User login
American Board of Internal Medicine Foundation's Choosing Wisely Campaign Promotes Evidence-Based Patient Care

The American Board of Internal Medicine (ABIM) established the ABIM Foundation to advance professionalism in improving healthcare. The foundation initiated the Choosing Wisely campaign [www.choosingwisely.org] in April 2012 to promote conversations that help physicians guide patients in selecting care that is supported by evidence, not duplicative of other tests or procedures, not harmful, and truly necessary. In order to achieve this, national organizations representing medical specialists were asked to identify five common tests or procedures whose necessity should be questioned.
John Bulger, DO, MBA, SFHM, chief quality officer at Geisinger Health System in Danville, Pa., chaired SHM’s Choosing Wisely recommendations committee. He says the “proximal concern over these tests may be the unnecessary cost of the test itself, [but] there are other unintended consequences.
“False positive or false negative results of unsupported testing may cause unwarranted emotional harm for the patient or may give a false sense of security,” he adds. “The latter may also be true for physicians who may fail to further investigate other ailments based on a previous false negative test. Tests ordered with little evidence tend to lead to more tests ordered with little evidence.”
To date, more than 60 specialty societies and 17 consumer groups have joined the Choosing Wisely effort, citing more than 300 potentially harmful tests and procedures that physicians should discuss with patients. New lists will be published throughout 2014.
—Karen Appold
The American Board of Internal Medicine (ABIM) established the ABIM Foundation to advance professionalism in improving healthcare. The foundation initiated the Choosing Wisely campaign [www.choosingwisely.org] in April 2012 to promote conversations that help physicians guide patients in selecting care that is supported by evidence, not duplicative of other tests or procedures, not harmful, and truly necessary. In order to achieve this, national organizations representing medical specialists were asked to identify five common tests or procedures whose necessity should be questioned.
John Bulger, DO, MBA, SFHM, chief quality officer at Geisinger Health System in Danville, Pa., chaired SHM’s Choosing Wisely recommendations committee. He says the “proximal concern over these tests may be the unnecessary cost of the test itself, [but] there are other unintended consequences.
“False positive or false negative results of unsupported testing may cause unwarranted emotional harm for the patient or may give a false sense of security,” he adds. “The latter may also be true for physicians who may fail to further investigate other ailments based on a previous false negative test. Tests ordered with little evidence tend to lead to more tests ordered with little evidence.”
To date, more than 60 specialty societies and 17 consumer groups have joined the Choosing Wisely effort, citing more than 300 potentially harmful tests and procedures that physicians should discuss with patients. New lists will be published throughout 2014.
—Karen Appold
The American Board of Internal Medicine (ABIM) established the ABIM Foundation to advance professionalism in improving healthcare. The foundation initiated the Choosing Wisely campaign [www.choosingwisely.org] in April 2012 to promote conversations that help physicians guide patients in selecting care that is supported by evidence, not duplicative of other tests or procedures, not harmful, and truly necessary. In order to achieve this, national organizations representing medical specialists were asked to identify five common tests or procedures whose necessity should be questioned.
John Bulger, DO, MBA, SFHM, chief quality officer at Geisinger Health System in Danville, Pa., chaired SHM’s Choosing Wisely recommendations committee. He says the “proximal concern over these tests may be the unnecessary cost of the test itself, [but] there are other unintended consequences.
“False positive or false negative results of unsupported testing may cause unwarranted emotional harm for the patient or may give a false sense of security,” he adds. “The latter may also be true for physicians who may fail to further investigate other ailments based on a previous false negative test. Tests ordered with little evidence tend to lead to more tests ordered with little evidence.”
To date, more than 60 specialty societies and 17 consumer groups have joined the Choosing Wisely effort, citing more than 300 potentially harmful tests and procedures that physicians should discuss with patients. New lists will be published throughout 2014.
—Karen Appold
When to Order Red Blood Cell Transfusion for Patients with Anemia
Background
Hospitalists commonly order red blood cell (RBC) transfusion as a therapy for patients with anemia resulting from a variety of clinical conditions. There has been lack of consensus on when to transfuse, because patients with anemia frequently have multiple co-morbidities, including coronary artery disease and congestive heart failure, which may influence their ability to tolerate a potentially ischemic state related to anemia or to accommodate volume fluctuations related to transfusion.
Furthermore, RBC transfusions are not without inherent risk. Life-threatening transfusion reactions occur in approximately seven per million transfused blood components, and transfusion-associated circulatory overload (TACO) can develop in one in 100 transfusions.1
Recently published guidelines provide recommendations for management of hemodynamically stable adults with anemia.
Guideline Update
The AABB published guidelines in the Annals of Internal Medicine in 2012 addressing RBC transfusion thresholds.1 The updated guideline makes a recommendation that clinicians utilize a restrictive transfusion strategy. Transfusion is strongly recommended for ICU patients with hemoglobin ≤7g/dL. In post-operative surgical patients and for post-operative patients with symptomatic anemia, transfusion is recommended for hemoglobin ≤8g/dL. The authors also made a weak recommendation to transfuse for hemoglobin ≤8g/dL or for symptoms in hospitalized hemodynamically stable patients with preexisting cardiovascular disease.
These recommendations draw from past literature, along with two more recent trials examining liberal or restrictive transfusion thresholds. The newer trials increased the total number of patients studied by nearly one third compared with prior reviews.2,3 The authors also incorporated recently published systematic reviews in their analysis.
Although the definition of a restrictive transfusion threshold varied across trials, including hemoglobin ≤7g/dL and ≤8g/dL, the authors used the pooled data to provide several recommendations in the new guideline. Of note, the pooled data was underpowered to detect up to a twofold increase in risk of myocardial infarction in patients in the restrictive strategy group.1
There were insufficient data for the authors to recommend for or against a restrictive transfusion strategy in patients with acute coronary syndrome, based on very low quality evidence.
Finally, the authors recommended that symptoms and hemoglobin level should both be used in determining transfusion criteria, based on low quality of evidence.
Analysis
The current AABB guidelines have two primary differences from earlier guidelines. First, the AABB authors used GRADE (Grading of Recommendations, Assessment, Development, and Evaluation) methodology to formalize evidence-based practice in their analysis of the literature. The authors purposely used the GRADE methodology to systematically evaluate the quality of the evidence base and explicitly state the strength of the recommendation for a particular transfusion threshold.4
Second, the AABB guidelines incorporated data from the more recently published FOCUS (Functional Outcomes in Cardiovascular patients Undergoing Surgical repair of hip fracture) and TRACS (Transfusion Requirements After Cardiac Surgery) trials, resulting in a stronger recommendation supporting the use of a restrictive transfusion strategy in non-ICU and post-operative patients. The findings of the FOCUS trial are especially applicable to hospitalists, because many patients who undergo hip fracture repair are directly cared for or are co-managed by hospitalists.
The current guidelines built upon previous guidelines that advocated a restrictive strategy (hemoglobin ≤7g/dL) in hemodynamically stable, critically ill adult patients.5 In general, restrictive transfusion strategy led to nearly 40% fewer patients receiving transfusion compared with the use of a liberal transfusion strategy.1 No additional harm to patients was evidenced in the restrictive transfusion group, though the trials were not designed to answer this question; moreover, there was no statistically significant difference in mortality or functional outcome between the two groups.
The authors of the current AABB guidelines recognized the importance of replicating the current findings in a more diverse patient population. An area where further study is indicated is in the use of specific transfusion thresholds in patients with acute coronary syndrome. These guidelines did not clarify whether or not there is a physiologic difference between use of different restrictive transfusion thresholds such as <8g/dL and <7g/dL.
The authors of the AABB guidelines also commented that performing a future trial to compare RBC transfusion for symptoms vs. hemoglobin “trigger” would be useful; however, they recognized that this may not be feasible due to the need to blind providers in the trial to hemoglobin values. Various society guidelines currently call for different transfusion thresholds or do not make a specific recommendation at all.1
Key Takeaways for Hospitalists
For the vast majority of medical patients, hospitalists can safely use a restrictive RBC transfusion threshold (≤7g/dL or ≤8g/dL), which can lead to a significant decrease in RBC transfusions without adversely affecting overall mortality.
Drs. Bortinger and Carbo are hospitalists at Beth Israel Deaconess Medical Center in Boston.
References
- Carson JL, Grossman BJ, Kleinman S, et al. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Inter Med. 2012;157(1):49-58.
- Carson AL, Terrin ML, Noveck H, et al. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;367(26):2453-2462.
- Hajjar LA, Vincent JL, Galas FR, et al. Transfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA. 2010;304(14):1559-1567.
- Carson JL, Carless PA, Herbert PC. Transfusion threshold and other strategies for guiding allogenic red blood cell transfusion. Cochrane Database Syst Rev. 2012;CD002042.
- Napolitano LM, Kurek S, Luchette FA, et al. Clinical practice guideline: red blood cell transfusion in adult trauma and critical care. Crit Care Med. 2009;37(12):3124-3157.
Background
Hospitalists commonly order red blood cell (RBC) transfusion as a therapy for patients with anemia resulting from a variety of clinical conditions. There has been lack of consensus on when to transfuse, because patients with anemia frequently have multiple co-morbidities, including coronary artery disease and congestive heart failure, which may influence their ability to tolerate a potentially ischemic state related to anemia or to accommodate volume fluctuations related to transfusion.
Furthermore, RBC transfusions are not without inherent risk. Life-threatening transfusion reactions occur in approximately seven per million transfused blood components, and transfusion-associated circulatory overload (TACO) can develop in one in 100 transfusions.1
Recently published guidelines provide recommendations for management of hemodynamically stable adults with anemia.
Guideline Update
The AABB published guidelines in the Annals of Internal Medicine in 2012 addressing RBC transfusion thresholds.1 The updated guideline makes a recommendation that clinicians utilize a restrictive transfusion strategy. Transfusion is strongly recommended for ICU patients with hemoglobin ≤7g/dL. In post-operative surgical patients and for post-operative patients with symptomatic anemia, transfusion is recommended for hemoglobin ≤8g/dL. The authors also made a weak recommendation to transfuse for hemoglobin ≤8g/dL or for symptoms in hospitalized hemodynamically stable patients with preexisting cardiovascular disease.
These recommendations draw from past literature, along with two more recent trials examining liberal or restrictive transfusion thresholds. The newer trials increased the total number of patients studied by nearly one third compared with prior reviews.2,3 The authors also incorporated recently published systematic reviews in their analysis.
Although the definition of a restrictive transfusion threshold varied across trials, including hemoglobin ≤7g/dL and ≤8g/dL, the authors used the pooled data to provide several recommendations in the new guideline. Of note, the pooled data was underpowered to detect up to a twofold increase in risk of myocardial infarction in patients in the restrictive strategy group.1
There were insufficient data for the authors to recommend for or against a restrictive transfusion strategy in patients with acute coronary syndrome, based on very low quality evidence.
Finally, the authors recommended that symptoms and hemoglobin level should both be used in determining transfusion criteria, based on low quality of evidence.
