User login
Woman, 32, With Crusty Red Blisters
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.

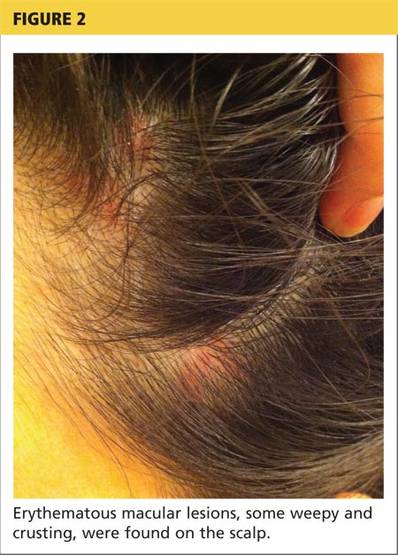
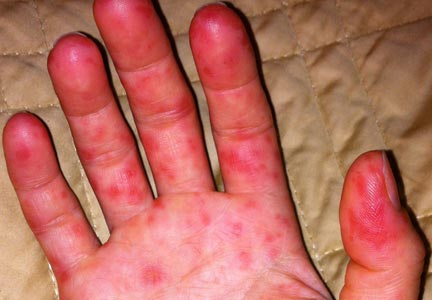
Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
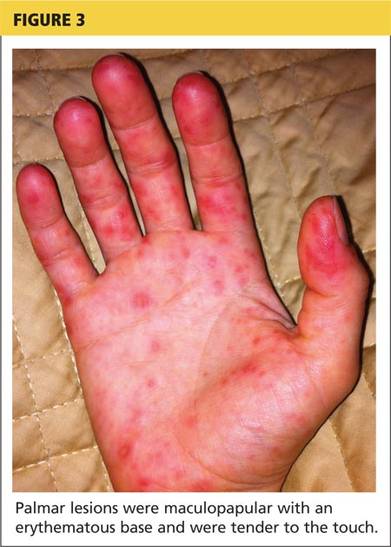
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
CLINICAL PRESENTATION
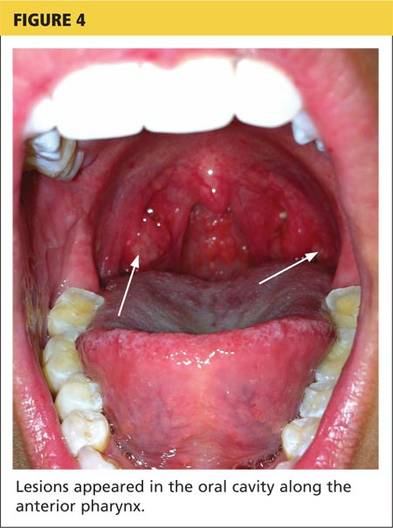
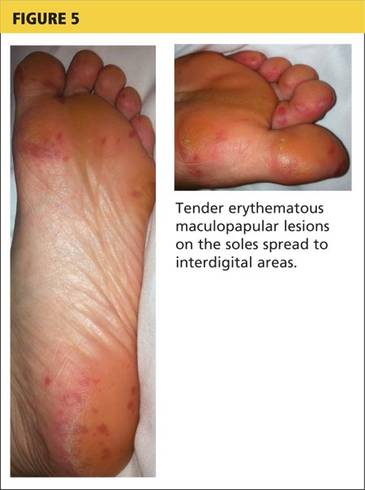
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.
Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
CLINICAL PRESENTATION
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.
Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
CLINICAL PRESENTATION
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
Second of 2 parts: The mysteries of psychiatry maintenance of certification, further unraveled
To recap what I discussed in Part 1 of this article (December 2014): As part of a trend across all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications, starting October 1, 1994.1 In 2000, the specialties that comprise the American Board of Medical Specialties (ABMS) agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what can be captured by a recertification examination. All ABMS member boards now use a 4-part process for recertification.
A great deal of professional and personal importance has been attached to maintaining one’s general and subspecialty certifications. To that end, the 2 parts of this article highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the self-assessment (SA) and performance-in-practice (PIP) components.
In this installment, I examine 3 components of MOC:
• continuing medical education (CME), including SA requirements
• improvement in medical practice (PIP)
• continuous maintenance of certification (C-MOC)
In addition to this review, all physicians who are subject to MOC should download and read the 20-page revised MOC Program booklet v. 2.1 (May 2014).2
Continuing medical education
The CME requirement is clear: All diplomate physicians must accrue, on average, 30 Category-1 CME credits a year; the CME must be relevant to the specialty or subspecialty in which the diplomate practices.3 For physicians who hold >1 ABPN certificates, the total CME requirement is the same; CME credits can be applied across each specialty and subspecialty.
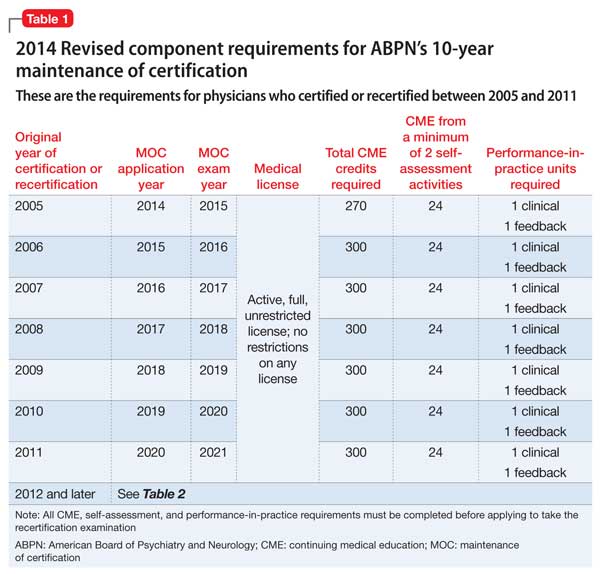
The May 2014 MOC revision states that, for physicians who certified or recertified between 2005 and 2011 and who applied for the 2015 examination in 2014, the required CME credit total is 270.2 For all subsequent years of certification or recertification, including 2012, diplomates are enrolled in C-MOC, which is described below.2
To even out the accrual of CME credits across the prior 10 years, ABPN mandates that, for diplomates who certified or recertified between 2005 and 2011, one hundred fifty of the CME credits be accrued in the 5 years before they apply for the examination. Diplomates in C-MOC should accrue, on average, 30 CME credits a year in each of the 3-year blocks (ie, 90 units in each block).2
Self-assessment
SA is a specific form of CME that is designed to provide comprehensive test-based feedback on knowledge acquired, to enhance the learning process.4 SA CME feedback must include:
• the correct answer to each test question
• recommended literature resources for each question
• performance compared to peers on each question.
Given the structured nature of SA activities, beginning January 1, 2014, one must use only ABPN-approved SA products (see Related Resources for a list of APBN-approved SA products).5
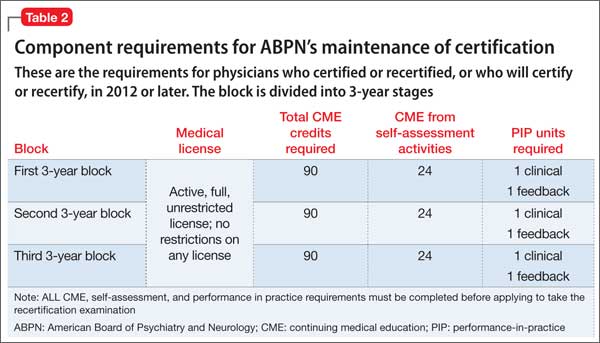
Table 1 and Table 2 outline SA requirements for, respectively, physicians who certified or recertified from 2005 through 2011, and those who certified or recertified in 2012 (and later). The SA requirement increases after 2011 to 24 credits in each 3-year block (8 credits a year, on average).2 Multiple SA activities can be used to fulfill the credit requirement of each 3-year block.
Note: Credits accrued by performing SA activities count toward the CME credit total.
Improvement in medical practice, or PIP
Physicians who are active clinically must complete PIP modules. Each module comprises peer or patient feedback plus a clinical aspect. The May 2014 MOC revision simplified the feedback process to mandate peer or patient feedback—but not both, as required previously.2 For the feedback PIP module, the physician selects 5 peers or patients to complete review forms, examines the results, and creates a plan of improvement. An exception to this “rule of 5” applies to diplomates who have a supervisor capable of evaluating all general competencies, defined below.
Related Resources provides a link to ABPN-created forms.
Within 24 months, but not sooner than 1 month, 5 peers or patients (or 1 applicable supervisor) are selected to complete review forms; changes in practice are noted. The same peers or patients might be selected for a second review. As noted in Table 1 and Table 2, the number of PIP modules is fewer for physicians who certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
There are 6 ABPN-approved feedback module options, of which the diplomate must choose 1 in any given block2:
• 5 patient surveys
• 5 peer evaluations of general competenciesa
• 5 resident evaluations of general competenciesa
• 360° evaluation of general competencies,a with 5 respondents
• institutional peer review of general competencies,a with 5 respondents
• 1 supervisor evaluation of general competencies.a
aGeneral competencies include patient care; practice-based learning and improvement; professionalism; medical knowledge; interpersonal and communication skills; and system-based practices.
Although many institutions have a quality improvement (QI) program, that program must be approved by the Multi-Specialty MOC Portfolio Approval Program sponsored by ABMS for a clinician to receive credit for 1 PIP clinical module. If the approved QI program includes patient or peer feedback (eg, a survey), the diplo mate can receive credit for 1 PIP feedback module.2
For the clinical PIP module, the physician selects 5 charts for review and examines them based on criteria found in an ABPN-approved (starting in 2014) PIP product. (Related Resources provides a link to this list.) After reviewing the initial 5 charts, a plan for improvement is created. Within 24 months, but no sooner than 1 month, 5 charts are again selected and reviewed, and changes in practice are noted. The same charts can be selected for the second review.
As noted in Table 1 and Table 2, the number of PIP modules is fewer for those who initially certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
The C-MOC process
Physicians who certified or recertified in 2012, or who will certify or recertify after that year, are enrolled automatically in C-MOC.6,7 The purpose of C-MOC is to keep diplomates on track to fulfill the higher level of SA requirements that began with this group; this is done by mandating use of the ABPN Physician Folios system. As shown in Table 2, there is no longer a 10-year cycle; instead, there are continuous 3-year stages, within which each diplomate must accrue 90 CME credits (on average, 30 credits a year), 24 SA credits (on average, 8 a year), 1 PIP clinical module, and 1 PIP feedback module.6,7
The first 3-year block of C-MOC requirements will be waived for physicians who complete Accreditation Council on Graduate Medical Education–accredited or ABPN-approved subspecialty training in 2012 or later—if they pass the corresponding ABPN subspecialty examination during the first 3-year block of enrollment in C-MOC.2 For diplomates enrolled in C-MOC, failure to track progress of each 3-year block, via the ABPN Physician Folios system, has significant consequences: Those who do not complete the first stage of the program by the end of 3 years will be listed on the ABPN Web site as “certified— not meeting MOC requirements.” Those who do not complete 2 stages by the end of 6 years will be listed as “not certified.”2
Cognitive exam still in place. The only remnant of the old 10-year cycle is the requirement to pass the cognitive examination every 10 years, although the exam can be taken earlier if the diplomate wishes. If all requirements are met and one does not sit for, or fails, the exam, the ABPN Web site will report the diplomate as “not meeting MOC requirements.” One can retake the exam within 1 year of the failed or missed exam, but a subsequent failure or missed exam will result in being listed as “not certified.”2
Fee structure. Instead of a single fee paid at the time of the exam(s), physicians in the C-MOC program pay an annual fee that covers participation in ABPN Physician Folios and 1 exam in a 10-year period. Fewer than 10 years of participation, or applying for a combined examination (for diplomates who hold multiple certifications), requires an additional fee.7
Bottom Line
Maintenance of certification (MOC) is manageable, although it requires you to be familiar with its various elements. Those elements include continuing medical education (CME requirements); the additional self-assessment component of CME; performance-in-practice modules; and continuous maintenance of certification. The MOC program booklet of the American Board of Psychiatry and Neurology provides all necessary details.
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Maintenance of Certification Program. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
5. Approved MOC Products. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/moc_products. asp. Accessed August 25, 2014.
6. Continuous MOC (C-MOC). American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/ContinuousCertificationApproach_0311.pdf. Accessed August 25, 2014.
7. C-MOC Program Overview. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/moc-handouts-CMOC-051314.pdf. Published May 13, 2014. Accessed August 25, 2014.
To recap what I discussed in Part 1 of this article (December 2014): As part of a trend across all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications, starting October 1, 1994.1 In 2000, the specialties that comprise the American Board of Medical Specialties (ABMS) agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what can be captured by a recertification examination. All ABMS member boards now use a 4-part process for recertification.
A great deal of professional and personal importance has been attached to maintaining one’s general and subspecialty certifications. To that end, the 2 parts of this article highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the self-assessment (SA) and performance-in-practice (PIP) components.
In this installment, I examine 3 components of MOC:
• continuing medical education (CME), including SA requirements
• improvement in medical practice (PIP)
• continuous maintenance of certification (C-MOC)
In addition to this review, all physicians who are subject to MOC should download and read the 20-page revised MOC Program booklet v. 2.1 (May 2014).2
Continuing medical education
The CME requirement is clear: All diplomate physicians must accrue, on average, 30 Category-1 CME credits a year; the CME must be relevant to the specialty or subspecialty in which the diplomate practices.3 For physicians who hold >1 ABPN certificates, the total CME requirement is the same; CME credits can be applied across each specialty and subspecialty.
The May 2014 MOC revision states that, for physicians who certified or recertified between 2005 and 2011 and who applied for the 2015 examination in 2014, the required CME credit total is 270.2 For all subsequent years of certification or recertification, including 2012, diplomates are enrolled in C-MOC, which is described below.2
To even out the accrual of CME credits across the prior 10 years, ABPN mandates that, for diplomates who certified or recertified between 2005 and 2011, one hundred fifty of the CME credits be accrued in the 5 years before they apply for the examination. Diplomates in C-MOC should accrue, on average, 30 CME credits a year in each of the 3-year blocks (ie, 90 units in each block).2
Self-assessment
SA is a specific form of CME that is designed to provide comprehensive test-based feedback on knowledge acquired, to enhance the learning process.4 SA CME feedback must include:
• the correct answer to each test question
• recommended literature resources for each question
• performance compared to peers on each question.
Given the structured nature of SA activities, beginning January 1, 2014, one must use only ABPN-approved SA products (see Related Resources for a list of APBN-approved SA products).5
Table 1 and Table 2 outline SA requirements for, respectively, physicians who certified or recertified from 2005 through 2011, and those who certified or recertified in 2012 (and later). The SA requirement increases after 2011 to 24 credits in each 3-year block (8 credits a year, on average).2 Multiple SA activities can be used to fulfill the credit requirement of each 3-year block.
Note: Credits accrued by performing SA activities count toward the CME credit total.
Improvement in medical practice, or PIP
Physicians who are active clinically must complete PIP modules. Each module comprises peer or patient feedback plus a clinical aspect. The May 2014 MOC revision simplified the feedback process to mandate peer or patient feedback—but not both, as required previously.2 For the feedback PIP module, the physician selects 5 peers or patients to complete review forms, examines the results, and creates a plan of improvement. An exception to this “rule of 5” applies to diplomates who have a supervisor capable of evaluating all general competencies, defined below.
Related Resources provides a link to ABPN-created forms.
Within 24 months, but not sooner than 1 month, 5 peers or patients (or 1 applicable supervisor) are selected to complete review forms; changes in practice are noted. The same peers or patients might be selected for a second review. As noted in Table 1 and Table 2, the number of PIP modules is fewer for physicians who certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
There are 6 ABPN-approved feedback module options, of which the diplomate must choose 1 in any given block2:
• 5 patient surveys
• 5 peer evaluations of general competenciesa
• 5 resident evaluations of general competenciesa
• 360° evaluation of general competencies,a with 5 respondents
• institutional peer review of general competencies,a with 5 respondents
• 1 supervisor evaluation of general competencies.a
aGeneral competencies include patient care; practice-based learning and improvement; professionalism; medical knowledge; interpersonal and communication skills; and system-based practices.
Although many institutions have a quality improvement (QI) program, that program must be approved by the Multi-Specialty MOC Portfolio Approval Program sponsored by ABMS for a clinician to receive credit for 1 PIP clinical module. If the approved QI program includes patient or peer feedback (eg, a survey), the diplo mate can receive credit for 1 PIP feedback module.2
For the clinical PIP module, the physician selects 5 charts for review and examines them based on criteria found in an ABPN-approved (starting in 2014) PIP product. (Related Resources provides a link to this list.) After reviewing the initial 5 charts, a plan for improvement is created. Within 24 months, but no sooner than 1 month, 5 charts are again selected and reviewed, and changes in practice are noted. The same charts can be selected for the second review.
