User login
High volume of noninvasive ventilation in hospitals doesn’t ensure good outcomes in acute COPD
Hospitals that frequently treat acute COPD exacerbations using noninvasive ventilation – a practice known to reduce mortality, length of stay, and the need for more invasive treatment – did not have better patient outcomes than did hospitals that used noninvasive ventilation less frequently, according to a report published in Annals of the American Thoracic Society.
Acute COPD exacerbations are “one of the few conditions with high-level evidence demonstrating the benefits of noninvasive ventilation in patients with respiratory distress,” and the treatment has been widely adopted for this patient population. However, for noninvasive ventilation to succeed, patients must be carefully selected and closely monitored, and a multidisciplinary team of nurses, respiratory therapists, and physicians must coordinate the treatment, often across multiple hospital settings, said Anuj B. Mehta, MD, of The Pulmonary Center, Boston University, and his associates.
Until now, it was not known whether hospitals with a high volume of noninvasive ventilation develop specialized expertise and thus deliver superior patient outcomes, or whether a high volume results from suboptimal patient selection or otherwise puts a strain on a hospital’s staff and thus produces poor outcomes. To examine this question, Dr. Mehta and his associates analyzed information in a database enrolling adults treated at 252 California hospitals for acute COPD exacerbation. They focused on 37,516 hospitalizations that occurred during a single year.
Overall, 9.3% of these patients received noninvasive ventilation. The median annual case volume of noninvasive ventilation for any indication was 64 per hospital. But rates of noninvasive ventilation varied widely across hospitals, with 40% of facilities significantly deviating from this median rate.
“Contrary to our hypothesis, we did not observe significantly lower COPD mortality” in hospitals with high volumes of noninvasive ventilation. For individual patients, admission to a hospital with a high volume of noninvasive ventilation was associated with significantly higher odds of treatment failure (adjusted OR, 1.95), and such failure was associated with significantly higher odds of death (adjusted OR, 1.81). In addition, at the hospital level, a high volume of noninvasive ventilation was associated with a significantly higher risk of treatment failure, which in turn was associated with higher patient mortality.
“Hospitals with higher total noninvasive ventilation case volume tended to use [it] in patients with more comorbidities and acute organ failures, suggesting potential overuse among patients at higher risk of treatment failure. ... [This] may partially explain why hospitals with high rates of using an evidence-based intervention did not achieve significant mortality benefits,” Dr. Mehta and his associates said (Ann Am Thorac Soc. 2016;13[10]:1752-9).
They added that the wide variation between hospitals in failure rates for noninvasive ventilation were likely attributable to unmeasured hospital factors, speculating that the site of treatment (regular ward vs. ICU); staffing ratios for nurses, respiratory therapists, and physicians; and the intensity of patient monitoring, such as the frequency of blood-gas measurement, may contribute.
“High rates of treatment failure at some hospitals suggest that further work is needed to maximize the real-world effectiveness of noninvasive ventilation, even for an indication [backed by] strong evidence,” the investigators said.
The National Institutes of Health; the National Heart, Lung, and Blood Institute; and Boston University supported the study. The investigators’ financial disclosures are available at www.atsjournals.org.
Eric Gartman, MD, FCCP, comments: It is unclear what conclusions can be drawn from this study given the likely heterogeneity between the included hospitals.

Eric Gartman, MD, FCCP, comments: It is unclear what conclusions can be drawn from this study given the likely heterogeneity between the included hospitals.
Eric Gartman, MD, FCCP, comments: It is unclear what conclusions can be drawn from this study given the likely heterogeneity between the included hospitals.
Hospitals that frequently treat acute COPD exacerbations using noninvasive ventilation – a practice known to reduce mortality, length of stay, and the need for more invasive treatment – did not have better patient outcomes than did hospitals that used noninvasive ventilation less frequently, according to a report published in Annals of the American Thoracic Society.
Acute COPD exacerbations are “one of the few conditions with high-level evidence demonstrating the benefits of noninvasive ventilation in patients with respiratory distress,” and the treatment has been widely adopted for this patient population. However, for noninvasive ventilation to succeed, patients must be carefully selected and closely monitored, and a multidisciplinary team of nurses, respiratory therapists, and physicians must coordinate the treatment, often across multiple hospital settings, said Anuj B. Mehta, MD, of The Pulmonary Center, Boston University, and his associates.
Until now, it was not known whether hospitals with a high volume of noninvasive ventilation develop specialized expertise and thus deliver superior patient outcomes, or whether a high volume results from suboptimal patient selection or otherwise puts a strain on a hospital’s staff and thus produces poor outcomes. To examine this question, Dr. Mehta and his associates analyzed information in a database enrolling adults treated at 252 California hospitals for acute COPD exacerbation. They focused on 37,516 hospitalizations that occurred during a single year.
Overall, 9.3% of these patients received noninvasive ventilation. The median annual case volume of noninvasive ventilation for any indication was 64 per hospital. But rates of noninvasive ventilation varied widely across hospitals, with 40% of facilities significantly deviating from this median rate.
“Contrary to our hypothesis, we did not observe significantly lower COPD mortality” in hospitals with high volumes of noninvasive ventilation. For individual patients, admission to a hospital with a high volume of noninvasive ventilation was associated with significantly higher odds of treatment failure (adjusted OR, 1.95), and such failure was associated with significantly higher odds of death (adjusted OR, 1.81). In addition, at the hospital level, a high volume of noninvasive ventilation was associated with a significantly higher risk of treatment failure, which in turn was associated with higher patient mortality.
“Hospitals with higher total noninvasive ventilation case volume tended to use [it] in patients with more comorbidities and acute organ failures, suggesting potential overuse among patients at higher risk of treatment failure. ... [This] may partially explain why hospitals with high rates of using an evidence-based intervention did not achieve significant mortality benefits,” Dr. Mehta and his associates said (Ann Am Thorac Soc. 2016;13[10]:1752-9).
They added that the wide variation between hospitals in failure rates for noninvasive ventilation were likely attributable to unmeasured hospital factors, speculating that the site of treatment (regular ward vs. ICU); staffing ratios for nurses, respiratory therapists, and physicians; and the intensity of patient monitoring, such as the frequency of blood-gas measurement, may contribute.
“High rates of treatment failure at some hospitals suggest that further work is needed to maximize the real-world effectiveness of noninvasive ventilation, even for an indication [backed by] strong evidence,” the investigators said.
The National Institutes of Health; the National Heart, Lung, and Blood Institute; and Boston University supported the study. The investigators’ financial disclosures are available at www.atsjournals.org.
Hospitals that frequently treat acute COPD exacerbations using noninvasive ventilation – a practice known to reduce mortality, length of stay, and the need for more invasive treatment – did not have better patient outcomes than did hospitals that used noninvasive ventilation less frequently, according to a report published in Annals of the American Thoracic Society.
Acute COPD exacerbations are “one of the few conditions with high-level evidence demonstrating the benefits of noninvasive ventilation in patients with respiratory distress,” and the treatment has been widely adopted for this patient population. However, for noninvasive ventilation to succeed, patients must be carefully selected and closely monitored, and a multidisciplinary team of nurses, respiratory therapists, and physicians must coordinate the treatment, often across multiple hospital settings, said Anuj B. Mehta, MD, of The Pulmonary Center, Boston University, and his associates.
Until now, it was not known whether hospitals with a high volume of noninvasive ventilation develop specialized expertise and thus deliver superior patient outcomes, or whether a high volume results from suboptimal patient selection or otherwise puts a strain on a hospital’s staff and thus produces poor outcomes. To examine this question, Dr. Mehta and his associates analyzed information in a database enrolling adults treated at 252 California hospitals for acute COPD exacerbation. They focused on 37,516 hospitalizations that occurred during a single year.
