User login
Losmapimod failed to beat placebo in acute MI trial
Twelve weeks of treatment with the anti-inflammatory drug losmapimod did not prevent cardiovascular death, myocardial infarction, or severe recurrent ischemia in patients hospitalized with acute MI, based on the results of LATITUDE-TIMI 60, a multicenter placebo-controlled phase 3 trial.
Losmapimod also did not significantly affect secondary outcomes such as all-cause mortality, said Dr. Michelle O’Donoghue at Brigham and Women’s Hospital in Boston and her associates.
“The results of this exploratory efficacy study did not justify proceeding to a larger efficacy trial in the existing patient population,” they concluded at the annual meeting of the American College of Cardiology and in a report published online April 4 in JAMA.
Losmapimod selectively inhibits pro-inflammatory p38 mitogen-activated protein kinase, which contributes to atherosclerosis and plaque destabilization and is activated by stressors such as oxidized low-density lipoprotein cholesterol, hypertension, ischemia, and volume overload. In a prior 12-week phase 2 study, losmapimod failed to reduce inflammatory biomarker levels or myonecrosis, but showed a trend toward lower rates of mortality, MI, recurrent ischemia, stroke, and heart failure, as well as improved left ventricular function.
Based on those observations, Dr. O’Donoghue and her associates randomly assigned 3,503 hospitalized patients with acute MI and at least one other cardiovascular risk factor to receive either losmapimod (7.5 mg twice daily) or placebo, along with guideline-recommended therapy for 12 weeks. Patients averaged 66 years old, about 90% were white, and about 30% were women. The primary endpoint was a composite of cardiovascular death, MI, or severe recurrent ischemia requiring urgent coronary revascularization (JAMA 2016 April 4 doi: 10.1001/jama.2016.3609).
The endpoint was met for 139 (8.1%) losmapimod patients and 123 (7%) placebo patients, for a non-significant hazard ratio of 1.16, said the researchers. Losmapimod was, however, associated with significant decreases in levels of high-sensitivity C-reactive protein and N-terminal pro-brain natriuretic peptide as compared with placebo, with durable effects at 12 weeks. Serious adverse events affected 16% of losmapimod patients and 14% of placebo patients, and rates of premature treatment discontinuation were 15.5% with losmapimod and nearly 14% with placebo. Losmapimod did not appear to increase the risk of infections compared with placebo at week 12, but did show a non-significant trend toward mildly increased hepatic transaminases.
“Because inflammation is believed to play a key role in atherogenesis, there remains intense interest to identify an anti-inflammatory therapeutic that will reduce the risk of cardiovascular events,” the researchers noted. “However, because inflammation acts along multiple redundant and interconnected pathways, the identification of an appropriate target may be difficult, and it is challenging to predict clinical efficacy prior to phase 3 testing. Ongoing clinical trials are currently evaluating additional anti-inflammatory therapeutics in patients with atherosclerosis, and will provide further insight into pathways that contribute to vascular disease.”
Pharmacodynamic data on losmapimod do not suggest that its anti-inflammatory effects increase at higher doses, the researchers also noted.
GlaxoSmithKline developed losmapimod and funded the study. Dr. O’Donoghue reported grant funding from Eisai, Merck, and AstraZeneca. The senior author and several coinvestigators disclosed relationships with numerous pharmaceutical companies.
Twelve weeks of treatment with the anti-inflammatory drug losmapimod did not prevent cardiovascular death, myocardial infarction, or severe recurrent ischemia in patients hospitalized with acute MI, based on the results of LATITUDE-TIMI 60, a multicenter placebo-controlled phase 3 trial.
Losmapimod also did not significantly affect secondary outcomes such as all-cause mortality, said Dr. Michelle O’Donoghue at Brigham and Women’s Hospital in Boston and her associates.
“The results of this exploratory efficacy study did not justify proceeding to a larger efficacy trial in the existing patient population,” they concluded at the annual meeting of the American College of Cardiology and in a report published online April 4 in JAMA.
Losmapimod selectively inhibits pro-inflammatory p38 mitogen-activated protein kinase, which contributes to atherosclerosis and plaque destabilization and is activated by stressors such as oxidized low-density lipoprotein cholesterol, hypertension, ischemia, and volume overload. In a prior 12-week phase 2 study, losmapimod failed to reduce inflammatory biomarker levels or myonecrosis, but showed a trend toward lower rates of mortality, MI, recurrent ischemia, stroke, and heart failure, as well as improved left ventricular function.
Based on those observations, Dr. O’Donoghue and her associates randomly assigned 3,503 hospitalized patients with acute MI and at least one other cardiovascular risk factor to receive either losmapimod (7.5 mg twice daily) or placebo, along with guideline-recommended therapy for 12 weeks. Patients averaged 66 years old, about 90% were white, and about 30% were women. The primary endpoint was a composite of cardiovascular death, MI, or severe recurrent ischemia requiring urgent coronary revascularization (JAMA 2016 April 4 doi: 10.1001/jama.2016.3609).
The endpoint was met for 139 (8.1%) losmapimod patients and 123 (7%) placebo patients, for a non-significant hazard ratio of 1.16, said the researchers. Losmapimod was, however, associated with significant decreases in levels of high-sensitivity C-reactive protein and N-terminal pro-brain natriuretic peptide as compared with placebo, with durable effects at 12 weeks. Serious adverse events affected 16% of losmapimod patients and 14% of placebo patients, and rates of premature treatment discontinuation were 15.5% with losmapimod and nearly 14% with placebo. Losmapimod did not appear to increase the risk of infections compared with placebo at week 12, but did show a non-significant trend toward mildly increased hepatic transaminases.
“Because inflammation is believed to play a key role in atherogenesis, there remains intense interest to identify an anti-inflammatory therapeutic that will reduce the risk of cardiovascular events,” the researchers noted. “However, because inflammation acts along multiple redundant and interconnected pathways, the identification of an appropriate target may be difficult, and it is challenging to predict clinical efficacy prior to phase 3 testing. Ongoing clinical trials are currently evaluating additional anti-inflammatory therapeutics in patients with atherosclerosis, and will provide further insight into pathways that contribute to vascular disease.”
Pharmacodynamic data on losmapimod do not suggest that its anti-inflammatory effects increase at higher doses, the researchers also noted.
GlaxoSmithKline developed losmapimod and funded the study. Dr. O’Donoghue reported grant funding from Eisai, Merck, and AstraZeneca. The senior author and several coinvestigators disclosed relationships with numerous pharmaceutical companies.
Twelve weeks of treatment with the anti-inflammatory drug losmapimod did not prevent cardiovascular death, myocardial infarction, or severe recurrent ischemia in patients hospitalized with acute MI, based on the results of LATITUDE-TIMI 60, a multicenter placebo-controlled phase 3 trial.
Losmapimod also did not significantly affect secondary outcomes such as all-cause mortality, said Dr. Michelle O’Donoghue at Brigham and Women’s Hospital in Boston and her associates.
“The results of this exploratory efficacy study did not justify proceeding to a larger efficacy trial in the existing patient population,” they concluded at the annual meeting of the American College of Cardiology and in a report published online April 4 in JAMA.
Losmapimod selectively inhibits pro-inflammatory p38 mitogen-activated protein kinase, which contributes to atherosclerosis and plaque destabilization and is activated by stressors such as oxidized low-density lipoprotein cholesterol, hypertension, ischemia, and volume overload. In a prior 12-week phase 2 study, losmapimod failed to reduce inflammatory biomarker levels or myonecrosis, but showed a trend toward lower rates of mortality, MI, recurrent ischemia, stroke, and heart failure, as well as improved left ventricular function.
Based on those observations, Dr. O’Donoghue and her associates randomly assigned 3,503 hospitalized patients with acute MI and at least one other cardiovascular risk factor to receive either losmapimod (7.5 mg twice daily) or placebo, along with guideline-recommended therapy for 12 weeks. Patients averaged 66 years old, about 90% were white, and about 30% were women. The primary endpoint was a composite of cardiovascular death, MI, or severe recurrent ischemia requiring urgent coronary revascularization (JAMA 2016 April 4 doi: 10.1001/jama.2016.3609).
The endpoint was met for 139 (8.1%) losmapimod patients and 123 (7%) placebo patients, for a non-significant hazard ratio of 1.16, said the researchers. Losmapimod was, however, associated with significant decreases in levels of high-sensitivity C-reactive protein and N-terminal pro-brain natriuretic peptide as compared with placebo, with durable effects at 12 weeks. Serious adverse events affected 16% of losmapimod patients and 14% of placebo patients, and rates of premature treatment discontinuation were 15.5% with losmapimod and nearly 14% with placebo. Losmapimod did not appear to increase the risk of infections compared with placebo at week 12, but did show a non-significant trend toward mildly increased hepatic transaminases.
“Because inflammation is believed to play a key role in atherogenesis, there remains intense interest to identify an anti-inflammatory therapeutic that will reduce the risk of cardiovascular events,” the researchers noted. “However, because inflammation acts along multiple redundant and interconnected pathways, the identification of an appropriate target may be difficult, and it is challenging to predict clinical efficacy prior to phase 3 testing. Ongoing clinical trials are currently evaluating additional anti-inflammatory therapeutics in patients with atherosclerosis, and will provide further insight into pathways that contribute to vascular disease.”
Pharmacodynamic data on losmapimod do not suggest that its anti-inflammatory effects increase at higher doses, the researchers also noted.
GlaxoSmithKline developed losmapimod and funded the study. Dr. O’Donoghue reported grant funding from Eisai, Merck, and AstraZeneca. The senior author and several coinvestigators disclosed relationships with numerous pharmaceutical companies.
FROM ACC 16
Key clinical point: The anti-inflammatory drug losmapimod missed its composite primary endpoint in a phase 3 study of patients with acute myocardial infarction.
Major finding: At week 12, there was no significant effect on cardiovascular death, MI, or severe recurrent ischemia requiring urgent coronary revascularization.
Data source: A multicenter randomized phase 3 trial of 3,503 hospitalized adults with acute MI and at least one other cardiovascular risk factor.
Disclosures: GlaxoSmithKline developed losmapimod and funded the study. Dr. O’Donoghue reported grant funding from Eisai, Merck, and AstraZeneca. The senior author and several coinvestigators disclosed relationships with numerous pharmaceutical companies.
Pump-delivered anesthetic reduces pain post-hernia repair
MONTREAL – An elastomeric pump delivering local anesthetic to the site of a ventral hernia repair can significantly reduce postoperative pain, according to a small, single-center study.
The prospective, randomized, double-blind placebo-controlled trial of the use of an elastomeric bupivacaine-containing pump for pain management after laparoscopic ventral hernia repair (LVHR) found that patients receiving bupivacaine reported significantly less postoperative pain than did those receiving saline through the pump.
“The idea is that the use of this technology, in combination with standard postoperative pain medication, would do two things: It would maintain the same level of postoperative pain control, as well as reducing the amount of narcotic and non-narcotic pain medication used,” said Francis DeAsis, a medical student at Midwestern University, Chicago.

“For our project, we focused on the specific anatomic location of the catheter,” Mr. DeAsis said in a presentation at the annual meeting of the Central Surgical Association. A previous study, he said, had shown no efficacy for pump-delivered bupivacaine when it was delivered directly into the hernia sac.
In the present study, the cannula for the pump was placed inferior to the costochondral margin, with catheters tunnelled bilaterally between the transversalis fascia and the parietal peritoneum. “The idea behind this was to knock out as many pain receptors as possible,” said Mr. DeAsis.
Primary outcome measures were the self-reported level of postoperative pain and the amount of postoperative medication use. Adult patients at a single site who were undergoing nonemergent ventral hernia repair and who had no history of opioid abuse or adverse reactions to opioids or local anesthetics were enrolled.
Patients were then prospectively randomized to the placebo arm, where they received 400 mL of saline via pump, or to the intervention arm, where they received 400 mL of 0.5% bupivacaine following LVHR. The pump delivered the study drug for a period of 4 days after placement in both groups.
Patients completed a pain diary and medication log for the 7 days after discharge. They rated their current pain level, reported how frequently they had pain, and reported satisfaction with pain control.
In-hospital use of opioid pain medication did not differ significantly between groups, but patients receiving bupivacaine through their pumps used significantly less ketorolac as inpatients (mean 30 mg, compared with 53 mg in the saline group, P = .01).
“Ketorolac is our first-line; we try to avoid narcotics for many different reasons ... Ketorolac PRN was ordered for everybody,” said Dr. Michael Ujiki, a general surgeon at NorthShore University HealthSystem, Evanston, Ill., and senior investigator for the study.
The median pain score on a 1-10 Likert scale at discharge was 2.0 (interquartile range, 0.0-3.0) for the bupivacaine group and 4.0 (interquartile range, 2.0-5.0) for the saline group (P = .06).
Once patients were discharged from the hospital, patients receiving bupivacaine had significantly less postoperative pain through postoperative day 4 and significantly less frequent pain through postoperative day 4. Finally, they reported being significantly more satisfied with their pain management scores on postoperative days 1-3.
The study was limited by the small cohort size. “The primary reason behind this was that during recruitment, we had a lot of patients voice concern about potentially receiving placebo,” thereby forgoing the chance for additional pain relief, said Mr. DeAsis. “A well-powered study should be conducted in the future.”
The investigators reported no relevant financial disclosures, but all study pumps were provided by the manufacturer, On-Q.
On Twitter @karioakes
MONTREAL – An elastomeric pump delivering local anesthetic to the site of a ventral hernia repair can significantly reduce postoperative pain, according to a small, single-center study.
The prospective, randomized, double-blind placebo-controlled trial of the use of an elastomeric bupivacaine-containing pump for pain management after laparoscopic ventral hernia repair (LVHR) found that patients receiving bupivacaine reported significantly less postoperative pain than did those receiving saline through the pump.
“The idea is that the use of this technology, in combination with standard postoperative pain medication, would do two things: It would maintain the same level of postoperative pain control, as well as reducing the amount of narcotic and non-narcotic pain medication used,” said Francis DeAsis, a medical student at Midwestern University, Chicago.
“For our project, we focused on the specific anatomic location of the catheter,” Mr. DeAsis said in a presentation at the annual meeting of the Central Surgical Association. A previous study, he said, had shown no efficacy for pump-delivered bupivacaine when it was delivered directly into the hernia sac.
In the present study, the cannula for the pump was placed inferior to the costochondral margin, with catheters tunnelled bilaterally between the transversalis fascia and the parietal peritoneum. “The idea behind this was to knock out as many pain receptors as possible,” said Mr. DeAsis.
Primary outcome measures were the self-reported level of postoperative pain and the amount of postoperative medication use. Adult patients at a single site who were undergoing nonemergent ventral hernia repair and who had no history of opioid abuse or adverse reactions to opioids or local anesthetics were enrolled.
Patients were then prospectively randomized to the placebo arm, where they received 400 mL of saline via pump, or to the intervention arm, where they received 400 mL of 0.5% bupivacaine following LVHR. The pump delivered the study drug for a period of 4 days after placement in both groups.
Patients completed a pain diary and medication log for the 7 days after discharge. They rated their current pain level, reported how frequently they had pain, and reported satisfaction with pain control.
In-hospital use of opioid pain medication did not differ significantly between groups, but patients receiving bupivacaine through their pumps used significantly less ketorolac as inpatients (mean 30 mg, compared with 53 mg in the saline group, P = .01).
“Ketorolac is our first-line; we try to avoid narcotics for many different reasons ... Ketorolac PRN was ordered for everybody,” said Dr. Michael Ujiki, a general surgeon at NorthShore University HealthSystem, Evanston, Ill., and senior investigator for the study.
The median pain score on a 1-10 Likert scale at discharge was 2.0 (interquartile range, 0.0-3.0) for the bupivacaine group and 4.0 (interquartile range, 2.0-5.0) for the saline group (P = .06).
Once patients were discharged from the hospital, patients receiving bupivacaine had significantly less postoperative pain through postoperative day 4 and significantly less frequent pain through postoperative day 4. Finally, they reported being significantly more satisfied with their pain management scores on postoperative days 1-3.
The study was limited by the small cohort size. “The primary reason behind this was that during recruitment, we had a lot of patients voice concern about potentially receiving placebo,” thereby forgoing the chance for additional pain relief, said Mr. DeAsis. “A well-powered study should be conducted in the future.”
The investigators reported no relevant financial disclosures, but all study pumps were provided by the manufacturer, On-Q.
On Twitter @karioakes
MONTREAL – An elastomeric pump delivering local anesthetic to the site of a ventral hernia repair can significantly reduce postoperative pain, according to a small, single-center study.
The prospective, randomized, double-blind placebo-controlled trial of the use of an elastomeric bupivacaine-containing pump for pain management after laparoscopic ventral hernia repair (LVHR) found that patients receiving bupivacaine reported significantly less postoperative pain than did those receiving saline through the pump.
“The idea is that the use of this technology, in combination with standard postoperative pain medication, would do two things: It would maintain the same level of postoperative pain control, as well as reducing the amount of narcotic and non-narcotic pain medication used,” said Francis DeAsis, a medical student at Midwestern University, Chicago.
“For our project, we focused on the specific anatomic location of the catheter,” Mr. DeAsis said in a presentation at the annual meeting of the Central Surgical Association. A previous study, he said, had shown no efficacy for pump-delivered bupivacaine when it was delivered directly into the hernia sac.
In the present study, the cannula for the pump was placed inferior to the costochondral margin, with catheters tunnelled bilaterally between the transversalis fascia and the parietal peritoneum. “The idea behind this was to knock out as many pain receptors as possible,” said Mr. DeAsis.
Primary outcome measures were the self-reported level of postoperative pain and the amount of postoperative medication use. Adult patients at a single site who were undergoing nonemergent ventral hernia repair and who had no history of opioid abuse or adverse reactions to opioids or local anesthetics were enrolled.
Patients were then prospectively randomized to the placebo arm, where they received 400 mL of saline via pump, or to the intervention arm, where they received 400 mL of 0.5% bupivacaine following LVHR. The pump delivered the study drug for a period of 4 days after placement in both groups.
Patients completed a pain diary and medication log for the 7 days after discharge. They rated their current pain level, reported how frequently they had pain, and reported satisfaction with pain control.
In-hospital use of opioid pain medication did not differ significantly between groups, but patients receiving bupivacaine through their pumps used significantly less ketorolac as inpatients (mean 30 mg, compared with 53 mg in the saline group, P = .01).
“Ketorolac is our first-line; we try to avoid narcotics for many different reasons ... Ketorolac PRN was ordered for everybody,” said Dr. Michael Ujiki, a general surgeon at NorthShore University HealthSystem, Evanston, Ill., and senior investigator for the study.
The median pain score on a 1-10 Likert scale at discharge was 2.0 (interquartile range, 0.0-3.0) for the bupivacaine group and 4.0 (interquartile range, 2.0-5.0) for the saline group (P = .06).
Once patients were discharged from the hospital, patients receiving bupivacaine had significantly less postoperative pain through postoperative day 4 and significantly less frequent pain through postoperative day 4. Finally, they reported being significantly more satisfied with their pain management scores on postoperative days 1-3.
The study was limited by the small cohort size. “The primary reason behind this was that during recruitment, we had a lot of patients voice concern about potentially receiving placebo,” thereby forgoing the chance for additional pain relief, said Mr. DeAsis. “A well-powered study should be conducted in the future.”
The investigators reported no relevant financial disclosures, but all study pumps were provided by the manufacturer, On-Q.
On Twitter @karioakes
AT THE ANNUAL MEETING OF THE CENTRAL SURGICAL ASSOCIATION
Key clinical point: Pain scores were reduced when patients had pump-delivered local anesthetic after hernia repair.
Major finding: Pain level and frequency of pain were reduced, and satisfaction with pain control was increased, when patients received local anesthetic via pump after laparoscopic ventral hernia repair.
Data source: Single-center double-blind, placebo-controlled study of elastomeric pump-delivered bupivacaine vs. placebo in 29 patients undergoing laparoscopic ventral hernia repair.
Disclosures: The pump was supplied by the manufacturer (On-Q); the investigators reported no other relevant disclosures.

Pump-delivered anesthetic reduces pain post-hernia repair
MONTREAL – An elastomeric pump delivering local anesthetic to the site of a ventral hernia repair can significantly reduce postoperative pain, according to a small, single-center study.
The prospective, randomized, double-blind placebo-controlled trial of the use of an elastomeric bupivacaine-containing pump for pain management after laparoscopic ventral hernia repair (LVHR) found that patients receiving bupivacaine reported significantly less postoperative pain than did those receiving saline through the pump.
“The idea is that the use of this technology, in combination with standard postoperative pain medication, would do two things: It would maintain the same level of postoperative pain control, as well as reducing the amount of narcotic and non-narcotic pain medication used,” said Francis DeAsis, a medical student at Midwestern University, Chicago.
