User login
Debunking Atopic Dermatitis Myths: Do Most Children Outgrow Atopic Dermatitis?
Myth: Children eventually outgrow atopic dermatitis and therefore do not need treatment
The negative impact of atopic dermatitis (AD) on quality of life in the pediatric population often prompts parents/guardians to inquire about whether a child with AD will ever outgrow their disease. If remission is expected as the child gets older, many may question if it is necessary to pursue treatment or just let the disease run its course. Although AD often is reported to resolve soon after the first decade of life, symptoms can persist well into the second decade and beyond, suggesting that AD may be a lifelong disease with periods of waxing and waning symptoms that require persistent treatment throughout the patient’s life.
A 2014 study included 7157 children with AD (mean age of disease onset, 1.7 years) who were enrolled in the Pediatric Eczema Elective Registry (PEER) program between the ages of 2 and 17 years with measurement of disease activity at regular 6-month intervals for up to 5 years. The study results indicated that more than 80% of patients at every age (age range, 2–26 years) had symptoms of AD and/or were using medication to treat their disease, and the majority (64%) of patients had never reported a 6-month period during which they achieved clearance of symptoms without medication. At the age of 20 years, 50% of patients reported at least 1 lifetime 6-month period during which they were both symptom and treatment free. In another study of adolescents with AD who also had AD in childhood (N=82), 48% of patients remained in the same AD severity grades and 13% deteriorated from childhood to adolescence; only 39% of patients showed improvement in disease severity from childhood to adolescence. The findings of these reports are contradictory to conventional clinical teaching, which indicates that AD generally resolves by age 12 in 50% to 70% of children.
Even though some children with AD may experience periods of disease clearance, these findings often do not persist and should not be confused with a permanent remission. Most patients require continued treatment with medications to achieve relief of symptoms. Therefore, physicians should not assure parents/guardians that a child can outgrow AD; rather, they should educate pediatric patients and their caregivers about the potentially lifelong disease course and encourage early intervention to mitigate symptoms and manage comorbidities as the patient ages.
Hon KL, Tsang YCK, Poon TCW, et al. Predicating eczema severity beyond childhood. World J Pediatr. 2016;12:44-48.
Margolis JS, Abuabara K, Bilker W, et al. Persistence of mild to moderate atopic dermatitis [published online April 2, 2014]. JAMA Dermatol. 2014;150:593-600.
Myth: Children eventually outgrow atopic dermatitis and therefore do not need treatment
The negative impact of atopic dermatitis (AD) on quality of life in the pediatric population often prompts parents/guardians to inquire about whether a child with AD will ever outgrow their disease. If remission is expected as the child gets older, many may question if it is necessary to pursue treatment or just let the disease run its course. Although AD often is reported to resolve soon after the first decade of life, symptoms can persist well into the second decade and beyond, suggesting that AD may be a lifelong disease with periods of waxing and waning symptoms that require persistent treatment throughout the patient’s life.
A 2014 study included 7157 children with AD (mean age of disease onset, 1.7 years) who were enrolled in the Pediatric Eczema Elective Registry (PEER) program between the ages of 2 and 17 years with measurement of disease activity at regular 6-month intervals for up to 5 years. The study results indicated that more than 80% of patients at every age (age range, 2–26 years) had symptoms of AD and/or were using medication to treat their disease, and the majority (64%) of patients had never reported a 6-month period during which they achieved clearance of symptoms without medication. At the age of 20 years, 50% of patients reported at least 1 lifetime 6-month period during which they were both symptom and treatment free. In another study of adolescents with AD who also had AD in childhood (N=82), 48% of patients remained in the same AD severity grades and 13% deteriorated from childhood to adolescence; only 39% of patients showed improvement in disease severity from childhood to adolescence. The findings of these reports are contradictory to conventional clinical teaching, which indicates that AD generally resolves by age 12 in 50% to 70% of children.
Even though some children with AD may experience periods of disease clearance, these findings often do not persist and should not be confused with a permanent remission. Most patients require continued treatment with medications to achieve relief of symptoms. Therefore, physicians should not assure parents/guardians that a child can outgrow AD; rather, they should educate pediatric patients and their caregivers about the potentially lifelong disease course and encourage early intervention to mitigate symptoms and manage comorbidities as the patient ages.
Myth: Children eventually outgrow atopic dermatitis and therefore do not need treatment
The negative impact of atopic dermatitis (AD) on quality of life in the pediatric population often prompts parents/guardians to inquire about whether a child with AD will ever outgrow their disease. If remission is expected as the child gets older, many may question if it is necessary to pursue treatment or just let the disease run its course. Although AD often is reported to resolve soon after the first decade of life, symptoms can persist well into the second decade and beyond, suggesting that AD may be a lifelong disease with periods of waxing and waning symptoms that require persistent treatment throughout the patient’s life.
A 2014 study included 7157 children with AD (mean age of disease onset, 1.7 years) who were enrolled in the Pediatric Eczema Elective Registry (PEER) program between the ages of 2 and 17 years with measurement of disease activity at regular 6-month intervals for up to 5 years. The study results indicated that more than 80% of patients at every age (age range, 2–26 years) had symptoms of AD and/or were using medication to treat their disease, and the majority (64%) of patients had never reported a 6-month period during which they achieved clearance of symptoms without medication. At the age of 20 years, 50% of patients reported at least 1 lifetime 6-month period during which they were both symptom and treatment free. In another study of adolescents with AD who also had AD in childhood (N=82), 48% of patients remained in the same AD severity grades and 13% deteriorated from childhood to adolescence; only 39% of patients showed improvement in disease severity from childhood to adolescence. The findings of these reports are contradictory to conventional clinical teaching, which indicates that AD generally resolves by age 12 in 50% to 70% of children.
Even though some children with AD may experience periods of disease clearance, these findings often do not persist and should not be confused with a permanent remission. Most patients require continued treatment with medications to achieve relief of symptoms. Therefore, physicians should not assure parents/guardians that a child can outgrow AD; rather, they should educate pediatric patients and their caregivers about the potentially lifelong disease course and encourage early intervention to mitigate symptoms and manage comorbidities as the patient ages.
Hon KL, Tsang YCK, Poon TCW, et al. Predicating eczema severity beyond childhood. World J Pediatr. 2016;12:44-48.
Margolis JS, Abuabara K, Bilker W, et al. Persistence of mild to moderate atopic dermatitis [published online April 2, 2014]. JAMA Dermatol. 2014;150:593-600.
Hon KL, Tsang YCK, Poon TCW, et al. Predicating eczema severity beyond childhood. World J Pediatr. 2016;12:44-48.
Margolis JS, Abuabara K, Bilker W, et al. Persistence of mild to moderate atopic dermatitis [published online April 2, 2014]. JAMA Dermatol. 2014;150:593-600.

A refined strategy for confirming diagnosis in suspected NSTEMI
ORLANDO – A novel diagnostic strategy of performing CT angiography or cardiovascular MRI first in patients with suspected non-ST-elevation MI safely improved appropriate selection for invasive coronary angiography in the Dutch randomized CARMENTA trial.
The strategy of using noninvasive imaging first significantly cut down on the high proportion of diagnostic invasive angiography procedures that end up showing no significant obstructive coronary artery disease in the current era of high-sensitivity cardiac troponin assays, Martijn W. Smulders, MD, reported at the annual meeting of the American College of Cardiology.

CARMENTA (Cardiovascular Magnetic Resonance Imaging and Computed Tomography Angiography) was a single-center, prospective, randomized trial including 207 patients with suspected NSTEMI on the basis of acute chest pain, an elevated high-sensitivity cardiac troponin level, and an inconclusive ECG. They were randomized to one of three diagnostic strategies: a routine invasive strategy in which they were sent straight to the cardiac catheterization lab for invasive coronary angiography, or either CTA- or CMR-first as gatekeeper strategies in which referral for invasive angiography was reserved for only those patients whose noninvasive imaging demonstrated myocardial ischemia, infarction, or obstructive CAD with at least a 70% stenosis.
The impetus for the trial was the investigators’ concern that widespread embrace of high-sensitivity cardiac troponin assays has resulted in a serious clinical problem: Although these assays offer very high sensitivity for rapid detection of acute MI, their positive predictive value is only 56%, compared with 76% for the older troponin assays.
“That means almost one out of two patients with acute chest pain and an elevated high-sensitivity troponin level does not have a type 1 MI. We see a twofold higher incidence of elevated troponin levels with these assays, so there has been a significant increase in referrals for invasive angiography – and up to one-third of these patients with suspected NSTEMI don’t have an obstructive stenosis. We need a strategy to improve patient selection,” explained Dr. Smulders of Maastricht (the Netherlands) University.
The CARMENTA strategy worked. The primary outcome – the proportion of patients with suspected NSTEMI who underwent invasive coronary angiography during their initial hospitalization – was 65% in the CTA-first group and 77% in the CMR group, compared with 100% in the routine invasive-strategy control group. Moreover, fully 38% of patients in the control group turned out not to have obstructive CAD, compared with 15% who were sent for invasive angiography only after CTA and 31% who first had CMR.
Procedure-related complications, a secondary outcome, occurred in 12% of the CMR-first group, 13% of the CTA-first group, and 16% of patients in the routine invasive strategy control group. Major adverse cardiac events during 1 year of follow-up, which was the other secondary outcome, occurred in 9% of the CMR group, 6% of the CTA group, and 9% of the control group.
A limitation of the CARMENTA trial was that, even though it was scheduled to enroll 288 patients to achieve strong statistical power, the study’s data safety monitoring committee recommended on the basis of an interim analysis that the trial be halted early. The reasoning was that the experience of the first 200 enrollees made it clear that the noninvasive-imaging-first strategy would achieve the goal of reducing the volume of referrals to invasive angiography for suspected NSTEMI.
Session cochair Stefan D. Anker, MD, was irked by the trial’s early termination, which weakened the strength of the conclusions, especially with regard to the safety of the novel strategy.
“I agree that this imaging-first strategy reduces procedures, but the use of the word ‘safely’ is premature,” said Dr. Anker, professor of homeostasis and cachexia at Charite Medical School in Berlin.
“I totally agree with you,” Dr. Smulders replied. “We need a bigger trial to confirm our results – preferably a multicenter trial.”
“How can you do a bigger trial when your data safety monitoring board didn’t allow you to complete even this trial? They killed your trial. That’s the way I see it,” Dr. Anker said.
The CARMENTA trial was funded by the Dutch Heart Foundation. Dr. Smulders reported having no financial conflicts of interest.
SOURCE: Smulders M. ACC 18.
ORLANDO – A novel diagnostic strategy of performing CT angiography or cardiovascular MRI first in patients with suspected non-ST-elevation MI safely improved appropriate selection for invasive coronary angiography in the Dutch randomized CARMENTA trial.
The strategy of using noninvasive imaging first significantly cut down on the high proportion of diagnostic invasive angiography procedures that end up showing no significant obstructive coronary artery disease in the current era of high-sensitivity cardiac troponin assays, Martijn W. Smulders, MD, reported at the annual meeting of the American College of Cardiology.
CARMENTA (Cardiovascular Magnetic Resonance Imaging and Computed Tomography Angiography) was a single-center, prospective, randomized trial including 207 patients with suspected NSTEMI on the basis of acute chest pain, an elevated high-sensitivity cardiac troponin level, and an inconclusive ECG. They were randomized to one of three diagnostic strategies: a routine invasive strategy in which they were sent straight to the cardiac catheterization lab for invasive coronary angiography, or either CTA- or CMR-first as gatekeeper strategies in which referral for invasive angiography was reserved for only those patients whose noninvasive imaging demonstrated myocardial ischemia, infarction, or obstructive CAD with at least a 70% stenosis.
The impetus for the trial was the investigators’ concern that widespread embrace of high-sensitivity cardiac troponin assays has resulted in a serious clinical problem: Although these assays offer very high sensitivity for rapid detection of acute MI, their positive predictive value is only 56%, compared with 76% for the older troponin assays.
“That means almost one out of two patients with acute chest pain and an elevated high-sensitivity troponin level does not have a type 1 MI. We see a twofold higher incidence of elevated troponin levels with these assays, so there has been a significant increase in referrals for invasive angiography – and up to one-third of these patients with suspected NSTEMI don’t have an obstructive stenosis. We need a strategy to improve patient selection,” explained Dr. Smulders of Maastricht (the Netherlands) University.
The CARMENTA strategy worked. The primary outcome – the proportion of patients with suspected NSTEMI who underwent invasive coronary angiography during their initial hospitalization – was 65% in the CTA-first group and 77% in the CMR group, compared with 100% in the routine invasive-strategy control group. Moreover, fully 38% of patients in the control group turned out not to have obstructive CAD, compared with 15% who were sent for invasive angiography only after CTA and 31% who first had CMR.
Procedure-related complications, a secondary outcome, occurred in 12% of the CMR-first group, 13% of the CTA-first group, and 16% of patients in the routine invasive strategy control group. Major adverse cardiac events during 1 year of follow-up, which was the other secondary outcome, occurred in 9% of the CMR group, 6% of the CTA group, and 9% of the control group.
A limitation of the CARMENTA trial was that, even though it was scheduled to enroll 288 patients to achieve strong statistical power, the study’s data safety monitoring committee recommended on the basis of an interim analysis that the trial be halted early. The reasoning was that the experience of the first 200 enrollees made it clear that the noninvasive-imaging-first strategy would achieve the goal of reducing the volume of referrals to invasive angiography for suspected NSTEMI.
Session cochair Stefan D. Anker, MD, was irked by the trial’s early termination, which weakened the strength of the conclusions, especially with regard to the safety of the novel strategy.
“I agree that this imaging-first strategy reduces procedures, but the use of the word ‘safely’ is premature,” said Dr. Anker, professor of homeostasis and cachexia at Charite Medical School in Berlin.
“I totally agree with you,” Dr. Smulders replied. “We need a bigger trial to confirm our results – preferably a multicenter trial.”
“How can you do a bigger trial when your data safety monitoring board didn’t allow you to complete even this trial? They killed your trial. That’s the way I see it,” Dr. Anker said.
The CARMENTA trial was funded by the Dutch Heart Foundation. Dr. Smulders reported having no financial conflicts of interest.
SOURCE: Smulders M. ACC 18.
ORLANDO – A novel diagnostic strategy of performing CT angiography or cardiovascular MRI first in patients with suspected non-ST-elevation MI safely improved appropriate selection for invasive coronary angiography in the Dutch randomized CARMENTA trial.
The strategy of using noninvasive imaging first significantly cut down on the high proportion of diagnostic invasive angiography procedures that end up showing no significant obstructive coronary artery disease in the current era of high-sensitivity cardiac troponin assays, Martijn W. Smulders, MD, reported at the annual meeting of the American College of Cardiology.
CARMENTA (Cardiovascular Magnetic Resonance Imaging and Computed Tomography Angiography) was a single-center, prospective, randomized trial including 207 patients with suspected NSTEMI on the basis of acute chest pain, an elevated high-sensitivity cardiac troponin level, and an inconclusive ECG. They were randomized to one of three diagnostic strategies: a routine invasive strategy in which they were sent straight to the cardiac catheterization lab for invasive coronary angiography, or either CTA- or CMR-first as gatekeeper strategies in which referral for invasive angiography was reserved for only those patients whose noninvasive imaging demonstrated myocardial ischemia, infarction, or obstructive CAD with at least a 70% stenosis.
The impetus for the trial was the investigators’ concern that widespread embrace of high-sensitivity cardiac troponin assays has resulted in a serious clinical problem: Although these assays offer very high sensitivity for rapid detection of acute MI, their positive predictive value is only 56%, compared with 76% for the older troponin assays.