Overall, 9.3% of these patients received noninvasive ventilation. The median annual case volume of noninvasive ventilation for any indication was 64 per hospital. But rates of noninvasive ventilation varied widely across hospitals, with 40% of facilities significantly deviating from this median rate.
“Contrary to our hypothesis, we did not observe significantly lower COPD mortality” in hospitals with high volumes of noninvasive ventilation. For individual patients, admission to a hospital with a high volume of noninvasive ventilation was associated with significantly higher odds of treatment failure (adjusted OR, 1.95), and such failure was associated with significantly higher odds of death (adjusted OR, 1.81). In addition, at the hospital level, a high volume of noninvasive ventilation was associated with a significantly higher risk of treatment failure, which in turn was associated with higher patient mortality.
“Hospitals with higher total noninvasive ventilation case volume tended to use [it] in patients with more comorbidities and acute organ failures, suggesting potential overuse among patients at higher risk of treatment failure. ... [This] may partially explain why hospitals with high rates of using an evidence-based intervention did not achieve significant mortality benefits,” Dr. Mehta and his associates said (Ann Am Thorac Soc. 2016;13[10]:1752-9).
They added that the wide variation between hospitals in failure rates for noninvasive ventilation were likely attributable to unmeasured hospital factors, speculating that the site of treatment (regular ward vs. ICU); staffing ratios for nurses, respiratory therapists, and physicians; and the intensity of patient monitoring, such as the frequency of blood-gas measurement, may contribute.
“High rates of treatment failure at some hospitals suggest that further work is needed to maximize the real-world effectiveness of noninvasive ventilation, even for an indication [backed by] strong evidence,” the investigators said.
The National Institutes of Health; the National Heart, Lung, and Blood Institute; and Boston University supported the study. The investigators’ financial disclosures are available at www.atsjournals.org.
FROM ANNALS OF THE AMERICAN THORACIC SOCIETY
Key clinical point: Outcomes for patients with acute COPD exacerbations were no better at hospitals where noninvasive ventilation was frequently used.
Major finding: For individual patients, admission to a hospital with a high volume of noninvasive ventilation was associated with significantly higher odds of treatment failure (adjusted OR, 1.95), and such failure was associated with significantly higher odds of death (adjusted OR, 1.81).
Data source: A multicenter observational study involving 37,516 hospitalizations for COPD exacerbation at 252 California medical centers during a 1-year period.
Disclosures: The National Institutes of Health; the National Heart, Lung, and Blood Institute; and Boston University supported the study. The investigators’ financial disclosures are available at www.atsjournals.org.

FDA, EPA clarify which fish pregnant women and young children should eat
The Food and Drug Administration and the Environmental Protection Agency have issued updated guidance about fish consumption for pregnant women and young children, clarifying which types of fish are recommended and what types of fish to avoid.
In guidance issued Jan. 18, the agencies sort 62 types of fish into three categories based on mercury level: best choices, good choices, and fish to avoid. They recommend that women who are pregnant, women who may become pregnant, breastfeeding mothers, and young children eat two to three servings of fish in the “best choices” category per week. Women and young children are advised to eat one serving per week of fish in the “good choices” category, according to the announcement. Fish in the “best choices” category make up nearly 90% of fish eaten in the United States, according to the FDA.
“Fish are an important source of protein and other nutrients for young children and women who are or may become pregnant or are breastfeeding,” Stephen Ostroff, MD, FDA’s deputy commissioner for Foods and Veterinary Medicine, said in a statement. “This advice clearly shows the great diversity of fish in the U.S. market that they can consume safely. This new, clear and concrete advice is an excellent tool for making safe and healthy choices when buying fish.”
The updated advice cautions pregnant women and others to avoid seven types of fish that generally have higher mercury levels. This includes tilefish from the Gulf of Mexico, shark; swordfish; orange roughy, bigeye tuna; marlin, and king mackerel. Meanwhile, recommended choices lower in mercury include such fish as shrimp, pollock, salmon, canned light tuna, tilapia, catfish, and cod.
Consumers are urged to check local advisories for fish caught recreationally and gauge their fish consumption based on any local and state advisories for those waters. If no information on fishing advisories is available, the FDA recommends eating just one fish meal a week from local waters and to avoid other fish that week.
[email protected]
On Twitter @legal_med
The Food and Drug Administration and the Environmental Protection Agency have issued updated guidance about fish consumption for pregnant women and young children, clarifying which types of fish are recommended and what types of fish to avoid.
In guidance issued Jan. 18, the agencies sort 62 types of fish into three categories based on mercury level: best choices, good choices, and fish to avoid. They recommend that women who are pregnant, women who may become pregnant, breastfeeding mothers, and young children eat two to three servings of fish in the “best choices” category per week. Women and young children are advised to eat one serving per week of fish in the “good choices” category, according to the announcement. Fish in the “best choices” category make up nearly 90% of fish eaten in the United States, according to the FDA.
“Fish are an important source of protein and other nutrients for young children and women who are or may become pregnant or are breastfeeding,” Stephen Ostroff, MD, FDA’s deputy commissioner for Foods and Veterinary Medicine, said in a statement. “This advice clearly shows the great diversity of fish in the U.S. market that they can consume safely. This new, clear and concrete advice is an excellent tool for making safe and healthy choices when buying fish.”
The updated advice cautions pregnant women and others to avoid seven types of fish that generally have higher mercury levels. This includes tilefish from the Gulf of Mexico, shark; swordfish; orange roughy, bigeye tuna; marlin, and king mackerel. Meanwhile, recommended choices lower in mercury include such fish as shrimp, pollock, salmon, canned light tuna, tilapia, catfish, and cod.
Consumers are urged to check local advisories for fish caught recreationally and gauge their fish consumption based on any local and state advisories for those waters. If no information on fishing advisories is available, the FDA recommends eating just one fish meal a week from local waters and to avoid other fish that week.
[email protected]
On Twitter @legal_med
The Food and Drug Administration and the Environmental Protection Agency have issued updated guidance about fish consumption for pregnant women and young children, clarifying which types of fish are recommended and what types of fish to avoid.
In guidance issued Jan. 18, the agencies sort 62 types of fish into three categories based on mercury level: best choices, good choices, and fish to avoid. They recommend that women who are pregnant, women who may become pregnant, breastfeeding mothers, and young children eat two to three servings of fish in the “best choices” category per week. Women and young children are advised to eat one serving per week of fish in the “good choices” category, according to the announcement. Fish in the “best choices” category make up nearly 90% of fish eaten in the United States, according to the FDA.
“Fish are an important source of protein and other nutrients for young children and women who are or may become pregnant or are breastfeeding,” Stephen Ostroff, MD, FDA’s deputy commissioner for Foods and Veterinary Medicine, said in a statement. “This advice clearly shows the great diversity of fish in the U.S. market that they can consume safely. This new, clear and concrete advice is an excellent tool for making safe and healthy choices when buying fish.”
The updated advice cautions pregnant women and others to avoid seven types of fish that generally have higher mercury levels. This includes tilefish from the Gulf of Mexico, shark; swordfish; orange roughy, bigeye tuna; marlin, and king mackerel. Meanwhile, recommended choices lower in mercury include such fish as shrimp, pollock, salmon, canned light tuna, tilapia, catfish, and cod.
Consumers are urged to check local advisories for fish caught recreationally and gauge their fish consumption based on any local and state advisories for those waters. If no information on fishing advisories is available, the FDA recommends eating just one fish meal a week from local waters and to avoid other fish that week.
