User login
AGA Clinical Practice Update: Best practice advice on EBT use released
The AGA Institute has released a series of new best practice statements that gastroenterologists should use when considering a patient for endoscopic bariatric treatments or surgeries (EBTs).
“There is a need for less-invasive weight loss therapies that are more effective and durable than lifestyle interventions alone, less invasive and risky than bariatric surgery, and easily performed at a lower expense than that of surgery, thereby allowing improved access and application to a larger segment of the population with moderate obesity,” wrote the authors of the expert review, led by Barham K. Abu Dayyeh, MD of the Mayo Clinic in Rochester, Minn. The report is in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.035). “[EBTs] potentially meet these criteria and may provide an effective treatment approach to obesity in selected patients.”

The best practice statements come from a review of relevant studies in the Ovid, MEDLINE, EMBASE, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus databases, among others, that were published between Jan. 1, 2000, and Sept. 30, 2016.
EBTs should be used on patients who have already been unable to lose weight despite lifestyle interventions and more traditional weight loss methods. However, patients that undergo EBTs should also be placed on a weight loss regimen that includes diet, exercise, and lifestyle changes.

In addition to being used for weight loss, they can also be used to transition a patient to traditional bariatric surgery, or to lower a patient’s weight so that they can undergo a different procedure unrelated to bariatric surgery. Anyone being considered for EBT, or a weight loss regimen involving EBT, should be thoroughly evaluated for comorbidities, behavior, or medical concerns that could lead to adverse effects.
Any patients who are placed on EBT regimens should be followed up regularly by their clinicians, to monitor their progress in terms of weight loss and the development of any adverse effects. Should any adverse outcomes arise, alternative therapies should be implemented as soon as possible. Clinicians are advised to know the ins and outs of risks, contraindications, and potential complications related to EBTs before ever implementing them in their practice, let alone recommending them to a patient.
Finally, it’s imperative that health care institutions with EBT programs make sure there are training protocols clinicians must stringently follow before being allowed to perform EBT procedures.
“Moving ahead, it will be important to better incorporate training in obesity management principles into the GI fellowship curriculum to have a more significant impact,” the authors wrote, adding that it’s important to study the “tandem and sequential use of a combination of EBTs and obesity pharmacotherapies in addition to a comprehensive life-style intervention program.”
Dr. Abu Dayyeh disclosed relationships with Apollo Endosurgery, Metamodix, Aspire Bariatric, and GI Dynamics. Other coauthors also disclosed potential conflicting interests.
The AGA Institute has released a series of new best practice statements that gastroenterologists should use when considering a patient for endoscopic bariatric treatments or surgeries (EBTs).
“There is a need for less-invasive weight loss therapies that are more effective and durable than lifestyle interventions alone, less invasive and risky than bariatric surgery, and easily performed at a lower expense than that of surgery, thereby allowing improved access and application to a larger segment of the population with moderate obesity,” wrote the authors of the expert review, led by Barham K. Abu Dayyeh, MD of the Mayo Clinic in Rochester, Minn. The report is in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.035). “[EBTs] potentially meet these criteria and may provide an effective treatment approach to obesity in selected patients.”
The best practice statements come from a review of relevant studies in the Ovid, MEDLINE, EMBASE, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus databases, among others, that were published between Jan. 1, 2000, and Sept. 30, 2016.
EBTs should be used on patients who have already been unable to lose weight despite lifestyle interventions and more traditional weight loss methods. However, patients that undergo EBTs should also be placed on a weight loss regimen that includes diet, exercise, and lifestyle changes.
In addition to being used for weight loss, they can also be used to transition a patient to traditional bariatric surgery, or to lower a patient’s weight so that they can undergo a different procedure unrelated to bariatric surgery. Anyone being considered for EBT, or a weight loss regimen involving EBT, should be thoroughly evaluated for comorbidities, behavior, or medical concerns that could lead to adverse effects.
Any patients who are placed on EBT regimens should be followed up regularly by their clinicians, to monitor their progress in terms of weight loss and the development of any adverse effects. Should any adverse outcomes arise, alternative therapies should be implemented as soon as possible. Clinicians are advised to know the ins and outs of risks, contraindications, and potential complications related to EBTs before ever implementing them in their practice, let alone recommending them to a patient.
Finally, it’s imperative that health care institutions with EBT programs make sure there are training protocols clinicians must stringently follow before being allowed to perform EBT procedures.
“Moving ahead, it will be important to better incorporate training in obesity management principles into the GI fellowship curriculum to have a more significant impact,” the authors wrote, adding that it’s important to study the “tandem and sequential use of a combination of EBTs and obesity pharmacotherapies in addition to a comprehensive life-style intervention program.”
Dr. Abu Dayyeh disclosed relationships with Apollo Endosurgery, Metamodix, Aspire Bariatric, and GI Dynamics. Other coauthors also disclosed potential conflicting interests.
The AGA Institute has released a series of new best practice statements that gastroenterologists should use when considering a patient for endoscopic bariatric treatments or surgeries (EBTs).
“There is a need for less-invasive weight loss therapies that are more effective and durable than lifestyle interventions alone, less invasive and risky than bariatric surgery, and easily performed at a lower expense than that of surgery, thereby allowing improved access and application to a larger segment of the population with moderate obesity,” wrote the authors of the expert review, led by Barham K. Abu Dayyeh, MD of the Mayo Clinic in Rochester, Minn. The report is in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2017.01.035). “[EBTs] potentially meet these criteria and may provide an effective treatment approach to obesity in selected patients.”
The best practice statements come from a review of relevant studies in the Ovid, MEDLINE, EMBASE, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus databases, among others, that were published between Jan. 1, 2000, and Sept. 30, 2016.
EBTs should be used on patients who have already been unable to lose weight despite lifestyle interventions and more traditional weight loss methods. However, patients that undergo EBTs should also be placed on a weight loss regimen that includes diet, exercise, and lifestyle changes.
In addition to being used for weight loss, they can also be used to transition a patient to traditional bariatric surgery, or to lower a patient’s weight so that they can undergo a different procedure unrelated to bariatric surgery. Anyone being considered for EBT, or a weight loss regimen involving EBT, should be thoroughly evaluated for comorbidities, behavior, or medical concerns that could lead to adverse effects.
Any patients who are placed on EBT regimens should be followed up regularly by their clinicians, to monitor their progress in terms of weight loss and the development of any adverse effects. Should any adverse outcomes arise, alternative therapies should be implemented as soon as possible. Clinicians are advised to know the ins and outs of risks, contraindications, and potential complications related to EBTs before ever implementing them in their practice, let alone recommending them to a patient.
Finally, it’s imperative that health care institutions with EBT programs make sure there are training protocols clinicians must stringently follow before being allowed to perform EBT procedures.
“Moving ahead, it will be important to better incorporate training in obesity management principles into the GI fellowship curriculum to have a more significant impact,” the authors wrote, adding that it’s important to study the “tandem and sequential use of a combination of EBTs and obesity pharmacotherapies in addition to a comprehensive life-style intervention program.”
Dr. Abu Dayyeh disclosed relationships with Apollo Endosurgery, Metamodix, Aspire Bariatric, and GI Dynamics. Other coauthors also disclosed potential conflicting interests.
FROM GASTROENTEROLOGY