Analysis
The current AABB guidelines have two primary differences from earlier guidelines. First, the AABB authors used GRADE (Grading of Recommendations, Assessment, Development, and Evaluation) methodology to formalize evidence-based practice in their analysis of the literature. The authors purposely used the GRADE methodology to systematically evaluate the quality of the evidence base and explicitly state the strength of the recommendation for a particular transfusion threshold.4
Second, the AABB guidelines incorporated data from the more recently published FOCUS (Functional Outcomes in Cardiovascular patients Undergoing Surgical repair of hip fracture) and TRACS (Transfusion Requirements After Cardiac Surgery) trials, resulting in a stronger recommendation supporting the use of a restrictive transfusion strategy in non-ICU and post-operative patients. The findings of the FOCUS trial are especially applicable to hospitalists, because many patients who undergo hip fracture repair are directly cared for or are co-managed by hospitalists.
The current guidelines built upon previous guidelines that advocated a restrictive strategy (hemoglobin ≤7g/dL) in hemodynamically stable, critically ill adult patients.5 In general, restrictive transfusion strategy led to nearly 40% fewer patients receiving transfusion compared with the use of a liberal transfusion strategy.1 No additional harm to patients was evidenced in the restrictive transfusion group, though the trials were not designed to answer this question; moreover, there was no statistically significant difference in mortality or functional outcome between the two groups.
The authors of the current AABB guidelines recognized the importance of replicating the current findings in a more diverse patient population. An area where further study is indicated is in the use of specific transfusion thresholds in patients with acute coronary syndrome. These guidelines did not clarify whether or not there is a physiologic difference between use of different restrictive transfusion thresholds such as <8g/dL and <7g/dL.
The authors of the AABB guidelines also commented that performing a future trial to compare RBC transfusion for symptoms vs. hemoglobin “trigger” would be useful; however, they recognized that this may not be feasible due to the need to blind providers in the trial to hemoglobin values. Various society guidelines currently call for different transfusion thresholds or do not make a specific recommendation at all.1
Key Takeaways for Hospitalists
For the vast majority of medical patients, hospitalists can safely use a restrictive RBC transfusion threshold (≤7g/dL or ≤8g/dL), which can lead to a significant decrease in RBC transfusions without adversely affecting overall mortality.
Drs. Bortinger and Carbo are hospitalists at Beth Israel Deaconess Medical Center in Boston.
References
- Carson JL, Grossman BJ, Kleinman S, et al. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Inter Med. 2012;157(1):49-58.
- Carson AL, Terrin ML, Noveck H, et al. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;367(26):2453-2462.
- Hajjar LA, Vincent JL, Galas FR, et al. Transfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA. 2010;304(14):1559-1567.
- Carson JL, Carless PA, Herbert PC. Transfusion threshold and other strategies for guiding allogenic red blood cell transfusion. Cochrane Database Syst Rev. 2012;CD002042.
- Napolitano LM, Kurek S, Luchette FA, et al. Clinical practice guideline: red blood cell transfusion in adult trauma and critical care. Crit Care Med. 2009;37(12):3124-3157.
Background
Hospitalists commonly order red blood cell (RBC) transfusion as a therapy for patients with anemia resulting from a variety of clinical conditions. There has been lack of consensus on when to transfuse, because patients with anemia frequently have multiple co-morbidities, including coronary artery disease and congestive heart failure, which may influence their ability to tolerate a potentially ischemic state related to anemia or to accommodate volume fluctuations related to transfusion.
Furthermore, RBC transfusions are not without inherent risk. Life-threatening transfusion reactions occur in approximately seven per million transfused blood components, and transfusion-associated circulatory overload (TACO) can develop in one in 100 transfusions.1
Recently published guidelines provide recommendations for management of hemodynamically stable adults with anemia.
Guideline Update
The AABB published guidelines in the Annals of Internal Medicine in 2012 addressing RBC transfusion thresholds.1 The updated guideline makes a recommendation that clinicians utilize a restrictive transfusion strategy. Transfusion is strongly recommended for ICU patients with hemoglobin ≤7g/dL. In post-operative surgical patients and for post-operative patients with symptomatic anemia, transfusion is recommended for hemoglobin ≤8g/dL. The authors also made a weak recommendation to transfuse for hemoglobin ≤8g/dL or for symptoms in hospitalized hemodynamically stable patients with preexisting cardiovascular disease.
These recommendations draw from past literature, along with two more recent trials examining liberal or restrictive transfusion thresholds. The newer trials increased the total number of patients studied by nearly one third compared with prior reviews.2,3 The authors also incorporated recently published systematic reviews in their analysis.
Although the definition of a restrictive transfusion threshold varied across trials, including hemoglobin ≤7g/dL and ≤8g/dL, the authors used the pooled data to provide several recommendations in the new guideline. Of note, the pooled data was underpowered to detect up to a twofold increase in risk of myocardial infarction in patients in the restrictive strategy group.1
There were insufficient data for the authors to recommend for or against a restrictive transfusion strategy in patients with acute coronary syndrome, based on very low quality evidence.
Finally, the authors recommended that symptoms and hemoglobin level should both be used in determining transfusion criteria, based on low quality of evidence.
Analysis
The current AABB guidelines have two primary differences from earlier guidelines. First, the AABB authors used GRADE (Grading of Recommendations, Assessment, Development, and Evaluation) methodology to formalize evidence-based practice in their analysis of the literature. The authors purposely used the GRADE methodology to systematically evaluate the quality of the evidence base and explicitly state the strength of the recommendation for a particular transfusion threshold.4
Second, the AABB guidelines incorporated data from the more recently published FOCUS (Functional Outcomes in Cardiovascular patients Undergoing Surgical repair of hip fracture) and TRACS (Transfusion Requirements After Cardiac Surgery) trials, resulting in a stronger recommendation supporting the use of a restrictive transfusion strategy in non-ICU and post-operative patients. The findings of the FOCUS trial are especially applicable to hospitalists, because many patients who undergo hip fracture repair are directly cared for or are co-managed by hospitalists.
The current guidelines built upon previous guidelines that advocated a restrictive strategy (hemoglobin ≤7g/dL) in hemodynamically stable, critically ill adult patients.5 In general, restrictive transfusion strategy led to nearly 40% fewer patients receiving transfusion compared with the use of a liberal transfusion strategy.1 No additional harm to patients was evidenced in the restrictive transfusion group, though the trials were not designed to answer this question; moreover, there was no statistically significant difference in mortality or functional outcome between the two groups.
The authors of the current AABB guidelines recognized the importance of replicating the current findings in a more diverse patient population. An area where further study is indicated is in the use of specific transfusion thresholds in patients with acute coronary syndrome. These guidelines did not clarify whether or not there is a physiologic difference between use of different restrictive transfusion thresholds such as <8g/dL and <7g/dL.
The authors of the AABB guidelines also commented that performing a future trial to compare RBC transfusion for symptoms vs. hemoglobin “trigger” would be useful; however, they recognized that this may not be feasible due to the need to blind providers in the trial to hemoglobin values. Various society guidelines currently call for different transfusion thresholds or do not make a specific recommendation at all.1
Key Takeaways for Hospitalists
For the vast majority of medical patients, hospitalists can safely use a restrictive RBC transfusion threshold (≤7g/dL or ≤8g/dL), which can lead to a significant decrease in RBC transfusions without adversely affecting overall mortality.
Drs. Bortinger and Carbo are hospitalists at Beth Israel Deaconess Medical Center in Boston.
References
- Carson JL, Grossman BJ, Kleinman S, et al. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Inter Med. 2012;157(1):49-58.
- Carson AL, Terrin ML, Noveck H, et al. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;367(26):2453-2462.
- Hajjar LA, Vincent JL, Galas FR, et al. Transfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA. 2010;304(14):1559-1567.
- Carson JL, Carless PA, Herbert PC. Transfusion threshold and other strategies for guiding allogenic red blood cell transfusion. Cochrane Database Syst Rev. 2012;CD002042.
- Napolitano LM, Kurek S, Luchette FA, et al. Clinical practice guideline: red blood cell transfusion in adult trauma and critical care. Crit Care Med. 2009;37(12):3124-3157.
Which Patients Should be Screened for Hepatitis C Virus Infection?
Case
A 65-year-old male with a history of a motor vehicle accident that required emergency surgery in 1982 is hospitalized for acute renal failure. He reports a distant history of IV heroin use and a brief incarceration. He does not currently use illicit drugs. He has no signs or symptoms of liver disease. Should this patient be screened for chronic hepatitis C virus (HCV) infection?
Brief Overview
HCV is a major public health concern in the United States and worldwide. It is estimated that more than 4.1 million people in the U.S. (1.6% prevalence) and more than 180 million worldwide (2.8% prevalence) are HCV antibody-positive.1,2 The acute infection is most often asymptomatic, and 80% to 100% of patients will remain HCV RNA-positive, 60% to 80% will have persistently elevated liver enzymes, and 16% will develop evidence of cirrhosis at 20 years after initial infection.3
A number of organizations in the United States have released HCV screening guidelines, including the CDC, the American Association for the Study of Liver Disease (AASLD), and the U.S. Preventive Services Task Force (USPSTF); however, despite these established recommendations, an estimated 50% of individuals with chronic HCV infection are unscreened and unaware of their infection status.4 Furthermore, in a recent study of one managed care network, even when one or more risk factors were present, only 29% of individuals underwent screening for HCV antibodies detection.5 The importance of detecting chronic HCV infection will have greater significance as newer and better-tolerated treatment options become available.6
Multiple organizations recommend screening for chronic HCV infection. This screening is recommended for patients with known risk factors and those in populations with a high prevalence of HCV infection.
Risk Factors and High-Prevalence Populations
IV or intranasal drug use. IV drug use is the main identifiable source of HCV infection in the U.S. It is estimated that 60% of new HCV infections occur in people who have injected drugs in the past six months.7 The prevalence of HCV antibodies in current IV drug users is between 72% and 96%.8 Intranasal cocaine use is also associated with a higher prevalence of HCV antibodies than the general population.8
Blood transfusion prior to July 1992. Testing of donor blood was not routinely done until 1990, and more sensitive testing was not implemented until July 1992.8 The prevalence of HCV antibodies in people who received blood transfusions prior to 1990 is 6%.8 Prior to 1990, the risk for transfusion-associated HCV infection was one in 526 units transfused.9 Since implementation of highly sensitive screening techniques, the risk of infection has dropped to less than one in 1.9 million units transfused.10
Clotting factors prior to 1987 or transplanted tissue prior to 1992. Individuals who have received clotting factors, other blood product transfusions, or transplanted tissue prior to 1987 are at an increased risk for developing HCV infection. For instance, individuals with hemophilia treated with clotting factors prior to 1987 had chronic HCV infection rates of up to 90%.8 In 1987, widespread use of protocols to inactivate HCV in clotting factors and other blood products was adopted.8 In addition, widespread screening of potential tissue donors and the use of HCV antibody-negative donors became routine.8
Alanine aminotransferase elevation. This can be considered screening or part of the diagnostic work-up of transaminitis. Regardless of the classification, this is a cohort of people with a high prevalence of HCV antibody. For individuals with one isolated alanine aminotransferase elevation, the prevalence is 3.2%.4 With two or more elevated aminotransferase results, the prevalence rises to 8.2%.4
Hemodialysis. Two major studies have estimated the prevalence of HCV antibody-positive in end-stage renal disease individuals on hemodialysis to be 7.8% and 10.4%.11,12 This prevalence can reach 64% at some dialysis centers.11 The risk of HCV infection has been associated with blood transfusions, longer duration of hemodialysis, and higher rates of HCV infection in the dialysis unit.13 With implementation of infection control practices in dialysis units, the incidence and prevalence of HCV infection are declining.13
Born in the U.S. between 1945 and 1965. The CDC and USPSTF recommend a one-time screening for HCV infection for people born in the U.S. between 1945 and 1965, regardless of the presence or absence of risk factors.6,14 This age group has an increased prevalence of HCV antibodies, at 3.25%.6
Human immunodeficiency virus (HIV). HCV has a prevalence of 30% in people infected with HIV.15 The rate of co-infection is likely secondary to shared routes of transmission. For example, 72.7% of HIV-infected individuals who used IV drugs had HCV antibodies, but only 3.5% of “low-risk” HIV-infected individuals had HCV antibodies.16
Born in a high prevalence country. In the U.S., a significant number of immigrants are from areas with a high endemic rate of HCV infection. High prevalence areas (greater than 3.5%) include Central Asia and East Asia, North Africa, and the Middle East.7 Of note, Egypt is thought to have the highest prevalence of chronic HCV infection in the world, with well over 10% of the population being antibody-positive.17 Although major guidelines do not currently recommend it, the high prevalence of chronic HCV infection in this population may warrant screening.
