User login
Practice management pearls: Advice from seasoned doctors for residents looking to start a practice
The notion that residency training falls short when it comes to preparing residents and doctors for starting their own practice is a common thread across the board, whether you’re just getting started or have been managing your own practice for years. I did a survey on LinkedIn and over 50 dermatology and plastic surgery colleagues generously provided their own personal insights and words of wisdom to help young doctors avoid common practice management problems.
I could not quote everyone, but here are some of the best tips that I received:
Choose your staff carefully – and invest in the right candidates
One of the biggest pieces of practice management advice that doctors had to offer was to hire the right employees from the beginning, even if that means spending a little more time in the hiring process. This will eliminate headaches and frustration later.

In his own practice in Palm Beach Gardens, Fla., Dr. Lickstein has chosen a stable group of staff members who are, “first and foremost, nice, compassionate, and mature,” he said. “They need to be able to relate to cosmetic and medical patients of all ages. My office manager screens them, and then we have potential candidates shadow us for at least a half-day in the office. Afterward, we seek feedback from the current staff. I also try and talk with the candidate for a while, because I’ve found that once you get them to loosen up, you can get an actual sense of how they really are.”
Along the same lines, Cincinnati plastic surgeon Alex Donath, MD, suggests incentivizing employees and giving them an active role in the hiring process. “Give everyone in the office a chance to meet new employee candidates,” he said, “as that will both give the employees a sense of involvement in the process and allow more opportunities to catch glimpses of poor interpersonal skills that could hurt your reputation.”
Many doctors stressed the importance of the interview process, detailed job descriptions, a 60-day trial period, and background checks prior to hiring. This advice goes along with the famous quote “Be slow to hire and quick to fire (in the first 60 days)” that I have seen in many business books.
Foster teamwork
Another important aspect of managing your practice is building a sense of teamwork and camaraderie among employees and other doctors. Sean Weiss, MD, a facial plastic surgeon in New Orleans, has a great team-building tip that he uses daily.
“I plan a daily morning huddle with my staff. During the huddle, we review the prior day’s performance, those patients that need following up on, and whether or not the prior day’s goals were met. We then review the patient list for the current day to identify patient needs. We look specifically for ways to improve efficiency and avoid slowing down the work flow. We also try to identify opportunities to cross-promote our offerings to increase awareness of our services. In about 10 minutes, the entire team becomes focused and ready for a productive day.”
For Lacey Elwyn, DO, making staff feel appreciated can be as simple as telling them thank you on a regular basis. “The success of a dermatology practice encompasses every staff member of the team,” Dr. Elwyn, a medical and cosmetic dermatologist in South Florida, said. “The physician should respect and value all staff members. Tell them when they are doing a great job and tell them that you appreciate them every day, but also let them know right away when something is wrong.”
Don’t forget about patient education
Janet Trowbridge, MD, PhD, who practices in Edmond, Wash., expressed a great point that not only do patients need to be educated about their medical or cosmetic concerns, but they also need to be educated about the way that health care works in general. “I would say that 50% of my time as a physician is spent educating patients not about their disease, but about how medicine works – or doesn’t work,” she said. “I am constantly amazed by how little the average person understands about how health care is delivered. I talk about copays, coinsurance, annual deductibles, and why their prescriptions are not being covered. Patients feel that the system has let them down.”
Play the dual role of doctor and businessperson
At the end of the day, if you are managing your own practice, you must be able to split your time and skill set between being a physician and being a businessperson. Having realized the importance of the business aspect of running a practice, Justin Bryant, DO, a plastic and reconstructive surgeon in Walled Lake, Mich., enrolled in a dual-degree program during medical school in order to obtain his MBA.
“That investment already has proven priceless, as I’ve helped attendings and colleagues with their practice in marketing, finance, technology, and simply in translating business terms and contracts with physicians,” he said. “Although I don’t think it’s necessary for all physicians to pursue an MBA, and it’s not the answer to every business problem in the field of medicine, when applied, it can be very powerful!”
Build and protect your online reputation
Now more than ever, it is imperative to build and protect your online reputation, as online reviews can make or break your business. For plastic surgeon Nirmal Nathan, MD, in Plantation, Fla., managing your reputation is one of the most important considerations when starting a practice. “I would tell residents to start early on reputation management,” he said. “Reviews are so important, even with patients referred by word of mouth. Good reputation management also allows you to quickly ramp up if you decide to move your practice location.”
A large portion of building your online reputation now as to do with what you post (and don’t post) on social media. For Haena Kim, MD, a facial plastic and reconstructive surgeon practicing in Walnut Creek, Calif., figuring out how you would like others to perceive you is the first step.
“In this day and age of social media,” she said, “it’s so hard not to feel the pressure to follow the crowd and be the loudest person out there, and it’s incredibly hard to be patient with your practice growth. It’s important to figure out how you want to present yourself and what you want patients to come away with.”
Sweat the small stuff
Seemingly small administrative and business-related tasks can quickly add up and create much larger problems if not addressed early on. Tito Vasquez, MD, who practices in Southport, Conn., summed this up with an excellent piece of advice to remember: “Sweat the small stuff now, so you don’t have to sweat over the big stuff later.”
In terms of the “small stuff” you’ll need to manage, Dr. Vasquez points to items such as learning local economics and politics, daily finances, office regulations, and documentation, investment and planning, internal and external marketing, and human resources. “While most of us would view this as mundane or at least secondary to the craft we learn,” he said, “it will actually take far greater importance to taking care of patients if you really want your business to succeed and thrive.”
Another essential aspect of business planning that may seem daunting or mundane to many doctors when first starting out is putting together the necessary training manuals to effectively run your practice. Robert Bader, MD, stressed the importance of creating manuals for the front office, back office, Material Safety Data Sheets, and Occupational Safety and Health Administration.
“This is the time, while you have some extra time, to take an active role in forming the foundation of your practice,” Dr. Bader of Deerfield Beach, Fla., said. “Set aside time every year to go over and make necessary changes to these manuals.”
Make decisions now that reflect long-term goals
When you start your practice, deciding on a location might seem like a secondary detail, but the fact of the matter is that location will ultimately play a large role in the future of your business and your life. Beverly Hills, Calif., plastic surgeon John Layke, DO, suggested “choosing where you would like to live, and then building a practice around that location. Being happy in the area you live will make a big difference,” he says. “No one will ultimately be happy making $1 million-plus per year if they are miserable living in the area. In the beginning, share office space with reputable people where you become ‘visible,’ then build the office of your dreams when you are ready.”
Summary
I was amazed at the number of responses that I received in response to this survey. It is my goal to help doctors mentor each other on these important issues so that we do not all have to recreate the wheel. Connect with me on LinkedIn if you want to participate in these surveys or if you want to see the results of them. I want to wish the residents who are graduating and going into their own practice the best of luck. My final advice is to reach out for help – it’s obvious that many people are willing to provide advice.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems. She is the author of the monthly “Cosmeceutical Critique” column in Dermatology News.
The notion that residency training falls short when it comes to preparing residents and doctors for starting their own practice is a common thread across the board, whether you’re just getting started or have been managing your own practice for years. I did a survey on LinkedIn and over 50 dermatology and plastic surgery colleagues generously provided their own personal insights and words of wisdom to help young doctors avoid common practice management problems.
I could not quote everyone, but here are some of the best tips that I received:
Choose your staff carefully – and invest in the right candidates
One of the biggest pieces of practice management advice that doctors had to offer was to hire the right employees from the beginning, even if that means spending a little more time in the hiring process. This will eliminate headaches and frustration later.
In his own practice in Palm Beach Gardens, Fla., Dr. Lickstein has chosen a stable group of staff members who are, “first and foremost, nice, compassionate, and mature,” he said. “They need to be able to relate to cosmetic and medical patients of all ages. My office manager screens them, and then we have potential candidates shadow us for at least a half-day in the office. Afterward, we seek feedback from the current staff. I also try and talk with the candidate for a while, because I’ve found that once you get them to loosen up, you can get an actual sense of how they really are.”
Along the same lines, Cincinnati plastic surgeon Alex Donath, MD, suggests incentivizing employees and giving them an active role in the hiring process. “Give everyone in the office a chance to meet new employee candidates,” he said, “as that will both give the employees a sense of involvement in the process and allow more opportunities to catch glimpses of poor interpersonal skills that could hurt your reputation.”
Many doctors stressed the importance of the interview process, detailed job descriptions, a 60-day trial period, and background checks prior to hiring. This advice goes along with the famous quote “Be slow to hire and quick to fire (in the first 60 days)” that I have seen in many business books.
Foster teamwork
Another important aspect of managing your practice is building a sense of teamwork and camaraderie among employees and other doctors. Sean Weiss, MD, a facial plastic surgeon in New Orleans, has a great team-building tip that he uses daily.
“I plan a daily morning huddle with my staff. During the huddle, we review the prior day’s performance, those patients that need following up on, and whether or not the prior day’s goals were met. We then review the patient list for the current day to identify patient needs. We look specifically for ways to improve efficiency and avoid slowing down the work flow. We also try to identify opportunities to cross-promote our offerings to increase awareness of our services. In about 10 minutes, the entire team becomes focused and ready for a productive day.”
For Lacey Elwyn, DO, making staff feel appreciated can be as simple as telling them thank you on a regular basis. “The success of a dermatology practice encompasses every staff member of the team,” Dr. Elwyn, a medical and cosmetic dermatologist in South Florida, said. “The physician should respect and value all staff members. Tell them when they are doing a great job and tell them that you appreciate them every day, but also let them know right away when something is wrong.”
Don’t forget about patient education
Janet Trowbridge, MD, PhD, who practices in Edmond, Wash., expressed a great point that not only do patients need to be educated about their medical or cosmetic concerns, but they also need to be educated about the way that health care works in general. “I would say that 50% of my time as a physician is spent educating patients not about their disease, but about how medicine works – or doesn’t work,” she said. “I am constantly amazed by how little the average person understands about how health care is delivered. I talk about copays, coinsurance, annual deductibles, and why their prescriptions are not being covered. Patients feel that the system has let them down.”
Play the dual role of doctor and businessperson
At the end of the day, if you are managing your own practice, you must be able to split your time and skill set between being a physician and being a businessperson. Having realized the importance of the business aspect of running a practice, Justin Bryant, DO, a plastic and reconstructive surgeon in Walled Lake, Mich., enrolled in a dual-degree program during medical school in order to obtain his MBA.
“That investment already has proven priceless, as I’ve helped attendings and colleagues with their practice in marketing, finance, technology, and simply in translating business terms and contracts with physicians,” he said. “Although I don’t think it’s necessary for all physicians to pursue an MBA, and it’s not the answer to every business problem in the field of medicine, when applied, it can be very powerful!”
Build and protect your online reputation
Now more than ever, it is imperative to build and protect your online reputation, as online reviews can make or break your business. For plastic surgeon Nirmal Nathan, MD, in Plantation, Fla., managing your reputation is one of the most important considerations when starting a practice. “I would tell residents to start early on reputation management,” he said. “Reviews are so important, even with patients referred by word of mouth. Good reputation management also allows you to quickly ramp up if you decide to move your practice location.”
A large portion of building your online reputation now as to do with what you post (and don’t post) on social media. For Haena Kim, MD, a facial plastic and reconstructive surgeon practicing in Walnut Creek, Calif., figuring out how you would like others to perceive you is the first step.
“In this day and age of social media,” she said, “it’s so hard not to feel the pressure to follow the crowd and be the loudest person out there, and it’s incredibly hard to be patient with your practice growth. It’s important to figure out how you want to present yourself and what you want patients to come away with.”
Sweat the small stuff
Seemingly small administrative and business-related tasks can quickly add up and create much larger problems if not addressed early on. Tito Vasquez, MD, who practices in Southport, Conn., summed this up with an excellent piece of advice to remember: “Sweat the small stuff now, so you don’t have to sweat over the big stuff later.”
In terms of the “small stuff” you’ll need to manage, Dr. Vasquez points to items such as learning local economics and politics, daily finances, office regulations, and documentation, investment and planning, internal and external marketing, and human resources. “While most of us would view this as mundane or at least secondary to the craft we learn,” he said, “it will actually take far greater importance to taking care of patients if you really want your business to succeed and thrive.”
Another essential aspect of business planning that may seem daunting or mundane to many doctors when first starting out is putting together the necessary training manuals to effectively run your practice. Robert Bader, MD, stressed the importance of creating manuals for the front office, back office, Material Safety Data Sheets, and Occupational Safety and Health Administration.
“This is the time, while you have some extra time, to take an active role in forming the foundation of your practice,” Dr. Bader of Deerfield Beach, Fla., said. “Set aside time every year to go over and make necessary changes to these manuals.”
Make decisions now that reflect long-term goals
When you start your practice, deciding on a location might seem like a secondary detail, but the fact of the matter is that location will ultimately play a large role in the future of your business and your life. Beverly Hills, Calif., plastic surgeon John Layke, DO, suggested “choosing where you would like to live, and then building a practice around that location. Being happy in the area you live will make a big difference,” he says. “No one will ultimately be happy making $1 million-plus per year if they are miserable living in the area. In the beginning, share office space with reputable people where you become ‘visible,’ then build the office of your dreams when you are ready.”
Summary
I was amazed at the number of responses that I received in response to this survey. It is my goal to help doctors mentor each other on these important issues so that we do not all have to recreate the wheel. Connect with me on LinkedIn if you want to participate in these surveys or if you want to see the results of them. I want to wish the residents who are graduating and going into their own practice the best of luck. My final advice is to reach out for help – it’s obvious that many people are willing to provide advice.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems. She is the author of the monthly “Cosmeceutical Critique” column in Dermatology News.
The notion that residency training falls short when it comes to preparing residents and doctors for starting their own practice is a common thread across the board, whether you’re just getting started or have been managing your own practice for years. I did a survey on LinkedIn and over 50 dermatology and plastic surgery colleagues generously provided their own personal insights and words of wisdom to help young doctors avoid common practice management problems.
I could not quote everyone, but here are some of the best tips that I received:
Choose your staff carefully – and invest in the right candidates
One of the biggest pieces of practice management advice that doctors had to offer was to hire the right employees from the beginning, even if that means spending a little more time in the hiring process. This will eliminate headaches and frustration later.
In his own practice in Palm Beach Gardens, Fla., Dr. Lickstein has chosen a stable group of staff members who are, “first and foremost, nice, compassionate, and mature,” he said. “They need to be able to relate to cosmetic and medical patients of all ages. My office manager screens them, and then we have potential candidates shadow us for at least a half-day in the office. Afterward, we seek feedback from the current staff. I also try and talk with the candidate for a while, because I’ve found that once you get them to loosen up, you can get an actual sense of how they really are.”
Along the same lines, Cincinnati plastic surgeon Alex Donath, MD, suggests incentivizing employees and giving them an active role in the hiring process. “Give everyone in the office a chance to meet new employee candidates,” he said, “as that will both give the employees a sense of involvement in the process and allow more opportunities to catch glimpses of poor interpersonal skills that could hurt your reputation.”
Many doctors stressed the importance of the interview process, detailed job descriptions, a 60-day trial period, and background checks prior to hiring. This advice goes along with the famous quote “Be slow to hire and quick to fire (in the first 60 days)” that I have seen in many business books.
Foster teamwork
Another important aspect of managing your practice is building a sense of teamwork and camaraderie among employees and other doctors. Sean Weiss, MD, a facial plastic surgeon in New Orleans, has a great team-building tip that he uses daily.
“I plan a daily morning huddle with my staff. During the huddle, we review the prior day’s performance, those patients that need following up on, and whether or not the prior day’s goals were met. We then review the patient list for the current day to identify patient needs. We look specifically for ways to improve efficiency and avoid slowing down the work flow. We also try to identify opportunities to cross-promote our offerings to increase awareness of our services. In about 10 minutes, the entire team becomes focused and ready for a productive day.”
For Lacey Elwyn, DO, making staff feel appreciated can be as simple as telling them thank you on a regular basis. “The success of a dermatology practice encompasses every staff member of the team,” Dr. Elwyn, a medical and cosmetic dermatologist in South Florida, said. “The physician should respect and value all staff members. Tell them when they are doing a great job and tell them that you appreciate them every day, but also let them know right away when something is wrong.”
Don’t forget about patient education
Janet Trowbridge, MD, PhD, who practices in Edmond, Wash., expressed a great point that not only do patients need to be educated about their medical or cosmetic concerns, but they also need to be educated about the way that health care works in general. “I would say that 50% of my time as a physician is spent educating patients not about their disease, but about how medicine works – or doesn’t work,” she said. “I am constantly amazed by how little the average person understands about how health care is delivered. I talk about copays, coinsurance, annual deductibles, and why their prescriptions are not being covered. Patients feel that the system has let them down.”
Play the dual role of doctor and businessperson
At the end of the day, if you are managing your own practice, you must be able to split your time and skill set between being a physician and being a businessperson. Having realized the importance of the business aspect of running a practice, Justin Bryant, DO, a plastic and reconstructive surgeon in Walled Lake, Mich., enrolled in a dual-degree program during medical school in order to obtain his MBA.
