User login
Shoulder pain: 3 cases to test your diagnostic skills
Shoulder pain is a common reason for visits to primary care physicians, who are most likely to diagnose it as rotator cuff tendinitis1,2—often erroneously. The complexity of the joint and the overlapping pathologies that may present as shoulder pain highlight the need to take a closer look when dealing with this diagnostic challenge.
Often, a targeted medical history—including the mechanism of injury and pain-provoking and pain-relieving factors—and a problem-based physical examination (incorporating many of the maneuvers highlighted in the text and tables that follow) will lead to an accurate diagnosis without the need for imaging studies. We recommend that imaging be reserved for patients who don’t respond to conventional treatments, cases in which the diagnosis is in doubt, and instances in which surgical intervention is being considered.
The 3 cases* that follow, and the take-away message incorporated in each, will give you an opportunity to test—and to hone—your shoulder pain diagnostic skill.
CASE 1 The history: Jesse, a 17-year-old student who’s active in football and track, came in during track season complaining of severe left shoulder pain. He denied any traumatic event or previous injury to the shoulder, but reported that any motion involving the shoulder caused pain. It hurt at night, the patient said, when he lay on his left side.
The physical: No muscle atrophy, redness, or swelling was evident, nor was there any indication of asymmetry or ecchymosis in the affected area. Jesse’s neck range of motion was normal; he had a very hard time with any active motion of the shoulder, however, because of the pain.
Evaluation of scapular motion demonstrated scapular dyskinesis3,4 without winging. Passive motion of the glenohumeral joint was much better than active motion. Strength testing appeared to be grossly intact but was limited by the pain. Shoulder impingement testing was positive. Sensation and deep tendon reflexes were intact.
Patients' names have been changed to protect their privacy
What’s the diagnosis?
Subacromial bursitis, suggested by the patient’s pain and altered scapular motion, was our working diagnosis, and we administered a subacromial injection of corticosteroid with lidocaine, for diagnostic as well as therapeutic purposes. Reexamination after the injection revealed immediate partial improvement in resting pain, range of motion, and strength. We referred Jesse to physical therapy with a focus on scapular stabilization and rotator cuff strength.
Three months later, Jesse returned to our office, complaining of weakness in his left shoulder. The pain had subsided a week after his first appointment, so he’d never gone to physical therapy. The weakness, which he had first noticed about 2 months after starting a lifting program in preparation for football season, was limited to resistance exercises, especially overhead shoulder presses and bench press. There were no other changes in his history, and he reported no reinjuries.
Physical examination revealed atrophy of the supraspinatus and infraspinatus muscles (FIGURE 1) and external rotation and shoulder abduction (in the scapular plane) resistance tests revealed weakness of these muscles. There was no scapular winging. The cervical spine exam was normal, and neurovascular status was intact in both upper extremities.
FIGURE 1
Severe shoulder pain, followed by weakness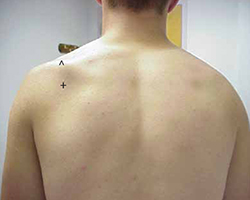
Physical examination reveals atrophy of the patient’s supraspinatus (^) and infraspinatus (+) muscles.
New evidence points to nerve injury. Based on Jesse’s current history and physical, nerve injury was our new working diagnosis. (We considered the possibility of a rotator cuff tear, but this was not corroborated by the history.)
We ordered an electromyogram/nerve conduction velocity study to localize the lesion. The test revealed a brachial plexitis/neuritis (also known as Parsonage-Turner syndrome or brachial amyotrophy). The etiology of most atraumatic brachial plexopathies is unknown, and most are thought to be viral or autoimmune in nature.5,6
A classic case of Parsonage-Turner syndrome. The typical presentation of Parsonage-Turner syndrome (like Jesse’s) is one of acute, intense shoulder pain for no known reason. After 1 to 3 weeks, the pain resolves and the patient is left with weakness, usually of the supraspinatus and infraspinatus muscles. The weakness typically resolves with time, but full resolution may take 6 to 9 months.5,6 (In Jesse’s case, it took about 6 months.)
The take-away message: Look beyond the shoulder
As this case illustrates, not all shoulder pain originates in the shoulder. When evaluating shoulder pain, it is essential to consider other causes. The differential diagnosis for shoulder pain includes cervical spine disorders, cholecystitis (right shoulder), diaphragmatic irritation (eg, in the case of splenic rupture, usually involving the left shoulder), cardiac disease, and thoracic outlet syndrome.7
Evaluation of the cervical spine should be part of a complete shoulder examination. It is vital to follow a systematic approach that carefully assesses the cervical region for the possibility of nerve root impingement and radicular dysfunction masquerading as a primary shoulder disorder. (TABLE 18,9 details pain and sensory distribution patterns, reflex involvement, and potential motor impairments associated with various spinal nerve root levels.)
TABLE 1
Assessing the cervical spine
| Nerve root | Pain distribution | Sensory distribution | Reflex changes | Motor involvement |
|---|---|---|---|---|
| C5 | Lateral neck/upper trapezius | Lateral arm | Biceps | Deltoid, biceps |
| C6 | Base of neck/upper trapezius to superior glenohumeral joint | Radial aspect of distal forearm, thenar eminence, and index finger | Brachioradialis | Biceps, extensor carpi radialis longus and brevis (wrist extension) |
| C7 | Base of neck, almost entire upper quadrant of the back | Third finger | Triceps | Triceps, wrist flexion, finger extension |
| C8 | No shoulder pain | 4th and 5th fingers, distal half of forearm (ulna side) | None | Finger flexion (grip strength) |
| Adapted from: Miller JD, et al. Am Fam Physician. 20008; Eubanks JD. Am Fam Physician. 2010.9 | ||||
Practitioners should develop their own approach to “clearing the neck.” A logical order is to note posture of the head/neck/shoulders, observe active motion, perform palpation and provocative tests, and then assess neurologic function with sensation/reflex/strength testing. Provocative tests that can help to identify cervical involvement relating to shoulder pain include Spurling’s maneuver, axial compression test, abduction relief sign, and Lhermitte’s sign.10,11
CASE 2 The history: Mark, a 17-year-old, right-handed volleyball player, presented with right shoulder pain, which he felt whenever he spiked or served the ball. The pain started last season, Mark said, diminished during the months when he wasn’t playing, then got progressively worse as his activity level increased. The pain was in the posterior aspect of the shoulder.
The physical: Physical examination revealed a well-developed, but thin (6’4”, 170 pounds) young man who was not in distress. The general examination was benign, and a joint-specific exam showed no asymmetry or atrophy on inspection and no tenderness to palpation over the posterior and anterior soft tissues of the right shoulder. Rotator cuff testing yielded intact strength for all 4 muscles, but external rotation and supraspinatus testing elicited pain. The crank test, drawer sign, load and shift test, relocation test, and sulcus sign, detailed in TABLE 2,12-14 were all positive for shoulder instability; the Clunk and O’Brien tests were negative, and the contralateral shoulder exam was within normal limits. General joint laxity was observed, with the ability to oppose the thumb to the volar forearm and hyperextension noted in both elbows and knees. There were no outward signs of connective tissue disease.
Because of the chronicity of Mark’s pain and the progressive nature of his symptoms, we ordered radiographs, including anterior-posterior, lateral axillary, and scapular Y views. These films showed a nearly skeletally mature male without bony abnormalities; the humeral head was well located in the glenoid.
TABLE 2
Testing for shoulder instability12-14
| Test | Procedure | Positive result/implication |
|---|---|---|
| Apprehension | Patient supine, arm abducted 90º, externally rotated with anteriorly directed force applied to humeral head | Pain/apprehension with force suggests anterior instability |
| Relocation* | Patient supine, posteriorly directed force applied to humeral head | Relief with force suggests anterior instability |
| Crank | Patient sitting, arm abducted 90º, elbow flexed to 90º, humerus supported with forced external rotation | Pain/apprehension with forced external rotation suggests anterior instability |
| Load and shift | Patient supine, arm held by examiner and abducted 90º, force applied along axis of humerus to "seat" the humerus within the glenoid, followed by anterior force directed to humeral head | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Drawer | Patient sitting, arm at side, proximal humeral shaft grasped by examiner, seating the humeral head within the glenoid then applying anterior translational force | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Sulcus | Patient sitting, arm at side, forearm grasped by examiner with an inferior/caudally directed force applied | Sulcus or depression seen inferior to acromion as humeral head subluxes posteriorly, pathognomonic for multidirectional instability |
| Clunk | Patient supine, examiner grasps at forearm and humeral shaft, with humeral head seated within the fossa, taking the arm through passive ROM from extension through forward flexion | Clunk sound or clicking sensation suggests labral tear |
| O’Brien | Patient sitting, arm is forward flexed to 90º and fully adducted and internally rotated; patient resists downward motion. If pain is elicited, the maneuver is repeated in external rotation | Pain elicited with resisted downward motion in internal rotation but relieved with external rotation suggests labral pathology |
| *Perform only if apprehension test is positive. ROM, range of motion | ||
What’s the diagnosis?
Multidirectional instability with recurrent subluxations and probable acute rotator cuff tendinitis was our provisional diagnosis. Treatment focused on physical therapy, with a concentration on scapular stabilization and rotator cuff strengthening.
Shoulder instability is relatively common and represents a spectrum of disorders ranging from dislocation to subluxation to simple laxity.12,13 A complete loss of humeral articulation within the glenoid fossa is evidence of dislocation, whereas subluxation includes approximation of the humeral head to the limits of the glenoid rim. Dislocation typically results from trauma, whereas subluxation can be the result of microtrauma and repetitive overuse injury. Anterior instability is the most common type and is reported in as many as 95% of all dislocations.13
The take-away message: Rule out instability
The shoulder is one of the most complex joints in the body. The rotator cuff structures, the glenoid labrum, and the collective capsular ligaments provide structural stability to the glenohumeral joint.12,13 The shoulder is vulnerable to instability because the shallow glenoid fossa offers little bony support for the humeral head. Thus, instability should always be included in an assessment of shoulder pain.
Key factors to consider in identifying shoulder instability include the location of the pain, the direction of traumatic force applied, the presence of a known complete dislocation vs apprehension with specific movement, the position of the arm in which pain is elicited, a previous occurrence of instability (subluxation or dislocation), and the presence of tingling or numbness.12-14 The maneuvers detailed in TABLE 212-14 can help identify instability, as they did in this case. Patients with hypermobility are at increased risk for shoulder instability, so a targeted exam and patient history aimed at identifying signs and symptoms of hypermobility is needed, as well.
Ask the patient to attempt to:
- bend the thumb to the volar forearm
- place hands to the floor with hyperextended knees
- perform maximal hyperextension of the fifth metacarpophalangeal joint (>90° is a positive result).
Findings from the medical history that indicate a predisposition to instability include generalized joint laxity, Ehlers-Danlos syndrome, Marfan syndrome, osteogenesis imperfecta, hyperhomocysteinuria, hyperlysinemia, benign joint hypermobility syndrome, juvenile rheumatoid arthritis, and previous shoulder or patellar dislocations.
Imaging tips: Scapular Y and/or axillary lateral views should always be included when ordering imaging studies for suspected instability/dislocations, as 50% of posterior dislocations are missed on standard shoulder x-rays that do not include them.12 In reviewing the x-rays, it is important to look for signs of a compression fracture of the posterior humeral head (known as a Hill-Sachs lesion) for anterior shoulder dislocations, and fractures to the anterior glenoid rim (known as a Bankart lesion).12-14
CASE 3 The history: Robert, a right-handed, 50-year-old motorcycle instructor, came to our office because of chronic right shoulder pain. The pain, located over the anterior portion of the glenohumeral joint, developed insidiously about 3 or 4 years ago, the patient reported. He had finally decided to seek help because he’d recently experienced an acute exacerbation of pain brought on by shoveling snow, after which he also noticed associated weakness, a clicking/popping on active motion, and mild loss of motion.
The physical: Robert’s cervical spine exam was unremarkable. He demonstrated full active range of motion (ROM) without exacerbation of right shoulder symptoms, and special tests for disc pathology at the neck were negative. Active ROM testing of the right shoulder revealed full abduction, with only minimal pain; full flexion, with moderate pain noted initially at 49°; full extension, with a painful arc noted at 50°; and full horizontal adduction, with a painful arc noted at the halfway point. The testing also revealed that his right thumb was 3 inches lower than the left on reaching for the opposite scapula. At the superior aspect of the acromioclavicular (AC) joint, 2+ tenderness was noted; 3+ tenderness was noted at the greater tubercle of the humerus.
After inspecting the shoulder region for alterations in bony landmarks, muscle bulk, carrying position, and movement characteristics, palpation of the region was performed.
When assessing shoulder strength, there are a variety of tests for each functioning component of the rotator cuff structures (TABLE 3).15-17 Manual muscle tests revealed: 4-/5 on external rotation (French horn test), 3+/5 on the lift-off test, and 5-/5 on all other tests for right shoulder function. Impingement testing was slightly positive, or pain producing, on Hawkins and Neer tests.18,19 For the Hawkins test, the examiner flexes the arm to 90° of shoulder flexion with the elbow flexed at 90°, then internally rotates the shoulder. For the Neer test, the arm is fully elevated in the scapular plane and internally rotated by the examiner.
The subscapularis muscle, which functions primarily in internal rotation, is tested by the French horn and lift-off tests. The teres minor muscle, which performs external rotation, is tested by the French horn test of external rotation. And the supraspinatus muscle, which performs abduction and external rotation, is tested by the empty can (also known as the Jobe) and full can tests. Some researchers suggest that the empty can test is better for diagnosing impingement, based on evidence showing that the full can test is better at diagnosing supraspinatus tears because it causes less pain during testing.20
TABLE 3
Suspect rotator cuff involvement?7,15-17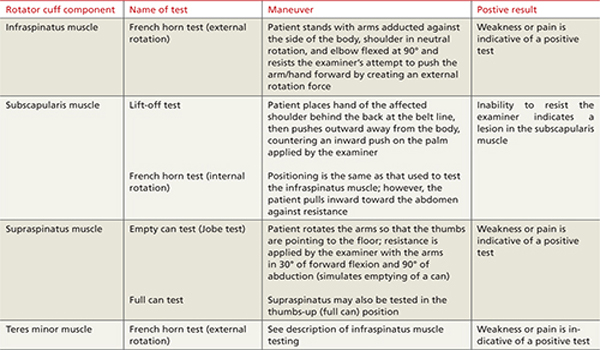
What’s the diagnosis?
Rotator cuff tear was suspected because Robert had positive elements of the “rotator cuff triad”—supraspinatus weakness (as indicated by a positive empty can test), external rotation weakness (revealed by the French horn test), and a positive Hawkins impingement test. We ordered diagnostic studies, including plain radiographs, which revealed degenerative changes at the acromioclavicular joint, decreased acromiohumeral interval, and no significant changes at the glenohumeral joint (FIGURE 2), and magnetic resonance imaging (MRI) of the right shoulder. The MRI revealed a large, full-thickness rotator cuff tear of the supraspinatus tendon with retraction. A torn and retracted biceps tendon and AC joint osteoarthritis were also shown, likely causing a mass effect on the supraspinatus. The patient underwent surgery to repair the torn rotator cuff, with excellent results.
FIGURE 2
Chronic right shoulder pain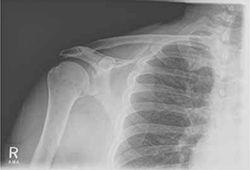
An AP view of the patient’s right shoulder shows acromioclavicular joint narrowing and degeneration and subtle narrowing of the acromiohumeral interval.
The take-away message: Keep the rotator-cuff triad in mind
Because none of the tests that comprise the triad is specific enough alone to diagnose a rotator cuff tear,15,20,21 Murrell and Walton16 suggested that the 3 tests be considered together for diagnostic purposes. If all 3 are positive, there is a 98% chance of a rotator cuff tear; if 2 tests are positive and the patient is older than 60 years, the findings are suggestive of a tear; and if all 3 tests (plus the drop arm test) are negative, there is less than a 5% chance of a major rotator cuff tear.16
CORRESPONDENCE Nilesh Shah, MD, Summa Center for Sports Health, 20 Olive Street, Suite 201, Akron, OH 44310; [email protected]
1. Van der Windt DA, Koes BW, De Jong BA, et al. Shoulder disorders in general practice: incidence, patient characteristics, and management. Ann Rheum Dis. 1995;54:959-964.
2. Johansson K, Adolfsson L, Foldevi M. Attitudes toward management of patients with subacromial pain in Swedish primary care. Fam Pract. 1999;16:233-237.
3. Kibler WB, McMullen J. Scapular dyskinesis and its relation to shoulder pain. J Am Acad Orthop Surg. 2003;11:142-151.
4. Kibler WB. The role of the scapula in athletic shoulder function. Am J Sports Med. 1998;26:325-337.
5. Vanermen B, Aertgeerts M, Hoogmartens M, et al. The syndrome of Parsonage and Turner. Discussion of clinical features with a review of 8 cases. Acta Orthop Belg. 1991;57:414-419.
6. Misamore GW, Lehman DE. Parsonage-Turner syndrome (acute brachial neuritis). J Bone Joint Surg. 1996;78:1405-1408.
7. Stevenson J, Trojian T. Evaluation of shoulder pain. J Fam Pract. 2002;51:605-611.
8. Miller JD, Pruitt RN, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
9. Eubanks JD. Cervical radiculopathy: nonoperative management of neck pain and radicular symptoms. Am Fam Physician. 2010;81:33-40.
10. Malanga GA, Landes P, Nadler SF. Provocative tests in cervical spine examination: historical basis and scientific analyses. Pain Physician. 2003;6:199-205.
11. Huston M, Ellis R. eds. Textbook of Musculoskeletal Medicine. Oxford, UK: Oxford University Press; 2005.
12. Mahaffey BL, Smith PA. Shoulder instability in young athletes. Am Fam Physician. 1999;59:2773-2782, 2787.
13. Petron DJ, Khan U. The shoulder and upper extremity. In: McKeag DB, Moeller JL. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Wolters Kluwer, Lippincott Williams & Wilkins; 2007:359–373.
14. Woodward TW, Best TM. The painful shoulder: part I. clinical evaluation. Am Fam Physician. 2000;61:3079-3088.
15. Kelly BT, Kadrmas WR, Speer KP. The manual muscle examination for rotator cuff strength: an electromyographic investigation. Am J Sports Med. 1996;24:581-588.
16. Murrell GA, Walton JR. Diagnosis of rotator cuff tears. Lancet. 2001;357:769-770.
17. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3:347-352.
18. Neer CS, Welsh RP. The shoulder in sports. Orthop Clin North Am. 1977;8:583-591.
19. Hawkins RJ, Kennedy JC. Impingement syndrome in athletics. Am J Sports Med. 1980;8:151-163.
20. Itoi E, Kido T, Sano A, et al. Which is more useful, the “full can test” or the “empty can test,” in detecting the torn supraspinatus tendon? Am J Sports Med. 1999;27:65-68.
21. Boettcher CE, Ginn KA, Cathers I. The ‘empty can’ and ‘full can’ tests do not selectively activate supraspinatus. J Sci Med Sport. 2009;12:435-439.
Shoulder pain is a common reason for visits to primary care physicians, who are most likely to diagnose it as rotator cuff tendinitis1,2—often erroneously. The complexity of the joint and the overlapping pathologies that may present as shoulder pain highlight the need to take a closer look when dealing with this diagnostic challenge.
Often, a targeted medical history—including the mechanism of injury and pain-provoking and pain-relieving factors—and a problem-based physical examination (incorporating many of the maneuvers highlighted in the text and tables that follow) will lead to an accurate diagnosis without the need for imaging studies. We recommend that imaging be reserved for patients who don’t respond to conventional treatments, cases in which the diagnosis is in doubt, and instances in which surgical intervention is being considered.
The 3 cases* that follow, and the take-away message incorporated in each, will give you an opportunity to test—and to hone—your shoulder pain diagnostic skill.
CASE 1 The history: Jesse, a 17-year-old student who’s active in football and track, came in during track season complaining of severe left shoulder pain. He denied any traumatic event or previous injury to the shoulder, but reported that any motion involving the shoulder caused pain. It hurt at night, the patient said, when he lay on his left side.
The physical: No muscle atrophy, redness, or swelling was evident, nor was there any indication of asymmetry or ecchymosis in the affected area. Jesse’s neck range of motion was normal; he had a very hard time with any active motion of the shoulder, however, because of the pain.
Evaluation of scapular motion demonstrated scapular dyskinesis3,4 without winging. Passive motion of the glenohumeral joint was much better than active motion. Strength testing appeared to be grossly intact but was limited by the pain. Shoulder impingement testing was positive. Sensation and deep tendon reflexes were intact.
Patients' names have been changed to protect their privacy
What’s the diagnosis?
Subacromial bursitis, suggested by the patient’s pain and altered scapular motion, was our working diagnosis, and we administered a subacromial injection of corticosteroid with lidocaine, for diagnostic as well as therapeutic purposes. Reexamination after the injection revealed immediate partial improvement in resting pain, range of motion, and strength. We referred Jesse to physical therapy with a focus on scapular stabilization and rotator cuff strength.
Three months later, Jesse returned to our office, complaining of weakness in his left shoulder. The pain had subsided a week after his first appointment, so he’d never gone to physical therapy. The weakness, which he had first noticed about 2 months after starting a lifting program in preparation for football season, was limited to resistance exercises, especially overhead shoulder presses and bench press. There were no other changes in his history, and he reported no reinjuries.
Physical examination revealed atrophy of the supraspinatus and infraspinatus muscles (FIGURE 1) and external rotation and shoulder abduction (in the scapular plane) resistance tests revealed weakness of these muscles. There was no scapular winging. The cervical spine exam was normal, and neurovascular status was intact in both upper extremities.
FIGURE 1
Severe shoulder pain, followed by weakness
Physical examination reveals atrophy of the patient’s supraspinatus (^) and infraspinatus (+) muscles.
New evidence points to nerve injury. Based on Jesse’s current history and physical, nerve injury was our new working diagnosis. (We considered the possibility of a rotator cuff tear, but this was not corroborated by the history.)
We ordered an electromyogram/nerve conduction velocity study to localize the lesion. The test revealed a brachial plexitis/neuritis (also known as Parsonage-Turner syndrome or brachial amyotrophy). The etiology of most atraumatic brachial plexopathies is unknown, and most are thought to be viral or autoimmune in nature.5,6
A classic case of Parsonage-Turner syndrome. The typical presentation of Parsonage-Turner syndrome (like Jesse’s) is one of acute, intense shoulder pain for no known reason. After 1 to 3 weeks, the pain resolves and the patient is left with weakness, usually of the supraspinatus and infraspinatus muscles. The weakness typically resolves with time, but full resolution may take 6 to 9 months.5,6 (In Jesse’s case, it took about 6 months.)
The take-away message: Look beyond the shoulder
As this case illustrates, not all shoulder pain originates in the shoulder. When evaluating shoulder pain, it is essential to consider other causes. The differential diagnosis for shoulder pain includes cervical spine disorders, cholecystitis (right shoulder), diaphragmatic irritation (eg, in the case of splenic rupture, usually involving the left shoulder), cardiac disease, and thoracic outlet syndrome.7
Evaluation of the cervical spine should be part of a complete shoulder examination. It is vital to follow a systematic approach that carefully assesses the cervical region for the possibility of nerve root impingement and radicular dysfunction masquerading as a primary shoulder disorder. (TABLE 18,9 details pain and sensory distribution patterns, reflex involvement, and potential motor impairments associated with various spinal nerve root levels.)
