User login
Erratum (2011;87:81-84)
Hodgkin Lymphoma Presenting as Generalized Pruritus in an Adolescent
What's Eating You? Hyalomma Ticks
Applying single-incision laparoscopic surgery to gyn practice: What’s involved
- Update on minimally invasive surgery
Amy Garcia, MD (April 2011) - 10 practical, evidence-based suggestions to improve your minimally invasive surgical skills now
Catherine A. Matthews, MD (April 2011)
The benefits of minimally invasive surgery—including less pain, faster recovery, and improved cosmesis—are well known.1,2 Standard laparotomy has been replaced by multiple-port operative laparoscopy for a great array of procedures, and advances in medical technology allow for a minimally invasive surgical approach even when a surgeon is faced with complex pathology.
Single-port laparoscopic surgery (SPLS) represents the latest advance in minimally invasive surgery. Using flexible endoscopes and articulating instruments, the surgeon can complete complex procedures through a single 2-cm incision in the abdomen. The incision is usually placed in the umbilicus, where it is easily hidden.3-8
Since the first laparoscopic hysterectomy through a single incision was performed 20 years ago, SPLS has been used successfully to perform nephrectomy, prostatectomy, hemicolectomy, cholecystectomy, splenectomy, intussusception reduction, gastrostomy tube placement, thoracoscopic lung biopsy, thoracoscopic decortication, and appendectomy.4-7
In gynecology, SPLS has been used to perform oophorectomy, salpingectomy, bilateral tubal ligation, ovarian cystectomy, surgical treatment of ectopic pregnancy, and both total and partial hysterectomy.7-11 At least two recent studies have concluded that SPLS is an acceptable way to treat many benign and malignant gynecologic conditions that are currently treated using multiport laparoscopy.3,11
This article outlines our approach to SPLS in the gynecologic patient and provides an overview of instrumentation, with the aim of allowing you to consider whether this approach might be feasible in your surgical practice, at your institution.
Unique setup required
When SPLS is performed through the umbilicus, the instruments must be held closer to the midline and more cephalad than during conventional laparoscopy to permit adequate visualization and manipulation. For this reason, the surgeon needs to assume a position higher over the torso and thorax of the patient, and both of the patient’s arms need to be tucked. Place the patient in a dorsal lithotomy position with a uterine manipulator in place to facilitate surgery—even when the uterus will be preserved and surgery involves only the adnexae.
With appropriate equipment and positioning, visualization and manipulation of anatomy are comparable to those of standard multiport laparoscopy.
New instruments simplify SPLS
Innovative surgical instruments allow for appropriate hand positioning outside the abdomen and minimize the internal collision of instruments brought through a single midline incision (FIGURE 1). A variety of single-port options are available, each with a unique patented design and method of insertion. In fact, the development of ports with multiple instrument channels has revolutionized SPLS.
FIGURE 1 Setup
Desired triangulation of instruments in SPLS setup.Before true single ports became available, it was necessary to place three 5-mm low-profile trocars in the fascia at three separate sites through a single skin incision. Pneumoperitoneum was established with a Veress needle, but the fascial incisions gradually merged with repeated cannula manipulation, producing air leaks.
Today, multiple-channel ports are placed using an open technique into a single skin and fascial incision. Trocars and instruments of varying size can be exchanged with ease without jeopardizing pneumoperitoneum.
Among the options:
- the SILS Port (Covidien) – a soft, flexible, three-channel port that allows for placement of blunt trocars ranging in size from 5 mm to 12 mm (FIGURE 2)
- the TriPort (Olympus America) – two flexible rings joined by a sleeve and multiple-channel port (FIGURE 3)
- GelPOINT Advanced Access Platform (Applied Medical) – a system constructed of synthetic gel material and consisting of a “GelSeal” cap, cannulas, and seals to accommodate 5-mm to 10-mm instrumentation (FIGURE 4).
We have found that all three devices allow for good range of motion while maintaining pneumoperitoneum.
(A recent article from Korea reports an inventive technique to perform single-incision laparoscopy using standard instrumentation: The authors fitted a self-retaining ring retractor with a surgical glove that had three of the fingers cut off and replaced by trocars.12)
FIGURE 2 SILS Port
The SILS Port is a soft, flexible, three-channel port that allows for placement of blunt trocars ranging in size from 5 mm to 12 mm.
FIGURE 3 TriPort
The TriPort system comprises two flexible rings joined by a sleeve and multiple-channel port.
FIGURE 4 GelPOINT
The GelPOINT Advanced Access Platform is constructed of synthetic gel material and accommodates 5-mm to 10-mm instrumentation.
A flexible laparoscope improves visualization
The ability to visualize the operative field is vital to any surgery, including SPLS. Use of a flexible laparoscope facilitates uncompromised visualization of the entire pelvis (FIGURE 5). Outside the abdomen, the flexible camera can be held laterally and away from the midline to help reduce the clashing of instruments and hands.
FIGURE 5 Flexible-tip laparoscope
A flexible-tip laparoscope ensures good visualization of the surgical field.If a flexible laparoscope is not available, a rigid 30-degree or 45-degree angled scope can be used, although visualization may be limited and adequate triangulation of instruments may be difficult to achieve.
When using a rigid laparoscope, a light cord that inserts into the back of the camera is necessary; otherwise, a 90-degree light cord adapter can be purchased.
Design enhancements facilitate coordination of instruments
One of the disadvantages of SPLS has been the restriction of movement that arises because of the close proximity of instruments and instrument handles. The latest designs have made articulation possible for tissue graspers, scissors, vessel sealers, and scopes.9,10 The value of articulation is apparent inside the abdomen, where it allows perfect positioning of the area of dissection. Outside the abdomen, the handles can be arranged in an angled pattern to allow the surgeon and assistant to operate comfortably (FIGURE 1).
In a four-handed procedure, one hand is on the camera, one on the uterine manipulator, and the remaining two hands operate the articulating grasper, vessel sealer, or needle driver, depending on the task.
Standard straight instruments can also be used for portions of the procedure.
Dissection and hemostasis are achieved in a manner similar to that of conventional laparoscopy. Our instrument of choice is an enhanced bipolar instrument, although harmonic and traditional biopolar energy can be used as well, depending on the preference of the surgeon.
The latest instruments are designed to dissect, cauterize, and cut, thereby decreasing the number of instrument exchanges necessary.
With the right aids, suturing can be simplified
Suturing through a single port can be a challenge. When possible, closure of the vaginal cuff following a total laparoscopic or laparoscopic-assisted vaginal hysterectomy should be performed from below. When endoscopic suturing is required, standard suturing using both intracorporeal and extracorporeal methods is possible.
Suturing aids such as the Endo Stitch (Covidien) or Lapra-Ty (Ethicon) are helpful. One author recommends Quill bidirectional, self-retaining suture with barbs (Angiotech) to avoid the need for knot-tying.6 (For more on self-retaining suture, see “Barbed suture, now in the toolbox of minimally invasive gyn surgery,” by Jon I. Einarsson, MD, MPH, and James A. Greenberg, MD, in the September 2009 issue at obgmanagement.com.)
The MiniLap (Stryker) is a 2.3-mm grasper that is inserted percutaneously directly through the abdominal wall without an incision. It can be used to set the needle on the needle driver or manipulate tissue while suturing. The resulting skin incision is barely visible and does not require closure.
Options are varied for specimen removal
Small specimens can be removed directly through a single-port system that has been opened, or they can be extracted after the system is removed, with rapid desufflation (FIGURE 6).
FIGURE 6 Single-incision oophorectomy
An ovary and tube removed through a single incision using the A) TriPort and B) SILS Port systems.Compared with conventional laparoscopy, the larger incision associated with single-port surgery facilitates specimen removal. Larger or potentially malignant specimens can be placed into an EndoCatch bag (Covidien) inserted through the single-incision 10-mm cannula (FIGURE 7).
FIGURE 7 Specimen removal
In this case, the specimen was placed in a 10-mm EndoCatch bag and removed through the 10-mm cannula of the SILS Port.In total laparoscopic hysterectomy or laparoscopic-assisted vaginal hysterectomy, the uterus is removed through the vagina. In supracervical hysterectomy, a small uterus can be removed through the cul-de-sac or directly through the single incision after placement in a bag.
When morcellation is required, the instrument can be placed through the cul-desac, cervix, or a single port. The morcellator can be placed directly through the SPLS port while utilizing the flexible scope in an angled direction (looking back toward the morcellator) for complete visualization (note: Covidien does not recommend this usage).
Transcervical tissue morcellation has also been described. In this approach, the cervix is dilated once the uterine body has been amputated, and the tissue morcellator is inserted through the cervix while the surgeon maintains visualization from above.6,13
How to master the technique
Many patients desire SPLS for its superior cosmetic outcome, but the approach may not always be appropriate. Depending on the procedure and characteristics of the patient (TABLE), multiple-port laparoscopy may be a better option. When a surgeon first attempts SPLS, we recommend that it be limited to the treatment of adnexal pathology only.
In your early SPlS cases, look for these patient characteristics
|
By using the techniques described in this article and selecting patients carefully, the surgeon can develop expertise in SPLS.11,14 During the learning process, the use of additional ports or Mini-Lap instruments (Stryker) can reduce the challenges of more difficult procedures and should be considered without reservation, as should the use of articulating accessory instruments and flexible or angled laparoscopes.
Although the clinical benefits of SPLS have yet to be determined, the cosmetic advantage of a single, hidden, umbilical incision likely increases patient satisfaction.
Clearly, the goal of SPLS is to use technology in a way that offers all of the benefits of traditional multiport laparoscopy without any of the limitations. Further study is required to determine whether SPLS meets this standard and, more important, whether it has any advantages over conventional techniques.
We want to hear from you! Tell us what you think.
1. Medeiros LR, Rosa DD, Bozzerri MC, et al. Laparoscopy versus laparotomy for benign ovarian tumour. Cochrane Database Syst Rev. 2009;(2):CD004751.-
2. Chapron C, Fauconnier A, Goffinet F, Breart G, Dubuisson JB. Laparoscopic surgery is not inherently dangerous for patients presenting with benign gynecological pathology. Results of a metaanalysis. Hum Reprod. 2002;17(5):1334-1342.
3. Fader AN, Escobar PF. Laparoendoscopic single-site surgery (LESS) in gynecologic oncology: technique and initial report. Gynecol Oncol. 2009;114(2):157-161.
4. Pelosi MA, Pelosi MA 3rd. Laparoscopic hysterectomy with bilateral salpingo-oophorectomy using a single umbilical puncture. N J Med. 1991;88(1):721-726.
5. Curcillo PG, King SA, Podolsky ER, Rottman SJ. Single port access (SPA) minimal access surgery through a single incision. Surg Technol Int. 2009;18:19-25.
6. Romanelli JR, Earle DB. Single-port laparoscopic surgery: an overview. Surg Endosc. 2009;23(7):1419-1427.
7. Ponsky TA. Single port laparoscopic cholecystectomy in adults and children: tools and techniques. J Am Coll Surg. 2009;209(5):e1-6.
8. Kim YW. Single port transumbilical myomectomy and ovarian cystectomy. J Minim Invasive Gynecol. 2009;16(6 suppl):S74.-
9. Ghezzi F, Cromi A, Fasola M, Bolis P. One-trocar salpingectomy for the treatment of tubal pregnancy: a “marionette-like” technique. BJOG. 2005;112(10):1417-1419.
10. Lim MC, Kim TJ, Kang S, Bae DS, Park SY, Seo SS. Embryonic natural orifice transumbilical endoscopic surgery (E-NOTES) for adnexal tumors. Surg Endosc. 2009;23(11):2445-2449.
11. Escobar PF, Starks DC, Fader AN, et al. Single-port risk-reducing salpingo-oophorectomy with and without hysterectomy: surgical outcomes and learning curve analysis. Gynecol Oncol. 2010;119(1):43-47.
12. Jeon HG, Jeong W, Oh CK. Initial experience with 50 laparoendoscopic single site surgeries using a homemade, single port device at a single center. J Urol. 2010;183(5):1866-1871.
13. Yoon G, Kim TJ, Lee YY, et al. Single port access subtotal hysterectomy with transcervical morcellation: a pilot study. J Minim Invasive Gynecol. 2010;17(1):78-81.
14. Ghomi A, Littman P, Prasad A, et al. Assessing the learning curve for laparoscopic supracervical hysterectomy. JSLS. 2007;11(2):190-194.
- Update on minimally invasive surgery
Amy Garcia, MD (April 2011) - 10 practical, evidence-based suggestions to improve your minimally invasive surgical skills now
Catherine A. Matthews, MD (April 2011)
The benefits of minimally invasive surgery—including less pain, faster recovery, and improved cosmesis—are well known.1,2 Standard laparotomy has been replaced by multiple-port operative laparoscopy for a great array of procedures, and advances in medical technology allow for a minimally invasive surgical approach even when a surgeon is faced with complex pathology.
Single-port laparoscopic surgery (SPLS) represents the latest advance in minimally invasive surgery. Using flexible endoscopes and articulating instruments, the surgeon can complete complex procedures through a single 2-cm incision in the abdomen. The incision is usually placed in the umbilicus, where it is easily hidden.3-8
Since the first laparoscopic hysterectomy through a single incision was performed 20 years ago, SPLS has been used successfully to perform nephrectomy, prostatectomy, hemicolectomy, cholecystectomy, splenectomy, intussusception reduction, gastrostomy tube placement, thoracoscopic lung biopsy, thoracoscopic decortication, and appendectomy.4-7
In gynecology, SPLS has been used to perform oophorectomy, salpingectomy, bilateral tubal ligation, ovarian cystectomy, surgical treatment of ectopic pregnancy, and both total and partial hysterectomy.7-11 At least two recent studies have concluded that SPLS is an acceptable way to treat many benign and malignant gynecologic conditions that are currently treated using multiport laparoscopy.3,11
This article outlines our approach to SPLS in the gynecologic patient and provides an overview of instrumentation, with the aim of allowing you to consider whether this approach might be feasible in your surgical practice, at your institution.
Unique setup required
When SPLS is performed through the umbilicus, the instruments must be held closer to the midline and more cephalad than during conventional laparoscopy to permit adequate visualization and manipulation. For this reason, the surgeon needs to assume a position higher over the torso and thorax of the patient, and both of the patient’s arms need to be tucked. Place the patient in a dorsal lithotomy position with a uterine manipulator in place to facilitate surgery—even when the uterus will be preserved and surgery involves only the adnexae.
With appropriate equipment and positioning, visualization and manipulation of anatomy are comparable to those of standard multiport laparoscopy.
New instruments simplify SPLS
Innovative surgical instruments allow for appropriate hand positioning outside the abdomen and minimize the internal collision of instruments brought through a single midline incision (FIGURE 1). A variety of single-port options are available, each with a unique patented design and method of insertion. In fact, the development of ports with multiple instrument channels has revolutionized SPLS.
FIGURE 1 Setup
Desired triangulation of instruments in SPLS setup.Before true single ports became available, it was necessary to place three 5-mm low-profile trocars in the fascia at three separate sites through a single skin incision. Pneumoperitoneum was established with a Veress needle, but the fascial incisions gradually merged with repeated cannula manipulation, producing air leaks.
Today, multiple-channel ports are placed using an open technique into a single skin and fascial incision. Trocars and instruments of varying size can be exchanged with ease without jeopardizing pneumoperitoneum.
Among the options:
- the SILS Port (Covidien) – a soft, flexible, three-channel port that allows for placement of blunt trocars ranging in size from 5 mm to 12 mm (FIGURE 2)
- the TriPort (Olympus America) – two flexible rings joined by a sleeve and multiple-channel port (FIGURE 3)
- GelPOINT Advanced Access Platform (Applied Medical) – a system constructed of synthetic gel material and consisting of a “GelSeal” cap, cannulas, and seals to accommodate 5-mm to 10-mm instrumentation (FIGURE 4).
We have found that all three devices allow for good range of motion while maintaining pneumoperitoneum.
(A recent article from Korea reports an inventive technique to perform single-incision laparoscopy using standard instrumentation: The authors fitted a self-retaining ring retractor with a surgical glove that had three of the fingers cut off and replaced by trocars.12)
FIGURE 2 SILS Port
The SILS Port is a soft, flexible, three-channel port that allows for placement of blunt trocars ranging in size from 5 mm to 12 mm.
FIGURE 3 TriPort
The TriPort system comprises two flexible rings joined by a sleeve and multiple-channel port.
FIGURE 4 GelPOINT
The GelPOINT Advanced Access Platform is constructed of synthetic gel material and accommodates 5-mm to 10-mm instrumentation.
A flexible laparoscope improves visualization
The ability to visualize the operative field is vital to any surgery, including SPLS. Use of a flexible laparoscope facilitates uncompromised visualization of the entire pelvis (FIGURE 5). Outside the abdomen, the flexible camera can be held laterally and away from the midline to help reduce the clashing of instruments and hands.
FIGURE 5 Flexible-tip laparoscope
A flexible-tip laparoscope ensures good visualization of the surgical field.If a flexible laparoscope is not available, a rigid 30-degree or 45-degree angled scope can be used, although visualization may be limited and adequate triangulation of instruments may be difficult to achieve.
When using a rigid laparoscope, a light cord that inserts into the back of the camera is necessary; otherwise, a 90-degree light cord adapter can be purchased.
Design enhancements facilitate coordination of instruments
One of the disadvantages of SPLS has been the restriction of movement that arises because of the close proximity of instruments and instrument handles. The latest designs have made articulation possible for tissue graspers, scissors, vessel sealers, and scopes.9,10 The value of articulation is apparent inside the abdomen, where it allows perfect positioning of the area of dissection. Outside the abdomen, the handles can be arranged in an angled pattern to allow the surgeon and assistant to operate comfortably (FIGURE 1).
In a four-handed procedure, one hand is on the camera, one on the uterine manipulator, and the remaining two hands operate the articulating grasper, vessel sealer, or needle driver, depending on the task.
Standard straight instruments can also be used for portions of the procedure.
Dissection and hemostasis are achieved in a manner similar to that of conventional laparoscopy. Our instrument of choice is an enhanced bipolar instrument, although harmonic and traditional biopolar energy can be used as well, depending on the preference of the surgeon.
The latest instruments are designed to dissect, cauterize, and cut, thereby decreasing the number of instrument exchanges necessary.
With the right aids, suturing can be simplified
Suturing through a single port can be a challenge. When possible, closure of the vaginal cuff following a total laparoscopic or laparoscopic-assisted vaginal hysterectomy should be performed from below. When endoscopic suturing is required, standard suturing using both intracorporeal and extracorporeal methods is possible.
Suturing aids such as the Endo Stitch (Covidien) or Lapra-Ty (Ethicon) are helpful. One author recommends Quill bidirectional, self-retaining suture with barbs (Angiotech) to avoid the need for knot-tying.6 (For more on self-retaining suture, see “Barbed suture, now in the toolbox of minimally invasive gyn surgery,” by Jon I. Einarsson, MD, MPH, and James A. Greenberg, MD, in the September 2009 issue at obgmanagement.com.)
The MiniLap (Stryker) is a 2.3-mm grasper that is inserted percutaneously directly through the abdominal wall without an incision. It can be used to set the needle on the needle driver or manipulate tissue while suturing. The resulting skin incision is barely visible and does not require closure.
Options are varied for specimen removal
Small specimens can be removed directly through a single-port system that has been opened, or they can be extracted after the system is removed, with rapid desufflation (FIGURE 6).
FIGURE 6 Single-incision oophorectomy
An ovary and tube removed through a single incision using the A) TriPort and B) SILS Port systems.Compared with conventional laparoscopy, the larger incision associated with single-port surgery facilitates specimen removal. Larger or potentially malignant specimens can be placed into an EndoCatch bag (Covidien) inserted through the single-incision 10-mm cannula (FIGURE 7).
FIGURE 7 Specimen removal
In this case, the specimen was placed in a 10-mm EndoCatch bag and removed through the 10-mm cannula of the SILS Port.In total laparoscopic hysterectomy or laparoscopic-assisted vaginal hysterectomy, the uterus is removed through the vagina. In supracervical hysterectomy, a small uterus can be removed through the cul-de-sac or directly through the single incision after placement in a bag.
When morcellation is required, the instrument can be placed through the cul-desac, cervix, or a single port. The morcellator can be placed directly through the SPLS port while utilizing the flexible scope in an angled direction (looking back toward the morcellator) for complete visualization (note: Covidien does not recommend this usage).
Transcervical tissue morcellation has also been described. In this approach, the cervix is dilated once the uterine body has been amputated, and the tissue morcellator is inserted through the cervix while the surgeon maintains visualization from above.6,13
How to master the technique
Many patients desire SPLS for its superior cosmetic outcome, but the approach may not always be appropriate. Depending on the procedure and characteristics of the patient (TABLE), multiple-port laparoscopy may be a better option. When a surgeon first attempts SPLS, we recommend that it be limited to the treatment of adnexal pathology only.
In your early SPlS cases, look for these patient characteristics
|
By using the techniques described in this article and selecting patients carefully, the surgeon can develop expertise in SPLS.11,14 During the learning process, the use of additional ports or Mini-Lap instruments (Stryker) can reduce the challenges of more difficult procedures and should be considered without reservation, as should the use of articulating accessory instruments and flexible or angled laparoscopes.
Although the clinical benefits of SPLS have yet to be determined, the cosmetic advantage of a single, hidden, umbilical incision likely increases patient satisfaction.
Clearly, the goal of SPLS is to use technology in a way that offers all of the benefits of traditional multiport laparoscopy without any of the limitations. Further study is required to determine whether SPLS meets this standard and, more important, whether it has any advantages over conventional techniques.
We want to hear from you! Tell us what you think.
- Update on minimally invasive surgery
Amy Garcia, MD (April 2011) - 10 practical, evidence-based suggestions to improve your minimally invasive surgical skills now
Catherine A. Matthews, MD (April 2011)
The benefits of minimally invasive surgery—including less pain, faster recovery, and improved cosmesis—are well known.1,2 Standard laparotomy has been replaced by multiple-port operative laparoscopy for a great array of procedures, and advances in medical technology allow for a minimally invasive surgical approach even when a surgeon is faced with complex pathology.
Single-port laparoscopic surgery (SPLS) represents the latest advance in minimally invasive surgery. Using flexible endoscopes and articulating instruments, the surgeon can complete complex procedures through a single 2-cm incision in the abdomen. The incision is usually placed in the umbilicus, where it is easily hidden.3-8
Since the first laparoscopic hysterectomy through a single incision was performed 20 years ago, SPLS has been used successfully to perform nephrectomy, prostatectomy, hemicolectomy, cholecystectomy, splenectomy, intussusception reduction, gastrostomy tube placement, thoracoscopic lung biopsy, thoracoscopic decortication, and appendectomy.4-7
In gynecology, SPLS has been used to perform oophorectomy, salpingectomy, bilateral tubal ligation, ovarian cystectomy, surgical treatment of ectopic pregnancy, and both total and partial hysterectomy.7-11 At least two recent studies have concluded that SPLS is an acceptable way to treat many benign and malignant gynecologic conditions that are currently treated using multiport laparoscopy.3,11
This article outlines our approach to SPLS in the gynecologic patient and provides an overview of instrumentation, with the aim of allowing you to consider whether this approach might be feasible in your surgical practice, at your institution.
Unique setup required
When SPLS is performed through the umbilicus, the instruments must be held closer to the midline and more cephalad than during conventional laparoscopy to permit adequate visualization and manipulation. For this reason, the surgeon needs to assume a position higher over the torso and thorax of the patient, and both of the patient’s arms need to be tucked. Place the patient in a dorsal lithotomy position with a uterine manipulator in place to facilitate surgery—even when the uterus will be preserved and surgery involves only the adnexae.
With appropriate equipment and positioning, visualization and manipulation of anatomy are comparable to those of standard multiport laparoscopy.
New instruments simplify SPLS
Innovative surgical instruments allow for appropriate hand positioning outside the abdomen and minimize the internal collision of instruments brought through a single midline incision (FIGURE 1). A variety of single-port options are available, each with a unique patented design and method of insertion. In fact, the development of ports with multiple instrument channels has revolutionized SPLS.
FIGURE 1 Setup
Desired triangulation of instruments in SPLS setup.Before true single ports became available, it was necessary to place three 5-mm low-profile trocars in the fascia at three separate sites through a single skin incision. Pneumoperitoneum was established with a Veress needle, but the fascial incisions gradually merged with repeated cannula manipulation, producing air leaks.
Today, multiple-channel ports are placed using an open technique into a single skin and fascial incision. Trocars and instruments of varying size can be exchanged with ease without jeopardizing pneumoperitoneum.
Among the options:
- the SILS Port (Covidien) – a soft, flexible, three-channel port that allows for placement of blunt trocars ranging in size from 5 mm to 12 mm (FIGURE 2)
- the TriPort (Olympus America) – two flexible rings joined by a sleeve and multiple-channel port (FIGURE 3)
- GelPOINT Advanced Access Platform (Applied Medical) – a system constructed of synthetic gel material and consisting of a “GelSeal” cap, cannulas, and seals to accommodate 5-mm to 10-mm instrumentation (FIGURE 4).
We have found that all three devices allow for good range of motion while maintaining pneumoperitoneum.