“For our project, we focused on the specific anatomic location of the catheter,” Mr. DeAsis said in a presentation at the annual meeting of the Central Surgical Association. A previous study, he said, had shown no efficacy for pump-delivered bupivacaine when it was delivered directly into the hernia sac.
In the present study, the cannula for the pump was placed inferior to the costochondral margin, with catheters tunnelled bilaterally between the transversalis fascia and the parietal peritoneum. “The idea behind this was to knock out as many pain receptors as possible,” said Mr. DeAsis.
Primary outcome measures were the self-reported level of postoperative pain and the amount of postoperative medication use. Adult patients at a single site who were undergoing nonemergent ventral hernia repair and who had no history of opioid abuse or adverse reactions to opioids or local anesthetics were enrolled.
Patients were then prospectively randomized to the placebo arm, where they received 400 mL of saline via pump, or to the intervention arm, where they received 400 mL of 0.5% bupivacaine following LVHR. The pump delivered the study drug for a period of 4 days after placement in both groups.
Patients completed a pain diary and medication log for the 7 days after discharge. They rated their current pain level, reported how frequently they had pain, and reported satisfaction with pain control.
In-hospital use of opioid pain medication did not differ significantly between groups, but patients receiving bupivacaine through their pumps used significantly less ketorolac as inpatients (mean 30 mg, compared with 53 mg in the saline group, P = .01).
“Ketorolac is our first-line; we try to avoid narcotics for many different reasons ... Ketorolac PRN was ordered for everybody,” said Dr. Michael Ujiki, a general surgeon at NorthShore University HealthSystem, Evanston, Ill., and senior investigator for the study.
The median pain score on a 1-10 Likert scale at discharge was 2.0 (interquartile range, 0.0-3.0) for the bupivacaine group and 4.0 (interquartile range, 2.0-5.0) for the saline group (P = .06).
Once patients were discharged from the hospital, patients receiving bupivacaine had significantly less postoperative pain through postoperative day 4 and significantly less frequent pain through postoperative day 4. Finally, they reported being significantly more satisfied with their pain management scores on postoperative days 1-3.
The study was limited by the small cohort size. “The primary reason behind this was that during recruitment, we had a lot of patients voice concern about potentially receiving placebo,” thereby forgoing the chance for additional pain relief, said Mr. DeAsis. “A well-powered study should be conducted in the future.”
The investigators reported no relevant financial disclosures, but all study pumps were provided by the manufacturer, On-Q.
On Twitter @karioakes
MONTREAL – An elastomeric pump delivering local anesthetic to the site of a ventral hernia repair can significantly reduce postoperative pain, according to a small, single-center study.
The prospective, randomized, double-blind placebo-controlled trial of the use of an elastomeric bupivacaine-containing pump for pain management after laparoscopic ventral hernia repair (LVHR) found that patients receiving bupivacaine reported significantly less postoperative pain than did those receiving saline through the pump.
“The idea is that the use of this technology, in combination with standard postoperative pain medication, would do two things: It would maintain the same level of postoperative pain control, as well as reducing the amount of narcotic and non-narcotic pain medication used,” said Francis DeAsis, a medical student at Midwestern University, Chicago.
“For our project, we focused on the specific anatomic location of the catheter,” Mr. DeAsis said in a presentation at the annual meeting of the Central Surgical Association. A previous study, he said, had shown no efficacy for pump-delivered bupivacaine when it was delivered directly into the hernia sac.
In the present study, the cannula for the pump was placed inferior to the costochondral margin, with catheters tunnelled bilaterally between the transversalis fascia and the parietal peritoneum. “The idea behind this was to knock out as many pain receptors as possible,” said Mr. DeAsis.
Primary outcome measures were the self-reported level of postoperative pain and the amount of postoperative medication use. Adult patients at a single site who were undergoing nonemergent ventral hernia repair and who had no history of opioid abuse or adverse reactions to opioids or local anesthetics were enrolled.
Patients were then prospectively randomized to the placebo arm, where they received 400 mL of saline via pump, or to the intervention arm, where they received 400 mL of 0.5% bupivacaine following LVHR. The pump delivered the study drug for a period of 4 days after placement in both groups.
Patients completed a pain diary and medication log for the 7 days after discharge. They rated their current pain level, reported how frequently they had pain, and reported satisfaction with pain control.
In-hospital use of opioid pain medication did not differ significantly between groups, but patients receiving bupivacaine through their pumps used significantly less ketorolac as inpatients (mean 30 mg, compared with 53 mg in the saline group, P = .01).
“Ketorolac is our first-line; we try to avoid narcotics for many different reasons ... Ketorolac PRN was ordered for everybody,” said Dr. Michael Ujiki, a general surgeon at NorthShore University HealthSystem, Evanston, Ill., and senior investigator for the study.
The median pain score on a 1-10 Likert scale at discharge was 2.0 (interquartile range, 0.0-3.0) for the bupivacaine group and 4.0 (interquartile range, 2.0-5.0) for the saline group (P = .06).
Once patients were discharged from the hospital, patients receiving bupivacaine had significantly less postoperative pain through postoperative day 4 and significantly less frequent pain through postoperative day 4. Finally, they reported being significantly more satisfied with their pain management scores on postoperative days 1-3.
The study was limited by the small cohort size. “The primary reason behind this was that during recruitment, we had a lot of patients voice concern about potentially receiving placebo,” thereby forgoing the chance for additional pain relief, said Mr. DeAsis. “A well-powered study should be conducted in the future.”
The investigators reported no relevant financial disclosures, but all study pumps were provided by the manufacturer, On-Q.
On Twitter @karioakes
MONTREAL – An elastomeric pump delivering local anesthetic to the site of a ventral hernia repair can significantly reduce postoperative pain, according to a small, single-center study.
The prospective, randomized, double-blind placebo-controlled trial of the use of an elastomeric bupivacaine-containing pump for pain management after laparoscopic ventral hernia repair (LVHR) found that patients receiving bupivacaine reported significantly less postoperative pain than did those receiving saline through the pump.
“The idea is that the use of this technology, in combination with standard postoperative pain medication, would do two things: It would maintain the same level of postoperative pain control, as well as reducing the amount of narcotic and non-narcotic pain medication used,” said Francis DeAsis, a medical student at Midwestern University, Chicago.
“For our project, we focused on the specific anatomic location of the catheter,” Mr. DeAsis said in a presentation at the annual meeting of the Central Surgical Association. A previous study, he said, had shown no efficacy for pump-delivered bupivacaine when it was delivered directly into the hernia sac.
In the present study, the cannula for the pump was placed inferior to the costochondral margin, with catheters tunnelled bilaterally between the transversalis fascia and the parietal peritoneum. “The idea behind this was to knock out as many pain receptors as possible,” said Mr. DeAsis.
Primary outcome measures were the self-reported level of postoperative pain and the amount of postoperative medication use. Adult patients at a single site who were undergoing nonemergent ventral hernia repair and who had no history of opioid abuse or adverse reactions to opioids or local anesthetics were enrolled.
Patients were then prospectively randomized to the placebo arm, where they received 400 mL of saline via pump, or to the intervention arm, where they received 400 mL of 0.5% bupivacaine following LVHR. The pump delivered the study drug for a period of 4 days after placement in both groups.
Patients completed a pain diary and medication log for the 7 days after discharge. They rated their current pain level, reported how frequently they had pain, and reported satisfaction with pain control.
In-hospital use of opioid pain medication did not differ significantly between groups, but patients receiving bupivacaine through their pumps used significantly less ketorolac as inpatients (mean 30 mg, compared with 53 mg in the saline group, P = .01).
“Ketorolac is our first-line; we try to avoid narcotics for many different reasons ... Ketorolac PRN was ordered for everybody,” said Dr. Michael Ujiki, a general surgeon at NorthShore University HealthSystem, Evanston, Ill., and senior investigator for the study.
The median pain score on a 1-10 Likert scale at discharge was 2.0 (interquartile range, 0.0-3.0) for the bupivacaine group and 4.0 (interquartile range, 2.0-5.0) for the saline group (P = .06).
Once patients were discharged from the hospital, patients receiving bupivacaine had significantly less postoperative pain through postoperative day 4 and significantly less frequent pain through postoperative day 4. Finally, they reported being significantly more satisfied with their pain management scores on postoperative days 1-3.
The study was limited by the small cohort size. “The primary reason behind this was that during recruitment, we had a lot of patients voice concern about potentially receiving placebo,” thereby forgoing the chance for additional pain relief, said Mr. DeAsis. “A well-powered study should be conducted in the future.”
The investigators reported no relevant financial disclosures, but all study pumps were provided by the manufacturer, On-Q.
On Twitter @karioakes
AT THE ANNUAL MEETING OF THE CENTRAL SURGICAL ASSOCIATION
Key clinical point: Pain scores were reduced when patients had pump-delivered local anesthetic after hernia repair.
Major finding: Pain level and frequency of pain were reduced, and satisfaction with pain control was increased, when patients received local anesthetic via pump after laparoscopic ventral hernia repair.
Data source: Single-center double-blind, placebo-controlled study of elastomeric pump-delivered bupivacaine vs. placebo in 29 patients undergoing laparoscopic ventral hernia repair.
Disclosures: The pump was supplied by the manufacturer (On-Q); the investigators reported no other relevant disclosures.

Staph aureus drives atopic dermatitis
LOS ANGELES – Evidence is building for the hypothesis that impairments in the skin’s microbiome promote Staphylococcus aureus colonization and drive atopic dermatitis, Dr. Donald Y.M. Leung said at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
The link between the bacteria and atopic dermatitis has long been discussed, but its role in pathogenesis still needs definition, he said.

“We’ve never been able to look at the total bacterial composition of the skin, but now with next-generation sequencing it’s finally possible to look at all the phylla and species. Other investigators have shown that during flares of atopic dermatitis there’s a reduction in bacterial diversity and an increase in staph, with S. aureus being particularly abundant. Then, post-flare, you see see a drop in S. aureus; this clearly suggests (it’s) important,” according to Dr. Leung, head of the division of pediatric allergy and immunology at National Jewish Health in Denver and professor of pediatrics at the University of Colorado.
Staphylococcus aureus is known to secrete virulence factors including cytotoxins, superantigens, lipases, and proteases that activate inflammatory cells and can cause significant skin barrier dysfunction.
The discovery that filaggrin mutations result in structural abnormalities in the skin barrier and are associated with sharply increased rates of atopic dermatitis and peanut allergy have strengthened the association, but filaggrin can’t be the whole story. Mutations in filaggrin are largely confined to individuals of Northern European ancestry; African Americans don’t have filaggrin mutations.
Yet atopic dermatitis is a global phenomenon. Further, a skin barrier defect is not enough to cause atopic dermatitis, Dr. Leung said. But such a defect, whether caused by a filaggrin mutation or something else, allows S. aureus to attach to and colonize the skin. Staph overgrowth or infection then activates an inflammatory cell cascade involving natural killer T cells, mast cells, cytokines, and Langerhans cells. That’s why the most effective treatments for atopic dermatitis address both the need to rebuild the skin barrier as well as the counterproductive immune response, he added.
Elsewhere at the AAAAI meeting, Dr. Andrea L. Jones, of National Jewish Health, presented an analysis of 718 children and adolescents with atopic dermatitis, all of whom had been cultured for S. aureus, in that institution’s database. Methicillin-resistant S. aureus (MRSA) was found in 19%; 57% were positive for methicillin-sensitive S. aureus (MSSA) and 23% lacked S. aureus. Of note, the prevalence of peanut allergy was highest at 78% in the group with MRSA; the prevalence was 39% in those with MSSA and 4% in those without S. aureus.
The prevalence of allergies to wheat, egg, milk, or soybeans in the youths with atopic dermatitis was unrelated to MRSA colonization.
“Our hypothesis – although we need to do a prospective study – is that staph colonization may lead to barrier dysfunction and thus allow environmental allergens to invade through the skin. Interestingly enough, people who weren’t colonized by staph had a very low level of sensitization to peanut,” said Dr. Leung, who was the senior investigator in the study.
Dr. Leung was a coauthor on another study that points to a potential new avenue of treatment in atopic dermatitis. Presented by investigators at the University of California, San Diego, at a recent meeting of the Society for Investigative Dermatology, the study showed that atopic dermatitis is marked by a defect in the commensal skin bacteria which normally keep S. aureus in check.
In that study, the amount of S. aureus growing on a defined area of lesional skin of atopic dermatitis patients was nearly 10-fold greater than that on nonlesional skin and the skin of controls without atopic dermatitis.
Commensal bacteria on lesional skin may possess markedly reduced antimicrobial activity. The NIH-sponsored Atopic Dermatitis Research Network plans to conduct clinical trials to see if transplanting beneficial commensal bacteria will reduce staph colonization in atopic dermatitis patients and thereby result in therapeutic benefit, Dr. Leung noted.
He reported serving on scientific advisory boards for more than half a dozen pharmaceutical companies and receiving numerous research grants from the NIH.
LOS ANGELES – Evidence is building for the hypothesis that impairments in the skin’s microbiome promote Staphylococcus aureus colonization and drive atopic dermatitis, Dr. Donald Y.M. Leung said at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
The link between the bacteria and atopic dermatitis has long been discussed, but its role in pathogenesis still needs definition, he said.
“We’ve never been able to look at the total bacterial composition of the skin, but now with next-generation sequencing it’s finally possible to look at all the phylla and species. Other investigators have shown that during flares of atopic dermatitis there’s a reduction in bacterial diversity and an increase in staph, with S. aureus being particularly abundant. Then, post-flare, you see see a drop in S. aureus; this clearly suggests (it’s) important,” according to Dr. Leung, head of the division of pediatric allergy and immunology at National Jewish Health in Denver and professor of pediatrics at the University of Colorado.
Staphylococcus aureus is known to secrete virulence factors including cytotoxins, superantigens, lipases, and proteases that activate inflammatory cells and can cause significant skin barrier dysfunction.
The discovery that filaggrin mutations result in structural abnormalities in the skin barrier and are associated with sharply increased rates of atopic dermatitis and peanut allergy have strengthened the association, but filaggrin can’t be the whole story. Mutations in filaggrin are largely confined to individuals of Northern European ancestry; African Americans don’t have filaggrin mutations.
Yet atopic dermatitis is a global phenomenon. Further, a skin barrier defect is not enough to cause atopic dermatitis, Dr. Leung said. But such a defect, whether caused by a filaggrin mutation or something else, allows S. aureus to attach to and colonize the skin. Staph overgrowth or infection then activates an inflammatory cell cascade involving natural killer T cells, mast cells, cytokines, and Langerhans cells. That’s why the most effective treatments for atopic dermatitis address both the need to rebuild the skin barrier as well as the counterproductive immune response, he added.
Elsewhere at the AAAAI meeting, Dr. Andrea L. Jones, of National Jewish Health, presented an analysis of 718 children and adolescents with atopic dermatitis, all of whom had been cultured for S. aureus, in that institution’s database. Methicillin-resistant S. aureus (MRSA) was found in 19%; 57% were positive for methicillin-sensitive S. aureus (MSSA) and 23% lacked S. aureus. Of note, the prevalence of peanut allergy was highest at 78% in the group with MRSA; the prevalence was 39% in those with MSSA and 4% in those without S. aureus.
The prevalence of allergies to wheat, egg, milk, or soybeans in the youths with atopic dermatitis was unrelated to MRSA colonization.
“Our hypothesis – although we need to do a prospective study – is that staph colonization may lead to barrier dysfunction and thus allow environmental allergens to invade through the skin. Interestingly enough, people who weren’t colonized by staph had a very low level of sensitization to peanut,” said Dr. Leung, who was the senior investigator in the study.
Dr. Leung was a coauthor on another study that points to a potential new avenue of treatment in atopic dermatitis. Presented by investigators at the University of California, San Diego, at a recent meeting of the Society for Investigative Dermatology, the study showed that atopic dermatitis is marked by a defect in the commensal skin bacteria which normally keep S. aureus in check.
In that study, the amount of S. aureus growing on a defined area of lesional skin of atopic dermatitis patients was nearly 10-fold greater than that on nonlesional skin and the skin of controls without atopic dermatitis.
Commensal bacteria on lesional skin may possess markedly reduced antimicrobial activity. The NIH-sponsored Atopic Dermatitis Research Network plans to conduct clinical trials to see if transplanting beneficial commensal bacteria will reduce staph colonization in atopic dermatitis patients and thereby result in therapeutic benefit, Dr. Leung noted.
He reported serving on scientific advisory boards for more than half a dozen pharmaceutical companies and receiving numerous research grants from the NIH.
LOS ANGELES – Evidence is building for the hypothesis that impairments in the skin’s microbiome promote Staphylococcus aureus colonization and drive atopic dermatitis, Dr. Donald Y.M. Leung said at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
The link between the bacteria and atopic dermatitis has long been discussed, but its role in pathogenesis still needs definition, he said.
“We’ve never been able to look at the total bacterial composition of the skin, but now with next-generation sequencing it’s finally possible to look at all the phylla and species. Other investigators have shown that during flares of atopic dermatitis there’s a reduction in bacterial diversity and an increase in staph, with S. aureus being particularly abundant. Then, post-flare, you see see a drop in S. aureus; this clearly suggests (it’s) important,” according to Dr. Leung, head of the division of pediatric allergy and immunology at National Jewish Health in Denver and professor of pediatrics at the University of Colorado.
Staphylococcus aureus is known to secrete virulence factors including cytotoxins, superantigens, lipases, and proteases that activate inflammatory cells and can cause significant skin barrier dysfunction.
The discovery that filaggrin mutations result in structural abnormalities in the skin barrier and are associated with sharply increased rates of atopic dermatitis and peanut allergy have strengthened the association, but filaggrin can’t be the whole story. Mutations in filaggrin are largely confined to individuals of Northern European ancestry; African Americans don’t have filaggrin mutations.
Yet atopic dermatitis is a global phenomenon. Further, a skin barrier defect is not enough to cause atopic dermatitis, Dr. Leung said. But such a defect, whether caused by a filaggrin mutation or something else, allows S. aureus to attach to and colonize the skin. Staph overgrowth or infection then activates an inflammatory cell cascade involving natural killer T cells, mast cells, cytokines, and Langerhans cells. That’s why the most effective treatments for atopic dermatitis address both the need to rebuild the skin barrier as well as the counterproductive immune response, he added.
Elsewhere at the AAAAI meeting, Dr. Andrea L. Jones, of National Jewish Health, presented an analysis of 718 children and adolescents with atopic dermatitis, all of whom had been cultured for S. aureus, in that institution’s database. Methicillin-resistant S. aureus (MRSA) was found in 19%; 57% were positive for methicillin-sensitive S. aureus (MSSA) and 23% lacked S. aureus. Of note, the prevalence of peanut allergy was highest at 78% in the group with MRSA; the prevalence was 39% in those with MSSA and 4% in those without S. aureus.
The prevalence of allergies to wheat, egg, milk, or soybeans in the youths with atopic dermatitis was unrelated to MRSA colonization.
“Our hypothesis – although we need to do a prospective study – is that staph colonization may lead to barrier dysfunction and thus allow environmental allergens to invade through the skin. Interestingly enough, people who weren’t colonized by staph had a very low level of sensitization to peanut,” said Dr. Leung, who was the senior investigator in the study.
Dr. Leung was a coauthor on another study that points to a potential new avenue of treatment in atopic dermatitis. Presented by investigators at the University of California, San Diego, at a recent meeting of the Society for Investigative Dermatology, the study showed that atopic dermatitis is marked by a defect in the commensal skin bacteria which normally keep S. aureus in check.
In that study, the amount of S. aureus growing on a defined area of lesional skin of atopic dermatitis patients was nearly 10-fold greater than that on nonlesional skin and the skin of controls without atopic dermatitis.
Commensal bacteria on lesional skin may possess markedly reduced antimicrobial activity. The NIH-sponsored Atopic Dermatitis Research Network plans to conduct clinical trials to see if transplanting beneficial commensal bacteria will reduce staph colonization in atopic dermatitis patients and thereby result in therapeutic benefit, Dr. Leung noted.
He reported serving on scientific advisory boards for more than half a dozen pharmaceutical companies and receiving numerous research grants from the NIH.
AT 2016 AAAAI ANNUAL MEETING

Ischemic Hepatitis Associated with High Inpatient Mortality
Clinical question: What is the incidence and outcome of patients with ischemic hepatitis?

Background: Ischemic hepatitis, or shock liver, is often diagnosed in patients with massive increase in aminotransferase levels most often exceeding 1000 IU/L in the setting of hepatic hypoperfusion. The data on overall incidence and mortality of these patients are limited.
Study Design: Systematic review and meta-analysis.
Setting: Variable.
Synopsis: Using a combination of PubMed, Embase, and Web of Science, the study included 24 papers on incidence and outcomes of ischemic hepatitis published between 1965 and 2015 with a combined total of 1,782 cases. The incidence of ischemic hepatitis varied based on patient location with incidence of 2/1000 in all inpatient admissions and 2.5/100 in ICU admissions. The majority of patients suffered from cardiac comorbidities and decompensation during their admission. Inpatient mortality with ischemic hepatitis was 49%.