“That means almost one out of two patients with acute chest pain and an elevated high-sensitivity troponin level does not have a type 1 MI. We see a twofold higher incidence of elevated troponin levels with these assays, so there has been a significant increase in referrals for invasive angiography – and up to one-third of these patients with suspected NSTEMI don’t have an obstructive stenosis. We need a strategy to improve patient selection,” explained Dr. Smulders of Maastricht (the Netherlands) University.
The CARMENTA strategy worked. The primary outcome – the proportion of patients with suspected NSTEMI who underwent invasive coronary angiography during their initial hospitalization – was 65% in the CTA-first group and 77% in the CMR group, compared with 100% in the routine invasive-strategy control group. Moreover, fully 38% of patients in the control group turned out not to have obstructive CAD, compared with 15% who were sent for invasive angiography only after CTA and 31% who first had CMR.
Procedure-related complications, a secondary outcome, occurred in 12% of the CMR-first group, 13% of the CTA-first group, and 16% of patients in the routine invasive strategy control group. Major adverse cardiac events during 1 year of follow-up, which was the other secondary outcome, occurred in 9% of the CMR group, 6% of the CTA group, and 9% of the control group.
A limitation of the CARMENTA trial was that, even though it was scheduled to enroll 288 patients to achieve strong statistical power, the study’s data safety monitoring committee recommended on the basis of an interim analysis that the trial be halted early. The reasoning was that the experience of the first 200 enrollees made it clear that the noninvasive-imaging-first strategy would achieve the goal of reducing the volume of referrals to invasive angiography for suspected NSTEMI.
Session cochair Stefan D. Anker, MD, was irked by the trial’s early termination, which weakened the strength of the conclusions, especially with regard to the safety of the novel strategy.
“I agree that this imaging-first strategy reduces procedures, but the use of the word ‘safely’ is premature,” said Dr. Anker, professor of homeostasis and cachexia at Charite Medical School in Berlin.
“I totally agree with you,” Dr. Smulders replied. “We need a bigger trial to confirm our results – preferably a multicenter trial.”
“How can you do a bigger trial when your data safety monitoring board didn’t allow you to complete even this trial? They killed your trial. That’s the way I see it,” Dr. Anker said.
The CARMENTA trial was funded by the Dutch Heart Foundation. Dr. Smulders reported having no financial conflicts of interest.
SOURCE: Smulders M. ACC 18.
REPORTING FROM ACC 18
Key clinical point: Dutch cardiologists have come up with a novel way to reduce the high rate of negative diagnostic coronary angiography in patients with suspected NSTEMI.
Major finding: Reserving invasive coronary angiography for only those patients with suspected NSTEMI who first showed positive findings on noninvasive CT angiography reduced invasive angiography volume by 35%.
Study details: This single-center, randomized, prospective, three-arm clinical trial included 207 patients with suspected NSTEMI.
Disclosures: The CARMENTA trial was funded by the Dutch Heart Foundation. The presenter reported having no financial conflicts of interest.
Source: Smulders M. ACC 18.
Commentary—Could Prazosin Play a Role in Treating Chronic Posttraumatic Headache?
Headache is a common symptom after any severity traumatic brain injury in the civilian and military populations. Currently, there is no evidence-based treatment protocol for posttraumatic headache, and management largely is based on therapies used in the primary headache disorders.
There is a complex interaction between mood disorders, posttraumatic stress disorder (PTSD), sleep disorders, and headache. Depression and PTSD are frequently seen in civilian and military populations accompanying chronic posttraumatic headache. In civilians, about one-third of patients with posttraumatic headache meet criteria for depression and PTSD. A longitudinal study of Iraq and Afghanistan veterans followed over three years found that co-occurrence of depression, PTSD, or both would increase the risk of chronic posttraumatic headache more than TBI alone. Another meta-analysis of civilian and military TBI found that, though PTSD could affect intensity and severity of chronic posttraumatic headache, TBI was an independent risk factor for chronic posttraumatic headache. PTSD and depression can cause sleep disruption and intensify pain syndromes, including headache.

Though prazosin had been shown to be effective in decreasing nightmares, improving sleep, or decreasing daytime sleepiness in many prior studies, the PACT trial, a randomized, double-blind controlled trial of 304 participants at Veterans Affairs medical centers, did not meet its primary end points of less frequent and less intense trauma-related nightmares, greater improvement in sleep quality, and overall clinical status among veterans assigned to prazosin, compared with veterans assigned to placebo. While disappointing, and surprising given the results of the preceding studies, do these results predict a similar failure in the use of prazosin for treatment of posttraumatic headache?
In an observational study of 126 veterans with blast-related mild TBI during Operation Iraqi Freedom or Operation Enduring Freedom, 82% of participants had co-occurring conditions, including frequent, severe headache, neurologic exam abnormalities, or cognitive disorders. This pilot study found that treatment with prazosin and sleep hygiene counseling improved sleep, but also decreased headache pain and frequency, as well as improved cognitive function over nine weeks. Improvements were maintained for six months. Though difficult to determine the interplay of sleep, posttraumatic headache, and depression, could prazosin independently reduce the burden of headache? Currently, a double-blind, randomized, controlled trial in veterans is examining the effectiveness of prazosin as a preventive agent in treating combat-related posttraumatic headache. This study was scheduled to enroll its last patient at the end of 2017, and results may be out soon.
There may be specific pharmacologic properties that make prazosin a useful drug for headache treatment. Prazosin is a very potent, selective alpha 1-adrenergic antagonist that passes through the blood–brain barrier. It is highly protein bound (97%), so absolute amounts in the CNS are likely to be low. Its use in the treatment of hypertension is based on decreased peripheral vascular resistance as a result of arteriolar and venous receptor blockade. It also can act in the CNS to decrease sympathetic outflow. While an effect on headache could be central, peripheral, or both, other drugs with alpha-adrenergic blocking effects have been used in the treatment of migraine for decades. The ergots, for example, were the first alpha-adrenergic agents to be discovered acting as partial agonists or antagonists at adrenergic, tryptaminergic, and dopaminergic receptors. The hydrogenated ergot alkaloids are among the most potent alpha-adrenergic blocking agents, but adverse effects prevent doses that can cause more than minimal blockade. Chlorpromazine and other dopamine (D2) receptor antagonists, which are highly effective in acute treatment of migraine, particularly with parenteral delivery, also produce significant alpha-adrenergic receptor blockade, while trazodone, amitriptyline, and the atypical antipsychotics, with various levels of alpha-adrenergic antagonism, have found some success in migraine prevention.
Clinical experience has shown that there is wide response variability to acute and chronic medication for migraine. Genetic studies of patients with migraine, though at an early stage, have identified genes involved with vascular and neuronal function. It is likely that clinical observation will be borne out by individual responses to drug classes based on individual genetic profiles, so that subtypes of patients in clinical trial populations may show efficacy based on these profiles. It is likely that prazosin will be useful in certain patient subtypes for the treatment of headache. Posttraumatic headache, which may share some similar pathways of headache physiology with primary headache disorders, adds another layer of response complexity.
—Sylvia Lucas, MD, PhD
Clinical Professor of Neurology and Neurological Surgery
University of Washington
Seattle
Suggested Reading
Nampiaparampil DE. Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA. 2008;300(6):711-719.
Peterlin BL, Nijjar SS, Tietjen GE. Post-traumatic stress disorder and migraine: epidemiology, sex differences, and potential mechanisms. Headache. 2011;51(6):860-868.
Ruff RL, Riechers RG, Wang XF, et al. For veterans with mild traumatic brain injury, improved posttraumatic stress disorder severity and sleep correlated with symptomatic improvement. J Rehabil Res Dev. 2012;49(9):1305-1320.
Headache is a common symptom after any severity traumatic brain injury in the civilian and military populations. Currently, there is no evidence-based treatment protocol for posttraumatic headache, and management largely is based on therapies used in the primary headache disorders.
There is a complex interaction between mood disorders, posttraumatic stress disorder (PTSD), sleep disorders, and headache. Depression and PTSD are frequently seen in civilian and military populations accompanying chronic posttraumatic headache. In civilians, about one-third of patients with posttraumatic headache meet criteria for depression and PTSD. A longitudinal study of Iraq and Afghanistan veterans followed over three years found that co-occurrence of depression, PTSD, or both would increase the risk of chronic posttraumatic headache more than TBI alone. Another meta-analysis of civilian and military TBI found that, though PTSD could affect intensity and severity of chronic posttraumatic headache, TBI was an independent risk factor for chronic posttraumatic headache. PTSD and depression can cause sleep disruption and intensify pain syndromes, including headache.
Though prazosin had been shown to be effective in decreasing nightmares, improving sleep, or decreasing daytime sleepiness in many prior studies, the PACT trial, a randomized, double-blind controlled trial of 304 participants at Veterans Affairs medical centers, did not meet its primary end points of less frequent and less intense trauma-related nightmares, greater improvement in sleep quality, and overall clinical status among veterans assigned to prazosin, compared with veterans assigned to placebo. While disappointing, and surprising given the results of the preceding studies, do these results predict a similar failure in the use of prazosin for treatment of posttraumatic headache?
In an observational study of 126 veterans with blast-related mild TBI during Operation Iraqi Freedom or Operation Enduring Freedom, 82% of participants had co-occurring conditions, including frequent, severe headache, neurologic exam abnormalities, or cognitive disorders. This pilot study found that treatment with prazosin and sleep hygiene counseling improved sleep, but also decreased headache pain and frequency, as well as improved cognitive function over nine weeks. Improvements were maintained for six months. Though difficult to determine the interplay of sleep, posttraumatic headache, and depression, could prazosin independently reduce the burden of headache? Currently, a double-blind, randomized, controlled trial in veterans is examining the effectiveness of prazosin as a preventive agent in treating combat-related posttraumatic headache. This study was scheduled to enroll its last patient at the end of 2017, and results may be out soon.
There may be specific pharmacologic properties that make prazosin a useful drug for headache treatment. Prazosin is a very potent, selective alpha 1-adrenergic antagonist that passes through the blood–brain barrier. It is highly protein bound (97%), so absolute amounts in the CNS are likely to be low. Its use in the treatment of hypertension is based on decreased peripheral vascular resistance as a result of arteriolar and venous receptor blockade. It also can act in the CNS to decrease sympathetic outflow. While an effect on headache could be central, peripheral, or both, other drugs with alpha-adrenergic blocking effects have been used in the treatment of migraine for decades. The ergots, for example, were the first alpha-adrenergic agents to be discovered acting as partial agonists or antagonists at adrenergic, tryptaminergic, and dopaminergic receptors. The hydrogenated ergot alkaloids are among the most potent alpha-adrenergic blocking agents, but adverse effects prevent doses that can cause more than minimal blockade. Chlorpromazine and other dopamine (D2) receptor antagonists, which are highly effective in acute treatment of migraine, particularly with parenteral delivery, also produce significant alpha-adrenergic receptor blockade, while trazodone, amitriptyline, and the atypical antipsychotics, with various levels of alpha-adrenergic antagonism, have found some success in migraine prevention.
Clinical experience has shown that there is wide response variability to acute and chronic medication for migraine. Genetic studies of patients with migraine, though at an early stage, have identified genes involved with vascular and neuronal function. It is likely that clinical observation will be borne out by individual responses to drug classes based on individual genetic profiles, so that subtypes of patients in clinical trial populations may show efficacy based on these profiles. It is likely that prazosin will be useful in certain patient subtypes for the treatment of headache. Posttraumatic headache, which may share some similar pathways of headache physiology with primary headache disorders, adds another layer of response complexity.
—Sylvia Lucas, MD, PhD
Clinical Professor of Neurology and Neurological Surgery
University of Washington
Seattle
Suggested Reading
Nampiaparampil DE. Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA. 2008;300(6):711-719.
Peterlin BL, Nijjar SS, Tietjen GE. Post-traumatic stress disorder and migraine: epidemiology, sex differences, and potential mechanisms. Headache. 2011;51(6):860-868.
Ruff RL, Riechers RG, Wang XF, et al. For veterans with mild traumatic brain injury, improved posttraumatic stress disorder severity and sleep correlated with symptomatic improvement. J Rehabil Res Dev. 2012;49(9):1305-1320.
Headache is a common symptom after any severity traumatic brain injury in the civilian and military populations. Currently, there is no evidence-based treatment protocol for posttraumatic headache, and management largely is based on therapies used in the primary headache disorders.
There is a complex interaction between mood disorders, posttraumatic stress disorder (PTSD), sleep disorders, and headache. Depression and PTSD are frequently seen in civilian and military populations accompanying chronic posttraumatic headache. In civilians, about one-third of patients with posttraumatic headache meet criteria for depression and PTSD. A longitudinal study of Iraq and Afghanistan veterans followed over three years found that co-occurrence of depression, PTSD, or both would increase the risk of chronic posttraumatic headache more than TBI alone. Another meta-analysis of civilian and military TBI found that, though PTSD could affect intensity and severity of chronic posttraumatic headache, TBI was an independent risk factor for chronic posttraumatic headache. PTSD and depression can cause sleep disruption and intensify pain syndromes, including headache.
Though prazosin had been shown to be effective in decreasing nightmares, improving sleep, or decreasing daytime sleepiness in many prior studies, the PACT trial, a randomized, double-blind controlled trial of 304 participants at Veterans Affairs medical centers, did not meet its primary end points of less frequent and less intense trauma-related nightmares, greater improvement in sleep quality, and overall clinical status among veterans assigned to prazosin, compared with veterans assigned to placebo. While disappointing, and surprising given the results of the preceding studies, do these results predict a similar failure in the use of prazosin for treatment of posttraumatic headache?
In an observational study of 126 veterans with blast-related mild TBI during Operation Iraqi Freedom or Operation Enduring Freedom, 82% of participants had co-occurring conditions, including frequent, severe headache, neurologic exam abnormalities, or cognitive disorders. This pilot study found that treatment with prazosin and sleep hygiene counseling improved sleep, but also decreased headache pain and frequency, as well as improved cognitive function over nine weeks. Improvements were maintained for six months. Though difficult to determine the interplay of sleep, posttraumatic headache, and depression, could prazosin independently reduce the burden of headache? Currently, a double-blind, randomized, controlled trial in veterans is examining the effectiveness of prazosin as a preventive agent in treating combat-related posttraumatic headache. This study was scheduled to enroll its last patient at the end of 2017, and results may be out soon.
There may be specific pharmacologic properties that make prazosin a useful drug for headache treatment. Prazosin is a very potent, selective alpha 1-adrenergic antagonist that passes through the blood–brain barrier. It is highly protein bound (97%), so absolute amounts in the CNS are likely to be low. Its use in the treatment of hypertension is based on decreased peripheral vascular resistance as a result of arteriolar and venous receptor blockade. It also can act in the CNS to decrease sympathetic outflow. While an effect on headache could be central, peripheral, or both, other drugs with alpha-adrenergic blocking effects have been used in the treatment of migraine for decades. The ergots, for example, were the first alpha-adrenergic agents to be discovered acting as partial agonists or antagonists at adrenergic, tryptaminergic, and dopaminergic receptors. The hydrogenated ergot alkaloids are among the most potent alpha-adrenergic blocking agents, but adverse effects prevent doses that can cause more than minimal blockade. Chlorpromazine and other dopamine (D2) receptor antagonists, which are highly effective in acute treatment of migraine, particularly with parenteral delivery, also produce significant alpha-adrenergic receptor blockade, while trazodone, amitriptyline, and the atypical antipsychotics, with various levels of alpha-adrenergic antagonism, have found some success in migraine prevention.
Clinical experience has shown that there is wide response variability to acute and chronic medication for migraine. Genetic studies of patients with migraine, though at an early stage, have identified genes involved with vascular and neuronal function. It is likely that clinical observation will be borne out by individual responses to drug classes based on individual genetic profiles, so that subtypes of patients in clinical trial populations may show efficacy based on these profiles. It is likely that prazosin will be useful in certain patient subtypes for the treatment of headache. Posttraumatic headache, which may share some similar pathways of headache physiology with primary headache disorders, adds another layer of response complexity.
