User login
Sofosbuvir and ribavirin critical to preventing posttransplantation HCV recurrence
Sofosbuvir and ribavirin treatments should be administered to patients with hepatitis C virus who undergo liver transplantations in order to significantly decrease the risks of posttransplant HCV recurrence, according to two new studies published in the January issue of Gastroenterology (10.1053/j.gastro.2014.09.023 and 10.1053/j.gastro.2014.10.001).
“In clinical trials, administration of sofosbuvir with ribavirin was associated with rapid decreases of HCV RNA to undetectable levels in patients with HCV genotype 1, 2, 3, 4, and 6 infections,” wrote lead author Dr. Michael P. Curry of the Beth Israel Deaconess Medical Center in Boston, and his coauthors on the first of these two studies. “In more than 3,000 patients treated to date, sofosbuvir has been shown to be safe, viral breakthrough during treatment has been rare (and associated with nonadherence), and few drug interactions have been observed.”
In a phase II, open-label study, Dr. Curry and his coinvestigators enrolled 61 patients with HCV of any genotype, and cirrhosis with a Child-Turcotte-Pugh score no greater than 7, who were all wait-listed to receive liver transplantations. Subjects received up to 48 weeks of treatment with 400 mg of sofosbuvir, and a separate dose of ribavirin prior to liver transplantation, while 43 patients received transplantations alone. The primary outcome sought by investigators was HCV-RNA levels less than 25 IU/mL at 12 weeks after transplantation among patients that had this level prior to the operation.
The investigators found that 43 subjects had the desired HCV-RNA levels; of that population, 49% had a posttransplantation virologic response, with the most frequent side effects reported by subjects being fatigue (38%), headache (23%), and anemia (21%). Of the 43 applicable subjects, 30 (70% of the population) had a posttransplantation virologic response at 12 weeks, 10 (23%) had recurrent infection, and 3 (7%) died.
“This study provides proof of concept that virologic suppression without interferon significantly can reduce the rate of recurrent HCV after liver transplantation,” the study says, adding that the results “compare favorably with those observed in other trials of pretransplantation antiviral therapy.”
In the second study, the authors ascertained that combination therapy consisting of sofosbuvir and ribavirin for 24 weeks is effective at preventing hepatitis C virus recurrence in patients who undergo liver transplantations.
“Recurrent HCV infection is the most common cause of mortality and graft loss following transplantation, and up to 30% of patients with recurrent infection develop cirrhosis within 5 years,” wrote the study’s authors, led by Dr. Michael Charlton of the Mayo Clinic in Rochester, Minn.
Using a prospective, multicenter, open-label pilot study, investigators enrolled and treated 40 patients with a 24-week regimen of 400 mg sofosbuvir and ribavirin starting at 400 mg, which was subsequently adjusted per patient based on individual creatinine clearance and hemoglobin levels. Subjects were 78% male and 85% white, with 83% having HCV genotype 1, 40% having cirrhosis, and 88% having been previously treated with interferon. The primary outcome investigators looked for was “sustained virologic response 12 weeks after treatment (SVR12).”
Data showed that SVR12 was achieved by 28 of the 40 subjects that received treatment, or 70%. The most commonly reported adverse effects were fatigue (30%), diarrhea (28%), headache (25%), and anemia (20%). No patients exhibited detectable viral resistance during or after treatment, and although two patients terminated their treatment because of adverse events, investigators reported no deaths, graft losses, or episodes of rejection.
“In contrast,” Dr. Charlton and his coauthors noted, “interferon-based treatments have been associated with posttreatment immunological dysfunction (particularly plasma cell hepatitis) and even hepatic decompensation in LT [liver transplant] recipients.”
The authors of the first study disclosed that Dr. Curry has received grants from and been affiliated with Gilead, which was a sponsor of the study. The authors of the second study reported no relevant financial disclosures.
Sofosbuvir and ribavirin treatments should be administered to patients with hepatitis C virus who undergo liver transplantations in order to significantly decrease the risks of posttransplant HCV recurrence, according to two new studies published in the January issue of Gastroenterology (10.1053/j.gastro.2014.09.023 and 10.1053/j.gastro.2014.10.001).
“In clinical trials, administration of sofosbuvir with ribavirin was associated with rapid decreases of HCV RNA to undetectable levels in patients with HCV genotype 1, 2, 3, 4, and 6 infections,” wrote lead author Dr. Michael P. Curry of the Beth Israel Deaconess Medical Center in Boston, and his coauthors on the first of these two studies. “In more than 3,000 patients treated to date, sofosbuvir has been shown to be safe, viral breakthrough during treatment has been rare (and associated with nonadherence), and few drug interactions have been observed.”
In a phase II, open-label study, Dr. Curry and his coinvestigators enrolled 61 patients with HCV of any genotype, and cirrhosis with a Child-Turcotte-Pugh score no greater than 7, who were all wait-listed to receive liver transplantations. Subjects received up to 48 weeks of treatment with 400 mg of sofosbuvir, and a separate dose of ribavirin prior to liver transplantation, while 43 patients received transplantations alone. The primary outcome sought by investigators was HCV-RNA levels less than 25 IU/mL at 12 weeks after transplantation among patients that had this level prior to the operation.
The investigators found that 43 subjects had the desired HCV-RNA levels; of that population, 49% had a posttransplantation virologic response, with the most frequent side effects reported by subjects being fatigue (38%), headache (23%), and anemia (21%). Of the 43 applicable subjects, 30 (70% of the population) had a posttransplantation virologic response at 12 weeks, 10 (23%) had recurrent infection, and 3 (7%) died.
“This study provides proof of concept that virologic suppression without interferon significantly can reduce the rate of recurrent HCV after liver transplantation,” the study says, adding that the results “compare favorably with those observed in other trials of pretransplantation antiviral therapy.”
In the second study, the authors ascertained that combination therapy consisting of sofosbuvir and ribavirin for 24 weeks is effective at preventing hepatitis C virus recurrence in patients who undergo liver transplantations.
“Recurrent HCV infection is the most common cause of mortality and graft loss following transplantation, and up to 30% of patients with recurrent infection develop cirrhosis within 5 years,” wrote the study’s authors, led by Dr. Michael Charlton of the Mayo Clinic in Rochester, Minn.
Using a prospective, multicenter, open-label pilot study, investigators enrolled and treated 40 patients with a 24-week regimen of 400 mg sofosbuvir and ribavirin starting at 400 mg, which was subsequently adjusted per patient based on individual creatinine clearance and hemoglobin levels. Subjects were 78% male and 85% white, with 83% having HCV genotype 1, 40% having cirrhosis, and 88% having been previously treated with interferon. The primary outcome investigators looked for was “sustained virologic response 12 weeks after treatment (SVR12).”
Data showed that SVR12 was achieved by 28 of the 40 subjects that received treatment, or 70%. The most commonly reported adverse effects were fatigue (30%), diarrhea (28%), headache (25%), and anemia (20%). No patients exhibited detectable viral resistance during or after treatment, and although two patients terminated their treatment because of adverse events, investigators reported no deaths, graft losses, or episodes of rejection.
“In contrast,” Dr. Charlton and his coauthors noted, “interferon-based treatments have been associated with posttreatment immunological dysfunction (particularly plasma cell hepatitis) and even hepatic decompensation in LT [liver transplant] recipients.”
The authors of the first study disclosed that Dr. Curry has received grants from and been affiliated with Gilead, which was a sponsor of the study. The authors of the second study reported no relevant financial disclosures.
Sofosbuvir and ribavirin treatments should be administered to patients with hepatitis C virus who undergo liver transplantations in order to significantly decrease the risks of posttransplant HCV recurrence, according to two new studies published in the January issue of Gastroenterology (10.1053/j.gastro.2014.09.023 and 10.1053/j.gastro.2014.10.001).
“In clinical trials, administration of sofosbuvir with ribavirin was associated with rapid decreases of HCV RNA to undetectable levels in patients with HCV genotype 1, 2, 3, 4, and 6 infections,” wrote lead author Dr. Michael P. Curry of the Beth Israel Deaconess Medical Center in Boston, and his coauthors on the first of these two studies. “In more than 3,000 patients treated to date, sofosbuvir has been shown to be safe, viral breakthrough during treatment has been rare (and associated with nonadherence), and few drug interactions have been observed.”
In a phase II, open-label study, Dr. Curry and his coinvestigators enrolled 61 patients with HCV of any genotype, and cirrhosis with a Child-Turcotte-Pugh score no greater than 7, who were all wait-listed to receive liver transplantations. Subjects received up to 48 weeks of treatment with 400 mg of sofosbuvir, and a separate dose of ribavirin prior to liver transplantation, while 43 patients received transplantations alone. The primary outcome sought by investigators was HCV-RNA levels less than 25 IU/mL at 12 weeks after transplantation among patients that had this level prior to the operation.
The investigators found that 43 subjects had the desired HCV-RNA levels; of that population, 49% had a posttransplantation virologic response, with the most frequent side effects reported by subjects being fatigue (38%), headache (23%), and anemia (21%). Of the 43 applicable subjects, 30 (70% of the population) had a posttransplantation virologic response at 12 weeks, 10 (23%) had recurrent infection, and 3 (7%) died.
“This study provides proof of concept that virologic suppression without interferon significantly can reduce the rate of recurrent HCV after liver transplantation,” the study says, adding that the results “compare favorably with those observed in other trials of pretransplantation antiviral therapy.”
In the second study, the authors ascertained that combination therapy consisting of sofosbuvir and ribavirin for 24 weeks is effective at preventing hepatitis C virus recurrence in patients who undergo liver transplantations.
“Recurrent HCV infection is the most common cause of mortality and graft loss following transplantation, and up to 30% of patients with recurrent infection develop cirrhosis within 5 years,” wrote the study’s authors, led by Dr. Michael Charlton of the Mayo Clinic in Rochester, Minn.
Using a prospective, multicenter, open-label pilot study, investigators enrolled and treated 40 patients with a 24-week regimen of 400 mg sofosbuvir and ribavirin starting at 400 mg, which was subsequently adjusted per patient based on individual creatinine clearance and hemoglobin levels. Subjects were 78% male and 85% white, with 83% having HCV genotype 1, 40% having cirrhosis, and 88% having been previously treated with interferon. The primary outcome investigators looked for was “sustained virologic response 12 weeks after treatment (SVR12).”
Data showed that SVR12 was achieved by 28 of the 40 subjects that received treatment, or 70%. The most commonly reported adverse effects were fatigue (30%), diarrhea (28%), headache (25%), and anemia (20%). No patients exhibited detectable viral resistance during or after treatment, and although two patients terminated their treatment because of adverse events, investigators reported no deaths, graft losses, or episodes of rejection.
“In contrast,” Dr. Charlton and his coauthors noted, “interferon-based treatments have been associated with posttreatment immunological dysfunction (particularly plasma cell hepatitis) and even hepatic decompensation in LT [liver transplant] recipients.”
The authors of the first study disclosed that Dr. Curry has received grants from and been affiliated with Gilead, which was a sponsor of the study. The authors of the second study reported no relevant financial disclosures.
FROM GASTROENTEROLOGY
Selecting antithrombotic therapy for patients with atrial fibrillation
Antithrombotic therapy reduces the risk of systemic embolism in patients with atrial fibrillation, but one approach does not suit all patients. The decision whether to start this therapy—and which agent to use—must take into account the patient’s risk of thromboembolism as well as bleeding.
Antithrombotic therapy encompasses antiplatelet drugs such as aspirin and clopidogrel and anticoagulants such as warfarin and the target-specific oral anticoagulants (TSOACs). Oral anticoagulation is more effective than antiplatelet therapy and is preferred in all but those at lowest risk, in whom either antiplatelet therapy or no therapy is deemed adequate.
Patients with valvular atrial fibrillation, specifically those who have rheumatic mitral stenosis or a prosthetic heart valve, are at significantly higher risk of systemic embolization. Their overall risk-benefit profile is nearly always in favor of anticoagulation. But the same is not necessarily true for patients with nonvalvular atrial fibrillation.
The following discussion sets forth our rationale for clinical decision-making, based on recommendations in the 2014 guidelines from the American Heart Association, American College of Cardiology, and Heart Rhythm Society.1 The second half of this review outlines the oral anticoagulants currently available.
ONE IN FOUR PEOPLE
Atrial fibrillation is common, with an incidence that increases with age. It affects more than 10% of people over age 80 and is often associated with cardiovascular disease.2 Based on Framingham Heart Study data, a person’s lifetime risk of developing it is about 25%.3
FIVEFOLD RISK OF STROKE
The most serious complication of atrial fibrillation is arterial thromboembolism, of which ischemic stroke is the most common and most feared manifestation. The risk of stroke is five times higher than normal in patients with atrial fibrillation.3 More than 15% of strokes may be attributable to atrial fibrillation, and the proportion increases with age.4
The risk of thromboembolism appears to be similar in patients with clinically manifest atrial fibrillation irrespective of the type (paroxysmal, persistent, or permanent). The Stroke Prevention in Atrial Fibrillation (SPAF) study5 and the Atrial Fibrillation Clopidogrel Trial With Irbesartan for Prevention of Vascular Events (ACTIVE W)6 showed that patients who had paroxysmal atrial fibrillation and at least one risk factor for thromboembolism had stroke rates comparable to those of their counterparts who had persistent and permanent atrial fibrillation.
Subclinical atrial fibrillation may be an important cause of stroke. Clinically silent episodes can be detected by implantable electronic devices, which record episodes of atrial tachyarrhythmia (atrial high-rate events). Subclinical episodes have been detected in 10% to 28% of monitored patients who did not have a history of atrial fibrillation.7,8 Patients who have atrial high-rate events detected by implantable devices have a higher risk of future clinically manifest atrial fibrillation, thromboembolic events, or both.7–9 Yet characteristics of atrial high-rate episodes that predict risk are not well defined and warrant further investigation.
CLINICAL RISK FACTORS FOR STROKE

To date, thousands of patients with nonvalvular atrial fibrillation have participated in randomized clinical trials of stroke prevention. The placebo groups from these trials provide a sizable database for retrospectively identifying clinical characteristics associated with thromboembolism. The Atrial Fibrillation Investigators10 pooled data from five large trials and found that risk factors consistently associated with stroke in multivariate analysis included diabetes mellitus, hypertension, prior systemic embolism, and advanced age.
Though the risk of stroke increases with age with no lower limit, most studies identify age 65 as a threshold, with further escalating risk after age 75. Moreover, women were observed to be at higher risk in some but not all studies. These risk factors have become components of commonly used risk-stratification schemes.
Hypertrophic cardiomyopathy. Maron et al11 reported that atrial fibrillation in patients with hypertrophic cardiomyopathy was independently associated with thromboembolism. In 900 patients with hypertrophic cardiomyopathy, the prevalence of systemic embolism was 6%. Patients with hypertrophic cardiomyopathy and a thromboembolic complication were seven times more likely to have atrial fibrillation than matched counterparts free of thromboembolism. A notable subset of patients experienced a stroke or embolic event before age 50, and the authors advised that the risk of thromboembolism should be considered in patients of any age with hypertrophic cardiomyopathy and atrial fibrillation.
Olivotto et al12 similarly found patients with hypertrophic cardiomyopathy and atrial fibrillation to be at significantly greater risk of stroke (odds ratio [OR] 17.7, 95% confidence interval [CI] 4.1–75.9, P < .001).
Chronic kidney disease is also associated with a higher risk of thromboembolism in patients with atrial fibrillation. A glomerular filtration rate of 60 mL/min or less is independently and inversely predictive of risk.13,14
While patients with end-stage renal disease have been largely excluded from stroke prevention trials, Vázquez et al15 prospectively followed 190 dialysis patients for 12 months. In multivariate analysis, compared with matched controls without documented atrial fibrillation, patients receiving renal replacement therapy and having any form of atrial fibrillation were eight times more likely to have systemic embolization.
IMAGING-BASED RISK FACTORS
In addition to clinical factors, several imaging-based factors have been associated with stroke risk in patients with atrial fibrillation.
Complex aortic atheroma or markers of blood stasis within the left atrium, such as reduced left atrial appendage emptying flow (< 20 cm/second), dense spontaneous echo contrast, or left atrial appendage thrombus, seen on transesophageal echocardiography, were independently associated with increased systemic embolic risk in the third SPAF substudy.16 Moreover, multivariate analysis of SPAF data found both left ventricular dysfunction of any severity and increased left atrial size (diameter corrected for body surface area by M-mode > 2.5 cm/m2) to be independent predictors of thromboembolism.17
Although enlargement of the left atrium has not been incorporated into traditional risk stratification schemes, data from Osranek et al18 further implicate it as a marker of risk. The cohort was small (N = 46), but consisted of patients with lone atrial fibrillation followed for nearly 30 years. Patients with normal left atrial size enjoyed a benign course, while those with left atrial enlargement (> 32 mL/m2) at diagnosis or later during follow-up had significantly worse event-free survival (hazard ratio [HR] 4.46, 95% CI 1.56–12.74, P < .01). All embolic strokes occurred in the group with left atrial enlargement.
RISK STRATIFICATION SCHEMES
Several models for predicting systemic embolism risk in patients with nonvalvular atrial fibrillation have been proposed and validated.
The CHADS2 score has been the most widely applied, being simple to use.19,20 It assigns 1 point each for Congestive heart failure, Hypertension, Age 75 or older, and Diabetes, and 2 points for prior Stroke or systemic thromboembolism.
In patients with chronic nonvalvular atrial fibrillation, Gage et al19 reported that the stroke rate was lowest in those with a score of 0, with an annual adjusted stroke rate of 1.9% per year, and highest in those with the maximal possible score (ie, 6), with a rate of 18.2%. The rate increased by a factor of 1.5 with each point in the CHADS2 score.
CHA2DS2-VASc. Endorsed for use in both the American and European guidelines,1,21 CHA2DS2-VASc is an extension of CHADS2. Points are assigned as follows:
- Congestive heart failure or left ventricular dysfunction (moderate to severe left ventricular dysfunction or recent heart failure exacerbation requiring hospitalization irrespective of ejection fraction): 1 point
- Hypertension: 1 point
- Age ≥ 75: 2 points; age 65–74: 1 point
- Diabetes mellitus: 1 point
- Stroke, transient ischemic attack, or thromboembolism: 2 points
- Vascular disease (prior myocardial infarction, peripheral arterial disease, or aortic plaque): 1 point
- Sex, female: 1 point
- Maximum score: 9 points.
Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk. Lip et al22 found that, in untreated patients with nonvalvular atrial fibrillation, rates of stroke ranged from 0 with a score of 0 to 15.2% per year with a score of 9 points.
In a large cohort with over 11,000 patient-years of follow-up, 98% of the thromboembolic events occurred in people with a CHA2DS2-VASc score of 2 or more. Moreover, more than 99% of patients with a score of less than 2 were free of stroke and thromboembolism.23
Compared with the CHADS2 score, CHA2DS2-VASc has superior negative predictive power. Of 1,084 patients from the European Heart Survey for Atrial Fibrillation, the newer scheme classified significantly fewer patients as being at either low risk (score of 0; 9% vs 20%) or intermediate risk (score of 1; 15% vs 35%).23 Though the overall rate of stroke was low, those categorized as being at low or intermediate risk by CHA2DS2-VASc had significantly fewer thromboembolic events than their counterparts according to CHADS2 (0.6% vs 3.3%).
Olesen et al24 similarly showed that in patients with a CHADS2 score of 0, reclassification by CHA2DS2-VASc yielded a range of annual stroke rates from 0.84% with a score of 0 up to 3.2% with a score of 3.
RISK-BASED ANTITHROMBOTIC THERAPY IN NONVALVULAR ATRIAL FIBRILLATION
The 2014 atrial fibrillation guidelines1 state that the decision to give antithrombotic therapy for atrial fibrillation should be individualized, based on the absolute and relative risks of stroke and bleeding, and ought to take into consideration the patient’s preferences. For patients with nonvalvular atrial fibrillation, selection of antithrombotic therapy should take into account the risk of thromboembolism determined by the CHA2DS2-VASc score and be irrespective of the pattern of atrial fibrillation (paroxysmal, persistent, or permanent). Antithrombotic therapy is similarly recommended for patients with atrial flutter, according to the same risk profile used for atrial fibrillation.
Studies have consistently shown24–27 that the risk of ischemic stroke without anticoagulation exceeds the risk of intracranial bleeding with anticoagulation in nearly all patients except those at lowest risk of thromboembolism. The CHA2DS2-VASc score better identified those at truly low risk, in whom treatment may offer more risk than benefit.24–27
The HAS-BLED score28 assigns points as follows:
- Hypertension (systolic blood pressure > 160 mm Hg): 1 point
- Abnormal renal function (dialysis, renal transplantation, or serum creatinine > 2.6 mg/mL) or liver function (cirrhosis, bilirubin more than two times the upper limit, or aminotransferase levels more than three times the upper limit): 1 or 2 points
- Stroke: 1 point
- Bleeding (prior major bleeding event or predisposition to bleeding): 1 point
- Labile international normalized ratio (INR) (supratherapeutic or time in therapeutic range < 60%): 1 point
- Elderly (age > 65): 1 point
- Drugs (antiplatelet, nonsteroidal anti-inflammatory) or alcohol (more than eight drinks per week): 1 or 2 points
- Maximum total: 9 points.
HAS-BLED is a practical and validated approach for estimating bleeding risk and is mentioned in the guidelines, but it is not recommended for use in guiding decisions about antithrombotic therapy. Specifically, it should not be used to exclude patients, but rather to identify those at high risk (score ≥ 3) who may require closer observation and more attentive monitoring of the INR.
ANTITHROMBOTIC THERAPY
Antithrombotic agents available for use in the United States include antiplatelet drugs (eg, aspirin and clopidogrel) and anticoagulants (unfractionated heparin and low-molecular-weight heparin, vitamin K antagonists such as warfarin, and direct thrombin and factor Xa inhibitors). Anticoagulation has been shown in randomized controlled trials to be superior to both placebo and antiplatelet agents used either alone or in combination.29
Aspirin has been downgraded
Aspirin has been compared with placebo in seven randomized controlled trials. Only the original SPAF study, in which aspirin 325 mg/day was used, found that it was beneficial. This result alone accounted for the 19% reduction in relative risk (95% CI 1%–35%, P < .05) in a meta-analysis performed by Hart et al.29 Even when combined with clopidogrel 75 mg/day, aspirin 75 to 100 mg/day is still inferior to warfarin.5 While dual antiplatelet therapy resulted in a 28% relative reduction in thromboembolism (95% CI 17%–38%, P < .01) compared with aspirin alone, major bleeding significantly increased by 57% (95% CI 29%–92%, P < .01).
Although aspirin may be beneficial, differences among patients may influence its efficacy. It may be more effective in preventing noncardioembolic stroke, particularly in diabetic and hypertensive patients.30,31 To date, aspirin has not been shown to be beneficial in low-risk populations.
The 2014 guidelines downgraded the recommendation for aspirin therapy. For patients at low risk and for some at intermediate risk, it is permissible to forgo therapy altogether, including aspirin.1
ORAL ANTICOAGULANTS
The rest of this paper reviews the oral anticoagulants that are approved for reducing the risk of thromboembolism in atrial fibrillation, focusing on each agent’s mechanism of action, pharmacokinetics, clinical efficacy, and safety.
WARFARIN, A VITAMIN K ANTAGONIST
Warfarin inhibits synthesis of vitamin K-dependent clotting factors (ie, factors II, VII, IX, and X) and proteins C and S by inhibiting the C1 subunit of vitamin K epoxide reductase, thereby interfering with production of vitamin K1 epoxide and consequent regeneration of vitamin K.
Pharmacokinetics. Warfarin is nearly completely absorbed after oral administration. Its anticoagulant effect can be seen within 24 hours of administration, but its peak effect is typically apparent only after 72 hours. Elimination occurs predominantly through metabolism by cytochrome P450 enzymes, principally CYP2C9. Its effective half-life ranges from 20 to 60 hours, with a mean of 40 hours.32
Warfarin’s effect, dosage, and bleeding risk are influenced by multiple factors, including vitamin K-containing foods such as green leafy vegetables, medications that either inhibit or induce hepatic cytochrome P450 enzymes, and polymorphisms in the VKORC1 and CYP2C9 genes.32
Reversal. Warfarin’s anticoagulant effect is reversed with vitamin K, but this reversal may not become apparent for 6 to 24 hours. In contrast, fresh-frozen plasma and prothrombin protein concentrate, which contain clotting factors, reverse warfarin immediately. Currently, a three-factor prothrombin protein concentrate (factors II, IX, and X) and a four-factor concentrate (factors II, VII, IX, and X plus proteins C and S) are available in the United States. Although prothrombin protein concentrate works rapidly and has a lower volume of administration, available data do not indicate it is clinically superior to fresh-frozen plasma.33,34 The ongoing randomized PROTECT trial (NCT00618098), comparing fresh-frozen plasma and four-factor prothrombin protein concentrate for reversal of vitamin K antagonist therapy, may provide further insight.
Efficacy and safety. Randomized controlled trials in patients with nonvalvular atrial fibrillation have shown that warfarin (in doses adjusted to maintain an INR greater than 2) is highly efficacious in preventing systemic embolism, with a relative risk reduction of 61% (95% CI 47%–71%, P < .05) compared with placebo.29,35 An INR of 2 to 3 is recommended for patients with nonvalvular atrial fibrillation, and those with atrial fibrillation and either a bioprosthetic valve or rheumatic heart disease. In contrast, an INR of 2.5 to 3.5 is recommended for patients with atrial fibrillation and mechanical valves in the aortic or mitral positions.1,36
Stroke prevention with warfarin is most effective when the achieved mean time in the therapeutic range is at least 70%. The risk of intracranial hemorrhage increases significantly at INRs higher than 3. An INR of 2 to 3 offers maximum protection with minimal risk of bleeding.37,38 Systematic follow-up of patients through anticoagulation clinics produces better compliance and control and is encouraged.
TARGET-SPECIFIC ORAL ANTICOAGULANTS
Although effective, warfarin requires frequent monitoring and dosage adjustment, has a delayed onset and protracted offset, and interacts with commonly consumed vitamin K–containing foods and frequently used drugs. These drawbacks prompted evaluation of existing antiplatelet agents, in combination or in conjunction with lower adjusted or fixed-dose warfarin. These regimens proved inferior,39–42 spurring interest in developing alternative oral anticoagulants.
TSOACs act by directly inhibiting thrombin (factor IIa) or by reducing thrombin production indirectly by inhibiting factor Xa. Three TSOACs are approved. Each was compared with adjusted-dose warfarin in randomized controlled trials.
Dabigatran
Dabigatran etexilate was the first TSOAC approved in the United States.
Pharmacokinetics. Dabigatran etexilate has a bioavailability of 3% to 7% after oral administration. Its absorption is enhanced in an acidic gastric environment and is limited by P-glycoprotein-facilitated transport out of enterocytes. Dabigatran etexilate is hydrolyzed to its active metabolite dabigatran, which directly inhibits thrombin. Maximal plasma drug concentration and peak anticoagulant effect are achieved within 0.5 to 2 hours after administration.
Dabigatran is predominantly excreted by the kidneys, and has a half-life of 12 to 17 hours in patients with normal renal function. The half-life extends to 27 hours in those with moderately severe renal impairment (creatinine clearance 15–30 mL/min). The recommended dose of 150 mg twice daily should be reduced to 75 mg twice daily in patients with a creatinine clearance of 15 to 30 mL/min. This drug is contraindicated in patients with a creatinine clearance less than 15 mL/min.43,44
Efficacy. The Randomized Evaluation of Long-term Anticoagulation Therapy (RE-LY) trial45 randomly assigned 18,113 patients with nonvalvular atrial fibrillation at risk of thromboembolism (mean CHADS2 score 2.1) to receive either dabigatran (either 150 mg twice daily or 110 mg twice daily) or warfarin (adjusted to an INR of 2.0 to 3.0). Of note, the lower approved dose of dabigatran (75 mg twice daily) was not tested in RE-LY.
At 2 years, higher-dose dabigatran was significantly more effective than both warfarin (RR 0.65, 95% CI 0.52–0.81, P < .05) and lower-dose dabigatran (RR 0.73, 95% CI 0.58–0.91, P < .05) in reducing the rate of systemic embolic events.
The risk of combined major bleeding events was no different with higher-dose dabigatran than with warfarin (RR 0.93, 95% CI 0.81–1.07, P < .05), but the rate of hemorrhagic stroke was significantly less with dabigatran than with warfarin (RR 0.26, 95% CI 0.14–0.49, P < .05). Higher rates of major gastrointestinal bleeding and dyspepsia occurred with dabigatran.
Concern about the safety of dabigatran was raised when post hoc evaluation of RE-LY found a higher incidence of myocardial infarction with dabigatran than with warfarin (RR 1.38, 95% CI 1–1.91, P = .048).46 Corroborating data were reported by Uchino and Hernandez,47 comparing dabigatran with either warfarin or low-molecular-weight heparin. However, without directly comparing dabigatran and placebo, it is unclear whether the small increase in myocardial infarction reflects a direct effect of dabigatran or absence of a protective effect of warfarin or low-molecular-weight heparin.
Rivaroxaban
Rivaroxaban is a direct factor Xa inhibitor that blocks the amplified burst of thrombin production and in turn inhibits platelet aggregation and thrombus formation.
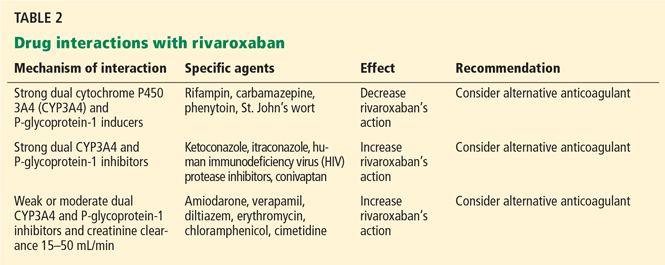
Pharmacokinetics. Rivaroxaban’s oral bioavailability is 80% to 100% after a single 15- or 20-mg dose taken with food. Its maximal anticoagulant effect is achieved within 2 hours. Two-thirds of the active drug is metabolized by either CYP450-dependent (CYP3A4, 2J2) or CYP-independent mechanisms; the inactive drug is then excreted in the urine and feces. The remaining, active drug is removed by the kidneys using the P-glycoprotein transporter.
The half-life of rivaroxaban is 5 to 9 hours. The recommended dosage of 20 mg daily should be reduced to 15 mg daily if the creatinine clearance rate is 30 to 50 mL/min, or to 10 mg if the creatinine clearance rate is 15 to 30 mL/min. Rivaroxaban is contraindicated in patients whose creatinine clearance rate is less than 15 mL/min.48–52
Efficacy and safety. In the Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET-AF),53 14,264 at-risk patients with nonvalvular atrial fibrillation (mean CHADS2 score 3.5) were randomly assigned to receive either rivaroxaban 20 mg daily (or 15 mg daily if their creatinine clearance was 30–49 mL/min; the lowest dose of rivaroxaban, 10 mg, was not studied in this trial) or warfarin (target INR 2.0–3.0). Outcomes with rivaroxaban compared with warfarin:
- Systemic embolism:
HR 0.79, 95% CI 0.66–0.96, P < .01, noninferiority - Total bleeding: no difference
- Intracranial bleeding:
HR 0.67, 95% CI 0.47–0.93, P = .02 - Fatal bleeding:
HR 0.50, 95% CI 0.31–0.79, P = .003 - Major gastrointestinal bleeding:
3.2% vs 2.2%, P < .001.
Apixaban
Apixaban is also a direct factor Xa inhibitor.
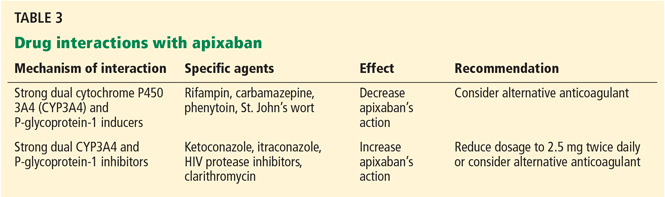
Pharmacokinetics. Apixaban’s oral bioavailability is 50%, with maximal blood concentration achieved at 3 to 4 hours. One-quarter of the drug is metabolized via CYP3A4. The remaining active drug is excreted by the kidneys and biliary/intestinal system via the P-glycoprotein transporter. Apixaban’s half-life is 9 to 14 hours.
The recommended dosage is 5 mg twice daily, but it should be reduced to 2.5 mg twice daily if at least two of the following characteristics are present: age 80 or older, weight 60 kg or less, and serum creatinine 1.5 mg/dL or more.54,55
Efficacy and safety. The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial56 enrolled 18,201 patients with nonvalvular atrial fibrillation (mean CHADS2 score 2.1) randomly assigned to receive either apixaban (5 mg twice daily with dosage reduction to 2.5 mg twice daily as noted above) or warfarin (target INR 2.0–3.0).
Compared with warfarin, apixaban was associated with lower risk of:
- Systemic embolism
(HR 0.79, 95% CI 0.66–0.95, P = .01) - Major bleeding
(HR 0.69, 95% CI 0.60–0.80, P < .001) - Intracranial hemorrhage
(HR 0.42, 95% CI 0.30–0.58, P < .001) - All-cause mortality
(HR 0.89 95% CI 0.80–0.99, P = .047).
Drug interactions with the novel oral anticoagulants
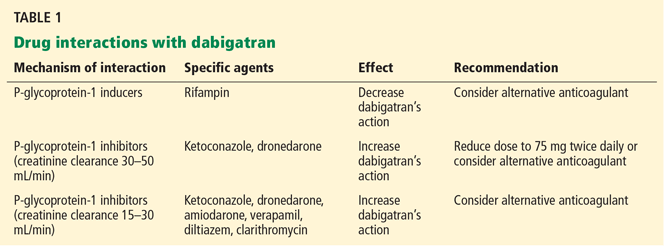
TSOACs were developed with the intent to avoid many of the shortcomings of warfarin. Each has a broader therapeutic window and a rapid onset of action, enabling fixed dosing without need for serial monitoring. Compared with warfarin, they have significantly fewer dietary and drug interactions.
Nonetheless, drug interactions do exist and are important to recognize (Tables 1–3). These primarily result from inhibition or induction of cytochrome P450 enzyme activity or P-glycoprotein transporter action, involved in metabolism and elimination of active drug.
Reversibility of the target-specific oral anticoagulants
While the anticoagulant effects of warfarin can be reversed by vitamin K, fresh-frozen plasma, and prothrombin complex concentrate, TSOACs have no currently approved antidotes. Management of bleeding due to these agents was recently reviewed in this journal by Fawole et al.57
Several nonspecific hemostatic agents have been suggested, including recombinant factor VIIa or prothrombin complex concentrates. The anticoagulant effect of rivaroxaban has been shown to be reversed by prothrombin complex concentrate in vitro; clinical effect has not been demonstrated.58 PRT06445 (andexanet alfa), a recombinant protein antidote specific for factor Xa inhibitors, has entered clinical studies, with a phase 2 trial reporting high reversing capability for apixaban.59
Unlike rivaroxaban and apixaban, which are highly bound to plasma protein, dabigatran can be effectively removed with hemodialysis. Liesenfeld et al60 showed that longer dialysis duration was the most relevant variable for reducing dabigatran plasma levels. Current clinical experience is limited, and standard recommendations and formal guidance are lacking.
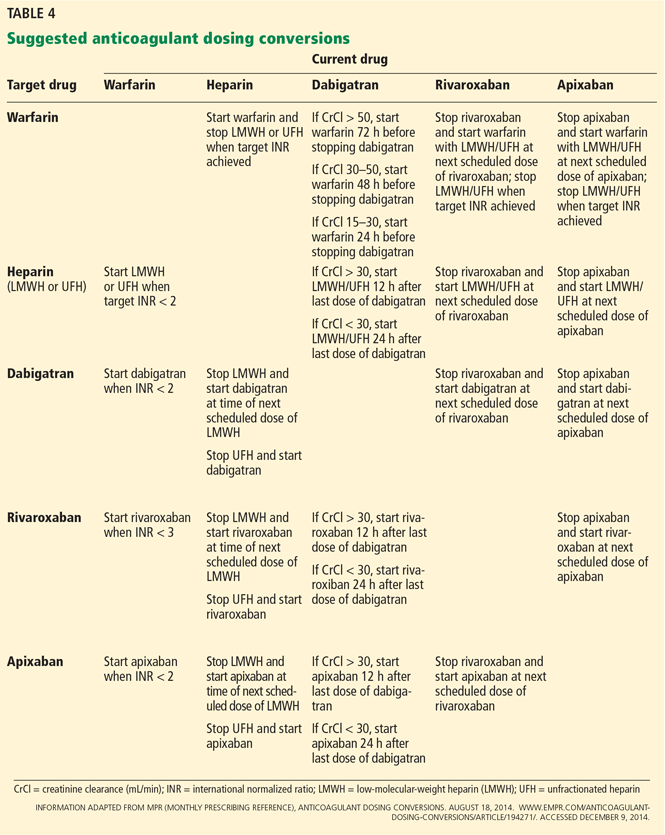
Switching oral anticoagulants
Suggested approaches for switching between anticoagulants are listed in Table 4.61
CHOOSING ANTITHROMBOTIC THERAPY
In valvular atrial fibrillation: warfarin
Anticoagulation with warfarin is advised for valvular atrial fibrillation. Patients with bioprosthetic heart valves or rheumatic valvular disease were not evaluated in randomized controlled trials of TSOACs. Dabigatran in particular is contraindicated in patients with mechanical heart valves, as the Randomized, Phase II Study to Evaluate the Safety and Pharmacokinetics of Oral Dabigatran Etexilate in Patients After Heart Valve Replacement (RE-ALIGN)62 found higher rates of stroke, valve-related thrombosis, and myocardial infarction in patients receiving dabigatran.
In nonvalvular atrial fibrillation
According to the 2014 guidelines,1 oral anticoagulation is preferred in all patients with nonvalvular atrial fibrillation but those at lowest risk (CHA2DS2-VASc = 0).
Experience with TSOACs is lacking in patients with end-stage kidney disease (creatinine clearance < 15 mL/min), and warfarin is advised in this group.
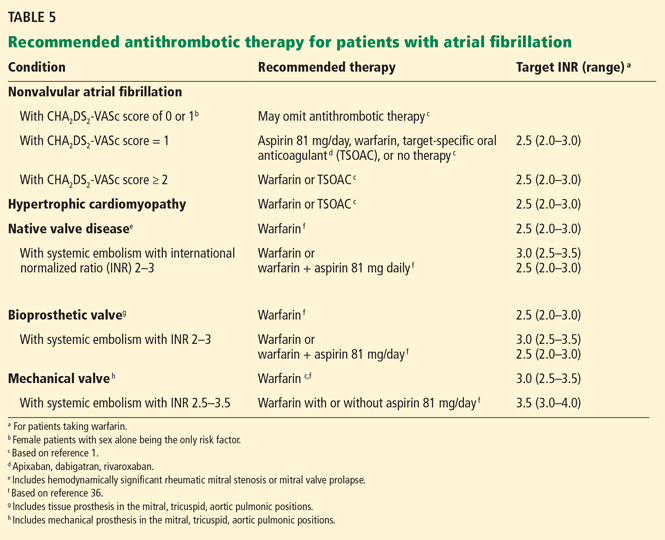
TSOACs are recommended in patients with nonvalvular atrial fibrillation in whom therapeutic INR levels cannot be maintained with warfarin. For most patients with nonvalvular atrial fibrillation, TSOACs are an option equivalent to warfarin. Anticoagulant choice is largely driven by dosing convenience, out-of-pocket cost for treatment with a TSOAC, and ready availability of antidotes for warfarin in case of bleeding (Tables 5 and 6).
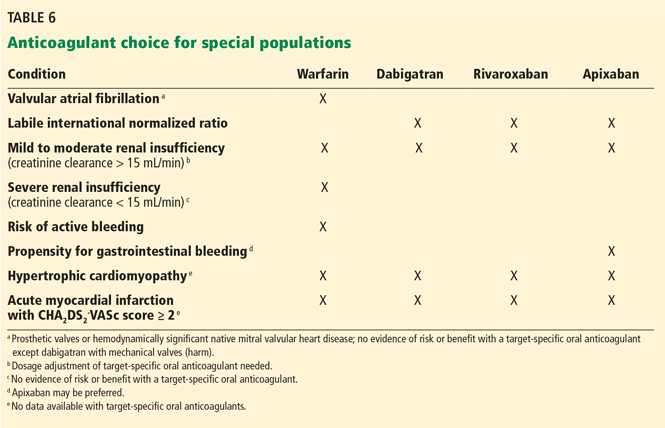
In patients with nonvalvular atrial fibrillation, TSOACs are as effective as warfarin in preventing systemic thromboembolism, and some of them have been shown to be superior in terms of lower rates of ischemic stroke (dabigatran), systemic embolism (apixaban), and mortality (apixaban; trend for dabigatran). All TSOACs demonstrate modestly favorable bleeding risk profiles compared with warfarin, with lower risk of intracranial hemorrhage. Potential differences in efficacy and safety among TSOACs are unknown since there have been no randomized direct comparisons between them. A summary of landmark trial results and assessment of the advantages and disadvantages of each are listed in Table 7.
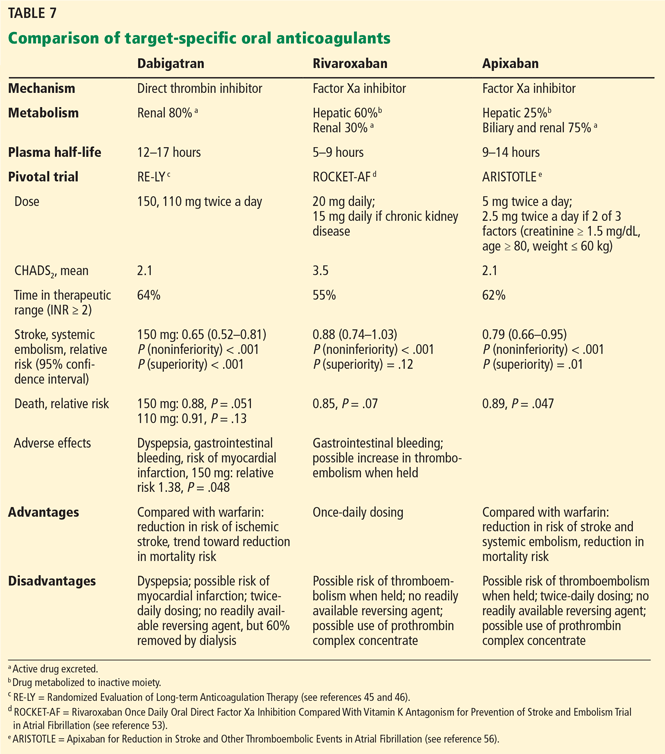
Two groups of patients with nonvalvular atrial fibrillation warrant special consideration:
Patients with hypertrophic cardiomyopathy. There are no randomized controlled trials of anticoagulation therapy in patients with hypertrophic cardiomyopathy; however, because of their high risk of thromboembolism, anticoagulation is indicated irrespective of the CHA2DS2-VASc score. TSOACs are an option as an alternative to warfarin.
Patients with coronary artery disease and an indication for antiplatelet therapy. In this group the decision for concurrent anticoagulation is guided by the CHA2DS2-VASc score. For patients who have intracoronary stents, dual antiplatelet therapy is the standard of care for reducing risk of cardiovascular events after stent implantation.63 When triple therapy (ie, two antiplatelet drugs and an anticoagulant) is indicated, such as after intracoronary stent placement, the guidelines suggest trying to minimize the duration of triple therapy. For instance, a bare-metal stent may be preferred. Alternatively, after coronary revascularization, it may be reasonable to use clopidogrel 75 mg daily with an oral anticoagulant and to omit aspirin.
Interrupting and bridging anticoagulation
Patients with atrial fibrillation often require suspension of anticoagulation, most commonly before an elective invasive procedure. The duration of interruption, timing of resumption, and need for bridging anticoagulation are guided by clinical judgment, which considers risk of thromboembolism and severity of procedure-related bleeding risk.
In general, if therapy needs to be interrupted, it should be restarted as soon as possible. Short-term interruption does not seem to be associated with clinically significant risk of thromboembolic events, whereas postoperative heparin bridging therapy increases the risk of hematoma with implantation of a cardiac electronic device.64,65
To date, evidence is lacking to advise upon periprocedure bridging anticoagulation. The Bridging Anticoagulation in Patients Who Require Temporary Interruption of Warfarin Therapy for an Elective Invasive Procedure or Surgery (BRIDGE) study (NCT00786474)— enrolling chronically anticoagulated patients undergoing an invasive procedure to randomly receive placebo or bridging low-molecular-weight heparin—may provide guidance.
Currently, it is common practice in low-risk patients undergoing an invasive procedure with significant bleeding risk to interrupt anticoagulation for up to 1 week without bridging. Warfarin is typically held 3 to 5 days, while TSOACs are held for 24 hours if renal function is preserved or up to 2 to 3 days if renal function is severely impaired (creatinine clearance 15–30 mL/min). If complete hemostasis is necessary, it could be confirmed by a normalized INR (for warfarin), activated partial thromboplastin time (dabigatran), or prothrombin time (apixaban or rivaroxaban).
For patients at high risk (valvular atrial fibrillation or CHA2DS2-VASc ≥ 2), bridging with unfractionated heparin or low-molecular-weight heparin during periods of subtherapeutic anticoagulation is common. Alternatively, it is becoming increasingly common to perform cardiac electronic device implantation, catheter ablation, and coronary angiography and intervention without interrupting anticoagulation.66–72
Recently, concern has been raised over a possible increase in thromboembolism upon discontinuation of rivaroxaban and apixaban. ROCKET-AF reported a spike in thrombotic events in the rivaroxaban-treated group at the end of the trial (HR 1.50, 95% CI 1.05–2.15, P = .026). This raised concern for a possible “rebound” effect upon drug cessation. Yet a post hoc analysis of ROCKET-AF demonstrated that events clustered in the rivaroxaban-treated cohort who completed the study and were transitioning to open-label warfarin, and this alone accounted for the rise in stroke occurrence. In contrast, there was no increase in the cohort of patients treated with rivaroxaban who either temporarily interrupted or permanently discontinued the drug.73 The authors concluded that increased stroke was the consequence of transiently interrupted anticoagulation, rather than a rebound prothrombotic effect. Similar results were reported in ARISTOTLE.
Another possibility is that, during the transition to warfarin therapy, transient hypercoagulability could be a function of warfarin. Azoulay et al74 observed in a large cohort that warfarin was associated with a 71% increased risk of stroke in the first 30 days after initiation, compared with decreased risk thereafter. Nevertheless, there is now a black- box warning recommendation for all three TSOACs that if discontinuation is required for a reason other than pathological bleeding, bridging with another anticoagulant should at least be considered.
The perioperative management of the TSOACs was recently reviewed in this journal by Anderson et al.75
WEIGHING THE RISKS OF STROKE AND BLEEDING
Stroke is the most feared complication in patients with atrial fibrillation. Risk reduction is an important goal in management, yet decisions for individuals must take into account both stroke and bleeding risks related to antithrombotic therapy.
The 2014 guidelines1 differ from past versions. First, they endorse the use of CHA2DS2-VASc for categorizing stroke risk in patients with nonvalvular atrial fibrillation. This in turn guides antithrombotic therapy. This scheme effectively identifies patients at very low risk of stroke (men with a score of 0, women with a score of 0 or 1), in whom it is reasonable to omit antithrombotic therapy. For all patients with valvular heart disease or hypertrophic cardiomyopathy, unless bleeding risk is prohibitive, anticoagulation is recommended irrespective of the CHA2DS2-VASc score. Second, they incorporate the TSOACs, which offer convenience and improved safety in select patients.
While the guidelines mention the potential relevance of subclinical atrial tachyarrhythmias as they pertain to stroke risk, there is no specific recommendation as to their management. We do take into consideration the finding of atrial high-rate events (≥ 180 bpm, ≥ 6 minutes in duration) diagnostically confirmed by cardiac implantable electronic devices or telemetric monitoring, particularly in patients with a clinical profile of high stroke risk. In addition, atriopathy with increased left atrial size and renal insufficiency, as discussed in this review, appear to correlate with greater risk of thromboembolism, yet neither is a component of the stroke risk scheme endorsed by the guidelines.
Other risk factors, some unknown to us, undoubtedly exist. Again, our empiric judgment is to at least consider these nontraditional risk factors while guided primarily by the CHA2DS2-VASc score when assessing stroke risk in patients with atrial fibrillation.
The goal in managing patients with atrial fibrillation is to balance thromboembolic risk reduction with the risk of bleeding associated with antithrombotic therapy.
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med 1982; 306:1018–1022.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Hart RG, Pearce LA, Rothbart RM, McAnulty JH, Asinger RW, Halperin JL. Stroke with intermittent atrial fibrillation: incidence and predictors during aspirin therapy. Stroke Prevention in Atrial Fibrillation Investigators. J Am Coll Cardiol 2000; 35:183–187.
- Hohnloser SH, Pajitnev D, Pogue J, et al; ACTIVE W Investigators. Incidence of stroke in paroxysmal versus sustained atrial fibrillation in patients taking oral anticoagulation or combined antiplatelet therapy: an ACTIVE W Substudy. J Am Coll Cardiol 2007; 50:2156–2161.
- Healey JS, Connolly SJ, Gold MR, et al; ASSERT Investigators. Subclinical atrial fibrillation and the risk of stroke. N Engl J Med 2012; 366:120–129.
- Glotzer TV, Hellkamp AS, Zimmerman J, et al; MOST Investigators. Atrial high rate episodes detected by pacemaker diagnostics predict death and stroke: report of the Atrial Diagnostics Ancillary Study of the MOde Selection Trial (MOST). Circulation 2003; 107:1614–1619.
- Glotzer TV, Daoud EG, Wyse DG, et al. The relationship between daily atrial tachyarrhythmia burden from implantable device diagnostics and stroke risk: the TRENDS study. Circ Arrhythm Electrophysiol 2009; 2:474–480.
- Atrial Fibrillation Investigators. Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Maron BJ, Olivotto I, Bellone P, et al. Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:301–307.
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104:2517–2524.
- Go AS, Fang MC, Udaltsova N, et al; ATRIA Study Investigators. Impact of proteinuria and glomerular filtration rate on risk of thromboembolism in atrial fibrillation: the anticoagulation and risk factors in atrial fibrillation (ATRIA) study. Circulation 2009; 119:1363–1369.
- Hart RG, Pearce LA, Asinger RW, Herzog CA. Warfarin in atrial fibrillation patients with moderate chronic kidney disease. Clin J Am Soc Nephrol 2011; 6:2599–2604.
- Vázquez E, Sánchez-Perales C, Borrego F, et al. Influence of atrial fibrillation on the morbido-mortality of patients on hemodialysis. Am Heart J 2000; 140:886–890.
- The Stroke Prevention in Atrial Fibrillation Investigators Committee on Echocardiography. Transesophageal echocardiographic correlates of thromboembolism in high-risk patients with nonvalvular atrial fibrillation. Ann Intern Med 1998; 128:639–647.
