User login
A Case of Streptococcus pyogenes Sepsis of Possible Oral Origin
Sepsis can be the result of single or multiple factors and sources of infection. Oral sources of sepsis and systemic infection are not commonly considered as the first potential source of infection when evaluating a septic patient. Oral infections of odontogenic or periodontal origin are frequently associated with localized or diffuse cellulitis of the head and neck region. 1 The patient’s health status and complicating problems, such as an immunocompromising condition, can further reduce the immune response for controlling chronic sources of infection. This in turn can lead to acute manifestations such as cellulitis, sepsis, or necrotizing fasciitis. Necrotizing fasciitis is caused by a polymicrobial or mixed aerobic-anaerobic infection from a variety of sources, including Streptococcus pyogenes .
Case
A 57-year-old woman with a history of major depressive disorder, paroxysmal atrial fibrillation, and opioid dependence that was in remission for more than 3 years was brought to the ED by a family member after the patient developed confusion and lethargy. She was primarily experiencing right breast pain and swelling. The breast pain was associated with high fevers, nausea, vomiting, and chills.
On examination, the patient’s vital signs were: blood pressure (BP), 109/58 mm Hg; heart rate, 160 beats/min; respiratory rate, 22 breaths/min; and temperature, 104°F. Laboratory evaluation was significant for a white blood cell count (WBC) of 8.7 x 103. There was a noted skin abrasion on the patient’s right hand. She was lethargic and confused. Blood cultures were positive for S pyogenes, and a swab of the right breast was negative for bacterial growth.
The patient was admitted to the medical intensive care unit (MICU) and placed on two vasopressors for control of low BP and assistance with low urine output. After a 6-L fluid resuscitation, the patient was started on vancomycin and piperacillin/tazobactam for possible cellulitis causing sepsis. An echocardiogram was negative for endocarditis. The patient continued to decline the following day with continuing tachycardia and tachypnea with hypotension and was intubated. Pulmonology services was consulted for possible acute respiratory distress syndrome secondary to sepsis; general surgery services was consulted for possible necrotizing fasciitis of the chest wall; and cardiology services was consulted for low-cardiac output.
On hospital day 4, the patient was taken to surgery for exploration, drainage, and debridement of the right axilla and breast; cultures were taken, and lack of organism growth was noted. While in the MICU, she was followed by the infectious disease service as her WBC remained elevated and peaking at 32.6 x 103, while blood cultures were negative for bacterial growth.
The dental service was consulted on hospital day 5 to evaluate for other possible sources of infection. Upon examination, the patient’s oral condition was noted as having advanced chronic periodontal disease that required full-mouth extraction. The patient remained hemodynamically unstable with platelet counts below 50 x 109/L until hospital day 7, at which time she was taken for surgery for full mouth extraction and associated alveoloplasty. Upon extraction, the patient continued to improve and was extubated on day 11 with platelets and WBC returning to normal levels by day 13 of her hospital stay. The patient remained hospitalized for a total MICU stay of 20 days and rehabilitation stay of more than 2 weeks.
Discussion
Oral infections most often present with acute onset and noted oral-facial cellulitis or abscess. Oral source of septicemia often are considered after ruling out most other potential sources. Although it is not certain that this case is directly related to the advanced chronic periodontal disease, S pyogenes has been noted to be a pathogen in periodontal disease progression.
According to the American Dental Association in 2012, dental visits to the ED cost the US health care system $1.6 billion and an average cost of $749 per visit. There are more than 2 million ED visits each year for dental pain and infection, and 39% return due to nonresolution of the dental problem. Patients return to the ED due to lack of access and resources to routine and emergent dental care. 2 The average daily cost of an MICU stay with mechanical ventilation was $2,193 in 2002. This particular case consisted of 11 days of mechanical ventilation, 20 MICU days, and an additional 20 days of inpatient rehabilitation which resulted in costs that exceeded $50,000. 3
1. Krishnan V, Johnson JV, Helfric JF. Management of maxillofacial infections: a review of 50 cases. J Oral Maxillofac Surg. 1993;51(8):868-873; discussion 873-874.
2. Wall T, Vujicic M. Emergency department use for dental conditions continues to increase. American Dental Association: Health Policy Institute. http://www.ada.org/~/media/ADA/Science%20and%20Research/HPI/Files/HPIBrief_0415_2.ashx. Published April 2015. Accessed November 6, 2017.
3. Dasta JF, McLaughlin TP, Mody SH, Piech CT. Daily cost of an intensive care unit day: the contribution of mechanical ventilation. Crit Care Med. 2005;33(6):1266-1271.
Sepsis can be the result of single or multiple factors and sources of infection. Oral sources of sepsis and systemic infection are not commonly considered as the first potential source of infection when evaluating a septic patient. Oral infections of odontogenic or periodontal origin are frequently associated with localized or diffuse cellulitis of the head and neck region. 1 The patient’s health status and complicating problems, such as an immunocompromising condition, can further reduce the immune response for controlling chronic sources of infection. This in turn can lead to acute manifestations such as cellulitis, sepsis, or necrotizing fasciitis. Necrotizing fasciitis is caused by a polymicrobial or mixed aerobic-anaerobic infection from a variety of sources, including Streptococcus pyogenes .
Case
A 57-year-old woman with a history of major depressive disorder, paroxysmal atrial fibrillation, and opioid dependence that was in remission for more than 3 years was brought to the ED by a family member after the patient developed confusion and lethargy. She was primarily experiencing right breast pain and swelling. The breast pain was associated with high fevers, nausea, vomiting, and chills.
On examination, the patient’s vital signs were: blood pressure (BP), 109/58 mm Hg; heart rate, 160 beats/min; respiratory rate, 22 breaths/min; and temperature, 104°F. Laboratory evaluation was significant for a white blood cell count (WBC) of 8.7 x 103. There was a noted skin abrasion on the patient’s right hand. She was lethargic and confused. Blood cultures were positive for S pyogenes, and a swab of the right breast was negative for bacterial growth.
The patient was admitted to the medical intensive care unit (MICU) and placed on two vasopressors for control of low BP and assistance with low urine output. After a 6-L fluid resuscitation, the patient was started on vancomycin and piperacillin/tazobactam for possible cellulitis causing sepsis. An echocardiogram was negative for endocarditis. The patient continued to decline the following day with continuing tachycardia and tachypnea with hypotension and was intubated. Pulmonology services was consulted for possible acute respiratory distress syndrome secondary to sepsis; general surgery services was consulted for possible necrotizing fasciitis of the chest wall; and cardiology services was consulted for low-cardiac output.
On hospital day 4, the patient was taken to surgery for exploration, drainage, and debridement of the right axilla and breast; cultures were taken, and lack of organism growth was noted. While in the MICU, she was followed by the infectious disease service as her WBC remained elevated and peaking at 32.6 x 103, while blood cultures were negative for bacterial growth.
The dental service was consulted on hospital day 5 to evaluate for other possible sources of infection. Upon examination, the patient’s oral condition was noted as having advanced chronic periodontal disease that required full-mouth extraction. The patient remained hemodynamically unstable with platelet counts below 50 x 109/L until hospital day 7, at which time she was taken for surgery for full mouth extraction and associated alveoloplasty. Upon extraction, the patient continued to improve and was extubated on day 11 with platelets and WBC returning to normal levels by day 13 of her hospital stay. The patient remained hospitalized for a total MICU stay of 20 days and rehabilitation stay of more than 2 weeks.
Discussion
Oral infections most often present with acute onset and noted oral-facial cellulitis or abscess. Oral source of septicemia often are considered after ruling out most other potential sources. Although it is not certain that this case is directly related to the advanced chronic periodontal disease, S pyogenes has been noted to be a pathogen in periodontal disease progression.
According to the American Dental Association in 2012, dental visits to the ED cost the US health care system $1.6 billion and an average cost of $749 per visit. There are more than 2 million ED visits each year for dental pain and infection, and 39% return due to nonresolution of the dental problem. Patients return to the ED due to lack of access and resources to routine and emergent dental care. 2 The average daily cost of an MICU stay with mechanical ventilation was $2,193 in 2002. This particular case consisted of 11 days of mechanical ventilation, 20 MICU days, and an additional 20 days of inpatient rehabilitation which resulted in costs that exceeded $50,000. 3
Sepsis can be the result of single or multiple factors and sources of infection. Oral sources of sepsis and systemic infection are not commonly considered as the first potential source of infection when evaluating a septic patient. Oral infections of odontogenic or periodontal origin are frequently associated with localized or diffuse cellulitis of the head and neck region. 1 The patient’s health status and complicating problems, such as an immunocompromising condition, can further reduce the immune response for controlling chronic sources of infection. This in turn can lead to acute manifestations such as cellulitis, sepsis, or necrotizing fasciitis. Necrotizing fasciitis is caused by a polymicrobial or mixed aerobic-anaerobic infection from a variety of sources, including Streptococcus pyogenes .
Case
A 57-year-old woman with a history of major depressive disorder, paroxysmal atrial fibrillation, and opioid dependence that was in remission for more than 3 years was brought to the ED by a family member after the patient developed confusion and lethargy. She was primarily experiencing right breast pain and swelling. The breast pain was associated with high fevers, nausea, vomiting, and chills.
On examination, the patient’s vital signs were: blood pressure (BP), 109/58 mm Hg; heart rate, 160 beats/min; respiratory rate, 22 breaths/min; and temperature, 104°F. Laboratory evaluation was significant for a white blood cell count (WBC) of 8.7 x 103. There was a noted skin abrasion on the patient’s right hand. She was lethargic and confused. Blood cultures were positive for S pyogenes, and a swab of the right breast was negative for bacterial growth.
The patient was admitted to the medical intensive care unit (MICU) and placed on two vasopressors for control of low BP and assistance with low urine output. After a 6-L fluid resuscitation, the patient was started on vancomycin and piperacillin/tazobactam for possible cellulitis causing sepsis. An echocardiogram was negative for endocarditis. The patient continued to decline the following day with continuing tachycardia and tachypnea with hypotension and was intubated. Pulmonology services was consulted for possible acute respiratory distress syndrome secondary to sepsis; general surgery services was consulted for possible necrotizing fasciitis of the chest wall; and cardiology services was consulted for low-cardiac output.
On hospital day 4, the patient was taken to surgery for exploration, drainage, and debridement of the right axilla and breast; cultures were taken, and lack of organism growth was noted. While in the MICU, she was followed by the infectious disease service as her WBC remained elevated and peaking at 32.6 x 103, while blood cultures were negative for bacterial growth.
The dental service was consulted on hospital day 5 to evaluate for other possible sources of infection. Upon examination, the patient’s oral condition was noted as having advanced chronic periodontal disease that required full-mouth extraction. The patient remained hemodynamically unstable with platelet counts below 50 x 109/L until hospital day 7, at which time she was taken for surgery for full mouth extraction and associated alveoloplasty. Upon extraction, the patient continued to improve and was extubated on day 11 with platelets and WBC returning to normal levels by day 13 of her hospital stay. The patient remained hospitalized for a total MICU stay of 20 days and rehabilitation stay of more than 2 weeks.
Discussion
Oral infections most often present with acute onset and noted oral-facial cellulitis or abscess. Oral source of septicemia often are considered after ruling out most other potential sources. Although it is not certain that this case is directly related to the advanced chronic periodontal disease, S pyogenes has been noted to be a pathogen in periodontal disease progression.
According to the American Dental Association in 2012, dental visits to the ED cost the US health care system $1.6 billion and an average cost of $749 per visit. There are more than 2 million ED visits each year for dental pain and infection, and 39% return due to nonresolution of the dental problem. Patients return to the ED due to lack of access and resources to routine and emergent dental care. 2 The average daily cost of an MICU stay with mechanical ventilation was $2,193 in 2002. This particular case consisted of 11 days of mechanical ventilation, 20 MICU days, and an additional 20 days of inpatient rehabilitation which resulted in costs that exceeded $50,000. 3
1. Krishnan V, Johnson JV, Helfric JF. Management of maxillofacial infections: a review of 50 cases. J Oral Maxillofac Surg. 1993;51(8):868-873; discussion 873-874.
2. Wall T, Vujicic M. Emergency department use for dental conditions continues to increase. American Dental Association: Health Policy Institute. http://www.ada.org/~/media/ADA/Science%20and%20Research/HPI/Files/HPIBrief_0415_2.ashx. Published April 2015. Accessed November 6, 2017.
3. Dasta JF, McLaughlin TP, Mody SH, Piech CT. Daily cost of an intensive care unit day: the contribution of mechanical ventilation. Crit Care Med. 2005;33(6):1266-1271.
1. Krishnan V, Johnson JV, Helfric JF. Management of maxillofacial infections: a review of 50 cases. J Oral Maxillofac Surg. 1993;51(8):868-873; discussion 873-874.
2. Wall T, Vujicic M. Emergency department use for dental conditions continues to increase. American Dental Association: Health Policy Institute. http://www.ada.org/~/media/ADA/Science%20and%20Research/HPI/Files/HPIBrief_0415_2.ashx. Published April 2015. Accessed November 6, 2017.
3. Dasta JF, McLaughlin TP, Mody SH, Piech CT. Daily cost of an intensive care unit day: the contribution of mechanical ventilation. Crit Care Med. 2005;33(6):1266-1271.
Case Studies in Toxicology: DILI Dally
Case
A 50-year-old Hispanic woman with a history of rheumatoid arthritis (RA), for which she was not currently taking medication, was referred to the ED by her primary care physician (PCP) for evaluation of generalized pruritus and jaundice, and an abnormal hepatic function panel.
The patient’s recent history was significant for a positive tuberculosis test (purified protein derivative [PPD], 13 mm), for which she had been on prophylactic medication. Laboratory evaluation taken during the patient’s recent follow-up visit with her PCP revealed the following significant hepatic abnormalities: total bilirubin, 20.0 mg/dL; direct bilirubin, 16.4 mg/dL; international normalized ratio, 2.9; aspartate aminotransferase, greater than 2,000 IU/L; and alanine aminotransferase, greater than 2,000 IU/L. The patient had no history of hepatic disease, and a hepatitis panel obtained in the ED was unremarkable.
Can this be drug-induced liver injury?
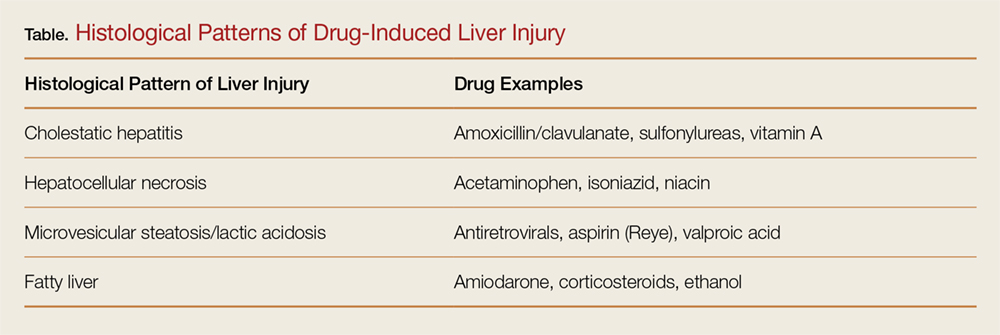
Drug-induced liver injury (DILI) accounts for nearly 50% of cases of acute liver failure in the United States.1 According to the National Institutes of Health database of drugs, supplements, and herbal medications acetaminophen is the most common drug associated with hepatotoxicity in the United States, whereas amoxicillin-clavulanate is the most common implicated drug worldwide.1,2 The histological pattern of DILI varies by drug (Table).3
Who is susceptible to drug-induced liver injury?
The factors that help predict DILI include drug pharmacokinetics and metabolism, as well as patient age, sex, and comorbidities. Although some patients are at an increased risk of DILI, it is extraordinarily difficult to accurately predict which patients will develop it. In general, there is a positive correlation between age and risk of developing DILI. For example, in a large US-based tuberculosis study, the incidence of isoniazid (INH)-induced hepatotoxicity was 4.4 per 1,000 patients aged 25 to 34 years. Patients older than age 50 years had a 20.83 per 1,000 incidence of DILI, and women also appear to be at increased risk.4
Pharmacogenetic factors affecting drug metabolism such as the specific cytochrome profile and acetylator status of an individual also influence a patient’s risk of developing DILI. Although our understanding of these issues is growing rapidly, our ability to apply this knowledge to the clinical venue is limited by the available technology, regulatory requirements, and cost.
Case Continuation
A detailed, careful history-taking in the ED revealed that, 2 months prior, the patient had been started on INH, rifampin, and pyridoxine for latent tuberculosis. She had been taking methotrexate for the RA but discontinued it 3 months ago because of the positive PPD. When routine outpatient laboratory testing results demonstrated significant hepatic dysfunction, the patient’s PCP advised her to immediately discontinue her medications and referred her to the ED for further evaluation and management.
By what mechanism does INH cause DILI?
