User login
Hospitalists Share Information, Insights Through RIV Posters at HM13

One of the busiest times of HM13—and, come to think of it, every recent annual meeting—is the poster session for the Research, Innovations, and Clinical Vignettes (RIV) competition. This year, more than 800 abstracts were submitted and reviewed, with nearly 600 being accepted for presentation at HM13. That meant thousands of hospitalists thumbtacking posters to rows and rows of portable bulletin boards in the Gaylord National Resort & Convention Center’s massive exhibit hall.
With all those posters and accompanying oral presentations, it’s impossible for RIV judges to chat with everybody, so they choose finalists based on the abstracts, then listen to quick-hit summaries before choosing a winner on site. And meeting attendees are just as strapped for time, so they do the best they can to see as many posters as they can, taking time to network with old connections and make new ones.
So with all the limitations on how many people will interact with your poster, the small chance of winning Best in Show, and the hundreds of work hours that go into a poster presentation, why do it?
“To share is what I think is really important,” says Todd Hecht, MD, FACP, SFHM, associate professor of clinical medicine at the Perelman School of Medicine at the University of Pennsylvania in Philadelphia. “If you don’t let other people know what you’re doing, they can’t bring it to their institutions, nor can you learn from others and bring their innovations to your own hospital.”
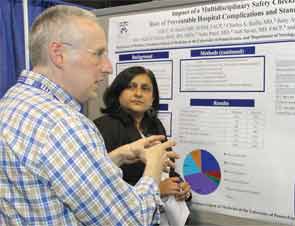
Dr. Hecht, director of the Anticoagulation Management Center and Anticoagulation Management Program at the Hospital of the University of Pennsylvania, takes the poster sessions very seriously. This year, he entered a poster in both the Innovations and Vignette categories. His Innovations poster, “Impact of a Multidisciplinary Safety Checklist on the Rate of Preventable Hospital Complications and Standardization of Care,” was a finalist.
That meant that, at the very least, he’d be able to explain to at least two judges what motivated his research team’s project. And what was the inspiration? A 90-year-old male patient with metastatic melanoma who, in the fall of 2011, refused to take medication for VTE prophylaxis, as lesions on his skin made the process rather painful. After refusing the doses for a bit, though, the high-risk patient unsurprisingly developed a pulmonary embolism (PE).
The man survived the PE, but Dr. Hecht and his colleagues began to wonder how many patients refuse VTE prophylaxis. So they investigated, and it turned out that from December 2010 to February 2011, 26.4% of the prescribed doses of prophylaxis on the medicine floors they studied were missed. Moreover, nearly 80% of all missed doses on the medicine floors were due to patient refusal.
“It was astonishing to me that it was that high,” Dr. Hecht says. “If there were 1,000 doses in a month, 260 of them were not being given—and 205 of them were not given because they were refused.”
Checklist Integration
So Dr. Hecht and colleagues set out to create a checklist that could be used daily on multidisciplinary rounds to help reduce the risk of VTE. First question on the list: Has prophylaxis been ordered, and if so, is the patient refusing it? Knowing that patients are “refusing” medication can lead to discussions about why that is happening, which in turn can lead to ways to convince the patient that the preventative measure is a good idea.
Dr. Hecht says the team also realized a checklist creates the opportunity to improve other quality metrics, such as hospital-associated infections (HAIs). Two questions on the checklist ask whether indwelling urinary catheters (IUCs) and central venous catheters (CVCs) can be removed. Two questions ask if telemetry can be stopped and whether there are any pain-management concerns. A final query asks whether there are any nursing, social work, or discharge-related questions—a step that, according to Dr. Hecht, loops the entire multidisciplinary team into the care-plan discussion.
“An ongoing challenge is making sure it’s not just questions being asked and being answered by rote,” Dr. Hecht says. “Just pause and think for just a second for each question. You can get through the checklist in 10 seconds, but you can’t go through the checklist in two seconds.”
The project’s results are what made it a finalist. After the checklist intervention, the number of missed doses of VTE prophylaxis plummeted 59% to just 10.9% (P<0.001) from September to November 2012; the number of “patient refused” doses dropped to 6.3% (P<0.001).
Not only was Dr. Hecht caught off guard by his findings, but so were the judges who visited his poster—Mangla Gulati, MD, FHM, of the University of Maryland School of Medicine and Rachel George, MD, MBA, FHM, of Cogent HMG.
“I wonder if it’s like that in every hospital,” Dr. George says. “I’d like to know.”
The positive reaction and feedback to Dr. Hecht’s poster, however, was not enough to win the Innovations category. That honor went to “SEPTRIS: Improving Sepsis Recognition and Management Through a Mobile Educational Game,” which was developed by a team of researchers at Stanford University in Palo Alto, Calif. The video game
(http://med.stanford.edu/septris/)—a mashup of sepsis and the once-popular Tetris puzzle game—already has been played 17,000 times and is on its way to being shared in other languages.
“Win or lose, it doesn’t matter,” Dr. Hecht says. “The goal is to share your information with other people and learn from them.”
Peter Watson, MD, FACP, FHM, sees it the same way. That’s why this year he was both judge and judged. The division head of hospital medicine for Henry Ford Medical Group in Detroit was part of a group presenting “Feasibility and Efficacy of a Specialized Pilot Training Program to Enhance Inpatient Communication Skills of Hospitalists.” He was a judge for the Research portion of the contest. He says he’s hard-pressed to say which process he enjoyed more, but one trick of the poster trade he passes along is that “judging actually makes you a better presenter on the back end,” especially when it comes to describing in less than five minutes a poster whose work may date back 12 to 18 months.
“In your brain,” he says, “you have a Tolstoy novel of information, but you have to break that down into a paragraph of CliffsNotes, and actually convince the people that are judging you that you have a really cool project that either is going to have a big impact in the field or may lead to other big studies or is going to impress somebody so much that they’re going to go back to their institution and say, ‘Hey, I’m going to do that.’”
Dr. Watson also urges people not to be discouraged by not winning the poster contest. First, all of the accepted abstracts get published online (www.shmabstracts.com) by the Journal of Hospital Medicine, a high point for medical students, residents, and early-career physicians looking to make a mark. Second, presenting information of value to one’s peers is the definition of a specialty that prides itself on collaboration.
“To see a second-year medical student presenting all the way up to a very senior division chief and everything in between is a really good example for our profession,” he says. “That’s really the magic of this meeting.”
Richard Quinn is a freelance writer in New Jersey.
One of the busiest times of HM13—and, come to think of it, every recent annual meeting—is the poster session for the Research, Innovations, and Clinical Vignettes (RIV) competition. This year, more than 800 abstracts were submitted and reviewed, with nearly 600 being accepted for presentation at HM13. That meant thousands of hospitalists thumbtacking posters to rows and rows of portable bulletin boards in the Gaylord National Resort & Convention Center’s massive exhibit hall.
With all those posters and accompanying oral presentations, it’s impossible for RIV judges to chat with everybody, so they choose finalists based on the abstracts, then listen to quick-hit summaries before choosing a winner on site. And meeting attendees are just as strapped for time, so they do the best they can to see as many posters as they can, taking time to network with old connections and make new ones.
So with all the limitations on how many people will interact with your poster, the small chance of winning Best in Show, and the hundreds of work hours that go into a poster presentation, why do it?
“To share is what I think is really important,” says Todd Hecht, MD, FACP, SFHM, associate professor of clinical medicine at the Perelman School of Medicine at the University of Pennsylvania in Philadelphia. “If you don’t let other people know what you’re doing, they can’t bring it to their institutions, nor can you learn from others and bring their innovations to your own hospital.”
Dr. Hecht, director of the Anticoagulation Management Center and Anticoagulation Management Program at the Hospital of the University of Pennsylvania, takes the poster sessions very seriously. This year, he entered a poster in both the Innovations and Vignette categories. His Innovations poster, “Impact of a Multidisciplinary Safety Checklist on the Rate of Preventable Hospital Complications and Standardization of Care,” was a finalist.
That meant that, at the very least, he’d be able to explain to at least two judges what motivated his research team’s project. And what was the inspiration? A 90-year-old male patient with metastatic melanoma who, in the fall of 2011, refused to take medication for VTE prophylaxis, as lesions on his skin made the process rather painful. After refusing the doses for a bit, though, the high-risk patient unsurprisingly developed a pulmonary embolism (PE).
The man survived the PE, but Dr. Hecht and his colleagues began to wonder how many patients refuse VTE prophylaxis. So they investigated, and it turned out that from December 2010 to February 2011, 26.4% of the prescribed doses of prophylaxis on the medicine floors they studied were missed. Moreover, nearly 80% of all missed doses on the medicine floors were due to patient refusal.
“It was astonishing to me that it was that high,” Dr. Hecht says. “If there were 1,000 doses in a month, 260 of them were not being given—and 205 of them were not given because they were refused.”
Checklist Integration
So Dr. Hecht and colleagues set out to create a checklist that could be used daily on multidisciplinary rounds to help reduce the risk of VTE. First question on the list: Has prophylaxis been ordered, and if so, is the patient refusing it? Knowing that patients are “refusing” medication can lead to discussions about why that is happening, which in turn can lead to ways to convince the patient that the preventative measure is a good idea.
Dr. Hecht says the team also realized a checklist creates the opportunity to improve other quality metrics, such as hospital-associated infections (HAIs). Two questions on the checklist ask whether indwelling urinary catheters (IUCs) and central venous catheters (CVCs) can be removed. Two questions ask if telemetry can be stopped and whether there are any pain-management concerns. A final query asks whether there are any nursing, social work, or discharge-related questions—a step that, according to Dr. Hecht, loops the entire multidisciplinary team into the care-plan discussion.
“An ongoing challenge is making sure it’s not just questions being asked and being answered by rote,” Dr. Hecht says. “Just pause and think for just a second for each question. You can get through the checklist in 10 seconds, but you can’t go through the checklist in two seconds.”
The project’s results are what made it a finalist. After the checklist intervention, the number of missed doses of VTE prophylaxis plummeted 59% to just 10.9% (P<0.001) from September to November 2012; the number of “patient refused” doses dropped to 6.3% (P<0.001).
Not only was Dr. Hecht caught off guard by his findings, but so were the judges who visited his poster—Mangla Gulati, MD, FHM, of the University of Maryland School of Medicine and Rachel George, MD, MBA, FHM, of Cogent HMG.
“I wonder if it’s like that in every hospital,” Dr. George says. “I’d like to know.”
The positive reaction and feedback to Dr. Hecht’s poster, however, was not enough to win the Innovations category. That honor went to “SEPTRIS: Improving Sepsis Recognition and Management Through a Mobile Educational Game,” which was developed by a team of researchers at Stanford University in Palo Alto, Calif. The video game
(http://med.stanford.edu/septris/)—a mashup of sepsis and the once-popular Tetris puzzle game—already has been played 17,000 times and is on its way to being shared in other languages.
“Win or lose, it doesn’t matter,” Dr. Hecht says. “The goal is to share your information with other people and learn from them.”
Peter Watson, MD, FACP, FHM, sees it the same way. That’s why this year he was both judge and judged. The division head of hospital medicine for Henry Ford Medical Group in Detroit was part of a group presenting “Feasibility and Efficacy of a Specialized Pilot Training Program to Enhance Inpatient Communication Skills of Hospitalists.” He was a judge for the Research portion of the contest. He says he’s hard-pressed to say which process he enjoyed more, but one trick of the poster trade he passes along is that “judging actually makes you a better presenter on the back end,” especially when it comes to describing in less than five minutes a poster whose work may date back 12 to 18 months.
“In your brain,” he says, “you have a Tolstoy novel of information, but you have to break that down into a paragraph of CliffsNotes, and actually convince the people that are judging you that you have a really cool project that either is going to have a big impact in the field or may lead to other big studies or is going to impress somebody so much that they’re going to go back to their institution and say, ‘Hey, I’m going to do that.’”
Dr. Watson also urges people not to be discouraged by not winning the poster contest. First, all of the accepted abstracts get published online (www.shmabstracts.com) by the Journal of Hospital Medicine, a high point for medical students, residents, and early-career physicians looking to make a mark. Second, presenting information of value to one’s peers is the definition of a specialty that prides itself on collaboration.
“To see a second-year medical student presenting all the way up to a very senior division chief and everything in between is a really good example for our profession,” he says. “That’s really the magic of this meeting.”
Richard Quinn is a freelance writer in New Jersey.
One of the busiest times of HM13—and, come to think of it, every recent annual meeting—is the poster session for the Research, Innovations, and Clinical Vignettes (RIV) competition. This year, more than 800 abstracts were submitted and reviewed, with nearly 600 being accepted for presentation at HM13. That meant thousands of hospitalists thumbtacking posters to rows and rows of portable bulletin boards in the Gaylord National Resort & Convention Center’s massive exhibit hall.
With all those posters and accompanying oral presentations, it’s impossible for RIV judges to chat with everybody, so they choose finalists based on the abstracts, then listen to quick-hit summaries before choosing a winner on site. And meeting attendees are just as strapped for time, so they do the best they can to see as many posters as they can, taking time to network with old connections and make new ones.
So with all the limitations on how many people will interact with your poster, the small chance of winning Best in Show, and the hundreds of work hours that go into a poster presentation, why do it?
“To share is what I think is really important,” says Todd Hecht, MD, FACP, SFHM, associate professor of clinical medicine at the Perelman School of Medicine at the University of Pennsylvania in Philadelphia. “If you don’t let other people know what you’re doing, they can’t bring it to their institutions, nor can you learn from others and bring their innovations to your own hospital.”
Dr. Hecht, director of the Anticoagulation Management Center and Anticoagulation Management Program at the Hospital of the University of Pennsylvania, takes the poster sessions very seriously. This year, he entered a poster in both the Innovations and Vignette categories. His Innovations poster, “Impact of a Multidisciplinary Safety Checklist on the Rate of Preventable Hospital Complications and Standardization of Care,” was a finalist.
That meant that, at the very least, he’d be able to explain to at least two judges what motivated his research team’s project. And what was the inspiration? A 90-year-old male patient with metastatic melanoma who, in the fall of 2011, refused to take medication for VTE prophylaxis, as lesions on his skin made the process rather painful. After refusing the doses for a bit, though, the high-risk patient unsurprisingly developed a pulmonary embolism (PE).
The man survived the PE, but Dr. Hecht and his colleagues began to wonder how many patients refuse VTE prophylaxis. So they investigated, and it turned out that from December 2010 to February 2011, 26.4% of the prescribed doses of prophylaxis on the medicine floors they studied were missed. Moreover, nearly 80% of all missed doses on the medicine floors were due to patient refusal.
“It was astonishing to me that it was that high,” Dr. Hecht says. “If there were 1,000 doses in a month, 260 of them were not being given—and 205 of them were not given because they were refused.”
Checklist Integration
So Dr. Hecht and colleagues set out to create a checklist that could be used daily on multidisciplinary rounds to help reduce the risk of VTE. First question on the list: Has prophylaxis been ordered, and if so, is the patient refusing it? Knowing that patients are “refusing” medication can lead to discussions about why that is happening, which in turn can lead to ways to convince the patient that the preventative measure is a good idea.