TABLE 1
Assessing the cervical spine
| Nerve root | Pain distribution | Sensory distribution | Reflex changes | Motor involvement |
|---|---|---|---|---|
| C5 | Lateral neck/upper trapezius | Lateral arm | Biceps | Deltoid, biceps |
| C6 | Base of neck/upper trapezius to superior glenohumeral joint | Radial aspect of distal forearm, thenar eminence, and index finger | Brachioradialis | Biceps, extensor carpi radialis longus and brevis (wrist extension) |
| C7 | Base of neck, almost entire upper quadrant of the back | Third finger | Triceps | Triceps, wrist flexion, finger extension |
| C8 | No shoulder pain | 4th and 5th fingers, distal half of forearm (ulna side) | None | Finger flexion (grip strength) |
| Adapted from: Miller JD, et al. Am Fam Physician. 20008; Eubanks JD. Am Fam Physician. 2010.9 | ||||
Practitioners should develop their own approach to “clearing the neck.” A logical order is to note posture of the head/neck/shoulders, observe active motion, perform palpation and provocative tests, and then assess neurologic function with sensation/reflex/strength testing. Provocative tests that can help to identify cervical involvement relating to shoulder pain include Spurling’s maneuver, axial compression test, abduction relief sign, and Lhermitte’s sign.10,11
CASE 2 The history: Mark, a 17-year-old, right-handed volleyball player, presented with right shoulder pain, which he felt whenever he spiked or served the ball. The pain started last season, Mark said, diminished during the months when he wasn’t playing, then got progressively worse as his activity level increased. The pain was in the posterior aspect of the shoulder.
The physical: Physical examination revealed a well-developed, but thin (6’4”, 170 pounds) young man who was not in distress. The general examination was benign, and a joint-specific exam showed no asymmetry or atrophy on inspection and no tenderness to palpation over the posterior and anterior soft tissues of the right shoulder. Rotator cuff testing yielded intact strength for all 4 muscles, but external rotation and supraspinatus testing elicited pain. The crank test, drawer sign, load and shift test, relocation test, and sulcus sign, detailed in TABLE 2,12-14 were all positive for shoulder instability; the Clunk and O’Brien tests were negative, and the contralateral shoulder exam was within normal limits. General joint laxity was observed, with the ability to oppose the thumb to the volar forearm and hyperextension noted in both elbows and knees. There were no outward signs of connective tissue disease.
Because of the chronicity of Mark’s pain and the progressive nature of his symptoms, we ordered radiographs, including anterior-posterior, lateral axillary, and scapular Y views. These films showed a nearly skeletally mature male without bony abnormalities; the humeral head was well located in the glenoid.
TABLE 2
Testing for shoulder instability12-14
| Test | Procedure | Positive result/implication |
|---|---|---|
| Apprehension | Patient supine, arm abducted 90º, externally rotated with anteriorly directed force applied to humeral head | Pain/apprehension with force suggests anterior instability |
| Relocation* | Patient supine, posteriorly directed force applied to humeral head | Relief with force suggests anterior instability |
| Crank | Patient sitting, arm abducted 90º, elbow flexed to 90º, humerus supported with forced external rotation | Pain/apprehension with forced external rotation suggests anterior instability |
| Load and shift | Patient supine, arm held by examiner and abducted 90º, force applied along axis of humerus to "seat" the humerus within the glenoid, followed by anterior force directed to humeral head | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Drawer | Patient sitting, arm at side, proximal humeral shaft grasped by examiner, seating the humeral head within the glenoid then applying anterior translational force | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Sulcus | Patient sitting, arm at side, forearm grasped by examiner with an inferior/caudally directed force applied | Sulcus or depression seen inferior to acromion as humeral head subluxes posteriorly, pathognomonic for multidirectional instability |
| Clunk | Patient supine, examiner grasps at forearm and humeral shaft, with humeral head seated within the fossa, taking the arm through passive ROM from extension through forward flexion | Clunk sound or clicking sensation suggests labral tear |
| O’Brien | Patient sitting, arm is forward flexed to 90º and fully adducted and internally rotated; patient resists downward motion. If pain is elicited, the maneuver is repeated in external rotation | Pain elicited with resisted downward motion in internal rotation but relieved with external rotation suggests labral pathology |
| *Perform only if apprehension test is positive. ROM, range of motion | ||
What’s the diagnosis?
Multidirectional instability with recurrent subluxations and probable acute rotator cuff tendinitis was our provisional diagnosis. Treatment focused on physical therapy, with a concentration on scapular stabilization and rotator cuff strengthening.
Shoulder instability is relatively common and represents a spectrum of disorders ranging from dislocation to subluxation to simple laxity.12,13 A complete loss of humeral articulation within the glenoid fossa is evidence of dislocation, whereas subluxation includes approximation of the humeral head to the limits of the glenoid rim. Dislocation typically results from trauma, whereas subluxation can be the result of microtrauma and repetitive overuse injury. Anterior instability is the most common type and is reported in as many as 95% of all dislocations.13
The take-away message: Rule out instability
The shoulder is one of the most complex joints in the body. The rotator cuff structures, the glenoid labrum, and the collective capsular ligaments provide structural stability to the glenohumeral joint.12,13 The shoulder is vulnerable to instability because the shallow glenoid fossa offers little bony support for the humeral head. Thus, instability should always be included in an assessment of shoulder pain.
Key factors to consider in identifying shoulder instability include the location of the pain, the direction of traumatic force applied, the presence of a known complete dislocation vs apprehension with specific movement, the position of the arm in which pain is elicited, a previous occurrence of instability (subluxation or dislocation), and the presence of tingling or numbness.12-14 The maneuvers detailed in TABLE 212-14 can help identify instability, as they did in this case. Patients with hypermobility are at increased risk for shoulder instability, so a targeted exam and patient history aimed at identifying signs and symptoms of hypermobility is needed, as well.
Ask the patient to attempt to:
- bend the thumb to the volar forearm
- place hands to the floor with hyperextended knees
- perform maximal hyperextension of the fifth metacarpophalangeal joint (>90° is a positive result).
Findings from the medical history that indicate a predisposition to instability include generalized joint laxity, Ehlers-Danlos syndrome, Marfan syndrome, osteogenesis imperfecta, hyperhomocysteinuria, hyperlysinemia, benign joint hypermobility syndrome, juvenile rheumatoid arthritis, and previous shoulder or patellar dislocations.
Imaging tips: Scapular Y and/or axillary lateral views should always be included when ordering imaging studies for suspected instability/dislocations, as 50% of posterior dislocations are missed on standard shoulder x-rays that do not include them.12 In reviewing the x-rays, it is important to look for signs of a compression fracture of the posterior humeral head (known as a Hill-Sachs lesion) for anterior shoulder dislocations, and fractures to the anterior glenoid rim (known as a Bankart lesion).12-14
CASE 3 The history: Robert, a right-handed, 50-year-old motorcycle instructor, came to our office because of chronic right shoulder pain. The pain, located over the anterior portion of the glenohumeral joint, developed insidiously about 3 or 4 years ago, the patient reported. He had finally decided to seek help because he’d recently experienced an acute exacerbation of pain brought on by shoveling snow, after which he also noticed associated weakness, a clicking/popping on active motion, and mild loss of motion.
The physical: Robert’s cervical spine exam was unremarkable. He demonstrated full active range of motion (ROM) without exacerbation of right shoulder symptoms, and special tests for disc pathology at the neck were negative. Active ROM testing of the right shoulder revealed full abduction, with only minimal pain; full flexion, with moderate pain noted initially at 49°; full extension, with a painful arc noted at 50°; and full horizontal adduction, with a painful arc noted at the halfway point. The testing also revealed that his right thumb was 3 inches lower than the left on reaching for the opposite scapula. At the superior aspect of the acromioclavicular (AC) joint, 2+ tenderness was noted; 3+ tenderness was noted at the greater tubercle of the humerus.
After inspecting the shoulder region for alterations in bony landmarks, muscle bulk, carrying position, and movement characteristics, palpation of the region was performed.
When assessing shoulder strength, there are a variety of tests for each functioning component of the rotator cuff structures (TABLE 3).15-17 Manual muscle tests revealed: 4-/5 on external rotation (French horn test), 3+/5 on the lift-off test, and 5-/5 on all other tests for right shoulder function. Impingement testing was slightly positive, or pain producing, on Hawkins and Neer tests.18,19 For the Hawkins test, the examiner flexes the arm to 90° of shoulder flexion with the elbow flexed at 90°, then internally rotates the shoulder. For the Neer test, the arm is fully elevated in the scapular plane and internally rotated by the examiner.
The subscapularis muscle, which functions primarily in internal rotation, is tested by the French horn and lift-off tests. The teres minor muscle, which performs external rotation, is tested by the French horn test of external rotation. And the supraspinatus muscle, which performs abduction and external rotation, is tested by the empty can (also known as the Jobe) and full can tests. Some researchers suggest that the empty can test is better for diagnosing impingement, based on evidence showing that the full can test is better at diagnosing supraspinatus tears because it causes less pain during testing.20
TABLE 3
Suspect rotator cuff involvement?7,15-17
What’s the diagnosis?
Rotator cuff tear was suspected because Robert had positive elements of the “rotator cuff triad”—supraspinatus weakness (as indicated by a positive empty can test), external rotation weakness (revealed by the French horn test), and a positive Hawkins impingement test. We ordered diagnostic studies, including plain radiographs, which revealed degenerative changes at the acromioclavicular joint, decreased acromiohumeral interval, and no significant changes at the glenohumeral joint (FIGURE 2), and magnetic resonance imaging (MRI) of the right shoulder. The MRI revealed a large, full-thickness rotator cuff tear of the supraspinatus tendon with retraction. A torn and retracted biceps tendon and AC joint osteoarthritis were also shown, likely causing a mass effect on the supraspinatus. The patient underwent surgery to repair the torn rotator cuff, with excellent results.
FIGURE 2
Chronic right shoulder pain
An AP view of the patient’s right shoulder shows acromioclavicular joint narrowing and degeneration and subtle narrowing of the acromiohumeral interval.
The take-away message: Keep the rotator-cuff triad in mind
Because none of the tests that comprise the triad is specific enough alone to diagnose a rotator cuff tear,15,20,21 Murrell and Walton16 suggested that the 3 tests be considered together for diagnostic purposes. If all 3 are positive, there is a 98% chance of a rotator cuff tear; if 2 tests are positive and the patient is older than 60 years, the findings are suggestive of a tear; and if all 3 tests (plus the drop arm test) are negative, there is less than a 5% chance of a major rotator cuff tear.16
CORRESPONDENCE Nilesh Shah, MD, Summa Center for Sports Health, 20 Olive Street, Suite 201, Akron, OH 44310; [email protected]
Shoulder pain is a common reason for visits to primary care physicians, who are most likely to diagnose it as rotator cuff tendinitis1,2—often erroneously. The complexity of the joint and the overlapping pathologies that may present as shoulder pain highlight the need to take a closer look when dealing with this diagnostic challenge.
Often, a targeted medical history—including the mechanism of injury and pain-provoking and pain-relieving factors—and a problem-based physical examination (incorporating many of the maneuvers highlighted in the text and tables that follow) will lead to an accurate diagnosis without the need for imaging studies. We recommend that imaging be reserved for patients who don’t respond to conventional treatments, cases in which the diagnosis is in doubt, and instances in which surgical intervention is being considered.
The 3 cases* that follow, and the take-away message incorporated in each, will give you an opportunity to test—and to hone—your shoulder pain diagnostic skill.
CASE 1 The history: Jesse, a 17-year-old student who’s active in football and track, came in during track season complaining of severe left shoulder pain. He denied any traumatic event or previous injury to the shoulder, but reported that any motion involving the shoulder caused pain. It hurt at night, the patient said, when he lay on his left side.
The physical: No muscle atrophy, redness, or swelling was evident, nor was there any indication of asymmetry or ecchymosis in the affected area. Jesse’s neck range of motion was normal; he had a very hard time with any active motion of the shoulder, however, because of the pain.
Evaluation of scapular motion demonstrated scapular dyskinesis3,4 without winging. Passive motion of the glenohumeral joint was much better than active motion. Strength testing appeared to be grossly intact but was limited by the pain. Shoulder impingement testing was positive. Sensation and deep tendon reflexes were intact.
Patients' names have been changed to protect their privacy
What’s the diagnosis?
Subacromial bursitis, suggested by the patient’s pain and altered scapular motion, was our working diagnosis, and we administered a subacromial injection of corticosteroid with lidocaine, for diagnostic as well as therapeutic purposes. Reexamination after the injection revealed immediate partial improvement in resting pain, range of motion, and strength. We referred Jesse to physical therapy with a focus on scapular stabilization and rotator cuff strength.
Three months later, Jesse returned to our office, complaining of weakness in his left shoulder. The pain had subsided a week after his first appointment, so he’d never gone to physical therapy. The weakness, which he had first noticed about 2 months after starting a lifting program in preparation for football season, was limited to resistance exercises, especially overhead shoulder presses and bench press. There were no other changes in his history, and he reported no reinjuries.
Physical examination revealed atrophy of the supraspinatus and infraspinatus muscles (FIGURE 1) and external rotation and shoulder abduction (in the scapular plane) resistance tests revealed weakness of these muscles. There was no scapular winging. The cervical spine exam was normal, and neurovascular status was intact in both upper extremities.
FIGURE 1
Severe shoulder pain, followed by weakness
Physical examination reveals atrophy of the patient’s supraspinatus (^) and infraspinatus (+) muscles.
New evidence points to nerve injury. Based on Jesse’s current history and physical, nerve injury was our new working diagnosis. (We considered the possibility of a rotator cuff tear, but this was not corroborated by the history.)
We ordered an electromyogram/nerve conduction velocity study to localize the lesion. The test revealed a brachial plexitis/neuritis (also known as Parsonage-Turner syndrome or brachial amyotrophy). The etiology of most atraumatic brachial plexopathies is unknown, and most are thought to be viral or autoimmune in nature.5,6
A classic case of Parsonage-Turner syndrome. The typical presentation of Parsonage-Turner syndrome (like Jesse’s) is one of acute, intense shoulder pain for no known reason. After 1 to 3 weeks, the pain resolves and the patient is left with weakness, usually of the supraspinatus and infraspinatus muscles. The weakness typically resolves with time, but full resolution may take 6 to 9 months.5,6 (In Jesse’s case, it took about 6 months.)
The take-away message: Look beyond the shoulder
As this case illustrates, not all shoulder pain originates in the shoulder. When evaluating shoulder pain, it is essential to consider other causes. The differential diagnosis for shoulder pain includes cervical spine disorders, cholecystitis (right shoulder), diaphragmatic irritation (eg, in the case of splenic rupture, usually involving the left shoulder), cardiac disease, and thoracic outlet syndrome.7
Evaluation of the cervical spine should be part of a complete shoulder examination. It is vital to follow a systematic approach that carefully assesses the cervical region for the possibility of nerve root impingement and radicular dysfunction masquerading as a primary shoulder disorder. (TABLE 18,9 details pain and sensory distribution patterns, reflex involvement, and potential motor impairments associated with various spinal nerve root levels.)
TABLE 1
Assessing the cervical spine
| Nerve root | Pain distribution | Sensory distribution | Reflex changes | Motor involvement |
|---|---|---|---|---|
| C5 | Lateral neck/upper trapezius | Lateral arm | Biceps | Deltoid, biceps |
| C6 | Base of neck/upper trapezius to superior glenohumeral joint | Radial aspect of distal forearm, thenar eminence, and index finger | Brachioradialis | Biceps, extensor carpi radialis longus and brevis (wrist extension) |
| C7 | Base of neck, almost entire upper quadrant of the back | Third finger | Triceps | Triceps, wrist flexion, finger extension |
| C8 | No shoulder pain | 4th and 5th fingers, distal half of forearm (ulna side) | None | Finger flexion (grip strength) |
| Adapted from: Miller JD, et al. Am Fam Physician. 20008; Eubanks JD. Am Fam Physician. 2010.9 | ||||
Practitioners should develop their own approach to “clearing the neck.” A logical order is to note posture of the head/neck/shoulders, observe active motion, perform palpation and provocative tests, and then assess neurologic function with sensation/reflex/strength testing. Provocative tests that can help to identify cervical involvement relating to shoulder pain include Spurling’s maneuver, axial compression test, abduction relief sign, and Lhermitte’s sign.10,11
CASE 2 The history: Mark, a 17-year-old, right-handed volleyball player, presented with right shoulder pain, which he felt whenever he spiked or served the ball. The pain started last season, Mark said, diminished during the months when he wasn’t playing, then got progressively worse as his activity level increased. The pain was in the posterior aspect of the shoulder.
The physical: Physical examination revealed a well-developed, but thin (6’4”, 170 pounds) young man who was not in distress. The general examination was benign, and a joint-specific exam showed no asymmetry or atrophy on inspection and no tenderness to palpation over the posterior and anterior soft tissues of the right shoulder. Rotator cuff testing yielded intact strength for all 4 muscles, but external rotation and supraspinatus testing elicited pain. The crank test, drawer sign, load and shift test, relocation test, and sulcus sign, detailed in TABLE 2,12-14 were all positive for shoulder instability; the Clunk and O’Brien tests were negative, and the contralateral shoulder exam was within normal limits. General joint laxity was observed, with the ability to oppose the thumb to the volar forearm and hyperextension noted in both elbows and knees. There were no outward signs of connective tissue disease.
Because of the chronicity of Mark’s pain and the progressive nature of his symptoms, we ordered radiographs, including anterior-posterior, lateral axillary, and scapular Y views. These films showed a nearly skeletally mature male without bony abnormalities; the humeral head was well located in the glenoid.
TABLE 2
Testing for shoulder instability12-14
| Test | Procedure | Positive result/implication |
|---|---|---|
| Apprehension | Patient supine, arm abducted 90º, externally rotated with anteriorly directed force applied to humeral head | Pain/apprehension with force suggests anterior instability |
| Relocation* | Patient supine, posteriorly directed force applied to humeral head | Relief with force suggests anterior instability |
| Crank | Patient sitting, arm abducted 90º, elbow flexed to 90º, humerus supported with forced external rotation | Pain/apprehension with forced external rotation suggests anterior instability |
| Load and shift | Patient supine, arm held by examiner and abducted 90º, force applied along axis of humerus to "seat" the humerus within the glenoid, followed by anterior force directed to humeral head | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Drawer | Patient sitting, arm at side, proximal humeral shaft grasped by examiner, seating the humeral head within the glenoid then applying anterior translational force | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Sulcus | Patient sitting, arm at side, forearm grasped by examiner with an inferior/caudally directed force applied | Sulcus or depression seen inferior to acromion as humeral head subluxes posteriorly, pathognomonic for multidirectional instability |
| Clunk | Patient supine, examiner grasps at forearm and humeral shaft, with humeral head seated within the fossa, taking the arm through passive ROM from extension through forward flexion | Clunk sound or clicking sensation suggests labral tear |
| O’Brien | Patient sitting, arm is forward flexed to 90º and fully adducted and internally rotated; patient resists downward motion. If pain is elicited, the maneuver is repeated in external rotation | Pain elicited with resisted downward motion in internal rotation but relieved with external rotation suggests labral pathology |
| *Perform only if apprehension test is positive. ROM, range of motion | ||
What’s the diagnosis?
Multidirectional instability with recurrent subluxations and probable acute rotator cuff tendinitis was our provisional diagnosis. Treatment focused on physical therapy, with a concentration on scapular stabilization and rotator cuff strengthening.
Shoulder instability is relatively common and represents a spectrum of disorders ranging from dislocation to subluxation to simple laxity.12,13 A complete loss of humeral articulation within the glenoid fossa is evidence of dislocation, whereas subluxation includes approximation of the humeral head to the limits of the glenoid rim. Dislocation typically results from trauma, whereas subluxation can be the result of microtrauma and repetitive overuse injury. Anterior instability is the most common type and is reported in as many as 95% of all dislocations.13
The take-away message: Rule out instability
The shoulder is one of the most complex joints in the body. The rotator cuff structures, the glenoid labrum, and the collective capsular ligaments provide structural stability to the glenohumeral joint.12,13 The shoulder is vulnerable to instability because the shallow glenoid fossa offers little bony support for the humeral head. Thus, instability should always be included in an assessment of shoulder pain.
Key factors to consider in identifying shoulder instability include the location of the pain, the direction of traumatic force applied, the presence of a known complete dislocation vs apprehension with specific movement, the position of the arm in which pain is elicited, a previous occurrence of instability (subluxation or dislocation), and the presence of tingling or numbness.12-14 The maneuvers detailed in TABLE 212-14 can help identify instability, as they did in this case. Patients with hypermobility are at increased risk for shoulder instability, so a targeted exam and patient history aimed at identifying signs and symptoms of hypermobility is needed, as well.
Ask the patient to attempt to:
- bend the thumb to the volar forearm
- place hands to the floor with hyperextended knees
- perform maximal hyperextension of the fifth metacarpophalangeal joint (>90° is a positive result).
Findings from the medical history that indicate a predisposition to instability include generalized joint laxity, Ehlers-Danlos syndrome, Marfan syndrome, osteogenesis imperfecta, hyperhomocysteinuria, hyperlysinemia, benign joint hypermobility syndrome, juvenile rheumatoid arthritis, and previous shoulder or patellar dislocations.
Imaging tips: Scapular Y and/or axillary lateral views should always be included when ordering imaging studies for suspected instability/dislocations, as 50% of posterior dislocations are missed on standard shoulder x-rays that do not include them.12 In reviewing the x-rays, it is important to look for signs of a compression fracture of the posterior humeral head (known as a Hill-Sachs lesion) for anterior shoulder dislocations, and fractures to the anterior glenoid rim (known as a Bankart lesion).12-14
CASE 3 The history: Robert, a right-handed, 50-year-old motorcycle instructor, came to our office because of chronic right shoulder pain. The pain, located over the anterior portion of the glenohumeral joint, developed insidiously about 3 or 4 years ago, the patient reported. He had finally decided to seek help because he’d recently experienced an acute exacerbation of pain brought on by shoveling snow, after which he also noticed associated weakness, a clicking/popping on active motion, and mild loss of motion.
The physical: Robert’s cervical spine exam was unremarkable. He demonstrated full active range of motion (ROM) without exacerbation of right shoulder symptoms, and special tests for disc pathology at the neck were negative. Active ROM testing of the right shoulder revealed full abduction, with only minimal pain; full flexion, with moderate pain noted initially at 49°; full extension, with a painful arc noted at 50°; and full horizontal adduction, with a painful arc noted at the halfway point. The testing also revealed that his right thumb was 3 inches lower than the left on reaching for the opposite scapula. At the superior aspect of the acromioclavicular (AC) joint, 2+ tenderness was noted; 3+ tenderness was noted at the greater tubercle of the humerus.
After inspecting the shoulder region for alterations in bony landmarks, muscle bulk, carrying position, and movement characteristics, palpation of the region was performed.