Interestingly, only 52.9% of patients had an episode of documented hypotension.
Hospitalists taking care of patients with massive rise in aminotransferases should consider ischemic hepatitis higher in their differential, even in the absence of documented hypotension.
There was significant variability in study design, sample size, and inclusion criteria among the studies, which reduces generalizability of this systematic review.
Bottom line: Ischemic hepatitis is associated with very high mortality and should be suspected in patients with high levels of alanine aminotransferase/aspartate aminotransferase even in the absence of documented hypotension.
Citation: Tapper EB, Sengupta N, Bonder A. The incidence and outcomes of ischemic hepatitis: a systematic review with meta-analysis. Am J Med. 2015;128(12):1314-1321.
Short Take
Music Can Help Ease Pain and Anxiety after Surgery
A systematic review and meta-analysis showed that music reduces pain and anxiety and decreases the need for pain medication in postoperative patients regardless of type of music or at what interval of the operative period the music was initiated.
Citation: Hole J, Hirsch M, Ball E, Meads C. Music as an aid for postoperative recovery in adults: a systematic review and meta-analysis. Lancet. 2015;386(10004):1659-1671
Clinical question: What is the incidence and outcome of patients with ischemic hepatitis?
Background: Ischemic hepatitis, or shock liver, is often diagnosed in patients with massive increase in aminotransferase levels most often exceeding 1000 IU/L in the setting of hepatic hypoperfusion. The data on overall incidence and mortality of these patients are limited.
Study Design: Systematic review and meta-analysis.
Setting: Variable.
Synopsis: Using a combination of PubMed, Embase, and Web of Science, the study included 24 papers on incidence and outcomes of ischemic hepatitis published between 1965 and 2015 with a combined total of 1,782 cases. The incidence of ischemic hepatitis varied based on patient location with incidence of 2/1000 in all inpatient admissions and 2.5/100 in ICU admissions. The majority of patients suffered from cardiac comorbidities and decompensation during their admission. Inpatient mortality with ischemic hepatitis was 49%.
Interestingly, only 52.9% of patients had an episode of documented hypotension.
Hospitalists taking care of patients with massive rise in aminotransferases should consider ischemic hepatitis higher in their differential, even in the absence of documented hypotension.
There was significant variability in study design, sample size, and inclusion criteria among the studies, which reduces generalizability of this systematic review.
Bottom line: Ischemic hepatitis is associated with very high mortality and should be suspected in patients with high levels of alanine aminotransferase/aspartate aminotransferase even in the absence of documented hypotension.
Citation: Tapper EB, Sengupta N, Bonder A. The incidence and outcomes of ischemic hepatitis: a systematic review with meta-analysis. Am J Med. 2015;128(12):1314-1321.
Short Take
Music Can Help Ease Pain and Anxiety after Surgery
A systematic review and meta-analysis showed that music reduces pain and anxiety and decreases the need for pain medication in postoperative patients regardless of type of music or at what interval of the operative period the music was initiated.
Citation: Hole J, Hirsch M, Ball E, Meads C. Music as an aid for postoperative recovery in adults: a systematic review and meta-analysis. Lancet. 2015;386(10004):1659-1671
Clinical question: What is the incidence and outcome of patients with ischemic hepatitis?
Background: Ischemic hepatitis, or shock liver, is often diagnosed in patients with massive increase in aminotransferase levels most often exceeding 1000 IU/L in the setting of hepatic hypoperfusion. The data on overall incidence and mortality of these patients are limited.
Study Design: Systematic review and meta-analysis.
Setting: Variable.
Synopsis: Using a combination of PubMed, Embase, and Web of Science, the study included 24 papers on incidence and outcomes of ischemic hepatitis published between 1965 and 2015 with a combined total of 1,782 cases. The incidence of ischemic hepatitis varied based on patient location with incidence of 2/1000 in all inpatient admissions and 2.5/100 in ICU admissions. The majority of patients suffered from cardiac comorbidities and decompensation during their admission. Inpatient mortality with ischemic hepatitis was 49%.
Interestingly, only 52.9% of patients had an episode of documented hypotension.
Hospitalists taking care of patients with massive rise in aminotransferases should consider ischemic hepatitis higher in their differential, even in the absence of documented hypotension.
There was significant variability in study design, sample size, and inclusion criteria among the studies, which reduces generalizability of this systematic review.
Bottom line: Ischemic hepatitis is associated with very high mortality and should be suspected in patients with high levels of alanine aminotransferase/aspartate aminotransferase even in the absence of documented hypotension.
Citation: Tapper EB, Sengupta N, Bonder A. The incidence and outcomes of ischemic hepatitis: a systematic review with meta-analysis. Am J Med. 2015;128(12):1314-1321.
Short Take
Music Can Help Ease Pain and Anxiety after Surgery
A systematic review and meta-analysis showed that music reduces pain and anxiety and decreases the need for pain medication in postoperative patients regardless of type of music or at what interval of the operative period the music was initiated.
Citation: Hole J, Hirsch M, Ball E, Meads C. Music as an aid for postoperative recovery in adults: a systematic review and meta-analysis. Lancet. 2015;386(10004):1659-1671

MicroRNA could be used to treat DLBCL, team says

A microRNA known as miR-181a dampens signals from the NF-κB pathway and affects the pathogenesis of diffuse large B-cell lymphoma (DLBCL), according to research published in Blood.
The study showed that, by reducing NF-κB signaling, miR-181a hinders tumor cell proliferation and survival.
And the effect is more pronounced in activated B-cell-like (ABC) DLBCL than in germinal center B-cell-like (GCB) DLBCL.
The researchers therefore believe miR-181a could be used to treat ABC DLBCL.
“The miR-181a microRNA is one of the first examples of a pathway that deactivates NF-κB at multiple levels, functioning as a master regulator,” said study author Izidore S. Lossos, MD, of the University of Miami Miller School of Medicine in Florida.
“In certain tumors, there is no expression of this microRNA, which allows cells to propagate. We believe miR-181a could eventually be used
therapeutically.”
To understand the role of miR-181a in the different types of DLBCL, Dr Lossos and his colleaguese studied both cell lines and mouse models.
The team found that miR-181a levels were significantly lower in ABC DLBCL than in GCB DLBCL.
When they increased miR-181a expression in DLBCL cell lines, the researchers observed a reduction in NF-κB activity and a decrease in cell proliferation and survival. These effects were more potent in ABC DLBCL than in GCB DLBCL.
When the researchers increased miR-181a expression in the mouse models, they observed a significant reduction in tumor growth and a significant increase in animal survival, but only in ABC DLBCL. In GCB DLBCL, there were no significant changes compared to controls.
The researchers said the ability of miR-181a to reduce NF-κB levels may be why the presence of miR-181a has been linked to better outcomes for certain DLBCL patients. Previous studies by Dr Lossos’s group have shown that DLBCL patients whose tumors contain more miR-181a have better prognoses.
With the current study, the team found that miR-181a is a master regulator, turning off a number of genes in the NF-κB pathway, including CARD11, a known DLBCL oncogene, and a number of transcription factors that drive NF-κB signaling.
“We knew that miR181a was biomarker for survival,” Dr Lossos said. “This explains the mechanisms behind it.”
In addition to providing a better understanding of the NF-κB pathway, these results provide hope that miR-181a could be used to improve treatment for patients with ABC DLBCL.
“We are trying to develop miR-181a as a potential therapy,” Dr Lossos said. “But we are only at the beginning. Much more work needs to be done. It will not be a simple journey, but we are sure it can be done and tested in humans eventually to see that it indeed will improve patients’ outcomes.” ![]()
A microRNA known as miR-181a dampens signals from the NF-κB pathway and affects the pathogenesis of diffuse large B-cell lymphoma (DLBCL), according to research published in Blood.
The study showed that, by reducing NF-κB signaling, miR-181a hinders tumor cell proliferation and survival.
And the effect is more pronounced in activated B-cell-like (ABC) DLBCL than in germinal center B-cell-like (GCB) DLBCL.
The researchers therefore believe miR-181a could be used to treat ABC DLBCL.
“The miR-181a microRNA is one of the first examples of a pathway that deactivates NF-κB at multiple levels, functioning as a master regulator,” said study author Izidore S. Lossos, MD, of the University of Miami Miller School of Medicine in Florida.
“In certain tumors, there is no expression of this microRNA, which allows cells to propagate. We believe miR-181a could eventually be used
therapeutically.”
To understand the role of miR-181a in the different types of DLBCL, Dr Lossos and his colleaguese studied both cell lines and mouse models.
The team found that miR-181a levels were significantly lower in ABC DLBCL than in GCB DLBCL.
When they increased miR-181a expression in DLBCL cell lines, the researchers observed a reduction in NF-κB activity and a decrease in cell proliferation and survival. These effects were more potent in ABC DLBCL than in GCB DLBCL.
When the researchers increased miR-181a expression in the mouse models, they observed a significant reduction in tumor growth and a significant increase in animal survival, but only in ABC DLBCL. In GCB DLBCL, there were no significant changes compared to controls.
The researchers said the ability of miR-181a to reduce NF-κB levels may be why the presence of miR-181a has been linked to better outcomes for certain DLBCL patients. Previous studies by Dr Lossos’s group have shown that DLBCL patients whose tumors contain more miR-181a have better prognoses.
With the current study, the team found that miR-181a is a master regulator, turning off a number of genes in the NF-κB pathway, including CARD11, a known DLBCL oncogene, and a number of transcription factors that drive NF-κB signaling.
“We knew that miR181a was biomarker for survival,” Dr Lossos said. “This explains the mechanisms behind it.”
In addition to providing a better understanding of the NF-κB pathway, these results provide hope that miR-181a could be used to improve treatment for patients with ABC DLBCL.
“We are trying to develop miR-181a as a potential therapy,” Dr Lossos said. “But we are only at the beginning. Much more work needs to be done. It will not be a simple journey, but we are sure it can be done and tested in humans eventually to see that it indeed will improve patients’ outcomes.” ![]()
A microRNA known as miR-181a dampens signals from the NF-κB pathway and affects the pathogenesis of diffuse large B-cell lymphoma (DLBCL), according to research published in Blood.
The study showed that, by reducing NF-κB signaling, miR-181a hinders tumor cell proliferation and survival.
And the effect is more pronounced in activated B-cell-like (ABC) DLBCL than in germinal center B-cell-like (GCB) DLBCL.
The researchers therefore believe miR-181a could be used to treat ABC DLBCL.
“The miR-181a microRNA is one of the first examples of a pathway that deactivates NF-κB at multiple levels, functioning as a master regulator,” said study author Izidore S. Lossos, MD, of the University of Miami Miller School of Medicine in Florida.
“In certain tumors, there is no expression of this microRNA, which allows cells to propagate. We believe miR-181a could eventually be used
therapeutically.”
To understand the role of miR-181a in the different types of DLBCL, Dr Lossos and his colleaguese studied both cell lines and mouse models.
The team found that miR-181a levels were significantly lower in ABC DLBCL than in GCB DLBCL.
When they increased miR-181a expression in DLBCL cell lines, the researchers observed a reduction in NF-κB activity and a decrease in cell proliferation and survival. These effects were more potent in ABC DLBCL than in GCB DLBCL.
When the researchers increased miR-181a expression in the mouse models, they observed a significant reduction in tumor growth and a significant increase in animal survival, but only in ABC DLBCL. In GCB DLBCL, there were no significant changes compared to controls.
The researchers said the ability of miR-181a to reduce NF-κB levels may be why the presence of miR-181a has been linked to better outcomes for certain DLBCL patients. Previous studies by Dr Lossos’s group have shown that DLBCL patients whose tumors contain more miR-181a have better prognoses.
With the current study, the team found that miR-181a is a master regulator, turning off a number of genes in the NF-κB pathway, including CARD11, a known DLBCL oncogene, and a number of transcription factors that drive NF-κB signaling.
“We knew that miR181a was biomarker for survival,” Dr Lossos said. “This explains the mechanisms behind it.”
In addition to providing a better understanding of the NF-κB pathway, these results provide hope that miR-181a could be used to improve treatment for patients with ABC DLBCL.
“We are trying to develop miR-181a as a potential therapy,” Dr Lossos said. “But we are only at the beginning. Much more work needs to be done. It will not be a simple journey, but we are sure it can be done and tested in humans eventually to see that it indeed will improve patients’ outcomes.” ![]()
Study provides new insight into blood vessel formation
A study published in Nature Cell Biology helps explain how hemodynamic forces contribute to the formation of new vascular lumens during blood vessel morphogenesis.
Investigators found that blood flow drives lumen expansion during sprouting angiogenesis in vivo by inducing spherical deformations of the apical
membrane of endothelial cells, in a process dubbed “inverse blebbing.”
“This work, combined with previous studies, highlights the importance of balanced endothelial cell contractility in allowing the expansion and maintenance of endothelial lumens during blood vessel development,” said study author Holger Gerhardt, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany.
These results challenge the previous idea that sprouting cells expand lumens independently of blood flow during angiogenesis through the generation and fusion of intracellular vacuoles.
The investigators showed that hemodynamic forces dynamically shape the apical membrane of single or groups of endothelial cells during angiogenesis to form and expand new lumenized vascular tubes.
“We find that this process relies on a tight balance between the forces applied on the membrane and the local contractile responses from the endothelial cells, as impairing this balance either way leads to lumen defects,” Dr Gerhardt said.
These findings suggest the process of blebbing does not require a specific polarity but is likely to be generally applicable to situations in which external versus internal pressure differences challenge the stability and elasticity of the actin cortex.
It more generally raises the question of the role of apical membrane contractility in the adaptation to varying hemodynamic environments, both during blood vessel morphogenesis, as connections form or remodel, and in pathological settings.
“Understanding whether and how this plasticity of the apical membrane and its underlying cortex is challenged in pathological conditions, where vessels exhibit altered perfusion and lack organized structure, has the potential to provide deeper insight into mechanisms of vascular adaptation and maladaptation,” Dr Gerhardt said. “We will definitely further investigate this.” ![]()
A study published in Nature Cell Biology helps explain how hemodynamic forces contribute to the formation of new vascular lumens during blood vessel morphogenesis.
Investigators found that blood flow drives lumen expansion during sprouting angiogenesis in vivo by inducing spherical deformations of the apical
membrane of endothelial cells, in a process dubbed “inverse blebbing.”
“This work, combined with previous studies, highlights the importance of balanced endothelial cell contractility in allowing the expansion and maintenance of endothelial lumens during blood vessel development,” said study author Holger Gerhardt, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany.
These results challenge the previous idea that sprouting cells expand lumens independently of blood flow during angiogenesis through the generation and fusion of intracellular vacuoles.
The investigators showed that hemodynamic forces dynamically shape the apical membrane of single or groups of endothelial cells during angiogenesis to form and expand new lumenized vascular tubes.
“We find that this process relies on a tight balance between the forces applied on the membrane and the local contractile responses from the endothelial cells, as impairing this balance either way leads to lumen defects,” Dr Gerhardt said.
These findings suggest the process of blebbing does not require a specific polarity but is likely to be generally applicable to situations in which external versus internal pressure differences challenge the stability and elasticity of the actin cortex.
It more generally raises the question of the role of apical membrane contractility in the adaptation to varying hemodynamic environments, both during blood vessel morphogenesis, as connections form or remodel, and in pathological settings.
“Understanding whether and how this plasticity of the apical membrane and its underlying cortex is challenged in pathological conditions, where vessels exhibit altered perfusion and lack organized structure, has the potential to provide deeper insight into mechanisms of vascular adaptation and maladaptation,” Dr Gerhardt said. “We will definitely further investigate this.” ![]()
A study published in Nature Cell Biology helps explain how hemodynamic forces contribute to the formation of new vascular lumens during blood vessel morphogenesis.
Investigators found that blood flow drives lumen expansion during sprouting angiogenesis in vivo by inducing spherical deformations of the apical
membrane of endothelial cells, in a process dubbed “inverse blebbing.”
“This work, combined with previous studies, highlights the importance of balanced endothelial cell contractility in allowing the expansion and maintenance of endothelial lumens during blood vessel development,” said study author Holger Gerhardt, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany.
These results challenge the previous idea that sprouting cells expand lumens independently of blood flow during angiogenesis through the generation and fusion of intracellular vacuoles.
The investigators showed that hemodynamic forces dynamically shape the apical membrane of single or groups of endothelial cells during angiogenesis to form and expand new lumenized vascular tubes.
“We find that this process relies on a tight balance between the forces applied on the membrane and the local contractile responses from the endothelial cells, as impairing this balance either way leads to lumen defects,” Dr Gerhardt said.
These findings suggest the process of blebbing does not require a specific polarity but is likely to be generally applicable to situations in which external versus internal pressure differences challenge the stability and elasticity of the actin cortex.
It more generally raises the question of the role of apical membrane contractility in the adaptation to varying hemodynamic environments, both during blood vessel morphogenesis, as connections form or remodel, and in pathological settings.
“Understanding whether and how this plasticity of the apical membrane and its underlying cortex is challenged in pathological conditions, where vessels exhibit altered perfusion and lack organized structure, has the potential to provide deeper insight into mechanisms of vascular adaptation and maladaptation,” Dr Gerhardt said. “We will definitely further investigate this.” ![]()
The Current State of PHM Fellowships
Pediatric hospital medicine (PHM) fellowship programs came into existence approximately 20 years ago in Canada,[1] and since that time the number of programs in North America has grown dramatically. The first 3 PHM fellowship programs in the United States were initiated in 2003, and by 2008 there were 7 active programs. Just 5 years later in 2013, there were 20 fellowship programs in existence. Now, in 2015, there are over 30 programs, with several more in development. The goal of postresidency training in PHM is to improve the care of hospitalized children by training future hospitalists to provide high‐quality, evidence‐based clinical care and to generate new knowledge and scholarship in areas such as clinical research, patient safety and quality improvement, medical education, practice management, and patient outcomes.[2] Many pediatric hospitalists want to be able to perform research or quality improvement, but feel that they lack the time, skills, resources, and mentorship to do so.[3] To date, fellowship‐trained hospitalists have a demonstrated track record of contributing to the body of literature that is shaping the care of hospitalized children.[4, 5]
At present, PHM is not a recognized subspecialty of the American Board of Pediatrics (ABP) and therefore does not fall under the purview of the Accreditation Council of Graduate Medical Education (ACGME), leading to concern from some about the variability in depth and breadth of training across programs.[1] The development and publication of the PHM Core Competencies in 2010 helped define the scope of practice of pediatric hospitalists and provide guidelines for training programs, specifically with respect to clinical and nonclinical areas for assessment of competency.[6] Furthermore, studies of early career hospitalists have identified areas for future fellowship curriculum development, such as core procedural skills, quality improvement, and practice management.[7]
In an effort to address training variability across programs, PHM fellowship directors (FDs) have come together as an organized group, first meeting in 2008, with the primary goal of defining training standards and sharing curricular resources. Annual meetings of the FDs, sponsored by the American Academy of Pediatrics Section on Hospital Medicine (AAP‐SOHM), began in 2012. A key objective of this annual meeting has been to develop a standardized fellowship curriculum for use across programs as well as to determine gaps in training that need to be addressed. During this process, we have received input from key stakeholders including community hospitalists, internal medicine‐pediatrics hospitalists, and the PHM Certification Steering Committee, which organized the application for subspecialty certification to the ABP. To inform this process of curriculum standardization, we fielded a survey of PHM fellowship directors. The purpose of this article is to summarize the current curricula, operations, and logistics of PHM fellowship programs.
METHODS
This was a cross‐sectional study of 31 PHM fellowship programs across the United States and Canada in April 2014. Inclusion criteria included all pediatric fellowships that were self‐identified to the AAP‐SOHM as providing a hospital medicine fellowship option. This included both PHM fellowships as well as academic general pediatric fellowships with a hospitalist track. A web‐based survey (SurveyMonkey, Inc.) was distributed by e‐mail to the FDs at the 31 training programs (see Supporting Information in the online version of this article). To enhance content validity of survey responses, survey questions were designed using an iterative consensus process among the authors, who included junior and senior FDs and represented the 2014 annual FD meeting planning committee. Items were created to gather feedback on the following key areas of PHM fellowships: program demographics, types of required and elective clinical rotations, graduate coursework offerings, amount of time spent in clinical activities, fellow billing practices, and description of fellows' research activities. The survey consisted of 30 multiple‐choice and short‐answer questions. Follow‐up e‐mail reminders were sent to all FDs 2 weeks and 4 weeks after the initial request was sent. Survey completion was voluntary, and no incentives were offered. The study was determined to be exempt by the Stanford University Institutional Review Board. Data were summarized using frequency distributions. No subgroup comparisons were made.
RESULTS
Program directors from 27/31 (87%) PHM fellowship programs responded to the survey; 25 were active programs, and 2 were under development. Responding programs represented all 4 major regions of the country and Canada, with varying program initiation dates, ranging from 1997 to 2013.