Dr. Hecht says the team also realized a checklist creates the opportunity to improve other quality metrics, such as hospital-associated infections (HAIs). Two questions on the checklist ask whether indwelling urinary catheters (IUCs) and central venous catheters (CVCs) can be removed. Two questions ask if telemetry can be stopped and whether there are any pain-management concerns. A final query asks whether there are any nursing, social work, or discharge-related questions—a step that, according to Dr. Hecht, loops the entire multidisciplinary team into the care-plan discussion.
“An ongoing challenge is making sure it’s not just questions being asked and being answered by rote,” Dr. Hecht says. “Just pause and think for just a second for each question. You can get through the checklist in 10 seconds, but you can’t go through the checklist in two seconds.”
The project’s results are what made it a finalist. After the checklist intervention, the number of missed doses of VTE prophylaxis plummeted 59% to just 10.9% (P<0.001) from September to November 2012; the number of “patient refused” doses dropped to 6.3% (P<0.001).
Not only was Dr. Hecht caught off guard by his findings, but so were the judges who visited his poster—Mangla Gulati, MD, FHM, of the University of Maryland School of Medicine and Rachel George, MD, MBA, FHM, of Cogent HMG.
“I wonder if it’s like that in every hospital,” Dr. George says. “I’d like to know.”
The positive reaction and feedback to Dr. Hecht’s poster, however, was not enough to win the Innovations category. That honor went to “SEPTRIS: Improving Sepsis Recognition and Management Through a Mobile Educational Game,” which was developed by a team of researchers at Stanford University in Palo Alto, Calif. The video game
(http://med.stanford.edu/septris/)—a mashup of sepsis and the once-popular Tetris puzzle game—already has been played 17,000 times and is on its way to being shared in other languages.
“Win or lose, it doesn’t matter,” Dr. Hecht says. “The goal is to share your information with other people and learn from them.”
Peter Watson, MD, FACP, FHM, sees it the same way. That’s why this year he was both judge and judged. The division head of hospital medicine for Henry Ford Medical Group in Detroit was part of a group presenting “Feasibility and Efficacy of a Specialized Pilot Training Program to Enhance Inpatient Communication Skills of Hospitalists.” He was a judge for the Research portion of the contest. He says he’s hard-pressed to say which process he enjoyed more, but one trick of the poster trade he passes along is that “judging actually makes you a better presenter on the back end,” especially when it comes to describing in less than five minutes a poster whose work may date back 12 to 18 months.
“In your brain,” he says, “you have a Tolstoy novel of information, but you have to break that down into a paragraph of CliffsNotes, and actually convince the people that are judging you that you have a really cool project that either is going to have a big impact in the field or may lead to other big studies or is going to impress somebody so much that they’re going to go back to their institution and say, ‘Hey, I’m going to do that.’”
Dr. Watson also urges people not to be discouraged by not winning the poster contest. First, all of the accepted abstracts get published online (www.shmabstracts.com) by the Journal of Hospital Medicine, a high point for medical students, residents, and early-career physicians looking to make a mark. Second, presenting information of value to one’s peers is the definition of a specialty that prides itself on collaboration.
“To see a second-year medical student presenting all the way up to a very senior division chief and everything in between is a really good example for our profession,” he says. “That’s really the magic of this meeting.”
Richard Quinn is a freelance writer in New Jersey.
Quality Improvement (QI) Remains a Central Theme at HM13
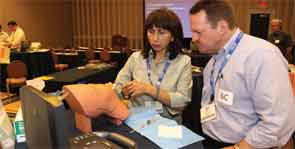

Like a grinning child at a carnival, Iqbal M. Binoj, MD, steps right up and gives it a try—except instead of tossing rings, he’s gripping an intraosseous infusion drill.
A tutor shows him how the device, which looks remarkably like a glue gun, inserts into the bones of the shoulder or knee and drills down until it hits the marrow. He is guided on using a steady speed to maintain the integrity of the cavity. He’s also taught about the maneuver’s low complication rates and ability to expedite workups.
“I’ve seen it used before, but I never did it,” says Dr. Binoj, a hospitalist with Cogent HMG at Genesis Medical Center in Davenport, Iowa.
Well, he never did it before a hands-on pre-course at HM13 that focused on improving hospitalists’ proficiency at such procedures as lumbar punctures and ultrasound-guided vascular access. Quality improvement (QI) is always a focus of SHM’s annual meeting, but sometimes the science of improving care is viewed from up on high.
Not everything needs to be a national imitative, an institution-wide project, or even a unit-based intervention. Sometimes, it’s as simple as teaching a room full of hospitalists how to use an intraosseous infusion drill, says Michelle Fox, RN, BSN, senior director of clinical affairs with Vidacare, which manufactures the drill used in the demonstration.
“Hospitalists have an increasing role in doing these procedures, not only in the environment they predominantly support but in other areas of the hospital,” Fox says, adding that “the primary goal of this course is to give them the opportunity to perfect those skills.”
Hospitalist Bradley Rosen, MD, MBA, FHM, medical of the inpatient specialty program at Cedars-Sinai in Los Angeles, says the point of hands-on demonstrations is to translate QI to the bedside. Take ultrasound devices, he says. In the past few years, the technology has become less expensive, better in resolution, more common, and more portable. Hospitalists must ensure hands-on training that keeps pace with that technology.
“We actually want people to get gloves on, hands on, learn where they may have challenges in terms of their own dexterity or workflow, which hand is dominant, and how to visualize on the ultrasound machine a three-dimensional structure in 2D,” he says. “We don’t want people watching from the sidelines. ... We try to get people in it and engaged.”
And once hospitalists master procedures or diagnostic maneuvers, they invariably are sought out by other physicians to pass that knowledge on to others, Dr. Rosen says.
“In so doing, we get involved in larger quality initiatives and systemwide changes that can go top-down,” he adds, “but from our perspective, it starts with the individual practitioner. And I think SHM has always advocated and preached the importance of the individual hospitalist doing the best possible job for your patient, and the group, and the institution.”
Shared Excellence
What’s best for individual institutions moving forward is what worries SHM immediate past president Shaun Frost, MD, SFHM. He fears CMS’ Value-Based Payment Modifier (VBPM) program could have the unintended consequence of spurring some hospitals to hang on to innovative ideas in order to keep a competitive business advantage.
In health care, where quality and affordability have long been viewed as valuable for nonmonetary reasons, “the medical profession willingly shares new information” to improve patient care, Dr. Frost said in his farewell speech. But he is concerned that commodification—imbuing monetary value into something that previously had none—could change that dynamic, a situation he says is “ethically not acceptable.”
“When somebody builds a better mousetrap, it should be freely shared so that all patients have the opportunity to benefit,” Dr. Frost said. “The pursuit of economic competitive advantage should not prevent us from collaborating and sharing new ideas that hopefully make the health system better.”
Kendall Rogers, MD, FACP, SFHM, chief of the division of hospital medicine at the University of New Mexico Health Sciences Center in Albuquerque, N.M., says part of that improvement in quality and patient safety will come via hospitalists pushing for improvements to health information technology (HIT), particularly to maximize computerized physician order entry (CPOE) and order sets. He empathizes with those who complain about the operability of existing systems but urges physicians to stop complaining and take action.
“We need to stop accepting what our existing limitations are, and we need to be the innovators,” he says. “Many of us aren’t even thinking about, ‘What are the products we need?’ We’re just reacting to the products we currently have and stating how they don’t meet our needs.”
He suggests people communally report safety or troubleshooting issues, in part via Hospital Medicine Exchange (HMX), an online community SHM launched last year to discuss HM issues (www.hmxchange.org). He also wants hospitalists to push HIT vendors to provide improved functionality, and for institutions to provide necessary training.
“We just need to be vocal,” says Dr. Rogers, chair of SHM’s IT Executive Committee. “I do believe this is all leading us to a good place, but there’s a dip down before we have a swing up.”
Frustration Surge
In the long run, hospitalist Anuj Mehta, MD, medical director of the adult hospitalist program at Nyack Hospital in New York, agrees with Dr. Rogers. But as a provider seeing patients day after day, he says it’s often easier to not engage HIT than it is to slog through it.
“We try to work around the system, and sometimes it’s a much longer workaround,” he says. “So what happens is loss of productivity, greater length of stay, poor patient satisfaction, more screen time, and less bedside time.”
Dr. Mehta says frustration is building as society—outside of medicine—moves rapidly through such technology as smartphones, tablets, and other intuitive devices that make actions easier. He notes that his toddler daughter could learn how to navigate an iPad in a fraction of the time it takes him to complete an HIT training course.
“You cannot have physicians going through learning for four hours, learning a system to do step one before step two before step three,” he laments. “It should flow naturally. I don’t think the IT people have realized that as of yet.”
Richard Quinn is a freelance writer in New Jersey.
Like a grinning child at a carnival, Iqbal M. Binoj, MD, steps right up and gives it a try—except instead of tossing rings, he’s gripping an intraosseous infusion drill.
A tutor shows him how the device, which looks remarkably like a glue gun, inserts into the bones of the shoulder or knee and drills down until it hits the marrow. He is guided on using a steady speed to maintain the integrity of the cavity. He’s also taught about the maneuver’s low complication rates and ability to expedite workups.
“I’ve seen it used before, but I never did it,” says Dr. Binoj, a hospitalist with Cogent HMG at Genesis Medical Center in Davenport, Iowa.
Well, he never did it before a hands-on pre-course at HM13 that focused on improving hospitalists’ proficiency at such procedures as lumbar punctures and ultrasound-guided vascular access. Quality improvement (QI) is always a focus of SHM’s annual meeting, but sometimes the science of improving care is viewed from up on high.
Not everything needs to be a national imitative, an institution-wide project, or even a unit-based intervention. Sometimes, it’s as simple as teaching a room full of hospitalists how to use an intraosseous infusion drill, says Michelle Fox, RN, BSN, senior director of clinical affairs with Vidacare, which manufactures the drill used in the demonstration.
“Hospitalists have an increasing role in doing these procedures, not only in the environment they predominantly support but in other areas of the hospital,” Fox says, adding that “the primary goal of this course is to give them the opportunity to perfect those skills.”
Hospitalist Bradley Rosen, MD, MBA, FHM, medical of the inpatient specialty program at Cedars-Sinai in Los Angeles, says the point of hands-on demonstrations is to translate QI to the bedside. Take ultrasound devices, he says. In the past few years, the technology has become less expensive, better in resolution, more common, and more portable. Hospitalists must ensure hands-on training that keeps pace with that technology.
“We actually want people to get gloves on, hands on, learn where they may have challenges in terms of their own dexterity or workflow, which hand is dominant, and how to visualize on the ultrasound machine a three-dimensional structure in 2D,” he says. “We don’t want people watching from the sidelines. ... We try to get people in it and engaged.”
And once hospitalists master procedures or diagnostic maneuvers, they invariably are sought out by other physicians to pass that knowledge on to others, Dr. Rosen says.
“In so doing, we get involved in larger quality initiatives and systemwide changes that can go top-down,” he adds, “but from our perspective, it starts with the individual practitioner. And I think SHM has always advocated and preached the importance of the individual hospitalist doing the best possible job for your patient, and the group, and the institution.”
Shared Excellence
What’s best for individual institutions moving forward is what worries SHM immediate past president Shaun Frost, MD, SFHM. He fears CMS’ Value-Based Payment Modifier (VBPM) program could have the unintended consequence of spurring some hospitals to hang on to innovative ideas in order to keep a competitive business advantage.
In health care, where quality and affordability have long been viewed as valuable for nonmonetary reasons, “the medical profession willingly shares new information” to improve patient care, Dr. Frost said in his farewell speech. But he is concerned that commodification—imbuing monetary value into something that previously had none—could change that dynamic, a situation he says is “ethically not acceptable.”
“When somebody builds a better mousetrap, it should be freely shared so that all patients have the opportunity to benefit,” Dr. Frost said. “The pursuit of economic competitive advantage should not prevent us from collaborating and sharing new ideas that hopefully make the health system better.”
Kendall Rogers, MD, FACP, SFHM, chief of the division of hospital medicine at the University of New Mexico Health Sciences Center in Albuquerque, N.M., says part of that improvement in quality and patient safety will come via hospitalists pushing for improvements to health information technology (HIT), particularly to maximize computerized physician order entry (CPOE) and order sets. He empathizes with those who complain about the operability of existing systems but urges physicians to stop complaining and take action.
“We need to stop accepting what our existing limitations are, and we need to be the innovators,” he says. “Many of us aren’t even thinking about, ‘What are the products we need?’ We’re just reacting to the products we currently have and stating how they don’t meet our needs.”
He suggests people communally report safety or troubleshooting issues, in part via Hospital Medicine Exchange (HMX), an online community SHM launched last year to discuss HM issues (www.hmxchange.org). He also wants hospitalists to push HIT vendors to provide improved functionality, and for institutions to provide necessary training.
“We just need to be vocal,” says Dr. Rogers, chair of SHM’s IT Executive Committee. “I do believe this is all leading us to a good place, but there’s a dip down before we have a swing up.”
Frustration Surge
In the long run, hospitalist Anuj Mehta, MD, medical director of the adult hospitalist program at Nyack Hospital in New York, agrees with Dr. Rogers. But as a provider seeing patients day after day, he says it’s often easier to not engage HIT than it is to slog through it.
“We try to work around the system, and sometimes it’s a much longer workaround,” he says. “So what happens is loss of productivity, greater length of stay, poor patient satisfaction, more screen time, and less bedside time.”
Dr. Mehta says frustration is building as society—outside of medicine—moves rapidly through such technology as smartphones, tablets, and other intuitive devices that make actions easier. He notes that his toddler daughter could learn how to navigate an iPad in a fraction of the time it takes him to complete an HIT training course.
“You cannot have physicians going through learning for four hours, learning a system to do step one before step two before step three,” he laments. “It should flow naturally. I don’t think the IT people have realized that as of yet.”
Richard Quinn is a freelance writer in New Jersey.
Like a grinning child at a carnival, Iqbal M. Binoj, MD, steps right up and gives it a try—except instead of tossing rings, he’s gripping an intraosseous infusion drill.
A tutor shows him how the device, which looks remarkably like a glue gun, inserts into the bones of the shoulder or knee and drills down until it hits the marrow. He is guided on using a steady speed to maintain the integrity of the cavity. He’s also taught about the maneuver’s low complication rates and ability to expedite workups.
“I’ve seen it used before, but I never did it,” says Dr. Binoj, a hospitalist with Cogent HMG at Genesis Medical Center in Davenport, Iowa.
Well, he never did it before a hands-on pre-course at HM13 that focused on improving hospitalists’ proficiency at such procedures as lumbar punctures and ultrasound-guided vascular access. Quality improvement (QI) is always a focus of SHM’s annual meeting, but sometimes the science of improving care is viewed from up on high.
Not everything needs to be a national imitative, an institution-wide project, or even a unit-based intervention. Sometimes, it’s as simple as teaching a room full of hospitalists how to use an intraosseous infusion drill, says Michelle Fox, RN, BSN, senior director of clinical affairs with Vidacare, which manufactures the drill used in the demonstration.