(A recent article from Korea reports an inventive technique to perform single-incision laparoscopy using standard instrumentation: The authors fitted a self-retaining ring retractor with a surgical glove that had three of the fingers cut off and replaced by trocars.12)
FIGURE 2 SILS Port
The SILS Port is a soft, flexible, three-channel port that allows for placement of blunt trocars ranging in size from 5 mm to 12 mm.
FIGURE 3 TriPort
The TriPort system comprises two flexible rings joined by a sleeve and multiple-channel port.
FIGURE 4 GelPOINT
The GelPOINT Advanced Access Platform is constructed of synthetic gel material and accommodates 5-mm to 10-mm instrumentation.
A flexible laparoscope improves visualization
The ability to visualize the operative field is vital to any surgery, including SPLS. Use of a flexible laparoscope facilitates uncompromised visualization of the entire pelvis (FIGURE 5). Outside the abdomen, the flexible camera can be held laterally and away from the midline to help reduce the clashing of instruments and hands.
FIGURE 5 Flexible-tip laparoscope
A flexible-tip laparoscope ensures good visualization of the surgical field.If a flexible laparoscope is not available, a rigid 30-degree or 45-degree angled scope can be used, although visualization may be limited and adequate triangulation of instruments may be difficult to achieve.
When using a rigid laparoscope, a light cord that inserts into the back of the camera is necessary; otherwise, a 90-degree light cord adapter can be purchased.
Design enhancements facilitate coordination of instruments
One of the disadvantages of SPLS has been the restriction of movement that arises because of the close proximity of instruments and instrument handles. The latest designs have made articulation possible for tissue graspers, scissors, vessel sealers, and scopes.9,10 The value of articulation is apparent inside the abdomen, where it allows perfect positioning of the area of dissection. Outside the abdomen, the handles can be arranged in an angled pattern to allow the surgeon and assistant to operate comfortably (FIGURE 1).
In a four-handed procedure, one hand is on the camera, one on the uterine manipulator, and the remaining two hands operate the articulating grasper, vessel sealer, or needle driver, depending on the task.
Standard straight instruments can also be used for portions of the procedure.
Dissection and hemostasis are achieved in a manner similar to that of conventional laparoscopy. Our instrument of choice is an enhanced bipolar instrument, although harmonic and traditional biopolar energy can be used as well, depending on the preference of the surgeon.
The latest instruments are designed to dissect, cauterize, and cut, thereby decreasing the number of instrument exchanges necessary.
With the right aids, suturing can be simplified
Suturing through a single port can be a challenge. When possible, closure of the vaginal cuff following a total laparoscopic or laparoscopic-assisted vaginal hysterectomy should be performed from below. When endoscopic suturing is required, standard suturing using both intracorporeal and extracorporeal methods is possible.
Suturing aids such as the Endo Stitch (Covidien) or Lapra-Ty (Ethicon) are helpful. One author recommends Quill bidirectional, self-retaining suture with barbs (Angiotech) to avoid the need for knot-tying.6 (For more on self-retaining suture, see “Barbed suture, now in the toolbox of minimally invasive gyn surgery,” by Jon I. Einarsson, MD, MPH, and James A. Greenberg, MD, in the September 2009 issue at obgmanagement.com.)
The MiniLap (Stryker) is a 2.3-mm grasper that is inserted percutaneously directly through the abdominal wall without an incision. It can be used to set the needle on the needle driver or manipulate tissue while suturing. The resulting skin incision is barely visible and does not require closure.
Options are varied for specimen removal
Small specimens can be removed directly through a single-port system that has been opened, or they can be extracted after the system is removed, with rapid desufflation (FIGURE 6).
FIGURE 6 Single-incision oophorectomy
An ovary and tube removed through a single incision using the A) TriPort and B) SILS Port systems.Compared with conventional laparoscopy, the larger incision associated with single-port surgery facilitates specimen removal. Larger or potentially malignant specimens can be placed into an EndoCatch bag (Covidien) inserted through the single-incision 10-mm cannula (FIGURE 7).
FIGURE 7 Specimen removal
In this case, the specimen was placed in a 10-mm EndoCatch bag and removed through the 10-mm cannula of the SILS Port.In total laparoscopic hysterectomy or laparoscopic-assisted vaginal hysterectomy, the uterus is removed through the vagina. In supracervical hysterectomy, a small uterus can be removed through the cul-de-sac or directly through the single incision after placement in a bag.
When morcellation is required, the instrument can be placed through the cul-desac, cervix, or a single port. The morcellator can be placed directly through the SPLS port while utilizing the flexible scope in an angled direction (looking back toward the morcellator) for complete visualization (note: Covidien does not recommend this usage).
Transcervical tissue morcellation has also been described. In this approach, the cervix is dilated once the uterine body has been amputated, and the tissue morcellator is inserted through the cervix while the surgeon maintains visualization from above.6,13
How to master the technique
Many patients desire SPLS for its superior cosmetic outcome, but the approach may not always be appropriate. Depending on the procedure and characteristics of the patient (TABLE), multiple-port laparoscopy may be a better option. When a surgeon first attempts SPLS, we recommend that it be limited to the treatment of adnexal pathology only.
In your early SPlS cases, look for these patient characteristics
|
By using the techniques described in this article and selecting patients carefully, the surgeon can develop expertise in SPLS.11,14 During the learning process, the use of additional ports or Mini-Lap instruments (Stryker) can reduce the challenges of more difficult procedures and should be considered without reservation, as should the use of articulating accessory instruments and flexible or angled laparoscopes.
Although the clinical benefits of SPLS have yet to be determined, the cosmetic advantage of a single, hidden, umbilical incision likely increases patient satisfaction.
Clearly, the goal of SPLS is to use technology in a way that offers all of the benefits of traditional multiport laparoscopy without any of the limitations. Further study is required to determine whether SPLS meets this standard and, more important, whether it has any advantages over conventional techniques.
We want to hear from you! Tell us what you think.
1. Medeiros LR, Rosa DD, Bozzerri MC, et al. Laparoscopy versus laparotomy for benign ovarian tumour. Cochrane Database Syst Rev. 2009;(2):CD004751.-
2. Chapron C, Fauconnier A, Goffinet F, Breart G, Dubuisson JB. Laparoscopic surgery is not inherently dangerous for patients presenting with benign gynecological pathology. Results of a metaanalysis. Hum Reprod. 2002;17(5):1334-1342.
3. Fader AN, Escobar PF. Laparoendoscopic single-site surgery (LESS) in gynecologic oncology: technique and initial report. Gynecol Oncol. 2009;114(2):157-161.
4. Pelosi MA, Pelosi MA 3rd. Laparoscopic hysterectomy with bilateral salpingo-oophorectomy using a single umbilical puncture. N J Med. 1991;88(1):721-726.
5. Curcillo PG, King SA, Podolsky ER, Rottman SJ. Single port access (SPA) minimal access surgery through a single incision. Surg Technol Int. 2009;18:19-25.
6. Romanelli JR, Earle DB. Single-port laparoscopic surgery: an overview. Surg Endosc. 2009;23(7):1419-1427.
7. Ponsky TA. Single port laparoscopic cholecystectomy in adults and children: tools and techniques. J Am Coll Surg. 2009;209(5):e1-6.
8. Kim YW. Single port transumbilical myomectomy and ovarian cystectomy. J Minim Invasive Gynecol. 2009;16(6 suppl):S74.-
9. Ghezzi F, Cromi A, Fasola M, Bolis P. One-trocar salpingectomy for the treatment of tubal pregnancy: a “marionette-like” technique. BJOG. 2005;112(10):1417-1419.
10. Lim MC, Kim TJ, Kang S, Bae DS, Park SY, Seo SS. Embryonic natural orifice transumbilical endoscopic surgery (E-NOTES) for adnexal tumors. Surg Endosc. 2009;23(11):2445-2449.
11. Escobar PF, Starks DC, Fader AN, et al. Single-port risk-reducing salpingo-oophorectomy with and without hysterectomy: surgical outcomes and learning curve analysis. Gynecol Oncol. 2010;119(1):43-47.
12. Jeon HG, Jeong W, Oh CK. Initial experience with 50 laparoendoscopic single site surgeries using a homemade, single port device at a single center. J Urol. 2010;183(5):1866-1871.
13. Yoon G, Kim TJ, Lee YY, et al. Single port access subtotal hysterectomy with transcervical morcellation: a pilot study. J Minim Invasive Gynecol. 2010;17(1):78-81.
14. Ghomi A, Littman P, Prasad A, et al. Assessing the learning curve for laparoscopic supracervical hysterectomy. JSLS. 2007;11(2):190-194.
1. Medeiros LR, Rosa DD, Bozzerri MC, et al. Laparoscopy versus laparotomy for benign ovarian tumour. Cochrane Database Syst Rev. 2009;(2):CD004751.-
2. Chapron C, Fauconnier A, Goffinet F, Breart G, Dubuisson JB. Laparoscopic surgery is not inherently dangerous for patients presenting with benign gynecological pathology. Results of a metaanalysis. Hum Reprod. 2002;17(5):1334-1342.
3. Fader AN, Escobar PF. Laparoendoscopic single-site surgery (LESS) in gynecologic oncology: technique and initial report. Gynecol Oncol. 2009;114(2):157-161.
4. Pelosi MA, Pelosi MA 3rd. Laparoscopic hysterectomy with bilateral salpingo-oophorectomy using a single umbilical puncture. N J Med. 1991;88(1):721-726.
5. Curcillo PG, King SA, Podolsky ER, Rottman SJ. Single port access (SPA) minimal access surgery through a single incision. Surg Technol Int. 2009;18:19-25.
6. Romanelli JR, Earle DB. Single-port laparoscopic surgery: an overview. Surg Endosc. 2009;23(7):1419-1427.
7. Ponsky TA. Single port laparoscopic cholecystectomy in adults and children: tools and techniques. J Am Coll Surg. 2009;209(5):e1-6.
8. Kim YW. Single port transumbilical myomectomy and ovarian cystectomy. J Minim Invasive Gynecol. 2009;16(6 suppl):S74.-
9. Ghezzi F, Cromi A, Fasola M, Bolis P. One-trocar salpingectomy for the treatment of tubal pregnancy: a “marionette-like” technique. BJOG. 2005;112(10):1417-1419.
10. Lim MC, Kim TJ, Kang S, Bae DS, Park SY, Seo SS. Embryonic natural orifice transumbilical endoscopic surgery (E-NOTES) for adnexal tumors. Surg Endosc. 2009;23(11):2445-2449.
11. Escobar PF, Starks DC, Fader AN, et al. Single-port risk-reducing salpingo-oophorectomy with and without hysterectomy: surgical outcomes and learning curve analysis. Gynecol Oncol. 2010;119(1):43-47.
12. Jeon HG, Jeong W, Oh CK. Initial experience with 50 laparoendoscopic single site surgeries using a homemade, single port device at a single center. J Urol. 2010;183(5):1866-1871.
13. Yoon G, Kim TJ, Lee YY, et al. Single port access subtotal hysterectomy with transcervical morcellation: a pilot study. J Minim Invasive Gynecol. 2010;17(1):78-81.
14. Ghomi A, Littman P, Prasad A, et al. Assessing the learning curve for laparoscopic supracervical hysterectomy. JSLS. 2007;11(2):190-194.
Overlooked diagnoses
“Not all mood swings are bipolar disorder” (Current Psychiatry, February 2011, p. 38-52) is a highly relevant and helpful article with a glaring omission. There is no mention of the emotional lability and behavioral dyscontrol associated with abuse, trauma, and invalidation. “Mood swing” symptoms are prominent in developmental trauma disorder and complex posttraumatic stress disorder, although these diagnoses are not yet in the DSM. Unfortunately, the effects of abuse, trauma, and invalidation often are unrecognized in the differential diagnoses of these children and too often the “kneejerk” diagnoses of bipolar disorder, oppositional defiant disorder, and attention-deficit/hyperactivity disorder are inappropriately assigned, delaying the implementation of trauma theory-informed therapy.
Bradford B. Schwartz, MD
Private Practice
York, PA
The authors respond
We thank Dr. Schwartz for his comments regarding emotional lability and behavioral dyscontrol associated with children who have experienced trauma, abuse, and invalidation. An assessment for possible trauma always is part of the initial assessment of each child referred to our program. None of the patients discussed in our article had a history of abuse or trauma. Referrals to our pediatric mood disorders program initially are screened through the Cincinnati Children’s Hospital Psychiatric Intake and Response Center, which functions as triage, gathering psychiatric history, including assessing trauma, and children with a history of abuse and trauma are referred to other clinicians specializing in this area. But Dr. Schwartz’s point is well taken—trauma or abuse always should be part of the differential diagnosis of children and adolescents referred for mood swings.
Robert A. Kowatch, MD, PhD
Professor of Psychiatry and Pediatrics
Erin Monroe, CNS
Clinical Nurse Specialist
Division of Psychiatry
Sergio V. Delgado, MD
Associate Professor of Psychiatry
and Pediatrics
Cincinnati Children’s Hospital Medical Center
Cincinnati, OH
“Not all mood swings are bipolar disorder” (Current Psychiatry, February 2011, p. 38-52) is a highly relevant and helpful article with a glaring omission. There is no mention of the emotional lability and behavioral dyscontrol associated with abuse, trauma, and invalidation. “Mood swing” symptoms are prominent in developmental trauma disorder and complex posttraumatic stress disorder, although these diagnoses are not yet in the DSM. Unfortunately, the effects of abuse, trauma, and invalidation often are unrecognized in the differential diagnoses of these children and too often the “kneejerk” diagnoses of bipolar disorder, oppositional defiant disorder, and attention-deficit/hyperactivity disorder are inappropriately assigned, delaying the implementation of trauma theory-informed therapy.
Bradford B. Schwartz, MD
Private Practice
York, PA
The authors respond
We thank Dr. Schwartz for his comments regarding emotional lability and behavioral dyscontrol associated with children who have experienced trauma, abuse, and invalidation. An assessment for possible trauma always is part of the initial assessment of each child referred to our program. None of the patients discussed in our article had a history of abuse or trauma. Referrals to our pediatric mood disorders program initially are screened through the Cincinnati Children’s Hospital Psychiatric Intake and Response Center, which functions as triage, gathering psychiatric history, including assessing trauma, and children with a history of abuse and trauma are referred to other clinicians specializing in this area. But Dr. Schwartz’s point is well taken—trauma or abuse always should be part of the differential diagnosis of children and adolescents referred for mood swings.
Robert A. Kowatch, MD, PhD
Professor of Psychiatry and Pediatrics
Erin Monroe, CNS
Clinical Nurse Specialist
Division of Psychiatry
Sergio V. Delgado, MD
Associate Professor of Psychiatry
and Pediatrics
Cincinnati Children’s Hospital Medical Center
Cincinnati, OH
“Not all mood swings are bipolar disorder” (Current Psychiatry, February 2011, p. 38-52) is a highly relevant and helpful article with a glaring omission. There is no mention of the emotional lability and behavioral dyscontrol associated with abuse, trauma, and invalidation. “Mood swing” symptoms are prominent in developmental trauma disorder and complex posttraumatic stress disorder, although these diagnoses are not yet in the DSM. Unfortunately, the effects of abuse, trauma, and invalidation often are unrecognized in the differential diagnoses of these children and too often the “kneejerk” diagnoses of bipolar disorder, oppositional defiant disorder, and attention-deficit/hyperactivity disorder are inappropriately assigned, delaying the implementation of trauma theory-informed therapy.
Bradford B. Schwartz, MD
Private Practice
York, PA
The authors respond
We thank Dr. Schwartz for his comments regarding emotional lability and behavioral dyscontrol associated with children who have experienced trauma, abuse, and invalidation. An assessment for possible trauma always is part of the initial assessment of each child referred to our program. None of the patients discussed in our article had a history of abuse or trauma. Referrals to our pediatric mood disorders program initially are screened through the Cincinnati Children’s Hospital Psychiatric Intake and Response Center, which functions as triage, gathering psychiatric history, including assessing trauma, and children with a history of abuse and trauma are referred to other clinicians specializing in this area. But Dr. Schwartz’s point is well taken—trauma or abuse always should be part of the differential diagnosis of children and adolescents referred for mood swings.
Robert A. Kowatch, MD, PhD
Professor of Psychiatry and Pediatrics
Erin Monroe, CNS
Clinical Nurse Specialist
Division of Psychiatry
Sergio V. Delgado, MD
Associate Professor of Psychiatry
and Pediatrics
Cincinnati Children’s Hospital Medical Center
Cincinnati, OH
Subjective cognitive impairment: When to be concerned about ‘senior moments’
MS. F, age 66, requests genetic testing because she is concerned about mild memory difficulties, such as forgetting names and where she puts her keys or checkbook, and fears she may be developing Alzheimer’s disease (AD). Her mother and sister were diagnosed with AD in their early 60s. Ms. F has 20 years of education and reports no problems with driving, managing her finances, remembering to take her medications, or maintaining social activities, which her husband confirms.
Detailed questioning about anxiety and depressive symptoms reveals substantial worries about future cognitive decline and some concerns about her finances and her husband’s health. Ms. F says she occasionally feels down and has low energy but denies other depressive symptoms. She reports no sleep disturbances—including snoring and daytime sleepiness, which could indicate obstructive sleep apnea—which her husband confirms. Ms. F takes levothyroxine for hypothyroidism, atenolol for hypertension, aspirin and clopidogrel for coronary artery disease, and atorvastatin for hyperlipidemia. In addition, she provides a long list of over-the-counter (OTC) supplements—ginkgo, huperzine, ginseng, phosphatidylserine, B1, B12, folate, vitamin D, alpha-lipoic acid, and vinpocetine—that she takes to “protect” her brain from AD.
Subjective cognitive impairment (SCI) in older persons is a common condition with a largely unclear prognosis. Many older adults (age ≥65) express concern about mild cognitive problems—“senior moments”—such as word-finding difficulties and forgetfulness.1 Individuals may wonder if walking into a room only to forget why might be the first sign of dementia. Some older adults try to counteract these memory problems by engaging in brain exercises—including costly computer games—and taking OTC “brain-enhancing” vitamins, herbal remedies, and other supplements.
Although some clinicians may view SCI as benign, that is not always true (Table l).2-5 This article discusses the clinical significance of these mild cognitive complaints by examining:
- age-related cognitive decline (ARCD)
- SCI
- how SCI can be differentiated from more serious conditions, such as mild cognitive impairment (MCI) and early stages of AD and other dementias.
We also will discuss assessing and treating cognitive complaints. Although distinctions between SCI and ARCD may be controversial, evidence suggests clinicians need to adopt a more nuanced clinical approach.
Table 1
Why SCI should be taken seriously
| SCI may create emotional distress because patients are aware of decline in their ‘mental sharpness’ |
| SCI patients might consume unnecessary and potentially harmful OTC supplements touted to promote memory |
| Patients might limit their driving and financial management to avoid making mistakes |
| SCI might impair medication adherence2 |
| SCI may be an early sign of dementia3 |
| Patients’ worry about their self-perceived memory loss might predict dementia4 |
| SCI may predict nursing home placement5 |
| Addressing SCI gives health care providers an opportunity to address anxiety or depression that often accompany SCI |
| Evaluation of potential causes of SCI may uncover reversible conditions that can be treated |
| OTC: over-the-counter; SCI: subjective cognitive impairment |
‘Normal’ cognitive decline
ARCD is subtle decline in cognitive abilities, such as episodic memory, attention, and time needed to complete complex activities.6,7 Individuals with ARCD might not have subjective memory complaints or objective cognitive deficits, and their ability to live independently may not be compromised.7 The degree of decline in ARCD may be smaller than previously thought.8 Park9 summarizes 4 main mechanisms thought to underlie age-related declines in cognition:
- reduced speed of processing
- decreased working memory capabilities
- declining inhibitory control (eg, impaired complex attentional capabilities)
- sensory changes (eg, visual and auditory deficits).
ARCD traditionally is thought to result from predictable changes in the brain associated with aging, such as reduced brain volume in the hippocampus and frontal lobes, loss of myelin, loss of synapses, and cytoskeletal changes.7 However, not all older adults experience ARCD. Some remain highly functional in their later years and continue to actively engage in life well into very old age.6,9
Subjective cognitive impairment
One-quarter to one-half of community-dwelling older adults report subjective cognitive complaints, such as forgetfulness and word-finding difficulties.10 Patients with SCI do not show objective evidence of cognitive impairment on neuropsychological tests and their cognitive problems cause no functional decline.10
Preliminary evidence indicates that SCI may be a harbinger of further cognitive decline. Reisberg et al3 found that compared with patients without SCI, patients with SCI were 4.5 times more likely to develop MCI—cognitive difficulties that can be detected by cognitive tests, but do not cause functional decline—or dementia within 7 years.3 Studies also have suggested that SCI may be a pre-MCI stage of subsequent dementia.11-13 AD generally has a long (10 to 12 years) and progressive prodromal phase before dementia onset and is characterized by successive emergence of cognitive deficits, memory complaints, depressive symptoms, and functional impairment.14
In light of this research, we believe patients with SCI and other risk factors for AD, such as a family history of AD, may be at higher risk of further cognitive and functional decline compared with individuals with ARCD and no AD risk factors. Therefore, patients with SCI and other risk factors for AD (Table 2)15-19 may benefit from annual follow-up to determine if cognitive problems have progressed to MCI or AD.
SCI may be a response to subclinical alterations in neurobiology—a phenomenon known as reverse causality.20 Biomarkers, such as cerebrospinal fluid levels of ß-amyloid and phosphorylated tau, and amyloid imaging using positron emission tomography may help identify AD in SCI patients.21 In these patients, SCI is a misnomer because the cognitive impairment is real—not “subjective”—but current tests are not sensitive enough to detect the cognitive decline the patient has recognized. This group of patients should be differentiated from individuals who may perceive typical cognitive aging (ARCD) as pathologic and complain about it. In the future, biomarkers may help differentiate these 2 groups.
Table 2
Factors that increase SCI patients’ risk for dementia
| Family history of Alzheimer’s disease |
| Mild behavioral impairment |
| Slow gait |
| Depression |
| Rapid weight loss |
| Multiple subtle neurologic abnormalities |
| Vascular disease (eg, peripheral vascular disease, coronary artery disease, cerebrovascular disease) |
| SCI: subjective cognitive impairment Source: References 15-19 |
Mild cognitive impairment
MCI is similar to SCI because MCI patients may present with complaints of memory decline and other cognitive difficulties22 but neither condition is associated with significant impairment of daily activities.23 The key difference is that patients with MCI demonstrate impaired performance on objective cognitive tests whereas SCI patients do not.24 In our experience, office-based tests do not reliably differentiate the 2 conditions because many patients with SCI may show mild impairment in tests such as the Mini-Mental State Exam (MMSE)25 but comprehensive neuropsychological testing reveals no objective cognitive deficits. Neuropsychological testing is essential to reliably differentiate SCI from MCI.
The distinction between SCI and MCI is clinically relevant because evidence suggests that MCI patients have a near-term risk of developing dementia, particularly AD.22,23 In a longitudinal study of 76 individuals with MCI, 12% of patients progressed to AD each year compared with 1% to 2% of healthy older adults.26 Patients with MCI are at increased risk of delirium (especially during hospitalization), falls, medication errors, and difficulty managing their finances.24 Older adults with MCI also have increased mortality compared with older adults with normal cognitive functioning.22 Both SCI and MCI should be differentiated from mild dementia. Common dementias in older adults include:
- AD dementia
- Vascular dementia (may occur with or without AD)
- Lewy body dementia
- Frontotemporal dementia
- Parkinson’s disease dementia.
By definition, all dementia types are associated with impaired ability to perform daily activities and cognitive decline.27
Assessing cognitive complaints
Evaluation of older adults’ cognitive complaints should begin with a thorough history to elicit symptoms of anxiety, depression, physical complaints, and any associated functional decline; a physical exam; and a comprehensive mental status examination. This initial evaluation should be followed by routine and specific investigations as indicated (Table 3).22,24,28,29
In a 6-year study of 100 older adults with and without objective evidence of memory decline, both groups showed similar rates of cognitive complaints.30 Also, researchers found no relationship between individuals’ perception of their cognitive functioning and performance on neuropsychological testing. Mood, education level, and apolipoprotein E epsilon 4 genotype status also did not correlate with participants’ subjective cognitive complaints. These findings highlight the need for objective test data to determine whether older adults’ memory complaints reflect pathologic changes in cognition. After a thorough diagnostic workup, some patients complaining of memory decline will have no detectable evidence of cognitive dysfunction or an identifiable cause. However, others may have identifiable causes of memory impairment (Table 4)28,29,31,32—which could be treated—some will have MCI, and others may be in an early stage of dementia.
