User login
EMA recommends orphan designation for cancer vaccine

The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
Haplo-HSCT approach appears safe, effective for nonmalignant disorders

Image courtesy of NIAID
VALENCIA, SPAIN—Interim results of a phase 1/2 trial suggest the adjunct T-cell therapy BPX-501 can safely accelerate immune recovery after haploidentical hematopoietic stem cell transplant (haplo-HSCT) in pediatric patients with nonmalignant disorders.
Twenty-four such patients received BPX-501 after haplo-HSCT on this trial.
At a median follow-up of 7 months, all 24 were still alive and disease-free.
In addition, the incidence of graft-versus-host disease (GVHD) was considered “very low.”
Pietro Merli, MD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results during the Presidential Symposium of the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT) as abstract O007.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, investigators reported results in 41 patients treated on this trial.
Dr Merli presented data on the 24 patients with nonmalignant disorders, including Fanconi anemia (n=5), beta-thalassemia major (n=5), severe combined immunodeficiency (n=5), Wiskott-Aldrich syndrome (n=4), Diamond-Blackfan anemia (n=1), hemophagocytic lymphohistiocytosis (n=1), immune deficiency due to mutation of XIAP gene (n=1), osteopetrosis (n=1), and sickle cell disease (n=1).
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only to be used in the event of uncontrollable GVHD.
Results
The median time to platelet recovery was 10 days (range, 7-16), and the median time to neutrophil recovery was 15 days (range, 10-33).
At a median follow-up of 220 days (range, 61-486), there were no reports of transplant-related mortality.
All 24 patients were still alive and disease-free. And none of the patients developed post-transplant lymphoproliferative disorder.
The cumulative incidence of skin-only acute GVHD was 16.6% (n=4), and the cumulative incidence of mild chronic GVHD was 5% (n=1).
This trial also included 17 patients with acute leukemias. Results in these patients were presented at the EBMT meeting as abstract WP16. ![]()
*Information in the abstract differs from that presented at the meeting.
Image courtesy of NIAID
VALENCIA, SPAIN—Interim results of a phase 1/2 trial suggest the adjunct T-cell therapy BPX-501 can safely accelerate immune recovery after haploidentical hematopoietic stem cell transplant (haplo-HSCT) in pediatric patients with nonmalignant disorders.
Twenty-four such patients received BPX-501 after haplo-HSCT on this trial.
At a median follow-up of 7 months, all 24 were still alive and disease-free.
In addition, the incidence of graft-versus-host disease (GVHD) was considered “very low.”
Pietro Merli, MD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results during the Presidential Symposium of the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT) as abstract O007.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, investigators reported results in 41 patients treated on this trial.
Dr Merli presented data on the 24 patients with nonmalignant disorders, including Fanconi anemia (n=5), beta-thalassemia major (n=5), severe combined immunodeficiency (n=5), Wiskott-Aldrich syndrome (n=4), Diamond-Blackfan anemia (n=1), hemophagocytic lymphohistiocytosis (n=1), immune deficiency due to mutation of XIAP gene (n=1), osteopetrosis (n=1), and sickle cell disease (n=1).
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only to be used in the event of uncontrollable GVHD.
Results
The median time to platelet recovery was 10 days (range, 7-16), and the median time to neutrophil recovery was 15 days (range, 10-33).
At a median follow-up of 220 days (range, 61-486), there were no reports of transplant-related mortality.
All 24 patients were still alive and disease-free. And none of the patients developed post-transplant lymphoproliferative disorder.
The cumulative incidence of skin-only acute GVHD was 16.6% (n=4), and the cumulative incidence of mild chronic GVHD was 5% (n=1).
This trial also included 17 patients with acute leukemias. Results in these patients were presented at the EBMT meeting as abstract WP16. ![]()
*Information in the abstract differs from that presented at the meeting.
Image courtesy of NIAID
VALENCIA, SPAIN—Interim results of a phase 1/2 trial suggest the adjunct T-cell therapy BPX-501 can safely accelerate immune recovery after haploidentical hematopoietic stem cell transplant (haplo-HSCT) in pediatric patients with nonmalignant disorders.
Twenty-four such patients received BPX-501 after haplo-HSCT on this trial.
At a median follow-up of 7 months, all 24 were still alive and disease-free.
In addition, the incidence of graft-versus-host disease (GVHD) was considered “very low.”
Pietro Merli, MD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results during the Presidential Symposium of the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT) as abstract O007.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, investigators reported results in 41 patients treated on this trial.
Dr Merli presented data on the 24 patients with nonmalignant disorders, including Fanconi anemia (n=5), beta-thalassemia major (n=5), severe combined immunodeficiency (n=5), Wiskott-Aldrich syndrome (n=4), Diamond-Blackfan anemia (n=1), hemophagocytic lymphohistiocytosis (n=1), immune deficiency due to mutation of XIAP gene (n=1), osteopetrosis (n=1), and sickle cell disease (n=1).
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only to be used in the event of uncontrollable GVHD.
Results
The median time to platelet recovery was 10 days (range, 7-16), and the median time to neutrophil recovery was 15 days (range, 10-33).
At a median follow-up of 220 days (range, 61-486), there were no reports of transplant-related mortality.
All 24 patients were still alive and disease-free. And none of the patients developed post-transplant lymphoproliferative disorder.
The cumulative incidence of skin-only acute GVHD was 16.6% (n=4), and the cumulative incidence of mild chronic GVHD was 5% (n=1).
This trial also included 17 patients with acute leukemias. Results in these patients were presented at the EBMT meeting as abstract WP16. ![]()
*Information in the abstract differs from that presented at the meeting.
Therapy may improve haplo-HSCT in leukemia patients

Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007. ![]()
*Information in the abstract differs from that presented at the meeting.
Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007. ![]()
*Information in the abstract differs from that presented at the meeting.
Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007. ![]()
*Information in the abstract differs from that presented at the meeting.
Interdisciplinary Rounds
Care of hospitalized patients requires effective teamwork within groups composed of physicians (eg, residents, hospitalists, specialists), advanced practice providers, nurses, patient‐care technicians, pharmacists, social workers, and therapists. Sadly, hospital‐based team members often fail to communicate. For example, 2 studies found that nurses and physicians communicated with one another on only 50% to 60% of their patients' hospital days, resulting in a lack of a mutual understanding of the plan of care.[1, 2]
Failure to communicate effectively may be because the hospital setting poses important challenges to teamwork, including the use of large teams with membership that changes frequently because of the need to provide care around the clock. Furthermore, individual team members often have high workloads, care for multiple patients simultaneously, and are seldom in the same place at the same time.
Interdisciplinary rounds (IDR) are a microsystem‐level solution with the goal to share information, achieve mutual understanding, and collaboratively revise the plan of care within care teams. Though common, IDR look very different across hospitals, making studies that evaluate novel strategies to improve IDR and measure their impact of great interest to hospital medicine.
In this issue of the Journal of Hospital Medicine, Bhamidipati and colleagues present a systematic review of published studies evaluating the effect of IDR on patient outcomes.[3] The systematic review included 22 studies, including 12 experimental/quasiexperimental and 10 observational studies. Overall, 13 studies were of low to medium quality, and 9 were high quality. Importantly, relatively few studies reported the degree to which IDR were implemented as planned. The investigators found evidence that IDR had a positive effect on length of stay (LOS) and staff satisfaction, but little evidence to support an effect on patient safety or satisfaction. Furthermore, the investigators found significant variability in IDR design and team composition. Some of this variation is to be expected, as IDR, like other interventions to improve quality and safety of patient care in complex settings, should be implemented with an expectation that the team may need to make adaptations based on local contextual factors such as workload (eg, daily census), environment (eg, open vs closed intensive care unit), local politics (eg, uniquely strong support for/against the intervention), and prior experience (eg, prior failed, similar interventions).[4, 5] Moreover, objectives for IDR may differ across settings. Some hospitals may have room (and a need) to improve LOS, whereas others may prioritize improving patient safety or patient experience metrics.
Bhamidipati and colleagues explain that their review did not reveal a causal pathway between IDR design and outcomes. We believe this lack of association is because most of the included studies did not propose a causal pathway between the IDR components implemented and the outcomes assessed. That is, few studies referred to conceptual models that explain how components of the IDR intervention might influence downstream patient outcomes.
IDR have the potential to influence a number of patient outcomes, including those reflecting efficiency (eg, length of stay), patient safety (eg, adverse events), and patient centeredness (eg, patient satisfaction). However, these outcomes are influenced by many factors, including patient characteristics and other efforts to improve care. As explained by the investigators, the results of many of the included studies may have been confounded due to relatively weak study designs and statistical analyses. Importantly, few of the studies included in this review report the more proximal measure of teamwork. If we hypothesize that IDR improve patient outcomes, they do so by improving teamwork. After all, the purpose of IDR is to assemble team members so they can communicate about and coordinate care. Measuring teamwork behaviors is difficult, especially on medical services. Measuring teamwork climate, the measurable aspects of team culture, is relatively easy. A recent systematic review of teamwork climate assessments in internal medicine identified the Safety Attitudes Questionnaire and the Team Climate Inventory as having substantial validity evidence and association with improved patient outcomes.[6]
Bhamidipati and colleagues proposed a definition for IDR and taxonomy for IDR design and reporting based on their systematic review. Although very useful, the IDR definition may be too limiting as evidenced by the fact that very few studies would be included in a systematic review using this definition as the inclusion criteria. Their proposed taxonomy should serve as a useful framework for future research efforts and appropriately recommends reporting of site characteristics, components of IDR design, and outcomes.
The systematic review by Bhamidipati et al. must also be interpreted in conjunction with another recently published systematic review by Pannick and colleagues assessing the effect of interdisciplinary team care interventions on general medical wards.[7] Contrary to the findings of the Bhamidipati et al. study, Pannick and colleagues found that most interdisciplinary team care interventions had no effect on LOS, but that half of the studies found an improvement in complications of care. Importantly, Pannick and colleagues included only experimental and quasiexperimental studies in their systematic review (ie, no observational studies).
There is clearly more work to be done in researching IDR and other interventions to improve teamwork in general medical settings. Larger studies are needed to provide sufficient power to detect improvement in outcomes. Future studies need to report the degree to which interventions are implemented as planned and need to use stronger study designs (eg, cluster randomized control or interrupted time series) to avoid the influence of confounders. Qualitative methods should be used to assess the influence of contextual factors on the success of interventions.[4] Most importantly, future studies should be based on conceptual models that explain how components of the intervention influence proximal measures of teamwork and downstream patient outcomes.
In the meantime, what is a hospital leader to do? We believe efforts to improve IDR are warranted, but that IDR program leaders need to first specify their primary objective(s). For example, in some hospitals, there may be little room to further reduce LOS, so another goalreducing preventable readmissions or reducing adverse eventsmight be specified as the key performance indicator. This crucial first step of creating a shared goal informs the design, implementation, and evaluation of IDR. We also believe that geographic localization of physicians to specific units is foundational to improving IDR. Physicians cannot feasibly attend IDR if their patients are spread across multiple units (or buildings). Finally, hospital leaders also need to view IDR as part of a larger set of interventions to improve teamwork. Leaders need to assess the adequacy of staffing levels, workflow, and team composition.[8] Unit‐based interdisciplinary leadership models should be used to help link efforts at various levels within a larger system.[9] These models designate a unit medical director and nurse manager who are jointly responsible for unit performance.
In conclusion, IDR play an important role in improving patient outcomes, but only do so by improving teamwork. In redesigning IDR, leaders need to be thoughtful about what outcomes IDR can affect, how IDR affect them, and how IDR fit into larger‐scale efforts to improve performance.
Disclosure
Nothing to report.
- , , , , , . Can we talk? Priorities for patient care differed among health care providers. In: Henriksen K, Battles JB, Marks ES, Lewin DI, eds. Advances in Patient Safety: From Research to Implementation. Volume 1: Research Findings. Rockville, MD: Agency for Healthcare Research and Quality; 2005.
- , , , et al. Patterns of nurse‐physician communication and agreement on the plan of care. Qual Saf Health Care. 2010;19(3):195–199.
- , , , , , . Structure and outcomes of inter‐disciplinary rounds in hospitalized medicine patients: a systematic review and suggested taxonomny. J Hosp Med. 2016;11:513–523.
- , , , et al. Advancing the science of patient safety. Ann Intern Med. 2011;154(10):693–696.
- . Improvement interventions are social treatments, not pills. Ann Intern Med. 2014;161(7):526–527.
- , , , et al. Teamwork assessment in internal medicine: a systematic review of validity evidence and outcomes. J Gen Intern Med. 2014;29(6):894–910.
- , , , et al. Effects of interdisciplinary team care interventions on general medical wards: a systematic review. JAMA Intern Med. 2015;175(8):1288–1298.
- , , , . Improving the quality and safety of care on the medical ward: A review and synthesis of the evidence base. Eur J Intern Med. 2014;25(10):874–887.
- , , , , , . Unit‐based interprofessional leadership models in six US hospitals. J Hosp Med. 2014;9(8):545–550.
Care of hospitalized patients requires effective teamwork within groups composed of physicians (eg, residents, hospitalists, specialists), advanced practice providers, nurses, patient‐care technicians, pharmacists, social workers, and therapists. Sadly, hospital‐based team members often fail to communicate. For example, 2 studies found that nurses and physicians communicated with one another on only 50% to 60% of their patients' hospital days, resulting in a lack of a mutual understanding of the plan of care.[1, 2]
Failure to communicate effectively may be because the hospital setting poses important challenges to teamwork, including the use of large teams with membership that changes frequently because of the need to provide care around the clock. Furthermore, individual team members often have high workloads, care for multiple patients simultaneously, and are seldom in the same place at the same time.
Interdisciplinary rounds (IDR) are a microsystem‐level solution with the goal to share information, achieve mutual understanding, and collaboratively revise the plan of care within care teams. Though common, IDR look very different across hospitals, making studies that evaluate novel strategies to improve IDR and measure their impact of great interest to hospital medicine.
In this issue of the Journal of Hospital Medicine, Bhamidipati and colleagues present a systematic review of published studies evaluating the effect of IDR on patient outcomes.[3] The systematic review included 22 studies, including 12 experimental/quasiexperimental and 10 observational studies. Overall, 13 studies were of low to medium quality, and 9 were high quality. Importantly, relatively few studies reported the degree to which IDR were implemented as planned. The investigators found evidence that IDR had a positive effect on length of stay (LOS) and staff satisfaction, but little evidence to support an effect on patient safety or satisfaction. Furthermore, the investigators found significant variability in IDR design and team composition. Some of this variation is to be expected, as IDR, like other interventions to improve quality and safety of patient care in complex settings, should be implemented with an expectation that the team may need to make adaptations based on local contextual factors such as workload (eg, daily census), environment (eg, open vs closed intensive care unit), local politics (eg, uniquely strong support for/against the intervention), and prior experience (eg, prior failed, similar interventions).[4, 5] Moreover, objectives for IDR may differ across settings. Some hospitals may have room (and a need) to improve LOS, whereas others may prioritize improving patient safety or patient experience metrics.
Bhamidipati and colleagues explain that their review did not reveal a causal pathway between IDR design and outcomes. We believe this lack of association is because most of the included studies did not propose a causal pathway between the IDR components implemented and the outcomes assessed. That is, few studies referred to conceptual models that explain how components of the IDR intervention might influence downstream patient outcomes.
IDR have the potential to influence a number of patient outcomes, including those reflecting efficiency (eg, length of stay), patient safety (eg, adverse events), and patient centeredness (eg, patient satisfaction). However, these outcomes are influenced by many factors, including patient characteristics and other efforts to improve care. As explained by the investigators, the results of many of the included studies may have been confounded due to relatively weak study designs and statistical analyses. Importantly, few of the studies included in this review report the more proximal measure of teamwork. If we hypothesize that IDR improve patient outcomes, they do so by improving teamwork. After all, the purpose of IDR is to assemble team members so they can communicate about and coordinate care. Measuring teamwork behaviors is difficult, especially on medical services. Measuring teamwork climate, the measurable aspects of team culture, is relatively easy. A recent systematic review of teamwork climate assessments in internal medicine identified the Safety Attitudes Questionnaire and the Team Climate Inventory as having substantial validity evidence and association with improved patient outcomes.[6]
Bhamidipati and colleagues proposed a definition for IDR and taxonomy for IDR design and reporting based on their systematic review. Although very useful, the IDR definition may be too limiting as evidenced by the fact that very few studies would be included in a systematic review using this definition as the inclusion criteria. Their proposed taxonomy should serve as a useful framework for future research efforts and appropriately recommends reporting of site characteristics, components of IDR design, and outcomes.
The systematic review by Bhamidipati et al. must also be interpreted in conjunction with another recently published systematic review by Pannick and colleagues assessing the effect of interdisciplinary team care interventions on general medical wards.[7] Contrary to the findings of the Bhamidipati et al. study, Pannick and colleagues found that most interdisciplinary team care interventions had no effect on LOS, but that half of the studies found an improvement in complications of care. Importantly, Pannick and colleagues included only experimental and quasiexperimental studies in their systematic review (ie, no observational studies).
There is clearly more work to be done in researching IDR and other interventions to improve teamwork in general medical settings. Larger studies are needed to provide sufficient power to detect improvement in outcomes. Future studies need to report the degree to which interventions are implemented as planned and need to use stronger study designs (eg, cluster randomized control or interrupted time series) to avoid the influence of confounders. Qualitative methods should be used to assess the influence of contextual factors on the success of interventions.[4] Most importantly, future studies should be based on conceptual models that explain how components of the intervention influence proximal measures of teamwork and downstream patient outcomes.
In the meantime, what is a hospital leader to do? We believe efforts to improve IDR are warranted, but that IDR program leaders need to first specify their primary objective(s). For example, in some hospitals, there may be little room to further reduce LOS, so another goalreducing preventable readmissions or reducing adverse eventsmight be specified as the key performance indicator. This crucial first step of creating a shared goal informs the design, implementation, and evaluation of IDR. We also believe that geographic localization of physicians to specific units is foundational to improving IDR. Physicians cannot feasibly attend IDR if their patients are spread across multiple units (or buildings). Finally, hospital leaders also need to view IDR as part of a larger set of interventions to improve teamwork. Leaders need to assess the adequacy of staffing levels, workflow, and team composition.[8] Unit‐based interdisciplinary leadership models should be used to help link efforts at various levels within a larger system.[9] These models designate a unit medical director and nurse manager who are jointly responsible for unit performance.
In conclusion, IDR play an important role in improving patient outcomes, but only do so by improving teamwork. In redesigning IDR, leaders need to be thoughtful about what outcomes IDR can affect, how IDR affect them, and how IDR fit into larger‐scale efforts to improve performance.
Disclosure
Nothing to report.
Care of hospitalized patients requires effective teamwork within groups composed of physicians (eg, residents, hospitalists, specialists), advanced practice providers, nurses, patient‐care technicians, pharmacists, social workers, and therapists. Sadly, hospital‐based team members often fail to communicate. For example, 2 studies found that nurses and physicians communicated with one another on only 50% to 60% of their patients' hospital days, resulting in a lack of a mutual understanding of the plan of care.[1, 2]
Failure to communicate effectively may be because the hospital setting poses important challenges to teamwork, including the use of large teams with membership that changes frequently because of the need to provide care around the clock. Furthermore, individual team members often have high workloads, care for multiple patients simultaneously, and are seldom in the same place at the same time.
Interdisciplinary rounds (IDR) are a microsystem‐level solution with the goal to share information, achieve mutual understanding, and collaboratively revise the plan of care within care teams. Though common, IDR look very different across hospitals, making studies that evaluate novel strategies to improve IDR and measure their impact of great interest to hospital medicine.
In this issue of the Journal of Hospital Medicine, Bhamidipati and colleagues present a systematic review of published studies evaluating the effect of IDR on patient outcomes.[3] The systematic review included 22 studies, including 12 experimental/quasiexperimental and 10 observational studies. Overall, 13 studies were of low to medium quality, and 9 were high quality. Importantly, relatively few studies reported the degree to which IDR were implemented as planned. The investigators found evidence that IDR had a positive effect on length of stay (LOS) and staff satisfaction, but little evidence to support an effect on patient safety or satisfaction. Furthermore, the investigators found significant variability in IDR design and team composition. Some of this variation is to be expected, as IDR, like other interventions to improve quality and safety of patient care in complex settings, should be implemented with an expectation that the team may need to make adaptations based on local contextual factors such as workload (eg, daily census), environment (eg, open vs closed intensive care unit), local politics (eg, uniquely strong support for/against the intervention), and prior experience (eg, prior failed, similar interventions).[4, 5] Moreover, objectives for IDR may differ across settings. Some hospitals may have room (and a need) to improve LOS, whereas others may prioritize improving patient safety or patient experience metrics.
Bhamidipati and colleagues explain that their review did not reveal a causal pathway between IDR design and outcomes. We believe this lack of association is because most of the included studies did not propose a causal pathway between the IDR components implemented and the outcomes assessed. That is, few studies referred to conceptual models that explain how components of the IDR intervention might influence downstream patient outcomes.