“Hospitalists have an increasing role in doing these procedures, not only in the environment they predominantly support but in other areas of the hospital,” Fox says, adding that “the primary goal of this course is to give them the opportunity to perfect those skills.”
Hospitalist Bradley Rosen, MD, MBA, FHM, medical of the inpatient specialty program at Cedars-Sinai in Los Angeles, says the point of hands-on demonstrations is to translate QI to the bedside. Take ultrasound devices, he says. In the past few years, the technology has become less expensive, better in resolution, more common, and more portable. Hospitalists must ensure hands-on training that keeps pace with that technology.
“We actually want people to get gloves on, hands on, learn where they may have challenges in terms of their own dexterity or workflow, which hand is dominant, and how to visualize on the ultrasound machine a three-dimensional structure in 2D,” he says. “We don’t want people watching from the sidelines. ... We try to get people in it and engaged.”
And once hospitalists master procedures or diagnostic maneuvers, they invariably are sought out by other physicians to pass that knowledge on to others, Dr. Rosen says.
“In so doing, we get involved in larger quality initiatives and systemwide changes that can go top-down,” he adds, “but from our perspective, it starts with the individual practitioner. And I think SHM has always advocated and preached the importance of the individual hospitalist doing the best possible job for your patient, and the group, and the institution.”
Shared Excellence
What’s best for individual institutions moving forward is what worries SHM immediate past president Shaun Frost, MD, SFHM. He fears CMS’ Value-Based Payment Modifier (VBPM) program could have the unintended consequence of spurring some hospitals to hang on to innovative ideas in order to keep a competitive business advantage.
In health care, where quality and affordability have long been viewed as valuable for nonmonetary reasons, “the medical profession willingly shares new information” to improve patient care, Dr. Frost said in his farewell speech. But he is concerned that commodification—imbuing monetary value into something that previously had none—could change that dynamic, a situation he says is “ethically not acceptable.”
“When somebody builds a better mousetrap, it should be freely shared so that all patients have the opportunity to benefit,” Dr. Frost said. “The pursuit of economic competitive advantage should not prevent us from collaborating and sharing new ideas that hopefully make the health system better.”
Kendall Rogers, MD, FACP, SFHM, chief of the division of hospital medicine at the University of New Mexico Health Sciences Center in Albuquerque, N.M., says part of that improvement in quality and patient safety will come via hospitalists pushing for improvements to health information technology (HIT), particularly to maximize computerized physician order entry (CPOE) and order sets. He empathizes with those who complain about the operability of existing systems but urges physicians to stop complaining and take action.
“We need to stop accepting what our existing limitations are, and we need to be the innovators,” he says. “Many of us aren’t even thinking about, ‘What are the products we need?’ We’re just reacting to the products we currently have and stating how they don’t meet our needs.”
He suggests people communally report safety or troubleshooting issues, in part via Hospital Medicine Exchange (HMX), an online community SHM launched last year to discuss HM issues (www.hmxchange.org). He also wants hospitalists to push HIT vendors to provide improved functionality, and for institutions to provide necessary training.
“We just need to be vocal,” says Dr. Rogers, chair of SHM’s IT Executive Committee. “I do believe this is all leading us to a good place, but there’s a dip down before we have a swing up.”
Frustration Surge
In the long run, hospitalist Anuj Mehta, MD, medical director of the adult hospitalist program at Nyack Hospital in New York, agrees with Dr. Rogers. But as a provider seeing patients day after day, he says it’s often easier to not engage HIT than it is to slog through it.
“We try to work around the system, and sometimes it’s a much longer workaround,” he says. “So what happens is loss of productivity, greater length of stay, poor patient satisfaction, more screen time, and less bedside time.”
Dr. Mehta says frustration is building as society—outside of medicine—moves rapidly through such technology as smartphones, tablets, and other intuitive devices that make actions easier. He notes that his toddler daughter could learn how to navigate an iPad in a fraction of the time it takes him to complete an HIT training course.
“You cannot have physicians going through learning for four hours, learning a system to do step one before step two before step three,” he laments. “It should flow naturally. I don’t think the IT people have realized that as of yet.”
Richard Quinn is a freelance writer in New Jersey.
Fix for Sustainable Growth Rate Formula a Top Priority
The Improving Access to Medicare Coverage Act isn’t the only legislative proposal on hospitalists’ radar right now. Republican members of the U.S. House of Representatives recently revised their plan to replace the sustainable growth rate (SGR) formula used to determine physician payments. A bill has not been introduced, but Beltway buzz hints one could be forthcoming this year.
“Fixing the flawed SGR physician payment is a top priority for the Committees on Energy and Commerce and Ways and Means,” GOP legislators said in an open letter to the “provider community.”2
The letter requested feedback from physicians and other stakeholders by April 15.
Any proposed fix would look to end the series of time-stamped delays that continue to delay a pending 27% cut to physician payments under Medicare. The latest delay was approved earlier this year, meaning that the new deadline for the SGR cut to be implemented is Dec. 31.
The SGR formula was created in 1997, but the pending cuts tied to the federal sequester were included in the Budget Control Act of 2011. At the time, the massive reduction to Medicare payments was tied to political brinksmanship over the country’s debt ceiling. But the cut also was considered a Draconian penalty that was never likely to actually happen.
Richard Quinn is a freelance writer in New Jersey.
References
- SHM. Letter to Congress members. SHM website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Letters_to_Congress_and_Regulatory_Agencies&Template=/CM/ContentDisplay.cfm&ContentID=33169. Accessed May 3, 2013.
- U.S. House of Representatives' Committee on Ways and Means, Energy and Commerce Committee, Health Subcommittee. Second draft of sustainable growth rate (SGR) repeal and reform proposal—request for feedback letter. U.S. House of Representatives' Committee on Ways and Means website. Available at: http://waysandmeans.house.gov/uploadedfiles/sgr_joint_release_document.pdf. Accessed May 3, 2013.
The Improving Access to Medicare Coverage Act isn’t the only legislative proposal on hospitalists’ radar right now. Republican members of the U.S. House of Representatives recently revised their plan to replace the sustainable growth rate (SGR) formula used to determine physician payments. A bill has not been introduced, but Beltway buzz hints one could be forthcoming this year.
“Fixing the flawed SGR physician payment is a top priority for the Committees on Energy and Commerce and Ways and Means,” GOP legislators said in an open letter to the “provider community.”2
The letter requested feedback from physicians and other stakeholders by April 15.
Any proposed fix would look to end the series of time-stamped delays that continue to delay a pending 27% cut to physician payments under Medicare. The latest delay was approved earlier this year, meaning that the new deadline for the SGR cut to be implemented is Dec. 31.
The SGR formula was created in 1997, but the pending cuts tied to the federal sequester were included in the Budget Control Act of 2011. At the time, the massive reduction to Medicare payments was tied to political brinksmanship over the country’s debt ceiling. But the cut also was considered a Draconian penalty that was never likely to actually happen.
Richard Quinn is a freelance writer in New Jersey.
References
- SHM. Letter to Congress members. SHM website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Letters_to_Congress_and_Regulatory_Agencies&Template=/CM/ContentDisplay.cfm&ContentID=33169. Accessed May 3, 2013.
- U.S. House of Representatives' Committee on Ways and Means, Energy and Commerce Committee, Health Subcommittee. Second draft of sustainable growth rate (SGR) repeal and reform proposal—request for feedback letter. U.S. House of Representatives' Committee on Ways and Means website. Available at: http://waysandmeans.house.gov/uploadedfiles/sgr_joint_release_document.pdf. Accessed May 3, 2013.
The Improving Access to Medicare Coverage Act isn’t the only legislative proposal on hospitalists’ radar right now. Republican members of the U.S. House of Representatives recently revised their plan to replace the sustainable growth rate (SGR) formula used to determine physician payments. A bill has not been introduced, but Beltway buzz hints one could be forthcoming this year.
“Fixing the flawed SGR physician payment is a top priority for the Committees on Energy and Commerce and Ways and Means,” GOP legislators said in an open letter to the “provider community.”2
The letter requested feedback from physicians and other stakeholders by April 15.
Any proposed fix would look to end the series of time-stamped delays that continue to delay a pending 27% cut to physician payments under Medicare. The latest delay was approved earlier this year, meaning that the new deadline for the SGR cut to be implemented is Dec. 31.
The SGR formula was created in 1997, but the pending cuts tied to the federal sequester were included in the Budget Control Act of 2011. At the time, the massive reduction to Medicare payments was tied to political brinksmanship over the country’s debt ceiling. But the cut also was considered a Draconian penalty that was never likely to actually happen.
Richard Quinn is a freelance writer in New Jersey.
References
- SHM. Letter to Congress members. SHM website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=Letters_to_Congress_and_Regulatory_Agencies&Template=/CM/ContentDisplay.cfm&ContentID=33169. Accessed May 3, 2013.
- U.S. House of Representatives' Committee on Ways and Means, Energy and Commerce Committee, Health Subcommittee. Second draft of sustainable growth rate (SGR) repeal and reform proposal—request for feedback letter. U.S. House of Representatives' Committee on Ways and Means website. Available at: http://waysandmeans.house.gov/uploadedfiles/sgr_joint_release_document.pdf. Accessed May 3, 2013.
Hospitalists Urge Congress to Reconsider Medicare’s “Observation Status” Rules


—Karim Godamunne, MD, MBA, SFHM
Hospitalists are pushing hard for a change to a Medicare rule requiring beneficiaries to accumulate at least three consecutive days of inpatient treatment at a hospital (not counting day of discharge) before it will cover care in a skilled nursing facility (SNF).
The issue was one of the talking points during last month’s Hospitalists on the Hill, SHM’s annual daylong advocacy campaign that this year coincided with the annual meeting in the nation’s capital. The issue gained attention from hospitalists and others in recent years, in part because of penalties hospitals face for readmissions—and also in part because hospitalists increasingly are providing care at SNFs and other post-acute-care facilities.
The spotlight is brighter now because a group of legislators is trying to identify Medicare beneficiaries previously given “observation status” as inpatients. The Improving Access to Medicare Coverage Act (H.R. 1179 and S. 569) also would establish a 90-day appeal period for those who have been denied the benefit.
SHM senior vice president Joe Miller says hospitalists used HM13 and the Hospitalists on the Hill advocacy day to discuss the issues and the proposed legislation with members of Congress, their staffs, and federal officials. He urges members to continue lobbying for changes. Although the topic might not have the resonance and impact of a fix to the sustainable growth rate (SGR), Miller says, “anybody that deals with admitting or discharging a patient will recognize the importance of this issue.”
The issue, according to Toby Edelman, a senior policy attorney for the Center for Medicare Advocacy in Washington, D.C, is that Medicare mandates that its program enrollees have at least three days of inpatient treatment before it will pay for SNF care. Medicare also covers the costs of post-acute care in other settings but does not require three days of inpatient treatment before doing so. The construct can be confusing to patients who spend time in a hospital but don’t realize that some or all of their stay is spent in “observation status,” meaning none of that time counts toward Medicare’s three-day threshold for reimbursement.
“Most people can’t believe you could be in a hospital bed for a week and then be told as you leave, ‘By the way, bring your checkbook to the nursing home because you weren’t an inpatient here and so now Medicare won’t pay for your stay in the nursing home,’” Edelman says. “This has been an issue for us for quite a while because the consequence for beneficiaries of being in observation is that people have to pay out of pocket for their nursing home care, and that cost is typically hundreds of dollars a day.”
The particular dilemma for hospitalists is managing transitions of care. Hospitalist Karim Godamunne, MD, MBA, SFHM, chief medical officer of North Fulton Hospital in Roswell, Ga., says hospitalists don’t want financial burdens to dictate care decisions, but they are caught in the middle of decisions that could saddle patients with uncovered costs.
He also worries that the issue will only grow in coming years as baby boomers put more pressure on the health-care system. “We have an aging population,” he adds. “This is not going to go away.”
That is one reason SHM is supporting the Improving Access to Medicare Coverage Act. SHM supported the bill when it was first introduced in March and it has been rapidly gaining cosponsors in recent weeks. This uptick in Congressional interest may be partly a response to the efforts of hospitalists during their time on the Hill. SHM staff and hospitalists are continuing their push now as society officials say hospitalists, who often handle both discharges from the hospital and care provided at SNFs, are in a position to lead discussions on how to sensibly fix the problem.
To that end, a recent SHM letter to the bill’s sponsors casts the issue as one of fiscal responsibility.1 Medicare not covering beneficiaries’ observation days cost patients out-of-pocket money and could cost hospitals in the long run.
“Patients who are admitted with observation status often choose to return home rather than paying out of pocket for a SNF stay,” SHM’s letter reads. “The resultant lack of appropriate post-acute SNF care can result in additional problems such as dehydration, falls, and many other avoidable complications. These complications can not only lead to otherwise preventable readmissions but also increase costs to Medicare for the treatment of conditions that were not present at the time of the original hospital stay.”
Given the debate on observation, Miller says, adopting the bill into law should be a no-brainer. The biggest sticking point likely is the perceived added cost to Medicare. Still, to streamline care and remove an added hurdle to coordinated care, Dr. Godamunne believes the bill should be embraced. He also says that many private insurers look to Medicare decisions to determine their own coverage approaches.
Basically, if Medicare changes its rules, that will carry a lot of weight in the private insurance world.
“This creates a lot of situations for the provider and the family,” Dr. Godamunne says. “You have to make a difficult decision, to try to help the family. You’re trying to provide good care, but on the other hand, there are rules and regulations and bylaws you work under. They don’t align that well, in this case.”
Richard Quinn is a freelance writer in New Jersey.
References
—Karim Godamunne, MD, MBA, SFHM
Hospitalists are pushing hard for a change to a Medicare rule requiring beneficiaries to accumulate at least three consecutive days of inpatient treatment at a hospital (not counting day of discharge) before it will cover care in a skilled nursing facility (SNF).
The issue was one of the talking points during last month’s Hospitalists on the Hill, SHM’s annual daylong advocacy campaign that this year coincided with the annual meeting in the nation’s capital. The issue gained attention from hospitalists and others in recent years, in part because of penalties hospitals face for readmissions—and also in part because hospitalists increasingly are providing care at SNFs and other post-acute-care facilities.
The spotlight is brighter now because a group of legislators is trying to identify Medicare beneficiaries previously given “observation status” as inpatients. The Improving Access to Medicare Coverage Act (H.R. 1179 and S. 569) also would establish a 90-day appeal period for those who have been denied the benefit.
SHM senior vice president Joe Miller says hospitalists used HM13 and the Hospitalists on the Hill advocacy day to discuss the issues and the proposed legislation with members of Congress, their staffs, and federal officials. He urges members to continue lobbying for changes. Although the topic might not have the resonance and impact of a fix to the sustainable growth rate (SGR), Miller says, “anybody that deals with admitting or discharging a patient will recognize the importance of this issue.”
The issue, according to Toby Edelman, a senior policy attorney for the Center for Medicare Advocacy in Washington, D.C, is that Medicare mandates that its program enrollees have at least three days of inpatient treatment before it will pay for SNF care. Medicare also covers the costs of post-acute care in other settings but does not require three days of inpatient treatment before doing so. The construct can be confusing to patients who spend time in a hospital but don’t realize that some or all of their stay is spent in “observation status,” meaning none of that time counts toward Medicare’s three-day threshold for reimbursement.