Other high-risk or high-prevalence populations. The prevalence of HCV infection in people who have had over 10 lifetime sexual partners (3% to 9%), those with a history of sexually transmitted disease (6%), men who have had sex with men (5%), and children born to HCV-infected mothers (5%) is increased compared with the general population.8 Incarcerated people in the U.S. have an HCV antibody prevalence of 16% to 41%.18 In addition, people who have sustained needle-stick injury or mucosal exposure, or those with potential exposures in unregulated tattoo or piercing salons, may also benefit from HCV antibody screening.14
Table 1 reviews HCV screening recommendations for the CDC, AASLD, and USPSTF.1,6,8,14
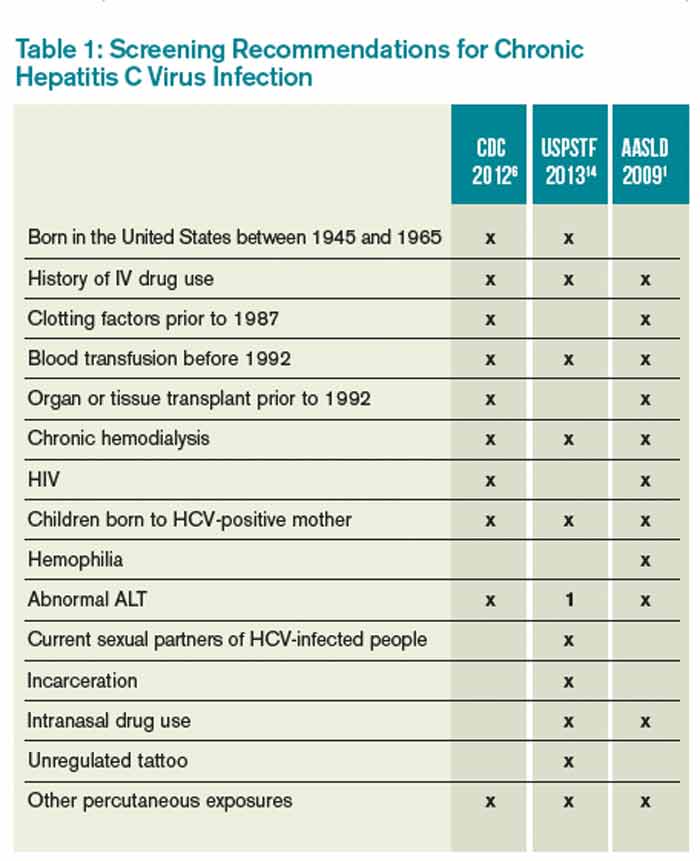
CDC=Centers for Disease Control and Prevention; USPSTF=U.S. Preventive Services Task Force; AASLD=American Association for the Study of Liver Disease; ALT=alanine aminotransferase; 1=considered diagnostic and not screening test
Screening Method
The most common initial screening test for the diagnosis of chronic HCV infection is the HCV antibody test. A positive antibody test should be followed by an HCV RNA test. In an individual with recent exposure, it takes between four and 10 weeks for the antibody to be detectable. HCV RNA testing can be positive as soon as two to three weeks after infection.8
Hospitalist Role in HCV Screening
None of the U.S.-based guidelines make recommendations on the preferred setting for HCV screening. According to the CDC, 60.4% of HCV screening was done in a physician office and 5.9% was done as a hospital inpatient.19 Traditionally, the PCP is responsible for screening for chronic diseases, including HCV infection; however, the current screening rate is insufficient, as 50% of people with chronic HCV infection remain unscreened.4
Given the insufficient rate of HCV screening at present, hospital medicine (HM) physicians have an opportunity to help improve this rate. Currently, there is no established standard of care for HCV screening in hospitalized patients. HM physicians could use the following strategies:
- Continue the current system and defer screening to outpatient providers;
- Offer screening to selected inpatients at high risk for chronic HCV infection; or
- Offer screening to all inpatients who meet screening criteria based on current guidelines.
Given the shortcomings of the current screening strategies, these authors would recommend widespread screening for chronic HCV infection in hospitalized people who meet screening criteria per current guidelines.
If HM physicians are to take an increased role in HCV screening, there are a number of important considerations. Because hospitalized patients have a limited length of stay, it would be unreasonable to expect HM physicians to test for HCV RNA viral load or genotype for all patients with a positive antibody test, because the duration of the inpatient stay may be shorter than the time it takes for these test results to return. These tests are often indicated after a positive HCV antibody test, however. Thus, communication of HCV antibody results to PCPs or other responsible providers is essential. If no follow-up is available or there are no responsible outpatient providers, HM physicians should continue with a limited screening strategy.
Back to the Case
This individual has multiple indications for chronic HCV infection screening. His risk factors include date of birth between 1945 and 1965, a history of IV drug use, and a history of incarceration. He also notes a history of emergency surgery, for which he may have received blood products prior to 1987. These factors significantly raise the likelihood of chronic HCV infection when compared with the general population. He was screened and found to be HCV antibody-positive. A follow-up HCV RNA viral load was also positive. He did not have any evidence of liver disease but did have a mild transaminitis. He has followed up as an outpatient with plans to start therapy.
Bottom Line
The current screening strategies for individuals with high prevalence of chronic HCV infection are insufficient. HM physicians have an opportunity to improve the rates of screening in this population.
Dr. Theisen-Toupal is an internist, Dr. Rosenthal is a clinical fellow in medicine, and Dr. Carbo is an assistant professor of medicine, all at Beth Israel Deaconess Medical Center in Boston. Dr. Li is an internist and associate professor of medicine at Harvard Medical School and director of the hospital medicine division at Beth Israel Deaconess Medical Center.
References
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335-1374.
- Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333-1342.
- Chopra S. Clinical manifestations and natural history of chronic hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/clinical-manifestations-and-natural-history-of-chronic-hepatitis-c-virus-infection. Accessed March 5, 2014.
- Spradling PR, Rupp L, Moorman AC, et al. Hepatitis B and C virus infection among 1.2 million people with access to care: factors associated with testing and infection prevalence. Clin Infect Dis. 2012;55(8):1047-1055.
- Roblin DW, Smith BD, Weinbaum CM, Sabin ME. HCV screening practices and prevalence in an MCO, 2000-2007. Am J Manag Care. 2011;17(8):548-555.
- Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among people born during 1945-1965. MMWR. August 17, 2012. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6104a1.htm. Accessed March 5, 2014.
- Chopra S. Epidemiology and transmission of hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/epidemiology-and-transmission-of-hepatitis-c-virus-infection?source=search_result&search=%22Epidemiology+and+transmission+of+hepatitis+C+virus+infection%22&selectedTitle=1~150. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR. October 16, 1998. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/00055154.htm. Accessed March 5, 2014.
- Donahue JG, Muñoz A, Ness PM, et al. The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992;327(6):369-373.
- Pomper GJ, Wu Y, Snyder EL. Risks of transfusion-transmitted infections: 2003. Curr Opin Hematol. 2003;10(6):412-418.
- Tokars JI, Miller ER, Alter MJ, Arduino MJ. National surveillance of dialysis associated diseases in the United States, 1995. ASAIO J. 1998;44(1):98-107.
- Finelli L, Miller JT, Tokars JI, Alter MJ, Arduino MJ. National surveillance of dialysis-associated diseases in the United States, 2002. Semin Dial. 2005;18(1):52-61.
- Natov S, Pereira BJG. Hepatitis C virus infection in patients on maintenance dialysis. UpToDate. Available at: http://www.uptodate.com/contents/hepatitis-c-virus-infection-in-patients-on-maintenance-dialysis?source=search_result&search=Hepatitis+C+virus+infection+in+patients+on+maintenance+dialysis.&selectedTitle=1~150. Accessed March 5, 2014.
- Moyer VA, U.S. Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
- Staples CT II, Rimland D, Dudas D. Hepatitis C in the HIV (human immunodeficiency virus) Atlanta V.A. (Veterans Affairs Medical Center) Cohort Study (HAVACS): the effect of coinfection on survival. Clin Infect Dis. 1999;29(1):150-154.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the U.S. adult AIDS clinical trials group. Clin Infect Dis. 2002;34(6):831-837.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 Suppl 1:S10-15.
- Centers for Disease Control and Prevention. Prevention and control of infections with hepatitis viruses in correctional settings. MMWR. January 24, 2003. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5201a1.htm. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Locations and reasons for initial testing for hepatitis C infection—chronic hepatitis cohort study, United States, 2006-2010. MMWR. August 16, 2013. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6232a3.htm?s_cid=mm6232a3_w. Accessed March 5, 2014.
Case
A 65-year-old male with a history of a motor vehicle accident that required emergency surgery in 1982 is hospitalized for acute renal failure. He reports a distant history of IV heroin use and a brief incarceration. He does not currently use illicit drugs. He has no signs or symptoms of liver disease. Should this patient be screened for chronic hepatitis C virus (HCV) infection?
Brief Overview
HCV is a major public health concern in the United States and worldwide. It is estimated that more than 4.1 million people in the U.S. (1.6% prevalence) and more than 180 million worldwide (2.8% prevalence) are HCV antibody-positive.1,2 The acute infection is most often asymptomatic, and 80% to 100% of patients will remain HCV RNA-positive, 60% to 80% will have persistently elevated liver enzymes, and 16% will develop evidence of cirrhosis at 20 years after initial infection.3
A number of organizations in the United States have released HCV screening guidelines, including the CDC, the American Association for the Study of Liver Disease (AASLD), and the U.S. Preventive Services Task Force (USPSTF); however, despite these established recommendations, an estimated 50% of individuals with chronic HCV infection are unscreened and unaware of their infection status.4 Furthermore, in a recent study of one managed care network, even when one or more risk factors were present, only 29% of individuals underwent screening for HCV antibodies detection.5 The importance of detecting chronic HCV infection will have greater significance as newer and better-tolerated treatment options become available.6
Multiple organizations recommend screening for chronic HCV infection. This screening is recommended for patients with known risk factors and those in populations with a high prevalence of HCV infection.