AGA Clinical Practice Update: PPIs should be prescribed sparingly, carefully
Updated best practice statements regarding the use of proton pump inhibitors first detail what types of patients should be using short and long-term PPIs.
“When PPIs are appropriately prescribed, their benefits are likely to outweigh their risks [but] when PPIs are inappropriately prescribed, modest risks become important because there is no potential benefit,” wrote the authors of the updated guidance, published in the March issue of Gastroenterology.
“There is currently insufficient evidence to recommend specific strategies for mitigating PPI adverse effects,” noted Daniel E. Freedberg, MD, of Columbia University, New York, and his colleagues.
PPIs should be used on a short-term basis for individuals with gastroesophageal reflux disease (GERD) or conditions such as erosive esophagitis. These patients can also use PPIs for maintenance and occasional symptom management, but those with uncomplicated GERD should be weaned off PPIs if they respond favorably to them.
If a patient is unable to be weaned off PPIs, then ambulatory esophageal pH and impedance monitoring should be done, as this will allow clinicians to determine if the patient has a functional syndrome or GERD. Lifelong PPI treatment should not be considered until this step is taken, according to the new best practice statements.
“Short-term PPIs are highly effective for uncomplicated GERD [but] because patients who cannot reduce PPIs face lifelong therapy, we would consider testing for an acid-related disorder in this situation,” the authors explained. “However, there is no high-quality evidence on which to base this recommendation.”
Patients who have symptomatic GERD or Barrett’s esophagus, either symptomatic or asymptomatic, should be on long-term PPI treatment. Patients who are at a higher risk for NSAID-induced ulcer bleeding should be taking PPIs if they continue to take NSAIDs.
When recommending long-term PPI treatment for a patient, the patient need not use probiotics on a regular basis; there appears to be no need to routinely check the patient’s bone mineral density, serum creatinine, magnesium, or vitamin B12 level on a regular basis. In addition, they need not consume more than the Recommended Dietary Allowance of calcium, magnesium, or vitamin B12.
Finally, the authors state that “specific PPI formulations should not be selected based on potential risks.” This is because no evidence has been found indicating that PPI formulations can be ranked in any way based on risk.
These recommendations come from the AGA’s Clinical Practice Updates Committee, which pored through studies published through July 2016 in the PubMed, EMbase, and Cochrane library databases. Expert opinions and quality assessments on each study contributed to forming these best practice statements.
“In sum, the best current strategies for mitigating the potential risks of long-term PPIs are to avoid prescribing them when they are not indicated and to reduce them to their minimum dose when they are indicated,” Dr. Freedberg and his colleagues concluded.
The researchers did not report any relevant financial disclosures.
Updated best practice statements regarding the use of proton pump inhibitors first detail what types of patients should be using short and long-term PPIs.
“When PPIs are appropriately prescribed, their benefits are likely to outweigh their risks [but] when PPIs are inappropriately prescribed, modest risks become important because there is no potential benefit,” wrote the authors of the updated guidance, published in the March issue of Gastroenterology.
“There is currently insufficient evidence to recommend specific strategies for mitigating PPI adverse effects,” noted Daniel E. Freedberg, MD, of Columbia University, New York, and his colleagues.
PPIs should be used on a short-term basis for individuals with gastroesophageal reflux disease (GERD) or conditions such as erosive esophagitis. These patients can also use PPIs for maintenance and occasional symptom management, but those with uncomplicated GERD should be weaned off PPIs if they respond favorably to them.
If a patient is unable to be weaned off PPIs, then ambulatory esophageal pH and impedance monitoring should be done, as this will allow clinicians to determine if the patient has a functional syndrome or GERD. Lifelong PPI treatment should not be considered until this step is taken, according to the new best practice statements.
“Short-term PPIs are highly effective for uncomplicated GERD [but] because patients who cannot reduce PPIs face lifelong therapy, we would consider testing for an acid-related disorder in this situation,” the authors explained. “However, there is no high-quality evidence on which to base this recommendation.”
Patients who have symptomatic GERD or Barrett’s esophagus, either symptomatic or asymptomatic, should be on long-term PPI treatment. Patients who are at a higher risk for NSAID-induced ulcer bleeding should be taking PPIs if they continue to take NSAIDs.
When recommending long-term PPI treatment for a patient, the patient need not use probiotics on a regular basis; there appears to be no need to routinely check the patient’s bone mineral density, serum creatinine, magnesium, or vitamin B12 level on a regular basis. In addition, they need not consume more than the Recommended Dietary Allowance of calcium, magnesium, or vitamin B12.
Finally, the authors state that “specific PPI formulations should not be selected based on potential risks.” This is because no evidence has been found indicating that PPI formulations can be ranked in any way based on risk.
These recommendations come from the AGA’s Clinical Practice Updates Committee, which pored through studies published through July 2016 in the PubMed, EMbase, and Cochrane library databases. Expert opinions and quality assessments on each study contributed to forming these best practice statements.
“In sum, the best current strategies for mitigating the potential risks of long-term PPIs are to avoid prescribing them when they are not indicated and to reduce them to their minimum dose when they are indicated,” Dr. Freedberg and his colleagues concluded.
The researchers did not report any relevant financial disclosures.
Updated best practice statements regarding the use of proton pump inhibitors first detail what types of patients should be using short and long-term PPIs.
“When PPIs are appropriately prescribed, their benefits are likely to outweigh their risks [but] when PPIs are inappropriately prescribed, modest risks become important because there is no potential benefit,” wrote the authors of the updated guidance, published in the March issue of Gastroenterology.
“There is currently insufficient evidence to recommend specific strategies for mitigating PPI adverse effects,” noted Daniel E. Freedberg, MD, of Columbia University, New York, and his colleagues.
PPIs should be used on a short-term basis for individuals with gastroesophageal reflux disease (GERD) or conditions such as erosive esophagitis. These patients can also use PPIs for maintenance and occasional symptom management, but those with uncomplicated GERD should be weaned off PPIs if they respond favorably to them.
If a patient is unable to be weaned off PPIs, then ambulatory esophageal pH and impedance monitoring should be done, as this will allow clinicians to determine if the patient has a functional syndrome or GERD. Lifelong PPI treatment should not be considered until this step is taken, according to the new best practice statements.
“Short-term PPIs are highly effective for uncomplicated GERD [but] because patients who cannot reduce PPIs face lifelong therapy, we would consider testing for an acid-related disorder in this situation,” the authors explained. “However, there is no high-quality evidence on which to base this recommendation.”
Patients who have symptomatic GERD or Barrett’s esophagus, either symptomatic or asymptomatic, should be on long-term PPI treatment. Patients who are at a higher risk for NSAID-induced ulcer bleeding should be taking PPIs if they continue to take NSAIDs.
When recommending long-term PPI treatment for a patient, the patient need not use probiotics on a regular basis; there appears to be no need to routinely check the patient’s bone mineral density, serum creatinine, magnesium, or vitamin B12 level on a regular basis. In addition, they need not consume more than the Recommended Dietary Allowance of calcium, magnesium, or vitamin B12.
Finally, the authors state that “specific PPI formulations should not be selected based on potential risks.” This is because no evidence has been found indicating that PPI formulations can be ranked in any way based on risk.
These recommendations come from the AGA’s Clinical Practice Updates Committee, which pored through studies published through July 2016 in the PubMed, EMbase, and Cochrane library databases. Expert opinions and quality assessments on each study contributed to forming these best practice statements.
“In sum, the best current strategies for mitigating the potential risks of long-term PPIs are to avoid prescribing them when they are not indicated and to reduce them to their minimum dose when they are indicated,” Dr. Freedberg and his colleagues concluded.
The researchers did not report any relevant financial disclosures.
FROM GASTROENTEROLOGY
March 2017 Quiz 2
Q2: Answer: B
This patient, with no imaging or laboratory findings to suggest cirrhosis, most likely has noncirrhotic portal hypertension (NCPH). There is now a well-described association between HIV and NCPH with the prevalence of NCPH in HIV estimated to be –0.5% to 1%. Patients typically are unaware of any underlying liver disease until presentation with variceal bleeding. Variceal bleeding is a much more common manifestation of NCPH than ascites. Clinical presentation with normal hepatic enzymes and normal hepatic synthetic function is a very typical feature in these patients. Although the exact etiology is not fully understood, NCPH in HIV is likely related to HAART, particularly didanosine use, hypercoagulability, microbial translocation from the gut, and direct effects of HIV. NCPH is a presinusoidal lesion, and liver biopsy may reveal paucity of portal vasculature and focal obliteration of small portal veins. Portal vein thrombosis in patients with HIV and NCPH is common and has been observed in 25%-75% of patients.
Reference
1. Vispo E., Morello J., Rodriguez-Novoa S., Soriano V. Noncirrhotic portal hypertension in HIV infection. Curr Opin Infect Dis. 2011;24:12-8.
2. Khanna R., Sarin S.K. Noncirrhotic portal hypertension – Diagnosis and management. J Hepatol. 2014;60:421-41.
Q2: Answer: B
This patient, with no imaging or laboratory findings to suggest cirrhosis, most likely has noncirrhotic portal hypertension (NCPH). There is now a well-described association between HIV and NCPH with the prevalence of NCPH in HIV estimated to be –0.5% to 1%. Patients typically are unaware of any underlying liver disease until presentation with variceal bleeding. Variceal bleeding is a much more common manifestation of NCPH than ascites. Clinical presentation with normal hepatic enzymes and normal hepatic synthetic function is a very typical feature in these patients. Although the exact etiology is not fully understood, NCPH in HIV is likely related to HAART, particularly didanosine use, hypercoagulability, microbial translocation from the gut, and direct effects of HIV. NCPH is a presinusoidal lesion, and liver biopsy may reveal paucity of portal vasculature and focal obliteration of small portal veins. Portal vein thrombosis in patients with HIV and NCPH is common and has been observed in 25%-75% of patients.
Reference
1. Vispo E., Morello J., Rodriguez-Novoa S., Soriano V. Noncirrhotic portal hypertension in HIV infection. Curr Opin Infect Dis. 2011;24:12-8.
2. Khanna R., Sarin S.K. Noncirrhotic portal hypertension – Diagnosis and management. J Hepatol. 2014;60:421-41.
Q2: Answer: B
This patient, with no imaging or laboratory findings to suggest cirrhosis, most likely has noncirrhotic portal hypertension (NCPH). There is now a well-described association between HIV and NCPH with the prevalence of NCPH in HIV estimated to be –0.5% to 1%. Patients typically are unaware of any underlying liver disease until presentation with variceal bleeding. Variceal bleeding is a much more common manifestation of NCPH than ascites. Clinical presentation with normal hepatic enzymes and normal hepatic synthetic function is a very typical feature in these patients. Although the exact etiology is not fully understood, NCPH in HIV is likely related to HAART, particularly didanosine use, hypercoagulability, microbial translocation from the gut, and direct effects of HIV. NCPH is a presinusoidal lesion, and liver biopsy may reveal paucity of portal vasculature and focal obliteration of small portal veins. Portal vein thrombosis in patients with HIV and NCPH is common and has been observed in 25%-75% of patients.
Reference
1. Vispo E., Morello J., Rodriguez-Novoa S., Soriano V. Noncirrhotic portal hypertension in HIV infection. Curr Opin Infect Dis. 2011;24:12-8.
2. Khanna R., Sarin S.K. Noncirrhotic portal hypertension – Diagnosis and management. J Hepatol. 2014;60:421-41.
Q2: A 52-year-old man with history of recurrent variceal bleeding presents for evaluation. He has an HIV infection that is controlled, with undetectable virus and CD4 count of 423 cells/mcL. He has no known underlying liver disease. He is currently on etravirine, emtricitabine, and tenofovir. He has previously taken didanosine. His physical exam is unremarkable and his laboratory data reveals a normal CBC, normal INR, and normal liver enzymes. Testing for hepatitis B and C and autoimmune liver disease, as well as iron overload and other etiologies of chronic liver disease are all negative. Ultrasound of the abdomen notes a normal-appearing liver and patent portal and hepatic veins. A liver biopsy demonstrates mildly dilated portal veins and mild fibrosis of the portal venous walls. There is no evidence of cirrhosis on the liver biopsy.
March 2017 Quiz 1
Q1: Answer: A
Critique: This is a classic presentation of eosinophilic esophagitis (EoE). As many as half of older children with food impactions suffer from EoE. EoE is characterized by a severe, eosinophilic infiltration of the esophagus that may respond to acid inhibition, systemic or topical steroid therapy, or removal of dietary allergens. Epidemiologic studies suggest a rising incidence in the United States in both children and adults, with at least one case occurring in every 10,000 children each year. Treatment is aimed at alleviating symptoms and healing esophageal inflammation. Allergy testing should be performed at the time of diagnosis; however, radioallergosorbent tests and skin-prick tests are often negative, and only half of affected children have a antecedent history of other allergic symptoms.
A five-food elimination diet can be helpful for many affected children and adults, although adherence to the diet can be difficult. There is a group of affected children who respond to high doses of proton pump inhibitors, and most patients respond to either systemic or topical steroid therapy. Even with therapy, some patients go on to develop esophageal strictures and may need serial or repeated dilatations.
While eosinophilic infiltration and inflammation may be present with gastroesophageal reflux disease and associated esophagitis, the number of eosinophils seen in this boy’s biopsies is much more consistent with EoE. Moreover, stricture formation as a result of peptic esophagitis in a child this age would be extremely rare. While inflammatory bowel disease may be associated with eosinophilic infiltration of the intestinal tract, isolated esophageal Crohn’s disease would be extraordinarily rare.Our patient has no history of any immune deficiency or steroid use that would predispose to fungal esophagitis. Achalasia typically presents with gradually worsening symptoms, and the obstruction would be at the lower esophageal sphincter, not in the mid-esophagus.
Reference
1. Liacouras C., Furuta G., Hirano I., et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128:3-20.
2. Furuta G., Liacouras C., Collins M., et al. Eosinophilic esophagitis in children and adults: A systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology. 2007;133:1342-63.
Q1: Answer: A
Critique: This is a classic presentation of eosinophilic esophagitis (EoE). As many as half of older children with food impactions suffer from EoE. EoE is characterized by a severe, eosinophilic infiltration of the esophagus that may respond to acid inhibition, systemic or topical steroid therapy, or removal of dietary allergens. Epidemiologic studies suggest a rising incidence in the United States in both children and adults, with at least one case occurring in every 10,000 children each year. Treatment is aimed at alleviating symptoms and healing esophageal inflammation. Allergy testing should be performed at the time of diagnosis; however, radioallergosorbent tests and skin-prick tests are often negative, and only half of affected children have a antecedent history of other allergic symptoms.
A five-food elimination diet can be helpful for many affected children and adults, although adherence to the diet can be difficult. There is a group of affected children who respond to high doses of proton pump inhibitors, and most patients respond to either systemic or topical steroid therapy. Even with therapy, some patients go on to develop esophageal strictures and may need serial or repeated dilatations.
While eosinophilic infiltration and inflammation may be present with gastroesophageal reflux disease and associated esophagitis, the number of eosinophils seen in this boy’s biopsies is much more consistent with EoE. Moreover, stricture formation as a result of peptic esophagitis in a child this age would be extremely rare. While inflammatory bowel disease may be associated with eosinophilic infiltration of the intestinal tract, isolated esophageal Crohn’s disease would be extraordinarily rare.Our patient has no history of any immune deficiency or steroid use that would predispose to fungal esophagitis. Achalasia typically presents with gradually worsening symptoms, and the obstruction would be at the lower esophageal sphincter, not in the mid-esophagus.
Reference
1. Liacouras C., Furuta G., Hirano I., et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128:3-20.
2. Furuta G., Liacouras C., Collins M., et al. Eosinophilic esophagitis in children and adults: A systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology. 2007;133:1342-63.
Q1: Answer: A
Critique: This is a classic presentation of eosinophilic esophagitis (EoE). As many as half of older children with food impactions suffer from EoE. EoE is characterized by a severe, eosinophilic infiltration of the esophagus that may respond to acid inhibition, systemic or topical steroid therapy, or removal of dietary allergens. Epidemiologic studies suggest a rising incidence in the United States in both children and adults, with at least one case occurring in every 10,000 children each year. Treatment is aimed at alleviating symptoms and healing esophageal inflammation. Allergy testing should be performed at the time of diagnosis; however, radioallergosorbent tests and skin-prick tests are often negative, and only half of affected children have a antecedent history of other allergic symptoms.
A five-food elimination diet can be helpful for many affected children and adults, although adherence to the diet can be difficult. There is a group of affected children who respond to high doses of proton pump inhibitors, and most patients respond to either systemic or topical steroid therapy. Even with therapy, some patients go on to develop esophageal strictures and may need serial or repeated dilatations.
While eosinophilic infiltration and inflammation may be present with gastroesophageal reflux disease and associated esophagitis, the number of eosinophils seen in this boy’s biopsies is much more consistent with EoE. Moreover, stricture formation as a result of peptic esophagitis in a child this age would be extremely rare. While inflammatory bowel disease may be associated with eosinophilic infiltration of the intestinal tract, isolated esophageal Crohn’s disease would be extraordinarily rare.Our patient has no history of any immune deficiency or steroid use that would predispose to fungal esophagitis. Achalasia typically presents with gradually worsening symptoms, and the obstruction would be at the lower esophageal sphincter, not in the mid-esophagus.
Reference
1. Liacouras C., Furuta G., Hirano I., et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128:3-20.
2. Furuta G., Liacouras C., Collins M., et al. Eosinophilic esophagitis in children and adults: A systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology. 2007;133:1342-63.
Q1: A 14-year-old boy with a history of mild seasonal allergies presents to the emergency room with chest pain and discomfort after eating a steak 2 hours ago. He is having trouble swallowing and feels there is a piece of food stuck in his chest, and he points to his mid-sternum. He tells you this has happened several other times over the past year, and he felt better after he vomited. His physical examination is entirely normal. He is taken to the operating room for emergency endoscopy where a large piece of steak is removed from his mid-esophagus, without complication. Biopsies of the mid-esophagus demonstrate acute and chronic inflammatory changes in the lamina propria with 35 eosinophils per high-powered field.
Clinical Challenges - March 2017: Gastrocardiac fistula with active bleeding
What's Your Diagnosis?
The Diagnosis
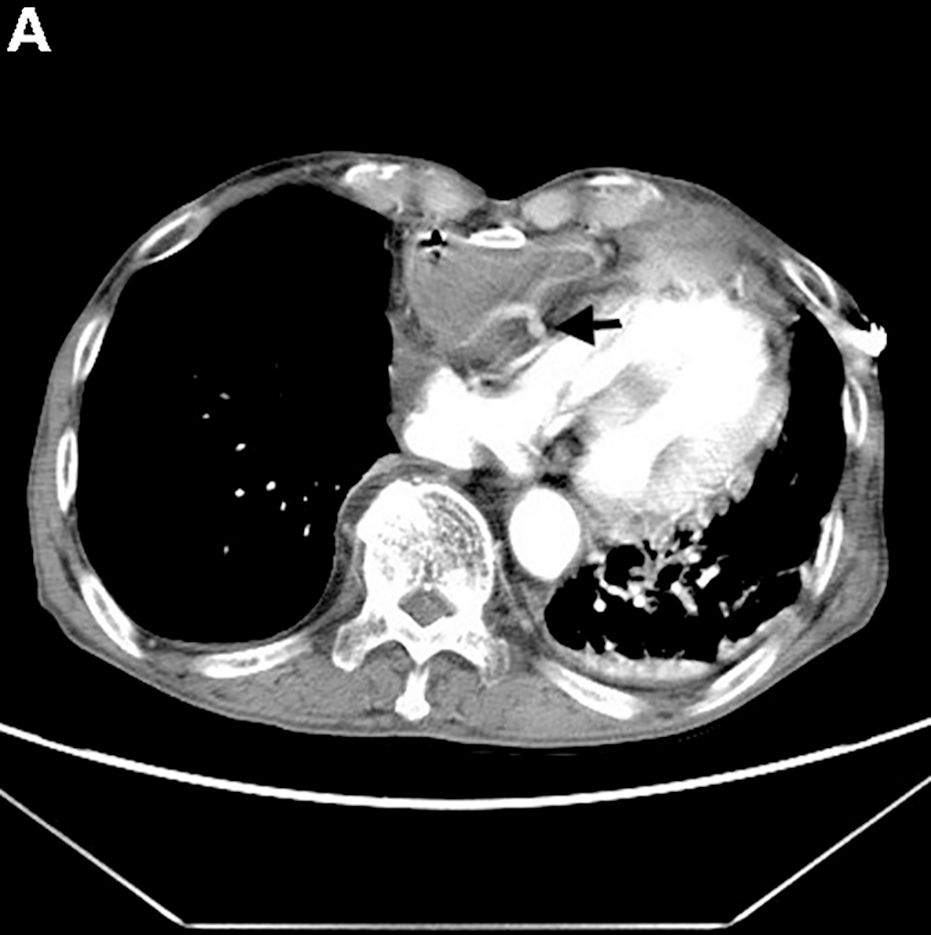
Answer: Gastrocardiac fistula with active bleeding
Active bleeding from a fistula between the right ventricle and reconstructed gastric conduit was identified after opening the gastric conduit (Figure B, black arrow). The surgeon decided to resect the gastric tube, create an esophagotomy and 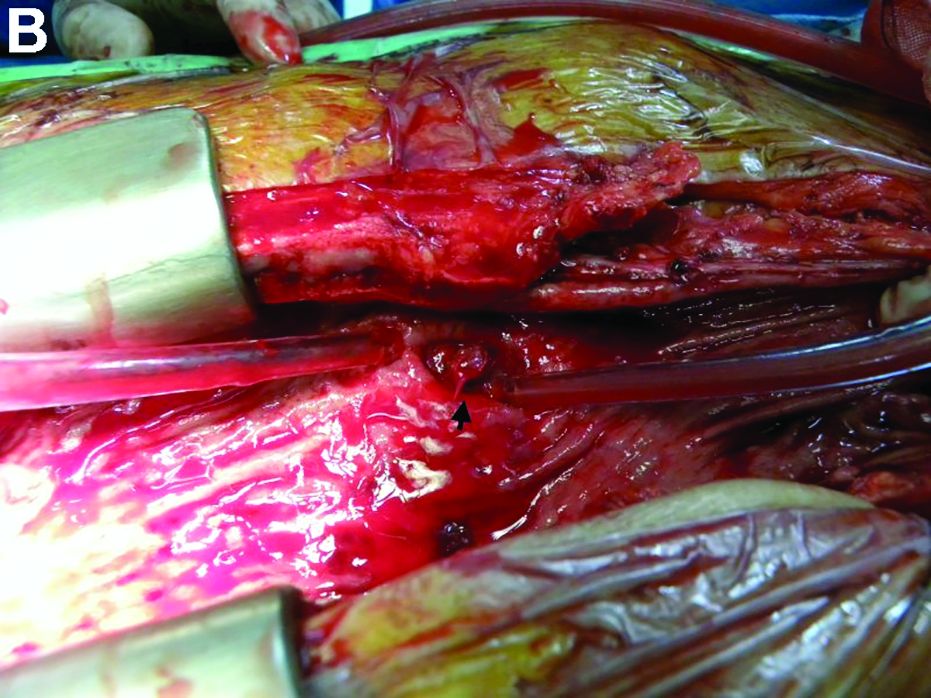
Only seven cases of fistula between postesophagectomy gastric conduits and cardiac chambers, including this case, have been reported in English literature. The disease mortality rate is as high as 60%.1 Several predisposing risk factors exist for gastrocardiac fistula, including malignancy, radiation, ischemia, and peptic ulcer disease.1 We surmised that the previous pericardiectomy was the predisposing factor in this case.
Fistula rarely develops between the upper gastrointestinal tract and adjacent structures, including the trachea, bronchi, pleura, aorta, pericardium, and heart.2,3 The symptoms differ depending on the location of the fistula, and recurrent bronchopneumonia, pleuritis, mediastinitis, pericarditis, and upper gastrointestinal bleeding may be present. Because of the high mortality rate, physicians should be alert to these fatal fistula. If fistula is suspected, a contrast radiological study and direct endoscopic visualization can be employed to establish a diagnosis.
Gastrocardiac fistula is a rare cause of upper gastrointestinal bleeding. The majority of diagnoses were made at autopsy. Only aggressive and emergent operative intervention can offer patients a chance of survival because they tend to deteriorate rapidly.1 This case of gastrocardiac fistula occurred after esophagectomy with gastric conduit reconstruction and a pericardiectomy. Immediate surgery is required for life-threatening upper gastrointestinal bleeding if gastrocardiac fistula is suspected. Patient survival is likely after immediate operation.
References
1. Pentiak, P., Seder, C.W., Chmielewski, G.W., et al. Benign post-esophagectomy gastrocardiac fistula. Interact Cardiovasc Thorac Surg. 2011;13(4):447-9.
2. Schouten van der Velden, A.P., Ruers, T.J., Bonenkamp, J.J. A cardiogastric fistula after gastric tube interposition (A case report and review of literature). J Surg Oncol. 2007;95(1):79-82.
3. Rana, Z.A., Hosmane, V.R., Rana, N.R., et al. Gastro-right ventricular fistula: a deadly complication of a gastric pull-through. Ann Thorac Surg. 2010;90(1):297-9.
The Diagnosis
Answer: Gastrocardiac fistula with active bleeding
Active bleeding from a fistula between the right ventricle and reconstructed gastric conduit was identified after opening the gastric conduit (Figure B, black arrow). The surgeon decided to resect the gastric tube, create an esophagotomy and
Only seven cases of fistula between postesophagectomy gastric conduits and cardiac chambers, including this case, have been reported in English literature. The disease mortality rate is as high as 60%.1 Several predisposing risk factors exist for gastrocardiac fistula, including malignancy, radiation, ischemia, and peptic ulcer disease.1 We surmised that the previous pericardiectomy was the predisposing factor in this case.
Fistula rarely develops between the upper gastrointestinal tract and adjacent structures, including the trachea, bronchi, pleura, aorta, pericardium, and heart.2,3 The symptoms differ depending on the location of the fistula, and recurrent bronchopneumonia, pleuritis, mediastinitis, pericarditis, and upper gastrointestinal bleeding may be present. Because of the high mortality rate, physicians should be alert to these fatal fistula. If fistula is suspected, a contrast radiological study and direct endoscopic visualization can be employed to establish a diagnosis.
Gastrocardiac fistula is a rare cause of upper gastrointestinal bleeding. The majority of diagnoses were made at autopsy. Only aggressive and emergent operative intervention can offer patients a chance of survival because they tend to deteriorate rapidly.1 This case of gastrocardiac fistula occurred after esophagectomy with gastric conduit reconstruction and a pericardiectomy. Immediate surgery is required for life-threatening upper gastrointestinal bleeding if gastrocardiac fistula is suspected. Patient survival is likely after immediate operation.
References
1. Pentiak, P., Seder, C.W., Chmielewski, G.W., et al. Benign post-esophagectomy gastrocardiac fistula. Interact Cardiovasc Thorac Surg. 2011;13(4):447-9.
2. Schouten van der Velden, A.P., Ruers, T.J., Bonenkamp, J.J. A cardiogastric fistula after gastric tube interposition (A case report and review of literature). J Surg Oncol. 2007;95(1):79-82.
3. Rana, Z.A., Hosmane, V.R., Rana, N.R., et al. Gastro-right ventricular fistula: a deadly complication of a gastric pull-through. Ann Thorac Surg. 2010;90(1):297-9.
The Diagnosis
Answer: Gastrocardiac fistula with active bleeding
Active bleeding from a fistula between the right ventricle and reconstructed gastric conduit was identified after opening the gastric conduit (Figure B, black arrow). The surgeon decided to resect the gastric tube, create an esophagotomy and
Only seven cases of fistula between postesophagectomy gastric conduits and cardiac chambers, including this case, have been reported in English literature. The disease mortality rate is as high as 60%.1 Several predisposing risk factors exist for gastrocardiac fistula, including malignancy, radiation, ischemia, and peptic ulcer disease.1 We surmised that the previous pericardiectomy was the predisposing factor in this case.
Fistula rarely develops between the upper gastrointestinal tract and adjacent structures, including the trachea, bronchi, pleura, aorta, pericardium, and heart.2,3 The symptoms differ depending on the location of the fistula, and recurrent bronchopneumonia, pleuritis, mediastinitis, pericarditis, and upper gastrointestinal bleeding may be present. Because of the high mortality rate, physicians should be alert to these fatal fistula. If fistula is suspected, a contrast radiological study and direct endoscopic visualization can be employed to establish a diagnosis.
Gastrocardiac fistula is a rare cause of upper gastrointestinal bleeding. The majority of diagnoses were made at autopsy. Only aggressive and emergent operative intervention can offer patients a chance of survival because they tend to deteriorate rapidly.1 This case of gastrocardiac fistula occurred after esophagectomy with gastric conduit reconstruction and a pericardiectomy. Immediate surgery is required for life-threatening upper gastrointestinal bleeding if gastrocardiac fistula is suspected. Patient survival is likely after immediate operation.
References
1. Pentiak, P., Seder, C.W., Chmielewski, G.W., et al. Benign post-esophagectomy gastrocardiac fistula. Interact Cardiovasc Thorac Surg. 2011;13(4):447-9.
2. Schouten van der Velden, A.P., Ruers, T.J., Bonenkamp, J.J. A cardiogastric fistula after gastric tube interposition (A case report and review of literature). J Surg Oncol. 2007;95(1):79-82.
3. Rana, Z.A., Hosmane, V.R., Rana, N.R., et al. Gastro-right ventricular fistula: a deadly complication of a gastric pull-through. Ann Thorac Surg. 2010;90(1):297-9.
What's Your Diagnosis?
What's Your Diagnosis?
By Chih-Ming Lin, PhD, Yang-Yuan Chen, MD, and Hsin-Yuan Fang, MD.
Published previously in Gastroenterology (2013;144:31,251-2).
What Makes an Excellent Gastroenterologist? IBD Patient Perspectives
We are a group of six adult Inflammatory bowel disease (IBD) patients who serve as the Patient Governance Committee for CCFA Partners – a patient powered research network that assists IBD patients, researchers, and healthcare providers to partner in finding the answers to questions patients care about and improving the health and lives of patients living with these conditions. To find out more about us, please visit our website at https://ccfa.med.unc.edu/ or send an email to [email protected].

