User login
Aspirin for CRC Prevention May Work Best in Adults With Unhealthy Lifestyles
TOPLINE:
METHODOLOGY:
- Aspirin is an established agent for CRC prevention. Whether individuals with more lifestyle risk factors might derive greater benefit from aspirin remains unclear.
- Researchers analyzed regular aspirin use (defined as taking two or more standard 325-mg tablets per week) using long-term follow-up data from 63,957 women in the Nurses’ Health Study and 43,698 men in the Health Professionals Follow-Up Study.
- They calculated a healthy lifestyle score for each participant based on body mass index (BMI), alcohol intake, physical activity, diet, and smoking, with higher scores corresponding to healthier lifestyles.
- Outcomes included multivariable-adjusted 10-year cumulative incidence of CRC, the absolute risk reduction (ARR) with aspirin use, and number needed to treat associated with regular aspirin use by lifestyle score.
TAKEAWAY:
- During more than 3 million person-years of follow-up, 2544 new cases of CRC were documented.
- The 10-year cumulative incidence of CRC was 1.98% among regular aspirin users compared with 2.95% among nonusers, corresponding to an ARR of 0.97%.
- The ARR associated with aspirin use was greatest among individuals with the unhealthiest lifestyle scores and progressively decreased with healthier lifestyle scores (P < .001 for additive interaction).
- Those with the unhealthiest lifestyle scores (0-1) had a 10-year ARR of 1.28% from aspirin use, whereas those with the healthiest lifestyle scores (4-5) had an ARR of 0.11%.
- The number needed to treat with aspirin for 10 years to prevent one CRC case was 78 for those with the unhealthiest lifestyles, compared with 909 for those with the healthiest lifestyles.
- Among the individual components of the healthy lifestyle score, higher BMI and smoking correlated with greater reductions in CRC risk from aspirin use.
IN PRACTICE:
“These results support the use of lifestyle risk factors to identify individuals who may have a more favorable risk-benefit profile for cancer prevention with aspirin,” the authors wrote.
SOURCE:
The study, with first author Daniel R. Sikavi, MD, from Massachusetts General Hospital and Harvard Medical School in Boston, was published online in JAMA Oncology.
LIMITATIONS:
The study population consisted of health professionals who were predominantly White, which may limit generalizability of the findings. Lifestyle factors and aspirin use were self-reported, which may introduce measurement errors. The study did not systematically assess adverse outcomes potentially due to aspirin use or the presence of a known hereditary cancer syndrome.
DISCLOSURES:
The study had no commercial funding. The authors had no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Aspirin is an established agent for CRC prevention. Whether individuals with more lifestyle risk factors might derive greater benefit from aspirin remains unclear.
- Researchers analyzed regular aspirin use (defined as taking two or more standard 325-mg tablets per week) using long-term follow-up data from 63,957 women in the Nurses’ Health Study and 43,698 men in the Health Professionals Follow-Up Study.
- They calculated a healthy lifestyle score for each participant based on body mass index (BMI), alcohol intake, physical activity, diet, and smoking, with higher scores corresponding to healthier lifestyles.
- Outcomes included multivariable-adjusted 10-year cumulative incidence of CRC, the absolute risk reduction (ARR) with aspirin use, and number needed to treat associated with regular aspirin use by lifestyle score.
TAKEAWAY:
- During more than 3 million person-years of follow-up, 2544 new cases of CRC were documented.
- The 10-year cumulative incidence of CRC was 1.98% among regular aspirin users compared with 2.95% among nonusers, corresponding to an ARR of 0.97%.
- The ARR associated with aspirin use was greatest among individuals with the unhealthiest lifestyle scores and progressively decreased with healthier lifestyle scores (P < .001 for additive interaction).
- Those with the unhealthiest lifestyle scores (0-1) had a 10-year ARR of 1.28% from aspirin use, whereas those with the healthiest lifestyle scores (4-5) had an ARR of 0.11%.
- The number needed to treat with aspirin for 10 years to prevent one CRC case was 78 for those with the unhealthiest lifestyles, compared with 909 for those with the healthiest lifestyles.
- Among the individual components of the healthy lifestyle score, higher BMI and smoking correlated with greater reductions in CRC risk from aspirin use.
IN PRACTICE:
“These results support the use of lifestyle risk factors to identify individuals who may have a more favorable risk-benefit profile for cancer prevention with aspirin,” the authors wrote.
SOURCE:
The study, with first author Daniel R. Sikavi, MD, from Massachusetts General Hospital and Harvard Medical School in Boston, was published online in JAMA Oncology.
LIMITATIONS:
The study population consisted of health professionals who were predominantly White, which may limit generalizability of the findings. Lifestyle factors and aspirin use were self-reported, which may introduce measurement errors. The study did not systematically assess adverse outcomes potentially due to aspirin use or the presence of a known hereditary cancer syndrome.
DISCLOSURES:
The study had no commercial funding. The authors had no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Aspirin is an established agent for CRC prevention. Whether individuals with more lifestyle risk factors might derive greater benefit from aspirin remains unclear.
- Researchers analyzed regular aspirin use (defined as taking two or more standard 325-mg tablets per week) using long-term follow-up data from 63,957 women in the Nurses’ Health Study and 43,698 men in the Health Professionals Follow-Up Study.
- They calculated a healthy lifestyle score for each participant based on body mass index (BMI), alcohol intake, physical activity, diet, and smoking, with higher scores corresponding to healthier lifestyles.
- Outcomes included multivariable-adjusted 10-year cumulative incidence of CRC, the absolute risk reduction (ARR) with aspirin use, and number needed to treat associated with regular aspirin use by lifestyle score.
TAKEAWAY:
- During more than 3 million person-years of follow-up, 2544 new cases of CRC were documented.
- The 10-year cumulative incidence of CRC was 1.98% among regular aspirin users compared with 2.95% among nonusers, corresponding to an ARR of 0.97%.
- The ARR associated with aspirin use was greatest among individuals with the unhealthiest lifestyle scores and progressively decreased with healthier lifestyle scores (P < .001 for additive interaction).
- Those with the unhealthiest lifestyle scores (0-1) had a 10-year ARR of 1.28% from aspirin use, whereas those with the healthiest lifestyle scores (4-5) had an ARR of 0.11%.
- The number needed to treat with aspirin for 10 years to prevent one CRC case was 78 for those with the unhealthiest lifestyles, compared with 909 for those with the healthiest lifestyles.
- Among the individual components of the healthy lifestyle score, higher BMI and smoking correlated with greater reductions in CRC risk from aspirin use.
IN PRACTICE:
“These results support the use of lifestyle risk factors to identify individuals who may have a more favorable risk-benefit profile for cancer prevention with aspirin,” the authors wrote.
SOURCE:
The study, with first author Daniel R. Sikavi, MD, from Massachusetts General Hospital and Harvard Medical School in Boston, was published online in JAMA Oncology.
LIMITATIONS:
The study population consisted of health professionals who were predominantly White, which may limit generalizability of the findings. Lifestyle factors and aspirin use were self-reported, which may introduce measurement errors. The study did not systematically assess adverse outcomes potentially due to aspirin use or the presence of a known hereditary cancer syndrome.
DISCLOSURES:
The study had no commercial funding. The authors had no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
The Prohibitive Price Tag
Earlier in 2024 the American Headache Society issued a position statement that CGRP (calcitonin gene-related peptide) agents are a first-line option for migraine prevention.
No Shinola, Sherlock.
Any of us working frontline neurology have figured that out, including me. And I was, honestly, pretty skeptical of them when they hit the pharmacy shelves. But these days, to quote The Monkees (and Neil Diamond), “I’m a Believer.”

Unfortunately, things don’t quite work out that way. Just because a drug is clearly successful doesn’t make it practical to use first line. Most insurances won’t even let family doctors prescribe them, so they have to send patients to a neurologist (which I’m not complaining about).
Then me and my neuro-brethren have to jump through hoops because of their cost. One month of any of these drugs costs the same as a few years (or more) of generic Topamax, Nortriptyline, Nadolol, etc. Granted, I shouldn’t complain about that, either. If everyone with migraines was getting them it would drive up insurance premiums across the board — including mine.
So, after patients have tried and failed at least two to four other options (depending on their plan) I can usually get a CGRP covered. This involves filling out some forms online and submitting them ... then waiting.
Even if the drug is approved, and successful, that’s still not the end of the story. Depending on the plan I have to get them reauthorized anywhere from every 3 to 12 months. There’s also the chance that in December I’ll get a letter saying the drug won’t be covered starting January, and to try one of the recommended alternatives, like generic Topamax, Nortriptyline, Nadolol, etc. Wash, rinse, repeat.
Having celebrities like Lady Gaga pushing them doesn’t help. The commercials never mention that getting the medication isn’t as easy as “ask your doctor.” Nor does it point out that Lady Gaga won’t have an issue with a CGRP agent’s price tag of $800-$1000 per month, while most of her fans need that money for rent and groceries.
The guidelines, in essence, are useful, but only apply to a perfect world where drug cost doesn’t matter. We aren’t in one. I’m not knocking the pharmaceutical companies — research and development take A LOT of money, and every drug that comes to market has to pay not only for itself, but for several others that failed. Innovation isn’t cheap.
That doesn’t make it any easier to explain to patients, who see ads, or news blurbs on Facebook, or whatever. I just wish the advertisements would have more transparency about how the pricing works.
After all, regardless of how good an automobile may be, don’t car ads show an MSRP?
Dr. Block has a solo neurology practice in Scottsdale, Arizona.
Earlier in 2024 the American Headache Society issued a position statement that CGRP (calcitonin gene-related peptide) agents are a first-line option for migraine prevention.
No Shinola, Sherlock.
Any of us working frontline neurology have figured that out, including me. And I was, honestly, pretty skeptical of them when they hit the pharmacy shelves. But these days, to quote The Monkees (and Neil Diamond), “I’m a Believer.”
Unfortunately, things don’t quite work out that way. Just because a drug is clearly successful doesn’t make it practical to use first line. Most insurances won’t even let family doctors prescribe them, so they have to send patients to a neurologist (which I’m not complaining about).
Then me and my neuro-brethren have to jump through hoops because of their cost. One month of any of these drugs costs the same as a few years (or more) of generic Topamax, Nortriptyline, Nadolol, etc. Granted, I shouldn’t complain about that, either. If everyone with migraines was getting them it would drive up insurance premiums across the board — including mine.
So, after patients have tried and failed at least two to four other options (depending on their plan) I can usually get a CGRP covered. This involves filling out some forms online and submitting them ... then waiting.
Even if the drug is approved, and successful, that’s still not the end of the story. Depending on the plan I have to get them reauthorized anywhere from every 3 to 12 months. There’s also the chance that in December I’ll get a letter saying the drug won’t be covered starting January, and to try one of the recommended alternatives, like generic Topamax, Nortriptyline, Nadolol, etc. Wash, rinse, repeat.
Having celebrities like Lady Gaga pushing them doesn’t help. The commercials never mention that getting the medication isn’t as easy as “ask your doctor.” Nor does it point out that Lady Gaga won’t have an issue with a CGRP agent’s price tag of $800-$1000 per month, while most of her fans need that money for rent and groceries.
The guidelines, in essence, are useful, but only apply to a perfect world where drug cost doesn’t matter. We aren’t in one. I’m not knocking the pharmaceutical companies — research and development take A LOT of money, and every drug that comes to market has to pay not only for itself, but for several others that failed. Innovation isn’t cheap.
That doesn’t make it any easier to explain to patients, who see ads, or news blurbs on Facebook, or whatever. I just wish the advertisements would have more transparency about how the pricing works.
After all, regardless of how good an automobile may be, don’t car ads show an MSRP?
Dr. Block has a solo neurology practice in Scottsdale, Arizona.
Earlier in 2024 the American Headache Society issued a position statement that CGRP (calcitonin gene-related peptide) agents are a first-line option for migraine prevention.
No Shinola, Sherlock.
Any of us working frontline neurology have figured that out, including me. And I was, honestly, pretty skeptical of them when they hit the pharmacy shelves. But these days, to quote The Monkees (and Neil Diamond), “I’m a Believer.”
Unfortunately, things don’t quite work out that way. Just because a drug is clearly successful doesn’t make it practical to use first line. Most insurances won’t even let family doctors prescribe them, so they have to send patients to a neurologist (which I’m not complaining about).
Then me and my neuro-brethren have to jump through hoops because of their cost. One month of any of these drugs costs the same as a few years (or more) of generic Topamax, Nortriptyline, Nadolol, etc. Granted, I shouldn’t complain about that, either. If everyone with migraines was getting them it would drive up insurance premiums across the board — including mine.
So, after patients have tried and failed at least two to four other options (depending on their plan) I can usually get a CGRP covered. This involves filling out some forms online and submitting them ... then waiting.
Even if the drug is approved, and successful, that’s still not the end of the story. Depending on the plan I have to get them reauthorized anywhere from every 3 to 12 months. There’s also the chance that in December I’ll get a letter saying the drug won’t be covered starting January, and to try one of the recommended alternatives, like generic Topamax, Nortriptyline, Nadolol, etc. Wash, rinse, repeat.
Having celebrities like Lady Gaga pushing them doesn’t help. The commercials never mention that getting the medication isn’t as easy as “ask your doctor.” Nor does it point out that Lady Gaga won’t have an issue with a CGRP agent’s price tag of $800-$1000 per month, while most of her fans need that money for rent and groceries.
The guidelines, in essence, are useful, but only apply to a perfect world where drug cost doesn’t matter. We aren’t in one. I’m not knocking the pharmaceutical companies — research and development take A LOT of money, and every drug that comes to market has to pay not only for itself, but for several others that failed. Innovation isn’t cheap.
That doesn’t make it any easier to explain to patients, who see ads, or news blurbs on Facebook, or whatever. I just wish the advertisements would have more transparency about how the pricing works.
After all, regardless of how good an automobile may be, don’t car ads show an MSRP?
Dr. Block has a solo neurology practice in Scottsdale, Arizona.
Closing the Gap: Priority Zones Identified for CRC Screening in Hispanic/Latino Populations
TOPLINE:
Researchers identified thousands of census tracts as priority zones where improving the screening of colorectal cancer (CRC) may benefit Hispanic or Latino communities.
METHODOLOGY:
- Hispanic or Latino individuals have the lowest rate of CRC screening among the six broader census-designated racial or ethnic groups in the United States, while they face a high proportion of cancer deaths due to CRC.
- Researchers performed a cross-sectional ecologic study using 2021 Centers for Disease Control and Prevention PLACES and 2019 American Community Survey data to identify priority zones for CRC screening where intervention programs may be targeted.
- They analyzed a total of 72,136 US census tracts, representing 98.7% of all US census tracts.
- Nine race and ethnic groups were selected on the basis of the population size and categorizations used in prior research on health or cancer disparity: non-Hispanic Black, non-Hispanic White, Asian, Mexican, Puerto Rican, Cuban, Dominican, Central or South American, and “other race.”
- Geographically weighted regression and Getis-Ord Gi* hot spot procedures were used to identify the screening priority zones for all Hispanic or Latino groups.
TAKEAWAY:
- The analysis identified 6519 hot spot tracts for Mexican, 3477 for Puerto Rican, 3522 for Central or South American, 1069 for Dominican, and 1424 for Cuban individuals. The average rates of screening for CRC were 57.2%, 59.9%, 59.3%, 58.9%, and 60.4%, respectively.
- The percentage of Cuban individuals showed a positive association with the CRC screening rate, while the percentage of Mexican, Puerto Rican, Dominican, and Central or South American Hispanic or Latino individuals and of the uninsured showed a negative association with the CRC screening rate.
- The priority zones for Mexican communities were primarily located in Texas and southwestern United States, while those for Puerto Rican, Central or South American, and other populations were located in southern Florida and the metro areas of New York City and Texas.