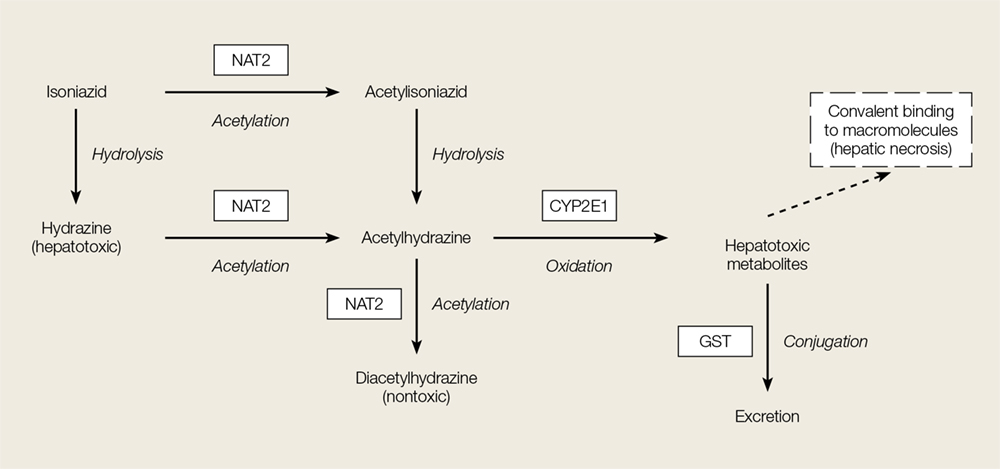
Acute INH-associated hepatitis primarily results from the direct hepatotoxic effects of INH metabolites. Isoniazid is metabolized in the liver via N-acetylation to acetylisoniazid (Figure). Oxidation of this compound in the liver leads to an accumulation of the hepatotoxic metabolites acetylhydrazine and hydrazine.5,6
Is there a role for N-acetylcysteine in INH hepatotoxicity?
No antidote is specifically designed to treat INH-induced hepatotoxicity, and management is largely supportive. Observation for progressive liver failure is indicated and evaluation for liver transplant may become necessary.
N-acetylcysteine (NAC) has a clear role in preventing hepatotoxicity from acetaminophen overdose through its ability to act as a precursor for the synthesis of glutathione—a compound that protects hepatocytes from oxidative damage. In advanced acetaminophen-toxic patients and those with non-acetaminophen toxicity, NAC has nonspecific effects that promote healing through several mechanisms, including anti-inflammatory effect and enhanced hepatic perfusion. Though there are no studies that specifically evaluate the role of NAC in patients with INH-induced hepatotoxicity, it is commonly and appropriately administered for its aforementioned nonspecific effects.8 Common side effects from NAC administration include nausea, vomiting, and diarrhea, which are generally treatable with symptomatic and supportive care.
Case Conclusion
The patient was admitted to the hepatology service for continued clinical care. Although she received NAC, hepatic function testing showed only mild improvement. Additional etiologies of liver failure were investigated, including a computed tomography scan of the abdomen/pelvis and an abdominal ultrasound with Doppler. Both studies were negative for any pathology, and autoimmune laboratory studies were likewise unremarkable.
The patient underwent a liver biopsy, which revealed inflammation and scattered eosinophils suggestive of drug-induced hepatic injury. Her clinical condition continued to deteriorate, and she was transferred to another hospital for transplant evaluation.
1. Lee WM. Drug-induced acute liver failure. Clin Liver Dis. 2013;17(4):575-586, viii. doi:10.1016/j.cld.2013.07.001.
2. National Institutes of Health Web site. LiverTox: Clinical and research information on drug-induced liver injury. https://livertox.nlm.nih.gov/. Updated February 10, 2017. Accessed October 12, 2017.
3. Ansari JA, Sayyed M, Sayeed F. Management of non alcoholic fatty liver diseases and their complications. Int J Pharmacol. 2011;7:579-588. doi:10.3923/ijp.2011.579.588.
4. Fountain FF, Tolley E, Chrisman CR, Self TH. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: a 7-year evaluation from a public health tuberculosis clinic. Chest. 2005;128(1):116-123. doi:10.1378/chest.128.1.116.
5. Hernon CH. Antituberculous medications. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:787-796.
6. Teixeira RL, Morato RG, Cabello PH, et al. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem Inst Oswaldo Cruz. 2011;106(6):716-724.
7. Mitchell JR, Thorgeirsson UP, Black M, et al. Increased incidence of isoniazid hepatitis in rapid acetylators: possible relation to hydranize metabolites. Clin Pharmacol Ther. 1975;18(1):70-79.
8. Lee WM, Hynan LS, Rossaro L, et al; Acute Liver Failure Study Group. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864. doi:10.1053/j.gastro.2009.06.006.
Case
A 50-year-old Hispanic woman with a history of rheumatoid arthritis (RA), for which she was not currently taking medication, was referred to the ED by her primary care physician (PCP) for evaluation of generalized pruritus and jaundice, and an abnormal hepatic function panel.
The patient’s recent history was significant for a positive tuberculosis test (purified protein derivative [PPD], 13 mm), for which she had been on prophylactic medication. Laboratory evaluation taken during the patient’s recent follow-up visit with her PCP revealed the following significant hepatic abnormalities: total bilirubin, 20.0 mg/dL; direct bilirubin, 16.4 mg/dL; international normalized ratio, 2.9; aspartate aminotransferase, greater than 2,000 IU/L; and alanine aminotransferase, greater than 2,000 IU/L. The patient had no history of hepatic disease, and a hepatitis panel obtained in the ED was unremarkable.
Can this be drug-induced liver injury?
Drug-induced liver injury (DILI) accounts for nearly 50% of cases of acute liver failure in the United States.1 According to the National Institutes of Health database of drugs, supplements, and herbal medications acetaminophen is the most common drug associated with hepatotoxicity in the United States, whereas amoxicillin-clavulanate is the most common implicated drug worldwide.1,2 The histological pattern of DILI varies by drug (Table).3
Who is susceptible to drug-induced liver injury?
The factors that help predict DILI include drug pharmacokinetics and metabolism, as well as patient age, sex, and comorbidities. Although some patients are at an increased risk of DILI, it is extraordinarily difficult to accurately predict which patients will develop it. In general, there is a positive correlation between age and risk of developing DILI. For example, in a large US-based tuberculosis study, the incidence of isoniazid (INH)-induced hepatotoxicity was 4.4 per 1,000 patients aged 25 to 34 years. Patients older than age 50 years had a 20.83 per 1,000 incidence of DILI, and women also appear to be at increased risk.4
Pharmacogenetic factors affecting drug metabolism such as the specific cytochrome profile and acetylator status of an individual also influence a patient’s risk of developing DILI. Although our understanding of these issues is growing rapidly, our ability to apply this knowledge to the clinical venue is limited by the available technology, regulatory requirements, and cost.
Case Continuation
A detailed, careful history-taking in the ED revealed that, 2 months prior, the patient had been started on INH, rifampin, and pyridoxine for latent tuberculosis. She had been taking methotrexate for the RA but discontinued it 3 months ago because of the positive PPD. When routine outpatient laboratory testing results demonstrated significant hepatic dysfunction, the patient’s PCP advised her to immediately discontinue her medications and referred her to the ED for further evaluation and management.
By what mechanism does INH cause DILI?
Acute INH-associated hepatitis primarily results from the direct hepatotoxic effects of INH metabolites. Isoniazid is metabolized in the liver via N-acetylation to acetylisoniazid (Figure). Oxidation of this compound in the liver leads to an accumulation of the hepatotoxic metabolites acetylhydrazine and hydrazine.5,6
Is there a role for N-acetylcysteine in INH hepatotoxicity?
No antidote is specifically designed to treat INH-induced hepatotoxicity, and management is largely supportive. Observation for progressive liver failure is indicated and evaluation for liver transplant may become necessary.
N-acetylcysteine (NAC) has a clear role in preventing hepatotoxicity from acetaminophen overdose through its ability to act as a precursor for the synthesis of glutathione—a compound that protects hepatocytes from oxidative damage. In advanced acetaminophen-toxic patients and those with non-acetaminophen toxicity, NAC has nonspecific effects that promote healing through several mechanisms, including anti-inflammatory effect and enhanced hepatic perfusion. Though there are no studies that specifically evaluate the role of NAC in patients with INH-induced hepatotoxicity, it is commonly and appropriately administered for its aforementioned nonspecific effects.8 Common side effects from NAC administration include nausea, vomiting, and diarrhea, which are generally treatable with symptomatic and supportive care.
Case Conclusion
The patient was admitted to the hepatology service for continued clinical care. Although she received NAC, hepatic function testing showed only mild improvement. Additional etiologies of liver failure were investigated, including a computed tomography scan of the abdomen/pelvis and an abdominal ultrasound with Doppler. Both studies were negative for any pathology, and autoimmune laboratory studies were likewise unremarkable.
The patient underwent a liver biopsy, which revealed inflammation and scattered eosinophils suggestive of drug-induced hepatic injury. Her clinical condition continued to deteriorate, and she was transferred to another hospital for transplant evaluation.
Case
A 50-year-old Hispanic woman with a history of rheumatoid arthritis (RA), for which she was not currently taking medication, was referred to the ED by her primary care physician (PCP) for evaluation of generalized pruritus and jaundice, and an abnormal hepatic function panel.
The patient’s recent history was significant for a positive tuberculosis test (purified protein derivative [PPD], 13 mm), for which she had been on prophylactic medication. Laboratory evaluation taken during the patient’s recent follow-up visit with her PCP revealed the following significant hepatic abnormalities: total bilirubin, 20.0 mg/dL; direct bilirubin, 16.4 mg/dL; international normalized ratio, 2.9; aspartate aminotransferase, greater than 2,000 IU/L; and alanine aminotransferase, greater than 2,000 IU/L. The patient had no history of hepatic disease, and a hepatitis panel obtained in the ED was unremarkable.
Can this be drug-induced liver injury?
Drug-induced liver injury (DILI) accounts for nearly 50% of cases of acute liver failure in the United States.1 According to the National Institutes of Health database of drugs, supplements, and herbal medications acetaminophen is the most common drug associated with hepatotoxicity in the United States, whereas amoxicillin-clavulanate is the most common implicated drug worldwide.1,2 The histological pattern of DILI varies by drug (Table).3
Who is susceptible to drug-induced liver injury?
The factors that help predict DILI include drug pharmacokinetics and metabolism, as well as patient age, sex, and comorbidities. Although some patients are at an increased risk of DILI, it is extraordinarily difficult to accurately predict which patients will develop it. In general, there is a positive correlation between age and risk of developing DILI. For example, in a large US-based tuberculosis study, the incidence of isoniazid (INH)-induced hepatotoxicity was 4.4 per 1,000 patients aged 25 to 34 years. Patients older than age 50 years had a 20.83 per 1,000 incidence of DILI, and women also appear to be at increased risk.4
Pharmacogenetic factors affecting drug metabolism such as the specific cytochrome profile and acetylator status of an individual also influence a patient’s risk of developing DILI. Although our understanding of these issues is growing rapidly, our ability to apply this knowledge to the clinical venue is limited by the available technology, regulatory requirements, and cost.
Case Continuation
A detailed, careful history-taking in the ED revealed that, 2 months prior, the patient had been started on INH, rifampin, and pyridoxine for latent tuberculosis. She had been taking methotrexate for the RA but discontinued it 3 months ago because of the positive PPD. When routine outpatient laboratory testing results demonstrated significant hepatic dysfunction, the patient’s PCP advised her to immediately discontinue her medications and referred her to the ED for further evaluation and management.
By what mechanism does INH cause DILI?
Acute INH-associated hepatitis primarily results from the direct hepatotoxic effects of INH metabolites. Isoniazid is metabolized in the liver via N-acetylation to acetylisoniazid (Figure). Oxidation of this compound in the liver leads to an accumulation of the hepatotoxic metabolites acetylhydrazine and hydrazine.5,6
Is there a role for N-acetylcysteine in INH hepatotoxicity?
No antidote is specifically designed to treat INH-induced hepatotoxicity, and management is largely supportive. Observation for progressive liver failure is indicated and evaluation for liver transplant may become necessary.
N-acetylcysteine (NAC) has a clear role in preventing hepatotoxicity from acetaminophen overdose through its ability to act as a precursor for the synthesis of glutathione—a compound that protects hepatocytes from oxidative damage. In advanced acetaminophen-toxic patients and those with non-acetaminophen toxicity, NAC has nonspecific effects that promote healing through several mechanisms, including anti-inflammatory effect and enhanced hepatic perfusion. Though there are no studies that specifically evaluate the role of NAC in patients with INH-induced hepatotoxicity, it is commonly and appropriately administered for its aforementioned nonspecific effects.8 Common side effects from NAC administration include nausea, vomiting, and diarrhea, which are generally treatable with symptomatic and supportive care.
Case Conclusion
The patient was admitted to the hepatology service for continued clinical care. Although she received NAC, hepatic function testing showed only mild improvement. Additional etiologies of liver failure were investigated, including a computed tomography scan of the abdomen/pelvis and an abdominal ultrasound with Doppler. Both studies were negative for any pathology, and autoimmune laboratory studies were likewise unremarkable.
The patient underwent a liver biopsy, which revealed inflammation and scattered eosinophils suggestive of drug-induced hepatic injury. Her clinical condition continued to deteriorate, and she was transferred to another hospital for transplant evaluation.
1. Lee WM. Drug-induced acute liver failure. Clin Liver Dis. 2013;17(4):575-586, viii. doi:10.1016/j.cld.2013.07.001.
2. National Institutes of Health Web site. LiverTox: Clinical and research information on drug-induced liver injury. https://livertox.nlm.nih.gov/. Updated February 10, 2017. Accessed October 12, 2017.
3. Ansari JA, Sayyed M, Sayeed F. Management of non alcoholic fatty liver diseases and their complications. Int J Pharmacol. 2011;7:579-588. doi:10.3923/ijp.2011.579.588.
4. Fountain FF, Tolley E, Chrisman CR, Self TH. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: a 7-year evaluation from a public health tuberculosis clinic. Chest. 2005;128(1):116-123. doi:10.1378/chest.128.1.116.
5. Hernon CH. Antituberculous medications. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:787-796.
6. Teixeira RL, Morato RG, Cabello PH, et al. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem Inst Oswaldo Cruz. 2011;106(6):716-724.
7. Mitchell JR, Thorgeirsson UP, Black M, et al. Increased incidence of isoniazid hepatitis in rapid acetylators: possible relation to hydranize metabolites. Clin Pharmacol Ther. 1975;18(1):70-79.
8. Lee WM, Hynan LS, Rossaro L, et al; Acute Liver Failure Study Group. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864. doi:10.1053/j.gastro.2009.06.006.
1. Lee WM. Drug-induced acute liver failure. Clin Liver Dis. 2013;17(4):575-586, viii. doi:10.1016/j.cld.2013.07.001.
2. National Institutes of Health Web site. LiverTox: Clinical and research information on drug-induced liver injury. https://livertox.nlm.nih.gov/. Updated February 10, 2017. Accessed October 12, 2017.
3. Ansari JA, Sayyed M, Sayeed F. Management of non alcoholic fatty liver diseases and their complications. Int J Pharmacol. 2011;7:579-588. doi:10.3923/ijp.2011.579.588.
4. Fountain FF, Tolley E, Chrisman CR, Self TH. Isoniazid hepatotoxicity associated with treatment of latent tuberculosis infection: a 7-year evaluation from a public health tuberculosis clinic. Chest. 2005;128(1):116-123. doi:10.1378/chest.128.1.116.
5. Hernon CH. Antituberculous medications. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:787-796.
6. Teixeira RL, Morato RG, Cabello PH, et al. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem Inst Oswaldo Cruz. 2011;106(6):716-724.
7. Mitchell JR, Thorgeirsson UP, Black M, et al. Increased incidence of isoniazid hepatitis in rapid acetylators: possible relation to hydranize metabolites. Clin Pharmacol Ther. 1975;18(1):70-79.
8. Lee WM, Hynan LS, Rossaro L, et al; Acute Liver Failure Study Group. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864. doi:10.1053/j.gastro.2009.06.006.
Carbon Monoxide: The Other Silent Killer
Case Presentations
Case 1: Smoke Inhalation (Carbon Monoxide and Cyanide)
A 50-year-old woman was pulled from the window of a burning building and found to be in cardiac arrest with pulseless electrical activity. Standard advanced cardiac life-support was started, and infusion of intra-osseous hydroxocobalamin (OHCob) was administered at the time of intubation because of the concern for cyanide (CN) gas exposure during smoke inhalation. Return of spontaneous circulation occurred before arrival at the hospital.
Upon presentation to the ED, the patient’s vital signs were: initial blood pressure (BP), 92/47 mm Hg; heart rate (HR), 112 beats/min; respiratory rate (RR), 31 breaths/min; and temperature (T), 99.7°F. Following intubation, the patient’s oxygen saturation (SaO2) on pulse oximetry (POX) was 93%, and her fraction of inspired oxygen (FiO2) was 100%.
On physical examination, the patient’s face was covered with soot. The lung sounds were equal and clear to auscultation bilaterally. The neurological examination was significant for a Glasgow Coma Scale of 3, without administered sedation, and there were no signs of dermal burns. Initial arterial blood gas (ABG) results were: pH, 7.06; carbon dioxide partial pressure (PCO2), 58 mm Hg; partial pressure of oxygen (PO2), 152 mm Hg; bicarbonate (HCO3), 17 mm Hg; SaO2, 98% (after intubation); FiO2, 100%; carboxyhemoglobin (COHb), 30%; and lactate, 14 mmol/L.
Case 2: Household Misadventure (Carbon Monoxide)
Several days after disabling the carbon monoxide (CO) detector in his home to silence the alarm that had continued to sound, a 67-year-old man developed weakness and called his local fire department. Upon arrival at the man’s home, the fire department confirmed an ambient air CO gas concentration over 200 ppm. Emergency medical services (EMS) promptly brought the patient to the local ED for evaluation and treatment.
