User login
January 2015 Quiz 2
ANSWER: D
Critique
Villous blunting is not specific to celiac disease. In this case, the patient’s history supports a diagnosis of common variable immunodeficiency (CVID), which is characterized by low levels of two Ig classes and recurrent infections. The infections most commonly involve the upper and lower respiratory tract.
Chronic diarrhea is seen in 40%-60% of patients and may lead to malabsorption. Diarrhea can be the result of infections (most commonly Salmonella, Campylobacter, Clostridium difficile, and Giardia lamblia), inflammatory disorders, or malignancy. Biopsies reveal villous blunting similar to that seen in celiac disease. Unlike celiac disease, however, the biopsies lack plasma cells. These patients also differ from those with celiac disease in that their celiac serologies are negative.
- Shah V.H., Rotterdam H., Kotler D.P., et al. All that scallops is not celiac disease. Gastrointest. Endoscopy 2000;51:717-20.
- Sperber K.F., Mayer L. Gastrointestinal manifestations of common variable immunodeficiency. Immunol. Allergy Clin. North Am. 1988;8:423-34.
ANSWER: D
Critique
Villous blunting is not specific to celiac disease. In this case, the patient’s history supports a diagnosis of common variable immunodeficiency (CVID), which is characterized by low levels of two Ig classes and recurrent infections. The infections most commonly involve the upper and lower respiratory tract.
Chronic diarrhea is seen in 40%-60% of patients and may lead to malabsorption. Diarrhea can be the result of infections (most commonly Salmonella, Campylobacter, Clostridium difficile, and Giardia lamblia), inflammatory disorders, or malignancy. Biopsies reveal villous blunting similar to that seen in celiac disease. Unlike celiac disease, however, the biopsies lack plasma cells. These patients also differ from those with celiac disease in that their celiac serologies are negative.
ANSWER: D
Critique
Villous blunting is not specific to celiac disease. In this case, the patient’s history supports a diagnosis of common variable immunodeficiency (CVID), which is characterized by low levels of two Ig classes and recurrent infections. The infections most commonly involve the upper and lower respiratory tract.
Chronic diarrhea is seen in 40%-60% of patients and may lead to malabsorption. Diarrhea can be the result of infections (most commonly Salmonella, Campylobacter, Clostridium difficile, and Giardia lamblia), inflammatory disorders, or malignancy. Biopsies reveal villous blunting similar to that seen in celiac disease. Unlike celiac disease, however, the biopsies lack plasma cells. These patients also differ from those with celiac disease in that their celiac serologies are negative.
- Shah V.H., Rotterdam H., Kotler D.P., et al. All that scallops is not celiac disease. Gastrointest. Endoscopy 2000;51:717-20.
- Sperber K.F., Mayer L. Gastrointestinal manifestations of common variable immunodeficiency. Immunol. Allergy Clin. North Am. 1988;8:423-34.
- Shah V.H., Rotterdam H., Kotler D.P., et al. All that scallops is not celiac disease. Gastrointest. Endoscopy 2000;51:717-20.
- Sperber K.F., Mayer L. Gastrointestinal manifestations of common variable immunodeficiency. Immunol. Allergy Clin. North Am. 1988;8:423-34.
Easy bruising • low platelet count • recent cold-like illness • Dx?
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
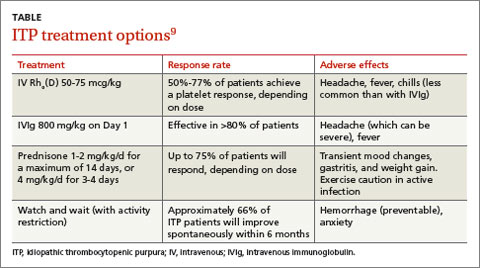
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
Hyperthyroidism • myalgia • rapidly progressing paralysis • Dx?
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
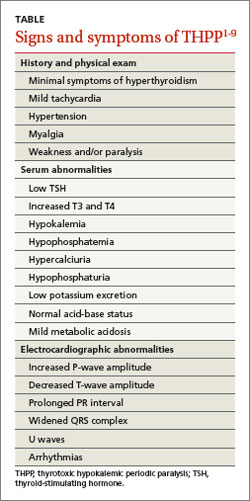
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.

Insulin therapy and cancer risk
To the Editor: We read with interest the article by Ching Sun et al1 on the relationship between diabetes therapy and cancer risk. We noted that there was no reference in the text to the long-acting insulins detemir and degludec, and we would like to add some relevant information.
With regard to detemir, a meta-analysis published in 2009 showed that patients treated with this insulin had a lower or similar rate of occurrence of a cancer compared with patients treated with neutral protamine Hagedorn insulin or insulin glargine.2 In addition, in a cohort study, no difference in cancer risk between insulin detemir users and nonusers was reported.3
Insulin detemir has a lower binding affinity for human insulin receptor isoform A (IR-A) relative to human insulin, and a much lower affinity for isoform B (IR-B). The binding affinity ratio of insulinlike growth factor-1 (IGF-1) receptor to insulin receptor for detemir is less than or equal to 1 relative to human insulin and displays a dissociation pattern from the insulin receptor that is similar to or faster than that of human insulin. Consequently, the relative mitogenic potency of detemir in cell types predominantly expressing either the IGF-1 receptor or the insulin receptor is low and corresponds to its IGF-1 receptor and insulin receptor affinities.4
Regarding insulin degludec, its affinity for both IR-A and IR-B, as well as for the IGF-1 receptor, has been found to be lower than human insulin. Its mitogenic response, in the absence of albumin, was reported to range from 4% to 14% relative to human insulin.5 Furthermore, in cellular assays, in which no albumin was added, the in vitro metabolic potency was determined to be in the range of 8% to 20%, resulting in a mitogenic-to-metabolic potency ratio of 1 or lower.5
It appears that insulins detemir and degludec have low mitogenic potential. However, additional studies are needed, especially with degludec, to further determine long-term safety.
- Ching Sun GE, Kashyap SR, Nasr C. Diabetes therapy and cancer risk: where do we stand when treating patients? Cleve Clin J Med 2014; 81:620–628.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Hansen BF, Glendorf T, Hegelund AC, et al. Molecular characterization of long-acting insulin analogues in comparison with human insulin, IGF-1 and insulin X10. PLoS One 2012; 7:e34274.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
To the Editor: We read with interest the article by Ching Sun et al1 on the relationship between diabetes therapy and cancer risk. We noted that there was no reference in the text to the long-acting insulins detemir and degludec, and we would like to add some relevant information.
With regard to detemir, a meta-analysis published in 2009 showed that patients treated with this insulin had a lower or similar rate of occurrence of a cancer compared with patients treated with neutral protamine Hagedorn insulin or insulin glargine.2 In addition, in a cohort study, no difference in cancer risk between insulin detemir users and nonusers was reported.3
Insulin detemir has a lower binding affinity for human insulin receptor isoform A (IR-A) relative to human insulin, and a much lower affinity for isoform B (IR-B). The binding affinity ratio of insulinlike growth factor-1 (IGF-1) receptor to insulin receptor for detemir is less than or equal to 1 relative to human insulin and displays a dissociation pattern from the insulin receptor that is similar to or faster than that of human insulin. Consequently, the relative mitogenic potency of detemir in cell types predominantly expressing either the IGF-1 receptor or the insulin receptor is low and corresponds to its IGF-1 receptor and insulin receptor affinities.4
Regarding insulin degludec, its affinity for both IR-A and IR-B, as well as for the IGF-1 receptor, has been found to be lower than human insulin. Its mitogenic response, in the absence of albumin, was reported to range from 4% to 14% relative to human insulin.5 Furthermore, in cellular assays, in which no albumin was added, the in vitro metabolic potency was determined to be in the range of 8% to 20%, resulting in a mitogenic-to-metabolic potency ratio of 1 or lower.5
It appears that insulins detemir and degludec have low mitogenic potential. However, additional studies are needed, especially with degludec, to further determine long-term safety.
To the Editor: We read with interest the article by Ching Sun et al1 on the relationship between diabetes therapy and cancer risk. We noted that there was no reference in the text to the long-acting insulins detemir and degludec, and we would like to add some relevant information.
With regard to detemir, a meta-analysis published in 2009 showed that patients treated with this insulin had a lower or similar rate of occurrence of a cancer compared with patients treated with neutral protamine Hagedorn insulin or insulin glargine.2 In addition, in a cohort study, no difference in cancer risk between insulin detemir users and nonusers was reported.3
Insulin detemir has a lower binding affinity for human insulin receptor isoform A (IR-A) relative to human insulin, and a much lower affinity for isoform B (IR-B). The binding affinity ratio of insulinlike growth factor-1 (IGF-1) receptor to insulin receptor for detemir is less than or equal to 1 relative to human insulin and displays a dissociation pattern from the insulin receptor that is similar to or faster than that of human insulin. Consequently, the relative mitogenic potency of detemir in cell types predominantly expressing either the IGF-1 receptor or the insulin receptor is low and corresponds to its IGF-1 receptor and insulin receptor affinities.4
Regarding insulin degludec, its affinity for both IR-A and IR-B, as well as for the IGF-1 receptor, has been found to be lower than human insulin. Its mitogenic response, in the absence of albumin, was reported to range from 4% to 14% relative to human insulin.5 Furthermore, in cellular assays, in which no albumin was added, the in vitro metabolic potency was determined to be in the range of 8% to 20%, resulting in a mitogenic-to-metabolic potency ratio of 1 or lower.5
It appears that insulins detemir and degludec have low mitogenic potential. However, additional studies are needed, especially with degludec, to further determine long-term safety.
- Ching Sun GE, Kashyap SR, Nasr C. Diabetes therapy and cancer risk: where do we stand when treating patients? Cleve Clin J Med 2014; 81:620–628.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Hansen BF, Glendorf T, Hegelund AC, et al. Molecular characterization of long-acting insulin analogues in comparison with human insulin, IGF-1 and insulin X10. PLoS One 2012; 7:e34274.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
- Ching Sun GE, Kashyap SR, Nasr C. Diabetes therapy and cancer risk: where do we stand when treating patients? Cleve Clin J Med 2014; 81:620–628.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Hansen BF, Glendorf T, Hegelund AC, et al. Molecular characterization of long-acting insulin analogues in comparison with human insulin, IGF-1 and insulin X10. PLoS One 2012; 7:e34274.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
In reply: Insulin therapy and cancer risk
In Reply: Dr. Fountas et al highlight further data on insulin therapy and cancer risk, specifically in regard to insulin detemir and insulin degludec. Detemir first gained US Food and Drug Administration (FDA) approval in 2005 as a basal insulin, dosed once or twice daily.1 Compared with regular human insulin, detemir has demonstrated proliferative and antiapoptotic activities in vitro in various cancer cell lines—eg, HCT-116 (colorectal cancer), PC-3 (prostate cancer), and MCF-7 (breast adenocarcinoma).2 But clinically, detemir has not demonstrated increased cancer risk compared with other basal insulins in randomized controlled trials or cohort studies.3–5
Degludec (U-200 insulin) is equal to twice the concentration of the usual U-100 insulin therapies presently available. In February 2013, the drug application for insulin degludec failed to obtain FDA approval, and the FDA requested additional data on cardiovascular safety. Thus, degludec is not currently available in the United States.6
Besides ameliorating nocturnal hypoglycemia,7 U-200 insulin may mitigate potential mitogenic effects.8 However, there are still very few data on degludec compared with the amount of data on insulin glargine. Insulin analogues with a decreased dissociation rate from the insulin receptor are associated with higher mitogenic potency than metabolic potency compared with human insulin.9,10 Degludec, like detemir, has an elevated dissociation rate from the insulin receptor, a low affinity for IGF-1 receptors, and a low mitogenic activity in vitro.8
At this juncture, neither detemir nor degludec has been associated with higher cancer risk, but these therapies are relatively new. And as Dr. Fountas et al indicated, their safety, particularly in regard to cancer risk in diabetes patients, should continue to be assessed.