“That investment already has proven priceless, as I’ve helped attendings and colleagues with their practice in marketing, finance, technology, and simply in translating business terms and contracts with physicians,” he said. “Although I don’t think it’s necessary for all physicians to pursue an MBA, and it’s not the answer to every business problem in the field of medicine, when applied, it can be very powerful!”
Build and protect your online reputation
Now more than ever, it is imperative to build and protect your online reputation, as online reviews can make or break your business. For plastic surgeon Nirmal Nathan, MD, in Plantation, Fla., managing your reputation is one of the most important considerations when starting a practice. “I would tell residents to start early on reputation management,” he said. “Reviews are so important, even with patients referred by word of mouth. Good reputation management also allows you to quickly ramp up if you decide to move your practice location.”
A large portion of building your online reputation now as to do with what you post (and don’t post) on social media. For Haena Kim, MD, a facial plastic and reconstructive surgeon practicing in Walnut Creek, Calif., figuring out how you would like others to perceive you is the first step.
“In this day and age of social media,” she said, “it’s so hard not to feel the pressure to follow the crowd and be the loudest person out there, and it’s incredibly hard to be patient with your practice growth. It’s important to figure out how you want to present yourself and what you want patients to come away with.”
Sweat the small stuff
Seemingly small administrative and business-related tasks can quickly add up and create much larger problems if not addressed early on. Tito Vasquez, MD, who practices in Southport, Conn., summed this up with an excellent piece of advice to remember: “Sweat the small stuff now, so you don’t have to sweat over the big stuff later.”
In terms of the “small stuff” you’ll need to manage, Dr. Vasquez points to items such as learning local economics and politics, daily finances, office regulations, and documentation, investment and planning, internal and external marketing, and human resources. “While most of us would view this as mundane or at least secondary to the craft we learn,” he said, “it will actually take far greater importance to taking care of patients if you really want your business to succeed and thrive.”
Another essential aspect of business planning that may seem daunting or mundane to many doctors when first starting out is putting together the necessary training manuals to effectively run your practice. Robert Bader, MD, stressed the importance of creating manuals for the front office, back office, Material Safety Data Sheets, and Occupational Safety and Health Administration.
“This is the time, while you have some extra time, to take an active role in forming the foundation of your practice,” Dr. Bader of Deerfield Beach, Fla., said. “Set aside time every year to go over and make necessary changes to these manuals.”
Make decisions now that reflect long-term goals
When you start your practice, deciding on a location might seem like a secondary detail, but the fact of the matter is that location will ultimately play a large role in the future of your business and your life. Beverly Hills, Calif., plastic surgeon John Layke, DO, suggested “choosing where you would like to live, and then building a practice around that location. Being happy in the area you live will make a big difference,” he says. “No one will ultimately be happy making $1 million-plus per year if they are miserable living in the area. In the beginning, share office space with reputable people where you become ‘visible,’ then build the office of your dreams when you are ready.”
Summary
I was amazed at the number of responses that I received in response to this survey. It is my goal to help doctors mentor each other on these important issues so that we do not all have to recreate the wheel. Connect with me on LinkedIn if you want to participate in these surveys or if you want to see the results of them. I want to wish the residents who are graduating and going into their own practice the best of luck. My final advice is to reach out for help – it’s obvious that many people are willing to provide advice.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems. She is the author of the monthly “Cosmeceutical Critique” column in Dermatology News.
Once-weekly Glucagon-like Peptide-1 Receptor Agonists

This supplement provides an overview of the role of once-weekly glucagon-like peptide-1 receptor agonist (GLP-1 RA) therapy in type 2 diabetes (T2D).
Topics include:
- Burden of illness in patients with T2D
- Pharmacokinetic properties and the mode and mechanism of action of GLP-1 RAs
- Safety and clinical efficacy of GLP-1 RAs
- Implications of cardiovascular outcomes trials in T2D
This supplement provides an overview of the role of once-weekly glucagon-like peptide-1 receptor agonist (GLP-1 RA) therapy in type 2 diabetes (T2D).
Topics include:
- Burden of illness in patients with T2D
- Pharmacokinetic properties and the mode and mechanism of action of GLP-1 RAs
- Safety and clinical efficacy of GLP-1 RAs
- Implications of cardiovascular outcomes trials in T2D
This supplement provides an overview of the role of once-weekly glucagon-like peptide-1 receptor agonist (GLP-1 RA) therapy in type 2 diabetes (T2D).
Topics include:
- Burden of illness in patients with T2D
- Pharmacokinetic properties and the mode and mechanism of action of GLP-1 RAs
- Safety and clinical efficacy of GLP-1 RAs
- Implications of cardiovascular outcomes trials in T2D

Emergency contraception ... and our duty to inform
In 2013, the emergency contraception containing levonorgestrel, most commonly known as Plan B, became available for purchase without prescription or age restriction. Yet, 5 years later, many adolescents and teens remain misinformed or uninformed completely. For the scope of this article, only levonorgestrel will be discussed, acknowledging that ulipristal acetate (Ella) is also an emergency contraception by prescription.
As providers we all recognize the challenges of engaging a teen patient long enough to have a meaningful conversation on health and wellness. There are even greater challenges when it comes to discussing sexual activity and sexually transmitted diseases. So the thought of discussing prevention of unwanted pregnancy may be daunting for most of us.
This topic has many layers. First and foremost, it touches on a hotly debated topic of where life begins, and emergency contraception may be thought to cross that line. Awareness of the option of emergency contraception is thought to give a free pass to promiscuous behavior. Some just feel there is not enough research to support the safe use of these products in adolescents. As with most things, taking the time to educate ourselves on the facts usually alleviates the conflicts.
Understanding levonorgestrel mechanism of action is important in clarifying its position in the prolife debate. The International Consortium for Emergency Contraception and the International Federation of Gynecologists and Obstetrics consider that inhibition or delay of ovulation is levonorgestrel’s mechanism of action, and that it does not prevent implantation of a fertilized egg. If taken after ovulation has occurred, it is ineffective in preventing pregnancy.1,2
Levonorgestrel emergency contraception was first approved by the Food and Drug Administration in 1999 under the brand name Plan B by Teva Women’s Health, then later Next Choice (Watson Pharma) was released. Initially, it was prescribed to be taken as a 0.75-mg tab within 72 hours of unprotected intercourse and repeated in 12 hours. Further studies revealed taking a 1.5-mg tab once was just as effective with no significant increase in adverse effects and Plan B One-Step was released.3
The Catholic Health Association presented a paper clarifying that levonorgestrel is not a postfertilization contraceptive (abortifacient), hopefully preventing delay of its use in victims of sexual assault seen in Catholic health care facilities.4
Safety for this product since its release has shown no deaths or serious complications.2 The most common side effect is nausea, usually without vomiting.2 Antinausea medication given 1 hour before can be helpful but is not routinely used. The length of menstrual cycle is shorter if given early in cycle but it may be lengthened by 2 days if taken post ovulation. It is not intended for repeated use, but 11 studies showed no adverse effects when it was used repeatedly in the same ovulatory cycle, and it was shown to be safe.2
For women whose emergency contraception failed, one study of 332 pregnant women who had used levonorgestrel found no teratogenic effect or risk of birth defects.5 Although it is not contraindicated in breastfeeding mothers, it was recommended that patients discontinue breastfeeding for 8 hours post ingestion. Recognized contraindications to oral contraceptives do not apply to levonorgestrel, given the temporary and relative low exposure to the hormone.3
As for efficacy, timing is of the essence. As stated previously, if not taken before ovulation has occurred, it is ineffective. If taken within 72 hours of unprotected intercourse, one study showed levonorgestrel would prevent 85% of pregnancies that otherwise might have occurred.3 Although the package insert says it must taken within 72 hours, studies have shown protection up to 120 hours post coitus, but that efficacy declines with every hour. Body mass index also may play a role in effectiveness, but the studies have been varied and more research is required before a determination is made.2

The annual well visit is the opportune time to educate parents and teens about abstinence, sex, sexually transmitted infections, and emergency contraception. Parents need to know the statistics of teen pregnancy and rates of STIs so they can be informed and further these conversations at home. should they find themselves in this dilemma. The websites not-2-late.com and bedsider.org are excellent sources of information on emergency contraception.
Keep in mind that 10% of all unintended pregnancies occur from nonconsensual intercourse so knowing what options are available is critical. Whether you give a handout with the information or undertake a more in-depth conversation during well visits, this is vital information that can change a person’s life.
Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. “Emergency Contraception: Questions And Answers For Decision-Makers,” International Consortium for Emergency Contraception, 2013.
2. Clin Obstet Gynecol. 2014 Dec;57(4):741-50.
3. Pediatrics 2012;130:1174-82.
4. Health Progress. 2010 Jan-Feb. 59-61.
5. Hum Reprod. 2009 Jul;24(7):1605-11.
6. Contraception. 2016 Feb;93(2):145-52.
In 2013, the emergency contraception containing levonorgestrel, most commonly known as Plan B, became available for purchase without prescription or age restriction. Yet, 5 years later, many adolescents and teens remain misinformed or uninformed completely. For the scope of this article, only levonorgestrel will be discussed, acknowledging that ulipristal acetate (Ella) is also an emergency contraception by prescription.
As providers we all recognize the challenges of engaging a teen patient long enough to have a meaningful conversation on health and wellness. There are even greater challenges when it comes to discussing sexual activity and sexually transmitted diseases. So the thought of discussing prevention of unwanted pregnancy may be daunting for most of us.
This topic has many layers. First and foremost, it touches on a hotly debated topic of where life begins, and emergency contraception may be thought to cross that line. Awareness of the option of emergency contraception is thought to give a free pass to promiscuous behavior. Some just feel there is not enough research to support the safe use of these products in adolescents. As with most things, taking the time to educate ourselves on the facts usually alleviates the conflicts.
Understanding levonorgestrel mechanism of action is important in clarifying its position in the prolife debate. The International Consortium for Emergency Contraception and the International Federation of Gynecologists and Obstetrics consider that inhibition or delay of ovulation is levonorgestrel’s mechanism of action, and that it does not prevent implantation of a fertilized egg. If taken after ovulation has occurred, it is ineffective in preventing pregnancy.1,2
Levonorgestrel emergency contraception was first approved by the Food and Drug Administration in 1999 under the brand name Plan B by Teva Women’s Health, then later Next Choice (Watson Pharma) was released. Initially, it was prescribed to be taken as a 0.75-mg tab within 72 hours of unprotected intercourse and repeated in 12 hours. Further studies revealed taking a 1.5-mg tab once was just as effective with no significant increase in adverse effects and Plan B One-Step was released.3
The Catholic Health Association presented a paper clarifying that levonorgestrel is not a postfertilization contraceptive (abortifacient), hopefully preventing delay of its use in victims of sexual assault seen in Catholic health care facilities.4
Safety for this product since its release has shown no deaths or serious complications.2 The most common side effect is nausea, usually without vomiting.2 Antinausea medication given 1 hour before can be helpful but is not routinely used. The length of menstrual cycle is shorter if given early in cycle but it may be lengthened by 2 days if taken post ovulation. It is not intended for repeated use, but 11 studies showed no adverse effects when it was used repeatedly in the same ovulatory cycle, and it was shown to be safe.2
For women whose emergency contraception failed, one study of 332 pregnant women who had used levonorgestrel found no teratogenic effect or risk of birth defects.5 Although it is not contraindicated in breastfeeding mothers, it was recommended that patients discontinue breastfeeding for 8 hours post ingestion. Recognized contraindications to oral contraceptives do not apply to levonorgestrel, given the temporary and relative low exposure to the hormone.3
As for efficacy, timing is of the essence. As stated previously, if not taken before ovulation has occurred, it is ineffective. If taken within 72 hours of unprotected intercourse, one study showed levonorgestrel would prevent 85% of pregnancies that otherwise might have occurred.3 Although the package insert says it must taken within 72 hours, studies have shown protection up to 120 hours post coitus, but that efficacy declines with every hour. Body mass index also may play a role in effectiveness, but the studies have been varied and more research is required before a determination is made.2
The annual well visit is the opportune time to educate parents and teens about abstinence, sex, sexually transmitted infections, and emergency contraception. Parents need to know the statistics of teen pregnancy and rates of STIs so they can be informed and further these conversations at home. should they find themselves in this dilemma. The websites not-2-late.com and bedsider.org are excellent sources of information on emergency contraception.
Keep in mind that 10% of all unintended pregnancies occur from nonconsensual intercourse so knowing what options are available is critical. Whether you give a handout with the information or undertake a more in-depth conversation during well visits, this is vital information that can change a person’s life.
Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. “Emergency Contraception: Questions And Answers For Decision-Makers,” International Consortium for Emergency Contraception, 2013.
2. Clin Obstet Gynecol. 2014 Dec;57(4):741-50.
3. Pediatrics 2012;130:1174-82.
4. Health Progress. 2010 Jan-Feb. 59-61.
5. Hum Reprod. 2009 Jul;24(7):1605-11.
6. Contraception. 2016 Feb;93(2):145-52.
In 2013, the emergency contraception containing levonorgestrel, most commonly known as Plan B, became available for purchase without prescription or age restriction. Yet, 5 years later, many adolescents and teens remain misinformed or uninformed completely. For the scope of this article, only levonorgestrel will be discussed, acknowledging that ulipristal acetate (Ella) is also an emergency contraception by prescription.
As providers we all recognize the challenges of engaging a teen patient long enough to have a meaningful conversation on health and wellness. There are even greater challenges when it comes to discussing sexual activity and sexually transmitted diseases. So the thought of discussing prevention of unwanted pregnancy may be daunting for most of us.
This topic has many layers. First and foremost, it touches on a hotly debated topic of where life begins, and emergency contraception may be thought to cross that line. Awareness of the option of emergency contraception is thought to give a free pass to promiscuous behavior. Some just feel there is not enough research to support the safe use of these products in adolescents. As with most things, taking the time to educate ourselves on the facts usually alleviates the conflicts.
Understanding levonorgestrel mechanism of action is important in clarifying its position in the prolife debate. The International Consortium for Emergency Contraception and the International Federation of Gynecologists and Obstetrics consider that inhibition or delay of ovulation is levonorgestrel’s mechanism of action, and that it does not prevent implantation of a fertilized egg. If taken after ovulation has occurred, it is ineffective in preventing pregnancy.1,2
Levonorgestrel emergency contraception was first approved by the Food and Drug Administration in 1999 under the brand name Plan B by Teva Women’s Health, then later Next Choice (Watson Pharma) was released. Initially, it was prescribed to be taken as a 0.75-mg tab within 72 hours of unprotected intercourse and repeated in 12 hours. Further studies revealed taking a 1.5-mg tab once was just as effective with no significant increase in adverse effects and Plan B One-Step was released.3
The Catholic Health Association presented a paper clarifying that levonorgestrel is not a postfertilization contraceptive (abortifacient), hopefully preventing delay of its use in victims of sexual assault seen in Catholic health care facilities.4
Safety for this product since its release has shown no deaths or serious complications.2 The most common side effect is nausea, usually without vomiting.2 Antinausea medication given 1 hour before can be helpful but is not routinely used. The length of menstrual cycle is shorter if given early in cycle but it may be lengthened by 2 days if taken post ovulation. It is not intended for repeated use, but 11 studies showed no adverse effects when it was used repeatedly in the same ovulatory cycle, and it was shown to be safe.2
For women whose emergency contraception failed, one study of 332 pregnant women who had used levonorgestrel found no teratogenic effect or risk of birth defects.5 Although it is not contraindicated in breastfeeding mothers, it was recommended that patients discontinue breastfeeding for 8 hours post ingestion. Recognized contraindications to oral contraceptives do not apply to levonorgestrel, given the temporary and relative low exposure to the hormone.3
As for efficacy, timing is of the essence. As stated previously, if not taken before ovulation has occurred, it is ineffective. If taken within 72 hours of unprotected intercourse, one study showed levonorgestrel would prevent 85% of pregnancies that otherwise might have occurred.3 Although the package insert says it must taken within 72 hours, studies have shown protection up to 120 hours post coitus, but that efficacy declines with every hour. Body mass index also may play a role in effectiveness, but the studies have been varied and more research is required before a determination is made.2
The annual well visit is the opportune time to educate parents and teens about abstinence, sex, sexually transmitted infections, and emergency contraception. Parents need to know the statistics of teen pregnancy and rates of STIs so they can be informed and further these conversations at home. should they find themselves in this dilemma. The websites not-2-late.com and bedsider.org are excellent sources of information on emergency contraception.
Keep in mind that 10% of all unintended pregnancies occur from nonconsensual intercourse so knowing what options are available is critical. Whether you give a handout with the information or undertake a more in-depth conversation during well visits, this is vital information that can change a person’s life.
Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. “Emergency Contraception: Questions And Answers For Decision-Makers,” International Consortium for Emergency Contraception, 2013.
2. Clin Obstet Gynecol. 2014 Dec;57(4):741-50.
3. Pediatrics 2012;130:1174-82.
4. Health Progress. 2010 Jan-Feb. 59-61.
5. Hum Reprod. 2009 Jul;24(7):1605-11.
6. Contraception. 2016 Feb;93(2):145-52.
Psoriasis therapy with biologics not linked to increased cancer risk
Biologic treatments were not associated with an increased risk of cancer among patients with psoriasis in the medium term, in a study that analyzed data from patient registries.