—Sylvia Lucas, MD, PhD
Clinical Professor of Neurology and Neurological Surgery
University of Washington
Seattle
Suggested Reading
Nampiaparampil DE. Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA. 2008;300(6):711-719.
Peterlin BL, Nijjar SS, Tietjen GE. Post-traumatic stress disorder and migraine: epidemiology, sex differences, and potential mechanisms. Headache. 2011;51(6):860-868.
Ruff RL, Riechers RG, Wang XF, et al. For veterans with mild traumatic brain injury, improved posttraumatic stress disorder severity and sleep correlated with symptomatic improvement. J Rehabil Res Dev. 2012;49(9):1305-1320.
Does Prazosin Benefit Patients With Posttraumatic Stress Disorder?
Among military veterans with chronic posttraumatic stress disorder (PTSD) and frequent nightmares, prazosin does not alleviate distressing dreams or improve sleep quality, according to trial results published in the February 8 issue of the New England Journal of Medicine.
Prior single-center trials found that prazosin, an alpha 1-adrenoreceptor antagonist, may alleviate nightmares associated with PTSD and improve overall clinical status. The present study’s eligibility criteria may have led to selection bias that contributed to its negative results, the researchers said.
The PACT Trial
To investigate the efficacy of prazosin in patients with chronic combat-related PTSD and frequent nightmares, Murray A. Raskind, MD, and colleagues conducted the Prazosin and Combat Trauma PTSD (PACT) trial. Dr. Raskind is the Director of the Veterans Affairs (VA) Northwest Network Mental Illness Research, Education, and Clinical Center and Professor and Vice Chair of Psychiatry and Behavioral Sciences at the University of Washington School of Medicine in Seattle.

The 26-week, double-blind, randomized, controlled trial included 304 veterans from 12 VA medical centers. Participants met DSM-IV criteria for PTSD; had a total score of at least 50 on the 17-item Clinician-Administered PTSD Scale (CAPS); had been exposed to one or more traumatic, life-threatening events in a war zone before the onset of recurrent nightmares; could recall combat-related nightmares; had a frequency score of at least 2 and a cumulative score of at least 5 on CAPS item B2 (ie, “recurrent distressing dreams”); and, for at least four weeks before randomization, were receiving a stable dose of nonexcluded medications or supportive psychotherapy. Exclusion criteria included unstable medical illness, a systolic blood pressure of less than 110 mm Hg in the supine position, active suicidal or homicidal ideation with plan or intent, and psychosocial instability.
Of 413 people screened, 304 underwent randomization (about 98% male; average age, 52); 152 patients were assigned to each treatment group. The two groups’ patient characteristics did not differ significantly at baseline. Researchers administered prazosin or placebo in escalating divided doses over five weeks to a daily maximum of 20 mg in men and 12 mg in women.
Primary Outcome Measures
The three primary outcome measures were change in score from baseline to 10 weeks on the CAPS item B2, change in score from baseline to 10 weeks on the Pittsburgh Sleep Quality Index, and the Clinical Global Impression of Change score at 10 weeks. None of the primary outcome measures significantly differed between the groups at 10 weeks. The groups’ outcome measures at 26 weeks and other secondary outcomes also were not significantly different.
The number of serious adverse events did not differ significantly by group. Of the adverse events, dizziness, lightheadedness, and urinary incontinence were significantly more common in the prazosin group, compared with the placebo group, whereas new or worsening suicidal ideation was significantly less common among participants who received prazosin, compared with patients who received placebo (8% vs 15%, respectively).
The investigators enrolled patients who were mainly in clinically stable condition, which may have led the trial to include patients whose distressing dreams were unlikely to respond to prazosin, Dr. Raskind and colleagues said.
Future Directions
“The failure of this new, large, multisite trial to replicate the previous studies is surprising and disappointing,” said Kerry J. Ressler, MD, PhD, Chief of the Division of Depression and Anxiety Disorders at McLean Hospital in Belmont, Massachusetts, and Professor of Psychiatry at Harvard Medical School in Boston, in an accompanying editorial. “PTSD remains a psychiatric malady that in some respects seems understandable and treatable on the basis of known neurobiologic pathways. Yet it is a complex syndrome with innumerable subtypes and variations…. There is a need to define clinical subtypes of PTSD on the basis of biologic markers.”
Studies of prazosin for the treatment of other conditions are under way. Investigators have initiated trials to examine whether prazosin reduces the frequency of chronic postconcussive headaches, compared with placebo.
—Jake Remaly
Suggested Reading
Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
Ressler KJ. Alpha-adrenergic receptors in PTSD - Failure or time for precision medicine? N Engl J Med. 2018; 378(6):575-576.
Among military veterans with chronic posttraumatic stress disorder (PTSD) and frequent nightmares, prazosin does not alleviate distressing dreams or improve sleep quality, according to trial results published in the February 8 issue of the New England Journal of Medicine.
Prior single-center trials found that prazosin, an alpha 1-adrenoreceptor antagonist, may alleviate nightmares associated with PTSD and improve overall clinical status. The present study’s eligibility criteria may have led to selection bias that contributed to its negative results, the researchers said.
The PACT Trial
To investigate the efficacy of prazosin in patients with chronic combat-related PTSD and frequent nightmares, Murray A. Raskind, MD, and colleagues conducted the Prazosin and Combat Trauma PTSD (PACT) trial. Dr. Raskind is the Director of the Veterans Affairs (VA) Northwest Network Mental Illness Research, Education, and Clinical Center and Professor and Vice Chair of Psychiatry and Behavioral Sciences at the University of Washington School of Medicine in Seattle.
The 26-week, double-blind, randomized, controlled trial included 304 veterans from 12 VA medical centers. Participants met DSM-IV criteria for PTSD; had a total score of at least 50 on the 17-item Clinician-Administered PTSD Scale (CAPS); had been exposed to one or more traumatic, life-threatening events in a war zone before the onset of recurrent nightmares; could recall combat-related nightmares; had a frequency score of at least 2 and a cumulative score of at least 5 on CAPS item B2 (ie, “recurrent distressing dreams”); and, for at least four weeks before randomization, were receiving a stable dose of nonexcluded medications or supportive psychotherapy. Exclusion criteria included unstable medical illness, a systolic blood pressure of less than 110 mm Hg in the supine position, active suicidal or homicidal ideation with plan or intent, and psychosocial instability.
Of 413 people screened, 304 underwent randomization (about 98% male; average age, 52); 152 patients were assigned to each treatment group. The two groups’ patient characteristics did not differ significantly at baseline. Researchers administered prazosin or placebo in escalating divided doses over five weeks to a daily maximum of 20 mg in men and 12 mg in women.
Primary Outcome Measures
The three primary outcome measures were change in score from baseline to 10 weeks on the CAPS item B2, change in score from baseline to 10 weeks on the Pittsburgh Sleep Quality Index, and the Clinical Global Impression of Change score at 10 weeks. None of the primary outcome measures significantly differed between the groups at 10 weeks. The groups’ outcome measures at 26 weeks and other secondary outcomes also were not significantly different.
The number of serious adverse events did not differ significantly by group. Of the adverse events, dizziness, lightheadedness, and urinary incontinence were significantly more common in the prazosin group, compared with the placebo group, whereas new or worsening suicidal ideation was significantly less common among participants who received prazosin, compared with patients who received placebo (8% vs 15%, respectively).
The investigators enrolled patients who were mainly in clinically stable condition, which may have led the trial to include patients whose distressing dreams were unlikely to respond to prazosin, Dr. Raskind and colleagues said.
Future Directions
“The failure of this new, large, multisite trial to replicate the previous studies is surprising and disappointing,” said Kerry J. Ressler, MD, PhD, Chief of the Division of Depression and Anxiety Disorders at McLean Hospital in Belmont, Massachusetts, and Professor of Psychiatry at Harvard Medical School in Boston, in an accompanying editorial. “PTSD remains a psychiatric malady that in some respects seems understandable and treatable on the basis of known neurobiologic pathways. Yet it is a complex syndrome with innumerable subtypes and variations…. There is a need to define clinical subtypes of PTSD on the basis of biologic markers.”
Studies of prazosin for the treatment of other conditions are under way. Investigators have initiated trials to examine whether prazosin reduces the frequency of chronic postconcussive headaches, compared with placebo.
—Jake Remaly
Suggested Reading
Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
Ressler KJ. Alpha-adrenergic receptors in PTSD - Failure or time for precision medicine? N Engl J Med. 2018; 378(6):575-576.
Among military veterans with chronic posttraumatic stress disorder (PTSD) and frequent nightmares, prazosin does not alleviate distressing dreams or improve sleep quality, according to trial results published in the February 8 issue of the New England Journal of Medicine.
Prior single-center trials found that prazosin, an alpha 1-adrenoreceptor antagonist, may alleviate nightmares associated with PTSD and improve overall clinical status. The present study’s eligibility criteria may have led to selection bias that contributed to its negative results, the researchers said.
The PACT Trial
To investigate the efficacy of prazosin in patients with chronic combat-related PTSD and frequent nightmares, Murray A. Raskind, MD, and colleagues conducted the Prazosin and Combat Trauma PTSD (PACT) trial. Dr. Raskind is the Director of the Veterans Affairs (VA) Northwest Network Mental Illness Research, Education, and Clinical Center and Professor and Vice Chair of Psychiatry and Behavioral Sciences at the University of Washington School of Medicine in Seattle.
The 26-week, double-blind, randomized, controlled trial included 304 veterans from 12 VA medical centers. Participants met DSM-IV criteria for PTSD; had a total score of at least 50 on the 17-item Clinician-Administered PTSD Scale (CAPS); had been exposed to one or more traumatic, life-threatening events in a war zone before the onset of recurrent nightmares; could recall combat-related nightmares; had a frequency score of at least 2 and a cumulative score of at least 5 on CAPS item B2 (ie, “recurrent distressing dreams”); and, for at least four weeks before randomization, were receiving a stable dose of nonexcluded medications or supportive psychotherapy. Exclusion criteria included unstable medical illness, a systolic blood pressure of less than 110 mm Hg in the supine position, active suicidal or homicidal ideation with plan or intent, and psychosocial instability.
Of 413 people screened, 304 underwent randomization (about 98% male; average age, 52); 152 patients were assigned to each treatment group. The two groups’ patient characteristics did not differ significantly at baseline. Researchers administered prazosin or placebo in escalating divided doses over five weeks to a daily maximum of 20 mg in men and 12 mg in women.
Primary Outcome Measures
The three primary outcome measures were change in score from baseline to 10 weeks on the CAPS item B2, change in score from baseline to 10 weeks on the Pittsburgh Sleep Quality Index, and the Clinical Global Impression of Change score at 10 weeks. None of the primary outcome measures significantly differed between the groups at 10 weeks. The groups’ outcome measures at 26 weeks and other secondary outcomes also were not significantly different.
The number of serious adverse events did not differ significantly by group. Of the adverse events, dizziness, lightheadedness, and urinary incontinence were significantly more common in the prazosin group, compared with the placebo group, whereas new or worsening suicidal ideation was significantly less common among participants who received prazosin, compared with patients who received placebo (8% vs 15%, respectively).
The investigators enrolled patients who were mainly in clinically stable condition, which may have led the trial to include patients whose distressing dreams were unlikely to respond to prazosin, Dr. Raskind and colleagues said.
Future Directions
“The failure of this new, large, multisite trial to replicate the previous studies is surprising and disappointing,” said Kerry J. Ressler, MD, PhD, Chief of the Division of Depression and Anxiety Disorders at McLean Hospital in Belmont, Massachusetts, and Professor of Psychiatry at Harvard Medical School in Boston, in an accompanying editorial. “PTSD remains a psychiatric malady that in some respects seems understandable and treatable on the basis of known neurobiologic pathways. Yet it is a complex syndrome with innumerable subtypes and variations…. There is a need to define clinical subtypes of PTSD on the basis of biologic markers.”
Studies of prazosin for the treatment of other conditions are under way. Investigators have initiated trials to examine whether prazosin reduces the frequency of chronic postconcussive headaches, compared with placebo.
—Jake Remaly
Suggested Reading
Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
Ressler KJ. Alpha-adrenergic receptors in PTSD - Failure or time for precision medicine? N Engl J Med. 2018; 378(6):575-576.