Risk Factors and High-Prevalence Populations
IV or intranasal drug use. IV drug use is the main identifiable source of HCV infection in the U.S. It is estimated that 60% of new HCV infections occur in people who have injected drugs in the past six months.7 The prevalence of HCV antibodies in current IV drug users is between 72% and 96%.8 Intranasal cocaine use is also associated with a higher prevalence of HCV antibodies than the general population.8
Blood transfusion prior to July 1992. Testing of donor blood was not routinely done until 1990, and more sensitive testing was not implemented until July 1992.8 The prevalence of HCV antibodies in people who received blood transfusions prior to 1990 is 6%.8 Prior to 1990, the risk for transfusion-associated HCV infection was one in 526 units transfused.9 Since implementation of highly sensitive screening techniques, the risk of infection has dropped to less than one in 1.9 million units transfused.10
Clotting factors prior to 1987 or transplanted tissue prior to 1992. Individuals who have received clotting factors, other blood product transfusions, or transplanted tissue prior to 1987 are at an increased risk for developing HCV infection. For instance, individuals with hemophilia treated with clotting factors prior to 1987 had chronic HCV infection rates of up to 90%.8 In 1987, widespread use of protocols to inactivate HCV in clotting factors and other blood products was adopted.8 In addition, widespread screening of potential tissue donors and the use of HCV antibody-negative donors became routine.8
Alanine aminotransferase elevation. This can be considered screening or part of the diagnostic work-up of transaminitis. Regardless of the classification, this is a cohort of people with a high prevalence of HCV antibody. For individuals with one isolated alanine aminotransferase elevation, the prevalence is 3.2%.4 With two or more elevated aminotransferase results, the prevalence rises to 8.2%.4
Hemodialysis. Two major studies have estimated the prevalence of HCV antibody-positive in end-stage renal disease individuals on hemodialysis to be 7.8% and 10.4%.11,12 This prevalence can reach 64% at some dialysis centers.11 The risk of HCV infection has been associated with blood transfusions, longer duration of hemodialysis, and higher rates of HCV infection in the dialysis unit.13 With implementation of infection control practices in dialysis units, the incidence and prevalence of HCV infection are declining.13
Born in the U.S. between 1945 and 1965. The CDC and USPSTF recommend a one-time screening for HCV infection for people born in the U.S. between 1945 and 1965, regardless of the presence or absence of risk factors.6,14 This age group has an increased prevalence of HCV antibodies, at 3.25%.6
Human immunodeficiency virus (HIV). HCV has a prevalence of 30% in people infected with HIV.15 The rate of co-infection is likely secondary to shared routes of transmission. For example, 72.7% of HIV-infected individuals who used IV drugs had HCV antibodies, but only 3.5% of “low-risk” HIV-infected individuals had HCV antibodies.16
Born in a high prevalence country. In the U.S., a significant number of immigrants are from areas with a high endemic rate of HCV infection. High prevalence areas (greater than 3.5%) include Central Asia and East Asia, North Africa, and the Middle East.7 Of note, Egypt is thought to have the highest prevalence of chronic HCV infection in the world, with well over 10% of the population being antibody-positive.17 Although major guidelines do not currently recommend it, the high prevalence of chronic HCV infection in this population may warrant screening.
Other high-risk or high-prevalence populations. The prevalence of HCV infection in people who have had over 10 lifetime sexual partners (3% to 9%), those with a history of sexually transmitted disease (6%), men who have had sex with men (5%), and children born to HCV-infected mothers (5%) is increased compared with the general population.8 Incarcerated people in the U.S. have an HCV antibody prevalence of 16% to 41%.18 In addition, people who have sustained needle-stick injury or mucosal exposure, or those with potential exposures in unregulated tattoo or piercing salons, may also benefit from HCV antibody screening.14
Table 1 reviews HCV screening recommendations for the CDC, AASLD, and USPSTF.1,6,8,14
CDC=Centers for Disease Control and Prevention; USPSTF=U.S. Preventive Services Task Force; AASLD=American Association for the Study of Liver Disease; ALT=alanine aminotransferase; 1=considered diagnostic and not screening test
Screening Method
The most common initial screening test for the diagnosis of chronic HCV infection is the HCV antibody test. A positive antibody test should be followed by an HCV RNA test. In an individual with recent exposure, it takes between four and 10 weeks for the antibody to be detectable. HCV RNA testing can be positive as soon as two to three weeks after infection.8
Hospitalist Role in HCV Screening
None of the U.S.-based guidelines make recommendations on the preferred setting for HCV screening. According to the CDC, 60.4% of HCV screening was done in a physician office and 5.9% was done as a hospital inpatient.19 Traditionally, the PCP is responsible for screening for chronic diseases, including HCV infection; however, the current screening rate is insufficient, as 50% of people with chronic HCV infection remain unscreened.4
Given the insufficient rate of HCV screening at present, hospital medicine (HM) physicians have an opportunity to help improve this rate. Currently, there is no established standard of care for HCV screening in hospitalized patients. HM physicians could use the following strategies:
- Continue the current system and defer screening to outpatient providers;
- Offer screening to selected inpatients at high risk for chronic HCV infection; or
- Offer screening to all inpatients who meet screening criteria based on current guidelines.
Given the shortcomings of the current screening strategies, these authors would recommend widespread screening for chronic HCV infection in hospitalized people who meet screening criteria per current guidelines.
If HM physicians are to take an increased role in HCV screening, there are a number of important considerations. Because hospitalized patients have a limited length of stay, it would be unreasonable to expect HM physicians to test for HCV RNA viral load or genotype for all patients with a positive antibody test, because the duration of the inpatient stay may be shorter than the time it takes for these test results to return. These tests are often indicated after a positive HCV antibody test, however. Thus, communication of HCV antibody results to PCPs or other responsible providers is essential. If no follow-up is available or there are no responsible outpatient providers, HM physicians should continue with a limited screening strategy.
Back to the Case
This individual has multiple indications for chronic HCV infection screening. His risk factors include date of birth between 1945 and 1965, a history of IV drug use, and a history of incarceration. He also notes a history of emergency surgery, for which he may have received blood products prior to 1987. These factors significantly raise the likelihood of chronic HCV infection when compared with the general population. He was screened and found to be HCV antibody-positive. A follow-up HCV RNA viral load was also positive. He did not have any evidence of liver disease but did have a mild transaminitis. He has followed up as an outpatient with plans to start therapy.
Bottom Line
The current screening strategies for individuals with high prevalence of chronic HCV infection are insufficient. HM physicians have an opportunity to improve the rates of screening in this population.
Dr. Theisen-Toupal is an internist, Dr. Rosenthal is a clinical fellow in medicine, and Dr. Carbo is an assistant professor of medicine, all at Beth Israel Deaconess Medical Center in Boston. Dr. Li is an internist and associate professor of medicine at Harvard Medical School and director of the hospital medicine division at Beth Israel Deaconess Medical Center.
References
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335-1374.
- Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333-1342.
- Chopra S. Clinical manifestations and natural history of chronic hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/clinical-manifestations-and-natural-history-of-chronic-hepatitis-c-virus-infection. Accessed March 5, 2014.
- Spradling PR, Rupp L, Moorman AC, et al. Hepatitis B and C virus infection among 1.2 million people with access to care: factors associated with testing and infection prevalence. Clin Infect Dis. 2012;55(8):1047-1055.
- Roblin DW, Smith BD, Weinbaum CM, Sabin ME. HCV screening practices and prevalence in an MCO, 2000-2007. Am J Manag Care. 2011;17(8):548-555.
- Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among people born during 1945-1965. MMWR. August 17, 2012. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6104a1.htm. Accessed March 5, 2014.
- Chopra S. Epidemiology and transmission of hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/epidemiology-and-transmission-of-hepatitis-c-virus-infection?source=search_result&search=%22Epidemiology+and+transmission+of+hepatitis+C+virus+infection%22&selectedTitle=1~150. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR. October 16, 1998. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/00055154.htm. Accessed March 5, 2014.
- Donahue JG, Muñoz A, Ness PM, et al. The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992;327(6):369-373.
- Pomper GJ, Wu Y, Snyder EL. Risks of transfusion-transmitted infections: 2003. Curr Opin Hematol. 2003;10(6):412-418.
- Tokars JI, Miller ER, Alter MJ, Arduino MJ. National surveillance of dialysis associated diseases in the United States, 1995. ASAIO J. 1998;44(1):98-107.
- Finelli L, Miller JT, Tokars JI, Alter MJ, Arduino MJ. National surveillance of dialysis-associated diseases in the United States, 2002. Semin Dial. 2005;18(1):52-61.
- Natov S, Pereira BJG. Hepatitis C virus infection in patients on maintenance dialysis. UpToDate. Available at: http://www.uptodate.com/contents/hepatitis-c-virus-infection-in-patients-on-maintenance-dialysis?source=search_result&search=Hepatitis+C+virus+infection+in+patients+on+maintenance+dialysis.&selectedTitle=1~150. Accessed March 5, 2014.
- Moyer VA, U.S. Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
- Staples CT II, Rimland D, Dudas D. Hepatitis C in the HIV (human immunodeficiency virus) Atlanta V.A. (Veterans Affairs Medical Center) Cohort Study (HAVACS): the effect of coinfection on survival. Clin Infect Dis. 1999;29(1):150-154.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the U.S. adult AIDS clinical trials group. Clin Infect Dis. 2002;34(6):831-837.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 Suppl 1:S10-15.
- Centers for Disease Control and Prevention. Prevention and control of infections with hepatitis viruses in correctional settings. MMWR. January 24, 2003. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5201a1.htm. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Locations and reasons for initial testing for hepatitis C infection—chronic hepatitis cohort study, United States, 2006-2010. MMWR. August 16, 2013. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6232a3.htm?s_cid=mm6232a3_w. Accessed March 5, 2014.
Case
A 65-year-old male with a history of a motor vehicle accident that required emergency surgery in 1982 is hospitalized for acute renal failure. He reports a distant history of IV heroin use and a brief incarceration. He does not currently use illicit drugs. He has no signs or symptoms of liver disease. Should this patient be screened for chronic hepatitis C virus (HCV) infection?
Brief Overview
HCV is a major public health concern in the United States and worldwide. It is estimated that more than 4.1 million people in the U.S. (1.6% prevalence) and more than 180 million worldwide (2.8% prevalence) are HCV antibody-positive.1,2 The acute infection is most often asymptomatic, and 80% to 100% of patients will remain HCV RNA-positive, 60% to 80% will have persistently elevated liver enzymes, and 16% will develop evidence of cirrhosis at 20 years after initial infection.3
A number of organizations in the United States have released HCV screening guidelines, including the CDC, the American Association for the Study of Liver Disease (AASLD), and the U.S. Preventive Services Task Force (USPSTF); however, despite these established recommendations, an estimated 50% of individuals with chronic HCV infection are unscreened and unaware of their infection status.4 Furthermore, in a recent study of one managed care network, even when one or more risk factors were present, only 29% of individuals underwent screening for HCV antibodies detection.5 The importance of detecting chronic HCV infection will have greater significance as newer and better-tolerated treatment options become available.6
Multiple organizations recommend screening for chronic HCV infection. This screening is recommended for patients with known risk factors and those in populations with a high prevalence of HCV infection.
