User login
Newer IgG4 testing proving effective in assessing patients
SANDESTIN, FLA. – New forms of IgG4 testing could be more helpful in making diagnoses of immunoglobulin G4-related disease, an expert said at the annual Congress of Clinical Rheumatology, while cautioning that the diagnosis is more about histology and pattern of involvement than antibody testing.
Arezou Khosroshahi, MD, of Emory University, Atlanta, said that the IgG4 levels found in serum using nephelometry can often be low in patients who otherwise show signs of the disease, which can affect a wide array of organs and typically involves elevated IgG4. Newer forms of testing – enzyme-linked ImmunoSpot (ELISPOT) and quantitative reverse transcription polymerase chain reaction (RT-qPCR) – could be more telling, she said.

But she found that, on flow cytometry, 88% of the woman’s circulating B cells were positive for IgG4, so the woman was treated with rituximab to deplete these cells.
“When the B cells were gone, we had release of the IgG4 in the serum and now we could pick it up with nephelometry,” she said.
This missed IgG4 with nephelometry prompted researchers to turn to ELISPOT, a sensitive method to count antibody-secreting cells. The test works well by capturing the antibodies’ presence right after they’re secreted, before they can become lost to receptor binding or in other ways.
“This was a better assay to measure the IgG4 antibodies rather than nephelometry,” she said.
Perhaps even better, studies in Europe have found that RT-qPCR testing for IgG4 RNA can be effective. This type of testing is easier than ELISPOT, and “they are finding the sensitivity to be much superior to nephelometry for immunoglobulin levels,” Dr. Khosroshahi said.
The higher the levels of IgG4, the more likely an IgG4-related disease diagnosis is warranted, and higher levels tend to lead to worse outcomes, she said.
She waved a caution flag, though: Other diseases can involve elevated IgG4, and even a normal IgG4 level does not necessarily rule out the disease. Other evaluations really form the cornerstone of the diagnosis of IgG4-related disease, she said.
“There should be characteristic histology and of course IgG4-staining, but more importantly, pattern of organ involvement. It’s very important,” Dr. Khosroshahi said. “If there is a mass in the pancreas and there is salivary gland and parotid gland swellings and other features of that going on, you are more concerned that that is a process going on.”
Dr. Khosroshahi had no relevant disclosures.
SANDESTIN, FLA. – New forms of IgG4 testing could be more helpful in making diagnoses of immunoglobulin G4-related disease, an expert said at the annual Congress of Clinical Rheumatology, while cautioning that the diagnosis is more about histology and pattern of involvement than antibody testing.
Arezou Khosroshahi, MD, of Emory University, Atlanta, said that the IgG4 levels found in serum using nephelometry can often be low in patients who otherwise show signs of the disease, which can affect a wide array of organs and typically involves elevated IgG4. Newer forms of testing – enzyme-linked ImmunoSpot (ELISPOT) and quantitative reverse transcription polymerase chain reaction (RT-qPCR) – could be more telling, she said.
But she found that, on flow cytometry, 88% of the woman’s circulating B cells were positive for IgG4, so the woman was treated with rituximab to deplete these cells.
“When the B cells were gone, we had release of the IgG4 in the serum and now we could pick it up with nephelometry,” she said.
This missed IgG4 with nephelometry prompted researchers to turn to ELISPOT, a sensitive method to count antibody-secreting cells. The test works well by capturing the antibodies’ presence right after they’re secreted, before they can become lost to receptor binding or in other ways.
“This was a better assay to measure the IgG4 antibodies rather than nephelometry,” she said.
Perhaps even better, studies in Europe have found that RT-qPCR testing for IgG4 RNA can be effective. This type of testing is easier than ELISPOT, and “they are finding the sensitivity to be much superior to nephelometry for immunoglobulin levels,” Dr. Khosroshahi said.
The higher the levels of IgG4, the more likely an IgG4-related disease diagnosis is warranted, and higher levels tend to lead to worse outcomes, she said.
She waved a caution flag, though: Other diseases can involve elevated IgG4, and even a normal IgG4 level does not necessarily rule out the disease. Other evaluations really form the cornerstone of the diagnosis of IgG4-related disease, she said.
“There should be characteristic histology and of course IgG4-staining, but more importantly, pattern of organ involvement. It’s very important,” Dr. Khosroshahi said. “If there is a mass in the pancreas and there is salivary gland and parotid gland swellings and other features of that going on, you are more concerned that that is a process going on.”
Dr. Khosroshahi had no relevant disclosures.
SANDESTIN, FLA. – New forms of IgG4 testing could be more helpful in making diagnoses of immunoglobulin G4-related disease, an expert said at the annual Congress of Clinical Rheumatology, while cautioning that the diagnosis is more about histology and pattern of involvement than antibody testing.
Arezou Khosroshahi, MD, of Emory University, Atlanta, said that the IgG4 levels found in serum using nephelometry can often be low in patients who otherwise show signs of the disease, which can affect a wide array of organs and typically involves elevated IgG4. Newer forms of testing – enzyme-linked ImmunoSpot (ELISPOT) and quantitative reverse transcription polymerase chain reaction (RT-qPCR) – could be more telling, she said.
But she found that, on flow cytometry, 88% of the woman’s circulating B cells were positive for IgG4, so the woman was treated with rituximab to deplete these cells.
“When the B cells were gone, we had release of the IgG4 in the serum and now we could pick it up with nephelometry,” she said.
This missed IgG4 with nephelometry prompted researchers to turn to ELISPOT, a sensitive method to count antibody-secreting cells. The test works well by capturing the antibodies’ presence right after they’re secreted, before they can become lost to receptor binding or in other ways.
“This was a better assay to measure the IgG4 antibodies rather than nephelometry,” she said.
Perhaps even better, studies in Europe have found that RT-qPCR testing for IgG4 RNA can be effective. This type of testing is easier than ELISPOT, and “they are finding the sensitivity to be much superior to nephelometry for immunoglobulin levels,” Dr. Khosroshahi said.
The higher the levels of IgG4, the more likely an IgG4-related disease diagnosis is warranted, and higher levels tend to lead to worse outcomes, she said.
She waved a caution flag, though: Other diseases can involve elevated IgG4, and even a normal IgG4 level does not necessarily rule out the disease. Other evaluations really form the cornerstone of the diagnosis of IgG4-related disease, she said.
“There should be characteristic histology and of course IgG4-staining, but more importantly, pattern of organ involvement. It’s very important,” Dr. Khosroshahi said. “If there is a mass in the pancreas and there is salivary gland and parotid gland swellings and other features of that going on, you are more concerned that that is a process going on.”
Dr. Khosroshahi had no relevant disclosures.
EXPERT ANALYSIS FROM CCR 18
I’M NOT A PROVIDER
I am not sure when it occurred. I don’t know how it happened. I don’t think anyone took a vote on it. It happened gradually over the last decade. I think it happened when the administrative staff became larger than the medical staff. In order to include everyone under the same umbrella, everyone became a provider.
All those patients in our waiting room suddenly became consumers or clients. My grandfather ran a grocery store and had a lot of customers, and my father was a lawyer and had a lot of clients. None of those customers or clients would come to see me as their doctor with their illnesses today if I were a provider. The use of the terminology of “providers” and “customers” lowers all health care staff to the lowest common denominator and demeans the concerns of my patients.

I do not mean to diminish the role of the auto mechanic and salesperson, but they know and I know that our roles and are different and we are not just providers of a medical commodity. They do not expect me to deal with them as though they were coming to buy a car. They understand that we are actually trying to cure and treat worried patients and not to sell to customers in a show room.
This change in nomenclature that has permeated health care has had significant effects on how medical care is provided. Hospital care has been depersonalized in order to expedite hospital stays and maximize reimbursement. Gone is the hospital visit of your doctors when you need them the most.
Part of it is the complexity of contemporary care that requires the input from varying levels of expertise. Patients are often shuttled from one doctor to another. Communication is carried out through the web and rarely doctor to doctor. Often doctors are dealt with both at the patient level and the administrative level as commodities off the shelf, like buying a pair of shoes. And doctors in the hospital and in the clinic can be replaced by another one as the shift changes or the schedule dictates with little regard to the patient’s – or customer’s – choice.
Can we return to the days of yore? Probably not. All we can do now is try to inject some level of humanity and empathy as we see our patients in today’s world of mechanized medicine.
Dr. Goldstein, medical editor of Cardiology News, is a professor of medicine at Wayne State University and the division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
I am not sure when it occurred. I don’t know how it happened. I don’t think anyone took a vote on it. It happened gradually over the last decade. I think it happened when the administrative staff became larger than the medical staff. In order to include everyone under the same umbrella, everyone became a provider.
All those patients in our waiting room suddenly became consumers or clients. My grandfather ran a grocery store and had a lot of customers, and my father was a lawyer and had a lot of clients. None of those customers or clients would come to see me as their doctor with their illnesses today if I were a provider. The use of the terminology of “providers” and “customers” lowers all health care staff to the lowest common denominator and demeans the concerns of my patients.
I do not mean to diminish the role of the auto mechanic and salesperson, but they know and I know that our roles and are different and we are not just providers of a medical commodity. They do not expect me to deal with them as though they were coming to buy a car. They understand that we are actually trying to cure and treat worried patients and not to sell to customers in a show room.
This change in nomenclature that has permeated health care has had significant effects on how medical care is provided. Hospital care has been depersonalized in order to expedite hospital stays and maximize reimbursement. Gone is the hospital visit of your doctors when you need them the most.
Part of it is the complexity of contemporary care that requires the input from varying levels of expertise. Patients are often shuttled from one doctor to another. Communication is carried out through the web and rarely doctor to doctor. Often doctors are dealt with both at the patient level and the administrative level as commodities off the shelf, like buying a pair of shoes. And doctors in the hospital and in the clinic can be replaced by another one as the shift changes or the schedule dictates with little regard to the patient’s – or customer’s – choice.
Can we return to the days of yore? Probably not. All we can do now is try to inject some level of humanity and empathy as we see our patients in today’s world of mechanized medicine.
Dr. Goldstein, medical editor of Cardiology News, is a professor of medicine at Wayne State University and the division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
I am not sure when it occurred. I don’t know how it happened. I don’t think anyone took a vote on it. It happened gradually over the last decade. I think it happened when the administrative staff became larger than the medical staff. In order to include everyone under the same umbrella, everyone became a provider.
All those patients in our waiting room suddenly became consumers or clients. My grandfather ran a grocery store and had a lot of customers, and my father was a lawyer and had a lot of clients. None of those customers or clients would come to see me as their doctor with their illnesses today if I were a provider. The use of the terminology of “providers” and “customers” lowers all health care staff to the lowest common denominator and demeans the concerns of my patients.
I do not mean to diminish the role of the auto mechanic and salesperson, but they know and I know that our roles and are different and we are not just providers of a medical commodity. They do not expect me to deal with them as though they were coming to buy a car. They understand that we are actually trying to cure and treat worried patients and not to sell to customers in a show room.
This change in nomenclature that has permeated health care has had significant effects on how medical care is provided. Hospital care has been depersonalized in order to expedite hospital stays and maximize reimbursement. Gone is the hospital visit of your doctors when you need them the most.
Part of it is the complexity of contemporary care that requires the input from varying levels of expertise. Patients are often shuttled from one doctor to another. Communication is carried out through the web and rarely doctor to doctor. Often doctors are dealt with both at the patient level and the administrative level as commodities off the shelf, like buying a pair of shoes. And doctors in the hospital and in the clinic can be replaced by another one as the shift changes or the schedule dictates with little regard to the patient’s – or customer’s – choice.
Can we return to the days of yore? Probably not. All we can do now is try to inject some level of humanity and empathy as we see our patients in today’s world of mechanized medicine.
Dr. Goldstein, medical editor of Cardiology News, is a professor of medicine at Wayne State University and the division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
Link between alcohol consumption, neuroinflammation has possible treatment implications
NEW YORK – Recent discoveries regarding the relationship between alcohol consumption and neuroinflammation suggest a possible role for adjunctive treatments and supplements in addiction treatment, according to Shram Shukla, MD.
For example, a qualitative review of the literature over the past 2-3 years showed that the “neuroinflammatory process in and of itself drives epigenetic changes, which ultimately upregulate neuroinflammation of the brain,” according to Dr. Shukla of Walter Reed National Military Medical Center, Bethesda, Md., who reported the findings in a poster at the annual meeting of the American Psychiatric Association.
In this video interview, he explained that this finding is important because “neuroinflammation leads to neurotoxicity, which leads to neuronal degeneration.”
“As we know, with patients who chronically abuse alcohol, they do have a level of cortical degeneration that we often see on imaging, so there is, perhaps, a role that this may play in that,” he added.
Dr. Shukla said he also found that the neuroinflammatory process, when it drives the epigenetic changes, affects the amygdala, which is known as a “high stress part of the brain.”
“What we found in animal models so far is that if we can impact where that epigenetic change occurs, we can prevent the anxiogenic behaviors we often see in alcohol withdrawal and abstinence; we associate that, in humans, to be the high-stress state we often see in patients when they ... are withdrawing from alcohol.”
This raised questions about whether certain medications and supplements, including vitamin C, pioglitazone, infliximab, and omega-3 fatty acids, could be of benefit, and it was shown that these do affect neuroinflammation and stop neurotoxicity from occurring – and also, in turn, prevent the epigenetic changes, he said.
The take-away is that
“Now we have something, potentially ... that can do some of the ancillary stuff, working on the withdrawal effects, helping with the behavioral response that we see in patients that are suffering from addiction,” he said.
Dr. Shukla reported having no disclosures.
SOURCE: Shukla S et al. APA Poster Session 4, Poster 3.
NEW YORK – Recent discoveries regarding the relationship between alcohol consumption and neuroinflammation suggest a possible role for adjunctive treatments and supplements in addiction treatment, according to Shram Shukla, MD.
For example, a qualitative review of the literature over the past 2-3 years showed that the “neuroinflammatory process in and of itself drives epigenetic changes, which ultimately upregulate neuroinflammation of the brain,” according to Dr. Shukla of Walter Reed National Military Medical Center, Bethesda, Md., who reported the findings in a poster at the annual meeting of the American Psychiatric Association.
In this video interview, he explained that this finding is important because “neuroinflammation leads to neurotoxicity, which leads to neuronal degeneration.”
“As we know, with patients who chronically abuse alcohol, they do have a level of cortical degeneration that we often see on imaging, so there is, perhaps, a role that this may play in that,” he added.
Dr. Shukla said he also found that the neuroinflammatory process, when it drives the epigenetic changes, affects the amygdala, which is known as a “high stress part of the brain.”
“What we found in animal models so far is that if we can impact where that epigenetic change occurs, we can prevent the anxiogenic behaviors we often see in alcohol withdrawal and abstinence; we associate that, in humans, to be the high-stress state we often see in patients when they ... are withdrawing from alcohol.”
This raised questions about whether certain medications and supplements, including vitamin C, pioglitazone, infliximab, and omega-3 fatty acids, could be of benefit, and it was shown that these do affect neuroinflammation and stop neurotoxicity from occurring – and also, in turn, prevent the epigenetic changes, he said.
The take-away is that
“Now we have something, potentially ... that can do some of the ancillary stuff, working on the withdrawal effects, helping with the behavioral response that we see in patients that are suffering from addiction,” he said.
Dr. Shukla reported having no disclosures.
SOURCE: Shukla S et al. APA Poster Session 4, Poster 3.
NEW YORK – Recent discoveries regarding the relationship between alcohol consumption and neuroinflammation suggest a possible role for adjunctive treatments and supplements in addiction treatment, according to Shram Shukla, MD.
For example, a qualitative review of the literature over the past 2-3 years showed that the “neuroinflammatory process in and of itself drives epigenetic changes, which ultimately upregulate neuroinflammation of the brain,” according to Dr. Shukla of Walter Reed National Military Medical Center, Bethesda, Md., who reported the findings in a poster at the annual meeting of the American Psychiatric Association.
In this video interview, he explained that this finding is important because “neuroinflammation leads to neurotoxicity, which leads to neuronal degeneration.”
“As we know, with patients who chronically abuse alcohol, they do have a level of cortical degeneration that we often see on imaging, so there is, perhaps, a role that this may play in that,” he added.
Dr. Shukla said he also found that the neuroinflammatory process, when it drives the epigenetic changes, affects the amygdala, which is known as a “high stress part of the brain.”
“What we found in animal models so far is that if we can impact where that epigenetic change occurs, we can prevent the anxiogenic behaviors we often see in alcohol withdrawal and abstinence; we associate that, in humans, to be the high-stress state we often see in patients when they ... are withdrawing from alcohol.”
This raised questions about whether certain medications and supplements, including vitamin C, pioglitazone, infliximab, and omega-3 fatty acids, could be of benefit, and it was shown that these do affect neuroinflammation and stop neurotoxicity from occurring – and also, in turn, prevent the epigenetic changes, he said.
The take-away is that
“Now we have something, potentially ... that can do some of the ancillary stuff, working on the withdrawal effects, helping with the behavioral response that we see in patients that are suffering from addiction,” he said.
Dr. Shukla reported having no disclosures.
SOURCE: Shukla S et al. APA Poster Session 4, Poster 3.
REPORTING FROM APA

New ‘immune checkpoint’ vaccine shows promise in treating colorectal cancer
A novel vaccine may provide a breakthrough treatment option in colorectal and other cancers.
“We [researchers] at the Peter MacCallum Cancer Centre in Melbourne, Australia, have developed a new, DNA-based vaccine, which we call TetMYB, for the treatment of MYB-overexpressing cancers such as colorectal, adenoid cystic carcinoma, and breast cancer, to name a few,” Toan Pham, MD, of the Peter MacCallum Cancer Centre, said in a media briefing in advance of the annual Digestive Disease Week® conference.
The immunotherapy approach described by Dr. Pham involves combining the TetMYB vaccine, which boosts the immune system, and an antibody, BGB-A317, that helps enhance the effectiveness of the vaccine.
“The research we are presenting at DDW involves testing this approach in mouse models, which we had previously published. In our studies, tumors in the mice responded very well to the treatment and cancer was cured in about half of them,” stated Dr. Pham.
The study is really composed of two smaller studies that looked at colonic adenoma, induced using tamoxifen-enriched feed, in a total of 22 mice. The mice were split into two study groups: 15 in a prophylactic study and 7 in a therapeutic pilot study.
In the prophylactic study, eight treatment mice received three TetMYB vaccinations at weeks 7, 9, and 11. Tamoxifen was given to the treatment and control mice at week 13.
The seven mice in the therapeutic pilot study were given tamoxifen at week 9 and subsequently monitored via colonoscopy weekly. Once the presence of adenoma was identified, mice received six doses of TetMYB and four doses of anti-PD1 antibody.
In both the prophylactic and therapeutic study, the mice survival rates were higher than expected. In the prophylactic study, the treatment groups’ median survival time was 356 days, nearly double the control group (183 days). More impressively, all mice in the therapeutic group were alive at 235 days.
“The mice were only expected to live for a couple of days or weeks. But, about 50% of them, they lived for more than 2 years,” said Dr. Pham. “Additionally, when the cured mice from the original study were later rechallenged with the same treatment, it was immediately rejected, thus proving there is an immune memory induced by the vaccine.”
With the positive results of the mouse trial, Dr. Pham spoke to the ongoing human clinical trial and the future of this vaccine.
“Currently, we are conducting a first-in-human phase 1 clinical trial,” stated Dr. Pham. “The next phase of evolution for our vaccine, which I will also be presenting at DDW, is testing our vaccine as an anti-adenoma vaccine.” Adenomas account for nearly 80% of bowel cancers, according to Dr. Pham.
Should the safety data from the phase 1 trial prove the vaccine to be safe, a follow-up clinical trial looking at high-risk populations would be the next step.
A novel vaccine may provide a breakthrough treatment option in colorectal and other cancers.
“We [researchers] at the Peter MacCallum Cancer Centre in Melbourne, Australia, have developed a new, DNA-based vaccine, which we call TetMYB, for the treatment of MYB-overexpressing cancers such as colorectal, adenoid cystic carcinoma, and breast cancer, to name a few,” Toan Pham, MD, of the Peter MacCallum Cancer Centre, said in a media briefing in advance of the annual Digestive Disease Week® conference.
The immunotherapy approach described by Dr. Pham involves combining the TetMYB vaccine, which boosts the immune system, and an antibody, BGB-A317, that helps enhance the effectiveness of the vaccine.
“The research we are presenting at DDW involves testing this approach in mouse models, which we had previously published. In our studies, tumors in the mice responded very well to the treatment and cancer was cured in about half of them,” stated Dr. Pham.
The study is really composed of two smaller studies that looked at colonic adenoma, induced using tamoxifen-enriched feed, in a total of 22 mice. The mice were split into two study groups: 15 in a prophylactic study and 7 in a therapeutic pilot study.
In the prophylactic study, eight treatment mice received three TetMYB vaccinations at weeks 7, 9, and 11. Tamoxifen was given to the treatment and control mice at week 13.
The seven mice in the therapeutic pilot study were given tamoxifen at week 9 and subsequently monitored via colonoscopy weekly. Once the presence of adenoma was identified, mice received six doses of TetMYB and four doses of anti-PD1 antibody.
In both the prophylactic and therapeutic study, the mice survival rates were higher than expected. In the prophylactic study, the treatment groups’ median survival time was 356 days, nearly double the control group (183 days). More impressively, all mice in the therapeutic group were alive at 235 days.
“The mice were only expected to live for a couple of days or weeks. But, about 50% of them, they lived for more than 2 years,” said Dr. Pham. “Additionally, when the cured mice from the original study were later rechallenged with the same treatment, it was immediately rejected, thus proving there is an immune memory induced by the vaccine.”
With the positive results of the mouse trial, Dr. Pham spoke to the ongoing human clinical trial and the future of this vaccine.
“Currently, we are conducting a first-in-human phase 1 clinical trial,” stated Dr. Pham. “The next phase of evolution for our vaccine, which I will also be presenting at DDW, is testing our vaccine as an anti-adenoma vaccine.” Adenomas account for nearly 80% of bowel cancers, according to Dr. Pham.
Should the safety data from the phase 1 trial prove the vaccine to be safe, a follow-up clinical trial looking at high-risk populations would be the next step.
A novel vaccine may provide a breakthrough treatment option in colorectal and other cancers.
“We [researchers] at the Peter MacCallum Cancer Centre in Melbourne, Australia, have developed a new, DNA-based vaccine, which we call TetMYB, for the treatment of MYB-overexpressing cancers such as colorectal, adenoid cystic carcinoma, and breast cancer, to name a few,” Toan Pham, MD, of the Peter MacCallum Cancer Centre, said in a media briefing in advance of the annual Digestive Disease Week® conference.
The immunotherapy approach described by Dr. Pham involves combining the TetMYB vaccine, which boosts the immune system, and an antibody, BGB-A317, that helps enhance the effectiveness of the vaccine.
“The research we are presenting at DDW involves testing this approach in mouse models, which we had previously published. In our studies, tumors in the mice responded very well to the treatment and cancer was cured in about half of them,” stated Dr. Pham.
The study is really composed of two smaller studies that looked at colonic adenoma, induced using tamoxifen-enriched feed, in a total of 22 mice. The mice were split into two study groups: 15 in a prophylactic study and 7 in a therapeutic pilot study.
In the prophylactic study, eight treatment mice received three TetMYB vaccinations at weeks 7, 9, and 11. Tamoxifen was given to the treatment and control mice at week 13.
The seven mice in the therapeutic pilot study were given tamoxifen at week 9 and subsequently monitored via colonoscopy weekly. Once the presence of adenoma was identified, mice received six doses of TetMYB and four doses of anti-PD1 antibody.
In both the prophylactic and therapeutic study, the mice survival rates were higher than expected. In the prophylactic study, the treatment groups’ median survival time was 356 days, nearly double the control group (183 days). More impressively, all mice in the therapeutic group were alive at 235 days.
“The mice were only expected to live for a couple of days or weeks. But, about 50% of them, they lived for more than 2 years,” said Dr. Pham. “Additionally, when the cured mice from the original study were later rechallenged with the same treatment, it was immediately rejected, thus proving there is an immune memory induced by the vaccine.”
With the positive results of the mouse trial, Dr. Pham spoke to the ongoing human clinical trial and the future of this vaccine.
“Currently, we are conducting a first-in-human phase 1 clinical trial,” stated Dr. Pham. “The next phase of evolution for our vaccine, which I will also be presenting at DDW, is testing our vaccine as an anti-adenoma vaccine.” Adenomas account for nearly 80% of bowel cancers, according to Dr. Pham.
Should the safety data from the phase 1 trial prove the vaccine to be safe, a follow-up clinical trial looking at high-risk populations would be the next step.
REPORTING FROM DDW
Tattoos: From Ancient Practice to Modern Treatment Dilemma
As dermatologists, we possess a vast knowledge of the epidermis. Some patients may choose to use the epidermis as a canvas for their art in the form of tattoos; however, tattoos can complicate dermatology visits in a myriad of ways. From patients seeking tattoo removal (a complicated task even with the most advanced laser treatments) to those whose native skin is obscured by a tattoo during melanoma screening, it is no wonder that many dermatologists become frustrated at the very mention of the word tattoo.
Tattoos have a long and complicated history entrenched in class divisions, gender identity, and culture. Although its origins are not well documented, many researchers believe that tattooing began in Egypt as early as 4000 BCE.1 From there, the practice spread east into South Asia and west to the British Isles and Scotland. The Iberians in the British Isles, the Picts in Scotland, the Gauls in Western Europe, and the Teutons in Germany all practiced tattooing, and the Romans were known to use tattooing to mark convicts and slaves.1 By 787 AD, tattooing was prevalent enough to warrant an official ban by Pope Hadrian I at the Second Ecumenical Council of Nicaea.2 The growing power of Christianity most likely contributed to the elimination of tattooing in the West, although many soldiers who fought in the Crusades received tattoos during their travels.3
Despite the long history of tattoos in both the East and West, Captain James Cook often is credited with discovering tattooing in the eighteenth century during his explorations in the Pacific.4 In Tahiti in 1769 and Hawaii in 1778, Cook encountered heavily tattooed populations who deposited dye into the skin by tapping sharpened instruments.3 These Polynesian tattoos, which were associated with healing and protective powers, often depicted genealogies and were composed of images of lines, stars, geometric designs, animals, and humans. Explorers in Polynesia who came after Cook noted that tattoo designs began to include rifles, cannons, and dates of chief’s deaths—an indication of the cultural exchange that occurred between Cook’s crew and the natives.3 The first tattooed peoples were displayed in the United States at the Centennial Exhibition in Philadelphia, Pennsylvania, in 1876.2 Later, at the 1901 World’s Fair in Buffalo, New York, the first full “freak show” emerged, and tattooed “natives” were displayed.5 Since they were introduced in the West, tattoos have been associated with an element of the exotic in the United States.
