User login
Point/Counterpoint: Is screening for asymptomatic carotid artery stenosis justified?
Introduction
The U.S. Preventive Services Task Force has just published its latest guidance on carotid screening for asymptomatic disease, basically stating that it should not be done (see story page 1). In this Point-Counterpoint Dr. Zierler and Dr. Berland provide their views on this controversial issue. From my perspective the debate must revolve around the following questions: The gist of the USPSTF statement seems to be that screening is being performed only to detect patients with critical carotid stenosis so that an intervention (CEA or CAS) can be performed. However, shouldn’t screening also be used to identify atherosclerotic burden in order to prevent cardiovascular morbidity? Whatever the reasons for screening, should national health systems pay for screening? If not, what about individual physicians charging for screenings on selected/nonselected patients? What about free screenings? And as Dr. Zierler and Dr. Berland suggest, is screening getting a bad rap just because screening and subsequent CEA or CAS are being poorly performed? Finally, I wouldn\'t be surprised if some of the task force members have had their own carotids screened despite their negative recommendation. We would be interested in your viewpoint, so please take our online, interactive poll on our home page (bottom right) to weigh in on this important issue.
Dr. Russell Samson is the Medical Editor of Vascular Specialist.
YES: Screen, but screen well.
By Todd Berland, M.D.

Every patient with symptomatic carotid artery stenosis was asymptomatic the day before. The impact of stroke can be devastating, with a 20% mortality from the acute event and 40%-50% survival over the next 5 years. Of those surviving the initial event, a significant percentage of patients are unable to return to work, and up to 25% over the age of 65 require long-term institutional care.1 There is no doubt that the emotional, financial, and societal burden of caring for stroke patients is significant. The Asymptomatic Carotid Atherosclerosis Study and the Asymptomatic Carotid Surgery Trial demonstrated a significant reduction in stroke in asymptomatic, high-grade carotid artery stenosis patients treated with carotid endarterectomy compared to medical management alone.2,3 So wouldn’t it seem as if carotid artery screening would be beneficial?
The U.S. Preventive Services Task Force recommended against routine carotid screening because of its "high risk" and low reward. I believe that when a certified lab screens the appropriate population such as individuals over 55 with cardiovascular risk factors that include hypertension, diabetes, smoking, and hypercholesterolemia, and combines this with an intervention that has a low stroke and morbidity rate, then the balance is tipped and carotid screening becomes both low risk and high reward.
One of the problems with carotid ultrasound is that too many entrepreneurs have made a business of it. Suboptimal equipment is being used by uncertified technicians in medical offices and church parking lots all across this country. It’s no surprise that the false positives are going to be high in these situations. Also, when one combines all of the above with the screening of those who aren’t at risk, where the general prevalence of disease is low, it can be a recipe for disaster. This is going to lead to additional studies such as CT angiograms or possibly even cerebral angiograms, both of which have inherent risks.
Even though it’s possible to understand why the USPSTF may have concluded against routine screening, I believe that at-risk patients should be screened by a certified lab, and that physicians performing the interventions should be able to do so with low morbidity and mortality. Vascular surgeons have been marginalized over recent years as many others have become interested in finding and treating carotid disease. Most of us are either registered vascular technologists or registered physicians in vascular interpretation, and our labs are certified by the Intersocietal Accreditation Commission. We go through hours of continuing medical education in regard to vascular ultrasound, and our labs’ results are tested and certified for accuracy.
We need to convince insurance companies and the Centers for Medicare & Medicaid Services that these studies should be permitted only in certified labs and the results interpreted by certified physicians such as vascular surgeons.
Moreover, when indicated, the interventions should be carried out by vascular surgeons who are trained to perform both carotid endarter- ectomy and carotid artery stenting to be able to offer the patient the best individualized treatment.
Dr. Berland is director of outpatient vascular interventions at NYU Langone Medical Center.
References
1. Circulation 2012;125:188.
2. JAMA 1995:273:1421-8.
3. Lancet 2004;363:1491-502.
NO: General screening is not appropriate.
By R. Eugene Zierler, M.D.

To most vascular specialists, the concept of detecting asymptomatic carotid stenosis and intervening to prevent stroke makes intuitive sense, but is it consistent with the evidence? In 2007, the USPSTF concluded that the general asymptomatic adult population should not be screened for carotid stenosis, and this recommendation has been reiterated in a 2014 draft recommendation statement.1,2 Other groups, including our own Society for Vascular Surgery, have published similar recommendations.3,4
Arguments in favor of screening for asymptomatic carotid stenosis are often based on the results of randomized controlled trials such as the Asymptomatic Carotid Atherosclerosis Study, which was reported in 1995.5 However, while these trials are historically important, they are no longer clinically relevant. Surgical and catheter-based interventions for carotid atherosclerosis have evolved significantly in the last 2 decades, but the outcomes associated with modern intensive medical therapy have also improved dramatically.6,7 It is not clear that carotid endarterectomy or stenting is superior to current medical management for asymptomatic carotid stenosis, and new trials such as the recently announced CREST-2 are designed to answer these important questions.
The relatively poor reliability and accuracy of duplex ultrasound as a screening test for carotid stenosis is a major theme in the USPSTF draft recommendations, but in spite of this criticism, carotid duplex scanning has served as a clinically valuable method for classifying the severity of carotid stenosis for more than 30 years.8 As pointed out by others, the best way to ensure quality in the vascular laboratory is to recognize the importance of certified vascular sonographers, accredited vascular laboratories, and qualified medical staff. But despite high-quality ultrasound testing, relying on carotid stenosis as a marker of stroke risk has significant limitations. While there is an association between the degree of carotid stenosis and risk of stroke, many patients with severe carotid stenosis do not have strokes and some with moderate stenosis do have strokes. For example, it was reported that 61% of the symptomatic patients in the North American Symptomatic Carotid Endarterectomy Trial had less than 50% carotid stenosis.9 This suggests that stenosis severity alone does not identify those patients who are at the highest risk for stroke. Fortunately, the concept of the "vulnerable plaque" is promising as a means for more accurate risk stratification, and features such as intraplaque hemorrhage and thin or ruptured fibrous caps do correlate with a high risk for stroke.10 Experience has shown that these features can be characterized by ultrasound.11
So although screening of the general population for asymptomatic carotid stenosis is not justified, there still may be subgroups of patients with specific risk factors and plaque features that could benefit from early intervention, and future clinical trials will establish whether or not this hypothesis has merit. Until more data are available the issue of screening for asymptomatic carotid stenosis will continue to provoke debate on multiple levels. Carotid disease screening is not covered by insurance, so cost and ability to pay are key factors to consider. In these discussions, the health of the patient and the population must always be the first priority, and clinical decision-making should be evidence based.
Dr. Zierler is professor of surgery at the University of Washington and medical director of the D.E. Strandness Jr. Vascular Laboratory at the university’s medical center and Harborview Medical Center, Seattle. He is also an associate medical editor of Vascular Specialist.
References
1. Ann. Intern. Med. 2007;147:860-70.
2. uspreventiveservicestaskforce.org/.htm.
3. JACC 2011;57:e16-94.
4. J. Vasc. Surg. 2011;54:e1-31.
5. JAMA 1995:273:1421-8.
6. Circulation 2013;127:739-42.
7. Stroke 2009;40:e573-83.
8. Vasc. Endovascular Surg. 2012;46:466-74.
9. N. Engl. J. Med. 1998;339:1415-25.
10. Imaging Med. 2010;2:63-75.
11. J. Vasc. Surg. 2010;52:1486-96.
Introduction
The U.S. Preventive Services Task Force has just published its latest guidance on carotid screening for asymptomatic disease, basically stating that it should not be done (see story page 1). In this Point-Counterpoint Dr. Zierler and Dr. Berland provide their views on this controversial issue. From my perspective the debate must revolve around the following questions: The gist of the USPSTF statement seems to be that screening is being performed only to detect patients with critical carotid stenosis so that an intervention (CEA or CAS) can be performed. However, shouldn’t screening also be used to identify atherosclerotic burden in order to prevent cardiovascular morbidity? Whatever the reasons for screening, should national health systems pay for screening? If not, what about individual physicians charging for screenings on selected/nonselected patients? What about free screenings? And as Dr. Zierler and Dr. Berland suggest, is screening getting a bad rap just because screening and subsequent CEA or CAS are being poorly performed? Finally, I wouldn\'t be surprised if some of the task force members have had their own carotids screened despite their negative recommendation. We would be interested in your viewpoint, so please take our online, interactive poll on our home page (bottom right) to weigh in on this important issue.
Dr. Russell Samson is the Medical Editor of Vascular Specialist.
YES: Screen, but screen well.
By Todd Berland, M.D.
Every patient with symptomatic carotid artery stenosis was asymptomatic the day before. The impact of stroke can be devastating, with a 20% mortality from the acute event and 40%-50% survival over the next 5 years. Of those surviving the initial event, a significant percentage of patients are unable to return to work, and up to 25% over the age of 65 require long-term institutional care.1 There is no doubt that the emotional, financial, and societal burden of caring for stroke patients is significant. The Asymptomatic Carotid Atherosclerosis Study and the Asymptomatic Carotid Surgery Trial demonstrated a significant reduction in stroke in asymptomatic, high-grade carotid artery stenosis patients treated with carotid endarterectomy compared to medical management alone.2,3 So wouldn’t it seem as if carotid artery screening would be beneficial?
The U.S. Preventive Services Task Force recommended against routine carotid screening because of its "high risk" and low reward. I believe that when a certified lab screens the appropriate population such as individuals over 55 with cardiovascular risk factors that include hypertension, diabetes, smoking, and hypercholesterolemia, and combines this with an intervention that has a low stroke and morbidity rate, then the balance is tipped and carotid screening becomes both low risk and high reward.
One of the problems with carotid ultrasound is that too many entrepreneurs have made a business of it. Suboptimal equipment is being used by uncertified technicians in medical offices and church parking lots all across this country. It’s no surprise that the false positives are going to be high in these situations. Also, when one combines all of the above with the screening of those who aren’t at risk, where the general prevalence of disease is low, it can be a recipe for disaster. This is going to lead to additional studies such as CT angiograms or possibly even cerebral angiograms, both of which have inherent risks.
Even though it’s possible to understand why the USPSTF may have concluded against routine screening, I believe that at-risk patients should be screened by a certified lab, and that physicians performing the interventions should be able to do so with low morbidity and mortality. Vascular surgeons have been marginalized over recent years as many others have become interested in finding and treating carotid disease. Most of us are either registered vascular technologists or registered physicians in vascular interpretation, and our labs are certified by the Intersocietal Accreditation Commission. We go through hours of continuing medical education in regard to vascular ultrasound, and our labs’ results are tested and certified for accuracy.
We need to convince insurance companies and the Centers for Medicare & Medicaid Services that these studies should be permitted only in certified labs and the results interpreted by certified physicians such as vascular surgeons.
Moreover, when indicated, the interventions should be carried out by vascular surgeons who are trained to perform both carotid endarter- ectomy and carotid artery stenting to be able to offer the patient the best individualized treatment.
Dr. Berland is director of outpatient vascular interventions at NYU Langone Medical Center.
References
1. Circulation 2012;125:188.
2. JAMA 1995:273:1421-8.
3. Lancet 2004;363:1491-502.
NO: General screening is not appropriate.
By R. Eugene Zierler, M.D.
To most vascular specialists, the concept of detecting asymptomatic carotid stenosis and intervening to prevent stroke makes intuitive sense, but is it consistent with the evidence? In 2007, the USPSTF concluded that the general asymptomatic adult population should not be screened for carotid stenosis, and this recommendation has been reiterated in a 2014 draft recommendation statement.1,2 Other groups, including our own Society for Vascular Surgery, have published similar recommendations.3,4
Arguments in favor of screening for asymptomatic carotid stenosis are often based on the results of randomized controlled trials such as the Asymptomatic Carotid Atherosclerosis Study, which was reported in 1995.5 However, while these trials are historically important, they are no longer clinically relevant. Surgical and catheter-based interventions for carotid atherosclerosis have evolved significantly in the last 2 decades, but the outcomes associated with modern intensive medical therapy have also improved dramatically.6,7 It is not clear that carotid endarterectomy or stenting is superior to current medical management for asymptomatic carotid stenosis, and new trials such as the recently announced CREST-2 are designed to answer these important questions.
The relatively poor reliability and accuracy of duplex ultrasound as a screening test for carotid stenosis is a major theme in the USPSTF draft recommendations, but in spite of this criticism, carotid duplex scanning has served as a clinically valuable method for classifying the severity of carotid stenosis for more than 30 years.8 As pointed out by others, the best way to ensure quality in the vascular laboratory is to recognize the importance of certified vascular sonographers, accredited vascular laboratories, and qualified medical staff. But despite high-quality ultrasound testing, relying on carotid stenosis as a marker of stroke risk has significant limitations. While there is an association between the degree of carotid stenosis and risk of stroke, many patients with severe carotid stenosis do not have strokes and some with moderate stenosis do have strokes. For example, it was reported that 61% of the symptomatic patients in the North American Symptomatic Carotid Endarterectomy Trial had less than 50% carotid stenosis.9 This suggests that stenosis severity alone does not identify those patients who are at the highest risk for stroke. Fortunately, the concept of the "vulnerable plaque" is promising as a means for more accurate risk stratification, and features such as intraplaque hemorrhage and thin or ruptured fibrous caps do correlate with a high risk for stroke.10 Experience has shown that these features can be characterized by ultrasound.11
So although screening of the general population for asymptomatic carotid stenosis is not justified, there still may be subgroups of patients with specific risk factors and plaque features that could benefit from early intervention, and future clinical trials will establish whether or not this hypothesis has merit. Until more data are available the issue of screening for asymptomatic carotid stenosis will continue to provoke debate on multiple levels. Carotid disease screening is not covered by insurance, so cost and ability to pay are key factors to consider. In these discussions, the health of the patient and the population must always be the first priority, and clinical decision-making should be evidence based.
Dr. Zierler is professor of surgery at the University of Washington and medical director of the D.E. Strandness Jr. Vascular Laboratory at the university’s medical center and Harborview Medical Center, Seattle. He is also an associate medical editor of Vascular Specialist.
References
1. Ann. Intern. Med. 2007;147:860-70.
2. uspreventiveservicestaskforce.org/.htm.
3. JACC 2011;57:e16-94.
4. J. Vasc. Surg. 2011;54:e1-31.
5. JAMA 1995:273:1421-8.
6. Circulation 2013;127:739-42.
7. Stroke 2009;40:e573-83.
8. Vasc. Endovascular Surg. 2012;46:466-74.
9. N. Engl. J. Med. 1998;339:1415-25.
10. Imaging Med. 2010;2:63-75.
11. J. Vasc. Surg. 2010;52:1486-96.
Introduction
The U.S. Preventive Services Task Force has just published its latest guidance on carotid screening for asymptomatic disease, basically stating that it should not be done (see story page 1). In this Point-Counterpoint Dr. Zierler and Dr. Berland provide their views on this controversial issue. From my perspective the debate must revolve around the following questions: The gist of the USPSTF statement seems to be that screening is being performed only to detect patients with critical carotid stenosis so that an intervention (CEA or CAS) can be performed. However, shouldn’t screening also be used to identify atherosclerotic burden in order to prevent cardiovascular morbidity? Whatever the reasons for screening, should national health systems pay for screening? If not, what about individual physicians charging for screenings on selected/nonselected patients? What about free screenings? And as Dr. Zierler and Dr. Berland suggest, is screening getting a bad rap just because screening and subsequent CEA or CAS are being poorly performed? Finally, I wouldn\'t be surprised if some of the task force members have had their own carotids screened despite their negative recommendation. We would be interested in your viewpoint, so please take our online, interactive poll on our home page (bottom right) to weigh in on this important issue.
Dr. Russell Samson is the Medical Editor of Vascular Specialist.
YES: Screen, but screen well.
By Todd Berland, M.D.
Every patient with symptomatic carotid artery stenosis was asymptomatic the day before. The impact of stroke can be devastating, with a 20% mortality from the acute event and 40%-50% survival over the next 5 years. Of those surviving the initial event, a significant percentage of patients are unable to return to work, and up to 25% over the age of 65 require long-term institutional care.1 There is no doubt that the emotional, financial, and societal burden of caring for stroke patients is significant. The Asymptomatic Carotid Atherosclerosis Study and the Asymptomatic Carotid Surgery Trial demonstrated a significant reduction in stroke in asymptomatic, high-grade carotid artery stenosis patients treated with carotid endarterectomy compared to medical management alone.2,3 So wouldn’t it seem as if carotid artery screening would be beneficial?
The U.S. Preventive Services Task Force recommended against routine carotid screening because of its "high risk" and low reward. I believe that when a certified lab screens the appropriate population such as individuals over 55 with cardiovascular risk factors that include hypertension, diabetes, smoking, and hypercholesterolemia, and combines this with an intervention that has a low stroke and morbidity rate, then the balance is tipped and carotid screening becomes both low risk and high reward.
One of the problems with carotid ultrasound is that too many entrepreneurs have made a business of it. Suboptimal equipment is being used by uncertified technicians in medical offices and church parking lots all across this country. It’s no surprise that the false positives are going to be high in these situations. Also, when one combines all of the above with the screening of those who aren’t at risk, where the general prevalence of disease is low, it can be a recipe for disaster. This is going to lead to additional studies such as CT angiograms or possibly even cerebral angiograms, both of which have inherent risks.
Even though it’s possible to understand why the USPSTF may have concluded against routine screening, I believe that at-risk patients should be screened by a certified lab, and that physicians performing the interventions should be able to do so with low morbidity and mortality. Vascular surgeons have been marginalized over recent years as many others have become interested in finding and treating carotid disease. Most of us are either registered vascular technologists or registered physicians in vascular interpretation, and our labs are certified by the Intersocietal Accreditation Commission. We go through hours of continuing medical education in regard to vascular ultrasound, and our labs’ results are tested and certified for accuracy.
