User login
New Medicare Rule Reduces Retroactive Billing Period
Medicare enrollment rules for retroactive billing implemented this month may catch some hospital medicine leaders by surprise—and cost them billing revenue.
The new rules from the Centers for Medicare and Medicaid Services (CMS), effective April 1, cut from 27 months to 30 days the window in which physicians can back-bill for services after successful enrollment or re-enrollment in Medicare. Most HM groups routinely allow new hospitalists to work prior to payor credentialing, then retroactively bill for those services once credentialing is completed, says Leslie Flores, MHA, a principal in Nelson/Flores Associates, an HM consulting firm in La Quinta, Calif., and director of SHM's Practice Management Institute.
Another provision of the rules states that practices must alert contractors of any changes in practice locations within 30 days, or risk expulsion from Medicare for as much as two years.
“This is likely to impact hospital medicine more than other specialties because of our rapid growth, the proportion of new graduates we hire, and the frequency with which hospitalists move around,” Flores says.
Marshall Maglothin, chief operating officer of Inpatient Specialists, which staffs 70 hospitalists at three Washington, D.C.-area hospitalists, suggests HM leaders read the new Medicare Provider Enrollment Toolkit, recently issued by the American Medical Association (AMA) and the Medical Group Management Association (MGMA). The resource includes an introduction to CMS’ Web-based version of the Provider Enrollment, Chain and Ownership System (PECOS), which became available this month for both HM groups and individual hospitalists. To download the toolkit, visit www.mgma.com.
“Until this gets worked over the next couple of months, there’s going to be a lot of missed revenue,” says Maglothin, who also runs HM advisory firm Blue Oak Consulting. “This is the ideal timeline, but it’s totally unrealistic when you’re dealing with over 600,000 physicians in the United States. There should have been an ease-in process.”
To help smooth the transition, CMS will hold a conference call to discuss provider issues at 2 p.m. (EST) Thursday. Capacity is limited, but to participate, call (800) 837-1935 and reference conference No. 94109369.
Medicare enrollment rules for retroactive billing implemented this month may catch some hospital medicine leaders by surprise—and cost them billing revenue.
The new rules from the Centers for Medicare and Medicaid Services (CMS), effective April 1, cut from 27 months to 30 days the window in which physicians can back-bill for services after successful enrollment or re-enrollment in Medicare. Most HM groups routinely allow new hospitalists to work prior to payor credentialing, then retroactively bill for those services once credentialing is completed, says Leslie Flores, MHA, a principal in Nelson/Flores Associates, an HM consulting firm in La Quinta, Calif., and director of SHM's Practice Management Institute.
Another provision of the rules states that practices must alert contractors of any changes in practice locations within 30 days, or risk expulsion from Medicare for as much as two years.
“This is likely to impact hospital medicine more than other specialties because of our rapid growth, the proportion of new graduates we hire, and the frequency with which hospitalists move around,” Flores says.
Marshall Maglothin, chief operating officer of Inpatient Specialists, which staffs 70 hospitalists at three Washington, D.C.-area hospitalists, suggests HM leaders read the new Medicare Provider Enrollment Toolkit, recently issued by the American Medical Association (AMA) and the Medical Group Management Association (MGMA). The resource includes an introduction to CMS’ Web-based version of the Provider Enrollment, Chain and Ownership System (PECOS), which became available this month for both HM groups and individual hospitalists. To download the toolkit, visit www.mgma.com.
“Until this gets worked over the next couple of months, there’s going to be a lot of missed revenue,” says Maglothin, who also runs HM advisory firm Blue Oak Consulting. “This is the ideal timeline, but it’s totally unrealistic when you’re dealing with over 600,000 physicians in the United States. There should have been an ease-in process.”
To help smooth the transition, CMS will hold a conference call to discuss provider issues at 2 p.m. (EST) Thursday. Capacity is limited, but to participate, call (800) 837-1935 and reference conference No. 94109369.
Medicare enrollment rules for retroactive billing implemented this month may catch some hospital medicine leaders by surprise—and cost them billing revenue.
The new rules from the Centers for Medicare and Medicaid Services (CMS), effective April 1, cut from 27 months to 30 days the window in which physicians can back-bill for services after successful enrollment or re-enrollment in Medicare. Most HM groups routinely allow new hospitalists to work prior to payor credentialing, then retroactively bill for those services once credentialing is completed, says Leslie Flores, MHA, a principal in Nelson/Flores Associates, an HM consulting firm in La Quinta, Calif., and director of SHM's Practice Management Institute.