Risk Factors and High-Prevalence Populations
IV or intranasal drug use. IV drug use is the main identifiable source of HCV infection in the U.S. It is estimated that 60% of new HCV infections occur in people who have injected drugs in the past six months.7 The prevalence of HCV antibodies in current IV drug users is between 72% and 96%.8 Intranasal cocaine use is also associated with a higher prevalence of HCV antibodies than the general population.8
Blood transfusion prior to July 1992. Testing of donor blood was not routinely done until 1990, and more sensitive testing was not implemented until July 1992.8 The prevalence of HCV antibodies in people who received blood transfusions prior to 1990 is 6%.8 Prior to 1990, the risk for transfusion-associated HCV infection was one in 526 units transfused.9 Since implementation of highly sensitive screening techniques, the risk of infection has dropped to less than one in 1.9 million units transfused.10
Clotting factors prior to 1987 or transplanted tissue prior to 1992. Individuals who have received clotting factors, other blood product transfusions, or transplanted tissue prior to 1987 are at an increased risk for developing HCV infection. For instance, individuals with hemophilia treated with clotting factors prior to 1987 had chronic HCV infection rates of up to 90%.8 In 1987, widespread use of protocols to inactivate HCV in clotting factors and other blood products was adopted.8 In addition, widespread screening of potential tissue donors and the use of HCV antibody-negative donors became routine.8
Alanine aminotransferase elevation. This can be considered screening or part of the diagnostic work-up of transaminitis. Regardless of the classification, this is a cohort of people with a high prevalence of HCV antibody. For individuals with one isolated alanine aminotransferase elevation, the prevalence is 3.2%.4 With two or more elevated aminotransferase results, the prevalence rises to 8.2%.4
Hemodialysis. Two major studies have estimated the prevalence of HCV antibody-positive in end-stage renal disease individuals on hemodialysis to be 7.8% and 10.4%.11,12 This prevalence can reach 64% at some dialysis centers.11 The risk of HCV infection has been associated with blood transfusions, longer duration of hemodialysis, and higher rates of HCV infection in the dialysis unit.13 With implementation of infection control practices in dialysis units, the incidence and prevalence of HCV infection are declining.13
Born in the U.S. between 1945 and 1965. The CDC and USPSTF recommend a one-time screening for HCV infection for people born in the U.S. between 1945 and 1965, regardless of the presence or absence of risk factors.6,14 This age group has an increased prevalence of HCV antibodies, at 3.25%.6
Human immunodeficiency virus (HIV). HCV has a prevalence of 30% in people infected with HIV.15 The rate of co-infection is likely secondary to shared routes of transmission. For example, 72.7% of HIV-infected individuals who used IV drugs had HCV antibodies, but only 3.5% of “low-risk” HIV-infected individuals had HCV antibodies.16
Born in a high prevalence country. In the U.S., a significant number of immigrants are from areas with a high endemic rate of HCV infection. High prevalence areas (greater than 3.5%) include Central Asia and East Asia, North Africa, and the Middle East.7 Of note, Egypt is thought to have the highest prevalence of chronic HCV infection in the world, with well over 10% of the population being antibody-positive.17 Although major guidelines do not currently recommend it, the high prevalence of chronic HCV infection in this population may warrant screening.
Other high-risk or high-prevalence populations. The prevalence of HCV infection in people who have had over 10 lifetime sexual partners (3% to 9%), those with a history of sexually transmitted disease (6%), men who have had sex with men (5%), and children born to HCV-infected mothers (5%) is increased compared with the general population.8 Incarcerated people in the U.S. have an HCV antibody prevalence of 16% to 41%.18 In addition, people who have sustained needle-stick injury or mucosal exposure, or those with potential exposures in unregulated tattoo or piercing salons, may also benefit from HCV antibody screening.14
Table 1 reviews HCV screening recommendations for the CDC, AASLD, and USPSTF.1,6,8,14
CDC=Centers for Disease Control and Prevention; USPSTF=U.S. Preventive Services Task Force; AASLD=American Association for the Study of Liver Disease; ALT=alanine aminotransferase; 1=considered diagnostic and not screening test
Screening Method
The most common initial screening test for the diagnosis of chronic HCV infection is the HCV antibody test. A positive antibody test should be followed by an HCV RNA test. In an individual with recent exposure, it takes between four and 10 weeks for the antibody to be detectable. HCV RNA testing can be positive as soon as two to three weeks after infection.8
Hospitalist Role in HCV Screening
None of the U.S.-based guidelines make recommendations on the preferred setting for HCV screening. According to the CDC, 60.4% of HCV screening was done in a physician office and 5.9% was done as a hospital inpatient.19 Traditionally, the PCP is responsible for screening for chronic diseases, including HCV infection; however, the current screening rate is insufficient, as 50% of people with chronic HCV infection remain unscreened.4
Given the insufficient rate of HCV screening at present, hospital medicine (HM) physicians have an opportunity to help improve this rate. Currently, there is no established standard of care for HCV screening in hospitalized patients. HM physicians could use the following strategies:
- Continue the current system and defer screening to outpatient providers;
- Offer screening to selected inpatients at high risk for chronic HCV infection; or
- Offer screening to all inpatients who meet screening criteria based on current guidelines.
Given the shortcomings of the current screening strategies, these authors would recommend widespread screening for chronic HCV infection in hospitalized people who meet screening criteria per current guidelines.
If HM physicians are to take an increased role in HCV screening, there are a number of important considerations. Because hospitalized patients have a limited length of stay, it would be unreasonable to expect HM physicians to test for HCV RNA viral load or genotype for all patients with a positive antibody test, because the duration of the inpatient stay may be shorter than the time it takes for these test results to return. These tests are often indicated after a positive HCV antibody test, however. Thus, communication of HCV antibody results to PCPs or other responsible providers is essential. If no follow-up is available or there are no responsible outpatient providers, HM physicians should continue with a limited screening strategy.
Back to the Case
This individual has multiple indications for chronic HCV infection screening. His risk factors include date of birth between 1945 and 1965, a history of IV drug use, and a history of incarceration. He also notes a history of emergency surgery, for which he may have received blood products prior to 1987. These factors significantly raise the likelihood of chronic HCV infection when compared with the general population. He was screened and found to be HCV antibody-positive. A follow-up HCV RNA viral load was also positive. He did not have any evidence of liver disease but did have a mild transaminitis. He has followed up as an outpatient with plans to start therapy.
Bottom Line
The current screening strategies for individuals with high prevalence of chronic HCV infection are insufficient. HM physicians have an opportunity to improve the rates of screening in this population.
Dr. Theisen-Toupal is an internist, Dr. Rosenthal is a clinical fellow in medicine, and Dr. Carbo is an assistant professor of medicine, all at Beth Israel Deaconess Medical Center in Boston. Dr. Li is an internist and associate professor of medicine at Harvard Medical School and director of the hospital medicine division at Beth Israel Deaconess Medical Center.
References
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335-1374.
- Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333-1342.
- Chopra S. Clinical manifestations and natural history of chronic hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/clinical-manifestations-and-natural-history-of-chronic-hepatitis-c-virus-infection. Accessed March 5, 2014.
- Spradling PR, Rupp L, Moorman AC, et al. Hepatitis B and C virus infection among 1.2 million people with access to care: factors associated with testing and infection prevalence. Clin Infect Dis. 2012;55(8):1047-1055.
- Roblin DW, Smith BD, Weinbaum CM, Sabin ME. HCV screening practices and prevalence in an MCO, 2000-2007. Am J Manag Care. 2011;17(8):548-555.
- Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among people born during 1945-1965. MMWR. August 17, 2012. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6104a1.htm. Accessed March 5, 2014.
- Chopra S. Epidemiology and transmission of hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/epidemiology-and-transmission-of-hepatitis-c-virus-infection?source=search_result&search=%22Epidemiology+and+transmission+of+hepatitis+C+virus+infection%22&selectedTitle=1~150. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR. October 16, 1998. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/00055154.htm. Accessed March 5, 2014.
- Donahue JG, Muñoz A, Ness PM, et al. The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992;327(6):369-373.
- Pomper GJ, Wu Y, Snyder EL. Risks of transfusion-transmitted infections: 2003. Curr Opin Hematol. 2003;10(6):412-418.
- Tokars JI, Miller ER, Alter MJ, Arduino MJ. National surveillance of dialysis associated diseases in the United States, 1995. ASAIO J. 1998;44(1):98-107.
- Finelli L, Miller JT, Tokars JI, Alter MJ, Arduino MJ. National surveillance of dialysis-associated diseases in the United States, 2002. Semin Dial. 2005;18(1):52-61.
- Natov S, Pereira BJG. Hepatitis C virus infection in patients on maintenance dialysis. UpToDate. Available at: http://www.uptodate.com/contents/hepatitis-c-virus-infection-in-patients-on-maintenance-dialysis?source=search_result&search=Hepatitis+C+virus+infection+in+patients+on+maintenance+dialysis.&selectedTitle=1~150. Accessed March 5, 2014.
- Moyer VA, U.S. Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
- Staples CT II, Rimland D, Dudas D. Hepatitis C in the HIV (human immunodeficiency virus) Atlanta V.A. (Veterans Affairs Medical Center) Cohort Study (HAVACS): the effect of coinfection on survival. Clin Infect Dis. 1999;29(1):150-154.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the U.S. adult AIDS clinical trials group. Clin Infect Dis. 2002;34(6):831-837.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 Suppl 1:S10-15.
- Centers for Disease Control and Prevention. Prevention and control of infections with hepatitis viruses in correctional settings. MMWR. January 24, 2003. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5201a1.htm. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Locations and reasons for initial testing for hepatitis C infection—chronic hepatitis cohort study, United States, 2006-2010. MMWR. August 16, 2013. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6232a3.htm?s_cid=mm6232a3_w. Accessed March 5, 2014.
Inhaled Corticosteroids Increase Risk of Serious Pneumonia in Patients with COPD

Clinical question: Does the risk of pneumonia vary for different inhaled agents?
Background: Inhaled corticosteroids (ICS) are known to increase the risk of pneumonia in COPD patients; duration, dosage, and various agents were investigated, especially fluticasone and budesonide.
Study design: Nested, case-control analysis.
Setting: Quebec health insurance database for new users with COPD, 1990-2005, with follow-up through 2007.
Synopsis: Investigators analyzed 163,514 patients, including 20,344 patients with serious pneumonia; current use of ICS was associated with a 69% increase in the rate of serious pneumonia (RR 1.69; 95% CI 1.63-1.75). The increased risk was sustained with long-term use but declined gradually to zero at six months after stopping ICS. The risk of serious pneumonia was higher with fluticasone (RR 2.01; 95% CI 1.93-2.10) than budesonide (RR 1.17; 95% CI 1.09-1.26).
Bottom line: Fluticasone was associated with an increased risk of pneumonia in COPD patients, consistent with earlier clinical trials, but the risk with budesonide was much lower.
Citation: Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax. 2013;68(11):1029-1036.
Clinical question: Does the risk of pneumonia vary for different inhaled agents?
Background: Inhaled corticosteroids (ICS) are known to increase the risk of pneumonia in COPD patients; duration, dosage, and various agents were investigated, especially fluticasone and budesonide.
Study design: Nested, case-control analysis.
Setting: Quebec health insurance database for new users with COPD, 1990-2005, with follow-up through 2007.
Synopsis: Investigators analyzed 163,514 patients, including 20,344 patients with serious pneumonia; current use of ICS was associated with a 69% increase in the rate of serious pneumonia (RR 1.69; 95% CI 1.63-1.75). The increased risk was sustained with long-term use but declined gradually to zero at six months after stopping ICS. The risk of serious pneumonia was higher with fluticasone (RR 2.01; 95% CI 1.93-2.10) than budesonide (RR 1.17; 95% CI 1.09-1.26).
Bottom line: Fluticasone was associated with an increased risk of pneumonia in COPD patients, consistent with earlier clinical trials, but the risk with budesonide was much lower.