We need to convince insurance companies and the Centers for Medicare & Medicaid Services that these studies should be permitted only in certified labs and the results interpreted by certified physicians such as vascular surgeons.
Moreover, when indicated, the interventions should be carried out by vascular surgeons who are trained to perform both carotid endarter- ectomy and carotid artery stenting to be able to offer the patient the best individualized treatment.
Dr. Berland is director of outpatient vascular interventions at NYU Langone Medical Center.
References
1. Circulation 2012;125:188.
2. JAMA 1995:273:1421-8.
3. Lancet 2004;363:1491-502.
NO: General screening is not appropriate.
By R. Eugene Zierler, M.D.
To most vascular specialists, the concept of detecting asymptomatic carotid stenosis and intervening to prevent stroke makes intuitive sense, but is it consistent with the evidence? In 2007, the USPSTF concluded that the general asymptomatic adult population should not be screened for carotid stenosis, and this recommendation has been reiterated in a 2014 draft recommendation statement.1,2 Other groups, including our own Society for Vascular Surgery, have published similar recommendations.3,4
Arguments in favor of screening for asymptomatic carotid stenosis are often based on the results of randomized controlled trials such as the Asymptomatic Carotid Atherosclerosis Study, which was reported in 1995.5 However, while these trials are historically important, they are no longer clinically relevant. Surgical and catheter-based interventions for carotid atherosclerosis have evolved significantly in the last 2 decades, but the outcomes associated with modern intensive medical therapy have also improved dramatically.6,7 It is not clear that carotid endarterectomy or stenting is superior to current medical management for asymptomatic carotid stenosis, and new trials such as the recently announced CREST-2 are designed to answer these important questions.
The relatively poor reliability and accuracy of duplex ultrasound as a screening test for carotid stenosis is a major theme in the USPSTF draft recommendations, but in spite of this criticism, carotid duplex scanning has served as a clinically valuable method for classifying the severity of carotid stenosis for more than 30 years.8 As pointed out by others, the best way to ensure quality in the vascular laboratory is to recognize the importance of certified vascular sonographers, accredited vascular laboratories, and qualified medical staff. But despite high-quality ultrasound testing, relying on carotid stenosis as a marker of stroke risk has significant limitations. While there is an association between the degree of carotid stenosis and risk of stroke, many patients with severe carotid stenosis do not have strokes and some with moderate stenosis do have strokes. For example, it was reported that 61% of the symptomatic patients in the North American Symptomatic Carotid Endarterectomy Trial had less than 50% carotid stenosis.9 This suggests that stenosis severity alone does not identify those patients who are at the highest risk for stroke. Fortunately, the concept of the "vulnerable plaque" is promising as a means for more accurate risk stratification, and features such as intraplaque hemorrhage and thin or ruptured fibrous caps do correlate with a high risk for stroke.10 Experience has shown that these features can be characterized by ultrasound.11
So although screening of the general population for asymptomatic carotid stenosis is not justified, there still may be subgroups of patients with specific risk factors and plaque features that could benefit from early intervention, and future clinical trials will establish whether or not this hypothesis has merit. Until more data are available the issue of screening for asymptomatic carotid stenosis will continue to provoke debate on multiple levels. Carotid disease screening is not covered by insurance, so cost and ability to pay are key factors to consider. In these discussions, the health of the patient and the population must always be the first priority, and clinical decision-making should be evidence based.
Dr. Zierler is professor of surgery at the University of Washington and medical director of the D.E. Strandness Jr. Vascular Laboratory at the university’s medical center and Harborview Medical Center, Seattle. He is also an associate medical editor of Vascular Specialist.
References
1. Ann. Intern. Med. 2007;147:860-70.
2. uspreventiveservicestaskforce.org/.htm.
3. JACC 2011;57:e16-94.
4. J. Vasc. Surg. 2011;54:e1-31.
5. JAMA 1995:273:1421-8.
6. Circulation 2013;127:739-42.
7. Stroke 2009;40:e573-83.
8. Vasc. Endovascular Surg. 2012;46:466-74.
9. N. Engl. J. Med. 1998;339:1415-25.
10. Imaging Med. 2010;2:63-75.
11. J. Vasc. Surg. 2010;52:1486-96.

Steps to incorporate business knowledge into the medical school curriculum
Declining reimbursements, health care law changes, and increased costs means the modern physician needs to have knowledge in business, law, and medicine. In today’s health care environment business acumen is becoming a necessity few practices can survive without. A recent CNN/Money article highlighted physicians going bankrupt. Lack of a basic understanding of running a business was a common theme for failed practices in the article. Another article, written in Forbes on "Doctors Going Broke" stated that failed business models along with the overly complex coding system were factors contributing to increased bankruptcy rates among physicians. As one physician stated, "Doctors are trained in medicine but not how to run a business." In bankruptcy, everyone loses: the physician, the patients, and the employees.
The need for medically related business and managerial training for medical students and physicians is an area of education that has been frequently discussed among physicians in private and academic practice. However, little has been done by physicians or in formal programs of medical or graduate medical education to address the need to introduce this type of training. Sure there are many programs offering dual MD/MBA degrees: Harvard, Dartmouth, and the University of Texas to name a few. But, is getting an MBA really necessary? This requires a minimum of an extra year of study. Also, one must be selected into such programs, not to mention more debt incurred for the student. The MD/MBA programs do not offer positions to all medical students; they can’t, and the logistics simply aren’t feasible. Why not make business knowledge available to all medical students by making it part of the medical curriculum?
The Business and Medicine Organization (BAM) was founded at the Florida State University College of Medicine on Nov. 1, 2011. The purpose of BAM is to educate our students about the practical and financial aspects of working in the medical field in the 21st century. BAM provides speakers with topics of interest relating to the business aspects of medicine. This includes employment, starting a practice, legal aspects, contracts, investments, insurance companies, and basic economic principles. The ultimate goal of BAM is to incorporate a business course into the medical school curriculum. The vision is for this course to be an elective at first in cooperation with the Florida State College of Business. Eventually, this will be a required course.
Since its inception, BAM has grown from 4 members to almost 100 members. This year it started a yearly scholarship given to those students interested in "the business aspect of medicine." We now have a working partnership with the College of Business. The next step is incorporating business into the curriculum. Last year’s BAM President, Aarian Afshari, met with the curriculum committee to push a business elective into the curriculum. We are still actively working with the curriculum committee on this issue. Since the current member count of BAM is close to 100 members, we will exert a strong influence on the curriculum committee’s decision. Basic business knowledge is a necessity for the physician in the economic reality of health care in the 21st century.
Mr. Hayson is currently a fourth-year medical student at Florida State University, Tallahassee. His goal is to become a vascular surgeon. Before entering medical school, he worked on Wall Street for 5 years as an equities and commodities trader. He has a BS in Finance from Lehigh University, Bethlehem, Penn., and a BS in Molecular and Microbiology from the University of Central Florida, Orlando.
Declining reimbursements, health care law changes, and increased costs means the modern physician needs to have knowledge in business, law, and medicine. In today’s health care environment business acumen is becoming a necessity few practices can survive without. A recent CNN/Money article highlighted physicians going bankrupt. Lack of a basic understanding of running a business was a common theme for failed practices in the article. Another article, written in Forbes on "Doctors Going Broke" stated that failed business models along with the overly complex coding system were factors contributing to increased bankruptcy rates among physicians. As one physician stated, "Doctors are trained in medicine but not how to run a business." In bankruptcy, everyone loses: the physician, the patients, and the employees.
The need for medically related business and managerial training for medical students and physicians is an area of education that has been frequently discussed among physicians in private and academic practice. However, little has been done by physicians or in formal programs of medical or graduate medical education to address the need to introduce this type of training. Sure there are many programs offering dual MD/MBA degrees: Harvard, Dartmouth, and the University of Texas to name a few. But, is getting an MBA really necessary? This requires a minimum of an extra year of study. Also, one must be selected into such programs, not to mention more debt incurred for the student. The MD/MBA programs do not offer positions to all medical students; they can’t, and the logistics simply aren’t feasible. Why not make business knowledge available to all medical students by making it part of the medical curriculum?
The Business and Medicine Organization (BAM) was founded at the Florida State University College of Medicine on Nov. 1, 2011. The purpose of BAM is to educate our students about the practical and financial aspects of working in the medical field in the 21st century. BAM provides speakers with topics of interest relating to the business aspects of medicine. This includes employment, starting a practice, legal aspects, contracts, investments, insurance companies, and basic economic principles. The ultimate goal of BAM is to incorporate a business course into the medical school curriculum. The vision is for this course to be an elective at first in cooperation with the Florida State College of Business. Eventually, this will be a required course.
Since its inception, BAM has grown from 4 members to almost 100 members. This year it started a yearly scholarship given to those students interested in "the business aspect of medicine." We now have a working partnership with the College of Business. The next step is incorporating business into the curriculum. Last year’s BAM President, Aarian Afshari, met with the curriculum committee to push a business elective into the curriculum. We are still actively working with the curriculum committee on this issue. Since the current member count of BAM is close to 100 members, we will exert a strong influence on the curriculum committee’s decision. Basic business knowledge is a necessity for the physician in the economic reality of health care in the 21st century.
Mr. Hayson is currently a fourth-year medical student at Florida State University, Tallahassee. His goal is to become a vascular surgeon. Before entering medical school, he worked on Wall Street for 5 years as an equities and commodities trader. He has a BS in Finance from Lehigh University, Bethlehem, Penn., and a BS in Molecular and Microbiology from the University of Central Florida, Orlando.
Declining reimbursements, health care law changes, and increased costs means the modern physician needs to have knowledge in business, law, and medicine. In today’s health care environment business acumen is becoming a necessity few practices can survive without. A recent CNN/Money article highlighted physicians going bankrupt. Lack of a basic understanding of running a business was a common theme for failed practices in the article. Another article, written in Forbes on "Doctors Going Broke" stated that failed business models along with the overly complex coding system were factors contributing to increased bankruptcy rates among physicians. As one physician stated, "Doctors are trained in medicine but not how to run a business." In bankruptcy, everyone loses: the physician, the patients, and the employees.
The need for medically related business and managerial training for medical students and physicians is an area of education that has been frequently discussed among physicians in private and academic practice. However, little has been done by physicians or in formal programs of medical or graduate medical education to address the need to introduce this type of training. Sure there are many programs offering dual MD/MBA degrees: Harvard, Dartmouth, and the University of Texas to name a few. But, is getting an MBA really necessary? This requires a minimum of an extra year of study. Also, one must be selected into such programs, not to mention more debt incurred for the student. The MD/MBA programs do not offer positions to all medical students; they can’t, and the logistics simply aren’t feasible. Why not make business knowledge available to all medical students by making it part of the medical curriculum?
The Business and Medicine Organization (BAM) was founded at the Florida State University College of Medicine on Nov. 1, 2011. The purpose of BAM is to educate our students about the practical and financial aspects of working in the medical field in the 21st century. BAM provides speakers with topics of interest relating to the business aspects of medicine. This includes employment, starting a practice, legal aspects, contracts, investments, insurance companies, and basic economic principles. The ultimate goal of BAM is to incorporate a business course into the medical school curriculum. The vision is for this course to be an elective at first in cooperation with the Florida State College of Business. Eventually, this will be a required course.
Since its inception, BAM has grown from 4 members to almost 100 members. This year it started a yearly scholarship given to those students interested in "the business aspect of medicine." We now have a working partnership with the College of Business. The next step is incorporating business into the curriculum. Last year’s BAM President, Aarian Afshari, met with the curriculum committee to push a business elective into the curriculum. We are still actively working with the curriculum committee on this issue. Since the current member count of BAM is close to 100 members, we will exert a strong influence on the curriculum committee’s decision. Basic business knowledge is a necessity for the physician in the economic reality of health care in the 21st century.
Mr. Hayson is currently a fourth-year medical student at Florida State University, Tallahassee. His goal is to become a vascular surgeon. Before entering medical school, he worked on Wall Street for 5 years as an equities and commodities trader. He has a BS in Finance from Lehigh University, Bethlehem, Penn., and a BS in Molecular and Microbiology from the University of Central Florida, Orlando.
Farewell to indigo carmine
Suddenly, indigo carmine is in short supply throughout the United States. One manufacturer has stopped production because of a raw materials shortage; another is experiencing manufacturing delays. Neither company can estimate a resupply or product return date.1
Indigo carmine is approved by the US Food and Drug Administration (FDA) to localize ureteral orifices during cystoscopy and is commonly used in obstetrics and gynecology as a marker dye in the following additional situations:
- administered in a dilute solution via a catheter to back fill the bladder and test for bladder injury
- administered via a cannula in the uterine cavity to test the patency of the fallopian tubes
- injected into the amniotic fluid compartment to test for premature rupture of the membranes (PROM)
- injected into the amniotic fluid of a twin gestation to mark the amniotic fluid of one twin.
With this agent in short supply, we need to identify alternative marker dyes to use in our clinical practice. In this editorial, I provide a list of possible options to replace indigo carmine. Evidence supporting the use and safety of marker dyes in obstetrics and gynecology is based on small cohorts or case reports. The available evidence is of modest to low quality, and it is especially challenging to identify uncommon adverse effects. Consequently, expert opinion guides most practice recommendations.
Options to test the function of the ureters at cystoscopy
Given the lack of availability of indigo carmine, I recommend one of the following three options to test the function of the ureters at cystoscopy.
Partially fill the bladder with a solution of either sterile water or a 10% dextrose solution. (Experienced surgeons may prefer to use saline.) The turbulence of the interaction between the ureteral urine jet and instilled fluid in the bladder may permit visualization of the urine stream exiting the ureteral orifices as it swirls through the sterile water or dextrose solution. Both sterile water and a 10% dextrose solution offer a contrast in viscosity between the urine and the cystoscopy fluid, which may enhance the ability to detect the urine jet leaving the ureter.2
Administer IV methylene blue. Methylene blue is FDA approved for methemoglobinemia treatment. For this indication, it is administered intravenously at a dose of 1 to 2 mg/kgover 5 to 10 minutes. Paradoxically, when administered at a dose of >7 mg/kg, methylene blue can cause methemoglobinemia.3
Methylene blue often is provided as a 1% solution of 10 mg/mL in 10-mL vials. To use intravenous (IV) methylene blue to test ureteral function at cystoscopy, administer IV methylene blue 50 mg over 5 minutes. Use the cystoscope to view the colored urine exiting the ureteral orifices.4,5 Administering a small dose of IV furosemide may accelerate the appearance of methylene blue in the urine.
When to use caution. Methylene blue blocks serotonin metabolism by inhibiting monoamine oxidase and may precipitate a serotonin syndrome in patients taking selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), or monoamine oxidase inhibitors (MAOIs).
Common findings in the serotonin syndrome include: temperature above 38° C (100.4° F), anxiety, agitation, delirium, clonus, tremor, and hypertonia.6 I recommend that you DO NOT use IV methylene blue in patients taking these medications.7,8
Great caution should be used before administering IV methylene blue in the presence of the following clinical situations:
- renal impairment
- G6PD deficiency
- pediatric patients.
Methylene blue never should be given by a subcutaneous or intrathecal route. In pregnant women it should never be injected into the amniotic fluid compartment.
Use preoperative oral phenazo-pyridine. If the preoperative plan includes a cystoscopy procedure to test ureteral function, administering oral phenazopyridine in the preoperative holding area will result in colorization of the urine within 30 minutes, and it will persist for approximately 4 to 5 hours. To use this approach, administer one dose of phenazopyridine (Pyridium, Azo-Gesic), 100 mg or 200 mg orally, 30 minutes to 1 hour before the planned surgical start time, in the preoperative holding area.9 During cystoscopy, the urine from the ureteral jet will be colored orange.
When to use caution. Phenazopyridine should not be administered to patients with G6PD deficiency.
Option to test for bladder injury
Methylene blue. If methylene blue is used to test the integrity of the bladder, I recommend diluting 10 mg methylene blue in 1 L normal saline and then instilling the dilute solution through a catheter into the bladder.10
It is unlikely that this technique will be associated with sufficient methylene blue absorption to cause a serotonin syndrome. Therefore, this technique can be used in patients taking SSRIs, SNRIs, and MAOIs.
Option to test patency of the fallopian tubes (chromopertubation)
Methylene blue. If methylene blue is used via a cannula in the uterine cavity to test the patency of the fallopian tubes, I recommend diluting 10 mg methylene blue in 150 mL normal saline and using the dilute solution to test tubal patency.
It is unlikely that this process will lead to sufficient methylene blue absorption to cause a serotonin syndrome. Therefore, this technique can be used in patients taking SSRIs, SNRIs and MAOIs. However, if intrauterine injection of the dilute dye solution results in extravasation of the dye into the pelvic veins, a significant amount of dye can enter the circulation.11 There are case reports of anaphylaxis following intrauterine injection of methylene blue to test tubal patency.12,13
Options to diagnose PROM and to use for twin amniocentesis
None. Most experts recommend against the intra-amniotic injection of methylene blue to diagnose PROM or in twin amniocentesis procedures. Methylene blue injected into the intra-amniotic fluid during amniocentesis in multiple gestations has been reported to cause fetal bowel obstruction or atresia.14,15 Fetal death also has been reported.16 Decades ago, indocyanine green, which is FDA approved to determine cardiac output, liver blood flow, and hepatic function, was reported to be useful to mark one sac of a twin gestation during amniocentesis.17 With modern ultrasonography technology, the need to rely on a dye to mark a sac of a twin has decreased significantly.
Our only option is to cope—effectively as possible Over decades, the medical community develops patterns of patient care that are critically dependent on the availability of key pharmaceuticals and devices. When a pharmaceutical or device suddenly becomes unavailable, it can disrupt important patterns of patient care. Imagine the impact on obstetrics practice if oxytocin became unavailable due to manufacturing shortages. Likely, both market forces and government regulation are the root cause of the shortfalls. Preventing the adverse consequences of the sudden loss of key pharmaceuticals and devices is an important priority to ensure optimal care of our patients.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Indigo carmine injection. American Society of Health-System Pharmacists (ASHP) Web site. http://www.ashp.org/menu/DrugShortages/CurrentShortages/Bulletin.aspx?id=861. Updated July 29, 2014. Accessed August 21, 2014.