Another provision of the rules states that practices must alert contractors of any changes in practice locations within 30 days, or risk expulsion from Medicare for as much as two years.
“This is likely to impact hospital medicine more than other specialties because of our rapid growth, the proportion of new graduates we hire, and the frequency with which hospitalists move around,” Flores says.
Marshall Maglothin, chief operating officer of Inpatient Specialists, which staffs 70 hospitalists at three Washington, D.C.-area hospitalists, suggests HM leaders read the new Medicare Provider Enrollment Toolkit, recently issued by the American Medical Association (AMA) and the Medical Group Management Association (MGMA). The resource includes an introduction to CMS’ Web-based version of the Provider Enrollment, Chain and Ownership System (PECOS), which became available this month for both HM groups and individual hospitalists. To download the toolkit, visit www.mgma.com.
“Until this gets worked over the next couple of months, there’s going to be a lot of missed revenue,” says Maglothin, who also runs HM advisory firm Blue Oak Consulting. “This is the ideal timeline, but it’s totally unrealistic when you’re dealing with over 600,000 physicians in the United States. There should have been an ease-in process.”
To help smooth the transition, CMS will hold a conference call to discuss provider issues at 2 p.m. (EST) Thursday. Capacity is limited, but to participate, call (800) 837-1935 and reference conference No. 94109369.
Pandemic Preparation
Now that more swine flu cases have been reported in New York City than in any other part of the U.S., local hospitalists are preparing to handle a potential influx of ill patients.
Dahlia Rizk, DO, FHM, director of the hospitalist program at Beth Israel Medical Center (BIMC) in New York City, says her 20-member team is receiving daily briefings from the hospital’s infection control expert. Hospitalists are learning about the latest confirmed cases and guidelines from the Centers for Disease Control and Prevention (CDC), the World Health Organization (WHO), and the NYC Department of Health and Mental Hygiene. Dr. Rizk is sharing the information with the rest of the hospital staff.
"This is our home; this is where we spend 90% of our day, if not 100%," Dr. Rizk says. “The staff know us, they rely on us, recognize us, and expect information and help from us when it comes to these kinds of situations."
John Novotny, MD, associate director of the hospitalist program at BIMC, says the hospital's strategy focuses on containing the virus and protecting other patients and staff from becoming infected by placing suspected swine flu patients in an isolation room that prevents the illness from being transmitted through droplets in the air. In addition, staff members will be expected to wear an N95 respirator facemask when entering the rooms of swine flu patients. Patients who are placed in the isolation room will be administered a PCR nasal swab test to confirm whether they are infected with influenza.
Dr. Rizk says patients showing mild to moderate symptoms will be asked to go home and remain there for seven days to reduce the chances of infecting others. “During this emergency, it is especially important to limit admissions of suspected influenza to those patients with more serious clinical conditions or significant comorbidities," she explains. "We need to focus on the priority of avoiding exposing other vulnerable inpatients to influenza, such as the elderly, the immune-compromised, or those with chronic heart and lung conditions.”
Drs. Novotny and Rizk suggest hospitalists follow these swine flu preparation tips:
- Communicate. Keep nurses and other staff up to date about the latest treatment and containment guidelines.
- Establish expectations. Be aware that staffers are looking to hospitalists for guidance during this emergency situation.
- Monitor for updates. Stay informed through your infectious disease division, the CDC, the WHO, and your local public health department.
For more information, visit the CDC website.
Now that more swine flu cases have been reported in New York City than in any other part of the U.S., local hospitalists are preparing to handle a potential influx of ill patients.
Dahlia Rizk, DO, FHM, director of the hospitalist program at Beth Israel Medical Center (BIMC) in New York City, says her 20-member team is receiving daily briefings from the hospital’s infection control expert. Hospitalists are learning about the latest confirmed cases and guidelines from the Centers for Disease Control and Prevention (CDC), the World Health Organization (WHO), and the NYC Department of Health and Mental Hygiene. Dr. Rizk is sharing the information with the rest of the hospital staff.
"This is our home; this is where we spend 90% of our day, if not 100%," Dr. Rizk says. “The staff know us, they rely on us, recognize us, and expect information and help from us when it comes to these kinds of situations."
John Novotny, MD, associate director of the hospitalist program at BIMC, says the hospital's strategy focuses on containing the virus and protecting other patients and staff from becoming infected by placing suspected swine flu patients in an isolation room that prevents the illness from being transmitted through droplets in the air. In addition, staff members will be expected to wear an N95 respirator facemask when entering the rooms of swine flu patients. Patients who are placed in the isolation room will be administered a PCR nasal swab test to confirm whether they are infected with influenza.