Citation: Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax. 2013;68(11):1029-1036.
Clinical question: Does the risk of pneumonia vary for different inhaled agents?
Background: Inhaled corticosteroids (ICS) are known to increase the risk of pneumonia in COPD patients; duration, dosage, and various agents were investigated, especially fluticasone and budesonide.
Study design: Nested, case-control analysis.
Setting: Quebec health insurance database for new users with COPD, 1990-2005, with follow-up through 2007.
Synopsis: Investigators analyzed 163,514 patients, including 20,344 patients with serious pneumonia; current use of ICS was associated with a 69% increase in the rate of serious pneumonia (RR 1.69; 95% CI 1.63-1.75). The increased risk was sustained with long-term use but declined gradually to zero at six months after stopping ICS. The risk of serious pneumonia was higher with fluticasone (RR 2.01; 95% CI 1.93-2.10) than budesonide (RR 1.17; 95% CI 1.09-1.26).
Bottom line: Fluticasone was associated with an increased risk of pneumonia in COPD patients, consistent with earlier clinical trials, but the risk with budesonide was much lower.
Citation: Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax. 2013;68(11):1029-1036.
Ambulatory Patients with COPD Exacerbations Can Be Managed Without Antibiotics in the Absence of Increased Sputum Purulence, Elevated C-Reactive Protein
Clinical question: Which criteria identify ambulatory patients with exacerbations of mild to moderate COPD who do not need antibiotics?
Background: The Anthonisen criteria (increased dyspnea, sputum volume, sputum purulence) are commonly used to identify which patients with COPD exacerbations would benefit from antibiotics. These criteria, however, were derived in patients with severe COPD. It is unknown whether these criteria are predictive in patients with mild to moderate COPD.
Study design: Multivariate logistic regression analysis of placebo group of a double-blinded RCT.
Setting: Multicenter, ambulatory, primary care clinics in Spain.
Synopsis: The original RCT enrolled 310 ambulatory patients with exacerbations of mild to moderate COPD and tested the efficacy of amoxicillin/clavulanate. Clinical failure without antibiotics was 19.9% compared to 9.5% with antibiotics (P=0.022). Here they analyzed the 152 patients from the placebo group to identify factors associated with increased risk of clinical failure. Only increased sputum purulence (OR 6.1, CI 1.5-25; P=0.005) or C-reactive protein (CRP) >40 mg/L (OR 13.4, CI 4.5-38.8, P<0.001) were independently associated with increased risk of failure. Presence of both predicted a 63.7% failure without antibiotics.
The study did not define “increased sputum purulence,” but this is similar to real-life clinical practice. Placebo effect cannot be ruled out, but correlation of the objective measures with the clinical assessments suggests that the clinical assessments were accurate. The study did not have a protocol for administering co-medications such as steroids and inhalers. Despite these limitations, the criteria of increased sputum purulence and CRP >40 mg/L identified COPD patients likely to have a clinical failure without antibiotics.
Bottom line: Patients with exacerbations of mild to moderate COPD who do not have increased sputum purulence or CRP >40 mg/L can be safely managed without antibiotics.
Citation: Maravitlles M, Moravas A, Hernandez S, Bayona C, Llor C. Is it possible to identify exacerbations of mild to moderate COPD that do not require antibiotic treatment? Chest. 2013;144(5):1571-1577.
Clinical question: Which criteria identify ambulatory patients with exacerbations of mild to moderate COPD who do not need antibiotics?
Background: The Anthonisen criteria (increased dyspnea, sputum volume, sputum purulence) are commonly used to identify which patients with COPD exacerbations would benefit from antibiotics. These criteria, however, were derived in patients with severe COPD. It is unknown whether these criteria are predictive in patients with mild to moderate COPD.
Study design: Multivariate logistic regression analysis of placebo group of a double-blinded RCT.
Setting: Multicenter, ambulatory, primary care clinics in Spain.
Synopsis: The original RCT enrolled 310 ambulatory patients with exacerbations of mild to moderate COPD and tested the efficacy of amoxicillin/clavulanate. Clinical failure without antibiotics was 19.9% compared to 9.5% with antibiotics (P=0.022). Here they analyzed the 152 patients from the placebo group to identify factors associated with increased risk of clinical failure. Only increased sputum purulence (OR 6.1, CI 1.5-25; P=0.005) or C-reactive protein (CRP) >40 mg/L (OR 13.4, CI 4.5-38.8, P<0.001) were independently associated with increased risk of failure. Presence of both predicted a 63.7% failure without antibiotics.
The study did not define “increased sputum purulence,” but this is similar to real-life clinical practice. Placebo effect cannot be ruled out, but correlation of the objective measures with the clinical assessments suggests that the clinical assessments were accurate. The study did not have a protocol for administering co-medications such as steroids and inhalers. Despite these limitations, the criteria of increased sputum purulence and CRP >40 mg/L identified COPD patients likely to have a clinical failure without antibiotics.
Bottom line: Patients with exacerbations of mild to moderate COPD who do not have increased sputum purulence or CRP >40 mg/L can be safely managed without antibiotics.
Citation: Maravitlles M, Moravas A, Hernandez S, Bayona C, Llor C. Is it possible to identify exacerbations of mild to moderate COPD that do not require antibiotic treatment? Chest. 2013;144(5):1571-1577.
Clinical question: Which criteria identify ambulatory patients with exacerbations of mild to moderate COPD who do not need antibiotics?
Background: The Anthonisen criteria (increased dyspnea, sputum volume, sputum purulence) are commonly used to identify which patients with COPD exacerbations would benefit from antibiotics. These criteria, however, were derived in patients with severe COPD. It is unknown whether these criteria are predictive in patients with mild to moderate COPD.
Study design: Multivariate logistic regression analysis of placebo group of a double-blinded RCT.
Setting: Multicenter, ambulatory, primary care clinics in Spain.
Synopsis: The original RCT enrolled 310 ambulatory patients with exacerbations of mild to moderate COPD and tested the efficacy of amoxicillin/clavulanate. Clinical failure without antibiotics was 19.9% compared to 9.5% with antibiotics (P=0.022). Here they analyzed the 152 patients from the placebo group to identify factors associated with increased risk of clinical failure. Only increased sputum purulence (OR 6.1, CI 1.5-25; P=0.005) or C-reactive protein (CRP) >40 mg/L (OR 13.4, CI 4.5-38.8, P<0.001) were independently associated with increased risk of failure. Presence of both predicted a 63.7% failure without antibiotics.
The study did not define “increased sputum purulence,” but this is similar to real-life clinical practice. Placebo effect cannot be ruled out, but correlation of the objective measures with the clinical assessments suggests that the clinical assessments were accurate. The study did not have a protocol for administering co-medications such as steroids and inhalers. Despite these limitations, the criteria of increased sputum purulence and CRP >40 mg/L identified COPD patients likely to have a clinical failure without antibiotics.
Bottom line: Patients with exacerbations of mild to moderate COPD who do not have increased sputum purulence or CRP >40 mg/L can be safely managed without antibiotics.
Citation: Maravitlles M, Moravas A, Hernandez S, Bayona C, Llor C. Is it possible to identify exacerbations of mild to moderate COPD that do not require antibiotic treatment? Chest. 2013;144(5):1571-1577.
Pre-Operative Angiotensin Axis Blockade Increases Risk of Hypotension, Acute Kidney Injury with Major Orthopedic Surgery
Clinical question: Do patients receiving pre-operative angiotensin axis blockade (AAB) prior to elective major orthopedic surgery have an increased risk of peri-operative hypotension and acute kidney injury (AKI)?
Background: Patients with pre-operative AAB from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers have an increased incidence of peri-operative hypotension. Patients undergoing cardiothoracic and vascular surgery with pre-operative AAB have increased incidence of post-operative AKI; however, there is scant literature evaluating the hypotensive and renal effects of pre-operative AAB prior to elective major orthopedic surgery.
Study design: Retrospective, cohort study.
Setting: Academic medical center.
Synopsis: Retrospective review of 922 patients undergoing spinal fusion, total knee arthroplasty, or total hip arthroplasty in one academic medical center in 2010 found that 37% received pre-operative AAB. Post-induction hypotension (systolic blood pressure ≤80 mm Hg for five minutes) was significantly higher in patients receiving AAB (12.2% vs. 6.7%; odds ratio [OR] 1.93, P=0.005). Post-operative AKI was significantly higher in patients receiving AAB (8.3% vs. 1.7%; OR 5.40, P<0.001), remaining significant after adjusting for intra-operative hypotension (OR 2.60, P=0.042). Developing AKI resulted in a significantly higher mean length of stay (5.76 vs. 3.28 days, P<0.001) but no difference in two-year mortality.
The findings suggest an association exists between pre-operative angiotensin-converting enzyme inhibitors/ARB, hypotension, and AKI following major orthopedic surgeries but does not demonstrate causality. A prospective, multi-center, randomized trial is needed to confirm that holding pre-operative AAB would decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia.
Bottom line: Patients who underwent elective major orthopedic surgery who received pre-operative AAB therapy had an associated increased risk of post-induction hypotension and post-operative AKI, resulting in a greater hospital length of stay.
Citation: Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288.
Clinical question: Do patients receiving pre-operative angiotensin axis blockade (AAB) prior to elective major orthopedic surgery have an increased risk of peri-operative hypotension and acute kidney injury (AKI)?
Background: Patients with pre-operative AAB from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers have an increased incidence of peri-operative hypotension. Patients undergoing cardiothoracic and vascular surgery with pre-operative AAB have increased incidence of post-operative AKI; however, there is scant literature evaluating the hypotensive and renal effects of pre-operative AAB prior to elective major orthopedic surgery.
Study design: Retrospective, cohort study.
Setting: Academic medical center.
Synopsis: Retrospective review of 922 patients undergoing spinal fusion, total knee arthroplasty, or total hip arthroplasty in one academic medical center in 2010 found that 37% received pre-operative AAB. Post-induction hypotension (systolic blood pressure ≤80 mm Hg for five minutes) was significantly higher in patients receiving AAB (12.2% vs. 6.7%; odds ratio [OR] 1.93, P=0.005). Post-operative AKI was significantly higher in patients receiving AAB (8.3% vs. 1.7%; OR 5.40, P<0.001), remaining significant after adjusting for intra-operative hypotension (OR 2.60, P=0.042). Developing AKI resulted in a significantly higher mean length of stay (5.76 vs. 3.28 days, P<0.001) but no difference in two-year mortality.
The findings suggest an association exists between pre-operative angiotensin-converting enzyme inhibitors/ARB, hypotension, and AKI following major orthopedic surgeries but does not demonstrate causality. A prospective, multi-center, randomized trial is needed to confirm that holding pre-operative AAB would decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia.
Bottom line: Patients who underwent elective major orthopedic surgery who received pre-operative AAB therapy had an associated increased risk of post-induction hypotension and post-operative AKI, resulting in a greater hospital length of stay.
Citation: Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288.
Clinical question: Do patients receiving pre-operative angiotensin axis blockade (AAB) prior to elective major orthopedic surgery have an increased risk of peri-operative hypotension and acute kidney injury (AKI)?
Background: Patients with pre-operative AAB from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers have an increased incidence of peri-operative hypotension. Patients undergoing cardiothoracic and vascular surgery with pre-operative AAB have increased incidence of post-operative AKI; however, there is scant literature evaluating the hypotensive and renal effects of pre-operative AAB prior to elective major orthopedic surgery.