“Cumulative length of exposure to biologics was not associated with the risk of developing cancers, even after controlling for the effect of age, gender, location,” as well as for previous exposure to methotrexate, cyclosporine, and phototherapy; duration of psoriasis; and comorbidities, reported Ignacio García-Doval, MD, of the Fundación Academia Española de Dermatología y Venereología, Madrid, and his associates.
The pooled adjusted odds ratio of cancer per year of biologic exposure was 1.02 (95% confidence interval, 0.92-1.13), demonstrating no significantly increased risk of cancer per cumulative year of biologic exposure for psoriasis therapy, Dr. García-Doval and his associates reported in the study, published in the British Journal of Dermatology. This was true even when broken down within the registries for comparison, and when analyzed by type of cancers, such as squamous cell carcinoma and basal cell carcinoma.
A limitation of the study was inadequate power to detect and compare risk between individual biologics, they said. Also, “as our data describe limited follow-up and latencies, it is still possible that a risk after longer periods of exposure and latencies exists.”
Most of the authors had numerous financial disclosures related to pharmaceutical companies. Psonet was supported with funds from the European Association of Venereology and Dermatology and the Italian Drug Agency. Funding for the individual registries includes support from pharmaceutical companies.
SOURCE: García-Doval I et al. Br J Dermatol. 2018 May 3. doi: 10.1111/bjd.16715.
Biologic treatments were not associated with an increased risk of cancer among patients with psoriasis in the medium term, in a study that analyzed data from patient registries.
“Cumulative length of exposure to biologics was not associated with the risk of developing cancers, even after controlling for the effect of age, gender, location,” as well as for previous exposure to methotrexate, cyclosporine, and phototherapy; duration of psoriasis; and comorbidities, reported Ignacio García-Doval, MD, of the Fundación Academia Española de Dermatología y Venereología, Madrid, and his associates.
The pooled adjusted odds ratio of cancer per year of biologic exposure was 1.02 (95% confidence interval, 0.92-1.13), demonstrating no significantly increased risk of cancer per cumulative year of biologic exposure for psoriasis therapy, Dr. García-Doval and his associates reported in the study, published in the British Journal of Dermatology. This was true even when broken down within the registries for comparison, and when analyzed by type of cancers, such as squamous cell carcinoma and basal cell carcinoma.
A limitation of the study was inadequate power to detect and compare risk between individual biologics, they said. Also, “as our data describe limited follow-up and latencies, it is still possible that a risk after longer periods of exposure and latencies exists.”
Most of the authors had numerous financial disclosures related to pharmaceutical companies. Psonet was supported with funds from the European Association of Venereology and Dermatology and the Italian Drug Agency. Funding for the individual registries includes support from pharmaceutical companies.
SOURCE: García-Doval I et al. Br J Dermatol. 2018 May 3. doi: 10.1111/bjd.16715.
Biologic treatments were not associated with an increased risk of cancer among patients with psoriasis in the medium term, in a study that analyzed data from patient registries.
“Cumulative length of exposure to biologics was not associated with the risk of developing cancers, even after controlling for the effect of age, gender, location,” as well as for previous exposure to methotrexate, cyclosporine, and phototherapy; duration of psoriasis; and comorbidities, reported Ignacio García-Doval, MD, of the Fundación Academia Española de Dermatología y Venereología, Madrid, and his associates.
The pooled adjusted odds ratio of cancer per year of biologic exposure was 1.02 (95% confidence interval, 0.92-1.13), demonstrating no significantly increased risk of cancer per cumulative year of biologic exposure for psoriasis therapy, Dr. García-Doval and his associates reported in the study, published in the British Journal of Dermatology. This was true even when broken down within the registries for comparison, and when analyzed by type of cancers, such as squamous cell carcinoma and basal cell carcinoma.
A limitation of the study was inadequate power to detect and compare risk between individual biologics, they said. Also, “as our data describe limited follow-up and latencies, it is still possible that a risk after longer periods of exposure and latencies exists.”
Most of the authors had numerous financial disclosures related to pharmaceutical companies. Psonet was supported with funds from the European Association of Venereology and Dermatology and the Italian Drug Agency. Funding for the individual registries includes support from pharmaceutical companies.
SOURCE: García-Doval I et al. Br J Dermatol. 2018 May 3. doi: 10.1111/bjd.16715.
FROM THE BRITISH JOURNAL OF DERMATOLOGY
Key clinical point:
Major finding: The pooled adjusted odds ratio of cancer per year of biologic exposure was 1.02.
Study details: Patient data were drawn from four national databases within Psonet, which included 579 cancer cases and 2,671 matched controls.
Disclosures: Most of the authors had numerous financial disclosures related to pharmaceutical companies. Psonet was supported with funds from the European Association of Venereology and Dermatology and the Italian Drug Agency. Funding for the individual registries includes support from pharmaceutical companies.
Source: García-Doval I et al. Br J Dermatol. 2018 May 3. doi: 10.1111/bjd.16715.
Uterine fibroids registry looks to provide comparative treatment data
Early findings from a uterine fibroids registry suggest more than half of enrolled women chose an alternative to hysterectomy, underscoring the need to study the efficacy of these uterine-sparing treatment options, said Elizabeth A. Stewart, MD, of the Mayo Clinic, Rochester, Minn., and her associates.
There is very little evidence on the comparative efficacy of uterine fibroid treatment options so the COMPARE-UF (Comparing Options for Management: Patient-Centered Results for Uterine Fibroids) registry was initiated for women who choose procedural therapy for symptomatic uterine fibroids. Data at the nine clinical centers – representing rural and urban populations – are being collected regarding hysterectomy, myomectomy (abdominal, hysteroscopic, vaginal, and laparoscopic/robotic), endometrial ablation, radiofrequency fibroid ablation, uterine artery embolization, magnetic resonance–guided focused ultrasound, and therapeutic use of progestin-releasing intrauterine devices.
A total of 16% of the women were under age 35, and 40% were aged 40 years or younger. “The fact that a sizable proportion of women are under age 40 suggests that with long-term follow-up, data on reproductive outcomes can be obtained as these women seek pregnancy,” Dr. Stewart and her associates noted.
Although African American women make up only 13% of the U.S. population, they constitute 42% of enrollment in COMPARE-UF, the investigators reported in the American Journal of Obstetrics and Gynecology. African American women chose similar or greater numbers of each type of myomectomy and uterine embolization as white women chose.
The study was supported by a grant from the Agency for Healthcare Research and Quality. The registry is supported by AHRQ and the Patient-Centered Outcomes Research Institute. Dr. Stewart reported personal fees from AbbVie, Allergan, Astellas, Pharma, Bayer, Gynesonics, and Myovant Sciences. Several researchers reported grants from pharmaceutical companies outside this study, and the remainder of the investigators reported no relevant financial disclosures.
SOURCE: Stewart EA et al. Am J Obstet Gynecol. 2018 May 8. doi: 10.1016/j.ajog.2018.05.004.
Early findings from a uterine fibroids registry suggest more than half of enrolled women chose an alternative to hysterectomy, underscoring the need to study the efficacy of these uterine-sparing treatment options, said Elizabeth A. Stewart, MD, of the Mayo Clinic, Rochester, Minn., and her associates.
There is very little evidence on the comparative efficacy of uterine fibroid treatment options so the COMPARE-UF (Comparing Options for Management: Patient-Centered Results for Uterine Fibroids) registry was initiated for women who choose procedural therapy for symptomatic uterine fibroids. Data at the nine clinical centers – representing rural and urban populations – are being collected regarding hysterectomy, myomectomy (abdominal, hysteroscopic, vaginal, and laparoscopic/robotic), endometrial ablation, radiofrequency fibroid ablation, uterine artery embolization, magnetic resonance–guided focused ultrasound, and therapeutic use of progestin-releasing intrauterine devices.
A total of 16% of the women were under age 35, and 40% were aged 40 years or younger. “The fact that a sizable proportion of women are under age 40 suggests that with long-term follow-up, data on reproductive outcomes can be obtained as these women seek pregnancy,” Dr. Stewart and her associates noted.
Although African American women make up only 13% of the U.S. population, they constitute 42% of enrollment in COMPARE-UF, the investigators reported in the American Journal of Obstetrics and Gynecology. African American women chose similar or greater numbers of each type of myomectomy and uterine embolization as white women chose.
The study was supported by a grant from the Agency for Healthcare Research and Quality. The registry is supported by AHRQ and the Patient-Centered Outcomes Research Institute. Dr. Stewart reported personal fees from AbbVie, Allergan, Astellas, Pharma, Bayer, Gynesonics, and Myovant Sciences. Several researchers reported grants from pharmaceutical companies outside this study, and the remainder of the investigators reported no relevant financial disclosures.
SOURCE: Stewart EA et al. Am J Obstet Gynecol. 2018 May 8. doi: 10.1016/j.ajog.2018.05.004.
Early findings from a uterine fibroids registry suggest more than half of enrolled women chose an alternative to hysterectomy, underscoring the need to study the efficacy of these uterine-sparing treatment options, said Elizabeth A. Stewart, MD, of the Mayo Clinic, Rochester, Minn., and her associates.
There is very little evidence on the comparative efficacy of uterine fibroid treatment options so the COMPARE-UF (Comparing Options for Management: Patient-Centered Results for Uterine Fibroids) registry was initiated for women who choose procedural therapy for symptomatic uterine fibroids. Data at the nine clinical centers – representing rural and urban populations – are being collected regarding hysterectomy, myomectomy (abdominal, hysteroscopic, vaginal, and laparoscopic/robotic), endometrial ablation, radiofrequency fibroid ablation, uterine artery embolization, magnetic resonance–guided focused ultrasound, and therapeutic use of progestin-releasing intrauterine devices.
A total of 16% of the women were under age 35, and 40% were aged 40 years or younger. “The fact that a sizable proportion of women are under age 40 suggests that with long-term follow-up, data on reproductive outcomes can be obtained as these women seek pregnancy,” Dr. Stewart and her associates noted.
Although African American women make up only 13% of the U.S. population, they constitute 42% of enrollment in COMPARE-UF, the investigators reported in the American Journal of Obstetrics and Gynecology. African American women chose similar or greater numbers of each type of myomectomy and uterine embolization as white women chose.
The study was supported by a grant from the Agency for Healthcare Research and Quality. The registry is supported by AHRQ and the Patient-Centered Outcomes Research Institute. Dr. Stewart reported personal fees from AbbVie, Allergan, Astellas, Pharma, Bayer, Gynesonics, and Myovant Sciences. Several researchers reported grants from pharmaceutical companies outside this study, and the remainder of the investigators reported no relevant financial disclosures.
SOURCE: Stewart EA et al. Am J Obstet Gynecol. 2018 May 8. doi: 10.1016/j.ajog.2018.05.004.
FROM THE AMERICAN JOURNAL OF OBSTETRICS AND GYNECOLOGY
Key clinical point:
Major finding: Of the initial 2,031 women enrolled in the registry, hysterectomy was chosen by 38%, and myomectomies were chosen by 46%.
Study details: Initial results from the COMPARE-UF registry.
Disclosures: The study was supported by a grant from the Agency for Healthcare Research and Quality. Dr. Stewart reported personal fees from AbbVie, Allergan, Astellas, Pharma, Bayer, Gynesonics, and Myovant Sciences. Several researchers reported grants from pharmaceutical companies outside this study, and the remainder of the investigators reported no relevant financial disclosures.
Source: Stewart EA et al. Am J Obstet Gynecol. 2018 May 8. doi: 10.1016/j.ajog.2018.05.004.
Dermatologic complaints prolong hospital stay for hematologic cancer
and are associated with an increased mean length of hospital stay, according to a large retrospective chart review from a major cancer center.
The substantial burden imposed by dermatologic complications in patients with cancer, particularly a hematologic malignancy, highlights the importance of greater collaboration between oncology and dermatology services to mitigate the impact of these events on both quality of life and outcome, wrote Gregory S. Phillips of Memorial Sloan Kettering Cancer Center, New York, and his associates. The study was published in the Journal of the American Academy of Dermatology.
The data that produced these conclusions were drawn from a retrospective chart review of 11,533 cancer patients treated at the center during 2015. Of these, 412 (3.6%) were referred for a dermatology consultation.
Those who received a dermatology consultation were comparable for median age (60 years) and gender (roughly 50:50 male:female), compared with those who did not. However, the odds ratio (OR) for a dermatology consultation was 6.56 among those with a hematologic malignancy compared with those who had a solid tumor. In those with leukemia, the proportion receiving a dermatologic consult was nearly ninefold greater.
Whether or not undertaken in a patient with a hematologic malignancy, dermatologic consults correlated with significantly greater morbidity, as well as mortality. This included a longer median length of stay (11 vs. 5 days; P less than .0001) and a higher in-hospital rate of death (9% vs. 2%; P less than .0001), compared with patients not needing a dermatology consultation.
Of dermatologic consultations in the total study population, the most common were for inflammatory conditions (27%), infections (24%), and drug reactions (17%). Neoplasm was the dermatologic diagnosis in 10% of the total population, but in 13% of those with hematologic malignancies.
Inpatient dermatology consultations were most frequently ordered by the hematology-oncology service, accounting for 44% of the total, followed by the solid tumor oncology service (27%) and the surgery service (15%). Multiple consultations were more likely in patients with leukemia or lymphoma than other forms of cancer.
In the dermatology consult, biopsy was employed for diagnosis in only 18%. As for treatment, 42% received topical therapy alone, and 38% received a systemic therapy. Dermatologic consultations that subsequently involved consultation with another service such as allergy and immunology, were rare, occurring in only 4% of cases.

Furthermore, they suggested that the data support increased attention to dermatologic complications in cancer. Although the impact of consultations on outcome was not evaluated in this study, the authors cited another recent study in which there was a more than 2-day reduction in hospital stay when a dermatology consultation was employed in noncancer patients with an inflammatory skin disease (JAMA Dermatol. 2017 Jun 1;153[6]:523-8). Moreover, they speculated that a prompt resolution of dermatologic complaints in cancer patients has implications for better outcomes if they result in fewer delays in anti-cancer therapy.
The study was partly funded by the a grant from the National Cancer Institute’s Cancer Centers Program. Dr. Lacouture reported financial relationships with AstraZeneca, Adgero Biopharmaceuticals, Berg, Bristol-Myers Squibb, Foamix, Janssen, Legacy Healthcare, NovoCure, and Quintiles. Another author reported ties with Amgen, Roche, Eaisi, and P Value Communications.
SOURCE: Phillips GS et al. J Am Acad Dermatol. 2018 Jun;78(6):1102-9.
and are associated with an increased mean length of hospital stay, according to a large retrospective chart review from a major cancer center.
The substantial burden imposed by dermatologic complications in patients with cancer, particularly a hematologic malignancy, highlights the importance of greater collaboration between oncology and dermatology services to mitigate the impact of these events on both quality of life and outcome, wrote Gregory S. Phillips of Memorial Sloan Kettering Cancer Center, New York, and his associates. The study was published in the Journal of the American Academy of Dermatology.
The data that produced these conclusions were drawn from a retrospective chart review of 11,533 cancer patients treated at the center during 2015. Of these, 412 (3.6%) were referred for a dermatology consultation.
Those who received a dermatology consultation were comparable for median age (60 years) and gender (roughly 50:50 male:female), compared with those who did not. However, the odds ratio (OR) for a dermatology consultation was 6.56 among those with a hematologic malignancy compared with those who had a solid tumor. In those with leukemia, the proportion receiving a dermatologic consult was nearly ninefold greater.
Whether or not undertaken in a patient with a hematologic malignancy, dermatologic consults correlated with significantly greater morbidity, as well as mortality. This included a longer median length of stay (11 vs. 5 days; P less than .0001) and a higher in-hospital rate of death (9% vs. 2%; P less than .0001), compared with patients not needing a dermatology consultation.
Of dermatologic consultations in the total study population, the most common were for inflammatory conditions (27%), infections (24%), and drug reactions (17%). Neoplasm was the dermatologic diagnosis in 10% of the total population, but in 13% of those with hematologic malignancies.
Inpatient dermatology consultations were most frequently ordered by the hematology-oncology service, accounting for 44% of the total, followed by the solid tumor oncology service (27%) and the surgery service (15%). Multiple consultations were more likely in patients with leukemia or lymphoma than other forms of cancer.
In the dermatology consult, biopsy was employed for diagnosis in only 18%. As for treatment, 42% received topical therapy alone, and 38% received a systemic therapy. Dermatologic consultations that subsequently involved consultation with another service such as allergy and immunology, were rare, occurring in only 4% of cases.
Furthermore, they suggested that the data support increased attention to dermatologic complications in cancer. Although the impact of consultations on outcome was not evaluated in this study, the authors cited another recent study in which there was a more than 2-day reduction in hospital stay when a dermatology consultation was employed in noncancer patients with an inflammatory skin disease (JAMA Dermatol. 2017 Jun 1;153[6]:523-8). Moreover, they speculated that a prompt resolution of dermatologic complaints in cancer patients has implications for better outcomes if they result in fewer delays in anti-cancer therapy.