As noted in Table 1 and Table 2, the number of PIP modules is fewer for those who initially certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
The C-MOC process
Physicians who certified or recertified in 2012, or who will certify or recertify after that year, are enrolled automatically in C-MOC.6,7 The purpose of C-MOC is to keep diplomates on track to fulfill the higher level of SA requirements that began with this group; this is done by mandating use of the ABPN Physician Folios system. As shown in Table 2, there is no longer a 10-year cycle; instead, there are continuous 3-year stages, within which each diplomate must accrue 90 CME credits (on average, 30 credits a year), 24 SA credits (on average, 8 a year), 1 PIP clinical module, and 1 PIP feedback module.6,7
The first 3-year block of C-MOC requirements will be waived for physicians who complete Accreditation Council on Graduate Medical Education–accredited or ABPN-approved subspecialty training in 2012 or later—if they pass the corresponding ABPN subspecialty examination during the first 3-year block of enrollment in C-MOC.2 For diplomates enrolled in C-MOC, failure to track progress of each 3-year block, via the ABPN Physician Folios system, has significant consequences: Those who do not complete the first stage of the program by the end of 3 years will be listed on the ABPN Web site as “certified— not meeting MOC requirements.” Those who do not complete 2 stages by the end of 6 years will be listed as “not certified.”2
Cognitive exam still in place. The only remnant of the old 10-year cycle is the requirement to pass the cognitive examination every 10 years, although the exam can be taken earlier if the diplomate wishes. If all requirements are met and one does not sit for, or fails, the exam, the ABPN Web site will report the diplomate as “not meeting MOC requirements.” One can retake the exam within 1 year of the failed or missed exam, but a subsequent failure or missed exam will result in being listed as “not certified.”2
Fee structure. Instead of a single fee paid at the time of the exam(s), physicians in the C-MOC program pay an annual fee that covers participation in ABPN Physician Folios and 1 exam in a 10-year period. Fewer than 10 years of participation, or applying for a combined examination (for diplomates who hold multiple certifications), requires an additional fee.7
Bottom Line
Maintenance of certification (MOC) is manageable, although it requires you to be familiar with its various elements. Those elements include continuing medical education (CME requirements); the additional self-assessment component of CME; performance-in-practice modules; and continuous maintenance of certification. The MOC program booklet of the American Board of Psychiatry and Neurology provides all necessary details.
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
To recap what I discussed in Part 1 of this article (December 2014): As part of a trend across all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications, starting October 1, 1994.1 In 2000, the specialties that comprise the American Board of Medical Specialties (ABMS) agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what can be captured by a recertification examination. All ABMS member boards now use a 4-part process for recertification.
A great deal of professional and personal importance has been attached to maintaining one’s general and subspecialty certifications. To that end, the 2 parts of this article highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the self-assessment (SA) and performance-in-practice (PIP) components.
In this installment, I examine 3 components of MOC:
• continuing medical education (CME), including SA requirements
• improvement in medical practice (PIP)
• continuous maintenance of certification (C-MOC)
In addition to this review, all physicians who are subject to MOC should download and read the 20-page revised MOC Program booklet v. 2.1 (May 2014).2
Continuing medical education
The CME requirement is clear: All diplomate physicians must accrue, on average, 30 Category-1 CME credits a year; the CME must be relevant to the specialty or subspecialty in which the diplomate practices.3 For physicians who hold >1 ABPN certificates, the total CME requirement is the same; CME credits can be applied across each specialty and subspecialty.
The May 2014 MOC revision states that, for physicians who certified or recertified between 2005 and 2011 and who applied for the 2015 examination in 2014, the required CME credit total is 270.2 For all subsequent years of certification or recertification, including 2012, diplomates are enrolled in C-MOC, which is described below.2
To even out the accrual of CME credits across the prior 10 years, ABPN mandates that, for diplomates who certified or recertified between 2005 and 2011, one hundred fifty of the CME credits be accrued in the 5 years before they apply for the examination. Diplomates in C-MOC should accrue, on average, 30 CME credits a year in each of the 3-year blocks (ie, 90 units in each block).2
Self-assessment
SA is a specific form of CME that is designed to provide comprehensive test-based feedback on knowledge acquired, to enhance the learning process.4 SA CME feedback must include:
• the correct answer to each test question
• recommended literature resources for each question
• performance compared to peers on each question.
Given the structured nature of SA activities, beginning January 1, 2014, one must use only ABPN-approved SA products (see Related Resources for a list of APBN-approved SA products).5
Table 1 and Table 2 outline SA requirements for, respectively, physicians who certified or recertified from 2005 through 2011, and those who certified or recertified in 2012 (and later). The SA requirement increases after 2011 to 24 credits in each 3-year block (8 credits a year, on average).2 Multiple SA activities can be used to fulfill the credit requirement of each 3-year block.
Note: Credits accrued by performing SA activities count toward the CME credit total.
Improvement in medical practice, or PIP
Physicians who are active clinically must complete PIP modules. Each module comprises peer or patient feedback plus a clinical aspect. The May 2014 MOC revision simplified the feedback process to mandate peer or patient feedback—but not both, as required previously.2 For the feedback PIP module, the physician selects 5 peers or patients to complete review forms, examines the results, and creates a plan of improvement. An exception to this “rule of 5” applies to diplomates who have a supervisor capable of evaluating all general competencies, defined below.
Related Resources provides a link to ABPN-created forms.
Within 24 months, but not sooner than 1 month, 5 peers or patients (or 1 applicable supervisor) are selected to complete review forms; changes in practice are noted. The same peers or patients might be selected for a second review. As noted in Table 1 and Table 2, the number of PIP modules is fewer for physicians who certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
There are 6 ABPN-approved feedback module options, of which the diplomate must choose 1 in any given block2:
• 5 patient surveys
• 5 peer evaluations of general competenciesa
• 5 resident evaluations of general competenciesa
• 360° evaluation of general competencies,a with 5 respondents
• institutional peer review of general competencies,a with 5 respondents
• 1 supervisor evaluation of general competencies.a
aGeneral competencies include patient care; practice-based learning and improvement; professionalism; medical knowledge; interpersonal and communication skills; and system-based practices.
Although many institutions have a quality improvement (QI) program, that program must be approved by the Multi-Specialty MOC Portfolio Approval Program sponsored by ABMS for a clinician to receive credit for 1 PIP clinical module. If the approved QI program includes patient or peer feedback (eg, a survey), the diplo mate can receive credit for 1 PIP feedback module.2
For the clinical PIP module, the physician selects 5 charts for review and examines them based on criteria found in an ABPN-approved (starting in 2014) PIP product. (Related Resources provides a link to this list.) After reviewing the initial 5 charts, a plan for improvement is created. Within 24 months, but no sooner than 1 month, 5 charts are again selected and reviewed, and changes in practice are noted. The same charts can be selected for the second review.
As noted in Table 1 and Table 2, the number of PIP modules is fewer for those who initially certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
The C-MOC process
Physicians who certified or recertified in 2012, or who will certify or recertify after that year, are enrolled automatically in C-MOC.6,7 The purpose of C-MOC is to keep diplomates on track to fulfill the higher level of SA requirements that began with this group; this is done by mandating use of the ABPN Physician Folios system. As shown in Table 2, there is no longer a 10-year cycle; instead, there are continuous 3-year stages, within which each diplomate must accrue 90 CME credits (on average, 30 credits a year), 24 SA credits (on average, 8 a year), 1 PIP clinical module, and 1 PIP feedback module.6,7
The first 3-year block of C-MOC requirements will be waived for physicians who complete Accreditation Council on Graduate Medical Education–accredited or ABPN-approved subspecialty training in 2012 or later—if they pass the corresponding ABPN subspecialty examination during the first 3-year block of enrollment in C-MOC.2 For diplomates enrolled in C-MOC, failure to track progress of each 3-year block, via the ABPN Physician Folios system, has significant consequences: Those who do not complete the first stage of the program by the end of 3 years will be listed on the ABPN Web site as “certified— not meeting MOC requirements.” Those who do not complete 2 stages by the end of 6 years will be listed as “not certified.”2
Cognitive exam still in place. The only remnant of the old 10-year cycle is the requirement to pass the cognitive examination every 10 years, although the exam can be taken earlier if the diplomate wishes. If all requirements are met and one does not sit for, or fails, the exam, the ABPN Web site will report the diplomate as “not meeting MOC requirements.” One can retake the exam within 1 year of the failed or missed exam, but a subsequent failure or missed exam will result in being listed as “not certified.”2
Fee structure. Instead of a single fee paid at the time of the exam(s), physicians in the C-MOC program pay an annual fee that covers participation in ABPN Physician Folios and 1 exam in a 10-year period. Fewer than 10 years of participation, or applying for a combined examination (for diplomates who hold multiple certifications), requires an additional fee.7
Bottom Line
Maintenance of certification (MOC) is manageable, although it requires you to be familiar with its various elements. Those elements include continuing medical education (CME requirements); the additional self-assessment component of CME; performance-in-practice modules; and continuous maintenance of certification. The MOC program booklet of the American Board of Psychiatry and Neurology provides all necessary details.
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Maintenance of Certification Program. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
5. Approved MOC Products. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/moc_products. asp. Accessed August 25, 2014.
6. Continuous MOC (C-MOC). American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/ContinuousCertificationApproach_0311.pdf. Accessed August 25, 2014.
7. C-MOC Program Overview. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/moc-handouts-CMOC-051314.pdf. Published May 13, 2014. Accessed August 25, 2014.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Maintenance of Certification Program. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
5. Approved MOC Products. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/moc_products. asp. Accessed August 25, 2014.
6. Continuous MOC (C-MOC). American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/ContinuousCertificationApproach_0311.pdf. Accessed August 25, 2014.
7. C-MOC Program Overview. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/moc-handouts-CMOC-051314.pdf. Published May 13, 2014. Accessed August 25, 2014.

Choosing a treatment for disruptive, impulse-control, and conduct disorders
Chronic disruptive and impulsive behaviors are significant concerns for psychiatric clinicians because of their persistence and potential legal ramifications. To date, few studies have assessed treatment options for pyromania, oppositional defiant disorder (ODD), intermittent explosive disorder (IED), kleptomania, and conduct disorder (CD).
This article reviews the literature on the treatment of these disorders, focusing primarily on randomized, controlled studies. Because of the lack of clinical studies for these disorders, however, case studies and open trials are mentioned for reference. Summaries of supported medication and psychological interventions are provided for each disorder.
Categorizing impulse-control disorders
The DSM-5 created a new chapter on disruptive, impulse control, and conduct disorders that brought together disorders previously classified as disorders usually first diagnosed in infancy, childhood, or adolescence (ODD, CD) and impulse-control disorders not elsewhere classified. These disorders are unified by the presence of difficult, disruptive, aggressive, or antisocial behavior. Disruptive, aggressive, or antisocial behavior usually is a multifaceted behavior, often associated with physical or verbal injury to self, others, or objects or with violating the rights of others. These behaviors can appear in several forms and can be defensive, premeditated, or impulsive.
Despite a high prevalence in the general population1 and in psychiatric cohorts,2 disruptive and impulse-control disorders have been relatively understudied. Controlled trials of treatments do not exist for many impulse-control disorders, and there are no FDA-approved medications for any of these disorders.
Oppositional defiant disorder
Irritability, anger, defiance, and temper are specific descriptors of ODD. ODD seems to be a developmental antecedent for some youth with CD, suggesting that these disorders could reflect different stages of a spectrum of disruptive behavior. Transient oppositional behavior is common among children and adolescents, but ODD occurs in 1% to 11% of youth.3 The disorder is more prevalent among boys before puberty and has an equal sex prevalence in young people after puberty.
Regrettably, most ODD research has included patients with comorbidities, most commonly attention-deficit/hyperactivity disorder (ADHD). Because of this limitation, the drugs and programs discussed below are drawn from meta-analyses and review articles.
Pharmacotherapy. No medications have been FDA-approved for ODD. Studies assessing ODD have employed a variety of methodologies, not all of which are double-blind. The meta-analyses and reviews cited in this section include both randomized and open trials, and should be interpreted as such.
Stimulants are commonly used to treat ODD because of a high comorbidity rate with ADHD, and these drugs have improved ODD symptoms in randomized trials.4 Methylphenidate and d-amphetamine have shown some efficacy in trials of ODD and CD.5-7 These medications are most commonly used when ODD is complicated by ADHD symptoms.
Antipsychotics also have been used to treat ODD, with the largest body of research suggesting that risperidone has some efficacy. Risperidone usually is considered a second- or third-line option because it has been associated with adverse effects in children and adolescents and requires caution in younger populations, despite its potential efficacy.4,8-10
Alpha-2 agonists—clonidine and guanfacine—have shown some efficacy in treating ODD but have not been studied extensively. Studies of clonidine, however, often have grouped ODD, CD, and ADHD, which limits our understanding of this medication for ODD alone.4,5,11
Atomoxetine has been studied for ODD, but its efficacy is limited, with different meta-analyses finding distinct results regarding efficacy. One explanation for these disparate findings is that improvements in oppositional symptoms may be secondary to improvement in ADHD symptoms.7,12-14
Psychological treatments. As noted for pharmacotherapy, this section provides general information on empirically studied therapies. A series of meta-analyses have been included for further review, but are not isolated to randomized, controlled studies.
Individual therapy has shown consistent improvements in ODD. Examples include behavior modification therapy and parent-child interaction therapy. These sessions emphasize skills to manage outbursts and erratic emotionality. Emotion regulation and behavior and social skills training have shown significant reductions in target measures. Some of these programs incorporate both patient and parent components.15-17
Family/teacher training programs such as “Helping the Noncompliant Child” and the “Triple P” have yielded significant improvements. These programs focus on ways to manage the child’s oppositional behavior at home and in the classroom, as well as strategies to limit positive reinforcement for problem behaviors.17-20
Group programs have shown some efficacy with ODD. These programs cover a wide number of needs and intents. Examples include the “Incredible Years” program and the Community Parent Education Program. Research has found that these programs show some efficacy as preemptive measures to reduce the rate of ODD among adolescents.
Conclusions. A number of treatment options for ODD have shown some efficacy. However, many of these options have only been studied in patients with comorbid ADHD, which limits current knowledge about ODD as a distinct disorder.
Intermittent explosive disorder
IED is defined by recurrent, significant outbursts of aggression, often leading to assaultive acts against people or property, which are disproportionate to outside stressors and are not better explained by another psychiatric diagnosis. Research suggests IED is common, with 6.3% of a community sample meeting criteria for lifetime IED.21
IED symptoms tend to start in adolescence and appear to be chronic.21,22 People with IED regard their behavior as distressing and problematic.22 Outbursts generally are short-lived (usually <30 minutes) and frequent (multiple times a month22). Legal and occupational difficulties are common.22
Pharmacotherapy. Data on drug treatment for IED comes for a small set of double-blind studies (Table). Although pharmacotherapies have been studied for treating aggression, impulsivity, and violent behavior, only 5 controlled studies are specific to IED.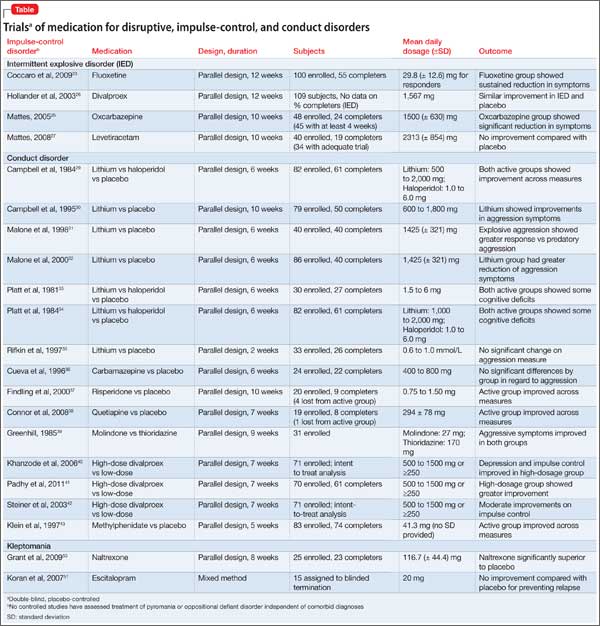
A double-blind, randomized, placebo-controlled trial of fluoxetine in 100 participants with IED found that fluoxetine produced a sustained reduction in aggression and irritability as early as the second week of treatment. Full or partial remission of impulsive aggressive behaviors occurred in 46% of fluoxetine-treated subjects. These findings have been supported by studies assessing other samples of aggressive patients, but not specifically IED.23,24 Another treatment study found that oxcarbazepine produced significant improvements in IED symptom severity, specifically on impulsive aggression.25
In a randomized, double-blind, placebo-controlled study, 96 participants with Cluster B personality disorders, 116 with IED, and 34 with posttraumatic stress disorder were assigned to divalproex sodium or placebo for 12 weeks. Using an intent-to-treat analysis, divalproex had no significant influence on aggression in patients with IED.26 Similarly, a study assessing levetiracetam for IED did not show any improvements to measures of impulsive aggression.27
Psychological treatments. The only available study on psychological treatments for IED found that patients receiving active cognitive-behavioral therapy (CBT) or group therapy showed significant improvements compared with waitlist controls. These improvements spanned several target symptoms of IED.28
Conclusions. Although there is a paucity of treatment studies for IED, fluoxetine may be an effective treatment based on available studies, and oxcarbazepine has shown some preliminary efficacy. CBT also has shown some initial efficacy in reducing symptom severity in IED.
Conduct disorder
The essential feature of CD is a repetitive and persistent pattern of behavior in which the basic rights of others or social norms are violated.3 These behaviors can entail:
• aggressive conduct that causes or threatens harm to others or to animals
• nonaggressive behavior resulting in property damage
• deceitfulness or theft
• serious violation of rules.
Prevalence among the general population is 2% to 10%. The disorder is more common among boys than girls.3
Pharmacotherapy. No medication is FDA-approved to treat CD. Fifteen controlled studies have examined medications in patients with CD (Table), although a number of these included a high rate of comorbid ADHD.
To date, 7 studies have shown efficacy with lithium for patients with CD.29-35 A number of trials assessing lithium also included a treatment condition with haloperidol, which showed significant improvement.29,30,33,34 Both lithium and haloperidol were associated with select deficits on cognitive tests, suggesting that there may be risks associated with these medications.
Preliminary double-blind results have indicated that methylphenidate, risperidone, quetiapine, molindone, thioridazine, and carbamazepine might be effective options for treating CD.36-43 The evidence for these medications is limited and additional studies are needed to replicate initial findings.
Three studies of divalproex sodium have shown some efficacy in randomized studies comparing high and low dosages of the drug.40-42 Because these studies did not include a placebo, additional studies are necessary to corroborate these findings.