- The Stroke Prevention in Atrial Fibrillation Investigators. Predictors of thromboembolism in atrial fibrillation: II. Echocardiographic features of patients at risk. Ann Intern Med 1992; 116:6–12.
- Osranek M, Bursi F, Bailey KR, et al. Left atrial volume predicts cardiovascular events in patients originally diagnosed with lone atrial fibrillation: three-decade follow-up. Eur Heart J 2005; 26:2556–2561.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- van Walraven C, Hart RG, Wells GA, et al. A clinical prediction rule to identify patients with atrial fibrillation and a low risk for stroke while taking aspirin. Arch Intern Med 2003; 163:936–943.
- Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J 2012; 33:2719–2747.
- Lip GY, Frison L, Halperin JL, Lane DA. Identifying patients at high risk for stroke despite anticoagulation: a comparison of contemporary stroke risk stratification schemes in an anticoagulated atrial fibrillation cohort. Stroke 2010; 41:2731–2738.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- Olesen JB, Torp-Pedersen C, Hansen ML, Lip GY. The value of the CHA2DS2-VASc score for refining stroke risk stratification in patients with atrial fibrillation with a CHADS2 score 0-1: a nationwide cohort study. Thromb Haemost 2012; 107:1172–1179.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Olesen JB, Lip GY, Lindhardsen J, et al. Risks of thromboembolism and bleeding with thromboprophylaxis in patients with atrial fibrillation: a net clinical benefit analysis using a ‘real world’ nationwide cohort study. Thromb Haemost 2011; 106:739–749.
- Friberg L, Rosenqvist M, Lip GY. Net clinical benefit of warfarin in patients with atrial fibrillation: a report from the Swedish atrial fibrillation cohort study. Circulation 2012; 125:2298–2307.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Hart RG, Pearce LA, Aguilar MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med 2007; 146:857–867.
- The Atrial Fibrillation Investigators. The efficacy of aspirin in patients with atrial fibrillation. Analysis of pooled data from 3 randomized trials. Arch Intern Med 1997; 157:1237–1240.
- Miller VT, Rothrock JF, Pearce LA, Feinberg WM, Hart RG, Anderson DC. Ischemic stroke in patients with atrial fibrillation: effect of aspirin according to stroke mechanism. Stroke Prevention in Atrial Fibrillation Investigators. Neurology 1993; 43:32–36.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery 1999; 45:1113–1119.
- Huttner HB, Schellinger PD, Hartmann M, et al. Hematoma growth and outcome in treated neurocritical care patients with intracerebral hemorrhage related to oral anticoagulant therapy: comparison of acute treatment strategies using vitamin K, fresh frozen plasma, and prothrombin complex concentrates. Stroke 2006; 37:1465–1470.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Gallagher AM, Setakis E, Plumb JM, Clemens A, van Staa TP. Risks of stroke and mortality associated with suboptimal anticoagulation in atrial fibrillation patients. Thromb Haemost 2011; 106:968–977.
- Odén A, Fahlén M, Hart RG. Optimal INR for prevention of stroke and death in atrial fibrillation: a critical appraisal. Thromb Res 2006; 117:493–499.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG, et al. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Adjusted-dose warfarin versus low-intensity, fixed-dose warfarin plus aspirin for high-risk patients with atrial fibrillation: Stroke Prevention in Atrial Fibrillation III randomised clinical trial. Lancet 1996; 348:633–638.
- Gulløv AL, Koefoed BG, Petersen P, et al. Fixed minidose warfarin and aspirin alone and in combination vs adjusted-dose warfarin for stroke prevention in atrial fibrillation: Second Copenhagen Atrial Fibrillation, Aspirin, and Anticoagulation Study. Arch Intern Med 1998; 158:1513–1521.
- Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos 2008; 36:386–399.
- Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 2007; 64:292–303.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Hohnloser SH, Oldgren J, Yang S, et al. Myocardial ischemic events in patients with atrial fibrillation treated with dabigatran or warfarin in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial. Circulation 2012; 125:669–676.
- Uchino K, Hernandez AV. Dabigatran association with higher risk of acute coronary events: meta-analysis of noninferiority randomized controlled trials. Arch Intern Med 2012; 172:397–402.
- Kubitza D, Becka M, Voith B, Zuehlsdorf M, Wensing G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther 2005; 78:412–421.
- Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct Factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharmacol 2005; 61:873–880.
- Kubitza D, Becka M, Roth A, Mueck W. Dose-escalation study of the pharmacokinetics and pharmacodynamics of rivaroxaban in healthy elderly subjects. Curr Med Res Opin 2008; 24:2757–2765.
- Weinz C, Schwarz T, Kubitza D, Mueck W, Lang D. Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab Dispos 2009; 37:1056–1064.
- Perzborn E, Roehrig S, Straub A, Kubitza D, Misselwitz F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat Rev Drug Discov 2011; 10:61–75.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Wong PC, Pinto DJ, Zhang D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis 2011; 31:478–492.
- Carreiro J, Ansell J. Apixaban, an oral direct Factor Xa inhibitor: awaiting the verdict. Expert Opin Investig Drugs 2008; 17:1937–1945.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Lu G, DeGuzman FR, Hollenbach SJ, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med 2013; 19:446–451.
- Liesenfeld KH, Staab A, Härtter S, Formella S, Clemens A, Lehr T. Pharmacometric characterization of dabigatran hemodialysis. Clin Pharmacokinet 2013; 52:453–462.
- MPR. Monthly Prescribing Reference. Anticoagulant dosing conversions. August 18, 2014. www.empr.com/anticoagulant-dosing-conversions/article/194271/. Accessed December 11, 2014.
- Eikelboom JW, Connolly SJ, Brueckmann M, et al; RE-ALIGN Investigators. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med 2013; 369:1206–1214.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Tischenko A, Gula LJ, Yee R, Klein GJ, Skanes AC, Krahn AD. Implantation of cardiac rhythm devices without interruption of oral anticoagulation compared with perioperative bridging with low-molecular weight heparin. Am Heart J 2009; 158:252–256.
- Robinson M, Healey JS, Eikelboom J, et al. Postoperative low-molecular-weight heparin bridging is associated with an increase in wound hematoma following surgery for pacemakers and implantable defibrillators. Pacing Clin Electrophysiol 2009; 32:378–382.
- Birnie DH, Healey JS, Wells GA, et al; BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
- Ahmed I, Gertner E, Nelson WB, et al. Continuing warfarin therapy is superior to interrupting warfarin with or without bridging anticoagulation therapy in patients undergoing pacemaker and defibrillator implantation. Heart Rhythm 2010; 7:745–749.
- Cheng A, Nazarian S, Brinker JA, et al. Continuation of warfarin during pacemaker or implantable cardioverter-defibrillator implantation: a randomized clinical trial. Heart Rhythm 2011; 8:536–540.
- Jamula E, Lloyd NS, Schwalm JD, Airaksinen KE, Douketis JD. Safety of uninterrupted anticoagulation in patients requiring elective coronary angiography with or without percutaneous coronary intervention: a systematic review and metaanalysis. Chest 2010; 138:840–847.
- Jamula E, Douketis JD, Schulman S. Perioperative anticoagulation in patients having implantation of a cardiac pacemaker or defibrillator: a systematic review and practical management guide. J Thromb Haemost 2008; 6:1615–1621.
- Korantzopoulos P, Letsas KP, Liu T, Fragakis N, Efremidis M, Goudevenos JA. Anticoagulation and antiplatelet therapy in implantation of electrophysiological devices. Europace 2011; 13:1669–1680.
- Di Biase L, Burkhardt JD, Mohanty P, et al. Periprocedural stroke and management of major bleeding complications in patients undergoing catheter ablation of atrial fibrillation: the impact of periprocedural therapeutic international normalized ratio. Circulation 2010; 121:2550–2556.
- Patel MR, Hellkamp AS, Lokhnygina Y, et al. Outcomes of discontinuing rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: analysis from the ROCKET AF trial (Rivaroxaban Once-Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation). J Am Coll Cardiol 2013; 61:651–658.
- Azoulay L, Dell’aniello S, Simon TA, Renoux C, Suissa S. Initiation of warfarin in patients with atrial fibrillation: early effects on ischaemic strokes. Eur Heart J 2013; Dec 18 [Epub ahead of print].
- Anderson M, Hassell KL, Trujillo TC, Wolfe B. When patients on target-specific oral anticoagulants need surgery. Cleve Clin J Med 2014; 81:629–639.
Antithrombotic therapy reduces the risk of systemic embolism in patients with atrial fibrillation, but one approach does not suit all patients. The decision whether to start this therapy—and which agent to use—must take into account the patient’s risk of thromboembolism as well as bleeding.
Antithrombotic therapy encompasses antiplatelet drugs such as aspirin and clopidogrel and anticoagulants such as warfarin and the target-specific oral anticoagulants (TSOACs). Oral anticoagulation is more effective than antiplatelet therapy and is preferred in all but those at lowest risk, in whom either antiplatelet therapy or no therapy is deemed adequate.
Patients with valvular atrial fibrillation, specifically those who have rheumatic mitral stenosis or a prosthetic heart valve, are at significantly higher risk of systemic embolization. Their overall risk-benefit profile is nearly always in favor of anticoagulation. But the same is not necessarily true for patients with nonvalvular atrial fibrillation.
The following discussion sets forth our rationale for clinical decision-making, based on recommendations in the 2014 guidelines from the American Heart Association, American College of Cardiology, and Heart Rhythm Society.1 The second half of this review outlines the oral anticoagulants currently available.
ONE IN FOUR PEOPLE
Atrial fibrillation is common, with an incidence that increases with age. It affects more than 10% of people over age 80 and is often associated with cardiovascular disease.2 Based on Framingham Heart Study data, a person’s lifetime risk of developing it is about 25%.3
FIVEFOLD RISK OF STROKE
The most serious complication of atrial fibrillation is arterial thromboembolism, of which ischemic stroke is the most common and most feared manifestation. The risk of stroke is five times higher than normal in patients with atrial fibrillation.3 More than 15% of strokes may be attributable to atrial fibrillation, and the proportion increases with age.4
The risk of thromboembolism appears to be similar in patients with clinically manifest atrial fibrillation irrespective of the type (paroxysmal, persistent, or permanent). The Stroke Prevention in Atrial Fibrillation (SPAF) study5 and the Atrial Fibrillation Clopidogrel Trial With Irbesartan for Prevention of Vascular Events (ACTIVE W)6 showed that patients who had paroxysmal atrial fibrillation and at least one risk factor for thromboembolism had stroke rates comparable to those of their counterparts who had persistent and permanent atrial fibrillation.
Subclinical atrial fibrillation may be an important cause of stroke. Clinically silent episodes can be detected by implantable electronic devices, which record episodes of atrial tachyarrhythmia (atrial high-rate events). Subclinical episodes have been detected in 10% to 28% of monitored patients who did not have a history of atrial fibrillation.7,8 Patients who have atrial high-rate events detected by implantable devices have a higher risk of future clinically manifest atrial fibrillation, thromboembolic events, or both.7–9 Yet characteristics of atrial high-rate episodes that predict risk are not well defined and warrant further investigation.
CLINICAL RISK FACTORS FOR STROKE
To date, thousands of patients with nonvalvular atrial fibrillation have participated in randomized clinical trials of stroke prevention. The placebo groups from these trials provide a sizable database for retrospectively identifying clinical characteristics associated with thromboembolism. The Atrial Fibrillation Investigators10 pooled data from five large trials and found that risk factors consistently associated with stroke in multivariate analysis included diabetes mellitus, hypertension, prior systemic embolism, and advanced age.
Though the risk of stroke increases with age with no lower limit, most studies identify age 65 as a threshold, with further escalating risk after age 75. Moreover, women were observed to be at higher risk in some but not all studies. These risk factors have become components of commonly used risk-stratification schemes.
Hypertrophic cardiomyopathy. Maron et al11 reported that atrial fibrillation in patients with hypertrophic cardiomyopathy was independently associated with thromboembolism. In 900 patients with hypertrophic cardiomyopathy, the prevalence of systemic embolism was 6%. Patients with hypertrophic cardiomyopathy and a thromboembolic complication were seven times more likely to have atrial fibrillation than matched counterparts free of thromboembolism. A notable subset of patients experienced a stroke or embolic event before age 50, and the authors advised that the risk of thromboembolism should be considered in patients of any age with hypertrophic cardiomyopathy and atrial fibrillation.
Olivotto et al12 similarly found patients with hypertrophic cardiomyopathy and atrial fibrillation to be at significantly greater risk of stroke (odds ratio [OR] 17.7, 95% confidence interval [CI] 4.1–75.9, P < .001).
Chronic kidney disease is also associated with a higher risk of thromboembolism in patients with atrial fibrillation. A glomerular filtration rate of 60 mL/min or less is independently and inversely predictive of risk.13,14
While patients with end-stage renal disease have been largely excluded from stroke prevention trials, Vázquez et al15 prospectively followed 190 dialysis patients for 12 months. In multivariate analysis, compared with matched controls without documented atrial fibrillation, patients receiving renal replacement therapy and having any form of atrial fibrillation were eight times more likely to have systemic embolization.
IMAGING-BASED RISK FACTORS
In addition to clinical factors, several imaging-based factors have been associated with stroke risk in patients with atrial fibrillation.
Complex aortic atheroma or markers of blood stasis within the left atrium, such as reduced left atrial appendage emptying flow (< 20 cm/second), dense spontaneous echo contrast, or left atrial appendage thrombus, seen on transesophageal echocardiography, were independently associated with increased systemic embolic risk in the third SPAF substudy.16 Moreover, multivariate analysis of SPAF data found both left ventricular dysfunction of any severity and increased left atrial size (diameter corrected for body surface area by M-mode > 2.5 cm/m2) to be independent predictors of thromboembolism.17
Although enlargement of the left atrium has not been incorporated into traditional risk stratification schemes, data from Osranek et al18 further implicate it as a marker of risk. The cohort was small (N = 46), but consisted of patients with lone atrial fibrillation followed for nearly 30 years. Patients with normal left atrial size enjoyed a benign course, while those with left atrial enlargement (> 32 mL/m2) at diagnosis or later during follow-up had significantly worse event-free survival (hazard ratio [HR] 4.46, 95% CI 1.56–12.74, P < .01). All embolic strokes occurred in the group with left atrial enlargement.
RISK STRATIFICATION SCHEMES
Several models for predicting systemic embolism risk in patients with nonvalvular atrial fibrillation have been proposed and validated.
The CHADS2 score has been the most widely applied, being simple to use.19,20 It assigns 1 point each for Congestive heart failure, Hypertension, Age 75 or older, and Diabetes, and 2 points for prior Stroke or systemic thromboembolism.
In patients with chronic nonvalvular atrial fibrillation, Gage et al19 reported that the stroke rate was lowest in those with a score of 0, with an annual adjusted stroke rate of 1.9% per year, and highest in those with the maximal possible score (ie, 6), with a rate of 18.2%. The rate increased by a factor of 1.5 with each point in the CHADS2 score.
CHA2DS2-VASc. Endorsed for use in both the American and European guidelines,1,21 CHA2DS2-VASc is an extension of CHADS2. Points are assigned as follows:
- Congestive heart failure or left ventricular dysfunction (moderate to severe left ventricular dysfunction or recent heart failure exacerbation requiring hospitalization irrespective of ejection fraction): 1 point
- Hypertension: 1 point
- Age ≥ 75: 2 points; age 65–74: 1 point
- Diabetes mellitus: 1 point
- Stroke, transient ischemic attack, or thromboembolism: 2 points
- Vascular disease (prior myocardial infarction, peripheral arterial disease, or aortic plaque): 1 point
- Sex, female: 1 point
- Maximum score: 9 points.
Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk. Lip et al22 found that, in untreated patients with nonvalvular atrial fibrillation, rates of stroke ranged from 0 with a score of 0 to 15.2% per year with a score of 9 points.
In a large cohort with over 11,000 patient-years of follow-up, 98% of the thromboembolic events occurred in people with a CHA2DS2-VASc score of 2 or more. Moreover, more than 99% of patients with a score of less than 2 were free of stroke and thromboembolism.23
Compared with the CHADS2 score, CHA2DS2-VASc has superior negative predictive power. Of 1,084 patients from the European Heart Survey for Atrial Fibrillation, the newer scheme classified significantly fewer patients as being at either low risk (score of 0; 9% vs 20%) or intermediate risk (score of 1; 15% vs 35%).23 Though the overall rate of stroke was low, those categorized as being at low or intermediate risk by CHA2DS2-VASc had significantly fewer thromboembolic events than their counterparts according to CHADS2 (0.6% vs 3.3%).
Olesen et al24 similarly showed that in patients with a CHADS2 score of 0, reclassification by CHA2DS2-VASc yielded a range of annual stroke rates from 0.84% with a score of 0 up to 3.2% with a score of 3.
RISK-BASED ANTITHROMBOTIC THERAPY IN NONVALVULAR ATRIAL FIBRILLATION
The 2014 atrial fibrillation guidelines1 state that the decision to give antithrombotic therapy for atrial fibrillation should be individualized, based on the absolute and relative risks of stroke and bleeding, and ought to take into consideration the patient’s preferences. For patients with nonvalvular atrial fibrillation, selection of antithrombotic therapy should take into account the risk of thromboembolism determined by the CHA2DS2-VASc score and be irrespective of the pattern of atrial fibrillation (paroxysmal, persistent, or permanent). Antithrombotic therapy is similarly recommended for patients with atrial flutter, according to the same risk profile used for atrial fibrillation.
Studies have consistently shown24–27 that the risk of ischemic stroke without anticoagulation exceeds the risk of intracranial bleeding with anticoagulation in nearly all patients except those at lowest risk of thromboembolism. The CHA2DS2-VASc score better identified those at truly low risk, in whom treatment may offer more risk than benefit.24–27
The HAS-BLED score28 assigns points as follows:
- Hypertension (systolic blood pressure > 160 mm Hg): 1 point
- Abnormal renal function (dialysis, renal transplantation, or serum creatinine > 2.6 mg/mL) or liver function (cirrhosis, bilirubin more than two times the upper limit, or aminotransferase levels more than three times the upper limit): 1 or 2 points
- Stroke: 1 point
- Bleeding (prior major bleeding event or predisposition to bleeding): 1 point
- Labile international normalized ratio (INR) (supratherapeutic or time in therapeutic range < 60%): 1 point
- Elderly (age > 65): 1 point
- Drugs (antiplatelet, nonsteroidal anti-inflammatory) or alcohol (more than eight drinks per week): 1 or 2 points
- Maximum total: 9 points.
HAS-BLED is a practical and validated approach for estimating bleeding risk and is mentioned in the guidelines, but it is not recommended for use in guiding decisions about antithrombotic therapy. Specifically, it should not be used to exclude patients, but rather to identify those at high risk (score ≥ 3) who may require closer observation and more attentive monitoring of the INR.
ANTITHROMBOTIC THERAPY
Antithrombotic agents available for use in the United States include antiplatelet drugs (eg, aspirin and clopidogrel) and anticoagulants (unfractionated heparin and low-molecular-weight heparin, vitamin K antagonists such as warfarin, and direct thrombin and factor Xa inhibitors). Anticoagulation has been shown in randomized controlled trials to be superior to both placebo and antiplatelet agents used either alone or in combination.29
Aspirin has been downgraded
Aspirin has been compared with placebo in seven randomized controlled trials. Only the original SPAF study, in which aspirin 325 mg/day was used, found that it was beneficial. This result alone accounted for the 19% reduction in relative risk (95% CI 1%–35%, P < .05) in a meta-analysis performed by Hart et al.29 Even when combined with clopidogrel 75 mg/day, aspirin 75 to 100 mg/day is still inferior to warfarin.5 While dual antiplatelet therapy resulted in a 28% relative reduction in thromboembolism (95% CI 17%–38%, P < .01) compared with aspirin alone, major bleeding significantly increased by 57% (95% CI 29%–92%, P < .01).
Although aspirin may be beneficial, differences among patients may influence its efficacy. It may be more effective in preventing noncardioembolic stroke, particularly in diabetic and hypertensive patients.30,31 To date, aspirin has not been shown to be beneficial in low-risk populations.
The 2014 guidelines downgraded the recommendation for aspirin therapy. For patients at low risk and for some at intermediate risk, it is permissible to forgo therapy altogether, including aspirin.1
ORAL ANTICOAGULANTS
The rest of this paper reviews the oral anticoagulants that are approved for reducing the risk of thromboembolism in atrial fibrillation, focusing on each agent’s mechanism of action, pharmacokinetics, clinical efficacy, and safety.
WARFARIN, A VITAMIN K ANTAGONIST
Warfarin inhibits synthesis of vitamin K-dependent clotting factors (ie, factors II, VII, IX, and X) and proteins C and S by inhibiting the C1 subunit of vitamin K epoxide reductase, thereby interfering with production of vitamin K1 epoxide and consequent regeneration of vitamin K.
Pharmacokinetics. Warfarin is nearly completely absorbed after oral administration. Its anticoagulant effect can be seen within 24 hours of administration, but its peak effect is typically apparent only after 72 hours. Elimination occurs predominantly through metabolism by cytochrome P450 enzymes, principally CYP2C9. Its effective half-life ranges from 20 to 60 hours, with a mean of 40 hours.32
Warfarin’s effect, dosage, and bleeding risk are influenced by multiple factors, including vitamin K-containing foods such as green leafy vegetables, medications that either inhibit or induce hepatic cytochrome P450 enzymes, and polymorphisms in the VKORC1 and CYP2C9 genes.32
Reversal. Warfarin’s anticoagulant effect is reversed with vitamin K, but this reversal may not become apparent for 6 to 24 hours. In contrast, fresh-frozen plasma and prothrombin protein concentrate, which contain clotting factors, reverse warfarin immediately. Currently, a three-factor prothrombin protein concentrate (factors II, IX, and X) and a four-factor concentrate (factors II, VII, IX, and X plus proteins C and S) are available in the United States. Although prothrombin protein concentrate works rapidly and has a lower volume of administration, available data do not indicate it is clinically superior to fresh-frozen plasma.33,34 The ongoing randomized PROTECT trial (NCT00618098), comparing fresh-frozen plasma and four-factor prothrombin protein concentrate for reversal of vitamin K antagonist therapy, may provide further insight.
Efficacy and safety. Randomized controlled trials in patients with nonvalvular atrial fibrillation have shown that warfarin (in doses adjusted to maintain an INR greater than 2) is highly efficacious in preventing systemic embolism, with a relative risk reduction of 61% (95% CI 47%–71%, P < .05) compared with placebo.29,35 An INR of 2 to 3 is recommended for patients with nonvalvular atrial fibrillation, and those with atrial fibrillation and either a bioprosthetic valve or rheumatic heart disease. In contrast, an INR of 2.5 to 3.5 is recommended for patients with atrial fibrillation and mechanical valves in the aortic or mitral positions.1,36
Stroke prevention with warfarin is most effective when the achieved mean time in the therapeutic range is at least 70%. The risk of intracranial hemorrhage increases significantly at INRs higher than 3. An INR of 2 to 3 offers maximum protection with minimal risk of bleeding.37,38 Systematic follow-up of patients through anticoagulation clinics produces better compliance and control and is encouraged.
TARGET-SPECIFIC ORAL ANTICOAGULANTS
Although effective, warfarin requires frequent monitoring and dosage adjustment, has a delayed onset and protracted offset, and interacts with commonly consumed vitamin K–containing foods and frequently used drugs. These drawbacks prompted evaluation of existing antiplatelet agents, in combination or in conjunction with lower adjusted or fixed-dose warfarin. These regimens proved inferior,39–42 spurring interest in developing alternative oral anticoagulants.
TSOACs act by directly inhibiting thrombin (factor IIa) or by reducing thrombin production indirectly by inhibiting factor Xa. Three TSOACs are approved. Each was compared with adjusted-dose warfarin in randomized controlled trials.
Dabigatran
Dabigatran etexilate was the first TSOAC approved in the United States.
Pharmacokinetics. Dabigatran etexilate has a bioavailability of 3% to 7% after oral administration. Its absorption is enhanced in an acidic gastric environment and is limited by P-glycoprotein-facilitated transport out of enterocytes. Dabigatran etexilate is hydrolyzed to its active metabolite dabigatran, which directly inhibits thrombin. Maximal plasma drug concentration and peak anticoagulant effect are achieved within 0.5 to 2 hours after administration.
Dabigatran is predominantly excreted by the kidneys, and has a half-life of 12 to 17 hours in patients with normal renal function. The half-life extends to 27 hours in those with moderately severe renal impairment (creatinine clearance 15–30 mL/min). The recommended dose of 150 mg twice daily should be reduced to 75 mg twice daily in patients with a creatinine clearance of 15 to 30 mL/min. This drug is contraindicated in patients with a creatinine clearance less than 15 mL/min.43,44
Efficacy. The Randomized Evaluation of Long-term Anticoagulation Therapy (RE-LY) trial45 randomly assigned 18,113 patients with nonvalvular atrial fibrillation at risk of thromboembolism (mean CHADS2 score 2.1) to receive either dabigatran (either 150 mg twice daily or 110 mg twice daily) or warfarin (adjusted to an INR of 2.0 to 3.0). Of note, the lower approved dose of dabigatran (75 mg twice daily) was not tested in RE-LY.
At 2 years, higher-dose dabigatran was significantly more effective than both warfarin (RR 0.65, 95% CI 0.52–0.81, P < .05) and lower-dose dabigatran (RR 0.73, 95% CI 0.58–0.91, P < .05) in reducing the rate of systemic embolic events.
The risk of combined major bleeding events was no different with higher-dose dabigatran than with warfarin (RR 0.93, 95% CI 0.81–1.07, P < .05), but the rate of hemorrhagic stroke was significantly less with dabigatran than with warfarin (RR 0.26, 95% CI 0.14–0.49, P < .05). Higher rates of major gastrointestinal bleeding and dyspepsia occurred with dabigatran.
Concern about the safety of dabigatran was raised when post hoc evaluation of RE-LY found a higher incidence of myocardial infarction with dabigatran than with warfarin (RR 1.38, 95% CI 1–1.91, P = .048).46 Corroborating data were reported by Uchino and Hernandez,47 comparing dabigatran with either warfarin or low-molecular-weight heparin. However, without directly comparing dabigatran and placebo, it is unclear whether the small increase in myocardial infarction reflects a direct effect of dabigatran or absence of a protective effect of warfarin or low-molecular-weight heparin.
Rivaroxaban
Rivaroxaban is a direct factor Xa inhibitor that blocks the amplified burst of thrombin production and in turn inhibits platelet aggregation and thrombus formation.
Pharmacokinetics. Rivaroxaban’s oral bioavailability is 80% to 100% after a single 15- or 20-mg dose taken with food. Its maximal anticoagulant effect is achieved within 2 hours. Two-thirds of the active drug is metabolized by either CYP450-dependent (CYP3A4, 2J2) or CYP-independent mechanisms; the inactive drug is then excreted in the urine and feces. The remaining, active drug is removed by the kidneys using the P-glycoprotein transporter.
The half-life of rivaroxaban is 5 to 9 hours. The recommended dosage of 20 mg daily should be reduced to 15 mg daily if the creatinine clearance rate is 30 to 50 mL/min, or to 10 mg if the creatinine clearance rate is 15 to 30 mL/min. Rivaroxaban is contraindicated in patients whose creatinine clearance rate is less than 15 mL/min.48–52
Efficacy and safety. In the Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET-AF),53 14,264 at-risk patients with nonvalvular atrial fibrillation (mean CHADS2 score 3.5) were randomly assigned to receive either rivaroxaban 20 mg daily (or 15 mg daily if their creatinine clearance was 30–49 mL/min; the lowest dose of rivaroxaban, 10 mg, was not studied in this trial) or warfarin (target INR 2.0–3.0). Outcomes with rivaroxaban compared with warfarin:
- Systemic embolism:
HR 0.79, 95% CI 0.66–0.96, P < .01, noninferiority - Total bleeding: no difference
- Intracranial bleeding:
HR 0.67, 95% CI 0.47–0.93, P = .02 - Fatal bleeding:
HR 0.50, 95% CI 0.31–0.79, P = .003 - Major gastrointestinal bleeding:
3.2% vs 2.2%, P < .001.
Apixaban
Apixaban is also a direct factor Xa inhibitor.
Pharmacokinetics. Apixaban’s oral bioavailability is 50%, with maximal blood concentration achieved at 3 to 4 hours. One-quarter of the drug is metabolized via CYP3A4. The remaining active drug is excreted by the kidneys and biliary/intestinal system via the P-glycoprotein transporter. Apixaban’s half-life is 9 to 14 hours.
The recommended dosage is 5 mg twice daily, but it should be reduced to 2.5 mg twice daily if at least two of the following characteristics are present: age 80 or older, weight 60 kg or less, and serum creatinine 1.5 mg/dL or more.54,55
Efficacy and safety. The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial56 enrolled 18,201 patients with nonvalvular atrial fibrillation (mean CHADS2 score 2.1) randomly assigned to receive either apixaban (5 mg twice daily with dosage reduction to 2.5 mg twice daily as noted above) or warfarin (target INR 2.0–3.0).
Compared with warfarin, apixaban was associated with lower risk of:
- Systemic embolism
(HR 0.79, 95% CI 0.66–0.95, P = .01) - Major bleeding
(HR 0.69, 95% CI 0.60–0.80, P < .001) - Intracranial hemorrhage
(HR 0.42, 95% CI 0.30–0.58, P < .001) - All-cause mortality
(HR 0.89 95% CI 0.80–0.99, P = .047).
Drug interactions with the novel oral anticoagulants
TSOACs were developed with the intent to avoid many of the shortcomings of warfarin. Each has a broader therapeutic window and a rapid onset of action, enabling fixed dosing without need for serial monitoring. Compared with warfarin, they have significantly fewer dietary and drug interactions.
Nonetheless, drug interactions do exist and are important to recognize (Tables 1–3). These primarily result from inhibition or induction of cytochrome P450 enzyme activity or P-glycoprotein transporter action, involved in metabolism and elimination of active drug.
Reversibility of the target-specific oral anticoagulants
While the anticoagulant effects of warfarin can be reversed by vitamin K, fresh-frozen plasma, and prothrombin complex concentrate, TSOACs have no currently approved antidotes. Management of bleeding due to these agents was recently reviewed in this journal by Fawole et al.57
Several nonspecific hemostatic agents have been suggested, including recombinant factor VIIa or prothrombin complex concentrates. The anticoagulant effect of rivaroxaban has been shown to be reversed by prothrombin complex concentrate in vitro; clinical effect has not been demonstrated.58 PRT06445 (andexanet alfa), a recombinant protein antidote specific for factor Xa inhibitors, has entered clinical studies, with a phase 2 trial reporting high reversing capability for apixaban.59
Unlike rivaroxaban and apixaban, which are highly bound to plasma protein, dabigatran can be effectively removed with hemodialysis. Liesenfeld et al60 showed that longer dialysis duration was the most relevant variable for reducing dabigatran plasma levels. Current clinical experience is limited, and standard recommendations and formal guidance are lacking.
Switching oral anticoagulants
Suggested approaches for switching between anticoagulants are listed in Table 4.61
CHOOSING ANTITHROMBOTIC THERAPY
In valvular atrial fibrillation: warfarin
Anticoagulation with warfarin is advised for valvular atrial fibrillation. Patients with bioprosthetic heart valves or rheumatic valvular disease were not evaluated in randomized controlled trials of TSOACs. Dabigatran in particular is contraindicated in patients with mechanical heart valves, as the Randomized, Phase II Study to Evaluate the Safety and Pharmacokinetics of Oral Dabigatran Etexilate in Patients After Heart Valve Replacement (RE-ALIGN)62 found higher rates of stroke, valve-related thrombosis, and myocardial infarction in patients receiving dabigatran.
In nonvalvular atrial fibrillation
According to the 2014 guidelines,1 oral anticoagulation is preferred in all patients with nonvalvular atrial fibrillation but those at lowest risk (CHA2DS2-VASc = 0).
Experience with TSOACs is lacking in patients with end-stage kidney disease (creatinine clearance < 15 mL/min), and warfarin is advised in this group.
TSOACs are recommended in patients with nonvalvular atrial fibrillation in whom therapeutic INR levels cannot be maintained with warfarin. For most patients with nonvalvular atrial fibrillation, TSOACs are an option equivalent to warfarin. Anticoagulant choice is largely driven by dosing convenience, out-of-pocket cost for treatment with a TSOAC, and ready availability of antidotes for warfarin in case of bleeding (Tables 5 and 6).
In patients with nonvalvular atrial fibrillation, TSOACs are as effective as warfarin in preventing systemic thromboembolism, and some of them have been shown to be superior in terms of lower rates of ischemic stroke (dabigatran), systemic embolism (apixaban), and mortality (apixaban; trend for dabigatran). All TSOACs demonstrate modestly favorable bleeding risk profiles compared with warfarin, with lower risk of intracranial hemorrhage. Potential differences in efficacy and safety among TSOACs are unknown since there have been no randomized direct comparisons between them. A summary of landmark trial results and assessment of the advantages and disadvantages of each are listed in Table 7.
Two groups of patients with nonvalvular atrial fibrillation warrant special consideration:
Patients with hypertrophic cardiomyopathy. There are no randomized controlled trials of anticoagulation therapy in patients with hypertrophic cardiomyopathy; however, because of their high risk of thromboembolism, anticoagulation is indicated irrespective of the CHA2DS2-VASc score. TSOACs are an option as an alternative to warfarin.
Patients with coronary artery disease and an indication for antiplatelet therapy. In this group the decision for concurrent anticoagulation is guided by the CHA2DS2-VASc score. For patients who have intracoronary stents, dual antiplatelet therapy is the standard of care for reducing risk of cardiovascular events after stent implantation.63 When triple therapy (ie, two antiplatelet drugs and an anticoagulant) is indicated, such as after intracoronary stent placement, the guidelines suggest trying to minimize the duration of triple therapy. For instance, a bare-metal stent may be preferred. Alternatively, after coronary revascularization, it may be reasonable to use clopidogrel 75 mg daily with an oral anticoagulant and to omit aspirin.
Interrupting and bridging anticoagulation
Patients with atrial fibrillation often require suspension of anticoagulation, most commonly before an elective invasive procedure. The duration of interruption, timing of resumption, and need for bridging anticoagulation are guided by clinical judgment, which considers risk of thromboembolism and severity of procedure-related bleeding risk.
In general, if therapy needs to be interrupted, it should be restarted as soon as possible. Short-term interruption does not seem to be associated with clinically significant risk of thromboembolic events, whereas postoperative heparin bridging therapy increases the risk of hematoma with implantation of a cardiac electronic device.64,65
To date, evidence is lacking to advise upon periprocedure bridging anticoagulation. The Bridging Anticoagulation in Patients Who Require Temporary Interruption of Warfarin Therapy for an Elective Invasive Procedure or Surgery (BRIDGE) study (NCT00786474)— enrolling chronically anticoagulated patients undergoing an invasive procedure to randomly receive placebo or bridging low-molecular-weight heparin—may provide guidance.
Currently, it is common practice in low-risk patients undergoing an invasive procedure with significant bleeding risk to interrupt anticoagulation for up to 1 week without bridging. Warfarin is typically held 3 to 5 days, while TSOACs are held for 24 hours if renal function is preserved or up to 2 to 3 days if renal function is severely impaired (creatinine clearance 15–30 mL/min). If complete hemostasis is necessary, it could be confirmed by a normalized INR (for warfarin), activated partial thromboplastin time (dabigatran), or prothrombin time (apixaban or rivaroxaban).
For patients at high risk (valvular atrial fibrillation or CHA2DS2-VASc ≥ 2), bridging with unfractionated heparin or low-molecular-weight heparin during periods of subtherapeutic anticoagulation is common. Alternatively, it is becoming increasingly common to perform cardiac electronic device implantation, catheter ablation, and coronary angiography and intervention without interrupting anticoagulation.66–72
Recently, concern has been raised over a possible increase in thromboembolism upon discontinuation of rivaroxaban and apixaban. ROCKET-AF reported a spike in thrombotic events in the rivaroxaban-treated group at the end of the trial (HR 1.50, 95% CI 1.05–2.15, P = .026). This raised concern for a possible “rebound” effect upon drug cessation. Yet a post hoc analysis of ROCKET-AF demonstrated that events clustered in the rivaroxaban-treated cohort who completed the study and were transitioning to open-label warfarin, and this alone accounted for the rise in stroke occurrence. In contrast, there was no increase in the cohort of patients treated with rivaroxaban who either temporarily interrupted or permanently discontinued the drug.73 The authors concluded that increased stroke was the consequence of transiently interrupted anticoagulation, rather than a rebound prothrombotic effect. Similar results were reported in ARISTOTLE.
Another possibility is that, during the transition to warfarin therapy, transient hypercoagulability could be a function of warfarin. Azoulay et al74 observed in a large cohort that warfarin was associated with a 71% increased risk of stroke in the first 30 days after initiation, compared with decreased risk thereafter. Nevertheless, there is now a black- box warning recommendation for all three TSOACs that if discontinuation is required for a reason other than pathological bleeding, bridging with another anticoagulant should at least be considered.
The perioperative management of the TSOACs was recently reviewed in this journal by Anderson et al.75
WEIGHING THE RISKS OF STROKE AND BLEEDING
Stroke is the most feared complication in patients with atrial fibrillation. Risk reduction is an important goal in management, yet decisions for individuals must take into account both stroke and bleeding risks related to antithrombotic therapy.
The 2014 guidelines1 differ from past versions. First, they endorse the use of CHA2DS2-VASc for categorizing stroke risk in patients with nonvalvular atrial fibrillation. This in turn guides antithrombotic therapy. This scheme effectively identifies patients at very low risk of stroke (men with a score of 0, women with a score of 0 or 1), in whom it is reasonable to omit antithrombotic therapy. For all patients with valvular heart disease or hypertrophic cardiomyopathy, unless bleeding risk is prohibitive, anticoagulation is recommended irrespective of the CHA2DS2-VASc score. Second, they incorporate the TSOACs, which offer convenience and improved safety in select patients.
While the guidelines mention the potential relevance of subclinical atrial tachyarrhythmias as they pertain to stroke risk, there is no specific recommendation as to their management. We do take into consideration the finding of atrial high-rate events (≥ 180 bpm, ≥ 6 minutes in duration) diagnostically confirmed by cardiac implantable electronic devices or telemetric monitoring, particularly in patients with a clinical profile of high stroke risk. In addition, atriopathy with increased left atrial size and renal insufficiency, as discussed in this review, appear to correlate with greater risk of thromboembolism, yet neither is a component of the stroke risk scheme endorsed by the guidelines.
Other risk factors, some unknown to us, undoubtedly exist. Again, our empiric judgment is to at least consider these nontraditional risk factors while guided primarily by the CHA2DS2-VASc score when assessing stroke risk in patients with atrial fibrillation.
The goal in managing patients with atrial fibrillation is to balance thromboembolic risk reduction with the risk of bleeding associated with antithrombotic therapy.
Antithrombotic therapy reduces the risk of systemic embolism in patients with atrial fibrillation, but one approach does not suit all patients. The decision whether to start this therapy—and which agent to use—must take into account the patient’s risk of thromboembolism as well as bleeding.
Antithrombotic therapy encompasses antiplatelet drugs such as aspirin and clopidogrel and anticoagulants such as warfarin and the target-specific oral anticoagulants (TSOACs). Oral anticoagulation is more effective than antiplatelet therapy and is preferred in all but those at lowest risk, in whom either antiplatelet therapy or no therapy is deemed adequate.
Patients with valvular atrial fibrillation, specifically those who have rheumatic mitral stenosis or a prosthetic heart valve, are at significantly higher risk of systemic embolization. Their overall risk-benefit profile is nearly always in favor of anticoagulation. But the same is not necessarily true for patients with nonvalvular atrial fibrillation.
The following discussion sets forth our rationale for clinical decision-making, based on recommendations in the 2014 guidelines from the American Heart Association, American College of Cardiology, and Heart Rhythm Society.1 The second half of this review outlines the oral anticoagulants currently available.
ONE IN FOUR PEOPLE
Atrial fibrillation is common, with an incidence that increases with age. It affects more than 10% of people over age 80 and is often associated with cardiovascular disease.2 Based on Framingham Heart Study data, a person’s lifetime risk of developing it is about 25%.3
FIVEFOLD RISK OF STROKE
The most serious complication of atrial fibrillation is arterial thromboembolism, of which ischemic stroke is the most common and most feared manifestation. The risk of stroke is five times higher than normal in patients with atrial fibrillation.3 More than 15% of strokes may be attributable to atrial fibrillation, and the proportion increases with age.4
The risk of thromboembolism appears to be similar in patients with clinically manifest atrial fibrillation irrespective of the type (paroxysmal, persistent, or permanent). The Stroke Prevention in Atrial Fibrillation (SPAF) study5 and the Atrial Fibrillation Clopidogrel Trial With Irbesartan for Prevention of Vascular Events (ACTIVE W)6 showed that patients who had paroxysmal atrial fibrillation and at least one risk factor for thromboembolism had stroke rates comparable to those of their counterparts who had persistent and permanent atrial fibrillation.
Subclinical atrial fibrillation may be an important cause of stroke. Clinically silent episodes can be detected by implantable electronic devices, which record episodes of atrial tachyarrhythmia (atrial high-rate events). Subclinical episodes have been detected in 10% to 28% of monitored patients who did not have a history of atrial fibrillation.7,8 Patients who have atrial high-rate events detected by implantable devices have a higher risk of future clinically manifest atrial fibrillation, thromboembolic events, or both.7–9 Yet characteristics of atrial high-rate episodes that predict risk are not well defined and warrant further investigation.
CLINICAL RISK FACTORS FOR STROKE
To date, thousands of patients with nonvalvular atrial fibrillation have participated in randomized clinical trials of stroke prevention. The placebo groups from these trials provide a sizable database for retrospectively identifying clinical characteristics associated with thromboembolism. The Atrial Fibrillation Investigators10 pooled data from five large trials and found that risk factors consistently associated with stroke in multivariate analysis included diabetes mellitus, hypertension, prior systemic embolism, and advanced age.
Though the risk of stroke increases with age with no lower limit, most studies identify age 65 as a threshold, with further escalating risk after age 75. Moreover, women were observed to be at higher risk in some but not all studies. These risk factors have become components of commonly used risk-stratification schemes.
Hypertrophic cardiomyopathy. Maron et al11 reported that atrial fibrillation in patients with hypertrophic cardiomyopathy was independently associated with thromboembolism. In 900 patients with hypertrophic cardiomyopathy, the prevalence of systemic embolism was 6%. Patients with hypertrophic cardiomyopathy and a thromboembolic complication were seven times more likely to have atrial fibrillation than matched counterparts free of thromboembolism. A notable subset of patients experienced a stroke or embolic event before age 50, and the authors advised that the risk of thromboembolism should be considered in patients of any age with hypertrophic cardiomyopathy and atrial fibrillation.
Olivotto et al12 similarly found patients with hypertrophic cardiomyopathy and atrial fibrillation to be at significantly greater risk of stroke (odds ratio [OR] 17.7, 95% confidence interval [CI] 4.1–75.9, P < .001).
Chronic kidney disease is also associated with a higher risk of thromboembolism in patients with atrial fibrillation. A glomerular filtration rate of 60 mL/min or less is independently and inversely predictive of risk.13,14
While patients with end-stage renal disease have been largely excluded from stroke prevention trials, Vázquez et al15 prospectively followed 190 dialysis patients for 12 months. In multivariate analysis, compared with matched controls without documented atrial fibrillation, patients receiving renal replacement therapy and having any form of atrial fibrillation were eight times more likely to have systemic embolization.
IMAGING-BASED RISK FACTORS
In addition to clinical factors, several imaging-based factors have been associated with stroke risk in patients with atrial fibrillation.
Complex aortic atheroma or markers of blood stasis within the left atrium, such as reduced left atrial appendage emptying flow (< 20 cm/second), dense spontaneous echo contrast, or left atrial appendage thrombus, seen on transesophageal echocardiography, were independently associated with increased systemic embolic risk in the third SPAF substudy.16 Moreover, multivariate analysis of SPAF data found both left ventricular dysfunction of any severity and increased left atrial size (diameter corrected for body surface area by M-mode > 2.5 cm/m2) to be independent predictors of thromboembolism.17
Although enlargement of the left atrium has not been incorporated into traditional risk stratification schemes, data from Osranek et al18 further implicate it as a marker of risk. The cohort was small (N = 46), but consisted of patients with lone atrial fibrillation followed for nearly 30 years. Patients with normal left atrial size enjoyed a benign course, while those with left atrial enlargement (> 32 mL/m2) at diagnosis or later during follow-up had significantly worse event-free survival (hazard ratio [HR] 4.46, 95% CI 1.56–12.74, P < .01). All embolic strokes occurred in the group with left atrial enlargement.
RISK STRATIFICATION SCHEMES
Several models for predicting systemic embolism risk in patients with nonvalvular atrial fibrillation have been proposed and validated.
The CHADS2 score has been the most widely applied, being simple to use.19,20 It assigns 1 point each for Congestive heart failure, Hypertension, Age 75 or older, and Diabetes, and 2 points for prior Stroke or systemic thromboembolism.
In patients with chronic nonvalvular atrial fibrillation, Gage et al19 reported that the stroke rate was lowest in those with a score of 0, with an annual adjusted stroke rate of 1.9% per year, and highest in those with the maximal possible score (ie, 6), with a rate of 18.2%. The rate increased by a factor of 1.5 with each point in the CHADS2 score.
CHA2DS2-VASc. Endorsed for use in both the American and European guidelines,1,21 CHA2DS2-VASc is an extension of CHADS2. Points are assigned as follows:
- Congestive heart failure or left ventricular dysfunction (moderate to severe left ventricular dysfunction or recent heart failure exacerbation requiring hospitalization irrespective of ejection fraction): 1 point
- Hypertension: 1 point
- Age ≥ 75: 2 points; age 65–74: 1 point
- Diabetes mellitus: 1 point
- Stroke, transient ischemic attack, or thromboembolism: 2 points
- Vascular disease (prior myocardial infarction, peripheral arterial disease, or aortic plaque): 1 point
- Sex, female: 1 point
- Maximum score: 9 points.
Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk. Lip et al22 found that, in untreated patients with nonvalvular atrial fibrillation, rates of stroke ranged from 0 with a score of 0 to 15.2% per year with a score of 9 points.
In a large cohort with over 11,000 patient-years of follow-up, 98% of the thromboembolic events occurred in people with a CHA2DS2-VASc score of 2 or more. Moreover, more than 99% of patients with a score of less than 2 were free of stroke and thromboembolism.23
Compared with the CHADS2 score, CHA2DS2-VASc has superior negative predictive power. Of 1,084 patients from the European Heart Survey for Atrial Fibrillation, the newer scheme classified significantly fewer patients as being at either low risk (score of 0; 9% vs 20%) or intermediate risk (score of 1; 15% vs 35%).23 Though the overall rate of stroke was low, those categorized as being at low or intermediate risk by CHA2DS2-VASc had significantly fewer thromboembolic events than their counterparts according to CHADS2 (0.6% vs 3.3%).
Olesen et al24 similarly showed that in patients with a CHADS2 score of 0, reclassification by CHA2DS2-VASc yielded a range of annual stroke rates from 0.84% with a score of 0 up to 3.2% with a score of 3.
RISK-BASED ANTITHROMBOTIC THERAPY IN NONVALVULAR ATRIAL FIBRILLATION
The 2014 atrial fibrillation guidelines1 state that the decision to give antithrombotic therapy for atrial fibrillation should be individualized, based on the absolute and relative risks of stroke and bleeding, and ought to take into consideration the patient’s preferences. For patients with nonvalvular atrial fibrillation, selection of antithrombotic therapy should take into account the risk of thromboembolism determined by the CHA2DS2-VASc score and be irrespective of the pattern of atrial fibrillation (paroxysmal, persistent, or permanent). Antithrombotic therapy is similarly recommended for patients with atrial flutter, according to the same risk profile used for atrial fibrillation.
Studies have consistently shown24–27 that the risk of ischemic stroke without anticoagulation exceeds the risk of intracranial bleeding with anticoagulation in nearly all patients except those at lowest risk of thromboembolism. The CHA2DS2-VASc score better identified those at truly low risk, in whom treatment may offer more risk than benefit.24–27
The HAS-BLED score28 assigns points as follows:
- Hypertension (systolic blood pressure > 160 mm Hg): 1 point
- Abnormal renal function (dialysis, renal transplantation, or serum creatinine > 2.6 mg/mL) or liver function (cirrhosis, bilirubin more than two times the upper limit, or aminotransferase levels more than three times the upper limit): 1 or 2 points
- Stroke: 1 point
- Bleeding (prior major bleeding event or predisposition to bleeding): 1 point
- Labile international normalized ratio (INR) (supratherapeutic or time in therapeutic range < 60%): 1 point
- Elderly (age > 65): 1 point
- Drugs (antiplatelet, nonsteroidal anti-inflammatory) or alcohol (more than eight drinks per week): 1 or 2 points
- Maximum total: 9 points.
HAS-BLED is a practical and validated approach for estimating bleeding risk and is mentioned in the guidelines, but it is not recommended for use in guiding decisions about antithrombotic therapy. Specifically, it should not be used to exclude patients, but rather to identify those at high risk (score ≥ 3) who may require closer observation and more attentive monitoring of the INR.
ANTITHROMBOTIC THERAPY
Antithrombotic agents available for use in the United States include antiplatelet drugs (eg, aspirin and clopidogrel) and anticoagulants (unfractionated heparin and low-molecular-weight heparin, vitamin K antagonists such as warfarin, and direct thrombin and factor Xa inhibitors). Anticoagulation has been shown in randomized controlled trials to be superior to both placebo and antiplatelet agents used either alone or in combination.29
Aspirin has been downgraded
Aspirin has been compared with placebo in seven randomized controlled trials. Only the original SPAF study, in which aspirin 325 mg/day was used, found that it was beneficial. This result alone accounted for the 19% reduction in relative risk (95% CI 1%–35%, P < .05) in a meta-analysis performed by Hart et al.29 Even when combined with clopidogrel 75 mg/day, aspirin 75 to 100 mg/day is still inferior to warfarin.5 While dual antiplatelet therapy resulted in a 28% relative reduction in thromboembolism (95% CI 17%–38%, P < .01) compared with aspirin alone, major bleeding significantly increased by 57% (95% CI 29%–92%, P < .01).
Although aspirin may be beneficial, differences among patients may influence its efficacy. It may be more effective in preventing noncardioembolic stroke, particularly in diabetic and hypertensive patients.30,31 To date, aspirin has not been shown to be beneficial in low-risk populations.
The 2014 guidelines downgraded the recommendation for aspirin therapy. For patients at low risk and for some at intermediate risk, it is permissible to forgo therapy altogether, including aspirin.1
ORAL ANTICOAGULANTS
The rest of this paper reviews the oral anticoagulants that are approved for reducing the risk of thromboembolism in atrial fibrillation, focusing on each agent’s mechanism of action, pharmacokinetics, clinical efficacy, and safety.
WARFARIN, A VITAMIN K ANTAGONIST
Warfarin inhibits synthesis of vitamin K-dependent clotting factors (ie, factors II, VII, IX, and X) and proteins C and S by inhibiting the C1 subunit of vitamin K epoxide reductase, thereby interfering with production of vitamin K1 epoxide and consequent regeneration of vitamin K.
