User login
PSA screens could be cost effective if low-risk cases went untreated
Given current treatment practices, prostate-specific antigen (PSA) screening is not cost effective unless performed every 4 years in men aged 55-69 years, and with a biopsy threshold of 10.0 ng/mL, researchers reported online in JAMA Oncology.
But several less conservative testing strategies could be cost effective if patients with Gleason scores under 7 and clinical T2a stage cancer or lower are not treated unless they clinically progress, said Joshua A. Roth, Ph.D., of Fred Hutchinson Cancer Research Center in Seattle and his associates.

The study has “clear implications for the future of PSA screening in the United States,” the investigators wrote (JAMA Oncol. Mar 24. doi: 10.1001/jamaoncol.2015.6275). “Rather than stopping PSA screening, as recommended by the U.S. Preventive Services Task Force, implementation of strategies that extend the screening interval and/or use higher PSA biopsy thresholds have the potential to preserve substantial benefit, while controlling harm and costs.”
The investigators constructed a hypothetical group of men in the United States who underwent 18 different PSA screening strategies starting at age 40. Under the current treatment paradigm, PSA screening increased years of life by 3%-6%, with a cost of $7,335-$21,649 for each extra year of life. Quality years of life rose only if the strategy included a narrower age range for testing or a biopsy threshold of 10.0 ng/mL.
If the more selective treatment model was used, screening 55- to 69-year-old men every 4 years and using a PSA biopsy threshold of 3.0 ng/mL was not only potentially cost effective, but also increased quality years of life. The same was true for quadrennial screening of men aged 50-74 years with a biopsy threshold of 4.0 ng/mL.
“Our work adds to a growing consensus that highly conservative use of the PSA test and biopsy referral is necessary if PSA screening is to be cost effective,” the researchers concluded. Less frequent screening and stricter biopsy criteria for biopsy were most likely to make screening cost effective, especially if physicians do not immediately treat low-risk cases, they added.
The study was supported by the National Cancer Institute, the Centers for Disease Control and Prevention, and the Agency for Healthcare Research and Quality. The investigators had no disclosures.
This study forces us to change the debate from “Should we screen?” to “How can we get physicians to follow best practice?” I have heard it said that the professional, financial, and malpractice incentives to screen and then treat low-risk cancer are too overwhelming to allow for significant practice change. But this is clearly disconfirmed by the literature. Use of active surveillance for low-risk prostate cancer has increased fourfold in the past few years; PSA testing in older men has also fallen recently. Rates of unnecessary treatment still remain much too high (about 60%) as does screening of older men (about 35% for those aged 75 years and older). More work needs to be done, and much more change needs to happen.

|
Andrew J. Vickers, Ph.D. |
Based on these results, if we follow the literature on how to screen with PSA and which screen-detected prostate cancers to treat, we will likely do more good than harm. If we simply carry on with common practice – screening older men, aggressively treating low-risk disease – then we should call for PSA screening to end.
Andrew J. Vickers, Ph.D., is at Memorial Sloan Kettering Cancer Center, New York. He reported being named on a patent application for a statistical method to detect prostate cancer; receiving royalties from sales of the test; and having stock options in OPKO Health, which commercialized the test. These comments are from his editorial (JAMA Oncol. 2016 Mar 24 doi: 10.1001/jamaoncol.2015.6276).
This study forces us to change the debate from “Should we screen?” to “How can we get physicians to follow best practice?” I have heard it said that the professional, financial, and malpractice incentives to screen and then treat low-risk cancer are too overwhelming to allow for significant practice change. But this is clearly disconfirmed by the literature. Use of active surveillance for low-risk prostate cancer has increased fourfold in the past few years; PSA testing in older men has also fallen recently. Rates of unnecessary treatment still remain much too high (about 60%) as does screening of older men (about 35% for those aged 75 years and older). More work needs to be done, and much more change needs to happen.
|
|
Andrew J. Vickers, Ph.D. |
Based on these results, if we follow the literature on how to screen with PSA and which screen-detected prostate cancers to treat, we will likely do more good than harm. If we simply carry on with common practice – screening older men, aggressively treating low-risk disease – then we should call for PSA screening to end.
Andrew J. Vickers, Ph.D., is at Memorial Sloan Kettering Cancer Center, New York. He reported being named on a patent application for a statistical method to detect prostate cancer; receiving royalties from sales of the test; and having stock options in OPKO Health, which commercialized the test. These comments are from his editorial (JAMA Oncol. 2016 Mar 24 doi: 10.1001/jamaoncol.2015.6276).
This study forces us to change the debate from “Should we screen?” to “How can we get physicians to follow best practice?” I have heard it said that the professional, financial, and malpractice incentives to screen and then treat low-risk cancer are too overwhelming to allow for significant practice change. But this is clearly disconfirmed by the literature. Use of active surveillance for low-risk prostate cancer has increased fourfold in the past few years; PSA testing in older men has also fallen recently. Rates of unnecessary treatment still remain much too high (about 60%) as does screening of older men (about 35% for those aged 75 years and older). More work needs to be done, and much more change needs to happen.
|
|
Andrew J. Vickers, Ph.D. |
Based on these results, if we follow the literature on how to screen with PSA and which screen-detected prostate cancers to treat, we will likely do more good than harm. If we simply carry on with common practice – screening older men, aggressively treating low-risk disease – then we should call for PSA screening to end.
Andrew J. Vickers, Ph.D., is at Memorial Sloan Kettering Cancer Center, New York. He reported being named on a patent application for a statistical method to detect prostate cancer; receiving royalties from sales of the test; and having stock options in OPKO Health, which commercialized the test. These comments are from his editorial (JAMA Oncol. 2016 Mar 24 doi: 10.1001/jamaoncol.2015.6276).