IDR have the potential to influence a number of patient outcomes, including those reflecting efficiency (eg, length of stay), patient safety (eg, adverse events), and patient centeredness (eg, patient satisfaction). However, these outcomes are influenced by many factors, including patient characteristics and other efforts to improve care. As explained by the investigators, the results of many of the included studies may have been confounded due to relatively weak study designs and statistical analyses. Importantly, few of the studies included in this review report the more proximal measure of teamwork. If we hypothesize that IDR improve patient outcomes, they do so by improving teamwork. After all, the purpose of IDR is to assemble team members so they can communicate about and coordinate care. Measuring teamwork behaviors is difficult, especially on medical services. Measuring teamwork climate, the measurable aspects of team culture, is relatively easy. A recent systematic review of teamwork climate assessments in internal medicine identified the Safety Attitudes Questionnaire and the Team Climate Inventory as having substantial validity evidence and association with improved patient outcomes.[6]
Bhamidipati and colleagues proposed a definition for IDR and taxonomy for IDR design and reporting based on their systematic review. Although very useful, the IDR definition may be too limiting as evidenced by the fact that very few studies would be included in a systematic review using this definition as the inclusion criteria. Their proposed taxonomy should serve as a useful framework for future research efforts and appropriately recommends reporting of site characteristics, components of IDR design, and outcomes.
The systematic review by Bhamidipati et al. must also be interpreted in conjunction with another recently published systematic review by Pannick and colleagues assessing the effect of interdisciplinary team care interventions on general medical wards.[7] Contrary to the findings of the Bhamidipati et al. study, Pannick and colleagues found that most interdisciplinary team care interventions had no effect on LOS, but that half of the studies found an improvement in complications of care. Importantly, Pannick and colleagues included only experimental and quasiexperimental studies in their systematic review (ie, no observational studies).
There is clearly more work to be done in researching IDR and other interventions to improve teamwork in general medical settings. Larger studies are needed to provide sufficient power to detect improvement in outcomes. Future studies need to report the degree to which interventions are implemented as planned and need to use stronger study designs (eg, cluster randomized control or interrupted time series) to avoid the influence of confounders. Qualitative methods should be used to assess the influence of contextual factors on the success of interventions.[4] Most importantly, future studies should be based on conceptual models that explain how components of the intervention influence proximal measures of teamwork and downstream patient outcomes.
In the meantime, what is a hospital leader to do? We believe efforts to improve IDR are warranted, but that IDR program leaders need to first specify their primary objective(s). For example, in some hospitals, there may be little room to further reduce LOS, so another goalreducing preventable readmissions or reducing adverse eventsmight be specified as the key performance indicator. This crucial first step of creating a shared goal informs the design, implementation, and evaluation of IDR. We also believe that geographic localization of physicians to specific units is foundational to improving IDR. Physicians cannot feasibly attend IDR if their patients are spread across multiple units (or buildings). Finally, hospital leaders also need to view IDR as part of a larger set of interventions to improve teamwork. Leaders need to assess the adequacy of staffing levels, workflow, and team composition.[8] Unit‐based interdisciplinary leadership models should be used to help link efforts at various levels within a larger system.[9] These models designate a unit medical director and nurse manager who are jointly responsible for unit performance.
In conclusion, IDR play an important role in improving patient outcomes, but only do so by improving teamwork. In redesigning IDR, leaders need to be thoughtful about what outcomes IDR can affect, how IDR affect them, and how IDR fit into larger‐scale efforts to improve performance.
Disclosure
Nothing to report.
- , , , , , . Can we talk? Priorities for patient care differed among health care providers. In: Henriksen K, Battles JB, Marks ES, Lewin DI, eds. Advances in Patient Safety: From Research to Implementation. Volume 1: Research Findings. Rockville, MD: Agency for Healthcare Research and Quality; 2005.
- , , , et al. Patterns of nurse‐physician communication and agreement on the plan of care. Qual Saf Health Care. 2010;19(3):195–199.
- , , , , , . Structure and outcomes of inter‐disciplinary rounds in hospitalized medicine patients: a systematic review and suggested taxonomny. J Hosp Med. 2016;11:513–523.
- , , , et al. Advancing the science of patient safety. Ann Intern Med. 2011;154(10):693–696.
- . Improvement interventions are social treatments, not pills. Ann Intern Med. 2014;161(7):526–527.
- , , , et al. Teamwork assessment in internal medicine: a systematic review of validity evidence and outcomes. J Gen Intern Med. 2014;29(6):894–910.
- , , , et al. Effects of interdisciplinary team care interventions on general medical wards: a systematic review. JAMA Intern Med. 2015;175(8):1288–1298.
- , , , . Improving the quality and safety of care on the medical ward: A review and synthesis of the evidence base. Eur J Intern Med. 2014;25(10):874–887.
- , , , , , . Unit‐based interprofessional leadership models in six US hospitals. J Hosp Med. 2014;9(8):545–550.
- , , , , , . Can we talk? Priorities for patient care differed among health care providers. In: Henriksen K, Battles JB, Marks ES, Lewin DI, eds. Advances in Patient Safety: From Research to Implementation. Volume 1: Research Findings. Rockville, MD: Agency for Healthcare Research and Quality; 2005.
- , , , et al. Patterns of nurse‐physician communication and agreement on the plan of care. Qual Saf Health Care. 2010;19(3):195–199.
- , , , , , . Structure and outcomes of inter‐disciplinary rounds in hospitalized medicine patients: a systematic review and suggested taxonomny. J Hosp Med. 2016;11:513–523.
- , , , et al. Advancing the science of patient safety. Ann Intern Med. 2011;154(10):693–696.
- . Improvement interventions are social treatments, not pills. Ann Intern Med. 2014;161(7):526–527.
- , , , et al. Teamwork assessment in internal medicine: a systematic review of validity evidence and outcomes. J Gen Intern Med. 2014;29(6):894–910.
- , , , et al. Effects of interdisciplinary team care interventions on general medical wards: a systematic review. JAMA Intern Med. 2015;175(8):1288–1298.
- , , , . Improving the quality and safety of care on the medical ward: A review and synthesis of the evidence base. Eur J Intern Med. 2014;25(10):874–887.
- , , , , , . Unit‐based interprofessional leadership models in six US hospitals. J Hosp Med. 2014;9(8):545–550.
Medicare Insurance Reduces Cost
Up to 10% of the acutely ill patients in hospitals today are not admitted, but cared for under outpatient observation.[1, 2] Hospitals use observation services to replace inpatient services for patients who do not meet inpatient illness standards. Use of this technique for Medicare beneficiaries has grown in recent years, and future policy changes may further increase the percentage of hospital stays classified as observation.[3, 4]
Increased use of observation services has cost‐sharing implications for Medicare beneficiaries. Because hospital observation stays are paid through Medicare's outpatient (Part B) rather than inpatient (Part A) benefit, beneficiaries do not have an out‐of‐pocket maximum per hospital stay.[3] Although prior analyses documented mean out‐of‐pocket costs of $400 to $600 per stay, out‐of‐pocket costs may exceed the Part A deductible and leave beneficiaries responsible for hospital costs that may have been reimbursed by Part A in an inpatient stay.[5, 6, 7] Approximately 6% to 10% of Medicare observation stays result in out‐of‐pocket costs exceeding the Part A deductible nationally, and recent work in this journal documented that 26.6% of beneficiaries with repeat observation stays within a 60‐day period may pay more than the 1‐time deductible‐based payment that beneficiaries are responsible for under Medicare Part A.[5, 6, 7]
However, prior analyses of beneficiary out‐of‐pocket costs did not account for supplemental insurance payments. Approximately 80% to 90% of fee‐for‐service Medicare beneficiaries who use either a private (Medigap or employer based) or state‐based (Medicaid or other plans) supplemental insurance plan.[8, 9, 10] The effect of supplemental insurance on out‐of‐pocket costs has been documented for inpatient stays, yet has not been explored for observation stays.[9] We sought to describe Medicare beneficiaries' out‐of‐pocket costs by accounting for payments from all insurers.
METHODS
We obtained payment data from 2 affiliated hospitals for all Medicare observation hospital stays between April 2013 and March 2014. Stays insured by Medicare Advantage plans (Part C) were excluded, and charges from skilled nursing facility (SNF) stays were not available. Although the exact origin of each stay was not available, the majority of stays at these hospitals came from emergency visits, direct placement from outpatient providers, and postoutpatient procedural monitoring. The dataset included charges, insurance adjustments and payments from all sources, diagnoses, and demographics. Insurance adjustments represented the reductions applied to total stay charges by each insurer in congruence with their contract with the health system. Our dataset included charges for all hospital materials and services usually billed under Part A, including medications. Out‐of‐pocket costs were calculated by subtracting insurance adjustments and payments from the total charges for each stay. To identify potential cost‐shifting from Medicare to beneficiaries, we compared out‐of‐pocket costs to the Part A deductible ($1184 for stays in 2013 and $1216 for 2014). Household income data were estimated using zip code and Internal Revenue Service data for 2013.[11]
The University of California Los Angeles institutional review board approved this study. Statistical significance was calculated using a 2‐tailed unpaired t test for means and 2 for percentages.
RESULTS
There were 2029 total observation stays during the study period, representing 5.0% of all discharges from both hospitals. Medicare beneficiaries accounted for 722 of those observation stays. Among the 498 finalized Medicare observation stays, the median patient age was 73 years, and median household income was $50,591. The median length of stay was 25 hours, with 1.8% of stays lasting longer than 2 midnights. Seventy percent of beneficiaries had private supplemental insurance, whereas 6% had state‐based supplemental plans. Table 1 presents detailed costs. Out‐of‐pocket costs ranged from $0 to $16,196. Stays without supplemental insurance had mean and median out of pocket costs of $537 and $286, respectively. The mean out‐of‐pocket costs for stays with private supplemental insurance decreased to $45 (P 0.01) and $168 (P = 0.21) for stays with state‐based supplemental insurance, with a median below $1 for both. On average, beneficiaries without supplemental insurance were responsible for $654 less than the Part A deductible. Thirteen beneficiaries had multiple finalized stays within 60 days, with a mean out‐of‐pocket cost of $119, median of $20, and 1 stay produced an out‐of‐pocket cost exceeding the Part A deductible. An additional 224 Medicare stays not finalized because of missing supplemental payments had a mean out‐of‐pocket cost of $125 (P 0.01) and median of $5 after applying supplemental insurer adjustments (not shown in Table 1).
| Medicare Stays, N = 498 | |||
|---|---|---|---|
| Mean (SD) | Median (IQR) | Range | |
| |||
| Payments by Medicare | $2,533 (2,883) | $1521 (1,898) | $4$29,633 |
| Payments by supplemental insurers | |||
| Private insurers (N = 351) | $454 (384) | $324 (332) | $0$2,590 |
| State‐based insurers (N = 29) | $222(402) | $43 (102) | $2$1,229 |
| Beneficiary out‐of‐pocket costs | |||
| Without supplemental insurer (N = 118) | $537 (1,557) | $286 (440) | $0$16,196* |
| With private supplemental insurer (N = 351) | $45 (414) | $0.39 (16) | $0$7,670* |
| With state‐based supplemental insurer (N = 29) | $168 (412) | $0.15 (27) | $01,870* |
A minority of observation stays produced out‐of‐pocket costs exceeding the Part A deductible. Those percentages were 7.6% for stays without a supplemental insurer, 3.5% (P 0.01) for stays with a state‐based supplemental insurer, and 0.3% (P 0.01) for stays with a private supplemental insurer. Of the 224 nonfinalized Medicare stays, 1.3% (P 0.01) exceeded the Part A deductible after supplemental insurer adjustments.
DISCUSSION
This study demonstrates that supplemental insurance can dramatically reduce Medicare beneficiaries' out‐of‐pocket costs in observation services. Mean out‐of‐pocket costs of $45 and $168 for stays with private and state‐based supplemental insurer plans are significantly lower than prior estimates calculated without supplemental insurance information, as are the percentages of stays with out‐of‐pocket costs exceeding the Part A deductible, at 0.3% and 3.5%, respectively.[5, 6, 7] Because the majority of Medicare beneficiaries use supplemental insurance, excessive out‐of‐pockets in observation services may occur less frequently than previously reported.[9, 10] Clinicians concerned about excessive out‐of‐pocket costs for Medicare beneficiaries can be reassured they are usually modest for beneficiaries with supplemental insurance.
This study's mean out‐of‐pocket cost and percentage of stays with out‐of‐pocket costs exceeding the Part A deductible for beneficiaries without supplemental insurance are similar to results from prior national analyses performed without supplemental insurer information.[5, 6, 7] But this study was limited by a small sample size from 2 affiliated hospitals, with few repeat observation stays within a 60‐day period. In addition, posthospitalization SNF fees were not included, which traditionally have been a significant source of out‐of‐pocket costs in observation services.[3, 7] Populations with supplemental insurance treated elsewhere may incur hospital out‐of‐pocket costs differing from these results due to dissimilarities in the presence and quality of supplemental insurance.
However, most Medigap plans are federally regulated to cover the majority of out‐of‐pockets unpaid by Medicare.[12] Medicaid plans usually place limits on out‐of‐pocket costs, and any other state or employer‐based supplemental plans will also reduce out‐of‐pocket costs.[13] Thus, it is likely accurate to assume mean observation services out‐of‐pocket costs for hospital fees are lower than previously reported by national analyses performed without supplemental insurance information. Attempts at estimating beneficiary out‐of‐pocket costs in the future should account for supplemental insurance adjustments and payments.
Acknowledgements
The authors thank Andrew Kaufman for his work in obtaining these data.
Disclosures: Dr. Doyle's time was supported by a National Research Service Award from the National Institutes of Health and administered through the University of California Los Angeles. Drs. Ettner and Nuckols received no support for this work. There are no conflicts of interest to report.
- , , , et al. Hospitalized but not admitted: characteristics of patients with “observation status” at an academic medical center. JAMA Intern Med. 2013;173(21):1991–1998.
- , , . Final report observation status related to hospital records. Available at: https://www.hcup‐us.ahrq.gov/reports/methods/FinalReportonObservationStatus_v2Final.pdf. Published September 27, 2002.
- . The two‐midnight rule. Health Policy Briefs. Health Affairs website. Available at: http://www.healthaffairs.org/healthpolicybriefs/brief.php?brief_id=133. Published January 22, 2015.
- , , . Sharp rise in Medicare enrollees being held in hospitals for observation raises concerns about causes and consequences. Health Aff (Millwood). 2012;31(6):1251–1259.
- , , , , . Patient financial responsibility for observation care. J Hosp Med. 2015;10(11):718–723.
- , , L. . Observation status: Financial Implications for Medicare Beneficiaries. AARP Public Policy Institute. Available at: http://www.aarp.org/content/dam/aarp/ppi/2015/Hosp Obs Financial Impact Paper.pdf. Published April 2015.
- . Hospitals' use of observation stays and short inpatient stays for Medicare beneficiaries. Department of Health and Human Services. Office of Inspector General. Available at: https://oig.hhs.gov/oei/reports/oei‐02‐12‐00040.pdf. Published July 29, 2013.
- . Trends in Medicare supplemental insurance and prescription drug benefits, 1996–2001: Data update. Available at: http://www.kff.org/medicare/upload/Trends‐in‐Medicare‐Supplemental‐Insurance‐and‐Prescription‐Drug‐Benefits‐1996–2001Data‐Update.pdf.
- . Medicare beneficiaries' out‐of‐pocket spending for health care. AARP Public Policy Institute. Available at: http://www.aarp.org/content/dam/aarp/research/public_policy_institute/health/medicare‐beneficiaries‐out‐of‐pocket‐spending‐AARP‐ppi‐health.pdf. Published May 2012.
- , , , et al. A primer on Medicare: key facts about the Medicare program and the people it covers. What types of supplemental insurance do beneficiaries have? Kaiser Family Foundation website. Available at: http://kff.org/report‐section/a‐primer‐on‐medicare‐what‐types‐of‐supplemental‐insurance‐do‐beneficiaries‐have. Published March 20, 2015.
- SOI tax stats—individual income tax statistics—ZIP code data (SOI). Available at: https://www.irs.gov/uac/SOI‐Tax‐Stats‐Individual‐Income‐Tax‐Statistics‐ZIP‐Code‐Data‐(SOI). Accessed January 1, 2016.
- How to compare Medigap policies. Medicare.gov website. Available at: https://www.medicare.gov/supplement‐other‐insurance/compare‐medigap/compare‐medigap.html.
- Cost sharing out of pocket costs. Medicaid.gov website. Available at: https://www.medicaid.gov/medicaid‐chip‐program‐information/by‐topics/cost‐sharing/cost‐sharing‐out‐of‐pocket‐costs.html. Accessed January 26, 2016.
- , , . Balancing margin and mission: hospitals alter billing and collection practices for uninsured patients. Issue Brief Cent Stud Health Syst Change. 2005;(99):1–4.
Up to 10% of the acutely ill patients in hospitals today are not admitted, but cared for under outpatient observation.[1, 2] Hospitals use observation services to replace inpatient services for patients who do not meet inpatient illness standards. Use of this technique for Medicare beneficiaries has grown in recent years, and future policy changes may further increase the percentage of hospital stays classified as observation.[3, 4]
Increased use of observation services has cost‐sharing implications for Medicare beneficiaries. Because hospital observation stays are paid through Medicare's outpatient (Part B) rather than inpatient (Part A) benefit, beneficiaries do not have an out‐of‐pocket maximum per hospital stay.[3] Although prior analyses documented mean out‐of‐pocket costs of $400 to $600 per stay, out‐of‐pocket costs may exceed the Part A deductible and leave beneficiaries responsible for hospital costs that may have been reimbursed by Part A in an inpatient stay.[5, 6, 7] Approximately 6% to 10% of Medicare observation stays result in out‐of‐pocket costs exceeding the Part A deductible nationally, and recent work in this journal documented that 26.6% of beneficiaries with repeat observation stays within a 60‐day period may pay more than the 1‐time deductible‐based payment that beneficiaries are responsible for under Medicare Part A.[5, 6, 7]
However, prior analyses of beneficiary out‐of‐pocket costs did not account for supplemental insurance payments. Approximately 80% to 90% of fee‐for‐service Medicare beneficiaries who use either a private (Medigap or employer based) or state‐based (Medicaid or other plans) supplemental insurance plan.[8, 9, 10] The effect of supplemental insurance on out‐of‐pocket costs has been documented for inpatient stays, yet has not been explored for observation stays.[9] We sought to describe Medicare beneficiaries' out‐of‐pocket costs by accounting for payments from all insurers.
METHODS
We obtained payment data from 2 affiliated hospitals for all Medicare observation hospital stays between April 2013 and March 2014. Stays insured by Medicare Advantage plans (Part C) were excluded, and charges from skilled nursing facility (SNF) stays were not available. Although the exact origin of each stay was not available, the majority of stays at these hospitals came from emergency visits, direct placement from outpatient providers, and postoutpatient procedural monitoring. The dataset included charges, insurance adjustments and payments from all sources, diagnoses, and demographics. Insurance adjustments represented the reductions applied to total stay charges by each insurer in congruence with their contract with the health system. Our dataset included charges for all hospital materials and services usually billed under Part A, including medications. Out‐of‐pocket costs were calculated by subtracting insurance adjustments and payments from the total charges for each stay. To identify potential cost‐shifting from Medicare to beneficiaries, we compared out‐of‐pocket costs to the Part A deductible ($1184 for stays in 2013 and $1216 for 2014). Household income data were estimated using zip code and Internal Revenue Service data for 2013.[11]
The University of California Los Angeles institutional review board approved this study. Statistical significance was calculated using a 2‐tailed unpaired t test for means and 2 for percentages.
RESULTS
There were 2029 total observation stays during the study period, representing 5.0% of all discharges from both hospitals. Medicare beneficiaries accounted for 722 of those observation stays. Among the 498 finalized Medicare observation stays, the median patient age was 73 years, and median household income was $50,591. The median length of stay was 25 hours, with 1.8% of stays lasting longer than 2 midnights. Seventy percent of beneficiaries had private supplemental insurance, whereas 6% had state‐based supplemental plans. Table 1 presents detailed costs. Out‐of‐pocket costs ranged from $0 to $16,196. Stays without supplemental insurance had mean and median out of pocket costs of $537 and $286, respectively. The mean out‐of‐pocket costs for stays with private supplemental insurance decreased to $45 (P 0.01) and $168 (P = 0.21) for stays with state‐based supplemental insurance, with a median below $1 for both. On average, beneficiaries without supplemental insurance were responsible for $654 less than the Part A deductible. Thirteen beneficiaries had multiple finalized stays within 60 days, with a mean out‐of‐pocket cost of $119, median of $20, and 1 stay produced an out‐of‐pocket cost exceeding the Part A deductible. An additional 224 Medicare stays not finalized because of missing supplemental payments had a mean out‐of‐pocket cost of $125 (P 0.01) and median of $5 after applying supplemental insurer adjustments (not shown in Table 1).
| Medicare Stays, N = 498 | |||
|---|---|---|---|
| Mean (SD) | Median (IQR) | Range | |
| |||
| Payments by Medicare | $2,533 (2,883) | $1521 (1,898) | $4$29,633 |
| Payments by supplemental insurers | |||
| Private insurers (N = 351) | $454 (384) | $324 (332) | $0$2,590 |
| State‐based insurers (N = 29) | $222(402) | $43 (102) | $2$1,229 |
| Beneficiary out‐of‐pocket costs | |||
| Without supplemental insurer (N = 118) | $537 (1,557) | $286 (440) | $0$16,196* |
| With private supplemental insurer (N = 351) | $45 (414) | $0.39 (16) | $0$7,670* |
| With state‐based supplemental insurer (N = 29) | $168 (412) | $0.15 (27) | $01,870* |
A minority of observation stays produced out‐of‐pocket costs exceeding the Part A deductible. Those percentages were 7.6% for stays without a supplemental insurer, 3.5% (P 0.01) for stays with a state‐based supplemental insurer, and 0.3% (P 0.01) for stays with a private supplemental insurer. Of the 224 nonfinalized Medicare stays, 1.3% (P 0.01) exceeded the Part A deductible after supplemental insurer adjustments.