Perianal Condyloma Acuminatum-like Plaque
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
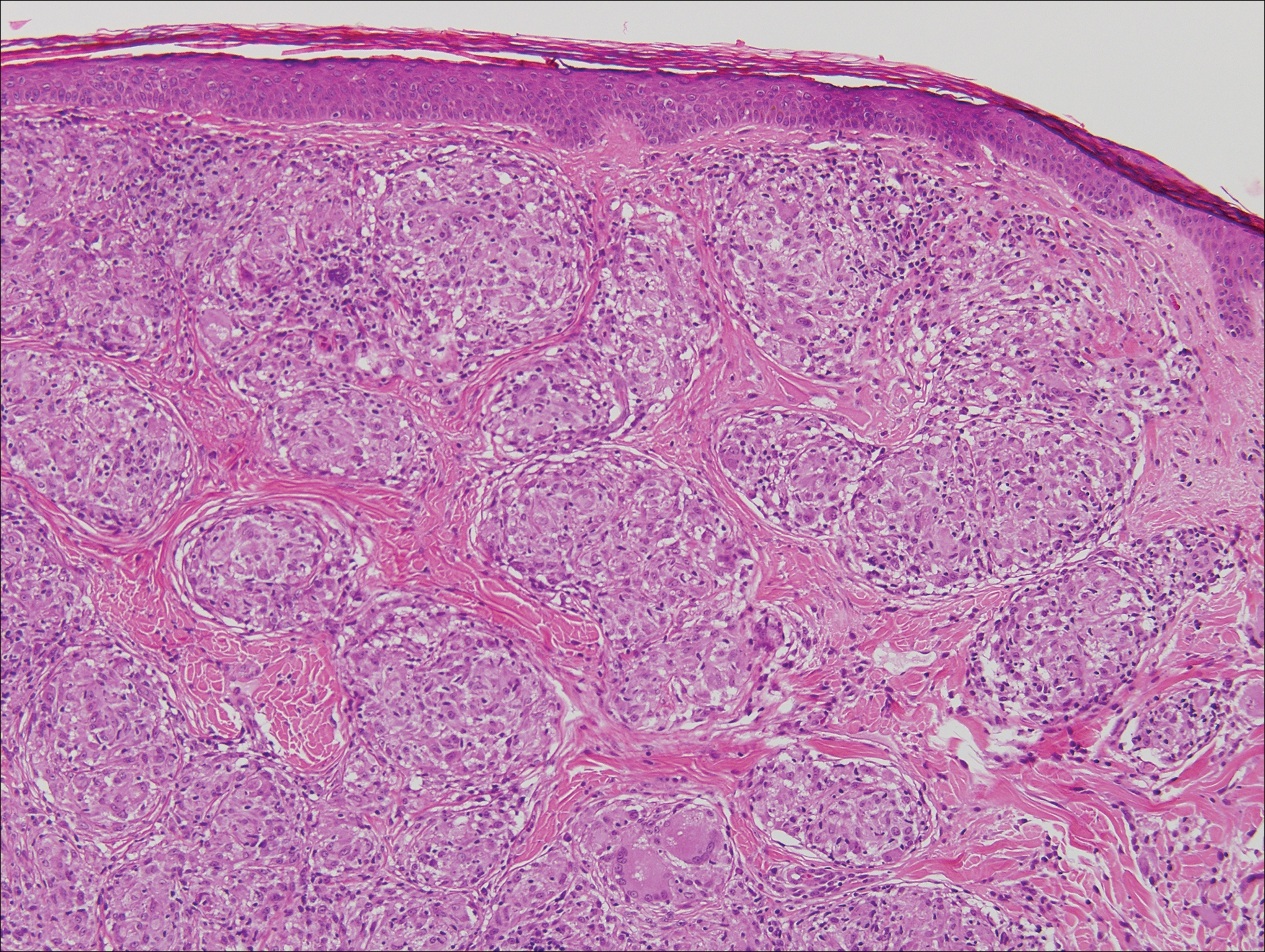
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
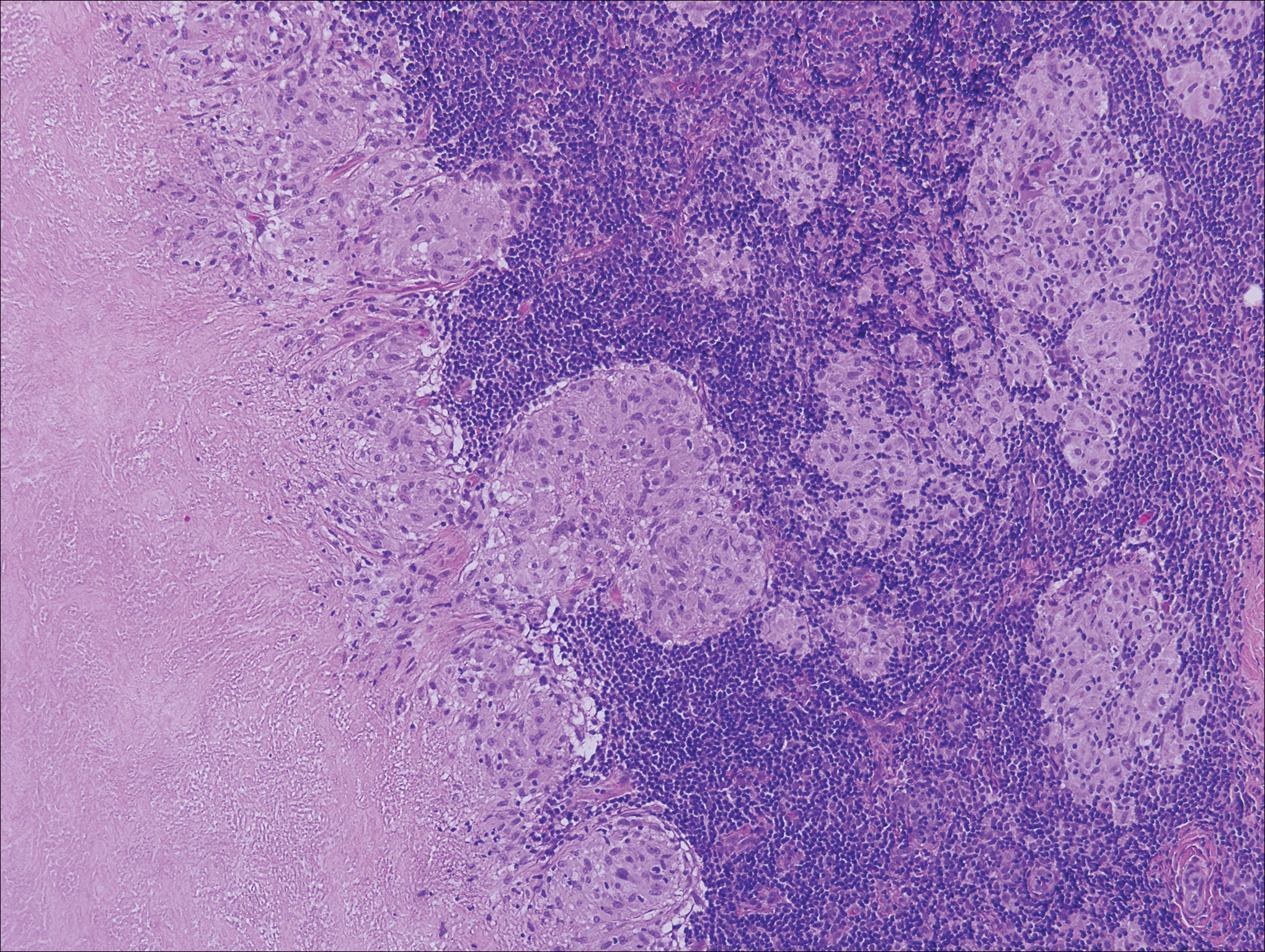
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
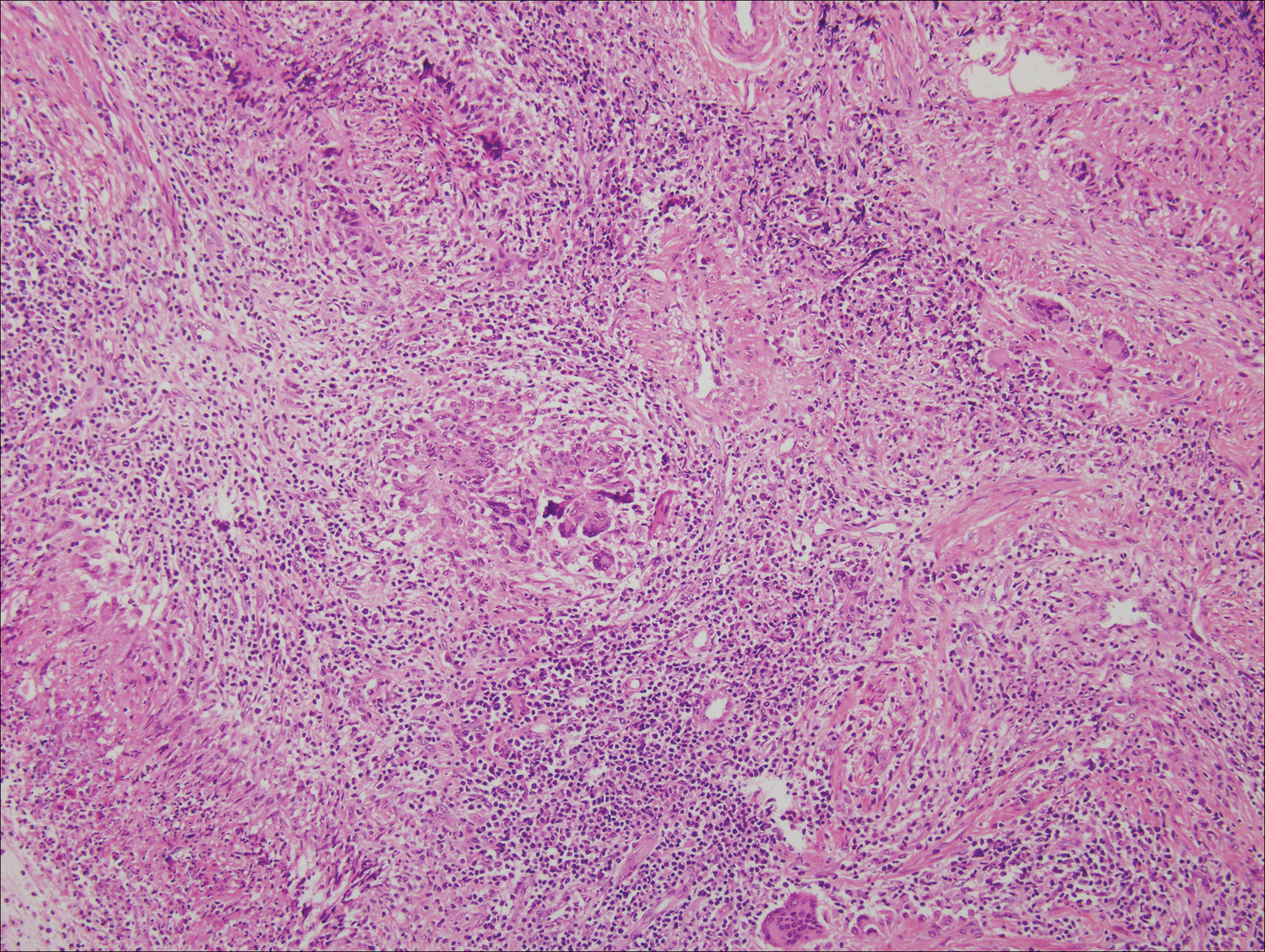
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
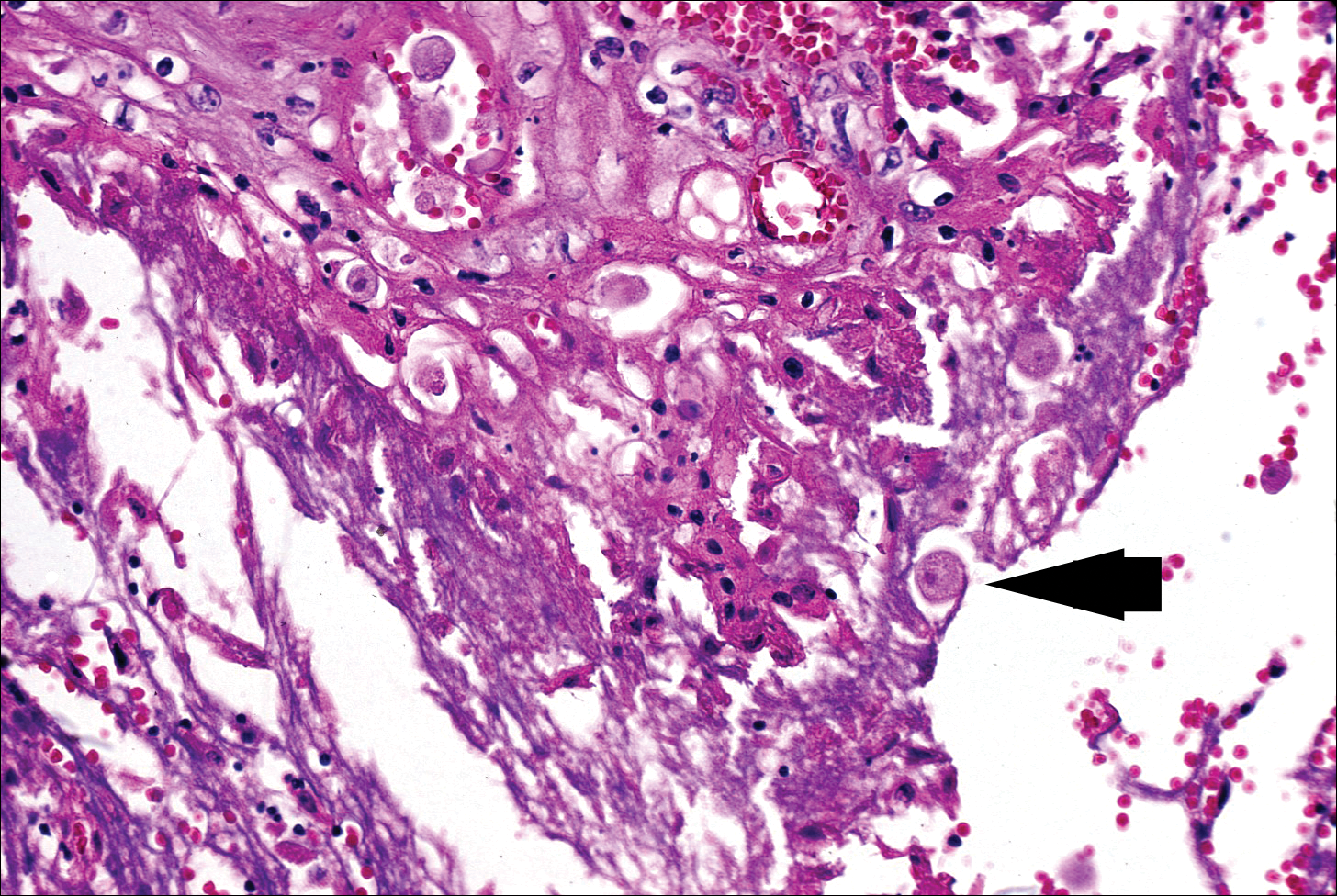
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
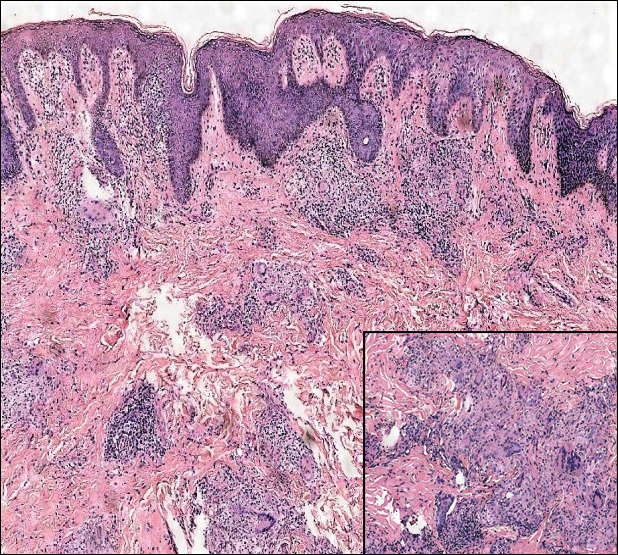
A 19-year-old man presented with a perianal condyloma acuminatum-like plaque of 2 years' duration and a 6-month history of diarrhea.
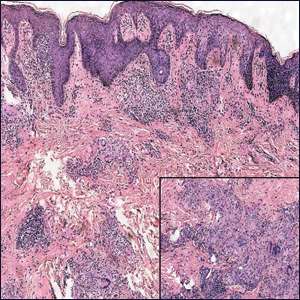
Aspirin Reduces Stroke Risk Associated With Preeclampsia
LOS ANGELES—Women with a history of preeclampsia have a significantly increased risk for early-onset stroke, but that risk is reduced in women taking aspirin, according to research described at the International Stroke Conference 2018.
The results came from an epidemiologic analysis of data for 83,790 women in the California Teachers Study. Among the 4,072 women with a history of preeclampsia, 3,003 were not on aspirin. During follow-up, these women had an incidence of ischemic and hemorrhagic stroke before age 60 of greater than 1%. Their incidence rate was 40% higher than that of the approximately 60,000 women without a history of preeclampsia who were not taking aspirin. The difference between groups remained statistically significant after the researchers adjusted the data for demographics, smoking, obesity, diabetes, and hypertension, said Eliza C. Miller, MD, a vascular neurologist at Columbia University in New York.

The findings suggest that an aspirin prevention trial in women at high risk for stroke, such as women with a history of preeclampsia, is warranted, said Dr. Miller. Cardiovascular risk prediction models such as the Framingham Risk Score could be modified to account for a history of preeclampsia, she added.
Dr. Miller’s analysis focused on women who entered the California Teachers Study when they were younger than 60, had no history of stroke, and provided data on their history of preeclampsia. The prevalence of a history of preeclampsia was 4.9% overall and 6.1% among women who had been pregnant at least once. This incidence rate was similar to those of other large populations of women, said Dr. Miller.
The average age was 44 among women with preeclampsia and 46 among women without preeclampsia. Women with a history of preeclampsia also had higher prevalence rates of obesity, hypertension, diabetes, and chronic kidney disease. About a quarter of all women regularly took aspirin.
After data adjustment, women with a history of preeclampsia had a 20% higher overall rate of stroke before age 60. The difference between groups was not significant in an analysis that included women taking aspirin and those not taking it. When the analysis examined only women not taking aspirin, the stroke rate in women with a history of preeclampsia was 40% higher than that in women without a history of preeclampsia, a statistically significant difference. In contrast, among women taking aspirin, women with and without a history of preeclampsia had similar rates of stroke.
—Mitchel L. Zoler
LOS ANGELES—Women with a history of preeclampsia have a significantly increased risk for early-onset stroke, but that risk is reduced in women taking aspirin, according to research described at the International Stroke Conference 2018.