Table 3
Investigation of older adults with SCI
| Investigation | Rationale |
|---|---|
| Routine | |
| Neuropsychological testing | Delineation of cognitive syndromes (SCI vs MCI vs AD*) |
| Hematology (full blood count) | Screen for anemia |
| Biochemistry (electrolytes, renal function, liver function, thyroid function, B12, and folate) | Screen for treatable causes of cognitive complaints |
| For specific indication suggested by history, physical exam, or neuropsychological testing | |
| Neuroimaging | Generalized and regional imaging (eg, hippocampal atrophy, space occupying lesions) |
| Electroencephalography | Epilepsy/seizures (especially absence and complex partial) |
| Cardiac (eg, echocardiography) | May reveal cardiac arrhythmia or sources of emboli |
| Inflammatory markers (eg, ESR) | Screen for inflammatory processes |
| Treponemal serology | Tertiary syphilis |
| *Alzheimer’s disease and other dementias AD: Alzheimer’s disease; ESR: erythrocyte sedimentation rate; MCI: mild cognitive impairment; SCI: subjective cognitive impairment Source: References 22,24,28,29 | |
Table 4
Differential diagnosis of SCI
| Cause of cognitive impairment | Potential mechanism |
|---|---|
| ARCD | Allostatic load, ‘wear and tear’ from a lifetime of physiological or psychological stresses and adaptations |
| Anemia | Neuronal hypoxia |
| Alzheimer’s disease | Amyloid and/or tau-mediated neurotoxicity, neuroinflammation |
| Cerebrovascular disease | Neuronal ischemia and hypoxia, neuroinflammation |
| Vitamin deficiencies (eg, B1, B12, folate, D) | Impaired neuronal and neurotransmitter function |
| Inadequate protein intake | Impaired neuronal function |
| Anticholinergic drug use | Decreased cholinergic neurotransmission |
| Alcohol use | Direct neurotoxicity and indirect causes such as malnutrition or head injury |
| Depression, anxiety | Hippocampal dysfunction with or without atrophy |
| Obstructive sleep apnea | Neuronal hypoxia, neuroinflammation |
| Head injury | Neuronal and synaptic loss |
| ARCD: age-related cognitive decline; SCI: subjective cognitive impairment Source: References 28,29,31,32 | |
CASE CONTINUED: No measurable deficits
Ms. F’s medical history is remarkable for coronary artery disease, hypothyroidism, hypertension, hyperlipidemia, cataracts, arthritis, back surgery (secondary to spondylosis), and foot surgery. Ms. F denies a history of alcohol or illicit substance abuse. She smoked tobacco for 30 years (2 packs per day), but quit 5 years ago after her heart attack. Physical exam is unremarkable except for mild obesity (body mass index = 31 kg/m2).
Ms. F’s mental status exam reveals anxious mood and affect. Her recall is 2 out of 3 items. Her MMSE score is 29/30 (1 point lost on recall) and her Geriatric Depression Scale33 score is 2/15, indicating minimal depressive symptoms. On neuropsychological testing, Ms. F demonstrates high average intellectual abilities; compared with others her age, she performs within expectations on all measures. That is, she performs within the above-average to low-average range on measures of attention, working memory, speed of processing, expressive language, learning, memory, visual spatial abilities, executive functioning, and knowledge of basic health and safety information.
Enhancing neuroplasticity
We recommend neuroplasticity-based interventions to treat SCI and promote healthy brain aging.20,29 For a checklist clinicians can use to promote healthy brain aging and thus improve patients’ cognitive health see this article at CurrentPsychiatry. com. Table 51,29 lists cognitive strategies to improve memory and maintain cognitive vitality.
Enhancing brain plasticity and neurogenesis requires engaging older adults in demanding sensory, cognitive, and motor activities on an intensive basis.34 Therapeutic stimulation of neuroplasticity and neurogenesis might contribute to functional “repair” of the diseased adult brain before damage to whole neuronal networks has ensued.29 An important treatment component is reassuring patients with SCI that they do not have AD or MCI. Treating comorbid anxiety and depression and reversible causes of cognitive complaints is key to successful outcomes.
Table 5
Strategies to improve memory and maintain cognitive vitality
| Strategy | Description |
|---|---|
| Mindfulness | Focus on 1 task at a time rather than trying to multitask. Research shows that cognition is more efficient in this manner |
| Cognitive strategies | Use mnemonics (such as ROY G BIV to remember the colors of the rainbow). Make associations for information, such as when meeting someone new, relate their name to someone else you know well. Use cues such as memory notebooks to prompt information recall. Engage in learning new and challenging cognitive activities, such as a new language, a music instrument, or dance. Consider computer-based brain exercises |
| Rehearsal | Practice information you want to remember, such as repeating the information several times or writing it down |
| Be patient | Getting frustrated when you have memory difficulties makes it more challenging to remember information |
| Exercise (mental and physical) | Engage in mental activities, such as reading and crossword puzzles. Do something that you are interested in, rather than making it a chore. Research has demonstrated that physical exercise also aids memory |
| Diet | What is good for the heart is good for the brain. Fruits, vegetables, food rich in omega-3 fatty acids (eg, fatty fish such as salmon), whole grains, spices (eg, turmeric), and small amounts of tree nuts (eg, walnuts) are recommended as part of a balanced diet |
| Source: References 1,29 | |
CASE CONTINUED: Reassurance and risk reduction
Ms. F’s psychiatrist reassures her that she does not have AD. She receives genetic counseling and decides to forgo genetic testing. Her psychiatrist educates Ms. F about the risks of OTC supplements—especially increased risk of bleeding because she takes aspirin and clopidogrel—and lack of data supporting their use. Ms. F is counseled that a healthy lifestyle, including regular exercise, Mediterranean diet with increased intake of omega-3 fatty acids, learning new things, and being socially active, is the safest way to promote brain health. Over 3 months, Ms. F discontinues all supplements except the vitamins and omega-3, starts exercising, resumes piano lessons that she stopped 10 years ago, and becomes a vegetarian. She continues to have mild SCI but she says she is not bothered by it and feels satisfied that she is doing all she can to promote her brain health.
Related Resources
- Desai AK. Healthy brain aging: evidence based methods to preserve brain function and prevent dementia. Philadelphia, PA: W. B. Saunders; 2010.
- Doidge N. The brain that changes itself. New York, NY: Penguin Books; 2007.
- Vance DE, Roberson AJ, McGuinness TM, et al. How neuroplasticity and cognitive reserve protect cognitive functioning. J Psychosoc Nurs Ment Health Serv. 2010; 48: 1-8.
Brain Training Resources
- Weil A, Small G. The healthy brain kit. Boulder, CO: Sounds True, Inc.; 2007. Audio CDs, brain-training cards and workbooks.
- Posit Science. www.positscience.com.
- Sharp Brains. www.sharpbrains.com.
Drug Brand Names
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Clopidogrel • Plavix
- Levothyroxine • Levoxyl, Synthroid
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
Abhilash K. Desai, MD, discusses emerging research on biomarkers that may help clarify diagnosis.
1. Small GW. What we need to know about age related memory loss. BMJ. 2002;324:1502-1505.
2. Hayes TL, Larimer N, Adami A, et al. Medication adherence in healthy elders. J Aging Health. 2009;21(4):567-580.
3. Reisberg B, Shulman MB, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
4. Jessen F, Wiese B, Bachmann C, et al. Prediction of dementia by subjective memory impairments: effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry. 2010;67:414-422.
5. Waldorff FB, Siersma V, Waldemar G. Association between subjective memory complaints and nursing home placement: a four-year follow-up. Int J Geriatr Psychiatry. 2009;24(6):602-609.
6. Salthouse TA. Selective review of cognitive aging. J Int Neuropsychol Soc. 2010;16:754-760.
7. Anderton B. Ageing of the brain. Mech Ageing Dev. 2002;23:811-817.
8. Salthouse TA. Influence of age on practice effects in longitudinal neurocognitive change. Neuropsychology. 2010;24(5):563-572.
9. Park D, Schwarz N. Cognitive aging: a primer. Philadelphia PA: Taylor and Francis Group; 2000.
10. Reisberg B, Shulman MB. Commentary on “a roadmap for the prevention of dementia II: Leon Thal Symposium 2008.” Subjective cognitive impairment as an antecedent of Alzheimer’s dementia: policy import. Alzheimers Dement. 2009;5:154-156.
11. Reisberg B, Gauthier S. Current evidence for subjective cognitive impairment (SCI) as the pre-mild cognitive impairment (MCI) stage of subsequently manifest Alzheimer’s disease. Int Psychogeriatr. 2008;20(1):1-16.
12. Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180-195.
13. Ramakers IH, Visser PJ, Aalten P, et al. Symptoms of preclinical dementia in general practice up to five years before dementia diagnosis. Dement Geriatr Cogn Disord. 2007;24(4):300-306.
14. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492-498.
15. Taragano FE, Allegri RF, Krupitzki H, et al. Mild behavioral impairment and risk of dementia: a prospective cohort study of 358 patients. J Clin Psychiatry. 2009;70(4):584-592.
16. Jayadev S, Steinbart EJ, Chi YY, et al. Conjugal Alzheimer disease: risk in children when both parents have Alzheimer disease. Arch Neurol. 2008;65(3):373-378.
17. Hajjar I, Yang F, Sorond F, et al. A novel aging phenotype of slow gait, impaired executive function, and depressive symptoms: relationship to blood pressure and other cardiovascular risks. J Gerontol A Biol Sci Med Sci. 2009;64(9):994-1001.
18. Yamamoto N, Yamanaka G, Ishikawa M, et al. Cardio-ankle vascular index as a predictor of cognitive impairment in community-dwelling elderly people: four-year follow-up. Dement Geriatr Cogn Disord. 2009;28(2):153-158.
19. Inzitari M, Pozzi C, Ferrucci L, et al. Subtle neurological abnormalities as risk factors for cognitive and functional decline, cerebrovascular events, and mortality in older community-dwelling adults. Arch Intern Med. 2008;168(12):1270-1276.
20. Shineman DW, Salthouse TA, Launer LJ, et al. Therapeutics of cognitive aging. Ann N Y Acad Sci. 2010;1191(suppl 1):E1-E10.
21. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118-1127.
22. Chertkow H, Massoud F, Nasreddine Z, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178(10):1273-1285.
23. Rosenberg PB, Lyketsos C. Mild cognitive impairment: searching for the prodrome of Alzheimer’s disease. World Psychiatry. 2008;7(2):72-78.
24. Rosenberg PB, Johnston D, Lyketsos CG. A clinical approach to mild cognitive impairment. Am J Psychiatry. 2006;163(11):1884-1890.
25. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
26. Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303-308.
27. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000:135–180.
28. Malhotra R, Desai AK. Healthy brain aging: what has sleep got to do with it? Clin Geriatr Med. 2010;26:45-56.
29. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26:1-16.
30. Weaver Cargin J, Collie A, Masters C, et al. The nature of cognitive complaints in healthy older adults with and without objective memory decline. J Clin Exp Neuropsychol. 2008;30:245-257.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med. 2007;69:47-53.
32. Deal JA, Carlson MC, Xue Q, et al. Anemia and 9-year domain-specific cognitive decline in community-dwelling older women: the Women’s Health and Aging Study II. J Am Geriatr Soc. 2009;57(9):1604-1611.
33. Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression scale: a preliminary report. J Psychiatr Res. 1983;17:37-49.
34. Mahncke HW, Bronstone A, Merzenich MM. Brain plasticity and functional losses in the aged: scientific bases for a novel intervention. Prog Brain Res. 2006;157:81-109.
MS. F, age 66, requests genetic testing because she is concerned about mild memory difficulties, such as forgetting names and where she puts her keys or checkbook, and fears she may be developing Alzheimer’s disease (AD). Her mother and sister were diagnosed with AD in their early 60s. Ms. F has 20 years of education and reports no problems with driving, managing her finances, remembering to take her medications, or maintaining social activities, which her husband confirms.
Detailed questioning about anxiety and depressive symptoms reveals substantial worries about future cognitive decline and some concerns about her finances and her husband’s health. Ms. F says she occasionally feels down and has low energy but denies other depressive symptoms. She reports no sleep disturbances—including snoring and daytime sleepiness, which could indicate obstructive sleep apnea—which her husband confirms. Ms. F takes levothyroxine for hypothyroidism, atenolol for hypertension, aspirin and clopidogrel for coronary artery disease, and atorvastatin for hyperlipidemia. In addition, she provides a long list of over-the-counter (OTC) supplements—ginkgo, huperzine, ginseng, phosphatidylserine, B1, B12, folate, vitamin D, alpha-lipoic acid, and vinpocetine—that she takes to “protect” her brain from AD.
Subjective cognitive impairment (SCI) in older persons is a common condition with a largely unclear prognosis. Many older adults (age ≥65) express concern about mild cognitive problems—“senior moments”—such as word-finding difficulties and forgetfulness.1 Individuals may wonder if walking into a room only to forget why might be the first sign of dementia. Some older adults try to counteract these memory problems by engaging in brain exercises—including costly computer games—and taking OTC “brain-enhancing” vitamins, herbal remedies, and other supplements.
Although some clinicians may view SCI as benign, that is not always true (Table l).2-5 This article discusses the clinical significance of these mild cognitive complaints by examining:
- age-related cognitive decline (ARCD)
- SCI
- how SCI can be differentiated from more serious conditions, such as mild cognitive impairment (MCI) and early stages of AD and other dementias.
We also will discuss assessing and treating cognitive complaints. Although distinctions between SCI and ARCD may be controversial, evidence suggests clinicians need to adopt a more nuanced clinical approach.
Table 1
Why SCI should be taken seriously
| SCI may create emotional distress because patients are aware of decline in their ‘mental sharpness’ |
| SCI patients might consume unnecessary and potentially harmful OTC supplements touted to promote memory |
| Patients might limit their driving and financial management to avoid making mistakes |
| SCI might impair medication adherence2 |
| SCI may be an early sign of dementia3 |
| Patients’ worry about their self-perceived memory loss might predict dementia4 |
| SCI may predict nursing home placement5 |
| Addressing SCI gives health care providers an opportunity to address anxiety or depression that often accompany SCI |
| Evaluation of potential causes of SCI may uncover reversible conditions that can be treated |
| OTC: over-the-counter; SCI: subjective cognitive impairment |
‘Normal’ cognitive decline
ARCD is subtle decline in cognitive abilities, such as episodic memory, attention, and time needed to complete complex activities.6,7 Individuals with ARCD might not have subjective memory complaints or objective cognitive deficits, and their ability to live independently may not be compromised.7 The degree of decline in ARCD may be smaller than previously thought.8 Park9 summarizes 4 main mechanisms thought to underlie age-related declines in cognition:
- reduced speed of processing
- decreased working memory capabilities
- declining inhibitory control (eg, impaired complex attentional capabilities)
- sensory changes (eg, visual and auditory deficits).
ARCD traditionally is thought to result from predictable changes in the brain associated with aging, such as reduced brain volume in the hippocampus and frontal lobes, loss of myelin, loss of synapses, and cytoskeletal changes.7 However, not all older adults experience ARCD. Some remain highly functional in their later years and continue to actively engage in life well into very old age.6,9
Subjective cognitive impairment
One-quarter to one-half of community-dwelling older adults report subjective cognitive complaints, such as forgetfulness and word-finding difficulties.10 Patients with SCI do not show objective evidence of cognitive impairment on neuropsychological tests and their cognitive problems cause no functional decline.10
Preliminary evidence indicates that SCI may be a harbinger of further cognitive decline. Reisberg et al3 found that compared with patients without SCI, patients with SCI were 4.5 times more likely to develop MCI—cognitive difficulties that can be detected by cognitive tests, but do not cause functional decline—or dementia within 7 years.3 Studies also have suggested that SCI may be a pre-MCI stage of subsequent dementia.11-13 AD generally has a long (10 to 12 years) and progressive prodromal phase before dementia onset and is characterized by successive emergence of cognitive deficits, memory complaints, depressive symptoms, and functional impairment.14
In light of this research, we believe patients with SCI and other risk factors for AD, such as a family history of AD, may be at higher risk of further cognitive and functional decline compared with individuals with ARCD and no AD risk factors. Therefore, patients with SCI and other risk factors for AD (Table 2)15-19 may benefit from annual follow-up to determine if cognitive problems have progressed to MCI or AD.
SCI may be a response to subclinical alterations in neurobiology—a phenomenon known as reverse causality.20 Biomarkers, such as cerebrospinal fluid levels of ß-amyloid and phosphorylated tau, and amyloid imaging using positron emission tomography may help identify AD in SCI patients.21 In these patients, SCI is a misnomer because the cognitive impairment is real—not “subjective”—but current tests are not sensitive enough to detect the cognitive decline the patient has recognized. This group of patients should be differentiated from individuals who may perceive typical cognitive aging (ARCD) as pathologic and complain about it. In the future, biomarkers may help differentiate these 2 groups.
Table 2
Factors that increase SCI patients’ risk for dementia
| Family history of Alzheimer’s disease |
| Mild behavioral impairment |
| Slow gait |
| Depression |
| Rapid weight loss |
| Multiple subtle neurologic abnormalities |
| Vascular disease (eg, peripheral vascular disease, coronary artery disease, cerebrovascular disease) |
| SCI: subjective cognitive impairment Source: References 15-19 |
Mild cognitive impairment
MCI is similar to SCI because MCI patients may present with complaints of memory decline and other cognitive difficulties22 but neither condition is associated with significant impairment of daily activities.23 The key difference is that patients with MCI demonstrate impaired performance on objective cognitive tests whereas SCI patients do not.24 In our experience, office-based tests do not reliably differentiate the 2 conditions because many patients with SCI may show mild impairment in tests such as the Mini-Mental State Exam (MMSE)25 but comprehensive neuropsychological testing reveals no objective cognitive deficits. Neuropsychological testing is essential to reliably differentiate SCI from MCI.
The distinction between SCI and MCI is clinically relevant because evidence suggests that MCI patients have a near-term risk of developing dementia, particularly AD.22,23 In a longitudinal study of 76 individuals with MCI, 12% of patients progressed to AD each year compared with 1% to 2% of healthy older adults.26 Patients with MCI are at increased risk of delirium (especially during hospitalization), falls, medication errors, and difficulty managing their finances.24 Older adults with MCI also have increased mortality compared with older adults with normal cognitive functioning.22 Both SCI and MCI should be differentiated from mild dementia. Common dementias in older adults include:
- AD dementia
- Vascular dementia (may occur with or without AD)
- Lewy body dementia
- Frontotemporal dementia
- Parkinson’s disease dementia.
By definition, all dementia types are associated with impaired ability to perform daily activities and cognitive decline.27
Assessing cognitive complaints
Evaluation of older adults’ cognitive complaints should begin with a thorough history to elicit symptoms of anxiety, depression, physical complaints, and any associated functional decline; a physical exam; and a comprehensive mental status examination. This initial evaluation should be followed by routine and specific investigations as indicated (Table 3).22,24,28,29
In a 6-year study of 100 older adults with and without objective evidence of memory decline, both groups showed similar rates of cognitive complaints.30 Also, researchers found no relationship between individuals’ perception of their cognitive functioning and performance on neuropsychological testing. Mood, education level, and apolipoprotein E epsilon 4 genotype status also did not correlate with participants’ subjective cognitive complaints. These findings highlight the need for objective test data to determine whether older adults’ memory complaints reflect pathologic changes in cognition. After a thorough diagnostic workup, some patients complaining of memory decline will have no detectable evidence of cognitive dysfunction or an identifiable cause. However, others may have identifiable causes of memory impairment (Table 4)28,29,31,32—which could be treated—some will have MCI, and others may be in an early stage of dementia.
Table 3
Investigation of older adults with SCI
| Investigation | Rationale |
|---|---|
| Routine | |
| Neuropsychological testing | Delineation of cognitive syndromes (SCI vs MCI vs AD*) |
| Hematology (full blood count) | Screen for anemia |
| Biochemistry (electrolytes, renal function, liver function, thyroid function, B12, and folate) | Screen for treatable causes of cognitive complaints |
| For specific indication suggested by history, physical exam, or neuropsychological testing | |
| Neuroimaging | Generalized and regional imaging (eg, hippocampal atrophy, space occupying lesions) |
| Electroencephalography | Epilepsy/seizures (especially absence and complex partial) |
| Cardiac (eg, echocardiography) | May reveal cardiac arrhythmia or sources of emboli |
| Inflammatory markers (eg, ESR) | Screen for inflammatory processes |
| Treponemal serology | Tertiary syphilis |
| *Alzheimer’s disease and other dementias AD: Alzheimer’s disease; ESR: erythrocyte sedimentation rate; MCI: mild cognitive impairment; SCI: subjective cognitive impairment Source: References 22,24,28,29 | |
Table 4
Differential diagnosis of SCI
| Cause of cognitive impairment | Potential mechanism |
|---|---|
| ARCD | Allostatic load, ‘wear and tear’ from a lifetime of physiological or psychological stresses and adaptations |
| Anemia | Neuronal hypoxia |
| Alzheimer’s disease | Amyloid and/or tau-mediated neurotoxicity, neuroinflammation |
| Cerebrovascular disease | Neuronal ischemia and hypoxia, neuroinflammation |
| Vitamin deficiencies (eg, B1, B12, folate, D) | Impaired neuronal and neurotransmitter function |
| Inadequate protein intake | Impaired neuronal function |
| Anticholinergic drug use | Decreased cholinergic neurotransmission |
| Alcohol use | Direct neurotoxicity and indirect causes such as malnutrition or head injury |
| Depression, anxiety | Hippocampal dysfunction with or without atrophy |
| Obstructive sleep apnea | Neuronal hypoxia, neuroinflammation |
| Head injury | Neuronal and synaptic loss |
| ARCD: age-related cognitive decline; SCI: subjective cognitive impairment Source: References 28,29,31,32 | |
CASE CONTINUED: No measurable deficits
Ms. F’s medical history is remarkable for coronary artery disease, hypothyroidism, hypertension, hyperlipidemia, cataracts, arthritis, back surgery (secondary to spondylosis), and foot surgery. Ms. F denies a history of alcohol or illicit substance abuse. She smoked tobacco for 30 years (2 packs per day), but quit 5 years ago after her heart attack. Physical exam is unremarkable except for mild obesity (body mass index = 31 kg/m2).
Ms. F’s mental status exam reveals anxious mood and affect. Her recall is 2 out of 3 items. Her MMSE score is 29/30 (1 point lost on recall) and her Geriatric Depression Scale33 score is 2/15, indicating minimal depressive symptoms. On neuropsychological testing, Ms. F demonstrates high average intellectual abilities; compared with others her age, she performs within expectations on all measures. That is, she performs within the above-average to low-average range on measures of attention, working memory, speed of processing, expressive language, learning, memory, visual spatial abilities, executive functioning, and knowledge of basic health and safety information.
Enhancing neuroplasticity
We recommend neuroplasticity-based interventions to treat SCI and promote healthy brain aging.20,29 For a checklist clinicians can use to promote healthy brain aging and thus improve patients’ cognitive health see this article at CurrentPsychiatry. com. Table 51,29 lists cognitive strategies to improve memory and maintain cognitive vitality.
Enhancing brain plasticity and neurogenesis requires engaging older adults in demanding sensory, cognitive, and motor activities on an intensive basis.34 Therapeutic stimulation of neuroplasticity and neurogenesis might contribute to functional “repair” of the diseased adult brain before damage to whole neuronal networks has ensued.29 An important treatment component is reassuring patients with SCI that they do not have AD or MCI. Treating comorbid anxiety and depression and reversible causes of cognitive complaints is key to successful outcomes.
Table 5
Strategies to improve memory and maintain cognitive vitality
| Strategy | Description |
|---|---|
| Mindfulness | Focus on 1 task at a time rather than trying to multitask. Research shows that cognition is more efficient in this manner |
| Cognitive strategies | Use mnemonics (such as ROY G BIV to remember the colors of the rainbow). Make associations for information, such as when meeting someone new, relate their name to someone else you know well. Use cues such as memory notebooks to prompt information recall. Engage in learning new and challenging cognitive activities, such as a new language, a music instrument, or dance. Consider computer-based brain exercises |
| Rehearsal | Practice information you want to remember, such as repeating the information several times or writing it down |
| Be patient | Getting frustrated when you have memory difficulties makes it more challenging to remember information |
| Exercise (mental and physical) | Engage in mental activities, such as reading and crossword puzzles. Do something that you are interested in, rather than making it a chore. Research has demonstrated that physical exercise also aids memory |
| Diet | What is good for the heart is good for the brain. Fruits, vegetables, food rich in omega-3 fatty acids (eg, fatty fish such as salmon), whole grains, spices (eg, turmeric), and small amounts of tree nuts (eg, walnuts) are recommended as part of a balanced diet |
| Source: References 1,29 | |
CASE CONTINUED: Reassurance and risk reduction
Ms. F’s psychiatrist reassures her that she does not have AD. She receives genetic counseling and decides to forgo genetic testing. Her psychiatrist educates Ms. F about the risks of OTC supplements—especially increased risk of bleeding because she takes aspirin and clopidogrel—and lack of data supporting their use. Ms. F is counseled that a healthy lifestyle, including regular exercise, Mediterranean diet with increased intake of omega-3 fatty acids, learning new things, and being socially active, is the safest way to promote brain health. Over 3 months, Ms. F discontinues all supplements except the vitamins and omega-3, starts exercising, resumes piano lessons that she stopped 10 years ago, and becomes a vegetarian. She continues to have mild SCI but she says she is not bothered by it and feels satisfied that she is doing all she can to promote her brain health.
Related Resources
- Desai AK. Healthy brain aging: evidence based methods to preserve brain function and prevent dementia. Philadelphia, PA: W. B. Saunders; 2010.
- Doidge N. The brain that changes itself. New York, NY: Penguin Books; 2007.
- Vance DE, Roberson AJ, McGuinness TM, et al. How neuroplasticity and cognitive reserve protect cognitive functioning. J Psychosoc Nurs Ment Health Serv. 2010; 48: 1-8.
Brain Training Resources
- Weil A, Small G. The healthy brain kit. Boulder, CO: Sounds True, Inc.; 2007. Audio CDs, brain-training cards and workbooks.
- Posit Science. www.positscience.com.
- Sharp Brains. www.sharpbrains.com.