[email protected]
On Twitter @legal_med
Repeal and replace: House bills offer potential road maps
As Republicans dig in to make good on their promise to repeal the Affordable Care Act, hints at what an eventual replacement may look like can be gleaned from legislation previously introduced in the House of Representatives.
One model could be the Empowering Patients First Act (H.R. 2300), sponsored by Rep. Tom Price (R-Ga.), a retired orthopedic surgeon and President-elect Trump’s nominee to head the Health and Human Services department. That legislation offers up a number of the usual GOP proposals related to health care, including refundable tax credits for low-income individuals buying insurance on the individual market; federal grants for states to provide health coverage through a high-risk pool, a reinsurance pool, or other mechanism to help subsidize the purchase of insurance; allowing individuals to purchase insurance through individual member associations; allowing small business owners to purchase insurance for their families and employees across state lines through their trade associations; and allowing insurance companies to sell coverage across state lines.

Another model can be found in House Republican’s health reform plan, called “A Better Way.”
The plan includes a number of provisions similar to those in Dr. Price’s plan, such as expanding consumer-directed health care options, allowing sale of insurance across state lines, expanding opportunities for pooling, and bringing about medical liability reform.
It also increases health insurance portability, helps to preserve the employer-sponsored insurance market, preserves wellness programs, promotes greater use of health savings accounts, and provides for greater opportunities to contribute and use them.
The Better Way plan maintains a few of the popular aspects of the Affordable Care Act, including a ban on coverage denial for preexisting conditions and the ability to keep adult children on parents’ health insurance up to age 26 years, in certain situations.
In addition, the plan would roll back a premium adjustment for older patients. The ACA mandates that premiums for older individuals could be no more than three times that of a younger enrollee. Prior to the ACA, the GOP plan notes that it was generally a limit of five times that of a younger enrollee, and the GOP vision is to bring that limit back.
“The ill-advised three-to-one policy is leading to artificially higher premiums for millions of Americans, especially younger and healthier patients,” according to the GOP plan.
The plan aims to repeal several ACA provisions including the Independent Payment Advisory Board, the Center for Medicare & Medicaid Innovations, and the ban on physician-owned hospitals. It would also extend value-based insurance design to Medicare Advantage, combine Medicare Parts A & B, and reform to uncompensated care.
President-elect Trump also gave hints as to what might be contained in a health reform plan he is formulating. In a Jan. 14 interview with the Washington Post, Mr. Trump said his plan aims to provide “insurance for all” while requiring drug manufacturers to negotiate with Medicare and Medicaid on pricing.
As Republicans dig in to make good on their promise to repeal the Affordable Care Act, hints at what an eventual replacement may look like can be gleaned from legislation previously introduced in the House of Representatives.
One model could be the Empowering Patients First Act (H.R. 2300), sponsored by Rep. Tom Price (R-Ga.), a retired orthopedic surgeon and President-elect Trump’s nominee to head the Health and Human Services department. That legislation offers up a number of the usual GOP proposals related to health care, including refundable tax credits for low-income individuals buying insurance on the individual market; federal grants for states to provide health coverage through a high-risk pool, a reinsurance pool, or other mechanism to help subsidize the purchase of insurance; allowing individuals to purchase insurance through individual member associations; allowing small business owners to purchase insurance for their families and employees across state lines through their trade associations; and allowing insurance companies to sell coverage across state lines.
Another model can be found in House Republican’s health reform plan, called “A Better Way.”
The plan includes a number of provisions similar to those in Dr. Price’s plan, such as expanding consumer-directed health care options, allowing sale of insurance across state lines, expanding opportunities for pooling, and bringing about medical liability reform.
It also increases health insurance portability, helps to preserve the employer-sponsored insurance market, preserves wellness programs, promotes greater use of health savings accounts, and provides for greater opportunities to contribute and use them.
The Better Way plan maintains a few of the popular aspects of the Affordable Care Act, including a ban on coverage denial for preexisting conditions and the ability to keep adult children on parents’ health insurance up to age 26 years, in certain situations.
In addition, the plan would roll back a premium adjustment for older patients. The ACA mandates that premiums for older individuals could be no more than three times that of a younger enrollee. Prior to the ACA, the GOP plan notes that it was generally a limit of five times that of a younger enrollee, and the GOP vision is to bring that limit back.
“The ill-advised three-to-one policy is leading to artificially higher premiums for millions of Americans, especially younger and healthier patients,” according to the GOP plan.
The plan aims to repeal several ACA provisions including the Independent Payment Advisory Board, the Center for Medicare & Medicaid Innovations, and the ban on physician-owned hospitals. It would also extend value-based insurance design to Medicare Advantage, combine Medicare Parts A & B, and reform to uncompensated care.
President-elect Trump also gave hints as to what might be contained in a health reform plan he is formulating. In a Jan. 14 interview with the Washington Post, Mr. Trump said his plan aims to provide “insurance for all” while requiring drug manufacturers to negotiate with Medicare and Medicaid on pricing.
As Republicans dig in to make good on their promise to repeal the Affordable Care Act, hints at what an eventual replacement may look like can be gleaned from legislation previously introduced in the House of Representatives.
One model could be the Empowering Patients First Act (H.R. 2300), sponsored by Rep. Tom Price (R-Ga.), a retired orthopedic surgeon and President-elect Trump’s nominee to head the Health and Human Services department. That legislation offers up a number of the usual GOP proposals related to health care, including refundable tax credits for low-income individuals buying insurance on the individual market; federal grants for states to provide health coverage through a high-risk pool, a reinsurance pool, or other mechanism to help subsidize the purchase of insurance; allowing individuals to purchase insurance through individual member associations; allowing small business owners to purchase insurance for their families and employees across state lines through their trade associations; and allowing insurance companies to sell coverage across state lines.
Another model can be found in House Republican’s health reform plan, called “A Better Way.”
The plan includes a number of provisions similar to those in Dr. Price’s plan, such as expanding consumer-directed health care options, allowing sale of insurance across state lines, expanding opportunities for pooling, and bringing about medical liability reform.
It also increases health insurance portability, helps to preserve the employer-sponsored insurance market, preserves wellness programs, promotes greater use of health savings accounts, and provides for greater opportunities to contribute and use them.
The Better Way plan maintains a few of the popular aspects of the Affordable Care Act, including a ban on coverage denial for preexisting conditions and the ability to keep adult children on parents’ health insurance up to age 26 years, in certain situations.
In addition, the plan would roll back a premium adjustment for older patients. The ACA mandates that premiums for older individuals could be no more than three times that of a younger enrollee. Prior to the ACA, the GOP plan notes that it was generally a limit of five times that of a younger enrollee, and the GOP vision is to bring that limit back.
“The ill-advised three-to-one policy is leading to artificially higher premiums for millions of Americans, especially younger and healthier patients,” according to the GOP plan.
The plan aims to repeal several ACA provisions including the Independent Payment Advisory Board, the Center for Medicare & Medicaid Innovations, and the ban on physician-owned hospitals. It would also extend value-based insurance design to Medicare Advantage, combine Medicare Parts A & B, and reform to uncompensated care.
President-elect Trump also gave hints as to what might be contained in a health reform plan he is formulating. In a Jan. 14 interview with the Washington Post, Mr. Trump said his plan aims to provide “insurance for all” while requiring drug manufacturers to negotiate with Medicare and Medicaid on pricing.
Prolonged work-related stress linked to NHL, other cancers in men

burning building in Quebec
Photo by Sylvain Pedneault
New research suggests that prolonged exposure to work-related stress may increase a man’s risk of several cancers.
The study showed a significant association between work-related stress lasting 15 years or more and non-Hodgkin lymphoma (NHL) as well as lung, colon, rectal, and stomach cancers.