Psychological treatments. Several forms of behavioral, family-based, and school-based therapies have been found effective in randomized trials. Specifically, behavioral therapy and parental skills training have shown consistent benefits for patients and their families. As with ODD, parental training programs for CD focus on parents’ skill acquisition to help manage outbursts and aggressive behavior. These treatments often follow a similar course to those used for other externalizing and disruptive disorders.44-46
Conclusions. Based on evidence, psychotherapy and some pharmacotherapies (eg, lithium) could be considered first-line treatment options for CD. Psychotherapy programs have shown efficacy in reducing aggression in high-risk groups.44 Lithium or antipsychotics could be useful for patients who do not respond sufficiently to psychotherapy. The risk of cognitive deficits with lithium and antipsychotics should be weighed against potential benefits of these medications.33,34
Kleptomania
Kleptomania is characterized by repetitive, poorly controlled stealing of items that are not needed for personal use. Kleptomania often begins in late adolescence or early adulthood.47 The course of the illness generally is chronic, with waxing and waning symptoms. Women are twice as likely as men to suffer from kleptomania.48 People with kleptomania frequently hoard, discard, or return stolen items.47
Most people with kleptomania try unsuccessfully to stop stealing, which often leads to feelings of shame and guilt.48 Many (64% to 87%) have been arrested because of their stealing behavior47; a smaller percentage (15% to 23%) have been incarcerated.48 Suicide attempts are common among these patients.49
Pharmacotherapy. There has been only 1 randomized, placebo-controlled study of pharmacotherapy for kleptomania (Table). An 8-week, double-blind, placebo-controlled trial was conducted to evaluate the safety and efficacy of oral naltrexone, 50 to 150 mg/d, in 25 patients with kleptomania. Those taking naltrexone had a significantly greater reduction in total score than those taking placebo on the Yale-Brown Obsessive Compulsive Scale Modified for Kleptomania; in stealing urges; and in stealing behavior. The mean effective dosage of naltrexone was 116.7 (± 44.4) mg/d.50
Naltrexone was well tolerated, with minimal nausea, and did not cause elevation of liver enzymes.
There is one available open-label study with a double-blind discontinuation phase assessing the efficacy of escitalopram for kleptomania. Continuation of escitalopram during the blinded discontinuation phase did produce lower relapse rates.51
Psychological treatments. There are no controlled studies of psychological treatments for kleptomania. Case reports suggest that cognitive and behavioral therapies might be effective:
• A young man who underwent 7 sessions of covert sensitization, combined with exposure and response prevention, over a 4-month period was able to reduce his stealing frequency.52
• In another case, a young woman underwent 5 weekly sessions when she was instructed to practice covert sensitization whenever she had an urge to steal. She remained in remission for 14 months with only a single lapse in behavior and with no reported urges to steal.53
• In 2 patients, imaginal desensitization in fourteen 15-minutes sessions over 5 days resulted in complete remission of symptoms for a 2-year period.54
Conclusions. The single controlled study of naltrexone for kleptomania suggests that naltrexone might be a beneficial treatment for this disorder. No controlled trials of psychosocial interventions have been reported. The current psychological research is based primarily on case reports.
This state of affairs likely is because of (1) the low prevalence of kleptomania and (2) clinical difficulties in treating patients involved in illegal activities. Nevertheless, there is a need for systematic studies of treating this disorder; such studies could involve collaboration across multiple treatment centers because of the disorder’s low prevalence.
Pyromania
Pyromania is characterized by (1) deliberate and purposeful fire setting on >1 occasion; (2) tension or affective arousal before the act; (3) fascination with, interest in, curiosity about, or attraction to fire and its situational contexts; and (4) pleasure, gratification, or relief when setting fires or when witnessing or participating in their aftermath.3
Although pyromania is thought to be a disorder primarily affecting men, recent research suggests that the sex ratio is equal among adults and may be slightly higher among adolescent females. Mean age of onset usually is late adolescence. Pyromania appears to be chronic if untreated.55
Urges to set fires are common and the fire setting is almost always pleasurable. Severe distress follows the fire setting, and persons with pyromania report significant functional impairment. High rates of co-occurring psychiatric disorders (depression, substance use disorders, other impulse-control disorders) are common among persons with pyromania.55
Pharmacotherapy. There are no randomized, controlled clinical trials examining pharmacotherapy for treating pyromania. There are no FDA-approved medications for pyromania.
In case reports, medications that have shown benefit in treating pyromania include topiramate, escitalopram, sertraline, fluoxetine, lithium, and a combination of olanzapine and sodium valproate. An equal number of medications have shown no benefit: fluoxetine, valproic acid, lithium, sertraline, olanzapine, escitalopram, citalopram, and clonazepam. A case report of an 18-year-old man with pyromania described successfully using a combination of topiramate with 3 weeks of daily CBT to achieve significant symptom improvement.56,57
Pyromania is a largely unrecognized disorder that causes significant psychological, social, and legal repercussions. Because few persons with pyromania volunteer information regarding fire-setting, it is important that clinicians recognize the disorder and screen patients appropriately. Various treatments have been helpful in case studies, but more research on the etiology and treatment of the disorder is needed.56,57
Conclusions based on the literature
In disruptive, impulse-control, and conduct disorders, the systematic study of treatment efficacy and tolerability is in its infancy. With few controlled studies published, it is not possible to make treatment recommendations with confidence. There are no FDA-approved drugs for treating any of these disorders.
Nonetheless, specific psychotherapies and drug therapies offer promising options, but often are based on small studies, often in patient populations with prominent comorbidities, and have not been replicated by independent investigators. For all of these disorders, issues such as which psychotherapy or medication to use and the ideal duration of treatment cannot be sufficiently addressed with the available data.
In conjunction with emerging epidemiological data supporting a relatively high prevalence of disruptive, impulse-control, and conduct disorders, the small amount of data regarding effective treatments highlights the clinical need for additional research.
Bottom Line
Empirically supported treatment options for impulse-control disorders currently are limited, because only select disorders have been studied across multiple trials. New research is needed to confirm possible treatment options and identify effective psychotherapeutic and pharmacological treatment alternatives.
Related Resources
• Grant JE. Impulse control disorders: a clinician’s guide to understanding and treating behavioral addictions. New York, NY: W. W. Norton & Company; 2008.
• Grant JE, Kim SW. Stop me because I can’t stop myself: taking control of impulsive behavior. New York, NY: McGraw- Hill; 2003.
• American Academy of Child and Adolescent Psychiatry. Conduct disorder resource center. http://www.aacap.org/AACAP/FamiliesandYouth/ResourceCenters/ConductDisorderResourceCenter/Home.aspx.
Drug Brand Names
Atomoxetine • Strattera Methylphenidate • Ritalin
Carbamazepine • Tegretol Molindone • Moban
Citalopram • Celexa Naltrexone • ReVia
Clonazepam • Klonopin Olanzapine • Zyprexa
Clonidine • Catapres Oxcarbazepine • Trileptal
D-amphetamine • Dexedrine Quetiapine • Seroquel
Divalproex sodium • Depakote Risperidone • Risperdal
Escitalopram • Lexapro Sertraline • Zoloft
Fluoxetine • Prozac Sodium valproate • Depacon
Guanfacine • Intuniv Thioridazine • Mellaril
Haloperidol • Haldol Topiramate • Topamax
Levetiracetam • Keppra Valproic acid • Depakote
Lithium • Eskalith, Lithobid
Disclosures
Dr. Grant receives grant or research support from Brainsway, Forest Pharmaceuticals, and Roche Pharmaceuticals. Mr. Leppink reports no financial relationship with any company whose products are mentioned in this article or with competing products.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Grant JE, Levine L, Kim D, et al. Impulse control disorders in adult psychiatric inpatients. Am J Psychiatry. 2005;162(11):2184-2188.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Turgay A. Psychopharmacological treatment of oppositional defiant disorder. CNS Drugs. 2009;23(1):1-17.
5. Hazell P. Review of attention-deficit/hyperactivity disorder comorbid with oppositional defiant disorder. Australas Psychiatry. 2010;18(6):556-559.
6. Burke JD, Loeber R, Birmaher B. Oppositional defiant disorder and conduct disorder: a review of the past 10 years, part II. J Am Acad Child Adolesc Psychiatry. 2002; 41(11):1275-1293.
7. Connor DF, Steeber J, McBurnett K. A review of attention-deficit/hyperactivity disorder complicated by symptoms of oppositional defiant disorder or conduct disorder. J Dev Behav Pediatr. 2010;31(5):427-440.
8. Aman MG, Bukstein OG, Gadow KD, et al. What does risperidone add to parent training and stimulant for severe aggression in child attention-deficit/hyperactivity disorder? J Am Acad Child Adolesc Psychiatry. 2014;53(1):47-60.e1.
9. Loy JH, Merry SN, Hetrick SE, et al. Atypical antipsychotics for disruptive behavior disorders in children and youths. Cochrane Database Syst Rev. 2012;9:CD008559.
10. Gadow KD, Arnold LE, Molina BS, et al. Risperidone added to parent training and stimulant medication: effects on attention-deficit/hyperactivity disorder, oppositional defiant disorder, conduct disorder, and peer aggression. J Am Acad Child Adolesc Psychiatry. 2014;53(9):948-959.e1.
12. Signorovitch J, Erder MH, Xie J, et al. Comparative effectiveness research using matching-adjusted indirect comparison: an application to treatment with guanfacine extended release or atomoxetine in children with attention-deficit/hyperactivity disorder and comorbid oppositional defiant disorder. Pharmacoepidemiol Drug Saf. 2012;21(suppl 2):130-137.
13. Bangs ME, Hazell P, Danckaerts M, et al; Atomoxetine ADHD/ODD Study Group. Atomoxetine for the treatment of attention-deficit/hyperactivity disorder and oppositional defiant disorder. Pediatrics. 2008;121(2):e314-e320.
14. Biederman J, Spencer TJ, Newcorn JH, et al. Effect of comorbid symptoms of oppositional defiant disorder on responses to atomoxetine in children with ADHD: a meta-analysis of controlled clinical trial data. Psychopharmacology (Berl). 2007;190(1):31-41.
15. Miller NV, Haas SM, Waschbusch DA, et al. Behavior therapy and callous-unemotional traits: effects of a pilot study examining modified behavioral contingencies on child behavior. Behav Ther. 2014;45(5):606-618.
16. Hamilton SS, Armando J. Oppositional defiant disorder. Am Fam Physician. 2008;78(7):861-866.
17. Steiner H, Remsing L; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with oppositional defiant disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(1):126-141.
18. Winther J, Carlsson A, Vance A. A pilot study of a school-based prevention and early intervention program to reduce oppositional defiant disorder/conduct disorder. Early Interv Psychiatry. 2014;8(2):181-189.
19. Plueck J, Eichelberger I, Hautmann C, et al. Effectiveness of a teacher-based indicated prevention program for preschool children with externalizing problem behavior [published online April 22, 2014]. Prev Sci. doi: 10.1007/s11121-014- 0487-x.
20. Dretzke J, Frew E, Davenport C, et al. The effectiveness and cost-effectiveness of parent training/education programmes for the treatment of conduct disorder, including oppositional defiant disorder, in children. Health Tech Assess. 2005;9(50):iii, ix-x, 1-233.
21. Coccaro EF, Schmidt CA, Samuels JF, et al. Lifetime and 1-month prevalence rates of intermittent explosive disorder in a community sample. J Clin Psychiatry. 2004;65(6):820-824.
22. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry. 1998;59(4):203-210; quiz 211.
23. Coccaro EF, Lee RJ, Kavoussi RJ. A double-blind, randomized, placebo-controlled trial of fluoxetine in patients with intermittent explosive disorder. J Clin Psychiatry. 2009;70(5):653-662.
24. Coccaro EF. Intermittent explosive disorder as a disorder of impulsive aggression for DSM-5. Am J Psychiatry. 2012;169(6):577-588.
25. Mattes JA. Oxcarbazepine in patients with impulsive aggression: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2005;25(6):575-579.
26. Hollander E, Tracy KA, Swann AC, et al. Divalproex in the treatment of impulsive aggression: efficacy in cluster B personality disorders. Neuropsychopharmacology. 2003;28(6):1186-1197.
27. Mattes JA. Levetiracetam in patients with impulsive aggression: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(2):310-315.
28. McCloskey MS, Noblett KL, Deffenbacher JL, et al. Cognitive-behavioral therapy for intermittent explosive disorder: a pilot randomized clinical trial. J Consult Clin Psychol. 2008;76(5):876-886.
29. Campbell M, Small AM, Green WH, et al. Behavioral efficacy of haloperidol and lithium carbonate. A comparison in hospitalized aggressive children with conduct disorder. Arch Gen Psychiatry. 1984;41(7):650-656.
30. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
31. Malone RP, Simpson GM. Psychopharmacology: use of placebos in clinical trials involving children and adolescents. Psychiatr Serv. 1998;49(11):1413-1414, 1417.
32. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
33. Platt JE, Campbell M, Green WH, et al. Effects of lithium carbonate and haloperidol on cognition in aggressive hospitalized school-age children. J Clin Psychopharmacol. 1981;1(1):8-13.
34. Platt JE, Campbell M, Green WH, et al. Cognitive effects of lithium carbonate and haloperidol in treatment-resistant aggressive children. Arch Gen Psychiatry. 1984;41(7):657-662.
35. Rifkin A, Karajgi B, Dicker R, et al. Lithium treatment of conduct disorders in adolescents. Am J Psychiatry. 1997;154(4):554-555.
36. Cueva JE, Overall JE, Small AM, et al. Carbamazepine in aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1996;35(4):480-490.
37. Findling RL, McNamara NK, Branicky LA, et al. A double-blind pilot study of risperidone in the treatment of conduct disorder. J Am Acad Child Adolesc Psychiatry. 2000;39(4):509-516.
38. Connor DF, McLaughlin TJ, Jeffers-Terry M. Randomized controlled pilot study of quetiapine in the treatment of adolescent conduct disorder. J Child Adolesc Psychopharmacol. 2008;18(2):140-156.
39. Greenhill LL, Solomon M, Pleak R, et al. Molindone hydrochloride treatment of hospitalized children with conduct disorder. J Clin Psychiatry. 1985;46(8 pt 2):20-25.
40. Khanzode LA, Saxena K, Kraemer H, et al. Efficacy profiles of psychopharmacology: divalproex sodium in conduct disorder. Child Psychiatry Hum Dev. 2006;37(1):55-64.
41. Padhy R, Saxena K, Remsing L, et al. Symptomatic response to divalproex in subtypes of conduct disorder. Child Psychiatry Hum Dev. 2011;42(5):584-593.
42. Steiner H, Petersen ML, Saxena K, et al. Divalproex sodium for the treatment of conduct disorder: a randomized controlled clinical trial. J Clin Psychiatry. 2003;64(10):1183-1191.
43. Klein RG, Abikoff H, Klass E, et al. Clinical efficacy of methylphenidate in conduct disorder with and without attention deficit hyperactivity disorder. Arch Gen Psychiatry. 1997;54(12):1073-1080.
44. Heneggeler SW, Sheidow AJ. Empirically supported family-based treatments for conduct disorder and delinquency in adolescents. J Marital Fam Ther. 2012;38(1):30-58.
45. Lochman JE, Powell NP, Boxmeyer CL, et al. Cognitive-behavioral therapy for externalizing disorder in children and adolescents. Child Adolesc Psychiatr Clin N Am. 2011;20(2):305-318.
46. Furlong M, McGilloway S, Bywater T, et al. Behavioural and cognitive-behavioural group-based parenting programmes for early-onset conduct problems in children aged 3 to 12 years. Cochrane Database Syst Rev. 2012;2:CD008225.
47. McElroy SL, Pope HG Jr, Hudson JI, et al. Kleptomania: a report of 20 cases. Am J Psychiatry. 1991;148(5):652-657.
48. Grant JE, Kim SW. Clinical characteristics and associated psychopathology of 22 patients with kleptomania. Compr Psychiatry. 2002;43(5):378-384.
49. Odlaug BL, Grant JE, Kim SW. Suicide attempts in 107 adolescents and adults with kleptomania. Arch Suicide Res. 2012;16(4):348-359.
50. Grant JE, Kim SW, Odlaug BL. A double-blind, placebo-controlled study of the opiate antagonist, naltrexone, in the treatment of kleptomania. Biol Psychiatry. 2009;65(7): 600-606.
51. Koran LM, Aboujaoude EN, Gamel NN. Escitalopram treatment of kleptomania: an open-label trial followed by double-blind discontinuation. J Clin Psychiatry. 2007;68(3):422-427.
52. Guidry LS. Use of a covert punishing contingency in compulsive stealing. J Behav Therapy Exp Psychiatry. 1975;6(2):169.
53. Gauthier J, Pellerin D. Management of compulsive shoplifting through covert sensitization. J Behav Therapy Exp Psychiatry. 1982;13(1):73-75.
54. McConaghy N, Blaszczynski A. Imaginal desensitization: a cost-effective treatment in two shop-lifters and a binge-eater resistant to previous therapy. Aus N Z J Psychiatry. 1988;22(1):78-82.
55. Grant JE, Won Kim S. Clinical characteristics and psychiatric comorbidity of pyromania. J Clin Psychiatry. 2007;68(11):1717-1722.
56. Grant JE, Odlaug B. Assessment and treatment of pyromania. In: Oxford handbook of impulse control disorders. Grant JE, Potenza MN, eds. Oxford, United Kingdom: Oxford University Press; 2012:353-359.
57. Dell’Osso B, Altamura AC, Allen A, et al. Epidemiologic and clinical updates on impulse control disorders: a critical review. Eur Arch Psychiatry Clin Neurosci. 2006;256(8):464-475.
Chronic disruptive and impulsive behaviors are significant concerns for psychiatric clinicians because of their persistence and potential legal ramifications. To date, few studies have assessed treatment options for pyromania, oppositional defiant disorder (ODD), intermittent explosive disorder (IED), kleptomania, and conduct disorder (CD).