The results came from an epidemiologic analysis of data for 83,790 women in the California Teachers Study. Among the 4,072 women with a history of preeclampsia, 3,003 were not on aspirin. During follow-up, these women had an incidence of ischemic and hemorrhagic stroke before age 60 of greater than 1%. Their incidence rate was 40% higher than that of the approximately 60,000 women without a history of preeclampsia who were not taking aspirin. The difference between groups remained statistically significant after the researchers adjusted the data for demographics, smoking, obesity, diabetes, and hypertension, said Eliza C. Miller, MD, a vascular neurologist at Columbia University in New York.
The findings suggest that an aspirin prevention trial in women at high risk for stroke, such as women with a history of preeclampsia, is warranted, said Dr. Miller. Cardiovascular risk prediction models such as the Framingham Risk Score could be modified to account for a history of preeclampsia, she added.
Dr. Miller’s analysis focused on women who entered the California Teachers Study when they were younger than 60, had no history of stroke, and provided data on their history of preeclampsia. The prevalence of a history of preeclampsia was 4.9% overall and 6.1% among women who had been pregnant at least once. This incidence rate was similar to those of other large populations of women, said Dr. Miller.
The average age was 44 among women with preeclampsia and 46 among women without preeclampsia. Women with a history of preeclampsia also had higher prevalence rates of obesity, hypertension, diabetes, and chronic kidney disease. About a quarter of all women regularly took aspirin.
After data adjustment, women with a history of preeclampsia had a 20% higher overall rate of stroke before age 60. The difference between groups was not significant in an analysis that included women taking aspirin and those not taking it. When the analysis examined only women not taking aspirin, the stroke rate in women with a history of preeclampsia was 40% higher than that in women without a history of preeclampsia, a statistically significant difference. In contrast, among women taking aspirin, women with and without a history of preeclampsia had similar rates of stroke.
—Mitchel L. Zoler
LOS ANGELES—Women with a history of preeclampsia have a significantly increased risk for early-onset stroke, but that risk is reduced in women taking aspirin, according to research described at the International Stroke Conference 2018.
The results came from an epidemiologic analysis of data for 83,790 women in the California Teachers Study. Among the 4,072 women with a history of preeclampsia, 3,003 were not on aspirin. During follow-up, these women had an incidence of ischemic and hemorrhagic stroke before age 60 of greater than 1%. Their incidence rate was 40% higher than that of the approximately 60,000 women without a history of preeclampsia who were not taking aspirin. The difference between groups remained statistically significant after the researchers adjusted the data for demographics, smoking, obesity, diabetes, and hypertension, said Eliza C. Miller, MD, a vascular neurologist at Columbia University in New York.
The findings suggest that an aspirin prevention trial in women at high risk for stroke, such as women with a history of preeclampsia, is warranted, said Dr. Miller. Cardiovascular risk prediction models such as the Framingham Risk Score could be modified to account for a history of preeclampsia, she added.
Dr. Miller’s analysis focused on women who entered the California Teachers Study when they were younger than 60, had no history of stroke, and provided data on their history of preeclampsia. The prevalence of a history of preeclampsia was 4.9% overall and 6.1% among women who had been pregnant at least once. This incidence rate was similar to those of other large populations of women, said Dr. Miller.
The average age was 44 among women with preeclampsia and 46 among women without preeclampsia. Women with a history of preeclampsia also had higher prevalence rates of obesity, hypertension, diabetes, and chronic kidney disease. About a quarter of all women regularly took aspirin.
After data adjustment, women with a history of preeclampsia had a 20% higher overall rate of stroke before age 60. The difference between groups was not significant in an analysis that included women taking aspirin and those not taking it. When the analysis examined only women not taking aspirin, the stroke rate in women with a history of preeclampsia was 40% higher than that in women without a history of preeclampsia, a statistically significant difference. In contrast, among women taking aspirin, women with and without a history of preeclampsia had similar rates of stroke.
—Mitchel L. Zoler
High Dose of Novel Compound Shows Promise for Relapsing-Remitting MS
SAN DIEGO—Patients with relapsing-remitting multiple sclerosis (MS) who received high doses of the novel human endogenous retrovirus-W antagonist GNbAC1 in a phase II trial had evidence of remyelination at week 24, according to research described at the ACTRIMS 2018 Forum. The treatment did not meet the primary end point of reduction in the number of active lesions seen on MRI, however.
GNbAC1 is a monoclonal antibody that targets and blocks the envelope protein pHER-W ENV, a potent agonist of Toll-like receptor 4 (TLR4), said study author Robert Glanzman, MD. It thereby inhibits TLR4-mediated pathogenicity, which includes activation of macrophages and microglia into proinflammatory phenotypes and direct inhibition of remyelination.

“Because we are not directly suppressing the immune system, what we think we are doing is taking away a driver of autoimmunity,” explained Dr. Glanzman, Chief Medical Officer of GeNeuro, a Geneva-based company that is developing GNbAC1.
In a study known as CHANGE-MS, 270 patients with relapsing-remitting MS were randomized to one of three doses of GNbAC1 (6 mg/kg, 12 mg/kg, or 18 mg/kg) or to placebo via monthly IV infusion over six months. The study was conducted at 70 centers in 13 European countries over three years. It had a 24-week, double-blind, placebo-controlled period, followed by a 24-week, dose-blind, active-only treatment period. During the latter period, controls were randomized to the three doses of GNbAC1. Brain MRI scans were performed at weeks 12, 16, 20, 24, and 48 to look for evidence of remyelination.
The mean age of patients was 38, and 65% of the population was female. The researchers observed no safety concerns and no significant effect on inflammatory measures during weeks 12 to 24, even though the absolute number of lesions was reduced by about 50%.
Although the primary end point of the cumulative number of gadolinium-enhancing lesions seen on brain MRI scans every four weeks during weeks 12 to 24 was not met, post hoc analyses suggest a decrease in neuroinflammation in the 18 mg/kg GNbAC1 group at week 24, compared with placebo. “We gained about a quarter or half of percent in normal-appearing white matter at the cerebral cortex at the high dose,” said Dr. Glanzman. “Normally, MS patients lose white matter over time, both in the cortex and in gray matter. We are actually showing evidence of remyelination, which is really exciting. If these data are replicated and confirmed at week 48, we think we really have an exciting compound.”
GeNeuro sponsored the study.
—Doug Brunk
SAN DIEGO—Patients with relapsing-remitting multiple sclerosis (MS) who received high doses of the novel human endogenous retrovirus-W antagonist GNbAC1 in a phase II trial had evidence of remyelination at week 24, according to research described at the ACTRIMS 2018 Forum. The treatment did not meet the primary end point of reduction in the number of active lesions seen on MRI, however.
GNbAC1 is a monoclonal antibody that targets and blocks the envelope protein pHER-W ENV, a potent agonist of Toll-like receptor 4 (TLR4), said study author Robert Glanzman, MD. It thereby inhibits TLR4-mediated pathogenicity, which includes activation of macrophages and microglia into proinflammatory phenotypes and direct inhibition of remyelination.
“Because we are not directly suppressing the immune system, what we think we are doing is taking away a driver of autoimmunity,” explained Dr. Glanzman, Chief Medical Officer of GeNeuro, a Geneva-based company that is developing GNbAC1.
In a study known as CHANGE-MS, 270 patients with relapsing-remitting MS were randomized to one of three doses of GNbAC1 (6 mg/kg, 12 mg/kg, or 18 mg/kg) or to placebo via monthly IV infusion over six months. The study was conducted at 70 centers in 13 European countries over three years. It had a 24-week, double-blind, placebo-controlled period, followed by a 24-week, dose-blind, active-only treatment period. During the latter period, controls were randomized to the three doses of GNbAC1. Brain MRI scans were performed at weeks 12, 16, 20, 24, and 48 to look for evidence of remyelination.
The mean age of patients was 38, and 65% of the population was female. The researchers observed no safety concerns and no significant effect on inflammatory measures during weeks 12 to 24, even though the absolute number of lesions was reduced by about 50%.
Although the primary end point of the cumulative number of gadolinium-enhancing lesions seen on brain MRI scans every four weeks during weeks 12 to 24 was not met, post hoc analyses suggest a decrease in neuroinflammation in the 18 mg/kg GNbAC1 group at week 24, compared with placebo. “We gained about a quarter or half of percent in normal-appearing white matter at the cerebral cortex at the high dose,” said Dr. Glanzman. “Normally, MS patients lose white matter over time, both in the cortex and in gray matter. We are actually showing evidence of remyelination, which is really exciting. If these data are replicated and confirmed at week 48, we think we really have an exciting compound.”
GeNeuro sponsored the study.
—Doug Brunk
SAN DIEGO—Patients with relapsing-remitting multiple sclerosis (MS) who received high doses of the novel human endogenous retrovirus-W antagonist GNbAC1 in a phase II trial had evidence of remyelination at week 24, according to research described at the ACTRIMS 2018 Forum. The treatment did not meet the primary end point of reduction in the number of active lesions seen on MRI, however.
GNbAC1 is a monoclonal antibody that targets and blocks the envelope protein pHER-W ENV, a potent agonist of Toll-like receptor 4 (TLR4), said study author Robert Glanzman, MD. It thereby inhibits TLR4-mediated pathogenicity, which includes activation of macrophages and microglia into proinflammatory phenotypes and direct inhibition of remyelination.
“Because we are not directly suppressing the immune system, what we think we are doing is taking away a driver of autoimmunity,” explained Dr. Glanzman, Chief Medical Officer of GeNeuro, a Geneva-based company that is developing GNbAC1.
In a study known as CHANGE-MS, 270 patients with relapsing-remitting MS were randomized to one of three doses of GNbAC1 (6 mg/kg, 12 mg/kg, or 18 mg/kg) or to placebo via monthly IV infusion over six months. The study was conducted at 70 centers in 13 European countries over three years. It had a 24-week, double-blind, placebo-controlled period, followed by a 24-week, dose-blind, active-only treatment period. During the latter period, controls were randomized to the three doses of GNbAC1. Brain MRI scans were performed at weeks 12, 16, 20, 24, and 48 to look for evidence of remyelination.
The mean age of patients was 38, and 65% of the population was female. The researchers observed no safety concerns and no significant effect on inflammatory measures during weeks 12 to 24, even though the absolute number of lesions was reduced by about 50%.
Although the primary end point of the cumulative number of gadolinium-enhancing lesions seen on brain MRI scans every four weeks during weeks 12 to 24 was not met, post hoc analyses suggest a decrease in neuroinflammation in the 18 mg/kg GNbAC1 group at week 24, compared with placebo. “We gained about a quarter or half of percent in normal-appearing white matter at the cerebral cortex at the high dose,” said Dr. Glanzman. “Normally, MS patients lose white matter over time, both in the cortex and in gray matter. We are actually showing evidence of remyelination, which is really exciting. If these data are replicated and confirmed at week 48, we think we really have an exciting compound.”
GeNeuro sponsored the study.
—Doug Brunk
Melanoma in US Hispanics: Recommended Strategies to Reduce Disparities in Outcomes
Cutaneous melanoma is a considerable public health concern. In the United States, an estimated 87,110 cases were diagnosed in 2017, and more than 9000 deaths are expected as result of this disease in 2018.1 Early diagnosis of melanoma is associated with favorable survival rates (5-year overall survival rates for melanoma in situ and stage IA melanoma, 99% and 97%, respectively).2 In contrast, the prognosis for advanced-stage melanoma is poor, with a 5-year survival rate of 16% for patients with stage IV disease. Therefore, early detection is critical to reducing mortality in melanoma patients.3
The term Hispanic refers to a panethnic category primarily encompassing Mexican-Americans, Cubans, and Puerto Ricans, as well as individuals from the Caribbean and Central and South America. These populations are diverse in birth origin, primary language, acculturation, distinct ethnic traditions, education level, and occupation. Hispanics in the United States are heterogeneous in many dimensions related to health risks, health care use, and health outcomes.4 Genetic predisposition, lifestyle risks, and access to and use of health care services can shape melanoma diagnosis, treatment, and progression across Hispanic populations differently than in other populations.
In this review, the epidemiology and clinical presentation of melanoma in US Hispanics is summarized, and recommendations for a research agenda to advance understanding of this disease in the most rapidly growing segment of the US population is provided.
In the period from 2008 to 2012, the age-adjusted incidence of melanoma in US Hispanics (4.6 per 100,00 men and 4.2 per 100,00 women) was lower than in NHWs.5 Garnett et al5 reported a decline in melanoma incidence in US Hispanics between 2003 and 2012—an observation that stands in contrast to state-level studies in California and Florida, in which small but substantial increases in melanoma incidence among Hispanics were reported.6,7 The rising incidence of melanomas thicker than 1.5 mm at presentation among Hispanic men living in California is particularly worrisome.6 Discrepancies in incidence trends might reflect changes in incidence over time or differences in state-level registry reporting of melanoma.5
Despite a lower overall incidence of melanoma in US Hispanics, those who do develop the disease are 2.4 times more likely (age-adjusted odds ratio) to present with stage III disease (confidence interval, 1.89-3.05)8 and are 3.64 times more likely to develop distant metastases (confidence interval, 2.65-5.0) than NHWs.3,7,9-13 Disparities also exist in the diagnosis of childhood melanoma: Hispanic children and adolescents who have a diagnosis of melanoma are 3 times more likely to present with advanced disease than NHW counterparts.14 Survival analyses by age and stage show considerably lower survival among Hispanic patients compared to NHW patients with stage I and II disease. In part, worse survival outcomes among Hispanics are the result of the pattern of more advanced disease at presentation.8,14,15
Late presentation for evaluation of melanomas in Hispanics has been attributed to a number of variables, including a lack of skin cancer awareness and knowledge,9,16 a lower rate of self- and physician-performed skin examinations,10 differences in tumor biology,9 and socioeconomic forces.7,17
In a previous study investigating the relationship between neighborhood characteristics and tumor stage at melanoma diagnosis in Hispanic men in California, Texas, and Florida, several key findings emerged.17 First, residency in a census tract with a high density of immigrants (California, Texas) and a high composition of Hispanics (California, Florida) was an important predictor of a late-stage melanoma diagnosis in fully adjusted models. Additionally, the strength of association between measures of socioeconomic status (ie, poverty and education) and tumor stage at melanoma diagnosis was attenuated in multivariate models when enclaves and availability of primary care resources were taken into account. Hispanic melanoma cases in areas with a low density of primary care physicians had an increased likelihood of late-stage diagnosis in California and Texas. The probability of late-stage diagnosis was concentrated in specific regions along the United States–Mexico border, in south central California, and along the southeastern coast of Florida. Lastly, in Texas, Hispanic men aged 18 to 34 years and 35 to 49 years were at an increased risk of late-stage melanoma diagnosis compared to men 65 years and older.17
Demographic and Clinical Characteristics of Melanoma in Hispanic Patients
Among Hispanics, white Hispanics comprise the majority of melanoma cases.5 Median age at diagnosis is younger in Hispanics compared to whites.5,6 Hispanic men typically are older (median age, 61 years) than Hispanic women (median age, 52 years) at diagnosis.5 Similar to what is seen in NHWs, young Hispanic women experience a higher melanoma incidence than young Hispanic men.5 Among older Hispanics, melanoma is more common in men.5,8
Melanomas located on the lower extremities and hips are more prevalent in Hispanics than in NHWs.5,8,18 Among Hispanics, there are age- and sex-based variations in the anatomic location of primary tumors: in Hispanic men, truncal tumors predominate, and in Hispanic women, tumors of the lower extremities are most common across all age groups.5 The incidence of melanomas located in the head and neck region increases with age for both Hispanic men and women.