Acknowledged by many to be the first professional tattooist in the United States, Martin Hildebrandt opened his shop in New York City, New York, in 1846.2 Initially, only sailors and soldiers were tattooed, which contributed to the concept of the so-called “tattooed serviceman.”5 However, after the Spanish-American War, tattoos became a fad among the high society in Europe. Tattooing at this time was still performed through the ancient Polynesian tapping method, making it both time-consuming and expensive. Tattoos generally were always placed in a private location, leading to popular speculation at the time about whom in the aristocracy possessed a tattoo, with some even speculating that Queen Victoria may have had a tattoo.1 However, this brief trend among the aristocracy came to an end when Samuel O’Reilly, an American tattoo artist, patented the first electric tattooing machine in 1891.6 His invention made tattooing faster, cheaper, and less painful, thereby making tattooing available to a much wider audience. In the United States, men in the military often were tattooed, especially during World Wars I and II, when patriotic themes and tattoos of important women in their lives (eg, the word Mom, the name of a sweetheart) became popular.
It is a popular belief that a tattoo renaissance occurred in the United States in the 1970s, sparked by an influx of Indonesian and Asian artistic styles. Today, tattoos are ubiquitous. A 2012 poll showed that 21% of adults in the United States have a tattoo.7 There are now 4 main types of tattoos: cosmetic (eg, permanent makeup), traumatic (eg, injury on asphalt), medical (eg, to mark radiation sites), and decorative—either amateur (often done by hand) or professional (done in tattoo parlors with electric tattooing needles).8
Laser Tattoo Removal
Today tattoos are easy and relatively cheap to get, and for most people they are not regarded as an important cultural milestone like they were in early Polynesian culture. As a result, dermatologists often may encounter patients seeking to have these permanent designs removed from their skin. Previously, tattoo removal was attempted using destructive processes such as scarification and cryotherapy and generally resulted in poor cosmetics outcomes. Today, lasers are at the forefront of tattoo removal. Traditional lasers use pulse durations in the nanosecond range, with newer generation lasers in the picosecond range delivering much shorter pulse durations, effectively delivering the same level of energy over less time. It is important to select the correct laser for optimal destruction of various tattoo ink colors (Table).8,9
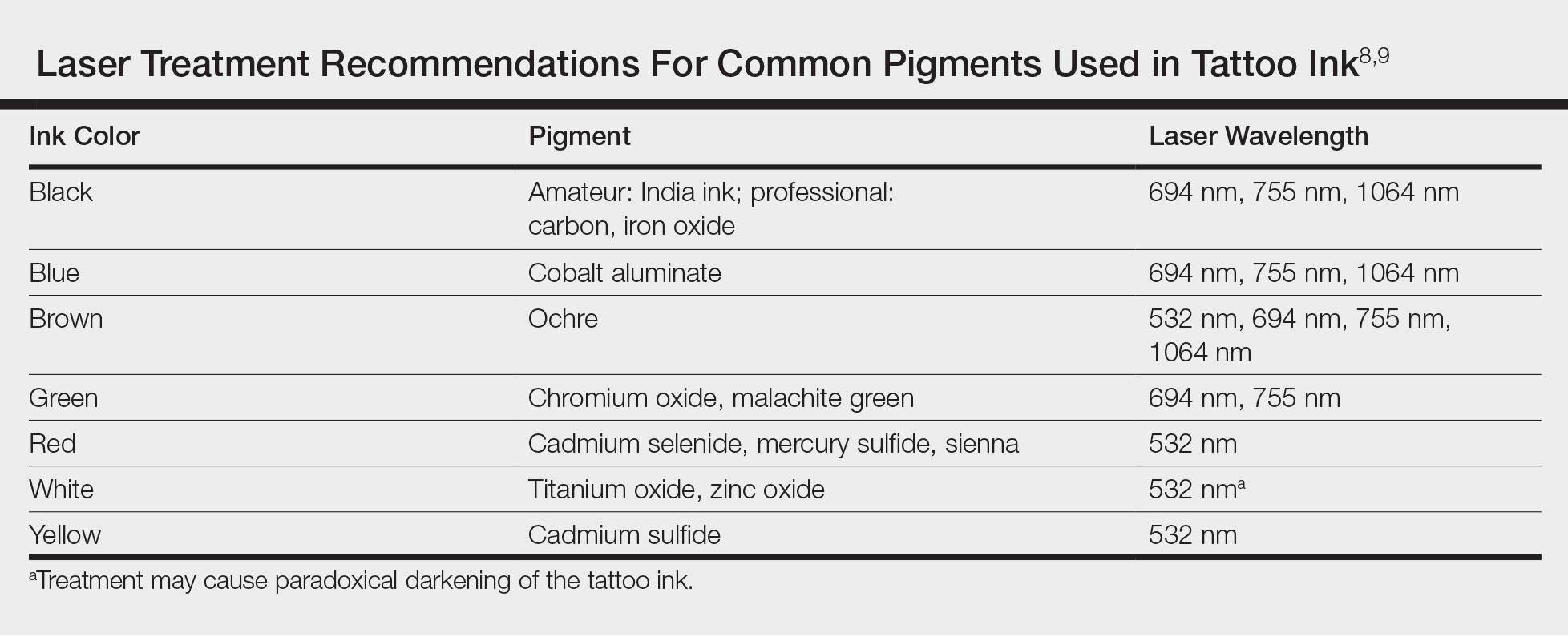
Controversy persists as to whether tattoo pigment destruction by lasers is caused by thermal or acoustic damage.10 It may be a combination of both, with rapid heating of the particles leading to a local shockwave as the energy collapses.11 The goal of tattoo removal is to create smaller granules of pigment that can be taken up by the patient’s lymphatic system. The largest granule that can be taken up by the lymphatic system is 0.4 μm.10
In laser treatment of any skin condition, the laser energy is delivered in a pulse duration that should be less than the thermal relaxation time of the chromophores (water, melanin, hemoglobin, or tattoo pigment are the main targets within the skin).12 Most tattoo chromophores are 30 nm to 300 nm, with a thermal relaxation time of less than 10 nanoseconds.10,12 As the number of treatments progresses, laser settings should be adjusted for smaller ink particles. Patients should be warned about pain, side effects, and the need for multiple treatments. Common side effects of laser tattoo removal include purpura, pinpoint bleeding, erythema, edema, crusting, and blistering.8
After laser treatment, cytoplasmic water in the cell is converted into steam leading to cavitation of the lysosome, which presents as whitening of the skin. The whitening causes optical scatter, thereby preventing immediate retreatment of the area.11 The R20 laser tattoo removal method discussed by Kossida et al,13 advises practitioners to wait 20 minutes between treatments to allow the air bubbles from the conversion of water to steam to disappear. Kossida et al13 demonstrated more effective removal in tattoos that were treated with this method compared to standard treatment. The recognition that trapped air bubbles delay multiple treatment cycles has led to the experimental use of perfluorodecalin, a fluorocarbon liquid capable of dissolving the air bubbles, for immediate retreatment.14 By dissolving the trapped air and eliminating the white color, multiple treatments can be completed during 1 session.
Risks of Laser Tattoo Removal
It is important to emphasize that there are potential risks associated with laser treatment for tattoo removal, many of which we are only just beginning to understand. Common side effects of laser treatment for tattoo removal include blisters, pain, bleeding, hyperpigmentation, or hypopigmentation; however, there also are rare potential risks. Tattoo ink can paradoxically darken when it contains metals such as titanium or zinc, as often is found in tan or white inks.15 The laser energy causes a shift of the metal from an oxidized to a reduced state, leading to a darker rather than lighter tattoo upon application of the laser. There also have been documented cases of intraprocedural anaphylaxis, delayed urticaria, as well as generalized eczematous reactions.16-18 In these cases, the patients had never experienced any allergic symptoms prior to the laser tattoo removal procedure.
Additionally, patients with active allergy to the pigments used in tattoo ink provide a therapeutic dilemma, as laser treatment may potentially systematize the tattoo ink, leading to a more widespread allergic reaction. A case of a generalized eczematous reaction after carbon dioxide laser therapy in a patient with documented tattoo allergy has been reported.19 More research is needed to fully understand the nature of immediate as well as delayed hypersensitivity reactions associated with laser tattoo removal.
Final Thoughts
With thousands of years of established traditions, it is unlikely that tattooing will go away anytime soon. Fortunately, lasers are providing us with an effective and safe method of removal.
- Caplan J, ed. Written on the Body: The Tattoo in European and American History. Princeton, NJ: Princeton University Press; 2000.
- DeMello M. Bodies of Inscription: Cultural History of the Modern Tattoo Community. Durham, NC: Duke University Press; 2000.
- DeMello M. “Not just for bikers anymore”: popular representations of american tattooing. J Popular Culture. 1995;29:37-52.
- Anastasia DJM. Living marked: tattooed women and perceptions of beauty and femininity. In: Segal MT, ed. Interactions and Intersections of Gendered Bodies at Work, at Home, and at Play. Bingly, UK: Emerald; 2010.
- Mifflin M. Bodies of Subversion: A Secret History of Women and Tattoo. New York: June Books; 1997.
- Atkinson M. Pretty in ink: conformity, resistance, and negotiation in women’s tattooing. Sex Roles. 2002;47:219-235.
- Braverman S. One in five US adults now has a tattoo. Harris Poll website. https://theharrispoll.com/new-york-n-y-february-23-2012-there-is-a-lot-of-culture-and-lore-associated-with-tattoos-from-ancient-art-to-modern-expressionism-and-there-are-many-reasons-people-choose-to-get-or-not-get-p/. Published February 23, 2012. Accessed May 25, 2018.
- Ho SG, Goh CL. Laser tattoo removal: a clinical update. J Cutan Aesthet Surg. 2015;8:9-15.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Sardana K, Ranjan R, Ghunawat S. Optimising laser tattoo removal. J Cutan Aesthet Surg. 2015;8:16-24.
- Shah SD, Aurangabadkar SJ. Newer trends in laser tattoo removal. J Cutan Aesthet Surg. 2015;8:25-29.
- Hsu VM, Aldahan AS, Mlacker S, et al. The picosecond laser for tattoo removal. Lasers Med Sci. 2016;31:1733-1737.
- Kossida T, Rigopoulos D, Katsambas A, et al. Optimal tattoo removal in a single laser session based on the method of repeated exposures.J Am Acad Dermatol. 2012;66:271-277.
- Biesman BS, O’Neil MP, Costner C. Rapid, high-fluence multipass Q-switched laser treatment of tattoos with a transparent perfluorodecalin-infused patch: a pilot study. Lasers Surg Med. 2015;47:613-618.
- Bernstein EF. Laser tattoo removal. Semin Plast Surg. 2007;21:175-192.
- Wilken R, Ho D, Petukhova T, et al. Intraoperative localized urticarial reaction during Q-switched Nd:YAG laser tattoo removal. J Drugs Dermatol. 2015;14:303-306.
- Hibler BP, Rossi AM. A case of delayed anaphylaxis after laser tattoo removal. JAAD Case Rep. 2015;1:80-81.
- Bernstein EF. A widespread allergic reaction to black tattoo ink caused by laser treatment. Lasers Surg Med. 2015;47:180-182.
- Meesters AA, De Rie MA, Wolkerstorfer A. Generalized eczematous reaction after fractional carbon dioxide laser therapy for tattoo allergy. J Cosmet Laser Ther. 2016;18:456-458.
As dermatologists, we possess a vast knowledge of the epidermis. Some patients may choose to use the epidermis as a canvas for their art in the form of tattoos; however, tattoos can complicate dermatology visits in a myriad of ways. From patients seeking tattoo removal (a complicated task even with the most advanced laser treatments) to those whose native skin is obscured by a tattoo during melanoma screening, it is no wonder that many dermatologists become frustrated at the very mention of the word tattoo.
Tattoos have a long and complicated history entrenched in class divisions, gender identity, and culture. Although its origins are not well documented, many researchers believe that tattooing began in Egypt as early as 4000 BCE.1 From there, the practice spread east into South Asia and west to the British Isles and Scotland. The Iberians in the British Isles, the Picts in Scotland, the Gauls in Western Europe, and the Teutons in Germany all practiced tattooing, and the Romans were known to use tattooing to mark convicts and slaves.1 By 787 AD, tattooing was prevalent enough to warrant an official ban by Pope Hadrian I at the Second Ecumenical Council of Nicaea.2 The growing power of Christianity most likely contributed to the elimination of tattooing in the West, although many soldiers who fought in the Crusades received tattoos during their travels.3
Despite the long history of tattoos in both the East and West, Captain James Cook often is credited with discovering tattooing in the eighteenth century during his explorations in the Pacific.4 In Tahiti in 1769 and Hawaii in 1778, Cook encountered heavily tattooed populations who deposited dye into the skin by tapping sharpened instruments.3 These Polynesian tattoos, which were associated with healing and protective powers, often depicted genealogies and were composed of images of lines, stars, geometric designs, animals, and humans. Explorers in Polynesia who came after Cook noted that tattoo designs began to include rifles, cannons, and dates of chief’s deaths—an indication of the cultural exchange that occurred between Cook’s crew and the natives.3 The first tattooed peoples were displayed in the United States at the Centennial Exhibition in Philadelphia, Pennsylvania, in 1876.2 Later, at the 1901 World’s Fair in Buffalo, New York, the first full “freak show” emerged, and tattooed “natives” were displayed.5 Since they were introduced in the West, tattoos have been associated with an element of the exotic in the United States.
Acknowledged by many to be the first professional tattooist in the United States, Martin Hildebrandt opened his shop in New York City, New York, in 1846.2 Initially, only sailors and soldiers were tattooed, which contributed to the concept of the so-called “tattooed serviceman.”5 However, after the Spanish-American War, tattoos became a fad among the high society in Europe. Tattooing at this time was still performed through the ancient Polynesian tapping method, making it both time-consuming and expensive. Tattoos generally were always placed in a private location, leading to popular speculation at the time about whom in the aristocracy possessed a tattoo, with some even speculating that Queen Victoria may have had a tattoo.1 However, this brief trend among the aristocracy came to an end when Samuel O’Reilly, an American tattoo artist, patented the first electric tattooing machine in 1891.6 His invention made tattooing faster, cheaper, and less painful, thereby making tattooing available to a much wider audience. In the United States, men in the military often were tattooed, especially during World Wars I and II, when patriotic themes and tattoos of important women in their lives (eg, the word Mom, the name of a sweetheart) became popular.
It is a popular belief that a tattoo renaissance occurred in the United States in the 1970s, sparked by an influx of Indonesian and Asian artistic styles. Today, tattoos are ubiquitous. A 2012 poll showed that 21% of adults in the United States have a tattoo.7 There are now 4 main types of tattoos: cosmetic (eg, permanent makeup), traumatic (eg, injury on asphalt), medical (eg, to mark radiation sites), and decorative—either amateur (often done by hand) or professional (done in tattoo parlors with electric tattooing needles).8
Laser Tattoo Removal
Today tattoos are easy and relatively cheap to get, and for most people they are not regarded as an important cultural milestone like they were in early Polynesian culture. As a result, dermatologists often may encounter patients seeking to have these permanent designs removed from their skin. Previously, tattoo removal was attempted using destructive processes such as scarification and cryotherapy and generally resulted in poor cosmetics outcomes. Today, lasers are at the forefront of tattoo removal. Traditional lasers use pulse durations in the nanosecond range, with newer generation lasers in the picosecond range delivering much shorter pulse durations, effectively delivering the same level of energy over less time. It is important to select the correct laser for optimal destruction of various tattoo ink colors (Table).8,9
Controversy persists as to whether tattoo pigment destruction by lasers is caused by thermal or acoustic damage.10 It may be a combination of both, with rapid heating of the particles leading to a local shockwave as the energy collapses.11 The goal of tattoo removal is to create smaller granules of pigment that can be taken up by the patient’s lymphatic system. The largest granule that can be taken up by the lymphatic system is 0.4 μm.10
In laser treatment of any skin condition, the laser energy is delivered in a pulse duration that should be less than the thermal relaxation time of the chromophores (water, melanin, hemoglobin, or tattoo pigment are the main targets within the skin).12 Most tattoo chromophores are 30 nm to 300 nm, with a thermal relaxation time of less than 10 nanoseconds.10,12 As the number of treatments progresses, laser settings should be adjusted for smaller ink particles. Patients should be warned about pain, side effects, and the need for multiple treatments. Common side effects of laser tattoo removal include purpura, pinpoint bleeding, erythema, edema, crusting, and blistering.8
After laser treatment, cytoplasmic water in the cell is converted into steam leading to cavitation of the lysosome, which presents as whitening of the skin. The whitening causes optical scatter, thereby preventing immediate retreatment of the area.11 The R20 laser tattoo removal method discussed by Kossida et al,13 advises practitioners to wait 20 minutes between treatments to allow the air bubbles from the conversion of water to steam to disappear. Kossida et al13 demonstrated more effective removal in tattoos that were treated with this method compared to standard treatment. The recognition that trapped air bubbles delay multiple treatment cycles has led to the experimental use of perfluorodecalin, a fluorocarbon liquid capable of dissolving the air bubbles, for immediate retreatment.14 By dissolving the trapped air and eliminating the white color, multiple treatments can be completed during 1 session.
Risks of Laser Tattoo Removal
It is important to emphasize that there are potential risks associated with laser treatment for tattoo removal, many of which we are only just beginning to understand. Common side effects of laser treatment for tattoo removal include blisters, pain, bleeding, hyperpigmentation, or hypopigmentation; however, there also are rare potential risks. Tattoo ink can paradoxically darken when it contains metals such as titanium or zinc, as often is found in tan or white inks.15 The laser energy causes a shift of the metal from an oxidized to a reduced state, leading to a darker rather than lighter tattoo upon application of the laser. There also have been documented cases of intraprocedural anaphylaxis, delayed urticaria, as well as generalized eczematous reactions.16-18 In these cases, the patients had never experienced any allergic symptoms prior to the laser tattoo removal procedure.
Additionally, patients with active allergy to the pigments used in tattoo ink provide a therapeutic dilemma, as laser treatment may potentially systematize the tattoo ink, leading to a more widespread allergic reaction. A case of a generalized eczematous reaction after carbon dioxide laser therapy in a patient with documented tattoo allergy has been reported.19 More research is needed to fully understand the nature of immediate as well as delayed hypersensitivity reactions associated with laser tattoo removal.
Final Thoughts
With thousands of years of established traditions, it is unlikely that tattooing will go away anytime soon. Fortunately, lasers are providing us with an effective and safe method of removal.
As dermatologists, we possess a vast knowledge of the epidermis. Some patients may choose to use the epidermis as a canvas for their art in the form of tattoos; however, tattoos can complicate dermatology visits in a myriad of ways. From patients seeking tattoo removal (a complicated task even with the most advanced laser treatments) to those whose native skin is obscured by a tattoo during melanoma screening, it is no wonder that many dermatologists become frustrated at the very mention of the word tattoo.
Tattoos have a long and complicated history entrenched in class divisions, gender identity, and culture. Although its origins are not well documented, many researchers believe that tattooing began in Egypt as early as 4000 BCE.1 From there, the practice spread east into South Asia and west to the British Isles and Scotland. The Iberians in the British Isles, the Picts in Scotland, the Gauls in Western Europe, and the Teutons in Germany all practiced tattooing, and the Romans were known to use tattooing to mark convicts and slaves.1 By 787 AD, tattooing was prevalent enough to warrant an official ban by Pope Hadrian I at the Second Ecumenical Council of Nicaea.2 The growing power of Christianity most likely contributed to the elimination of tattooing in the West, although many soldiers who fought in the Crusades received tattoos during their travels.3
Despite the long history of tattoos in both the East and West, Captain James Cook often is credited with discovering tattooing in the eighteenth century during his explorations in the Pacific.4 In Tahiti in 1769 and Hawaii in 1778, Cook encountered heavily tattooed populations who deposited dye into the skin by tapping sharpened instruments.3 These Polynesian tattoos, which were associated with healing and protective powers, often depicted genealogies and were composed of images of lines, stars, geometric designs, animals, and humans. Explorers in Polynesia who came after Cook noted that tattoo designs began to include rifles, cannons, and dates of chief’s deaths—an indication of the cultural exchange that occurred between Cook’s crew and the natives.3 The first tattooed peoples were displayed in the United States at the Centennial Exhibition in Philadelphia, Pennsylvania, in 1876.2 Later, at the 1901 World’s Fair in Buffalo, New York, the first full “freak show” emerged, and tattooed “natives” were displayed.5 Since they were introduced in the West, tattoos have been associated with an element of the exotic in the United States.
Acknowledged by many to be the first professional tattooist in the United States, Martin Hildebrandt opened his shop in New York City, New York, in 1846.2 Initially, only sailors and soldiers were tattooed, which contributed to the concept of the so-called “tattooed serviceman.”5 However, after the Spanish-American War, tattoos became a fad among the high society in Europe. Tattooing at this time was still performed through the ancient Polynesian tapping method, making it both time-consuming and expensive. Tattoos generally were always placed in a private location, leading to popular speculation at the time about whom in the aristocracy possessed a tattoo, with some even speculating that Queen Victoria may have had a tattoo.1 However, this brief trend among the aristocracy came to an end when Samuel O’Reilly, an American tattoo artist, patented the first electric tattooing machine in 1891.6 His invention made tattooing faster, cheaper, and less painful, thereby making tattooing available to a much wider audience. In the United States, men in the military often were tattooed, especially during World Wars I and II, when patriotic themes and tattoos of important women in their lives (eg, the word Mom, the name of a sweetheart) became popular.
It is a popular belief that a tattoo renaissance occurred in the United States in the 1970s, sparked by an influx of Indonesian and Asian artistic styles. Today, tattoos are ubiquitous. A 2012 poll showed that 21% of adults in the United States have a tattoo.7 There are now 4 main types of tattoos: cosmetic (eg, permanent makeup), traumatic (eg, injury on asphalt), medical (eg, to mark radiation sites), and decorative—either amateur (often done by hand) or professional (done in tattoo parlors with electric tattooing needles).8
Laser Tattoo Removal
Today tattoos are easy and relatively cheap to get, and for most people they are not regarded as an important cultural milestone like they were in early Polynesian culture. As a result, dermatologists often may encounter patients seeking to have these permanent designs removed from their skin. Previously, tattoo removal was attempted using destructive processes such as scarification and cryotherapy and generally resulted in poor cosmetics outcomes. Today, lasers are at the forefront of tattoo removal. Traditional lasers use pulse durations in the nanosecond range, with newer generation lasers in the picosecond range delivering much shorter pulse durations, effectively delivering the same level of energy over less time. It is important to select the correct laser for optimal destruction of various tattoo ink colors (Table).8,9
Controversy persists as to whether tattoo pigment destruction by lasers is caused by thermal or acoustic damage.10 It may be a combination of both, with rapid heating of the particles leading to a local shockwave as the energy collapses.11 The goal of tattoo removal is to create smaller granules of pigment that can be taken up by the patient’s lymphatic system. The largest granule that can be taken up by the lymphatic system is 0.4 μm.10
In laser treatment of any skin condition, the laser energy is delivered in a pulse duration that should be less than the thermal relaxation time of the chromophores (water, melanin, hemoglobin, or tattoo pigment are the main targets within the skin).12 Most tattoo chromophores are 30 nm to 300 nm, with a thermal relaxation time of less than 10 nanoseconds.10,12 As the number of treatments progresses, laser settings should be adjusted for smaller ink particles. Patients should be warned about pain, side effects, and the need for multiple treatments. Common side effects of laser tattoo removal include purpura, pinpoint bleeding, erythema, edema, crusting, and blistering.8
After laser treatment, cytoplasmic water in the cell is converted into steam leading to cavitation of the lysosome, which presents as whitening of the skin. The whitening causes optical scatter, thereby preventing immediate retreatment of the area.11 The R20 laser tattoo removal method discussed by Kossida et al,13 advises practitioners to wait 20 minutes between treatments to allow the air bubbles from the conversion of water to steam to disappear. Kossida et al13 demonstrated more effective removal in tattoos that were treated with this method compared to standard treatment. The recognition that trapped air bubbles delay multiple treatment cycles has led to the experimental use of perfluorodecalin, a fluorocarbon liquid capable of dissolving the air bubbles, for immediate retreatment.14 By dissolving the trapped air and eliminating the white color, multiple treatments can be completed during 1 session.
Risks of Laser Tattoo Removal
It is important to emphasize that there are potential risks associated with laser treatment for tattoo removal, many of which we are only just beginning to understand. Common side effects of laser treatment for tattoo removal include blisters, pain, bleeding, hyperpigmentation, or hypopigmentation; however, there also are rare potential risks. Tattoo ink can paradoxically darken when it contains metals such as titanium or zinc, as often is found in tan or white inks.15 The laser energy causes a shift of the metal from an oxidized to a reduced state, leading to a darker rather than lighter tattoo upon application of the laser. There also have been documented cases of intraprocedural anaphylaxis, delayed urticaria, as well as generalized eczematous reactions.16-18 In these cases, the patients had never experienced any allergic symptoms prior to the laser tattoo removal procedure.
Additionally, patients with active allergy to the pigments used in tattoo ink provide a therapeutic dilemma, as laser treatment may potentially systematize the tattoo ink, leading to a more widespread allergic reaction. A case of a generalized eczematous reaction after carbon dioxide laser therapy in a patient with documented tattoo allergy has been reported.19 More research is needed to fully understand the nature of immediate as well as delayed hypersensitivity reactions associated with laser tattoo removal.
Final Thoughts
With thousands of years of established traditions, it is unlikely that tattooing will go away anytime soon. Fortunately, lasers are providing us with an effective and safe method of removal.
- Caplan J, ed. Written on the Body: The Tattoo in European and American History. Princeton, NJ: Princeton University Press; 2000.
- DeMello M. Bodies of Inscription: Cultural History of the Modern Tattoo Community. Durham, NC: Duke University Press; 2000.
- DeMello M. “Not just for bikers anymore”: popular representations of american tattooing. J Popular Culture. 1995;29:37-52.
- Anastasia DJM. Living marked: tattooed women and perceptions of beauty and femininity. In: Segal MT, ed. Interactions and Intersections of Gendered Bodies at Work, at Home, and at Play. Bingly, UK: Emerald; 2010.
- Mifflin M. Bodies of Subversion: A Secret History of Women and Tattoo. New York: June Books; 1997.
- Atkinson M. Pretty in ink: conformity, resistance, and negotiation in women’s tattooing. Sex Roles. 2002;47:219-235.
- Braverman S. One in five US adults now has a tattoo. Harris Poll website. https://theharrispoll.com/new-york-n-y-february-23-2012-there-is-a-lot-of-culture-and-lore-associated-with-tattoos-from-ancient-art-to-modern-expressionism-and-there-are-many-reasons-people-choose-to-get-or-not-get-p/. Published February 23, 2012. Accessed May 25, 2018.
- Ho SG, Goh CL. Laser tattoo removal: a clinical update. J Cutan Aesthet Surg. 2015;8:9-15.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Sardana K, Ranjan R, Ghunawat S. Optimising laser tattoo removal. J Cutan Aesthet Surg. 2015;8:16-24.
- Shah SD, Aurangabadkar SJ. Newer trends in laser tattoo removal. J Cutan Aesthet Surg. 2015;8:25-29.