“Most people can’t believe you could be in a hospital bed for a week and then be told as you leave, ‘By the way, bring your checkbook to the nursing home because you weren’t an inpatient here and so now Medicare won’t pay for your stay in the nursing home,’” Edelman says. “This has been an issue for us for quite a while because the consequence for beneficiaries of being in observation is that people have to pay out of pocket for their nursing home care, and that cost is typically hundreds of dollars a day.”
The particular dilemma for hospitalists is managing transitions of care. Hospitalist Karim Godamunne, MD, MBA, SFHM, chief medical officer of North Fulton Hospital in Roswell, Ga., says hospitalists don’t want financial burdens to dictate care decisions, but they are caught in the middle of decisions that could saddle patients with uncovered costs.
He also worries that the issue will only grow in coming years as baby boomers put more pressure on the health-care system. “We have an aging population,” he adds. “This is not going to go away.”
That is one reason SHM is supporting the Improving Access to Medicare Coverage Act. SHM supported the bill when it was first introduced in March and it has been rapidly gaining cosponsors in recent weeks. This uptick in Congressional interest may be partly a response to the efforts of hospitalists during their time on the Hill. SHM staff and hospitalists are continuing their push now as society officials say hospitalists, who often handle both discharges from the hospital and care provided at SNFs, are in a position to lead discussions on how to sensibly fix the problem.
To that end, a recent SHM letter to the bill’s sponsors casts the issue as one of fiscal responsibility.1 Medicare not covering beneficiaries’ observation days cost patients out-of-pocket money and could cost hospitals in the long run.
“Patients who are admitted with observation status often choose to return home rather than paying out of pocket for a SNF stay,” SHM’s letter reads. “The resultant lack of appropriate post-acute SNF care can result in additional problems such as dehydration, falls, and many other avoidable complications. These complications can not only lead to otherwise preventable readmissions but also increase costs to Medicare for the treatment of conditions that were not present at the time of the original hospital stay.”
Given the debate on observation, Miller says, adopting the bill into law should be a no-brainer. The biggest sticking point likely is the perceived added cost to Medicare. Still, to streamline care and remove an added hurdle to coordinated care, Dr. Godamunne believes the bill should be embraced. He also says that many private insurers look to Medicare decisions to determine their own coverage approaches.
Basically, if Medicare changes its rules, that will carry a lot of weight in the private insurance world.
“This creates a lot of situations for the provider and the family,” Dr. Godamunne says. “You have to make a difficult decision, to try to help the family. You’re trying to provide good care, but on the other hand, there are rules and regulations and bylaws you work under. They don’t align that well, in this case.”
Richard Quinn is a freelance writer in New Jersey.
References
—Karim Godamunne, MD, MBA, SFHM
Hospitalists are pushing hard for a change to a Medicare rule requiring beneficiaries to accumulate at least three consecutive days of inpatient treatment at a hospital (not counting day of discharge) before it will cover care in a skilled nursing facility (SNF).
The issue was one of the talking points during last month’s Hospitalists on the Hill, SHM’s annual daylong advocacy campaign that this year coincided with the annual meeting in the nation’s capital. The issue gained attention from hospitalists and others in recent years, in part because of penalties hospitals face for readmissions—and also in part because hospitalists increasingly are providing care at SNFs and other post-acute-care facilities.
The spotlight is brighter now because a group of legislators is trying to identify Medicare beneficiaries previously given “observation status” as inpatients. The Improving Access to Medicare Coverage Act (H.R. 1179 and S. 569) also would establish a 90-day appeal period for those who have been denied the benefit.
SHM senior vice president Joe Miller says hospitalists used HM13 and the Hospitalists on the Hill advocacy day to discuss the issues and the proposed legislation with members of Congress, their staffs, and federal officials. He urges members to continue lobbying for changes. Although the topic might not have the resonance and impact of a fix to the sustainable growth rate (SGR), Miller says, “anybody that deals with admitting or discharging a patient will recognize the importance of this issue.”
The issue, according to Toby Edelman, a senior policy attorney for the Center for Medicare Advocacy in Washington, D.C, is that Medicare mandates that its program enrollees have at least three days of inpatient treatment before it will pay for SNF care. Medicare also covers the costs of post-acute care in other settings but does not require three days of inpatient treatment before doing so. The construct can be confusing to patients who spend time in a hospital but don’t realize that some or all of their stay is spent in “observation status,” meaning none of that time counts toward Medicare’s three-day threshold for reimbursement.
“Most people can’t believe you could be in a hospital bed for a week and then be told as you leave, ‘By the way, bring your checkbook to the nursing home because you weren’t an inpatient here and so now Medicare won’t pay for your stay in the nursing home,’” Edelman says. “This has been an issue for us for quite a while because the consequence for beneficiaries of being in observation is that people have to pay out of pocket for their nursing home care, and that cost is typically hundreds of dollars a day.”
The particular dilemma for hospitalists is managing transitions of care. Hospitalist Karim Godamunne, MD, MBA, SFHM, chief medical officer of North Fulton Hospital in Roswell, Ga., says hospitalists don’t want financial burdens to dictate care decisions, but they are caught in the middle of decisions that could saddle patients with uncovered costs.
He also worries that the issue will only grow in coming years as baby boomers put more pressure on the health-care system. “We have an aging population,” he adds. “This is not going to go away.”
That is one reason SHM is supporting the Improving Access to Medicare Coverage Act. SHM supported the bill when it was first introduced in March and it has been rapidly gaining cosponsors in recent weeks. This uptick in Congressional interest may be partly a response to the efforts of hospitalists during their time on the Hill. SHM staff and hospitalists are continuing their push now as society officials say hospitalists, who often handle both discharges from the hospital and care provided at SNFs, are in a position to lead discussions on how to sensibly fix the problem.
To that end, a recent SHM letter to the bill’s sponsors casts the issue as one of fiscal responsibility.1 Medicare not covering beneficiaries’ observation days cost patients out-of-pocket money and could cost hospitals in the long run.
“Patients who are admitted with observation status often choose to return home rather than paying out of pocket for a SNF stay,” SHM’s letter reads. “The resultant lack of appropriate post-acute SNF care can result in additional problems such as dehydration, falls, and many other avoidable complications. These complications can not only lead to otherwise preventable readmissions but also increase costs to Medicare for the treatment of conditions that were not present at the time of the original hospital stay.”
Given the debate on observation, Miller says, adopting the bill into law should be a no-brainer. The biggest sticking point likely is the perceived added cost to Medicare. Still, to streamline care and remove an added hurdle to coordinated care, Dr. Godamunne believes the bill should be embraced. He also says that many private insurers look to Medicare decisions to determine their own coverage approaches.
Basically, if Medicare changes its rules, that will carry a lot of weight in the private insurance world.
“This creates a lot of situations for the provider and the family,” Dr. Godamunne says. “You have to make a difficult decision, to try to help the family. You’re trying to provide good care, but on the other hand, there are rules and regulations and bylaws you work under. They don’t align that well, in this case.”
Richard Quinn is a freelance writer in New Jersey.
References
Pediatric Hospitalist Charts Decade-Long Journey in Health Care

Dear Mark,
I am pleased and excited that you are willing to abandon your plan for being a vagabond and will give serious consideration to joining the faculty of the Department of Pediatrics to become a core member of a new [general pediatric inpatient] program that I believe has exciting potential.
So reads the first line of my very first job offer letter. Obviously, my chairman had a sense of humor. But he also was not off target, as before May 21 of my third year of residency, I had no meaningful work lined up. Dreams of locum tenens work in Hawaii or a California coastal town quickly disappeared as I received only offers for work in small-town Mississippi and Oklahoma. Eleven years later, I don’t think I could have planned a more fulfilling early career, particularly when the alternative might have been surfing on the Mississippi River.
I would like this opportunity, in my final column as The Hospitalist’s pediatric editor, to reflect on this odyssey from vagabond to hospitalist.
The Early Years
As a new attending, I was appropriately terrified of how much I didn’t know. I also had ambitious goals at first, wanting to emulate my two favorite role models from residency, Charles Ginsburg and Heinz Eichenwald. We might call them hospitalists now, but back then they were old-fashioned, generalist inpatient clinician-educators, even while chairing the department of pediatrics over their separate tenures. They were the smartest and wisest teachers that I have ever met. These early years were a pseudo-fellowship of sorts; under their tutelage, I soaked up more than I ever had during residency.
Despite all of this learning, I remained sheltered in my clinician-educator bubble. The path to excellence for me was defined through frequent trips to the library (where journals used to be stored) and trying to teach as well as my mentors did. I largely was ignorant of the national hospitalist movement, until the 2007 SHM annual meeting was held in my backyard in Dallas. Listening to Bob Wachter that year, and then Don Berwick the following year, I suddenly realized the tremendous and intertwined importance of the quality movement and hospitalists. We were going to fix medicine. OK, maybe not all of medicine, but it happened to be the perfect time for me to learn about our health-care crisis, quality, and the role of hospital medicine.
If my first five years were about clinical medicine, the next five years were all about lessons in leadership. I had a new role, directing 8 15 20 25 hospitalists—and now was accountable for the group’s results. I’ve often said that an explicit leadership role is like stepping behind a curtain, where your own previous n=1 perspective is now the challenge of herding a group of n=25. And let’s be clear that it’s one thing to manage the group and keep the ship afloat, but it’s entirely another thing to lead the group toward success.
A Path for Me
Although the cacophony of managing that many voices was deafening early on, I found solace in the lessons of quality improvement (QI), where no project lives without a team that is all going the same direction. Between the national opportunities for collaborative improvement and the day-to-day experiences within my group, I found two simple principles worked well: 1) engage the team and 2) deliver objective results.
And just as I had craved a clinical learning environment early on, I now found myself learning from local and national peers putting their leadership skills in action to produce quality outcomes. The beauty of collaborative teamwork is that it creates self-sustaining capacity for more positive results.
Looking forward, the opportunities seem limitless for pediatric hospital medicine. From the inherent fulfillment of our day-to-day bedside work to the explicit leadership that we offer the complex hospital system, our family of pediatric hospitalists has blazed career paths in all directions. We are program directors. We are directors of quality and safety. We are division directors and section chiefs. We are professors. We are fellowship-trained. We are CEOs, of entire hospitals and the CMO of CMS. There has never been a better time to be a pediatric hospitalist.
This rapid ascent has to be the fastest in the history of medicine and might surprise the unsuspecting, but these career paths really should have been expected. Residents and students still identify the most with their ward months—we always will be leaders in education. Hospitals and health-care systems recognize the value of hospitalists as systems improvers and will forever need enlightened physicians to guide safer, better care. But we also remain generalists, perched over the exact intersection of acute illness and health. From this vantage point, we have the perfect perspective from which to lead the transformation of our health-care system. I’m not sure there is a leadership position in health care that a hospitalist will not fill in the near future.
A New Frontier
With all of this opportunity before us, there exists an imperative for true leadership. And unlike all of our past requirements for achievement, relying on our quantitative abilities will no longer be enough. Rather, we will need to focus on the qualitative “soft” skills, whether you call this emotional intelligence, interpersonal communication, or behavioral economics. The creation of value-based, care-delivery systems requires high-functioning units. We will need to design and lead teams from the bedside to the boardroom.
In the coming years, this leadership imperative will only intensify, as we all will be pressured to do more with less. We will be asked to improve quality and decrease costs. We will need to broaden our focus to health in addition to acute illness. Doing more with less will require courage and leadership. If you look at our growth curve to date, we have an abundance of both.
Dr. Shen is medical director of hospital medicine at Dell Children's Medical Center in Austin, Texas. He served as The Hospitalist's pediatric editor since 2010 and this marks his last column in his role as editor. In his newfound spare time, he looks forward to defining value in health care.
Dear Mark,
I am pleased and excited that you are willing to abandon your plan for being a vagabond and will give serious consideration to joining the faculty of the Department of Pediatrics to become a core member of a new [general pediatric inpatient] program that I believe has exciting potential.
So reads the first line of my very first job offer letter. Obviously, my chairman had a sense of humor. But he also was not off target, as before May 21 of my third year of residency, I had no meaningful work lined up. Dreams of locum tenens work in Hawaii or a California coastal town quickly disappeared as I received only offers for work in small-town Mississippi and Oklahoma. Eleven years later, I don’t think I could have planned a more fulfilling early career, particularly when the alternative might have been surfing on the Mississippi River.
I would like this opportunity, in my final column as The Hospitalist’s pediatric editor, to reflect on this odyssey from vagabond to hospitalist.
The Early Years
As a new attending, I was appropriately terrified of how much I didn’t know. I also had ambitious goals at first, wanting to emulate my two favorite role models from residency, Charles Ginsburg and Heinz Eichenwald. We might call them hospitalists now, but back then they were old-fashioned, generalist inpatient clinician-educators, even while chairing the department of pediatrics over their separate tenures. They were the smartest and wisest teachers that I have ever met. These early years were a pseudo-fellowship of sorts; under their tutelage, I soaked up more than I ever had during residency.
Despite all of this learning, I remained sheltered in my clinician-educator bubble. The path to excellence for me was defined through frequent trips to the library (where journals used to be stored) and trying to teach as well as my mentors did. I largely was ignorant of the national hospitalist movement, until the 2007 SHM annual meeting was held in my backyard in Dallas. Listening to Bob Wachter that year, and then Don Berwick the following year, I suddenly realized the tremendous and intertwined importance of the quality movement and hospitalists. We were going to fix medicine. OK, maybe not all of medicine, but it happened to be the perfect time for me to learn about our health-care crisis, quality, and the role of hospital medicine.
If my first five years were about clinical medicine, the next five years were all about lessons in leadership. I had a new role, directing 8 15 20 25 hospitalists—and now was accountable for the group’s results. I’ve often said that an explicit leadership role is like stepping behind a curtain, where your own previous n=1 perspective is now the challenge of herding a group of n=25. And let’s be clear that it’s one thing to manage the group and keep the ship afloat, but it’s entirely another thing to lead the group toward success.
A Path for Me
Although the cacophony of managing that many voices was deafening early on, I found solace in the lessons of quality improvement (QI), where no project lives without a team that is all going the same direction. Between the national opportunities for collaborative improvement and the day-to-day experiences within my group, I found two simple principles worked well: 1) engage the team and 2) deliver objective results.
And just as I had craved a clinical learning environment early on, I now found myself learning from local and national peers putting their leadership skills in action to produce quality outcomes. The beauty of collaborative teamwork is that it creates self-sustaining capacity for more positive results.
Looking forward, the opportunities seem limitless for pediatric hospital medicine. From the inherent fulfillment of our day-to-day bedside work to the explicit leadership that we offer the complex hospital system, our family of pediatric hospitalists has blazed career paths in all directions. We are program directors. We are directors of quality and safety. We are division directors and section chiefs. We are professors. We are fellowship-trained. We are CEOs, of entire hospitals and the CMO of CMS. There has never been a better time to be a pediatric hospitalist.