Program Demographics
The duration of most programs (17/27) was 2 years (63%), with 6 (22%) 1‐year programs and 4 (15%) 3‐year programs making up the remainder. Four programs described variable lengths, which could be tailored based on the fellow's individual interest. Two of the programs are 2 years in length, but offer a 1‐year option for fellows who wish to focus on enhancing clinical skills without an academic focus. The other 2 programs are 2 years in length, but will offer an extension to a third year for those pursuing a graduate degree.
Fellow Clinical Activities
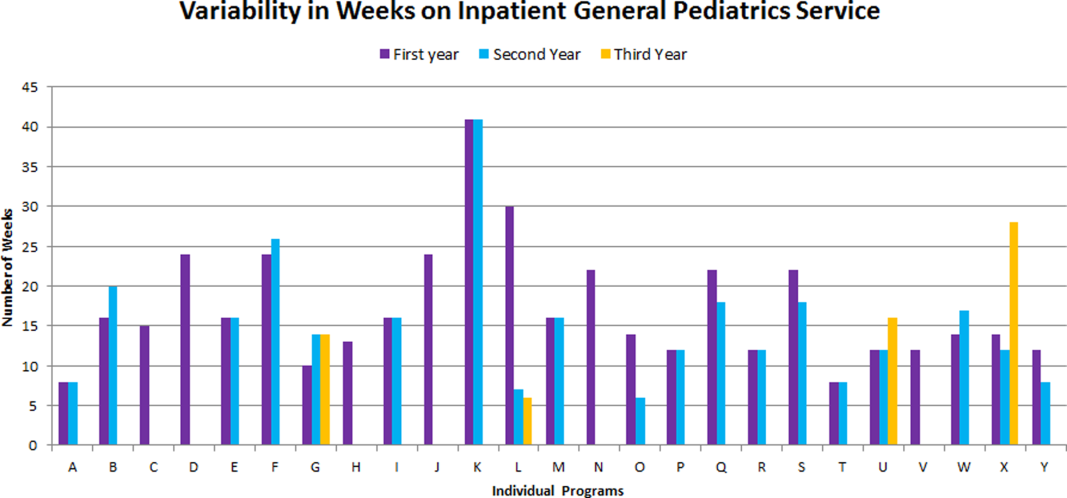
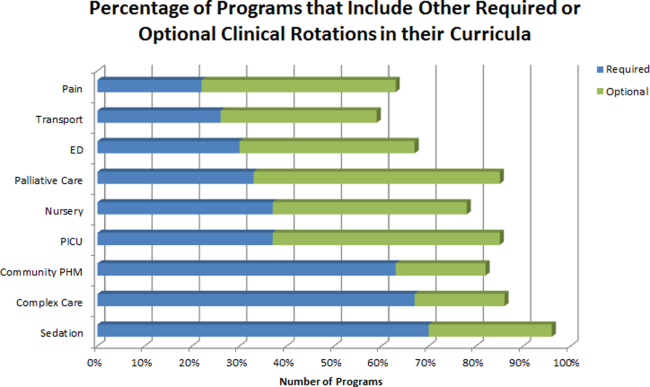
The average amount of total clinical time (weeks on service) across responding programs was 50% (range, 20%65%). When looking specifically at time on the inpatient general pediatric service, number of weeks varied by year of training and by institution, with 12 to 41 weeks in the first year of fellowship, 6 to 41 weeks in the second year of fellowship, and 6 to 28 weeks in the third year of fellowship (Figure 1). Though the range is large, on average, fellows spend 17 weeks on inpatient general pediatrics service during each year of training. Of note, the median number of weeks on inpatient general pediatrics service by year of training was 15 weeks, 16 weeks, and 16.5 weeks, respectively. In addition to inpatient general pediatrics service time, most programs require other clinical rotations, with sedation, complex care, and inpatient pediatrics at community sites being the most frequent (Figure 2). Of the 6 responding 1‐year programs, 5 (83%) allow fellows to bill/generate clinical revenue at some point during their training. Of the 15 responding 2‐year programs, 11 (73%) allow fellows to bill/generate clinical revenue at some point during their training. Of the 4 responding 3‐year programs, 2 (50%) allow their fellows to bill/generate clinical revenue at some point during their training.
Fellow Scholarly Activities
With respect to time dedicated to research, the majority of programs offer coursework such as courses for credit, noncredit courses, or certificate courses. In addition, 11 programs offer fellows a masters' degree in areas including public health, clinical science, epidemiology, education, academic sciences, healthcare quality, clinical and translational research, or health services administration. The majority of these degrees are paid for by departmental funds, with tuition reimbursement, university support, training grants, and personal funds making up the remainder. Twenty‐one (81%) programs provide a scholarship oversight committee for their fellows. Current fellows' (n = 63) primary areas of research are varied and include clinical research (36%), quality‐improvement research (22%), medical education research (20%), health services research (16%), and other areas (6%).
DISCUSSION
This is the most comprehensive description of pediatric hospital medicine fellowship curricula to date. Understanding the scope of these programs is an important first step in developing a standardized curriculum that can be used by all. The results of this survey indicate that although there is variability among PHM fellowship curricular content, several common themes exist.
The number of clinical weeks on the inpatient general pediatrics service varied from program to program, though the majority of programs require fellows to spend 15 to 16 weeks each year of training. The variability may be due in part to the way in which respondents defined the term week on clinical service. For example, if the fellow is primarily on a shift schedule, then he/she may only work 2 to 3 shifts in 1 week, which may have been viewed similarly to daily presence on a more traditional inpatient teaching service with 5 to 7 consecutive days of service. The current study did not explore the details of inpatient general pediatric clinical activities or exposure to opportunities to hone procedural skills, areas that are worth investigating as we move forward to better understand the needs of trainees.
Most residency training programs in general pediatrics require a significant amount of inpatient clinical time, specifically a minimum of 10 units or months, though only half of this time is required to be in inpatient general pediatrics.[8] Although nonfellowship trained early career hospitalists may feel adequately prepared to manage the clinical care of some hospitalized children, perceived competency is significantly lower than their fellowship‐trained colleagues with regard to care of the child with medical complexity and technology‐dependence, and with regard to provision of sedation for procedures.[7] The majority of FDs surveyed in our study indicated that additional clinical experience with sedation, complex care, and inpatient pediatrics at community sites were required of their fellows. Of note, many of these rotations are not commonly required in pediatric residency training programs; however, the PHM core competencies suggest that hospitalists should demonstrate proficiency in these areas to provide optimal care for hospitalized children. Our results suggest that current PHM fellowship curricula help address these clinical gaps. The requirement of these particular specialized experiences may reflect the clinical scope of practice that is expected from potential employers or may be related to staffing needs. It is well documented that the inpatient demographic of large pediatric tertiary care referral centers has changed over the past decade, with an increasing prevalence of children with medical complexity.[9, 10] In both tertiary referral centers and community hospitals, the expansion of the role of the hospitalist in providing specialized clinical services, such as sedation or surgical comanagement, has been significantly driven by financial factors, though a more recent focus on improvement of efficiency and quality of care within the hospital system has relied heavily on hospitalist input.[11, 12, 13] Important next steps in curriculum standardization include ensuring that training programs allow for adequate clinical exposure and proper assessment of competency in these areas, and determining the full complement of clinical training experiences that will produce hospitalists with a well‐defined scope of practice that adequately addresses the needs of hospitalized children.
Most fellowship‐trained hospitalists work primarily in university‐affiliated institutions with expectations for scholarly productivity.[5, 7] Fellowship‐trained hospitalists have made large contributions to the growing body of PHM literature, specifically in the realms of medical education, healthcare quality, clinical pediatrics, and healthcare outcomes.[4] Many PHM fellowship‐trained hospitalists have educational or administrative leadership roles.[2] Our results indicate that current PHM fellows continue to be active in a variety of research activities. In addition, FDs reported that the vast majority of programs included scholarship oversight committees, which ensure a mentored and structured research experience. Finally, most programs require or offer additional coursework, and many programs with university affiliations allow for attainment of graduate degrees. Inclusion of robust research training and infrastructure in all programs is a paramount goal of PHM fellowship training. This will allow graduates to be successful researchers, generating new knowledge and supporting the provision of high‐quality, evidence‐based, and value‐driven care for hospitalized children.
A unique feature of several PHM fellowship programs is that fellows are allowed to bill for clinical encounters. Many programs rely on clinical revenue to support fellow salaries.[14] For some programs, a portion of this clinical revenue comes from fellows billing for clinical encounters.[15] Programs that allow fellows to bill/generate clinical revenue have fellows working in attending roles without direct supervision, whereas nonbilling fellows have direct supervision by an attending.[15] In the current ABP training model, subspecialty fellows cannot independently bill for clinical encounters within their own subspecialty, though they can moonlight as long as they meet the duty hour requirements set forth by the ACGME.[16] FDs will need to consider the impact of this requirement on fellow autonomy and on financial revenue for funding fellow salaries if the field achieves ABP subspecialty status.
Regardless of whether or not PHM becomes a designated subspecialty of the ABP, FDs will continue to work together to develop a standard core curriculum that incorporates elements of clinical and nonclinical training to ensure that graduates not only provide high‐quality care for hospitalized children, but also generate new knowledge that advances the field in care delivery and quality of care in any setting. The results of this study will not only help to inform curriculum standardization, but also assessment and evaluation methods. Currently, PHM FDs meet annually and are nearing consensus on a standard 2‐year curriculum based on the PHM Core Competencies that incorporates core clinical, systems, and scholarly domains. We continue to solicit the input of stakeholders, including new FDs, community hospitalist leaders, internal medicine‐pediatrics hospitalist leaders, the Joint Council of Pediatric Hospital Medicine, and leaders of national organizations, such as the American Academy of Pediatrics, Academic Pediatrics Association, and Society of Hospital Medicine. Additional work around standardizing the fellowship application and recruitment process has resulted in our recent acceptance into the Fall Subspecialty Match through the National Residency Match Program, as well as development and implementation of a common fellowship application form. The FD group has recently formalized, voting into place an executive steering committee, which is responsible for the development and execution of long‐term goals that include finalizing a standardized curriculum, refining program and fellow assessment methods through critical evaluation of fellow metrics and outcomes, and standardization of evaluation methods.
Adopting a standard 2‐year curriculum may affect some programs, specifically those that are currently 1 year in duration. These programs would need to extend the length of their fellowship to allow for the breadth of experiences expected with a standardized 2‐year curriculum. This could result in significant financial challenges, effectively increasing the cost to administer the program. In addition, at present, programs have the flexibility to highlight individual areas of strength to attract candidates, allowing fellows to gain an in‐depth experience in domains such as clinical research, quality improvement, medical education, or health services research. With a standardized curriculum, some programs may have to assemble specific clinical and nonclinical experiences to meet the agreed‐upon expectations for PHM fellowship training. If these resources are not available, programs may need to seek relationships with other institutions to complete their offerings, a possibility that is being actively explored by this group. FDs continue to work with each other to share resources, identify training opportunities, and partner with each other to ensure that the requirements of a standard curriculum can be met.
This study has several limitations. First, it was a voluntary survey of program directors, and though we captured over 80% of programs at the time of the survey, there are currently more programs that have come into existence and more still that are in the development stage, leading to potential sampling error. Second, variable effort or accuracy by participants may have led to some degree of response error, such as content error or nonreporting error. Third, the survey questions focused on high‐level information, making it difficult to make nuanced comparisons between curricular elements or determine best curricular practice. In addition, this survey did not explore medical education and quality improvement activities of fellows, 2 major areas in which hospitalists play a major role in the inpatient setting.[1, 17, 18, 19, 20]
CONCLUSION
PHM fellowship programs have grown and continue to grow at a rapid rate. Variability in training is evident, both in clinical experiences and research experiences, though several common elements were identified in this study. The majority of programs are 2 years, and clinical experience comprises approximately 50% of training time, often including key rotations such as sedation, complex care, and rotations at community hospitals. Future directions include standardizing clinical training and expectations for scholarship, formulating appropriate methods for assessment of competency that can be used across programs, and seeking sustainable sources of funding.
Disclosure
Nothing to report.
- , . Characteristics of pediatric hospital medicine fellowships and training programs. J Hosp Med. 2009;4(3):157–163.
- , . Pediatric hospitalists in medical education: current roles and future directions. Curr Probl Pediatr Adolesc Health Care. 2012;42(5):120–126.
- , , . Research needs of pediatric hospitalists. Hosp Pediatr. 2011;1(1):38–44.
- , , , . Pediatric hospital medicine fellowships: outcomes and future directions. Paper presented at: Pediatric Hospital Medicine 2014; July 26, 2014; Orlando, FL.
- , , . Pediatric hospitalist research productivity: predictors of success at presenting abstracts and publishing peer‐reviewed manuscripts among pediatric hospitalists. Hosp Pediatr. 2012;2(3):149–160.
- , , . Pediatric hospital medicine core competencies: development and methodology. J Hosp Med. 2010;5:339–343.
- , , , . Perceived core competency achievements of fellowship and non‐fellowship early career pediatric hospitalists. J Hosp Med. 2015;10(6):373–389.
- Accreditation Council of Graduate Medical Education. ACGME program requirements for graduate medical education in pediatrics. Available at: https://www.acgme.org/acgmeweb/Portals/0/PFAssets/2013‐PR‐FAQ‐PIF/320_pediatrics_07012013.pdf. Published September 30, 2012. Accessed July 7, 2015.
- , , , , , . Increasing prevalence of medically complex children in US hospitals. Pediatrics. 2010;126(4):638–646.
- , , , et al. Children with complex chronic conditions in inpatient hospital settings in the United States. Pediatrics. 2010;126(4):647–655.
- , . The expanding role of hospitalists in the United States. Swiss Med Wkly. 2006;136:591–596.
- , , , , , . Pediatric hospitalist comanagement of spinal fusion surgery patients. J Hosp Med. 2007;2(1):23–30.
- , , , , . Development of a pediatric hospitalist sedation service: training and implementation. J Hosp Med. 2012;7(4):335–339.
- , . Sources of funding and support for pediatric hospital medicine fellowship programs. Poster presented at: Pediatric Hospital Medicine 2014; July 27, 2014; Orlando, FL.
- Council of Pediatric Hospital Medicine Fellowship Directors. Pediatric Hospital Medicine Fellowship Directors Annual Meeting: funding and return on investment. July 24, 2014.
- Accreditation Council of Graduate Medical Education. Frequently asked questions: ACGME common duty hour requirements. Available at: https://www.acgme.org/acgmeweb/Portals/0/PDFs/dh‐faqs2011.pdf. Updated June 18, 2014. Accessed July 7, 2015.
- , . Pediatric hospitalists: training, current practice and career goals. J Hosp Med. 2009;4(3):179–186.
- , . The hospitalist movement and its implications for the care of hospitalized children. Pediatrics. 1999;103:473–477.
- . Pediatric hospitalists and medical education. Pediatr Ann. 2014;43(7):e151–e156
- , , , et al. Quality improvement research in pediatric hospital medicine and the role of the Pediatric Research in Inpatient Settings (PRIS) network. Acad Pediatr. 2013;13(6):S54–S60.
Pediatric hospital medicine (PHM) fellowship programs came into existence approximately 20 years ago in Canada,[1] and since that time the number of programs in North America has grown dramatically. The first 3 PHM fellowship programs in the United States were initiated in 2003, and by 2008 there were 7 active programs. Just 5 years later in 2013, there were 20 fellowship programs in existence. Now, in 2015, there are over 30 programs, with several more in development. The goal of postresidency training in PHM is to improve the care of hospitalized children by training future hospitalists to provide high‐quality, evidence‐based clinical care and to generate new knowledge and scholarship in areas such as clinical research, patient safety and quality improvement, medical education, practice management, and patient outcomes.[2] Many pediatric hospitalists want to be able to perform research or quality improvement, but feel that they lack the time, skills, resources, and mentorship to do so.[3] To date, fellowship‐trained hospitalists have a demonstrated track record of contributing to the body of literature that is shaping the care of hospitalized children.[4, 5]
At present, PHM is not a recognized subspecialty of the American Board of Pediatrics (ABP) and therefore does not fall under the purview of the Accreditation Council of Graduate Medical Education (ACGME), leading to concern from some about the variability in depth and breadth of training across programs.[1] The development and publication of the PHM Core Competencies in 2010 helped define the scope of practice of pediatric hospitalists and provide guidelines for training programs, specifically with respect to clinical and nonclinical areas for assessment of competency.[6] Furthermore, studies of early career hospitalists have identified areas for future fellowship curriculum development, such as core procedural skills, quality improvement, and practice management.[7]
In an effort to address training variability across programs, PHM fellowship directors (FDs) have come together as an organized group, first meeting in 2008, with the primary goal of defining training standards and sharing curricular resources. Annual meetings of the FDs, sponsored by the American Academy of Pediatrics Section on Hospital Medicine (AAP‐SOHM), began in 2012. A key objective of this annual meeting has been to develop a standardized fellowship curriculum for use across programs as well as to determine gaps in training that need to be addressed. During this process, we have received input from key stakeholders including community hospitalists, internal medicine‐pediatrics hospitalists, and the PHM Certification Steering Committee, which organized the application for subspecialty certification to the ABP. To inform this process of curriculum standardization, we fielded a survey of PHM fellowship directors. The purpose of this article is to summarize the current curricula, operations, and logistics of PHM fellowship programs.
METHODS
This was a cross‐sectional study of 31 PHM fellowship programs across the United States and Canada in April 2014. Inclusion criteria included all pediatric fellowships that were self‐identified to the AAP‐SOHM as providing a hospital medicine fellowship option. This included both PHM fellowships as well as academic general pediatric fellowships with a hospitalist track. A web‐based survey (SurveyMonkey, Inc.) was distributed by e‐mail to the FDs at the 31 training programs (see Supporting Information in the online version of this article). To enhance content validity of survey responses, survey questions were designed using an iterative consensus process among the authors, who included junior and senior FDs and represented the 2014 annual FD meeting planning committee. Items were created to gather feedback on the following key areas of PHM fellowships: program demographics, types of required and elective clinical rotations, graduate coursework offerings, amount of time spent in clinical activities, fellow billing practices, and description of fellows' research activities. The survey consisted of 30 multiple‐choice and short‐answer questions. Follow‐up e‐mail reminders were sent to all FDs 2 weeks and 4 weeks after the initial request was sent. Survey completion was voluntary, and no incentives were offered. The study was determined to be exempt by the Stanford University Institutional Review Board. Data were summarized using frequency distributions. No subgroup comparisons were made.
RESULTS
Program directors from 27/31 (87%) PHM fellowship programs responded to the survey; 25 were active programs, and 2 were under development. Responding programs represented all 4 major regions of the country and Canada, with varying program initiation dates, ranging from 1997 to 2013.
Program Demographics
The duration of most programs (17/27) was 2 years (63%), with 6 (22%) 1‐year programs and 4 (15%) 3‐year programs making up the remainder. Four programs described variable lengths, which could be tailored based on the fellow's individual interest. Two of the programs are 2 years in length, but offer a 1‐year option for fellows who wish to focus on enhancing clinical skills without an academic focus. The other 2 programs are 2 years in length, but will offer an extension to a third year for those pursuing a graduate degree.
Fellow Clinical Activities
The average amount of total clinical time (weeks on service) across responding programs was 50% (range, 20%65%). When looking specifically at time on the inpatient general pediatric service, number of weeks varied by year of training and by institution, with 12 to 41 weeks in the first year of fellowship, 6 to 41 weeks in the second year of fellowship, and 6 to 28 weeks in the third year of fellowship (Figure 1). Though the range is large, on average, fellows spend 17 weeks on inpatient general pediatrics service during each year of training. Of note, the median number of weeks on inpatient general pediatrics service by year of training was 15 weeks, 16 weeks, and 16.5 weeks, respectively. In addition to inpatient general pediatrics service time, most programs require other clinical rotations, with sedation, complex care, and inpatient pediatrics at community sites being the most frequent (Figure 2). Of the 6 responding 1‐year programs, 5 (83%) allow fellows to bill/generate clinical revenue at some point during their training. Of the 15 responding 2‐year programs, 11 (73%) allow fellows to bill/generate clinical revenue at some point during their training. Of the 4 responding 3‐year programs, 2 (50%) allow their fellows to bill/generate clinical revenue at some point during their training.
Fellow Scholarly Activities
With respect to time dedicated to research, the majority of programs offer coursework such as courses for credit, noncredit courses, or certificate courses. In addition, 11 programs offer fellows a masters' degree in areas including public health, clinical science, epidemiology, education, academic sciences, healthcare quality, clinical and translational research, or health services administration. The majority of these degrees are paid for by departmental funds, with tuition reimbursement, university support, training grants, and personal funds making up the remainder. Twenty‐one (81%) programs provide a scholarship oversight committee for their fellows. Current fellows' (n = 63) primary areas of research are varied and include clinical research (36%), quality‐improvement research (22%), medical education research (20%), health services research (16%), and other areas (6%).