Open communication between patient and physician
Perhaps the single most important quality of a physician is a willingness to listen. IBD patients often don’t feel like they are being heard. Starting with a conversation about the patient’s goals in terms of managing the disease as well as their goals in life will help the physician understand the patient’s unique situation and concerns. This is really a twofold proposition: what are the patient’s short-term and long-term goals? What is the most effective treatment plan to help them? How do the physician and the patient define treatment success?

At times, physicians and patients might disagree on treatment goals and patients will want their decisions respected, even if they differ from the physician’s preference. Patients want the ability to be unreservedly open with their doctors and for their doctors to listen without being defensive. Having a chronic, incurable illness is a lifelong journey, and they need someone who will respect their autonomy as well as help them weather the ups and downs of a life with IBD.

Coordinating care and transitions

When a patient needs to transfer to a new physician it’s important to help them find the right fit for their particular circumstances. Ask what is most important to patients. Is it the distance between their residence and their provider? Is it ability to manage complex disease? Is the physician in-network? All of these are important factors in helping the patient find the right care.

Holistic approach to treatment
Treating an IBD patient means treating the patient as a whole, not only their symptoms. IBD can lead to many challenges for patients and that is why treatment plans must consider not only physical, but also emotional and mental health, needs. One underserved area is pain management. While the dangers of opiates have been well documented, it seems the pendulum has swung too far in the opposite direction: some doctors are ignoring the topic of pain management altogether or establishing policies against prescribing any narcotic pain medications. This trend is troubling. Pain management is not an issue that goes away by ignoring it and remains a very important part of overall care needs. Doctors should be encouraged to take the time to learn about the many different approaches to pain management, including nonnarcotic and nonmedication therapies.

Conclusion
The mark of a high-functioning patient/physician relationship is that the patient feels empowered to be engaged with the management of their disease. An empowered patient is one who feels comfortable asking about new therapeutic options, explores new approaches to managing their disease without fear of being judged, and sticks with a treatment plan. By treating patients as partners in the fight against IBD, you can help patients accomplish their goals through a relationship based on mutual trust.

Patient Accounts
Since my diagnosis 15 years ago, the gastroenterologists who have cared for me were all effective clinicians who improved my quality of life. However, the best physicians asked me directly what aspects of my life I found most important.
My answer to this “life priority” question has changed over time. As a teenager, I wanted to fit in with my peer group as much as I could. In my early 20s, I wanted to take part in physical activity and reduce my pain as much as possible. Today, I prioritize being mentally sharp and reliable for those who depend on me professionally and maintaining empathy for those who depend on me emotionally.
I can imagine that my priorities are more easily relatable to an adult physician now than when I was in my teens, but the best gastroenterologists have empathetically listened and respected my wishes, within reason, throughout my entire experience of illness.
To me, what makes an excellent gastroenterologist is the ability to understand a patient’s greatest priorities, the activities or feelings or connections that make that person feel most whole, and, whenever possible, to direct treatment strategy according to these priorities.
– Jessica Burris
As young physicians, you may feel the need to know the answers to all our questions or a thorny diagnostic problem we present. The truth is we don’t expect you to know all the answers in the moment, it’s OK to stay you don’t know, but stay curious in finding a solution.
Also, at times there is a third presence in the room with you and your patient: the electronic medical record. It can be easy to become distracted and not make eye contact with us, which can seem as if you aren’t paying attention. Remember to always be fully present with your patient. Your patient will truly appreciate it.
– David Walter
We are a group of six adult Inflammatory bowel disease (IBD) patients who serve as the Patient Governance Committee for CCFA Partners – a patient powered research network that assists IBD patients, researchers, and healthcare providers to partner in finding the answers to questions patients care about and improving the health and lives of patients living with these conditions. To find out more about us, please visit our website at https://ccfa.med.unc.edu/ or send an email to [email protected].
Open communication between patient and physician
Perhaps the single most important quality of a physician is a willingness to listen. IBD patients often don’t feel like they are being heard. Starting with a conversation about the patient’s goals in terms of managing the disease as well as their goals in life will help the physician understand the patient’s unique situation and concerns. This is really a twofold proposition: what are the patient’s short-term and long-term goals? What is the most effective treatment plan to help them? How do the physician and the patient define treatment success?
At times, physicians and patients might disagree on treatment goals and patients will want their decisions respected, even if they differ from the physician’s preference. Patients want the ability to be unreservedly open with their doctors and for their doctors to listen without being defensive. Having a chronic, incurable illness is a lifelong journey, and they need someone who will respect their autonomy as well as help them weather the ups and downs of a life with IBD.
Coordinating care and transitions
When a patient needs to transfer to a new physician it’s important to help them find the right fit for their particular circumstances. Ask what is most important to patients. Is it the distance between their residence and their provider? Is it ability to manage complex disease? Is the physician in-network? All of these are important factors in helping the patient find the right care.
Holistic approach to treatment
Treating an IBD patient means treating the patient as a whole, not only their symptoms. IBD can lead to many challenges for patients and that is why treatment plans must consider not only physical, but also emotional and mental health, needs. One underserved area is pain management. While the dangers of opiates have been well documented, it seems the pendulum has swung too far in the opposite direction: some doctors are ignoring the topic of pain management altogether or establishing policies against prescribing any narcotic pain medications. This trend is troubling. Pain management is not an issue that goes away by ignoring it and remains a very important part of overall care needs. Doctors should be encouraged to take the time to learn about the many different approaches to pain management, including nonnarcotic and nonmedication therapies.
Conclusion
The mark of a high-functioning patient/physician relationship is that the patient feels empowered to be engaged with the management of their disease. An empowered patient is one who feels comfortable asking about new therapeutic options, explores new approaches to managing their disease without fear of being judged, and sticks with a treatment plan. By treating patients as partners in the fight against IBD, you can help patients accomplish their goals through a relationship based on mutual trust.
Patient Accounts
Since my diagnosis 15 years ago, the gastroenterologists who have cared for me were all effective clinicians who improved my quality of life. However, the best physicians asked me directly what aspects of my life I found most important.
My answer to this “life priority” question has changed over time. As a teenager, I wanted to fit in with my peer group as much as I could. In my early 20s, I wanted to take part in physical activity and reduce my pain as much as possible. Today, I prioritize being mentally sharp and reliable for those who depend on me professionally and maintaining empathy for those who depend on me emotionally.
I can imagine that my priorities are more easily relatable to an adult physician now than when I was in my teens, but the best gastroenterologists have empathetically listened and respected my wishes, within reason, throughout my entire experience of illness.
To me, what makes an excellent gastroenterologist is the ability to understand a patient’s greatest priorities, the activities or feelings or connections that make that person feel most whole, and, whenever possible, to direct treatment strategy according to these priorities.
– Jessica Burris
As young physicians, you may feel the need to know the answers to all our questions or a thorny diagnostic problem we present. The truth is we don’t expect you to know all the answers in the moment, it’s OK to stay you don’t know, but stay curious in finding a solution.
Also, at times there is a third presence in the room with you and your patient: the electronic medical record. It can be easy to become distracted and not make eye contact with us, which can seem as if you aren’t paying attention. Remember to always be fully present with your patient. Your patient will truly appreciate it.
– David Walter
We are a group of six adult Inflammatory bowel disease (IBD) patients who serve as the Patient Governance Committee for CCFA Partners – a patient powered research network that assists IBD patients, researchers, and healthcare providers to partner in finding the answers to questions patients care about and improving the health and lives of patients living with these conditions. To find out more about us, please visit our website at https://ccfa.med.unc.edu/ or send an email to [email protected].
Open communication between patient and physician
Perhaps the single most important quality of a physician is a willingness to listen. IBD patients often don’t feel like they are being heard. Starting with a conversation about the patient’s goals in terms of managing the disease as well as their goals in life will help the physician understand the patient’s unique situation and concerns. This is really a twofold proposition: what are the patient’s short-term and long-term goals? What is the most effective treatment plan to help them? How do the physician and the patient define treatment success?
At times, physicians and patients might disagree on treatment goals and patients will want their decisions respected, even if they differ from the physician’s preference. Patients want the ability to be unreservedly open with their doctors and for their doctors to listen without being defensive. Having a chronic, incurable illness is a lifelong journey, and they need someone who will respect their autonomy as well as help them weather the ups and downs of a life with IBD.
Coordinating care and transitions
When a patient needs to transfer to a new physician it’s important to help them find the right fit for their particular circumstances. Ask what is most important to patients. Is it the distance between their residence and their provider? Is it ability to manage complex disease? Is the physician in-network? All of these are important factors in helping the patient find the right care.
Holistic approach to treatment
Treating an IBD patient means treating the patient as a whole, not only their symptoms. IBD can lead to many challenges for patients and that is why treatment plans must consider not only physical, but also emotional and mental health, needs. One underserved area is pain management. While the dangers of opiates have been well documented, it seems the pendulum has swung too far in the opposite direction: some doctors are ignoring the topic of pain management altogether or establishing policies against prescribing any narcotic pain medications. This trend is troubling. Pain management is not an issue that goes away by ignoring it and remains a very important part of overall care needs. Doctors should be encouraged to take the time to learn about the many different approaches to pain management, including nonnarcotic and nonmedication therapies.
Conclusion
The mark of a high-functioning patient/physician relationship is that the patient feels empowered to be engaged with the management of their disease. An empowered patient is one who feels comfortable asking about new therapeutic options, explores new approaches to managing their disease without fear of being judged, and sticks with a treatment plan. By treating patients as partners in the fight against IBD, you can help patients accomplish their goals through a relationship based on mutual trust.
Patient Accounts
Since my diagnosis 15 years ago, the gastroenterologists who have cared for me were all effective clinicians who improved my quality of life. However, the best physicians asked me directly what aspects of my life I found most important.
My answer to this “life priority” question has changed over time. As a teenager, I wanted to fit in with my peer group as much as I could. In my early 20s, I wanted to take part in physical activity and reduce my pain as much as possible. Today, I prioritize being mentally sharp and reliable for those who depend on me professionally and maintaining empathy for those who depend on me emotionally.
I can imagine that my priorities are more easily relatable to an adult physician now than when I was in my teens, but the best gastroenterologists have empathetically listened and respected my wishes, within reason, throughout my entire experience of illness.
To me, what makes an excellent gastroenterologist is the ability to understand a patient’s greatest priorities, the activities or feelings or connections that make that person feel most whole, and, whenever possible, to direct treatment strategy according to these priorities.
– Jessica Burris
As young physicians, you may feel the need to know the answers to all our questions or a thorny diagnostic problem we present. The truth is we don’t expect you to know all the answers in the moment, it’s OK to stay you don’t know, but stay curious in finding a solution.
Also, at times there is a third presence in the room with you and your patient: the electronic medical record. It can be easy to become distracted and not make eye contact with us, which can seem as if you aren’t paying attention. Remember to always be fully present with your patient. Your patient will truly appreciate it.
– David Walter
The Vanishing Tide: As MACRA Moves In, IBD Quality Measures Move Out
Your next patient is a 67-year-old Medicare beneficiary with corticosteroid-dependent ulcerative colitis. Despite 4 months of maximally dosed mesalamine, his colitis flares with prednisone taper below 20 mg daily. Hepatitis B serologies and tuberculin skin test were negative 10 months ago. Which of the following do you recommend?
A. Steroid-sparing therapy initiation
B. Repeat latent tuberculosis screening in anticipation of anti–tumor necrosis factor (TNF) therapy
C. Bone loss assessment
D. Pneumococcal vaccination
E. Tobacco use screening

Quality measure reporting is a costly undertaking, with medical practices spending an average of 15.1 hours per physician per week ($40,069 per physician annually) dealing with external quality measures.2 How did this expensive alphabet soup of quality measure reporting arise and how does it impact inflammatory bowel disease (IBD) care?
Why are IBD quality measures needed?