This rapid ascent has to be the fastest in the history of medicine and might surprise the unsuspecting, but these career paths really should have been expected. Residents and students still identify the most with their ward months—we always will be leaders in education. Hospitals and health-care systems recognize the value of hospitalists as systems improvers and will forever need enlightened physicians to guide safer, better care. But we also remain generalists, perched over the exact intersection of acute illness and health. From this vantage point, we have the perfect perspective from which to lead the transformation of our health-care system. I’m not sure there is a leadership position in health care that a hospitalist will not fill in the near future.
A New Frontier
With all of this opportunity before us, there exists an imperative for true leadership. And unlike all of our past requirements for achievement, relying on our quantitative abilities will no longer be enough. Rather, we will need to focus on the qualitative “soft” skills, whether you call this emotional intelligence, interpersonal communication, or behavioral economics. The creation of value-based, care-delivery systems requires high-functioning units. We will need to design and lead teams from the bedside to the boardroom.
In the coming years, this leadership imperative will only intensify, as we all will be pressured to do more with less. We will be asked to improve quality and decrease costs. We will need to broaden our focus to health in addition to acute illness. Doing more with less will require courage and leadership. If you look at our growth curve to date, we have an abundance of both.
Dr. Shen is medical director of hospital medicine at Dell Children's Medical Center in Austin, Texas. He served as The Hospitalist's pediatric editor since 2010 and this marks his last column in his role as editor. In his newfound spare time, he looks forward to defining value in health care.
Dear Mark,
I am pleased and excited that you are willing to abandon your plan for being a vagabond and will give serious consideration to joining the faculty of the Department of Pediatrics to become a core member of a new [general pediatric inpatient] program that I believe has exciting potential.
So reads the first line of my very first job offer letter. Obviously, my chairman had a sense of humor. But he also was not off target, as before May 21 of my third year of residency, I had no meaningful work lined up. Dreams of locum tenens work in Hawaii or a California coastal town quickly disappeared as I received only offers for work in small-town Mississippi and Oklahoma. Eleven years later, I don’t think I could have planned a more fulfilling early career, particularly when the alternative might have been surfing on the Mississippi River.
I would like this opportunity, in my final column as The Hospitalist’s pediatric editor, to reflect on this odyssey from vagabond to hospitalist.
The Early Years
As a new attending, I was appropriately terrified of how much I didn’t know. I also had ambitious goals at first, wanting to emulate my two favorite role models from residency, Charles Ginsburg and Heinz Eichenwald. We might call them hospitalists now, but back then they were old-fashioned, generalist inpatient clinician-educators, even while chairing the department of pediatrics over their separate tenures. They were the smartest and wisest teachers that I have ever met. These early years were a pseudo-fellowship of sorts; under their tutelage, I soaked up more than I ever had during residency.
Despite all of this learning, I remained sheltered in my clinician-educator bubble. The path to excellence for me was defined through frequent trips to the library (where journals used to be stored) and trying to teach as well as my mentors did. I largely was ignorant of the national hospitalist movement, until the 2007 SHM annual meeting was held in my backyard in Dallas. Listening to Bob Wachter that year, and then Don Berwick the following year, I suddenly realized the tremendous and intertwined importance of the quality movement and hospitalists. We were going to fix medicine. OK, maybe not all of medicine, but it happened to be the perfect time for me to learn about our health-care crisis, quality, and the role of hospital medicine.
If my first five years were about clinical medicine, the next five years were all about lessons in leadership. I had a new role, directing 8 15 20 25 hospitalists—and now was accountable for the group’s results. I’ve often said that an explicit leadership role is like stepping behind a curtain, where your own previous n=1 perspective is now the challenge of herding a group of n=25. And let’s be clear that it’s one thing to manage the group and keep the ship afloat, but it’s entirely another thing to lead the group toward success.
A Path for Me
Although the cacophony of managing that many voices was deafening early on, I found solace in the lessons of quality improvement (QI), where no project lives without a team that is all going the same direction. Between the national opportunities for collaborative improvement and the day-to-day experiences within my group, I found two simple principles worked well: 1) engage the team and 2) deliver objective results.
And just as I had craved a clinical learning environment early on, I now found myself learning from local and national peers putting their leadership skills in action to produce quality outcomes. The beauty of collaborative teamwork is that it creates self-sustaining capacity for more positive results.
Looking forward, the opportunities seem limitless for pediatric hospital medicine. From the inherent fulfillment of our day-to-day bedside work to the explicit leadership that we offer the complex hospital system, our family of pediatric hospitalists has blazed career paths in all directions. We are program directors. We are directors of quality and safety. We are division directors and section chiefs. We are professors. We are fellowship-trained. We are CEOs, of entire hospitals and the CMO of CMS. There has never been a better time to be a pediatric hospitalist.
This rapid ascent has to be the fastest in the history of medicine and might surprise the unsuspecting, but these career paths really should have been expected. Residents and students still identify the most with their ward months—we always will be leaders in education. Hospitals and health-care systems recognize the value of hospitalists as systems improvers and will forever need enlightened physicians to guide safer, better care. But we also remain generalists, perched over the exact intersection of acute illness and health. From this vantage point, we have the perfect perspective from which to lead the transformation of our health-care system. I’m not sure there is a leadership position in health care that a hospitalist will not fill in the near future.
A New Frontier
With all of this opportunity before us, there exists an imperative for true leadership. And unlike all of our past requirements for achievement, relying on our quantitative abilities will no longer be enough. Rather, we will need to focus on the qualitative “soft” skills, whether you call this emotional intelligence, interpersonal communication, or behavioral economics. The creation of value-based, care-delivery systems requires high-functioning units. We will need to design and lead teams from the bedside to the boardroom.
In the coming years, this leadership imperative will only intensify, as we all will be pressured to do more with less. We will be asked to improve quality and decrease costs. We will need to broaden our focus to health in addition to acute illness. Doing more with less will require courage and leadership. If you look at our growth curve to date, we have an abundance of both.
Dr. Shen is medical director of hospital medicine at Dell Children's Medical Center in Austin, Texas. He served as The Hospitalist's pediatric editor since 2010 and this marks his last column in his role as editor. In his newfound spare time, he looks forward to defining value in health care.
Hospital Medicine Leaders Flock to HM13 for Answers, Encouragement

Ibe Mbanu, MD, MBA, MPH, became medical director of the adult hospitalist department at Bon Secours St. Mary’s Hospital in Richmond, Va., about six months ago. Since then, he’s been besieged by a torrent of reform-based challenges he says make his job exponentially more difficult than that of medical directors just a few years ago.
Accountable-care organizations (ACOs), value-based purchasing, and discussions about bundled payments for episodic care are changing rapidly, and as a new administrator in a group with 24 hospitalists and three nonphysician providers (NPPs), he felt he needed to attend his first SHM annual meeting to keep up.
“The landscape in health care is rapidly evolving, at a frantic pace,” Dr. Mbanu says. “I essentially came here to just try to get a condensed source of information on how to manage the changes that are coming through the pipeline, and how to effectively run my department.”
Managing a practice is a challenge, and many of the more than 2,700 attendees at HM13 said the four-day confab’s focus on the topic was a major draw. From a rebooted continuing medical education (CME) pre-course appropriately named “What Keeps You Awake at Night? Hot Topics in Hospitalist Practice Management” to dozens of breakout sessions on the topic, it’s clear that successful practice management is a concern for many hospitalists.
“Before, the drivers were pretty clear,” Dr. Mbanu says. “Volume, productivity. Now we’re switching more toward a business model that’s changing from volume to value. Trying to adapt to that change is pretty challenging.
“Now it’s critical to really understand the environment.”
Comanagement Conundrum
One particularly hot topic this year was the trend of hospitalists taking on more comanagement responsibilities for patients previously managed by other specialties, including neurology, surgery, and others. Frank Volpicelli, MD, a first-year hospitalist and instructor at New York University (NYU) Langone Medical Center in New York, was one of three members of his HM group that attended the “Perioperative Medicine: Medical Consultation and Co-Management” pre-course. This summer, his group is going to establish a presence in the preoperative clinic.
“We hope very strongly that we can prevent some complications, identify patients that we should be following when they come into the hospital, and help the surgeons out,” he says. “No. 1, keep them in the [operating room] more, and No. 2, get in front of some of the complications that they are less comfortable managing.”
Ralph Velazquez, MD, senior vice president of care management for OSF Healthcare System in Peoria, Ill., isn’t so sure comanagement of more and more patients is the best practice-management model moving forward. For example, as physician compensation is tied more to how much their care costs to deliver, a hospitalist comanaging a surgical patient’s elective knee replacement could be financially penalized for the cost of that person’s stay, despite having nothing to do with the most expensive portion of the bill.
“You have a financial model that says do more billings, but as you start developing analytics … you may see there is no difference between the model that’s doing more billing, in terms of improving quality, and the one that is doing less,” Dr. Velazquez says. “So if you’re getting the same amount of quality, and the only thing you’re doing is generating more cost by doing more billing, you need to reevaluate your strategy.”
He believes some patients benefit from comanagement, but HM groups have to be diligent in seeking them out.
“We look for simple solutions and one-size-fits-all,” he adds. “Comanagement in complex patients—definitely there’s a need for that. Comanagement in noncomplex patients, elective patients—there’s no need for that. It’s just additional cost. I don’t think it’s going to produce any value.”
Startup Academy
John Colombo, MD, FACP, a 30-year veteran of internal medicine who moved to HM a few years ago when one of the hospitals he worked at asked him to launch a hospitalist group, thinks bundled payments might alleviate that value conundrum. Then again, he’s not quite sure. That’s why attended his first annual meeting.
“I found it difficult starting a new program from scratch,” says Dr. Colombo, of Crozer Keystone Health System in Drexel Hill, Pa. “Even with the materials available, there’s not a lot of ‘how to do it’ out there. There’s no ‘Starting Hospitals for Dummies’ book.”
Dr. Colombo spent much of his meeting focused on recruiting, compensation, bonus structures, and scheduling concerns. He said all are important in the hospital-heavy metropolitan Philadelphia region where he works. Plus, with departures and retirements at other programs in his health system, Dr. Colombo went from no HM experience three years ago to being in charge of four HM programs.
“The biggest thing is, I wanted to make sure I hadn’t stepped in something that I shouldn’t have already,” he adds. “There’s many different ways to do things. So I’ve learned a few different ways. I found value.”
Demonstrate Value
Another way to discover value in running a practice is looking at the business side of the house, says Denice Cora-Bramble, MD, MBA, chief medical officer and executive vice president of Ambulatory & Community Health Services at the Children’s National Medical Center in Washington, D.C.
Dr. Bramble says many hospitalists need to understand that while clinical care is what brought them to medicine, their future paychecks depend on recognizing how to provide that care in a way that demonstrates business value.
“When you finish residency, if you have not intentionally sought out those courses or those seminars, you need to recognize that as a blind spot,” she says. “You need to fill that toolkit as it relates to the business side of medicine.
“You don’t necessarily have to know all the answers, but you need to know the right questions to ask,” she says.
Dr. Bramble adds that hospitalist leaders should take advantage of certificate programs, leadership courses, basic budgeting classes, or anything that gives them added education about the economics of healthcare.
“It all comes down to demonstrating your outcomes, demonstrating the value that you bring to that institution,” she says. “And with health-care reform, I think hospitalists are uniquely positioned to be able to partner with other areas of the hospital to look at this value-based approach.”
Gary Gammon, MD, FHM, the newly named medical director of the Hospitalist Service at FirstHealth Moore Regional Hospital in Pinehurst, N.C., is doing his part to demonstrate value to his administrators. While his group does multidisciplinary rounds on patients, one of his questions for the pre-course faculty was to make sure that system of rounding is an evidence-based practice. He’s also looking for ways to establish more hegemony to his practice to ensure the rounds are effective, regardless of which physicians and others are participating.
The feedback he received was that most people view multidisciplinary rounds as a best practice. Now, Dr. Gammon can feel more authoritative that he and his 32 hospitalists and 12 extenders are practicing HM the way it should be practiced.
“I wanted to hear just what I heard, which is the leaders in the community feel that it’s helping, feel that it’s the right thing to do, feel that there’s objective data,” he says. “This is the stuff that makes me say, ‘OK, I’ve got the same problems everybody else has.’”
Richard Quinn is a freelance writer in New Jersey.
Ibe Mbanu, MD, MBA, MPH, became medical director of the adult hospitalist department at Bon Secours St. Mary’s Hospital in Richmond, Va., about six months ago. Since then, he’s been besieged by a torrent of reform-based challenges he says make his job exponentially more difficult than that of medical directors just a few years ago.
Accountable-care organizations (ACOs), value-based purchasing, and discussions about bundled payments for episodic care are changing rapidly, and as a new administrator in a group with 24 hospitalists and three nonphysician providers (NPPs), he felt he needed to attend his first SHM annual meeting to keep up.
“The landscape in health care is rapidly evolving, at a frantic pace,” Dr. Mbanu says. “I essentially came here to just try to get a condensed source of information on how to manage the changes that are coming through the pipeline, and how to effectively run my department.”
Managing a practice is a challenge, and many of the more than 2,700 attendees at HM13 said the four-day confab’s focus on the topic was a major draw. From a rebooted continuing medical education (CME) pre-course appropriately named “What Keeps You Awake at Night? Hot Topics in Hospitalist Practice Management” to dozens of breakout sessions on the topic, it’s clear that successful practice management is a concern for many hospitalists.
“Before, the drivers were pretty clear,” Dr. Mbanu says. “Volume, productivity. Now we’re switching more toward a business model that’s changing from volume to value. Trying to adapt to that change is pretty challenging.
“Now it’s critical to really understand the environment.”
Comanagement Conundrum
One particularly hot topic this year was the trend of hospitalists taking on more comanagement responsibilities for patients previously managed by other specialties, including neurology, surgery, and others. Frank Volpicelli, MD, a first-year hospitalist and instructor at New York University (NYU) Langone Medical Center in New York, was one of three members of his HM group that attended the “Perioperative Medicine: Medical Consultation and Co-Management” pre-course. This summer, his group is going to establish a presence in the preoperative clinic.
“We hope very strongly that we can prevent some complications, identify patients that we should be following when they come into the hospital, and help the surgeons out,” he says. “No. 1, keep them in the [operating room] more, and No. 2, get in front of some of the complications that they are less comfortable managing.”
Ralph Velazquez, MD, senior vice president of care management for OSF Healthcare System in Peoria, Ill., isn’t so sure comanagement of more and more patients is the best practice-management model moving forward. For example, as physician compensation is tied more to how much their care costs to deliver, a hospitalist comanaging a surgical patient’s elective knee replacement could be financially penalized for the cost of that person’s stay, despite having nothing to do with the most expensive portion of the bill.
“You have a financial model that says do more billings, but as you start developing analytics … you may see there is no difference between the model that’s doing more billing, in terms of improving quality, and the one that is doing less,” Dr. Velazquez says. “So if you’re getting the same amount of quality, and the only thing you’re doing is generating more cost by doing more billing, you need to reevaluate your strategy.”
He believes some patients benefit from comanagement, but HM groups have to be diligent in seeking them out.