DISCUSSION
This study demonstrates that supplemental insurance can dramatically reduce Medicare beneficiaries' out‐of‐pocket costs in observation services. Mean out‐of‐pocket costs of $45 and $168 for stays with private and state‐based supplemental insurer plans are significantly lower than prior estimates calculated without supplemental insurance information, as are the percentages of stays with out‐of‐pocket costs exceeding the Part A deductible, at 0.3% and 3.5%, respectively.[5, 6, 7] Because the majority of Medicare beneficiaries use supplemental insurance, excessive out‐of‐pockets in observation services may occur less frequently than previously reported.[9, 10] Clinicians concerned about excessive out‐of‐pocket costs for Medicare beneficiaries can be reassured they are usually modest for beneficiaries with supplemental insurance.
This study's mean out‐of‐pocket cost and percentage of stays with out‐of‐pocket costs exceeding the Part A deductible for beneficiaries without supplemental insurance are similar to results from prior national analyses performed without supplemental insurer information.[5, 6, 7] But this study was limited by a small sample size from 2 affiliated hospitals, with few repeat observation stays within a 60‐day period. In addition, posthospitalization SNF fees were not included, which traditionally have been a significant source of out‐of‐pocket costs in observation services.[3, 7] Populations with supplemental insurance treated elsewhere may incur hospital out‐of‐pocket costs differing from these results due to dissimilarities in the presence and quality of supplemental insurance.
However, most Medigap plans are federally regulated to cover the majority of out‐of‐pockets unpaid by Medicare.[12] Medicaid plans usually place limits on out‐of‐pocket costs, and any other state or employer‐based supplemental plans will also reduce out‐of‐pocket costs.[13] Thus, it is likely accurate to assume mean observation services out‐of‐pocket costs for hospital fees are lower than previously reported by national analyses performed without supplemental insurance information. Attempts at estimating beneficiary out‐of‐pocket costs in the future should account for supplemental insurance adjustments and payments.
Acknowledgements
The authors thank Andrew Kaufman for his work in obtaining these data.
Disclosures: Dr. Doyle's time was supported by a National Research Service Award from the National Institutes of Health and administered through the University of California Los Angeles. Drs. Ettner and Nuckols received no support for this work. There are no conflicts of interest to report.
Up to 10% of the acutely ill patients in hospitals today are not admitted, but cared for under outpatient observation.[1, 2] Hospitals use observation services to replace inpatient services for patients who do not meet inpatient illness standards. Use of this technique for Medicare beneficiaries has grown in recent years, and future policy changes may further increase the percentage of hospital stays classified as observation.[3, 4]
Increased use of observation services has cost‐sharing implications for Medicare beneficiaries. Because hospital observation stays are paid through Medicare's outpatient (Part B) rather than inpatient (Part A) benefit, beneficiaries do not have an out‐of‐pocket maximum per hospital stay.[3] Although prior analyses documented mean out‐of‐pocket costs of $400 to $600 per stay, out‐of‐pocket costs may exceed the Part A deductible and leave beneficiaries responsible for hospital costs that may have been reimbursed by Part A in an inpatient stay.[5, 6, 7] Approximately 6% to 10% of Medicare observation stays result in out‐of‐pocket costs exceeding the Part A deductible nationally, and recent work in this journal documented that 26.6% of beneficiaries with repeat observation stays within a 60‐day period may pay more than the 1‐time deductible‐based payment that beneficiaries are responsible for under Medicare Part A.[5, 6, 7]
However, prior analyses of beneficiary out‐of‐pocket costs did not account for supplemental insurance payments. Approximately 80% to 90% of fee‐for‐service Medicare beneficiaries who use either a private (Medigap or employer based) or state‐based (Medicaid or other plans) supplemental insurance plan.[8, 9, 10] The effect of supplemental insurance on out‐of‐pocket costs has been documented for inpatient stays, yet has not been explored for observation stays.[9] We sought to describe Medicare beneficiaries' out‐of‐pocket costs by accounting for payments from all insurers.
METHODS
We obtained payment data from 2 affiliated hospitals for all Medicare observation hospital stays between April 2013 and March 2014. Stays insured by Medicare Advantage plans (Part C) were excluded, and charges from skilled nursing facility (SNF) stays were not available. Although the exact origin of each stay was not available, the majority of stays at these hospitals came from emergency visits, direct placement from outpatient providers, and postoutpatient procedural monitoring. The dataset included charges, insurance adjustments and payments from all sources, diagnoses, and demographics. Insurance adjustments represented the reductions applied to total stay charges by each insurer in congruence with their contract with the health system. Our dataset included charges for all hospital materials and services usually billed under Part A, including medications. Out‐of‐pocket costs were calculated by subtracting insurance adjustments and payments from the total charges for each stay. To identify potential cost‐shifting from Medicare to beneficiaries, we compared out‐of‐pocket costs to the Part A deductible ($1184 for stays in 2013 and $1216 for 2014). Household income data were estimated using zip code and Internal Revenue Service data for 2013.[11]
The University of California Los Angeles institutional review board approved this study. Statistical significance was calculated using a 2‐tailed unpaired t test for means and 2 for percentages.
RESULTS
There were 2029 total observation stays during the study period, representing 5.0% of all discharges from both hospitals. Medicare beneficiaries accounted for 722 of those observation stays. Among the 498 finalized Medicare observation stays, the median patient age was 73 years, and median household income was $50,591. The median length of stay was 25 hours, with 1.8% of stays lasting longer than 2 midnights. Seventy percent of beneficiaries had private supplemental insurance, whereas 6% had state‐based supplemental plans. Table 1 presents detailed costs. Out‐of‐pocket costs ranged from $0 to $16,196. Stays without supplemental insurance had mean and median out of pocket costs of $537 and $286, respectively. The mean out‐of‐pocket costs for stays with private supplemental insurance decreased to $45 (P 0.01) and $168 (P = 0.21) for stays with state‐based supplemental insurance, with a median below $1 for both. On average, beneficiaries without supplemental insurance were responsible for $654 less than the Part A deductible. Thirteen beneficiaries had multiple finalized stays within 60 days, with a mean out‐of‐pocket cost of $119, median of $20, and 1 stay produced an out‐of‐pocket cost exceeding the Part A deductible. An additional 224 Medicare stays not finalized because of missing supplemental payments had a mean out‐of‐pocket cost of $125 (P 0.01) and median of $5 after applying supplemental insurer adjustments (not shown in Table 1).
| Medicare Stays, N = 498 | |||
|---|---|---|---|
| Mean (SD) | Median (IQR) | Range | |
| |||
| Payments by Medicare | $2,533 (2,883) | $1521 (1,898) | $4$29,633 |
| Payments by supplemental insurers | |||
| Private insurers (N = 351) | $454 (384) | $324 (332) | $0$2,590 |
| State‐based insurers (N = 29) | $222(402) | $43 (102) | $2$1,229 |
| Beneficiary out‐of‐pocket costs | |||
| Without supplemental insurer (N = 118) | $537 (1,557) | $286 (440) | $0$16,196* |
| With private supplemental insurer (N = 351) | $45 (414) | $0.39 (16) | $0$7,670* |
| With state‐based supplemental insurer (N = 29) | $168 (412) | $0.15 (27) | $01,870* |
A minority of observation stays produced out‐of‐pocket costs exceeding the Part A deductible. Those percentages were 7.6% for stays without a supplemental insurer, 3.5% (P 0.01) for stays with a state‐based supplemental insurer, and 0.3% (P 0.01) for stays with a private supplemental insurer. Of the 224 nonfinalized Medicare stays, 1.3% (P 0.01) exceeded the Part A deductible after supplemental insurer adjustments.
DISCUSSION
This study demonstrates that supplemental insurance can dramatically reduce Medicare beneficiaries' out‐of‐pocket costs in observation services. Mean out‐of‐pocket costs of $45 and $168 for stays with private and state‐based supplemental insurer plans are significantly lower than prior estimates calculated without supplemental insurance information, as are the percentages of stays with out‐of‐pocket costs exceeding the Part A deductible, at 0.3% and 3.5%, respectively.[5, 6, 7] Because the majority of Medicare beneficiaries use supplemental insurance, excessive out‐of‐pockets in observation services may occur less frequently than previously reported.[9, 10] Clinicians concerned about excessive out‐of‐pocket costs for Medicare beneficiaries can be reassured they are usually modest for beneficiaries with supplemental insurance.
This study's mean out‐of‐pocket cost and percentage of stays with out‐of‐pocket costs exceeding the Part A deductible for beneficiaries without supplemental insurance are similar to results from prior national analyses performed without supplemental insurer information.[5, 6, 7] But this study was limited by a small sample size from 2 affiliated hospitals, with few repeat observation stays within a 60‐day period. In addition, posthospitalization SNF fees were not included, which traditionally have been a significant source of out‐of‐pocket costs in observation services.[3, 7] Populations with supplemental insurance treated elsewhere may incur hospital out‐of‐pocket costs differing from these results due to dissimilarities in the presence and quality of supplemental insurance.
However, most Medigap plans are federally regulated to cover the majority of out‐of‐pockets unpaid by Medicare.[12] Medicaid plans usually place limits on out‐of‐pocket costs, and any other state or employer‐based supplemental plans will also reduce out‐of‐pocket costs.[13] Thus, it is likely accurate to assume mean observation services out‐of‐pocket costs for hospital fees are lower than previously reported by national analyses performed without supplemental insurance information. Attempts at estimating beneficiary out‐of‐pocket costs in the future should account for supplemental insurance adjustments and payments.
Acknowledgements
The authors thank Andrew Kaufman for his work in obtaining these data.
Disclosures: Dr. Doyle's time was supported by a National Research Service Award from the National Institutes of Health and administered through the University of California Los Angeles. Drs. Ettner and Nuckols received no support for this work. There are no conflicts of interest to report.
- , , , et al. Hospitalized but not admitted: characteristics of patients with “observation status” at an academic medical center. JAMA Intern Med. 2013;173(21):1991–1998.
- , , . Final report observation status related to hospital records. Available at: https://www.hcup‐us.ahrq.gov/reports/methods/FinalReportonObservationStatus_v2Final.pdf. Published September 27, 2002.
- . The two‐midnight rule. Health Policy Briefs. Health Affairs website. Available at: http://www.healthaffairs.org/healthpolicybriefs/brief.php?brief_id=133. Published January 22, 2015.
- , , . Sharp rise in Medicare enrollees being held in hospitals for observation raises concerns about causes and consequences. Health Aff (Millwood). 2012;31(6):1251–1259.
- , , , , . Patient financial responsibility for observation care. J Hosp Med. 2015;10(11):718–723.
- , , L. . Observation status: Financial Implications for Medicare Beneficiaries. AARP Public Policy Institute. Available at: http://www.aarp.org/content/dam/aarp/ppi/2015/Hosp Obs Financial Impact Paper.pdf. Published April 2015.
- . Hospitals' use of observation stays and short inpatient stays for Medicare beneficiaries. Department of Health and Human Services. Office of Inspector General. Available at: https://oig.hhs.gov/oei/reports/oei‐02‐12‐00040.pdf. Published July 29, 2013.
- . Trends in Medicare supplemental insurance and prescription drug benefits, 1996–2001: Data update. Available at: http://www.kff.org/medicare/upload/Trends‐in‐Medicare‐Supplemental‐Insurance‐and‐Prescription‐Drug‐Benefits‐1996–2001Data‐Update.pdf.
- . Medicare beneficiaries' out‐of‐pocket spending for health care. AARP Public Policy Institute. Available at: http://www.aarp.org/content/dam/aarp/research/public_policy_institute/health/medicare‐beneficiaries‐out‐of‐pocket‐spending‐AARP‐ppi‐health.pdf. Published May 2012.
- , , , et al. A primer on Medicare: key facts about the Medicare program and the people it covers. What types of supplemental insurance do beneficiaries have? Kaiser Family Foundation website. Available at: http://kff.org/report‐section/a‐primer‐on‐medicare‐what‐types‐of‐supplemental‐insurance‐do‐beneficiaries‐have. Published March 20, 2015.
- SOI tax stats—individual income tax statistics—ZIP code data (SOI). Available at: https://www.irs.gov/uac/SOI‐Tax‐Stats‐Individual‐Income‐Tax‐Statistics‐ZIP‐Code‐Data‐(SOI). Accessed January 1, 2016.
- How to compare Medigap policies. Medicare.gov website. Available at: https://www.medicare.gov/supplement‐other‐insurance/compare‐medigap/compare‐medigap.html.
- Cost sharing out of pocket costs. Medicaid.gov website. Available at: https://www.medicaid.gov/medicaid‐chip‐program‐information/by‐topics/cost‐sharing/cost‐sharing‐out‐of‐pocket‐costs.html. Accessed January 26, 2016.
- , , . Balancing margin and mission: hospitals alter billing and collection practices for uninsured patients. Issue Brief Cent Stud Health Syst Change. 2005;(99):1–4.
- , , , et al. Hospitalized but not admitted: characteristics of patients with “observation status” at an academic medical center. JAMA Intern Med. 2013;173(21):1991–1998.
- , , . Final report observation status related to hospital records. Available at: https://www.hcup‐us.ahrq.gov/reports/methods/FinalReportonObservationStatus_v2Final.pdf. Published September 27, 2002.
- . The two‐midnight rule. Health Policy Briefs. Health Affairs website. Available at: http://www.healthaffairs.org/healthpolicybriefs/brief.php?brief_id=133. Published January 22, 2015.
- , , . Sharp rise in Medicare enrollees being held in hospitals for observation raises concerns about causes and consequences. Health Aff (Millwood). 2012;31(6):1251–1259.
- , , , , . Patient financial responsibility for observation care. J Hosp Med. 2015;10(11):718–723.
- , , L. . Observation status: Financial Implications for Medicare Beneficiaries. AARP Public Policy Institute. Available at: http://www.aarp.org/content/dam/aarp/ppi/2015/Hosp Obs Financial Impact Paper.pdf. Published April 2015.
- . Hospitals' use of observation stays and short inpatient stays for Medicare beneficiaries. Department of Health and Human Services. Office of Inspector General. Available at: https://oig.hhs.gov/oei/reports/oei‐02‐12‐00040.pdf. Published July 29, 2013.
- . Trends in Medicare supplemental insurance and prescription drug benefits, 1996–2001: Data update. Available at: http://www.kff.org/medicare/upload/Trends‐in‐Medicare‐Supplemental‐Insurance‐and‐Prescription‐Drug‐Benefits‐1996–2001Data‐Update.pdf.
- . Medicare beneficiaries' out‐of‐pocket spending for health care. AARP Public Policy Institute. Available at: http://www.aarp.org/content/dam/aarp/research/public_policy_institute/health/medicare‐beneficiaries‐out‐of‐pocket‐spending‐AARP‐ppi‐health.pdf. Published May 2012.
- , , , et al. A primer on Medicare: key facts about the Medicare program and the people it covers. What types of supplemental insurance do beneficiaries have? Kaiser Family Foundation website. Available at: http://kff.org/report‐section/a‐primer‐on‐medicare‐what‐types‐of‐supplemental‐insurance‐do‐beneficiaries‐have. Published March 20, 2015.
- SOI tax stats—individual income tax statistics—ZIP code data (SOI). Available at: https://www.irs.gov/uac/SOI‐Tax‐Stats‐Individual‐Income‐Tax‐Statistics‐ZIP‐Code‐Data‐(SOI). Accessed January 1, 2016.
- How to compare Medigap policies. Medicare.gov website. Available at: https://www.medicare.gov/supplement‐other‐insurance/compare‐medigap/compare‐medigap.html.
- Cost sharing out of pocket costs. Medicaid.gov website. Available at: https://www.medicaid.gov/medicaid‐chip‐program‐information/by‐topics/cost‐sharing/cost‐sharing‐out‐of‐pocket‐costs.html. Accessed January 26, 2016.
- , , . Balancing margin and mission: hospitals alter billing and collection practices for uninsured patients. Issue Brief Cent Stud Health Syst Change. 2005;(99):1–4.
Long‐term Antipsychotics in Elders
Delirium, a clinical syndrome characterized by inattention and acute cognitive dysfunction, is very common in older hospitalized patients, with a reported incidence of 18% to 35% at time of admission and overall occurrence rates of 29% to 64%.[1] Previous studies have reported that a diagnosis of delirium is not benign and is associated with other adverse outcomes including prolonged hospitalization, institutionalization, increased cost, and mortality. These outcomes occurred independent of age, prior cognitive functioning, and comorbidities.[2] Guidelines recommend that management of inpatient delirium should be focused on addressing the underlying etiology and managed with nonpharmacological interventions whenever possible.[3, 4, 5] However, implementing these recommendations can prove to be very challenging in hospital settings. Providers frequently have to resort to medical therapies, including antipsychotics (APs). Although these medications are commonly used to treat delirium in elderly patients, there is limited evidence to support their efficacy, and there are currently no proven pharmacological alternatives to these medications.[6] Furthermore, previous studies have demonstrated an increased risk of stroke, infection, cognitive impairment, and mortality in elders with dementia who receive long‐term AP therapy.[7, 8, 9] Yet as many as 48% of hospitalized elders who were newly started on APs had these drugs continued at time of discharge.[10]
There have been few studies describing the long‐term outcomes of elderly patient who are started on APs in the hospital. Most information on outcomes comes from patients with dementia. Therefore, we studied the 1‐year outcomes of a cohort of patients with and without dementia who were started on APs in the hospital and then discharged on these medications. In this cohort, we aimed to describe the number of readmissions, reasons for readmissions, duration of AP therapy, use of other sedating medications such as anxiolytics, hypnotics, and antihistamines as well as the incidence of readmission and death 1 year after the index hospital discharge.
METHODS
We previously described a retrospective cohort of 300 elders (65 years old) admitted to a tertiary care hospital between October 1, 2012 and September 31, 2013 who were newly prescribed APs while hospitalized.[10] Of patients alive at the time of discharge (260), 56% (146 patients) were discharged on APs. Two investigators extracted these 148 patient charts independently to identify and quantify the number of readmissions to the index hospital. We then limited the sample to only the first readmission per patient following the index admission and extracted this readmission for each patient. We first determined if APs were present on the admission medication reconciliation. If APs were not present on admission, we examined whether they were resumed during the hospitalization using the electronic medication administration summary. If they were present on admission, we looked to see if they were discontinued during the readmission and if additional new APs were started during the hospitalizations. We documented the circumstances around APs use and identified patients who died during their hospitalizations. We identified delirium using the same terms that were described in our prior study on the same cohort of patients.[10] We determined if patients were delirious using a predetermined algorithm (Figure 1). Briefly, we first determined delirium was documented. We then examined whether there was a Confusion Assessment Method (CAM) instrument included in the record. If a CAM instrument was not documented, we then looked for documentation using specific terms (eg, disorientation, confusions). We identified patients with dementia by determining whether dementia was documented along with other admission medical comorbidities. If it was not, we determined whether dementia was newly diagnosed during the hospital stay using progress notes or consultation notes. We did not objectively define criteria for diagnosis of dementia. We used the National Death Index (NDI) to determine mortality for all patients 1 year after discharge from the index hospitalization. The NDI is a national database of death records maintained by the National Center for Health Statistics. It has shown consistently high sensitivity and specificity for detection of death.[11]

We used descriptive statistics (means, standard deviations, range, and percents as appropriate to the scale of measurement) to describe the patient sample. We then used multiple logistic regression to identify significant predictors of death within 1 year of discharge.[12] Univariate analysis was used to select candidates for the logistic model (t tests for continuous factors and 2 for discrete factors). All factors with a significance level 0.2 on univariate analysis were included in the logistic regression, in addition to age and sex (regardless of significance). A maximum likelihood procedure was used to calculate the regression coefficients for the logistic model. The likelihood ratio criterion was used to determine the significance of individual factors in the regression model.[13] Factors with a significance level of 0.15 or less were retained in the final model, in addition to age and sex.
