Lineberger Comprehensive Cancer Center pointed out that this subgroup comprised only 82 GLP-1 RA users and 106 people on diet or exercise.
“Issues related to structural inequality and access to care often affect this kind of study,” he said, adding that further work on social and structural factors is needed “before we posit any biological underpinnings for this observed difference.”
What Questions Remain?
Hsu and colleagues called for long-term prospective trials and postmarketing surveillance to track cancer outcomes among GLP-1 RA users, particularly younger patients.
Underlying mechanisms remain a key unknown, the investigators noted, as it remains unclear whether GLP-1 RAs lower cancer risk through weight loss, direct effects on cancer cells, or some combination thereof.
“Knowing the mechanism behind the association shown in this paper would be important for understanding which cancer types may be treated best with GLP-1 RAs,” Iglesia said.
The investigators and Iglesia reported having no conflicts of interest.
A version of this article first appeared on Medscape.com.
Individuals with obesity without diabetes taking GLP-1 receptor agonists (RAs) may have a reduced risk for certain cancers, a new study suggests.
The target trial emulation of more than 160,000 patients found that individuals receiving the medications had a 41% lower risk for obesity-associated cancers (OACs), with an even more substantial 68% risk reduction among men.
“If confirmed in prospective studies, GLP-1 RAs may be associated with a broader clinical profile that extends beyond obesity management to include potential effects on cancer risk,” wrote lead author A.H.-C. Hsu, of Houston Methodist Neal Cancer Center, Houston, and colleagues in Annals of Oncology.
How Are GLP-1 RAs Linked to Reduced Cancer Risk?
According to the investigators, obesity is a recognized risk factor for 13 malignancies: breast, colorectal, endometrial, kidney, pancreatic, thyroid, ovarian, esophageal, gastric, liver, and gallbladder cancers, as well as multiple myeloma and meningioma. These conditions account for about 40% of cancers diagnosed in high-income countries, with incidence rising fastest among younger adults.
Preclinical work suggests GLP-1 receptor activation can suppress proliferation in cancer cells that express the receptor, although these mechanisms remain poorly understood. Observational clinical data have linked GLP-1 RAs to lower cancer risk, mainly in people with type 2 diabetes, but it has remained unclear whether the same association would hold among patients with obesity without diabetes.
How Was the New Study Designed?
The investigators first identified 229,467 adults with obesity without diabetes or a prior OAC in the TriNetX federated database. These patients were entered into a target trial emulation framework, which uses trial-like eligibility, treatment, and follow-up rules to approximate a randomized comparison from observational data.
The population was divided into two cohorts; the first comprised patients who filled at least two prescriptions for a GLP-1 RA and the second included patients who received only diet or exercise counseling. Groups were balanced using 1:1 propensity score matching, yielding 80,899 patients per group. The mean age was 47 years, and about 72% of the population were women.
The primary outcome was the cumulative incidence of OACs over 2 years. Further analyses characterized the same outcome based on sex, BMI, race, and the two most-used drugs, semaglutide and tirzepatide.
What Were the Key Findings?
GLP-1 RA use was associated with a 41% lower incidence of OACs overall (hazard ratio [HR], 0.59). The association held across most subgroups, with a more pronounced benefit among men (HR, 0.32) compared with women (HR, 0.65).
Taking tirzepatide (HR, 0.31) was also associated with a greater risk reduction than taking semaglutide (HR, 0.80); however, the investigators encouraged a cautious interpretation of these findings because the study was not designed for a head-to-head comparison between the agents.
In contrast with the above positive findings, no significant risk reduction was observed among Black patients (HR, 0.77; 95% CI, 0.58-1.03) vs a roughly 50% risk reduction among White patients (HR, 0.54; 95% CI, 0.47-0.62).
The reason for the lack of benefit among Black patients is unclear.
Among specific OACs, greatest risk reductions were reported for multiple myeloma (HR, 0.37), pancreatic cancer (HR, 0.40), endometrial cancer (HR, 0.42), and colorectal cancer (HR, 0.49).