Shortly after arrival at the ED, the patient’s weakness had resolved. His vital signs at examination were: BP, 154/85 mm Hg; HR, 79 beats/min; RR, 15 breaths/min; and T, 98.8°F. The patient’s COHb level was 28% with administration of 100% oxygen (O2) via a nonrebreather mask (NRBM).
Carbon Monoxide Toxicity
Carbon monoxide is a toxin of considerable importance to emergency physicians (EPs). The diagnosis at times can be challenging, the interpretation of COHb can be confusing, and the role of hyperbaric oxygen (HBO) therapy in the treatment of CO poisoning remains controversial.
Natural Sources
Carbon monoxide is formed from the incomplete combustion of organic (carbonaceous) fuels, such as charcoal, wood, petroleum distillates (gasoline, kerosene, diesel fuel), and natural gas. Though the majority of atmospheric CO comes from natural sources (eg, volcanoes, forest fires, marsh gases), poisoning exposures are primarily due to man-made CO.
Man-Made Sources
Motor vehicle exhaust is the most abundant source of man-made CO, and exposures to exhaust fumes are common causes of both intentional and unintentional poisonings and death. Other frequent sources of CO poisoning include smoke inhalation from house fires; inadequate ventilation during use of kerosene space heaters; charcoal grills or hibachis; burning wood or charcoal; fuel-powered tools such as generators, fork lifts, and chain saws; or faulty (natural or bottled) gas appliances, such as stoves, furnaces, or water heaters (Table 1). Though propane is known to burn more cleanly than natural gas (ie, less harmful to the environment), it still can produce CO.

Though neither electrical appliances nor “gas leaks” are sources of CO, like CO, natural gas (mostly methane) and bottle gas (propane) are odorless, tasteless, and colorless. Utility companies add sulfur containing mercaptans to natural gas so that leaks can be detected, but CO is only formed when the fuel is burned in a gas-powered appliance.
Endogenous Carbon Monoxide
Endogenous CO production can occur from catabolism of heme or from hepatic metabolism of methylene chloride, but exposures to this solvent are unlikely to generate COHb concentrations above 10%.
Epidemiology
The incidence of CO poisoning is likely more frequent than documented since many cases of minor exposures are unreported due to self-limiting effects and/or the vague, nonspecific nature of symptoms associated with minor exposures. In 2015, US Poison Control Centers reported over 14,000 cases of CO poisoning, only 43% of which were treated in a health care facility.1 The vast majority of exposures (97%) were unintentional and resulted in 52 deaths (0.398%).1
Data from hospitalized patients in 2007 revealed that over 200,000 ED visits and 22,000 hospitalizations were possibly associated with unintentional, non-fire-related CO exposures.2 Approximately 10% of the exposures in each of these populations were confirmed by specific International Classification of Diseases Medical E codes.2
Regardless of dataset, ED visits due to CO exposure are most common in young adults and women, occur in winter months from exposure in and around homes, and result in discharge from the ED. Elderly patients have the highest rate of hospital admission.
Carbon monoxide poisoning has long been considered a leading cause of poisoning death, though numbers appear to be declining, and CO was responsible for fewer deaths than opioids in 2017.2 The National Center for Health Statistics reported 56,133 CO-related deaths from 1979 through 1988—an average of 5,600 per year.3,4 Of these, 46% were from suicide; 28% were related to burns or house fires; and 21% (11,547) were characterized as unintentional. Motor vehicle exhaust was associated with 57% of the unintentional deaths. A more recent analysis of unintentional exposures reported 2,244 deaths during the period of 2010 to 2015—an average of 374 deaths per year (393 in 2015).5
Preventive measures are likely responsible for the significant decline in non-fire-related CO poisoning deaths from the early 1970s through the 1990s. The introduction of catalytic converters in automobiles in 1975 and O2 sensors in 1981 eventually reduced automotive CO emissions by 95% compared to pre-1975 vehicles.6 Both unintentional death and suicide rates associated with CO from motor vehicles subsequently declined by 81% and 43%, respectively. The lower decline in suicidal deaths serves as a reminder that intentional exposure to motor vehicles remains dangerous and potentially lethal.
Pathophysiology/Mechanisms of Toxicity
Carbon monoxide is a colorless, odorless gas that readily reaches the bloodstream during alveolar gas exchange. Since absorption is rapid, exposures to high CO concentrations can produce toxicity within minutes, though exposure severity is related to both inspired CO concentration and duration of exposure.
Endogenous Elimination
Carbon monoxide is eliminated from the body in expired air, with an elimination half-life dependent on FiO2 and atmospheric pressure. Accordingly, COHb decreases with a half-life (all approximate) of 4 to 6 hours when patients are breathing room air (21% O2), 60 to 90 minutes with O2 delivery at 95% to 100%, and 20 to 40 minutes under hyperbaric conditions (2.5-3.0 atmospheres absolute [ATA]).
Effect on Hemoglobin
Once absorbed, CO has an affinity for hemoglobin (Hb) that is over 200 times greater than does O2.7 The formation of COHb results in both a decreased O2-carrying capacity of Hb at the sites where O2 would have been, and because of its new configuration, COHb does not allow currently bound O2 to be offloaded. This is graphically represented by a shift of the O2-Hb dissociation curve to the left. In addition, CO continues to be bound by other intracellular heme molecules in myoglobin of skeletal and myocardial muscle, and the cytochrome oxidase system in mitochondria.8
Immunologic and Inflammatory Effects
Carbon monoxide poisoning results in a cascade of immunologic and inflammatory effects, such as generation of nitric oxide, lipid peroxidation from neutrophils, mitochondrial oxidative stress, and apoptosis. These effects result in cellular asphyxia in all organs, but the most emergent life-threatening concerns are ischemia to the brain and heart.
Severity of Toxicity and Exposure
As previously noted, the severity of CO poisoning is dose-dependent, meaning that it is related to the concentration of CO in inspired air and the duration of the exposure. Carbon monoxide is typically absent in fresh air, but levels may approach 2 to 5 ppm due to cooking, wood burning, mild air pollution, etc. The source of levels above 5 ppm should generally be investigated.
Maximum safe exposure levels for workers over an 8-hour period range from 25 to 50 ppm. Exposures to CO levels above 50 to 100 ppm are likely to elicit symptoms in most patients, depending on duration of the exposure. Carbon monoxide levels of 200 ppm may result in a mild headache after 2 to 3 hours of exposure, and a more severe headache and nausea after 1 to 2 hours of exposures to 400 ppm of CO.
Accordingly, home CO detectors use a combination of ppm and time for alarms, and they may not sound an alarm at 40 ppm until the level persists for 8 or so hours. Home CO detectors, however, will sound an alarm immediately when a level of 80 to 100 ppm is reached.
Clinical Presentation
Acute Exposure
Acute exposure to CO causes a variety of effects that are largely nonspecific, as there is no toxic syndrome (toxidrome) considered pathognomonic for CO poisoning. Ambient CO levels, duration of exposure, minute ventilation, presence of other toxic gases, and patient comorbidities can all contribute to the severity of exposure and presenting signs and symptoms. Effects associated with mild poisoning include headache, dizziness, blurred vision, fatigue or weakness, nausea, and shortness of breath. Patients with pre-existing respiratory, cardiovascular (CV), or neurological compromise are likely to present with more pronounced symptoms. In either case, these complaints may easily be confused with a viral illness, emphasizing the importance of eliciting a history of potential exposure to CO, particularly when multiple patients are involved.
As the concentration of COHb increases, more significant clinical effects can be expected, including tachycardia, chest pain, hypotension, dysrhythmias, lethargy, coma, apnea, and seizures. Hypoxia can result in myocardial injury, cerebral edema, stroke, and acute pulmonary and kidney injury.
Following acute exposure, the severity of effects correlates with the peak pretreatment COHb concentration. However, the peak concentration is usually unknown, since most patients with significant exposures will have some time period elapsed between the exposure and the determination of COHb, and the COHb will have declined at a rate depending on FiO2 and minute ventilation. In these circumstances, COHb is a poor indicator for HBO therapy and outcome.
Delayed Neurological Sequelae
Persistent, recurrent, or delayed (following period of no symptoms) neurological effects can occur in up to 40% of cases, and patients with significant exposures (eg, loss of consciousness) appear to be at greatest risk. These effects most often occur within the first 3 weeks following exposure, and have been known to persist for months to years. Such effects include headache, dizziness, impaired memory or cognition, and emotional lability. Predicting which factors in CO exposure and/or treatments can be modified to prevent neurological sequelae remains challenging.
Diagnostic Testing
Pulse Co-oximetry
Prehospital care POX typically reads COHb as oxyhemoglobin, thereby displaying a normal SaO2.9 Noninvasive CO pulse co-oximetry using a pulse oximeter (Rad-57, Masimo Corporation) provides a reading between –6 to +4 of the true COHb with a false-positive rate of 11% and false-negative rate of 46%.10 This high false-negative rate makes noninvasive CO pulse co-oximetry a poor tool to rule out a CO exposure.11 If OHCob has been administered due to concerns for CN poisoning (smoke inhalation), concentrations of COHb detected by a co-oximeter medical device may be decreased, as noted by a mean decrease of 1% in healthy volunteers exposed to OHCob only.12
Venous and Arterial Blood Gas Testing
For a patient in the hospital, exposure to CO can rapidly be determined using co-oximetry to measure COHb in a venous or arterial sample. Obtaining a venous sample may be a more practical approach, as other venous measurements will likely also be obtained. Baseline “normal” COHb levels should be less than 5%, but may be up to 8% to 10% in tobacco smokers.
Other Laboratory Studies
Other important laboratory tests that should be obtained are a complete blood count, lactate level, venous blood gas, and basic metabolic panel (to assess acid/base status). In two retrospective studies of patients exposed to CO, elevated lactate levels were associated with altered mental status.13,14 However, elevated lactate levels were not seen in a majority of patients with CO poisoning.
In addition, CN exposure should be considered when the lactate level is greater than 8 mmol/L, particularly in patients with smoke inhalation.15 Troponin I and creatinine phosphokinase tests can be used to screen for myocardial or skeletal muscle injury.
Effect of Hydroxocobalamin on Laboratory Evaluation
It is important to be cautious when interpreting the results of laboratory studies in patients who have been given OHCob due to the potential co-exposure to CN (smoke inhalation). The red discoloration of body fluids after OHCob administration makes laboratory evaluation by spectrophotometric techniques erroneous.16 Of greatest concern is the accuracy of COHb concentrations.17 In a study using rabbit models by Lee et al,17 OHCob administration was shown to falsely increase COHb concentrations.

Livshits et al18 reported conflicting effects on COHb in two human cases. In the first case, the patient’s true COHb was 93% lower (2.5% vs 34.9%) following administration of 5 g of OHCob, as measured with a rapid blood gas analyzer. In the second patient, COHb was 76% lower (10.7% vs 44%) following OHCob administration, which was also measured by a blood gas analyzer. Both of these cases illustrate lower true COHb concentrations than would be expected following the administration of only supplemental O2.
In a controlled experiment by Pace et al19 examining the effects of OHCob on measurement of COHb at both physiological (3%) and pathological (30% and 50%) concentrations in human blood samples, the degree of interference depended on the type of co-oximeter used, the degree of COHb elevation (at pathological levels only), and the concentration of OHCob added. Other studies, including an evaluation of OHCob interference by Carlsson et al20 using nine different analyzers have confirmed the interference of OHCob on photometric assays. Of particular clinical importance, a falsely increased lactate level was seen after true lactate levels were found to be below 4.8 mmol/L (but not greater) using spectrophotometric or electrochemical detection.21 This increase in the false-positive assessment of the degree of toxicity could lead unnecessary escalation of care.
These studies emphasize the need to exercise caution when interpreting laboratory test results following OHCob administration. Ideally, it would be best if blood samples were obtained prior to OHCob administration by EMS or in the ED, if the clinical scenario allows it.
Imaging Studies
For patients presenting after a closed-space fire, a chest radiograph will help assess for pulmonary injury. The classic finding of CO poisoning on head computed tomography (CT) and magnetic resonance imaging scans is evidence of ischemia in the basal ganglia. The radiographic findings may help determine the diagnosis of the altered mental status patient who presents without a history. An electrocardiogram (ECG) is also useful for detection of myocardial ischemia or dysrhythmias, when signs or symptoms of either are possible from history and physical or cardiac monitoring.
Intentional Inhalation
Intentional inhalation of fumes containing CO is a relatively common mechanism for suicide. In patients who survive, it is important for EPs and other providers to suspect additional means of self-harm. For example, at our institution, we have encountered several patients with self-inflicted trauma after remaining conscious following a medication overdose. Accordingly, patients who have intentionally inhaled CO should also be evaluated for occult medication poisoning (and trauma).
Treatment
The first step in treating a patient with CO poisoning occurs prior to arrival at the ED, when he or she is removed from continued exposure. The second step is assessing whether this is only a CO exposure, or a mixture of gases from combustion in a closed space, that might also contain CN. When CO and CN are combined OHCob is indicated to treat CN toxicity.
Additionally, if the patient is brought to the ED via EMS, O2 therapy will most likely have been initiated en route. In either case, the concentration of COHb may not accurately reflect the magnitude of exposure or prognosis, and should not be used to dictate the level of therapy or disposition. The patient’s vital signs and clinical findings of end organ toxicity should guide the appropriate supportive care.
Supplemental Oxygen Therapy
Initial administration of 100% O2 during assessment of airway, breathing, and circulation is the first step in accelerating the removal of CO from Hb. For patients suffering from smoke inhalation, assessment and establishment of a secure airway when there are signs of soot or burns in the airway must always take precedence over other actions. Continuous cardiac monitoring, POX, observation, and establishment of intravenous access are often needed for detection and management of CV instability or change in mental status in cases of moderate-to-severe CO exposures. Mild exposures with headache, nausea, and flu-like symptoms can be managed with symptomatic treatment and normobaric O2 until resolution of symptoms and improvement in COHb occur.
Hyperbaric Oxygen Therapy
Hyperbaric oxygen therapy involves the delivery of high-flow O2 (typically at 100%) under increased atmospheric pressure (2.5 -3.0 ATA). Oxygen delivered at ambient air pressure (1.0 ATA) is often referred to as normobaric oxygen. Although HBO is best known for its ability to enhance CO elimination, research points to a much more eloquent mitigation of CO toxicity on the molecular level. These mechanisms include an increased amount of dissolved O2 in blood, regeneration of cytochrome oxidase, decreased leukocyte adhesion to microvascular endothelium in the brain, decreased lipid peroxidation in the brain after loss of consciousness, and preservation of adenosine triphosphate.22
For most patients, the majority—if not all—of COHb will be eliminated by the time they present to a suitable HBO chamber. Despite the knowledge that HBO therapy has a positive toxicokinetic effect by increasing the elimination of CO, all of the major, prospective studies on the usefulness of HBO are related to prevention of neuropsychiatric sequelae mediated by immunological and inflammatory effects. The role of HBO in the treatment of CO poisoning has been debated for decades. Multiple studies that differ in methodology, patient populations, delivery of HBO treatments, and assessment of benefits fail to provide a consensus on the role of HBO therapy (Table 2).23-30
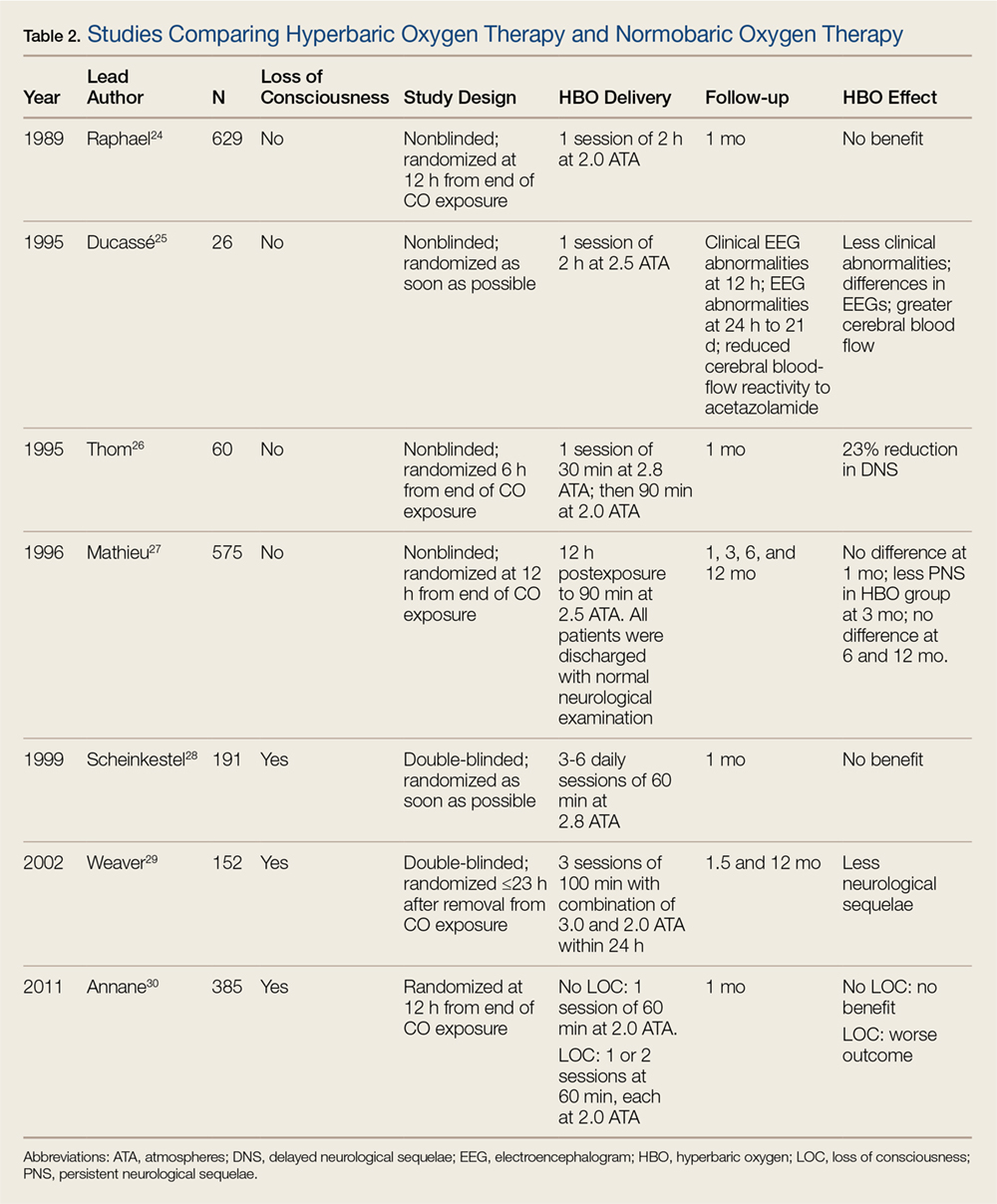
Before transferring a patient to a facility for HBO therapy, the potential risks and benefits of transport must be considered. In a 10-year retrospective study by Sloan et al31 of 297 CO-poisoned patients (mean COHb, 38%) 46% of patients had cardiopulmonary and neurological complications prior to HBO therapy at some point in the transfer pathway. During HBO therapy, 18% of patients had complications that included emesis, agitation requiring sedation, seizures, hypotension, tension pneumothorax, cardiac arrest, cardiac arrhythmias, and myocardial ischemia. It is therefore incumbent that personnel attending patients undergoing HBO therapy for CO poisoning be aware of, and able to manage, this variety of serious effects.