IN PRACTICE:
“Our findings and interactive web map may serve as a translational tool for public health authorities, policymakers, clinicians, and other stakeholders to target investment and interventions to increase guideline-concordant CRC screening uptake benefiting specific H/L [Hispanic or Latino] communities in the United States,” the authors wrote. “These data can inform more precise neighborhood-level interventions to increase CRC screening considering unique characteristics important for these H/L [Hispanic or Latino] groups.”
SOURCE:
The study, led by R. Blake Buchalter, PhD, MPH, Center for Populations Health Research, Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, was published online in the American Journal of Public Health.
LIMITATIONS:
The study’s cross-sectional design limited the ability to infer causality. The use of census tract-level data did not capture individual-level screening behaviors. The study did not account for nativity status or years of migration owing to the lack of data. The Centers for Disease Control and Prevention PLACES dataset may not represent the actual screening delivered as it is based on survey data.
DISCLOSURES:
The National Cancer Institute partially supported this study. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Researchers identified thousands of census tracts as priority zones where improving the screening of colorectal cancer (CRC) may benefit Hispanic or Latino communities.
METHODOLOGY:
- Hispanic or Latino individuals have the lowest rate of CRC screening among the six broader census-designated racial or ethnic groups in the United States, while they face a high proportion of cancer deaths due to CRC.
- Researchers performed a cross-sectional ecologic study using 2021 Centers for Disease Control and Prevention PLACES and 2019 American Community Survey data to identify priority zones for CRC screening where intervention programs may be targeted.
- They analyzed a total of 72,136 US census tracts, representing 98.7% of all US census tracts.
- Nine race and ethnic groups were selected on the basis of the population size and categorizations used in prior research on health or cancer disparity: non-Hispanic Black, non-Hispanic White, Asian, Mexican, Puerto Rican, Cuban, Dominican, Central or South American, and “other race.”
- Geographically weighted regression and Getis-Ord Gi* hot spot procedures were used to identify the screening priority zones for all Hispanic or Latino groups.
TAKEAWAY:
- The analysis identified 6519 hot spot tracts for Mexican, 3477 for Puerto Rican, 3522 for Central or South American, 1069 for Dominican, and 1424 for Cuban individuals. The average rates of screening for CRC were 57.2%, 59.9%, 59.3%, 58.9%, and 60.4%, respectively.
- The percentage of Cuban individuals showed a positive association with the CRC screening rate, while the percentage of Mexican, Puerto Rican, Dominican, and Central or South American Hispanic or Latino individuals and of the uninsured showed a negative association with the CRC screening rate.
- The priority zones for Mexican communities were primarily located in Texas and southwestern United States, while those for Puerto Rican, Central or South American, and other populations were located in southern Florida and the metro areas of New York City and Texas.
IN PRACTICE:
“Our findings and interactive web map may serve as a translational tool for public health authorities, policymakers, clinicians, and other stakeholders to target investment and interventions to increase guideline-concordant CRC screening uptake benefiting specific H/L [Hispanic or Latino] communities in the United States,” the authors wrote. “These data can inform more precise neighborhood-level interventions to increase CRC screening considering unique characteristics important for these H/L [Hispanic or Latino] groups.”
SOURCE:
The study, led by R. Blake Buchalter, PhD, MPH, Center for Populations Health Research, Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, was published online in the American Journal of Public Health.
LIMITATIONS:
The study’s cross-sectional design limited the ability to infer causality. The use of census tract-level data did not capture individual-level screening behaviors. The study did not account for nativity status or years of migration owing to the lack of data. The Centers for Disease Control and Prevention PLACES dataset may not represent the actual screening delivered as it is based on survey data.
DISCLOSURES:
The National Cancer Institute partially supported this study. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Researchers identified thousands of census tracts as priority zones where improving the screening of colorectal cancer (CRC) may benefit Hispanic or Latino communities.
METHODOLOGY:
- Hispanic or Latino individuals have the lowest rate of CRC screening among the six broader census-designated racial or ethnic groups in the United States, while they face a high proportion of cancer deaths due to CRC.
- Researchers performed a cross-sectional ecologic study using 2021 Centers for Disease Control and Prevention PLACES and 2019 American Community Survey data to identify priority zones for CRC screening where intervention programs may be targeted.
- They analyzed a total of 72,136 US census tracts, representing 98.7% of all US census tracts.
- Nine race and ethnic groups were selected on the basis of the population size and categorizations used in prior research on health or cancer disparity: non-Hispanic Black, non-Hispanic White, Asian, Mexican, Puerto Rican, Cuban, Dominican, Central or South American, and “other race.”
- Geographically weighted regression and Getis-Ord Gi* hot spot procedures were used to identify the screening priority zones for all Hispanic or Latino groups.
TAKEAWAY:
- The analysis identified 6519 hot spot tracts for Mexican, 3477 for Puerto Rican, 3522 for Central or South American, 1069 for Dominican, and 1424 for Cuban individuals. The average rates of screening for CRC were 57.2%, 59.9%, 59.3%, 58.9%, and 60.4%, respectively.
- The percentage of Cuban individuals showed a positive association with the CRC screening rate, while the percentage of Mexican, Puerto Rican, Dominican, and Central or South American Hispanic or Latino individuals and of the uninsured showed a negative association with the CRC screening rate.
- The priority zones for Mexican communities were primarily located in Texas and southwestern United States, while those for Puerto Rican, Central or South American, and other populations were located in southern Florida and the metro areas of New York City and Texas.
IN PRACTICE:
“Our findings and interactive web map may serve as a translational tool for public health authorities, policymakers, clinicians, and other stakeholders to target investment and interventions to increase guideline-concordant CRC screening uptake benefiting specific H/L [Hispanic or Latino] communities in the United States,” the authors wrote. “These data can inform more precise neighborhood-level interventions to increase CRC screening considering unique characteristics important for these H/L [Hispanic or Latino] groups.”
SOURCE:
The study, led by R. Blake Buchalter, PhD, MPH, Center for Populations Health Research, Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, was published online in the American Journal of Public Health.
LIMITATIONS:
The study’s cross-sectional design limited the ability to infer causality. The use of census tract-level data did not capture individual-level screening behaviors. The study did not account for nativity status or years of migration owing to the lack of data. The Centers for Disease Control and Prevention PLACES dataset may not represent the actual screening delivered as it is based on survey data.
DISCLOSURES:
The National Cancer Institute partially supported this study. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Blaschkolinear Lupus Erythematosus: Strategies for Early Detection and Management
To the Editor:
Chronic cutaneous lupus erythematosus (CCLE) is an inflammatory condition with myriad cutaneous manifestations. Most forms of CCLE have the potential to progress to systemic lupus erythematosus (SLE).1
Blaschkolinear lupus erythematosus (BLE) is an exceedingly rare subtype of cutaneous lupus erythematosus that usually manifests during childhood as linear plaques along the lines of Blaschko.2,3 Under normal conditions, Blaschko lines are not noticeable; they correspond to the direction of ectodermal cell migration during cutaneous embryogenesis.4,5 The embryonic cells travel ventrolaterally, forming a V-shaped pattern on the back, an S-shaped pattern on the trunk, and an hourglass-shaped pattern on the face with several perpendicular intersections near the mouth and nose.6 During their migration, the cells are susceptible to somatic mutations and clonal expansion, resulting in a monoclonal population of genetically heterogenous cells. This phenomenon is known as somatic mosaicism and may lead to an increased susceptibility to an array of congenital and inflammatory dermatoses, such as cutaneous lupus erythematosus.4 Blaschkolinear entities tend to manifest in a unilateral distribution following exposure to a certain environmental trigger, such as trauma, viral illness, or UV radiation, although a trigger is not always present.7 We report a case of BLE manifesting on the head and neck in an adult patient.
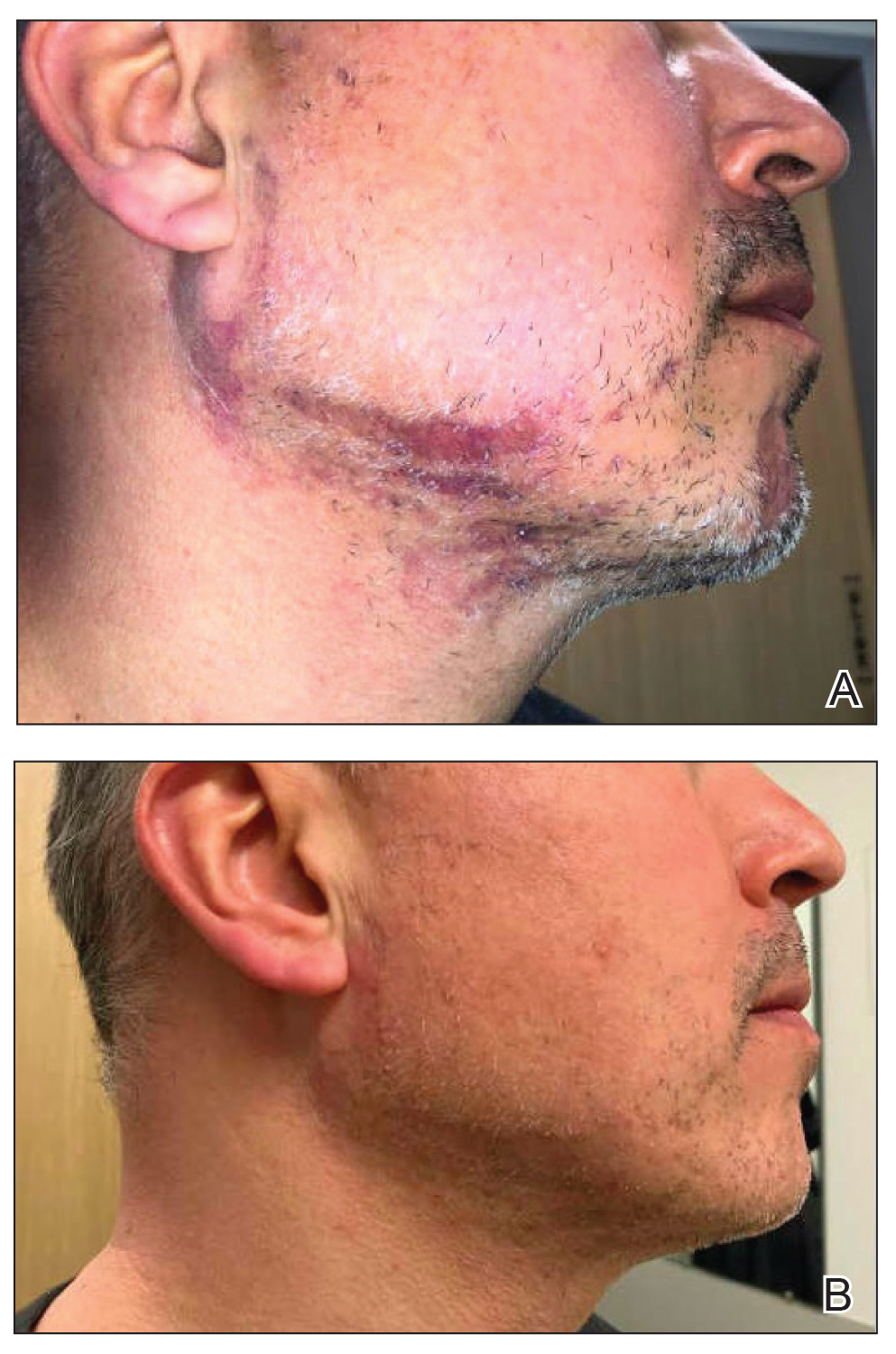
A 46-year-old man presented with a pruritic rash of 3 months’ duration on the right cheek that extended inferiorly to the right upper chest. He had a medical history of well-controlled psoriasis, and he denied any antecedent trauma, fevers, chills, arthralgia, or night sweats. There had been no improvement with mometasone ointment 0.1% applied daily for 2 months as prescribed by his primary care provider. Physical examination revealed indurated, red-brown, atrophic plaques in a blaschkolinear distribution around the nose, right upper jaw, right side of the neck, and right upper chest (Figure, A).
Histopathology of punch biopsies from the right jaw and right upper chest showed an atrophic epidermis with scattered dyskeratotic keratinocytes and vacuolar alteration of the basal cell layer. A superficial and deep perivascular and periadnexal lymphocytic infiltrate was observed in both biopsies. Staining with Verhoeff-van Gieson elastin and periodic acid–Schiff highlighted prominent basement membrane thickening and loss of elastic fibers in the superficial dermis. These findings favored a diagnosis of CCLE, and the clinical blaschkolinear distribution of the rash led to our specific diagnosis of BLE. Laboratory workup for SLE including a complete blood cell count; urine analysis; and testing for liver and kidney function, antinuclearantibodies, complement levels, and erythrocyte sedimentation rate revealed no abnormalities.
The patient started hydroxychloroquine 200 mg twice daily and methotrexate 25 mg weekly along with strict photoprotection measures, including wearing photoprotective clothing and avoiding sunlight during the most intense hours of the day.
Linear lichen planus is an important differential diagnosis to consider in patients with a blaschkolinear eruption.7 Although the clinical manifestations of BLE and linear lichen planus are similar, they differ histopathologically. One study found that only 33.3% of patients (6/18) who clinically presented with blaschkolinear eruptions were correctly diagnosed before histologic examination.7 Visualization of the adnexa as well as the superficial and deep vascular plexuses is paramount in distinguishing between linear lichen planus and BLE; linear lichen planus does not have perivascular and periadnexal infiltration, while BLE does. Thus, in our experience, a punch biopsy—rather than a shave biopsy—should be performed to access the deeper layers of the skin.
Because these 2 entities have noteworthy differences in their management, prognosis, and long-term follow-up, accurate diagnosis is critical. To start, BLE is treated with the use of photoprotection, whereas linear lichen planus is commonly treated with phototherapy. Given the potential for forms of CCLE to progress to SLE, serial monitoring is indicated in patients with BLE. As the risk for progression to SLE is highest in the first 3 years after diagnosis, a review of systems and laboratory testing should occur every 2 to 3 months in the first year after diagnosis (sooner if the disease presentation is more severe).9 Also, treatment with hydroxychloroquine likely delays transformation to SLE and is important in the early management of BLE.10 On the other hand, linear lichen planus tends to self-resolve without progression to systemic involvement, warranting limited follow-up.9
Blaschkolinear lupus erythematosus typically manifests in childhood, but it also can be seen in adults, such as in our patient. Adult-onset BLE is rare but may be underrecognized or underreported in the literature.11 However, dermatologists should consider it in the differential diagnosis for any patient with a blaschkolinear eruption, as establishing the correct diagnosis is key to ensuring prompt and effective treatment for this rare inflammatory condition.