Given current treatment practices, prostate-specific antigen (PSA) screening is not cost effective unless performed every 4 years in men aged 55-69 years, and with a biopsy threshold of 10.0 ng/mL, researchers reported online in JAMA Oncology.
But several less conservative testing strategies could be cost effective if patients with Gleason scores under 7 and clinical T2a stage cancer or lower are not treated unless they clinically progress, said Joshua A. Roth, Ph.D., of Fred Hutchinson Cancer Research Center in Seattle and his associates.
The study has “clear implications for the future of PSA screening in the United States,” the investigators wrote (JAMA Oncol. Mar 24. doi: 10.1001/jamaoncol.2015.6275). “Rather than stopping PSA screening, as recommended by the U.S. Preventive Services Task Force, implementation of strategies that extend the screening interval and/or use higher PSA biopsy thresholds have the potential to preserve substantial benefit, while controlling harm and costs.”
The investigators constructed a hypothetical group of men in the United States who underwent 18 different PSA screening strategies starting at age 40. Under the current treatment paradigm, PSA screening increased years of life by 3%-6%, with a cost of $7,335-$21,649 for each extra year of life. Quality years of life rose only if the strategy included a narrower age range for testing or a biopsy threshold of 10.0 ng/mL.
If the more selective treatment model was used, screening 55- to 69-year-old men every 4 years and using a PSA biopsy threshold of 3.0 ng/mL was not only potentially cost effective, but also increased quality years of life. The same was true for quadrennial screening of men aged 50-74 years with a biopsy threshold of 4.0 ng/mL.
“Our work adds to a growing consensus that highly conservative use of the PSA test and biopsy referral is necessary if PSA screening is to be cost effective,” the researchers concluded. Less frequent screening and stricter biopsy criteria for biopsy were most likely to make screening cost effective, especially if physicians do not immediately treat low-risk cases, they added.
The study was supported by the National Cancer Institute, the Centers for Disease Control and Prevention, and the Agency for Healthcare Research and Quality. The investigators had no disclosures.
Given current treatment practices, prostate-specific antigen (PSA) screening is not cost effective unless performed every 4 years in men aged 55-69 years, and with a biopsy threshold of 10.0 ng/mL, researchers reported online in JAMA Oncology.
But several less conservative testing strategies could be cost effective if patients with Gleason scores under 7 and clinical T2a stage cancer or lower are not treated unless they clinically progress, said Joshua A. Roth, Ph.D., of Fred Hutchinson Cancer Research Center in Seattle and his associates.
The study has “clear implications for the future of PSA screening in the United States,” the investigators wrote (JAMA Oncol. Mar 24. doi: 10.1001/jamaoncol.2015.6275). “Rather than stopping PSA screening, as recommended by the U.S. Preventive Services Task Force, implementation of strategies that extend the screening interval and/or use higher PSA biopsy thresholds have the potential to preserve substantial benefit, while controlling harm and costs.”
The investigators constructed a hypothetical group of men in the United States who underwent 18 different PSA screening strategies starting at age 40. Under the current treatment paradigm, PSA screening increased years of life by 3%-6%, with a cost of $7,335-$21,649 for each extra year of life. Quality years of life rose only if the strategy included a narrower age range for testing or a biopsy threshold of 10.0 ng/mL.
If the more selective treatment model was used, screening 55- to 69-year-old men every 4 years and using a PSA biopsy threshold of 3.0 ng/mL was not only potentially cost effective, but also increased quality years of life. The same was true for quadrennial screening of men aged 50-74 years with a biopsy threshold of 4.0 ng/mL.
“Our work adds to a growing consensus that highly conservative use of the PSA test and biopsy referral is necessary if PSA screening is to be cost effective,” the researchers concluded. Less frequent screening and stricter biopsy criteria for biopsy were most likely to make screening cost effective, especially if physicians do not immediately treat low-risk cases, they added.
The study was supported by the National Cancer Institute, the Centers for Disease Control and Prevention, and the Agency for Healthcare Research and Quality. The investigators had no disclosures.
FROM JAMA ONCOLOGY
Key clinical point: A modeling study found that screening for prostate-specific antigen could be cost effective if low-risk cases are not treated unless they progress.
Major finding: Screening 55- to 69-year-old men every 4 years and using a PSA biopsy threshold of 3.0 ng/mL was potentially cost effective and also increased quality years of life. The same was true for quadrennial screening of men aged 50-74 years with a biopsy threshold of 4.0 ng/mL.
Data source: A microsimulation model of prostate cancer incidence and mortality.
Disclosures: The study was supported by the National Cancer Institute, the Centers for Disease Control and Prevention, and the Agency for Healthcare Research and Quality. The investigators had no disclosures.

U.S. flu activity continues downward trend
Influenza-like illness (ILI) activity in the United States continued to drop during the week ending March 26, 2016, with only one state still at the highest level, according to the Centers for Disease Control and Prevention.
That one state was New Jersey, which was at level 10 on the CDC’s 1-10 scale of ILI activity. One state at level 10 was down from three states the week before and seven states 2 weeks earlier. Also down for a second consecutive week was the proportion of outpatient visits for ILI, which was 2.9% for the most recent week, compared with 3.2% the previous week and a season high of 3.7% for the week ending March 12, the CDC’s Influenza-like Illness Surveillance Network (ILINet) reported.