“We look for simple solutions and one-size-fits-all,” he adds. “Comanagement in complex patients—definitely there’s a need for that. Comanagement in noncomplex patients, elective patients—there’s no need for that. It’s just additional cost. I don’t think it’s going to produce any value.”
Startup Academy
John Colombo, MD, FACP, a 30-year veteran of internal medicine who moved to HM a few years ago when one of the hospitals he worked at asked him to launch a hospitalist group, thinks bundled payments might alleviate that value conundrum. Then again, he’s not quite sure. That’s why attended his first annual meeting.
“I found it difficult starting a new program from scratch,” says Dr. Colombo, of Crozer Keystone Health System in Drexel Hill, Pa. “Even with the materials available, there’s not a lot of ‘how to do it’ out there. There’s no ‘Starting Hospitals for Dummies’ book.”
Dr. Colombo spent much of his meeting focused on recruiting, compensation, bonus structures, and scheduling concerns. He said all are important in the hospital-heavy metropolitan Philadelphia region where he works. Plus, with departures and retirements at other programs in his health system, Dr. Colombo went from no HM experience three years ago to being in charge of four HM programs.
“The biggest thing is, I wanted to make sure I hadn’t stepped in something that I shouldn’t have already,” he adds. “There’s many different ways to do things. So I’ve learned a few different ways. I found value.”
Demonstrate Value
Another way to discover value in running a practice is looking at the business side of the house, says Denice Cora-Bramble, MD, MBA, chief medical officer and executive vice president of Ambulatory & Community Health Services at the Children’s National Medical Center in Washington, D.C.
Dr. Bramble says many hospitalists need to understand that while clinical care is what brought them to medicine, their future paychecks depend on recognizing how to provide that care in a way that demonstrates business value.
“When you finish residency, if you have not intentionally sought out those courses or those seminars, you need to recognize that as a blind spot,” she says. “You need to fill that toolkit as it relates to the business side of medicine.
“You don’t necessarily have to know all the answers, but you need to know the right questions to ask,” she says.
Dr. Bramble adds that hospitalist leaders should take advantage of certificate programs, leadership courses, basic budgeting classes, or anything that gives them added education about the economics of healthcare.
“It all comes down to demonstrating your outcomes, demonstrating the value that you bring to that institution,” she says. “And with health-care reform, I think hospitalists are uniquely positioned to be able to partner with other areas of the hospital to look at this value-based approach.”
Gary Gammon, MD, FHM, the newly named medical director of the Hospitalist Service at FirstHealth Moore Regional Hospital in Pinehurst, N.C., is doing his part to demonstrate value to his administrators. While his group does multidisciplinary rounds on patients, one of his questions for the pre-course faculty was to make sure that system of rounding is an evidence-based practice. He’s also looking for ways to establish more hegemony to his practice to ensure the rounds are effective, regardless of which physicians and others are participating.
The feedback he received was that most people view multidisciplinary rounds as a best practice. Now, Dr. Gammon can feel more authoritative that he and his 32 hospitalists and 12 extenders are practicing HM the way it should be practiced.
“I wanted to hear just what I heard, which is the leaders in the community feel that it’s helping, feel that it’s the right thing to do, feel that there’s objective data,” he says. “This is the stuff that makes me say, ‘OK, I’ve got the same problems everybody else has.’”
Richard Quinn is a freelance writer in New Jersey.
Ibe Mbanu, MD, MBA, MPH, became medical director of the adult hospitalist department at Bon Secours St. Mary’s Hospital in Richmond, Va., about six months ago. Since then, he’s been besieged by a torrent of reform-based challenges he says make his job exponentially more difficult than that of medical directors just a few years ago.
Accountable-care organizations (ACOs), value-based purchasing, and discussions about bundled payments for episodic care are changing rapidly, and as a new administrator in a group with 24 hospitalists and three nonphysician providers (NPPs), he felt he needed to attend his first SHM annual meeting to keep up.
“The landscape in health care is rapidly evolving, at a frantic pace,” Dr. Mbanu says. “I essentially came here to just try to get a condensed source of information on how to manage the changes that are coming through the pipeline, and how to effectively run my department.”
Managing a practice is a challenge, and many of the more than 2,700 attendees at HM13 said the four-day confab’s focus on the topic was a major draw. From a rebooted continuing medical education (CME) pre-course appropriately named “What Keeps You Awake at Night? Hot Topics in Hospitalist Practice Management” to dozens of breakout sessions on the topic, it’s clear that successful practice management is a concern for many hospitalists.
“Before, the drivers were pretty clear,” Dr. Mbanu says. “Volume, productivity. Now we’re switching more toward a business model that’s changing from volume to value. Trying to adapt to that change is pretty challenging.
“Now it’s critical to really understand the environment.”
Comanagement Conundrum
One particularly hot topic this year was the trend of hospitalists taking on more comanagement responsibilities for patients previously managed by other specialties, including neurology, surgery, and others. Frank Volpicelli, MD, a first-year hospitalist and instructor at New York University (NYU) Langone Medical Center in New York, was one of three members of his HM group that attended the “Perioperative Medicine: Medical Consultation and Co-Management” pre-course. This summer, his group is going to establish a presence in the preoperative clinic.
“We hope very strongly that we can prevent some complications, identify patients that we should be following when they come into the hospital, and help the surgeons out,” he says. “No. 1, keep them in the [operating room] more, and No. 2, get in front of some of the complications that they are less comfortable managing.”
Ralph Velazquez, MD, senior vice president of care management for OSF Healthcare System in Peoria, Ill., isn’t so sure comanagement of more and more patients is the best practice-management model moving forward. For example, as physician compensation is tied more to how much their care costs to deliver, a hospitalist comanaging a surgical patient’s elective knee replacement could be financially penalized for the cost of that person’s stay, despite having nothing to do with the most expensive portion of the bill.
“You have a financial model that says do more billings, but as you start developing analytics … you may see there is no difference between the model that’s doing more billing, in terms of improving quality, and the one that is doing less,” Dr. Velazquez says. “So if you’re getting the same amount of quality, and the only thing you’re doing is generating more cost by doing more billing, you need to reevaluate your strategy.”
He believes some patients benefit from comanagement, but HM groups have to be diligent in seeking them out.
“We look for simple solutions and one-size-fits-all,” he adds. “Comanagement in complex patients—definitely there’s a need for that. Comanagement in noncomplex patients, elective patients—there’s no need for that. It’s just additional cost. I don’t think it’s going to produce any value.”
Startup Academy
John Colombo, MD, FACP, a 30-year veteran of internal medicine who moved to HM a few years ago when one of the hospitals he worked at asked him to launch a hospitalist group, thinks bundled payments might alleviate that value conundrum. Then again, he’s not quite sure. That’s why attended his first annual meeting.
“I found it difficult starting a new program from scratch,” says Dr. Colombo, of Crozer Keystone Health System in Drexel Hill, Pa. “Even with the materials available, there’s not a lot of ‘how to do it’ out there. There’s no ‘Starting Hospitals for Dummies’ book.”
Dr. Colombo spent much of his meeting focused on recruiting, compensation, bonus structures, and scheduling concerns. He said all are important in the hospital-heavy metropolitan Philadelphia region where he works. Plus, with departures and retirements at other programs in his health system, Dr. Colombo went from no HM experience three years ago to being in charge of four HM programs.
“The biggest thing is, I wanted to make sure I hadn’t stepped in something that I shouldn’t have already,” he adds. “There’s many different ways to do things. So I’ve learned a few different ways. I found value.”
Demonstrate Value
Another way to discover value in running a practice is looking at the business side of the house, says Denice Cora-Bramble, MD, MBA, chief medical officer and executive vice president of Ambulatory & Community Health Services at the Children’s National Medical Center in Washington, D.C.
Dr. Bramble says many hospitalists need to understand that while clinical care is what brought them to medicine, their future paychecks depend on recognizing how to provide that care in a way that demonstrates business value.
“When you finish residency, if you have not intentionally sought out those courses or those seminars, you need to recognize that as a blind spot,” she says. “You need to fill that toolkit as it relates to the business side of medicine.
“You don’t necessarily have to know all the answers, but you need to know the right questions to ask,” she says.
Dr. Bramble adds that hospitalist leaders should take advantage of certificate programs, leadership courses, basic budgeting classes, or anything that gives them added education about the economics of healthcare.
“It all comes down to demonstrating your outcomes, demonstrating the value that you bring to that institution,” she says. “And with health-care reform, I think hospitalists are uniquely positioned to be able to partner with other areas of the hospital to look at this value-based approach.”
Gary Gammon, MD, FHM, the newly named medical director of the Hospitalist Service at FirstHealth Moore Regional Hospital in Pinehurst, N.C., is doing his part to demonstrate value to his administrators. While his group does multidisciplinary rounds on patients, one of his questions for the pre-course faculty was to make sure that system of rounding is an evidence-based practice. He’s also looking for ways to establish more hegemony to his practice to ensure the rounds are effective, regardless of which physicians and others are participating.
The feedback he received was that most people view multidisciplinary rounds as a best practice. Now, Dr. Gammon can feel more authoritative that he and his 32 hospitalists and 12 extenders are practicing HM the way it should be practiced.
“I wanted to hear just what I heard, which is the leaders in the community feel that it’s helping, feel that it’s the right thing to do, feel that there’s objective data,” he says. “This is the stuff that makes me say, ‘OK, I’ve got the same problems everybody else has.’”
Richard Quinn is a freelance writer in New Jersey.
A Future in Hospital Medicine Comes into Focus
Last month, I attended SHM’s annual meeting and met dozens of hospitalists.
In fact, I had the pleasure of sharing the stage with my brother, SHM president Eric Howell, MD, SFHM, as he announced his goal of recruiting 1,000 medical students and residents into SHM in the next year. As he counts down toward that goal, he now calls me “No. 1,000.”
In just a few months, I’ll be applying for residency. And I’m grateful for those hospitalists—including my brother—who have helped guide my decision.
Unlike many medical students, I have had the benefit of knowing about HM for years. Now, as I start to make decisions that will guide my career—my subinternship for fourth year, my residency applications, and even the overarching goals for my life as a physician—the benefits of being a hospitalist come into even sharper focus.
I want to be the doctor that I’ve always envisioned a doctor to be: taking care of any problem, working with patients directly, and being the “quarterback” of a team of caregivers. That’s why an HM career is appealing to me.
Plus, hospitalists often have the chance to explore other interests. For me, I’d like to pursue interests in women’s health. For my brother, it was the chance to lead within the hospital administration.
I believe I’ll also get the chance to have the lifestyle I’ve been working for, one that gives me the balance between life inside and outside of the hospital.
Making career choices as a medical student can be tough. They say “you have so much time to decide” until they say “you have to decide right now.” I’m happy that, when I do decide, there’s a specialty out there that gives me options.
—Lesley Sutherland, medical student
Last month, I attended SHM’s annual meeting and met dozens of hospitalists.
In fact, I had the pleasure of sharing the stage with my brother, SHM president Eric Howell, MD, SFHM, as he announced his goal of recruiting 1,000 medical students and residents into SHM in the next year. As he counts down toward that goal, he now calls me “No. 1,000.”
In just a few months, I’ll be applying for residency. And I’m grateful for those hospitalists—including my brother—who have helped guide my decision.
Unlike many medical students, I have had the benefit of knowing about HM for years. Now, as I start to make decisions that will guide my career—my subinternship for fourth year, my residency applications, and even the overarching goals for my life as a physician—the benefits of being a hospitalist come into even sharper focus.
I want to be the doctor that I’ve always envisioned a doctor to be: taking care of any problem, working with patients directly, and being the “quarterback” of a team of caregivers. That’s why an HM career is appealing to me.
Plus, hospitalists often have the chance to explore other interests. For me, I’d like to pursue interests in women’s health. For my brother, it was the chance to lead within the hospital administration.
I believe I’ll also get the chance to have the lifestyle I’ve been working for, one that gives me the balance between life inside and outside of the hospital.
Making career choices as a medical student can be tough. They say “you have so much time to decide” until they say “you have to decide right now.” I’m happy that, when I do decide, there’s a specialty out there that gives me options.
—Lesley Sutherland, medical student
Last month, I attended SHM’s annual meeting and met dozens of hospitalists.
In fact, I had the pleasure of sharing the stage with my brother, SHM president Eric Howell, MD, SFHM, as he announced his goal of recruiting 1,000 medical students and residents into SHM in the next year. As he counts down toward that goal, he now calls me “No. 1,000.”
In just a few months, I’ll be applying for residency. And I’m grateful for those hospitalists—including my brother—who have helped guide my decision.
Unlike many medical students, I have had the benefit of knowing about HM for years. Now, as I start to make decisions that will guide my career—my subinternship for fourth year, my residency applications, and even the overarching goals for my life as a physician—the benefits of being a hospitalist come into even sharper focus.
I want to be the doctor that I’ve always envisioned a doctor to be: taking care of any problem, working with patients directly, and being the “quarterback” of a team of caregivers. That’s why an HM career is appealing to me.
Plus, hospitalists often have the chance to explore other interests. For me, I’d like to pursue interests in women’s health. For my brother, it was the chance to lead within the hospital administration.
I believe I’ll also get the chance to have the lifestyle I’ve been working for, one that gives me the balance between life inside and outside of the hospital.
Making career choices as a medical student can be tough. They say “you have so much time to decide” until they say “you have to decide right now.” I’m happy that, when I do decide, there’s a specialty out there that gives me options.
—Lesley Sutherland, medical student
Speakers at HM13 Stress Overarching Reform, Day-to-Day Implementation

To some HM13 attendees, the keynote speakers might have seemed to be talking about different things.
Patrick Conway, MD, MSc, FAAP, SFHM, chief medical officer and director of the Center for Clinical Standards and Quality at the Centers for Medicare & Medicaid Services (CMS), hinted at promising results from the first accountable-care organizations (ACOs) and noted a meaningful reduction in 30-day readmission rates for the first time in years.
David Feinberg, MD, MBA, president of UCLA Health System in Los Angeles, told hospitalists that unless they’re getting patient care right every time, they’re not getting it right enough. And nothing would make him happier than seeing fewer hospitalists at SHM’s annual meeting—because that would mean fewer hospitalized patients.
HM pioneer Bob Wachter, MD, MHM, said it’s time for hospitalists to link their quality-improvement (QI) efforts and safety acumen to projects focused on cutting costs and reducing waste in the health-care system.
So while each made their points in a different way, each plenary speaker left many meeting-goers with a similar thought: Hospitalists are positioned at the nexus of big-picture reform and day-to-day implementation. So if hospitalists as a specialty continue to embrace teamwork, evidence-based practice, quality, safety, and a sense that the patient comes first, they will cement themselves as leaders in the next iteration of health-care delivery.
“There is enormous change going on in the healthcare system,” says SHM CEO Larry Wellikson. “And we are right in the middle of this. We are essential. If we are bad, we are going to sink it. And if we’re great, we are going to take it to another level.”
Needle Movement
Dr. Conway said some of that progress already is evident. He disclosed that initial findings from the first data sets coming from the first ACOs are showing promising results, though he can’t go into detail until the information is publicly released. However, he did boast that after decades of Medicare readmission rates hovering around 19%, data from late 2012 and early 2013 show that figure has dropped to below 18%.
