User login
Conservative early approach likely best, RA expert says
SANDESTIN, FLA. – A conservative approach to early rheumatoid arthritis treatment has carried the day in the practice of Gerd R. Burmester, MD.
In a talk at the annual Congress of Clinical Rheumatology, Dr. Burmester said that, although there is room to argue for a more aggressive approach, with more intense treatment early, a less aggressive philosophy has worked well in his clinic.

Dr. Burmester, director of rheumatology and clinical immunology at Charite-University in Berlin and a past president of the European League Against Rheumatism (EULAR), said he drew inspiration from the results of the 2015 study CARE-RA, in which patients were treated with initial therapy of methotrexate plus sulfasalazine and a fairly high dose of 60 mg of prednisolone; methotrexate plus leflunomide plus 30 mg of prednisolone; or just methotrexate plus 30 mg of prednisolone that is quickly tapered down (Ann Rheum Dis. 2015 Jan;74[1]:27-34).
“Everyone would say, ‘Okay, this is quite easy – the more intensive drug regimen should give you better results,’ ” Dr. Burmester said. “But if you look at the data, there’s no difference.” And after just 8 weeks, the patients’ corticosteroid dose was down to 5 mg.
This, he said, “has changed my daily typical practice, quite a bit.”
“I start with, usually, 15 mg of methotrexate subcutaneously,” because of better efficacy and less liver toxicity than oral administration, he said, or an oral dose if a patient resists the subcutaneous administration or there is another reason to avoid it. “And I add 30 mg of prednisone and taper it down – 30, 20, 12.5 mg, and then down to 5 and eventually discontinued altogether.”
“This is an interesting scheme,” he said. “And this is exactly what I do with my patients.”
His approach might be worth noting not only for his leadership roles, but because of his fastidious approach to being a clinician – he said he still, personally, takes every patient’s 28-joint Disease Activity Score and Simple Disease Activity Index at every visit.
In a recent paper, he argued, along with prominent Canadian rheumatologist Janet Pope, both sides of the debate, for and against more aggressive treatment – methotrexate combined with conventional synthetic or biologic DMARDs – very early in the disease course (Lancet. 2017 Jun 10;389[10086]:2338-48).
“If you use a combination treatment with a biologic right away, what might be the advantages?” he said. “More patients would achieve rapid remission. It might result in long-term benefits, less joint damage, higher chance of reducing therapy in the future.”
On the other hand, he said, there are disadvantages.
“This is, of course, more expensive, if you use a biologic up front in early RA,” he said. “Not all patients of course need it, and some have also side effects.” He added that little time is lost if a treat-to-target principle is followed. Plus, patients tend to be more accepting of monotherapy than combination therapy at the start of treatment, and combination therapy might require more time spent in the clinic.
Data from German databases, dating back to 1997, show that far more patients are reaching remission today after several years of treatment (Z Rheumatol. 2017 Feb;76[1]:50-7). But, he added, “It’s not yet perfect. ... We still have quite a few patients who are in moderate disease activity” despite the availability of so many treatment options.
“There’s still, of course, a huge unmet need in this devastating disease if you don’t treat it correctly.”
Dr. Burmester reports receiving clinical trial support and/or honoraria for lectures and consulting from AbbVie, Bristol-Myers Squibb, Lilly, Roche, MedImmune, Merck Sharpe & Dohme, Pfizer, Sanofi, and UCB.
SANDESTIN, FLA. – A conservative approach to early rheumatoid arthritis treatment has carried the day in the practice of Gerd R. Burmester, MD.
In a talk at the annual Congress of Clinical Rheumatology, Dr. Burmester said that, although there is room to argue for a more aggressive approach, with more intense treatment early, a less aggressive philosophy has worked well in his clinic.
Dr. Burmester, director of rheumatology and clinical immunology at Charite-University in Berlin and a past president of the European League Against Rheumatism (EULAR), said he drew inspiration from the results of the 2015 study CARE-RA, in which patients were treated with initial therapy of methotrexate plus sulfasalazine and a fairly high dose of 60 mg of prednisolone; methotrexate plus leflunomide plus 30 mg of prednisolone; or just methotrexate plus 30 mg of prednisolone that is quickly tapered down (Ann Rheum Dis. 2015 Jan;74[1]:27-34).
“Everyone would say, ‘Okay, this is quite easy – the more intensive drug regimen should give you better results,’ ” Dr. Burmester said. “But if you look at the data, there’s no difference.” And after just 8 weeks, the patients’ corticosteroid dose was down to 5 mg.
This, he said, “has changed my daily typical practice, quite a bit.”
“I start with, usually, 15 mg of methotrexate subcutaneously,” because of better efficacy and less liver toxicity than oral administration, he said, or an oral dose if a patient resists the subcutaneous administration or there is another reason to avoid it. “And I add 30 mg of prednisone and taper it down – 30, 20, 12.5 mg, and then down to 5 and eventually discontinued altogether.”
“This is an interesting scheme,” he said. “And this is exactly what I do with my patients.”
His approach might be worth noting not only for his leadership roles, but because of his fastidious approach to being a clinician – he said he still, personally, takes every patient’s 28-joint Disease Activity Score and Simple Disease Activity Index at every visit.
In a recent paper, he argued, along with prominent Canadian rheumatologist Janet Pope, both sides of the debate, for and against more aggressive treatment – methotrexate combined with conventional synthetic or biologic DMARDs – very early in the disease course (Lancet. 2017 Jun 10;389[10086]:2338-48).
“If you use a combination treatment with a biologic right away, what might be the advantages?” he said. “More patients would achieve rapid remission. It might result in long-term benefits, less joint damage, higher chance of reducing therapy in the future.”
On the other hand, he said, there are disadvantages.
“This is, of course, more expensive, if you use a biologic up front in early RA,” he said. “Not all patients of course need it, and some have also side effects.” He added that little time is lost if a treat-to-target principle is followed. Plus, patients tend to be more accepting of monotherapy than combination therapy at the start of treatment, and combination therapy might require more time spent in the clinic.
Data from German databases, dating back to 1997, show that far more patients are reaching remission today after several years of treatment (Z Rheumatol. 2017 Feb;76[1]:50-7). But, he added, “It’s not yet perfect. ... We still have quite a few patients who are in moderate disease activity” despite the availability of so many treatment options.
“There’s still, of course, a huge unmet need in this devastating disease if you don’t treat it correctly.”
Dr. Burmester reports receiving clinical trial support and/or honoraria for lectures and consulting from AbbVie, Bristol-Myers Squibb, Lilly, Roche, MedImmune, Merck Sharpe & Dohme, Pfizer, Sanofi, and UCB.
SANDESTIN, FLA. – A conservative approach to early rheumatoid arthritis treatment has carried the day in the practice of Gerd R. Burmester, MD.
In a talk at the annual Congress of Clinical Rheumatology, Dr. Burmester said that, although there is room to argue for a more aggressive approach, with more intense treatment early, a less aggressive philosophy has worked well in his clinic.
Dr. Burmester, director of rheumatology and clinical immunology at Charite-University in Berlin and a past president of the European League Against Rheumatism (EULAR), said he drew inspiration from the results of the 2015 study CARE-RA, in which patients were treated with initial therapy of methotrexate plus sulfasalazine and a fairly high dose of 60 mg of prednisolone; methotrexate plus leflunomide plus 30 mg of prednisolone; or just methotrexate plus 30 mg of prednisolone that is quickly tapered down (Ann Rheum Dis. 2015 Jan;74[1]:27-34).
“Everyone would say, ‘Okay, this is quite easy – the more intensive drug regimen should give you better results,’ ” Dr. Burmester said. “But if you look at the data, there’s no difference.” And after just 8 weeks, the patients’ corticosteroid dose was down to 5 mg.
This, he said, “has changed my daily typical practice, quite a bit.”
“I start with, usually, 15 mg of methotrexate subcutaneously,” because of better efficacy and less liver toxicity than oral administration, he said, or an oral dose if a patient resists the subcutaneous administration or there is another reason to avoid it. “And I add 30 mg of prednisone and taper it down – 30, 20, 12.5 mg, and then down to 5 and eventually discontinued altogether.”
“This is an interesting scheme,” he said. “And this is exactly what I do with my patients.”
His approach might be worth noting not only for his leadership roles, but because of his fastidious approach to being a clinician – he said he still, personally, takes every patient’s 28-joint Disease Activity Score and Simple Disease Activity Index at every visit.
In a recent paper, he argued, along with prominent Canadian rheumatologist Janet Pope, both sides of the debate, for and against more aggressive treatment – methotrexate combined with conventional synthetic or biologic DMARDs – very early in the disease course (Lancet. 2017 Jun 10;389[10086]:2338-48).
“If you use a combination treatment with a biologic right away, what might be the advantages?” he said. “More patients would achieve rapid remission. It might result in long-term benefits, less joint damage, higher chance of reducing therapy in the future.”
On the other hand, he said, there are disadvantages.
“This is, of course, more expensive, if you use a biologic up front in early RA,” he said. “Not all patients of course need it, and some have also side effects.” He added that little time is lost if a treat-to-target principle is followed. Plus, patients tend to be more accepting of monotherapy than combination therapy at the start of treatment, and combination therapy might require more time spent in the clinic.
Data from German databases, dating back to 1997, show that far more patients are reaching remission today after several years of treatment (Z Rheumatol. 2017 Feb;76[1]:50-7). But, he added, “It’s not yet perfect. ... We still have quite a few patients who are in moderate disease activity” despite the availability of so many treatment options.
“There’s still, of course, a huge unmet need in this devastating disease if you don’t treat it correctly.”
Dr. Burmester reports receiving clinical trial support and/or honoraria for lectures and consulting from AbbVie, Bristol-Myers Squibb, Lilly, Roche, MedImmune, Merck Sharpe & Dohme, Pfizer, Sanofi, and UCB.
EXPERT ANALYSIS FROM CCR 18
NIH launches early Ebola treatment trial
A study of a potential new Ebola treatment has begun at the National Institutes of Health Clinical Center in Bethesda, Md. The small phase 1 clinical trial will examine the safety and tolerability of a single monoclonal antibody (mAb114), which was developed by scientists at the National Institute of Allergy and Infectious Diseases (NIAID) and their collaborators. Investigators plan to enroll between 18 and 30 healthy volunteers aged 18-60. The trial will not expose participants to Ebola virus, according to the NIH announcement.
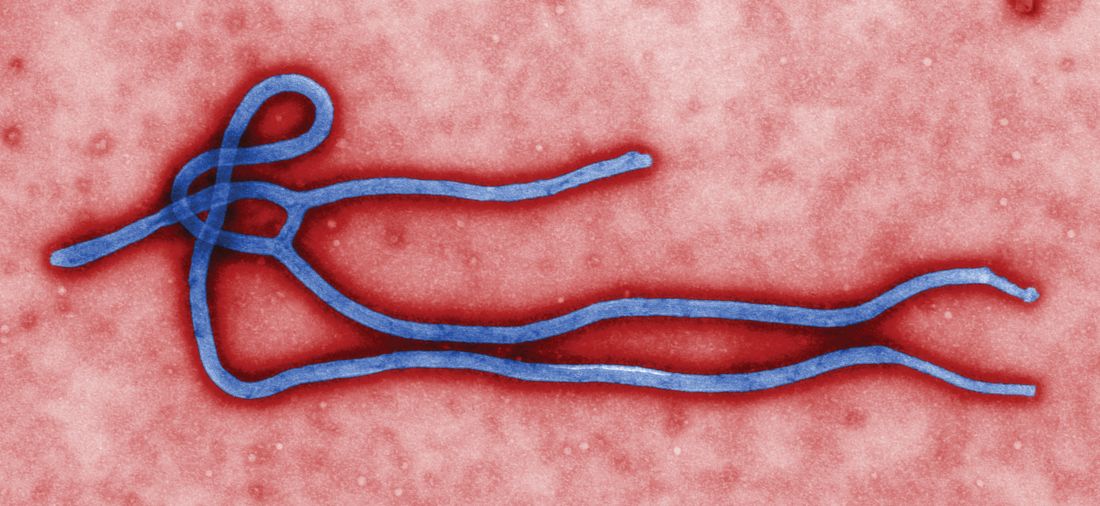
MAb114 is a monoclonal antibody – a protein that binds to a single target on a pathogen — isolated from a human survivor of the 1995 Ebola outbreak in Kikwit, Democratic Republic of the Congo. Nancy Sullivan, PhD, chief of the Biodefense Research Section in NIAID’s Vaccine Research Center, and her team, in collaboration with researchers from the National Institute of Biomedical Research in the Democratic Republic of the Congo and the Institute for Biomedical Research in Switzerland, discovered that the survivor retained antibodies against Ebola 11 years after infection. They isolated and tested the antibodies and selected mAb114 as the most promising.
Although rVSV-ZEBOV, an experimental vaccine, is now available and in use in Africa during the current outbreak, specific treatment modalities are lacking.
More information can be found at www.clinicaltrials.gov, trial # NCT03478891.
A study of a potential new Ebola treatment has begun at the National Institutes of Health Clinical Center in Bethesda, Md. The small phase 1 clinical trial will examine the safety and tolerability of a single monoclonal antibody (mAb114), which was developed by scientists at the National Institute of Allergy and Infectious Diseases (NIAID) and their collaborators. Investigators plan to enroll between 18 and 30 healthy volunteers aged 18-60. The trial will not expose participants to Ebola virus, according to the NIH announcement.
MAb114 is a monoclonal antibody – a protein that binds to a single target on a pathogen — isolated from a human survivor of the 1995 Ebola outbreak in Kikwit, Democratic Republic of the Congo. Nancy Sullivan, PhD, chief of the Biodefense Research Section in NIAID’s Vaccine Research Center, and her team, in collaboration with researchers from the National Institute of Biomedical Research in the Democratic Republic of the Congo and the Institute for Biomedical Research in Switzerland, discovered that the survivor retained antibodies against Ebola 11 years after infection. They isolated and tested the antibodies and selected mAb114 as the most promising.
Although rVSV-ZEBOV, an experimental vaccine, is now available and in use in Africa during the current outbreak, specific treatment modalities are lacking.
More information can be found at www.clinicaltrials.gov, trial # NCT03478891.
A study of a potential new Ebola treatment has begun at the National Institutes of Health Clinical Center in Bethesda, Md. The small phase 1 clinical trial will examine the safety and tolerability of a single monoclonal antibody (mAb114), which was developed by scientists at the National Institute of Allergy and Infectious Diseases (NIAID) and their collaborators. Investigators plan to enroll between 18 and 30 healthy volunteers aged 18-60. The trial will not expose participants to Ebola virus, according to the NIH announcement.