This article reviews the literature on the treatment of these disorders, focusing primarily on randomized, controlled studies. Because of the lack of clinical studies for these disorders, however, case studies and open trials are mentioned for reference. Summaries of supported medication and psychological interventions are provided for each disorder.
Categorizing impulse-control disorders
The DSM-5 created a new chapter on disruptive, impulse control, and conduct disorders that brought together disorders previously classified as disorders usually first diagnosed in infancy, childhood, or adolescence (ODD, CD) and impulse-control disorders not elsewhere classified. These disorders are unified by the presence of difficult, disruptive, aggressive, or antisocial behavior. Disruptive, aggressive, or antisocial behavior usually is a multifaceted behavior, often associated with physical or verbal injury to self, others, or objects or with violating the rights of others. These behaviors can appear in several forms and can be defensive, premeditated, or impulsive.
Despite a high prevalence in the general population1 and in psychiatric cohorts,2 disruptive and impulse-control disorders have been relatively understudied. Controlled trials of treatments do not exist for many impulse-control disorders, and there are no FDA-approved medications for any of these disorders.
Oppositional defiant disorder
Irritability, anger, defiance, and temper are specific descriptors of ODD. ODD seems to be a developmental antecedent for some youth with CD, suggesting that these disorders could reflect different stages of a spectrum of disruptive behavior. Transient oppositional behavior is common among children and adolescents, but ODD occurs in 1% to 11% of youth.3 The disorder is more prevalent among boys before puberty and has an equal sex prevalence in young people after puberty.
Regrettably, most ODD research has included patients with comorbidities, most commonly attention-deficit/hyperactivity disorder (ADHD). Because of this limitation, the drugs and programs discussed below are drawn from meta-analyses and review articles.
Pharmacotherapy. No medications have been FDA-approved for ODD. Studies assessing ODD have employed a variety of methodologies, not all of which are double-blind. The meta-analyses and reviews cited in this section include both randomized and open trials, and should be interpreted as such.
Stimulants are commonly used to treat ODD because of a high comorbidity rate with ADHD, and these drugs have improved ODD symptoms in randomized trials.4 Methylphenidate and d-amphetamine have shown some efficacy in trials of ODD and CD.5-7 These medications are most commonly used when ODD is complicated by ADHD symptoms.
Antipsychotics also have been used to treat ODD, with the largest body of research suggesting that risperidone has some efficacy. Risperidone usually is considered a second- or third-line option because it has been associated with adverse effects in children and adolescents and requires caution in younger populations, despite its potential efficacy.4,8-10
Alpha-2 agonists—clonidine and guanfacine—have shown some efficacy in treating ODD but have not been studied extensively. Studies of clonidine, however, often have grouped ODD, CD, and ADHD, which limits our understanding of this medication for ODD alone.4,5,11
Atomoxetine has been studied for ODD, but its efficacy is limited, with different meta-analyses finding distinct results regarding efficacy. One explanation for these disparate findings is that improvements in oppositional symptoms may be secondary to improvement in ADHD symptoms.7,12-14
Psychological treatments. As noted for pharmacotherapy, this section provides general information on empirically studied therapies. A series of meta-analyses have been included for further review, but are not isolated to randomized, controlled studies.
Individual therapy has shown consistent improvements in ODD. Examples include behavior modification therapy and parent-child interaction therapy. These sessions emphasize skills to manage outbursts and erratic emotionality. Emotion regulation and behavior and social skills training have shown significant reductions in target measures. Some of these programs incorporate both patient and parent components.15-17
Family/teacher training programs such as “Helping the Noncompliant Child” and the “Triple P” have yielded significant improvements. These programs focus on ways to manage the child’s oppositional behavior at home and in the classroom, as well as strategies to limit positive reinforcement for problem behaviors.17-20
Group programs have shown some efficacy with ODD. These programs cover a wide number of needs and intents. Examples include the “Incredible Years” program and the Community Parent Education Program. Research has found that these programs show some efficacy as preemptive measures to reduce the rate of ODD among adolescents.
Conclusions. A number of treatment options for ODD have shown some efficacy. However, many of these options have only been studied in patients with comorbid ADHD, which limits current knowledge about ODD as a distinct disorder.
Intermittent explosive disorder
IED is defined by recurrent, significant outbursts of aggression, often leading to assaultive acts against people or property, which are disproportionate to outside stressors and are not better explained by another psychiatric diagnosis. Research suggests IED is common, with 6.3% of a community sample meeting criteria for lifetime IED.21
IED symptoms tend to start in adolescence and appear to be chronic.21,22 People with IED regard their behavior as distressing and problematic.22 Outbursts generally are short-lived (usually <30 minutes) and frequent (multiple times a month22). Legal and occupational difficulties are common.22
Pharmacotherapy. Data on drug treatment for IED comes for a small set of double-blind studies (Table). Although pharmacotherapies have been studied for treating aggression, impulsivity, and violent behavior, only 5 controlled studies are specific to IED.
A double-blind, randomized, placebo-controlled trial of fluoxetine in 100 participants with IED found that fluoxetine produced a sustained reduction in aggression and irritability as early as the second week of treatment. Full or partial remission of impulsive aggressive behaviors occurred in 46% of fluoxetine-treated subjects. These findings have been supported by studies assessing other samples of aggressive patients, but not specifically IED.23,24 Another treatment study found that oxcarbazepine produced significant improvements in IED symptom severity, specifically on impulsive aggression.25
In a randomized, double-blind, placebo-controlled study, 96 participants with Cluster B personality disorders, 116 with IED, and 34 with posttraumatic stress disorder were assigned to divalproex sodium or placebo for 12 weeks. Using an intent-to-treat analysis, divalproex had no significant influence on aggression in patients with IED.26 Similarly, a study assessing levetiracetam for IED did not show any improvements to measures of impulsive aggression.27
Psychological treatments. The only available study on psychological treatments for IED found that patients receiving active cognitive-behavioral therapy (CBT) or group therapy showed significant improvements compared with waitlist controls. These improvements spanned several target symptoms of IED.28
Conclusions. Although there is a paucity of treatment studies for IED, fluoxetine may be an effective treatment based on available studies, and oxcarbazepine has shown some preliminary efficacy. CBT also has shown some initial efficacy in reducing symptom severity in IED.
Conduct disorder
The essential feature of CD is a repetitive and persistent pattern of behavior in which the basic rights of others or social norms are violated.3 These behaviors can entail:
• aggressive conduct that causes or threatens harm to others or to animals
• nonaggressive behavior resulting in property damage
• deceitfulness or theft
• serious violation of rules.
Prevalence among the general population is 2% to 10%. The disorder is more common among boys than girls.3
Pharmacotherapy. No medication is FDA-approved to treat CD. Fifteen controlled studies have examined medications in patients with CD (Table), although a number of these included a high rate of comorbid ADHD.
To date, 7 studies have shown efficacy with lithium for patients with CD.29-35 A number of trials assessing lithium also included a treatment condition with haloperidol, which showed significant improvement.29,30,33,34 Both lithium and haloperidol were associated with select deficits on cognitive tests, suggesting that there may be risks associated with these medications.
Preliminary double-blind results have indicated that methylphenidate, risperidone, quetiapine, molindone, thioridazine, and carbamazepine might be effective options for treating CD.36-43 The evidence for these medications is limited and additional studies are needed to replicate initial findings.
Three studies of divalproex sodium have shown some efficacy in randomized studies comparing high and low dosages of the drug.40-42 Because these studies did not include a placebo, additional studies are necessary to corroborate these findings.
Psychological treatments. Several forms of behavioral, family-based, and school-based therapies have been found effective in randomized trials. Specifically, behavioral therapy and parental skills training have shown consistent benefits for patients and their families. As with ODD, parental training programs for CD focus on parents’ skill acquisition to help manage outbursts and aggressive behavior. These treatments often follow a similar course to those used for other externalizing and disruptive disorders.44-46
Conclusions. Based on evidence, psychotherapy and some pharmacotherapies (eg, lithium) could be considered first-line treatment options for CD. Psychotherapy programs have shown efficacy in reducing aggression in high-risk groups.44 Lithium or antipsychotics could be useful for patients who do not respond sufficiently to psychotherapy. The risk of cognitive deficits with lithium and antipsychotics should be weighed against potential benefits of these medications.33,34
Kleptomania
Kleptomania is characterized by repetitive, poorly controlled stealing of items that are not needed for personal use. Kleptomania often begins in late adolescence or early adulthood.47 The course of the illness generally is chronic, with waxing and waning symptoms. Women are twice as likely as men to suffer from kleptomania.48 People with kleptomania frequently hoard, discard, or return stolen items.47
Most people with kleptomania try unsuccessfully to stop stealing, which often leads to feelings of shame and guilt.48 Many (64% to 87%) have been arrested because of their stealing behavior47; a smaller percentage (15% to 23%) have been incarcerated.48 Suicide attempts are common among these patients.49
Pharmacotherapy. There has been only 1 randomized, placebo-controlled study of pharmacotherapy for kleptomania (Table). An 8-week, double-blind, placebo-controlled trial was conducted to evaluate the safety and efficacy of oral naltrexone, 50 to 150 mg/d, in 25 patients with kleptomania. Those taking naltrexone had a significantly greater reduction in total score than those taking placebo on the Yale-Brown Obsessive Compulsive Scale Modified for Kleptomania; in stealing urges; and in stealing behavior. The mean effective dosage of naltrexone was 116.7 (± 44.4) mg/d.50
Naltrexone was well tolerated, with minimal nausea, and did not cause elevation of liver enzymes.
There is one available open-label study with a double-blind discontinuation phase assessing the efficacy of escitalopram for kleptomania. Continuation of escitalopram during the blinded discontinuation phase did produce lower relapse rates.51
Psychological treatments. There are no controlled studies of psychological treatments for kleptomania. Case reports suggest that cognitive and behavioral therapies might be effective:
• A young man who underwent 7 sessions of covert sensitization, combined with exposure and response prevention, over a 4-month period was able to reduce his stealing frequency.52
• In another case, a young woman underwent 5 weekly sessions when she was instructed to practice covert sensitization whenever she had an urge to steal. She remained in remission for 14 months with only a single lapse in behavior and with no reported urges to steal.53
• In 2 patients, imaginal desensitization in fourteen 15-minutes sessions over 5 days resulted in complete remission of symptoms for a 2-year period.54
Conclusions. The single controlled study of naltrexone for kleptomania suggests that naltrexone might be a beneficial treatment for this disorder. No controlled trials of psychosocial interventions have been reported. The current psychological research is based primarily on case reports.
This state of affairs likely is because of (1) the low prevalence of kleptomania and (2) clinical difficulties in treating patients involved in illegal activities. Nevertheless, there is a need for systematic studies of treating this disorder; such studies could involve collaboration across multiple treatment centers because of the disorder’s low prevalence.
Pyromania
Pyromania is characterized by (1) deliberate and purposeful fire setting on >1 occasion; (2) tension or affective arousal before the act; (3) fascination with, interest in, curiosity about, or attraction to fire and its situational contexts; and (4) pleasure, gratification, or relief when setting fires or when witnessing or participating in their aftermath.3
Although pyromania is thought to be a disorder primarily affecting men, recent research suggests that the sex ratio is equal among adults and may be slightly higher among adolescent females. Mean age of onset usually is late adolescence. Pyromania appears to be chronic if untreated.55
Urges to set fires are common and the fire setting is almost always pleasurable. Severe distress follows the fire setting, and persons with pyromania report significant functional impairment. High rates of co-occurring psychiatric disorders (depression, substance use disorders, other impulse-control disorders) are common among persons with pyromania.55
Pharmacotherapy. There are no randomized, controlled clinical trials examining pharmacotherapy for treating pyromania. There are no FDA-approved medications for pyromania.
In case reports, medications that have shown benefit in treating pyromania include topiramate, escitalopram, sertraline, fluoxetine, lithium, and a combination of olanzapine and sodium valproate. An equal number of medications have shown no benefit: fluoxetine, valproic acid, lithium, sertraline, olanzapine, escitalopram, citalopram, and clonazepam. A case report of an 18-year-old man with pyromania described successfully using a combination of topiramate with 3 weeks of daily CBT to achieve significant symptom improvement.56,57
Pyromania is a largely unrecognized disorder that causes significant psychological, social, and legal repercussions. Because few persons with pyromania volunteer information regarding fire-setting, it is important that clinicians recognize the disorder and screen patients appropriately. Various treatments have been helpful in case studies, but more research on the etiology and treatment of the disorder is needed.56,57
Conclusions based on the literature
In disruptive, impulse-control, and conduct disorders, the systematic study of treatment efficacy and tolerability is in its infancy. With few controlled studies published, it is not possible to make treatment recommendations with confidence. There are no FDA-approved drugs for treating any of these disorders.
Nonetheless, specific psychotherapies and drug therapies offer promising options, but often are based on small studies, often in patient populations with prominent comorbidities, and have not been replicated by independent investigators. For all of these disorders, issues such as which psychotherapy or medication to use and the ideal duration of treatment cannot be sufficiently addressed with the available data.
In conjunction with emerging epidemiological data supporting a relatively high prevalence of disruptive, impulse-control, and conduct disorders, the small amount of data regarding effective treatments highlights the clinical need for additional research.
Bottom Line
Empirically supported treatment options for impulse-control disorders currently are limited, because only select disorders have been studied across multiple trials. New research is needed to confirm possible treatment options and identify effective psychotherapeutic and pharmacological treatment alternatives.
Related Resources
• Grant JE. Impulse control disorders: a clinician’s guide to understanding and treating behavioral addictions. New York, NY: W. W. Norton & Company; 2008.
• Grant JE, Kim SW. Stop me because I can’t stop myself: taking control of impulsive behavior. New York, NY: McGraw- Hill; 2003.
• American Academy of Child and Adolescent Psychiatry. Conduct disorder resource center. http://www.aacap.org/AACAP/FamiliesandYouth/ResourceCenters/ConductDisorderResourceCenter/Home.aspx.
Drug Brand Names
Atomoxetine • Strattera Methylphenidate • Ritalin
Carbamazepine • Tegretol Molindone • Moban
Citalopram • Celexa Naltrexone • ReVia
Clonazepam • Klonopin Olanzapine • Zyprexa
Clonidine • Catapres Oxcarbazepine • Trileptal
D-amphetamine • Dexedrine Quetiapine • Seroquel
Divalproex sodium • Depakote Risperidone • Risperdal
Escitalopram • Lexapro Sertraline • Zoloft
Fluoxetine • Prozac Sodium valproate • Depacon
Guanfacine • Intuniv Thioridazine • Mellaril
Haloperidol • Haldol Topiramate • Topamax
Levetiracetam • Keppra Valproic acid • Depakote
Lithium • Eskalith, Lithobid
Disclosures
Dr. Grant receives grant or research support from Brainsway, Forest Pharmaceuticals, and Roche Pharmaceuticals. Mr. Leppink reports no financial relationship with any company whose products are mentioned in this article or with competing products.
Chronic disruptive and impulsive behaviors are significant concerns for psychiatric clinicians because of their persistence and potential legal ramifications. To date, few studies have assessed treatment options for pyromania, oppositional defiant disorder (ODD), intermittent explosive disorder (IED), kleptomania, and conduct disorder (CD).
This article reviews the literature on the treatment of these disorders, focusing primarily on randomized, controlled studies. Because of the lack of clinical studies for these disorders, however, case studies and open trials are mentioned for reference. Summaries of supported medication and psychological interventions are provided for each disorder.
Categorizing impulse-control disorders
The DSM-5 created a new chapter on disruptive, impulse control, and conduct disorders that brought together disorders previously classified as disorders usually first diagnosed in infancy, childhood, or adolescence (ODD, CD) and impulse-control disorders not elsewhere classified. These disorders are unified by the presence of difficult, disruptive, aggressive, or antisocial behavior. Disruptive, aggressive, or antisocial behavior usually is a multifaceted behavior, often associated with physical or verbal injury to self, others, or objects or with violating the rights of others. These behaviors can appear in several forms and can be defensive, premeditated, or impulsive.
Despite a high prevalence in the general population1 and in psychiatric cohorts,2 disruptive and impulse-control disorders have been relatively understudied. Controlled trials of treatments do not exist for many impulse-control disorders, and there are no FDA-approved medications for any of these disorders.
Oppositional defiant disorder
Irritability, anger, defiance, and temper are specific descriptors of ODD. ODD seems to be a developmental antecedent for some youth with CD, suggesting that these disorders could reflect different stages of a spectrum of disruptive behavior. Transient oppositional behavior is common among children and adolescents, but ODD occurs in 1% to 11% of youth.3 The disorder is more prevalent among boys before puberty and has an equal sex prevalence in young people after puberty.
Regrettably, most ODD research has included patients with comorbidities, most commonly attention-deficit/hyperactivity disorder (ADHD). Because of this limitation, the drugs and programs discussed below are drawn from meta-analyses and review articles.
Pharmacotherapy. No medications have been FDA-approved for ODD. Studies assessing ODD have employed a variety of methodologies, not all of which are double-blind. The meta-analyses and reviews cited in this section include both randomized and open trials, and should be interpreted as such.
Stimulants are commonly used to treat ODD because of a high comorbidity rate with ADHD, and these drugs have improved ODD symptoms in randomized trials.4 Methylphenidate and d-amphetamine have shown some efficacy in trials of ODD and CD.5-7 These medications are most commonly used when ODD is complicated by ADHD symptoms.
Antipsychotics also have been used to treat ODD, with the largest body of research suggesting that risperidone has some efficacy. Risperidone usually is considered a second- or third-line option because it has been associated with adverse effects in children and adolescents and requires caution in younger populations, despite its potential efficacy.4,8-10
Alpha-2 agonists—clonidine and guanfacine—have shown some efficacy in treating ODD but have not been studied extensively. Studies of clonidine, however, often have grouped ODD, CD, and ADHD, which limits our understanding of this medication for ODD alone.4,5,11
Atomoxetine has been studied for ODD, but its efficacy is limited, with different meta-analyses finding distinct results regarding efficacy. One explanation for these disparate findings is that improvements in oppositional symptoms may be secondary to improvement in ADHD symptoms.7,12-14
Psychological treatments. As noted for pharmacotherapy, this section provides general information on empirically studied therapies. A series of meta-analyses have been included for further review, but are not isolated to randomized, controlled studies.