Pharmacokinetics. Warfarin is nearly completely absorbed after oral administration. Its anticoagulant effect can be seen within 24 hours of administration, but its peak effect is typically apparent only after 72 hours. Elimination occurs predominantly through metabolism by cytochrome P450 enzymes, principally CYP2C9. Its effective half-life ranges from 20 to 60 hours, with a mean of 40 hours.32
Warfarin’s effect, dosage, and bleeding risk are influenced by multiple factors, including vitamin K-containing foods such as green leafy vegetables, medications that either inhibit or induce hepatic cytochrome P450 enzymes, and polymorphisms in the VKORC1 and CYP2C9 genes.32
Reversal. Warfarin’s anticoagulant effect is reversed with vitamin K, but this reversal may not become apparent for 6 to 24 hours. In contrast, fresh-frozen plasma and prothrombin protein concentrate, which contain clotting factors, reverse warfarin immediately. Currently, a three-factor prothrombin protein concentrate (factors II, IX, and X) and a four-factor concentrate (factors II, VII, IX, and X plus proteins C and S) are available in the United States. Although prothrombin protein concentrate works rapidly and has a lower volume of administration, available data do not indicate it is clinically superior to fresh-frozen plasma.33,34 The ongoing randomized PROTECT trial (NCT00618098), comparing fresh-frozen plasma and four-factor prothrombin protein concentrate for reversal of vitamin K antagonist therapy, may provide further insight.
Efficacy and safety. Randomized controlled trials in patients with nonvalvular atrial fibrillation have shown that warfarin (in doses adjusted to maintain an INR greater than 2) is highly efficacious in preventing systemic embolism, with a relative risk reduction of 61% (95% CI 47%–71%, P < .05) compared with placebo.29,35 An INR of 2 to 3 is recommended for patients with nonvalvular atrial fibrillation, and those with atrial fibrillation and either a bioprosthetic valve or rheumatic heart disease. In contrast, an INR of 2.5 to 3.5 is recommended for patients with atrial fibrillation and mechanical valves in the aortic or mitral positions.1,36
Stroke prevention with warfarin is most effective when the achieved mean time in the therapeutic range is at least 70%. The risk of intracranial hemorrhage increases significantly at INRs higher than 3. An INR of 2 to 3 offers maximum protection with minimal risk of bleeding.37,38 Systematic follow-up of patients through anticoagulation clinics produces better compliance and control and is encouraged.
TARGET-SPECIFIC ORAL ANTICOAGULANTS
Although effective, warfarin requires frequent monitoring and dosage adjustment, has a delayed onset and protracted offset, and interacts with commonly consumed vitamin K–containing foods and frequently used drugs. These drawbacks prompted evaluation of existing antiplatelet agents, in combination or in conjunction with lower adjusted or fixed-dose warfarin. These regimens proved inferior,39–42 spurring interest in developing alternative oral anticoagulants.
TSOACs act by directly inhibiting thrombin (factor IIa) or by reducing thrombin production indirectly by inhibiting factor Xa. Three TSOACs are approved. Each was compared with adjusted-dose warfarin in randomized controlled trials.
Dabigatran
Dabigatran etexilate was the first TSOAC approved in the United States.
Pharmacokinetics. Dabigatran etexilate has a bioavailability of 3% to 7% after oral administration. Its absorption is enhanced in an acidic gastric environment and is limited by P-glycoprotein-facilitated transport out of enterocytes. Dabigatran etexilate is hydrolyzed to its active metabolite dabigatran, which directly inhibits thrombin. Maximal plasma drug concentration and peak anticoagulant effect are achieved within 0.5 to 2 hours after administration.
Dabigatran is predominantly excreted by the kidneys, and has a half-life of 12 to 17 hours in patients with normal renal function. The half-life extends to 27 hours in those with moderately severe renal impairment (creatinine clearance 15–30 mL/min). The recommended dose of 150 mg twice daily should be reduced to 75 mg twice daily in patients with a creatinine clearance of 15 to 30 mL/min. This drug is contraindicated in patients with a creatinine clearance less than 15 mL/min.43,44
Efficacy. The Randomized Evaluation of Long-term Anticoagulation Therapy (RE-LY) trial45 randomly assigned 18,113 patients with nonvalvular atrial fibrillation at risk of thromboembolism (mean CHADS2 score 2.1) to receive either dabigatran (either 150 mg twice daily or 110 mg twice daily) or warfarin (adjusted to an INR of 2.0 to 3.0). Of note, the lower approved dose of dabigatran (75 mg twice daily) was not tested in RE-LY.
At 2 years, higher-dose dabigatran was significantly more effective than both warfarin (RR 0.65, 95% CI 0.52–0.81, P < .05) and lower-dose dabigatran (RR 0.73, 95% CI 0.58–0.91, P < .05) in reducing the rate of systemic embolic events.
The risk of combined major bleeding events was no different with higher-dose dabigatran than with warfarin (RR 0.93, 95% CI 0.81–1.07, P < .05), but the rate of hemorrhagic stroke was significantly less with dabigatran than with warfarin (RR 0.26, 95% CI 0.14–0.49, P < .05). Higher rates of major gastrointestinal bleeding and dyspepsia occurred with dabigatran.
Concern about the safety of dabigatran was raised when post hoc evaluation of RE-LY found a higher incidence of myocardial infarction with dabigatran than with warfarin (RR 1.38, 95% CI 1–1.91, P = .048).46 Corroborating data were reported by Uchino and Hernandez,47 comparing dabigatran with either warfarin or low-molecular-weight heparin. However, without directly comparing dabigatran and placebo, it is unclear whether the small increase in myocardial infarction reflects a direct effect of dabigatran or absence of a protective effect of warfarin or low-molecular-weight heparin.
Rivaroxaban
Rivaroxaban is a direct factor Xa inhibitor that blocks the amplified burst of thrombin production and in turn inhibits platelet aggregation and thrombus formation.
Pharmacokinetics. Rivaroxaban’s oral bioavailability is 80% to 100% after a single 15- or 20-mg dose taken with food. Its maximal anticoagulant effect is achieved within 2 hours. Two-thirds of the active drug is metabolized by either CYP450-dependent (CYP3A4, 2J2) or CYP-independent mechanisms; the inactive drug is then excreted in the urine and feces. The remaining, active drug is removed by the kidneys using the P-glycoprotein transporter.
The half-life of rivaroxaban is 5 to 9 hours. The recommended dosage of 20 mg daily should be reduced to 15 mg daily if the creatinine clearance rate is 30 to 50 mL/min, or to 10 mg if the creatinine clearance rate is 15 to 30 mL/min. Rivaroxaban is contraindicated in patients whose creatinine clearance rate is less than 15 mL/min.48–52
Efficacy and safety. In the Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET-AF),53 14,264 at-risk patients with nonvalvular atrial fibrillation (mean CHADS2 score 3.5) were randomly assigned to receive either rivaroxaban 20 mg daily (or 15 mg daily if their creatinine clearance was 30–49 mL/min; the lowest dose of rivaroxaban, 10 mg, was not studied in this trial) or warfarin (target INR 2.0–3.0). Outcomes with rivaroxaban compared with warfarin:
- Systemic embolism:
HR 0.79, 95% CI 0.66–0.96, P < .01, noninferiority - Total bleeding: no difference
- Intracranial bleeding:
HR 0.67, 95% CI 0.47–0.93, P = .02 - Fatal bleeding:
HR 0.50, 95% CI 0.31–0.79, P = .003 - Major gastrointestinal bleeding:
3.2% vs 2.2%, P < .001.
Apixaban
Apixaban is also a direct factor Xa inhibitor.
Pharmacokinetics. Apixaban’s oral bioavailability is 50%, with maximal blood concentration achieved at 3 to 4 hours. One-quarter of the drug is metabolized via CYP3A4. The remaining active drug is excreted by the kidneys and biliary/intestinal system via the P-glycoprotein transporter. Apixaban’s half-life is 9 to 14 hours.
The recommended dosage is 5 mg twice daily, but it should be reduced to 2.5 mg twice daily if at least two of the following characteristics are present: age 80 or older, weight 60 kg or less, and serum creatinine 1.5 mg/dL or more.54,55
Efficacy and safety. The Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial56 enrolled 18,201 patients with nonvalvular atrial fibrillation (mean CHADS2 score 2.1) randomly assigned to receive either apixaban (5 mg twice daily with dosage reduction to 2.5 mg twice daily as noted above) or warfarin (target INR 2.0–3.0).
Compared with warfarin, apixaban was associated with lower risk of:
- Systemic embolism
(HR 0.79, 95% CI 0.66–0.95, P = .01) - Major bleeding
(HR 0.69, 95% CI 0.60–0.80, P < .001) - Intracranial hemorrhage
(HR 0.42, 95% CI 0.30–0.58, P < .001) - All-cause mortality
(HR 0.89 95% CI 0.80–0.99, P = .047).
Drug interactions with the novel oral anticoagulants
TSOACs were developed with the intent to avoid many of the shortcomings of warfarin. Each has a broader therapeutic window and a rapid onset of action, enabling fixed dosing without need for serial monitoring. Compared with warfarin, they have significantly fewer dietary and drug interactions.
Nonetheless, drug interactions do exist and are important to recognize (Tables 1–3). These primarily result from inhibition or induction of cytochrome P450 enzyme activity or P-glycoprotein transporter action, involved in metabolism and elimination of active drug.
Reversibility of the target-specific oral anticoagulants
While the anticoagulant effects of warfarin can be reversed by vitamin K, fresh-frozen plasma, and prothrombin complex concentrate, TSOACs have no currently approved antidotes. Management of bleeding due to these agents was recently reviewed in this journal by Fawole et al.57
Several nonspecific hemostatic agents have been suggested, including recombinant factor VIIa or prothrombin complex concentrates. The anticoagulant effect of rivaroxaban has been shown to be reversed by prothrombin complex concentrate in vitro; clinical effect has not been demonstrated.58 PRT06445 (andexanet alfa), a recombinant protein antidote specific for factor Xa inhibitors, has entered clinical studies, with a phase 2 trial reporting high reversing capability for apixaban.59
Unlike rivaroxaban and apixaban, which are highly bound to plasma protein, dabigatran can be effectively removed with hemodialysis. Liesenfeld et al60 showed that longer dialysis duration was the most relevant variable for reducing dabigatran plasma levels. Current clinical experience is limited, and standard recommendations and formal guidance are lacking.
Switching oral anticoagulants
Suggested approaches for switching between anticoagulants are listed in Table 4.61
CHOOSING ANTITHROMBOTIC THERAPY
In valvular atrial fibrillation: warfarin
Anticoagulation with warfarin is advised for valvular atrial fibrillation. Patients with bioprosthetic heart valves or rheumatic valvular disease were not evaluated in randomized controlled trials of TSOACs. Dabigatran in particular is contraindicated in patients with mechanical heart valves, as the Randomized, Phase II Study to Evaluate the Safety and Pharmacokinetics of Oral Dabigatran Etexilate in Patients After Heart Valve Replacement (RE-ALIGN)62 found higher rates of stroke, valve-related thrombosis, and myocardial infarction in patients receiving dabigatran.
In nonvalvular atrial fibrillation
According to the 2014 guidelines,1 oral anticoagulation is preferred in all patients with nonvalvular atrial fibrillation but those at lowest risk (CHA2DS2-VASc = 0).
Experience with TSOACs is lacking in patients with end-stage kidney disease (creatinine clearance < 15 mL/min), and warfarin is advised in this group.
TSOACs are recommended in patients with nonvalvular atrial fibrillation in whom therapeutic INR levels cannot be maintained with warfarin. For most patients with nonvalvular atrial fibrillation, TSOACs are an option equivalent to warfarin. Anticoagulant choice is largely driven by dosing convenience, out-of-pocket cost for treatment with a TSOAC, and ready availability of antidotes for warfarin in case of bleeding (Tables 5 and 6).
In patients with nonvalvular atrial fibrillation, TSOACs are as effective as warfarin in preventing systemic thromboembolism, and some of them have been shown to be superior in terms of lower rates of ischemic stroke (dabigatran), systemic embolism (apixaban), and mortality (apixaban; trend for dabigatran). All TSOACs demonstrate modestly favorable bleeding risk profiles compared with warfarin, with lower risk of intracranial hemorrhage. Potential differences in efficacy and safety among TSOACs are unknown since there have been no randomized direct comparisons between them. A summary of landmark trial results and assessment of the advantages and disadvantages of each are listed in Table 7.
Two groups of patients with nonvalvular atrial fibrillation warrant special consideration:
Patients with hypertrophic cardiomyopathy. There are no randomized controlled trials of anticoagulation therapy in patients with hypertrophic cardiomyopathy; however, because of their high risk of thromboembolism, anticoagulation is indicated irrespective of the CHA2DS2-VASc score. TSOACs are an option as an alternative to warfarin.
Patients with coronary artery disease and an indication for antiplatelet therapy. In this group the decision for concurrent anticoagulation is guided by the CHA2DS2-VASc score. For patients who have intracoronary stents, dual antiplatelet therapy is the standard of care for reducing risk of cardiovascular events after stent implantation.63 When triple therapy (ie, two antiplatelet drugs and an anticoagulant) is indicated, such as after intracoronary stent placement, the guidelines suggest trying to minimize the duration of triple therapy. For instance, a bare-metal stent may be preferred. Alternatively, after coronary revascularization, it may be reasonable to use clopidogrel 75 mg daily with an oral anticoagulant and to omit aspirin.
Interrupting and bridging anticoagulation
Patients with atrial fibrillation often require suspension of anticoagulation, most commonly before an elective invasive procedure. The duration of interruption, timing of resumption, and need for bridging anticoagulation are guided by clinical judgment, which considers risk of thromboembolism and severity of procedure-related bleeding risk.
In general, if therapy needs to be interrupted, it should be restarted as soon as possible. Short-term interruption does not seem to be associated with clinically significant risk of thromboembolic events, whereas postoperative heparin bridging therapy increases the risk of hematoma with implantation of a cardiac electronic device.64,65
To date, evidence is lacking to advise upon periprocedure bridging anticoagulation. The Bridging Anticoagulation in Patients Who Require Temporary Interruption of Warfarin Therapy for an Elective Invasive Procedure or Surgery (BRIDGE) study (NCT00786474)— enrolling chronically anticoagulated patients undergoing an invasive procedure to randomly receive placebo or bridging low-molecular-weight heparin—may provide guidance.
Currently, it is common practice in low-risk patients undergoing an invasive procedure with significant bleeding risk to interrupt anticoagulation for up to 1 week without bridging. Warfarin is typically held 3 to 5 days, while TSOACs are held for 24 hours if renal function is preserved or up to 2 to 3 days if renal function is severely impaired (creatinine clearance 15–30 mL/min). If complete hemostasis is necessary, it could be confirmed by a normalized INR (for warfarin), activated partial thromboplastin time (dabigatran), or prothrombin time (apixaban or rivaroxaban).
For patients at high risk (valvular atrial fibrillation or CHA2DS2-VASc ≥ 2), bridging with unfractionated heparin or low-molecular-weight heparin during periods of subtherapeutic anticoagulation is common. Alternatively, it is becoming increasingly common to perform cardiac electronic device implantation, catheter ablation, and coronary angiography and intervention without interrupting anticoagulation.66–72
Recently, concern has been raised over a possible increase in thromboembolism upon discontinuation of rivaroxaban and apixaban. ROCKET-AF reported a spike in thrombotic events in the rivaroxaban-treated group at the end of the trial (HR 1.50, 95% CI 1.05–2.15, P = .026). This raised concern for a possible “rebound” effect upon drug cessation. Yet a post hoc analysis of ROCKET-AF demonstrated that events clustered in the rivaroxaban-treated cohort who completed the study and were transitioning to open-label warfarin, and this alone accounted for the rise in stroke occurrence. In contrast, there was no increase in the cohort of patients treated with rivaroxaban who either temporarily interrupted or permanently discontinued the drug.73 The authors concluded that increased stroke was the consequence of transiently interrupted anticoagulation, rather than a rebound prothrombotic effect. Similar results were reported in ARISTOTLE.
Another possibility is that, during the transition to warfarin therapy, transient hypercoagulability could be a function of warfarin. Azoulay et al74 observed in a large cohort that warfarin was associated with a 71% increased risk of stroke in the first 30 days after initiation, compared with decreased risk thereafter. Nevertheless, there is now a black- box warning recommendation for all three TSOACs that if discontinuation is required for a reason other than pathological bleeding, bridging with another anticoagulant should at least be considered.
The perioperative management of the TSOACs was recently reviewed in this journal by Anderson et al.75
WEIGHING THE RISKS OF STROKE AND BLEEDING
Stroke is the most feared complication in patients with atrial fibrillation. Risk reduction is an important goal in management, yet decisions for individuals must take into account both stroke and bleeding risks related to antithrombotic therapy.
The 2014 guidelines1 differ from past versions. First, they endorse the use of CHA2DS2-VASc for categorizing stroke risk in patients with nonvalvular atrial fibrillation. This in turn guides antithrombotic therapy. This scheme effectively identifies patients at very low risk of stroke (men with a score of 0, women with a score of 0 or 1), in whom it is reasonable to omit antithrombotic therapy. For all patients with valvular heart disease or hypertrophic cardiomyopathy, unless bleeding risk is prohibitive, anticoagulation is recommended irrespective of the CHA2DS2-VASc score. Second, they incorporate the TSOACs, which offer convenience and improved safety in select patients.
While the guidelines mention the potential relevance of subclinical atrial tachyarrhythmias as they pertain to stroke risk, there is no specific recommendation as to their management. We do take into consideration the finding of atrial high-rate events (≥ 180 bpm, ≥ 6 minutes in duration) diagnostically confirmed by cardiac implantable electronic devices or telemetric monitoring, particularly in patients with a clinical profile of high stroke risk. In addition, atriopathy with increased left atrial size and renal insufficiency, as discussed in this review, appear to correlate with greater risk of thromboembolism, yet neither is a component of the stroke risk scheme endorsed by the guidelines.
Other risk factors, some unknown to us, undoubtedly exist. Again, our empiric judgment is to at least consider these nontraditional risk factors while guided primarily by the CHA2DS2-VASc score when assessing stroke risk in patients with atrial fibrillation.
The goal in managing patients with atrial fibrillation is to balance thromboembolic risk reduction with the risk of bleeding associated with antithrombotic therapy.
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med 1982; 306:1018–1022.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Hart RG, Pearce LA, Rothbart RM, McAnulty JH, Asinger RW, Halperin JL. Stroke with intermittent atrial fibrillation: incidence and predictors during aspirin therapy. Stroke Prevention in Atrial Fibrillation Investigators. J Am Coll Cardiol 2000; 35:183–187.
- Hohnloser SH, Pajitnev D, Pogue J, et al; ACTIVE W Investigators. Incidence of stroke in paroxysmal versus sustained atrial fibrillation in patients taking oral anticoagulation or combined antiplatelet therapy: an ACTIVE W Substudy. J Am Coll Cardiol 2007; 50:2156–2161.
- Healey JS, Connolly SJ, Gold MR, et al; ASSERT Investigators. Subclinical atrial fibrillation and the risk of stroke. N Engl J Med 2012; 366:120–129.
- Glotzer TV, Hellkamp AS, Zimmerman J, et al; MOST Investigators. Atrial high rate episodes detected by pacemaker diagnostics predict death and stroke: report of the Atrial Diagnostics Ancillary Study of the MOde Selection Trial (MOST). Circulation 2003; 107:1614–1619.
- Glotzer TV, Daoud EG, Wyse DG, et al. The relationship between daily atrial tachyarrhythmia burden from implantable device diagnostics and stroke risk: the TRENDS study. Circ Arrhythm Electrophysiol 2009; 2:474–480.
- Atrial Fibrillation Investigators. Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Maron BJ, Olivotto I, Bellone P, et al. Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:301–307.
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104:2517–2524.
- Go AS, Fang MC, Udaltsova N, et al; ATRIA Study Investigators. Impact of proteinuria and glomerular filtration rate on risk of thromboembolism in atrial fibrillation: the anticoagulation and risk factors in atrial fibrillation (ATRIA) study. Circulation 2009; 119:1363–1369.
- Hart RG, Pearce LA, Asinger RW, Herzog CA. Warfarin in atrial fibrillation patients with moderate chronic kidney disease. Clin J Am Soc Nephrol 2011; 6:2599–2604.
- Vázquez E, Sánchez-Perales C, Borrego F, et al. Influence of atrial fibrillation on the morbido-mortality of patients on hemodialysis. Am Heart J 2000; 140:886–890.
- The Stroke Prevention in Atrial Fibrillation Investigators Committee on Echocardiography. Transesophageal echocardiographic correlates of thromboembolism in high-risk patients with nonvalvular atrial fibrillation. Ann Intern Med 1998; 128:639–647.
- The Stroke Prevention in Atrial Fibrillation Investigators. Predictors of thromboembolism in atrial fibrillation: II. Echocardiographic features of patients at risk. Ann Intern Med 1992; 116:6–12.
- Osranek M, Bursi F, Bailey KR, et al. Left atrial volume predicts cardiovascular events in patients originally diagnosed with lone atrial fibrillation: three-decade follow-up. Eur Heart J 2005; 26:2556–2561.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- van Walraven C, Hart RG, Wells GA, et al. A clinical prediction rule to identify patients with atrial fibrillation and a low risk for stroke while taking aspirin. Arch Intern Med 2003; 163:936–943.
- Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J 2012; 33:2719–2747.
- Lip GY, Frison L, Halperin JL, Lane DA. Identifying patients at high risk for stroke despite anticoagulation: a comparison of contemporary stroke risk stratification schemes in an anticoagulated atrial fibrillation cohort. Stroke 2010; 41:2731–2738.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- Olesen JB, Torp-Pedersen C, Hansen ML, Lip GY. The value of the CHA2DS2-VASc score for refining stroke risk stratification in patients with atrial fibrillation with a CHADS2 score 0-1: a nationwide cohort study. Thromb Haemost 2012; 107:1172–1179.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Olesen JB, Lip GY, Lindhardsen J, et al. Risks of thromboembolism and bleeding with thromboprophylaxis in patients with atrial fibrillation: a net clinical benefit analysis using a ‘real world’ nationwide cohort study. Thromb Haemost 2011; 106:739–749.
- Friberg L, Rosenqvist M, Lip GY. Net clinical benefit of warfarin in patients with atrial fibrillation: a report from the Swedish atrial fibrillation cohort study. Circulation 2012; 125:2298–2307.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Hart RG, Pearce LA, Aguilar MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med 2007; 146:857–867.
- The Atrial Fibrillation Investigators. The efficacy of aspirin in patients with atrial fibrillation. Analysis of pooled data from 3 randomized trials. Arch Intern Med 1997; 157:1237–1240.
- Miller VT, Rothrock JF, Pearce LA, Feinberg WM, Hart RG, Anderson DC. Ischemic stroke in patients with atrial fibrillation: effect of aspirin according to stroke mechanism. Stroke Prevention in Atrial Fibrillation Investigators. Neurology 1993; 43:32–36.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery 1999; 45:1113–1119.
- Huttner HB, Schellinger PD, Hartmann M, et al. Hematoma growth and outcome in treated neurocritical care patients with intracerebral hemorrhage related to oral anticoagulant therapy: comparison of acute treatment strategies using vitamin K, fresh frozen plasma, and prothrombin complex concentrates. Stroke 2006; 37:1465–1470.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Gallagher AM, Setakis E, Plumb JM, Clemens A, van Staa TP. Risks of stroke and mortality associated with suboptimal anticoagulation in atrial fibrillation patients. Thromb Haemost 2011; 106:968–977.
- Odén A, Fahlén M, Hart RG. Optimal INR for prevention of stroke and death in atrial fibrillation: a critical appraisal. Thromb Res 2006; 117:493–499.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG, et al. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Adjusted-dose warfarin versus low-intensity, fixed-dose warfarin plus aspirin for high-risk patients with atrial fibrillation: Stroke Prevention in Atrial Fibrillation III randomised clinical trial. Lancet 1996; 348:633–638.
- Gulløv AL, Koefoed BG, Petersen P, et al. Fixed minidose warfarin and aspirin alone and in combination vs adjusted-dose warfarin for stroke prevention in atrial fibrillation: Second Copenhagen Atrial Fibrillation, Aspirin, and Anticoagulation Study. Arch Intern Med 1998; 158:1513–1521.
- Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos 2008; 36:386–399.
- Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 2007; 64:292–303.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Hohnloser SH, Oldgren J, Yang S, et al. Myocardial ischemic events in patients with atrial fibrillation treated with dabigatran or warfarin in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial. Circulation 2012; 125:669–676.
- Uchino K, Hernandez AV. Dabigatran association with higher risk of acute coronary events: meta-analysis of noninferiority randomized controlled trials. Arch Intern Med 2012; 172:397–402.
- Kubitza D, Becka M, Voith B, Zuehlsdorf M, Wensing G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther 2005; 78:412–421.
- Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct Factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharmacol 2005; 61:873–880.
- Kubitza D, Becka M, Roth A, Mueck W. Dose-escalation study of the pharmacokinetics and pharmacodynamics of rivaroxaban in healthy elderly subjects. Curr Med Res Opin 2008; 24:2757–2765.
- Weinz C, Schwarz T, Kubitza D, Mueck W, Lang D. Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab Dispos 2009; 37:1056–1064.
- Perzborn E, Roehrig S, Straub A, Kubitza D, Misselwitz F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat Rev Drug Discov 2011; 10:61–75.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Wong PC, Pinto DJ, Zhang D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis 2011; 31:478–492.
- Carreiro J, Ansell J. Apixaban, an oral direct Factor Xa inhibitor: awaiting the verdict. Expert Opin Investig Drugs 2008; 17:1937–1945.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Lu G, DeGuzman FR, Hollenbach SJ, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med 2013; 19:446–451.
- Liesenfeld KH, Staab A, Härtter S, Formella S, Clemens A, Lehr T. Pharmacometric characterization of dabigatran hemodialysis. Clin Pharmacokinet 2013; 52:453–462.
- MPR. Monthly Prescribing Reference. Anticoagulant dosing conversions. August 18, 2014. www.empr.com/anticoagulant-dosing-conversions/article/194271/. Accessed December 11, 2014.
- Eikelboom JW, Connolly SJ, Brueckmann M, et al; RE-ALIGN Investigators. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med 2013; 369:1206–1214.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Tischenko A, Gula LJ, Yee R, Klein GJ, Skanes AC, Krahn AD. Implantation of cardiac rhythm devices without interruption of oral anticoagulation compared with perioperative bridging with low-molecular weight heparin. Am Heart J 2009; 158:252–256.
- Robinson M, Healey JS, Eikelboom J, et al. Postoperative low-molecular-weight heparin bridging is associated with an increase in wound hematoma following surgery for pacemakers and implantable defibrillators. Pacing Clin Electrophysiol 2009; 32:378–382.
- Birnie DH, Healey JS, Wells GA, et al; BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
- Ahmed I, Gertner E, Nelson WB, et al. Continuing warfarin therapy is superior to interrupting warfarin with or without bridging anticoagulation therapy in patients undergoing pacemaker and defibrillator implantation. Heart Rhythm 2010; 7:745–749.
- Cheng A, Nazarian S, Brinker JA, et al. Continuation of warfarin during pacemaker or implantable cardioverter-defibrillator implantation: a randomized clinical trial. Heart Rhythm 2011; 8:536–540.
- Jamula E, Lloyd NS, Schwalm JD, Airaksinen KE, Douketis JD. Safety of uninterrupted anticoagulation in patients requiring elective coronary angiography with or without percutaneous coronary intervention: a systematic review and metaanalysis. Chest 2010; 138:840–847.
- Jamula E, Douketis JD, Schulman S. Perioperative anticoagulation in patients having implantation of a cardiac pacemaker or defibrillator: a systematic review and practical management guide. J Thromb Haemost 2008; 6:1615–1621.
- Korantzopoulos P, Letsas KP, Liu T, Fragakis N, Efremidis M, Goudevenos JA. Anticoagulation and antiplatelet therapy in implantation of electrophysiological devices. Europace 2011; 13:1669–1680.
- Di Biase L, Burkhardt JD, Mohanty P, et al. Periprocedural stroke and management of major bleeding complications in patients undergoing catheter ablation of atrial fibrillation: the impact of periprocedural therapeutic international normalized ratio. Circulation 2010; 121:2550–2556.
- Patel MR, Hellkamp AS, Lokhnygina Y, et al. Outcomes of discontinuing rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: analysis from the ROCKET AF trial (Rivaroxaban Once-Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation). J Am Coll Cardiol 2013; 61:651–658.
- Azoulay L, Dell’aniello S, Simon TA, Renoux C, Suissa S. Initiation of warfarin in patients with atrial fibrillation: early effects on ischaemic strokes. Eur Heart J 2013; Dec 18 [Epub ahead of print].
- Anderson M, Hassell KL, Trujillo TC, Wolfe B. When patients on target-specific oral anticoagulants need surgery. Cleve Clin J Med 2014; 81:629–639.
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med 1982; 306:1018–1022.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Hart RG, Pearce LA, Rothbart RM, McAnulty JH, Asinger RW, Halperin JL. Stroke with intermittent atrial fibrillation: incidence and predictors during aspirin therapy. Stroke Prevention in Atrial Fibrillation Investigators. J Am Coll Cardiol 2000; 35:183–187.
- Hohnloser SH, Pajitnev D, Pogue J, et al; ACTIVE W Investigators. Incidence of stroke in paroxysmal versus sustained atrial fibrillation in patients taking oral anticoagulation or combined antiplatelet therapy: an ACTIVE W Substudy. J Am Coll Cardiol 2007; 50:2156–2161.
- Healey JS, Connolly SJ, Gold MR, et al; ASSERT Investigators. Subclinical atrial fibrillation and the risk of stroke. N Engl J Med 2012; 366:120–129.
- Glotzer TV, Hellkamp AS, Zimmerman J, et al; MOST Investigators. Atrial high rate episodes detected by pacemaker diagnostics predict death and stroke: report of the Atrial Diagnostics Ancillary Study of the MOde Selection Trial (MOST). Circulation 2003; 107:1614–1619.
- Glotzer TV, Daoud EG, Wyse DG, et al. The relationship between daily atrial tachyarrhythmia burden from implantable device diagnostics and stroke risk: the TRENDS study. Circ Arrhythm Electrophysiol 2009; 2:474–480.
- Atrial Fibrillation Investigators. Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Maron BJ, Olivotto I, Bellone P, et al. Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39:301–307.
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104:2517–2524.
- Go AS, Fang MC, Udaltsova N, et al; ATRIA Study Investigators. Impact of proteinuria and glomerular filtration rate on risk of thromboembolism in atrial fibrillation: the anticoagulation and risk factors in atrial fibrillation (ATRIA) study. Circulation 2009; 119:1363–1369.
- Hart RG, Pearce LA, Asinger RW, Herzog CA. Warfarin in atrial fibrillation patients with moderate chronic kidney disease. Clin J Am Soc Nephrol 2011; 6:2599–2604.
- Vázquez E, Sánchez-Perales C, Borrego F, et al. Influence of atrial fibrillation on the morbido-mortality of patients on hemodialysis. Am Heart J 2000; 140:886–890.
- The Stroke Prevention in Atrial Fibrillation Investigators Committee on Echocardiography. Transesophageal echocardiographic correlates of thromboembolism in high-risk patients with nonvalvular atrial fibrillation. Ann Intern Med 1998; 128:639–647.
- The Stroke Prevention in Atrial Fibrillation Investigators. Predictors of thromboembolism in atrial fibrillation: II. Echocardiographic features of patients at risk. Ann Intern Med 1992; 116:6–12.
- Osranek M, Bursi F, Bailey KR, et al. Left atrial volume predicts cardiovascular events in patients originally diagnosed with lone atrial fibrillation: three-decade follow-up. Eur Heart J 2005; 26:2556–2561.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- van Walraven C, Hart RG, Wells GA, et al. A clinical prediction rule to identify patients with atrial fibrillation and a low risk for stroke while taking aspirin. Arch Intern Med 2003; 163:936–943.
- Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J 2012; 33:2719–2747.
- Lip GY, Frison L, Halperin JL, Lane DA. Identifying patients at high risk for stroke despite anticoagulation: a comparison of contemporary stroke risk stratification schemes in an anticoagulated atrial fibrillation cohort. Stroke 2010; 41:2731–2738.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- Olesen JB, Torp-Pedersen C, Hansen ML, Lip GY. The value of the CHA2DS2-VASc score for refining stroke risk stratification in patients with atrial fibrillation with a CHADS2 score 0-1: a nationwide cohort study. Thromb Haemost 2012; 107:1172–1179.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Olesen JB, Lip GY, Lindhardsen J, et al. Risks of thromboembolism and bleeding with thromboprophylaxis in patients with atrial fibrillation: a net clinical benefit analysis using a ‘real world’ nationwide cohort study. Thromb Haemost 2011; 106:739–749.
- Friberg L, Rosenqvist M, Lip GY. Net clinical benefit of warfarin in patients with atrial fibrillation: a report from the Swedish atrial fibrillation cohort study. Circulation 2012; 125:2298–2307.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Hart RG, Pearce LA, Aguilar MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med 2007; 146:857–867.
- The Atrial Fibrillation Investigators. The efficacy of aspirin in patients with atrial fibrillation. Analysis of pooled data from 3 randomized trials. Arch Intern Med 1997; 157:1237–1240.
- Miller VT, Rothrock JF, Pearce LA, Feinberg WM, Hart RG, Anderson DC. Ischemic stroke in patients with atrial fibrillation: effect of aspirin according to stroke mechanism. Stroke Prevention in Atrial Fibrillation Investigators. Neurology 1993; 43:32–36.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery 1999; 45:1113–1119.
- Huttner HB, Schellinger PD, Hartmann M, et al. Hematoma growth and outcome in treated neurocritical care patients with intracerebral hemorrhage related to oral anticoagulant therapy: comparison of acute treatment strategies using vitamin K, fresh frozen plasma, and prothrombin complex concentrates. Stroke 2006; 37:1465–1470.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Gallagher AM, Setakis E, Plumb JM, Clemens A, van Staa TP. Risks of stroke and mortality associated with suboptimal anticoagulation in atrial fibrillation patients. Thromb Haemost 2011; 106:968–977.
- Odén A, Fahlén M, Hart RG. Optimal INR for prevention of stroke and death in atrial fibrillation: a critical appraisal. Thromb Res 2006; 117:493–499.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- ACTIVE Investigators; Connolly SJ, Pogue J, Hart RG, et al. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N Engl J Med 2009; 360:2066–2078.
- Adjusted-dose warfarin versus low-intensity, fixed-dose warfarin plus aspirin for high-risk patients with atrial fibrillation: Stroke Prevention in Atrial Fibrillation III randomised clinical trial. Lancet 1996; 348:633–638.
- Gulløv AL, Koefoed BG, Petersen P, et al. Fixed minidose warfarin and aspirin alone and in combination vs adjusted-dose warfarin for stroke prevention in atrial fibrillation: Second Copenhagen Atrial Fibrillation, Aspirin, and Anticoagulation Study. Arch Intern Med 1998; 158:1513–1521.
- Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos 2008; 36:386–399.
- Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 2007; 64:292–303.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Hohnloser SH, Oldgren J, Yang S, et al. Myocardial ischemic events in patients with atrial fibrillation treated with dabigatran or warfarin in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial. Circulation 2012; 125:669–676.
- Uchino K, Hernandez AV. Dabigatran association with higher risk of acute coronary events: meta-analysis of noninferiority randomized controlled trials. Arch Intern Med 2012; 172:397–402.
- Kubitza D, Becka M, Voith B, Zuehlsdorf M, Wensing G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther 2005; 78:412–421.
- Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct Factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharmacol 2005; 61:873–880.
- Kubitza D, Becka M, Roth A, Mueck W. Dose-escalation study of the pharmacokinetics and pharmacodynamics of rivaroxaban in healthy elderly subjects. Curr Med Res Opin 2008; 24:2757–2765.
- Weinz C, Schwarz T, Kubitza D, Mueck W, Lang D. Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab Dispos 2009; 37:1056–1064.
- Perzborn E, Roehrig S, Straub A, Kubitza D, Misselwitz F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat Rev Drug Discov 2011; 10:61–75.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Wong PC, Pinto DJ, Zhang D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis 2011; 31:478–492.
- Carreiro J, Ansell J. Apixaban, an oral direct Factor Xa inhibitor: awaiting the verdict. Expert Opin Investig Drugs 2008; 17:1937–1945.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Lu G, DeGuzman FR, Hollenbach SJ, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med 2013; 19:446–451.
- Liesenfeld KH, Staab A, Härtter S, Formella S, Clemens A, Lehr T. Pharmacometric characterization of dabigatran hemodialysis. Clin Pharmacokinet 2013; 52:453–462.
- MPR. Monthly Prescribing Reference. Anticoagulant dosing conversions. August 18, 2014. www.empr.com/anticoagulant-dosing-conversions/article/194271/. Accessed December 11, 2014.
- Eikelboom JW, Connolly SJ, Brueckmann M, et al; RE-ALIGN Investigators. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med 2013; 369:1206–1214.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Tischenko A, Gula LJ, Yee R, Klein GJ, Skanes AC, Krahn AD. Implantation of cardiac rhythm devices without interruption of oral anticoagulation compared with perioperative bridging with low-molecular weight heparin. Am Heart J 2009; 158:252–256.
- Robinson M, Healey JS, Eikelboom J, et al. Postoperative low-molecular-weight heparin bridging is associated with an increase in wound hematoma following surgery for pacemakers and implantable defibrillators. Pacing Clin Electrophysiol 2009; 32:378–382.
- Birnie DH, Healey JS, Wells GA, et al; BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
- Ahmed I, Gertner E, Nelson WB, et al. Continuing warfarin therapy is superior to interrupting warfarin with or without bridging anticoagulation therapy in patients undergoing pacemaker and defibrillator implantation. Heart Rhythm 2010; 7:745–749.
- Cheng A, Nazarian S, Brinker JA, et al. Continuation of warfarin during pacemaker or implantable cardioverter-defibrillator implantation: a randomized clinical trial. Heart Rhythm 2011; 8:536–540.
- Jamula E, Lloyd NS, Schwalm JD, Airaksinen KE, Douketis JD. Safety of uninterrupted anticoagulation in patients requiring elective coronary angiography with or without percutaneous coronary intervention: a systematic review and metaanalysis. Chest 2010; 138:840–847.
- Jamula E, Douketis JD, Schulman S. Perioperative anticoagulation in patients having implantation of a cardiac pacemaker or defibrillator: a systematic review and practical management guide. J Thromb Haemost 2008; 6:1615–1621.
- Korantzopoulos P, Letsas KP, Liu T, Fragakis N, Efremidis M, Goudevenos JA. Anticoagulation and antiplatelet therapy in implantation of electrophysiological devices. Europace 2011; 13:1669–1680.
- Di Biase L, Burkhardt JD, Mohanty P, et al. Periprocedural stroke and management of major bleeding complications in patients undergoing catheter ablation of atrial fibrillation: the impact of periprocedural therapeutic international normalized ratio. Circulation 2010; 121:2550–2556.
- Patel MR, Hellkamp AS, Lokhnygina Y, et al. Outcomes of discontinuing rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: analysis from the ROCKET AF trial (Rivaroxaban Once-Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation). J Am Coll Cardiol 2013; 61:651–658.
- Azoulay L, Dell’aniello S, Simon TA, Renoux C, Suissa S. Initiation of warfarin in patients with atrial fibrillation: early effects on ischaemic strokes. Eur Heart J 2013; Dec 18 [Epub ahead of print].
- Anderson M, Hassell KL, Trujillo TC, Wolfe B. When patients on target-specific oral anticoagulants need surgery. Cleve Clin J Med 2014; 81:629–639.
KEY POINTS
- Valvular atrial fibrillation poses a high risk of systemic embolization, particularly stroke, and nearly all patients who have valvular atrial fibrillation need anticoagulation therapy with warfarin.
- Nonvalvular atrial fibrillation poses a somewhat lower risk. The new guidelines propose a new risk-classification scheme, called CHA2DS2-VASc; patients at very low risk of stroke may be able to forgo anticoagulation.
- The new guidelines downplay the role of aspirin, although it is still an option in some situations.
- Several novel oral anticoagulants have been approved in the past few years for thromboprophylaxis in patients with nonvalvular atrial fibrillation.
Quitting smoking: Still a challenge, but newer tools show promise
Tobacco is a dirty weed,
I like it.
It satisfies no normal need,
I like it.
It makes you thin, it makes you lean,
It takes the hair right off your bean.
It’s the worst darn stuff I’ve ever seen.
I like it.
Graham Lee Hemminger. The Penn State Froth, November 1915: 19. Courtesy of Paul J. Dzyak, Jr., Paterno Library, Pennsylvania State University, State College, PA.
All physicians recognize the harm in tobacco smoking and try to convince patients to quit for health reasons, but quitting is challenging and frustrating for both doctor and patient. Physicians can improve quitting outcomes by applying their knowledge of the physiologic basis of nicotine addiction and newer tools that are making a real difference in smoking cessation.
THE NO. 1 PREVENTABLE CAUSE OF DEATH
Tobacco use remains the single largest preventable cause of death and disease in the United States: 443,000 US adults die of smoking-related illnesses each year, or one every 8 seconds.1 Tobacco smoking is currently responsible for 18% of all deaths and 37% of all preventable deaths. One-third of all smokers die early, with men losing 13 years of life and women losing 15 years. (See The rise and partial fall of smoking for a historical overview.)
Smoking, the leading cause of lung cancer, is also implicated in cancers of the mouth, larynx, esophagus, stomach, kidney, bladder, and cervix and has been linked to leukemia. (Though nicotine is responsible for the addictive properties of tobacco, it does not cause cancer itself: other substances in tobacco smoke, many of them byproducts of combustion, are carcinogenic.)

Running a close second to cancer as a smoking-related cause of death is cardiovascular disease, including stroke, myocardial infarction, microvascular dementia, peripheral vascular disease, and aortic aneurysm. Pulmonary and respiratory diseases, including chronic obstructive pulmonary disease, pneumonia, and asthma, are the third most common fatal smoking-related ailments.
Other medical consequences include erectile dysfunction, infertility, pregnancy complications, and low birth weight. Smoking also causes adverse surgical outcomes, poor wound healing, hip fractures, low bone density, peptic ulcer disease, and cataracts.
Smoking is estimated to cost the United States $96 billion in direct medical expenses and $97 billion in lost productivity annually.2
On the positive side, quitting smoking has health benefits at any age, and smokers who quit before age 35 have death rates similar to those in people who have never smoked.1,3
WHY IS IT SO HARD TO QUIT?
Most smokers want to quit, and many try to—but few succeed. In the 2010 National Health Interview Surveys, 68.8% of adult smokers said they wanted to stop smoking, and 52.4% had tried to in the past year, but only 6.2% had succeeded.4 Many recovering alcoholics and drug addicts say that quitting tobacco was much harder than abstaining from other substances of choice.
Why is it so hard to quit?
Smoking is a classic addiction
Addictions are usually diagnosed by behavioral signs, and nicotine addiction has many of the clinical hallmarks, eg:
- Tolerance, with a trend toward increasing the potency of the dose and the frequency of smoking over time
- Mental preoccupation with smoking, as it often becomes woven into one’s daily schedule and is associated with almost everything the smoker does throughout the day. Having no cigarettes in the house can generate anxiety that is relieved only by obtaining more
- Squandering scarce financial resources on nicotine products, over time amounting to substantial sums, and since smoking rates are higher in poor people than in the affluent, these are people who can least afford it
- Withdrawal symptoms, characterized by jitteriness, irritability, headache, insomnia, anxiety, and increased appetite.
People continue to smoke despite adverse consequences such as falling asleep while smoking and setting fire to the bed or to the house, or losing digits to peripheral vascular disease. Being unable to quit and to stay off smoking is a hallmark of tobacco dependence. Relapses are often triggered by being near other smokers or seeing a billboard advertising cigarettes. Eventually, the nicotine addict comes to value and crave nicotine more than health or life itself.
Nicotine stimulates ‘reward’ centers in the brain
Nicotine is an alkaloid found in many plants (including potatoes) but in especially high concentrations in tobacco. In mammals, it is a stimulant, rapidly producing dependence and addiction.
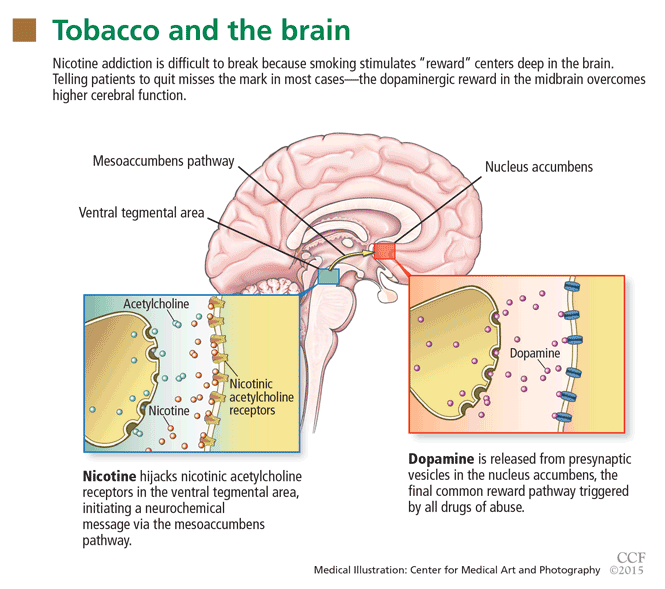
Inhaled by smoking, nicotine is absorbed across the large alveolar surface, avoids first-pass metabolism, and is transported rapidly to the brain (Figure 1). In fact, nicotine reaches the brain less than 20 seconds after inhalation, which is slightly faster even than when drugs are injected intravenously.5
Tobacco smoke contains approximately 4,800 compounds, many of which activate neurotransmitter systems such as dopamine, norepinephrine, acetylcholine, glutamate, serotonin, beta-endorphin, and gamma-aminobutyric acid. The most significant of these is the dopamine reward system known as the mesoaccumbens pathway. This system is activated within seconds of smoking and produces a sense of pleasure.
Nicotine binds to nicotinic acetylcholine receptors, primarily to alpha-4, beta-2 receptors in the ventral tegmental area of the midbrain. Once this binding occurs, a neurochemical message is conveyed to the nucleus accumbens via the release of dopamine in the mesoaccumbens pathway—the final common reward pathway triggered by all drugs of abuse. Since these structures and pathways of the brain are anatomically central, the addiction is driven by the basal ganglia and midbrain, the phylogenetically oldest parts of the brain. Nicotine therefore drives its addicts to continue smoking by producing strong neurochemical rewards and by causing strongly negative reactions when discontinued.
Genetically mediated susceptibility probably contributes to addiction. People whose neurochemical pathways are easily stimulated by this drug are probably at far greater risk of addiction. Paradoxically, people who are rapid metabolizers of nicotine are at greater risk than slow metabolizers.6 (Nicotine is metabolized by cytochrome P450 2A6 in the liver.)
Tolerance and withdrawal
Tolerance develops with long-term use, mediated by up-regulation (increased numbers) of alpha-4, beta-2 cholinergic receptors in the ventral tegmental area. Any reduction in nicotine level causes distress because receptors are unoccupied; with more receptors, nicotine intake must increase to keep physiologic balance and avoid withdrawal. Since the half-life of nicotine is only about 2 hours, the smoker must smoke almost constantly to satisfy receptors hungry for the stimulating drug. If drug levels drop, withdrawal occurs very quickly.
Eventually, smokers use nicotine less for pleasure and more as a way to avoid withdrawal. The cycle of pleasure, eventual tolerance, withdrawal, craving, and compulsion is biologically driven, like the drives of thirst, reproduction, and hunger. Nicotine hijacks species-sustaining reward mechanisms, leading to the malignant, compulsive disease of nicotine addiction.
Treatment doomed to fail?
Because nicotine addiction involves the midbrain, cessation strategies that rely on higher cerebral function are not likely to succeed. Counseling, common sense, and willpower simply cannot overcome the dopaminergic stimulating power or assuage the withdrawal sickness of nicotine dependence. Telling patients that smoking is bad for them misses the mark in most cases. Patients want to quit, but the drive to smoke is too powerful. Attempts to cut down rather than abstain from smoking also fail.
Nicotine is a formidable adversary for the patient and for the doctor or other health professional. Until recently, treatment was usually ineffective.
So, what does work against nicotine addiction?
PHARMACOTHERAPIES FOR SMOKING CESSATION
Nicotine replacement therapy
The oldest of the pharmacotherapies for nicotine addiction is nicotine replacement, in the form of patch, gum, lozenge, or nasal spray.
Advantages:
- Nicotine replacement therapy eliminates exposure to the other harmful compounds in tobacco, with few to none of the health risks associated with smoking.
- By delivering nicotine by a different route, nicotine replacement therapy breaks the association between smoking and feeling good. The addict is already dopamine-stimulated before putting a cigarette in the mouth, merely by association and suggestion. Using a different route of nicotine administration avoids that associative stimulation from the act of smoking, so that quitting becomes easier.
- The dose of nicotine is lower with replacement therapy than with smoking. The cigarette is the most efficient delivery mechanism for getting nicotine into the body. A smoked cigarette produces a rapid spike in plasma nicotine levels, far higher and faster than nicotine gum, nasal spray, or transdermal patch. Peak levels of plasma nicotine from nicotine replacement therapy are only 30% to 50% as high as those achieved by smoking.7–9
- It is inexpensive.
Disadvantages:
- Nicotine replacement therapy maintains the addiction to nicotine, with its neurophysiologic distortions.
- Some patients continue nicotine replacement therapy for years.
Use of nicotine gum can be a problem because of the need for frequent administration. The gum is chewed until the user feels a tingling or peppery taste in the mouth, after which the gum must be placed inside the cheek to allow for maximal absorption of the nicotine. Once the tingling has faded, the user is to chew another piece and repeat the cycle as long as craving is perceived. On the other hand, the nicotine patch is applied once daily. Both of these products are available over-the-counter.
Caution is indicated when starting nicotine replacement therapy in those with recent myocardial infarction, angina, or arrhythmia.
Effectiveness. Nicotine replacement therapy has been shown to be as effective as bupropion (see below) but not as effective as varenicline when used in single administration form (patch, gum, lozenge, or inhaler alone). The four single-administration forms of nicotine replacement therapy are all equally efficacious. Combinations of nicotine replacement formulations have been reported to be as effective as varenicline and superior to single formulations.10
How about electronic cigarettes? Electronic cigarettes, or e-cigarettes, supply nicotine in a noncombustion vapor and are advertised as an alternative to smoking. No claim is made for reducing smoking, so the products, including the liquids involved, are not regulated by the US Food and Drug Administration (FDA). Controversy exists as to whether they actually increase the number of smokers by introducing young people to “vaping” to get nicotine. Since nicotine is still inhaled, the addictive potential remains unabated. E-cigarettes are unregulated vehicles for supplying nicotine and may pose other health risks, and there is very limited evidence to support the efficacy of e-cigarettes as aids to smoking cessation. Since no controlled study has demonstrated successful cessation of smoking with e-cigarettes, they are best regarded for now as merely another way to introduce nicotine into the body.