When an HBO chamber is at a clinical site with experts in the field and staff available 24 hours a day, the decision to utilize HBO may easily be made without obstacles. For most EPs, however, this is not the case. Locating and transferring a patient to an HBO center is typically a considerable logistical challenge. For many rural facilities, HBO is just not a timely therapeutic option. Two studies state the benefit of HBO therapy is greatest when starting within 6 hours from the end of the CO exposure.24,26
Identifying those CO-poisoned patients who meet evidence-based criteria for HBO is difficult. Patients with mild CO poisoning will do well without HBO, and critically ill patients will probably not consistently benefit from HBO. However, a pragmatic solution must be considered when efficacy studies are incongruent with conflicting results. When signs of end-organ toxicity from CO are present, but cardiac arrest has not yet occurred and the logistics are streamlined, the benefit of HBO may outweigh the risk.
Signs of end-organ toxicity include syncope, seizures, coma, ischemic changes on ECG, and pregnancy with unresolved maternal distress or fetal distress. Although a COHb level greater than 25% or 15% (pregnant) alone is commonly used as an indication for HBO, this is largely based on opinion. Conversely, HBO is unlikely to be helpful in patients who have been resuscitated after CO-related cardiac arrest.32
Treatment Guidelines
The American College of Emergency Physicians recently developed a position statement regarding the management and treatment of CO poisoning.33 The clinical policy addresses several of the controversies discussed in this review, and provides a level of evidence for each response (Table 3).

Case Conclusions
Case 1 (Smoke Inhalation Due to CO and Cyanide Poisoning)
The patient in this case suffered severe CO and CN toxicity. A head CT scan revealed diffuse edema consistent with anoxic brain injury. After conferring with the family regarding the patient’s condition and prognosis, the decision was made to withdraw life-sustaining therapy and support, and the patient died.
Case 2 (Household Misadventure)
The patient in this case was successfully treated with 100% O2 via a NRBM and was subsequently discharged home within 4 hours from presentation.
Conclusion
Exposures to CO are ubiquitous due to our heavy reliance on carbon combustion, and the manifestations of CO toxicity are protean. Therefore, CO poisoning must be considered more frequently in the differential diagnosis of indiscriminant symptoms affecting the neurological, cardiac, pulmonary, and gastrointestinal systems, especially when multiple patients have similar symptoms.
The diagnosis of CO poisoning is straightforward when a serum COHb level is obtained on a venous or arterial blood sample. Treatment starts when the patient is removed from further CO exposure and breaths normobaric oxygen at ambient levels or supplemented. Because there is no clear evidenced-based indication for HBO therapy, further treatment with HBO is naturally limited by rational constraints.
1. Mowry JB, Spyker DA, Brooks DE, Zimmerman A, Schauben JL. 2015 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 33rd Annual Report. Clin Toxicol. 2016;54(10):924-1109. doi:10.1080/15563650.2016.1245421.
2. Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664. doi:10.1016/j.ajem.2011.03.003.
3. Cobb N, Etzel RA. Unintentional carbon monoxide-related deaths in the United States, 1979 through 1988. JAMA. 1991;266(5):659-663.
4. Sircar K, Clower J, Shin MK, Bailey C, King M, Yip F. Carbon monoxide poisoning deaths in the United States, 1999 to 2012. Am J Emerg Med. 2015;33(9):1140-1145. doi:10.1016/j.ajem.2015.05.002.
5. Centers for Disease Control and Prevention. Environmental Public Health Tracking Network. Carbon monoxide poisoning emergency department visits. https://ephtracking.cdc.gov/showHome.action. Updated September 8, 2017. Accessed October 18, 2017.
6. Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
7. Buckley NA, Juurlink DN, Isbister G, Bennett MH, Lavonas EJ. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2011;13(4):CD002041. doi:10.1002/14651858.CD002041.pub3.
8. Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med. 2012;186(11):1095-1101. doi:10.1164/rccm.201207-1284CI.
9. Bozeman WP, Myers RA, Barish RA. Confirmation of the pulse oximetry gap in carbon monoxide poisoning. Ann of Emerg Med. 1997;30(5):608-611.
10. Zaouter C, Zavorsky GS. The measurement of carboxyhemoglobin and methemoglobin using a non-invasive pulse CO-oximeter. Respir Physiol Neurobiol. 2012;182(2-3):88-92. doi:10.1016/j.resp.2012.05.010.
11. Shamir MY, Avramovich A, Smaka T. The current status of continuous noninvasive measurement of total, carboxy, and methemoglobin concentration. Anesth Analg. 2012;114(5);972-978. doi:10.1213/ANE.0b013e318233041a.
12. Cashin BV, Matlock AG, Kang C, Reynolds PS, Wills BK. Effect of hydroxocobalamin on surface oximetry in nonexposed humans. Prehosp Disaster Med. 2013;28(4):367-369. doi:10.1017/S1049023X13003518.
13. Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011;30(8):836-843. doi:10.1177/0960327110384527.
14. Doğan NÖ, Savrun A, Levent S, et al. Can initial lactate levels predict the severity of unintentional carbon monoxide poisoning? Hum Exp Toxicol. 2015;34(3):324-329. doi:10.1177/0960327114538986.
15. Baud FJ, Borron SW, Mégarbane B, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044-2050. doi:10.1097/01.CCM.0000026325.65944.7D.
16. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5):e22089. doi:10.1002/jcla.22089.
17. Lee J, Mukai D, Kreuter K, Mahon S, Tromberg B, Brenner M. Potential interference by hydroxocobalamin on cooximetry hemoglobin measurements during cyanide and smoke inhalation treatments. Ann Emerg Med. 2007;49(6):802-805. doi:10.
1016/j.annemergmed.2006.11.016.
18. Livshits Z, Lugassy DM, Shawn LK, Hoffman RS. Falsely Low Carboxyhemoglobin after Hydroxocobalamin Therapy [Letter]. N Engl J Med. 2012;367(13):1270-1271. doi:10.1056/NEJMc1114820.
19. Pace R, Bon Homme M, Hoffman RS, Lugassy D. Effects of hydroxocobalamin on carboxyhemoglobin measured under physiologic and pathologic conditions. Clin Toxicol (Phila). 2014;52(7):647-650. doi:10.3109/15563650.2014.939659.
20. Carlsson CJ, Hansen HE, Hilsted L, Malm J, Ødum L, Szecsi PB. An evaluation of the interference of hydroxycobalamin with chemistry and co-oximetry tests on nine commonly used instruments. Scand J Clin Lab Invest. 2011;71(5):378-386. doi:10.3109/00365513.2011.573573.
21. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5). doi:10.1002/jcla.22089.
22. Tomaszewski C. Carbon monoxide. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:1581-1593.
23. Hampson NB, Mathieu D, Piantodosi CA et al. Carbon monoxide poisoning: interpretation of randomized clinical trials and unresolved treatment issues. Undersea Hyperb Med. 2001;28(3):157-164.
24. Raphael JC, Elkharrat D, Jars-Guincestre MC, et al. Trial of normobaric and hyperbaric oxygen for acute carbon monoxide intoxication. Lancet. 1989;2(8660):414-419.
25. Ducassé JL, Celsis P, Marc-Vergnes JP. Non-comatose patients with acute carbon monoxide poisoning: hyperbaric or normobaric oxygenation? Undersea Hyperb Med. 1995;22(1):9-15.
26. Thom SR, Taber RL, Mendiguren II, Clark JM, Hardy KR, Fisher AB. Delayed neuropsychologic sequelae after carbon monoxide poisoning: prevention by treatment with hyperbaric oxygen. Ann Emerg Med. 1995;25(4):474-480.
27. Mathieu D, Wattel F, Mathieu-Nolf M, et al. Randomized prospective study comparing the effects of HBO versus 12 hours of nbp in non comatose CO poisoned patients: results of the interim analysis. Undersea Hyperb Med. 1996;23(Suppl:7-8).
28. Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomized controlled clinical trial. Med J Aust. 1999;170(5):203-210.
29. Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347(14):1057-1067. doi:10.1056/NEJMoa013121.
30. Annane D, Chadda K, Gajdos P, Jars-Guincestre MC, Chevret S, Raphael JC. Hyperbaric oxygen therapy for acute domestic carbon monoxide poisoning: two randomized controlled trials. Intensive Care Med. 2011;37(3):486-492. doi:10.1007/s00134-010-2093-0.
31. Sloan EP, Murphy DG, Hart R, et al. Complications and protocol considerations in carbon monoxide-poisoned patients who require hyperbaric oxygen therapy: report from a ten-year experience. Ann Emerg Med. 1989;18(6):629-634.
32. Hampson NB, Zmaeff JL. Outcome of patients experiencing cardiac arrest with carbon monoxide poisoning treated with hyperbaric oxygen. Ann Emerg Med. 2001;38(1):36-41. doi:10.1067/mem.2001.115532.
33. Wolf SJ, Maloney GE, Shih RD, Shy BD, Brown MD; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute carbon monoxide poisoning. Ann Emerg Med. pii:S0196-0644(17)30332-3. doi:10.1016/j.annemergmed.2017.03.036.
Case Presentations
Case 1: Smoke Inhalation (Carbon Monoxide and Cyanide)
A 50-year-old woman was pulled from the window of a burning building and found to be in cardiac arrest with pulseless electrical activity. Standard advanced cardiac life-support was started, and infusion of intra-osseous hydroxocobalamin (OHCob) was administered at the time of intubation because of the concern for cyanide (CN) gas exposure during smoke inhalation. Return of spontaneous circulation occurred before arrival at the hospital.
Upon presentation to the ED, the patient’s vital signs were: initial blood pressure (BP), 92/47 mm Hg; heart rate (HR), 112 beats/min; respiratory rate (RR), 31 breaths/min; and temperature (T), 99.7°F. Following intubation, the patient’s oxygen saturation (SaO2) on pulse oximetry (POX) was 93%, and her fraction of inspired oxygen (FiO2) was 100%.
On physical examination, the patient’s face was covered with soot. The lung sounds were equal and clear to auscultation bilaterally. The neurological examination was significant for a Glasgow Coma Scale of 3, without administered sedation, and there were no signs of dermal burns. Initial arterial blood gas (ABG) results were: pH, 7.06; carbon dioxide partial pressure (PCO2), 58 mm Hg; partial pressure of oxygen (PO2), 152 mm Hg; bicarbonate (HCO3), 17 mm Hg; SaO2, 98% (after intubation); FiO2, 100%; carboxyhemoglobin (COHb), 30%; and lactate, 14 mmol/L.
Case 2: Household Misadventure (Carbon Monoxide)
Several days after disabling the carbon monoxide (CO) detector in his home to silence the alarm that had continued to sound, a 67-year-old man developed weakness and called his local fire department. Upon arrival at the man’s home, the fire department confirmed an ambient air CO gas concentration over 200 ppm. Emergency medical services (EMS) promptly brought the patient to the local ED for evaluation and treatment.
Shortly after arrival at the ED, the patient’s weakness had resolved. His vital signs at examination were: BP, 154/85 mm Hg; HR, 79 beats/min; RR, 15 breaths/min; and T, 98.8°F. The patient’s COHb level was 28% with administration of 100% oxygen (O2) via a nonrebreather mask (NRBM).
Carbon Monoxide Toxicity
Carbon monoxide is a toxin of considerable importance to emergency physicians (EPs). The diagnosis at times can be challenging, the interpretation of COHb can be confusing, and the role of hyperbaric oxygen (HBO) therapy in the treatment of CO poisoning remains controversial.
Natural Sources
Carbon monoxide is formed from the incomplete combustion of organic (carbonaceous) fuels, such as charcoal, wood, petroleum distillates (gasoline, kerosene, diesel fuel), and natural gas. Though the majority of atmospheric CO comes from natural sources (eg, volcanoes, forest fires, marsh gases), poisoning exposures are primarily due to man-made CO.
Man-Made Sources
Motor vehicle exhaust is the most abundant source of man-made CO, and exposures to exhaust fumes are common causes of both intentional and unintentional poisonings and death. Other frequent sources of CO poisoning include smoke inhalation from house fires; inadequate ventilation during use of kerosene space heaters; charcoal grills or hibachis; burning wood or charcoal; fuel-powered tools such as generators, fork lifts, and chain saws; or faulty (natural or bottled) gas appliances, such as stoves, furnaces, or water heaters (Table 1). Though propane is known to burn more cleanly than natural gas (ie, less harmful to the environment), it still can produce CO.
Though neither electrical appliances nor “gas leaks” are sources of CO, like CO, natural gas (mostly methane) and bottle gas (propane) are odorless, tasteless, and colorless. Utility companies add sulfur containing mercaptans to natural gas so that leaks can be detected, but CO is only formed when the fuel is burned in a gas-powered appliance.
Endogenous Carbon Monoxide
Endogenous CO production can occur from catabolism of heme or from hepatic metabolism of methylene chloride, but exposures to this solvent are unlikely to generate COHb concentrations above 10%.
Epidemiology
The incidence of CO poisoning is likely more frequent than documented since many cases of minor exposures are unreported due to self-limiting effects and/or the vague, nonspecific nature of symptoms associated with minor exposures. In 2015, US Poison Control Centers reported over 14,000 cases of CO poisoning, only 43% of which were treated in a health care facility.1 The vast majority of exposures (97%) were unintentional and resulted in 52 deaths (0.398%).1
Data from hospitalized patients in 2007 revealed that over 200,000 ED visits and 22,000 hospitalizations were possibly associated with unintentional, non-fire-related CO exposures.2 Approximately 10% of the exposures in each of these populations were confirmed by specific International Classification of Diseases Medical E codes.2
Regardless of dataset, ED visits due to CO exposure are most common in young adults and women, occur in winter months from exposure in and around homes, and result in discharge from the ED. Elderly patients have the highest rate of hospital admission.
Carbon monoxide poisoning has long been considered a leading cause of poisoning death, though numbers appear to be declining, and CO was responsible for fewer deaths than opioids in 2017.2 The National Center for Health Statistics reported 56,133 CO-related deaths from 1979 through 1988—an average of 5,600 per year.3,4 Of these, 46% were from suicide; 28% were related to burns or house fires; and 21% (11,547) were characterized as unintentional. Motor vehicle exhaust was associated with 57% of the unintentional deaths. A more recent analysis of unintentional exposures reported 2,244 deaths during the period of 2010 to 2015—an average of 374 deaths per year (393 in 2015).5
Preventive measures are likely responsible for the significant decline in non-fire-related CO poisoning deaths from the early 1970s through the 1990s. The introduction of catalytic converters in automobiles in 1975 and O2 sensors in 1981 eventually reduced automotive CO emissions by 95% compared to pre-1975 vehicles.6 Both unintentional death and suicide rates associated with CO from motor vehicles subsequently declined by 81% and 43%, respectively. The lower decline in suicidal deaths serves as a reminder that intentional exposure to motor vehicles remains dangerous and potentially lethal.
Pathophysiology/Mechanisms of Toxicity
Carbon monoxide is a colorless, odorless gas that readily reaches the bloodstream during alveolar gas exchange. Since absorption is rapid, exposures to high CO concentrations can produce toxicity within minutes, though exposure severity is related to both inspired CO concentration and duration of exposure.