Study design: Retrospective, cohort study.
Setting: Academic medical center.
Synopsis: Retrospective review of 922 patients undergoing spinal fusion, total knee arthroplasty, or total hip arthroplasty in one academic medical center in 2010 found that 37% received pre-operative AAB. Post-induction hypotension (systolic blood pressure ≤80 mm Hg for five minutes) was significantly higher in patients receiving AAB (12.2% vs. 6.7%; odds ratio [OR] 1.93, P=0.005). Post-operative AKI was significantly higher in patients receiving AAB (8.3% vs. 1.7%; OR 5.40, P<0.001), remaining significant after adjusting for intra-operative hypotension (OR 2.60, P=0.042). Developing AKI resulted in a significantly higher mean length of stay (5.76 vs. 3.28 days, P<0.001) but no difference in two-year mortality.
The findings suggest an association exists between pre-operative angiotensin-converting enzyme inhibitors/ARB, hypotension, and AKI following major orthopedic surgeries but does not demonstrate causality. A prospective, multi-center, randomized trial is needed to confirm that holding pre-operative AAB would decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia.
Bottom line: Patients who underwent elective major orthopedic surgery who received pre-operative AAB therapy had an associated increased risk of post-induction hypotension and post-operative AKI, resulting in a greater hospital length of stay.
Citation: Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288.
American College of Physicians Releases Clinical Practice Guideline for Treating Anemia in Heart Disease Patients
Clinical question: What is the recommended threshold for red blood cell (RBC) transfusion and erythropoiesis-stimulating agents (ESA) in anemic hospitalized patients with coronary heart disease?
Background: Anemia can worsen cardiac function and is associated with increased risk of hospitalization and death in patients with coronary heart disease (CHD) or congestive heart failure (CHF). It is unclear if treatments such as RBC transfusion, ESA, or iron replacement improve outcomes in patients with heart disease.
Study design: Systematic review.
Setting: Studies of hospitalized medical and surgical patients.
Synopsis: The guideline was developed by reviewing studies evaluating anemia treatment outcomes, including mortality, hospitalization, exercise tolerance, quality of life, and cardiovascular events. Six studies evaluated the benefits and harms resulting from RBC transfusion, each determined to be low-quality evidence. The current evidence showed no benefit when comparing liberal (hemoglobin (Hgb) >10 g/dL) versus restrictive (Hgb <10 g/dL) transfusion thresholds. Potential harms of transfusion included fever, transfusion-related acute lung injury, and CHF.
Given the low-quality evidence, the American College of Physicians (ACP) makes a weak recommendation for a restrictive transfusion strategy of Hgb 7-8 g/dL in patients with CHD.
A review of 16 RCTs (moderate-quality evidence) evaluated the effects of ESAs in mild to moderate anemia and showed no difference in outcomes for patients with CHD and CHF. Harms associated with ESA therapy included hypertension and venous thrombosis. The ACP makes a strong recommendation not to use ESAs in patients with heart disease.
Bottom line: The ACP recommends restrictive transfusion with hemoglobin threshold of 7-8 g/dL in hospitalized patients with CHD (weak recommendation, low-quality evidence) and recommends against using erythropoiesis-stimulating agents for mild to moderate anemia in patients with CHF or CHD (strong recommendation, moderate-quality evidence).
Citation: Qaseem A, Humphrey LL, Fitterman N, Starkey M, Shekelle P. Treatment of anemia in patients with heart disease: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2013;159(11):770-779.
Clinical question: What is the recommended threshold for red blood cell (RBC) transfusion and erythropoiesis-stimulating agents (ESA) in anemic hospitalized patients with coronary heart disease?
Background: Anemia can worsen cardiac function and is associated with increased risk of hospitalization and death in patients with coronary heart disease (CHD) or congestive heart failure (CHF). It is unclear if treatments such as RBC transfusion, ESA, or iron replacement improve outcomes in patients with heart disease.
Study design: Systematic review.
Setting: Studies of hospitalized medical and surgical patients.
Synopsis: The guideline was developed by reviewing studies evaluating anemia treatment outcomes, including mortality, hospitalization, exercise tolerance, quality of life, and cardiovascular events. Six studies evaluated the benefits and harms resulting from RBC transfusion, each determined to be low-quality evidence. The current evidence showed no benefit when comparing liberal (hemoglobin (Hgb) >10 g/dL) versus restrictive (Hgb <10 g/dL) transfusion thresholds. Potential harms of transfusion included fever, transfusion-related acute lung injury, and CHF.
Given the low-quality evidence, the American College of Physicians (ACP) makes a weak recommendation for a restrictive transfusion strategy of Hgb 7-8 g/dL in patients with CHD.
A review of 16 RCTs (moderate-quality evidence) evaluated the effects of ESAs in mild to moderate anemia and showed no difference in outcomes for patients with CHD and CHF. Harms associated with ESA therapy included hypertension and venous thrombosis. The ACP makes a strong recommendation not to use ESAs in patients with heart disease.
Bottom line: The ACP recommends restrictive transfusion with hemoglobin threshold of 7-8 g/dL in hospitalized patients with CHD (weak recommendation, low-quality evidence) and recommends against using erythropoiesis-stimulating agents for mild to moderate anemia in patients with CHF or CHD (strong recommendation, moderate-quality evidence).
Citation: Qaseem A, Humphrey LL, Fitterman N, Starkey M, Shekelle P. Treatment of anemia in patients with heart disease: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2013;159(11):770-779.
Clinical question: What is the recommended threshold for red blood cell (RBC) transfusion and erythropoiesis-stimulating agents (ESA) in anemic hospitalized patients with coronary heart disease?
Background: Anemia can worsen cardiac function and is associated with increased risk of hospitalization and death in patients with coronary heart disease (CHD) or congestive heart failure (CHF). It is unclear if treatments such as RBC transfusion, ESA, or iron replacement improve outcomes in patients with heart disease.
Study design: Systematic review.
Setting: Studies of hospitalized medical and surgical patients.
Synopsis: The guideline was developed by reviewing studies evaluating anemia treatment outcomes, including mortality, hospitalization, exercise tolerance, quality of life, and cardiovascular events. Six studies evaluated the benefits and harms resulting from RBC transfusion, each determined to be low-quality evidence. The current evidence showed no benefit when comparing liberal (hemoglobin (Hgb) >10 g/dL) versus restrictive (Hgb <10 g/dL) transfusion thresholds. Potential harms of transfusion included fever, transfusion-related acute lung injury, and CHF.
Given the low-quality evidence, the American College of Physicians (ACP) makes a weak recommendation for a restrictive transfusion strategy of Hgb 7-8 g/dL in patients with CHD.
A review of 16 RCTs (moderate-quality evidence) evaluated the effects of ESAs in mild to moderate anemia and showed no difference in outcomes for patients with CHD and CHF. Harms associated with ESA therapy included hypertension and venous thrombosis. The ACP makes a strong recommendation not to use ESAs in patients with heart disease.
Bottom line: The ACP recommends restrictive transfusion with hemoglobin threshold of 7-8 g/dL in hospitalized patients with CHD (weak recommendation, low-quality evidence) and recommends against using erythropoiesis-stimulating agents for mild to moderate anemia in patients with CHF or CHD (strong recommendation, moderate-quality evidence).
Citation: Qaseem A, Humphrey LL, Fitterman N, Starkey M, Shekelle P. Treatment of anemia in patients with heart disease: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2013;159(11):770-779.
Risk Stratification Model Predicts Continued Stability in Patients with Pulmonary Embolism
Clinical question: Can right ventricular (RV) dysfunction and troponin levels be used to risk stratify hemodynamically stable patients with acute pulmonary embolism (PE)?
Background: PE can present with varying degrees of clinical severity, necessitating appropriate risk stratification for decision making and management. Observational studies have demonstrated that RV dysfunction and injury provide important prognostic information in patients with acute PE, but this has not been confirmed in large prospective studies.
Study design: Prospective, cohort study.
Setting: Multicenter registry of academic and community hospitals in Italy.
Synopsis: Using the Italian Pulmonary Embolism Registry database of 860 hemodynamically stable patients with PE who underwent troponin measurement and echocardiogram, this study demonstrated that a model including RV dysfunction and injury has an incremental prognostic value for risk stratification of in-hospital death or clinical deterioration. The negative predictive value of negative echocardiography with negative troponin was 100% for in-hospital death and 99% for in-hospital clinical deterioration.
This study was limited by its observational approach; in addition, it did not provide its metrics for defining hemodynamic stability and did not standardize or report treatment strategies for these patients.
The predictive value of this study is impressive and could lead to the identification of patients in whom early discharge or brief hospitalization is appropriate; however, its application could be challenging in clinical environments where timely echocardiography resources are not readily available to a hemodynamically stable patient population.
Bottom line: Negative troponin and absence of RV dysfunction predict clinically stable pulmonary embolism.
Citation: Becattini C, Casazza F, Forgione C, et al. Acute pulmonary embolism: external validation of an integrated risk stratification model. Chest. 2013;144(5):1539-1545.
Clinical question: Can right ventricular (RV) dysfunction and troponin levels be used to risk stratify hemodynamically stable patients with acute pulmonary embolism (PE)?
Background: PE can present with varying degrees of clinical severity, necessitating appropriate risk stratification for decision making and management. Observational studies have demonstrated that RV dysfunction and injury provide important prognostic information in patients with acute PE, but this has not been confirmed in large prospective studies.
Study design: Prospective, cohort study.
Setting: Multicenter registry of academic and community hospitals in Italy.
Synopsis: Using the Italian Pulmonary Embolism Registry database of 860 hemodynamically stable patients with PE who underwent troponin measurement and echocardiogram, this study demonstrated that a model including RV dysfunction and injury has an incremental prognostic value for risk stratification of in-hospital death or clinical deterioration. The negative predictive value of negative echocardiography with negative troponin was 100% for in-hospital death and 99% for in-hospital clinical deterioration.
This study was limited by its observational approach; in addition, it did not provide its metrics for defining hemodynamic stability and did not standardize or report treatment strategies for these patients.
The predictive value of this study is impressive and could lead to the identification of patients in whom early discharge or brief hospitalization is appropriate; however, its application could be challenging in clinical environments where timely echocardiography resources are not readily available to a hemodynamically stable patient population.
Bottom line: Negative troponin and absence of RV dysfunction predict clinically stable pulmonary embolism.
Citation: Becattini C, Casazza F, Forgione C, et al. Acute pulmonary embolism: external validation of an integrated risk stratification model. Chest. 2013;144(5):1539-1545.
Clinical question: Can right ventricular (RV) dysfunction and troponin levels be used to risk stratify hemodynamically stable patients with acute pulmonary embolism (PE)?
Background: PE can present with varying degrees of clinical severity, necessitating appropriate risk stratification for decision making and management. Observational studies have demonstrated that RV dysfunction and injury provide important prognostic information in patients with acute PE, but this has not been confirmed in large prospective studies.
Study design: Prospective, cohort study.
Setting: Multicenter registry of academic and community hospitals in Italy.
Synopsis: Using the Italian Pulmonary Embolism Registry database of 860 hemodynamically stable patients with PE who underwent troponin measurement and echocardiogram, this study demonstrated that a model including RV dysfunction and injury has an incremental prognostic value for risk stratification of in-hospital death or clinical deterioration. The negative predictive value of negative echocardiography with negative troponin was 100% for in-hospital death and 99% for in-hospital clinical deterioration.