2. Lin BL, Iwata Y. Modified cystoscopy to evaluate unilateral traumatic injury of the ureter during pelvic surgery. Am J Obstet Gynecol. 1990;162(5):1343–1344.
3. Lee M, Sharifi R. Methylene blue versus indigo carmine. Urology. 1996;47(5):783–784.
4. Thompson JD. Operative injuries to the ureter: prevention, recognition and management. In Rock JA, Thompson JD, eds. TeLinde’s Operative Gynecology. Philadelphia, PA: Lippincott Williams & Wilkins; 1997:1155.
5. Wang AC. The techniques of trocar insertion and intraoperative urethroscopy in tension-free vaginal taping: an experience of 600 cases. Acta Obstet Gynecol Scand. 2004;83(3):293–298.
6. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112–1120.
7. Shah-Khan MG, Lovely J, Degnim AC. Safety of methylene blue dye for lymphatic mapping in patients taking selective serotonin reuptake inhibitors. Am J Surg. 2012;204(5):798–799.
8. Ng BK, Cameron AJ. The role of methylene blue in serotonin syndrome: a systematic review. Psychosomatics. 2010;51(3):194–200.
9. Hui JY, Harvey MA, Johnston SL. Confirmation of ureteric patency during cystoscopy using phenazopyridine HCl: a low-cost approach. J Obstet Gynaecol Can. 2009;31(9):845–849.
10. Moore CR, Shirodkar SP, Avallone MA, et al. Intravesical methylene blue facilitates precise identification of the diverticular neck during robot-assisted laparoscopic bladder diverticulectomy. J Laparoendosc Adv Surg Tech A. 2012;22(5):492–495.
11. Mhaskar R, Mhaskar AM. Methemoglobinemia following chromopertubation in treated pelvic tuberculosis. Int J Gynaecol Obstet. 2002;77(1):41–42.
12. Rzymski P, Wozniak J, Opala T, Wilczak M, Sajdak S. Anaphylactic reaction to methylene blue dye after laparoscopic chromopertubation. Int J Gynaecol Obstet. 2003;81(1):71–72.
13. Dewachter P, Mouton-Faivre C, Trechot P, Lieu JC, Mertes PM. Severe anaphylactic shock with methylene blue instillation. Anesth Analg. 2005;101(1):149–150.
14. McFadyen I. The dangers of intra-amniotic methylene blue. Br J Obstet Gynaecol. 1992;99(2):89–90.
15. Van der Pol JG, Wolf H, Boer K, et al. Jejunal atresia related to the use of methylene blue in genetic amniocentesis in twins. Br J Obstet Gynaecol. 1992;99(2):141–143.
16. Kidd SA, Lancaster PA, Anderson JC, et al. Fetal death after exposure to methylene blue dye during mid-trimester amniocentesis in twin pregnancy. Prenat Diagn. 1996;16(1):39–47.
17. Hobbins JC, Winsberg F, Blanchett M, et al. Section 5: fetal imaging. Prenat Diagn. 1981;1(5):35–38.

Robert L. Barbieri, MD
Dr. Barbieri is Editor in Chief, OBG Management; Chair, Obstetrics and Gynecology at Brigham and Women’s Hospital, Boston, Massachusetts; and Kate Macy Ladd Professor of Obstetrics, Gynecology, and Reproductive Biology at Harvard Medical School, Boston.
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Dr. Barbieri is Editor in Chief, OBG Management; Chair, Obstetrics and Gynecology at Brigham and Women’s Hospital, Boston, Massachusetts; and Kate Macy Ladd Professor of Obstetrics, Gynecology, and Reproductive Biology at Harvard Medical School, Boston.
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Dr. Barbieri is Editor in Chief, OBG Management; Chair, Obstetrics and Gynecology at Brigham and Women’s Hospital, Boston, Massachusetts; and Kate Macy Ladd Professor of Obstetrics, Gynecology, and Reproductive Biology at Harvard Medical School, Boston.
Dr. Barbieri reports no financial relationships relevant to this article.
Suddenly, indigo carmine is in short supply throughout the United States. One manufacturer has stopped production because of a raw materials shortage; another is experiencing manufacturing delays. Neither company can estimate a resupply or product return date.1
Indigo carmine is approved by the US Food and Drug Administration (FDA) to localize ureteral orifices during cystoscopy and is commonly used in obstetrics and gynecology as a marker dye in the following additional situations:
- administered in a dilute solution via a catheter to back fill the bladder and test for bladder injury
- administered via a cannula in the uterine cavity to test the patency of the fallopian tubes
- injected into the amniotic fluid compartment to test for premature rupture of the membranes (PROM)
- injected into the amniotic fluid of a twin gestation to mark the amniotic fluid of one twin.
With this agent in short supply, we need to identify alternative marker dyes to use in our clinical practice. In this editorial, I provide a list of possible options to replace indigo carmine. Evidence supporting the use and safety of marker dyes in obstetrics and gynecology is based on small cohorts or case reports. The available evidence is of modest to low quality, and it is especially challenging to identify uncommon adverse effects. Consequently, expert opinion guides most practice recommendations.
Options to test the function of the ureters at cystoscopy
Given the lack of availability of indigo carmine, I recommend one of the following three options to test the function of the ureters at cystoscopy.
Partially fill the bladder with a solution of either sterile water or a 10% dextrose solution. (Experienced surgeons may prefer to use saline.) The turbulence of the interaction between the ureteral urine jet and instilled fluid in the bladder may permit visualization of the urine stream exiting the ureteral orifices as it swirls through the sterile water or dextrose solution. Both sterile water and a 10% dextrose solution offer a contrast in viscosity between the urine and the cystoscopy fluid, which may enhance the ability to detect the urine jet leaving the ureter.2
Administer IV methylene blue. Methylene blue is FDA approved for methemoglobinemia treatment. For this indication, it is administered intravenously at a dose of 1 to 2 mg/kgover 5 to 10 minutes. Paradoxically, when administered at a dose of >7 mg/kg, methylene blue can cause methemoglobinemia.3
Methylene blue often is provided as a 1% solution of 10 mg/mL in 10-mL vials. To use intravenous (IV) methylene blue to test ureteral function at cystoscopy, administer IV methylene blue 50 mg over 5 minutes. Use the cystoscope to view the colored urine exiting the ureteral orifices.4,5 Administering a small dose of IV furosemide may accelerate the appearance of methylene blue in the urine.
When to use caution. Methylene blue blocks serotonin metabolism by inhibiting monoamine oxidase and may precipitate a serotonin syndrome in patients taking selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), or monoamine oxidase inhibitors (MAOIs).
Common findings in the serotonin syndrome include: temperature above 38° C (100.4° F), anxiety, agitation, delirium, clonus, tremor, and hypertonia.6 I recommend that you DO NOT use IV methylene blue in patients taking these medications.7,8
Great caution should be used before administering IV methylene blue in the presence of the following clinical situations:
- renal impairment
- G6PD deficiency
- pediatric patients.
Methylene blue never should be given by a subcutaneous or intrathecal route. In pregnant women it should never be injected into the amniotic fluid compartment.
Use preoperative oral phenazo-pyridine. If the preoperative plan includes a cystoscopy procedure to test ureteral function, administering oral phenazopyridine in the preoperative holding area will result in colorization of the urine within 30 minutes, and it will persist for approximately 4 to 5 hours. To use this approach, administer one dose of phenazopyridine (Pyridium, Azo-Gesic), 100 mg or 200 mg orally, 30 minutes to 1 hour before the planned surgical start time, in the preoperative holding area.9 During cystoscopy, the urine from the ureteral jet will be colored orange.
When to use caution. Phenazopyridine should not be administered to patients with G6PD deficiency.
Option to test for bladder injury
Methylene blue. If methylene blue is used to test the integrity of the bladder, I recommend diluting 10 mg methylene blue in 1 L normal saline and then instilling the dilute solution through a catheter into the bladder.10
It is unlikely that this technique will be associated with sufficient methylene blue absorption to cause a serotonin syndrome. Therefore, this technique can be used in patients taking SSRIs, SNRIs, and MAOIs.
Option to test patency of the fallopian tubes (chromopertubation)
Methylene blue. If methylene blue is used via a cannula in the uterine cavity to test the patency of the fallopian tubes, I recommend diluting 10 mg methylene blue in 150 mL normal saline and using the dilute solution to test tubal patency.
It is unlikely that this process will lead to sufficient methylene blue absorption to cause a serotonin syndrome. Therefore, this technique can be used in patients taking SSRIs, SNRIs and MAOIs. However, if intrauterine injection of the dilute dye solution results in extravasation of the dye into the pelvic veins, a significant amount of dye can enter the circulation.11 There are case reports of anaphylaxis following intrauterine injection of methylene blue to test tubal patency.12,13
Options to diagnose PROM and to use for twin amniocentesis
None. Most experts recommend against the intra-amniotic injection of methylene blue to diagnose PROM or in twin amniocentesis procedures. Methylene blue injected into the intra-amniotic fluid during amniocentesis in multiple gestations has been reported to cause fetal bowel obstruction or atresia.14,15 Fetal death also has been reported.16 Decades ago, indocyanine green, which is FDA approved to determine cardiac output, liver blood flow, and hepatic function, was reported to be useful to mark one sac of a twin gestation during amniocentesis.17 With modern ultrasonography technology, the need to rely on a dye to mark a sac of a twin has decreased significantly.
Our only option is to cope—effectively as possible Over decades, the medical community develops patterns of patient care that are critically dependent on the availability of key pharmaceuticals and devices. When a pharmaceutical or device suddenly becomes unavailable, it can disrupt important patterns of patient care. Imagine the impact on obstetrics practice if oxytocin became unavailable due to manufacturing shortages. Likely, both market forces and government regulation are the root cause of the shortfalls. Preventing the adverse consequences of the sudden loss of key pharmaceuticals and devices is an important priority to ensure optimal care of our patients.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Suddenly, indigo carmine is in short supply throughout the United States. One manufacturer has stopped production because of a raw materials shortage; another is experiencing manufacturing delays. Neither company can estimate a resupply or product return date.1
Indigo carmine is approved by the US Food and Drug Administration (FDA) to localize ureteral orifices during cystoscopy and is commonly used in obstetrics and gynecology as a marker dye in the following additional situations:
- administered in a dilute solution via a catheter to back fill the bladder and test for bladder injury
- administered via a cannula in the uterine cavity to test the patency of the fallopian tubes
- injected into the amniotic fluid compartment to test for premature rupture of the membranes (PROM)
- injected into the amniotic fluid of a twin gestation to mark the amniotic fluid of one twin.
With this agent in short supply, we need to identify alternative marker dyes to use in our clinical practice. In this editorial, I provide a list of possible options to replace indigo carmine. Evidence supporting the use and safety of marker dyes in obstetrics and gynecology is based on small cohorts or case reports. The available evidence is of modest to low quality, and it is especially challenging to identify uncommon adverse effects. Consequently, expert opinion guides most practice recommendations.
Options to test the function of the ureters at cystoscopy
Given the lack of availability of indigo carmine, I recommend one of the following three options to test the function of the ureters at cystoscopy.
Partially fill the bladder with a solution of either sterile water or a 10% dextrose solution. (Experienced surgeons may prefer to use saline.) The turbulence of the interaction between the ureteral urine jet and instilled fluid in the bladder may permit visualization of the urine stream exiting the ureteral orifices as it swirls through the sterile water or dextrose solution. Both sterile water and a 10% dextrose solution offer a contrast in viscosity between the urine and the cystoscopy fluid, which may enhance the ability to detect the urine jet leaving the ureter.2
Administer IV methylene blue. Methylene blue is FDA approved for methemoglobinemia treatment. For this indication, it is administered intravenously at a dose of 1 to 2 mg/kgover 5 to 10 minutes. Paradoxically, when administered at a dose of >7 mg/kg, methylene blue can cause methemoglobinemia.3
Methylene blue often is provided as a 1% solution of 10 mg/mL in 10-mL vials. To use intravenous (IV) methylene blue to test ureteral function at cystoscopy, administer IV methylene blue 50 mg over 5 minutes. Use the cystoscope to view the colored urine exiting the ureteral orifices.4,5 Administering a small dose of IV furosemide may accelerate the appearance of methylene blue in the urine.
When to use caution. Methylene blue blocks serotonin metabolism by inhibiting monoamine oxidase and may precipitate a serotonin syndrome in patients taking selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), or monoamine oxidase inhibitors (MAOIs).
Common findings in the serotonin syndrome include: temperature above 38° C (100.4° F), anxiety, agitation, delirium, clonus, tremor, and hypertonia.6 I recommend that you DO NOT use IV methylene blue in patients taking these medications.7,8
Great caution should be used before administering IV methylene blue in the presence of the following clinical situations:
- renal impairment
- G6PD deficiency
- pediatric patients.
Methylene blue never should be given by a subcutaneous or intrathecal route. In pregnant women it should never be injected into the amniotic fluid compartment.
Use preoperative oral phenazo-pyridine. If the preoperative plan includes a cystoscopy procedure to test ureteral function, administering oral phenazopyridine in the preoperative holding area will result in colorization of the urine within 30 minutes, and it will persist for approximately 4 to 5 hours. To use this approach, administer one dose of phenazopyridine (Pyridium, Azo-Gesic), 100 mg or 200 mg orally, 30 minutes to 1 hour before the planned surgical start time, in the preoperative holding area.9 During cystoscopy, the urine from the ureteral jet will be colored orange.
When to use caution. Phenazopyridine should not be administered to patients with G6PD deficiency.
Option to test for bladder injury
Methylene blue. If methylene blue is used to test the integrity of the bladder, I recommend diluting 10 mg methylene blue in 1 L normal saline and then instilling the dilute solution through a catheter into the bladder.10
It is unlikely that this technique will be associated with sufficient methylene blue absorption to cause a serotonin syndrome. Therefore, this technique can be used in patients taking SSRIs, SNRIs, and MAOIs.
Option to test patency of the fallopian tubes (chromopertubation)
Methylene blue. If methylene blue is used via a cannula in the uterine cavity to test the patency of the fallopian tubes, I recommend diluting 10 mg methylene blue in 150 mL normal saline and using the dilute solution to test tubal patency.
It is unlikely that this process will lead to sufficient methylene blue absorption to cause a serotonin syndrome. Therefore, this technique can be used in patients taking SSRIs, SNRIs and MAOIs. However, if intrauterine injection of the dilute dye solution results in extravasation of the dye into the pelvic veins, a significant amount of dye can enter the circulation.11 There are case reports of anaphylaxis following intrauterine injection of methylene blue to test tubal patency.12,13
Options to diagnose PROM and to use for twin amniocentesis
None. Most experts recommend against the intra-amniotic injection of methylene blue to diagnose PROM or in twin amniocentesis procedures. Methylene blue injected into the intra-amniotic fluid during amniocentesis in multiple gestations has been reported to cause fetal bowel obstruction or atresia.14,15 Fetal death also has been reported.16 Decades ago, indocyanine green, which is FDA approved to determine cardiac output, liver blood flow, and hepatic function, was reported to be useful to mark one sac of a twin gestation during amniocentesis.17 With modern ultrasonography technology, the need to rely on a dye to mark a sac of a twin has decreased significantly.
Our only option is to cope—effectively as possible Over decades, the medical community develops patterns of patient care that are critically dependent on the availability of key pharmaceuticals and devices. When a pharmaceutical or device suddenly becomes unavailable, it can disrupt important patterns of patient care. Imagine the impact on obstetrics practice if oxytocin became unavailable due to manufacturing shortages. Likely, both market forces and government regulation are the root cause of the shortfalls. Preventing the adverse consequences of the sudden loss of key pharmaceuticals and devices is an important priority to ensure optimal care of our patients.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Indigo carmine injection. American Society of Health-System Pharmacists (ASHP) Web site. http://www.ashp.org/menu/DrugShortages/CurrentShortages/Bulletin.aspx?id=861. Updated July 29, 2014. Accessed August 21, 2014.
2. Lin BL, Iwata Y. Modified cystoscopy to evaluate unilateral traumatic injury of the ureter during pelvic surgery. Am J Obstet Gynecol. 1990;162(5):1343–1344.
3. Lee M, Sharifi R. Methylene blue versus indigo carmine. Urology. 1996;47(5):783–784.
4. Thompson JD. Operative injuries to the ureter: prevention, recognition and management. In Rock JA, Thompson JD, eds. TeLinde’s Operative Gynecology. Philadelphia, PA: Lippincott Williams & Wilkins; 1997:1155.
5. Wang AC. The techniques of trocar insertion and intraoperative urethroscopy in tension-free vaginal taping: an experience of 600 cases. Acta Obstet Gynecol Scand. 2004;83(3):293–298.
6. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112–1120.
7. Shah-Khan MG, Lovely J, Degnim AC. Safety of methylene blue dye for lymphatic mapping in patients taking selective serotonin reuptake inhibitors. Am J Surg. 2012;204(5):798–799.
8. Ng BK, Cameron AJ. The role of methylene blue in serotonin syndrome: a systematic review. Psychosomatics. 2010;51(3):194–200.
9. Hui JY, Harvey MA, Johnston SL. Confirmation of ureteric patency during cystoscopy using phenazopyridine HCl: a low-cost approach. J Obstet Gynaecol Can. 2009;31(9):845–849.
10. Moore CR, Shirodkar SP, Avallone MA, et al. Intravesical methylene blue facilitates precise identification of the diverticular neck during robot-assisted laparoscopic bladder diverticulectomy. J Laparoendosc Adv Surg Tech A. 2012;22(5):492–495.
11. Mhaskar R, Mhaskar AM. Methemoglobinemia following chromopertubation in treated pelvic tuberculosis. Int J Gynaecol Obstet. 2002;77(1):41–42.