- Levemir [package insert]. Plainsboro, NJ: Novo Nordisk Inc; 2013.
- Weinstein D, Simon M, Yehezkel E, Laron Z, Werner H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab Res Rev 2009; 25:41–49.
- Simó R, Plana-Ripoll O, Puente D, et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS One. 2013; 8:e79968.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Novo Nordisk. 2013. Novo Nordisk receives Complete Response Letter in the US for Tresiba® and Ryzodeg®. [Press release]. www.novonordisk.com/include/asp/exe_news_attachment.asp?sAttachmentGUID=83700060-0ce3-4577-a35a-f3e57801637d. Accessed December 1, 2014.
- Heller S, Buse J, Fisher M, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012; 379:1489–1497.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
- Hansen BF, Danielsen GM, Drejer K, et al. Sustained signaling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochem J 1996; 315:271–279.
- Kurtzhals P, Schäffer L, Sørensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000; 49:999–1005.
In Reply: Dr. Fountas et al highlight further data on insulin therapy and cancer risk, specifically in regard to insulin detemir and insulin degludec. Detemir first gained US Food and Drug Administration (FDA) approval in 2005 as a basal insulin, dosed once or twice daily.1 Compared with regular human insulin, detemir has demonstrated proliferative and antiapoptotic activities in vitro in various cancer cell lines—eg, HCT-116 (colorectal cancer), PC-3 (prostate cancer), and MCF-7 (breast adenocarcinoma).2 But clinically, detemir has not demonstrated increased cancer risk compared with other basal insulins in randomized controlled trials or cohort studies.3–5
Degludec (U-200 insulin) is equal to twice the concentration of the usual U-100 insulin therapies presently available. In February 2013, the drug application for insulin degludec failed to obtain FDA approval, and the FDA requested additional data on cardiovascular safety. Thus, degludec is not currently available in the United States.6
Besides ameliorating nocturnal hypoglycemia,7 U-200 insulin may mitigate potential mitogenic effects.8 However, there are still very few data on degludec compared with the amount of data on insulin glargine. Insulin analogues with a decreased dissociation rate from the insulin receptor are associated with higher mitogenic potency than metabolic potency compared with human insulin.9,10 Degludec, like detemir, has an elevated dissociation rate from the insulin receptor, a low affinity for IGF-1 receptors, and a low mitogenic activity in vitro.8
At this juncture, neither detemir nor degludec has been associated with higher cancer risk, but these therapies are relatively new. And as Dr. Fountas et al indicated, their safety, particularly in regard to cancer risk in diabetes patients, should continue to be assessed.
In Reply: Dr. Fountas et al highlight further data on insulin therapy and cancer risk, specifically in regard to insulin detemir and insulin degludec. Detemir first gained US Food and Drug Administration (FDA) approval in 2005 as a basal insulin, dosed once or twice daily.1 Compared with regular human insulin, detemir has demonstrated proliferative and antiapoptotic activities in vitro in various cancer cell lines—eg, HCT-116 (colorectal cancer), PC-3 (prostate cancer), and MCF-7 (breast adenocarcinoma).2 But clinically, detemir has not demonstrated increased cancer risk compared with other basal insulins in randomized controlled trials or cohort studies.3–5
Degludec (U-200 insulin) is equal to twice the concentration of the usual U-100 insulin therapies presently available. In February 2013, the drug application for insulin degludec failed to obtain FDA approval, and the FDA requested additional data on cardiovascular safety. Thus, degludec is not currently available in the United States.6
Besides ameliorating nocturnal hypoglycemia,7 U-200 insulin may mitigate potential mitogenic effects.8 However, there are still very few data on degludec compared with the amount of data on insulin glargine. Insulin analogues with a decreased dissociation rate from the insulin receptor are associated with higher mitogenic potency than metabolic potency compared with human insulin.9,10 Degludec, like detemir, has an elevated dissociation rate from the insulin receptor, a low affinity for IGF-1 receptors, and a low mitogenic activity in vitro.8
At this juncture, neither detemir nor degludec has been associated with higher cancer risk, but these therapies are relatively new. And as Dr. Fountas et al indicated, their safety, particularly in regard to cancer risk in diabetes patients, should continue to be assessed.
- Levemir [package insert]. Plainsboro, NJ: Novo Nordisk Inc; 2013.
- Weinstein D, Simon M, Yehezkel E, Laron Z, Werner H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab Res Rev 2009; 25:41–49.
- Simó R, Plana-Ripoll O, Puente D, et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS One. 2013; 8:e79968.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Novo Nordisk. 2013. Novo Nordisk receives Complete Response Letter in the US for Tresiba® and Ryzodeg®. [Press release]. www.novonordisk.com/include/asp/exe_news_attachment.asp?sAttachmentGUID=83700060-0ce3-4577-a35a-f3e57801637d. Accessed December 1, 2014.
- Heller S, Buse J, Fisher M, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012; 379:1489–1497.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
- Hansen BF, Danielsen GM, Drejer K, et al. Sustained signaling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochem J 1996; 315:271–279.
- Kurtzhals P, Schäffer L, Sørensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000; 49:999–1005.
- Levemir [package insert]. Plainsboro, NJ: Novo Nordisk Inc; 2013.
- Weinstein D, Simon M, Yehezkel E, Laron Z, Werner H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab Res Rev 2009; 25:41–49.
- Simó R, Plana-Ripoll O, Puente D, et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS One. 2013; 8:e79968.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Novo Nordisk. 2013. Novo Nordisk receives Complete Response Letter in the US for Tresiba® and Ryzodeg®. [Press release]. www.novonordisk.com/include/asp/exe_news_attachment.asp?sAttachmentGUID=83700060-0ce3-4577-a35a-f3e57801637d. Accessed December 1, 2014.
- Heller S, Buse J, Fisher M, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012; 379:1489–1497.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
- Hansen BF, Danielsen GM, Drejer K, et al. Sustained signaling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochem J 1996; 315:271–279.
- Kurtzhals P, Schäffer L, Sørensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000; 49:999–1005.
Management of Plasma Cell Disorders
The plasma cell disorders are a spectrum of conditions that include asymptomatic precursor conditions—monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM)—as well as symptomatic multiple myeloma (MM) and solitary plasmacytoma. Other plasma cell disorders include immunoglobulin light chain amyloidosis and POEMS syndrome, which are characterized by a unique set of end-organ manifestations. There are other related plasma cell and B-cell proliferations, such as light chain deposition disease and cryoglobulinemia, that are beyond the scope of this review but are relevant to the hematologist/oncologist and have been reviewed in detail elsewhere.
To read the full article in PDF:
The plasma cell disorders are a spectrum of conditions that include asymptomatic precursor conditions—monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM)—as well as symptomatic multiple myeloma (MM) and solitary plasmacytoma. Other plasma cell disorders include immunoglobulin light chain amyloidosis and POEMS syndrome, which are characterized by a unique set of end-organ manifestations. There are other related plasma cell and B-cell proliferations, such as light chain deposition disease and cryoglobulinemia, that are beyond the scope of this review but are relevant to the hematologist/oncologist and have been reviewed in detail elsewhere.
To read the full article in PDF:
The plasma cell disorders are a spectrum of conditions that include asymptomatic precursor conditions—monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM)—as well as symptomatic multiple myeloma (MM) and solitary plasmacytoma. Other plasma cell disorders include immunoglobulin light chain amyloidosis and POEMS syndrome, which are characterized by a unique set of end-organ manifestations. There are other related plasma cell and B-cell proliferations, such as light chain deposition disease and cryoglobulinemia, that are beyond the scope of this review but are relevant to the hematologist/oncologist and have been reviewed in detail elsewhere.
To read the full article in PDF:
Combo shows promise for rel/ref MM

Credit: Linda Bartlett
SAN FRANCISCO—Combination therapy involving a novel monoclonal antibody (mAb) produces encouraging activity in relapsed or refractory multiple myeloma (MM), according to researchers.
The team conducted a phase 1b trial testing the IgG1 mAb SAR650984 in combination with lenalidomide and dexamethasone (SAR-len-dex).
The treatment produced an overall response rate (ORR) of 58% and a higher ORR among patients who received the highest dose of SAR.
Furthermore, the combination had a “very manageable safety profile,” according to study investigator Thomas Martin, MD, of the University of California at San Francisco.
“The safety findings are really consistent with those of the individual drugs,” he said.
Dr Martin presented these findings at the 2014 ASH Annual Meeting as abstract 83.* The trial was sponsored by Sanofi (the company developing SAR), but investigators also received research funding from Karyopharm, Bristol Myers Squibb, Millennium, and Celgene.
Dr Martin explained that SAR is a humanized IgG1 mAb that binds selectively to a unique epitope on the human CD38 receptor, and it has 4 potential mechanisms of action: antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, direct apoptosis without crosslinking, and inhibition of CD38 enzyme activity.
He said there is “ample evidence” to suggest that SAR-len-dex would be active in MM. First, both lenalidomide and SAR have demonstrated single-agent activity in MM. Second, lenalidomide can increase IL-2 production, which leads to enhanced antibody-dependent cellular cytotoxicity. And finally, SAR and lenalidomide showed additive effects in a mouse model of MM.
With that in mind, Dr Martin and his colleagues decided to test SAR-len-dex in patients with relapsed or refractory MM.
The team enrolled 31 patients and administered SAR at 3 different dose levels. Patients received 3 mg/kg (n=4), 5 mg/kg (n=3), or 10 mg/kg (n=24) every 2 weeks. They received lenalidomide at 25 mg on days 1-21 per 28-day cycle and dexamethasone at 40 mg once a week on days 1, 8, 15, and 22.
The patients’ median age was 59 (range, 45-74), the median time since diagnosis was 4 years (range, 1-12), the median number of prior treatment regimens was 7 (range, 2-14), and the median number of prior lines of therapy was 4 (range, 1-11).
“The median time from the last lenalidomide-containing regimen was 9 months,” Dr Martin noted. “Ninety-four percent of the patients had prior lenalidomide, and 74% of these patients were lenalidomide refractory.”