Drug Brand Names
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Clopidogrel • Plavix
- Levothyroxine • Levoxyl, Synthroid
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
Abhilash K. Desai, MD, discusses emerging research on biomarkers that may help clarify diagnosis.
MS. F, age 66, requests genetic testing because she is concerned about mild memory difficulties, such as forgetting names and where she puts her keys or checkbook, and fears she may be developing Alzheimer’s disease (AD). Her mother and sister were diagnosed with AD in their early 60s. Ms. F has 20 years of education and reports no problems with driving, managing her finances, remembering to take her medications, or maintaining social activities, which her husband confirms.
Detailed questioning about anxiety and depressive symptoms reveals substantial worries about future cognitive decline and some concerns about her finances and her husband’s health. Ms. F says she occasionally feels down and has low energy but denies other depressive symptoms. She reports no sleep disturbances—including snoring and daytime sleepiness, which could indicate obstructive sleep apnea—which her husband confirms. Ms. F takes levothyroxine for hypothyroidism, atenolol for hypertension, aspirin and clopidogrel for coronary artery disease, and atorvastatin for hyperlipidemia. In addition, she provides a long list of over-the-counter (OTC) supplements—ginkgo, huperzine, ginseng, phosphatidylserine, B1, B12, folate, vitamin D, alpha-lipoic acid, and vinpocetine—that she takes to “protect” her brain from AD.
Subjective cognitive impairment (SCI) in older persons is a common condition with a largely unclear prognosis. Many older adults (age ≥65) express concern about mild cognitive problems—“senior moments”—such as word-finding difficulties and forgetfulness.1 Individuals may wonder if walking into a room only to forget why might be the first sign of dementia. Some older adults try to counteract these memory problems by engaging in brain exercises—including costly computer games—and taking OTC “brain-enhancing” vitamins, herbal remedies, and other supplements.
Although some clinicians may view SCI as benign, that is not always true (Table l).2-5 This article discusses the clinical significance of these mild cognitive complaints by examining:
- age-related cognitive decline (ARCD)
- SCI
- how SCI can be differentiated from more serious conditions, such as mild cognitive impairment (MCI) and early stages of AD and other dementias.
We also will discuss assessing and treating cognitive complaints. Although distinctions between SCI and ARCD may be controversial, evidence suggests clinicians need to adopt a more nuanced clinical approach.
Table 1
Why SCI should be taken seriously
| SCI may create emotional distress because patients are aware of decline in their ‘mental sharpness’ |
| SCI patients might consume unnecessary and potentially harmful OTC supplements touted to promote memory |
| Patients might limit their driving and financial management to avoid making mistakes |
| SCI might impair medication adherence2 |
| SCI may be an early sign of dementia3 |
| Patients’ worry about their self-perceived memory loss might predict dementia4 |
| SCI may predict nursing home placement5 |
| Addressing SCI gives health care providers an opportunity to address anxiety or depression that often accompany SCI |
| Evaluation of potential causes of SCI may uncover reversible conditions that can be treated |
| OTC: over-the-counter; SCI: subjective cognitive impairment |
‘Normal’ cognitive decline
ARCD is subtle decline in cognitive abilities, such as episodic memory, attention, and time needed to complete complex activities.6,7 Individuals with ARCD might not have subjective memory complaints or objective cognitive deficits, and their ability to live independently may not be compromised.7 The degree of decline in ARCD may be smaller than previously thought.8 Park9 summarizes 4 main mechanisms thought to underlie age-related declines in cognition:
- reduced speed of processing
- decreased working memory capabilities
- declining inhibitory control (eg, impaired complex attentional capabilities)
- sensory changes (eg, visual and auditory deficits).
ARCD traditionally is thought to result from predictable changes in the brain associated with aging, such as reduced brain volume in the hippocampus and frontal lobes, loss of myelin, loss of synapses, and cytoskeletal changes.7 However, not all older adults experience ARCD. Some remain highly functional in their later years and continue to actively engage in life well into very old age.6,9
Subjective cognitive impairment
One-quarter to one-half of community-dwelling older adults report subjective cognitive complaints, such as forgetfulness and word-finding difficulties.10 Patients with SCI do not show objective evidence of cognitive impairment on neuropsychological tests and their cognitive problems cause no functional decline.10
Preliminary evidence indicates that SCI may be a harbinger of further cognitive decline. Reisberg et al3 found that compared with patients without SCI, patients with SCI were 4.5 times more likely to develop MCI—cognitive difficulties that can be detected by cognitive tests, but do not cause functional decline—or dementia within 7 years.3 Studies also have suggested that SCI may be a pre-MCI stage of subsequent dementia.11-13 AD generally has a long (10 to 12 years) and progressive prodromal phase before dementia onset and is characterized by successive emergence of cognitive deficits, memory complaints, depressive symptoms, and functional impairment.14
In light of this research, we believe patients with SCI and other risk factors for AD, such as a family history of AD, may be at higher risk of further cognitive and functional decline compared with individuals with ARCD and no AD risk factors. Therefore, patients with SCI and other risk factors for AD (Table 2)15-19 may benefit from annual follow-up to determine if cognitive problems have progressed to MCI or AD.
SCI may be a response to subclinical alterations in neurobiology—a phenomenon known as reverse causality.20 Biomarkers, such as cerebrospinal fluid levels of ß-amyloid and phosphorylated tau, and amyloid imaging using positron emission tomography may help identify AD in SCI patients.21 In these patients, SCI is a misnomer because the cognitive impairment is real—not “subjective”—but current tests are not sensitive enough to detect the cognitive decline the patient has recognized. This group of patients should be differentiated from individuals who may perceive typical cognitive aging (ARCD) as pathologic and complain about it. In the future, biomarkers may help differentiate these 2 groups.
Table 2
Factors that increase SCI patients’ risk for dementia
| Family history of Alzheimer’s disease |
| Mild behavioral impairment |
| Slow gait |
| Depression |
| Rapid weight loss |
| Multiple subtle neurologic abnormalities |
| Vascular disease (eg, peripheral vascular disease, coronary artery disease, cerebrovascular disease) |
| SCI: subjective cognitive impairment Source: References 15-19 |
Mild cognitive impairment
MCI is similar to SCI because MCI patients may present with complaints of memory decline and other cognitive difficulties22 but neither condition is associated with significant impairment of daily activities.23 The key difference is that patients with MCI demonstrate impaired performance on objective cognitive tests whereas SCI patients do not.24 In our experience, office-based tests do not reliably differentiate the 2 conditions because many patients with SCI may show mild impairment in tests such as the Mini-Mental State Exam (MMSE)25 but comprehensive neuropsychological testing reveals no objective cognitive deficits. Neuropsychological testing is essential to reliably differentiate SCI from MCI.
The distinction between SCI and MCI is clinically relevant because evidence suggests that MCI patients have a near-term risk of developing dementia, particularly AD.22,23 In a longitudinal study of 76 individuals with MCI, 12% of patients progressed to AD each year compared with 1% to 2% of healthy older adults.26 Patients with MCI are at increased risk of delirium (especially during hospitalization), falls, medication errors, and difficulty managing their finances.24 Older adults with MCI also have increased mortality compared with older adults with normal cognitive functioning.22 Both SCI and MCI should be differentiated from mild dementia. Common dementias in older adults include:
- AD dementia
- Vascular dementia (may occur with or without AD)
- Lewy body dementia
- Frontotemporal dementia
- Parkinson’s disease dementia.
By definition, all dementia types are associated with impaired ability to perform daily activities and cognitive decline.27
Assessing cognitive complaints
Evaluation of older adults’ cognitive complaints should begin with a thorough history to elicit symptoms of anxiety, depression, physical complaints, and any associated functional decline; a physical exam; and a comprehensive mental status examination. This initial evaluation should be followed by routine and specific investigations as indicated (Table 3).22,24,28,29
In a 6-year study of 100 older adults with and without objective evidence of memory decline, both groups showed similar rates of cognitive complaints.30 Also, researchers found no relationship between individuals’ perception of their cognitive functioning and performance on neuropsychological testing. Mood, education level, and apolipoprotein E epsilon 4 genotype status also did not correlate with participants’ subjective cognitive complaints. These findings highlight the need for objective test data to determine whether older adults’ memory complaints reflect pathologic changes in cognition. After a thorough diagnostic workup, some patients complaining of memory decline will have no detectable evidence of cognitive dysfunction or an identifiable cause. However, others may have identifiable causes of memory impairment (Table 4)28,29,31,32—which could be treated—some will have MCI, and others may be in an early stage of dementia.
Table 3
Investigation of older adults with SCI
| Investigation | Rationale |
|---|---|
| Routine | |
| Neuropsychological testing | Delineation of cognitive syndromes (SCI vs MCI vs AD*) |
| Hematology (full blood count) | Screen for anemia |
| Biochemistry (electrolytes, renal function, liver function, thyroid function, B12, and folate) | Screen for treatable causes of cognitive complaints |
| For specific indication suggested by history, physical exam, or neuropsychological testing | |
| Neuroimaging | Generalized and regional imaging (eg, hippocampal atrophy, space occupying lesions) |
| Electroencephalography | Epilepsy/seizures (especially absence and complex partial) |
| Cardiac (eg, echocardiography) | May reveal cardiac arrhythmia or sources of emboli |
| Inflammatory markers (eg, ESR) | Screen for inflammatory processes |
| Treponemal serology | Tertiary syphilis |
| *Alzheimer’s disease and other dementias AD: Alzheimer’s disease; ESR: erythrocyte sedimentation rate; MCI: mild cognitive impairment; SCI: subjective cognitive impairment Source: References 22,24,28,29 | |
Table 4
Differential diagnosis of SCI
| Cause of cognitive impairment | Potential mechanism |
|---|---|
| ARCD | Allostatic load, ‘wear and tear’ from a lifetime of physiological or psychological stresses and adaptations |
| Anemia | Neuronal hypoxia |
| Alzheimer’s disease | Amyloid and/or tau-mediated neurotoxicity, neuroinflammation |
| Cerebrovascular disease | Neuronal ischemia and hypoxia, neuroinflammation |
| Vitamin deficiencies (eg, B1, B12, folate, D) | Impaired neuronal and neurotransmitter function |
| Inadequate protein intake | Impaired neuronal function |
| Anticholinergic drug use | Decreased cholinergic neurotransmission |
| Alcohol use | Direct neurotoxicity and indirect causes such as malnutrition or head injury |
| Depression, anxiety | Hippocampal dysfunction with or without atrophy |
| Obstructive sleep apnea | Neuronal hypoxia, neuroinflammation |
| Head injury | Neuronal and synaptic loss |
| ARCD: age-related cognitive decline; SCI: subjective cognitive impairment Source: References 28,29,31,32 | |
CASE CONTINUED: No measurable deficits
Ms. F’s medical history is remarkable for coronary artery disease, hypothyroidism, hypertension, hyperlipidemia, cataracts, arthritis, back surgery (secondary to spondylosis), and foot surgery. Ms. F denies a history of alcohol or illicit substance abuse. She smoked tobacco for 30 years (2 packs per day), but quit 5 years ago after her heart attack. Physical exam is unremarkable except for mild obesity (body mass index = 31 kg/m2).
Ms. F’s mental status exam reveals anxious mood and affect. Her recall is 2 out of 3 items. Her MMSE score is 29/30 (1 point lost on recall) and her Geriatric Depression Scale33 score is 2/15, indicating minimal depressive symptoms. On neuropsychological testing, Ms. F demonstrates high average intellectual abilities; compared with others her age, she performs within expectations on all measures. That is, she performs within the above-average to low-average range on measures of attention, working memory, speed of processing, expressive language, learning, memory, visual spatial abilities, executive functioning, and knowledge of basic health and safety information.
Enhancing neuroplasticity
We recommend neuroplasticity-based interventions to treat SCI and promote healthy brain aging.20,29 For a checklist clinicians can use to promote healthy brain aging and thus improve patients’ cognitive health see this article at CurrentPsychiatry. com. Table 51,29 lists cognitive strategies to improve memory and maintain cognitive vitality.
Enhancing brain plasticity and neurogenesis requires engaging older adults in demanding sensory, cognitive, and motor activities on an intensive basis.34 Therapeutic stimulation of neuroplasticity and neurogenesis might contribute to functional “repair” of the diseased adult brain before damage to whole neuronal networks has ensued.29 An important treatment component is reassuring patients with SCI that they do not have AD or MCI. Treating comorbid anxiety and depression and reversible causes of cognitive complaints is key to successful outcomes.
Table 5
Strategies to improve memory and maintain cognitive vitality
| Strategy | Description |
|---|---|
| Mindfulness | Focus on 1 task at a time rather than trying to multitask. Research shows that cognition is more efficient in this manner |
| Cognitive strategies | Use mnemonics (such as ROY G BIV to remember the colors of the rainbow). Make associations for information, such as when meeting someone new, relate their name to someone else you know well. Use cues such as memory notebooks to prompt information recall. Engage in learning new and challenging cognitive activities, such as a new language, a music instrument, or dance. Consider computer-based brain exercises |
| Rehearsal | Practice information you want to remember, such as repeating the information several times or writing it down |
| Be patient | Getting frustrated when you have memory difficulties makes it more challenging to remember information |
| Exercise (mental and physical) | Engage in mental activities, such as reading and crossword puzzles. Do something that you are interested in, rather than making it a chore. Research has demonstrated that physical exercise also aids memory |
| Diet | What is good for the heart is good for the brain. Fruits, vegetables, food rich in omega-3 fatty acids (eg, fatty fish such as salmon), whole grains, spices (eg, turmeric), and small amounts of tree nuts (eg, walnuts) are recommended as part of a balanced diet |
| Source: References 1,29 | |
CASE CONTINUED: Reassurance and risk reduction
Ms. F’s psychiatrist reassures her that she does not have AD. She receives genetic counseling and decides to forgo genetic testing. Her psychiatrist educates Ms. F about the risks of OTC supplements—especially increased risk of bleeding because she takes aspirin and clopidogrel—and lack of data supporting their use. Ms. F is counseled that a healthy lifestyle, including regular exercise, Mediterranean diet with increased intake of omega-3 fatty acids, learning new things, and being socially active, is the safest way to promote brain health. Over 3 months, Ms. F discontinues all supplements except the vitamins and omega-3, starts exercising, resumes piano lessons that she stopped 10 years ago, and becomes a vegetarian. She continues to have mild SCI but she says she is not bothered by it and feels satisfied that she is doing all she can to promote her brain health.
Related Resources
- Desai AK. Healthy brain aging: evidence based methods to preserve brain function and prevent dementia. Philadelphia, PA: W. B. Saunders; 2010.
- Doidge N. The brain that changes itself. New York, NY: Penguin Books; 2007.
- Vance DE, Roberson AJ, McGuinness TM, et al. How neuroplasticity and cognitive reserve protect cognitive functioning. J Psychosoc Nurs Ment Health Serv. 2010; 48: 1-8.
Brain Training Resources
- Weil A, Small G. The healthy brain kit. Boulder, CO: Sounds True, Inc.; 2007. Audio CDs, brain-training cards and workbooks.
- Posit Science. www.positscience.com.
- Sharp Brains. www.sharpbrains.com.
Drug Brand Names
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Clopidogrel • Plavix
- Levothyroxine • Levoxyl, Synthroid
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
Abhilash K. Desai, MD, discusses emerging research on biomarkers that may help clarify diagnosis.
1. Small GW. What we need to know about age related memory loss. BMJ. 2002;324:1502-1505.
2. Hayes TL, Larimer N, Adami A, et al. Medication adherence in healthy elders. J Aging Health. 2009;21(4):567-580.
3. Reisberg B, Shulman MB, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
4. Jessen F, Wiese B, Bachmann C, et al. Prediction of dementia by subjective memory impairments: effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry. 2010;67:414-422.
5. Waldorff FB, Siersma V, Waldemar G. Association between subjective memory complaints and nursing home placement: a four-year follow-up. Int J Geriatr Psychiatry. 2009;24(6):602-609.
6. Salthouse TA. Selective review of cognitive aging. J Int Neuropsychol Soc. 2010;16:754-760.
7. Anderton B. Ageing of the brain. Mech Ageing Dev. 2002;23:811-817.
8. Salthouse TA. Influence of age on practice effects in longitudinal neurocognitive change. Neuropsychology. 2010;24(5):563-572.
9. Park D, Schwarz N. Cognitive aging: a primer. Philadelphia PA: Taylor and Francis Group; 2000.
10. Reisberg B, Shulman MB. Commentary on “a roadmap for the prevention of dementia II: Leon Thal Symposium 2008.” Subjective cognitive impairment as an antecedent of Alzheimer’s dementia: policy import. Alzheimers Dement. 2009;5:154-156.
11. Reisberg B, Gauthier S. Current evidence for subjective cognitive impairment (SCI) as the pre-mild cognitive impairment (MCI) stage of subsequently manifest Alzheimer’s disease. Int Psychogeriatr. 2008;20(1):1-16.
12. Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180-195.
13. Ramakers IH, Visser PJ, Aalten P, et al. Symptoms of preclinical dementia in general practice up to five years before dementia diagnosis. Dement Geriatr Cogn Disord. 2007;24(4):300-306.
14. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492-498.
15. Taragano FE, Allegri RF, Krupitzki H, et al. Mild behavioral impairment and risk of dementia: a prospective cohort study of 358 patients. J Clin Psychiatry. 2009;70(4):584-592.
16. Jayadev S, Steinbart EJ, Chi YY, et al. Conjugal Alzheimer disease: risk in children when both parents have Alzheimer disease. Arch Neurol. 2008;65(3):373-378.
17. Hajjar I, Yang F, Sorond F, et al. A novel aging phenotype of slow gait, impaired executive function, and depressive symptoms: relationship to blood pressure and other cardiovascular risks. J Gerontol A Biol Sci Med Sci. 2009;64(9):994-1001.
18. Yamamoto N, Yamanaka G, Ishikawa M, et al. Cardio-ankle vascular index as a predictor of cognitive impairment in community-dwelling elderly people: four-year follow-up. Dement Geriatr Cogn Disord. 2009;28(2):153-158.
19. Inzitari M, Pozzi C, Ferrucci L, et al. Subtle neurological abnormalities as risk factors for cognitive and functional decline, cerebrovascular events, and mortality in older community-dwelling adults. Arch Intern Med. 2008;168(12):1270-1276.
20. Shineman DW, Salthouse TA, Launer LJ, et al. Therapeutics of cognitive aging. Ann N Y Acad Sci. 2010;1191(suppl 1):E1-E10.
21. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118-1127.
22. Chertkow H, Massoud F, Nasreddine Z, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178(10):1273-1285.
23. Rosenberg PB, Lyketsos C. Mild cognitive impairment: searching for the prodrome of Alzheimer’s disease. World Psychiatry. 2008;7(2):72-78.
24. Rosenberg PB, Johnston D, Lyketsos CG. A clinical approach to mild cognitive impairment. Am J Psychiatry. 2006;163(11):1884-1890.
25. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
26. Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303-308.
27. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000:135–180.
28. Malhotra R, Desai AK. Healthy brain aging: what has sleep got to do with it? Clin Geriatr Med. 2010;26:45-56.
29. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26:1-16.
30. Weaver Cargin J, Collie A, Masters C, et al. The nature of cognitive complaints in healthy older adults with and without objective memory decline. J Clin Exp Neuropsychol. 2008;30:245-257.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med. 2007;69:47-53.
32. Deal JA, Carlson MC, Xue Q, et al. Anemia and 9-year domain-specific cognitive decline in community-dwelling older women: the Women’s Health and Aging Study II. J Am Geriatr Soc. 2009;57(9):1604-1611.
33. Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression scale: a preliminary report. J Psychiatr Res. 1983;17:37-49.
34. Mahncke HW, Bronstone A, Merzenich MM. Brain plasticity and functional losses in the aged: scientific bases for a novel intervention. Prog Brain Res. 2006;157:81-109.
1. Small GW. What we need to know about age related memory loss. BMJ. 2002;324:1502-1505.
2. Hayes TL, Larimer N, Adami A, et al. Medication adherence in healthy elders. J Aging Health. 2009;21(4):567-580.
3. Reisberg B, Shulman MB, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
4. Jessen F, Wiese B, Bachmann C, et al. Prediction of dementia by subjective memory impairments: effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry. 2010;67:414-422.
5. Waldorff FB, Siersma V, Waldemar G. Association between subjective memory complaints and nursing home placement: a four-year follow-up. Int J Geriatr Psychiatry. 2009;24(6):602-609.
6. Salthouse TA. Selective review of cognitive aging. J Int Neuropsychol Soc. 2010;16:754-760.
7. Anderton B. Ageing of the brain. Mech Ageing Dev. 2002;23:811-817.
8. Salthouse TA. Influence of age on practice effects in longitudinal neurocognitive change. Neuropsychology. 2010;24(5):563-572.
9. Park D, Schwarz N. Cognitive aging: a primer. Philadelphia PA: Taylor and Francis Group; 2000.
10. Reisberg B, Shulman MB. Commentary on “a roadmap for the prevention of dementia II: Leon Thal Symposium 2008.” Subjective cognitive impairment as an antecedent of Alzheimer’s dementia: policy import. Alzheimers Dement. 2009;5:154-156.
11. Reisberg B, Gauthier S. Current evidence for subjective cognitive impairment (SCI) as the pre-mild cognitive impairment (MCI) stage of subsequently manifest Alzheimer’s disease. Int Psychogeriatr. 2008;20(1):1-16.
12. Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180-195.
13. Ramakers IH, Visser PJ, Aalten P, et al. Symptoms of preclinical dementia in general practice up to five years before dementia diagnosis. Dement Geriatr Cogn Disord. 2007;24(4):300-306.
14. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492-498.
15. Taragano FE, Allegri RF, Krupitzki H, et al. Mild behavioral impairment and risk of dementia: a prospective cohort study of 358 patients. J Clin Psychiatry. 2009;70(4):584-592.
16. Jayadev S, Steinbart EJ, Chi YY, et al. Conjugal Alzheimer disease: risk in children when both parents have Alzheimer disease. Arch Neurol. 2008;65(3):373-378.
17. Hajjar I, Yang F, Sorond F, et al. A novel aging phenotype of slow gait, impaired executive function, and depressive symptoms: relationship to blood pressure and other cardiovascular risks. J Gerontol A Biol Sci Med Sci. 2009;64(9):994-1001.
18. Yamamoto N, Yamanaka G, Ishikawa M, et al. Cardio-ankle vascular index as a predictor of cognitive impairment in community-dwelling elderly people: four-year follow-up. Dement Geriatr Cogn Disord. 2009;28(2):153-158.
19. Inzitari M, Pozzi C, Ferrucci L, et al. Subtle neurological abnormalities as risk factors for cognitive and functional decline, cerebrovascular events, and mortality in older community-dwelling adults. Arch Intern Med. 2008;168(12):1270-1276.
20. Shineman DW, Salthouse TA, Launer LJ, et al. Therapeutics of cognitive aging. Ann N Y Acad Sci. 2010;1191(suppl 1):E1-E10.
21. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118-1127.
22. Chertkow H, Massoud F, Nasreddine Z, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178(10):1273-1285.
23. Rosenberg PB, Lyketsos C. Mild cognitive impairment: searching for the prodrome of Alzheimer’s disease. World Psychiatry. 2008;7(2):72-78.
24. Rosenberg PB, Johnston D, Lyketsos CG. A clinical approach to mild cognitive impairment. Am J Psychiatry. 2006;163(11):1884-1890.
25. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
26. Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303-308.
27. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000:135–180.
28. Malhotra R, Desai AK. Healthy brain aging: what has sleep got to do with it? Clin Geriatr Med. 2010;26:45-56.
29. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26:1-16.
30. Weaver Cargin J, Collie A, Masters C, et al. The nature of cognitive complaints in healthy older adults with and without objective memory decline. J Clin Exp Neuropsychol. 2008;30:245-257.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med. 2007;69:47-53.
32. Deal JA, Carlson MC, Xue Q, et al. Anemia and 9-year domain-specific cognitive decline in community-dwelling older women: the Women’s Health and Aging Study II. J Am Geriatr Soc. 2009;57(9):1604-1611.
33. Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression scale: a preliminary report. J Psychiatr Res. 1983;17:37-49.
34. Mahncke HW, Bronstone A, Merzenich MM. Brain plasticity and functional losses in the aged: scientific bases for a novel intervention. Prog Brain Res. 2006;157:81-109.
Managing ADHD in children: Are you doing enough?
• Side effects of psychostimulants can often be managed with monitoring, dose adjustment, a switch to another drug, or adjunctive therapy. A
• Weigh and measure a child being treated for ADHD twice a year; aberrant growth may indicate a need for a change in medication regimen. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Untreated attention deficit hyperactivity disorder (ADHD) can have serious academic, social, and psychological consequences, both for young patients and their parents. Diagnosis is based on criteria detailed in the Diagnostic and Statistical Manual of Mental Health Disorders, Fourth Edition Text Revision (DSM-IV-TR), with observations of the child’s behavior obtained from more than one setting.
Physicians should also consider the possibility of coexisting conditions, which could complicate diagnosis and subsequent attempts to treat the signs and symptoms of ADHD. Treatment is multifaceted, and will vary depending on severity, comorbidities, and the degree of compliance with nonpharmacologic modalities.
