User login
Blood Aspergillus RNA a promising biomarker for invasive aspergillosis
Elevated Aspergillus RNA blood levels after 4-6 weeks of antifungal treatment predict poor response at week 12 in patients with proven or probable invasive aspergillosis, according to results of a small observational study of 41 evaluable patients.
The investigators are working to address the need for reliable biomarkers of early invasive aspergillosis (IA) treatment response. Standard clinical and radiological criteria are somewhat subjective, and serial biopsies and bronchoalveolar lavage are often impractical, reported Yanan Zhao, PhD, of the New Jersey Medical School–Rutgers Biomedical and Health Sciences, Newark, and her associates.
Study participants’ blood was checked for serum galactomannan (GM), 1, 3-beta-D-glucan (BG), and Aspergillus RNA within 24 hours of starting antifungal therapy, then twice per week during the first 2 weeks, then once during weeks 4, 6, and 12, the investigators reported (Med Mycol. 2016 Jun 22. pii: myw043).
Ribosomal Aspergillus RNA – like GM and BG, a marker of fungal load – was measured by nucleic acid sequence-based amplification (NASBA), a robust isothermal amplification technique more sensitive than polymerase chain reaction due largely “to increased starting target numbers (RNA versus DNA) and more robust amplification.” Although NASBA has been used before to diagnose IA, using it to monitor treatment “is still in its infancy,” the authors noted.
Eleven of 14 patients who did not respond to treatment at 12 weeks (79%) had Aspergillus RNA in their blood after 4 weeks of treatment, and 12 (86%) were positive at 6 weeks.
Among patients who did respond at 12 weeks, 11 of 27 (41%) had RNA in their blood at 4 weeks, and 14 (52%) at 6 weeks. The findings were statistically significant.
There was no correlation between Aspergillus RNA and serum GM levels in terms of outcomes, but the kinetics of circulating Aspergillus RNA correlated with BG in some patients, with an excellent match in three.
Serum GM responds fairly soon if treatment is working. Aspergillus RNA, however, responds more slowly, like BG. “This may explain ... the correlation between Aspergillus RNA and BG ... Therefore, the combination of Aspergillus RNA and BG might be useful to assess therapeutic response, particularly in GM negative cases,” the investigators said.
This work was funded by Merck. Four investigators are current or former employees.
Elevated Aspergillus RNA blood levels after 4-6 weeks of antifungal treatment predict poor response at week 12 in patients with proven or probable invasive aspergillosis, according to results of a small observational study of 41 evaluable patients.
The investigators are working to address the need for reliable biomarkers of early invasive aspergillosis (IA) treatment response. Standard clinical and radiological criteria are somewhat subjective, and serial biopsies and bronchoalveolar lavage are often impractical, reported Yanan Zhao, PhD, of the New Jersey Medical School–Rutgers Biomedical and Health Sciences, Newark, and her associates.
Study participants’ blood was checked for serum galactomannan (GM), 1, 3-beta-D-glucan (BG), and Aspergillus RNA within 24 hours of starting antifungal therapy, then twice per week during the first 2 weeks, then once during weeks 4, 6, and 12, the investigators reported (Med Mycol. 2016 Jun 22. pii: myw043).
Ribosomal Aspergillus RNA – like GM and BG, a marker of fungal load – was measured by nucleic acid sequence-based amplification (NASBA), a robust isothermal amplification technique more sensitive than polymerase chain reaction due largely “to increased starting target numbers (RNA versus DNA) and more robust amplification.” Although NASBA has been used before to diagnose IA, using it to monitor treatment “is still in its infancy,” the authors noted.
Eleven of 14 patients who did not respond to treatment at 12 weeks (79%) had Aspergillus RNA in their blood after 4 weeks of treatment, and 12 (86%) were positive at 6 weeks.
Among patients who did respond at 12 weeks, 11 of 27 (41%) had RNA in their blood at 4 weeks, and 14 (52%) at 6 weeks. The findings were statistically significant.
There was no correlation between Aspergillus RNA and serum GM levels in terms of outcomes, but the kinetics of circulating Aspergillus RNA correlated with BG in some patients, with an excellent match in three.
Serum GM responds fairly soon if treatment is working. Aspergillus RNA, however, responds more slowly, like BG. “This may explain ... the correlation between Aspergillus RNA and BG ... Therefore, the combination of Aspergillus RNA and BG might be useful to assess therapeutic response, particularly in GM negative cases,” the investigators said.
This work was funded by Merck. Four investigators are current or former employees.
Elevated Aspergillus RNA blood levels after 4-6 weeks of antifungal treatment predict poor response at week 12 in patients with proven or probable invasive aspergillosis, according to results of a small observational study of 41 evaluable patients.
The investigators are working to address the need for reliable biomarkers of early invasive aspergillosis (IA) treatment response. Standard clinical and radiological criteria are somewhat subjective, and serial biopsies and bronchoalveolar lavage are often impractical, reported Yanan Zhao, PhD, of the New Jersey Medical School–Rutgers Biomedical and Health Sciences, Newark, and her associates.
Study participants’ blood was checked for serum galactomannan (GM), 1, 3-beta-D-glucan (BG), and Aspergillus RNA within 24 hours of starting antifungal therapy, then twice per week during the first 2 weeks, then once during weeks 4, 6, and 12, the investigators reported (Med Mycol. 2016 Jun 22. pii: myw043).
Ribosomal Aspergillus RNA – like GM and BG, a marker of fungal load – was measured by nucleic acid sequence-based amplification (NASBA), a robust isothermal amplification technique more sensitive than polymerase chain reaction due largely “to increased starting target numbers (RNA versus DNA) and more robust amplification.” Although NASBA has been used before to diagnose IA, using it to monitor treatment “is still in its infancy,” the authors noted.
Eleven of 14 patients who did not respond to treatment at 12 weeks (79%) had Aspergillus RNA in their blood after 4 weeks of treatment, and 12 (86%) were positive at 6 weeks.
Among patients who did respond at 12 weeks, 11 of 27 (41%) had RNA in their blood at 4 weeks, and 14 (52%) at 6 weeks. The findings were statistically significant.
There was no correlation between Aspergillus RNA and serum GM levels in terms of outcomes, but the kinetics of circulating Aspergillus RNA correlated with BG in some patients, with an excellent match in three.
Serum GM responds fairly soon if treatment is working. Aspergillus RNA, however, responds more slowly, like BG. “This may explain ... the correlation between Aspergillus RNA and BG ... Therefore, the combination of Aspergillus RNA and BG might be useful to assess therapeutic response, particularly in GM negative cases,” the investigators said.
This work was funded by Merck. Four investigators are current or former employees.
FROM MEDICAL MYCOLOGY
Key clinical point: Elevated Aspergillus RNA blood levels during the first 4-6 weeks of antifungal treatment predicts poor response at week 12.
Major finding: Eleven of 14 patients who did not respond to antifungals at 12 weeks (79%) had Aspergillus RNA in their blood after 4 weeks of treatment, versus 11 of 27 (41%) who did respond (P = .046).
Data source: Small observational study of patients with proven or probable invasive aspergillosis.
Disclosures: This work was funded by Merck. Four investigators are current or former employees.
Expert advocates single pass CO2 laser resurfacing
NEWPORT BEACH, CALIF. – Facial CO2 laser resurfacing deserves a comeback, according to Victor Ross, MD, director of laser and cosmetic dermatology at the Scripps Clinic in San Diego.
The practice fell out of favor in the late 1990s because of long recovery times, hypopigmentation, and the risk of keloid scars, among other issues. Physicians were just being too aggressive, doing multiple full-field passes in one session, he said at the Summit in Aesthetic Medicine, held by Global Academy for Medical Education.

It turns out that doing one pass very conservatively – with maybe a second pass around the mouth for deeper wrinkles – delivers a lot of the benefits with none of the downsides. It takes maybe 45 minutes, and “we get very nice results, I think better than fractional [laser] results,” Dr. Ross said. The wrinkle-smoothing effect may not be as potent or durable as the old-school approach, “but it’s a more natural look and [there’s] much faster recovery. Patients go pink instead of red,” and can wear makeup sooner. “You don’t get delayed hypopigmentation.”
The general trend in cosmetic dermatology is to do multiple procedures in one office visit instead of spacing them out over several appointments. For instance, lentigines on the hand could be targeted with intense pulsed light and then hand crepiness could be treated with a fractional laser. You “can get a lot done and a very nice result in one” session, but it makes sense to dial settings back maybe 15%-20% when different devices are used on the same area, he said.
I keep several lasers in one room” and sometimes “go back and forth, back and forth like a mad chef,” he said.
Companies are helping further the approach. One company, for instance, has added a nonablative fractional laser to its intense pulsed light platform. Others are combining ablative and nonablative fractional lasers. It’s all about “allowing you to have more flexibility in how you deliver energy,” Dr. Ross said.
Meanwhile, another newer kid on the block, the picosecond laser, has helped a bit with tattoo removal, but “we are still not really hitting a home run. There’s no doubt that picosecond lasers are more efficient than nanosecond lasers, but they’re maybe 20%-30% better,” he said. The problem is that most commercially available picosecond lasers have pulse durations on the order of 500-800 picoseconds, which is not optimal for the smallest pigment particles. If pulse durations are too short and energies too high, “you get this kind of white plasma in the skin, which doesn’t” help.
Industry hasn’t solved the problem yet; for now “I would give us a C-plus on tattoos,” he said.
Dr. Ross had some advice for dermatologists looking to outfit a new cosmetic practice. Right off the bat, “you will need something for red and brown spots, because you are going to see a lot of that.” An intense pulsed light device or a large-spot potassium titanyl phosphate (KTP) laser fills the bill, he said.
He added that he would have a device for resurfacing, and as the third device, “I would get maybe a Q-switch laser for tattoos,” he said.
So armed, a dermatologist could handle much of what’s likely to come through the door.
Dr. Ross works with a number of companies, including Lutronic, Cynosure, and Ellipse. Global Academy and this news organization are owned by the same company.
NEWPORT BEACH, CALIF. – Facial CO2 laser resurfacing deserves a comeback, according to Victor Ross, MD, director of laser and cosmetic dermatology at the Scripps Clinic in San Diego.
The practice fell out of favor in the late 1990s because of long recovery times, hypopigmentation, and the risk of keloid scars, among other issues. Physicians were just being too aggressive, doing multiple full-field passes in one session, he said at the Summit in Aesthetic Medicine, held by Global Academy for Medical Education.
It turns out that doing one pass very conservatively – with maybe a second pass around the mouth for deeper wrinkles – delivers a lot of the benefits with none of the downsides. It takes maybe 45 minutes, and “we get very nice results, I think better than fractional [laser] results,” Dr. Ross said. The wrinkle-smoothing effect may not be as potent or durable as the old-school approach, “but it’s a more natural look and [there’s] much faster recovery. Patients go pink instead of red,” and can wear makeup sooner. “You don’t get delayed hypopigmentation.”
The general trend in cosmetic dermatology is to do multiple procedures in one office visit instead of spacing them out over several appointments. For instance, lentigines on the hand could be targeted with intense pulsed light and then hand crepiness could be treated with a fractional laser. You “can get a lot done and a very nice result in one” session, but it makes sense to dial settings back maybe 15%-20% when different devices are used on the same area, he said.
I keep several lasers in one room” and sometimes “go back and forth, back and forth like a mad chef,” he said.
Companies are helping further the approach. One company, for instance, has added a nonablative fractional laser to its intense pulsed light platform. Others are combining ablative and nonablative fractional lasers. It’s all about “allowing you to have more flexibility in how you deliver energy,” Dr. Ross said.
Meanwhile, another newer kid on the block, the picosecond laser, has helped a bit with tattoo removal, but “we are still not really hitting a home run. There’s no doubt that picosecond lasers are more efficient than nanosecond lasers, but they’re maybe 20%-30% better,” he said. The problem is that most commercially available picosecond lasers have pulse durations on the order of 500-800 picoseconds, which is not optimal for the smallest pigment particles. If pulse durations are too short and energies too high, “you get this kind of white plasma in the skin, which doesn’t” help.
Industry hasn’t solved the problem yet; for now “I would give us a C-plus on tattoos,” he said.
Dr. Ross had some advice for dermatologists looking to outfit a new cosmetic practice. Right off the bat, “you will need something for red and brown spots, because you are going to see a lot of that.” An intense pulsed light device or a large-spot potassium titanyl phosphate (KTP) laser fills the bill, he said.
He added that he would have a device for resurfacing, and as the third device, “I would get maybe a Q-switch laser for tattoos,” he said.
So armed, a dermatologist could handle much of what’s likely to come through the door.