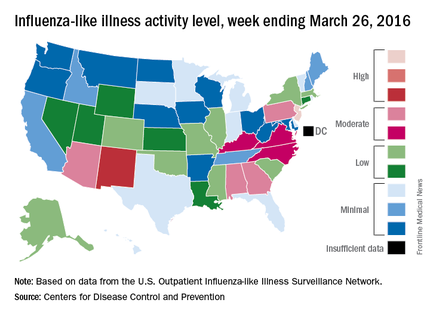
The only other state in the “high” range for the week ending March 26 was New Mexico at level 8. States in the “moderate” range were Alabama, Arizona, Georgia, and Pennsylvania at level 7 and Kentucky, North Carolina, and Virginia at level 6, according to data from ILINet.
Three flu-related pediatric deaths were reported to CDC during the week, but two occurred the previous week and one occurred in February. That brings the total of flu-related pediatric deaths to 33 for the 2015-2016 influenza season, the CDC said. However, 7.7% of all deaths reported through the 122 Cities Mortality Reporting System were due to pneumonia and influenza. This percentage was above the epidemic threshold of 7.2%.
The CDC also reported a cumulative rate for the season of 21.4 laboratory-confirmed influenza-associated hospitalizations per 100,000 population. The highest rate of hospitalization was among adults aged 65 years or older (54.5 per 100,000 population), followed by adults aged 50-64 (31.4 per 100,000 population) and children aged 0-4 years (29.3 per 100,000 population).
Influenza-like illness (ILI) activity in the United States continued to drop during the week ending March 26, 2016, with only one state still at the highest level, according to the Centers for Disease Control and Prevention.
That one state was New Jersey, which was at level 10 on the CDC’s 1-10 scale of ILI activity. One state at level 10 was down from three states the week before and seven states 2 weeks earlier. Also down for a second consecutive week was the proportion of outpatient visits for ILI, which was 2.9% for the most recent week, compared with 3.2% the previous week and a season high of 3.7% for the week ending March 12, the CDC’s Influenza-like Illness Surveillance Network (ILINet) reported.
The only other state in the “high” range for the week ending March 26 was New Mexico at level 8. States in the “moderate” range were Alabama, Arizona, Georgia, and Pennsylvania at level 7 and Kentucky, North Carolina, and Virginia at level 6, according to data from ILINet.
Three flu-related pediatric deaths were reported to CDC during the week, but two occurred the previous week and one occurred in February. That brings the total of flu-related pediatric deaths to 33 for the 2015-2016 influenza season, the CDC said. However, 7.7% of all deaths reported through the 122 Cities Mortality Reporting System were due to pneumonia and influenza. This percentage was above the epidemic threshold of 7.2%.
The CDC also reported a cumulative rate for the season of 21.4 laboratory-confirmed influenza-associated hospitalizations per 100,000 population. The highest rate of hospitalization was among adults aged 65 years or older (54.5 per 100,000 population), followed by adults aged 50-64 (31.4 per 100,000 population) and children aged 0-4 years (29.3 per 100,000 population).
Influenza-like illness (ILI) activity in the United States continued to drop during the week ending March 26, 2016, with only one state still at the highest level, according to the Centers for Disease Control and Prevention.
That one state was New Jersey, which was at level 10 on the CDC’s 1-10 scale of ILI activity. One state at level 10 was down from three states the week before and seven states 2 weeks earlier. Also down for a second consecutive week was the proportion of outpatient visits for ILI, which was 2.9% for the most recent week, compared with 3.2% the previous week and a season high of 3.7% for the week ending March 12, the CDC’s Influenza-like Illness Surveillance Network (ILINet) reported.
The only other state in the “high” range for the week ending March 26 was New Mexico at level 8. States in the “moderate” range were Alabama, Arizona, Georgia, and Pennsylvania at level 7 and Kentucky, North Carolina, and Virginia at level 6, according to data from ILINet.
Three flu-related pediatric deaths were reported to CDC during the week, but two occurred the previous week and one occurred in February. That brings the total of flu-related pediatric deaths to 33 for the 2015-2016 influenza season, the CDC said. However, 7.7% of all deaths reported through the 122 Cities Mortality Reporting System were due to pneumonia and influenza. This percentage was above the epidemic threshold of 7.2%.
The CDC also reported a cumulative rate for the season of 21.4 laboratory-confirmed influenza-associated hospitalizations per 100,000 population. The highest rate of hospitalization was among adults aged 65 years or older (54.5 per 100,000 population), followed by adults aged 50-64 (31.4 per 100,000 population) and children aged 0-4 years (29.3 per 100,000 population).
Rituximab bests fingolimod in MS patients switching from natalizumab
For patients with relapsing-remitting multiple sclerosis (RRMS) switching to other therapies due to positive JC virus serology, treatment with rituximab resulted in a lower rate of clinical relapse as compared with fingolimod in a retrospective outcomes study of registry data from three Swedish MS centers, according to a report published online March 31 in Annals of Neurology.
Although natalizumab (Tysabri) is approved for the treatment of highly active RRMS, its long-term use can increase the risk of developing progressive multifocal leukoencephalopathy. Because this is a serious and potentially lethal condition associated with opportunistic brain infection with the JC virus, those patients testing positive for JC virus antibodies may require a switch to an alternative treatment, such as fingolimod (Gilenya), or off-label use of rituximab (Rituxan). The efficacy, safety, and tolerability associated with switching to either of these alternative therapies were compared by Peter Alping of the department of clinical neuroscience, Karolinska Institutet, Stockholm, and his associates (Ann Neurol. 2016 Mar 31. doi: 10.1002/ana.24651).