Individual therapy has shown consistent improvements in ODD. Examples include behavior modification therapy and parent-child interaction therapy. These sessions emphasize skills to manage outbursts and erratic emotionality. Emotion regulation and behavior and social skills training have shown significant reductions in target measures. Some of these programs incorporate both patient and parent components.15-17
Family/teacher training programs such as “Helping the Noncompliant Child” and the “Triple P” have yielded significant improvements. These programs focus on ways to manage the child’s oppositional behavior at home and in the classroom, as well as strategies to limit positive reinforcement for problem behaviors.17-20
Group programs have shown some efficacy with ODD. These programs cover a wide number of needs and intents. Examples include the “Incredible Years” program and the Community Parent Education Program. Research has found that these programs show some efficacy as preemptive measures to reduce the rate of ODD among adolescents.
Conclusions. A number of treatment options for ODD have shown some efficacy. However, many of these options have only been studied in patients with comorbid ADHD, which limits current knowledge about ODD as a distinct disorder.
Intermittent explosive disorder
IED is defined by recurrent, significant outbursts of aggression, often leading to assaultive acts against people or property, which are disproportionate to outside stressors and are not better explained by another psychiatric diagnosis. Research suggests IED is common, with 6.3% of a community sample meeting criteria for lifetime IED.21
IED symptoms tend to start in adolescence and appear to be chronic.21,22 People with IED regard their behavior as distressing and problematic.22 Outbursts generally are short-lived (usually <30 minutes) and frequent (multiple times a month22). Legal and occupational difficulties are common.22
Pharmacotherapy. Data on drug treatment for IED comes for a small set of double-blind studies (Table). Although pharmacotherapies have been studied for treating aggression, impulsivity, and violent behavior, only 5 controlled studies are specific to IED.
A double-blind, randomized, placebo-controlled trial of fluoxetine in 100 participants with IED found that fluoxetine produced a sustained reduction in aggression and irritability as early as the second week of treatment. Full or partial remission of impulsive aggressive behaviors occurred in 46% of fluoxetine-treated subjects. These findings have been supported by studies assessing other samples of aggressive patients, but not specifically IED.23,24 Another treatment study found that oxcarbazepine produced significant improvements in IED symptom severity, specifically on impulsive aggression.25
In a randomized, double-blind, placebo-controlled study, 96 participants with Cluster B personality disorders, 116 with IED, and 34 with posttraumatic stress disorder were assigned to divalproex sodium or placebo for 12 weeks. Using an intent-to-treat analysis, divalproex had no significant influence on aggression in patients with IED.26 Similarly, a study assessing levetiracetam for IED did not show any improvements to measures of impulsive aggression.27
Psychological treatments. The only available study on psychological treatments for IED found that patients receiving active cognitive-behavioral therapy (CBT) or group therapy showed significant improvements compared with waitlist controls. These improvements spanned several target symptoms of IED.28
Conclusions. Although there is a paucity of treatment studies for IED, fluoxetine may be an effective treatment based on available studies, and oxcarbazepine has shown some preliminary efficacy. CBT also has shown some initial efficacy in reducing symptom severity in IED.
Conduct disorder
The essential feature of CD is a repetitive and persistent pattern of behavior in which the basic rights of others or social norms are violated.3 These behaviors can entail:
• aggressive conduct that causes or threatens harm to others or to animals
• nonaggressive behavior resulting in property damage
• deceitfulness or theft
• serious violation of rules.
Prevalence among the general population is 2% to 10%. The disorder is more common among boys than girls.3
Pharmacotherapy. No medication is FDA-approved to treat CD. Fifteen controlled studies have examined medications in patients with CD (Table), although a number of these included a high rate of comorbid ADHD.
To date, 7 studies have shown efficacy with lithium for patients with CD.29-35 A number of trials assessing lithium also included a treatment condition with haloperidol, which showed significant improvement.29,30,33,34 Both lithium and haloperidol were associated with select deficits on cognitive tests, suggesting that there may be risks associated with these medications.
Preliminary double-blind results have indicated that methylphenidate, risperidone, quetiapine, molindone, thioridazine, and carbamazepine might be effective options for treating CD.36-43 The evidence for these medications is limited and additional studies are needed to replicate initial findings.
Three studies of divalproex sodium have shown some efficacy in randomized studies comparing high and low dosages of the drug.40-42 Because these studies did not include a placebo, additional studies are necessary to corroborate these findings.
Psychological treatments. Several forms of behavioral, family-based, and school-based therapies have been found effective in randomized trials. Specifically, behavioral therapy and parental skills training have shown consistent benefits for patients and their families. As with ODD, parental training programs for CD focus on parents’ skill acquisition to help manage outbursts and aggressive behavior. These treatments often follow a similar course to those used for other externalizing and disruptive disorders.44-46
Conclusions. Based on evidence, psychotherapy and some pharmacotherapies (eg, lithium) could be considered first-line treatment options for CD. Psychotherapy programs have shown efficacy in reducing aggression in high-risk groups.44 Lithium or antipsychotics could be useful for patients who do not respond sufficiently to psychotherapy. The risk of cognitive deficits with lithium and antipsychotics should be weighed against potential benefits of these medications.33,34
Kleptomania
Kleptomania is characterized by repetitive, poorly controlled stealing of items that are not needed for personal use. Kleptomania often begins in late adolescence or early adulthood.47 The course of the illness generally is chronic, with waxing and waning symptoms. Women are twice as likely as men to suffer from kleptomania.48 People with kleptomania frequently hoard, discard, or return stolen items.47
Most people with kleptomania try unsuccessfully to stop stealing, which often leads to feelings of shame and guilt.48 Many (64% to 87%) have been arrested because of their stealing behavior47; a smaller percentage (15% to 23%) have been incarcerated.48 Suicide attempts are common among these patients.49
Pharmacotherapy. There has been only 1 randomized, placebo-controlled study of pharmacotherapy for kleptomania (Table). An 8-week, double-blind, placebo-controlled trial was conducted to evaluate the safety and efficacy of oral naltrexone, 50 to 150 mg/d, in 25 patients with kleptomania. Those taking naltrexone had a significantly greater reduction in total score than those taking placebo on the Yale-Brown Obsessive Compulsive Scale Modified for Kleptomania; in stealing urges; and in stealing behavior. The mean effective dosage of naltrexone was 116.7 (± 44.4) mg/d.50
Naltrexone was well tolerated, with minimal nausea, and did not cause elevation of liver enzymes.
There is one available open-label study with a double-blind discontinuation phase assessing the efficacy of escitalopram for kleptomania. Continuation of escitalopram during the blinded discontinuation phase did produce lower relapse rates.51
Psychological treatments. There are no controlled studies of psychological treatments for kleptomania. Case reports suggest that cognitive and behavioral therapies might be effective:
• A young man who underwent 7 sessions of covert sensitization, combined with exposure and response prevention, over a 4-month period was able to reduce his stealing frequency.52
• In another case, a young woman underwent 5 weekly sessions when she was instructed to practice covert sensitization whenever she had an urge to steal. She remained in remission for 14 months with only a single lapse in behavior and with no reported urges to steal.53
• In 2 patients, imaginal desensitization in fourteen 15-minutes sessions over 5 days resulted in complete remission of symptoms for a 2-year period.54
Conclusions. The single controlled study of naltrexone for kleptomania suggests that naltrexone might be a beneficial treatment for this disorder. No controlled trials of psychosocial interventions have been reported. The current psychological research is based primarily on case reports.
This state of affairs likely is because of (1) the low prevalence of kleptomania and (2) clinical difficulties in treating patients involved in illegal activities. Nevertheless, there is a need for systematic studies of treating this disorder; such studies could involve collaboration across multiple treatment centers because of the disorder’s low prevalence.
Pyromania
Pyromania is characterized by (1) deliberate and purposeful fire setting on >1 occasion; (2) tension or affective arousal before the act; (3) fascination with, interest in, curiosity about, or attraction to fire and its situational contexts; and (4) pleasure, gratification, or relief when setting fires or when witnessing or participating in their aftermath.3
Although pyromania is thought to be a disorder primarily affecting men, recent research suggests that the sex ratio is equal among adults and may be slightly higher among adolescent females. Mean age of onset usually is late adolescence. Pyromania appears to be chronic if untreated.55
Urges to set fires are common and the fire setting is almost always pleasurable. Severe distress follows the fire setting, and persons with pyromania report significant functional impairment. High rates of co-occurring psychiatric disorders (depression, substance use disorders, other impulse-control disorders) are common among persons with pyromania.55
Pharmacotherapy. There are no randomized, controlled clinical trials examining pharmacotherapy for treating pyromania. There are no FDA-approved medications for pyromania.
In case reports, medications that have shown benefit in treating pyromania include topiramate, escitalopram, sertraline, fluoxetine, lithium, and a combination of olanzapine and sodium valproate. An equal number of medications have shown no benefit: fluoxetine, valproic acid, lithium, sertraline, olanzapine, escitalopram, citalopram, and clonazepam. A case report of an 18-year-old man with pyromania described successfully using a combination of topiramate with 3 weeks of daily CBT to achieve significant symptom improvement.56,57
Pyromania is a largely unrecognized disorder that causes significant psychological, social, and legal repercussions. Because few persons with pyromania volunteer information regarding fire-setting, it is important that clinicians recognize the disorder and screen patients appropriately. Various treatments have been helpful in case studies, but more research on the etiology and treatment of the disorder is needed.56,57
Conclusions based on the literature
In disruptive, impulse-control, and conduct disorders, the systematic study of treatment efficacy and tolerability is in its infancy. With few controlled studies published, it is not possible to make treatment recommendations with confidence. There are no FDA-approved drugs for treating any of these disorders.
Nonetheless, specific psychotherapies and drug therapies offer promising options, but often are based on small studies, often in patient populations with prominent comorbidities, and have not been replicated by independent investigators. For all of these disorders, issues such as which psychotherapy or medication to use and the ideal duration of treatment cannot be sufficiently addressed with the available data.
In conjunction with emerging epidemiological data supporting a relatively high prevalence of disruptive, impulse-control, and conduct disorders, the small amount of data regarding effective treatments highlights the clinical need for additional research.
Bottom Line
Empirically supported treatment options for impulse-control disorders currently are limited, because only select disorders have been studied across multiple trials. New research is needed to confirm possible treatment options and identify effective psychotherapeutic and pharmacological treatment alternatives.
Related Resources
• Grant JE. Impulse control disorders: a clinician’s guide to understanding and treating behavioral addictions. New York, NY: W. W. Norton & Company; 2008.
• Grant JE, Kim SW. Stop me because I can’t stop myself: taking control of impulsive behavior. New York, NY: McGraw- Hill; 2003.
• American Academy of Child and Adolescent Psychiatry. Conduct disorder resource center. http://www.aacap.org/AACAP/FamiliesandYouth/ResourceCenters/ConductDisorderResourceCenter/Home.aspx.
Drug Brand Names
Atomoxetine • Strattera Methylphenidate • Ritalin
Carbamazepine • Tegretol Molindone • Moban
Citalopram • Celexa Naltrexone • ReVia
Clonazepam • Klonopin Olanzapine • Zyprexa
Clonidine • Catapres Oxcarbazepine • Trileptal
D-amphetamine • Dexedrine Quetiapine • Seroquel
Divalproex sodium • Depakote Risperidone • Risperdal
Escitalopram • Lexapro Sertraline • Zoloft
Fluoxetine • Prozac Sodium valproate • Depacon
Guanfacine • Intuniv Thioridazine • Mellaril
Haloperidol • Haldol Topiramate • Topamax
Levetiracetam • Keppra Valproic acid • Depakote
Lithium • Eskalith, Lithobid
Disclosures
Dr. Grant receives grant or research support from Brainsway, Forest Pharmaceuticals, and Roche Pharmaceuticals. Mr. Leppink reports no financial relationship with any company whose products are mentioned in this article or with competing products.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Grant JE, Levine L, Kim D, et al. Impulse control disorders in adult psychiatric inpatients. Am J Psychiatry. 2005;162(11):2184-2188.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Turgay A. Psychopharmacological treatment of oppositional defiant disorder. CNS Drugs. 2009;23(1):1-17.
5. Hazell P. Review of attention-deficit/hyperactivity disorder comorbid with oppositional defiant disorder. Australas Psychiatry. 2010;18(6):556-559.
6. Burke JD, Loeber R, Birmaher B. Oppositional defiant disorder and conduct disorder: a review of the past 10 years, part II. J Am Acad Child Adolesc Psychiatry. 2002; 41(11):1275-1293.
7. Connor DF, Steeber J, McBurnett K. A review of attention-deficit/hyperactivity disorder complicated by symptoms of oppositional defiant disorder or conduct disorder. J Dev Behav Pediatr. 2010;31(5):427-440.
8. Aman MG, Bukstein OG, Gadow KD, et al. What does risperidone add to parent training and stimulant for severe aggression in child attention-deficit/hyperactivity disorder? J Am Acad Child Adolesc Psychiatry. 2014;53(1):47-60.e1.
9. Loy JH, Merry SN, Hetrick SE, et al. Atypical antipsychotics for disruptive behavior disorders in children and youths. Cochrane Database Syst Rev. 2012;9:CD008559.
10. Gadow KD, Arnold LE, Molina BS, et al. Risperidone added to parent training and stimulant medication: effects on attention-deficit/hyperactivity disorder, oppositional defiant disorder, conduct disorder, and peer aggression. J Am Acad Child Adolesc Psychiatry. 2014;53(9):948-959.e1.
12. Signorovitch J, Erder MH, Xie J, et al. Comparative effectiveness research using matching-adjusted indirect comparison: an application to treatment with guanfacine extended release or atomoxetine in children with attention-deficit/hyperactivity disorder and comorbid oppositional defiant disorder. Pharmacoepidemiol Drug Saf. 2012;21(suppl 2):130-137.
13. Bangs ME, Hazell P, Danckaerts M, et al; Atomoxetine ADHD/ODD Study Group. Atomoxetine for the treatment of attention-deficit/hyperactivity disorder and oppositional defiant disorder. Pediatrics. 2008;121(2):e314-e320.
14. Biederman J, Spencer TJ, Newcorn JH, et al. Effect of comorbid symptoms of oppositional defiant disorder on responses to atomoxetine in children with ADHD: a meta-analysis of controlled clinical trial data. Psychopharmacology (Berl). 2007;190(1):31-41.
15. Miller NV, Haas SM, Waschbusch DA, et al. Behavior therapy and callous-unemotional traits: effects of a pilot study examining modified behavioral contingencies on child behavior. Behav Ther. 2014;45(5):606-618.
16. Hamilton SS, Armando J. Oppositional defiant disorder. Am Fam Physician. 2008;78(7):861-866.
17. Steiner H, Remsing L; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with oppositional defiant disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(1):126-141.
18. Winther J, Carlsson A, Vance A. A pilot study of a school-based prevention and early intervention program to reduce oppositional defiant disorder/conduct disorder. Early Interv Psychiatry. 2014;8(2):181-189.
19. Plueck J, Eichelberger I, Hautmann C, et al. Effectiveness of a teacher-based indicated prevention program for preschool children with externalizing problem behavior [published online April 22, 2014]. Prev Sci. doi: 10.1007/s11121-014- 0487-x.
20. Dretzke J, Frew E, Davenport C, et al. The effectiveness and cost-effectiveness of parent training/education programmes for the treatment of conduct disorder, including oppositional defiant disorder, in children. Health Tech Assess. 2005;9(50):iii, ix-x, 1-233.
21. Coccaro EF, Schmidt CA, Samuels JF, et al. Lifetime and 1-month prevalence rates of intermittent explosive disorder in a community sample. J Clin Psychiatry. 2004;65(6):820-824.
22. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry. 1998;59(4):203-210; quiz 211.
23. Coccaro EF, Lee RJ, Kavoussi RJ. A double-blind, randomized, placebo-controlled trial of fluoxetine in patients with intermittent explosive disorder. J Clin Psychiatry. 2009;70(5):653-662.
24. Coccaro EF. Intermittent explosive disorder as a disorder of impulsive aggression for DSM-5. Am J Psychiatry. 2012;169(6):577-588.
25. Mattes JA. Oxcarbazepine in patients with impulsive aggression: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2005;25(6):575-579.
26. Hollander E, Tracy KA, Swann AC, et al. Divalproex in the treatment of impulsive aggression: efficacy in cluster B personality disorders. Neuropsychopharmacology. 2003;28(6):1186-1197.
27. Mattes JA. Levetiracetam in patients with impulsive aggression: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(2):310-315.
28. McCloskey MS, Noblett KL, Deffenbacher JL, et al. Cognitive-behavioral therapy for intermittent explosive disorder: a pilot randomized clinical trial. J Consult Clin Psychol. 2008;76(5):876-886.
29. Campbell M, Small AM, Green WH, et al. Behavioral efficacy of haloperidol and lithium carbonate. A comparison in hospitalized aggressive children with conduct disorder. Arch Gen Psychiatry. 1984;41(7):650-656.
30. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
31. Malone RP, Simpson GM. Psychopharmacology: use of placebos in clinical trials involving children and adolescents. Psychiatr Serv. 1998;49(11):1413-1414, 1417.
32. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
33. Platt JE, Campbell M, Green WH, et al. Effects of lithium carbonate and haloperidol on cognition in aggressive hospitalized school-age children. J Clin Psychopharmacol. 1981;1(1):8-13.