Dr. Ross works with a number of companies, including Lutronic, Cynosure, and Ellipse. Global Academy and this news organization are owned by the same company.
NEWPORT BEACH, CALIF. – Facial CO2 laser resurfacing deserves a comeback, according to Victor Ross, MD, director of laser and cosmetic dermatology at the Scripps Clinic in San Diego.
The practice fell out of favor in the late 1990s because of long recovery times, hypopigmentation, and the risk of keloid scars, among other issues. Physicians were just being too aggressive, doing multiple full-field passes in one session, he said at the Summit in Aesthetic Medicine, held by Global Academy for Medical Education.
It turns out that doing one pass very conservatively – with maybe a second pass around the mouth for deeper wrinkles – delivers a lot of the benefits with none of the downsides. It takes maybe 45 minutes, and “we get very nice results, I think better than fractional [laser] results,” Dr. Ross said. The wrinkle-smoothing effect may not be as potent or durable as the old-school approach, “but it’s a more natural look and [there’s] much faster recovery. Patients go pink instead of red,” and can wear makeup sooner. “You don’t get delayed hypopigmentation.”
The general trend in cosmetic dermatology is to do multiple procedures in one office visit instead of spacing them out over several appointments. For instance, lentigines on the hand could be targeted with intense pulsed light and then hand crepiness could be treated with a fractional laser. You “can get a lot done and a very nice result in one” session, but it makes sense to dial settings back maybe 15%-20% when different devices are used on the same area, he said.
I keep several lasers in one room” and sometimes “go back and forth, back and forth like a mad chef,” he said.
Companies are helping further the approach. One company, for instance, has added a nonablative fractional laser to its intense pulsed light platform. Others are combining ablative and nonablative fractional lasers. It’s all about “allowing you to have more flexibility in how you deliver energy,” Dr. Ross said.
Meanwhile, another newer kid on the block, the picosecond laser, has helped a bit with tattoo removal, but “we are still not really hitting a home run. There’s no doubt that picosecond lasers are more efficient than nanosecond lasers, but they’re maybe 20%-30% better,” he said. The problem is that most commercially available picosecond lasers have pulse durations on the order of 500-800 picoseconds, which is not optimal for the smallest pigment particles. If pulse durations are too short and energies too high, “you get this kind of white plasma in the skin, which doesn’t” help.
Industry hasn’t solved the problem yet; for now “I would give us a C-plus on tattoos,” he said.
Dr. Ross had some advice for dermatologists looking to outfit a new cosmetic practice. Right off the bat, “you will need something for red and brown spots, because you are going to see a lot of that.” An intense pulsed light device or a large-spot potassium titanyl phosphate (KTP) laser fills the bill, he said.
He added that he would have a device for resurfacing, and as the third device, “I would get maybe a Q-switch laser for tattoos,” he said.
So armed, a dermatologist could handle much of what’s likely to come through the door.
Dr. Ross works with a number of companies, including Lutronic, Cynosure, and Ellipse. Global Academy and this news organization are owned by the same company.
EXPERT ANALYSIS FROM THE SUMMIT IN AESTHETIC MEDICINE

Automated cell counter approved for use in US

The US Food and Drug Administration has granted 510(k) clearance for the GloCyte Automated Cell Counter System and GloCyte Low and High Level Controls.
The GloCyte Automated Cell Counter System is intended for use by trained healthcare professionals in clinical laboratories to quantify red blood cells (RBCs) and total nucleated cells (TNCs) in cerebrospinal fluid collected from adult and pediatric patients.
Low and High Level Controls are assayed hematology controls designed to monitor the performance of the GloCyte system.
The GloCyte system is designed to provide accurate cell counts at clinically relevant low levels to assist the diagnosis of infections, inflammatory processes, hemorrhage, leukemia, and malignancies that may involve the central nervous system.
“To date, there has not been a way to provide dependable, low cell counts,” said John Coughlin, president and CEO of Advanced Instruments, Inc., makers of the GloCyte system.
“We use a novel combination of fluorescence, microscopy with digital image analysis principles, highly specific reagents, and an intelligent counting algorithm to provide accurate and precise cell counts.”
The GloCyte system includes the GloCyte instrument, computer (hardware and software), vacuum station, sample preparation tray, barcode reader, pipettes (10 and 30 μL), test cartridge, TNC and RBC reagents, and Low and High Level Controls.
The test method uses 1 of 2 reagents to stain TNCs (propidium iodide with detergent) or RBCs (fluorochrome labeled anti-human RBC antibody in buffer with stabilizers) and a digital imaging system to count the cells. The image is captured by a digital CCD camera, and the fluorescent stained cells are counted via digital image processing.
The GloCyte system requires 30 μL of sample per test, and it uses disposable test cartridges to ensure no sample carryover. The system also includes Levey-Jennings charts and an audit table.
Advanced Instruments expects to begin shipping the GloCyte system in September. For more information about the system, visit aicompanies.com/glocyte. ![]()
The US Food and Drug Administration has granted 510(k) clearance for the GloCyte Automated Cell Counter System and GloCyte Low and High Level Controls.
The GloCyte Automated Cell Counter System is intended for use by trained healthcare professionals in clinical laboratories to quantify red blood cells (RBCs) and total nucleated cells (TNCs) in cerebrospinal fluid collected from adult and pediatric patients.
Low and High Level Controls are assayed hematology controls designed to monitor the performance of the GloCyte system.
The GloCyte system is designed to provide accurate cell counts at clinically relevant low levels to assist the diagnosis of infections, inflammatory processes, hemorrhage, leukemia, and malignancies that may involve the central nervous system.
“To date, there has not been a way to provide dependable, low cell counts,” said John Coughlin, president and CEO of Advanced Instruments, Inc., makers of the GloCyte system.
“We use a novel combination of fluorescence, microscopy with digital image analysis principles, highly specific reagents, and an intelligent counting algorithm to provide accurate and precise cell counts.”
The GloCyte system includes the GloCyte instrument, computer (hardware and software), vacuum station, sample preparation tray, barcode reader, pipettes (10 and 30 μL), test cartridge, TNC and RBC reagents, and Low and High Level Controls.
The test method uses 1 of 2 reagents to stain TNCs (propidium iodide with detergent) or RBCs (fluorochrome labeled anti-human RBC antibody in buffer with stabilizers) and a digital imaging system to count the cells. The image is captured by a digital CCD camera, and the fluorescent stained cells are counted via digital image processing.
The GloCyte system requires 30 μL of sample per test, and it uses disposable test cartridges to ensure no sample carryover. The system also includes Levey-Jennings charts and an audit table.
Advanced Instruments expects to begin shipping the GloCyte system in September. For more information about the system, visit aicompanies.com/glocyte. ![]()
The US Food and Drug Administration has granted 510(k) clearance for the GloCyte Automated Cell Counter System and GloCyte Low and High Level Controls.
The GloCyte Automated Cell Counter System is intended for use by trained healthcare professionals in clinical laboratories to quantify red blood cells (RBCs) and total nucleated cells (TNCs) in cerebrospinal fluid collected from adult and pediatric patients.
Low and High Level Controls are assayed hematology controls designed to monitor the performance of the GloCyte system.
The GloCyte system is designed to provide accurate cell counts at clinically relevant low levels to assist the diagnosis of infections, inflammatory processes, hemorrhage, leukemia, and malignancies that may involve the central nervous system.
“To date, there has not been a way to provide dependable, low cell counts,” said John Coughlin, president and CEO of Advanced Instruments, Inc., makers of the GloCyte system.
“We use a novel combination of fluorescence, microscopy with digital image analysis principles, highly specific reagents, and an intelligent counting algorithm to provide accurate and precise cell counts.”
The GloCyte system includes the GloCyte instrument, computer (hardware and software), vacuum station, sample preparation tray, barcode reader, pipettes (10 and 30 μL), test cartridge, TNC and RBC reagents, and Low and High Level Controls.
The test method uses 1 of 2 reagents to stain TNCs (propidium iodide with detergent) or RBCs (fluorochrome labeled anti-human RBC antibody in buffer with stabilizers) and a digital imaging system to count the cells. The image is captured by a digital CCD camera, and the fluorescent stained cells are counted via digital image processing.
The GloCyte system requires 30 μL of sample per test, and it uses disposable test cartridges to ensure no sample carryover. The system also includes Levey-Jennings charts and an audit table.
Advanced Instruments expects to begin shipping the GloCyte system in September. For more information about the system, visit aicompanies.com/glocyte. ![]()
From the Washington Office: The operationalization of MACRA
On April 27, 2016, the Centers for Medicare and Medicaid Services (CMS) released its proposed rule on the Medicare Access and CHIP Reauthorization Act (MACRA). Fellows will remember that the MACRA legislation, passed in April of 2015, permanently repealed the Sustainable Growth Rate (SGR) formula and thus, represents the greatest sea change in Medicare physician payment since the establishment of the RBRVS (Resource-Based Relative Value Scale) in 1992.
In broadest policy terms, the law continues to advance the CMS policy goal of basing payment on quality and value over volume. From a granular perspective, the law combines Medicare’s three current quality programs into one new system. CMS published this 982-page proposed rule after reviewing the comments submitted by ACS and other interested parties in response to its request for information last fall. As I write, staff of the Division of Advocacy and Health Policy are in the process of crafting the ACS response to the proposed rule. Comments were due on June 27, 2016. It is anticipated that CMS will publish the final rule later this year, likely in late October or early November. Accordingly, the following description of the implementation of MACRA is based on the current understanding from the proposed rule, and is likely to change in some respects with the final rule.

CMS has designated the payment program operationalizing the MACRA law as the Quality Payment Program (QPP). The QPP has two tracks, the Merit-based Incentive Payment System (MIPS) and advanced Alternative Payment Models (APMs). For the first several years of the program, it is widely expected that the vast majority of physicians will participate in the QPP via the MIPS track. As such, I will direct the remainder of the text this month to the MIPS program.
MIPS: Merit-Based Incentive Payment System
The Merit-based Incentive Payment System, MIPS, consists of four components. They are: Quality, Resource Use, Advancing Care Information (ACI) and the Clinical Practice Improvement Activities (CPIA). Though the names have changed, Fellows are familiar with the substance of three of the components. For example, the Quality component replaces the Physician Quality Reporting System (PQRS); the Resource Use component replaces the Value-Based Modifier (VBM); and the Advancing Care Information component modifies and replaces the Electronic Health Record Meaningful Use (EHR-MU) program. The fourth component of MIPS is the new Clinical Practice Improvement Activities, which the legislation designates as intended to provide “credit for work to improve practice and facilitates future participation in alternative payment models.”
Composite Performance Score: MIPS participants will be assigned a Composite Performance Score based on their performance in all four components. For 2017, the first year for assessment under the QPP, 50 percent of the score will be based on performance in the Quality component, 10 percent will be based on the Resource Use component, 25 percent will be based on the Advancing Care Information component, and 15 percent will be based on the Clinical Practice Improvement Activities. In future years, there will be a gradual increase in the relative value of the Resource Use component with an equal and accompanying decrease in the value of the Quality component. As proposed, by the third assessment year (2019), the Quality and Resource Use components are expected to each account for 30 percent of the Composite Performance Score.
MIPS Quality Component: Though the Quality component of MIPS replaces the PQRS, CMS is proposing some changes that Fellows will welcome. As opposed to the previous PQRS requirement to report nine measures, the MIPS Quality component requires providers to report only six measures. One of these six must be an “Outcome” measure and another must be a “Cross-cutting” measure. While the reporting threshold for the percentage of patients on which reports will be required is proposed to increase substantially, ACS and other physician groups will be advocating that the required percentage published in the final rule be close to the 50 percent level found in current law.
Resources Use Component: There is also some good news relative to the Resource Use component in that there are NO reporting requirements. CMS will calculate this component from Medicare claims data and base its assessment of individual provider performance on the resource measures currently used for the Value-Based Modifier. Namely, those are the VBM Total per Capita Cost measure and the VBM Medicare Spending per Beneficiary measure. In addition, CMS will also be taking into account measures that specifically focus on episodes of care, something for which the College has previously advocated. Beginning in 2018, CMS plans to also take into consideration factors of patient condition and patient relation in order to address physician concerns about risk adjustment and attribution.
Advancing Care Information Component: This modifies and replaces the Electronic Health Record Meaningful Use program. The score for this component is derived in two parts, a Base score (50 percent) and a Performance score (up to an additional 50 percent). The threshold for achieving the Base score remains “all or nothing.” Only after meeting the requirements for the Base score is one eligible to receive additional Performance score credit based on the level of performance on a subset of the same measures required to achieve the Base score. Assessment in 2017 will be based on the EHR-MU requirements published in the 2015 Final Rule for the EHR-MU program.