When assessing shoulder strength, there are a variety of tests for each functioning component of the rotator cuff structures (TABLE 3).15-17 Manual muscle tests revealed: 4-/5 on external rotation (French horn test), 3+/5 on the lift-off test, and 5-/5 on all other tests for right shoulder function. Impingement testing was slightly positive, or pain producing, on Hawkins and Neer tests.18,19 For the Hawkins test, the examiner flexes the arm to 90° of shoulder flexion with the elbow flexed at 90°, then internally rotates the shoulder. For the Neer test, the arm is fully elevated in the scapular plane and internally rotated by the examiner.
The subscapularis muscle, which functions primarily in internal rotation, is tested by the French horn and lift-off tests. The teres minor muscle, which performs external rotation, is tested by the French horn test of external rotation. And the supraspinatus muscle, which performs abduction and external rotation, is tested by the empty can (also known as the Jobe) and full can tests. Some researchers suggest that the empty can test is better for diagnosing impingement, based on evidence showing that the full can test is better at diagnosing supraspinatus tears because it causes less pain during testing.20
TABLE 3
Suspect rotator cuff involvement?7,15-17
What’s the diagnosis?
Rotator cuff tear was suspected because Robert had positive elements of the “rotator cuff triad”—supraspinatus weakness (as indicated by a positive empty can test), external rotation weakness (revealed by the French horn test), and a positive Hawkins impingement test. We ordered diagnostic studies, including plain radiographs, which revealed degenerative changes at the acromioclavicular joint, decreased acromiohumeral interval, and no significant changes at the glenohumeral joint (FIGURE 2), and magnetic resonance imaging (MRI) of the right shoulder. The MRI revealed a large, full-thickness rotator cuff tear of the supraspinatus tendon with retraction. A torn and retracted biceps tendon and AC joint osteoarthritis were also shown, likely causing a mass effect on the supraspinatus. The patient underwent surgery to repair the torn rotator cuff, with excellent results.
FIGURE 2
Chronic right shoulder pain
An AP view of the patient’s right shoulder shows acromioclavicular joint narrowing and degeneration and subtle narrowing of the acromiohumeral interval.
The take-away message: Keep the rotator-cuff triad in mind
Because none of the tests that comprise the triad is specific enough alone to diagnose a rotator cuff tear,15,20,21 Murrell and Walton16 suggested that the 3 tests be considered together for diagnostic purposes. If all 3 are positive, there is a 98% chance of a rotator cuff tear; if 2 tests are positive and the patient is older than 60 years, the findings are suggestive of a tear; and if all 3 tests (plus the drop arm test) are negative, there is less than a 5% chance of a major rotator cuff tear.16
CORRESPONDENCE Nilesh Shah, MD, Summa Center for Sports Health, 20 Olive Street, Suite 201, Akron, OH 44310; [email protected]
1. Van der Windt DA, Koes BW, De Jong BA, et al. Shoulder disorders in general practice: incidence, patient characteristics, and management. Ann Rheum Dis. 1995;54:959-964.
2. Johansson K, Adolfsson L, Foldevi M. Attitudes toward management of patients with subacromial pain in Swedish primary care. Fam Pract. 1999;16:233-237.
3. Kibler WB, McMullen J. Scapular dyskinesis and its relation to shoulder pain. J Am Acad Orthop Surg. 2003;11:142-151.
4. Kibler WB. The role of the scapula in athletic shoulder function. Am J Sports Med. 1998;26:325-337.
5. Vanermen B, Aertgeerts M, Hoogmartens M, et al. The syndrome of Parsonage and Turner. Discussion of clinical features with a review of 8 cases. Acta Orthop Belg. 1991;57:414-419.
6. Misamore GW, Lehman DE. Parsonage-Turner syndrome (acute brachial neuritis). J Bone Joint Surg. 1996;78:1405-1408.
7. Stevenson J, Trojian T. Evaluation of shoulder pain. J Fam Pract. 2002;51:605-611.
8. Miller JD, Pruitt RN, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
9. Eubanks JD. Cervical radiculopathy: nonoperative management of neck pain and radicular symptoms. Am Fam Physician. 2010;81:33-40.
10. Malanga GA, Landes P, Nadler SF. Provocative tests in cervical spine examination: historical basis and scientific analyses. Pain Physician. 2003;6:199-205.
11. Huston M, Ellis R. eds. Textbook of Musculoskeletal Medicine. Oxford, UK: Oxford University Press; 2005.
12. Mahaffey BL, Smith PA. Shoulder instability in young athletes. Am Fam Physician. 1999;59:2773-2782, 2787.
13. Petron DJ, Khan U. The shoulder and upper extremity. In: McKeag DB, Moeller JL. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Wolters Kluwer, Lippincott Williams & Wilkins; 2007:359–373.
14. Woodward TW, Best TM. The painful shoulder: part I. clinical evaluation. Am Fam Physician. 2000;61:3079-3088.
15. Kelly BT, Kadrmas WR, Speer KP. The manual muscle examination for rotator cuff strength: an electromyographic investigation. Am J Sports Med. 1996;24:581-588.
16. Murrell GA, Walton JR. Diagnosis of rotator cuff tears. Lancet. 2001;357:769-770.
17. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3:347-352.
18. Neer CS, Welsh RP. The shoulder in sports. Orthop Clin North Am. 1977;8:583-591.
19. Hawkins RJ, Kennedy JC. Impingement syndrome in athletics. Am J Sports Med. 1980;8:151-163.
20. Itoi E, Kido T, Sano A, et al. Which is more useful, the “full can test” or the “empty can test,” in detecting the torn supraspinatus tendon? Am J Sports Med. 1999;27:65-68.
21. Boettcher CE, Ginn KA, Cathers I. The ‘empty can’ and ‘full can’ tests do not selectively activate supraspinatus. J Sci Med Sport. 2009;12:435-439.
1. Van der Windt DA, Koes BW, De Jong BA, et al. Shoulder disorders in general practice: incidence, patient characteristics, and management. Ann Rheum Dis. 1995;54:959-964.
2. Johansson K, Adolfsson L, Foldevi M. Attitudes toward management of patients with subacromial pain in Swedish primary care. Fam Pract. 1999;16:233-237.
3. Kibler WB, McMullen J. Scapular dyskinesis and its relation to shoulder pain. J Am Acad Orthop Surg. 2003;11:142-151.
4. Kibler WB. The role of the scapula in athletic shoulder function. Am J Sports Med. 1998;26:325-337.
5. Vanermen B, Aertgeerts M, Hoogmartens M, et al. The syndrome of Parsonage and Turner. Discussion of clinical features with a review of 8 cases. Acta Orthop Belg. 1991;57:414-419.
6. Misamore GW, Lehman DE. Parsonage-Turner syndrome (acute brachial neuritis). J Bone Joint Surg. 1996;78:1405-1408.
7. Stevenson J, Trojian T. Evaluation of shoulder pain. J Fam Pract. 2002;51:605-611.
8. Miller JD, Pruitt RN, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
9. Eubanks JD. Cervical radiculopathy: nonoperative management of neck pain and radicular symptoms. Am Fam Physician. 2010;81:33-40.
10. Malanga GA, Landes P, Nadler SF. Provocative tests in cervical spine examination: historical basis and scientific analyses. Pain Physician. 2003;6:199-205.
11. Huston M, Ellis R. eds. Textbook of Musculoskeletal Medicine. Oxford, UK: Oxford University Press; 2005.
12. Mahaffey BL, Smith PA. Shoulder instability in young athletes. Am Fam Physician. 1999;59:2773-2782, 2787.
13. Petron DJ, Khan U. The shoulder and upper extremity. In: McKeag DB, Moeller JL. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Wolters Kluwer, Lippincott Williams & Wilkins; 2007:359–373.
14. Woodward TW, Best TM. The painful shoulder: part I. clinical evaluation. Am Fam Physician. 2000;61:3079-3088.
15. Kelly BT, Kadrmas WR, Speer KP. The manual muscle examination for rotator cuff strength: an electromyographic investigation. Am J Sports Med. 1996;24:581-588.
16. Murrell GA, Walton JR. Diagnosis of rotator cuff tears. Lancet. 2001;357:769-770.
17. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3:347-352.
18. Neer CS, Welsh RP. The shoulder in sports. Orthop Clin North Am. 1977;8:583-591.
19. Hawkins RJ, Kennedy JC. Impingement syndrome in athletics. Am J Sports Med. 1980;8:151-163.
20. Itoi E, Kido T, Sano A, et al. Which is more useful, the “full can test” or the “empty can test,” in detecting the torn supraspinatus tendon? Am J Sports Med. 1999;27:65-68.
21. Boettcher CE, Ginn KA, Cathers I. The ‘empty can’ and ‘full can’ tests do not selectively activate supraspinatus. J Sci Med Sport. 2009;12:435-439.
Suicide assessment: Targeting acute risk factors
At his wife’s urging, Mr. L, age 34, presents to the local emergency room (ER). Approximately 1 week ago, he woke up in the middle of the night and told her he was afraid he would die because he had heart palpitations, a choking sensation, dizziness, and shortness of breath.
The ER physician rules out an acute medical illness and requests a psychiatric consultation. Mr. L is reluctant to talk to the psychiatrist, saying he has just had a difficult couple of weeks because of problems at work. With Mr. L’s permission, the psychiatrist speaks with his wife and learns that for several weeks Mr. L has been having problems falling asleep and has been waking up early. Mrs. L noticed her husband is unable to sit still, not enjoying his favorite television shows, and drinking more alcohol at night.
The clinical picture became clearer after Mr. L tells the psychiatrist that approximately 1 month ago, he lost his appetite, had low energy and concentration, and began to feel depressed. He denies having suicidal thoughts or plans, but says his suffering is increasing and he doesn’t know what to do.
Suicide is our worst outcome; at times it can seem like we are helpless to change its frequency or evaluate its likelihood. As clinicians, we are not expected to predict who will commit suicide, but are expected to perform an adequate suicide risk assessment and determine who is at high risk. We need to clearly document a patient’s suicide risk level in his or her chart, and our subsequent actions need to be consistent with that assessment. For instance, arranging for additional supports—including psychiatric hospitalization when necessary—for a patient deemed to be at high risk for suicide is considered the standard of care. In this article, I:
- discuss demographic factors related to suicide
- explore the importance of time-related suicide risk factors and the few treatments shown to reduce suicide risk
- review protective and preventive factors.
Sobering statistics
Over the past decade, suicide rates in the United States have remained fixed at slightly more than 30,000 per year. In 2009—the most recent year for which statistics are available from the Centers for Disease Control and Prevention—there were 36,547 suicides in the United States, making it the 10th leading cause of death.1 The rates of suicide completions and attempts vary by sex and age. Males complete suicide 4 times more often than females, whereas females attempt suicide 3 times more often. Among individuals age 15 to 24, 86% of those who completed suicide were male; in older persons (age >65), 85% were male.2 Although rates of completed suicide are highest among older adults, rates of suicide attempts are greatest among young persons. The ratio of attempted-to-completed suicide is 100 to 200:1 in individuals age 15 to 24 but 4:1 in those age >65.2
Whites and Native Americans have the highest suicide rates (12.3 and 12.9 per 100,000, respectively).2 Guns are the most common method of completed suicide in all age groups in the United States: they are used in 53% of all suicides and 76% of those among persons age >70.3 In >90% of completed suicides, the decedent had been diagnosed with ≥1 psychiatric disorder.3 By far, the most common psychiatric illness is major depressive disorder, present in 75% of those who commit suicide.3
Understanding intent
Many physicians believe that patients will tell them if they are feeling worse and are starting to think more seriously about suicide. There is no better example of this than the “contract for safety” or “no-harm contract,” in which a patient signs a paper agreeing to notify a clinician if he begins to develop more intense suicidal feelings. Studies have shown that these “no-harm contracts” do not prevent suicide; this makes sense because if a patient decides to kill himself, telling a clinician puts up an obstacle.4-6
Patients who commit suicide often communicate their suicidal intent, but usually tell family members rather than clinicians. In 1 study, 78% of patients who committed suicide on an inpatient unit denied suicidal ideation at their last communication with staff; although 60% told their spouse and 50% told other relatives, only 18% told their physician.7 In this study, precautions provided a false sense of security: 51% of patients were receiving 15-minute suicide checks or 1-to-1 observation at the time of suicide.7
Who is at risk?
The most recent American Psychiatric Association Task Force Report on Suicide identified 57 risk factors for suicide.8,9 This has led to confusion among clinicians and may have led some clinicians to repeatedly ask patients about suicidal ideation rather than conduct a suicide risk assessment.
Although a history of suicide attempts and a family history of suicide are well-established risk factors,9 these are not acute factors. It is important to differentiate between suicide attempts and suicide completions. Although many suicide attempts are accurate substitutes for actual suicides, there is a spectrum of intent in suicide attempts that differentiates them in terms of lethality.10 Clinicians need a more thorough understanding of who is at acute risk for suicide, which will help them make decisions about patients’ imminent risk to themselves.
In the only study that examined time-related predictors of suicide, Fawcett et al11 used the Schedule for Affective Disorders and Schizophrenia (SADS) to evaluate 954 patients with major affective disorders over 10 years. Raters were blinded to treatment, and clinicians could use any combination of psychotherapy or pharmacotherapy. These researchers found that acute risk factors—those associated with suicide within 1 year—were psychic anxiety, anhedonia, diminished concentration, insomnia, panic attacks, and active alcohol abuse (Table 1).11 These factors were present in the context of an underlying depressive disorder. Hopelessness, suicidal ideation, and a history of suicide attempts were linked to suicide between 2 and 10 years.
Busch et al12 performed a retrospective study on an inpatient unit using the SADS to evaluate symptoms present the week before patients’ suicides. They found that 79% of patients had extreme psychic anxiety, agitation, or both, and that 54% had active psychosis. The same authors studied an additional 12 cases of inpatient suicide and found 9 patients had severe anxiety, agitation, or both, and insomnia. The median time to suicide from admission was 3.5 days and none of the 12 patients had been started on an antidepressant, antipsychotic, or anxiolytic. This underscores the need to initiate symptomatic treatment quickly, even before reaching a definitive diagnosis.
The Columbia Suicide Severity Rating Scale (C-SSRS), which evaluates suicide ideation and behavior in the past week and lifetime, has predictive validity in determining those at highest risk for making a suicide attempt within up to 24 weeks of follow-up.13 A limitation of the C-SSRS is that it has predictive validity for suicide attempts only, and not suicide completions.
Table 1
Acute suicide risk factors: 3 A’s + 3 P’s
| Alcohol abuse |
| Attention (or concentration) impairment |
| Awake (insomnia) |
| Panic attacks |
| Pleasure (diminished) |
| Psychic anxiety |
| Source: Reference 11 |
Treatments to lower risk
Although identifying risk factors such as older age, being unmarried, male sex, experiencing a recent loss, a family history of completed suicide, and being white or Native American are helpful in evaluating a patient’s suicide risk, they are not time-sensitive or modifiable, which limits their value.
In contrast, most of the acute risk factors identified by Fawcett et al potentially are treatable. Psychic anxiety, insomnia, and panic attacks can be treated with benzodiazepines or other anxiolytics and sedative/hypnotics. Active psychosis, which Busch et al identified as a risk factor for inpatient suicide, may respond to antipsychotics.
Other medications have been identified as modifying suicide risk (Table 2).14-20 Among patients with major affective disorders, lithium has been shown to reduce suicidal acts by 93%, suicide attempts by 93%, and suicide completions by 82%.14 Lithium produces the largest suicide risk reduction in unipolar depression, at 100%, followed by bipolar II disorder (82%) and bipolar I disorder (67%).15 Several studies have demonstrated that lithium can reduce the mortality rate from suicide for patients with affective disorders, and that this effect persists.16,17
Clozapine has been associated with reduced rates of suicide attempts and completed suicides in patients with chronic psychosis. In a meta-analysis, long-term clozapine treatment was associated with an approximately 3-fold overall reduction of risk of suicidal behaviors,18 although a prospective study found no reduction in risk of completed suicide in patients with schizophrenia treated with clozapine.19
In one study, electroconvulsive therapy (ECT) reduced suicidal thoughts and acts by 38% after 1 week and 80% overall.20 There have been reports of amelioration of suicidal thoughts after just 1 ECT treatment.21 There are no published studies that show a reduction in suicide completions with ECT; however, this may be due to the relatively small number of patients who receive ECT and the infrequency of completed suicides.
Protective factors. The balance between protective factors and risk factors determines appropriate clinical decision making when attempting to evaluate a patient’s suicide risk. Perhaps the best measure of protective factors is the Reasons for Living Inventory, developed by Linehan et al,22 which has been validated in some populations, including adolescents and young adults.23 This inventory delineates protective factors against suicidal ideation and behavior rather than completed suicides.
Similar to suicide protection, suicide prevention focuses on factors that can serve as obstacles to a patient’s desire or ability to commit suicide. A large systematic literature review by Mann et al24 found that only primary care physician education and restricting access to lethal means prevented suicide. When working to remove lethal means from a suicidal patient’s home, it is critical to verify that this has been done rather than merely making a suggestion to a family member. It is necessary to follow up with a phone call and document the completion of this task.
When a patient commits suicide, it is common for psychiatrists to feel like there must have been something they could have done to prevent such a tragedy. Although typically that is not the case, there is more we can do to improve our suicide risk assessment skills. Focusing on acute, modifiable suicide risk factors may help us lower a patient’s risk. Also, shortening the time frame now considered acute (within 1 year) to hours and days and looking for additional risk factors may improve mental health professionals’ ability to accurately assess acute suicide risk.
Table 2
Treatments to lower suicide risk
| Acute |
| Benzodiazepines—to diminish panic, anxiety, insomnia |
| Antipsychotics—if acute psychosis is present |
| Trazodone (or non-benzodiazepine hypnotics)—if insomnia is present without daytime anxiety |
| Diagnosis–specific |
| Clozapine—for patients with schizophrenia and high suicide risk |
| Lithium—for patients with bipolar disorder (if not contraindicated); consider for patients with refractory unipolar depression at high suicide risk |
| Electroconvulsive therapy—for patients with severe depression and high suicide risk |
| Source: References 14-20 |
CASE CONTINUED: Hospitalization and improvement
The psychiatrist determines Mr. L is at high risk for suicide and recommends psychiatric hospitalization. She starts him on citalopram, 10 mg/d, and clonazepam, 0.5 mg twice daily and 1 mg at bedtime, to help with anxiety and insomnia. After 3 days, Mr. L tolerates the medications, sleeps better, and feels more hopeful about the future. The psychiatrist increases citalopram to 20 mg/d.
Four days later, Mr. L is eating better, can concentrate, and denies further episodes of dizziness or anxiety. The inpatient psychiatrist assesses his acute suicide risk as low and discharges him to a week-long partial hospitalization program.
Related Resources
- American Association of Suicidology. www.suicidology.org.
- Harvard School of Public Health. Means Matter. www.hsph.harvard.edu/means-matter.
- Simon RI. Preventing patient suicide: clinical assessment and management. Arlington, VA: American Psychiatric Publishing; 2011.
Drug Brand Names
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Lithium • Eskalith, Lithobid
- Trazodone • Desyrel, Oleptro
Disclosure
Dr. Freeman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention. US death rate falls for 10th straight year. http://www.cdc.gov/media/releases/2011/p0316_deathrate.html. Published March 16 2011. Accessed November 22, 2011.
2. Centers for Disease Control and Prevention. Suicide: facts at a glance. http://www.cdc.gov/violenceprevention/suicide. Published Summer 2010. Accessed November 17 2011.
3. Karch DL, Dahlberg LL, Patel N. Surveillance for violent deaths—National Violent Death Reporting System 16 states, 2007. MMWR Surveill Summ. 2010;59(4):1-50.
4. Resnick PJ. Recognizing that the suicidal patient views you as an ‘adversary.’ Current Psychiatry. 2002;1(1):8.-
5. Stanford EJ, Goetz RR, Bloom JD. The No Harm Contract in the emergency assessment of suicidal risk. J Clin Psychiatry. 1994;55(8):344-348.
6. Edwards SJ, Sachmann MD. No-suicide contracts no-suicide agreements, and no-suicide assurances: a study of their nature, utilization, perceived effectiveness, and potential to cause harm. Crisis. 2010;31(6):290-302.
7. Busch KA, Fawcett J, Jacobs DG. Clinical correlates of inpatient suicide. J Clin Psychiatry. 2003;64(1):14-19.
8. American Psychiatric Association. Practice guideline for the assessment and treatment of patients with suicidal behaviors. http://psychiatryonline.org/content.aspx?bookid=28§ionid=1673332#56008. Published November 2003. Accessed November 22 2011.
9. Jacobs DG, Brewer ML. Application of the APA practice guidelines on suicide to clinical practice. CNS Spectr. 2006;11(6):447-454.
10. Nasser EH, Overholser JC. Assessing varying degrees of lethality in depressed adolescent suicide attempters. Acta Psychiatr Scand. 1999;99(6):423-431.
11. Fawcett J, Scheftner WA, Fogg L, et al. Time-related predictors of suicide in major affective disorder. Am J Psychiatry. 1990;147(9):1189-1194.
12. Busch KA, Fawcett J. A fine-grained study of patients who commit suicide. Psychiatric Ann. 2004;34(5):357-364.
13. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale (C-SSRS): initial validity and internal consistency findings from three multi-site studies with adolescents and adults. Am J Psychiatry. 2011;168:1266-1277.
14. Müller-Oerlinghausen B, Berghöfer A, Ahrens B. The antisuicidal and mortality-reducing effect of lithium prophylaxis: consequences for guidelines in clinical psychiatry. Can J Psychiatry. 2003;48(7):433-439.
15. Tondo L, Hennen J, Baldessarini RJ. Lower suicide risk with long-term lithium treatment in major affective illness: a meta-analysis. Acta Psychiatr Scand. 2001;104(3):163-172.
16. Kessing LV, Søndergård L, Kvist K, et al. Suicide risk in patients treated with lithium. Arch Gen Psychiatry. 2005;62(8):860-866.
17. Baldessarini RJ, Tondo L, Hennen J. Lithium treatment and suicide risk in major affective disorders: update and new findings. J Clin Psychiatry. 2003;64(suppl 5):44-52.
18. Hennen J, Baldessarini RJ. Suicidal risk during treatment with clozapine: a meta-analysis. Schizophr Res. 2005;73(2-3):139-145.
19. Kuo CJ, Tsai SY, Lo CH, et al. Risk factors for completed suicide in schizophrenia. J Clin Psychiatry. 2005;66(5):579-585.
20. Kobeissi J, Aloysi A, Tobias K, et al. Resolution of severe suicidality with a single electroconvulsive therapy. J ECT. 2011;27(1):86-88.
21. Kellner CH, Fink M, Knapp R, et al. Relief of expressed suicidal intent by ECT: a consortium for research in ECT study. Am J Psychiatry. 2005;162(5):977-982.
22. Linehan MM, Goodstein JL, Nielsen SL, et al. Reasons for staying alive when you are thinking of killing yourself: the reasons for living inventory. J Consult Clin Psychol. 1983;51(2):276-286.
23. Cole DA. Validation of the reasons for living inventory in general and delinquent adolescent samples. J Abnorm Child Psychol. 1989;17(1):13-27.
24. Mann JJ, Apter A, Bertolote J, et al. Suicide prevention strategies: a systematic review. JAMA. 2005;294(16):2064-2074.