Dr. Rizk says patients showing mild to moderate symptoms will be asked to go home and remain there for seven days to reduce the chances of infecting others. “During this emergency, it is especially important to limit admissions of suspected influenza to those patients with more serious clinical conditions or significant comorbidities," she explains. "We need to focus on the priority of avoiding exposing other vulnerable inpatients to influenza, such as the elderly, the immune-compromised, or those with chronic heart and lung conditions.”
Drs. Novotny and Rizk suggest hospitalists follow these swine flu preparation tips:
- Communicate. Keep nurses and other staff up to date about the latest treatment and containment guidelines.
- Establish expectations. Be aware that staffers are looking to hospitalists for guidance during this emergency situation.
- Monitor for updates. Stay informed through your infectious disease division, the CDC, the WHO, and your local public health department.
For more information, visit the CDC website.
Now that more swine flu cases have been reported in New York City than in any other part of the U.S., local hospitalists are preparing to handle a potential influx of ill patients.
Dahlia Rizk, DO, FHM, director of the hospitalist program at Beth Israel Medical Center (BIMC) in New York City, says her 20-member team is receiving daily briefings from the hospital’s infection control expert. Hospitalists are learning about the latest confirmed cases and guidelines from the Centers for Disease Control and Prevention (CDC), the World Health Organization (WHO), and the NYC Department of Health and Mental Hygiene. Dr. Rizk is sharing the information with the rest of the hospital staff.
"This is our home; this is where we spend 90% of our day, if not 100%," Dr. Rizk says. “The staff know us, they rely on us, recognize us, and expect information and help from us when it comes to these kinds of situations."
John Novotny, MD, associate director of the hospitalist program at BIMC, says the hospital's strategy focuses on containing the virus and protecting other patients and staff from becoming infected by placing suspected swine flu patients in an isolation room that prevents the illness from being transmitted through droplets in the air. In addition, staff members will be expected to wear an N95 respirator facemask when entering the rooms of swine flu patients. Patients who are placed in the isolation room will be administered a PCR nasal swab test to confirm whether they are infected with influenza.
Dr. Rizk says patients showing mild to moderate symptoms will be asked to go home and remain there for seven days to reduce the chances of infecting others. “During this emergency, it is especially important to limit admissions of suspected influenza to those patients with more serious clinical conditions or significant comorbidities," she explains. "We need to focus on the priority of avoiding exposing other vulnerable inpatients to influenza, such as the elderly, the immune-compromised, or those with chronic heart and lung conditions.”
Drs. Novotny and Rizk suggest hospitalists follow these swine flu preparation tips:
- Communicate. Keep nurses and other staff up to date about the latest treatment and containment guidelines.
- Establish expectations. Be aware that staffers are looking to hospitalists for guidance during this emergency situation.
- Monitor for updates. Stay informed through your infectious disease division, the CDC, the WHO, and your local public health department.
For more information, visit the CDC website.
SHM New Members
z
z
z
The latest research you need to know
In This Edition
- Perioperative smoking cessation reduces postoperative complications.
- Quality of life is not diminished in heart failure patients receiving defibrillator therapy.
- Simplification of the revised Geneva score may be useful in assessing for pulmonary embolism.
- Non-cardiac surgery after drug eluting stents not associated with major cardiac risk.
- Major cardiac risk is lowest 90 days after bare metal stent percutaneous coronary intervention.
- Extensive cancer screening in unexplained VTE detects more malignancies, but does not affect cancer related mortality.
- Preadmission use of statins decreases after hospitalization mortality in pneumonia.
- Drug-eluting stents are better than bare-metal stents for PCI in acute MI, but additional randomized study is needed.
Does a short duration of perioperative smoking cessation lead to a reduction in postoperative complications?
Background: Prior studies have demonstrated a reduction in postoperative complications when patients stop smoking in the perioperative period. However, they have not clearly shown what effect a fairly short duration of cessation, such as a period of only four weeks, has on the frequency of complications.
Study design: Randomized controlled trial.
Setting: Four university-affiliated hospitals in Sweden.
Synopsis: Using 117 patients who were daily smokers for less than one year between the ages of 18-79 who were scheduled for elective general or orthopedic surgery, this study showed that a smoking-cessation intervention initiated as little as four weeks prior to surgery resulted in fewer postoperative complications. The complication rate was reduced from 41% in the control group to 21% in the intervention group, which received cessation counseling and nicotine-replacement therapy. The relative risk reduction was 49% (95% confidence interval, 3-40) with a number needed to treat of five.