No significant risk reductions were observed for breast cancer, ovarian cancer, or meningioma.
What Are the Clinical Implications?
The investigators emphasized that the study, being observational, is insufficient to confirm that GLP-1 RAs prevent cancer in this patient population. Still, the findings may be worth mentioning to patients considering GLP-1 RAs.
While using GLP-1 RAs specifically to lower cancer risk is “not ready for prime time yet,” the data may offer some reassurance for patients concerned about OACs who already have reason to be prescribed GLP-1 RAs, Michael D. Iglesia, MD, PhD, told Medscape Medical News.
Iglesia, who was not involved in the study, took a more cautious stance on the subgroup findings, first pointing out that women report higher rates of gastrointestinal side effects with GLP-1 RAs, so “the men in this study may have received more consistent GLP-1 RA exposure.”
Commenting on the negative findings among Black patients in the study, Iglesia, who is a medical oncologist at the UNC Lineberger Comprehensive Cancer Center pointed out that this subgroup comprised only 82 GLP-1 RA users and 106 people on diet or exercise.
“Issues related to structural inequality and access to care often affect this kind of study,” he said, adding that further work on social and structural factors is needed “before we posit any biological underpinnings for this observed difference.”
What Questions Remain?
Hsu and colleagues called for long-term prospective trials and postmarketing surveillance to track cancer outcomes among GLP-1 RA users, particularly younger patients.
Underlying mechanisms remain a key unknown, the investigators noted, as it remains unclear whether GLP-1 RAs lower cancer risk through weight loss, direct effects on cancer cells, or some combination thereof.
“Knowing the mechanism behind the association shown in this paper would be important for understanding which cancer types may be treated best with GLP-1 RAs,” Iglesia said.
The investigators and Iglesia reported having no conflicts of interest.
A version of this article first appeared on Medscape.com.
GLP-1s Tied to Lower Cancer Risk in Patients With Obesity
GLP-1s Tied to Lower Cancer Risk in Patients With Obesity
RAS Drug Nearly Doubles Survival in Metastatic Pancreatic Cancer
RAS Drug Nearly Doubles Survival in Metastatic Pancreatic Cancer
An investigational oral drug that targets RAS, the dominant oncogenic driver in pancreatic cancer, nearly doubled overall survival compared with common second-line chemotherapy regimens in patients with previously treated metastatic disease.
In the 500-patient phase 3 RASolute 302 trial, patients who received daraxonrasib, a RAS(ON) multi-selective inhibitor, lived a median of 13.2 months compared with 6.7 months for those who received chemotherapy. The drug also doubled progression-free survival, tripled the response rate, and delayed deterioration in both pain and quality of life.
“Results from RASolute 302 support daraxonrasib as a new standard of care for patients with previously treated metastatic pancreatic cancer,” said lead investigator Brian Wolpin, MD, MPH, director of the Hale Family Center for Pancreatic Cancer Research, Dana-Farber Cancer Institute in Boston, who presented the findings at the American Society of Clinical Oncology (ASCO) 2026. The study was also published simultaneously on May 31 in The New England Journal of Medicine.
Pancreatic ductal adenocarcinoma is one of the most lethal cancers, with most patients presenting with advanced disease at diagnosis. For those whose cancer progresses after first-line chemotherapy, no single standard second-line treatment has been established. Available regimens typically produce median progression-free survival of 3-4 months and a median overall survival of 6-7 months.
More than 90% of pancreatic cancers harbor activating RAS mutations — most commonly KRAS G12D and G12V — that drive tumor growth. The first approved KRAS inhibitors — sotorasib and adagrasib for certain lung and colorectal cancers — target KRAS G12C, which is rare in pancreatic cancer. So far, no RAS-targeted therapy has been approved for pancreatic cancer.
Daraxonrasib takes a broader approach. The oral agent can target a range of RAS variants, including mutant and wild-type RAS, by binding the active form of RAS and blocking downstream signaling.