Clinical Practice Improvement Component: The fourth component of MIPS is the Clinical Practice Improvement Activities component. As mentioned previously, this is a new requirement with no prior analogous program requirement. As such, it is very much in evolution. In the first year of MIPS assessment (2017), achieving full credit for the CPIA component should not pose much additional administrative burden as reporting will be by simple attestation. Physicians will chose from a list of 94 activities assigned two different weighted values. In order to receive full credit for the CPIA component, most providers will need to attest that they have participated in a minimum of three and a maximum of six of the 94 activities, depending on the weight of the activities selected, for 90 days.
As outlined above, the final rule on the Quality Payment Program is expected to be released in late October or early November. As proposed, assessment under the provisions of that final rule would begin in January of 2017. This leaves all providers with a very short time window in which to become familiar with the program that will impact their Medicare payment beginning in 2019.
Accordingly, in the next several editions of this column, I will provide more specific information about each of the four MIPS components, the scoring mechanism for MIPS assessment and the aforementioned alternate track to MIPS, the Alternative Payment Models. While it is easy to understand how many Fellows could initially find this change daunting and overwhelming, I am confident that with a minimal investment of time all can develop adequate working knowledge of the MIPS and APMs to participate successfully in the QPP.
Until next month...
Dr. Bailey is an ACS Fellow, a pediatric surgeon, and Medical Director, Advocacy, for the Division of Advocacy and Health Policy, in the ACS offices in Washington.
On April 27, 2016, the Centers for Medicare and Medicaid Services (CMS) released its proposed rule on the Medicare Access and CHIP Reauthorization Act (MACRA). Fellows will remember that the MACRA legislation, passed in April of 2015, permanently repealed the Sustainable Growth Rate (SGR) formula and thus, represents the greatest sea change in Medicare physician payment since the establishment of the RBRVS (Resource-Based Relative Value Scale) in 1992.
In broadest policy terms, the law continues to advance the CMS policy goal of basing payment on quality and value over volume. From a granular perspective, the law combines Medicare’s three current quality programs into one new system. CMS published this 982-page proposed rule after reviewing the comments submitted by ACS and other interested parties in response to its request for information last fall. As I write, staff of the Division of Advocacy and Health Policy are in the process of crafting the ACS response to the proposed rule. Comments were due on June 27, 2016. It is anticipated that CMS will publish the final rule later this year, likely in late October or early November. Accordingly, the following description of the implementation of MACRA is based on the current understanding from the proposed rule, and is likely to change in some respects with the final rule.
CMS has designated the payment program operationalizing the MACRA law as the Quality Payment Program (QPP). The QPP has two tracks, the Merit-based Incentive Payment System (MIPS) and advanced Alternative Payment Models (APMs). For the first several years of the program, it is widely expected that the vast majority of physicians will participate in the QPP via the MIPS track. As such, I will direct the remainder of the text this month to the MIPS program.
MIPS: Merit-Based Incentive Payment System
The Merit-based Incentive Payment System, MIPS, consists of four components. They are: Quality, Resource Use, Advancing Care Information (ACI) and the Clinical Practice Improvement Activities (CPIA). Though the names have changed, Fellows are familiar with the substance of three of the components. For example, the Quality component replaces the Physician Quality Reporting System (PQRS); the Resource Use component replaces the Value-Based Modifier (VBM); and the Advancing Care Information component modifies and replaces the Electronic Health Record Meaningful Use (EHR-MU) program. The fourth component of MIPS is the new Clinical Practice Improvement Activities, which the legislation designates as intended to provide “credit for work to improve practice and facilitates future participation in alternative payment models.”
Composite Performance Score: MIPS participants will be assigned a Composite Performance Score based on their performance in all four components. For 2017, the first year for assessment under the QPP, 50 percent of the score will be based on performance in the Quality component, 10 percent will be based on the Resource Use component, 25 percent will be based on the Advancing Care Information component, and 15 percent will be based on the Clinical Practice Improvement Activities. In future years, there will be a gradual increase in the relative value of the Resource Use component with an equal and accompanying decrease in the value of the Quality component. As proposed, by the third assessment year (2019), the Quality and Resource Use components are expected to each account for 30 percent of the Composite Performance Score.
MIPS Quality Component: Though the Quality component of MIPS replaces the PQRS, CMS is proposing some changes that Fellows will welcome. As opposed to the previous PQRS requirement to report nine measures, the MIPS Quality component requires providers to report only six measures. One of these six must be an “Outcome” measure and another must be a “Cross-cutting” measure. While the reporting threshold for the percentage of patients on which reports will be required is proposed to increase substantially, ACS and other physician groups will be advocating that the required percentage published in the final rule be close to the 50 percent level found in current law.
Resources Use Component: There is also some good news relative to the Resource Use component in that there are NO reporting requirements. CMS will calculate this component from Medicare claims data and base its assessment of individual provider performance on the resource measures currently used for the Value-Based Modifier. Namely, those are the VBM Total per Capita Cost measure and the VBM Medicare Spending per Beneficiary measure. In addition, CMS will also be taking into account measures that specifically focus on episodes of care, something for which the College has previously advocated. Beginning in 2018, CMS plans to also take into consideration factors of patient condition and patient relation in order to address physician concerns about risk adjustment and attribution.
Advancing Care Information Component: This modifies and replaces the Electronic Health Record Meaningful Use program. The score for this component is derived in two parts, a Base score (50 percent) and a Performance score (up to an additional 50 percent). The threshold for achieving the Base score remains “all or nothing.” Only after meeting the requirements for the Base score is one eligible to receive additional Performance score credit based on the level of performance on a subset of the same measures required to achieve the Base score. Assessment in 2017 will be based on the EHR-MU requirements published in the 2015 Final Rule for the EHR-MU program.
Clinical Practice Improvement Component: The fourth component of MIPS is the Clinical Practice Improvement Activities component. As mentioned previously, this is a new requirement with no prior analogous program requirement. As such, it is very much in evolution. In the first year of MIPS assessment (2017), achieving full credit for the CPIA component should not pose much additional administrative burden as reporting will be by simple attestation. Physicians will chose from a list of 94 activities assigned two different weighted values. In order to receive full credit for the CPIA component, most providers will need to attest that they have participated in a minimum of three and a maximum of six of the 94 activities, depending on the weight of the activities selected, for 90 days.
As outlined above, the final rule on the Quality Payment Program is expected to be released in late October or early November. As proposed, assessment under the provisions of that final rule would begin in January of 2017. This leaves all providers with a very short time window in which to become familiar with the program that will impact their Medicare payment beginning in 2019.
Accordingly, in the next several editions of this column, I will provide more specific information about each of the four MIPS components, the scoring mechanism for MIPS assessment and the aforementioned alternate track to MIPS, the Alternative Payment Models. While it is easy to understand how many Fellows could initially find this change daunting and overwhelming, I am confident that with a minimal investment of time all can develop adequate working knowledge of the MIPS and APMs to participate successfully in the QPP.
Until next month...
Dr. Bailey is an ACS Fellow, a pediatric surgeon, and Medical Director, Advocacy, for the Division of Advocacy and Health Policy, in the ACS offices in Washington.
On April 27, 2016, the Centers for Medicare and Medicaid Services (CMS) released its proposed rule on the Medicare Access and CHIP Reauthorization Act (MACRA). Fellows will remember that the MACRA legislation, passed in April of 2015, permanently repealed the Sustainable Growth Rate (SGR) formula and thus, represents the greatest sea change in Medicare physician payment since the establishment of the RBRVS (Resource-Based Relative Value Scale) in 1992.
In broadest policy terms, the law continues to advance the CMS policy goal of basing payment on quality and value over volume. From a granular perspective, the law combines Medicare’s three current quality programs into one new system. CMS published this 982-page proposed rule after reviewing the comments submitted by ACS and other interested parties in response to its request for information last fall. As I write, staff of the Division of Advocacy and Health Policy are in the process of crafting the ACS response to the proposed rule. Comments were due on June 27, 2016. It is anticipated that CMS will publish the final rule later this year, likely in late October or early November. Accordingly, the following description of the implementation of MACRA is based on the current understanding from the proposed rule, and is likely to change in some respects with the final rule.
CMS has designated the payment program operationalizing the MACRA law as the Quality Payment Program (QPP). The QPP has two tracks, the Merit-based Incentive Payment System (MIPS) and advanced Alternative Payment Models (APMs). For the first several years of the program, it is widely expected that the vast majority of physicians will participate in the QPP via the MIPS track. As such, I will direct the remainder of the text this month to the MIPS program.
MIPS: Merit-Based Incentive Payment System
The Merit-based Incentive Payment System, MIPS, consists of four components. They are: Quality, Resource Use, Advancing Care Information (ACI) and the Clinical Practice Improvement Activities (CPIA). Though the names have changed, Fellows are familiar with the substance of three of the components. For example, the Quality component replaces the Physician Quality Reporting System (PQRS); the Resource Use component replaces the Value-Based Modifier (VBM); and the Advancing Care Information component modifies and replaces the Electronic Health Record Meaningful Use (EHR-MU) program. The fourth component of MIPS is the new Clinical Practice Improvement Activities, which the legislation designates as intended to provide “credit for work to improve practice and facilitates future participation in alternative payment models.”
Composite Performance Score: MIPS participants will be assigned a Composite Performance Score based on their performance in all four components. For 2017, the first year for assessment under the QPP, 50 percent of the score will be based on performance in the Quality component, 10 percent will be based on the Resource Use component, 25 percent will be based on the Advancing Care Information component, and 15 percent will be based on the Clinical Practice Improvement Activities. In future years, there will be a gradual increase in the relative value of the Resource Use component with an equal and accompanying decrease in the value of the Quality component. As proposed, by the third assessment year (2019), the Quality and Resource Use components are expected to each account for 30 percent of the Composite Performance Score.
MIPS Quality Component: Though the Quality component of MIPS replaces the PQRS, CMS is proposing some changes that Fellows will welcome. As opposed to the previous PQRS requirement to report nine measures, the MIPS Quality component requires providers to report only six measures. One of these six must be an “Outcome” measure and another must be a “Cross-cutting” measure. While the reporting threshold for the percentage of patients on which reports will be required is proposed to increase substantially, ACS and other physician groups will be advocating that the required percentage published in the final rule be close to the 50 percent level found in current law.
Resources Use Component: There is also some good news relative to the Resource Use component in that there are NO reporting requirements. CMS will calculate this component from Medicare claims data and base its assessment of individual provider performance on the resource measures currently used for the Value-Based Modifier. Namely, those are the VBM Total per Capita Cost measure and the VBM Medicare Spending per Beneficiary measure. In addition, CMS will also be taking into account measures that specifically focus on episodes of care, something for which the College has previously advocated. Beginning in 2018, CMS plans to also take into consideration factors of patient condition and patient relation in order to address physician concerns about risk adjustment and attribution.
Advancing Care Information Component: This modifies and replaces the Electronic Health Record Meaningful Use program. The score for this component is derived in two parts, a Base score (50 percent) and a Performance score (up to an additional 50 percent). The threshold for achieving the Base score remains “all or nothing.” Only after meeting the requirements for the Base score is one eligible to receive additional Performance score credit based on the level of performance on a subset of the same measures required to achieve the Base score. Assessment in 2017 will be based on the EHR-MU requirements published in the 2015 Final Rule for the EHR-MU program.
Clinical Practice Improvement Component: The fourth component of MIPS is the Clinical Practice Improvement Activities component. As mentioned previously, this is a new requirement with no prior analogous program requirement. As such, it is very much in evolution. In the first year of MIPS assessment (2017), achieving full credit for the CPIA component should not pose much additional administrative burden as reporting will be by simple attestation. Physicians will chose from a list of 94 activities assigned two different weighted values. In order to receive full credit for the CPIA component, most providers will need to attest that they have participated in a minimum of three and a maximum of six of the 94 activities, depending on the weight of the activities selected, for 90 days.
As outlined above, the final rule on the Quality Payment Program is expected to be released in late October or early November. As proposed, assessment under the provisions of that final rule would begin in January of 2017. This leaves all providers with a very short time window in which to become familiar with the program that will impact their Medicare payment beginning in 2019.
Accordingly, in the next several editions of this column, I will provide more specific information about each of the four MIPS components, the scoring mechanism for MIPS assessment and the aforementioned alternate track to MIPS, the Alternative Payment Models. While it is easy to understand how many Fellows could initially find this change daunting and overwhelming, I am confident that with a minimal investment of time all can develop adequate working knowledge of the MIPS and APMs to participate successfully in the QPP.
Until next month...
Dr. Bailey is an ACS Fellow, a pediatric surgeon, and Medical Director, Advocacy, for the Division of Advocacy and Health Policy, in the ACS offices in Washington.