Of the 29% of patients who had received prior pomalidomide, all were refractory to it. The same was true of the 48% of patients who received carfilzomib. And of the 94% of patients who received prior bortezomib, 52% were refractory to it.
Adverse events
The maximum-tolerated dose of SAR was not reached. Treatment-emergent adverse events occurring in 30% of patients or more included anemia, neutropenia, thrombocytopenia, febrile neutropenia, diarrhea, fatigue, insomnia, muscle spasms, nausea, pneumonia, pyrexia, and upper respiratory tract infections.
Grade 3/4 events occurring in 5% of patients or more included anemia, neutropenia, thrombocytopenia, febrile neutropenia, fatigue, insomnia, and pneumonia.
“All of these events are commonly associated with the backbone treatment of lenalidomide and dexamethasone, and no unexpected or untoward adverse events were seen,” Dr Martin noted.
The most common SAR-associated adverse events were infusion reactions. About 35% of patients experienced an infusion reaction in cycle 1, and 10% did so in cycle 2.
Most reactions were grade 1 and 2 and did not lead to treatment discontinuation. Two patients did discontinue treatment due to grade 3 infusion reactions, but both events were ultimately resolved.
Response and survival
The ORR was 58% (n=18), and the clinical benefit rate was 65% (n=20). Two patients had a stringent complete response, 7 had a very good partial response, 9 had a partial response, 2 had a minimal response, 6 had stable disease, 4 progressed, and 1 was not evaluable.
Responses were seen at all dose levels, but the best responses occurred in patients who received the highest dose of SAR. Among patients who received the highest dose, the ORR was 68%, and the clinical benefit rate was 65%.
The ORR was 50% in patients who were refractory to prior treatment with an immunomodulatory drug, 40% in patients who were refractory to carfilzomib, and 33% in patients who were refractory to pomalidomide.
At 9 months of follow-up, the median progression-free survival was 6.2 months. The median progression-free survival was not reached for patients who had received 1 to 2 prior lines of therapy, and it was 5.8 months for patients who had received 3 or more prior lines of therapy.
“SAR in combination with lenalidomide/dexamethasone showed encouraging activity in this heavily pretreated population,” Dr Martin said in closing, adding that the combination compares favorably to other treatments tested in patients who received the same number of prior lines of therapy. ![]()
*Information in the abstract differs from that presented at the meeting.
Credit: Linda Bartlett
SAN FRANCISCO—Combination therapy involving a novel monoclonal antibody (mAb) produces encouraging activity in relapsed or refractory multiple myeloma (MM), according to researchers.
The team conducted a phase 1b trial testing the IgG1 mAb SAR650984 in combination with lenalidomide and dexamethasone (SAR-len-dex).
The treatment produced an overall response rate (ORR) of 58% and a higher ORR among patients who received the highest dose of SAR.
Furthermore, the combination had a “very manageable safety profile,” according to study investigator Thomas Martin, MD, of the University of California at San Francisco.
“The safety findings are really consistent with those of the individual drugs,” he said.
Dr Martin presented these findings at the 2014 ASH Annual Meeting as abstract 83.* The trial was sponsored by Sanofi (the company developing SAR), but investigators also received research funding from Karyopharm, Bristol Myers Squibb, Millennium, and Celgene.
Dr Martin explained that SAR is a humanized IgG1 mAb that binds selectively to a unique epitope on the human CD38 receptor, and it has 4 potential mechanisms of action: antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, direct apoptosis without crosslinking, and inhibition of CD38 enzyme activity.
He said there is “ample evidence” to suggest that SAR-len-dex would be active in MM. First, both lenalidomide and SAR have demonstrated single-agent activity in MM. Second, lenalidomide can increase IL-2 production, which leads to enhanced antibody-dependent cellular cytotoxicity. And finally, SAR and lenalidomide showed additive effects in a mouse model of MM.
With that in mind, Dr Martin and his colleagues decided to test SAR-len-dex in patients with relapsed or refractory MM.
The team enrolled 31 patients and administered SAR at 3 different dose levels. Patients received 3 mg/kg (n=4), 5 mg/kg (n=3), or 10 mg/kg (n=24) every 2 weeks. They received lenalidomide at 25 mg on days 1-21 per 28-day cycle and dexamethasone at 40 mg once a week on days 1, 8, 15, and 22.
The patients’ median age was 59 (range, 45-74), the median time since diagnosis was 4 years (range, 1-12), the median number of prior treatment regimens was 7 (range, 2-14), and the median number of prior lines of therapy was 4 (range, 1-11).
“The median time from the last lenalidomide-containing regimen was 9 months,” Dr Martin noted. “Ninety-four percent of the patients had prior lenalidomide, and 74% of these patients were lenalidomide refractory.”
Of the 29% of patients who had received prior pomalidomide, all were refractory to it. The same was true of the 48% of patients who received carfilzomib. And of the 94% of patients who received prior bortezomib, 52% were refractory to it.
Adverse events
The maximum-tolerated dose of SAR was not reached. Treatment-emergent adverse events occurring in 30% of patients or more included anemia, neutropenia, thrombocytopenia, febrile neutropenia, diarrhea, fatigue, insomnia, muscle spasms, nausea, pneumonia, pyrexia, and upper respiratory tract infections.
Grade 3/4 events occurring in 5% of patients or more included anemia, neutropenia, thrombocytopenia, febrile neutropenia, fatigue, insomnia, and pneumonia.
“All of these events are commonly associated with the backbone treatment of lenalidomide and dexamethasone, and no unexpected or untoward adverse events were seen,” Dr Martin noted.
The most common SAR-associated adverse events were infusion reactions. About 35% of patients experienced an infusion reaction in cycle 1, and 10% did so in cycle 2.
Most reactions were grade 1 and 2 and did not lead to treatment discontinuation. Two patients did discontinue treatment due to grade 3 infusion reactions, but both events were ultimately resolved.
Response and survival
The ORR was 58% (n=18), and the clinical benefit rate was 65% (n=20). Two patients had a stringent complete response, 7 had a very good partial response, 9 had a partial response, 2 had a minimal response, 6 had stable disease, 4 progressed, and 1 was not evaluable.
Responses were seen at all dose levels, but the best responses occurred in patients who received the highest dose of SAR. Among patients who received the highest dose, the ORR was 68%, and the clinical benefit rate was 65%.
The ORR was 50% in patients who were refractory to prior treatment with an immunomodulatory drug, 40% in patients who were refractory to carfilzomib, and 33% in patients who were refractory to pomalidomide.
At 9 months of follow-up, the median progression-free survival was 6.2 months. The median progression-free survival was not reached for patients who had received 1 to 2 prior lines of therapy, and it was 5.8 months for patients who had received 3 or more prior lines of therapy.
“SAR in combination with lenalidomide/dexamethasone showed encouraging activity in this heavily pretreated population,” Dr Martin said in closing, adding that the combination compares favorably to other treatments tested in patients who received the same number of prior lines of therapy. ![]()
*Information in the abstract differs from that presented at the meeting.
Credit: Linda Bartlett
SAN FRANCISCO—Combination therapy involving a novel monoclonal antibody (mAb) produces encouraging activity in relapsed or refractory multiple myeloma (MM), according to researchers.
The team conducted a phase 1b trial testing the IgG1 mAb SAR650984 in combination with lenalidomide and dexamethasone (SAR-len-dex).
The treatment produced an overall response rate (ORR) of 58% and a higher ORR among patients who received the highest dose of SAR.
Furthermore, the combination had a “very manageable safety profile,” according to study investigator Thomas Martin, MD, of the University of California at San Francisco.
“The safety findings are really consistent with those of the individual drugs,” he said.
Dr Martin presented these findings at the 2014 ASH Annual Meeting as abstract 83.* The trial was sponsored by Sanofi (the company developing SAR), but investigators also received research funding from Karyopharm, Bristol Myers Squibb, Millennium, and Celgene.
Dr Martin explained that SAR is a humanized IgG1 mAb that binds selectively to a unique epitope on the human CD38 receptor, and it has 4 potential mechanisms of action: antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, direct apoptosis without crosslinking, and inhibition of CD38 enzyme activity.
He said there is “ample evidence” to suggest that SAR-len-dex would be active in MM. First, both lenalidomide and SAR have demonstrated single-agent activity in MM. Second, lenalidomide can increase IL-2 production, which leads to enhanced antibody-dependent cellular cytotoxicity. And finally, SAR and lenalidomide showed additive effects in a mouse model of MM.
With that in mind, Dr Martin and his colleagues decided to test SAR-len-dex in patients with relapsed or refractory MM.
The team enrolled 31 patients and administered SAR at 3 different dose levels. Patients received 3 mg/kg (n=4), 5 mg/kg (n=3), or 10 mg/kg (n=24) every 2 weeks. They received lenalidomide at 25 mg on days 1-21 per 28-day cycle and dexamethasone at 40 mg once a week on days 1, 8, 15, and 22.
The patients’ median age was 59 (range, 45-74), the median time since diagnosis was 4 years (range, 1-12), the median number of prior treatment regimens was 7 (range, 2-14), and the median number of prior lines of therapy was 4 (range, 1-11).
“The median time from the last lenalidomide-containing regimen was 9 months,” Dr Martin noted. “Ninety-four percent of the patients had prior lenalidomide, and 74% of these patients were lenalidomide refractory.”
Of the 29% of patients who had received prior pomalidomide, all were refractory to it. The same was true of the 48% of patients who received carfilzomib. And of the 94% of patients who received prior bortezomib, 52% were refractory to it.
Adverse events
The maximum-tolerated dose of SAR was not reached. Treatment-emergent adverse events occurring in 30% of patients or more included anemia, neutropenia, thrombocytopenia, febrile neutropenia, diarrhea, fatigue, insomnia, muscle spasms, nausea, pneumonia, pyrexia, and upper respiratory tract infections.
Grade 3/4 events occurring in 5% of patients or more included anemia, neutropenia, thrombocytopenia, febrile neutropenia, fatigue, insomnia, and pneumonia.
“All of these events are commonly associated with the backbone treatment of lenalidomide and dexamethasone, and no unexpected or untoward adverse events were seen,” Dr Martin noted.
The most common SAR-associated adverse events were infusion reactions. About 35% of patients experienced an infusion reaction in cycle 1, and 10% did so in cycle 2.
Most reactions were grade 1 and 2 and did not lead to treatment discontinuation. Two patients did discontinue treatment due to grade 3 infusion reactions, but both events were ultimately resolved.
Response and survival
The ORR was 58% (n=18), and the clinical benefit rate was 65% (n=20). Two patients had a stringent complete response, 7 had a very good partial response, 9 had a partial response, 2 had a minimal response, 6 had stable disease, 4 progressed, and 1 was not evaluable.
Responses were seen at all dose levels, but the best responses occurred in patients who received the highest dose of SAR. Among patients who received the highest dose, the ORR was 68%, and the clinical benefit rate was 65%.
The ORR was 50% in patients who were refractory to prior treatment with an immunomodulatory drug, 40% in patients who were refractory to carfilzomib, and 33% in patients who were refractory to pomalidomide.