Because this was a randomized controlled trial with a large observed benefit, it appears to be reasonable to endorse perioperative smoking cessation as late as four weeks before an elective surgery. The study was limited in its ability to detect a difference in wound infections by the small sample size and the possibility patients might have unblinded themselves to outcome assessors, causing an overestimation of the effect of the intervention on the primary outcome of all complications.
Bottom line: Perioperative smoking cessation reduces postoperative complications even when started just four weeks prior to surgery.
Citation: Lindstrom D, Azodi OS, Wladis A, et al. Effects of a perioperative smoking cessation intervention on postoperative complications. Ann Surg. 2008;248(5):739-745.
Does implantable-defibrillator therapy cause deterioration in quality of life for patients with heart failure?
Background: Patients with depressed left-ventricular function are known to have improved survival after receiving implantable cardioverter defibrillators (ICDs). However, there is concern ICD therapy can prolong survival at the expense of a diminished quality of life.
Study design: Randomized placebo-controlled trial.
Setting: Multiple centers in the U.S., Canada, and New Zealand.
Synopsis: Using 2,479 patients from the Sudden Cardiac Death in Heart Failure trial who were 18 and older and had stable heart failure and depressed left-ventricular function, this study demonstrated no significant quality-of-life difference at 30 months when compared with patients who received ICD, amiodarone, and state-of-the-art medical therapy or an amiodarone placebo and state-of-the-art medical therapy. While functional status did not differ at any time between the three groups, psychological well-being was improved in the ICD group at three months (p=0.01) and 12 months (p=0.03) when compared with the placebo group, but at 30 months there was no difference between the groups.
While the trial was randomized and placebo-controlled, the investigators were unable to blind patients or outcome assessors. Nevertheless, the lack of deterioration of quality of life in ICD patients is reassuring.
Bottom line: Placement of ICDs in heart failure patients with a high risk of sudden cardiac death does not appear to decrease quality of life.
Citation: Mark DB, Anstrom KJ, Sun JL, et al. Quality of life with defibrillator therapy or amiodarone in heart failure. N Engl J Med. 2008;359:999-1008.
Can a simplified, revised Geneva score retain diagnostic accuracy and clinical utility?
Background: The revised Geneva score is a validated and objective clinical decision rule, but has multiple variables with different weights. This can make the tool cumbersome and difficult to remember, and could lead to inaccurate calculations and misjudgments in patient care.
Study design: Retrospective cohort study.
Setting: Four university-affiliated European hospitals.
Synopsis: Using data from two prior prospective trials involving patients with suspected pulmonary embolism (PE), this study showed re-analysis of these patients with a simplified, revised Geneva score, which gives only one point to each clinical factor, resulted in the same level of diagnostic accuracy. Specifically, data from 1,049 patients was used to construct a receiver-operating characteristic curve analysis comparing the standardized and simplified Geneva score, which showed areas under the curve of 0.75 (95% confidence interval 0.71-0.78) and 0.74 (0.70-0.77), respectively. Additionally, the safety of using this clinical tool to rule out PE was demonstrated when using both a three-level (low-intermediate probability) and a dichotomized scheme (PE unlikely) in combination with a negative D-dimer test.
The retrospective nature of the study was its major limitation. The authors suggest a prospective study to complete validation of the simplified, revised Geneva score.
Bottom line: With prospective analysis, it might be possible to further validate a simplified, revised Geneva score.
Citation: Klok FA, Mos ICM, Nijkeuter M, et al. Simplification of the revised Geneva score for assessing clinical probability of pulmonary embolism. Arch Intern Med. 2008;168(19):2131-2136.
Is the rate of postoperative major adverse cardiac events (MACEs) inversely related to time after percutaneous coronary intervention (PCI) with a drug-eluting stent (DES)?
Background: The American College of Cardiology and the American Heart Association recently released an advisory that included a recommendation to delay elective noncardiac surgery (NCS) for one year after DES placement. However, no large study addresses the timing of NCS after PCI with DES.
Study design: Retrospective observational study.
Setting: Mayo Clinic, Rochester, Minn.
Synopsis: Looking at 520 patients who had NCS after DES at the Mayo Clinic, 5.4% experienced MACEs, but the rate of MACEs was not significantly associated with the time after stent placement to surgery (p=0.337). However, observed rates of MACEs were lower after one year. Elderly patients and those going for emergent surgery are at the highest risk for MACE. Bleeding complications were not associated with antiplatelet use.