This study was limited by its observational approach; in addition, it did not provide its metrics for defining hemodynamic stability and did not standardize or report treatment strategies for these patients.
The predictive value of this study is impressive and could lead to the identification of patients in whom early discharge or brief hospitalization is appropriate; however, its application could be challenging in clinical environments where timely echocardiography resources are not readily available to a hemodynamically stable patient population.
Bottom line: Negative troponin and absence of RV dysfunction predict clinically stable pulmonary embolism.
Citation: Becattini C, Casazza F, Forgione C, et al. Acute pulmonary embolism: external validation of an integrated risk stratification model. Chest. 2013;144(5):1539-1545.
Accelerated Diagnostic Protocol for Chest Pain Results in Earlier Discharge of Low-Risk Patients
Clinical question: In patients with possible cardiac chest pain, does an accelerated diagnostic protocol result in higher rates of successful early discharge when compared with the standard care pathway?
Background: An accelerated diagnostic pathway (ADP), which combines TIMI [thrombolysis in myocardial infarction] score, EKG, and zero- and two-hour troponin levels, can identify low-risk patients presenting with chest pain; however, little is known about its effect on the rates of early, successful discharge in a real-world population.
Study design: RCT.
Setting: An academic general and tertiary care hospital in Christchurch, New Zealand.
Synopsis: This study of 542 patients ages 18 years and older who presented with chest pain demonstrated that when compared with a conventional pathway, an ADP results in nearly twice the proportion of patients (19.3% vs. 11.0%; P=0.008) achieving safe, early discharge from the ED, defined as discharge within six hours and no major adverse cardiac events within 30 days. This was a single-center trial in New Zealand, which limits its sample size and its generalizability; however, if larger studies led to the implementation of ADP by ED physicians, there would likely be fewer low-risk patients admitted to hospitalist services for evaluation of chest pain.
Bottom line: An accelerated diagnostic chest pain protocol results in earlier discharge of low-risk patients.
Citation: Than M, Aldous S, Lord SJ, et al. A 2-hour diagnostic protocol for possible cardiac chest pain in the emergency department: a randomized clinical trial. JAMA Intern Med. 2014;174(1):51-58.
Clinical question: In patients with possible cardiac chest pain, does an accelerated diagnostic protocol result in higher rates of successful early discharge when compared with the standard care pathway?
Background: An accelerated diagnostic pathway (ADP), which combines TIMI [thrombolysis in myocardial infarction] score, EKG, and zero- and two-hour troponin levels, can identify low-risk patients presenting with chest pain; however, little is known about its effect on the rates of early, successful discharge in a real-world population.
Study design: RCT.
Setting: An academic general and tertiary care hospital in Christchurch, New Zealand.
Synopsis: This study of 542 patients ages 18 years and older who presented with chest pain demonstrated that when compared with a conventional pathway, an ADP results in nearly twice the proportion of patients (19.3% vs. 11.0%; P=0.008) achieving safe, early discharge from the ED, defined as discharge within six hours and no major adverse cardiac events within 30 days. This was a single-center trial in New Zealand, which limits its sample size and its generalizability; however, if larger studies led to the implementation of ADP by ED physicians, there would likely be fewer low-risk patients admitted to hospitalist services for evaluation of chest pain.
Bottom line: An accelerated diagnostic chest pain protocol results in earlier discharge of low-risk patients.
Citation: Than M, Aldous S, Lord SJ, et al. A 2-hour diagnostic protocol for possible cardiac chest pain in the emergency department: a randomized clinical trial. JAMA Intern Med. 2014;174(1):51-58.
Clinical question: In patients with possible cardiac chest pain, does an accelerated diagnostic protocol result in higher rates of successful early discharge when compared with the standard care pathway?
Background: An accelerated diagnostic pathway (ADP), which combines TIMI [thrombolysis in myocardial infarction] score, EKG, and zero- and two-hour troponin levels, can identify low-risk patients presenting with chest pain; however, little is known about its effect on the rates of early, successful discharge in a real-world population.
Study design: RCT.
Setting: An academic general and tertiary care hospital in Christchurch, New Zealand.
Synopsis: This study of 542 patients ages 18 years and older who presented with chest pain demonstrated that when compared with a conventional pathway, an ADP results in nearly twice the proportion of patients (19.3% vs. 11.0%; P=0.008) achieving safe, early discharge from the ED, defined as discharge within six hours and no major adverse cardiac events within 30 days. This was a single-center trial in New Zealand, which limits its sample size and its generalizability; however, if larger studies led to the implementation of ADP by ED physicians, there would likely be fewer low-risk patients admitted to hospitalist services for evaluation of chest pain.
Bottom line: An accelerated diagnostic chest pain protocol results in earlier discharge of low-risk patients.
Citation: Than M, Aldous S, Lord SJ, et al. A 2-hour diagnostic protocol for possible cardiac chest pain in the emergency department: a randomized clinical trial. JAMA Intern Med. 2014;174(1):51-58.
Restrictive Blood Transfusion Strategy with Trigger Hemoglobin
Clinical question: What effect does a restrictive hemoglobin transfusion trigger of <7 g/dL have on clinical outcomes compared to more liberal transfusion triggers?
Background: Blood transfusions are standard of care in managing anemia despite sparse evidence that they improve clinical outcomes. As more evidence mounts that blood transfusions are associated with worse outcomes, the threshold hemoglobin level for transfusion has decreased. The safest hemoglobin trigger for blood transfusion remains unknown.
Study design: Meta-analysis of randomized control trials (RCTs) and systematic review of observational studies.
Setting: Data from MEDLINE.
Synopsis: Pooled data from three RCTs, including 2364 patients, compared outcomes between a restrictive hemoglobin transfusion trigger of <7 g/dL with a more liberal trigger. The restrictive strategy resulted in decreased in-hospital mortality (RR 0.74, CI 0.6-0.92), total mortality (RR 0.8, CI 0.65-0.98), rebleeding (RR 0.64, CI 0.45-0.9), acute coronary syndrome (RR 0.44, CI 0.22-0.89), and pulmonary edema (RR 0.48, CI 0.33-0.72). Using the restrictive strategy, the number needed to treat to prevent one death was 33.
A second meta-analysis of 16 RCTs found that less restrictive transfusion strategies (triggers of 7.5 to 10 g/dL) were not effective in improving outcomes. A systematic review of observational studies suggested that normovolemic patients can tolerate hemoglobins of 5-6 g/dL without affecting oxygen delivery.
The primary meta-analysis was limited by the inclusion of adult and pediatric patients and different indications for transfusion (critical illness, gastrointestinal bleed), which could have introduced some bias. Although the safest transfusion trigger remains unknown, this study demonstrates that <7 g/dL is safer than more liberal triggers.
Bottom line: A restrictive blood transfusion strategy with hemoglobin trigger of <7 g/dL in patients with critical illness or bleeding significantly decreases rebleeding, cardiac events, and total mortality.
Citation: Salpeter SR, Buckley JS, Chatterjee S. Impact of more restrictive blood transfusion strategies on clinical outcomes: a meta-analysis and systematic review. Am J Med. 2014;127(2): 124-131.
Clinical question: What effect does a restrictive hemoglobin transfusion trigger of <7 g/dL have on clinical outcomes compared to more liberal transfusion triggers?
Background: Blood transfusions are standard of care in managing anemia despite sparse evidence that they improve clinical outcomes. As more evidence mounts that blood transfusions are associated with worse outcomes, the threshold hemoglobin level for transfusion has decreased. The safest hemoglobin trigger for blood transfusion remains unknown.
Study design: Meta-analysis of randomized control trials (RCTs) and systematic review of observational studies.
Setting: Data from MEDLINE.
Synopsis: Pooled data from three RCTs, including 2364 patients, compared outcomes between a restrictive hemoglobin transfusion trigger of <7 g/dL with a more liberal trigger. The restrictive strategy resulted in decreased in-hospital mortality (RR 0.74, CI 0.6-0.92), total mortality (RR 0.8, CI 0.65-0.98), rebleeding (RR 0.64, CI 0.45-0.9), acute coronary syndrome (RR 0.44, CI 0.22-0.89), and pulmonary edema (RR 0.48, CI 0.33-0.72). Using the restrictive strategy, the number needed to treat to prevent one death was 33.
A second meta-analysis of 16 RCTs found that less restrictive transfusion strategies (triggers of 7.5 to 10 g/dL) were not effective in improving outcomes. A systematic review of observational studies suggested that normovolemic patients can tolerate hemoglobins of 5-6 g/dL without affecting oxygen delivery.
The primary meta-analysis was limited by the inclusion of adult and pediatric patients and different indications for transfusion (critical illness, gastrointestinal bleed), which could have introduced some bias. Although the safest transfusion trigger remains unknown, this study demonstrates that <7 g/dL is safer than more liberal triggers.
Bottom line: A restrictive blood transfusion strategy with hemoglobin trigger of <7 g/dL in patients with critical illness or bleeding significantly decreases rebleeding, cardiac events, and total mortality.
Citation: Salpeter SR, Buckley JS, Chatterjee S. Impact of more restrictive blood transfusion strategies on clinical outcomes: a meta-analysis and systematic review. Am J Med. 2014;127(2): 124-131.
Clinical question: What effect does a restrictive hemoglobin transfusion trigger of <7 g/dL have on clinical outcomes compared to more liberal transfusion triggers?
Background: Blood transfusions are standard of care in managing anemia despite sparse evidence that they improve clinical outcomes. As more evidence mounts that blood transfusions are associated with worse outcomes, the threshold hemoglobin level for transfusion has decreased. The safest hemoglobin trigger for blood transfusion remains unknown.
Study design: Meta-analysis of randomized control trials (RCTs) and systematic review of observational studies.
Setting: Data from MEDLINE.
Synopsis: Pooled data from three RCTs, including 2364 patients, compared outcomes between a restrictive hemoglobin transfusion trigger of <7 g/dL with a more liberal trigger. The restrictive strategy resulted in decreased in-hospital mortality (RR 0.74, CI 0.6-0.92), total mortality (RR 0.8, CI 0.65-0.98), rebleeding (RR 0.64, CI 0.45-0.9), acute coronary syndrome (RR 0.44, CI 0.22-0.89), and pulmonary edema (RR 0.48, CI 0.33-0.72). Using the restrictive strategy, the number needed to treat to prevent one death was 33.
A second meta-analysis of 16 RCTs found that less restrictive transfusion strategies (triggers of 7.5 to 10 g/dL) were not effective in improving outcomes. A systematic review of observational studies suggested that normovolemic patients can tolerate hemoglobins of 5-6 g/dL without affecting oxygen delivery.
The primary meta-analysis was limited by the inclusion of adult and pediatric patients and different indications for transfusion (critical illness, gastrointestinal bleed), which could have introduced some bias. Although the safest transfusion trigger remains unknown, this study demonstrates that <7 g/dL is safer than more liberal triggers.
Bottom line: A restrictive blood transfusion strategy with hemoglobin trigger of <7 g/dL in patients with critical illness or bleeding significantly decreases rebleeding, cardiac events, and total mortality.
Citation: Salpeter SR, Buckley JS, Chatterjee S. Impact of more restrictive blood transfusion strategies on clinical outcomes: a meta-analysis and systematic review. Am J Med. 2014;127(2): 124-131.