Pembrolizumab-ipilimumab combo is highly active in advanced melanoma
CHICAGO – The combination of pembrolizumab, an antibody to the human cell surface receptor programmed death-1 (PD-1), and ipilimumab, an antibody to the human T-cell receptor cytotoxic T-lymphocyte-associated antigen 4 (CTLA4), is highly active against advanced melanoma and has acceptable safety, finds the KEYNOTE 029 trial’s expansion cohort.
The 153 patients in the cohort received a standard dose of pembrolizumab (2 mg/kg every 3 weeks) with a reduced dose of ipilimumab (1 mg/kg every 3 weeks for four doses) on the basis of earlier data showing substantial toxicity when a standard dose of ipilimumab was combined with other immune checkpoint inhibitors.
Results reported at the annual meeting of the American Society of Clinical Oncology showed that the overall response rate was 57%, and the disease control rate was 78%. Although 42% of patients experienced grade 3 or 4 treatment-related adverse events, most of these events resolved, and there were no treatment-related deaths.
“Pembrolizumab 2 mg/kg in combination with four doses of ipilimumab 1 mg/kg has a manageable toxicity profile and provides robust antitumor activity in patients with advanced melanoma,” concluded the investigators, who were led by Georgina Long, PhD, MBBS, chair of Melanoma Medical Oncology and Translational Research at the Melanoma Institute Australia and Royal North Shore Hospital, University of Sydney.
The response rate seen in KEYNOTE 029 was almost identical to that seen in the CheckMate 067 trial with the combination of nivolumab and standard-dose ipilimumab (3 mg/kg every 3 weeks for four doses), noted invited discussant Marc S. Ernstoff, MD, professor and chair of the department of medicine at the Roswell Park Cancer Institute in Buffalo, N.Y. It was also “remarkably comparable” to the 69% seen in the COMBI-d melanoma trial with the combination of dabrafenib (a BRAF inhibitor) and trametinib (an inhibitor of MEK MAPK/ERK kinase).
“There is a significant amount of grade 3 and 4 toxicity, but the dose of ipilimumab appeared to decrease this in the pembrolizumab-ipilimumab study compared to the nivolumab-ipilimumab study,” he noted. “There was a high percent of low-grade toxicities reported in all of these studies, and I would argue that as we are seeing patients survive longer, these low-grade toxicities are going to become more of an issue for us as oncologists to be able to deal with in terms of quality of life for patients surviving.”
There is good rationale for combining CTLA4 blockade and PD1 (or PD-L1) blockade in melanoma, Dr. Long maintained when introducing the research. “We know that CTLA inhibition at the priming phase in the periphery, at antigen presentation, is effective, as is PD-1 or PD-L1 [inhibition] at the effector phase down in the tumor bed,” she elaborated.
The patients with advanced melanoma enrolled in the expansion cohort could have received any number of prior therapies other than immune checkpoint inhibitors. However, in 87%, the study regimen was their first therapy.
At the time of data cutoff, 72% of patients had received all four planned doses of ipilimumab (Yervoy), and 56% were continuing on pembrolizumab (Keytruda).
The rates of any-grade and grade 3 or 4 treatment-related adverse events were 95% and 42%, respectively. The corresponding rates of immune-mediated adverse events were 58% and 25%.
The most common grade 3 or 4 treatment-related adverse events were lipase elevation (14%) and rash (3%). The former was asymptomatic and had no sequelae in the majority of cases, Dr. Long reported.
Hepatitis, colitis, and skin reactions were the most common grade 3 or 4 immune-mediated adverse events. The majority of immune-mediated adverse events were managed with systemic treatment, usually corticosteroids, and resolved.
When it came to efficacy, the overall response rate with the combination was similar across subgroups of patients stratified by PD-L1 status in the tumor and adjacent immune tissue, treatment history, baseline lactate dehydrogenase level, and BRAF mutational status.
Responses were ongoing in 98% of patients at data cutoff, with the duration of response ranging from about 6 weeks to 43 weeks, Dr. Long said. The disease control rate was 78%.
With a median follow-up of 10.0 months, median progression-free survival and overall survival were not yet reached. However, the 6-month rates of these outcomes were 70% and 93%, respectively.
Dr. Long disclosed that she is a consultant/adviser to Amgen, Bristol-Myers Squibb, Merck, Novartis, Provectus, and Roche, and that she has received honoraria from Bristol-Myers Squibb, Merck, and Novartis. The trial was supported by Merck.
CHICAGO – The combination of pembrolizumab, an antibody to the human cell surface receptor programmed death-1 (PD-1), and ipilimumab, an antibody to the human T-cell receptor cytotoxic T-lymphocyte-associated antigen 4 (CTLA4), is highly active against advanced melanoma and has acceptable safety, finds the KEYNOTE 029 trial’s expansion cohort.
The 153 patients in the cohort received a standard dose of pembrolizumab (2 mg/kg every 3 weeks) with a reduced dose of ipilimumab (1 mg/kg every 3 weeks for four doses) on the basis of earlier data showing substantial toxicity when a standard dose of ipilimumab was combined with other immune checkpoint inhibitors.
Results reported at the annual meeting of the American Society of Clinical Oncology showed that the overall response rate was 57%, and the disease control rate was 78%. Although 42% of patients experienced grade 3 or 4 treatment-related adverse events, most of these events resolved, and there were no treatment-related deaths.
“Pembrolizumab 2 mg/kg in combination with four doses of ipilimumab 1 mg/kg has a manageable toxicity profile and provides robust antitumor activity in patients with advanced melanoma,” concluded the investigators, who were led by Georgina Long, PhD, MBBS, chair of Melanoma Medical Oncology and Translational Research at the Melanoma Institute Australia and Royal North Shore Hospital, University of Sydney.
The response rate seen in KEYNOTE 029 was almost identical to that seen in the CheckMate 067 trial with the combination of nivolumab and standard-dose ipilimumab (3 mg/kg every 3 weeks for four doses), noted invited discussant Marc S. Ernstoff, MD, professor and chair of the department of medicine at the Roswell Park Cancer Institute in Buffalo, N.Y. It was also “remarkably comparable” to the 69% seen in the COMBI-d melanoma trial with the combination of dabrafenib (a BRAF inhibitor) and trametinib (an inhibitor of MEK MAPK/ERK kinase).
“There is a significant amount of grade 3 and 4 toxicity, but the dose of ipilimumab appeared to decrease this in the pembrolizumab-ipilimumab study compared to the nivolumab-ipilimumab study,” he noted. “There was a high percent of low-grade toxicities reported in all of these studies, and I would argue that as we are seeing patients survive longer, these low-grade toxicities are going to become more of an issue for us as oncologists to be able to deal with in terms of quality of life for patients surviving.”
There is good rationale for combining CTLA4 blockade and PD1 (or PD-L1) blockade in melanoma, Dr. Long maintained when introducing the research. “We know that CTLA inhibition at the priming phase in the periphery, at antigen presentation, is effective, as is PD-1 or PD-L1 [inhibition] at the effector phase down in the tumor bed,” she elaborated.
The patients with advanced melanoma enrolled in the expansion cohort could have received any number of prior therapies other than immune checkpoint inhibitors. However, in 87%, the study regimen was their first therapy.
At the time of data cutoff, 72% of patients had received all four planned doses of ipilimumab (Yervoy), and 56% were continuing on pembrolizumab (Keytruda).
The rates of any-grade and grade 3 or 4 treatment-related adverse events were 95% and 42%, respectively. The corresponding rates of immune-mediated adverse events were 58% and 25%.
The most common grade 3 or 4 treatment-related adverse events were lipase elevation (14%) and rash (3%). The former was asymptomatic and had no sequelae in the majority of cases, Dr. Long reported.
Hepatitis, colitis, and skin reactions were the most common grade 3 or 4 immune-mediated adverse events. The majority of immune-mediated adverse events were managed with systemic treatment, usually corticosteroids, and resolved.
When it came to efficacy, the overall response rate with the combination was similar across subgroups of patients stratified by PD-L1 status in the tumor and adjacent immune tissue, treatment history, baseline lactate dehydrogenase level, and BRAF mutational status.
Responses were ongoing in 98% of patients at data cutoff, with the duration of response ranging from about 6 weeks to 43 weeks, Dr. Long said. The disease control rate was 78%.
With a median follow-up of 10.0 months, median progression-free survival and overall survival were not yet reached. However, the 6-month rates of these outcomes were 70% and 93%, respectively.
Dr. Long disclosed that she is a consultant/adviser to Amgen, Bristol-Myers Squibb, Merck, Novartis, Provectus, and Roche, and that she has received honoraria from Bristol-Myers Squibb, Merck, and Novartis. The trial was supported by Merck.
CHICAGO – The combination of pembrolizumab, an antibody to the human cell surface receptor programmed death-1 (PD-1), and ipilimumab, an antibody to the human T-cell receptor cytotoxic T-lymphocyte-associated antigen 4 (CTLA4), is highly active against advanced melanoma and has acceptable safety, finds the KEYNOTE 029 trial’s expansion cohort.
The 153 patients in the cohort received a standard dose of pembrolizumab (2 mg/kg every 3 weeks) with a reduced dose of ipilimumab (1 mg/kg every 3 weeks for four doses) on the basis of earlier data showing substantial toxicity when a standard dose of ipilimumab was combined with other immune checkpoint inhibitors.
Results reported at the annual meeting of the American Society of Clinical Oncology showed that the overall response rate was 57%, and the disease control rate was 78%. Although 42% of patients experienced grade 3 or 4 treatment-related adverse events, most of these events resolved, and there were no treatment-related deaths.
“Pembrolizumab 2 mg/kg in combination with four doses of ipilimumab 1 mg/kg has a manageable toxicity profile and provides robust antitumor activity in patients with advanced melanoma,” concluded the investigators, who were led by Georgina Long, PhD, MBBS, chair of Melanoma Medical Oncology and Translational Research at the Melanoma Institute Australia and Royal North Shore Hospital, University of Sydney.
The response rate seen in KEYNOTE 029 was almost identical to that seen in the CheckMate 067 trial with the combination of nivolumab and standard-dose ipilimumab (3 mg/kg every 3 weeks for four doses), noted invited discussant Marc S. Ernstoff, MD, professor and chair of the department of medicine at the Roswell Park Cancer Institute in Buffalo, N.Y. It was also “remarkably comparable” to the 69% seen in the COMBI-d melanoma trial with the combination of dabrafenib (a BRAF inhibitor) and trametinib (an inhibitor of MEK MAPK/ERK kinase).
“There is a significant amount of grade 3 and 4 toxicity, but the dose of ipilimumab appeared to decrease this in the pembrolizumab-ipilimumab study compared to the nivolumab-ipilimumab study,” he noted. “There was a high percent of low-grade toxicities reported in all of these studies, and I would argue that as we are seeing patients survive longer, these low-grade toxicities are going to become more of an issue for us as oncologists to be able to deal with in terms of quality of life for patients surviving.”
There is good rationale for combining CTLA4 blockade and PD1 (or PD-L1) blockade in melanoma, Dr. Long maintained when introducing the research. “We know that CTLA inhibition at the priming phase in the periphery, at antigen presentation, is effective, as is PD-1 or PD-L1 [inhibition] at the effector phase down in the tumor bed,” she elaborated.
The patients with advanced melanoma enrolled in the expansion cohort could have received any number of prior therapies other than immune checkpoint inhibitors. However, in 87%, the study regimen was their first therapy.
At the time of data cutoff, 72% of patients had received all four planned doses of ipilimumab (Yervoy), and 56% were continuing on pembrolizumab (Keytruda).
The rates of any-grade and grade 3 or 4 treatment-related adverse events were 95% and 42%, respectively. The corresponding rates of immune-mediated adverse events were 58% and 25%.
The most common grade 3 or 4 treatment-related adverse events were lipase elevation (14%) and rash (3%). The former was asymptomatic and had no sequelae in the majority of cases, Dr. Long reported.
Hepatitis, colitis, and skin reactions were the most common grade 3 or 4 immune-mediated adverse events. The majority of immune-mediated adverse events were managed with systemic treatment, usually corticosteroids, and resolved.
When it came to efficacy, the overall response rate with the combination was similar across subgroups of patients stratified by PD-L1 status in the tumor and adjacent immune tissue, treatment history, baseline lactate dehydrogenase level, and BRAF mutational status.
Responses were ongoing in 98% of patients at data cutoff, with the duration of response ranging from about 6 weeks to 43 weeks, Dr. Long said. The disease control rate was 78%.