Endogenous Elimination
Carbon monoxide is eliminated from the body in expired air, with an elimination half-life dependent on FiO2 and atmospheric pressure. Accordingly, COHb decreases with a half-life (all approximate) of 4 to 6 hours when patients are breathing room air (21% O2), 60 to 90 minutes with O2 delivery at 95% to 100%, and 20 to 40 minutes under hyperbaric conditions (2.5-3.0 atmospheres absolute [ATA]).
Effect on Hemoglobin
Once absorbed, CO has an affinity for hemoglobin (Hb) that is over 200 times greater than does O2.7 The formation of COHb results in both a decreased O2-carrying capacity of Hb at the sites where O2 would have been, and because of its new configuration, COHb does not allow currently bound O2 to be offloaded. This is graphically represented by a shift of the O2-Hb dissociation curve to the left. In addition, CO continues to be bound by other intracellular heme molecules in myoglobin of skeletal and myocardial muscle, and the cytochrome oxidase system in mitochondria.8
Immunologic and Inflammatory Effects
Carbon monoxide poisoning results in a cascade of immunologic and inflammatory effects, such as generation of nitric oxide, lipid peroxidation from neutrophils, mitochondrial oxidative stress, and apoptosis. These effects result in cellular asphyxia in all organs, but the most emergent life-threatening concerns are ischemia to the brain and heart.
Severity of Toxicity and Exposure
As previously noted, the severity of CO poisoning is dose-dependent, meaning that it is related to the concentration of CO in inspired air and the duration of the exposure. Carbon monoxide is typically absent in fresh air, but levels may approach 2 to 5 ppm due to cooking, wood burning, mild air pollution, etc. The source of levels above 5 ppm should generally be investigated.
Maximum safe exposure levels for workers over an 8-hour period range from 25 to 50 ppm. Exposures to CO levels above 50 to 100 ppm are likely to elicit symptoms in most patients, depending on duration of the exposure. Carbon monoxide levels of 200 ppm may result in a mild headache after 2 to 3 hours of exposure, and a more severe headache and nausea after 1 to 2 hours of exposures to 400 ppm of CO.
Accordingly, home CO detectors use a combination of ppm and time for alarms, and they may not sound an alarm at 40 ppm until the level persists for 8 or so hours. Home CO detectors, however, will sound an alarm immediately when a level of 80 to 100 ppm is reached.
Clinical Presentation
Acute Exposure
Acute exposure to CO causes a variety of effects that are largely nonspecific, as there is no toxic syndrome (toxidrome) considered pathognomonic for CO poisoning. Ambient CO levels, duration of exposure, minute ventilation, presence of other toxic gases, and patient comorbidities can all contribute to the severity of exposure and presenting signs and symptoms. Effects associated with mild poisoning include headache, dizziness, blurred vision, fatigue or weakness, nausea, and shortness of breath. Patients with pre-existing respiratory, cardiovascular (CV), or neurological compromise are likely to present with more pronounced symptoms. In either case, these complaints may easily be confused with a viral illness, emphasizing the importance of eliciting a history of potential exposure to CO, particularly when multiple patients are involved.
As the concentration of COHb increases, more significant clinical effects can be expected, including tachycardia, chest pain, hypotension, dysrhythmias, lethargy, coma, apnea, and seizures. Hypoxia can result in myocardial injury, cerebral edema, stroke, and acute pulmonary and kidney injury.
Following acute exposure, the severity of effects correlates with the peak pretreatment COHb concentration. However, the peak concentration is usually unknown, since most patients with significant exposures will have some time period elapsed between the exposure and the determination of COHb, and the COHb will have declined at a rate depending on FiO2 and minute ventilation. In these circumstances, COHb is a poor indicator for HBO therapy and outcome.
Delayed Neurological Sequelae
Persistent, recurrent, or delayed (following period of no symptoms) neurological effects can occur in up to 40% of cases, and patients with significant exposures (eg, loss of consciousness) appear to be at greatest risk. These effects most often occur within the first 3 weeks following exposure, and have been known to persist for months to years. Such effects include headache, dizziness, impaired memory or cognition, and emotional lability. Predicting which factors in CO exposure and/or treatments can be modified to prevent neurological sequelae remains challenging.
Diagnostic Testing
Pulse Co-oximetry
Prehospital care POX typically reads COHb as oxyhemoglobin, thereby displaying a normal SaO2.9 Noninvasive CO pulse co-oximetry using a pulse oximeter (Rad-57, Masimo Corporation) provides a reading between –6 to +4 of the true COHb with a false-positive rate of 11% and false-negative rate of 46%.10 This high false-negative rate makes noninvasive CO pulse co-oximetry a poor tool to rule out a CO exposure.11 If OHCob has been administered due to concerns for CN poisoning (smoke inhalation), concentrations of COHb detected by a co-oximeter medical device may be decreased, as noted by a mean decrease of 1% in healthy volunteers exposed to OHCob only.12
Venous and Arterial Blood Gas Testing
For a patient in the hospital, exposure to CO can rapidly be determined using co-oximetry to measure COHb in a venous or arterial sample. Obtaining a venous sample may be a more practical approach, as other venous measurements will likely also be obtained. Baseline “normal” COHb levels should be less than 5%, but may be up to 8% to 10% in tobacco smokers.
Other Laboratory Studies
Other important laboratory tests that should be obtained are a complete blood count, lactate level, venous blood gas, and basic metabolic panel (to assess acid/base status). In two retrospective studies of patients exposed to CO, elevated lactate levels were associated with altered mental status.13,14 However, elevated lactate levels were not seen in a majority of patients with CO poisoning.
In addition, CN exposure should be considered when the lactate level is greater than 8 mmol/L, particularly in patients with smoke inhalation.15 Troponin I and creatinine phosphokinase tests can be used to screen for myocardial or skeletal muscle injury.
Effect of Hydroxocobalamin on Laboratory Evaluation
It is important to be cautious when interpreting the results of laboratory studies in patients who have been given OHCob due to the potential co-exposure to CN (smoke inhalation). The red discoloration of body fluids after OHCob administration makes laboratory evaluation by spectrophotometric techniques erroneous.16 Of greatest concern is the accuracy of COHb concentrations.17 In a study using rabbit models by Lee et al,17 OHCob administration was shown to falsely increase COHb concentrations.
Livshits et al18 reported conflicting effects on COHb in two human cases. In the first case, the patient’s true COHb was 93% lower (2.5% vs 34.9%) following administration of 5 g of OHCob, as measured with a rapid blood gas analyzer. In the second patient, COHb was 76% lower (10.7% vs 44%) following OHCob administration, which was also measured by a blood gas analyzer. Both of these cases illustrate lower true COHb concentrations than would be expected following the administration of only supplemental O2.
In a controlled experiment by Pace et al19 examining the effects of OHCob on measurement of COHb at both physiological (3%) and pathological (30% and 50%) concentrations in human blood samples, the degree of interference depended on the type of co-oximeter used, the degree of COHb elevation (at pathological levels only), and the concentration of OHCob added. Other studies, including an evaluation of OHCob interference by Carlsson et al20 using nine different analyzers have confirmed the interference of OHCob on photometric assays. Of particular clinical importance, a falsely increased lactate level was seen after true lactate levels were found to be below 4.8 mmol/L (but not greater) using spectrophotometric or electrochemical detection.21 This increase in the false-positive assessment of the degree of toxicity could lead unnecessary escalation of care.
These studies emphasize the need to exercise caution when interpreting laboratory test results following OHCob administration. Ideally, it would be best if blood samples were obtained prior to OHCob administration by EMS or in the ED, if the clinical scenario allows it.
Imaging Studies
For patients presenting after a closed-space fire, a chest radiograph will help assess for pulmonary injury. The classic finding of CO poisoning on head computed tomography (CT) and magnetic resonance imaging scans is evidence of ischemia in the basal ganglia. The radiographic findings may help determine the diagnosis of the altered mental status patient who presents without a history. An electrocardiogram (ECG) is also useful for detection of myocardial ischemia or dysrhythmias, when signs or symptoms of either are possible from history and physical or cardiac monitoring.
Intentional Inhalation
Intentional inhalation of fumes containing CO is a relatively common mechanism for suicide. In patients who survive, it is important for EPs and other providers to suspect additional means of self-harm. For example, at our institution, we have encountered several patients with self-inflicted trauma after remaining conscious following a medication overdose. Accordingly, patients who have intentionally inhaled CO should also be evaluated for occult medication poisoning (and trauma).
Treatment
The first step in treating a patient with CO poisoning occurs prior to arrival at the ED, when he or she is removed from continued exposure. The second step is assessing whether this is only a CO exposure, or a mixture of gases from combustion in a closed space, that might also contain CN. When CO and CN are combined OHCob is indicated to treat CN toxicity.
Additionally, if the patient is brought to the ED via EMS, O2 therapy will most likely have been initiated en route. In either case, the concentration of COHb may not accurately reflect the magnitude of exposure or prognosis, and should not be used to dictate the level of therapy or disposition. The patient’s vital signs and clinical findings of end organ toxicity should guide the appropriate supportive care.
Supplemental Oxygen Therapy
Initial administration of 100% O2 during assessment of airway, breathing, and circulation is the first step in accelerating the removal of CO from Hb. For patients suffering from smoke inhalation, assessment and establishment of a secure airway when there are signs of soot or burns in the airway must always take precedence over other actions. Continuous cardiac monitoring, POX, observation, and establishment of intravenous access are often needed for detection and management of CV instability or change in mental status in cases of moderate-to-severe CO exposures. Mild exposures with headache, nausea, and flu-like symptoms can be managed with symptomatic treatment and normobaric O2 until resolution of symptoms and improvement in COHb occur.
Hyperbaric Oxygen Therapy
Hyperbaric oxygen therapy involves the delivery of high-flow O2 (typically at 100%) under increased atmospheric pressure (2.5 -3.0 ATA). Oxygen delivered at ambient air pressure (1.0 ATA) is often referred to as normobaric oxygen. Although HBO is best known for its ability to enhance CO elimination, research points to a much more eloquent mitigation of CO toxicity on the molecular level. These mechanisms include an increased amount of dissolved O2 in blood, regeneration of cytochrome oxidase, decreased leukocyte adhesion to microvascular endothelium in the brain, decreased lipid peroxidation in the brain after loss of consciousness, and preservation of adenosine triphosphate.22
For most patients, the majority—if not all—of COHb will be eliminated by the time they present to a suitable HBO chamber. Despite the knowledge that HBO therapy has a positive toxicokinetic effect by increasing the elimination of CO, all of the major, prospective studies on the usefulness of HBO are related to prevention of neuropsychiatric sequelae mediated by immunological and inflammatory effects. The role of HBO in the treatment of CO poisoning has been debated for decades. Multiple studies that differ in methodology, patient populations, delivery of HBO treatments, and assessment of benefits fail to provide a consensus on the role of HBO therapy (Table 2).23-30
Before transferring a patient to a facility for HBO therapy, the potential risks and benefits of transport must be considered. In a 10-year retrospective study by Sloan et al31 of 297 CO-poisoned patients (mean COHb, 38%) 46% of patients had cardiopulmonary and neurological complications prior to HBO therapy at some point in the transfer pathway. During HBO therapy, 18% of patients had complications that included emesis, agitation requiring sedation, seizures, hypotension, tension pneumothorax, cardiac arrest, cardiac arrhythmias, and myocardial ischemia. It is therefore incumbent that personnel attending patients undergoing HBO therapy for CO poisoning be aware of, and able to manage, this variety of serious effects.
When an HBO chamber is at a clinical site with experts in the field and staff available 24 hours a day, the decision to utilize HBO may easily be made without obstacles. For most EPs, however, this is not the case. Locating and transferring a patient to an HBO center is typically a considerable logistical challenge. For many rural facilities, HBO is just not a timely therapeutic option. Two studies state the benefit of HBO therapy is greatest when starting within 6 hours from the end of the CO exposure.24,26
Identifying those CO-poisoned patients who meet evidence-based criteria for HBO is difficult. Patients with mild CO poisoning will do well without HBO, and critically ill patients will probably not consistently benefit from HBO. However, a pragmatic solution must be considered when efficacy studies are incongruent with conflicting results. When signs of end-organ toxicity from CO are present, but cardiac arrest has not yet occurred and the logistics are streamlined, the benefit of HBO may outweigh the risk.
Signs of end-organ toxicity include syncope, seizures, coma, ischemic changes on ECG, and pregnancy with unresolved maternal distress or fetal distress. Although a COHb level greater than 25% or 15% (pregnant) alone is commonly used as an indication for HBO, this is largely based on opinion. Conversely, HBO is unlikely to be helpful in patients who have been resuscitated after CO-related cardiac arrest.32
Treatment Guidelines
The American College of Emergency Physicians recently developed a position statement regarding the management and treatment of CO poisoning.33 The clinical policy addresses several of the controversies discussed in this review, and provides a level of evidence for each response (Table 3).
Case Conclusions
Case 1 (Smoke Inhalation Due to CO and Cyanide Poisoning)
The patient in this case suffered severe CO and CN toxicity. A head CT scan revealed diffuse edema consistent with anoxic brain injury. After conferring with the family regarding the patient’s condition and prognosis, the decision was made to withdraw life-sustaining therapy and support, and the patient died.
Case 2 (Household Misadventure)
The patient in this case was successfully treated with 100% O2 via a NRBM and was subsequently discharged home within 4 hours from presentation.
Conclusion
Exposures to CO are ubiquitous due to our heavy reliance on carbon combustion, and the manifestations of CO toxicity are protean. Therefore, CO poisoning must be considered more frequently in the differential diagnosis of indiscriminant symptoms affecting the neurological, cardiac, pulmonary, and gastrointestinal systems, especially when multiple patients have similar symptoms.
The diagnosis of CO poisoning is straightforward when a serum COHb level is obtained on a venous or arterial blood sample. Treatment starts when the patient is removed from further CO exposure and breaths normobaric oxygen at ambient levels or supplemented. Because there is no clear evidenced-based indication for HBO therapy, further treatment with HBO is naturally limited by rational constraints.
Case Presentations
Case 1: Smoke Inhalation (Carbon Monoxide and Cyanide)
A 50-year-old woman was pulled from the window of a burning building and found to be in cardiac arrest with pulseless electrical activity. Standard advanced cardiac life-support was started, and infusion of intra-osseous hydroxocobalamin (OHCob) was administered at the time of intubation because of the concern for cyanide (CN) gas exposure during smoke inhalation. Return of spontaneous circulation occurred before arrival at the hospital.
Upon presentation to the ED, the patient’s vital signs were: initial blood pressure (BP), 92/47 mm Hg; heart rate (HR), 112 beats/min; respiratory rate (RR), 31 breaths/min; and temperature (T), 99.7°F. Following intubation, the patient’s oxygen saturation (SaO2) on pulse oximetry (POX) was 93%, and her fraction of inspired oxygen (FiO2) was 100%.
On physical examination, the patient’s face was covered with soot. The lung sounds were equal and clear to auscultation bilaterally. The neurological examination was significant for a Glasgow Coma Scale of 3, without administered sedation, and there were no signs of dermal burns. Initial arterial blood gas (ABG) results were: pH, 7.06; carbon dioxide partial pressure (PCO2), 58 mm Hg; partial pressure of oxygen (PO2), 152 mm Hg; bicarbonate (HCO3), 17 mm Hg; SaO2, 98% (after intubation); FiO2, 100%; carboxyhemoglobin (COHb), 30%; and lactate, 14 mmol/L.
Case 2: Household Misadventure (Carbon Monoxide)
Several days after disabling the carbon monoxide (CO) detector in his home to silence the alarm that had continued to sound, a 67-year-old man developed weakness and called his local fire department. Upon arrival at the man’s home, the fire department confirmed an ambient air CO gas concentration over 200 ppm. Emergency medical services (EMS) promptly brought the patient to the local ED for evaluation and treatment.
Shortly after arrival at the ED, the patient’s weakness had resolved. His vital signs at examination were: BP, 154/85 mm Hg; HR, 79 beats/min; RR, 15 breaths/min; and T, 98.8°F. The patient’s COHb level was 28% with administration of 100% oxygen (O2) via a nonrebreather mask (NRBM).
Carbon Monoxide Toxicity
Carbon monoxide is a toxin of considerable importance to emergency physicians (EPs). The diagnosis at times can be challenging, the interpretation of COHb can be confusing, and the role of hyperbaric oxygen (HBO) therapy in the treatment of CO poisoning remains controversial.
Natural Sources
Carbon monoxide is formed from the incomplete combustion of organic (carbonaceous) fuels, such as charcoal, wood, petroleum distillates (gasoline, kerosene, diesel fuel), and natural gas. Though the majority of atmospheric CO comes from natural sources (eg, volcanoes, forest fires, marsh gases), poisoning exposures are primarily due to man-made CO.
Man-Made Sources
Motor vehicle exhaust is the most abundant source of man-made CO, and exposures to exhaust fumes are common causes of both intentional and unintentional poisonings and death. Other frequent sources of CO poisoning include smoke inhalation from house fires; inadequate ventilation during use of kerosene space heaters; charcoal grills or hibachis; burning wood or charcoal; fuel-powered tools such as generators, fork lifts, and chain saws; or faulty (natural or bottled) gas appliances, such as stoves, furnaces, or water heaters (Table 1). Though propane is known to burn more cleanly than natural gas (ie, less harmful to the environment), it still can produce CO.