RESULTS
The 260 patients discharged alive from their index admissions had a 1‐year mortality rate of 29% (75/260). Of the 146/260 patients discharged on APs, 60 (41%) patients experienced at least 1 readmission (mean = 2 readmissions per patient; range, 18, with 111 total readmissions for 60 patients) within 1 year from discharge (Figure 2). Most common diagnoses at the time of readmissions were related neurological and psychiatric disorders (14%), cardiovascular and circulation disorders (13%), renal injury and electrolyte disorders (11%), and infections (6%). Among patients with at least 1 readmission, the mean age was 81.3 (range, 65.599.7), 60% were male, and 45% were admitted from a skilled nursing facility or rehabilitation facility (Table 1). Median time to readmission was 43.5 days (range, 1343 days), and 79% were readmitted to a medical service. The remaining 20% were admitted to a surgical service. Inpatient mortality during first readmissions was 8% (5/60). At the time of first readmission, 39/60 (65%) of patients were still on the same APs on which they had been discharged, and the APs were continued during the hospitalization in 79% of the patients (61% quetiapine, 19% olanzapine, and 13% risperidone). About half of patients whose APs were discontinued prior to readmission received a new AP during their hospital stays (9/20; 45%). One patient had been started on quetiapine in the outpatient setting. No patients were found to have new benzodiazepines, nonbenzodiazepine hypnotic, or antihistamines on their admission medication list.
| Variables | Value* |
|---|---|
| |
| Age, mean (range), yr | 81.3 (65.599.7) |
| Gender, no. (%) | |
| Male | 36 (60) |
| Female | 24 (40) |
| Admitted from, no. (%) | |
| Home | 33 (55) |
| Rehabilitation facilities | 5 (8) |
| SNF | 22 (37) |
| Services, no. (%) | |
| Medicine | 48 (80) |
| Surgery | 12 (20) |
| Types of APs continued on readmission (from index admission), no. (%) | |
| Quetiapine | 19 (61) |
| Olanzapine | 6 (19) |
| Risperidone | 4 (13) |
| Haloperidol | 2 (7) |
| Types of APs started during readmission, no. (%) | |
| Quetiapine | 7 (39) |
| Risperidone | 2 (11) |
| Haloperidol | 16 (89) |
| Indications for AP use, no. (%) | |
| Delirium | 14 (77) |
| Undocumented | 3 (17) |
| Other | 1 (6) |
| ECG, no. (%) | |
| Prior to APs administration | 17 (94) |
| After APs administration | 4 (22) |
| QTc prolongation >500 ms, no. (%) | |
| Prior to APs administration | 3 (18) |
| After APs administration∥ | 2 (50) |
| Discharge destination, no. (%) | |
| Home | 23 (38) |
| Rehabilitation facilities | 4 (7) |
| SNF | 28 (47) |
| Death | 5 (8) |
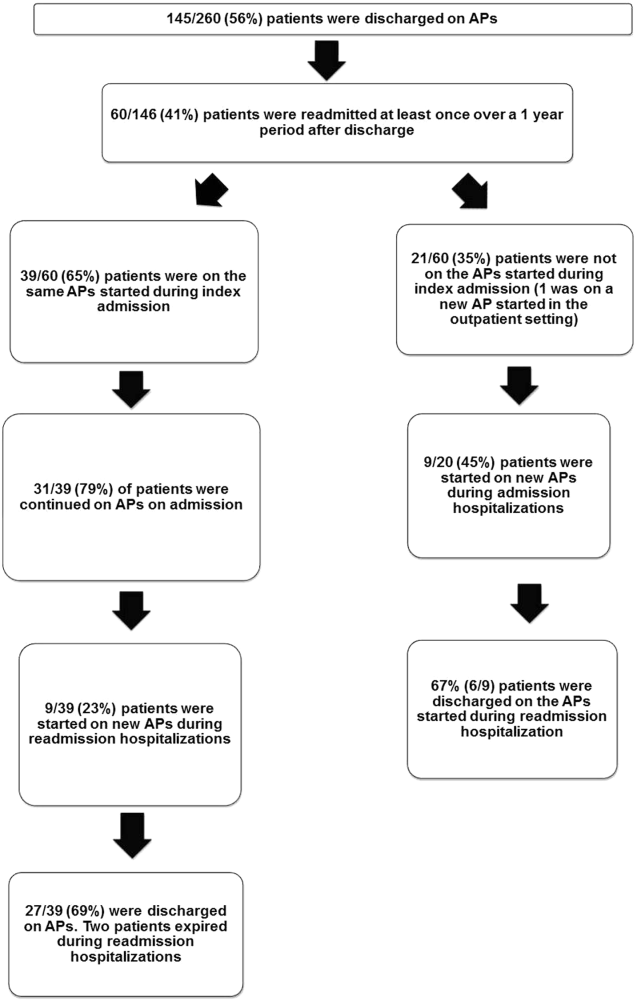
Eighteen patients received 1 or more new APs during the readmission hospitalizations. These included haloperidol (89%) and quetiapine (39%). Delirium was the main reported indication for starting APs (78%), but in 17% of cases no indication was documented. An electrocardiogram (ECG) was performed in 94% prior to APs administration and for 22% after APs administration. Corrected QT interval (QTc) of >500 ms was present in 18% of patients in pretreatment ECG and 50% of patients in post‐AP ECG. Of patients who survived readmission, 58% (32/55) were discharged to postacute facilities. Of the 39 patients who were on the same APs from index admission, 27 (69%) patients were eventually discharged on the same APs or new APs started during the readmission.
In the multivariable model (Table 2), predictors of death at 1 year included discharge to postacute facilities after index admission (odds ratio [OR]: 2.28; 95% confidence interval [CI]: 1.10‐4.73, P = 0.03) and QTc prolongation >500 ms during index admission (OR: 3.41; 95% CI: 1.34‐8.67, P = 0.01). Age and gender were not associated with 1‐year mortality.
| Odds Ratio | 95% Confidence Interval | P Value | |
|---|---|---|---|
| |||
| Age | 1.03 | 0.991.06 | 0.13 |
| Male sex | 0.87 | 0.501.52 | 0.63 |
| Risperdal | 3.53 | 0.6419.40 | 0.15 |
| QTc prolongation after AP administration* | 3.41 | 1.348.67 | 0.01 |
| Presence of geriatric psychiatry consult | 0.30 | 0.091.04 | 0.06 |
| Discharged to postacute facilities vs home | 2.28 | 1.104.73 | 0.03 |
DISCUSSION
In a cohort of elderly patients who were discharged on APs, nearly one‐third (29%) died within 1 year of the hospitalization in which APs were initiated. Nearly half of the survivors from the index admission (41%) experienced at least 1 admission within 1 year from discharge. Of readmitted patients, two‐thirds were taking the same APs that had been started during the index hospitalization. Half of the patients not on APs on readmission were started on an AP during the hospitalization, most often because they became delirious on return to the acute care setting. Compared to patients discharged home after an index admission, patients who were discharged to postacute facilities were almost 4 times as likely to die during the year subsequent to the admission. These data suggest that once patients are started on APs, most are continued on them until the next admission or are restarted during that readmission. Moreover, hospitalized elders who require an AP are at high risk for mortality in the coming year.
Prior studies have reported that patients with delirium have elevated 1‐year mortality rates.[14, 15, 16, 17, 18, 19] A secondary analysis of the Delirium Prevention Trial, which included 437 hospitalized older patients, revealed a 1‐year mortality rate of 20% in those who were never delirious during hospitalization, compared to 26% to 38% in patients with delirium.[19] Additionally, 1‐year mortality in hospitalized older patients with delirium (36%) was shown to be higher than patients with dementia (29%) or depression (26%).[17] Unlike these studies, not all of the patients in our study had documented delirium, but all received an AP. Still, it is notable that the 1‐year mortality rate for delirium in general is similar to what we found in this study.
The literature has also reported that long‐term AP use is associated with excess mortality in elder patients, especially those with dementia.[20, 21, 22] In a retrospective cohort study, older patients with dementia who were taking antipsychotics had significantly higher 1‐year mortality rates (23%29%) than patients not taking antipsychotic medications (15%). In a large Canadian propensity score‐matched cohort study that included over 13,000 demented older adults, the mortality was higher in the community‐dwelling elders who received atypical APs compared to no APs, with a difference of 1.1% in 180‐day mortality rate after initiation of APs.[21] The absolute mortality rate was 2.6% higher in patients who received typical compared to atypical APs. Unlike these studies, not every patient in our cohort had a diagnosis of dementia, but again, mortality rates in these studies appear similar to our cohort.
In contrast, other observational studies have not found an increased risk associated with receipt of APs. For example, a prospective study that enrolled approximately 950 patients with probable dementia showed that AP use was not associated with time to death after adjustment for comorbidities, demographic and cognitive variables.[23] These conflicting results highlight the difficulties of attributing outcomes in high‐risk populations. Although the excess mortality observed in patients taking APs may be related to the risks of APs, it is quite possible that patients who require APs (most often for delirium or agitated dementia) are at higher risk of death. This confounding by indication may be nearly impossible to adjust for retrospectively, even using techniques such as propensity matching.
Our report adds to the literature; we know of no studies to date describing a cohort of patients, most with delirium, who were started on APs in the hospital. We also attempted to identify the reasons that patients were started on APs, which have been infrequently reported. As noted above, our 1‐year mortality rate of 29% among older patients prescribed APs in the hospital was quite similar to mortality rates both for patients with delirium who were not necessarily treated with APs and patients with dementia who were treated with APs. This finding further supports the argument that risk factors for mortality, including dementia, delirium, and AP use are very difficult to tease apart. It is possible that the reasons that APs are prescribed (agitated delirium or dementia) have as much to do with the excess mortality reported in observational studies of APs as the use of APs themselves.
The high rate of continued AP use we observed (two‐thirds of readmitted patients) may reflect limited pharmacological alternatives to these medications with little evidence to support treating the symptoms of delirium with other drug classes, along with suboptimal environmental and behavioral modifications in postacute facilities and hospitals. This is unfortunate given that delirium is often preventable. Systematic implementation of well‐documented strategies to decrease delirium in hospitals and postacute facilities would likely reduce the prescription of APs and has the potential to slow the decline in this vulnerable population. A meta‐analysis incorporating both randomized and nonrandomized trials of medical and surgical patients showed that multicomponent nonpharmacologic interventions decreased delirium by 50%.[24] Thus, simple interventions such as reorientation, early mobilization, optimizing vision and hearing, sleepwake cycle preservation, and hydration might avoid roughly 1 million cases of delirium in hospitalized older adults annually.[24] The Hospital Elder Life Program and Acute Care for Elders units are examples of programs that have been shown to decrease the incidence of delirium.[25, 26]
Despite vigorous efforts to prevent delirium, a subgroup of patients still will become delirious. These patients are at high risk for death. Our mortality prediction model revealed that patients who were discharged to postacute facilities were 4 times more likely to die during the subsequent year compared to patients who were discharged home. Patients discharged to postacute facilities are likely to have a higher burden of disease, greater functional and cognitive impairment, and more frailty than those who are able to return to the community. Very ill and/or frail patients receiving APs in the hospital and requiring APs on discharge to postacute care facilities have limited survival and may benefit from expedited palliative care interventions to clarify prognosis and goals, and relieve suffering. At a minimum, our study identifies a need for further study to identify this very high‐risk group of elders. It is notable that 50% of patients were found to have a post‐treatment ECG with a QTc of >500 ms, a finding that has not been previously described. This would put these patients at higher risk of mortality, and as such we suggest that current guidelines should continue to emphasize the importance of post‐treatment ECGs and set clear criteria for discontinuation in elderly patients.
Our study is limited by its retrospective, single‐center design and small sample size, therefore limiting the interpretation and generalizability of the results to other hospitals. Quetiapine was the most common AP medication used in our hospital; therefore, our findings cannot be generalized to hospitals that utilize other AP agents. Future studies should examine antipsychotic use across hospitals to determine variation in prescribing patterns and outcomes. Nevertheless, the care of these patients were transitioned to a large number of geriatricians and primary care and nursing home physicians after discharge, and the reflected practice patterns extended beyond our hospital. Additionally, we were unable to determine when and why APs were discontinued or started in the outpatient setting. We were only able to detect readmissions to the 3 hospitals within our health system and therefore may have missed some readmissions to other institutions, although the majority of patients in our region tend to return to the same hospital. For patients who were not readmitted, we were also unable to identify whether they remained on the APs initiated during their index hospitalizations. Any retrospective study is limited by the difficulty of distinguishing delirium from the behavioral and psychiatric symptoms of dementia, but we identified delirium using standard terms described in previous literature.[10] We were unable to determine the types of delirium (hyperactive vs hypoactive) given that the documentations on behavioral symptoms were largely missing from the charts. The number of patients with preexisting diagnosis of dementia was likely underestimated, as we were only able to verify the diagnosis from the medical history. Additionally, the retrospective design based on chart review limited the factors that we could detect and grade accurately for inclusion in our mortality prediction model. Of note, our model did not contain objective measures of cognition, agitation, function, and markers for frailty such as walking speed, weak grip strength, weight loss, and low physical activity.
CONCLUSION
Initiating an AP (eg, haloperidol, quetiapine, olanzapine, and risperidone) in the hospital is likely to result in long‐term use of these medications despite the fact that AP use has been associated with multiple risks including falls, fractures, stroke, cardiovascular disease, and increased mortality in those with underlying dementia.[27] When possible, behavioral interventions to prevent delirium and slow the trajectory of decline should be implemented to reduce AP use. If patients with delirium are started on antipsychotics, it is important to monitor for prolonged QTc given the associated risk of mortality. In a subgroup of patients at high risk for death in the upcoming year, occurrence of delirium or use of APs during a hospitalization should both be considered triggers for early advance care planning and possibly palliative care and end‐of‐life discussions, with an emphasis on quality of life.
Disclosures: The research was supported by the Department of Medicine, Baystate Medical Center/Tufts University School of Medicine. Dr. Lagu is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number K01HL114745. Drs. Lagu and Loh had full access to all of the data in the study. They take responsibility for the integrity of the data and the accuracy of the analysis. Drs. Loh, Brennan, Lindenauer, and Lagu conceived of the study. Drs. Loh, Ramdass, and Ms. Garb acquired the data. Ms Garb analyzed and interpreted the data. Drs. Loh, Ramdass, and Thim drafted the manuscript. Drs. Brennan, Lindenauer, and Lagu and Ms. Garb critically reviewed the manuscript for important intellectual content. Dr. Lagu has received consulting fees from the Institute for Healthcare Improvement, under contract to the Centers for Medicare and Medicaid Services, for her work on a project to help health systems achieve disability competence. Dr. Brennan is supported by a Geriatric Work Force Enhancement Grant from the US Department of Health and Human Services award number 1 U1QHP287020100. The authors report no conflicts of interest.
- , , . Delirium in elderly people. Lancet. 2014;383:911–922.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156:848–856, W296.
- American Geriatrics Society Expert Panel on Postoperative Delirium in Older Adults. American Geriatrics Society abstracted clinical practice guideline for postoperative delirium in older adults. J Am Geriatr Soc. 2015;63:142–150.
- Practice guideline for the treatment of patients with delirium. American Psychiatric Association. Am J Psychiatry. 1999;156:1–20.
- , ; Guideline Development Group. The prevention, diagnosis and management of delirium in older people: concise guidelines. Clin Med (Lond). 2006;6:303–308.
- , , . Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11–21.
- , , , et al. Antipsychotics, other psychotropics, and the risk of death in patients with dementia: number needed to harm. JAMA Psychiatry. 2015;72:438–445.
- , . Safety and efficacy of antipsychotic drugs for the behavioral and psychological symptoms of dementia. Indian J Psychiatry. 2009;51(suppl 1):S87–S92.
- , , , . Use and safety of antipsychotics in behavioral disorders in elderly people with dementia. J Clin Psychopharmacol. 2014;34:109–123.
- , , , , , . From hospital to community: use of antipsychotics in hospitalized elders. J Hosp Med. 2014;9:802–804.
- , , . Comparison of National Death Index and World Wide Web Death Searches. Am J Epidemiol. 2000;152:107–111.
- . Analysis of Binary Data. London, United Kingdom: Methuen; 1970:76–99.
- . Statistical Methods for Survival Data Analysis. New York, NY: John Wiley 1992:233–236.
- , , , . The risk of adverse outcomes in hospitalized older patients in relation to a frailty index based on a comprehensive geriatric assessment. Age Ageing. 2014;43:127–132.
- , , , et al. Risk factors for delirium and inpatient mortality with delirium. J Postgrad Med. 2013;59:263–270.
- , , , , , . Comprehensive geriatric assessment predicts mortality and adverse outcomes in hospitalized older adults. BMC Geriatr. 2014;14:129.
- , , , , , . One‐year mortality of elderly inpatients with delirium, dementia, or depression seen by a consultation‐liaison service. Psychosomatics. 2012;53:433–438.
- , , , . Excess mortality in general hospital patients with delirium: a 5‐year follow‐up of 519 patients seen in psychiatric consultation. J Psychosom Res. 1994;38:339–346.
- , , , et al. Older adults discharged from the hospital with delirium: 1‐year outcomes. J Am Geriatr Soc. 2006;54:1245–1250.
- , , , et al. The dementia antipsychotic withdrawal trial (DART‐AD): long‐term follow‐up of a randomised placebo‐controlled trial. Lancet Neurol. 2009;8:151–157.
- , , , et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775–786.
- , , , et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335–2341.
- , , , et al. The long‐term effects of conventional and atypical antipsychotics in patients with probable Alzheimer's disease. Am J Psychiatry. 2013;170:1051–1058.
- , , , et al. Effectiveness of multicomponent nonpharmacological delirium interventions: a meta‐analysis. JAMA Intern Med. 2015;175:512–520.
- , , , , . Sustainability and scalability of the hospital elder life program at a community hospital. J Am Geriatr Soc. 2011;59:359–365.
- , , , et al. Effectiveness of acute geriatric unit care using acute care for elders components: a systematic review and meta‐analysis. J Am Geriatr Soc. 2012;60:2237–2245.
- , . Adverse effects of antipsychotic medications. Am Fam Physician. 2010;81:617–622.
Delirium, a clinical syndrome characterized by inattention and acute cognitive dysfunction, is very common in older hospitalized patients, with a reported incidence of 18% to 35% at time of admission and overall occurrence rates of 29% to 64%.[1] Previous studies have reported that a diagnosis of delirium is not benign and is associated with other adverse outcomes including prolonged hospitalization, institutionalization, increased cost, and mortality. These outcomes occurred independent of age, prior cognitive functioning, and comorbidities.[2] Guidelines recommend that management of inpatient delirium should be focused on addressing the underlying etiology and managed with nonpharmacological interventions whenever possible.[3, 4, 5] However, implementing these recommendations can prove to be very challenging in hospital settings. Providers frequently have to resort to medical therapies, including antipsychotics (APs). Although these medications are commonly used to treat delirium in elderly patients, there is limited evidence to support their efficacy, and there are currently no proven pharmacological alternatives to these medications.[6] Furthermore, previous studies have demonstrated an increased risk of stroke, infection, cognitive impairment, and mortality in elders with dementia who receive long‐term AP therapy.[7, 8, 9] Yet as many as 48% of hospitalized elders who were newly started on APs had these drugs continued at time of discharge.[10]
There have been few studies describing the long‐term outcomes of elderly patient who are started on APs in the hospital. Most information on outcomes comes from patients with dementia. Therefore, we studied the 1‐year outcomes of a cohort of patients with and without dementia who were started on APs in the hospital and then discharged on these medications. In this cohort, we aimed to describe the number of readmissions, reasons for readmissions, duration of AP therapy, use of other sedating medications such as anxiolytics, hypnotics, and antihistamines as well as the incidence of readmission and death 1 year after the index hospital discharge.
METHODS
We previously described a retrospective cohort of 300 elders (65 years old) admitted to a tertiary care hospital between October 1, 2012 and September 31, 2013 who were newly prescribed APs while hospitalized.[10] Of patients alive at the time of discharge (260), 56% (146 patients) were discharged on APs. Two investigators extracted these 148 patient charts independently to identify and quantify the number of readmissions to the index hospital. We then limited the sample to only the first readmission per patient following the index admission and extracted this readmission for each patient. We first determined if APs were present on the admission medication reconciliation. If APs were not present on admission, we examined whether they were resumed during the hospitalization using the electronic medication administration summary. If they were present on admission, we looked to see if they were discontinued during the readmission and if additional new APs were started during the hospitalizations. We documented the circumstances around APs use and identified patients who died during their hospitalizations. We identified delirium using the same terms that were described in our prior study on the same cohort of patients.[10] We determined if patients were delirious using a predetermined algorithm (Figure 1). Briefly, we first determined delirium was documented. We then examined whether there was a Confusion Assessment Method (CAM) instrument included in the record. If a CAM instrument was not documented, we then looked for documentation using specific terms (eg, disorientation, confusions). We identified patients with dementia by determining whether dementia was documented along with other admission medical comorbidities. If it was not, we determined whether dementia was newly diagnosed during the hospital stay using progress notes or consultation notes. We did not objectively define criteria for diagnosis of dementia. We used the National Death Index (NDI) to determine mortality for all patients 1 year after discharge from the index hospitalization. The NDI is a national database of death records maintained by the National Center for Health Statistics. It has shown consistently high sensitivity and specificity for detection of death.[11]
We used descriptive statistics (means, standard deviations, range, and percents as appropriate to the scale of measurement) to describe the patient sample. We then used multiple logistic regression to identify significant predictors of death within 1 year of discharge.[12] Univariate analysis was used to select candidates for the logistic model (t tests for continuous factors and 2 for discrete factors). All factors with a significance level 0.2 on univariate analysis were included in the logistic regression, in addition to age and sex (regardless of significance). A maximum likelihood procedure was used to calculate the regression coefficients for the logistic model. The likelihood ratio criterion was used to determine the significance of individual factors in the regression model.[13] Factors with a significance level of 0.15 or less were retained in the final model, in addition to age and sex.