12. Rzymski P, Wozniak J, Opala T, Wilczak M, Sajdak S. Anaphylactic reaction to methylene blue dye after laparoscopic chromopertubation. Int J Gynaecol Obstet. 2003;81(1):71–72.
13. Dewachter P, Mouton-Faivre C, Trechot P, Lieu JC, Mertes PM. Severe anaphylactic shock with methylene blue instillation. Anesth Analg. 2005;101(1):149–150.
14. McFadyen I. The dangers of intra-amniotic methylene blue. Br J Obstet Gynaecol. 1992;99(2):89–90.
15. Van der Pol JG, Wolf H, Boer K, et al. Jejunal atresia related to the use of methylene blue in genetic amniocentesis in twins. Br J Obstet Gynaecol. 1992;99(2):141–143.
16. Kidd SA, Lancaster PA, Anderson JC, et al. Fetal death after exposure to methylene blue dye during mid-trimester amniocentesis in twin pregnancy. Prenat Diagn. 1996;16(1):39–47.
17. Hobbins JC, Winsberg F, Blanchett M, et al. Section 5: fetal imaging. Prenat Diagn. 1981;1(5):35–38.
1. Indigo carmine injection. American Society of Health-System Pharmacists (ASHP) Web site. http://www.ashp.org/menu/DrugShortages/CurrentShortages/Bulletin.aspx?id=861. Updated July 29, 2014. Accessed August 21, 2014.
2. Lin BL, Iwata Y. Modified cystoscopy to evaluate unilateral traumatic injury of the ureter during pelvic surgery. Am J Obstet Gynecol. 1990;162(5):1343–1344.
3. Lee M, Sharifi R. Methylene blue versus indigo carmine. Urology. 1996;47(5):783–784.
4. Thompson JD. Operative injuries to the ureter: prevention, recognition and management. In Rock JA, Thompson JD, eds. TeLinde’s Operative Gynecology. Philadelphia, PA: Lippincott Williams & Wilkins; 1997:1155.
5. Wang AC. The techniques of trocar insertion and intraoperative urethroscopy in tension-free vaginal taping: an experience of 600 cases. Acta Obstet Gynecol Scand. 2004;83(3):293–298.
6. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112–1120.
7. Shah-Khan MG, Lovely J, Degnim AC. Safety of methylene blue dye for lymphatic mapping in patients taking selective serotonin reuptake inhibitors. Am J Surg. 2012;204(5):798–799.
8. Ng BK, Cameron AJ. The role of methylene blue in serotonin syndrome: a systematic review. Psychosomatics. 2010;51(3):194–200.
9. Hui JY, Harvey MA, Johnston SL. Confirmation of ureteric patency during cystoscopy using phenazopyridine HCl: a low-cost approach. J Obstet Gynaecol Can. 2009;31(9):845–849.
10. Moore CR, Shirodkar SP, Avallone MA, et al. Intravesical methylene blue facilitates precise identification of the diverticular neck during robot-assisted laparoscopic bladder diverticulectomy. J Laparoendosc Adv Surg Tech A. 2012;22(5):492–495.
11. Mhaskar R, Mhaskar AM. Methemoglobinemia following chromopertubation in treated pelvic tuberculosis. Int J Gynaecol Obstet. 2002;77(1):41–42.
12. Rzymski P, Wozniak J, Opala T, Wilczak M, Sajdak S. Anaphylactic reaction to methylene blue dye after laparoscopic chromopertubation. Int J Gynaecol Obstet. 2003;81(1):71–72.
13. Dewachter P, Mouton-Faivre C, Trechot P, Lieu JC, Mertes PM. Severe anaphylactic shock with methylene blue instillation. Anesth Analg. 2005;101(1):149–150.
14. McFadyen I. The dangers of intra-amniotic methylene blue. Br J Obstet Gynaecol. 1992;99(2):89–90.
15. Van der Pol JG, Wolf H, Boer K, et al. Jejunal atresia related to the use of methylene blue in genetic amniocentesis in twins. Br J Obstet Gynaecol. 1992;99(2):141–143.
16. Kidd SA, Lancaster PA, Anderson JC, et al. Fetal death after exposure to methylene blue dye during mid-trimester amniocentesis in twin pregnancy. Prenat Diagn. 1996;16(1):39–47.
17. Hobbins JC, Winsberg F, Blanchett M, et al. Section 5: fetal imaging. Prenat Diagn. 1981;1(5):35–38.

Laparoscopic dual-port contained power morcellation: An offered solution
Minimally invasive surgery utilizing laparoscopy for hysterectomy and myomectomy has become more common in women with gynecologic pathology. The benefits of this approach compared with laparotomy include decreased hospital stay, shorter recovery and, in experienced hands, significantly decreased morbidity.1–3
Approximately 600,000 hysterectomies are performed annually in the United States—30% of which are performed laparoscopically.4 The primary indication for surgical intervention is uterine leiomyoma. This pathology accounts for 40% of procedures.5 During these surgeries, electromechanical morcellation (EMM), or open “power” morcellation, is commonly used to cut large tissue specimens into small pieces for removal and thereby avoid a larger incision. Concerns have been raised regarding the use of open power morcellation because of the risk of spreading an unrecognized malignancy.
Based on case reports and retrospective studies, the FDA issued a statement in April of this year discouraging the use of EMM for hysterectomy and myomectomy in women with uterine fibroids.6 The concern for inadvertent spread of an occult malignancy was the reasoning for the communication. Since that time, the FDA’s Obstetrics and Gynecology Devices Panel of the Medical Devices Advisory Committee held a public meeting in which the panel heard comments from patients, societies, and industry regarding their positions on the safety of laparoscopic power morcellation. The panel made several recommendations to the FDA but, at the time of this writing, the FDA has yet to issue a final decision.
Reaction to FDA’s action/inaction
The FDA’s “safety” communication was in response to the concern of a few who experienced a bad outcome believed to be secondary to open power morcellation of enlarged uteri or fibroid tumors. In its statement, the FDA estimated the risk of an occult sarcoma to be about 1 in 350 and stated that the risk of disseminating a sarcoma with morcellation is substantial. The FDA discouraged the use of the power morcellator during hysterectomy or myomectomy for uterine fibroids.
Many organizations, including the Society of Gynecologic Oncology, The American Association of Gynecologic Laparoscopists (AAGL), and the American College of Obstetricians and Gynecologists, issued less stringent statements regarding this technology.7–9 These organizations stated generally that there were too few data to make a statement at that time, advocated the collection of more data, and encouraged detailed informed consent to be given to patients undergoing these procedures.
However, the FDA’s statement, and lack of a timely follow-up to clarify the role of the laparoscopic power morcellator in gynecologic surgery, has effectively stopped the use of this technology in its current form. In fact, in response to the statement, Ethicon Endosurgery has discontinued the distribution and sales of its power morcellator and many institutions have severely or completely restricted the use of this technology. The reason for these restrictions is that the medicolegal consequences of an adverse outcome would be very difficult to defend given the current, albeit premature, recommendations of the FDA. This statement makes it difficult to defend any adverse outcome that may occur in association with the use of the laparoscopic power morcellator. Furthermore, this statement by the FDA has largely prevented the medical community at large from collecting additional useful information to allow for a data-driven determination.
Power morcellation is not without risks. In fact, we outline them in this article. However, we believe that minimally invasive surgery should be allowed to continue to advance. In that vein, here we describe a technique of dual-port contained EMM. This surgical approach is performed under direct visualization—which solves the problem of poor visualization that hinders other contained EMM techniques.
Risks of power morcellation
The potential for inadvertent spread of occult malignancy is not the only risk of open EMM. Reports of disseminated leiomyomatosis, adenomyosis, and endometriosis also have been described from inadvertent tissue dispersion during open EMM with resulting ectopic reperitonealization.10–12
The procedure itself is not without risks. A recent systematic review documented 55 major and minor complications from EMM.13 Multiple organ systems were injured including bowel, urinary, vascular, and others, resulting in six deaths from these complications. The investigators concluded that “laparoscopic morcellator–related injuries continue to increase and short- and long-term complications are emerging in both the medical literature and device-related databases. Surgeon inexperience is descriptively identified as one of the most common contributing factors.”
All of the above risks must be weighed against the known benefits of laparoscopic surgery and presented to each patient to assist in deciding which route of surgery should be performed.
Tissue extraction options for large specimens
Large specimen extraction options during gynecologic surgery include:
Vaginal coring. Delivery through the vagina or colpotomy during vaginal or laparoscopic hysterectomy uses the technique of coring, which has long been established in our field.
Manual morcellation through a single incision. Mini-laparotomy or laparoendoscopic single-site surgery (LESS) incisions provide another option of removal with manual morcellation after laparoscopic hysterectomy or myomectomy. One study revealed that specimens up to 22 weeks in size can be placed in a large EndoCatch bag and morcellated extracorporeally by circumferentially coring with a scalpel.14
Contained power morcellation through a single port. Finally, the technique of contained EMM was recently described.15 This technique uses a large containment bag placed through a LESS incision with EMM being performed in an artificially created pneumoperitoneum. This technique isolates the specimen so that it can be morcellated without risk of exposing the patient to any malignant cells that might be unrecognized within the specimen.
Each of these techniques allows many patients to consider a minimally invasive option for their surgery. However, the ability to safely morcellate a very large uterus or myoma may be limited by visualization, and the experience of the surgeon is often critical in the successful performance of these procedures.16
Therefore, at Washington Universitywe have developed a technique using dual ports, with isolation of the uterus or myomas to improve visualization and prevent spillage of malignant tumor or dispersion of other benign tissue.
Dual-port EMM: Technique, tips, and tricks
Our technique of dual-port contained EMM allows the removal of large fibroids or uteri much larger than 20 weeks in size safely under direct visualization through a 15-mm incision. The technique uses:
- Karl Storz Rotocut tissue morcellator with spacers (FIGURE 1)
- 15-mm trocar
- 5-mm balloon trocar
- 20320-inch containment bag (FIGURE 2).


Containment bag placement
Once the specimen is free, we place it to the right or left side of the abdomen. The 15-mm trocar is placed through the umbilicus while visualizing from a lateral trocar site. We then fan-fold the containment bag and introduce it through the 15-mm trocar, keeping the bag oriented with the opening anterior (FIGURE 3). The bag is then grasped at the opening along the drawstring with an atraumatic grasper.
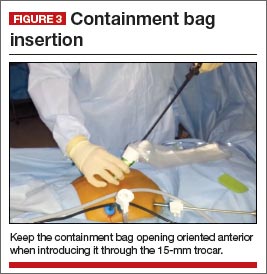
Tip: Care must be taken when introducing the bag in order to avoid tearing or making a small hole in it.
The leading edge is then introduced into the deepest part of the pelvis, and the remainder of the bag (left outside of the abdomen) is then fed cephalad into the abdomen.
Once the bag is completely in the abdomen, we orient the bag with the opening as wide as possible. This allows placement of a very large specimen. Once the specimen is within the containment bag, the drawstring is pulled tight and the mouth of the bag is removed through the 15-mm trocar site at the umbilicus.
The abdominal lateral gas port is opened to allow the intra-abdominal pneumoperitoneum to escape. A 5-mm trocar is placed into the bag through the opening at the umbilicus and the containment bag is insufflated with carbon dioxide and the insufflation pressure is set to 30 mm. The laparoscope placed through this trocar allows the artificial pneumoperitoneum being created to be observed (VIDEO).

Tip: The containment bag covers the entire abdominal cavity and should be fully distended. If it does not distend fully, a hole in the bag may be present and the bag must be replaced.
At this point, we place a balloon trocar at the lateral trocar site and into the bag under direct visualization. The balloon tip is inflated and pulled up tightly against the bag and abdominal wall (FIGURE 4). This allows a tight seal so there is no gas leak or spillage of the morcellated specimen. The laparoscope is placed through this trocar and the insufflation tubing is moved to this port.
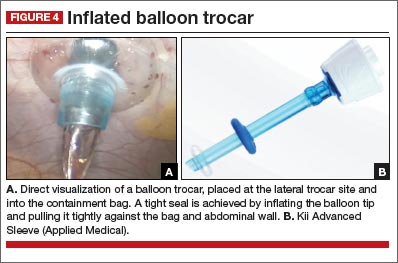
Morcellator insertion
The morcellator is introduced through the umbilicus under direct visualization using the short morcellator blade in most instances. Spacers are used to set the length of the morcellator within the containment bag. The tip of the morcellator should be approximately 3 cm to 4 cm within the bag but well away from the retroperitoneum. Remember, any bag will be cut easily by the morcellator and should be thought of as peritoneum only and not a tough barrier. Serious injuries could otherwise develop.
At this point, place the patient flat or out of Trendelenburg position. Morcellation may now proceed.
Tip: Morcellation is best performed with the morcellator perpendicular to the abdomen under direct visualization using a 30° laparoscope to optimize the view. Morcellation in this position uses gravity to facilitate “peeling” of the specimen during morcellation and allows for faster removal.
Before removing the morcellator, inspect the containment bag for any large pieces that may have been dispersed during the morcellation process and remove them. Once there are only small fragments remaining, remove the morcellator, allowing the carbon dioxide to escape. Deflate the balloon tip on the trocar.
Now the containment bag with the remaining specimen may be removed through the umbilicus, while simultaneously removing the balloon-tip trocar from the bag.
A safe minimally invasive approach
This technique has allowed us to safely remove specimens larger than 1,500 g while keeping them in a contained environment with no spill of tissue within the abdomen.
Tracking and adaptation needed
The FDA safety communication has severely limited the practice of morcellation in the minimally invasive gynecologic surgical setting. Many hospitals around the country have reacted by placing significant restrictions on the use of EMM or banned it outright. This action may reverse the national trend of increasing rates of laparoscopic hysterectomy and force many practitioners to return to open surgery.
Currently, it is unclear what the true risk of tissue extraction is whether it is performed via EMM or manually. Large national databases including the BOLD database from the Surgical Review Corporation, as well as AAGL, must be utilized to track these cases and their outcomes to guide therapy. In the meantime, in order to continue to offer a minimally invasive approach to gynecologic surgery, new techniques and instrumentation in the operating room will need to be modified to adapt to these new guidelines. This is vital to maintain or even reduce the rates of open hysterectomy and associated morbidity while diminishing the potential risks of inadvertent benign as well as malignant tissue dispersion with tissue extraction.
Share your thoughts on this article! Send your Letter to the Editor to: [email protected]
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677. doi:10.1002/14651858.CD003677.pub4.
2. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
3. Wiser A, Holcroft CA, Tolandi T, Abenhaim HA. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10(2):117–122.
4. Wright JD, Ananth CV, Lewin SN, et al. Robotically assisted vs laparoscopic hysterectomy among women with benign gynecologic disease. JAMA. 2013;309(7):689–698.
5. Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000-2004. Am J Obstet Gynecol. 2008;198(1):34.e1–e7.
6. U S Food and Drug Administration. Quantitative assessment of the prevalence of unsuspected uterine sarcoma in women undergoing treatment of uterine fibroids: Summary and key findings. Silver Spring, Maryland: FDA. http://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM393589.pdf. Published April 17, 2014. Accessed August 19, 2014.
7. Society of Gynecologic Oncology (SGO). SGO Position Statement: Morcellation. https://www.sgo.org/newsroom/position-statements-2/morcellation. Published December 2013. Accessed March 1, 2014.
8. AAGL. Member Update: Disseminated leiomyosarcoma with power morcellation (Update #2). https://www.aagl.org/aaglnews/member-update-disseminated-leiomyosarcoma-with-power-morcellation-update-2/. Published July 11, 2014. Accessed August 19, 2014.
9. American College of Obstetricians and Gynecologists. Power morcellation and occult malignancy in gynecologic surgery. http://www.acog.org/Resources_And_Publications/Task_Force_and_Work_Group_Reports/Power_Morcellation_and_Occult_Malignancy_in_Gynecologic_Surgery. Published May 2014. Accessed August 19, 2014.
10. Sepilian V, Della Badia C. Iatrogenic endometriosis caused by uterine morcellation during a supracervical hysterectomy. Obstet Gynecol. 2003;102(5 Pt 2):1125–1127.
11. Takeda A, Mori M, Sakai K, Mitsui T, Nakamura H. Parasitic peritoneal leiomyomatosis diagnosed 6 years after laparoscopic myomectomy with electric tissue morcellation: report of a case and review of the literature. J Minim Invasive Gynecol. 2007;14(6):770–775.
12. Donnez O, Squifflet J, Leconte I, Jadoul P, Donnez J. Posthysterectomy pelvic adenomyotic masses observed in 8 cases out of a series of 1405 laparoscopic subtotal hysterectomies. J Minim Invasive Gynecol. 2007;14(2):156–160.
13. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21(3):486–491.
14. Serur E, Lakhi N. Laparoscopic hysterectomy with manual morcellation of the uterus: an original technique that permits the safe and quick removal of a large uterus. Am J. Obstet Gynecol. 2011;204(6):566.e1–e2.
15. Shibley KA. Feasibilty of intra-abdominal tissue isolation and extraction with an artificially created pneumoperitoneum, at laparoscopy for gynecologic procedures. J Min Invasive Gynecol. 2012;19(6):S75.
Scott W. Biest, MD, and David G. Mutch, MD

Dr. Biest is Assistant Professor, Department of Obstetrics and Gynecology, and Director, Division of Minimally Invasive Gynecology, Washington University School of Medicine in St Louis, Missouri.

Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Scott W. Biest, MD, and David G. Mutch, MD
Dr. Biest is Assistant Professor, Department of Obstetrics and Gynecology, and Director, Division of Minimally Invasive Gynecology, Washington University School of Medicine in St Louis, Missouri.
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Scott W. Biest, MD, and David G. Mutch, MD
Dr. Biest is Assistant Professor, Department of Obstetrics and Gynecology, and Director, Division of Minimally Invasive Gynecology, Washington University School of Medicine in St Louis, Missouri.