The study was partly funded by the a grant from the National Cancer Institute’s Cancer Centers Program. Dr. Lacouture reported financial relationships with AstraZeneca, Adgero Biopharmaceuticals, Berg, Bristol-Myers Squibb, Foamix, Janssen, Legacy Healthcare, NovoCure, and Quintiles. Another author reported ties with Amgen, Roche, Eaisi, and P Value Communications.
SOURCE: Phillips GS et al. J Am Acad Dermatol. 2018 Jun;78(6):1102-9.
and are associated with an increased mean length of hospital stay, according to a large retrospective chart review from a major cancer center.
The substantial burden imposed by dermatologic complications in patients with cancer, particularly a hematologic malignancy, highlights the importance of greater collaboration between oncology and dermatology services to mitigate the impact of these events on both quality of life and outcome, wrote Gregory S. Phillips of Memorial Sloan Kettering Cancer Center, New York, and his associates. The study was published in the Journal of the American Academy of Dermatology.
The data that produced these conclusions were drawn from a retrospective chart review of 11,533 cancer patients treated at the center during 2015. Of these, 412 (3.6%) were referred for a dermatology consultation.
Those who received a dermatology consultation were comparable for median age (60 years) and gender (roughly 50:50 male:female), compared with those who did not. However, the odds ratio (OR) for a dermatology consultation was 6.56 among those with a hematologic malignancy compared with those who had a solid tumor. In those with leukemia, the proportion receiving a dermatologic consult was nearly ninefold greater.
Whether or not undertaken in a patient with a hematologic malignancy, dermatologic consults correlated with significantly greater morbidity, as well as mortality. This included a longer median length of stay (11 vs. 5 days; P less than .0001) and a higher in-hospital rate of death (9% vs. 2%; P less than .0001), compared with patients not needing a dermatology consultation.
Of dermatologic consultations in the total study population, the most common were for inflammatory conditions (27%), infections (24%), and drug reactions (17%). Neoplasm was the dermatologic diagnosis in 10% of the total population, but in 13% of those with hematologic malignancies.
Inpatient dermatology consultations were most frequently ordered by the hematology-oncology service, accounting for 44% of the total, followed by the solid tumor oncology service (27%) and the surgery service (15%). Multiple consultations were more likely in patients with leukemia or lymphoma than other forms of cancer.
In the dermatology consult, biopsy was employed for diagnosis in only 18%. As for treatment, 42% received topical therapy alone, and 38% received a systemic therapy. Dermatologic consultations that subsequently involved consultation with another service such as allergy and immunology, were rare, occurring in only 4% of cases.
Furthermore, they suggested that the data support increased attention to dermatologic complications in cancer. Although the impact of consultations on outcome was not evaluated in this study, the authors cited another recent study in which there was a more than 2-day reduction in hospital stay when a dermatology consultation was employed in noncancer patients with an inflammatory skin disease (JAMA Dermatol. 2017 Jun 1;153[6]:523-8). Moreover, they speculated that a prompt resolution of dermatologic complaints in cancer patients has implications for better outcomes if they result in fewer delays in anti-cancer therapy.
The study was partly funded by the a grant from the National Cancer Institute’s Cancer Centers Program. Dr. Lacouture reported financial relationships with AstraZeneca, Adgero Biopharmaceuticals, Berg, Bristol-Myers Squibb, Foamix, Janssen, Legacy Healthcare, NovoCure, and Quintiles. Another author reported ties with Amgen, Roche, Eaisi, and P Value Communications.
SOURCE: Phillips GS et al. J Am Acad Dermatol. 2018 Jun;78(6):1102-9.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: Dermatologic complications are more common in hematologic than solid tumor cancers and correlate with adverse outcomes.
Major finding: In cancer patients, dermatologic consults are associated with longer hospital stay (11 vs. 5 days) and death (9% vs. 2%).
Study details: Retrospective chart review of inpatient dermatology consultations during 2015.
Disclosures: The study was partly funded by the a grant from the National Cancer Institute’s Cancer Centers Program. Dr. Lacouture reported financial relationships with AstraZeneca, Adgero Biopharmaceuticals, Berg, Bristol-Myers Squibb, Foamix, Janssen, Legacy Healthcare, NovoCure, and Quintiles. Another author reported ties with Amgen, Roche, Eaisi, and P Value Communications.
Source: Phillips et al. J Am Acad Dermatol. 2018;78:1102-9.
MDedge Psychcast: Episode 107
Dr. Worley uses a nautical metaphor to describe the phenomenon and possible ways to combat it.
Dr. Worley uses a nautical metaphor to describe the phenomenon and possible ways to combat it.
Dr. Worley uses a nautical metaphor to describe the phenomenon and possible ways to combat it.
An MRI Analysis of the Pelvis to Determine the Ideal Method for Ultrasound-Guided Bone Marrow Aspiration from the Iliac Crest
ABSTRACT
Use of mesenchymal stem cells from bone marrow has gained significant popularity. The iliac crest has been determined to be an effective site for harvesting mesenchymal stem cells. Review of the literature reveals that multiple techniques are used to harvest bone marrow aspirate from the iliac crest, but the descriptions are based on the experience of various authors as opposed to studied anatomy. A safe, reliable, and reproducible method for aspiration has yet to be studied and described. We hypothesized that there would be an ideal angle and distance for aspiration that would be the safest, most consistent, and most reliable. Using magnetic resonance imaging (MRI), we reviewed 26 total lumbar spine MRI scans (13 males, 13 females) and found that an angle of 24° should be used when entering the most medial aspect of the posterior superior iliac spine (PSIS) and that this angle did not differ between the sexes. The distance that the trocar can advance after entry before hitting the anterior ilium wall varied significantly between males and females, being 7.53 cm in males and 6.74 cm in females. In addition, the size of the PSIS table was significantly different between males and females (1.20 cm and 0.96 cm, respectively). No other significant differences in the measurements gathered were found. Using the data gleaned from this study, we developed an aspiration technique. This method uses ultrasound to determine the location of the PSIS and the entry point on the PSIS. This contrasts with most techniques that use landmark palpation, which is known to be unreliable and inaccurate. The described technique for aspiration from the PSIS is safe, reliable, reproducible, and substantiated by data.
The iliac crest is an effective site for harvesting bone marrow stem cells. It allows for easy access and is superficial in most individuals, allowing for a relatively quick and simple procedure. Use of mesenchymal stem cells (MSCs) for treatment of orthopedic injuries has grown recently. Whereas overall use has increased, review of the literature reveals very few techniques for iliac crest aspiration,1 but these are not based on anatomic relationships or studies. Hernigou and colleagues2,3 attempted to quantitatively evaluate potential “sectors” allowing for safe aspiration using cadaver and computed tomographic reconstruction imaging. We used magnetic resonance imaging (MRI) to analyze aspiration parameters. Owing to the ilium’s anatomy, improper positioning or aspiration technique during aspiration can result in serious injury.2,4-6 We hypothesized that there is an ideal angle and positioning for bone marrow aspiration from the posterior superior iliac spine (PSIS) that is safe, consistent, and reproducible. Although most aspiration techniques use landmark palpation, this is unreliable and inaccurate, especially when compared with ultrasound-guided injections7-16 and procedures.9,12,17-19 We describe our technique using ultrasound to visualize patient anatomy and accurately determine anatomic entry with the trocar.
METHODS
MRI scans of 26 patients (13 males, 13 females) were reviewed to determine average angles and distances. Axial T2-weighted views of the lumbar spine were used in all analyses. The sacroiliac (SI) joint angle was defined as the angle formed between the vector through the midline of the pelvis and the vector that is parallel to the SI joint. The approach angle was defined as the angle formed between the vector of the most medial aspect of the PSIS through the ilium to the anterior wall and the vector through the midline of the pelvis (Figure 1). 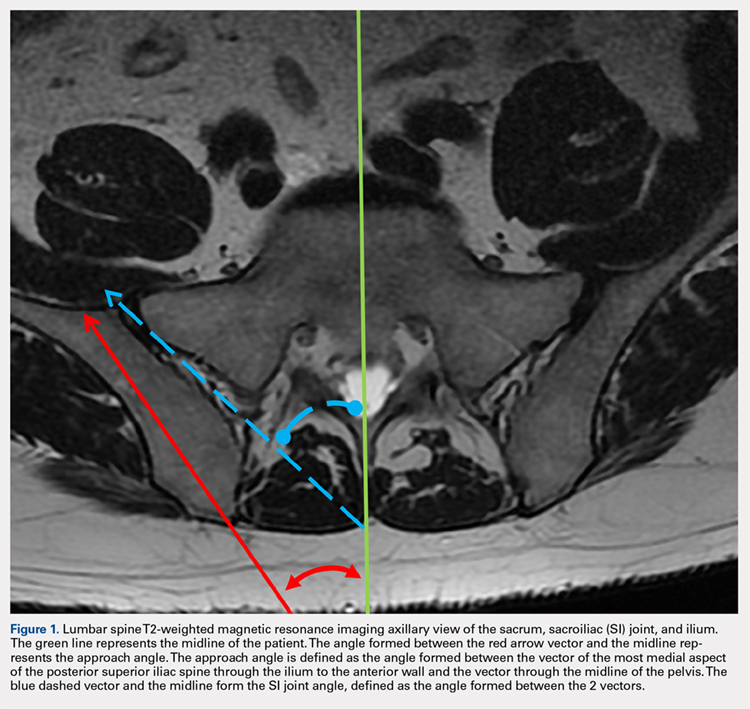
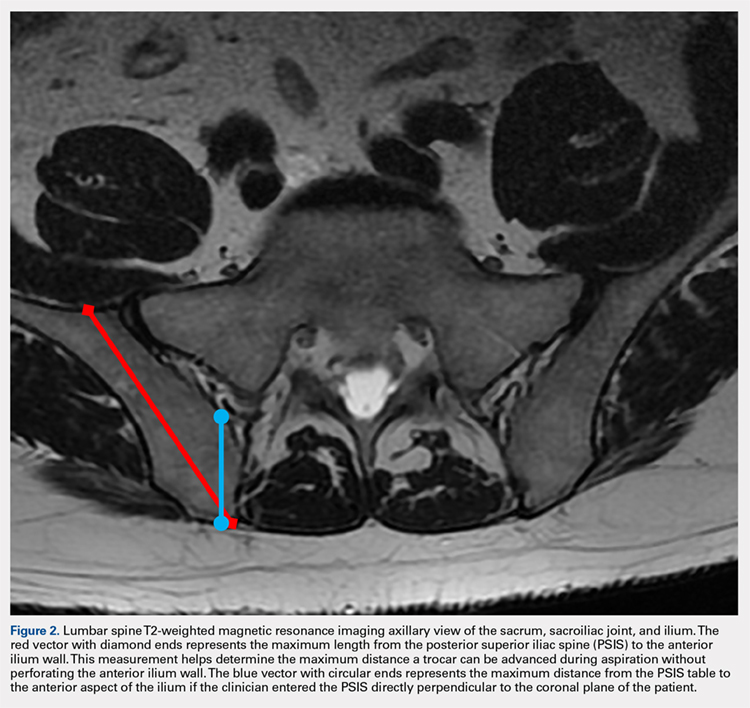
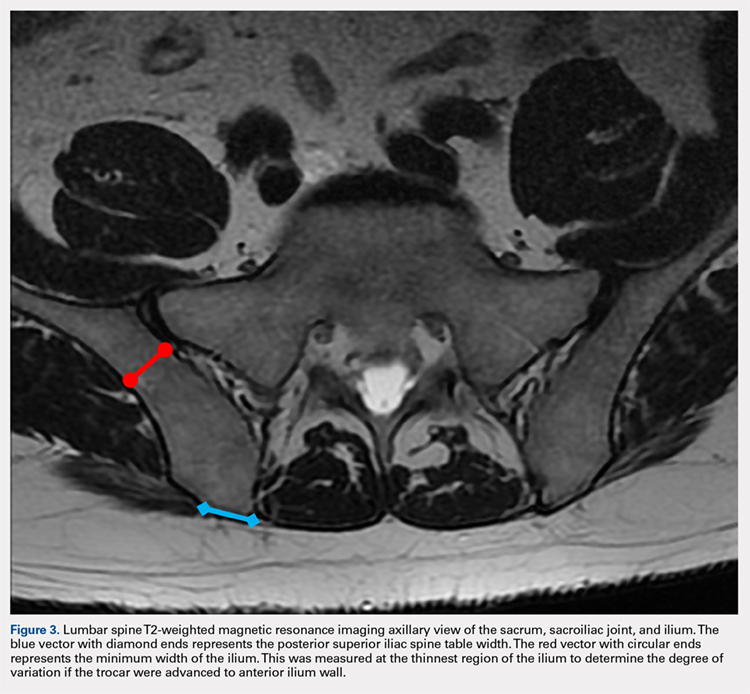
Continue to: For the 13 males, the mean SI joint...
RESULTS
The results are reported in the Table.
Table. Measurements of Patients Taken on Axial T2-Weighted Views of Lumbosacral MRI Scansa
Patient | SI Joint Angle (°) | Approach Angle (°) | PSIS Table Width (cm) | PSIS to Anterior Ilium Wall (cm) | Perpendicular Distance PSIS to Anterior Joint (cm) | Post Ilium Wall to SI Joint Width (cm) |
Males | ||||||
1 | 28.80 | 19.50 | 1.24 | 8.80 | 4.16 | 1.52 |
2 | 31.80 | 27.60 | 1.70 | 7.89 | 3.49 | 1.02 |
3 | 33.70 | 27.70 | 1.12 | 8.14 | 3.15 | 1.28 |
4 | 23.70 | 26.40 | 0.95 | 6.66 | 3.22 | 0.65 |
5 | 35.90 | 28.40 | 0.84 | 7.60 | 2.57 | 0.95 |
6 | 33.80 | 29.30 | 1.20 | 7.73 | 2.34 | 0.90 |
7 | 30.30 | 21.20 | 1.36 | 8.44 | 3.95 | 1.18 |
8 | 34.50 | 20.40 | 1.53 | 7.08 | 3.98 | 1.56 |
9 | 28.70 | 24.00 | 1.34 | 8.19 | 3.51 | 1.31 |
10 | 22.40 | 20.10 | 1.37 | 7.30 | 3.87 | 1.28 |
11 | 33.60 | 20.80 | 0.88 | 6.43 | 3.26 | 0.94 |
12 | 48.50 | 31.00 | 1.15 | 6.69 | 2.97 | 1.38 |
13 | 20.20 | 20.90 | 0.94 | 6.95 | 3.79 | 1.05 |
Averages | 31.22 | 24.41 | 1.20 | 7.53 | 3.40 | 1.16 |
Standard Deviation | 7.18 | 4.11 | 0.26 | 0.75 | 0.56 | 0.26 |
Females | ||||||
14 | 22.80 | 23.20 | 1.54 | 7.21 | 3.45 | 1.39 |
15 | 33.30 | 21.40 | 1.09 | 7.26 | 3.57 | 0.98 |
16 | 19.70 | 15.60 | 0.78 | 8.32 | 3.76 | 0.86 |
17 | 17.50 | 15.60 | 0.61 | 7.57 | 3.37 | 1.03 |
18 | 48.20 | 26.60 | 0.94 | 6.62 | 3.16 | 0.71 |
19 | 38.20 | 28.30 | 0.90 | 6.32 | 2.23 | 0.91 |
20 | 44.50 | 31.70 | 0.99 | 6.19 | 3.06 | 0.76 |
21 | 24.10 | 18.00 | 0.92 | 6.99 | 3.23 | 0.71 |
22 | 17.20 | 14.80 | 0.81 | 6.00 | 2.81 | 1.13 |
23 | 42.00 | 38.50 | 1.00 | 5.33 | 2.47 | 1.42 |
24 | 32.00 | 25.50 | 0.98 | 6.01 | 2.79 | 1.21 |
25 | 24.70 | 24.80 | 0.87 | 6.09 | 2.79 | 1.02 |
26 | 19.80 | 22.30 | 1.04 | 7.71 | 2.37 | 1.36 |
Averages | 29.54 | 23.56 | 0.96 | 6.74 | 3.00 | 1.04 |
Standard Deviation | 10.84 | 6.88 | 0.21 | 0.85 | 0.48 | 0.25 |
All patients Averages | 30.38 | 23.98 | 1.08 | 7.14 | 3.20 | 1.10 |
Standard Deviation | 9.05 | 5.57 | 0.26 | 0.88 | 0.55 | 0.26 |
aStatistical significance is denoted as P < .02.
Abbreviations: MRI, magnetic resonance imaging; PSIS, posterior iliac spine; SI, sacroiliac.