- Grönhagen CM, Fored CM, Granath F, et al. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164:1335-1341. doi:10.1111/j.1365-2133.2011.10272.x
- Requena C, Torrelo A, de Prada I, et al. Linear childhood cutaneous lupus erythematosus following Blaschko lines. J Eur Acad Dermatol Venereol. 2002;16:618-620. doi:10.1046/j.1468-3083.2002.00588.x
- Lim D, Hatami A, Kokta V, et al. Linear cutaneous lupus erythematosus in children-report of two cases and review of the literature: a case report. SAGE Open Med Case Rep. 2020;8:2050313x20979206. doi:10.1177/2050313X20979206
- Jin H, Zhang G, Zhou Y, et al. Old lines tell new tales: Blaschko linear lupus erythematosus. Autoimmun Rev. 2016;15:291-306. doi:10.1016/j.autrev.2015.11.014
- Yu S, Yu H-S. A patient with subacute cutaneous lupus erythematosus along Blaschko lines: implications for the role of keratinocytes in lupus erythematosus. Dermatologica Sinica. 2016;34:144-147. doi:10.1016/j.dsi.2015.12.002
- Kouzak SS, Mendes MST, Costa IMC. Cutaneous mosaicisms: concepts, patterns and classifications. An Bras Dermatol. 2013;88:507-517. doi:10.1590/abd1806-4841.20132015
- Liu W, Vano-Galvan S, Liu J-W, et al. Pigmented linear discoid lupus erythematosus following the lines of Blaschko: a retrospective study of a Chinese series. Indian J Dermatol Venereol Leprol. 2020;86:359-365. doi:10.4103/ijdvl.IJDVL_341_19
- O’Brien JC, Chong BF. Not just skin deep: systemic disease involvement in patients with cutaneous lupus. J Invest Dermatol Symp Proc. 2017;18:S69-S74. doi:10.1016/j.jisp.2016.09.001
- Curtiss P, Walker AM, Chong BF. A systematic review of the progression of cutaneous lupus to systemic lupus erythematosus. Front Immunol. 2022:13:866319. doi:10.3389/fimmu.2022.866319
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404. doi:10.1016/j.berh.2013.07.008
- Milosavljevic K, Fibeger E, Virata AR. A case of linear cutaneous lupus erythematosus in a 55-year-old woman. Am J Case Rep. 2020;21:E921495. doi:10.12659/AJCR.921495
To the Editor:
Chronic cutaneous lupus erythematosus (CCLE) is an inflammatory condition with myriad cutaneous manifestations. Most forms of CCLE have the potential to progress to systemic lupus erythematosus (SLE).1
Blaschkolinear lupus erythematosus (BLE) is an exceedingly rare subtype of cutaneous lupus erythematosus that usually manifests during childhood as linear plaques along the lines of Blaschko.2,3 Under normal conditions, Blaschko lines are not noticeable; they correspond to the direction of ectodermal cell migration during cutaneous embryogenesis.4,5 The embryonic cells travel ventrolaterally, forming a V-shaped pattern on the back, an S-shaped pattern on the trunk, and an hourglass-shaped pattern on the face with several perpendicular intersections near the mouth and nose.6 During their migration, the cells are susceptible to somatic mutations and clonal expansion, resulting in a monoclonal population of genetically heterogenous cells. This phenomenon is known as somatic mosaicism and may lead to an increased susceptibility to an array of congenital and inflammatory dermatoses, such as cutaneous lupus erythematosus.4 Blaschkolinear entities tend to manifest in a unilateral distribution following exposure to a certain environmental trigger, such as trauma, viral illness, or UV radiation, although a trigger is not always present.7 We report a case of BLE manifesting on the head and neck in an adult patient.
A 46-year-old man presented with a pruritic rash of 3 months’ duration on the right cheek that extended inferiorly to the right upper chest. He had a medical history of well-controlled psoriasis, and he denied any antecedent trauma, fevers, chills, arthralgia, or night sweats. There had been no improvement with mometasone ointment 0.1% applied daily for 2 months as prescribed by his primary care provider. Physical examination revealed indurated, red-brown, atrophic plaques in a blaschkolinear distribution around the nose, right upper jaw, right side of the neck, and right upper chest (Figure, A).
Histopathology of punch biopsies from the right jaw and right upper chest showed an atrophic epidermis with scattered dyskeratotic keratinocytes and vacuolar alteration of the basal cell layer. A superficial and deep perivascular and periadnexal lymphocytic infiltrate was observed in both biopsies. Staining with Verhoeff-van Gieson elastin and periodic acid–Schiff highlighted prominent basement membrane thickening and loss of elastic fibers in the superficial dermis. These findings favored a diagnosis of CCLE, and the clinical blaschkolinear distribution of the rash led to our specific diagnosis of BLE. Laboratory workup for SLE including a complete blood cell count; urine analysis; and testing for liver and kidney function, antinuclearantibodies, complement levels, and erythrocyte sedimentation rate revealed no abnormalities.
The patient started hydroxychloroquine 200 mg twice daily and methotrexate 25 mg weekly along with strict photoprotection measures, including wearing photoprotective clothing and avoiding sunlight during the most intense hours of the day.
Linear lichen planus is an important differential diagnosis to consider in patients with a blaschkolinear eruption.7 Although the clinical manifestations of BLE and linear lichen planus are similar, they differ histopathologically. One study found that only 33.3% of patients (6/18) who clinically presented with blaschkolinear eruptions were correctly diagnosed before histologic examination.7 Visualization of the adnexa as well as the superficial and deep vascular plexuses is paramount in distinguishing between linear lichen planus and BLE; linear lichen planus does not have perivascular and periadnexal infiltration, while BLE does. Thus, in our experience, a punch biopsy—rather than a shave biopsy—should be performed to access the deeper layers of the skin.
Because these 2 entities have noteworthy differences in their management, prognosis, and long-term follow-up, accurate diagnosis is critical. To start, BLE is treated with the use of photoprotection, whereas linear lichen planus is commonly treated with phototherapy. Given the potential for forms of CCLE to progress to SLE, serial monitoring is indicated in patients with BLE. As the risk for progression to SLE is highest in the first 3 years after diagnosis, a review of systems and laboratory testing should occur every 2 to 3 months in the first year after diagnosis (sooner if the disease presentation is more severe).9 Also, treatment with hydroxychloroquine likely delays transformation to SLE and is important in the early management of BLE.10 On the other hand, linear lichen planus tends to self-resolve without progression to systemic involvement, warranting limited follow-up.9
Blaschkolinear lupus erythematosus typically manifests in childhood, but it also can be seen in adults, such as in our patient. Adult-onset BLE is rare but may be underrecognized or underreported in the literature.11 However, dermatologists should consider it in the differential diagnosis for any patient with a blaschkolinear eruption, as establishing the correct diagnosis is key to ensuring prompt and effective treatment for this rare inflammatory condition.
To the Editor:
Chronic cutaneous lupus erythematosus (CCLE) is an inflammatory condition with myriad cutaneous manifestations. Most forms of CCLE have the potential to progress to systemic lupus erythematosus (SLE).1
Blaschkolinear lupus erythematosus (BLE) is an exceedingly rare subtype of cutaneous lupus erythematosus that usually manifests during childhood as linear plaques along the lines of Blaschko.2,3 Under normal conditions, Blaschko lines are not noticeable; they correspond to the direction of ectodermal cell migration during cutaneous embryogenesis.4,5 The embryonic cells travel ventrolaterally, forming a V-shaped pattern on the back, an S-shaped pattern on the trunk, and an hourglass-shaped pattern on the face with several perpendicular intersections near the mouth and nose.6 During their migration, the cells are susceptible to somatic mutations and clonal expansion, resulting in a monoclonal population of genetically heterogenous cells. This phenomenon is known as somatic mosaicism and may lead to an increased susceptibility to an array of congenital and inflammatory dermatoses, such as cutaneous lupus erythematosus.4 Blaschkolinear entities tend to manifest in a unilateral distribution following exposure to a certain environmental trigger, such as trauma, viral illness, or UV radiation, although a trigger is not always present.7 We report a case of BLE manifesting on the head and neck in an adult patient.
A 46-year-old man presented with a pruritic rash of 3 months’ duration on the right cheek that extended inferiorly to the right upper chest. He had a medical history of well-controlled psoriasis, and he denied any antecedent trauma, fevers, chills, arthralgia, or night sweats. There had been no improvement with mometasone ointment 0.1% applied daily for 2 months as prescribed by his primary care provider. Physical examination revealed indurated, red-brown, atrophic plaques in a blaschkolinear distribution around the nose, right upper jaw, right side of the neck, and right upper chest (Figure, A).
Histopathology of punch biopsies from the right jaw and right upper chest showed an atrophic epidermis with scattered dyskeratotic keratinocytes and vacuolar alteration of the basal cell layer. A superficial and deep perivascular and periadnexal lymphocytic infiltrate was observed in both biopsies. Staining with Verhoeff-van Gieson elastin and periodic acid–Schiff highlighted prominent basement membrane thickening and loss of elastic fibers in the superficial dermis. These findings favored a diagnosis of CCLE, and the clinical blaschkolinear distribution of the rash led to our specific diagnosis of BLE. Laboratory workup for SLE including a complete blood cell count; urine analysis; and testing for liver and kidney function, antinuclearantibodies, complement levels, and erythrocyte sedimentation rate revealed no abnormalities.
The patient started hydroxychloroquine 200 mg twice daily and methotrexate 25 mg weekly along with strict photoprotection measures, including wearing photoprotective clothing and avoiding sunlight during the most intense hours of the day.
Linear lichen planus is an important differential diagnosis to consider in patients with a blaschkolinear eruption.7 Although the clinical manifestations of BLE and linear lichen planus are similar, they differ histopathologically. One study found that only 33.3% of patients (6/18) who clinically presented with blaschkolinear eruptions were correctly diagnosed before histologic examination.7 Visualization of the adnexa as well as the superficial and deep vascular plexuses is paramount in distinguishing between linear lichen planus and BLE; linear lichen planus does not have perivascular and periadnexal infiltration, while BLE does. Thus, in our experience, a punch biopsy—rather than a shave biopsy—should be performed to access the deeper layers of the skin.
Because these 2 entities have noteworthy differences in their management, prognosis, and long-term follow-up, accurate diagnosis is critical. To start, BLE is treated with the use of photoprotection, whereas linear lichen planus is commonly treated with phototherapy. Given the potential for forms of CCLE to progress to SLE, serial monitoring is indicated in patients with BLE. As the risk for progression to SLE is highest in the first 3 years after diagnosis, a review of systems and laboratory testing should occur every 2 to 3 months in the first year after diagnosis (sooner if the disease presentation is more severe).9 Also, treatment with hydroxychloroquine likely delays transformation to SLE and is important in the early management of BLE.10 On the other hand, linear lichen planus tends to self-resolve without progression to systemic involvement, warranting limited follow-up.9
Blaschkolinear lupus erythematosus typically manifests in childhood, but it also can be seen in adults, such as in our patient. Adult-onset BLE is rare but may be underrecognized or underreported in the literature.11 However, dermatologists should consider it in the differential diagnosis for any patient with a blaschkolinear eruption, as establishing the correct diagnosis is key to ensuring prompt and effective treatment for this rare inflammatory condition.
- Grönhagen CM, Fored CM, Granath F, et al. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164:1335-1341. doi:10.1111/j.1365-2133.2011.10272.x
- Requena C, Torrelo A, de Prada I, et al. Linear childhood cutaneous lupus erythematosus following Blaschko lines. J Eur Acad Dermatol Venereol. 2002;16:618-620. doi:10.1046/j.1468-3083.2002.00588.x
- Lim D, Hatami A, Kokta V, et al. Linear cutaneous lupus erythematosus in children-report of two cases and review of the literature: a case report. SAGE Open Med Case Rep. 2020;8:2050313x20979206. doi:10.1177/2050313X20979206
- Jin H, Zhang G, Zhou Y, et al. Old lines tell new tales: Blaschko linear lupus erythematosus. Autoimmun Rev. 2016;15:291-306. doi:10.1016/j.autrev.2015.11.014
- Yu S, Yu H-S. A patient with subacute cutaneous lupus erythematosus along Blaschko lines: implications for the role of keratinocytes in lupus erythematosus. Dermatologica Sinica. 2016;34:144-147. doi:10.1016/j.dsi.2015.12.002
- Kouzak SS, Mendes MST, Costa IMC. Cutaneous mosaicisms: concepts, patterns and classifications. An Bras Dermatol. 2013;88:507-517. doi:10.1590/abd1806-4841.20132015
- Liu W, Vano-Galvan S, Liu J-W, et al. Pigmented linear discoid lupus erythematosus following the lines of Blaschko: a retrospective study of a Chinese series. Indian J Dermatol Venereol Leprol. 2020;86:359-365. doi:10.4103/ijdvl.IJDVL_341_19
- O’Brien JC, Chong BF. Not just skin deep: systemic disease involvement in patients with cutaneous lupus. J Invest Dermatol Symp Proc. 2017;18:S69-S74. doi:10.1016/j.jisp.2016.09.001
- Curtiss P, Walker AM, Chong BF. A systematic review of the progression of cutaneous lupus to systemic lupus erythematosus. Front Immunol. 2022:13:866319. doi:10.3389/fimmu.2022.866319
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404. doi:10.1016/j.berh.2013.07.008
- Milosavljevic K, Fibeger E, Virata AR. A case of linear cutaneous lupus erythematosus in a 55-year-old woman. Am J Case Rep. 2020;21:E921495. doi:10.12659/AJCR.921495
- Grönhagen CM, Fored CM, Granath F, et al. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164:1335-1341. doi:10.1111/j.1365-2133.2011.10272.x
- Requena C, Torrelo A, de Prada I, et al. Linear childhood cutaneous lupus erythematosus following Blaschko lines. J Eur Acad Dermatol Venereol. 2002;16:618-620. doi:10.1046/j.1468-3083.2002.00588.x
- Lim D, Hatami A, Kokta V, et al. Linear cutaneous lupus erythematosus in children-report of two cases and review of the literature: a case report. SAGE Open Med Case Rep. 2020;8:2050313x20979206. doi:10.1177/2050313X20979206
- Jin H, Zhang G, Zhou Y, et al. Old lines tell new tales: Blaschko linear lupus erythematosus. Autoimmun Rev. 2016;15:291-306. doi:10.1016/j.autrev.2015.11.014
- Yu S, Yu H-S. A patient with subacute cutaneous lupus erythematosus along Blaschko lines: implications for the role of keratinocytes in lupus erythematosus. Dermatologica Sinica. 2016;34:144-147. doi:10.1016/j.dsi.2015.12.002
- Kouzak SS, Mendes MST, Costa IMC. Cutaneous mosaicisms: concepts, patterns and classifications. An Bras Dermatol. 2013;88:507-517. doi:10.1590/abd1806-4841.20132015
- Liu W, Vano-Galvan S, Liu J-W, et al. Pigmented linear discoid lupus erythematosus following the lines of Blaschko: a retrospective study of a Chinese series. Indian J Dermatol Venereol Leprol. 2020;86:359-365. doi:10.4103/ijdvl.IJDVL_341_19
- O’Brien JC, Chong BF. Not just skin deep: systemic disease involvement in patients with cutaneous lupus. J Invest Dermatol Symp Proc. 2017;18:S69-S74. doi:10.1016/j.jisp.2016.09.001
- Curtiss P, Walker AM, Chong BF. A systematic review of the progression of cutaneous lupus to systemic lupus erythematosus. Front Immunol. 2022:13:866319. doi:10.3389/fimmu.2022.866319
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404. doi:10.1016/j.berh.2013.07.008
- Milosavljevic K, Fibeger E, Virata AR. A case of linear cutaneous lupus erythematosus in a 55-year-old woman. Am J Case Rep. 2020;21:E921495. doi:10.12659/AJCR.921495
Practice Points
- Blaschkolinear lupus erythematosus (BLE), an exceedingly rare subtype of chronic cutaneous lupus erythematosus, usually presents during childhood as linear plaques along the lines of Blaschko.
- It is important to consider linear lichen planus in patients with a blaschkolinear eruption, as the clinical manifestations are similar but there are differences in histopathology, management, prognosis, and long-term follow-up.
- Serial monitoring is indicated in patients with BLE given the potential for progression to systemic lupus erythematosus, which may be delayed with early use of hydroxychloroquine.
COVID-19 Booster Vaccine Shortens Menstrual Cycles in Teens
TOPLINE:
The vaccine did not appear to be associated with shifts in menstrual flow, pain, or other symptoms.
METHODOLOGY:
- Reports of menstrual cycle changes following the COVID-19 vaccination began to emerge in early 2021, raising concerns about the impact of the vaccine on menstrual health.
- Researchers conducted a prospective study including 65 adolescent girls (mean age, 17.3 years), of whom 47 had received an initial series of COVID-19 vaccination at least 6 months prior to receiving a booster dose (booster group), and 18 had not received the booster vaccine (control group), two of whom had never received any COVID-19 vaccine, four who had received an initial vaccine but not a booster, and 12 who had received an initial vaccine and booster but more than 6 months prior to the study.
- Menstrual cycle length was measured for three cycles prior to and four cycles after vaccination in the booster group and for seven cycles in the control group.