With a median follow-up of 10.0 months, median progression-free survival and overall survival were not yet reached. However, the 6-month rates of these outcomes were 70% and 93%, respectively.
Dr. Long disclosed that she is a consultant/adviser to Amgen, Bristol-Myers Squibb, Merck, Novartis, Provectus, and Roche, and that she has received honoraria from Bristol-Myers Squibb, Merck, and Novartis. The trial was supported by Merck.
AT THE 2016 ASCO ANNUAL MEETING
Key clinical point: Dual immune checkpoint blockade with pembrolizumab and ipilimumab is efficacious in advanced melanoma.
Major finding: The overall response rate was 57%, and the disease control rate was 78%.
Data source: An expansion cohort from a phase I/II trial among 153 patients with advanced melanoma (KEYNOTE 029).
Disclosures: Dr. Long disclosed that she is a consultant/advisor to Amgen, Bristol-Myers Squibb, Merck, Novartis, Provectus, and Roche, and that she has received honoraria from Bristol-Myers Squibb, Merck, and Novartis. The trial was supported by Merck.
FDA advisory panel backs biosimilar
The Food and Drug Administration’s Arthritis Advisory Committee, together with an added complement of dermatologists and gastroenterologists, unanimously recommended during meetings on July 12 and 13 that the agency license a biosimilar Humira (adalimumab) that is made by Amgen and a biosimilar Enbrel (etanercept) that is made by Sandoz for many of the same indications held by the reference drugs.
The FDA advisory panel that endorsed biosimilar Humira recommended the agent’s approval in a 26-0 vote for: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis in patients at least 4 years old, plaque psoriasis, adult Crohn’s disease, and adult ulcerative colitis. Humira itself is approved for 10 indications.
A slightly different group of 20 advisory panel members (without any gastroenterologists) voted 20-0 in favor of the FDA granting biosimilar Enbrel all five of the indications now held by Enbrel: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis, and plaque psoriasis.
The biosimilar Humira and the biosimilar Enbrel are, respectively, the third and fourth candidate biosimilars to emerge from the FDA’s development program and receive advisory committee scrutiny and support. The first agent through the process, biosimilar filgrastim (Zarxio) received FDA approval in 2015 and is available in the United States. Although the second biosimilar through the process, the tumor necrosis factor inhibitor Inflectra that is biosimilar Remicade (infliximab), received FDA approval in April of this year, it has not yet become available for sale,
A spokeswoman for the company that will market it, Pfizer, said that the company expects to start U.S. sales of Inflectra before the end of 2016.
While the Arthritis Advisory Committee ended each of its daylong deliberations for each of the two candidate biosimilars with unanimous support, the panelists’ discussions among themselves and with FDA staffers reflected some uncertainty with the biosimilar concept, especially during the first day when they focused on biosimilar Humira. The major sticking point revolved around the regulatory pathway to approval first established by the Biologics Price Competition and Innovation Act of 2009 and subsequently refined by the FDA that allows a candidate biosimilar to establish its biosimilarity primarily though the results of analytical studies that establish that the candidate molecule is highly similar to the reference molecule. This approval scheme uses clinical trials in a confirmatory role to establish biosimilarity rather than as the linchpin of approval.
It also means that the FDA can grant clinical indications to the biosimilar drug based not on the results from clinical trials, but based entirely on what have already been demonstrated as safe and effective clinical applications for the reference drug. For example, the biosimilar Humira underwent testing in two clinical studies showing similar efficacy and safety as Humira in patients with rheumatoid arthritis and in patients with plaque psoriasis, but received endorsements based on extrapolations for an additional five indications. Biosimilar Enbrel was compared with Enbrel in patients with plaque psoriasis only and still received extrapolated indications for the additional four rheumatologic conditions.

“This is a new level of extrapolation, across indications,” noted Sarah E. Streett, MD, a gastroenterologist at Stanford (Calif.) University, one of several panelists who initially voiced uncertainty about the concept.
But FDA staffer Nikolay P. Nikolov, MD, who led the agency’s presentation, assured the panelists that the concept of extrapolation was at the heart of biosimilar development and regulatory assessment.
“We have confidence from the data that the two molecules [the reference drug and biosimilar drug] are so similar that we can rely on the safety and efficacy of the reference product. The premise of our approach to biosimilars is that this is not a new molecule that we know nothing about.”
The other uncertainty about biosimilar Humira and biosimilar Enbrel that raised concerns of many panelists were the prospects for nonmedical switching once these drugs reach the market. Nonmedical switching refers to when an insurance company or pharmacy benefit manager substitutes a biosimilar for a reference drug without approval from or even the knowledge of the prescribing physician or the patient. Many of the people who spoke during the public forum period on both days of hearings voiced their concerns about this prospect.

“Nonmedical switching is a major concern of clinicians and policy makers, and we need greater clarification from the FDA,” said committee chair Daniel H. Solomon, MD, a rheumatologist and professor of medicine at Harvard Medical School in Boston.
“I see a remarkable disconnect between the public’s concerns [about nonmedical switching] and the charge to the committee. These are essential issues that need a forum to be aired out,” said panelist Steven F. Solga, MD, chief of gastroenterology at St. Luke’s Hospital in Bethlehem, Pa.
Dr. Nikolov assured committee members that the FDA recognized this concern and was working on it. “We appreciate the disconnect between the charge and the concerns of the community. I assure you that the issues brought up will be part of our discussions so we can get this [biosimilar pathway] implemented the right way.”
According to the FDA’s regulations, a biosimilar designation does not allow for nonmedical switching, something that could only happen under a related but distinct designation known as interchangeability. During the committee meeting on July 13, a FDA staffer said that the agency is currently developing guidance for an “interchangeable” designation and plans to have it available before then end of 2016.
On Twitter @mitchelzoler
The Food and Drug Administration’s Arthritis Advisory Committee, together with an added complement of dermatologists and gastroenterologists, unanimously recommended during meetings on July 12 and 13 that the agency license a biosimilar Humira (adalimumab) that is made by Amgen and a biosimilar Enbrel (etanercept) that is made by Sandoz for many of the same indications held by the reference drugs.
The FDA advisory panel that endorsed biosimilar Humira recommended the agent’s approval in a 26-0 vote for: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis in patients at least 4 years old, plaque psoriasis, adult Crohn’s disease, and adult ulcerative colitis. Humira itself is approved for 10 indications.
A slightly different group of 20 advisory panel members (without any gastroenterologists) voted 20-0 in favor of the FDA granting biosimilar Enbrel all five of the indications now held by Enbrel: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis, and plaque psoriasis.
The biosimilar Humira and the biosimilar Enbrel are, respectively, the third and fourth candidate biosimilars to emerge from the FDA’s development program and receive advisory committee scrutiny and support. The first agent through the process, biosimilar filgrastim (Zarxio) received FDA approval in 2015 and is available in the United States. Although the second biosimilar through the process, the tumor necrosis factor inhibitor Inflectra that is biosimilar Remicade (infliximab), received FDA approval in April of this year, it has not yet become available for sale,
A spokeswoman for the company that will market it, Pfizer, said that the company expects to start U.S. sales of Inflectra before the end of 2016.
While the Arthritis Advisory Committee ended each of its daylong deliberations for each of the two candidate biosimilars with unanimous support, the panelists’ discussions among themselves and with FDA staffers reflected some uncertainty with the biosimilar concept, especially during the first day when they focused on biosimilar Humira. The major sticking point revolved around the regulatory pathway to approval first established by the Biologics Price Competition and Innovation Act of 2009 and subsequently refined by the FDA that allows a candidate biosimilar to establish its biosimilarity primarily though the results of analytical studies that establish that the candidate molecule is highly similar to the reference molecule. This approval scheme uses clinical trials in a confirmatory role to establish biosimilarity rather than as the linchpin of approval.
It also means that the FDA can grant clinical indications to the biosimilar drug based not on the results from clinical trials, but based entirely on what have already been demonstrated as safe and effective clinical applications for the reference drug. For example, the biosimilar Humira underwent testing in two clinical studies showing similar efficacy and safety as Humira in patients with rheumatoid arthritis and in patients with plaque psoriasis, but received endorsements based on extrapolations for an additional five indications. Biosimilar Enbrel was compared with Enbrel in patients with plaque psoriasis only and still received extrapolated indications for the additional four rheumatologic conditions.
“This is a new level of extrapolation, across indications,” noted Sarah E. Streett, MD, a gastroenterologist at Stanford (Calif.) University, one of several panelists who initially voiced uncertainty about the concept.
But FDA staffer Nikolay P. Nikolov, MD, who led the agency’s presentation, assured the panelists that the concept of extrapolation was at the heart of biosimilar development and regulatory assessment.
“We have confidence from the data that the two molecules [the reference drug and biosimilar drug] are so similar that we can rely on the safety and efficacy of the reference product. The premise of our approach to biosimilars is that this is not a new molecule that we know nothing about.”
The other uncertainty about biosimilar Humira and biosimilar Enbrel that raised concerns of many panelists were the prospects for nonmedical switching once these drugs reach the market. Nonmedical switching refers to when an insurance company or pharmacy benefit manager substitutes a biosimilar for a reference drug without approval from or even the knowledge of the prescribing physician or the patient. Many of the people who spoke during the public forum period on both days of hearings voiced their concerns about this prospect.
“Nonmedical switching is a major concern of clinicians and policy makers, and we need greater clarification from the FDA,” said committee chair Daniel H. Solomon, MD, a rheumatologist and professor of medicine at Harvard Medical School in Boston.
“I see a remarkable disconnect between the public’s concerns [about nonmedical switching] and the charge to the committee. These are essential issues that need a forum to be aired out,” said panelist Steven F. Solga, MD, chief of gastroenterology at St. Luke’s Hospital in Bethlehem, Pa.
Dr. Nikolov assured committee members that the FDA recognized this concern and was working on it. “We appreciate the disconnect between the charge and the concerns of the community. I assure you that the issues brought up will be part of our discussions so we can get this [biosimilar pathway] implemented the right way.”
According to the FDA’s regulations, a biosimilar designation does not allow for nonmedical switching, something that could only happen under a related but distinct designation known as interchangeability. During the committee meeting on July 13, a FDA staffer said that the agency is currently developing guidance for an “interchangeable” designation and plans to have it available before then end of 2016.
On Twitter @mitchelzoler
The Food and Drug Administration’s Arthritis Advisory Committee, together with an added complement of dermatologists and gastroenterologists, unanimously recommended during meetings on July 12 and 13 that the agency license a biosimilar Humira (adalimumab) that is made by Amgen and a biosimilar Enbrel (etanercept) that is made by Sandoz for many of the same indications held by the reference drugs.
The FDA advisory panel that endorsed biosimilar Humira recommended the agent’s approval in a 26-0 vote for: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis in patients at least 4 years old, plaque psoriasis, adult Crohn’s disease, and adult ulcerative colitis. Humira itself is approved for 10 indications.
A slightly different group of 20 advisory panel members (without any gastroenterologists) voted 20-0 in favor of the FDA granting biosimilar Enbrel all five of the indications now held by Enbrel: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, juvenile idiopathic arthritis, and plaque psoriasis.
The biosimilar Humira and the biosimilar Enbrel are, respectively, the third and fourth candidate biosimilars to emerge from the FDA’s development program and receive advisory committee scrutiny and support. The first agent through the process, biosimilar filgrastim (Zarxio) received FDA approval in 2015 and is available in the United States. Although the second biosimilar through the process, the tumor necrosis factor inhibitor Inflectra that is biosimilar Remicade (infliximab), received FDA approval in April of this year, it has not yet become available for sale,
A spokeswoman for the company that will market it, Pfizer, said that the company expects to start U.S. sales of Inflectra before the end of 2016.
While the Arthritis Advisory Committee ended each of its daylong deliberations for each of the two candidate biosimilars with unanimous support, the panelists’ discussions among themselves and with FDA staffers reflected some uncertainty with the biosimilar concept, especially during the first day when they focused on biosimilar Humira. The major sticking point revolved around the regulatory pathway to approval first established by the Biologics Price Competition and Innovation Act of 2009 and subsequently refined by the FDA that allows a candidate biosimilar to establish its biosimilarity primarily though the results of analytical studies that establish that the candidate molecule is highly similar to the reference molecule. This approval scheme uses clinical trials in a confirmatory role to establish biosimilarity rather than as the linchpin of approval.
It also means that the FDA can grant clinical indications to the biosimilar drug based not on the results from clinical trials, but based entirely on what have already been demonstrated as safe and effective clinical applications for the reference drug. For example, the biosimilar Humira underwent testing in two clinical studies showing similar efficacy and safety as Humira in patients with rheumatoid arthritis and in patients with plaque psoriasis, but received endorsements based on extrapolations for an additional five indications. Biosimilar Enbrel was compared with Enbrel in patients with plaque psoriasis only and still received extrapolated indications for the additional four rheumatologic conditions.