At his wife’s urging, Mr. L, age 34, presents to the local emergency room (ER). Approximately 1 week ago, he woke up in the middle of the night and told her he was afraid he would die because he had heart palpitations, a choking sensation, dizziness, and shortness of breath.
The ER physician rules out an acute medical illness and requests a psychiatric consultation. Mr. L is reluctant to talk to the psychiatrist, saying he has just had a difficult couple of weeks because of problems at work. With Mr. L’s permission, the psychiatrist speaks with his wife and learns that for several weeks Mr. L has been having problems falling asleep and has been waking up early. Mrs. L noticed her husband is unable to sit still, not enjoying his favorite television shows, and drinking more alcohol at night.
The clinical picture became clearer after Mr. L tells the psychiatrist that approximately 1 month ago, he lost his appetite, had low energy and concentration, and began to feel depressed. He denies having suicidal thoughts or plans, but says his suffering is increasing and he doesn’t know what to do.
Suicide is our worst outcome; at times it can seem like we are helpless to change its frequency or evaluate its likelihood. As clinicians, we are not expected to predict who will commit suicide, but are expected to perform an adequate suicide risk assessment and determine who is at high risk. We need to clearly document a patient’s suicide risk level in his or her chart, and our subsequent actions need to be consistent with that assessment. For instance, arranging for additional supports—including psychiatric hospitalization when necessary—for a patient deemed to be at high risk for suicide is considered the standard of care. In this article, I:
- discuss demographic factors related to suicide
- explore the importance of time-related suicide risk factors and the few treatments shown to reduce suicide risk
- review protective and preventive factors.
Sobering statistics
Over the past decade, suicide rates in the United States have remained fixed at slightly more than 30,000 per year. In 2009—the most recent year for which statistics are available from the Centers for Disease Control and Prevention—there were 36,547 suicides in the United States, making it the 10th leading cause of death.1 The rates of suicide completions and attempts vary by sex and age. Males complete suicide 4 times more often than females, whereas females attempt suicide 3 times more often. Among individuals age 15 to 24, 86% of those who completed suicide were male; in older persons (age >65), 85% were male.2 Although rates of completed suicide are highest among older adults, rates of suicide attempts are greatest among young persons. The ratio of attempted-to-completed suicide is 100 to 200:1 in individuals age 15 to 24 but 4:1 in those age >65.2
Whites and Native Americans have the highest suicide rates (12.3 and 12.9 per 100,000, respectively).2 Guns are the most common method of completed suicide in all age groups in the United States: they are used in 53% of all suicides and 76% of those among persons age >70.3 In >90% of completed suicides, the decedent had been diagnosed with ≥1 psychiatric disorder.3 By far, the most common psychiatric illness is major depressive disorder, present in 75% of those who commit suicide.3
Understanding intent
Many physicians believe that patients will tell them if they are feeling worse and are starting to think more seriously about suicide. There is no better example of this than the “contract for safety” or “no-harm contract,” in which a patient signs a paper agreeing to notify a clinician if he begins to develop more intense suicidal feelings. Studies have shown that these “no-harm contracts” do not prevent suicide; this makes sense because if a patient decides to kill himself, telling a clinician puts up an obstacle.4-6
Patients who commit suicide often communicate their suicidal intent, but usually tell family members rather than clinicians. In 1 study, 78% of patients who committed suicide on an inpatient unit denied suicidal ideation at their last communication with staff; although 60% told their spouse and 50% told other relatives, only 18% told their physician.7 In this study, precautions provided a false sense of security: 51% of patients were receiving 15-minute suicide checks or 1-to-1 observation at the time of suicide.7
Who is at risk?
The most recent American Psychiatric Association Task Force Report on Suicide identified 57 risk factors for suicide.8,9 This has led to confusion among clinicians and may have led some clinicians to repeatedly ask patients about suicidal ideation rather than conduct a suicide risk assessment.
Although a history of suicide attempts and a family history of suicide are well-established risk factors,9 these are not acute factors. It is important to differentiate between suicide attempts and suicide completions. Although many suicide attempts are accurate substitutes for actual suicides, there is a spectrum of intent in suicide attempts that differentiates them in terms of lethality.10 Clinicians need a more thorough understanding of who is at acute risk for suicide, which will help them make decisions about patients’ imminent risk to themselves.
In the only study that examined time-related predictors of suicide, Fawcett et al11 used the Schedule for Affective Disorders and Schizophrenia (SADS) to evaluate 954 patients with major affective disorders over 10 years. Raters were blinded to treatment, and clinicians could use any combination of psychotherapy or pharmacotherapy. These researchers found that acute risk factors—those associated with suicide within 1 year—were psychic anxiety, anhedonia, diminished concentration, insomnia, panic attacks, and active alcohol abuse (Table 1).11 These factors were present in the context of an underlying depressive disorder. Hopelessness, suicidal ideation, and a history of suicide attempts were linked to suicide between 2 and 10 years.
Busch et al12 performed a retrospective study on an inpatient unit using the SADS to evaluate symptoms present the week before patients’ suicides. They found that 79% of patients had extreme psychic anxiety, agitation, or both, and that 54% had active psychosis. The same authors studied an additional 12 cases of inpatient suicide and found 9 patients had severe anxiety, agitation, or both, and insomnia. The median time to suicide from admission was 3.5 days and none of the 12 patients had been started on an antidepressant, antipsychotic, or anxiolytic. This underscores the need to initiate symptomatic treatment quickly, even before reaching a definitive diagnosis.
The Columbia Suicide Severity Rating Scale (C-SSRS), which evaluates suicide ideation and behavior in the past week and lifetime, has predictive validity in determining those at highest risk for making a suicide attempt within up to 24 weeks of follow-up.13 A limitation of the C-SSRS is that it has predictive validity for suicide attempts only, and not suicide completions.
Table 1
Acute suicide risk factors: 3 A’s + 3 P’s
| Alcohol abuse |
| Attention (or concentration) impairment |
| Awake (insomnia) |
| Panic attacks |
| Pleasure (diminished) |
| Psychic anxiety |
| Source: Reference 11 |
Treatments to lower risk
Although identifying risk factors such as older age, being unmarried, male sex, experiencing a recent loss, a family history of completed suicide, and being white or Native American are helpful in evaluating a patient’s suicide risk, they are not time-sensitive or modifiable, which limits their value.
In contrast, most of the acute risk factors identified by Fawcett et al potentially are treatable. Psychic anxiety, insomnia, and panic attacks can be treated with benzodiazepines or other anxiolytics and sedative/hypnotics. Active psychosis, which Busch et al identified as a risk factor for inpatient suicide, may respond to antipsychotics.
Other medications have been identified as modifying suicide risk (Table 2).14-20 Among patients with major affective disorders, lithium has been shown to reduce suicidal acts by 93%, suicide attempts by 93%, and suicide completions by 82%.14 Lithium produces the largest suicide risk reduction in unipolar depression, at 100%, followed by bipolar II disorder (82%) and bipolar I disorder (67%).15 Several studies have demonstrated that lithium can reduce the mortality rate from suicide for patients with affective disorders, and that this effect persists.16,17
Clozapine has been associated with reduced rates of suicide attempts and completed suicides in patients with chronic psychosis. In a meta-analysis, long-term clozapine treatment was associated with an approximately 3-fold overall reduction of risk of suicidal behaviors,18 although a prospective study found no reduction in risk of completed suicide in patients with schizophrenia treated with clozapine.19
In one study, electroconvulsive therapy (ECT) reduced suicidal thoughts and acts by 38% after 1 week and 80% overall.20 There have been reports of amelioration of suicidal thoughts after just 1 ECT treatment.21 There are no published studies that show a reduction in suicide completions with ECT; however, this may be due to the relatively small number of patients who receive ECT and the infrequency of completed suicides.
Protective factors. The balance between protective factors and risk factors determines appropriate clinical decision making when attempting to evaluate a patient’s suicide risk. Perhaps the best measure of protective factors is the Reasons for Living Inventory, developed by Linehan et al,22 which has been validated in some populations, including adolescents and young adults.23 This inventory delineates protective factors against suicidal ideation and behavior rather than completed suicides.
Similar to suicide protection, suicide prevention focuses on factors that can serve as obstacles to a patient’s desire or ability to commit suicide. A large systematic literature review by Mann et al24 found that only primary care physician education and restricting access to lethal means prevented suicide. When working to remove lethal means from a suicidal patient’s home, it is critical to verify that this has been done rather than merely making a suggestion to a family member. It is necessary to follow up with a phone call and document the completion of this task.
When a patient commits suicide, it is common for psychiatrists to feel like there must have been something they could have done to prevent such a tragedy. Although typically that is not the case, there is more we can do to improve our suicide risk assessment skills. Focusing on acute, modifiable suicide risk factors may help us lower a patient’s risk. Also, shortening the time frame now considered acute (within 1 year) to hours and days and looking for additional risk factors may improve mental health professionals’ ability to accurately assess acute suicide risk.
Table 2
Treatments to lower suicide risk
| Acute |
| Benzodiazepines—to diminish panic, anxiety, insomnia |
| Antipsychotics—if acute psychosis is present |
| Trazodone (or non-benzodiazepine hypnotics)—if insomnia is present without daytime anxiety |
| Diagnosis–specific |
| Clozapine—for patients with schizophrenia and high suicide risk |
| Lithium—for patients with bipolar disorder (if not contraindicated); consider for patients with refractory unipolar depression at high suicide risk |
| Electroconvulsive therapy—for patients with severe depression and high suicide risk |
| Source: References 14-20 |
CASE CONTINUED: Hospitalization and improvement
The psychiatrist determines Mr. L is at high risk for suicide and recommends psychiatric hospitalization. She starts him on citalopram, 10 mg/d, and clonazepam, 0.5 mg twice daily and 1 mg at bedtime, to help with anxiety and insomnia. After 3 days, Mr. L tolerates the medications, sleeps better, and feels more hopeful about the future. The psychiatrist increases citalopram to 20 mg/d.
Four days later, Mr. L is eating better, can concentrate, and denies further episodes of dizziness or anxiety. The inpatient psychiatrist assesses his acute suicide risk as low and discharges him to a week-long partial hospitalization program.
Related Resources
- American Association of Suicidology. www.suicidology.org.
- Harvard School of Public Health. Means Matter. www.hsph.harvard.edu/means-matter.
- Simon RI. Preventing patient suicide: clinical assessment and management. Arlington, VA: American Psychiatric Publishing; 2011.
Drug Brand Names
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Lithium • Eskalith, Lithobid
- Trazodone • Desyrel, Oleptro
Disclosure
Dr. Freeman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
At his wife’s urging, Mr. L, age 34, presents to the local emergency room (ER). Approximately 1 week ago, he woke up in the middle of the night and told her he was afraid he would die because he had heart palpitations, a choking sensation, dizziness, and shortness of breath.
The ER physician rules out an acute medical illness and requests a psychiatric consultation. Mr. L is reluctant to talk to the psychiatrist, saying he has just had a difficult couple of weeks because of problems at work. With Mr. L’s permission, the psychiatrist speaks with his wife and learns that for several weeks Mr. L has been having problems falling asleep and has been waking up early. Mrs. L noticed her husband is unable to sit still, not enjoying his favorite television shows, and drinking more alcohol at night.
The clinical picture became clearer after Mr. L tells the psychiatrist that approximately 1 month ago, he lost his appetite, had low energy and concentration, and began to feel depressed. He denies having suicidal thoughts or plans, but says his suffering is increasing and he doesn’t know what to do.
Suicide is our worst outcome; at times it can seem like we are helpless to change its frequency or evaluate its likelihood. As clinicians, we are not expected to predict who will commit suicide, but are expected to perform an adequate suicide risk assessment and determine who is at high risk. We need to clearly document a patient’s suicide risk level in his or her chart, and our subsequent actions need to be consistent with that assessment. For instance, arranging for additional supports—including psychiatric hospitalization when necessary—for a patient deemed to be at high risk for suicide is considered the standard of care. In this article, I:
- discuss demographic factors related to suicide
- explore the importance of time-related suicide risk factors and the few treatments shown to reduce suicide risk
- review protective and preventive factors.
Sobering statistics
Over the past decade, suicide rates in the United States have remained fixed at slightly more than 30,000 per year. In 2009—the most recent year for which statistics are available from the Centers for Disease Control and Prevention—there were 36,547 suicides in the United States, making it the 10th leading cause of death.1 The rates of suicide completions and attempts vary by sex and age. Males complete suicide 4 times more often than females, whereas females attempt suicide 3 times more often. Among individuals age 15 to 24, 86% of those who completed suicide were male; in older persons (age >65), 85% were male.2 Although rates of completed suicide are highest among older adults, rates of suicide attempts are greatest among young persons. The ratio of attempted-to-completed suicide is 100 to 200:1 in individuals age 15 to 24 but 4:1 in those age >65.2
Whites and Native Americans have the highest suicide rates (12.3 and 12.9 per 100,000, respectively).2 Guns are the most common method of completed suicide in all age groups in the United States: they are used in 53% of all suicides and 76% of those among persons age >70.3 In >90% of completed suicides, the decedent had been diagnosed with ≥1 psychiatric disorder.3 By far, the most common psychiatric illness is major depressive disorder, present in 75% of those who commit suicide.3
Understanding intent
Many physicians believe that patients will tell them if they are feeling worse and are starting to think more seriously about suicide. There is no better example of this than the “contract for safety” or “no-harm contract,” in which a patient signs a paper agreeing to notify a clinician if he begins to develop more intense suicidal feelings. Studies have shown that these “no-harm contracts” do not prevent suicide; this makes sense because if a patient decides to kill himself, telling a clinician puts up an obstacle.4-6
Patients who commit suicide often communicate their suicidal intent, but usually tell family members rather than clinicians. In 1 study, 78% of patients who committed suicide on an inpatient unit denied suicidal ideation at their last communication with staff; although 60% told their spouse and 50% told other relatives, only 18% told their physician.7 In this study, precautions provided a false sense of security: 51% of patients were receiving 15-minute suicide checks or 1-to-1 observation at the time of suicide.7
Who is at risk?
The most recent American Psychiatric Association Task Force Report on Suicide identified 57 risk factors for suicide.8,9 This has led to confusion among clinicians and may have led some clinicians to repeatedly ask patients about suicidal ideation rather than conduct a suicide risk assessment.
Although a history of suicide attempts and a family history of suicide are well-established risk factors,9 these are not acute factors. It is important to differentiate between suicide attempts and suicide completions. Although many suicide attempts are accurate substitutes for actual suicides, there is a spectrum of intent in suicide attempts that differentiates them in terms of lethality.10 Clinicians need a more thorough understanding of who is at acute risk for suicide, which will help them make decisions about patients’ imminent risk to themselves.
In the only study that examined time-related predictors of suicide, Fawcett et al11 used the Schedule for Affective Disorders and Schizophrenia (SADS) to evaluate 954 patients with major affective disorders over 10 years. Raters were blinded to treatment, and clinicians could use any combination of psychotherapy or pharmacotherapy. These researchers found that acute risk factors—those associated with suicide within 1 year—were psychic anxiety, anhedonia, diminished concentration, insomnia, panic attacks, and active alcohol abuse (Table 1).11 These factors were present in the context of an underlying depressive disorder. Hopelessness, suicidal ideation, and a history of suicide attempts were linked to suicide between 2 and 10 years.
Busch et al12 performed a retrospective study on an inpatient unit using the SADS to evaluate symptoms present the week before patients’ suicides. They found that 79% of patients had extreme psychic anxiety, agitation, or both, and that 54% had active psychosis. The same authors studied an additional 12 cases of inpatient suicide and found 9 patients had severe anxiety, agitation, or both, and insomnia. The median time to suicide from admission was 3.5 days and none of the 12 patients had been started on an antidepressant, antipsychotic, or anxiolytic. This underscores the need to initiate symptomatic treatment quickly, even before reaching a definitive diagnosis.
The Columbia Suicide Severity Rating Scale (C-SSRS), which evaluates suicide ideation and behavior in the past week and lifetime, has predictive validity in determining those at highest risk for making a suicide attempt within up to 24 weeks of follow-up.13 A limitation of the C-SSRS is that it has predictive validity for suicide attempts only, and not suicide completions.
Table 1
Acute suicide risk factors: 3 A’s + 3 P’s
| Alcohol abuse |
| Attention (or concentration) impairment |
| Awake (insomnia) |
| Panic attacks |
| Pleasure (diminished) |
| Psychic anxiety |
| Source: Reference 11 |
Treatments to lower risk
Although identifying risk factors such as older age, being unmarried, male sex, experiencing a recent loss, a family history of completed suicide, and being white or Native American are helpful in evaluating a patient’s suicide risk, they are not time-sensitive or modifiable, which limits their value.
In contrast, most of the acute risk factors identified by Fawcett et al potentially are treatable. Psychic anxiety, insomnia, and panic attacks can be treated with benzodiazepines or other anxiolytics and sedative/hypnotics. Active psychosis, which Busch et al identified as a risk factor for inpatient suicide, may respond to antipsychotics.
Other medications have been identified as modifying suicide risk (Table 2).14-20 Among patients with major affective disorders, lithium has been shown to reduce suicidal acts by 93%, suicide attempts by 93%, and suicide completions by 82%.14 Lithium produces the largest suicide risk reduction in unipolar depression, at 100%, followed by bipolar II disorder (82%) and bipolar I disorder (67%).15 Several studies have demonstrated that lithium can reduce the mortality rate from suicide for patients with affective disorders, and that this effect persists.16,17
Clozapine has been associated with reduced rates of suicide attempts and completed suicides in patients with chronic psychosis. In a meta-analysis, long-term clozapine treatment was associated with an approximately 3-fold overall reduction of risk of suicidal behaviors,18 although a prospective study found no reduction in risk of completed suicide in patients with schizophrenia treated with clozapine.19
In one study, electroconvulsive therapy (ECT) reduced suicidal thoughts and acts by 38% after 1 week and 80% overall.20 There have been reports of amelioration of suicidal thoughts after just 1 ECT treatment.21 There are no published studies that show a reduction in suicide completions with ECT; however, this may be due to the relatively small number of patients who receive ECT and the infrequency of completed suicides.
Protective factors. The balance between protective factors and risk factors determines appropriate clinical decision making when attempting to evaluate a patient’s suicide risk. Perhaps the best measure of protective factors is the Reasons for Living Inventory, developed by Linehan et al,22 which has been validated in some populations, including adolescents and young adults.23 This inventory delineates protective factors against suicidal ideation and behavior rather than completed suicides.
Similar to suicide protection, suicide prevention focuses on factors that can serve as obstacles to a patient’s desire or ability to commit suicide. A large systematic literature review by Mann et al24 found that only primary care physician education and restricting access to lethal means prevented suicide. When working to remove lethal means from a suicidal patient’s home, it is critical to verify that this has been done rather than merely making a suggestion to a family member. It is necessary to follow up with a phone call and document the completion of this task.
When a patient commits suicide, it is common for psychiatrists to feel like there must have been something they could have done to prevent such a tragedy. Although typically that is not the case, there is more we can do to improve our suicide risk assessment skills. Focusing on acute, modifiable suicide risk factors may help us lower a patient’s risk. Also, shortening the time frame now considered acute (within 1 year) to hours and days and looking for additional risk factors may improve mental health professionals’ ability to accurately assess acute suicide risk.
Table 2
Treatments to lower suicide risk
| Acute |
| Benzodiazepines—to diminish panic, anxiety, insomnia |
| Antipsychotics—if acute psychosis is present |
| Trazodone (or non-benzodiazepine hypnotics)—if insomnia is present without daytime anxiety |
| Diagnosis–specific |
| Clozapine—for patients with schizophrenia and high suicide risk |
| Lithium—for patients with bipolar disorder (if not contraindicated); consider for patients with refractory unipolar depression at high suicide risk |
| Electroconvulsive therapy—for patients with severe depression and high suicide risk |
| Source: References 14-20 |
CASE CONTINUED: Hospitalization and improvement
The psychiatrist determines Mr. L is at high risk for suicide and recommends psychiatric hospitalization. She starts him on citalopram, 10 mg/d, and clonazepam, 0.5 mg twice daily and 1 mg at bedtime, to help with anxiety and insomnia. After 3 days, Mr. L tolerates the medications, sleeps better, and feels more hopeful about the future. The psychiatrist increases citalopram to 20 mg/d.
Four days later, Mr. L is eating better, can concentrate, and denies further episodes of dizziness or anxiety. The inpatient psychiatrist assesses his acute suicide risk as low and discharges him to a week-long partial hospitalization program.
Related Resources
- American Association of Suicidology. www.suicidology.org.
- Harvard School of Public Health. Means Matter. www.hsph.harvard.edu/means-matter.
- Simon RI. Preventing patient suicide: clinical assessment and management. Arlington, VA: American Psychiatric Publishing; 2011.
Drug Brand Names
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Lithium • Eskalith, Lithobid
- Trazodone • Desyrel, Oleptro
Disclosure
Dr. Freeman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention. US death rate falls for 10th straight year. http://www.cdc.gov/media/releases/2011/p0316_deathrate.html. Published March 16 2011. Accessed November 22, 2011.
2. Centers for Disease Control and Prevention. Suicide: facts at a glance. http://www.cdc.gov/violenceprevention/suicide. Published Summer 2010. Accessed November 17 2011.
3. Karch DL, Dahlberg LL, Patel N. Surveillance for violent deaths—National Violent Death Reporting System 16 states, 2007. MMWR Surveill Summ. 2010;59(4):1-50.
4. Resnick PJ. Recognizing that the suicidal patient views you as an ‘adversary.’ Current Psychiatry. 2002;1(1):8.-
5. Stanford EJ, Goetz RR, Bloom JD. The No Harm Contract in the emergency assessment of suicidal risk. J Clin Psychiatry. 1994;55(8):344-348.
6. Edwards SJ, Sachmann MD. No-suicide contracts no-suicide agreements, and no-suicide assurances: a study of their nature, utilization, perceived effectiveness, and potential to cause harm. Crisis. 2010;31(6):290-302.
7. Busch KA, Fawcett J, Jacobs DG. Clinical correlates of inpatient suicide. J Clin Psychiatry. 2003;64(1):14-19.
8. American Psychiatric Association. Practice guideline for the assessment and treatment of patients with suicidal behaviors. http://psychiatryonline.org/content.aspx?bookid=28§ionid=1673332#56008. Published November 2003. Accessed November 22 2011.
9. Jacobs DG, Brewer ML. Application of the APA practice guidelines on suicide to clinical practice. CNS Spectr. 2006;11(6):447-454.
10. Nasser EH, Overholser JC. Assessing varying degrees of lethality in depressed adolescent suicide attempters. Acta Psychiatr Scand. 1999;99(6):423-431.
11. Fawcett J, Scheftner WA, Fogg L, et al. Time-related predictors of suicide in major affective disorder. Am J Psychiatry. 1990;147(9):1189-1194.
12. Busch KA, Fawcett J. A fine-grained study of patients who commit suicide. Psychiatric Ann. 2004;34(5):357-364.
13. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale (C-SSRS): initial validity and internal consistency findings from three multi-site studies with adolescents and adults. Am J Psychiatry. 2011;168:1266-1277.