RESULTS
The 260 patients discharged alive from their index admissions had a 1‐year mortality rate of 29% (75/260). Of the 146/260 patients discharged on APs, 60 (41%) patients experienced at least 1 readmission (mean = 2 readmissions per patient; range, 18, with 111 total readmissions for 60 patients) within 1 year from discharge (Figure 2). Most common diagnoses at the time of readmissions were related neurological and psychiatric disorders (14%), cardiovascular and circulation disorders (13%), renal injury and electrolyte disorders (11%), and infections (6%). Among patients with at least 1 readmission, the mean age was 81.3 (range, 65.599.7), 60% were male, and 45% were admitted from a skilled nursing facility or rehabilitation facility (Table 1). Median time to readmission was 43.5 days (range, 1343 days), and 79% were readmitted to a medical service. The remaining 20% were admitted to a surgical service. Inpatient mortality during first readmissions was 8% (5/60). At the time of first readmission, 39/60 (65%) of patients were still on the same APs on which they had been discharged, and the APs were continued during the hospitalization in 79% of the patients (61% quetiapine, 19% olanzapine, and 13% risperidone). About half of patients whose APs were discontinued prior to readmission received a new AP during their hospital stays (9/20; 45%). One patient had been started on quetiapine in the outpatient setting. No patients were found to have new benzodiazepines, nonbenzodiazepine hypnotic, or antihistamines on their admission medication list.
| Variables | Value* |
|---|---|
| |
| Age, mean (range), yr | 81.3 (65.599.7) |
| Gender, no. (%) | |
| Male | 36 (60) |
| Female | 24 (40) |
| Admitted from, no. (%) | |
| Home | 33 (55) |
| Rehabilitation facilities | 5 (8) |
| SNF | 22 (37) |
| Services, no. (%) | |
| Medicine | 48 (80) |
| Surgery | 12 (20) |
| Types of APs continued on readmission (from index admission), no. (%) | |
| Quetiapine | 19 (61) |
| Olanzapine | 6 (19) |
| Risperidone | 4 (13) |
| Haloperidol | 2 (7) |
| Types of APs started during readmission, no. (%) | |
| Quetiapine | 7 (39) |
| Risperidone | 2 (11) |
| Haloperidol | 16 (89) |
| Indications for AP use, no. (%) | |
| Delirium | 14 (77) |
| Undocumented | 3 (17) |
| Other | 1 (6) |
| ECG, no. (%) | |
| Prior to APs administration | 17 (94) |
| After APs administration | 4 (22) |
| QTc prolongation >500 ms, no. (%) | |
| Prior to APs administration | 3 (18) |
| After APs administration∥ | 2 (50) |
| Discharge destination, no. (%) | |
| Home | 23 (38) |
| Rehabilitation facilities | 4 (7) |
| SNF | 28 (47) |
| Death | 5 (8) |
Eighteen patients received 1 or more new APs during the readmission hospitalizations. These included haloperidol (89%) and quetiapine (39%). Delirium was the main reported indication for starting APs (78%), but in 17% of cases no indication was documented. An electrocardiogram (ECG) was performed in 94% prior to APs administration and for 22% after APs administration. Corrected QT interval (QTc) of >500 ms was present in 18% of patients in pretreatment ECG and 50% of patients in post‐AP ECG. Of patients who survived readmission, 58% (32/55) were discharged to postacute facilities. Of the 39 patients who were on the same APs from index admission, 27 (69%) patients were eventually discharged on the same APs or new APs started during the readmission.
In the multivariable model (Table 2), predictors of death at 1 year included discharge to postacute facilities after index admission (odds ratio [OR]: 2.28; 95% confidence interval [CI]: 1.10‐4.73, P = 0.03) and QTc prolongation >500 ms during index admission (OR: 3.41; 95% CI: 1.34‐8.67, P = 0.01). Age and gender were not associated with 1‐year mortality.
| Odds Ratio | 95% Confidence Interval | P Value | |
|---|---|---|---|
| |||
| Age | 1.03 | 0.991.06 | 0.13 |
| Male sex | 0.87 | 0.501.52 | 0.63 |
| Risperdal | 3.53 | 0.6419.40 | 0.15 |
| QTc prolongation after AP administration* | 3.41 | 1.348.67 | 0.01 |
| Presence of geriatric psychiatry consult | 0.30 | 0.091.04 | 0.06 |
| Discharged to postacute facilities vs home | 2.28 | 1.104.73 | 0.03 |
DISCUSSION
In a cohort of elderly patients who were discharged on APs, nearly one‐third (29%) died within 1 year of the hospitalization in which APs were initiated. Nearly half of the survivors from the index admission (41%) experienced at least 1 admission within 1 year from discharge. Of readmitted patients, two‐thirds were taking the same APs that had been started during the index hospitalization. Half of the patients not on APs on readmission were started on an AP during the hospitalization, most often because they became delirious on return to the acute care setting. Compared to patients discharged home after an index admission, patients who were discharged to postacute facilities were almost 4 times as likely to die during the year subsequent to the admission. These data suggest that once patients are started on APs, most are continued on them until the next admission or are restarted during that readmission. Moreover, hospitalized elders who require an AP are at high risk for mortality in the coming year.
Prior studies have reported that patients with delirium have elevated 1‐year mortality rates.[14, 15, 16, 17, 18, 19] A secondary analysis of the Delirium Prevention Trial, which included 437 hospitalized older patients, revealed a 1‐year mortality rate of 20% in those who were never delirious during hospitalization, compared to 26% to 38% in patients with delirium.[19] Additionally, 1‐year mortality in hospitalized older patients with delirium (36%) was shown to be higher than patients with dementia (29%) or depression (26%).[17] Unlike these studies, not all of the patients in our study had documented delirium, but all received an AP. Still, it is notable that the 1‐year mortality rate for delirium in general is similar to what we found in this study.
The literature has also reported that long‐term AP use is associated with excess mortality in elder patients, especially those with dementia.[20, 21, 22] In a retrospective cohort study, older patients with dementia who were taking antipsychotics had significantly higher 1‐year mortality rates (23%29%) than patients not taking antipsychotic medications (15%). In a large Canadian propensity score‐matched cohort study that included over 13,000 demented older adults, the mortality was higher in the community‐dwelling elders who received atypical APs compared to no APs, with a difference of 1.1% in 180‐day mortality rate after initiation of APs.[21] The absolute mortality rate was 2.6% higher in patients who received typical compared to atypical APs. Unlike these studies, not every patient in our cohort had a diagnosis of dementia, but again, mortality rates in these studies appear similar to our cohort.
In contrast, other observational studies have not found an increased risk associated with receipt of APs. For example, a prospective study that enrolled approximately 950 patients with probable dementia showed that AP use was not associated with time to death after adjustment for comorbidities, demographic and cognitive variables.[23] These conflicting results highlight the difficulties of attributing outcomes in high‐risk populations. Although the excess mortality observed in patients taking APs may be related to the risks of APs, it is quite possible that patients who require APs (most often for delirium or agitated dementia) are at higher risk of death. This confounding by indication may be nearly impossible to adjust for retrospectively, even using techniques such as propensity matching.
Our report adds to the literature; we know of no studies to date describing a cohort of patients, most with delirium, who were started on APs in the hospital. We also attempted to identify the reasons that patients were started on APs, which have been infrequently reported. As noted above, our 1‐year mortality rate of 29% among older patients prescribed APs in the hospital was quite similar to mortality rates both for patients with delirium who were not necessarily treated with APs and patients with dementia who were treated with APs. This finding further supports the argument that risk factors for mortality, including dementia, delirium, and AP use are very difficult to tease apart. It is possible that the reasons that APs are prescribed (agitated delirium or dementia) have as much to do with the excess mortality reported in observational studies of APs as the use of APs themselves.
The high rate of continued AP use we observed (two‐thirds of readmitted patients) may reflect limited pharmacological alternatives to these medications with little evidence to support treating the symptoms of delirium with other drug classes, along with suboptimal environmental and behavioral modifications in postacute facilities and hospitals. This is unfortunate given that delirium is often preventable. Systematic implementation of well‐documented strategies to decrease delirium in hospitals and postacute facilities would likely reduce the prescription of APs and has the potential to slow the decline in this vulnerable population. A meta‐analysis incorporating both randomized and nonrandomized trials of medical and surgical patients showed that multicomponent nonpharmacologic interventions decreased delirium by 50%.[24] Thus, simple interventions such as reorientation, early mobilization, optimizing vision and hearing, sleepwake cycle preservation, and hydration might avoid roughly 1 million cases of delirium in hospitalized older adults annually.[24] The Hospital Elder Life Program and Acute Care for Elders units are examples of programs that have been shown to decrease the incidence of delirium.[25, 26]
Despite vigorous efforts to prevent delirium, a subgroup of patients still will become delirious. These patients are at high risk for death. Our mortality prediction model revealed that patients who were discharged to postacute facilities were 4 times more likely to die during the subsequent year compared to patients who were discharged home. Patients discharged to postacute facilities are likely to have a higher burden of disease, greater functional and cognitive impairment, and more frailty than those who are able to return to the community. Very ill and/or frail patients receiving APs in the hospital and requiring APs on discharge to postacute care facilities have limited survival and may benefit from expedited palliative care interventions to clarify prognosis and goals, and relieve suffering. At a minimum, our study identifies a need for further study to identify this very high‐risk group of elders. It is notable that 50% of patients were found to have a post‐treatment ECG with a QTc of >500 ms, a finding that has not been previously described. This would put these patients at higher risk of mortality, and as such we suggest that current guidelines should continue to emphasize the importance of post‐treatment ECGs and set clear criteria for discontinuation in elderly patients.
Our study is limited by its retrospective, single‐center design and small sample size, therefore limiting the interpretation and generalizability of the results to other hospitals. Quetiapine was the most common AP medication used in our hospital; therefore, our findings cannot be generalized to hospitals that utilize other AP agents. Future studies should examine antipsychotic use across hospitals to determine variation in prescribing patterns and outcomes. Nevertheless, the care of these patients were transitioned to a large number of geriatricians and primary care and nursing home physicians after discharge, and the reflected practice patterns extended beyond our hospital. Additionally, we were unable to determine when and why APs were discontinued or started in the outpatient setting. We were only able to detect readmissions to the 3 hospitals within our health system and therefore may have missed some readmissions to other institutions, although the majority of patients in our region tend to return to the same hospital. For patients who were not readmitted, we were also unable to identify whether they remained on the APs initiated during their index hospitalizations. Any retrospective study is limited by the difficulty of distinguishing delirium from the behavioral and psychiatric symptoms of dementia, but we identified delirium using standard terms described in previous literature.[10] We were unable to determine the types of delirium (hyperactive vs hypoactive) given that the documentations on behavioral symptoms were largely missing from the charts. The number of patients with preexisting diagnosis of dementia was likely underestimated, as we were only able to verify the diagnosis from the medical history. Additionally, the retrospective design based on chart review limited the factors that we could detect and grade accurately for inclusion in our mortality prediction model. Of note, our model did not contain objective measures of cognition, agitation, function, and markers for frailty such as walking speed, weak grip strength, weight loss, and low physical activity.
CONCLUSION
Initiating an AP (eg, haloperidol, quetiapine, olanzapine, and risperidone) in the hospital is likely to result in long‐term use of these medications despite the fact that AP use has been associated with multiple risks including falls, fractures, stroke, cardiovascular disease, and increased mortality in those with underlying dementia.[27] When possible, behavioral interventions to prevent delirium and slow the trajectory of decline should be implemented to reduce AP use. If patients with delirium are started on antipsychotics, it is important to monitor for prolonged QTc given the associated risk of mortality. In a subgroup of patients at high risk for death in the upcoming year, occurrence of delirium or use of APs during a hospitalization should both be considered triggers for early advance care planning and possibly palliative care and end‐of‐life discussions, with an emphasis on quality of life.
Disclosures: The research was supported by the Department of Medicine, Baystate Medical Center/Tufts University School of Medicine. Dr. Lagu is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number K01HL114745. Drs. Lagu and Loh had full access to all of the data in the study. They take responsibility for the integrity of the data and the accuracy of the analysis. Drs. Loh, Brennan, Lindenauer, and Lagu conceived of the study. Drs. Loh, Ramdass, and Ms. Garb acquired the data. Ms Garb analyzed and interpreted the data. Drs. Loh, Ramdass, and Thim drafted the manuscript. Drs. Brennan, Lindenauer, and Lagu and Ms. Garb critically reviewed the manuscript for important intellectual content. Dr. Lagu has received consulting fees from the Institute for Healthcare Improvement, under contract to the Centers for Medicare and Medicaid Services, for her work on a project to help health systems achieve disability competence. Dr. Brennan is supported by a Geriatric Work Force Enhancement Grant from the US Department of Health and Human Services award number 1 U1QHP287020100. The authors report no conflicts of interest.
Delirium, a clinical syndrome characterized by inattention and acute cognitive dysfunction, is very common in older hospitalized patients, with a reported incidence of 18% to 35% at time of admission and overall occurrence rates of 29% to 64%.[1] Previous studies have reported that a diagnosis of delirium is not benign and is associated with other adverse outcomes including prolonged hospitalization, institutionalization, increased cost, and mortality. These outcomes occurred independent of age, prior cognitive functioning, and comorbidities.[2] Guidelines recommend that management of inpatient delirium should be focused on addressing the underlying etiology and managed with nonpharmacological interventions whenever possible.[3, 4, 5] However, implementing these recommendations can prove to be very challenging in hospital settings. Providers frequently have to resort to medical therapies, including antipsychotics (APs). Although these medications are commonly used to treat delirium in elderly patients, there is limited evidence to support their efficacy, and there are currently no proven pharmacological alternatives to these medications.[6] Furthermore, previous studies have demonstrated an increased risk of stroke, infection, cognitive impairment, and mortality in elders with dementia who receive long‐term AP therapy.[7, 8, 9] Yet as many as 48% of hospitalized elders who were newly started on APs had these drugs continued at time of discharge.[10]
There have been few studies describing the long‐term outcomes of elderly patient who are started on APs in the hospital. Most information on outcomes comes from patients with dementia. Therefore, we studied the 1‐year outcomes of a cohort of patients with and without dementia who were started on APs in the hospital and then discharged on these medications. In this cohort, we aimed to describe the number of readmissions, reasons for readmissions, duration of AP therapy, use of other sedating medications such as anxiolytics, hypnotics, and antihistamines as well as the incidence of readmission and death 1 year after the index hospital discharge.
METHODS
We previously described a retrospective cohort of 300 elders (65 years old) admitted to a tertiary care hospital between October 1, 2012 and September 31, 2013 who were newly prescribed APs while hospitalized.[10] Of patients alive at the time of discharge (260), 56% (146 patients) were discharged on APs. Two investigators extracted these 148 patient charts independently to identify and quantify the number of readmissions to the index hospital. We then limited the sample to only the first readmission per patient following the index admission and extracted this readmission for each patient. We first determined if APs were present on the admission medication reconciliation. If APs were not present on admission, we examined whether they were resumed during the hospitalization using the electronic medication administration summary. If they were present on admission, we looked to see if they were discontinued during the readmission and if additional new APs were started during the hospitalizations. We documented the circumstances around APs use and identified patients who died during their hospitalizations. We identified delirium using the same terms that were described in our prior study on the same cohort of patients.[10] We determined if patients were delirious using a predetermined algorithm (Figure 1). Briefly, we first determined delirium was documented. We then examined whether there was a Confusion Assessment Method (CAM) instrument included in the record. If a CAM instrument was not documented, we then looked for documentation using specific terms (eg, disorientation, confusions). We identified patients with dementia by determining whether dementia was documented along with other admission medical comorbidities. If it was not, we determined whether dementia was newly diagnosed during the hospital stay using progress notes or consultation notes. We did not objectively define criteria for diagnosis of dementia. We used the National Death Index (NDI) to determine mortality for all patients 1 year after discharge from the index hospitalization. The NDI is a national database of death records maintained by the National Center for Health Statistics. It has shown consistently high sensitivity and specificity for detection of death.[11]
We used descriptive statistics (means, standard deviations, range, and percents as appropriate to the scale of measurement) to describe the patient sample. We then used multiple logistic regression to identify significant predictors of death within 1 year of discharge.[12] Univariate analysis was used to select candidates for the logistic model (t tests for continuous factors and 2 for discrete factors). All factors with a significance level 0.2 on univariate analysis were included in the logistic regression, in addition to age and sex (regardless of significance). A maximum likelihood procedure was used to calculate the regression coefficients for the logistic model. The likelihood ratio criterion was used to determine the significance of individual factors in the regression model.[13] Factors with a significance level of 0.15 or less were retained in the final model, in addition to age and sex.
RESULTS
The 260 patients discharged alive from their index admissions had a 1‐year mortality rate of 29% (75/260). Of the 146/260 patients discharged on APs, 60 (41%) patients experienced at least 1 readmission (mean = 2 readmissions per patient; range, 18, with 111 total readmissions for 60 patients) within 1 year from discharge (Figure 2). Most common diagnoses at the time of readmissions were related neurological and psychiatric disorders (14%), cardiovascular and circulation disorders (13%), renal injury and electrolyte disorders (11%), and infections (6%). Among patients with at least 1 readmission, the mean age was 81.3 (range, 65.599.7), 60% were male, and 45% were admitted from a skilled nursing facility or rehabilitation facility (Table 1). Median time to readmission was 43.5 days (range, 1343 days), and 79% were readmitted to a medical service. The remaining 20% were admitted to a surgical service. Inpatient mortality during first readmissions was 8% (5/60). At the time of first readmission, 39/60 (65%) of patients were still on the same APs on which they had been discharged, and the APs were continued during the hospitalization in 79% of the patients (61% quetiapine, 19% olanzapine, and 13% risperidone). About half of patients whose APs were discontinued prior to readmission received a new AP during their hospital stays (9/20; 45%). One patient had been started on quetiapine in the outpatient setting. No patients were found to have new benzodiazepines, nonbenzodiazepine hypnotic, or antihistamines on their admission medication list.
| Variables | Value* |
|---|---|
| |
| Age, mean (range), yr | 81.3 (65.599.7) |
| Gender, no. (%) | |
| Male | 36 (60) |
| Female | 24 (40) |
| Admitted from, no. (%) | |
| Home | 33 (55) |
| Rehabilitation facilities | 5 (8) |
| SNF | 22 (37) |
| Services, no. (%) | |
| Medicine | 48 (80) |
| Surgery | 12 (20) |
| Types of APs continued on readmission (from index admission), no. (%) | |
| Quetiapine | 19 (61) |
| Olanzapine | 6 (19) |
| Risperidone | 4 (13) |
| Haloperidol | 2 (7) |
| Types of APs started during readmission, no. (%) | |
| Quetiapine | 7 (39) |
| Risperidone | 2 (11) |
| Haloperidol | 16 (89) |
| Indications for AP use, no. (%) | |
| Delirium | 14 (77) |
| Undocumented | 3 (17) |
| Other | 1 (6) |
| ECG, no. (%) | |
| Prior to APs administration | 17 (94) |
| After APs administration | 4 (22) |
| QTc prolongation >500 ms, no. (%) | |
| Prior to APs administration | 3 (18) |
| After APs administration∥ | 2 (50) |
| Discharge destination, no. (%) | |
| Home | 23 (38) |
| Rehabilitation facilities | 4 (7) |
| SNF | 28 (47) |
| Death | 5 (8) |
Eighteen patients received 1 or more new APs during the readmission hospitalizations. These included haloperidol (89%) and quetiapine (39%). Delirium was the main reported indication for starting APs (78%), but in 17% of cases no indication was documented. An electrocardiogram (ECG) was performed in 94% prior to APs administration and for 22% after APs administration. Corrected QT interval (QTc) of >500 ms was present in 18% of patients in pretreatment ECG and 50% of patients in post‐AP ECG. Of patients who survived readmission, 58% (32/55) were discharged to postacute facilities. Of the 39 patients who were on the same APs from index admission, 27 (69%) patients were eventually discharged on the same APs or new APs started during the readmission.
In the multivariable model (Table 2), predictors of death at 1 year included discharge to postacute facilities after index admission (odds ratio [OR]: 2.28; 95% confidence interval [CI]: 1.10‐4.73, P = 0.03) and QTc prolongation >500 ms during index admission (OR: 3.41; 95% CI: 1.34‐8.67, P = 0.01). Age and gender were not associated with 1‐year mortality.
| Odds Ratio | 95% Confidence Interval | P Value | |
|---|---|---|---|
| |||
| Age | 1.03 | 0.991.06 | 0.13 |
| Male sex | 0.87 | 0.501.52 | 0.63 |
| Risperdal | 3.53 | 0.6419.40 | 0.15 |
| QTc prolongation after AP administration* | 3.41 | 1.348.67 | 0.01 |
| Presence of geriatric psychiatry consult | 0.30 | 0.091.04 | 0.06 |
| Discharged to postacute facilities vs home | 2.28 | 1.104.73 | 0.03 |
DISCUSSION
In a cohort of elderly patients who were discharged on APs, nearly one‐third (29%) died within 1 year of the hospitalization in which APs were initiated. Nearly half of the survivors from the index admission (41%) experienced at least 1 admission within 1 year from discharge. Of readmitted patients, two‐thirds were taking the same APs that had been started during the index hospitalization. Half of the patients not on APs on readmission were started on an AP during the hospitalization, most often because they became delirious on return to the acute care setting. Compared to patients discharged home after an index admission, patients who were discharged to postacute facilities were almost 4 times as likely to die during the year subsequent to the admission. These data suggest that once patients are started on APs, most are continued on them until the next admission or are restarted during that readmission. Moreover, hospitalized elders who require an AP are at high risk for mortality in the coming year.