For the 13 males, the mean SI joint angle was 31.22° ± 7.18° (range, 20.20° to 48.50°). The mean approach angle was 24.41° ± 4.11° (range, 19.50° to 31.00°). The mean PSIS table width was 1.20 cm ± 0.26 cm (range, 0.84 cm to 1.70 cm). The mean distance from the PSIS to the anterior ilium wall was 7.53 cm ± 0.75 cm (range, 6.43 cm to 8.80 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.40 cm ± 0.56 cm (range, 2.34 cm to 4.16 cm). The mean minimum width of the ilium to the SI joint was 1.16 cm ± 0.26 cm (range, 0.65 cm to 1.56 cm).
For the 13 females, the mean SI joint angle was 29.54° ± 10.84° (range, 17.20° to 48.20°). The mean approach angle was 23.56° ± 6.88° (range, 14.80° to 38.50°). The mean PSIS table width was 0.96 cm ± 0.21 cm (range, 0.61 cm to 1.54 cm). The mean distance from the PSIS to the anterior ilium wall was 6.74 cm ± 0.85 cm (range, 5.33 cm to 8.32 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.00 cm ± 0.48 cm (range, 2.23 cm to 3.76 cm). The mean minimum width of the ilium to the SI joint was 1.04 cm ± 0.25 cm (range, 0.71 cm to 1.42 cm).
For the 26 total patients, the mean SI joint angle was 30.38° ± 9.05° (range, 17.20° to 48.50°). The mean approach angle was 23.98° ± 5.57° (range, 14.80° to 38.50°). The mean PSIS table width was 1.08 cm ± 0.26 cm (range, 0.61 cm to 1.70 cm). The mean distance from the PSIS to the anterior ilium wall was 7.14 cm ± 0.88 cm (range, 5.33 cm to 8.80 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.20 cm ± 0.55 cm (range, 2.23 cm to 4.16 cm). The mean minimum width of the ilium to the SI joint was 1.10 cm ± 0.26 cm (range, 0.65 cm to 1.56 cm).
There was a statistically significant difference between the male and female groups for the maximum distance the trocar can be advanced from the PSIS to the anterior ilium wall (P < .02), and a statistically significant difference for the PSIS table width (P < .02). There were no significant differences between the male and female groups for the approach angle, the SI joint angle, the perpendicular distance from the PSIS to the anterior ilium, and the minimum width of the ilium to the SI joint.
Continue to: The patient is brought to the procedure...
TECHNIQUE: ILIAC CREST (PSIS) BONE MARROW ASPIRATION
The patient is brought to the procedure room and placed in a prone position. The donor site is prepared and draped in the usual sterile manner. Ultrasound is used to identify the median sacral crest in a short-axis view. The probe is then moved laterally to identify the PSIS (Figures 4A, 4B). 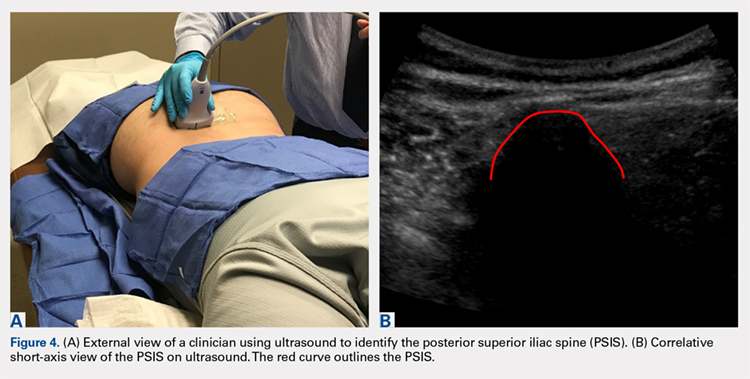
The crosshairs on the ultrasound probe are used to mark the center lines of each plane. The central point marks the location of the PSIS. Alternatively, an in-plane technique can be used to place a spinal needle on the exact entry point on the PSIS. Once the PSIS and entry point are identified, the site is blocked with 10 mL of 0.5% ropivacaine.
Prior to introduction of the trocar, all instrumentation is primed with heparin and syringes are prepped with anticoagulant citrate dextrose solution, solution A. A stab incision is made at the site. The trocar is placed at the entry point, which should be centered in a superior-inferior plane and at the most medial point of the PSIS. Starting with the trocar vertical, the trocar is angled laterally 24° by dropping the hand medially toward the midline. No angulation cephalad or caudad is necessary, but cephalad must be avoided so as not to skive superiorly. This angle, which is recommended for both males and females, allows for the greatest distance the trocar can travel in bone before hitting the anterior ilium wall. A standard deviation of 5.57° is present, which should be considered. Steady pressure should be applied with a slight twisting motion on the PSIS. If advancement of the trocar is too difficult, a mallet or drill can be used to assist in penetration.
With the trocar advanced into the bone 1 cm, the trocar needle is removed while the cannula remains in place. The syringe is attached to the top of the cannula. The syringe plunger is pulled back to aspirate 20 mL of bone marrow. The cannula and syringe assembly are advanced 2 cm farther into the bone to allow for aspiration of a new location within the bone marrow cavity, and 20 mL of bone marrow are again aspirated. This is done a final time, advancing the trocar another 2 cm and aspirating a final 20 mL of bone marrow. The entire process should yield roughly 60 mL of bone marrow from one side. If desired, the same process can be repeated for the contralateral PSIS to yield a total of 120 mL of bone marrow from the 2 sites.
Based on our data, the average distance to the anterior ilium wall was 7 cm, but the shortest distance noted in this study was 5 cm. On the basis of the data presented, this technique allows for safe advancement based on even the shortest measured distance, without fear of puncturing the anterior ilium wall. Perforation could damage the femoral nerve and the internal or external iliac artery or vein that lie anterior to the ilium.
Continue to: We hypothesized that there...
DISCUSSION
We hypothesized that there would be an optimal angle of entry and maximal safe distance the trocar could advance through the ilium when aspirating. Because male and female pelvic anatomy differs, we also hypothesized that there would be differences in distance and size measurements for males and females. Our results supported our hypothesis that there is an ideal approach angle. The results also showed that the maximum distance the trocar can advance and the width of the PSIS table differ significantly between males and females.
Although pelvic anatomy differs between males and females, there should be an ideal entry angle that would allow maximum advancement into the ilium without perforating the anterior wall, which we defined as the approach angle. In our comparison of 26 MRI scans, we found that the approach angle did not differ significantly between the 2 groups (13 males, 13 females). This allows clinicians to enter the PSIS at roughly 24° medial to the parasagittal line, maximizing the space before puncturing into the anterior pelvis in either males or females.
If clinicians were to enter perpendicular to the patient’s PSIS, they would, on average, be able to advance only 3.20 cm before encountering the SI joint. When entering at 24° as we recommend, the average distance increases to 7.14 cm. Although the angle did not differ significantly, there was a significant difference between males and females in the length from the PSIS to the anterior wall, with males having 7.53 cm distance and females 6.74 cm. This is an important measurement because if the anterior ilium wall is punctured, the femoral nerve and the common, internal and external iliac arteries and veins could be damaged, resulting in retroperitoneal hemorrhage.
A fatality in 2001 in the United Kingdom led to a national audit of bone marrow aspiration and biopsies.4-6 Although these procedures were done primarily for patients with cancer, hemorrhagic events were the most frequent and serious events. This audit led to the identification of many risk factors. Bain4-6 conducted reviews of bone marrow aspirations and biopsies in the United Kingdom from 2002 to 2004. Of a total of 53,088 procedures conducted during that time frame, 48 (0.09%) adverse events occurred, with 29 (0.05%) being hemorrhagic events. Although infrequent, hemorrhagic adverse events represent significant morbidity. Reviews such as those conducted by Bain4-6 highlight the importance of a study that helps determine the optimal parameters for aspiration to ensure safety and reliability.
Hernigou and colleagues2,3 conducted studies analyzing different “sectors” in an attempt to develop a safe aspiration technique. They found that obese patients were at higher risk, and some sites of aspiration (sectors 1, 4, 5) had increased risk for perforation and damage to surrounding structures. Their sector 6, which incorporated the entirety of the PSIS table, was considered the safest, most reliable site for trocar introduction.2,3 Hernigou and colleagues,2 in comparing the bone mass of the sectors, also noted that sector 6 has the greatest bone thickness close to the entry point, making it the most favorable site. The PSIS is not just a point; it is more a “table.” The PSIS can be palpated posteriorly, but this is inaccurate and unreliable, particularly in larger individuals. The PSIS table can be identified on ultrasound before introducing the trocar, which is a more reliable method of landmark identification than palpation guidance, just as in ultrasound-guided injections7-16 and procedures.9,12,17-19
Continue to: If the PSIS is not accurately...
If the PSIS is not accurately identified, penetration laterally will result in entering the ilium wing, where it is quite narrow. We found the distance between the posterior ilium wall and the SI joint to be only 1.10 cm wide (Figure 3); we defined this area as the narrow corridor. Superior and lateral entry could damage the superior cluneal nerves coming over the iliac crest, which are located 6 cm lateral to the SI joint. Inferior and lateral entry 6 cm below the PSIS could reach the greater sciatic foramen, damaging the sacral plexus and superior gluteal artery and vein. If the entry slips above the PSIS over the pelvis, the trocar could enter the retroperitoneal space and damage the femoral nerve and common iliac artery and vein, leading to a retroperitoneal hemorrhage.4-6,20
MSCs are found as perivascular cells and lie in the cortices of bones.21 Following the approach angle and directed line from the PSIS to the anterior ilium wall described in this study (Figures 1 and 2), the trocar would pass through the narrow corridor as it advances farther into the ilium. The minimum width of this corridor was measured in this study and, on average, was 1.10 cm wide from cortex to cortex (Figure 3). As the bone marrow is aspirated from this narrow corridor, the clinician is gathering MSCs from both the lateral and medial cortices of the ilium. By aspirating from a greater surface area of the cortices, it is believed that this will increase the total collection of MSCs.
CONCLUSION
Although there are reports in the literature that describe techniques for bone marrow aspiration from the iliac crest, the techniques are very general and vague regarding the ideal angles and methods. Studies have attempted to quantify the safest entry sites for aspiration but have not detailed ideal parameters for collection. Blind aspiration from the iliac crest can have serious implications if adverse events occur, and thus there is a need for a safe and reliable method of aspiration from the iliac crest. Ultrasound guidance to identify anatomy, as opposed to palpation guidance, ensures anatomic placement of the trocar while minimizing the risk of aspiration. Based on the measurements gathered in this study, an optimal angle of entry and safe distance of penetration have been identified. Using our data and relevant literature, we developed a technique for a safe, consistent, and reliable method of bone marrow aspiration out of the iliac crest.
1. Chahla J, Mannava S, Cinque ME, Geeslin AG, Codina D, LaPrade RF. Bone marrow aspirate concentrate harvesting and processing technique. Arthrosc Tech. 2017;6(2):e441-e445. doi:10.1016/j.eats.2016.10.024.
2. Hernigou J, Alves A, Homma Y, Guissou I, Hernigou P. Anatomy of the ilium for bone marrow aspiration: map of sectors and implication for safe trocar placement. Int Orthop. 2014;38(12):2585-2590. doi:10.1007/s00264-014-2353-7.
3. Hernigou J, Picard L, Alves A, Silvera J, Homma Y, Hernigou P. Understanding bone safety zones during bone marrow aspiration from the iliac crest: the sector rule. Int Orthop. 2014;38(11):2377-2384. doi:10.1007/s00264-014-2343-9.
4. Bain BJ. Bone marrow biopsy morbidity: review of 2003. J Clin Pathol. 2005;58(4):406-408. doi:10.1136/jcp.2004.022178.
5. Bain BJ. Bone marrow biopsy morbidity and mortality: 2002 data. Clin Lab Haematol. 2004;26(5):315-318. doi:10.1111/j.1365-2257.2004.00630.x.
6. Bain BJ. Morbidity associated with bone marrow aspiration and trephine biopsy - a review of UK data for 2004. Haematologica. 2006;91(9):1293-1294.
7. Berkoff DJ, Miller LE, Block JE. Clinical utility of ultrasound guidance for intra-articular knee injections: a review. Clin Interv Aging. 2012;7:89-95. doi:10.2147/CIA.S29265.
8. Henkus HE, Cobben LP, Coerkamp EG, Nelissen RG, van Arkel ER. The accuracy of subacromial injections: a prospective randomized magnetic resonance imaging study. Arthroscopy. 2006;22(3):277-282. doi:10.1016/j.arthro.2005.12.019.
9. Hirahara AM, Panero AJ. A guide to ultrasound of the shoulder, part 3: interventional and procedural uses. Am J Orthop. 2016;45(7):440-445.
10. Jackson DW, Evans NA, Thomas BM. Accuracy of needle placement into the intra-articular space of the knee. J Bone Joint Surg Am. 2002;84-A(9):1522-1527.
11. Naredo E, Cabero F, Beneyto P, et al. A randomized comparative study of short term response to blind versus sonographic-guided injection of local corticosteroids in patients with painful shoulder. J Rheumatol. 2004;31(2):308-314.
12. Panero AJ, Hirahara AM. A guide to ultrasound of the shoulder, part 2: the diagnostic evaluation. Am J Orthop. 2016;45(4):233-238.
13. Sethi PM, El Attrache N. Accuracy of intra-articular injection of the glenohumeral joint: a cadaveric study. Orthopedics. 2006;29(2):149-152.
14. Sibbit WL Jr, Peisajovich A, Michael AA, et al. Does sonographic needle guidance affect the clinical outcome of intraarticular injections? J Rheumatol. 2009;36(9):1892-1902. doi:10.3899/jrheum.090013.
15. Smith J, Brault JS, Rizzo M, Sayeed YA, Finnoff JT. Accuracy of sonographically guided and palpation guided scaphotrapeziotrapezoid joint injections. J Ultrasound Med. 2011;30(11):1509-1515. doi:10.7863/jum.2011.30.11.1509.
16. Yamakado K. The targeting accuracy of subacromial injection to the shoulder: an arthrographic evaluation. Arthroscopy. 2002;18(8):887-891.
17. Hirahara AM, Andersen WJ. Ultrasound-guided percutaneous reconstruction of the anterolateral ligament: surgical technique and case report. Am J Orthop. 2016;45(7):418-422, 460.
18. Hirahara AM, Andersen WJ. Ultrasound-guided percutaneous repair of medial patellofemoral ligament: surgical technique and outcomes. Am J Orthop. 2017;46(3):152-157.
19. Hirahara AM, Mackay G, Andersen WJ. Ultrasound-guided InternalBrace of the medial collateral ligament. Arthrosc Tech. Submitted.
20. Jamaludin WFW, Mukari SAM, Wahid SFA. Retroperitoneal hemorrhage associated with bone marrow trephine biopsy. Am J Case Rep. 2013;14:489-493. doi:10.12659/AJCR.889274.
21. Bianco P, Cao X, Frenette PS, et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19(1):35-42. doi:10.1038/nm.3028.
ABSTRACT
Use of mesenchymal stem cells from bone marrow has gained significant popularity. The iliac crest has been determined to be an effective site for harvesting mesenchymal stem cells. Review of the literature reveals that multiple techniques are used to harvest bone marrow aspirate from the iliac crest, but the descriptions are based on the experience of various authors as opposed to studied anatomy. A safe, reliable, and reproducible method for aspiration has yet to be studied and described. We hypothesized that there would be an ideal angle and distance for aspiration that would be the safest, most consistent, and most reliable. Using magnetic resonance imaging (MRI), we reviewed 26 total lumbar spine MRI scans (13 males, 13 females) and found that an angle of 24° should be used when entering the most medial aspect of the posterior superior iliac spine (PSIS) and that this angle did not differ between the sexes. The distance that the trocar can advance after entry before hitting the anterior ilium wall varied significantly between males and females, being 7.53 cm in males and 6.74 cm in females. In addition, the size of the PSIS table was significantly different between males and females (1.20 cm and 0.96 cm, respectively). No other significant differences in the measurements gathered were found. Using the data gleaned from this study, we developed an aspiration technique. This method uses ultrasound to determine the location of the PSIS and the entry point on the PSIS. This contrasts with most techniques that use landmark palpation, which is known to be unreliable and inaccurate. The described technique for aspiration from the PSIS is safe, reliable, reproducible, and substantiated by data.
The iliac crest is an effective site for harvesting bone marrow stem cells. It allows for easy access and is superficial in most individuals, allowing for a relatively quick and simple procedure. Use of mesenchymal stem cells (MSCs) for treatment of orthopedic injuries has grown recently. Whereas overall use has increased, review of the literature reveals very few techniques for iliac crest aspiration,1 but these are not based on anatomic relationships or studies. Hernigou and colleagues2,3 attempted to quantitatively evaluate potential “sectors” allowing for safe aspiration using cadaver and computed tomographic reconstruction imaging. We used magnetic resonance imaging (MRI) to analyze aspiration parameters. Owing to the ilium’s anatomy, improper positioning or aspiration technique during aspiration can result in serious injury.2,4-6 We hypothesized that there is an ideal angle and positioning for bone marrow aspiration from the posterior superior iliac spine (PSIS) that is safe, consistent, and reproducible. Although most aspiration techniques use landmark palpation, this is unreliable and inaccurate, especially when compared with ultrasound-guided injections7-16 and procedures.9,12,17-19 We describe our technique using ultrasound to visualize patient anatomy and accurately determine anatomic entry with the trocar.