At 9 months of follow-up, the median progression-free survival was 6.2 months. The median progression-free survival was not reached for patients who had received 1 to 2 prior lines of therapy, and it was 5.8 months for patients who had received 3 or more prior lines of therapy.
“SAR in combination with lenalidomide/dexamethasone showed encouraging activity in this heavily pretreated population,” Dr Martin said in closing, adding that the combination compares favorably to other treatments tested in patients who received the same number of prior lines of therapy. ![]()
*Information in the abstract differs from that presented at the meeting.
New data added to obinutuzumab label

Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved a supplemental biologics license application for obinutuzumab (Gazyva) in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
The approval adds to the drug’s label data from stage 2 of the CLL11 study, which showed that obinutuzumab plus chlorambucil offers significant clinical improvements when compared head-to-head with rituximab plus chlorambucil.
This includes progression-free survival (PFS), complete response (CR), and minimal residual disease (MRD) data from stage 2 of the study. In addition, overall survival data was added from stage 1, in which researchers compared obinutuzumab plus chlorambucil to chlorambucil alone.
The label now reflects that obinutuzumab plus chlorambucil improved PFS compared to rituximab plus chlorambucil. The median PFS was 26.7 months and 14.9 months, respectively (hazard ratio=0.42, P<0.0001).
Additionally, obinutuzumab plus chlorambucil nearly tripled the CR rate when compared to rituximab plus chlorambucil. The CR rates were 26.1% and 8.8%, respectively.
Of the patients who achieved a CR with or without complete recovery from abnormal blood cell counts, 19% (18/94) of patients in the obinutuzumab arm and 6% (2/34) in the rituximab arm were MRD negative in the bone marrow.
Forty-one percent (39/94) of patients in the obinutuzumab arm and 12% (4/34) in the rituximab arm were MRD-negative in the peripheral blood.
At nearly 2 years, the rate of death was 9% (22/238) for patients who received obinutuzumab plus chlorambucil and 20% (24/118) for those who received chlorambucil alone (hazard ratio=0.41). The median overall survival has not yet been reached.
About obinutuzumab
Obinutuzumab is an engineered monoclonal antibody designed to attach to CD20 on B cells. The drug attacks targeted cells both directly and together with the body’s immune system.
The prescribing information for obinutuzumab includes warnings that the drug can cause serious or life-threatening side effects. These include hepatitis B reactivation, progressive multifocal leukoencephalopathy, infusion reactions, tumor lysis syndrome, infections, and neutropenia.
The most common side effects of the drug are infusion reactions, neutropenia, thrombocytopenia, anemia, fever, cough, nausea, and diarrhea.
Obinutuzumab was FDA-approved for use in combination with chlorambucil to treat previously untreated CLL in November 2013. The drug (which is known as Gazyvaro in Europe) was approved by the European Commission for the same indication in July 2014.
Obinutuzumab was discovered by Roche Glycart AG, an independent research unit of Roche. In the US, the drug is part of a collaboration between Genentech and Biogen Idec.![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved a supplemental biologics license application for obinutuzumab (Gazyva) in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
The approval adds to the drug’s label data from stage 2 of the CLL11 study, which showed that obinutuzumab plus chlorambucil offers significant clinical improvements when compared head-to-head with rituximab plus chlorambucil.
This includes progression-free survival (PFS), complete response (CR), and minimal residual disease (MRD) data from stage 2 of the study. In addition, overall survival data was added from stage 1, in which researchers compared obinutuzumab plus chlorambucil to chlorambucil alone.
The label now reflects that obinutuzumab plus chlorambucil improved PFS compared to rituximab plus chlorambucil. The median PFS was 26.7 months and 14.9 months, respectively (hazard ratio=0.42, P<0.0001).
Additionally, obinutuzumab plus chlorambucil nearly tripled the CR rate when compared to rituximab plus chlorambucil. The CR rates were 26.1% and 8.8%, respectively.
Of the patients who achieved a CR with or without complete recovery from abnormal blood cell counts, 19% (18/94) of patients in the obinutuzumab arm and 6% (2/34) in the rituximab arm were MRD negative in the bone marrow.
Forty-one percent (39/94) of patients in the obinutuzumab arm and 12% (4/34) in the rituximab arm were MRD-negative in the peripheral blood.
At nearly 2 years, the rate of death was 9% (22/238) for patients who received obinutuzumab plus chlorambucil and 20% (24/118) for those who received chlorambucil alone (hazard ratio=0.41). The median overall survival has not yet been reached.
About obinutuzumab
Obinutuzumab is an engineered monoclonal antibody designed to attach to CD20 on B cells. The drug attacks targeted cells both directly and together with the body’s immune system.
The prescribing information for obinutuzumab includes warnings that the drug can cause serious or life-threatening side effects. These include hepatitis B reactivation, progressive multifocal leukoencephalopathy, infusion reactions, tumor lysis syndrome, infections, and neutropenia.
The most common side effects of the drug are infusion reactions, neutropenia, thrombocytopenia, anemia, fever, cough, nausea, and diarrhea.
Obinutuzumab was FDA-approved for use in combination with chlorambucil to treat previously untreated CLL in November 2013. The drug (which is known as Gazyvaro in Europe) was approved by the European Commission for the same indication in July 2014.
Obinutuzumab was discovered by Roche Glycart AG, an independent research unit of Roche. In the US, the drug is part of a collaboration between Genentech and Biogen Idec.![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved a supplemental biologics license application for obinutuzumab (Gazyva) in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
The approval adds to the drug’s label data from stage 2 of the CLL11 study, which showed that obinutuzumab plus chlorambucil offers significant clinical improvements when compared head-to-head with rituximab plus chlorambucil.
This includes progression-free survival (PFS), complete response (CR), and minimal residual disease (MRD) data from stage 2 of the study. In addition, overall survival data was added from stage 1, in which researchers compared obinutuzumab plus chlorambucil to chlorambucil alone.
The label now reflects that obinutuzumab plus chlorambucil improved PFS compared to rituximab plus chlorambucil. The median PFS was 26.7 months and 14.9 months, respectively (hazard ratio=0.42, P<0.0001).
Additionally, obinutuzumab plus chlorambucil nearly tripled the CR rate when compared to rituximab plus chlorambucil. The CR rates were 26.1% and 8.8%, respectively.
Of the patients who achieved a CR with or without complete recovery from abnormal blood cell counts, 19% (18/94) of patients in the obinutuzumab arm and 6% (2/34) in the rituximab arm were MRD negative in the bone marrow.
Forty-one percent (39/94) of patients in the obinutuzumab arm and 12% (4/34) in the rituximab arm were MRD-negative in the peripheral blood.
At nearly 2 years, the rate of death was 9% (22/238) for patients who received obinutuzumab plus chlorambucil and 20% (24/118) for those who received chlorambucil alone (hazard ratio=0.41). The median overall survival has not yet been reached.
About obinutuzumab
Obinutuzumab is an engineered monoclonal antibody designed to attach to CD20 on B cells. The drug attacks targeted cells both directly and together with the body’s immune system.
The prescribing information for obinutuzumab includes warnings that the drug can cause serious or life-threatening side effects. These include hepatitis B reactivation, progressive multifocal leukoencephalopathy, infusion reactions, tumor lysis syndrome, infections, and neutropenia.
The most common side effects of the drug are infusion reactions, neutropenia, thrombocytopenia, anemia, fever, cough, nausea, and diarrhea.
Obinutuzumab was FDA-approved for use in combination with chlorambucil to treat previously untreated CLL in November 2013. The drug (which is known as Gazyvaro in Europe) was approved by the European Commission for the same indication in July 2014.
Obinutuzumab was discovered by Roche Glycart AG, an independent research unit of Roche. In the US, the drug is part of a collaboration between Genentech and Biogen Idec.![]()
Malpractice Counsel
Stroke in a Young Man
A 26-year-old man presented to the ED with the chief complaint of mild right-sided weakness, paresthesias, and slurred speech. He stated the onset was sudden—approximately 30 minutes prior to arrival to the ED. The patient denied any previous similar symptoms and was otherwise in good health; he denied taking any medications. He drank alcohol socially, but denied smoking or illicit drug use.
On physical examination, his vital signs and oxygen saturation were normal. Pulmonary, cardiovascular, and abdominal examinations were also normal. The patient thought his speech was somewhat slurred, but the triage nurse and treating emergency physician (EP) had difficulty detecting any altered speech. He was noted to have mild (4+/5) right upper and lower extremity weakness; no facial droop was detected. The patient did have a mild pronator drift of the right upper extremity. Gait testing revealed a mild limp of the right lower extremity.
The EP consulted the hospitalist, and the patient was admitted to a monitored bed. The following morning, a brain magnetic resonance image revealed an ischemic stroke in the distribution of the left middle cerebral artery. The patient’s hospital course was uncomplicated, but at the time of discharge, he continued to have mild right-sided weakness and required the use of a cane.
The patient sued the hospital and the EP for negligence in failing to treat his condition in a timely manner and for not consulting a neurologist. The plaintiff’s attorneys argued the patient should have been given tissue plasminogen activator (tPA), which would have avoided the residual right-sided weakness. The defense denied negligence and argued the patient’s symptoms could have been due to several things for which tPA would have been an inappropriate treatment. A defense verdict was returned.
Discussion
Stroke in young patients is relatively rare. With “young” defined as aged 18 to 45 years, this population accounts for approximately 2% to 12% of cerebral infarcts.1 In one nationwide US study of stroke in young adults, Ellis2 found that 4.9% of individuals experiencing a stroke in 2007 were between ages 18 and 44 years. Among this group, 78% experienced an ischemic stroke; 11.2% experienced a subarachnoid hemorrhage (SAH); and 10.8% had an intracerebral hemorrhage.2
While the clinical presentation of stroke in young adults is similar to that of older patients, the etiologies and risk factors are very different. In older patients, atherosclerosis is the major cause of ischemic stroke. In studies of young adults with ischemic stroke, cardioembolism was found to be the leading cause. Under this category, a patent foramen ovale (PFO) was considered a common cause, followed by atrial fibrillation, bacterial endocarditis, rheumatic heart disease, and atrial myxoma. There is, however, increasing controversy over the role of PFO as an etiology of stroke. Many investigators think its role has been overstated and is probably more of an incidental finding than a causal relationship.3 Patients with a suspected cardioembolic etiology will usually require an echocardiogram (with saline contrast or a “bubble study” for suspected PFO), cardiac monitoring, and a possible Holter monitor at the time of discharge (to detect paroxysmal arrhythmias).