Although this study does not provide a clear cutoff time for when it is safe to proceed to NCS after DES, it is somewhat reassuring to see the relatively small number of MACEs and the lack of bleeding complications associated with antiplatelet use. However, careful coordination between hospitalists, cardiologists, anesthesiologists, and surgeons is still needed when coordinating NCS after DES, especially in the elderly or during emergent situations.
Bottom line: While time to noncardiac surgery after drug-eluting stent placement is not associated with major adverse cardiac events, observed rates of events are lower after one year.
Citation: Rabbitts JA, Nuttall GA, Brown MJ, et al. Cardiac risk of non-cardiac surgery after percutaneous coronary intervention with drug-eluting stents. Anesthesiology.2008;109: 596-604.
Is the risk of MACEs and bleeding events for patients undergoing NCS related to the time interval between PCI with bare-metal stent?
Background: In order to prevent thrombosis of bare-metal stents (BMS) placed during percutaneous coronary intervention (PCI), antiplatelet therapy is used. This poses a risk of bleeding, if surgery is needed during the antiplatelet therapy. The American College of Cardiology and the American Heart Association recommends delaying NCS for at least six weeks after PCI with BMS.
Study design: Retrospective observational study.
Setting: Mayo Clinic, Rochester, Minn.
Synopsis: Looking at 899 patients who had NCS within one year of PCI with BMS at the Mayo Clinic between Jan. 1, 1990, and Jan. 1, 2005, this study found that when NCS was done 30 days or less after PCI with BMS, the MACEs rate was 10.5%, compared with 2.8% when NCS was done 91 or more days after PCI with BMS. After a multivariable analysis, it also was shown bleeding events were not associated with time between PCI with BMS and NCS.
While the American College of Cardiology and the American Heart Association recommends delaying NCS for at least six weeks after PCI with BMS, waiting at least 90 days would permit completion of antiplatelet therapy and re-endothelialization of the stent.
Bottom line: The risk of MACEs with noncardiac surgery is lowest when performed at least 90 days after PCI with bare-metal stent.
Citation: Nuttall GA, Brown MJ, Stombaugh JW, et al. Time and cardiac risk of surgery after bare-metal stent, percutaneous coronary intervention. Anesthesiology. 2008;109: 588-595.
Should we screen extensively for cancer in patients with newly diagnosed venous thromboembolism (VTE)?
Background: It is well known VTE can be the first manifestation of previously undiagnosed cancer. Retrospective studies have suggested “limited” cancer screening, including a history and physical examination, along with basic blood work, adequately identifies malignancy in patients with unexplained VTE. However, more recent prospective studies have suggested more extensive screening, which includes imaging studies or tumor-marker measurement, can increase the rate of cancer detection.
Study design: Systematic review.
Setting: Literature search using MEDLINE, EMBASE, the Cochrane Register of Controlled Trials, and evidence-based medicine reviews.
Synopsis: Thirty-six studies of 9,516 patients with VTE reported the period prevalence of previously undiagnosed cancer from baseline to 12 months was 6.3% (95% confidence interval (CI) of 5.6% to 6.9%) in all patients with VTE, and was even higher in patients with unprovoked VTE, 10% (95% CI 8.6% to 11.3%). Of the 34 articles used for prevalence assessment, an extensive screening strategy using CT scans of the abdomen and pelvis increased the proportion of previously undiagnosed cancer detection from 49.4% (CI, 40.2% to 58.5%; limited screening) to 69.7% (CI, 61.1% to 77.8%) in patients with unprovoked VTE. Ultrasonography of the abdomen and pelvis and tumor-marker screening did not result in a clinically significant increase in the frequency of cancer detection.
Four studies compared the rate of detection of early-stage, previously undiagnosed cancer between the limited and extensive screening strategies. Extensive screening led to an absolute decrease in cancer-related mortality of 1.9%, but this difference was not statistically significant.
In this systematic review, there is a great deal of heterogeneity in the studies. Most of the studies did not look at whether an increase in detection of new malignant conditions resulted in a change in the detection rate of early-stage cancer, or a decrease in cancer-related morbidity, cancer-related mortality, or overall mortality. Furthermore, the studies did not assess the consequences of extensive screening, such as patient anxiety and discomfort, testing complications, burden of additional tests for false-positive results, or cost-effectiveness. However, it is important for hospitalists to recognize undiagnosed cancer is common in unexplained VTE and warrants at least a limited-screening approach with more extensive screening.
Bottom line: Although the prevalence of undiagnosed cancer is common in VTE, extensive screening did not offer a cancer-related mortality benefit. CT of the abdomen and pelvis did, however, lead to a greater number of cancer diagnoses in patients with unexplained VTE.
Citation: Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149: 323-333.