DISCUSSION
This is the most comprehensive description of pediatric hospital medicine fellowship curricula to date. Understanding the scope of these programs is an important first step in developing a standardized curriculum that can be used by all. The results of this survey indicate that although there is variability among PHM fellowship curricular content, several common themes exist.
The number of clinical weeks on the inpatient general pediatrics service varied from program to program, though the majority of programs require fellows to spend 15 to 16 weeks each year of training. The variability may be due in part to the way in which respondents defined the term week on clinical service. For example, if the fellow is primarily on a shift schedule, then he/she may only work 2 to 3 shifts in 1 week, which may have been viewed similarly to daily presence on a more traditional inpatient teaching service with 5 to 7 consecutive days of service. The current study did not explore the details of inpatient general pediatric clinical activities or exposure to opportunities to hone procedural skills, areas that are worth investigating as we move forward to better understand the needs of trainees.
Most residency training programs in general pediatrics require a significant amount of inpatient clinical time, specifically a minimum of 10 units or months, though only half of this time is required to be in inpatient general pediatrics.[8] Although nonfellowship trained early career hospitalists may feel adequately prepared to manage the clinical care of some hospitalized children, perceived competency is significantly lower than their fellowship‐trained colleagues with regard to care of the child with medical complexity and technology‐dependence, and with regard to provision of sedation for procedures.[7] The majority of FDs surveyed in our study indicated that additional clinical experience with sedation, complex care, and inpatient pediatrics at community sites were required of their fellows. Of note, many of these rotations are not commonly required in pediatric residency training programs; however, the PHM core competencies suggest that hospitalists should demonstrate proficiency in these areas to provide optimal care for hospitalized children. Our results suggest that current PHM fellowship curricula help address these clinical gaps. The requirement of these particular specialized experiences may reflect the clinical scope of practice that is expected from potential employers or may be related to staffing needs. It is well documented that the inpatient demographic of large pediatric tertiary care referral centers has changed over the past decade, with an increasing prevalence of children with medical complexity.[9, 10] In both tertiary referral centers and community hospitals, the expansion of the role of the hospitalist in providing specialized clinical services, such as sedation or surgical comanagement, has been significantly driven by financial factors, though a more recent focus on improvement of efficiency and quality of care within the hospital system has relied heavily on hospitalist input.[11, 12, 13] Important next steps in curriculum standardization include ensuring that training programs allow for adequate clinical exposure and proper assessment of competency in these areas, and determining the full complement of clinical training experiences that will produce hospitalists with a well‐defined scope of practice that adequately addresses the needs of hospitalized children.
Most fellowship‐trained hospitalists work primarily in university‐affiliated institutions with expectations for scholarly productivity.[5, 7] Fellowship‐trained hospitalists have made large contributions to the growing body of PHM literature, specifically in the realms of medical education, healthcare quality, clinical pediatrics, and healthcare outcomes.[4] Many PHM fellowship‐trained hospitalists have educational or administrative leadership roles.[2] Our results indicate that current PHM fellows continue to be active in a variety of research activities. In addition, FDs reported that the vast majority of programs included scholarship oversight committees, which ensure a mentored and structured research experience. Finally, most programs require or offer additional coursework, and many programs with university affiliations allow for attainment of graduate degrees. Inclusion of robust research training and infrastructure in all programs is a paramount goal of PHM fellowship training. This will allow graduates to be successful researchers, generating new knowledge and supporting the provision of high‐quality, evidence‐based, and value‐driven care for hospitalized children.
A unique feature of several PHM fellowship programs is that fellows are allowed to bill for clinical encounters. Many programs rely on clinical revenue to support fellow salaries.[14] For some programs, a portion of this clinical revenue comes from fellows billing for clinical encounters.[15] Programs that allow fellows to bill/generate clinical revenue have fellows working in attending roles without direct supervision, whereas nonbilling fellows have direct supervision by an attending.[15] In the current ABP training model, subspecialty fellows cannot independently bill for clinical encounters within their own subspecialty, though they can moonlight as long as they meet the duty hour requirements set forth by the ACGME.[16] FDs will need to consider the impact of this requirement on fellow autonomy and on financial revenue for funding fellow salaries if the field achieves ABP subspecialty status.
Regardless of whether or not PHM becomes a designated subspecialty of the ABP, FDs will continue to work together to develop a standard core curriculum that incorporates elements of clinical and nonclinical training to ensure that graduates not only provide high‐quality care for hospitalized children, but also generate new knowledge that advances the field in care delivery and quality of care in any setting. The results of this study will not only help to inform curriculum standardization, but also assessment and evaluation methods. Currently, PHM FDs meet annually and are nearing consensus on a standard 2‐year curriculum based on the PHM Core Competencies that incorporates core clinical, systems, and scholarly domains. We continue to solicit the input of stakeholders, including new FDs, community hospitalist leaders, internal medicine‐pediatrics hospitalist leaders, the Joint Council of Pediatric Hospital Medicine, and leaders of national organizations, such as the American Academy of Pediatrics, Academic Pediatrics Association, and Society of Hospital Medicine. Additional work around standardizing the fellowship application and recruitment process has resulted in our recent acceptance into the Fall Subspecialty Match through the National Residency Match Program, as well as development and implementation of a common fellowship application form. The FD group has recently formalized, voting into place an executive steering committee, which is responsible for the development and execution of long‐term goals that include finalizing a standardized curriculum, refining program and fellow assessment methods through critical evaluation of fellow metrics and outcomes, and standardization of evaluation methods.
Adopting a standard 2‐year curriculum may affect some programs, specifically those that are currently 1 year in duration. These programs would need to extend the length of their fellowship to allow for the breadth of experiences expected with a standardized 2‐year curriculum. This could result in significant financial challenges, effectively increasing the cost to administer the program. In addition, at present, programs have the flexibility to highlight individual areas of strength to attract candidates, allowing fellows to gain an in‐depth experience in domains such as clinical research, quality improvement, medical education, or health services research. With a standardized curriculum, some programs may have to assemble specific clinical and nonclinical experiences to meet the agreed‐upon expectations for PHM fellowship training. If these resources are not available, programs may need to seek relationships with other institutions to complete their offerings, a possibility that is being actively explored by this group. FDs continue to work with each other to share resources, identify training opportunities, and partner with each other to ensure that the requirements of a standard curriculum can be met.
This study has several limitations. First, it was a voluntary survey of program directors, and though we captured over 80% of programs at the time of the survey, there are currently more programs that have come into existence and more still that are in the development stage, leading to potential sampling error. Second, variable effort or accuracy by participants may have led to some degree of response error, such as content error or nonreporting error. Third, the survey questions focused on high‐level information, making it difficult to make nuanced comparisons between curricular elements or determine best curricular practice. In addition, this survey did not explore medical education and quality improvement activities of fellows, 2 major areas in which hospitalists play a major role in the inpatient setting.[1, 17, 18, 19, 20]
CONCLUSION
PHM fellowship programs have grown and continue to grow at a rapid rate. Variability in training is evident, both in clinical experiences and research experiences, though several common elements were identified in this study. The majority of programs are 2 years, and clinical experience comprises approximately 50% of training time, often including key rotations such as sedation, complex care, and rotations at community hospitals. Future directions include standardizing clinical training and expectations for scholarship, formulating appropriate methods for assessment of competency that can be used across programs, and seeking sustainable sources of funding.
Disclosure
Nothing to report.
Pediatric hospital medicine (PHM) fellowship programs came into existence approximately 20 years ago in Canada,[1] and since that time the number of programs in North America has grown dramatically. The first 3 PHM fellowship programs in the United States were initiated in 2003, and by 2008 there were 7 active programs. Just 5 years later in 2013, there were 20 fellowship programs in existence. Now, in 2015, there are over 30 programs, with several more in development. The goal of postresidency training in PHM is to improve the care of hospitalized children by training future hospitalists to provide high‐quality, evidence‐based clinical care and to generate new knowledge and scholarship in areas such as clinical research, patient safety and quality improvement, medical education, practice management, and patient outcomes.[2] Many pediatric hospitalists want to be able to perform research or quality improvement, but feel that they lack the time, skills, resources, and mentorship to do so.[3] To date, fellowship‐trained hospitalists have a demonstrated track record of contributing to the body of literature that is shaping the care of hospitalized children.[4, 5]
At present, PHM is not a recognized subspecialty of the American Board of Pediatrics (ABP) and therefore does not fall under the purview of the Accreditation Council of Graduate Medical Education (ACGME), leading to concern from some about the variability in depth and breadth of training across programs.[1] The development and publication of the PHM Core Competencies in 2010 helped define the scope of practice of pediatric hospitalists and provide guidelines for training programs, specifically with respect to clinical and nonclinical areas for assessment of competency.[6] Furthermore, studies of early career hospitalists have identified areas for future fellowship curriculum development, such as core procedural skills, quality improvement, and practice management.[7]
In an effort to address training variability across programs, PHM fellowship directors (FDs) have come together as an organized group, first meeting in 2008, with the primary goal of defining training standards and sharing curricular resources. Annual meetings of the FDs, sponsored by the American Academy of Pediatrics Section on Hospital Medicine (AAP‐SOHM), began in 2012. A key objective of this annual meeting has been to develop a standardized fellowship curriculum for use across programs as well as to determine gaps in training that need to be addressed. During this process, we have received input from key stakeholders including community hospitalists, internal medicine‐pediatrics hospitalists, and the PHM Certification Steering Committee, which organized the application for subspecialty certification to the ABP. To inform this process of curriculum standardization, we fielded a survey of PHM fellowship directors. The purpose of this article is to summarize the current curricula, operations, and logistics of PHM fellowship programs.
METHODS
This was a cross‐sectional study of 31 PHM fellowship programs across the United States and Canada in April 2014. Inclusion criteria included all pediatric fellowships that were self‐identified to the AAP‐SOHM as providing a hospital medicine fellowship option. This included both PHM fellowships as well as academic general pediatric fellowships with a hospitalist track. A web‐based survey (SurveyMonkey, Inc.) was distributed by e‐mail to the FDs at the 31 training programs (see Supporting Information in the online version of this article). To enhance content validity of survey responses, survey questions were designed using an iterative consensus process among the authors, who included junior and senior FDs and represented the 2014 annual FD meeting planning committee. Items were created to gather feedback on the following key areas of PHM fellowships: program demographics, types of required and elective clinical rotations, graduate coursework offerings, amount of time spent in clinical activities, fellow billing practices, and description of fellows' research activities. The survey consisted of 30 multiple‐choice and short‐answer questions. Follow‐up e‐mail reminders were sent to all FDs 2 weeks and 4 weeks after the initial request was sent. Survey completion was voluntary, and no incentives were offered. The study was determined to be exempt by the Stanford University Institutional Review Board. Data were summarized using frequency distributions. No subgroup comparisons were made.
RESULTS
Program directors from 27/31 (87%) PHM fellowship programs responded to the survey; 25 were active programs, and 2 were under development. Responding programs represented all 4 major regions of the country and Canada, with varying program initiation dates, ranging from 1997 to 2013.
Program Demographics
The duration of most programs (17/27) was 2 years (63%), with 6 (22%) 1‐year programs and 4 (15%) 3‐year programs making up the remainder. Four programs described variable lengths, which could be tailored based on the fellow's individual interest. Two of the programs are 2 years in length, but offer a 1‐year option for fellows who wish to focus on enhancing clinical skills without an academic focus. The other 2 programs are 2 years in length, but will offer an extension to a third year for those pursuing a graduate degree.
Fellow Clinical Activities
The average amount of total clinical time (weeks on service) across responding programs was 50% (range, 20%65%). When looking specifically at time on the inpatient general pediatric service, number of weeks varied by year of training and by institution, with 12 to 41 weeks in the first year of fellowship, 6 to 41 weeks in the second year of fellowship, and 6 to 28 weeks in the third year of fellowship (Figure 1). Though the range is large, on average, fellows spend 17 weeks on inpatient general pediatrics service during each year of training. Of note, the median number of weeks on inpatient general pediatrics service by year of training was 15 weeks, 16 weeks, and 16.5 weeks, respectively. In addition to inpatient general pediatrics service time, most programs require other clinical rotations, with sedation, complex care, and inpatient pediatrics at community sites being the most frequent (Figure 2). Of the 6 responding 1‐year programs, 5 (83%) allow fellows to bill/generate clinical revenue at some point during their training. Of the 15 responding 2‐year programs, 11 (73%) allow fellows to bill/generate clinical revenue at some point during their training. Of the 4 responding 3‐year programs, 2 (50%) allow their fellows to bill/generate clinical revenue at some point during their training.
Fellow Scholarly Activities
With respect to time dedicated to research, the majority of programs offer coursework such as courses for credit, noncredit courses, or certificate courses. In addition, 11 programs offer fellows a masters' degree in areas including public health, clinical science, epidemiology, education, academic sciences, healthcare quality, clinical and translational research, or health services administration. The majority of these degrees are paid for by departmental funds, with tuition reimbursement, university support, training grants, and personal funds making up the remainder. Twenty‐one (81%) programs provide a scholarship oversight committee for their fellows. Current fellows' (n = 63) primary areas of research are varied and include clinical research (36%), quality‐improvement research (22%), medical education research (20%), health services research (16%), and other areas (6%).
DISCUSSION
This is the most comprehensive description of pediatric hospital medicine fellowship curricula to date. Understanding the scope of these programs is an important first step in developing a standardized curriculum that can be used by all. The results of this survey indicate that although there is variability among PHM fellowship curricular content, several common themes exist.
The number of clinical weeks on the inpatient general pediatrics service varied from program to program, though the majority of programs require fellows to spend 15 to 16 weeks each year of training. The variability may be due in part to the way in which respondents defined the term week on clinical service. For example, if the fellow is primarily on a shift schedule, then he/she may only work 2 to 3 shifts in 1 week, which may have been viewed similarly to daily presence on a more traditional inpatient teaching service with 5 to 7 consecutive days of service. The current study did not explore the details of inpatient general pediatric clinical activities or exposure to opportunities to hone procedural skills, areas that are worth investigating as we move forward to better understand the needs of trainees.
Most residency training programs in general pediatrics require a significant amount of inpatient clinical time, specifically a minimum of 10 units or months, though only half of this time is required to be in inpatient general pediatrics.[8] Although nonfellowship trained early career hospitalists may feel adequately prepared to manage the clinical care of some hospitalized children, perceived competency is significantly lower than their fellowship‐trained colleagues with regard to care of the child with medical complexity and technology‐dependence, and with regard to provision of sedation for procedures.[7] The majority of FDs surveyed in our study indicated that additional clinical experience with sedation, complex care, and inpatient pediatrics at community sites were required of their fellows. Of note, many of these rotations are not commonly required in pediatric residency training programs; however, the PHM core competencies suggest that hospitalists should demonstrate proficiency in these areas to provide optimal care for hospitalized children. Our results suggest that current PHM fellowship curricula help address these clinical gaps. The requirement of these particular specialized experiences may reflect the clinical scope of practice that is expected from potential employers or may be related to staffing needs. It is well documented that the inpatient demographic of large pediatric tertiary care referral centers has changed over the past decade, with an increasing prevalence of children with medical complexity.[9, 10] In both tertiary referral centers and community hospitals, the expansion of the role of the hospitalist in providing specialized clinical services, such as sedation or surgical comanagement, has been significantly driven by financial factors, though a more recent focus on improvement of efficiency and quality of care within the hospital system has relied heavily on hospitalist input.[11, 12, 13] Important next steps in curriculum standardization include ensuring that training programs allow for adequate clinical exposure and proper assessment of competency in these areas, and determining the full complement of clinical training experiences that will produce hospitalists with a well‐defined scope of practice that adequately addresses the needs of hospitalized children.
Most fellowship‐trained hospitalists work primarily in university‐affiliated institutions with expectations for scholarly productivity.[5, 7] Fellowship‐trained hospitalists have made large contributions to the growing body of PHM literature, specifically in the realms of medical education, healthcare quality, clinical pediatrics, and healthcare outcomes.[4] Many PHM fellowship‐trained hospitalists have educational or administrative leadership roles.[2] Our results indicate that current PHM fellows continue to be active in a variety of research activities. In addition, FDs reported that the vast majority of programs included scholarship oversight committees, which ensure a mentored and structured research experience. Finally, most programs require or offer additional coursework, and many programs with university affiliations allow for attainment of graduate degrees. Inclusion of robust research training and infrastructure in all programs is a paramount goal of PHM fellowship training. This will allow graduates to be successful researchers, generating new knowledge and supporting the provision of high‐quality, evidence‐based, and value‐driven care for hospitalized children.
A unique feature of several PHM fellowship programs is that fellows are allowed to bill for clinical encounters. Many programs rely on clinical revenue to support fellow salaries.[14] For some programs, a portion of this clinical revenue comes from fellows billing for clinical encounters.[15] Programs that allow fellows to bill/generate clinical revenue have fellows working in attending roles without direct supervision, whereas nonbilling fellows have direct supervision by an attending.[15] In the current ABP training model, subspecialty fellows cannot independently bill for clinical encounters within their own subspecialty, though they can moonlight as long as they meet the duty hour requirements set forth by the ACGME.[16] FDs will need to consider the impact of this requirement on fellow autonomy and on financial revenue for funding fellow salaries if the field achieves ABP subspecialty status.
Regardless of whether or not PHM becomes a designated subspecialty of the ABP, FDs will continue to work together to develop a standard core curriculum that incorporates elements of clinical and nonclinical training to ensure that graduates not only provide high‐quality care for hospitalized children, but also generate new knowledge that advances the field in care delivery and quality of care in any setting. The results of this study will not only help to inform curriculum standardization, but also assessment and evaluation methods. Currently, PHM FDs meet annually and are nearing consensus on a standard 2‐year curriculum based on the PHM Core Competencies that incorporates core clinical, systems, and scholarly domains. We continue to solicit the input of stakeholders, including new FDs, community hospitalist leaders, internal medicine‐pediatrics hospitalist leaders, the Joint Council of Pediatric Hospital Medicine, and leaders of national organizations, such as the American Academy of Pediatrics, Academic Pediatrics Association, and Society of Hospital Medicine. Additional work around standardizing the fellowship application and recruitment process has resulted in our recent acceptance into the Fall Subspecialty Match through the National Residency Match Program, as well as development and implementation of a common fellowship application form. The FD group has recently formalized, voting into place an executive steering committee, which is responsible for the development and execution of long‐term goals that include finalizing a standardized curriculum, refining program and fellow assessment methods through critical evaluation of fellow metrics and outcomes, and standardization of evaluation methods.
Adopting a standard 2‐year curriculum may affect some programs, specifically those that are currently 1 year in duration. These programs would need to extend the length of their fellowship to allow for the breadth of experiences expected with a standardized 2‐year curriculum. This could result in significant financial challenges, effectively increasing the cost to administer the program. In addition, at present, programs have the flexibility to highlight individual areas of strength to attract candidates, allowing fellows to gain an in‐depth experience in domains such as clinical research, quality improvement, medical education, or health services research. With a standardized curriculum, some programs may have to assemble specific clinical and nonclinical experiences to meet the agreed‐upon expectations for PHM fellowship training. If these resources are not available, programs may need to seek relationships with other institutions to complete their offerings, a possibility that is being actively explored by this group. FDs continue to work with each other to share resources, identify training opportunities, and partner with each other to ensure that the requirements of a standard curriculum can be met.
This study has several limitations. First, it was a voluntary survey of program directors, and though we captured over 80% of programs at the time of the survey, there are currently more programs that have come into existence and more still that are in the development stage, leading to potential sampling error. Second, variable effort or accuracy by participants may have led to some degree of response error, such as content error or nonreporting error. Third, the survey questions focused on high‐level information, making it difficult to make nuanced comparisons between curricular elements or determine best curricular practice. In addition, this survey did not explore medical education and quality improvement activities of fellows, 2 major areas in which hospitalists play a major role in the inpatient setting.[1, 17, 18, 19, 20]
CONCLUSION
PHM fellowship programs have grown and continue to grow at a rapid rate. Variability in training is evident, both in clinical experiences and research experiences, though several common elements were identified in this study. The majority of programs are 2 years, and clinical experience comprises approximately 50% of training time, often including key rotations such as sedation, complex care, and rotations at community hospitals. Future directions include standardizing clinical training and expectations for scholarship, formulating appropriate methods for assessment of competency that can be used across programs, and seeking sustainable sources of funding.
Disclosure
Nothing to report.
- , . Characteristics of pediatric hospital medicine fellowships and training programs. J Hosp Med. 2009;4(3):157–163.
- , . Pediatric hospitalists in medical education: current roles and future directions. Curr Probl Pediatr Adolesc Health Care. 2012;42(5):120–126.
- , , . Research needs of pediatric hospitalists. Hosp Pediatr. 2011;1(1):38–44.