Prior studies have reported that patients with delirium have elevated 1‐year mortality rates.[14, 15, 16, 17, 18, 19] A secondary analysis of the Delirium Prevention Trial, which included 437 hospitalized older patients, revealed a 1‐year mortality rate of 20% in those who were never delirious during hospitalization, compared to 26% to 38% in patients with delirium.[19] Additionally, 1‐year mortality in hospitalized older patients with delirium (36%) was shown to be higher than patients with dementia (29%) or depression (26%).[17] Unlike these studies, not all of the patients in our study had documented delirium, but all received an AP. Still, it is notable that the 1‐year mortality rate for delirium in general is similar to what we found in this study.
The literature has also reported that long‐term AP use is associated with excess mortality in elder patients, especially those with dementia.[20, 21, 22] In a retrospective cohort study, older patients with dementia who were taking antipsychotics had significantly higher 1‐year mortality rates (23%29%) than patients not taking antipsychotic medications (15%). In a large Canadian propensity score‐matched cohort study that included over 13,000 demented older adults, the mortality was higher in the community‐dwelling elders who received atypical APs compared to no APs, with a difference of 1.1% in 180‐day mortality rate after initiation of APs.[21] The absolute mortality rate was 2.6% higher in patients who received typical compared to atypical APs. Unlike these studies, not every patient in our cohort had a diagnosis of dementia, but again, mortality rates in these studies appear similar to our cohort.
In contrast, other observational studies have not found an increased risk associated with receipt of APs. For example, a prospective study that enrolled approximately 950 patients with probable dementia showed that AP use was not associated with time to death after adjustment for comorbidities, demographic and cognitive variables.[23] These conflicting results highlight the difficulties of attributing outcomes in high‐risk populations. Although the excess mortality observed in patients taking APs may be related to the risks of APs, it is quite possible that patients who require APs (most often for delirium or agitated dementia) are at higher risk of death. This confounding by indication may be nearly impossible to adjust for retrospectively, even using techniques such as propensity matching.
Our report adds to the literature; we know of no studies to date describing a cohort of patients, most with delirium, who were started on APs in the hospital. We also attempted to identify the reasons that patients were started on APs, which have been infrequently reported. As noted above, our 1‐year mortality rate of 29% among older patients prescribed APs in the hospital was quite similar to mortality rates both for patients with delirium who were not necessarily treated with APs and patients with dementia who were treated with APs. This finding further supports the argument that risk factors for mortality, including dementia, delirium, and AP use are very difficult to tease apart. It is possible that the reasons that APs are prescribed (agitated delirium or dementia) have as much to do with the excess mortality reported in observational studies of APs as the use of APs themselves.
The high rate of continued AP use we observed (two‐thirds of readmitted patients) may reflect limited pharmacological alternatives to these medications with little evidence to support treating the symptoms of delirium with other drug classes, along with suboptimal environmental and behavioral modifications in postacute facilities and hospitals. This is unfortunate given that delirium is often preventable. Systematic implementation of well‐documented strategies to decrease delirium in hospitals and postacute facilities would likely reduce the prescription of APs and has the potential to slow the decline in this vulnerable population. A meta‐analysis incorporating both randomized and nonrandomized trials of medical and surgical patients showed that multicomponent nonpharmacologic interventions decreased delirium by 50%.[24] Thus, simple interventions such as reorientation, early mobilization, optimizing vision and hearing, sleepwake cycle preservation, and hydration might avoid roughly 1 million cases of delirium in hospitalized older adults annually.[24] The Hospital Elder Life Program and Acute Care for Elders units are examples of programs that have been shown to decrease the incidence of delirium.[25, 26]
Despite vigorous efforts to prevent delirium, a subgroup of patients still will become delirious. These patients are at high risk for death. Our mortality prediction model revealed that patients who were discharged to postacute facilities were 4 times more likely to die during the subsequent year compared to patients who were discharged home. Patients discharged to postacute facilities are likely to have a higher burden of disease, greater functional and cognitive impairment, and more frailty than those who are able to return to the community. Very ill and/or frail patients receiving APs in the hospital and requiring APs on discharge to postacute care facilities have limited survival and may benefit from expedited palliative care interventions to clarify prognosis and goals, and relieve suffering. At a minimum, our study identifies a need for further study to identify this very high‐risk group of elders. It is notable that 50% of patients were found to have a post‐treatment ECG with a QTc of >500 ms, a finding that has not been previously described. This would put these patients at higher risk of mortality, and as such we suggest that current guidelines should continue to emphasize the importance of post‐treatment ECGs and set clear criteria for discontinuation in elderly patients.
Our study is limited by its retrospective, single‐center design and small sample size, therefore limiting the interpretation and generalizability of the results to other hospitals. Quetiapine was the most common AP medication used in our hospital; therefore, our findings cannot be generalized to hospitals that utilize other AP agents. Future studies should examine antipsychotic use across hospitals to determine variation in prescribing patterns and outcomes. Nevertheless, the care of these patients were transitioned to a large number of geriatricians and primary care and nursing home physicians after discharge, and the reflected practice patterns extended beyond our hospital. Additionally, we were unable to determine when and why APs were discontinued or started in the outpatient setting. We were only able to detect readmissions to the 3 hospitals within our health system and therefore may have missed some readmissions to other institutions, although the majority of patients in our region tend to return to the same hospital. For patients who were not readmitted, we were also unable to identify whether they remained on the APs initiated during their index hospitalizations. Any retrospective study is limited by the difficulty of distinguishing delirium from the behavioral and psychiatric symptoms of dementia, but we identified delirium using standard terms described in previous literature.[10] We were unable to determine the types of delirium (hyperactive vs hypoactive) given that the documentations on behavioral symptoms were largely missing from the charts. The number of patients with preexisting diagnosis of dementia was likely underestimated, as we were only able to verify the diagnosis from the medical history. Additionally, the retrospective design based on chart review limited the factors that we could detect and grade accurately for inclusion in our mortality prediction model. Of note, our model did not contain objective measures of cognition, agitation, function, and markers for frailty such as walking speed, weak grip strength, weight loss, and low physical activity.
CONCLUSION
Initiating an AP (eg, haloperidol, quetiapine, olanzapine, and risperidone) in the hospital is likely to result in long‐term use of these medications despite the fact that AP use has been associated with multiple risks including falls, fractures, stroke, cardiovascular disease, and increased mortality in those with underlying dementia.[27] When possible, behavioral interventions to prevent delirium and slow the trajectory of decline should be implemented to reduce AP use. If patients with delirium are started on antipsychotics, it is important to monitor for prolonged QTc given the associated risk of mortality. In a subgroup of patients at high risk for death in the upcoming year, occurrence of delirium or use of APs during a hospitalization should both be considered triggers for early advance care planning and possibly palliative care and end‐of‐life discussions, with an emphasis on quality of life.
Disclosures: The research was supported by the Department of Medicine, Baystate Medical Center/Tufts University School of Medicine. Dr. Lagu is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number K01HL114745. Drs. Lagu and Loh had full access to all of the data in the study. They take responsibility for the integrity of the data and the accuracy of the analysis. Drs. Loh, Brennan, Lindenauer, and Lagu conceived of the study. Drs. Loh, Ramdass, and Ms. Garb acquired the data. Ms Garb analyzed and interpreted the data. Drs. Loh, Ramdass, and Thim drafted the manuscript. Drs. Brennan, Lindenauer, and Lagu and Ms. Garb critically reviewed the manuscript for important intellectual content. Dr. Lagu has received consulting fees from the Institute for Healthcare Improvement, under contract to the Centers for Medicare and Medicaid Services, for her work on a project to help health systems achieve disability competence. Dr. Brennan is supported by a Geriatric Work Force Enhancement Grant from the US Department of Health and Human Services award number 1 U1QHP287020100. The authors report no conflicts of interest.
- , , . Delirium in elderly people. Lancet. 2014;383:911–922.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156:848–856, W296.
- American Geriatrics Society Expert Panel on Postoperative Delirium in Older Adults. American Geriatrics Society abstracted clinical practice guideline for postoperative delirium in older adults. J Am Geriatr Soc. 2015;63:142–150.
- Practice guideline for the treatment of patients with delirium. American Psychiatric Association. Am J Psychiatry. 1999;156:1–20.
- , ; Guideline Development Group. The prevention, diagnosis and management of delirium in older people: concise guidelines. Clin Med (Lond). 2006;6:303–308.
- , , . Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11–21.
- , , , et al. Antipsychotics, other psychotropics, and the risk of death in patients with dementia: number needed to harm. JAMA Psychiatry. 2015;72:438–445.
- , . Safety and efficacy of antipsychotic drugs for the behavioral and psychological symptoms of dementia. Indian J Psychiatry. 2009;51(suppl 1):S87–S92.
- , , , . Use and safety of antipsychotics in behavioral disorders in elderly people with dementia. J Clin Psychopharmacol. 2014;34:109–123.
- , , , , , . From hospital to community: use of antipsychotics in hospitalized elders. J Hosp Med. 2014;9:802–804.
- , , . Comparison of National Death Index and World Wide Web Death Searches. Am J Epidemiol. 2000;152:107–111.
- . Analysis of Binary Data. London, United Kingdom: Methuen; 1970:76–99.
- . Statistical Methods for Survival Data Analysis. New York, NY: John Wiley 1992:233–236.
- , , , . The risk of adverse outcomes in hospitalized older patients in relation to a frailty index based on a comprehensive geriatric assessment. Age Ageing. 2014;43:127–132.
- , , , et al. Risk factors for delirium and inpatient mortality with delirium. J Postgrad Med. 2013;59:263–270.
- , , , , , . Comprehensive geriatric assessment predicts mortality and adverse outcomes in hospitalized older adults. BMC Geriatr. 2014;14:129.
- , , , , , . One‐year mortality of elderly inpatients with delirium, dementia, or depression seen by a consultation‐liaison service. Psychosomatics. 2012;53:433–438.
- , , , . Excess mortality in general hospital patients with delirium: a 5‐year follow‐up of 519 patients seen in psychiatric consultation. J Psychosom Res. 1994;38:339–346.
- , , , et al. Older adults discharged from the hospital with delirium: 1‐year outcomes. J Am Geriatr Soc. 2006;54:1245–1250.
- , , , et al. The dementia antipsychotic withdrawal trial (DART‐AD): long‐term follow‐up of a randomised placebo‐controlled trial. Lancet Neurol. 2009;8:151–157.
- , , , et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775–786.
- , , , et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335–2341.
- , , , et al. The long‐term effects of conventional and atypical antipsychotics in patients with probable Alzheimer's disease. Am J Psychiatry. 2013;170:1051–1058.
- , , , et al. Effectiveness of multicomponent nonpharmacological delirium interventions: a meta‐analysis. JAMA Intern Med. 2015;175:512–520.
- , , , , . Sustainability and scalability of the hospital elder life program at a community hospital. J Am Geriatr Soc. 2011;59:359–365.
- , , , et al. Effectiveness of acute geriatric unit care using acute care for elders components: a systematic review and meta‐analysis. J Am Geriatr Soc. 2012;60:2237–2245.
- , . Adverse effects of antipsychotic medications. Am Fam Physician. 2010;81:617–622.
- , , . Delirium in elderly people. Lancet. 2014;383:911–922.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156:848–856, W296.
- American Geriatrics Society Expert Panel on Postoperative Delirium in Older Adults. American Geriatrics Society abstracted clinical practice guideline for postoperative delirium in older adults. J Am Geriatr Soc. 2015;63:142–150.
- Practice guideline for the treatment of patients with delirium. American Psychiatric Association. Am J Psychiatry. 1999;156:1–20.
- , ; Guideline Development Group. The prevention, diagnosis and management of delirium in older people: concise guidelines. Clin Med (Lond). 2006;6:303–308.
- , , . Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11–21.
- , , , et al. Antipsychotics, other psychotropics, and the risk of death in patients with dementia: number needed to harm. JAMA Psychiatry. 2015;72:438–445.
- , . Safety and efficacy of antipsychotic drugs for the behavioral and psychological symptoms of dementia. Indian J Psychiatry. 2009;51(suppl 1):S87–S92.
- , , , . Use and safety of antipsychotics in behavioral disorders in elderly people with dementia. J Clin Psychopharmacol. 2014;34:109–123.
- , , , , , . From hospital to community: use of antipsychotics in hospitalized elders. J Hosp Med. 2014;9:802–804.
- , , . Comparison of National Death Index and World Wide Web Death Searches. Am J Epidemiol. 2000;152:107–111.
- . Analysis of Binary Data. London, United Kingdom: Methuen; 1970:76–99.
- . Statistical Methods for Survival Data Analysis. New York, NY: John Wiley 1992:233–236.
- , , , . The risk of adverse outcomes in hospitalized older patients in relation to a frailty index based on a comprehensive geriatric assessment. Age Ageing. 2014;43:127–132.
- , , , et al. Risk factors for delirium and inpatient mortality with delirium. J Postgrad Med. 2013;59:263–270.
- , , , , , . Comprehensive geriatric assessment predicts mortality and adverse outcomes in hospitalized older adults. BMC Geriatr. 2014;14:129.
- , , , , , . One‐year mortality of elderly inpatients with delirium, dementia, or depression seen by a consultation‐liaison service. Psychosomatics. 2012;53:433–438.
- , , , . Excess mortality in general hospital patients with delirium: a 5‐year follow‐up of 519 patients seen in psychiatric consultation. J Psychosom Res. 1994;38:339–346.
- , , , et al. Older adults discharged from the hospital with delirium: 1‐year outcomes. J Am Geriatr Soc. 2006;54:1245–1250.
- , , , et al. The dementia antipsychotic withdrawal trial (DART‐AD): long‐term follow‐up of a randomised placebo‐controlled trial. Lancet Neurol. 2009;8:151–157.
- , , , et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775–786.
- , , , et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335–2341.
- , , , et al. The long‐term effects of conventional and atypical antipsychotics in patients with probable Alzheimer's disease. Am J Psychiatry. 2013;170:1051–1058.
- , , , et al. Effectiveness of multicomponent nonpharmacological delirium interventions: a meta‐analysis. JAMA Intern Med. 2015;175:512–520.
- , , , , . Sustainability and scalability of the hospital elder life program at a community hospital. J Am Geriatr Soc. 2011;59:359–365.
- , , , et al. Effectiveness of acute geriatric unit care using acute care for elders components: a systematic review and meta‐analysis. J Am Geriatr Soc. 2012;60:2237–2245.
- , . Adverse effects of antipsychotic medications. Am Fam Physician. 2010;81:617–622.

Inflectra becomes first FDA-approved biosimilar for inflammatory diseases
A biosimilar version of the anti–tumor necrosis factor–alpha agent Remicade has been approved by the Food and Drug Administration, making it the first biosimilar drug approved by the agency for inflammatory diseases and just the second biosimilar it has approved.
The agency said in its April 5 announcement that the biosimilar drug, to be marketed as Inflectra, will have the same indications as Remicade: moderately to severely active Crohn’s disease in patients aged 6 years and older who have had an inadequate response to conventional therapy; moderately to severely active ulcerative colitis that has inadequately responded to conventional therapy; moderately to severely active rheumatoid arthritis in combination with methotrexate; active ankylosing spondylitis; active psoriatic arthritis; and chronic, severe plaque psoriasis.

The drug, given the generic name of infliximab-dyyb under the agency’s nomenclature for biosimilar products, earned its approval as a biosimilar by showing it has no clinically meaningful differences in terms of safety and effectiveness from Remicade. According to FDA regulations, biosimilar products can have only minor differences in clinically inactive components and must have the same mechanism(s) of action (to the extent that it is known) and route(s) of administration, dosage form(s), and strength(s) as the reference product; and can be approved only for the indication(s) and condition(s) of use that have been approved for the reference product.
Inflectra’s approval is only as a biosimilar, not as an interchangeable product. The agency has yet to define the regulatory requirements for interchangeability that are necessary to meet the requirements of the Biologics Price Competition and Innovation Act of 2009. That Act states that an approved biosimilar “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” A statement about implementation of the Act on the FDA website explains that for interchangeability, “a sponsor must demonstrate that the biosimilar product can be expected to produce the same clinical result as the reference product in any given patient and, for a biological product that is administered more than once, that the risk of alternating or switching between use of the biosimilar product and the reference product is not greater than the risk of maintaining the patient on the reference product.”
Like Remicade, Inflectra will come with a boxed warning and a Medication Guide that describes important information about its uses and risks, which include serious infections (tuberculosis, bacterial sepsis, invasive fungal infections, and others), lymphoma and other malignancies, liver injury, blood problems, lupuslike syndrome, psoriasis, and in rare cases, nervous system disorders.
Inflectra is manufactured by Celltrion, based in South Korea, for Illinois-based Hospira. Inflectra’s label can be found here.
A biosimilar version of the anti–tumor necrosis factor–alpha agent Remicade has been approved by the Food and Drug Administration, making it the first biosimilar drug approved by the agency for inflammatory diseases and just the second biosimilar it has approved.
The agency said in its April 5 announcement that the biosimilar drug, to be marketed as Inflectra, will have the same indications as Remicade: moderately to severely active Crohn’s disease in patients aged 6 years and older who have had an inadequate response to conventional therapy; moderately to severely active ulcerative colitis that has inadequately responded to conventional therapy; moderately to severely active rheumatoid arthritis in combination with methotrexate; active ankylosing spondylitis; active psoriatic arthritis; and chronic, severe plaque psoriasis.
The drug, given the generic name of infliximab-dyyb under the agency’s nomenclature for biosimilar products, earned its approval as a biosimilar by showing it has no clinically meaningful differences in terms of safety and effectiveness from Remicade. According to FDA regulations, biosimilar products can have only minor differences in clinically inactive components and must have the same mechanism(s) of action (to the extent that it is known) and route(s) of administration, dosage form(s), and strength(s) as the reference product; and can be approved only for the indication(s) and condition(s) of use that have been approved for the reference product.
Inflectra’s approval is only as a biosimilar, not as an interchangeable product. The agency has yet to define the regulatory requirements for interchangeability that are necessary to meet the requirements of the Biologics Price Competition and Innovation Act of 2009. That Act states that an approved biosimilar “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” A statement about implementation of the Act on the FDA website explains that for interchangeability, “a sponsor must demonstrate that the biosimilar product can be expected to produce the same clinical result as the reference product in any given patient and, for a biological product that is administered more than once, that the risk of alternating or switching between use of the biosimilar product and the reference product is not greater than the risk of maintaining the patient on the reference product.”
Like Remicade, Inflectra will come with a boxed warning and a Medication Guide that describes important information about its uses and risks, which include serious infections (tuberculosis, bacterial sepsis, invasive fungal infections, and others), lymphoma and other malignancies, liver injury, blood problems, lupuslike syndrome, psoriasis, and in rare cases, nervous system disorders.
Inflectra is manufactured by Celltrion, based in South Korea, for Illinois-based Hospira. Inflectra’s label can be found here.
A biosimilar version of the anti–tumor necrosis factor–alpha agent Remicade has been approved by the Food and Drug Administration, making it the first biosimilar drug approved by the agency for inflammatory diseases and just the second biosimilar it has approved.
The agency said in its April 5 announcement that the biosimilar drug, to be marketed as Inflectra, will have the same indications as Remicade: moderately to severely active Crohn’s disease in patients aged 6 years and older who have had an inadequate response to conventional therapy; moderately to severely active ulcerative colitis that has inadequately responded to conventional therapy; moderately to severely active rheumatoid arthritis in combination with methotrexate; active ankylosing spondylitis; active psoriatic arthritis; and chronic, severe plaque psoriasis.
The drug, given the generic name of infliximab-dyyb under the agency’s nomenclature for biosimilar products, earned its approval as a biosimilar by showing it has no clinically meaningful differences in terms of safety and effectiveness from Remicade. According to FDA regulations, biosimilar products can have only minor differences in clinically inactive components and must have the same mechanism(s) of action (to the extent that it is known) and route(s) of administration, dosage form(s), and strength(s) as the reference product; and can be approved only for the indication(s) and condition(s) of use that have been approved for the reference product.
Inflectra’s approval is only as a biosimilar, not as an interchangeable product. The agency has yet to define the regulatory requirements for interchangeability that are necessary to meet the requirements of the Biologics Price Competition and Innovation Act of 2009. That Act states that an approved biosimilar “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” A statement about implementation of the Act on the FDA website explains that for interchangeability, “a sponsor must demonstrate that the biosimilar product can be expected to produce the same clinical result as the reference product in any given patient and, for a biological product that is administered more than once, that the risk of alternating or switching between use of the biosimilar product and the reference product is not greater than the risk of maintaining the patient on the reference product.”
Like Remicade, Inflectra will come with a boxed warning and a Medication Guide that describes important information about its uses and risks, which include serious infections (tuberculosis, bacterial sepsis, invasive fungal infections, and others), lymphoma and other malignancies, liver injury, blood problems, lupuslike syndrome, psoriasis, and in rare cases, nervous system disorders.
Inflectra is manufactured by Celltrion, based in South Korea, for Illinois-based Hospira. Inflectra’s label can be found here.

Cesarean scar defect: What is it and how should it be treated?