Does the use of preadmission statins decrease the risk of death, bacteremia, and pulmonary complications in patients admitted with pneumonia?
Background: Both experimental and clinical studies have suggested statins improve outcomes in severe infections, such as sepsis. This is thought to be due to the antithrombotic, anti-inflammatory, and immunomodulatory effects of statins. However, previous studies on the effect of statins on pneumonia have conflicting outcomes.
Study design: Population-based cohort study of 29,900 patients.
Setting: Danish Health Registry.
Synopsis: Researchers studied patients ages 15 years and older hospitalized with pneumonia for the first time between January 1997 and December 2004. While statin users had more co-morbidities than nonusers, the 30-day mortality was 10.3% in users, compared with 15.7% in nonusers, corresponding to an adjusted 30-day mortality rate ratio of 0.69 (95% CI of 0.58-0.82). The 90-day mortality ratio was 16.8% in users, compared with 22.4% in nonusers, corresponding to an adjusted 90-day mortality ratio of 0.75 (95% CI of 0.65-0.86). Former use of statins was not associated with a decreased risk of death. The adjusted risk for bacteremia and pulmonary complications was not significantly different between nonusers and users.
Because this was an observational study, a causal relationship cannot be determined. Hospitalists should be alerted to the possibility statins might, in the future, prove to be a standard treatment modality in pneumonia. A randomized, double-blind trial might help further determine the effect of the acute use of statins on pneumonia outcomes.
Bottom line: Preadmission statin use is associated with a decrease in 30- and 90-day mortality in pneumonia.
Citation: Thomsen RW, Riis A, Kornum JB, Christensen S, Johnsen SP, Sorensen HT. Preadmission use of statins and outcomes after hospitalization with pneumonia. Arch Intern Med. 2008;168(19):2081-2087.
Do outcomes differ when patients with acute myocardial infarction (MI) undergo PCI with drug-eluting stents (DES) compared with bare-metal stents?
Background: Randomized trials comparing drug-eluting stents with bare-metal stents in acute MI have been limited in size and duration. Concern exists regarding higher mortality among patients with ST-elevation MI treated with DES.
Study design: Observational, cohort study.
Setting: Patients were identified from a state-mandated database, in which all PCI performed in Massachusetts are reported.
Synopsis: Between April 2003 and September 2004, 7,217 eligible patients underwent stenting for acute MI. They were assigned to either the DES group or the bare-metal stent (BMS) group using propensity score matching. Patients in the DES group had lower mortality at two years, compared to a matched cohort of patients in the BMS group with MI (10.7% vs. 12.8%; absolute risk difference, -2.1%, CI, -3.8% to -0.4%). A statistically significant difference was noted among patients with or without ST-elevation MI.
The rates of target vessel revascularization at two years with MI were significantly lower among patients receiving DES than among those receiving BMS (9.6% vs. 14.5%; risk difference, -4.9%; CI, -6.1% to -3.1%).
The study is limited by its observational nature and residual confounding bias after matching. Importantly, this study was performed to determine if DESs were harmful, and the finding of reduced mortality was unanticipated.
Bottom line: Although patients with acute MI treated with drug-eluting stents had lower mortality and repeat revascularization rates compared with bare-metal stents, this outcome merits confirmation in randomized trials.
Citation: Mauri L, Silbaugh TS, Garg P, et al. Drug-eluting or bare-metal stents for acute myocardial infarction. N Engl J Med. 2008;359 (13):1330-1342.
In This Edition
- Perioperative smoking cessation reduces postoperative complications.
- Quality of life is not diminished in heart failure patients receiving defibrillator therapy.
- Simplification of the revised Geneva score may be useful in assessing for pulmonary embolism.
- Non-cardiac surgery after drug eluting stents not associated with major cardiac risk.
- Major cardiac risk is lowest 90 days after bare metal stent percutaneous coronary intervention.
- Extensive cancer screening in unexplained VTE detects more malignancies, but does not affect cancer related mortality.
- Preadmission use of statins decreases after hospitalization mortality in pneumonia.
- Drug-eluting stents are better than bare-metal stents for PCI in acute MI, but additional randomized study is needed.
Does a short duration of perioperative smoking cessation lead to a reduction in postoperative complications?
Background: Prior studies have demonstrated a reduction in postoperative complications when patients stop smoking in the perioperative period. However, they have not clearly shown what effect a fairly short duration of cessation, such as a period of only four weeks, has on the frequency of complications.
Study design: Randomized controlled trial.
Setting: Four university-affiliated hospitals in Sweden.