In the phase 3 open-label RASolute 302 trial, researchers randomized 500 patients with previously treated metastatic pancreatic cancer 1:1 to receive daraxonrasib 300 mg orally once daily or investigator’s choice of chemotherapy. The most used regimens in the control arm were gemcitabine plus nab-paclitaxel (56.5%) and liposomal irinotecan plus fluorouracil and leucovorin (32.7%).
Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1. Documented RAS mutational status was required, and nearly 92% of patients had RAS G12 mutations.
The dual primary endpoints were overall survival and progression-free survival in the RAS G12 population. Key secondary endpoints included the same measures in the overall population as well as objective response rate and patient-reported quality of life. The median follow-up was 8.5 months.
In the RAS G12 population, daraxonrasib reduced the risk for death by 60% (hazard ratio [HR], 0.40; P < .001). The median overall survival was 13.2 months with daraxonrasib vs 6.6 months with chemotherapy. Results were nearly identical in the overall population: 13.2 vs 6.7 months (HR, 0.40; P < .001).
Progression-free survival in the RAS G12 population was 7.3 vs 3.5 months (HR, 0.45; P < .001). The objective response rate was 33.2% vs 11.8%.
Daraxonrasib significantly delayed deterioration in pain (median, 9.0 vs 3.7 months; HR, 0.51; P < .001) and global quality of life (5.6 vs 2.4 months; HR, 0.60; P < .001).
Treatment-related adverse events of grade ≥ 3 occurred in 43.6% of patients on daraxonrasib vs 57.5% of patients on chemotherapy. The most common high-grade events with daraxonrasib were rash (13.7%) and stomatitis (12.0%), whereas neutropenia (27.6%) and anemia (16.4%) were most common with chemotherapy.
One patient in the daraxonrasib arm died from treatment-related pneumonitis. Discontinuation due to adverse events occurred in 1.2% of patients on daraxonrasib vs 11.2% of patients on chemotherapy.
The median duration of treatment was 6.2 months with daraxonrasib vs 1.5-3.2 months across chemotherapy regimens, and 42% of patients on daraxonrasib remained on treatment at data cutoff, Wolpin said.
The open-label design is a limitation. About 15% of patients randomized to chemotherapy never received treatment, largely after learning their treatment assignment, although all were included in the intention-to-treat analysis.
Invited discussant Jennifer Knox, MD, a medical oncologist at Princess Margaret Cancer Center and professor of medicine at the University of Toronto in Toronto, Ontario, Canada, called daraxonrasib a game changer, describing the drug as “probably the most exciting strategy in five decades” for pancreatic cancer.
“It is an absolutely beautiful curve,” Knox said of the overall survival data, noting that the Kaplan-Meier curves separated early and continued to widen over time — a pattern rarely seen in pancreatic cancer trials where initial separation between treatment arms typically narrows as patients in both groups deteriorate.
Knox highlighted the pain and quality-of-life data as “the most important endpoints for our patients,” given the severity of the disease.
She pointed out, however, that the combination of rash and stomatitis will be “challenging” in practice, and called for improved supportive care and closer collaboration with dermatology as oncologists gain experience with this first-in-class drug.
Knox said RAS-targeted therapy “should dominate trials across the full spectrum of pancreatic cancer clinical presentations,” pointing to first-line combination trials already underway and noting that treating a larger, earlier population would have the greatest impact.
“This is the first glimpse at the real power of targeting RAS in pancreas cancer,” Knox said.
The study was funded by Revolution Medicines. Wolpin reported consulting or advisory roles with Revolution Medicines, Mirati Therapeutics, Ipsen, and others, and institutional research funding from Revolution Medicines, Agios, Amgen, AstraZeneca, Lilly, and Novartis. Knox reported receiving honoraria from Astellas Pharma, AstraZeneca, Incyte, and Ipsen, and consulting or advisory roles with AstraZeneca/MedImmune, Incyte, and Ipsen.