“This is a new level of extrapolation, across indications,” noted Sarah E. Streett, MD, a gastroenterologist at Stanford (Calif.) University, one of several panelists who initially voiced uncertainty about the concept.
But FDA staffer Nikolay P. Nikolov, MD, who led the agency’s presentation, assured the panelists that the concept of extrapolation was at the heart of biosimilar development and regulatory assessment.
“We have confidence from the data that the two molecules [the reference drug and biosimilar drug] are so similar that we can rely on the safety and efficacy of the reference product. The premise of our approach to biosimilars is that this is not a new molecule that we know nothing about.”
The other uncertainty about biosimilar Humira and biosimilar Enbrel that raised concerns of many panelists were the prospects for nonmedical switching once these drugs reach the market. Nonmedical switching refers to when an insurance company or pharmacy benefit manager substitutes a biosimilar for a reference drug without approval from or even the knowledge of the prescribing physician or the patient. Many of the people who spoke during the public forum period on both days of hearings voiced their concerns about this prospect.
“Nonmedical switching is a major concern of clinicians and policy makers, and we need greater clarification from the FDA,” said committee chair Daniel H. Solomon, MD, a rheumatologist and professor of medicine at Harvard Medical School in Boston.
“I see a remarkable disconnect between the public’s concerns [about nonmedical switching] and the charge to the committee. These are essential issues that need a forum to be aired out,” said panelist Steven F. Solga, MD, chief of gastroenterology at St. Luke’s Hospital in Bethlehem, Pa.
Dr. Nikolov assured committee members that the FDA recognized this concern and was working on it. “We appreciate the disconnect between the charge and the concerns of the community. I assure you that the issues brought up will be part of our discussions so we can get this [biosimilar pathway] implemented the right way.”
According to the FDA’s regulations, a biosimilar designation does not allow for nonmedical switching, something that could only happen under a related but distinct designation known as interchangeability. During the committee meeting on July 13, a FDA staffer said that the agency is currently developing guidance for an “interchangeable” designation and plans to have it available before then end of 2016.
On Twitter @mitchelzoler
Study finds emergence of azithromycin-resistant gonorrhea
Resistance to azithromycin and to a lesser degree cephalosporin antibiotics was observed in patients with gonorrhea, according to an analysis of 2014 data from a national surveillance system,
“It is unclear whether these increases mark the beginning of trends, but emergence of cephalosporin and azithromycin resistance would complicate gonorrhea treatment substantially,” reported Robert D. Kirkcaldy, MD, and his colleagues. The results were published July 15 in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report.
Dr. Kirkcaldy of the CDC’s Division of STD Prevention, Atlanta, and his coinvestigators evaluated 2014 data from the Gonococcal Isolate Surveillance Project (GISP), which the CDC established in 1986 to monitor trends in antimicrobial susceptibilities of Neisseri. gonorrhoeae strains in the United States. The N. gonorrhoeae isolates are collected at 27 participating STD clinics each month from up to the first 25 men with gonococcal urethritis who present to the clinics.
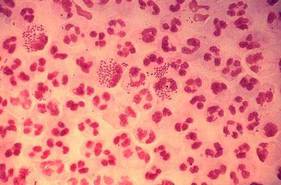
In 2014, a total of 5,093 isolates were collected at the 27 sites. Among these, 25% demonstrated resistance to tetracycline, 19% to ciprofloxacin, and 16% to penicillin. At the same time, resistance to azithromycin increased from 0.6% in 2013 to 2.5% in 2014, predominantly in the Midwest.
Resistance to the cephalosporin antibiotic cefixime, meanwhile, increased from 0.1% in 2006 to 1.4% in 2010 and 2011, fell to 0.4% in 2013, and increased to 0.8% in 2014.
Resistance to the cephalosporin antibiotic ceftriaxone increased from 0.1% in 2008 to 0.4% in 2011, and decreased to 0.1% in 2013 and 2014).
“Local and state health departments can use GISP data to determine allocation of STD prevention services and resources, guide prevention planning, and communicate best treatment practices to health care providers,” the researchers wrote. “Continued surveillance, appropriate treatment, development of new antibiotics, and prevention of transmission remain the best strategies to reduce gonorrhea incidence and morbidity.”
The researchers reported having no financial disclosures.
Resistance to azithromycin and to a lesser degree cephalosporin antibiotics was observed in patients with gonorrhea, according to an analysis of 2014 data from a national surveillance system,
“It is unclear whether these increases mark the beginning of trends, but emergence of cephalosporin and azithromycin resistance would complicate gonorrhea treatment substantially,” reported Robert D. Kirkcaldy, MD, and his colleagues. The results were published July 15 in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report.
Dr. Kirkcaldy of the CDC’s Division of STD Prevention, Atlanta, and his coinvestigators evaluated 2014 data from the Gonococcal Isolate Surveillance Project (GISP), which the CDC established in 1986 to monitor trends in antimicrobial susceptibilities of Neisseri. gonorrhoeae strains in the United States. The N. gonorrhoeae isolates are collected at 27 participating STD clinics each month from up to the first 25 men with gonococcal urethritis who present to the clinics.
In 2014, a total of 5,093 isolates were collected at the 27 sites. Among these, 25% demonstrated resistance to tetracycline, 19% to ciprofloxacin, and 16% to penicillin. At the same time, resistance to azithromycin increased from 0.6% in 2013 to 2.5% in 2014, predominantly in the Midwest.
Resistance to the cephalosporin antibiotic cefixime, meanwhile, increased from 0.1% in 2006 to 1.4% in 2010 and 2011, fell to 0.4% in 2013, and increased to 0.8% in 2014.
Resistance to the cephalosporin antibiotic ceftriaxone increased from 0.1% in 2008 to 0.4% in 2011, and decreased to 0.1% in 2013 and 2014).
“Local and state health departments can use GISP data to determine allocation of STD prevention services and resources, guide prevention planning, and communicate best treatment practices to health care providers,” the researchers wrote. “Continued surveillance, appropriate treatment, development of new antibiotics, and prevention of transmission remain the best strategies to reduce gonorrhea incidence and morbidity.”
The researchers reported having no financial disclosures.
Resistance to azithromycin and to a lesser degree cephalosporin antibiotics was observed in patients with gonorrhea, according to an analysis of 2014 data from a national surveillance system,
“It is unclear whether these increases mark the beginning of trends, but emergence of cephalosporin and azithromycin resistance would complicate gonorrhea treatment substantially,” reported Robert D. Kirkcaldy, MD, and his colleagues. The results were published July 15 in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report.
Dr. Kirkcaldy of the CDC’s Division of STD Prevention, Atlanta, and his coinvestigators evaluated 2014 data from the Gonococcal Isolate Surveillance Project (GISP), which the CDC established in 1986 to monitor trends in antimicrobial susceptibilities of Neisseri. gonorrhoeae strains in the United States. The N. gonorrhoeae isolates are collected at 27 participating STD clinics each month from up to the first 25 men with gonococcal urethritis who present to the clinics.
In 2014, a total of 5,093 isolates were collected at the 27 sites. Among these, 25% demonstrated resistance to tetracycline, 19% to ciprofloxacin, and 16% to penicillin. At the same time, resistance to azithromycin increased from 0.6% in 2013 to 2.5% in 2014, predominantly in the Midwest.
Resistance to the cephalosporin antibiotic cefixime, meanwhile, increased from 0.1% in 2006 to 1.4% in 2010 and 2011, fell to 0.4% in 2013, and increased to 0.8% in 2014.
Resistance to the cephalosporin antibiotic ceftriaxone increased from 0.1% in 2008 to 0.4% in 2011, and decreased to 0.1% in 2013 and 2014).
“Local and state health departments can use GISP data to determine allocation of STD prevention services and resources, guide prevention planning, and communicate best treatment practices to health care providers,” the researchers wrote. “Continued surveillance, appropriate treatment, development of new antibiotics, and prevention of transmission remain the best strategies to reduce gonorrhea incidence and morbidity.”
The researchers reported having no financial disclosures.
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
Key clinical point: Resistance to azithromycin is emerging among patients diagnosed with gonorrhea.
Major finding: Among patients with gonorrhea, resistance to azithromycin increased from 0.6% in 2013 to 2.5% in 2014, predominantly in the Midwest.
Data source: An analysis of 5,093 Neisseria gonorrhoeae isolates from 27 clinics as part of the CDC’s Gonococcal Isolate Surveillance Project.
Disclosures: The researchers had no financial disclosures.
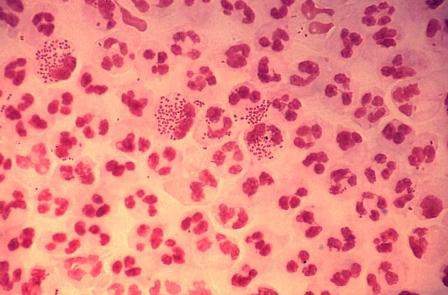
Infections, Antibiotics More Common in Type 2 Diabetes
People with type 2 diabetes are up to 55% more likely to experience hospital-treated infections and up to 30% more likely to receive an antibiotic prescription in the community setting, compared with the general population, but these associations moderated over an 8-year period – a phenomenon that could be at least partly related to better treatment of diabetes and an overall improvement in mean blood glucose levels, according to results of a large population-based study.
“These findings may be driven by earlier detection and treatment of milder type 2 diabetes cases over time,” or by improved therapy of hyperglycemia and other risk factors, wrote Anil Mor, MD, of Aarhus University Hospital, Denmark, and his associates (Clin Infect Dis. 2016 June 26. doi: 10.1093/cid/ciw345).
The study ran from 2004 to 2012 and used data from the Danish National Patient Registry. It tracked community- and hospital-treated infections in approximately 774,017 controls; of these, 155,158 had type 2 diabetes. Patients with diabetes were more likely to have serious medical comorbidities compared with controls (29% vs. 21%). These included myocardial infarction (5% vs. 3%), heart failure (4% vs. 2%), cerebrovascular diseases (7% vs. 5%), peripheral vascular diseases (4% vs. 2%), and chronic pulmonary disease (6% vs. 2%).
Over the study period, 62% of the diabetes patients received an antibiotic, compared with 55% of the controls – a 24% increased relative risk in a model that adjusted for factors such as alcohol use, Charlson comorbidity index, and cardiovascular and renal comorbidities. Cephalosporins were the most commonly prescribed drugs, followed by antimycobacterial agents, quinolones, and antibiotics commonly used for urinary tract and Staphylococcus aureus infections.
Hospital-treated infections were significantly more common among patients with diabetes, with 19% having at least one such infection compared with 13% of controls (RR 1.55). On a larger scale, at a median follow-up of 2.8 years, the hospital-treated rate among diabetes patients was 58 per 1,000 person/years vs. 39 per 1,000 person/years among controls – a relative risk of 1.49.
Patients were at highest risk for emphysematous cholecystitis (adjusted rate ratio 1.74) and abscesses, tuberculosis, and meningococcal infections. Pneumonia was approximately 30% more likely among patients.
The risk of a hospital-treated infection was highest among younger patients aged 40-50 years (RR 1.77) and lowest among those older than 80 years (RR 1.29). It was also higher among those with higher comorbidity scores. Statin use appeared to attenuate some of the risk, the authors noted. The authors did not discuss the possible cause of this association.
The annual risk of a hospital-treated infection among patients declined from a high of 1.89 in 2004 to 1.59 in 2011. The risk of receiving a community-based antibiotic prescription declined as well, from 1.31 in 2004 to 1.26 in 2011.
These changes were not seen in the control group, suggesting that patients with diabetes were experiencing a unique improvement in infections – earlier detection and treatment of type 2 diabetes, and better comorbidity management could be explanations, the investigators speculated.
The study was sponsored by the Danish Centre for Strategic Research in Type 2 Diabetes and the Program for Clinical Research Infrastructure established by the Lundbeck Foundation and the Novo Nordisk Foundation. Several of the coauthors reported financial ties with various pharmaceutical companies.
People with type 2 diabetes are up to 55% more likely to experience hospital-treated infections and up to 30% more likely to receive an antibiotic prescription in the community setting, compared with the general population, but these associations moderated over an 8-year period – a phenomenon that could be at least partly related to better treatment of diabetes and an overall improvement in mean blood glucose levels, according to results of a large population-based study.
“These findings may be driven by earlier detection and treatment of milder type 2 diabetes cases over time,” or by improved therapy of hyperglycemia and other risk factors, wrote Anil Mor, MD, of Aarhus University Hospital, Denmark, and his associates (Clin Infect Dis. 2016 June 26. doi: 10.1093/cid/ciw345).