Though neither electrical appliances nor “gas leaks” are sources of CO, like CO, natural gas (mostly methane) and bottle gas (propane) are odorless, tasteless, and colorless. Utility companies add sulfur containing mercaptans to natural gas so that leaks can be detected, but CO is only formed when the fuel is burned in a gas-powered appliance.
Endogenous Carbon Monoxide
Endogenous CO production can occur from catabolism of heme or from hepatic metabolism of methylene chloride, but exposures to this solvent are unlikely to generate COHb concentrations above 10%.
Epidemiology
The incidence of CO poisoning is likely more frequent than documented since many cases of minor exposures are unreported due to self-limiting effects and/or the vague, nonspecific nature of symptoms associated with minor exposures. In 2015, US Poison Control Centers reported over 14,000 cases of CO poisoning, only 43% of which were treated in a health care facility.1 The vast majority of exposures (97%) were unintentional and resulted in 52 deaths (0.398%).1
Data from hospitalized patients in 2007 revealed that over 200,000 ED visits and 22,000 hospitalizations were possibly associated with unintentional, non-fire-related CO exposures.2 Approximately 10% of the exposures in each of these populations were confirmed by specific International Classification of Diseases Medical E codes.2
Regardless of dataset, ED visits due to CO exposure are most common in young adults and women, occur in winter months from exposure in and around homes, and result in discharge from the ED. Elderly patients have the highest rate of hospital admission.
Carbon monoxide poisoning has long been considered a leading cause of poisoning death, though numbers appear to be declining, and CO was responsible for fewer deaths than opioids in 2017.2 The National Center for Health Statistics reported 56,133 CO-related deaths from 1979 through 1988—an average of 5,600 per year.3,4 Of these, 46% were from suicide; 28% were related to burns or house fires; and 21% (11,547) were characterized as unintentional. Motor vehicle exhaust was associated with 57% of the unintentional deaths. A more recent analysis of unintentional exposures reported 2,244 deaths during the period of 2010 to 2015—an average of 374 deaths per year (393 in 2015).5
Preventive measures are likely responsible for the significant decline in non-fire-related CO poisoning deaths from the early 1970s through the 1990s. The introduction of catalytic converters in automobiles in 1975 and O2 sensors in 1981 eventually reduced automotive CO emissions by 95% compared to pre-1975 vehicles.6 Both unintentional death and suicide rates associated with CO from motor vehicles subsequently declined by 81% and 43%, respectively. The lower decline in suicidal deaths serves as a reminder that intentional exposure to motor vehicles remains dangerous and potentially lethal.
Pathophysiology/Mechanisms of Toxicity
Carbon monoxide is a colorless, odorless gas that readily reaches the bloodstream during alveolar gas exchange. Since absorption is rapid, exposures to high CO concentrations can produce toxicity within minutes, though exposure severity is related to both inspired CO concentration and duration of exposure.
Endogenous Elimination
Carbon monoxide is eliminated from the body in expired air, with an elimination half-life dependent on FiO2 and atmospheric pressure. Accordingly, COHb decreases with a half-life (all approximate) of 4 to 6 hours when patients are breathing room air (21% O2), 60 to 90 minutes with O2 delivery at 95% to 100%, and 20 to 40 minutes under hyperbaric conditions (2.5-3.0 atmospheres absolute [ATA]).
Effect on Hemoglobin
Once absorbed, CO has an affinity for hemoglobin (Hb) that is over 200 times greater than does O2.7 The formation of COHb results in both a decreased O2-carrying capacity of Hb at the sites where O2 would have been, and because of its new configuration, COHb does not allow currently bound O2 to be offloaded. This is graphically represented by a shift of the O2-Hb dissociation curve to the left. In addition, CO continues to be bound by other intracellular heme molecules in myoglobin of skeletal and myocardial muscle, and the cytochrome oxidase system in mitochondria.8
Immunologic and Inflammatory Effects
Carbon monoxide poisoning results in a cascade of immunologic and inflammatory effects, such as generation of nitric oxide, lipid peroxidation from neutrophils, mitochondrial oxidative stress, and apoptosis. These effects result in cellular asphyxia in all organs, but the most emergent life-threatening concerns are ischemia to the brain and heart.
Severity of Toxicity and Exposure
As previously noted, the severity of CO poisoning is dose-dependent, meaning that it is related to the concentration of CO in inspired air and the duration of the exposure. Carbon monoxide is typically absent in fresh air, but levels may approach 2 to 5 ppm due to cooking, wood burning, mild air pollution, etc. The source of levels above 5 ppm should generally be investigated.
Maximum safe exposure levels for workers over an 8-hour period range from 25 to 50 ppm. Exposures to CO levels above 50 to 100 ppm are likely to elicit symptoms in most patients, depending on duration of the exposure. Carbon monoxide levels of 200 ppm may result in a mild headache after 2 to 3 hours of exposure, and a more severe headache and nausea after 1 to 2 hours of exposures to 400 ppm of CO.
Accordingly, home CO detectors use a combination of ppm and time for alarms, and they may not sound an alarm at 40 ppm until the level persists for 8 or so hours. Home CO detectors, however, will sound an alarm immediately when a level of 80 to 100 ppm is reached.
Clinical Presentation
Acute Exposure
Acute exposure to CO causes a variety of effects that are largely nonspecific, as there is no toxic syndrome (toxidrome) considered pathognomonic for CO poisoning. Ambient CO levels, duration of exposure, minute ventilation, presence of other toxic gases, and patient comorbidities can all contribute to the severity of exposure and presenting signs and symptoms. Effects associated with mild poisoning include headache, dizziness, blurred vision, fatigue or weakness, nausea, and shortness of breath. Patients with pre-existing respiratory, cardiovascular (CV), or neurological compromise are likely to present with more pronounced symptoms. In either case, these complaints may easily be confused with a viral illness, emphasizing the importance of eliciting a history of potential exposure to CO, particularly when multiple patients are involved.
As the concentration of COHb increases, more significant clinical effects can be expected, including tachycardia, chest pain, hypotension, dysrhythmias, lethargy, coma, apnea, and seizures. Hypoxia can result in myocardial injury, cerebral edema, stroke, and acute pulmonary and kidney injury.
Following acute exposure, the severity of effects correlates with the peak pretreatment COHb concentration. However, the peak concentration is usually unknown, since most patients with significant exposures will have some time period elapsed between the exposure and the determination of COHb, and the COHb will have declined at a rate depending on FiO2 and minute ventilation. In these circumstances, COHb is a poor indicator for HBO therapy and outcome.
Delayed Neurological Sequelae
Persistent, recurrent, or delayed (following period of no symptoms) neurological effects can occur in up to 40% of cases, and patients with significant exposures (eg, loss of consciousness) appear to be at greatest risk. These effects most often occur within the first 3 weeks following exposure, and have been known to persist for months to years. Such effects include headache, dizziness, impaired memory or cognition, and emotional lability. Predicting which factors in CO exposure and/or treatments can be modified to prevent neurological sequelae remains challenging.
Diagnostic Testing
Pulse Co-oximetry
Prehospital care POX typically reads COHb as oxyhemoglobin, thereby displaying a normal SaO2.9 Noninvasive CO pulse co-oximetry using a pulse oximeter (Rad-57, Masimo Corporation) provides a reading between –6 to +4 of the true COHb with a false-positive rate of 11% and false-negative rate of 46%.10 This high false-negative rate makes noninvasive CO pulse co-oximetry a poor tool to rule out a CO exposure.11 If OHCob has been administered due to concerns for CN poisoning (smoke inhalation), concentrations of COHb detected by a co-oximeter medical device may be decreased, as noted by a mean decrease of 1% in healthy volunteers exposed to OHCob only.12
Venous and Arterial Blood Gas Testing
For a patient in the hospital, exposure to CO can rapidly be determined using co-oximetry to measure COHb in a venous or arterial sample. Obtaining a venous sample may be a more practical approach, as other venous measurements will likely also be obtained. Baseline “normal” COHb levels should be less than 5%, but may be up to 8% to 10% in tobacco smokers.
Other Laboratory Studies
Other important laboratory tests that should be obtained are a complete blood count, lactate level, venous blood gas, and basic metabolic panel (to assess acid/base status). In two retrospective studies of patients exposed to CO, elevated lactate levels were associated with altered mental status.13,14 However, elevated lactate levels were not seen in a majority of patients with CO poisoning.
In addition, CN exposure should be considered when the lactate level is greater than 8 mmol/L, particularly in patients with smoke inhalation.15 Troponin I and creatinine phosphokinase tests can be used to screen for myocardial or skeletal muscle injury.
Effect of Hydroxocobalamin on Laboratory Evaluation
It is important to be cautious when interpreting the results of laboratory studies in patients who have been given OHCob due to the potential co-exposure to CN (smoke inhalation). The red discoloration of body fluids after OHCob administration makes laboratory evaluation by spectrophotometric techniques erroneous.16 Of greatest concern is the accuracy of COHb concentrations.17 In a study using rabbit models by Lee et al,17 OHCob administration was shown to falsely increase COHb concentrations.
Livshits et al18 reported conflicting effects on COHb in two human cases. In the first case, the patient’s true COHb was 93% lower (2.5% vs 34.9%) following administration of 5 g of OHCob, as measured with a rapid blood gas analyzer. In the second patient, COHb was 76% lower (10.7% vs 44%) following OHCob administration, which was also measured by a blood gas analyzer. Both of these cases illustrate lower true COHb concentrations than would be expected following the administration of only supplemental O2.
In a controlled experiment by Pace et al19 examining the effects of OHCob on measurement of COHb at both physiological (3%) and pathological (30% and 50%) concentrations in human blood samples, the degree of interference depended on the type of co-oximeter used, the degree of COHb elevation (at pathological levels only), and the concentration of OHCob added. Other studies, including an evaluation of OHCob interference by Carlsson et al20 using nine different analyzers have confirmed the interference of OHCob on photometric assays. Of particular clinical importance, a falsely increased lactate level was seen after true lactate levels were found to be below 4.8 mmol/L (but not greater) using spectrophotometric or electrochemical detection.21 This increase in the false-positive assessment of the degree of toxicity could lead unnecessary escalation of care.
These studies emphasize the need to exercise caution when interpreting laboratory test results following OHCob administration. Ideally, it would be best if blood samples were obtained prior to OHCob administration by EMS or in the ED, if the clinical scenario allows it.
Imaging Studies
For patients presenting after a closed-space fire, a chest radiograph will help assess for pulmonary injury. The classic finding of CO poisoning on head computed tomography (CT) and magnetic resonance imaging scans is evidence of ischemia in the basal ganglia. The radiographic findings may help determine the diagnosis of the altered mental status patient who presents without a history. An electrocardiogram (ECG) is also useful for detection of myocardial ischemia or dysrhythmias, when signs or symptoms of either are possible from history and physical or cardiac monitoring.
Intentional Inhalation
Intentional inhalation of fumes containing CO is a relatively common mechanism for suicide. In patients who survive, it is important for EPs and other providers to suspect additional means of self-harm. For example, at our institution, we have encountered several patients with self-inflicted trauma after remaining conscious following a medication overdose. Accordingly, patients who have intentionally inhaled CO should also be evaluated for occult medication poisoning (and trauma).
Treatment
The first step in treating a patient with CO poisoning occurs prior to arrival at the ED, when he or she is removed from continued exposure. The second step is assessing whether this is only a CO exposure, or a mixture of gases from combustion in a closed space, that might also contain CN. When CO and CN are combined OHCob is indicated to treat CN toxicity.
Additionally, if the patient is brought to the ED via EMS, O2 therapy will most likely have been initiated en route. In either case, the concentration of COHb may not accurately reflect the magnitude of exposure or prognosis, and should not be used to dictate the level of therapy or disposition. The patient’s vital signs and clinical findings of end organ toxicity should guide the appropriate supportive care.
Supplemental Oxygen Therapy
Initial administration of 100% O2 during assessment of airway, breathing, and circulation is the first step in accelerating the removal of CO from Hb. For patients suffering from smoke inhalation, assessment and establishment of a secure airway when there are signs of soot or burns in the airway must always take precedence over other actions. Continuous cardiac monitoring, POX, observation, and establishment of intravenous access are often needed for detection and management of CV instability or change in mental status in cases of moderate-to-severe CO exposures. Mild exposures with headache, nausea, and flu-like symptoms can be managed with symptomatic treatment and normobaric O2 until resolution of symptoms and improvement in COHb occur.
Hyperbaric Oxygen Therapy
Hyperbaric oxygen therapy involves the delivery of high-flow O2 (typically at 100%) under increased atmospheric pressure (2.5 -3.0 ATA). Oxygen delivered at ambient air pressure (1.0 ATA) is often referred to as normobaric oxygen. Although HBO is best known for its ability to enhance CO elimination, research points to a much more eloquent mitigation of CO toxicity on the molecular level. These mechanisms include an increased amount of dissolved O2 in blood, regeneration of cytochrome oxidase, decreased leukocyte adhesion to microvascular endothelium in the brain, decreased lipid peroxidation in the brain after loss of consciousness, and preservation of adenosine triphosphate.22
For most patients, the majority—if not all—of COHb will be eliminated by the time they present to a suitable HBO chamber. Despite the knowledge that HBO therapy has a positive toxicokinetic effect by increasing the elimination of CO, all of the major, prospective studies on the usefulness of HBO are related to prevention of neuropsychiatric sequelae mediated by immunological and inflammatory effects. The role of HBO in the treatment of CO poisoning has been debated for decades. Multiple studies that differ in methodology, patient populations, delivery of HBO treatments, and assessment of benefits fail to provide a consensus on the role of HBO therapy (Table 2).23-30
Before transferring a patient to a facility for HBO therapy, the potential risks and benefits of transport must be considered. In a 10-year retrospective study by Sloan et al31 of 297 CO-poisoned patients (mean COHb, 38%) 46% of patients had cardiopulmonary and neurological complications prior to HBO therapy at some point in the transfer pathway. During HBO therapy, 18% of patients had complications that included emesis, agitation requiring sedation, seizures, hypotension, tension pneumothorax, cardiac arrest, cardiac arrhythmias, and myocardial ischemia. It is therefore incumbent that personnel attending patients undergoing HBO therapy for CO poisoning be aware of, and able to manage, this variety of serious effects.
When an HBO chamber is at a clinical site with experts in the field and staff available 24 hours a day, the decision to utilize HBO may easily be made without obstacles. For most EPs, however, this is not the case. Locating and transferring a patient to an HBO center is typically a considerable logistical challenge. For many rural facilities, HBO is just not a timely therapeutic option. Two studies state the benefit of HBO therapy is greatest when starting within 6 hours from the end of the CO exposure.24,26
Identifying those CO-poisoned patients who meet evidence-based criteria for HBO is difficult. Patients with mild CO poisoning will do well without HBO, and critically ill patients will probably not consistently benefit from HBO. However, a pragmatic solution must be considered when efficacy studies are incongruent with conflicting results. When signs of end-organ toxicity from CO are present, but cardiac arrest has not yet occurred and the logistics are streamlined, the benefit of HBO may outweigh the risk.
Signs of end-organ toxicity include syncope, seizures, coma, ischemic changes on ECG, and pregnancy with unresolved maternal distress or fetal distress. Although a COHb level greater than 25% or 15% (pregnant) alone is commonly used as an indication for HBO, this is largely based on opinion. Conversely, HBO is unlikely to be helpful in patients who have been resuscitated after CO-related cardiac arrest.32
Treatment Guidelines
The American College of Emergency Physicians recently developed a position statement regarding the management and treatment of CO poisoning.33 The clinical policy addresses several of the controversies discussed in this review, and provides a level of evidence for each response (Table 3).
Case Conclusions
Case 1 (Smoke Inhalation Due to CO and Cyanide Poisoning)
The patient in this case suffered severe CO and CN toxicity. A head CT scan revealed diffuse edema consistent with anoxic brain injury. After conferring with the family regarding the patient’s condition and prognosis, the decision was made to withdraw life-sustaining therapy and support, and the patient died.
Case 2 (Household Misadventure)
The patient in this case was successfully treated with 100% O2 via a NRBM and was subsequently discharged home within 4 hours from presentation.
Conclusion
Exposures to CO are ubiquitous due to our heavy reliance on carbon combustion, and the manifestations of CO toxicity are protean. Therefore, CO poisoning must be considered more frequently in the differential diagnosis of indiscriminant symptoms affecting the neurological, cardiac, pulmonary, and gastrointestinal systems, especially when multiple patients have similar symptoms.
The diagnosis of CO poisoning is straightforward when a serum COHb level is obtained on a venous or arterial blood sample. Treatment starts when the patient is removed from further CO exposure and breaths normobaric oxygen at ambient levels or supplemented. Because there is no clear evidenced-based indication for HBO therapy, further treatment with HBO is naturally limited by rational constraints.
1. Mowry JB, Spyker DA, Brooks DE, Zimmerman A, Schauben JL. 2015 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 33rd Annual Report. Clin Toxicol. 2016;54(10):924-1109. doi:10.1080/15563650.2016.1245421.
2. Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664. doi:10.1016/j.ajem.2011.03.003.
3. Cobb N, Etzel RA. Unintentional carbon monoxide-related deaths in the United States, 1979 through 1988. JAMA. 1991;266(5):659-663.