MAb114 is a monoclonal antibody – a protein that binds to a single target on a pathogen — isolated from a human survivor of the 1995 Ebola outbreak in Kikwit, Democratic Republic of the Congo. Nancy Sullivan, PhD, chief of the Biodefense Research Section in NIAID’s Vaccine Research Center, and her team, in collaboration with researchers from the National Institute of Biomedical Research in the Democratic Republic of the Congo and the Institute for Biomedical Research in Switzerland, discovered that the survivor retained antibodies against Ebola 11 years after infection. They isolated and tested the antibodies and selected mAb114 as the most promising.
Although rVSV-ZEBOV, an experimental vaccine, is now available and in use in Africa during the current outbreak, specific treatment modalities are lacking.
More information can be found at www.clinicaltrials.gov, trial # NCT03478891.
MDR Candida auris is on the move
MADRID – The anticipated global emergence of multidrug resistant Candida auris is now an established fact, but a case study presented at the European Society of Clinical Microbiology and Infectious Diseases annual congress demonstrates just how devastating an outbreak can be to a medical facility and its surgical ICU patients.
The dangerous invasive infection is spreading through Asia, Europe, and the Americas, causing potentially fatal candidemias and proving devilishly difficult to eradicate in health care facilities once it becomes established.
Several multidrug resistant (MDR) C. auris outbreaks were reported at the ECCMID meeting. Most troubling: a continuing outbreak in a hospital in Valencia, Spain, in which 17 patients have died – a 41% fatality rate among those who developed a fulminant C. auris candidemia, Javier Pemán, MD, said at the meeting. The strain appeared to be a clonal population not previously identified in published reports.
“C. auris is hard to remove from the hospital environment,” once it becomes established, said Dr. Pemán of La Fe University and Polytechnic Hospital, Valencia. “When an outbreak lasts for months, as ours has, it is difficult, but necessary, to maintain control measures, identify it early in the lab, and isolate and treat patients early with combination therapy.”

He and his team have relied primarily on a combination of amphotericin B and echinocandin (AMB+ECN), although, he added, the optimal dosing and treatment time aren’t known, and many C. auris isolates are echinocandin resistant.
MDR C. auris first appearedin Tokyo in 2009. It then spread to South Korea around 2011, and then appeared across Asia and Western Europe. Its first appearance in Spain was the 2016 Le Fe outbreak.
According to the Centers for Disease Control and Prevention, single cases have appeared in Austria, Belgium, Malaysia, Norway, and the United Arab Emirates. Canada, Colombia, France, Germany, India, Israel, Japan, Kenya, Kuwait, Oman, Pakistan, Panama, South Korea, South Africa, Spain, the United Arab Emirates, the United Kingdom, and Venezuela have experienced multiple outbreaks.
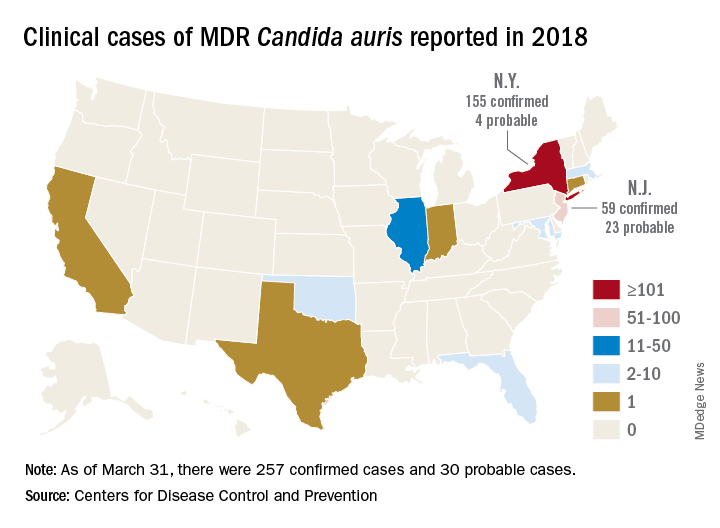
The CDC has recorded 257 confirmed and 30 probable cases of MDR C. auris in the United States as of March 31, 2018. Most of these occurred in New York City and New Jersey; a number of patients had recent stays in hospitals in India, Pakistan, South Africa, the UAE, and Venezuela.
Jacques Meis, MD, of the department of medical microbiology and infectious diseases at Canisius Wilhelmina Hospital, Nijmegen, the Netherlands, set the stage for an extended discussion of C. auris at the meeting.
“This is a multidrug resistant yeast that has emerged in the last decade. Some rare isolates are resistant to all three major antifungal classes. Unlike other Candida species, it seems to persist for prolonged periods in health care environments and to colonize patients’ skin. It behaves rather like resistant bacteria.”

Once established in a health care setting – often an intensive care ward – C. auris poses major infection controls challenges and can be very hard to identify and eradicate, said Dr. Meis.
The identification problem is well known. The 2016 CDC alert noted that “commercially available biochemical-based tests, including API strips and VITEK-2, used in many U.S. laboratories to identify fungi, cannot differentiate C. auris from related species. Because of these challenges, clinical laboratories have misidentified the organism as C. haemulonii and Saccharomyces cerevisiae.”
“It’s often misidentified as other Candida species or as Saccharomyces when we investigate with biochemical methods. C. auris is best identified using Matrix Assisted Laser Desorption/Ionization time of flight mass spectrometry (MALDI-TOF),” said Dr. Meis.
Among the presentations at ECCMID were a report of a U.K. outbreak that affected 70 patients in a neuroscience ICU. It was traced to axillary skin-surface temperature probes, and eradicated only after those probes were removed. More than 90% of the isolates were resistant to fluconazole, voriconazole, and posaconazole; 18% were amphotericin resistant.
A poster described the microbiological characteristics of 50 C. auris isolates taken from 11 hospitals in Korea.
Dr. Pemán described the outbreak in Valencia, which began in April 2016; the report was simultaneously published in the online journal Mycoses (2018 Apr 14. doi: 10.1111/myc.12781).
The index case was a 66-year-old man with hepatocellular carcinoma who underwent a liver resection at Hospital Le Fe in April 2016. During his stay in the surgical ICU (SICU), he developed a fungal infection from an unknown, highly fluconazole-resistant yeast. The pathogen was twice misidentified, first as C. haemulonii and then as S. cerevisiae.
Three weeks later, the patient in the adjacent bed developed a similar infection. Sequencing of the internal transcribed spacer confirmed both as a Candida isolate – an organism previously unknown in Spain.
The SICU setup was apparently very conducive to the C. auris life cycle, Dr. Pemán said. It’s a relatively open ward divided into three rooms with 12 beds in each. There are no isolation beds, and dozens of workers have access to the ward every day, including clinical and cleaning staff.
After identifying the second isolate, Dr. Pemán said, infection control staff went into action. They instituted contact precautions in the SICU, and took regular cultures from newly admitted patients and cultures of every SICU patient every 7 days.
“We also started an intense search for more cases throughout the hospital and in 101 SICU workers. Of 305 samples from hands and ears, we found nothing.” They reviewed all the prior fluconazole-resistant Candida isolates; C. auris was not present in the hospital before the index case.
Three weeks after case 2, six new SICU patients tested positive for C. auris (two blood cultures, one vascular line, one respiratory specimen, two rectal swabs, and one urinary tract sample).
“We reinforced contact precautions in colonized and infected patients and started a twice-daily environmental cleaning practice with quaternary ammonium around them,” said Dr. Pemán. They instituted a proactive hospital-wide hand hygiene campaign and spread the word about the outbreak.
By July, there were 11 new colonized patients, 3 of whom developed candidemia. These patients were grouped in the same SICU ward and underwent daily skin treatments with 4% aqueous chlorhexidine wipes.
The environmental inspection found C. auris on beds, tables, walls, and the floor all around infected patients. The pathogen also was living on IV pumps, computer keyboards, and bedside tables. Blood pressure cuffs were a favorite haunt: 19 of 36 samples in the adjacent ICU were positive. These data were separately reported at ECCMID.
Despite all of these efforts at eradication, infections continued to rise. By November, there were 24 newly colonized patients and nine new candidemia episodes in SICU and regular ICU patients. In December, a new infection control bundle began: A surveillance nurse in the C. auris SICU ward was in charge of compliance; any patient with any yeast growth in culture was isolated, and staff used 2% alcohol chlorhexidine wipes before and after IV catheter handling. Staff also washed down all surfaces three times daily with a disinfectant.
Patients could leave isolation after three consecutive C. auris–negative cultures. After discharge, an ultraviolet light decontamination procedure disinfected each patient room.
The pathogen was almost unbelievably resilient, Dr. Pemán noted in the Mycoses article. “In some cases, C. auris was recovered from walls after cleaning with cationic surface–active products ... it was not known until very recently that these products, as well as quaternary ammonium disinfectants, cannot effectively remove C. auris from surfaces.”
As a result of the previous measures, the outbreak slowed down during December 2016, with two new candidemia cases, but by February, the outbreak resumed with 50 new cases and 18 candidemias detected. Cases continued to emerge throughout 2017.
By September 2017, 250 patients had been colonized; 116 of these were included in the Mycoses report. There were 30 episodes of candidemia (26%); of these, 17 died by 30 days (41.4%). Spondylodiscitis and endocarditis each developed in two patients and one developed ventriculitis.
A separate poster by Dr. Pemán and his colleagues gave more details:
• A 52-year-old woman with C. auris–induced endocarditis died after 4 weeks of treatment with AMB+ECN and flucytosine. She had undergone a prosthetic heart valve placement for Ebstein’s anomaly.
• A 71-year-old man with hydrocephalus developed a C. auris–induced infection of his ventriculoperitoneal shunt; he also had undergone cardiovascular surgery and had an ischemic cardiomyopathy. He died despite shunt removal and 8 weeks of AMB+ECN.
• A 71-year-old man who underwent cardiovascular surgery and received a prosthetic heart valve developed endocarditis. He is alive and at last report, on week 26 of AMB+ECN, flucytosine, and isavuconazole.
• A 68-year-old man who underwent abdominal surgery for hepatocellular carcinoma developed spondylodiscitis and is alive after 24 weeks of AMB+ECN.
• A 48-year old female multiple trauma patient developed spondylodiscitis and is alive after 48 weeks of treatment with AMB+ECN.
A multivariate analysis determined that antibacterial treatment increased the risk of candidemia by almost 30 times (odds ratio, 29.59). The next highest risk was neutropenia (OR, 20.7) and then simply being a hospital and SICU patient. Dr. Pemán’s poster said, “In the 16 months before the index case, La Fe recorded 89 candidemias, none caused by C. auris. In the 16 months afterward, there were 154 candidemias, largely C. auris. Before April 2016, C. parapsilosis accounted for the largest portion of candidemias (46%) followed by C. albicans. After the index case, C. auris accounted for 42%, followed by C. parapsilosis (21%) and C. albicans (18%).”
Because of its fluconazole resistance, patients with C. auris received a combined antifungal treatment of liposomal amphotericin B 3 mg/kg per day for 5 days, and a standard dose of echinocandin for 3 weeks. Many C. auris strains are echinocandin resistant, Dr. Pemán noted. This particular strain was clonal, different from any other previously reported, he said.
“Our results confirm those previously reported by other authors, that C. auris is grouped in different independent clusters according to its geographical origin. Although all Spanish isolates were genotypically distinct from Indian, Omani, U.K., and Venezuelan isolates, there seems to be some connection with South African isolates.”
Hospital Le Fe continues to struggle with C. auris. As of March, 335 patients have tested positive for the pathogen, and 80 have developed candidemias.
“We feel we may be approaching the end of this episode, but it’s really not possible to be sure,” he said.
Dr. Pemán had no relevant financial disclosures.
SOURCE: ECCMID 2018 Peman et al. S0067.
MADRID – The anticipated global emergence of multidrug resistant Candida auris is now an established fact, but a case study presented at the European Society of Clinical Microbiology and Infectious Diseases annual congress demonstrates just how devastating an outbreak can be to a medical facility and its surgical ICU patients.
The dangerous invasive infection is spreading through Asia, Europe, and the Americas, causing potentially fatal candidemias and proving devilishly difficult to eradicate in health care facilities once it becomes established.
Several multidrug resistant (MDR) C. auris outbreaks were reported at the ECCMID meeting. Most troubling: a continuing outbreak in a hospital in Valencia, Spain, in which 17 patients have died – a 41% fatality rate among those who developed a fulminant C. auris candidemia, Javier Pemán, MD, said at the meeting. The strain appeared to be a clonal population not previously identified in published reports.
“C. auris is hard to remove from the hospital environment,” once it becomes established, said Dr. Pemán of La Fe University and Polytechnic Hospital, Valencia. “When an outbreak lasts for months, as ours has, it is difficult, but necessary, to maintain control measures, identify it early in the lab, and isolate and treat patients early with combination therapy.”
He and his team have relied primarily on a combination of amphotericin B and echinocandin (AMB+ECN), although, he added, the optimal dosing and treatment time aren’t known, and many C. auris isolates are echinocandin resistant.
MDR C. auris first appearedin Tokyo in 2009. It then spread to South Korea around 2011, and then appeared across Asia and Western Europe. Its first appearance in Spain was the 2016 Le Fe outbreak.
According to the Centers for Disease Control and Prevention, single cases have appeared in Austria, Belgium, Malaysia, Norway, and the United Arab Emirates. Canada, Colombia, France, Germany, India, Israel, Japan, Kenya, Kuwait, Oman, Pakistan, Panama, South Korea, South Africa, Spain, the United Arab Emirates, the United Kingdom, and Venezuela have experienced multiple outbreaks.
The CDC has recorded 257 confirmed and 30 probable cases of MDR C. auris in the United States as of March 31, 2018. Most of these occurred in New York City and New Jersey; a number of patients had recent stays in hospitals in India, Pakistan, South Africa, the UAE, and Venezuela.
Jacques Meis, MD, of the department of medical microbiology and infectious diseases at Canisius Wilhelmina Hospital, Nijmegen, the Netherlands, set the stage for an extended discussion of C. auris at the meeting.
“This is a multidrug resistant yeast that has emerged in the last decade. Some rare isolates are resistant to all three major antifungal classes. Unlike other Candida species, it seems to persist for prolonged periods in health care environments and to colonize patients’ skin. It behaves rather like resistant bacteria.”