Of the 256 patients included in the study, 142 (55%) were switched to fingolimod. The efficacy outcomes comparison showed a statistically significant difference in favor of rituximab, with which 2% of patients had a clinical relapse in the first 1.5 years of treatment after natalizumab cessation as opposed to 18% for fingolimod. This corresponded to an annual relapse rate of 0.02 and 0.16, respectively. Additionally, a review of patients’ contrast-enhancing lesions on MRI scans after at least 3 months of treatment indicated that disease progression was detected less frequently in patients switched to rituximab (1%) as opposed to those switched to fingolimod (16%).
The safety and tolerability data also indicated more favorable results for those patients switched to rituximab. For example, a significantly lower rate of adverse events was noted in the rituximab group (5%) when compared with the fingolimod group (21%). Similarly, a lower rate of treatment discontinuation was observed for those switched to rituximab (2%) when compared with those switched to fingolimod (28%).
The statistically significant differences found in the efficacy, safety, and tolerability data persisted even after adjusting for possible confounding factors including patient age, sex, disability status, time on natalizumab, washout time, follow-up time, and study center. Despite the differences noted in the safety findings, the authors said that both treatments seemed to be safe in general.
In most cases, natalizumab had been given 300 mg IV every 4 weeks. Fingolimod was given orally 0.5 mg once daily. Nearly all patients who received rituximab got a single IV infusion of 500 or 1,000 mg every 6 months, but in some cases the first infusion had been repeated after 2 weeks.
In the absence of formal randomized clinical trial data, the authors said that these findings support the use of rituximab over fingolimod in this particular population of MS patients.
This study was supported by the Swedish Medical Research council and the Stockholm County. First author Peter Alping declared no competing interests; several of his associates reported ties to numerous industry sources.
For patients with relapsing-remitting multiple sclerosis (RRMS) switching to other therapies due to positive JC virus serology, treatment with rituximab resulted in a lower rate of clinical relapse as compared with fingolimod in a retrospective outcomes study of registry data from three Swedish MS centers, according to a report published online March 31 in Annals of Neurology.
Although natalizumab (Tysabri) is approved for the treatment of highly active RRMS, its long-term use can increase the risk of developing progressive multifocal leukoencephalopathy. Because this is a serious and potentially lethal condition associated with opportunistic brain infection with the JC virus, those patients testing positive for JC virus antibodies may require a switch to an alternative treatment, such as fingolimod (Gilenya), or off-label use of rituximab (Rituxan). The efficacy, safety, and tolerability associated with switching to either of these alternative therapies were compared by Peter Alping of the department of clinical neuroscience, Karolinska Institutet, Stockholm, and his associates (Ann Neurol. 2016 Mar 31. doi: 10.1002/ana.24651).
Of the 256 patients included in the study, 142 (55%) were switched to fingolimod. The efficacy outcomes comparison showed a statistically significant difference in favor of rituximab, with which 2% of patients had a clinical relapse in the first 1.5 years of treatment after natalizumab cessation as opposed to 18% for fingolimod. This corresponded to an annual relapse rate of 0.02 and 0.16, respectively. Additionally, a review of patients’ contrast-enhancing lesions on MRI scans after at least 3 months of treatment indicated that disease progression was detected less frequently in patients switched to rituximab (1%) as opposed to those switched to fingolimod (16%).
The safety and tolerability data also indicated more favorable results for those patients switched to rituximab. For example, a significantly lower rate of adverse events was noted in the rituximab group (5%) when compared with the fingolimod group (21%). Similarly, a lower rate of treatment discontinuation was observed for those switched to rituximab (2%) when compared with those switched to fingolimod (28%).
The statistically significant differences found in the efficacy, safety, and tolerability data persisted even after adjusting for possible confounding factors including patient age, sex, disability status, time on natalizumab, washout time, follow-up time, and study center. Despite the differences noted in the safety findings, the authors said that both treatments seemed to be safe in general.
In most cases, natalizumab had been given 300 mg IV every 4 weeks. Fingolimod was given orally 0.5 mg once daily. Nearly all patients who received rituximab got a single IV infusion of 500 or 1,000 mg every 6 months, but in some cases the first infusion had been repeated after 2 weeks.
In the absence of formal randomized clinical trial data, the authors said that these findings support the use of rituximab over fingolimod in this particular population of MS patients.
This study was supported by the Swedish Medical Research council and the Stockholm County. First author Peter Alping declared no competing interests; several of his associates reported ties to numerous industry sources.
For patients with relapsing-remitting multiple sclerosis (RRMS) switching to other therapies due to positive JC virus serology, treatment with rituximab resulted in a lower rate of clinical relapse as compared with fingolimod in a retrospective outcomes study of registry data from three Swedish MS centers, according to a report published online March 31 in Annals of Neurology.
Although natalizumab (Tysabri) is approved for the treatment of highly active RRMS, its long-term use can increase the risk of developing progressive multifocal leukoencephalopathy. Because this is a serious and potentially lethal condition associated with opportunistic brain infection with the JC virus, those patients testing positive for JC virus antibodies may require a switch to an alternative treatment, such as fingolimod (Gilenya), or off-label use of rituximab (Rituxan). The efficacy, safety, and tolerability associated with switching to either of these alternative therapies were compared by Peter Alping of the department of clinical neuroscience, Karolinska Institutet, Stockholm, and his associates (Ann Neurol. 2016 Mar 31. doi: 10.1002/ana.24651).
Of the 256 patients included in the study, 142 (55%) were switched to fingolimod. The efficacy outcomes comparison showed a statistically significant difference in favor of rituximab, with which 2% of patients had a clinical relapse in the first 1.5 years of treatment after natalizumab cessation as opposed to 18% for fingolimod. This corresponded to an annual relapse rate of 0.02 and 0.16, respectively. Additionally, a review of patients’ contrast-enhancing lesions on MRI scans after at least 3 months of treatment indicated that disease progression was detected less frequently in patients switched to rituximab (1%) as opposed to those switched to fingolimod (16%).