14. Müller-Oerlinghausen B, Berghöfer A, Ahrens B. The antisuicidal and mortality-reducing effect of lithium prophylaxis: consequences for guidelines in clinical psychiatry. Can J Psychiatry. 2003;48(7):433-439.
15. Tondo L, Hennen J, Baldessarini RJ. Lower suicide risk with long-term lithium treatment in major affective illness: a meta-analysis. Acta Psychiatr Scand. 2001;104(3):163-172.
16. Kessing LV, Søndergård L, Kvist K, et al. Suicide risk in patients treated with lithium. Arch Gen Psychiatry. 2005;62(8):860-866.
17. Baldessarini RJ, Tondo L, Hennen J. Lithium treatment and suicide risk in major affective disorders: update and new findings. J Clin Psychiatry. 2003;64(suppl 5):44-52.
18. Hennen J, Baldessarini RJ. Suicidal risk during treatment with clozapine: a meta-analysis. Schizophr Res. 2005;73(2-3):139-145.
19. Kuo CJ, Tsai SY, Lo CH, et al. Risk factors for completed suicide in schizophrenia. J Clin Psychiatry. 2005;66(5):579-585.
20. Kobeissi J, Aloysi A, Tobias K, et al. Resolution of severe suicidality with a single electroconvulsive therapy. J ECT. 2011;27(1):86-88.
21. Kellner CH, Fink M, Knapp R, et al. Relief of expressed suicidal intent by ECT: a consortium for research in ECT study. Am J Psychiatry. 2005;162(5):977-982.
22. Linehan MM, Goodstein JL, Nielsen SL, et al. Reasons for staying alive when you are thinking of killing yourself: the reasons for living inventory. J Consult Clin Psychol. 1983;51(2):276-286.
23. Cole DA. Validation of the reasons for living inventory in general and delinquent adolescent samples. J Abnorm Child Psychol. 1989;17(1):13-27.
24. Mann JJ, Apter A, Bertolote J, et al. Suicide prevention strategies: a systematic review. JAMA. 2005;294(16):2064-2074.
1. Centers for Disease Control and Prevention. US death rate falls for 10th straight year. http://www.cdc.gov/media/releases/2011/p0316_deathrate.html. Published March 16 2011. Accessed November 22, 2011.
2. Centers for Disease Control and Prevention. Suicide: facts at a glance. http://www.cdc.gov/violenceprevention/suicide. Published Summer 2010. Accessed November 17 2011.
3. Karch DL, Dahlberg LL, Patel N. Surveillance for violent deaths—National Violent Death Reporting System 16 states, 2007. MMWR Surveill Summ. 2010;59(4):1-50.
4. Resnick PJ. Recognizing that the suicidal patient views you as an ‘adversary.’ Current Psychiatry. 2002;1(1):8.-
5. Stanford EJ, Goetz RR, Bloom JD. The No Harm Contract in the emergency assessment of suicidal risk. J Clin Psychiatry. 1994;55(8):344-348.
6. Edwards SJ, Sachmann MD. No-suicide contracts no-suicide agreements, and no-suicide assurances: a study of their nature, utilization, perceived effectiveness, and potential to cause harm. Crisis. 2010;31(6):290-302.
7. Busch KA, Fawcett J, Jacobs DG. Clinical correlates of inpatient suicide. J Clin Psychiatry. 2003;64(1):14-19.
8. American Psychiatric Association. Practice guideline for the assessment and treatment of patients with suicidal behaviors. http://psychiatryonline.org/content.aspx?bookid=28§ionid=1673332#56008. Published November 2003. Accessed November 22 2011.
9. Jacobs DG, Brewer ML. Application of the APA practice guidelines on suicide to clinical practice. CNS Spectr. 2006;11(6):447-454.
10. Nasser EH, Overholser JC. Assessing varying degrees of lethality in depressed adolescent suicide attempters. Acta Psychiatr Scand. 1999;99(6):423-431.
11. Fawcett J, Scheftner WA, Fogg L, et al. Time-related predictors of suicide in major affective disorder. Am J Psychiatry. 1990;147(9):1189-1194.
12. Busch KA, Fawcett J. A fine-grained study of patients who commit suicide. Psychiatric Ann. 2004;34(5):357-364.
13. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale (C-SSRS): initial validity and internal consistency findings from three multi-site studies with adolescents and adults. Am J Psychiatry. 2011;168:1266-1277.
14. Müller-Oerlinghausen B, Berghöfer A, Ahrens B. The antisuicidal and mortality-reducing effect of lithium prophylaxis: consequences for guidelines in clinical psychiatry. Can J Psychiatry. 2003;48(7):433-439.
15. Tondo L, Hennen J, Baldessarini RJ. Lower suicide risk with long-term lithium treatment in major affective illness: a meta-analysis. Acta Psychiatr Scand. 2001;104(3):163-172.
16. Kessing LV, Søndergård L, Kvist K, et al. Suicide risk in patients treated with lithium. Arch Gen Psychiatry. 2005;62(8):860-866.
17. Baldessarini RJ, Tondo L, Hennen J. Lithium treatment and suicide risk in major affective disorders: update and new findings. J Clin Psychiatry. 2003;64(suppl 5):44-52.
18. Hennen J, Baldessarini RJ. Suicidal risk during treatment with clozapine: a meta-analysis. Schizophr Res. 2005;73(2-3):139-145.
19. Kuo CJ, Tsai SY, Lo CH, et al. Risk factors for completed suicide in schizophrenia. J Clin Psychiatry. 2005;66(5):579-585.
20. Kobeissi J, Aloysi A, Tobias K, et al. Resolution of severe suicidality with a single electroconvulsive therapy. J ECT. 2011;27(1):86-88.
21. Kellner CH, Fink M, Knapp R, et al. Relief of expressed suicidal intent by ECT: a consortium for research in ECT study. Am J Psychiatry. 2005;162(5):977-982.
22. Linehan MM, Goodstein JL, Nielsen SL, et al. Reasons for staying alive when you are thinking of killing yourself: the reasons for living inventory. J Consult Clin Psychol. 1983;51(2):276-286.
23. Cole DA. Validation of the reasons for living inventory in general and delinquent adolescent samples. J Abnorm Child Psychol. 1989;17(1):13-27.
24. Mann JJ, Apter A, Bertolote J, et al. Suicide prevention strategies: a systematic review. JAMA. 2005;294(16):2064-2074.
Community-Based Surveillance in Clinical Stage I Germ Cell Tumors
Objective: Although depression is prevalent among cancer patients, it remains underdiagnosed and undertreated. Quality of life is an important outcome in cancer patients and can be measured by questionnaires such as the Functional Assessment of Cancer Therapy-General version (FACT-G). The purpose of our study was to establish whether or not a group of items in FACT-G could be used as a screening tool for depression as well as for assessing quality of life.
Methods: A total of 62 chemotherapy patients (median age, 62 years [range, 22-81 years]; 55% women) completed Zung Self-Rating Depression Scale (ZSDS) and FACT-G questionnaires. Patients with ZSDS scores of 40 or more underwent clinical interviews for major depression. Pearson’s correlation was used to examine the relationship between the ZSDS and FACT-G scores. FACT-G score results were then analyzed to evaluate if subsets of the FACT-G can be used as a screening tool for major depression.
Results: In all, 30 of 62 patients (48%) had ZSDS scores 40 and were ruled out for major depression, and 30 of the 32 patients with ZSDS scores 40 participated clinical interviews. Of those who were interviewed, 7 patients (23%) were confirmed to have major depression. Overall, the prevalence of major depression was 7 of 60 patients (12%; 95% CI: 5%-23%). The ZSDS and FACT-G scores had strong correlation (r -0.75). The composite score of six statements in FACT-G were found to have sensitivity of 100% and specificity of 81% in predicting major depression, using a cut-off value of 12 (range, 0-24). The six statements were, I have a lack of energy; I feel sad; I feel nervous; I am able to enjoy life; I am sleeping well; and I am enjoying the things I usually do for fun.
Conclusions: The prevalence of major depression among all participants was 12%. The ZSDS score and FACT-G score had strong correlation; the subsets of FACT-G may be useful as a screening tool for depression.
*For a PDF of the full article, click on the link to the left of this introduction.
Objective: Although depression is prevalent among cancer patients, it remains underdiagnosed and undertreated. Quality of life is an important outcome in cancer patients and can be measured by questionnaires such as the Functional Assessment of Cancer Therapy-General version (FACT-G). The purpose of our study was to establish whether or not a group of items in FACT-G could be used as a screening tool for depression as well as for assessing quality of life.
Methods: A total of 62 chemotherapy patients (median age, 62 years [range, 22-81 years]; 55% women) completed Zung Self-Rating Depression Scale (ZSDS) and FACT-G questionnaires. Patients with ZSDS scores of 40 or more underwent clinical interviews for major depression. Pearson’s correlation was used to examine the relationship between the ZSDS and FACT-G scores. FACT-G score results were then analyzed to evaluate if subsets of the FACT-G can be used as a screening tool for major depression.
Results: In all, 30 of 62 patients (48%) had ZSDS scores 40 and were ruled out for major depression, and 30 of the 32 patients with ZSDS scores 40 participated clinical interviews. Of those who were interviewed, 7 patients (23%) were confirmed to have major depression. Overall, the prevalence of major depression was 7 of 60 patients (12%; 95% CI: 5%-23%). The ZSDS and FACT-G scores had strong correlation (r -0.75). The composite score of six statements in FACT-G were found to have sensitivity of 100% and specificity of 81% in predicting major depression, using a cut-off value of 12 (range, 0-24). The six statements were, I have a lack of energy; I feel sad; I feel nervous; I am able to enjoy life; I am sleeping well; and I am enjoying the things I usually do for fun.
Conclusions: The prevalence of major depression among all participants was 12%. The ZSDS score and FACT-G score had strong correlation; the subsets of FACT-G may be useful as a screening tool for depression.
*For a PDF of the full article, click on the link to the left of this introduction.
Objective: Although depression is prevalent among cancer patients, it remains underdiagnosed and undertreated. Quality of life is an important outcome in cancer patients and can be measured by questionnaires such as the Functional Assessment of Cancer Therapy-General version (FACT-G). The purpose of our study was to establish whether or not a group of items in FACT-G could be used as a screening tool for depression as well as for assessing quality of life.
Methods: A total of 62 chemotherapy patients (median age, 62 years [range, 22-81 years]; 55% women) completed Zung Self-Rating Depression Scale (ZSDS) and FACT-G questionnaires. Patients with ZSDS scores of 40 or more underwent clinical interviews for major depression. Pearson’s correlation was used to examine the relationship between the ZSDS and FACT-G scores. FACT-G score results were then analyzed to evaluate if subsets of the FACT-G can be used as a screening tool for major depression.
Results: In all, 30 of 62 patients (48%) had ZSDS scores 40 and were ruled out for major depression, and 30 of the 32 patients with ZSDS scores 40 participated clinical interviews. Of those who were interviewed, 7 patients (23%) were confirmed to have major depression. Overall, the prevalence of major depression was 7 of 60 patients (12%; 95% CI: 5%-23%). The ZSDS and FACT-G scores had strong correlation (r -0.75). The composite score of six statements in FACT-G were found to have sensitivity of 100% and specificity of 81% in predicting major depression, using a cut-off value of 12 (range, 0-24). The six statements were, I have a lack of energy; I feel sad; I feel nervous; I am able to enjoy life; I am sleeping well; and I am enjoying the things I usually do for fun.
Conclusions: The prevalence of major depression among all participants was 12%. The ZSDS score and FACT-G score had strong correlation; the subsets of FACT-G may be useful as a screening tool for depression.
*For a PDF of the full article, click on the link to the left of this introduction.
The Hoopla Over Mesh: What It Means for Practice
The Food and Drug Administration's warning last summer of the risks associated with transvaginal placement of mesh for repair of pelvic organ prolapse and stress urinary incontinence – and its overall, ongoing review of how mesh products are cleared for use–have changed the climate for ob.gyns. and patients. It has upped the ante for comprehensive patient counseling and brought to the fore the fact that pelvic floor repair is a combination of art, science, judgment, skill, training, and experience.
In July 2011, the FDA issued a “safety communication” to physicians and patients, which was based on an analysis of adverse event reports and a systematic literature review, warning that the transvaginal placement of mesh to treat pelvic organ prolapse (POP) appears to be riskier than traditional repairs without any evidence of greater effectiveness. While an earlier FDA notice issued in 2008 had said in essence that there may be a problem with transvaginal mesh, the most recent warning said there is a problem – that serious complications associated with surgical mesh used for transvaginal repair of POP are not rare.
The agency made a distinction between apical and posterior repair, and anterior repair, concluding that there is no evidence that either apical or posterior repair done with mesh provides any added benefit compared with traditional surgery without mesh.
With regard to anterior repair, the FDA concluded that mesh augmentation may provide an anatomic benefit compared with traditional nonmesh repair, although this anatomic benefit may not necessarily lead to better symptomatic results.
The FDA also reviewed all types of midurethral sling (MUS) devices used to treat stress urinary incontinence (SUI), grouping retropubic and transobturator slings as first-generation and mini-slings as second-generation devices.
Whereas these devices were deemed to be as effective as or better than traditional repairs, the FDA stated its concerns about the potential for long-term problems including mesh erosion and pelvic pain. Moreover, the agency stated the need for more data to better evaluate mini-slings for comparative efficacy and complications.
More broadly, the FDA is reevaluating how transvaginal mesh products should be regulated and brought to market. Unlike other devices that are widely used by ob.gyns., not one of the pelvic floor mesh kits for POP or midurethral slings for SUI has been evaluated by way of an independent, FDA-mandated randomized clinical trial. This is because transvaginal meshes are currently classified as class II devices and, as such, have been cleared for market by the less rigorous 510(k) notification process rather than a more rigorous premarket approval (PMA) process.
While the FDA considers the 510(k) pathway still suitable for MUS devices used to treat SUI, the agency is taking a harder look at transvaginal mesh used to repair POP and has recommended reclassification of these devices into class III. This switch would require the more onerous PMA process and allow the FDA to require clinical trials comparing procedures that involve mesh with those in which mesh is not used.
How the FDA Regulates Devices
That transvaginal mesh devices are embroiled in a broader and ongoing controversy over how best to regulate or approve medical devices is important to understand. Innovation and potential market share continue to drive a steady stream of new medical devices for gynecologic surgery.
Until 36 years ago there was no federal regulation of medical devices. The Medical Device Amendments of 1976 established three device classes, based on risk levels and the ability of postmarketing controls to manage those risks. The law then identified pathways, based largely on this classification system, for bringing devices to the market.
Class I devices are generally those for which general postmarketing controls such as good manufacturing processes and record keeping are deemed sufficient to provide reasonable assurance of safety and effectiveness. Devices in class II, which are “moderate risk,” need special controls such as performance standards and postmarketing surveillance to provide reasonable assurance of safety and effectiveness. In class III are life-sustaining or life-supporting “high-risk” devices that cannot be placed in class I or II because there is insufficient information to establish requisite assurance with postmarketing controls.
While FDA-approved randomized and controlled clinical trials are required for class III devices as part of the standard PMA process, class II devices are cleared for the market based on the substantially less rigorous 510(k) Premarket Notification Program process, which requires manufacturers to demonstrate safety and effectiveness by proving “substantial equivalence” to another device that is already cleared by the FDA based on intended use and product design.
Whereas clinical data are not required, this review of substantial equivalence requires labeling and performance data, including material safety, mechanical performance, and animal testing. Approval of the first surgical mesh for repair of POP was judged to be substantially equivalent to surgical mesh used for hernia repair.
In recent years there has been growing concern about this process of clearing medical devices based simply on substantial equivalence with a predicate. New products should not necessarily be assumed to have equal or improved safety and efficacy. The Institute of Medicine weighed in this past summer with a report on the 510(k) clearance process, calling it flawed in its ability to provide determinations about each device's safety and effectiveness.
The future of transvaginal mesh products is now entangled in these concerns. Unlike devices for endometrial ablation and transcervical hysteroscopic sterilization, which are justifiably classified as class III devices, all transvaginal mesh devices to date have been cleared as class II devices.
Since 2001, the FDA has cleared via the 510(k) approval process more than 100 synthetic mesh devices or kits indicated for POP repair, and more than 75 mesh devices to treat SUI (including 7 second-generation mini-slings), using the 510(k) notification process. None of the clearances were based on clinical data.
While there have indeed been some randomized clinical trials (in its recent review, FDA officials reported having looked at 22 randomized controlled trials and 38 observational studies on the use of mesh to treat POP), many of these trials have been designed and conducted with industry sponsorship.
The FDA typically calls upon its advisory panels to provide independent expert advice when specific issues or problems arise and when regulatory decisions need to be made both before and after approval of medical devices.
After issuing its “safety communication” last July, the FDA convened the Obstetrics and Gynecology Devices Advisory Panel in September to make recommendations regarding the safety and effectiveness of surgical mesh for repair of POP and SUI. Ironically, transvaginal mesh devices had previously been regulated by the FDA's Plastic Surgery Devices Panel.
The 2-day public hearing included presentations regarding adverse events and effectiveness of transvaginal mesh for POP and then SUI by FDA staff reviewers, key medical organizations, related industry as a consortium, and public advocacy groups as well as personal testimony by patients having undergone these procedures.
After hearing the testimony and an exhaustive discussion, the majority of panel members supported reclassifying mesh devices for POP from class II to class III. On the other hand, while the majority did not recommend the reclassification of devices for SUI, the panel concurred that more clinical data was warranted to establish the safety and efficacy of second-generation mini-slings.
The FDA's final regulatory decisions will slowly evolve as the issues of safety and effectiveness are balanced with reducing the burden for industry and continuing to foster a hospitable climate for medical innovation.
Adverse Event Reports
The FDA's safety communication released in July, which updated the 2008 FDA Public Health Notification, was generated by continuing concerns raised by rising reports of adverse events as well as concern voiced by the American Urogynecologic Society.
The adverse event reports have been compiled via the FDA's Manufacturer and User Facility Device Experience (MAUDE) database, which collects both mandated reporting by manufacturers and voluntary reports by physicians, patients, and any interested party. It is presumed that complications are generally underreported.
From 2008 to 2010, the FDA received 2,874 adverse event reports associated with urogynecologic mesh – about three times the number of reports filed from 2005 to 2007. Of these, 1,503 were associated with products for POP, and 1,371 were associated with products for SUI.
It is unclear, of course, how much of this increase reflects an increase in actual adverse events and how much stems from the increased use of mesh, an increased awareness of adverse events, possible duplication of reporting, and other factors that are inherent limitations of the reporting process. Moreover, the complication rate is not known because the total number of adverse events and the total number of implanted delivery systems are not known.
Erosion, exposure, and extrusion continue to be the most frequent and concerning adverse events associated with mesh used for POP and SUI. With its more recent review, the FDA has new concerns about the delayed appearances of erosion and mesh exposure. While there are few treatment cohorts to evaluate after 36 months, there have been a number of reports of long-term adverse outcomes – some at time points up to 60 months post procedure.
Moreover, the FDA is concerned about the risk for later development of dyspareunia and new pelvic pain from mesh contraction, retraction, vaginal shrinkage, and subsequent reoperation – problems not identified or flagged when the agency completed its last comprehensive review before issuing the 2008 notification.
Current State of Transvaginal Mesh
In the most recent safety communication, the FDA instructs patients to be aware of the risks associated with surgical mesh for transvaginal repair of POP and SUI. It warns patients that having transvaginal mesh surgery may increase their risk of needing additional surgery due to mesh-related complications, and it advises patients to ask their surgeons about all POP treatment options.
The alert also tells patients to notify their physicians regarding vaginal or pain symptoms after surgery with transvaginal mesh, and to let their health care providers know they have implanted mesh – advice that, in and of itself, can create fear. Any patient doing diligent research will see the statement and related discussion.
In issuing the communication, the FDA has set the bar at a higher level of expectation for patient counseling and informed consent.
While the FDA does not regulate the practice of medicine by regulating how or which physicians can use devices, the agency indirectly is regulating the use of transvaginal mesh devices through its alerts.
And without question, the probability for medical-legal conflict has been substantially heightened. Propelled by the FDA warnings, a cursory Internet search for “pelvic mesh lawyers” or “vaginal mesh lawsuit attorneys” yields a list of firms encouraging free case reviews.
Patients should be counseled that transvaginal mesh procedures are considered innovative techniques for pelvic floor repair that demonstrate high rates of anatomic cure in shorter-term series.
Preoperative counseling should cover the following principles and guidelines:
▸ There are potential adverse sequelae of transvaginal mesh repairs.
▸ There are limited data comparing transvaginal mesh systems with traditional vaginal prolapse repairs or with traditional use of graft material in the form of augmented colporrhaphy and sacrocolpopexy.
▸ The placement of surgical mesh for POP by sacrocolpopexy for apical prolapse is a well established clinical practice and may result in lower rates of mesh complications.
▸ Transvaginal apical or posterior repair with mesh does not appear to provide any added benefit compared with traditional surgery without mesh.
The main role for mesh with POP repair is in the anterior compartment, where a higher risk of recurrence with traditional repairs has been documented.
Overall, transvaginal mesh repair of POP is best suited to women who are high risk due to medical conditions and in those with recurrent prolapse, particularly of the anterior compartment.
▸ The effectiveness of retropubic and transobturator suburethral slings for SUI has been demonstrated, while the safety and effectiveness of single-incision mini-slings is less well established.
Rather than the fault of the device or method, the failure or success of transvaginal mesh repairs may rely far more on the skill and judgment of the surgeon.
All surgery incorporates an intricate blend of art and science. We must be realistic in evaluating our skills, experience, and expertise in performing transvaginal mesh procedures.
Even in the best of circumstances, factors such as obesity, hypoestrogenism, advanced age, poor nutrition, extreme life activity, multiparity, Northern European descent, smoking, prior reparative surgery, and diabetes may reduce the success of transvaginal mesh procedures and increase complications.
While patient concerns will be heightened, the decision to perform a particular type of restorative or reparative surgery for POP, with or without mesh, should always favor reduced risk along with optimal and durable outcome that is both anatomic and functional in nature. And clinical decision making, as always, must be guided by our Hippocratic vow “primum non nocere”!
{kind=link}
Vitals
Source Elsevier Global Medical News
{kind=link}
Source Elsevier Global Medical News
To Mesh or Not to Mesh?
On July 13, 2011, the Food and Drug Administration issued a safety communication, “Update on Serious Complications Associated with Transvaginal Placement of Surgical Mesh for Pelvic Organ Prolapse,” intended for health care providers and patients. Previously, on Oct. 20, 2008, the FDA issued a Public Health Notification and Additional Patient Information statement on serious complications associated with surgical mesh placed transvaginally to treat pelvic organ prolapse (POP) and stress urinary incontinence (SUI).
In the July 2011 bulletin, the FDA stated that “serious complications associated with surgical mesh for transvaginal repair of pelvic organ prolapse are not rare. … Furthermore, it is not clear that transvaginal pelvic organ prolapse repair with mesh is more effective than traditional nonmesh repair in all patients with pelvic organ prolapse and it may expose patients to greater risk.”