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of Gynecologic Oncology at Washington University School of Medicine in St. Louis. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Minimally invasive surgery utilizing laparoscopy for hysterectomy and myomectomy has become more common in women with gynecologic pathology. The benefits of this approach compared with laparotomy include decreased hospital stay, shorter recovery and, in experienced hands, significantly decreased morbidity.1–3
Approximately 600,000 hysterectomies are performed annually in the United States—30% of which are performed laparoscopically.4 The primary indication for surgical intervention is uterine leiomyoma. This pathology accounts for 40% of procedures.5 During these surgeries, electromechanical morcellation (EMM), or open “power” morcellation, is commonly used to cut large tissue specimens into small pieces for removal and thereby avoid a larger incision. Concerns have been raised regarding the use of open power morcellation because of the risk of spreading an unrecognized malignancy.
Based on case reports and retrospective studies, the FDA issued a statement in April of this year discouraging the use of EMM for hysterectomy and myomectomy in women with uterine fibroids.6 The concern for inadvertent spread of an occult malignancy was the reasoning for the communication. Since that time, the FDA’s Obstetrics and Gynecology Devices Panel of the Medical Devices Advisory Committee held a public meeting in which the panel heard comments from patients, societies, and industry regarding their positions on the safety of laparoscopic power morcellation. The panel made several recommendations to the FDA but, at the time of this writing, the FDA has yet to issue a final decision.
Reaction to FDA’s action/inaction
The FDA’s “safety” communication was in response to the concern of a few who experienced a bad outcome believed to be secondary to open power morcellation of enlarged uteri or fibroid tumors. In its statement, the FDA estimated the risk of an occult sarcoma to be about 1 in 350 and stated that the risk of disseminating a sarcoma with morcellation is substantial. The FDA discouraged the use of the power morcellator during hysterectomy or myomectomy for uterine fibroids.
Many organizations, including the Society of Gynecologic Oncology, The American Association of Gynecologic Laparoscopists (AAGL), and the American College of Obstetricians and Gynecologists, issued less stringent statements regarding this technology.7–9 These organizations stated generally that there were too few data to make a statement at that time, advocated the collection of more data, and encouraged detailed informed consent to be given to patients undergoing these procedures.
However, the FDA’s statement, and lack of a timely follow-up to clarify the role of the laparoscopic power morcellator in gynecologic surgery, has effectively stopped the use of this technology in its current form. In fact, in response to the statement, Ethicon Endosurgery has discontinued the distribution and sales of its power morcellator and many institutions have severely or completely restricted the use of this technology. The reason for these restrictions is that the medicolegal consequences of an adverse outcome would be very difficult to defend given the current, albeit premature, recommendations of the FDA. This statement makes it difficult to defend any adverse outcome that may occur in association with the use of the laparoscopic power morcellator. Furthermore, this statement by the FDA has largely prevented the medical community at large from collecting additional useful information to allow for a data-driven determination.
Power morcellation is not without risks. In fact, we outline them in this article. However, we believe that minimally invasive surgery should be allowed to continue to advance. In that vein, here we describe a technique of dual-port contained EMM. This surgical approach is performed under direct visualization—which solves the problem of poor visualization that hinders other contained EMM techniques.
Risks of power morcellation
The potential for inadvertent spread of occult malignancy is not the only risk of open EMM. Reports of disseminated leiomyomatosis, adenomyosis, and endometriosis also have been described from inadvertent tissue dispersion during open EMM with resulting ectopic reperitonealization.10–12
The procedure itself is not without risks. A recent systematic review documented 55 major and minor complications from EMM.13 Multiple organ systems were injured including bowel, urinary, vascular, and others, resulting in six deaths from these complications. The investigators concluded that “laparoscopic morcellator–related injuries continue to increase and short- and long-term complications are emerging in both the medical literature and device-related databases. Surgeon inexperience is descriptively identified as one of the most common contributing factors.”
All of the above risks must be weighed against the known benefits of laparoscopic surgery and presented to each patient to assist in deciding which route of surgery should be performed.
Tissue extraction options for large specimens
Large specimen extraction options during gynecologic surgery include:
Vaginal coring. Delivery through the vagina or colpotomy during vaginal or laparoscopic hysterectomy uses the technique of coring, which has long been established in our field.
Manual morcellation through a single incision. Mini-laparotomy or laparoendoscopic single-site surgery (LESS) incisions provide another option of removal with manual morcellation after laparoscopic hysterectomy or myomectomy. One study revealed that specimens up to 22 weeks in size can be placed in a large EndoCatch bag and morcellated extracorporeally by circumferentially coring with a scalpel.14
Contained power morcellation through a single port. Finally, the technique of contained EMM was recently described.15 This technique uses a large containment bag placed through a LESS incision with EMM being performed in an artificially created pneumoperitoneum. This technique isolates the specimen so that it can be morcellated without risk of exposing the patient to any malignant cells that might be unrecognized within the specimen.
Each of these techniques allows many patients to consider a minimally invasive option for their surgery. However, the ability to safely morcellate a very large uterus or myoma may be limited by visualization, and the experience of the surgeon is often critical in the successful performance of these procedures.16
Therefore, at Washington Universitywe have developed a technique using dual ports, with isolation of the uterus or myomas to improve visualization and prevent spillage of malignant tumor or dispersion of other benign tissue.
Dual-port EMM: Technique, tips, and tricks
Our technique of dual-port contained EMM allows the removal of large fibroids or uteri much larger than 20 weeks in size safely under direct visualization through a 15-mm incision. The technique uses:
- Karl Storz Rotocut tissue morcellator with spacers (FIGURE 1)
- 15-mm trocar
- 5-mm balloon trocar
- 20320-inch containment bag (FIGURE 2).
Containment bag placement
Once the specimen is free, we place it to the right or left side of the abdomen. The 15-mm trocar is placed through the umbilicus while visualizing from a lateral trocar site. We then fan-fold the containment bag and introduce it through the 15-mm trocar, keeping the bag oriented with the opening anterior (FIGURE 3). The bag is then grasped at the opening along the drawstring with an atraumatic grasper.
Tip: Care must be taken when introducing the bag in order to avoid tearing or making a small hole in it.
The leading edge is then introduced into the deepest part of the pelvis, and the remainder of the bag (left outside of the abdomen) is then fed cephalad into the abdomen.
Once the bag is completely in the abdomen, we orient the bag with the opening as wide as possible. This allows placement of a very large specimen. Once the specimen is within the containment bag, the drawstring is pulled tight and the mouth of the bag is removed through the 15-mm trocar site at the umbilicus.
The abdominal lateral gas port is opened to allow the intra-abdominal pneumoperitoneum to escape. A 5-mm trocar is placed into the bag through the opening at the umbilicus and the containment bag is insufflated with carbon dioxide and the insufflation pressure is set to 30 mm. The laparoscope placed through this trocar allows the artificial pneumoperitoneum being created to be observed (VIDEO).
Tip: The containment bag covers the entire abdominal cavity and should be fully distended. If it does not distend fully, a hole in the bag may be present and the bag must be replaced.
At this point, we place a balloon trocar at the lateral trocar site and into the bag under direct visualization. The balloon tip is inflated and pulled up tightly against the bag and abdominal wall (FIGURE 4). This allows a tight seal so there is no gas leak or spillage of the morcellated specimen. The laparoscope is placed through this trocar and the insufflation tubing is moved to this port.
Morcellator insertion
The morcellator is introduced through the umbilicus under direct visualization using the short morcellator blade in most instances. Spacers are used to set the length of the morcellator within the containment bag. The tip of the morcellator should be approximately 3 cm to 4 cm within the bag but well away from the retroperitoneum. Remember, any bag will be cut easily by the morcellator and should be thought of as peritoneum only and not a tough barrier. Serious injuries could otherwise develop.
At this point, place the patient flat or out of Trendelenburg position. Morcellation may now proceed.
Tip: Morcellation is best performed with the morcellator perpendicular to the abdomen under direct visualization using a 30° laparoscope to optimize the view. Morcellation in this position uses gravity to facilitate “peeling” of the specimen during morcellation and allows for faster removal.
Before removing the morcellator, inspect the containment bag for any large pieces that may have been dispersed during the morcellation process and remove them. Once there are only small fragments remaining, remove the morcellator, allowing the carbon dioxide to escape. Deflate the balloon tip on the trocar.
Now the containment bag with the remaining specimen may be removed through the umbilicus, while simultaneously removing the balloon-tip trocar from the bag.
A safe minimally invasive approach
This technique has allowed us to safely remove specimens larger than 1,500 g while keeping them in a contained environment with no spill of tissue within the abdomen.
Tracking and adaptation needed
The FDA safety communication has severely limited the practice of morcellation in the minimally invasive gynecologic surgical setting. Many hospitals around the country have reacted by placing significant restrictions on the use of EMM or banned it outright. This action may reverse the national trend of increasing rates of laparoscopic hysterectomy and force many practitioners to return to open surgery.
Currently, it is unclear what the true risk of tissue extraction is whether it is performed via EMM or manually. Large national databases including the BOLD database from the Surgical Review Corporation, as well as AAGL, must be utilized to track these cases and their outcomes to guide therapy. In the meantime, in order to continue to offer a minimally invasive approach to gynecologic surgery, new techniques and instrumentation in the operating room will need to be modified to adapt to these new guidelines. This is vital to maintain or even reduce the rates of open hysterectomy and associated morbidity while diminishing the potential risks of inadvertent benign as well as malignant tissue dispersion with tissue extraction.
Share your thoughts on this article! Send your Letter to the Editor to: [email protected]
Minimally invasive surgery utilizing laparoscopy for hysterectomy and myomectomy has become more common in women with gynecologic pathology. The benefits of this approach compared with laparotomy include decreased hospital stay, shorter recovery and, in experienced hands, significantly decreased morbidity.1–3
Approximately 600,000 hysterectomies are performed annually in the United States—30% of which are performed laparoscopically.4 The primary indication for surgical intervention is uterine leiomyoma. This pathology accounts for 40% of procedures.5 During these surgeries, electromechanical morcellation (EMM), or open “power” morcellation, is commonly used to cut large tissue specimens into small pieces for removal and thereby avoid a larger incision. Concerns have been raised regarding the use of open power morcellation because of the risk of spreading an unrecognized malignancy.
Based on case reports and retrospective studies, the FDA issued a statement in April of this year discouraging the use of EMM for hysterectomy and myomectomy in women with uterine fibroids.6 The concern for inadvertent spread of an occult malignancy was the reasoning for the communication. Since that time, the FDA’s Obstetrics and Gynecology Devices Panel of the Medical Devices Advisory Committee held a public meeting in which the panel heard comments from patients, societies, and industry regarding their positions on the safety of laparoscopic power morcellation. The panel made several recommendations to the FDA but, at the time of this writing, the FDA has yet to issue a final decision.
Reaction to FDA’s action/inaction
The FDA’s “safety” communication was in response to the concern of a few who experienced a bad outcome believed to be secondary to open power morcellation of enlarged uteri or fibroid tumors. In its statement, the FDA estimated the risk of an occult sarcoma to be about 1 in 350 and stated that the risk of disseminating a sarcoma with morcellation is substantial. The FDA discouraged the use of the power morcellator during hysterectomy or myomectomy for uterine fibroids.
Many organizations, including the Society of Gynecologic Oncology, The American Association of Gynecologic Laparoscopists (AAGL), and the American College of Obstetricians and Gynecologists, issued less stringent statements regarding this technology.7–9 These organizations stated generally that there were too few data to make a statement at that time, advocated the collection of more data, and encouraged detailed informed consent to be given to patients undergoing these procedures.
However, the FDA’s statement, and lack of a timely follow-up to clarify the role of the laparoscopic power morcellator in gynecologic surgery, has effectively stopped the use of this technology in its current form. In fact, in response to the statement, Ethicon Endosurgery has discontinued the distribution and sales of its power morcellator and many institutions have severely or completely restricted the use of this technology. The reason for these restrictions is that the medicolegal consequences of an adverse outcome would be very difficult to defend given the current, albeit premature, recommendations of the FDA. This statement makes it difficult to defend any adverse outcome that may occur in association with the use of the laparoscopic power morcellator. Furthermore, this statement by the FDA has largely prevented the medical community at large from collecting additional useful information to allow for a data-driven determination.
Power morcellation is not without risks. In fact, we outline them in this article. However, we believe that minimally invasive surgery should be allowed to continue to advance. In that vein, here we describe a technique of dual-port contained EMM. This surgical approach is performed under direct visualization—which solves the problem of poor visualization that hinders other contained EMM techniques.
Risks of power morcellation
The potential for inadvertent spread of occult malignancy is not the only risk of open EMM. Reports of disseminated leiomyomatosis, adenomyosis, and endometriosis also have been described from inadvertent tissue dispersion during open EMM with resulting ectopic reperitonealization.10–12
The procedure itself is not without risks. A recent systematic review documented 55 major and minor complications from EMM.13 Multiple organ systems were injured including bowel, urinary, vascular, and others, resulting in six deaths from these complications. The investigators concluded that “laparoscopic morcellator–related injuries continue to increase and short- and long-term complications are emerging in both the medical literature and device-related databases. Surgeon inexperience is descriptively identified as one of the most common contributing factors.”
All of the above risks must be weighed against the known benefits of laparoscopic surgery and presented to each patient to assist in deciding which route of surgery should be performed.
Tissue extraction options for large specimens
Large specimen extraction options during gynecologic surgery include:
Vaginal coring. Delivery through the vagina or colpotomy during vaginal or laparoscopic hysterectomy uses the technique of coring, which has long been established in our field.
Manual morcellation through a single incision. Mini-laparotomy or laparoendoscopic single-site surgery (LESS) incisions provide another option of removal with manual morcellation after laparoscopic hysterectomy or myomectomy. One study revealed that specimens up to 22 weeks in size can be placed in a large EndoCatch bag and morcellated extracorporeally by circumferentially coring with a scalpel.14
Contained power morcellation through a single port. Finally, the technique of contained EMM was recently described.15 This technique uses a large containment bag placed through a LESS incision with EMM being performed in an artificially created pneumoperitoneum. This technique isolates the specimen so that it can be morcellated without risk of exposing the patient to any malignant cells that might be unrecognized within the specimen.
Each of these techniques allows many patients to consider a minimally invasive option for their surgery. However, the ability to safely morcellate a very large uterus or myoma may be limited by visualization, and the experience of the surgeon is often critical in the successful performance of these procedures.16
Therefore, at Washington Universitywe have developed a technique using dual ports, with isolation of the uterus or myomas to improve visualization and prevent spillage of malignant tumor or dispersion of other benign tissue.
Dual-port EMM: Technique, tips, and tricks
Our technique of dual-port contained EMM allows the removal of large fibroids or uteri much larger than 20 weeks in size safely under direct visualization through a 15-mm incision. The technique uses:
- Karl Storz Rotocut tissue morcellator with spacers (FIGURE 1)
- 15-mm trocar
- 5-mm balloon trocar
- 20320-inch containment bag (FIGURE 2).
Containment bag placement
Once the specimen is free, we place it to the right or left side of the abdomen. The 15-mm trocar is placed through the umbilicus while visualizing from a lateral trocar site. We then fan-fold the containment bag and introduce it through the 15-mm trocar, keeping the bag oriented with the opening anterior (FIGURE 3). The bag is then grasped at the opening along the drawstring with an atraumatic grasper.
Tip: Care must be taken when introducing the bag in order to avoid tearing or making a small hole in it.
The leading edge is then introduced into the deepest part of the pelvis, and the remainder of the bag (left outside of the abdomen) is then fed cephalad into the abdomen.
Once the bag is completely in the abdomen, we orient the bag with the opening as wide as possible. This allows placement of a very large specimen. Once the specimen is within the containment bag, the drawstring is pulled tight and the mouth of the bag is removed through the 15-mm trocar site at the umbilicus.
The abdominal lateral gas port is opened to allow the intra-abdominal pneumoperitoneum to escape. A 5-mm trocar is placed into the bag through the opening at the umbilicus and the containment bag is insufflated with carbon dioxide and the insufflation pressure is set to 30 mm. The laparoscope placed through this trocar allows the artificial pneumoperitoneum being created to be observed (VIDEO).
Tip: The containment bag covers the entire abdominal cavity and should be fully distended. If it does not distend fully, a hole in the bag may be present and the bag must be replaced.
At this point, we place a balloon trocar at the lateral trocar site and into the bag under direct visualization. The balloon tip is inflated and pulled up tightly against the bag and abdominal wall (FIGURE 4). This allows a tight seal so there is no gas leak or spillage of the morcellated specimen. The laparoscope is placed through this trocar and the insufflation tubing is moved to this port.
Morcellator insertion
The morcellator is introduced through the umbilicus under direct visualization using the short morcellator blade in most instances. Spacers are used to set the length of the morcellator within the containment bag. The tip of the morcellator should be approximately 3 cm to 4 cm within the bag but well away from the retroperitoneum. Remember, any bag will be cut easily by the morcellator and should be thought of as peritoneum only and not a tough barrier. Serious injuries could otherwise develop.
At this point, place the patient flat or out of Trendelenburg position. Morcellation may now proceed.
Tip: Morcellation is best performed with the morcellator perpendicular to the abdomen under direct visualization using a 30° laparoscope to optimize the view. Morcellation in this position uses gravity to facilitate “peeling” of the specimen during morcellation and allows for faster removal.
Before removing the morcellator, inspect the containment bag for any large pieces that may have been dispersed during the morcellation process and remove them. Once there are only small fragments remaining, remove the morcellator, allowing the carbon dioxide to escape. Deflate the balloon tip on the trocar.
Now the containment bag with the remaining specimen may be removed through the umbilicus, while simultaneously removing the balloon-tip trocar from the bag.
A safe minimally invasive approach
This technique has allowed us to safely remove specimens larger than 1,500 g while keeping them in a contained environment with no spill of tissue within the abdomen.
Tracking and adaptation needed
The FDA safety communication has severely limited the practice of morcellation in the minimally invasive gynecologic surgical setting. Many hospitals around the country have reacted by placing significant restrictions on the use of EMM or banned it outright. This action may reverse the national trend of increasing rates of laparoscopic hysterectomy and force many practitioners to return to open surgery.