34. Platt JE, Campbell M, Green WH, et al. Cognitive effects of lithium carbonate and haloperidol in treatment-resistant aggressive children. Arch Gen Psychiatry. 1984;41(7):657-662.
35. Rifkin A, Karajgi B, Dicker R, et al. Lithium treatment of conduct disorders in adolescents. Am J Psychiatry. 1997;154(4):554-555.
36. Cueva JE, Overall JE, Small AM, et al. Carbamazepine in aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1996;35(4):480-490.
37. Findling RL, McNamara NK, Branicky LA, et al. A double-blind pilot study of risperidone in the treatment of conduct disorder. J Am Acad Child Adolesc Psychiatry. 2000;39(4):509-516.
38. Connor DF, McLaughlin TJ, Jeffers-Terry M. Randomized controlled pilot study of quetiapine in the treatment of adolescent conduct disorder. J Child Adolesc Psychopharmacol. 2008;18(2):140-156.
39. Greenhill LL, Solomon M, Pleak R, et al. Molindone hydrochloride treatment of hospitalized children with conduct disorder. J Clin Psychiatry. 1985;46(8 pt 2):20-25.
40. Khanzode LA, Saxena K, Kraemer H, et al. Efficacy profiles of psychopharmacology: divalproex sodium in conduct disorder. Child Psychiatry Hum Dev. 2006;37(1):55-64.
41. Padhy R, Saxena K, Remsing L, et al. Symptomatic response to divalproex in subtypes of conduct disorder. Child Psychiatry Hum Dev. 2011;42(5):584-593.
42. Steiner H, Petersen ML, Saxena K, et al. Divalproex sodium for the treatment of conduct disorder: a randomized controlled clinical trial. J Clin Psychiatry. 2003;64(10):1183-1191.
43. Klein RG, Abikoff H, Klass E, et al. Clinical efficacy of methylphenidate in conduct disorder with and without attention deficit hyperactivity disorder. Arch Gen Psychiatry. 1997;54(12):1073-1080.
44. Heneggeler SW, Sheidow AJ. Empirically supported family-based treatments for conduct disorder and delinquency in adolescents. J Marital Fam Ther. 2012;38(1):30-58.
45. Lochman JE, Powell NP, Boxmeyer CL, et al. Cognitive-behavioral therapy for externalizing disorder in children and adolescents. Child Adolesc Psychiatr Clin N Am. 2011;20(2):305-318.
46. Furlong M, McGilloway S, Bywater T, et al. Behavioural and cognitive-behavioural group-based parenting programmes for early-onset conduct problems in children aged 3 to 12 years. Cochrane Database Syst Rev. 2012;2:CD008225.
47. McElroy SL, Pope HG Jr, Hudson JI, et al. Kleptomania: a report of 20 cases. Am J Psychiatry. 1991;148(5):652-657.
48. Grant JE, Kim SW. Clinical characteristics and associated psychopathology of 22 patients with kleptomania. Compr Psychiatry. 2002;43(5):378-384.
49. Odlaug BL, Grant JE, Kim SW. Suicide attempts in 107 adolescents and adults with kleptomania. Arch Suicide Res. 2012;16(4):348-359.
50. Grant JE, Kim SW, Odlaug BL. A double-blind, placebo-controlled study of the opiate antagonist, naltrexone, in the treatment of kleptomania. Biol Psychiatry. 2009;65(7): 600-606.
51. Koran LM, Aboujaoude EN, Gamel NN. Escitalopram treatment of kleptomania: an open-label trial followed by double-blind discontinuation. J Clin Psychiatry. 2007;68(3):422-427.
52. Guidry LS. Use of a covert punishing contingency in compulsive stealing. J Behav Therapy Exp Psychiatry. 1975;6(2):169.
53. Gauthier J, Pellerin D. Management of compulsive shoplifting through covert sensitization. J Behav Therapy Exp Psychiatry. 1982;13(1):73-75.
54. McConaghy N, Blaszczynski A. Imaginal desensitization: a cost-effective treatment in two shop-lifters and a binge-eater resistant to previous therapy. Aus N Z J Psychiatry. 1988;22(1):78-82.
55. Grant JE, Won Kim S. Clinical characteristics and psychiatric comorbidity of pyromania. J Clin Psychiatry. 2007;68(11):1717-1722.
56. Grant JE, Odlaug B. Assessment and treatment of pyromania. In: Oxford handbook of impulse control disorders. Grant JE, Potenza MN, eds. Oxford, United Kingdom: Oxford University Press; 2012:353-359.
57. Dell’Osso B, Altamura AC, Allen A, et al. Epidemiologic and clinical updates on impulse control disorders: a critical review. Eur Arch Psychiatry Clin Neurosci. 2006;256(8):464-475.
1. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
2. Grant JE, Levine L, Kim D, et al. Impulse control disorders in adult psychiatric inpatients. Am J Psychiatry. 2005;162(11):2184-2188.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Turgay A. Psychopharmacological treatment of oppositional defiant disorder. CNS Drugs. 2009;23(1):1-17.
5. Hazell P. Review of attention-deficit/hyperactivity disorder comorbid with oppositional defiant disorder. Australas Psychiatry. 2010;18(6):556-559.
6. Burke JD, Loeber R, Birmaher B. Oppositional defiant disorder and conduct disorder: a review of the past 10 years, part II. J Am Acad Child Adolesc Psychiatry. 2002; 41(11):1275-1293.
7. Connor DF, Steeber J, McBurnett K. A review of attention-deficit/hyperactivity disorder complicated by symptoms of oppositional defiant disorder or conduct disorder. J Dev Behav Pediatr. 2010;31(5):427-440.
8. Aman MG, Bukstein OG, Gadow KD, et al. What does risperidone add to parent training and stimulant for severe aggression in child attention-deficit/hyperactivity disorder? J Am Acad Child Adolesc Psychiatry. 2014;53(1):47-60.e1.
9. Loy JH, Merry SN, Hetrick SE, et al. Atypical antipsychotics for disruptive behavior disorders in children and youths. Cochrane Database Syst Rev. 2012;9:CD008559.
10. Gadow KD, Arnold LE, Molina BS, et al. Risperidone added to parent training and stimulant medication: effects on attention-deficit/hyperactivity disorder, oppositional defiant disorder, conduct disorder, and peer aggression. J Am Acad Child Adolesc Psychiatry. 2014;53(9):948-959.e1.
12. Signorovitch J, Erder MH, Xie J, et al. Comparative effectiveness research using matching-adjusted indirect comparison: an application to treatment with guanfacine extended release or atomoxetine in children with attention-deficit/hyperactivity disorder and comorbid oppositional defiant disorder. Pharmacoepidemiol Drug Saf. 2012;21(suppl 2):130-137.
13. Bangs ME, Hazell P, Danckaerts M, et al; Atomoxetine ADHD/ODD Study Group. Atomoxetine for the treatment of attention-deficit/hyperactivity disorder and oppositional defiant disorder. Pediatrics. 2008;121(2):e314-e320.
14. Biederman J, Spencer TJ, Newcorn JH, et al. Effect of comorbid symptoms of oppositional defiant disorder on responses to atomoxetine in children with ADHD: a meta-analysis of controlled clinical trial data. Psychopharmacology (Berl). 2007;190(1):31-41.
15. Miller NV, Haas SM, Waschbusch DA, et al. Behavior therapy and callous-unemotional traits: effects of a pilot study examining modified behavioral contingencies on child behavior. Behav Ther. 2014;45(5):606-618.
16. Hamilton SS, Armando J. Oppositional defiant disorder. Am Fam Physician. 2008;78(7):861-866.
17. Steiner H, Remsing L; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with oppositional defiant disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(1):126-141.
18. Winther J, Carlsson A, Vance A. A pilot study of a school-based prevention and early intervention program to reduce oppositional defiant disorder/conduct disorder. Early Interv Psychiatry. 2014;8(2):181-189.
19. Plueck J, Eichelberger I, Hautmann C, et al. Effectiveness of a teacher-based indicated prevention program for preschool children with externalizing problem behavior [published online April 22, 2014]. Prev Sci. doi: 10.1007/s11121-014- 0487-x.
20. Dretzke J, Frew E, Davenport C, et al. The effectiveness and cost-effectiveness of parent training/education programmes for the treatment of conduct disorder, including oppositional defiant disorder, in children. Health Tech Assess. 2005;9(50):iii, ix-x, 1-233.
21. Coccaro EF, Schmidt CA, Samuels JF, et al. Lifetime and 1-month prevalence rates of intermittent explosive disorder in a community sample. J Clin Psychiatry. 2004;65(6):820-824.
22. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry. 1998;59(4):203-210; quiz 211.
23. Coccaro EF, Lee RJ, Kavoussi RJ. A double-blind, randomized, placebo-controlled trial of fluoxetine in patients with intermittent explosive disorder. J Clin Psychiatry. 2009;70(5):653-662.
24. Coccaro EF. Intermittent explosive disorder as a disorder of impulsive aggression for DSM-5. Am J Psychiatry. 2012;169(6):577-588.
25. Mattes JA. Oxcarbazepine in patients with impulsive aggression: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2005;25(6):575-579.
26. Hollander E, Tracy KA, Swann AC, et al. Divalproex in the treatment of impulsive aggression: efficacy in cluster B personality disorders. Neuropsychopharmacology. 2003;28(6):1186-1197.
27. Mattes JA. Levetiracetam in patients with impulsive aggression: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(2):310-315.
28. McCloskey MS, Noblett KL, Deffenbacher JL, et al. Cognitive-behavioral therapy for intermittent explosive disorder: a pilot randomized clinical trial. J Consult Clin Psychol. 2008;76(5):876-886.
29. Campbell M, Small AM, Green WH, et al. Behavioral efficacy of haloperidol and lithium carbonate. A comparison in hospitalized aggressive children with conduct disorder. Arch Gen Psychiatry. 1984;41(7):650-656.
30. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
31. Malone RP, Simpson GM. Psychopharmacology: use of placebos in clinical trials involving children and adolescents. Psychiatr Serv. 1998;49(11):1413-1414, 1417.
32. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
33. Platt JE, Campbell M, Green WH, et al. Effects of lithium carbonate and haloperidol on cognition in aggressive hospitalized school-age children. J Clin Psychopharmacol. 1981;1(1):8-13.
34. Platt JE, Campbell M, Green WH, et al. Cognitive effects of lithium carbonate and haloperidol in treatment-resistant aggressive children. Arch Gen Psychiatry. 1984;41(7):657-662.
35. Rifkin A, Karajgi B, Dicker R, et al. Lithium treatment of conduct disorders in adolescents. Am J Psychiatry. 1997;154(4):554-555.
36. Cueva JE, Overall JE, Small AM, et al. Carbamazepine in aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1996;35(4):480-490.
37. Findling RL, McNamara NK, Branicky LA, et al. A double-blind pilot study of risperidone in the treatment of conduct disorder. J Am Acad Child Adolesc Psychiatry. 2000;39(4):509-516.
38. Connor DF, McLaughlin TJ, Jeffers-Terry M. Randomized controlled pilot study of quetiapine in the treatment of adolescent conduct disorder. J Child Adolesc Psychopharmacol. 2008;18(2):140-156.
39. Greenhill LL, Solomon M, Pleak R, et al. Molindone hydrochloride treatment of hospitalized children with conduct disorder. J Clin Psychiatry. 1985;46(8 pt 2):20-25.
40. Khanzode LA, Saxena K, Kraemer H, et al. Efficacy profiles of psychopharmacology: divalproex sodium in conduct disorder. Child Psychiatry Hum Dev. 2006;37(1):55-64.
41. Padhy R, Saxena K, Remsing L, et al. Symptomatic response to divalproex in subtypes of conduct disorder. Child Psychiatry Hum Dev. 2011;42(5):584-593.
42. Steiner H, Petersen ML, Saxena K, et al. Divalproex sodium for the treatment of conduct disorder: a randomized controlled clinical trial. J Clin Psychiatry. 2003;64(10):1183-1191.
43. Klein RG, Abikoff H, Klass E, et al. Clinical efficacy of methylphenidate in conduct disorder with and without attention deficit hyperactivity disorder. Arch Gen Psychiatry. 1997;54(12):1073-1080.
44. Heneggeler SW, Sheidow AJ. Empirically supported family-based treatments for conduct disorder and delinquency in adolescents. J Marital Fam Ther. 2012;38(1):30-58.
45. Lochman JE, Powell NP, Boxmeyer CL, et al. Cognitive-behavioral therapy for externalizing disorder in children and adolescents. Child Adolesc Psychiatr Clin N Am. 2011;20(2):305-318.
46. Furlong M, McGilloway S, Bywater T, et al. Behavioural and cognitive-behavioural group-based parenting programmes for early-onset conduct problems in children aged 3 to 12 years. Cochrane Database Syst Rev. 2012;2:CD008225.
47. McElroy SL, Pope HG Jr, Hudson JI, et al. Kleptomania: a report of 20 cases. Am J Psychiatry. 1991;148(5):652-657.
48. Grant JE, Kim SW. Clinical characteristics and associated psychopathology of 22 patients with kleptomania. Compr Psychiatry. 2002;43(5):378-384.
49. Odlaug BL, Grant JE, Kim SW. Suicide attempts in 107 adolescents and adults with kleptomania. Arch Suicide Res. 2012;16(4):348-359.
50. Grant JE, Kim SW, Odlaug BL. A double-blind, placebo-controlled study of the opiate antagonist, naltrexone, in the treatment of kleptomania. Biol Psychiatry. 2009;65(7): 600-606.
51. Koran LM, Aboujaoude EN, Gamel NN. Escitalopram treatment of kleptomania: an open-label trial followed by double-blind discontinuation. J Clin Psychiatry. 2007;68(3):422-427.
52. Guidry LS. Use of a covert punishing contingency in compulsive stealing. J Behav Therapy Exp Psychiatry. 1975;6(2):169.
53. Gauthier J, Pellerin D. Management of compulsive shoplifting through covert sensitization. J Behav Therapy Exp Psychiatry. 1982;13(1):73-75.
54. McConaghy N, Blaszczynski A. Imaginal desensitization: a cost-effective treatment in two shop-lifters and a binge-eater resistant to previous therapy. Aus N Z J Psychiatry. 1988;22(1):78-82.
55. Grant JE, Won Kim S. Clinical characteristics and psychiatric comorbidity of pyromania. J Clin Psychiatry. 2007;68(11):1717-1722.
56. Grant JE, Odlaug B. Assessment and treatment of pyromania. In: Oxford handbook of impulse control disorders. Grant JE, Potenza MN, eds. Oxford, United Kingdom: Oxford University Press; 2012:353-359.
57. Dell’Osso B, Altamura AC, Allen A, et al. Epidemiologic and clinical updates on impulse control disorders: a critical review. Eur Arch Psychiatry Clin Neurosci. 2006;256(8):464-475.

Suvorexant for sleep-onset insomnia or sleep-maintenance insomnia, or both
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2

Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2
Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2
Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
Akathisia: Is restlessness a primary condition or an adverse drug effect?
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112). 
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
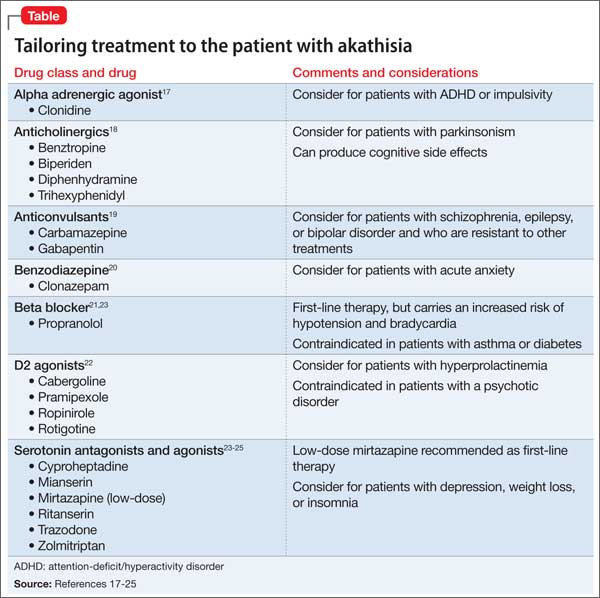
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112).
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112).
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.

Man With Diverticulitis Undergoes Precolonoscopy Evaluation
ANSWER
This ECG shows probable ectopic rhythm with second-degree atrioventricular (AV) block and a nonspecific intraventricular conduction block.
The P-wave morphology is unusual; rather than being upright and positive in leads II and aVF, the P waves are biphasic and prolonged, suggesting they originate from an atrial source other than the sinus node.
The rhythm strip of lead I in this ECG isn’t of much help in determining the atrial rhythm, as the P waves are small. However, if you look at the strip beginning with either lead II or III and keep in mind that the strip is continuous even though the leads change (eg, lead III becomes lead aVF which becomes V3, etc), you can see an atrial complex immediately following the T wave that is very similar to the P wave prior to the QRS complex. The rate of the P waves is 84 beats/min, which is twice that of the QRS complex (42 beats/min) and therefore consistent with a 2:1 heart block.
A nonspecific AV conduction block is evidenced by a QRS duration > 120 ms that does not have the appearance of a right or left bundle branch block.
Finally, while the QT interval of 514 ms is worrisome for long QT interval, it is normal when corrected for rate.
ANSWER
This ECG shows probable ectopic rhythm with second-degree atrioventricular (AV) block and a nonspecific intraventricular conduction block.
The P-wave morphology is unusual; rather than being upright and positive in leads II and aVF, the P waves are biphasic and prolonged, suggesting they originate from an atrial source other than the sinus node.
The rhythm strip of lead I in this ECG isn’t of much help in determining the atrial rhythm, as the P waves are small. However, if you look at the strip beginning with either lead II or III and keep in mind that the strip is continuous even though the leads change (eg, lead III becomes lead aVF which becomes V3, etc), you can see an atrial complex immediately following the T wave that is very similar to the P wave prior to the QRS complex. The rate of the P waves is 84 beats/min, which is twice that of the QRS complex (42 beats/min) and therefore consistent with a 2:1 heart block.