Men who had worked as firefighters, engineers, mechanics, and repair workers were most likely to report work-related stress.
Marie-Élise Parent, PhD, of Institut national de la recherche scientifique (INRS) in Laval, Quebec, Canada, and her colleagues conducted this research and published
the results in Preventive Medicine.
The researchers studied 3103 men with 11 different types of cancer who were diagnosed from 1979 to 1985. The team compared these men to 512 control subjects from the general population.
Both cases and controls were interviewed and asked to describe each job they had during their lifetime, including the occurrence of stress related to a job and the reason for that stress.
The researchers then calculated odds ratios (OR) for the association between perceived workplace stress and its duration, and each cancer site. The analyses were adjusted for lifestyle and occupational factors.
The team found that having at least one stressful job in a lifetime was associated with increased odds of 5 cancers:
- Lung—OR=1.33
- Colon—OR=1.51
- Bladder—OR=1.37
- Rectal—OR=1.52
- Stomach—OR=1.53.
When the researchers looked at the duration of stress, they found no significant association between any of the cancers and work-related stress lasting less than 15 years.
However, there were significant associations for several cancers and work-related stress lasting 15 to 30 years or more than 30 years. These included:
- NHL—15-30 years, OR=1.47; >30 years, OR=1.69 (P=0.02)
- Lung cancer—15-30 years, OR=1.47; >30 years, OR=1.51 (P=0.01)
- Colon cancer—15-30 years, OR=1.32; >30 years, OR=1.64 (P<0.01)
- Rectal cancer—15-30 years, OR=1.84; >30 years, OR=1.48 (P=0.01)
- Stomach cancer—15-30 years, OR=2.15; >30 years, OR=1.48 (P=0.01).
The occupations with the highest prevalence of work-related stress were firefighter (40% of firefighting jobs reported as stressful), industrial and aerospace engineer (31%), and motor vehicle and rail transport mechanic/repair worker (28%).
For the same individual, stress varied depending on the job held.
The study also showed that perceived stress was not limited to high work load and time constraints. Customer service, sales commissions, responsibilities, having an anxious temperament, job insecurity, financial problems, challenging or dangerous work conditions, employee supervision, interpersonal conflict, and a difficult commute were all sources of stress listed by study participants.
The researchers said one of the biggest flaws in previous studies of this kind is that none of them assessed work-related stress over a full working lifetime. The team said this made it impossible to determine how the duration of exposure to work-related stress affects cancer development.
This study, on the other hand, shows the importance of measuring stress at different points in an individual’s working life, the researchers said. They added that their results raise the question of whether chronic psychological stress should be viewed as a public health issue.
However, the team also pointed out that these results are unsubstantiated because they are based on a summary assessment of work-related stress for a given job. There is a need for epidemiological studies based on reliable stress measurements, repeated over time, and that take all sources of stress into account. ![]()
burning building in Quebec
Photo by Sylvain Pedneault
New research suggests that prolonged exposure to work-related stress may increase a man’s risk of several cancers.
The study showed a significant association between work-related stress lasting 15 years or more and non-Hodgkin lymphoma (NHL) as well as lung, colon, rectal, and stomach cancers.
Men who had worked as firefighters, engineers, mechanics, and repair workers were most likely to report work-related stress.
Marie-Élise Parent, PhD, of Institut national de la recherche scientifique (INRS) in Laval, Quebec, Canada, and her colleagues conducted this research and published
the results in Preventive Medicine.
The researchers studied 3103 men with 11 different types of cancer who were diagnosed from 1979 to 1985. The team compared these men to 512 control subjects from the general population.
Both cases and controls were interviewed and asked to describe each job they had during their lifetime, including the occurrence of stress related to a job and the reason for that stress.
The researchers then calculated odds ratios (OR) for the association between perceived workplace stress and its duration, and each cancer site. The analyses were adjusted for lifestyle and occupational factors.
The team found that having at least one stressful job in a lifetime was associated with increased odds of 5 cancers:
- Lung—OR=1.33
- Colon—OR=1.51
- Bladder—OR=1.37
- Rectal—OR=1.52
- Stomach—OR=1.53.
When the researchers looked at the duration of stress, they found no significant association between any of the cancers and work-related stress lasting less than 15 years.
However, there were significant associations for several cancers and work-related stress lasting 15 to 30 years or more than 30 years. These included:
- NHL—15-30 years, OR=1.47; >30 years, OR=1.69 (P=0.02)
- Lung cancer—15-30 years, OR=1.47; >30 years, OR=1.51 (P=0.01)
- Colon cancer—15-30 years, OR=1.32; >30 years, OR=1.64 (P<0.01)
- Rectal cancer—15-30 years, OR=1.84; >30 years, OR=1.48 (P=0.01)
- Stomach cancer—15-30 years, OR=2.15; >30 years, OR=1.48 (P=0.01).
The occupations with the highest prevalence of work-related stress were firefighter (40% of firefighting jobs reported as stressful), industrial and aerospace engineer (31%), and motor vehicle and rail transport mechanic/repair worker (28%).
For the same individual, stress varied depending on the job held.
The study also showed that perceived stress was not limited to high work load and time constraints. Customer service, sales commissions, responsibilities, having an anxious temperament, job insecurity, financial problems, challenging or dangerous work conditions, employee supervision, interpersonal conflict, and a difficult commute were all sources of stress listed by study participants.
The researchers said one of the biggest flaws in previous studies of this kind is that none of them assessed work-related stress over a full working lifetime. The team said this made it impossible to determine how the duration of exposure to work-related stress affects cancer development.
This study, on the other hand, shows the importance of measuring stress at different points in an individual’s working life, the researchers said. They added that their results raise the question of whether chronic psychological stress should be viewed as a public health issue.
However, the team also pointed out that these results are unsubstantiated because they are based on a summary assessment of work-related stress for a given job. There is a need for epidemiological studies based on reliable stress measurements, repeated over time, and that take all sources of stress into account. ![]()
burning building in Quebec
Photo by Sylvain Pedneault
New research suggests that prolonged exposure to work-related stress may increase a man’s risk of several cancers.
The study showed a significant association between work-related stress lasting 15 years or more and non-Hodgkin lymphoma (NHL) as well as lung, colon, rectal, and stomach cancers.
Men who had worked as firefighters, engineers, mechanics, and repair workers were most likely to report work-related stress.
Marie-Élise Parent, PhD, of Institut national de la recherche scientifique (INRS) in Laval, Quebec, Canada, and her colleagues conducted this research and published
the results in Preventive Medicine.
The researchers studied 3103 men with 11 different types of cancer who were diagnosed from 1979 to 1985. The team compared these men to 512 control subjects from the general population.
Both cases and controls were interviewed and asked to describe each job they had during their lifetime, including the occurrence of stress related to a job and the reason for that stress.
The researchers then calculated odds ratios (OR) for the association between perceived workplace stress and its duration, and each cancer site. The analyses were adjusted for lifestyle and occupational factors.
The team found that having at least one stressful job in a lifetime was associated with increased odds of 5 cancers:
- Lung—OR=1.33
- Colon—OR=1.51
- Bladder—OR=1.37
- Rectal—OR=1.52
- Stomach—OR=1.53.
When the researchers looked at the duration of stress, they found no significant association between any of the cancers and work-related stress lasting less than 15 years.
However, there were significant associations for several cancers and work-related stress lasting 15 to 30 years or more than 30 years. These included:
- NHL—15-30 years, OR=1.47; >30 years, OR=1.69 (P=0.02)
- Lung cancer—15-30 years, OR=1.47; >30 years, OR=1.51 (P=0.01)
- Colon cancer—15-30 years, OR=1.32; >30 years, OR=1.64 (P<0.01)
- Rectal cancer—15-30 years, OR=1.84; >30 years, OR=1.48 (P=0.01)
- Stomach cancer—15-30 years, OR=2.15; >30 years, OR=1.48 (P=0.01).