- , , , . Pediatric hospital medicine fellowships: outcomes and future directions. Paper presented at: Pediatric Hospital Medicine 2014; July 26, 2014; Orlando, FL.
- , , . Pediatric hospitalist research productivity: predictors of success at presenting abstracts and publishing peer‐reviewed manuscripts among pediatric hospitalists. Hosp Pediatr. 2012;2(3):149–160.
- , , . Pediatric hospital medicine core competencies: development and methodology. J Hosp Med. 2010;5:339–343.
- , , , . Perceived core competency achievements of fellowship and non‐fellowship early career pediatric hospitalists. J Hosp Med. 2015;10(6):373–389.
- Accreditation Council of Graduate Medical Education. ACGME program requirements for graduate medical education in pediatrics. Available at: https://www.acgme.org/acgmeweb/Portals/0/PFAssets/2013‐PR‐FAQ‐PIF/320_pediatrics_07012013.pdf. Published September 30, 2012. Accessed July 7, 2015.
- , , , , , . Increasing prevalence of medically complex children in US hospitals. Pediatrics. 2010;126(4):638–646.
- , , , et al. Children with complex chronic conditions in inpatient hospital settings in the United States. Pediatrics. 2010;126(4):647–655.
- , . The expanding role of hospitalists in the United States. Swiss Med Wkly. 2006;136:591–596.
- , , , , , . Pediatric hospitalist comanagement of spinal fusion surgery patients. J Hosp Med. 2007;2(1):23–30.
- , , , , . Development of a pediatric hospitalist sedation service: training and implementation. J Hosp Med. 2012;7(4):335–339.
- , . Sources of funding and support for pediatric hospital medicine fellowship programs. Poster presented at: Pediatric Hospital Medicine 2014; July 27, 2014; Orlando, FL.
- Council of Pediatric Hospital Medicine Fellowship Directors. Pediatric Hospital Medicine Fellowship Directors Annual Meeting: funding and return on investment. July 24, 2014.
- Accreditation Council of Graduate Medical Education. Frequently asked questions: ACGME common duty hour requirements. Available at: https://www.acgme.org/acgmeweb/Portals/0/PDFs/dh‐faqs2011.pdf. Updated June 18, 2014. Accessed July 7, 2015.
- , . Pediatric hospitalists: training, current practice and career goals. J Hosp Med. 2009;4(3):179–186.
- , . The hospitalist movement and its implications for the care of hospitalized children. Pediatrics. 1999;103:473–477.
- . Pediatric hospitalists and medical education. Pediatr Ann. 2014;43(7):e151–e156
- , , , et al. Quality improvement research in pediatric hospital medicine and the role of the Pediatric Research in Inpatient Settings (PRIS) network. Acad Pediatr. 2013;13(6):S54–S60.
- , . Characteristics of pediatric hospital medicine fellowships and training programs. J Hosp Med. 2009;4(3):157–163.
- , . Pediatric hospitalists in medical education: current roles and future directions. Curr Probl Pediatr Adolesc Health Care. 2012;42(5):120–126.
- , , . Research needs of pediatric hospitalists. Hosp Pediatr. 2011;1(1):38–44.
- , , , . Pediatric hospital medicine fellowships: outcomes and future directions. Paper presented at: Pediatric Hospital Medicine 2014; July 26, 2014; Orlando, FL.
- , , . Pediatric hospitalist research productivity: predictors of success at presenting abstracts and publishing peer‐reviewed manuscripts among pediatric hospitalists. Hosp Pediatr. 2012;2(3):149–160.
- , , . Pediatric hospital medicine core competencies: development and methodology. J Hosp Med. 2010;5:339–343.
- , , , . Perceived core competency achievements of fellowship and non‐fellowship early career pediatric hospitalists. J Hosp Med. 2015;10(6):373–389.
- Accreditation Council of Graduate Medical Education. ACGME program requirements for graduate medical education in pediatrics. Available at: https://www.acgme.org/acgmeweb/Portals/0/PFAssets/2013‐PR‐FAQ‐PIF/320_pediatrics_07012013.pdf. Published September 30, 2012. Accessed July 7, 2015.
- , , , , , . Increasing prevalence of medically complex children in US hospitals. Pediatrics. 2010;126(4):638–646.
- , , , et al. Children with complex chronic conditions in inpatient hospital settings in the United States. Pediatrics. 2010;126(4):647–655.
- , . The expanding role of hospitalists in the United States. Swiss Med Wkly. 2006;136:591–596.
- , , , , , . Pediatric hospitalist comanagement of spinal fusion surgery patients. J Hosp Med. 2007;2(1):23–30.
- , , , , . Development of a pediatric hospitalist sedation service: training and implementation. J Hosp Med. 2012;7(4):335–339.
- , . Sources of funding and support for pediatric hospital medicine fellowship programs. Poster presented at: Pediatric Hospital Medicine 2014; July 27, 2014; Orlando, FL.
- Council of Pediatric Hospital Medicine Fellowship Directors. Pediatric Hospital Medicine Fellowship Directors Annual Meeting: funding and return on investment. July 24, 2014.
- Accreditation Council of Graduate Medical Education. Frequently asked questions: ACGME common duty hour requirements. Available at: https://www.acgme.org/acgmeweb/Portals/0/PDFs/dh‐faqs2011.pdf. Updated June 18, 2014. Accessed July 7, 2015.
- , . Pediatric hospitalists: training, current practice and career goals. J Hosp Med. 2009;4(3):179–186.
- , . The hospitalist movement and its implications for the care of hospitalized children. Pediatrics. 1999;103:473–477.
- . Pediatric hospitalists and medical education. Pediatr Ann. 2014;43(7):e151–e156
- , , , et al. Quality improvement research in pediatric hospital medicine and the role of the Pediatric Research in Inpatient Settings (PRIS) network. Acad Pediatr. 2013;13(6):S54–S60.
© 2016 Society of Hospital Medicine

Antibiotics in Persons Who Inject Drugs
In the United States, there are an estimated 744,000 individuals who have engaged in recent injection drug use (IDU) and 6.6 million individuals who have ever injected a drug.[1] The practice of IDU predisposes individuals to serious bacterial and fungal infections that often require long‐term intravenous antibiotics. In individuals without IDU, these serious infections are often treated with outpatient parenteral antibiotic therapy (OPAT). However, a different standard exists for many persons who inject drugs (PWID)the mandated completion of antibiotics in an inpatient setting.
Though mandating inpatient antibiotic therapy for PWID is a widely adopted standard, this practice is not evidence based and may increase overall costs to the healthcare system. In 2012, in a quality‐improvement initiative, UKHealthCare established a protocol for treating appropriate PWID with OPAT.[2] They found very few inpatient providers willing to discharge PWID on OPAT, even with an established protocol.
To better understand the reasons for the low adoption of this protocol, Fanucchi and colleagues developed a survey designed to assess attitudes, practices, and mediating factors impacting the decision making about discharging PWID on OPAT.[2] The results of this survey are reported in this issue of the Journal of Hospital Medicine.
The study found that 95% of inpatient providers use OPAT for patients without IDU, but only 29% would even consider OPAT in PWID. The most common barriers to discharging a patient with IDU on OPAT were socioeconomic factors, willingness of infectious diseases physicians to follow as an outpatient, and concerns for misuse of peripherally inserted central catheters and adherence with antibiotic treatment.
At first glance, these reservations seem very reasonable. The presence of socioeconomic factors such as homelessness or lack of infectious diseases specialist follow‐up would make the risks of discharge on OPAT significant. The concerns for misuse of peripherally inserted central catheters and adherence to antibiotic treatment suggest that inpatient providers have an overall goal of reducing drug misuse and improving treatment outcomes.
Unfortunately, there are no data to suggest that completion of antibiotics in an inpatient setting reduces drug misuse or improves adherence to antibiotic treatments. Studies have found that at least 16% of PWID will misuse drugs during their hospitalization,[3] and 25% to 30% will be discharged against medical advice.[3, 4] This may be in large part due to the fact that inpatient providers are historically poor at addressing substance use disorders, even in patients with serious infections associated with IDU.[5] Yet the provision of methadone during hospitalization has been associated with a significant reduction in discharges against medical advice.[4] Rather than focusing on placing restrictions on individuals with risky behaviors, patients may benefit more from minimizing these risks through prompt recognition and management of substance use disorder.
Although limited, there is also evidence to support the feasibility of safe and effective OPAT in some PWID. A study by Ho et al. used OPAT to treat 29 PWID hospitalized with serious infections.[6] The study population had adequate housing, a reliable guardian, and signed a contract agreeing to abstain from drug misuse. In addition, all patients received substance use counseling and novel tamper‐proof security seals to prevent misuse of peripherally inserted central catheters, and antibiotics were delivered daily at an infusion center. They found no evidence of line tampering, excess readmissions, or excess line infections. Of note, the study population included 2 patients who were discharged against medical advice but successfully completed OPAT without issue. Although we do not believe that all individuals are appropriate for OPAT, this study suggests that OPAT can be considered in select PWID.
The study by Fanucchi et al. also reinforces the importance of making individualized risk assessments of persons with a history of IDU rather than assuming uniformity among the population. Of particular note is the lack of agreed‐upon definition of remote history of IDU (range, 2120 months; median, 12 months). The idea that individuals with a decade of sobriety could be subject to the same restrictions as a patient injecting multiple times a day speaks to providers' discomfort with assessing the individual risk of a person who has suffered from substance use disorder. Further, the fact that so few providers felt substance use disorder treatment was a critical component of a decision to allow OPAT raises concerns that providers are not aware of effective means to treat addiction. In particular, it is crucial for providers to understand that medication‐assisted treatment, such as methadone or buprenorphine for opioid use disorder, has significant evidence to support efficacy in decreasing drug misuse and improving outcomes.
This study suggests more work will need to be done before inpatient providers will be comfortable discharging any PWID with OPAT. This includes improved outpatient services (enhanced case management and home health services, and better access to outpatient physicians including infectious diseases specialists), the development of tamper‐evident devices to deter misuse of peripherally inserted central catheters, and defined legal protection for providers.
In addition, more research needs to be done on this population to objectively stratify risk for PWID and assess outcomes for PWID treated with OPAT versus the current standard of care. This research should have a particular focus on the long‐term financial and societal costs associated with PWID leaving against medical advice or receiving potentially unnecessary inpatient services. Minimizing the length of stay may defray inpatient costs and afford investment into more robust, effective outpatient services. It is essential that we develop a system to provide antibiotics in a way that optimizes outcomes and is cost‐effective.
Regardless of the decision to mandate antibiotic treatment in an inpatient setting or to discharge with OPAT, it is clear that more needs to be done to address addiction in hospitalized patients. All hospitalized PWID should receive safe injection education and a referral to a substance use disorder specialist. In addition, individuals with opioid‐misuse or opioid use disorder should receive opioid overdose education and naloxone distribution. Hospitalizations serve as important opportunities to engage individuals in the treatment of their addiction. It is essential that hospitalists begin utilizing these opportunities.
Disclosures: Nothing to report.
- , , , et al. Estimating the number of persons who inject drugs in the United States by meta‐analysis to calculate national rates of HIV and hepatitis C virus infections. PLoS One. 2014;9:e97596.
- , , , . Perceptions and practices of physicians regarding outpatient parenteral antibiotic therapy in persons who inject drugs. J Hosp Med. 2016;11(8):581–582.
- , , , et al. Needles and the damage done: reasons for admission and financial costs associated with injecting drug use in a Central London teaching hospital. J Infect. 2012;66:95–102.
- , , , et al. HIV‐positive injection drug users who leave the hospital against medical advice: the mitigating role of methadone and social support. J Acquir Immune Defic Syndr. 2004;35:56–59.
- , , , , . Suboptimal addiction interventions for patients hospitalized with injection drug use‐associated infective endocarditis [published online November 18, 2015]. Am J Med. doi: 10.1016/j.amjmed.2015.09.024.
- , , , . Safe and successful treatment of intravenous drug users with a peripherally inserted central catheter in an outpatient parenteral antibiotic treatment service. J Antimicrob Chemother. 2010;65:2641–2644.
In the United States, there are an estimated 744,000 individuals who have engaged in recent injection drug use (IDU) and 6.6 million individuals who have ever injected a drug.[1] The practice of IDU predisposes individuals to serious bacterial and fungal infections that often require long‐term intravenous antibiotics. In individuals without IDU, these serious infections are often treated with outpatient parenteral antibiotic therapy (OPAT). However, a different standard exists for many persons who inject drugs (PWID)the mandated completion of antibiotics in an inpatient setting.
Though mandating inpatient antibiotic therapy for PWID is a widely adopted standard, this practice is not evidence based and may increase overall costs to the healthcare system. In 2012, in a quality‐improvement initiative, UKHealthCare established a protocol for treating appropriate PWID with OPAT.[2] They found very few inpatient providers willing to discharge PWID on OPAT, even with an established protocol.
To better understand the reasons for the low adoption of this protocol, Fanucchi and colleagues developed a survey designed to assess attitudes, practices, and mediating factors impacting the decision making about discharging PWID on OPAT.[2] The results of this survey are reported in this issue of the Journal of Hospital Medicine.
The study found that 95% of inpatient providers use OPAT for patients without IDU, but only 29% would even consider OPAT in PWID. The most common barriers to discharging a patient with IDU on OPAT were socioeconomic factors, willingness of infectious diseases physicians to follow as an outpatient, and concerns for misuse of peripherally inserted central catheters and adherence with antibiotic treatment.
At first glance, these reservations seem very reasonable. The presence of socioeconomic factors such as homelessness or lack of infectious diseases specialist follow‐up would make the risks of discharge on OPAT significant. The concerns for misuse of peripherally inserted central catheters and adherence to antibiotic treatment suggest that inpatient providers have an overall goal of reducing drug misuse and improving treatment outcomes.
Unfortunately, there are no data to suggest that completion of antibiotics in an inpatient setting reduces drug misuse or improves adherence to antibiotic treatments. Studies have found that at least 16% of PWID will misuse drugs during their hospitalization,[3] and 25% to 30% will be discharged against medical advice.[3, 4] This may be in large part due to the fact that inpatient providers are historically poor at addressing substance use disorders, even in patients with serious infections associated with IDU.[5] Yet the provision of methadone during hospitalization has been associated with a significant reduction in discharges against medical advice.[4] Rather than focusing on placing restrictions on individuals with risky behaviors, patients may benefit more from minimizing these risks through prompt recognition and management of substance use disorder.
Although limited, there is also evidence to support the feasibility of safe and effective OPAT in some PWID. A study by Ho et al. used OPAT to treat 29 PWID hospitalized with serious infections.[6] The study population had adequate housing, a reliable guardian, and signed a contract agreeing to abstain from drug misuse. In addition, all patients received substance use counseling and novel tamper‐proof security seals to prevent misuse of peripherally inserted central catheters, and antibiotics were delivered daily at an infusion center. They found no evidence of line tampering, excess readmissions, or excess line infections. Of note, the study population included 2 patients who were discharged against medical advice but successfully completed OPAT without issue. Although we do not believe that all individuals are appropriate for OPAT, this study suggests that OPAT can be considered in select PWID.
The study by Fanucchi et al. also reinforces the importance of making individualized risk assessments of persons with a history of IDU rather than assuming uniformity among the population. Of particular note is the lack of agreed‐upon definition of remote history of IDU (range, 2120 months; median, 12 months). The idea that individuals with a decade of sobriety could be subject to the same restrictions as a patient injecting multiple times a day speaks to providers' discomfort with assessing the individual risk of a person who has suffered from substance use disorder. Further, the fact that so few providers felt substance use disorder treatment was a critical component of a decision to allow OPAT raises concerns that providers are not aware of effective means to treat addiction. In particular, it is crucial for providers to understand that medication‐assisted treatment, such as methadone or buprenorphine for opioid use disorder, has significant evidence to support efficacy in decreasing drug misuse and improving outcomes.
This study suggests more work will need to be done before inpatient providers will be comfortable discharging any PWID with OPAT. This includes improved outpatient services (enhanced case management and home health services, and better access to outpatient physicians including infectious diseases specialists), the development of tamper‐evident devices to deter misuse of peripherally inserted central catheters, and defined legal protection for providers.
In addition, more research needs to be done on this population to objectively stratify risk for PWID and assess outcomes for PWID treated with OPAT versus the current standard of care. This research should have a particular focus on the long‐term financial and societal costs associated with PWID leaving against medical advice or receiving potentially unnecessary inpatient services. Minimizing the length of stay may defray inpatient costs and afford investment into more robust, effective outpatient services. It is essential that we develop a system to provide antibiotics in a way that optimizes outcomes and is cost‐effective.
Regardless of the decision to mandate antibiotic treatment in an inpatient setting or to discharge with OPAT, it is clear that more needs to be done to address addiction in hospitalized patients. All hospitalized PWID should receive safe injection education and a referral to a substance use disorder specialist. In addition, individuals with opioid‐misuse or opioid use disorder should receive opioid overdose education and naloxone distribution. Hospitalizations serve as important opportunities to engage individuals in the treatment of their addiction. It is essential that hospitalists begin utilizing these opportunities.
Disclosures: Nothing to report.
In the United States, there are an estimated 744,000 individuals who have engaged in recent injection drug use (IDU) and 6.6 million individuals who have ever injected a drug.[1] The practice of IDU predisposes individuals to serious bacterial and fungal infections that often require long‐term intravenous antibiotics. In individuals without IDU, these serious infections are often treated with outpatient parenteral antibiotic therapy (OPAT). However, a different standard exists for many persons who inject drugs (PWID)the mandated completion of antibiotics in an inpatient setting.
Though mandating inpatient antibiotic therapy for PWID is a widely adopted standard, this practice is not evidence based and may increase overall costs to the healthcare system. In 2012, in a quality‐improvement initiative, UKHealthCare established a protocol for treating appropriate PWID with OPAT.[2] They found very few inpatient providers willing to discharge PWID on OPAT, even with an established protocol.
To better understand the reasons for the low adoption of this protocol, Fanucchi and colleagues developed a survey designed to assess attitudes, practices, and mediating factors impacting the decision making about discharging PWID on OPAT.[2] The results of this survey are reported in this issue of the Journal of Hospital Medicine.
The study found that 95% of inpatient providers use OPAT for patients without IDU, but only 29% would even consider OPAT in PWID. The most common barriers to discharging a patient with IDU on OPAT were socioeconomic factors, willingness of infectious diseases physicians to follow as an outpatient, and concerns for misuse of peripherally inserted central catheters and adherence with antibiotic treatment.
At first glance, these reservations seem very reasonable. The presence of socioeconomic factors such as homelessness or lack of infectious diseases specialist follow‐up would make the risks of discharge on OPAT significant. The concerns for misuse of peripherally inserted central catheters and adherence to antibiotic treatment suggest that inpatient providers have an overall goal of reducing drug misuse and improving treatment outcomes.
Unfortunately, there are no data to suggest that completion of antibiotics in an inpatient setting reduces drug misuse or improves adherence to antibiotic treatments. Studies have found that at least 16% of PWID will misuse drugs during their hospitalization,[3] and 25% to 30% will be discharged against medical advice.[3, 4] This may be in large part due to the fact that inpatient providers are historically poor at addressing substance use disorders, even in patients with serious infections associated with IDU.[5] Yet the provision of methadone during hospitalization has been associated with a significant reduction in discharges against medical advice.[4] Rather than focusing on placing restrictions on individuals with risky behaviors, patients may benefit more from minimizing these risks through prompt recognition and management of substance use disorder.
Although limited, there is also evidence to support the feasibility of safe and effective OPAT in some PWID. A study by Ho et al. used OPAT to treat 29 PWID hospitalized with serious infections.[6] The study population had adequate housing, a reliable guardian, and signed a contract agreeing to abstain from drug misuse. In addition, all patients received substance use counseling and novel tamper‐proof security seals to prevent misuse of peripherally inserted central catheters, and antibiotics were delivered daily at an infusion center. They found no evidence of line tampering, excess readmissions, or excess line infections. Of note, the study population included 2 patients who were discharged against medical advice but successfully completed OPAT without issue. Although we do not believe that all individuals are appropriate for OPAT, this study suggests that OPAT can be considered in select PWID.
The study by Fanucchi et al. also reinforces the importance of making individualized risk assessments of persons with a history of IDU rather than assuming uniformity among the population. Of particular note is the lack of agreed‐upon definition of remote history of IDU (range, 2120 months; median, 12 months). The idea that individuals with a decade of sobriety could be subject to the same restrictions as a patient injecting multiple times a day speaks to providers' discomfort with assessing the individual risk of a person who has suffered from substance use disorder. Further, the fact that so few providers felt substance use disorder treatment was a critical component of a decision to allow OPAT raises concerns that providers are not aware of effective means to treat addiction. In particular, it is crucial for providers to understand that medication‐assisted treatment, such as methadone or buprenorphine for opioid use disorder, has significant evidence to support efficacy in decreasing drug misuse and improving outcomes.