A version of this article was previously published on Medscape.com.
An investigational oral drug that targets RAS, the dominant oncogenic driver in pancreatic cancer, nearly doubled overall survival compared with common second-line chemotherapy regimens in patients with previously treated metastatic disease.
In the 500-patient phase 3 RASolute 302 trial, patients who received daraxonrasib, a RAS(ON) multi-selective inhibitor, lived a median of 13.2 months compared with 6.7 months for those who received chemotherapy. The drug also doubled progression-free survival, tripled the response rate, and delayed deterioration in both pain and quality of life.
“Results from RASolute 302 support daraxonrasib as a new standard of care for patients with previously treated metastatic pancreatic cancer,” said lead investigator Brian Wolpin, MD, MPH, director of the Hale Family Center for Pancreatic Cancer Research, Dana-Farber Cancer Institute in Boston, who presented the findings at the American Society of Clinical Oncology (ASCO) 2026. The study was also published simultaneously on May 31 in The New England Journal of Medicine.
Pancreatic ductal adenocarcinoma is one of the most lethal cancers, with most patients presenting with advanced disease at diagnosis. For those whose cancer progresses after first-line chemotherapy, no single standard second-line treatment has been established. Available regimens typically produce median progression-free survival of 3-4 months and a median overall survival of 6-7 months.
More than 90% of pancreatic cancers harbor activating RAS mutations — most commonly KRAS G12D and G12V — that drive tumor growth. The first approved KRAS inhibitors — sotorasib and adagrasib for certain lung and colorectal cancers — target KRAS G12C, which is rare in pancreatic cancer. So far, no RAS-targeted therapy has been approved for pancreatic cancer.
Daraxonrasib takes a broader approach. The oral agent can target a range of RAS variants, including mutant and wild-type RAS, by binding the active form of RAS and blocking downstream signaling.
In the phase 3 open-label RASolute 302 trial, researchers randomized 500 patients with previously treated metastatic pancreatic cancer 1:1 to receive daraxonrasib 300 mg orally once daily or investigator’s choice of chemotherapy. The most used regimens in the control arm were gemcitabine plus nab-paclitaxel (56.5%) and liposomal irinotecan plus fluorouracil and leucovorin (32.7%).
Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1. Documented RAS mutational status was required, and nearly 92% of patients had RAS G12 mutations.
The dual primary endpoints were overall survival and progression-free survival in the RAS G12 population. Key secondary endpoints included the same measures in the overall population as well as objective response rate and patient-reported quality of life. The median follow-up was 8.5 months.
In the RAS G12 population, daraxonrasib reduced the risk for death by 60% (hazard ratio [HR], 0.40; P < .001). The median overall survival was 13.2 months with daraxonrasib vs 6.6 months with chemotherapy. Results were nearly identical in the overall population: 13.2 vs 6.7 months (HR, 0.40; P < .001).
Progression-free survival in the RAS G12 population was 7.3 vs 3.5 months (HR, 0.45; P < .001). The objective response rate was 33.2% vs 11.8%.
Daraxonrasib significantly delayed deterioration in pain (median, 9.0 vs 3.7 months; HR, 0.51; P < .001) and global quality of life (5.6 vs 2.4 months; HR, 0.60; P < .001).
Treatment-related adverse events of grade ≥ 3 occurred in 43.6% of patients on daraxonrasib vs 57.5% of patients on chemotherapy. The most common high-grade events with daraxonrasib were rash (13.7%) and stomatitis (12.0%), whereas neutropenia (27.6%) and anemia (16.4%) were most common with chemotherapy.
One patient in the daraxonrasib arm died from treatment-related pneumonitis. Discontinuation due to adverse events occurred in 1.2% of patients on daraxonrasib vs 11.2% of patients on chemotherapy.
The median duration of treatment was 6.2 months with daraxonrasib vs 1.5-3.2 months across chemotherapy regimens, and 42% of patients on daraxonrasib remained on treatment at data cutoff, Wolpin said.