What makes a good quality measure?
Quality must be defined and measured before it can be improved. This is easier said than done, especially for IBD where a gold standard in “ideal care” is ill defined and continually evolving as new research emerges. Nonetheless, hundreds of health care quality measures have been proposed. Desirable quality measure attributes should satisfy three broad categories: importance, scientific soundness, and feasibility.10 Quality measures should address relevant and important aspects of health that are highly prevalent and for which evidence indicates a need for improvement. There should be strong evidence supporting the beneficial impact of adhering to a given measure.
Quality measures are commonly classified as process measures or outcome measures. Process measures (“doing the right thing”) are steps taken by providers in the care of an individual patient. These often derive from evidence-based best practices. Outcome measures (“having the desired result”) identify what happens to patients as a result of care received.8 Outcome measures may be more meaningful, but there are limitations in using them to study quality of IBD care. For example, factors beyond physician control affect patient outcomes and long delays may exist between care decisions and subsequent outcomes (e.g., surgery, malnutrition).8
What IBD quality measures already exist?
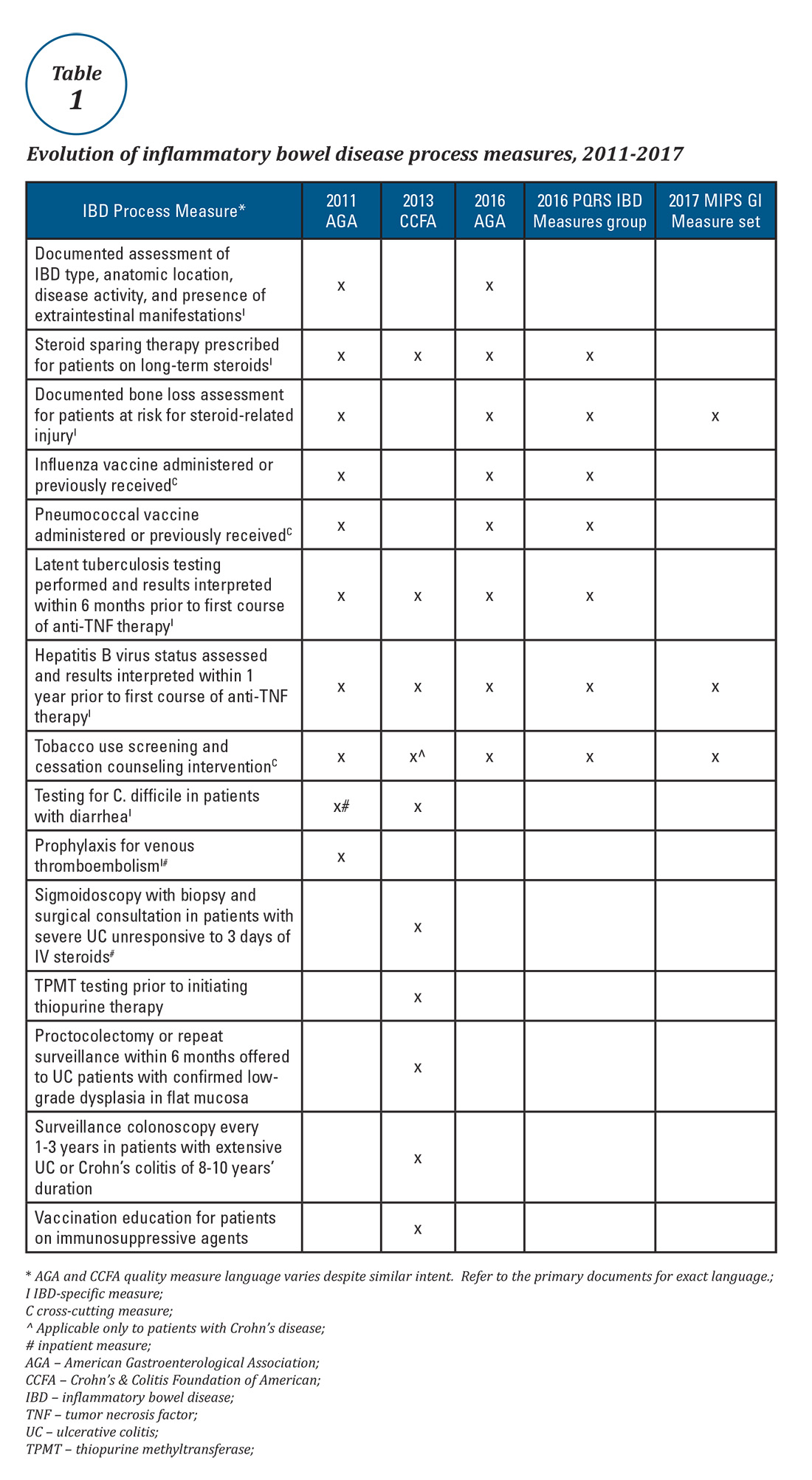
Expert panels from the AGA and the Crohn’s & Colitis Foundation of America (CCFA) produced IBD quality measure sets comprising mostly process measures (Table 1). The original 10 AGA measures released in 2011 address aspects of disease assessment, treatment, complication prevention, and health care maintenance.12 They include seven IBD-specific measures, three cross-cutting measures – defined by Centers for Medicare & Medicaid Services (CMS) as being broadly applicable across multiple clinical settings – and two inpatient measures. A major goal of the AGA measures was to facilitate quality reporting to the former PQRS program.
What are some quality measure limitations?
Quality measure development has an evidence base but designing an optimal measure and demonstrating impact can be challenging. Few IBD process measures are validated and thus there is often logic but not data linking process measure adherence to improved outcomes. The denominator (number of eligible patients) and potential impact of broad adherence vary for each quality measure. For example, only a small fraction of IBD patients are infected with hepatitis B and fewer than 10% will experience viral reactivation during anti-TNF therapy.17,18 Even with optimal adherence to the hepatitis B measure, few reactivations will be prevented. The wording of some measures lacks precision, allowing physicians to potentially claim credit without improving care. For example, ordering a bone density scan satisfies the bone loss assessment measure, even if osteoporosis goes unrecognized and untreated. Finally, some measures relate to actions that may not be under the control of the gastroenterologist whose performance is being measured (e.g., administering vaccinations).
IBD quality measures under MIPS
Table 1 depicts the evolution of IBD process measures from 2011 to 2017. Rather than building upon initial experience to revise and refine IBD quality measures, the measures have instead been progressively culled with the changing pay-for-performance landscape. In 2016, AGA eliminated the two inpatient measures.19 Seven of the remaining eight measures formed the IBD Measures Group which was reportable under PQRS. In 2017, MIPS brought a seismic shift in quality measure focus. The PQRS IBD Measures Group was abolished – as were all Measures Groups – and replaced by a 16-item GI Measures Set. Although AGA advocated for all of the IBD measures to be included, the new GI Measures Set deemphasized the IBD-specific measures in favor of expanded cross-cutting measures (e.g., screening for abnormal body mass index, documenting current medications, sending specialist report to referring provider).20 This reflected a previously observed trend that gastroenterologists more often reported on cross-cutting measures than specialist-specific measures.21 However, there was no evidence-based justification for dropping certain IBD-specific measures (especially the steroid-sparing therapy measure) in favor of retaining the two chosen IBD-specific measures – bone loss assessment and hepatitis B screening – which apply to only a subset of IBD patients and have limited potential to impact clinical outcomes. Although it is not mandatory to report using the GI Measures Set, we suspect that many gastroenterologists will use this set to guide their initial reporting.
There are formidable regulatory obstacles to improving the IBD quality measures included in MIPS. CMS requires that new quality measures proposed for inclusion in MIPS be fully specified and tested for validity and reliability by the individual measure developers (such as AGA). This is a costly and time-intensive process that has complicated efforts to successfully advocate for inclusion of GI-specific quality measures in MIPS, as there is no existing infrastructure for quality measure testing.
A word about Alternative Payment Models (APMs)
APMs represent the non-MIPS pathway for participating in the QPP. APMs focus on chronic disease care coordination and qualify for lump-sum incentive payments by adhering to stringent standards and financial risk-sharing requirements. A detailed overview of APMs is beyond the scope of this discussion, as the vast majority of MACRA-eligible gastroenterologists will participate in MIPS and there are currently no GI-specific APMs. However, this is an evolving area and Project Sonar has been submitted to the Physician-Focused Payment Model Technical Advisory Committee for consideration as an APM for Crohn’s disease.23
Conclusion
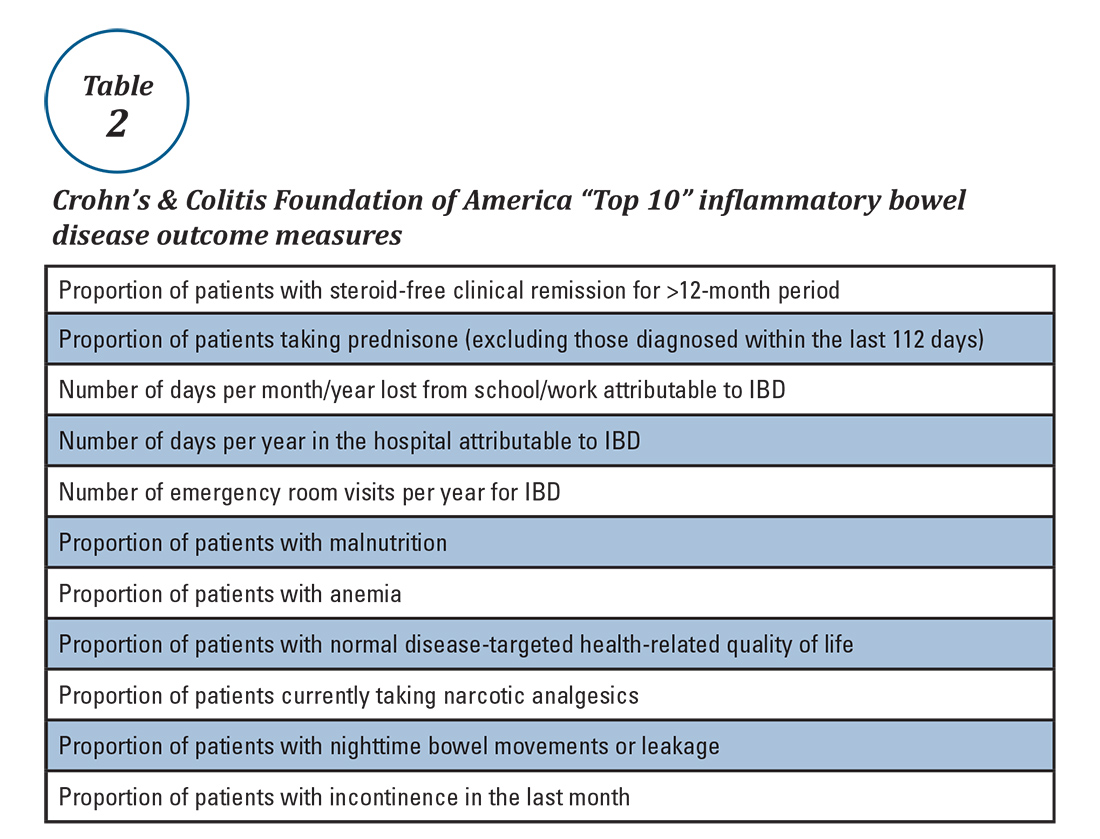
Quality measurement and reporting are at a crossroads. Ideally, performance improvement should be an internally driven process that addresses specific local priorities and needs. Most medical practices (73%) believe that current externally driven quality measures do not represent care quality and only 28% use their quality scores to focus their internal quality improvement activities.2 The burden and cost of external quality reporting demand better alignment with local priorities as resources are currently being diverted away from internally driven efforts that might have the greatest potential to improve patient outcomes.24 The dawn of the MACRA era presents an opportunity to shape the future of the IBD quality movement. Through validating and prioritizing existing measures and developing novel, precisely stated, and high-value metrics, there remains vast (and measurable) potential to enhance patient outcomes.
Dr. McConnell is a fellow in gastroenterology and advanced inflammatory bowel disease, division of gastroenterology, University of California, San Francisco. Dr. Velayos is professor of medicine, co–medical director, Center for Crohn’s and Colitis, University of California, San Francisco.
References
1. September 2016 Medscape survey summary. Available at http://www.healthcaredive.com/news/survey-29-of-physicians-still-havent-heard-of-macra/429322/. Accessed March 23, 2017.
2. Casalino L.P., et al. Health Aff. 2016;35:401-6.
3. Rubin D.T., et al. Curr Med Res Opin. 2017;33:529-36.
4. David G., et al. Gastroenterology. 2013;144:S-647.
5. Nguyen G.C., et al. Clin Gastroenterol Hepatol. 2006;4:1507-13.
6. Esrailian E., et al. Aliment Pharmacol Ther. 2007;26:1005-18.
7. Spiegel B.M., et al. Clin Gastroenterol Hepatol. 2009;7:68-74.
8. Kappelman M.D., et al. Inflamm Bowel Dis. 2010;16:125-133.
9. Reddy S.I., et al. Am J Gastroenterol. 2005;100:1357-61.
10. National Quality Measures Clearinghouse. Available at https://www.qualitymeasures.ahrq.gov/help-and-about/quality-measure-tutorials/desirable-attributes-of-a-quality-measure. Accessed March 23, 2017.
11. McGlynn E.A. Med Care. 2003;41(1 Suppl):139-47.
12. American Gastroenterological Association. Available at https://www.gastro.org/practice/quality-initiatives/IBD_Measures.pdf. Accessed March 23, 2017.
13. Melmed G.Y., et al. Inflamm Bowel Dis. 2013;19:662-8.
14. Feuerstein J.D., et al. Clin Gastroenterol Hepatol. 2016;14:421-8.
15. Sapir T., et al. Dig Dis Sci. 2016;61:1862-9.
16. Crohn’s & Colitis Foundation of America. IBD Qorus. Available at http://www.ccfa.org/science-and-professionals/ibdqorus/. Accessed March 23, 2017.
17. Hou J.K., et al. Gastroenterology. 2015;148(Suppl 1):S-61.
18. Reddy K.R., et al. Gastroenterology. 2015;48:215-9.
19. American Gastroenterological Association. Available at http://www.gastro.org/practice-management/measures/2016_AGA_Measures_-_IBD.pdf. Accessed March 23, 2017.
20. American Gastroenterological Association. Available at http://www.gastro.org/news_items/gi-quality-measures-for-2017-are-released-in-macra-final-rule. Accessed March 23, 2017.
21. Centers for Medicare & Medicaid Services. Available at https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/PQRS/Downloads/2014_PQRS_Experience_Rpt.pdf. Accessed March 23, 2017.
22. Dahlhamer J.M., et al. MMWR. 2016;65:1166-9.
23. U.S. Department of Health & Human Services Office of the Assistant Secretary for Planning and Evaluation. Available at https://aspe.hhs.gov/system/files/pdf/253406/ProjectSonarSonarMD.pdf. Accessed March 23, 2017.
24. Meyer G.S., et al. BMJ Qual Saf. 2012;21:964-8.
Your next patient is a 67-year-old Medicare beneficiary with corticosteroid-dependent ulcerative colitis. Despite 4 months of maximally dosed mesalamine, his colitis flares with prednisone taper below 20 mg daily. Hepatitis B serologies and tuberculin skin test were negative 10 months ago. Which of the following do you recommend?
A. Steroid-sparing therapy initiation
B. Repeat latent tuberculosis screening in anticipation of anti–tumor necrosis factor (TNF) therapy
C. Bone loss assessment
D. Pneumococcal vaccination
E. Tobacco use screening
Quality measure reporting is a costly undertaking, with medical practices spending an average of 15.1 hours per physician per week ($40,069 per physician annually) dealing with external quality measures.2 How did this expensive alphabet soup of quality measure reporting arise and how does it impact inflammatory bowel disease (IBD) care?
Why are IBD quality measures needed?
What makes a good quality measure?
Quality must be defined and measured before it can be improved. This is easier said than done, especially for IBD where a gold standard in “ideal care” is ill defined and continually evolving as new research emerges. Nonetheless, hundreds of health care quality measures have been proposed. Desirable quality measure attributes should satisfy three broad categories: importance, scientific soundness, and feasibility.10 Quality measures should address relevant and important aspects of health that are highly prevalent and for which evidence indicates a need for improvement. There should be strong evidence supporting the beneficial impact of adhering to a given measure.
Quality measures are commonly classified as process measures or outcome measures. Process measures (“doing the right thing”) are steps taken by providers in the care of an individual patient. These often derive from evidence-based best practices. Outcome measures (“having the desired result”) identify what happens to patients as a result of care received.8 Outcome measures may be more meaningful, but there are limitations in using them to study quality of IBD care. For example, factors beyond physician control affect patient outcomes and long delays may exist between care decisions and subsequent outcomes (e.g., surgery, malnutrition).8
What IBD quality measures already exist?
Expert panels from the AGA and the Crohn’s & Colitis Foundation of America (CCFA) produced IBD quality measure sets comprising mostly process measures (Table 1). The original 10 AGA measures released in 2011 address aspects of disease assessment, treatment, complication prevention, and health care maintenance.12 They include seven IBD-specific measures, three cross-cutting measures – defined by Centers for Medicare & Medicaid Services (CMS) as being broadly applicable across multiple clinical settings – and two inpatient measures. A major goal of the AGA measures was to facilitate quality reporting to the former PQRS program.
What are some quality measure limitations?
Quality measure development has an evidence base but designing an optimal measure and demonstrating impact can be challenging. Few IBD process measures are validated and thus there is often logic but not data linking process measure adherence to improved outcomes. The denominator (number of eligible patients) and potential impact of broad adherence vary for each quality measure. For example, only a small fraction of IBD patients are infected with hepatitis B and fewer than 10% will experience viral reactivation during anti-TNF therapy.17,18 Even with optimal adherence to the hepatitis B measure, few reactivations will be prevented. The wording of some measures lacks precision, allowing physicians to potentially claim credit without improving care. For example, ordering a bone density scan satisfies the bone loss assessment measure, even if osteoporosis goes unrecognized and untreated. Finally, some measures relate to actions that may not be under the control of the gastroenterologist whose performance is being measured (e.g., administering vaccinations).
IBD quality measures under MIPS
Table 1 depicts the evolution of IBD process measures from 2011 to 2017. Rather than building upon initial experience to revise and refine IBD quality measures, the measures have instead been progressively culled with the changing pay-for-performance landscape. In 2016, AGA eliminated the two inpatient measures.19 Seven of the remaining eight measures formed the IBD Measures Group which was reportable under PQRS. In 2017, MIPS brought a seismic shift in quality measure focus. The PQRS IBD Measures Group was abolished – as were all Measures Groups – and replaced by a 16-item GI Measures Set. Although AGA advocated for all of the IBD measures to be included, the new GI Measures Set deemphasized the IBD-specific measures in favor of expanded cross-cutting measures (e.g., screening for abnormal body mass index, documenting current medications, sending specialist report to referring provider).20 This reflected a previously observed trend that gastroenterologists more often reported on cross-cutting measures than specialist-specific measures.21 However, there was no evidence-based justification for dropping certain IBD-specific measures (especially the steroid-sparing therapy measure) in favor of retaining the two chosen IBD-specific measures – bone loss assessment and hepatitis B screening – which apply to only a subset of IBD patients and have limited potential to impact clinical outcomes. Although it is not mandatory to report using the GI Measures Set, we suspect that many gastroenterologists will use this set to guide their initial reporting.
There are formidable regulatory obstacles to improving the IBD quality measures included in MIPS. CMS requires that new quality measures proposed for inclusion in MIPS be fully specified and tested for validity and reliability by the individual measure developers (such as AGA). This is a costly and time-intensive process that has complicated efforts to successfully advocate for inclusion of GI-specific quality measures in MIPS, as there is no existing infrastructure for quality measure testing.
A word about Alternative Payment Models (APMs)
APMs represent the non-MIPS pathway for participating in the QPP. APMs focus on chronic disease care coordination and qualify for lump-sum incentive payments by adhering to stringent standards and financial risk-sharing requirements. A detailed overview of APMs is beyond the scope of this discussion, as the vast majority of MACRA-eligible gastroenterologists will participate in MIPS and there are currently no GI-specific APMs. However, this is an evolving area and Project Sonar has been submitted to the Physician-Focused Payment Model Technical Advisory Committee for consideration as an APM for Crohn’s disease.23
Conclusion
Quality measurement and reporting are at a crossroads. Ideally, performance improvement should be an internally driven process that addresses specific local priorities and needs. Most medical practices (73%) believe that current externally driven quality measures do not represent care quality and only 28% use their quality scores to focus their internal quality improvement activities.2 The burden and cost of external quality reporting demand better alignment with local priorities as resources are currently being diverted away from internally driven efforts that might have the greatest potential to improve patient outcomes.24 The dawn of the MACRA era presents an opportunity to shape the future of the IBD quality movement. Through validating and prioritizing existing measures and developing novel, precisely stated, and high-value metrics, there remains vast (and measurable) potential to enhance patient outcomes.
Dr. McConnell is a fellow in gastroenterology and advanced inflammatory bowel disease, division of gastroenterology, University of California, San Francisco. Dr. Velayos is professor of medicine, co–medical director, Center for Crohn’s and Colitis, University of California, San Francisco.
References
1. September 2016 Medscape survey summary. Available at http://www.healthcaredive.com/news/survey-29-of-physicians-still-havent-heard-of-macra/429322/. Accessed March 23, 2017.
2. Casalino L.P., et al. Health Aff. 2016;35:401-6.
3. Rubin D.T., et al. Curr Med Res Opin. 2017;33:529-36.
4. David G., et al. Gastroenterology. 2013;144:S-647.
5. Nguyen G.C., et al. Clin Gastroenterol Hepatol. 2006;4:1507-13.
6. Esrailian E., et al. Aliment Pharmacol Ther. 2007;26:1005-18.
7. Spiegel B.M., et al. Clin Gastroenterol Hepatol. 2009;7:68-74.
8. Kappelman M.D., et al. Inflamm Bowel Dis. 2010;16:125-133.
9. Reddy S.I., et al. Am J Gastroenterol. 2005;100:1357-61.
10. National Quality Measures Clearinghouse. Available at https://www.qualitymeasures.ahrq.gov/help-and-about/quality-measure-tutorials/desirable-attributes-of-a-quality-measure. Accessed March 23, 2017.
11. McGlynn E.A. Med Care. 2003;41(1 Suppl):139-47.
12. American Gastroenterological Association. Available at https://www.gastro.org/practice/quality-initiatives/IBD_Measures.pdf. Accessed March 23, 2017.
13. Melmed G.Y., et al. Inflamm Bowel Dis. 2013;19:662-8.
14. Feuerstein J.D., et al. Clin Gastroenterol Hepatol. 2016;14:421-8.
15. Sapir T., et al. Dig Dis Sci. 2016;61:1862-9.
16. Crohn’s & Colitis Foundation of America. IBD Qorus. Available at http://www.ccfa.org/science-and-professionals/ibdqorus/. Accessed March 23, 2017.
17. Hou J.K., et al. Gastroenterology. 2015;148(Suppl 1):S-61.
18. Reddy K.R., et al. Gastroenterology. 2015;48:215-9.
19. American Gastroenterological Association. Available at http://www.gastro.org/practice-management/measures/2016_AGA_Measures_-_IBD.pdf. Accessed March 23, 2017.
20. American Gastroenterological Association. Available at http://www.gastro.org/news_items/gi-quality-measures-for-2017-are-released-in-macra-final-rule. Accessed March 23, 2017.
21. Centers for Medicare & Medicaid Services. Available at https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/PQRS/Downloads/2014_PQRS_Experience_Rpt.pdf. Accessed March 23, 2017.
22. Dahlhamer J.M., et al. MMWR. 2016;65:1166-9.
23. U.S. Department of Health & Human Services Office of the Assistant Secretary for Planning and Evaluation. Available at https://aspe.hhs.gov/system/files/pdf/253406/ProjectSonarSonarMD.pdf. Accessed March 23, 2017.
24. Meyer G.S., et al. BMJ Qual Saf. 2012;21:964-8.
Your next patient is a 67-year-old Medicare beneficiary with corticosteroid-dependent ulcerative colitis. Despite 4 months of maximally dosed mesalamine, his colitis flares with prednisone taper below 20 mg daily. Hepatitis B serologies and tuberculin skin test were negative 10 months ago. Which of the following do you recommend?
A. Steroid-sparing therapy initiation
B. Repeat latent tuberculosis screening in anticipation of anti–tumor necrosis factor (TNF) therapy
C. Bone loss assessment
D. Pneumococcal vaccination
E. Tobacco use screening
Quality measure reporting is a costly undertaking, with medical practices spending an average of 15.1 hours per physician per week ($40,069 per physician annually) dealing with external quality measures.2 How did this expensive alphabet soup of quality measure reporting arise and how does it impact inflammatory bowel disease (IBD) care?
Why are IBD quality measures needed?
What makes a good quality measure?
Quality must be defined and measured before it can be improved. This is easier said than done, especially for IBD where a gold standard in “ideal care” is ill defined and continually evolving as new research emerges. Nonetheless, hundreds of health care quality measures have been proposed. Desirable quality measure attributes should satisfy three broad categories: importance, scientific soundness, and feasibility.10 Quality measures should address relevant and important aspects of health that are highly prevalent and for which evidence indicates a need for improvement. There should be strong evidence supporting the beneficial impact of adhering to a given measure.
Quality measures are commonly classified as process measures or outcome measures. Process measures (“doing the right thing”) are steps taken by providers in the care of an individual patient. These often derive from evidence-based best practices. Outcome measures (“having the desired result”) identify what happens to patients as a result of care received.8 Outcome measures may be more meaningful, but there are limitations in using them to study quality of IBD care. For example, factors beyond physician control affect patient outcomes and long delays may exist between care decisions and subsequent outcomes (e.g., surgery, malnutrition).8
What IBD quality measures already exist?
Expert panels from the AGA and the Crohn’s & Colitis Foundation of America (CCFA) produced IBD quality measure sets comprising mostly process measures (Table 1). The original 10 AGA measures released in 2011 address aspects of disease assessment, treatment, complication prevention, and health care maintenance.12 They include seven IBD-specific measures, three cross-cutting measures – defined by Centers for Medicare & Medicaid Services (CMS) as being broadly applicable across multiple clinical settings – and two inpatient measures. A major goal of the AGA measures was to facilitate quality reporting to the former PQRS program.
What are some quality measure limitations?
Quality measure development has an evidence base but designing an optimal measure and demonstrating impact can be challenging. Few IBD process measures are validated and thus there is often logic but not data linking process measure adherence to improved outcomes. The denominator (number of eligible patients) and potential impact of broad adherence vary for each quality measure. For example, only a small fraction of IBD patients are infected with hepatitis B and fewer than 10% will experience viral reactivation during anti-TNF therapy.17,18 Even with optimal adherence to the hepatitis B measure, few reactivations will be prevented. The wording of some measures lacks precision, allowing physicians to potentially claim credit without improving care. For example, ordering a bone density scan satisfies the bone loss assessment measure, even if osteoporosis goes unrecognized and untreated. Finally, some measures relate to actions that may not be under the control of the gastroenterologist whose performance is being measured (e.g., administering vaccinations).
IBD quality measures under MIPS
Table 1 depicts the evolution of IBD process measures from 2011 to 2017. Rather than building upon initial experience to revise and refine IBD quality measures, the measures have instead been progressively culled with the changing pay-for-performance landscape. In 2016, AGA eliminated the two inpatient measures.19 Seven of the remaining eight measures formed the IBD Measures Group which was reportable under PQRS. In 2017, MIPS brought a seismic shift in quality measure focus. The PQRS IBD Measures Group was abolished – as were all Measures Groups – and replaced by a 16-item GI Measures Set. Although AGA advocated for all of the IBD measures to be included, the new GI Measures Set deemphasized the IBD-specific measures in favor of expanded cross-cutting measures (e.g., screening for abnormal body mass index, documenting current medications, sending specialist report to referring provider).20 This reflected a previously observed trend that gastroenterologists more often reported on cross-cutting measures than specialist-specific measures.21 However, there was no evidence-based justification for dropping certain IBD-specific measures (especially the steroid-sparing therapy measure) in favor of retaining the two chosen IBD-specific measures – bone loss assessment and hepatitis B screening – which apply to only a subset of IBD patients and have limited potential to impact clinical outcomes. Although it is not mandatory to report using the GI Measures Set, we suspect that many gastroenterologists will use this set to guide their initial reporting.
There are formidable regulatory obstacles to improving the IBD quality measures included in MIPS. CMS requires that new quality measures proposed for inclusion in MIPS be fully specified and tested for validity and reliability by the individual measure developers (such as AGA). This is a costly and time-intensive process that has complicated efforts to successfully advocate for inclusion of GI-specific quality measures in MIPS, as there is no existing infrastructure for quality measure testing.
A word about Alternative Payment Models (APMs)
APMs represent the non-MIPS pathway for participating in the QPP. APMs focus on chronic disease care coordination and qualify for lump-sum incentive payments by adhering to stringent standards and financial risk-sharing requirements. A detailed overview of APMs is beyond the scope of this discussion, as the vast majority of MACRA-eligible gastroenterologists will participate in MIPS and there are currently no GI-specific APMs. However, this is an evolving area and Project Sonar has been submitted to the Physician-Focused Payment Model Technical Advisory Committee for consideration as an APM for Crohn’s disease.23
Conclusion
Quality measurement and reporting are at a crossroads. Ideally, performance improvement should be an internally driven process that addresses specific local priorities and needs. Most medical practices (73%) believe that current externally driven quality measures do not represent care quality and only 28% use their quality scores to focus their internal quality improvement activities.2 The burden and cost of external quality reporting demand better alignment with local priorities as resources are currently being diverted away from internally driven efforts that might have the greatest potential to improve patient outcomes.24 The dawn of the MACRA era presents an opportunity to shape the future of the IBD quality movement. Through validating and prioritizing existing measures and developing novel, precisely stated, and high-value metrics, there remains vast (and measurable) potential to enhance patient outcomes.
Dr. McConnell is a fellow in gastroenterology and advanced inflammatory bowel disease, division of gastroenterology, University of California, San Francisco. Dr. Velayos is professor of medicine, co–medical director, Center for Crohn’s and Colitis, University of California, San Francisco.
References
1. September 2016 Medscape survey summary. Available at http://www.healthcaredive.com/news/survey-29-of-physicians-still-havent-heard-of-macra/429322/. Accessed March 23, 2017.
2. Casalino L.P., et al. Health Aff. 2016;35:401-6.
3. Rubin D.T., et al. Curr Med Res Opin. 2017;33:529-36.
4. David G., et al. Gastroenterology. 2013;144:S-647.
5. Nguyen G.C., et al. Clin Gastroenterol Hepatol. 2006;4:1507-13.
6. Esrailian E., et al. Aliment Pharmacol Ther. 2007;26:1005-18.
7. Spiegel B.M., et al. Clin Gastroenterol Hepatol. 2009;7:68-74.
8. Kappelman M.D., et al. Inflamm Bowel Dis. 2010;16:125-133.
9. Reddy S.I., et al. Am J Gastroenterol. 2005;100:1357-61.
10. National Quality Measures Clearinghouse. Available at https://www.qualitymeasures.ahrq.gov/help-and-about/quality-measure-tutorials/desirable-attributes-of-a-quality-measure. Accessed March 23, 2017.
11. McGlynn E.A. Med Care. 2003;41(1 Suppl):139-47.
12. American Gastroenterological Association. Available at https://www.gastro.org/practice/quality-initiatives/IBD_Measures.pdf. Accessed March 23, 2017.
13. Melmed G.Y., et al. Inflamm Bowel Dis. 2013;19:662-8.
14. Feuerstein J.D., et al. Clin Gastroenterol Hepatol. 2016;14:421-8.
15. Sapir T., et al. Dig Dis Sci. 2016;61:1862-9.
16. Crohn’s & Colitis Foundation of America. IBD Qorus. Available at http://www.ccfa.org/science-and-professionals/ibdqorus/. Accessed March 23, 2017.
17. Hou J.K., et al. Gastroenterology. 2015;148(Suppl 1):S-61.
18. Reddy K.R., et al. Gastroenterology. 2015;48:215-9.
19. American Gastroenterological Association. Available at http://www.gastro.org/practice-management/measures/2016_AGA_Measures_-_IBD.pdf. Accessed March 23, 2017.
20. American Gastroenterological Association. Available at http://www.gastro.org/news_items/gi-quality-measures-for-2017-are-released-in-macra-final-rule. Accessed March 23, 2017.
21. Centers for Medicare & Medicaid Services. Available at https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/PQRS/Downloads/2014_PQRS_Experience_Rpt.pdf. Accessed March 23, 2017.
22. Dahlhamer J.M., et al. MMWR. 2016;65:1166-9.
23. U.S. Department of Health & Human Services Office of the Assistant Secretary for Planning and Evaluation. Available at https://aspe.hhs.gov/system/files/pdf/253406/ProjectSonarSonarMD.pdf. Accessed March 23, 2017.
24. Meyer G.S., et al. BMJ Qual Saf. 2012;21:964-8.
A Practical Guide for Developing a Relationship with the Pharmaceutical, Biotech, and Device Industries
The primary goal of the biotechnology and pharmaceutical industry is to develop medications and medical devices for the treatment of patients, while earning financial gain for investors. An important component of achieving this goal is the role physicians play in the drug and medical device development process. In particular, a physician’s role is to combine their clinical expertise with their knowledge of industry products to better diagnose and treat the ailments of their patients. Thus, medicine and industry have a dependent relationship. In recent times this relationship has been fraught with turmoil as the public, scientific community, and federal government have discovered real and perceived conflicts of interest.
For example, there has been public outrage in the past with reports of doctors receiving gifts, money, and lavish trips in return for prescribing medications or using certain medical devices. Because of this, Congress passed the Sunshine Act, deeming it necessary to report all physician and industry engagements that have any perceived financial value. The passage of this act was in addition to local policies set forth by academic institutions, hospitals, and private practices.