- Menstrual flow, pain, and stress were measured at baseline and monthly for 3 months post vaccination.
TAKEAWAY:
- Participants in the booster group experienced shorter cycles by an average of 5.35 days after receiving the COVID-19 booster vaccine (P = .03), particularly during the second cycle. In contrast, those in the control group did not experience any changes in the menstrual cycle length.
- Receiving the booster dose in the follicular phase was associated with significantly shorter menstrual cycles, compared with pre-booster cycles (P = .0157).
- Menstrual flow, pain, and other symptoms remained unaffected after the COVID-19 booster vaccination.
- Higher stress levels at baseline were also associated with a shorter length of the menstrual cycle (P = .03) in both groups, regardless of the booster vaccination status.
IN PRACTICE:
“These data are potentially important for counseling parents regarding potential vaccine refusal in the future for their teen daughters,” the authors wrote.
SOURCE:
This study was led by Laura A. Payne, PhD, from McLean Hospital in Boston, and was published online in the Journal of Adolescent Health.
LIMITATIONS:
The sample size for the booster and control groups was relatively small and homogeneous. The study did not include the height, weight, birth control use, or other chronic conditions of the participants, which may have influenced the functioning of the menstrual cycle. The control group included a majority of teens who had previously received a vaccine and even a booster, which could have affected results.
DISCLOSURES:
This study was supported by grants from the Eunice Kennedy Shriver National Institute for Child Health and Human Development. Some authors received consulting fees, travel reimbursements, honoraria, research funding, and royalties from Bayer Healthcare, Mahana Therapeutics, Gates, and Merck, among others.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
The vaccine did not appear to be associated with shifts in menstrual flow, pain, or other symptoms.
METHODOLOGY:
- Reports of menstrual cycle changes following the COVID-19 vaccination began to emerge in early 2021, raising concerns about the impact of the vaccine on menstrual health.
- Researchers conducted a prospective study including 65 adolescent girls (mean age, 17.3 years), of whom 47 had received an initial series of COVID-19 vaccination at least 6 months prior to receiving a booster dose (booster group), and 18 had not received the booster vaccine (control group), two of whom had never received any COVID-19 vaccine, four who had received an initial vaccine but not a booster, and 12 who had received an initial vaccine and booster but more than 6 months prior to the study.
- Menstrual cycle length was measured for three cycles prior to and four cycles after vaccination in the booster group and for seven cycles in the control group.
- Menstrual flow, pain, and stress were measured at baseline and monthly for 3 months post vaccination.
TAKEAWAY:
- Participants in the booster group experienced shorter cycles by an average of 5.35 days after receiving the COVID-19 booster vaccine (P = .03), particularly during the second cycle. In contrast, those in the control group did not experience any changes in the menstrual cycle length.
- Receiving the booster dose in the follicular phase was associated with significantly shorter menstrual cycles, compared with pre-booster cycles (P = .0157).
- Menstrual flow, pain, and other symptoms remained unaffected after the COVID-19 booster vaccination.
- Higher stress levels at baseline were also associated with a shorter length of the menstrual cycle (P = .03) in both groups, regardless of the booster vaccination status.
IN PRACTICE:
“These data are potentially important for counseling parents regarding potential vaccine refusal in the future for their teen daughters,” the authors wrote.
SOURCE:
This study was led by Laura A. Payne, PhD, from McLean Hospital in Boston, and was published online in the Journal of Adolescent Health.
LIMITATIONS:
The sample size for the booster and control groups was relatively small and homogeneous. The study did not include the height, weight, birth control use, or other chronic conditions of the participants, which may have influenced the functioning of the menstrual cycle. The control group included a majority of teens who had previously received a vaccine and even a booster, which could have affected results.
DISCLOSURES:
This study was supported by grants from the Eunice Kennedy Shriver National Institute for Child Health and Human Development. Some authors received consulting fees, travel reimbursements, honoraria, research funding, and royalties from Bayer Healthcare, Mahana Therapeutics, Gates, and Merck, among others.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
The vaccine did not appear to be associated with shifts in menstrual flow, pain, or other symptoms.
METHODOLOGY:
- Reports of menstrual cycle changes following the COVID-19 vaccination began to emerge in early 2021, raising concerns about the impact of the vaccine on menstrual health.
- Researchers conducted a prospective study including 65 adolescent girls (mean age, 17.3 years), of whom 47 had received an initial series of COVID-19 vaccination at least 6 months prior to receiving a booster dose (booster group), and 18 had not received the booster vaccine (control group), two of whom had never received any COVID-19 vaccine, four who had received an initial vaccine but not a booster, and 12 who had received an initial vaccine and booster but more than 6 months prior to the study.
- Menstrual cycle length was measured for three cycles prior to and four cycles after vaccination in the booster group and for seven cycles in the control group.
- Menstrual flow, pain, and stress were measured at baseline and monthly for 3 months post vaccination.
TAKEAWAY:
- Participants in the booster group experienced shorter cycles by an average of 5.35 days after receiving the COVID-19 booster vaccine (P = .03), particularly during the second cycle. In contrast, those in the control group did not experience any changes in the menstrual cycle length.
- Receiving the booster dose in the follicular phase was associated with significantly shorter menstrual cycles, compared with pre-booster cycles (P = .0157).
- Menstrual flow, pain, and other symptoms remained unaffected after the COVID-19 booster vaccination.
- Higher stress levels at baseline were also associated with a shorter length of the menstrual cycle (P = .03) in both groups, regardless of the booster vaccination status.
IN PRACTICE:
“These data are potentially important for counseling parents regarding potential vaccine refusal in the future for their teen daughters,” the authors wrote.
SOURCE:
This study was led by Laura A. Payne, PhD, from McLean Hospital in Boston, and was published online in the Journal of Adolescent Health.
LIMITATIONS:
The sample size for the booster and control groups was relatively small and homogeneous. The study did not include the height, weight, birth control use, or other chronic conditions of the participants, which may have influenced the functioning of the menstrual cycle. The control group included a majority of teens who had previously received a vaccine and even a booster, which could have affected results.
DISCLOSURES:
This study was supported by grants from the Eunice Kennedy Shriver National Institute for Child Health and Human Development. Some authors received consulting fees, travel reimbursements, honoraria, research funding, and royalties from Bayer Healthcare, Mahana Therapeutics, Gates, and Merck, among others.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Support for Laser Treatment to Reduce NMSC Risk is Increasing
CARLSBAD, CALIFORNIA — and a key 2017 publication laid the groundwork for current approaches, according to Elizabeth Tanzi, MD.
In the article, which was published in Molecules, Mike Kemp, PhD, and Jeffrey Bryant Travers, MD, PhD, at Wright State University, Dayton, Ohio, and Dan F. Spandau, PhD, at Indiana University School of Medicine, Indianapolis, demonstrated that geriatric skin responds to ultraviolet B (UVB) differently than young skin because of differences in insulin-like growth factor 1 (IGF-1) levels produced by dermal fibroblasts.

“As we age, our fibroblasts become senescent, inactive,” Dr. Tanzi, associate clinical professor of dermatology at George Washington University, Washington, DC, said at the Controversies and Conversations in Laser and Cosmetic Surgery symposium. “They don’t make as many growth factors, particularly IGF-1, and therefore we don’t stimulate the responses. We need more of our growth factors.”
In later, separate work, Dr. Travers, Dr. Spandau, and colleagues found that using dermabrasion or fractionated laser resurfacing to wound the skin can result in increased dermal IGF-1 levels and normalization of the abnormal pro-carcinogenic UV response associated with geriatric skin — a treatment that has the potential to prevent NMSC. That study “was the epiphany” for fostering interest among researchers in the field of lasers and medicine, Dr. Tanzi said.
In a retrospective cohort study, Mathew Avram, MD, JD, and colleagues reviewed patients with a history of facial keratinocyte carcinoma (KC) who were treated at Massachusetts General Hospital in Boston between 2005 and 2021. The study population included 43 patients treated with either the 1927- or the 1550-nm nonablative fractional laser (NAFL) and 52 matched controls. The rate of subsequent facial KC development was 20.9% in NAFL-treated patients and 40.4% in controls (relative risk, 0.52, P = .049).

During a separate presentation at the meeting, Dr. Avram, director of lasers and cosmetics at Massachusetts General Hospital, Boston, said that, when he and his colleagues controlled for age, gender, and skin type, controls were 2.65 times more likely to develop new facial KC, compared with those treated with NAFL (P = .0169). “This enhanced effect was seen with the 1550-nm device, compared with the 1927-nm device. The study shows us that 1550-nm/1927-nm NAFL may have a protective effect for patients with a history of KC, but the role of each wavelength is to be determined. We also need a prospective, controlled study to verify the results.”
In an ongoing study first presented at the 2023 annual meeting of the American Society for Dermatologic Surgery, Dr. Tanzi and colleagues enrolled 15 patients aged ≥ 55 years to evaluate the restoration of physiologic features and biomarkers in skin treated with 25% trichloroacetic acid (TCA), plus the 1550-nm or 1927-nm NAFL. Four sites on the back were treated and biopsies were taken at baseline and at 3 months post treatment. The protocol involved TCA 25% to speckled frost, with the 1550-nm device set to level 6 at 70 mJ and the 1927-nm device set to level 8 at 20 mJ. Immunohistochemical stains are still pending; however, physiologic changes were noted.
Three months after a single treatment, the 1927-nm treated areas showed statistically significant elongation of fibroblasts (consistent with younger fibroblasts) on histology. “Although not a large study, it supports the growing body of research that demonstrates we are improving the health of our patients’ skin with certain types of laser treatments, not just beautifying it,” Dr. Tanzi said.
Dr. Tanzi disclosed being a member of the advisory board for AbbVie/Allergan and Sciton, and is a consultant for Alastin/Galderma, Candesant Biomedical, Cytrellis, Revance, and Solta Medical. Dr. Avram disclosed that he receives intellectual property royalties from and holds stock options in Cytrellis, and is a consultant to Allergan and holds stock options in BAI Biosciences, Sofwave, and La Jolla NanoMedical.
A version of this article first appeared on Medscape.com.
CARLSBAD, CALIFORNIA — and a key 2017 publication laid the groundwork for current approaches, according to Elizabeth Tanzi, MD.
In the article, which was published in Molecules, Mike Kemp, PhD, and Jeffrey Bryant Travers, MD, PhD, at Wright State University, Dayton, Ohio, and Dan F. Spandau, PhD, at Indiana University School of Medicine, Indianapolis, demonstrated that geriatric skin responds to ultraviolet B (UVB) differently than young skin because of differences in insulin-like growth factor 1 (IGF-1) levels produced by dermal fibroblasts.
“As we age, our fibroblasts become senescent, inactive,” Dr. Tanzi, associate clinical professor of dermatology at George Washington University, Washington, DC, said at the Controversies and Conversations in Laser and Cosmetic Surgery symposium. “They don’t make as many growth factors, particularly IGF-1, and therefore we don’t stimulate the responses. We need more of our growth factors.”
In later, separate work, Dr. Travers, Dr. Spandau, and colleagues found that using dermabrasion or fractionated laser resurfacing to wound the skin can result in increased dermal IGF-1 levels and normalization of the abnormal pro-carcinogenic UV response associated with geriatric skin — a treatment that has the potential to prevent NMSC. That study “was the epiphany” for fostering interest among researchers in the field of lasers and medicine, Dr. Tanzi said.
In a retrospective cohort study, Mathew Avram, MD, JD, and colleagues reviewed patients with a history of facial keratinocyte carcinoma (KC) who were treated at Massachusetts General Hospital in Boston between 2005 and 2021. The study population included 43 patients treated with either the 1927- or the 1550-nm nonablative fractional laser (NAFL) and 52 matched controls. The rate of subsequent facial KC development was 20.9% in NAFL-treated patients and 40.4% in controls (relative risk, 0.52, P = .049).
During a separate presentation at the meeting, Dr. Avram, director of lasers and cosmetics at Massachusetts General Hospital, Boston, said that, when he and his colleagues controlled for age, gender, and skin type, controls were 2.65 times more likely to develop new facial KC, compared with those treated with NAFL (P = .0169). “This enhanced effect was seen with the 1550-nm device, compared with the 1927-nm device. The study shows us that 1550-nm/1927-nm NAFL may have a protective effect for patients with a history of KC, but the role of each wavelength is to be determined. We also need a prospective, controlled study to verify the results.”
In an ongoing study first presented at the 2023 annual meeting of the American Society for Dermatologic Surgery, Dr. Tanzi and colleagues enrolled 15 patients aged ≥ 55 years to evaluate the restoration of physiologic features and biomarkers in skin treated with 25% trichloroacetic acid (TCA), plus the 1550-nm or 1927-nm NAFL. Four sites on the back were treated and biopsies were taken at baseline and at 3 months post treatment. The protocol involved TCA 25% to speckled frost, with the 1550-nm device set to level 6 at 70 mJ and the 1927-nm device set to level 8 at 20 mJ. Immunohistochemical stains are still pending; however, physiologic changes were noted.
Three months after a single treatment, the 1927-nm treated areas showed statistically significant elongation of fibroblasts (consistent with younger fibroblasts) on histology. “Although not a large study, it supports the growing body of research that demonstrates we are improving the health of our patients’ skin with certain types of laser treatments, not just beautifying it,” Dr. Tanzi said.
Dr. Tanzi disclosed being a member of the advisory board for AbbVie/Allergan and Sciton, and is a consultant for Alastin/Galderma, Candesant Biomedical, Cytrellis, Revance, and Solta Medical. Dr. Avram disclosed that he receives intellectual property royalties from and holds stock options in Cytrellis, and is a consultant to Allergan and holds stock options in BAI Biosciences, Sofwave, and La Jolla NanoMedical.
A version of this article first appeared on Medscape.com.
CARLSBAD, CALIFORNIA — and a key 2017 publication laid the groundwork for current approaches, according to Elizabeth Tanzi, MD.
In the article, which was published in Molecules, Mike Kemp, PhD, and Jeffrey Bryant Travers, MD, PhD, at Wright State University, Dayton, Ohio, and Dan F. Spandau, PhD, at Indiana University School of Medicine, Indianapolis, demonstrated that geriatric skin responds to ultraviolet B (UVB) differently than young skin because of differences in insulin-like growth factor 1 (IGF-1) levels produced by dermal fibroblasts.
“As we age, our fibroblasts become senescent, inactive,” Dr. Tanzi, associate clinical professor of dermatology at George Washington University, Washington, DC, said at the Controversies and Conversations in Laser and Cosmetic Surgery symposium. “They don’t make as many growth factors, particularly IGF-1, and therefore we don’t stimulate the responses. We need more of our growth factors.”
In later, separate work, Dr. Travers, Dr. Spandau, and colleagues found that using dermabrasion or fractionated laser resurfacing to wound the skin can result in increased dermal IGF-1 levels and normalization of the abnormal pro-carcinogenic UV response associated with geriatric skin — a treatment that has the potential to prevent NMSC. That study “was the epiphany” for fostering interest among researchers in the field of lasers and medicine, Dr. Tanzi said.
In a retrospective cohort study, Mathew Avram, MD, JD, and colleagues reviewed patients with a history of facial keratinocyte carcinoma (KC) who were treated at Massachusetts General Hospital in Boston between 2005 and 2021. The study population included 43 patients treated with either the 1927- or the 1550-nm nonablative fractional laser (NAFL) and 52 matched controls. The rate of subsequent facial KC development was 20.9% in NAFL-treated patients and 40.4% in controls (relative risk, 0.52, P = .049).
During a separate presentation at the meeting, Dr. Avram, director of lasers and cosmetics at Massachusetts General Hospital, Boston, said that, when he and his colleagues controlled for age, gender, and skin type, controls were 2.65 times more likely to develop new facial KC, compared with those treated with NAFL (P = .0169). “This enhanced effect was seen with the 1550-nm device, compared with the 1927-nm device. The study shows us that 1550-nm/1927-nm NAFL may have a protective effect for patients with a history of KC, but the role of each wavelength is to be determined. We also need a prospective, controlled study to verify the results.”