Once established in a health care setting – often an intensive care ward – C. auris poses major infection controls challenges and can be very hard to identify and eradicate, said Dr. Meis.
The identification problem is well known. The 2016 CDC alert noted that “commercially available biochemical-based tests, including API strips and VITEK-2, used in many U.S. laboratories to identify fungi, cannot differentiate C. auris from related species. Because of these challenges, clinical laboratories have misidentified the organism as C. haemulonii and Saccharomyces cerevisiae.”
“It’s often misidentified as other Candida species or as Saccharomyces when we investigate with biochemical methods. C. auris is best identified using Matrix Assisted Laser Desorption/Ionization time of flight mass spectrometry (MALDI-TOF),” said Dr. Meis.
Among the presentations at ECCMID were a report of a U.K. outbreak that affected 70 patients in a neuroscience ICU. It was traced to axillary skin-surface temperature probes, and eradicated only after those probes were removed. More than 90% of the isolates were resistant to fluconazole, voriconazole, and posaconazole; 18% were amphotericin resistant.
A poster described the microbiological characteristics of 50 C. auris isolates taken from 11 hospitals in Korea.
Dr. Pemán described the outbreak in Valencia, which began in April 2016; the report was simultaneously published in the online journal Mycoses (2018 Apr 14. doi: 10.1111/myc.12781).
The index case was a 66-year-old man with hepatocellular carcinoma who underwent a liver resection at Hospital Le Fe in April 2016. During his stay in the surgical ICU (SICU), he developed a fungal infection from an unknown, highly fluconazole-resistant yeast. The pathogen was twice misidentified, first as C. haemulonii and then as S. cerevisiae.
Three weeks later, the patient in the adjacent bed developed a similar infection. Sequencing of the internal transcribed spacer confirmed both as a Candida isolate – an organism previously unknown in Spain.
The SICU setup was apparently very conducive to the C. auris life cycle, Dr. Pemán said. It’s a relatively open ward divided into three rooms with 12 beds in each. There are no isolation beds, and dozens of workers have access to the ward every day, including clinical and cleaning staff.
After identifying the second isolate, Dr. Pemán said, infection control staff went into action. They instituted contact precautions in the SICU, and took regular cultures from newly admitted patients and cultures of every SICU patient every 7 days.
“We also started an intense search for more cases throughout the hospital and in 101 SICU workers. Of 305 samples from hands and ears, we found nothing.” They reviewed all the prior fluconazole-resistant Candida isolates; C. auris was not present in the hospital before the index case.
Three weeks after case 2, six new SICU patients tested positive for C. auris (two blood cultures, one vascular line, one respiratory specimen, two rectal swabs, and one urinary tract sample).
“We reinforced contact precautions in colonized and infected patients and started a twice-daily environmental cleaning practice with quaternary ammonium around them,” said Dr. Pemán. They instituted a proactive hospital-wide hand hygiene campaign and spread the word about the outbreak.
By July, there were 11 new colonized patients, 3 of whom developed candidemia. These patients were grouped in the same SICU ward and underwent daily skin treatments with 4% aqueous chlorhexidine wipes.
The environmental inspection found C. auris on beds, tables, walls, and the floor all around infected patients. The pathogen also was living on IV pumps, computer keyboards, and bedside tables. Blood pressure cuffs were a favorite haunt: 19 of 36 samples in the adjacent ICU were positive. These data were separately reported at ECCMID.
Despite all of these efforts at eradication, infections continued to rise. By November, there were 24 newly colonized patients and nine new candidemia episodes in SICU and regular ICU patients. In December, a new infection control bundle began: A surveillance nurse in the C. auris SICU ward was in charge of compliance; any patient with any yeast growth in culture was isolated, and staff used 2% alcohol chlorhexidine wipes before and after IV catheter handling. Staff also washed down all surfaces three times daily with a disinfectant.
Patients could leave isolation after three consecutive C. auris–negative cultures. After discharge, an ultraviolet light decontamination procedure disinfected each patient room.
The pathogen was almost unbelievably resilient, Dr. Pemán noted in the Mycoses article. “In some cases, C. auris was recovered from walls after cleaning with cationic surface–active products ... it was not known until very recently that these products, as well as quaternary ammonium disinfectants, cannot effectively remove C. auris from surfaces.”
As a result of the previous measures, the outbreak slowed down during December 2016, with two new candidemia cases, but by February, the outbreak resumed with 50 new cases and 18 candidemias detected. Cases continued to emerge throughout 2017.
By September 2017, 250 patients had been colonized; 116 of these were included in the Mycoses report. There were 30 episodes of candidemia (26%); of these, 17 died by 30 days (41.4%). Spondylodiscitis and endocarditis each developed in two patients and one developed ventriculitis.
A separate poster by Dr. Pemán and his colleagues gave more details:
• A 52-year-old woman with C. auris–induced endocarditis died after 4 weeks of treatment with AMB+ECN and flucytosine. She had undergone a prosthetic heart valve placement for Ebstein’s anomaly.
• A 71-year-old man with hydrocephalus developed a C. auris–induced infection of his ventriculoperitoneal shunt; he also had undergone cardiovascular surgery and had an ischemic cardiomyopathy. He died despite shunt removal and 8 weeks of AMB+ECN.
• A 71-year-old man who underwent cardiovascular surgery and received a prosthetic heart valve developed endocarditis. He is alive and at last report, on week 26 of AMB+ECN, flucytosine, and isavuconazole.
• A 68-year-old man who underwent abdominal surgery for hepatocellular carcinoma developed spondylodiscitis and is alive after 24 weeks of AMB+ECN.
• A 48-year old female multiple trauma patient developed spondylodiscitis and is alive after 48 weeks of treatment with AMB+ECN.
A multivariate analysis determined that antibacterial treatment increased the risk of candidemia by almost 30 times (odds ratio, 29.59). The next highest risk was neutropenia (OR, 20.7) and then simply being a hospital and SICU patient. Dr. Pemán’s poster said, “In the 16 months before the index case, La Fe recorded 89 candidemias, none caused by C. auris. In the 16 months afterward, there were 154 candidemias, largely C. auris. Before April 2016, C. parapsilosis accounted for the largest portion of candidemias (46%) followed by C. albicans. After the index case, C. auris accounted for 42%, followed by C. parapsilosis (21%) and C. albicans (18%).”
Because of its fluconazole resistance, patients with C. auris received a combined antifungal treatment of liposomal amphotericin B 3 mg/kg per day for 5 days, and a standard dose of echinocandin for 3 weeks. Many C. auris strains are echinocandin resistant, Dr. Pemán noted. This particular strain was clonal, different from any other previously reported, he said.
“Our results confirm those previously reported by other authors, that C. auris is grouped in different independent clusters according to its geographical origin. Although all Spanish isolates were genotypically distinct from Indian, Omani, U.K., and Venezuelan isolates, there seems to be some connection with South African isolates.”
Hospital Le Fe continues to struggle with C. auris. As of March, 335 patients have tested positive for the pathogen, and 80 have developed candidemias.
“We feel we may be approaching the end of this episode, but it’s really not possible to be sure,” he said.
Dr. Pemán had no relevant financial disclosures.
SOURCE: ECCMID 2018 Peman et al. S0067.
MADRID – The anticipated global emergence of multidrug resistant Candida auris is now an established fact, but a case study presented at the European Society of Clinical Microbiology and Infectious Diseases annual congress demonstrates just how devastating an outbreak can be to a medical facility and its surgical ICU patients.
The dangerous invasive infection is spreading through Asia, Europe, and the Americas, causing potentially fatal candidemias and proving devilishly difficult to eradicate in health care facilities once it becomes established.
Several multidrug resistant (MDR) C. auris outbreaks were reported at the ECCMID meeting. Most troubling: a continuing outbreak in a hospital in Valencia, Spain, in which 17 patients have died – a 41% fatality rate among those who developed a fulminant C. auris candidemia, Javier Pemán, MD, said at the meeting. The strain appeared to be a clonal population not previously identified in published reports.
“C. auris is hard to remove from the hospital environment,” once it becomes established, said Dr. Pemán of La Fe University and Polytechnic Hospital, Valencia. “When an outbreak lasts for months, as ours has, it is difficult, but necessary, to maintain control measures, identify it early in the lab, and isolate and treat patients early with combination therapy.”
He and his team have relied primarily on a combination of amphotericin B and echinocandin (AMB+ECN), although, he added, the optimal dosing and treatment time aren’t known, and many C. auris isolates are echinocandin resistant.
MDR C. auris first appearedin Tokyo in 2009. It then spread to South Korea around 2011, and then appeared across Asia and Western Europe. Its first appearance in Spain was the 2016 Le Fe outbreak.
According to the Centers for Disease Control and Prevention, single cases have appeared in Austria, Belgium, Malaysia, Norway, and the United Arab Emirates. Canada, Colombia, France, Germany, India, Israel, Japan, Kenya, Kuwait, Oman, Pakistan, Panama, South Korea, South Africa, Spain, the United Arab Emirates, the United Kingdom, and Venezuela have experienced multiple outbreaks.
The CDC has recorded 257 confirmed and 30 probable cases of MDR C. auris in the United States as of March 31, 2018. Most of these occurred in New York City and New Jersey; a number of patients had recent stays in hospitals in India, Pakistan, South Africa, the UAE, and Venezuela.
Jacques Meis, MD, of the department of medical microbiology and infectious diseases at Canisius Wilhelmina Hospital, Nijmegen, the Netherlands, set the stage for an extended discussion of C. auris at the meeting.
“This is a multidrug resistant yeast that has emerged in the last decade. Some rare isolates are resistant to all three major antifungal classes. Unlike other Candida species, it seems to persist for prolonged periods in health care environments and to colonize patients’ skin. It behaves rather like resistant bacteria.”
Once established in a health care setting – often an intensive care ward – C. auris poses major infection controls challenges and can be very hard to identify and eradicate, said Dr. Meis.
The identification problem is well known. The 2016 CDC alert noted that “commercially available biochemical-based tests, including API strips and VITEK-2, used in many U.S. laboratories to identify fungi, cannot differentiate C. auris from related species. Because of these challenges, clinical laboratories have misidentified the organism as C. haemulonii and Saccharomyces cerevisiae.”
“It’s often misidentified as other Candida species or as Saccharomyces when we investigate with biochemical methods. C. auris is best identified using Matrix Assisted Laser Desorption/Ionization time of flight mass spectrometry (MALDI-TOF),” said Dr. Meis.
Among the presentations at ECCMID were a report of a U.K. outbreak that affected 70 patients in a neuroscience ICU. It was traced to axillary skin-surface temperature probes, and eradicated only after those probes were removed. More than 90% of the isolates were resistant to fluconazole, voriconazole, and posaconazole; 18% were amphotericin resistant.
A poster described the microbiological characteristics of 50 C. auris isolates taken from 11 hospitals in Korea.
Dr. Pemán described the outbreak in Valencia, which began in April 2016; the report was simultaneously published in the online journal Mycoses (2018 Apr 14. doi: 10.1111/myc.12781).
The index case was a 66-year-old man with hepatocellular carcinoma who underwent a liver resection at Hospital Le Fe in April 2016. During his stay in the surgical ICU (SICU), he developed a fungal infection from an unknown, highly fluconazole-resistant yeast. The pathogen was twice misidentified, first as C. haemulonii and then as S. cerevisiae.
Three weeks later, the patient in the adjacent bed developed a similar infection. Sequencing of the internal transcribed spacer confirmed both as a Candida isolate – an organism previously unknown in Spain.
The SICU setup was apparently very conducive to the C. auris life cycle, Dr. Pemán said. It’s a relatively open ward divided into three rooms with 12 beds in each. There are no isolation beds, and dozens of workers have access to the ward every day, including clinical and cleaning staff.
After identifying the second isolate, Dr. Pemán said, infection control staff went into action. They instituted contact precautions in the SICU, and took regular cultures from newly admitted patients and cultures of every SICU patient every 7 days.
“We also started an intense search for more cases throughout the hospital and in 101 SICU workers. Of 305 samples from hands and ears, we found nothing.” They reviewed all the prior fluconazole-resistant Candida isolates; C. auris was not present in the hospital before the index case.
Three weeks after case 2, six new SICU patients tested positive for C. auris (two blood cultures, one vascular line, one respiratory specimen, two rectal swabs, and one urinary tract sample).
“We reinforced contact precautions in colonized and infected patients and started a twice-daily environmental cleaning practice with quaternary ammonium around them,” said Dr. Pemán. They instituted a proactive hospital-wide hand hygiene campaign and spread the word about the outbreak.
By July, there were 11 new colonized patients, 3 of whom developed candidemia. These patients were grouped in the same SICU ward and underwent daily skin treatments with 4% aqueous chlorhexidine wipes.
The environmental inspection found C. auris on beds, tables, walls, and the floor all around infected patients. The pathogen also was living on IV pumps, computer keyboards, and bedside tables. Blood pressure cuffs were a favorite haunt: 19 of 36 samples in the adjacent ICU were positive. These data were separately reported at ECCMID.
Despite all of these efforts at eradication, infections continued to rise. By November, there were 24 newly colonized patients and nine new candidemia episodes in SICU and regular ICU patients. In December, a new infection control bundle began: A surveillance nurse in the C. auris SICU ward was in charge of compliance; any patient with any yeast growth in culture was isolated, and staff used 2% alcohol chlorhexidine wipes before and after IV catheter handling. Staff also washed down all surfaces three times daily with a disinfectant.
Patients could leave isolation after three consecutive C. auris–negative cultures. After discharge, an ultraviolet light decontamination procedure disinfected each patient room.
The pathogen was almost unbelievably resilient, Dr. Pemán noted in the Mycoses article. “In some cases, C. auris was recovered from walls after cleaning with cationic surface–active products ... it was not known until very recently that these products, as well as quaternary ammonium disinfectants, cannot effectively remove C. auris from surfaces.”
As a result of the previous measures, the outbreak slowed down during December 2016, with two new candidemia cases, but by February, the outbreak resumed with 50 new cases and 18 candidemias detected. Cases continued to emerge throughout 2017.
By September 2017, 250 patients had been colonized; 116 of these were included in the Mycoses report. There were 30 episodes of candidemia (26%); of these, 17 died by 30 days (41.4%). Spondylodiscitis and endocarditis each developed in two patients and one developed ventriculitis.
A separate poster by Dr. Pemán and his colleagues gave more details:
• A 52-year-old woman with C. auris–induced endocarditis died after 4 weeks of treatment with AMB+ECN and flucytosine. She had undergone a prosthetic heart valve placement for Ebstein’s anomaly.