Bupropion
Bupropion, an antidepressant also sold as Wellbutrin SR, was approved in 1997 for use in smoking cessation under the trade name Zyban. The manufacturer, Glaxo SmithKline, learned serendipitously that depressive patients taking bupropion were able to quit smoking. After some field trials, this “new” medication was born. It was the first nonnicotine drug for tobacco dependence to gain FDA approval.
Its mechanism of action in combating smoking is unknown but is thought to be related to mild inhibition of dopamine re-uptake in the midbrain.
The drug is approved for smokers over age 18 who are smoking at least nine cigarettes daily. It requires a prescription, and the typical dose is 150 mg twice daily for 8 to 12 weeks, up to 12 months. Smoking is allowed for the first 7 days of drug use.
Contraindications include a history of seizures, concurrent use of bupropion, bulimia, anorexia, detoxification from alcohol or sedatives, use of monoamine oxidase inhibitors, and allergy to bupropion. Warnings are noted for diseases of heart, liver, or kidney; for use with selective serotonin reuptake inhibitors or tricyclic antidepressants; for pregnancy; and for adolescents because of heightened suicide risk.
Side effects. Seizure risk has been estimated at 1 in 1,000 bupropion users at dosages of up to 300 mg daily and is 10 times greater at dosages of 450 to 600 mg/day.11
The most common side effect reported is insomnia, which occurs in about one-third of people who take the medication. Less common side effects include dry mouth, anxiety, and hypertension. Pretreatment screening should include a history of seizure, closed head trauma, brain surgery, stroke, and the eating disorders anorexia nervosa and bulimia. The FDA has required a boxed warning regarding the association of bupropion with psychiatric symptoms.12
Effectiveness. Compared with placebo, bupropion reduces withdrawal symptoms such as irritability, frustration, anger, restlessness, depression, craving, poor concentration, and urge to smoke. Bupropion SR, 150 or 300 mg per day, has been reported to lead to substantial abstinence rates when used with intensive telephone counseling. In a randomized trial,13 side effects were common, especially at the higher dose, but there were no serious adverse effects such as deaths or seizures.13
Buproprion has been found to be as efficacious in improving the odds of quitting as single forms of nicotine replacement therapy, but not as efficacious as nicotine replacement therapy forms used in combination. Bupropion does not appear to be as effective as varenicline.9 US Public Health Service guidelines since 2000 have included nicotine replacement therapy and sustained-release bupropion in combination.
Disadvantages. Bupropion is significantly more expensive than nicotine replacement therapy, but it is often covered by insurance when it is used for smoking cessation. Bupropion has many contraindications, produces drug-drug interactions, is often poorly tolerated, and has many side effects. Some deaths have been reported. Zyban is available by prescription only, an indicator of its relative risk, with the added drawback of higher cost to patients.
Varenicline
Varenicline (Chantix, Champix) was granted a priority review by the FDA in 2005, as it showed significantly better results than other current therapies. It was approved in 2006 and added as a first-line agent in the 2008 guidelines.12
Mechanism of action. A synthetic “designer” drug made for its specific purpose, the varenicline molecule is a modified version of cytisine, a naturally occurring alkaloid previously marketed as Tabex in Eastern Europe. Cytisine is a selective alpha-4, beta-2 nicotinic acetylcholine receptor partial agonist. The high-affinity alpha-4, beta-2 nicotinic acetylcholine receptors exist in the mesolimbic dopaminergic system, the reward center of the brain.14
Varenicline has the same mechanism of action as cytisine but penetrates the central nervous system better. This mechanism of action allows varenicline to block the attachment of the nicotine molecule to this receptor, preventing nicotine’s dominant effect. Varenicline, however, is a partial agonist, so that when it attaches itself to the receptor, it causes a partial agonist effect, which is an opening of the receptor channel to sodium ions, causing partial stimulation of the cells in the ventral tegmental area, and ultimately causing a mild release of dopamine in the nucleus accumbens.15,16 Thus, varenicline effectively stimulates the receptor partially, while at the same time blocking the effects of nicotine.
Pharmacokinetics. After oral intake, the maximal plasma concentration of varenicline is reached in 3 to 4 hours. Food does not inhibit absorption. There is minimal hepatic metabolism, with 92% of the drug excreted unchanged in the urine. There are no known drug-drug interactions. The 24-hour half-life of varenicline allows for once-daily dosing.
Effectiveness. Several phase 2 and phase 3 studies compared varenicline with placebo and other drugs in terms of efficacy, dosing, and safety in 3,600 smokers. The initial phase 2 study, lasting 7 weeks, showed a 4-week abstinence rate of 48% with varenicline compared with 17% with placebo.17
Two phase 3 trials with 2,052 participants demonstrated that, at 12 weeks, abstinence rates were 44% with varenicline, 17% with bupropion, and 17% with placebo. At the end of 1 year, those groups again demonstrated significant differences in nicotine abstinence—22% in the varenicline group vs 15% with bupropion and 9% with placebo. Also, varenicline was superior to bupropion and placebo in reducing craving.18,19 For those who were nicotine-free after 12 weeks of treatment, continuing varenicline for another 12 weeks boosted nicotine abstinence rates from 36% to 44% at 1 year.20
Though varenicline produces a mild physiologic dependence, it is not addictive and does not produce tolerance to itself. There is no need to increase the dose over time. Three percent of patients have reported mild irritability on stopping varenicline.
In sum, varenicline has been shown to be more effective than bupropion and any of the four single formulations of nicotine replacement when they are used alone. It has not been shown to be more effective than combinations of nicotine replacement therapy.10
Safety considerations with varenicline. Psychiatric adverse events associated with varenicline have included severe depression, agitation, and suicidal behavior—including completed suicide. Motor vehicle accidents and erratic behaviors have led to a ban on varenicline use by airline pilots, truck drivers, and maritime workers. Skin rashes (including Stevens-Johnson syndrome), renal failure, and cataracts have also been reported. Safety has not been established with schizophrenia, bipolar disorder, or major depression. The physician should ask about prior psychiatric history, illnesses, and reactions before prescribing varenicline. Generally, it is prudent to avoid varenicline in patients with a significant psychiatric history.
Nausea and sleep disturbances such as vivid dreams and insomnia are the most frequently reported side effects.
Black box warnings with bupropion and varenicline. In July 2009, the FDA issued boxed warnings for bupropion SR and for varenicline for smoking cessation because of reports of neuropsychiatric symptoms, including changes in behavior, hostility, agitation, depressed mood, suicidal thoughts and behavior, attempted suicide, and completed suicide.21 These can occur in people with or without a history of mental illness, and whether the patient has stopped smoking or not. Providers should inform patients, family members, and caregivers about the potential for these symptoms and what to do if symptoms develop—ie, stop the medication immediately and contact the health care provider.
Patients should also be told to use caution when driving, operating machinery, or performing hazardous activities until they know how the medication will affect them.21
When prescribing varenicline. Advise patients to set a “quit date” 7 days after starting varenicline—they can continue smoking for the first 7 days on the drug. The starter packet for varenicline comes as 0.5 mg daily for 3 days, then twice daily for 2 days; the dose increases to 1 mg twice daily thereafter. Smokers report that it is much easier to quit after 7 days on varenicline.
Maintenance packs are available for 1 month of daily dosing. Generally, one starter pack is prescribed, with a second prescription for continuing packs for 2 to 5 more months. Varenicline is best taken with a full glass of water. If the smoker abstains for the first 3 months of therapy, it is best to prescribe an additional 3 months of medication to improve long-term abstinence from nicotine. With nausea or renal disease, lower the dose. Avoid prescribing varenicline for the elderly, teens, and pregnant women.
Varenicline is available only by prescription, and no generic equivalent is available.
WHEN IT’S TIME TO QUIT
A useful prescribing plan is:
- For most people, begin with nicotine patches plus gum
- If nicotine replacement therapy fails, prescribe varenicline
- Prescribe bupropion for patients with depression or if varenicline fails.
According to the US Public Health Service guideline,12 in a meta-analysis comparing various tobacco cessation medications with placebo and nicotine patch, the combination of nicotine patch (> 14 weeks) plus gum was 3.6 times as effective as placebo and 1.9 times as effective as nicotine patch alone. Varenicline at 2 mg per day was 3.1 times as effective as placebo and 1.6 times as effective as nicotine patch alone. Therefore, the combination of nicotine patch and gum is an inexpensive yet effective way to begin a course of smoking cessation therapy.
Behavioral counseling
Timing is important to successful quitting. Patients generally know when it’s a good time to quit—and when it’s not. Avoid trying to get patients to quit when they are stressed, overly busy, fatigued, or anxious. Try to get the patient to set a time to quit that’s ideal, and then encourage the patient to stick to it. For example, scheduling the quit day on a celebration, anniversary, or birthday gives that date added significance and enhances motivation. Follow the patient frequently for 6 to 12 months with intense monitoring and encouragement, and to assess for any adverse effects of medication.
The 2008 update to the Public Health Service Clinical Practice Guidelines on treating tobacco use and dependence concludes that counseling and medication are each effective alone in increasing smoking cessation and are even more effective when used together.12 Even very brief, 3-minute discussions and encouragement have been shown to be helpful. The Public Health Service evidence-based clinical practice guideline on cessation states that brief advice by medical providers to quit smoking is an effective intervention.12

Doctors who show great interest in smoking cessation seem to be more effective in persuading patients to quit. They should take note of smoking rather than ignoring it. A modified version of the CAGE questionnaire to assess problem drinking is recommended as a tool to assess patients’ smoking behavior and initiate a discussion about it (Table 1).22 Emphasize the health and financial costs to the patient. Try to form a therapeutic alliance with the patient against smoking: “Let’s see what we can do about this problem.” Be positive and optimistic in offering help with counseling, support, and medications.
Caution smokers against switching to “light” tar and nicotine cigarettes, as controlled experiments have failed to show consistent reductions in the amounts of tar and nicotine these products deliver into the lungs. Smokers also appear to compensate or adapt their smoking habits to increase the yield from these products. There is insufficient evidence to support the supposed health benefits of such low-yield smoking products.23
Always refer the patient for counseling with the pharmaceutical company help line or with a supported quit line. Some manufacturers of smoking cessation medications offer counseling or web-based support for patients trying to quit. For example, patients who are prescribed varenicline are offered the GETQUIT Plan, a free program that includes online education, tracking of progress, and “check-ins with slip-up support.” These services are often underused yet represent a ready source of helpful support.
If relapses occur, encourage the patient to keep trying again and again, as it may take several attempts to succeed.
Quit lines
To help smokers and other tobacco users quit, all states now have a toll-free cessation quit line, a telephone service accessible through a national toll-free number (1-800-QUIT-NOW). Quit lines also can be a referral source for health care providers who might not have the time or staff to provide all of the steps in the recommended “five-A” cessation counseling model,12 ie:
- Ask about tobacco use
- Advise to quit
- Assess willingness to make a quit attempt
- Assist in quit attempt
- Arrange follow-up.
Quit lines have been shown to improve outcomes when compared with people trying to stop on their own.12 Quit line services have evolved from their modest beginnings as providers of information and counseling to a level at which in many states, evidence-based medications are provided through quit lines.13,24 Medication use, coupled with quit line counseling intervention, increases the likelihood of tobacco abstinence and is consistent with US Public Health Service guideline recommendations that all tobacco users should be offered at least one medication as part of their quit attempt.12
WOMEN SMOKERS HAVE UNIQUE HEALTH RISKS
Women have unique health risks arising from smoking: low-birth-weight babies, sudden infant death syndrome, cervical cancer, and an increasing rate of lung cancer. In general, women have poorer responses to nicotine replacement therapy, are more concerned about gaining weight after quitting, and demonstrate more mood lability after quitting. Women seem more energized by the taste, smell, and overall sensations involved in smoking.
Weight gain will occur when quitting smoking; this is hard to overcome. More exercise may help, and a trial of bupropion with nicotine replacement therapy may mitigate weight gain.
Women who are pregnant present a special challenge when it comes to weighing the benefit of medications against continued smoking. For pregnant women who want to quit smoking, the best treatment is counseling without nicotine replacement or other pharmacotherapy. There are inadequate data for the use of varenicline or bupropion in pregnancy. If medication is needed, start nicotine replacement therapy early in pregnancy, as its risk is the same as or less than the smoking risk to the fetus.
The US Public Health Service guideline provides a useful discussion and bibliography related to this topic.12 All of the FDA-approved medications for tobacco cessation carry an FDA pregnancy category designation of C or D—ie, not recommended for use by pregnant women. These designations are not absolute contraindications and do allow for use in life-threatening situations or when other treatment modalities have failed. Some clinicians and their patients may decide that the potential for fetal harm, including fetal death, with continued smoking is high enough to warrant use of medications.
A careful and thorough discussion of the risks and benefits is recommended between the patient and her physician regarding this issue.
A CALL TO ARMS
The statistics are incontrovertible but do not tell the whole story. The day-to-day practices of physicians bear witness to the suffering that compulsive smoking creates for the smoker. As in all addictions, those around the addict suffer as well, from secondary smoke but also from fear and anxiety about premature loss of their loved ones. Smoking causes suffering and early death, and it is vitally important that doctors—the front-line troops—take up the fight against it as America’s number-one preventable cause of health problems and death.
To be effective champions in the public health fight against smoking, doctors must develop an understanding of compulsive smoking as a biologically driven process of addiction. The smoker attempting to quit is literally in the fight of his or her life and needs emotional support, cognitive-behavioral tools, and state-of-the-art pharmacology to overcome the slow destruction caused by the “dirty weed.”
- US Department of Health and Human Services. How tobacco smoke causes disease: the biology and behavioral basis for smoking-attributable disease: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2010.
- Centers for Disease Control and Prevention (CDC). Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000-2004. MMWR Morb Mortal Wkly Rep 2008; 57:1226–1228.
- Doll R, Peto R, Boreham J, Sutherland I. Mortality in relation to smoking. Fifty-years’ observations on male British doctors. BMJ 2004; 328:1519-1528.
- Centers for Disease Control and Prevention (CDC). Quitting smoking among adults—United States, 2001-2010. MMWR Morb Mortal Wkly Rep 2011; 60:1513–1519.
- Benowitz NL, Hukkanen J, Jacob P 3rd. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol 2009; 192:29–60.
- Rubinstein ML, Shiffman S, Moscicki AB, Rait MA, Sen S, Benowitz NL. Nicotine metabolism and addiction among adolescent smokers. Addiction 2013; 108:406–412.
- Benowitz NL, Porchet H, Sheiner L, Jacob P 3rd. Nicotine absorption and cardiovascular effects with smokeless tobacco use: comparison with cigarettes and nicotine gum. Clin Pharmacol Ther 1988; 44:23–28.
- Schneider NG, Lunell E, Olmstead RE, Fagerström KO. Clinical pharmacokinetics of nasal nicotine delivery. A review and comparison to other nicotine systems. Clin Pharmacokinet 1996; 31:65–80.
- Benowitz NL. Nicotine replacement therapy. What has been accomplished—can we do better? Drugs 1993; 45:157–170.
- Cahill K, Stevens S, Perera R, Lancaster T. Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev 2013; 5:CD009329.
- Committee on Safety in Medicines and the Medicines Control Agency. Zyban safety reminder. Current Problems in Pharmacovigilance 2001; 27:5.
- Clinical Practice Guideline Treating Tobacco Use and Dependence 2008 Update Panel, Liaisons, and Staff. A clinical practice guideline for treating tobacco use and dependence: 2008 update. A US public health service report. Am J Prev Med 2008; 35:158–176.
- Swan GE, McAfee T, Curry SJ, et al. Effectiveness of bupropion sustained release for smoking cessation in a health care setting: a randomized trial. Arch Intern Med 2003; 163:2337–2344.
- Watkins SS, Koob GF, Markou A. Neural mechanisms underlying nicotine addiction: acute positive reinforcement and withdrawal. Nicotine Tob Res 2000; 2:19–37.
- Coe JW, Brooks PR, Wirtz MC, et al. 3,5-Bicyclic aryl piperidines: a novel class of alpha4beta2 neuronal nicotinic receptor partial agonists for smoking cessation. Bioorg Med Chem Lett 2005; 15:4889–4897.
- Picciotto MR, Zoli M, Changeux JP. Use of knock-out mice to determine the molecular basis for the actions of nicotine. Nicotine Tob Res 1999; 1(suppl 2):S121–S125.
- Nides M, Oncken C, Gonzales D, et al. Smoking cessation with varenicline, a selective alpha4beta2 nicotinic receptor partial agonist: results from a 7-week, randomized, placebo- and bupropion-controlled trial with 1-year follow-up. Arch Intern Med 2006; 166:1561–1568.
- Jorenby DE, Hays JT, Rigotti NA, et al; Varenicline Phase 3 Study Group. Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA 2006; 296:56–63.
- Gonzales D, Rennard SI, Nides M, et al; Varenicline Phase 3 Study Group. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA 2006; 296:47–55.
- Tonstad S, Tønnesen P, Hajek P, Williams KE, Billing CB, Reeves KR; Varenicline Phase 3 Study Group. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA 2006; 296:64–71.
- US Food and Drug Administration (FDA). Public health advisory: FDA requires new boxed warnings for the smoking cessation drugs Chantix and Zyban. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/PublicHealthAdvisories/ucm169988.htm. Accessed October 8, 2014.
- Rustin TA. Assessing nicotine dependence. Am Fam Physician 2000; 62:579–592.
- Centers for Disease Control and Prevention (CDC). Smoking & tobacco use. Low-yield cigarettes. www.cdc.gov/tobacco/data_statistics/fact_sheets/tobacco_industry/low_yield_cigarettes/index.htm. Accessed October 8, 2014.
- Biazzo LL, Froshaug DB, Harwell TS, et al. Characteristics and abstinence outcomes among tobacco quitline enrollees using varenicline or nicotine replacement therapy. Nicotine Tob Res 2010; 12:567–573.
- US Department of Health and Human Services. The health consequences of smoking—nicotine addiction; a report of the Surgeon General. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, 1988.
- Agaku I, King B, Dube SR, Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion, CDC. Current cigarette smoking among adults—United States, 2011. MMWR, 2012; 61(44):889–894.
Tobacco is a dirty weed,
I like it.
It satisfies no normal need,
I like it.
It makes you thin, it makes you lean,
It takes the hair right off your bean.
It’s the worst darn stuff I’ve ever seen.
I like it.
Graham Lee Hemminger. The Penn State Froth, November 1915: 19. Courtesy of Paul J. Dzyak, Jr., Paterno Library, Pennsylvania State University, State College, PA.
All physicians recognize the harm in tobacco smoking and try to convince patients to quit for health reasons, but quitting is challenging and frustrating for both doctor and patient. Physicians can improve quitting outcomes by applying their knowledge of the physiologic basis of nicotine addiction and newer tools that are making a real difference in smoking cessation.
THE NO. 1 PREVENTABLE CAUSE OF DEATH
Tobacco use remains the single largest preventable cause of death and disease in the United States: 443,000 US adults die of smoking-related illnesses each year, or one every 8 seconds.1 Tobacco smoking is currently responsible for 18% of all deaths and 37% of all preventable deaths. One-third of all smokers die early, with men losing 13 years of life and women losing 15 years. (See The rise and partial fall of smoking for a historical overview.)
Smoking, the leading cause of lung cancer, is also implicated in cancers of the mouth, larynx, esophagus, stomach, kidney, bladder, and cervix and has been linked to leukemia. (Though nicotine is responsible for the addictive properties of tobacco, it does not cause cancer itself: other substances in tobacco smoke, many of them byproducts of combustion, are carcinogenic.)
Running a close second to cancer as a smoking-related cause of death is cardiovascular disease, including stroke, myocardial infarction, microvascular dementia, peripheral vascular disease, and aortic aneurysm. Pulmonary and respiratory diseases, including chronic obstructive pulmonary disease, pneumonia, and asthma, are the third most common fatal smoking-related ailments.
Other medical consequences include erectile dysfunction, infertility, pregnancy complications, and low birth weight. Smoking also causes adverse surgical outcomes, poor wound healing, hip fractures, low bone density, peptic ulcer disease, and cataracts.
Smoking is estimated to cost the United States $96 billion in direct medical expenses and $97 billion in lost productivity annually.2
On the positive side, quitting smoking has health benefits at any age, and smokers who quit before age 35 have death rates similar to those in people who have never smoked.1,3
WHY IS IT SO HARD TO QUIT?
Most smokers want to quit, and many try to—but few succeed. In the 2010 National Health Interview Surveys, 68.8% of adult smokers said they wanted to stop smoking, and 52.4% had tried to in the past year, but only 6.2% had succeeded.4 Many recovering alcoholics and drug addicts say that quitting tobacco was much harder than abstaining from other substances of choice.
Why is it so hard to quit?
Smoking is a classic addiction
Addictions are usually diagnosed by behavioral signs, and nicotine addiction has many of the clinical hallmarks, eg:
- Tolerance, with a trend toward increasing the potency of the dose and the frequency of smoking over time
- Mental preoccupation with smoking, as it often becomes woven into one’s daily schedule and is associated with almost everything the smoker does throughout the day. Having no cigarettes in the house can generate anxiety that is relieved only by obtaining more
- Squandering scarce financial resources on nicotine products, over time amounting to substantial sums, and since smoking rates are higher in poor people than in the affluent, these are people who can least afford it
- Withdrawal symptoms, characterized by jitteriness, irritability, headache, insomnia, anxiety, and increased appetite.
People continue to smoke despite adverse consequences such as falling asleep while smoking and setting fire to the bed or to the house, or losing digits to peripheral vascular disease. Being unable to quit and to stay off smoking is a hallmark of tobacco dependence. Relapses are often triggered by being near other smokers or seeing a billboard advertising cigarettes. Eventually, the nicotine addict comes to value and crave nicotine more than health or life itself.
Nicotine stimulates ‘reward’ centers in the brain
Nicotine is an alkaloid found in many plants (including potatoes) but in especially high concentrations in tobacco. In mammals, it is a stimulant, rapidly producing dependence and addiction.
Inhaled by smoking, nicotine is absorbed across the large alveolar surface, avoids first-pass metabolism, and is transported rapidly to the brain (Figure 1). In fact, nicotine reaches the brain less than 20 seconds after inhalation, which is slightly faster even than when drugs are injected intravenously.5
Tobacco smoke contains approximately 4,800 compounds, many of which activate neurotransmitter systems such as dopamine, norepinephrine, acetylcholine, glutamate, serotonin, beta-endorphin, and gamma-aminobutyric acid. The most significant of these is the dopamine reward system known as the mesoaccumbens pathway. This system is activated within seconds of smoking and produces a sense of pleasure.
Nicotine binds to nicotinic acetylcholine receptors, primarily to alpha-4, beta-2 receptors in the ventral tegmental area of the midbrain. Once this binding occurs, a neurochemical message is conveyed to the nucleus accumbens via the release of dopamine in the mesoaccumbens pathway—the final common reward pathway triggered by all drugs of abuse. Since these structures and pathways of the brain are anatomically central, the addiction is driven by the basal ganglia and midbrain, the phylogenetically oldest parts of the brain. Nicotine therefore drives its addicts to continue smoking by producing strong neurochemical rewards and by causing strongly negative reactions when discontinued.
Genetically mediated susceptibility probably contributes to addiction. People whose neurochemical pathways are easily stimulated by this drug are probably at far greater risk of addiction. Paradoxically, people who are rapid metabolizers of nicotine are at greater risk than slow metabolizers.6 (Nicotine is metabolized by cytochrome P450 2A6 in the liver.)
Tolerance and withdrawal
Tolerance develops with long-term use, mediated by up-regulation (increased numbers) of alpha-4, beta-2 cholinergic receptors in the ventral tegmental area. Any reduction in nicotine level causes distress because receptors are unoccupied; with more receptors, nicotine intake must increase to keep physiologic balance and avoid withdrawal. Since the half-life of nicotine is only about 2 hours, the smoker must smoke almost constantly to satisfy receptors hungry for the stimulating drug. If drug levels drop, withdrawal occurs very quickly.
Eventually, smokers use nicotine less for pleasure and more as a way to avoid withdrawal. The cycle of pleasure, eventual tolerance, withdrawal, craving, and compulsion is biologically driven, like the drives of thirst, reproduction, and hunger. Nicotine hijacks species-sustaining reward mechanisms, leading to the malignant, compulsive disease of nicotine addiction.
Treatment doomed to fail?
Because nicotine addiction involves the midbrain, cessation strategies that rely on higher cerebral function are not likely to succeed. Counseling, common sense, and willpower simply cannot overcome the dopaminergic stimulating power or assuage the withdrawal sickness of nicotine dependence. Telling patients that smoking is bad for them misses the mark in most cases. Patients want to quit, but the drive to smoke is too powerful. Attempts to cut down rather than abstain from smoking also fail.
Nicotine is a formidable adversary for the patient and for the doctor or other health professional. Until recently, treatment was usually ineffective.
So, what does work against nicotine addiction?
PHARMACOTHERAPIES FOR SMOKING CESSATION
Nicotine replacement therapy
The oldest of the pharmacotherapies for nicotine addiction is nicotine replacement, in the form of patch, gum, lozenge, or nasal spray.
Advantages:
- Nicotine replacement therapy eliminates exposure to the other harmful compounds in tobacco, with few to none of the health risks associated with smoking.
- By delivering nicotine by a different route, nicotine replacement therapy breaks the association between smoking and feeling good. The addict is already dopamine-stimulated before putting a cigarette in the mouth, merely by association and suggestion. Using a different route of nicotine administration avoids that associative stimulation from the act of smoking, so that quitting becomes easier.
- The dose of nicotine is lower with replacement therapy than with smoking. The cigarette is the most efficient delivery mechanism for getting nicotine into the body. A smoked cigarette produces a rapid spike in plasma nicotine levels, far higher and faster than nicotine gum, nasal spray, or transdermal patch. Peak levels of plasma nicotine from nicotine replacement therapy are only 30% to 50% as high as those achieved by smoking.7–9
- It is inexpensive.
Disadvantages:
- Nicotine replacement therapy maintains the addiction to nicotine, with its neurophysiologic distortions.
- Some patients continue nicotine replacement therapy for years.
Use of nicotine gum can be a problem because of the need for frequent administration. The gum is chewed until the user feels a tingling or peppery taste in the mouth, after which the gum must be placed inside the cheek to allow for maximal absorption of the nicotine. Once the tingling has faded, the user is to chew another piece and repeat the cycle as long as craving is perceived. On the other hand, the nicotine patch is applied once daily. Both of these products are available over-the-counter.
Caution is indicated when starting nicotine replacement therapy in those with recent myocardial infarction, angina, or arrhythmia.
Effectiveness. Nicotine replacement therapy has been shown to be as effective as bupropion (see below) but not as effective as varenicline when used in single administration form (patch, gum, lozenge, or inhaler alone). The four single-administration forms of nicotine replacement therapy are all equally efficacious. Combinations of nicotine replacement formulations have been reported to be as effective as varenicline and superior to single formulations.10
How about electronic cigarettes? Electronic cigarettes, or e-cigarettes, supply nicotine in a noncombustion vapor and are advertised as an alternative to smoking. No claim is made for reducing smoking, so the products, including the liquids involved, are not regulated by the US Food and Drug Administration (FDA). Controversy exists as to whether they actually increase the number of smokers by introducing young people to “vaping” to get nicotine. Since nicotine is still inhaled, the addictive potential remains unabated. E-cigarettes are unregulated vehicles for supplying nicotine and may pose other health risks, and there is very limited evidence to support the efficacy of e-cigarettes as aids to smoking cessation. Since no controlled study has demonstrated successful cessation of smoking with e-cigarettes, they are best regarded for now as merely another way to introduce nicotine into the body.
Bupropion
Bupropion, an antidepressant also sold as Wellbutrin SR, was approved in 1997 for use in smoking cessation under the trade name Zyban. The manufacturer, Glaxo SmithKline, learned serendipitously that depressive patients taking bupropion were able to quit smoking. After some field trials, this “new” medication was born. It was the first nonnicotine drug for tobacco dependence to gain FDA approval.
Its mechanism of action in combating smoking is unknown but is thought to be related to mild inhibition of dopamine re-uptake in the midbrain.
The drug is approved for smokers over age 18 who are smoking at least nine cigarettes daily. It requires a prescription, and the typical dose is 150 mg twice daily for 8 to 12 weeks, up to 12 months. Smoking is allowed for the first 7 days of drug use.
Contraindications include a history of seizures, concurrent use of bupropion, bulimia, anorexia, detoxification from alcohol or sedatives, use of monoamine oxidase inhibitors, and allergy to bupropion. Warnings are noted for diseases of heart, liver, or kidney; for use with selective serotonin reuptake inhibitors or tricyclic antidepressants; for pregnancy; and for adolescents because of heightened suicide risk.
Side effects. Seizure risk has been estimated at 1 in 1,000 bupropion users at dosages of up to 300 mg daily and is 10 times greater at dosages of 450 to 600 mg/day.11
The most common side effect reported is insomnia, which occurs in about one-third of people who take the medication. Less common side effects include dry mouth, anxiety, and hypertension. Pretreatment screening should include a history of seizure, closed head trauma, brain surgery, stroke, and the eating disorders anorexia nervosa and bulimia. The FDA has required a boxed warning regarding the association of bupropion with psychiatric symptoms.12
Effectiveness. Compared with placebo, bupropion reduces withdrawal symptoms such as irritability, frustration, anger, restlessness, depression, craving, poor concentration, and urge to smoke. Bupropion SR, 150 or 300 mg per day, has been reported to lead to substantial abstinence rates when used with intensive telephone counseling. In a randomized trial,13 side effects were common, especially at the higher dose, but there were no serious adverse effects such as deaths or seizures.13
Buproprion has been found to be as efficacious in improving the odds of quitting as single forms of nicotine replacement therapy, but not as efficacious as nicotine replacement therapy forms used in combination. Bupropion does not appear to be as effective as varenicline.9 US Public Health Service guidelines since 2000 have included nicotine replacement therapy and sustained-release bupropion in combination.
Disadvantages. Bupropion is significantly more expensive than nicotine replacement therapy, but it is often covered by insurance when it is used for smoking cessation. Bupropion has many contraindications, produces drug-drug interactions, is often poorly tolerated, and has many side effects. Some deaths have been reported. Zyban is available by prescription only, an indicator of its relative risk, with the added drawback of higher cost to patients.
Varenicline
Varenicline (Chantix, Champix) was granted a priority review by the FDA in 2005, as it showed significantly better results than other current therapies. It was approved in 2006 and added as a first-line agent in the 2008 guidelines.12
Mechanism of action. A synthetic “designer” drug made for its specific purpose, the varenicline molecule is a modified version of cytisine, a naturally occurring alkaloid previously marketed as Tabex in Eastern Europe. Cytisine is a selective alpha-4, beta-2 nicotinic acetylcholine receptor partial agonist. The high-affinity alpha-4, beta-2 nicotinic acetylcholine receptors exist in the mesolimbic dopaminergic system, the reward center of the brain.14
Varenicline has the same mechanism of action as cytisine but penetrates the central nervous system better. This mechanism of action allows varenicline to block the attachment of the nicotine molecule to this receptor, preventing nicotine’s dominant effect. Varenicline, however, is a partial agonist, so that when it attaches itself to the receptor, it causes a partial agonist effect, which is an opening of the receptor channel to sodium ions, causing partial stimulation of the cells in the ventral tegmental area, and ultimately causing a mild release of dopamine in the nucleus accumbens.15,16 Thus, varenicline effectively stimulates the receptor partially, while at the same time blocking the effects of nicotine.
Pharmacokinetics. After oral intake, the maximal plasma concentration of varenicline is reached in 3 to 4 hours. Food does not inhibit absorption. There is minimal hepatic metabolism, with 92% of the drug excreted unchanged in the urine. There are no known drug-drug interactions. The 24-hour half-life of varenicline allows for once-daily dosing.
Effectiveness. Several phase 2 and phase 3 studies compared varenicline with placebo and other drugs in terms of efficacy, dosing, and safety in 3,600 smokers. The initial phase 2 study, lasting 7 weeks, showed a 4-week abstinence rate of 48% with varenicline compared with 17% with placebo.17
Two phase 3 trials with 2,052 participants demonstrated that, at 12 weeks, abstinence rates were 44% with varenicline, 17% with bupropion, and 17% with placebo. At the end of 1 year, those groups again demonstrated significant differences in nicotine abstinence—22% in the varenicline group vs 15% with bupropion and 9% with placebo. Also, varenicline was superior to bupropion and placebo in reducing craving.18,19 For those who were nicotine-free after 12 weeks of treatment, continuing varenicline for another 12 weeks boosted nicotine abstinence rates from 36% to 44% at 1 year.20
Though varenicline produces a mild physiologic dependence, it is not addictive and does not produce tolerance to itself. There is no need to increase the dose over time. Three percent of patients have reported mild irritability on stopping varenicline.
In sum, varenicline has been shown to be more effective than bupropion and any of the four single formulations of nicotine replacement when they are used alone. It has not been shown to be more effective than combinations of nicotine replacement therapy.10
Safety considerations with varenicline. Psychiatric adverse events associated with varenicline have included severe depression, agitation, and suicidal behavior—including completed suicide. Motor vehicle accidents and erratic behaviors have led to a ban on varenicline use by airline pilots, truck drivers, and maritime workers. Skin rashes (including Stevens-Johnson syndrome), renal failure, and cataracts have also been reported. Safety has not been established with schizophrenia, bipolar disorder, or major depression. The physician should ask about prior psychiatric history, illnesses, and reactions before prescribing varenicline. Generally, it is prudent to avoid varenicline in patients with a significant psychiatric history.
Nausea and sleep disturbances such as vivid dreams and insomnia are the most frequently reported side effects.
Black box warnings with bupropion and varenicline. In July 2009, the FDA issued boxed warnings for bupropion SR and for varenicline for smoking cessation because of reports of neuropsychiatric symptoms, including changes in behavior, hostility, agitation, depressed mood, suicidal thoughts and behavior, attempted suicide, and completed suicide.21 These can occur in people with or without a history of mental illness, and whether the patient has stopped smoking or not. Providers should inform patients, family members, and caregivers about the potential for these symptoms and what to do if symptoms develop—ie, stop the medication immediately and contact the health care provider.
Patients should also be told to use caution when driving, operating machinery, or performing hazardous activities until they know how the medication will affect them.21
When prescribing varenicline. Advise patients to set a “quit date” 7 days after starting varenicline—they can continue smoking for the first 7 days on the drug. The starter packet for varenicline comes as 0.5 mg daily for 3 days, then twice daily for 2 days; the dose increases to 1 mg twice daily thereafter. Smokers report that it is much easier to quit after 7 days on varenicline.
Maintenance packs are available for 1 month of daily dosing. Generally, one starter pack is prescribed, with a second prescription for continuing packs for 2 to 5 more months. Varenicline is best taken with a full glass of water. If the smoker abstains for the first 3 months of therapy, it is best to prescribe an additional 3 months of medication to improve long-term abstinence from nicotine. With nausea or renal disease, lower the dose. Avoid prescribing varenicline for the elderly, teens, and pregnant women.
Varenicline is available only by prescription, and no generic equivalent is available.
WHEN IT’S TIME TO QUIT
A useful prescribing plan is:
- For most people, begin with nicotine patches plus gum
- If nicotine replacement therapy fails, prescribe varenicline
- Prescribe bupropion for patients with depression or if varenicline fails.
According to the US Public Health Service guideline,12 in a meta-analysis comparing various tobacco cessation medications with placebo and nicotine patch, the combination of nicotine patch (> 14 weeks) plus gum was 3.6 times as effective as placebo and 1.9 times as effective as nicotine patch alone. Varenicline at 2 mg per day was 3.1 times as effective as placebo and 1.6 times as effective as nicotine patch alone. Therefore, the combination of nicotine patch and gum is an inexpensive yet effective way to begin a course of smoking cessation therapy.
Behavioral counseling
Timing is important to successful quitting. Patients generally know when it’s a good time to quit—and when it’s not. Avoid trying to get patients to quit when they are stressed, overly busy, fatigued, or anxious. Try to get the patient to set a time to quit that’s ideal, and then encourage the patient to stick to it. For example, scheduling the quit day on a celebration, anniversary, or birthday gives that date added significance and enhances motivation. Follow the patient frequently for 6 to 12 months with intense monitoring and encouragement, and to assess for any adverse effects of medication.
The 2008 update to the Public Health Service Clinical Practice Guidelines on treating tobacco use and dependence concludes that counseling and medication are each effective alone in increasing smoking cessation and are even more effective when used together.12 Even very brief, 3-minute discussions and encouragement have been shown to be helpful. The Public Health Service evidence-based clinical practice guideline on cessation states that brief advice by medical providers to quit smoking is an effective intervention.12
Doctors who show great interest in smoking cessation seem to be more effective in persuading patients to quit. They should take note of smoking rather than ignoring it. A modified version of the CAGE questionnaire to assess problem drinking is recommended as a tool to assess patients’ smoking behavior and initiate a discussion about it (Table 1).22 Emphasize the health and financial costs to the patient. Try to form a therapeutic alliance with the patient against smoking: “Let’s see what we can do about this problem.” Be positive and optimistic in offering help with counseling, support, and medications.
Caution smokers against switching to “light” tar and nicotine cigarettes, as controlled experiments have failed to show consistent reductions in the amounts of tar and nicotine these products deliver into the lungs. Smokers also appear to compensate or adapt their smoking habits to increase the yield from these products. There is insufficient evidence to support the supposed health benefits of such low-yield smoking products.23
Always refer the patient for counseling with the pharmaceutical company help line or with a supported quit line. Some manufacturers of smoking cessation medications offer counseling or web-based support for patients trying to quit. For example, patients who are prescribed varenicline are offered the GETQUIT Plan, a free program that includes online education, tracking of progress, and “check-ins with slip-up support.” These services are often underused yet represent a ready source of helpful support.
If relapses occur, encourage the patient to keep trying again and again, as it may take several attempts to succeed.
Quit lines
To help smokers and other tobacco users quit, all states now have a toll-free cessation quit line, a telephone service accessible through a national toll-free number (1-800-QUIT-NOW). Quit lines also can be a referral source for health care providers who might not have the time or staff to provide all of the steps in the recommended “five-A” cessation counseling model,12 ie:
- Ask about tobacco use
- Advise to quit
- Assess willingness to make a quit attempt
- Assist in quit attempt
- Arrange follow-up.
Quit lines have been shown to improve outcomes when compared with people trying to stop on their own.12 Quit line services have evolved from their modest beginnings as providers of information and counseling to a level at which in many states, evidence-based medications are provided through quit lines.13,24 Medication use, coupled with quit line counseling intervention, increases the likelihood of tobacco abstinence and is consistent with US Public Health Service guideline recommendations that all tobacco users should be offered at least one medication as part of their quit attempt.12
WOMEN SMOKERS HAVE UNIQUE HEALTH RISKS
Women have unique health risks arising from smoking: low-birth-weight babies, sudden infant death syndrome, cervical cancer, and an increasing rate of lung cancer. In general, women have poorer responses to nicotine replacement therapy, are more concerned about gaining weight after quitting, and demonstrate more mood lability after quitting. Women seem more energized by the taste, smell, and overall sensations involved in smoking.
Weight gain will occur when quitting smoking; this is hard to overcome. More exercise may help, and a trial of bupropion with nicotine replacement therapy may mitigate weight gain.
Women who are pregnant present a special challenge when it comes to weighing the benefit of medications against continued smoking. For pregnant women who want to quit smoking, the best treatment is counseling without nicotine replacement or other pharmacotherapy. There are inadequate data for the use of varenicline or bupropion in pregnancy. If medication is needed, start nicotine replacement therapy early in pregnancy, as its risk is the same as or less than the smoking risk to the fetus.
The US Public Health Service guideline provides a useful discussion and bibliography related to this topic.12 All of the FDA-approved medications for tobacco cessation carry an FDA pregnancy category designation of C or D—ie, not recommended for use by pregnant women. These designations are not absolute contraindications and do allow for use in life-threatening situations or when other treatment modalities have failed. Some clinicians and their patients may decide that the potential for fetal harm, including fetal death, with continued smoking is high enough to warrant use of medications.
A careful and thorough discussion of the risks and benefits is recommended between the patient and her physician regarding this issue.
A CALL TO ARMS
The statistics are incontrovertible but do not tell the whole story. The day-to-day practices of physicians bear witness to the suffering that compulsive smoking creates for the smoker. As in all addictions, those around the addict suffer as well, from secondary smoke but also from fear and anxiety about premature loss of their loved ones. Smoking causes suffering and early death, and it is vitally important that doctors—the front-line troops—take up the fight against it as America’s number-one preventable cause of health problems and death.
To be effective champions in the public health fight against smoking, doctors must develop an understanding of compulsive smoking as a biologically driven process of addiction. The smoker attempting to quit is literally in the fight of his or her life and needs emotional support, cognitive-behavioral tools, and state-of-the-art pharmacology to overcome the slow destruction caused by the “dirty weed.”
Tobacco is a dirty weed,
I like it.
It satisfies no normal need,
I like it.
It makes you thin, it makes you lean,
It takes the hair right off your bean.
It’s the worst darn stuff I’ve ever seen.
I like it.
Graham Lee Hemminger. The Penn State Froth, November 1915: 19. Courtesy of Paul J. Dzyak, Jr., Paterno Library, Pennsylvania State University, State College, PA.
All physicians recognize the harm in tobacco smoking and try to convince patients to quit for health reasons, but quitting is challenging and frustrating for both doctor and patient. Physicians can improve quitting outcomes by applying their knowledge of the physiologic basis of nicotine addiction and newer tools that are making a real difference in smoking cessation.
THE NO. 1 PREVENTABLE CAUSE OF DEATH
Tobacco use remains the single largest preventable cause of death and disease in the United States: 443,000 US adults die of smoking-related illnesses each year, or one every 8 seconds.1 Tobacco smoking is currently responsible for 18% of all deaths and 37% of all preventable deaths. One-third of all smokers die early, with men losing 13 years of life and women losing 15 years. (See The rise and partial fall of smoking for a historical overview.)
Smoking, the leading cause of lung cancer, is also implicated in cancers of the mouth, larynx, esophagus, stomach, kidney, bladder, and cervix and has been linked to leukemia. (Though nicotine is responsible for the addictive properties of tobacco, it does not cause cancer itself: other substances in tobacco smoke, many of them byproducts of combustion, are carcinogenic.)
Running a close second to cancer as a smoking-related cause of death is cardiovascular disease, including stroke, myocardial infarction, microvascular dementia, peripheral vascular disease, and aortic aneurysm. Pulmonary and respiratory diseases, including chronic obstructive pulmonary disease, pneumonia, and asthma, are the third most common fatal smoking-related ailments.
Other medical consequences include erectile dysfunction, infertility, pregnancy complications, and low birth weight. Smoking also causes adverse surgical outcomes, poor wound healing, hip fractures, low bone density, peptic ulcer disease, and cataracts.
Smoking is estimated to cost the United States $96 billion in direct medical expenses and $97 billion in lost productivity annually.2
On the positive side, quitting smoking has health benefits at any age, and smokers who quit before age 35 have death rates similar to those in people who have never smoked.1,3
WHY IS IT SO HARD TO QUIT?
Most smokers want to quit, and many try to—but few succeed. In the 2010 National Health Interview Surveys, 68.8% of adult smokers said they wanted to stop smoking, and 52.4% had tried to in the past year, but only 6.2% had succeeded.4 Many recovering alcoholics and drug addicts say that quitting tobacco was much harder than abstaining from other substances of choice.
Why is it so hard to quit?
Smoking is a classic addiction
Addictions are usually diagnosed by behavioral signs, and nicotine addiction has many of the clinical hallmarks, eg:
- Tolerance, with a trend toward increasing the potency of the dose and the frequency of smoking over time
- Mental preoccupation with smoking, as it often becomes woven into one’s daily schedule and is associated with almost everything the smoker does throughout the day. Having no cigarettes in the house can generate anxiety that is relieved only by obtaining more
- Squandering scarce financial resources on nicotine products, over time amounting to substantial sums, and since smoking rates are higher in poor people than in the affluent, these are people who can least afford it
- Withdrawal symptoms, characterized by jitteriness, irritability, headache, insomnia, anxiety, and increased appetite.
People continue to smoke despite adverse consequences such as falling asleep while smoking and setting fire to the bed or to the house, or losing digits to peripheral vascular disease. Being unable to quit and to stay off smoking is a hallmark of tobacco dependence. Relapses are often triggered by being near other smokers or seeing a billboard advertising cigarettes. Eventually, the nicotine addict comes to value and crave nicotine more than health or life itself.
Nicotine stimulates ‘reward’ centers in the brain
Nicotine is an alkaloid found in many plants (including potatoes) but in especially high concentrations in tobacco. In mammals, it is a stimulant, rapidly producing dependence and addiction.
Inhaled by smoking, nicotine is absorbed across the large alveolar surface, avoids first-pass metabolism, and is transported rapidly to the brain (Figure 1). In fact, nicotine reaches the brain less than 20 seconds after inhalation, which is slightly faster even than when drugs are injected intravenously.5
Tobacco smoke contains approximately 4,800 compounds, many of which activate neurotransmitter systems such as dopamine, norepinephrine, acetylcholine, glutamate, serotonin, beta-endorphin, and gamma-aminobutyric acid. The most significant of these is the dopamine reward system known as the mesoaccumbens pathway. This system is activated within seconds of smoking and produces a sense of pleasure.
Nicotine binds to nicotinic acetylcholine receptors, primarily to alpha-4, beta-2 receptors in the ventral tegmental area of the midbrain. Once this binding occurs, a neurochemical message is conveyed to the nucleus accumbens via the release of dopamine in the mesoaccumbens pathway—the final common reward pathway triggered by all drugs of abuse. Since these structures and pathways of the brain are anatomically central, the addiction is driven by the basal ganglia and midbrain, the phylogenetically oldest parts of the brain. Nicotine therefore drives its addicts to continue smoking by producing strong neurochemical rewards and by causing strongly negative reactions when discontinued.
Genetically mediated susceptibility probably contributes to addiction. People whose neurochemical pathways are easily stimulated by this drug are probably at far greater risk of addiction. Paradoxically, people who are rapid metabolizers of nicotine are at greater risk than slow metabolizers.6 (Nicotine is metabolized by cytochrome P450 2A6 in the liver.)
Tolerance and withdrawal
Tolerance develops with long-term use, mediated by up-regulation (increased numbers) of alpha-4, beta-2 cholinergic receptors in the ventral tegmental area. Any reduction in nicotine level causes distress because receptors are unoccupied; with more receptors, nicotine intake must increase to keep physiologic balance and avoid withdrawal. Since the half-life of nicotine is only about 2 hours, the smoker must smoke almost constantly to satisfy receptors hungry for the stimulating drug. If drug levels drop, withdrawal occurs very quickly.
Eventually, smokers use nicotine less for pleasure and more as a way to avoid withdrawal. The cycle of pleasure, eventual tolerance, withdrawal, craving, and compulsion is biologically driven, like the drives of thirst, reproduction, and hunger. Nicotine hijacks species-sustaining reward mechanisms, leading to the malignant, compulsive disease of nicotine addiction.
Treatment doomed to fail?
Because nicotine addiction involves the midbrain, cessation strategies that rely on higher cerebral function are not likely to succeed. Counseling, common sense, and willpower simply cannot overcome the dopaminergic stimulating power or assuage the withdrawal sickness of nicotine dependence. Telling patients that smoking is bad for them misses the mark in most cases. Patients want to quit, but the drive to smoke is too powerful. Attempts to cut down rather than abstain from smoking also fail.
Nicotine is a formidable adversary for the patient and for the doctor or other health professional. Until recently, treatment was usually ineffective.
So, what does work against nicotine addiction?
PHARMACOTHERAPIES FOR SMOKING CESSATION
Nicotine replacement therapy
The oldest of the pharmacotherapies for nicotine addiction is nicotine replacement, in the form of patch, gum, lozenge, or nasal spray.
Advantages:
- Nicotine replacement therapy eliminates exposure to the other harmful compounds in tobacco, with few to none of the health risks associated with smoking.
- By delivering nicotine by a different route, nicotine replacement therapy breaks the association between smoking and feeling good. The addict is already dopamine-stimulated before putting a cigarette in the mouth, merely by association and suggestion. Using a different route of nicotine administration avoids that associative stimulation from the act of smoking, so that quitting becomes easier.
- The dose of nicotine is lower with replacement therapy than with smoking. The cigarette is the most efficient delivery mechanism for getting nicotine into the body. A smoked cigarette produces a rapid spike in plasma nicotine levels, far higher and faster than nicotine gum, nasal spray, or transdermal patch. Peak levels of plasma nicotine from nicotine replacement therapy are only 30% to 50% as high as those achieved by smoking.7–9
- It is inexpensive.
Disadvantages:
- Nicotine replacement therapy maintains the addiction to nicotine, with its neurophysiologic distortions.
- Some patients continue nicotine replacement therapy for years.
Use of nicotine gum can be a problem because of the need for frequent administration. The gum is chewed until the user feels a tingling or peppery taste in the mouth, after which the gum must be placed inside the cheek to allow for maximal absorption of the nicotine. Once the tingling has faded, the user is to chew another piece and repeat the cycle as long as craving is perceived. On the other hand, the nicotine patch is applied once daily. Both of these products are available over-the-counter.
Caution is indicated when starting nicotine replacement therapy in those with recent myocardial infarction, angina, or arrhythmia.
Effectiveness. Nicotine replacement therapy has been shown to be as effective as bupropion (see below) but not as effective as varenicline when used in single administration form (patch, gum, lozenge, or inhaler alone). The four single-administration forms of nicotine replacement therapy are all equally efficacious. Combinations of nicotine replacement formulations have been reported to be as effective as varenicline and superior to single formulations.10
How about electronic cigarettes? Electronic cigarettes, or e-cigarettes, supply nicotine in a noncombustion vapor and are advertised as an alternative to smoking. No claim is made for reducing smoking, so the products, including the liquids involved, are not regulated by the US Food and Drug Administration (FDA). Controversy exists as to whether they actually increase the number of smokers by introducing young people to “vaping” to get nicotine. Since nicotine is still inhaled, the addictive potential remains unabated. E-cigarettes are unregulated vehicles for supplying nicotine and may pose other health risks, and there is very limited evidence to support the efficacy of e-cigarettes as aids to smoking cessation. Since no controlled study has demonstrated successful cessation of smoking with e-cigarettes, they are best regarded for now as merely another way to introduce nicotine into the body.
Bupropion
Bupropion, an antidepressant also sold as Wellbutrin SR, was approved in 1997 for use in smoking cessation under the trade name Zyban. The manufacturer, Glaxo SmithKline, learned serendipitously that depressive patients taking bupropion were able to quit smoking. After some field trials, this “new” medication was born. It was the first nonnicotine drug for tobacco dependence to gain FDA approval.
Its mechanism of action in combating smoking is unknown but is thought to be related to mild inhibition of dopamine re-uptake in the midbrain.