METHODS
MRI scans of 26 patients (13 males, 13 females) were reviewed to determine average angles and distances. Axial T2-weighted views of the lumbar spine were used in all analyses. The sacroiliac (SI) joint angle was defined as the angle formed between the vector through the midline of the pelvis and the vector that is parallel to the SI joint. The approach angle was defined as the angle formed between the vector of the most medial aspect of the PSIS through the ilium to the anterior wall and the vector through the midline of the pelvis (Figure 1).
Continue to: For the 13 males, the mean SI joint...
RESULTS
The results are reported in the Table.
Table. Measurements of Patients Taken on Axial T2-Weighted Views of Lumbosacral MRI Scansa
Patient | SI Joint Angle (°) | Approach Angle (°) | PSIS Table Width (cm) | PSIS to Anterior Ilium Wall (cm) | Perpendicular Distance PSIS to Anterior Joint (cm) | Post Ilium Wall to SI Joint Width (cm) |
Males | ||||||
1 | 28.80 | 19.50 | 1.24 | 8.80 | 4.16 | 1.52 |
2 | 31.80 | 27.60 | 1.70 | 7.89 | 3.49 | 1.02 |
3 | 33.70 | 27.70 | 1.12 | 8.14 | 3.15 | 1.28 |
4 | 23.70 | 26.40 | 0.95 | 6.66 | 3.22 | 0.65 |
5 | 35.90 | 28.40 | 0.84 | 7.60 | 2.57 | 0.95 |
6 | 33.80 | 29.30 | 1.20 | 7.73 | 2.34 | 0.90 |
7 | 30.30 | 21.20 | 1.36 | 8.44 | 3.95 | 1.18 |
8 | 34.50 | 20.40 | 1.53 | 7.08 | 3.98 | 1.56 |
9 | 28.70 | 24.00 | 1.34 | 8.19 | 3.51 | 1.31 |
10 | 22.40 | 20.10 | 1.37 | 7.30 | 3.87 | 1.28 |
11 | 33.60 | 20.80 | 0.88 | 6.43 | 3.26 | 0.94 |
12 | 48.50 | 31.00 | 1.15 | 6.69 | 2.97 | 1.38 |
13 | 20.20 | 20.90 | 0.94 | 6.95 | 3.79 | 1.05 |
Averages | 31.22 | 24.41 | 1.20 | 7.53 | 3.40 | 1.16 |
Standard Deviation | 7.18 | 4.11 | 0.26 | 0.75 | 0.56 | 0.26 |
Females | ||||||
14 | 22.80 | 23.20 | 1.54 | 7.21 | 3.45 | 1.39 |
15 | 33.30 | 21.40 | 1.09 | 7.26 | 3.57 | 0.98 |
16 | 19.70 | 15.60 | 0.78 | 8.32 | 3.76 | 0.86 |
17 | 17.50 | 15.60 | 0.61 | 7.57 | 3.37 | 1.03 |
18 | 48.20 | 26.60 | 0.94 | 6.62 | 3.16 | 0.71 |
19 | 38.20 | 28.30 | 0.90 | 6.32 | 2.23 | 0.91 |
20 | 44.50 | 31.70 | 0.99 | 6.19 | 3.06 | 0.76 |
21 | 24.10 | 18.00 | 0.92 | 6.99 | 3.23 | 0.71 |
22 | 17.20 | 14.80 | 0.81 | 6.00 | 2.81 | 1.13 |
23 | 42.00 | 38.50 | 1.00 | 5.33 | 2.47 | 1.42 |
24 | 32.00 | 25.50 | 0.98 | 6.01 | 2.79 | 1.21 |
25 | 24.70 | 24.80 | 0.87 | 6.09 | 2.79 | 1.02 |
26 | 19.80 | 22.30 | 1.04 | 7.71 | 2.37 | 1.36 |
Averages | 29.54 | 23.56 | 0.96 | 6.74 | 3.00 | 1.04 |
Standard Deviation | 10.84 | 6.88 | 0.21 | 0.85 | 0.48 | 0.25 |
All patients Averages | 30.38 | 23.98 | 1.08 | 7.14 | 3.20 | 1.10 |
Standard Deviation | 9.05 | 5.57 | 0.26 | 0.88 | 0.55 | 0.26 |
aStatistical significance is denoted as P < .02.
Abbreviations: MRI, magnetic resonance imaging; PSIS, posterior iliac spine; SI, sacroiliac.
For the 13 males, the mean SI joint angle was 31.22° ± 7.18° (range, 20.20° to 48.50°). The mean approach angle was 24.41° ± 4.11° (range, 19.50° to 31.00°). The mean PSIS table width was 1.20 cm ± 0.26 cm (range, 0.84 cm to 1.70 cm). The mean distance from the PSIS to the anterior ilium wall was 7.53 cm ± 0.75 cm (range, 6.43 cm to 8.80 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.40 cm ± 0.56 cm (range, 2.34 cm to 4.16 cm). The mean minimum width of the ilium to the SI joint was 1.16 cm ± 0.26 cm (range, 0.65 cm to 1.56 cm).
For the 13 females, the mean SI joint angle was 29.54° ± 10.84° (range, 17.20° to 48.20°). The mean approach angle was 23.56° ± 6.88° (range, 14.80° to 38.50°). The mean PSIS table width was 0.96 cm ± 0.21 cm (range, 0.61 cm to 1.54 cm). The mean distance from the PSIS to the anterior ilium wall was 6.74 cm ± 0.85 cm (range, 5.33 cm to 8.32 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.00 cm ± 0.48 cm (range, 2.23 cm to 3.76 cm). The mean minimum width of the ilium to the SI joint was 1.04 cm ± 0.25 cm (range, 0.71 cm to 1.42 cm).
For the 26 total patients, the mean SI joint angle was 30.38° ± 9.05° (range, 17.20° to 48.50°). The mean approach angle was 23.98° ± 5.57° (range, 14.80° to 38.50°). The mean PSIS table width was 1.08 cm ± 0.26 cm (range, 0.61 cm to 1.70 cm). The mean distance from the PSIS to the anterior ilium wall was 7.14 cm ± 0.88 cm (range, 5.33 cm to 8.80 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.20 cm ± 0.55 cm (range, 2.23 cm to 4.16 cm). The mean minimum width of the ilium to the SI joint was 1.10 cm ± 0.26 cm (range, 0.65 cm to 1.56 cm).
There was a statistically significant difference between the male and female groups for the maximum distance the trocar can be advanced from the PSIS to the anterior ilium wall (P < .02), and a statistically significant difference for the PSIS table width (P < .02). There were no significant differences between the male and female groups for the approach angle, the SI joint angle, the perpendicular distance from the PSIS to the anterior ilium, and the minimum width of the ilium to the SI joint.
Continue to: The patient is brought to the procedure...
TECHNIQUE: ILIAC CREST (PSIS) BONE MARROW ASPIRATION
The patient is brought to the procedure room and placed in a prone position. The donor site is prepared and draped in the usual sterile manner. Ultrasound is used to identify the median sacral crest in a short-axis view. The probe is then moved laterally to identify the PSIS (Figures 4A, 4B).
The crosshairs on the ultrasound probe are used to mark the center lines of each plane. The central point marks the location of the PSIS. Alternatively, an in-plane technique can be used to place a spinal needle on the exact entry point on the PSIS. Once the PSIS and entry point are identified, the site is blocked with 10 mL of 0.5% ropivacaine.
Prior to introduction of the trocar, all instrumentation is primed with heparin and syringes are prepped with anticoagulant citrate dextrose solution, solution A. A stab incision is made at the site. The trocar is placed at the entry point, which should be centered in a superior-inferior plane and at the most medial point of the PSIS. Starting with the trocar vertical, the trocar is angled laterally 24° by dropping the hand medially toward the midline. No angulation cephalad or caudad is necessary, but cephalad must be avoided so as not to skive superiorly. This angle, which is recommended for both males and females, allows for the greatest distance the trocar can travel in bone before hitting the anterior ilium wall. A standard deviation of 5.57° is present, which should be considered. Steady pressure should be applied with a slight twisting motion on the PSIS. If advancement of the trocar is too difficult, a mallet or drill can be used to assist in penetration.
With the trocar advanced into the bone 1 cm, the trocar needle is removed while the cannula remains in place. The syringe is attached to the top of the cannula. The syringe plunger is pulled back to aspirate 20 mL of bone marrow. The cannula and syringe assembly are advanced 2 cm farther into the bone to allow for aspiration of a new location within the bone marrow cavity, and 20 mL of bone marrow are again aspirated. This is done a final time, advancing the trocar another 2 cm and aspirating a final 20 mL of bone marrow. The entire process should yield roughly 60 mL of bone marrow from one side. If desired, the same process can be repeated for the contralateral PSIS to yield a total of 120 mL of bone marrow from the 2 sites.
Based on our data, the average distance to the anterior ilium wall was 7 cm, but the shortest distance noted in this study was 5 cm. On the basis of the data presented, this technique allows for safe advancement based on even the shortest measured distance, without fear of puncturing the anterior ilium wall. Perforation could damage the femoral nerve and the internal or external iliac artery or vein that lie anterior to the ilium.
Continue to: We hypothesized that there...
DISCUSSION
We hypothesized that there would be an optimal angle of entry and maximal safe distance the trocar could advance through the ilium when aspirating. Because male and female pelvic anatomy differs, we also hypothesized that there would be differences in distance and size measurements for males and females. Our results supported our hypothesis that there is an ideal approach angle. The results also showed that the maximum distance the trocar can advance and the width of the PSIS table differ significantly between males and females.
Although pelvic anatomy differs between males and females, there should be an ideal entry angle that would allow maximum advancement into the ilium without perforating the anterior wall, which we defined as the approach angle. In our comparison of 26 MRI scans, we found that the approach angle did not differ significantly between the 2 groups (13 males, 13 females). This allows clinicians to enter the PSIS at roughly 24° medial to the parasagittal line, maximizing the space before puncturing into the anterior pelvis in either males or females.
If clinicians were to enter perpendicular to the patient’s PSIS, they would, on average, be able to advance only 3.20 cm before encountering the SI joint. When entering at 24° as we recommend, the average distance increases to 7.14 cm. Although the angle did not differ significantly, there was a significant difference between males and females in the length from the PSIS to the anterior wall, with males having 7.53 cm distance and females 6.74 cm. This is an important measurement because if the anterior ilium wall is punctured, the femoral nerve and the common, internal and external iliac arteries and veins could be damaged, resulting in retroperitoneal hemorrhage.
A fatality in 2001 in the United Kingdom led to a national audit of bone marrow aspiration and biopsies.4-6 Although these procedures were done primarily for patients with cancer, hemorrhagic events were the most frequent and serious events. This audit led to the identification of many risk factors. Bain4-6 conducted reviews of bone marrow aspirations and biopsies in the United Kingdom from 2002 to 2004. Of a total of 53,088 procedures conducted during that time frame, 48 (0.09%) adverse events occurred, with 29 (0.05%) being hemorrhagic events. Although infrequent, hemorrhagic adverse events represent significant morbidity. Reviews such as those conducted by Bain4-6 highlight the importance of a study that helps determine the optimal parameters for aspiration to ensure safety and reliability.
Hernigou and colleagues2,3 conducted studies analyzing different “sectors” in an attempt to develop a safe aspiration technique. They found that obese patients were at higher risk, and some sites of aspiration (sectors 1, 4, 5) had increased risk for perforation and damage to surrounding structures. Their sector 6, which incorporated the entirety of the PSIS table, was considered the safest, most reliable site for trocar introduction.2,3 Hernigou and colleagues,2 in comparing the bone mass of the sectors, also noted that sector 6 has the greatest bone thickness close to the entry point, making it the most favorable site. The PSIS is not just a point; it is more a “table.” The PSIS can be palpated posteriorly, but this is inaccurate and unreliable, particularly in larger individuals. The PSIS table can be identified on ultrasound before introducing the trocar, which is a more reliable method of landmark identification than palpation guidance, just as in ultrasound-guided injections7-16 and procedures.9,12,17-19
Continue to: If the PSIS is not accurately...
If the PSIS is not accurately identified, penetration laterally will result in entering the ilium wing, where it is quite narrow. We found the distance between the posterior ilium wall and the SI joint to be only 1.10 cm wide (Figure 3); we defined this area as the narrow corridor. Superior and lateral entry could damage the superior cluneal nerves coming over the iliac crest, which are located 6 cm lateral to the SI joint. Inferior and lateral entry 6 cm below the PSIS could reach the greater sciatic foramen, damaging the sacral plexus and superior gluteal artery and vein. If the entry slips above the PSIS over the pelvis, the trocar could enter the retroperitoneal space and damage the femoral nerve and common iliac artery and vein, leading to a retroperitoneal hemorrhage.4-6,20
MSCs are found as perivascular cells and lie in the cortices of bones.21 Following the approach angle and directed line from the PSIS to the anterior ilium wall described in this study (Figures 1 and 2), the trocar would pass through the narrow corridor as it advances farther into the ilium. The minimum width of this corridor was measured in this study and, on average, was 1.10 cm wide from cortex to cortex (Figure 3). As the bone marrow is aspirated from this narrow corridor, the clinician is gathering MSCs from both the lateral and medial cortices of the ilium. By aspirating from a greater surface area of the cortices, it is believed that this will increase the total collection of MSCs.
CONCLUSION
Although there are reports in the literature that describe techniques for bone marrow aspiration from the iliac crest, the techniques are very general and vague regarding the ideal angles and methods. Studies have attempted to quantify the safest entry sites for aspiration but have not detailed ideal parameters for collection. Blind aspiration from the iliac crest can have serious implications if adverse events occur, and thus there is a need for a safe and reliable method of aspiration from the iliac crest. Ultrasound guidance to identify anatomy, as opposed to palpation guidance, ensures anatomic placement of the trocar while minimizing the risk of aspiration. Based on the measurements gathered in this study, an optimal angle of entry and safe distance of penetration have been identified. Using our data and relevant literature, we developed a technique for a safe, consistent, and reliable method of bone marrow aspiration out of the iliac crest.
ABSTRACT
Use of mesenchymal stem cells from bone marrow has gained significant popularity. The iliac crest has been determined to be an effective site for harvesting mesenchymal stem cells. Review of the literature reveals that multiple techniques are used to harvest bone marrow aspirate from the iliac crest, but the descriptions are based on the experience of various authors as opposed to studied anatomy. A safe, reliable, and reproducible method for aspiration has yet to be studied and described. We hypothesized that there would be an ideal angle and distance for aspiration that would be the safest, most consistent, and most reliable. Using magnetic resonance imaging (MRI), we reviewed 26 total lumbar spine MRI scans (13 males, 13 females) and found that an angle of 24° should be used when entering the most medial aspect of the posterior superior iliac spine (PSIS) and that this angle did not differ between the sexes. The distance that the trocar can advance after entry before hitting the anterior ilium wall varied significantly between males and females, being 7.53 cm in males and 6.74 cm in females. In addition, the size of the PSIS table was significantly different between males and females (1.20 cm and 0.96 cm, respectively). No other significant differences in the measurements gathered were found. Using the data gleaned from this study, we developed an aspiration technique. This method uses ultrasound to determine the location of the PSIS and the entry point on the PSIS. This contrasts with most techniques that use landmark palpation, which is known to be unreliable and inaccurate. The described technique for aspiration from the PSIS is safe, reliable, reproducible, and substantiated by data.
The iliac crest is an effective site for harvesting bone marrow stem cells. It allows for easy access and is superficial in most individuals, allowing for a relatively quick and simple procedure. Use of mesenchymal stem cells (MSCs) for treatment of orthopedic injuries has grown recently. Whereas overall use has increased, review of the literature reveals very few techniques for iliac crest aspiration,1 but these are not based on anatomic relationships or studies. Hernigou and colleagues2,3 attempted to quantitatively evaluate potential “sectors” allowing for safe aspiration using cadaver and computed tomographic reconstruction imaging. We used magnetic resonance imaging (MRI) to analyze aspiration parameters. Owing to the ilium’s anatomy, improper positioning or aspiration technique during aspiration can result in serious injury.2,4-6 We hypothesized that there is an ideal angle and positioning for bone marrow aspiration from the posterior superior iliac spine (PSIS) that is safe, consistent, and reproducible. Although most aspiration techniques use landmark palpation, this is unreliable and inaccurate, especially when compared with ultrasound-guided injections7-16 and procedures.9,12,17-19 We describe our technique using ultrasound to visualize patient anatomy and accurately determine anatomic entry with the trocar.
METHODS
MRI scans of 26 patients (13 males, 13 females) were reviewed to determine average angles and distances. Axial T2-weighted views of the lumbar spine were used in all analyses. The sacroiliac (SI) joint angle was defined as the angle formed between the vector through the midline of the pelvis and the vector that is parallel to the SI joint. The approach angle was defined as the angle formed between the vector of the most medial aspect of the PSIS through the ilium to the anterior wall and the vector through the midline of the pelvis (Figure 1).
Continue to: For the 13 males, the mean SI joint...
RESULTS
The results are reported in the Table.