The safety and tolerability data also indicated more favorable results for those patients switched to rituximab. For example, a significantly lower rate of adverse events was noted in the rituximab group (5%) when compared with the fingolimod group (21%). Similarly, a lower rate of treatment discontinuation was observed for those switched to rituximab (2%) when compared with those switched to fingolimod (28%).
The statistically significant differences found in the efficacy, safety, and tolerability data persisted even after adjusting for possible confounding factors including patient age, sex, disability status, time on natalizumab, washout time, follow-up time, and study center. Despite the differences noted in the safety findings, the authors said that both treatments seemed to be safe in general.
In most cases, natalizumab had been given 300 mg IV every 4 weeks. Fingolimod was given orally 0.5 mg once daily. Nearly all patients who received rituximab got a single IV infusion of 500 or 1,000 mg every 6 months, but in some cases the first infusion had been repeated after 2 weeks.
In the absence of formal randomized clinical trial data, the authors said that these findings support the use of rituximab over fingolimod in this particular population of MS patients.
This study was supported by the Swedish Medical Research council and the Stockholm County. First author Peter Alping declared no competing interests; several of his associates reported ties to numerous industry sources.
FROM ANNALS OF NEUROLOGY
Key clinical point: Patients with RRMS switching from natalizumab to rituximab due to JC virus antibody positivity achieved better efficacy outcomes than did those switched to fingolimod.
Major finding: A significantly higher percentage of study participants experienced clinical relapse when switched to fingolimod (18%) as opposed to rituximab (2%) within 1.5 years of natalizumab cessation.
Data source: A retrospective outcomes study of registry data from three Swedish MS centers involving 256 JC virus antibody–positive RRMS patients who had switched therapies.
Disclosures: This study was supported by the Swedish Medical Research council and the Stockholm County. First author Peter Alping declared no competing interests; several of his associates reported ties to numerous industry sources.

VIDEO: Low thyroid function increases odds of type 2 diabetes
BOSTON – Results of a population-based study involving more than 8,000 adults from the Netherlands who were diabetes free at baseline has implicated low thyroid function with a 13% increased likelihood of developing type 2 diabetes, and up to 40% higher in individuals with prediabetes.
The heightened risk exists even for individuals with subclinical hypothyroidism, in whom thyroid-stimulating hormone (TSH) in the blood is still in the normal concentration range.
“These findings suggest we should consider screening people with prediabetes for low thyroid function,” Dr. Layal Chaker of Erasmus Medical Center, Rotterdam, the Netherlands, said at the annual meeting of the Endocrine Society.
Thyroid screening is recommended for patients with type 1 diabetes, since they are at increased risk of thyroid disease. An association between thyroid dysfunction in the form of hypothyroidism and type 2 diabetes has been surmised, since type 2 diabetes and hypothyroidism tend to be more prevalent in older adults, and since hypothyroidism has been linked with weight gain and reduced sensitivity to insulin.
To further study the link between thyroid function and diabetes, Dr. Chaker and her colleagues studied data from 8,452 participants aged 45 years and above (mean age 62 years, 58% female) from the Rotterdam Study, a prospective, longitudinal cohort study in the Ommoord district of Rotterdam that was undertaken to investigate the risk factors of cardiovascular, neurological, ophthalmologic, and endocrine diseases in the elderly. The cohort was considered representative of the general population in the Netherlands. All participants had blood tests to measure blood glucose, TSH, and free thyroxine (FT4). Normal blood glucose was considered to be under 5.9 mmol/L, prediabetes as over 5.9 to less than 7.0 mmol/L glucose, and diabetes as above 7.0 mmol/L.
Prediabetes and type 2 diabetes developed in 1,100 and 798 subjects, respectively, during a mean follow-up of 7.9 years. Higher TSH levels increased the risk of development of type 2 diabetes risk (hazard ratio [HR] 1.13, 95% confidence interval [CI], 1.08-1.18, per logTSH). This risk held even for subjects whose TSH levels were at the lower end of the reference range of thyroid function (HR 1.24, CI, 1.06-1.45). The risk of diabetes was reduced in subjects with FT4 levels that were elevated (HR 0.96, CI, 0.93-0.99, per pmol/L) and for those whose FT4 levels were in the reference range (HR 0.96, CI, 0.92-0.99). Low thyroid function, even within the normal range, was associated with a 1.4 times risk of progression from prediabetes to type 2 diabetes (P = .002).
“Low and, surprisingly, low-normal thyroid function are risk factors for incident diabetes, especially in individuals with prediabetes,” said Dr. Chaker.
The data point to the need to clarify whether screening for and treatment of subclinical hypothyroidism can help curb the development of diabetes, she added.
Dr. Chaker had no disclosures.
BOSTON – Results of a population-based study involving more than 8,000 adults from the Netherlands who were diabetes free at baseline has implicated low thyroid function with a 13% increased likelihood of developing type 2 diabetes, and up to 40% higher in individuals with prediabetes.
The heightened risk exists even for individuals with subclinical hypothyroidism, in whom thyroid-stimulating hormone (TSH) in the blood is still in the normal concentration range.
“These findings suggest we should consider screening people with prediabetes for low thyroid function,” Dr. Layal Chaker of Erasmus Medical Center, Rotterdam, the Netherlands, said at the annual meeting of the Endocrine Society.