A nonspecific AV conduction block is evidenced by a QRS duration > 120 ms that does not have the appearance of a right or left bundle branch block.
Finally, while the QT interval of 514 ms is worrisome for long QT interval, it is normal when corrected for rate.
ANSWER
This ECG shows probable ectopic rhythm with second-degree atrioventricular (AV) block and a nonspecific intraventricular conduction block.
The P-wave morphology is unusual; rather than being upright and positive in leads II and aVF, the P waves are biphasic and prolonged, suggesting they originate from an atrial source other than the sinus node.
The rhythm strip of lead I in this ECG isn’t of much help in determining the atrial rhythm, as the P waves are small. However, if you look at the strip beginning with either lead II or III and keep in mind that the strip is continuous even though the leads change (eg, lead III becomes lead aVF which becomes V3, etc), you can see an atrial complex immediately following the T wave that is very similar to the P wave prior to the QRS complex. The rate of the P waves is 84 beats/min, which is twice that of the QRS complex (42 beats/min) and therefore consistent with a 2:1 heart block.
A nonspecific AV conduction block is evidenced by a QRS duration > 120 ms that does not have the appearance of a right or left bundle branch block.
Finally, while the QT interval of 514 ms is worrisome for long QT interval, it is normal when corrected for rate.
A 74-year-old man with recurring episodes of melena presents for a preoperative evaluation prior to colonoscopy. He has had three such procedures in the past five years, all of which indicated diverticulitis. The current episode began about a month ago, but the patient delayed seeking care until last week due to work obligations. The patient reports feeling more lethargic and becoming more easily tired than he has with previous episodes, which concerns him. He doesn’t think he has lost more blood than before but admits he’s been “too busy” to notice. He denies chest pain, shortness of breath, palpitations, peripheral extremity swelling, or recent weight change (gain or loss). He has not experienced loss of appetite or abdominal pain. Medical history is remarkable for hypertension, cholecystitis, and diverticulitis. There is no history of coronary artery disease, diabetes, or chronic obstructive pulmonary disease. Surgical history is remarkable for cholecystectomy and surgical repair of a high fracture of the left ankle. The patient owns a 475-acre farm, where he has lived his entire life. He is a widower who relies on his four sons to help with chores, although he insists on driving the combine himself during harvest (which is why he delayed seeking care this time). He does not smoke or drink. His current medications include hydrochlorothiazide and naproxen as needed for musculoskeletal discomfort. The review of systems is remarkable for fatigue and “the usual aches and pains of working on a farm.” The remainder of the review is noncontributory. The physical exam reveals a thin, weather-worn male in no distress. His height is 76 in and his weight, 172 lb. Both are unchanged from his previous clinic visit (six months ago). Vital signs include a blood pressure of 138/78 mm Hg; pulse, 46 beats/min; O2 saturation, 96%; and temperature, 98.2°F. Pertinent findings include normal breath sounds, a regular (albeit slow at 46 beats/min) rhythm, an early grade II/VI systolic murmur heard at the left upper sternal border, and a soft, nontender abdomen. There is no peripheral edema and no femoral or carotid bruits. The neurologic exam is intact. While the patient is undergoing preoperative laboratory tests and ECG, you review his medical record. Of note, the bradycardia found during today’s physical was not present six months ago. Laboratory data include a normal chemistry panel and a hematocrit of 38%. The ECG reveals a ventricular rate of 42 beats/min; PR interval, not reported; QRS duration, 130 ms; QT/QTc interval, 514/429 ms; P axis, 83°; R axis, 84°; and T axis, –43°. What is your interpretation of this ECG?
Man Unresponsive After Being Struck by Car
ANSWER
The radiograph demonstrates bilateral patchy, fluffy infiltrates as well as what is sometimes referred to as ground-glass opacities. In the setting of trauma and respiratory compromise, these areas are most suggestive of pulmonary contusions and early acute respiratory distress syndrome. Other possibilities in the differential diagnosis include pulmonary edema, atypical pneumonia, and pulmonary metastases.
ANSWER
The radiograph demonstrates bilateral patchy, fluffy infiltrates as well as what is sometimes referred to as ground-glass opacities. In the setting of trauma and respiratory compromise, these areas are most suggestive of pulmonary contusions and early acute respiratory distress syndrome. Other possibilities in the differential diagnosis include pulmonary edema, atypical pneumonia, and pulmonary metastases.
ANSWER
The radiograph demonstrates bilateral patchy, fluffy infiltrates as well as what is sometimes referred to as ground-glass opacities. In the setting of trauma and respiratory compromise, these areas are most suggestive of pulmonary contusions and early acute respiratory distress syndrome. Other possibilities in the differential diagnosis include pulmonary edema, atypical pneumonia, and pulmonary metastases.
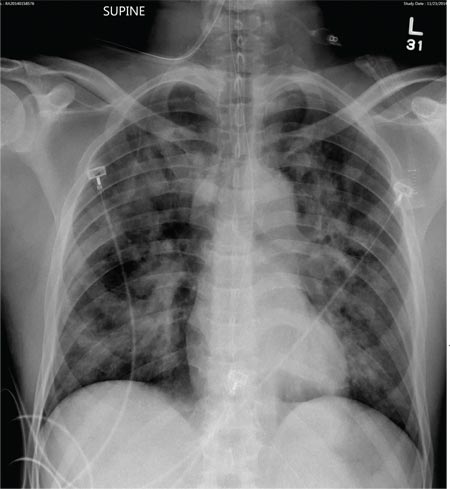
A 50-year-old man is transferred to your facility from an outlying community hospital. He is purportedly a pedestrian who was struck by a car. EMS personnel reported him to be unresponsive at the scene. He was intubated for airway protection and stabilized at the outside facility prior to transfer. Upon arrival at your facility, he is still intubated and unresponsive, and his Glasgow Coma Scale score is 3T. His heart rate is 150 beats/min and his blood pressure, 105/56 mm Hg. No additional history is available. Primary survey reveals a large scalp laceration with currently controlled bleeding. His pupils are nonreactive bilaterally. The patient is tachycardic with bilateral crackles. He also has a laceration and deformity of his right lower extremity. No imaging was provided in the transfer, so you obtain a portable chest radiograph. What is your impression?
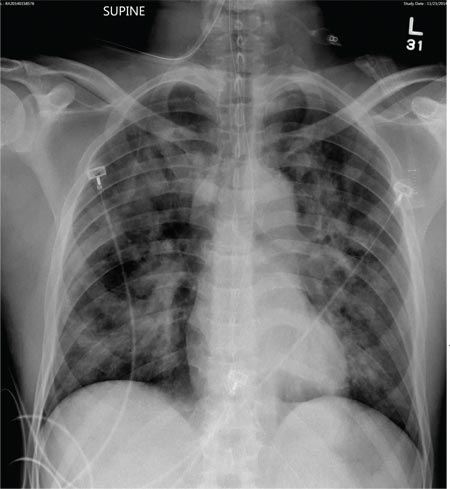
Approach can cure even high-risk FL, study suggests
SAN FRANCISCO—Follicular lymphoma (FL) patients who receive high-dose therapy with autologous stem cell transplant (HDT/ASCT) after they’ve responded to chemotherapy can achieve long-term cancer-free survival, new research suggests.
The study showed that many patients transplanted in complete remission (CR) did not relapse and could be considered cured.
Patients transplanted in their first CR fared the best, as median progression-free survival (PFS) and overall survival (OS) times were not reached.
But even patients transplanted in their second/third CR or in their first partial remission (PR) survived a median of 15 years or more, although their PFS times were shorter, at about 14 years and 3 years, respectively.
Carlos Grande García, MD, of Hospital Universitario 12 de Octubre in Madrid, Spain, presented these results at the 2014 ASH Annual Meeting (abstract 675.)*
“In follicular lymphoma patients, intensification with high-dose therapy and autologous stem cell support offers an advantage in terms of progression-free survival in comparison with conventional chemo,” he said. “But, so far, no randomized studies have yet shown any overall survival advantage.”
“Follicular lymphoma has a long natural course, and most patients have received different salvage therapies. Probably, this is why the available phase 3 studies have had insufficient time to confirm the impact on OS.”
To investigate the impact of HDT/ASCT on OS, Dr Grande García and his colleagues conducted a retrospective study of 655 FL patients who received HDT/ASCT from 1989 to 2007. Patients with histological transformation, those undergoing a second transplant, and those with a follow-up of less than 7 years were excluded.
Patient characteristics
The median follow-up was 12 years from HDT/ASCT and 14.4 years from diagnosis. At diagnosis, the median patient age was 47, 49.6% of patients were male, and 90% had stage III/IV disease.
According to FLIPI, 33% of patients were good risk, 36% were intermediate risk, and 31% were poor risk. According to FLIPI-2, the percentages were 22%, 38%, and 40%, respectively. Thirty percent of patients had received rituximab prior to HDT/ASCT.
Thirty-one percent of patients (n=203) were in their first CR at the time of transplant, 43% of whom required more than one line of therapy to reach first CR.
Thirty-one percent of patients (n=202) were in second or third CR, 21.5% (n=149) were in first PR, 12.5% (n=81) were in sensitive relapse (defined as a response other than CR or first PR), and 5% (n=29) had overt disease (which included untreated relapsed disease, first refractory disease, and second refractory disease).
Patients received a variety of conditioning regimens, including total-body irradiation plus cyclophosphamide, BEAM (carmustine, etoposide, cytarabine, and melphalan), BEAC (carmustine, etoposide, cytarabine, and cyclophosphamide), and other regimens. They received stem cells from peripheral blood (81%), bone marrow (14%), or both sources (5%).
There were 4 graft failures and 17 early toxic deaths. Thirty-one percent of patients experienced grade 3/4 hematologic toxicities.
PFS and OS
In all patients, the median PFS was 9.25 years, and the median OS was 19.5 years.
When the researchers looked at outcomes according to patients’ status at transplant, they found the median OS and PFS were not reached among patients in first CR. At a median follow-up of 12.75 years, the OS rate was 72%, and the PFS rate was 68%.
“Beginning at 10 years from transplantation, only 6 patients have died,” Dr Grande García noted, “one from disease progression, 3 from second malignancy, [and] 2 from unrelated causes.”
For patients in second or third CR, the median OS was not reached, and the median PFS was 13.9 years. For those in first PR, the median OS was 15 years, and the median PFS was 2.6 years.
For patients with sensitive disease, the median OS was 5.1 years, and the median PFS was 2 years. For those with overt disease, the median OS was 4.4 years, and the median PFS was 0.5 years.
In multivariate analysis, the following characteristics were significant predictors of OS: being older than 47 years of age (hazard ratio [HR]=1.74, P=0.0001), female sex (HR=0.58, P=0.00004), status at HDT/ASCT (HR=2.06, P<10-5), and receipt of rituximab prior to HDT/ASCT (HR=0.61, P=0.004).
Significant predictors of PFS included age (HR=1.34, P=0.01), sex (HR=0.64, P<10-5), status at HDT/ASCT (HR=2.15, P<10-5), and rituximab use (HR=0.67, P=0.003).
For patients transplanted in first CR, only sex was a significant predictor of PFS (HR=0.48, P=0.008) and OS (HR=0.43, P=0.007).
Secondary malignancies
Overall, 13% of patients developed secondary malignancies, of which 46% were solid neoplasias, 44% were myelodysplastic syndromes/acute myeloid leukemias, and 10% were other malignancies.
The incidence of secondary malignancies at 10 years was 3.5%, and the median time from HDT/ASCT to diagnosis was 16 years. There were no significant differences in the rate of secondary malignancy according to a patient’s status at HDT/ASCT or according to the use of rituximab.
“The incidence of second malignancies is not higher than that reported in other series without transplantation,” Dr Grande García noted.
“[HDT/ASCT] is highly effective, even for patients with poor initial features. A significant number of patients transplanted in CR never relapse and may be considered cured.” ![]()
*Information in the abstract differs from that presented at the meeting.
SAN FRANCISCO—Follicular lymphoma (FL) patients who receive high-dose therapy with autologous stem cell transplant (HDT/ASCT) after they’ve responded to chemotherapy can achieve long-term cancer-free survival, new research suggests.
The study showed that many patients transplanted in complete remission (CR) did not relapse and could be considered cured.
Patients transplanted in their first CR fared the best, as median progression-free survival (PFS) and overall survival (OS) times were not reached.
But even patients transplanted in their second/third CR or in their first partial remission (PR) survived a median of 15 years or more, although their PFS times were shorter, at about 14 years and 3 years, respectively.
Carlos Grande García, MD, of Hospital Universitario 12 de Octubre in Madrid, Spain, presented these results at the 2014 ASH Annual Meeting (abstract 675.)*
“In follicular lymphoma patients, intensification with high-dose therapy and autologous stem cell support offers an advantage in terms of progression-free survival in comparison with conventional chemo,” he said. “But, so far, no randomized studies have yet shown any overall survival advantage.”
“Follicular lymphoma has a long natural course, and most patients have received different salvage therapies. Probably, this is why the available phase 3 studies have had insufficient time to confirm the impact on OS.”
To investigate the impact of HDT/ASCT on OS, Dr Grande García and his colleagues conducted a retrospective study of 655 FL patients who received HDT/ASCT from 1989 to 2007. Patients with histological transformation, those undergoing a second transplant, and those with a follow-up of less than 7 years were excluded.
Patient characteristics
The median follow-up was 12 years from HDT/ASCT and 14.4 years from diagnosis. At diagnosis, the median patient age was 47, 49.6% of patients were male, and 90% had stage III/IV disease.
According to FLIPI, 33% of patients were good risk, 36% were intermediate risk, and 31% were poor risk. According to FLIPI-2, the percentages were 22%, 38%, and 40%, respectively. Thirty percent of patients had received rituximab prior to HDT/ASCT.
Thirty-one percent of patients (n=203) were in their first CR at the time of transplant, 43% of whom required more than one line of therapy to reach first CR.
Thirty-one percent of patients (n=202) were in second or third CR, 21.5% (n=149) were in first PR, 12.5% (n=81) were in sensitive relapse (defined as a response other than CR or first PR), and 5% (n=29) had overt disease (which included untreated relapsed disease, first refractory disease, and second refractory disease).
Patients received a variety of conditioning regimens, including total-body irradiation plus cyclophosphamide, BEAM (carmustine, etoposide, cytarabine, and melphalan), BEAC (carmustine, etoposide, cytarabine, and cyclophosphamide), and other regimens. They received stem cells from peripheral blood (81%), bone marrow (14%), or both sources (5%).
There were 4 graft failures and 17 early toxic deaths. Thirty-one percent of patients experienced grade 3/4 hematologic toxicities.
PFS and OS
In all patients, the median PFS was 9.25 years, and the median OS was 19.5 years.
When the researchers looked at outcomes according to patients’ status at transplant, they found the median OS and PFS were not reached among patients in first CR. At a median follow-up of 12.75 years, the OS rate was 72%, and the PFS rate was 68%.
“Beginning at 10 years from transplantation, only 6 patients have died,” Dr Grande García noted, “one from disease progression, 3 from second malignancy, [and] 2 from unrelated causes.”
For patients in second or third CR, the median OS was not reached, and the median PFS was 13.9 years. For those in first PR, the median OS was 15 years, and the median PFS was 2.6 years.
For patients with sensitive disease, the median OS was 5.1 years, and the median PFS was 2 years. For those with overt disease, the median OS was 4.4 years, and the median PFS was 0.5 years.
In multivariate analysis, the following characteristics were significant predictors of OS: being older than 47 years of age (hazard ratio [HR]=1.74, P=0.0001), female sex (HR=0.58, P=0.00004), status at HDT/ASCT (HR=2.06, P<10-5), and receipt of rituximab prior to HDT/ASCT (HR=0.61, P=0.004).
Significant predictors of PFS included age (HR=1.34, P=0.01), sex (HR=0.64, P<10-5), status at HDT/ASCT (HR=2.15, P<10-5), and rituximab use (HR=0.67, P=0.003).
For patients transplanted in first CR, only sex was a significant predictor of PFS (HR=0.48, P=0.008) and OS (HR=0.43, P=0.007).
Secondary malignancies
Overall, 13% of patients developed secondary malignancies, of which 46% were solid neoplasias, 44% were myelodysplastic syndromes/acute myeloid leukemias, and 10% were other malignancies.
The incidence of secondary malignancies at 10 years was 3.5%, and the median time from HDT/ASCT to diagnosis was 16 years. There were no significant differences in the rate of secondary malignancy according to a patient’s status at HDT/ASCT or according to the use of rituximab.
“The incidence of second malignancies is not higher than that reported in other series without transplantation,” Dr Grande García noted.
“[HDT/ASCT] is highly effective, even for patients with poor initial features. A significant number of patients transplanted in CR never relapse and may be considered cured.” ![]()
*Information in the abstract differs from that presented at the meeting.
SAN FRANCISCO—Follicular lymphoma (FL) patients who receive high-dose therapy with autologous stem cell transplant (HDT/ASCT) after they’ve responded to chemotherapy can achieve long-term cancer-free survival, new research suggests.
The study showed that many patients transplanted in complete remission (CR) did not relapse and could be considered cured.
Patients transplanted in their first CR fared the best, as median progression-free survival (PFS) and overall survival (OS) times were not reached.
But even patients transplanted in their second/third CR or in their first partial remission (PR) survived a median of 15 years or more, although their PFS times were shorter, at about 14 years and 3 years, respectively.
Carlos Grande García, MD, of Hospital Universitario 12 de Octubre in Madrid, Spain, presented these results at the 2014 ASH Annual Meeting (abstract 675.)*
“In follicular lymphoma patients, intensification with high-dose therapy and autologous stem cell support offers an advantage in terms of progression-free survival in comparison with conventional chemo,” he said. “But, so far, no randomized studies have yet shown any overall survival advantage.”