The occupations with the highest prevalence of work-related stress were firefighter (40% of firefighting jobs reported as stressful), industrial and aerospace engineer (31%), and motor vehicle and rail transport mechanic/repair worker (28%).
For the same individual, stress varied depending on the job held.
The study also showed that perceived stress was not limited to high work load and time constraints. Customer service, sales commissions, responsibilities, having an anxious temperament, job insecurity, financial problems, challenging or dangerous work conditions, employee supervision, interpersonal conflict, and a difficult commute were all sources of stress listed by study participants.
The researchers said one of the biggest flaws in previous studies of this kind is that none of them assessed work-related stress over a full working lifetime. The team said this made it impossible to determine how the duration of exposure to work-related stress affects cancer development.
This study, on the other hand, shows the importance of measuring stress at different points in an individual’s working life, the researchers said. They added that their results raise the question of whether chronic psychological stress should be viewed as a public health issue.
However, the team also pointed out that these results are unsubstantiated because they are based on a summary assessment of work-related stress for a given job. There is a need for epidemiological studies based on reliable stress measurements, repeated over time, and that take all sources of stress into account. ![]()
Pharma is gaming the system for orphan drugs, investigation suggests

Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs. ![]()
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs. ![]()
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs. ![]()
FDA releases draft guidances on biosimilars, medical communications

Photo by Bill Branson
The US Food and Drug Administration (FDA) has released 3 new draft guidance documents for industry.
One document provides an overview of scientific considerations when attempting to demonstrate that a biosimilar product is interchangeable with a reference product.
The other 2 guidance documents are intended to clarify the FDA’s position regarding communications about medical products.
The first draft guidance, “Considerations in Demonstrating Interchangeability With a Reference Product,” is intended to assist applicants in demonstrating that a proposed therapeutic protein product (eg, monoclonal antibodies) is interchangeable with a reference product under section 351(k) of the Public Health Service Act.
An interchangeable biological product is biosimilar to the reference product and can be expected to produce the same clinical result as the reference product in any given patient.
For a biological product that is administered more than once, the risk in terms of safety or diminished efficacy of alternating or switching between the biological product and the reference product must not be greater than the risk of using the reference product without such alternating/switching.
The approval pathway for biosimilar and interchangeable products was established by the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the Affordable Care Act in March 2010.
The FDA’s draft guidance on biosimilars contains information on:
- Factors impacting the type and amount of data and information needed to support a demonstration of interchangeability
- The data and information needed to support a demonstration of interchangeability
- Considerations for the design and analysis of a switching study or studies to support a demonstration of interchangeability
- Recommendations regarding the use of US-licensed reference products in a switching study or studies
- Considerations for developing presentations (eg, container closure systems) for proposed interchangeable products.
In addition to soliciting comments on this draft guidance, the FDA is inviting comments on questions posed in the notice of availability about interchangeability in general, the regulation of interchangeable products over their life cycle, and questions about considerations regarding post-approval manufacturing changes.
The FDA also released a Center for Drug Evaluation and Research “From Our Perspective,” by Leah Christl, describing aspects of this draft guidance.
For more information on how to submit comments on the draft guidance and questions posed in the notice of availability, see the Federal Register notice.
Medical communications
The FDA also released 2 draft guidance documents that, the agency says, will each help provide clarity for medical product companies, as well as other interested parties, on the FDA’s current thinking and recommendations for a few different types of communications about medical products.
The first draft guidance, “Drug and Device Manufacturer Communications with Payors, Formulary Committees, and Similar Entities,” explains the FDA’s current thinking and recommendations on firms’ communication of healthcare economic information about approved drugs under section 502(a) of the Federal Food, Drug, and Cosmetic Act, which was recently amended by the 21st Century Cures Act.
The guidance also answers common questions and provides the FDA’s recommendations regarding firms’ communications to payors about investigational drugs and devices that are not yet approved or cleared for any use.
The second draft guidance, “Medical Product Communications That Are Consistent With the FDA-Required Labeling,” explains the FDA’s current thinking about firms’ medical product communications that include data and information that are not contained in their products’ FDA-required labeling, but that concern the approved or cleared uses of their products.
The FDA has opened a public comment period for each draft guidance.
The agency is also asking for stakeholder input on another, distinct topic—communications about unapproved uses of approved or cleared medical products.
The FDA held a Part 15 hearing in November 2016 to hear from a broad range of stakeholders regarding this topic. The agency has now reopened the comment period for the docket opened in connection with that public hearing for an additional 90 days (until April 10, 2017) to allow interested parties an opportunity to review the 2 draft guidances before submitting comments to any of the relevant dockets.
The FDA also added a document to the docket for the public hearing titled, “Memorandum: Public Health Interests and First Amendment Considerations Related to Manufacturer Communications Regarding Unapproved Uses of Approved or Cleared Medical Products.”
This document provides additional background on the issues the FDA is considering as part of its review of the agency’s rules and policies relating to firm communications regarding unapproved uses of approved or cleared medical products, including a discussion of First Amendment considerations.
The FDA is requesting input on the memorandum as it relates to the questions set forth in the initial notice of public hearing. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has released 3 new draft guidance documents for industry.
One document provides an overview of scientific considerations when attempting to demonstrate that a biosimilar product is interchangeable with a reference product.
The other 2 guidance documents are intended to clarify the FDA’s position regarding communications about medical products.
The first draft guidance, “Considerations in Demonstrating Interchangeability With a Reference Product,” is intended to assist applicants in demonstrating that a proposed therapeutic protein product (eg, monoclonal antibodies) is interchangeable with a reference product under section 351(k) of the Public Health Service Act.
An interchangeable biological product is biosimilar to the reference product and can be expected to produce the same clinical result as the reference product in any given patient.
For a biological product that is administered more than once, the risk in terms of safety or diminished efficacy of alternating or switching between the biological product and the reference product must not be greater than the risk of using the reference product without such alternating/switching.
The approval pathway for biosimilar and interchangeable products was established by the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the Affordable Care Act in March 2010.
The FDA’s draft guidance on biosimilars contains information on:
- Factors impacting the type and amount of data and information needed to support a demonstration of interchangeability
- The data and information needed to support a demonstration of interchangeability
- Considerations for the design and analysis of a switching study or studies to support a demonstration of interchangeability
- Recommendations regarding the use of US-licensed reference products in a switching study or studies
- Considerations for developing presentations (eg, container closure systems) for proposed interchangeable products.
In addition to soliciting comments on this draft guidance, the FDA is inviting comments on questions posed in the notice of availability about interchangeability in general, the regulation of interchangeable products over their life cycle, and questions about considerations regarding post-approval manufacturing changes.
The FDA also released a Center for Drug Evaluation and Research “From Our Perspective,” by Leah Christl, describing aspects of this draft guidance.
For more information on how to submit comments on the draft guidance and questions posed in the notice of availability, see the Federal Register notice.
Medical communications
The FDA also released 2 draft guidance documents that, the agency says, will each help provide clarity for medical product companies, as well as other interested parties, on the FDA’s current thinking and recommendations for a few different types of communications about medical products.
The first draft guidance, “Drug and Device Manufacturer Communications with Payors, Formulary Committees, and Similar Entities,” explains the FDA’s current thinking and recommendations on firms’ communication of healthcare economic information about approved drugs under section 502(a) of the Federal Food, Drug, and Cosmetic Act, which was recently amended by the 21st Century Cures Act.