In an ongoing study first presented at the 2023 annual meeting of the American Society for Dermatologic Surgery, Dr. Tanzi and colleagues enrolled 15 patients aged ≥ 55 years to evaluate the restoration of physiologic features and biomarkers in skin treated with 25% trichloroacetic acid (TCA), plus the 1550-nm or 1927-nm NAFL. Four sites on the back were treated and biopsies were taken at baseline and at 3 months post treatment. The protocol involved TCA 25% to speckled frost, with the 1550-nm device set to level 6 at 70 mJ and the 1927-nm device set to level 8 at 20 mJ. Immunohistochemical stains are still pending; however, physiologic changes were noted.
Three months after a single treatment, the 1927-nm treated areas showed statistically significant elongation of fibroblasts (consistent with younger fibroblasts) on histology. “Although not a large study, it supports the growing body of research that demonstrates we are improving the health of our patients’ skin with certain types of laser treatments, not just beautifying it,” Dr. Tanzi said.
Dr. Tanzi disclosed being a member of the advisory board for AbbVie/Allergan and Sciton, and is a consultant for Alastin/Galderma, Candesant Biomedical, Cytrellis, Revance, and Solta Medical. Dr. Avram disclosed that he receives intellectual property royalties from and holds stock options in Cytrellis, and is a consultant to Allergan and holds stock options in BAI Biosciences, Sofwave, and La Jolla NanoMedical.
A version of this article first appeared on Medscape.com.
Hidradenitis Suppurativa Risk Reduced After Patients Quit Smoking
TOPLINE:
, with this reduction becoming evident 3-4 years after cessation, in a cohort study from Korea.
METHODOLOGY:
- Researchers conducted a population-based cohort study using the Korean National Health Insurance Service database.
- A total of 6,230,189 participants in South Korea who underwent two consecutive biennial health examinations from 2004 to 2005 and 2006 to 2007 were included.
- Participants were categorized into six groups on the basis of their smoking status at both checkups: Sustained smokers, relapsed smokers, new smokers, smoking quitters, sustained ex-smokers, and never smokers.
- The primary outcome was the development of HS.
TAKEAWAY:
- A total of 3761 HS cases were detected during the 84,457,025 person-years of observation.
- Smoking quitters (adjusted hazard ratio [AHR], 0.68; 95% CI, 0.56-0.83), sustained ex-smokers (AHR, 0.67; 95% CI, 0.57-0.77), and never smokers (AHR, 0.57; 95% CI, 0.52-0.63) exhibited a reduced risk of developing HS compared with sustained smokers.
- The risk of developing HS varied over time, with smoking quitters showing no significant risk reduction compared with sustained smokers in the first 3 years. After 3 years, a statistically significant decrease in HS risk was observed among quitters, which persisted over time.
- At 3-6 years, the risk reduction in sustained quitters was comparable with that of never smokers (AHR, 0.58 and 0.63, respectively).
IN PRACTICE:
“Smoking cessation and maintaining a smoke-free lifestyle may be important preventive measures against the development of HS,” the authors concluded. In an accompanying editorial, Alexandra Charrow, MD, and Leandra A. Barnes, MD, of the departments of dermatology at Brigham and Women’s Hospital, Boston, and Stanford University, Palo Alto, California, respectively, wrote that while the study “importantly contributes to the understanding of the association of smoking tobacco and HS onset, prospective cohort studies in large, diverse cohorts of patients with HS may help dermatologists better understand the causal relationship between smoking and the onset or exacerbation of HS.” For now, they added, “dermatologists must continue to use comprehensive HS treatment strategies, including lifestyle modifications that promote overall health like smoking cessation, to improve the lives of those enduring HS.”
SOURCE:
The study was led by Seong Rae Kim, MD, Department of Dermatology, Seoul National University College of Medicine, Seoul, Republic of Korea, and was published online, along with the editorial, on August 21 in JAMA Dermatology.
LIMITATIONS:
The study limitations include the potential for unexamined confounding factors like hereditary background, reliance on self-reported smoking status, and the exclusion of electronic cigarette use and nicotine replacement therapy. The predominantly male smoker population may limit generalizability, and delayed diagnosis of HS may not reflect the actual time of onset.
DISCLOSURES:
The study funding source was not disclosed. One study author reported various financial ties with pharmaceutical companies outside this work; other authors had no disclosures. Dr. Charrow’s disclosures included receiving personal fees from several pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
, with this reduction becoming evident 3-4 years after cessation, in a cohort study from Korea.
METHODOLOGY:
- Researchers conducted a population-based cohort study using the Korean National Health Insurance Service database.
- A total of 6,230,189 participants in South Korea who underwent two consecutive biennial health examinations from 2004 to 2005 and 2006 to 2007 were included.
- Participants were categorized into six groups on the basis of their smoking status at both checkups: Sustained smokers, relapsed smokers, new smokers, smoking quitters, sustained ex-smokers, and never smokers.
- The primary outcome was the development of HS.
TAKEAWAY:
- A total of 3761 HS cases were detected during the 84,457,025 person-years of observation.
- Smoking quitters (adjusted hazard ratio [AHR], 0.68; 95% CI, 0.56-0.83), sustained ex-smokers (AHR, 0.67; 95% CI, 0.57-0.77), and never smokers (AHR, 0.57; 95% CI, 0.52-0.63) exhibited a reduced risk of developing HS compared with sustained smokers.
- The risk of developing HS varied over time, with smoking quitters showing no significant risk reduction compared with sustained smokers in the first 3 years. After 3 years, a statistically significant decrease in HS risk was observed among quitters, which persisted over time.
- At 3-6 years, the risk reduction in sustained quitters was comparable with that of never smokers (AHR, 0.58 and 0.63, respectively).
IN PRACTICE:
“Smoking cessation and maintaining a smoke-free lifestyle may be important preventive measures against the development of HS,” the authors concluded. In an accompanying editorial, Alexandra Charrow, MD, and Leandra A. Barnes, MD, of the departments of dermatology at Brigham and Women’s Hospital, Boston, and Stanford University, Palo Alto, California, respectively, wrote that while the study “importantly contributes to the understanding of the association of smoking tobacco and HS onset, prospective cohort studies in large, diverse cohorts of patients with HS may help dermatologists better understand the causal relationship between smoking and the onset or exacerbation of HS.” For now, they added, “dermatologists must continue to use comprehensive HS treatment strategies, including lifestyle modifications that promote overall health like smoking cessation, to improve the lives of those enduring HS.”
SOURCE:
The study was led by Seong Rae Kim, MD, Department of Dermatology, Seoul National University College of Medicine, Seoul, Republic of Korea, and was published online, along with the editorial, on August 21 in JAMA Dermatology.
LIMITATIONS:
The study limitations include the potential for unexamined confounding factors like hereditary background, reliance on self-reported smoking status, and the exclusion of electronic cigarette use and nicotine replacement therapy. The predominantly male smoker population may limit generalizability, and delayed diagnosis of HS may not reflect the actual time of onset.
DISCLOSURES:
The study funding source was not disclosed. One study author reported various financial ties with pharmaceutical companies outside this work; other authors had no disclosures. Dr. Charrow’s disclosures included receiving personal fees from several pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
, with this reduction becoming evident 3-4 years after cessation, in a cohort study from Korea.
METHODOLOGY:
- Researchers conducted a population-based cohort study using the Korean National Health Insurance Service database.
- A total of 6,230,189 participants in South Korea who underwent two consecutive biennial health examinations from 2004 to 2005 and 2006 to 2007 were included.
- Participants were categorized into six groups on the basis of their smoking status at both checkups: Sustained smokers, relapsed smokers, new smokers, smoking quitters, sustained ex-smokers, and never smokers.
- The primary outcome was the development of HS.
TAKEAWAY:
- A total of 3761 HS cases were detected during the 84,457,025 person-years of observation.
- Smoking quitters (adjusted hazard ratio [AHR], 0.68; 95% CI, 0.56-0.83), sustained ex-smokers (AHR, 0.67; 95% CI, 0.57-0.77), and never smokers (AHR, 0.57; 95% CI, 0.52-0.63) exhibited a reduced risk of developing HS compared with sustained smokers.
- The risk of developing HS varied over time, with smoking quitters showing no significant risk reduction compared with sustained smokers in the first 3 years. After 3 years, a statistically significant decrease in HS risk was observed among quitters, which persisted over time.
- At 3-6 years, the risk reduction in sustained quitters was comparable with that of never smokers (AHR, 0.58 and 0.63, respectively).
IN PRACTICE:
“Smoking cessation and maintaining a smoke-free lifestyle may be important preventive measures against the development of HS,” the authors concluded. In an accompanying editorial, Alexandra Charrow, MD, and Leandra A. Barnes, MD, of the departments of dermatology at Brigham and Women’s Hospital, Boston, and Stanford University, Palo Alto, California, respectively, wrote that while the study “importantly contributes to the understanding of the association of smoking tobacco and HS onset, prospective cohort studies in large, diverse cohorts of patients with HS may help dermatologists better understand the causal relationship between smoking and the onset or exacerbation of HS.” For now, they added, “dermatologists must continue to use comprehensive HS treatment strategies, including lifestyle modifications that promote overall health like smoking cessation, to improve the lives of those enduring HS.”
SOURCE:
The study was led by Seong Rae Kim, MD, Department of Dermatology, Seoul National University College of Medicine, Seoul, Republic of Korea, and was published online, along with the editorial, on August 21 in JAMA Dermatology.
LIMITATIONS:
The study limitations include the potential for unexamined confounding factors like hereditary background, reliance on self-reported smoking status, and the exclusion of electronic cigarette use and nicotine replacement therapy. The predominantly male smoker population may limit generalizability, and delayed diagnosis of HS may not reflect the actual time of onset.
DISCLOSURES:
The study funding source was not disclosed. One study author reported various financial ties with pharmaceutical companies outside this work; other authors had no disclosures. Dr. Charrow’s disclosures included receiving personal fees from several pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Beyond One-Size-Fits-All: Precision Psychiatry Is Here
The field of psychiatry is experiencing a transformative shift toward precision medicine, a paradigm that tailors treatment to the unique characteristics of individual patients. This approach echoes advances in fields like oncology and cardiology, where precision tools have already revolutionized patient care.
But what exactly is precision psychiatry? How does it differ from traditional psychiatry? What will it look like in clinical practice? And are we there yet?
Beyond One-Size-Fits-All
The prevailing “one-size-fits-all” approach in psychiatry, which relies heavily on subjective symptom reporting, often proves ineffective due to the broad heterogeneity of diagnostic categories. This can lead to a “trial-and-error” cycle in treatment, which is time-consuming, costly, and frustrating for both doctors and patients.
In contrast, precision psychiatry has the potential to identify subtypes of psychiatric disorders and tailor treatments using measurable, objective data.
“The data supporting the use of precision psychiatry are very promising, particularly for treatment-resistant depression,” Leanne Williams, PhD, professor in the Department of Psychiatry and Behavioral Sciences at Stanford University, Stanford, and director of the Stanford Center for Precision Mental Health and Wellness, Palo Alto, California, said in an interview with this news organization.
Using functional MRI (fMRI), Dr. Williams and her team have mapped and measured patients’ brain circuitry to identify eight “biotypes” of depression that reflect combinations of dysfunction in six different circuits of the brain.
They are using these biotypes to guide treatment decisions in the clinic, matching individual patients to more targeted and effective therapies.
“We’re offering functional MRI to directly assess brain function along with other measures, so precision psychiatry is happening, and it’s really wanted by patients and their families. And the data suggest that we can double the rate of good outcomes,” said Dr. Williams.
“Neuroimaging techniques, particularly fMRI, have revolutionized our ability to map and quantify circuit abnormalities. Neural circuit measurements potentially offer the most direct window into the neural bases of psychiatric symptoms and, crucially, their modulation by treatment,” Teddy Akiki, MD, clinical scholar, Department of Psychiatry and Behavioral Sciences at Stanford, California, who works with Dr. Williams, told this news organization.
Blood-based biomarkers can complement brain imaging by providing additional information to better target treatment, help predict side effects, and guide dosage adjustments.
Precision Tools
A team led by Alexander B. Niculescu, III, MD, PhD, has found that a panel of blood-based biomarkers can distinguish between depression and bipolar disorder, predict a person’s future risk for these disorders, and inform more tailored medication choices.
Dr. Niculescu is currently a professor of psychiatry and medical neuroscience at the Indiana University School of Medicine, Indianapolis. He will head west in September to direct the newly created Center for Precision Psychiatry at the University of Arizona College of Medicine–Phoenix.
MindX Sciences, the start-up company Dr. Niculescu cofounded, has been providing blood biomarker reports to “early adopting” doctors and patients.
“We are in the process of collecting and writing up the outcome data on the first 100 cases. The feedback we have received so far from the doctors and patients who have used it, as well as biopharma companies who have used it, has been very positive,” Dr. Niculescu told this news organization.
Another benefit of precision psychiatry lies in its potential to significantly accelerate drug development.
“By identifying specific neural circuits involved in subtypes of psychiatric conditions, we can repurpose or develop drugs that target these circuits more precisely. This approach allows for smaller, more focused trials with potentially higher success rates, which could speed up the typically slow and costly process of psychiatric drug development,” said Dr. Akiki.
Dr. Niculescu agreed. With precision psychiatry tools, “psychiatric drug development will become faster, cheaper, and more successful with the use of biomarkers and other precision tools,” he said.
The Future Is Already Here
The implementation and widespread adoption of precision psychiatry have several challenges.
It requires sophisticated technology and expertise, which may not be readily available in all clinical settings. Moreover, while evidence supports its use in conditions like major depression, there are fewer data on its efficacy in other psychiatric disorders, like schizophrenia.
Dr. Williams said future research should focus on expanding the evidence base for precision psychiatry across a broader range of psychiatric conditions.
Efforts to make precision tools more accessible and scalable, such as developing portable imaging technologies or more readily available biomarker tests, are also critical.
Integrating these precision tools into routine psychiatric practice will also require training and education for clinicians, as well as cost-effective solutions to make these approaches widely available.
“Mental health clinicians throughout the country are starting to employ semi-objective and objective measures in their practices, particularly self-report symptom questionnaires and pharmacogenomic assessment,” Laura Hack, MD, PhD, assistant professor, Department of Psychiatry and Behavioral Sciences, Stanford University, told this news organization.
“For precision psychiatry measures to be widely implemented, it is essential to demonstrate their reliability, clinical validity, clinical utility, and cost-effectiveness. Additionally, there is a need to develop clinical guidelines for their use, ensure that measurement tools are accessible, and educate all relevant stakeholders,” said Dr. Hack.
Right now, functional neuroimaging is used “only on a very limited basis in current clinical psychiatric practice,” Dr. Hack noted.
“We are developing standardized systems that will require less specialized expertise in functional neuroimaging and can be readily integrated into routine clinical care,” Dr. Akiki added.
Quoting William Gibson, “The future [of precision psychiatry] is already here; it’s just not evenly distributed,” said Dr. Niculescu.
Dr. Williams has disclosed relationships with One Mind PsyberGuide, Laureate Institute for Brain Research, and Et Cere Inc. Dr. Niculescu is a cofounder of MindX Sciences and is listed as inventor on a patent application filed by Indiana University. Dr. Akiki and Dr. Hack had no relevant disclosures.
A version of this article first appeared on Medscape.com.
The field of psychiatry is experiencing a transformative shift toward precision medicine, a paradigm that tailors treatment to the unique characteristics of individual patients. This approach echoes advances in fields like oncology and cardiology, where precision tools have already revolutionized patient care.
But what exactly is precision psychiatry? How does it differ from traditional psychiatry? What will it look like in clinical practice? And are we there yet?