Following cardioembolic etiologies, arterial dissection is the next most common category.4 In one study of patients aged 31 to 45 years old, arterial dissection was the most common cause of ischemic stroke.4 Clinical features suggesting dissection include a history of head or neck trauma (even minor trauma), headache or neck pain, and local neurological findings (eg, cranial nerve palsy or Horner syndrome).3 Unfortunately, only about 25% of patients volunteer a history of recent neck trauma. If a cervical or vertebral artery dissection is suspected, contrast enhanced magnetic resonance angiography (MRA) is the most sensitive and specific test, followed by carotid ultrasound and CT angiography.3
Traditional risk factors for stroke include hypertension and diabetes mellitus (DM). This is not true for younger adults that experience an ischemic stroke. Cigarette smoking is a very important risk factor for cerebrovascular accident in young adults; in addition, the more one smokes, the greater the risk. Other risk factors in young adults include history of migraine headaches (especially migraine with aura), pregnancy and the postpartum period, and illicit drug use.3
The defense’s argument that there are many causes of stroke in young adults that would be inappropriate for treatment with tPA, such as a PFO, carotid dissection or bacterial endocarditis, is absolutely true. Young patients need to be aggressively worked up for the etiology of their stroke, and may require additional testing, such as an MRA, echocardiogram, or Holter monitoring to determine the underlying cause of their stroke.
Obstruction Following Gastric Bypass Surgery
A 47-year-old woman presented to the ED complaining of severe back and abdominal pain. Onset had been gradual and began approximately 4 hours prior to arrival. She described the pain as crampy and constant. The patient had vomited twice; she denied diarrhea and had a normal bowel movement the previous day. She denied any vaginal or urinary complaints. Her past medical history was significant for hypertension and status post gastric bypass surgery 6 months prior. She had lost 42 pounds to date. She denied smoking or alcohol use.
The patient’s vital signs on physical examination were: blood pressure, 154/92 mm Hg; pulse, 106 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 99˚F. Oxygen saturation was 96% on room air. The patient’s lungs were clear to auscultation bilaterally. The heart was mildly tachycardic, with a regular rhythm and without murmurs, rubs, or gallops. The abdominal examination revealed diffuse tenderness and involuntary guarding. There was no distention or rebound. Bowel sounds were present but hypoactive. Examination of the back revealed bilateral paraspinal muscle tenderness without costovertebral angle tenderness.
The EP ordered a CBC, BMP, serum lipase, and a urinalysis. The patient was given an intravenous (IV) bolus of 250 cc normal saline in addition to IV morphine 4 mg and IV ondansetron 4 mg. Her white blood cell (WBC) count was slightly elevated at 12.2 g/dL, with a normal differential. The remainder of the laboratory studies were normal, except for a serum bicarbonate of 22 mmol/L.
The patient stated she felt somewhat improved, but continued to have abdominal and back pain. The EP admitted her to the hospital for observation and pain control. She died the following day from a bowel obstruction. The family sued the EP for negligence in failing to order appropriate testing and for not consulting with specialists to diagnose the bowel obstruction, which is a known complication of gastric bypass surgery. The jury returned a verdict of $2.4 million against the EP.
Discussion
The frequency of bariatric surgery in the United States continues to increase, primarily due to its success with regard to weight loss, but also because of its demonstrated improvement in hypertension, obstructive sleep apnea, hyperlipidemia, and type 2 DM.1

Frequently, the term “gastric bypass surgery” is used interchangeably with bariatric surgery. However, the EP must realize these terms encompass multiple different operations. The four most common types of bariatric surgery in the United Stated are (1) adjustable gastric banding (AGB); (2) the Roux-en-Y gastric bypass (RYGB); (3) biliopancreatic diversion with duodenal switch (BPD-DS); and (4) vertical sleeve gastrectomy (VSG).2 (See the Table for a brief explanation of each type of procedure.)
Since each procedure has its own respective associated complications, it is important for the EP to know which the type of gastric bypass surgery the patient had. For example, leakage is much more frequent following RYGB than in gastric banding, while slippage and obstruction are the most common complications of gastric banding.3,4 It is also very helpful to know the specific type of procedure when discussing the case with the surgical consultant.
Based on a recent review of over 800,000 bariatric surgery patients, seven serious common complications following the surgery were identified.3 These included bleeding, leakage, obstruction, stomal ulceration, pulmonary embolism and respiratory complications, blood sugar disturbances (usually hypoglycemia and/or metabolic acidosis), and nutritional disturbances. While not all-inclusive, this list represents the most common serious complications of gastric bypass surgery.
The complaint of abdominal pain in a patient that has undergone bariatric surgery should be taken very seriously. In addition to determining the specific procedure performed and date, the patient should be questioned about vomiting, bowel movements, and the presence of blood in stool or vomit. Depending upon the degree of pain present, the patient may need to be given IV opioid analgesia to facilitate a thorough abdominal examination. A rectal examination should be performed to identify occult gastrointestinal bleeding.
These patients require laboratory testing, including CBC, BMP, and other laboratory evaluation as indicated by the history and physical examination. Early consultation with the bariatric surgeon is recommended. Many, if not most, patients with abdominal pain and vomiting will require imaging, usually a CT scan with contrast of the abdomen and pelvis. Because of the difficulty in interpreting the CT scan results in these patients, the bariatric surgeon will often want to personally review the films rather than rely solely on the interpretation by radiology services.
Unfortunately, the EP in this case did not appreciate the seriousness of the situation. The presence of severe abdominal pain, tenderness, guarding, mild tachycardia with leukocytosis, and metabolic acidosis all pointed to a more serious etiology than muscle spasm. This patient required IV fluids, analgesia, and imaging, as well as consultation with the bariatric surgeon.
- Chatzikonstantinou A, Wolf ME, Hennerici MG. Ischemic stroke in young adults: classification and risk factors. J Neurol. 2012;259(4):653-659.
- Ellis C. Stroke in young adults. Disabil Health J. 2010;3(3):222-224.
- Ferro JM, Massaro AR, Mas JL. Aetiological diagnosis of ischemic stroke in young adults. Lancet Neurol. 2010;9(11):1085-1096.
- Chan MT, Nadareishvili ZG, Norris JW; Canadian Stroke Consortium. Diagnostic strategies in young patients with ischemic stroke in Canada. Can J Neurol Sci. 2000;27(2):120-124.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004;292(14):1724-1737.
- Livingston EH. Patient guide: Endocrine and nutritional management after bariatric surgery: A patient’s guide. Hormone Health Network Web site. http://www.hormone.org/~/media/Hormone/Files/Patient%20Guides/Mens%20Health/PGBariatricSurgery_2014.pdf. Accessed December 17, 2014.
- Hussain A, El-Hasani S. Bariatric emergencies: current evidence and strategies of management. World J Emerg Surg. 2013;8(1):58.
- Campanille FC, Boru C, Rizzello M, et al. Acute complications after laparoscopic bariatric procedures: update for the general surgeon. Langenbecks Arch Surg. 2013;398(5):669-686
Stroke in a Young Man
A 26-year-old man presented to the ED with the chief complaint of mild right-sided weakness, paresthesias, and slurred speech. He stated the onset was sudden—approximately 30 minutes prior to arrival to the ED. The patient denied any previous similar symptoms and was otherwise in good health; he denied taking any medications. He drank alcohol socially, but denied smoking or illicit drug use.
On physical examination, his vital signs and oxygen saturation were normal. Pulmonary, cardiovascular, and abdominal examinations were also normal. The patient thought his speech was somewhat slurred, but the triage nurse and treating emergency physician (EP) had difficulty detecting any altered speech. He was noted to have mild (4+/5) right upper and lower extremity weakness; no facial droop was detected. The patient did have a mild pronator drift of the right upper extremity. Gait testing revealed a mild limp of the right lower extremity.
The EP consulted the hospitalist, and the patient was admitted to a monitored bed. The following morning, a brain magnetic resonance image revealed an ischemic stroke in the distribution of the left middle cerebral artery. The patient’s hospital course was uncomplicated, but at the time of discharge, he continued to have mild right-sided weakness and required the use of a cane.
The patient sued the hospital and the EP for negligence in failing to treat his condition in a timely manner and for not consulting a neurologist. The plaintiff’s attorneys argued the patient should have been given tissue plasminogen activator (tPA), which would have avoided the residual right-sided weakness. The defense denied negligence and argued the patient’s symptoms could have been due to several things for which tPA would have been an inappropriate treatment. A defense verdict was returned.
Discussion
Stroke in young patients is relatively rare. With “young” defined as aged 18 to 45 years, this population accounts for approximately 2% to 12% of cerebral infarcts.1 In one nationwide US study of stroke in young adults, Ellis2 found that 4.9% of individuals experiencing a stroke in 2007 were between ages 18 and 44 years. Among this group, 78% experienced an ischemic stroke; 11.2% experienced a subarachnoid hemorrhage (SAH); and 10.8% had an intracerebral hemorrhage.2
While the clinical presentation of stroke in young adults is similar to that of older patients, the etiologies and risk factors are very different. In older patients, atherosclerosis is the major cause of ischemic stroke. In studies of young adults with ischemic stroke, cardioembolism was found to be the leading cause. Under this category, a patent foramen ovale (PFO) was considered a common cause, followed by atrial fibrillation, bacterial endocarditis, rheumatic heart disease, and atrial myxoma. There is, however, increasing controversy over the role of PFO as an etiology of stroke. Many investigators think its role has been overstated and is probably more of an incidental finding than a causal relationship.3 Patients with a suspected cardioembolic etiology will usually require an echocardiogram (with saline contrast or a “bubble study” for suspected PFO), cardiac monitoring, and a possible Holter monitor at the time of discharge (to detect paroxysmal arrhythmias).
Following cardioembolic etiologies, arterial dissection is the next most common category.4 In one study of patients aged 31 to 45 years old, arterial dissection was the most common cause of ischemic stroke.4 Clinical features suggesting dissection include a history of head or neck trauma (even minor trauma), headache or neck pain, and local neurological findings (eg, cranial nerve palsy or Horner syndrome).3 Unfortunately, only about 25% of patients volunteer a history of recent neck trauma. If a cervical or vertebral artery dissection is suspected, contrast enhanced magnetic resonance angiography (MRA) is the most sensitive and specific test, followed by carotid ultrasound and CT angiography.3
Traditional risk factors for stroke include hypertension and diabetes mellitus (DM). This is not true for younger adults that experience an ischemic stroke. Cigarette smoking is a very important risk factor for cerebrovascular accident in young adults; in addition, the more one smokes, the greater the risk. Other risk factors in young adults include history of migraine headaches (especially migraine with aura), pregnancy and the postpartum period, and illicit drug use.3
The defense’s argument that there are many causes of stroke in young adults that would be inappropriate for treatment with tPA, such as a PFO, carotid dissection or bacterial endocarditis, is absolutely true. Young patients need to be aggressively worked up for the etiology of their stroke, and may require additional testing, such as an MRA, echocardiogram, or Holter monitoring to determine the underlying cause of their stroke.
Obstruction Following Gastric Bypass Surgery
A 47-year-old woman presented to the ED complaining of severe back and abdominal pain. Onset had been gradual and began approximately 4 hours prior to arrival. She described the pain as crampy and constant. The patient had vomited twice; she denied diarrhea and had a normal bowel movement the previous day. She denied any vaginal or urinary complaints. Her past medical history was significant for hypertension and status post gastric bypass surgery 6 months prior. She had lost 42 pounds to date. She denied smoking or alcohol use.