4. Sircar K, Clower J, Shin MK, Bailey C, King M, Yip F. Carbon monoxide poisoning deaths in the United States, 1999 to 2012. Am J Emerg Med. 2015;33(9):1140-1145. doi:10.1016/j.ajem.2015.05.002.
5. Centers for Disease Control and Prevention. Environmental Public Health Tracking Network. Carbon monoxide poisoning emergency department visits. https://ephtracking.cdc.gov/showHome.action. Updated September 8, 2017. Accessed October 18, 2017.
6. Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
7. Buckley NA, Juurlink DN, Isbister G, Bennett MH, Lavonas EJ. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2011;13(4):CD002041. doi:10.1002/14651858.CD002041.pub3.
8. Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med. 2012;186(11):1095-1101. doi:10.1164/rccm.201207-1284CI.
9. Bozeman WP, Myers RA, Barish RA. Confirmation of the pulse oximetry gap in carbon monoxide poisoning. Ann of Emerg Med. 1997;30(5):608-611.
10. Zaouter C, Zavorsky GS. The measurement of carboxyhemoglobin and methemoglobin using a non-invasive pulse CO-oximeter. Respir Physiol Neurobiol. 2012;182(2-3):88-92. doi:10.1016/j.resp.2012.05.010.
11. Shamir MY, Avramovich A, Smaka T. The current status of continuous noninvasive measurement of total, carboxy, and methemoglobin concentration. Anesth Analg. 2012;114(5);972-978. doi:10.1213/ANE.0b013e318233041a.
12. Cashin BV, Matlock AG, Kang C, Reynolds PS, Wills BK. Effect of hydroxocobalamin on surface oximetry in nonexposed humans. Prehosp Disaster Med. 2013;28(4):367-369. doi:10.1017/S1049023X13003518.
13. Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011;30(8):836-843. doi:10.1177/0960327110384527.
14. Doğan NÖ, Savrun A, Levent S, et al. Can initial lactate levels predict the severity of unintentional carbon monoxide poisoning? Hum Exp Toxicol. 2015;34(3):324-329. doi:10.1177/0960327114538986.
15. Baud FJ, Borron SW, Mégarbane B, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044-2050. doi:10.1097/01.CCM.0000026325.65944.7D.
16. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5):e22089. doi:10.1002/jcla.22089.
17. Lee J, Mukai D, Kreuter K, Mahon S, Tromberg B, Brenner M. Potential interference by hydroxocobalamin on cooximetry hemoglobin measurements during cyanide and smoke inhalation treatments. Ann Emerg Med. 2007;49(6):802-805. doi:10.
1016/j.annemergmed.2006.11.016.
18. Livshits Z, Lugassy DM, Shawn LK, Hoffman RS. Falsely Low Carboxyhemoglobin after Hydroxocobalamin Therapy [Letter]. N Engl J Med. 2012;367(13):1270-1271. doi:10.1056/NEJMc1114820.
19. Pace R, Bon Homme M, Hoffman RS, Lugassy D. Effects of hydroxocobalamin on carboxyhemoglobin measured under physiologic and pathologic conditions. Clin Toxicol (Phila). 2014;52(7):647-650. doi:10.3109/15563650.2014.939659.
20. Carlsson CJ, Hansen HE, Hilsted L, Malm J, Ødum L, Szecsi PB. An evaluation of the interference of hydroxycobalamin with chemistry and co-oximetry tests on nine commonly used instruments. Scand J Clin Lab Invest. 2011;71(5):378-386. doi:10.3109/00365513.2011.573573.
21. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5). doi:10.1002/jcla.22089.
22. Tomaszewski C. Carbon monoxide. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:1581-1593.
23. Hampson NB, Mathieu D, Piantodosi CA et al. Carbon monoxide poisoning: interpretation of randomized clinical trials and unresolved treatment issues. Undersea Hyperb Med. 2001;28(3):157-164.
24. Raphael JC, Elkharrat D, Jars-Guincestre MC, et al. Trial of normobaric and hyperbaric oxygen for acute carbon monoxide intoxication. Lancet. 1989;2(8660):414-419.
25. Ducassé JL, Celsis P, Marc-Vergnes JP. Non-comatose patients with acute carbon monoxide poisoning: hyperbaric or normobaric oxygenation? Undersea Hyperb Med. 1995;22(1):9-15.
26. Thom SR, Taber RL, Mendiguren II, Clark JM, Hardy KR, Fisher AB. Delayed neuropsychologic sequelae after carbon monoxide poisoning: prevention by treatment with hyperbaric oxygen. Ann Emerg Med. 1995;25(4):474-480.
27. Mathieu D, Wattel F, Mathieu-Nolf M, et al. Randomized prospective study comparing the effects of HBO versus 12 hours of nbp in non comatose CO poisoned patients: results of the interim analysis. Undersea Hyperb Med. 1996;23(Suppl:7-8).
28. Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomized controlled clinical trial. Med J Aust. 1999;170(5):203-210.
29. Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347(14):1057-1067. doi:10.1056/NEJMoa013121.
30. Annane D, Chadda K, Gajdos P, Jars-Guincestre MC, Chevret S, Raphael JC. Hyperbaric oxygen therapy for acute domestic carbon monoxide poisoning: two randomized controlled trials. Intensive Care Med. 2011;37(3):486-492. doi:10.1007/s00134-010-2093-0.
31. Sloan EP, Murphy DG, Hart R, et al. Complications and protocol considerations in carbon monoxide-poisoned patients who require hyperbaric oxygen therapy: report from a ten-year experience. Ann Emerg Med. 1989;18(6):629-634.
32. Hampson NB, Zmaeff JL. Outcome of patients experiencing cardiac arrest with carbon monoxide poisoning treated with hyperbaric oxygen. Ann Emerg Med. 2001;38(1):36-41. doi:10.1067/mem.2001.115532.
33. Wolf SJ, Maloney GE, Shih RD, Shy BD, Brown MD; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute carbon monoxide poisoning. Ann Emerg Med. pii:S0196-0644(17)30332-3. doi:10.1016/j.annemergmed.2017.03.036.
1. Mowry JB, Spyker DA, Brooks DE, Zimmerman A, Schauben JL. 2015 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 33rd Annual Report. Clin Toxicol. 2016;54(10):924-1109. doi:10.1080/15563650.2016.1245421.
2. Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664. doi:10.1016/j.ajem.2011.03.003.
3. Cobb N, Etzel RA. Unintentional carbon monoxide-related deaths in the United States, 1979 through 1988. JAMA. 1991;266(5):659-663.
4. Sircar K, Clower J, Shin MK, Bailey C, King M, Yip F. Carbon monoxide poisoning deaths in the United States, 1999 to 2012. Am J Emerg Med. 2015;33(9):1140-1145. doi:10.1016/j.ajem.2015.05.002.
5. Centers for Disease Control and Prevention. Environmental Public Health Tracking Network. Carbon monoxide poisoning emergency department visits. https://ephtracking.cdc.gov/showHome.action. Updated September 8, 2017. Accessed October 18, 2017.
6. Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
7. Buckley NA, Juurlink DN, Isbister G, Bennett MH, Lavonas EJ. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2011;13(4):CD002041. doi:10.1002/14651858.CD002041.pub3.
8. Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med. 2012;186(11):1095-1101. doi:10.1164/rccm.201207-1284CI.
9. Bozeman WP, Myers RA, Barish RA. Confirmation of the pulse oximetry gap in carbon monoxide poisoning. Ann of Emerg Med. 1997;30(5):608-611.
10. Zaouter C, Zavorsky GS. The measurement of carboxyhemoglobin and methemoglobin using a non-invasive pulse CO-oximeter. Respir Physiol Neurobiol. 2012;182(2-3):88-92. doi:10.1016/j.resp.2012.05.010.
11. Shamir MY, Avramovich A, Smaka T. The current status of continuous noninvasive measurement of total, carboxy, and methemoglobin concentration. Anesth Analg. 2012;114(5);972-978. doi:10.1213/ANE.0b013e318233041a.
12. Cashin BV, Matlock AG, Kang C, Reynolds PS, Wills BK. Effect of hydroxocobalamin on surface oximetry in nonexposed humans. Prehosp Disaster Med. 2013;28(4):367-369. doi:10.1017/S1049023X13003518.
13. Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011;30(8):836-843. doi:10.1177/0960327110384527.
14. Doğan NÖ, Savrun A, Levent S, et al. Can initial lactate levels predict the severity of unintentional carbon monoxide poisoning? Hum Exp Toxicol. 2015;34(3):324-329. doi:10.1177/0960327114538986.
15. Baud FJ, Borron SW, Mégarbane B, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044-2050. doi:10.1097/01.CCM.0000026325.65944.7D.
16. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5):e22089. doi:10.1002/jcla.22089.
17. Lee J, Mukai D, Kreuter K, Mahon S, Tromberg B, Brenner M. Potential interference by hydroxocobalamin on cooximetry hemoglobin measurements during cyanide and smoke inhalation treatments. Ann Emerg Med. 2007;49(6):802-805. doi:10.
1016/j.annemergmed.2006.11.016.
18. Livshits Z, Lugassy DM, Shawn LK, Hoffman RS. Falsely Low Carboxyhemoglobin after Hydroxocobalamin Therapy [Letter]. N Engl J Med. 2012;367(13):1270-1271. doi:10.1056/NEJMc1114820.
19. Pace R, Bon Homme M, Hoffman RS, Lugassy D. Effects of hydroxocobalamin on carboxyhemoglobin measured under physiologic and pathologic conditions. Clin Toxicol (Phila). 2014;52(7):647-650. doi:10.3109/15563650.2014.939659.
20. Carlsson CJ, Hansen HE, Hilsted L, Malm J, Ødum L, Szecsi PB. An evaluation of the interference of hydroxycobalamin with chemistry and co-oximetry tests on nine commonly used instruments. Scand J Clin Lab Invest. 2011;71(5):378-386. doi:10.3109/00365513.2011.573573.
21. Fueyo L, Robles J, Aguilar I, Yáñez AM, Socias M, Parera M. Hemolysis index to detect degree of hydroxocobalamin interference with common laboratory tests. J Clin Lab Anal. 2017;31(5). doi:10.1002/jcla.22089.
22. Tomaszewski C. Carbon monoxide. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:1581-1593.
23. Hampson NB, Mathieu D, Piantodosi CA et al. Carbon monoxide poisoning: interpretation of randomized clinical trials and unresolved treatment issues. Undersea Hyperb Med. 2001;28(3):157-164.
24. Raphael JC, Elkharrat D, Jars-Guincestre MC, et al. Trial of normobaric and hyperbaric oxygen for acute carbon monoxide intoxication. Lancet. 1989;2(8660):414-419.
25. Ducassé JL, Celsis P, Marc-Vergnes JP. Non-comatose patients with acute carbon monoxide poisoning: hyperbaric or normobaric oxygenation? Undersea Hyperb Med. 1995;22(1):9-15.
26. Thom SR, Taber RL, Mendiguren II, Clark JM, Hardy KR, Fisher AB. Delayed neuropsychologic sequelae after carbon monoxide poisoning: prevention by treatment with hyperbaric oxygen. Ann Emerg Med. 1995;25(4):474-480.
27. Mathieu D, Wattel F, Mathieu-Nolf M, et al. Randomized prospective study comparing the effects of HBO versus 12 hours of nbp in non comatose CO poisoned patients: results of the interim analysis. Undersea Hyperb Med. 1996;23(Suppl:7-8).
28. Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomized controlled clinical trial. Med J Aust. 1999;170(5):203-210.
29. Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347(14):1057-1067. doi:10.1056/NEJMoa013121.
30. Annane D, Chadda K, Gajdos P, Jars-Guincestre MC, Chevret S, Raphael JC. Hyperbaric oxygen therapy for acute domestic carbon monoxide poisoning: two randomized controlled trials. Intensive Care Med. 2011;37(3):486-492. doi:10.1007/s00134-010-2093-0.
31. Sloan EP, Murphy DG, Hart R, et al. Complications and protocol considerations in carbon monoxide-poisoned patients who require hyperbaric oxygen therapy: report from a ten-year experience. Ann Emerg Med. 1989;18(6):629-634.
32. Hampson NB, Zmaeff JL. Outcome of patients experiencing cardiac arrest with carbon monoxide poisoning treated with hyperbaric oxygen. Ann Emerg Med. 2001;38(1):36-41. doi:10.1067/mem.2001.115532.
33. Wolf SJ, Maloney GE, Shih RD, Shy BD, Brown MD; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute carbon monoxide poisoning. Ann Emerg Med. pii:S0196-0644(17)30332-3. doi:10.1016/j.annemergmed.2017.03.036.

Natural and Unnatural Disasters
Between late August and early November of this year, three strong Gulf Coast and Atlantic hurricanes and several intense, fast-moving northern California forest fires claimed more than 285 lives and caused countless additional injuries and illnesses. During the same period, three unnatural disasters—in Las Vegas, New York City (NYC), and now Sutherland Springs, Texas—were responsible for a total of 84 deaths and 558 injuries. Emergency physicians (EPs) and our colleagues helped deal with the aftermath of all of these incidents, saving lives and ameliorating survivors’ pain and suffering. But ironically, preventing future deaths and injuries from natural disasters may be easier than preventing loss of life from depraved human behavior.
An October 9, 2017 Wall Street Journal (WSJ) article by Jeanne Whalen entitled “Training Ground for Military Trauma Experts: U.S. Gun Violence,” describes how military surgeons helped treat victims of the Las Vegas shooting, one of several arrangements across the United States where steady gun violence provides a training ground that experts can then use on the battlefield. The article includes a photograph of Tom Scalea, MD, Chief of the R. Adams Cowley (Maryland) Shock Trauma Center and EM board member, operating with the assistance of an Air Force surgeon “embedded” at the hospital.
Before September 11, 2001, US hospitals looked to military surgeons experienced in treating combat injuries to direct and staff their trauma centers. Now, the military looks to US hospitals to provide their surgeons with experience treating victims of gun violence, explosives, and high-speed vehicular injuries prior to sending them into war zones! In the week before this issue of EM went to press, a terrorist driving a rental truck down an NYC bicycle path killed 8 people and injured 11 near the site of the 1993 and 2001 World Trade Center attacks. Five days later, 26 church worshipers near Austin, Texas lost their lives and 20 more were seriously injured when a lone gunman shot them with an assault rifle.
The gun violence statistics in this country are staggering. According to the nonprofit Gun Violence Archive (GVA; http://www.gunviolencearchive.org/), from January 1 through November 8, 2017 there have been 52,719 incidents resulting in 13,245 deaths and 27,111
The pervasiveness of the gun culture in this country offers little hope of eliminating such incidents in the future, which makes it especially important for all EPs to be skilled in state-of-the-art trauma management. (See parts I and II of “The changing landscape of trauma care” in the July and August 2017 issues of EM [www.mdedge.com/emed-journal]). As Baltimore trauma surgeon Tom Scalea notes in the WSJ article cited earlier, “Mass shooting? That’s every weekend.…it makes me despondent….I don’t have the ability to make that go away. I have the ability to keep as many alive as I can, and we’re pretty good at it.”
As for preventing deaths from natural disasters, more accurate weather forecasting and newer technology offer more hope. Among the 134 storm-related deaths from Hurricane Irma in September, 14 were heat-related after the storm disabled a transformer supplying power to the air conditioning system of a Hollywood, Florida nursing home. A new state law will now require all nursing homes to have adequate backup generators. But for the increasing numbers of older persons with comorbidities, taking multiple medications, and living in hot climates, air conditioning must be considered life support equipment that requires immediate repair or replacement when it fails—or transfer of the residents to a cool facility.
If only we could someday also prevent terrorism and other acts of senseless violence.
*The GVA defines a mass shooting as a single incident resulting in 4 or more people (not including the shooter) shot and/or killed at the same general time and location.
Between late August and early November of this year, three strong Gulf Coast and Atlantic hurricanes and several intense, fast-moving northern California forest fires claimed more than 285 lives and caused countless additional injuries and illnesses. During the same period, three unnatural disasters—in Las Vegas, New York City (NYC), and now Sutherland Springs, Texas—were responsible for a total of 84 deaths and 558 injuries. Emergency physicians (EPs) and our colleagues helped deal with the aftermath of all of these incidents, saving lives and ameliorating survivors’ pain and suffering. But ironically, preventing future deaths and injuries from natural disasters may be easier than preventing loss of life from depraved human behavior.
An October 9, 2017 Wall Street Journal (WSJ) article by Jeanne Whalen entitled “Training Ground for Military Trauma Experts: U.S. Gun Violence,” describes how military surgeons helped treat victims of the Las Vegas shooting, one of several arrangements across the United States where steady gun violence provides a training ground that experts can then use on the battlefield. The article includes a photograph of Tom Scalea, MD, Chief of the R. Adams Cowley (Maryland) Shock Trauma Center and EM board member, operating with the assistance of an Air Force surgeon “embedded” at the hospital.
Before September 11, 2001, US hospitals looked to military surgeons experienced in treating combat injuries to direct and staff their trauma centers. Now, the military looks to US hospitals to provide their surgeons with experience treating victims of gun violence, explosives, and high-speed vehicular injuries prior to sending them into war zones! In the week before this issue of EM went to press, a terrorist driving a rental truck down an NYC bicycle path killed 8 people and injured 11 near the site of the 1993 and 2001 World Trade Center attacks. Five days later, 26 church worshipers near Austin, Texas lost their lives and 20 more were seriously injured when a lone gunman shot them with an assault rifle.