Beyond One-Size-Fits-All
The prevailing “one-size-fits-all” approach in psychiatry, which relies heavily on subjective symptom reporting, often proves ineffective due to the broad heterogeneity of diagnostic categories. This can lead to a “trial-and-error” cycle in treatment, which is time-consuming, costly, and frustrating for both doctors and patients.
In contrast, precision psychiatry has the potential to identify subtypes of psychiatric disorders and tailor treatments using measurable, objective data.
“The data supporting the use of precision psychiatry are very promising, particularly for treatment-resistant depression,” Leanne Williams, PhD, professor in the Department of Psychiatry and Behavioral Sciences at Stanford University, Stanford, and director of the Stanford Center for Precision Mental Health and Wellness, Palo Alto, California, said in an interview with this news organization.
Using functional MRI (fMRI), Dr. Williams and her team have mapped and measured patients’ brain circuitry to identify eight “biotypes” of depression that reflect combinations of dysfunction in six different circuits of the brain.
They are using these biotypes to guide treatment decisions in the clinic, matching individual patients to more targeted and effective therapies.
“We’re offering functional MRI to directly assess brain function along with other measures, so precision psychiatry is happening, and it’s really wanted by patients and their families. And the data suggest that we can double the rate of good outcomes,” said Dr. Williams.
“Neuroimaging techniques, particularly fMRI, have revolutionized our ability to map and quantify circuit abnormalities. Neural circuit measurements potentially offer the most direct window into the neural bases of psychiatric symptoms and, crucially, their modulation by treatment,” Teddy Akiki, MD, clinical scholar, Department of Psychiatry and Behavioral Sciences at Stanford, California, who works with Dr. Williams, told this news organization.
Blood-based biomarkers can complement brain imaging by providing additional information to better target treatment, help predict side effects, and guide dosage adjustments.
Precision Tools
A team led by Alexander B. Niculescu, III, MD, PhD, has found that a panel of blood-based biomarkers can distinguish between depression and bipolar disorder, predict a person’s future risk for these disorders, and inform more tailored medication choices.
Dr. Niculescu is currently a professor of psychiatry and medical neuroscience at the Indiana University School of Medicine, Indianapolis. He will head west in September to direct the newly created Center for Precision Psychiatry at the University of Arizona College of Medicine–Phoenix.
MindX Sciences, the start-up company Dr. Niculescu cofounded, has been providing blood biomarker reports to “early adopting” doctors and patients.
“We are in the process of collecting and writing up the outcome data on the first 100 cases. The feedback we have received so far from the doctors and patients who have used it, as well as biopharma companies who have used it, has been very positive,” Dr. Niculescu told this news organization.
Another benefit of precision psychiatry lies in its potential to significantly accelerate drug development.
“By identifying specific neural circuits involved in subtypes of psychiatric conditions, we can repurpose or develop drugs that target these circuits more precisely. This approach allows for smaller, more focused trials with potentially higher success rates, which could speed up the typically slow and costly process of psychiatric drug development,” said Dr. Akiki.
Dr. Niculescu agreed. With precision psychiatry tools, “psychiatric drug development will become faster, cheaper, and more successful with the use of biomarkers and other precision tools,” he said.
The Future Is Already Here
The implementation and widespread adoption of precision psychiatry have several challenges.
It requires sophisticated technology and expertise, which may not be readily available in all clinical settings. Moreover, while evidence supports its use in conditions like major depression, there are fewer data on its efficacy in other psychiatric disorders, like schizophrenia.
Dr. Williams said future research should focus on expanding the evidence base for precision psychiatry across a broader range of psychiatric conditions.
Efforts to make precision tools more accessible and scalable, such as developing portable imaging technologies or more readily available biomarker tests, are also critical.
Integrating these precision tools into routine psychiatric practice will also require training and education for clinicians, as well as cost-effective solutions to make these approaches widely available.
“Mental health clinicians throughout the country are starting to employ semi-objective and objective measures in their practices, particularly self-report symptom questionnaires and pharmacogenomic assessment,” Laura Hack, MD, PhD, assistant professor, Department of Psychiatry and Behavioral Sciences, Stanford University, told this news organization.
“For precision psychiatry measures to be widely implemented, it is essential to demonstrate their reliability, clinical validity, clinical utility, and cost-effectiveness. Additionally, there is a need to develop clinical guidelines for their use, ensure that measurement tools are accessible, and educate all relevant stakeholders,” said Dr. Hack.
Right now, functional neuroimaging is used “only on a very limited basis in current clinical psychiatric practice,” Dr. Hack noted.
“We are developing standardized systems that will require less specialized expertise in functional neuroimaging and can be readily integrated into routine clinical care,” Dr. Akiki added.
Quoting William Gibson, “The future [of precision psychiatry] is already here; it’s just not evenly distributed,” said Dr. Niculescu.
Dr. Williams has disclosed relationships with One Mind PsyberGuide, Laureate Institute for Brain Research, and Et Cere Inc. Dr. Niculescu is a cofounder of MindX Sciences and is listed as inventor on a patent application filed by Indiana University. Dr. Akiki and Dr. Hack had no relevant disclosures.
A version of this article first appeared on Medscape.com.
The field of psychiatry is experiencing a transformative shift toward precision medicine, a paradigm that tailors treatment to the unique characteristics of individual patients. This approach echoes advances in fields like oncology and cardiology, where precision tools have already revolutionized patient care.
But what exactly is precision psychiatry? How does it differ from traditional psychiatry? What will it look like in clinical practice? And are we there yet?
Beyond One-Size-Fits-All
The prevailing “one-size-fits-all” approach in psychiatry, which relies heavily on subjective symptom reporting, often proves ineffective due to the broad heterogeneity of diagnostic categories. This can lead to a “trial-and-error” cycle in treatment, which is time-consuming, costly, and frustrating for both doctors and patients.
In contrast, precision psychiatry has the potential to identify subtypes of psychiatric disorders and tailor treatments using measurable, objective data.
“The data supporting the use of precision psychiatry are very promising, particularly for treatment-resistant depression,” Leanne Williams, PhD, professor in the Department of Psychiatry and Behavioral Sciences at Stanford University, Stanford, and director of the Stanford Center for Precision Mental Health and Wellness, Palo Alto, California, said in an interview with this news organization.
Using functional MRI (fMRI), Dr. Williams and her team have mapped and measured patients’ brain circuitry to identify eight “biotypes” of depression that reflect combinations of dysfunction in six different circuits of the brain.
They are using these biotypes to guide treatment decisions in the clinic, matching individual patients to more targeted and effective therapies.
“We’re offering functional MRI to directly assess brain function along with other measures, so precision psychiatry is happening, and it’s really wanted by patients and their families. And the data suggest that we can double the rate of good outcomes,” said Dr. Williams.
“Neuroimaging techniques, particularly fMRI, have revolutionized our ability to map and quantify circuit abnormalities. Neural circuit measurements potentially offer the most direct window into the neural bases of psychiatric symptoms and, crucially, their modulation by treatment,” Teddy Akiki, MD, clinical scholar, Department of Psychiatry and Behavioral Sciences at Stanford, California, who works with Dr. Williams, told this news organization.
Blood-based biomarkers can complement brain imaging by providing additional information to better target treatment, help predict side effects, and guide dosage adjustments.
Precision Tools
A team led by Alexander B. Niculescu, III, MD, PhD, has found that a panel of blood-based biomarkers can distinguish between depression and bipolar disorder, predict a person’s future risk for these disorders, and inform more tailored medication choices.
Dr. Niculescu is currently a professor of psychiatry and medical neuroscience at the Indiana University School of Medicine, Indianapolis. He will head west in September to direct the newly created Center for Precision Psychiatry at the University of Arizona College of Medicine–Phoenix.
MindX Sciences, the start-up company Dr. Niculescu cofounded, has been providing blood biomarker reports to “early adopting” doctors and patients.
“We are in the process of collecting and writing up the outcome data on the first 100 cases. The feedback we have received so far from the doctors and patients who have used it, as well as biopharma companies who have used it, has been very positive,” Dr. Niculescu told this news organization.
Another benefit of precision psychiatry lies in its potential to significantly accelerate drug development.
“By identifying specific neural circuits involved in subtypes of psychiatric conditions, we can repurpose or develop drugs that target these circuits more precisely. This approach allows for smaller, more focused trials with potentially higher success rates, which could speed up the typically slow and costly process of psychiatric drug development,” said Dr. Akiki.
Dr. Niculescu agreed. With precision psychiatry tools, “psychiatric drug development will become faster, cheaper, and more successful with the use of biomarkers and other precision tools,” he said.
The Future Is Already Here
The implementation and widespread adoption of precision psychiatry have several challenges.
It requires sophisticated technology and expertise, which may not be readily available in all clinical settings. Moreover, while evidence supports its use in conditions like major depression, there are fewer data on its efficacy in other psychiatric disorders, like schizophrenia.
Dr. Williams said future research should focus on expanding the evidence base for precision psychiatry across a broader range of psychiatric conditions.
Efforts to make precision tools more accessible and scalable, such as developing portable imaging technologies or more readily available biomarker tests, are also critical.
Integrating these precision tools into routine psychiatric practice will also require training and education for clinicians, as well as cost-effective solutions to make these approaches widely available.
“Mental health clinicians throughout the country are starting to employ semi-objective and objective measures in their practices, particularly self-report symptom questionnaires and pharmacogenomic assessment,” Laura Hack, MD, PhD, assistant professor, Department of Psychiatry and Behavioral Sciences, Stanford University, told this news organization.
“For precision psychiatry measures to be widely implemented, it is essential to demonstrate their reliability, clinical validity, clinical utility, and cost-effectiveness. Additionally, there is a need to develop clinical guidelines for their use, ensure that measurement tools are accessible, and educate all relevant stakeholders,” said Dr. Hack.
Right now, functional neuroimaging is used “only on a very limited basis in current clinical psychiatric practice,” Dr. Hack noted.
“We are developing standardized systems that will require less specialized expertise in functional neuroimaging and can be readily integrated into routine clinical care,” Dr. Akiki added.
Quoting William Gibson, “The future [of precision psychiatry] is already here; it’s just not evenly distributed,” said Dr. Niculescu.
Dr. Williams has disclosed relationships with One Mind PsyberGuide, Laureate Institute for Brain Research, and Et Cere Inc. Dr. Niculescu is a cofounder of MindX Sciences and is listed as inventor on a patent application filed by Indiana University. Dr. Akiki and Dr. Hack had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Do Neurology Patient Advocacy Groups Wield Too Much Power?
Advocacy groups for patients with neurologic disorders have become a common feature in the landscape of drug and device development and federal research funding allocation.
On Capitol Hill, advocates have racked up some impressive legislative wins that aim to set a federal agenda for developing new medications.
At the Food and Drug Administration (FDA), advocacy groups played a significant role in several recent high-profile and controversial approvals for drugs for Alzheimer’s disease, Duchenne muscular dystrophy (DMD), and amyotrophic lateral sclerosis (ALS).
Such gains suggest these groups are growing in power. But with these wins come questions about whether large advocacy organizations — some of which receive significant industry funding — wield too much influence.
“You need to think very carefully about how you open these processes up to greater patient involvement,” Matthew S. McCoy, PhD, assistant professor of medical ethics and health policy at the University of Pennsylvania, Philadelphia, told this news organization. It’s important not to “end up with a situation where it’s the best-connected, the most well-resourced, the most-savvy patient organizations that are able to exercise outsize influence.”
Just because a group has deep pockets does not mean that its priorities align with the disease burden. And not every patient population is represented by a professionalized patient advocacy organization, Dr. McCoy noted. “There is the potential for the rich to get richer.”
A Seat at the Table
Long ago, the FDA and the National Institutes of Health (NIH) began giving patients a seat at the table, in part because of the path blazed by AIDS activists in the late 1980s and early 1990s, said Dr. McCoy.
Patient advocacy is often visible during FDA advisory committee meetings. The agency usually allows an hour, sometimes more, for members of the public to express support or concerns about the product being reviewed. Patients and caregivers — often aided by advocacy organizations — also submit hundreds, sometimes thousands, of letters before a product review.
The Alzheimer’s Association spent years advocating for approval of the anti-amyloid agent aducanumab (Aduhelm, Biogen/Eisai). In 2020, the organization urged patients and caregivers to submit written and oral testimony to the FDA advisory panel that was reviewing the drug. Despite patients’ pleas, the panel ultimately declined to support the drug’s approval, citing safety concerns and limited evidence of efficacy.
As controversy swirled around the medication — which had the potential for life-threatening brain swelling — advocates continued to apply pressure. Going against the expert panel’s recommendation, in June 2021, the FDA granted accelerated approval prompting three of the panelists to resign in protest.
Aducanumab’s initial price — $56,000 a year — was seen as a major threat to the viability of Medicare. Still, the Alzheimer’s Association stood behind the decision to approve the drug. But by early 2024, Biogen/Eisai said they would stop selling aducanumab, citing other priorities.
Once again patient advocates showed up in March 2022 when the FDA advisers were reviewing Amylyx Pharmaceuticals’ ALS drug Relyvrio (sodium phenylbutyrate and taurursodiol). Trials had showed limited efficacy, but patients testified they would accept greater risk for a chance to be treated with the drug. The committee ultimately voted against approval; 6 months later, the FDA approved Relyvrio anyway.
In April 2024, Amylyx removed Relyvrio from the market following phase 3 trial results that showed no difference between the treatment and placebo.
The drug manufacturer Sarepta Therapeutics, which develops treatments for genetic conditions such as DMD, has a history of working with — and funding — patient advocacy groups. The company encourages nonprofits to apply for grants or sponsorship on its website. At a 2016 advisory committee, when Sarepta was seeking approval of its first DMD therapy eteplirsen (Exondys 51), 52 speakers, most from patient advocacy groups, pleaded for the drug’s approval. When the panel voted no, Sarepta mobilized families to pressure the agency. Exondys was eventually approved.
In June, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, unilaterally gave final expanded approval to Sarepta Therapeutics’ gene therapy Elevidys for DMD. Dr. Marks overrode his own FDA reviewers, who said the product lacked substantial evidence of efficacy. He acknowledged the drug had not met its primary endpoint but said he found secondary and exploratory endpoints “compelling” and cited an unmet medical need.
In an opinion piece in The Washington Post, Aaron Kesselheim, MD, JD, MPH, the director of the Program on Regulation, Therapeutics, and Law at Brigham and Women’s Hospital, Boston, Massachusetts, and a former member of the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee, questioned the approval stating that it undermined both public trust and manufacturers’ incentives to do the hard work of proving effectiveness.
Patient Voices the ‘Secret Sauce’
Drugmakers aren’t alone in seeing the value of having patients speak directly to government entities. When the Michael J. Fox Foundation wanted to gather cosponsors for the National Plan to End Parkinson’s Act, which President Joe Biden signed into law in July, it recruited and trained patients and caregivers for congressional meetings, said Ted Thompson, senior vice president of public policy at the foundation.
Having those individuals “making the personal case for how this disease affects their families ... was really the secret sauce,” in garnering a large number of cosponsors and getting legislation signed into law within 2 years of its introduction, Mr. Thompson told this news organization.
ALS advocacy groups launched a similar campaign to secure passage of the Accelerating Access to Critical Therapies for ALS Act in 2021.
Both pieces of legislation seek to set a federal agenda for developing new therapies in neurodegenerative diseases, in part by directing the FDA and NIH to fund research, engage patients more directly, and form public-private partnerships and councils to spur innovation.
But some said patient advocates are still coming far too late to the party.
“By the time you hear from patient groups at the meetings at the FDA, often the best opportunities for their input are long past,” Leah Zoe Gibson Rand, DPhil, a research scientist with the Program on Regulation, Therapeutics, and Law at Brigham and Women’s Hospital, told this news organization. There should be more focus on the patient perspective earlier in drug development and trial design.
“There are some things that the patient voice could uniquely tell the agency,” said Holly Fernandez Lynch, JD, associate professor of medical ethics and health policy at the University of Pennsylvania. Patients can give insight on what it means to live with a disease, what symptoms are particularly burdensome, and which endpoints matter.