- Hsu VM, Aldahan AS, Mlacker S, et al. The picosecond laser for tattoo removal. Lasers Med Sci. 2016;31:1733-1737.
- Kossida T, Rigopoulos D, Katsambas A, et al. Optimal tattoo removal in a single laser session based on the method of repeated exposures.J Am Acad Dermatol. 2012;66:271-277.
- Biesman BS, O’Neil MP, Costner C. Rapid, high-fluence multipass Q-switched laser treatment of tattoos with a transparent perfluorodecalin-infused patch: a pilot study. Lasers Surg Med. 2015;47:613-618.
- Bernstein EF. Laser tattoo removal. Semin Plast Surg. 2007;21:175-192.
- Wilken R, Ho D, Petukhova T, et al. Intraoperative localized urticarial reaction during Q-switched Nd:YAG laser tattoo removal. J Drugs Dermatol. 2015;14:303-306.
- Hibler BP, Rossi AM. A case of delayed anaphylaxis after laser tattoo removal. JAAD Case Rep. 2015;1:80-81.
- Bernstein EF. A widespread allergic reaction to black tattoo ink caused by laser treatment. Lasers Surg Med. 2015;47:180-182.
- Meesters AA, De Rie MA, Wolkerstorfer A. Generalized eczematous reaction after fractional carbon dioxide laser therapy for tattoo allergy. J Cosmet Laser Ther. 2016;18:456-458.
- Caplan J, ed. Written on the Body: The Tattoo in European and American History. Princeton, NJ: Princeton University Press; 2000.
- DeMello M. Bodies of Inscription: Cultural History of the Modern Tattoo Community. Durham, NC: Duke University Press; 2000.
- DeMello M. “Not just for bikers anymore”: popular representations of american tattooing. J Popular Culture. 1995;29:37-52.
- Anastasia DJM. Living marked: tattooed women and perceptions of beauty and femininity. In: Segal MT, ed. Interactions and Intersections of Gendered Bodies at Work, at Home, and at Play. Bingly, UK: Emerald; 2010.
- Mifflin M. Bodies of Subversion: A Secret History of Women and Tattoo. New York: June Books; 1997.
- Atkinson M. Pretty in ink: conformity, resistance, and negotiation in women’s tattooing. Sex Roles. 2002;47:219-235.
- Braverman S. One in five US adults now has a tattoo. Harris Poll website. https://theharrispoll.com/new-york-n-y-february-23-2012-there-is-a-lot-of-culture-and-lore-associated-with-tattoos-from-ancient-art-to-modern-expressionism-and-there-are-many-reasons-people-choose-to-get-or-not-get-p/. Published February 23, 2012. Accessed May 25, 2018.
- Ho SG, Goh CL. Laser tattoo removal: a clinical update. J Cutan Aesthet Surg. 2015;8:9-15.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Sardana K, Ranjan R, Ghunawat S. Optimising laser tattoo removal. J Cutan Aesthet Surg. 2015;8:16-24.
- Shah SD, Aurangabadkar SJ. Newer trends in laser tattoo removal. J Cutan Aesthet Surg. 2015;8:25-29.
- Hsu VM, Aldahan AS, Mlacker S, et al. The picosecond laser for tattoo removal. Lasers Med Sci. 2016;31:1733-1737.
- Kossida T, Rigopoulos D, Katsambas A, et al. Optimal tattoo removal in a single laser session based on the method of repeated exposures.J Am Acad Dermatol. 2012;66:271-277.
- Biesman BS, O’Neil MP, Costner C. Rapid, high-fluence multipass Q-switched laser treatment of tattoos with a transparent perfluorodecalin-infused patch: a pilot study. Lasers Surg Med. 2015;47:613-618.
- Bernstein EF. Laser tattoo removal. Semin Plast Surg. 2007;21:175-192.
- Wilken R, Ho D, Petukhova T, et al. Intraoperative localized urticarial reaction during Q-switched Nd:YAG laser tattoo removal. J Drugs Dermatol. 2015;14:303-306.
- Hibler BP, Rossi AM. A case of delayed anaphylaxis after laser tattoo removal. JAAD Case Rep. 2015;1:80-81.
- Bernstein EF. A widespread allergic reaction to black tattoo ink caused by laser treatment. Lasers Surg Med. 2015;47:180-182.
- Meesters AA, De Rie MA, Wolkerstorfer A. Generalized eczematous reaction after fractional carbon dioxide laser therapy for tattoo allergy. J Cosmet Laser Ther. 2016;18:456-458.

Catching Up With Our Past CHEST Presidents
Where are they now? What have they been up to? CHEST’s Past Presidents each forged the way for the many successes of the American College of Chest Physicians, leading to enhanced patient care around the globe. Their outstanding leadership and vision are evidenced today in many of CHEST’s strategic initiatives.
D. Robert McCaffree, MD, MSHA, Master FCCP
CHEST President 1997 - 1998
I received the chain of office (yes, there is an actual chain) from Dr. Bart Chernow in New Orleans during CHEST 1997. I remember this time as being a time of beginnings, challenges, and changes. Bart had been the stimulus for the CHEST Foundation and the form and function of this foundation was being developed. The women’s caucus (probably not the official name) was becoming more organized and more of a force under the leadership of Dr. Diane Stover and Dr. Deborah Shure and others, and the Woman, Girls, Tobacco, and Lung Cancer educational program was being refined. It was this program that got my wife, Mary Anne, involved with the CHEST, and she became a Fellow (FCCP). The American College of Chest Physicians was in the midst of the national tobacco settlement efforts at this time. Our involvement began when Mike Moore, Attorney-General of Mississippi, filed the first suit against the tobacco industry in 1994. Under the stimulus of Dr. John Studdard, our current President, the college was the only medical organization to file an amicus curiae brief supporting this, thus thrusting us into the midst of the tobacco settlement debates and in a leadership position. During the time I was President-elect and President, I was fortunate to represent us both in the ENACT Coalition (composed of national health groups, such as the American Cancer Society), as well as on the Koop-Kessler Congressional Advisory Committee. I also testified before Congress on the tobacco issues and met at the White House with DHHS Secretary Donna Shalala. On a different front, our international activities were not as developed as now, but we did make two memorable trips to India. Many thanks to Dr. Kay Guntupalli for helping make those trips so memorable. After this absolutely wonderful year, I passed the chain to Dr. Allen Goldberg in Toronto.

Among other activities, I was Chief of Staff at the Oklahoma City VAMC for 18 years, retiring from that position in 2009. I was honored by having the MICU at the VA named after me. In the community, I helped start the Hospice of Oklahoma County and then the Hospice Foundation of Oklahoma, both of which I served as first chairman. I also helped start Palliative Care Week on the OUHSC campus. I am currently the vice-chair of the Health Alliance for the Uninsured in Oklahoma City, which helps support the many free clinics in our city. My wonderful wife, Mary Anne, is also involved in many community activities. On a personal level, we try to see our two children and two grandchildren as often as possible, which is not often enough. My free time activities include reading, playing the piano, fly fishing (not often enough), and exercise.
My time as President of the American College of Chest Physicians was one of the best and most important experiences of my life. My memories of working with Al Lever, David Eubanks, Marilyn Lederer, Lynne Marcus, Steve Welch, and all the other administrative and physician leaders during that time remain very dear to me. The influence of CHEST continues to this very day. I can never repay all that I have gained from this experience. I wish I had the space allowance to expand on my experiences. But while my word allowance is limited, my gratitude is unlimited.
Where are they now? What have they been up to? CHEST’s Past Presidents each forged the way for the many successes of the American College of Chest Physicians, leading to enhanced patient care around the globe. Their outstanding leadership and vision are evidenced today in many of CHEST’s strategic initiatives.
D. Robert McCaffree, MD, MSHA, Master FCCP
CHEST President 1997 - 1998
I received the chain of office (yes, there is an actual chain) from Dr. Bart Chernow in New Orleans during CHEST 1997. I remember this time as being a time of beginnings, challenges, and changes. Bart had been the stimulus for the CHEST Foundation and the form and function of this foundation was being developed. The women’s caucus (probably not the official name) was becoming more organized and more of a force under the leadership of Dr. Diane Stover and Dr. Deborah Shure and others, and the Woman, Girls, Tobacco, and Lung Cancer educational program was being refined. It was this program that got my wife, Mary Anne, involved with the CHEST, and she became a Fellow (FCCP). The American College of Chest Physicians was in the midst of the national tobacco settlement efforts at this time. Our involvement began when Mike Moore, Attorney-General of Mississippi, filed the first suit against the tobacco industry in 1994. Under the stimulus of Dr. John Studdard, our current President, the college was the only medical organization to file an amicus curiae brief supporting this, thus thrusting us into the midst of the tobacco settlement debates and in a leadership position. During the time I was President-elect and President, I was fortunate to represent us both in the ENACT Coalition (composed of national health groups, such as the American Cancer Society), as well as on the Koop-Kessler Congressional Advisory Committee. I also testified before Congress on the tobacco issues and met at the White House with DHHS Secretary Donna Shalala. On a different front, our international activities were not as developed as now, but we did make two memorable trips to India. Many thanks to Dr. Kay Guntupalli for helping make those trips so memorable. After this absolutely wonderful year, I passed the chain to Dr. Allen Goldberg in Toronto.
Among other activities, I was Chief of Staff at the Oklahoma City VAMC for 18 years, retiring from that position in 2009. I was honored by having the MICU at the VA named after me. In the community, I helped start the Hospice of Oklahoma County and then the Hospice Foundation of Oklahoma, both of which I served as first chairman. I also helped start Palliative Care Week on the OUHSC campus. I am currently the vice-chair of the Health Alliance for the Uninsured in Oklahoma City, which helps support the many free clinics in our city. My wonderful wife, Mary Anne, is also involved in many community activities. On a personal level, we try to see our two children and two grandchildren as often as possible, which is not often enough. My free time activities include reading, playing the piano, fly fishing (not often enough), and exercise.
My time as President of the American College of Chest Physicians was one of the best and most important experiences of my life. My memories of working with Al Lever, David Eubanks, Marilyn Lederer, Lynne Marcus, Steve Welch, and all the other administrative and physician leaders during that time remain very dear to me. The influence of CHEST continues to this very day. I can never repay all that I have gained from this experience. I wish I had the space allowance to expand on my experiences. But while my word allowance is limited, my gratitude is unlimited.
Where are they now? What have they been up to? CHEST’s Past Presidents each forged the way for the many successes of the American College of Chest Physicians, leading to enhanced patient care around the globe. Their outstanding leadership and vision are evidenced today in many of CHEST’s strategic initiatives.
D. Robert McCaffree, MD, MSHA, Master FCCP
CHEST President 1997 - 1998
I received the chain of office (yes, there is an actual chain) from Dr. Bart Chernow in New Orleans during CHEST 1997. I remember this time as being a time of beginnings, challenges, and changes. Bart had been the stimulus for the CHEST Foundation and the form and function of this foundation was being developed. The women’s caucus (probably not the official name) was becoming more organized and more of a force under the leadership of Dr. Diane Stover and Dr. Deborah Shure and others, and the Woman, Girls, Tobacco, and Lung Cancer educational program was being refined. It was this program that got my wife, Mary Anne, involved with the CHEST, and she became a Fellow (FCCP). The American College of Chest Physicians was in the midst of the national tobacco settlement efforts at this time. Our involvement began when Mike Moore, Attorney-General of Mississippi, filed the first suit against the tobacco industry in 1994. Under the stimulus of Dr. John Studdard, our current President, the college was the only medical organization to file an amicus curiae brief supporting this, thus thrusting us into the midst of the tobacco settlement debates and in a leadership position. During the time I was President-elect and President, I was fortunate to represent us both in the ENACT Coalition (composed of national health groups, such as the American Cancer Society), as well as on the Koop-Kessler Congressional Advisory Committee. I also testified before Congress on the tobacco issues and met at the White House with DHHS Secretary Donna Shalala. On a different front, our international activities were not as developed as now, but we did make two memorable trips to India. Many thanks to Dr. Kay Guntupalli for helping make those trips so memorable. After this absolutely wonderful year, I passed the chain to Dr. Allen Goldberg in Toronto.
Among other activities, I was Chief of Staff at the Oklahoma City VAMC for 18 years, retiring from that position in 2009. I was honored by having the MICU at the VA named after me. In the community, I helped start the Hospice of Oklahoma County and then the Hospice Foundation of Oklahoma, both of which I served as first chairman. I also helped start Palliative Care Week on the OUHSC campus. I am currently the vice-chair of the Health Alliance for the Uninsured in Oklahoma City, which helps support the many free clinics in our city. My wonderful wife, Mary Anne, is also involved in many community activities. On a personal level, we try to see our two children and two grandchildren as often as possible, which is not often enough. My free time activities include reading, playing the piano, fly fishing (not often enough), and exercise.
My time as President of the American College of Chest Physicians was one of the best and most important experiences of my life. My memories of working with Al Lever, David Eubanks, Marilyn Lederer, Lynne Marcus, Steve Welch, and all the other administrative and physician leaders during that time remain very dear to me. The influence of CHEST continues to this very day. I can never repay all that I have gained from this experience. I wish I had the space allowance to expand on my experiences. But while my word allowance is limited, my gratitude is unlimited.
Is Posthospital Syndrome a Result of Hospitalization-Induced Allostatic Overload?
After discharge from the hospital, patients have a significantly elevated risk for adverse events, including emergency department use, hospital readmission, and death. More than 1 in 3 patients discharged from the hospital require acute care in the month after hospital discharge, and more than 1 in 6 require readmission, with readmission diagnoses frequently differing from those of the preceding hospitalization.1-4 This heightened susceptibility to adverse events persists beyond 30 days but levels off by 7 weeks after discharge, suggesting that the period of increased risk is transient and dynamic.5
The term posthospital syndrome (PHS) describes this period of vulnerability to major adverse events following hospitalization.6 In addition to increased risk for readmission and mortality, patients in this period often show evidence of generalized dysfunction with new cognitive impairment, mobility disability, or functional decline.7-12 To date, the etiology of this vulnerability is neither well understood nor effectively addressed by transitional care interventions.13
One hypothesis to explain PHS is that stressors associated with the experience of hospitalization contribute to transient multisystem dysfunction that induces susceptibility to a broad range of medical maladies. These stressors include frequent sleep disruption, noxious sounds, painful stimuli, mobility restrictions, and poor nutrition.12 The stress hypothesis as a cause of PHS is therefore based, in large part, on evidence about allostasis and the deleterious effects of allostatic overload.
Allostasis defines a system functioning within normal stress-response parameters to promote adaptation and survival.14 In allostasis, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic and parasympathetic branches of the autonomic nervous system (ANS) exist in homeostatic balance and respond to environmental stimuli within a range of healthy physiologic parameters. The hallmark of a system in allostasis is the ability to rapidly activate, then successfully deactivate, a stress response once the stressor (ie, threat) has resolved.14,15 To promote survival and potentiate “fight or flight” mechanisms, an appropriate stress response necessarily impacts multiple physiologic systems that result in hemodynamic augmentation and gluconeogenesis to support the anticipated action of large muscle groups, heightened vigilance and memory capabilities to improve rapid decision-making, and enhancement of innate and adaptive immune capabilities to prepare for wound repair and infection defense.14-16 The stress response is subsequently terminated by negative feedback mechanisms of glucocorticoids as well as a shift of the ANS from sympathetic to parasympathetic tone.17,18
Extended or repetitive stress exposure, however, leads to dysregulation of allostatic mechanisms responsible for stress adaptation and hinders an efficient and effective stress response. After extended stress exposure, baseline (ie, resting) HPA activity resets, causing a disruption of normal diurnal cortisol rhythm and an increase in total cortisol concentration. Moreover, in response to stress, HPA and ANS system excitation becomes impaired, and negative feedback properties are undermined.14,15 This maladaptive state, known as allostatic overload, disrupts the finely tuned mechanisms that are the foundation of mind-body balance and yields pathophysiologic consequences to multiple organ systems. Downstream ramifications of allostatic overload include cognitive deterioration, cardiovascular and immune system dysfunction, and functional decline.14,15,19
Although a stress response is an expected and necessary aspect of acute illness that promotes survival, the central thesis of this work is that additional environmental and social stressors inherent in hospitalization may unnecessarily compound stress and increase the risk of HPA axis dysfunction, allostatic overload, and subsequent multisystem dysfunction, predisposing individuals to adverse outcomes after hospital discharge. Based on data from both human subjects and animal models, we present a possible pathophysiologic mechanism for the postdischarge vulnerability of PHS, encourage critical contemplation of traditional hospitalization, and suggest interventions that might improve outcomes.
POSTHOSPITAL SYNDROME
Posthospital syndrome (PHS) describes a transient period of vulnerability after hospitalization during which patients are at elevated risk for adverse events from a broad range of conditions. In support of this characterization, epidemiologic data have demonstrated high rates of adverse outcomes following hospitalization. For example, data have shown that more than 1 in 6 older adults is readmitted to the hospital within 30 days of discharge.20 Death is also common in this first month, during which rates of postdischarge mortality may exceed initial inpatient mortality.21,22 Elevated vulnerability after hospitalization is not restricted to older adults, as readmission risk among younger patients 18 to 64 years of age may be even higher for selected conditions, such as heart failure.3,23
Vulnerability after hospitalization is broad. In patients over age 65 initially admitted for heart failure or acute myocardial infarction, only 35% and 10% of readmissions are for recurrent heart failure or reinfarction, respectively.1 Nearly half of readmissions are for noncardiovascular causes.1 Similarly, following hospitalization for pneumonia, more than 60 percent of readmissions are for nonpulmonary etiologies. Moreover, the risk for all these causes of readmission is much higher than baseline risk, indicating an extended period of lack of resilience to many types of illness.24 These patterns of broad susceptibility also extend to younger adults hospitalized with common medical conditions.3
Accumulating evidence suggests that hospitalized patients face functional decline, debility, and risk for adverse events despite resolution of the presenting illness, implying perhaps that the hospital environment itself is hazardous to patients’ health. In 1993, Creditor hypothesized that the “hazards of hospitalization,” including enforced bed-rest, sensory deprivation, social isolation, and malnutrition lead to a “cascade of dependency” in which a collection of small insults to multiple organ systems precipitates loss of function and debility despite cure or resolution of presenting illness.12 Covinsky (2011) later defined hospitalization-associated disability as an iatrogenic hospital-related “disorder” characterized by new impairments in abilities to perform basic activities of daily living such as bathing, feeding, toileting, dressing, transferring, and walking at the time of hospital discharge.11 Others have described a postintensive-care syndrome (PICS),25 characterized by cognitive, psychiatric, and physical impairments acquired during hospitalization for critical illness that persist postdischarge and increase the long-term risk for adverse outcomes, including elevated mortality rates,26,27 readmission rates,28 and physical disabilities.29 Similar to the “hazards of hospitalization,” PICS is thought to be related to common experiences of ICU stays, including mobility restriction, sensory deprivation, sleep disruption, sedation, malnutrition, and polypharmacy.30-33
Taken together, these data suggest that adverse health consequences attributable to hospitalization extend across the spectrum of age, presenting disease severity, and hospital treatment location. As detailed below, the PHS hypothesis is rooted in a mechanistic understanding of the role of exogenous stressors in producing physiologic dysregulation and subsequent adverse health effects across multiple organ systems.
Nature of Stress in the Hospital
Compounding the stress of acute illness, hospitalized patients are routinely and repetitively exposed to a wide variety of environmental stressors that may have downstream adverse consequences (Table 1). In the absence of overt clinical manifestations of harm, the possible subclinical physiologic dysfunction generated by the following stress exposures may increase patients’ susceptibility to the manifestations of PHS.
Sleep Disruption
Sleep disruptions trigger potent stress responses,34,35 yet they are common occurrences during hospitalization. In surveys, about half of patients report poor sleep quality during hospitalization that persists for many months after discharge.36 In a simulated hospital setting, test subjects exposed to typical hospital sounds (paging system, machine alarms, etc.) experienced significant sleep-wake cycle abnormalities.37 Although no work has yet focused specifically on the physiologic consequences of sleep disruption and stress in hospitalized patients, in healthy humans, mild sleep disruption has clear effects on allostasis by disrupting HPA activity, raising cortisol levels, diminishing parasympathetic tone, and impairing cognitive performance.18,34,35,38,39
Malnourishment
Malnourishment in hospitalized patients is common, with one-fifth of hospitalized patients receiving nothing per mouth or clear liquid diets for more than 3 continuous days,40 and one-fifth of hospitalized elderly patients receiving less than half of their calculated nutrition requirements.41 Although the relationship between food restriction, cortisol levels, and postdischarge outcomes has not been fully explored, in healthy humans, meal anticipation, meal withdrawal (withholding an expected meal), and self-reported dietary restraint are known to generate stress responses.42,43 Furthermore, malnourishment during hospitalization is associated with increased 90-day and 1-year mortality after discharge,44 adding malnourishment to the list of plausible components of hospital-related stress.
Mobility Restriction
Physical activity counterbalances stress responses and minimizes downstream consequences of allostatic load,15 yet mobility limitations via physical and chemical restraints are common in hospitalized patients, particularly among the elderly.45-47 Many patients are tethered to devices that make ambulation hazardous, such as urinary catheters and infusion pumps. Even without physical or chemical restraints or a limited mobility order, patients may be hesitant to leave the room so as not to miss transport to a diagnostic study or an unscheduled physician’s visit. Indeed, mobility limitations of hospitalized patients increase the risk for adverse events after discharge, while interventions designed to encourage mobility are associated with improved postdischarge outcomes.47,48
Other Stressors
Other hospital-related aversive stimuli are less commonly quantified, but clearly exist. According to surveys of hospitalized patients, sources of emotional stress include social isolation; loss of autonomy and privacy; fear of serious illness; lack of control over activities of daily living; lack of clear communication between treatment team and patients; and death of a patient roommate.49,50 Furthermore, consider the physical discomfort and emotional distress of patients with urinary incontinence awaiting assistance for a diaper or bedding change or the pain of repetitive blood draws or other invasive testing. Although individualized, the subjective discomfort and emotional distress associated with these experiences undoubtedly contribute to the stress of hospitalization.
IMPACT OF ALLOSTATIC OVERLOAD ON PHYSIOLOGIC FUNCTION
Animal Models of Stress
Laboratory techniques reminiscent of the numerous environmental stressors associated with hospitalization have been used to reliably trigger allostatic overload in healthy young animals.51 These techniques include sequential exposure to aversive stimuli, including food and water deprivation, continuous overnight illumination, paired housing with known and unknown cagemates, mobility restriction, soiled cage conditions, and continuous noise. All of these techniques have been shown to cause HPA axis and ANS dysfunction, allostatic overload, and subsequent stress-mediated consequences to multiple organ systems.19,52-54 Given the remarkable similarity of these protocols to common experiences during hospitalization, animal models of stress may be useful in understanding the spectrum of maladaptive consequences experienced by patients within the hospital (Figure 1).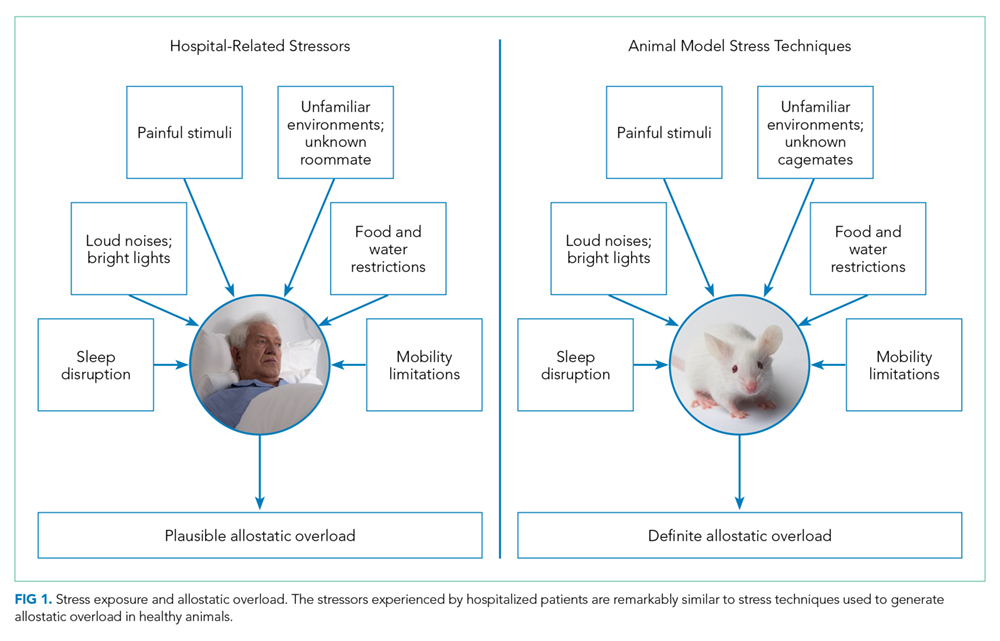
These animal models of stress have resulted in a number of instructive findings. For example, in rodents, extended stress exposure induces structural and functional remodeling of neuronal networks that precipitate learning and memory, working memory, and attention impairments.55-57 These exposures also result in cardiovascular abnormalities, including dyslipidemia, progressive atherosclerosis,58,59 and enhanced inflammatory cytokine expression,60 all of which increase both atherosclerotic burden and susceptibility to plaque rupture, leading to elevated risk for major cardiovascular adverse events. Moreover, these extended stress exposures in animals increase susceptibility to both bacterial and viral infections and increase their severity.16,61 This outcome appears to be driven by a stress-induced elevation of glucocorticoid levels, decreased leukocyte proliferation, altered leukocyte trafficking, and a transition to a proinflammatory cytokine environment.16, 61 Allostatic overload has also been shown to contribute to metabolic dysregulation involving insulin resistance, persistence of hyperglycemia, dyslipidemia, catabolism of lean muscle, and visceral adipose tissue deposition.62-64 In addition to cardiovascular, immune, and metabolic consequences of allostatic overload, the spectrum of physiologic dysfunction in animal models is broad and includes mood disorder symptoms,65 intestinal barrier abnormalities,66 airway reactivity exacerbation,67 and enhanced tumor growth.68
Although the majority of this research highlights the multisystem effects of variable stress exposure in healthy animals, preliminary evidence suggests that aged or diseased animals subjected to additional stressors display a heightened inflammatory cytokine response that contributes to exaggerated sickness behavior and greater and prolonged cognitive deficits.69 Future studies exploring the consequences of extended stress exposure in animals with existing disease or debility may therefore more closely simulate the experience of hospitalized patients and perhaps further our understanding of PHS.