In its bulletin, the FDA noted a marked increase in reported adverse events related to surgical mesh devices used to repair POP and SUI in reporting years 2005-2007 vs. 2008-2010. The most frequent complications reported to the FDA regarding transvaginal mesh placement for POP were mesh erosion through the vagina, pain, infection, bleeding, dyspareunia, organ perforation, and urinary problems. Also noted were recurrent prolapse, neuromuscular problems, vaginal scarring/shrinkage, and emotional problems. Moreover, men may experience irritation and pain to the penis during intercourse secondary to exposed mesh.
The FDA also reported on its systematic review of literature from the period of 1996-2011 to evaluate transvaginal mesh safety and effectiveness. In particular, the FDA noted the following:
▸ Potential for additional risk when mesh is utilized in POP surgery.
▸ Greater rate of complications in POP surgery when mesh placed transvaginally, rather than transabdominally.
▸ No advantage of mesh for either apical or posterior repair, compared with traditional surgery without mesh.
▸ Although mesh may be beneficial anatomically for anterior repair, symptoms may not improve over conventional anterior repair.
The FDA then went on to make recommendations to both health care workers and patients.
Health care workers are advised to obtain specialized training for each mesh placement technique. Mesh should be considered only after weighing the risks and benefits, as well as considering other nonsurgical and surgical options including nonmesh and transabdominal mesh techniques.
Patients must be made aware that surgical mesh is a permanent implant, which may make future surgical repair more challenging.
Moreover, mesh may place the patient at greater risk for requiring additional surgery for the development of additional complications. Removal of mesh when complications arise may involve multiple surgeries and may negatively impact the patient's quality of life. Complete removal of the mesh may not be possible, and even if it is removed, symptoms may continue. Patients also must realize the lack of long-term data.
To understand how this latest FDA bulletin will impact the surgical treatment of POP and SUI, I have called upon Dr. Andrew I. Brill, director of minimally invasive surgery and reparative pelvic surgery at California Pacific Medical Center, San Francisco. He also is a voting member of the FDA Obstetrics and Gynecology Device Panel. Prior to moving to the Bay Area in 2006, Dr. Brill was professor of obstetrics and gynecology at the University of Illinois at Chicago, where he directed one of the first accredited fellowships in minimally invasive gynecology. Dr. Brill is a past president of both the AAGL and the board of directors of the AAGL/Society of Reproductive Surgeons Fellowship in Minimally Invasive Gynecology. Widely recognized in the United States and abroad as a leading educator in the field of minimally invasive gynecology, Dr. Brill is a frequent lecturer and telesurgeon, and he continues to be a regular contributor to peer literature and textbooks, having coauthored a leading textbook and more than 50 articles and book chapters.
The Food and Drug Administration's warning last summer of the risks associated with transvaginal placement of mesh for repair of pelvic organ prolapse and stress urinary incontinence – and its overall, ongoing review of how mesh products are cleared for use–have changed the climate for ob.gyns. and patients. It has upped the ante for comprehensive patient counseling and brought to the fore the fact that pelvic floor repair is a combination of art, science, judgment, skill, training, and experience.
In July 2011, the FDA issued a “safety communication” to physicians and patients, which was based on an analysis of adverse event reports and a systematic literature review, warning that the transvaginal placement of mesh to treat pelvic organ prolapse (POP) appears to be riskier than traditional repairs without any evidence of greater effectiveness. While an earlier FDA notice issued in 2008 had said in essence that there may be a problem with transvaginal mesh, the most recent warning said there is a problem – that serious complications associated with surgical mesh used for transvaginal repair of POP are not rare.
The agency made a distinction between apical and posterior repair, and anterior repair, concluding that there is no evidence that either apical or posterior repair done with mesh provides any added benefit compared with traditional surgery without mesh.
With regard to anterior repair, the FDA concluded that mesh augmentation may provide an anatomic benefit compared with traditional nonmesh repair, although this anatomic benefit may not necessarily lead to better symptomatic results.
The FDA also reviewed all types of midurethral sling (MUS) devices used to treat stress urinary incontinence (SUI), grouping retropubic and transobturator slings as first-generation and mini-slings as second-generation devices.
Whereas these devices were deemed to be as effective as or better than traditional repairs, the FDA stated its concerns about the potential for long-term problems including mesh erosion and pelvic pain. Moreover, the agency stated the need for more data to better evaluate mini-slings for comparative efficacy and complications.
More broadly, the FDA is reevaluating how transvaginal mesh products should be regulated and brought to market. Unlike other devices that are widely used by ob.gyns., not one of the pelvic floor mesh kits for POP or midurethral slings for SUI has been evaluated by way of an independent, FDA-mandated randomized clinical trial. This is because transvaginal meshes are currently classified as class II devices and, as such, have been cleared for market by the less rigorous 510(k) notification process rather than a more rigorous premarket approval (PMA) process.
While the FDA considers the 510(k) pathway still suitable for MUS devices used to treat SUI, the agency is taking a harder look at transvaginal mesh used to repair POP and has recommended reclassification of these devices into class III. This switch would require the more onerous PMA process and allow the FDA to require clinical trials comparing procedures that involve mesh with those in which mesh is not used.
How the FDA Regulates Devices
That transvaginal mesh devices are embroiled in a broader and ongoing controversy over how best to regulate or approve medical devices is important to understand. Innovation and potential market share continue to drive a steady stream of new medical devices for gynecologic surgery.
Until 36 years ago there was no federal regulation of medical devices. The Medical Device Amendments of 1976 established three device classes, based on risk levels and the ability of postmarketing controls to manage those risks. The law then identified pathways, based largely on this classification system, for bringing devices to the market.
Class I devices are generally those for which general postmarketing controls such as good manufacturing processes and record keeping are deemed sufficient to provide reasonable assurance of safety and effectiveness. Devices in class II, which are “moderate risk,” need special controls such as performance standards and postmarketing surveillance to provide reasonable assurance of safety and effectiveness. In class III are life-sustaining or life-supporting “high-risk” devices that cannot be placed in class I or II because there is insufficient information to establish requisite assurance with postmarketing controls.
While FDA-approved randomized and controlled clinical trials are required for class III devices as part of the standard PMA process, class II devices are cleared for the market based on the substantially less rigorous 510(k) Premarket Notification Program process, which requires manufacturers to demonstrate safety and effectiveness by proving “substantial equivalence” to another device that is already cleared by the FDA based on intended use and product design.
Whereas clinical data are not required, this review of substantial equivalence requires labeling and performance data, including material safety, mechanical performance, and animal testing. Approval of the first surgical mesh for repair of POP was judged to be substantially equivalent to surgical mesh used for hernia repair.
In recent years there has been growing concern about this process of clearing medical devices based simply on substantial equivalence with a predicate. New products should not necessarily be assumed to have equal or improved safety and efficacy. The Institute of Medicine weighed in this past summer with a report on the 510(k) clearance process, calling it flawed in its ability to provide determinations about each device's safety and effectiveness.
The future of transvaginal mesh products is now entangled in these concerns. Unlike devices for endometrial ablation and transcervical hysteroscopic sterilization, which are justifiably classified as class III devices, all transvaginal mesh devices to date have been cleared as class II devices.
Since 2001, the FDA has cleared via the 510(k) approval process more than 100 synthetic mesh devices or kits indicated for POP repair, and more than 75 mesh devices to treat SUI (including 7 second-generation mini-slings), using the 510(k) notification process. None of the clearances were based on clinical data.
While there have indeed been some randomized clinical trials (in its recent review, FDA officials reported having looked at 22 randomized controlled trials and 38 observational studies on the use of mesh to treat POP), many of these trials have been designed and conducted with industry sponsorship.
The FDA typically calls upon its advisory panels to provide independent expert advice when specific issues or problems arise and when regulatory decisions need to be made both before and after approval of medical devices.
After issuing its “safety communication” last July, the FDA convened the Obstetrics and Gynecology Devices Advisory Panel in September to make recommendations regarding the safety and effectiveness of surgical mesh for repair of POP and SUI. Ironically, transvaginal mesh devices had previously been regulated by the FDA's Plastic Surgery Devices Panel.
The 2-day public hearing included presentations regarding adverse events and effectiveness of transvaginal mesh for POP and then SUI by FDA staff reviewers, key medical organizations, related industry as a consortium, and public advocacy groups as well as personal testimony by patients having undergone these procedures.
After hearing the testimony and an exhaustive discussion, the majority of panel members supported reclassifying mesh devices for POP from class II to class III. On the other hand, while the majority did not recommend the reclassification of devices for SUI, the panel concurred that more clinical data was warranted to establish the safety and efficacy of second-generation mini-slings.
The FDA's final regulatory decisions will slowly evolve as the issues of safety and effectiveness are balanced with reducing the burden for industry and continuing to foster a hospitable climate for medical innovation.
Adverse Event Reports
The FDA's safety communication released in July, which updated the 2008 FDA Public Health Notification, was generated by continuing concerns raised by rising reports of adverse events as well as concern voiced by the American Urogynecologic Society.
The adverse event reports have been compiled via the FDA's Manufacturer and User Facility Device Experience (MAUDE) database, which collects both mandated reporting by manufacturers and voluntary reports by physicians, patients, and any interested party. It is presumed that complications are generally underreported.
From 2008 to 2010, the FDA received 2,874 adverse event reports associated with urogynecologic mesh – about three times the number of reports filed from 2005 to 2007. Of these, 1,503 were associated with products for POP, and 1,371 were associated with products for SUI.
It is unclear, of course, how much of this increase reflects an increase in actual adverse events and how much stems from the increased use of mesh, an increased awareness of adverse events, possible duplication of reporting, and other factors that are inherent limitations of the reporting process. Moreover, the complication rate is not known because the total number of adverse events and the total number of implanted delivery systems are not known.
Erosion, exposure, and extrusion continue to be the most frequent and concerning adverse events associated with mesh used for POP and SUI. With its more recent review, the FDA has new concerns about the delayed appearances of erosion and mesh exposure. While there are few treatment cohorts to evaluate after 36 months, there have been a number of reports of long-term adverse outcomes – some at time points up to 60 months post procedure.
Moreover, the FDA is concerned about the risk for later development of dyspareunia and new pelvic pain from mesh contraction, retraction, vaginal shrinkage, and subsequent reoperation – problems not identified or flagged when the agency completed its last comprehensive review before issuing the 2008 notification.
Current State of Transvaginal Mesh
In the most recent safety communication, the FDA instructs patients to be aware of the risks associated with surgical mesh for transvaginal repair of POP and SUI. It warns patients that having transvaginal mesh surgery may increase their risk of needing additional surgery due to mesh-related complications, and it advises patients to ask their surgeons about all POP treatment options.
The alert also tells patients to notify their physicians regarding vaginal or pain symptoms after surgery with transvaginal mesh, and to let their health care providers know they have implanted mesh – advice that, in and of itself, can create fear. Any patient doing diligent research will see the statement and related discussion.
In issuing the communication, the FDA has set the bar at a higher level of expectation for patient counseling and informed consent.
While the FDA does not regulate the practice of medicine by regulating how or which physicians can use devices, the agency indirectly is regulating the use of transvaginal mesh devices through its alerts.
And without question, the probability for medical-legal conflict has been substantially heightened. Propelled by the FDA warnings, a cursory Internet search for “pelvic mesh lawyers” or “vaginal mesh lawsuit attorneys” yields a list of firms encouraging free case reviews.
Patients should be counseled that transvaginal mesh procedures are considered innovative techniques for pelvic floor repair that demonstrate high rates of anatomic cure in shorter-term series.
Preoperative counseling should cover the following principles and guidelines:
▸ There are potential adverse sequelae of transvaginal mesh repairs.
▸ There are limited data comparing transvaginal mesh systems with traditional vaginal prolapse repairs or with traditional use of graft material in the form of augmented colporrhaphy and sacrocolpopexy.
▸ The placement of surgical mesh for POP by sacrocolpopexy for apical prolapse is a well established clinical practice and may result in lower rates of mesh complications.
▸ Transvaginal apical or posterior repair with mesh does not appear to provide any added benefit compared with traditional surgery without mesh.
The main role for mesh with POP repair is in the anterior compartment, where a higher risk of recurrence with traditional repairs has been documented.
Overall, transvaginal mesh repair of POP is best suited to women who are high risk due to medical conditions and in those with recurrent prolapse, particularly of the anterior compartment.
▸ The effectiveness of retropubic and transobturator suburethral slings for SUI has been demonstrated, while the safety and effectiveness of single-incision mini-slings is less well established.
Rather than the fault of the device or method, the failure or success of transvaginal mesh repairs may rely far more on the skill and judgment of the surgeon.
All surgery incorporates an intricate blend of art and science. We must be realistic in evaluating our skills, experience, and expertise in performing transvaginal mesh procedures.
Even in the best of circumstances, factors such as obesity, hypoestrogenism, advanced age, poor nutrition, extreme life activity, multiparity, Northern European descent, smoking, prior reparative surgery, and diabetes may reduce the success of transvaginal mesh procedures and increase complications.
While patient concerns will be heightened, the decision to perform a particular type of restorative or reparative surgery for POP, with or without mesh, should always favor reduced risk along with optimal and durable outcome that is both anatomic and functional in nature. And clinical decision making, as always, must be guided by our Hippocratic vow “primum non nocere”!
Vitals
Source Elsevier Global Medical News
Source Elsevier Global Medical News
To Mesh or Not to Mesh?
On July 13, 2011, the Food and Drug Administration issued a safety communication, “Update on Serious Complications Associated with Transvaginal Placement of Surgical Mesh for Pelvic Organ Prolapse,” intended for health care providers and patients. Previously, on Oct. 20, 2008, the FDA issued a Public Health Notification and Additional Patient Information statement on serious complications associated with surgical mesh placed transvaginally to treat pelvic organ prolapse (POP) and stress urinary incontinence (SUI).
In the July 2011 bulletin, the FDA stated that “serious complications associated with surgical mesh for transvaginal repair of pelvic organ prolapse are not rare. … Furthermore, it is not clear that transvaginal pelvic organ prolapse repair with mesh is more effective than traditional nonmesh repair in all patients with pelvic organ prolapse and it may expose patients to greater risk.”
In its bulletin, the FDA noted a marked increase in reported adverse events related to surgical mesh devices used to repair POP and SUI in reporting years 2005-2007 vs. 2008-2010. The most frequent complications reported to the FDA regarding transvaginal mesh placement for POP were mesh erosion through the vagina, pain, infection, bleeding, dyspareunia, organ perforation, and urinary problems. Also noted were recurrent prolapse, neuromuscular problems, vaginal scarring/shrinkage, and emotional problems. Moreover, men may experience irritation and pain to the penis during intercourse secondary to exposed mesh.
The FDA also reported on its systematic review of literature from the period of 1996-2011 to evaluate transvaginal mesh safety and effectiveness. In particular, the FDA noted the following:
▸ Potential for additional risk when mesh is utilized in POP surgery.
▸ Greater rate of complications in POP surgery when mesh placed transvaginally, rather than transabdominally.
▸ No advantage of mesh for either apical or posterior repair, compared with traditional surgery without mesh.
▸ Although mesh may be beneficial anatomically for anterior repair, symptoms may not improve over conventional anterior repair.
The FDA then went on to make recommendations to both health care workers and patients.
Health care workers are advised to obtain specialized training for each mesh placement technique. Mesh should be considered only after weighing the risks and benefits, as well as considering other nonsurgical and surgical options including nonmesh and transabdominal mesh techniques.
Patients must be made aware that surgical mesh is a permanent implant, which may make future surgical repair more challenging.
Moreover, mesh may place the patient at greater risk for requiring additional surgery for the development of additional complications. Removal of mesh when complications arise may involve multiple surgeries and may negatively impact the patient's quality of life. Complete removal of the mesh may not be possible, and even if it is removed, symptoms may continue. Patients also must realize the lack of long-term data.
To understand how this latest FDA bulletin will impact the surgical treatment of POP and SUI, I have called upon Dr. Andrew I. Brill, director of minimally invasive surgery and reparative pelvic surgery at California Pacific Medical Center, San Francisco. He also is a voting member of the FDA Obstetrics and Gynecology Device Panel. Prior to moving to the Bay Area in 2006, Dr. Brill was professor of obstetrics and gynecology at the University of Illinois at Chicago, where he directed one of the first accredited fellowships in minimally invasive gynecology. Dr. Brill is a past president of both the AAGL and the board of directors of the AAGL/Society of Reproductive Surgeons Fellowship in Minimally Invasive Gynecology. Widely recognized in the United States and abroad as a leading educator in the field of minimally invasive gynecology, Dr. Brill is a frequent lecturer and telesurgeon, and he continues to be a regular contributor to peer literature and textbooks, having coauthored a leading textbook and more than 50 articles and book chapters.
The Food and Drug Administration's warning last summer of the risks associated with transvaginal placement of mesh for repair of pelvic organ prolapse and stress urinary incontinence – and its overall, ongoing review of how mesh products are cleared for use–have changed the climate for ob.gyns. and patients. It has upped the ante for comprehensive patient counseling and brought to the fore the fact that pelvic floor repair is a combination of art, science, judgment, skill, training, and experience.
In July 2011, the FDA issued a “safety communication” to physicians and patients, which was based on an analysis of adverse event reports and a systematic literature review, warning that the transvaginal placement of mesh to treat pelvic organ prolapse (POP) appears to be riskier than traditional repairs without any evidence of greater effectiveness. While an earlier FDA notice issued in 2008 had said in essence that there may be a problem with transvaginal mesh, the most recent warning said there is a problem – that serious complications associated with surgical mesh used for transvaginal repair of POP are not rare.
The agency made a distinction between apical and posterior repair, and anterior repair, concluding that there is no evidence that either apical or posterior repair done with mesh provides any added benefit compared with traditional surgery without mesh.
With regard to anterior repair, the FDA concluded that mesh augmentation may provide an anatomic benefit compared with traditional nonmesh repair, although this anatomic benefit may not necessarily lead to better symptomatic results.
The FDA also reviewed all types of midurethral sling (MUS) devices used to treat stress urinary incontinence (SUI), grouping retropubic and transobturator slings as first-generation and mini-slings as second-generation devices.
Whereas these devices were deemed to be as effective as or better than traditional repairs, the FDA stated its concerns about the potential for long-term problems including mesh erosion and pelvic pain. Moreover, the agency stated the need for more data to better evaluate mini-slings for comparative efficacy and complications.
More broadly, the FDA is reevaluating how transvaginal mesh products should be regulated and brought to market. Unlike other devices that are widely used by ob.gyns., not one of the pelvic floor mesh kits for POP or midurethral slings for SUI has been evaluated by way of an independent, FDA-mandated randomized clinical trial. This is because transvaginal meshes are currently classified as class II devices and, as such, have been cleared for market by the less rigorous 510(k) notification process rather than a more rigorous premarket approval (PMA) process.
While the FDA considers the 510(k) pathway still suitable for MUS devices used to treat SUI, the agency is taking a harder look at transvaginal mesh used to repair POP and has recommended reclassification of these devices into class III. This switch would require the more onerous PMA process and allow the FDA to require clinical trials comparing procedures that involve mesh with those in which mesh is not used.
How the FDA Regulates Devices
That transvaginal mesh devices are embroiled in a broader and ongoing controversy over how best to regulate or approve medical devices is important to understand. Innovation and potential market share continue to drive a steady stream of new medical devices for gynecologic surgery.
Until 36 years ago there was no federal regulation of medical devices. The Medical Device Amendments of 1976 established three device classes, based on risk levels and the ability of postmarketing controls to manage those risks. The law then identified pathways, based largely on this classification system, for bringing devices to the market.
Class I devices are generally those for which general postmarketing controls such as good manufacturing processes and record keeping are deemed sufficient to provide reasonable assurance of safety and effectiveness. Devices in class II, which are “moderate risk,” need special controls such as performance standards and postmarketing surveillance to provide reasonable assurance of safety and effectiveness. In class III are life-sustaining or life-supporting “high-risk” devices that cannot be placed in class I or II because there is insufficient information to establish requisite assurance with postmarketing controls.
While FDA-approved randomized and controlled clinical trials are required for class III devices as part of the standard PMA process, class II devices are cleared for the market based on the substantially less rigorous 510(k) Premarket Notification Program process, which requires manufacturers to demonstrate safety and effectiveness by proving “substantial equivalence” to another device that is already cleared by the FDA based on intended use and product design.
Whereas clinical data are not required, this review of substantial equivalence requires labeling and performance data, including material safety, mechanical performance, and animal testing. Approval of the first surgical mesh for repair of POP was judged to be substantially equivalent to surgical mesh used for hernia repair.
In recent years there has been growing concern about this process of clearing medical devices based simply on substantial equivalence with a predicate. New products should not necessarily be assumed to have equal or improved safety and efficacy. The Institute of Medicine weighed in this past summer with a report on the 510(k) clearance process, calling it flawed in its ability to provide determinations about each device's safety and effectiveness.
The future of transvaginal mesh products is now entangled in these concerns. Unlike devices for endometrial ablation and transcervical hysteroscopic sterilization, which are justifiably classified as class III devices, all transvaginal mesh devices to date have been cleared as class II devices.
Since 2001, the FDA has cleared via the 510(k) approval process more than 100 synthetic mesh devices or kits indicated for POP repair, and more than 75 mesh devices to treat SUI (including 7 second-generation mini-slings), using the 510(k) notification process. None of the clearances were based on clinical data.
While there have indeed been some randomized clinical trials (in its recent review, FDA officials reported having looked at 22 randomized controlled trials and 38 observational studies on the use of mesh to treat POP), many of these trials have been designed and conducted with industry sponsorship.
The FDA typically calls upon its advisory panels to provide independent expert advice when specific issues or problems arise and when regulatory decisions need to be made both before and after approval of medical devices.
After issuing its “safety communication” last July, the FDA convened the Obstetrics and Gynecology Devices Advisory Panel in September to make recommendations regarding the safety and effectiveness of surgical mesh for repair of POP and SUI. Ironically, transvaginal mesh devices had previously been regulated by the FDA's Plastic Surgery Devices Panel.
The 2-day public hearing included presentations regarding adverse events and effectiveness of transvaginal mesh for POP and then SUI by FDA staff reviewers, key medical organizations, related industry as a consortium, and public advocacy groups as well as personal testimony by patients having undergone these procedures.
After hearing the testimony and an exhaustive discussion, the majority of panel members supported reclassifying mesh devices for POP from class II to class III. On the other hand, while the majority did not recommend the reclassification of devices for SUI, the panel concurred that more clinical data was warranted to establish the safety and efficacy of second-generation mini-slings.
The FDA's final regulatory decisions will slowly evolve as the issues of safety and effectiveness are balanced with reducing the burden for industry and continuing to foster a hospitable climate for medical innovation.
Adverse Event Reports
The FDA's safety communication released in July, which updated the 2008 FDA Public Health Notification, was generated by continuing concerns raised by rising reports of adverse events as well as concern voiced by the American Urogynecologic Society.
The adverse event reports have been compiled via the FDA's Manufacturer and User Facility Device Experience (MAUDE) database, which collects both mandated reporting by manufacturers and voluntary reports by physicians, patients, and any interested party. It is presumed that complications are generally underreported.
From 2008 to 2010, the FDA received 2,874 adverse event reports associated with urogynecologic mesh – about three times the number of reports filed from 2005 to 2007. Of these, 1,503 were associated with products for POP, and 1,371 were associated with products for SUI.