The drug is approved for smokers over age 18 who are smoking at least nine cigarettes daily. It requires a prescription, and the typical dose is 150 mg twice daily for 8 to 12 weeks, up to 12 months. Smoking is allowed for the first 7 days of drug use.
Contraindications include a history of seizures, concurrent use of bupropion, bulimia, anorexia, detoxification from alcohol or sedatives, use of monoamine oxidase inhibitors, and allergy to bupropion. Warnings are noted for diseases of heart, liver, or kidney; for use with selective serotonin reuptake inhibitors or tricyclic antidepressants; for pregnancy; and for adolescents because of heightened suicide risk.
Side effects. Seizure risk has been estimated at 1 in 1,000 bupropion users at dosages of up to 300 mg daily and is 10 times greater at dosages of 450 to 600 mg/day.11
The most common side effect reported is insomnia, which occurs in about one-third of people who take the medication. Less common side effects include dry mouth, anxiety, and hypertension. Pretreatment screening should include a history of seizure, closed head trauma, brain surgery, stroke, and the eating disorders anorexia nervosa and bulimia. The FDA has required a boxed warning regarding the association of bupropion with psychiatric symptoms.12
Effectiveness. Compared with placebo, bupropion reduces withdrawal symptoms such as irritability, frustration, anger, restlessness, depression, craving, poor concentration, and urge to smoke. Bupropion SR, 150 or 300 mg per day, has been reported to lead to substantial abstinence rates when used with intensive telephone counseling. In a randomized trial,13 side effects were common, especially at the higher dose, but there were no serious adverse effects such as deaths or seizures.13
Buproprion has been found to be as efficacious in improving the odds of quitting as single forms of nicotine replacement therapy, but not as efficacious as nicotine replacement therapy forms used in combination. Bupropion does not appear to be as effective as varenicline.9 US Public Health Service guidelines since 2000 have included nicotine replacement therapy and sustained-release bupropion in combination.
Disadvantages. Bupropion is significantly more expensive than nicotine replacement therapy, but it is often covered by insurance when it is used for smoking cessation. Bupropion has many contraindications, produces drug-drug interactions, is often poorly tolerated, and has many side effects. Some deaths have been reported. Zyban is available by prescription only, an indicator of its relative risk, with the added drawback of higher cost to patients.
Varenicline
Varenicline (Chantix, Champix) was granted a priority review by the FDA in 2005, as it showed significantly better results than other current therapies. It was approved in 2006 and added as a first-line agent in the 2008 guidelines.12
Mechanism of action. A synthetic “designer” drug made for its specific purpose, the varenicline molecule is a modified version of cytisine, a naturally occurring alkaloid previously marketed as Tabex in Eastern Europe. Cytisine is a selective alpha-4, beta-2 nicotinic acetylcholine receptor partial agonist. The high-affinity alpha-4, beta-2 nicotinic acetylcholine receptors exist in the mesolimbic dopaminergic system, the reward center of the brain.14
Varenicline has the same mechanism of action as cytisine but penetrates the central nervous system better. This mechanism of action allows varenicline to block the attachment of the nicotine molecule to this receptor, preventing nicotine’s dominant effect. Varenicline, however, is a partial agonist, so that when it attaches itself to the receptor, it causes a partial agonist effect, which is an opening of the receptor channel to sodium ions, causing partial stimulation of the cells in the ventral tegmental area, and ultimately causing a mild release of dopamine in the nucleus accumbens.15,16 Thus, varenicline effectively stimulates the receptor partially, while at the same time blocking the effects of nicotine.
Pharmacokinetics. After oral intake, the maximal plasma concentration of varenicline is reached in 3 to 4 hours. Food does not inhibit absorption. There is minimal hepatic metabolism, with 92% of the drug excreted unchanged in the urine. There are no known drug-drug interactions. The 24-hour half-life of varenicline allows for once-daily dosing.
Effectiveness. Several phase 2 and phase 3 studies compared varenicline with placebo and other drugs in terms of efficacy, dosing, and safety in 3,600 smokers. The initial phase 2 study, lasting 7 weeks, showed a 4-week abstinence rate of 48% with varenicline compared with 17% with placebo.17
Two phase 3 trials with 2,052 participants demonstrated that, at 12 weeks, abstinence rates were 44% with varenicline, 17% with bupropion, and 17% with placebo. At the end of 1 year, those groups again demonstrated significant differences in nicotine abstinence—22% in the varenicline group vs 15% with bupropion and 9% with placebo. Also, varenicline was superior to bupropion and placebo in reducing craving.18,19 For those who were nicotine-free after 12 weeks of treatment, continuing varenicline for another 12 weeks boosted nicotine abstinence rates from 36% to 44% at 1 year.20
Though varenicline produces a mild physiologic dependence, it is not addictive and does not produce tolerance to itself. There is no need to increase the dose over time. Three percent of patients have reported mild irritability on stopping varenicline.
In sum, varenicline has been shown to be more effective than bupropion and any of the four single formulations of nicotine replacement when they are used alone. It has not been shown to be more effective than combinations of nicotine replacement therapy.10
Safety considerations with varenicline. Psychiatric adverse events associated with varenicline have included severe depression, agitation, and suicidal behavior—including completed suicide. Motor vehicle accidents and erratic behaviors have led to a ban on varenicline use by airline pilots, truck drivers, and maritime workers. Skin rashes (including Stevens-Johnson syndrome), renal failure, and cataracts have also been reported. Safety has not been established with schizophrenia, bipolar disorder, or major depression. The physician should ask about prior psychiatric history, illnesses, and reactions before prescribing varenicline. Generally, it is prudent to avoid varenicline in patients with a significant psychiatric history.
Nausea and sleep disturbances such as vivid dreams and insomnia are the most frequently reported side effects.
Black box warnings with bupropion and varenicline. In July 2009, the FDA issued boxed warnings for bupropion SR and for varenicline for smoking cessation because of reports of neuropsychiatric symptoms, including changes in behavior, hostility, agitation, depressed mood, suicidal thoughts and behavior, attempted suicide, and completed suicide.21 These can occur in people with or without a history of mental illness, and whether the patient has stopped smoking or not. Providers should inform patients, family members, and caregivers about the potential for these symptoms and what to do if symptoms develop—ie, stop the medication immediately and contact the health care provider.
Patients should also be told to use caution when driving, operating machinery, or performing hazardous activities until they know how the medication will affect them.21
When prescribing varenicline. Advise patients to set a “quit date” 7 days after starting varenicline—they can continue smoking for the first 7 days on the drug. The starter packet for varenicline comes as 0.5 mg daily for 3 days, then twice daily for 2 days; the dose increases to 1 mg twice daily thereafter. Smokers report that it is much easier to quit after 7 days on varenicline.
Maintenance packs are available for 1 month of daily dosing. Generally, one starter pack is prescribed, with a second prescription for continuing packs for 2 to 5 more months. Varenicline is best taken with a full glass of water. If the smoker abstains for the first 3 months of therapy, it is best to prescribe an additional 3 months of medication to improve long-term abstinence from nicotine. With nausea or renal disease, lower the dose. Avoid prescribing varenicline for the elderly, teens, and pregnant women.
Varenicline is available only by prescription, and no generic equivalent is available.
WHEN IT’S TIME TO QUIT
A useful prescribing plan is:
- For most people, begin with nicotine patches plus gum
- If nicotine replacement therapy fails, prescribe varenicline
- Prescribe bupropion for patients with depression or if varenicline fails.
According to the US Public Health Service guideline,12 in a meta-analysis comparing various tobacco cessation medications with placebo and nicotine patch, the combination of nicotine patch (> 14 weeks) plus gum was 3.6 times as effective as placebo and 1.9 times as effective as nicotine patch alone. Varenicline at 2 mg per day was 3.1 times as effective as placebo and 1.6 times as effective as nicotine patch alone. Therefore, the combination of nicotine patch and gum is an inexpensive yet effective way to begin a course of smoking cessation therapy.
Behavioral counseling
Timing is important to successful quitting. Patients generally know when it’s a good time to quit—and when it’s not. Avoid trying to get patients to quit when they are stressed, overly busy, fatigued, or anxious. Try to get the patient to set a time to quit that’s ideal, and then encourage the patient to stick to it. For example, scheduling the quit day on a celebration, anniversary, or birthday gives that date added significance and enhances motivation. Follow the patient frequently for 6 to 12 months with intense monitoring and encouragement, and to assess for any adverse effects of medication.
The 2008 update to the Public Health Service Clinical Practice Guidelines on treating tobacco use and dependence concludes that counseling and medication are each effective alone in increasing smoking cessation and are even more effective when used together.12 Even very brief, 3-minute discussions and encouragement have been shown to be helpful. The Public Health Service evidence-based clinical practice guideline on cessation states that brief advice by medical providers to quit smoking is an effective intervention.12
Doctors who show great interest in smoking cessation seem to be more effective in persuading patients to quit. They should take note of smoking rather than ignoring it. A modified version of the CAGE questionnaire to assess problem drinking is recommended as a tool to assess patients’ smoking behavior and initiate a discussion about it (Table 1).22 Emphasize the health and financial costs to the patient. Try to form a therapeutic alliance with the patient against smoking: “Let’s see what we can do about this problem.” Be positive and optimistic in offering help with counseling, support, and medications.
Caution smokers against switching to “light” tar and nicotine cigarettes, as controlled experiments have failed to show consistent reductions in the amounts of tar and nicotine these products deliver into the lungs. Smokers also appear to compensate or adapt their smoking habits to increase the yield from these products. There is insufficient evidence to support the supposed health benefits of such low-yield smoking products.23
Always refer the patient for counseling with the pharmaceutical company help line or with a supported quit line. Some manufacturers of smoking cessation medications offer counseling or web-based support for patients trying to quit. For example, patients who are prescribed varenicline are offered the GETQUIT Plan, a free program that includes online education, tracking of progress, and “check-ins with slip-up support.” These services are often underused yet represent a ready source of helpful support.
If relapses occur, encourage the patient to keep trying again and again, as it may take several attempts to succeed.
Quit lines
To help smokers and other tobacco users quit, all states now have a toll-free cessation quit line, a telephone service accessible through a national toll-free number (1-800-QUIT-NOW). Quit lines also can be a referral source for health care providers who might not have the time or staff to provide all of the steps in the recommended “five-A” cessation counseling model,12 ie:
- Ask about tobacco use
- Advise to quit
- Assess willingness to make a quit attempt
- Assist in quit attempt
- Arrange follow-up.
Quit lines have been shown to improve outcomes when compared with people trying to stop on their own.12 Quit line services have evolved from their modest beginnings as providers of information and counseling to a level at which in many states, evidence-based medications are provided through quit lines.13,24 Medication use, coupled with quit line counseling intervention, increases the likelihood of tobacco abstinence and is consistent with US Public Health Service guideline recommendations that all tobacco users should be offered at least one medication as part of their quit attempt.12
WOMEN SMOKERS HAVE UNIQUE HEALTH RISKS
Women have unique health risks arising from smoking: low-birth-weight babies, sudden infant death syndrome, cervical cancer, and an increasing rate of lung cancer. In general, women have poorer responses to nicotine replacement therapy, are more concerned about gaining weight after quitting, and demonstrate more mood lability after quitting. Women seem more energized by the taste, smell, and overall sensations involved in smoking.
Weight gain will occur when quitting smoking; this is hard to overcome. More exercise may help, and a trial of bupropion with nicotine replacement therapy may mitigate weight gain.
Women who are pregnant present a special challenge when it comes to weighing the benefit of medications against continued smoking. For pregnant women who want to quit smoking, the best treatment is counseling without nicotine replacement or other pharmacotherapy. There are inadequate data for the use of varenicline or bupropion in pregnancy. If medication is needed, start nicotine replacement therapy early in pregnancy, as its risk is the same as or less than the smoking risk to the fetus.
The US Public Health Service guideline provides a useful discussion and bibliography related to this topic.12 All of the FDA-approved medications for tobacco cessation carry an FDA pregnancy category designation of C or D—ie, not recommended for use by pregnant women. These designations are not absolute contraindications and do allow for use in life-threatening situations or when other treatment modalities have failed. Some clinicians and their patients may decide that the potential for fetal harm, including fetal death, with continued smoking is high enough to warrant use of medications.
A careful and thorough discussion of the risks and benefits is recommended between the patient and her physician regarding this issue.
A CALL TO ARMS
The statistics are incontrovertible but do not tell the whole story. The day-to-day practices of physicians bear witness to the suffering that compulsive smoking creates for the smoker. As in all addictions, those around the addict suffer as well, from secondary smoke but also from fear and anxiety about premature loss of their loved ones. Smoking causes suffering and early death, and it is vitally important that doctors—the front-line troops—take up the fight against it as America’s number-one preventable cause of health problems and death.
To be effective champions in the public health fight against smoking, doctors must develop an understanding of compulsive smoking as a biologically driven process of addiction. The smoker attempting to quit is literally in the fight of his or her life and needs emotional support, cognitive-behavioral tools, and state-of-the-art pharmacology to overcome the slow destruction caused by the “dirty weed.”
- US Department of Health and Human Services. How tobacco smoke causes disease: the biology and behavioral basis for smoking-attributable disease: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2010.
- Centers for Disease Control and Prevention (CDC). Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000-2004. MMWR Morb Mortal Wkly Rep 2008; 57:1226–1228.
- Doll R, Peto R, Boreham J, Sutherland I. Mortality in relation to smoking. Fifty-years’ observations on male British doctors. BMJ 2004; 328:1519-1528.
- Centers for Disease Control and Prevention (CDC). Quitting smoking among adults—United States, 2001-2010. MMWR Morb Mortal Wkly Rep 2011; 60:1513–1519.
- Benowitz NL, Hukkanen J, Jacob P 3rd. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol 2009; 192:29–60.
- Rubinstein ML, Shiffman S, Moscicki AB, Rait MA, Sen S, Benowitz NL. Nicotine metabolism and addiction among adolescent smokers. Addiction 2013; 108:406–412.
- Benowitz NL, Porchet H, Sheiner L, Jacob P 3rd. Nicotine absorption and cardiovascular effects with smokeless tobacco use: comparison with cigarettes and nicotine gum. Clin Pharmacol Ther 1988; 44:23–28.
- Schneider NG, Lunell E, Olmstead RE, Fagerström KO. Clinical pharmacokinetics of nasal nicotine delivery. A review and comparison to other nicotine systems. Clin Pharmacokinet 1996; 31:65–80.
- Benowitz NL. Nicotine replacement therapy. What has been accomplished—can we do better? Drugs 1993; 45:157–170.
- Cahill K, Stevens S, Perera R, Lancaster T. Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev 2013; 5:CD009329.
- Committee on Safety in Medicines and the Medicines Control Agency. Zyban safety reminder. Current Problems in Pharmacovigilance 2001; 27:5.
- Clinical Practice Guideline Treating Tobacco Use and Dependence 2008 Update Panel, Liaisons, and Staff. A clinical practice guideline for treating tobacco use and dependence: 2008 update. A US public health service report. Am J Prev Med 2008; 35:158–176.
- Swan GE, McAfee T, Curry SJ, et al. Effectiveness of bupropion sustained release for smoking cessation in a health care setting: a randomized trial. Arch Intern Med 2003; 163:2337–2344.
- Watkins SS, Koob GF, Markou A. Neural mechanisms underlying nicotine addiction: acute positive reinforcement and withdrawal. Nicotine Tob Res 2000; 2:19–37.
- Coe JW, Brooks PR, Wirtz MC, et al. 3,5-Bicyclic aryl piperidines: a novel class of alpha4beta2 neuronal nicotinic receptor partial agonists for smoking cessation. Bioorg Med Chem Lett 2005; 15:4889–4897.
- Picciotto MR, Zoli M, Changeux JP. Use of knock-out mice to determine the molecular basis for the actions of nicotine. Nicotine Tob Res 1999; 1(suppl 2):S121–S125.
- Nides M, Oncken C, Gonzales D, et al. Smoking cessation with varenicline, a selective alpha4beta2 nicotinic receptor partial agonist: results from a 7-week, randomized, placebo- and bupropion-controlled trial with 1-year follow-up. Arch Intern Med 2006; 166:1561–1568.
- Jorenby DE, Hays JT, Rigotti NA, et al; Varenicline Phase 3 Study Group. Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA 2006; 296:56–63.
- Gonzales D, Rennard SI, Nides M, et al; Varenicline Phase 3 Study Group. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA 2006; 296:47–55.
- Tonstad S, Tønnesen P, Hajek P, Williams KE, Billing CB, Reeves KR; Varenicline Phase 3 Study Group. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA 2006; 296:64–71.
- US Food and Drug Administration (FDA). Public health advisory: FDA requires new boxed warnings for the smoking cessation drugs Chantix and Zyban. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/PublicHealthAdvisories/ucm169988.htm. Accessed October 8, 2014.
- Rustin TA. Assessing nicotine dependence. Am Fam Physician 2000; 62:579–592.
- Centers for Disease Control and Prevention (CDC). Smoking & tobacco use. Low-yield cigarettes. www.cdc.gov/tobacco/data_statistics/fact_sheets/tobacco_industry/low_yield_cigarettes/index.htm. Accessed October 8, 2014.
- Biazzo LL, Froshaug DB, Harwell TS, et al. Characteristics and abstinence outcomes among tobacco quitline enrollees using varenicline or nicotine replacement therapy. Nicotine Tob Res 2010; 12:567–573.
- US Department of Health and Human Services. The health consequences of smoking—nicotine addiction; a report of the Surgeon General. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, 1988.
- Agaku I, King B, Dube SR, Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion, CDC. Current cigarette smoking among adults—United States, 2011. MMWR, 2012; 61(44):889–894.
- US Department of Health and Human Services. How tobacco smoke causes disease: the biology and behavioral basis for smoking-attributable disease: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2010.
- Centers for Disease Control and Prevention (CDC). Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000-2004. MMWR Morb Mortal Wkly Rep 2008; 57:1226–1228.
- Doll R, Peto R, Boreham J, Sutherland I. Mortality in relation to smoking. Fifty-years’ observations on male British doctors. BMJ 2004; 328:1519-1528.
- Centers for Disease Control and Prevention (CDC). Quitting smoking among adults—United States, 2001-2010. MMWR Morb Mortal Wkly Rep 2011; 60:1513–1519.
- Benowitz NL, Hukkanen J, Jacob P 3rd. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol 2009; 192:29–60.
- Rubinstein ML, Shiffman S, Moscicki AB, Rait MA, Sen S, Benowitz NL. Nicotine metabolism and addiction among adolescent smokers. Addiction 2013; 108:406–412.
- Benowitz NL, Porchet H, Sheiner L, Jacob P 3rd. Nicotine absorption and cardiovascular effects with smokeless tobacco use: comparison with cigarettes and nicotine gum. Clin Pharmacol Ther 1988; 44:23–28.
- Schneider NG, Lunell E, Olmstead RE, Fagerström KO. Clinical pharmacokinetics of nasal nicotine delivery. A review and comparison to other nicotine systems. Clin Pharmacokinet 1996; 31:65–80.
- Benowitz NL. Nicotine replacement therapy. What has been accomplished—can we do better? Drugs 1993; 45:157–170.
- Cahill K, Stevens S, Perera R, Lancaster T. Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev 2013; 5:CD009329.
- Committee on Safety in Medicines and the Medicines Control Agency. Zyban safety reminder. Current Problems in Pharmacovigilance 2001; 27:5.
- Clinical Practice Guideline Treating Tobacco Use and Dependence 2008 Update Panel, Liaisons, and Staff. A clinical practice guideline for treating tobacco use and dependence: 2008 update. A US public health service report. Am J Prev Med 2008; 35:158–176.
- Swan GE, McAfee T, Curry SJ, et al. Effectiveness of bupropion sustained release for smoking cessation in a health care setting: a randomized trial. Arch Intern Med 2003; 163:2337–2344.
- Watkins SS, Koob GF, Markou A. Neural mechanisms underlying nicotine addiction: acute positive reinforcement and withdrawal. Nicotine Tob Res 2000; 2:19–37.
- Coe JW, Brooks PR, Wirtz MC, et al. 3,5-Bicyclic aryl piperidines: a novel class of alpha4beta2 neuronal nicotinic receptor partial agonists for smoking cessation. Bioorg Med Chem Lett 2005; 15:4889–4897.
- Picciotto MR, Zoli M, Changeux JP. Use of knock-out mice to determine the molecular basis for the actions of nicotine. Nicotine Tob Res 1999; 1(suppl 2):S121–S125.
- Nides M, Oncken C, Gonzales D, et al. Smoking cessation with varenicline, a selective alpha4beta2 nicotinic receptor partial agonist: results from a 7-week, randomized, placebo- and bupropion-controlled trial with 1-year follow-up. Arch Intern Med 2006; 166:1561–1568.
- Jorenby DE, Hays JT, Rigotti NA, et al; Varenicline Phase 3 Study Group. Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA 2006; 296:56–63.
- Gonzales D, Rennard SI, Nides M, et al; Varenicline Phase 3 Study Group. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA 2006; 296:47–55.
- Tonstad S, Tønnesen P, Hajek P, Williams KE, Billing CB, Reeves KR; Varenicline Phase 3 Study Group. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA 2006; 296:64–71.
- US Food and Drug Administration (FDA). Public health advisory: FDA requires new boxed warnings for the smoking cessation drugs Chantix and Zyban. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/PublicHealthAdvisories/ucm169988.htm. Accessed October 8, 2014.
- Rustin TA. Assessing nicotine dependence. Am Fam Physician 2000; 62:579–592.
- Centers for Disease Control and Prevention (CDC). Smoking & tobacco use. Low-yield cigarettes. www.cdc.gov/tobacco/data_statistics/fact_sheets/tobacco_industry/low_yield_cigarettes/index.htm. Accessed October 8, 2014.
- Biazzo LL, Froshaug DB, Harwell TS, et al. Characteristics and abstinence outcomes among tobacco quitline enrollees using varenicline or nicotine replacement therapy. Nicotine Tob Res 2010; 12:567–573.
- US Department of Health and Human Services. The health consequences of smoking—nicotine addiction; a report of the Surgeon General. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, 1988.
- Agaku I, King B, Dube SR, Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion, CDC. Current cigarette smoking among adults—United States, 2011. MMWR, 2012; 61(44):889–894.
KEY POINTS
- Nicotine dependence is a life-threatening, biochemically based disease, driven by changes in midbrain receptors and reward mechanisms.
- The state of the art in smoking cessation involves encouragement, persistence, and evidence-based pharmacotherapy.
- Physicians should be assertive in addressing nicotine dependence, approaching patients with encouragement to quit, consistent monitoring and support, telephone “quit lines,” and counseling, as well as persistence and optimism. The combination of proactive, engaged, brief counseling and pharmacotherapy will yield the best results.
Rule out pulmonary tuberculosis: Clinical and radiographic clues for the internist
Tuberculosis rates in the United States are at an all-time low, which is good news for public health. However, as clinicians see fewer cases of tuberculosis, their skill at making this diagnosis rapidly diminishes.
In 2012, for the first time, fewer than 10,000 tuberculosis cases were reported in the United States to the Centers for Disease Control and Prevention (CDC),1 for a case rate of 3.2 per 100,000. This is in sharp contrast to the worldwide burden of tuberculosis: the World Health Organization2 estimated that there were 8.6 million new cases of tuberculosis in 2012. As a result of travel and immigration, clinicians in the United States will continue to see sporadic cases of active tuberculosis in their hospitals and clinics.
This review describes the clinical and radiographic clues to the diagnosis of pulmonary tuberculosis, discusses the use and discontinuation of respiratory isolation, and reviews the use of new diagnostic technologies.
CASE 1: A COLLEGE STUDENT WITH FATIGUE
A 23-year-old graduate student presents to the student health clinic with vague symptoms of fatigue and several pounds of weight loss over the past 3 months. When asked about coughing, he says he thinks he has had a mild, nonproductive cough for about a month. On examination he is thin, appears comfortable, and has faint rales in the right middle lung zone.
The clinician thinks that the symptoms are likely related to stress, lack of sleep, and difficulty adapting to graduate school life. However, in view of the pulmonary finding on examination, the physician obtains a complete blood cell count (CBC) and a chest radiograph. The CBC is normal. The radiograph (Figure 1) reveals a patchy, somewhat nodular infiltrate in the right upper lobe. The radiologist reviews the results, noting that tuberculosis is high on the list of possible diagnoses. The clinician calls the student and obtains the following additional history.
The patient was born in Thailand and arrived in the United States 3 months ago. Soon after his arrival, he had a tuberculin skin test with purified protein derivative in the student health department, which produced an induration 18 mm in diameter. The patient dismissed this finding as a false-positive result, attributing it to having received BCG vaccine in his native country, and he therefore did not follow up as recommended for a chest radiograph. He denies having fever, night sweats, or hemoptysis.
Since the patient lives in a college dormitory and has four roommates, the clinician admits him to the hospital for further evaluation and for airborne infection isolation. Sputum smears are positive for acid-fast bacilli, and samples ultimately grow Mycobacterium tuberculosis. He is started on standard antituberculosis treatment with isoniazid, rifampin, ethambutol, and pyrazinamide and discharged about 1 week later. He does well. Approximately 50 of his classmates are tested for possible exposure to tuberculosis.
CASE 2: A MAN WITH ACUTE-ONSET SYMPTOMS
A 35-year-old man presents to the emergency department for evaluation of cough with sputum production, fever, nausea, vomiting, and diarrhea. The symptoms began suddenly 1 week previously. He has no medical history, was born in the United States, and works in computer sales. On examination he looks uncomfortable, is slightly tachypneic, and has a temperature of 101°F (38.3°C).
Given his complaint of cough, chest radiography and a CBC are ordered. The white blood cell count is 18.0 × 109/L (reference range 4.5–11.0), with 50% bands (reference range 3%–5%). The chest radiograph (Figure 2) shows a dense infiltrate in the right upper lobe, with air bronchograms and possible right hilar fullness.
The patient is diagnosed with community-acquired pneumonia, and because his oral intake is poor, he is admitted to the hospital and started on azithromycin and ceftriaxone. Blood cultures the next day grow Streptococcus pneumoniae. He fully recovers. A follow-up radiograph is performed 6 weeks later because of the right hilar fullness, and it is normal.
COMMENT
These two cases demonstrate the importance of clinical, demographic, laboratory, and radiographic clues to raise or lower our suspicion for pulmonary tuberculosis. Both patients had right-upper-lobe infiltrates on radiography, yet the diagnosis of tuberculosis was considered only in the first patient.
CLINICAL CLUES TO PULMONARY TUBERCULOSIS
Symptoms of tuberculosis are generally indolent in onset, often so much so that the patient does not realize that he or she is sick until after starting treatment and beginning to improve. In addition, the symptoms can be vague, including only mild fatigue and cough. The classic symptoms of prolonged nonproductive cough, hemoptysis, weight loss, and fever often do not appear until the disease is quite advanced in the lung and the patient has been sick for months.
Since the symptoms of tuberculosis can be nonspecific, the patient’s social and demographic characteristics are important in assessing the likelihood that his or her current illness is tuberculosis.
Foreign birth
Almost two-thirds of all reported tuberculosis cases in the United States are in people who were born outside of the United States.1 The highest risk of reactivation appears to be within the first 5 years after immigration to the United States.3
Other risk factors
- Extensive travel to tuberculosis-endemic regions of the world
- Previous incarceration
- Intravenous drug use
- Work in health care
- Homelessness
- Known exposure to tuberculosis in the past.
Certain medical conditions predispose to reactivation of tuberculosis and should be considered when evaluating someone for active tuberculosis. These include human immunodeficiency virus infection and immunosuppression from tumor necrosis factor inhibitors, steroids, and medications used in organ transplantation. Other risk factors include diabetes requiring insulin, end-stage renal disease, and hematologic malignancies.4 Absence of these risk factors does not exclude tuberculosis, but it decreases the likelihood.
Findings on physical examination and laboratory testing are generally nonspecific in active tuberculosis. In particular, fever is present in 40% to 80% of patients. The white blood cell count is generally normal or only slightly elevated.
Radiographic signs
While the presenting symptoms and physical findings can be nonspecific, there are definite clues to the diagnosis of tuberculosis on chest radiography. In adults, most cases of tuberculosis are reactivation-type, which means the patient was exposed to tuberculosis many months to years in the past.
Reactivation-type tuberculosis usually occurs in the upper lobes, classically in the apical and posterior segments. The infiltrates tend to be patchy rather than densely consolidated. Cavitation, when present, increases the likelihood of tuberculosis. Intrathoracic lymphadenopathy, which occurs in primary tuberculosis, is generally not seen in adults with typical reactivation pulmonary tuberculosis.
However, adults who are highly immunosuppressed, such as those with advanced human immunodeficiency virus infection, organ transplant recipients, or those taking tumor necrosis factor inhibitors, may have radiographic features that are atypical for tuberculosis. For example, they may present with hilar adenopathy or lower-lobe infiltrates.5
Are there clinical prediction rules for tuberculosis?
Because tuberculosis rates have been declining and most hospitals have a limited number of rooms for airborne infection isolation, several studies have evaluated clinical prediction rules for diagnosing pulmonary tuberculosis.
In general, the signs and symptoms that predict tuberculosis are similar to those discussed above, including chronic symptoms, immunosuppression, birth in a region with a high incidence of the disease, a chest radiograph showing upper-zone findings, a positive tuberculin skin test, and fever.6–8 The studies that identified these factors are limited in generalizability as they were performed and validated in single institutions, and the prediction rules have not been widely adopted. Yet they provide a straightforward way to determine which patients should be prioritized for isolation.
RETURN TO THE CASES
The student in case 1 had several features suggesting tuberculosis: indolent and nonspecific symptoms, normal CBC, patchy upper-lobe infiltrates, birth in a country that has a high incidence of tuberculosis, and a positive skin test.
In contrast, the man in case 2 had features that made tuberculosis much less likely: acute symptoms, markedly elevated white blood cell count, and densely consolidated infiltrate. He was also born in the United States and had no additional risk factors for tuberculosis exposure.
EVALUATION OF SUSPECTED TUBERCULOSIS
Who should be admitted to the hospital for evaluation?
In general, a patient with suspected tuberculosis can be evaluated as an outpatient. However, there are a number of reasons to consider hospital admission for the initial workup and for starting treatment:
- Clinical instability requiring inpatient care: eg, hypoxia, unstable vital signs, inability to tolerate oral intake
- Residence in a congregate setting such as a homeless shelter, nursing home, or college dormitory, where there is an ongoing risk of transmitting the infection to others
- Concern that the patient might be lost to follow-up if discharged from the emergency department or clinic
- Vulnerable contacts living in the home with the patient (eg, newborn infants, severely immunosuppressed people)
- Lack of resources in the community to provide prompt evaluation and initiation of treatment; most urban areas have tuberculosis clinics with outreach staff available to provide support for patients, but these resources are scarcer in rural regions.
When should a patient be placed in airborne infection isolation?
Patients with suspected active pulmonary tuberculosis should be placed in airborne infection isolation (also called respiratory or negative-pressure isolation). The purpose of this isolation method is to prevent transmission to other patients and to health care workers. Isolation and other environmental and personal controls such as ultraviolet light and N-95 masks are highly effective in preventing transmission.9
However, there are disadvantages to placing patients in isolation. Only 1 out of every 10 to 25 patients isolated actually has tuberculosis,10 and patients typically remain in isolation for 4 to 7 days. Therefore, unnecessary isolation can delay diagnostic testing for other illnesses and may waste already-limited health care resources. In addition, isolation carries the potential for decreased contact with providers.
When can a patient be released from isolation?
One of the problems with airborne infection isolation is determining when it is safe to discontinue it, especially when the diagnosis of tuberculosis appears less likely.
Traditionally, we have used the requirement of three negative sputum smears for acid-fast bacilli on 3 separate days, as well as low clinical suspicion for tuberculosis. The use of three sputum smears for acid-fast bacilli is based on studies11,12 in populations that have a high prevalence of tuberculosis. These studies found that after three sputum smears were obtained, additional sputum smears were unlikely to improve the sensitivity of the test. The studies focused on maximizing the sensitivity of the test and detecting all potential cases.11,12 However, in US hospitals today, the focus is on rapidly excluding the diagnosis of tuberculosis to minimize hospital length of stay and to allow evaluation for alternative diagnoses.
Several studies have called into question the need for three negative sputum smears to discontinue isolation.13–16 Mathew et al13 found a negative predictive value of 97.8% with a single negative sputum smear for the diagnosis of culture-positive tuberculosis. Each additional sputum increased the negative predictive value by only 0.2%. The authors suggested that one or two negative sputum smears are sufficient to discontinue isolation in a region that has a low incidence of tuberculosis. These studies were all performed at single institutions in the United States and Canada, and their findings are relevant to regions that have a low incidence of tuberculosis.
The CDC continues to recommend that airborne infection isolation be discontinued only when either another diagnosis is made that explains the clinical syndrome or the patient has three negative acid-fast bacilli sputum smear results or two negative acid-fast bacilli smears and one negative nucleic amplification test (discussed below). These should be done at least 8 hours apart and should include at least one early-morning specimen.9
In a minority of cases, empiric treatment for tuberculosis is indicated despite negative sputum smears, based on clinical and radiographic manifestations. Patients receiving empiric treatment for pulmonary tuberculosis should remain in airborne infection isolation during the initiation of treatment (if a hospital stay is required) until cleared by a specialist in infectious disease or tuberculosis.
Can molecular techniques help in rapidly diagnosing tuberculosis?
Additional tests for tuberculosis that are performed on clinical specimens have been available for the past 10 years.
Nucleic acid amplification can detect tuberculosis directly in sputum, bronchoscopy specimens, or other clinical specimens. It is available at reference laboratories, large hospitals, and many state laboratories, often with 24-hour turnaround. Both commercial and in-house tests are performed. The CDC considers nucleic acid amplification to be very helpful and underutilized. An important limitation of the test is that it performs best in smear-positive specimens, with a sensitivity of 96.8%, whereas its sensitivity in smear-negative samples is only 73%.17 For this reason, nucleic acid amplification is still not widely used in US hospitals.
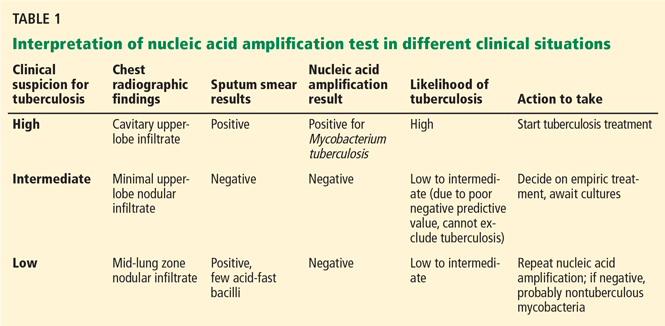
The CDC recommends nucleic acid amplification testing in all patients in whom the diagnosis of tuberculosis is being considered but is not yet confirmed.18 Table 1 outlines the use of nucleic acid amplification in several clinical situations. Use and interpretation of this test in suspected tuberculosis often requires consultation with clinicians who are experienced in the diagnosis of this disease.
How should an interferon-gamma-release assay or a tuberculin skin test be used in evaluating suspected tuberculosis?
Patients who have never tested positive for tuberculosis on a skin test should be tested by tuberculin skin testing or with an interferon-gamma-release assay during an evaluation for suspected pulmonary tuberculosis. The interferon-gamma-release assays available in the United States are the QuantiFERON-TB Gold In-Tube test and the T-Spot TB test. Most larger hospitals have one of the two available.
Of note: up to 25% of patients with active tuberculosis can have a negative skin test or interferon-gamma-release assay at the time of initial diagnosis, the number being higher in those who are immunosuppressed.

An interferon-gamma-release assay, which is performed on the patient’s serum, is preferred in those who have previously received the BCG vaccine, as there is no cross-reactivity between the vaccine and the antigens in the assay. In patients with active tuberculosis, the interferon-gamma-release assay does not perform any better than the skin test, so the choice of test should be determined by availability. Table 2 compares the characteristics of tuberculin skin testing and the interferon- gamma-release assay.19
In evaluating for active tuberculosis, a positive skin test or interferon-gamma-release assay can be helpful in increasing the likelihood of tuberculosis, but a negative result does not exclude active tuberculosis.
Is computed tomography necessary in patients suspected of having active pulmonary tuberculosis?
Additional imaging is often performed in patients with suspected pulmonary tuberculosis, or before the diagnosis of tuberculosis is considered. Computed tomography provides more detailed images of pulmonary infiltrates and may reveal more extensive disease than plain radiography, but the images are not diagnostic. Ultimately, sputum and sometimes tissue are required. Far too often, a sputum smear for acid-fast bacilli is the last test to be performed, after both computed tomography and bronchoscopy have been done. In addition, in order to undergo computed tomography, the patient must be removed from airborne infection isolation.
The decision to perform computed tomography must be individualized to the patient and to the clinical situation. It is certainly not a necessary test for the diagnosis of pulmonary tuberculosis.
When should the diagnosis be reported?
Tuberculosis is a reportable illness in the United States. Although each state varies in its specific requirements, if tuberculosis treatment is being initiated or tuberculosis is strongly suspected, a report should be made to the local public health authority for tuberculosis within 24 hours.
This report allows for outreach services to be offered to the patient, often including directly observed therapy in which doses of antituberculosis treatment are provided and observed to ensure completion of treatment. In addition, public health authorities bear the responsibility for contact investigation to determine if transmission of tuberculosis has occurred in the community.
- Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2012. MMWR Morb Mortal Wkly Rep 2013; 62:201–205.
- World Health Organization (WHO). Tuberculosis. WHO Global Tuberculosis Report 2013. www.who.int/tb/publications/factsheet_global.pdf. Accessed November 13, 2014.
- McKenna MT, McCray E, Onorato I. The epidemiology of tuberculosis among foreign-born persons in the United States, 1986 to 1993. N Engl J Med 1995; 332:1071–1076.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med 2000; 161:S221–S247.
- Pitchenik AE, Rubinson HA. The radiographic appearance of tuberculosis in patients with the acquired immune deficiency syndrome (AIDS) and pre-AIDS. Am Rev Respir Dis 1985; 131:393–396.
- Wisnivesky JP, Kaplan J, Henschke C, McGinn TG, Crystal RG. Evaluation of clinical parameters to predict Mycobacterium tuberculosis in inpatients. Arch Intern Med 2000; 160:2471–2476.
- Wisnivesky JP, Henschke C, Balentine J, Willner C, Deloire AM, McGinn TG. Prospective validation of a prediction model for isolating inpatients with suspected pulmonary tuberculosis. Arch Intern Med 2005; 165:453–457.
- Rakoczy KS, Cohen SH, Nguyen HH. Derivation and validation of a clinical prediction score for isolation of inpatients with suspected pulmonary tuberculosis. Infect Control Hosp Epidemiol 2008; 29:927–932.
- Jensen PA, Lambert LA, Iademarco MF, Ridzon R; Centers for Disease Control and Prevention (CDC). Guidelines for preventing the transmission of Mycobacterium tuberculosis in health-care settings, 2005. MMWR Recomm Rep 2005; 54(RR-17):1–141.
- Campos M, Quartin A, Mendes E, et al. Feasibility of shortening respiratory isolation with a single sputum nucleic acid amplification test. Am J Respir Crit Care Med 2008; 178:300–305.
- MacGregor RR. A year’s experience with tuberculosis in a private urban teaching hospital in the postsanatorium era. Am J Med 1975; 58:221–228.
- Greenbaum M, Beyt BE Jr, Murray PR. The accuracy of diagnosing pulmonary tuberculosis at a teaching hospital. Am Rev Respir Dis 1980; 121:477–481.
- Mathew P, Kuo YH, Vazirani B, Eng RH, Weinstein MP. Are three sputum acid-fast bacillus smears necessary for discontinuing tuberculosis isolation? J Clin Microbiol 2002; 40:3482–3484.
- Bryan CS, Rapp DJ, Brown CA. Discontinuation of respiratory isolation for possible tuberculosis: do two negative sputum smear results suffice? Infect Control Hosp Epidemiol 2006; 27:515–516.
- Nelson SM, Deike MA, Cartwright CP. Value of examining multiple sputum specimens in the diagnosis of pulmonary tuberculosis. J Clin Microbiol 1998; 36:467–469.
- Wilmer A, Bryce E, Grant J. The role of the third acid-fast bacillus smear in tuberculosis screening for infection control purposes: a controversial topic revisited. Can J Infect Dis Med Microbiol 2011; 22:e1–e3.
- Dinnes J, Deeks J, Kunst H, et al. A systematic review of rapid diagnostic tests for the detection of tuberculosis infection. Health Technol Assess 2007; 11:1–196.
- Centers for Disease Control and Prevention (CDC). Updated guidelines for the use of nucleic acid amplification tests in the diagnosis of tuberculosis. MMWR Morb Mortal Wkly Rep 2009; 58:7–10.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee; Centers for Disease Control and Prevention (CDC). Updated guidelines for using interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
Tuberculosis rates in the United States are at an all-time low, which is good news for public health. However, as clinicians see fewer cases of tuberculosis, their skill at making this diagnosis rapidly diminishes.
In 2012, for the first time, fewer than 10,000 tuberculosis cases were reported in the United States to the Centers for Disease Control and Prevention (CDC),1 for a case rate of 3.2 per 100,000. This is in sharp contrast to the worldwide burden of tuberculosis: the World Health Organization2 estimated that there were 8.6 million new cases of tuberculosis in 2012. As a result of travel and immigration, clinicians in the United States will continue to see sporadic cases of active tuberculosis in their hospitals and clinics.
This review describes the clinical and radiographic clues to the diagnosis of pulmonary tuberculosis, discusses the use and discontinuation of respiratory isolation, and reviews the use of new diagnostic technologies.
CASE 1: A COLLEGE STUDENT WITH FATIGUE
A 23-year-old graduate student presents to the student health clinic with vague symptoms of fatigue and several pounds of weight loss over the past 3 months. When asked about coughing, he says he thinks he has had a mild, nonproductive cough for about a month. On examination he is thin, appears comfortable, and has faint rales in the right middle lung zone.
The clinician thinks that the symptoms are likely related to stress, lack of sleep, and difficulty adapting to graduate school life. However, in view of the pulmonary finding on examination, the physician obtains a complete blood cell count (CBC) and a chest radiograph. The CBC is normal. The radiograph (Figure 1) reveals a patchy, somewhat nodular infiltrate in the right upper lobe. The radiologist reviews the results, noting that tuberculosis is high on the list of possible diagnoses. The clinician calls the student and obtains the following additional history.
The patient was born in Thailand and arrived in the United States 3 months ago. Soon after his arrival, he had a tuberculin skin test with purified protein derivative in the student health department, which produced an induration 18 mm in diameter. The patient dismissed this finding as a false-positive result, attributing it to having received BCG vaccine in his native country, and he therefore did not follow up as recommended for a chest radiograph. He denies having fever, night sweats, or hemoptysis.
Since the patient lives in a college dormitory and has four roommates, the clinician admits him to the hospital for further evaluation and for airborne infection isolation. Sputum smears are positive for acid-fast bacilli, and samples ultimately grow Mycobacterium tuberculosis. He is started on standard antituberculosis treatment with isoniazid, rifampin, ethambutol, and pyrazinamide and discharged about 1 week later. He does well. Approximately 50 of his classmates are tested for possible exposure to tuberculosis.
CASE 2: A MAN WITH ACUTE-ONSET SYMPTOMS
A 35-year-old man presents to the emergency department for evaluation of cough with sputum production, fever, nausea, vomiting, and diarrhea. The symptoms began suddenly 1 week previously. He has no medical history, was born in the United States, and works in computer sales. On examination he looks uncomfortable, is slightly tachypneic, and has a temperature of 101°F (38.3°C).
Given his complaint of cough, chest radiography and a CBC are ordered. The white blood cell count is 18.0 × 109/L (reference range 4.5–11.0), with 50% bands (reference range 3%–5%). The chest radiograph (Figure 2) shows a dense infiltrate in the right upper lobe, with air bronchograms and possible right hilar fullness.
The patient is diagnosed with community-acquired pneumonia, and because his oral intake is poor, he is admitted to the hospital and started on azithromycin and ceftriaxone. Blood cultures the next day grow Streptococcus pneumoniae. He fully recovers. A follow-up radiograph is performed 6 weeks later because of the right hilar fullness, and it is normal.
COMMENT
These two cases demonstrate the importance of clinical, demographic, laboratory, and radiographic clues to raise or lower our suspicion for pulmonary tuberculosis. Both patients had right-upper-lobe infiltrates on radiography, yet the diagnosis of tuberculosis was considered only in the first patient.
CLINICAL CLUES TO PULMONARY TUBERCULOSIS
Symptoms of tuberculosis are generally indolent in onset, often so much so that the patient does not realize that he or she is sick until after starting treatment and beginning to improve. In addition, the symptoms can be vague, including only mild fatigue and cough. The classic symptoms of prolonged nonproductive cough, hemoptysis, weight loss, and fever often do not appear until the disease is quite advanced in the lung and the patient has been sick for months.
Since the symptoms of tuberculosis can be nonspecific, the patient’s social and demographic characteristics are important in assessing the likelihood that his or her current illness is tuberculosis.
Foreign birth
Almost two-thirds of all reported tuberculosis cases in the United States are in people who were born outside of the United States.1 The highest risk of reactivation appears to be within the first 5 years after immigration to the United States.3
Other risk factors
- Extensive travel to tuberculosis-endemic regions of the world
- Previous incarceration
- Intravenous drug use
- Work in health care
- Homelessness
- Known exposure to tuberculosis in the past.
Certain medical conditions predispose to reactivation of tuberculosis and should be considered when evaluating someone for active tuberculosis. These include human immunodeficiency virus infection and immunosuppression from tumor necrosis factor inhibitors, steroids, and medications used in organ transplantation. Other risk factors include diabetes requiring insulin, end-stage renal disease, and hematologic malignancies.4 Absence of these risk factors does not exclude tuberculosis, but it decreases the likelihood.
Findings on physical examination and laboratory testing are generally nonspecific in active tuberculosis. In particular, fever is present in 40% to 80% of patients. The white blood cell count is generally normal or only slightly elevated.
Radiographic signs
While the presenting symptoms and physical findings can be nonspecific, there are definite clues to the diagnosis of tuberculosis on chest radiography. In adults, most cases of tuberculosis are reactivation-type, which means the patient was exposed to tuberculosis many months to years in the past.
Reactivation-type tuberculosis usually occurs in the upper lobes, classically in the apical and posterior segments. The infiltrates tend to be patchy rather than densely consolidated. Cavitation, when present, increases the likelihood of tuberculosis. Intrathoracic lymphadenopathy, which occurs in primary tuberculosis, is generally not seen in adults with typical reactivation pulmonary tuberculosis.
However, adults who are highly immunosuppressed, such as those with advanced human immunodeficiency virus infection, organ transplant recipients, or those taking tumor necrosis factor inhibitors, may have radiographic features that are atypical for tuberculosis. For example, they may present with hilar adenopathy or lower-lobe infiltrates.5
Are there clinical prediction rules for tuberculosis?
Because tuberculosis rates have been declining and most hospitals have a limited number of rooms for airborne infection isolation, several studies have evaluated clinical prediction rules for diagnosing pulmonary tuberculosis.
In general, the signs and symptoms that predict tuberculosis are similar to those discussed above, including chronic symptoms, immunosuppression, birth in a region with a high incidence of the disease, a chest radiograph showing upper-zone findings, a positive tuberculin skin test, and fever.6–8 The studies that identified these factors are limited in generalizability as they were performed and validated in single institutions, and the prediction rules have not been widely adopted. Yet they provide a straightforward way to determine which patients should be prioritized for isolation.
RETURN TO THE CASES
The student in case 1 had several features suggesting tuberculosis: indolent and nonspecific symptoms, normal CBC, patchy upper-lobe infiltrates, birth in a country that has a high incidence of tuberculosis, and a positive skin test.
In contrast, the man in case 2 had features that made tuberculosis much less likely: acute symptoms, markedly elevated white blood cell count, and densely consolidated infiltrate. He was also born in the United States and had no additional risk factors for tuberculosis exposure.
EVALUATION OF SUSPECTED TUBERCULOSIS
Who should be admitted to the hospital for evaluation?
In general, a patient with suspected tuberculosis can be evaluated as an outpatient. However, there are a number of reasons to consider hospital admission for the initial workup and for starting treatment:
- Clinical instability requiring inpatient care: eg, hypoxia, unstable vital signs, inability to tolerate oral intake
- Residence in a congregate setting such as a homeless shelter, nursing home, or college dormitory, where there is an ongoing risk of transmitting the infection to others
- Concern that the patient might be lost to follow-up if discharged from the emergency department or clinic
- Vulnerable contacts living in the home with the patient (eg, newborn infants, severely immunosuppressed people)
- Lack of resources in the community to provide prompt evaluation and initiation of treatment; most urban areas have tuberculosis clinics with outreach staff available to provide support for patients, but these resources are scarcer in rural regions.
When should a patient be placed in airborne infection isolation?
Patients with suspected active pulmonary tuberculosis should be placed in airborne infection isolation (also called respiratory or negative-pressure isolation). The purpose of this isolation method is to prevent transmission to other patients and to health care workers. Isolation and other environmental and personal controls such as ultraviolet light and N-95 masks are highly effective in preventing transmission.9
However, there are disadvantages to placing patients in isolation. Only 1 out of every 10 to 25 patients isolated actually has tuberculosis,10 and patients typically remain in isolation for 4 to 7 days. Therefore, unnecessary isolation can delay diagnostic testing for other illnesses and may waste already-limited health care resources. In addition, isolation carries the potential for decreased contact with providers.
When can a patient be released from isolation?
One of the problems with airborne infection isolation is determining when it is safe to discontinue it, especially when the diagnosis of tuberculosis appears less likely.
Traditionally, we have used the requirement of three negative sputum smears for acid-fast bacilli on 3 separate days, as well as low clinical suspicion for tuberculosis. The use of three sputum smears for acid-fast bacilli is based on studies11,12 in populations that have a high prevalence of tuberculosis. These studies found that after three sputum smears were obtained, additional sputum smears were unlikely to improve the sensitivity of the test. The studies focused on maximizing the sensitivity of the test and detecting all potential cases.11,12 However, in US hospitals today, the focus is on rapidly excluding the diagnosis of tuberculosis to minimize hospital length of stay and to allow evaluation for alternative diagnoses.
Several studies have called into question the need for three negative sputum smears to discontinue isolation.13–16 Mathew et al13 found a negative predictive value of 97.8% with a single negative sputum smear for the diagnosis of culture-positive tuberculosis. Each additional sputum increased the negative predictive value by only 0.2%. The authors suggested that one or two negative sputum smears are sufficient to discontinue isolation in a region that has a low incidence of tuberculosis. These studies were all performed at single institutions in the United States and Canada, and their findings are relevant to regions that have a low incidence of tuberculosis.
The CDC continues to recommend that airborne infection isolation be discontinued only when either another diagnosis is made that explains the clinical syndrome or the patient has three negative acid-fast bacilli sputum smear results or two negative acid-fast bacilli smears and one negative nucleic amplification test (discussed below). These should be done at least 8 hours apart and should include at least one early-morning specimen.9
In a minority of cases, empiric treatment for tuberculosis is indicated despite negative sputum smears, based on clinical and radiographic manifestations. Patients receiving empiric treatment for pulmonary tuberculosis should remain in airborne infection isolation during the initiation of treatment (if a hospital stay is required) until cleared by a specialist in infectious disease or tuberculosis.