Currently, it is unclear what the true risk of tissue extraction is whether it is performed via EMM or manually. Large national databases including the BOLD database from the Surgical Review Corporation, as well as AAGL, must be utilized to track these cases and their outcomes to guide therapy. In the meantime, in order to continue to offer a minimally invasive approach to gynecologic surgery, new techniques and instrumentation in the operating room will need to be modified to adapt to these new guidelines. This is vital to maintain or even reduce the rates of open hysterectomy and associated morbidity while diminishing the potential risks of inadvertent benign as well as malignant tissue dispersion with tissue extraction.
Share your thoughts on this article! Send your Letter to the Editor to: [email protected]
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677. doi:10.1002/14651858.CD003677.pub4.
2. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
3. Wiser A, Holcroft CA, Tolandi T, Abenhaim HA. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10(2):117–122.
4. Wright JD, Ananth CV, Lewin SN, et al. Robotically assisted vs laparoscopic hysterectomy among women with benign gynecologic disease. JAMA. 2013;309(7):689–698.
5. Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000-2004. Am J Obstet Gynecol. 2008;198(1):34.e1–e7.
6. U S Food and Drug Administration. Quantitative assessment of the prevalence of unsuspected uterine sarcoma in women undergoing treatment of uterine fibroids: Summary and key findings. Silver Spring, Maryland: FDA. http://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM393589.pdf. Published April 17, 2014. Accessed August 19, 2014.
7. Society of Gynecologic Oncology (SGO). SGO Position Statement: Morcellation. https://www.sgo.org/newsroom/position-statements-2/morcellation. Published December 2013. Accessed March 1, 2014.
8. AAGL. Member Update: Disseminated leiomyosarcoma with power morcellation (Update #2). https://www.aagl.org/aaglnews/member-update-disseminated-leiomyosarcoma-with-power-morcellation-update-2/. Published July 11, 2014. Accessed August 19, 2014.
9. American College of Obstetricians and Gynecologists. Power morcellation and occult malignancy in gynecologic surgery. http://www.acog.org/Resources_And_Publications/Task_Force_and_Work_Group_Reports/Power_Morcellation_and_Occult_Malignancy_in_Gynecologic_Surgery. Published May 2014. Accessed August 19, 2014.
10. Sepilian V, Della Badia C. Iatrogenic endometriosis caused by uterine morcellation during a supracervical hysterectomy. Obstet Gynecol. 2003;102(5 Pt 2):1125–1127.
11. Takeda A, Mori M, Sakai K, Mitsui T, Nakamura H. Parasitic peritoneal leiomyomatosis diagnosed 6 years after laparoscopic myomectomy with electric tissue morcellation: report of a case and review of the literature. J Minim Invasive Gynecol. 2007;14(6):770–775.
12. Donnez O, Squifflet J, Leconte I, Jadoul P, Donnez J. Posthysterectomy pelvic adenomyotic masses observed in 8 cases out of a series of 1405 laparoscopic subtotal hysterectomies. J Minim Invasive Gynecol. 2007;14(2):156–160.
13. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21(3):486–491.
14. Serur E, Lakhi N. Laparoscopic hysterectomy with manual morcellation of the uterus: an original technique that permits the safe and quick removal of a large uterus. Am J. Obstet Gynecol. 2011;204(6):566.e1–e2.
15. Shibley KA. Feasibilty of intra-abdominal tissue isolation and extraction with an artificially created pneumoperitoneum, at laparoscopy for gynecologic procedures. J Min Invasive Gynecol. 2012;19(6):S75.
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677. doi:10.1002/14651858.CD003677.pub4.
2. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
3. Wiser A, Holcroft CA, Tolandi T, Abenhaim HA. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10(2):117–122.
4. Wright JD, Ananth CV, Lewin SN, et al. Robotically assisted vs laparoscopic hysterectomy among women with benign gynecologic disease. JAMA. 2013;309(7):689–698.
5. Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000-2004. Am J Obstet Gynecol. 2008;198(1):34.e1–e7.
6. U S Food and Drug Administration. Quantitative assessment of the prevalence of unsuspected uterine sarcoma in women undergoing treatment of uterine fibroids: Summary and key findings. Silver Spring, Maryland: FDA. http://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM393589.pdf. Published April 17, 2014. Accessed August 19, 2014.
7. Society of Gynecologic Oncology (SGO). SGO Position Statement: Morcellation. https://www.sgo.org/newsroom/position-statements-2/morcellation. Published December 2013. Accessed March 1, 2014.
8. AAGL. Member Update: Disseminated leiomyosarcoma with power morcellation (Update #2). https://www.aagl.org/aaglnews/member-update-disseminated-leiomyosarcoma-with-power-morcellation-update-2/. Published July 11, 2014. Accessed August 19, 2014.
9. American College of Obstetricians and Gynecologists. Power morcellation and occult malignancy in gynecologic surgery. http://www.acog.org/Resources_And_Publications/Task_Force_and_Work_Group_Reports/Power_Morcellation_and_Occult_Malignancy_in_Gynecologic_Surgery. Published May 2014. Accessed August 19, 2014.
10. Sepilian V, Della Badia C. Iatrogenic endometriosis caused by uterine morcellation during a supracervical hysterectomy. Obstet Gynecol. 2003;102(5 Pt 2):1125–1127.
11. Takeda A, Mori M, Sakai K, Mitsui T, Nakamura H. Parasitic peritoneal leiomyomatosis diagnosed 6 years after laparoscopic myomectomy with electric tissue morcellation: report of a case and review of the literature. J Minim Invasive Gynecol. 2007;14(6):770–775.
12. Donnez O, Squifflet J, Leconte I, Jadoul P, Donnez J. Posthysterectomy pelvic adenomyotic masses observed in 8 cases out of a series of 1405 laparoscopic subtotal hysterectomies. J Minim Invasive Gynecol. 2007;14(2):156–160.
13. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21(3):486–491.
14. Serur E, Lakhi N. Laparoscopic hysterectomy with manual morcellation of the uterus: an original technique that permits the safe and quick removal of a large uterus. Am J. Obstet Gynecol. 2011;204(6):566.e1–e2.
15. Shibley KA. Feasibilty of intra-abdominal tissue isolation and extraction with an artificially created pneumoperitoneum, at laparoscopy for gynecologic procedures. J Min Invasive Gynecol. 2012;19(6):S75.
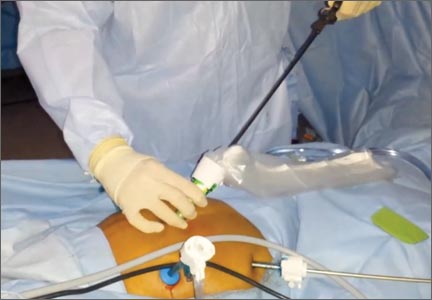
Aspirin can prevent major vascular events after VTE

Credit: Sage Ross
Low-dose aspirin can decrease the risk of major vascular events among patients with venous thromboembolism (VTE), according to a study published in Circulation.
The study included more than 1000 patients who had completed initial anticoagulant therapy after their first VTE.
Patients who went on to receive a daily dose of aspirin had a lower incidence of recurrent VTE, myocardial infarction, stroke, and cardiovascular death, compared
to those who received placebo.
The effects were not as substantial as those typically observed with warfarin or newer anticoagulants, but the results suggest low-dose aspirin is a feasible option for patients who cannot receive long-term anticoagulant therapy.
“The study provides evidence that, after a first venous thrombosis or embolism, daily aspirin reduces the risk of another event, without causing undue bleeding,” said study author John Simes, MD, of the University of Sydney in New South Wales, Australia.
“This treatment is an alternative to long-term anticoagulation and will be especially useful for patients who do not want the inconvenience of close medical monitoring or the risk of bleeding. [Additionally,] aspirin will be ideal in the many countries where prolonged anticoagulant treatment is too expensive.”
For this study, Dr Simes and his colleagues evaluated data from the ASPIRE and WARFASA trials. ASPIRE included 822 patients who were followed for an average of 3 years. And WARFASA included 402 patients who were followed for at least 2 years.
The patients had completed initial treatment with heparin and warfarin or an equivalent anticoagulant regimen following a first, unprovoked VTE. They were randomized to receive placebo (n=608) or a 100 mg daily dose of aspirin (n=616).
The researchers evaluated the differences between these 2 groups with regard to bleeding, recurrent VTE, and major vascular events, which includes recurrent VTE, myocardial infarction, stroke, and cardiovascular death.
VTE recurred in 18.4% of patients in the placebo arm and 13.1% of patients in the aspirin arm. The annual rate of recurrent VTE was 7.5% and 5.1%, respectively. So aspirin reduced the risk of VTE by 32% (hazard ratio [HR]=0.68, P=0.008).
Aspirin reduced the risk of deep vein thrombosis by 34% (HR=0.66, P=0.01) and the risk of pulmonary embolism by 34% (HR=0.66, P=0.08).
Aspirin also reduce the risk of major vascular events by 34%. The annual rate of these events was 5.7% in the aspirin arm and 8.7% in the placebo arm (HR=0.66, P=0.002).
There was no significant difference between the treatment arms with regard to bleeding. The annual rate of bleeding was 0.7% in the placebo arm and 1.1% in the aspirin arm. The annual rate of major bleeding was 0.4% and 0.5%, respectively.
“Aspirin does not require laboratory monitoring and is associated with about a 10-fold lower incidence of bleeding compared with oral anticoagulants,” said study author Cecilia Becattini, MD, of the University of Perugia in Italy.
“We are convinced that it will be an alternative for extended prevention of venous thromboembolism after 6 to 12 months of anticoagulant treatment.”
The researchers stressed that aspirin will not be a substitute for anticoagulant therapy in all patients. But it is an option for patients who are stopping anticoagulant therapy or for patients in whom long-term anticoagulant therapy is unsuitable.
“Although less effective, aspirin is inexpensive, easily obtainable, safe, and familiar to patients and clinicians worldwide,” Dr Simes noted. “If cost is the main consideration, aspirin is a particularly useful therapy. The costs of treating future thromboembolic events is greater than the cost of the preventive treatment.” ![]()
Credit: Sage Ross
Low-dose aspirin can decrease the risk of major vascular events among patients with venous thromboembolism (VTE), according to a study published in Circulation.
The study included more than 1000 patients who had completed initial anticoagulant therapy after their first VTE.
Patients who went on to receive a daily dose of aspirin had a lower incidence of recurrent VTE, myocardial infarction, stroke, and cardiovascular death, compared
to those who received placebo.
The effects were not as substantial as those typically observed with warfarin or newer anticoagulants, but the results suggest low-dose aspirin is a feasible option for patients who cannot receive long-term anticoagulant therapy.
“The study provides evidence that, after a first venous thrombosis or embolism, daily aspirin reduces the risk of another event, without causing undue bleeding,” said study author John Simes, MD, of the University of Sydney in New South Wales, Australia.
“This treatment is an alternative to long-term anticoagulation and will be especially useful for patients who do not want the inconvenience of close medical monitoring or the risk of bleeding. [Additionally,] aspirin will be ideal in the many countries where prolonged anticoagulant treatment is too expensive.”
For this study, Dr Simes and his colleagues evaluated data from the ASPIRE and WARFASA trials. ASPIRE included 822 patients who were followed for an average of 3 years. And WARFASA included 402 patients who were followed for at least 2 years.
The patients had completed initial treatment with heparin and warfarin or an equivalent anticoagulant regimen following a first, unprovoked VTE. They were randomized to receive placebo (n=608) or a 100 mg daily dose of aspirin (n=616).
The researchers evaluated the differences between these 2 groups with regard to bleeding, recurrent VTE, and major vascular events, which includes recurrent VTE, myocardial infarction, stroke, and cardiovascular death.
VTE recurred in 18.4% of patients in the placebo arm and 13.1% of patients in the aspirin arm. The annual rate of recurrent VTE was 7.5% and 5.1%, respectively. So aspirin reduced the risk of VTE by 32% (hazard ratio [HR]=0.68, P=0.008).
Aspirin reduced the risk of deep vein thrombosis by 34% (HR=0.66, P=0.01) and the risk of pulmonary embolism by 34% (HR=0.66, P=0.08).
Aspirin also reduce the risk of major vascular events by 34%. The annual rate of these events was 5.7% in the aspirin arm and 8.7% in the placebo arm (HR=0.66, P=0.002).
There was no significant difference between the treatment arms with regard to bleeding. The annual rate of bleeding was 0.7% in the placebo arm and 1.1% in the aspirin arm. The annual rate of major bleeding was 0.4% and 0.5%, respectively.
“Aspirin does not require laboratory monitoring and is associated with about a 10-fold lower incidence of bleeding compared with oral anticoagulants,” said study author Cecilia Becattini, MD, of the University of Perugia in Italy.
“We are convinced that it will be an alternative for extended prevention of venous thromboembolism after 6 to 12 months of anticoagulant treatment.”
The researchers stressed that aspirin will not be a substitute for anticoagulant therapy in all patients. But it is an option for patients who are stopping anticoagulant therapy or for patients in whom long-term anticoagulant therapy is unsuitable.
“Although less effective, aspirin is inexpensive, easily obtainable, safe, and familiar to patients and clinicians worldwide,” Dr Simes noted. “If cost is the main consideration, aspirin is a particularly useful therapy. The costs of treating future thromboembolic events is greater than the cost of the preventive treatment.” ![]()
Credit: Sage Ross
Low-dose aspirin can decrease the risk of major vascular events among patients with venous thromboembolism (VTE), according to a study published in Circulation.
The study included more than 1000 patients who had completed initial anticoagulant therapy after their first VTE.
Patients who went on to receive a daily dose of aspirin had a lower incidence of recurrent VTE, myocardial infarction, stroke, and cardiovascular death, compared
to those who received placebo.
The effects were not as substantial as those typically observed with warfarin or newer anticoagulants, but the results suggest low-dose aspirin is a feasible option for patients who cannot receive long-term anticoagulant therapy.
“The study provides evidence that, after a first venous thrombosis or embolism, daily aspirin reduces the risk of another event, without causing undue bleeding,” said study author John Simes, MD, of the University of Sydney in New South Wales, Australia.
“This treatment is an alternative to long-term anticoagulation and will be especially useful for patients who do not want the inconvenience of close medical monitoring or the risk of bleeding. [Additionally,] aspirin will be ideal in the many countries where prolonged anticoagulant treatment is too expensive.”
For this study, Dr Simes and his colleagues evaluated data from the ASPIRE and WARFASA trials. ASPIRE included 822 patients who were followed for an average of 3 years. And WARFASA included 402 patients who were followed for at least 2 years.
The patients had completed initial treatment with heparin and warfarin or an equivalent anticoagulant regimen following a first, unprovoked VTE. They were randomized to receive placebo (n=608) or a 100 mg daily dose of aspirin (n=616).
The researchers evaluated the differences between these 2 groups with regard to bleeding, recurrent VTE, and major vascular events, which includes recurrent VTE, myocardial infarction, stroke, and cardiovascular death.
VTE recurred in 18.4% of patients in the placebo arm and 13.1% of patients in the aspirin arm. The annual rate of recurrent VTE was 7.5% and 5.1%, respectively. So aspirin reduced the risk of VTE by 32% (hazard ratio [HR]=0.68, P=0.008).
Aspirin reduced the risk of deep vein thrombosis by 34% (HR=0.66, P=0.01) and the risk of pulmonary embolism by 34% (HR=0.66, P=0.08).
Aspirin also reduce the risk of major vascular events by 34%. The annual rate of these events was 5.7% in the aspirin arm and 8.7% in the placebo arm (HR=0.66, P=0.002).
There was no significant difference between the treatment arms with regard to bleeding. The annual rate of bleeding was 0.7% in the placebo arm and 1.1% in the aspirin arm. The annual rate of major bleeding was 0.4% and 0.5%, respectively.
“Aspirin does not require laboratory monitoring and is associated with about a 10-fold lower incidence of bleeding compared with oral anticoagulants,” said study author Cecilia Becattini, MD, of the University of Perugia in Italy.
“We are convinced that it will be an alternative for extended prevention of venous thromboembolism after 6 to 12 months of anticoagulant treatment.”
The researchers stressed that aspirin will not be a substitute for anticoagulant therapy in all patients. But it is an option for patients who are stopping anticoagulant therapy or for patients in whom long-term anticoagulant therapy is unsuitable.
“Although less effective, aspirin is inexpensive, easily obtainable, safe, and familiar to patients and clinicians worldwide,” Dr Simes noted. “If cost is the main consideration, aspirin is a particularly useful therapy. The costs of treating future thromboembolic events is greater than the cost of the preventive treatment.” ![]()
Study links gene dysfunction to Fanconi anemia, AML
Credit: Tom Ellenberger
Researchers say they’ve discovered “an intimate link” between RUNX genes and Fanconi anemia, a finding that also has implications for treating acute myeloid leukemia (AML).
The group found that RUNX1 and RUNX3 interact with Fanconi anemia group D2 (FANCD2), a protein involved in the repair of DNA damage.
The RUNX proteins facilitate the recruitment of FANCD2 to sites of DNA damage in both Fanconi anemia and AML.
Motomi Osato, MD, PhD, of the Cancer Science Institute of Singapore, and his colleagues recounted these findings in Cell Reports.
The researchers began by studying RUNX deficiency in mice. They found that knockdown of both RUNX1 and RUNX3 led to bone marrow failure or myeloproliferative disorders in the mice.
These clinical manifestations are seen in inherited bone marrow failure syndromes such as Fanconi anemia, which is caused by the disruption of gene products that participate in the repair of DNA interstrand crosslinks (ICLs).
With this in mind, the researchers decided to investigate RUNX function in the ICL repair pathway. And they found RUNX proteins play a critical role in the pathway by facilitating the recruitment of FANCD2 to sites of DNA damage.