How Do I Get Started?
After checking with your institution, hospital or private practice administrator, the first step is to reach out to a local representative (“rep”) of a pharmaceutical or biotech company in which you are interested. You can accomplish this via the website of the company or by visiting the booth at major gastrointestinal conferences such as Digestive Disease Week (DDW®).
Pharma and device reps are quite knowledgeable about the latest clinical studies regarding their products, disease states, and various competing products in the market. In addition to being a source of valuable medical knowledge and disease-specific practice guidelines, they also can connect you with their medical science liaison (MSL). MSLs often have a background in pharmacy and/or research. Thus, they can provide insights into mechanisms of disease treatments and go beyond discussion of the product label, which pharmaceutical reps adhere to. They also know what therapies or diagnostic tools are in the phases of development and could be available for a clinical trial.
MSLs are also the gatekeepers for Investigator Initiated Studies (IIS). An IIS is a research project that is industry-funded and is solely designed and executed by the clinician. The application process is rigorous but awards may be easier to obtain for non-research-based clinicians who want to develop a disease-specific project that needs funding. Their grant application process can be brief, ideas may not require prior data, and turnaround time to funding may be shorter. IISs often lead to exploratory findings that may facilitate publications or lay groundwork for large-scale grants or even clinical trials. In some instances, you may be granted access to internal data and prescribing patterns, which can answer interesting clinical and research questions.
How Do I Get Started with Clinical Trials?
Being a primary investigator on a clinical trial is a big responsibility. You are responsible to the trial sponsor in addition to your patients. For young clinicians who lack experience with clinical trials, the first thing to do is to find a clinician in your department or another department, who has expertise in performing an industry-sponsored study. These individuals can be invaluable for you in terms of guiding you through the study feasibility process, study startup, and possibly being the lead or co-investigator with you. Partnering with someone with expertise in industry-sponsored clinical trials will help you gain the trust of the industry sponsor, which may be a requirement for some.
There are many additional requirements that need to be fulfilled aside from just having an appropriate and adequate patient population to pull from. You will need to have a coordinator for the study who will help you with patient care, data entry, and study- specific issues. Clinical trials require a significant amount of documentation and reporting that has to be performed within a timely manner. There is no degree prerequisite of the coordinator but it can simplify things for the clinician if they have a RN or LPN degree. Having such a degree will facilitate dual roles of patient care, lab draws, drug administration, medical charting, and other patient care matters.
In addition, you will need to have approval from either your local or central institutional review board (IRB). Also, you will have to review budget and study-specific requirements for equipment and infrastructure with your department manager. You will need to demonstrate adequate ancillary support to process, store, and ship biological specimens. In some instances, you will need a dedicated pharmacist to mix or dispense study drugs.
The process is lengthy and involved, but rewarding in terms of being involved in the drug development process. You will have opportunities to attend meetings at which you can network with other clinicians and provide the sponsor feedback on how the study is going.
How Do I Develop a Consulting Role with Industry?
It is important to check with your institution, hospital, or practice if there are any limitations in becoming a consultant for a pharmaceutical or device company. If it is allowed and will not interfere with your clinical duties, it is important to note that this role takes time to develop. It often comes about after years of experience doing research, clinical and/or basic science, with publications to support expertise. Working on an IIS is a good way to work hand-in-hand with expert industry researchers and facilitate the consulting relationship. Being a primary investigator of clinical trials with successful enrollment of patients and meeting attendance will provide you with insight into the drug development process.
What if None of This Works Out for Me?
Do not give up! Persistence, experience, and hard work are the keys to developing relationships with industry. Remember, industry has a vast network of clinicians and researchers they already work with. The overall pool of companies and experts is limited and can be difficult to break into. But it can be done. Some rely on their research experience, clinical training, and mentors to develop the necessary contacts. Others can develop the contacts via IIS applications. Industry lacks access to the physician-patient experience; this can be your greatest asset and key to your success if leveraged properly. You can consider applying for mentorship with experts in your field via AGA-sponsored events held annually at DDW® to get additional guidance.
Final Thoughts
It is important to remember that all industry relationships require time to develop. They also come at an opportunity cost of time away from your clinical practice and your family, friends and hobbies. However, these relationships also offer a way to increase your insight into new and old treatment and diagnostic paradigms. It is also a way to remain excited about your field and prevent the feeling that your day-to-day clinical practice is becoming routine.
Dr. Nitin Gupta is an Assistant Professor of Medicine, Director of Inflammatory Bowel Disease, and Program Director for the Gastroenterology Fellowship at University of Mississippi Medical Center in Jackson, MS. He has worked in basic science, translational and clinical research and continues projects in these areas. He has experience working with industry via roles of being a primary investigator in several clinical trials and consulting relationships.
The primary goal of the biotechnology and pharmaceutical industry is to develop medications and medical devices for the treatment of patients, while earning financial gain for investors. An important component of achieving this goal is the role physicians play in the drug and medical device development process. In particular, a physician’s role is to combine their clinical expertise with their knowledge of industry products to better diagnose and treat the ailments of their patients. Thus, medicine and industry have a dependent relationship. In recent times this relationship has been fraught with turmoil as the public, scientific community, and federal government have discovered real and perceived conflicts of interest.
For example, there has been public outrage in the past with reports of doctors receiving gifts, money, and lavish trips in return for prescribing medications or using certain medical devices. Because of this, Congress passed the Sunshine Act, deeming it necessary to report all physician and industry engagements that have any perceived financial value. The passage of this act was in addition to local policies set forth by academic institutions, hospitals, and private practices.
How Do I Get Started?
After checking with your institution, hospital or private practice administrator, the first step is to reach out to a local representative (“rep”) of a pharmaceutical or biotech company in which you are interested. You can accomplish this via the website of the company or by visiting the booth at major gastrointestinal conferences such as Digestive Disease Week (DDW®).
Pharma and device reps are quite knowledgeable about the latest clinical studies regarding their products, disease states, and various competing products in the market. In addition to being a source of valuable medical knowledge and disease-specific practice guidelines, they also can connect you with their medical science liaison (MSL). MSLs often have a background in pharmacy and/or research. Thus, they can provide insights into mechanisms of disease treatments and go beyond discussion of the product label, which pharmaceutical reps adhere to. They also know what therapies or diagnostic tools are in the phases of development and could be available for a clinical trial.
MSLs are also the gatekeepers for Investigator Initiated Studies (IIS). An IIS is a research project that is industry-funded and is solely designed and executed by the clinician. The application process is rigorous but awards may be easier to obtain for non-research-based clinicians who want to develop a disease-specific project that needs funding. Their grant application process can be brief, ideas may not require prior data, and turnaround time to funding may be shorter. IISs often lead to exploratory findings that may facilitate publications or lay groundwork for large-scale grants or even clinical trials. In some instances, you may be granted access to internal data and prescribing patterns, which can answer interesting clinical and research questions.
How Do I Get Started with Clinical Trials?
Being a primary investigator on a clinical trial is a big responsibility. You are responsible to the trial sponsor in addition to your patients. For young clinicians who lack experience with clinical trials, the first thing to do is to find a clinician in your department or another department, who has expertise in performing an industry-sponsored study. These individuals can be invaluable for you in terms of guiding you through the study feasibility process, study startup, and possibly being the lead or co-investigator with you. Partnering with someone with expertise in industry-sponsored clinical trials will help you gain the trust of the industry sponsor, which may be a requirement for some.
There are many additional requirements that need to be fulfilled aside from just having an appropriate and adequate patient population to pull from. You will need to have a coordinator for the study who will help you with patient care, data entry, and study- specific issues. Clinical trials require a significant amount of documentation and reporting that has to be performed within a timely manner. There is no degree prerequisite of the coordinator but it can simplify things for the clinician if they have a RN or LPN degree. Having such a degree will facilitate dual roles of patient care, lab draws, drug administration, medical charting, and other patient care matters.
In addition, you will need to have approval from either your local or central institutional review board (IRB). Also, you will have to review budget and study-specific requirements for equipment and infrastructure with your department manager. You will need to demonstrate adequate ancillary support to process, store, and ship biological specimens. In some instances, you will need a dedicated pharmacist to mix or dispense study drugs.
The process is lengthy and involved, but rewarding in terms of being involved in the drug development process. You will have opportunities to attend meetings at which you can network with other clinicians and provide the sponsor feedback on how the study is going.
How Do I Develop a Consulting Role with Industry?
It is important to check with your institution, hospital, or practice if there are any limitations in becoming a consultant for a pharmaceutical or device company. If it is allowed and will not interfere with your clinical duties, it is important to note that this role takes time to develop. It often comes about after years of experience doing research, clinical and/or basic science, with publications to support expertise. Working on an IIS is a good way to work hand-in-hand with expert industry researchers and facilitate the consulting relationship. Being a primary investigator of clinical trials with successful enrollment of patients and meeting attendance will provide you with insight into the drug development process.
What if None of This Works Out for Me?
Do not give up! Persistence, experience, and hard work are the keys to developing relationships with industry. Remember, industry has a vast network of clinicians and researchers they already work with. The overall pool of companies and experts is limited and can be difficult to break into. But it can be done. Some rely on their research experience, clinical training, and mentors to develop the necessary contacts. Others can develop the contacts via IIS applications. Industry lacks access to the physician-patient experience; this can be your greatest asset and key to your success if leveraged properly. You can consider applying for mentorship with experts in your field via AGA-sponsored events held annually at DDW® to get additional guidance.
Final Thoughts
It is important to remember that all industry relationships require time to develop. They also come at an opportunity cost of time away from your clinical practice and your family, friends and hobbies. However, these relationships also offer a way to increase your insight into new and old treatment and diagnostic paradigms. It is also a way to remain excited about your field and prevent the feeling that your day-to-day clinical practice is becoming routine.
Dr. Nitin Gupta is an Assistant Professor of Medicine, Director of Inflammatory Bowel Disease, and Program Director for the Gastroenterology Fellowship at University of Mississippi Medical Center in Jackson, MS. He has worked in basic science, translational and clinical research and continues projects in these areas. He has experience working with industry via roles of being a primary investigator in several clinical trials and consulting relationships.
The primary goal of the biotechnology and pharmaceutical industry is to develop medications and medical devices for the treatment of patients, while earning financial gain for investors. An important component of achieving this goal is the role physicians play in the drug and medical device development process. In particular, a physician’s role is to combine their clinical expertise with their knowledge of industry products to better diagnose and treat the ailments of their patients. Thus, medicine and industry have a dependent relationship. In recent times this relationship has been fraught with turmoil as the public, scientific community, and federal government have discovered real and perceived conflicts of interest.
For example, there has been public outrage in the past with reports of doctors receiving gifts, money, and lavish trips in return for prescribing medications or using certain medical devices. Because of this, Congress passed the Sunshine Act, deeming it necessary to report all physician and industry engagements that have any perceived financial value. The passage of this act was in addition to local policies set forth by academic institutions, hospitals, and private practices.
How Do I Get Started?
After checking with your institution, hospital or private practice administrator, the first step is to reach out to a local representative (“rep”) of a pharmaceutical or biotech company in which you are interested. You can accomplish this via the website of the company or by visiting the booth at major gastrointestinal conferences such as Digestive Disease Week (DDW®).
Pharma and device reps are quite knowledgeable about the latest clinical studies regarding their products, disease states, and various competing products in the market. In addition to being a source of valuable medical knowledge and disease-specific practice guidelines, they also can connect you with their medical science liaison (MSL). MSLs often have a background in pharmacy and/or research. Thus, they can provide insights into mechanisms of disease treatments and go beyond discussion of the product label, which pharmaceutical reps adhere to. They also know what therapies or diagnostic tools are in the phases of development and could be available for a clinical trial.
MSLs are also the gatekeepers for Investigator Initiated Studies (IIS). An IIS is a research project that is industry-funded and is solely designed and executed by the clinician. The application process is rigorous but awards may be easier to obtain for non-research-based clinicians who want to develop a disease-specific project that needs funding. Their grant application process can be brief, ideas may not require prior data, and turnaround time to funding may be shorter. IISs often lead to exploratory findings that may facilitate publications or lay groundwork for large-scale grants or even clinical trials. In some instances, you may be granted access to internal data and prescribing patterns, which can answer interesting clinical and research questions.
How Do I Get Started with Clinical Trials?
Being a primary investigator on a clinical trial is a big responsibility. You are responsible to the trial sponsor in addition to your patients. For young clinicians who lack experience with clinical trials, the first thing to do is to find a clinician in your department or another department, who has expertise in performing an industry-sponsored study. These individuals can be invaluable for you in terms of guiding you through the study feasibility process, study startup, and possibly being the lead or co-investigator with you. Partnering with someone with expertise in industry-sponsored clinical trials will help you gain the trust of the industry sponsor, which may be a requirement for some.
There are many additional requirements that need to be fulfilled aside from just having an appropriate and adequate patient population to pull from. You will need to have a coordinator for the study who will help you with patient care, data entry, and study- specific issues. Clinical trials require a significant amount of documentation and reporting that has to be performed within a timely manner. There is no degree prerequisite of the coordinator but it can simplify things for the clinician if they have a RN or LPN degree. Having such a degree will facilitate dual roles of patient care, lab draws, drug administration, medical charting, and other patient care matters.
In addition, you will need to have approval from either your local or central institutional review board (IRB). Also, you will have to review budget and study-specific requirements for equipment and infrastructure with your department manager. You will need to demonstrate adequate ancillary support to process, store, and ship biological specimens. In some instances, you will need a dedicated pharmacist to mix or dispense study drugs.
The process is lengthy and involved, but rewarding in terms of being involved in the drug development process. You will have opportunities to attend meetings at which you can network with other clinicians and provide the sponsor feedback on how the study is going.
How Do I Develop a Consulting Role with Industry?
It is important to check with your institution, hospital, or practice if there are any limitations in becoming a consultant for a pharmaceutical or device company. If it is allowed and will not interfere with your clinical duties, it is important to note that this role takes time to develop. It often comes about after years of experience doing research, clinical and/or basic science, with publications to support expertise. Working on an IIS is a good way to work hand-in-hand with expert industry researchers and facilitate the consulting relationship. Being a primary investigator of clinical trials with successful enrollment of patients and meeting attendance will provide you with insight into the drug development process.
What if None of This Works Out for Me?
Do not give up! Persistence, experience, and hard work are the keys to developing relationships with industry. Remember, industry has a vast network of clinicians and researchers they already work with. The overall pool of companies and experts is limited and can be difficult to break into. But it can be done. Some rely on their research experience, clinical training, and mentors to develop the necessary contacts. Others can develop the contacts via IIS applications. Industry lacks access to the physician-patient experience; this can be your greatest asset and key to your success if leveraged properly. You can consider applying for mentorship with experts in your field via AGA-sponsored events held annually at DDW® to get additional guidance.
Final Thoughts
It is important to remember that all industry relationships require time to develop. They also come at an opportunity cost of time away from your clinical practice and your family, friends and hobbies. However, these relationships also offer a way to increase your insight into new and old treatment and diagnostic paradigms. It is also a way to remain excited about your field and prevent the feeling that your day-to-day clinical practice is becoming routine.
Dr. Nitin Gupta is an Assistant Professor of Medicine, Director of Inflammatory Bowel Disease, and Program Director for the Gastroenterology Fellowship at University of Mississippi Medical Center in Jackson, MS. He has worked in basic science, translational and clinical research and continues projects in these areas. He has experience working with industry via roles of being a primary investigator in several clinical trials and consulting relationships.
Building and Maintaining a Successful Inflammatory Bowel Disease Practice
Anyone can build a successful inflammatory bowel disease (IBD) practice. To do so requires commitment and focus in the area of IBD including both Crohn’s disease and ulcerative colitis. It also requires a fundamental knowledge of medicine as well as a desire to excel and learn all that one can in these areas. Given the high number of stakeholders, good interpersonal skills are vital. Establishing an IBD practice provides an opportunity to make a big difference in peoples’ lives and the age range of impact is about the broadest in all of medical practice. The more resources you have, the greater the potential impact of your care. Table 1 lists resources that are useful to provide optimal IBD patient care.