Can molecular techniques help in rapidly diagnosing tuberculosis?
Additional tests for tuberculosis that are performed on clinical specimens have been available for the past 10 years.
Nucleic acid amplification can detect tuberculosis directly in sputum, bronchoscopy specimens, or other clinical specimens. It is available at reference laboratories, large hospitals, and many state laboratories, often with 24-hour turnaround. Both commercial and in-house tests are performed. The CDC considers nucleic acid amplification to be very helpful and underutilized. An important limitation of the test is that it performs best in smear-positive specimens, with a sensitivity of 96.8%, whereas its sensitivity in smear-negative samples is only 73%.17 For this reason, nucleic acid amplification is still not widely used in US hospitals.
The CDC recommends nucleic acid amplification testing in all patients in whom the diagnosis of tuberculosis is being considered but is not yet confirmed.18 Table 1 outlines the use of nucleic acid amplification in several clinical situations. Use and interpretation of this test in suspected tuberculosis often requires consultation with clinicians who are experienced in the diagnosis of this disease.
How should an interferon-gamma-release assay or a tuberculin skin test be used in evaluating suspected tuberculosis?
Patients who have never tested positive for tuberculosis on a skin test should be tested by tuberculin skin testing or with an interferon-gamma-release assay during an evaluation for suspected pulmonary tuberculosis. The interferon-gamma-release assays available in the United States are the QuantiFERON-TB Gold In-Tube test and the T-Spot TB test. Most larger hospitals have one of the two available.
Of note: up to 25% of patients with active tuberculosis can have a negative skin test or interferon-gamma-release assay at the time of initial diagnosis, the number being higher in those who are immunosuppressed.
An interferon-gamma-release assay, which is performed on the patient’s serum, is preferred in those who have previously received the BCG vaccine, as there is no cross-reactivity between the vaccine and the antigens in the assay. In patients with active tuberculosis, the interferon-gamma-release assay does not perform any better than the skin test, so the choice of test should be determined by availability. Table 2 compares the characteristics of tuberculin skin testing and the interferon- gamma-release assay.19
In evaluating for active tuberculosis, a positive skin test or interferon-gamma-release assay can be helpful in increasing the likelihood of tuberculosis, but a negative result does not exclude active tuberculosis.
Is computed tomography necessary in patients suspected of having active pulmonary tuberculosis?
Additional imaging is often performed in patients with suspected pulmonary tuberculosis, or before the diagnosis of tuberculosis is considered. Computed tomography provides more detailed images of pulmonary infiltrates and may reveal more extensive disease than plain radiography, but the images are not diagnostic. Ultimately, sputum and sometimes tissue are required. Far too often, a sputum smear for acid-fast bacilli is the last test to be performed, after both computed tomography and bronchoscopy have been done. In addition, in order to undergo computed tomography, the patient must be removed from airborne infection isolation.
The decision to perform computed tomography must be individualized to the patient and to the clinical situation. It is certainly not a necessary test for the diagnosis of pulmonary tuberculosis.
When should the diagnosis be reported?
Tuberculosis is a reportable illness in the United States. Although each state varies in its specific requirements, if tuberculosis treatment is being initiated or tuberculosis is strongly suspected, a report should be made to the local public health authority for tuberculosis within 24 hours.
This report allows for outreach services to be offered to the patient, often including directly observed therapy in which doses of antituberculosis treatment are provided and observed to ensure completion of treatment. In addition, public health authorities bear the responsibility for contact investigation to determine if transmission of tuberculosis has occurred in the community.
Tuberculosis rates in the United States are at an all-time low, which is good news for public health. However, as clinicians see fewer cases of tuberculosis, their skill at making this diagnosis rapidly diminishes.
In 2012, for the first time, fewer than 10,000 tuberculosis cases were reported in the United States to the Centers for Disease Control and Prevention (CDC),1 for a case rate of 3.2 per 100,000. This is in sharp contrast to the worldwide burden of tuberculosis: the World Health Organization2 estimated that there were 8.6 million new cases of tuberculosis in 2012. As a result of travel and immigration, clinicians in the United States will continue to see sporadic cases of active tuberculosis in their hospitals and clinics.
This review describes the clinical and radiographic clues to the diagnosis of pulmonary tuberculosis, discusses the use and discontinuation of respiratory isolation, and reviews the use of new diagnostic technologies.
CASE 1: A COLLEGE STUDENT WITH FATIGUE
A 23-year-old graduate student presents to the student health clinic with vague symptoms of fatigue and several pounds of weight loss over the past 3 months. When asked about coughing, he says he thinks he has had a mild, nonproductive cough for about a month. On examination he is thin, appears comfortable, and has faint rales in the right middle lung zone.
The clinician thinks that the symptoms are likely related to stress, lack of sleep, and difficulty adapting to graduate school life. However, in view of the pulmonary finding on examination, the physician obtains a complete blood cell count (CBC) and a chest radiograph. The CBC is normal. The radiograph (Figure 1) reveals a patchy, somewhat nodular infiltrate in the right upper lobe. The radiologist reviews the results, noting that tuberculosis is high on the list of possible diagnoses. The clinician calls the student and obtains the following additional history.
The patient was born in Thailand and arrived in the United States 3 months ago. Soon after his arrival, he had a tuberculin skin test with purified protein derivative in the student health department, which produced an induration 18 mm in diameter. The patient dismissed this finding as a false-positive result, attributing it to having received BCG vaccine in his native country, and he therefore did not follow up as recommended for a chest radiograph. He denies having fever, night sweats, or hemoptysis.
Since the patient lives in a college dormitory and has four roommates, the clinician admits him to the hospital for further evaluation and for airborne infection isolation. Sputum smears are positive for acid-fast bacilli, and samples ultimately grow Mycobacterium tuberculosis. He is started on standard antituberculosis treatment with isoniazid, rifampin, ethambutol, and pyrazinamide and discharged about 1 week later. He does well. Approximately 50 of his classmates are tested for possible exposure to tuberculosis.
CASE 2: A MAN WITH ACUTE-ONSET SYMPTOMS
A 35-year-old man presents to the emergency department for evaluation of cough with sputum production, fever, nausea, vomiting, and diarrhea. The symptoms began suddenly 1 week previously. He has no medical history, was born in the United States, and works in computer sales. On examination he looks uncomfortable, is slightly tachypneic, and has a temperature of 101°F (38.3°C).
Given his complaint of cough, chest radiography and a CBC are ordered. The white blood cell count is 18.0 × 109/L (reference range 4.5–11.0), with 50% bands (reference range 3%–5%). The chest radiograph (Figure 2) shows a dense infiltrate in the right upper lobe, with air bronchograms and possible right hilar fullness.
The patient is diagnosed with community-acquired pneumonia, and because his oral intake is poor, he is admitted to the hospital and started on azithromycin and ceftriaxone. Blood cultures the next day grow Streptococcus pneumoniae. He fully recovers. A follow-up radiograph is performed 6 weeks later because of the right hilar fullness, and it is normal.
COMMENT
These two cases demonstrate the importance of clinical, demographic, laboratory, and radiographic clues to raise or lower our suspicion for pulmonary tuberculosis. Both patients had right-upper-lobe infiltrates on radiography, yet the diagnosis of tuberculosis was considered only in the first patient.
CLINICAL CLUES TO PULMONARY TUBERCULOSIS
Symptoms of tuberculosis are generally indolent in onset, often so much so that the patient does not realize that he or she is sick until after starting treatment and beginning to improve. In addition, the symptoms can be vague, including only mild fatigue and cough. The classic symptoms of prolonged nonproductive cough, hemoptysis, weight loss, and fever often do not appear until the disease is quite advanced in the lung and the patient has been sick for months.
Since the symptoms of tuberculosis can be nonspecific, the patient’s social and demographic characteristics are important in assessing the likelihood that his or her current illness is tuberculosis.
Foreign birth
Almost two-thirds of all reported tuberculosis cases in the United States are in people who were born outside of the United States.1 The highest risk of reactivation appears to be within the first 5 years after immigration to the United States.3
Other risk factors
- Extensive travel to tuberculosis-endemic regions of the world
- Previous incarceration
- Intravenous drug use
- Work in health care
- Homelessness
- Known exposure to tuberculosis in the past.
Certain medical conditions predispose to reactivation of tuberculosis and should be considered when evaluating someone for active tuberculosis. These include human immunodeficiency virus infection and immunosuppression from tumor necrosis factor inhibitors, steroids, and medications used in organ transplantation. Other risk factors include diabetes requiring insulin, end-stage renal disease, and hematologic malignancies.4 Absence of these risk factors does not exclude tuberculosis, but it decreases the likelihood.
Findings on physical examination and laboratory testing are generally nonspecific in active tuberculosis. In particular, fever is present in 40% to 80% of patients. The white blood cell count is generally normal or only slightly elevated.
Radiographic signs
While the presenting symptoms and physical findings can be nonspecific, there are definite clues to the diagnosis of tuberculosis on chest radiography. In adults, most cases of tuberculosis are reactivation-type, which means the patient was exposed to tuberculosis many months to years in the past.
Reactivation-type tuberculosis usually occurs in the upper lobes, classically in the apical and posterior segments. The infiltrates tend to be patchy rather than densely consolidated. Cavitation, when present, increases the likelihood of tuberculosis. Intrathoracic lymphadenopathy, which occurs in primary tuberculosis, is generally not seen in adults with typical reactivation pulmonary tuberculosis.
However, adults who are highly immunosuppressed, such as those with advanced human immunodeficiency virus infection, organ transplant recipients, or those taking tumor necrosis factor inhibitors, may have radiographic features that are atypical for tuberculosis. For example, they may present with hilar adenopathy or lower-lobe infiltrates.5
Are there clinical prediction rules for tuberculosis?
Because tuberculosis rates have been declining and most hospitals have a limited number of rooms for airborne infection isolation, several studies have evaluated clinical prediction rules for diagnosing pulmonary tuberculosis.
In general, the signs and symptoms that predict tuberculosis are similar to those discussed above, including chronic symptoms, immunosuppression, birth in a region with a high incidence of the disease, a chest radiograph showing upper-zone findings, a positive tuberculin skin test, and fever.6–8 The studies that identified these factors are limited in generalizability as they were performed and validated in single institutions, and the prediction rules have not been widely adopted. Yet they provide a straightforward way to determine which patients should be prioritized for isolation.
RETURN TO THE CASES
The student in case 1 had several features suggesting tuberculosis: indolent and nonspecific symptoms, normal CBC, patchy upper-lobe infiltrates, birth in a country that has a high incidence of tuberculosis, and a positive skin test.
In contrast, the man in case 2 had features that made tuberculosis much less likely: acute symptoms, markedly elevated white blood cell count, and densely consolidated infiltrate. He was also born in the United States and had no additional risk factors for tuberculosis exposure.
EVALUATION OF SUSPECTED TUBERCULOSIS
Who should be admitted to the hospital for evaluation?
In general, a patient with suspected tuberculosis can be evaluated as an outpatient. However, there are a number of reasons to consider hospital admission for the initial workup and for starting treatment:
- Clinical instability requiring inpatient care: eg, hypoxia, unstable vital signs, inability to tolerate oral intake
- Residence in a congregate setting such as a homeless shelter, nursing home, or college dormitory, where there is an ongoing risk of transmitting the infection to others
- Concern that the patient might be lost to follow-up if discharged from the emergency department or clinic
- Vulnerable contacts living in the home with the patient (eg, newborn infants, severely immunosuppressed people)
- Lack of resources in the community to provide prompt evaluation and initiation of treatment; most urban areas have tuberculosis clinics with outreach staff available to provide support for patients, but these resources are scarcer in rural regions.
When should a patient be placed in airborne infection isolation?
Patients with suspected active pulmonary tuberculosis should be placed in airborne infection isolation (also called respiratory or negative-pressure isolation). The purpose of this isolation method is to prevent transmission to other patients and to health care workers. Isolation and other environmental and personal controls such as ultraviolet light and N-95 masks are highly effective in preventing transmission.9
However, there are disadvantages to placing patients in isolation. Only 1 out of every 10 to 25 patients isolated actually has tuberculosis,10 and patients typically remain in isolation for 4 to 7 days. Therefore, unnecessary isolation can delay diagnostic testing for other illnesses and may waste already-limited health care resources. In addition, isolation carries the potential for decreased contact with providers.
When can a patient be released from isolation?
One of the problems with airborne infection isolation is determining when it is safe to discontinue it, especially when the diagnosis of tuberculosis appears less likely.
Traditionally, we have used the requirement of three negative sputum smears for acid-fast bacilli on 3 separate days, as well as low clinical suspicion for tuberculosis. The use of three sputum smears for acid-fast bacilli is based on studies11,12 in populations that have a high prevalence of tuberculosis. These studies found that after three sputum smears were obtained, additional sputum smears were unlikely to improve the sensitivity of the test. The studies focused on maximizing the sensitivity of the test and detecting all potential cases.11,12 However, in US hospitals today, the focus is on rapidly excluding the diagnosis of tuberculosis to minimize hospital length of stay and to allow evaluation for alternative diagnoses.
Several studies have called into question the need for three negative sputum smears to discontinue isolation.13–16 Mathew et al13 found a negative predictive value of 97.8% with a single negative sputum smear for the diagnosis of culture-positive tuberculosis. Each additional sputum increased the negative predictive value by only 0.2%. The authors suggested that one or two negative sputum smears are sufficient to discontinue isolation in a region that has a low incidence of tuberculosis. These studies were all performed at single institutions in the United States and Canada, and their findings are relevant to regions that have a low incidence of tuberculosis.
The CDC continues to recommend that airborne infection isolation be discontinued only when either another diagnosis is made that explains the clinical syndrome or the patient has three negative acid-fast bacilli sputum smear results or two negative acid-fast bacilli smears and one negative nucleic amplification test (discussed below). These should be done at least 8 hours apart and should include at least one early-morning specimen.9
In a minority of cases, empiric treatment for tuberculosis is indicated despite negative sputum smears, based on clinical and radiographic manifestations. Patients receiving empiric treatment for pulmonary tuberculosis should remain in airborne infection isolation during the initiation of treatment (if a hospital stay is required) until cleared by a specialist in infectious disease or tuberculosis.
Can molecular techniques help in rapidly diagnosing tuberculosis?
Additional tests for tuberculosis that are performed on clinical specimens have been available for the past 10 years.
Nucleic acid amplification can detect tuberculosis directly in sputum, bronchoscopy specimens, or other clinical specimens. It is available at reference laboratories, large hospitals, and many state laboratories, often with 24-hour turnaround. Both commercial and in-house tests are performed. The CDC considers nucleic acid amplification to be very helpful and underutilized. An important limitation of the test is that it performs best in smear-positive specimens, with a sensitivity of 96.8%, whereas its sensitivity in smear-negative samples is only 73%.17 For this reason, nucleic acid amplification is still not widely used in US hospitals.
The CDC recommends nucleic acid amplification testing in all patients in whom the diagnosis of tuberculosis is being considered but is not yet confirmed.18 Table 1 outlines the use of nucleic acid amplification in several clinical situations. Use and interpretation of this test in suspected tuberculosis often requires consultation with clinicians who are experienced in the diagnosis of this disease.
How should an interferon-gamma-release assay or a tuberculin skin test be used in evaluating suspected tuberculosis?
Patients who have never tested positive for tuberculosis on a skin test should be tested by tuberculin skin testing or with an interferon-gamma-release assay during an evaluation for suspected pulmonary tuberculosis. The interferon-gamma-release assays available in the United States are the QuantiFERON-TB Gold In-Tube test and the T-Spot TB test. Most larger hospitals have one of the two available.
Of note: up to 25% of patients with active tuberculosis can have a negative skin test or interferon-gamma-release assay at the time of initial diagnosis, the number being higher in those who are immunosuppressed.
An interferon-gamma-release assay, which is performed on the patient’s serum, is preferred in those who have previously received the BCG vaccine, as there is no cross-reactivity between the vaccine and the antigens in the assay. In patients with active tuberculosis, the interferon-gamma-release assay does not perform any better than the skin test, so the choice of test should be determined by availability. Table 2 compares the characteristics of tuberculin skin testing and the interferon- gamma-release assay.19
In evaluating for active tuberculosis, a positive skin test or interferon-gamma-release assay can be helpful in increasing the likelihood of tuberculosis, but a negative result does not exclude active tuberculosis.
Is computed tomography necessary in patients suspected of having active pulmonary tuberculosis?
Additional imaging is often performed in patients with suspected pulmonary tuberculosis, or before the diagnosis of tuberculosis is considered. Computed tomography provides more detailed images of pulmonary infiltrates and may reveal more extensive disease than plain radiography, but the images are not diagnostic. Ultimately, sputum and sometimes tissue are required. Far too often, a sputum smear for acid-fast bacilli is the last test to be performed, after both computed tomography and bronchoscopy have been done. In addition, in order to undergo computed tomography, the patient must be removed from airborne infection isolation.
The decision to perform computed tomography must be individualized to the patient and to the clinical situation. It is certainly not a necessary test for the diagnosis of pulmonary tuberculosis.
When should the diagnosis be reported?
Tuberculosis is a reportable illness in the United States. Although each state varies in its specific requirements, if tuberculosis treatment is being initiated or tuberculosis is strongly suspected, a report should be made to the local public health authority for tuberculosis within 24 hours.
This report allows for outreach services to be offered to the patient, often including directly observed therapy in which doses of antituberculosis treatment are provided and observed to ensure completion of treatment. In addition, public health authorities bear the responsibility for contact investigation to determine if transmission of tuberculosis has occurred in the community.
- Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2012. MMWR Morb Mortal Wkly Rep 2013; 62:201–205.
- World Health Organization (WHO). Tuberculosis. WHO Global Tuberculosis Report 2013. www.who.int/tb/publications/factsheet_global.pdf. Accessed November 13, 2014.
- McKenna MT, McCray E, Onorato I. The epidemiology of tuberculosis among foreign-born persons in the United States, 1986 to 1993. N Engl J Med 1995; 332:1071–1076.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med 2000; 161:S221–S247.
- Pitchenik AE, Rubinson HA. The radiographic appearance of tuberculosis in patients with the acquired immune deficiency syndrome (AIDS) and pre-AIDS. Am Rev Respir Dis 1985; 131:393–396.
- Wisnivesky JP, Kaplan J, Henschke C, McGinn TG, Crystal RG. Evaluation of clinical parameters to predict Mycobacterium tuberculosis in inpatients. Arch Intern Med 2000; 160:2471–2476.
- Wisnivesky JP, Henschke C, Balentine J, Willner C, Deloire AM, McGinn TG. Prospective validation of a prediction model for isolating inpatients with suspected pulmonary tuberculosis. Arch Intern Med 2005; 165:453–457.
- Rakoczy KS, Cohen SH, Nguyen HH. Derivation and validation of a clinical prediction score for isolation of inpatients with suspected pulmonary tuberculosis. Infect Control Hosp Epidemiol 2008; 29:927–932.
- Jensen PA, Lambert LA, Iademarco MF, Ridzon R; Centers for Disease Control and Prevention (CDC). Guidelines for preventing the transmission of Mycobacterium tuberculosis in health-care settings, 2005. MMWR Recomm Rep 2005; 54(RR-17):1–141.
- Campos M, Quartin A, Mendes E, et al. Feasibility of shortening respiratory isolation with a single sputum nucleic acid amplification test. Am J Respir Crit Care Med 2008; 178:300–305.
- MacGregor RR. A year’s experience with tuberculosis in a private urban teaching hospital in the postsanatorium era. Am J Med 1975; 58:221–228.
- Greenbaum M, Beyt BE Jr, Murray PR. The accuracy of diagnosing pulmonary tuberculosis at a teaching hospital. Am Rev Respir Dis 1980; 121:477–481.
- Mathew P, Kuo YH, Vazirani B, Eng RH, Weinstein MP. Are three sputum acid-fast bacillus smears necessary for discontinuing tuberculosis isolation? J Clin Microbiol 2002; 40:3482–3484.
- Bryan CS, Rapp DJ, Brown CA. Discontinuation of respiratory isolation for possible tuberculosis: do two negative sputum smear results suffice? Infect Control Hosp Epidemiol 2006; 27:515–516.
- Nelson SM, Deike MA, Cartwright CP. Value of examining multiple sputum specimens in the diagnosis of pulmonary tuberculosis. J Clin Microbiol 1998; 36:467–469.
- Wilmer A, Bryce E, Grant J. The role of the third acid-fast bacillus smear in tuberculosis screening for infection control purposes: a controversial topic revisited. Can J Infect Dis Med Microbiol 2011; 22:e1–e3.
- Dinnes J, Deeks J, Kunst H, et al. A systematic review of rapid diagnostic tests for the detection of tuberculosis infection. Health Technol Assess 2007; 11:1–196.
- Centers for Disease Control and Prevention (CDC). Updated guidelines for the use of nucleic acid amplification tests in the diagnosis of tuberculosis. MMWR Morb Mortal Wkly Rep 2009; 58:7–10.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee; Centers for Disease Control and Prevention (CDC). Updated guidelines for using interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
- Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2012. MMWR Morb Mortal Wkly Rep 2013; 62:201–205.
- World Health Organization (WHO). Tuberculosis. WHO Global Tuberculosis Report 2013. www.who.int/tb/publications/factsheet_global.pdf. Accessed November 13, 2014.
- McKenna MT, McCray E, Onorato I. The epidemiology of tuberculosis among foreign-born persons in the United States, 1986 to 1993. N Engl J Med 1995; 332:1071–1076.
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med 2000; 161:S221–S247.
- Pitchenik AE, Rubinson HA. The radiographic appearance of tuberculosis in patients with the acquired immune deficiency syndrome (AIDS) and pre-AIDS. Am Rev Respir Dis 1985; 131:393–396.
- Wisnivesky JP, Kaplan J, Henschke C, McGinn TG, Crystal RG. Evaluation of clinical parameters to predict Mycobacterium tuberculosis in inpatients. Arch Intern Med 2000; 160:2471–2476.
- Wisnivesky JP, Henschke C, Balentine J, Willner C, Deloire AM, McGinn TG. Prospective validation of a prediction model for isolating inpatients with suspected pulmonary tuberculosis. Arch Intern Med 2005; 165:453–457.
- Rakoczy KS, Cohen SH, Nguyen HH. Derivation and validation of a clinical prediction score for isolation of inpatients with suspected pulmonary tuberculosis. Infect Control Hosp Epidemiol 2008; 29:927–932.
- Jensen PA, Lambert LA, Iademarco MF, Ridzon R; Centers for Disease Control and Prevention (CDC). Guidelines for preventing the transmission of Mycobacterium tuberculosis in health-care settings, 2005. MMWR Recomm Rep 2005; 54(RR-17):1–141.
- Campos M, Quartin A, Mendes E, et al. Feasibility of shortening respiratory isolation with a single sputum nucleic acid amplification test. Am J Respir Crit Care Med 2008; 178:300–305.
- MacGregor RR. A year’s experience with tuberculosis in a private urban teaching hospital in the postsanatorium era. Am J Med 1975; 58:221–228.
- Greenbaum M, Beyt BE Jr, Murray PR. The accuracy of diagnosing pulmonary tuberculosis at a teaching hospital. Am Rev Respir Dis 1980; 121:477–481.
- Mathew P, Kuo YH, Vazirani B, Eng RH, Weinstein MP. Are three sputum acid-fast bacillus smears necessary for discontinuing tuberculosis isolation? J Clin Microbiol 2002; 40:3482–3484.
- Bryan CS, Rapp DJ, Brown CA. Discontinuation of respiratory isolation for possible tuberculosis: do two negative sputum smear results suffice? Infect Control Hosp Epidemiol 2006; 27:515–516.
- Nelson SM, Deike MA, Cartwright CP. Value of examining multiple sputum specimens in the diagnosis of pulmonary tuberculosis. J Clin Microbiol 1998; 36:467–469.
- Wilmer A, Bryce E, Grant J. The role of the third acid-fast bacillus smear in tuberculosis screening for infection control purposes: a controversial topic revisited. Can J Infect Dis Med Microbiol 2011; 22:e1–e3.
- Dinnes J, Deeks J, Kunst H, et al. A systematic review of rapid diagnostic tests for the detection of tuberculosis infection. Health Technol Assess 2007; 11:1–196.
- Centers for Disease Control and Prevention (CDC). Updated guidelines for the use of nucleic acid amplification tests in the diagnosis of tuberculosis. MMWR Morb Mortal Wkly Rep 2009; 58:7–10.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee; Centers for Disease Control and Prevention (CDC). Updated guidelines for using interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
KEY POINTS
- Tuberculosis continues to be in the differential diagnosis for patients hospitalized in the United States.
- Clinical, demographic, and radiologic data obtained during the patient’s initial evaluation are helpful in determining the likelihood of tuberculosis.
- Sputum smears for acid-fast bacilli and either skin testing with purified protein derivative or blood testing with an interferon-gamma-release assay continue to be the mainstays of the initial evaluation for pulmonary tuberculosis.
- Nucleic acid amplification testing of sputum or bronchoscopy specimens can provide additional information and should be considered when pulmonary tuberculosis is part of the differential diagnosis.
Enterovirus D68: A clinically important respiratory enterovirus
In the fall of 2014, the United States experienced an outbreak of severe respiratory illness due to a virus of emerging importance, enterovirus D68 (EV-D68). Here, we review the features of this virus and related viruses, the clinical syndromes this virus causes, the epidemiology of the recent outbreak, and its diagnosis and treatment.
THE ENTEROVIRUSES: AN OVERVIEW
Originally identified in 1962 from the throat swab of a child with pneumonia, human EV-D68 has unique genetic and clinical features that blur the typical division between human enteroviruses and rhinoviruses.1–4 Enteroviruses and rhinoviruses are closely related species within the Picornaviridae family that are now classified together within the genus Enterovirus.5 Picornaviruses are small, nonenveloped, positive-stranded RNA viruses of medical significance.
Poliovirus: The first enterovirus discovered
The first human enterovirus to be discovered was poliovirus.6 Although sporadic cases of “infantile paralysis” occurred before the late 19th century, epidemic poliomyelitis abruptly appeared in Europe and the United States beginning around 1880. Before the introduction in 1955 of the inactivated poliovirus vaccine and then the oral poliovirus vaccine, polio was one of the most feared illnesses in the developed world. Outbreaks occurred primarily in cities during summer months. At its peak, epidemic polio killed or paralyzed more than half a million people a year.
One hypothesis to explain the sudden emergence of epidemic polio is that improved personal hygiene and public sanitation delayed the age at which children acquired this enteric infection.7 Infections acquired after infancy occurred in the absence of maternal antibodies that may have protected against the virus’s propensity to invade the nervous system.
Nonpolio human enteroviruses
In the decades since poliovirus was discovered, more than 100 nonpolio human enteroviruses have been recognized.8 This group includes the coxsackieviruses, echoviruses, and the newer numbered nonpolio human enteroviruses classified into four species, designated Human enterovirus A, B, C, and D. The last of these, Human enterovirus D, includes three serotypes known to cause disease in humans: EV-D68, EV-D70, and EV-D94.9
As with poliovirus infection, most people infected with a nonpolio human enterovirus have a mild illness without distinctive features.5 In temperate climates, enteroviral infections are most common during the summer and fall and are an important cause of the “summer cold.” In tropical climates, the seasonal pattern is absent, and infections may occur throughout the year.
The clinical syndromes associated with a nonpolio human enterovirus can include nonspecific febrile illness; upper respiratory tract infection; pharyngitis; herpangina; hand, foot, and mouth syndrome; various skin exanthems; bronchiolitis; asthma exacerbation; gastrointestinal manifestations such as diarrhea and vomiting (which are especially common); more serious clinical syndromes such as hepatitis, pancreatitis, and cardiomyopathy; and neurologic illness, including aseptic meningitis, encephalitis, and polio-like paralytic disease.
Outbreaks caused by nonpolio human enteroviruses occur on a regular basis, may vary by strain from year to year, and often occur within a geographic region; multiple strains may circulate simultaneously. Occasionally, as with EV-D68 in August 2014 in the United States, epidemics can emerge suddenly and spread rapidly across the world, causing disease in hundreds or thousands of people, demonstrating the breadth of illness associated with particular strains.10
ENTEROVIRUS D68: AN EMERGING PATHOGEN
EV-D68 was first isolated in the United States from four children in Berkeley, California, who had lower respiratory tract symptoms (bronchiolitis and pneumonia) in 1962. The finding was published in the medical literature in 1967.1 Since its initial identification, EV-D68 was infrequently reported as a cause of human disease, with the US Centers for Disease Control and Prevention (CDC) listing only 26 cases in the 36 years from 1970 through 2005.11
However, the past decade has seen EV-D68 emerge as a significant respiratory pathogen, with more reports of acute respiratory illness associated with it in North America, Europe, and Asia, especially in children.12–17 A seasonal pattern may exist; a longitudinal survey of samples collected from New York City detected a focal outbreak in the fall of 2009.18
The observation that recent EV-D68 outbreaks have primarily been in children suggests that most adults have immunity to it. In this regard, seroepidemiologic studies from Finland demonstrated that most adults have neutralizing antibodies from previous infection.9
The blurred line between enteroviruses and rhinoviruses
Enteroviruses and rhinoviruses are typically distinguished on the basis of the temperature at which they grow best (rhinoviruses grow better at lower temperatures, allowing them to replicate in the nose) and their sensitivity to acidity (enteroviruses are more resistant, enabling them to survive in the stomach).
The original (“Fermon”) strain of EV-D68 isolated in 1962 was first classified as an enterovirus because it was resistant to low pH.1 However, when molecular sequencing became available, EV-D68 was found to be identical to human rhinovirus 87 (HRV87), a phylogenetic outlier among the rhinoviruses that binds to cells at a receptor site distinct from that of other human rhinoviruses.19
Thereafter, further testing showed that both EV-D68 and HRV87 isolates were sensitive to acid treatment by two different methods.4 Moreover, unlike most enteroviruses, EV-D68 behaves like a rhinovirus and grows preferentially at 33°C, the temperature of the nose.2
How enterovirus D68 enters cells
Viral surface proteins, including hemagglutinin, from certain respiratory viruses have the ability to bind sugars on cells in the nose and lungs, which facilitates viral entry and replication. EV-D68 binds specifically to alpha 2-6 sialic acid, the predominant sialic acid found in the human upper respiratory tract.19,20 The absence of EV-D68 binding affinity for alpha 2-3 sialic acid, present in ciliated epithelial cells of the lower tract, suggests that alternative mechanisms may be responsible for the severe lower respiratory disease associated with this virus.
Entry of EV-D68 into cells requires additional mediators. EV-D70 belongs to the same genetic cluster as EV-D68 and enters HeLa cells using decay-accelerating factor (DAF).21 Evidence that EV-D68 also uses DAF for cell entry comes from experiments showing that monoclonal antibodies against DAF inhibit the cytopathic effects of this virus.4 Virus-receptor interactions have been more thoroughly characterized for other enteroviruses.22 In this regard, coxsackieviruses of group B use DAF as a coreceptor. Since DAF is expressed at high levels in both epithelial and endothelial cells, it may play an important role in the induction of the viremia that precedes the infection of specific tissues such as the heart or pancreas.
Different strains exist
EV-D68 strains can be divided into three genetic groups based on the sequence of the capsid-coding VP1 region, the most variable genome region of enteroviruses.23
Investigators have explored whether emergent EV-D68 strains differ in their anti-
genicity and receptor-binding properties in comparison to the Fermon strain isolated in 1962.20 Using antisera generated from various strains of EV-D68, significant differences were observed in terms of hemagglutination inhibition and neutralization titers both between emergent strains and the original Fermon strain and among the emergent strains.
Viremia in systemic disease
Like other enteroviruses, EV-D68 has the ability to infect lymphocytes.9 This may provide a mechanism by which the virus is transported during the viremic phase to secondary target organs. Indeed, EV-D68 was detected in the serum of 12 (43%) of 28 pediatric patients with pneumonia and positive nasopharyngeal swabs.24
Interestingly, whether EV-D68 was detected in the serum varied with age. Viremia was not detected in the serum of children younger than 1 year, an observation suggesting that maternal antibodies protect against viremia.
The role of viremia in systemic disease associated with EV-D68 is intriguing, especially since delayed acquisition of polio infection beyond infancy is hypothesized to have contributed to disease severity.7
ENTEROVIRUS D68 CAUSES SEVERE LOWER RESPIRATORY DISEASE
While identification of large numbers of patients with respiratory illnesses due to EV-D68 in a single season is unique to 2014, clusters of EV-D68-related respiratory illnesses have previously been recognized.25,26
As with EV-D68 outbreaks in other parts of the world, the outbreak in the US Midwest in August 2014 primarily involved children, many of whom needed to be admitted to the hospital because of severe lower respiratory symptoms.10 In the 30 children admitted to two children’s hospitals described in the initial report, difficulty breathing, hypoxemia, and wheezing were common. A minority of patients (23%) presented with fever. Of hospitalized children, 67% required admission to the intensive care unit. Two patients required intubation, including one who required extracorporeal membrane oxygenation. Six required bilevel positive airway pressure therapy.
Cleveland Clinic experience
At Cleveland Clinic during the same time, nearly 45% of patients identified with a respiratory enterovirus infection required intensive care.
For patients previously diagnosed with asthma, chronic lung disease, or wheezing, essential supportive care measures included continuing the inhaled steroids the patients were already taking, early use of short-acting beta agonists, and, in those with previously diagnosed asthma, consideration of a systemic steroid. Many of our patients with previously diagnosed asthma had an unusually long prodrome of an increase in mild symptoms, followed by a rapid and severe decline in respiratory status.
At the later phase, supportive care measures that were needed included maintenance of hydration and monitoring of oxyhemoglobin saturation with use of supplemental oxygen as necessary, as well as close observation of clinical indicators of respiratory distress, such as development of crackles, asymmetric air exchange, and progression in wheezing or in use of accessory muscles. In an attempt to avoid invasive ventilatory support in patients with asthma or other comorbid conditions, some patients were treated with aerosolized epinephrine, ipratropium, heliox, and noninvasive positive pressure ventilatory support.
NEUROLOGIC DISEASE: ACUTE FLACCID PARALYSIS
Although EV-D68 causes primarily respiratory illness, systemic disease occurs, especially neurologic involvement.
Before the recent outbreak of EV-D68, two cases of neurologic involvement from EV-D68 were reported. The first of these, mentioned in a 2006 enterovirus surveillance report issued by the CDC, was in a young adult with acute flaccid paralysis and EV-D68 isolated from the cerebral spinal fluid.11 In the second case, from 2010, a 5-year-old boy developed fatal meningomyeloencephalitis. The child had presented with pneumonia and acute flaccid paralysis. EV-D68 was identified in his cerebral spinal fluid by polymerase chain reaction (PCR), and histopathologic study of the meninges, cerebellum, midbrain, pons, medulla, and cervical cord demonstrated extensive T-cell lymphocytic meningomyelitis and encephalitis, characterized by prominent neuronophagia in motor nuclei.27
At the same time as the recent outbreak of EV-D68 respiratory disease, neurologists throughout the United States observed an increase in the number of children with polio-like acute flaccid paralysis. On September 26, 2014, the CDC issued an alert describing acute neurologic illness with focal limb weakness of unknown etiology in children, possibly associated with EV-D68.28 The report described nine cases of an acute neurologic illness in children ages 1 through 18 years (median age, 10) hospitalized in Colorado between August 9 and September 17, 2014. Common clinical features included acute focal limb weakness and paralysis and acute cranial nerve dysfunction, with no altered mental status or seizures. Pain before the onset of weakness was also identified as a common complaint.
Specific findings on magnetic resonance imaging of the spinal cord consisted of nonenhancing lesions largely restricted to the gray matter and in most cases spanning more than one level of the spinal cord. In patients with cranial nerve dysfunction, correlating nonenhancing brainstem lesions were observed.
Most children experienced a febrile respiratory illness in the 2 weeks preceding the onset of neurologic symptoms. In most cases, cerebrospinal fluid analyses demonstrated mild or moderate pleocytosis consistent with an inflammatory or infectious process, with normal to mildly elevated protein and normal glucose levels. In six of the eight patients tested, nasopharyngeal specimens were positive for rhinovirus-enterovirus. Of the six positive specimens, at least four were typed as EV-D68.
The CDC also reported a second cluster of cases of acute flaccid paralysis with anterior myelitis on magnetic resonance imaging, in 23 children (mean age 10 years) in California from June 2012 to June 2014.29 No common cause was identified, although clinical and laboratory findings supported a viral etiology. Two patients tested positive for EV-D68 from upper respiratory tract specimens. Common features among the clinical presentations included an upper respiratory or gastrointestinal prodrome less than 10 days before the onset of the paralysis (83%), cerebrospinal fluid pleocytosis (83%), and absence of sensory deficits (78%). Ten patients (43%) also had concomitant mental status changes, and eight (34%) had cranial nerve abnormalities.
Details regarding outcomes from these paralytic illnesses remain unclear, although it would appear that time to recovery has been prolonged in many cases, and the degree of recovery remains uncertain.
TREATMENT IS SUPPORTIVE
The treatment of EV-D68 infection is mainly supportive, as no specific antiviral therapy is currently available for any of the enteroviruses. Critically ill patients require organ-specific supportive care.
Potential targets for novel antienteroviral therapies exist; some of the experimental compounds were initially evaluated for their activity against polioviruses or rhinoviruses.30
TESTING MAY HAVE A ROLE
In general, testing does not play a role in the management of patients with mild disease, but it may be indicated for epidemiologic purposes or for specific diagnosis in critically ill patients. Molecular techniques are commonly used to detect respiratory viruses from clinical samples, either as discrete tests or as a multiplex viral panel.
Since patients with EV-D68 infection typically have respiratory symptoms, the virus is generally tested for in nasal wash samples. However, depending on the clinical presentation, it may be appropriate to attempt to detect the virus from other sites using either PCR or culture.
Many clinical laboratories use real-time PCR assays designed to detect both rhinoviruses and enteroviruses, but these tests do not distinguish between the species. While more specific real-time PCR assays are available that generally distinguish rhinoviruses from enteroviruses,31 during the recent outbreak our laboratory observed that confirmed EV-D68 samples cross-reacted with rhinovirus. Most clinical laboratories do not routinely perform viral sequence analysis to specifically identify EV-D68, but this test may be obtained through state health departments and the CDC on a case-by-case basis.
Recently, the CDC’s enterovirus laboratory announced the development of a real-time PCR assay specifically for EV-D68, which may make specific detection more readily available.
INFECTION PREVENTION
The routes by which EV-D68 is transmitted are not fully understood. In contrast to most enteroviruses, which are spread in a fecal-oral manner, it is possible that EV-D68 is also spread through close respiratory or mucous contact.
For this reason, interim infection prevention guidelines issued by the CDC recommend that hospitals use droplet precautions along with contact or standard precautions, depending on the scenario.32 In our children’s hospital, we use droplet and contact precautions for hospitalized patients.
- Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol 1967; 85:297–310.
- Oberste MS, Maher K, Schnurr D, et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J Gen Virol 2004; 85:2577–2584.
- Ishiko H, Miura R, Shimada Y, et al. Human rhinovirus 87 identified as human enterovirus 68 by VP4-based molecular diagnosis. Intervirology 2002; 45:136–141.
- Blomqvist S, Savolainen C, Raman L, Roivainen M, Hovi T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J Clin Microbiol 2002; 40:4218–4223.
- Cherry JD, Krogstad P. Enterovirus, parechoviruses, and Saffold viruses. In: Cherry JD, Harrison GJ, Kaplan SL, Steinbach WJ, Hoetez PJ, editors. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases. Vol 2. Seventh ed. Philadelphia: Elsevier Saunders; 2014:2051–2109.
- Rotbart HA. Enteroviral infections of the central nervous system. Clin Infect Dis 1995; 20:971–981.
- Nathanson N, Kew OM. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 2010; 172:1213–1229.
- Santti J, Vainionpää R, Hyypiä T. Molecular detection and typing of human picornaviruses. Virus Res 1999; 62:177–183.
- Smura T, Ylipaasto P, Klemola P, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol 2010; 82:1940–1949.
- Midgley CM, Jackson MA, Selvarangan R, et al. Severe respiratory illness associated with enterovirus d68 - Missouri and Illinois, 2014. MMWR Morb Mortal Wkly Rep 2014; 63:798–799.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill Summ 2006; 55:1–20.
- Tokarz R, Firth C, Madhi SA, et al. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol 2012; 93:1952–1958.
- Clusters of acute respiratory illness associated with human enterovirus 68—Asia, Europe, and United States, 2008–2010. MMWR Morb Mortal Wkly Rep 2011; 60:1301–1304.
- Lauinger IL, Bible JM, Halligan EP, Aarons EJ, MacMahon E, Tong CY. Lineages, sub-lineages and variants of enterovirus 68 in recent outbreaks. PLoS One 2012; 7:e36005.
- Lu QB, Wo Y, Wang HY, et al. Detection of enterovirus 68 as one of the commonest types of enterovirus found in patients with acute respiratory tract infection in China. J Med Microbiol 2014; 63:408–414.
- Meijer A, van der Sanden S, Snijders BE, et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in The Netherlands in 2010. Virology 2012; 423:49–57.
- Rahamat-Langendoen J, Riezebos-Brilman A, Borger R, et al. Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J Clin Virol 2011; 52:103–106.
- Tokarz R, Kapoor V, Wu W, et al. Longitudinal molecular microbial analysis of influenza-like illness in New York City, May 2009 through May 2010. Virol J 2011; 8:288.
- Uncapher CR, DeWitt CM, Colonno RJ. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991; 180:814–817.
- Imamura T, Okamoto M, Nakakita S, et al. Antigenic and receptor binding properties of enterovirus 68. J Virol 2014; 88:2374–2384.
- Karnauchow TM, Tolson DL, Harrison BA, Altman E, Lublin DM, Dimock K. The HeLa cell receptor for enterovirus 70 is decay-accelerating factor (CD55). J Virol 1996; 70:5143–5152.
- Selinka HC, Wolde A, Sauter M, Kandolf R, Klingel K. Virus-receptor interactions of coxsackie B viruses and their putative influence on cardiotropism. Med Microbiol Immunol 2004; 193:127–131.
- Piralla A, Girello A, Grignani M, et al. Phylogenetic characterization of enterovirus 68 strains in patients with respiratory syndromes in Italy. J Med Virol 2014; 86:1590–1593.
- Imamura T, Suzuki A, Lupisan S, et al. Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza Other Respir Viruses 2014; 8:21–24.
- Imamura T, Fuji N, Suzuki A, et al. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis 2011; 17:1430–1435.
- Kaida A, Kubo H, Sekiguchi J, et al. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg Infect Dis 2011; 17:1494–1497.
- Kreuter JD, Barnes A, McCarthy JE, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med 2011; 135:793–796.
- Pastula DM, Aliabadi N, Haynes AK, et al. Acute neurologic illness of unknown etiology in children—Colorado, August-September 2014. MMWR Morb Mortal Wkly Rep 2014; 63:901–902.
- Ayscue P, Haren KV, Sheriff H, et al. Acute flaccid paralysis with anterior myelitis—California, June 2012–June 2014. MMWR Morb Mortal Wkly Rep 2014; 63:903–906.
- Abzug MJ. The enteroviruses: problems in need of treatments. J Infect 2014; 68(suppl 1):S108–S114.
- Pierce VM, Hodinka RL. Comparison of the GenMark Diagnostics eSensor respiratory viral panel to real-time PCR for detection of respiratory viruses in children. J Clin Microbiol 2012; 50:3458–3465.
- Non-polio enterovirus infection: enterovirus D68 (EV-D68)-CDC. 2014; www.cdc.gov/non-polio-enterovirus/about/ev-d68.html. Accessed November 21, 2014.
In the fall of 2014, the United States experienced an outbreak of severe respiratory illness due to a virus of emerging importance, enterovirus D68 (EV-D68). Here, we review the features of this virus and related viruses, the clinical syndromes this virus causes, the epidemiology of the recent outbreak, and its diagnosis and treatment.
THE ENTEROVIRUSES: AN OVERVIEW
Originally identified in 1962 from the throat swab of a child with pneumonia, human EV-D68 has unique genetic and clinical features that blur the typical division between human enteroviruses and rhinoviruses.1–4 Enteroviruses and rhinoviruses are closely related species within the Picornaviridae family that are now classified together within the genus Enterovirus.5 Picornaviruses are small, nonenveloped, positive-stranded RNA viruses of medical significance.
Poliovirus: The first enterovirus discovered
The first human enterovirus to be discovered was poliovirus.6 Although sporadic cases of “infantile paralysis” occurred before the late 19th century, epidemic poliomyelitis abruptly appeared in Europe and the United States beginning around 1880. Before the introduction in 1955 of the inactivated poliovirus vaccine and then the oral poliovirus vaccine, polio was one of the most feared illnesses in the developed world. Outbreaks occurred primarily in cities during summer months. At its peak, epidemic polio killed or paralyzed more than half a million people a year.
One hypothesis to explain the sudden emergence of epidemic polio is that improved personal hygiene and public sanitation delayed the age at which children acquired this enteric infection.7 Infections acquired after infancy occurred in the absence of maternal antibodies that may have protected against the virus’s propensity to invade the nervous system.
Nonpolio human enteroviruses
In the decades since poliovirus was discovered, more than 100 nonpolio human enteroviruses have been recognized.8 This group includes the coxsackieviruses, echoviruses, and the newer numbered nonpolio human enteroviruses classified into four species, designated Human enterovirus A, B, C, and D. The last of these, Human enterovirus D, includes three serotypes known to cause disease in humans: EV-D68, EV-D70, and EV-D94.9
As with poliovirus infection, most people infected with a nonpolio human enterovirus have a mild illness without distinctive features.5 In temperate climates, enteroviral infections are most common during the summer and fall and are an important cause of the “summer cold.” In tropical climates, the seasonal pattern is absent, and infections may occur throughout the year.
The clinical syndromes associated with a nonpolio human enterovirus can include nonspecific febrile illness; upper respiratory tract infection; pharyngitis; herpangina; hand, foot, and mouth syndrome; various skin exanthems; bronchiolitis; asthma exacerbation; gastrointestinal manifestations such as diarrhea and vomiting (which are especially common); more serious clinical syndromes such as hepatitis, pancreatitis, and cardiomyopathy; and neurologic illness, including aseptic meningitis, encephalitis, and polio-like paralytic disease.
Outbreaks caused by nonpolio human enteroviruses occur on a regular basis, may vary by strain from year to year, and often occur within a geographic region; multiple strains may circulate simultaneously. Occasionally, as with EV-D68 in August 2014 in the United States, epidemics can emerge suddenly and spread rapidly across the world, causing disease in hundreds or thousands of people, demonstrating the breadth of illness associated with particular strains.10
ENTEROVIRUS D68: AN EMERGING PATHOGEN
EV-D68 was first isolated in the United States from four children in Berkeley, California, who had lower respiratory tract symptoms (bronchiolitis and pneumonia) in 1962. The finding was published in the medical literature in 1967.1 Since its initial identification, EV-D68 was infrequently reported as a cause of human disease, with the US Centers for Disease Control and Prevention (CDC) listing only 26 cases in the 36 years from 1970 through 2005.11
However, the past decade has seen EV-D68 emerge as a significant respiratory pathogen, with more reports of acute respiratory illness associated with it in North America, Europe, and Asia, especially in children.12–17 A seasonal pattern may exist; a longitudinal survey of samples collected from New York City detected a focal outbreak in the fall of 2009.18
The observation that recent EV-D68 outbreaks have primarily been in children suggests that most adults have immunity to it. In this regard, seroepidemiologic studies from Finland demonstrated that most adults have neutralizing antibodies from previous infection.9
The blurred line between enteroviruses and rhinoviruses
Enteroviruses and rhinoviruses are typically distinguished on the basis of the temperature at which they grow best (rhinoviruses grow better at lower temperatures, allowing them to replicate in the nose) and their sensitivity to acidity (enteroviruses are more resistant, enabling them to survive in the stomach).
The original (“Fermon”) strain of EV-D68 isolated in 1962 was first classified as an enterovirus because it was resistant to low pH.1 However, when molecular sequencing became available, EV-D68 was found to be identical to human rhinovirus 87 (HRV87), a phylogenetic outlier among the rhinoviruses that binds to cells at a receptor site distinct from that of other human rhinoviruses.19
Thereafter, further testing showed that both EV-D68 and HRV87 isolates were sensitive to acid treatment by two different methods.4 Moreover, unlike most enteroviruses, EV-D68 behaves like a rhinovirus and grows preferentially at 33°C, the temperature of the nose.2
How enterovirus D68 enters cells
Viral surface proteins, including hemagglutinin, from certain respiratory viruses have the ability to bind sugars on cells in the nose and lungs, which facilitates viral entry and replication. EV-D68 binds specifically to alpha 2-6 sialic acid, the predominant sialic acid found in the human upper respiratory tract.19,20 The absence of EV-D68 binding affinity for alpha 2-3 sialic acid, present in ciliated epithelial cells of the lower tract, suggests that alternative mechanisms may be responsible for the severe lower respiratory disease associated with this virus.
Entry of EV-D68 into cells requires additional mediators. EV-D70 belongs to the same genetic cluster as EV-D68 and enters HeLa cells using decay-accelerating factor (DAF).21 Evidence that EV-D68 also uses DAF for cell entry comes from experiments showing that monoclonal antibodies against DAF inhibit the cytopathic effects of this virus.4 Virus-receptor interactions have been more thoroughly characterized for other enteroviruses.22 In this regard, coxsackieviruses of group B use DAF as a coreceptor. Since DAF is expressed at high levels in both epithelial and endothelial cells, it may play an important role in the induction of the viremia that precedes the infection of specific tissues such as the heart or pancreas.
Different strains exist
EV-D68 strains can be divided into three genetic groups based on the sequence of the capsid-coding VP1 region, the most variable genome region of enteroviruses.23
Investigators have explored whether emergent EV-D68 strains differ in their anti-
genicity and receptor-binding properties in comparison to the Fermon strain isolated in 1962.20 Using antisera generated from various strains of EV-D68, significant differences were observed in terms of hemagglutination inhibition and neutralization titers both between emergent strains and the original Fermon strain and among the emergent strains.
Viremia in systemic disease
Like other enteroviruses, EV-D68 has the ability to infect lymphocytes.9 This may provide a mechanism by which the virus is transported during the viremic phase to secondary target organs. Indeed, EV-D68 was detected in the serum of 12 (43%) of 28 pediatric patients with pneumonia and positive nasopharyngeal swabs.24
Interestingly, whether EV-D68 was detected in the serum varied with age. Viremia was not detected in the serum of children younger than 1 year, an observation suggesting that maternal antibodies protect against viremia.