The guidance also answers common questions and provides the FDA’s recommendations regarding firms’ communications to payors about investigational drugs and devices that are not yet approved or cleared for any use.
The second draft guidance, “Medical Product Communications That Are Consistent With the FDA-Required Labeling,” explains the FDA’s current thinking about firms’ medical product communications that include data and information that are not contained in their products’ FDA-required labeling, but that concern the approved or cleared uses of their products.
The FDA has opened a public comment period for each draft guidance.
The agency is also asking for stakeholder input on another, distinct topic—communications about unapproved uses of approved or cleared medical products.
The FDA held a Part 15 hearing in November 2016 to hear from a broad range of stakeholders regarding this topic. The agency has now reopened the comment period for the docket opened in connection with that public hearing for an additional 90 days (until April 10, 2017) to allow interested parties an opportunity to review the 2 draft guidances before submitting comments to any of the relevant dockets.
The FDA also added a document to the docket for the public hearing titled, “Memorandum: Public Health Interests and First Amendment Considerations Related to Manufacturer Communications Regarding Unapproved Uses of Approved or Cleared Medical Products.”
This document provides additional background on the issues the FDA is considering as part of its review of the agency’s rules and policies relating to firm communications regarding unapproved uses of approved or cleared medical products, including a discussion of First Amendment considerations.
The FDA is requesting input on the memorandum as it relates to the questions set forth in the initial notice of public hearing. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has released 3 new draft guidance documents for industry.
One document provides an overview of scientific considerations when attempting to demonstrate that a biosimilar product is interchangeable with a reference product.
The other 2 guidance documents are intended to clarify the FDA’s position regarding communications about medical products.
The first draft guidance, “Considerations in Demonstrating Interchangeability With a Reference Product,” is intended to assist applicants in demonstrating that a proposed therapeutic protein product (eg, monoclonal antibodies) is interchangeable with a reference product under section 351(k) of the Public Health Service Act.
An interchangeable biological product is biosimilar to the reference product and can be expected to produce the same clinical result as the reference product in any given patient.
For a biological product that is administered more than once, the risk in terms of safety or diminished efficacy of alternating or switching between the biological product and the reference product must not be greater than the risk of using the reference product without such alternating/switching.
The approval pathway for biosimilar and interchangeable products was established by the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the Affordable Care Act in March 2010.
The FDA’s draft guidance on biosimilars contains information on:
- Factors impacting the type and amount of data and information needed to support a demonstration of interchangeability
- The data and information needed to support a demonstration of interchangeability
- Considerations for the design and analysis of a switching study or studies to support a demonstration of interchangeability
- Recommendations regarding the use of US-licensed reference products in a switching study or studies
- Considerations for developing presentations (eg, container closure systems) for proposed interchangeable products.
In addition to soliciting comments on this draft guidance, the FDA is inviting comments on questions posed in the notice of availability about interchangeability in general, the regulation of interchangeable products over their life cycle, and questions about considerations regarding post-approval manufacturing changes.
The FDA also released a Center for Drug Evaluation and Research “From Our Perspective,” by Leah Christl, describing aspects of this draft guidance.
For more information on how to submit comments on the draft guidance and questions posed in the notice of availability, see the Federal Register notice.
Medical communications
The FDA also released 2 draft guidance documents that, the agency says, will each help provide clarity for medical product companies, as well as other interested parties, on the FDA’s current thinking and recommendations for a few different types of communications about medical products.
The first draft guidance, “Drug and Device Manufacturer Communications with Payors, Formulary Committees, and Similar Entities,” explains the FDA’s current thinking and recommendations on firms’ communication of healthcare economic information about approved drugs under section 502(a) of the Federal Food, Drug, and Cosmetic Act, which was recently amended by the 21st Century Cures Act.
The guidance also answers common questions and provides the FDA’s recommendations regarding firms’ communications to payors about investigational drugs and devices that are not yet approved or cleared for any use.
The second draft guidance, “Medical Product Communications That Are Consistent With the FDA-Required Labeling,” explains the FDA’s current thinking about firms’ medical product communications that include data and information that are not contained in their products’ FDA-required labeling, but that concern the approved or cleared uses of their products.
The FDA has opened a public comment period for each draft guidance.
The agency is also asking for stakeholder input on another, distinct topic—communications about unapproved uses of approved or cleared medical products.
The FDA held a Part 15 hearing in November 2016 to hear from a broad range of stakeholders regarding this topic. The agency has now reopened the comment period for the docket opened in connection with that public hearing for an additional 90 days (until April 10, 2017) to allow interested parties an opportunity to review the 2 draft guidances before submitting comments to any of the relevant dockets.
The FDA also added a document to the docket for the public hearing titled, “Memorandum: Public Health Interests and First Amendment Considerations Related to Manufacturer Communications Regarding Unapproved Uses of Approved or Cleared Medical Products.”
This document provides additional background on the issues the FDA is considering as part of its review of the agency’s rules and policies relating to firm communications regarding unapproved uses of approved or cleared medical products, including a discussion of First Amendment considerations.
The FDA is requesting input on the memorandum as it relates to the questions set forth in the initial notice of public hearing. ![]()
Enzyme derived from yeast kills ALL cells

as it buds before dividing
Image by Carolyn Larabell
L-asparaginase derived from baker’s yeast has shown early promise for treating acute lymphoblastic leukemia (ALL), according to researchers.
The team isolated L-asparaginase from Saccharomyces cerevisiae and found the enzyme could kill ALL cells in vitro, while largely sparing healthy control cells.
Gisele Monteiro de Souza, PhD, of the University of São Paulo in São Paulo, Brazil, and her colleagues detailed these findings in Scientific Reports.
“In this study, we characterized the enzyme L-asparaginase from S cerevisiae,” Dr Souza said. “The results show this protein can efficiently annihilate leukemia cells with low cytotoxicity to healthy cells.”
She and her colleagues conducted this study in search of alternatives to L-asparaginase extracted from bacteria (Escherichia coli and Erwinia chrysanthemi).
“Our goal in this project wasn’t to produce the enzyme, but rather to find a new source of the biodrug in microorganisms for use in patients who develop resistance to the bacterial enzyme,” said study author Marcos Antonio de Oliveira, of São Paulo State University in São Vicente, Brazil.
To this end, the researchers isolated fungi from several different Brazilian environments as well as marine and land environments in Antarctica. According to Oliveira, these organisms often secrete asparaginase into the extracellular medium in response to a shortage of nitrogen.
“This lowers the cost of purifying the molecule for drug production, an important factor from an industrial standpoint,” he said.
The group also used bioinformatics tools to mine information on the genomes of several microorganisms from international databases.
In this way, they identified a gene responsible for producing an enzyme that closely resembles the enzymes found in E coli and E chrysanthemi, but with a number of advantages, in the genome of S cerevisiae.
The gene of interest from L-asparaginase was cloned, and the researchers used genetic engineering to make E coli express large amounts of the enzyme originally found in yeast.
“We were able to obtain the recombinant protein,” said study author Iris Munhoz Costa, of the University of São Paulo.
“We then performed studies to characterize its secondary structure and identify important regions called catalytic sites. Finally, we evaluated its efficacy in vitro.”
The enzyme was tested in 3 different cell lines: ALL cells incapable of producing asparagine at normal levels (MOLT4), another ALL cell line capable of producing asparagine at normal levels (REH), and non-malignant control cells (HUVECs).
These 3 cell lines were subdivided into 2 groups. One was treated with L-asparaginase derived from E coli enzyme, and the other was treated with L-asparaginase from yeast.