It is unclear, of course, how much of this increase reflects an increase in actual adverse events and how much stems from the increased use of mesh, an increased awareness of adverse events, possible duplication of reporting, and other factors that are inherent limitations of the reporting process. Moreover, the complication rate is not known because the total number of adverse events and the total number of implanted delivery systems are not known.
Erosion, exposure, and extrusion continue to be the most frequent and concerning adverse events associated with mesh used for POP and SUI. With its more recent review, the FDA has new concerns about the delayed appearances of erosion and mesh exposure. While there are few treatment cohorts to evaluate after 36 months, there have been a number of reports of long-term adverse outcomes – some at time points up to 60 months post procedure.
Moreover, the FDA is concerned about the risk for later development of dyspareunia and new pelvic pain from mesh contraction, retraction, vaginal shrinkage, and subsequent reoperation – problems not identified or flagged when the agency completed its last comprehensive review before issuing the 2008 notification.
Current State of Transvaginal Mesh
In the most recent safety communication, the FDA instructs patients to be aware of the risks associated with surgical mesh for transvaginal repair of POP and SUI. It warns patients that having transvaginal mesh surgery may increase their risk of needing additional surgery due to mesh-related complications, and it advises patients to ask their surgeons about all POP treatment options.
The alert also tells patients to notify their physicians regarding vaginal or pain symptoms after surgery with transvaginal mesh, and to let their health care providers know they have implanted mesh – advice that, in and of itself, can create fear. Any patient doing diligent research will see the statement and related discussion.
In issuing the communication, the FDA has set the bar at a higher level of expectation for patient counseling and informed consent.
While the FDA does not regulate the practice of medicine by regulating how or which physicians can use devices, the agency indirectly is regulating the use of transvaginal mesh devices through its alerts.
And without question, the probability for medical-legal conflict has been substantially heightened. Propelled by the FDA warnings, a cursory Internet search for “pelvic mesh lawyers” or “vaginal mesh lawsuit attorneys” yields a list of firms encouraging free case reviews.
Patients should be counseled that transvaginal mesh procedures are considered innovative techniques for pelvic floor repair that demonstrate high rates of anatomic cure in shorter-term series.
Preoperative counseling should cover the following principles and guidelines:
▸ There are potential adverse sequelae of transvaginal mesh repairs.
▸ There are limited data comparing transvaginal mesh systems with traditional vaginal prolapse repairs or with traditional use of graft material in the form of augmented colporrhaphy and sacrocolpopexy.
▸ The placement of surgical mesh for POP by sacrocolpopexy for apical prolapse is a well established clinical practice and may result in lower rates of mesh complications.
▸ Transvaginal apical or posterior repair with mesh does not appear to provide any added benefit compared with traditional surgery without mesh.
The main role for mesh with POP repair is in the anterior compartment, where a higher risk of recurrence with traditional repairs has been documented.
Overall, transvaginal mesh repair of POP is best suited to women who are high risk due to medical conditions and in those with recurrent prolapse, particularly of the anterior compartment.
▸ The effectiveness of retropubic and transobturator suburethral slings for SUI has been demonstrated, while the safety and effectiveness of single-incision mini-slings is less well established.
Rather than the fault of the device or method, the failure or success of transvaginal mesh repairs may rely far more on the skill and judgment of the surgeon.
All surgery incorporates an intricate blend of art and science. We must be realistic in evaluating our skills, experience, and expertise in performing transvaginal mesh procedures.
Even in the best of circumstances, factors such as obesity, hypoestrogenism, advanced age, poor nutrition, extreme life activity, multiparity, Northern European descent, smoking, prior reparative surgery, and diabetes may reduce the success of transvaginal mesh procedures and increase complications.
While patient concerns will be heightened, the decision to perform a particular type of restorative or reparative surgery for POP, with or without mesh, should always favor reduced risk along with optimal and durable outcome that is both anatomic and functional in nature. And clinical decision making, as always, must be guided by our Hippocratic vow “primum non nocere”!
Vitals
Source Elsevier Global Medical News
Source Elsevier Global Medical News
To Mesh or Not to Mesh?
On July 13, 2011, the Food and Drug Administration issued a safety communication, “Update on Serious Complications Associated with Transvaginal Placement of Surgical Mesh for Pelvic Organ Prolapse,” intended for health care providers and patients. Previously, on Oct. 20, 2008, the FDA issued a Public Health Notification and Additional Patient Information statement on serious complications associated with surgical mesh placed transvaginally to treat pelvic organ prolapse (POP) and stress urinary incontinence (SUI).
In the July 2011 bulletin, the FDA stated that “serious complications associated with surgical mesh for transvaginal repair of pelvic organ prolapse are not rare. … Furthermore, it is not clear that transvaginal pelvic organ prolapse repair with mesh is more effective than traditional nonmesh repair in all patients with pelvic organ prolapse and it may expose patients to greater risk.”
In its bulletin, the FDA noted a marked increase in reported adverse events related to surgical mesh devices used to repair POP and SUI in reporting years 2005-2007 vs. 2008-2010. The most frequent complications reported to the FDA regarding transvaginal mesh placement for POP were mesh erosion through the vagina, pain, infection, bleeding, dyspareunia, organ perforation, and urinary problems. Also noted were recurrent prolapse, neuromuscular problems, vaginal scarring/shrinkage, and emotional problems. Moreover, men may experience irritation and pain to the penis during intercourse secondary to exposed mesh.
The FDA also reported on its systematic review of literature from the period of 1996-2011 to evaluate transvaginal mesh safety and effectiveness. In particular, the FDA noted the following:
▸ Potential for additional risk when mesh is utilized in POP surgery.
▸ Greater rate of complications in POP surgery when mesh placed transvaginally, rather than transabdominally.
▸ No advantage of mesh for either apical or posterior repair, compared with traditional surgery without mesh.
▸ Although mesh may be beneficial anatomically for anterior repair, symptoms may not improve over conventional anterior repair.
The FDA then went on to make recommendations to both health care workers and patients.
Health care workers are advised to obtain specialized training for each mesh placement technique. Mesh should be considered only after weighing the risks and benefits, as well as considering other nonsurgical and surgical options including nonmesh and transabdominal mesh techniques.
Patients must be made aware that surgical mesh is a permanent implant, which may make future surgical repair more challenging.
Moreover, mesh may place the patient at greater risk for requiring additional surgery for the development of additional complications. Removal of mesh when complications arise may involve multiple surgeries and may negatively impact the patient's quality of life. Complete removal of the mesh may not be possible, and even if it is removed, symptoms may continue. Patients also must realize the lack of long-term data.
To understand how this latest FDA bulletin will impact the surgical treatment of POP and SUI, I have called upon Dr. Andrew I. Brill, director of minimally invasive surgery and reparative pelvic surgery at California Pacific Medical Center, San Francisco. He also is a voting member of the FDA Obstetrics and Gynecology Device Panel. Prior to moving to the Bay Area in 2006, Dr. Brill was professor of obstetrics and gynecology at the University of Illinois at Chicago, where he directed one of the first accredited fellowships in minimally invasive gynecology. Dr. Brill is a past president of both the AAGL and the board of directors of the AAGL/Society of Reproductive Surgeons Fellowship in Minimally Invasive Gynecology. Widely recognized in the United States and abroad as a leading educator in the field of minimally invasive gynecology, Dr. Brill is a frequent lecturer and telesurgeon, and he continues to be a regular contributor to peer literature and textbooks, having coauthored a leading textbook and more than 50 articles and book chapters.
Brentuximab vedotin ushers in a new era in treating lymphomas
Hodgkin lymphoma represents one of the major successes of modern oncology. Several decades ago, it was fatal in most patients. With the development of the combination therapy mechlorethamine, vincristine, prednisone, and procarbazine (MOPP), many patients were cured of this disease. However, the regimen was associated with an unacceptable risk of acute toxicities, infertility, and secondary malignancies.1 Several subsequent studies established adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD) as the standard treatment because of its greater efficacy and less toxicity compared with MOPP.2 As a result, about 90% of patients with limited-stage disease are now cured, as are 60% of those with advanced disease. Newer regimens such as bleomycin, etoposide, adriamycin, cyclophosphamide, prednisone, and procarbazine (BEACOPP) seem to prolong time to treatment failure, but with considerably greater toxicity,3 and with no clear improvement in overall survival. A minority of patients who are either refractory to initial treatment or who subsequently relapse can be cured with such modalities as stem-cell transplantation. However, few effective options are available for the remainder of patients...
*For a PDF of the full article, click in the link to the left of this article.
(See Community Translations, “Bretuximab vedotin in Hodgkin lymphoma and systemic anaplastic large-cell lymphoma”)
Hodgkin lymphoma represents one of the major successes of modern oncology. Several decades ago, it was fatal in most patients. With the development of the combination therapy mechlorethamine, vincristine, prednisone, and procarbazine (MOPP), many patients were cured of this disease. However, the regimen was associated with an unacceptable risk of acute toxicities, infertility, and secondary malignancies.1 Several subsequent studies established adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD) as the standard treatment because of its greater efficacy and less toxicity compared with MOPP.2 As a result, about 90% of patients with limited-stage disease are now cured, as are 60% of those with advanced disease. Newer regimens such as bleomycin, etoposide, adriamycin, cyclophosphamide, prednisone, and procarbazine (BEACOPP) seem to prolong time to treatment failure, but with considerably greater toxicity,3 and with no clear improvement in overall survival. A minority of patients who are either refractory to initial treatment or who subsequently relapse can be cured with such modalities as stem-cell transplantation. However, few effective options are available for the remainder of patients...
*For a PDF of the full article, click in the link to the left of this article.
(See Community Translations, “Bretuximab vedotin in Hodgkin lymphoma and systemic anaplastic large-cell lymphoma”)
Hodgkin lymphoma represents one of the major successes of modern oncology. Several decades ago, it was fatal in most patients. With the development of the combination therapy mechlorethamine, vincristine, prednisone, and procarbazine (MOPP), many patients were cured of this disease. However, the regimen was associated with an unacceptable risk of acute toxicities, infertility, and secondary malignancies.1 Several subsequent studies established adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD) as the standard treatment because of its greater efficacy and less toxicity compared with MOPP.2 As a result, about 90% of patients with limited-stage disease are now cured, as are 60% of those with advanced disease. Newer regimens such as bleomycin, etoposide, adriamycin, cyclophosphamide, prednisone, and procarbazine (BEACOPP) seem to prolong time to treatment failure, but with considerably greater toxicity,3 and with no clear improvement in overall survival. A minority of patients who are either refractory to initial treatment or who subsequently relapse can be cured with such modalities as stem-cell transplantation. However, few effective options are available for the remainder of patients...
*For a PDF of the full article, click in the link to the left of this article.
(See Community Translations, “Bretuximab vedotin in Hodgkin lymphoma and systemic anaplastic large-cell lymphoma”)
SURVIVORSHIP Embracing the ‘new normal’
Since 1971, when President Richard M. Nixon announced the “war on cancer” with the signing of the National Cancer Act, we have seen an increase of 300% in the number of survivors, which is now reaching more than 12 million in the United States, according to the Centers for Disease Control. By 2020, that number will likely approach 20 million. Investment in research, early detection, and prevention has contributed to making these numbers a reality, and community-based oncology centers have played a critical role in delivering quality care and improved survival numbers based on the findings of that research. Therefore, it is logical that these same networks of community-based providers that have helped create survivors now help take the next step in addressing the needs of cancer patients on their journey to a life beyond cancer.
*For a PDF of the full article, click in the link to the left of this introduction.
Since 1971, when President Richard M. Nixon announced the “war on cancer” with the signing of the National Cancer Act, we have seen an increase of 300% in the number of survivors, which is now reaching more than 12 million in the United States, according to the Centers for Disease Control. By 2020, that number will likely approach 20 million. Investment in research, early detection, and prevention has contributed to making these numbers a reality, and community-based oncology centers have played a critical role in delivering quality care and improved survival numbers based on the findings of that research. Therefore, it is logical that these same networks of community-based providers that have helped create survivors now help take the next step in addressing the needs of cancer patients on their journey to a life beyond cancer.
*For a PDF of the full article, click in the link to the left of this introduction.
Since 1971, when President Richard M. Nixon announced the “war on cancer” with the signing of the National Cancer Act, we have seen an increase of 300% in the number of survivors, which is now reaching more than 12 million in the United States, according to the Centers for Disease Control. By 2020, that number will likely approach 20 million. Investment in research, early detection, and prevention has contributed to making these numbers a reality, and community-based oncology centers have played a critical role in delivering quality care and improved survival numbers based on the findings of that research. Therefore, it is logical that these same networks of community-based providers that have helped create survivors now help take the next step in addressing the needs of cancer patients on their journey to a life beyond cancer.
*For a PDF of the full article, click in the link to the left of this introduction.
Retrospective Study of Patients with Metastatic Triple-Negative Breast Cancer: Survival, Health Care Utilization, and Cost
Background: Triple-negative breast cancer (TNBC) is a subset of breast cancer. Health care cost and utilization data for TNBC are lacking.
Objective: We examined differences between metastatic TNBC and metastatic non-TNBC in survival and health care costs and utilization.
Methods: This retrospective analysis of metastatic TNBC (134 patients) and metastatic non-TNBC (445 patients) used a proprietary oncology registry, the Impact Intelligence Oncology Management registry database, linked with health insurance claims and social security mortality data.
Results: We found metastatic patients whose breast cancer is triple negative to be younger (56.49 vs 59.24 years), to be more likely to have recurrent disease (64.93 vs 45.39%), and to have greater mortality vs metastatic non-TNBC patients (67.16 vs 50.79%) (all P less than .05). Recurrent patients with metastatic TNBC have the highest risk of death (HR, 1.9; P less than .001), whereas survival was greatest for de novo metastatic non-TNBC. Patients with metastatic TNBC had more all-cause annual hospitalizations, more hospitalized days, and higher total costs vs metastatic non-TNBC. Annual payer’s total costs, annual payer’s inpatient costs, cancer-related hospitalizations, and cancer-related inpatient costs also were greater among patients with metastatic TNBC.
Limitations: While the study spans slightly more than 2 years, 5-10 years would have been preferable to achieve a full clinical profile of indexed patients. The database also omitted factors that potentially confound the results, such as race and socioeconomic status.
Conclusions: Metastatic TNBC is associated with significant burden of disease and higher health care utilization vs metastatic non-TNBC, which may be due in part to the aggressive clinical course of the disease...
* For a PDF of the full article, click in the link to the left of this introduction.
Background: Triple-negative breast cancer (TNBC) is a subset of breast cancer. Health care cost and utilization data for TNBC are lacking.
Objective: We examined differences between metastatic TNBC and metastatic non-TNBC in survival and health care costs and utilization.
Methods: This retrospective analysis of metastatic TNBC (134 patients) and metastatic non-TNBC (445 patients) used a proprietary oncology registry, the Impact Intelligence Oncology Management registry database, linked with health insurance claims and social security mortality data.
Results: We found metastatic patients whose breast cancer is triple negative to be younger (56.49 vs 59.24 years), to be more likely to have recurrent disease (64.93 vs 45.39%), and to have greater mortality vs metastatic non-TNBC patients (67.16 vs 50.79%) (all P less than .05). Recurrent patients with metastatic TNBC have the highest risk of death (HR, 1.9; P less than .001), whereas survival was greatest for de novo metastatic non-TNBC. Patients with metastatic TNBC had more all-cause annual hospitalizations, more hospitalized days, and higher total costs vs metastatic non-TNBC. Annual payer’s total costs, annual payer’s inpatient costs, cancer-related hospitalizations, and cancer-related inpatient costs also were greater among patients with metastatic TNBC.
Limitations: While the study spans slightly more than 2 years, 5-10 years would have been preferable to achieve a full clinical profile of indexed patients. The database also omitted factors that potentially confound the results, such as race and socioeconomic status.
Conclusions: Metastatic TNBC is associated with significant burden of disease and higher health care utilization vs metastatic non-TNBC, which may be due in part to the aggressive clinical course of the disease...
* For a PDF of the full article, click in the link to the left of this introduction.
Background: Triple-negative breast cancer (TNBC) is a subset of breast cancer. Health care cost and utilization data for TNBC are lacking.
Objective: We examined differences between metastatic TNBC and metastatic non-TNBC in survival and health care costs and utilization.
Methods: This retrospective analysis of metastatic TNBC (134 patients) and metastatic non-TNBC (445 patients) used a proprietary oncology registry, the Impact Intelligence Oncology Management registry database, linked with health insurance claims and social security mortality data.
Results: We found metastatic patients whose breast cancer is triple negative to be younger (56.49 vs 59.24 years), to be more likely to have recurrent disease (64.93 vs 45.39%), and to have greater mortality vs metastatic non-TNBC patients (67.16 vs 50.79%) (all P less than .05). Recurrent patients with metastatic TNBC have the highest risk of death (HR, 1.9; P less than .001), whereas survival was greatest for de novo metastatic non-TNBC. Patients with metastatic TNBC had more all-cause annual hospitalizations, more hospitalized days, and higher total costs vs metastatic non-TNBC. Annual payer’s total costs, annual payer’s inpatient costs, cancer-related hospitalizations, and cancer-related inpatient costs also were greater among patients with metastatic TNBC.
Limitations: While the study spans slightly more than 2 years, 5-10 years would have been preferable to achieve a full clinical profile of indexed patients. The database also omitted factors that potentially confound the results, such as race and socioeconomic status.
Conclusions: Metastatic TNBC is associated with significant burden of disease and higher health care utilization vs metastatic non-TNBC, which may be due in part to the aggressive clinical course of the disease...
* For a PDF of the full article, click in the link to the left of this introduction.
Put Medical School Debt in Perspective As You Enter Job Market
According to the Association of American Medical Colleges, the average debt for a young physician graduating from medical school in 2010 was $158,000. How do you keep that debt in perspective as you plot your career path?
Look at the Bigger Picture
Making plans for repaying medical school debt should start before your job search, says Danielle Salovich, president of the American Medical Student Association in Sterling, Va. “Part of your medical school exit interview should include campus financial advisors who give you options for loan repayment,” she says.
—Danielle Salovich, president, American Medical Student Association, Sterling, Va.
Consider what payment schedule will work best with your projected budget. Many medical students do not have financial or business backgrounds, so she advises enlisting the services of a trusted financial advisor.
Variety of Options
Recruitment packages for early-career hospitalists vary from region to region, based on an area’s appeal and marketplace pressures, says Kent McMackin, senior vice president of Physician Services for Cogent HMG in Brentwood, Tenn. Hospitals are looking to control costs in the wake of regulatory and reimbursement pressures. One alternative is to investigate loan repayment and scholarship programs.
Bonuses and Benefits
Many young physicians are especially interested in the size of signing bonuses, which can be a plus, Salovich says, if they’re planning on making a down payment on a house or taking a chunk out of their school loan principal. Joel Greenwald, MD, CFP, partner at Sterling Retirement Resources in St. Louis Park, Minn., cautions against the “lure” of a big signing bonus. More important, he says, is the total compensation and benefits package. Comprehensive disability insurance should be at the top of the list, he says, since your ability to work is your most important asset.
Another factor to consider, McMackin says, is the type of mentoring available when you start that first job.
“Physicians work so hard in medical school, where they are not taught about how to develop and understand the inter-relationships between executives in the C-suite, X-ray, dietary, and other services in the hospital,” he says. “We need to make sure that physicians have access to national networks of mentors and to best practices and information-sharing.”
Those principles—taught at Cogent HMG Academy, SHM’s Leadership Academy, and others—help build career sustainability, he says.
Above all, keep your individual goals in mind and make a decision that works best with your particular situation. “I still believe,” Salovich says, “that a medical education is a wise investment.”
Gretchen Henkel is a freelance writer based in California.
Know What You Owe
Debt management “starts with getting a handle on your loan obligations,” Dr. Greenwald says. Start with a spreadsheet listing all your lenders, principal balances, and interest rates. Then arrange those obligations in descending order, with the highest-interest balances at the top. It’s more important to chisel down the high-interest loans first (e.g. credit-card debt) and to keep paying the minimum amounts on the lower ones. In addition, some interest is tax-deductible, so it is not as toxic as a high-interest credit card balance.
Here are some resources to help:
- Loan Repayment and Scholarship Program Resources.
- The National Health Service Corps offers student loan assistance for providers serving in communities with limited access to healthcare.
- AMSA offers a variety of information and tools to help organize a student-loan payoff plan.
According to the Association of American Medical Colleges, the average debt for a young physician graduating from medical school in 2010 was $158,000. How do you keep that debt in perspective as you plot your career path?
Look at the Bigger Picture
Making plans for repaying medical school debt should start before your job search, says Danielle Salovich, president of the American Medical Student Association in Sterling, Va. “Part of your medical school exit interview should include campus financial advisors who give you options for loan repayment,” she says.
—Danielle Salovich, president, American Medical Student Association, Sterling, Va.
Consider what payment schedule will work best with your projected budget. Many medical students do not have financial or business backgrounds, so she advises enlisting the services of a trusted financial advisor.
Variety of Options
Recruitment packages for early-career hospitalists vary from region to region, based on an area’s appeal and marketplace pressures, says Kent McMackin, senior vice president of Physician Services for Cogent HMG in Brentwood, Tenn. Hospitals are looking to control costs in the wake of regulatory and reimbursement pressures. One alternative is to investigate loan repayment and scholarship programs.
Bonuses and Benefits
Many young physicians are especially interested in the size of signing bonuses, which can be a plus, Salovich says, if they’re planning on making a down payment on a house or taking a chunk out of their school loan principal. Joel Greenwald, MD, CFP, partner at Sterling Retirement Resources in St. Louis Park, Minn., cautions against the “lure” of a big signing bonus. More important, he says, is the total compensation and benefits package. Comprehensive disability insurance should be at the top of the list, he says, since your ability to work is your most important asset.
Another factor to consider, McMackin says, is the type of mentoring available when you start that first job.
“Physicians work so hard in medical school, where they are not taught about how to develop and understand the inter-relationships between executives in the C-suite, X-ray, dietary, and other services in the hospital,” he says. “We need to make sure that physicians have access to national networks of mentors and to best practices and information-sharing.”
Those principles—taught at Cogent HMG Academy, SHM’s Leadership Academy, and others—help build career sustainability, he says.
Above all, keep your individual goals in mind and make a decision that works best with your particular situation. “I still believe,” Salovich says, “that a medical education is a wise investment.”
Gretchen Henkel is a freelance writer based in California.
Know What You Owe
Debt management “starts with getting a handle on your loan obligations,” Dr. Greenwald says. Start with a spreadsheet listing all your lenders, principal balances, and interest rates. Then arrange those obligations in descending order, with the highest-interest balances at the top. It’s more important to chisel down the high-interest loans first (e.g. credit-card debt) and to keep paying the minimum amounts on the lower ones. In addition, some interest is tax-deductible, so it is not as toxic as a high-interest credit card balance.
Here are some resources to help:
- Loan Repayment and Scholarship Program Resources.
- The National Health Service Corps offers student loan assistance for providers serving in communities with limited access to healthcare.
- AMSA offers a variety of information and tools to help organize a student-loan payoff plan.
According to the Association of American Medical Colleges, the average debt for a young physician graduating from medical school in 2010 was $158,000. How do you keep that debt in perspective as you plot your career path?
Look at the Bigger Picture
Making plans for repaying medical school debt should start before your job search, says Danielle Salovich, president of the American Medical Student Association in Sterling, Va. “Part of your medical school exit interview should include campus financial advisors who give you options for loan repayment,” she says.