Synopsis: Using 117 patients who were daily smokers for less than one year between the ages of 18-79 who were scheduled for elective general or orthopedic surgery, this study showed that a smoking-cessation intervention initiated as little as four weeks prior to surgery resulted in fewer postoperative complications. The complication rate was reduced from 41% in the control group to 21% in the intervention group, which received cessation counseling and nicotine-replacement therapy. The relative risk reduction was 49% (95% confidence interval, 3-40) with a number needed to treat of five.
Because this was a randomized controlled trial with a large observed benefit, it appears to be reasonable to endorse perioperative smoking cessation as late as four weeks before an elective surgery. The study was limited in its ability to detect a difference in wound infections by the small sample size and the possibility patients might have unblinded themselves to outcome assessors, causing an overestimation of the effect of the intervention on the primary outcome of all complications.
Bottom line: Perioperative smoking cessation reduces postoperative complications even when started just four weeks prior to surgery.
Citation: Lindstrom D, Azodi OS, Wladis A, et al. Effects of a perioperative smoking cessation intervention on postoperative complications. Ann Surg. 2008;248(5):739-745.
Does implantable-defibrillator therapy cause deterioration in quality of life for patients with heart failure?
Background: Patients with depressed left-ventricular function are known to have improved survival after receiving implantable cardioverter defibrillators (ICDs). However, there is concern ICD therapy can prolong survival at the expense of a diminished quality of life.
Study design: Randomized placebo-controlled trial.
Setting: Multiple centers in the U.S., Canada, and New Zealand.
Synopsis: Using 2,479 patients from the Sudden Cardiac Death in Heart Failure trial who were 18 and older and had stable heart failure and depressed left-ventricular function, this study demonstrated no significant quality-of-life difference at 30 months when compared with patients who received ICD, amiodarone, and state-of-the-art medical therapy or an amiodarone placebo and state-of-the-art medical therapy. While functional status did not differ at any time between the three groups, psychological well-being was improved in the ICD group at three months (p=0.01) and 12 months (p=0.03) when compared with the placebo group, but at 30 months there was no difference between the groups.
While the trial was randomized and placebo-controlled, the investigators were unable to blind patients or outcome assessors. Nevertheless, the lack of deterioration of quality of life in ICD patients is reassuring.
Bottom line: Placement of ICDs in heart failure patients with a high risk of sudden cardiac death does not appear to decrease quality of life.
Citation: Mark DB, Anstrom KJ, Sun JL, et al. Quality of life with defibrillator therapy or amiodarone in heart failure. N Engl J Med. 2008;359:999-1008.
Can a simplified, revised Geneva score retain diagnostic accuracy and clinical utility?
Background: The revised Geneva score is a validated and objective clinical decision rule, but has multiple variables with different weights. This can make the tool cumbersome and difficult to remember, and could lead to inaccurate calculations and misjudgments in patient care.
Study design: Retrospective cohort study.
Setting: Four university-affiliated European hospitals.
Synopsis: Using data from two prior prospective trials involving patients with suspected pulmonary embolism (PE), this study showed re-analysis of these patients with a simplified, revised Geneva score, which gives only one point to each clinical factor, resulted in the same level of diagnostic accuracy. Specifically, data from 1,049 patients was used to construct a receiver-operating characteristic curve analysis comparing the standardized and simplified Geneva score, which showed areas under the curve of 0.75 (95% confidence interval 0.71-0.78) and 0.74 (0.70-0.77), respectively. Additionally, the safety of using this clinical tool to rule out PE was demonstrated when using both a three-level (low-intermediate probability) and a dichotomized scheme (PE unlikely) in combination with a negative D-dimer test.
The retrospective nature of the study was its major limitation. The authors suggest a prospective study to complete validation of the simplified, revised Geneva score.
Bottom line: With prospective analysis, it might be possible to further validate a simplified, revised Geneva score.
Citation: Klok FA, Mos ICM, Nijkeuter M, et al. Simplification of the revised Geneva score for assessing clinical probability of pulmonary embolism. Arch Intern Med. 2008;168(19):2131-2136.
Is the rate of postoperative major adverse cardiac events (MACEs) inversely related to time after percutaneous coronary intervention (PCI) with a drug-eluting stent (DES)?
Background: The American College of Cardiology and the American Heart Association recently released an advisory that included a recommendation to delay elective noncardiac surgery (NCS) for one year after DES placement. However, no large study addresses the timing of NCS after PCI with DES.
Study design: Retrospective observational study.
Setting: Mayo Clinic, Rochester, Minn.