“That is a 1.5% to 2% shift in readmissions nationally,” he said. “It is a credit to the work you and others are doing in the field. That’s hundreds of thousands of Medicare beneficiaries that are not readmitted every year, that stay home healthy. … It’s a tremendous example of moving a national needle.”
He dismissed those who attribute the initial readmission progress solely to penalties instituted on readmissions, though he acknowledged that CMS is using both carrots and sticks to push change.

“It’s a combination of interventions,” he said.
And all of those initiatives must be aimed jointly at improving the patient experience, said Dr. Feinberg, a child psychiatrist by training whose mantra is “patient-centeredness.” Dr. Feinberg’s reputation is that of a physician-administrator who puts patients first. For example, even though his health system (www.uclahealth.org) is in the 99th percentile for patient satisfaction, he is unhappy. That’s because the top ranking means roughly 85 out of every 100 patients served are pretty happy with their experience.
“It means that we’re the cream of the crap,” he said. “Of the last 100 people we took care of, 15 of them—and, by definition, those 15 people are someone’s mom, someone’s brother, someone’s coworker—would not refer us to a friend, or rate us a 9 or 10. So, I think, while we’ve really moved the needle, we’re really not done until we get it right with every patient, every time.”
He added that those who argue against difficult or time-consuming innovations and improvements that better patient care are arguing against the moral high ground of how they would want a family member to be treated in the hospital.
“The pushback I hear is, ‘Some of this stuff is unpreventable,’” Dr. Feinberg said. “Well, maybe it’s unpreventable the way we’re doing it now. But maybe we need to think differently. Maybe it is unpreventable, but if this decreases the prevalence, or makes it better, then to me, it’s important to do.”
Dr. Feinberg, who took over as UCLA Health System’s president in 2011, says he still spends several hours every day talking to patients. For those who say there’s not enough time to stay connected to patients and that all the time spent making sure patients are happy takes away from other activities, he says they’re forgetting what brought them into medicine in the first place: healing. He blames the delivery system for stifling what he believes is a provider’s desire to help people.
“We haven’t allowed the culture to come out,” he said. “I think it’s there.”

Dr. Wachter has a similar faith in the hospitalist culture—although his is based in the pluripotent nature of the specialty. Hospitalists have worked hard to be viewed as “generalists, able to solve all kinds of problems,” and that means the specialty is poised to adapt and thrive.
“We will morph into what is needed,” said Dr. Wachter, a past president of SHM whose titles include chief of the division of hospital medicine at the University of California at San Francisco and chair of the American Board of Internal Medicine. “That will be all sorts of things: comanagement, dealing with the residency limits in teaching hospitals, systems improvement, cost reductions, transitions, working in skilled nursing facilities, all the specialty hospitalists.
“We will fill new niches,” he said.

What Dr. Wachter does not want to see is that the field grows “fat and happy,” as it is now firmly entrenched in the U.S. health-care delivery system. In fact, he urged hospitalists to welcome change, particularly initiatives that improve quality and safety, reduce costs and waste, and, ultimately, improve the patient experience.
But he cautioned against conceptually separating QI and cost reduction. Instead, they should be viewed as equally meaningful parts of his oft-quoted value equation, which, viewed from the health-care consumer’s point of view, is quality divided by cost.
“You can’t survive and thrive in a world with the kinds of pressures that we have to improve performance if you do business the same old way,” he added. “It’s no longer possible to achieve the things you need to achieve handling these as single projects. You need to transform the way you think about care.”
Richard Quinn is a freelance writer in New Jersey.
To some HM13 attendees, the keynote speakers might have seemed to be talking about different things.
Patrick Conway, MD, MSc, FAAP, SFHM, chief medical officer and director of the Center for Clinical Standards and Quality at the Centers for Medicare & Medicaid Services (CMS), hinted at promising results from the first accountable-care organizations (ACOs) and noted a meaningful reduction in 30-day readmission rates for the first time in years.
David Feinberg, MD, MBA, president of UCLA Health System in Los Angeles, told hospitalists that unless they’re getting patient care right every time, they’re not getting it right enough. And nothing would make him happier than seeing fewer hospitalists at SHM’s annual meeting—because that would mean fewer hospitalized patients.
HM pioneer Bob Wachter, MD, MHM, said it’s time for hospitalists to link their quality-improvement (QI) efforts and safety acumen to projects focused on cutting costs and reducing waste in the health-care system.
So while each made their points in a different way, each plenary speaker left many meeting-goers with a similar thought: Hospitalists are positioned at the nexus of big-picture reform and day-to-day implementation. So if hospitalists as a specialty continue to embrace teamwork, evidence-based practice, quality, safety, and a sense that the patient comes first, they will cement themselves as leaders in the next iteration of health-care delivery.
“There is enormous change going on in the healthcare system,” says SHM CEO Larry Wellikson. “And we are right in the middle of this. We are essential. If we are bad, we are going to sink it. And if we’re great, we are going to take it to another level.”
Needle Movement
Dr. Conway said some of that progress already is evident. He disclosed that initial findings from the first data sets coming from the first ACOs are showing promising results, though he can’t go into detail until the information is publicly released. However, he did boast that after decades of Medicare readmission rates hovering around 19%, data from late 2012 and early 2013 show that figure has dropped to below 18%.
“That is a 1.5% to 2% shift in readmissions nationally,” he said. “It is a credit to the work you and others are doing in the field. That’s hundreds of thousands of Medicare beneficiaries that are not readmitted every year, that stay home healthy. … It’s a tremendous example of moving a national needle.”
He dismissed those who attribute the initial readmission progress solely to penalties instituted on readmissions, though he acknowledged that CMS is using both carrots and sticks to push change.
“It’s a combination of interventions,” he said.
And all of those initiatives must be aimed jointly at improving the patient experience, said Dr. Feinberg, a child psychiatrist by training whose mantra is “patient-centeredness.” Dr. Feinberg’s reputation is that of a physician-administrator who puts patients first. For example, even though his health system (www.uclahealth.org) is in the 99th percentile for patient satisfaction, he is unhappy. That’s because the top ranking means roughly 85 out of every 100 patients served are pretty happy with their experience.
“It means that we’re the cream of the crap,” he said. “Of the last 100 people we took care of, 15 of them—and, by definition, those 15 people are someone’s mom, someone’s brother, someone’s coworker—would not refer us to a friend, or rate us a 9 or 10. So, I think, while we’ve really moved the needle, we’re really not done until we get it right with every patient, every time.”
He added that those who argue against difficult or time-consuming innovations and improvements that better patient care are arguing against the moral high ground of how they would want a family member to be treated in the hospital.
“The pushback I hear is, ‘Some of this stuff is unpreventable,’” Dr. Feinberg said. “Well, maybe it’s unpreventable the way we’re doing it now. But maybe we need to think differently. Maybe it is unpreventable, but if this decreases the prevalence, or makes it better, then to me, it’s important to do.”
Dr. Feinberg, who took over as UCLA Health System’s president in 2011, says he still spends several hours every day talking to patients. For those who say there’s not enough time to stay connected to patients and that all the time spent making sure patients are happy takes away from other activities, he says they’re forgetting what brought them into medicine in the first place: healing. He blames the delivery system for stifling what he believes is a provider’s desire to help people.
“We haven’t allowed the culture to come out,” he said. “I think it’s there.”
Dr. Wachter has a similar faith in the hospitalist culture—although his is based in the pluripotent nature of the specialty. Hospitalists have worked hard to be viewed as “generalists, able to solve all kinds of problems,” and that means the specialty is poised to adapt and thrive.
“We will morph into what is needed,” said Dr. Wachter, a past president of SHM whose titles include chief of the division of hospital medicine at the University of California at San Francisco and chair of the American Board of Internal Medicine. “That will be all sorts of things: comanagement, dealing with the residency limits in teaching hospitals, systems improvement, cost reductions, transitions, working in skilled nursing facilities, all the specialty hospitalists.
“We will fill new niches,” he said.
What Dr. Wachter does not want to see is that the field grows “fat and happy,” as it is now firmly entrenched in the U.S. health-care delivery system. In fact, he urged hospitalists to welcome change, particularly initiatives that improve quality and safety, reduce costs and waste, and, ultimately, improve the patient experience.
But he cautioned against conceptually separating QI and cost reduction. Instead, they should be viewed as equally meaningful parts of his oft-quoted value equation, which, viewed from the health-care consumer’s point of view, is quality divided by cost.
“You can’t survive and thrive in a world with the kinds of pressures that we have to improve performance if you do business the same old way,” he added. “It’s no longer possible to achieve the things you need to achieve handling these as single projects. You need to transform the way you think about care.”
Richard Quinn is a freelance writer in New Jersey.
To some HM13 attendees, the keynote speakers might have seemed to be talking about different things.
Patrick Conway, MD, MSc, FAAP, SFHM, chief medical officer and director of the Center for Clinical Standards and Quality at the Centers for Medicare & Medicaid Services (CMS), hinted at promising results from the first accountable-care organizations (ACOs) and noted a meaningful reduction in 30-day readmission rates for the first time in years.
David Feinberg, MD, MBA, president of UCLA Health System in Los Angeles, told hospitalists that unless they’re getting patient care right every time, they’re not getting it right enough. And nothing would make him happier than seeing fewer hospitalists at SHM’s annual meeting—because that would mean fewer hospitalized patients.
HM pioneer Bob Wachter, MD, MHM, said it’s time for hospitalists to link their quality-improvement (QI) efforts and safety acumen to projects focused on cutting costs and reducing waste in the health-care system.
So while each made their points in a different way, each plenary speaker left many meeting-goers with a similar thought: Hospitalists are positioned at the nexus of big-picture reform and day-to-day implementation. So if hospitalists as a specialty continue to embrace teamwork, evidence-based practice, quality, safety, and a sense that the patient comes first, they will cement themselves as leaders in the next iteration of health-care delivery.
“There is enormous change going on in the healthcare system,” says SHM CEO Larry Wellikson. “And we are right in the middle of this. We are essential. If we are bad, we are going to sink it. And if we’re great, we are going to take it to another level.”
Needle Movement
Dr. Conway said some of that progress already is evident. He disclosed that initial findings from the first data sets coming from the first ACOs are showing promising results, though he can’t go into detail until the information is publicly released. However, he did boast that after decades of Medicare readmission rates hovering around 19%, data from late 2012 and early 2013 show that figure has dropped to below 18%.
“That is a 1.5% to 2% shift in readmissions nationally,” he said. “It is a credit to the work you and others are doing in the field. That’s hundreds of thousands of Medicare beneficiaries that are not readmitted every year, that stay home healthy. … It’s a tremendous example of moving a national needle.”
He dismissed those who attribute the initial readmission progress solely to penalties instituted on readmissions, though he acknowledged that CMS is using both carrots and sticks to push change.
“It’s a combination of interventions,” he said.
And all of those initiatives must be aimed jointly at improving the patient experience, said Dr. Feinberg, a child psychiatrist by training whose mantra is “patient-centeredness.” Dr. Feinberg’s reputation is that of a physician-administrator who puts patients first. For example, even though his health system (www.uclahealth.org) is in the 99th percentile for patient satisfaction, he is unhappy. That’s because the top ranking means roughly 85 out of every 100 patients served are pretty happy with their experience.
“It means that we’re the cream of the crap,” he said. “Of the last 100 people we took care of, 15 of them—and, by definition, those 15 people are someone’s mom, someone’s brother, someone’s coworker—would not refer us to a friend, or rate us a 9 or 10. So, I think, while we’ve really moved the needle, we’re really not done until we get it right with every patient, every time.”
He added that those who argue against difficult or time-consuming innovations and improvements that better patient care are arguing against the moral high ground of how they would want a family member to be treated in the hospital.
“The pushback I hear is, ‘Some of this stuff is unpreventable,’” Dr. Feinberg said. “Well, maybe it’s unpreventable the way we’re doing it now. But maybe we need to think differently. Maybe it is unpreventable, but if this decreases the prevalence, or makes it better, then to me, it’s important to do.”
Dr. Feinberg, who took over as UCLA Health System’s president in 2011, says he still spends several hours every day talking to patients. For those who say there’s not enough time to stay connected to patients and that all the time spent making sure patients are happy takes away from other activities, he says they’re forgetting what brought them into medicine in the first place: healing. He blames the delivery system for stifling what he believes is a provider’s desire to help people.
“We haven’t allowed the culture to come out,” he said. “I think it’s there.”
Dr. Wachter has a similar faith in the hospitalist culture—although his is based in the pluripotent nature of the specialty. Hospitalists have worked hard to be viewed as “generalists, able to solve all kinds of problems,” and that means the specialty is poised to adapt and thrive.
“We will morph into what is needed,” said Dr. Wachter, a past president of SHM whose titles include chief of the division of hospital medicine at the University of California at San Francisco and chair of the American Board of Internal Medicine. “That will be all sorts of things: comanagement, dealing with the residency limits in teaching hospitals, systems improvement, cost reductions, transitions, working in skilled nursing facilities, all the specialty hospitalists.
“We will fill new niches,” he said.
What Dr. Wachter does not want to see is that the field grows “fat and happy,” as it is now firmly entrenched in the U.S. health-care delivery system. In fact, he urged hospitalists to welcome change, particularly initiatives that improve quality and safety, reduce costs and waste, and, ultimately, improve the patient experience.
But he cautioned against conceptually separating QI and cost reduction. Instead, they should be viewed as equally meaningful parts of his oft-quoted value equation, which, viewed from the health-care consumer’s point of view, is quality divided by cost.
“You can’t survive and thrive in a world with the kinds of pressures that we have to improve performance if you do business the same old way,” he added. “It’s no longer possible to achieve the things you need to achieve handling these as single projects. You need to transform the way you think about care.”
Richard Quinn is a freelance writer in New Jersey.
SHM Challenges Hospitalists to Recruit Medical Students, House Staff

By the time you read this, SHM will have completed another amazing annual meeting, very likely smashing some records in the process. Pre-courses have been taught, Washington’s Capitol Hill “visited,” lectures communicated, Bob Wachter’s update … updated. Staff at SHM will be busy crunching numbers and analyzing data so they can quantify the success and uniqueness of HM13.
It was at HM13 that I was lucky enough to meet many of you who are hospitalists just like me. Between Bob Wachter and Larry Wellikson, I also was able to muscle in on stage for a few minutes and share a glimpse of what I am passionate about. If you were there, you know I challenged our society to double the number of student and house staff members to 1,000. I launched the effort by inducting a special medical student (at least to me!), my sister, Lesley Sutherland (see “I Am No. 1,000,” below), bringing the new total number needed down to 999. I plan to repeatedly induct students and housestaff over the next year, and I hope many of you will, too.
As a society, we have had phenomenal membership growth over the past 15 years, expanding from a few hundred members to more than 11,000. SHM’s growth is a tremendous success story; in all of health care’s history, no other medical specialty’s ranks have grown as quickly as HM has.
But virtually all of our growth has come from board-certified (BC) or board-eligible (BE) physicians; very little has come from house officers or students. Over the last four years alone, the society has gone from 9,850 to 11,731 total members, an impressive 16% increase. However, during that same period, housestaff members have remained at about 400. This year, student members barely number 100.