—Danielle Salovich, president, American Medical Student Association, Sterling, Va.
Consider what payment schedule will work best with your projected budget. Many medical students do not have financial or business backgrounds, so she advises enlisting the services of a trusted financial advisor.
Variety of Options
Recruitment packages for early-career hospitalists vary from region to region, based on an area’s appeal and marketplace pressures, says Kent McMackin, senior vice president of Physician Services for Cogent HMG in Brentwood, Tenn. Hospitals are looking to control costs in the wake of regulatory and reimbursement pressures. One alternative is to investigate loan repayment and scholarship programs.
Bonuses and Benefits
Many young physicians are especially interested in the size of signing bonuses, which can be a plus, Salovich says, if they’re planning on making a down payment on a house or taking a chunk out of their school loan principal. Joel Greenwald, MD, CFP, partner at Sterling Retirement Resources in St. Louis Park, Minn., cautions against the “lure” of a big signing bonus. More important, he says, is the total compensation and benefits package. Comprehensive disability insurance should be at the top of the list, he says, since your ability to work is your most important asset.
Another factor to consider, McMackin says, is the type of mentoring available when you start that first job.
“Physicians work so hard in medical school, where they are not taught about how to develop and understand the inter-relationships between executives in the C-suite, X-ray, dietary, and other services in the hospital,” he says. “We need to make sure that physicians have access to national networks of mentors and to best practices and information-sharing.”
Those principles—taught at Cogent HMG Academy, SHM’s Leadership Academy, and others—help build career sustainability, he says.
Above all, keep your individual goals in mind and make a decision that works best with your particular situation. “I still believe,” Salovich says, “that a medical education is a wise investment.”
Gretchen Henkel is a freelance writer based in California.
Know What You Owe
Debt management “starts with getting a handle on your loan obligations,” Dr. Greenwald says. Start with a spreadsheet listing all your lenders, principal balances, and interest rates. Then arrange those obligations in descending order, with the highest-interest balances at the top. It’s more important to chisel down the high-interest loans first (e.g. credit-card debt) and to keep paying the minimum amounts on the lower ones. In addition, some interest is tax-deductible, so it is not as toxic as a high-interest credit card balance.
Here are some resources to help:
- Loan Repayment and Scholarship Program Resources.
- The National Health Service Corps offers student loan assistance for providers serving in communities with limited access to healthcare.
- AMSA offers a variety of information and tools to help organize a student-loan payoff plan.
Relationship Building From the Lab to an Island
"In the comfort of the Gottmans’ own island home, learn research-based tools for building and maintaining relationship intimacy. Have you ever planned a wonderful romantic vacation, complete with a lovely room, candle-lit dinners, and white sand beaches, only to have your dream become a nightmare when tense quarrels and silence spoil the sunset? We have been listening to painful stories like these for nearly 35 years.
"Now you can restore your relationship while having a world-class romantic holiday. In an exclusive, small group two-day couples retreat (there will be no more than six couples in each retreat), we offer you research-based skills for deepening intimacy, managing conflicts, and enhancing your sense of shared meaning in a private, comfortable, and beautiful setting."
That is a description from the brochure and website of the Gottman Relationship Institute, a marital therapy retreat conducted on Orcas Island off the coast of Washington, and run by an academic who has crossed over to the public sector: John M. Gottman, Ph.D.

Dr. Gottman is a psychologist who has received National Institute of Mental Health grants and numerous awards, and has authored or coauthored 119 academic articles and 37 books. In addition, Dr. Gottman is cofounder with his wife, Julie Gottman, also a Ph.D psychologist, of the Gottman Relationship Institute in Seattle. (Another personalized marriage counseling retreat is called Marriage Quest.* This one is held in Cabot, Vt., and is run by Israel Helfand, Ph.D., and his wife, Cathie Helfand, M.S. The Helfands are marriage counselors and sex therapists who have been working together with couples since 1983.)
We are all familiar with Dr. Gottman’s relationship interaction ratios. In happy couples, there are 20 positive interactions to 1 negative interaction, in conflicted couples the ratio is 5 to 1, and in soon-to-divorce couples the ratio is 0.8 to 1. Unhappy couples have a filter that screens out positive events and makes even neutral events negative. Happy couples have a filter that screens out negative events and makes neutral events positive.
Dr. Gottman’s research involves a quantitative mathematical approach to understanding the changes in the marital dyad and the relative influence that each has on the other. This model predicts changes in the relationship of a couple in his research laboratory.
His relational ratios and equations are now being applied to the dynamics of psychotherapy (Cogn. Neurodyn. 2011;5:265-75). The influence that the client has on the therapist and the influence that the therapist has on the client are mathematically mapped. Without going into the mathematical computations, I can say that the findings are that the therapist or client who is most responsive to the other ends up being the most positive, often through a series of emotional ups and downs. In other words, being positive, either on the part of the therapist or the client, yields positive outcomes. However, being too responsive to clients produces mediocre outcomes, and being negative to patients produces negative outcomes.
The authors do not claim any major theoretical advances. But they do believe that understanding the dynamic emotional states that exist between the therapist and the patient holds clues to the good therapeutic relationship.
For their part, Dr. Gottman and his wife certainly have balanced out the positives with the negatives when it comes to work and lifestyle balance. They’ve got an island home, the gratification of a successful practice, the avoidance of the health care system, and a continued ability to influence the field of relational psychology. In fact, he has developed a Relationship Vision Quest based on extensive research laboratory findings. One critical question has always been: Does this research translate into clinical practice?
Dr. Gottman now has his utopian island where he can put his research to the test – and he is looking for volunteers.
This column, Families in Psychiatry, appears in Clinical Psychiatry News. Dr. Heru is an associate professor of psychiatry at the University of Colorado at Denver, Aurora. She has been a member of the Association of Family Psychiatrists since 2002 and currently serves as the organization’s treasurer. In addition, she is the coauthor of two books on working with families and author of numerous articles on this topic.
*Correction, 1/9/2012: An earlier version of this column inadvertently referred to the Gottmans' retreat as Marriage Quest.
"In the comfort of the Gottmans’ own island home, learn research-based tools for building and maintaining relationship intimacy. Have you ever planned a wonderful romantic vacation, complete with a lovely room, candle-lit dinners, and white sand beaches, only to have your dream become a nightmare when tense quarrels and silence spoil the sunset? We have been listening to painful stories like these for nearly 35 years.
"Now you can restore your relationship while having a world-class romantic holiday. In an exclusive, small group two-day couples retreat (there will be no more than six couples in each retreat), we offer you research-based skills for deepening intimacy, managing conflicts, and enhancing your sense of shared meaning in a private, comfortable, and beautiful setting."
That is a description from the brochure and website of the Gottman Relationship Institute, a marital therapy retreat conducted on Orcas Island off the coast of Washington, and run by an academic who has crossed over to the public sector: John M. Gottman, Ph.D.
Dr. Gottman is a psychologist who has received National Institute of Mental Health grants and numerous awards, and has authored or coauthored 119 academic articles and 37 books. In addition, Dr. Gottman is cofounder with his wife, Julie Gottman, also a Ph.D psychologist, of the Gottman Relationship Institute in Seattle. (Another personalized marriage counseling retreat is called Marriage Quest.* This one is held in Cabot, Vt., and is run by Israel Helfand, Ph.D., and his wife, Cathie Helfand, M.S. The Helfands are marriage counselors and sex therapists who have been working together with couples since 1983.)
We are all familiar with Dr. Gottman’s relationship interaction ratios. In happy couples, there are 20 positive interactions to 1 negative interaction, in conflicted couples the ratio is 5 to 1, and in soon-to-divorce couples the ratio is 0.8 to 1. Unhappy couples have a filter that screens out positive events and makes even neutral events negative. Happy couples have a filter that screens out negative events and makes neutral events positive.
Dr. Gottman’s research involves a quantitative mathematical approach to understanding the changes in the marital dyad and the relative influence that each has on the other. This model predicts changes in the relationship of a couple in his research laboratory.
His relational ratios and equations are now being applied to the dynamics of psychotherapy (Cogn. Neurodyn. 2011;5:265-75). The influence that the client has on the therapist and the influence that the therapist has on the client are mathematically mapped. Without going into the mathematical computations, I can say that the findings are that the therapist or client who is most responsive to the other ends up being the most positive, often through a series of emotional ups and downs. In other words, being positive, either on the part of the therapist or the client, yields positive outcomes. However, being too responsive to clients produces mediocre outcomes, and being negative to patients produces negative outcomes.
The authors do not claim any major theoretical advances. But they do believe that understanding the dynamic emotional states that exist between the therapist and the patient holds clues to the good therapeutic relationship.
For their part, Dr. Gottman and his wife certainly have balanced out the positives with the negatives when it comes to work and lifestyle balance. They’ve got an island home, the gratification of a successful practice, the avoidance of the health care system, and a continued ability to influence the field of relational psychology. In fact, he has developed a Relationship Vision Quest based on extensive research laboratory findings. One critical question has always been: Does this research translate into clinical practice?
Dr. Gottman now has his utopian island where he can put his research to the test – and he is looking for volunteers.
This column, Families in Psychiatry, appears in Clinical Psychiatry News. Dr. Heru is an associate professor of psychiatry at the University of Colorado at Denver, Aurora. She has been a member of the Association of Family Psychiatrists since 2002 and currently serves as the organization’s treasurer. In addition, she is the coauthor of two books on working with families and author of numerous articles on this topic.
*Correction, 1/9/2012: An earlier version of this column inadvertently referred to the Gottmans' retreat as Marriage Quest.
"In the comfort of the Gottmans’ own island home, learn research-based tools for building and maintaining relationship intimacy. Have you ever planned a wonderful romantic vacation, complete with a lovely room, candle-lit dinners, and white sand beaches, only to have your dream become a nightmare when tense quarrels and silence spoil the sunset? We have been listening to painful stories like these for nearly 35 years.
"Now you can restore your relationship while having a world-class romantic holiday. In an exclusive, small group two-day couples retreat (there will be no more than six couples in each retreat), we offer you research-based skills for deepening intimacy, managing conflicts, and enhancing your sense of shared meaning in a private, comfortable, and beautiful setting."
That is a description from the brochure and website of the Gottman Relationship Institute, a marital therapy retreat conducted on Orcas Island off the coast of Washington, and run by an academic who has crossed over to the public sector: John M. Gottman, Ph.D.
Dr. Gottman is a psychologist who has received National Institute of Mental Health grants and numerous awards, and has authored or coauthored 119 academic articles and 37 books. In addition, Dr. Gottman is cofounder with his wife, Julie Gottman, also a Ph.D psychologist, of the Gottman Relationship Institute in Seattle. (Another personalized marriage counseling retreat is called Marriage Quest.* This one is held in Cabot, Vt., and is run by Israel Helfand, Ph.D., and his wife, Cathie Helfand, M.S. The Helfands are marriage counselors and sex therapists who have been working together with couples since 1983.)
We are all familiar with Dr. Gottman’s relationship interaction ratios. In happy couples, there are 20 positive interactions to 1 negative interaction, in conflicted couples the ratio is 5 to 1, and in soon-to-divorce couples the ratio is 0.8 to 1. Unhappy couples have a filter that screens out positive events and makes even neutral events negative. Happy couples have a filter that screens out negative events and makes neutral events positive.
Dr. Gottman’s research involves a quantitative mathematical approach to understanding the changes in the marital dyad and the relative influence that each has on the other. This model predicts changes in the relationship of a couple in his research laboratory.
His relational ratios and equations are now being applied to the dynamics of psychotherapy (Cogn. Neurodyn. 2011;5:265-75). The influence that the client has on the therapist and the influence that the therapist has on the client are mathematically mapped. Without going into the mathematical computations, I can say that the findings are that the therapist or client who is most responsive to the other ends up being the most positive, often through a series of emotional ups and downs. In other words, being positive, either on the part of the therapist or the client, yields positive outcomes. However, being too responsive to clients produces mediocre outcomes, and being negative to patients produces negative outcomes.
The authors do not claim any major theoretical advances. But they do believe that understanding the dynamic emotional states that exist between the therapist and the patient holds clues to the good therapeutic relationship.
For their part, Dr. Gottman and his wife certainly have balanced out the positives with the negatives when it comes to work and lifestyle balance. They’ve got an island home, the gratification of a successful practice, the avoidance of the health care system, and a continued ability to influence the field of relational psychology. In fact, he has developed a Relationship Vision Quest based on extensive research laboratory findings. One critical question has always been: Does this research translate into clinical practice?
Dr. Gottman now has his utopian island where he can put his research to the test – and he is looking for volunteers.
This column, Families in Psychiatry, appears in Clinical Psychiatry News. Dr. Heru is an associate professor of psychiatry at the University of Colorado at Denver, Aurora. She has been a member of the Association of Family Psychiatrists since 2002 and currently serves as the organization’s treasurer. In addition, she is the coauthor of two books on working with families and author of numerous articles on this topic.
*Correction, 1/9/2012: An earlier version of this column inadvertently referred to the Gottmans' retreat as Marriage Quest.
Hematologists, Hospitalists Differ in Care of Sickle Cell Patients
SAN DIEGO – When caring for patients with sickle cell disease who were hospitalized for vaso-occlusive crisis, hospitalists more often used demand-only patient-controlled analgesia, compared with hematologists.
In addition, symptoms of acute chest syndrome were reported more often among patients cared for by hospitalists than by those cared for by hematologists.
The findings, reported during a poster session at the annual meeting of the American Society of Hematology, underscore the need for improved collaboration between hematologists and hospitalists in the care of patients with sickle cell disease.

These patients have been "increasingly managed by hospitalists rather than adult hematology specialists," the researchers, led by Dr. Nirmish Shah, wrote in their abstract. "Hospitalists provide efficient inpatient management; however, many possess knowledge gaps and carry biases into their relationship with patients with sickle cell disease."
In an effort to better understand the differences between hematologists and hospitalists in the care practices of this patient population, Dr. Shah and his associates reviewed the records of 298 patients with sickle cell disease who were hospitalized for vaso-occlusive crisis at Duke University Medical Center, Durham, N.C., between Sept. 1, 2008, and Jan. 31, 2011. In November of 2009, all inpatient care for patients with sickle cell disease was moved from the hematologists to the hospitalist service, said Dr. Shah, who directs the medical center’s sickle cell transition program.
Of the 298 patients, 120 were cared for by hematologists over 13 months while 178 were cared for by hospitalists over 13 months. The mean age of patients was 32 years.
There were no significant differences between hematologists and hospitalists in standard ordering practices including incentive spirometry, a bowel regimen, an anti-itching medication, and DVT prophylaxis. However, patients cared for by a hematologist had a higher total number of hours on a patient-controlled analgesia device (212 vs. 171 hours).
Dr. Shah also reported that, compared with hospitalists, hematologists were significantly less likely to use demand-only patient-controlled analgesia (23% vs. 42%) and significantly more likely to use both continuous and demand patient-controlled analgesia (67% vs. 54%).
Transfusions were similarly used by clinicians in both specialties. However, the number of units transfused was higher for hematologists, compared with hospitalists (a mean of 2.6 units vs. 1.9 units).
Erythrocytapheresis was used more often by hematologists than by hospitalists, but this difference did not reach statistical significance (6.7% vs. 2.2%).
No patients cared for by hematologists were diagnosed with acute chest syndrome, but 5.6% of those cared for by hospitalists were. In addition, patients cared for by hematologists had a significantly longer hospitalization, compared with those cared for by hospitalists (a mean of 10 days vs. 8.4 days, respectively) and a nonsignificantly lower 7-day readmission rate (6.7% vs. 7.2%) and 30-day readmission rate (35% vs. 40%).
"There often is a knowledge gap in hospitalist care of sickle cell patients," Dr. Shah said in an interview. "Because they may have a gap, it is our job as hematologists to be available as consultants. In addition, we should decide on the right method for controlling pain in this population of patients. I’m not trying to say that [hospitalists are] doing something in a wrong fashion. I think it brings up the idea that we need to collaborate more. At our hospital we’re assigning a nurse practitioner to round independently and help assist hospitalists now, in an attempt to improve our care for patients with sickle cell disease."
Dr. Shah said that he had no relevant financial disclosures.
(**Corrected: An earlier version of this story referred to "anesthesia" rather than "analgesia.")
SAN DIEGO – When caring for patients with sickle cell disease who were hospitalized for vaso-occlusive crisis, hospitalists more often used demand-only patient-controlled analgesia, compared with hematologists.
In addition, symptoms of acute chest syndrome were reported more often among patients cared for by hospitalists than by those cared for by hematologists.
The findings, reported during a poster session at the annual meeting of the American Society of Hematology, underscore the need for improved collaboration between hematologists and hospitalists in the care of patients with sickle cell disease.
These patients have been "increasingly managed by hospitalists rather than adult hematology specialists," the researchers, led by Dr. Nirmish Shah, wrote in their abstract. "Hospitalists provide efficient inpatient management; however, many possess knowledge gaps and carry biases into their relationship with patients with sickle cell disease."
In an effort to better understand the differences between hematologists and hospitalists in the care practices of this patient population, Dr. Shah and his associates reviewed the records of 298 patients with sickle cell disease who were hospitalized for vaso-occlusive crisis at Duke University Medical Center, Durham, N.C., between Sept. 1, 2008, and Jan. 31, 2011. In November of 2009, all inpatient care for patients with sickle cell disease was moved from the hematologists to the hospitalist service, said Dr. Shah, who directs the medical center’s sickle cell transition program.
Of the 298 patients, 120 were cared for by hematologists over 13 months while 178 were cared for by hospitalists over 13 months. The mean age of patients was 32 years.
There were no significant differences between hematologists and hospitalists in standard ordering practices including incentive spirometry, a bowel regimen, an anti-itching medication, and DVT prophylaxis. However, patients cared for by a hematologist had a higher total number of hours on a patient-controlled analgesia device (212 vs. 171 hours).
Dr. Shah also reported that, compared with hospitalists, hematologists were significantly less likely to use demand-only patient-controlled analgesia (23% vs. 42%) and significantly more likely to use both continuous and demand patient-controlled analgesia (67% vs. 54%).
Transfusions were similarly used by clinicians in both specialties. However, the number of units transfused was higher for hematologists, compared with hospitalists (a mean of 2.6 units vs. 1.9 units).
Erythrocytapheresis was used more often by hematologists than by hospitalists, but this difference did not reach statistical significance (6.7% vs. 2.2%).
No patients cared for by hematologists were diagnosed with acute chest syndrome, but 5.6% of those cared for by hospitalists were. In addition, patients cared for by hematologists had a significantly longer hospitalization, compared with those cared for by hospitalists (a mean of 10 days vs. 8.4 days, respectively) and a nonsignificantly lower 7-day readmission rate (6.7% vs. 7.2%) and 30-day readmission rate (35% vs. 40%).
"There often is a knowledge gap in hospitalist care of sickle cell patients," Dr. Shah said in an interview. "Because they may have a gap, it is our job as hematologists to be available as consultants. In addition, we should decide on the right method for controlling pain in this population of patients. I’m not trying to say that [hospitalists are] doing something in a wrong fashion. I think it brings up the idea that we need to collaborate more. At our hospital we’re assigning a nurse practitioner to round independently and help assist hospitalists now, in an attempt to improve our care for patients with sickle cell disease."
Dr. Shah said that he had no relevant financial disclosures.
(**Corrected: An earlier version of this story referred to "anesthesia" rather than "analgesia.")
SAN DIEGO – When caring for patients with sickle cell disease who were hospitalized for vaso-occlusive crisis, hospitalists more often used demand-only patient-controlled analgesia, compared with hematologists.
In addition, symptoms of acute chest syndrome were reported more often among patients cared for by hospitalists than by those cared for by hematologists.
The findings, reported during a poster session at the annual meeting of the American Society of Hematology, underscore the need for improved collaboration between hematologists and hospitalists in the care of patients with sickle cell disease.
These patients have been "increasingly managed by hospitalists rather than adult hematology specialists," the researchers, led by Dr. Nirmish Shah, wrote in their abstract. "Hospitalists provide efficient inpatient management; however, many possess knowledge gaps and carry biases into their relationship with patients with sickle cell disease."
In an effort to better understand the differences between hematologists and hospitalists in the care practices of this patient population, Dr. Shah and his associates reviewed the records of 298 patients with sickle cell disease who were hospitalized for vaso-occlusive crisis at Duke University Medical Center, Durham, N.C., between Sept. 1, 2008, and Jan. 31, 2011. In November of 2009, all inpatient care for patients with sickle cell disease was moved from the hematologists to the hospitalist service, said Dr. Shah, who directs the medical center’s sickle cell transition program.
Of the 298 patients, 120 were cared for by hematologists over 13 months while 178 were cared for by hospitalists over 13 months. The mean age of patients was 32 years.
There were no significant differences between hematologists and hospitalists in standard ordering practices including incentive spirometry, a bowel regimen, an anti-itching medication, and DVT prophylaxis. However, patients cared for by a hematologist had a higher total number of hours on a patient-controlled analgesia device (212 vs. 171 hours).
Dr. Shah also reported that, compared with hospitalists, hematologists were significantly less likely to use demand-only patient-controlled analgesia (23% vs. 42%) and significantly more likely to use both continuous and demand patient-controlled analgesia (67% vs. 54%).
Transfusions were similarly used by clinicians in both specialties. However, the number of units transfused was higher for hematologists, compared with hospitalists (a mean of 2.6 units vs. 1.9 units).
Erythrocytapheresis was used more often by hematologists than by hospitalists, but this difference did not reach statistical significance (6.7% vs. 2.2%).
No patients cared for by hematologists were diagnosed with acute chest syndrome, but 5.6% of those cared for by hospitalists were. In addition, patients cared for by hematologists had a significantly longer hospitalization, compared with those cared for by hospitalists (a mean of 10 days vs. 8.4 days, respectively) and a nonsignificantly lower 7-day readmission rate (6.7% vs. 7.2%) and 30-day readmission rate (35% vs. 40%).
"There often is a knowledge gap in hospitalist care of sickle cell patients," Dr. Shah said in an interview. "Because they may have a gap, it is our job as hematologists to be available as consultants. In addition, we should decide on the right method for controlling pain in this population of patients. I’m not trying to say that [hospitalists are] doing something in a wrong fashion. I think it brings up the idea that we need to collaborate more. At our hospital we’re assigning a nurse practitioner to round independently and help assist hospitalists now, in an attempt to improve our care for patients with sickle cell disease."
Dr. Shah said that he had no relevant financial disclosures.
(**Corrected: An earlier version of this story referred to "anesthesia" rather than "analgesia.")
FROM THE ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY
Major Finding: Compared with sickle cell patients cared for by a hospitalist, those cared for by a hematologist had more hours on a patient-controlled analgesia device (212 vs. 171 hours). In addition, hematologists were significantly less likely than were hospitalists to use demand-only patient-controlled analgesia (23% vs. 42%) and more likely to use both continuous and demand patient-controlled analgesia (67% vs. 54%).
Data Source: A study of 298 patients with sickle cell disease who were hospitalized for vaso-occlusive crisis at Duke University Medical Center between Sept. 1, 2008, and Jan. 31, 2011.
Disclosures: Dr. Shah said that he had no relevant financial disclosures.