But, she said, “listening to the patient voice cannot mean that FDA just steps aside and lets anything on the market that patients are willing to try.” Individuals “who lack good treatment options have a very good reason to want to try things that haven’t yet been proven.”
If the FDA allows drugs on the market just because patients are willing to try, “5 or 10 years down the road, it’s not at all clear that we would end up with drugs that are better, or drugs that work, or drugs that we know anything more about,” said Dr. Lynch.
Does Taking Industry Money Equal Conflicts of Interest?
Many patient advocacy organizations receive funding from drug companies, medical device makers, or other industry sources, but they aren’t always transparent about how much or from which companies, according to studies.
The Alzheimer’s Association continued to push for the approval of aducanumab, even as the group received millions of dollars from the drugmakers. The association was accused of failing to disclose the potential conflict. It still lobbied for approval, even after the FDA advisers in 2020 voted against the drug.
It is not uncommon for individuals who speak in favor of a product’s approval to receive money for transportation and/or lodging from the drug’s manufacturer. In 2018, Dr. McCoy and colleagues reported in JAMA Internal Medicine that, between 2009 and 2017, a quarter of the speakers at the Anesthetic and Analgesic Drug Products Advisory Committee had conflicts of interest (COIs), mostly from industry, and that they were not disclosed in approximately 20% of the instances.
In a 2017 study of 104 large patient advocacy organizations published in The New England Journal of Medicine, Dr. McCoy and colleagues reported that 83% had received funds from industry. At least 39% had a current or former industry executive on the governing board, and 12% had a current or former industry executive in a board leadership position. Of the 104, 38 were focused on cancer and 13 on neurologic conditions. Of these, only 12% had published policies for managing institutional COIs.
Dr. McCoy emphasized the industry’s reliance on partnering with patient groups, particularly during FDA advisory committee meetings. “The sponsors wouldn’t be paying for patients to show up and give these testimonies if they didn’t think it made a difference. The audience isn’t just panel members; it’s also agency officials and maybe elected officials as well.”
“The Fox Foundation, with a $300 million-plus budget, gets about $5-$6 million a year from industry,” said Mr. Thompson. The money is earmarked for the organization’s Parkinson’s Disease Education Consortium; none goes toward advocacy. And, “the foundation has never specifically endorsed a product or device.”
When organizations that receive industry funding back a particular product, “it does appear to be [a conflict], and whether it is an actual one or not, appearances sometimes are all that matter,” said Mr. Thompson.
Dr. Lynch said accepting industry money “is a really significant conflict.” While advocates might need that money to fund advocacy efforts or make grants to advance research priorities, the acceptance might hinder willingness to demand evidence or to complain about a product’s price tag. “You don’t want to bite the hand that feeds you, right?”
Both Dr. McCoy and Dr. Lynch said patient groups — and individual patients — should at a minimum disclose industry funding, especially when speaking at an advisory committee.
Federal agencies and members of Congress actively seek patient input when considering legislation and funding priorities. But the individuals testifying at an advisory committee aren’t likely to represent all patients, and there’s a danger that they are just the loudest voices, said Dr. McCoy.
“We need to think more carefully about how we actually understand the preferences of a big, diverse patient population,” he said.
Dr. Lynch agreed.
Within the ALS community, “a lot of people who take different perspectives than some of those that are the leading voices get shouted down, and their voices get drowned out, and they get attacked on social media,” she said.
The group may be at the table, “but they’re just one voice at the table,” she said.
Dr. McCoy reported that his wife works for the Leukemia & Lymphoma Society, a patient advocacy organization. Dr. Rand reported no relevant financial relationships. Dr. Lynch received funding from Arnold Ventures and the Greenwall Foundation for work related to the FDA and patient advocacy.
A version of this article first appeared on Medscape.com.
Advocacy groups for patients with neurologic disorders have become a common feature in the landscape of drug and device development and federal research funding allocation.
On Capitol Hill, advocates have racked up some impressive legislative wins that aim to set a federal agenda for developing new medications.
At the Food and Drug Administration (FDA), advocacy groups played a significant role in several recent high-profile and controversial approvals for drugs for Alzheimer’s disease, Duchenne muscular dystrophy (DMD), and amyotrophic lateral sclerosis (ALS).
Such gains suggest these groups are growing in power. But with these wins come questions about whether large advocacy organizations — some of which receive significant industry funding — wield too much influence.
“You need to think very carefully about how you open these processes up to greater patient involvement,” Matthew S. McCoy, PhD, assistant professor of medical ethics and health policy at the University of Pennsylvania, Philadelphia, told this news organization. It’s important not to “end up with a situation where it’s the best-connected, the most well-resourced, the most-savvy patient organizations that are able to exercise outsize influence.”
Just because a group has deep pockets does not mean that its priorities align with the disease burden. And not every patient population is represented by a professionalized patient advocacy organization, Dr. McCoy noted. “There is the potential for the rich to get richer.”
A Seat at the Table
Long ago, the FDA and the National Institutes of Health (NIH) began giving patients a seat at the table, in part because of the path blazed by AIDS activists in the late 1980s and early 1990s, said Dr. McCoy.
Patient advocacy is often visible during FDA advisory committee meetings. The agency usually allows an hour, sometimes more, for members of the public to express support or concerns about the product being reviewed. Patients and caregivers — often aided by advocacy organizations — also submit hundreds, sometimes thousands, of letters before a product review.
The Alzheimer’s Association spent years advocating for approval of the anti-amyloid agent aducanumab (Aduhelm, Biogen/Eisai). In 2020, the organization urged patients and caregivers to submit written and oral testimony to the FDA advisory panel that was reviewing the drug. Despite patients’ pleas, the panel ultimately declined to support the drug’s approval, citing safety concerns and limited evidence of efficacy.
As controversy swirled around the medication — which had the potential for life-threatening brain swelling — advocates continued to apply pressure. Going against the expert panel’s recommendation, in June 2021, the FDA granted accelerated approval prompting three of the panelists to resign in protest.
Aducanumab’s initial price — $56,000 a year — was seen as a major threat to the viability of Medicare. Still, the Alzheimer’s Association stood behind the decision to approve the drug. But by early 2024, Biogen/Eisai said they would stop selling aducanumab, citing other priorities.
Once again patient advocates showed up in March 2022 when the FDA advisers were reviewing Amylyx Pharmaceuticals’ ALS drug Relyvrio (sodium phenylbutyrate and taurursodiol). Trials had showed limited efficacy, but patients testified they would accept greater risk for a chance to be treated with the drug. The committee ultimately voted against approval; 6 months later, the FDA approved Relyvrio anyway.
In April 2024, Amylyx removed Relyvrio from the market following phase 3 trial results that showed no difference between the treatment and placebo.
The drug manufacturer Sarepta Therapeutics, which develops treatments for genetic conditions such as DMD, has a history of working with — and funding — patient advocacy groups. The company encourages nonprofits to apply for grants or sponsorship on its website. At a 2016 advisory committee, when Sarepta was seeking approval of its first DMD therapy eteplirsen (Exondys 51), 52 speakers, most from patient advocacy groups, pleaded for the drug’s approval. When the panel voted no, Sarepta mobilized families to pressure the agency. Exondys was eventually approved.
In June, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, unilaterally gave final expanded approval to Sarepta Therapeutics’ gene therapy Elevidys for DMD. Dr. Marks overrode his own FDA reviewers, who said the product lacked substantial evidence of efficacy. He acknowledged the drug had not met its primary endpoint but said he found secondary and exploratory endpoints “compelling” and cited an unmet medical need.
In an opinion piece in The Washington Post, Aaron Kesselheim, MD, JD, MPH, the director of the Program on Regulation, Therapeutics, and Law at Brigham and Women’s Hospital, Boston, Massachusetts, and a former member of the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee, questioned the approval stating that it undermined both public trust and manufacturers’ incentives to do the hard work of proving effectiveness.
Patient Voices the ‘Secret Sauce’
Drugmakers aren’t alone in seeing the value of having patients speak directly to government entities. When the Michael J. Fox Foundation wanted to gather cosponsors for the National Plan to End Parkinson’s Act, which President Joe Biden signed into law in July, it recruited and trained patients and caregivers for congressional meetings, said Ted Thompson, senior vice president of public policy at the foundation.
Having those individuals “making the personal case for how this disease affects their families ... was really the secret sauce,” in garnering a large number of cosponsors and getting legislation signed into law within 2 years of its introduction, Mr. Thompson told this news organization.
ALS advocacy groups launched a similar campaign to secure passage of the Accelerating Access to Critical Therapies for ALS Act in 2021.
Both pieces of legislation seek to set a federal agenda for developing new therapies in neurodegenerative diseases, in part by directing the FDA and NIH to fund research, engage patients more directly, and form public-private partnerships and councils to spur innovation.
But some said patient advocates are still coming far too late to the party.
“By the time you hear from patient groups at the meetings at the FDA, often the best opportunities for their input are long past,” Leah Zoe Gibson Rand, DPhil, a research scientist with the Program on Regulation, Therapeutics, and Law at Brigham and Women’s Hospital, told this news organization. There should be more focus on the patient perspective earlier in drug development and trial design.
“There are some things that the patient voice could uniquely tell the agency,” said Holly Fernandez Lynch, JD, associate professor of medical ethics and health policy at the University of Pennsylvania. Patients can give insight on what it means to live with a disease, what symptoms are particularly burdensome, and which endpoints matter.
But, she said, “listening to the patient voice cannot mean that FDA just steps aside and lets anything on the market that patients are willing to try.” Individuals “who lack good treatment options have a very good reason to want to try things that haven’t yet been proven.”
If the FDA allows drugs on the market just because patients are willing to try, “5 or 10 years down the road, it’s not at all clear that we would end up with drugs that are better, or drugs that work, or drugs that we know anything more about,” said Dr. Lynch.
Does Taking Industry Money Equal Conflicts of Interest?
Many patient advocacy organizations receive funding from drug companies, medical device makers, or other industry sources, but they aren’t always transparent about how much or from which companies, according to studies.
The Alzheimer’s Association continued to push for the approval of aducanumab, even as the group received millions of dollars from the drugmakers. The association was accused of failing to disclose the potential conflict. It still lobbied for approval, even after the FDA advisers in 2020 voted against the drug.
It is not uncommon for individuals who speak in favor of a product’s approval to receive money for transportation and/or lodging from the drug’s manufacturer. In 2018, Dr. McCoy and colleagues reported in JAMA Internal Medicine that, between 2009 and 2017, a quarter of the speakers at the Anesthetic and Analgesic Drug Products Advisory Committee had conflicts of interest (COIs), mostly from industry, and that they were not disclosed in approximately 20% of the instances.
In a 2017 study of 104 large patient advocacy organizations published in The New England Journal of Medicine, Dr. McCoy and colleagues reported that 83% had received funds from industry. At least 39% had a current or former industry executive on the governing board, and 12% had a current or former industry executive in a board leadership position. Of the 104, 38 were focused on cancer and 13 on neurologic conditions. Of these, only 12% had published policies for managing institutional COIs.
Dr. McCoy emphasized the industry’s reliance on partnering with patient groups, particularly during FDA advisory committee meetings. “The sponsors wouldn’t be paying for patients to show up and give these testimonies if they didn’t think it made a difference. The audience isn’t just panel members; it’s also agency officials and maybe elected officials as well.”
“The Fox Foundation, with a $300 million-plus budget, gets about $5-$6 million a year from industry,” said Mr. Thompson. The money is earmarked for the organization’s Parkinson’s Disease Education Consortium; none goes toward advocacy. And, “the foundation has never specifically endorsed a product or device.”
When organizations that receive industry funding back a particular product, “it does appear to be [a conflict], and whether it is an actual one or not, appearances sometimes are all that matter,” said Mr. Thompson.
Dr. Lynch said accepting industry money “is a really significant conflict.” While advocates might need that money to fund advocacy efforts or make grants to advance research priorities, the acceptance might hinder willingness to demand evidence or to complain about a product’s price tag. “You don’t want to bite the hand that feeds you, right?”
Both Dr. McCoy and Dr. Lynch said patient groups — and individual patients — should at a minimum disclose industry funding, especially when speaking at an advisory committee.
Federal agencies and members of Congress actively seek patient input when considering legislation and funding priorities. But the individuals testifying at an advisory committee aren’t likely to represent all patients, and there’s a danger that they are just the loudest voices, said Dr. McCoy.
“We need to think more carefully about how we actually understand the preferences of a big, diverse patient population,” he said.
Dr. Lynch agreed.
Within the ALS community, “a lot of people who take different perspectives than some of those that are the leading voices get shouted down, and their voices get drowned out, and they get attacked on social media,” she said.
The group may be at the table, “but they’re just one voice at the table,” she said.
Dr. McCoy reported that his wife works for the Leukemia & Lymphoma Society, a patient advocacy organization. Dr. Rand reported no relevant financial relationships. Dr. Lynch received funding from Arnold Ventures and the Greenwall Foundation for work related to the FDA and patient advocacy.
A version of this article first appeared on Medscape.com.
Advocacy groups for patients with neurologic disorders have become a common feature in the landscape of drug and device development and federal research funding allocation.
On Capitol Hill, advocates have racked up some impressive legislative wins that aim to set a federal agenda for developing new medications.
At the Food and Drug Administration (FDA), advocacy groups played a significant role in several recent high-profile and controversial approvals for drugs for Alzheimer’s disease, Duchenne muscular dystrophy (DMD), and amyotrophic lateral sclerosis (ALS).
Such gains suggest these groups are growing in power. But with these wins come questions about whether large advocacy organizations — some of which receive significant industry funding — wield too much influence.
“You need to think very carefully about how you open these processes up to greater patient involvement,” Matthew S. McCoy, PhD, assistant professor of medical ethics and health policy at the University of Pennsylvania, Philadelphia, told this news organization. It’s important not to “end up with a situation where it’s the best-connected, the most well-resourced, the most-savvy patient organizations that are able to exercise outsize influence.”
Just because a group has deep pockets does not mean that its priorities align with the disease burden. And not every patient population is represented by a professionalized patient advocacy organization, Dr. McCoy noted. “There is the potential for the rich to get richer.”
A Seat at the Table
Long ago, the FDA and the National Institutes of Health (NIH) began giving patients a seat at the table, in part because of the path blazed by AIDS activists in the late 1980s and early 1990s, said Dr. McCoy.
Patient advocacy is often visible during FDA advisory committee meetings. The agency usually allows an hour, sometimes more, for members of the public to express support or concerns about the product being reviewed. Patients and caregivers — often aided by advocacy organizations — also submit hundreds, sometimes thousands, of letters before a product review.
The Alzheimer’s Association spent years advocating for approval of the anti-amyloid agent aducanumab (Aduhelm, Biogen/Eisai). In 2020, the organization urged patients and caregivers to submit written and oral testimony to the FDA advisory panel that was reviewing the drug. Despite patients’ pleas, the panel ultimately declined to support the drug’s approval, citing safety concerns and limited evidence of efficacy.
As controversy swirled around the medication — which had the potential for life-threatening brain swelling — advocates continued to apply pressure. Going against the expert panel’s recommendation, in June 2021, the FDA granted accelerated approval prompting three of the panelists to resign in protest.
Aducanumab’s initial price — $56,000 a year — was seen as a major threat to the viability of Medicare. Still, the Alzheimer’s Association stood behind the decision to approve the drug. But by early 2024, Biogen/Eisai said they would stop selling aducanumab, citing other priorities.
Once again patient advocates showed up in March 2022 when the FDA advisers were reviewing Amylyx Pharmaceuticals’ ALS drug Relyvrio (sodium phenylbutyrate and taurursodiol). Trials had showed limited efficacy, but patients testified they would accept greater risk for a chance to be treated with the drug. The committee ultimately voted against approval; 6 months later, the FDA approved Relyvrio anyway.