The open-label design is a limitation. About 15% of patients randomized to chemotherapy never received treatment, largely after learning their treatment assignment, although all were included in the intention-to-treat analysis.
Invited discussant Jennifer Knox, MD, a medical oncologist at Princess Margaret Cancer Center and professor of medicine at the University of Toronto in Toronto, Ontario, Canada, called daraxonrasib a game changer, describing the drug as “probably the most exciting strategy in five decades” for pancreatic cancer.
“It is an absolutely beautiful curve,” Knox said of the overall survival data, noting that the Kaplan-Meier curves separated early and continued to widen over time — a pattern rarely seen in pancreatic cancer trials where initial separation between treatment arms typically narrows as patients in both groups deteriorate.
Knox highlighted the pain and quality-of-life data as “the most important endpoints for our patients,” given the severity of the disease.
She pointed out, however, that the combination of rash and stomatitis will be “challenging” in practice, and called for improved supportive care and closer collaboration with dermatology as oncologists gain experience with this first-in-class drug.
Knox said RAS-targeted therapy “should dominate trials across the full spectrum of pancreatic cancer clinical presentations,” pointing to first-line combination trials already underway and noting that treating a larger, earlier population would have the greatest impact.
“This is the first glimpse at the real power of targeting RAS in pancreas cancer,” Knox said.
The study was funded by Revolution Medicines. Wolpin reported consulting or advisory roles with Revolution Medicines, Mirati Therapeutics, Ipsen, and others, and institutional research funding from Revolution Medicines, Agios, Amgen, AstraZeneca, Lilly, and Novartis. Knox reported receiving honoraria from Astellas Pharma, AstraZeneca, Incyte, and Ipsen, and consulting or advisory roles with AstraZeneca/MedImmune, Incyte, and Ipsen.
A version of this article was previously published on Medscape.com.
An investigational oral drug that targets RAS, the dominant oncogenic driver in pancreatic cancer, nearly doubled overall survival compared with common second-line chemotherapy regimens in patients with previously treated metastatic disease.
In the 500-patient phase 3 RASolute 302 trial, patients who received daraxonrasib, a RAS(ON) multi-selective inhibitor, lived a median of 13.2 months compared with 6.7 months for those who received chemotherapy. The drug also doubled progression-free survival, tripled the response rate, and delayed deterioration in both pain and quality of life.
“Results from RASolute 302 support daraxonrasib as a new standard of care for patients with previously treated metastatic pancreatic cancer,” said lead investigator Brian Wolpin, MD, MPH, director of the Hale Family Center for Pancreatic Cancer Research, Dana-Farber Cancer Institute in Boston, who presented the findings at the American Society of Clinical Oncology (ASCO) 2026. The study was also published simultaneously on May 31 in The New England Journal of Medicine.
Pancreatic ductal adenocarcinoma is one of the most lethal cancers, with most patients presenting with advanced disease at diagnosis. For those whose cancer progresses after first-line chemotherapy, no single standard second-line treatment has been established. Available regimens typically produce median progression-free survival of 3-4 months and a median overall survival of 6-7 months.
More than 90% of pancreatic cancers harbor activating RAS mutations — most commonly KRAS G12D and G12V — that drive tumor growth. The first approved KRAS inhibitors — sotorasib and adagrasib for certain lung and colorectal cancers — target KRAS G12C, which is rare in pancreatic cancer. So far, no RAS-targeted therapy has been approved for pancreatic cancer.
Daraxonrasib takes a broader approach. The oral agent can target a range of RAS variants, including mutant and wild-type RAS, by binding the active form of RAS and blocking downstream signaling.
In the phase 3 open-label RASolute 302 trial, researchers randomized 500 patients with previously treated metastatic pancreatic cancer 1:1 to receive daraxonrasib 300 mg orally once daily or investigator’s choice of chemotherapy. The most used regimens in the control arm were gemcitabine plus nab-paclitaxel (56.5%) and liposomal irinotecan plus fluorouracil and leucovorin (32.7%).
Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1. Documented RAS mutational status was required, and nearly 92% of patients had RAS G12 mutations.
The dual primary endpoints were overall survival and progression-free survival in the RAS G12 population. Key secondary endpoints included the same measures in the overall population as well as objective response rate and patient-reported quality of life. The median follow-up was 8.5 months.
In the RAS G12 population, daraxonrasib reduced the risk for death by 60% (hazard ratio [HR], 0.40; P < .001). The median overall survival was 13.2 months with daraxonrasib vs 6.6 months with chemotherapy. Results were nearly identical in the overall population: 13.2 vs 6.7 months (HR, 0.40; P < .001).
Progression-free survival in the RAS G12 population was 7.3 vs 3.5 months (HR, 0.45; P < .001). The objective response rate was 33.2% vs 11.8%.
Daraxonrasib significantly delayed deterioration in pain (median, 9.0 vs 3.7 months; HR, 0.51; P < .001) and global quality of life (5.6 vs 2.4 months; HR, 0.60; P < .001).
Treatment-related adverse events of grade ≥ 3 occurred in 43.6% of patients on daraxonrasib vs 57.5% of patients on chemotherapy. The most common high-grade events with daraxonrasib were rash (13.7%) and stomatitis (12.0%), whereas neutropenia (27.6%) and anemia (16.4%) were most common with chemotherapy.
One patient in the daraxonrasib arm died from treatment-related pneumonitis. Discontinuation due to adverse events occurred in 1.2% of patients on daraxonrasib vs 11.2% of patients on chemotherapy.
The median duration of treatment was 6.2 months with daraxonrasib vs 1.5-3.2 months across chemotherapy regimens, and 42% of patients on daraxonrasib remained on treatment at data cutoff, Wolpin said.
The open-label design is a limitation. About 15% of patients randomized to chemotherapy never received treatment, largely after learning their treatment assignment, although all were included in the intention-to-treat analysis.
Invited discussant Jennifer Knox, MD, a medical oncologist at Princess Margaret Cancer Center and professor of medicine at the University of Toronto in Toronto, Ontario, Canada, called daraxonrasib a game changer, describing the drug as “probably the most exciting strategy in five decades” for pancreatic cancer.
“It is an absolutely beautiful curve,” Knox said of the overall survival data, noting that the Kaplan-Meier curves separated early and continued to widen over time — a pattern rarely seen in pancreatic cancer trials where initial separation between treatment arms typically narrows as patients in both groups deteriorate.
Knox highlighted the pain and quality-of-life data as “the most important endpoints for our patients,” given the severity of the disease.
She pointed out, however, that the combination of rash and stomatitis will be “challenging” in practice, and called for improved supportive care and closer collaboration with dermatology as oncologists gain experience with this first-in-class drug.
Knox said RAS-targeted therapy “should dominate trials across the full spectrum of pancreatic cancer clinical presentations,” pointing to first-line combination trials already underway and noting that treating a larger, earlier population would have the greatest impact.
“This is the first glimpse at the real power of targeting RAS in pancreas cancer,” Knox said.
The study was funded by Revolution Medicines. Wolpin reported consulting or advisory roles with Revolution Medicines, Mirati Therapeutics, Ipsen, and others, and institutional research funding from Revolution Medicines, Agios, Amgen, AstraZeneca, Lilly, and Novartis. Knox reported receiving honoraria from Astellas Pharma, AstraZeneca, Incyte, and Ipsen, and consulting or advisory roles with AstraZeneca/MedImmune, Incyte, and Ipsen.
A version of this article was previously published on Medscape.com.
RAS Drug Nearly Doubles Survival in Metastatic Pancreatic Cancer
RAS Drug Nearly Doubles Survival in Metastatic Pancreatic Cancer
Rare Blood Diseases the Focus of AVAHO Virtual Session
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.