• A 71-year-old man with hydrocephalus developed a C. auris–induced infection of his ventriculoperitoneal shunt; he also had undergone cardiovascular surgery and had an ischemic cardiomyopathy. He died despite shunt removal and 8 weeks of AMB+ECN.
• A 71-year-old man who underwent cardiovascular surgery and received a prosthetic heart valve developed endocarditis. He is alive and at last report, on week 26 of AMB+ECN, flucytosine, and isavuconazole.
• A 68-year-old man who underwent abdominal surgery for hepatocellular carcinoma developed spondylodiscitis and is alive after 24 weeks of AMB+ECN.
• A 48-year old female multiple trauma patient developed spondylodiscitis and is alive after 48 weeks of treatment with AMB+ECN.
A multivariate analysis determined that antibacterial treatment increased the risk of candidemia by almost 30 times (odds ratio, 29.59). The next highest risk was neutropenia (OR, 20.7) and then simply being a hospital and SICU patient. Dr. Pemán’s poster said, “In the 16 months before the index case, La Fe recorded 89 candidemias, none caused by C. auris. In the 16 months afterward, there were 154 candidemias, largely C. auris. Before April 2016, C. parapsilosis accounted for the largest portion of candidemias (46%) followed by C. albicans. After the index case, C. auris accounted for 42%, followed by C. parapsilosis (21%) and C. albicans (18%).”
Because of its fluconazole resistance, patients with C. auris received a combined antifungal treatment of liposomal amphotericin B 3 mg/kg per day for 5 days, and a standard dose of echinocandin for 3 weeks. Many C. auris strains are echinocandin resistant, Dr. Pemán noted. This particular strain was clonal, different from any other previously reported, he said.
“Our results confirm those previously reported by other authors, that C. auris is grouped in different independent clusters according to its geographical origin. Although all Spanish isolates were genotypically distinct from Indian, Omani, U.K., and Venezuelan isolates, there seems to be some connection with South African isolates.”
Hospital Le Fe continues to struggle with C. auris. As of March, 335 patients have tested positive for the pathogen, and 80 have developed candidemias.
“We feel we may be approaching the end of this episode, but it’s really not possible to be sure,” he said.
Dr. Pemán had no relevant financial disclosures.
SOURCE: ECCMID 2018 Peman et al. S0067.
REPORTING FROM ECCMID 2018
Time Won't Heal This Wound
An 85-year-old black man presents with a nonhealing, asymptomatic lesion on his cheek. He says the problem began several months ago, during a fishing trip, when some fishing line got caught in his beard, pulling out a few hairs in the process.
An accompanying relative, however, is quite certain that the lesion predates the fishing incident (for which he was present). He believes the lesion has been there for two years. He also advises that the patient’s memory is “not what it used to be.”
The patient has a significant history of sun exposure from his job as a stonemason, which kept him outdoors most of the time. He has been seen by a variety of providers and diagnosed with several infections, including pyoderma—but antibiotics have had no effect on the lesion.
EXAMINATION
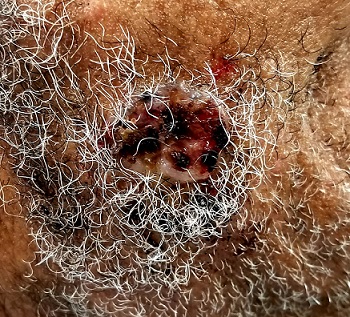
Located on the right lateral cheek is a 2.4-cm, full-thickness ulceration that penetrates well into adipose tissue. Little if any redness can be seen around the lesion, and no adjacent nodes are palpable. A shave biopsy of the lesion is obtained.
What is the diagnosis?
DISCUSSION
The pathology report showed evidence of a basosquamous cell carcinoma.
This case effectively illustrates a key message: Nonhealing lesions should be considered cancerous until proven otherwise (via biopsy). This remains true even in individuals with darker skin; they may have lower risk for skin cancer than do fair-skinned individuals, but they do not have no risk—especially if there is a lifetime history of sun exposure.
The depth and width of the lesion suggest it had been present for many years, slowing growing. This timeframe, along with the lack of response to antibiotics, made infection unlikely. Furthermore, an infection serious enough to cause ulceration would be red and painful.
The mixed picture on the pathology report is unusual but not at all unknown; it just means the lesion had features of both basal and squamous cell carcinoma. Unfortunately, the biopsy results, in conjunction with the lesion’s dimensions, indicate an increased risk for metastasis (or at least spread to local nodes). There could also be perineural involvement if the cancer cells spread to deeper structures through the penetrating nerves.
The entire clinical picture in this case made the patient a candidate for Mohs surgery, which would ensure two things: clear excision margins and optimal wound closure. Should the surgeon find perineural involvement, he or she might advise postoperative radiation therapy to guarantee complete eradication of the cancer.
TAKE-HOME LEARNING POINTS
- Nonhealing lesions should be considered cancerous until proven otherwise by biopsy.
- Even though dark-skinned individuals have far less risk for skin cancer than those with fair skin, a lifetime of sun exposure can overcome the odds.
- Size and depth of the lesion increases risk for metastasis or perineural involvement (spreading to deeper structures through the nerves).
- Mohs surgery, as well as postoperative radiation therapy, can be used to completely eradicate the cancer.
An 85-year-old black man presents with a nonhealing, asymptomatic lesion on his cheek. He says the problem began several months ago, during a fishing trip, when some fishing line got caught in his beard, pulling out a few hairs in the process.
An accompanying relative, however, is quite certain that the lesion predates the fishing incident (for which he was present). He believes the lesion has been there for two years. He also advises that the patient’s memory is “not what it used to be.”
The patient has a significant history of sun exposure from his job as a stonemason, which kept him outdoors most of the time. He has been seen by a variety of providers and diagnosed with several infections, including pyoderma—but antibiotics have had no effect on the lesion.
EXAMINATION
Located on the right lateral cheek is a 2.4-cm, full-thickness ulceration that penetrates well into adipose tissue. Little if any redness can be seen around the lesion, and no adjacent nodes are palpable. A shave biopsy of the lesion is obtained.
What is the diagnosis?
DISCUSSION
The pathology report showed evidence of a basosquamous cell carcinoma.
This case effectively illustrates a key message: Nonhealing lesions should be considered cancerous until proven otherwise (via biopsy). This remains true even in individuals with darker skin; they may have lower risk for skin cancer than do fair-skinned individuals, but they do not have no risk—especially if there is a lifetime history of sun exposure.
The depth and width of the lesion suggest it had been present for many years, slowing growing. This timeframe, along with the lack of response to antibiotics, made infection unlikely. Furthermore, an infection serious enough to cause ulceration would be red and painful.
The mixed picture on the pathology report is unusual but not at all unknown; it just means the lesion had features of both basal and squamous cell carcinoma. Unfortunately, the biopsy results, in conjunction with the lesion’s dimensions, indicate an increased risk for metastasis (or at least spread to local nodes). There could also be perineural involvement if the cancer cells spread to deeper structures through the penetrating nerves.
The entire clinical picture in this case made the patient a candidate for Mohs surgery, which would ensure two things: clear excision margins and optimal wound closure. Should the surgeon find perineural involvement, he or she might advise postoperative radiation therapy to guarantee complete eradication of the cancer.
TAKE-HOME LEARNING POINTS
- Nonhealing lesions should be considered cancerous until proven otherwise by biopsy.
- Even though dark-skinned individuals have far less risk for skin cancer than those with fair skin, a lifetime of sun exposure can overcome the odds.
- Size and depth of the lesion increases risk for metastasis or perineural involvement (spreading to deeper structures through the nerves).
- Mohs surgery, as well as postoperative radiation therapy, can be used to completely eradicate the cancer.
An 85-year-old black man presents with a nonhealing, asymptomatic lesion on his cheek. He says the problem began several months ago, during a fishing trip, when some fishing line got caught in his beard, pulling out a few hairs in the process.
An accompanying relative, however, is quite certain that the lesion predates the fishing incident (for which he was present). He believes the lesion has been there for two years. He also advises that the patient’s memory is “not what it used to be.”
The patient has a significant history of sun exposure from his job as a stonemason, which kept him outdoors most of the time. He has been seen by a variety of providers and diagnosed with several infections, including pyoderma—but antibiotics have had no effect on the lesion.
EXAMINATION
Located on the right lateral cheek is a 2.4-cm, full-thickness ulceration that penetrates well into adipose tissue. Little if any redness can be seen around the lesion, and no adjacent nodes are palpable. A shave biopsy of the lesion is obtained.
What is the diagnosis?
DISCUSSION
The pathology report showed evidence of a basosquamous cell carcinoma.
This case effectively illustrates a key message: Nonhealing lesions should be considered cancerous until proven otherwise (via biopsy). This remains true even in individuals with darker skin; they may have lower risk for skin cancer than do fair-skinned individuals, but they do not have no risk—especially if there is a lifetime history of sun exposure.
The depth and width of the lesion suggest it had been present for many years, slowing growing. This timeframe, along with the lack of response to antibiotics, made infection unlikely. Furthermore, an infection serious enough to cause ulceration would be red and painful.
The mixed picture on the pathology report is unusual but not at all unknown; it just means the lesion had features of both basal and squamous cell carcinoma. Unfortunately, the biopsy results, in conjunction with the lesion’s dimensions, indicate an increased risk for metastasis (or at least spread to local nodes). There could also be perineural involvement if the cancer cells spread to deeper structures through the penetrating nerves.
The entire clinical picture in this case made the patient a candidate for Mohs surgery, which would ensure two things: clear excision margins and optimal wound closure. Should the surgeon find perineural involvement, he or she might advise postoperative radiation therapy to guarantee complete eradication of the cancer.
TAKE-HOME LEARNING POINTS
- Nonhealing lesions should be considered cancerous until proven otherwise by biopsy.
- Even though dark-skinned individuals have far less risk for skin cancer than those with fair skin, a lifetime of sun exposure can overcome the odds.
- Size and depth of the lesion increases risk for metastasis or perineural involvement (spreading to deeper structures through the nerves).
- Mohs surgery, as well as postoperative radiation therapy, can be used to completely eradicate the cancer.
Oral diabetes drugs linked to lower levels of bone formation marker
in a Danish clinical trial.
Procollagen type 1 N-terminal peptide (P1NP) plasma concentrations were lower in patients who received either metformin or metformin plus rosiglitazone, Tore Bjerregaard Stage, PhD, a specialist in clinical pharmacology and pharmacy at the University of Southern Denmark, Odense, and his coauthors wrote in Bone.
By contrast, insulin did not appear to influence markers of bone turnover.
Improving glycemic control was associated with increased plasma concentrations of C-terminal telopeptide of collagen (CTx), a marker of bone resorption. However, this finding might reflect “normalization, rather than an abnormal increase in bone resorption,” Dr. Stage and his colleagues wrote.
These findings come from an analysis of the South Danish Diabetes Study, a 2-year, multicenter, randomized, controlled trial including 371 patients with type 2 diabetes. Patients were first randomized to receive short- or long-acting human insulin, then further randomized to metformin plus rosiglitazone, metformin plus placebo, rosiglitazone plus placebo, or two placebos.
Bone turnover markers were assessed at baseline and at 3-, 12-, and 24-month follow-ups.
Dr. Stage and his coinvestigators hoped the analysis would provide insights into how antidiabetic medication might influence bone turnover, potentially helping explain the increased risk of fracture found in patients with type 2 diabetes. “Alterations in bone metabolism due to antidiabetic medication may influence bone metabolism both directly, e.g., by insulin promoting bone formation, and indirectly by improvement of glycemic control,” they wrote.
Overall, levels of both bone turnover markers increased over time in the study. However, investigators found that concentrations of the bone formation marker P1NP were 13% lower in patients randomized to metformin alone, and 21% lower in patients randomized to metformin and rosiglitazone; no such association was found between P1NP concentrations and treatment with rosiglitazone alone. By contrast, the type of oral antidiabetic drug treatment had no effect on concentrations of CTx concentrations, the investigators said.
Type of insulin treatment received in the trial did not appear to have an impact on concentrations of either bone turnover marker, they added.
HbA1c had no influence on concentrations of P1NP; but it was inversely correlated with levels of CTx, a finding that the investigators said merits more study.
“Further clinical trials investigating the effects of improved glycemic control on bone remodeling including other biochemical markers of bone turnover are needed to confirm if lowering of glucose levels solely changes bone resorption and not formation,” Dr. Stage and his coauthors wrote.
This study was funded with grants from the Danish Council for Independent Research and the Region of Southern Denmark. Dr. Stage and coauthors reported no conflicts of interest related to the report.
SOURCE: Stage TB et al. Bone. 2018 Apr 12;112:35-41.
in a Danish clinical trial.
Procollagen type 1 N-terminal peptide (P1NP) plasma concentrations were lower in patients who received either metformin or metformin plus rosiglitazone, Tore Bjerregaard Stage, PhD, a specialist in clinical pharmacology and pharmacy at the University of Southern Denmark, Odense, and his coauthors wrote in Bone.
By contrast, insulin did not appear to influence markers of bone turnover.
Improving glycemic control was associated with increased plasma concentrations of C-terminal telopeptide of collagen (CTx), a marker of bone resorption. However, this finding might reflect “normalization, rather than an abnormal increase in bone resorption,” Dr. Stage and his colleagues wrote.
These findings come from an analysis of the South Danish Diabetes Study, a 2-year, multicenter, randomized, controlled trial including 371 patients with type 2 diabetes. Patients were first randomized to receive short- or long-acting human insulin, then further randomized to metformin plus rosiglitazone, metformin plus placebo, rosiglitazone plus placebo, or two placebos.
Bone turnover markers were assessed at baseline and at 3-, 12-, and 24-month follow-ups.
Dr. Stage and his coinvestigators hoped the analysis would provide insights into how antidiabetic medication might influence bone turnover, potentially helping explain the increased risk of fracture found in patients with type 2 diabetes. “Alterations in bone metabolism due to antidiabetic medication may influence bone metabolism both directly, e.g., by insulin promoting bone formation, and indirectly by improvement of glycemic control,” they wrote.
Overall, levels of both bone turnover markers increased over time in the study. However, investigators found that concentrations of the bone formation marker P1NP were 13% lower in patients randomized to metformin alone, and 21% lower in patients randomized to metformin and rosiglitazone; no such association was found between P1NP concentrations and treatment with rosiglitazone alone. By contrast, the type of oral antidiabetic drug treatment had no effect on concentrations of CTx concentrations, the investigators said.