The role of viremia in systemic disease associated with EV-D68 is intriguing, especially since delayed acquisition of polio infection beyond infancy is hypothesized to have contributed to disease severity.7
ENTEROVIRUS D68 CAUSES SEVERE LOWER RESPIRATORY DISEASE
While identification of large numbers of patients with respiratory illnesses due to EV-D68 in a single season is unique to 2014, clusters of EV-D68-related respiratory illnesses have previously been recognized.25,26
As with EV-D68 outbreaks in other parts of the world, the outbreak in the US Midwest in August 2014 primarily involved children, many of whom needed to be admitted to the hospital because of severe lower respiratory symptoms.10 In the 30 children admitted to two children’s hospitals described in the initial report, difficulty breathing, hypoxemia, and wheezing were common. A minority of patients (23%) presented with fever. Of hospitalized children, 67% required admission to the intensive care unit. Two patients required intubation, including one who required extracorporeal membrane oxygenation. Six required bilevel positive airway pressure therapy.
Cleveland Clinic experience
At Cleveland Clinic during the same time, nearly 45% of patients identified with a respiratory enterovirus infection required intensive care.
For patients previously diagnosed with asthma, chronic lung disease, or wheezing, essential supportive care measures included continuing the inhaled steroids the patients were already taking, early use of short-acting beta agonists, and, in those with previously diagnosed asthma, consideration of a systemic steroid. Many of our patients with previously diagnosed asthma had an unusually long prodrome of an increase in mild symptoms, followed by a rapid and severe decline in respiratory status.
At the later phase, supportive care measures that were needed included maintenance of hydration and monitoring of oxyhemoglobin saturation with use of supplemental oxygen as necessary, as well as close observation of clinical indicators of respiratory distress, such as development of crackles, asymmetric air exchange, and progression in wheezing or in use of accessory muscles. In an attempt to avoid invasive ventilatory support in patients with asthma or other comorbid conditions, some patients were treated with aerosolized epinephrine, ipratropium, heliox, and noninvasive positive pressure ventilatory support.
NEUROLOGIC DISEASE: ACUTE FLACCID PARALYSIS
Although EV-D68 causes primarily respiratory illness, systemic disease occurs, especially neurologic involvement.
Before the recent outbreak of EV-D68, two cases of neurologic involvement from EV-D68 were reported. The first of these, mentioned in a 2006 enterovirus surveillance report issued by the CDC, was in a young adult with acute flaccid paralysis and EV-D68 isolated from the cerebral spinal fluid.11 In the second case, from 2010, a 5-year-old boy developed fatal meningomyeloencephalitis. The child had presented with pneumonia and acute flaccid paralysis. EV-D68 was identified in his cerebral spinal fluid by polymerase chain reaction (PCR), and histopathologic study of the meninges, cerebellum, midbrain, pons, medulla, and cervical cord demonstrated extensive T-cell lymphocytic meningomyelitis and encephalitis, characterized by prominent neuronophagia in motor nuclei.27
At the same time as the recent outbreak of EV-D68 respiratory disease, neurologists throughout the United States observed an increase in the number of children with polio-like acute flaccid paralysis. On September 26, 2014, the CDC issued an alert describing acute neurologic illness with focal limb weakness of unknown etiology in children, possibly associated with EV-D68.28 The report described nine cases of an acute neurologic illness in children ages 1 through 18 years (median age, 10) hospitalized in Colorado between August 9 and September 17, 2014. Common clinical features included acute focal limb weakness and paralysis and acute cranial nerve dysfunction, with no altered mental status or seizures. Pain before the onset of weakness was also identified as a common complaint.
Specific findings on magnetic resonance imaging of the spinal cord consisted of nonenhancing lesions largely restricted to the gray matter and in most cases spanning more than one level of the spinal cord. In patients with cranial nerve dysfunction, correlating nonenhancing brainstem lesions were observed.
Most children experienced a febrile respiratory illness in the 2 weeks preceding the onset of neurologic symptoms. In most cases, cerebrospinal fluid analyses demonstrated mild or moderate pleocytosis consistent with an inflammatory or infectious process, with normal to mildly elevated protein and normal glucose levels. In six of the eight patients tested, nasopharyngeal specimens were positive for rhinovirus-enterovirus. Of the six positive specimens, at least four were typed as EV-D68.
The CDC also reported a second cluster of cases of acute flaccid paralysis with anterior myelitis on magnetic resonance imaging, in 23 children (mean age 10 years) in California from June 2012 to June 2014.29 No common cause was identified, although clinical and laboratory findings supported a viral etiology. Two patients tested positive for EV-D68 from upper respiratory tract specimens. Common features among the clinical presentations included an upper respiratory or gastrointestinal prodrome less than 10 days before the onset of the paralysis (83%), cerebrospinal fluid pleocytosis (83%), and absence of sensory deficits (78%). Ten patients (43%) also had concomitant mental status changes, and eight (34%) had cranial nerve abnormalities.
Details regarding outcomes from these paralytic illnesses remain unclear, although it would appear that time to recovery has been prolonged in many cases, and the degree of recovery remains uncertain.
TREATMENT IS SUPPORTIVE
The treatment of EV-D68 infection is mainly supportive, as no specific antiviral therapy is currently available for any of the enteroviruses. Critically ill patients require organ-specific supportive care.
Potential targets for novel antienteroviral therapies exist; some of the experimental compounds were initially evaluated for their activity against polioviruses or rhinoviruses.30
TESTING MAY HAVE A ROLE
In general, testing does not play a role in the management of patients with mild disease, but it may be indicated for epidemiologic purposes or for specific diagnosis in critically ill patients. Molecular techniques are commonly used to detect respiratory viruses from clinical samples, either as discrete tests or as a multiplex viral panel.
Since patients with EV-D68 infection typically have respiratory symptoms, the virus is generally tested for in nasal wash samples. However, depending on the clinical presentation, it may be appropriate to attempt to detect the virus from other sites using either PCR or culture.
Many clinical laboratories use real-time PCR assays designed to detect both rhinoviruses and enteroviruses, but these tests do not distinguish between the species. While more specific real-time PCR assays are available that generally distinguish rhinoviruses from enteroviruses,31 during the recent outbreak our laboratory observed that confirmed EV-D68 samples cross-reacted with rhinovirus. Most clinical laboratories do not routinely perform viral sequence analysis to specifically identify EV-D68, but this test may be obtained through state health departments and the CDC on a case-by-case basis.
Recently, the CDC’s enterovirus laboratory announced the development of a real-time PCR assay specifically for EV-D68, which may make specific detection more readily available.
INFECTION PREVENTION
The routes by which EV-D68 is transmitted are not fully understood. In contrast to most enteroviruses, which are spread in a fecal-oral manner, it is possible that EV-D68 is also spread through close respiratory or mucous contact.
For this reason, interim infection prevention guidelines issued by the CDC recommend that hospitals use droplet precautions along with contact or standard precautions, depending on the scenario.32 In our children’s hospital, we use droplet and contact precautions for hospitalized patients.
In the fall of 2014, the United States experienced an outbreak of severe respiratory illness due to a virus of emerging importance, enterovirus D68 (EV-D68). Here, we review the features of this virus and related viruses, the clinical syndromes this virus causes, the epidemiology of the recent outbreak, and its diagnosis and treatment.
THE ENTEROVIRUSES: AN OVERVIEW
Originally identified in 1962 from the throat swab of a child with pneumonia, human EV-D68 has unique genetic and clinical features that blur the typical division between human enteroviruses and rhinoviruses.1–4 Enteroviruses and rhinoviruses are closely related species within the Picornaviridae family that are now classified together within the genus Enterovirus.5 Picornaviruses are small, nonenveloped, positive-stranded RNA viruses of medical significance.
Poliovirus: The first enterovirus discovered
The first human enterovirus to be discovered was poliovirus.6 Although sporadic cases of “infantile paralysis” occurred before the late 19th century, epidemic poliomyelitis abruptly appeared in Europe and the United States beginning around 1880. Before the introduction in 1955 of the inactivated poliovirus vaccine and then the oral poliovirus vaccine, polio was one of the most feared illnesses in the developed world. Outbreaks occurred primarily in cities during summer months. At its peak, epidemic polio killed or paralyzed more than half a million people a year.
One hypothesis to explain the sudden emergence of epidemic polio is that improved personal hygiene and public sanitation delayed the age at which children acquired this enteric infection.7 Infections acquired after infancy occurred in the absence of maternal antibodies that may have protected against the virus’s propensity to invade the nervous system.
Nonpolio human enteroviruses
In the decades since poliovirus was discovered, more than 100 nonpolio human enteroviruses have been recognized.8 This group includes the coxsackieviruses, echoviruses, and the newer numbered nonpolio human enteroviruses classified into four species, designated Human enterovirus A, B, C, and D. The last of these, Human enterovirus D, includes three serotypes known to cause disease in humans: EV-D68, EV-D70, and EV-D94.9
As with poliovirus infection, most people infected with a nonpolio human enterovirus have a mild illness without distinctive features.5 In temperate climates, enteroviral infections are most common during the summer and fall and are an important cause of the “summer cold.” In tropical climates, the seasonal pattern is absent, and infections may occur throughout the year.
The clinical syndromes associated with a nonpolio human enterovirus can include nonspecific febrile illness; upper respiratory tract infection; pharyngitis; herpangina; hand, foot, and mouth syndrome; various skin exanthems; bronchiolitis; asthma exacerbation; gastrointestinal manifestations such as diarrhea and vomiting (which are especially common); more serious clinical syndromes such as hepatitis, pancreatitis, and cardiomyopathy; and neurologic illness, including aseptic meningitis, encephalitis, and polio-like paralytic disease.
Outbreaks caused by nonpolio human enteroviruses occur on a regular basis, may vary by strain from year to year, and often occur within a geographic region; multiple strains may circulate simultaneously. Occasionally, as with EV-D68 in August 2014 in the United States, epidemics can emerge suddenly and spread rapidly across the world, causing disease in hundreds or thousands of people, demonstrating the breadth of illness associated with particular strains.10
ENTEROVIRUS D68: AN EMERGING PATHOGEN
EV-D68 was first isolated in the United States from four children in Berkeley, California, who had lower respiratory tract symptoms (bronchiolitis and pneumonia) in 1962. The finding was published in the medical literature in 1967.1 Since its initial identification, EV-D68 was infrequently reported as a cause of human disease, with the US Centers for Disease Control and Prevention (CDC) listing only 26 cases in the 36 years from 1970 through 2005.11
However, the past decade has seen EV-D68 emerge as a significant respiratory pathogen, with more reports of acute respiratory illness associated with it in North America, Europe, and Asia, especially in children.12–17 A seasonal pattern may exist; a longitudinal survey of samples collected from New York City detected a focal outbreak in the fall of 2009.18
The observation that recent EV-D68 outbreaks have primarily been in children suggests that most adults have immunity to it. In this regard, seroepidemiologic studies from Finland demonstrated that most adults have neutralizing antibodies from previous infection.9
The blurred line between enteroviruses and rhinoviruses
Enteroviruses and rhinoviruses are typically distinguished on the basis of the temperature at which they grow best (rhinoviruses grow better at lower temperatures, allowing them to replicate in the nose) and their sensitivity to acidity (enteroviruses are more resistant, enabling them to survive in the stomach).
The original (“Fermon”) strain of EV-D68 isolated in 1962 was first classified as an enterovirus because it was resistant to low pH.1 However, when molecular sequencing became available, EV-D68 was found to be identical to human rhinovirus 87 (HRV87), a phylogenetic outlier among the rhinoviruses that binds to cells at a receptor site distinct from that of other human rhinoviruses.19
Thereafter, further testing showed that both EV-D68 and HRV87 isolates were sensitive to acid treatment by two different methods.4 Moreover, unlike most enteroviruses, EV-D68 behaves like a rhinovirus and grows preferentially at 33°C, the temperature of the nose.2
How enterovirus D68 enters cells
Viral surface proteins, including hemagglutinin, from certain respiratory viruses have the ability to bind sugars on cells in the nose and lungs, which facilitates viral entry and replication. EV-D68 binds specifically to alpha 2-6 sialic acid, the predominant sialic acid found in the human upper respiratory tract.19,20 The absence of EV-D68 binding affinity for alpha 2-3 sialic acid, present in ciliated epithelial cells of the lower tract, suggests that alternative mechanisms may be responsible for the severe lower respiratory disease associated with this virus.
Entry of EV-D68 into cells requires additional mediators. EV-D70 belongs to the same genetic cluster as EV-D68 and enters HeLa cells using decay-accelerating factor (DAF).21 Evidence that EV-D68 also uses DAF for cell entry comes from experiments showing that monoclonal antibodies against DAF inhibit the cytopathic effects of this virus.4 Virus-receptor interactions have been more thoroughly characterized for other enteroviruses.22 In this regard, coxsackieviruses of group B use DAF as a coreceptor. Since DAF is expressed at high levels in both epithelial and endothelial cells, it may play an important role in the induction of the viremia that precedes the infection of specific tissues such as the heart or pancreas.
Different strains exist
EV-D68 strains can be divided into three genetic groups based on the sequence of the capsid-coding VP1 region, the most variable genome region of enteroviruses.23
Investigators have explored whether emergent EV-D68 strains differ in their anti-
genicity and receptor-binding properties in comparison to the Fermon strain isolated in 1962.20 Using antisera generated from various strains of EV-D68, significant differences were observed in terms of hemagglutination inhibition and neutralization titers both between emergent strains and the original Fermon strain and among the emergent strains.
Viremia in systemic disease
Like other enteroviruses, EV-D68 has the ability to infect lymphocytes.9 This may provide a mechanism by which the virus is transported during the viremic phase to secondary target organs. Indeed, EV-D68 was detected in the serum of 12 (43%) of 28 pediatric patients with pneumonia and positive nasopharyngeal swabs.24
Interestingly, whether EV-D68 was detected in the serum varied with age. Viremia was not detected in the serum of children younger than 1 year, an observation suggesting that maternal antibodies protect against viremia.
The role of viremia in systemic disease associated with EV-D68 is intriguing, especially since delayed acquisition of polio infection beyond infancy is hypothesized to have contributed to disease severity.7
ENTEROVIRUS D68 CAUSES SEVERE LOWER RESPIRATORY DISEASE
While identification of large numbers of patients with respiratory illnesses due to EV-D68 in a single season is unique to 2014, clusters of EV-D68-related respiratory illnesses have previously been recognized.25,26
As with EV-D68 outbreaks in other parts of the world, the outbreak in the US Midwest in August 2014 primarily involved children, many of whom needed to be admitted to the hospital because of severe lower respiratory symptoms.10 In the 30 children admitted to two children’s hospitals described in the initial report, difficulty breathing, hypoxemia, and wheezing were common. A minority of patients (23%) presented with fever. Of hospitalized children, 67% required admission to the intensive care unit. Two patients required intubation, including one who required extracorporeal membrane oxygenation. Six required bilevel positive airway pressure therapy.
Cleveland Clinic experience
At Cleveland Clinic during the same time, nearly 45% of patients identified with a respiratory enterovirus infection required intensive care.
For patients previously diagnosed with asthma, chronic lung disease, or wheezing, essential supportive care measures included continuing the inhaled steroids the patients were already taking, early use of short-acting beta agonists, and, in those with previously diagnosed asthma, consideration of a systemic steroid. Many of our patients with previously diagnosed asthma had an unusually long prodrome of an increase in mild symptoms, followed by a rapid and severe decline in respiratory status.
At the later phase, supportive care measures that were needed included maintenance of hydration and monitoring of oxyhemoglobin saturation with use of supplemental oxygen as necessary, as well as close observation of clinical indicators of respiratory distress, such as development of crackles, asymmetric air exchange, and progression in wheezing or in use of accessory muscles. In an attempt to avoid invasive ventilatory support in patients with asthma or other comorbid conditions, some patients were treated with aerosolized epinephrine, ipratropium, heliox, and noninvasive positive pressure ventilatory support.
NEUROLOGIC DISEASE: ACUTE FLACCID PARALYSIS
Although EV-D68 causes primarily respiratory illness, systemic disease occurs, especially neurologic involvement.
Before the recent outbreak of EV-D68, two cases of neurologic involvement from EV-D68 were reported. The first of these, mentioned in a 2006 enterovirus surveillance report issued by the CDC, was in a young adult with acute flaccid paralysis and EV-D68 isolated from the cerebral spinal fluid.11 In the second case, from 2010, a 5-year-old boy developed fatal meningomyeloencephalitis. The child had presented with pneumonia and acute flaccid paralysis. EV-D68 was identified in his cerebral spinal fluid by polymerase chain reaction (PCR), and histopathologic study of the meninges, cerebellum, midbrain, pons, medulla, and cervical cord demonstrated extensive T-cell lymphocytic meningomyelitis and encephalitis, characterized by prominent neuronophagia in motor nuclei.27
At the same time as the recent outbreak of EV-D68 respiratory disease, neurologists throughout the United States observed an increase in the number of children with polio-like acute flaccid paralysis. On September 26, 2014, the CDC issued an alert describing acute neurologic illness with focal limb weakness of unknown etiology in children, possibly associated with EV-D68.28 The report described nine cases of an acute neurologic illness in children ages 1 through 18 years (median age, 10) hospitalized in Colorado between August 9 and September 17, 2014. Common clinical features included acute focal limb weakness and paralysis and acute cranial nerve dysfunction, with no altered mental status or seizures. Pain before the onset of weakness was also identified as a common complaint.
Specific findings on magnetic resonance imaging of the spinal cord consisted of nonenhancing lesions largely restricted to the gray matter and in most cases spanning more than one level of the spinal cord. In patients with cranial nerve dysfunction, correlating nonenhancing brainstem lesions were observed.
Most children experienced a febrile respiratory illness in the 2 weeks preceding the onset of neurologic symptoms. In most cases, cerebrospinal fluid analyses demonstrated mild or moderate pleocytosis consistent with an inflammatory or infectious process, with normal to mildly elevated protein and normal glucose levels. In six of the eight patients tested, nasopharyngeal specimens were positive for rhinovirus-enterovirus. Of the six positive specimens, at least four were typed as EV-D68.
The CDC also reported a second cluster of cases of acute flaccid paralysis with anterior myelitis on magnetic resonance imaging, in 23 children (mean age 10 years) in California from June 2012 to June 2014.29 No common cause was identified, although clinical and laboratory findings supported a viral etiology. Two patients tested positive for EV-D68 from upper respiratory tract specimens. Common features among the clinical presentations included an upper respiratory or gastrointestinal prodrome less than 10 days before the onset of the paralysis (83%), cerebrospinal fluid pleocytosis (83%), and absence of sensory deficits (78%). Ten patients (43%) also had concomitant mental status changes, and eight (34%) had cranial nerve abnormalities.
Details regarding outcomes from these paralytic illnesses remain unclear, although it would appear that time to recovery has been prolonged in many cases, and the degree of recovery remains uncertain.
TREATMENT IS SUPPORTIVE
The treatment of EV-D68 infection is mainly supportive, as no specific antiviral therapy is currently available for any of the enteroviruses. Critically ill patients require organ-specific supportive care.
Potential targets for novel antienteroviral therapies exist; some of the experimental compounds were initially evaluated for their activity against polioviruses or rhinoviruses.30
TESTING MAY HAVE A ROLE
In general, testing does not play a role in the management of patients with mild disease, but it may be indicated for epidemiologic purposes or for specific diagnosis in critically ill patients. Molecular techniques are commonly used to detect respiratory viruses from clinical samples, either as discrete tests or as a multiplex viral panel.
Since patients with EV-D68 infection typically have respiratory symptoms, the virus is generally tested for in nasal wash samples. However, depending on the clinical presentation, it may be appropriate to attempt to detect the virus from other sites using either PCR or culture.
Many clinical laboratories use real-time PCR assays designed to detect both rhinoviruses and enteroviruses, but these tests do not distinguish between the species. While more specific real-time PCR assays are available that generally distinguish rhinoviruses from enteroviruses,31 during the recent outbreak our laboratory observed that confirmed EV-D68 samples cross-reacted with rhinovirus. Most clinical laboratories do not routinely perform viral sequence analysis to specifically identify EV-D68, but this test may be obtained through state health departments and the CDC on a case-by-case basis.
Recently, the CDC’s enterovirus laboratory announced the development of a real-time PCR assay specifically for EV-D68, which may make specific detection more readily available.
INFECTION PREVENTION
The routes by which EV-D68 is transmitted are not fully understood. In contrast to most enteroviruses, which are spread in a fecal-oral manner, it is possible that EV-D68 is also spread through close respiratory or mucous contact.
For this reason, interim infection prevention guidelines issued by the CDC recommend that hospitals use droplet precautions along with contact or standard precautions, depending on the scenario.32 In our children’s hospital, we use droplet and contact precautions for hospitalized patients.
- Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol 1967; 85:297–310.
- Oberste MS, Maher K, Schnurr D, et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J Gen Virol 2004; 85:2577–2584.
- Ishiko H, Miura R, Shimada Y, et al. Human rhinovirus 87 identified as human enterovirus 68 by VP4-based molecular diagnosis. Intervirology 2002; 45:136–141.
- Blomqvist S, Savolainen C, Raman L, Roivainen M, Hovi T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J Clin Microbiol 2002; 40:4218–4223.
- Cherry JD, Krogstad P. Enterovirus, parechoviruses, and Saffold viruses. In: Cherry JD, Harrison GJ, Kaplan SL, Steinbach WJ, Hoetez PJ, editors. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases. Vol 2. Seventh ed. Philadelphia: Elsevier Saunders; 2014:2051–2109.
- Rotbart HA. Enteroviral infections of the central nervous system. Clin Infect Dis 1995; 20:971–981.
- Nathanson N, Kew OM. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 2010; 172:1213–1229.
- Santti J, Vainionpää R, Hyypiä T. Molecular detection and typing of human picornaviruses. Virus Res 1999; 62:177–183.
- Smura T, Ylipaasto P, Klemola P, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol 2010; 82:1940–1949.
- Midgley CM, Jackson MA, Selvarangan R, et al. Severe respiratory illness associated with enterovirus d68 - Missouri and Illinois, 2014. MMWR Morb Mortal Wkly Rep 2014; 63:798–799.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill Summ 2006; 55:1–20.
- Tokarz R, Firth C, Madhi SA, et al. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol 2012; 93:1952–1958.
- Clusters of acute respiratory illness associated with human enterovirus 68—Asia, Europe, and United States, 2008–2010. MMWR Morb Mortal Wkly Rep 2011; 60:1301–1304.
- Lauinger IL, Bible JM, Halligan EP, Aarons EJ, MacMahon E, Tong CY. Lineages, sub-lineages and variants of enterovirus 68 in recent outbreaks. PLoS One 2012; 7:e36005.
- Lu QB, Wo Y, Wang HY, et al. Detection of enterovirus 68 as one of the commonest types of enterovirus found in patients with acute respiratory tract infection in China. J Med Microbiol 2014; 63:408–414.
- Meijer A, van der Sanden S, Snijders BE, et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in The Netherlands in 2010. Virology 2012; 423:49–57.
- Rahamat-Langendoen J, Riezebos-Brilman A, Borger R, et al. Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J Clin Virol 2011; 52:103–106.
- Tokarz R, Kapoor V, Wu W, et al. Longitudinal molecular microbial analysis of influenza-like illness in New York City, May 2009 through May 2010. Virol J 2011; 8:288.
- Uncapher CR, DeWitt CM, Colonno RJ. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991; 180:814–817.
- Imamura T, Okamoto M, Nakakita S, et al. Antigenic and receptor binding properties of enterovirus 68. J Virol 2014; 88:2374–2384.
- Karnauchow TM, Tolson DL, Harrison BA, Altman E, Lublin DM, Dimock K. The HeLa cell receptor for enterovirus 70 is decay-accelerating factor (CD55). J Virol 1996; 70:5143–5152.
- Selinka HC, Wolde A, Sauter M, Kandolf R, Klingel K. Virus-receptor interactions of coxsackie B viruses and their putative influence on cardiotropism. Med Microbiol Immunol 2004; 193:127–131.
- Piralla A, Girello A, Grignani M, et al. Phylogenetic characterization of enterovirus 68 strains in patients with respiratory syndromes in Italy. J Med Virol 2014; 86:1590–1593.
- Imamura T, Suzuki A, Lupisan S, et al. Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza Other Respir Viruses 2014; 8:21–24.
- Imamura T, Fuji N, Suzuki A, et al. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis 2011; 17:1430–1435.
- Kaida A, Kubo H, Sekiguchi J, et al. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg Infect Dis 2011; 17:1494–1497.
- Kreuter JD, Barnes A, McCarthy JE, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med 2011; 135:793–796.
- Pastula DM, Aliabadi N, Haynes AK, et al. Acute neurologic illness of unknown etiology in children—Colorado, August-September 2014. MMWR Morb Mortal Wkly Rep 2014; 63:901–902.
- Ayscue P, Haren KV, Sheriff H, et al. Acute flaccid paralysis with anterior myelitis—California, June 2012–June 2014. MMWR Morb Mortal Wkly Rep 2014; 63:903–906.
- Abzug MJ. The enteroviruses: problems in need of treatments. J Infect 2014; 68(suppl 1):S108–S114.
- Pierce VM, Hodinka RL. Comparison of the GenMark Diagnostics eSensor respiratory viral panel to real-time PCR for detection of respiratory viruses in children. J Clin Microbiol 2012; 50:3458–3465.
- Non-polio enterovirus infection: enterovirus D68 (EV-D68)-CDC. 2014; www.cdc.gov/non-polio-enterovirus/about/ev-d68.html. Accessed November 21, 2014.
- Schieble JH, Fox VL, Lennette EH. A probable new human picornavirus associated with respiratory diseases. Am J Epidemiol 1967; 85:297–310.
- Oberste MS, Maher K, Schnurr D, et al. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J Gen Virol 2004; 85:2577–2584.
- Ishiko H, Miura R, Shimada Y, et al. Human rhinovirus 87 identified as human enterovirus 68 by VP4-based molecular diagnosis. Intervirology 2002; 45:136–141.
- Blomqvist S, Savolainen C, Raman L, Roivainen M, Hovi T. Human rhinovirus 87 and enterovirus 68 represent a unique serotype with rhinovirus and enterovirus features. J Clin Microbiol 2002; 40:4218–4223.
- Cherry JD, Krogstad P. Enterovirus, parechoviruses, and Saffold viruses. In: Cherry JD, Harrison GJ, Kaplan SL, Steinbach WJ, Hoetez PJ, editors. Feigin and Cherry’s Textbook of Pediatric Infectious Diseases. Vol 2. Seventh ed. Philadelphia: Elsevier Saunders; 2014:2051–2109.
- Rotbart HA. Enteroviral infections of the central nervous system. Clin Infect Dis 1995; 20:971–981.
- Nathanson N, Kew OM. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 2010; 172:1213–1229.
- Santti J, Vainionpää R, Hyypiä T. Molecular detection and typing of human picornaviruses. Virus Res 1999; 62:177–183.
- Smura T, Ylipaasto P, Klemola P, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol 2010; 82:1940–1949.
- Midgley CM, Jackson MA, Selvarangan R, et al. Severe respiratory illness associated with enterovirus d68 - Missouri and Illinois, 2014. MMWR Morb Mortal Wkly Rep 2014; 63:798–799.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill Summ 2006; 55:1–20.
- Tokarz R, Firth C, Madhi SA, et al. Worldwide emergence of multiple clades of enterovirus 68. J Gen Virol 2012; 93:1952–1958.
- Clusters of acute respiratory illness associated with human enterovirus 68—Asia, Europe, and United States, 2008–2010. MMWR Morb Mortal Wkly Rep 2011; 60:1301–1304.
- Lauinger IL, Bible JM, Halligan EP, Aarons EJ, MacMahon E, Tong CY. Lineages, sub-lineages and variants of enterovirus 68 in recent outbreaks. PLoS One 2012; 7:e36005.
- Lu QB, Wo Y, Wang HY, et al. Detection of enterovirus 68 as one of the commonest types of enterovirus found in patients with acute respiratory tract infection in China. J Med Microbiol 2014; 63:408–414.
- Meijer A, van der Sanden S, Snijders BE, et al. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in The Netherlands in 2010. Virology 2012; 423:49–57.
- Rahamat-Langendoen J, Riezebos-Brilman A, Borger R, et al. Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J Clin Virol 2011; 52:103–106.
- Tokarz R, Kapoor V, Wu W, et al. Longitudinal molecular microbial analysis of influenza-like illness in New York City, May 2009 through May 2010. Virol J 2011; 8:288.
- Uncapher CR, DeWitt CM, Colonno RJ. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991; 180:814–817.
- Imamura T, Okamoto M, Nakakita S, et al. Antigenic and receptor binding properties of enterovirus 68. J Virol 2014; 88:2374–2384.
- Karnauchow TM, Tolson DL, Harrison BA, Altman E, Lublin DM, Dimock K. The HeLa cell receptor for enterovirus 70 is decay-accelerating factor (CD55). J Virol 1996; 70:5143–5152.
- Selinka HC, Wolde A, Sauter M, Kandolf R, Klingel K. Virus-receptor interactions of coxsackie B viruses and their putative influence on cardiotropism. Med Microbiol Immunol 2004; 193:127–131.
- Piralla A, Girello A, Grignani M, et al. Phylogenetic characterization of enterovirus 68 strains in patients with respiratory syndromes in Italy. J Med Virol 2014; 86:1590–1593.
- Imamura T, Suzuki A, Lupisan S, et al. Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza Other Respir Viruses 2014; 8:21–24.
- Imamura T, Fuji N, Suzuki A, et al. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis 2011; 17:1430–1435.
- Kaida A, Kubo H, Sekiguchi J, et al. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg Infect Dis 2011; 17:1494–1497.
- Kreuter JD, Barnes A, McCarthy JE, et al. A fatal central nervous system enterovirus 68 infection. Arch Pathol Lab Med 2011; 135:793–796.
- Pastula DM, Aliabadi N, Haynes AK, et al. Acute neurologic illness of unknown etiology in children—Colorado, August-September 2014. MMWR Morb Mortal Wkly Rep 2014; 63:901–902.
- Ayscue P, Haren KV, Sheriff H, et al. Acute flaccid paralysis with anterior myelitis—California, June 2012–June 2014. MMWR Morb Mortal Wkly Rep 2014; 63:903–906.
- Abzug MJ. The enteroviruses: problems in need of treatments. J Infect 2014; 68(suppl 1):S108–S114.
- Pierce VM, Hodinka RL. Comparison of the GenMark Diagnostics eSensor respiratory viral panel to real-time PCR for detection of respiratory viruses in children. J Clin Microbiol 2012; 50:3458–3465.
- Non-polio enterovirus infection: enterovirus D68 (EV-D68)-CDC. 2014; www.cdc.gov/non-polio-enterovirus/about/ev-d68.html. Accessed November 21, 2014.
KEY POINTS
- EV-D68 is a respiratory virus that has genetic and biologic features that blur the distinction between the rhinoviruses and enteroviruses.
- Recognition of EV-D68 as an important cause of viral lower respiratory tract illness in children underscores the role of specific strain typing in advancing our understanding of the epidemiology of respiratory virus infections.
- Given the inability of commonly used clinical tests for rhinovirus to distinguish EV-D68 in the absence of strain-specific sequence data, caution needs to be used in attributing severe or acute lower respiratory illness to rhinovirus and in interpreting epidemiologic associations between asthma and rhinovirus.
- Emerging data suggest that, in addition to its important role in pediatric respiratory illness, EV-D68 may cause systemic disease, especially acute neurologic disease.
When should I discuss driving with my older patients?
Most older drivers are safe drivers and are less likely than younger people to drive recklessly, at high speeds, or under the influence of alcohol.1 However, motor vehicle injuries are the second leading cause of injury-related deaths among older adults. Very old adults (80 years and over) have higher rates of fatality and injury in motor vehicle crashes per million miles driven than any other age group except for teenagers.1 Therefore, consider safety screening of all very old drivers plus any older adult with certain high-risk medical conditions, including the following.
NEUROCOGNITIVE DISORDERS
Drivers with Alzheimer disease—the most common type of major neurocognitive disorder (dementia) in older adults in the United States—are at high risk for adverse driving events due to impaired memory, attentiveness, problem-solving skills, multitasking, orientation, judgment, and reaction speed. Even in amnesic mild cognitive impairment—a mild neurocognitive disorder without functional decline—driving skills such as lane control may be impaired.2
Frontotemporal dementia, a less common cause of dementia in older adults, is associated with profound impairments in reasoning, task flexibility, planning, and execution. Persons with frontotemporal dementia are more likely to speed, run stop signs, and suffer more off-road crashes and collisions.3
The diagnosis of dementia, however, is less predictive of driving risk than the stage of dementia. The American Academy of Neurology recommends that health care providers clinically “stage” all demented individuals using a validated tool at diagnosis and periodically afterwards. The Clinical Dementia Rating (CDR) scale is appropriate for staging dementia in the office. The CDR has also been shown to identify people with dementia who are at an increased risk of unsafe driving, with strong evidence (level of evidence A) relating dementia stage to driving risk.4 The CDR assigns a score of 1 for mild dementia (function impaired in at least one complex activity); 2 for moderate dementia (function impaired in at least one basic activity); and 3 for severe dementia. Individuals with a CDR score of 2 or higher are considered to be at very high risk if still driving. These persons should be encouraged to surrender their driving privileges.4 Even with mild dementia (CDR score of 1), as few as 41% of drivers may drive safely.4 Most persons with mild cognitive impairment (CDR score of 0.5) are safe drivers.

Patients often have poor insight into their driving safety. However, a caregiver’s rating of driving skills as marginal or unsafe is useful in identifying unsafe drivers (level of evidence B) and can be considered a red flag.4 Predictors with less support in the literature (level of evidence C) include recent traffic citations, motor vehicle accidents, and self-reported situational avoidance, such as limiting driving to familiar roadways. Additional predictors include Mini-Mental State Examination scores of 24 or less, and/or the emergence of an aggressive or impulsive personality (Table 1). A driver evaluation is helpful when there is mild cognitive impairment or mild dementia with at least one red flag.
Clinicians who are not comfortable with staging dementia as mild, moderate, or severe may consider referring to a neurologist or geriatrician.
There is no evidence to support or refute the benefit of interventional strategies such as driver rehabilitation for drivers with dementia.
PARKINSON DISEASE
Individuals with mild motor disability from Parkinson disease may be fit drivers. As the disease progresses, drivers with Parkinson disease may make more errors than healthy elders in visual scanning, signaling, vehicle positioning, and velocity regulation (eg, traveling so slowly that it may be unsafe).5 Clinicians can consider referring a patient with Parkinson disease for a baseline driving evaluation upon diagnosis, and then every 1 to 2 years for reassessment. Alternate transportation should be arranged as the disease progresses.
EPISODIC INCAPACITATION
Approximately 1% to 3% of all motor vehicle accidents are due to sudden incapacitation of an otherwise safe driver.
Syncope. Neurally mediated (vasovagal) syncope accounts for 30% to 35% of syncopal episodes while driving.6 Cardiac arrhythmias are the next most common cause and include bradyarrhythmias (7%), supraventricular tachyarrhythmias (2%–15%), and ventricular tachyarrhythmias (5%–17%). Because neurocardiogenic syncope often recurs, consider restricting driving for those with recurrent or severe neurocardiogenic syncopal episodes until symptoms are controlled.
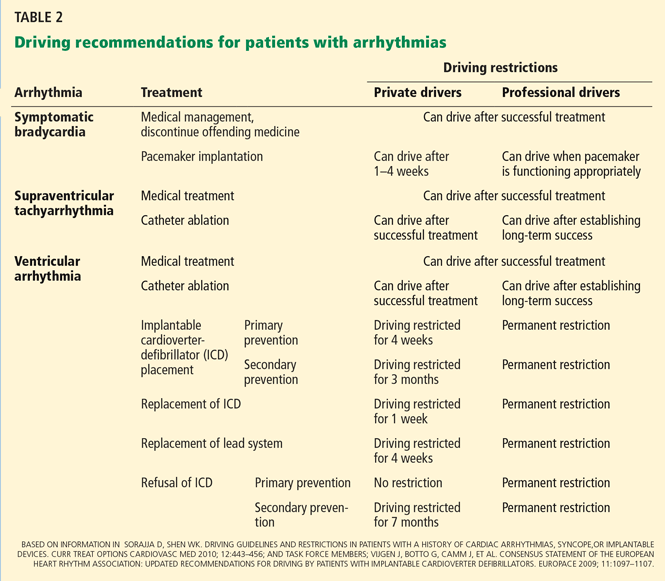
Arrhythmias. Driving recommendations for various arrhythmias7,8 are listed in Table 2.
Many patients who have an implantable cardioverter-defibrillator (ICD) device experience an unexpected shock. For individuals with a history of ventricular tachycardia or fibrillation, the 5-year actuarial incidence of appropriate ICD shocks ranges between 55% and 70%. However, data indicate that 90% to 100% of drivers who received ICD discharges while driving continued to drive without causing motor vehicle accidents.9,10
Seizures. States differ in their rules for reporting drivers who have epilepsy or breakthrough seizures. Physicians should refer to their state regulations when counseling these patients.
POLYPHARMACY
Polypharmacy is common in older adults. Many take psychoactive drugs that can impair tracking, alertness, coordination, and reaction time. With the “Roadwise Rx” tool (www.roadwiserx.com), health care providers and patients can enter the names of medicines to check if they affect driving ability. Nonproprietary on-line tools such as “START” (Screening Tool to Alert doctors to Right Treatment) and “STOPP” (Screening Tool of Older Persons’ Potentially Inappropriate Prescriptions) can be used to prune medication lists.
DRIVING EVALUATION
America is a nation of highways overflowing with cars. Cars provide transportation but also reflect wealth and personality, particularly for men. Practically, the ability to drive a car allows older men and women to socialize in the community, shop for essentials, and take care of themselves without being a burden. Driving cessation can cause social isolation and depressive symptoms and can strain caregiver resources.
It is therefore understandable for health care providers to feel reluctant or uncomfortable counseling older adults to give up their driving privileges. A health care provider who identifies driving safety concerns can refer a patient to a geriatrician for further risk assessment or to a certified driver rehabilitation specialist (CDRS) for a driving evaluation. A CDRS will also offer the patient and caregiver information on local resources for transportation alternatives. A list of local CDRSs can be found on the Association for Driver Rehabilitation Specialists website (www.aded.net). Many hospitals have occupational therapists who are CDRSs.
The evaluation typically involves an assessment of the driver’s knowledge of traffic signs and laws, a cognitive assessment, possibly a simulation, and finally an on-road driving evaluation if deemed appropriate. Medicare coverage depends on diagnosis and the state carrier.
- Williams AF. Teenage drivers: patterns of risk. J Safety Res 2003; 34:5–15.
- Griffith HR, Okonkwo OC, Stewart CC, et al. Lower hippocampal volume predicts decrements in lane control among drivers with amnestic mild cognitive impairment. J Geriatr Psychiatry Neurol 2013; 26:259–266.
- de Simone V, Kaplan L, Patronas N, Wassermann EM, Grafman J. Driving abilities in frontotemporal dementia patients. Dement Geriatr Cogn Disord 2007; 23:1–7.
- Iverson DJ, Gronseth GS, Reger MA, Classen S, Dubinsky RM, Rizzo M; Quality Standards Subcomittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Classen S, Brumback B, Monahan M, et al. Driving errors in Parkinson’s disease: moving closer to predicting on-road outcomes. Am J Occup Ther 2014; 68:77–85.
- Blitzer ML, Saliba BC, Ghantous AE, Marieb MA, Schoenfeld MH. Causes of impaired consciousness while driving a motorized vehicle. Am J Cardiol 2003; 91:1373–1374.
- Sorajja D, Shen WK. Driving guidelines and restrictions in patients with a history of cardiac arrhythmias, syncope,or implantable devices. Curr Treat Options Cardiovasc Med 2010; 12:443–456.
- Task force members; Vijgen J, Botto G, Camm J, et al. Consensus statement of the European Heart Rhythm Association: updated recommendations for driving by patients with implantable cardioverter defibrillators. Europace 2009; 11:1097–1107.
- Conti JB, Woodard DA, Tucker KJ, Bryant B, King LC, Curtis AB. Modification of patient driving behavior after implantation of a cardioverter defibrillator. Pacing Clin Electrophysiol 1997; 20:2200–2204.
- Lerecouvreux M, Aït Saïd M, Paziaud O, et al. Automobile driving and implantable defibrillators. Arch Mal Coeur Vaiss 2005; 98:288–293. Article in French.
Most older drivers are safe drivers and are less likely than younger people to drive recklessly, at high speeds, or under the influence of alcohol.1 However, motor vehicle injuries are the second leading cause of injury-related deaths among older adults. Very old adults (80 years and over) have higher rates of fatality and injury in motor vehicle crashes per million miles driven than any other age group except for teenagers.1 Therefore, consider safety screening of all very old drivers plus any older adult with certain high-risk medical conditions, including the following.
NEUROCOGNITIVE DISORDERS
Drivers with Alzheimer disease—the most common type of major neurocognitive disorder (dementia) in older adults in the United States—are at high risk for adverse driving events due to impaired memory, attentiveness, problem-solving skills, multitasking, orientation, judgment, and reaction speed. Even in amnesic mild cognitive impairment—a mild neurocognitive disorder without functional decline—driving skills such as lane control may be impaired.2
Frontotemporal dementia, a less common cause of dementia in older adults, is associated with profound impairments in reasoning, task flexibility, planning, and execution. Persons with frontotemporal dementia are more likely to speed, run stop signs, and suffer more off-road crashes and collisions.3
The diagnosis of dementia, however, is less predictive of driving risk than the stage of dementia. The American Academy of Neurology recommends that health care providers clinically “stage” all demented individuals using a validated tool at diagnosis and periodically afterwards. The Clinical Dementia Rating (CDR) scale is appropriate for staging dementia in the office. The CDR has also been shown to identify people with dementia who are at an increased risk of unsafe driving, with strong evidence (level of evidence A) relating dementia stage to driving risk.4 The CDR assigns a score of 1 for mild dementia (function impaired in at least one complex activity); 2 for moderate dementia (function impaired in at least one basic activity); and 3 for severe dementia. Individuals with a CDR score of 2 or higher are considered to be at very high risk if still driving. These persons should be encouraged to surrender their driving privileges.4 Even with mild dementia (CDR score of 1), as few as 41% of drivers may drive safely.4 Most persons with mild cognitive impairment (CDR score of 0.5) are safe drivers.
Patients often have poor insight into their driving safety. However, a caregiver’s rating of driving skills as marginal or unsafe is useful in identifying unsafe drivers (level of evidence B) and can be considered a red flag.4 Predictors with less support in the literature (level of evidence C) include recent traffic citations, motor vehicle accidents, and self-reported situational avoidance, such as limiting driving to familiar roadways. Additional predictors include Mini-Mental State Examination scores of 24 or less, and/or the emergence of an aggressive or impulsive personality (Table 1). A driver evaluation is helpful when there is mild cognitive impairment or mild dementia with at least one red flag.
Clinicians who are not comfortable with staging dementia as mild, moderate, or severe may consider referring to a neurologist or geriatrician.
There is no evidence to support or refute the benefit of interventional strategies such as driver rehabilitation for drivers with dementia.
PARKINSON DISEASE
Individuals with mild motor disability from Parkinson disease may be fit drivers. As the disease progresses, drivers with Parkinson disease may make more errors than healthy elders in visual scanning, signaling, vehicle positioning, and velocity regulation (eg, traveling so slowly that it may be unsafe).5 Clinicians can consider referring a patient with Parkinson disease for a baseline driving evaluation upon diagnosis, and then every 1 to 2 years for reassessment. Alternate transportation should be arranged as the disease progresses.
EPISODIC INCAPACITATION
Approximately 1% to 3% of all motor vehicle accidents are due to sudden incapacitation of an otherwise safe driver.
Syncope. Neurally mediated (vasovagal) syncope accounts for 30% to 35% of syncopal episodes while driving.6 Cardiac arrhythmias are the next most common cause and include bradyarrhythmias (7%), supraventricular tachyarrhythmias (2%–15%), and ventricular tachyarrhythmias (5%–17%). Because neurocardiogenic syncope often recurs, consider restricting driving for those with recurrent or severe neurocardiogenic syncopal episodes until symptoms are controlled.
Arrhythmias. Driving recommendations for various arrhythmias7,8 are listed in Table 2.
Many patients who have an implantable cardioverter-defibrillator (ICD) device experience an unexpected shock. For individuals with a history of ventricular tachycardia or fibrillation, the 5-year actuarial incidence of appropriate ICD shocks ranges between 55% and 70%. However, data indicate that 90% to 100% of drivers who received ICD discharges while driving continued to drive without causing motor vehicle accidents.9,10
Seizures. States differ in their rules for reporting drivers who have epilepsy or breakthrough seizures. Physicians should refer to their state regulations when counseling these patients.
POLYPHARMACY
Polypharmacy is common in older adults. Many take psychoactive drugs that can impair tracking, alertness, coordination, and reaction time. With the “Roadwise Rx” tool (www.roadwiserx.com), health care providers and patients can enter the names of medicines to check if they affect driving ability. Nonproprietary on-line tools such as “START” (Screening Tool to Alert doctors to Right Treatment) and “STOPP” (Screening Tool of Older Persons’ Potentially Inappropriate Prescriptions) can be used to prune medication lists.
DRIVING EVALUATION
America is a nation of highways overflowing with cars. Cars provide transportation but also reflect wealth and personality, particularly for men. Practically, the ability to drive a car allows older men and women to socialize in the community, shop for essentials, and take care of themselves without being a burden. Driving cessation can cause social isolation and depressive symptoms and can strain caregiver resources.
It is therefore understandable for health care providers to feel reluctant or uncomfortable counseling older adults to give up their driving privileges. A health care provider who identifies driving safety concerns can refer a patient to a geriatrician for further risk assessment or to a certified driver rehabilitation specialist (CDRS) for a driving evaluation. A CDRS will also offer the patient and caregiver information on local resources for transportation alternatives. A list of local CDRSs can be found on the Association for Driver Rehabilitation Specialists website (www.aded.net). Many hospitals have occupational therapists who are CDRSs.
The evaluation typically involves an assessment of the driver’s knowledge of traffic signs and laws, a cognitive assessment, possibly a simulation, and finally an on-road driving evaluation if deemed appropriate. Medicare coverage depends on diagnosis and the state carrier.
Most older drivers are safe drivers and are less likely than younger people to drive recklessly, at high speeds, or under the influence of alcohol.1 However, motor vehicle injuries are the second leading cause of injury-related deaths among older adults. Very old adults (80 years and over) have higher rates of fatality and injury in motor vehicle crashes per million miles driven than any other age group except for teenagers.1 Therefore, consider safety screening of all very old drivers plus any older adult with certain high-risk medical conditions, including the following.
NEUROCOGNITIVE DISORDERS
Drivers with Alzheimer disease—the most common type of major neurocognitive disorder (dementia) in older adults in the United States—are at high risk for adverse driving events due to impaired memory, attentiveness, problem-solving skills, multitasking, orientation, judgment, and reaction speed. Even in amnesic mild cognitive impairment—a mild neurocognitive disorder without functional decline—driving skills such as lane control may be impaired.2
Frontotemporal dementia, a less common cause of dementia in older adults, is associated with profound impairments in reasoning, task flexibility, planning, and execution. Persons with frontotemporal dementia are more likely to speed, run stop signs, and suffer more off-road crashes and collisions.3
The diagnosis of dementia, however, is less predictive of driving risk than the stage of dementia. The American Academy of Neurology recommends that health care providers clinically “stage” all demented individuals using a validated tool at diagnosis and periodically afterwards. The Clinical Dementia Rating (CDR) scale is appropriate for staging dementia in the office. The CDR has also been shown to identify people with dementia who are at an increased risk of unsafe driving, with strong evidence (level of evidence A) relating dementia stage to driving risk.4 The CDR assigns a score of 1 for mild dementia (function impaired in at least one complex activity); 2 for moderate dementia (function impaired in at least one basic activity); and 3 for severe dementia. Individuals with a CDR score of 2 or higher are considered to be at very high risk if still driving. These persons should be encouraged to surrender their driving privileges.4 Even with mild dementia (CDR score of 1), as few as 41% of drivers may drive safely.4 Most persons with mild cognitive impairment (CDR score of 0.5) are safe drivers.
Patients often have poor insight into their driving safety. However, a caregiver’s rating of driving skills as marginal or unsafe is useful in identifying unsafe drivers (level of evidence B) and can be considered a red flag.4 Predictors with less support in the literature (level of evidence C) include recent traffic citations, motor vehicle accidents, and self-reported situational avoidance, such as limiting driving to familiar roadways. Additional predictors include Mini-Mental State Examination scores of 24 or less, and/or the emergence of an aggressive or impulsive personality (Table 1). A driver evaluation is helpful when there is mild cognitive impairment or mild dementia with at least one red flag.
Clinicians who are not comfortable with staging dementia as mild, moderate, or severe may consider referring to a neurologist or geriatrician.
There is no evidence to support or refute the benefit of interventional strategies such as driver rehabilitation for drivers with dementia.
PARKINSON DISEASE
Individuals with mild motor disability from Parkinson disease may be fit drivers. As the disease progresses, drivers with Parkinson disease may make more errors than healthy elders in visual scanning, signaling, vehicle positioning, and velocity regulation (eg, traveling so slowly that it may be unsafe).5 Clinicians can consider referring a patient with Parkinson disease for a baseline driving evaluation upon diagnosis, and then every 1 to 2 years for reassessment. Alternate transportation should be arranged as the disease progresses.
EPISODIC INCAPACITATION
Approximately 1% to 3% of all motor vehicle accidents are due to sudden incapacitation of an otherwise safe driver.
Syncope. Neurally mediated (vasovagal) syncope accounts for 30% to 35% of syncopal episodes while driving.6 Cardiac arrhythmias are the next most common cause and include bradyarrhythmias (7%), supraventricular tachyarrhythmias (2%–15%), and ventricular tachyarrhythmias (5%–17%). Because neurocardiogenic syncope often recurs, consider restricting driving for those with recurrent or severe neurocardiogenic syncopal episodes until symptoms are controlled.
Arrhythmias. Driving recommendations for various arrhythmias7,8 are listed in Table 2.
Many patients who have an implantable cardioverter-defibrillator (ICD) device experience an unexpected shock. For individuals with a history of ventricular tachycardia or fibrillation, the 5-year actuarial incidence of appropriate ICD shocks ranges between 55% and 70%. However, data indicate that 90% to 100% of drivers who received ICD discharges while driving continued to drive without causing motor vehicle accidents.9,10
Seizures. States differ in their rules for reporting drivers who have epilepsy or breakthrough seizures. Physicians should refer to their state regulations when counseling these patients.
POLYPHARMACY
Polypharmacy is common in older adults. Many take psychoactive drugs that can impair tracking, alertness, coordination, and reaction time. With the “Roadwise Rx” tool (www.roadwiserx.com), health care providers and patients can enter the names of medicines to check if they affect driving ability. Nonproprietary on-line tools such as “START” (Screening Tool to Alert doctors to Right Treatment) and “STOPP” (Screening Tool of Older Persons’ Potentially Inappropriate Prescriptions) can be used to prune medication lists.
DRIVING EVALUATION
America is a nation of highways overflowing with cars. Cars provide transportation but also reflect wealth and personality, particularly for men. Practically, the ability to drive a car allows older men and women to socialize in the community, shop for essentials, and take care of themselves without being a burden. Driving cessation can cause social isolation and depressive symptoms and can strain caregiver resources.