Thyroid screening is recommended for patients with type 1 diabetes, since they are at increased risk of thyroid disease. An association between thyroid dysfunction in the form of hypothyroidism and type 2 diabetes has been surmised, since type 2 diabetes and hypothyroidism tend to be more prevalent in older adults, and since hypothyroidism has been linked with weight gain and reduced sensitivity to insulin.
To further study the link between thyroid function and diabetes, Dr. Chaker and her colleagues studied data from 8,452 participants aged 45 years and above (mean age 62 years, 58% female) from the Rotterdam Study, a prospective, longitudinal cohort study in the Ommoord district of Rotterdam that was undertaken to investigate the risk factors of cardiovascular, neurological, ophthalmologic, and endocrine diseases in the elderly. The cohort was considered representative of the general population in the Netherlands. All participants had blood tests to measure blood glucose, TSH, and free thyroxine (FT4). Normal blood glucose was considered to be under 5.9 mmol/L, prediabetes as over 5.9 to less than 7.0 mmol/L glucose, and diabetes as above 7.0 mmol/L.
Prediabetes and type 2 diabetes developed in 1,100 and 798 subjects, respectively, during a mean follow-up of 7.9 years. Higher TSH levels increased the risk of development of type 2 diabetes risk (hazard ratio [HR] 1.13, 95% confidence interval [CI], 1.08-1.18, per logTSH). This risk held even for subjects whose TSH levels were at the lower end of the reference range of thyroid function (HR 1.24, CI, 1.06-1.45). The risk of diabetes was reduced in subjects with FT4 levels that were elevated (HR 0.96, CI, 0.93-0.99, per pmol/L) and for those whose FT4 levels were in the reference range (HR 0.96, CI, 0.92-0.99). Low thyroid function, even within the normal range, was associated with a 1.4 times risk of progression from prediabetes to type 2 diabetes (P = .002).
“Low and, surprisingly, low-normal thyroid function are risk factors for incident diabetes, especially in individuals with prediabetes,” said Dr. Chaker.
The data point to the need to clarify whether screening for and treatment of subclinical hypothyroidism can help curb the development of diabetes, she added.
Dr. Chaker had no disclosures.
BOSTON – Results of a population-based study involving more than 8,000 adults from the Netherlands who were diabetes free at baseline has implicated low thyroid function with a 13% increased likelihood of developing type 2 diabetes, and up to 40% higher in individuals with prediabetes.
The heightened risk exists even for individuals with subclinical hypothyroidism, in whom thyroid-stimulating hormone (TSH) in the blood is still in the normal concentration range.
“These findings suggest we should consider screening people with prediabetes for low thyroid function,” Dr. Layal Chaker of Erasmus Medical Center, Rotterdam, the Netherlands, said at the annual meeting of the Endocrine Society.
Thyroid screening is recommended for patients with type 1 diabetes, since they are at increased risk of thyroid disease. An association between thyroid dysfunction in the form of hypothyroidism and type 2 diabetes has been surmised, since type 2 diabetes and hypothyroidism tend to be more prevalent in older adults, and since hypothyroidism has been linked with weight gain and reduced sensitivity to insulin.
To further study the link between thyroid function and diabetes, Dr. Chaker and her colleagues studied data from 8,452 participants aged 45 years and above (mean age 62 years, 58% female) from the Rotterdam Study, a prospective, longitudinal cohort study in the Ommoord district of Rotterdam that was undertaken to investigate the risk factors of cardiovascular, neurological, ophthalmologic, and endocrine diseases in the elderly. The cohort was considered representative of the general population in the Netherlands. All participants had blood tests to measure blood glucose, TSH, and free thyroxine (FT4). Normal blood glucose was considered to be under 5.9 mmol/L, prediabetes as over 5.9 to less than 7.0 mmol/L glucose, and diabetes as above 7.0 mmol/L.
Prediabetes and type 2 diabetes developed in 1,100 and 798 subjects, respectively, during a mean follow-up of 7.9 years. Higher TSH levels increased the risk of development of type 2 diabetes risk (hazard ratio [HR] 1.13, 95% confidence interval [CI], 1.08-1.18, per logTSH). This risk held even for subjects whose TSH levels were at the lower end of the reference range of thyroid function (HR 1.24, CI, 1.06-1.45). The risk of diabetes was reduced in subjects with FT4 levels that were elevated (HR 0.96, CI, 0.93-0.99, per pmol/L) and for those whose FT4 levels were in the reference range (HR 0.96, CI, 0.92-0.99). Low thyroid function, even within the normal range, was associated with a 1.4 times risk of progression from prediabetes to type 2 diabetes (P = .002).
“Low and, surprisingly, low-normal thyroid function are risk factors for incident diabetes, especially in individuals with prediabetes,” said Dr. Chaker.
The data point to the need to clarify whether screening for and treatment of subclinical hypothyroidism can help curb the development of diabetes, she added.
Dr. Chaker had no disclosures.
AT ENDO 2016
Key clinical point: Hypothyroidism increases the risk of developing type 2 diabetes.
Major finding: Higher TSH levels increased the risk of development of type 2 diabetes risk (hazard ratio 1.13, 95% confidence interval, 1.08-1.18, per logTSH).
Data source: Population-based study of 8,452 adult from the Netherlands
Disclosures: Dr. Chaker had no disclosures.

VIDEO: Management of difficult-to-treat IBD cases
PHILADELPHIA – The lack of evidence-based management strategies for the treatment of difficult inflammatory bowel disease cases, such as with Crohn’s disease, can lead to confusion for some clinicians.
Dr. Mark T. Osterman, assistant professor of medicine at the University of Pennsylvania, Philadelphia, discusses when to use antibiotics in Crohn’s disease and when to consider antibiotics in combination with draining fistulae. He also discusses available pharmacotherapies, and the value of bowel rest.