Table. Measurements of Patients Taken on Axial T2-Weighted Views of Lumbosacral MRI Scansa
Patient | SI Joint Angle (°) | Approach Angle (°) | PSIS Table Width (cm) | PSIS to Anterior Ilium Wall (cm) | Perpendicular Distance PSIS to Anterior Joint (cm) | Post Ilium Wall to SI Joint Width (cm) |
Males | ||||||
1 | 28.80 | 19.50 | 1.24 | 8.80 | 4.16 | 1.52 |
2 | 31.80 | 27.60 | 1.70 | 7.89 | 3.49 | 1.02 |
3 | 33.70 | 27.70 | 1.12 | 8.14 | 3.15 | 1.28 |
4 | 23.70 | 26.40 | 0.95 | 6.66 | 3.22 | 0.65 |
5 | 35.90 | 28.40 | 0.84 | 7.60 | 2.57 | 0.95 |
6 | 33.80 | 29.30 | 1.20 | 7.73 | 2.34 | 0.90 |
7 | 30.30 | 21.20 | 1.36 | 8.44 | 3.95 | 1.18 |
8 | 34.50 | 20.40 | 1.53 | 7.08 | 3.98 | 1.56 |
9 | 28.70 | 24.00 | 1.34 | 8.19 | 3.51 | 1.31 |
10 | 22.40 | 20.10 | 1.37 | 7.30 | 3.87 | 1.28 |
11 | 33.60 | 20.80 | 0.88 | 6.43 | 3.26 | 0.94 |
12 | 48.50 | 31.00 | 1.15 | 6.69 | 2.97 | 1.38 |
13 | 20.20 | 20.90 | 0.94 | 6.95 | 3.79 | 1.05 |
Averages | 31.22 | 24.41 | 1.20 | 7.53 | 3.40 | 1.16 |
Standard Deviation | 7.18 | 4.11 | 0.26 | 0.75 | 0.56 | 0.26 |
Females | ||||||
14 | 22.80 | 23.20 | 1.54 | 7.21 | 3.45 | 1.39 |
15 | 33.30 | 21.40 | 1.09 | 7.26 | 3.57 | 0.98 |
16 | 19.70 | 15.60 | 0.78 | 8.32 | 3.76 | 0.86 |
17 | 17.50 | 15.60 | 0.61 | 7.57 | 3.37 | 1.03 |
18 | 48.20 | 26.60 | 0.94 | 6.62 | 3.16 | 0.71 |
19 | 38.20 | 28.30 | 0.90 | 6.32 | 2.23 | 0.91 |
20 | 44.50 | 31.70 | 0.99 | 6.19 | 3.06 | 0.76 |
21 | 24.10 | 18.00 | 0.92 | 6.99 | 3.23 | 0.71 |
22 | 17.20 | 14.80 | 0.81 | 6.00 | 2.81 | 1.13 |
23 | 42.00 | 38.50 | 1.00 | 5.33 | 2.47 | 1.42 |
24 | 32.00 | 25.50 | 0.98 | 6.01 | 2.79 | 1.21 |
25 | 24.70 | 24.80 | 0.87 | 6.09 | 2.79 | 1.02 |
26 | 19.80 | 22.30 | 1.04 | 7.71 | 2.37 | 1.36 |
Averages | 29.54 | 23.56 | 0.96 | 6.74 | 3.00 | 1.04 |
Standard Deviation | 10.84 | 6.88 | 0.21 | 0.85 | 0.48 | 0.25 |
All patients Averages | 30.38 | 23.98 | 1.08 | 7.14 | 3.20 | 1.10 |
Standard Deviation | 9.05 | 5.57 | 0.26 | 0.88 | 0.55 | 0.26 |
aStatistical significance is denoted as P < .02.
Abbreviations: MRI, magnetic resonance imaging; PSIS, posterior iliac spine; SI, sacroiliac.
For the 13 males, the mean SI joint angle was 31.22° ± 7.18° (range, 20.20° to 48.50°). The mean approach angle was 24.41° ± 4.11° (range, 19.50° to 31.00°). The mean PSIS table width was 1.20 cm ± 0.26 cm (range, 0.84 cm to 1.70 cm). The mean distance from the PSIS to the anterior ilium wall was 7.53 cm ± 0.75 cm (range, 6.43 cm to 8.80 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.40 cm ± 0.56 cm (range, 2.34 cm to 4.16 cm). The mean minimum width of the ilium to the SI joint was 1.16 cm ± 0.26 cm (range, 0.65 cm to 1.56 cm).
For the 13 females, the mean SI joint angle was 29.54° ± 10.84° (range, 17.20° to 48.20°). The mean approach angle was 23.56° ± 6.88° (range, 14.80° to 38.50°). The mean PSIS table width was 0.96 cm ± 0.21 cm (range, 0.61 cm to 1.54 cm). The mean distance from the PSIS to the anterior ilium wall was 6.74 cm ± 0.85 cm (range, 5.33 cm to 8.32 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.00 cm ± 0.48 cm (range, 2.23 cm to 3.76 cm). The mean minimum width of the ilium to the SI joint was 1.04 cm ± 0.25 cm (range, 0.71 cm to 1.42 cm).
For the 26 total patients, the mean SI joint angle was 30.38° ± 9.05° (range, 17.20° to 48.50°). The mean approach angle was 23.98° ± 5.57° (range, 14.80° to 38.50°). The mean PSIS table width was 1.08 cm ± 0.26 cm (range, 0.61 cm to 1.70 cm). The mean distance from the PSIS to the anterior ilium wall was 7.14 cm ± 0.88 cm (range, 5.33 cm to 8.80 cm). The mean perpendicular distance from the PSIS table to the anterior ilium was 3.20 cm ± 0.55 cm (range, 2.23 cm to 4.16 cm). The mean minimum width of the ilium to the SI joint was 1.10 cm ± 0.26 cm (range, 0.65 cm to 1.56 cm).
There was a statistically significant difference between the male and female groups for the maximum distance the trocar can be advanced from the PSIS to the anterior ilium wall (P < .02), and a statistically significant difference for the PSIS table width (P < .02). There were no significant differences between the male and female groups for the approach angle, the SI joint angle, the perpendicular distance from the PSIS to the anterior ilium, and the minimum width of the ilium to the SI joint.
Continue to: The patient is brought to the procedure...
TECHNIQUE: ILIAC CREST (PSIS) BONE MARROW ASPIRATION
The patient is brought to the procedure room and placed in a prone position. The donor site is prepared and draped in the usual sterile manner. Ultrasound is used to identify the median sacral crest in a short-axis view. The probe is then moved laterally to identify the PSIS (Figures 4A, 4B).
The crosshairs on the ultrasound probe are used to mark the center lines of each plane. The central point marks the location of the PSIS. Alternatively, an in-plane technique can be used to place a spinal needle on the exact entry point on the PSIS. Once the PSIS and entry point are identified, the site is blocked with 10 mL of 0.5% ropivacaine.
Prior to introduction of the trocar, all instrumentation is primed with heparin and syringes are prepped with anticoagulant citrate dextrose solution, solution A. A stab incision is made at the site. The trocar is placed at the entry point, which should be centered in a superior-inferior plane and at the most medial point of the PSIS. Starting with the trocar vertical, the trocar is angled laterally 24° by dropping the hand medially toward the midline. No angulation cephalad or caudad is necessary, but cephalad must be avoided so as not to skive superiorly. This angle, which is recommended for both males and females, allows for the greatest distance the trocar can travel in bone before hitting the anterior ilium wall. A standard deviation of 5.57° is present, which should be considered. Steady pressure should be applied with a slight twisting motion on the PSIS. If advancement of the trocar is too difficult, a mallet or drill can be used to assist in penetration.
With the trocar advanced into the bone 1 cm, the trocar needle is removed while the cannula remains in place. The syringe is attached to the top of the cannula. The syringe plunger is pulled back to aspirate 20 mL of bone marrow. The cannula and syringe assembly are advanced 2 cm farther into the bone to allow for aspiration of a new location within the bone marrow cavity, and 20 mL of bone marrow are again aspirated. This is done a final time, advancing the trocar another 2 cm and aspirating a final 20 mL of bone marrow. The entire process should yield roughly 60 mL of bone marrow from one side. If desired, the same process can be repeated for the contralateral PSIS to yield a total of 120 mL of bone marrow from the 2 sites.
Based on our data, the average distance to the anterior ilium wall was 7 cm, but the shortest distance noted in this study was 5 cm. On the basis of the data presented, this technique allows for safe advancement based on even the shortest measured distance, without fear of puncturing the anterior ilium wall. Perforation could damage the femoral nerve and the internal or external iliac artery or vein that lie anterior to the ilium.
Continue to: We hypothesized that there...
DISCUSSION
We hypothesized that there would be an optimal angle of entry and maximal safe distance the trocar could advance through the ilium when aspirating. Because male and female pelvic anatomy differs, we also hypothesized that there would be differences in distance and size measurements for males and females. Our results supported our hypothesis that there is an ideal approach angle. The results also showed that the maximum distance the trocar can advance and the width of the PSIS table differ significantly between males and females.
Although pelvic anatomy differs between males and females, there should be an ideal entry angle that would allow maximum advancement into the ilium without perforating the anterior wall, which we defined as the approach angle. In our comparison of 26 MRI scans, we found that the approach angle did not differ significantly between the 2 groups (13 males, 13 females). This allows clinicians to enter the PSIS at roughly 24° medial to the parasagittal line, maximizing the space before puncturing into the anterior pelvis in either males or females.
If clinicians were to enter perpendicular to the patient’s PSIS, they would, on average, be able to advance only 3.20 cm before encountering the SI joint. When entering at 24° as we recommend, the average distance increases to 7.14 cm. Although the angle did not differ significantly, there was a significant difference between males and females in the length from the PSIS to the anterior wall, with males having 7.53 cm distance and females 6.74 cm. This is an important measurement because if the anterior ilium wall is punctured, the femoral nerve and the common, internal and external iliac arteries and veins could be damaged, resulting in retroperitoneal hemorrhage.
A fatality in 2001 in the United Kingdom led to a national audit of bone marrow aspiration and biopsies.4-6 Although these procedures were done primarily for patients with cancer, hemorrhagic events were the most frequent and serious events. This audit led to the identification of many risk factors. Bain4-6 conducted reviews of bone marrow aspirations and biopsies in the United Kingdom from 2002 to 2004. Of a total of 53,088 procedures conducted during that time frame, 48 (0.09%) adverse events occurred, with 29 (0.05%) being hemorrhagic events. Although infrequent, hemorrhagic adverse events represent significant morbidity. Reviews such as those conducted by Bain4-6 highlight the importance of a study that helps determine the optimal parameters for aspiration to ensure safety and reliability.
Hernigou and colleagues2,3 conducted studies analyzing different “sectors” in an attempt to develop a safe aspiration technique. They found that obese patients were at higher risk, and some sites of aspiration (sectors 1, 4, 5) had increased risk for perforation and damage to surrounding structures. Their sector 6, which incorporated the entirety of the PSIS table, was considered the safest, most reliable site for trocar introduction.2,3 Hernigou and colleagues,2 in comparing the bone mass of the sectors, also noted that sector 6 has the greatest bone thickness close to the entry point, making it the most favorable site. The PSIS is not just a point; it is more a “table.” The PSIS can be palpated posteriorly, but this is inaccurate and unreliable, particularly in larger individuals. The PSIS table can be identified on ultrasound before introducing the trocar, which is a more reliable method of landmark identification than palpation guidance, just as in ultrasound-guided injections7-16 and procedures.9,12,17-19
Continue to: If the PSIS is not accurately...
If the PSIS is not accurately identified, penetration laterally will result in entering the ilium wing, where it is quite narrow. We found the distance between the posterior ilium wall and the SI joint to be only 1.10 cm wide (Figure 3); we defined this area as the narrow corridor. Superior and lateral entry could damage the superior cluneal nerves coming over the iliac crest, which are located 6 cm lateral to the SI joint. Inferior and lateral entry 6 cm below the PSIS could reach the greater sciatic foramen, damaging the sacral plexus and superior gluteal artery and vein. If the entry slips above the PSIS over the pelvis, the trocar could enter the retroperitoneal space and damage the femoral nerve and common iliac artery and vein, leading to a retroperitoneal hemorrhage.4-6,20
MSCs are found as perivascular cells and lie in the cortices of bones.21 Following the approach angle and directed line from the PSIS to the anterior ilium wall described in this study (Figures 1 and 2), the trocar would pass through the narrow corridor as it advances farther into the ilium. The minimum width of this corridor was measured in this study and, on average, was 1.10 cm wide from cortex to cortex (Figure 3). As the bone marrow is aspirated from this narrow corridor, the clinician is gathering MSCs from both the lateral and medial cortices of the ilium. By aspirating from a greater surface area of the cortices, it is believed that this will increase the total collection of MSCs.
CONCLUSION
Although there are reports in the literature that describe techniques for bone marrow aspiration from the iliac crest, the techniques are very general and vague regarding the ideal angles and methods. Studies have attempted to quantify the safest entry sites for aspiration but have not detailed ideal parameters for collection. Blind aspiration from the iliac crest can have serious implications if adverse events occur, and thus there is a need for a safe and reliable method of aspiration from the iliac crest. Ultrasound guidance to identify anatomy, as opposed to palpation guidance, ensures anatomic placement of the trocar while minimizing the risk of aspiration. Based on the measurements gathered in this study, an optimal angle of entry and safe distance of penetration have been identified. Using our data and relevant literature, we developed a technique for a safe, consistent, and reliable method of bone marrow aspiration out of the iliac crest.
1. Chahla J, Mannava S, Cinque ME, Geeslin AG, Codina D, LaPrade RF. Bone marrow aspirate concentrate harvesting and processing technique. Arthrosc Tech. 2017;6(2):e441-e445. doi:10.1016/j.eats.2016.10.024.
2. Hernigou J, Alves A, Homma Y, Guissou I, Hernigou P. Anatomy of the ilium for bone marrow aspiration: map of sectors and implication for safe trocar placement. Int Orthop. 2014;38(12):2585-2590. doi:10.1007/s00264-014-2353-7.
3. Hernigou J, Picard L, Alves A, Silvera J, Homma Y, Hernigou P. Understanding bone safety zones during bone marrow aspiration from the iliac crest: the sector rule. Int Orthop. 2014;38(11):2377-2384. doi:10.1007/s00264-014-2343-9.
4. Bain BJ. Bone marrow biopsy morbidity: review of 2003. J Clin Pathol. 2005;58(4):406-408. doi:10.1136/jcp.2004.022178.
5. Bain BJ. Bone marrow biopsy morbidity and mortality: 2002 data. Clin Lab Haematol. 2004;26(5):315-318. doi:10.1111/j.1365-2257.2004.00630.x.
6. Bain BJ. Morbidity associated with bone marrow aspiration and trephine biopsy - a review of UK data for 2004. Haematologica. 2006;91(9):1293-1294.
7. Berkoff DJ, Miller LE, Block JE. Clinical utility of ultrasound guidance for intra-articular knee injections: a review. Clin Interv Aging. 2012;7:89-95. doi:10.2147/CIA.S29265.
8. Henkus HE, Cobben LP, Coerkamp EG, Nelissen RG, van Arkel ER. The accuracy of subacromial injections: a prospective randomized magnetic resonance imaging study. Arthroscopy. 2006;22(3):277-282. doi:10.1016/j.arthro.2005.12.019.
9. Hirahara AM, Panero AJ. A guide to ultrasound of the shoulder, part 3: interventional and procedural uses. Am J Orthop. 2016;45(7):440-445.
10. Jackson DW, Evans NA, Thomas BM. Accuracy of needle placement into the intra-articular space of the knee. J Bone Joint Surg Am. 2002;84-A(9):1522-1527.
11. Naredo E, Cabero F, Beneyto P, et al. A randomized comparative study of short term response to blind versus sonographic-guided injection of local corticosteroids in patients with painful shoulder. J Rheumatol. 2004;31(2):308-314.
12. Panero AJ, Hirahara AM. A guide to ultrasound of the shoulder, part 2: the diagnostic evaluation. Am J Orthop. 2016;45(4):233-238.
13. Sethi PM, El Attrache N. Accuracy of intra-articular injection of the glenohumeral joint: a cadaveric study. Orthopedics. 2006;29(2):149-152.
14. Sibbit WL Jr, Peisajovich A, Michael AA, et al. Does sonographic needle guidance affect the clinical outcome of intraarticular injections? J Rheumatol. 2009;36(9):1892-1902. doi:10.3899/jrheum.090013.
15. Smith J, Brault JS, Rizzo M, Sayeed YA, Finnoff JT. Accuracy of sonographically guided and palpation guided scaphotrapeziotrapezoid joint injections. J Ultrasound Med. 2011;30(11):1509-1515. doi:10.7863/jum.2011.30.11.1509.
16. Yamakado K. The targeting accuracy of subacromial injection to the shoulder: an arthrographic evaluation. Arthroscopy. 2002;18(8):887-891.
17. Hirahara AM, Andersen WJ. Ultrasound-guided percutaneous reconstruction of the anterolateral ligament: surgical technique and case report. Am J Orthop. 2016;45(7):418-422, 460.