It is therefore understandable for health care providers to feel reluctant or uncomfortable counseling older adults to give up their driving privileges. A health care provider who identifies driving safety concerns can refer a patient to a geriatrician for further risk assessment or to a certified driver rehabilitation specialist (CDRS) for a driving evaluation. A CDRS will also offer the patient and caregiver information on local resources for transportation alternatives. A list of local CDRSs can be found on the Association for Driver Rehabilitation Specialists website (www.aded.net). Many hospitals have occupational therapists who are CDRSs.
The evaluation typically involves an assessment of the driver’s knowledge of traffic signs and laws, a cognitive assessment, possibly a simulation, and finally an on-road driving evaluation if deemed appropriate. Medicare coverage depends on diagnosis and the state carrier.
- Williams AF. Teenage drivers: patterns of risk. J Safety Res 2003; 34:5–15.
- Griffith HR, Okonkwo OC, Stewart CC, et al. Lower hippocampal volume predicts decrements in lane control among drivers with amnestic mild cognitive impairment. J Geriatr Psychiatry Neurol 2013; 26:259–266.
- de Simone V, Kaplan L, Patronas N, Wassermann EM, Grafman J. Driving abilities in frontotemporal dementia patients. Dement Geriatr Cogn Disord 2007; 23:1–7.
- Iverson DJ, Gronseth GS, Reger MA, Classen S, Dubinsky RM, Rizzo M; Quality Standards Subcomittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Classen S, Brumback B, Monahan M, et al. Driving errors in Parkinson’s disease: moving closer to predicting on-road outcomes. Am J Occup Ther 2014; 68:77–85.
- Blitzer ML, Saliba BC, Ghantous AE, Marieb MA, Schoenfeld MH. Causes of impaired consciousness while driving a motorized vehicle. Am J Cardiol 2003; 91:1373–1374.
- Sorajja D, Shen WK. Driving guidelines and restrictions in patients with a history of cardiac arrhythmias, syncope,or implantable devices. Curr Treat Options Cardiovasc Med 2010; 12:443–456.
- Task force members; Vijgen J, Botto G, Camm J, et al. Consensus statement of the European Heart Rhythm Association: updated recommendations for driving by patients with implantable cardioverter defibrillators. Europace 2009; 11:1097–1107.
- Conti JB, Woodard DA, Tucker KJ, Bryant B, King LC, Curtis AB. Modification of patient driving behavior after implantation of a cardioverter defibrillator. Pacing Clin Electrophysiol 1997; 20:2200–2204.
- Lerecouvreux M, Aït Saïd M, Paziaud O, et al. Automobile driving and implantable defibrillators. Arch Mal Coeur Vaiss 2005; 98:288–293. Article in French.
- Williams AF. Teenage drivers: patterns of risk. J Safety Res 2003; 34:5–15.
- Griffith HR, Okonkwo OC, Stewart CC, et al. Lower hippocampal volume predicts decrements in lane control among drivers with amnestic mild cognitive impairment. J Geriatr Psychiatry Neurol 2013; 26:259–266.
- de Simone V, Kaplan L, Patronas N, Wassermann EM, Grafman J. Driving abilities in frontotemporal dementia patients. Dement Geriatr Cogn Disord 2007; 23:1–7.
- Iverson DJ, Gronseth GS, Reger MA, Classen S, Dubinsky RM, Rizzo M; Quality Standards Subcomittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Classen S, Brumback B, Monahan M, et al. Driving errors in Parkinson’s disease: moving closer to predicting on-road outcomes. Am J Occup Ther 2014; 68:77–85.
- Blitzer ML, Saliba BC, Ghantous AE, Marieb MA, Schoenfeld MH. Causes of impaired consciousness while driving a motorized vehicle. Am J Cardiol 2003; 91:1373–1374.
- Sorajja D, Shen WK. Driving guidelines and restrictions in patients with a history of cardiac arrhythmias, syncope,or implantable devices. Curr Treat Options Cardiovasc Med 2010; 12:443–456.
- Task force members; Vijgen J, Botto G, Camm J, et al. Consensus statement of the European Heart Rhythm Association: updated recommendations for driving by patients with implantable cardioverter defibrillators. Europace 2009; 11:1097–1107.
- Conti JB, Woodard DA, Tucker KJ, Bryant B, King LC, Curtis AB. Modification of patient driving behavior after implantation of a cardioverter defibrillator. Pacing Clin Electrophysiol 1997; 20:2200–2204.
- Lerecouvreux M, Aït Saïd M, Paziaud O, et al. Automobile driving and implantable defibrillators. Arch Mal Coeur Vaiss 2005; 98:288–293. Article in French.

Retroperitoneal cyst hemorrhage in polycystic kidney disease
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
Joint pain in a man with lung cancer
A 46-year-old man with a 50-pack-year smoking history, hepatitis C, and polysubstance abuse presented with worsening joint pain over the past 2 months involving both knees, ankles, wrists, and hands. On examination, along with clubbing of the fingers and toes, he had tenderness and restriction in range of motion in all the affected joints. A fixed, nontender, hard mass was noted in the left axilla extending into the infraclavicular and supraclavicular regions.
Computed tomography (CT) with contrast revealed a mass in the left upper lobe of the lung, eroding the ribs and extending into the axilla, consistent with a suspected diagnosis of lung cancer (Figure 1). Radiography of the hands and feet showed thickening of the periosteal membrane and periosteal formation of new bone (Figure 2). A bone scan revealed diffuse increased uptake in the affected joints with no metastases (Figure 3). A workup for brain and adrenal metastases was negative. CT-guided needle biopsy confirmed adenocarcinoma of the lung.
The final diagnosis was hypertrophic pulmonary osteoarthropathy (HPOA) secondary to stage IIIB: T4N2M0 adenocarcinoma of the lung. Resection of the lung mass was deemed difficult, so the patient was scheduled for neoadjuvant concurrent chemoradiation therapy before resection. Ibuprofen and intravenous pamidronate provided symptom relief.
HYPERTROPHIC PULMONARY OSTEOARTHROPATHY
HPOA is characterized by abnormal proliferation of the skin and periosteal formation of new bone at the distal ends of long bones, metatarsals, metacarpals, and proximal phalanges.
Primary HPOA is genetically inherited and accounts for 5% of cases, while secondary HPOA, accounting for the other 95%, is a paraneoplastic syndrome.1 Secondary HPOA is most commonly associated with lung cancer, usually stage III or IV adenocarcinoma.1 However, only 1% of lung cancer patients develop HPOA.1
Other conditions linked to secondary HPOA are mesothelioma, renal cell carcinoma, gastrointestinal cancers, melanoma, thymic cancer, nasopharyngeal cancers, and Hodgkin lymphoma. HPOA is also associated with pulmonary infection, cystic fibrosis, and right-to-left cardiac shunt. Risk factors associated with secondary HPOA are smoking, male sex, and age greater than 55.
Patients often present with unremitting pain, edema, and erythema in the extremities. Radiography reveals periosteal membrane thickening and periosteal new bone formation.1 Findings on bone scintigraphy that confirm the diagnosis include diffuse increased uptake and bracelet-like uptake.
The prognosis and treatment of secondary HPOA are directly related to the underlying disease. Symptoms can be relieved with nonsteroidal anti-inflammatory drugs, bisphosphonates, and octreotide. Unilateral vagotomy is an option in refractory cases.1,2
- Ito T, Goto K, Yoh K, et al. Hypertrophic pulmonary osteoarthropathy as a paraneoplastic manifestation of lung cancer. J Thorac Oncol 2010; 5:976–980.
- Davis MC, Sherry V. Hypertrophic osteoarthropathy as a clinical manifestation of lung cancer. Clin J Oncol Nurs 2011; 15:561–563.
A 46-year-old man with a 50-pack-year smoking history, hepatitis C, and polysubstance abuse presented with worsening joint pain over the past 2 months involving both knees, ankles, wrists, and hands. On examination, along with clubbing of the fingers and toes, he had tenderness and restriction in range of motion in all the affected joints. A fixed, nontender, hard mass was noted in the left axilla extending into the infraclavicular and supraclavicular regions.
Computed tomography (CT) with contrast revealed a mass in the left upper lobe of the lung, eroding the ribs and extending into the axilla, consistent with a suspected diagnosis of lung cancer (Figure 1). Radiography of the hands and feet showed thickening of the periosteal membrane and periosteal formation of new bone (Figure 2). A bone scan revealed diffuse increased uptake in the affected joints with no metastases (Figure 3). A workup for brain and adrenal metastases was negative. CT-guided needle biopsy confirmed adenocarcinoma of the lung.
The final diagnosis was hypertrophic pulmonary osteoarthropathy (HPOA) secondary to stage IIIB: T4N2M0 adenocarcinoma of the lung. Resection of the lung mass was deemed difficult, so the patient was scheduled for neoadjuvant concurrent chemoradiation therapy before resection. Ibuprofen and intravenous pamidronate provided symptom relief.
HYPERTROPHIC PULMONARY OSTEOARTHROPATHY
HPOA is characterized by abnormal proliferation of the skin and periosteal formation of new bone at the distal ends of long bones, metatarsals, metacarpals, and proximal phalanges.
Primary HPOA is genetically inherited and accounts for 5% of cases, while secondary HPOA, accounting for the other 95%, is a paraneoplastic syndrome.1 Secondary HPOA is most commonly associated with lung cancer, usually stage III or IV adenocarcinoma.1 However, only 1% of lung cancer patients develop HPOA.1
Other conditions linked to secondary HPOA are mesothelioma, renal cell carcinoma, gastrointestinal cancers, melanoma, thymic cancer, nasopharyngeal cancers, and Hodgkin lymphoma. HPOA is also associated with pulmonary infection, cystic fibrosis, and right-to-left cardiac shunt. Risk factors associated with secondary HPOA are smoking, male sex, and age greater than 55.
Patients often present with unremitting pain, edema, and erythema in the extremities. Radiography reveals periosteal membrane thickening and periosteal new bone formation.1 Findings on bone scintigraphy that confirm the diagnosis include diffuse increased uptake and bracelet-like uptake.
The prognosis and treatment of secondary HPOA are directly related to the underlying disease. Symptoms can be relieved with nonsteroidal anti-inflammatory drugs, bisphosphonates, and octreotide. Unilateral vagotomy is an option in refractory cases.1,2
A 46-year-old man with a 50-pack-year smoking history, hepatitis C, and polysubstance abuse presented with worsening joint pain over the past 2 months involving both knees, ankles, wrists, and hands. On examination, along with clubbing of the fingers and toes, he had tenderness and restriction in range of motion in all the affected joints. A fixed, nontender, hard mass was noted in the left axilla extending into the infraclavicular and supraclavicular regions.
Computed tomography (CT) with contrast revealed a mass in the left upper lobe of the lung, eroding the ribs and extending into the axilla, consistent with a suspected diagnosis of lung cancer (Figure 1). Radiography of the hands and feet showed thickening of the periosteal membrane and periosteal formation of new bone (Figure 2). A bone scan revealed diffuse increased uptake in the affected joints with no metastases (Figure 3). A workup for brain and adrenal metastases was negative. CT-guided needle biopsy confirmed adenocarcinoma of the lung.
The final diagnosis was hypertrophic pulmonary osteoarthropathy (HPOA) secondary to stage IIIB: T4N2M0 adenocarcinoma of the lung. Resection of the lung mass was deemed difficult, so the patient was scheduled for neoadjuvant concurrent chemoradiation therapy before resection. Ibuprofen and intravenous pamidronate provided symptom relief.
HYPERTROPHIC PULMONARY OSTEOARTHROPATHY
HPOA is characterized by abnormal proliferation of the skin and periosteal formation of new bone at the distal ends of long bones, metatarsals, metacarpals, and proximal phalanges.
Primary HPOA is genetically inherited and accounts for 5% of cases, while secondary HPOA, accounting for the other 95%, is a paraneoplastic syndrome.1 Secondary HPOA is most commonly associated with lung cancer, usually stage III or IV adenocarcinoma.1 However, only 1% of lung cancer patients develop HPOA.1
Other conditions linked to secondary HPOA are mesothelioma, renal cell carcinoma, gastrointestinal cancers, melanoma, thymic cancer, nasopharyngeal cancers, and Hodgkin lymphoma. HPOA is also associated with pulmonary infection, cystic fibrosis, and right-to-left cardiac shunt. Risk factors associated with secondary HPOA are smoking, male sex, and age greater than 55.
Patients often present with unremitting pain, edema, and erythema in the extremities. Radiography reveals periosteal membrane thickening and periosteal new bone formation.1 Findings on bone scintigraphy that confirm the diagnosis include diffuse increased uptake and bracelet-like uptake.
The prognosis and treatment of secondary HPOA are directly related to the underlying disease. Symptoms can be relieved with nonsteroidal anti-inflammatory drugs, bisphosphonates, and octreotide. Unilateral vagotomy is an option in refractory cases.1,2
- Ito T, Goto K, Yoh K, et al. Hypertrophic pulmonary osteoarthropathy as a paraneoplastic manifestation of lung cancer. J Thorac Oncol 2010; 5:976–980.
- Davis MC, Sherry V. Hypertrophic osteoarthropathy as a clinical manifestation of lung cancer. Clin J Oncol Nurs 2011; 15:561–563.
- Ito T, Goto K, Yoh K, et al. Hypertrophic pulmonary osteoarthropathy as a paraneoplastic manifestation of lung cancer. J Thorac Oncol 2010; 5:976–980.
- Davis MC, Sherry V. Hypertrophic osteoarthropathy as a clinical manifestation of lung cancer. Clin J Oncol Nurs 2011; 15:561–563.
Short and sweet: Writing better consult notes in the era of the electronic medical record
After 4 decades of clinical practice in a teaching hospital, I believe that the notes we write to document medical consultations are too long. When I review them for my own patients, the only part I read is the consultant’s assessment and diagnostic and therapeutic recommendations. Many of my colleagues and trainees do the same.
In the old days, when medical records were handwritten, the first three pages of my hospital’s four-page consultation form were for the history, review of systems, physical examination, and test results. The top two-thirds of the last page was for diagnostic impressions and recommendations for additional testing and treatment, to be completed by the trainee performing the consultation.
This left only the bottom third of this page for attestation and additional remarks from the senior consultant. Often, this last (but most used) page was just a bullet list of diagnostic possibilities and suggested tests and treatments, with nothing about the critical reasoning underlying the differential diagnosis and recommendations. This was probably the result of fatigue from having to fill in the first three pages by hand, and then having only limited space on the final page.
Even though the written record has been replaced by the electronic medical record in my hospital, consult notes continue to be at least as long as before, without any change in the length of the assessment and recommendations section. I would guess this is true in most institutions and practices that have switched to an electronic record system.
WHY ARE CONSULT NOTES SO LONG?
The main factor contributing to the lengthy consultation document is that the Center for Medicare and Medicaid Services, with other third-party payers following suit, ties the level of reimbursement to detailed documentation of the history (present, past medical, past surgical, medications, allergies, social, and family), review of systems, and physical examination in the consultation.1 Physicians are under constant pressure from professional fee-coders to meet these requirements.
Since most of this information is already in the medical record, to require that it be documented again in the consultation note is unnecessary duplication. I believe that consultants comply with this requirement mainly to ensure adequate reimbursement, even though they know that the referring medical team will probably not read the repeated information.
Electronic medical record systems, which focus disproportionately on meeting insurers’ requirements governing reimbursement,2–5 have made it easier to create a lengthy consult note by checking boxes in templates and copying and pasting from other parts of the electronic record.2,6–12 Although verbatim copying and pasting may result in punitive audits by insurers, this practice remains common,13 including, in my experience, in consultations.
WHAT ARE THE NEGATIVE EFFECTS OF A NEEDLESSLY LONG CONSULT NOTE?
Time spent on repeating information—even if less time is required when using an electronic system—is clearly time wasted, since this part of the consult note is hardly ever read. Equally bad, the assessment and recommendations section in consult notes continues to be very short, probably because long-standing physician practices change slowly.
An ideal consult note has been described as one that, in addition to addressing the patient care issues, is as brief as possible, avoids duplication of already documented information, and has educational value to the person requesting it.14,15 The educational value of the consultation is especially important in teaching hospitals.
If the only part of the consultation perused in depth consists merely of lists of diagnoses, recommended tests, and therapy and does not include the consultant’s critical reasoning underlying them, the educational value of the consultation is lost.
HOW CAN THE FORMAT BE MADE SHORTER, YET MORE USEFUL?
The note should begin by briefly documenting the reason the consultation was requested. Ideally, institutions should train their staff to state this very specifically. For example, instead of “clearance for surgery,” it is better to ask, “Please identify risks involved in proposed surgery and suggest ways to reduce them.” The former steers the consultant to merely say “cleared for surgery, but with increased risk,” whereas the latter ensures a more specific and detailed response.
The consulting team must review in detail and verify the accuracy of all available information in the patient’s record. Once this is done, instead of repeating it, a statement that all existing information has been thoroughly reviewed should suffice, with mention in a separate paragraph of only the additional relevant positive or negative points in the history related to the issue the consultant has been asked to address.
The consultant shares with all users of the medical record the responsibility of pointing out and correcting any errors in the previously recorded information, thereby decreasing perpetuation of erroneous “chart lore,” an undesirable consequence of copying and pasting. If only previously unrecorded data and corrections to existing information are documented, the referring team is more likely to read the note because it points out relevant information that has been overlooked.
The main part of the document should consist of a detailed assessment and recommendations section, which should include not only a list of diagnoses and recommendations for testing and treatment, but also the consultant’s reasoning behind them, the results of tests already obtained that support the consultant’s conclusions, and information of value for teaching and cost-effective practice. A critically reasoned assessment and recommendation section not only will prove very educational, but by challenging the consultant to justify his or her choices, may discourage unnecessary testing and questionable therapy4,14 and thereby contribute to cost-saving.
My suggestions would not shorten the time spent by the consulting team in evaluating the patient, but only eliminate redundant documentation. I believe the consultation document will be shorter but adequate for patient care, the referring team will read and use the entire document, its educational value will be enhanced, and the time spent on redundant documentation will be saved.
A CASE VIGNETTE
The following vignette (from my own subspecialty) of a patient with acute kidney injury illustrates how a consult note can be made shorter but more useful and educational.
A 78-year-old man had a history of long-standing insulin-requiring diabetes mellitus, hypertension (treated with lisinopril and amlodipine), and benign prostatic hypertrophy. One month earlier, his blood urea nitrogen level had been 15 mg/dL and his serum creatinine had been 1.2 mg/dL.
He presented with a 3-day history of vomiting, diarrhea, and fever, presumed to be viral gastroenteritis. His blood urea nitrogen level was 100 mg, serum creatinine 2.5 mg, and blood glucose 450 mg/dL. Urinalysis revealed 2+ albuminuria, 3+ glucosuria, and 6 red blood cells per high-power field.
In the emergency department he received 2 L of normal saline and regular insulin intravenously, and an indwelling bladder catheter was inserted. He was admitted after 6 hours.
Tests obtained on arrival on the inpatient floor revealed a urinary fractional excretion of sodium of 2.5% and a blood glucose level of 295 mg/dL. His admission history and physical listed his home medications as insulin glargine, amlodipine, lisinopril, and tamsulosin. It also listed the differential diagnosis for acute kidney injury as:
- Prerenal azotemia due to volume depletion
- Rapidly progressive glomerulonephritis to be ruled out in view of proteinuria and microhematuria
- Obstructive uropathy to be ruled out.
Ultrasonography the morning after admission showed normal kidneys and no hydronephrosis. The absence of hydronephrosis was interpreted by the primary team as ruling out obstruction secondary to benign prostatic hypertrophy. The nephrology team saw the patient in consultation the day after admission and discovered the following additional information: urinalysis done 6 months earlier had also shown albuminuria and microhematuria, and the patient had been taking over-the-counter ibuprofen 400 mg three times daily for several days prior to admission.

Table 1 compares consultation documentation in the usual format and in the format I am suggesting. The revised format has much more information of educational value (eg, the importance of reviewing past urinalysis results, asking about over-the-counter medications, factors affecting fractional excretion of sodium, effect of bladder catheterization on hydronephrosis due to benign prostatic hypertrophy, and measuring urine protein only after acute kidney injury resolves). It also encourages cost-effective care (ultrasonography could have been delayed or avoided, and the patient could have been cautioned about ibuprofen-like drugs to decrease the risk of recurrent acute kidney injury).
FINAL THOUGHTS
The modifications I have suggested in consult notes will be accepted only if they are reimbursement-neutral. I hope insurers will not equate a shorter note with an opportunity to lower reimbursement and will see the value in not paying for things almost never read. I hope they will recognize and pay for the effort that went into creating a shorter document that contributes adequately to patient care, provides greater educational value, and may promote cost-effective medical practice. Also, not requiring redundant documentation may reduce or even eliminate undesirable copying and pasting.
Accountable-care organizations are an important part of the Affordable Care Act,16 which went into effect in 2014. Many organizations had already come into existence in the United States before the act became effective, and their numbers and the number of patients covered by them are projected to grow enormously over the next few years.17
Since the accountable-care organization model will rely heavily on capitated reimbursement to contain costs, these organizations are likely to scrutinize and curtail the use of consultations. I believe that a shorter consultation note—yet one that is more useful for patient care, education, and cost-containment—is more likely to pass such scrutiny, especially if it decreases time spent on documentation. Furthermore, unlike the fee-for-service model, in a capitated-payment system it may not be necessary to lengthen consultation documentation just to ensure adequate reimbursement.
- Department of Health and Human Services; Office of Inspector General. Consultations in Medicare: coding and reimbursement. http://oig.hhs.gov/oei/reports/oei-09-02-00030.pdf. Accessed November 24, 2014.
- Hartzband P, Groopman J. Off the record—avoiding the pitfalls of going electronic. N Engl J Med 2008; 358:1656–1658.
- O’Malley AS, Grossman JM, Cohen GR, Kemper NM, Pham HH. Are electronic medical records helpful for care coordination? Experiences of physician practices. J Gen Intern Med 2010; 25:177–185.
- The Center for Public Integrity; Schulte F. Electronic medical records probed for over-billing. Critics question credibility of federal panel charged with investigating. www.publicintegrity.org/2013/02/14/12208/electronic-medical-records-probed-over-billing. Accessed November 24, 2014.
- Li B. Cracking the codes: do electronic medical records facilitate hospital revenue enhancement? www.kellogg.northwestern.edu/faculty/b-li/JMP.pdf. Accessed November 24, 2014.
- Hirschtick RE. A piece of my mind. Copy-and-paste. JAMA 2006; 295:2335–2336.
- Thielke S, Hammond K, Helbig S. Copying and pasting of examinations within the electronic medical record. Int J Med Inform 2007; 76(suppl 1):S122–S128.
- Hanlon JT. The electronic medical record: diving into a shallow pool? Cleve Clin J Med 2010; 77:408–411.
- Fitzgerald FT. The emperor’s new clothes. Ann Intern Med 2012; 156:396–397.
- Bernat JL. Ethical and quality pitfalls in electronic health records. Neurology 2013; 80:1057–1061.
- Thornton JD, Schold JD, Venkateshaiah L, Lander B. Prevalence of copied information by attendings and residents in critical care progress notes. Crit Care Med 2013; 41:382–388.
- Foote RS. The challenge to the medical record. JAMA Intern Med 2013; 173:1171–1172.
- Tamburello LM. The road to EMR noncompliance and fraud is paved with cut and paste. MD Advis 2013; 6:24–30.
- Goldman L, Lee T, Rudd P. Ten commandments for effective consultations. Arch Intern Med 1983; 143:1753–1755.
- Salerno SM, Hurst FP, Halvorson S, Mercado DL. Principles of effective consultation: an update for the 21st-century consultant. Arch Intern Med 2007; 167:271–275.
- Longworth DL. Accountable care organizations, the patient-centered medical home, and health care reform: what does it all mean? Cleve Clin J Med 2011; 78:571–582.
- Meyer H. Many accountable care organizations are now up and running, if not off to the races. Health Aff (Millwood) 2012; 31:2363–2367.
After 4 decades of clinical practice in a teaching hospital, I believe that the notes we write to document medical consultations are too long. When I review them for my own patients, the only part I read is the consultant’s assessment and diagnostic and therapeutic recommendations. Many of my colleagues and trainees do the same.
In the old days, when medical records were handwritten, the first three pages of my hospital’s four-page consultation form were for the history, review of systems, physical examination, and test results. The top two-thirds of the last page was for diagnostic impressions and recommendations for additional testing and treatment, to be completed by the trainee performing the consultation.
This left only the bottom third of this page for attestation and additional remarks from the senior consultant. Often, this last (but most used) page was just a bullet list of diagnostic possibilities and suggested tests and treatments, with nothing about the critical reasoning underlying the differential diagnosis and recommendations. This was probably the result of fatigue from having to fill in the first three pages by hand, and then having only limited space on the final page.
Even though the written record has been replaced by the electronic medical record in my hospital, consult notes continue to be at least as long as before, without any change in the length of the assessment and recommendations section. I would guess this is true in most institutions and practices that have switched to an electronic record system.
WHY ARE CONSULT NOTES SO LONG?
The main factor contributing to the lengthy consultation document is that the Center for Medicare and Medicaid Services, with other third-party payers following suit, ties the level of reimbursement to detailed documentation of the history (present, past medical, past surgical, medications, allergies, social, and family), review of systems, and physical examination in the consultation.1 Physicians are under constant pressure from professional fee-coders to meet these requirements.
Since most of this information is already in the medical record, to require that it be documented again in the consultation note is unnecessary duplication. I believe that consultants comply with this requirement mainly to ensure adequate reimbursement, even though they know that the referring medical team will probably not read the repeated information.
Electronic medical record systems, which focus disproportionately on meeting insurers’ requirements governing reimbursement,2–5 have made it easier to create a lengthy consult note by checking boxes in templates and copying and pasting from other parts of the electronic record.2,6–12 Although verbatim copying and pasting may result in punitive audits by insurers, this practice remains common,13 including, in my experience, in consultations.
WHAT ARE THE NEGATIVE EFFECTS OF A NEEDLESSLY LONG CONSULT NOTE?
Time spent on repeating information—even if less time is required when using an electronic system—is clearly time wasted, since this part of the consult note is hardly ever read. Equally bad, the assessment and recommendations section in consult notes continues to be very short, probably because long-standing physician practices change slowly.
An ideal consult note has been described as one that, in addition to addressing the patient care issues, is as brief as possible, avoids duplication of already documented information, and has educational value to the person requesting it.14,15 The educational value of the consultation is especially important in teaching hospitals.
If the only part of the consultation perused in depth consists merely of lists of diagnoses, recommended tests, and therapy and does not include the consultant’s critical reasoning underlying them, the educational value of the consultation is lost.
HOW CAN THE FORMAT BE MADE SHORTER, YET MORE USEFUL?
The note should begin by briefly documenting the reason the consultation was requested. Ideally, institutions should train their staff to state this very specifically. For example, instead of “clearance for surgery,” it is better to ask, “Please identify risks involved in proposed surgery and suggest ways to reduce them.” The former steers the consultant to merely say “cleared for surgery, but with increased risk,” whereas the latter ensures a more specific and detailed response.
The consulting team must review in detail and verify the accuracy of all available information in the patient’s record. Once this is done, instead of repeating it, a statement that all existing information has been thoroughly reviewed should suffice, with mention in a separate paragraph of only the additional relevant positive or negative points in the history related to the issue the consultant has been asked to address.
The consultant shares with all users of the medical record the responsibility of pointing out and correcting any errors in the previously recorded information, thereby decreasing perpetuation of erroneous “chart lore,” an undesirable consequence of copying and pasting. If only previously unrecorded data and corrections to existing information are documented, the referring team is more likely to read the note because it points out relevant information that has been overlooked.
The main part of the document should consist of a detailed assessment and recommendations section, which should include not only a list of diagnoses and recommendations for testing and treatment, but also the consultant’s reasoning behind them, the results of tests already obtained that support the consultant’s conclusions, and information of value for teaching and cost-effective practice. A critically reasoned assessment and recommendation section not only will prove very educational, but by challenging the consultant to justify his or her choices, may discourage unnecessary testing and questionable therapy4,14 and thereby contribute to cost-saving.
My suggestions would not shorten the time spent by the consulting team in evaluating the patient, but only eliminate redundant documentation. I believe the consultation document will be shorter but adequate for patient care, the referring team will read and use the entire document, its educational value will be enhanced, and the time spent on redundant documentation will be saved.
A CASE VIGNETTE
The following vignette (from my own subspecialty) of a patient with acute kidney injury illustrates how a consult note can be made shorter but more useful and educational.
A 78-year-old man had a history of long-standing insulin-requiring diabetes mellitus, hypertension (treated with lisinopril and amlodipine), and benign prostatic hypertrophy. One month earlier, his blood urea nitrogen level had been 15 mg/dL and his serum creatinine had been 1.2 mg/dL.
He presented with a 3-day history of vomiting, diarrhea, and fever, presumed to be viral gastroenteritis. His blood urea nitrogen level was 100 mg, serum creatinine 2.5 mg, and blood glucose 450 mg/dL. Urinalysis revealed 2+ albuminuria, 3+ glucosuria, and 6 red blood cells per high-power field.
In the emergency department he received 2 L of normal saline and regular insulin intravenously, and an indwelling bladder catheter was inserted. He was admitted after 6 hours.
Tests obtained on arrival on the inpatient floor revealed a urinary fractional excretion of sodium of 2.5% and a blood glucose level of 295 mg/dL. His admission history and physical listed his home medications as insulin glargine, amlodipine, lisinopril, and tamsulosin. It also listed the differential diagnosis for acute kidney injury as:
- Prerenal azotemia due to volume depletion
- Rapidly progressive glomerulonephritis to be ruled out in view of proteinuria and microhematuria
- Obstructive uropathy to be ruled out.
Ultrasonography the morning after admission showed normal kidneys and no hydronephrosis. The absence of hydronephrosis was interpreted by the primary team as ruling out obstruction secondary to benign prostatic hypertrophy. The nephrology team saw the patient in consultation the day after admission and discovered the following additional information: urinalysis done 6 months earlier had also shown albuminuria and microhematuria, and the patient had been taking over-the-counter ibuprofen 400 mg three times daily for several days prior to admission.
Table 1 compares consultation documentation in the usual format and in the format I am suggesting. The revised format has much more information of educational value (eg, the importance of reviewing past urinalysis results, asking about over-the-counter medications, factors affecting fractional excretion of sodium, effect of bladder catheterization on hydronephrosis due to benign prostatic hypertrophy, and measuring urine protein only after acute kidney injury resolves). It also encourages cost-effective care (ultrasonography could have been delayed or avoided, and the patient could have been cautioned about ibuprofen-like drugs to decrease the risk of recurrent acute kidney injury).
FINAL THOUGHTS
The modifications I have suggested in consult notes will be accepted only if they are reimbursement-neutral. I hope insurers will not equate a shorter note with an opportunity to lower reimbursement and will see the value in not paying for things almost never read. I hope they will recognize and pay for the effort that went into creating a shorter document that contributes adequately to patient care, provides greater educational value, and may promote cost-effective medical practice. Also, not requiring redundant documentation may reduce or even eliminate undesirable copying and pasting.
Accountable-care organizations are an important part of the Affordable Care Act,16 which went into effect in 2014. Many organizations had already come into existence in the United States before the act became effective, and their numbers and the number of patients covered by them are projected to grow enormously over the next few years.17
Since the accountable-care organization model will rely heavily on capitated reimbursement to contain costs, these organizations are likely to scrutinize and curtail the use of consultations. I believe that a shorter consultation note—yet one that is more useful for patient care, education, and cost-containment—is more likely to pass such scrutiny, especially if it decreases time spent on documentation. Furthermore, unlike the fee-for-service model, in a capitated-payment system it may not be necessary to lengthen consultation documentation just to ensure adequate reimbursement.
After 4 decades of clinical practice in a teaching hospital, I believe that the notes we write to document medical consultations are too long. When I review them for my own patients, the only part I read is the consultant’s assessment and diagnostic and therapeutic recommendations. Many of my colleagues and trainees do the same.
In the old days, when medical records were handwritten, the first three pages of my hospital’s four-page consultation form were for the history, review of systems, physical examination, and test results. The top two-thirds of the last page was for diagnostic impressions and recommendations for additional testing and treatment, to be completed by the trainee performing the consultation.
This left only the bottom third of this page for attestation and additional remarks from the senior consultant. Often, this last (but most used) page was just a bullet list of diagnostic possibilities and suggested tests and treatments, with nothing about the critical reasoning underlying the differential diagnosis and recommendations. This was probably the result of fatigue from having to fill in the first three pages by hand, and then having only limited space on the final page.
Even though the written record has been replaced by the electronic medical record in my hospital, consult notes continue to be at least as long as before, without any change in the length of the assessment and recommendations section. I would guess this is true in most institutions and practices that have switched to an electronic record system.
WHY ARE CONSULT NOTES SO LONG?
The main factor contributing to the lengthy consultation document is that the Center for Medicare and Medicaid Services, with other third-party payers following suit, ties the level of reimbursement to detailed documentation of the history (present, past medical, past surgical, medications, allergies, social, and family), review of systems, and physical examination in the consultation.1 Physicians are under constant pressure from professional fee-coders to meet these requirements.
Since most of this information is already in the medical record, to require that it be documented again in the consultation note is unnecessary duplication. I believe that consultants comply with this requirement mainly to ensure adequate reimbursement, even though they know that the referring medical team will probably not read the repeated information.
Electronic medical record systems, which focus disproportionately on meeting insurers’ requirements governing reimbursement,2–5 have made it easier to create a lengthy consult note by checking boxes in templates and copying and pasting from other parts of the electronic record.2,6–12 Although verbatim copying and pasting may result in punitive audits by insurers, this practice remains common,13 including, in my experience, in consultations.
WHAT ARE THE NEGATIVE EFFECTS OF A NEEDLESSLY LONG CONSULT NOTE?
Time spent on repeating information—even if less time is required when using an electronic system—is clearly time wasted, since this part of the consult note is hardly ever read. Equally bad, the assessment and recommendations section in consult notes continues to be very short, probably because long-standing physician practices change slowly.
An ideal consult note has been described as one that, in addition to addressing the patient care issues, is as brief as possible, avoids duplication of already documented information, and has educational value to the person requesting it.14,15 The educational value of the consultation is especially important in teaching hospitals.
If the only part of the consultation perused in depth consists merely of lists of diagnoses, recommended tests, and therapy and does not include the consultant’s critical reasoning underlying them, the educational value of the consultation is lost.
HOW CAN THE FORMAT BE MADE SHORTER, YET MORE USEFUL?
The note should begin by briefly documenting the reason the consultation was requested. Ideally, institutions should train their staff to state this very specifically. For example, instead of “clearance for surgery,” it is better to ask, “Please identify risks involved in proposed surgery and suggest ways to reduce them.” The former steers the consultant to merely say “cleared for surgery, but with increased risk,” whereas the latter ensures a more specific and detailed response.
The consulting team must review in detail and verify the accuracy of all available information in the patient’s record. Once this is done, instead of repeating it, a statement that all existing information has been thoroughly reviewed should suffice, with mention in a separate paragraph of only the additional relevant positive or negative points in the history related to the issue the consultant has been asked to address.
The consultant shares with all users of the medical record the responsibility of pointing out and correcting any errors in the previously recorded information, thereby decreasing perpetuation of erroneous “chart lore,” an undesirable consequence of copying and pasting. If only previously unrecorded data and corrections to existing information are documented, the referring team is more likely to read the note because it points out relevant information that has been overlooked.
The main part of the document should consist of a detailed assessment and recommendations section, which should include not only a list of diagnoses and recommendations for testing and treatment, but also the consultant’s reasoning behind them, the results of tests already obtained that support the consultant’s conclusions, and information of value for teaching and cost-effective practice. A critically reasoned assessment and recommendation section not only will prove very educational, but by challenging the consultant to justify his or her choices, may discourage unnecessary testing and questionable therapy4,14 and thereby contribute to cost-saving.
My suggestions would not shorten the time spent by the consulting team in evaluating the patient, but only eliminate redundant documentation. I believe the consultation document will be shorter but adequate for patient care, the referring team will read and use the entire document, its educational value will be enhanced, and the time spent on redundant documentation will be saved.
A CASE VIGNETTE
The following vignette (from my own subspecialty) of a patient with acute kidney injury illustrates how a consult note can be made shorter but more useful and educational.
A 78-year-old man had a history of long-standing insulin-requiring diabetes mellitus, hypertension (treated with lisinopril and amlodipine), and benign prostatic hypertrophy. One month earlier, his blood urea nitrogen level had been 15 mg/dL and his serum creatinine had been 1.2 mg/dL.
He presented with a 3-day history of vomiting, diarrhea, and fever, presumed to be viral gastroenteritis. His blood urea nitrogen level was 100 mg, serum creatinine 2.5 mg, and blood glucose 450 mg/dL. Urinalysis revealed 2+ albuminuria, 3+ glucosuria, and 6 red blood cells per high-power field.
In the emergency department he received 2 L of normal saline and regular insulin intravenously, and an indwelling bladder catheter was inserted. He was admitted after 6 hours.
Tests obtained on arrival on the inpatient floor revealed a urinary fractional excretion of sodium of 2.5% and a blood glucose level of 295 mg/dL. His admission history and physical listed his home medications as insulin glargine, amlodipine, lisinopril, and tamsulosin. It also listed the differential diagnosis for acute kidney injury as:
- Prerenal azotemia due to volume depletion
- Rapidly progressive glomerulonephritis to be ruled out in view of proteinuria and microhematuria
- Obstructive uropathy to be ruled out.
Ultrasonography the morning after admission showed normal kidneys and no hydronephrosis. The absence of hydronephrosis was interpreted by the primary team as ruling out obstruction secondary to benign prostatic hypertrophy. The nephrology team saw the patient in consultation the day after admission and discovered the following additional information: urinalysis done 6 months earlier had also shown albuminuria and microhematuria, and the patient had been taking over-the-counter ibuprofen 400 mg three times daily for several days prior to admission.
Table 1 compares consultation documentation in the usual format and in the format I am suggesting. The revised format has much more information of educational value (eg, the importance of reviewing past urinalysis results, asking about over-the-counter medications, factors affecting fractional excretion of sodium, effect of bladder catheterization on hydronephrosis due to benign prostatic hypertrophy, and measuring urine protein only after acute kidney injury resolves). It also encourages cost-effective care (ultrasonography could have been delayed or avoided, and the patient could have been cautioned about ibuprofen-like drugs to decrease the risk of recurrent acute kidney injury).
FINAL THOUGHTS
The modifications I have suggested in consult notes will be accepted only if they are reimbursement-neutral. I hope insurers will not equate a shorter note with an opportunity to lower reimbursement and will see the value in not paying for things almost never read. I hope they will recognize and pay for the effort that went into creating a shorter document that contributes adequately to patient care, provides greater educational value, and may promote cost-effective medical practice. Also, not requiring redundant documentation may reduce or even eliminate undesirable copying and pasting.
Accountable-care organizations are an important part of the Affordable Care Act,16 which went into effect in 2014. Many organizations had already come into existence in the United States before the act became effective, and their numbers and the number of patients covered by them are projected to grow enormously over the next few years.17
Since the accountable-care organization model will rely heavily on capitated reimbursement to contain costs, these organizations are likely to scrutinize and curtail the use of consultations. I believe that a shorter consultation note—yet one that is more useful for patient care, education, and cost-containment—is more likely to pass such scrutiny, especially if it decreases time spent on documentation. Furthermore, unlike the fee-for-service model, in a capitated-payment system it may not be necessary to lengthen consultation documentation just to ensure adequate reimbursement.
- Department of Health and Human Services; Office of Inspector General. Consultations in Medicare: coding and reimbursement. http://oig.hhs.gov/oei/reports/oei-09-02-00030.pdf. Accessed November 24, 2014.
- Hartzband P, Groopman J. Off the record—avoiding the pitfalls of going electronic. N Engl J Med 2008; 358:1656–1658.
- O’Malley AS, Grossman JM, Cohen GR, Kemper NM, Pham HH. Are electronic medical records helpful for care coordination? Experiences of physician practices. J Gen Intern Med 2010; 25:177–185.
- The Center for Public Integrity; Schulte F. Electronic medical records probed for over-billing. Critics question credibility of federal panel charged with investigating. www.publicintegrity.org/2013/02/14/12208/electronic-medical-records-probed-over-billing. Accessed November 24, 2014.
- Li B. Cracking the codes: do electronic medical records facilitate hospital revenue enhancement? www.kellogg.northwestern.edu/faculty/b-li/JMP.pdf. Accessed November 24, 2014.
- Hirschtick RE. A piece of my mind. Copy-and-paste. JAMA 2006; 295:2335–2336.
- Thielke S, Hammond K, Helbig S. Copying and pasting of examinations within the electronic medical record. Int J Med Inform 2007; 76(suppl 1):S122–S128.
- Hanlon JT. The electronic medical record: diving into a shallow pool? Cleve Clin J Med 2010; 77:408–411.
- Fitzgerald FT. The emperor’s new clothes. Ann Intern Med 2012; 156:396–397.
- Bernat JL. Ethical and quality pitfalls in electronic health records. Neurology 2013; 80:1057–1061.
- Thornton JD, Schold JD, Venkateshaiah L, Lander B. Prevalence of copied information by attendings and residents in critical care progress notes. Crit Care Med 2013; 41:382–388.
- Foote RS. The challenge to the medical record. JAMA Intern Med 2013; 173:1171–1172.
- Tamburello LM. The road to EMR noncompliance and fraud is paved with cut and paste. MD Advis 2013; 6:24–30.
- Goldman L, Lee T, Rudd P. Ten commandments for effective consultations. Arch Intern Med 1983; 143:1753–1755.
- Salerno SM, Hurst FP, Halvorson S, Mercado DL. Principles of effective consultation: an update for the 21st-century consultant. Arch Intern Med 2007; 167:271–275.
- Longworth DL. Accountable care organizations, the patient-centered medical home, and health care reform: what does it all mean? Cleve Clin J Med 2011; 78:571–582.
- Meyer H. Many accountable care organizations are now up and running, if not off to the races. Health Aff (Millwood) 2012; 31:2363–2367.
- Department of Health and Human Services; Office of Inspector General. Consultations in Medicare: coding and reimbursement. http://oig.hhs.gov/oei/reports/oei-09-02-00030.pdf. Accessed November 24, 2014.
- Hartzband P, Groopman J. Off the record—avoiding the pitfalls of going electronic. N Engl J Med 2008; 358:1656–1658.
- O’Malley AS, Grossman JM, Cohen GR, Kemper NM, Pham HH. Are electronic medical records helpful for care coordination? Experiences of physician practices. J Gen Intern Med 2010; 25:177–185.
- The Center for Public Integrity; Schulte F. Electronic medical records probed for over-billing. Critics question credibility of federal panel charged with investigating. www.publicintegrity.org/2013/02/14/12208/electronic-medical-records-probed-over-billing. Accessed November 24, 2014.
- Li B. Cracking the codes: do electronic medical records facilitate hospital revenue enhancement? www.kellogg.northwestern.edu/faculty/b-li/JMP.pdf. Accessed November 24, 2014.
- Hirschtick RE. A piece of my mind. Copy-and-paste. JAMA 2006; 295:2335–2336.
- Thielke S, Hammond K, Helbig S. Copying and pasting of examinations within the electronic medical record. Int J Med Inform 2007; 76(suppl 1):S122–S128.
- Hanlon JT. The electronic medical record: diving into a shallow pool? Cleve Clin J Med 2010; 77:408–411.
- Fitzgerald FT. The emperor’s new clothes. Ann Intern Med 2012; 156:396–397.
- Bernat JL. Ethical and quality pitfalls in electronic health records. Neurology 2013; 80:1057–1061.
- Thornton JD, Schold JD, Venkateshaiah L, Lander B. Prevalence of copied information by attendings and residents in critical care progress notes. Crit Care Med 2013; 41:382–388.
- Foote RS. The challenge to the medical record. JAMA Intern Med 2013; 173:1171–1172.
- Tamburello LM. The road to EMR noncompliance and fraud is paved with cut and paste. MD Advis 2013; 6:24–30.
- Goldman L, Lee T, Rudd P. Ten commandments for effective consultations. Arch Intern Med 1983; 143:1753–1755.
- Salerno SM, Hurst FP, Halvorson S, Mercado DL. Principles of effective consultation: an update for the 21st-century consultant. Arch Intern Med 2007; 167:271–275.
- Longworth DL. Accountable care organizations, the patient-centered medical home, and health care reform: what does it all mean? Cleve Clin J Med 2011; 78:571–582.
- Meyer H. Many accountable care organizations are now up and running, if not off to the races. Health Aff (Millwood) 2012; 31:2363–2367.
A new year and a new face for www.ccjm.org

Bob Dylan’s song “The Times They Are a-Changin’” was released in January 1964. As with many things Dylan, the song’s true intent is a bit unclear, but it remains one of the most invoked lyrical symbols of change 51 years later. In 2015, the Journal, planning to “heed the call,” is changing its online visage. I hope that our intent will not be viewed as unclear.
Our mission is unchanged: to provide our readers with free access to credible, relevant, readable information, and the opportunity to earn free CME credit. So why change the website? Innovations in digital publishing, the ability to offer a broader landscape of medical information—and the chance to more effectively solicit advertising to pay for it all—prompted us to collaborate with another publisher, Frontline Medical Communications.
Frontline describes itself as health care’s largest medical communications company and as a leader in digital, print, and live events. You likely have encountered their products, which include Internal Medicine News, Cardiology News, and Clinical Endocrinology News, and CME courses such as Perspectives in Rheumatic Diseases, which I codirect. Our collaboration will allow us to offer you links to new and, we hope, interesting material. For example, our online readers will have access to MD-IQ, a popular interactive self-test, as well as brief reports and timely commentaries from specialty scientific meetings.
But even though www.ccjm.org has a new look, everything on it remains open to all and free of charge. You will still have easy access to other educational and clinical information offered by Cleveland Clinic, including information about Clinic authors. At your first visit to our revamped site you will be asked to register, but your subsequent visits will be unencumbered except for a request to sign in using your e-mail address if you log in from a different device. The e-mail address is used for identification purposes only, as site sponsors want to know the (depersonalized) demographics of our readership. You will receive occasional e-mails with links to clinical content that may interest you. If you do not wish to receive these e-mails, just follow the instructions to opt out of them. Our goal is to be unobtrusive.
Our free CME process is the same. Each CME article includes a link to the Cleveland Clinic Center for Continuing Education site with instructions on how to complete the activity. Plus, the CME pull-down menu at the top of our home page will provide easy access to all currently active journal CME offerings. We hope the transition glitches will be few and the benefits many. And the option remains for you to read, download, and print our articles in PDF format, just as you always have.
As we start the new year, we at the Journal wish you our readers a happy, healthy, peaceful, and educational 2015.
Bob Dylan’s song “The Times They Are a-Changin’” was released in January 1964. As with many things Dylan, the song’s true intent is a bit unclear, but it remains one of the most invoked lyrical symbols of change 51 years later. In 2015, the Journal, planning to “heed the call,” is changing its online visage. I hope that our intent will not be viewed as unclear.
Our mission is unchanged: to provide our readers with free access to credible, relevant, readable information, and the opportunity to earn free CME credit. So why change the website? Innovations in digital publishing, the ability to offer a broader landscape of medical information—and the chance to more effectively solicit advertising to pay for it all—prompted us to collaborate with another publisher, Frontline Medical Communications.
Frontline describes itself as health care’s largest medical communications company and as a leader in digital, print, and live events. You likely have encountered their products, which include Internal Medicine News, Cardiology News, and Clinical Endocrinology News, and CME courses such as Perspectives in Rheumatic Diseases, which I codirect. Our collaboration will allow us to offer you links to new and, we hope, interesting material. For example, our online readers will have access to MD-IQ, a popular interactive self-test, as well as brief reports and timely commentaries from specialty scientific meetings.
But even though www.ccjm.org has a new look, everything on it remains open to all and free of charge. You will still have easy access to other educational and clinical information offered by Cleveland Clinic, including information about Clinic authors. At your first visit to our revamped site you will be asked to register, but your subsequent visits will be unencumbered except for a request to sign in using your e-mail address if you log in from a different device. The e-mail address is used for identification purposes only, as site sponsors want to know the (depersonalized) demographics of our readership. You will receive occasional e-mails with links to clinical content that may interest you. If you do not wish to receive these e-mails, just follow the instructions to opt out of them. Our goal is to be unobtrusive.
Our free CME process is the same. Each CME article includes a link to the Cleveland Clinic Center for Continuing Education site with instructions on how to complete the activity. Plus, the CME pull-down menu at the top of our home page will provide easy access to all currently active journal CME offerings. We hope the transition glitches will be few and the benefits many. And the option remains for you to read, download, and print our articles in PDF format, just as you always have.
As we start the new year, we at the Journal wish you our readers a happy, healthy, peaceful, and educational 2015.
Bob Dylan’s song “The Times They Are a-Changin’” was released in January 1964. As with many things Dylan, the song’s true intent is a bit unclear, but it remains one of the most invoked lyrical symbols of change 51 years later. In 2015, the Journal, planning to “heed the call,” is changing its online visage. I hope that our intent will not be viewed as unclear.
Our mission is unchanged: to provide our readers with free access to credible, relevant, readable information, and the opportunity to earn free CME credit. So why change the website? Innovations in digital publishing, the ability to offer a broader landscape of medical information—and the chance to more effectively solicit advertising to pay for it all—prompted us to collaborate with another publisher, Frontline Medical Communications.
Frontline describes itself as health care’s largest medical communications company and as a leader in digital, print, and live events. You likely have encountered their products, which include Internal Medicine News, Cardiology News, and Clinical Endocrinology News, and CME courses such as Perspectives in Rheumatic Diseases, which I codirect. Our collaboration will allow us to offer you links to new and, we hope, interesting material. For example, our online readers will have access to MD-IQ, a popular interactive self-test, as well as brief reports and timely commentaries from specialty scientific meetings.
But even though www.ccjm.org has a new look, everything on it remains open to all and free of charge. You will still have easy access to other educational and clinical information offered by Cleveland Clinic, including information about Clinic authors. At your first visit to our revamped site you will be asked to register, but your subsequent visits will be unencumbered except for a request to sign in using your e-mail address if you log in from a different device. The e-mail address is used for identification purposes only, as site sponsors want to know the (depersonalized) demographics of our readership. You will receive occasional e-mails with links to clinical content that may interest you. If you do not wish to receive these e-mails, just follow the instructions to opt out of them. Our goal is to be unobtrusive.
Our free CME process is the same. Each CME article includes a link to the Cleveland Clinic Center for Continuing Education site with instructions on how to complete the activity. Plus, the CME pull-down menu at the top of our home page will provide easy access to all currently active journal CME offerings. We hope the transition glitches will be few and the benefits many. And the option remains for you to read, download, and print our articles in PDF format, just as you always have.
As we start the new year, we at the Journal wish you our readers a happy, healthy, peaceful, and educational 2015.