To explore the clinical relevance of RUNX participation in DNA damage repair, the team conducted experiments in Kasumi-1 and SKNO-1 cells. These AML cell lines express RUNX1-ETO, which is thought to suppress the expression and/or function of RUNX1 and RUNX3 simultaneously.
The researchers showed that Kasumi-1 and SKNO-1 cells were sensitive to the ICL-inducing agent mytomycin C, and knocking down RUNX1-ETO reduced this sensitivity. Depleting RUNX1-ETO also led to increased FANCD2 recruitment to chromatin.
The team said these results suggest that RUNX1-ETO might repress FANCD2 foci formation and ICL repair in AML cells. And they predicted that RUNX dysfunction would sensitize the cells to PARP inhibitors.
So they tested 2 PARP inhibitors—olaparib and rucaparib—in Kasumi-1 cells and observed sensitivity to both drugs. Knocking down RUNX1-ETO partially reduced this sensitivity, while adding mytomycin C increased sensitivity.
“PARP inhibitors have been with us for quite some time, but nobody has realized their application for leukemia,” Dr Osato said. “Our study has shed light on the possibility of a more effective treatment using a combined therapy with PARP inhibitors, which can potentially be extended to other types of common cancers.”
The researchers are now conducting additional drug testing in xenograft models. ![]()
Credit: Tom Ellenberger
Researchers say they’ve discovered “an intimate link” between RUNX genes and Fanconi anemia, a finding that also has implications for treating acute myeloid leukemia (AML).
The group found that RUNX1 and RUNX3 interact with Fanconi anemia group D2 (FANCD2), a protein involved in the repair of DNA damage.
The RUNX proteins facilitate the recruitment of FANCD2 to sites of DNA damage in both Fanconi anemia and AML.
Motomi Osato, MD, PhD, of the Cancer Science Institute of Singapore, and his colleagues recounted these findings in Cell Reports.
The researchers began by studying RUNX deficiency in mice. They found that knockdown of both RUNX1 and RUNX3 led to bone marrow failure or myeloproliferative disorders in the mice.
These clinical manifestations are seen in inherited bone marrow failure syndromes such as Fanconi anemia, which is caused by the disruption of gene products that participate in the repair of DNA interstrand crosslinks (ICLs).
With this in mind, the researchers decided to investigate RUNX function in the ICL repair pathway. And they found RUNX proteins play a critical role in the pathway by facilitating the recruitment of FANCD2 to sites of DNA damage.
To explore the clinical relevance of RUNX participation in DNA damage repair, the team conducted experiments in Kasumi-1 and SKNO-1 cells. These AML cell lines express RUNX1-ETO, which is thought to suppress the expression and/or function of RUNX1 and RUNX3 simultaneously.
The researchers showed that Kasumi-1 and SKNO-1 cells were sensitive to the ICL-inducing agent mytomycin C, and knocking down RUNX1-ETO reduced this sensitivity. Depleting RUNX1-ETO also led to increased FANCD2 recruitment to chromatin.
The team said these results suggest that RUNX1-ETO might repress FANCD2 foci formation and ICL repair in AML cells. And they predicted that RUNX dysfunction would sensitize the cells to PARP inhibitors.
So they tested 2 PARP inhibitors—olaparib and rucaparib—in Kasumi-1 cells and observed sensitivity to both drugs. Knocking down RUNX1-ETO partially reduced this sensitivity, while adding mytomycin C increased sensitivity.
“PARP inhibitors have been with us for quite some time, but nobody has realized their application for leukemia,” Dr Osato said. “Our study has shed light on the possibility of a more effective treatment using a combined therapy with PARP inhibitors, which can potentially be extended to other types of common cancers.”
The researchers are now conducting additional drug testing in xenograft models. ![]()
Credit: Tom Ellenberger
Researchers say they’ve discovered “an intimate link” between RUNX genes and Fanconi anemia, a finding that also has implications for treating acute myeloid leukemia (AML).
The group found that RUNX1 and RUNX3 interact with Fanconi anemia group D2 (FANCD2), a protein involved in the repair of DNA damage.
The RUNX proteins facilitate the recruitment of FANCD2 to sites of DNA damage in both Fanconi anemia and AML.
Motomi Osato, MD, PhD, of the Cancer Science Institute of Singapore, and his colleagues recounted these findings in Cell Reports.
The researchers began by studying RUNX deficiency in mice. They found that knockdown of both RUNX1 and RUNX3 led to bone marrow failure or myeloproliferative disorders in the mice.
These clinical manifestations are seen in inherited bone marrow failure syndromes such as Fanconi anemia, which is caused by the disruption of gene products that participate in the repair of DNA interstrand crosslinks (ICLs).
With this in mind, the researchers decided to investigate RUNX function in the ICL repair pathway. And they found RUNX proteins play a critical role in the pathway by facilitating the recruitment of FANCD2 to sites of DNA damage.
To explore the clinical relevance of RUNX participation in DNA damage repair, the team conducted experiments in Kasumi-1 and SKNO-1 cells. These AML cell lines express RUNX1-ETO, which is thought to suppress the expression and/or function of RUNX1 and RUNX3 simultaneously.
The researchers showed that Kasumi-1 and SKNO-1 cells were sensitive to the ICL-inducing agent mytomycin C, and knocking down RUNX1-ETO reduced this sensitivity. Depleting RUNX1-ETO also led to increased FANCD2 recruitment to chromatin.
The team said these results suggest that RUNX1-ETO might repress FANCD2 foci formation and ICL repair in AML cells. And they predicted that RUNX dysfunction would sensitize the cells to PARP inhibitors.
So they tested 2 PARP inhibitors—olaparib and rucaparib—in Kasumi-1 cells and observed sensitivity to both drugs. Knocking down RUNX1-ETO partially reduced this sensitivity, while adding mytomycin C increased sensitivity.
“PARP inhibitors have been with us for quite some time, but nobody has realized their application for leukemia,” Dr Osato said. “Our study has shed light on the possibility of a more effective treatment using a combined therapy with PARP inhibitors, which can potentially be extended to other types of common cancers.”
The researchers are now conducting additional drug testing in xenograft models. ![]()
Chemo and CAR T cells prompt responses in NHL
An infusion of chimeric antigen receptor (CAR) T-cell therapy following chemotherapy can elicit responses in patients with non-Hodgkin lymphoma, a small study suggests.
However, patients also experienced significant acute toxicities, including fever, low blood pressure, focal neurological deficits, and delirium.
James N. Kochenderfer, MD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues reported these results in the Journal of Clinical Oncology.
The trial was sponsored by the National Cancer Institute, but the CAR T-cell therapy being tested uses the same CAR construct as KTE-C19, which is being developed by Kite Pharma, Inc.
The study included 15 patients with advanced B-cell malignancies. The patients first received conditioning with cyclophosphamide and fludarabine.
A day later, they received a single infusion of the CAR T-cell therapy, which consists of T cells taken from each patient’s peripheral blood and modified to target CD19.
The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Response rates
Thirteen patients were evaluable for response. One patient was lost to follow-up because of noncompliance, and 1 died soon after treatment. The researchers said the cause of death was likely cardiac arrhythmia.
The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs are ongoing, with the duration ranging from 9 months to 22 months.
Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. All 3 CRs are ongoing, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the ongoing CR is 11 months.
Toxicity
As seen in other studies, the CAR T-cell therapy was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or IL-6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
One of these patients developed aphasia on day 5 after CAR T-cell infusion. It occurred intermittently for 7 days before resolving. The patient also experienced right-sided facial paresis that lasted approximately 20 minutes on day 8 after infusion.
Another patient developed aphasia 5 days after CAR T-cell infusion, but this was followed by confusion and severe generalized myoclonus. All symptoms resolved by 11 days after the infusion, except for a mild tremor that resolved over the next month.
A third patient developed aphasia 5 days after CAR T-cell infusion. This was followed by confusion, hemifacial spasms, apraxia, and gait disturbances. These effects varied in severity but dramatically improved 20 days after the infusion, according to the researchers. ![]()
An infusion of chimeric antigen receptor (CAR) T-cell therapy following chemotherapy can elicit responses in patients with non-Hodgkin lymphoma, a small study suggests.
However, patients also experienced significant acute toxicities, including fever, low blood pressure, focal neurological deficits, and delirium.
James N. Kochenderfer, MD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues reported these results in the Journal of Clinical Oncology.
The trial was sponsored by the National Cancer Institute, but the CAR T-cell therapy being tested uses the same CAR construct as KTE-C19, which is being developed by Kite Pharma, Inc.
The study included 15 patients with advanced B-cell malignancies. The patients first received conditioning with cyclophosphamide and fludarabine.
A day later, they received a single infusion of the CAR T-cell therapy, which consists of T cells taken from each patient’s peripheral blood and modified to target CD19.
The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Response rates
Thirteen patients were evaluable for response. One patient was lost to follow-up because of noncompliance, and 1 died soon after treatment. The researchers said the cause of death was likely cardiac arrhythmia.
The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs are ongoing, with the duration ranging from 9 months to 22 months.
Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. All 3 CRs are ongoing, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the ongoing CR is 11 months.
Toxicity
As seen in other studies, the CAR T-cell therapy was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or IL-6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
One of these patients developed aphasia on day 5 after CAR T-cell infusion. It occurred intermittently for 7 days before resolving. The patient also experienced right-sided facial paresis that lasted approximately 20 minutes on day 8 after infusion.
Another patient developed aphasia 5 days after CAR T-cell infusion, but this was followed by confusion and severe generalized myoclonus. All symptoms resolved by 11 days after the infusion, except for a mild tremor that resolved over the next month.
A third patient developed aphasia 5 days after CAR T-cell infusion. This was followed by confusion, hemifacial spasms, apraxia, and gait disturbances. These effects varied in severity but dramatically improved 20 days after the infusion, according to the researchers. ![]()
An infusion of chimeric antigen receptor (CAR) T-cell therapy following chemotherapy can elicit responses in patients with non-Hodgkin lymphoma, a small study suggests.
However, patients also experienced significant acute toxicities, including fever, low blood pressure, focal neurological deficits, and delirium.
James N. Kochenderfer, MD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues reported these results in the Journal of Clinical Oncology.
The trial was sponsored by the National Cancer Institute, but the CAR T-cell therapy being tested uses the same CAR construct as KTE-C19, which is being developed by Kite Pharma, Inc.
The study included 15 patients with advanced B-cell malignancies. The patients first received conditioning with cyclophosphamide and fludarabine.
A day later, they received a single infusion of the CAR T-cell therapy, which consists of T cells taken from each patient’s peripheral blood and modified to target CD19.
The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Response rates
Thirteen patients were evaluable for response. One patient was lost to follow-up because of noncompliance, and 1 died soon after treatment. The researchers said the cause of death was likely cardiac arrhythmia.
The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs are ongoing, with the duration ranging from 9 months to 22 months.
Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. All 3 CRs are ongoing, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the ongoing CR is 11 months.
Toxicity
As seen in other studies, the CAR T-cell therapy was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or IL-6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
One of these patients developed aphasia on day 5 after CAR T-cell infusion. It occurred intermittently for 7 days before resolving. The patient also experienced right-sided facial paresis that lasted approximately 20 minutes on day 8 after infusion.
Another patient developed aphasia 5 days after CAR T-cell infusion, but this was followed by confusion and severe generalized myoclonus. All symptoms resolved by 11 days after the infusion, except for a mild tremor that resolved over the next month.
A third patient developed aphasia 5 days after CAR T-cell infusion. This was followed by confusion, hemifacial spasms, apraxia, and gait disturbances. These effects varied in severity but dramatically improved 20 days after the infusion, according to the researchers. ![]()
Tool mines scientific literature, generates hypotheses

Credit: NIH
A new tool may help researchers sift through the scientific literature to discover hypothesis-generating information relevant to their own research.
The resource, called the Knowledge Integration Toolkit (KnIT), extracts relevant information from the literature, includes it in a network that can be queried, and
then attempts to use these data to generate reasonable and testable hypotheses that can help direct lab studies.
Researchers tested KnIT in a retrospective case study involving published data on p53 and found the tool could accurately predict the existence of proteins that modify p53.
Details from this study were published in the Association for Computing Machinery’s digital library.
Olivier Lichtarge, MD, PhD, of the Baylor College of Medicine in Houston, Texas, is scheduled to discuss the study on August 27 at the 20th Annual Association for Computing Machinery’s Special Interest Group on Knowledge Discovery and Data Mining Conference in New York, New York.
“On average, a scientist might read between 1 and 5 research papers on a good day,” Dr Lichtarge noted.
“But, to put this in perspective with p53, there are over 70,000 papers published on this protein. Even if a scientist reads 5 papers a day, it could take nearly 38 years to completely understand all of the research already available today on this protein.”
Scientists formulate hypotheses based on what they read and know, but because they cannot read everything, their hypotheses may be biased, according to Dr Lichtarge.
“A computer certainly may not reason as well as a scientist,” he said, “but the little it can, logically and objectively, may contribute greatly when applied to our entire body of knowledge.”
With that in mind, Dr Lichtarge and his colleagues initiated a project to develop a knowledge integration tool that took advantage of existing text mining capabilities, such as those used by IBM’s Watson technology—cognitive technology that processes information more like a human than a computer.
And the team came up with KnIT. In the first test using KnIT, they sought to identify new protein kinases that phosphorylate p53.
There are more than 500 known human kinases and tens of thousands of possible proteins they can target. Thirty-three are currently known to modify p53.
The researchers used KnIT to mine the scientific literature up to 2003, when only half of the 33 phosphorylating protein kinases had been discovered.
Seventy-four kinases were extracted as potential modifiers. Of these, prior to 2003, 10 were known to phosphorylate p53, and 9 were discovered at a later date.
Of the 10 already known, KnIT accounted for them in reasoning as well as ranking the likelihood that the other 64 kinases targeted p53. Of the 9 found nearly a decade later, KnIT accurately predicted 7.
“This study showed that, in a very narrow field of study regarding p53, we can, in fact, suggest new relationships and new functions associated with p53, which can later be directly validated in the laboratory,” Dr Lichtarge said.
“Our long-term hope is to systematically extract knowledge directly from the totality of the public medical literature. For this, we need technological advances to read text, extract facts from every sentence, and to integrate this information into a network that describes the relationship between all of the objects and entities discussed in the literature.”
“This first study is promising, because it suggests a proof of principle for a small step towards this type of knowledge discovery. With more research, we hope to get closer to clinical and therapeutic applications.” ![]()
Credit: NIH
A new tool may help researchers sift through the scientific literature to discover hypothesis-generating information relevant to their own research.
The resource, called the Knowledge Integration Toolkit (KnIT), extracts relevant information from the literature, includes it in a network that can be queried, and
then attempts to use these data to generate reasonable and testable hypotheses that can help direct lab studies.
Researchers tested KnIT in a retrospective case study involving published data on p53 and found the tool could accurately predict the existence of proteins that modify p53.
Details from this study were published in the Association for Computing Machinery’s digital library.
Olivier Lichtarge, MD, PhD, of the Baylor College of Medicine in Houston, Texas, is scheduled to discuss the study on August 27 at the 20th Annual Association for Computing Machinery’s Special Interest Group on Knowledge Discovery and Data Mining Conference in New York, New York.
“On average, a scientist might read between 1 and 5 research papers on a good day,” Dr Lichtarge noted.
“But, to put this in perspective with p53, there are over 70,000 papers published on this protein. Even if a scientist reads 5 papers a day, it could take nearly 38 years to completely understand all of the research already available today on this protein.”
Scientists formulate hypotheses based on what they read and know, but because they cannot read everything, their hypotheses may be biased, according to Dr Lichtarge.
“A computer certainly may not reason as well as a scientist,” he said, “but the little it can, logically and objectively, may contribute greatly when applied to our entire body of knowledge.”
With that in mind, Dr Lichtarge and his colleagues initiated a project to develop a knowledge integration tool that took advantage of existing text mining capabilities, such as those used by IBM’s Watson technology—cognitive technology that processes information more like a human than a computer.
And the team came up with KnIT. In the first test using KnIT, they sought to identify new protein kinases that phosphorylate p53.
There are more than 500 known human kinases and tens of thousands of possible proteins they can target. Thirty-three are currently known to modify p53.
The researchers used KnIT to mine the scientific literature up to 2003, when only half of the 33 phosphorylating protein kinases had been discovered.
Seventy-four kinases were extracted as potential modifiers. Of these, prior to 2003, 10 were known to phosphorylate p53, and 9 were discovered at a later date.
Of the 10 already known, KnIT accounted for them in reasoning as well as ranking the likelihood that the other 64 kinases targeted p53. Of the 9 found nearly a decade later, KnIT accurately predicted 7.
“This study showed that, in a very narrow field of study regarding p53, we can, in fact, suggest new relationships and new functions associated with p53, which can later be directly validated in the laboratory,” Dr Lichtarge said.
“Our long-term hope is to systematically extract knowledge directly from the totality of the public medical literature. For this, we need technological advances to read text, extract facts from every sentence, and to integrate this information into a network that describes the relationship between all of the objects and entities discussed in the literature.”
“This first study is promising, because it suggests a proof of principle for a small step towards this type of knowledge discovery. With more research, we hope to get closer to clinical and therapeutic applications.” ![]()
Credit: NIH
A new tool may help researchers sift through the scientific literature to discover hypothesis-generating information relevant to their own research.
The resource, called the Knowledge Integration Toolkit (KnIT), extracts relevant information from the literature, includes it in a network that can be queried, and
then attempts to use these data to generate reasonable and testable hypotheses that can help direct lab studies.
Researchers tested KnIT in a retrospective case study involving published data on p53 and found the tool could accurately predict the existence of proteins that modify p53.
Details from this study were published in the Association for Computing Machinery’s digital library.
Olivier Lichtarge, MD, PhD, of the Baylor College of Medicine in Houston, Texas, is scheduled to discuss the study on August 27 at the 20th Annual Association for Computing Machinery’s Special Interest Group on Knowledge Discovery and Data Mining Conference in New York, New York.
“On average, a scientist might read between 1 and 5 research papers on a good day,” Dr Lichtarge noted.