The study ran from 2004 to 2012 and used data from the Danish National Patient Registry. It tracked community- and hospital-treated infections in approximately 774,017 controls; of these, 155,158 had type 2 diabetes. Patients with diabetes were more likely to have serious medical comorbidities compared with controls (29% vs. 21%). These included myocardial infarction (5% vs. 3%), heart failure (4% vs. 2%), cerebrovascular diseases (7% vs. 5%), peripheral vascular diseases (4% vs. 2%), and chronic pulmonary disease (6% vs. 2%).
Over the study period, 62% of the diabetes patients received an antibiotic, compared with 55% of the controls – a 24% increased relative risk in a model that adjusted for factors such as alcohol use, Charlson comorbidity index, and cardiovascular and renal comorbidities. Cephalosporins were the most commonly prescribed drugs, followed by antimycobacterial agents, quinolones, and antibiotics commonly used for urinary tract and Staphylococcus aureus infections.
Hospital-treated infections were significantly more common among patients with diabetes, with 19% having at least one such infection compared with 13% of controls (RR 1.55). On a larger scale, at a median follow-up of 2.8 years, the hospital-treated rate among diabetes patients was 58 per 1,000 person/years vs. 39 per 1,000 person/years among controls – a relative risk of 1.49.
Patients were at highest risk for emphysematous cholecystitis (adjusted rate ratio 1.74) and abscesses, tuberculosis, and meningococcal infections. Pneumonia was approximately 30% more likely among patients.
The risk of a hospital-treated infection was highest among younger patients aged 40-50 years (RR 1.77) and lowest among those older than 80 years (RR 1.29). It was also higher among those with higher comorbidity scores. Statin use appeared to attenuate some of the risk, the authors noted. The authors did not discuss the possible cause of this association.
The annual risk of a hospital-treated infection among patients declined from a high of 1.89 in 2004 to 1.59 in 2011. The risk of receiving a community-based antibiotic prescription declined as well, from 1.31 in 2004 to 1.26 in 2011.
These changes were not seen in the control group, suggesting that patients with diabetes were experiencing a unique improvement in infections – earlier detection and treatment of type 2 diabetes, and better comorbidity management could be explanations, the investigators speculated.
The study was sponsored by the Danish Centre for Strategic Research in Type 2 Diabetes and the Program for Clinical Research Infrastructure established by the Lundbeck Foundation and the Novo Nordisk Foundation. Several of the coauthors reported financial ties with various pharmaceutical companies.
People with type 2 diabetes are up to 55% more likely to experience hospital-treated infections and up to 30% more likely to receive an antibiotic prescription in the community setting, compared with the general population, but these associations moderated over an 8-year period – a phenomenon that could be at least partly related to better treatment of diabetes and an overall improvement in mean blood glucose levels, according to results of a large population-based study.
“These findings may be driven by earlier detection and treatment of milder type 2 diabetes cases over time,” or by improved therapy of hyperglycemia and other risk factors, wrote Anil Mor, MD, of Aarhus University Hospital, Denmark, and his associates (Clin Infect Dis. 2016 June 26. doi: 10.1093/cid/ciw345).
The study ran from 2004 to 2012 and used data from the Danish National Patient Registry. It tracked community- and hospital-treated infections in approximately 774,017 controls; of these, 155,158 had type 2 diabetes. Patients with diabetes were more likely to have serious medical comorbidities compared with controls (29% vs. 21%). These included myocardial infarction (5% vs. 3%), heart failure (4% vs. 2%), cerebrovascular diseases (7% vs. 5%), peripheral vascular diseases (4% vs. 2%), and chronic pulmonary disease (6% vs. 2%).
Over the study period, 62% of the diabetes patients received an antibiotic, compared with 55% of the controls – a 24% increased relative risk in a model that adjusted for factors such as alcohol use, Charlson comorbidity index, and cardiovascular and renal comorbidities. Cephalosporins were the most commonly prescribed drugs, followed by antimycobacterial agents, quinolones, and antibiotics commonly used for urinary tract and Staphylococcus aureus infections.
Hospital-treated infections were significantly more common among patients with diabetes, with 19% having at least one such infection compared with 13% of controls (RR 1.55). On a larger scale, at a median follow-up of 2.8 years, the hospital-treated rate among diabetes patients was 58 per 1,000 person/years vs. 39 per 1,000 person/years among controls – a relative risk of 1.49.
Patients were at highest risk for emphysematous cholecystitis (adjusted rate ratio 1.74) and abscesses, tuberculosis, and meningococcal infections. Pneumonia was approximately 30% more likely among patients.
The risk of a hospital-treated infection was highest among younger patients aged 40-50 years (RR 1.77) and lowest among those older than 80 years (RR 1.29). It was also higher among those with higher comorbidity scores. Statin use appeared to attenuate some of the risk, the authors noted. The authors did not discuss the possible cause of this association.
The annual risk of a hospital-treated infection among patients declined from a high of 1.89 in 2004 to 1.59 in 2011. The risk of receiving a community-based antibiotic prescription declined as well, from 1.31 in 2004 to 1.26 in 2011.
These changes were not seen in the control group, suggesting that patients with diabetes were experiencing a unique improvement in infections – earlier detection and treatment of type 2 diabetes, and better comorbidity management could be explanations, the investigators speculated.
The study was sponsored by the Danish Centre for Strategic Research in Type 2 Diabetes and the Program for Clinical Research Infrastructure established by the Lundbeck Foundation and the Novo Nordisk Foundation. Several of the coauthors reported financial ties with various pharmaceutical companies.
FROM CLINICAL INFECTIOUS DISEASES
Infections, antibiotics more common in type 2 diabetes
People with type 2 diabetes are up to 55% more likely to experience hospital-treated infections and up to 30% more likely to receive an antibiotic prescription in the community setting, compared with the general population, but these associations moderated over an 8-year period – a phenomenon that could be at least partly related to better treatment of diabetes and an overall improvement in mean blood glucose levels, according to results of a large population-based study.
“These findings may be driven by earlier detection and treatment of milder type 2 diabetes cases over time,” or by improved therapy of hyperglycemia and other risk factors, wrote Anil Mor, MD, of Aarhus University Hospital, Denmark, and his associates (Clin Infect Dis. 2016 June 26. doi: 10.1093/cid/ciw345).

The study ran from 2004 to 2012 and used data from the Danish National Patient Registry. It tracked community- and hospital-treated infections in approximately 774,017 controls; of these, 155,158 had type 2 diabetes. Patients with diabetes were more likely to have serious medical comorbidities compared with controls (29% vs. 21%). These included myocardial infarction (5% vs. 3%), heart failure (4% vs. 2%), cerebrovascular diseases (7% vs. 5%), peripheral vascular diseases (4% vs. 2%), and chronic pulmonary disease (6% vs. 2%).
Over the study period, 62% of the diabetes patients received an antibiotic, compared with 55% of the controls – a 24% increased relative risk in a model that adjusted for factors such as alcohol use, Charlson comorbidity index, and cardiovascular and renal comorbidities. Cephalosporins were the most commonly prescribed drugs, followed by antimycobacterial agents, quinolones, and antibiotics commonly used for urinary tract and Staphylococcus aureus infections.
Hospital-treated infections were significantly more common among patients with diabetes, with 19% having at least one such infection compared with 13% of controls (RR 1.55). On a larger scale, at a median follow-up of 2.8 years, the hospital-treated rate among diabetes patients was 58 per 1,000 person/years vs. 39 per 1,000 person/years among controls – a relative risk of 1.49.
Patients were at highest risk for emphysematous cholecystitis (adjusted rate ratio 1.74) and abscesses, tuberculosis, and meningococcal infections. Pneumonia was approximately 30% more likely among patients.
The risk of a hospital-treated infection was highest among younger patients aged 40-50 years (RR 1.77) and lowest among those older than 80 years (RR 1.29). It was also higher among those with higher comorbidity scores. Statin use appeared to attenuate some of the risk, the authors noted. The authors did not discuss the possible cause of this association.
The annual risk of a hospital-treated infection among patients declined from a high of 1.89 in 2004 to 1.59 in 2011. The risk of receiving a community-based antibiotic prescription declined as well, from 1.31 in 2004 to 1.26 in 2011.
These changes were not seen in the control group, suggesting that patients with diabetes were experiencing a unique improvement in infections – earlier detection and treatment of type 2 diabetes, and better comorbidity management could be explanations, the investigators speculated.
The study was sponsored by the Danish Centre for Strategic Research in Type 2 Diabetes and the Program for Clinical Research Infrastructure established by the Lundbeck Foundation and the Novo Nordisk Foundation. Several of the coauthors reported financial ties with various pharmaceutical companies.
On Twitter @Alz_Gal
People with type 2 diabetes are up to 55% more likely to experience hospital-treated infections and up to 30% more likely to receive an antibiotic prescription in the community setting, compared with the general population, but these associations moderated over an 8-year period – a phenomenon that could be at least partly related to better treatment of diabetes and an overall improvement in mean blood glucose levels, according to results of a large population-based study.
“These findings may be driven by earlier detection and treatment of milder type 2 diabetes cases over time,” or by improved therapy of hyperglycemia and other risk factors, wrote Anil Mor, MD, of Aarhus University Hospital, Denmark, and his associates (Clin Infect Dis. 2016 June 26. doi: 10.1093/cid/ciw345).
The study ran from 2004 to 2012 and used data from the Danish National Patient Registry. It tracked community- and hospital-treated infections in approximately 774,017 controls; of these, 155,158 had type 2 diabetes. Patients with diabetes were more likely to have serious medical comorbidities compared with controls (29% vs. 21%). These included myocardial infarction (5% vs. 3%), heart failure (4% vs. 2%), cerebrovascular diseases (7% vs. 5%), peripheral vascular diseases (4% vs. 2%), and chronic pulmonary disease (6% vs. 2%).
Over the study period, 62% of the diabetes patients received an antibiotic, compared with 55% of the controls – a 24% increased relative risk in a model that adjusted for factors such as alcohol use, Charlson comorbidity index, and cardiovascular and renal comorbidities. Cephalosporins were the most commonly prescribed drugs, followed by antimycobacterial agents, quinolones, and antibiotics commonly used for urinary tract and Staphylococcus aureus infections.
Hospital-treated infections were significantly more common among patients with diabetes, with 19% having at least one such infection compared with 13% of controls (RR 1.55). On a larger scale, at a median follow-up of 2.8 years, the hospital-treated rate among diabetes patients was 58 per 1,000 person/years vs. 39 per 1,000 person/years among controls – a relative risk of 1.49.
Patients were at highest risk for emphysematous cholecystitis (adjusted rate ratio 1.74) and abscesses, tuberculosis, and meningococcal infections. Pneumonia was approximately 30% more likely among patients.
The risk of a hospital-treated infection was highest among younger patients aged 40-50 years (RR 1.77) and lowest among those older than 80 years (RR 1.29). It was also higher among those with higher comorbidity scores. Statin use appeared to attenuate some of the risk, the authors noted. The authors did not discuss the possible cause of this association.
The annual risk of a hospital-treated infection among patients declined from a high of 1.89 in 2004 to 1.59 in 2011. The risk of receiving a community-based antibiotic prescription declined as well, from 1.31 in 2004 to 1.26 in 2011.
These changes were not seen in the control group, suggesting that patients with diabetes were experiencing a unique improvement in infections – earlier detection and treatment of type 2 diabetes, and better comorbidity management could be explanations, the investigators speculated.
The study was sponsored by the Danish Centre for Strategic Research in Type 2 Diabetes and the Program for Clinical Research Infrastructure established by the Lundbeck Foundation and the Novo Nordisk Foundation. Several of the coauthors reported financial ties with various pharmaceutical companies.
On Twitter @Alz_Gal
People with type 2 diabetes are up to 55% more likely to experience hospital-treated infections and up to 30% more likely to receive an antibiotic prescription in the community setting, compared with the general population, but these associations moderated over an 8-year period – a phenomenon that could be at least partly related to better treatment of diabetes and an overall improvement in mean blood glucose levels, according to results of a large population-based study.
“These findings may be driven by earlier detection and treatment of milder type 2 diabetes cases over time,” or by improved therapy of hyperglycemia and other risk factors, wrote Anil Mor, MD, of Aarhus University Hospital, Denmark, and his associates (Clin Infect Dis. 2016 June 26. doi: 10.1093/cid/ciw345).