Hospitalized Patients
While no intervention studies have examined the effects of potential hospital stressors on the development of allostatic overload, there is evidence from small studies that dysregulated stress responses during hospitalization are associated with adverse events. For example, high serum cortisol, catecholamine, and proinflammatory cytokine levels during hospitalization have individually been associated with the development of cognitive dysfunction,70-72 increased risk of cardiovascular events such as myocardial infarction and stroke in the year following discharge,73-76 and the development of wound infections after discharge.77 Moreover, elevated plasma glucose during admission for myocardial infarction in patients with or without diabetes has been associated with greater in-hospital and 1-year mortality,78 with a similar relationship seen between elevated plasma glucose and survival after admission for stroke79 and pneumonia.80 Furthermore, in addition to atherothrombosis, stress may contribute to the risk for venous thromboembolism,81 resulting in readmissions for deep vein thrombosis or pulmonary embolism posthospitalization. Although potentially surrogate markers of illness acuity, a handful of studies have shown that these stress biomarkers are actually only weakly correlated with,82 or independent of,72,76 disease severity. As discussed in detail below, future studies utilizing a summative measure of multisystem physiologic dysfunction as opposed to individual biomarkers may more accurately reflect the cumulative stress effects of hospitalization and subsequent risk for adverse events.
Additional Considerations
Elderly patients, in particular, may have heightened susceptibility to the consequences of allostatic overload due to common geriatric issues such as multimorbidity and frailty. Patients with chronic diseases display both baseline HPA axis abnormalities as well as dysregulated stress responses and may therefore be more vulnerable to hospitalization-related stress. For example, when subjected to psychosocial stress, patients with chronic conditions such as diabetes, heart failure, or atherosclerosis demonstrate elevated cortisol levels, increased circulating markers of inflammation, as well as prolonged hemodynamic recovery after stress resolution compared with normal controls.83-85 Additionally, frailty may affect an individual’s susceptibility to exogenous stress. Indeed, frailty identified on hospital admission increases the risk for adverse outcomes during hospitalization and postdischarge.86 Although the specific etiology of this relationship is unclear, persons with frailty are known to have elevated levels of cortisol and other inflammatory markers,87,88 which may contribute to adverse outcomes in the face of additional stressors.
IMPLICATIONS AND NEXT STEPS
A large body of evidence stretching from bench to bedside suggests that environmental stressors associated with hospitalization are toxic. Understanding PHS within the context of hospital-induced allostatic overload presents a unifying theory for the interrelated multisystem dysfunction and increased susceptibility to adverse events that patients experience after discharge (Figure 2). Furthermore, it defines a potential pathophysiological mechanism for the cognitive impairment, elevated cardiovascular risk, immune system dysfunction, metabolic derangements, and functional decline associated with PHS. Additionally, this theory highlights environmental interventions to limit PHS development and suggests mechanisms to promote stress resilience. Although it is difficult to disentangle the consequences of the endogenous stress triggered by an acute illness from the exogenous stressors related to hospitalization, it is likely that the 2 simultaneous exposures compound risk for stress system dysregulation and allostatic overload. Moreover, hospitalized patients with preexisting HPA axis dysfunction at baseline from chronic disease or advancing age may be even more susceptible to these adverse outcomes. If this hypothesis is true, a reduction in PHS would require mitigation of the modifiable environmental stressors encountered by patients during hospitalization. Directed efforts to diminish ambient noise, limit nighttime disruptions, thoughtfully plan procedures, consider ongoing nutritional status, and promote opportunities for patients to exert some control over their environment may diminish the burden of extrinsic stressors encountered by all patients in the hospital and improve outcomes after discharge.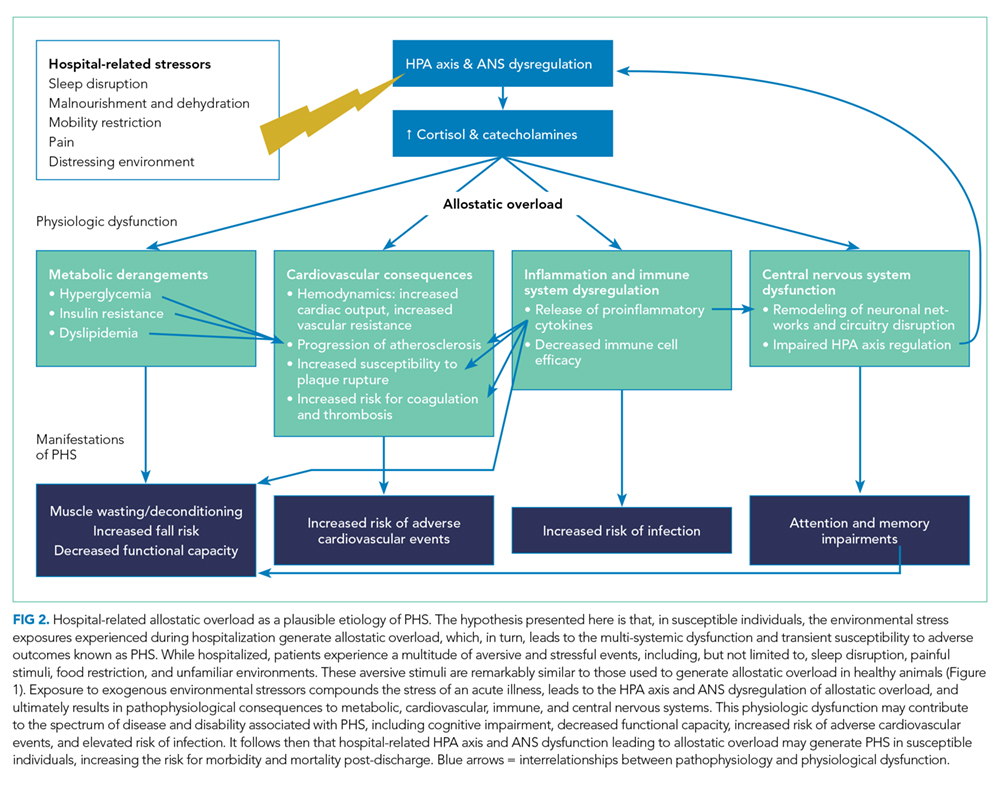
Hospitals are increasingly recognizing the importance of improving patients’ experience of hospitalization by reducing exposure to potential toxicities. For example, many hospitals are now attempting to reduce sleep disturbances and sleep latency through reduced nighttime noise and light levels, fewer nighttime interruptions for vital signs checks and medication administration, and commonsensical interventions like massages, herbal teas, and warm milk prior to bedtime.89 Likewise, intensive care units are targeting environmental and physical stressors with a multifaceted approach to decrease sedative use, promote healthy sleep cycles, and encourage exercise and ambulation even in those patients who are mechanically ventilated.30 Another promising development has been the increase of Hospital at Home programs. In these programs, patients who meet the criteria for inpatient admission are instead comprehensively managed at home for their acute illness through a multidisciplinary effort between physicians, nurses, social workers, physical therapists, and others. Patients hospitalized at home report higher levels of satisfaction and have modest functional gains, improved health-related quality of life, and decreased risk of mortality at 6 months compared with hospitalized patients.90,91 With some admitting diagnoses (eg, heart failure), hospitalization at home may be associated with decreased readmission risk.92 Although not yet investigated on a physiologic level, perhaps the benefits of hospital at home are partially due to the dramatic difference in exposure to environmental stressors.
A tool that quantifies hospital-associated stress may help health providers appreciate the experience of patients and better target interventions to aspects of their structure and process that contribute to allostatic overload. Importantly, allostatic overload cannot be identified by one biomarker of stress but instead requires evidence of dysregulation across inflammatory, neuroendocrine, hormonal, and cardiometabolic systems. Future studies to address the burden of stress faced by hospitalized patients should consider a summative measure of multisystem dysregulation as opposed to isolated assessments of individual biomarkers. Allostatic load has previously been operationalized as the summation of a variety of hemodynamic, hormonal, and metabolic factors, including blood pressure, lipid profile, glycosylated hemoglobin, cortisol, catecholamine levels, and inflammatory markers.93 To develop a hospital-associated allostatic load index, models should ideally be adjusted for acute illness severity, patient-reported stress, and capacity for stress resilience. This tool could then be used to quantify hospitalization-related allostatic load and identify those at greatest risk for adverse events after discharge, as well as measure the effectiveness of strategic environmental interventions (Table 2). A natural first experiment may be a comparison of the allostatic load of hospitalized patients versus those hospitalized at home.
The risk of adverse outcomes after discharge is likely a function of the vulnerability of the patient and the degree to which the patient’s healthcare team and social support network mitigates this vulnerability. That is, there is a risk that a person struggles in the postdischarge period and, in many circumstances, a strong healthcare team and social network can identify health problems early and prevent them from progressing to the point that they require hospitalization.13,94-96 There are also hospital occurrences, outside of allostatic load, that can lead to complications that lengthen the stay, weaken the patient, and directly contribute to subsequent vulnerability.94,97 Our contention is that the allostatic load of hospitalization, which may also vary by patient depending on the circumstances of hospitalization, is just one contributor, albeit potentially an important one, to vulnerability to medical problems after discharge.
In conclusion, a plausible etiology of PHS is the maladaptive mind-body consequences of common stressors during hospitalization that compound the stress of acute illness and produce allostatic overload. This stress-induced dysfunction potentially contributes to a spectrum of generalized disease susceptibility and risk of adverse outcomes after discharge. Focused efforts to diminish patient exposure to hospital-related stressors during and after hospitalization might diminish the presence or severity of PHS. Viewing PHS from this perspective enables the development of hypothesis-driven risk-prediction models, encourages critical contemplation of traditional hospitalization, and suggests that targeted environmental interventions may significantly reduce adverse outcomes.
1. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. http://dx.doi.org/10.1001/jama.2012.216476.
2. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. http://dx.doi.org/10.1056/NEJMsa0803563.
3. Ranasinghe I, Wang Y, Dharmarajan K, Hsieh AF, Bernheim SM, Krumholz HM. Readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia among young and middle-aged adults: a retrospective observational cohort study. PLoS Med. 2014;11(9):e1001737. http://dx.doi.org/10.1371/journal.pmed.1001737.
4. Vashi AA, Fox JP, Carr BG, et al. Use of hospital-based acute care among patients recently discharged from the hospital. JAMA. 2013;309(4):364-371. http://dx.doi.org/10.1001/jama.2012.216219.
5. Dharmarajan K, Hsieh AF, Kulkarni VT, et al. Trajectories of risk after hospitalization for heart failure, acute myocardial infarction, or pneumonia: retrospective cohort study. BMJ. 2015;350:h411. http://dx.doi.org/10.1136/bmj.h411.
6. Krumholz HM. Post-hospital syndrome--an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. http://dx.doi.org/10.1056/NEJMp1212324.
7. Ferrante LE, Pisani MA, Murphy TE, Gahbauer EA, Leo-Summers LS, Gill TM. Functional trajectories among older persons before and after critical illness. JAMA Intern Med. 2015;175(4):523-529. http://dx.doi.org/10.1001/jamainternmed.2014.7889.
8. Gill TM, Allore HG, Holford TR, Guo Z. Hospitalization, restricted activity, and the development of disability among older persons. JAMA. 2004;292(17):2115-2124. http://dx.doi.org/10.1001/jama.292.17.2115.
9. Inouye SK, Zhang Y, Han L, Leo-Summers L, Jones R, Marcantonio E. Recoverable cognitive dysfunction at hospital admission in older persons during acute illness. J Gen Intern Med. 2006;21(12):1276-1281. http://dx.doi.org/10.1111/j.1525-1497.2006.00613.x.
10. Lindquist LA, Go L, Fleisher J, Jain N, Baker D. Improvements in cognition following hospital discharge of community dwelling seniors. J Gen Intern Med. 2011;26(7):765-770. http://dx.doi.org/10.1007/s11606-011-1681-1.
11. Covinsky KE, Pierluissi E, Johnston CB. Hospitalization-associated disability: “She was probably able to ambulate, but I’m not sure.” JAMA. 2011;306(16):1782-1793. http://dx.doi.org/10.1001/jama.2011.1556.
12. Creditor MC. Hazards of hospitalization of the elderly. Ann Intern Med. 1993;118(3):219-223. http://dx.doi.org/10.7326/0003-4819-118-3-199302010-00011.
13. Dharmarajan K, Krumholz HM. Strategies to reduce 30-day readmissions in older patients hospitalized with heart failure and acute myocardial infarction. Curr Geriatr Rep. 2014;3(4):306-315. http://dx.doi.org/10.1007/s13670-014-0103-8.
14. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338(3):171-179. http://dx.doi.org/10.1056/NEJM199801153380307.
15. McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431-445. http://dx.doi.org/10.1146/annurev-med-052209-100430.
16. Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. http://dx.doi.org/10.1159/000216188.
17. Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361-372. http://dx.doi.org/10.1196/annals.1366.014.
18. Jacobson L, Akana SF, Cascio CS, Shinsako J, Dallman MF. Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology. 1988;122(4):1343-1348. http://dx.doi.org/10.1210/endo-122-4-1343.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873-904. http://dx.doi.org/10.1152/physrev.00041.2006.
20. Medicare Hospital Quality Chartbook 2014: Performance Report on Outcome Measures. Prepared by Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation for Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/quality-initiatives-patient-assessment-instruments/hospitalqualityinits/downloads/medicare-hospital-quality-chartbook-2014.pdf. Accessed February 26, 2018.
21. Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993-2006. JAMA. 2010;303(21):2141-2147. http://dx.doi.org/10.1001/jama.2010.748.
22. Drye EE, Normand SL, Wang Y, et al. Comparison of hospital risk-standardized mortality rates calculated by using in-hospital and 30-day models: an observational study with implications for hospital profiling. Ann Intern Med. 2012;156(1 Pt 1):19-26. http://dx.doi.org/10.7326/0003-4819-156-1-201201030-00004.
23. Dharmarajan K, Hsieh A, Dreyer RP, Welsh J, Qin L, Krumholz HM. Relationship between age and trajectories of rehospitalization risk in older adults. J Am Geriatr Soc. 2017;65(2):421-426. http://dx.doi.org/10.1111/jgs.14583.
24. Krumholz HM, Hsieh A, Dreyer RP, Welsh J, Desai NR, Dharmarajan K. Trajectories of risk for specific readmission diagnoses after hospitalization for heart failure, acute myocardial infarction, or pneumonia. PLoS One. 2016;11(10):e0160492. http://dx.doi.org/10.1371/journal.pone.0160492.
25. Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med. 2012;40(2):502-509. http://dx.doi.org/10.1097/CCM.0b013e318232da75.
26. Brinkman S, de Jonge E, Abu-Hanna A, Arbous MS, de Lange DW, de Keizer NF. Mortality after hospital discharge in ICU patients. Crit Care Med. 2013;41(5):1229-1236. http://dx.doi.org/10.1097/CCM.0b013e31827ca4e1.
27. Steenbergen S, Rijkenberg S, Adonis T, Kroeze G, van Stijn I, Endeman H. Long-term treated intensive care patients outcomes: the one-year mortality rate, quality of life, health care use and long-term complications as reported by general practitioners. BMC Anesthesiol. 2015;15:142. http://dx.doi.org/10.1186/s12871-015-0121-x.
28. Hill AD, Fowler RA, Pinto R, Herridge MS, Cuthbertson BH, Scales DC. Long-term outcomes and healthcare utilization following critical illness--a population-based study. Crit Care. 2016;20:76. http://dx.doi.org/10.1186/s13054-016-1248-y.
29. Jackson JC, Pandharipande PP, Girard TD, et al. Depression, post-traumatic stress disorder, and functional disability in survivors of critical illness in the BRAIN-ICU study: a longitudinal cohort study. Lancet Respir Med. 2014;2(5):369-379. http://dx.doi.org/10.1016/S2213-2600(14)70051-7.
30. Balas MC, Vasilevskis EE, Olsen KM, et al. Effectiveness and safety of the awakening and breathing coordination, delirium monitoring/management, and early exercise/mobility bundle. Crit Care Med. 2014;42(5):1024-1036. http://dx.doi.org/10.1097/CCM.0000000000000129.
31. Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;370(17):1626-1635. http://dx.doi.org/10.1056/NEJMra1209390.
32. Mendez-Tellez PA, Needham DM. Early physical rehabilitation in the ICU and ventilator liberation. Respir Care. 2012;57(10):1663-1669. http://dx.doi.org/10.4187/respcare.01931.
33. Schweickert WD, Hall J. ICU-acquired weakness. Chest. 2007;131(5):1541-1549. http://dx.doi.org/10.1378/chest.06-2065.
34. Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Int J Endocrinol. 2010;2010:759234. http://dx.doi.org/10.1155/2010/759234.
35. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354(9188):1435-1439. http://dx.doi.org/10.1016/S0140-6736(99)01376-8.
36. Orwelius L, Nordlund A, Nordlund P, Edell-Gustafsson U, Sjoberg F. Prevalence of sleep disturbances and long-term reduced health-related quality of life after critical care: a prospective multicenter cohort study. Crit Care. 2008;12(4):R97. http://dx.doi.org/10.1186/cc6973.
37. Buxton OM, Ellenbogen JM, Wang W, et al. Sleep disruption due to hospital noises: a prospective evaluation. Ann Intern Med. 2012;157(3):170-179. http://dx.doi.org/10.7326/0003-4819-157-3-201208070-00472.
38. Sunbul M, Kanar BG, Durmus E, Kivrak T, Sari I. Acute sleep deprivation is associated with increased arterial stiffness in healthy young adults. Sleep Breath. 2014;18(1):215-220. http://dx.doi.org/10.1007/s11325-013-0873-9.
39. Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26(2):117-126. http://dx.doi.org/10.1093/sleep/26.2.117.
40. Franklin GA, McClave SA, Hurt RT, et al. Physician-delivered malnutrition: why do patients receive nothing by mouth or a clear liquid diet in a university hospital setting? JPEN J Parenter Enteral Nutr. 2011;35(3):337-342. http://dx.doi.org/10.1177/0148607110374060.
41. Sullivan DH, Sun S, Walls RC. Protein-energy undernutrition among elderly hospitalized patients: a prospective study. JAMA. 1999;281(21):2013-2019. http://dx.doi.org/10.1001/jama.281.21.2013.
42. Anderson DA, Shapiro JR, Lundgren JD, Spataro LE, Frye CA. Self-reported dietary restraint is associated with elevated levels of salivary cortisol. Appetite. 2002;38(1):13-17. http://dx.doi.org/10.1006/appe.2001.0459.
43. Ott V, Friedrich M, Prilop S, et al. Food anticipation and subsequent food withdrawal increase serum cortisol in healthy men. Physiol Behav. 2011;103(5):594-599. http://dx.doi.org/10.1016/j.physbeh.2011.04.020.
44. Covinsky KE, Martin GE, Beyth RJ, Justice AC, Sehgal AR, Landefeld CS. The relationship between clinical assessments of nutritional status and adverse outcomes in older hospitalized medical patients. J Am Geriatr Soc. 1999;47(5):532-538. http://dx.doi.org/10.1111/j.1532-5415.1999.tb02566.x.
45. Lazarus BA, Murphy JB, Coletta EM, McQuade WH, Culpepper L. The provision of physical activity to hospitalized elderly patients. Arch Intern Med. 1991;151(12):2452-2456.
46. Minnick AF, Mion LC, Johnson ME, Catrambone C, Leipzig R. Prevalence and variation of physical restraint use in acute care settings in the US. J Nurs Scholarsh. 2007;39(1):30-37. http://dx.doi.org/10.1111/j.1547-5069.2007.00140.x.
47. Zisberg A, Shadmi E, Sinoff G, Gur-Yaish N, Srulovici E, Admi H. Low mobility during hospitalization and functional decline in older adults. J Am Geriatr Soc. 2011;59(2):266-273. http://dx.doi.org/10.1111/j.1532-5415.2010.03276.x.
48. Landefeld CS, Palmer RM, Kresevic DM, Fortinsky RH, Kowal J. A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338-1344. http://dx.doi.org/10.1056/NEJM199505183322006.
49. Douglas CH, Douglas MR. Patient-friendly hospital environments: exploring the patients’ perspective. Health Expectations: an international journal of public participation in health care and health policy. 2004;7(1):61-73. http://dx.doi.org/10.1046/j.1369-6513.2003.00251.x.
50. Volicer BJ. Hospital stress and patient reports of pain and physical status. Journal Human Stress. 1978;4(2):28-37. http://dx.doi.org/10.1080/0097840X.1978.9934984.
51. Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl). 1987;93(3):358-364. http://dx.doi.org/10.1007/BF00187257.
52. Grippo AJ, Francis J, Beltz TG, Felder RB, Johnson AK. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol Behav. 2005;84(5):697-706. http://dx.doi.org/10.1016/j.physbeh.2005.02.011.
53. Krishnan V, Nestler EJ. Animal models of depression: molecular perspectives. Curr Top Behav Neurosci. 2011;7:121-147. http://dx.doi.org/10.1007/7854_2010_108.
54. Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69(1):83-88. http://dx.doi.org/10.1016/0306-4522(95)00256-I.
55. McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265-277. http://dx.doi.org/10.1111/j.1749-6632.2001.tb05830.x.
56. McEwen BS. The brain on stress: toward an integrative approach to brain, body, and behavior. Perspect Psychol Sci. 2013;8(6):673-675. http://dx.doi.org/10.1177/1745691613506907.
57. McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16-29. http://dx.doi.org/10.1016/j.neuron.2013.06.028.
58. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325-329. http://dx.doi.org/10.1038/nature11260.
59. Lu XT, Liu YF, Zhang L, et al. Unpredictable chronic mild stress promotes atherosclerosis in high cholesterol-fed rabbits. Psychosom Med. 2012;74(6):604-611. http://dx.doi.org/10.1097/PSY.0b013e31825d0b71.
60. Heidt T, Sager HB, Courties G, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20(7):754-758. http://dx.doi.org/10.1038/nm.3589.
61. Sheridan JF, Feng NG, Bonneau RH, Allen CM, Huneycutt BS, Glaser R. Restraint stress differentially affects anti-viral cellular and humoral immune responses in mice. J Neuroimmunol. 1991;31(3):245-255. http://dx.doi.org/10.1016/0165-5728(91)90046-A.
62. Kyrou I, Tsigos C. Stress hormones: physiological stress and regulation of metabolism. Curr Opin Pharmacol. 2009;9(6):787-793. http://dx.doi.org/10.1016/j.coph.2009.08.007.
63. Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30(1):1-10. http://dx.doi.org/10.1016/j.psyneuen.2004.05.007.
64. Tamashiro KL, Sakai RR, Shively CA, Karatsoreos IN, Reagan LP. Chronic stress, metabolism, and metabolic syndrome. Stress. 2011;14(5):468-474. http://dx.doi.org/10.3109/10253890.2011.606341.
65. McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54(3):200-207. http://dx.doi.org/10.1016/S0006-3223(03)00177-X.
66. Zareie M, Johnson-Henry K, Jury J, et al. Probiotics prevent bacterial translocation and improve intestinal barrier function in rats following chronic psychological stress. Gut. 2006;55(11):1553-1560. http://dx.doi.org/10.1136/gut.2005.080739.
67. Joachim RA, Quarcoo D, Arck PC, Herz U, Renz H, Klapp BF. Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom Med. 2003;65(5):811-815. http://dx.doi.org/10.1097/01.PSY.0000088582.50468.A3.
68. Thaker PH, Han LY, Kamat AA, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939-944. http://dx.doi.org/10.1038/nm1447.
69. Schreuder L, Eggen BJ, Biber K, Schoemaker RG, Laman JD, de Rooij SE. Pathophysiological and behavioral effects of systemic inflammation in aged and diseased rodents with relevance to delirium: A systematic review. Brain Behav Immun. 2017;62:362-381. http://dx.doi.org/10.1016/j.bbi.2017.01.010.
70. Mu DL, Li LH, Wang DX, et al. High postoperative serum cortisol level is associated with increased risk of cognitive dysfunction early after coronary artery bypass graft surgery: a prospective cohort study. PLoS One. 2013;8(10):e77637. http://dx.doi.org/10.1371/journal.pone.0077637.
71. Mu DL, Wang DX, Li LH, et al. High serum cortisol level is associated with increased risk of delirium after coronary artery bypass graft surgery: a prospective cohort study. Crit Care. 2010;14(6):R238. http://dx.doi.org/10.1186/cc9393.
72. Nguyen DN, Huyghens L, Zhang H, Schiettecatte J, Smitz J, Vincent JL. Cortisol is an associated-risk factor of brain dysfunction in patients with severe sepsis and septic shock. Biomed Res Int. 2014;2014:712742. http://dx.doi.org/10.1155/2014/712742.
73. Elkind MS, Carty CL, O’Meara ES, et al. Hospitalization for infection and risk of acute ischemic stroke: the Cardiovascular Health Study. Stroke. 2011;42(7):1851-1856. http://dx.doi.org/10.1161/STROKEAHA.110.608588.
74. Feibel JH, Hardy PM, Campbell RG, Goldstein MN, Joynt RJ. Prognostic value of the stress response following stroke. JAMA. 1977;238(13):1374-1376.
75. Jutla SK, Yuyun MF, Quinn PA, Ng LL. Plasma cortisol and prognosis of patients with acute myocardial infarction. J Cardiovasc Med (Hagerstown). 2014;15(1):33-41. http://dx.doi.org/10.2459/JCM.0b013e328364100b.
76. Yende S, D’Angelo G, Kellum JA, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med. 2008;177(11):1242-1247. http://dx.doi.org/10.1164/rccm.200712-1777OC.
77. Gouin JP, Kiecolt-Glaser JK. The impact of psychological stress on wound healing: methods and mechanisms. Immunol Allergy Clin North Am. 2011;31(1):81-93. http://dx.doi.org/10.1016/j.iac.2010.09.010.
78. Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355(9206):773-778. http://dx.doi.org/10.1016/S0140-6736(99)08415-9.
79. O’Neill PA, Davies I, Fullerton KJ, Bennett D. Stress hormone and blood glucose response following acute stroke in the elderly. Stroke. 1991;22(7):842-847. http://dx.doi.org/10.1161/01.STR.22.7.842.
80. Waterer GW, Kessler LA, Wunderink RG. Medium-term survival after hospitalization with community-acquired pneumonia. Am J Respir Crit Care Med. 2004;169(8):910-914. http://dx.doi.org/10.1164/rccm.200310-1448OC.
81. Rosengren A, Freden M, Hansson PO, Wilhelmsen L, Wedel H, Eriksson H. Psychosocial factors and venous thromboembolism: a long-term follow-up study of Swedish men. J Thrombosis Haemostasis. 2008;6(4):558-564. http://dx.doi.org/10.1111/j.1538-7836.2007.02857.x.
82. Oswald GA, Smith CC, Betteridge DJ, Yudkin JS. Determinants and importance of stress hyperglycaemia in non-diabetic patients with myocardial infarction. BMJ. 1986;293(6552):917-922. http://dx.doi.org/10.1136/bmj.293.6552.917.
83. Middlekauff HR, Nguyen AH, Negrao CE, et al. Impact of acute mental stress on sympathetic nerve activity and regional blood flow in advanced heart failure: implications for ‘triggering’ adverse cardiac events. Circulation. 1997;96(6):1835-1842. http://dx.doi.org/10.1161/01.CIR.96.6.1835.
84. Nijm J, Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41(3):224-233. http://dx.doi.org/10.1080/07853890802508934.
85. Steptoe A, Hackett RA, Lazzarino AI, et al. Disruption of multisystem responses to stress in type 2 diabetes: investigating the dynamics of allostatic load. Proc Natl Acad Sci U S A. 2014;111(44):15693-15698. http://dx.doi.org/10.1073/pnas.1410401111.