“The best treatment of all for difficult inflammatory bowel disease is aggressive and early, so that the condition doesn’t go from bad to worse,” Dr. Osterman said.
The video was recorded at this year’s meeting of Digestive Diseases: New Advances, a meeting held by Global Academy for Medical Education and Rutgers, the State University of New Jersey. Global Academy and this news organization are owned by the same company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
PHILADELPHIA – The lack of evidence-based management strategies for the treatment of difficult inflammatory bowel disease cases, such as with Crohn’s disease, can lead to confusion for some clinicians.
Dr. Mark T. Osterman, assistant professor of medicine at the University of Pennsylvania, Philadelphia, discusses when to use antibiotics in Crohn’s disease and when to consider antibiotics in combination with draining fistulae. He also discusses available pharmacotherapies, and the value of bowel rest.
“The best treatment of all for difficult inflammatory bowel disease is aggressive and early, so that the condition doesn’t go from bad to worse,” Dr. Osterman said.
The video was recorded at this year’s meeting of Digestive Diseases: New Advances, a meeting held by Global Academy for Medical Education and Rutgers, the State University of New Jersey. Global Academy and this news organization are owned by the same company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
PHILADELPHIA – The lack of evidence-based management strategies for the treatment of difficult inflammatory bowel disease cases, such as with Crohn’s disease, can lead to confusion for some clinicians.
Dr. Mark T. Osterman, assistant professor of medicine at the University of Pennsylvania, Philadelphia, discusses when to use antibiotics in Crohn’s disease and when to consider antibiotics in combination with draining fistulae. He also discusses available pharmacotherapies, and the value of bowel rest.
“The best treatment of all for difficult inflammatory bowel disease is aggressive and early, so that the condition doesn’t go from bad to worse,” Dr. Osterman said.
The video was recorded at this year’s meeting of Digestive Diseases: New Advances, a meeting held by Global Academy for Medical Education and Rutgers, the State University of New Jersey. Global Academy and this news organization are owned by the same company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
EXPERT ANALYSIS FROM DIGESTIVE DISEASES: NEW ADVANCES

How to make your patient with sleep apnea a “super user” of positive airway pressure therapy
Adherence to positive airway pressure (PAP) therapy is a difficult patient management issue. Recent studies confirm widely varying PAP therapy adherence rates (30%-84%). Clinicians at the John D. Dingell VA Medical Center in Detroit developed a set of "super user" criteria, a 5-point method for encouraging patients to maximize adherence to PAP therapy. All 5 criteria, which are discussed in detail in this article from Federal Practitioner, must be satisfied to attain “super user” status. For more, go to: http://www.fedprac.com/the-publication/issue-single-view/how-to-make-your-patient-with-sleep-apnea-a-super-user-of-positive-airway-pressure-therapy/bbd97c064c5f91688f04b82c8b04b054/ocregister.html.
Adherence to positive airway pressure (PAP) therapy is a difficult patient management issue. Recent studies confirm widely varying PAP therapy adherence rates (30%-84%). Clinicians at the John D. Dingell VA Medical Center in Detroit developed a set of "super user" criteria, a 5-point method for encouraging patients to maximize adherence to PAP therapy. All 5 criteria, which are discussed in detail in this article from Federal Practitioner, must be satisfied to attain “super user” status. For more, go to: http://www.fedprac.com/the-publication/issue-single-view/how-to-make-your-patient-with-sleep-apnea-a-super-user-of-positive-airway-pressure-therapy/bbd97c064c5f91688f04b82c8b04b054/ocregister.html.
Adherence to positive airway pressure (PAP) therapy is a difficult patient management issue. Recent studies confirm widely varying PAP therapy adherence rates (30%-84%). Clinicians at the John D. Dingell VA Medical Center in Detroit developed a set of "super user" criteria, a 5-point method for encouraging patients to maximize adherence to PAP therapy. All 5 criteria, which are discussed in detail in this article from Federal Practitioner, must be satisfied to attain “super user” status. For more, go to: http://www.fedprac.com/the-publication/issue-single-view/how-to-make-your-patient-with-sleep-apnea-a-super-user-of-positive-airway-pressure-therapy/bbd97c064c5f91688f04b82c8b04b054/ocregister.html.
Chronic pain and depression: Treatment of 2 culprits in common
Because pain and depression share common neurobiological pathways and clinical manifestations, you can use similar strategies and, often, the same agents to treat both conditions when they occur together. This article from Current Psychiatry, available at http://www.currentpsychiatry.com/specialty-focus/depressive-disorders/article/chronic-pain-and-depression-treatment-of-2-culprits-in-common/6bc388cf05b07e8dc6bbc1b86f50f0bf.html, reviews different treatment options (including non-drug interventions) that can help patients with both pain and depression, as well as drug-drug interactions that can occur.
Because pain and depression share common neurobiological pathways and clinical manifestations, you can use similar strategies and, often, the same agents to treat both conditions when they occur together. This article from Current Psychiatry, available at http://www.currentpsychiatry.com/specialty-focus/depressive-disorders/article/chronic-pain-and-depression-treatment-of-2-culprits-in-common/6bc388cf05b07e8dc6bbc1b86f50f0bf.html, reviews different treatment options (including non-drug interventions) that can help patients with both pain and depression, as well as drug-drug interactions that can occur.