Type of insulin treatment received in the trial did not appear to have an impact on concentrations of either bone turnover marker, they added.
HbA1c had no influence on concentrations of P1NP; but it was inversely correlated with levels of CTx, a finding that the investigators said merits more study.
“Further clinical trials investigating the effects of improved glycemic control on bone remodeling including other biochemical markers of bone turnover are needed to confirm if lowering of glucose levels solely changes bone resorption and not formation,” Dr. Stage and his coauthors wrote.
This study was funded with grants from the Danish Council for Independent Research and the Region of Southern Denmark. Dr. Stage and coauthors reported no conflicts of interest related to the report.
SOURCE: Stage TB et al. Bone. 2018 Apr 12;112:35-41.
in a Danish clinical trial.
Procollagen type 1 N-terminal peptide (P1NP) plasma concentrations were lower in patients who received either metformin or metformin plus rosiglitazone, Tore Bjerregaard Stage, PhD, a specialist in clinical pharmacology and pharmacy at the University of Southern Denmark, Odense, and his coauthors wrote in Bone.
By contrast, insulin did not appear to influence markers of bone turnover.
Improving glycemic control was associated with increased plasma concentrations of C-terminal telopeptide of collagen (CTx), a marker of bone resorption. However, this finding might reflect “normalization, rather than an abnormal increase in bone resorption,” Dr. Stage and his colleagues wrote.
These findings come from an analysis of the South Danish Diabetes Study, a 2-year, multicenter, randomized, controlled trial including 371 patients with type 2 diabetes. Patients were first randomized to receive short- or long-acting human insulin, then further randomized to metformin plus rosiglitazone, metformin plus placebo, rosiglitazone plus placebo, or two placebos.
Bone turnover markers were assessed at baseline and at 3-, 12-, and 24-month follow-ups.
Dr. Stage and his coinvestigators hoped the analysis would provide insights into how antidiabetic medication might influence bone turnover, potentially helping explain the increased risk of fracture found in patients with type 2 diabetes. “Alterations in bone metabolism due to antidiabetic medication may influence bone metabolism both directly, e.g., by insulin promoting bone formation, and indirectly by improvement of glycemic control,” they wrote.
Overall, levels of both bone turnover markers increased over time in the study. However, investigators found that concentrations of the bone formation marker P1NP were 13% lower in patients randomized to metformin alone, and 21% lower in patients randomized to metformin and rosiglitazone; no such association was found between P1NP concentrations and treatment with rosiglitazone alone. By contrast, the type of oral antidiabetic drug treatment had no effect on concentrations of CTx concentrations, the investigators said.
Type of insulin treatment received in the trial did not appear to have an impact on concentrations of either bone turnover marker, they added.
HbA1c had no influence on concentrations of P1NP; but it was inversely correlated with levels of CTx, a finding that the investigators said merits more study.
“Further clinical trials investigating the effects of improved glycemic control on bone remodeling including other biochemical markers of bone turnover are needed to confirm if lowering of glucose levels solely changes bone resorption and not formation,” Dr. Stage and his coauthors wrote.
This study was funded with grants from the Danish Council for Independent Research and the Region of Southern Denmark. Dr. Stage and coauthors reported no conflicts of interest related to the report.
SOURCE: Stage TB et al. Bone. 2018 Apr 12;112:35-41.
FROM BONE
Key clinical point: Treatment with oral antidiabetic drugs was associated with reductions in levels of P1NP, a marker of bone formation.
Major finding: Concentrations of P1NP were 13% lower in patients randomized to metformin and 21% lower in patients randomized to metformin and rosiglitazone.
Study details: An analysis of the South Danish Diabetes Study, a 2-year, multicenter, randomized, controlled trial of 371 patients with type 2 diabetes.
Disclosures: The authors reported no conflicts of interest related to the study.
Source: Stage TB et al. Bone. 2018 Apr 12;112:35-41.
USPSTF: Fall prevention in the elderly? Think exercise
The United States Preventive Services Task Force (USPSTF) commissioned a systematic evidence review of 62 randomized clinical trials with a total of 35,058 patients to gather evidence on the effectiveness and harms of primary care–relevant interventions to prevent falls in community-dwelling adults 65 years or older.1 It thereby has updated its 2012 statement, in which exercise or physical therapy and vitamin D supplementation were recommended to prevent falls.
Importance

Scope of review
Out of the 62 randomized clinical trials, 65% of intervention studies targeted patients at high risk of falls; they were most commonly identified by history of prior falls, but mobility, gait, and balance impairment were often also considered. Specific medical diagnoses that could affect fall-related outcomes (osteoporosis, visual impairment, neurocognitive disorders) were excluded. This review did not look at the outcome of studies in populations who were vitamin D deficient because, in this population, vitamin D supplementation would be considered treatment rather than prevention. Of note, women constituted the majority in most studies.
Exercise interventions
USPSTF found five good-quality and 16 fair-quality studies, which altogether included a total of 7,297 patients, that reported on various exercise interventions to prevent falls; altogether, these studies included a total of 7,297 patients. Of the studies, 57% recruited populations at high risk for falls with a mean age ranging from 68 to 88 years. Exercise interventions included supervised individual classes, group classes, and physical therapy. The most common exercise component was gait, balance, and functional training; other common components included, in order of frequency, were resistance training, flexibility training, and endurance training. Most common frequency and duration were three sessions per week for 12 months. Exercise interventions reduced the number of persons experiencing a fall (relative risk 0.89; 95% confidence interval, 0.81-0.97), reduced the number of injurious falls (incidence rate ratio, 0.81; 95% CI, 0.73-0.90), and revealed a statistically insignificant reduction in the number of falls. Reported adverse events were minor and most commonly included pain or bruising related to exercise.
Multifactorial interventions
USPSTF found seven good-quality and 19 fair-quality studies that reported on multifactorial interventions; altogether, these studies included a total of 15,506 patients. Of the studies, 73% recruited populations at high risk for falls, and the mean age ranged from 71.9 to 85 years. Multifactorial interventions had two components:
- Initial assessment to screen for modifiable risk factors for falls (multidisciplinary comprehensive geriatric assessment or specific assessment that evaluated various factors, such as balance, gait, vision, cardiovascular health, medication, environment, cognition, and psychological health).
- Subsequent customized interventions (group or individual exercise, cognitive-behavioral therapy, nutrition, environmental modification, physical or occupational therapy, social or community services, and referral to specialists).
While studies found that multifactorial interventions reduced the number of falls (IRR, 0.79; 95% CI, 0.68-0.91), they did not reduce the number of people who experienced a fall (RR, 0.95; 95% CI, 0.89-1.01) or an injurious fall (RR, 0.94; 95% CI, 0.85-1.03). Four studies reported minor harm, mostly bruising, from exercise. Therefore, USPSTF has recommended that clinicians take into consideration patient’s medical history (including prior falls and comorbidities) to selectively offer multifactorial interventions.
Vitamin D supplementation
USPSTF found four good-quality and three fair-quality studies that reported on the effect of vitamin D supplementation on the prevention of falls; altogether, these studies included a total of 7,531 patients. Of the studies, 43% recruited populations at high risk for falls. The mean age ranged from 71 to 76.8 years, and mean serum 25-OH vitamin D levels ranged from 26.4 to 31.8 ng/mL. Vitamin D formulations and dosages varied among trials from 700 IU/day to 150,000 IU/3 months to 500,000 IU/year. Pooled analyses did not show a significant reduction in falls (IRR, 0.97; 95% CI, 0.79-1.20) or the number of persons experiencing a fall (RR, 0.97; 95% CI, 0.88-1.08). Only two trials reported on injurious falls; one reported an increase and the other reported no statistically significant difference. One study using high doses of Vitamin D supplementation (500,000 IU per year) showed statistically significant increase in all three endpoints.
Recommendation of others for fall prevention
The National Institution of Aging has emphasized exercise for strength and balance, monitoring for environmental hazards, and hearing and vision care, as well as medication management. The American Geriatric Society (AGS) has recommended asking about prior falls annually and assessing gait and balance on those who have experienced a fall. The AGS also has recommended strength and gait training, environmental modification, medication management, and vitamin D supplementation of at least 800 IU/day for those vitamin D deficient or at increased risk of falls. The Center for Disease Control and Prevention recommends STEADI (Stopping Elderly Accidents, Deaths & Injuries), a coordinated approach to implement the AGS’s clinical practice guidelines. The American Academy of Family Physicians recommends exercise or physical therapy and vitamin D supplementation.
The bottom line
Regarding reduction of falls, the USPSTF found adequate evidence that exercise interventions confer a moderate net benefit, multifactorial interventions have a small net benefit, and vitamin D supplementation offers no net benefit in preventing falls.
References
1. Guirquis-Blake JM et al. JAMA. 2018 Apr 24;319(16):1705-16.
2. U.S. Preventive Services Task Force et al. JAMA. 2018 Apr 24;319(16):1696-1704.
Dr. Shrestha is a first-year resident in the family medicine residency program at Abington (Pa.) Jefferson Health. Dr. Skolnik is a professor of family and community medicine at Jefferson Medical College, Philadelphia, and an associate director of the family medicine residency program at Abington Jefferson Health.
.
The United States Preventive Services Task Force (USPSTF) commissioned a systematic evidence review of 62 randomized clinical trials with a total of 35,058 patients to gather evidence on the effectiveness and harms of primary care–relevant interventions to prevent falls in community-dwelling adults 65 years or older.1 It thereby has updated its 2012 statement, in which exercise or physical therapy and vitamin D supplementation were recommended to prevent falls.
Importance
Scope of review
Out of the 62 randomized clinical trials, 65% of intervention studies targeted patients at high risk of falls; they were most commonly identified by history of prior falls, but mobility, gait, and balance impairment were often also considered. Specific medical diagnoses that could affect fall-related outcomes (osteoporosis, visual impairment, neurocognitive disorders) were excluded. This review did not look at the outcome of studies in populations who were vitamin D deficient because, in this population, vitamin D supplementation would be considered treatment rather than prevention. Of note, women constituted the majority in most studies.
Exercise interventions
USPSTF found five good-quality and 16 fair-quality studies, which altogether included a total of 7,297 patients, that reported on various exercise interventions to prevent falls; altogether, these studies included a total of 7,297 patients. Of the studies, 57% recruited populations at high risk for falls with a mean age ranging from 68 to 88 years. Exercise interventions included supervised individual classes, group classes, and physical therapy. The most common exercise component was gait, balance, and functional training; other common components included, in order of frequency, were resistance training, flexibility training, and endurance training. Most common frequency and duration were three sessions per week for 12 months. Exercise interventions reduced the number of persons experiencing a fall (relative risk 0.89; 95% confidence interval, 0.81-0.97), reduced the number of injurious falls (incidence rate ratio, 0.81; 95% CI, 0.73-0.90), and revealed a statistically insignificant reduction in the number of falls. Reported adverse events were minor and most commonly included pain or bruising related to exercise.
Multifactorial interventions
USPSTF found seven good-quality and 19 fair-quality studies that reported on multifactorial interventions; altogether, these studies included a total of 15,506 patients. Of the studies, 73% recruited populations at high risk for falls, and the mean age ranged from 71.9 to 85 years. Multifactorial interventions had two components:
- Initial assessment to screen for modifiable risk factors for falls (multidisciplinary comprehensive geriatric assessment or specific assessment that evaluated various factors, such as balance, gait, vision, cardiovascular health, medication, environment, cognition, and psychological health).
- Subsequent customized interventions (group or individual exercise, cognitive-behavioral therapy, nutrition, environmental modification, physical or occupational therapy, social or community services, and referral to specialists).
While studies found that multifactorial interventions reduced the number of falls (IRR, 0.79; 95% CI, 0.68-0.91), they did not reduce the number of people who experienced a fall (RR, 0.95; 95% CI, 0.89-1.01) or an injurious fall (RR, 0.94; 95% CI, 0.85-1.03). Four studies reported minor harm, mostly bruising, from exercise. Therefore, USPSTF has recommended that clinicians take into consideration patient’s medical history (including prior falls and comorbidities) to selectively offer multifactorial interventions.
Vitamin D supplementation
USPSTF found four good-quality and three fair-quality studies that reported on the effect of vitamin D supplementation on the prevention of falls; altogether, these studies included a total of 7,531 patients. Of the studies, 43% recruited populations at high risk for falls. The mean age ranged from 71 to 76.8 years, and mean serum 25-OH vitamin D levels ranged from 26.4 to 31.8 ng/mL. Vitamin D formulations and dosages varied among trials from 700 IU/day to 150,000 IU/3 months to 500,000 IU/year. Pooled analyses did not show a significant reduction in falls (IRR, 0.97; 95% CI, 0.79-1.20) or the number of persons experiencing a fall (RR, 0.97; 95% CI, 0.88-1.08). Only two trials reported on injurious falls; one reported an increase and the other reported no statistically significant difference. One study using high doses of Vitamin D supplementation (500,000 IU per year) showed statistically significant increase in all three endpoints.
Recommendation of others for fall prevention
The National Institution of Aging has emphasized exercise for strength and balance, monitoring for environmental hazards, and hearing and vision care, as well as medication management. The American Geriatric Society (AGS) has recommended asking about prior falls annually and assessing gait and balance on those who have experienced a fall. The AGS also has recommended strength and gait training, environmental modification, medication management, and vitamin D supplementation of at least 800 IU/day for those vitamin D deficient or at increased risk of falls. The Center for Disease Control and Prevention recommends STEADI (Stopping Elderly Accidents, Deaths & Injuries), a coordinated approach to implement the AGS’s clinical practice guidelines. The American Academy of Family Physicians recommends exercise or physical therapy and vitamin D supplementation.
The bottom line
Regarding reduction of falls, the USPSTF found adequate evidence that exercise interventions confer a moderate net benefit, multifactorial interventions have a small net benefit, and vitamin D supplementation offers no net benefit in preventing falls.
References
1. Guirquis-Blake JM et al. JAMA. 2018 Apr 24;319(16):1705-16.
2. U.S. Preventive Services Task Force et al. JAMA. 2018 Apr 24;319(16):1696-1704.
Dr. Shrestha is a first-year resident in the family medicine residency program at Abington (Pa.) Jefferson Health. Dr. Skolnik is a professor of family and community medicine at Jefferson Medical College, Philadelphia, and an associate director of the family medicine residency program at Abington Jefferson Health.
.