86. Sepehri A, Beggs T, Hassan A, et al. The impact of frailty on outcomes after cardiac surgery: a systematic review. J Thorac Cardiovasc Surg. 2014;148(6):3110-3117. http://dx.doi.org/10.1016/j.jtcvs.2014.07.087.
87. Johar H, Emeny RT, Bidlingmaier M, et al. Blunted diurnal cortisol pattern is associated with frailty: a cross-sectional study of 745 participants aged 65 to 90 years. J Clin Endocrinol Metab. 2014;99(3):E464-468. http://dx.doi.org/10.1210/jc.2013-3079.
88. Yao X, Li H, Leng SX. Inflammation and immune system alterations in frailty. Clin Geriatr Med. 2011;27(1):79-87. http://dx.doi.org/10.1016/j.cger.2010.08.002.
89. Hospital Elder Life Program (HELP) for Prevention of Delirium. 2017; http://www.hospitalelderlifeprogram.org/. Accessed February 16, 2018.
90. Shepperd S, Doll H, Angus RM, et al. Admission avoidance hospital at home. Cochrane Database of System Rev. 2008;(4):CD007491. http://dx.doi.org/10.1002/14651858.CD007491.pub2’
91. Leff B, Burton L, Mader SL, et al. Comparison of functional outcomes associated with hospital at home care and traditional acute hospital care. J Am Geriatrics Soc. 2009;57(2):273-278. http://dx.doi.org/10.1111/j.1532-5415.2008.02103.x.
92. Qaddoura A, Yazdan-Ashoori P, Kabali C, et al. Efficacy of hospital at home in patients with heart failure: a systematic review and meta-analysis. PloS One. 2015;10(6):e0129282. http://dx.doi.org/10.1371/journal.pone.0129282.
93. Seeman T, Gruenewald T, Karlamangla A, et al. Modeling multisystem biological risk in young adults: The Coronary Artery Risk Development in Young Adults Study. Am J Hum Biol. 2010;22(4):463-472. http://dx.doi.org/10.1002/ajhb.21018.
94. Auerbach AD, Kripalani S, Vasilevskis EE, et al. Preventability and causes of readmissions in a national cohort of general medicine patients. JAMA Intern Med. 2016;176(4):484-493. http://dx.doi.org/10.1001/jamainternmed.2015.7863.
95. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. http://dx.doi.org/10.7326/0003-4819-155-8-201110180-00008.
96. Takahashi PY, Naessens JM, Peterson SM, et al. Short-term and long-term effectiveness of a post-hospital care transitions program in an older, medically complex population. Healthcare. 2016;4(1):30-35. http://dx.doi.org/10.1016/j.hjdsi.2015.06.006.
 97. Dharmarajan K, Swami S, Gou RY, Jones RN, Inouye SK. Pathway from delirium to death: potential in-hospital mediators of excess mortality. J Am Geriatr Soc. 2017;65(5):1026-1033. http://dx.doi.org/10.1111/jgs.14743.
97. Dharmarajan K, Swami S, Gou RY, Jones RN, Inouye SK. Pathway from delirium to death: potential in-hospital mediators of excess mortality. J Am Geriatr Soc. 2017;65(5):1026-1033. http://dx.doi.org/10.1111/jgs.14743.
After discharge from the hospital, patients have a significantly elevated risk for adverse events, including emergency department use, hospital readmission, and death. More than 1 in 3 patients discharged from the hospital require acute care in the month after hospital discharge, and more than 1 in 6 require readmission, with readmission diagnoses frequently differing from those of the preceding hospitalization.1-4 This heightened susceptibility to adverse events persists beyond 30 days but levels off by 7 weeks after discharge, suggesting that the period of increased risk is transient and dynamic.5
The term posthospital syndrome (PHS) describes this period of vulnerability to major adverse events following hospitalization.6 In addition to increased risk for readmission and mortality, patients in this period often show evidence of generalized dysfunction with new cognitive impairment, mobility disability, or functional decline.7-12 To date, the etiology of this vulnerability is neither well understood nor effectively addressed by transitional care interventions.13
One hypothesis to explain PHS is that stressors associated with the experience of hospitalization contribute to transient multisystem dysfunction that induces susceptibility to a broad range of medical maladies. These stressors include frequent sleep disruption, noxious sounds, painful stimuli, mobility restrictions, and poor nutrition.12 The stress hypothesis as a cause of PHS is therefore based, in large part, on evidence about allostasis and the deleterious effects of allostatic overload.
Allostasis defines a system functioning within normal stress-response parameters to promote adaptation and survival.14 In allostasis, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic and parasympathetic branches of the autonomic nervous system (ANS) exist in homeostatic balance and respond to environmental stimuli within a range of healthy physiologic parameters. The hallmark of a system in allostasis is the ability to rapidly activate, then successfully deactivate, a stress response once the stressor (ie, threat) has resolved.14,15 To promote survival and potentiate “fight or flight” mechanisms, an appropriate stress response necessarily impacts multiple physiologic systems that result in hemodynamic augmentation and gluconeogenesis to support the anticipated action of large muscle groups, heightened vigilance and memory capabilities to improve rapid decision-making, and enhancement of innate and adaptive immune capabilities to prepare for wound repair and infection defense.14-16 The stress response is subsequently terminated by negative feedback mechanisms of glucocorticoids as well as a shift of the ANS from sympathetic to parasympathetic tone.17,18
Extended or repetitive stress exposure, however, leads to dysregulation of allostatic mechanisms responsible for stress adaptation and hinders an efficient and effective stress response. After extended stress exposure, baseline (ie, resting) HPA activity resets, causing a disruption of normal diurnal cortisol rhythm and an increase in total cortisol concentration. Moreover, in response to stress, HPA and ANS system excitation becomes impaired, and negative feedback properties are undermined.14,15 This maladaptive state, known as allostatic overload, disrupts the finely tuned mechanisms that are the foundation of mind-body balance and yields pathophysiologic consequences to multiple organ systems. Downstream ramifications of allostatic overload include cognitive deterioration, cardiovascular and immune system dysfunction, and functional decline.14,15,19
Although a stress response is an expected and necessary aspect of acute illness that promotes survival, the central thesis of this work is that additional environmental and social stressors inherent in hospitalization may unnecessarily compound stress and increase the risk of HPA axis dysfunction, allostatic overload, and subsequent multisystem dysfunction, predisposing individuals to adverse outcomes after hospital discharge. Based on data from both human subjects and animal models, we present a possible pathophysiologic mechanism for the postdischarge vulnerability of PHS, encourage critical contemplation of traditional hospitalization, and suggest interventions that might improve outcomes.
POSTHOSPITAL SYNDROME
Posthospital syndrome (PHS) describes a transient period of vulnerability after hospitalization during which patients are at elevated risk for adverse events from a broad range of conditions. In support of this characterization, epidemiologic data have demonstrated high rates of adverse outcomes following hospitalization. For example, data have shown that more than 1 in 6 older adults is readmitted to the hospital within 30 days of discharge.20 Death is also common in this first month, during which rates of postdischarge mortality may exceed initial inpatient mortality.21,22 Elevated vulnerability after hospitalization is not restricted to older adults, as readmission risk among younger patients 18 to 64 years of age may be even higher for selected conditions, such as heart failure.3,23
Vulnerability after hospitalization is broad. In patients over age 65 initially admitted for heart failure or acute myocardial infarction, only 35% and 10% of readmissions are for recurrent heart failure or reinfarction, respectively.1 Nearly half of readmissions are for noncardiovascular causes.1 Similarly, following hospitalization for pneumonia, more than 60 percent of readmissions are for nonpulmonary etiologies. Moreover, the risk for all these causes of readmission is much higher than baseline risk, indicating an extended period of lack of resilience to many types of illness.24 These patterns of broad susceptibility also extend to younger adults hospitalized with common medical conditions.3
Accumulating evidence suggests that hospitalized patients face functional decline, debility, and risk for adverse events despite resolution of the presenting illness, implying perhaps that the hospital environment itself is hazardous to patients’ health. In 1993, Creditor hypothesized that the “hazards of hospitalization,” including enforced bed-rest, sensory deprivation, social isolation, and malnutrition lead to a “cascade of dependency” in which a collection of small insults to multiple organ systems precipitates loss of function and debility despite cure or resolution of presenting illness.12 Covinsky (2011) later defined hospitalization-associated disability as an iatrogenic hospital-related “disorder” characterized by new impairments in abilities to perform basic activities of daily living such as bathing, feeding, toileting, dressing, transferring, and walking at the time of hospital discharge.11 Others have described a postintensive-care syndrome (PICS),25 characterized by cognitive, psychiatric, and physical impairments acquired during hospitalization for critical illness that persist postdischarge and increase the long-term risk for adverse outcomes, including elevated mortality rates,26,27 readmission rates,28 and physical disabilities.29 Similar to the “hazards of hospitalization,” PICS is thought to be related to common experiences of ICU stays, including mobility restriction, sensory deprivation, sleep disruption, sedation, malnutrition, and polypharmacy.30-33
Taken together, these data suggest that adverse health consequences attributable to hospitalization extend across the spectrum of age, presenting disease severity, and hospital treatment location. As detailed below, the PHS hypothesis is rooted in a mechanistic understanding of the role of exogenous stressors in producing physiologic dysregulation and subsequent adverse health effects across multiple organ systems.
Nature of Stress in the Hospital
Compounding the stress of acute illness, hospitalized patients are routinely and repetitively exposed to a wide variety of environmental stressors that may have downstream adverse consequences (Table 1). In the absence of overt clinical manifestations of harm, the possible subclinical physiologic dysfunction generated by the following stress exposures may increase patients’ susceptibility to the manifestations of PHS.
Sleep Disruption
Sleep disruptions trigger potent stress responses,34,35 yet they are common occurrences during hospitalization. In surveys, about half of patients report poor sleep quality during hospitalization that persists for many months after discharge.36 In a simulated hospital setting, test subjects exposed to typical hospital sounds (paging system, machine alarms, etc.) experienced significant sleep-wake cycle abnormalities.37 Although no work has yet focused specifically on the physiologic consequences of sleep disruption and stress in hospitalized patients, in healthy humans, mild sleep disruption has clear effects on allostasis by disrupting HPA activity, raising cortisol levels, diminishing parasympathetic tone, and impairing cognitive performance.18,34,35,38,39
Malnourishment
Malnourishment in hospitalized patients is common, with one-fifth of hospitalized patients receiving nothing per mouth or clear liquid diets for more than 3 continuous days,40 and one-fifth of hospitalized elderly patients receiving less than half of their calculated nutrition requirements.41 Although the relationship between food restriction, cortisol levels, and postdischarge outcomes has not been fully explored, in healthy humans, meal anticipation, meal withdrawal (withholding an expected meal), and self-reported dietary restraint are known to generate stress responses.42,43 Furthermore, malnourishment during hospitalization is associated with increased 90-day and 1-year mortality after discharge,44 adding malnourishment to the list of plausible components of hospital-related stress.
Mobility Restriction
Physical activity counterbalances stress responses and minimizes downstream consequences of allostatic load,15 yet mobility limitations via physical and chemical restraints are common in hospitalized patients, particularly among the elderly.45-47 Many patients are tethered to devices that make ambulation hazardous, such as urinary catheters and infusion pumps. Even without physical or chemical restraints or a limited mobility order, patients may be hesitant to leave the room so as not to miss transport to a diagnostic study or an unscheduled physician’s visit. Indeed, mobility limitations of hospitalized patients increase the risk for adverse events after discharge, while interventions designed to encourage mobility are associated with improved postdischarge outcomes.47,48
Other Stressors
Other hospital-related aversive stimuli are less commonly quantified, but clearly exist. According to surveys of hospitalized patients, sources of emotional stress include social isolation; loss of autonomy and privacy; fear of serious illness; lack of control over activities of daily living; lack of clear communication between treatment team and patients; and death of a patient roommate.49,50 Furthermore, consider the physical discomfort and emotional distress of patients with urinary incontinence awaiting assistance for a diaper or bedding change or the pain of repetitive blood draws or other invasive testing. Although individualized, the subjective discomfort and emotional distress associated with these experiences undoubtedly contribute to the stress of hospitalization.
IMPACT OF ALLOSTATIC OVERLOAD ON PHYSIOLOGIC FUNCTION
Animal Models of Stress
Laboratory techniques reminiscent of the numerous environmental stressors associated with hospitalization have been used to reliably trigger allostatic overload in healthy young animals.51 These techniques include sequential exposure to aversive stimuli, including food and water deprivation, continuous overnight illumination, paired housing with known and unknown cagemates, mobility restriction, soiled cage conditions, and continuous noise. All of these techniques have been shown to cause HPA axis and ANS dysfunction, allostatic overload, and subsequent stress-mediated consequences to multiple organ systems.19,52-54 Given the remarkable similarity of these protocols to common experiences during hospitalization, animal models of stress may be useful in understanding the spectrum of maladaptive consequences experienced by patients within the hospital (Figure 1).
These animal models of stress have resulted in a number of instructive findings. For example, in rodents, extended stress exposure induces structural and functional remodeling of neuronal networks that precipitate learning and memory, working memory, and attention impairments.55-57 These exposures also result in cardiovascular abnormalities, including dyslipidemia, progressive atherosclerosis,58,59 and enhanced inflammatory cytokine expression,60 all of which increase both atherosclerotic burden and susceptibility to plaque rupture, leading to elevated risk for major cardiovascular adverse events. Moreover, these extended stress exposures in animals increase susceptibility to both bacterial and viral infections and increase their severity.16,61 This outcome appears to be driven by a stress-induced elevation of glucocorticoid levels, decreased leukocyte proliferation, altered leukocyte trafficking, and a transition to a proinflammatory cytokine environment.16, 61 Allostatic overload has also been shown to contribute to metabolic dysregulation involving insulin resistance, persistence of hyperglycemia, dyslipidemia, catabolism of lean muscle, and visceral adipose tissue deposition.62-64 In addition to cardiovascular, immune, and metabolic consequences of allostatic overload, the spectrum of physiologic dysfunction in animal models is broad and includes mood disorder symptoms,65 intestinal barrier abnormalities,66 airway reactivity exacerbation,67 and enhanced tumor growth.68
Although the majority of this research highlights the multisystem effects of variable stress exposure in healthy animals, preliminary evidence suggests that aged or diseased animals subjected to additional stressors display a heightened inflammatory cytokine response that contributes to exaggerated sickness behavior and greater and prolonged cognitive deficits.69 Future studies exploring the consequences of extended stress exposure in animals with existing disease or debility may therefore more closely simulate the experience of hospitalized patients and perhaps further our understanding of PHS.
Hospitalized Patients
While no intervention studies have examined the effects of potential hospital stressors on the development of allostatic overload, there is evidence from small studies that dysregulated stress responses during hospitalization are associated with adverse events. For example, high serum cortisol, catecholamine, and proinflammatory cytokine levels during hospitalization have individually been associated with the development of cognitive dysfunction,70-72 increased risk of cardiovascular events such as myocardial infarction and stroke in the year following discharge,73-76 and the development of wound infections after discharge.77 Moreover, elevated plasma glucose during admission for myocardial infarction in patients with or without diabetes has been associated with greater in-hospital and 1-year mortality,78 with a similar relationship seen between elevated plasma glucose and survival after admission for stroke79 and pneumonia.80 Furthermore, in addition to atherothrombosis, stress may contribute to the risk for venous thromboembolism,81 resulting in readmissions for deep vein thrombosis or pulmonary embolism posthospitalization. Although potentially surrogate markers of illness acuity, a handful of studies have shown that these stress biomarkers are actually only weakly correlated with,82 or independent of,72,76 disease severity. As discussed in detail below, future studies utilizing a summative measure of multisystem physiologic dysfunction as opposed to individual biomarkers may more accurately reflect the cumulative stress effects of hospitalization and subsequent risk for adverse events.
Additional Considerations
Elderly patients, in particular, may have heightened susceptibility to the consequences of allostatic overload due to common geriatric issues such as multimorbidity and frailty. Patients with chronic diseases display both baseline HPA axis abnormalities as well as dysregulated stress responses and may therefore be more vulnerable to hospitalization-related stress. For example, when subjected to psychosocial stress, patients with chronic conditions such as diabetes, heart failure, or atherosclerosis demonstrate elevated cortisol levels, increased circulating markers of inflammation, as well as prolonged hemodynamic recovery after stress resolution compared with normal controls.83-85 Additionally, frailty may affect an individual’s susceptibility to exogenous stress. Indeed, frailty identified on hospital admission increases the risk for adverse outcomes during hospitalization and postdischarge.86 Although the specific etiology of this relationship is unclear, persons with frailty are known to have elevated levels of cortisol and other inflammatory markers,87,88 which may contribute to adverse outcomes in the face of additional stressors.
IMPLICATIONS AND NEXT STEPS
A large body of evidence stretching from bench to bedside suggests that environmental stressors associated with hospitalization are toxic. Understanding PHS within the context of hospital-induced allostatic overload presents a unifying theory for the interrelated multisystem dysfunction and increased susceptibility to adverse events that patients experience after discharge (Figure 2). Furthermore, it defines a potential pathophysiological mechanism for the cognitive impairment, elevated cardiovascular risk, immune system dysfunction, metabolic derangements, and functional decline associated with PHS. Additionally, this theory highlights environmental interventions to limit PHS development and suggests mechanisms to promote stress resilience. Although it is difficult to disentangle the consequences of the endogenous stress triggered by an acute illness from the exogenous stressors related to hospitalization, it is likely that the 2 simultaneous exposures compound risk for stress system dysregulation and allostatic overload. Moreover, hospitalized patients with preexisting HPA axis dysfunction at baseline from chronic disease or advancing age may be even more susceptible to these adverse outcomes. If this hypothesis is true, a reduction in PHS would require mitigation of the modifiable environmental stressors encountered by patients during hospitalization. Directed efforts to diminish ambient noise, limit nighttime disruptions, thoughtfully plan procedures, consider ongoing nutritional status, and promote opportunities for patients to exert some control over their environment may diminish the burden of extrinsic stressors encountered by all patients in the hospital and improve outcomes after discharge.
Hospitals are increasingly recognizing the importance of improving patients’ experience of hospitalization by reducing exposure to potential toxicities. For example, many hospitals are now attempting to reduce sleep disturbances and sleep latency through reduced nighttime noise and light levels, fewer nighttime interruptions for vital signs checks and medication administration, and commonsensical interventions like massages, herbal teas, and warm milk prior to bedtime.89 Likewise, intensive care units are targeting environmental and physical stressors with a multifaceted approach to decrease sedative use, promote healthy sleep cycles, and encourage exercise and ambulation even in those patients who are mechanically ventilated.30 Another promising development has been the increase of Hospital at Home programs. In these programs, patients who meet the criteria for inpatient admission are instead comprehensively managed at home for their acute illness through a multidisciplinary effort between physicians, nurses, social workers, physical therapists, and others. Patients hospitalized at home report higher levels of satisfaction and have modest functional gains, improved health-related quality of life, and decreased risk of mortality at 6 months compared with hospitalized patients.90,91 With some admitting diagnoses (eg, heart failure), hospitalization at home may be associated with decreased readmission risk.92 Although not yet investigated on a physiologic level, perhaps the benefits of hospital at home are partially due to the dramatic difference in exposure to environmental stressors.
A tool that quantifies hospital-associated stress may help health providers appreciate the experience of patients and better target interventions to aspects of their structure and process that contribute to allostatic overload. Importantly, allostatic overload cannot be identified by one biomarker of stress but instead requires evidence of dysregulation across inflammatory, neuroendocrine, hormonal, and cardiometabolic systems. Future studies to address the burden of stress faced by hospitalized patients should consider a summative measure of multisystem dysregulation as opposed to isolated assessments of individual biomarkers. Allostatic load has previously been operationalized as the summation of a variety of hemodynamic, hormonal, and metabolic factors, including blood pressure, lipid profile, glycosylated hemoglobin, cortisol, catecholamine levels, and inflammatory markers.93 To develop a hospital-associated allostatic load index, models should ideally be adjusted for acute illness severity, patient-reported stress, and capacity for stress resilience. This tool could then be used to quantify hospitalization-related allostatic load and identify those at greatest risk for adverse events after discharge, as well as measure the effectiveness of strategic environmental interventions (Table 2). A natural first experiment may be a comparison of the allostatic load of hospitalized patients versus those hospitalized at home.
The risk of adverse outcomes after discharge is likely a function of the vulnerability of the patient and the degree to which the patient’s healthcare team and social support network mitigates this vulnerability. That is, there is a risk that a person struggles in the postdischarge period and, in many circumstances, a strong healthcare team and social network can identify health problems early and prevent them from progressing to the point that they require hospitalization.13,94-96 There are also hospital occurrences, outside of allostatic load, that can lead to complications that lengthen the stay, weaken the patient, and directly contribute to subsequent vulnerability.94,97 Our contention is that the allostatic load of hospitalization, which may also vary by patient depending on the circumstances of hospitalization, is just one contributor, albeit potentially an important one, to vulnerability to medical problems after discharge.
In conclusion, a plausible etiology of PHS is the maladaptive mind-body consequences of common stressors during hospitalization that compound the stress of acute illness and produce allostatic overload. This stress-induced dysfunction potentially contributes to a spectrum of generalized disease susceptibility and risk of adverse outcomes after discharge. Focused efforts to diminish patient exposure to hospital-related stressors during and after hospitalization might diminish the presence or severity of PHS. Viewing PHS from this perspective enables the development of hypothesis-driven risk-prediction models, encourages critical contemplation of traditional hospitalization, and suggests that targeted environmental interventions may significantly reduce adverse outcomes.
After discharge from the hospital, patients have a significantly elevated risk for adverse events, including emergency department use, hospital readmission, and death. More than 1 in 3 patients discharged from the hospital require acute care in the month after hospital discharge, and more than 1 in 6 require readmission, with readmission diagnoses frequently differing from those of the preceding hospitalization.1-4 This heightened susceptibility to adverse events persists beyond 30 days but levels off by 7 weeks after discharge, suggesting that the period of increased risk is transient and dynamic.5
The term posthospital syndrome (PHS) describes this period of vulnerability to major adverse events following hospitalization.6 In addition to increased risk for readmission and mortality, patients in this period often show evidence of generalized dysfunction with new cognitive impairment, mobility disability, or functional decline.7-12 To date, the etiology of this vulnerability is neither well understood nor effectively addressed by transitional care interventions.13
One hypothesis to explain PHS is that stressors associated with the experience of hospitalization contribute to transient multisystem dysfunction that induces susceptibility to a broad range of medical maladies. These stressors include frequent sleep disruption, noxious sounds, painful stimuli, mobility restrictions, and poor nutrition.12 The stress hypothesis as a cause of PHS is therefore based, in large part, on evidence about allostasis and the deleterious effects of allostatic overload.
Allostasis defines a system functioning within normal stress-response parameters to promote adaptation and survival.14 In allostasis, the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic and parasympathetic branches of the autonomic nervous system (ANS) exist in homeostatic balance and respond to environmental stimuli within a range of healthy physiologic parameters. The hallmark of a system in allostasis is the ability to rapidly activate, then successfully deactivate, a stress response once the stressor (ie, threat) has resolved.14,15 To promote survival and potentiate “fight or flight” mechanisms, an appropriate stress response necessarily impacts multiple physiologic systems that result in hemodynamic augmentation and gluconeogenesis to support the anticipated action of large muscle groups, heightened vigilance and memory capabilities to improve rapid decision-making, and enhancement of innate and adaptive immune capabilities to prepare for wound repair and infection defense.14-16 The stress response is subsequently terminated by negative feedback mechanisms of glucocorticoids as well as a shift of the ANS from sympathetic to parasympathetic tone.17,18
Extended or repetitive stress exposure, however, leads to dysregulation of allostatic mechanisms responsible for stress adaptation and hinders an efficient and effective stress response. After extended stress exposure, baseline (ie, resting) HPA activity resets, causing a disruption of normal diurnal cortisol rhythm and an increase in total cortisol concentration. Moreover, in response to stress, HPA and ANS system excitation becomes impaired, and negative feedback properties are undermined.14,15 This maladaptive state, known as allostatic overload, disrupts the finely tuned mechanisms that are the foundation of mind-body balance and yields pathophysiologic consequences to multiple organ systems. Downstream ramifications of allostatic overload include cognitive deterioration, cardiovascular and immune system dysfunction, and functional decline.14,15,19
Although a stress response is an expected and necessary aspect of acute illness that promotes survival, the central thesis of this work is that additional environmental and social stressors inherent in hospitalization may unnecessarily compound stress and increase the risk of HPA axis dysfunction, allostatic overload, and subsequent multisystem dysfunction, predisposing individuals to adverse outcomes after hospital discharge. Based on data from both human subjects and animal models, we present a possible pathophysiologic mechanism for the postdischarge vulnerability of PHS, encourage critical contemplation of traditional hospitalization, and suggest interventions that might improve outcomes.
POSTHOSPITAL SYNDROME
Posthospital syndrome (PHS) describes a transient period of vulnerability after hospitalization during which patients are at elevated risk for adverse events from a broad range of conditions. In support of this characterization, epidemiologic data have demonstrated high rates of adverse outcomes following hospitalization. For example, data have shown that more than 1 in 6 older adults is readmitted to the hospital within 30 days of discharge.20 Death is also common in this first month, during which rates of postdischarge mortality may exceed initial inpatient mortality.21,22 Elevated vulnerability after hospitalization is not restricted to older adults, as readmission risk among younger patients 18 to 64 years of age may be even higher for selected conditions, such as heart failure.3,23
Vulnerability after hospitalization is broad. In patients over age 65 initially admitted for heart failure or acute myocardial infarction, only 35% and 10% of readmissions are for recurrent heart failure or reinfarction, respectively.1 Nearly half of readmissions are for noncardiovascular causes.1 Similarly, following hospitalization for pneumonia, more than 60 percent of readmissions are for nonpulmonary etiologies. Moreover, the risk for all these causes of readmission is much higher than baseline risk, indicating an extended period of lack of resilience to many types of illness.24 These patterns of broad susceptibility also extend to younger adults hospitalized with common medical conditions.3
Accumulating evidence suggests that hospitalized patients face functional decline, debility, and risk for adverse events despite resolution of the presenting illness, implying perhaps that the hospital environment itself is hazardous to patients’ health. In 1993, Creditor hypothesized that the “hazards of hospitalization,” including enforced bed-rest, sensory deprivation, social isolation, and malnutrition lead to a “cascade of dependency” in which a collection of small insults to multiple organ systems precipitates loss of function and debility despite cure or resolution of presenting illness.12 Covinsky (2011) later defined hospitalization-associated disability as an iatrogenic hospital-related “disorder” characterized by new impairments in abilities to perform basic activities of daily living such as bathing, feeding, toileting, dressing, transferring, and walking at the time of hospital discharge.11 Others have described a postintensive-care syndrome (PICS),25 characterized by cognitive, psychiatric, and physical impairments acquired during hospitalization for critical illness that persist postdischarge and increase the long-term risk for adverse outcomes, including elevated mortality rates,26,27 readmission rates,28 and physical disabilities.29 Similar to the “hazards of hospitalization,” PICS is thought to be related to common experiences of ICU stays, including mobility restriction, sensory deprivation, sleep disruption, sedation, malnutrition, and polypharmacy.30-33
Taken together, these data suggest that adverse health consequences attributable to hospitalization extend across the spectrum of age, presenting disease severity, and hospital treatment location. As detailed below, the PHS hypothesis is rooted in a mechanistic understanding of the role of exogenous stressors in producing physiologic dysregulation and subsequent adverse health effects across multiple organ systems.