“The bacterial enzyme killed about 90% of the MOLT4 human leukemia cells and displayed low toxicity to the healthy HUVEC cells, killing only 10%,” Dr Souza said.
“The yeast enzyme killed between 70% and 80% of the MOLT4 cells and displayed less than 10% toxicity for HUVEC cells. Neither was significantly effective against REH cells.”
In her view, the results are encouraging, in contrast with those of studies performed with the same enzyme in the 1970s. At that time, the tests involved a version of the protein extracted directly from yeast and containing many impurities.
The group’s next step is to perform new in vitro trials with different cell types to evaluate the immune response and toxicity. If the results are positive, the first tests in animals may be next.
The researchers are also studying possible modifications that could be made to the molecule’s structure to increase antitumor activity and extend the enzyme’s half-life. ![]()
as it buds before dividing
Image by Carolyn Larabell
L-asparaginase derived from baker’s yeast has shown early promise for treating acute lymphoblastic leukemia (ALL), according to researchers.
The team isolated L-asparaginase from Saccharomyces cerevisiae and found the enzyme could kill ALL cells in vitro, while largely sparing healthy control cells.
Gisele Monteiro de Souza, PhD, of the University of São Paulo in São Paulo, Brazil, and her colleagues detailed these findings in Scientific Reports.
“In this study, we characterized the enzyme L-asparaginase from S cerevisiae,” Dr Souza said. “The results show this protein can efficiently annihilate leukemia cells with low cytotoxicity to healthy cells.”
She and her colleagues conducted this study in search of alternatives to L-asparaginase extracted from bacteria (Escherichia coli and Erwinia chrysanthemi).
“Our goal in this project wasn’t to produce the enzyme, but rather to find a new source of the biodrug in microorganisms for use in patients who develop resistance to the bacterial enzyme,” said study author Marcos Antonio de Oliveira, of São Paulo State University in São Vicente, Brazil.
To this end, the researchers isolated fungi from several different Brazilian environments as well as marine and land environments in Antarctica. According to Oliveira, these organisms often secrete asparaginase into the extracellular medium in response to a shortage of nitrogen.
“This lowers the cost of purifying the molecule for drug production, an important factor from an industrial standpoint,” he said.
The group also used bioinformatics tools to mine information on the genomes of several microorganisms from international databases.
In this way, they identified a gene responsible for producing an enzyme that closely resembles the enzymes found in E coli and E chrysanthemi, but with a number of advantages, in the genome of S cerevisiae.
The gene of interest from L-asparaginase was cloned, and the researchers used genetic engineering to make E coli express large amounts of the enzyme originally found in yeast.
“We were able to obtain the recombinant protein,” said study author Iris Munhoz Costa, of the University of São Paulo.
“We then performed studies to characterize its secondary structure and identify important regions called catalytic sites. Finally, we evaluated its efficacy in vitro.”
The enzyme was tested in 3 different cell lines: ALL cells incapable of producing asparagine at normal levels (MOLT4), another ALL cell line capable of producing asparagine at normal levels (REH), and non-malignant control cells (HUVECs).
These 3 cell lines were subdivided into 2 groups. One was treated with L-asparaginase derived from E coli enzyme, and the other was treated with L-asparaginase from yeast.
“The bacterial enzyme killed about 90% of the MOLT4 human leukemia cells and displayed low toxicity to the healthy HUVEC cells, killing only 10%,” Dr Souza said.
“The yeast enzyme killed between 70% and 80% of the MOLT4 cells and displayed less than 10% toxicity for HUVEC cells. Neither was significantly effective against REH cells.”
In her view, the results are encouraging, in contrast with those of studies performed with the same enzyme in the 1970s. At that time, the tests involved a version of the protein extracted directly from yeast and containing many impurities.
The group’s next step is to perform new in vitro trials with different cell types to evaluate the immune response and toxicity. If the results are positive, the first tests in animals may be next.
The researchers are also studying possible modifications that could be made to the molecule’s structure to increase antitumor activity and extend the enzyme’s half-life. ![]()
as it buds before dividing
Image by Carolyn Larabell
L-asparaginase derived from baker’s yeast has shown early promise for treating acute lymphoblastic leukemia (ALL), according to researchers.
The team isolated L-asparaginase from Saccharomyces cerevisiae and found the enzyme could kill ALL cells in vitro, while largely sparing healthy control cells.
Gisele Monteiro de Souza, PhD, of the University of São Paulo in São Paulo, Brazil, and her colleagues detailed these findings in Scientific Reports.
“In this study, we characterized the enzyme L-asparaginase from S cerevisiae,” Dr Souza said. “The results show this protein can efficiently annihilate leukemia cells with low cytotoxicity to healthy cells.”
She and her colleagues conducted this study in search of alternatives to L-asparaginase extracted from bacteria (Escherichia coli and Erwinia chrysanthemi).
“Our goal in this project wasn’t to produce the enzyme, but rather to find a new source of the biodrug in microorganisms for use in patients who develop resistance to the bacterial enzyme,” said study author Marcos Antonio de Oliveira, of São Paulo State University in São Vicente, Brazil.
To this end, the researchers isolated fungi from several different Brazilian environments as well as marine and land environments in Antarctica. According to Oliveira, these organisms often secrete asparaginase into the extracellular medium in response to a shortage of nitrogen.
“This lowers the cost of purifying the molecule for drug production, an important factor from an industrial standpoint,” he said.
The group also used bioinformatics tools to mine information on the genomes of several microorganisms from international databases.
In this way, they identified a gene responsible for producing an enzyme that closely resembles the enzymes found in E coli and E chrysanthemi, but with a number of advantages, in the genome of S cerevisiae.
The gene of interest from L-asparaginase was cloned, and the researchers used genetic engineering to make E coli express large amounts of the enzyme originally found in yeast.
“We were able to obtain the recombinant protein,” said study author Iris Munhoz Costa, of the University of São Paulo.
“We then performed studies to characterize its secondary structure and identify important regions called catalytic sites. Finally, we evaluated its efficacy in vitro.”
The enzyme was tested in 3 different cell lines: ALL cells incapable of producing asparagine at normal levels (MOLT4), another ALL cell line capable of producing asparagine at normal levels (REH), and non-malignant control cells (HUVECs).
These 3 cell lines were subdivided into 2 groups. One was treated with L-asparaginase derived from E coli enzyme, and the other was treated with L-asparaginase from yeast.
“The bacterial enzyme killed about 90% of the MOLT4 human leukemia cells and displayed low toxicity to the healthy HUVEC cells, killing only 10%,” Dr Souza said.
“The yeast enzyme killed between 70% and 80% of the MOLT4 cells and displayed less than 10% toxicity for HUVEC cells. Neither was significantly effective against REH cells.”
In her view, the results are encouraging, in contrast with those of studies performed with the same enzyme in the 1970s. At that time, the tests involved a version of the protein extracted directly from yeast and containing many impurities.
The group’s next step is to perform new in vitro trials with different cell types to evaluate the immune response and toxicity. If the results are positive, the first tests in animals may be next.