The study ran from 2004 to 2012 and used data from the Danish National Patient Registry. It tracked community- and hospital-treated infections in approximately 774,017 controls; of these, 155,158 had type 2 diabetes. Patients with diabetes were more likely to have serious medical comorbidities compared with controls (29% vs. 21%). These included myocardial infarction (5% vs. 3%), heart failure (4% vs. 2%), cerebrovascular diseases (7% vs. 5%), peripheral vascular diseases (4% vs. 2%), and chronic pulmonary disease (6% vs. 2%).
Over the study period, 62% of the diabetes patients received an antibiotic, compared with 55% of the controls – a 24% increased relative risk in a model that adjusted for factors such as alcohol use, Charlson comorbidity index, and cardiovascular and renal comorbidities. Cephalosporins were the most commonly prescribed drugs, followed by antimycobacterial agents, quinolones, and antibiotics commonly used for urinary tract and Staphylococcus aureus infections.
Hospital-treated infections were significantly more common among patients with diabetes, with 19% having at least one such infection compared with 13% of controls (RR 1.55). On a larger scale, at a median follow-up of 2.8 years, the hospital-treated rate among diabetes patients was 58 per 1,000 person/years vs. 39 per 1,000 person/years among controls – a relative risk of 1.49.
Patients were at highest risk for emphysematous cholecystitis (adjusted rate ratio 1.74) and abscesses, tuberculosis, and meningococcal infections. Pneumonia was approximately 30% more likely among patients.
The risk of a hospital-treated infection was highest among younger patients aged 40-50 years (RR 1.77) and lowest among those older than 80 years (RR 1.29). It was also higher among those with higher comorbidity scores. Statin use appeared to attenuate some of the risk, the authors noted. The authors did not discuss the possible cause of this association.
The annual risk of a hospital-treated infection among patients declined from a high of 1.89 in 2004 to 1.59 in 2011. The risk of receiving a community-based antibiotic prescription declined as well, from 1.31 in 2004 to 1.26 in 2011.
These changes were not seen in the control group, suggesting that patients with diabetes were experiencing a unique improvement in infections – earlier detection and treatment of type 2 diabetes, and better comorbidity management could be explanations, the investigators speculated.
The study was sponsored by the Danish Centre for Strategic Research in Type 2 Diabetes and the Program for Clinical Research Infrastructure established by the Lundbeck Foundation and the Novo Nordisk Foundation. Several of the coauthors reported financial ties with various pharmaceutical companies.
On Twitter @Alz_Gal
FROM CLINICAL INFECTIOUS DISEASES
Key clinical point: Both hospital-treated infections and community-acquired antibiotics are more common among people with type 2 diabetes
Major finding: Hospital-treated infections were 55% more likely; community-dispensed antibiotics, 30% more common.
Data source: An observational study comprising almost 900,000 people in Denmark.
Disclosures: The study was sponsored by the Danish Centre for Strategic Research in Type 2 Diabetes and the Program for Clinical Research Infrastructure established by the Lundbeck Foundation and the Novo Nordisk Foundation. Several coauthors reported financial ties with various pharmaceutical companies.
PTSD, Eating Disorders Tied to Suppressing Negative Emotions
A pathway from posttraumatic stress disorder to eating disorders might be through the maladaptive coping mechanism of expressive suppression, a study of 860 older veterans shows.
“Expressive suppression reflects attempts to reduce outward expression of emotion and may be adaptive in the short term; however, this strategy becomes less effective over the long term,” wrote Karen S. Mitchell, PhD, and Erika J. Wolf, PhD., both of the National Center for PTSD, VA Boston Healthcare System. “This finding aligns with previous ... work suggesting that in some individuals, [eating disorder] symptoms may be used to cope with negative affect.”
“We weren’t surprised by the findings,” Dr. Mitchell said in an interview. “They were consistent with our hypothesis that for some people, disordered eating may be used to cope with PTSD symptoms.”
She said the clinical implications of the study are direct. “It is important to assess eating habits and other potentially harmful coping strategies among patients with trauma histories and PTSD. On the other hand, it would be helpful to assess trauma histories in patients with eating disorders to determine if trauma reminders or other PTSD symptoms serve as a maintaining factor for the eating disorder.”
Dr. Mitchell and Dr. Wolf randomly selected 1,126 veterans who had reported trauma exposure in the GfK Knowledge Networks study (Depress Anxiety. 2013 May;30[5]:432-4), which looked at psychological resilience in a sample of U.S. veterans aged 60 and older. The veterans in the randomly selected group were asked to participate in a survey about PTSD, dissociation, and disordered eating, and 860 responded, 787 of whom were men (Psychiatry Res. 2016;243:23-9).
The participants’ age range was 22-96 years; mean age was 63. Most were white (85%) and married (77%), and had attended some college (87%).
Participants were asked to complete several surveys, including the Eating Disorder Diagnostic Scale (EDDS), the Yale Food Addiction Scale, the Emotion Regulation Questionnaire, and the National Stressful Events Survey (NSES).
The investigators said the sudden, unexpected death of a loved one (17.5%), and the aftermath of combat (13.6%) were cited as the worst traumatic experiences most cited by the participants. They also cited the violent death of a loved one (7.3%), the witnessing of dead bodies or body parts (6.3%), and physical or sexual assault (5.5%) as traumatic experiences.
Meanwhile, 23 participants (18 men [2.8%] and 5 women [9.1%]) met the criteria for bulimia nervosa; 20 met the criteria for binge eating disorder (16 men [2.5%] and 4 women [7.3%]); and 16 people (12 men [1.9%] and 4 women [7.3%]) met the proposed criteria for food addiction, reported Dr. Mitchell and Dr. Wolf, who also both are affiliated with the department of psychiatry at Boston University.
After analyzing the data, the researchers found that “the indirect path from PTSD to expressive suppression to [eating disorder] symptoms was significant in the full sample (P = .002; 95% confidence interval, 0.069-0.314) and in the male subsample (P = .029; 95% CI, 0.014-0.255).”
The researchers cited several limitations. For example, the participants’ diagnoses were not confirmed by interviews. In addition, “the validity of DSM-5 diagnoses using the EDDS and NSES has not yet been established,” they wrote.
Nevertheless, Dr. Mitchell and Dr. Wolf wrote, their findings highlight the importance of looking at eating disorders and food addiction in populations that traditionally have been underserved.
The study was funded by National Institutes of Health grant and an award from the Department of Veterans Affairs.
A pathway from posttraumatic stress disorder to eating disorders might be through the maladaptive coping mechanism of expressive suppression, a study of 860 older veterans shows.
“Expressive suppression reflects attempts to reduce outward expression of emotion and may be adaptive in the short term; however, this strategy becomes less effective over the long term,” wrote Karen S. Mitchell, PhD, and Erika J. Wolf, PhD., both of the National Center for PTSD, VA Boston Healthcare System. “This finding aligns with previous ... work suggesting that in some individuals, [eating disorder] symptoms may be used to cope with negative affect.”
“We weren’t surprised by the findings,” Dr. Mitchell said in an interview. “They were consistent with our hypothesis that for some people, disordered eating may be used to cope with PTSD symptoms.”
She said the clinical implications of the study are direct. “It is important to assess eating habits and other potentially harmful coping strategies among patients with trauma histories and PTSD. On the other hand, it would be helpful to assess trauma histories in patients with eating disorders to determine if trauma reminders or other PTSD symptoms serve as a maintaining factor for the eating disorder.”
Dr. Mitchell and Dr. Wolf randomly selected 1,126 veterans who had reported trauma exposure in the GfK Knowledge Networks study (Depress Anxiety. 2013 May;30[5]:432-4), which looked at psychological resilience in a sample of U.S. veterans aged 60 and older. The veterans in the randomly selected group were asked to participate in a survey about PTSD, dissociation, and disordered eating, and 860 responded, 787 of whom were men (Psychiatry Res. 2016;243:23-9).
The participants’ age range was 22-96 years; mean age was 63. Most were white (85%) and married (77%), and had attended some college (87%).
Participants were asked to complete several surveys, including the Eating Disorder Diagnostic Scale (EDDS), the Yale Food Addiction Scale, the Emotion Regulation Questionnaire, and the National Stressful Events Survey (NSES).
The investigators said the sudden, unexpected death of a loved one (17.5%), and the aftermath of combat (13.6%) were cited as the worst traumatic experiences most cited by the participants. They also cited the violent death of a loved one (7.3%), the witnessing of dead bodies or body parts (6.3%), and physical or sexual assault (5.5%) as traumatic experiences.
Meanwhile, 23 participants (18 men [2.8%] and 5 women [9.1%]) met the criteria for bulimia nervosa; 20 met the criteria for binge eating disorder (16 men [2.5%] and 4 women [7.3%]); and 16 people (12 men [1.9%] and 4 women [7.3%]) met the proposed criteria for food addiction, reported Dr. Mitchell and Dr. Wolf, who also both are affiliated with the department of psychiatry at Boston University.
After analyzing the data, the researchers found that “the indirect path from PTSD to expressive suppression to [eating disorder] symptoms was significant in the full sample (P = .002; 95% confidence interval, 0.069-0.314) and in the male subsample (P = .029; 95% CI, 0.014-0.255).”
The researchers cited several limitations. For example, the participants’ diagnoses were not confirmed by interviews. In addition, “the validity of DSM-5 diagnoses using the EDDS and NSES has not yet been established,” they wrote.
Nevertheless, Dr. Mitchell and Dr. Wolf wrote, their findings highlight the importance of looking at eating disorders and food addiction in populations that traditionally have been underserved.
The study was funded by National Institutes of Health grant and an award from the Department of Veterans Affairs.
A pathway from posttraumatic stress disorder to eating disorders might be through the maladaptive coping mechanism of expressive suppression, a study of 860 older veterans shows.
“Expressive suppression reflects attempts to reduce outward expression of emotion and may be adaptive in the short term; however, this strategy becomes less effective over the long term,” wrote Karen S. Mitchell, PhD, and Erika J. Wolf, PhD., both of the National Center for PTSD, VA Boston Healthcare System. “This finding aligns with previous ... work suggesting that in some individuals, [eating disorder] symptoms may be used to cope with negative affect.”
“We weren’t surprised by the findings,” Dr. Mitchell said in an interview. “They were consistent with our hypothesis that for some people, disordered eating may be used to cope with PTSD symptoms.”
She said the clinical implications of the study are direct. “It is important to assess eating habits and other potentially harmful coping strategies among patients with trauma histories and PTSD. On the other hand, it would be helpful to assess trauma histories in patients with eating disorders to determine if trauma reminders or other PTSD symptoms serve as a maintaining factor for the eating disorder.”
Dr. Mitchell and Dr. Wolf randomly selected 1,126 veterans who had reported trauma exposure in the GfK Knowledge Networks study (Depress Anxiety. 2013 May;30[5]:432-4), which looked at psychological resilience in a sample of U.S. veterans aged 60 and older. The veterans in the randomly selected group were asked to participate in a survey about PTSD, dissociation, and disordered eating, and 860 responded, 787 of whom were men (Psychiatry Res. 2016;243:23-9).
The participants’ age range was 22-96 years; mean age was 63. Most were white (85%) and married (77%), and had attended some college (87%).
Participants were asked to complete several surveys, including the Eating Disorder Diagnostic Scale (EDDS), the Yale Food Addiction Scale, the Emotion Regulation Questionnaire, and the National Stressful Events Survey (NSES).
The investigators said the sudden, unexpected death of a loved one (17.5%), and the aftermath of combat (13.6%) were cited as the worst traumatic experiences most cited by the participants. They also cited the violent death of a loved one (7.3%), the witnessing of dead bodies or body parts (6.3%), and physical or sexual assault (5.5%) as traumatic experiences.
Meanwhile, 23 participants (18 men [2.8%] and 5 women [9.1%]) met the criteria for bulimia nervosa; 20 met the criteria for binge eating disorder (16 men [2.5%] and 4 women [7.3%]); and 16 people (12 men [1.9%] and 4 women [7.3%]) met the proposed criteria for food addiction, reported Dr. Mitchell and Dr. Wolf, who also both are affiliated with the department of psychiatry at Boston University.
After analyzing the data, the researchers found that “the indirect path from PTSD to expressive suppression to [eating disorder] symptoms was significant in the full sample (P = .002; 95% confidence interval, 0.069-0.314) and in the male subsample (P = .029; 95% CI, 0.014-0.255).”
The researchers cited several limitations. For example, the participants’ diagnoses were not confirmed by interviews. In addition, “the validity of DSM-5 diagnoses using the EDDS and NSES has not yet been established,” they wrote.
Nevertheless, Dr. Mitchell and Dr. Wolf wrote, their findings highlight the importance of looking at eating disorders and food addiction in populations that traditionally have been underserved.
The study was funded by National Institutes of Health grant and an award from the Department of Veterans Affairs.
FROM PSYCHIATRY RESEARCH