Nature of Stress in the Hospital
Compounding the stress of acute illness, hospitalized patients are routinely and repetitively exposed to a wide variety of environmental stressors that may have downstream adverse consequences (Table 1). In the absence of overt clinical manifestations of harm, the possible subclinical physiologic dysfunction generated by the following stress exposures may increase patients’ susceptibility to the manifestations of PHS.
Sleep Disruption
Sleep disruptions trigger potent stress responses,34,35 yet they are common occurrences during hospitalization. In surveys, about half of patients report poor sleep quality during hospitalization that persists for many months after discharge.36 In a simulated hospital setting, test subjects exposed to typical hospital sounds (paging system, machine alarms, etc.) experienced significant sleep-wake cycle abnormalities.37 Although no work has yet focused specifically on the physiologic consequences of sleep disruption and stress in hospitalized patients, in healthy humans, mild sleep disruption has clear effects on allostasis by disrupting HPA activity, raising cortisol levels, diminishing parasympathetic tone, and impairing cognitive performance.18,34,35,38,39
Malnourishment
Malnourishment in hospitalized patients is common, with one-fifth of hospitalized patients receiving nothing per mouth or clear liquid diets for more than 3 continuous days,40 and one-fifth of hospitalized elderly patients receiving less than half of their calculated nutrition requirements.41 Although the relationship between food restriction, cortisol levels, and postdischarge outcomes has not been fully explored, in healthy humans, meal anticipation, meal withdrawal (withholding an expected meal), and self-reported dietary restraint are known to generate stress responses.42,43 Furthermore, malnourishment during hospitalization is associated with increased 90-day and 1-year mortality after discharge,44 adding malnourishment to the list of plausible components of hospital-related stress.
Mobility Restriction
Physical activity counterbalances stress responses and minimizes downstream consequences of allostatic load,15 yet mobility limitations via physical and chemical restraints are common in hospitalized patients, particularly among the elderly.45-47 Many patients are tethered to devices that make ambulation hazardous, such as urinary catheters and infusion pumps. Even without physical or chemical restraints or a limited mobility order, patients may be hesitant to leave the room so as not to miss transport to a diagnostic study or an unscheduled physician’s visit. Indeed, mobility limitations of hospitalized patients increase the risk for adverse events after discharge, while interventions designed to encourage mobility are associated with improved postdischarge outcomes.47,48
Other Stressors
Other hospital-related aversive stimuli are less commonly quantified, but clearly exist. According to surveys of hospitalized patients, sources of emotional stress include social isolation; loss of autonomy and privacy; fear of serious illness; lack of control over activities of daily living; lack of clear communication between treatment team and patients; and death of a patient roommate.49,50 Furthermore, consider the physical discomfort and emotional distress of patients with urinary incontinence awaiting assistance for a diaper or bedding change or the pain of repetitive blood draws or other invasive testing. Although individualized, the subjective discomfort and emotional distress associated with these experiences undoubtedly contribute to the stress of hospitalization.
IMPACT OF ALLOSTATIC OVERLOAD ON PHYSIOLOGIC FUNCTION
Animal Models of Stress
Laboratory techniques reminiscent of the numerous environmental stressors associated with hospitalization have been used to reliably trigger allostatic overload in healthy young animals.51 These techniques include sequential exposure to aversive stimuli, including food and water deprivation, continuous overnight illumination, paired housing with known and unknown cagemates, mobility restriction, soiled cage conditions, and continuous noise. All of these techniques have been shown to cause HPA axis and ANS dysfunction, allostatic overload, and subsequent stress-mediated consequences to multiple organ systems.19,52-54 Given the remarkable similarity of these protocols to common experiences during hospitalization, animal models of stress may be useful in understanding the spectrum of maladaptive consequences experienced by patients within the hospital (Figure 1).
These animal models of stress have resulted in a number of instructive findings. For example, in rodents, extended stress exposure induces structural and functional remodeling of neuronal networks that precipitate learning and memory, working memory, and attention impairments.55-57 These exposures also result in cardiovascular abnormalities, including dyslipidemia, progressive atherosclerosis,58,59 and enhanced inflammatory cytokine expression,60 all of which increase both atherosclerotic burden and susceptibility to plaque rupture, leading to elevated risk for major cardiovascular adverse events. Moreover, these extended stress exposures in animals increase susceptibility to both bacterial and viral infections and increase their severity.16,61 This outcome appears to be driven by a stress-induced elevation of glucocorticoid levels, decreased leukocyte proliferation, altered leukocyte trafficking, and a transition to a proinflammatory cytokine environment.16, 61 Allostatic overload has also been shown to contribute to metabolic dysregulation involving insulin resistance, persistence of hyperglycemia, dyslipidemia, catabolism of lean muscle, and visceral adipose tissue deposition.62-64 In addition to cardiovascular, immune, and metabolic consequences of allostatic overload, the spectrum of physiologic dysfunction in animal models is broad and includes mood disorder symptoms,65 intestinal barrier abnormalities,66 airway reactivity exacerbation,67 and enhanced tumor growth.68
Although the majority of this research highlights the multisystem effects of variable stress exposure in healthy animals, preliminary evidence suggests that aged or diseased animals subjected to additional stressors display a heightened inflammatory cytokine response that contributes to exaggerated sickness behavior and greater and prolonged cognitive deficits.69 Future studies exploring the consequences of extended stress exposure in animals with existing disease or debility may therefore more closely simulate the experience of hospitalized patients and perhaps further our understanding of PHS.
Hospitalized Patients
While no intervention studies have examined the effects of potential hospital stressors on the development of allostatic overload, there is evidence from small studies that dysregulated stress responses during hospitalization are associated with adverse events. For example, high serum cortisol, catecholamine, and proinflammatory cytokine levels during hospitalization have individually been associated with the development of cognitive dysfunction,70-72 increased risk of cardiovascular events such as myocardial infarction and stroke in the year following discharge,73-76 and the development of wound infections after discharge.77 Moreover, elevated plasma glucose during admission for myocardial infarction in patients with or without diabetes has been associated with greater in-hospital and 1-year mortality,78 with a similar relationship seen between elevated plasma glucose and survival after admission for stroke79 and pneumonia.80 Furthermore, in addition to atherothrombosis, stress may contribute to the risk for venous thromboembolism,81 resulting in readmissions for deep vein thrombosis or pulmonary embolism posthospitalization. Although potentially surrogate markers of illness acuity, a handful of studies have shown that these stress biomarkers are actually only weakly correlated with,82 or independent of,72,76 disease severity. As discussed in detail below, future studies utilizing a summative measure of multisystem physiologic dysfunction as opposed to individual biomarkers may more accurately reflect the cumulative stress effects of hospitalization and subsequent risk for adverse events.
Additional Considerations
Elderly patients, in particular, may have heightened susceptibility to the consequences of allostatic overload due to common geriatric issues such as multimorbidity and frailty. Patients with chronic diseases display both baseline HPA axis abnormalities as well as dysregulated stress responses and may therefore be more vulnerable to hospitalization-related stress. For example, when subjected to psychosocial stress, patients with chronic conditions such as diabetes, heart failure, or atherosclerosis demonstrate elevated cortisol levels, increased circulating markers of inflammation, as well as prolonged hemodynamic recovery after stress resolution compared with normal controls.83-85 Additionally, frailty may affect an individual’s susceptibility to exogenous stress. Indeed, frailty identified on hospital admission increases the risk for adverse outcomes during hospitalization and postdischarge.86 Although the specific etiology of this relationship is unclear, persons with frailty are known to have elevated levels of cortisol and other inflammatory markers,87,88 which may contribute to adverse outcomes in the face of additional stressors.
IMPLICATIONS AND NEXT STEPS
A large body of evidence stretching from bench to bedside suggests that environmental stressors associated with hospitalization are toxic. Understanding PHS within the context of hospital-induced allostatic overload presents a unifying theory for the interrelated multisystem dysfunction and increased susceptibility to adverse events that patients experience after discharge (Figure 2). Furthermore, it defines a potential pathophysiological mechanism for the cognitive impairment, elevated cardiovascular risk, immune system dysfunction, metabolic derangements, and functional decline associated with PHS. Additionally, this theory highlights environmental interventions to limit PHS development and suggests mechanisms to promote stress resilience. Although it is difficult to disentangle the consequences of the endogenous stress triggered by an acute illness from the exogenous stressors related to hospitalization, it is likely that the 2 simultaneous exposures compound risk for stress system dysregulation and allostatic overload. Moreover, hospitalized patients with preexisting HPA axis dysfunction at baseline from chronic disease or advancing age may be even more susceptible to these adverse outcomes. If this hypothesis is true, a reduction in PHS would require mitigation of the modifiable environmental stressors encountered by patients during hospitalization. Directed efforts to diminish ambient noise, limit nighttime disruptions, thoughtfully plan procedures, consider ongoing nutritional status, and promote opportunities for patients to exert some control over their environment may diminish the burden of extrinsic stressors encountered by all patients in the hospital and improve outcomes after discharge.
Hospitals are increasingly recognizing the importance of improving patients’ experience of hospitalization by reducing exposure to potential toxicities. For example, many hospitals are now attempting to reduce sleep disturbances and sleep latency through reduced nighttime noise and light levels, fewer nighttime interruptions for vital signs checks and medication administration, and commonsensical interventions like massages, herbal teas, and warm milk prior to bedtime.89 Likewise, intensive care units are targeting environmental and physical stressors with a multifaceted approach to decrease sedative use, promote healthy sleep cycles, and encourage exercise and ambulation even in those patients who are mechanically ventilated.30 Another promising development has been the increase of Hospital at Home programs. In these programs, patients who meet the criteria for inpatient admission are instead comprehensively managed at home for their acute illness through a multidisciplinary effort between physicians, nurses, social workers, physical therapists, and others. Patients hospitalized at home report higher levels of satisfaction and have modest functional gains, improved health-related quality of life, and decreased risk of mortality at 6 months compared with hospitalized patients.90,91 With some admitting diagnoses (eg, heart failure), hospitalization at home may be associated with decreased readmission risk.92 Although not yet investigated on a physiologic level, perhaps the benefits of hospital at home are partially due to the dramatic difference in exposure to environmental stressors.
A tool that quantifies hospital-associated stress may help health providers appreciate the experience of patients and better target interventions to aspects of their structure and process that contribute to allostatic overload. Importantly, allostatic overload cannot be identified by one biomarker of stress but instead requires evidence of dysregulation across inflammatory, neuroendocrine, hormonal, and cardiometabolic systems. Future studies to address the burden of stress faced by hospitalized patients should consider a summative measure of multisystem dysregulation as opposed to isolated assessments of individual biomarkers. Allostatic load has previously been operationalized as the summation of a variety of hemodynamic, hormonal, and metabolic factors, including blood pressure, lipid profile, glycosylated hemoglobin, cortisol, catecholamine levels, and inflammatory markers.93 To develop a hospital-associated allostatic load index, models should ideally be adjusted for acute illness severity, patient-reported stress, and capacity for stress resilience. This tool could then be used to quantify hospitalization-related allostatic load and identify those at greatest risk for adverse events after discharge, as well as measure the effectiveness of strategic environmental interventions (Table 2). A natural first experiment may be a comparison of the allostatic load of hospitalized patients versus those hospitalized at home.
The risk of adverse outcomes after discharge is likely a function of the vulnerability of the patient and the degree to which the patient’s healthcare team and social support network mitigates this vulnerability. That is, there is a risk that a person struggles in the postdischarge period and, in many circumstances, a strong healthcare team and social network can identify health problems early and prevent them from progressing to the point that they require hospitalization.13,94-96 There are also hospital occurrences, outside of allostatic load, that can lead to complications that lengthen the stay, weaken the patient, and directly contribute to subsequent vulnerability.94,97 Our contention is that the allostatic load of hospitalization, which may also vary by patient depending on the circumstances of hospitalization, is just one contributor, albeit potentially an important one, to vulnerability to medical problems after discharge.
In conclusion, a plausible etiology of PHS is the maladaptive mind-body consequences of common stressors during hospitalization that compound the stress of acute illness and produce allostatic overload. This stress-induced dysfunction potentially contributes to a spectrum of generalized disease susceptibility and risk of adverse outcomes after discharge. Focused efforts to diminish patient exposure to hospital-related stressors during and after hospitalization might diminish the presence or severity of PHS. Viewing PHS from this perspective enables the development of hypothesis-driven risk-prediction models, encourages critical contemplation of traditional hospitalization, and suggests that targeted environmental interventions may significantly reduce adverse outcomes.
1. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. http://dx.doi.org/10.1001/jama.2012.216476.
2. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. http://dx.doi.org/10.1056/NEJMsa0803563.
3. Ranasinghe I, Wang Y, Dharmarajan K, Hsieh AF, Bernheim SM, Krumholz HM. Readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia among young and middle-aged adults: a retrospective observational cohort study. PLoS Med. 2014;11(9):e1001737. http://dx.doi.org/10.1371/journal.pmed.1001737.
4. Vashi AA, Fox JP, Carr BG, et al. Use of hospital-based acute care among patients recently discharged from the hospital. JAMA. 2013;309(4):364-371. http://dx.doi.org/10.1001/jama.2012.216219.
5. Dharmarajan K, Hsieh AF, Kulkarni VT, et al. Trajectories of risk after hospitalization for heart failure, acute myocardial infarction, or pneumonia: retrospective cohort study. BMJ. 2015;350:h411. http://dx.doi.org/10.1136/bmj.h411.
6. Krumholz HM. Post-hospital syndrome--an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. http://dx.doi.org/10.1056/NEJMp1212324.
7. Ferrante LE, Pisani MA, Murphy TE, Gahbauer EA, Leo-Summers LS, Gill TM. Functional trajectories among older persons before and after critical illness. JAMA Intern Med. 2015;175(4):523-529. http://dx.doi.org/10.1001/jamainternmed.2014.7889.
8. Gill TM, Allore HG, Holford TR, Guo Z. Hospitalization, restricted activity, and the development of disability among older persons. JAMA. 2004;292(17):2115-2124. http://dx.doi.org/10.1001/jama.292.17.2115.
9. Inouye SK, Zhang Y, Han L, Leo-Summers L, Jones R, Marcantonio E. Recoverable cognitive dysfunction at hospital admission in older persons during acute illness. J Gen Intern Med. 2006;21(12):1276-1281. http://dx.doi.org/10.1111/j.1525-1497.2006.00613.x.
10. Lindquist LA, Go L, Fleisher J, Jain N, Baker D. Improvements in cognition following hospital discharge of community dwelling seniors. J Gen Intern Med. 2011;26(7):765-770. http://dx.doi.org/10.1007/s11606-011-1681-1.
11. Covinsky KE, Pierluissi E, Johnston CB. Hospitalization-associated disability: “She was probably able to ambulate, but I’m not sure.” JAMA. 2011;306(16):1782-1793. http://dx.doi.org/10.1001/jama.2011.1556.
12. Creditor MC. Hazards of hospitalization of the elderly. Ann Intern Med. 1993;118(3):219-223. http://dx.doi.org/10.7326/0003-4819-118-3-199302010-00011.
13. Dharmarajan K, Krumholz HM. Strategies to reduce 30-day readmissions in older patients hospitalized with heart failure and acute myocardial infarction. Curr Geriatr Rep. 2014;3(4):306-315. http://dx.doi.org/10.1007/s13670-014-0103-8.
14. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338(3):171-179. http://dx.doi.org/10.1056/NEJM199801153380307.
15. McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431-445. http://dx.doi.org/10.1146/annurev-med-052209-100430.
16. Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. http://dx.doi.org/10.1159/000216188.
17. Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361-372. http://dx.doi.org/10.1196/annals.1366.014.
18. Jacobson L, Akana SF, Cascio CS, Shinsako J, Dallman MF. Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology. 1988;122(4):1343-1348. http://dx.doi.org/10.1210/endo-122-4-1343.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873-904. http://dx.doi.org/10.1152/physrev.00041.2006.
20. Medicare Hospital Quality Chartbook 2014: Performance Report on Outcome Measures. Prepared by Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation for Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/quality-initiatives-patient-assessment-instruments/hospitalqualityinits/downloads/medicare-hospital-quality-chartbook-2014.pdf. Accessed February 26, 2018.
21. Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993-2006. JAMA. 2010;303(21):2141-2147. http://dx.doi.org/10.1001/jama.2010.748.
22. Drye EE, Normand SL, Wang Y, et al. Comparison of hospital risk-standardized mortality rates calculated by using in-hospital and 30-day models: an observational study with implications for hospital profiling. Ann Intern Med. 2012;156(1 Pt 1):19-26. http://dx.doi.org/10.7326/0003-4819-156-1-201201030-00004.
23. Dharmarajan K, Hsieh A, Dreyer RP, Welsh J, Qin L, Krumholz HM. Relationship between age and trajectories of rehospitalization risk in older adults. J Am Geriatr Soc. 2017;65(2):421-426. http://dx.doi.org/10.1111/jgs.14583.
24. Krumholz HM, Hsieh A, Dreyer RP, Welsh J, Desai NR, Dharmarajan K. Trajectories of risk for specific readmission diagnoses after hospitalization for heart failure, acute myocardial infarction, or pneumonia. PLoS One. 2016;11(10):e0160492. http://dx.doi.org/10.1371/journal.pone.0160492.
25. Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med. 2012;40(2):502-509. http://dx.doi.org/10.1097/CCM.0b013e318232da75.
26. Brinkman S, de Jonge E, Abu-Hanna A, Arbous MS, de Lange DW, de Keizer NF. Mortality after hospital discharge in ICU patients. Crit Care Med. 2013;41(5):1229-1236. http://dx.doi.org/10.1097/CCM.0b013e31827ca4e1.
27. Steenbergen S, Rijkenberg S, Adonis T, Kroeze G, van Stijn I, Endeman H. Long-term treated intensive care patients outcomes: the one-year mortality rate, quality of life, health care use and long-term complications as reported by general practitioners. BMC Anesthesiol. 2015;15:142. http://dx.doi.org/10.1186/s12871-015-0121-x.
28. Hill AD, Fowler RA, Pinto R, Herridge MS, Cuthbertson BH, Scales DC. Long-term outcomes and healthcare utilization following critical illness--a population-based study. Crit Care. 2016;20:76. http://dx.doi.org/10.1186/s13054-016-1248-y.
29. Jackson JC, Pandharipande PP, Girard TD, et al. Depression, post-traumatic stress disorder, and functional disability in survivors of critical illness in the BRAIN-ICU study: a longitudinal cohort study. Lancet Respir Med. 2014;2(5):369-379. http://dx.doi.org/10.1016/S2213-2600(14)70051-7.
30. Balas MC, Vasilevskis EE, Olsen KM, et al. Effectiveness and safety of the awakening and breathing coordination, delirium monitoring/management, and early exercise/mobility bundle. Crit Care Med. 2014;42(5):1024-1036. http://dx.doi.org/10.1097/CCM.0000000000000129.
31. Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;370(17):1626-1635. http://dx.doi.org/10.1056/NEJMra1209390.
32. Mendez-Tellez PA, Needham DM. Early physical rehabilitation in the ICU and ventilator liberation. Respir Care. 2012;57(10):1663-1669. http://dx.doi.org/10.4187/respcare.01931.
33. Schweickert WD, Hall J. ICU-acquired weakness. Chest. 2007;131(5):1541-1549. http://dx.doi.org/10.1378/chest.06-2065.
34. Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Int J Endocrinol. 2010;2010:759234. http://dx.doi.org/10.1155/2010/759234.
35. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354(9188):1435-1439. http://dx.doi.org/10.1016/S0140-6736(99)01376-8.
36. Orwelius L, Nordlund A, Nordlund P, Edell-Gustafsson U, Sjoberg F. Prevalence of sleep disturbances and long-term reduced health-related quality of life after critical care: a prospective multicenter cohort study. Crit Care. 2008;12(4):R97. http://dx.doi.org/10.1186/cc6973.
37. Buxton OM, Ellenbogen JM, Wang W, et al. Sleep disruption due to hospital noises: a prospective evaluation. Ann Intern Med. 2012;157(3):170-179. http://dx.doi.org/10.7326/0003-4819-157-3-201208070-00472.
38. Sunbul M, Kanar BG, Durmus E, Kivrak T, Sari I. Acute sleep deprivation is associated with increased arterial stiffness in healthy young adults. Sleep Breath. 2014;18(1):215-220. http://dx.doi.org/10.1007/s11325-013-0873-9.
39. Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26(2):117-126. http://dx.doi.org/10.1093/sleep/26.2.117.
40. Franklin GA, McClave SA, Hurt RT, et al. Physician-delivered malnutrition: why do patients receive nothing by mouth or a clear liquid diet in a university hospital setting? JPEN J Parenter Enteral Nutr. 2011;35(3):337-342. http://dx.doi.org/10.1177/0148607110374060.
41. Sullivan DH, Sun S, Walls RC. Protein-energy undernutrition among elderly hospitalized patients: a prospective study. JAMA. 1999;281(21):2013-2019. http://dx.doi.org/10.1001/jama.281.21.2013.
42. Anderson DA, Shapiro JR, Lundgren JD, Spataro LE, Frye CA. Self-reported dietary restraint is associated with elevated levels of salivary cortisol. Appetite. 2002;38(1):13-17. http://dx.doi.org/10.1006/appe.2001.0459.
43. Ott V, Friedrich M, Prilop S, et al. Food anticipation and subsequent food withdrawal increase serum cortisol in healthy men. Physiol Behav. 2011;103(5):594-599. http://dx.doi.org/10.1016/j.physbeh.2011.04.020.
44. Covinsky KE, Martin GE, Beyth RJ, Justice AC, Sehgal AR, Landefeld CS. The relationship between clinical assessments of nutritional status and adverse outcomes in older hospitalized medical patients. J Am Geriatr Soc. 1999;47(5):532-538. http://dx.doi.org/10.1111/j.1532-5415.1999.tb02566.x.
45. Lazarus BA, Murphy JB, Coletta EM, McQuade WH, Culpepper L. The provision of physical activity to hospitalized elderly patients. Arch Intern Med. 1991;151(12):2452-2456.
46. Minnick AF, Mion LC, Johnson ME, Catrambone C, Leipzig R. Prevalence and variation of physical restraint use in acute care settings in the US. J Nurs Scholarsh. 2007;39(1):30-37. http://dx.doi.org/10.1111/j.1547-5069.2007.00140.x.
47. Zisberg A, Shadmi E, Sinoff G, Gur-Yaish N, Srulovici E, Admi H. Low mobility during hospitalization and functional decline in older adults. J Am Geriatr Soc. 2011;59(2):266-273. http://dx.doi.org/10.1111/j.1532-5415.2010.03276.x.
48. Landefeld CS, Palmer RM, Kresevic DM, Fortinsky RH, Kowal J. A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338-1344. http://dx.doi.org/10.1056/NEJM199505183322006.
49. Douglas CH, Douglas MR. Patient-friendly hospital environments: exploring the patients’ perspective. Health Expectations: an international journal of public participation in health care and health policy. 2004;7(1):61-73. http://dx.doi.org/10.1046/j.1369-6513.2003.00251.x.
50. Volicer BJ. Hospital stress and patient reports of pain and physical status. Journal Human Stress. 1978;4(2):28-37. http://dx.doi.org/10.1080/0097840X.1978.9934984.
51. Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl). 1987;93(3):358-364. http://dx.doi.org/10.1007/BF00187257.
52. Grippo AJ, Francis J, Beltz TG, Felder RB, Johnson AK. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol Behav. 2005;84(5):697-706. http://dx.doi.org/10.1016/j.physbeh.2005.02.011.
53. Krishnan V, Nestler EJ. Animal models of depression: molecular perspectives. Curr Top Behav Neurosci. 2011;7:121-147. http://dx.doi.org/10.1007/7854_2010_108.
54. Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69(1):83-88. http://dx.doi.org/10.1016/0306-4522(95)00256-I.
55. McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265-277. http://dx.doi.org/10.1111/j.1749-6632.2001.tb05830.x.
56. McEwen BS. The brain on stress: toward an integrative approach to brain, body, and behavior. Perspect Psychol Sci. 2013;8(6):673-675. http://dx.doi.org/10.1177/1745691613506907.
57. McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16-29. http://dx.doi.org/10.1016/j.neuron.2013.06.028.
58. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325-329. http://dx.doi.org/10.1038/nature11260.
59. Lu XT, Liu YF, Zhang L, et al. Unpredictable chronic mild stress promotes atherosclerosis in high cholesterol-fed rabbits. Psychosom Med. 2012;74(6):604-611. http://dx.doi.org/10.1097/PSY.0b013e31825d0b71.
60. Heidt T, Sager HB, Courties G, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20(7):754-758. http://dx.doi.org/10.1038/nm.3589.
61. Sheridan JF, Feng NG, Bonneau RH, Allen CM, Huneycutt BS, Glaser R. Restraint stress differentially affects anti-viral cellular and humoral immune responses in mice. J Neuroimmunol. 1991;31(3):245-255. http://dx.doi.org/10.1016/0165-5728(91)90046-A.
62. Kyrou I, Tsigos C. Stress hormones: physiological stress and regulation of metabolism. Curr Opin Pharmacol. 2009;9(6):787-793. http://dx.doi.org/10.1016/j.coph.2009.08.007.
63. Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30(1):1-10. http://dx.doi.org/10.1016/j.psyneuen.2004.05.007.
64. Tamashiro KL, Sakai RR, Shively CA, Karatsoreos IN, Reagan LP. Chronic stress, metabolism, and metabolic syndrome. Stress. 2011;14(5):468-474. http://dx.doi.org/10.3109/10253890.2011.606341.
65. McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54(3):200-207. http://dx.doi.org/10.1016/S0006-3223(03)00177-X.
66. Zareie M, Johnson-Henry K, Jury J, et al. Probiotics prevent bacterial translocation and improve intestinal barrier function in rats following chronic psychological stress. Gut. 2006;55(11):1553-1560. http://dx.doi.org/10.1136/gut.2005.080739.
67. Joachim RA, Quarcoo D, Arck PC, Herz U, Renz H, Klapp BF. Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom Med. 2003;65(5):811-815. http://dx.doi.org/10.1097/01.PSY.0000088582.50468.A3.
68. Thaker PH, Han LY, Kamat AA, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939-944. http://dx.doi.org/10.1038/nm1447.