Because pain and depression share common neurobiological pathways and clinical manifestations, you can use similar strategies and, often, the same agents to treat both conditions when they occur together. This article from Current Psychiatry, available at http://www.currentpsychiatry.com/specialty-focus/depressive-disorders/article/chronic-pain-and-depression-treatment-of-2-culprits-in-common/6bc388cf05b07e8dc6bbc1b86f50f0bf.html, reviews different treatment options (including non-drug interventions) that can help patients with both pain and depression, as well as drug-drug interactions that can occur.
Homelessness, HIV, and HCV
Homelessness and unstable housing situations are associated with higher rates of human immunodeficiency virus (HIV) and hepatitis C infection (HCV), according to researchers from Columbia University in New York City, McMaster University in Hamilton, Ontario, Canada, and the Ontario HIV Treatment Network in Canada. The researchers reviewed 152 studies involving 139,757 individuals who had HIV or were co-infected with HCV. The researchers found “strong evidence” that the lack of stable, secure, and adequate housing is a significant barrier to consistent and appropriate medical care, as well as the reduction of risk behaviors. For more on this research, see the Federal Practitioner article at: http://www.fedprac.com/the-publication/issue-single-view/homelessness-hiv-and-hcv/6a66b2b7db3f0299caa7aaf050129fb4/ocregister.html.
Homelessness and unstable housing situations are associated with higher rates of human immunodeficiency virus (HIV) and hepatitis C infection (HCV), according to researchers from Columbia University in New York City, McMaster University in Hamilton, Ontario, Canada, and the Ontario HIV Treatment Network in Canada. The researchers reviewed 152 studies involving 139,757 individuals who had HIV or were co-infected with HCV. The researchers found “strong evidence” that the lack of stable, secure, and adequate housing is a significant barrier to consistent and appropriate medical care, as well as the reduction of risk behaviors. For more on this research, see the Federal Practitioner article at: http://www.fedprac.com/the-publication/issue-single-view/homelessness-hiv-and-hcv/6a66b2b7db3f0299caa7aaf050129fb4/ocregister.html.
Homelessness and unstable housing situations are associated with higher rates of human immunodeficiency virus (HIV) and hepatitis C infection (HCV), according to researchers from Columbia University in New York City, McMaster University in Hamilton, Ontario, Canada, and the Ontario HIV Treatment Network in Canada. The researchers reviewed 152 studies involving 139,757 individuals who had HIV or were co-infected with HCV. The researchers found “strong evidence” that the lack of stable, secure, and adequate housing is a significant barrier to consistent and appropriate medical care, as well as the reduction of risk behaviors. For more on this research, see the Federal Practitioner article at: http://www.fedprac.com/the-publication/issue-single-view/homelessness-hiv-and-hcv/6a66b2b7db3f0299caa7aaf050129fb4/ocregister.html.
Incretin-based diabetes drugs don’t raise heart failure risk
Incretin-based antidiabetic drugs didn’t raise the risk of hospitalization for heart failure, according to an international observational study involving 1.5 million patients reported online in The New England Journal of Medicine. “With 3.2 million person-years of observations, we had the statistical power to robustly assess this important drug safety issue,” the investigators said. Read more on the study at Cardiology News: http://www.ecardiologynews.com/specialty-focus/heart-failure/single-article-page/incretin-based-diabetes-drugs-dont-raise-heart-failure-risk/72ea7cb26766fc17483ad005269c5da2.html.
Incretin-based antidiabetic drugs didn’t raise the risk of hospitalization for heart failure, according to an international observational study involving 1.5 million patients reported online in The New England Journal of Medicine. “With 3.2 million person-years of observations, we had the statistical power to robustly assess this important drug safety issue,” the investigators said. Read more on the study at Cardiology News: http://www.ecardiologynews.com/specialty-focus/heart-failure/single-article-page/incretin-based-diabetes-drugs-dont-raise-heart-failure-risk/72ea7cb26766fc17483ad005269c5da2.html.
Incretin-based antidiabetic drugs didn’t raise the risk of hospitalization for heart failure, according to an international observational study involving 1.5 million patients reported online in The New England Journal of Medicine. “With 3.2 million person-years of observations, we had the statistical power to robustly assess this important drug safety issue,” the investigators said. Read more on the study at Cardiology News: http://www.ecardiologynews.com/specialty-focus/heart-failure/single-article-page/incretin-based-diabetes-drugs-dont-raise-heart-failure-risk/72ea7cb26766fc17483ad005269c5da2.html.
The challenges of type 1 diabetes: A case-based review
The prevalence of type 1 diabetes in people younger than age 20 increased by 23% from 2001 to 2009, and the disease now affects as many as 1.25 million Americans. This case from Clinician Reviews provides a brief overview of the diagnosis and management considerations required for successful care of these patients. The article is available at: http://www.clinicianreviews.com/the-publication/issue-single-view/the-challenges-of-type-1-diabetes-a-case-based-review/a5a6fab4483946a829d60880ec2e3a74.html.
The prevalence of type 1 diabetes in people younger than age 20 increased by 23% from 2001 to 2009, and the disease now affects as many as 1.25 million Americans. This case from Clinician Reviews provides a brief overview of the diagnosis and management considerations required for successful care of these patients. The article is available at: http://www.clinicianreviews.com/the-publication/issue-single-view/the-challenges-of-type-1-diabetes-a-case-based-review/a5a6fab4483946a829d60880ec2e3a74.html.
The prevalence of type 1 diabetes in people younger than age 20 increased by 23% from 2001 to 2009, and the disease now affects as many as 1.25 million Americans. This case from Clinician Reviews provides a brief overview of the diagnosis and management considerations required for successful care of these patients. The article is available at: http://www.clinicianreviews.com/the-publication/issue-single-view/the-challenges-of-type-1-diabetes-a-case-based-review/a5a6fab4483946a829d60880ec2e3a74.html.