This study suggests more work will need to be done before inpatient providers will be comfortable discharging any PWID with OPAT. This includes improved outpatient services (enhanced case management and home health services, and better access to outpatient physicians including infectious diseases specialists), the development of tamper‐evident devices to deter misuse of peripherally inserted central catheters, and defined legal protection for providers.
In addition, more research needs to be done on this population to objectively stratify risk for PWID and assess outcomes for PWID treated with OPAT versus the current standard of care. This research should have a particular focus on the long‐term financial and societal costs associated with PWID leaving against medical advice or receiving potentially unnecessary inpatient services. Minimizing the length of stay may defray inpatient costs and afford investment into more robust, effective outpatient services. It is essential that we develop a system to provide antibiotics in a way that optimizes outcomes and is cost‐effective.
Regardless of the decision to mandate antibiotic treatment in an inpatient setting or to discharge with OPAT, it is clear that more needs to be done to address addiction in hospitalized patients. All hospitalized PWID should receive safe injection education and a referral to a substance use disorder specialist. In addition, individuals with opioid‐misuse or opioid use disorder should receive opioid overdose education and naloxone distribution. Hospitalizations serve as important opportunities to engage individuals in the treatment of their addiction. It is essential that hospitalists begin utilizing these opportunities.
Disclosures: Nothing to report.
- , , , et al. Estimating the number of persons who inject drugs in the United States by meta‐analysis to calculate national rates of HIV and hepatitis C virus infections. PLoS One. 2014;9:e97596.
- , , , . Perceptions and practices of physicians regarding outpatient parenteral antibiotic therapy in persons who inject drugs. J Hosp Med. 2016;11(8):581–582.
- , , , et al. Needles and the damage done: reasons for admission and financial costs associated with injecting drug use in a Central London teaching hospital. J Infect. 2012;66:95–102.
- , , , et al. HIV‐positive injection drug users who leave the hospital against medical advice: the mitigating role of methadone and social support. J Acquir Immune Defic Syndr. 2004;35:56–59.
- , , , , . Suboptimal addiction interventions for patients hospitalized with injection drug use‐associated infective endocarditis [published online November 18, 2015]. Am J Med. doi: 10.1016/j.amjmed.2015.09.024.
- , , , . Safe and successful treatment of intravenous drug users with a peripherally inserted central catheter in an outpatient parenteral antibiotic treatment service. J Antimicrob Chemother. 2010;65:2641–2644.
- , , , et al. Estimating the number of persons who inject drugs in the United States by meta‐analysis to calculate national rates of HIV and hepatitis C virus infections. PLoS One. 2014;9:e97596.
- , , , . Perceptions and practices of physicians regarding outpatient parenteral antibiotic therapy in persons who inject drugs. J Hosp Med. 2016;11(8):581–582.
- , , , et al. Needles and the damage done: reasons for admission and financial costs associated with injecting drug use in a Central London teaching hospital. J Infect. 2012;66:95–102.
- , , , et al. HIV‐positive injection drug users who leave the hospital against medical advice: the mitigating role of methadone and social support. J Acquir Immune Defic Syndr. 2004;35:56–59.
- , , , , . Suboptimal addiction interventions for patients hospitalized with injection drug use‐associated infective endocarditis [published online November 18, 2015]. Am J Med. doi: 10.1016/j.amjmed.2015.09.024.
- , , , . Safe and successful treatment of intravenous drug users with a peripherally inserted central catheter in an outpatient parenteral antibiotic treatment service. J Antimicrob Chemother. 2010;65:2641–2644.
Caught red‐handed
A previously healthy 58‐year‐old man presented to a community hospital's emergency department 1 day after the sudden onset of a severe headache, fever, diffuse abdominal pain, nausea, vomiting, and disorientation. The patient had a history of allergic rhinitis and his only medication was a daily multivitamin.
Key features of this patient's presentation include the abrupt onset of severe headache, disorientation, fever, and abdominal pain. The list of entities likely to make a previously healthy individual this ill this quickly is typically circumscribed. His presentation raises the possibility of bacterial meningitis (including Listeria, given his age), viral encephalitis, or other extraneural etiologies of sepsis. Noninfectious explanations seem much less likely given the rapid tempo of illness.
His proclivity for gardening and apparent tick exposure raise the question of tick‐borne illnesses. This would constitute a rather explosive onset for any of these; however, babesiosis, Rocky Mountain spotted fever (RMSF), ehrlichiosis, and anaplasmosis could present this abruptly, with dog exposure linked to RMSF.
The potential causes of fever and rash are myriad, although the severity and acuity of this patient's illness narrow the differential considerably, likely to an infectious cause. Diagnoses that typically include a generalized exanthem involving the palms and soles are meningococcal meningitis, overwhelming Staphylococcus aureus sepsis, RMSF (realizing that this disease is not common in the upper Midwest), and toxic shock syndrome. The rash described is not the classic and/or fully developed rash typical of any of these; subsequent evolution to a petechial appearance would lend further support to the first 3 diagnoses. Ehrlichiosis is still a possibility, although the palm and sole involvement would be unusual. The presence of a rash makes anaplasmosis very unlikely, although not entirely excluded. The finding of modest splenomegaly does not help further distinguish between these possibilities.
Empiric antimicrobials should be immediately administered after blood cultures, a complete blood count, and coagulation studies are obtained. Doxycycline would be appropriate to treat the possible tick‐borne diseases already mentioned, whereas antimicrobials appropriate to cover community‐acquired bacterial meningitis in a 58‐year‐old (ie, vancomycin, ampicillin, and a third‐generation cephalosporin) should also be empirically administered. Given the patient's altered mentation, a brain computed tomography (CT) should be urgently obtained. Provided this did not show evidence of increased intracranial pressure and that coagulation studies and a platelet count did not suggest a contraindication, a lumbar puncture should then be performed promptly. The patient should be placed in droplet precautions until meningococcal disease is excluded. Although most patients with bacterial meningitis will exhibit meningismus, a substantial minority will not.
These laboratory results do not significantly affect the differential diagnosis. Although nonspecific, moderate thrombocytopenia and modest elevation of hepatic transaminases are typical for tick‐borne diseases, whereas leukocytosis is somewhat atypical for these entities. Marked elevation of the C‐reactive protein with a less striking increase in the erythrocyte sedimentation rate, along with significant hypoalbuminemia, are commonly encountered early in the course of critical infectious illnesses. The elevated troponin likely reflects severe sepsis and demand ischemia, and is associated with a less favorable prognosis; an electrocardiogram and serial cardiac biomarkers are appropriate to help exclude an acute coronary syndrome. As already noted, blood cultures need to be obtained and a lumbar puncture should be performed, provided this can be safely accomplished.
Results of the lumbar puncture exclude bacterial meningitis as the explanation of this patient's illness; the mildly elevated protein is nonspecific. These studies do not otherwise change the differential diagnosis.
Supporting data for a diagnosis of pneumonia, such as pulmonary infiltrates or supplemental oxygen requirement, are lacking. Given his critical illness, broad spectrum antimicrobial coverage is indicated, and as a primary central nervous system (CNS) infection now appears unlikely, piperacillin/tazobactam (which does not have adequate CNS penetration) and vancomycin are reasonable. Empiric treatment for RMSF is appropriate, and should have been initiated earlier in the patient's course, despite the upper Midwest being out of the typical range for this disease. Doxycycline will also provide excellent coverage for ehrlichiosis and anaplasmosis.
Given the patient's deterioration, it is important to stop and reconsider the differential diagnosis in an attempt to avoid anchoring bias and premature closure. The patient's illness is almost certainly infectious in nature, and the differential is not substantially altered by the most recent information. A skin biopsy should be performed in an attempt to secure the diagnosis.
The patient's overall course, including rapid onset of severe illness and especially the apparent dramatic response to doxycycline, make tick‐borne illness very likely. Completing a course of doxycycline is certainly appropriate, typically for 7 to 14 days. The acute serologies drawn prior to discharge may well reveal the causative agent, but convalescent serology should also be obtained at the time of an outpatient follow‐up visit as immunoglobulin G has a delayed rise. Without hyponatremia or respiratory symptoms, Legionella seems unlikely.
The appearance of late desquamation of the palms and soles is an unexpected and important sign. Desquamation in this pattern following an illness of this nature strongly suggests a diagnosis of staphylococcal toxic shock syndrome (TSS), and in conjunction with the negative serologies, argues that tick‐borne disease is unlikely. The list of other entities that might lead to desquamation in this setting is very short, namely adult Kawasaki disease and drug reaction. The former seems reasonably excluded based on details of the case, whereas a doxycycline‐related drug reaction, although not entirely implausible, seems quite unlikely as this medication was started after the onset of the initial rash. This patient most likely had staphylococcal TSS secondary to a minor and unappreciated skin lesion.
DISCUSSION
TSS is a systemic illness resulting in multiorgan dysfunction.[1] Infection by S aureus or Streptococcus pyogenes causes TSS by stimulating maladaptive T‐cell proliferation and cytokine release resulting in shock.[1, 2] A definitive diagnosis requires fever, a diffuse macular erythematous rash (often resembling a sunburn), with subsequent desquamation, hypotension, and involvement of at least 3 organ systems. Blood cultures, cerebrospinal cultures, and serologies for other organisms should be negative; although Staphylococcus and Streptococcus species may be isolated, they frequently are not (Table 1).[3]
| Diagnostic Criteria* | This Case |
|---|---|
| |
| Fever: Temperature 102.0F | Fever: 105.3F on admission |
| Rash: Diffuse macular erythroderma | Diffuse morbilliform rash with progression to confluent erythroderma |
| Desquamation of rash: occurs 12 weeks following rash onset | Desquamation 12 days after discharge |
| Hypotension: SBP 90 mm Hg for adults | Intermittent |
| Multisystem involvement, 3 of the following: | 4 organ systems definitively involved |
| GI: vomiting or diarrhea at disease onset | Vomiting and abdominal pain |
| Muscular: severe myalgias, or creatine phosphokinase >2 times the upper limit of normal | |
| Mucous membranes: vaginal, oropharyngeal, or conjunctival hyperemia | |
| Renal: BUN or Cr >2 times the upper limit of normal, or pyuria without evidence of infection | |
| Hepatic: total bilirubin, AST, or ALT levels >2 times the upper limit of normal | AST and ALT peaked at 128IU/L and 94 IU/L |
| Hematologic: platelets 100,000/mm3 | Platelet nadir of 80,000/mm3 |
| CNS: disorientation or altered consciousness without focal neurologic signs | Disorientation and somnolence |
| Probable case: 4 out of 5 clinical criteria present | |
| Confirmed case: 5 out of 5 clinical criteria present, or patient dies before desquamation can occur | |
A rare cause of shock, TSS is most associated with a surge of menstruation‐related cases linked to tampon use in young women in the 1980s.[4] However, in Centers for Disease Control and Prevention (CDC) surveillance between 1987 and 1996, only 59% of the 1069 cases identified were noted to be menstruation‐related, as compared to nearly 80% of all cases earlier in the decade.[4, 5] Today, the syndrome is more likely to present after musculoskeletal and cutaneous trauma, oropharyngeal infections, surgical procedures, and device implantation.[1, 6] Despite the disease's evolving epidemiology, the illness script used by physicians likely continues to focus on young women as the primary at risk population for TSS, causing physicians to neglect the diagnosis in other populations.[1, 6, 7, 8, 9] Given this change in risk factors, it is imperative that clinicians rewrite their scripts and recognize the early signs of TSS in all patients to enable quick and effective treatment.
In addition to its shifting epidemiology and rarity, the diagnosis of TSS vexes clinicians for several reasons. First, TSS cannot be quickly and definitively diagnosed because 2 diagnostic criteria cannot be fulfilled during the acute illness. The disease's hallmarka desquamative rashoccurs only if the patient survives.[3] Serologies often take weeks to return, further delaying diagnosis. During this period of diagnostic delay, the illness has usually already resolved or resulted in death. In addition, the presenting symptoms of rash, fever, and shock are nonspecific. Alternative etiologies include meningococcal meningitis, which can also present dramatically as with this patient; RMSF, which can occasionally have a fulminant presentation; bacterial sepsis, usually from Staphylococcus or Streptococcus species; acute viral syndromes; and severe drug reactions.[6, 10, 11, 12] Palmoplantar desquamation, as in this case, can further narrow the differential as this presentation is uncommon but characteristic of TSS, RMSF, and secondary syphilis.[11] Other diagnostic clues offered by the pattern of the rash may be limited by physician discomfort with diagnosing and describing rashes. Because of this lack of a definitive diagnostic test in the acute setting, it is imperative that the clinician include TSS in the differential of fever, shock, and rash, as mortality from TSS can exceed 20% in patients who are untreated.[13]
Treatment of TSS is straightforward once considered and includes the administration of antibiotics that cover both Staphylococcus and Streptococcus species, in addition to aggressive hydration and supportive care.[14] The final critical detail in this case was the appropriate arrangement of follow‐up. Given the patient's drastic improvement, the complicated process of arranging follow‐up for a transferred patient, and the current model where the hospitalists providing inpatient care do not typically follow their patients in clinic, patients such as these can easily be lost to follow‐up. Had this occurred, the desquamation would have been missed, and the patient's diagnosis would have been incomplete.
This patient was eventually diagnosed with TSS by fulfilling all 5 CDC criteria (Table 1).[3] He made a full recovery, likely aided by the administration of broad‐spectrum antibiotics (followed by doxycycline, which provided community‐acquired methicillin‐resistant S aureus coverage) and his lack of serious comorbidities. This case should serve as a reminder to hospitalists that with a discerning eye, a careful assessment of the clinical facts, and appropriate follow‐up, perhaps the next case of TSS can be caught red‐handed.
KEY POINTS
- When presented with a patient with fever, rash, and shock, hospitalists should consider meningococcal meningitis, RMSF bacterial sepsis, acute viral illness, severe drug reaction, and TSS.
- TSS, caused by S aureus or S pyogenes, is no longer predominantly associated with tampon use. Postsurgical infection and cutaneous trauma have become important present‐day risk factors.
- The initial presentation of TSS is nonspecific. Definitive diagnosis requires proper follow‐up, allowing time for infectious serologies to return negative and for the disease's hallmark desquamation to occur.
Disclosure
Nothing to report.
- . Toxic shock syndrome: major advances in pathogenesis, but not treatment. Crit Care Clin. 2013;29:651–675.
- . The toxic shock syndromes. Infect Dis Clin North Am. 1996;10(4):727–746.
- Centers for Disease Control and Prevention. National Notifiable Diseases Surveillance System. Toxic shock syndrome (other than Streptococcal) (TSS) 2011 Case Definition. Available at: http://wwwn.cdc.gov/nndss/conditions/toxic‐shock‐syndrome‐other‐than‐streptococcal/case‐definition/2011. Accessed June 4, 2015.
- Centers for Disease Control and Prevention. Update: toxic‐shock syndrome—United States. MMWR Morb Mortal Wkly Rep. 1983;32(30):398–400.
- , , , , , . Toxic shock syndrome in the United States: surveillance update, 1979–1996. Emerg Infect Dis. 1999;5(6):807–810.
- . Fever and rash. Infect Dis Clin North Am. 1996;10(1):101–110.
- , , , et al. Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS One. 2011;6(8):e22997.
- , , , et al. Toxic‐shock syndrome in menstruating women: association with tampon use and staphylococcus aureus and clinical features in 52 cases. N Engl J Med. 1980;303(25):1436–1442.
- , , , . Toxic‐shock syndrome—epidemiologic features, recurrence, risk factors, and prevention. N Engl J Med. 1980;303:1429–1435.
- , . Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62(4):804–816.
- . Toxic shock syndrome: broadening the differential diagnosis. J Am Board Fam Pract. 2001;14(2):131–136.
- , , , . Spatial clustering by disease severity among reported Rocky Mountain spotted fever cases in the United States, 2001–2005. Am J Trop Med Hyg. 2009;80(1):72–77.
- , , , et al. One in five mortality in non‐menstrual toxic shock syndrome versus no mortality in menstrual cases in a balanced French series of 55 cases. Eur J Clin Microbio Infect Dis. 2008;27(1):37–43.
- , . Gram‐positive toxic shock syndromes. Lancet Infect Dis. 2009;9(5):281–290.
A previously healthy 58‐year‐old man presented to a community hospital's emergency department 1 day after the sudden onset of a severe headache, fever, diffuse abdominal pain, nausea, vomiting, and disorientation. The patient had a history of allergic rhinitis and his only medication was a daily multivitamin.
Key features of this patient's presentation include the abrupt onset of severe headache, disorientation, fever, and abdominal pain. The list of entities likely to make a previously healthy individual this ill this quickly is typically circumscribed. His presentation raises the possibility of bacterial meningitis (including Listeria, given his age), viral encephalitis, or other extraneural etiologies of sepsis. Noninfectious explanations seem much less likely given the rapid tempo of illness.
His proclivity for gardening and apparent tick exposure raise the question of tick‐borne illnesses. This would constitute a rather explosive onset for any of these; however, babesiosis, Rocky Mountain spotted fever (RMSF), ehrlichiosis, and anaplasmosis could present this abruptly, with dog exposure linked to RMSF.
The potential causes of fever and rash are myriad, although the severity and acuity of this patient's illness narrow the differential considerably, likely to an infectious cause. Diagnoses that typically include a generalized exanthem involving the palms and soles are meningococcal meningitis, overwhelming Staphylococcus aureus sepsis, RMSF (realizing that this disease is not common in the upper Midwest), and toxic shock syndrome. The rash described is not the classic and/or fully developed rash typical of any of these; subsequent evolution to a petechial appearance would lend further support to the first 3 diagnoses. Ehrlichiosis is still a possibility, although the palm and sole involvement would be unusual. The presence of a rash makes anaplasmosis very unlikely, although not entirely excluded. The finding of modest splenomegaly does not help further distinguish between these possibilities.
Empiric antimicrobials should be immediately administered after blood cultures, a complete blood count, and coagulation studies are obtained. Doxycycline would be appropriate to treat the possible tick‐borne diseases already mentioned, whereas antimicrobials appropriate to cover community‐acquired bacterial meningitis in a 58‐year‐old (ie, vancomycin, ampicillin, and a third‐generation cephalosporin) should also be empirically administered. Given the patient's altered mentation, a brain computed tomography (CT) should be urgently obtained. Provided this did not show evidence of increased intracranial pressure and that coagulation studies and a platelet count did not suggest a contraindication, a lumbar puncture should then be performed promptly. The patient should be placed in droplet precautions until meningococcal disease is excluded. Although most patients with bacterial meningitis will exhibit meningismus, a substantial minority will not.
These laboratory results do not significantly affect the differential diagnosis. Although nonspecific, moderate thrombocytopenia and modest elevation of hepatic transaminases are typical for tick‐borne diseases, whereas leukocytosis is somewhat atypical for these entities. Marked elevation of the C‐reactive protein with a less striking increase in the erythrocyte sedimentation rate, along with significant hypoalbuminemia, are commonly encountered early in the course of critical infectious illnesses. The elevated troponin likely reflects severe sepsis and demand ischemia, and is associated with a less favorable prognosis; an electrocardiogram and serial cardiac biomarkers are appropriate to help exclude an acute coronary syndrome. As already noted, blood cultures need to be obtained and a lumbar puncture should be performed, provided this can be safely accomplished.
Results of the lumbar puncture exclude bacterial meningitis as the explanation of this patient's illness; the mildly elevated protein is nonspecific. These studies do not otherwise change the differential diagnosis.
Supporting data for a diagnosis of pneumonia, such as pulmonary infiltrates or supplemental oxygen requirement, are lacking. Given his critical illness, broad spectrum antimicrobial coverage is indicated, and as a primary central nervous system (CNS) infection now appears unlikely, piperacillin/tazobactam (which does not have adequate CNS penetration) and vancomycin are reasonable. Empiric treatment for RMSF is appropriate, and should have been initiated earlier in the patient's course, despite the upper Midwest being out of the typical range for this disease. Doxycycline will also provide excellent coverage for ehrlichiosis and anaplasmosis.
Given the patient's deterioration, it is important to stop and reconsider the differential diagnosis in an attempt to avoid anchoring bias and premature closure. The patient's illness is almost certainly infectious in nature, and the differential is not substantially altered by the most recent information. A skin biopsy should be performed in an attempt to secure the diagnosis.
The patient's overall course, including rapid onset of severe illness and especially the apparent dramatic response to doxycycline, make tick‐borne illness very likely. Completing a course of doxycycline is certainly appropriate, typically for 7 to 14 days. The acute serologies drawn prior to discharge may well reveal the causative agent, but convalescent serology should also be obtained at the time of an outpatient follow‐up visit as immunoglobulin G has a delayed rise. Without hyponatremia or respiratory symptoms, Legionella seems unlikely.