69. Schreuder L, Eggen BJ, Biber K, Schoemaker RG, Laman JD, de Rooij SE. Pathophysiological and behavioral effects of systemic inflammation in aged and diseased rodents with relevance to delirium: A systematic review. Brain Behav Immun. 2017;62:362-381. http://dx.doi.org/10.1016/j.bbi.2017.01.010.
70. Mu DL, Li LH, Wang DX, et al. High postoperative serum cortisol level is associated with increased risk of cognitive dysfunction early after coronary artery bypass graft surgery: a prospective cohort study. PLoS One. 2013;8(10):e77637. http://dx.doi.org/10.1371/journal.pone.0077637.
71. Mu DL, Wang DX, Li LH, et al. High serum cortisol level is associated with increased risk of delirium after coronary artery bypass graft surgery: a prospective cohort study. Crit Care. 2010;14(6):R238. http://dx.doi.org/10.1186/cc9393.
72. Nguyen DN, Huyghens L, Zhang H, Schiettecatte J, Smitz J, Vincent JL. Cortisol is an associated-risk factor of brain dysfunction in patients with severe sepsis and septic shock. Biomed Res Int. 2014;2014:712742. http://dx.doi.org/10.1155/2014/712742.
73. Elkind MS, Carty CL, O’Meara ES, et al. Hospitalization for infection and risk of acute ischemic stroke: the Cardiovascular Health Study. Stroke. 2011;42(7):1851-1856. http://dx.doi.org/10.1161/STROKEAHA.110.608588.
74. Feibel JH, Hardy PM, Campbell RG, Goldstein MN, Joynt RJ. Prognostic value of the stress response following stroke. JAMA. 1977;238(13):1374-1376.
75. Jutla SK, Yuyun MF, Quinn PA, Ng LL. Plasma cortisol and prognosis of patients with acute myocardial infarction. J Cardiovasc Med (Hagerstown). 2014;15(1):33-41. http://dx.doi.org/10.2459/JCM.0b013e328364100b.
76. Yende S, D’Angelo G, Kellum JA, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med. 2008;177(11):1242-1247. http://dx.doi.org/10.1164/rccm.200712-1777OC.
77. Gouin JP, Kiecolt-Glaser JK. The impact of psychological stress on wound healing: methods and mechanisms. Immunol Allergy Clin North Am. 2011;31(1):81-93. http://dx.doi.org/10.1016/j.iac.2010.09.010.
78. Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355(9206):773-778. http://dx.doi.org/10.1016/S0140-6736(99)08415-9.
79. O’Neill PA, Davies I, Fullerton KJ, Bennett D. Stress hormone and blood glucose response following acute stroke in the elderly. Stroke. 1991;22(7):842-847. http://dx.doi.org/10.1161/01.STR.22.7.842.
80. Waterer GW, Kessler LA, Wunderink RG. Medium-term survival after hospitalization with community-acquired pneumonia. Am J Respir Crit Care Med. 2004;169(8):910-914. http://dx.doi.org/10.1164/rccm.200310-1448OC.
81. Rosengren A, Freden M, Hansson PO, Wilhelmsen L, Wedel H, Eriksson H. Psychosocial factors and venous thromboembolism: a long-term follow-up study of Swedish men. J Thrombosis Haemostasis. 2008;6(4):558-564. http://dx.doi.org/10.1111/j.1538-7836.2007.02857.x.
82. Oswald GA, Smith CC, Betteridge DJ, Yudkin JS. Determinants and importance of stress hyperglycaemia in non-diabetic patients with myocardial infarction. BMJ. 1986;293(6552):917-922. http://dx.doi.org/10.1136/bmj.293.6552.917.
83. Middlekauff HR, Nguyen AH, Negrao CE, et al. Impact of acute mental stress on sympathetic nerve activity and regional blood flow in advanced heart failure: implications for ‘triggering’ adverse cardiac events. Circulation. 1997;96(6):1835-1842. http://dx.doi.org/10.1161/01.CIR.96.6.1835.
84. Nijm J, Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41(3):224-233. http://dx.doi.org/10.1080/07853890802508934.
85. Steptoe A, Hackett RA, Lazzarino AI, et al. Disruption of multisystem responses to stress in type 2 diabetes: investigating the dynamics of allostatic load. Proc Natl Acad Sci U S A. 2014;111(44):15693-15698. http://dx.doi.org/10.1073/pnas.1410401111.
86. Sepehri A, Beggs T, Hassan A, et al. The impact of frailty on outcomes after cardiac surgery: a systematic review. J Thorac Cardiovasc Surg. 2014;148(6):3110-3117. http://dx.doi.org/10.1016/j.jtcvs.2014.07.087.
87. Johar H, Emeny RT, Bidlingmaier M, et al. Blunted diurnal cortisol pattern is associated with frailty: a cross-sectional study of 745 participants aged 65 to 90 years. J Clin Endocrinol Metab. 2014;99(3):E464-468. http://dx.doi.org/10.1210/jc.2013-3079.
88. Yao X, Li H, Leng SX. Inflammation and immune system alterations in frailty. Clin Geriatr Med. 2011;27(1):79-87. http://dx.doi.org/10.1016/j.cger.2010.08.002.
89. Hospital Elder Life Program (HELP) for Prevention of Delirium. 2017; http://www.hospitalelderlifeprogram.org/. Accessed February 16, 2018.
90. Shepperd S, Doll H, Angus RM, et al. Admission avoidance hospital at home. Cochrane Database of System Rev. 2008;(4):CD007491. http://dx.doi.org/10.1002/14651858.CD007491.pub2’
91. Leff B, Burton L, Mader SL, et al. Comparison of functional outcomes associated with hospital at home care and traditional acute hospital care. J Am Geriatrics Soc. 2009;57(2):273-278. http://dx.doi.org/10.1111/j.1532-5415.2008.02103.x.
92. Qaddoura A, Yazdan-Ashoori P, Kabali C, et al. Efficacy of hospital at home in patients with heart failure: a systematic review and meta-analysis. PloS One. 2015;10(6):e0129282. http://dx.doi.org/10.1371/journal.pone.0129282.
93. Seeman T, Gruenewald T, Karlamangla A, et al. Modeling multisystem biological risk in young adults: The Coronary Artery Risk Development in Young Adults Study. Am J Hum Biol. 2010;22(4):463-472. http://dx.doi.org/10.1002/ajhb.21018.
94. Auerbach AD, Kripalani S, Vasilevskis EE, et al. Preventability and causes of readmissions in a national cohort of general medicine patients. JAMA Intern Med. 2016;176(4):484-493. http://dx.doi.org/10.1001/jamainternmed.2015.7863.
95. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. http://dx.doi.org/10.7326/0003-4819-155-8-201110180-00008.
96. Takahashi PY, Naessens JM, Peterson SM, et al. Short-term and long-term effectiveness of a post-hospital care transitions program in an older, medically complex population. Healthcare. 2016;4(1):30-35. http://dx.doi.org/10.1016/j.hjdsi.2015.06.006.
97. Dharmarajan K, Swami S, Gou RY, Jones RN, Inouye SK. Pathway from delirium to death: potential in-hospital mediators of excess mortality. J Am Geriatr Soc. 2017;65(5):1026-1033. http://dx.doi.org/10.1111/jgs.14743.
1. Dharmarajan K, Hsieh AF, Lin Z, et al. Diagnoses and timing of 30-day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA. 2013;309(4):355-363. http://dx.doi.org/10.1001/jama.2012.216476.
2. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428. http://dx.doi.org/10.1056/NEJMsa0803563.
3. Ranasinghe I, Wang Y, Dharmarajan K, Hsieh AF, Bernheim SM, Krumholz HM. Readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia among young and middle-aged adults: a retrospective observational cohort study. PLoS Med. 2014;11(9):e1001737. http://dx.doi.org/10.1371/journal.pmed.1001737.
4. Vashi AA, Fox JP, Carr BG, et al. Use of hospital-based acute care among patients recently discharged from the hospital. JAMA. 2013;309(4):364-371. http://dx.doi.org/10.1001/jama.2012.216219.
5. Dharmarajan K, Hsieh AF, Kulkarni VT, et al. Trajectories of risk after hospitalization for heart failure, acute myocardial infarction, or pneumonia: retrospective cohort study. BMJ. 2015;350:h411. http://dx.doi.org/10.1136/bmj.h411.
6. Krumholz HM. Post-hospital syndrome--an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. http://dx.doi.org/10.1056/NEJMp1212324.
7. Ferrante LE, Pisani MA, Murphy TE, Gahbauer EA, Leo-Summers LS, Gill TM. Functional trajectories among older persons before and after critical illness. JAMA Intern Med. 2015;175(4):523-529. http://dx.doi.org/10.1001/jamainternmed.2014.7889.
8. Gill TM, Allore HG, Holford TR, Guo Z. Hospitalization, restricted activity, and the development of disability among older persons. JAMA. 2004;292(17):2115-2124. http://dx.doi.org/10.1001/jama.292.17.2115.
9. Inouye SK, Zhang Y, Han L, Leo-Summers L, Jones R, Marcantonio E. Recoverable cognitive dysfunction at hospital admission in older persons during acute illness. J Gen Intern Med. 2006;21(12):1276-1281. http://dx.doi.org/10.1111/j.1525-1497.2006.00613.x.
10. Lindquist LA, Go L, Fleisher J, Jain N, Baker D. Improvements in cognition following hospital discharge of community dwelling seniors. J Gen Intern Med. 2011;26(7):765-770. http://dx.doi.org/10.1007/s11606-011-1681-1.
11. Covinsky KE, Pierluissi E, Johnston CB. Hospitalization-associated disability: “She was probably able to ambulate, but I’m not sure.” JAMA. 2011;306(16):1782-1793. http://dx.doi.org/10.1001/jama.2011.1556.
12. Creditor MC. Hazards of hospitalization of the elderly. Ann Intern Med. 1993;118(3):219-223. http://dx.doi.org/10.7326/0003-4819-118-3-199302010-00011.
13. Dharmarajan K, Krumholz HM. Strategies to reduce 30-day readmissions in older patients hospitalized with heart failure and acute myocardial infarction. Curr Geriatr Rep. 2014;3(4):306-315. http://dx.doi.org/10.1007/s13670-014-0103-8.
14. McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338(3):171-179. http://dx.doi.org/10.1056/NEJM199801153380307.
15. McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431-445. http://dx.doi.org/10.1146/annurev-med-052209-100430.
16. Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16(5):300-317. http://dx.doi.org/10.1159/000216188.
17. Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361-372. http://dx.doi.org/10.1196/annals.1366.014.
18. Jacobson L, Akana SF, Cascio CS, Shinsako J, Dallman MF. Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology. 1988;122(4):1343-1348. http://dx.doi.org/10.1210/endo-122-4-1343.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873-904. http://dx.doi.org/10.1152/physrev.00041.2006.
20. Medicare Hospital Quality Chartbook 2014: Performance Report on Outcome Measures. Prepared by Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation for Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/quality-initiatives-patient-assessment-instruments/hospitalqualityinits/downloads/medicare-hospital-quality-chartbook-2014.pdf. Accessed February 26, 2018.
21. Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993-2006. JAMA. 2010;303(21):2141-2147. http://dx.doi.org/10.1001/jama.2010.748.
22. Drye EE, Normand SL, Wang Y, et al. Comparison of hospital risk-standardized mortality rates calculated by using in-hospital and 30-day models: an observational study with implications for hospital profiling. Ann Intern Med. 2012;156(1 Pt 1):19-26. http://dx.doi.org/10.7326/0003-4819-156-1-201201030-00004.
23. Dharmarajan K, Hsieh A, Dreyer RP, Welsh J, Qin L, Krumholz HM. Relationship between age and trajectories of rehospitalization risk in older adults. J Am Geriatr Soc. 2017;65(2):421-426. http://dx.doi.org/10.1111/jgs.14583.
24. Krumholz HM, Hsieh A, Dreyer RP, Welsh J, Desai NR, Dharmarajan K. Trajectories of risk for specific readmission diagnoses after hospitalization for heart failure, acute myocardial infarction, or pneumonia. PLoS One. 2016;11(10):e0160492. http://dx.doi.org/10.1371/journal.pone.0160492.
25. Needham DM, Davidson J, Cohen H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med. 2012;40(2):502-509. http://dx.doi.org/10.1097/CCM.0b013e318232da75.
26. Brinkman S, de Jonge E, Abu-Hanna A, Arbous MS, de Lange DW, de Keizer NF. Mortality after hospital discharge in ICU patients. Crit Care Med. 2013;41(5):1229-1236. http://dx.doi.org/10.1097/CCM.0b013e31827ca4e1.
27. Steenbergen S, Rijkenberg S, Adonis T, Kroeze G, van Stijn I, Endeman H. Long-term treated intensive care patients outcomes: the one-year mortality rate, quality of life, health care use and long-term complications as reported by general practitioners. BMC Anesthesiol. 2015;15:142. http://dx.doi.org/10.1186/s12871-015-0121-x.
28. Hill AD, Fowler RA, Pinto R, Herridge MS, Cuthbertson BH, Scales DC. Long-term outcomes and healthcare utilization following critical illness--a population-based study. Crit Care. 2016;20:76. http://dx.doi.org/10.1186/s13054-016-1248-y.
29. Jackson JC, Pandharipande PP, Girard TD, et al. Depression, post-traumatic stress disorder, and functional disability in survivors of critical illness in the BRAIN-ICU study: a longitudinal cohort study. Lancet Respir Med. 2014;2(5):369-379. http://dx.doi.org/10.1016/S2213-2600(14)70051-7.
30. Balas MC, Vasilevskis EE, Olsen KM, et al. Effectiveness and safety of the awakening and breathing coordination, delirium monitoring/management, and early exercise/mobility bundle. Crit Care Med. 2014;42(5):1024-1036. http://dx.doi.org/10.1097/CCM.0000000000000129.
31. Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;370(17):1626-1635. http://dx.doi.org/10.1056/NEJMra1209390.
32. Mendez-Tellez PA, Needham DM. Early physical rehabilitation in the ICU and ventilator liberation. Respir Care. 2012;57(10):1663-1669. http://dx.doi.org/10.4187/respcare.01931.
33. Schweickert WD, Hall J. ICU-acquired weakness. Chest. 2007;131(5):1541-1549. http://dx.doi.org/10.1378/chest.06-2065.
34. Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Int J Endocrinol. 2010;2010:759234. http://dx.doi.org/10.1155/2010/759234.
35. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354(9188):1435-1439. http://dx.doi.org/10.1016/S0140-6736(99)01376-8.
36. Orwelius L, Nordlund A, Nordlund P, Edell-Gustafsson U, Sjoberg F. Prevalence of sleep disturbances and long-term reduced health-related quality of life after critical care: a prospective multicenter cohort study. Crit Care. 2008;12(4):R97. http://dx.doi.org/10.1186/cc6973.
37. Buxton OM, Ellenbogen JM, Wang W, et al. Sleep disruption due to hospital noises: a prospective evaluation. Ann Intern Med. 2012;157(3):170-179. http://dx.doi.org/10.7326/0003-4819-157-3-201208070-00472.
38. Sunbul M, Kanar BG, Durmus E, Kivrak T, Sari I. Acute sleep deprivation is associated with increased arterial stiffness in healthy young adults. Sleep Breath. 2014;18(1):215-220. http://dx.doi.org/10.1007/s11325-013-0873-9.
39. Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26(2):117-126. http://dx.doi.org/10.1093/sleep/26.2.117.
40. Franklin GA, McClave SA, Hurt RT, et al. Physician-delivered malnutrition: why do patients receive nothing by mouth or a clear liquid diet in a university hospital setting? JPEN J Parenter Enteral Nutr. 2011;35(3):337-342. http://dx.doi.org/10.1177/0148607110374060.
41. Sullivan DH, Sun S, Walls RC. Protein-energy undernutrition among elderly hospitalized patients: a prospective study. JAMA. 1999;281(21):2013-2019. http://dx.doi.org/10.1001/jama.281.21.2013.
42. Anderson DA, Shapiro JR, Lundgren JD, Spataro LE, Frye CA. Self-reported dietary restraint is associated with elevated levels of salivary cortisol. Appetite. 2002;38(1):13-17. http://dx.doi.org/10.1006/appe.2001.0459.
43. Ott V, Friedrich M, Prilop S, et al. Food anticipation and subsequent food withdrawal increase serum cortisol in healthy men. Physiol Behav. 2011;103(5):594-599. http://dx.doi.org/10.1016/j.physbeh.2011.04.020.
44. Covinsky KE, Martin GE, Beyth RJ, Justice AC, Sehgal AR, Landefeld CS. The relationship between clinical assessments of nutritional status and adverse outcomes in older hospitalized medical patients. J Am Geriatr Soc. 1999;47(5):532-538. http://dx.doi.org/10.1111/j.1532-5415.1999.tb02566.x.
45. Lazarus BA, Murphy JB, Coletta EM, McQuade WH, Culpepper L. The provision of physical activity to hospitalized elderly patients. Arch Intern Med. 1991;151(12):2452-2456.
46. Minnick AF, Mion LC, Johnson ME, Catrambone C, Leipzig R. Prevalence and variation of physical restraint use in acute care settings in the US. J Nurs Scholarsh. 2007;39(1):30-37. http://dx.doi.org/10.1111/j.1547-5069.2007.00140.x.
47. Zisberg A, Shadmi E, Sinoff G, Gur-Yaish N, Srulovici E, Admi H. Low mobility during hospitalization and functional decline in older adults. J Am Geriatr Soc. 2011;59(2):266-273. http://dx.doi.org/10.1111/j.1532-5415.2010.03276.x.
48. Landefeld CS, Palmer RM, Kresevic DM, Fortinsky RH, Kowal J. A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338-1344. http://dx.doi.org/10.1056/NEJM199505183322006.
49. Douglas CH, Douglas MR. Patient-friendly hospital environments: exploring the patients’ perspective. Health Expectations: an international journal of public participation in health care and health policy. 2004;7(1):61-73. http://dx.doi.org/10.1046/j.1369-6513.2003.00251.x.
50. Volicer BJ. Hospital stress and patient reports of pain and physical status. Journal Human Stress. 1978;4(2):28-37. http://dx.doi.org/10.1080/0097840X.1978.9934984.
51. Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl). 1987;93(3):358-364. http://dx.doi.org/10.1007/BF00187257.
52. Grippo AJ, Francis J, Beltz TG, Felder RB, Johnson AK. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol Behav. 2005;84(5):697-706. http://dx.doi.org/10.1016/j.physbeh.2005.02.011.
53. Krishnan V, Nestler EJ. Animal models of depression: molecular perspectives. Curr Top Behav Neurosci. 2011;7:121-147. http://dx.doi.org/10.1007/7854_2010_108.
54. Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69(1):83-88. http://dx.doi.org/10.1016/0306-4522(95)00256-I.
55. McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265-277. http://dx.doi.org/10.1111/j.1749-6632.2001.tb05830.x.
56. McEwen BS. The brain on stress: toward an integrative approach to brain, body, and behavior. Perspect Psychol Sci. 2013;8(6):673-675. http://dx.doi.org/10.1177/1745691613506907.
57. McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16-29. http://dx.doi.org/10.1016/j.neuron.2013.06.028.
58. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325-329. http://dx.doi.org/10.1038/nature11260.
59. Lu XT, Liu YF, Zhang L, et al. Unpredictable chronic mild stress promotes atherosclerosis in high cholesterol-fed rabbits. Psychosom Med. 2012;74(6):604-611. http://dx.doi.org/10.1097/PSY.0b013e31825d0b71.
60. Heidt T, Sager HB, Courties G, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20(7):754-758. http://dx.doi.org/10.1038/nm.3589.
61. Sheridan JF, Feng NG, Bonneau RH, Allen CM, Huneycutt BS, Glaser R. Restraint stress differentially affects anti-viral cellular and humoral immune responses in mice. J Neuroimmunol. 1991;31(3):245-255. http://dx.doi.org/10.1016/0165-5728(91)90046-A.
62. Kyrou I, Tsigos C. Stress hormones: physiological stress and regulation of metabolism. Curr Opin Pharmacol. 2009;9(6):787-793. http://dx.doi.org/10.1016/j.coph.2009.08.007.
63. Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30(1):1-10. http://dx.doi.org/10.1016/j.psyneuen.2004.05.007.
64. Tamashiro KL, Sakai RR, Shively CA, Karatsoreos IN, Reagan LP. Chronic stress, metabolism, and metabolic syndrome. Stress. 2011;14(5):468-474. http://dx.doi.org/10.3109/10253890.2011.606341.
65. McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54(3):200-207. http://dx.doi.org/10.1016/S0006-3223(03)00177-X.
66. Zareie M, Johnson-Henry K, Jury J, et al. Probiotics prevent bacterial translocation and improve intestinal barrier function in rats following chronic psychological stress. Gut. 2006;55(11):1553-1560. http://dx.doi.org/10.1136/gut.2005.080739.
67. Joachim RA, Quarcoo D, Arck PC, Herz U, Renz H, Klapp BF. Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom Med. 2003;65(5):811-815. http://dx.doi.org/10.1097/01.PSY.0000088582.50468.A3.
68. Thaker PH, Han LY, Kamat AA, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939-944. http://dx.doi.org/10.1038/nm1447.
69. Schreuder L, Eggen BJ, Biber K, Schoemaker RG, Laman JD, de Rooij SE. Pathophysiological and behavioral effects of systemic inflammation in aged and diseased rodents with relevance to delirium: A systematic review. Brain Behav Immun. 2017;62:362-381. http://dx.doi.org/10.1016/j.bbi.2017.01.010.
70. Mu DL, Li LH, Wang DX, et al. High postoperative serum cortisol level is associated with increased risk of cognitive dysfunction early after coronary artery bypass graft surgery: a prospective cohort study. PLoS One. 2013;8(10):e77637. http://dx.doi.org/10.1371/journal.pone.0077637.
71. Mu DL, Wang DX, Li LH, et al. High serum cortisol level is associated with increased risk of delirium after coronary artery bypass graft surgery: a prospective cohort study. Crit Care. 2010;14(6):R238. http://dx.doi.org/10.1186/cc9393.
72. Nguyen DN, Huyghens L, Zhang H, Schiettecatte J, Smitz J, Vincent JL. Cortisol is an associated-risk factor of brain dysfunction in patients with severe sepsis and septic shock. Biomed Res Int. 2014;2014:712742. http://dx.doi.org/10.1155/2014/712742.
73. Elkind MS, Carty CL, O’Meara ES, et al. Hospitalization for infection and risk of acute ischemic stroke: the Cardiovascular Health Study. Stroke. 2011;42(7):1851-1856. http://dx.doi.org/10.1161/STROKEAHA.110.608588.
74. Feibel JH, Hardy PM, Campbell RG, Goldstein MN, Joynt RJ. Prognostic value of the stress response following stroke. JAMA. 1977;238(13):1374-1376.
75. Jutla SK, Yuyun MF, Quinn PA, Ng LL. Plasma cortisol and prognosis of patients with acute myocardial infarction. J Cardiovasc Med (Hagerstown). 2014;15(1):33-41. http://dx.doi.org/10.2459/JCM.0b013e328364100b.
76. Yende S, D’Angelo G, Kellum JA, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med. 2008;177(11):1242-1247. http://dx.doi.org/10.1164/rccm.200712-1777OC.
77. Gouin JP, Kiecolt-Glaser JK. The impact of psychological stress on wound healing: methods and mechanisms. Immunol Allergy Clin North Am. 2011;31(1):81-93. http://dx.doi.org/10.1016/j.iac.2010.09.010.
78. Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355(9206):773-778. http://dx.doi.org/10.1016/S0140-6736(99)08415-9.
79. O’Neill PA, Davies I, Fullerton KJ, Bennett D. Stress hormone and blood glucose response following acute stroke in the elderly. Stroke. 1991;22(7):842-847. http://dx.doi.org/10.1161/01.STR.22.7.842.
80. Waterer GW, Kessler LA, Wunderink RG. Medium-term survival after hospitalization with community-acquired pneumonia. Am J Respir Crit Care Med. 2004;169(8):910-914. http://dx.doi.org/10.1164/rccm.200310-1448OC.
81. Rosengren A, Freden M, Hansson PO, Wilhelmsen L, Wedel H, Eriksson H. Psychosocial factors and venous thromboembolism: a long-term follow-up study of Swedish men. J Thrombosis Haemostasis. 2008;6(4):558-564. http://dx.doi.org/10.1111/j.1538-7836.2007.02857.x.
82. Oswald GA, Smith CC, Betteridge DJ, Yudkin JS. Determinants and importance of stress hyperglycaemia in non-diabetic patients with myocardial infarction. BMJ. 1986;293(6552):917-922. http://dx.doi.org/10.1136/bmj.293.6552.917.
83. Middlekauff HR, Nguyen AH, Negrao CE, et al. Impact of acute mental stress on sympathetic nerve activity and regional blood flow in advanced heart failure: implications for ‘triggering’ adverse cardiac events. Circulation. 1997;96(6):1835-1842. http://dx.doi.org/10.1161/01.CIR.96.6.1835.
84. Nijm J, Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41(3):224-233. http://dx.doi.org/10.1080/07853890802508934.
85. Steptoe A, Hackett RA, Lazzarino AI, et al. Disruption of multisystem responses to stress in type 2 diabetes: investigating the dynamics of allostatic load. Proc Natl Acad Sci U S A. 2014;111(44):15693-15698. http://dx.doi.org/10.1073/pnas.1410401111.
86. Sepehri A, Beggs T, Hassan A, et al. The impact of frailty on outcomes after cardiac surgery: a systematic review. J Thorac Cardiovasc Surg. 2014;148(6):3110-3117. http://dx.doi.org/10.1016/j.jtcvs.2014.07.087.
87. Johar H, Emeny RT, Bidlingmaier M, et al. Blunted diurnal cortisol pattern is associated with frailty: a cross-sectional study of 745 participants aged 65 to 90 years. J Clin Endocrinol Metab. 2014;99(3):E464-468. http://dx.doi.org/10.1210/jc.2013-3079.
88. Yao X, Li H, Leng SX. Inflammation and immune system alterations in frailty. Clin Geriatr Med. 2011;27(1):79-87. http://dx.doi.org/10.1016/j.cger.2010.08.002.
89. Hospital Elder Life Program (HELP) for Prevention of Delirium. 2017; http://www.hospitalelderlifeprogram.org/. Accessed February 16, 2018.
90. Shepperd S, Doll H, Angus RM, et al. Admission avoidance hospital at home. Cochrane Database of System Rev. 2008;(4):CD007491. http://dx.doi.org/10.1002/14651858.CD007491.pub2’
91. Leff B, Burton L, Mader SL, et al. Comparison of functional outcomes associated with hospital at home care and traditional acute hospital care. J Am Geriatrics Soc. 2009;57(2):273-278. http://dx.doi.org/10.1111/j.1532-5415.2008.02103.x.
92. Qaddoura A, Yazdan-Ashoori P, Kabali C, et al. Efficacy of hospital at home in patients with heart failure: a systematic review and meta-analysis. PloS One. 2015;10(6):e0129282. http://dx.doi.org/10.1371/journal.pone.0129282.