“But, to put this in perspective with p53, there are over 70,000 papers published on this protein. Even if a scientist reads 5 papers a day, it could take nearly 38 years to completely understand all of the research already available today on this protein.”
Scientists formulate hypotheses based on what they read and know, but because they cannot read everything, their hypotheses may be biased, according to Dr Lichtarge.
“A computer certainly may not reason as well as a scientist,” he said, “but the little it can, logically and objectively, may contribute greatly when applied to our entire body of knowledge.”
With that in mind, Dr Lichtarge and his colleagues initiated a project to develop a knowledge integration tool that took advantage of existing text mining capabilities, such as those used by IBM’s Watson technology—cognitive technology that processes information more like a human than a computer.
And the team came up with KnIT. In the first test using KnIT, they sought to identify new protein kinases that phosphorylate p53.
There are more than 500 known human kinases and tens of thousands of possible proteins they can target. Thirty-three are currently known to modify p53.
The researchers used KnIT to mine the scientific literature up to 2003, when only half of the 33 phosphorylating protein kinases had been discovered.
Seventy-four kinases were extracted as potential modifiers. Of these, prior to 2003, 10 were known to phosphorylate p53, and 9 were discovered at a later date.
Of the 10 already known, KnIT accounted for them in reasoning as well as ranking the likelihood that the other 64 kinases targeted p53. Of the 9 found nearly a decade later, KnIT accurately predicted 7.
“This study showed that, in a very narrow field of study regarding p53, we can, in fact, suggest new relationships and new functions associated with p53, which can later be directly validated in the laboratory,” Dr Lichtarge said.
“Our long-term hope is to systematically extract knowledge directly from the totality of the public medical literature. For this, we need technological advances to read text, extract facts from every sentence, and to integrate this information into a network that describes the relationship between all of the objects and entities discussed in the literature.”
“This first study is promising, because it suggests a proof of principle for a small step towards this type of knowledge discovery. With more research, we hope to get closer to clinical and therapeutic applications.” ![]()
USPSTF: Offer behavioral counseling to prevent cardiovascular disease
Overweight or obese adults at risk for cardiovascular disease should receive intensive behavioral counseling interventions, according to a recommendation statement by the U. S. Preventive Services Task Force.
After a comprehensive review of the current literature, the USPSTF concluded "with moderate certainty" that interventions promoting a healthful diet and increased physical activity have a moderate net benefit in this patient population, said Dr. Michael L. LeFevre, chair of the task force at the time the recommendation was finalized, and professor of family medicine at the University of Missouri, Columbia.
The recommendation statement issued Aug. 25 is "an update and refinement" of the 2003 USPSTF recommendation on dietary counseling for at-risk adults, this time targeting overweight or obese patients who have additional CVD risk factors such as hypertension, dyslipidemia, impaired fasting glucose, or metabolic syndrome.
The group reviewed 74 trials assessing the effectiveness of behavioral counseling interventions of various intensities. Only 16 reported on direct health outcomes such as CVD events, mortality, or quality of life, the task force noted, so there is inadequate evidence about intensive behavioral counseling’s effect on such outcomes.
A total of 71 trials involving more than 32,000 participants focused on intermediate health outcomes such as lipid levels, blood pressure, glucose levels, weight, and medication use. Overall, intensive counseling interventions made "small but important changes" in these outcomes, with total cholesterol levels decreasing approximately 3-6 mg/dL, LDL cholesterol decreasing by 1.5-5.0 mg/dL, systolic blood pressure decreasing by 1-3 mm Hg, diastolic blood pressure decreasing by 1-2 mm Hg, and fasting glucose levels decreasing by 1-3 mg/dL.
In addition, weight decreased by a mean of approximately 3 kg, and the proportion of patients who participated in moderate-intensity exercise for 150 minutes per week rose from 10% to 25%. However, few of these studies followed patients for more than 1-2 years, so the evidence was inadequate to assess longer-term benefits.
Because very few and mostly minor adverse events were associated with these interventions, they yielded a moderate net benefit.
Most of the intensive behavioral counseling interventions that were assessed averaged 5-16 individual or group sessions during a period of 9-12 months. All included didactic education; in most programs, specially trained professionals (dietitians, nutritionists, physiotherapists, exercise professionals, health educators, and psychologists) provided monitoring and feedback for the participants, devised individualized care plans, and taught problem-solving skills. Many types and combinations of interventions were effective, and almost all were delivered outside the primary care setting.
The recommendation statement is in line with others published by the American Heart Association, the American College of Sports Medicine, and the American Academy of Family Physicians. The AAFP is updating its recommendations regarding behavioral counseling to prevent CVD, Dr. LeFevre and his associates noted.
The USPSTF, funded by but independent of the federal government, makes recommendations about the effectiveness of specific preventive-care services based on evidence of benefits and harms, without considering costs.
Do you refer overweight or obese patients with cardiovascular risk factors for intensive behavioral counseling? Take our Quick Poll on the Family Practice News homepage.
Overweight or obese adults at risk for cardiovascular disease should receive intensive behavioral counseling interventions, according to a recommendation statement by the U. S. Preventive Services Task Force.
After a comprehensive review of the current literature, the USPSTF concluded "with moderate certainty" that interventions promoting a healthful diet and increased physical activity have a moderate net benefit in this patient population, said Dr. Michael L. LeFevre, chair of the task force at the time the recommendation was finalized, and professor of family medicine at the University of Missouri, Columbia.
The recommendation statement issued Aug. 25 is "an update and refinement" of the 2003 USPSTF recommendation on dietary counseling for at-risk adults, this time targeting overweight or obese patients who have additional CVD risk factors such as hypertension, dyslipidemia, impaired fasting glucose, or metabolic syndrome.
The group reviewed 74 trials assessing the effectiveness of behavioral counseling interventions of various intensities. Only 16 reported on direct health outcomes such as CVD events, mortality, or quality of life, the task force noted, so there is inadequate evidence about intensive behavioral counseling’s effect on such outcomes.
A total of 71 trials involving more than 32,000 participants focused on intermediate health outcomes such as lipid levels, blood pressure, glucose levels, weight, and medication use. Overall, intensive counseling interventions made "small but important changes" in these outcomes, with total cholesterol levels decreasing approximately 3-6 mg/dL, LDL cholesterol decreasing by 1.5-5.0 mg/dL, systolic blood pressure decreasing by 1-3 mm Hg, diastolic blood pressure decreasing by 1-2 mm Hg, and fasting glucose levels decreasing by 1-3 mg/dL.
In addition, weight decreased by a mean of approximately 3 kg, and the proportion of patients who participated in moderate-intensity exercise for 150 minutes per week rose from 10% to 25%. However, few of these studies followed patients for more than 1-2 years, so the evidence was inadequate to assess longer-term benefits.
Because very few and mostly minor adverse events were associated with these interventions, they yielded a moderate net benefit.
Most of the intensive behavioral counseling interventions that were assessed averaged 5-16 individual or group sessions during a period of 9-12 months. All included didactic education; in most programs, specially trained professionals (dietitians, nutritionists, physiotherapists, exercise professionals, health educators, and psychologists) provided monitoring and feedback for the participants, devised individualized care plans, and taught problem-solving skills. Many types and combinations of interventions were effective, and almost all were delivered outside the primary care setting.
The recommendation statement is in line with others published by the American Heart Association, the American College of Sports Medicine, and the American Academy of Family Physicians. The AAFP is updating its recommendations regarding behavioral counseling to prevent CVD, Dr. LeFevre and his associates noted.
The USPSTF, funded by but independent of the federal government, makes recommendations about the effectiveness of specific preventive-care services based on evidence of benefits and harms, without considering costs.
Do you refer overweight or obese patients with cardiovascular risk factors for intensive behavioral counseling? Take our Quick Poll on the Family Practice News homepage.
Overweight or obese adults at risk for cardiovascular disease should receive intensive behavioral counseling interventions, according to a recommendation statement by the U. S. Preventive Services Task Force.
After a comprehensive review of the current literature, the USPSTF concluded "with moderate certainty" that interventions promoting a healthful diet and increased physical activity have a moderate net benefit in this patient population, said Dr. Michael L. LeFevre, chair of the task force at the time the recommendation was finalized, and professor of family medicine at the University of Missouri, Columbia.
The recommendation statement issued Aug. 25 is "an update and refinement" of the 2003 USPSTF recommendation on dietary counseling for at-risk adults, this time targeting overweight or obese patients who have additional CVD risk factors such as hypertension, dyslipidemia, impaired fasting glucose, or metabolic syndrome.
The group reviewed 74 trials assessing the effectiveness of behavioral counseling interventions of various intensities. Only 16 reported on direct health outcomes such as CVD events, mortality, or quality of life, the task force noted, so there is inadequate evidence about intensive behavioral counseling’s effect on such outcomes.
A total of 71 trials involving more than 32,000 participants focused on intermediate health outcomes such as lipid levels, blood pressure, glucose levels, weight, and medication use. Overall, intensive counseling interventions made "small but important changes" in these outcomes, with total cholesterol levels decreasing approximately 3-6 mg/dL, LDL cholesterol decreasing by 1.5-5.0 mg/dL, systolic blood pressure decreasing by 1-3 mm Hg, diastolic blood pressure decreasing by 1-2 mm Hg, and fasting glucose levels decreasing by 1-3 mg/dL.
In addition, weight decreased by a mean of approximately 3 kg, and the proportion of patients who participated in moderate-intensity exercise for 150 minutes per week rose from 10% to 25%. However, few of these studies followed patients for more than 1-2 years, so the evidence was inadequate to assess longer-term benefits.
Because very few and mostly minor adverse events were associated with these interventions, they yielded a moderate net benefit.
Most of the intensive behavioral counseling interventions that were assessed averaged 5-16 individual or group sessions during a period of 9-12 months. All included didactic education; in most programs, specially trained professionals (dietitians, nutritionists, physiotherapists, exercise professionals, health educators, and psychologists) provided monitoring and feedback for the participants, devised individualized care plans, and taught problem-solving skills. Many types and combinations of interventions were effective, and almost all were delivered outside the primary care setting.
The recommendation statement is in line with others published by the American Heart Association, the American College of Sports Medicine, and the American Academy of Family Physicians. The AAFP is updating its recommendations regarding behavioral counseling to prevent CVD, Dr. LeFevre and his associates noted.
The USPSTF, funded by but independent of the federal government, makes recommendations about the effectiveness of specific preventive-care services based on evidence of benefits and harms, without considering costs.
Do you refer overweight or obese patients with cardiovascular risk factors for intensive behavioral counseling? Take our Quick Poll on the Family Practice News homepage.
FROM ANNALS OF INTERNAL MEDICINE
Key clinical point: Refer overweight or obese patients at risk of cardiovascular disease for intensive behavioral counseling.
Major Finding: Intensive behavioral counseling interventions made small but important changes in several intermediate health outcomes, with total cholesterol levels decreasing by about 3-6 mg/dL, LDL cholesterol decreasing by 1.5-5.0 mg/dL, systolic blood pressure decreasing by 1-3 mm Hg, diastolic blood pressure decreasing by 1-2 mm Hg, fasting glucose levels decreasing by 1-3 mg/dL, and weight decreasing by a mean of 3 kg.
Data Source: An update and refinement of a 2003 USPSTF recommendation on dietary counseling for overweight/obese adults who have additional risk factors for CVD, based on a comprehensive review of the literature.
Disclosures: The USPSTF, funded by but independent of the federal government, makes recommendations about the effectiveness of specific preventive-care services based on evidence of benefits and harms, without considering costs.

AHA wants e-cigarettes regulated but notes they help some smokers quit
The American Heart Association has released its recommendations regarding the sale and usage of e-cigarettes, cautioning that the devices should be regulated to avoid enticing children to smoke while also saying that e-cigarettes could be used as a way to help some current smokers quit.
The guidelines, published online Aug. 24 in Circulation, state that clinicians "should not discourage" their patients from resorting to e-cigarettes if other approved forms of smoking cessation, such as the nicotine patch, have been exhausted. The AHA warned, however, that further studies are needed to fully understand the effects of e-cigarette usage, stressing that e-cigarettes have not been approved by the Food and Drug Administration as an acceptable smoking cessation device and could contain low amounts of toxic chemicals that would prove more harmful than helpful to patients in the long run.

"E-cigarettes have caused a major shift in the tobacco-control landscape," Aruni Bhatnagar, Ph.D., lead author of the guidelines and chair of cardiovascular medicine at the University of Louisville, Ky., said in a statement. "It’s critical that we rigorously examine the long-term impact of this new technology on public health, cardiovascular disease, and stroke, and pay careful attention to the effect of e-cigarettes on adolescents."
For now, the AHA says that clinicians should tell their patients who use e-cigarettes to set a firm quit date, warning that any device that delivers nicotine into the body is harmful and likely lethal.
"Nicotine is a dangerous and highly addictive chemical no matter what form it takes – conventional cigarettes or some other tobacco product," Dr. Elliott Antman, AHA president, said in the statement. "Every life that has been lost to tobacco addiction" could have been saved, he noted.
To that end, the AHA also called for strong regulations regarding the potential marketability of e-cigarettes to youngsters. The group recommended a federal ban on the sale of e-cigarettes to minors, warning that the devices could become a gateway to actual cigarettes for young people who see e-cigarettes as "high-tech, accessible, and convenient," according to a JAMA Pediatrics study of 40,000 middle and high school students. The AHA also recommended that all existing rules and regulations in place for tobacco-related products should apply to e-cigarettes.
"We must protect future generations from any potential smokescreens in the tobacco product landscape that will cause us to lose precious ground in the fight to make our nation 100% tobacco-free," Dr. Antman said.
According to the AHA, 20 million Americans have lost their lives to tobacco over the last 50 years.
The American Heart Association has released its recommendations regarding the sale and usage of e-cigarettes, cautioning that the devices should be regulated to avoid enticing children to smoke while also saying that e-cigarettes could be used as a way to help some current smokers quit.
The guidelines, published online Aug. 24 in Circulation, state that clinicians "should not discourage" their patients from resorting to e-cigarettes if other approved forms of smoking cessation, such as the nicotine patch, have been exhausted. The AHA warned, however, that further studies are needed to fully understand the effects of e-cigarette usage, stressing that e-cigarettes have not been approved by the Food and Drug Administration as an acceptable smoking cessation device and could contain low amounts of toxic chemicals that would prove more harmful than helpful to patients in the long run.
"E-cigarettes have caused a major shift in the tobacco-control landscape," Aruni Bhatnagar, Ph.D., lead author of the guidelines and chair of cardiovascular medicine at the University of Louisville, Ky., said in a statement. "It’s critical that we rigorously examine the long-term impact of this new technology on public health, cardiovascular disease, and stroke, and pay careful attention to the effect of e-cigarettes on adolescents."
For now, the AHA says that clinicians should tell their patients who use e-cigarettes to set a firm quit date, warning that any device that delivers nicotine into the body is harmful and likely lethal.
"Nicotine is a dangerous and highly addictive chemical no matter what form it takes – conventional cigarettes or some other tobacco product," Dr. Elliott Antman, AHA president, said in the statement. "Every life that has been lost to tobacco addiction" could have been saved, he noted.
To that end, the AHA also called for strong regulations regarding the potential marketability of e-cigarettes to youngsters. The group recommended a federal ban on the sale of e-cigarettes to minors, warning that the devices could become a gateway to actual cigarettes for young people who see e-cigarettes as "high-tech, accessible, and convenient," according to a JAMA Pediatrics study of 40,000 middle and high school students. The AHA also recommended that all existing rules and regulations in place for tobacco-related products should apply to e-cigarettes.
"We must protect future generations from any potential smokescreens in the tobacco product landscape that will cause us to lose precious ground in the fight to make our nation 100% tobacco-free," Dr. Antman said.
According to the AHA, 20 million Americans have lost their lives to tobacco over the last 50 years.
The American Heart Association has released its recommendations regarding the sale and usage of e-cigarettes, cautioning that the devices should be regulated to avoid enticing children to smoke while also saying that e-cigarettes could be used as a way to help some current smokers quit.
The guidelines, published online Aug. 24 in Circulation, state that clinicians "should not discourage" their patients from resorting to e-cigarettes if other approved forms of smoking cessation, such as the nicotine patch, have been exhausted. The AHA warned, however, that further studies are needed to fully understand the effects of e-cigarette usage, stressing that e-cigarettes have not been approved by the Food and Drug Administration as an acceptable smoking cessation device and could contain low amounts of toxic chemicals that would prove more harmful than helpful to patients in the long run.
"E-cigarettes have caused a major shift in the tobacco-control landscape," Aruni Bhatnagar, Ph.D., lead author of the guidelines and chair of cardiovascular medicine at the University of Louisville, Ky., said in a statement. "It’s critical that we rigorously examine the long-term impact of this new technology on public health, cardiovascular disease, and stroke, and pay careful attention to the effect of e-cigarettes on adolescents."
For now, the AHA says that clinicians should tell their patients who use e-cigarettes to set a firm quit date, warning that any device that delivers nicotine into the body is harmful and likely lethal.
"Nicotine is a dangerous and highly addictive chemical no matter what form it takes – conventional cigarettes or some other tobacco product," Dr. Elliott Antman, AHA president, said in the statement. "Every life that has been lost to tobacco addiction" could have been saved, he noted.
To that end, the AHA also called for strong regulations regarding the potential marketability of e-cigarettes to youngsters. The group recommended a federal ban on the sale of e-cigarettes to minors, warning that the devices could become a gateway to actual cigarettes for young people who see e-cigarettes as "high-tech, accessible, and convenient," according to a JAMA Pediatrics study of 40,000 middle and high school students. The AHA also recommended that all existing rules and regulations in place for tobacco-related products should apply to e-cigarettes.
"We must protect future generations from any potential smokescreens in the tobacco product landscape that will cause us to lose precious ground in the fight to make our nation 100% tobacco-free," Dr. Antman said.
According to the AHA, 20 million Americans have lost their lives to tobacco over the last 50 years.
FROM CIRCULATION