Cesarean delivery is one of the most common surgical procedures in women, with rates of 30% or more in the United States.1 As a result, the rate is rising for cesarean scar defect—the presence of a “niche” at the site of cesarean delivery scar—with the reported prevalence between 24% and 70% in a random population of women with at least one cesarean delivery.2 Other terms for cesarean scar defect include a niche, isthmocele, uteroperitoneal fistula, and diverticulum.1–9
Formation of cesarean scar defect
Cesarean scar defect forms after cesarean delivery, at the site of hysterotomy, on the anterior wall of the uterine isthmus (FIGURE 1). While this is the typical location, the defect has also been found at the endocervical canal and mid-uterine body. Improper healing of the cesarean incision leads to thinning of the anterior uterine wall, which creates an indentation and fluid-filled pouch at the cesarean scar site. The exact reason why a niche develops has not yet been determined; however, there are several hypotheses, broken down by pregnancy-related and patient-related factors. Surgical techniques that may increase the chance of niche development include low (cervical) hysterotomy, single-layer uterine wall closure, use of locking sutures, closure of hysterotomy with endometrial-sparing technique, and multiple cesarean deliveries.3,4 Patients with medical conditions that may impact wound healing (such as diabetes and smoking) may be at increased risk for niche formation.

Viewed hysteroscopically, the defect appears as a concave shape in the anterior uterine wall; to the inexperienced eye, it may resemble a second cavity (FIGURE 2).
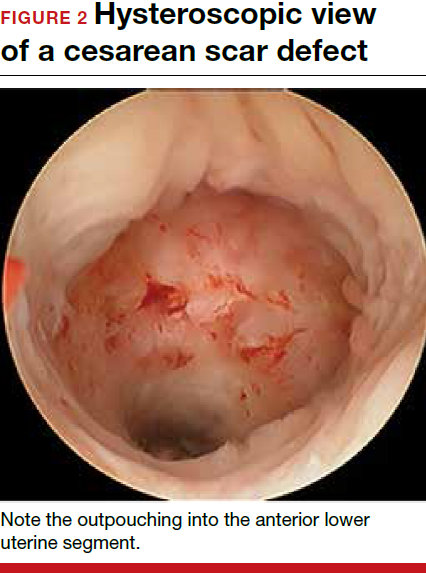
Pelvic pain and other serious consequences
The presence of fibrotic tissue in the niche acts like a valve, leading to the accumulation of blood in this reservoir-like area. A niche thus can cause delayed menstruation through the cervix, resulting in abnormal bleeding, pelvic pain, vaginal discharge, dysmenorrhea, dyspareunia, and infertility. Accumulated blood in this area can ultimately degrade cervical mucus and sperm quality, as well as inhibit sperm transport, a proposed mechanism of infertility.5,6 Women with a niche who conceive are at potential risk for cesarean scar ectopic pregnancy, with the embryo implanting in the pouch and subsequently growing and developing improperly.
Read about evaluation and treatment.
Evaluation and treatment
Patients presenting with the symptoms de-scribed above who have had a prior cesarean delivery should be evaluated for a cesarean scar defect.9 The best time to assess for the abnormality is after the patient’s menstrual cycle, when the endometrial lining is at its thinnest and recently menstruated blood has collected in the defect (this can highlight the niche on imaging). Transvaginal ultrasonography (FIGURE 3) or saline-infusion sonohysterogram serve as a first-line test for in-office diagnosis.7 Magnetic resonance imaging (MRI), 3-D ultrasonography, and hysteroscopy are additional useful imaging modalities that can aid in the diagnosis.
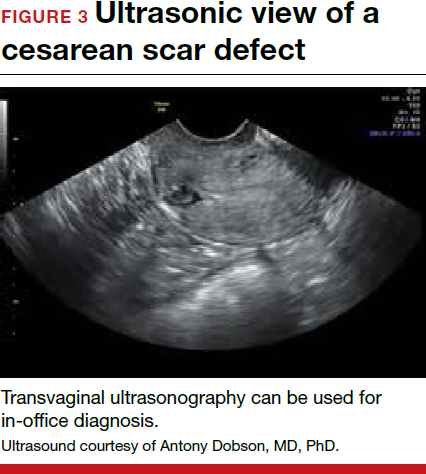
Treatments for cesarean scar defect vary dramatically and include hormonal therapy, hysteroscopic resection, vaginal or laparoscopic repair, and hysterectomy. Nonsurgical treatment should be reserved for women who desire a noninvasive approach, as the evidence for symptom resolution is limited.8
To promote fertility and decrease symptoms, the abnormal, fibrotic tissue must be removed. In our experience, since 2003, we have found that use of a laparoscopic approach is best for women desiring future fertility and that hysteroscopic resection is best for women whose childbearing is completed.9 Our management is dictated by the patient’s fertility plans, since there is concern that cesarean scar defect in a gravid uterus presents a risk for uterine rupture. The laparoscopic approach allows the defect to be repaired and the integrity of the myometrium restored.9
What are the coding options for cesarean scar defect repair?
Melanie Witt, RN, CPC, COBGC, MA
As the accompanying article discusses, the primary treatment for a cesarean scar defect depends on whether the patient wishes to preserve fertility, but assigning a procedure code for either surgical option will entail reporting an unlisted procedure code.
Under Current Procedural Terminology (CPT) guidelines (which are developed and copyrighted by the American Medical Association), procedure code selected must accurately describe the service/procedure performed rather than just approximate the service. This means that when a procedure-specific code does not exist, an unlisted procedure code that represents the type of surgery, the approach, and the anatomic site needs to be selected.
When an unlisted CPT code is reported, payment is based on the complexity of the surgery, and one way to communicate this to a payer is to provide additional documentation that not only includes the operative report but also suggests one or more existing CPT codes that have a published relative value unit (RVU) that approximates the work involved for the unlisted procedure.
The coding options for hysteroscopic and laparoscopic treatment options are listed below. The comparison codes offered will give the surgeon a range to look at, but the ultimate decision to use one of those suggested, or to choose an entirely different comparison code, is entirely within the control of the physician.
ICD-10-CM diagnostic coding
While the cesarean scar defect is a sequela of cesarean delivery, which is always reported as a secondary code, the choice of a primary diagnosis code can be either a gynecologic and/or an obstetric complication code. The choice may be determined by payer policy, as the use of an obstetric complication may not be accepted with a gynecologic procedure code. From a coding perspective, however, use of all 3 of these codes from the International Classification of Diseases, 10th Revision, Clinical Modification (ICD-10-CM) paints the most accurate description of the defect and its cause:
- N85.8 Other specified noninflammatory disorders of uterus versus
- O34.21 Maternal care for scar from previous cesarean delivery plus
- O94 Sequelae of complication of pregnancy, childbirth, and the puerperium.
Hysteroscopic resection codes:
- 58579 Unlisted hysteroscopy procedure, uterus
- The codes that may most closely approximate the physician work include 58561 (Hysteroscopy, surgical; with removal of leiomyomata) with 15.48 RVUs or 58560 (Hysteroscopy, surgical; with division or resection of intrauterine septum [any method]) with 10.92 RVUs.
Laparoscopic repair codes:
- 58578 Unlisted laparoscopy procedure, uterus
- The codes that may most closely approximate the physician work include 58520 (Hysterorrhaphy, repair of ruptured uterus [nonobstetrical] 24.25 RVUs or 58662 (Laparoscopy, surgical; with fulguration or excision of lesions of the ovary, pelvic viscera, or peritoneal surface by any method) with 20.14 RVUs.
You may also want to report a diagnostic hysteroscopy (code 58555), but keep in mind that payment will depend on documentation that clearly indicates that the use of the hysteroscope was for diagnostic purposes. Use of the hysteroscope to simply identify the surgical site to be repaired via the laparoscope will usually not be reimbursed separately.
Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Read about techniques for repair.
Techniques for repairing cesarean scar defect
For hysteroscopic resection of a niche, the uterus is distended and the intrauterine defect is visualized hysteroscopically, as seen in FIGURE 2. Using a bipolar or unipolar resectoscope, resect the fibrotic tissue of the defect and endometrial-like glands present within the niche. The goal of this relatively quick procedure is to open up the reservoir and facilitate the complete drainage of menstrual blood, thus alleviating the patient’s symptoms.Postoperatively, follow the patient for symptom resolution, and evaluate for defect resolution with transvaginal ultrasonography.
For a laparoscopic repair, first identify the niche hysteroscopically. At the same time as hysteroscopic examination of the cavity, the defect can be evaluated laparoscopically (FIGURE 4). The light from the hysteroscope can be visualized easily laparoscopically because of the thinned myometrium in the area of the defect. Map out the niche by transvaginally passing a cervical dilator into the defect in the uterine cavity (FIGURE 5). Again, given the thinning of this segment of the uterus, the dilator can be easily visualized laparoscopically. Be cautious when placing this dilator, as there is often overlying bladder. Prevent incidental cystotomy by gently advancing the dilator into the defect only until the niche can be adequately detected.9At this point, develop a bladder flap by opening the vesicovaginal and vesicocervical space, mobilizing the bladder inferiorly (FIGURE 6). With the guide of the dilator mapping out the defect (FIGURE 7), excise the fibrotic edges of the niche with thermal energy (monopolar cautery or CO2 laser) or sharp dissection (FIGURE 8). This leaves healthy myometrial tissue margins. Reapproximate these margins with absorbable suture (2-0 polyglactin 910 [Vicryl]) in an interrupted or running fashion, in 2 layers9 (FIGURE 9). Following the laparoscopic repair, perform hysteroscopic evaluation of the uterine cavity to assure complete resolution of the defect (FIGURE 10). With the hysteroscope in place, perform concurrent laparoscopic assessment of the repair. Check for impermeability by assuring no hysteroscopic fluid escapes at the site of repaired hysterotomy.9
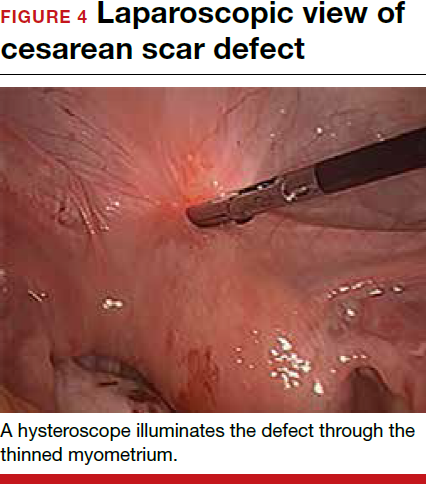
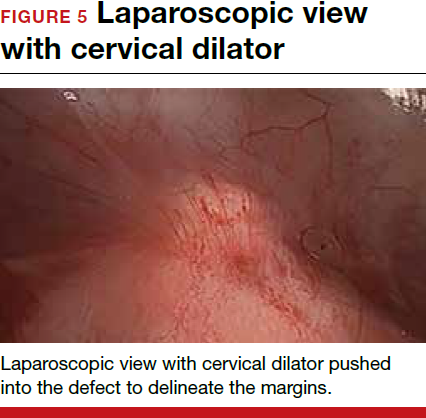
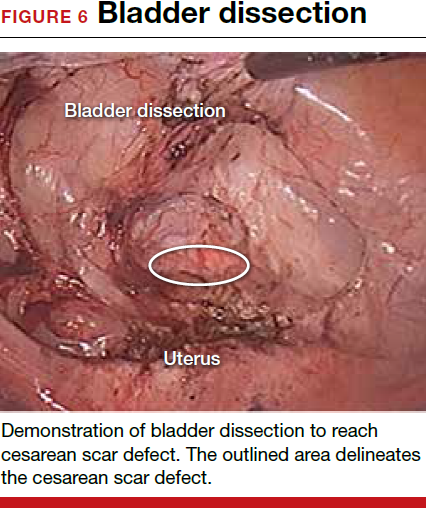
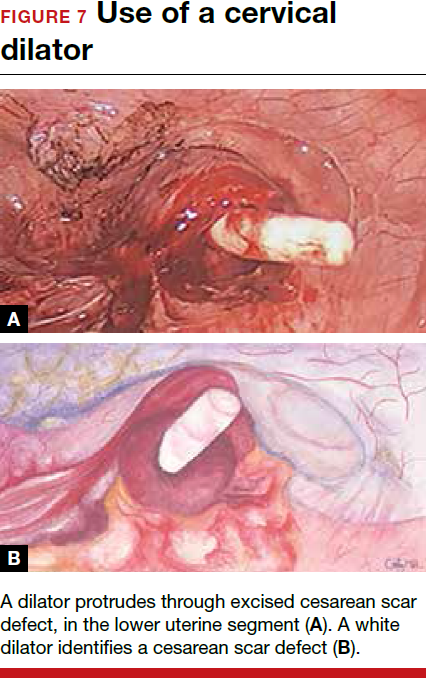
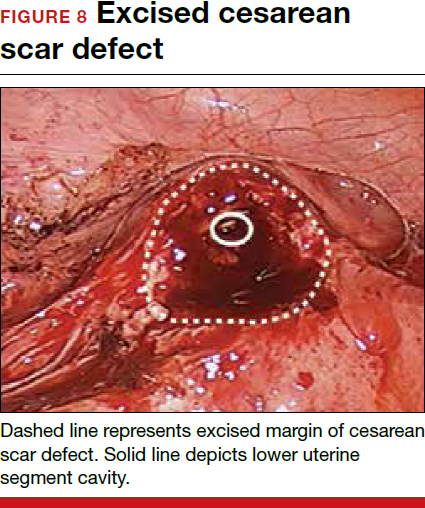
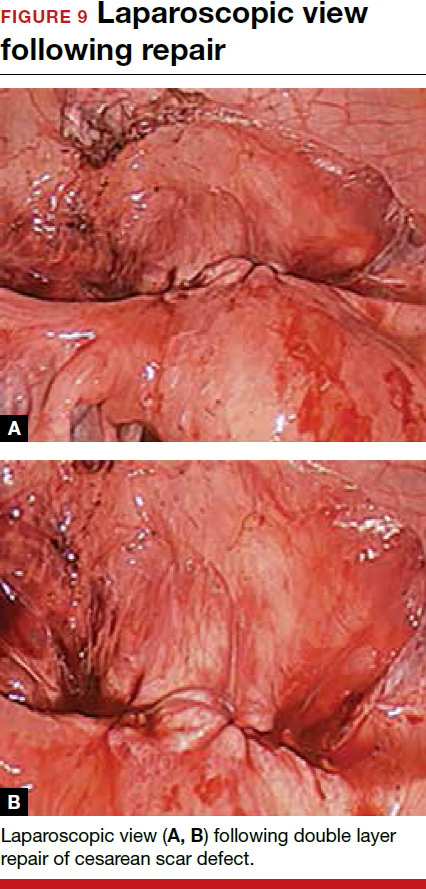
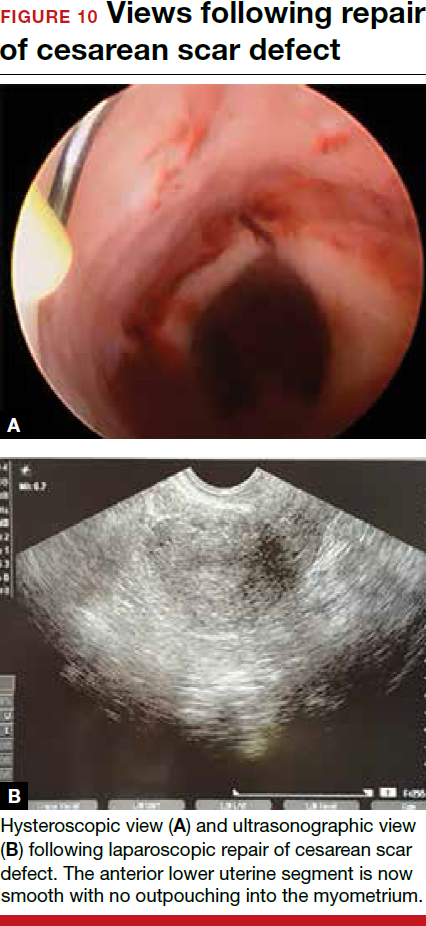
Postoperative care requires following the patient for symptom resolution and counseling regarding future fertility plans. We recommend that patients wait 6 months following the procedure before attempting conception.
When it comes to recommendations regarding preventing cesarean scar defects, additional randomized controlled trials need to be performed to evaluate various surgical techniques. At this time, there is no conclusive evidence that one method of hysterotomy closure is superior to another in preventing cesarean scar defect.
Symptoms often resolve with repair
When a patient with a prior cesarean delivery presents with symptoms of abnormal uterine bleeding, vaginal discharge, dysmenorrhea, dyspareunia, pelvic pain, or infertility that remain unexplained, consider cesarean scar defect as the culprit. Once a diagnosis of niche has been confirmed, the treatment approach should be dictated by the patient’s plans for future fertility. Hysteroscopic resection has been reported to have a 92% to 100% success rate for resolving symptoms of pain and bleeding, while 75% of patients undergoing laparoscopic niche repair for infertility achieved pregnancy.10,11 In our practice, a majority of patients experience symptom relief and go on to carry healthy pregnancies.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.

Dr. Camran Nezhat is Director of the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto, California.

Dr. Grace is a Fellow of the Society of Laparoendoscopic Surgeons at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.

Ms. Soliemannjad is an Intern at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.

Dr. Meshkat Razavi is Fellow at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.

Dr. Azadeh Nezhat is Co-Director of the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
The authors report no financial relationships relevant to this article.
Dr. Camran Nezhat is Director of the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto, California.
Dr. Grace is a Fellow of the Society of Laparoendoscopic Surgeons at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
Ms. Soliemannjad is an Intern at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
Dr. Meshkat Razavi is Fellow at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
Dr. Azadeh Nezhat is Co-Director of the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
The authors report no financial relationships relevant to this article.
Dr. Camran Nezhat is Director of the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto, California.
Dr. Grace is a Fellow of the Society of Laparoendoscopic Surgeons at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
Ms. Soliemannjad is an Intern at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
Dr. Meshkat Razavi is Fellow at the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
Dr. Azadeh Nezhat is Co-Director of the Center for Special Minimally Invasive and Robotic Surgery, Palo Alto.
The authors report no financial relationships relevant to this article.
Cesarean delivery is one of the most common surgical procedures in women, with rates of 30% or more in the United States.1 As a result, the rate is rising for cesarean scar defect—the presence of a “niche” at the site of cesarean delivery scar—with the reported prevalence between 24% and 70% in a random population of women with at least one cesarean delivery.2 Other terms for cesarean scar defect include a niche, isthmocele, uteroperitoneal fistula, and diverticulum.1–9
Formation of cesarean scar defect
Cesarean scar defect forms after cesarean delivery, at the site of hysterotomy, on the anterior wall of the uterine isthmus (FIGURE 1). While this is the typical location, the defect has also been found at the endocervical canal and mid-uterine body. Improper healing of the cesarean incision leads to thinning of the anterior uterine wall, which creates an indentation and fluid-filled pouch at the cesarean scar site. The exact reason why a niche develops has not yet been determined; however, there are several hypotheses, broken down by pregnancy-related and patient-related factors. Surgical techniques that may increase the chance of niche development include low (cervical) hysterotomy, single-layer uterine wall closure, use of locking sutures, closure of hysterotomy with endometrial-sparing technique, and multiple cesarean deliveries.3,4 Patients with medical conditions that may impact wound healing (such as diabetes and smoking) may be at increased risk for niche formation.
Viewed hysteroscopically, the defect appears as a concave shape in the anterior uterine wall; to the inexperienced eye, it may resemble a second cavity (FIGURE 2).
Pelvic pain and other serious consequences
The presence of fibrotic tissue in the niche acts like a valve, leading to the accumulation of blood in this reservoir-like area. A niche thus can cause delayed menstruation through the cervix, resulting in abnormal bleeding, pelvic pain, vaginal discharge, dysmenorrhea, dyspareunia, and infertility. Accumulated blood in this area can ultimately degrade cervical mucus and sperm quality, as well as inhibit sperm transport, a proposed mechanism of infertility.5,6 Women with a niche who conceive are at potential risk for cesarean scar ectopic pregnancy, with the embryo implanting in the pouch and subsequently growing and developing improperly.
Read about evaluation and treatment.
Evaluation and treatment
Patients presenting with the symptoms de-scribed above who have had a prior cesarean delivery should be evaluated for a cesarean scar defect.9 The best time to assess for the abnormality is after the patient’s menstrual cycle, when the endometrial lining is at its thinnest and recently menstruated blood has collected in the defect (this can highlight the niche on imaging). Transvaginal ultrasonography (FIGURE 3) or saline-infusion sonohysterogram serve as a first-line test for in-office diagnosis.7 Magnetic resonance imaging (MRI), 3-D ultrasonography, and hysteroscopy are additional useful imaging modalities that can aid in the diagnosis.
Treatments for cesarean scar defect vary dramatically and include hormonal therapy, hysteroscopic resection, vaginal or laparoscopic repair, and hysterectomy. Nonsurgical treatment should be reserved for women who desire a noninvasive approach, as the evidence for symptom resolution is limited.8
To promote fertility and decrease symptoms, the abnormal, fibrotic tissue must be removed. In our experience, since 2003, we have found that use of a laparoscopic approach is best for women desiring future fertility and that hysteroscopic resection is best for women whose childbearing is completed.9 Our management is dictated by the patient’s fertility plans, since there is concern that cesarean scar defect in a gravid uterus presents a risk for uterine rupture. The laparoscopic approach allows the defect to be repaired and the integrity of the myometrium restored.9
What are the coding options for cesarean scar defect repair?