“Follicular lymphoma has a long natural course, and most patients have received different salvage therapies. Probably, this is why the available phase 3 studies have had insufficient time to confirm the impact on OS.”
To investigate the impact of HDT/ASCT on OS, Dr Grande García and his colleagues conducted a retrospective study of 655 FL patients who received HDT/ASCT from 1989 to 2007. Patients with histological transformation, those undergoing a second transplant, and those with a follow-up of less than 7 years were excluded.
Patient characteristics
The median follow-up was 12 years from HDT/ASCT and 14.4 years from diagnosis. At diagnosis, the median patient age was 47, 49.6% of patients were male, and 90% had stage III/IV disease.
According to FLIPI, 33% of patients were good risk, 36% were intermediate risk, and 31% were poor risk. According to FLIPI-2, the percentages were 22%, 38%, and 40%, respectively. Thirty percent of patients had received rituximab prior to HDT/ASCT.
Thirty-one percent of patients (n=203) were in their first CR at the time of transplant, 43% of whom required more than one line of therapy to reach first CR.
Thirty-one percent of patients (n=202) were in second or third CR, 21.5% (n=149) were in first PR, 12.5% (n=81) were in sensitive relapse (defined as a response other than CR or first PR), and 5% (n=29) had overt disease (which included untreated relapsed disease, first refractory disease, and second refractory disease).
Patients received a variety of conditioning regimens, including total-body irradiation plus cyclophosphamide, BEAM (carmustine, etoposide, cytarabine, and melphalan), BEAC (carmustine, etoposide, cytarabine, and cyclophosphamide), and other regimens. They received stem cells from peripheral blood (81%), bone marrow (14%), or both sources (5%).
There were 4 graft failures and 17 early toxic deaths. Thirty-one percent of patients experienced grade 3/4 hematologic toxicities.
PFS and OS
In all patients, the median PFS was 9.25 years, and the median OS was 19.5 years.
When the researchers looked at outcomes according to patients’ status at transplant, they found the median OS and PFS were not reached among patients in first CR. At a median follow-up of 12.75 years, the OS rate was 72%, and the PFS rate was 68%.
“Beginning at 10 years from transplantation, only 6 patients have died,” Dr Grande García noted, “one from disease progression, 3 from second malignancy, [and] 2 from unrelated causes.”
For patients in second or third CR, the median OS was not reached, and the median PFS was 13.9 years. For those in first PR, the median OS was 15 years, and the median PFS was 2.6 years.
For patients with sensitive disease, the median OS was 5.1 years, and the median PFS was 2 years. For those with overt disease, the median OS was 4.4 years, and the median PFS was 0.5 years.
In multivariate analysis, the following characteristics were significant predictors of OS: being older than 47 years of age (hazard ratio [HR]=1.74, P=0.0001), female sex (HR=0.58, P=0.00004), status at HDT/ASCT (HR=2.06, P<10-5), and receipt of rituximab prior to HDT/ASCT (HR=0.61, P=0.004).
Significant predictors of PFS included age (HR=1.34, P=0.01), sex (HR=0.64, P<10-5), status at HDT/ASCT (HR=2.15, P<10-5), and rituximab use (HR=0.67, P=0.003).
For patients transplanted in first CR, only sex was a significant predictor of PFS (HR=0.48, P=0.008) and OS (HR=0.43, P=0.007).
Secondary malignancies
Overall, 13% of patients developed secondary malignancies, of which 46% were solid neoplasias, 44% were myelodysplastic syndromes/acute myeloid leukemias, and 10% were other malignancies.
The incidence of secondary malignancies at 10 years was 3.5%, and the median time from HDT/ASCT to diagnosis was 16 years. There were no significant differences in the rate of secondary malignancy according to a patient’s status at HDT/ASCT or according to the use of rituximab.
“The incidence of second malignancies is not higher than that reported in other series without transplantation,” Dr Grande García noted.
“[HDT/ASCT] is highly effective, even for patients with poor initial features. A significant number of patients transplanted in CR never relapse and may be considered cured.” ![]()
*Information in the abstract differs from that presented at the meeting.
Age-adjusted D-dimer is ‘probably safe,’ team says

Credit: Medical College
of Georgia
A new study shows that, although age-adjusted D-dimer testing produces fewer false-positive results than conventional D-dimer testing, some cases of pulmonary embolism (PE) slip through the cracks.
Researchers compared the two testing methods in patients older than 50 and found that using an age-adjusted D-dimer threshold reduced the need for additional imaging.
Unfortunately, it also had a false-negative rate of 1.5%, failing to catch PE in 4 patients.
Scott Woller, MD, of Intermountain Medical Center in Salt Lake City, Utah, and his colleagues reported these findings in CHEST.
The team conducted this study with the goal of eliminating false-positive D-dimer results and reducing the need for additional imaging, which can be detrimental to older patients.
“A CT scan is most often used to ultimately rule out a pulmonary embolism,” Dr Woller said. “However, it delivers radiation to the patient and contrast dye.”
“Elderly patients are at greater risk for inadvertent harm related to the CT scan, and the contrast dye may also impact kidney function. Plus, the scan adds to the cost of the patient’s care. If we can safely and accurately diagnose the patient’s risk of a pulmonary embolism using [age-adjusted D-dimer], we can eliminate the need for additional imaging tests.”
With this in mind, the researchers evaluated 923 patients older than 50 years of age who presented to the emergency department at Intermountain Medical Center with a suspected PE, a calculated Revised Geneva Score (RGS), and a D-dimer test.
All of the patients underwent CT pulmonary angiography (CTPA), and the researchers compared the false-negative rate of a conventional D-dimer threshold with an age-adjusted D-dimer threshold.
The team found that age-adjusted D-dimer reduced the need for CTPA by 18.3% (95% CI, 15.9%-21.0%), compared to conventional D-dimer.
However, in the 273 patients with a negative age-adjusted D-dimer result and an RGS of 10 or greater, 4 PEs occurred within 90 days. This translates to a false-negative result rate of 1.5% (95% CI, 0.4%-3.7%).
In comparison, the false-negative rate for conventional D-dimer was 0% (95% CI, 0%-2.8%). Among the 104 patients who had a negative test result and an RGS of 10 or greater, there were no PEs within 90 days.
These results suggest an age-adjusted D-dimer threshold does reduce the need for imaging in patients older than 50, the researchers said. They added that this method is probably safe for these patients, but a prospective trial is needed to more thoroughly investigate safety. ![]()
Credit: Medical College
of Georgia
A new study shows that, although age-adjusted D-dimer testing produces fewer false-positive results than conventional D-dimer testing, some cases of pulmonary embolism (PE) slip through the cracks.
Researchers compared the two testing methods in patients older than 50 and found that using an age-adjusted D-dimer threshold reduced the need for additional imaging.
Unfortunately, it also had a false-negative rate of 1.5%, failing to catch PE in 4 patients.
Scott Woller, MD, of Intermountain Medical Center in Salt Lake City, Utah, and his colleagues reported these findings in CHEST.
The team conducted this study with the goal of eliminating false-positive D-dimer results and reducing the need for additional imaging, which can be detrimental to older patients.
“A CT scan is most often used to ultimately rule out a pulmonary embolism,” Dr Woller said. “However, it delivers radiation to the patient and contrast dye.”
“Elderly patients are at greater risk for inadvertent harm related to the CT scan, and the contrast dye may also impact kidney function. Plus, the scan adds to the cost of the patient’s care. If we can safely and accurately diagnose the patient’s risk of a pulmonary embolism using [age-adjusted D-dimer], we can eliminate the need for additional imaging tests.”
With this in mind, the researchers evaluated 923 patients older than 50 years of age who presented to the emergency department at Intermountain Medical Center with a suspected PE, a calculated Revised Geneva Score (RGS), and a D-dimer test.
All of the patients underwent CT pulmonary angiography (CTPA), and the researchers compared the false-negative rate of a conventional D-dimer threshold with an age-adjusted D-dimer threshold.
The team found that age-adjusted D-dimer reduced the need for CTPA by 18.3% (95% CI, 15.9%-21.0%), compared to conventional D-dimer.
However, in the 273 patients with a negative age-adjusted D-dimer result and an RGS of 10 or greater, 4 PEs occurred within 90 days. This translates to a false-negative result rate of 1.5% (95% CI, 0.4%-3.7%).
In comparison, the false-negative rate for conventional D-dimer was 0% (95% CI, 0%-2.8%). Among the 104 patients who had a negative test result and an RGS of 10 or greater, there were no PEs within 90 days.
These results suggest an age-adjusted D-dimer threshold does reduce the need for imaging in patients older than 50, the researchers said. They added that this method is probably safe for these patients, but a prospective trial is needed to more thoroughly investigate safety. ![]()
Credit: Medical College
of Georgia
A new study shows that, although age-adjusted D-dimer testing produces fewer false-positive results than conventional D-dimer testing, some cases of pulmonary embolism (PE) slip through the cracks.
Researchers compared the two testing methods in patients older than 50 and found that using an age-adjusted D-dimer threshold reduced the need for additional imaging.
Unfortunately, it also had a false-negative rate of 1.5%, failing to catch PE in 4 patients.
Scott Woller, MD, of Intermountain Medical Center in Salt Lake City, Utah, and his colleagues reported these findings in CHEST.
The team conducted this study with the goal of eliminating false-positive D-dimer results and reducing the need for additional imaging, which can be detrimental to older patients.
“A CT scan is most often used to ultimately rule out a pulmonary embolism,” Dr Woller said. “However, it delivers radiation to the patient and contrast dye.”
“Elderly patients are at greater risk for inadvertent harm related to the CT scan, and the contrast dye may also impact kidney function. Plus, the scan adds to the cost of the patient’s care. If we can safely and accurately diagnose the patient’s risk of a pulmonary embolism using [age-adjusted D-dimer], we can eliminate the need for additional imaging tests.”
With this in mind, the researchers evaluated 923 patients older than 50 years of age who presented to the emergency department at Intermountain Medical Center with a suspected PE, a calculated Revised Geneva Score (RGS), and a D-dimer test.
All of the patients underwent CT pulmonary angiography (CTPA), and the researchers compared the false-negative rate of a conventional D-dimer threshold with an age-adjusted D-dimer threshold.
The team found that age-adjusted D-dimer reduced the need for CTPA by 18.3% (95% CI, 15.9%-21.0%), compared to conventional D-dimer.
However, in the 273 patients with a negative age-adjusted D-dimer result and an RGS of 10 or greater, 4 PEs occurred within 90 days. This translates to a false-negative result rate of 1.5% (95% CI, 0.4%-3.7%).
In comparison, the false-negative rate for conventional D-dimer was 0% (95% CI, 0%-2.8%). Among the 104 patients who had a negative test result and an RGS of 10 or greater, there were no PEs within 90 days.
These results suggest an age-adjusted D-dimer threshold does reduce the need for imaging in patients older than 50, the researchers said. They added that this method is probably safe for these patients, but a prospective trial is needed to more thoroughly investigate safety. ![]()
FDA approves new formulation of drug for ALL

The US Food and Drug Administration (FDA) has approved the intravenous administration of asparaginase Erwinia chrysanthemi (Erwinaze).
The product is indicated as a component of a multi-agent chemotherapy regimen to treat patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E coli-derived asparaginase.
Previously, the only FDA-approved route of administration for asparaginase Erwinia chrysanthemi was through intramuscular injection.
The FDA’s decision to expand the drug’s use was based on a pharmacokinetic study (published in Blood in 2013) of intravenous asparaginase Erwinia chrysanthemi.
The trial included 30 patients with ALL or lymphoblastic lymphoma who developed hypersensitivity (grade ≥ 2) to E coli–derived asparaginase. The patients’ median age was 6.5 years (range, 1-17), 63% were male, and 83% were Caucasian.
Patients received intravenous asparaginase Erwinia chrysanthemi at 25,000 IU/m2/dose, on a Monday/Wednesday/Friday schedule for 2 consecutive weeks (6 doses=1 cycle) for each dose of pegaspargase remaining in their original treatment plan. All other chemotherapy was continued per the original treatment plan.
Before the first dose of intravenous asparaginase Erwinia chrysanthemi, nadir serum asparaginase activity (NSAA) levels were below the limit of quantification (defined as 0.0129 IU/mL) for 91% of patients.
The study’s primary endpoint was the proportion of patients who achieved NSAA ≥ 0.1 IU/mL, which has been associated with complete asparagine depletion, at 48 hours after dose 5 in cycle 1. Nineteen of the 23 evaluable patients (83%) achieved this endpoint.
A secondary objective of the study was to determine the proportion of patients who achieved NSAA ≥ 0.1 IU/mL at 72 hours after dose 6 in cycle 1. Nine patients (45%) achieved this endpoint.
In all 30 patients, the most common asparaginase-related toxicities reported during cycle 1 were hypersensitivity (23%), vomiting (20%), nausea (20%), and hyperglycemia (13%). Pancreatitis and thrombosis each occurred in 3% of patients. One patient experienced a transient ischemic attack.
The most common grade 3 or 4 adverse event was febrile neutropenia (7%). Four patients discontinued treatment before completing cycle 1—3 of them due to hypersensitivity and 1 due to pancreatitis. There were no deaths.
This study was funded by Jazz Pharmaceuticals, the company developing asparaginase Erwinia chrysanthemi. ![]()
The US Food and Drug Administration (FDA) has approved the intravenous administration of asparaginase Erwinia chrysanthemi (Erwinaze).
The product is indicated as a component of a multi-agent chemotherapy regimen to treat patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E coli-derived asparaginase.
Previously, the only FDA-approved route of administration for asparaginase Erwinia chrysanthemi was through intramuscular injection.
The FDA’s decision to expand the drug’s use was based on a pharmacokinetic study (published in Blood in 2013) of intravenous asparaginase Erwinia chrysanthemi.
The trial included 30 patients with ALL or lymphoblastic lymphoma who developed hypersensitivity (grade ≥ 2) to E coli–derived asparaginase. The patients’ median age was 6.5 years (range, 1-17), 63% were male, and 83% were Caucasian.
Patients received intravenous asparaginase Erwinia chrysanthemi at 25,000 IU/m2/dose, on a Monday/Wednesday/Friday schedule for 2 consecutive weeks (6 doses=1 cycle) for each dose of pegaspargase remaining in their original treatment plan. All other chemotherapy was continued per the original treatment plan.
Before the first dose of intravenous asparaginase Erwinia chrysanthemi, nadir serum asparaginase activity (NSAA) levels were below the limit of quantification (defined as 0.0129 IU/mL) for 91% of patients.
The study’s primary endpoint was the proportion of patients who achieved NSAA ≥ 0.1 IU/mL, which has been associated with complete asparagine depletion, at 48 hours after dose 5 in cycle 1. Nineteen of the 23 evaluable patients (83%) achieved this endpoint.
A secondary objective of the study was to determine the proportion of patients who achieved NSAA ≥ 0.1 IU/mL at 72 hours after dose 6 in cycle 1. Nine patients (45%) achieved this endpoint.
In all 30 patients, the most common asparaginase-related toxicities reported during cycle 1 were hypersensitivity (23%), vomiting (20%), nausea (20%), and hyperglycemia (13%). Pancreatitis and thrombosis each occurred in 3% of patients. One patient experienced a transient ischemic attack.
The most common grade 3 or 4 adverse event was febrile neutropenia (7%). Four patients discontinued treatment before completing cycle 1—3 of them due to hypersensitivity and 1 due to pancreatitis. There were no deaths.
This study was funded by Jazz Pharmaceuticals, the company developing asparaginase Erwinia chrysanthemi. ![]()
The US Food and Drug Administration (FDA) has approved the intravenous administration of asparaginase Erwinia chrysanthemi (Erwinaze).
The product is indicated as a component of a multi-agent chemotherapy regimen to treat patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E coli-derived asparaginase.
Previously, the only FDA-approved route of administration for asparaginase Erwinia chrysanthemi was through intramuscular injection.
The FDA’s decision to expand the drug’s use was based on a pharmacokinetic study (published in Blood in 2013) of intravenous asparaginase Erwinia chrysanthemi.
The trial included 30 patients with ALL or lymphoblastic lymphoma who developed hypersensitivity (grade ≥ 2) to E coli–derived asparaginase. The patients’ median age was 6.5 years (range, 1-17), 63% were male, and 83% were Caucasian.
Patients received intravenous asparaginase Erwinia chrysanthemi at 25,000 IU/m2/dose, on a Monday/Wednesday/Friday schedule for 2 consecutive weeks (6 doses=1 cycle) for each dose of pegaspargase remaining in their original treatment plan. All other chemotherapy was continued per the original treatment plan.
Before the first dose of intravenous asparaginase Erwinia chrysanthemi, nadir serum asparaginase activity (NSAA) levels were below the limit of quantification (defined as 0.0129 IU/mL) for 91% of patients.
The study’s primary endpoint was the proportion of patients who achieved NSAA ≥ 0.1 IU/mL, which has been associated with complete asparagine depletion, at 48 hours after dose 5 in cycle 1. Nineteen of the 23 evaluable patients (83%) achieved this endpoint.
A secondary objective of the study was to determine the proportion of patients who achieved NSAA ≥ 0.1 IU/mL at 72 hours after dose 6 in cycle 1. Nine patients (45%) achieved this endpoint.
In all 30 patients, the most common asparaginase-related toxicities reported during cycle 1 were hypersensitivity (23%), vomiting (20%), nausea (20%), and hyperglycemia (13%). Pancreatitis and thrombosis each occurred in 3% of patients. One patient experienced a transient ischemic attack.
The most common grade 3 or 4 adverse event was febrile neutropenia (7%). Four patients discontinued treatment before completing cycle 1—3 of them due to hypersensitivity and 1 due to pancreatitis. There were no deaths.
This study was funded by Jazz Pharmaceuticals, the company developing asparaginase Erwinia chrysanthemi. ![]()