A comprehensive approach is called for
Managing pediatric ADHD in a primary care setting requires a comprehensive, goal-oriented treatment plan. The primary goal, as noted in the American Academy of Child and Adolescent Psychiatry (AACAP)’s ADHD guideline,1 is to maximize the child’s functioning, both in terms of an improvement in relationships and academic performance and a reduction of disruptive behavior. Parents and children should be integrated into community supports and school resources, the guideline recommends1 (strength of recommendation [SOR]: A).
Additional recommendations focus on patient (and parental) education, and on medication, monitoring, and follow-up (SOR: A). Physicians should:
Educate parents and patients about common ADHD symptoms and treatment strategies.
Initiate pharmacotherapy. Select an agent that is approved by the US Food and Drug Administration (FDA) for ADHD. These include the psychostimulants dextroamphetamine, D- and DL-methylphenidate, and mixed salts amphetamine; and atomoxetine, a noradrenergic reuptake inhibitor. (Central nervous system stimulants should be avoided in children with cardiac abnormalities, who are at increased risk of experiencing sympathomimetic effects.)
Familiarize themselves with medication side effects. Decreased appetite, insomnia, headache, abdominal pain, and irritable mood are the most common side effects of psychostimulants. Common side effects of atomoxetine include somnolence, anorexia, nausea, skin rash, and a mild increase in blood pressure or heart rate. Notably, there is a small risk of suicide associated with atomoxetine.
Monitor patients for the emergence and severity of side effects. Many of the side effects of stimulants are transient and can be managed through monitoring, as long as it does not compromise the patient’s health or interfere with daily living. Side effects can also be managed with dose adjustment, change of drug treatment, or adjunctive therapy.
Measure height and weight of the patient twice yearly. If a child’s height or weight crosses 2 percentiles on his or her growth curve, it may be an indication of aberrant growth—and a drug holiday or switching to a different medication should be considered.
Evaluate treatment success several times a year. The review should include behavior, academic progress, emergence of comorbid disorders, and the need for behavioral therapy and continuing pharmacotherapy. A lack of response to one psychostimulant is not predictive of the patient’s response to another, the AACAP emphasizes, and it is important to keep trying to find another medication until treatment goals are reached.1
If none of the FDA-approved ADHD medications has the desired results, the AACAP recommends (SOR: B):
- a referral to a cognitive behavioral therapist or child psychologist
- a trial with a medication that is not FDA-approved for ADHD, such as bupropion, a tricyclic antidepressant, or an alpha-agonist
- a reevaluation of the ADHD diagnosis, adherence to the treatment plan, and the presence of comorbid conditions.1
AAP stresses hands-on behavioral intervention
The American Academy of Pediatrics (AAP) also has a clinical practice guideline for the treatment of ADHD, issued in 2001.2 Its recommendations are similar to those of the AACAP. But AAP puts additional emphasis on parental training in behavioral therapy and classroom behavioral interventions, and considers both to be more effective than cognitive behavioral therapy (CBT).2
Virtual reality: A viable option?
Although conventional treatment of childhood ADHD has had considerable clinical success, other forms of treatment may be needed in some cases—if a child’s parents reject psychopharmacologic treatment, for example, or medication trials and traditional behavioral therapies, such as CBT, fail to bring the desired results.
Virtual reality (VR), a computer-generated 3-dimensional interactive system, is an emerging clinical tool. VR programs such as The Virtual Classroom3,4—in which a child is “immersed” in a simulated classroom setting—have shown promise for ADHD assessment and treatment.
Perhaps the biggest benefit of VR as an ADHD intervention is the opportunity for a clinician to place a patient in a virtual classroom, with tasks that require the child’s attention as well as distractors, such as conversation, ambient noise, and moving objects. Another advantage is the ability to integrate traditional assessment tools (Continuous Performance Tasks, for example) and treatment modalities, such as CBT.5 This can be accomplished through a graphic display of a child’s performance during a VR session, which the therapist can use as part of the therapeutic process.3 And VR has no side effects.
Several facilities are either using or experimenting with VR for ADHD. More information is available from the Virtual Reality Medical Center at http://www.vrphobia.com/adhd.htm.
CORRESPONDENCE
Keith B. Holten, MD, Berger Health System, 600 North Pickaway Street, Circleville, OH 43113; [email protected]
1. Pliszka S. AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46:894-921.
2. American Academy of Pediatrics. Subcommittee on Attention-Deficit/Hyperactivity Disorder. Clinical practice guideline: treatment of the school-aged child with attention-deficit/hyperactivity disorder. Pediatrics. 2001;108:1033-1044.
3. Rizzo AA, Buckwalter JG, Humphrey L, et al. The virtual classroom: a virtual environment for the assessment and rehabilitation of attention deficits. CyberPsych Behav. 2000;3:483-499.
4. Rizzo AA, Klimchuk D, Mitura R, et al. A virtual reality scenario for all seasons: the virtual classroom. CNS Spectr. 2006;11:35-44.
5. Pollak Y, Weiss PL, Rizzo AA, et al. The utility of a continuous performance test embedded in virtual reality in measuring ADHD-related deficits. J Dev Behav Pediatr. 2009;30:2-6.
• Side effects of psychostimulants can often be managed with monitoring, dose adjustment, a switch to another drug, or adjunctive therapy. A
• Weigh and measure a child being treated for ADHD twice a year; aberrant growth may indicate a need for a change in medication regimen. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Untreated attention deficit hyperactivity disorder (ADHD) can have serious academic, social, and psychological consequences, both for young patients and their parents. Diagnosis is based on criteria detailed in the Diagnostic and Statistical Manual of Mental Health Disorders, Fourth Edition Text Revision (DSM-IV-TR), with observations of the child’s behavior obtained from more than one setting.
Physicians should also consider the possibility of coexisting conditions, which could complicate diagnosis and subsequent attempts to treat the signs and symptoms of ADHD. Treatment is multifaceted, and will vary depending on severity, comorbidities, and the degree of compliance with nonpharmacologic modalities.
A comprehensive approach is called for
Managing pediatric ADHD in a primary care setting requires a comprehensive, goal-oriented treatment plan. The primary goal, as noted in the American Academy of Child and Adolescent Psychiatry (AACAP)’s ADHD guideline,1 is to maximize the child’s functioning, both in terms of an improvement in relationships and academic performance and a reduction of disruptive behavior. Parents and children should be integrated into community supports and school resources, the guideline recommends1 (strength of recommendation [SOR]: A).
Additional recommendations focus on patient (and parental) education, and on medication, monitoring, and follow-up (SOR: A). Physicians should:
Educate parents and patients about common ADHD symptoms and treatment strategies.
Initiate pharmacotherapy. Select an agent that is approved by the US Food and Drug Administration (FDA) for ADHD. These include the psychostimulants dextroamphetamine, D- and DL-methylphenidate, and mixed salts amphetamine; and atomoxetine, a noradrenergic reuptake inhibitor. (Central nervous system stimulants should be avoided in children with cardiac abnormalities, who are at increased risk of experiencing sympathomimetic effects.)
Familiarize themselves with medication side effects. Decreased appetite, insomnia, headache, abdominal pain, and irritable mood are the most common side effects of psychostimulants. Common side effects of atomoxetine include somnolence, anorexia, nausea, skin rash, and a mild increase in blood pressure or heart rate. Notably, there is a small risk of suicide associated with atomoxetine.
Monitor patients for the emergence and severity of side effects. Many of the side effects of stimulants are transient and can be managed through monitoring, as long as it does not compromise the patient’s health or interfere with daily living. Side effects can also be managed with dose adjustment, change of drug treatment, or adjunctive therapy.
Measure height and weight of the patient twice yearly. If a child’s height or weight crosses 2 percentiles on his or her growth curve, it may be an indication of aberrant growth—and a drug holiday or switching to a different medication should be considered.
Evaluate treatment success several times a year. The review should include behavior, academic progress, emergence of comorbid disorders, and the need for behavioral therapy and continuing pharmacotherapy. A lack of response to one psychostimulant is not predictive of the patient’s response to another, the AACAP emphasizes, and it is important to keep trying to find another medication until treatment goals are reached.1
If none of the FDA-approved ADHD medications has the desired results, the AACAP recommends (SOR: B):
- a referral to a cognitive behavioral therapist or child psychologist
- a trial with a medication that is not FDA-approved for ADHD, such as bupropion, a tricyclic antidepressant, or an alpha-agonist
- a reevaluation of the ADHD diagnosis, adherence to the treatment plan, and the presence of comorbid conditions.1
AAP stresses hands-on behavioral intervention
The American Academy of Pediatrics (AAP) also has a clinical practice guideline for the treatment of ADHD, issued in 2001.2 Its recommendations are similar to those of the AACAP. But AAP puts additional emphasis on parental training in behavioral therapy and classroom behavioral interventions, and considers both to be more effective than cognitive behavioral therapy (CBT).2
Virtual reality: A viable option?
Although conventional treatment of childhood ADHD has had considerable clinical success, other forms of treatment may be needed in some cases—if a child’s parents reject psychopharmacologic treatment, for example, or medication trials and traditional behavioral therapies, such as CBT, fail to bring the desired results.
Virtual reality (VR), a computer-generated 3-dimensional interactive system, is an emerging clinical tool. VR programs such as The Virtual Classroom3,4—in which a child is “immersed” in a simulated classroom setting—have shown promise for ADHD assessment and treatment.
Perhaps the biggest benefit of VR as an ADHD intervention is the opportunity for a clinician to place a patient in a virtual classroom, with tasks that require the child’s attention as well as distractors, such as conversation, ambient noise, and moving objects. Another advantage is the ability to integrate traditional assessment tools (Continuous Performance Tasks, for example) and treatment modalities, such as CBT.5 This can be accomplished through a graphic display of a child’s performance during a VR session, which the therapist can use as part of the therapeutic process.3 And VR has no side effects.
Several facilities are either using or experimenting with VR for ADHD. More information is available from the Virtual Reality Medical Center at http://www.vrphobia.com/adhd.htm.
CORRESPONDENCE
Keith B. Holten, MD, Berger Health System, 600 North Pickaway Street, Circleville, OH 43113; [email protected]
• Side effects of psychostimulants can often be managed with monitoring, dose adjustment, a switch to another drug, or adjunctive therapy. A
• Weigh and measure a child being treated for ADHD twice a year; aberrant growth may indicate a need for a change in medication regimen. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Untreated attention deficit hyperactivity disorder (ADHD) can have serious academic, social, and psychological consequences, both for young patients and their parents. Diagnosis is based on criteria detailed in the Diagnostic and Statistical Manual of Mental Health Disorders, Fourth Edition Text Revision (DSM-IV-TR), with observations of the child’s behavior obtained from more than one setting.
Physicians should also consider the possibility of coexisting conditions, which could complicate diagnosis and subsequent attempts to treat the signs and symptoms of ADHD. Treatment is multifaceted, and will vary depending on severity, comorbidities, and the degree of compliance with nonpharmacologic modalities.
A comprehensive approach is called for
Managing pediatric ADHD in a primary care setting requires a comprehensive, goal-oriented treatment plan. The primary goal, as noted in the American Academy of Child and Adolescent Psychiatry (AACAP)’s ADHD guideline,1 is to maximize the child’s functioning, both in terms of an improvement in relationships and academic performance and a reduction of disruptive behavior. Parents and children should be integrated into community supports and school resources, the guideline recommends1 (strength of recommendation [SOR]: A).
Additional recommendations focus on patient (and parental) education, and on medication, monitoring, and follow-up (SOR: A). Physicians should:
Educate parents and patients about common ADHD symptoms and treatment strategies.
Initiate pharmacotherapy. Select an agent that is approved by the US Food and Drug Administration (FDA) for ADHD. These include the psychostimulants dextroamphetamine, D- and DL-methylphenidate, and mixed salts amphetamine; and atomoxetine, a noradrenergic reuptake inhibitor. (Central nervous system stimulants should be avoided in children with cardiac abnormalities, who are at increased risk of experiencing sympathomimetic effects.)
Familiarize themselves with medication side effects. Decreased appetite, insomnia, headache, abdominal pain, and irritable mood are the most common side effects of psychostimulants. Common side effects of atomoxetine include somnolence, anorexia, nausea, skin rash, and a mild increase in blood pressure or heart rate. Notably, there is a small risk of suicide associated with atomoxetine.
Monitor patients for the emergence and severity of side effects. Many of the side effects of stimulants are transient and can be managed through monitoring, as long as it does not compromise the patient’s health or interfere with daily living. Side effects can also be managed with dose adjustment, change of drug treatment, or adjunctive therapy.
Measure height and weight of the patient twice yearly. If a child’s height or weight crosses 2 percentiles on his or her growth curve, it may be an indication of aberrant growth—and a drug holiday or switching to a different medication should be considered.
Evaluate treatment success several times a year. The review should include behavior, academic progress, emergence of comorbid disorders, and the need for behavioral therapy and continuing pharmacotherapy. A lack of response to one psychostimulant is not predictive of the patient’s response to another, the AACAP emphasizes, and it is important to keep trying to find another medication until treatment goals are reached.1
If none of the FDA-approved ADHD medications has the desired results, the AACAP recommends (SOR: B):
- a referral to a cognitive behavioral therapist or child psychologist
- a trial with a medication that is not FDA-approved for ADHD, such as bupropion, a tricyclic antidepressant, or an alpha-agonist
- a reevaluation of the ADHD diagnosis, adherence to the treatment plan, and the presence of comorbid conditions.1
AAP stresses hands-on behavioral intervention
The American Academy of Pediatrics (AAP) also has a clinical practice guideline for the treatment of ADHD, issued in 2001.2 Its recommendations are similar to those of the AACAP. But AAP puts additional emphasis on parental training in behavioral therapy and classroom behavioral interventions, and considers both to be more effective than cognitive behavioral therapy (CBT).2
Virtual reality: A viable option?
Although conventional treatment of childhood ADHD has had considerable clinical success, other forms of treatment may be needed in some cases—if a child’s parents reject psychopharmacologic treatment, for example, or medication trials and traditional behavioral therapies, such as CBT, fail to bring the desired results.
Virtual reality (VR), a computer-generated 3-dimensional interactive system, is an emerging clinical tool. VR programs such as The Virtual Classroom3,4—in which a child is “immersed” in a simulated classroom setting—have shown promise for ADHD assessment and treatment.
Perhaps the biggest benefit of VR as an ADHD intervention is the opportunity for a clinician to place a patient in a virtual classroom, with tasks that require the child’s attention as well as distractors, such as conversation, ambient noise, and moving objects. Another advantage is the ability to integrate traditional assessment tools (Continuous Performance Tasks, for example) and treatment modalities, such as CBT.5 This can be accomplished through a graphic display of a child’s performance during a VR session, which the therapist can use as part of the therapeutic process.3 And VR has no side effects.
Several facilities are either using or experimenting with VR for ADHD. More information is available from the Virtual Reality Medical Center at http://www.vrphobia.com/adhd.htm.
CORRESPONDENCE
Keith B. Holten, MD, Berger Health System, 600 North Pickaway Street, Circleville, OH 43113; [email protected]
1. Pliszka S. AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46:894-921.
2. American Academy of Pediatrics. Subcommittee on Attention-Deficit/Hyperactivity Disorder. Clinical practice guideline: treatment of the school-aged child with attention-deficit/hyperactivity disorder. Pediatrics. 2001;108:1033-1044.
3. Rizzo AA, Buckwalter JG, Humphrey L, et al. The virtual classroom: a virtual environment for the assessment and rehabilitation of attention deficits. CyberPsych Behav. 2000;3:483-499.
4. Rizzo AA, Klimchuk D, Mitura R, et al. A virtual reality scenario for all seasons: the virtual classroom. CNS Spectr. 2006;11:35-44.
5. Pollak Y, Weiss PL, Rizzo AA, et al. The utility of a continuous performance test embedded in virtual reality in measuring ADHD-related deficits. J Dev Behav Pediatr. 2009;30:2-6.
1. Pliszka S. AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46:894-921.
2. American Academy of Pediatrics. Subcommittee on Attention-Deficit/Hyperactivity Disorder. Clinical practice guideline: treatment of the school-aged child with attention-deficit/hyperactivity disorder. Pediatrics. 2001;108:1033-1044.
3. Rizzo AA, Buckwalter JG, Humphrey L, et al. The virtual classroom: a virtual environment for the assessment and rehabilitation of attention deficits. CyberPsych Behav. 2000;3:483-499.
4. Rizzo AA, Klimchuk D, Mitura R, et al. A virtual reality scenario for all seasons: the virtual classroom. CNS Spectr. 2006;11:35-44.
5. Pollak Y, Weiss PL, Rizzo AA, et al. The utility of a continuous performance test embedded in virtual reality in measuring ADHD-related deficits. J Dev Behav Pediatr. 2009;30:2-6.
Irregularly shaped abdominal mass
A 46-year-old man sought care at our clinic for an abdominal mass, fatigue, and shortness of breath. He also indicated that he was feeling depressed.
Four years earlier, he’d had a prolonged hospitalization for severe cor pulmonale, during which he suffered a perforated cecum. He had multiple abdominal surgeries, including a right hemicolectomy. His postoperative course was complicated by multi-system organ failure and several nosocomial infections.
In the wake of his recovery, he developed an anterior midline abdominal mass that slowly enlarged over the following years (FIGURE 1). He sought a surgical consultation, but was deferred because of his high-risk operative profile.
Our examination of the patient revealed an anterior, midline, irregularly shaped mass measuring 14 × 20 in. The nontender mass was hollow to percussion and was not as prominent when the patient was supine.
FIGURE 1
Abdominal mass measuring 14 × 20 in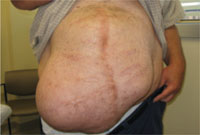
Four years earlier, this 46-year-old patient had undergone multiple abdominal surgeries. On this visit, he sought care for a nontender mass that was hollow to percussion.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Ventral hernia
An abdominal ultrasound revealed subcutaneous, peristalsing bowel loops consistent with a ventral hernia (FIGURE 2). A small amount of ascites was also found.
Most abdominal wall hernias occur in the inguinal region, but in 2003 there were 360,000 ventral hernia repairs performed in the United States.1 Ventral hernias can be further classified as primary or incisional (depending on patient history) and according to their location—midline (epigastric and umbilical) or lateral (Spighelian and lumbar).2
An abdominal hernia typically presents as a nontender, protruding mass that is either stable in size or gradually expands. The mass may be pulsatile, depending on the contents of the hernia and their activity. Hernias may be reducible, meaning that the contents are able to return to the abdominal cavity with external pressure or if the patient is supine. If a hernia is not reducible, then incarceration becomes a significant risk. Compromised blood supply to the incarcerated organ(s) can lead to tissue necrosis and viscous perforation. Epigastric hernias, in particular, carry a high risk of incarceration.3
FIGURE 2
Another view of the ventral hernia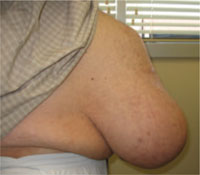
3 conditions comprise the differential
The differential diagnosis includes diastasis recti, ascites, and lipoma.
Diastasis recti is a separation of the rectus abdominus muscles at the linea alba. It is seen almost exclusively in pregnant women and newborns. In this condition, the flat abdominal wall muscles remain intact, and thus abdominal contents would not protrude.
Ascites is the collection of fluid in the abdominal cavity, secondary to conditions such as cirrhosis or congestive heart failure. In ascites, the abdomen is dull to percussion, with no discrete, irregular mass.
Lipoma is a solid benign tumor composed of fatty tissue. A lipoma of this size is rare, and would be solid to percussion. Also, it would not be reducible with the patient supine.
Ultrasound or CT scan is diagnostic
After a thorough history and physical examination, ultrasonography or CT often helps differentiate a ventral hernia from other abdominal wall defects. In patients with a ventral hernia, either imaging modality will demonstrate prolapsed loops of hollow viscus.
A CT scan was not an option for our patient because none of the local machines could accommodate the size and shape of his body. He had an abdominal ultrasound instead.
Surgery sets things right
Treatment of a ventral hernia involves either an open or laparoscopic surgical correction, often with the placement of a supportive mesh4 (SOR: B, inconsistent or limited-quality patient-oriented evidence). Repair of epigastric hernias is crucial even in asymptomatic patients due to the high rate of incarceration.3
Our patient was referred to a hernia specialty clinic at a nationally recognized medical center. He moved out of state shortly thereafter and was lost to follow-up.
CORRESPONDENCE
William Murdoch, MD, WSU/Crittenton Family Medicine Residency, 1135 West University Drive, Suite #250, Rochester Hills, MI 48307; [email protected]
1. Park AE, et al. Abdominal wall hernia. Curr Probl Surg. 2006;43:326-375.
2. Muysoms FE, et al. Classification of primary and incisional abdominal wall hernias. Hernia. 2009;13:407-414.
3. Salameh JR. Primary and unusual abdominal wall hernias. Surg Clin North Am. 2008;88:45-60.
4. Bencini L, et al. Comparison of laparoscopic and open repair for primary ventral hernias. Surg Laparosc Endosc Percutan Tech. 2009;19:341-344.
| 5 common derm mistakes and how to avoid them Richard P. Usatine, MD |
William Murdoch, MD
Pierre A. Morris, MD
Department of Family Medicine and Public Health Sciences, Wayne State University School of Medicine, Detroit, Mich
[email protected]
DEPARTMENT EDITOR
Richard P. Usatine, MD
University of Texas Health Science Center at San Antonio
The authors reported no potential conflict of interest relevant to this article.
| 5 common derm mistakes and how to avoid them Richard P. Usatine, MD |
William Murdoch, MD
Pierre A. Morris, MD
Department of Family Medicine and Public Health Sciences, Wayne State University School of Medicine, Detroit, Mich
[email protected]
DEPARTMENT EDITOR
Richard P. Usatine, MD
University of Texas Health Science Center at San Antonio
The authors reported no potential conflict of interest relevant to this article.
| 5 common derm mistakes and how to avoid them Richard P. Usatine, MD |
William Murdoch, MD
Pierre A. Morris, MD
Department of Family Medicine and Public Health Sciences, Wayne State University School of Medicine, Detroit, Mich
[email protected]
DEPARTMENT EDITOR
Richard P. Usatine, MD
University of Texas Health Science Center at San Antonio
The authors reported no potential conflict of interest relevant to this article.
A 46-year-old man sought care at our clinic for an abdominal mass, fatigue, and shortness of breath. He also indicated that he was feeling depressed.
Four years earlier, he’d had a prolonged hospitalization for severe cor pulmonale, during which he suffered a perforated cecum. He had multiple abdominal surgeries, including a right hemicolectomy. His postoperative course was complicated by multi-system organ failure and several nosocomial infections.
In the wake of his recovery, he developed an anterior midline abdominal mass that slowly enlarged over the following years (FIGURE 1). He sought a surgical consultation, but was deferred because of his high-risk operative profile.
Our examination of the patient revealed an anterior, midline, irregularly shaped mass measuring 14 × 20 in. The nontender mass was hollow to percussion and was not as prominent when the patient was supine.
FIGURE 1
Abdominal mass measuring 14 × 20 in
Four years earlier, this 46-year-old patient had undergone multiple abdominal surgeries. On this visit, he sought care for a nontender mass that was hollow to percussion.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Ventral hernia
An abdominal ultrasound revealed subcutaneous, peristalsing bowel loops consistent with a ventral hernia (FIGURE 2). A small amount of ascites was also found.
Most abdominal wall hernias occur in the inguinal region, but in 2003 there were 360,000 ventral hernia repairs performed in the United States.1 Ventral hernias can be further classified as primary or incisional (depending on patient history) and according to their location—midline (epigastric and umbilical) or lateral (Spighelian and lumbar).2
An abdominal hernia typically presents as a nontender, protruding mass that is either stable in size or gradually expands. The mass may be pulsatile, depending on the contents of the hernia and their activity. Hernias may be reducible, meaning that the contents are able to return to the abdominal cavity with external pressure or if the patient is supine. If a hernia is not reducible, then incarceration becomes a significant risk. Compromised blood supply to the incarcerated organ(s) can lead to tissue necrosis and viscous perforation. Epigastric hernias, in particular, carry a high risk of incarceration.3
FIGURE 2
Another view of the ventral hernia
3 conditions comprise the differential
The differential diagnosis includes diastasis recti, ascites, and lipoma.
Diastasis recti is a separation of the rectus abdominus muscles at the linea alba. It is seen almost exclusively in pregnant women and newborns. In this condition, the flat abdominal wall muscles remain intact, and thus abdominal contents would not protrude.
Ascites is the collection of fluid in the abdominal cavity, secondary to conditions such as cirrhosis or congestive heart failure. In ascites, the abdomen is dull to percussion, with no discrete, irregular mass.
Lipoma is a solid benign tumor composed of fatty tissue. A lipoma of this size is rare, and would be solid to percussion. Also, it would not be reducible with the patient supine.
Ultrasound or CT scan is diagnostic
After a thorough history and physical examination, ultrasonography or CT often helps differentiate a ventral hernia from other abdominal wall defects. In patients with a ventral hernia, either imaging modality will demonstrate prolapsed loops of hollow viscus.
A CT scan was not an option for our patient because none of the local machines could accommodate the size and shape of his body. He had an abdominal ultrasound instead.
Surgery sets things right
Treatment of a ventral hernia involves either an open or laparoscopic surgical correction, often with the placement of a supportive mesh4 (SOR: B, inconsistent or limited-quality patient-oriented evidence). Repair of epigastric hernias is crucial even in asymptomatic patients due to the high rate of incarceration.3
Our patient was referred to a hernia specialty clinic at a nationally recognized medical center. He moved out of state shortly thereafter and was lost to follow-up.