You, the gastroenterologist, is the most important resource for the patient. Medical school, residency, fellowship, and “postgraduate” training serves as the foundation for your wealth of knowledge. Maximizing your training is of value, and this can be done by being part of an academic program, keeping abreast of current literature, and attending meetings and post-graduate courses. AGA offers a variety of publications (http://www.gastro.org/journals-and-publications) and continued training opportunities (http://www.gastro.org/education).
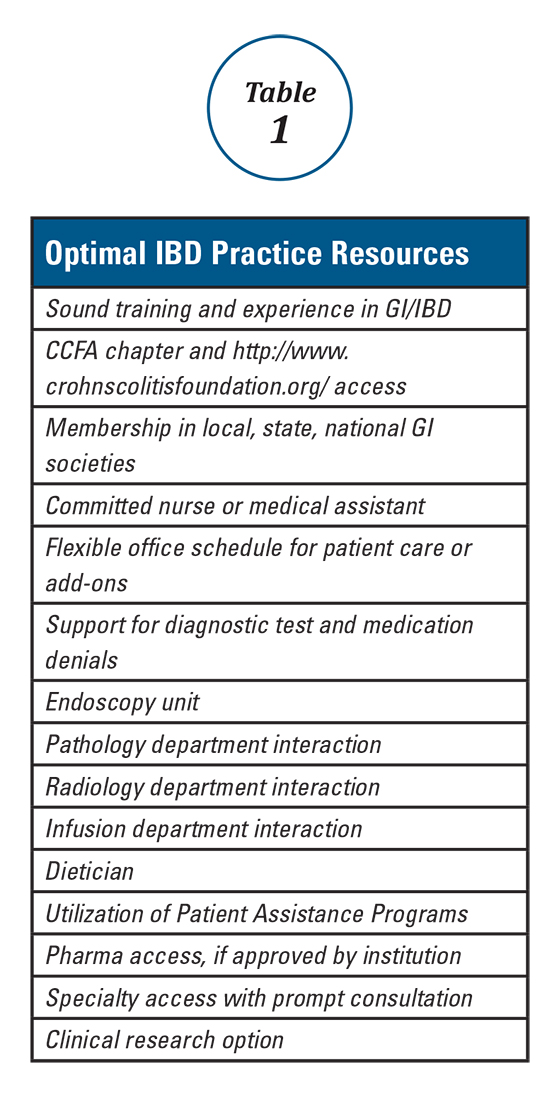
One further point regarding scheduling is that one must be willing and able to see patients urgently, rather than sending them to the emergency room. ERs are appropriate for true emergencies, but are not an ideal place for care when an IBD patient has a flare and requires prompt follow-up. I try to avoid ER visits for my patients unless they are vomiting, have severe abdominal pain, significant bleeding or have clear signs of toxicity. In an ER, abdominal pain equals a CT scan; one should consider seeing these patients in the office and triaging accordingly.
With the increasing requirements of managed care and restrictive medical plans, there has been a similar rise in the frequency of diagnostic test as well as procedure and medication denials. Re-approval and recertification of biologics and other medications have become common, which can add a great deal to your workload and that of your staff. Integration of endoscopy, pathology, and imaging (e.g., ultrasound, CT/CTE) improves response time, dialogue, and can have a positive impact on care. Office infusion allows for a better integration of this service into your practice. There is typically better communication with the infusion nurse(s) and better expedited care as well as fewer cancellations for minor infections. This all helps avoid infusion procedure delays. Infliximab, vedolizumab, ustekinumab, and lyophilized certolizumab pegol as well as intravenous iron administration can also expand services and enhance quality.
Having a medical assistant, nurse, and others in your practice to assist with patient services and care is a must. There will be many phone calls, emails, and other interactions regarding appointments, consults, routine lab testing, radiology testing, standard medications, biologics, and other treatments that necessitate an effective team-approach. For this role, either a nurse or an experienced medical assistant would be well-suited. Additional support staff and services can also aid our IBD patients. A dietitian knowledgeable in IBD and practical dietary options can, in many instances, prove invaluable. Understanding and utilizing pharma-sponsored “Patient Assistance Programs” provides drug access for the 10-20% (or more) of patients who do not have insurance or biologic coverage. Having specialty access and collegiality with colorectal surgeons, general surgeons, OB/GYNs, dermatologists, hematologists, oncologists, and others is important to expedite consults and provide collaborative care. Finally, offering clinical research options improves access for patients with limited and no coverage and also helps provide needed options for all IBD patients.
This brief overview has hopefully given you some insight into how to provide a higher level of evaluation and care for our IBD patients. These approaches have allowed me to build and maintain a successful IBD practice, and I hope that the integration of some or all of these strategies help you to build and sustain a successful IBD practice.
Dr. Wolf is director of IBD research, Atlanta Gastroenterology Associates.
Anyone can build a successful inflammatory bowel disease (IBD) practice. To do so requires commitment and focus in the area of IBD including both Crohn’s disease and ulcerative colitis. It also requires a fundamental knowledge of medicine as well as a desire to excel and learn all that one can in these areas. Given the high number of stakeholders, good interpersonal skills are vital. Establishing an IBD practice provides an opportunity to make a big difference in peoples’ lives and the age range of impact is about the broadest in all of medical practice. The more resources you have, the greater the potential impact of your care. Table 1 lists resources that are useful to provide optimal IBD patient care.
You, the gastroenterologist, is the most important resource for the patient. Medical school, residency, fellowship, and “postgraduate” training serves as the foundation for your wealth of knowledge. Maximizing your training is of value, and this can be done by being part of an academic program, keeping abreast of current literature, and attending meetings and post-graduate courses. AGA offers a variety of publications (http://www.gastro.org/journals-and-publications) and continued training opportunities (http://www.gastro.org/education).
One further point regarding scheduling is that one must be willing and able to see patients urgently, rather than sending them to the emergency room. ERs are appropriate for true emergencies, but are not an ideal place for care when an IBD patient has a flare and requires prompt follow-up. I try to avoid ER visits for my patients unless they are vomiting, have severe abdominal pain, significant bleeding or have clear signs of toxicity. In an ER, abdominal pain equals a CT scan; one should consider seeing these patients in the office and triaging accordingly.
With the increasing requirements of managed care and restrictive medical plans, there has been a similar rise in the frequency of diagnostic test as well as procedure and medication denials. Re-approval and recertification of biologics and other medications have become common, which can add a great deal to your workload and that of your staff. Integration of endoscopy, pathology, and imaging (e.g., ultrasound, CT/CTE) improves response time, dialogue, and can have a positive impact on care. Office infusion allows for a better integration of this service into your practice. There is typically better communication with the infusion nurse(s) and better expedited care as well as fewer cancellations for minor infections. This all helps avoid infusion procedure delays. Infliximab, vedolizumab, ustekinumab, and lyophilized certolizumab pegol as well as intravenous iron administration can also expand services and enhance quality.
Having a medical assistant, nurse, and others in your practice to assist with patient services and care is a must. There will be many phone calls, emails, and other interactions regarding appointments, consults, routine lab testing, radiology testing, standard medications, biologics, and other treatments that necessitate an effective team-approach. For this role, either a nurse or an experienced medical assistant would be well-suited. Additional support staff and services can also aid our IBD patients. A dietitian knowledgeable in IBD and practical dietary options can, in many instances, prove invaluable. Understanding and utilizing pharma-sponsored “Patient Assistance Programs” provides drug access for the 10-20% (or more) of patients who do not have insurance or biologic coverage. Having specialty access and collegiality with colorectal surgeons, general surgeons, OB/GYNs, dermatologists, hematologists, oncologists, and others is important to expedite consults and provide collaborative care. Finally, offering clinical research options improves access for patients with limited and no coverage and also helps provide needed options for all IBD patients.
This brief overview has hopefully given you some insight into how to provide a higher level of evaluation and care for our IBD patients. These approaches have allowed me to build and maintain a successful IBD practice, and I hope that the integration of some or all of these strategies help you to build and sustain a successful IBD practice.
Dr. Wolf is director of IBD research, Atlanta Gastroenterology Associates.
Anyone can build a successful inflammatory bowel disease (IBD) practice. To do so requires commitment and focus in the area of IBD including both Crohn’s disease and ulcerative colitis. It also requires a fundamental knowledge of medicine as well as a desire to excel and learn all that one can in these areas. Given the high number of stakeholders, good interpersonal skills are vital. Establishing an IBD practice provides an opportunity to make a big difference in peoples’ lives and the age range of impact is about the broadest in all of medical practice. The more resources you have, the greater the potential impact of your care. Table 1 lists resources that are useful to provide optimal IBD patient care.
You, the gastroenterologist, is the most important resource for the patient. Medical school, residency, fellowship, and “postgraduate” training serves as the foundation for your wealth of knowledge. Maximizing your training is of value, and this can be done by being part of an academic program, keeping abreast of current literature, and attending meetings and post-graduate courses. AGA offers a variety of publications (http://www.gastro.org/journals-and-publications) and continued training opportunities (http://www.gastro.org/education).
One further point regarding scheduling is that one must be willing and able to see patients urgently, rather than sending them to the emergency room. ERs are appropriate for true emergencies, but are not an ideal place for care when an IBD patient has a flare and requires prompt follow-up. I try to avoid ER visits for my patients unless they are vomiting, have severe abdominal pain, significant bleeding or have clear signs of toxicity. In an ER, abdominal pain equals a CT scan; one should consider seeing these patients in the office and triaging accordingly.
With the increasing requirements of managed care and restrictive medical plans, there has been a similar rise in the frequency of diagnostic test as well as procedure and medication denials. Re-approval and recertification of biologics and other medications have become common, which can add a great deal to your workload and that of your staff. Integration of endoscopy, pathology, and imaging (e.g., ultrasound, CT/CTE) improves response time, dialogue, and can have a positive impact on care. Office infusion allows for a better integration of this service into your practice. There is typically better communication with the infusion nurse(s) and better expedited care as well as fewer cancellations for minor infections. This all helps avoid infusion procedure delays. Infliximab, vedolizumab, ustekinumab, and lyophilized certolizumab pegol as well as intravenous iron administration can also expand services and enhance quality.
Having a medical assistant, nurse, and others in your practice to assist with patient services and care is a must. There will be many phone calls, emails, and other interactions regarding appointments, consults, routine lab testing, radiology testing, standard medications, biologics, and other treatments that necessitate an effective team-approach. For this role, either a nurse or an experienced medical assistant would be well-suited. Additional support staff and services can also aid our IBD patients. A dietitian knowledgeable in IBD and practical dietary options can, in many instances, prove invaluable. Understanding and utilizing pharma-sponsored “Patient Assistance Programs” provides drug access for the 10-20% (or more) of patients who do not have insurance or biologic coverage. Having specialty access and collegiality with colorectal surgeons, general surgeons, OB/GYNs, dermatologists, hematologists, oncologists, and others is important to expedite consults and provide collaborative care. Finally, offering clinical research options improves access for patients with limited and no coverage and also helps provide needed options for all IBD patients.
This brief overview has hopefully given you some insight into how to provide a higher level of evaluation and care for our IBD patients. These approaches have allowed me to build and maintain a successful IBD practice, and I hope that the integration of some or all of these strategies help you to build and sustain a successful IBD practice.
Dr. Wolf is director of IBD research, Atlanta Gastroenterology Associates.
Health Maintenance and Preventive Care in Patients with Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) consists of two chronic inflammatory diseases, Crohn’s disease (CD) and ulcerative colitis (UC), as well as a small category of patients (~10%) who have atypical features called IBD-unclassified (IBD-U) or indeterminate colitis. The prevalence of IBD ranges from 0.3% to 0.5% overall in North America and Europe.1 In North America, the incidences of CD and UC are estimated to be 3.1 to 14.6 per 100,000 person-years and 2.2 to 14.3 cases per 100,000 person-years, respectively; similar rates are seen in Europe.2 However, incidences up to 19.2 and 20.2 per 100,000 for UC and CD, respectively, have been reported in Canada.3,4 The incidences of both UC and CD are increasing over time in Western countries and in rapidly industrializing countries throughout Asia and South America.5-8