93. Seeman T, Gruenewald T, Karlamangla A, et al. Modeling multisystem biological risk in young adults: The Coronary Artery Risk Development in Young Adults Study. Am J Hum Biol. 2010;22(4):463-472. http://dx.doi.org/10.1002/ajhb.21018.
94. Auerbach AD, Kripalani S, Vasilevskis EE, et al. Preventability and causes of readmissions in a national cohort of general medicine patients. JAMA Intern Med. 2016;176(4):484-493. http://dx.doi.org/10.1001/jamainternmed.2015.7863.
95. Hansen LO, Young RS, Hinami K, Leung A, Williams MV. Interventions to reduce 30-day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520-528. http://dx.doi.org/10.7326/0003-4819-155-8-201110180-00008.
96. Takahashi PY, Naessens JM, Peterson SM, et al. Short-term and long-term effectiveness of a post-hospital care transitions program in an older, medically complex population. Healthcare. 2016;4(1):30-35. http://dx.doi.org/10.1016/j.hjdsi.2015.06.006.
97. Dharmarajan K, Swami S, Gou RY, Jones RN, Inouye SK. Pathway from delirium to death: potential in-hospital mediators of excess mortality. J Am Geriatr Soc. 2017;65(5):1026-1033. http://dx.doi.org/10.1111/jgs.14743.
© 2018 Society of Hospital Medicine
Ketorolac may reduce breast cancer recurrence risk, particularly in overweight patients
Ketorolac administered during primary tumor surgery may cut risk of distant recurrences in patients with breast cancer, results of a retrospective study show.
Overweight patients appeared most likely to benefit from interoperative treatment with this nonsteroidal anti-inflammatory drug, study investigators reported.
“This approach could be extremely appealing for parts of the globe where obesity has been strongly increasing during the last decade and where resources for cancer treatment are scarce,” they wrote. The report was published in the Journal of the National Cancer Institute.
Ketorolac inhibits enzymes upregulated by leptin, a hormone abnormally secreted in the setting of overweight or obesity, which might explain the concentration of benefit in high–body mass index individuals, noted Christine Desmedt, PhD, of the Breast Cancer Translational Research Laboratory, Institut Jules Bordet, Brussels, and her coauthors.
Indeed, the study also showed no benefit to intraoperative administration of another NSAID, diclofenac, which does not appear to have the same enzyme-inhibitory effects as ketorolac, the investigators said.
This recently published analysis by Dr. Desmedt and her colleagues was based on two retrospective series of patients: one evaluating intraoperative ketorolac in 529 patients versus 298 patients who received no ketorolac, and one evaluating intraoperative diclofenac in 787 patients, versus 220 who did not receive that NSAID.
The investigators found a significant association between ketorolac given during surgery and decreased incidence of distant metastasis (adjusted hazard ratio [aHR], 0.59, 95% confidence interval, 0.37-0.96, P = .03). Reduced recurrence was most evident in patients with high BMI (aHR, 0.55; 95% CI, 0.31-0.96; P = .04).
Further evaluation revealed that the benefit of ketorolac was “clearly associated” with a reduction in early metastases, both overall and in the high-BMI subgroup, the investigators said.
By contrast, intraoperative diclofenac was not associated with a decrease in distant recurrences, overall (adjusted HR, 1.04; 95% CI, 0.58-1.87, P = .88) or in BMI subgroup analysis, investigators said.
While some might be surprised that a single dose of ketorolac could reduce distant recurrence, it might be explained by the timing of NSAID delivery, they noted. In previous studies, primary tumor removal has been shown to disturb disease homeostasis, and thus might trigger early recurrences.
“Complex system dynamics are exquisitely sensitive on initial conditions, and, therefore, changes occurring in critical early times may be able to cause major changes in system evolution,” the investigators wrote in a discussion of the results.
The finding is also not without precedent. The authors cited one Scandinavian randomized trial in which a single course of perioperative cyclophosphamide significantly improved disease-free survival at more than 17 years of follow-up; by contrast, giving the treatment 2-4 weeks after mastectomy provided no such benefit.
In addition, ketorolac’s potential perioperative benefit has been shown in other tumor types, including improved disease-free survival in one institutional series of lung cancer patients, and reduced disease-specific mortality in a retrospective study of ovarian cancer patients.
The present breast cancer study is limited because it is retrospective, and does not address questions regarding optimal timing or duration of dose. However, “it suggests a potentially important repositioning of ketorolac in the intraoperative treatment of breast cancer patients with elevated BMI, and points to the need for a prospective confirmatory randomized trial,” the authors said.
Dr. Desmedt and her colleagues reported no conflicts of interest related to the study.
SOURCE: Desmedt C et al. J Natl Cancer Inst. 2018 Apr 30. doi: 10.1093/jnci/djy042.
Ketorolac administered during primary tumor surgery may cut risk of distant recurrences in patients with breast cancer, results of a retrospective study show.
Overweight patients appeared most likely to benefit from interoperative treatment with this nonsteroidal anti-inflammatory drug, study investigators reported.
“This approach could be extremely appealing for parts of the globe where obesity has been strongly increasing during the last decade and where resources for cancer treatment are scarce,” they wrote. The report was published in the Journal of the National Cancer Institute.
Ketorolac inhibits enzymes upregulated by leptin, a hormone abnormally secreted in the setting of overweight or obesity, which might explain the concentration of benefit in high–body mass index individuals, noted Christine Desmedt, PhD, of the Breast Cancer Translational Research Laboratory, Institut Jules Bordet, Brussels, and her coauthors.
Indeed, the study also showed no benefit to intraoperative administration of another NSAID, diclofenac, which does not appear to have the same enzyme-inhibitory effects as ketorolac, the investigators said.
This recently published analysis by Dr. Desmedt and her colleagues was based on two retrospective series of patients: one evaluating intraoperative ketorolac in 529 patients versus 298 patients who received no ketorolac, and one evaluating intraoperative diclofenac in 787 patients, versus 220 who did not receive that NSAID.
The investigators found a significant association between ketorolac given during surgery and decreased incidence of distant metastasis (adjusted hazard ratio [aHR], 0.59, 95% confidence interval, 0.37-0.96, P = .03). Reduced recurrence was most evident in patients with high BMI (aHR, 0.55; 95% CI, 0.31-0.96; P = .04).
Further evaluation revealed that the benefit of ketorolac was “clearly associated” with a reduction in early metastases, both overall and in the high-BMI subgroup, the investigators said.
By contrast, intraoperative diclofenac was not associated with a decrease in distant recurrences, overall (adjusted HR, 1.04; 95% CI, 0.58-1.87, P = .88) or in BMI subgroup analysis, investigators said.
While some might be surprised that a single dose of ketorolac could reduce distant recurrence, it might be explained by the timing of NSAID delivery, they noted. In previous studies, primary tumor removal has been shown to disturb disease homeostasis, and thus might trigger early recurrences.
“Complex system dynamics are exquisitely sensitive on initial conditions, and, therefore, changes occurring in critical early times may be able to cause major changes in system evolution,” the investigators wrote in a discussion of the results.
The finding is also not without precedent. The authors cited one Scandinavian randomized trial in which a single course of perioperative cyclophosphamide significantly improved disease-free survival at more than 17 years of follow-up; by contrast, giving the treatment 2-4 weeks after mastectomy provided no such benefit.
In addition, ketorolac’s potential perioperative benefit has been shown in other tumor types, including improved disease-free survival in one institutional series of lung cancer patients, and reduced disease-specific mortality in a retrospective study of ovarian cancer patients.
The present breast cancer study is limited because it is retrospective, and does not address questions regarding optimal timing or duration of dose. However, “it suggests a potentially important repositioning of ketorolac in the intraoperative treatment of breast cancer patients with elevated BMI, and points to the need for a prospective confirmatory randomized trial,” the authors said.
Dr. Desmedt and her colleagues reported no conflicts of interest related to the study.
SOURCE: Desmedt C et al. J Natl Cancer Inst. 2018 Apr 30. doi: 10.1093/jnci/djy042.
Ketorolac administered during primary tumor surgery may cut risk of distant recurrences in patients with breast cancer, results of a retrospective study show.
Overweight patients appeared most likely to benefit from interoperative treatment with this nonsteroidal anti-inflammatory drug, study investigators reported.
“This approach could be extremely appealing for parts of the globe where obesity has been strongly increasing during the last decade and where resources for cancer treatment are scarce,” they wrote. The report was published in the Journal of the National Cancer Institute.
Ketorolac inhibits enzymes upregulated by leptin, a hormone abnormally secreted in the setting of overweight or obesity, which might explain the concentration of benefit in high–body mass index individuals, noted Christine Desmedt, PhD, of the Breast Cancer Translational Research Laboratory, Institut Jules Bordet, Brussels, and her coauthors.
Indeed, the study also showed no benefit to intraoperative administration of another NSAID, diclofenac, which does not appear to have the same enzyme-inhibitory effects as ketorolac, the investigators said.
This recently published analysis by Dr. Desmedt and her colleagues was based on two retrospective series of patients: one evaluating intraoperative ketorolac in 529 patients versus 298 patients who received no ketorolac, and one evaluating intraoperative diclofenac in 787 patients, versus 220 who did not receive that NSAID.
The investigators found a significant association between ketorolac given during surgery and decreased incidence of distant metastasis (adjusted hazard ratio [aHR], 0.59, 95% confidence interval, 0.37-0.96, P = .03). Reduced recurrence was most evident in patients with high BMI (aHR, 0.55; 95% CI, 0.31-0.96; P = .04).
Further evaluation revealed that the benefit of ketorolac was “clearly associated” with a reduction in early metastases, both overall and in the high-BMI subgroup, the investigators said.
By contrast, intraoperative diclofenac was not associated with a decrease in distant recurrences, overall (adjusted HR, 1.04; 95% CI, 0.58-1.87, P = .88) or in BMI subgroup analysis, investigators said.
While some might be surprised that a single dose of ketorolac could reduce distant recurrence, it might be explained by the timing of NSAID delivery, they noted. In previous studies, primary tumor removal has been shown to disturb disease homeostasis, and thus might trigger early recurrences.
“Complex system dynamics are exquisitely sensitive on initial conditions, and, therefore, changes occurring in critical early times may be able to cause major changes in system evolution,” the investigators wrote in a discussion of the results.
The finding is also not without precedent. The authors cited one Scandinavian randomized trial in which a single course of perioperative cyclophosphamide significantly improved disease-free survival at more than 17 years of follow-up; by contrast, giving the treatment 2-4 weeks after mastectomy provided no such benefit.
In addition, ketorolac’s potential perioperative benefit has been shown in other tumor types, including improved disease-free survival in one institutional series of lung cancer patients, and reduced disease-specific mortality in a retrospective study of ovarian cancer patients.
The present breast cancer study is limited because it is retrospective, and does not address questions regarding optimal timing or duration of dose. However, “it suggests a potentially important repositioning of ketorolac in the intraoperative treatment of breast cancer patients with elevated BMI, and points to the need for a prospective confirmatory randomized trial,” the authors said.
Dr. Desmedt and her colleagues reported no conflicts of interest related to the study.
SOURCE: Desmedt C et al. J Natl Cancer Inst. 2018 Apr 30. doi: 10.1093/jnci/djy042.
FROM THE JOURNAL OF THE NATIONAL CANCER INSTITUTE
Key clinical point: Administration of ketorolac during primary tumor surgery was associated with a reduction of distant recurrences, particularly in overweight patients.
Major finding: Reduced recurrence was most evident in patients with high BMI (adjusted hazard ratio, 0.55; 95% CI, 0.31-0.96; P = .04).
Study details: Analysis of two retrospective series, including a total of 1,834 patients with breast cancer, evaluating intraoperative administration of ketorolac or diclofenac.
Disclosures: The authors declared no conflicts of interest.
Source: Desmedt C et al. J Natl Cancer Inst. 2018 Apr 30. doi: 10.1093/jnci/djy042.
Hospital safety program curbs surgical site infections
The Agency for Healthcare Research and Quality (AHRQ) designed the program to reduce surgical site infections (SSIs), which are harmful to patients and expensive for the health care system, wrote Della M. Lin, MD, of Johns Hopkins University, Baltimore, and the department of surgery at the University of Hawaii, Honolulu, and her colleagues.
In a study published in the Journal of the American College of Surgeons, the researchers reviewed data from a statewide intervention conducted at 15 hospitals in Hawaii from January 2013 to June 2015. The intervention included the Comprehensive Unit-based Safety Program and individualized interventions for each hospital to help reduce SSIs. The primary outcome was the number of colorectal SSIs. A secondary outcome of hospital safety culture was assessed using the AHRQ Hospital Survey on Patient Safety Culture. The participating hospitals ranged from a 25-bed critical-access hospital to a 533-bed academic medical center.
Overall, the colorectal SSI rate decreased significantly (from 12% to 5%) from the first quarter of 2013 to the second quarter of 2015, with a significant linear decrease over the study period. The rate of superficial SSIs decreased significantly, falling from 8% to 3%. However, the rate of deep SSIs was not significantly different before and after the intervention program (2% vs. 0%), nor was the organ space SSI rate (3% vs. 2%). The standardized infection ratio decreased from 1.83 to 0.92.
The culture of safety in the hospitals improved, but more modestly, in 10 of 12 areas that were measured over the study period.
The overall perception of patient safety improved from 49% to 53%, teamwork across different units improved from 49% to 54%, management and support for patient safety improved from 53% to 60%, and nonpunitive response to errors improved from 36% to 40%.
In addition, communication and openness improved from 50% to 53%, frequency of reported events improved from 51% to 60%, feedback and communication about errors improved from 52% to 59%, organizational learning and continuous improvement increased from 59% to 70%, teamwork within units improved from 68% to 75%, and expectations and actions by supervisors and managers to promote safety improved from 58% to 64%. Staff responses reflect agreement on improvement in the areas of issues of communication, feedback mechanisms, and teamwork, but the change in culture was not on the order of the SSI change.
The most common interventions to reduce SSIs were the use of reliable chlorhexidine wash or wipe before surgery/surgical prep; appropriate use of antibiotics with respect to selection, dosage, and timing; standardized postsurgical debriefing; and differentiating clean/dirty/clean in the use of anastomosis trays and closing trays.
One bundle component, the implementation of the standard operating room debrief, was found to be of particular value to participants. The investigators noted that debrief questions such as “What went well?” and “What needs to be improved?” had “encouraged new processes of thinking beyond first-order problem solving. The debrief challenge embraced by the teams emphasized that ‘bundles’ did not consist of only technical interventions [e.g. clean/dirty trays, chlorhexidine gluconate wipes in preop], but embedded culture interventions—new processes for problem solving.”
The study findings were limited by several factors, such as the use of public SSI data that were not audited for accuracy and the inability to monitor the reliability of the implementation of the various interventions, the researchers said. In addition, “In this current study, there was a change in SSI rates and a change in safety culture, but correlations between the two were negligible or weak for most domains of safety culture,” they noted. The question of sustainability of the SSI improvement without the concomitant staff support of culture change was not addressed by the investigators.
However, the results suggest that a 62% decrease is robust, and that for some hospitals with a low volume of colorectal cases, “teams could attend to iteratively reduce surgical harm beyond SSI,” the researchers wrote.
The study was supported in part by the AHRQ. Dr. Lin disclosed serving as a board member and as a paid independent contractor to the Hawaii Medical Service Association. Her coauthors had no financial conflicts to disclose.
SOURCE: Lin DM et al. J Am Coll Surg. 2018 May 18. doi: 10.1016/j.jamcollsurg.2018.04.031.
The Agency for Healthcare Research and Quality (AHRQ) designed the program to reduce surgical site infections (SSIs), which are harmful to patients and expensive for the health care system, wrote Della M. Lin, MD, of Johns Hopkins University, Baltimore, and the department of surgery at the University of Hawaii, Honolulu, and her colleagues.
In a study published in the Journal of the American College of Surgeons, the researchers reviewed data from a statewide intervention conducted at 15 hospitals in Hawaii from January 2013 to June 2015. The intervention included the Comprehensive Unit-based Safety Program and individualized interventions for each hospital to help reduce SSIs. The primary outcome was the number of colorectal SSIs. A secondary outcome of hospital safety culture was assessed using the AHRQ Hospital Survey on Patient Safety Culture. The participating hospitals ranged from a 25-bed critical-access hospital to a 533-bed academic medical center.
Overall, the colorectal SSI rate decreased significantly (from 12% to 5%) from the first quarter of 2013 to the second quarter of 2015, with a significant linear decrease over the study period. The rate of superficial SSIs decreased significantly, falling from 8% to 3%. However, the rate of deep SSIs was not significantly different before and after the intervention program (2% vs. 0%), nor was the organ space SSI rate (3% vs. 2%). The standardized infection ratio decreased from 1.83 to 0.92.
The culture of safety in the hospitals improved, but more modestly, in 10 of 12 areas that were measured over the study period.
The overall perception of patient safety improved from 49% to 53%, teamwork across different units improved from 49% to 54%, management and support for patient safety improved from 53% to 60%, and nonpunitive response to errors improved from 36% to 40%.
In addition, communication and openness improved from 50% to 53%, frequency of reported events improved from 51% to 60%, feedback and communication about errors improved from 52% to 59%, organizational learning and continuous improvement increased from 59% to 70%, teamwork within units improved from 68% to 75%, and expectations and actions by supervisors and managers to promote safety improved from 58% to 64%. Staff responses reflect agreement on improvement in the areas of issues of communication, feedback mechanisms, and teamwork, but the change in culture was not on the order of the SSI change.
The most common interventions to reduce SSIs were the use of reliable chlorhexidine wash or wipe before surgery/surgical prep; appropriate use of antibiotics with respect to selection, dosage, and timing; standardized postsurgical debriefing; and differentiating clean/dirty/clean in the use of anastomosis trays and closing trays.
One bundle component, the implementation of the standard operating room debrief, was found to be of particular value to participants. The investigators noted that debrief questions such as “What went well?” and “What needs to be improved?” had “encouraged new processes of thinking beyond first-order problem solving. The debrief challenge embraced by the teams emphasized that ‘bundles’ did not consist of only technical interventions [e.g. clean/dirty trays, chlorhexidine gluconate wipes in preop], but embedded culture interventions—new processes for problem solving.”
The study findings were limited by several factors, such as the use of public SSI data that were not audited for accuracy and the inability to monitor the reliability of the implementation of the various interventions, the researchers said. In addition, “In this current study, there was a change in SSI rates and a change in safety culture, but correlations between the two were negligible or weak for most domains of safety culture,” they noted. The question of sustainability of the SSI improvement without the concomitant staff support of culture change was not addressed by the investigators.
However, the results suggest that a 62% decrease is robust, and that for some hospitals with a low volume of colorectal cases, “teams could attend to iteratively reduce surgical harm beyond SSI,” the researchers wrote.
The study was supported in part by the AHRQ. Dr. Lin disclosed serving as a board member and as a paid independent contractor to the Hawaii Medical Service Association. Her coauthors had no financial conflicts to disclose.
SOURCE: Lin DM et al. J Am Coll Surg. 2018 May 18. doi: 10.1016/j.jamcollsurg.2018.04.031.
The Agency for Healthcare Research and Quality (AHRQ) designed the program to reduce surgical site infections (SSIs), which are harmful to patients and expensive for the health care system, wrote Della M. Lin, MD, of Johns Hopkins University, Baltimore, and the department of surgery at the University of Hawaii, Honolulu, and her colleagues.
In a study published in the Journal of the American College of Surgeons, the researchers reviewed data from a statewide intervention conducted at 15 hospitals in Hawaii from January 2013 to June 2015. The intervention included the Comprehensive Unit-based Safety Program and individualized interventions for each hospital to help reduce SSIs. The primary outcome was the number of colorectal SSIs. A secondary outcome of hospital safety culture was assessed using the AHRQ Hospital Survey on Patient Safety Culture. The participating hospitals ranged from a 25-bed critical-access hospital to a 533-bed academic medical center.
Overall, the colorectal SSI rate decreased significantly (from 12% to 5%) from the first quarter of 2013 to the second quarter of 2015, with a significant linear decrease over the study period. The rate of superficial SSIs decreased significantly, falling from 8% to 3%. However, the rate of deep SSIs was not significantly different before and after the intervention program (2% vs. 0%), nor was the organ space SSI rate (3% vs. 2%). The standardized infection ratio decreased from 1.83 to 0.92.
The culture of safety in the hospitals improved, but more modestly, in 10 of 12 areas that were measured over the study period.
The overall perception of patient safety improved from 49% to 53%, teamwork across different units improved from 49% to 54%, management and support for patient safety improved from 53% to 60%, and nonpunitive response to errors improved from 36% to 40%.
In addition, communication and openness improved from 50% to 53%, frequency of reported events improved from 51% to 60%, feedback and communication about errors improved from 52% to 59%, organizational learning and continuous improvement increased from 59% to 70%, teamwork within units improved from 68% to 75%, and expectations and actions by supervisors and managers to promote safety improved from 58% to 64%. Staff responses reflect agreement on improvement in the areas of issues of communication, feedback mechanisms, and teamwork, but the change in culture was not on the order of the SSI change.
The most common interventions to reduce SSIs were the use of reliable chlorhexidine wash or wipe before surgery/surgical prep; appropriate use of antibiotics with respect to selection, dosage, and timing; standardized postsurgical debriefing; and differentiating clean/dirty/clean in the use of anastomosis trays and closing trays.
One bundle component, the implementation of the standard operating room debrief, was found to be of particular value to participants. The investigators noted that debrief questions such as “What went well?” and “What needs to be improved?” had “encouraged new processes of thinking beyond first-order problem solving. The debrief challenge embraced by the teams emphasized that ‘bundles’ did not consist of only technical interventions [e.g. clean/dirty trays, chlorhexidine gluconate wipes in preop], but embedded culture interventions—new processes for problem solving.”
The study findings were limited by several factors, such as the use of public SSI data that were not audited for accuracy and the inability to monitor the reliability of the implementation of the various interventions, the researchers said. In addition, “In this current study, there was a change in SSI rates and a change in safety culture, but correlations between the two were negligible or weak for most domains of safety culture,” they noted. The question of sustainability of the SSI improvement without the concomitant staff support of culture change was not addressed by the investigators.
However, the results suggest that a 62% decrease is robust, and that for some hospitals with a low volume of colorectal cases, “teams could attend to iteratively reduce surgical harm beyond SSI,” the researchers wrote.
The study was supported in part by the AHRQ. Dr. Lin disclosed serving as a board member and as a paid independent contractor to the Hawaii Medical Service Association. Her coauthors had no financial conflicts to disclose.
SOURCE: Lin DM et al. J Am Coll Surg. 2018 May 18. doi: 10.1016/j.jamcollsurg.2018.04.031.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF SURGEONS
Key clinical point: Hospital participation in an Agency for Healthcare Research and Quality safety program improved safety culture and reduced surgical site infections.
Major finding: Surgical site infections among colorectal surgery patients decreased by 61.7% after the intervention.
Study details: The data come from a cohort study of 15 hospitals in Hawaii from January 2013 to June 2015.
Disclosures: The study was supported in part by the AHRQ. Dr. Lin disclosed serving as a board member and as a paid independent contractor to the Hawaii Medical Service Association. Her coauthors had no financial conflicts to disclose.
Source: Lin DM et al. J Am Coll Surg. 2018 May 18. doi: 10.1016/j.jamcollsurg.2018.04.031.
Clinician denial of some patient requests decrease patient satisfaction
Background: Literature regarding patient satisfaction often focuses on nonspecific recommendations to improve patient-centered communication. There is lack of guidance on concrete advice for clinicians, particularly with regard to how a provider’s responses to different patient requests are received.
Study design: Cross-sectional study.
Setting: An outpatient family medicine clinic.
Synopsis: Patient requests from 1,141 patients visiting the University of California, Davis, Family Medicine Clinic were sampled. The study examined clinician’s approval or denial of patients’ requests for referrals, pain medications, other new medicines, laboratory testing, radiology testing, or other testing and the patients’ reported satisfaction of the clinician.
Clinician denial of particular requests was associated with decreased patient satisfaction. Specifically, a 19.75% drop for referral, 10.72% drop for pain medication, 20.36% drop for other new medications, and 9.19% drop for laboratory test. This study did not examine other potential reasons for decreased satisfaction.
Bottom line: Clinicians can better understand how to communicate in a patient-centered manner by understanding that not all patient requests are perceived as equal.
Citation: Jerant A et al. Association of clinical denial of patient requests with patient satisfaction. JAMA Intern Med. 2018 Jan 1;178(1):85-91.
Dr. Shaffie is a hospitalist at Denver Health Medical Center and an assistant professor of medicine at the University of Colorado at Denver, Aurora.
Background: Literature regarding patient satisfaction often focuses on nonspecific recommendations to improve patient-centered communication. There is lack of guidance on concrete advice for clinicians, particularly with regard to how a provider’s responses to different patient requests are received.
Study design: Cross-sectional study.
Setting: An outpatient family medicine clinic.
Synopsis: Patient requests from 1,141 patients visiting the University of California, Davis, Family Medicine Clinic were sampled. The study examined clinician’s approval or denial of patients’ requests for referrals, pain medications, other new medicines, laboratory testing, radiology testing, or other testing and the patients’ reported satisfaction of the clinician.
Clinician denial of particular requests was associated with decreased patient satisfaction. Specifically, a 19.75% drop for referral, 10.72% drop for pain medication, 20.36% drop for other new medications, and 9.19% drop for laboratory test. This study did not examine other potential reasons for decreased satisfaction.
Bottom line: Clinicians can better understand how to communicate in a patient-centered manner by understanding that not all patient requests are perceived as equal.
Citation: Jerant A et al. Association of clinical denial of patient requests with patient satisfaction. JAMA Intern Med. 2018 Jan 1;178(1):85-91.
Dr. Shaffie is a hospitalist at Denver Health Medical Center and an assistant professor of medicine at the University of Colorado at Denver, Aurora.
Background: Literature regarding patient satisfaction often focuses on nonspecific recommendations to improve patient-centered communication. There is lack of guidance on concrete advice for clinicians, particularly with regard to how a provider’s responses to different patient requests are received.
Study design: Cross-sectional study.
Setting: An outpatient family medicine clinic.
Synopsis: Patient requests from 1,141 patients visiting the University of California, Davis, Family Medicine Clinic were sampled. The study examined clinician’s approval or denial of patients’ requests for referrals, pain medications, other new medicines, laboratory testing, radiology testing, or other testing and the patients’ reported satisfaction of the clinician.
Clinician denial of particular requests was associated with decreased patient satisfaction. Specifically, a 19.75% drop for referral, 10.72% drop for pain medication, 20.36% drop for other new medications, and 9.19% drop for laboratory test. This study did not examine other potential reasons for decreased satisfaction.
Bottom line: Clinicians can better understand how to communicate in a patient-centered manner by understanding that not all patient requests are perceived as equal.
Citation: Jerant A et al. Association of clinical denial of patient requests with patient satisfaction. JAMA Intern Med. 2018 Jan 1;178(1):85-91.
Dr. Shaffie is a hospitalist at Denver Health Medical Center and an assistant professor of medicine at the University of Colorado at Denver, Aurora.