The appearance of late desquamation of the palms and soles is an unexpected and important sign. Desquamation in this pattern following an illness of this nature strongly suggests a diagnosis of staphylococcal toxic shock syndrome (TSS), and in conjunction with the negative serologies, argues that tick‐borne disease is unlikely. The list of other entities that might lead to desquamation in this setting is very short, namely adult Kawasaki disease and drug reaction. The former seems reasonably excluded based on details of the case, whereas a doxycycline‐related drug reaction, although not entirely implausible, seems quite unlikely as this medication was started after the onset of the initial rash. This patient most likely had staphylococcal TSS secondary to a minor and unappreciated skin lesion.
DISCUSSION
TSS is a systemic illness resulting in multiorgan dysfunction.[1] Infection by S aureus or Streptococcus pyogenes causes TSS by stimulating maladaptive T‐cell proliferation and cytokine release resulting in shock.[1, 2] A definitive diagnosis requires fever, a diffuse macular erythematous rash (often resembling a sunburn), with subsequent desquamation, hypotension, and involvement of at least 3 organ systems. Blood cultures, cerebrospinal cultures, and serologies for other organisms should be negative; although Staphylococcus and Streptococcus species may be isolated, they frequently are not (Table 1).[3]
| Diagnostic Criteria* | This Case |
|---|---|
| |
| Fever: Temperature 102.0F | Fever: 105.3F on admission |
| Rash: Diffuse macular erythroderma | Diffuse morbilliform rash with progression to confluent erythroderma |
| Desquamation of rash: occurs 12 weeks following rash onset | Desquamation 12 days after discharge |
| Hypotension: SBP 90 mm Hg for adults | Intermittent |
| Multisystem involvement, 3 of the following: | 4 organ systems definitively involved |
| GI: vomiting or diarrhea at disease onset | Vomiting and abdominal pain |
| Muscular: severe myalgias, or creatine phosphokinase >2 times the upper limit of normal | |
| Mucous membranes: vaginal, oropharyngeal, or conjunctival hyperemia | |
| Renal: BUN or Cr >2 times the upper limit of normal, or pyuria without evidence of infection | |
| Hepatic: total bilirubin, AST, or ALT levels >2 times the upper limit of normal | AST and ALT peaked at 128IU/L and 94 IU/L |
| Hematologic: platelets 100,000/mm3 | Platelet nadir of 80,000/mm3 |
| CNS: disorientation or altered consciousness without focal neurologic signs | Disorientation and somnolence |
| Probable case: 4 out of 5 clinical criteria present | |
| Confirmed case: 5 out of 5 clinical criteria present, or patient dies before desquamation can occur | |
A rare cause of shock, TSS is most associated with a surge of menstruation‐related cases linked to tampon use in young women in the 1980s.[4] However, in Centers for Disease Control and Prevention (CDC) surveillance between 1987 and 1996, only 59% of the 1069 cases identified were noted to be menstruation‐related, as compared to nearly 80% of all cases earlier in the decade.[4, 5] Today, the syndrome is more likely to present after musculoskeletal and cutaneous trauma, oropharyngeal infections, surgical procedures, and device implantation.[1, 6] Despite the disease's evolving epidemiology, the illness script used by physicians likely continues to focus on young women as the primary at risk population for TSS, causing physicians to neglect the diagnosis in other populations.[1, 6, 7, 8, 9] Given this change in risk factors, it is imperative that clinicians rewrite their scripts and recognize the early signs of TSS in all patients to enable quick and effective treatment.
In addition to its shifting epidemiology and rarity, the diagnosis of TSS vexes clinicians for several reasons. First, TSS cannot be quickly and definitively diagnosed because 2 diagnostic criteria cannot be fulfilled during the acute illness. The disease's hallmarka desquamative rashoccurs only if the patient survives.[3] Serologies often take weeks to return, further delaying diagnosis. During this period of diagnostic delay, the illness has usually already resolved or resulted in death. In addition, the presenting symptoms of rash, fever, and shock are nonspecific. Alternative etiologies include meningococcal meningitis, which can also present dramatically as with this patient; RMSF, which can occasionally have a fulminant presentation; bacterial sepsis, usually from Staphylococcus or Streptococcus species; acute viral syndromes; and severe drug reactions.[6, 10, 11, 12] Palmoplantar desquamation, as in this case, can further narrow the differential as this presentation is uncommon but characteristic of TSS, RMSF, and secondary syphilis.[11] Other diagnostic clues offered by the pattern of the rash may be limited by physician discomfort with diagnosing and describing rashes. Because of this lack of a definitive diagnostic test in the acute setting, it is imperative that the clinician include TSS in the differential of fever, shock, and rash, as mortality from TSS can exceed 20% in patients who are untreated.[13]
Treatment of TSS is straightforward once considered and includes the administration of antibiotics that cover both Staphylococcus and Streptococcus species, in addition to aggressive hydration and supportive care.[14] The final critical detail in this case was the appropriate arrangement of follow‐up. Given the patient's drastic improvement, the complicated process of arranging follow‐up for a transferred patient, and the current model where the hospitalists providing inpatient care do not typically follow their patients in clinic, patients such as these can easily be lost to follow‐up. Had this occurred, the desquamation would have been missed, and the patient's diagnosis would have been incomplete.
This patient was eventually diagnosed with TSS by fulfilling all 5 CDC criteria (Table 1).[3] He made a full recovery, likely aided by the administration of broad‐spectrum antibiotics (followed by doxycycline, which provided community‐acquired methicillin‐resistant S aureus coverage) and his lack of serious comorbidities. This case should serve as a reminder to hospitalists that with a discerning eye, a careful assessment of the clinical facts, and appropriate follow‐up, perhaps the next case of TSS can be caught red‐handed.
KEY POINTS
- When presented with a patient with fever, rash, and shock, hospitalists should consider meningococcal meningitis, RMSF bacterial sepsis, acute viral illness, severe drug reaction, and TSS.
- TSS, caused by S aureus or S pyogenes, is no longer predominantly associated with tampon use. Postsurgical infection and cutaneous trauma have become important present‐day risk factors.
- The initial presentation of TSS is nonspecific. Definitive diagnosis requires proper follow‐up, allowing time for infectious serologies to return negative and for the disease's hallmark desquamation to occur.
Disclosure
Nothing to report.
A previously healthy 58‐year‐old man presented to a community hospital's emergency department 1 day after the sudden onset of a severe headache, fever, diffuse abdominal pain, nausea, vomiting, and disorientation. The patient had a history of allergic rhinitis and his only medication was a daily multivitamin.
Key features of this patient's presentation include the abrupt onset of severe headache, disorientation, fever, and abdominal pain. The list of entities likely to make a previously healthy individual this ill this quickly is typically circumscribed. His presentation raises the possibility of bacterial meningitis (including Listeria, given his age), viral encephalitis, or other extraneural etiologies of sepsis. Noninfectious explanations seem much less likely given the rapid tempo of illness.
His proclivity for gardening and apparent tick exposure raise the question of tick‐borne illnesses. This would constitute a rather explosive onset for any of these; however, babesiosis, Rocky Mountain spotted fever (RMSF), ehrlichiosis, and anaplasmosis could present this abruptly, with dog exposure linked to RMSF.
The potential causes of fever and rash are myriad, although the severity and acuity of this patient's illness narrow the differential considerably, likely to an infectious cause. Diagnoses that typically include a generalized exanthem involving the palms and soles are meningococcal meningitis, overwhelming Staphylococcus aureus sepsis, RMSF (realizing that this disease is not common in the upper Midwest), and toxic shock syndrome. The rash described is not the classic and/or fully developed rash typical of any of these; subsequent evolution to a petechial appearance would lend further support to the first 3 diagnoses. Ehrlichiosis is still a possibility, although the palm and sole involvement would be unusual. The presence of a rash makes anaplasmosis very unlikely, although not entirely excluded. The finding of modest splenomegaly does not help further distinguish between these possibilities.
Empiric antimicrobials should be immediately administered after blood cultures, a complete blood count, and coagulation studies are obtained. Doxycycline would be appropriate to treat the possible tick‐borne diseases already mentioned, whereas antimicrobials appropriate to cover community‐acquired bacterial meningitis in a 58‐year‐old (ie, vancomycin, ampicillin, and a third‐generation cephalosporin) should also be empirically administered. Given the patient's altered mentation, a brain computed tomography (CT) should be urgently obtained. Provided this did not show evidence of increased intracranial pressure and that coagulation studies and a platelet count did not suggest a contraindication, a lumbar puncture should then be performed promptly. The patient should be placed in droplet precautions until meningococcal disease is excluded. Although most patients with bacterial meningitis will exhibit meningismus, a substantial minority will not.
These laboratory results do not significantly affect the differential diagnosis. Although nonspecific, moderate thrombocytopenia and modest elevation of hepatic transaminases are typical for tick‐borne diseases, whereas leukocytosis is somewhat atypical for these entities. Marked elevation of the C‐reactive protein with a less striking increase in the erythrocyte sedimentation rate, along with significant hypoalbuminemia, are commonly encountered early in the course of critical infectious illnesses. The elevated troponin likely reflects severe sepsis and demand ischemia, and is associated with a less favorable prognosis; an electrocardiogram and serial cardiac biomarkers are appropriate to help exclude an acute coronary syndrome. As already noted, blood cultures need to be obtained and a lumbar puncture should be performed, provided this can be safely accomplished.
Results of the lumbar puncture exclude bacterial meningitis as the explanation of this patient's illness; the mildly elevated protein is nonspecific. These studies do not otherwise change the differential diagnosis.
Supporting data for a diagnosis of pneumonia, such as pulmonary infiltrates or supplemental oxygen requirement, are lacking. Given his critical illness, broad spectrum antimicrobial coverage is indicated, and as a primary central nervous system (CNS) infection now appears unlikely, piperacillin/tazobactam (which does not have adequate CNS penetration) and vancomycin are reasonable. Empiric treatment for RMSF is appropriate, and should have been initiated earlier in the patient's course, despite the upper Midwest being out of the typical range for this disease. Doxycycline will also provide excellent coverage for ehrlichiosis and anaplasmosis.
Given the patient's deterioration, it is important to stop and reconsider the differential diagnosis in an attempt to avoid anchoring bias and premature closure. The patient's illness is almost certainly infectious in nature, and the differential is not substantially altered by the most recent information. A skin biopsy should be performed in an attempt to secure the diagnosis.
The patient's overall course, including rapid onset of severe illness and especially the apparent dramatic response to doxycycline, make tick‐borne illness very likely. Completing a course of doxycycline is certainly appropriate, typically for 7 to 14 days. The acute serologies drawn prior to discharge may well reveal the causative agent, but convalescent serology should also be obtained at the time of an outpatient follow‐up visit as immunoglobulin G has a delayed rise. Without hyponatremia or respiratory symptoms, Legionella seems unlikely.
The appearance of late desquamation of the palms and soles is an unexpected and important sign. Desquamation in this pattern following an illness of this nature strongly suggests a diagnosis of staphylococcal toxic shock syndrome (TSS), and in conjunction with the negative serologies, argues that tick‐borne disease is unlikely. The list of other entities that might lead to desquamation in this setting is very short, namely adult Kawasaki disease and drug reaction. The former seems reasonably excluded based on details of the case, whereas a doxycycline‐related drug reaction, although not entirely implausible, seems quite unlikely as this medication was started after the onset of the initial rash. This patient most likely had staphylococcal TSS secondary to a minor and unappreciated skin lesion.
DISCUSSION
TSS is a systemic illness resulting in multiorgan dysfunction.[1] Infection by S aureus or Streptococcus pyogenes causes TSS by stimulating maladaptive T‐cell proliferation and cytokine release resulting in shock.[1, 2] A definitive diagnosis requires fever, a diffuse macular erythematous rash (often resembling a sunburn), with subsequent desquamation, hypotension, and involvement of at least 3 organ systems. Blood cultures, cerebrospinal cultures, and serologies for other organisms should be negative; although Staphylococcus and Streptococcus species may be isolated, they frequently are not (Table 1).[3]
| Diagnostic Criteria* | This Case |
|---|---|
| |
| Fever: Temperature 102.0F | Fever: 105.3F on admission |
| Rash: Diffuse macular erythroderma | Diffuse morbilliform rash with progression to confluent erythroderma |
| Desquamation of rash: occurs 12 weeks following rash onset | Desquamation 12 days after discharge |
| Hypotension: SBP 90 mm Hg for adults | Intermittent |
| Multisystem involvement, 3 of the following: | 4 organ systems definitively involved |
| GI: vomiting or diarrhea at disease onset | Vomiting and abdominal pain |
| Muscular: severe myalgias, or creatine phosphokinase >2 times the upper limit of normal | |
| Mucous membranes: vaginal, oropharyngeal, or conjunctival hyperemia | |
| Renal: BUN or Cr >2 times the upper limit of normal, or pyuria without evidence of infection | |
| Hepatic: total bilirubin, AST, or ALT levels >2 times the upper limit of normal | AST and ALT peaked at 128IU/L and 94 IU/L |
| Hematologic: platelets 100,000/mm3 | Platelet nadir of 80,000/mm3 |
| CNS: disorientation or altered consciousness without focal neurologic signs | Disorientation and somnolence |
| Probable case: 4 out of 5 clinical criteria present | |
| Confirmed case: 5 out of 5 clinical criteria present, or patient dies before desquamation can occur | |
A rare cause of shock, TSS is most associated with a surge of menstruation‐related cases linked to tampon use in young women in the 1980s.[4] However, in Centers for Disease Control and Prevention (CDC) surveillance between 1987 and 1996, only 59% of the 1069 cases identified were noted to be menstruation‐related, as compared to nearly 80% of all cases earlier in the decade.[4, 5] Today, the syndrome is more likely to present after musculoskeletal and cutaneous trauma, oropharyngeal infections, surgical procedures, and device implantation.[1, 6] Despite the disease's evolving epidemiology, the illness script used by physicians likely continues to focus on young women as the primary at risk population for TSS, causing physicians to neglect the diagnosis in other populations.[1, 6, 7, 8, 9] Given this change in risk factors, it is imperative that clinicians rewrite their scripts and recognize the early signs of TSS in all patients to enable quick and effective treatment.
In addition to its shifting epidemiology and rarity, the diagnosis of TSS vexes clinicians for several reasons. First, TSS cannot be quickly and definitively diagnosed because 2 diagnostic criteria cannot be fulfilled during the acute illness. The disease's hallmarka desquamative rashoccurs only if the patient survives.[3] Serologies often take weeks to return, further delaying diagnosis. During this period of diagnostic delay, the illness has usually already resolved or resulted in death. In addition, the presenting symptoms of rash, fever, and shock are nonspecific. Alternative etiologies include meningococcal meningitis, which can also present dramatically as with this patient; RMSF, which can occasionally have a fulminant presentation; bacterial sepsis, usually from Staphylococcus or Streptococcus species; acute viral syndromes; and severe drug reactions.[6, 10, 11, 12] Palmoplantar desquamation, as in this case, can further narrow the differential as this presentation is uncommon but characteristic of TSS, RMSF, and secondary syphilis.[11] Other diagnostic clues offered by the pattern of the rash may be limited by physician discomfort with diagnosing and describing rashes. Because of this lack of a definitive diagnostic test in the acute setting, it is imperative that the clinician include TSS in the differential of fever, shock, and rash, as mortality from TSS can exceed 20% in patients who are untreated.[13]
Treatment of TSS is straightforward once considered and includes the administration of antibiotics that cover both Staphylococcus and Streptococcus species, in addition to aggressive hydration and supportive care.[14] The final critical detail in this case was the appropriate arrangement of follow‐up. Given the patient's drastic improvement, the complicated process of arranging follow‐up for a transferred patient, and the current model where the hospitalists providing inpatient care do not typically follow their patients in clinic, patients such as these can easily be lost to follow‐up. Had this occurred, the desquamation would have been missed, and the patient's diagnosis would have been incomplete.
This patient was eventually diagnosed with TSS by fulfilling all 5 CDC criteria (Table 1).[3] He made a full recovery, likely aided by the administration of broad‐spectrum antibiotics (followed by doxycycline, which provided community‐acquired methicillin‐resistant S aureus coverage) and his lack of serious comorbidities. This case should serve as a reminder to hospitalists that with a discerning eye, a careful assessment of the clinical facts, and appropriate follow‐up, perhaps the next case of TSS can be caught red‐handed.
KEY POINTS
- When presented with a patient with fever, rash, and shock, hospitalists should consider meningococcal meningitis, RMSF bacterial sepsis, acute viral illness, severe drug reaction, and TSS.
- TSS, caused by S aureus or S pyogenes, is no longer predominantly associated with tampon use. Postsurgical infection and cutaneous trauma have become important present‐day risk factors.
- The initial presentation of TSS is nonspecific. Definitive diagnosis requires proper follow‐up, allowing time for infectious serologies to return negative and for the disease's hallmark desquamation to occur.
Disclosure
Nothing to report.
- . Toxic shock syndrome: major advances in pathogenesis, but not treatment. Crit Care Clin. 2013;29:651–675.
- . The toxic shock syndromes. Infect Dis Clin North Am. 1996;10(4):727–746.
- Centers for Disease Control and Prevention. National Notifiable Diseases Surveillance System. Toxic shock syndrome (other than Streptococcal) (TSS) 2011 Case Definition. Available at: http://wwwn.cdc.gov/nndss/conditions/toxic‐shock‐syndrome‐other‐than‐streptococcal/case‐definition/2011. Accessed June 4, 2015.
- Centers for Disease Control and Prevention. Update: toxic‐shock syndrome—United States. MMWR Morb Mortal Wkly Rep. 1983;32(30):398–400.
- , , , , , . Toxic shock syndrome in the United States: surveillance update, 1979–1996. Emerg Infect Dis. 1999;5(6):807–810.
- . Fever and rash. Infect Dis Clin North Am. 1996;10(1):101–110.
- , , , et al. Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS One. 2011;6(8):e22997.
- , , , et al. Toxic‐shock syndrome in menstruating women: association with tampon use and staphylococcus aureus and clinical features in 52 cases. N Engl J Med. 1980;303(25):1436–1442.
- , , , . Toxic‐shock syndrome—epidemiologic features, recurrence, risk factors, and prevention. N Engl J Med. 1980;303:1429–1435.
- , . Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62(4):804–816.
- . Toxic shock syndrome: broadening the differential diagnosis. J Am Board Fam Pract. 2001;14(2):131–136.
- , , , . Spatial clustering by disease severity among reported Rocky Mountain spotted fever cases in the United States, 2001–2005. Am J Trop Med Hyg. 2009;80(1):72–77.
- , , , et al. One in five mortality in non‐menstrual toxic shock syndrome versus no mortality in menstrual cases in a balanced French series of 55 cases. Eur J Clin Microbio Infect Dis. 2008;27(1):37–43.
- , . Gram‐positive toxic shock syndromes. Lancet Infect Dis. 2009;9(5):281–290.
- . Toxic shock syndrome: major advances in pathogenesis, but not treatment. Crit Care Clin. 2013;29:651–675.
- . The toxic shock syndromes. Infect Dis Clin North Am. 1996;10(4):727–746.
- Centers for Disease Control and Prevention. National Notifiable Diseases Surveillance System. Toxic shock syndrome (other than Streptococcal) (TSS) 2011 Case Definition. Available at: http://wwwn.cdc.gov/nndss/conditions/toxic‐shock‐syndrome‐other‐than‐streptococcal/case‐definition/2011. Accessed June 4, 2015.
- Centers for Disease Control and Prevention. Update: toxic‐shock syndrome—United States. MMWR Morb Mortal Wkly Rep. 1983;32(30):398–400.
- , , , , , . Toxic shock syndrome in the United States: surveillance update, 1979–1996. Emerg Infect Dis. 1999;5(6):807–810.
- . Fever and rash. Infect Dis Clin North Am. 1996;10(1):101–110.
- , , , et al. Staphylococcal toxic shock syndrome 2000–2006: epidemiology, clinical features, and molecular characteristics. PLoS One. 2011;6(8):e22997.
- , , , et al. Toxic‐shock syndrome in menstruating women: association with tampon use and staphylococcus aureus and clinical features in 52 cases. N Engl J Med. 1980;303(25):1436–1442.
- , , , . Toxic‐shock syndrome—epidemiologic features, recurrence, risk factors, and prevention. N Engl J Med. 1980;303:1429–1435.
- , . Evaluating the febrile patient with a rash. Am Fam Physician. 2000;62(4):804–816.
- . Toxic shock syndrome: broadening the differential diagnosis. J Am Board Fam Pract. 2001;14(2):131–136.
- , , , . Spatial clustering by disease severity among reported Rocky Mountain spotted fever cases in the United States, 2001–2005. Am J Trop Med Hyg. 2009;80(1):72–77.
- , , , et al. One in five mortality in non‐menstrual toxic shock syndrome versus no mortality in menstrual cases in a balanced French series of 55 cases. Eur J Clin Microbio Infect Dis. 2008;27(1):37–43.
- , . Gram‐positive toxic shock syndromes. Lancet Infect Dis. 2009;9(5):281–290.