This surprises me.
The Connection: Students and House Officers
It surprises me because, as best I can tell, HM is a career path that meets many of the interests of the new generation of students and house officers. Based on my totally unscientific analysis (I asked my sister, her colleagues, and the house officers with whom I work), many are interested in shorter training, flexible schedules, work-life balance, excitement, and a decent salary. Some report wanting to focus on patient safety, teaching, leadership, and teamwork. If those aren’t what drew the “BC/BE” physicians to HM in droves, I don’t know what did.
That leads me to believe that SHM and, more broadly, HM have exactly what students are looking for.
But HM isn’t just good for medical students and house officers. More students and house officers are also good for the specialty. There continues to be a constant demand for hospitalists in hospitals across the country, and growing SHM’s ranks clearly has a positive benefit for all of our members.
Most important, though: Attracting more students and house officers to HM is good for health care and patients. Hospitalists have proven their value as trusted caregivers for patients and stewards of the hospital. And more hospitalists can only help to achieve our common goal of truly transforming health care and revolutionizing patient care.
All we need to do is to connect students and house officers to our society. Fortunately, many in SHM already are working on just that.
How SHM Members are Connecting, and How You Can, Too
The Physicians in Training (PIT) Committee has been focusing on this topic for the past year. Through the leadership of Drs. Vineet “Vinny” Arora and Darlene Tad-y, PIT has developed a multistep approach to increase student and house officer involvement, including outreach, educational programs, and trainee-specific SHM offerings (e.g. a student/resident section).
Some regional chapters, such as the Boston-area chapter of SHM, have begun to provide awards to trainees, complete with money to travel to the annual meeting. I also know that the Greater Baltimore-area chapter has put on a job fair each year for the past two years. SHM, the staff, and PIT are expanding these ideas, with plans to make SHM a professional home for students and house officers alike.
But local chapters, SHM staff, and even the PIT Committee likely cannot meet the challenge to increase student and resident membership to 1,000 by HM14 alone. We will need the broader participation of the SHM membership—and that means you!
If you’re a hospitalist with teaching responsibilities, make sure your team knows that you are a hospitalist! If you have contact with residents or students, invite them to a local chapter meeting. At the very least, email them a link to SHM and the ZDoggMD video shown at HM13—they’ll love it.
Tell them that student membership is free, and the resident membership fee is the lowest it has ever been: $100 annually, one of the lowest fees for residents of a professional society. With that membership comes a world of networking, opportunities for professional growth, and the opportunity to be a part of something special.
There are more than 64,000 students and 25,000 house staff across the country. Help me connect just 999 more of them to SHM.
By the time you read this, SHM will have completed another amazing annual meeting, very likely smashing some records in the process. Pre-courses have been taught, Washington’s Capitol Hill “visited,” lectures communicated, Bob Wachter’s update … updated. Staff at SHM will be busy crunching numbers and analyzing data so they can quantify the success and uniqueness of HM13.
It was at HM13 that I was lucky enough to meet many of you who are hospitalists just like me. Between Bob Wachter and Larry Wellikson, I also was able to muscle in on stage for a few minutes and share a glimpse of what I am passionate about. If you were there, you know I challenged our society to double the number of student and house staff members to 1,000. I launched the effort by inducting a special medical student (at least to me!), my sister, Lesley Sutherland (see “I Am No. 1,000,” below), bringing the new total number needed down to 999. I plan to repeatedly induct students and housestaff over the next year, and I hope many of you will, too.
As a society, we have had phenomenal membership growth over the past 15 years, expanding from a few hundred members to more than 11,000. SHM’s growth is a tremendous success story; in all of health care’s history, no other medical specialty’s ranks have grown as quickly as HM has.
But virtually all of our growth has come from board-certified (BC) or board-eligible (BE) physicians; very little has come from house officers or students. Over the last four years alone, the society has gone from 9,850 to 11,731 total members, an impressive 16% increase. However, during that same period, housestaff members have remained at about 400. This year, student members barely number 100.
This surprises me.
The Connection: Students and House Officers
It surprises me because, as best I can tell, HM is a career path that meets many of the interests of the new generation of students and house officers. Based on my totally unscientific analysis (I asked my sister, her colleagues, and the house officers with whom I work), many are interested in shorter training, flexible schedules, work-life balance, excitement, and a decent salary. Some report wanting to focus on patient safety, teaching, leadership, and teamwork. If those aren’t what drew the “BC/BE” physicians to HM in droves, I don’t know what did.
That leads me to believe that SHM and, more broadly, HM have exactly what students are looking for.
But HM isn’t just good for medical students and house officers. More students and house officers are also good for the specialty. There continues to be a constant demand for hospitalists in hospitals across the country, and growing SHM’s ranks clearly has a positive benefit for all of our members.
Most important, though: Attracting more students and house officers to HM is good for health care and patients. Hospitalists have proven their value as trusted caregivers for patients and stewards of the hospital. And more hospitalists can only help to achieve our common goal of truly transforming health care and revolutionizing patient care.
All we need to do is to connect students and house officers to our society. Fortunately, many in SHM already are working on just that.
How SHM Members are Connecting, and How You Can, Too
The Physicians in Training (PIT) Committee has been focusing on this topic for the past year. Through the leadership of Drs. Vineet “Vinny” Arora and Darlene Tad-y, PIT has developed a multistep approach to increase student and house officer involvement, including outreach, educational programs, and trainee-specific SHM offerings (e.g. a student/resident section).
Some regional chapters, such as the Boston-area chapter of SHM, have begun to provide awards to trainees, complete with money to travel to the annual meeting. I also know that the Greater Baltimore-area chapter has put on a job fair each year for the past two years. SHM, the staff, and PIT are expanding these ideas, with plans to make SHM a professional home for students and house officers alike.
But local chapters, SHM staff, and even the PIT Committee likely cannot meet the challenge to increase student and resident membership to 1,000 by HM14 alone. We will need the broader participation of the SHM membership—and that means you!
If you’re a hospitalist with teaching responsibilities, make sure your team knows that you are a hospitalist! If you have contact with residents or students, invite them to a local chapter meeting. At the very least, email them a link to SHM and the ZDoggMD video shown at HM13—they’ll love it.
Tell them that student membership is free, and the resident membership fee is the lowest it has ever been: $100 annually, one of the lowest fees for residents of a professional society. With that membership comes a world of networking, opportunities for professional growth, and the opportunity to be a part of something special.
There are more than 64,000 students and 25,000 house staff across the country. Help me connect just 999 more of them to SHM.
By the time you read this, SHM will have completed another amazing annual meeting, very likely smashing some records in the process. Pre-courses have been taught, Washington’s Capitol Hill “visited,” lectures communicated, Bob Wachter’s update … updated. Staff at SHM will be busy crunching numbers and analyzing data so they can quantify the success and uniqueness of HM13.
It was at HM13 that I was lucky enough to meet many of you who are hospitalists just like me. Between Bob Wachter and Larry Wellikson, I also was able to muscle in on stage for a few minutes and share a glimpse of what I am passionate about. If you were there, you know I challenged our society to double the number of student and house staff members to 1,000. I launched the effort by inducting a special medical student (at least to me!), my sister, Lesley Sutherland (see “I Am No. 1,000,” below), bringing the new total number needed down to 999. I plan to repeatedly induct students and housestaff over the next year, and I hope many of you will, too.
As a society, we have had phenomenal membership growth over the past 15 years, expanding from a few hundred members to more than 11,000. SHM’s growth is a tremendous success story; in all of health care’s history, no other medical specialty’s ranks have grown as quickly as HM has.
But virtually all of our growth has come from board-certified (BC) or board-eligible (BE) physicians; very little has come from house officers or students. Over the last four years alone, the society has gone from 9,850 to 11,731 total members, an impressive 16% increase. However, during that same period, housestaff members have remained at about 400. This year, student members barely number 100.
This surprises me.
The Connection: Students and House Officers
It surprises me because, as best I can tell, HM is a career path that meets many of the interests of the new generation of students and house officers. Based on my totally unscientific analysis (I asked my sister, her colleagues, and the house officers with whom I work), many are interested in shorter training, flexible schedules, work-life balance, excitement, and a decent salary. Some report wanting to focus on patient safety, teaching, leadership, and teamwork. If those aren’t what drew the “BC/BE” physicians to HM in droves, I don’t know what did.
That leads me to believe that SHM and, more broadly, HM have exactly what students are looking for.
But HM isn’t just good for medical students and house officers. More students and house officers are also good for the specialty. There continues to be a constant demand for hospitalists in hospitals across the country, and growing SHM’s ranks clearly has a positive benefit for all of our members.
Most important, though: Attracting more students and house officers to HM is good for health care and patients. Hospitalists have proven their value as trusted caregivers for patients and stewards of the hospital. And more hospitalists can only help to achieve our common goal of truly transforming health care and revolutionizing patient care.
All we need to do is to connect students and house officers to our society. Fortunately, many in SHM already are working on just that.
How SHM Members are Connecting, and How You Can, Too
The Physicians in Training (PIT) Committee has been focusing on this topic for the past year. Through the leadership of Drs. Vineet “Vinny” Arora and Darlene Tad-y, PIT has developed a multistep approach to increase student and house officer involvement, including outreach, educational programs, and trainee-specific SHM offerings (e.g. a student/resident section).
Some regional chapters, such as the Boston-area chapter of SHM, have begun to provide awards to trainees, complete with money to travel to the annual meeting. I also know that the Greater Baltimore-area chapter has put on a job fair each year for the past two years. SHM, the staff, and PIT are expanding these ideas, with plans to make SHM a professional home for students and house officers alike.
But local chapters, SHM staff, and even the PIT Committee likely cannot meet the challenge to increase student and resident membership to 1,000 by HM14 alone. We will need the broader participation of the SHM membership—and that means you!
If you’re a hospitalist with teaching responsibilities, make sure your team knows that you are a hospitalist! If you have contact with residents or students, invite them to a local chapter meeting. At the very least, email them a link to SHM and the ZDoggMD video shown at HM13—they’ll love it.
Tell them that student membership is free, and the resident membership fee is the lowest it has ever been: $100 annually, one of the lowest fees for residents of a professional society. With that membership comes a world of networking, opportunities for professional growth, and the opportunity to be a part of something special.
There are more than 64,000 students and 25,000 house staff across the country. Help me connect just 999 more of them to SHM.
Most Health-Care Professionals Use Personal Smartphones for Work
Proportion of U.S. health-care workers who used their personal smartphones for work in the past year.5 The survey, conducted by Cisco Systems Inc., found that 36% of workers believe their employers are ready for “bring your own device” policies, while 41% say their devices are not password-protected and 53% access unsecure wi-fi networks at work. Additionally, 9 out of 10 workers receive no financial support from employers for using their smartphones at work.
Larry Beresford is a freelance writer in San Francisco
References
- Weigel C, Suen W, Gupta G. Using Lean methodology to teach quality improvement to internal medicine residents at a safety net hospital. Am J Med Qual. 2013 Feb 4 [Epub ahead of print].
- Morganti KG, Lovejoy S, Beckjord EB, Haviland AM, Haas AC, Farley DO. A retrospective evaluation of the Perfecting Patient Care University training program for health care organizations. Am J Med Qual. 2013 Apr 9 [Epub ahead of print].
- Myers JS, Tess A, Glasheen JJ, et al. The Quality and Safety Educators’ Academy: fulfilling an unmet need for faculty development. Am J Med Qual. 2013 Apr 11 [Epub ahead of print].
- Dong XQ, Simon MA. Elder abuse as a risk factor for hospitalization in older persons. JAMA Intern Med. 2013 Apr 8:1-7. doi: 10.1001/jamainternmed.2013.238 [Epub ahead of print].
- Cisco mConcierge. 90% American workers use their own smartphones for work. Cisco mConcierge website. Available at: http://www.ciscomcon.com/sw/swchannel/registration/internet/registrationcfm?SWAPPID=91&RegPageID=350200&SWTHEMEID=12949. Accessed
Proportion of U.S. health-care workers who used their personal smartphones for work in the past year.5 The survey, conducted by Cisco Systems Inc., found that 36% of workers believe their employers are ready for “bring your own device” policies, while 41% say their devices are not password-protected and 53% access unsecure wi-fi networks at work. Additionally, 9 out of 10 workers receive no financial support from employers for using their smartphones at work.
Larry Beresford is a freelance writer in San Francisco
References
- Weigel C, Suen W, Gupta G. Using Lean methodology to teach quality improvement to internal medicine residents at a safety net hospital. Am J Med Qual. 2013 Feb 4 [Epub ahead of print].
- Morganti KG, Lovejoy S, Beckjord EB, Haviland AM, Haas AC, Farley DO. A retrospective evaluation of the Perfecting Patient Care University training program for health care organizations. Am J Med Qual. 2013 Apr 9 [Epub ahead of print].
- Myers JS, Tess A, Glasheen JJ, et al. The Quality and Safety Educators’ Academy: fulfilling an unmet need for faculty development. Am J Med Qual. 2013 Apr 11 [Epub ahead of print].
- Dong XQ, Simon MA. Elder abuse as a risk factor for hospitalization in older persons. JAMA Intern Med. 2013 Apr 8:1-7. doi: 10.1001/jamainternmed.2013.238 [Epub ahead of print].
- Cisco mConcierge. 90% American workers use their own smartphones for work. Cisco mConcierge website. Available at: http://www.ciscomcon.com/sw/swchannel/registration/internet/registrationcfm?SWAPPID=91&RegPageID=350200&SWTHEMEID=12949. Accessed
Proportion of U.S. health-care workers who used their personal smartphones for work in the past year.5 The survey, conducted by Cisco Systems Inc., found that 36% of workers believe their employers are ready for “bring your own device” policies, while 41% say their devices are not password-protected and 53% access unsecure wi-fi networks at work. Additionally, 9 out of 10 workers receive no financial support from employers for using their smartphones at work.
Larry Beresford is a freelance writer in San Francisco
References
- Weigel C, Suen W, Gupta G. Using Lean methodology to teach quality improvement to internal medicine residents at a safety net hospital. Am J Med Qual. 2013 Feb 4 [Epub ahead of print].
- Morganti KG, Lovejoy S, Beckjord EB, Haviland AM, Haas AC, Farley DO. A retrospective evaluation of the Perfecting Patient Care University training program for health care organizations. Am J Med Qual. 2013 Apr 9 [Epub ahead of print].
- Myers JS, Tess A, Glasheen JJ, et al. The Quality and Safety Educators’ Academy: fulfilling an unmet need for faculty development. Am J Med Qual. 2013 Apr 11 [Epub ahead of print].
- Dong XQ, Simon MA. Elder abuse as a risk factor for hospitalization in older persons. JAMA Intern Med. 2013 Apr 8:1-7. doi: 10.1001/jamainternmed.2013.238 [Epub ahead of print].
- Cisco mConcierge. 90% American workers use their own smartphones for work. Cisco mConcierge website. Available at: http://www.ciscomcon.com/sw/swchannel/registration/internet/registrationcfm?SWAPPID=91&RegPageID=350200&SWTHEMEID=12949. Accessed