For melanomas in which the histologic type is known, superficial spreading melanoma is the most common subtype among Hispanics.5,17,19 Acral lentiginous melanomas and nodular melanomas are more common among Hispanics than among NHWs.5,17,19
The observation that Hispanics with melanoma are more prone to lower-extremity tumors and nodular and acral lentiginous melanoma subtypes than NHWs suggests that UV exposure history may be of less importance in this population. Although numerous studies have explored melanoma risk factors in NHWs, there is a striking paucity of such studies in Hispanics. For example, there are conflicting data regarding the role of UV exposure in melanoma risk among Hispanics. Hu et al20 found that UV index and latitude correlated with melanoma risk in this population, whereas Eide et al21 found no association between UV exposure and melanoma incidence in Hispanics. A prospective study involving a multiethnic cohort (of whom 40 of the 107 participants were Hispanic) found no clear association between a history of sunburn and melanoma risk in Hispanics.18
Strategies for Reducing Disparities in Outcomes
Our knowledge of melanoma epidemiology in Hispanics derives mainly from secondary analyses of state-level and national cancer registry data sets.5-8,13-15,17,19,20 These administrative data sources often are limited by missing data (eg, tumor thickness, histologic subtype) or lack important patient-level information (eg, self-identified race and ethnicity, health insurance status). Additionally, the manner in which data are collected and integrated into research varies; for example, socioeconomic measures often are reported as either area-based or composite measures.
The host phenotypic characteristics of melanoma in NHWs are well understood, but the biological and environmental determinants of melanoma risk in Hispanics and other minorities are unknown. For example, fair complexion, red hair, blue eyes, increased freckling density, and the presence of numerous dysplastic and common melanocytic nevi indicate a propensity toward cutaneous melanoma.23,24 However, the relevance of such risk factors in Hispanics is unknown and has not been widely investigated in this patient population. Park et al18 found that a person’s sunburn susceptibility phenotype (defined as hair and eye color, ability to tan, and skin reaction to sunlight) was associated with an increased risk of melanoma among nonwhite, multiracial individuals. However, this study was limited by a small number of minority cases, which included only 40 Hispanic participants with melanoma.18 There is a need for rigorous observational studies to clearly define the phenotypic characteristics, sun-exposure behavior patterns, and genetic contributors to melanoma genesis in Hispanics.
The biologic determinants of postdiagnosis survival in Hispanics with melanoma are not well understood. It is unknown if genetic predisposition modifies melanoma risk in Hispanics. For example, the frequency of BRAF gene mutation or other driver mutations in US Hispanics has been understudied. It is important to know if mutation frequency patterns differ in Hispanics patients compared to NHWs because this knowledge could have considerable implications for treatment. Several recommendations should be considered to address these knowledge gaps. First, there is a need for development or enhancement of melanoma biorepositories, which should include tumor and nontumor specimens from a diverse sample of melanoma patients.
Conclusion
- American Cancer Society. Key statistics for melanoma skin cancer. www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html. Accessed January 13, 2018.
- Balch CM, Gershenwald JE, Soong S, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199-6206.
- Katalinic A, Waldmann A, Weinstock MA, et al. Does skin cancer screening save lives? Cancer. 2012;118:5395-5402.
- Bergad LW, Klein HS. Hispanics in the United States: A Demographic, Social, and Economic History, 1980-2005. New York, NY: Cambridge University Press; 2010.
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Pollitt RA, Clarke CA, Swetter SM, et al. The expanding melanoma burden in California Hispanics: importance of socioeconomic distribution, histologic subtype, and anatomic location. Cancer. 2011;117:152-161.
- Hu S, Parmet, Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites,Hispanics, and blacks in Florida. JAMA Dermatology. 2010;145:1369-1374.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Pollitt RA, Swetter SM, Johnson TM, et al. Examining the pathways linking lower socioeconomic status and advanced melanoma. Cancer. 2012;118:4004-4013.
- Ortiz CA, Goodwin JS, Freeman JL. The effect of socioeconomic factors on incidence, stage at diagnosis and survival of cutaneous melanoma. Med Sci Monit. 2005;11:RA163-RA172.
- Singh SD, Ajani UA, Johnson CJ, et al. Association of cutaneous melanoma incidence with area-based socioeconomic indicators-United States, 2004-2006. J Am Acad Dermatol. 2011;65(5 suppl 1):S58-S68.
- Pollitt RA, Clarke CA, Shema SJ, et al. California Medicaid enrollment and melanoma stage at diagnosis: a population-based study. Am J Prev Med. 2008;35:7-13.
- Clairwood M, Ricketts J, Grant-Kels J, et al. Melanoma in skin of color in Connecticut: an analysis of melanoma incidence and stage at diagnosis in non-Hispanic blacks, non-Hispanic whites, and Hispanics. Int J Dermatol. 2014;53:425-433.
- Hamilton EC, Nguyen HT, Chang YC, et al. Health disparities influence childhood melanoma stage at diagnosis and outcome. J Pediatr. 2016;175:182-187.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Imahiyerobo-Ip J, Ip I, Jamal S, et al. Skin cancer awareness in communities of color. J Am Acad Dermatol. 2011;64:198-200.
- Harvey VM, Enos CW, Chen JT, et al. The role of neighborhood characteristics in late stage melanoma diagnosis among Hispanic men in California, Texas, and Florida, 1996-2012 [published online June 18, 2017]. J Cancer Epidemiol. 2017;2017:8418904.
- Park SL, Le Marchand L, Wilkens LR, et al. Risk factors for malignant melanoma in white and non-white/non-African American populations: the multiethnic cohort. Cancer Prev Res. 2012;5:423-434.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5 suppl 1):S26-S37.
- Hu S, Ma F, Collado-Mesa F, et al. UV radiation, latitude, and melanoma in US Hispanics and blacks. Arch Dermatol. 2004;140:819-824.
- Eide MJ, Weinstock MA. Association of UV index, latitude, and melanoma incidence in nonwhite populations—US Surveillance, Epidemiology, and End Results (SEER) program, 1992 to 2001. Arch Dermatol. 2005;141:477-481.
- Polite BN, Adams-Campbell LL, Brawley OW, et al. Charting the future of cancer health disparities research: a position statement from the American Association for Cancer Research, the American Cancer Society, the American Society of Clinical Oncology, and the National Cancer Institute. Cancer Res. 2017;77:4548-4555.
- Gandini S, Sera F, Cattaruzza MS, et al. Meta-analysis of risk factors for cutaneous melanoma: III. family history, actinic damage and phenotypic factors. Eur J Cancer. 2005;41:2040-2059.
- Chang YM, Newton-Bishop JA, Bishop DT, et al. A pooled analysis of melanocytic nevus phenotype and the risk of cutaneous melanoma at different latitudes. Int J Cancer. 2009;124:420-428.
- Palmer JR, Ambrosone CB, Olshan AF. A collaborative study of the etiology of breast cancer subtypes in African American women: the AMBER consortium. Cancer Causes Control. 2014;25:309-319.
- Rapkin BD, Weiss E, Lounsbury D, et al. Reducing disparities in cancer screening and prevention through community-based participatory research partnerships with local libraries: a comprehensive dynamic trial. Am J Community Psychol. 2017;60:145-159.
Cutaneous melanoma is a considerable public health concern. In the United States, an estimated 87,110 cases were diagnosed in 2017, and more than 9000 deaths are expected as result of this disease in 2018.1 Early diagnosis of melanoma is associated with favorable survival rates (5-year overall survival rates for melanoma in situ and stage IA melanoma, 99% and 97%, respectively).2 In contrast, the prognosis for advanced-stage melanoma is poor, with a 5-year survival rate of 16% for patients with stage IV disease. Therefore, early detection is critical to reducing mortality in melanoma patients.3
The term Hispanic refers to a panethnic category primarily encompassing Mexican-Americans, Cubans, and Puerto Ricans, as well as individuals from the Caribbean and Central and South America. These populations are diverse in birth origin, primary language, acculturation, distinct ethnic traditions, education level, and occupation. Hispanics in the United States are heterogeneous in many dimensions related to health risks, health care use, and health outcomes.4 Genetic predisposition, lifestyle risks, and access to and use of health care services can shape melanoma diagnosis, treatment, and progression across Hispanic populations differently than in other populations.
In this review, the epidemiology and clinical presentation of melanoma in US Hispanics is summarized, and recommendations for a research agenda to advance understanding of this disease in the most rapidly growing segment of the US population is provided.
In the period from 2008 to 2012, the age-adjusted incidence of melanoma in US Hispanics (4.6 per 100,00 men and 4.2 per 100,00 women) was lower than in NHWs.5 Garnett et al5 reported a decline in melanoma incidence in US Hispanics between 2003 and 2012—an observation that stands in contrast to state-level studies in California and Florida, in which small but substantial increases in melanoma incidence among Hispanics were reported.6,7 The rising incidence of melanomas thicker than 1.5 mm at presentation among Hispanic men living in California is particularly worrisome.6 Discrepancies in incidence trends might reflect changes in incidence over time or differences in state-level registry reporting of melanoma.5
Despite a lower overall incidence of melanoma in US Hispanics, those who do develop the disease are 2.4 times more likely (age-adjusted odds ratio) to present with stage III disease (confidence interval, 1.89-3.05)8 and are 3.64 times more likely to develop distant metastases (confidence interval, 2.65-5.0) than NHWs.3,7,9-13 Disparities also exist in the diagnosis of childhood melanoma: Hispanic children and adolescents who have a diagnosis of melanoma are 3 times more likely to present with advanced disease than NHW counterparts.14 Survival analyses by age and stage show considerably lower survival among Hispanic patients compared to NHW patients with stage I and II disease. In part, worse survival outcomes among Hispanics are the result of the pattern of more advanced disease at presentation.8,14,15
Late presentation for evaluation of melanomas in Hispanics has been attributed to a number of variables, including a lack of skin cancer awareness and knowledge,9,16 a lower rate of self- and physician-performed skin examinations,10 differences in tumor biology,9 and socioeconomic forces.7,17
In a previous study investigating the relationship between neighborhood characteristics and tumor stage at melanoma diagnosis in Hispanic men in California, Texas, and Florida, several key findings emerged.17 First, residency in a census tract with a high density of immigrants (California, Texas) and a high composition of Hispanics (California, Florida) was an important predictor of a late-stage melanoma diagnosis in fully adjusted models. Additionally, the strength of association between measures of socioeconomic status (ie, poverty and education) and tumor stage at melanoma diagnosis was attenuated in multivariate models when enclaves and availability of primary care resources were taken into account. Hispanic melanoma cases in areas with a low density of primary care physicians had an increased likelihood of late-stage diagnosis in California and Texas. The probability of late-stage diagnosis was concentrated in specific regions along the United States–Mexico border, in south central California, and along the southeastern coast of Florida. Lastly, in Texas, Hispanic men aged 18 to 34 years and 35 to 49 years were at an increased risk of late-stage melanoma diagnosis compared to men 65 years and older.17
Demographic and Clinical Characteristics of Melanoma in Hispanic Patients
Among Hispanics, white Hispanics comprise the majority of melanoma cases.5 Median age at diagnosis is younger in Hispanics compared to whites.5,6 Hispanic men typically are older (median age, 61 years) than Hispanic women (median age, 52 years) at diagnosis.5 Similar to what is seen in NHWs, young Hispanic women experience a higher melanoma incidence than young Hispanic men.5 Among older Hispanics, melanoma is more common in men.5,8
Melanomas located on the lower extremities and hips are more prevalent in Hispanics than in NHWs.5,8,18 Among Hispanics, there are age- and sex-based variations in the anatomic location of primary tumors: in Hispanic men, truncal tumors predominate, and in Hispanic women, tumors of the lower extremities are most common across all age groups.5 The incidence of melanomas located in the head and neck region increases with age for both Hispanic men and women.
For melanomas in which the histologic type is known, superficial spreading melanoma is the most common subtype among Hispanics.5,17,19 Acral lentiginous melanomas and nodular melanomas are more common among Hispanics than among NHWs.5,17,19
The observation that Hispanics with melanoma are more prone to lower-extremity tumors and nodular and acral lentiginous melanoma subtypes than NHWs suggests that UV exposure history may be of less importance in this population. Although numerous studies have explored melanoma risk factors in NHWs, there is a striking paucity of such studies in Hispanics. For example, there are conflicting data regarding the role of UV exposure in melanoma risk among Hispanics. Hu et al20 found that UV index and latitude correlated with melanoma risk in this population, whereas Eide et al21 found no association between UV exposure and melanoma incidence in Hispanics. A prospective study involving a multiethnic cohort (of whom 40 of the 107 participants were Hispanic) found no clear association between a history of sunburn and melanoma risk in Hispanics.18
Strategies for Reducing Disparities in Outcomes
Our knowledge of melanoma epidemiology in Hispanics derives mainly from secondary analyses of state-level and national cancer registry data sets.5-8,13-15,17,19,20 These administrative data sources often are limited by missing data (eg, tumor thickness, histologic subtype) or lack important patient-level information (eg, self-identified race and ethnicity, health insurance status). Additionally, the manner in which data are collected and integrated into research varies; for example, socioeconomic measures often are reported as either area-based or composite measures.
The host phenotypic characteristics of melanoma in NHWs are well understood, but the biological and environmental determinants of melanoma risk in Hispanics and other minorities are unknown. For example, fair complexion, red hair, blue eyes, increased freckling density, and the presence of numerous dysplastic and common melanocytic nevi indicate a propensity toward cutaneous melanoma.23,24 However, the relevance of such risk factors in Hispanics is unknown and has not been widely investigated in this patient population. Park et al18 found that a person’s sunburn susceptibility phenotype (defined as hair and eye color, ability to tan, and skin reaction to sunlight) was associated with an increased risk of melanoma among nonwhite, multiracial individuals. However, this study was limited by a small number of minority cases, which included only 40 Hispanic participants with melanoma.18 There is a need for rigorous observational studies to clearly define the phenotypic characteristics, sun-exposure behavior patterns, and genetic contributors to melanoma genesis in Hispanics.
The biologic determinants of postdiagnosis survival in Hispanics with melanoma are not well understood. It is unknown if genetic predisposition modifies melanoma risk in Hispanics. For example, the frequency of BRAF gene mutation or other driver mutations in US Hispanics has been understudied. It is important to know if mutation frequency patterns differ in Hispanics patients compared to NHWs because this knowledge could have considerable implications for treatment. Several recommendations should be considered to address these knowledge gaps. First, there is a need for development or enhancement of melanoma biorepositories, which should include tumor and nontumor specimens from a diverse sample of melanoma patients.
Conclusion
Cutaneous melanoma is a considerable public health concern. In the United States, an estimated 87,110 cases were diagnosed in 2017, and more than 9000 deaths are expected as result of this disease in 2018.1 Early diagnosis of melanoma is associated with favorable survival rates (5-year overall survival rates for melanoma in situ and stage IA melanoma, 99% and 97%, respectively).2 In contrast, the prognosis for advanced-stage melanoma is poor, with a 5-year survival rate of 16% for patients with stage IV disease. Therefore, early detection is critical to reducing mortality in melanoma patients.3
The term Hispanic refers to a panethnic category primarily encompassing Mexican-Americans, Cubans, and Puerto Ricans, as well as individuals from the Caribbean and Central and South America. These populations are diverse in birth origin, primary language, acculturation, distinct ethnic traditions, education level, and occupation. Hispanics in the United States are heterogeneous in many dimensions related to health risks, health care use, and health outcomes.4 Genetic predisposition, lifestyle risks, and access to and use of health care services can shape melanoma diagnosis, treatment, and progression across Hispanic populations differently than in other populations.
In this review, the epidemiology and clinical presentation of melanoma in US Hispanics is summarized, and recommendations for a research agenda to advance understanding of this disease in the most rapidly growing segment of the US population is provided.