In April 2024, Amylyx removed Relyvrio from the market following phase 3 trial results that showed no difference between the treatment and placebo.
The drug manufacturer Sarepta Therapeutics, which develops treatments for genetic conditions such as DMD, has a history of working with — and funding — patient advocacy groups. The company encourages nonprofits to apply for grants or sponsorship on its website. At a 2016 advisory committee, when Sarepta was seeking approval of its first DMD therapy eteplirsen (Exondys 51), 52 speakers, most from patient advocacy groups, pleaded for the drug’s approval. When the panel voted no, Sarepta mobilized families to pressure the agency. Exondys was eventually approved.
In June, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, unilaterally gave final expanded approval to Sarepta Therapeutics’ gene therapy Elevidys for DMD. Dr. Marks overrode his own FDA reviewers, who said the product lacked substantial evidence of efficacy. He acknowledged the drug had not met its primary endpoint but said he found secondary and exploratory endpoints “compelling” and cited an unmet medical need.
In an opinion piece in The Washington Post, Aaron Kesselheim, MD, JD, MPH, the director of the Program on Regulation, Therapeutics, and Law at Brigham and Women’s Hospital, Boston, Massachusetts, and a former member of the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee, questioned the approval stating that it undermined both public trust and manufacturers’ incentives to do the hard work of proving effectiveness.
Patient Voices the ‘Secret Sauce’
Drugmakers aren’t alone in seeing the value of having patients speak directly to government entities. When the Michael J. Fox Foundation wanted to gather cosponsors for the National Plan to End Parkinson’s Act, which President Joe Biden signed into law in July, it recruited and trained patients and caregivers for congressional meetings, said Ted Thompson, senior vice president of public policy at the foundation.
Having those individuals “making the personal case for how this disease affects their families ... was really the secret sauce,” in garnering a large number of cosponsors and getting legislation signed into law within 2 years of its introduction, Mr. Thompson told this news organization.
ALS advocacy groups launched a similar campaign to secure passage of the Accelerating Access to Critical Therapies for ALS Act in 2021.
Both pieces of legislation seek to set a federal agenda for developing new therapies in neurodegenerative diseases, in part by directing the FDA and NIH to fund research, engage patients more directly, and form public-private partnerships and councils to spur innovation.
But some said patient advocates are still coming far too late to the party.
“By the time you hear from patient groups at the meetings at the FDA, often the best opportunities for their input are long past,” Leah Zoe Gibson Rand, DPhil, a research scientist with the Program on Regulation, Therapeutics, and Law at Brigham and Women’s Hospital, told this news organization. There should be more focus on the patient perspective earlier in drug development and trial design.
“There are some things that the patient voice could uniquely tell the agency,” said Holly Fernandez Lynch, JD, associate professor of medical ethics and health policy at the University of Pennsylvania. Patients can give insight on what it means to live with a disease, what symptoms are particularly burdensome, and which endpoints matter.
But, she said, “listening to the patient voice cannot mean that FDA just steps aside and lets anything on the market that patients are willing to try.” Individuals “who lack good treatment options have a very good reason to want to try things that haven’t yet been proven.”
If the FDA allows drugs on the market just because patients are willing to try, “5 or 10 years down the road, it’s not at all clear that we would end up with drugs that are better, or drugs that work, or drugs that we know anything more about,” said Dr. Lynch.
Does Taking Industry Money Equal Conflicts of Interest?
Many patient advocacy organizations receive funding from drug companies, medical device makers, or other industry sources, but they aren’t always transparent about how much or from which companies, according to studies.
The Alzheimer’s Association continued to push for the approval of aducanumab, even as the group received millions of dollars from the drugmakers. The association was accused of failing to disclose the potential conflict. It still lobbied for approval, even after the FDA advisers in 2020 voted against the drug.
It is not uncommon for individuals who speak in favor of a product’s approval to receive money for transportation and/or lodging from the drug’s manufacturer. In 2018, Dr. McCoy and colleagues reported in JAMA Internal Medicine that, between 2009 and 2017, a quarter of the speakers at the Anesthetic and Analgesic Drug Products Advisory Committee had conflicts of interest (COIs), mostly from industry, and that they were not disclosed in approximately 20% of the instances.
In a 2017 study of 104 large patient advocacy organizations published in The New England Journal of Medicine, Dr. McCoy and colleagues reported that 83% had received funds from industry. At least 39% had a current or former industry executive on the governing board, and 12% had a current or former industry executive in a board leadership position. Of the 104, 38 were focused on cancer and 13 on neurologic conditions. Of these, only 12% had published policies for managing institutional COIs.
Dr. McCoy emphasized the industry’s reliance on partnering with patient groups, particularly during FDA advisory committee meetings. “The sponsors wouldn’t be paying for patients to show up and give these testimonies if they didn’t think it made a difference. The audience isn’t just panel members; it’s also agency officials and maybe elected officials as well.”
“The Fox Foundation, with a $300 million-plus budget, gets about $5-$6 million a year from industry,” said Mr. Thompson. The money is earmarked for the organization’s Parkinson’s Disease Education Consortium; none goes toward advocacy. And, “the foundation has never specifically endorsed a product or device.”
When organizations that receive industry funding back a particular product, “it does appear to be [a conflict], and whether it is an actual one or not, appearances sometimes are all that matter,” said Mr. Thompson.
Dr. Lynch said accepting industry money “is a really significant conflict.” While advocates might need that money to fund advocacy efforts or make grants to advance research priorities, the acceptance might hinder willingness to demand evidence or to complain about a product’s price tag. “You don’t want to bite the hand that feeds you, right?”
Both Dr. McCoy and Dr. Lynch said patient groups — and individual patients — should at a minimum disclose industry funding, especially when speaking at an advisory committee.
Federal agencies and members of Congress actively seek patient input when considering legislation and funding priorities. But the individuals testifying at an advisory committee aren’t likely to represent all patients, and there’s a danger that they are just the loudest voices, said Dr. McCoy.
“We need to think more carefully about how we actually understand the preferences of a big, diverse patient population,” he said.
Dr. Lynch agreed.
Within the ALS community, “a lot of people who take different perspectives than some of those that are the leading voices get shouted down, and their voices get drowned out, and they get attacked on social media,” she said.
The group may be at the table, “but they’re just one voice at the table,” she said.
Dr. McCoy reported that his wife works for the Leukemia & Lymphoma Society, a patient advocacy organization. Dr. Rand reported no relevant financial relationships. Dr. Lynch received funding from Arnold Ventures and the Greenwall Foundation for work related to the FDA and patient advocacy.
A version of this article first appeared on Medscape.com.
More Protein Is Advantageous for Elderly Patients With CKD
In older individuals with chronic kidney disease (CKD), a higher intake of animal or plant protein is associated with reduced mortality. This finding comes from an analysis of three cohorts from Spain and Sweden, the results of which were published in JAMA Network Open.
In old age, our protein requirement increases. The recommended protein intake is between 1.0 and 1.2 g per kg of actual body weight per day. For elderly patients with acute and chronic illnesses, injuries, or malnutrition, the requirement may be higher.
“While older adults may need more protein than younger persons, higher protein intake could accelerate disease progression among those with CKD, a prevalent condition in older adults that often has no cure and high morbidity and mortality,” wrote Dr. Adrián Carballo-Casla of the Aging Research Center at the Karolinska Institutet in Stockholm, Sweden, and his colleagues.
Protein Restriction
The current Kidney Disease: Improving Global Outcomes guideline recommends that patients with mild CKD (ie, stages 1 and 2) not consume more than 1.3 g/kg/day of protein. In stages 3-5 (without dialysis) of CKD, protein intake should be limited to 0.6-0.8 g/kg/day. “Such a regimen of lower protein intake has been shown to slow CKD progression rates and improve metabolic derangements in persons with CKD stages 4 and 5 not receiving dialysis,” the researchers wrote. “Insufficient evidence of the overall health impact of limiting protein intake in older persons with mild or moderate CKD, and whether this impact is different in older adults without CKD, is available.”
The authors analyzed data from three cohorts from Spain and Sweden that included 8543 participants aged at least 60 years. A total of 14,399 observations were analyzed, including 4789 participants with CKD stages 1-3 and 9610 without CKD. To capture protein intake over a longer period and minimize variations among individual study participants, the researchers arranged the data so that there was one observation per time interval for each participant. During the 10-year follow-up, 1468 deaths were documented.
“We observed an inverse association between total protein intake and mortality among participants with CKD but a somewhat weaker one than among those without CKD,” the researchers wrote.
Slightly Weaker Association
Compared with participants with a protein intake of 0.8 g/kg/day, participants with CKD who consumed 1.0 g/kg/day of protein had a 12% reduced risk for death. At an intake of 1.2 g/kg/day, the mortality risk decreased by 21%. It decreased by 27% at a protein intake of 1.4 g/kg/day. In patients without CKD, the corresponding risk reductions were 23%, 37%, and 44%.
While in participants without CKD, mortality decreased by 15% with each increase in protein intake of 0.2 g/kg/day, in patients with CKD, the decrease was only 8%.
The association did not change according to whether the protein was of animal or plant origin. The age of the study participants (ie, whether they were under or over age 75 years) also did not play a role.
Benefits Outweigh Drawbacks
The researchers pointed out that the biological effects of protein sources could depend on the total intake, as well as the proportion of plant protein in the diet. “Not only did 68% of total protein come from animal sources in our study, but also the mean protein intake was well above the current recommendations for persons with moderate CKD,” they wrote. It is therefore unclear whether the results could be extrapolated to older patients who follow a plant-based or low-protein diet.
“The stronger associations in participants without CKD suggest that the benefits of proteins may outweigh the downsides in older persons with mild or moderate CKD,” the researchers concluded.
This story was translated from the Medscape German edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
In older individuals with chronic kidney disease (CKD), a higher intake of animal or plant protein is associated with reduced mortality. This finding comes from an analysis of three cohorts from Spain and Sweden, the results of which were published in JAMA Network Open.
In old age, our protein requirement increases. The recommended protein intake is between 1.0 and 1.2 g per kg of actual body weight per day. For elderly patients with acute and chronic illnesses, injuries, or malnutrition, the requirement may be higher.
“While older adults may need more protein than younger persons, higher protein intake could accelerate disease progression among those with CKD, a prevalent condition in older adults that often has no cure and high morbidity and mortality,” wrote Dr. Adrián Carballo-Casla of the Aging Research Center at the Karolinska Institutet in Stockholm, Sweden, and his colleagues.
Protein Restriction
The current Kidney Disease: Improving Global Outcomes guideline recommends that patients with mild CKD (ie, stages 1 and 2) not consume more than 1.3 g/kg/day of protein. In stages 3-5 (without dialysis) of CKD, protein intake should be limited to 0.6-0.8 g/kg/day. “Such a regimen of lower protein intake has been shown to slow CKD progression rates and improve metabolic derangements in persons with CKD stages 4 and 5 not receiving dialysis,” the researchers wrote. “Insufficient evidence of the overall health impact of limiting protein intake in older persons with mild or moderate CKD, and whether this impact is different in older adults without CKD, is available.”
The authors analyzed data from three cohorts from Spain and Sweden that included 8543 participants aged at least 60 years. A total of 14,399 observations were analyzed, including 4789 participants with CKD stages 1-3 and 9610 without CKD. To capture protein intake over a longer period and minimize variations among individual study participants, the researchers arranged the data so that there was one observation per time interval for each participant. During the 10-year follow-up, 1468 deaths were documented.
“We observed an inverse association between total protein intake and mortality among participants with CKD but a somewhat weaker one than among those without CKD,” the researchers wrote.
Slightly Weaker Association
Compared with participants with a protein intake of 0.8 g/kg/day, participants with CKD who consumed 1.0 g/kg/day of protein had a 12% reduced risk for death. At an intake of 1.2 g/kg/day, the mortality risk decreased by 21%. It decreased by 27% at a protein intake of 1.4 g/kg/day. In patients without CKD, the corresponding risk reductions were 23%, 37%, and 44%.
While in participants without CKD, mortality decreased by 15% with each increase in protein intake of 0.2 g/kg/day, in patients with CKD, the decrease was only 8%.
The association did not change according to whether the protein was of animal or plant origin. The age of the study participants (ie, whether they were under or over age 75 years) also did not play a role.
Benefits Outweigh Drawbacks
The researchers pointed out that the biological effects of protein sources could depend on the total intake, as well as the proportion of plant protein in the diet. “Not only did 68% of total protein come from animal sources in our study, but also the mean protein intake was well above the current recommendations for persons with moderate CKD,” they wrote. It is therefore unclear whether the results could be extrapolated to older patients who follow a plant-based or low-protein diet.
“The stronger associations in participants without CKD suggest that the benefits of proteins may outweigh the downsides in older persons with mild or moderate CKD,” the researchers concluded.
This story was translated from the Medscape German edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
In older individuals with chronic kidney disease (CKD), a higher intake of animal or plant protein is associated with reduced mortality. This finding comes from an analysis of three cohorts from Spain and Sweden, the results of which were published in JAMA Network Open.
In old age, our protein requirement increases. The recommended protein intake is between 1.0 and 1.2 g per kg of actual body weight per day. For elderly patients with acute and chronic illnesses, injuries, or malnutrition, the requirement may be higher.
“While older adults may need more protein than younger persons, higher protein intake could accelerate disease progression among those with CKD, a prevalent condition in older adults that often has no cure and high morbidity and mortality,” wrote Dr. Adrián Carballo-Casla of the Aging Research Center at the Karolinska Institutet in Stockholm, Sweden, and his colleagues.
Protein Restriction
The current Kidney Disease: Improving Global Outcomes guideline recommends that patients with mild CKD (ie, stages 1 and 2) not consume more than 1.3 g/kg/day of protein. In stages 3-5 (without dialysis) of CKD, protein intake should be limited to 0.6-0.8 g/kg/day. “Such a regimen of lower protein intake has been shown to slow CKD progression rates and improve metabolic derangements in persons with CKD stages 4 and 5 not receiving dialysis,” the researchers wrote. “Insufficient evidence of the overall health impact of limiting protein intake in older persons with mild or moderate CKD, and whether this impact is different in older adults without CKD, is available.”
The authors analyzed data from three cohorts from Spain and Sweden that included 8543 participants aged at least 60 years. A total of 14,399 observations were analyzed, including 4789 participants with CKD stages 1-3 and 9610 without CKD. To capture protein intake over a longer period and minimize variations among individual study participants, the researchers arranged the data so that there was one observation per time interval for each participant. During the 10-year follow-up, 1468 deaths were documented.
“We observed an inverse association between total protein intake and mortality among participants with CKD but a somewhat weaker one than among those without CKD,” the researchers wrote.
Slightly Weaker Association
Compared with participants with a protein intake of 0.8 g/kg/day, participants with CKD who consumed 1.0 g/kg/day of protein had a 12% reduced risk for death. At an intake of 1.2 g/kg/day, the mortality risk decreased by 21%. It decreased by 27% at a protein intake of 1.4 g/kg/day. In patients without CKD, the corresponding risk reductions were 23%, 37%, and 44%.
While in participants without CKD, mortality decreased by 15% with each increase in protein intake of 0.2 g/kg/day, in patients with CKD, the decrease was only 8%.
The association did not change according to whether the protein was of animal or plant origin. The age of the study participants (ie, whether they were under or over age 75 years) also did not play a role.
Benefits Outweigh Drawbacks
The researchers pointed out that the biological effects of protein sources could depend on the total intake, as well as the proportion of plant protein in the diet. “Not only did 68% of total protein come from animal sources in our study, but also the mean protein intake was well above the current recommendations for persons with moderate CKD,” they wrote. It is therefore unclear whether the results could be extrapolated to older patients who follow a plant-based or low-protein diet.
“The stronger associations in participants without CKD suggest that the benefits of proteins may outweigh the downsides in older persons with mild or moderate CKD,” the researchers concluded.
This story was translated from the Medscape German edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
FROM JAMA NETWORK OPEN