The patient’s vital signs on physical examination were: blood pressure, 154/92 mm Hg; pulse, 106 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 99˚F. Oxygen saturation was 96% on room air. The patient’s lungs were clear to auscultation bilaterally. The heart was mildly tachycardic, with a regular rhythm and without murmurs, rubs, or gallops. The abdominal examination revealed diffuse tenderness and involuntary guarding. There was no distention or rebound. Bowel sounds were present but hypoactive. Examination of the back revealed bilateral paraspinal muscle tenderness without costovertebral angle tenderness.
The EP ordered a CBC, BMP, serum lipase, and a urinalysis. The patient was given an intravenous (IV) bolus of 250 cc normal saline in addition to IV morphine 4 mg and IV ondansetron 4 mg. Her white blood cell (WBC) count was slightly elevated at 12.2 g/dL, with a normal differential. The remainder of the laboratory studies were normal, except for a serum bicarbonate of 22 mmol/L.
The patient stated she felt somewhat improved, but continued to have abdominal and back pain. The EP admitted her to the hospital for observation and pain control. She died the following day from a bowel obstruction. The family sued the EP for negligence in failing to order appropriate testing and for not consulting with specialists to diagnose the bowel obstruction, which is a known complication of gastric bypass surgery. The jury returned a verdict of $2.4 million against the EP.
Discussion
The frequency of bariatric surgery in the United States continues to increase, primarily due to its success with regard to weight loss, but also because of its demonstrated improvement in hypertension, obstructive sleep apnea, hyperlipidemia, and type 2 DM.1
Frequently, the term “gastric bypass surgery” is used interchangeably with bariatric surgery. However, the EP must realize these terms encompass multiple different operations. The four most common types of bariatric surgery in the United Stated are (1) adjustable gastric banding (AGB); (2) the Roux-en-Y gastric bypass (RYGB); (3) biliopancreatic diversion with duodenal switch (BPD-DS); and (4) vertical sleeve gastrectomy (VSG).2 (See the Table for a brief explanation of each type of procedure.)
Since each procedure has its own respective associated complications, it is important for the EP to know which the type of gastric bypass surgery the patient had. For example, leakage is much more frequent following RYGB than in gastric banding, while slippage and obstruction are the most common complications of gastric banding.3,4 It is also very helpful to know the specific type of procedure when discussing the case with the surgical consultant.
Based on a recent review of over 800,000 bariatric surgery patients, seven serious common complications following the surgery were identified.3 These included bleeding, leakage, obstruction, stomal ulceration, pulmonary embolism and respiratory complications, blood sugar disturbances (usually hypoglycemia and/or metabolic acidosis), and nutritional disturbances. While not all-inclusive, this list represents the most common serious complications of gastric bypass surgery.
The complaint of abdominal pain in a patient that has undergone bariatric surgery should be taken very seriously. In addition to determining the specific procedure performed and date, the patient should be questioned about vomiting, bowel movements, and the presence of blood in stool or vomit. Depending upon the degree of pain present, the patient may need to be given IV opioid analgesia to facilitate a thorough abdominal examination. A rectal examination should be performed to identify occult gastrointestinal bleeding.
These patients require laboratory testing, including CBC, BMP, and other laboratory evaluation as indicated by the history and physical examination. Early consultation with the bariatric surgeon is recommended. Many, if not most, patients with abdominal pain and vomiting will require imaging, usually a CT scan with contrast of the abdomen and pelvis. Because of the difficulty in interpreting the CT scan results in these patients, the bariatric surgeon will often want to personally review the films rather than rely solely on the interpretation by radiology services.
Unfortunately, the EP in this case did not appreciate the seriousness of the situation. The presence of severe abdominal pain, tenderness, guarding, mild tachycardia with leukocytosis, and metabolic acidosis all pointed to a more serious etiology than muscle spasm. This patient required IV fluids, analgesia, and imaging, as well as consultation with the bariatric surgeon.
Stroke in a Young Man
A 26-year-old man presented to the ED with the chief complaint of mild right-sided weakness, paresthesias, and slurred speech. He stated the onset was sudden—approximately 30 minutes prior to arrival to the ED. The patient denied any previous similar symptoms and was otherwise in good health; he denied taking any medications. He drank alcohol socially, but denied smoking or illicit drug use.
On physical examination, his vital signs and oxygen saturation were normal. Pulmonary, cardiovascular, and abdominal examinations were also normal. The patient thought his speech was somewhat slurred, but the triage nurse and treating emergency physician (EP) had difficulty detecting any altered speech. He was noted to have mild (4+/5) right upper and lower extremity weakness; no facial droop was detected. The patient did have a mild pronator drift of the right upper extremity. Gait testing revealed a mild limp of the right lower extremity.
The EP consulted the hospitalist, and the patient was admitted to a monitored bed. The following morning, a brain magnetic resonance image revealed an ischemic stroke in the distribution of the left middle cerebral artery. The patient’s hospital course was uncomplicated, but at the time of discharge, he continued to have mild right-sided weakness and required the use of a cane.
The patient sued the hospital and the EP for negligence in failing to treat his condition in a timely manner and for not consulting a neurologist. The plaintiff’s attorneys argued the patient should have been given tissue plasminogen activator (tPA), which would have avoided the residual right-sided weakness. The defense denied negligence and argued the patient’s symptoms could have been due to several things for which tPA would have been an inappropriate treatment. A defense verdict was returned.
Discussion
Stroke in young patients is relatively rare. With “young” defined as aged 18 to 45 years, this population accounts for approximately 2% to 12% of cerebral infarcts.1 In one nationwide US study of stroke in young adults, Ellis2 found that 4.9% of individuals experiencing a stroke in 2007 were between ages 18 and 44 years. Among this group, 78% experienced an ischemic stroke; 11.2% experienced a subarachnoid hemorrhage (SAH); and 10.8% had an intracerebral hemorrhage.2
While the clinical presentation of stroke in young adults is similar to that of older patients, the etiologies and risk factors are very different. In older patients, atherosclerosis is the major cause of ischemic stroke. In studies of young adults with ischemic stroke, cardioembolism was found to be the leading cause. Under this category, a patent foramen ovale (PFO) was considered a common cause, followed by atrial fibrillation, bacterial endocarditis, rheumatic heart disease, and atrial myxoma. There is, however, increasing controversy over the role of PFO as an etiology of stroke. Many investigators think its role has been overstated and is probably more of an incidental finding than a causal relationship.3 Patients with a suspected cardioembolic etiology will usually require an echocardiogram (with saline contrast or a “bubble study” for suspected PFO), cardiac monitoring, and a possible Holter monitor at the time of discharge (to detect paroxysmal arrhythmias).
Following cardioembolic etiologies, arterial dissection is the next most common category.4 In one study of patients aged 31 to 45 years old, arterial dissection was the most common cause of ischemic stroke.4 Clinical features suggesting dissection include a history of head or neck trauma (even minor trauma), headache or neck pain, and local neurological findings (eg, cranial nerve palsy or Horner syndrome).3 Unfortunately, only about 25% of patients volunteer a history of recent neck trauma. If a cervical or vertebral artery dissection is suspected, contrast enhanced magnetic resonance angiography (MRA) is the most sensitive and specific test, followed by carotid ultrasound and CT angiography.3
Traditional risk factors for stroke include hypertension and diabetes mellitus (DM). This is not true for younger adults that experience an ischemic stroke. Cigarette smoking is a very important risk factor for cerebrovascular accident in young adults; in addition, the more one smokes, the greater the risk. Other risk factors in young adults include history of migraine headaches (especially migraine with aura), pregnancy and the postpartum period, and illicit drug use.3
The defense’s argument that there are many causes of stroke in young adults that would be inappropriate for treatment with tPA, such as a PFO, carotid dissection or bacterial endocarditis, is absolutely true. Young patients need to be aggressively worked up for the etiology of their stroke, and may require additional testing, such as an MRA, echocardiogram, or Holter monitoring to determine the underlying cause of their stroke.
Obstruction Following Gastric Bypass Surgery
A 47-year-old woman presented to the ED complaining of severe back and abdominal pain. Onset had been gradual and began approximately 4 hours prior to arrival. She described the pain as crampy and constant. The patient had vomited twice; she denied diarrhea and had a normal bowel movement the previous day. She denied any vaginal or urinary complaints. Her past medical history was significant for hypertension and status post gastric bypass surgery 6 months prior. She had lost 42 pounds to date. She denied smoking or alcohol use.
The patient’s vital signs on physical examination were: blood pressure, 154/92 mm Hg; pulse, 106 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 99˚F. Oxygen saturation was 96% on room air. The patient’s lungs were clear to auscultation bilaterally. The heart was mildly tachycardic, with a regular rhythm and without murmurs, rubs, or gallops. The abdominal examination revealed diffuse tenderness and involuntary guarding. There was no distention or rebound. Bowel sounds were present but hypoactive. Examination of the back revealed bilateral paraspinal muscle tenderness without costovertebral angle tenderness.
The EP ordered a CBC, BMP, serum lipase, and a urinalysis. The patient was given an intravenous (IV) bolus of 250 cc normal saline in addition to IV morphine 4 mg and IV ondansetron 4 mg. Her white blood cell (WBC) count was slightly elevated at 12.2 g/dL, with a normal differential. The remainder of the laboratory studies were normal, except for a serum bicarbonate of 22 mmol/L.
The patient stated she felt somewhat improved, but continued to have abdominal and back pain. The EP admitted her to the hospital for observation and pain control. She died the following day from a bowel obstruction. The family sued the EP for negligence in failing to order appropriate testing and for not consulting with specialists to diagnose the bowel obstruction, which is a known complication of gastric bypass surgery. The jury returned a verdict of $2.4 million against the EP.
Discussion
The frequency of bariatric surgery in the United States continues to increase, primarily due to its success with regard to weight loss, but also because of its demonstrated improvement in hypertension, obstructive sleep apnea, hyperlipidemia, and type 2 DM.1
Frequently, the term “gastric bypass surgery” is used interchangeably with bariatric surgery. However, the EP must realize these terms encompass multiple different operations. The four most common types of bariatric surgery in the United Stated are (1) adjustable gastric banding (AGB); (2) the Roux-en-Y gastric bypass (RYGB); (3) biliopancreatic diversion with duodenal switch (BPD-DS); and (4) vertical sleeve gastrectomy (VSG).2 (See the Table for a brief explanation of each type of procedure.)
Since each procedure has its own respective associated complications, it is important for the EP to know which the type of gastric bypass surgery the patient had. For example, leakage is much more frequent following RYGB than in gastric banding, while slippage and obstruction are the most common complications of gastric banding.3,4 It is also very helpful to know the specific type of procedure when discussing the case with the surgical consultant.
Based on a recent review of over 800,000 bariatric surgery patients, seven serious common complications following the surgery were identified.3 These included bleeding, leakage, obstruction, stomal ulceration, pulmonary embolism and respiratory complications, blood sugar disturbances (usually hypoglycemia and/or metabolic acidosis), and nutritional disturbances. While not all-inclusive, this list represents the most common serious complications of gastric bypass surgery.