18. Hirahara AM, Andersen WJ. Ultrasound-guided percutaneous repair of medial patellofemoral ligament: surgical technique and outcomes. Am J Orthop. 2017;46(3):152-157.
19. Hirahara AM, Mackay G, Andersen WJ. Ultrasound-guided InternalBrace of the medial collateral ligament. Arthrosc Tech. Submitted.
20. Jamaludin WFW, Mukari SAM, Wahid SFA. Retroperitoneal hemorrhage associated with bone marrow trephine biopsy. Am J Case Rep. 2013;14:489-493. doi:10.12659/AJCR.889274.
21. Bianco P, Cao X, Frenette PS, et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19(1):35-42. doi:10.1038/nm.3028.
1. Chahla J, Mannava S, Cinque ME, Geeslin AG, Codina D, LaPrade RF. Bone marrow aspirate concentrate harvesting and processing technique. Arthrosc Tech. 2017;6(2):e441-e445. doi:10.1016/j.eats.2016.10.024.
2. Hernigou J, Alves A, Homma Y, Guissou I, Hernigou P. Anatomy of the ilium for bone marrow aspiration: map of sectors and implication for safe trocar placement. Int Orthop. 2014;38(12):2585-2590. doi:10.1007/s00264-014-2353-7.
3. Hernigou J, Picard L, Alves A, Silvera J, Homma Y, Hernigou P. Understanding bone safety zones during bone marrow aspiration from the iliac crest: the sector rule. Int Orthop. 2014;38(11):2377-2384. doi:10.1007/s00264-014-2343-9.
4. Bain BJ. Bone marrow biopsy morbidity: review of 2003. J Clin Pathol. 2005;58(4):406-408. doi:10.1136/jcp.2004.022178.
5. Bain BJ. Bone marrow biopsy morbidity and mortality: 2002 data. Clin Lab Haematol. 2004;26(5):315-318. doi:10.1111/j.1365-2257.2004.00630.x.
6. Bain BJ. Morbidity associated with bone marrow aspiration and trephine biopsy - a review of UK data for 2004. Haematologica. 2006;91(9):1293-1294.
7. Berkoff DJ, Miller LE, Block JE. Clinical utility of ultrasound guidance for intra-articular knee injections: a review. Clin Interv Aging. 2012;7:89-95. doi:10.2147/CIA.S29265.
8. Henkus HE, Cobben LP, Coerkamp EG, Nelissen RG, van Arkel ER. The accuracy of subacromial injections: a prospective randomized magnetic resonance imaging study. Arthroscopy. 2006;22(3):277-282. doi:10.1016/j.arthro.2005.12.019.
9. Hirahara AM, Panero AJ. A guide to ultrasound of the shoulder, part 3: interventional and procedural uses. Am J Orthop. 2016;45(7):440-445.
10. Jackson DW, Evans NA, Thomas BM. Accuracy of needle placement into the intra-articular space of the knee. J Bone Joint Surg Am. 2002;84-A(9):1522-1527.
11. Naredo E, Cabero F, Beneyto P, et al. A randomized comparative study of short term response to blind versus sonographic-guided injection of local corticosteroids in patients with painful shoulder. J Rheumatol. 2004;31(2):308-314.
12. Panero AJ, Hirahara AM. A guide to ultrasound of the shoulder, part 2: the diagnostic evaluation. Am J Orthop. 2016;45(4):233-238.
13. Sethi PM, El Attrache N. Accuracy of intra-articular injection of the glenohumeral joint: a cadaveric study. Orthopedics. 2006;29(2):149-152.
14. Sibbit WL Jr, Peisajovich A, Michael AA, et al. Does sonographic needle guidance affect the clinical outcome of intraarticular injections? J Rheumatol. 2009;36(9):1892-1902. doi:10.3899/jrheum.090013.
15. Smith J, Brault JS, Rizzo M, Sayeed YA, Finnoff JT. Accuracy of sonographically guided and palpation guided scaphotrapeziotrapezoid joint injections. J Ultrasound Med. 2011;30(11):1509-1515. doi:10.7863/jum.2011.30.11.1509.
16. Yamakado K. The targeting accuracy of subacromial injection to the shoulder: an arthrographic evaluation. Arthroscopy. 2002;18(8):887-891.
17. Hirahara AM, Andersen WJ. Ultrasound-guided percutaneous reconstruction of the anterolateral ligament: surgical technique and case report. Am J Orthop. 2016;45(7):418-422, 460.
18. Hirahara AM, Andersen WJ. Ultrasound-guided percutaneous repair of medial patellofemoral ligament: surgical technique and outcomes. Am J Orthop. 2017;46(3):152-157.
19. Hirahara AM, Mackay G, Andersen WJ. Ultrasound-guided InternalBrace of the medial collateral ligament. Arthrosc Tech. Submitted.
20. Jamaludin WFW, Mukari SAM, Wahid SFA. Retroperitoneal hemorrhage associated with bone marrow trephine biopsy. Am J Case Rep. 2013;14:489-493. doi:10.12659/AJCR.889274.
21. Bianco P, Cao X, Frenette PS, et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19(1):35-42. doi:10.1038/nm.3028.
TAKE-HOME POINTS
- There is an ideal angle and distance for optimization of a bone marrow harvest from the iliac crest.
- Ultrasound is a reliable technology that allows clinicians to accurately and consistently identify the PSIS and avoid neurovascular structures.
- This safe, reliable bone marrow aspiration technique can lower the risk of serious potential complications.
- The ideal angle does not differ significantly between sexes, but the safe distance a clinician can advance does.
- The PSIS should be considered a “table” as opposed to a protuberance.
FP can perform the circumcision
FP can perform the circumcision
I enjoyed Dr. Lerner's brief review but was puzzled by the question of who should perform the circumcision. The most obvious and one of the most frequent answers was ignored. It should be done by the family physician who delivered the baby, is caring for him in the nursery, and will be caring for him another couple of decades.
John R. Carroll, MD
Corpus Christi, Texas
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
FP can perform the circumcision
I enjoyed Dr. Lerner's brief review but was puzzled by the question of who should perform the circumcision. The most obvious and one of the most frequent answers was ignored. It should be done by the family physician who delivered the baby, is caring for him in the nursery, and will be caring for him another couple of decades.
John R. Carroll, MD
Corpus Christi, Texas
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
FP can perform the circumcision
I enjoyed Dr. Lerner's brief review but was puzzled by the question of who should perform the circumcision. The most obvious and one of the most frequent answers was ignored. It should be done by the family physician who delivered the baby, is caring for him in the nursery, and will be caring for him another couple of decades.
John R. Carroll, MD
Corpus Christi, Texas
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.

Does Age of Exposure to Tackle Football Affect CTE Severity?
Younger age of exposure to tackle football is not associated with chronic traumatic encephalopathy (CTE) pathologic severity, Alzheimer’s disease pathology, or Lewy body pathology, according to data published online ahead of print April 30 in Annals of Neurology. Younger age of exposure does appear to predict earlier neurobehavioral symptom onset, however, the authors said.

“These findings suggest that exposure to repetitive head impacts from tackle football as a youth may reduce resiliency to diseases, including, but not limited to, CTE, that affect the brain in later life,” said Michael L. Alosco, PhD, Assistant Professor of Neurology at the the Boston University Alzheimer’s Disease and CTE Center. “This study adds to growing research suggesting that incurring repeated head impacts through tackle football in earlier life can lead to both short-term and long-term effects on the brain.”
Repetitive Head Impacts and Neurodevelopment
Previous research has linked younger age of first exposure to tackle football with smaller thalamic volume in former National Football League players. A recent study of 214 former and amateur football players found that age of first exposure to tackle football—before age 12, in particular—predicted increased odds of self-reported neuropsychiatric and executive impairment.
“Youth exposure to repetitive head impacts may disrupt neurodevelopment to lower the threshold for later clinical dysfunction,” said the researchers.
To examine the effect of age of first exposure to tackle football on CTE pathologic severity and age of neurobehavioral symptom onset in tackle football players with neuropathologically confirmed CTE, Dr. Alosco and colleagues analyzed a sample of 246 amateur and professional tackle football players whose brains had been donated to the Veteran’s Affairs–Boston University–Concussion Legacy Foundation Brain Bank. The researchers interviewed informants to ascertain players’ age of first exposure and age of onset of cognitive, behavioral, or mood symptoms. A total of 211 football players were diagnosed with CTE; 35 did not have CTE. Of the 211 participants with CTE, 126 had CTE only, and the other participants had comorbid neurodegenerative diseases.
Onset of Cognitive, Behavioral, and Mood Symptoms
Of the 211 participants with CTE, 183 developed cognitive and behavioral or mood symptoms prior to death, eight had only cognitive symptoms, 12 had only behavioral or mood symptoms, and seven did not endorse any symptoms examined in the study. Clinical data for one participant were not available.
Among tackle football players with CTE, every one year younger that they began to play tackle football predicted earlier onset of cognitive symptoms by 2.44 years and of behavioral or mood symptoms by 2.50 years. Exposure before age 12 predicted earlier cognitive and behavioral or mood symptom onset by 13.39 years and 13.28 years, respectively.
Secondary subset analyses indicated that younger age of exposure to tackle football was associated with earlier onset of functional impairment in participants who were determined to have had dementia. Researchers observed nearly identical effects in participants with CTE only.
Study limitations include the lack of an appropriate control or comparison group, the researchers noted. In addition, the results may not be generalizable to a broader tackle football population.
“Given the growing public health concerns for participation in tackle football, prospective studies of former tackle football players that include objective clinical assessments are needed to better understand the relationship between youth tackle football exposure and long-term neurobehavioral outcomes,” said the researchers.
“More research on this topic is needed before any clinical recommendations, as well as recommendations on policy or rule changes, can be made,” said Dr. Alosco.
“Boston University and sites across the country are currently conducting longitudinal studies on former football players, which will allow us to begin to study cognition and behavior and mood functioning over time.”
—Erica Tricarico
Suggested Reading
Alosco ML, Mez J, Tripodis Y, et al. Age of first exposure to tackle football and chronic traumatic encephalopathy. Ann Neurol. 2018 Apr 30 [Epub ahead of print].
Younger age of exposure to tackle football is not associated with chronic traumatic encephalopathy (CTE) pathologic severity, Alzheimer’s disease pathology, or Lewy body pathology, according to data published online ahead of print April 30 in Annals of Neurology. Younger age of exposure does appear to predict earlier neurobehavioral symptom onset, however, the authors said.
“These findings suggest that exposure to repetitive head impacts from tackle football as a youth may reduce resiliency to diseases, including, but not limited to, CTE, that affect the brain in later life,” said Michael L. Alosco, PhD, Assistant Professor of Neurology at the the Boston University Alzheimer’s Disease and CTE Center. “This study adds to growing research suggesting that incurring repeated head impacts through tackle football in earlier life can lead to both short-term and long-term effects on the brain.”
Repetitive Head Impacts and Neurodevelopment
Previous research has linked younger age of first exposure to tackle football with smaller thalamic volume in former National Football League players. A recent study of 214 former and amateur football players found that age of first exposure to tackle football—before age 12, in particular—predicted increased odds of self-reported neuropsychiatric and executive impairment.
“Youth exposure to repetitive head impacts may disrupt neurodevelopment to lower the threshold for later clinical dysfunction,” said the researchers.
To examine the effect of age of first exposure to tackle football on CTE pathologic severity and age of neurobehavioral symptom onset in tackle football players with neuropathologically confirmed CTE, Dr. Alosco and colleagues analyzed a sample of 246 amateur and professional tackle football players whose brains had been donated to the Veteran’s Affairs–Boston University–Concussion Legacy Foundation Brain Bank. The researchers interviewed informants to ascertain players’ age of first exposure and age of onset of cognitive, behavioral, or mood symptoms. A total of 211 football players were diagnosed with CTE; 35 did not have CTE. Of the 211 participants with CTE, 126 had CTE only, and the other participants had comorbid neurodegenerative diseases.
Onset of Cognitive, Behavioral, and Mood Symptoms
Of the 211 participants with CTE, 183 developed cognitive and behavioral or mood symptoms prior to death, eight had only cognitive symptoms, 12 had only behavioral or mood symptoms, and seven did not endorse any symptoms examined in the study. Clinical data for one participant were not available.
Among tackle football players with CTE, every one year younger that they began to play tackle football predicted earlier onset of cognitive symptoms by 2.44 years and of behavioral or mood symptoms by 2.50 years. Exposure before age 12 predicted earlier cognitive and behavioral or mood symptom onset by 13.39 years and 13.28 years, respectively.
Secondary subset analyses indicated that younger age of exposure to tackle football was associated with earlier onset of functional impairment in participants who were determined to have had dementia. Researchers observed nearly identical effects in participants with CTE only.
Study limitations include the lack of an appropriate control or comparison group, the researchers noted. In addition, the results may not be generalizable to a broader tackle football population.
“Given the growing public health concerns for participation in tackle football, prospective studies of former tackle football players that include objective clinical assessments are needed to better understand the relationship between youth tackle football exposure and long-term neurobehavioral outcomes,” said the researchers.
“More research on this topic is needed before any clinical recommendations, as well as recommendations on policy or rule changes, can be made,” said Dr. Alosco.
“Boston University and sites across the country are currently conducting longitudinal studies on former football players, which will allow us to begin to study cognition and behavior and mood functioning over time.”
—Erica Tricarico
Suggested Reading
Alosco ML, Mez J, Tripodis Y, et al. Age of first exposure to tackle football and chronic traumatic encephalopathy. Ann Neurol. 2018 Apr 30 [Epub ahead of print].
Younger age of exposure to tackle football is not associated with chronic traumatic encephalopathy (CTE) pathologic severity, Alzheimer’s disease pathology, or Lewy body pathology, according to data published online ahead of print April 30 in Annals of Neurology. Younger age of exposure does appear to predict earlier neurobehavioral symptom onset, however, the authors said.
“These findings suggest that exposure to repetitive head impacts from tackle football as a youth may reduce resiliency to diseases, including, but not limited to, CTE, that affect the brain in later life,” said Michael L. Alosco, PhD, Assistant Professor of Neurology at the the Boston University Alzheimer’s Disease and CTE Center. “This study adds to growing research suggesting that incurring repeated head impacts through tackle football in earlier life can lead to both short-term and long-term effects on the brain.”
Repetitive Head Impacts and Neurodevelopment
Previous research has linked younger age of first exposure to tackle football with smaller thalamic volume in former National Football League players. A recent study of 214 former and amateur football players found that age of first exposure to tackle football—before age 12, in particular—predicted increased odds of self-reported neuropsychiatric and executive impairment.
“Youth exposure to repetitive head impacts may disrupt neurodevelopment to lower the threshold for later clinical dysfunction,” said the researchers.
To examine the effect of age of first exposure to tackle football on CTE pathologic severity and age of neurobehavioral symptom onset in tackle football players with neuropathologically confirmed CTE, Dr. Alosco and colleagues analyzed a sample of 246 amateur and professional tackle football players whose brains had been donated to the Veteran’s Affairs–Boston University–Concussion Legacy Foundation Brain Bank. The researchers interviewed informants to ascertain players’ age of first exposure and age of onset of cognitive, behavioral, or mood symptoms. A total of 211 football players were diagnosed with CTE; 35 did not have CTE. Of the 211 participants with CTE, 126 had CTE only, and the other participants had comorbid neurodegenerative diseases.
Onset of Cognitive, Behavioral, and Mood Symptoms
Of the 211 participants with CTE, 183 developed cognitive and behavioral or mood symptoms prior to death, eight had only cognitive symptoms, 12 had only behavioral or mood symptoms, and seven did not endorse any symptoms examined in the study. Clinical data for one participant were not available.
Among tackle football players with CTE, every one year younger that they began to play tackle football predicted earlier onset of cognitive symptoms by 2.44 years and of behavioral or mood symptoms by 2.50 years. Exposure before age 12 predicted earlier cognitive and behavioral or mood symptom onset by 13.39 years and 13.28 years, respectively.
Secondary subset analyses indicated that younger age of exposure to tackle football was associated with earlier onset of functional impairment in participants who were determined to have had dementia. Researchers observed nearly identical effects in participants with CTE only.
Study limitations include the lack of an appropriate control or comparison group, the researchers noted. In addition, the results may not be generalizable to a broader tackle football population.
“Given the growing public health concerns for participation in tackle football, prospective studies of former tackle football players that include objective clinical assessments are needed to better understand the relationship between youth tackle football exposure and long-term neurobehavioral outcomes,” said the researchers.
“More research on this topic is needed before any clinical recommendations, as well as recommendations on policy or rule changes, can be made,” said Dr. Alosco.
“Boston University and sites across the country are currently conducting longitudinal studies on former football players, which will allow us to begin to study cognition and behavior and mood functioning over time.”
—Erica Tricarico
Suggested Reading
Alosco ML, Mez J, Tripodis Y, et al. Age of first exposure to tackle football and chronic traumatic encephalopathy. Ann Neurol. 2018 Apr 30 [Epub ahead of print].