The United States Preventive Services Task Force (USPSTF) commissioned a systematic evidence review of 62 randomized clinical trials with a total of 35,058 patients to gather evidence on the effectiveness and harms of primary care–relevant interventions to prevent falls in community-dwelling adults 65 years or older.1 It thereby has updated its 2012 statement, in which exercise or physical therapy and vitamin D supplementation were recommended to prevent falls.
Importance
Scope of review
Out of the 62 randomized clinical trials, 65% of intervention studies targeted patients at high risk of falls; they were most commonly identified by history of prior falls, but mobility, gait, and balance impairment were often also considered. Specific medical diagnoses that could affect fall-related outcomes (osteoporosis, visual impairment, neurocognitive disorders) were excluded. This review did not look at the outcome of studies in populations who were vitamin D deficient because, in this population, vitamin D supplementation would be considered treatment rather than prevention. Of note, women constituted the majority in most studies.
Exercise interventions
USPSTF found five good-quality and 16 fair-quality studies, which altogether included a total of 7,297 patients, that reported on various exercise interventions to prevent falls; altogether, these studies included a total of 7,297 patients. Of the studies, 57% recruited populations at high risk for falls with a mean age ranging from 68 to 88 years. Exercise interventions included supervised individual classes, group classes, and physical therapy. The most common exercise component was gait, balance, and functional training; other common components included, in order of frequency, were resistance training, flexibility training, and endurance training. Most common frequency and duration were three sessions per week for 12 months. Exercise interventions reduced the number of persons experiencing a fall (relative risk 0.89; 95% confidence interval, 0.81-0.97), reduced the number of injurious falls (incidence rate ratio, 0.81; 95% CI, 0.73-0.90), and revealed a statistically insignificant reduction in the number of falls. Reported adverse events were minor and most commonly included pain or bruising related to exercise.
Multifactorial interventions
USPSTF found seven good-quality and 19 fair-quality studies that reported on multifactorial interventions; altogether, these studies included a total of 15,506 patients. Of the studies, 73% recruited populations at high risk for falls, and the mean age ranged from 71.9 to 85 years. Multifactorial interventions had two components:
- Initial assessment to screen for modifiable risk factors for falls (multidisciplinary comprehensive geriatric assessment or specific assessment that evaluated various factors, such as balance, gait, vision, cardiovascular health, medication, environment, cognition, and psychological health).
- Subsequent customized interventions (group or individual exercise, cognitive-behavioral therapy, nutrition, environmental modification, physical or occupational therapy, social or community services, and referral to specialists).
While studies found that multifactorial interventions reduced the number of falls (IRR, 0.79; 95% CI, 0.68-0.91), they did not reduce the number of people who experienced a fall (RR, 0.95; 95% CI, 0.89-1.01) or an injurious fall (RR, 0.94; 95% CI, 0.85-1.03). Four studies reported minor harm, mostly bruising, from exercise. Therefore, USPSTF has recommended that clinicians take into consideration patient’s medical history (including prior falls and comorbidities) to selectively offer multifactorial interventions.
Vitamin D supplementation
USPSTF found four good-quality and three fair-quality studies that reported on the effect of vitamin D supplementation on the prevention of falls; altogether, these studies included a total of 7,531 patients. Of the studies, 43% recruited populations at high risk for falls. The mean age ranged from 71 to 76.8 years, and mean serum 25-OH vitamin D levels ranged from 26.4 to 31.8 ng/mL. Vitamin D formulations and dosages varied among trials from 700 IU/day to 150,000 IU/3 months to 500,000 IU/year. Pooled analyses did not show a significant reduction in falls (IRR, 0.97; 95% CI, 0.79-1.20) or the number of persons experiencing a fall (RR, 0.97; 95% CI, 0.88-1.08). Only two trials reported on injurious falls; one reported an increase and the other reported no statistically significant difference. One study using high doses of Vitamin D supplementation (500,000 IU per year) showed statistically significant increase in all three endpoints.
Recommendation of others for fall prevention
The National Institution of Aging has emphasized exercise for strength and balance, monitoring for environmental hazards, and hearing and vision care, as well as medication management. The American Geriatric Society (AGS) has recommended asking about prior falls annually and assessing gait and balance on those who have experienced a fall. The AGS also has recommended strength and gait training, environmental modification, medication management, and vitamin D supplementation of at least 800 IU/day for those vitamin D deficient or at increased risk of falls. The Center for Disease Control and Prevention recommends STEADI (Stopping Elderly Accidents, Deaths & Injuries), a coordinated approach to implement the AGS’s clinical practice guidelines. The American Academy of Family Physicians recommends exercise or physical therapy and vitamin D supplementation.
The bottom line
Regarding reduction of falls, the USPSTF found adequate evidence that exercise interventions confer a moderate net benefit, multifactorial interventions have a small net benefit, and vitamin D supplementation offers no net benefit in preventing falls.
References
1. Guirquis-Blake JM et al. JAMA. 2018 Apr 24;319(16):1705-16.
2. U.S. Preventive Services Task Force et al. JAMA. 2018 Apr 24;319(16):1696-1704.
Dr. Shrestha is a first-year resident in the family medicine residency program at Abington (Pa.) Jefferson Health. Dr. Skolnik is a professor of family and community medicine at Jefferson Medical College, Philadelphia, and an associate director of the family medicine residency program at Abington Jefferson Health.
.
Spotlight on nonmelanoma skin cancer’s true burden
CHICAGO – The true extent of the burden imposed by nonmelanoma skin cancer remains widely underappreciated by health policy makers, the public, employers, and nondermatologist physicians, Marta J. Van Beek, MD, asserted at the annual meeting of the American College of Mohs Surgery.
It’s very much in the interest of Mohs surgeons, as the experts in cutaneous malignancies, to get the accurate message out, she added.

Abundant evidence indicates there is an ongoing epidemic of nonmelanoma skin cancer (NMSC) in the United States – and it is associated with a surprising amount of morbidity and mortality, the dermatologic surgeon observed.
For example, while the American Academy of Dermatology’s 93-page Burden of Skin Disease report identified melanoma as the No. 1 cause of mortality because of skin disease – no surprise there – what may come as news to many is that NMSC was No. 2, accounting for 4,376 deaths in 2013, or 19% of the total. That’s more deaths than occurred because of wounds and burns.
And while the number of cases of NMSC is going up year after year as the population ages, it’s also the case that patients with complex NMSC are developing it at a younger and younger age. As documented in the AAD’s DataDerm registry encompassing more than 6 million patients seen by dermatologists during 2015-2017, well over 20,000 patients who underwent Mohs micrographic surgery for NMSC were aged 45-55 years, and another 60,000 were aged 55-65 years. That being said, Mohs surgery was used to treat 477,365 NMSCs in 318,933 patients included in DataDerm during 2015-2017, and in that population, basal cell carcinomas outnumbered squamous cell carcinomas 2:1.
An interesting aspect of the burden imparted by NMSC is that patients with NMSC have a higher risk of other types of cancer, and when they develop those other primary cancers they tend to do more poorly than cancer patients without a history of NMSC, Dr. Van Beek continued.
She cited a comprehensive study by investigators at the Medical University of South Carolina, Charleston, who concluded that the odds of developing a noncutaneous second primary malignancy were 27% greater in individuals with a history of NMSC than in those without such a history. The increased risk was statistically significant for 26 types of noncutaneous cancer, consistent in both men and women, and the younger a patient’s age at onset of NMSC, the stronger the association with noncutaneous cancers (Adv Cancer Res. 2016;130:257-91).
In a separate systematic review by some of the same investigators, patients with a history of squamous cell carcinoma were at a 30% increased risk of all-cause mortality and 117% greater cancer-specific mortality than those without a history of the disease. The associations were less potent for basal cell carcinoma (Arch Dermatol Res. 2017 May;309[4]:243-51).
“You are more likely to die of your nonskin cancer if you’ve ever had a skin cancer, regardless of what that other cancer is. This may mean that once you have a skin cancer, maybe that proves you have poor protoplasm that makes you more prone to other cancers, but even if that’s the case I think it demonstrates that nonmelanoma skin cancer has a substantial contribution to morbidity and mortality outside of what we normally think about,” Dr. Van Beek said.
Another underappreciated aspect of the burden of NMSC is what economists call lost opportunity cost. This isn’t the direct medical cost, but work time missed because of disease. In 2013, according to the AAD Burden of Skin Disease report, melanoma was responsible for $88 million worth of lost productivity, while for NMSC, the figure was $376 million.
“When you’re talking about the burden of disease, it’s important to actually talk to employers about how important it is to pay for the treatment of skin cancer because that keeps people at work and productive,” the dermatologist said.
Investigators for the World Health Organization’s Global Burden of Disease project estimate that the total years lost to disability for patients with NMSC are comparable with the figures for patients with thyroid, esophageal, or ovarian cancer, Dr. Van Beek noted.
Payers and health policy makers are unnerved by the growing utilization of Mohs surgery, she warned.
“This is really important: If you want to substantiate our utilization, you have to make policy makers understand that we are doing this because more people have skin cancer,” she emphasized.
Dr. Van Beek reported no financial conflicts regarding her presentation.
CHICAGO – The true extent of the burden imposed by nonmelanoma skin cancer remains widely underappreciated by health policy makers, the public, employers, and nondermatologist physicians, Marta J. Van Beek, MD, asserted at the annual meeting of the American College of Mohs Surgery.
It’s very much in the interest of Mohs surgeons, as the experts in cutaneous malignancies, to get the accurate message out, she added.
Abundant evidence indicates there is an ongoing epidemic of nonmelanoma skin cancer (NMSC) in the United States – and it is associated with a surprising amount of morbidity and mortality, the dermatologic surgeon observed.
For example, while the American Academy of Dermatology’s 93-page Burden of Skin Disease report identified melanoma as the No. 1 cause of mortality because of skin disease – no surprise there – what may come as news to many is that NMSC was No. 2, accounting for 4,376 deaths in 2013, or 19% of the total. That’s more deaths than occurred because of wounds and burns.
And while the number of cases of NMSC is going up year after year as the population ages, it’s also the case that patients with complex NMSC are developing it at a younger and younger age. As documented in the AAD’s DataDerm registry encompassing more than 6 million patients seen by dermatologists during 2015-2017, well over 20,000 patients who underwent Mohs micrographic surgery for NMSC were aged 45-55 years, and another 60,000 were aged 55-65 years. That being said, Mohs surgery was used to treat 477,365 NMSCs in 318,933 patients included in DataDerm during 2015-2017, and in that population, basal cell carcinomas outnumbered squamous cell carcinomas 2:1.
An interesting aspect of the burden imparted by NMSC is that patients with NMSC have a higher risk of other types of cancer, and when they develop those other primary cancers they tend to do more poorly than cancer patients without a history of NMSC, Dr. Van Beek continued.
She cited a comprehensive study by investigators at the Medical University of South Carolina, Charleston, who concluded that the odds of developing a noncutaneous second primary malignancy were 27% greater in individuals with a history of NMSC than in those without such a history. The increased risk was statistically significant for 26 types of noncutaneous cancer, consistent in both men and women, and the younger a patient’s age at onset of NMSC, the stronger the association with noncutaneous cancers (Adv Cancer Res. 2016;130:257-91).
In a separate systematic review by some of the same investigators, patients with a history of squamous cell carcinoma were at a 30% increased risk of all-cause mortality and 117% greater cancer-specific mortality than those without a history of the disease. The associations were less potent for basal cell carcinoma (Arch Dermatol Res. 2017 May;309[4]:243-51).
“You are more likely to die of your nonskin cancer if you’ve ever had a skin cancer, regardless of what that other cancer is. This may mean that once you have a skin cancer, maybe that proves you have poor protoplasm that makes you more prone to other cancers, but even if that’s the case I think it demonstrates that nonmelanoma skin cancer has a substantial contribution to morbidity and mortality outside of what we normally think about,” Dr. Van Beek said.
Another underappreciated aspect of the burden of NMSC is what economists call lost opportunity cost. This isn’t the direct medical cost, but work time missed because of disease. In 2013, according to the AAD Burden of Skin Disease report, melanoma was responsible for $88 million worth of lost productivity, while for NMSC, the figure was $376 million.
“When you’re talking about the burden of disease, it’s important to actually talk to employers about how important it is to pay for the treatment of skin cancer because that keeps people at work and productive,” the dermatologist said.
Investigators for the World Health Organization’s Global Burden of Disease project estimate that the total years lost to disability for patients with NMSC are comparable with the figures for patients with thyroid, esophageal, or ovarian cancer, Dr. Van Beek noted.
Payers and health policy makers are unnerved by the growing utilization of Mohs surgery, she warned.
“This is really important: If you want to substantiate our utilization, you have to make policy makers understand that we are doing this because more people have skin cancer,” she emphasized.
Dr. Van Beek reported no financial conflicts regarding her presentation.
CHICAGO – The true extent of the burden imposed by nonmelanoma skin cancer remains widely underappreciated by health policy makers, the public, employers, and nondermatologist physicians, Marta J. Van Beek, MD, asserted at the annual meeting of the American College of Mohs Surgery.
It’s very much in the interest of Mohs surgeons, as the experts in cutaneous malignancies, to get the accurate message out, she added.
Abundant evidence indicates there is an ongoing epidemic of nonmelanoma skin cancer (NMSC) in the United States – and it is associated with a surprising amount of morbidity and mortality, the dermatologic surgeon observed.
For example, while the American Academy of Dermatology’s 93-page Burden of Skin Disease report identified melanoma as the No. 1 cause of mortality because of skin disease – no surprise there – what may come as news to many is that NMSC was No. 2, accounting for 4,376 deaths in 2013, or 19% of the total. That’s more deaths than occurred because of wounds and burns.
And while the number of cases of NMSC is going up year after year as the population ages, it’s also the case that patients with complex NMSC are developing it at a younger and younger age. As documented in the AAD’s DataDerm registry encompassing more than 6 million patients seen by dermatologists during 2015-2017, well over 20,000 patients who underwent Mohs micrographic surgery for NMSC were aged 45-55 years, and another 60,000 were aged 55-65 years. That being said, Mohs surgery was used to treat 477,365 NMSCs in 318,933 patients included in DataDerm during 2015-2017, and in that population, basal cell carcinomas outnumbered squamous cell carcinomas 2:1.
An interesting aspect of the burden imparted by NMSC is that patients with NMSC have a higher risk of other types of cancer, and when they develop those other primary cancers they tend to do more poorly than cancer patients without a history of NMSC, Dr. Van Beek continued.