The researchers are also studying possible modifications that could be made to the molecule’s structure to increase antitumor activity and extend the enzyme’s half-life. ![]()
A New Narrative for Diabetes Management
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
![]()
Clinical Professor and Academic Coordinator, Pace University, College of Health Professions, Department of Physician Assistant Studies, New York, NY
![]()
Clinical Professor and Academic Coordinator, Pace University, College of Health Professions, Department of Physician Assistant Studies, New York, NY
![]()
Clinical Professor and Academic Coordinator, Pace University, College of Health Professions, Department of Physician Assistant Studies, New York, NY
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Crusty rash from head to toe
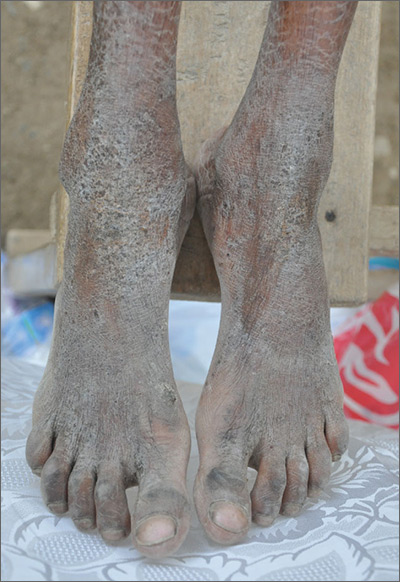
The FP diagnosed crusted scabies based on the patient’s clinical appearance and the distribution of crusting. He happened to have a dermatoscope and was able to see scabies mites moving and creating burrows under the skin. The mites generally appeared as dark triangles with light oval bodies. Crusted scabies is also known as "Norwegian scabies," but the preferred term is crusted scabies. Crusted scabies is more common in individuals who are immunosuppressed, chronically ill (for any reason), or who live in nursing homes.
The child's HIV test was negative and the FP never found out the cause of the cachexia. The diagnosis of crusted scabies was important for everyone involved in the transportation and care of this child. Once the diagnosis was known, everyone was careful to use gloves when caring for her so as to avoid acquiring scabies. The mother was almost certainly infested, along with other family members who’d been in direct contact with the child.
The recommended treatment for crusted scabies is oral ivermectin at a dose of 0.2 mg/kg (maximum dose, 12 mg) once for each infested person to be repeated in 10 days. Fortunately, the global health team had oral ivermectin to give to the child and the family. The FP also told the family to wash all of their clothes and bedclothes.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Chanoine P, Smith M. Scabies. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:575-580.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP diagnosed crusted scabies based on the patient’s clinical appearance and the distribution of crusting. He happened to have a dermatoscope and was able to see scabies mites moving and creating burrows under the skin. The mites generally appeared as dark triangles with light oval bodies. Crusted scabies is also known as "Norwegian scabies," but the preferred term is crusted scabies. Crusted scabies is more common in individuals who are immunosuppressed, chronically ill (for any reason), or who live in nursing homes.
The child's HIV test was negative and the FP never found out the cause of the cachexia. The diagnosis of crusted scabies was important for everyone involved in the transportation and care of this child. Once the diagnosis was known, everyone was careful to use gloves when caring for her so as to avoid acquiring scabies. The mother was almost certainly infested, along with other family members who’d been in direct contact with the child.
The recommended treatment for crusted scabies is oral ivermectin at a dose of 0.2 mg/kg (maximum dose, 12 mg) once for each infested person to be repeated in 10 days. Fortunately, the global health team had oral ivermectin to give to the child and the family. The FP also told the family to wash all of their clothes and bedclothes.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Chanoine P, Smith M. Scabies. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:575-580.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP diagnosed crusted scabies based on the patient’s clinical appearance and the distribution of crusting. He happened to have a dermatoscope and was able to see scabies mites moving and creating burrows under the skin. The mites generally appeared as dark triangles with light oval bodies. Crusted scabies is also known as "Norwegian scabies," but the preferred term is crusted scabies. Crusted scabies is more common in individuals who are immunosuppressed, chronically ill (for any reason), or who live in nursing homes.
The child's HIV test was negative and the FP never found out the cause of the cachexia. The diagnosis of crusted scabies was important for everyone involved in the transportation and care of this child. Once the diagnosis was known, everyone was careful to use gloves when caring for her so as to avoid acquiring scabies. The mother was almost certainly infested, along with other family members who’d been in direct contact with the child.
The recommended treatment for crusted scabies is oral ivermectin at a dose of 0.2 mg/kg (maximum dose, 12 mg) once for each infested person to be repeated in 10 days. Fortunately, the global health team had oral ivermectin to give to the child and the family. The FP also told the family to wash all of their clothes and bedclothes.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Chanoine P, Smith M. Scabies. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:575-580.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
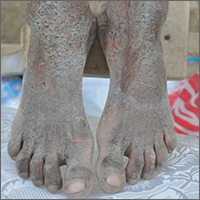
My declining enthusiasm for inpatient work
I’m getting old, and as I age, my desire to do inpatient work seems to diminish each year.
When I started out 20 years ago, I thrived on it. There was excitement in an urgent ED call: the chance to go in and give tissue plasminogen activator, do an emergent lumbar puncture, or break status epilepticus.
Back when I was fresh out of training, I hustled. I walked through the ED to make sure they knew I was around. I hung out in the doctors’ lounge, shaking hands and introducing myself. I was trying to build my practice and get my name out. Other neurologists, closer to retirement than I, were more than happy to let me move in and take the hospital’s late-night and weekend calls.

With time, hospital medicine becomes more of a young person’s game. As I move from being a newly minted attending to an old fogy, I’m happy to leave the work to the next generation.
My hospital work is now down to one-in-3 weekends and no weekdays. I’m not quite ready to give it up entirely and don’t want to turn my back on my call partners of 15 years. I still enjoy the challenge of the cases, the joking around with the nurses, and the family meetings to bring comfort and explain things as best I can.
But I can live without the late night runs, the driving back and forth, and the unpredictable hours. These days, my time at home is more valuable than it was when I started, and I envy those who keep banker’s hours.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I’m getting old, and as I age, my desire to do inpatient work seems to diminish each year.
When I started out 20 years ago, I thrived on it. There was excitement in an urgent ED call: the chance to go in and give tissue plasminogen activator, do an emergent lumbar puncture, or break status epilepticus.
Back when I was fresh out of training, I hustled. I walked through the ED to make sure they knew I was around. I hung out in the doctors’ lounge, shaking hands and introducing myself. I was trying to build my practice and get my name out. Other neurologists, closer to retirement than I, were more than happy to let me move in and take the hospital’s late-night and weekend calls.
With time, hospital medicine becomes more of a young person’s game. As I move from being a newly minted attending to an old fogy, I’m happy to leave the work to the next generation.
My hospital work is now down to one-in-3 weekends and no weekdays. I’m not quite ready to give it up entirely and don’t want to turn my back on my call partners of 15 years. I still enjoy the challenge of the cases, the joking around with the nurses, and the family meetings to bring comfort and explain things as best I can.
But I can live without the late night runs, the driving back and forth, and the unpredictable hours. These days, my time at home is more valuable than it was when I started, and I envy those who keep banker’s hours.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I’m getting old, and as I age, my desire to do inpatient work seems to diminish each year.
When I started out 20 years ago, I thrived on it. There was excitement in an urgent ED call: the chance to go in and give tissue plasminogen activator, do an emergent lumbar puncture, or break status epilepticus.
Back when I was fresh out of training, I hustled. I walked through the ED to make sure they knew I was around. I hung out in the doctors’ lounge, shaking hands and introducing myself. I was trying to build my practice and get my name out. Other neurologists, closer to retirement than I, were more than happy to let me move in and take the hospital’s late-night and weekend calls.
With time, hospital medicine becomes more of a young person’s game. As I move from being a newly minted attending to an old fogy, I’m happy to leave the work to the next generation.
My hospital work is now down to one-in-3 weekends and no weekdays. I’m not quite ready to give it up entirely and don’t want to turn my back on my call partners of 15 years. I still enjoy the challenge of the cases, the joking around with the nurses, and the family meetings to bring comfort and explain things as best I can.
But I can live without the late night runs, the driving back and forth, and the unpredictable hours. These days, my time at home is more valuable than it was when I started, and I envy those who keep banker’s hours.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.