The complaint of abdominal pain in a patient that has undergone bariatric surgery should be taken very seriously. In addition to determining the specific procedure performed and date, the patient should be questioned about vomiting, bowel movements, and the presence of blood in stool or vomit. Depending upon the degree of pain present, the patient may need to be given IV opioid analgesia to facilitate a thorough abdominal examination. A rectal examination should be performed to identify occult gastrointestinal bleeding.
These patients require laboratory testing, including CBC, BMP, and other laboratory evaluation as indicated by the history and physical examination. Early consultation with the bariatric surgeon is recommended. Many, if not most, patients with abdominal pain and vomiting will require imaging, usually a CT scan with contrast of the abdomen and pelvis. Because of the difficulty in interpreting the CT scan results in these patients, the bariatric surgeon will often want to personally review the films rather than rely solely on the interpretation by radiology services.
Unfortunately, the EP in this case did not appreciate the seriousness of the situation. The presence of severe abdominal pain, tenderness, guarding, mild tachycardia with leukocytosis, and metabolic acidosis all pointed to a more serious etiology than muscle spasm. This patient required IV fluids, analgesia, and imaging, as well as consultation with the bariatric surgeon.
- Chatzikonstantinou A, Wolf ME, Hennerici MG. Ischemic stroke in young adults: classification and risk factors. J Neurol. 2012;259(4):653-659.
- Ellis C. Stroke in young adults. Disabil Health J. 2010;3(3):222-224.
- Ferro JM, Massaro AR, Mas JL. Aetiological diagnosis of ischemic stroke in young adults. Lancet Neurol. 2010;9(11):1085-1096.
- Chan MT, Nadareishvili ZG, Norris JW; Canadian Stroke Consortium. Diagnostic strategies in young patients with ischemic stroke in Canada. Can J Neurol Sci. 2000;27(2):120-124.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004;292(14):1724-1737.
- Livingston EH. Patient guide: Endocrine and nutritional management after bariatric surgery: A patient’s guide. Hormone Health Network Web site. http://www.hormone.org/~/media/Hormone/Files/Patient%20Guides/Mens%20Health/PGBariatricSurgery_2014.pdf. Accessed December 17, 2014.
- Hussain A, El-Hasani S. Bariatric emergencies: current evidence and strategies of management. World J Emerg Surg. 2013;8(1):58.
- Campanille FC, Boru C, Rizzello M, et al. Acute complications after laparoscopic bariatric procedures: update for the general surgeon. Langenbecks Arch Surg. 2013;398(5):669-686
- Chatzikonstantinou A, Wolf ME, Hennerici MG. Ischemic stroke in young adults: classification and risk factors. J Neurol. 2012;259(4):653-659.
- Ellis C. Stroke in young adults. Disabil Health J. 2010;3(3):222-224.
- Ferro JM, Massaro AR, Mas JL. Aetiological diagnosis of ischemic stroke in young adults. Lancet Neurol. 2010;9(11):1085-1096.
- Chan MT, Nadareishvili ZG, Norris JW; Canadian Stroke Consortium. Diagnostic strategies in young patients with ischemic stroke in Canada. Can J Neurol Sci. 2000;27(2):120-124.
- Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004;292(14):1724-1737.
- Livingston EH. Patient guide: Endocrine and nutritional management after bariatric surgery: A patient’s guide. Hormone Health Network Web site. http://www.hormone.org/~/media/Hormone/Files/Patient%20Guides/Mens%20Health/PGBariatricSurgery_2014.pdf. Accessed December 17, 2014.
- Hussain A, El-Hasani S. Bariatric emergencies: current evidence and strategies of management. World J Emerg Surg. 2013;8(1):58.
- Campanille FC, Boru C, Rizzello M, et al. Acute complications after laparoscopic bariatric procedures: update for the general surgeon. Langenbecks Arch Surg. 2013;398(5):669-686
Curbside Consult: A new column for Clinical Psychiatry News
We are pleased to introduce Curbside Consult with the Group for the Advancement of Psychiatry's (GAP) Family and Cultural committees. The column is inspired by the DSM-5's emphasis on developing a cultural formulation of patients' illnesses and addressing family dynamics and resilience in promoting care that fosters prevention and recovery.
What is GAP?
GAP was formed in 1946 under the leadership of Dr. William Menninger by a group of young psychiatrists who had served in World War II. They returned to the United States to find an inadequate system of civilian care. They were eager to professionalize the field and collaboratively develop new and creative thinking. They developed an organization that met as a whole twice a year, organized into committees of particular interest to the members and crucial to the needs of psychiatric care. The committees wrote monographs that formed a crucial role in the development of modern psychiatric thought.
Mission of GAP
- Bring together top psychiatrists of all disciplines
- Offer an objective, critical perspective on current issues facing psychiatry
- Develop smart analysis and recommendations
- Shape psychiatric thinking, clinical practice, and mental health programs
- Advocate for necessary changes in the psychiatric field
- Inspire the next generation of leading psychiatric thinkers
The Family and Cultural Psychiatry committees want to focus psychiatrists on the resilience inherent in the families and cultures of our patients in order to promote psychiatric care that focuses on prevention and recovery.

The Family and Cultural Psychiatry committees see every patient as connected to family members and belonging to a network of cultures that might include their national origin, race/ethnicity, religion/spirituality, language, occupation, age, sexual orientation and gender identity, or any other element of the person's background and collective life.
Over time, these family and cultural influences have shaped all aspects of the person’s response to adversity, experience of illness, and expectations of help seeking, even among patients who are currently living alone or do not recognize their background as explicitly cultural. This influence is highly individual; each person has his/her own combination of family and cultural experiences. How does the psychiatrist access this experience and use it to help develop resilience in our patients? How do we encourage them to use strengths/support from their family and culture, and to identify narratives that are helpful?
We see this column as one way to help answer these questions. We will bring to bear both family and cultural perspectives on the care of patients in everyday clinical practice through our comments on case vignettes sent in by readers. It can be challenging to integrate an understanding of family and culture into each patient encounter.

Our committees will work together to develop a coherent response that integrates both family and cultural perspectives and can be applied in real-world patient situations by clinicians who might not have access to specialized consultation. We aim to contribute to the growing awareness in our field of the cultural complexity of our patients, as developed and transmitted in the nexus of their families, which requires from us as clinicians a more inclusive and holistic approach to care.
This column helps to meet the goals of accreditation bodies such as The Joint Commission and the Accreditation Council for Graduate Medical Education (ACGME) for cultural and linguistic competence and patient- and family-centered care. Understanding how to think about, assess, and engage in treatment with the diversity of our patients’ cultural and family backgrounds constitutes important educational topics for all psychiatric trainees. In conjunction with formal didactics, these cases can be used as a focus for discussion in psychiatric residency training programs, ACGME Clinical Learning Environment Review (CLER), health care quality improvement activities, and faculty development programs.

Practicing clinicians also will find the DSM-5 Outline for Cultural Formulation and Cultural Formulation Interview to be a helpful clinical tool for eliciting and organizing cultural information, and in differential diagnosis and treatment planning.
The following is a list of the guiding principles we will use for assessment:
1. Heterogeneity and diversity exists within all families, cultures, and societies.
2. Avoid stereotyping, essentializing, and overgeneralizing.
3. Individualize and tailor diagnostic assessment, treatment, and care.
4. Address any language access barriers through the use of qualified medical interpreters and appropriately translated educational and informational materials.
5. Employ plain language in communicating with patients with limited health and mental health literacy.
6. Recognize the impact on both the patient and the clinician of our families of origin.
7. Engage in reflective, mindful practice and attend to cultural countertransference to provide insight into one’s own values, beliefs, and behaviors.
8. Cultivate cultural humility – the realization that our understanding of the other person’s background is always limited and incomplete.
9. Every encounter is a cross-cultural one.
10. Developing cultural competence is a lifelong journey and not a final destination.
Guidelines for Case Submission

We are requesting that you submit cases to [email protected] in which your understanding and treatment are affected by challenging cultural and family issues. We will then write back with our best answers about how one might proceed in such a case. Your case and our response will be published in Clinical Psychiatry News. Please limit your case description to 250 words and please include the following details:
1. Patient’s presenting problem or reason for the visit.
2. Patient’s age and gender.
3. Indicators of the patient’s identity – self-identified race/ethnicity, culture, religion/spirituality, socioeconomic status, education, among other variables.
4. Patient’s living situation, family composition, and genogram information (if available).
5. Patient’s geographic location (rural, suburban, urban) and occupation.
6. Patient’s and family’s degree of participation in their identified culture.
7. Questions of the individual submitting the case, including concerns about the role of the family and culture in the case, diagnosis, and treatment planning.
8. Please follow local ethical requirements, disguise the case to protect confidentiality and attend to HIPAA requirements, so that patients or family members reading the article would not recognize themselves.
Additional information might be requested, and editing of the case, questions, and commentary might be needed prior to final publication.
Please note that the opinions expressed in the case commentaries should not be seen as formal medical consultations and do not represent the opinions of GAP, CPN, or the institutions where the authors are employed or with which they are affiliated.
Contributors:
Michael S. Ascher, M.D. – University of Pennsylvania, Perelman School of Medicine
Alison M. Heru, M.D. – University of Colorado at Denver, Aurora
Roberto Lewis-Fernández, M.D. – Columbia University and New York State Psychiatric Institute
Robert C. Like, M.D., M.S. – Rutgers University, Robert Wood Johnson Medical School
Resources:
DSM-5 – Outline for Cultural Formulation and Cultural Formulation Interview: http://www.psychiatry.org/practice/dsm/dsm5/online-assessment-measures#Cultural
Clinical Manual of Couples and Family Therapy, Washington: American Psychiatric Publishing Inc., 2009.
Thinking Through Cultures: Expeditions in Cultural Psychology. Cambridge, Mass.: Harvard University Press, 1991.
Clinical Manual of Cultural Psychiatry, 2nd Edition, Washington: American Psychiatric Publishing Inc., 2015.
We are pleased to introduce Curbside Consult with the Group for the Advancement of Psychiatry's (GAP) Family and Cultural committees. The column is inspired by the DSM-5's emphasis on developing a cultural formulation of patients' illnesses and addressing family dynamics and resilience in promoting care that fosters prevention and recovery.
What is GAP?
GAP was formed in 1946 under the leadership of Dr. William Menninger by a group of young psychiatrists who had served in World War II. They returned to the United States to find an inadequate system of civilian care. They were eager to professionalize the field and collaboratively develop new and creative thinking. They developed an organization that met as a whole twice a year, organized into committees of particular interest to the members and crucial to the needs of psychiatric care. The committees wrote monographs that formed a crucial role in the development of modern psychiatric thought.
Mission of GAP
- Bring together top psychiatrists of all disciplines
- Offer an objective, critical perspective on current issues facing psychiatry
- Develop smart analysis and recommendations
- Shape psychiatric thinking, clinical practice, and mental health programs
- Advocate for necessary changes in the psychiatric field
- Inspire the next generation of leading psychiatric thinkers
The Family and Cultural Psychiatry committees want to focus psychiatrists on the resilience inherent in the families and cultures of our patients in order to promote psychiatric care that focuses on prevention and recovery.