CORRESPONDENCE
William Murdoch, MD, WSU/Crittenton Family Medicine Residency, 1135 West University Drive, Suite #250, Rochester Hills, MI 48307; [email protected]
A 46-year-old man sought care at our clinic for an abdominal mass, fatigue, and shortness of breath. He also indicated that he was feeling depressed.
Four years earlier, he’d had a prolonged hospitalization for severe cor pulmonale, during which he suffered a perforated cecum. He had multiple abdominal surgeries, including a right hemicolectomy. His postoperative course was complicated by multi-system organ failure and several nosocomial infections.
In the wake of his recovery, he developed an anterior midline abdominal mass that slowly enlarged over the following years (FIGURE 1). He sought a surgical consultation, but was deferred because of his high-risk operative profile.
Our examination of the patient revealed an anterior, midline, irregularly shaped mass measuring 14 × 20 in. The nontender mass was hollow to percussion and was not as prominent when the patient was supine.
FIGURE 1
Abdominal mass measuring 14 × 20 in
Four years earlier, this 46-year-old patient had undergone multiple abdominal surgeries. On this visit, he sought care for a nontender mass that was hollow to percussion.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Ventral hernia
An abdominal ultrasound revealed subcutaneous, peristalsing bowel loops consistent with a ventral hernia (FIGURE 2). A small amount of ascites was also found.
Most abdominal wall hernias occur in the inguinal region, but in 2003 there were 360,000 ventral hernia repairs performed in the United States.1 Ventral hernias can be further classified as primary or incisional (depending on patient history) and according to their location—midline (epigastric and umbilical) or lateral (Spighelian and lumbar).2
An abdominal hernia typically presents as a nontender, protruding mass that is either stable in size or gradually expands. The mass may be pulsatile, depending on the contents of the hernia and their activity. Hernias may be reducible, meaning that the contents are able to return to the abdominal cavity with external pressure or if the patient is supine. If a hernia is not reducible, then incarceration becomes a significant risk. Compromised blood supply to the incarcerated organ(s) can lead to tissue necrosis and viscous perforation. Epigastric hernias, in particular, carry a high risk of incarceration.3
FIGURE 2
Another view of the ventral hernia
3 conditions comprise the differential
The differential diagnosis includes diastasis recti, ascites, and lipoma.
Diastasis recti is a separation of the rectus abdominus muscles at the linea alba. It is seen almost exclusively in pregnant women and newborns. In this condition, the flat abdominal wall muscles remain intact, and thus abdominal contents would not protrude.
Ascites is the collection of fluid in the abdominal cavity, secondary to conditions such as cirrhosis or congestive heart failure. In ascites, the abdomen is dull to percussion, with no discrete, irregular mass.
Lipoma is a solid benign tumor composed of fatty tissue. A lipoma of this size is rare, and would be solid to percussion. Also, it would not be reducible with the patient supine.
Ultrasound or CT scan is diagnostic
After a thorough history and physical examination, ultrasonography or CT often helps differentiate a ventral hernia from other abdominal wall defects. In patients with a ventral hernia, either imaging modality will demonstrate prolapsed loops of hollow viscus.
A CT scan was not an option for our patient because none of the local machines could accommodate the size and shape of his body. He had an abdominal ultrasound instead.
Surgery sets things right
Treatment of a ventral hernia involves either an open or laparoscopic surgical correction, often with the placement of a supportive mesh4 (SOR: B, inconsistent or limited-quality patient-oriented evidence). Repair of epigastric hernias is crucial even in asymptomatic patients due to the high rate of incarceration.3
Our patient was referred to a hernia specialty clinic at a nationally recognized medical center. He moved out of state shortly thereafter and was lost to follow-up.
CORRESPONDENCE
William Murdoch, MD, WSU/Crittenton Family Medicine Residency, 1135 West University Drive, Suite #250, Rochester Hills, MI 48307; [email protected]
1. Park AE, et al. Abdominal wall hernia. Curr Probl Surg. 2006;43:326-375.
2. Muysoms FE, et al. Classification of primary and incisional abdominal wall hernias. Hernia. 2009;13:407-414.
3. Salameh JR. Primary and unusual abdominal wall hernias. Surg Clin North Am. 2008;88:45-60.
4. Bencini L, et al. Comparison of laparoscopic and open repair for primary ventral hernias. Surg Laparosc Endosc Percutan Tech. 2009;19:341-344.
1. Park AE, et al. Abdominal wall hernia. Curr Probl Surg. 2006;43:326-375.
2. Muysoms FE, et al. Classification of primary and incisional abdominal wall hernias. Hernia. 2009;13:407-414.
3. Salameh JR. Primary and unusual abdominal wall hernias. Surg Clin North Am. 2008;88:45-60.
4. Bencini L, et al. Comparison of laparoscopic and open repair for primary ventral hernias. Surg Laparosc Endosc Percutan Tech. 2009;19:341-344.
Ureteral calculi: What should you consider before intervening?
THE SIZE OF THE CALCULI, their location, and complicating factors such as infection should all be considered.
Most ureteral calculi smaller than 5 mm pass spontaneously, as do approximately half of calculi between 5 and 10 mm. Calculi larger than 10 mm are unlikely to pass without intervention. Distal calculi are more likely to pass spontaneously than calculi in mid- or proximal ureteral locations; most spontaneous passage occurs within 4 to 6 weeks (strength of recommendation [SOR]: A, prospective cohort studies).
All patients with calculi complicated by such factors as obstruction, infection, renal injury, or a single kidney require surgical consultation (SOR: C, expert opinion).
Medical expulsion therapy with alpha-blockers (usually tamsulosin) and nifedipine improves passage rates, including for some calculi larger than 10 mm (SOR: A, meta-analysis of prospective cohort studies).
Evidence summary
A meta-analysis of 5 prospective cohort studies evaluated the rate of spontaneous passage of ureteral calculi according to size. Calculi smaller than 5 mm passed spontaneously in 68% of patients (5 studies, N=224). Calculi between 5 and 10 mm passed spontaneously in 47% of patients (3 studies, N=104).1
A prospective cohort study evaluated spontaneous passage rates of ureteral calculi by size in 172 patients who were diagnosed by unenhanced helical computed tomography.2 Investigators found spontaneous passage rates of 87% for 1-mm calculi, 76% for 2- to 4-mm calculi, 60% for 5- to 7-mm calculi, 48% for 7- to 9-mm calculi, and 25% for calculi larger than 9 mm.
Spontaneous passage rates differed significantly for calculi 1 to 4 mm in size compared with calculi 5 to 7 mm in size (P<.001) and for calculi 5 to 7 mm in size compared with calculi 8 mm or larger (P<.001). Calculi in either the distal ureter or ureterovesicular junction were more likely to pass that those in the mid- or proximal ureter (75% to 79% vs 48% to 60%; P<.001).
Most smaller calculi pass in 4 to 6 weeks
Another prospective cohort study (N=75) found that most calculi pass spontaneously within 4 to 6 weeks. In 95% of patients, calculi passed within 31 days (2 mm or smaller), 40 days (2-4 mm), or 39 days (4-6 mm).3
Some cases require prompt surgery
The American Urological Association (AUA) expert panel recommends early surgical intervention, regardless of calculus size, under the following circumstances: obstruction with high-grade hydronephrosis, infection, impending renal deterioration, intractable pain, nausea and vomiting, or obstruction in a solitary or transplanted kidney.1
Medical expulsion therapy trumps waiting for distal calculi to pass
A meta-analysis comparing rates of calculus passage found that medical expulsion therapy was more effective than expectant management for patients with distal ureteral calculi. Sixteen RCTs (N=1235) evaluated alpha-antagonists (mostly tamsulosin), and 9 RCTs (N=686) evaluated nifedipine. Treat ment periods for medical expulsion therapy ranged from 30 to 60 days.
Alpha-antagonists increased expulsion rates over expectant management for calculi ranging in size from 3 to 18 mm with a mean diameter greater than 5 mm (relative risk [RR]=1.59; 95% confidence interval [CI], 1.44-1.75; number needed to treat [NNT]=3). The mean time until passage ranged from 2.7 to 14.2 days. Nifedipine also increased expulsion rates for calculi with a mean diameter larger than 5 mm, ranging in size from 3.9 to 12.8 mm (RR=1.50; 95% CI, 1.34-1.68; NNT=4).4
Recommendations
The Joint European Association of Urology/ AUA Nephrolithiasis Guideline Panel recommends observation with periodic evaluation for patients newly diagnosed with ureteral calculi smaller than 10 mm.1 Patients may be offered medical expulsion therapy to facilitate calculus passage. Surveillance should be maintained until calculi pass; intervention should be considered if calculi don’t pass spontaneously within about 30 days.
The Panel states that patients with ureteral calculi larger than 10 mm could be observed (with or without medical expulsion therapy); however, most cases will require surgical intervention.1
Acknowledgements
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Medical Department of the United States Army or the US Army Service at large.
1. European Association of Urology/American Urology Association Nephrolithiasis Guideline Panel. 2007 Guideline for the management of ureteral calculi. Available at: www.auanet.org/content/guidelines-and-quality-care/clinical-guidelines.cfm?sub=uc. Accessed August 16, 2010.
2. Coll DM, Varanelli MJ, Smith RC. Relationship of spontaneous passage of ureteral calculi to calculus size and location as revealed by unenhanced helical CT. Am J Roentgenol. 2002;178:101-103.
3. Miller OF, Kane CJ. Time to calculus passage for observed ureteral calculi: a guide for patient education. J Urol. 1999;162:688-691.
4. Singh A, Alter HJ, Littlepage A. A systematic review of medical therapy to facilitate passage of ureteral calculi. Ann Emerg Med. 2007;50:552-563.
THE SIZE OF THE CALCULI, their location, and complicating factors such as infection should all be considered.
Most ureteral calculi smaller than 5 mm pass spontaneously, as do approximately half of calculi between 5 and 10 mm. Calculi larger than 10 mm are unlikely to pass without intervention. Distal calculi are more likely to pass spontaneously than calculi in mid- or proximal ureteral locations; most spontaneous passage occurs within 4 to 6 weeks (strength of recommendation [SOR]: A, prospective cohort studies).
All patients with calculi complicated by such factors as obstruction, infection, renal injury, or a single kidney require surgical consultation (SOR: C, expert opinion).
Medical expulsion therapy with alpha-blockers (usually tamsulosin) and nifedipine improves passage rates, including for some calculi larger than 10 mm (SOR: A, meta-analysis of prospective cohort studies).
Evidence summary
A meta-analysis of 5 prospective cohort studies evaluated the rate of spontaneous passage of ureteral calculi according to size. Calculi smaller than 5 mm passed spontaneously in 68% of patients (5 studies, N=224). Calculi between 5 and 10 mm passed spontaneously in 47% of patients (3 studies, N=104).1
A prospective cohort study evaluated spontaneous passage rates of ureteral calculi by size in 172 patients who were diagnosed by unenhanced helical computed tomography.2 Investigators found spontaneous passage rates of 87% for 1-mm calculi, 76% for 2- to 4-mm calculi, 60% for 5- to 7-mm calculi, 48% for 7- to 9-mm calculi, and 25% for calculi larger than 9 mm.
Spontaneous passage rates differed significantly for calculi 1 to 4 mm in size compared with calculi 5 to 7 mm in size (P<.001) and for calculi 5 to 7 mm in size compared with calculi 8 mm or larger (P<.001). Calculi in either the distal ureter or ureterovesicular junction were more likely to pass that those in the mid- or proximal ureter (75% to 79% vs 48% to 60%; P<.001).
Most smaller calculi pass in 4 to 6 weeks
Another prospective cohort study (N=75) found that most calculi pass spontaneously within 4 to 6 weeks. In 95% of patients, calculi passed within 31 days (2 mm or smaller), 40 days (2-4 mm), or 39 days (4-6 mm).3
Some cases require prompt surgery
The American Urological Association (AUA) expert panel recommends early surgical intervention, regardless of calculus size, under the following circumstances: obstruction with high-grade hydronephrosis, infection, impending renal deterioration, intractable pain, nausea and vomiting, or obstruction in a solitary or transplanted kidney.1
Medical expulsion therapy trumps waiting for distal calculi to pass
A meta-analysis comparing rates of calculus passage found that medical expulsion therapy was more effective than expectant management for patients with distal ureteral calculi. Sixteen RCTs (N=1235) evaluated alpha-antagonists (mostly tamsulosin), and 9 RCTs (N=686) evaluated nifedipine. Treat ment periods for medical expulsion therapy ranged from 30 to 60 days.
Alpha-antagonists increased expulsion rates over expectant management for calculi ranging in size from 3 to 18 mm with a mean diameter greater than 5 mm (relative risk [RR]=1.59; 95% confidence interval [CI], 1.44-1.75; number needed to treat [NNT]=3). The mean time until passage ranged from 2.7 to 14.2 days. Nifedipine also increased expulsion rates for calculi with a mean diameter larger than 5 mm, ranging in size from 3.9 to 12.8 mm (RR=1.50; 95% CI, 1.34-1.68; NNT=4).4
Recommendations
The Joint European Association of Urology/ AUA Nephrolithiasis Guideline Panel recommends observation with periodic evaluation for patients newly diagnosed with ureteral calculi smaller than 10 mm.1 Patients may be offered medical expulsion therapy to facilitate calculus passage. Surveillance should be maintained until calculi pass; intervention should be considered if calculi don’t pass spontaneously within about 30 days.
The Panel states that patients with ureteral calculi larger than 10 mm could be observed (with or without medical expulsion therapy); however, most cases will require surgical intervention.1
Acknowledgements
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Medical Department of the United States Army or the US Army Service at large.
THE SIZE OF THE CALCULI, their location, and complicating factors such as infection should all be considered.
Most ureteral calculi smaller than 5 mm pass spontaneously, as do approximately half of calculi between 5 and 10 mm. Calculi larger than 10 mm are unlikely to pass without intervention. Distal calculi are more likely to pass spontaneously than calculi in mid- or proximal ureteral locations; most spontaneous passage occurs within 4 to 6 weeks (strength of recommendation [SOR]: A, prospective cohort studies).
All patients with calculi complicated by such factors as obstruction, infection, renal injury, or a single kidney require surgical consultation (SOR: C, expert opinion).
Medical expulsion therapy with alpha-blockers (usually tamsulosin) and nifedipine improves passage rates, including for some calculi larger than 10 mm (SOR: A, meta-analysis of prospective cohort studies).
Evidence summary
A meta-analysis of 5 prospective cohort studies evaluated the rate of spontaneous passage of ureteral calculi according to size. Calculi smaller than 5 mm passed spontaneously in 68% of patients (5 studies, N=224). Calculi between 5 and 10 mm passed spontaneously in 47% of patients (3 studies, N=104).1
A prospective cohort study evaluated spontaneous passage rates of ureteral calculi by size in 172 patients who were diagnosed by unenhanced helical computed tomography.2 Investigators found spontaneous passage rates of 87% for 1-mm calculi, 76% for 2- to 4-mm calculi, 60% for 5- to 7-mm calculi, 48% for 7- to 9-mm calculi, and 25% for calculi larger than 9 mm.
Spontaneous passage rates differed significantly for calculi 1 to 4 mm in size compared with calculi 5 to 7 mm in size (P<.001) and for calculi 5 to 7 mm in size compared with calculi 8 mm or larger (P<.001). Calculi in either the distal ureter or ureterovesicular junction were more likely to pass that those in the mid- or proximal ureter (75% to 79% vs 48% to 60%; P<.001).
Most smaller calculi pass in 4 to 6 weeks
Another prospective cohort study (N=75) found that most calculi pass spontaneously within 4 to 6 weeks. In 95% of patients, calculi passed within 31 days (2 mm or smaller), 40 days (2-4 mm), or 39 days (4-6 mm).3
Some cases require prompt surgery
The American Urological Association (AUA) expert panel recommends early surgical intervention, regardless of calculus size, under the following circumstances: obstruction with high-grade hydronephrosis, infection, impending renal deterioration, intractable pain, nausea and vomiting, or obstruction in a solitary or transplanted kidney.1
Medical expulsion therapy trumps waiting for distal calculi to pass
A meta-analysis comparing rates of calculus passage found that medical expulsion therapy was more effective than expectant management for patients with distal ureteral calculi. Sixteen RCTs (N=1235) evaluated alpha-antagonists (mostly tamsulosin), and 9 RCTs (N=686) evaluated nifedipine. Treat ment periods for medical expulsion therapy ranged from 30 to 60 days.
Alpha-antagonists increased expulsion rates over expectant management for calculi ranging in size from 3 to 18 mm with a mean diameter greater than 5 mm (relative risk [RR]=1.59; 95% confidence interval [CI], 1.44-1.75; number needed to treat [NNT]=3). The mean time until passage ranged from 2.7 to 14.2 days. Nifedipine also increased expulsion rates for calculi with a mean diameter larger than 5 mm, ranging in size from 3.9 to 12.8 mm (RR=1.50; 95% CI, 1.34-1.68; NNT=4).4
Recommendations
The Joint European Association of Urology/ AUA Nephrolithiasis Guideline Panel recommends observation with periodic evaluation for patients newly diagnosed with ureteral calculi smaller than 10 mm.1 Patients may be offered medical expulsion therapy to facilitate calculus passage. Surveillance should be maintained until calculi pass; intervention should be considered if calculi don’t pass spontaneously within about 30 days.
The Panel states that patients with ureteral calculi larger than 10 mm could be observed (with or without medical expulsion therapy); however, most cases will require surgical intervention.1
Acknowledgements
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Medical Department of the United States Army or the US Army Service at large.
1. European Association of Urology/American Urology Association Nephrolithiasis Guideline Panel. 2007 Guideline for the management of ureteral calculi. Available at: www.auanet.org/content/guidelines-and-quality-care/clinical-guidelines.cfm?sub=uc. Accessed August 16, 2010.
2. Coll DM, Varanelli MJ, Smith RC. Relationship of spontaneous passage of ureteral calculi to calculus size and location as revealed by unenhanced helical CT. Am J Roentgenol. 2002;178:101-103.
3. Miller OF, Kane CJ. Time to calculus passage for observed ureteral calculi: a guide for patient education. J Urol. 1999;162:688-691.
4. Singh A, Alter HJ, Littlepage A. A systematic review of medical therapy to facilitate passage of ureteral calculi. Ann Emerg Med. 2007;50:552-563.
1. European Association of Urology/American Urology Association Nephrolithiasis Guideline Panel. 2007 Guideline for the management of ureteral calculi. Available at: www.auanet.org/content/guidelines-and-quality-care/clinical-guidelines.cfm?sub=uc. Accessed August 16, 2010.
2. Coll DM, Varanelli MJ, Smith RC. Relationship of spontaneous passage of ureteral calculi to calculus size and location as revealed by unenhanced helical CT. Am J Roentgenol. 2002;178:101-103.
3. Miller OF, Kane CJ. Time to calculus passage for observed ureteral calculi: a guide for patient education. J Urol. 1999;162:688-691.
4. Singh A, Alter HJ, Littlepage A. A systematic review of medical therapy to facilitate passage of ureteral calculi. Ann Emerg Med. 2007;50:552-563.
Evidence-based answers from the Family Physicians Inquiries Network
Weight-loss talks: What works (and what doesn’t)
Background In primary care encounters, it is unknown whether physician advice on weight-related matters leads to patient weight loss. To examine this issue, we analyzed physician weight loss advice and measured corresponding changes in patients’ dietary intake, physical activity, and weight.
Methods Using audio-recorded primary care encounters between 40 physicians and 461 of their overweight or obese patients, we coded weight-related advice as nonspecific, specific nutritional, specific exercise, or specific weight. Physicians and patients were told the study was about preventive health, not weight. We used mixed models (SAS Proc Mixed), controlled for physician clustering and baseline covariates, to assess changes in diet, exercise, and measured weight, both pre-encounter and 3 months post-encounter.
Results When discussing weight, physicians typically provided a combination of specific weight, nutrition, and physical activity advice to their patients (34%). Combined advice resulted in patients reducing their dietary fat intake (P=.02). However, when physicians provided physical activity advice only, patients were significantly (P=.02) more likely to gain weight (+1.41 kg) compared with those who received no advice.
Conclusion When giving weight-related advice, most physicians provided a combination of lifestyle recommendations. Combining advice may help patients reduce their fat in-take. Physical activity advice alone may not be particularly helpful.
The US Preventive Services Task Force (USPSTF) recommends that physicians screen patients for obesity and offer intensive counseling and behavioral interventions to promote sustained weight loss.1 Evidence suggests that physician counseling, including advice, can help patients to lose weight, increase physical activity, and improve diet.2-9 However, little is known about what specific types of weight loss advice physicians give to patients, and whether some types are more effective than others at influencing behavior change.
We analyzed physician weight loss advice delivered in primary care visits and measured changes in patients’ dietary intake, physical activity, and body weight. We examined both the type of weight loss advice delivered and the impact of type of advice on weight and behavior change.
Methods
This study analyzed audio recordings from Project CHAT – Communicating Health: Analyzing Talk. The project was approved by the Duke University Medical Center Institutional Review Board.
Recruitment Physicians. We obtained consent from 40 primary care physicians in community-based practices and told them the study would examine communication around preventive health topics, not weight specifically.
Patients. We identified potential participants by reviewing scheduled appointments 3 weeks in advance. Eligible participants were at least 18 years of age, English-speaking, overweight or obese (body mass index [BMI] ≥25 kg/m2), cognitively competent, and not pregnant. After we obtained consent, a remotely located research assistant started a digital audio recorder as the patient entered the exam room. Immediately after the encounter, the research assistant administered a post-encounter survey to the patient and recorded the patient’s vital signs (N=461). Three months later, the research assistant met with the participant to record vital signs and administer a survey assessing changes in dietary fat intake and exercise (N=426).
Data coding
We coded advice into 4 broad categories: (1) nutrition advice, (2) physical activity advice, (3) specific weight loss advice, and (4) nonspecific weight loss/weight-related advice. We transcribed each piece of advice verbatim.
Nutrition advice consisted of 9 sub-categories: calorie/portion control, meal timing/planning, commercial diet plans, negative diet plans, increase fruits/vegetables, reduce sugar/carbohydrates, reduce fat/cholesterol, other micronutrient recommendations, and specific food items from multiple categories.
Physical activity advice consisted of 6 subcategories: walking, aerobic exercise, anaerobic exercise, exercise intensity, exercise duration, and exercise for comorbid conditions.
Specific weight loss advice consisted of 3 categories: weight loss behavior, weight loss for comorbid conditions, and referrals.
Nonspecific weight loss advice also consisted of 3 subcategories in which physicians provided no details about the general topics of nutrition, physical activity, or weight loss.
Two independent coders (CBT and MEC) assessed each piece of advice and double coded 20% of conversations for reliability. Cohen’s kappa was used to calculate inter-rater reliability for each code using Landis and Koch’s classification (0.21-0.40=fair agreement; 0.41-0.60=moderate agreement; 0.61-0.80=substantial agreement; 0.81-1.0=near-perfect agreement).10 Three advice categories achieved near perfect agreement: nutrition (kappa= 0.94; 95% confidence interval [CI] 0.82-1.0; 99.2% agreement), physical activity (kappa=0.91; 95% CI, 0.84-0.99; 98.6% agreement), and weight loss (kappa=0.95; 95% CI, 0.82-1.0; 99.7% agreement). The nonspecific weight loss advice category had slightly lower agreement but still achieved near-perfect agreement (kappa=0.82; 95% CI, 0.62-1.0; 99.2% agreement).
After all advice was coded, we placed conversations into 1 of 6 categories: (1) no advice given; (2) nonspecific advice only; (3) nutrition only; (4) physical activity only; (5) weight loss only; or (6) combination of nutrition, physical activity, and/or weight loss.
Measures
Dietary fat and fiber intake. We assessed dietary fat intake at baseline and at 3 months using the 22-item Fat- and Fiber-Related Diet Behavior Questionnaire.11,12 Questions about frequency of food selections included, “When you ate dessert, how often did you eat only fruit?” and “When you ate chicken, how often did you take off the skin?” We averaged responses into a total score wherein 1 reflected higher fiber, lower fat food choices; a score of 4 reflected lower fiber, higher fat choices (α=0.74 at baseline and α=0.77 at 3-month follow-up).
Physical activity. We measured physical activity (baseline, 3 months) using the Framingham Physical Activity Index.13 Participants recalled the average number of hours spent engaged in various daily activities (sleeping, working, leisure) and the level of activity for each (sedentary, slight, moderate, or heavy). The composite score accounts for activity duration and intensity.
Anthropometrics. We measured patient weight (baseline, 3 months) and height (baseline only) using a calibrated scale and portable stadiometer. Patients removed shoes, outerwear, and belongings from their pockets before being weighed.