In the period from 2008 to 2012, the age-adjusted incidence of melanoma in US Hispanics (4.6 per 100,00 men and 4.2 per 100,00 women) was lower than in NHWs.5 Garnett et al5 reported a decline in melanoma incidence in US Hispanics between 2003 and 2012—an observation that stands in contrast to state-level studies in California and Florida, in which small but substantial increases in melanoma incidence among Hispanics were reported.6,7 The rising incidence of melanomas thicker than 1.5 mm at presentation among Hispanic men living in California is particularly worrisome.6 Discrepancies in incidence trends might reflect changes in incidence over time or differences in state-level registry reporting of melanoma.5
Despite a lower overall incidence of melanoma in US Hispanics, those who do develop the disease are 2.4 times more likely (age-adjusted odds ratio) to present with stage III disease (confidence interval, 1.89-3.05)8 and are 3.64 times more likely to develop distant metastases (confidence interval, 2.65-5.0) than NHWs.3,7,9-13 Disparities also exist in the diagnosis of childhood melanoma: Hispanic children and adolescents who have a diagnosis of melanoma are 3 times more likely to present with advanced disease than NHW counterparts.14 Survival analyses by age and stage show considerably lower survival among Hispanic patients compared to NHW patients with stage I and II disease. In part, worse survival outcomes among Hispanics are the result of the pattern of more advanced disease at presentation.8,14,15
Late presentation for evaluation of melanomas in Hispanics has been attributed to a number of variables, including a lack of skin cancer awareness and knowledge,9,16 a lower rate of self- and physician-performed skin examinations,10 differences in tumor biology,9 and socioeconomic forces.7,17
In a previous study investigating the relationship between neighborhood characteristics and tumor stage at melanoma diagnosis in Hispanic men in California, Texas, and Florida, several key findings emerged.17 First, residency in a census tract with a high density of immigrants (California, Texas) and a high composition of Hispanics (California, Florida) was an important predictor of a late-stage melanoma diagnosis in fully adjusted models. Additionally, the strength of association between measures of socioeconomic status (ie, poverty and education) and tumor stage at melanoma diagnosis was attenuated in multivariate models when enclaves and availability of primary care resources were taken into account. Hispanic melanoma cases in areas with a low density of primary care physicians had an increased likelihood of late-stage diagnosis in California and Texas. The probability of late-stage diagnosis was concentrated in specific regions along the United States–Mexico border, in south central California, and along the southeastern coast of Florida. Lastly, in Texas, Hispanic men aged 18 to 34 years and 35 to 49 years were at an increased risk of late-stage melanoma diagnosis compared to men 65 years and older.17
Demographic and Clinical Characteristics of Melanoma in Hispanic Patients
Among Hispanics, white Hispanics comprise the majority of melanoma cases.5 Median age at diagnosis is younger in Hispanics compared to whites.5,6 Hispanic men typically are older (median age, 61 years) than Hispanic women (median age, 52 years) at diagnosis.5 Similar to what is seen in NHWs, young Hispanic women experience a higher melanoma incidence than young Hispanic men.5 Among older Hispanics, melanoma is more common in men.5,8
Melanomas located on the lower extremities and hips are more prevalent in Hispanics than in NHWs.5,8,18 Among Hispanics, there are age- and sex-based variations in the anatomic location of primary tumors: in Hispanic men, truncal tumors predominate, and in Hispanic women, tumors of the lower extremities are most common across all age groups.5 The incidence of melanomas located in the head and neck region increases with age for both Hispanic men and women.
For melanomas in which the histologic type is known, superficial spreading melanoma is the most common subtype among Hispanics.5,17,19 Acral lentiginous melanomas and nodular melanomas are more common among Hispanics than among NHWs.5,17,19
The observation that Hispanics with melanoma are more prone to lower-extremity tumors and nodular and acral lentiginous melanoma subtypes than NHWs suggests that UV exposure history may be of less importance in this population. Although numerous studies have explored melanoma risk factors in NHWs, there is a striking paucity of such studies in Hispanics. For example, there are conflicting data regarding the role of UV exposure in melanoma risk among Hispanics. Hu et al20 found that UV index and latitude correlated with melanoma risk in this population, whereas Eide et al21 found no association between UV exposure and melanoma incidence in Hispanics. A prospective study involving a multiethnic cohort (of whom 40 of the 107 participants were Hispanic) found no clear association between a history of sunburn and melanoma risk in Hispanics.18
Strategies for Reducing Disparities in Outcomes
Our knowledge of melanoma epidemiology in Hispanics derives mainly from secondary analyses of state-level and national cancer registry data sets.5-8,13-15,17,19,20 These administrative data sources often are limited by missing data (eg, tumor thickness, histologic subtype) or lack important patient-level information (eg, self-identified race and ethnicity, health insurance status). Additionally, the manner in which data are collected and integrated into research varies; for example, socioeconomic measures often are reported as either area-based or composite measures.
The host phenotypic characteristics of melanoma in NHWs are well understood, but the biological and environmental determinants of melanoma risk in Hispanics and other minorities are unknown. For example, fair complexion, red hair, blue eyes, increased freckling density, and the presence of numerous dysplastic and common melanocytic nevi indicate a propensity toward cutaneous melanoma.23,24 However, the relevance of such risk factors in Hispanics is unknown and has not been widely investigated in this patient population. Park et al18 found that a person’s sunburn susceptibility phenotype (defined as hair and eye color, ability to tan, and skin reaction to sunlight) was associated with an increased risk of melanoma among nonwhite, multiracial individuals. However, this study was limited by a small number of minority cases, which included only 40 Hispanic participants with melanoma.18 There is a need for rigorous observational studies to clearly define the phenotypic characteristics, sun-exposure behavior patterns, and genetic contributors to melanoma genesis in Hispanics.
The biologic determinants of postdiagnosis survival in Hispanics with melanoma are not well understood. It is unknown if genetic predisposition modifies melanoma risk in Hispanics. For example, the frequency of BRAF gene mutation or other driver mutations in US Hispanics has been understudied. It is important to know if mutation frequency patterns differ in Hispanics patients compared to NHWs because this knowledge could have considerable implications for treatment. Several recommendations should be considered to address these knowledge gaps. First, there is a need for development or enhancement of melanoma biorepositories, which should include tumor and nontumor specimens from a diverse sample of melanoma patients.
Conclusion
- American Cancer Society. Key statistics for melanoma skin cancer. www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html. Accessed January 13, 2018.
- Balch CM, Gershenwald JE, Soong S, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199-6206.
- Katalinic A, Waldmann A, Weinstock MA, et al. Does skin cancer screening save lives? Cancer. 2012;118:5395-5402.
- Bergad LW, Klein HS. Hispanics in the United States: A Demographic, Social, and Economic History, 1980-2005. New York, NY: Cambridge University Press; 2010.
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Pollitt RA, Clarke CA, Swetter SM, et al. The expanding melanoma burden in California Hispanics: importance of socioeconomic distribution, histologic subtype, and anatomic location. Cancer. 2011;117:152-161.
- Hu S, Parmet, Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites,Hispanics, and blacks in Florida. JAMA Dermatology. 2010;145:1369-1374.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Pollitt RA, Swetter SM, Johnson TM, et al. Examining the pathways linking lower socioeconomic status and advanced melanoma. Cancer. 2012;118:4004-4013.
- Ortiz CA, Goodwin JS, Freeman JL. The effect of socioeconomic factors on incidence, stage at diagnosis and survival of cutaneous melanoma. Med Sci Monit. 2005;11:RA163-RA172.
- Singh SD, Ajani UA, Johnson CJ, et al. Association of cutaneous melanoma incidence with area-based socioeconomic indicators-United States, 2004-2006. J Am Acad Dermatol. 2011;65(5 suppl 1):S58-S68.
- Pollitt RA, Clarke CA, Shema SJ, et al. California Medicaid enrollment and melanoma stage at diagnosis: a population-based study. Am J Prev Med. 2008;35:7-13.
- Clairwood M, Ricketts J, Grant-Kels J, et al. Melanoma in skin of color in Connecticut: an analysis of melanoma incidence and stage at diagnosis in non-Hispanic blacks, non-Hispanic whites, and Hispanics. Int J Dermatol. 2014;53:425-433.
- Hamilton EC, Nguyen HT, Chang YC, et al. Health disparities influence childhood melanoma stage at diagnosis and outcome. J Pediatr. 2016;175:182-187.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Imahiyerobo-Ip J, Ip I, Jamal S, et al. Skin cancer awareness in communities of color. J Am Acad Dermatol. 2011;64:198-200.
- Harvey VM, Enos CW, Chen JT, et al. The role of neighborhood characteristics in late stage melanoma diagnosis among Hispanic men in California, Texas, and Florida, 1996-2012 [published online June 18, 2017]. J Cancer Epidemiol. 2017;2017:8418904.
- Park SL, Le Marchand L, Wilkens LR, et al. Risk factors for malignant melanoma in white and non-white/non-African American populations: the multiethnic cohort. Cancer Prev Res. 2012;5:423-434.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5 suppl 1):S26-S37.
- Hu S, Ma F, Collado-Mesa F, et al. UV radiation, latitude, and melanoma in US Hispanics and blacks. Arch Dermatol. 2004;140:819-824.
- Eide MJ, Weinstock MA. Association of UV index, latitude, and melanoma incidence in nonwhite populations—US Surveillance, Epidemiology, and End Results (SEER) program, 1992 to 2001. Arch Dermatol. 2005;141:477-481.
- Polite BN, Adams-Campbell LL, Brawley OW, et al. Charting the future of cancer health disparities research: a position statement from the American Association for Cancer Research, the American Cancer Society, the American Society of Clinical Oncology, and the National Cancer Institute. Cancer Res. 2017;77:4548-4555.
- Gandini S, Sera F, Cattaruzza MS, et al. Meta-analysis of risk factors for cutaneous melanoma: III. family history, actinic damage and phenotypic factors. Eur J Cancer. 2005;41:2040-2059.
- Chang YM, Newton-Bishop JA, Bishop DT, et al. A pooled analysis of melanocytic nevus phenotype and the risk of cutaneous melanoma at different latitudes. Int J Cancer. 2009;124:420-428.
- Palmer JR, Ambrosone CB, Olshan AF. A collaborative study of the etiology of breast cancer subtypes in African American women: the AMBER consortium. Cancer Causes Control. 2014;25:309-319.
- Rapkin BD, Weiss E, Lounsbury D, et al. Reducing disparities in cancer screening and prevention through community-based participatory research partnerships with local libraries: a comprehensive dynamic trial. Am J Community Psychol. 2017;60:145-159.
- American Cancer Society. Key statistics for melanoma skin cancer. www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html. Accessed January 13, 2018.
- Balch CM, Gershenwald JE, Soong S, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199-6206.
- Katalinic A, Waldmann A, Weinstock MA, et al. Does skin cancer screening save lives? Cancer. 2012;118:5395-5402.
- Bergad LW, Klein HS. Hispanics in the United States: A Demographic, Social, and Economic History, 1980-2005. New York, NY: Cambridge University Press; 2010.
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Pollitt RA, Clarke CA, Swetter SM, et al. The expanding melanoma burden in California Hispanics: importance of socioeconomic distribution, histologic subtype, and anatomic location. Cancer. 2011;117:152-161.
- Hu S, Parmet, Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites,Hispanics, and blacks in Florida. JAMA Dermatology. 2010;145:1369-1374.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Pollitt RA, Swetter SM, Johnson TM, et al. Examining the pathways linking lower socioeconomic status and advanced melanoma. Cancer. 2012;118:4004-4013.
- Ortiz CA, Goodwin JS, Freeman JL. The effect of socioeconomic factors on incidence, stage at diagnosis and survival of cutaneous melanoma. Med Sci Monit. 2005;11:RA163-RA172.
- Singh SD, Ajani UA, Johnson CJ, et al. Association of cutaneous melanoma incidence with area-based socioeconomic indicators-United States, 2004-2006. J Am Acad Dermatol. 2011;65(5 suppl 1):S58-S68.
- Pollitt RA, Clarke CA, Shema SJ, et al. California Medicaid enrollment and melanoma stage at diagnosis: a population-based study. Am J Prev Med. 2008;35:7-13.
- Clairwood M, Ricketts J, Grant-Kels J, et al. Melanoma in skin of color in Connecticut: an analysis of melanoma incidence and stage at diagnosis in non-Hispanic blacks, non-Hispanic whites, and Hispanics. Int J Dermatol. 2014;53:425-433.
- Hamilton EC, Nguyen HT, Chang YC, et al. Health disparities influence childhood melanoma stage at diagnosis and outcome. J Pediatr. 2016;175:182-187.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Imahiyerobo-Ip J, Ip I, Jamal S, et al. Skin cancer awareness in communities of color. J Am Acad Dermatol. 2011;64:198-200.
- Harvey VM, Enos CW, Chen JT, et al. The role of neighborhood characteristics in late stage melanoma diagnosis among Hispanic men in California, Texas, and Florida, 1996-2012 [published online June 18, 2017]. J Cancer Epidemiol. 2017;2017:8418904.
- Park SL, Le Marchand L, Wilkens LR, et al. Risk factors for malignant melanoma in white and non-white/non-African American populations: the multiethnic cohort. Cancer Prev Res. 2012;5:423-434.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5 suppl 1):S26-S37.
- Hu S, Ma F, Collado-Mesa F, et al. UV radiation, latitude, and melanoma in US Hispanics and blacks. Arch Dermatol. 2004;140:819-824.
- Eide MJ, Weinstock MA. Association of UV index, latitude, and melanoma incidence in nonwhite populations—US Surveillance, Epidemiology, and End Results (SEER) program, 1992 to 2001. Arch Dermatol. 2005;141:477-481.
- Polite BN, Adams-Campbell LL, Brawley OW, et al. Charting the future of cancer health disparities research: a position statement from the American Association for Cancer Research, the American Cancer Society, the American Society of Clinical Oncology, and the National Cancer Institute. Cancer Res. 2017;77:4548-4555.
- Gandini S, Sera F, Cattaruzza MS, et al. Meta-analysis of risk factors for cutaneous melanoma: III. family history, actinic damage and phenotypic factors. Eur J Cancer. 2005;41:2040-2059.
- Chang YM, Newton-Bishop JA, Bishop DT, et al. A pooled analysis of melanocytic nevus phenotype and the risk of cutaneous melanoma at different latitudes. Int J Cancer. 2009;124:420-428.
- Palmer JR, Ambrosone CB, Olshan AF. A collaborative study of the etiology of breast cancer subtypes in African American women: the AMBER consortium. Cancer Causes Control. 2014;25:309-319.
- Rapkin BD, Weiss E, Lounsbury D, et al. Reducing disparities in cancer screening and prevention through community-based participatory research partnerships with local libraries: a comprehensive dynamic trial. Am J Community Psychol. 2017;60:145-159.
Practice Points
- Although the age-adjusted incidence of melanoma among US Hispanics is lower than among non-Hispanic whites, Hispanics with melanoma are more likely to present with stage III disease and have distant metastases.
- Late presentation of melanoma in Hispanics is not completely understood but may be attributed to socioeconomic factors, lack of skin cancer awareness and knowledge, lower rate of self- and physician-performed skin examinations, and differences in tumor biology, among other variables.
- Research is needed to address gaps in knowledge about the risk of melanoma and comparatively poor outcomes among Hispanics so interventional efforts for prevention, early detection, and treatment can be implemented.

What Is the Mechanism of Alemtuzumab-Induced Autoimmunity?
SAN DIEGO—The monoclonal antibody alemtuzumab can be an effective treatment for people living with multiple sclerosis (MS), but the agent is also associated with an increased risk for developing other autoimmune diseases, leaving clinicians with a conundrum.
Alemtuzumab is an efficacious treatment in MS that can slow the rate of brain atrophy over the long term, said Alasdair Coles, MD, Professor of Neuroimmunology at the University of Cambridge, United Kingdom, at ACTRIMS 2018 Forum. “But one or two years after each cycle of alemtuzumab, patients are at high risk of autoimmune diseases. This is the not-too-worrying thyroid disease, but there are some troubling and potentially serious complications at lower frequency.”

Autoimmune thyroid disease can affect as much as 40% of patients treated with alemtuzumab, but immune thrombocytopenia (3%) and autoimmune renal disease (0.1%) are also reported. About one in 10 people treated with the monoclonal antibody for MS can also develop de novo asymptomatic autoantibodies.