The Family and Cultural Psychiatry committees see every patient as connected to family members and belonging to a network of cultures that might include their national origin, race/ethnicity, religion/spirituality, language, occupation, age, sexual orientation and gender identity, or any other element of the person's background and collective life.
Over time, these family and cultural influences have shaped all aspects of the person’s response to adversity, experience of illness, and expectations of help seeking, even among patients who are currently living alone or do not recognize their background as explicitly cultural. This influence is highly individual; each person has his/her own combination of family and cultural experiences. How does the psychiatrist access this experience and use it to help develop resilience in our patients? How do we encourage them to use strengths/support from their family and culture, and to identify narratives that are helpful?
We see this column as one way to help answer these questions. We will bring to bear both family and cultural perspectives on the care of patients in everyday clinical practice through our comments on case vignettes sent in by readers. It can be challenging to integrate an understanding of family and culture into each patient encounter.
Our committees will work together to develop a coherent response that integrates both family and cultural perspectives and can be applied in real-world patient situations by clinicians who might not have access to specialized consultation. We aim to contribute to the growing awareness in our field of the cultural complexity of our patients, as developed and transmitted in the nexus of their families, which requires from us as clinicians a more inclusive and holistic approach to care.
This column helps to meet the goals of accreditation bodies such as The Joint Commission and the Accreditation Council for Graduate Medical Education (ACGME) for cultural and linguistic competence and patient- and family-centered care. Understanding how to think about, assess, and engage in treatment with the diversity of our patients’ cultural and family backgrounds constitutes important educational topics for all psychiatric trainees. In conjunction with formal didactics, these cases can be used as a focus for discussion in psychiatric residency training programs, ACGME Clinical Learning Environment Review (CLER), health care quality improvement activities, and faculty development programs.
Practicing clinicians also will find the DSM-5 Outline for Cultural Formulation and Cultural Formulation Interview to be a helpful clinical tool for eliciting and organizing cultural information, and in differential diagnosis and treatment planning.
The following is a list of the guiding principles we will use for assessment:
1. Heterogeneity and diversity exists within all families, cultures, and societies.
2. Avoid stereotyping, essentializing, and overgeneralizing.
3. Individualize and tailor diagnostic assessment, treatment, and care.
4. Address any language access barriers through the use of qualified medical interpreters and appropriately translated educational and informational materials.
5. Employ plain language in communicating with patients with limited health and mental health literacy.
6. Recognize the impact on both the patient and the clinician of our families of origin.
7. Engage in reflective, mindful practice and attend to cultural countertransference to provide insight into one’s own values, beliefs, and behaviors.
8. Cultivate cultural humility – the realization that our understanding of the other person’s background is always limited and incomplete.
9. Every encounter is a cross-cultural one.
10. Developing cultural competence is a lifelong journey and not a final destination.
Guidelines for Case Submission
We are requesting that you submit cases to [email protected] in which your understanding and treatment are affected by challenging cultural and family issues. We will then write back with our best answers about how one might proceed in such a case. Your case and our response will be published in Clinical Psychiatry News. Please limit your case description to 250 words and please include the following details:
1. Patient’s presenting problem or reason for the visit.
2. Patient’s age and gender.
3. Indicators of the patient’s identity – self-identified race/ethnicity, culture, religion/spirituality, socioeconomic status, education, among other variables.
4. Patient’s living situation, family composition, and genogram information (if available).
5. Patient’s geographic location (rural, suburban, urban) and occupation.
6. Patient’s and family’s degree of participation in their identified culture.
7. Questions of the individual submitting the case, including concerns about the role of the family and culture in the case, diagnosis, and treatment planning.
8. Please follow local ethical requirements, disguise the case to protect confidentiality and attend to HIPAA requirements, so that patients or family members reading the article would not recognize themselves.
Additional information might be requested, and editing of the case, questions, and commentary might be needed prior to final publication.
Please note that the opinions expressed in the case commentaries should not be seen as formal medical consultations and do not represent the opinions of GAP, CPN, or the institutions where the authors are employed or with which they are affiliated.
Contributors:
Michael S. Ascher, M.D. – University of Pennsylvania, Perelman School of Medicine
Alison M. Heru, M.D. – University of Colorado at Denver, Aurora
Roberto Lewis-Fernández, M.D. – Columbia University and New York State Psychiatric Institute
Robert C. Like, M.D., M.S. – Rutgers University, Robert Wood Johnson Medical School
Resources:
DSM-5 – Outline for Cultural Formulation and Cultural Formulation Interview: http://www.psychiatry.org/practice/dsm/dsm5/online-assessment-measures#Cultural
Clinical Manual of Couples and Family Therapy, Washington: American Psychiatric Publishing Inc., 2009.
Thinking Through Cultures: Expeditions in Cultural Psychology. Cambridge, Mass.: Harvard University Press, 1991.
Clinical Manual of Cultural Psychiatry, 2nd Edition, Washington: American Psychiatric Publishing Inc., 2015.
We are pleased to introduce Curbside Consult with the Group for the Advancement of Psychiatry's (GAP) Family and Cultural committees. The column is inspired by the DSM-5's emphasis on developing a cultural formulation of patients' illnesses and addressing family dynamics and resilience in promoting care that fosters prevention and recovery.
What is GAP?
GAP was formed in 1946 under the leadership of Dr. William Menninger by a group of young psychiatrists who had served in World War II. They returned to the United States to find an inadequate system of civilian care. They were eager to professionalize the field and collaboratively develop new and creative thinking. They developed an organization that met as a whole twice a year, organized into committees of particular interest to the members and crucial to the needs of psychiatric care. The committees wrote monographs that formed a crucial role in the development of modern psychiatric thought.
Mission of GAP
- Bring together top psychiatrists of all disciplines
- Offer an objective, critical perspective on current issues facing psychiatry
- Develop smart analysis and recommendations
- Shape psychiatric thinking, clinical practice, and mental health programs
- Advocate for necessary changes in the psychiatric field
- Inspire the next generation of leading psychiatric thinkers
The Family and Cultural Psychiatry committees want to focus psychiatrists on the resilience inherent in the families and cultures of our patients in order to promote psychiatric care that focuses on prevention and recovery.
The Family and Cultural Psychiatry committees see every patient as connected to family members and belonging to a network of cultures that might include their national origin, race/ethnicity, religion/spirituality, language, occupation, age, sexual orientation and gender identity, or any other element of the person's background and collective life.
Over time, these family and cultural influences have shaped all aspects of the person’s response to adversity, experience of illness, and expectations of help seeking, even among patients who are currently living alone or do not recognize their background as explicitly cultural. This influence is highly individual; each person has his/her own combination of family and cultural experiences. How does the psychiatrist access this experience and use it to help develop resilience in our patients? How do we encourage them to use strengths/support from their family and culture, and to identify narratives that are helpful?
We see this column as one way to help answer these questions. We will bring to bear both family and cultural perspectives on the care of patients in everyday clinical practice through our comments on case vignettes sent in by readers. It can be challenging to integrate an understanding of family and culture into each patient encounter.
Our committees will work together to develop a coherent response that integrates both family and cultural perspectives and can be applied in real-world patient situations by clinicians who might not have access to specialized consultation. We aim to contribute to the growing awareness in our field of the cultural complexity of our patients, as developed and transmitted in the nexus of their families, which requires from us as clinicians a more inclusive and holistic approach to care.
This column helps to meet the goals of accreditation bodies such as The Joint Commission and the Accreditation Council for Graduate Medical Education (ACGME) for cultural and linguistic competence and patient- and family-centered care. Understanding how to think about, assess, and engage in treatment with the diversity of our patients’ cultural and family backgrounds constitutes important educational topics for all psychiatric trainees. In conjunction with formal didactics, these cases can be used as a focus for discussion in psychiatric residency training programs, ACGME Clinical Learning Environment Review (CLER), health care quality improvement activities, and faculty development programs.
Practicing clinicians also will find the DSM-5 Outline for Cultural Formulation and Cultural Formulation Interview to be a helpful clinical tool for eliciting and organizing cultural information, and in differential diagnosis and treatment planning.
The following is a list of the guiding principles we will use for assessment:
1. Heterogeneity and diversity exists within all families, cultures, and societies.
2. Avoid stereotyping, essentializing, and overgeneralizing.
3. Individualize and tailor diagnostic assessment, treatment, and care.
4. Address any language access barriers through the use of qualified medical interpreters and appropriately translated educational and informational materials.
5. Employ plain language in communicating with patients with limited health and mental health literacy.
6. Recognize the impact on both the patient and the clinician of our families of origin.
7. Engage in reflective, mindful practice and attend to cultural countertransference to provide insight into one’s own values, beliefs, and behaviors.
8. Cultivate cultural humility – the realization that our understanding of the other person’s background is always limited and incomplete.
9. Every encounter is a cross-cultural one.
10. Developing cultural competence is a lifelong journey and not a final destination.
Guidelines for Case Submission
We are requesting that you submit cases to [email protected] in which your understanding and treatment are affected by challenging cultural and family issues. We will then write back with our best answers about how one might proceed in such a case. Your case and our response will be published in Clinical Psychiatry News. Please limit your case description to 250 words and please include the following details:
1. Patient’s presenting problem or reason for the visit.
2. Patient’s age and gender.
3. Indicators of the patient’s identity – self-identified race/ethnicity, culture, religion/spirituality, socioeconomic status, education, among other variables.
4. Patient’s living situation, family composition, and genogram information (if available).
5. Patient’s geographic location (rural, suburban, urban) and occupation.
6. Patient’s and family’s degree of participation in their identified culture.
7. Questions of the individual submitting the case, including concerns about the role of the family and culture in the case, diagnosis, and treatment planning.
8. Please follow local ethical requirements, disguise the case to protect confidentiality and attend to HIPAA requirements, so that patients or family members reading the article would not recognize themselves.
Additional information might be requested, and editing of the case, questions, and commentary might be needed prior to final publication.
Please note that the opinions expressed in the case commentaries should not be seen as formal medical consultations and do not represent the opinions of GAP, CPN, or the institutions where the authors are employed or with which they are affiliated.
Contributors:
Michael S. Ascher, M.D. – University of Pennsylvania, Perelman School of Medicine
Alison M. Heru, M.D. – University of Colorado at Denver, Aurora
Roberto Lewis-Fernández, M.D. – Columbia University and New York State Psychiatric Institute
Robert C. Like, M.D., M.S. – Rutgers University, Robert Wood Johnson Medical School
Resources:
DSM-5 – Outline for Cultural Formulation and Cultural Formulation Interview: http://www.psychiatry.org/practice/dsm/dsm5/online-assessment-measures#Cultural
Clinical Manual of Couples and Family Therapy, Washington: American Psychiatric Publishing Inc., 2009.
Thinking Through Cultures: Expeditions in Cultural Psychology. Cambridge, Mass.: Harvard University Press, 1991.
Clinical Manual of Cultural Psychiatry, 2nd Edition, Washington: American Psychiatric Publishing Inc., 2015.