Analysis
We analyzed data using SAS (SAS Institute, Inc., Cary, NC). We assessed the association between type of advice and weight loss, improvement in dietary fat intake behaviors, and increase in physical activity between baseline and the 3-month follow-up visit. We used PROC MIXED to fit general linear models; we incorporated responses into these models from all participants who provided measurements for at least one time point. This modeling framework yields unbiased estimates when missing data are unrelated to the observed variable.14
Primary predictors: (1) type of advice (none, nonspecific, nutrition, physical activity, weight loss, and combination), (2) time since baseline visit, and (3) time by type of advice interaction. All models included a priori defined patient, physician, and visit-related covariates that were theoretically or empirically related to changes in the outcomes (weight, physical activity, or dietary fat in-take). The 14 patient covariates were sex; age; race; high school education; economic security (enough money to pay monthly bills); over-weight (BMI, 25-29.9 kg/m2) or obese (BMI ≥30 kg/m2); actively trying to lose weight (yes/ no); motivated to lose weight (Likert scale 1-7); comfortable discussing weight (Likert scale 1-5); confident about losing weight (Likert scale 1-5); and patient-reported comorbid conditions of diabetes, hypertension, arthritis, and hyperlipidemia.
The 9 physician covariates were sex; race; years since medical school graduation; specialty (family vs internal medicine); self-efficacy (Likert scale 1-5); barriers for weight counseling (Likert scale 1-5); comfort discussing weight (Likert scale 1-5); insurance reimbursement concerns (Likert scale 1-5); and prior training in behavioral counseling (yes/no). Finally, 2 visit-level covariates were included: minutes spent addressing weight issues and visit type (preventive vs chronic).
Results
Sample characteristics
Of the 40 physicians, 19 were family physicians and 21 were internists. More than half of the physicians were female (60%), and 85% were white. Mean age was 47.2 years and mean BMI was 24.9 kg/m2. Of the 461 patients, 66% were female, 65% were white, 35% were African American, and two-thirds had post-high school education (TABLE 1). Mean patient age was 59.8 years; only 4% of the patients were new to their physicians.
TABLE 1
Patient characteristics (N=461)
| % or mean (SD) | |
|---|---|
| Race | |
| White/Asian/Pacific Islander | 65% |
| African American | 35% |
| Female | 66% |
| Age, y (missing=1)* | 59.8 (13.9) |
| BMI, kg/m2 (missing=1)* | 33.1 (7.1) |
| Education (missing=1)* | |
| Post-high school | 67% |
| Income (missing=37)* | |
| $45,000 or less | 48% |
| High financial burden (missing=13)* | |
| Pay bills with trouble | 14% |
| Diagnosed with: | |
| Diabetes | 31% |
| Hypertension (missing=1)* | 69% |
| Hyperlipidemia (missing=1)* | 56% |
| Arthritis | 47% |
| New patient | 4% |
| BMI, body mass index; SD, standard deviation. * Missing data at baseline. | |
Frequency of advice
Physicians gave some type of weight-related advice in 63% of the encounters. They combined types of advice in 34% of all conversations, provided physical activity advice only in 13%, nutrition advice only in 8%, nonspecific advice in 5%, and weight loss advice only in 3%. Many times when physicians gave advice, it was centered on self (eg, “I need you to do X” or “What will it take for me to get you to do Y?”).
Nutrition advice most commonly pertained to specific food items from multiple categories (27% of conversations). Physicians also advised patients to reduce sugar/carbohydrates, control calories and portions, add other micronutrients, eat more fruits/vegetables, and eat meals more frequently.
Walking was the physical activity topic discussed most frequently, followed by exercise duration, exercise for comorbidities, aerobic activities, exercise intensity, and anaerobic exercise. The most common specific weight loss topic was weight loss behavioral advice, followed by weight loss for comorbid conditions. Physicians rarely provided referrals to weight-loss programs.
Effect of type of advice on fat and fiber diet behavior score
Receipt of nutrition advice only was not associated with reduction in fat intake (P=.43, TABLE 2). However, those who received combined types of advice exhibited a significantly greater reduction of fat intake compared with those who received no advice (Fat- and Fiber-Related Diet Behavior Questionnaire score reduction of 0.15 vs 0.05; P=.02).
TABLE 2
How types of physician advice affected dietary fat intake, physical activity, and weight
| Type of advice | ||||||
|---|---|---|---|---|---|---|
| None | Nutrition only | Physical activity only | Weight loss only | Combined advice | Nonspecific | |
| Dietary fat change in Fat- and Fiber-Related Diet Behavior Questionnaire score differences | ||||||
| At 3 months from baseline (95% CI) | -0.05 (-0.11 to 0.004) | -0.10 (-0.22 to 0.01) | -0.07 (-0.16 to 0.02) | -0.08 (-0.26 to 0.09) | -0.15 (-0.20 to -0.09) | 0.03 (0.11 to 0.18) |
| P value* | .43 | .75 | .73 | .02 | .31 | |
| Physical activity score (change in MET hours) | ||||||
| At 3 months from baseline (95% CI) | 0.48 (-0.17 to 1.11) | 0.83 (-0.51 to 2.14) | 0.69 (-0.33 to 1.69) | -0.72 (-2.66 to 1.21) | 0.24 (-0.40 to 0.86) | -0.07 (-1.74 to 1.59) |
| P value* | .64 | .73 | .25 | .60 | .55 | |
| Weight change (kg) | ||||||
| At 3 months from baseline (95% CI) | -0.18 (-0.39 to 0.75) | -0.18 (-1.38 to 1.02) | 1.41 (0.51 to 2.31) | -0.26 (-1.99 to 1.47) | -0.55 (-1.12 to 0.02) | -0.62 (-2.11 to 0.87) |
| P value* | .59 | .02 | .63 | .08 | .32 | |
| CI, confidence interval; MET, metabolic equivalent tasks. *Test of difference between advice given and no advice given. | ||||||
Effect of type of advice on Framingham Physical Activity score
No type of advice, including physical activity advice, led to a change in Framingham Physical Activity scores at the 3-month visit (overall P=.76; TABLE 2).
Effect of type of advice on weight loss
Patients who received physical activity advice gained significantly more weight than patients who received no advice (1.41 kg gained vs 0.18 kg lost; P=.02). Patients who received combined advice lost more weight than patients who received no advice, but the difference did not reach statistical significance (0.55 kg lost vs 0.18 kg lost; P=.08).
Discussion
Physicians typically took an “all or nothing” approach to weight-related issues, giving no advice (37%) or a combination of nutrition, physical activity, and weight loss advice (34%). It seems when physicians do give advice, most of them follow the USPSTF guidelines by addressing nutrition and physical activity together.15
Providing advice alone did not predict a change in patient behavior. For instance, we found no significant association between dietary fat reduction and having received only nutrition advice. Possible explanations include the following:
- Although physicians advised patients to reduce fat/cholesterol intake in 28% of conversations, they did so mostly in combination with other types of advice. Nutrition-only advice occurred in only 8% of conversations. Thus, there may have been insufficient power to detect the impact of this specific type of advice.
- With nutrition-only advice, the most common recommendation was to reduce carbohydrates/sweets, which should not affect fat intake.
Advising patients solely on physical activity led to unintended weight gain overall. Other data have shown that exercise without dietary changes, though beneficial in many ways, is not substantially effective for weight loss.15 People may eat more when they exercise, either to reward themselves or to satiate increased appetite from increased energy expenditure. Or, if physicians recommend the standard goal of 150 minutes of intensive physical activity per week, normally sedentary patients may see that as unattainable and become too discouraged to try.1,16,17
Combining types of advice seemed to help patients reduce their fat intake. Overall, however, simple, brief advice from a physician may not be enough to promote healthy lifestyle changes.
Also notable was that physicians rarely provided referrals, even though this is a strong recommendation from the National Institutes of Health, the American Diabetes Association, and the USPSTF.1,16,17 It could be that many physicians believe referrals are not covered by insurance. Yet, the low frequency of referrals may suggest an important missing component of weight loss therapy, especially given that physician advice alone seems an inadequate intervention.
Avoid physician-centered appeals. Advice was often given in a physician-centered way. There are 3 possible explanations for such phrasing:
- In the absence of clear evidence about how to deliver weight loss advice, physicians may be formulating advice based on their personal or clinical experiences.
- Physicians either assume or sense that patients lack internal motivation to make lifestyle changes for themselves and instead request that patients make changes for the doctor-patient relationship.
- Physicians might be trying to invoke authority in the hope that patients will respond accordingly.
Whatever the reason, the literature on self-centered physician talk indicates that patients are less satisfied when physicians make the visit more about themselves than about patients.18 A better strategy might be to use Motivational Interviewing19 that supports patient autonomy and attempts to elicit and build on internal motivation.
The take-away message is that behavior change is complex and that knowledge is a necessary but insufficient agent for change. Following the tenets of Social Cognitive Theory,20 physicians might also need to address patient motivation, confidence, outcome expectations, and skills to help promote behavior change.
Strengths and limitations of this study
We recorded conversations rather than relying on physician or patient recall. Additionally, these primary care patients were not enrolled in a weight-loss trial and, therefore, were not self-selected to be highly motivated to lose weight. Because of this, and the large and ethnically diverse sample, our results should be generalizable to many clinical settings.
One limitation is that few younger, lower-income patients were included in the sample, which limits generalizability to those populations. Also, the study was observational. Although we adjusted for a broad set of patient, physician, and visit covariates, unmeasured confounding variables may still account for at least part of the observed associations. The analysis is limited by the use of self-reported dietary fat intake and physical activity measures. A food diary and accelerometer would have been more accurate; however, such involved measures could invoke changes in behavior, which would have confounded our ability to assess the effect of physician advice on weight loss.
Acknowledgements
The authors thank all of the physicians and patients who participated in this study, the study project managers Gretchen Yonish and Iguehi Esoimeme, and research assistant Justin Manusov.
CORRESPONDENCE
Stewart C. Alexander, PhD, Department of Medicine, Duke University School of Medicine, P.O. Box 3140 Medical Center, Durham, NC 27710; [email protected]
1. US Preventive Services Task Force. Screening for obesity in adults: recommendations and rationale. Ann Intern Med. 2003;139:930-932.
2. Nawaz H, Adams ML, Katz DL. Physician-patient interactions regarding diet, exercise, and smoking. Prev Med. 2000;31:652-657.
3. Sciamanna CN, Tate DF, Lang W, et al. Who reports receiving advice to lose weight? Results from a multistate survey. Arch Intern Med. 2000;160:2334-2339.
4. Galuska DA, Will JC, Serdula MK, et al. Are health care professionals advising obese patients to lose weight? JAMA. 1999;282:1576-1578.
5. Evans E. Why should obesity be managed? The obese individual’s perspective. Int J Obes Relat Metab Disord. 1999;23(suppl 4):S3-S6.
6. Mehrotra C, Naimi TS, Serdula M, et al. Arthritis, body mass index, and professional advice to lose weight: implications for clinical medicine and public health. Am J Prev Med. 2004;27:16-21.
7. Loureiro ML, Nayga RM Jr. Obesity, weight loss, and physician’s advice. Soc Sci Med. 2006;62:2458-2468.
8. Flocke SA, Clark A, Schlessman K, et al. Exercise, diet, and weight loss advice in the family medicine outpatient setting. Fam Med. 2005;37:415-421.
9. Alexander SC, Cox ME, Østbye T, et al. Do the 5 A’s work for weight-loss counseling? Presented at: International Conference on Communication in Healthcare; October 4–7, 2009; Miami, Fla.
10. McGinn T, Wyer PC, Newman TB, et al. Tips for learners of evidence-based medicine: 3. Measures of observer variability (kappa statistic). CMAJ. 2004;171:1369-1373.
11. Shannon J, Kristal AR, Curry SJ, et al. Application of a behavioral approach to measuring dietary change: the fat- and fiber-related diet behavior questionnaire. Cancer Epidemiol Biomarkers Prev. 1997;6:355-361.
12. Kristal AR, Shattuck AL, Henry HJ. Patterns of dietary behavior associated with selecting diets low in fat: reliability and validity of a behavioral approach to dietary assessment. J Am Diet Assoc. 1990;90:214-220.
13. Kannel WB, Sorlie P. Some health benefits of physical activity: the Framingham study. Arch Intern Med. 1979;139:857-861.
14. Little RJA, Rubin DB. Statistical Analysis With Missing Data. New York, NY: John Wiley & Sons; 2002.
15. Franz MJ, VanWormer JJ, Crain AL, et al. Weight-loss outcomes: a systematic review and meta-analysis of weight-loss clinical trials with a minimum 1-year follow-up. J Am Diet Assoc. 2007;107:1755-1767.
16. National Institutes of Health. The practical guide: identification, evaluation, and treatment of overweight and obesity in adults. NIH publication 00-4084. 2000. Available at: http://www.nhlbi.nih.gov/guidelines/obesity/prctgd_c.pdf. Accessed August 17, 2009.
17. American Diabetes Association. Standards of medical care in diabetes–2010. Diabetes Care. 2010;33(suppl 1):S11-S61.
18. Beach MC, Roter DL. Interpersonal expectations in the patient-physician relationship. J Gen Intern Med. 2000;15:825-827.
19. Miller WR, Rollnick SP, Miller WR. Motivational Interviewing: Preparing People for Change. 2nd ed. New York, NY: Guilford Press; 2002.
20. Bandura A. Social Foundations of Thought and Action: A Social Cognitive Theory. Englewood Cliffs, NJ: Prentice-Hall; 1986.
Background In primary care encounters, it is unknown whether physician advice on weight-related matters leads to patient weight loss. To examine this issue, we analyzed physician weight loss advice and measured corresponding changes in patients’ dietary intake, physical activity, and weight.
Methods Using audio-recorded primary care encounters between 40 physicians and 461 of their overweight or obese patients, we coded weight-related advice as nonspecific, specific nutritional, specific exercise, or specific weight. Physicians and patients were told the study was about preventive health, not weight. We used mixed models (SAS Proc Mixed), controlled for physician clustering and baseline covariates, to assess changes in diet, exercise, and measured weight, both pre-encounter and 3 months post-encounter.
Results When discussing weight, physicians typically provided a combination of specific weight, nutrition, and physical activity advice to their patients (34%). Combined advice resulted in patients reducing their dietary fat intake (P=.02). However, when physicians provided physical activity advice only, patients were significantly (P=.02) more likely to gain weight (+1.41 kg) compared with those who received no advice.
Conclusion When giving weight-related advice, most physicians provided a combination of lifestyle recommendations. Combining advice may help patients reduce their fat in-take. Physical activity advice alone may not be particularly helpful.
The US Preventive Services Task Force (USPSTF) recommends that physicians screen patients for obesity and offer intensive counseling and behavioral interventions to promote sustained weight loss.1 Evidence suggests that physician counseling, including advice, can help patients to lose weight, increase physical activity, and improve diet.2-9 However, little is known about what specific types of weight loss advice physicians give to patients, and whether some types are more effective than others at influencing behavior change.
We analyzed physician weight loss advice delivered in primary care visits and measured changes in patients’ dietary intake, physical activity, and body weight. We examined both the type of weight loss advice delivered and the impact of type of advice on weight and behavior change.
Methods
This study analyzed audio recordings from Project CHAT – Communicating Health: Analyzing Talk. The project was approved by the Duke University Medical Center Institutional Review Board.
Recruitment Physicians. We obtained consent from 40 primary care physicians in community-based practices and told them the study would examine communication around preventive health topics, not weight specifically.
Patients. We identified potential participants by reviewing scheduled appointments 3 weeks in advance. Eligible participants were at least 18 years of age, English-speaking, overweight or obese (body mass index [BMI] ≥25 kg/m2), cognitively competent, and not pregnant. After we obtained consent, a remotely located research assistant started a digital audio recorder as the patient entered the exam room. Immediately after the encounter, the research assistant administered a post-encounter survey to the patient and recorded the patient’s vital signs (N=461). Three months later, the research assistant met with the participant to record vital signs and administer a survey assessing changes in dietary fat intake and exercise (N=426).
Data coding
We coded advice into 4 broad categories: (1) nutrition advice, (2) physical activity advice, (3) specific weight loss advice, and (4) nonspecific weight loss/weight-related advice. We transcribed each piece of advice verbatim.
Nutrition advice consisted of 9 sub-categories: calorie/portion control, meal timing/planning, commercial diet plans, negative diet plans, increase fruits/vegetables, reduce sugar/carbohydrates, reduce fat/cholesterol, other micronutrient recommendations, and specific food items from multiple categories.
Physical activity advice consisted of 6 subcategories: walking, aerobic exercise, anaerobic exercise, exercise intensity, exercise duration, and exercise for comorbid conditions.
Specific weight loss advice consisted of 3 categories: weight loss behavior, weight loss for comorbid conditions, and referrals.
Nonspecific weight loss advice also consisted of 3 subcategories in which physicians provided no details about the general topics of nutrition, physical activity, or weight loss.
Two independent coders (CBT and MEC) assessed each piece of advice and double coded 20% of conversations for reliability. Cohen’s kappa was used to calculate inter-rater reliability for each code using Landis and Koch’s classification (0.21-0.40=fair agreement; 0.41-0.60=moderate agreement; 0.61-0.80=substantial agreement; 0.81-1.0=near-perfect agreement).10 Three advice categories achieved near perfect agreement: nutrition (kappa= 0.94; 95% confidence interval [CI] 0.82-1.0; 99.2% agreement), physical activity (kappa=0.91; 95% CI, 0.84-0.99; 98.6% agreement), and weight loss (kappa=0.95; 95% CI, 0.82-1.0; 99.7% agreement). The nonspecific weight loss advice category had slightly lower agreement but still achieved near-perfect agreement (kappa=0.82; 95% CI, 0.62-1.0; 99.2% agreement).
After all advice was coded, we placed conversations into 1 of 6 categories: (1) no advice given; (2) nonspecific advice only; (3) nutrition only; (4) physical activity only; (5) weight loss only; or (6) combination of nutrition, physical activity, and/or weight loss.
Measures
Dietary fat and fiber intake. We assessed dietary fat intake at baseline and at 3 months using the 22-item Fat- and Fiber-Related Diet Behavior Questionnaire.11,12 Questions about frequency of food selections included, “When you ate dessert, how often did you eat only fruit?” and “When you ate chicken, how often did you take off the skin?” We averaged responses into a total score wherein 1 reflected higher fiber, lower fat food choices; a score of 4 reflected lower fiber, higher fat choices (α=0.74 at baseline and α=0.77 at 3-month follow-up).
Physical activity. We measured physical activity (baseline, 3 months) using the Framingham Physical Activity Index.13 Participants recalled the average number of hours spent engaged in various daily activities (sleeping, working, leisure) and the level of activity for each (sedentary, slight, moderate, or heavy). The composite score accounts for activity duration and intensity.
Anthropometrics. We measured patient weight (baseline, 3 months) and height (baseline only) using a calibrated scale and portable stadiometer. Patients removed shoes, outerwear, and belongings from their pockets before being weighed.
Analysis
We analyzed data using SAS (SAS Institute, Inc., Cary, NC). We assessed the association between type of advice and weight loss, improvement in dietary fat intake behaviors, and increase in physical activity between baseline and the 3-month follow-up visit. We used PROC MIXED to fit general linear models; we incorporated responses into these models from all participants who provided measurements for at least one time point. This modeling framework yields unbiased estimates when missing data are unrelated to the observed variable.14
Primary predictors: (1) type of advice (none, nonspecific, nutrition, physical activity, weight loss, and combination), (2) time since baseline visit, and (3) time by type of advice interaction. All models included a priori defined patient, physician, and visit-related covariates that were theoretically or empirically related to changes in the outcomes (weight, physical activity, or dietary fat in-take). The 14 patient covariates were sex; age; race; high school education; economic security (enough money to pay monthly bills); over-weight (BMI, 25-29.9 kg/m2) or obese (BMI ≥30 kg/m2); actively trying to lose weight (yes/ no); motivated to lose weight (Likert scale 1-7); comfortable discussing weight (Likert scale 1-5); confident about losing weight (Likert scale 1-5); and patient-reported comorbid conditions of diabetes, hypertension, arthritis, and hyperlipidemia.
The 9 physician covariates were sex; race; years since medical school graduation; specialty (family vs internal medicine); self-efficacy (Likert scale 1-5); barriers for weight counseling (Likert scale 1-5); comfort discussing weight (Likert scale 1-5); insurance reimbursement concerns (Likert scale 1-5); and prior training in behavioral counseling (yes/no). Finally, 2 visit-level covariates were included: minutes spent addressing weight issues and visit type (preventive vs chronic).
Results
Sample characteristics
Of the 40 physicians, 19 were family physicians and 21 were internists. More than half of the physicians were female (60%), and 85% were white. Mean age was 47.2 years and mean BMI was 24.9 kg/m2. Of the 461 patients, 66% were female, 65% were white, 35% were African American, and two-thirds had post-high school education (TABLE 1). Mean patient age was 59.8 years; only 4% of the patients were new to their physicians.
TABLE 1
Patient characteristics (N=461)
| % or mean (SD) | |
|---|---|
| Race | |
| White/Asian/Pacific Islander | 65% |
| African American | 35% |
| Female | 66% |
| Age, y (missing=1)* | 59.8 (13.9) |
| BMI, kg/m2 (missing=1)* | 33.1 (7.1) |
| Education (missing=1)* | |
| Post-high school | 67% |
| Income (missing=37)* | |
| $45,000 or less | 48% |
| High financial burden (missing=13)* | |
| Pay bills with trouble | 14% |
| Diagnosed with: | |
| Diabetes | 31% |
| Hypertension (missing=1)* | 69% |
| Hyperlipidemia (missing=1)* | 56% |
| Arthritis | 47% |
| New patient | 4% |
| BMI, body mass index; SD, standard deviation. * Missing data at baseline. | |
Frequency of advice
Physicians gave some type of weight-related advice in 63% of the encounters. They combined types of advice in 34% of all conversations, provided physical activity advice only in 13%, nutrition advice only in 8%, nonspecific advice in 5%, and weight loss advice only in 3%. Many times when physicians gave advice, it was centered on self (eg, “I need you to do X” or “What will it take for me to get you to do Y?”).
Nutrition advice most commonly pertained to specific food items from multiple categories (27% of conversations). Physicians also advised patients to reduce sugar/carbohydrates, control calories and portions, add other micronutrients, eat more fruits/vegetables, and eat meals more frequently.
Walking was the physical activity topic discussed most frequently, followed by exercise duration, exercise for comorbidities, aerobic activities, exercise intensity, and anaerobic exercise. The most common specific weight loss topic was weight loss behavioral advice, followed by weight loss for comorbid conditions. Physicians rarely provided referrals to weight-loss programs.
Effect of type of advice on fat and fiber diet behavior score
Receipt of nutrition advice only was not associated with reduction in fat intake (P=.43, TABLE 2). However, those who received combined types of advice exhibited a significantly greater reduction of fat intake compared with those who received no advice (Fat- and Fiber-Related Diet Behavior Questionnaire score reduction of 0.15 vs 0.05; P=.02).
TABLE 2
How types of physician advice affected dietary fat intake, physical activity, and weight
| Type of advice | ||||||
|---|---|---|---|---|---|---|
| None | Nutrition only | Physical activity only | Weight loss only | Combined advice | Nonspecific | |
| Dietary fat change in Fat- and Fiber-Related Diet Behavior Questionnaire score differences | ||||||
| At 3 months from baseline (95% CI) | -0.05 (-0.11 to 0.004) | -0.10 (-0.22 to 0.01) | -0.07 (-0.16 to 0.02) | -0.08 (-0.26 to 0.09) | -0.15 (-0.20 to -0.09) | 0.03 (0.11 to 0.18) |
| P value* | .43 | .75 | .73 | .02 | .31 | |
| Physical activity score (change in MET hours) | ||||||
| At 3 months from baseline (95% CI) | 0.48 (-0.17 to 1.11) | 0.83 (-0.51 to 2.14) | 0.69 (-0.33 to 1.69) | -0.72 (-2.66 to 1.21) | 0.24 (-0.40 to 0.86) | -0.07 (-1.74 to 1.59) |
| P value* | .64 | .73 | .25 | .60 | .55 | |
| Weight change (kg) | ||||||
| At 3 months from baseline (95% CI) | -0.18 (-0.39 to 0.75) | -0.18 (-1.38 to 1.02) | 1.41 (0.51 to 2.31) | -0.26 (-1.99 to 1.47) | -0.55 (-1.12 to 0.02) | -0.62 (-2.11 to 0.87) |
| P value* | .59 | .02 | .63 | .08 | .32 | |
| CI, confidence interval; MET, metabolic equivalent tasks. *Test of difference between advice given and no advice given. | ||||||
Effect of type of advice on Framingham Physical Activity score
No type of advice, including physical activity advice, led to a change in Framingham Physical Activity scores at the 3-month visit (overall P=.76; TABLE 2).
Effect of type of advice on weight loss
Patients who received physical activity advice gained significantly more weight than patients who received no advice (1.41 kg gained vs 0.18 kg lost; P=.02). Patients who received combined advice lost more weight than patients who received no advice, but the difference did not reach statistical significance (0.55 kg lost vs 0.18 kg lost; P=.08).
Discussion
Physicians typically took an “all or nothing” approach to weight-related issues, giving no advice (37%) or a combination of nutrition, physical activity, and weight loss advice (34%). It seems when physicians do give advice, most of them follow the USPSTF guidelines by addressing nutrition and physical activity together.15
Providing advice alone did not predict a change in patient behavior. For instance, we found no significant association between dietary fat reduction and having received only nutrition advice. Possible explanations include the following:
- Although physicians advised patients to reduce fat/cholesterol intake in 28% of conversations, they did so mostly in combination with other types of advice. Nutrition-only advice occurred in only 8% of conversations. Thus, there may have been insufficient power to detect the impact of this specific type of advice.