She cited a comprehensive study by investigators at the Medical University of South Carolina, Charleston, who concluded that the odds of developing a noncutaneous second primary malignancy were 27% greater in individuals with a history of NMSC than in those without such a history. The increased risk was statistically significant for 26 types of noncutaneous cancer, consistent in both men and women, and the younger a patient’s age at onset of NMSC, the stronger the association with noncutaneous cancers (Adv Cancer Res. 2016;130:257-91).
In a separate systematic review by some of the same investigators, patients with a history of squamous cell carcinoma were at a 30% increased risk of all-cause mortality and 117% greater cancer-specific mortality than those without a history of the disease. The associations were less potent for basal cell carcinoma (Arch Dermatol Res. 2017 May;309[4]:243-51).
“You are more likely to die of your nonskin cancer if you’ve ever had a skin cancer, regardless of what that other cancer is. This may mean that once you have a skin cancer, maybe that proves you have poor protoplasm that makes you more prone to other cancers, but even if that’s the case I think it demonstrates that nonmelanoma skin cancer has a substantial contribution to morbidity and mortality outside of what we normally think about,” Dr. Van Beek said.
Another underappreciated aspect of the burden of NMSC is what economists call lost opportunity cost. This isn’t the direct medical cost, but work time missed because of disease. In 2013, according to the AAD Burden of Skin Disease report, melanoma was responsible for $88 million worth of lost productivity, while for NMSC, the figure was $376 million.
“When you’re talking about the burden of disease, it’s important to actually talk to employers about how important it is to pay for the treatment of skin cancer because that keeps people at work and productive,” the dermatologist said.
Investigators for the World Health Organization’s Global Burden of Disease project estimate that the total years lost to disability for patients with NMSC are comparable with the figures for patients with thyroid, esophageal, or ovarian cancer, Dr. Van Beek noted.
Payers and health policy makers are unnerved by the growing utilization of Mohs surgery, she warned.
“This is really important: If you want to substantiate our utilization, you have to make policy makers understand that we are doing this because more people have skin cancer,” she emphasized.
Dr. Van Beek reported no financial conflicts regarding her presentation.
EXPERT ANALYSIS FROM THE ACMS ANNUAL MEETING
CMS proposes inpatient payment model for CAR T therapies
Physicians may finally have some clarity on payment for inpatient administration of two chimeric antigen receptor–T-cell therapies if a proposed rule from the Centers of Medicare & Medicaid Services becomes final.
The agency is seeking to assign ICD-10-PCS codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019. The agency is also considering the creation of a new Medicare Severity–Diagnosis Related Group (MS-DRG) code for procedures involving the use of chimeric antigen receptor (CAR) T-cell therapy drugs. The proposal was published in May in the Federal Register.
The proposal demonstrates that CMS is listening to physicians’ concerns about CAR T payments and working to provide a more reasonable framework, said Stephanie Farnia, director of health policy and strategic relations for the American Society for Blood and Marrow Transplantation.
“The primary point of significance is that CAR-T care episodes should be assigned to a specific MS-DRG in FY2019, which will give physicians a clearer sense of inpatient reimbursement in advance,” she said in an interview.
Uncertainty about inpatient payment for administration of the two approved CAR-T therapies have been a lingering concern of specialists using, or interested in using, the therapies. In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
However, physicians noted that even if the drugs are first administered in the outpatient setting, inpatient care is likely to occur with CAR T-cell therapies because some patients will need to be admitted in order to be monitored for serious side effects. In such cases, all payments will become part of the inpatient stay per CMS’ 3-day payment window rule.
In outlining the most recent payment proposal, CMS stated that its clinical advisers believe that patients receiving treatment with CAR T-cell therapy would have similar clinical characteristics and comorbidities as patients treated with autologous bone marrow transplant therapy, who are currently assigned to MS-DRG 016 “Autologous Bone Marrow Transplant with CC/MCC”. Therefore, CMS officials said they are suggesting ICD-10-PCS procedure codes XW033C3 and XW043C3 to pre-MDC MS-DRG 016. Additionally, the agency is proposing to revise the title of MS-DRG 016 from “Autologous Bone Marrow Transplant with CC/MCC” to “Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy.”
However, the agency emphasized that it invites public comment on alternative payment approaches for CAR T-cell therapies in the context of the pending, new technology add-on payment applications by the CAR-T drugmakers Novartis Pharmaceuticals and Kite Pharma. If approved in the final rule, the technology add-on payments would provide an additional and separate payment equivalent to up to 50% of the product cost plus the MS-DRG payment received for the episode of care.
Other items in the proposed rule include approximate base payments for hematopoietic stem cell transplantation for fiscal 2019, which starts Oct. 1, 2018. The approximate per unit payment is $5,498, which translates to a total base payment of $64,790 for allogeneic bone marrow transplant (MS-DRG 014) for instance. In an American Society for Blood and Marrow Transplantation blog, Ms. Farnia outlined the approximate base weights and estimated reimbursements calculated from the rule.
CMS did not make any changes to the payment model for allogeneic hematopoietic cell transplantation despite continued requests by physicians for donor acquisition charges. Ms. Farnia encourages doctors to express to CMS the importance of such reimbursement during the comment period.
Public comments on the inpatient payment proposal are due by June 25, 2018, and can be submitted at https://www.regulations.gov. Additionally, CMS will convene a meeting of the Medicare Evidence Development & Coverage Advisory Committee on August 22, 2018, to consider whether to issue a national coverage policy for CAR T-cell therapies that would set consistent parameters for patient access.
Physicians may finally have some clarity on payment for inpatient administration of two chimeric antigen receptor–T-cell therapies if a proposed rule from the Centers of Medicare & Medicaid Services becomes final.
The agency is seeking to assign ICD-10-PCS codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019. The agency is also considering the creation of a new Medicare Severity–Diagnosis Related Group (MS-DRG) code for procedures involving the use of chimeric antigen receptor (CAR) T-cell therapy drugs. The proposal was published in May in the Federal Register.
The proposal demonstrates that CMS is listening to physicians’ concerns about CAR T payments and working to provide a more reasonable framework, said Stephanie Farnia, director of health policy and strategic relations for the American Society for Blood and Marrow Transplantation.
“The primary point of significance is that CAR-T care episodes should be assigned to a specific MS-DRG in FY2019, which will give physicians a clearer sense of inpatient reimbursement in advance,” she said in an interview.
Uncertainty about inpatient payment for administration of the two approved CAR-T therapies have been a lingering concern of specialists using, or interested in using, the therapies. In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
However, physicians noted that even if the drugs are first administered in the outpatient setting, inpatient care is likely to occur with CAR T-cell therapies because some patients will need to be admitted in order to be monitored for serious side effects. In such cases, all payments will become part of the inpatient stay per CMS’ 3-day payment window rule.
In outlining the most recent payment proposal, CMS stated that its clinical advisers believe that patients receiving treatment with CAR T-cell therapy would have similar clinical characteristics and comorbidities as patients treated with autologous bone marrow transplant therapy, who are currently assigned to MS-DRG 016 “Autologous Bone Marrow Transplant with CC/MCC”. Therefore, CMS officials said they are suggesting ICD-10-PCS procedure codes XW033C3 and XW043C3 to pre-MDC MS-DRG 016. Additionally, the agency is proposing to revise the title of MS-DRG 016 from “Autologous Bone Marrow Transplant with CC/MCC” to “Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy.”
However, the agency emphasized that it invites public comment on alternative payment approaches for CAR T-cell therapies in the context of the pending, new technology add-on payment applications by the CAR-T drugmakers Novartis Pharmaceuticals and Kite Pharma. If approved in the final rule, the technology add-on payments would provide an additional and separate payment equivalent to up to 50% of the product cost plus the MS-DRG payment received for the episode of care.
Other items in the proposed rule include approximate base payments for hematopoietic stem cell transplantation for fiscal 2019, which starts Oct. 1, 2018. The approximate per unit payment is $5,498, which translates to a total base payment of $64,790 for allogeneic bone marrow transplant (MS-DRG 014) for instance. In an American Society for Blood and Marrow Transplantation blog, Ms. Farnia outlined the approximate base weights and estimated reimbursements calculated from the rule.
CMS did not make any changes to the payment model for allogeneic hematopoietic cell transplantation despite continued requests by physicians for donor acquisition charges. Ms. Farnia encourages doctors to express to CMS the importance of such reimbursement during the comment period.
Public comments on the inpatient payment proposal are due by June 25, 2018, and can be submitted at https://www.regulations.gov. Additionally, CMS will convene a meeting of the Medicare Evidence Development & Coverage Advisory Committee on August 22, 2018, to consider whether to issue a national coverage policy for CAR T-cell therapies that would set consistent parameters for patient access.
Physicians may finally have some clarity on payment for inpatient administration of two chimeric antigen receptor–T-cell therapies if a proposed rule from the Centers of Medicare & Medicaid Services becomes final.
The agency is seeking to assign ICD-10-PCS codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019. The agency is also considering the creation of a new Medicare Severity–Diagnosis Related Group (MS-DRG) code for procedures involving the use of chimeric antigen receptor (CAR) T-cell therapy drugs. The proposal was published in May in the Federal Register.
The proposal demonstrates that CMS is listening to physicians’ concerns about CAR T payments and working to provide a more reasonable framework, said Stephanie Farnia, director of health policy and strategic relations for the American Society for Blood and Marrow Transplantation.
“The primary point of significance is that CAR-T care episodes should be assigned to a specific MS-DRG in FY2019, which will give physicians a clearer sense of inpatient reimbursement in advance,” she said in an interview.
Uncertainty about inpatient payment for administration of the two approved CAR-T therapies have been a lingering concern of specialists using, or interested in using, the therapies. In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
However, physicians noted that even if the drugs are first administered in the outpatient setting, inpatient care is likely to occur with CAR T-cell therapies because some patients will need to be admitted in order to be monitored for serious side effects. In such cases, all payments will become part of the inpatient stay per CMS’ 3-day payment window rule.
In outlining the most recent payment proposal, CMS stated that its clinical advisers believe that patients receiving treatment with CAR T-cell therapy would have similar clinical characteristics and comorbidities as patients treated with autologous bone marrow transplant therapy, who are currently assigned to MS-DRG 016 “Autologous Bone Marrow Transplant with CC/MCC”. Therefore, CMS officials said they are suggesting ICD-10-PCS procedure codes XW033C3 and XW043C3 to pre-MDC MS-DRG 016. Additionally, the agency is proposing to revise the title of MS-DRG 016 from “Autologous Bone Marrow Transplant with CC/MCC” to “Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy.”
However, the agency emphasized that it invites public comment on alternative payment approaches for CAR T-cell therapies in the context of the pending, new technology add-on payment applications by the CAR-T drugmakers Novartis Pharmaceuticals and Kite Pharma. If approved in the final rule, the technology add-on payments would provide an additional and separate payment equivalent to up to 50% of the product cost plus the MS-DRG payment received for the episode of care.
Other items in the proposed rule include approximate base payments for hematopoietic stem cell transplantation for fiscal 2019, which starts Oct. 1, 2018. The approximate per unit payment is $5,498, which translates to a total base payment of $64,790 for allogeneic bone marrow transplant (MS-DRG 014) for instance. In an American Society for Blood and Marrow Transplantation blog, Ms. Farnia outlined the approximate base weights and estimated reimbursements calculated from the rule.
CMS did not make any changes to the payment model for allogeneic hematopoietic cell transplantation despite continued requests by physicians for donor acquisition charges. Ms. Farnia encourages doctors to express to CMS the importance of such reimbursement during the comment period.
Public comments on the inpatient payment proposal are due by June 25, 2018, and can be submitted at https://www.regulations.gov. Additionally, CMS will convene a meeting of the Medicare Evidence Development & Coverage Advisory Committee on August 22, 2018, to consider whether to issue a national coverage policy for CAR T-cell therapies that would set consistent parameters for patient access.
Secondary Syphilis: An Atypical Presentation Complicated by a False Negative Rapid Plasma Reagin Test
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
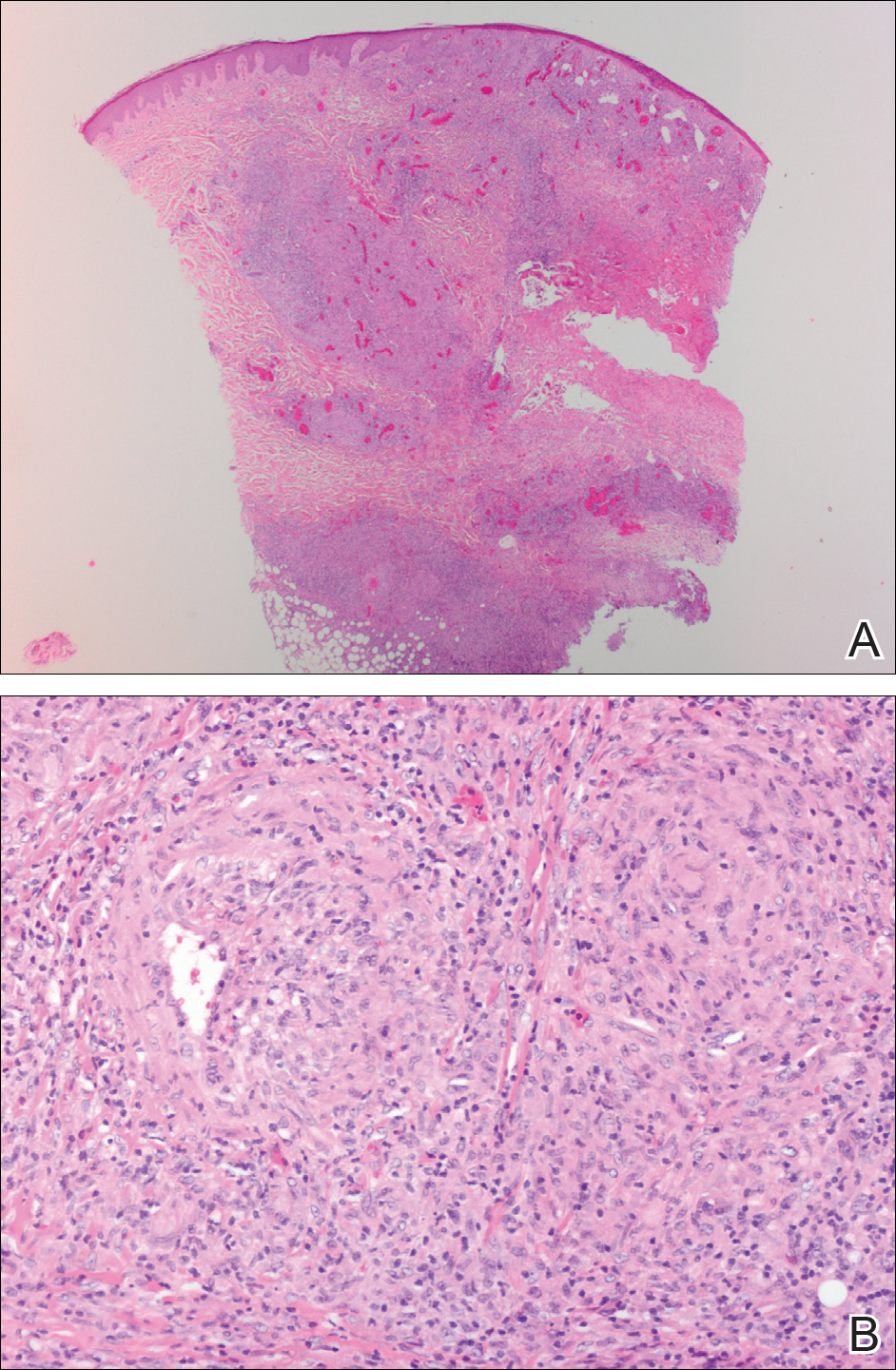
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.

Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.
Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.
Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
Practice Points
- Fluorescent treponemal antibody absorption testing more accurately detects syphilis than rapid plasma reagin (RPR).
- Rapid plasma reagin testing is more useful in monitoring serum response once treatment has been initiated.
- If only RPR is being performed at your institution, ensure the laboratory is performing serial dilutions to negate the prozone phenomenon.
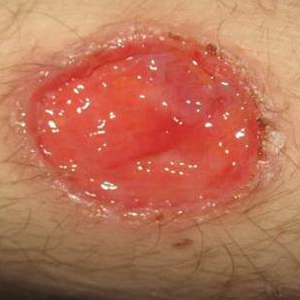
Noninvasive Neurostimulator Shows Efficacy in Episodic Migraine
LOS ANGELES—Noninvasive vagus nerve stimulation (nVNS) is a rapidly effective, well-tolerated, and practical option for the acute treatment of episodic migraine, according to the results of PRESTO, a multicenter, randomized, sham-controlled, double-blind trial presented at the 70th Annual Meeting of the American Academy of Neurology. “There are multiple options for nVNS in the acute treatment of migraine,” said Cristina Tassorelli, MD, PhD, Professor of Neurology at the University of Pavia and Director of the Headache Science Centre at the Casimiro Mondino National Neurological Institute of Pavia in Italy. “It can be used alone because it is effective, it can be used in combination with drugs because we do not expect any significant interactions, and it is also indicated in patients who have developed medication overuse.”

Neuromodulation for Migraine
The aim of the PRESTO trial was to evaluate the efficacy, safety, and tolerability of gammaCore, an nVNS device, for the acute treatment of migraine. The handheld gammaCore device is already FDA cleared for the acute treatment of pain associated with episodic cluster headache and migraine headache in adults. PRESTO was the trial that supported the FDA clearance of gammaCore for migraine.
Following an observational period of four weeks, patients enrolled in PRESTO were randomized to nVNS or sham stimulation for four weeks or until five attacks were treated. Following this period, patients entered the open-label period for another four weeks or five additional attacks.
Low-intensity current, which induced a tingling sensation on the skin that was similar to the sensation of the active stimulation, was used for the sham stimulation. Patients were instructed to treat their attack early with two two-minute stimulations, one for each side of the neck. At 15 minutes, patients assessed the intensity of their pain. If pain was still present, another set of two stimulations was self-administered. A second assessment of pain occurred at 120 minutes, with the possibility of administering another stimulation. Rescue medication use before 120 minutes was considered to indicate treatment failure.
Patients with episodic migraine with or without aura were recruited from 10 Italian tertiary headache centers. A total of 285 patients were enrolled, and 248 randomized: 122 to the active arm and 126 to the sham arm. A total of 117 patients completed the double-blind period in the active arm, and 123 patients in the sham arm. “We had a few discontinuations in both arms, one of which was due to the device,” Dr. Tassorelli and colleagues noted. “Demographic and baseline characteristics show typical pictures of patients with episodic migraine,” Dr. Tassorelli said. Study subjects were mostly young, mostly female, and mostly experiencing migraine without aura. Subjects had a mean of five attacks of migraine per month, a mean of six days of headache per month, and a mean monthly intake of five acute migraine medications. One-third of the patients were on stable prophylactic medications. About 40% of the patients were having moderate pain when they treated their first attack. One-third of them were experiencing mild pain, while 23% were experiencing severe pain in the active group and 15% were having severe pain in the sham group.
Stimulation Reduced Pain
The primary end point of the study was pain freedom at 120 minutes for the first attack. The investigators found a significant difference between the two arms at 30 minutes that became more evident at 60 minutes. At 120 minutes, there was a difference, “but we lost the statistical significance,” Dr. Tassorelli reported. “However, a more refined, post hoc repeated-measure analysis showed that the group treated with the active stimulation had significantly more pain relief than the sham stimulation over the 120-minute period.”
One of the secondary outcome measures was headache
The rate of participants who had a therapeutic response for 50% of attacks or more at 120 minutes speaks to the consistency of the response. Almost half of the patients responded to nVNS at 120 minutes for 50% or more of all treated attacks—47.6% achieved mild or no pain at 120 minutes in the treatment group versus 32.3% in the sham group; 32.4% achieved pain freedom at 120 minutes in the treatment group versus 18.2% in the sham group.
“We did not have many adverse events in this study,” Dr. Tassorelli said. Adverse effects of nVNS, mainly application site discomfort, were infrequent, mild, and transitory. No serious adverse events were recorded, and none of the patients in the active treatment group discontinued treatment due to adverse events.
In summary, nVNS “proved superior to sham for pain freedom at 30 minutes and at 60 minutes, but not at 120 minutes, which was our primary end point,” said Dr. Tassorelli and colleagues. “However, repeated-measures analysis validated the primary end point, indicating the superiority of active stimulation over sham through 120 minutes. This study provides class one evidence for the efficacy of nVNS in the acute treatment of episodic migraine.”
This study was sponsored by electroCore.
—Glenn S. Williams
LOS ANGELES—Noninvasive vagus nerve stimulation (nVNS) is a rapidly effective, well-tolerated, and practical option for the acute treatment of episodic migraine, according to the results of PRESTO, a multicenter, randomized, sham-controlled, double-blind trial presented at the 70th Annual Meeting of the American Academy of Neurology. “There are multiple options for nVNS in the acute treatment of migraine,” said Cristina Tassorelli, MD, PhD, Professor of Neurology at the University of Pavia and Director of the Headache Science Centre at the Casimiro Mondino National Neurological Institute of Pavia in Italy. “It can be used alone because it is effective, it can be used in combination with drugs because we do not expect any significant interactions, and it is also indicated in patients who have developed medication overuse.”
Neuromodulation for Migraine
The aim of the PRESTO trial was to evaluate the efficacy, safety, and tolerability of gammaCore, an nVNS device, for the acute treatment of migraine. The handheld gammaCore device is already FDA cleared for the acute treatment of pain associated with episodic cluster headache and migraine headache in adults. PRESTO was the trial that supported the FDA clearance of gammaCore for migraine.
Following an observational period of four weeks, patients enrolled in PRESTO were randomized to nVNS or sham stimulation for four weeks or until five attacks were treated. Following this period, patients entered the open-label period for another four weeks or five additional attacks.
Low-intensity current, which induced a tingling sensation on the skin that was similar to the sensation of the active stimulation, was used for the sham stimulation. Patients were instructed to treat their attack early with two two-minute stimulations, one for each side of the neck. At 15 minutes, patients assessed the intensity of their pain. If pain was still present, another set of two stimulations was self-administered. A second assessment of pain occurred at 120 minutes, with the possibility of administering another stimulation. Rescue medication use before 120 minutes was considered to indicate treatment failure.
Patients with episodic migraine with or without aura were recruited from 10 Italian tertiary headache centers. A total of 285 patients were enrolled, and 248 randomized: 122 to the active arm and 126 to the sham arm. A total of 117 patients completed the double-blind period in the active arm, and 123 patients in the sham arm. “We had a few discontinuations in both arms, one of which was due to the device,” Dr. Tassorelli and colleagues noted. “Demographic and baseline characteristics show typical pictures of patients with episodic migraine,” Dr. Tassorelli said. Study subjects were mostly young, mostly female, and mostly experiencing migraine without aura. Subjects had a mean of five attacks of migraine per month, a mean of six days of headache per month, and a mean monthly intake of five acute migraine medications. One-third of the patients were on stable prophylactic medications. About 40% of the patients were having moderate pain when they treated their first attack. One-third of them were experiencing mild pain, while 23% were experiencing severe pain in the active group and 15% were having severe pain in the sham group.
Stimulation Reduced Pain
The primary end point of the study was pain freedom at 120 minutes for the first attack. The investigators found a significant difference between the two arms at 30 minutes that became more evident at 60 minutes. At 120 minutes, there was a difference, “but we lost the statistical significance,” Dr. Tassorelli reported. “However, a more refined, post hoc repeated-measure analysis showed that the group treated with the active stimulation had significantly more pain relief than the sham stimulation over the 120-minute period.”
One of the secondary outcome measures was headache
The rate of participants who had a therapeutic response for 50% of attacks or more at 120 minutes speaks to the consistency of the response. Almost half of the patients responded to nVNS at 120 minutes for 50% or more of all treated attacks—47.6% achieved mild or no pain at 120 minutes in the treatment group versus 32.3% in the sham group; 32.4% achieved pain freedom at 120 minutes in the treatment group versus 18.2% in the sham group.
“We did not have many adverse events in this study,” Dr. Tassorelli said. Adverse effects of nVNS, mainly application site discomfort, were infrequent, mild, and transitory. No serious adverse events were recorded, and none of the patients in the active treatment group discontinued treatment due to adverse events.
In summary, nVNS “proved superior to sham for pain freedom at 30 minutes and at 60 minutes, but not at 120 minutes, which was our primary end point,” said Dr. Tassorelli and colleagues. “However, repeated-measures analysis validated the primary end point, indicating the superiority of active stimulation over sham through 120 minutes. This study provides class one evidence for the efficacy of nVNS in the acute treatment of episodic migraine.”
This study was sponsored by electroCore.
—Glenn S. Williams
LOS ANGELES—Noninvasive vagus nerve stimulation (nVNS) is a rapidly effective, well-tolerated, and practical option for the acute treatment of episodic migraine, according to the results of PRESTO, a multicenter, randomized, sham-controlled, double-blind trial presented at the 70th Annual Meeting of the American Academy of Neurology. “There are multiple options for nVNS in the acute treatment of migraine,” said Cristina Tassorelli, MD, PhD, Professor of Neurology at the University of Pavia and Director of the Headache Science Centre at the Casimiro Mondino National Neurological Institute of Pavia in Italy. “It can be used alone because it is effective, it can be used in combination with drugs because we do not expect any significant interactions, and it is also indicated in patients who have developed medication overuse.”
Neuromodulation for Migraine
The aim of the PRESTO trial was to evaluate the efficacy, safety, and tolerability of gammaCore, an nVNS device, for the acute treatment of migraine. The handheld gammaCore device is already FDA cleared for the acute treatment of pain associated with episodic cluster headache and migraine headache in adults. PRESTO was the trial that supported the FDA clearance of gammaCore for migraine.
Following an observational period of four weeks, patients enrolled in PRESTO were randomized to nVNS or sham stimulation for four weeks or until five attacks were treated. Following this period, patients entered the open-label period for another four weeks or five additional attacks.
Low-intensity current, which induced a tingling sensation on the skin that was similar to the sensation of the active stimulation, was used for the sham stimulation. Patients were instructed to treat their attack early with two two-minute stimulations, one for each side of the neck. At 15 minutes, patients assessed the intensity of their pain. If pain was still present, another set of two stimulations was self-administered. A second assessment of pain occurred at 120 minutes, with the possibility of administering another stimulation. Rescue medication use before 120 minutes was considered to indicate treatment failure.
Patients with episodic migraine with or without aura were recruited from 10 Italian tertiary headache centers. A total of 285 patients were enrolled, and 248 randomized: 122 to the active arm and 126 to the sham arm. A total of 117 patients completed the double-blind period in the active arm, and 123 patients in the sham arm. “We had a few discontinuations in both arms, one of which was due to the device,” Dr. Tassorelli and colleagues noted. “Demographic and baseline characteristics show typical pictures of patients with episodic migraine,” Dr. Tassorelli said. Study subjects were mostly young, mostly female, and mostly experiencing migraine without aura. Subjects had a mean of five attacks of migraine per month, a mean of six days of headache per month, and a mean monthly intake of five acute migraine medications. One-third of the patients were on stable prophylactic medications. About 40% of the patients were having moderate pain when they treated their first attack. One-third of them were experiencing mild pain, while 23% were experiencing severe pain in the active group and 15% were having severe pain in the sham group.
Stimulation Reduced Pain
The primary end point of the study was pain freedom at 120 minutes for the first attack. The investigators found a significant difference between the two arms at 30 minutes that became more evident at 60 minutes. At 120 minutes, there was a difference, “but we lost the statistical significance,” Dr. Tassorelli reported. “However, a more refined, post hoc repeated-measure analysis showed that the group treated with the active stimulation had significantly more pain relief than the sham stimulation over the 120-minute period.”
One of the secondary outcome measures was headache
The rate of participants who had a therapeutic response for 50% of attacks or more at 120 minutes speaks to the consistency of the response. Almost half of the patients responded to nVNS at 120 minutes for 50% or more of all treated attacks—47.6% achieved mild or no pain at 120 minutes in the treatment group versus 32.3% in the sham group; 32.4% achieved pain freedom at 120 minutes in the treatment group versus 18.2% in the sham group.
“We did not have many adverse events in this study,” Dr. Tassorelli said. Adverse effects of nVNS, mainly application site discomfort, were infrequent, mild, and transitory. No serious adverse events were recorded, and none of the patients in the active treatment group discontinued treatment due to adverse events.
In summary, nVNS “proved superior to sham for pain freedom at 30 minutes and at 60 minutes, but not at 120 minutes, which was our primary end point,” said Dr. Tassorelli and colleagues. “However, repeated-measures analysis validated the primary end point, indicating the superiority of active stimulation over sham through 120 minutes. This study provides class one evidence for the efficacy of nVNS in the acute treatment of episodic migraine.”
This study was sponsored by electroCore.
—Glenn S. Williams