Influenza vaccine and pneumococcal vaccine
Influenza A and B outbreaks are commonly seen during the fall and early spring and risk factors for pneumonia and hospitalization include older age, chronic medical conditions, and immunosuppression. The CDC now recommend annual influenza vaccination for all individuals older than six months. For patients on immunosuppression, the vaccine administered should be the inactivated vaccine, as live attenuated vaccines should not be administered to these patients.

In IBD patients, the influenza and pneumococcal vaccines are both well tolerated without an increased rate of adverse effects over the general population and without an increased risk of IBD flares after vaccination.12 A common question for patients on biologic therapy is whether the vaccine should be timed at a specific point in the dose cycle. For infliximab, and likely other biologics, the timing does not change the vaccine immunogenicity and patients should be given these vaccines regardless of where they are in the cycle of administration of their biologic.13 In addition, there is significant response to influenza and pneumococcal vaccines in patients on combination therapy with immunomodulators and anti-TNFs and concerns about a lack of response to vaccines should not discourage vaccination since benefits are still acquired by patients even if immunogenicity is somewhat decreased.14,15
Other vaccinations
In addition to the influenza and pneumococcal vaccines, adult and pediatric patients with IBD should follow the ACIP recommendations for tetanus, diphtheria, pertussis (Tdap), Td boosters, hepatitis A, hepatitis B, human papilloma virus (HPV), and meningococcal vaccinations.16,17
Live vaccines including measles mumps rubella (MMR), varicella, and zoster vaccines are in general contraindicated in immunosuppressed patients on corticosteroids, azathioprine/6-mercaptopurine, methotrexate, anti-TNF, and anti-integrin biologics. An inactive varicella-zoster vaccine will likely be available in the near future and may obviate the need for the live vaccine, which is an important development given the increased risk of zoster in patients with IBD on immunosuppression.18
Osteoporosis screening