- With nutrition-only advice, the most common recommendation was to reduce carbohydrates/sweets, which should not affect fat intake.
Advising patients solely on physical activity led to unintended weight gain overall. Other data have shown that exercise without dietary changes, though beneficial in many ways, is not substantially effective for weight loss.15 People may eat more when they exercise, either to reward themselves or to satiate increased appetite from increased energy expenditure. Or, if physicians recommend the standard goal of 150 minutes of intensive physical activity per week, normally sedentary patients may see that as unattainable and become too discouraged to try.1,16,17
Combining types of advice seemed to help patients reduce their fat intake. Overall, however, simple, brief advice from a physician may not be enough to promote healthy lifestyle changes.
Also notable was that physicians rarely provided referrals, even though this is a strong recommendation from the National Institutes of Health, the American Diabetes Association, and the USPSTF.1,16,17 It could be that many physicians believe referrals are not covered by insurance. Yet, the low frequency of referrals may suggest an important missing component of weight loss therapy, especially given that physician advice alone seems an inadequate intervention.
Avoid physician-centered appeals. Advice was often given in a physician-centered way. There are 3 possible explanations for such phrasing:
- In the absence of clear evidence about how to deliver weight loss advice, physicians may be formulating advice based on their personal or clinical experiences.
- Physicians either assume or sense that patients lack internal motivation to make lifestyle changes for themselves and instead request that patients make changes for the doctor-patient relationship.
- Physicians might be trying to invoke authority in the hope that patients will respond accordingly.
Whatever the reason, the literature on self-centered physician talk indicates that patients are less satisfied when physicians make the visit more about themselves than about patients.18 A better strategy might be to use Motivational Interviewing19 that supports patient autonomy and attempts to elicit and build on internal motivation.
The take-away message is that behavior change is complex and that knowledge is a necessary but insufficient agent for change. Following the tenets of Social Cognitive Theory,20 physicians might also need to address patient motivation, confidence, outcome expectations, and skills to help promote behavior change.
Strengths and limitations of this study
We recorded conversations rather than relying on physician or patient recall. Additionally, these primary care patients were not enrolled in a weight-loss trial and, therefore, were not self-selected to be highly motivated to lose weight. Because of this, and the large and ethnically diverse sample, our results should be generalizable to many clinical settings.
One limitation is that few younger, lower-income patients were included in the sample, which limits generalizability to those populations. Also, the study was observational. Although we adjusted for a broad set of patient, physician, and visit covariates, unmeasured confounding variables may still account for at least part of the observed associations. The analysis is limited by the use of self-reported dietary fat intake and physical activity measures. A food diary and accelerometer would have been more accurate; however, such involved measures could invoke changes in behavior, which would have confounded our ability to assess the effect of physician advice on weight loss.
Acknowledgements
The authors thank all of the physicians and patients who participated in this study, the study project managers Gretchen Yonish and Iguehi Esoimeme, and research assistant Justin Manusov.
CORRESPONDENCE
Stewart C. Alexander, PhD, Department of Medicine, Duke University School of Medicine, P.O. Box 3140 Medical Center, Durham, NC 27710; [email protected]
Background In primary care encounters, it is unknown whether physician advice on weight-related matters leads to patient weight loss. To examine this issue, we analyzed physician weight loss advice and measured corresponding changes in patients’ dietary intake, physical activity, and weight.
Methods Using audio-recorded primary care encounters between 40 physicians and 461 of their overweight or obese patients, we coded weight-related advice as nonspecific, specific nutritional, specific exercise, or specific weight. Physicians and patients were told the study was about preventive health, not weight. We used mixed models (SAS Proc Mixed), controlled for physician clustering and baseline covariates, to assess changes in diet, exercise, and measured weight, both pre-encounter and 3 months post-encounter.
Results When discussing weight, physicians typically provided a combination of specific weight, nutrition, and physical activity advice to their patients (34%). Combined advice resulted in patients reducing their dietary fat intake (P=.02). However, when physicians provided physical activity advice only, patients were significantly (P=.02) more likely to gain weight (+1.41 kg) compared with those who received no advice.
Conclusion When giving weight-related advice, most physicians provided a combination of lifestyle recommendations. Combining advice may help patients reduce their fat in-take. Physical activity advice alone may not be particularly helpful.
The US Preventive Services Task Force (USPSTF) recommends that physicians screen patients for obesity and offer intensive counseling and behavioral interventions to promote sustained weight loss.1 Evidence suggests that physician counseling, including advice, can help patients to lose weight, increase physical activity, and improve diet.2-9 However, little is known about what specific types of weight loss advice physicians give to patients, and whether some types are more effective than others at influencing behavior change.
We analyzed physician weight loss advice delivered in primary care visits and measured changes in patients’ dietary intake, physical activity, and body weight. We examined both the type of weight loss advice delivered and the impact of type of advice on weight and behavior change.
Methods
This study analyzed audio recordings from Project CHAT – Communicating Health: Analyzing Talk. The project was approved by the Duke University Medical Center Institutional Review Board.
Recruitment Physicians. We obtained consent from 40 primary care physicians in community-based practices and told them the study would examine communication around preventive health topics, not weight specifically.
Patients. We identified potential participants by reviewing scheduled appointments 3 weeks in advance. Eligible participants were at least 18 years of age, English-speaking, overweight or obese (body mass index [BMI] ≥25 kg/m2), cognitively competent, and not pregnant. After we obtained consent, a remotely located research assistant started a digital audio recorder as the patient entered the exam room. Immediately after the encounter, the research assistant administered a post-encounter survey to the patient and recorded the patient’s vital signs (N=461). Three months later, the research assistant met with the participant to record vital signs and administer a survey assessing changes in dietary fat intake and exercise (N=426).
Data coding
We coded advice into 4 broad categories: (1) nutrition advice, (2) physical activity advice, (3) specific weight loss advice, and (4) nonspecific weight loss/weight-related advice. We transcribed each piece of advice verbatim.
Nutrition advice consisted of 9 sub-categories: calorie/portion control, meal timing/planning, commercial diet plans, negative diet plans, increase fruits/vegetables, reduce sugar/carbohydrates, reduce fat/cholesterol, other micronutrient recommendations, and specific food items from multiple categories.
Physical activity advice consisted of 6 subcategories: walking, aerobic exercise, anaerobic exercise, exercise intensity, exercise duration, and exercise for comorbid conditions.
Specific weight loss advice consisted of 3 categories: weight loss behavior, weight loss for comorbid conditions, and referrals.
Nonspecific weight loss advice also consisted of 3 subcategories in which physicians provided no details about the general topics of nutrition, physical activity, or weight loss.
Two independent coders (CBT and MEC) assessed each piece of advice and double coded 20% of conversations for reliability. Cohen’s kappa was used to calculate inter-rater reliability for each code using Landis and Koch’s classification (0.21-0.40=fair agreement; 0.41-0.60=moderate agreement; 0.61-0.80=substantial agreement; 0.81-1.0=near-perfect agreement).10 Three advice categories achieved near perfect agreement: nutrition (kappa= 0.94; 95% confidence interval [CI] 0.82-1.0; 99.2% agreement), physical activity (kappa=0.91; 95% CI, 0.84-0.99; 98.6% agreement), and weight loss (kappa=0.95; 95% CI, 0.82-1.0; 99.7% agreement). The nonspecific weight loss advice category had slightly lower agreement but still achieved near-perfect agreement (kappa=0.82; 95% CI, 0.62-1.0; 99.2% agreement).
After all advice was coded, we placed conversations into 1 of 6 categories: (1) no advice given; (2) nonspecific advice only; (3) nutrition only; (4) physical activity only; (5) weight loss only; or (6) combination of nutrition, physical activity, and/or weight loss.
Measures
Dietary fat and fiber intake. We assessed dietary fat intake at baseline and at 3 months using the 22-item Fat- and Fiber-Related Diet Behavior Questionnaire.11,12 Questions about frequency of food selections included, “When you ate dessert, how often did you eat only fruit?” and “When you ate chicken, how often did you take off the skin?” We averaged responses into a total score wherein 1 reflected higher fiber, lower fat food choices; a score of 4 reflected lower fiber, higher fat choices (α=0.74 at baseline and α=0.77 at 3-month follow-up).
Physical activity. We measured physical activity (baseline, 3 months) using the Framingham Physical Activity Index.13 Participants recalled the average number of hours spent engaged in various daily activities (sleeping, working, leisure) and the level of activity for each (sedentary, slight, moderate, or heavy). The composite score accounts for activity duration and intensity.
Anthropometrics. We measured patient weight (baseline, 3 months) and height (baseline only) using a calibrated scale and portable stadiometer. Patients removed shoes, outerwear, and belongings from their pockets before being weighed.
Analysis
We analyzed data using SAS (SAS Institute, Inc., Cary, NC). We assessed the association between type of advice and weight loss, improvement in dietary fat intake behaviors, and increase in physical activity between baseline and the 3-month follow-up visit. We used PROC MIXED to fit general linear models; we incorporated responses into these models from all participants who provided measurements for at least one time point. This modeling framework yields unbiased estimates when missing data are unrelated to the observed variable.14
Primary predictors: (1) type of advice (none, nonspecific, nutrition, physical activity, weight loss, and combination), (2) time since baseline visit, and (3) time by type of advice interaction. All models included a priori defined patient, physician, and visit-related covariates that were theoretically or empirically related to changes in the outcomes (weight, physical activity, or dietary fat in-take). The 14 patient covariates were sex; age; race; high school education; economic security (enough money to pay monthly bills); over-weight (BMI, 25-29.9 kg/m2) or obese (BMI ≥30 kg/m2); actively trying to lose weight (yes/ no); motivated to lose weight (Likert scale 1-7); comfortable discussing weight (Likert scale 1-5); confident about losing weight (Likert scale 1-5); and patient-reported comorbid conditions of diabetes, hypertension, arthritis, and hyperlipidemia.
The 9 physician covariates were sex; race; years since medical school graduation; specialty (family vs internal medicine); self-efficacy (Likert scale 1-5); barriers for weight counseling (Likert scale 1-5); comfort discussing weight (Likert scale 1-5); insurance reimbursement concerns (Likert scale 1-5); and prior training in behavioral counseling (yes/no). Finally, 2 visit-level covariates were included: minutes spent addressing weight issues and visit type (preventive vs chronic).
Results
Sample characteristics
Of the 40 physicians, 19 were family physicians and 21 were internists. More than half of the physicians were female (60%), and 85% were white. Mean age was 47.2 years and mean BMI was 24.9 kg/m2. Of the 461 patients, 66% were female, 65% were white, 35% were African American, and two-thirds had post-high school education (TABLE 1). Mean patient age was 59.8 years; only 4% of the patients were new to their physicians.
TABLE 1
Patient characteristics (N=461)
| % or mean (SD) | |
|---|---|
| Race | |
| White/Asian/Pacific Islander | 65% |
| African American | 35% |
| Female | 66% |
| Age, y (missing=1)* | 59.8 (13.9) |
| BMI, kg/m2 (missing=1)* | 33.1 (7.1) |
| Education (missing=1)* | |
| Post-high school | 67% |
| Income (missing=37)* | |
| $45,000 or less | 48% |
| High financial burden (missing=13)* | |
| Pay bills with trouble | 14% |
| Diagnosed with: | |
| Diabetes | 31% |
| Hypertension (missing=1)* | 69% |
| Hyperlipidemia (missing=1)* | 56% |
| Arthritis | 47% |
| New patient | 4% |
| BMI, body mass index; SD, standard deviation. * Missing data at baseline. | |
Frequency of advice
Physicians gave some type of weight-related advice in 63% of the encounters. They combined types of advice in 34% of all conversations, provided physical activity advice only in 13%, nutrition advice only in 8%, nonspecific advice in 5%, and weight loss advice only in 3%. Many times when physicians gave advice, it was centered on self (eg, “I need you to do X” or “What will it take for me to get you to do Y?”).
Nutrition advice most commonly pertained to specific food items from multiple categories (27% of conversations). Physicians also advised patients to reduce sugar/carbohydrates, control calories and portions, add other micronutrients, eat more fruits/vegetables, and eat meals more frequently.
Walking was the physical activity topic discussed most frequently, followed by exercise duration, exercise for comorbidities, aerobic activities, exercise intensity, and anaerobic exercise. The most common specific weight loss topic was weight loss behavioral advice, followed by weight loss for comorbid conditions. Physicians rarely provided referrals to weight-loss programs.
Effect of type of advice on fat and fiber diet behavior score
Receipt of nutrition advice only was not associated with reduction in fat intake (P=.43, TABLE 2). However, those who received combined types of advice exhibited a significantly greater reduction of fat intake compared with those who received no advice (Fat- and Fiber-Related Diet Behavior Questionnaire score reduction of 0.15 vs 0.05; P=.02).
TABLE 2
How types of physician advice affected dietary fat intake, physical activity, and weight
| Type of advice | ||||||
|---|---|---|---|---|---|---|
| None | Nutrition only | Physical activity only | Weight loss only | Combined advice | Nonspecific | |
| Dietary fat change in Fat- and Fiber-Related Diet Behavior Questionnaire score differences | ||||||
| At 3 months from baseline (95% CI) | -0.05 (-0.11 to 0.004) | -0.10 (-0.22 to 0.01) | -0.07 (-0.16 to 0.02) | -0.08 (-0.26 to 0.09) | -0.15 (-0.20 to -0.09) | 0.03 (0.11 to 0.18) |
| P value* | .43 | .75 | .73 | .02 | .31 | |
| Physical activity score (change in MET hours) | ||||||
| At 3 months from baseline (95% CI) | 0.48 (-0.17 to 1.11) | 0.83 (-0.51 to 2.14) | 0.69 (-0.33 to 1.69) | -0.72 (-2.66 to 1.21) | 0.24 (-0.40 to 0.86) | -0.07 (-1.74 to 1.59) |
| P value* | .64 | .73 | .25 | .60 | .55 | |
| Weight change (kg) | ||||||
| At 3 months from baseline (95% CI) | -0.18 (-0.39 to 0.75) | -0.18 (-1.38 to 1.02) | 1.41 (0.51 to 2.31) | -0.26 (-1.99 to 1.47) | -0.55 (-1.12 to 0.02) | -0.62 (-2.11 to 0.87) |
| P value* | .59 | .02 | .63 | .08 | .32 | |
| CI, confidence interval; MET, metabolic equivalent tasks. *Test of difference between advice given and no advice given. | ||||||
Effect of type of advice on Framingham Physical Activity score
No type of advice, including physical activity advice, led to a change in Framingham Physical Activity scores at the 3-month visit (overall P=.76; TABLE 2).
Effect of type of advice on weight loss
Patients who received physical activity advice gained significantly more weight than patients who received no advice (1.41 kg gained vs 0.18 kg lost; P=.02). Patients who received combined advice lost more weight than patients who received no advice, but the difference did not reach statistical significance (0.55 kg lost vs 0.18 kg lost; P=.08).
Discussion
Physicians typically took an “all or nothing” approach to weight-related issues, giving no advice (37%) or a combination of nutrition, physical activity, and weight loss advice (34%). It seems when physicians do give advice, most of them follow the USPSTF guidelines by addressing nutrition and physical activity together.15
Providing advice alone did not predict a change in patient behavior. For instance, we found no significant association between dietary fat reduction and having received only nutrition advice. Possible explanations include the following:
- Although physicians advised patients to reduce fat/cholesterol intake in 28% of conversations, they did so mostly in combination with other types of advice. Nutrition-only advice occurred in only 8% of conversations. Thus, there may have been insufficient power to detect the impact of this specific type of advice.
- With nutrition-only advice, the most common recommendation was to reduce carbohydrates/sweets, which should not affect fat intake.
Advising patients solely on physical activity led to unintended weight gain overall. Other data have shown that exercise without dietary changes, though beneficial in many ways, is not substantially effective for weight loss.15 People may eat more when they exercise, either to reward themselves or to satiate increased appetite from increased energy expenditure. Or, if physicians recommend the standard goal of 150 minutes of intensive physical activity per week, normally sedentary patients may see that as unattainable and become too discouraged to try.1,16,17
Combining types of advice seemed to help patients reduce their fat intake. Overall, however, simple, brief advice from a physician may not be enough to promote healthy lifestyle changes.
Also notable was that physicians rarely provided referrals, even though this is a strong recommendation from the National Institutes of Health, the American Diabetes Association, and the USPSTF.1,16,17 It could be that many physicians believe referrals are not covered by insurance. Yet, the low frequency of referrals may suggest an important missing component of weight loss therapy, especially given that physician advice alone seems an inadequate intervention.
Avoid physician-centered appeals. Advice was often given in a physician-centered way. There are 3 possible explanations for such phrasing:
- In the absence of clear evidence about how to deliver weight loss advice, physicians may be formulating advice based on their personal or clinical experiences.
- Physicians either assume or sense that patients lack internal motivation to make lifestyle changes for themselves and instead request that patients make changes for the doctor-patient relationship.
- Physicians might be trying to invoke authority in the hope that patients will respond accordingly.
Whatever the reason, the literature on self-centered physician talk indicates that patients are less satisfied when physicians make the visit more about themselves than about patients.18 A better strategy might be to use Motivational Interviewing19 that supports patient autonomy and attempts to elicit and build on internal motivation.
The take-away message is that behavior change is complex and that knowledge is a necessary but insufficient agent for change. Following the tenets of Social Cognitive Theory,20 physicians might also need to address patient motivation, confidence, outcome expectations, and skills to help promote behavior change.
Strengths and limitations of this study
We recorded conversations rather than relying on physician or patient recall. Additionally, these primary care patients were not enrolled in a weight-loss trial and, therefore, were not self-selected to be highly motivated to lose weight. Because of this, and the large and ethnically diverse sample, our results should be generalizable to many clinical settings.
One limitation is that few younger, lower-income patients were included in the sample, which limits generalizability to those populations. Also, the study was observational. Although we adjusted for a broad set of patient, physician, and visit covariates, unmeasured confounding variables may still account for at least part of the observed associations. The analysis is limited by the use of self-reported dietary fat intake and physical activity measures. A food diary and accelerometer would have been more accurate; however, such involved measures could invoke changes in behavior, which would have confounded our ability to assess the effect of physician advice on weight loss.
Acknowledgements
The authors thank all of the physicians and patients who participated in this study, the study project managers Gretchen Yonish and Iguehi Esoimeme, and research assistant Justin Manusov.
CORRESPONDENCE
Stewart C. Alexander, PhD, Department of Medicine, Duke University School of Medicine, P.O. Box 3140 Medical Center, Durham, NC 27710; [email protected]
1. US Preventive Services Task Force. Screening for obesity in adults: recommendations and rationale. Ann Intern Med. 2003;139:930-932.
2. Nawaz H, Adams ML, Katz DL. Physician-patient interactions regarding diet, exercise, and smoking. Prev Med. 2000;31:652-657.
3. Sciamanna CN, Tate DF, Lang W, et al. Who reports receiving advice to lose weight? Results from a multistate survey. Arch Intern Med. 2000;160:2334-2339.
4. Galuska DA, Will JC, Serdula MK, et al. Are health care professionals advising obese patients to lose weight? JAMA. 1999;282:1576-1578.
5. Evans E. Why should obesity be managed? The obese individual’s perspective. Int J Obes Relat Metab Disord. 1999;23(suppl 4):S3-S6.
6. Mehrotra C, Naimi TS, Serdula M, et al. Arthritis, body mass index, and professional advice to lose weight: implications for clinical medicine and public health. Am J Prev Med. 2004;27:16-21.
7. Loureiro ML, Nayga RM Jr. Obesity, weight loss, and physician’s advice. Soc Sci Med. 2006;62:2458-2468.
8. Flocke SA, Clark A, Schlessman K, et al. Exercise, diet, and weight loss advice in the family medicine outpatient setting. Fam Med. 2005;37:415-421.
9. Alexander SC, Cox ME, Østbye T, et al. Do the 5 A’s work for weight-loss counseling? Presented at: International Conference on Communication in Healthcare; October 4–7, 2009; Miami, Fla.
10. McGinn T, Wyer PC, Newman TB, et al. Tips for learners of evidence-based medicine: 3. Measures of observer variability (kappa statistic). CMAJ. 2004;171:1369-1373.
11. Shannon J, Kristal AR, Curry SJ, et al. Application of a behavioral approach to measuring dietary change: the fat- and fiber-related diet behavior questionnaire. Cancer Epidemiol Biomarkers Prev. 1997;6:355-361.
12. Kristal AR, Shattuck AL, Henry HJ. Patterns of dietary behavior associated with selecting diets low in fat: reliability and validity of a behavioral approach to dietary assessment. J Am Diet Assoc. 1990;90:214-220.
13. Kannel WB, Sorlie P. Some health benefits of physical activity: the Framingham study. Arch Intern Med. 1979;139:857-861.
14. Little RJA, Rubin DB. Statistical Analysis With Missing Data. New York, NY: John Wiley & Sons; 2002.
15. Franz MJ, VanWormer JJ, Crain AL, et al. Weight-loss outcomes: a systematic review and meta-analysis of weight-loss clinical trials with a minimum 1-year follow-up. J Am Diet Assoc. 2007;107:1755-1767.
16. National Institutes of Health. The practical guide: identification, evaluation, and treatment of overweight and obesity in adults. NIH publication 00-4084. 2000. Available at: http://www.nhlbi.nih.gov/guidelines/obesity/prctgd_c.pdf. Accessed August 17, 2009.
17. American Diabetes Association. Standards of medical care in diabetes–2010. Diabetes Care. 2010;33(suppl 1):S11-S61.
18. Beach MC, Roter DL. Interpersonal expectations in the patient-physician relationship. J Gen Intern Med. 2000;15:825-827.
19. Miller WR, Rollnick SP, Miller WR. Motivational Interviewing: Preparing People for Change. 2nd ed. New York, NY: Guilford Press; 2002.
20. Bandura A. Social Foundations of Thought and Action: A Social Cognitive Theory. Englewood Cliffs, NJ: Prentice-Hall; 1986.
1. US Preventive Services Task Force. Screening for obesity in adults: recommendations and rationale. Ann Intern Med. 2003;139:930-932.
2. Nawaz H, Adams ML, Katz DL. Physician-patient interactions regarding diet, exercise, and smoking. Prev Med. 2000;31:652-657.
3. Sciamanna CN, Tate DF, Lang W, et al. Who reports receiving advice to lose weight? Results from a multistate survey. Arch Intern Med. 2000;160:2334-2339.
4. Galuska DA, Will JC, Serdula MK, et al. Are health care professionals advising obese patients to lose weight? JAMA. 1999;282:1576-1578.
5. Evans E. Why should obesity be managed? The obese individual’s perspective. Int J Obes Relat Metab Disord. 1999;23(suppl 4):S3-S6.
6. Mehrotra C, Naimi TS, Serdula M, et al. Arthritis, body mass index, and professional advice to lose weight: implications for clinical medicine and public health. Am J Prev Med. 2004;27:16-21.
7. Loureiro ML, Nayga RM Jr. Obesity, weight loss, and physician’s advice. Soc Sci Med. 2006;62:2458-2468.
8. Flocke SA, Clark A, Schlessman K, et al. Exercise, diet, and weight loss advice in the family medicine outpatient setting. Fam Med. 2005;37:415-421.
9. Alexander SC, Cox ME, Østbye T, et al. Do the 5 A’s work for weight-loss counseling? Presented at: International Conference on Communication in Healthcare; October 4–7, 2009; Miami, Fla.
10. McGinn T, Wyer PC, Newman TB, et al. Tips for learners of evidence-based medicine: 3. Measures of observer variability (kappa statistic). CMAJ. 2004;171:1369-1373.
11. Shannon J, Kristal AR, Curry SJ, et al. Application of a behavioral approach to measuring dietary change: the fat- and fiber-related diet behavior questionnaire. Cancer Epidemiol Biomarkers Prev. 1997;6:355-361.
12. Kristal AR, Shattuck AL, Henry HJ. Patterns of dietary behavior associated with selecting diets low in fat: reliability and validity of a behavioral approach to dietary assessment. J Am Diet Assoc. 1990;90:214-220.
13. Kannel WB, Sorlie P. Some health benefits of physical activity: the Framingham study. Arch Intern Med. 1979;139:857-861.
14. Little RJA, Rubin DB. Statistical Analysis With Missing Data. New York, NY: John Wiley & Sons; 2002.
15. Franz MJ, VanWormer JJ, Crain AL, et al. Weight-loss outcomes: a systematic review and meta-analysis of weight-loss clinical trials with a minimum 1-year follow-up. J Am Diet Assoc. 2007;107:1755-1767.
16. National Institutes of Health. The practical guide: identification, evaluation, and treatment of overweight and obesity in adults. NIH publication 00-4084. 2000. Available at: http://www.nhlbi.nih.gov/guidelines/obesity/prctgd_c.pdf. Accessed August 17, 2009.
17. American Diabetes Association. Standards of medical care in diabetes–2010. Diabetes Care. 2010;33(suppl 1):S11-S61.
18. Beach MC, Roter DL. Interpersonal expectations in the patient-physician relationship. J Gen Intern Med. 2000;15:825-827.
19. Miller WR, Rollnick SP, Miller WR. Motivational Interviewing: Preparing People for Change. 2nd ed. New York, NY: Guilford Press; 2002.
20. Bandura A. Social Foundations of Thought and Action: A Social Cognitive Theory. Englewood Cliffs, NJ: Prentice-Hall; 1986.