“People ask, ‘Why doesn’t MS come back as part of this generic mechanism?’ I don’t know the answer to that question,” said Dr. Coles.
In the United States, alemtuzumab is indicated for treatment of relapsing-remitting MS in adults who have failed to respond adequately to two or more previous therapies. In contrast, “this has become a first-line treatment in the UK,” said Dr. Coles. “Unfortunately, we can offer no proven treatment to prevent this autoimmunity.”
Considering Proposed Mechanisms
Dr. Coles and other researchers are investigating the cellular mechanism underlying the paradoxical autoimmunity associated with alemtuzumab. Some have suggested that faulty immune B cells could be the culprit, but “there is no difference in B cell reconstitution between those who do and do not get autoimmunity,” said Dr. Coles. “So, we do not think that autoimmunity after alemtuzumab is primarily a B cell problem.” Other investigators have suggested that the mechanism is the depletion of a key immune regulatory cell associated with alemtuzumab. One such example is depletion in T cells as part of an autoimmune cascade that involve CD52-high expressing cells and sialic acid-binding immunoglobulin-like lectin 10. “We do not believe this,” said Dr. Coles. “We cannot replicate the finding of reduced CD52 high cells in type 1 diabetes or MS, nor the binding of SIGLEC-10 to CD52.”
Along with his colleague Joanne Jones, MD, PhD, also at the University of Cambridge, Dr. Coles and his team instead propose that autoimmunity after alemtuzumab therapy is associated with a homeostatic proliferation of T cells in the context of a defective thymus. “We see thymic function reduced after alemtuzumab for a few months. We do not know if alemtuzumab is having a direct impact on the thymus or if it is an indirect effect through a cytokine storm at the time of administering alemtuzumab.”
In addition, in contrast with B cells, both CD4-positive and CD8-positive T cells are clonally restricted after alemtuzumab treatment, said Dr. Coles. “These are the only changes that distinguish patients who do and do not develop autoimmunity,” he said. “Those who develop autoimmunity have reduced clonality and impaired thymic function, compared with those who do not.”
The theory is that the reconstitution of T cells after alemtuzumab comes preferentially from expansion of peripheral T cells, rather than naïve T cells from the thymus, which leads to a higher representation of autoreactive T cells and thus leads to B-cell- and antibody-mediated autoimmunity.
The Bigger Picture
The autoimmune phenomenon is not unique to alemtuzumab or MS. “This turns out to be one of a family of clinical situations where the reconstitution of the depleted lymphocyte repertoire leads to autoimmunity,” Dr. Coles said. A similar effect was seen years ago when very lymphopenic patients with HIV were given antiviral therapy. About 10% of treated patients had this effect. Furthermore, about 10% of patients who undergo bone marrow transplant may experience similar autoimmune concerns.
“What we do think is true is that we have tapped into a classical expression of autoimmunity,” Dr. Coles said. “Alemtuzumab is a fantastic opportunity to study the mechanisms underlying lymphopenia-associated autoimmunity.”
A Tantalizing Prospect
“It is a tantalizing prospect that susceptible individuals might be identified in the future prior to treatment,” Dr. Coles said. “We looked at IL-21. We showed that after treatment, and perhaps more interestingly, before treatment with alemtuzumab, serum levels of IL-21 are greater in those who subsequently develop autoimmune disease. This [finding] suggests [that] some individuals are prone to developing autoimmune disease and could be identified potentially prior to treatment with alemtuzumab.”
More research is needed, including the development of more sensitive IL-21 assays for use in this population, Dr. Coles said. “Please do not attempt to predict the risk of autoimmunity after alemtuzumab using the current commercial assays. This is a source of some frustration for me.” A potential route of lymphocyte repertoire reconstitution after alemtuzumab is thymic reconstitution, which could lead to a more diverse immune repertoire, Dr. Coles said. “If we can direct reconstitution through the thymic reconstitution, we should be able to prevent autoimmunity.”
Dr. Coles receives honoraria for travel and speaking from Sanofi Genzyme, which markets alemtuzumab.
—Damian McNamara
SAN DIEGO—The monoclonal antibody alemtuzumab can be an effective treatment for people living with multiple sclerosis (MS), but the agent is also associated with an increased risk for developing other autoimmune diseases, leaving clinicians with a conundrum.
Alemtuzumab is an efficacious treatment in MS that can slow the rate of brain atrophy over the long term, said Alasdair Coles, MD, Professor of Neuroimmunology at the University of Cambridge, United Kingdom, at ACTRIMS 2018 Forum. “But one or two years after each cycle of alemtuzumab, patients are at high risk of autoimmune diseases. This is the not-too-worrying thyroid disease, but there are some troubling and potentially serious complications at lower frequency.”
Autoimmune thyroid disease can affect as much as 40% of patients treated with alemtuzumab, but immune thrombocytopenia (3%) and autoimmune renal disease (0.1%) are also reported. About one in 10 people treated with the monoclonal antibody for MS can also develop de novo asymptomatic autoantibodies.
“People ask, ‘Why doesn’t MS come back as part of this generic mechanism?’ I don’t know the answer to that question,” said Dr. Coles.
In the United States, alemtuzumab is indicated for treatment of relapsing-remitting MS in adults who have failed to respond adequately to two or more previous therapies. In contrast, “this has become a first-line treatment in the UK,” said Dr. Coles. “Unfortunately, we can offer no proven treatment to prevent this autoimmunity.”
Considering Proposed Mechanisms
Dr. Coles and other researchers are investigating the cellular mechanism underlying the paradoxical autoimmunity associated with alemtuzumab. Some have suggested that faulty immune B cells could be the culprit, but “there is no difference in B cell reconstitution between those who do and do not get autoimmunity,” said Dr. Coles. “So, we do not think that autoimmunity after alemtuzumab is primarily a B cell problem.” Other investigators have suggested that the mechanism is the depletion of a key immune regulatory cell associated with alemtuzumab. One such example is depletion in T cells as part of an autoimmune cascade that involve CD52-high expressing cells and sialic acid-binding immunoglobulin-like lectin 10. “We do not believe this,” said Dr. Coles. “We cannot replicate the finding of reduced CD52 high cells in type 1 diabetes or MS, nor the binding of SIGLEC-10 to CD52.”
Along with his colleague Joanne Jones, MD, PhD, also at the University of Cambridge, Dr. Coles and his team instead propose that autoimmunity after alemtuzumab therapy is associated with a homeostatic proliferation of T cells in the context of a defective thymus. “We see thymic function reduced after alemtuzumab for a few months. We do not know if alemtuzumab is having a direct impact on the thymus or if it is an indirect effect through a cytokine storm at the time of administering alemtuzumab.”
In addition, in contrast with B cells, both CD4-positive and CD8-positive T cells are clonally restricted after alemtuzumab treatment, said Dr. Coles. “These are the only changes that distinguish patients who do and do not develop autoimmunity,” he said. “Those who develop autoimmunity have reduced clonality and impaired thymic function, compared with those who do not.”
The theory is that the reconstitution of T cells after alemtuzumab comes preferentially from expansion of peripheral T cells, rather than naïve T cells from the thymus, which leads to a higher representation of autoreactive T cells and thus leads to B-cell- and antibody-mediated autoimmunity.
The Bigger Picture
The autoimmune phenomenon is not unique to alemtuzumab or MS. “This turns out to be one of a family of clinical situations where the reconstitution of the depleted lymphocyte repertoire leads to autoimmunity,” Dr. Coles said. A similar effect was seen years ago when very lymphopenic patients with HIV were given antiviral therapy. About 10% of treated patients had this effect. Furthermore, about 10% of patients who undergo bone marrow transplant may experience similar autoimmune concerns.
“What we do think is true is that we have tapped into a classical expression of autoimmunity,” Dr. Coles said. “Alemtuzumab is a fantastic opportunity to study the mechanisms underlying lymphopenia-associated autoimmunity.”
A Tantalizing Prospect
“It is a tantalizing prospect that susceptible individuals might be identified in the future prior to treatment,” Dr. Coles said. “We looked at IL-21. We showed that after treatment, and perhaps more interestingly, before treatment with alemtuzumab, serum levels of IL-21 are greater in those who subsequently develop autoimmune disease. This [finding] suggests [that] some individuals are prone to developing autoimmune disease and could be identified potentially prior to treatment with alemtuzumab.”
More research is needed, including the development of more sensitive IL-21 assays for use in this population, Dr. Coles said. “Please do not attempt to predict the risk of autoimmunity after alemtuzumab using the current commercial assays. This is a source of some frustration for me.” A potential route of lymphocyte repertoire reconstitution after alemtuzumab is thymic reconstitution, which could lead to a more diverse immune repertoire, Dr. Coles said. “If we can direct reconstitution through the thymic reconstitution, we should be able to prevent autoimmunity.”
Dr. Coles receives honoraria for travel and speaking from Sanofi Genzyme, which markets alemtuzumab.
—Damian McNamara
SAN DIEGO—The monoclonal antibody alemtuzumab can be an effective treatment for people living with multiple sclerosis (MS), but the agent is also associated with an increased risk for developing other autoimmune diseases, leaving clinicians with a conundrum.
Alemtuzumab is an efficacious treatment in MS that can slow the rate of brain atrophy over the long term, said Alasdair Coles, MD, Professor of Neuroimmunology at the University of Cambridge, United Kingdom, at ACTRIMS 2018 Forum. “But one or two years after each cycle of alemtuzumab, patients are at high risk of autoimmune diseases. This is the not-too-worrying thyroid disease, but there are some troubling and potentially serious complications at lower frequency.”
Autoimmune thyroid disease can affect as much as 40% of patients treated with alemtuzumab, but immune thrombocytopenia (3%) and autoimmune renal disease (0.1%) are also reported. About one in 10 people treated with the monoclonal antibody for MS can also develop de novo asymptomatic autoantibodies.
“People ask, ‘Why doesn’t MS come back as part of this generic mechanism?’ I don’t know the answer to that question,” said Dr. Coles.
In the United States, alemtuzumab is indicated for treatment of relapsing-remitting MS in adults who have failed to respond adequately to two or more previous therapies. In contrast, “this has become a first-line treatment in the UK,” said Dr. Coles. “Unfortunately, we can offer no proven treatment to prevent this autoimmunity.”
Considering Proposed Mechanisms
Dr. Coles and other researchers are investigating the cellular mechanism underlying the paradoxical autoimmunity associated with alemtuzumab. Some have suggested that faulty immune B cells could be the culprit, but “there is no difference in B cell reconstitution between those who do and do not get autoimmunity,” said Dr. Coles. “So, we do not think that autoimmunity after alemtuzumab is primarily a B cell problem.” Other investigators have suggested that the mechanism is the depletion of a key immune regulatory cell associated with alemtuzumab. One such example is depletion in T cells as part of an autoimmune cascade that involve CD52-high expressing cells and sialic acid-binding immunoglobulin-like lectin 10. “We do not believe this,” said Dr. Coles. “We cannot replicate the finding of reduced CD52 high cells in type 1 diabetes or MS, nor the binding of SIGLEC-10 to CD52.”
Along with his colleague Joanne Jones, MD, PhD, also at the University of Cambridge, Dr. Coles and his team instead propose that autoimmunity after alemtuzumab therapy is associated with a homeostatic proliferation of T cells in the context of a defective thymus. “We see thymic function reduced after alemtuzumab for a few months. We do not know if alemtuzumab is having a direct impact on the thymus or if it is an indirect effect through a cytokine storm at the time of administering alemtuzumab.”
In addition, in contrast with B cells, both CD4-positive and CD8-positive T cells are clonally restricted after alemtuzumab treatment, said Dr. Coles. “These are the only changes that distinguish patients who do and do not develop autoimmunity,” he said. “Those who develop autoimmunity have reduced clonality and impaired thymic function, compared with those who do not.”
The theory is that the reconstitution of T cells after alemtuzumab comes preferentially from expansion of peripheral T cells, rather than naïve T cells from the thymus, which leads to a higher representation of autoreactive T cells and thus leads to B-cell- and antibody-mediated autoimmunity.
The Bigger Picture
The autoimmune phenomenon is not unique to alemtuzumab or MS. “This turns out to be one of a family of clinical situations where the reconstitution of the depleted lymphocyte repertoire leads to autoimmunity,” Dr. Coles said. A similar effect was seen years ago when very lymphopenic patients with HIV were given antiviral therapy. About 10% of treated patients had this effect. Furthermore, about 10% of patients who undergo bone marrow transplant may experience similar autoimmune concerns.
“What we do think is true is that we have tapped into a classical expression of autoimmunity,” Dr. Coles said. “Alemtuzumab is a fantastic opportunity to study the mechanisms underlying lymphopenia-associated autoimmunity.”
A Tantalizing Prospect
“It is a tantalizing prospect that susceptible individuals might be identified in the future prior to treatment,” Dr. Coles said. “We looked at IL-21. We showed that after treatment, and perhaps more interestingly, before treatment with alemtuzumab, serum levels of IL-21 are greater in those who subsequently develop autoimmune disease. This [finding] suggests [that] some individuals are prone to developing autoimmune disease and could be identified potentially prior to treatment with alemtuzumab.”
More research is needed, including the development of more sensitive IL-21 assays for use in this population, Dr. Coles said. “Please do not attempt to predict the risk of autoimmunity after alemtuzumab using the current commercial assays. This is a source of some frustration for me.” A potential route of lymphocyte repertoire reconstitution after alemtuzumab is thymic reconstitution, which could lead to a more diverse immune repertoire, Dr. Coles said. “If we can direct reconstitution through the thymic reconstitution, we should be able to prevent autoimmunity.”
Dr. Coles receives honoraria for travel and speaking from Sanofi Genzyme, which markets alemtuzumab.
—Damian McNamara
European Commission expands denosumab indication
The , making it is available for the prevention of skeletal-related events in adults with multiple myeloma and other advanced malignancies involving bone.
The European approval is based on the monoclonal antibody’s strong performance in a phase 3, international trial looking specifically at prevention of skeletal-related events in multiple myeloma patients.
During the trial, the drug demonstrated noninferiority to zoledronic acid in delaying the time to first skeletal-related event (hazard ratio, 0.98, 95% confidence interval: 0.85-1.14), according to Amgen, which markets denosumab. The median time to first skeletal-related event was 22.8 months for denosumab versus 24.0 months for zoledronic acid.
The denosumab indication was expanded to include prevention of skeletal-related events by the Food and Drug Administration in the United States in January 2018.
The , making it is available for the prevention of skeletal-related events in adults with multiple myeloma and other advanced malignancies involving bone.
The European approval is based on the monoclonal antibody’s strong performance in a phase 3, international trial looking specifically at prevention of skeletal-related events in multiple myeloma patients.
During the trial, the drug demonstrated noninferiority to zoledronic acid in delaying the time to first skeletal-related event (hazard ratio, 0.98, 95% confidence interval: 0.85-1.14), according to Amgen, which markets denosumab. The median time to first skeletal-related event was 22.8 months for denosumab versus 24.0 months for zoledronic acid.
The denosumab indication was expanded to include prevention of skeletal-related events by the Food and Drug Administration in the United States in January 2018.
The , making it is available for the prevention of skeletal-related events in adults with multiple myeloma and other advanced malignancies involving bone.
The European approval is based on the monoclonal antibody’s strong performance in a phase 3, international trial looking specifically at prevention of skeletal-related events in multiple myeloma patients.
During the trial, the drug demonstrated noninferiority to zoledronic acid in delaying the time to first skeletal-related event (hazard ratio, 0.98, 95% confidence interval: 0.85-1.14), according to Amgen, which markets denosumab. The median time to first skeletal-related event was 22.8 months for denosumab versus 24.0 months for zoledronic acid.
The denosumab indication was expanded to include prevention of skeletal-related events by the Food and Drug Administration in the United States in January 2018.