Melanie Witt, RN, CPC, COBGC, MA
As the accompanying article discusses, the primary treatment for a cesarean scar defect depends on whether the patient wishes to preserve fertility, but assigning a procedure code for either surgical option will entail reporting an unlisted procedure code.
Under Current Procedural Terminology (CPT) guidelines (which are developed and copyrighted by the American Medical Association), procedure code selected must accurately describe the service/procedure performed rather than just approximate the service. This means that when a procedure-specific code does not exist, an unlisted procedure code that represents the type of surgery, the approach, and the anatomic site needs to be selected.
When an unlisted CPT code is reported, payment is based on the complexity of the surgery, and one way to communicate this to a payer is to provide additional documentation that not only includes the operative report but also suggests one or more existing CPT codes that have a published relative value unit (RVU) that approximates the work involved for the unlisted procedure.
The coding options for hysteroscopic and laparoscopic treatment options are listed below. The comparison codes offered will give the surgeon a range to look at, but the ultimate decision to use one of those suggested, or to choose an entirely different comparison code, is entirely within the control of the physician.
ICD-10-CM diagnostic coding
While the cesarean scar defect is a sequela of cesarean delivery, which is always reported as a secondary code, the choice of a primary diagnosis code can be either a gynecologic and/or an obstetric complication code. The choice may be determined by payer policy, as the use of an obstetric complication may not be accepted with a gynecologic procedure code. From a coding perspective, however, use of all 3 of these codes from the International Classification of Diseases, 10th Revision, Clinical Modification (ICD-10-CM) paints the most accurate description of the defect and its cause:
- N85.8 Other specified noninflammatory disorders of uterus versus
- O34.21 Maternal care for scar from previous cesarean delivery plus
- O94 Sequelae of complication of pregnancy, childbirth, and the puerperium.
Hysteroscopic resection codes:
- 58579 Unlisted hysteroscopy procedure, uterus
- The codes that may most closely approximate the physician work include 58561 (Hysteroscopy, surgical; with removal of leiomyomata) with 15.48 RVUs or 58560 (Hysteroscopy, surgical; with division or resection of intrauterine septum [any method]) with 10.92 RVUs.
Laparoscopic repair codes:
- 58578 Unlisted laparoscopy procedure, uterus
- The codes that may most closely approximate the physician work include 58520 (Hysterorrhaphy, repair of ruptured uterus [nonobstetrical] 24.25 RVUs or 58662 (Laparoscopy, surgical; with fulguration or excision of lesions of the ovary, pelvic viscera, or peritoneal surface by any method) with 20.14 RVUs.
You may also want to report a diagnostic hysteroscopy (code 58555), but keep in mind that payment will depend on documentation that clearly indicates that the use of the hysteroscope was for diagnostic purposes. Use of the hysteroscope to simply identify the surgical site to be repaired via the laparoscope will usually not be reimbursed separately.
Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Read about techniques for repair.
Techniques for repairing cesarean scar defect
For hysteroscopic resection of a niche, the uterus is distended and the intrauterine defect is visualized hysteroscopically, as seen in FIGURE 2. Using a bipolar or unipolar resectoscope, resect the fibrotic tissue of the defect and endometrial-like glands present within the niche. The goal of this relatively quick procedure is to open up the reservoir and facilitate the complete drainage of menstrual blood, thus alleviating the patient’s symptoms.Postoperatively, follow the patient for symptom resolution, and evaluate for defect resolution with transvaginal ultrasonography.
For a laparoscopic repair, first identify the niche hysteroscopically. At the same time as hysteroscopic examination of the cavity, the defect can be evaluated laparoscopically (FIGURE 4). The light from the hysteroscope can be visualized easily laparoscopically because of the thinned myometrium in the area of the defect. Map out the niche by transvaginally passing a cervical dilator into the defect in the uterine cavity (FIGURE 5). Again, given the thinning of this segment of the uterus, the dilator can be easily visualized laparoscopically. Be cautious when placing this dilator, as there is often overlying bladder. Prevent incidental cystotomy by gently advancing the dilator into the defect only until the niche can be adequately detected.9At this point, develop a bladder flap by opening the vesicovaginal and vesicocervical space, mobilizing the bladder inferiorly (FIGURE 6). With the guide of the dilator mapping out the defect (FIGURE 7), excise the fibrotic edges of the niche with thermal energy (monopolar cautery or CO2 laser) or sharp dissection (FIGURE 8). This leaves healthy myometrial tissue margins. Reapproximate these margins with absorbable suture (2-0 polyglactin 910 [Vicryl]) in an interrupted or running fashion, in 2 layers9 (FIGURE 9). Following the laparoscopic repair, perform hysteroscopic evaluation of the uterine cavity to assure complete resolution of the defect (FIGURE 10). With the hysteroscope in place, perform concurrent laparoscopic assessment of the repair. Check for impermeability by assuring no hysteroscopic fluid escapes at the site of repaired hysterotomy.9
Postoperative care requires following the patient for symptom resolution and counseling regarding future fertility plans. We recommend that patients wait 6 months following the procedure before attempting conception.
When it comes to recommendations regarding preventing cesarean scar defects, additional randomized controlled trials need to be performed to evaluate various surgical techniques. At this time, there is no conclusive evidence that one method of hysterotomy closure is superior to another in preventing cesarean scar defect.
Symptoms often resolve with repair
When a patient with a prior cesarean delivery presents with symptoms of abnormal uterine bleeding, vaginal discharge, dysmenorrhea, dyspareunia, pelvic pain, or infertility that remain unexplained, consider cesarean scar defect as the culprit. Once a diagnosis of niche has been confirmed, the treatment approach should be dictated by the patient’s plans for future fertility. Hysteroscopic resection has been reported to have a 92% to 100% success rate for resolving symptoms of pain and bleeding, while 75% of patients undergoing laparoscopic niche repair for infertility achieved pregnancy.10,11 In our practice, a majority of patients experience symptom relief and go on to carry healthy pregnancies.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Cesarean delivery is one of the most common surgical procedures in women, with rates of 30% or more in the United States.1 As a result, the rate is rising for cesarean scar defect—the presence of a “niche” at the site of cesarean delivery scar—with the reported prevalence between 24% and 70% in a random population of women with at least one cesarean delivery.2 Other terms for cesarean scar defect include a niche, isthmocele, uteroperitoneal fistula, and diverticulum.1–9
Formation of cesarean scar defect
Cesarean scar defect forms after cesarean delivery, at the site of hysterotomy, on the anterior wall of the uterine isthmus (FIGURE 1). While this is the typical location, the defect has also been found at the endocervical canal and mid-uterine body. Improper healing of the cesarean incision leads to thinning of the anterior uterine wall, which creates an indentation and fluid-filled pouch at the cesarean scar site. The exact reason why a niche develops has not yet been determined; however, there are several hypotheses, broken down by pregnancy-related and patient-related factors. Surgical techniques that may increase the chance of niche development include low (cervical) hysterotomy, single-layer uterine wall closure, use of locking sutures, closure of hysterotomy with endometrial-sparing technique, and multiple cesarean deliveries.3,4 Patients with medical conditions that may impact wound healing (such as diabetes and smoking) may be at increased risk for niche formation.
Viewed hysteroscopically, the defect appears as a concave shape in the anterior uterine wall; to the inexperienced eye, it may resemble a second cavity (FIGURE 2).
Pelvic pain and other serious consequences
The presence of fibrotic tissue in the niche acts like a valve, leading to the accumulation of blood in this reservoir-like area. A niche thus can cause delayed menstruation through the cervix, resulting in abnormal bleeding, pelvic pain, vaginal discharge, dysmenorrhea, dyspareunia, and infertility. Accumulated blood in this area can ultimately degrade cervical mucus and sperm quality, as well as inhibit sperm transport, a proposed mechanism of infertility.5,6 Women with a niche who conceive are at potential risk for cesarean scar ectopic pregnancy, with the embryo implanting in the pouch and subsequently growing and developing improperly.
Read about evaluation and treatment.
Evaluation and treatment
Patients presenting with the symptoms de-scribed above who have had a prior cesarean delivery should be evaluated for a cesarean scar defect.9 The best time to assess for the abnormality is after the patient’s menstrual cycle, when the endometrial lining is at its thinnest and recently menstruated blood has collected in the defect (this can highlight the niche on imaging). Transvaginal ultrasonography (FIGURE 3) or saline-infusion sonohysterogram serve as a first-line test for in-office diagnosis.7 Magnetic resonance imaging (MRI), 3-D ultrasonography, and hysteroscopy are additional useful imaging modalities that can aid in the diagnosis.
Treatments for cesarean scar defect vary dramatically and include hormonal therapy, hysteroscopic resection, vaginal or laparoscopic repair, and hysterectomy. Nonsurgical treatment should be reserved for women who desire a noninvasive approach, as the evidence for symptom resolution is limited.8
To promote fertility and decrease symptoms, the abnormal, fibrotic tissue must be removed. In our experience, since 2003, we have found that use of a laparoscopic approach is best for women desiring future fertility and that hysteroscopic resection is best for women whose childbearing is completed.9 Our management is dictated by the patient’s fertility plans, since there is concern that cesarean scar defect in a gravid uterus presents a risk for uterine rupture. The laparoscopic approach allows the defect to be repaired and the integrity of the myometrium restored.9
What are the coding options for cesarean scar defect repair?
Melanie Witt, RN, CPC, COBGC, MA
As the accompanying article discusses, the primary treatment for a cesarean scar defect depends on whether the patient wishes to preserve fertility, but assigning a procedure code for either surgical option will entail reporting an unlisted procedure code.
Under Current Procedural Terminology (CPT) guidelines (which are developed and copyrighted by the American Medical Association), procedure code selected must accurately describe the service/procedure performed rather than just approximate the service. This means that when a procedure-specific code does not exist, an unlisted procedure code that represents the type of surgery, the approach, and the anatomic site needs to be selected.
When an unlisted CPT code is reported, payment is based on the complexity of the surgery, and one way to communicate this to a payer is to provide additional documentation that not only includes the operative report but also suggests one or more existing CPT codes that have a published relative value unit (RVU) that approximates the work involved for the unlisted procedure.
The coding options for hysteroscopic and laparoscopic treatment options are listed below. The comparison codes offered will give the surgeon a range to look at, but the ultimate decision to use one of those suggested, or to choose an entirely different comparison code, is entirely within the control of the physician.
ICD-10-CM diagnostic coding
While the cesarean scar defect is a sequela of cesarean delivery, which is always reported as a secondary code, the choice of a primary diagnosis code can be either a gynecologic and/or an obstetric complication code. The choice may be determined by payer policy, as the use of an obstetric complication may not be accepted with a gynecologic procedure code. From a coding perspective, however, use of all 3 of these codes from the International Classification of Diseases, 10th Revision, Clinical Modification (ICD-10-CM) paints the most accurate description of the defect and its cause:
- N85.8 Other specified noninflammatory disorders of uterus versus
- O34.21 Maternal care for scar from previous cesarean delivery plus
- O94 Sequelae of complication of pregnancy, childbirth, and the puerperium.
Hysteroscopic resection codes:
- 58579 Unlisted hysteroscopy procedure, uterus
- The codes that may most closely approximate the physician work include 58561 (Hysteroscopy, surgical; with removal of leiomyomata) with 15.48 RVUs or 58560 (Hysteroscopy, surgical; with division or resection of intrauterine septum [any method]) with 10.92 RVUs.
Laparoscopic repair codes:
- 58578 Unlisted laparoscopy procedure, uterus
- The codes that may most closely approximate the physician work include 58520 (Hysterorrhaphy, repair of ruptured uterus [nonobstetrical] 24.25 RVUs or 58662 (Laparoscopy, surgical; with fulguration or excision of lesions of the ovary, pelvic viscera, or peritoneal surface by any method) with 20.14 RVUs.
You may also want to report a diagnostic hysteroscopy (code 58555), but keep in mind that payment will depend on documentation that clearly indicates that the use of the hysteroscope was for diagnostic purposes. Use of the hysteroscope to simply identify the surgical site to be repaired via the laparoscope will usually not be reimbursed separately.
Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Read about techniques for repair.
Techniques for repairing cesarean scar defect
For hysteroscopic resection of a niche, the uterus is distended and the intrauterine defect is visualized hysteroscopically, as seen in FIGURE 2. Using a bipolar or unipolar resectoscope, resect the fibrotic tissue of the defect and endometrial-like glands present within the niche. The goal of this relatively quick procedure is to open up the reservoir and facilitate the complete drainage of menstrual blood, thus alleviating the patient’s symptoms.Postoperatively, follow the patient for symptom resolution, and evaluate for defect resolution with transvaginal ultrasonography.
For a laparoscopic repair, first identify the niche hysteroscopically. At the same time as hysteroscopic examination of the cavity, the defect can be evaluated laparoscopically (FIGURE 4). The light from the hysteroscope can be visualized easily laparoscopically because of the thinned myometrium in the area of the defect. Map out the niche by transvaginally passing a cervical dilator into the defect in the uterine cavity (FIGURE 5). Again, given the thinning of this segment of the uterus, the dilator can be easily visualized laparoscopically. Be cautious when placing this dilator, as there is often overlying bladder. Prevent incidental cystotomy by gently advancing the dilator into the defect only until the niche can be adequately detected.9At this point, develop a bladder flap by opening the vesicovaginal and vesicocervical space, mobilizing the bladder inferiorly (FIGURE 6). With the guide of the dilator mapping out the defect (FIGURE 7), excise the fibrotic edges of the niche with thermal energy (monopolar cautery or CO2 laser) or sharp dissection (FIGURE 8). This leaves healthy myometrial tissue margins. Reapproximate these margins with absorbable suture (2-0 polyglactin 910 [Vicryl]) in an interrupted or running fashion, in 2 layers9 (FIGURE 9). Following the laparoscopic repair, perform hysteroscopic evaluation of the uterine cavity to assure complete resolution of the defect (FIGURE 10). With the hysteroscope in place, perform concurrent laparoscopic assessment of the repair. Check for impermeability by assuring no hysteroscopic fluid escapes at the site of repaired hysterotomy.9
Postoperative care requires following the patient for symptom resolution and counseling regarding future fertility plans. We recommend that patients wait 6 months following the procedure before attempting conception.
When it comes to recommendations regarding preventing cesarean scar defects, additional randomized controlled trials need to be performed to evaluate various surgical techniques. At this time, there is no conclusive evidence that one method of hysterotomy closure is superior to another in preventing cesarean scar defect.
Symptoms often resolve with repair
When a patient with a prior cesarean delivery presents with symptoms of abnormal uterine bleeding, vaginal discharge, dysmenorrhea, dyspareunia, pelvic pain, or infertility that remain unexplained, consider cesarean scar defect as the culprit. Once a diagnosis of niche has been confirmed, the treatment approach should be dictated by the patient’s plans for future fertility. Hysteroscopic resection has been reported to have a 92% to 100% success rate for resolving symptoms of pain and bleeding, while 75% of patients undergoing laparoscopic niche repair for infertility achieved pregnancy.10,11 In our practice, a majority of patients experience symptom relief and go on to carry healthy pregnancies.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In this Article
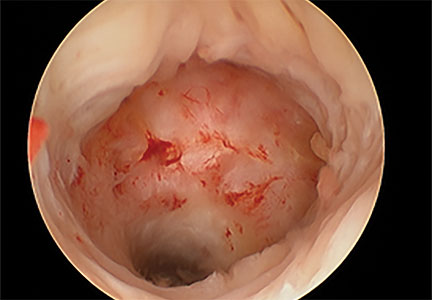
FDA adds safety warnings to certain type 2 diabetes medications
Type 2 diabetes medicines that contain saxagliptin and alogliptin may increase the risk of heart failure, especially in patients who already have heart or kidney disease, according to results from an Food and Drug Administration safety review.
The development, which was announced by MedWatch on April 5, 2016, means that the FDA will add new warnings to the drug labels about this safety issue. “Health care professionals should consider discontinuing medications containing saxagliptin and alogliptin in patients who develop heart failure and monitor their diabetes control,” the communication states. “If a patient’s blood sugar level is not well-controlled with their current treatment, other diabetes medicines may be required.”
The medications of concern include Onglyza (saxagliptin); Kombiglyze XR (saxagliptin and metformin extended release); Nesina (alogliptin); Kazano (alogliptin and metformin), and Oseni (alogliptin and pioglitazone). The move comes after two clinical trials showed that more patients who received saxagliptin- or alogliptin-containing medicines were hospitalized for heart failure, compared with patients who received placebo (for specifics, see the data summary section in the FDA Drug Safety Communication).
The communication noted that patients taking these medicines should contact their health care clinician if they develop signs and symptoms of heart failure such as: unusual shortness of breath during daily activities; trouble breathing when lying down; tiredness, weakness, or fatigue; and weight gain with swelling in the ankles, feet, legs, or stomach.
Clinicians and patients can report adverse events or side effects related to the use of these products at www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home.
Type 2 diabetes medicines that contain saxagliptin and alogliptin may increase the risk of heart failure, especially in patients who already have heart or kidney disease, according to results from an Food and Drug Administration safety review.
The development, which was announced by MedWatch on April 5, 2016, means that the FDA will add new warnings to the drug labels about this safety issue. “Health care professionals should consider discontinuing medications containing saxagliptin and alogliptin in patients who develop heart failure and monitor their diabetes control,” the communication states. “If a patient’s blood sugar level is not well-controlled with their current treatment, other diabetes medicines may be required.”
The medications of concern include Onglyza (saxagliptin); Kombiglyze XR (saxagliptin and metformin extended release); Nesina (alogliptin); Kazano (alogliptin and metformin), and Oseni (alogliptin and pioglitazone). The move comes after two clinical trials showed that more patients who received saxagliptin- or alogliptin-containing medicines were hospitalized for heart failure, compared with patients who received placebo (for specifics, see the data summary section in the FDA Drug Safety Communication).
The communication noted that patients taking these medicines should contact their health care clinician if they develop signs and symptoms of heart failure such as: unusual shortness of breath during daily activities; trouble breathing when lying down; tiredness, weakness, or fatigue; and weight gain with swelling in the ankles, feet, legs, or stomach.
Clinicians and patients can report adverse events or side effects related to the use of these products at www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home.
Type 2 diabetes medicines that contain saxagliptin and alogliptin may increase the risk of heart failure, especially in patients who already have heart or kidney disease, according to results from an Food and Drug Administration safety review.
The development, which was announced by MedWatch on April 5, 2016, means that the FDA will add new warnings to the drug labels about this safety issue. “Health care professionals should consider discontinuing medications containing saxagliptin and alogliptin in patients who develop heart failure and monitor their diabetes control,” the communication states. “If a patient’s blood sugar level is not well-controlled with their current treatment, other diabetes medicines may be required.”
The medications of concern include Onglyza (saxagliptin); Kombiglyze XR (saxagliptin and metformin extended release); Nesina (alogliptin); Kazano (alogliptin and metformin), and Oseni (alogliptin and pioglitazone). The move comes after two clinical trials showed that more patients who received saxagliptin- or alogliptin-containing medicines were hospitalized for heart failure, compared with patients who received placebo (for specifics, see the data summary section in the FDA Drug Safety Communication).
The communication noted that patients taking these medicines should contact their health care clinician if they develop signs and symptoms of heart failure such as: unusual shortness of breath during daily activities; trouble breathing when lying down; tiredness, weakness, or fatigue; and weight gain with swelling in the ankles, feet, legs, or stomach.
Clinicians and patients can report adverse events or side effects related to the use of these products at www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home.
VIDEO: Fire and Ice - Which catheter ablation approach is best in AF?
CHICAGO – The largest-ever randomized trial of catheter ablation for atrial fibrillation ended in a draw, but there may be a clear winner for some patients.
Safety and 1-year efficacy of radiofrequency ablation and cryoballoon ablation were roughly 65% in both treatment arms of the 769-patient Fire and Ice trial.
However, in an interview at the annual meeting of the American College of Cardiology, principal investigator Dr. Karl-Heinz Kuck of Asklepios Klinik St. Georg, Hamburg, Germany, explains why the results are actually a victory for cryoablation.
CHICAGO – The largest-ever randomized trial of catheter ablation for atrial fibrillation ended in a draw, but there may be a clear winner for some patients.
Safety and 1-year efficacy of radiofrequency ablation and cryoballoon ablation were roughly 65% in both treatment arms of the 769-patient Fire and Ice trial.
However, in an interview at the annual meeting of the American College of Cardiology, principal investigator Dr. Karl-Heinz Kuck of Asklepios Klinik St. Georg, Hamburg, Germany, explains why the results are actually a victory for cryoablation.
CHICAGO – The largest-ever randomized trial of catheter ablation for atrial fibrillation ended in a draw, but there may be a clear winner for some patients.
Safety and 1-year efficacy of radiofrequency ablation and cryoballoon ablation were roughly 65% in both treatment arms of the 769-patient Fire and Ice trial.
However, in an interview at the annual meeting of the American College of Cardiology, principal investigator Dr. Karl-Heinz Kuck of Asklepios Klinik St. Georg, Hamburg, Germany, explains why the results are actually a victory for cryoablation.
AT ACC 16