Skin cancer screening
Multiple studies have demonstrated that immunosuppression, especially with methotrexate and azathioprine/6-mercaptopurine (6MP) is a risk factor for the development of initial and recurrent non-melanoma skin cancer (NMSC) in IBD patients, the data for biologics are less definitive.23-25 In addition, biologics are associated with increased risk of melanoma in IBD.26 The elevated risk of skin cancer begins in the first year of treatment with thiopurines and may continue after discontinuation. On the basis of this data, screening for melanoma and NMSC is recommended in IBD patients on immunosuppression. Especially for patients on thiopurines it is reasonable for the initial dermatologist visit to occur in the first year of treatment and thereafter with at least annual visits for a full body skin examination. In addition, it is reasonable to recommend regular sunscreen use and protective clothing such as hats.
Cervical cancer screening
A recent meta-analysis shows that women with IBD on immunosuppression have an increased risk of cervical high grade dysplasia and cervical cancer.27 HPV is the major risk factor for cervical cancer and is necessary for its development. The current American College of Gynecology guidelines for women on immunosuppression are to start cervical cancer screening at 21 and annual screening thereafter with Pap and HPV testing.28
Smoking
Smoking has well known associations with poor outcomes in the general population such as increased risk of lung and pancreatic cancers, as well as high risk of cardiovascular disease. In addition, smoking has risks specific to IBD. In CD, smoking is associated with increased disease activity, increased risk of post-operative recurrence, and increased severity of disease.29 Smoking cessation is associated with improved long-term disease outcomes and less risk.30 Making it a point to regularly discuss smoking cessation and partnering with PCPs to offer evidence-based quitting aids may be one of our most significant and beneficial interventions.
Depression and anxiety
Several studies have shown high levels of depression and anxiety in IBD patients and higher levels of depression are associated with increased symptoms, clinical recurrence, poor quality of life and decreased social support.31-33 A recent systematic review of several studies suggested that antidepressants use in IBD patients benefits their mental health and may improve their clinical course as well.34 As such, screening for depression and anxiety regularly and either offering treatment or referral to psychiatrists and psychologists for further management is recommended.10
Conclusion
Patients with IBD frequently develop long-term relationships with their gastroenterologists due to their lifelong chronic disease. It is therefore incumbent on us to be attentive to issues related to IBD patients’ preventive care and collaborate with PCPs to coordinate care for our patients since many of these interventions have both short-term and long-term benefits.
Dr. Chachu is assistant professor and gastroenterologist at Duke University, Durham, N.C.
References
1. Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-21.e2.
2. Loftus EV, Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504-17.
3. Bernstein CN, Wajda A, Svenson LW, et al. The Epidemiology of Inflammatory Bowel Disease in Canada: A Population-Based Study. The American journal of gastroenterology. 2006;101(7):1559-68.
4. Lowe AM, Roy PO, M BP, et al. Epidemiology of Crohn’s disease in Quebec, Canada. Inflammatory bowel diseases. 2009;15(3):429-35.
5. Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2007;5(12):1424-9.
6. Kappelman MD, Moore KR, Allen JK, et al. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Digestive diseases and sciences. 2013;58(2):519-25.
7. Ng SC, Kaplan G, Banerjee R, et al. 78 Incidence and Phenotype of Inflammatory Bowel Disease From 13 Countries in Asia-Pacific: Results From the Asia-Pacific Crohn’s and Colitis Epidemiologic Study 2011-2013. Gastroenterology.150(4):S21.
8. Parente JML, Coy CSR, Campelo V, et al. Inflammatory bowel disease in an underdeveloped region of Northeastern Brazil. World Journal of Gastroenterology : WJG. 2015;21(4):1197-206.
9. Selby L, Kane S, Wilson J, et al. Receipt of preventive health services by IBD patients is significantly lower than by primary care patients. Inflammatory bowel diseases. 2008;14(2):253-8.
10. Farraye FA, Melmed GY, Lichtenstein GR, et al. ACG Clinical Guideline: Preventive Care in Inflammatory Bowel Disease. The American journal of gastroenterology. 2017;112(2):241-58.
11. Long MD, Martin C, Sandler RS, et al. Increased risk of pneumonia among patients with inflammatory bowel disease. The American journal of gastroenterology. 2013;108(2):240-8.
12. Rahier JF, Papay P, Salleron J, et al. H1N1 vaccines in a large observational cohort of patients with inflammatory bowel disease treated with immunomodulators and biological therapy. Gut. 2011;60(4):456-62.
13. deBruyn J, Fonseca K, Ghosh S, et al. Immunogenicity of Influenza Vaccine for Patients with Inflammatory Bowel Disease on Maintenance Infliximab Therapy: A Randomized Trial. Inflammatory bowel diseases. 2016;22(3):638-47.
14. Brezinschek HP, Hofstaetter T, Leeb BF, et al. Immunization of patients with rheumatoid arthritis with antitumor necrosis factor alpha therapy and methotrexate. Current opinion in rheumatology. 2008;20(3):295-9.
15. Kaine JL, Kivitz AJ, Birbara C, et al. Immune responses following administration of influenza and pneumococcal vaccines to patients with rheumatoid arthritis receiving adalimumab. J Rheumatol. 2007;34(2):272-9.
16. Kim DK, Riley LE, Harriman KH, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Adults Aged 19 Years or Older - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):136-8.
17. Robinson CL, Romero JR, Kempe A, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Children and Adolescents Aged 18 Years or Younger - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):134-5.
18. Cullen G, Baden RP, Cheifetz AS. Varicella zoster virus infection in inflammatory bowel disease. Inflammatory bowel diseases. 2012;18(12):2392-403.
19. Card T, West J, Hubbard R, et al. Hip fractures in patients with inflammatory bowel disease and their relationship to corticosteroid use: a population based cohort study. Gut. 2004;53(2):251-5.
20. Casals-Seoane F, Chaparro M, Mate J, et al. Clinical Course of Bone Metabolism Disorders in Patients with Inflammatory Bowel Disease: A 5-Year Prospective Study. Inflammatory bowel diseases. 2016;22(8):1929-36.
21. Melek J, Sakuraba A. Efficacy and safety of medical therapy for low bone mineral density in patients with inflammatory bowel disease: a meta-analysis and systematic review. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2014;12(1):32-44.e5.
22. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporosis International. 2014;25(10):2359-81.
23. Peyrin-Biroulet L, Khosrotehrani K, Carrat F, et al. Increased risk for nonmelanoma skin cancers in patients who receive thiopurines for inflammatory bowel disease. Gastroenterology. 2011;141(5):1621-28.e1-5.
24. Long MD, Herfarth HH, Pipkin CA, et al. Increased risk for non-melanoma skin cancer in patients with inflammatory bowel disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2010;8(3):268-74.
25. Scott FI, Mamtani R, Brensinger CM, et al. Risk of Nonmelanoma Skin Cancer Associated With the Use of Immunosuppressant and Biologic Agents in Patients With a History of Autoimmune Disease and Nonmelanoma Skin Cancer. JAMA dermatology. 2016;152(2):164-72.
26. Long MD, Martin CF, Pipkin CA, et al. Risk of melanoma and nonmelanoma skin cancer among patients with inflammatory bowel disease. Gastroenterology. 2012;143(2):390-9.e1.
27. Allegretti JR, Barnes EL, Cameron A. Are patients with inflammatory bowel disease on chronic immunosuppressive therapy at increased risk of cervical high-grade dysplasia/cancer? A meta-analysis. Inflammatory bowel diseases. 2015;21(5):1089-97.
28. Practice Bulletin No. 168: Cervical Cancer Screening and Prevention. Obstetrics and gynecology. 2016;128(4):e111-30.
29. Ryan WR, Allan RN, Yamamoto T, et al. Crohn’s disease patients who quit smoking have a reduced risk of reoperation for recurrence. American journal of surgery. 2004;187(2):219-25.
30. Cosnes J, Beaugerie L, Carbonnel F, et al. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120(5):1093-9.
31. Fuller-Thomson E, Sulman J. Depression and inflammatory bowel disease: findings from two nationally representative Canadian surveys. Inflammatory bowel diseases. 2006;12(8):697-707.
32. Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. General hospital psychiatry. 1996;18(4):220-9.
33. Mikocka-Walus A, Pittet V, Rossel J-B, et al. Symptoms of Depression and Anxiety Are Independently Associated With Clinical Recurrence of Inflammatory Bowel Disease. Clinical Gastroenterology and Hepatology.14(6):829-35.e1.
34. Macer BJD, Prady SL, Mikocka-Walus A. Antidepressants in Inflammatory Bowel Disease: A Systematic Review. Inflammatory bowel diseases. 2017;23(4):534-50.
Inflammatory bowel disease (IBD) consists of two chronic inflammatory diseases, Crohn’s disease (CD) and ulcerative colitis (UC), as well as a small category of patients (~10%) who have atypical features called IBD-unclassified (IBD-U) or indeterminate colitis. The prevalence of IBD ranges from 0.3% to 0.5% overall in North America and Europe.1 In North America, the incidences of CD and UC are estimated to be 3.1 to 14.6 per 100,000 person-years and 2.2 to 14.3 cases per 100,000 person-years, respectively; similar rates are seen in Europe.2 However, incidences up to 19.2 and 20.2 per 100,000 for UC and CD, respectively, have been reported in Canada.3,4 The incidences of both UC and CD are increasing over time in Western countries and in rapidly industrializing countries throughout Asia and South America.5-8
Influenza vaccine and pneumococcal vaccine
Influenza A and B outbreaks are commonly seen during the fall and early spring and risk factors for pneumonia and hospitalization include older age, chronic medical conditions, and immunosuppression. The CDC now recommend annual influenza vaccination for all individuals older than six months. For patients on immunosuppression, the vaccine administered should be the inactivated vaccine, as live attenuated vaccines should not be administered to these patients.
In IBD patients, the influenza and pneumococcal vaccines are both well tolerated without an increased rate of adverse effects over the general population and without an increased risk of IBD flares after vaccination.12 A common question for patients on biologic therapy is whether the vaccine should be timed at a specific point in the dose cycle. For infliximab, and likely other biologics, the timing does not change the vaccine immunogenicity and patients should be given these vaccines regardless of where they are in the cycle of administration of their biologic.13 In addition, there is significant response to influenza and pneumococcal vaccines in patients on combination therapy with immunomodulators and anti-TNFs and concerns about a lack of response to vaccines should not discourage vaccination since benefits are still acquired by patients even if immunogenicity is somewhat decreased.14,15
Other vaccinations
In addition to the influenza and pneumococcal vaccines, adult and pediatric patients with IBD should follow the ACIP recommendations for tetanus, diphtheria, pertussis (Tdap), Td boosters, hepatitis A, hepatitis B, human papilloma virus (HPV), and meningococcal vaccinations.16,17
Live vaccines including measles mumps rubella (MMR), varicella, and zoster vaccines are in general contraindicated in immunosuppressed patients on corticosteroids, azathioprine/6-mercaptopurine, methotrexate, anti-TNF, and anti-integrin biologics. An inactive varicella-zoster vaccine will likely be available in the near future and may obviate the need for the live vaccine, which is an important development given the increased risk of zoster in patients with IBD on immunosuppression.18
Osteoporosis screening
Skin cancer screening
Multiple studies have demonstrated that immunosuppression, especially with methotrexate and azathioprine/6-mercaptopurine (6MP) is a risk factor for the development of initial and recurrent non-melanoma skin cancer (NMSC) in IBD patients, the data for biologics are less definitive.23-25 In addition, biologics are associated with increased risk of melanoma in IBD.26 The elevated risk of skin cancer begins in the first year of treatment with thiopurines and may continue after discontinuation. On the basis of this data, screening for melanoma and NMSC is recommended in IBD patients on immunosuppression. Especially for patients on thiopurines it is reasonable for the initial dermatologist visit to occur in the first year of treatment and thereafter with at least annual visits for a full body skin examination. In addition, it is reasonable to recommend regular sunscreen use and protective clothing such as hats.
Cervical cancer screening
A recent meta-analysis shows that women with IBD on immunosuppression have an increased risk of cervical high grade dysplasia and cervical cancer.27 HPV is the major risk factor for cervical cancer and is necessary for its development. The current American College of Gynecology guidelines for women on immunosuppression are to start cervical cancer screening at 21 and annual screening thereafter with Pap and HPV testing.28
Smoking
Smoking has well known associations with poor outcomes in the general population such as increased risk of lung and pancreatic cancers, as well as high risk of cardiovascular disease. In addition, smoking has risks specific to IBD. In CD, smoking is associated with increased disease activity, increased risk of post-operative recurrence, and increased severity of disease.29 Smoking cessation is associated with improved long-term disease outcomes and less risk.30 Making it a point to regularly discuss smoking cessation and partnering with PCPs to offer evidence-based quitting aids may be one of our most significant and beneficial interventions.
Depression and anxiety
Several studies have shown high levels of depression and anxiety in IBD patients and higher levels of depression are associated with increased symptoms, clinical recurrence, poor quality of life and decreased social support.31-33 A recent systematic review of several studies suggested that antidepressants use in IBD patients benefits their mental health and may improve their clinical course as well.34 As such, screening for depression and anxiety regularly and either offering treatment or referral to psychiatrists and psychologists for further management is recommended.10
Conclusion
Patients with IBD frequently develop long-term relationships with their gastroenterologists due to their lifelong chronic disease. It is therefore incumbent on us to be attentive to issues related to IBD patients’ preventive care and collaborate with PCPs to coordinate care for our patients since many of these interventions have both short-term and long-term benefits.
Dr. Chachu is assistant professor and gastroenterologist at Duke University, Durham, N.C.
References
1. Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-21.e2.
2. Loftus EV, Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504-17.
3. Bernstein CN, Wajda A, Svenson LW, et al. The Epidemiology of Inflammatory Bowel Disease in Canada: A Population-Based Study. The American journal of gastroenterology. 2006;101(7):1559-68.
4. Lowe AM, Roy PO, M BP, et al. Epidemiology of Crohn’s disease in Quebec, Canada. Inflammatory bowel diseases. 2009;15(3):429-35.
5. Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2007;5(12):1424-9.
6. Kappelman MD, Moore KR, Allen JK, et al. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Digestive diseases and sciences. 2013;58(2):519-25.
7. Ng SC, Kaplan G, Banerjee R, et al. 78 Incidence and Phenotype of Inflammatory Bowel Disease From 13 Countries in Asia-Pacific: Results From the Asia-Pacific Crohn’s and Colitis Epidemiologic Study 2011-2013. Gastroenterology.150(4):S21.
8. Parente JML, Coy CSR, Campelo V, et al. Inflammatory bowel disease in an underdeveloped region of Northeastern Brazil. World Journal of Gastroenterology : WJG. 2015;21(4):1197-206.
9. Selby L, Kane S, Wilson J, et al. Receipt of preventive health services by IBD patients is significantly lower than by primary care patients. Inflammatory bowel diseases. 2008;14(2):253-8.
10. Farraye FA, Melmed GY, Lichtenstein GR, et al. ACG Clinical Guideline: Preventive Care in Inflammatory Bowel Disease. The American journal of gastroenterology. 2017;112(2):241-58.
11. Long MD, Martin C, Sandler RS, et al. Increased risk of pneumonia among patients with inflammatory bowel disease. The American journal of gastroenterology. 2013;108(2):240-8.
12. Rahier JF, Papay P, Salleron J, et al. H1N1 vaccines in a large observational cohort of patients with inflammatory bowel disease treated with immunomodulators and biological therapy. Gut. 2011;60(4):456-62.
13. deBruyn J, Fonseca K, Ghosh S, et al. Immunogenicity of Influenza Vaccine for Patients with Inflammatory Bowel Disease on Maintenance Infliximab Therapy: A Randomized Trial. Inflammatory bowel diseases. 2016;22(3):638-47.
14. Brezinschek HP, Hofstaetter T, Leeb BF, et al. Immunization of patients with rheumatoid arthritis with antitumor necrosis factor alpha therapy and methotrexate. Current opinion in rheumatology. 2008;20(3):295-9.
15. Kaine JL, Kivitz AJ, Birbara C, et al. Immune responses following administration of influenza and pneumococcal vaccines to patients with rheumatoid arthritis receiving adalimumab. J Rheumatol. 2007;34(2):272-9.
16. Kim DK, Riley LE, Harriman KH, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Adults Aged 19 Years or Older - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):136-8.
17. Robinson CL, Romero JR, Kempe A, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Children and Adolescents Aged 18 Years or Younger - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):134-5.
18. Cullen G, Baden RP, Cheifetz AS. Varicella zoster virus infection in inflammatory bowel disease. Inflammatory bowel diseases. 2012;18(12):2392-403.
19. Card T, West J, Hubbard R, et al. Hip fractures in patients with inflammatory bowel disease and their relationship to corticosteroid use: a population based cohort study. Gut. 2004;53(2):251-5.
20. Casals-Seoane F, Chaparro M, Mate J, et al. Clinical Course of Bone Metabolism Disorders in Patients with Inflammatory Bowel Disease: A 5-Year Prospective Study. Inflammatory bowel diseases. 2016;22(8):1929-36.
21. Melek J, Sakuraba A. Efficacy and safety of medical therapy for low bone mineral density in patients with inflammatory bowel disease: a meta-analysis and systematic review. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2014;12(1):32-44.e5.
22. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporosis International. 2014;25(10):2359-81.
23. Peyrin-Biroulet L, Khosrotehrani K, Carrat F, et al. Increased risk for nonmelanoma skin cancers in patients who receive thiopurines for inflammatory bowel disease. Gastroenterology. 2011;141(5):1621-28.e1-5.
24. Long MD, Herfarth HH, Pipkin CA, et al. Increased risk for non-melanoma skin cancer in patients with inflammatory bowel disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2010;8(3):268-74.
25. Scott FI, Mamtani R, Brensinger CM, et al. Risk of Nonmelanoma Skin Cancer Associated With the Use of Immunosuppressant and Biologic Agents in Patients With a History of Autoimmune Disease and Nonmelanoma Skin Cancer. JAMA dermatology. 2016;152(2):164-72.
26. Long MD, Martin CF, Pipkin CA, et al. Risk of melanoma and nonmelanoma skin cancer among patients with inflammatory bowel disease. Gastroenterology. 2012;143(2):390-9.e1.
27. Allegretti JR, Barnes EL, Cameron A. Are patients with inflammatory bowel disease on chronic immunosuppressive therapy at increased risk of cervical high-grade dysplasia/cancer? A meta-analysis. Inflammatory bowel diseases. 2015;21(5):1089-97.
28. Practice Bulletin No. 168: Cervical Cancer Screening and Prevention. Obstetrics and gynecology. 2016;128(4):e111-30.
29. Ryan WR, Allan RN, Yamamoto T, et al. Crohn’s disease patients who quit smoking have a reduced risk of reoperation for recurrence. American journal of surgery. 2004;187(2):219-25.
30. Cosnes J, Beaugerie L, Carbonnel F, et al. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120(5):1093-9.
31. Fuller-Thomson E, Sulman J. Depression and inflammatory bowel disease: findings from two nationally representative Canadian surveys. Inflammatory bowel diseases. 2006;12(8):697-707.
32. Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. General hospital psychiatry. 1996;18(4):220-9.
33. Mikocka-Walus A, Pittet V, Rossel J-B, et al. Symptoms of Depression and Anxiety Are Independently Associated With Clinical Recurrence of Inflammatory Bowel Disease. Clinical Gastroenterology and Hepatology.14(6):829-35.e1.
34. Macer BJD, Prady SL, Mikocka-Walus A. Antidepressants in Inflammatory Bowel Disease: A Systematic Review. Inflammatory bowel diseases. 2017;23(4):534-50.
Inflammatory bowel disease (IBD) consists of two chronic inflammatory diseases, Crohn’s disease (CD) and ulcerative colitis (UC), as well as a small category of patients (~10%) who have atypical features called IBD-unclassified (IBD-U) or indeterminate colitis. The prevalence of IBD ranges from 0.3% to 0.5% overall in North America and Europe.1 In North America, the incidences of CD and UC are estimated to be 3.1 to 14.6 per 100,000 person-years and 2.2 to 14.3 cases per 100,000 person-years, respectively; similar rates are seen in Europe.2 However, incidences up to 19.2 and 20.2 per 100,000 for UC and CD, respectively, have been reported in Canada.3,4 The incidences of both UC and CD are increasing over time in Western countries and in rapidly industrializing countries throughout Asia and South America.5-8
Influenza vaccine and pneumococcal vaccine
Influenza A and B outbreaks are commonly seen during the fall and early spring and risk factors for pneumonia and hospitalization include older age, chronic medical conditions, and immunosuppression. The CDC now recommend annual influenza vaccination for all individuals older than six months. For patients on immunosuppression, the vaccine administered should be the inactivated vaccine, as live attenuated vaccines should not be administered to these patients.
In IBD patients, the influenza and pneumococcal vaccines are both well tolerated without an increased rate of adverse effects over the general population and without an increased risk of IBD flares after vaccination.12 A common question for patients on biologic therapy is whether the vaccine should be timed at a specific point in the dose cycle. For infliximab, and likely other biologics, the timing does not change the vaccine immunogenicity and patients should be given these vaccines regardless of where they are in the cycle of administration of their biologic.13 In addition, there is significant response to influenza and pneumococcal vaccines in patients on combination therapy with immunomodulators and anti-TNFs and concerns about a lack of response to vaccines should not discourage vaccination since benefits are still acquired by patients even if immunogenicity is somewhat decreased.14,15
Other vaccinations
In addition to the influenza and pneumococcal vaccines, adult and pediatric patients with IBD should follow the ACIP recommendations for tetanus, diphtheria, pertussis (Tdap), Td boosters, hepatitis A, hepatitis B, human papilloma virus (HPV), and meningococcal vaccinations.16,17
Live vaccines including measles mumps rubella (MMR), varicella, and zoster vaccines are in general contraindicated in immunosuppressed patients on corticosteroids, azathioprine/6-mercaptopurine, methotrexate, anti-TNF, and anti-integrin biologics. An inactive varicella-zoster vaccine will likely be available in the near future and may obviate the need for the live vaccine, which is an important development given the increased risk of zoster in patients with IBD on immunosuppression.18
Osteoporosis screening
Skin cancer screening
Multiple studies have demonstrated that immunosuppression, especially with methotrexate and azathioprine/6-mercaptopurine (6MP) is a risk factor for the development of initial and recurrent non-melanoma skin cancer (NMSC) in IBD patients, the data for biologics are less definitive.23-25 In addition, biologics are associated with increased risk of melanoma in IBD.26 The elevated risk of skin cancer begins in the first year of treatment with thiopurines and may continue after discontinuation. On the basis of this data, screening for melanoma and NMSC is recommended in IBD patients on immunosuppression. Especially for patients on thiopurines it is reasonable for the initial dermatologist visit to occur in the first year of treatment and thereafter with at least annual visits for a full body skin examination. In addition, it is reasonable to recommend regular sunscreen use and protective clothing such as hats.
Cervical cancer screening
A recent meta-analysis shows that women with IBD on immunosuppression have an increased risk of cervical high grade dysplasia and cervical cancer.27 HPV is the major risk factor for cervical cancer and is necessary for its development. The current American College of Gynecology guidelines for women on immunosuppression are to start cervical cancer screening at 21 and annual screening thereafter with Pap and HPV testing.28
Smoking
Smoking has well known associations with poor outcomes in the general population such as increased risk of lung and pancreatic cancers, as well as high risk of cardiovascular disease. In addition, smoking has risks specific to IBD. In CD, smoking is associated with increased disease activity, increased risk of post-operative recurrence, and increased severity of disease.29 Smoking cessation is associated with improved long-term disease outcomes and less risk.30 Making it a point to regularly discuss smoking cessation and partnering with PCPs to offer evidence-based quitting aids may be one of our most significant and beneficial interventions.
Depression and anxiety
Several studies have shown high levels of depression and anxiety in IBD patients and higher levels of depression are associated with increased symptoms, clinical recurrence, poor quality of life and decreased social support.31-33 A recent systematic review of several studies suggested that antidepressants use in IBD patients benefits their mental health and may improve their clinical course as well.34 As such, screening for depression and anxiety regularly and either offering treatment or referral to psychiatrists and psychologists for further management is recommended.10
Conclusion
Patients with IBD frequently develop long-term relationships with their gastroenterologists due to their lifelong chronic disease. It is therefore incumbent on us to be attentive to issues related to IBD patients’ preventive care and collaborate with PCPs to coordinate care for our patients since many of these interventions have both short-term and long-term benefits.
Dr. Chachu is assistant professor and gastroenterologist at Duke University, Durham, N.C.
References
1. Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-21.e2.
2. Loftus EV, Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504-17.
3. Bernstein CN, Wajda A, Svenson LW, et al. The Epidemiology of Inflammatory Bowel Disease in Canada: A Population-Based Study. The American journal of gastroenterology. 2006;101(7):1559-68.
4. Lowe AM, Roy PO, M BP, et al. Epidemiology of Crohn’s disease in Quebec, Canada. Inflammatory bowel diseases. 2009;15(3):429-35.
5. Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2007;5(12):1424-9.
6. Kappelman MD, Moore KR, Allen JK, et al. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Digestive diseases and sciences. 2013;58(2):519-25.
7. Ng SC, Kaplan G, Banerjee R, et al. 78 Incidence and Phenotype of Inflammatory Bowel Disease From 13 Countries in Asia-Pacific: Results From the Asia-Pacific Crohn’s and Colitis Epidemiologic Study 2011-2013. Gastroenterology.150(4):S21.
8. Parente JML, Coy CSR, Campelo V, et al. Inflammatory bowel disease in an underdeveloped region of Northeastern Brazil. World Journal of Gastroenterology : WJG. 2015;21(4):1197-206.
9. Selby L, Kane S, Wilson J, et al. Receipt of preventive health services by IBD patients is significantly lower than by primary care patients. Inflammatory bowel diseases. 2008;14(2):253-8.
10. Farraye FA, Melmed GY, Lichtenstein GR, et al. ACG Clinical Guideline: Preventive Care in Inflammatory Bowel Disease. The American journal of gastroenterology. 2017;112(2):241-58.
11. Long MD, Martin C, Sandler RS, et al. Increased risk of pneumonia among patients with inflammatory bowel disease. The American journal of gastroenterology. 2013;108(2):240-8.
12. Rahier JF, Papay P, Salleron J, et al. H1N1 vaccines in a large observational cohort of patients with inflammatory bowel disease treated with immunomodulators and biological therapy. Gut. 2011;60(4):456-62.
13. deBruyn J, Fonseca K, Ghosh S, et al. Immunogenicity of Influenza Vaccine for Patients with Inflammatory Bowel Disease on Maintenance Infliximab Therapy: A Randomized Trial. Inflammatory bowel diseases. 2016;22(3):638-47.
14. Brezinschek HP, Hofstaetter T, Leeb BF, et al. Immunization of patients with rheumatoid arthritis with antitumor necrosis factor alpha therapy and methotrexate. Current opinion in rheumatology. 2008;20(3):295-9.
15. Kaine JL, Kivitz AJ, Birbara C, et al. Immune responses following administration of influenza and pneumococcal vaccines to patients with rheumatoid arthritis receiving adalimumab. J Rheumatol. 2007;34(2):272-9.
16. Kim DK, Riley LE, Harriman KH, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Adults Aged 19 Years or Older - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):136-8.
17. Robinson CL, Romero JR, Kempe A, et al. Advisory Committee on Immunization Practices Recommended Immunization Schedule for Children and Adolescents Aged 18 Years or Younger - United States, 2017. MMWR Morbidity and mortality weekly report. 2017;66(5):134-5.
18. Cullen G, Baden RP, Cheifetz AS. Varicella zoster virus infection in inflammatory bowel disease. Inflammatory bowel diseases. 2012;18(12):2392-403.
19. Card T, West J, Hubbard R, et al. Hip fractures in patients with inflammatory bowel disease and their relationship to corticosteroid use: a population based cohort study. Gut. 2004;53(2):251-5.
20. Casals-Seoane F, Chaparro M, Mate J, et al. Clinical Course of Bone Metabolism Disorders in Patients with Inflammatory Bowel Disease: A 5-Year Prospective Study. Inflammatory bowel diseases. 2016;22(8):1929-36.
21. Melek J, Sakuraba A. Efficacy and safety of medical therapy for low bone mineral density in patients with inflammatory bowel disease: a meta-analysis and systematic review. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2014;12(1):32-44.e5.
22. Cosman F, de Beur SJ, LeBoff MS, et al. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporosis International. 2014;25(10):2359-81.
23. Peyrin-Biroulet L, Khosrotehrani K, Carrat F, et al. Increased risk for nonmelanoma skin cancers in patients who receive thiopurines for inflammatory bowel disease. Gastroenterology. 2011;141(5):1621-28.e1-5.
24. Long MD, Herfarth HH, Pipkin CA, et al. Increased risk for non-melanoma skin cancer in patients with inflammatory bowel disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2010;8(3):268-74.
25. Scott FI, Mamtani R, Brensinger CM, et al. Risk of Nonmelanoma Skin Cancer Associated With the Use of Immunosuppressant and Biologic Agents in Patients With a History of Autoimmune Disease and Nonmelanoma Skin Cancer. JAMA dermatology. 2016;152(2):164-72.
26. Long MD, Martin CF, Pipkin CA, et al. Risk of melanoma and nonmelanoma skin cancer among patients with inflammatory bowel disease. Gastroenterology. 2012;143(2):390-9.e1.
27. Allegretti JR, Barnes EL, Cameron A. Are patients with inflammatory bowel disease on chronic immunosuppressive therapy at increased risk of cervical high-grade dysplasia/cancer? A meta-analysis. Inflammatory bowel diseases. 2015;21(5):1089-97.
28. Practice Bulletin No. 168: Cervical Cancer Screening and Prevention. Obstetrics and gynecology. 2016;128(4):e111-30.
29. Ryan WR, Allan RN, Yamamoto T, et al. Crohn’s disease patients who quit smoking have a reduced risk of reoperation for recurrence. American journal of surgery. 2004;187(2):219-25.
30. Cosnes J, Beaugerie L, Carbonnel F, et al. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120(5):1093-9.
31. Fuller-Thomson E, Sulman J. Depression and inflammatory bowel disease: findings from two nationally representative Canadian surveys. Inflammatory bowel diseases. 2006;12(8):697-707.
32. Walker EA, Gelfand MD, Gelfand AN, et al. The relationship of current psychiatric disorder to functional disability and distress in patients with inflammatory bowel disease. General hospital psychiatry. 1996;18(4):220-9.
33. Mikocka-Walus A, Pittet V, Rossel J-B, et al. Symptoms of Depression and Anxiety Are Independently Associated With Clinical Recurrence of Inflammatory Bowel Disease. Clinical Gastroenterology and Hepatology.14(6):829-35.e1.
34. Macer BJD, Prady SL, Mikocka-Walus A. Antidepressants in Inflammatory Bowel Disease: A Systematic Review. Inflammatory bowel diseases. 2017;23(4):534-50.