The gun violence statistics in this country are staggering. According to the nonprofit Gun Violence Archive (GVA; http://www.gunviolencearchive.org/), from January 1 through November 8, 2017 there have been 52,719 incidents resulting in 13,245 deaths and 27,111
The pervasiveness of the gun culture in this country offers little hope of eliminating such incidents in the future, which makes it especially important for all EPs to be skilled in state-of-the-art trauma management. (See parts I and II of “The changing landscape of trauma care” in the July and August 2017 issues of EM [www.mdedge.com/emed-journal]). As Baltimore trauma surgeon Tom Scalea notes in the WSJ article cited earlier, “Mass shooting? That’s every weekend.…it makes me despondent….I don’t have the ability to make that go away. I have the ability to keep as many alive as I can, and we’re pretty good at it.”
As for preventing deaths from natural disasters, more accurate weather forecasting and newer technology offer more hope. Among the 134 storm-related deaths from Hurricane Irma in September, 14 were heat-related after the storm disabled a transformer supplying power to the air conditioning system of a Hollywood, Florida nursing home. A new state law will now require all nursing homes to have adequate backup generators. But for the increasing numbers of older persons with comorbidities, taking multiple medications, and living in hot climates, air conditioning must be considered life support equipment that requires immediate repair or replacement when it fails—or transfer of the residents to a cool facility.
If only we could someday also prevent terrorism and other acts of senseless violence.
*The GVA defines a mass shooting as a single incident resulting in 4 or more people (not including the shooter) shot and/or killed at the same general time and location.
Between late August and early November of this year, three strong Gulf Coast and Atlantic hurricanes and several intense, fast-moving northern California forest fires claimed more than 285 lives and caused countless additional injuries and illnesses. During the same period, three unnatural disasters—in Las Vegas, New York City (NYC), and now Sutherland Springs, Texas—were responsible for a total of 84 deaths and 558 injuries. Emergency physicians (EPs) and our colleagues helped deal with the aftermath of all of these incidents, saving lives and ameliorating survivors’ pain and suffering. But ironically, preventing future deaths and injuries from natural disasters may be easier than preventing loss of life from depraved human behavior.
An October 9, 2017 Wall Street Journal (WSJ) article by Jeanne Whalen entitled “Training Ground for Military Trauma Experts: U.S. Gun Violence,” describes how military surgeons helped treat victims of the Las Vegas shooting, one of several arrangements across the United States where steady gun violence provides a training ground that experts can then use on the battlefield. The article includes a photograph of Tom Scalea, MD, Chief of the R. Adams Cowley (Maryland) Shock Trauma Center and EM board member, operating with the assistance of an Air Force surgeon “embedded” at the hospital.
Before September 11, 2001, US hospitals looked to military surgeons experienced in treating combat injuries to direct and staff their trauma centers. Now, the military looks to US hospitals to provide their surgeons with experience treating victims of gun violence, explosives, and high-speed vehicular injuries prior to sending them into war zones! In the week before this issue of EM went to press, a terrorist driving a rental truck down an NYC bicycle path killed 8 people and injured 11 near the site of the 1993 and 2001 World Trade Center attacks. Five days later, 26 church worshipers near Austin, Texas lost their lives and 20 more were seriously injured when a lone gunman shot them with an assault rifle.
The gun violence statistics in this country are staggering. According to the nonprofit Gun Violence Archive (GVA; http://www.gunviolencearchive.org/), from January 1 through November 8, 2017 there have been 52,719 incidents resulting in 13,245 deaths and 27,111
The pervasiveness of the gun culture in this country offers little hope of eliminating such incidents in the future, which makes it especially important for all EPs to be skilled in state-of-the-art trauma management. (See parts I and II of “The changing landscape of trauma care” in the July and August 2017 issues of EM [www.mdedge.com/emed-journal]). As Baltimore trauma surgeon Tom Scalea notes in the WSJ article cited earlier, “Mass shooting? That’s every weekend.…it makes me despondent….I don’t have the ability to make that go away. I have the ability to keep as many alive as I can, and we’re pretty good at it.”
As for preventing deaths from natural disasters, more accurate weather forecasting and newer technology offer more hope. Among the 134 storm-related deaths from Hurricane Irma in September, 14 were heat-related after the storm disabled a transformer supplying power to the air conditioning system of a Hollywood, Florida nursing home. A new state law will now require all nursing homes to have adequate backup generators. But for the increasing numbers of older persons with comorbidities, taking multiple medications, and living in hot climates, air conditioning must be considered life support equipment that requires immediate repair or replacement when it fails—or transfer of the residents to a cool facility.
If only we could someday also prevent terrorism and other acts of senseless violence.
*The GVA defines a mass shooting as a single incident resulting in 4 or more people (not including the shooter) shot and/or killed at the same general time and location.

Back to Basics: An Uncommon, Unrelated Presentation of a Common Disease
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
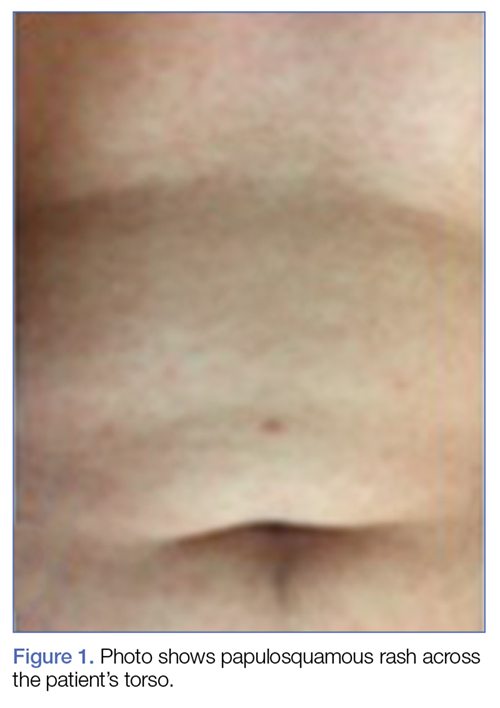
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
Home noninvasive ventilation reduces COPD readmissions
Clinical question: Is there a benefit to home noninvasive ventilation (NIV) following a hospital admission for chronic obstructive pulmonary disease (COPD) exacerbation?
Background: Preventing hospital readmission following a COPD exacerbation is a priority; however, the role of NIV in this situation remains uncertain.

Setting: 13 medical centers in the United Kingdom.
Synopsis: Investigators randomized 116 patients with COPD and persistent hypercapnia (paCO2 less than 53) 2-4 weeks following a COPD exacerbation to either home oxygen therapy with NIV or to home oxygen therapy alone. The study’s primary endpoint was a composite of time to readmission or death within 12 months. They found that the median time to this endpoint was significantly longer in the intervention group (1.4 vs. 4.3 months; 95% CI, 0.31-0.77; P = .002) and that the absolute risk reduction was 17.0% (80.4% vs. 63.4%; 95% CI, 0.1%-34.0%). The differences were driven by readmissions, as the mortality rate did not differ significantly between groups, although the study was not powered to evaluate this. Of note, the median NIV settings were 24/4, which constitutes a “high-pressure strategy” which may account for the benefits seen in this study that have been absent in some other trials.
Bottom line: NIV reduced readmissions in patients with COPD and persistent hypercapnia several weeks following an acute exacerbation.
Citation: Murphy PB, Rehal S, Arbane G, et al. Effect of home noninvasive ventilation with oxygen therapy vs. oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation, a randomized clinical trial. JAMA. 2017;317(21):2177-86.
Dr. Herscher is assistant professor, division of hospital medicine, Icahn School of Medicine of the Mount Sinai Health System.
Clinical question: Is there a benefit to home noninvasive ventilation (NIV) following a hospital admission for chronic obstructive pulmonary disease (COPD) exacerbation?
Background: Preventing hospital readmission following a COPD exacerbation is a priority; however, the role of NIV in this situation remains uncertain.
Setting: 13 medical centers in the United Kingdom.
Synopsis: Investigators randomized 116 patients with COPD and persistent hypercapnia (paCO2 less than 53) 2-4 weeks following a COPD exacerbation to either home oxygen therapy with NIV or to home oxygen therapy alone. The study’s primary endpoint was a composite of time to readmission or death within 12 months. They found that the median time to this endpoint was significantly longer in the intervention group (1.4 vs. 4.3 months; 95% CI, 0.31-0.77; P = .002) and that the absolute risk reduction was 17.0% (80.4% vs. 63.4%; 95% CI, 0.1%-34.0%). The differences were driven by readmissions, as the mortality rate did not differ significantly between groups, although the study was not powered to evaluate this. Of note, the median NIV settings were 24/4, which constitutes a “high-pressure strategy” which may account for the benefits seen in this study that have been absent in some other trials.
Bottom line: NIV reduced readmissions in patients with COPD and persistent hypercapnia several weeks following an acute exacerbation.
Citation: Murphy PB, Rehal S, Arbane G, et al. Effect of home noninvasive ventilation with oxygen therapy vs. oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation, a randomized clinical trial. JAMA. 2017;317(21):2177-86.
Dr. Herscher is assistant professor, division of hospital medicine, Icahn School of Medicine of the Mount Sinai Health System.
Clinical question: Is there a benefit to home noninvasive ventilation (NIV) following a hospital admission for chronic obstructive pulmonary disease (COPD) exacerbation?
Background: Preventing hospital readmission following a COPD exacerbation is a priority; however, the role of NIV in this situation remains uncertain.
Setting: 13 medical centers in the United Kingdom.
Synopsis: Investigators randomized 116 patients with COPD and persistent hypercapnia (paCO2 less than 53) 2-4 weeks following a COPD exacerbation to either home oxygen therapy with NIV or to home oxygen therapy alone. The study’s primary endpoint was a composite of time to readmission or death within 12 months. They found that the median time to this endpoint was significantly longer in the intervention group (1.4 vs. 4.3 months; 95% CI, 0.31-0.77; P = .002) and that the absolute risk reduction was 17.0% (80.4% vs. 63.4%; 95% CI, 0.1%-34.0%). The differences were driven by readmissions, as the mortality rate did not differ significantly between groups, although the study was not powered to evaluate this. Of note, the median NIV settings were 24/4, which constitutes a “high-pressure strategy” which may account for the benefits seen in this study that have been absent in some other trials.
Bottom line: NIV reduced readmissions in patients with COPD and persistent hypercapnia several weeks following an acute exacerbation.
Citation: Murphy PB, Rehal S, Arbane G, et al. Effect of home noninvasive ventilation with oxygen therapy vs. oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation, a randomized clinical trial. JAMA. 2017;317(21):2177-86.
Dr. Herscher is assistant professor, division of hospital medicine, Icahn School of Medicine of the Mount Sinai Health System.
Duodenal Perforation After Endoscopic Procedure
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure). 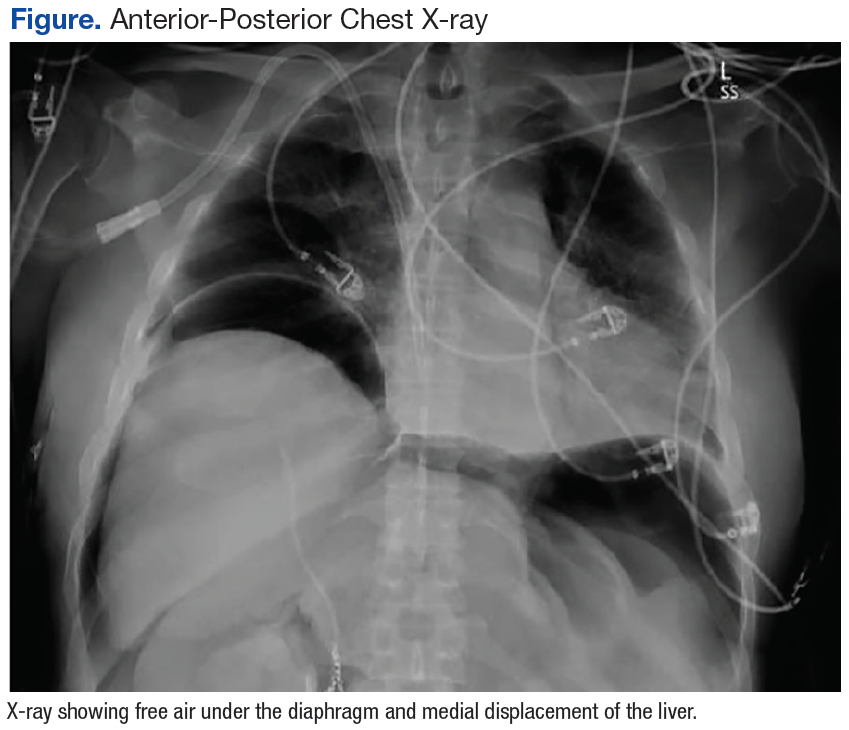
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure).
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure).
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
FDA approves letermovir as CMV prophylaxis

The US Food and Drug Administration (FDA) has approved the oral and intravenous formulations of letermovir (PREVYMIS™).
Letermovir is a member of a new class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The FDA approved letermovir as prophylaxis for cytomegalovirus (CMV) infection and disease in adult recipients of allogeneic hematopoietic stem cell transplants (HSCTs) who are CMV-seropositive.
“PREVYMIS is the first new medicine for CMV infection approved in the US in 15 years,” said Roy Baynes, senior vice president, head of clinical development, and chief medical officer of Merck Research Laboratories, the company marketing letermovir.
Letermovir is expected to be available in December. The list price (wholesaler acquisition cost) per day is $195.00 for letermovir tablets and $270.00 for letermovir injections. (Wholesaler acquisition costs do not include discounts that may be paid on the product.)
The recommended dosage of letermovir is 480 mg once daily, initiated as early as day 0 and up to day 28 post-transplant (before or after engraftment) and continued through day 100. If letermovir is co-administered with cyclosporine, the dosage of letermovir should be decreased to 240 mg once daily.
Letermovir is available as 240 mg and 480 mg tablets, which may be administered with or without food. Letermovir is also available as a 240 mg and 480 mg injection for intravenous infusion via a peripheral catheter or central venous line at a constant rate over 1 hour.
For more details on letermovir, see the full prescribing information.
Trial results
The FDA’s approval of letermovir was supported by results of a phase 3 trial of adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%).
Results from this trial were presented at the 2017 BMT Tandem Meetings. ![]()
The US Food and Drug Administration (FDA) has approved the oral and intravenous formulations of letermovir (PREVYMIS™).
Letermovir is a member of a new class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The FDA approved letermovir as prophylaxis for cytomegalovirus (CMV) infection and disease in adult recipients of allogeneic hematopoietic stem cell transplants (HSCTs) who are CMV-seropositive.
“PREVYMIS is the first new medicine for CMV infection approved in the US in 15 years,” said Roy Baynes, senior vice president, head of clinical development, and chief medical officer of Merck Research Laboratories, the company marketing letermovir.
Letermovir is expected to be available in December. The list price (wholesaler acquisition cost) per day is $195.00 for letermovir tablets and $270.00 for letermovir injections. (Wholesaler acquisition costs do not include discounts that may be paid on the product.)
The recommended dosage of letermovir is 480 mg once daily, initiated as early as day 0 and up to day 28 post-transplant (before or after engraftment) and continued through day 100. If letermovir is co-administered with cyclosporine, the dosage of letermovir should be decreased to 240 mg once daily.
Letermovir is available as 240 mg and 480 mg tablets, which may be administered with or without food. Letermovir is also available as a 240 mg and 480 mg injection for intravenous infusion via a peripheral catheter or central venous line at a constant rate over 1 hour.
For more details on letermovir, see the full prescribing information.
Trial results
The FDA’s approval of letermovir was supported by results of a phase 3 trial of adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%).
Results from this trial were presented at the 2017 BMT Tandem Meetings. ![]()
The US Food and Drug Administration (FDA) has approved the oral and intravenous formulations of letermovir (PREVYMIS™).
Letermovir is a member of a new class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The FDA approved letermovir as prophylaxis for cytomegalovirus (CMV) infection and disease in adult recipients of allogeneic hematopoietic stem cell transplants (HSCTs) who are CMV-seropositive.
“PREVYMIS is the first new medicine for CMV infection approved in the US in 15 years,” said Roy Baynes, senior vice president, head of clinical development, and chief medical officer of Merck Research Laboratories, the company marketing letermovir.
Letermovir is expected to be available in December. The list price (wholesaler acquisition cost) per day is $195.00 for letermovir tablets and $270.00 for letermovir injections. (Wholesaler acquisition costs do not include discounts that may be paid on the product.)
The recommended dosage of letermovir is 480 mg once daily, initiated as early as day 0 and up to day 28 post-transplant (before or after engraftment) and continued through day 100. If letermovir is co-administered with cyclosporine, the dosage of letermovir should be decreased to 240 mg once daily.
Letermovir is available as 240 mg and 480 mg tablets, which may be administered with or without food. Letermovir is also available as a 240 mg and 480 mg injection for intravenous infusion via a peripheral catheter or central venous line at a constant rate over 1 hour.
For more details on letermovir, see the full prescribing information.
Trial results
The FDA’s approval of letermovir was supported by results of a phase 3 trial of adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%).
Results from this trial were presented at the 2017 BMT Tandem Meetings. ![]()
FDA approves brentuximab vedotin for pcALCL, MF

The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
FDA approves IV formulation of aprepitant for CINV

The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()