Synopsis: Looking at 520 patients who had NCS after DES at the Mayo Clinic, 5.4% experienced MACEs, but the rate of MACEs was not significantly associated with the time after stent placement to surgery (p=0.337). However, observed rates of MACEs were lower after one year. Elderly patients and those going for emergent surgery are at the highest risk for MACE. Bleeding complications were not associated with antiplatelet use.
Although this study does not provide a clear cutoff time for when it is safe to proceed to NCS after DES, it is somewhat reassuring to see the relatively small number of MACEs and the lack of bleeding complications associated with antiplatelet use. However, careful coordination between hospitalists, cardiologists, anesthesiologists, and surgeons is still needed when coordinating NCS after DES, especially in the elderly or during emergent situations.
Bottom line: While time to noncardiac surgery after drug-eluting stent placement is not associated with major adverse cardiac events, observed rates of events are lower after one year.
Citation: Rabbitts JA, Nuttall GA, Brown MJ, et al. Cardiac risk of non-cardiac surgery after percutaneous coronary intervention with drug-eluting stents. Anesthesiology.2008;109: 596-604.
Is the risk of MACEs and bleeding events for patients undergoing NCS related to the time interval between PCI with bare-metal stent?
Background: In order to prevent thrombosis of bare-metal stents (BMS) placed during percutaneous coronary intervention (PCI), antiplatelet therapy is used. This poses a risk of bleeding, if surgery is needed during the antiplatelet therapy. The American College of Cardiology and the American Heart Association recommends delaying NCS for at least six weeks after PCI with BMS.
Study design: Retrospective observational study.
Setting: Mayo Clinic, Rochester, Minn.
Synopsis: Looking at 899 patients who had NCS within one year of PCI with BMS at the Mayo Clinic between Jan. 1, 1990, and Jan. 1, 2005, this study found that when NCS was done 30 days or less after PCI with BMS, the MACEs rate was 10.5%, compared with 2.8% when NCS was done 91 or more days after PCI with BMS. After a multivariable analysis, it also was shown bleeding events were not associated with time between PCI with BMS and NCS.
While the American College of Cardiology and the American Heart Association recommends delaying NCS for at least six weeks after PCI with BMS, waiting at least 90 days would permit completion of antiplatelet therapy and re-endothelialization of the stent.
Bottom line: The risk of MACEs with noncardiac surgery is lowest when performed at least 90 days after PCI with bare-metal stent.
Citation: Nuttall GA, Brown MJ, Stombaugh JW, et al. Time and cardiac risk of surgery after bare-metal stent, percutaneous coronary intervention. Anesthesiology. 2008;109: 588-595.
Should we screen extensively for cancer in patients with newly diagnosed venous thromboembolism (VTE)?
Background: It is well known VTE can be the first manifestation of previously undiagnosed cancer. Retrospective studies have suggested “limited” cancer screening, including a history and physical examination, along with basic blood work, adequately identifies malignancy in patients with unexplained VTE. However, more recent prospective studies have suggested more extensive screening, which includes imaging studies or tumor-marker measurement, can increase the rate of cancer detection.
Study design: Systematic review.
Setting: Literature search using MEDLINE, EMBASE, the Cochrane Register of Controlled Trials, and evidence-based medicine reviews.
Synopsis: Thirty-six studies of 9,516 patients with VTE reported the period prevalence of previously undiagnosed cancer from baseline to 12 months was 6.3% (95% confidence interval (CI) of 5.6% to 6.9%) in all patients with VTE, and was even higher in patients with unprovoked VTE, 10% (95% CI 8.6% to 11.3%). Of the 34 articles used for prevalence assessment, an extensive screening strategy using CT scans of the abdomen and pelvis increased the proportion of previously undiagnosed cancer detection from 49.4% (CI, 40.2% to 58.5%; limited screening) to 69.7% (CI, 61.1% to 77.8%) in patients with unprovoked VTE. Ultrasonography of the abdomen and pelvis and tumor-marker screening did not result in a clinically significant increase in the frequency of cancer detection.
Four studies compared the rate of detection of early-stage, previously undiagnosed cancer between the limited and extensive screening strategies. Extensive screening led to an absolute decrease in cancer-related mortality of 1.9%, but this difference was not statistically significant.
In this systematic review, there is a great deal of heterogeneity in the studies. Most of the studies did not look at whether an increase in detection of new malignant conditions resulted in a change in the detection rate of early-stage cancer, or a decrease in cancer-related morbidity, cancer-related mortality, or overall mortality. Furthermore, the studies did not assess the consequences of extensive screening, such as patient anxiety and discomfort, testing complications, burden of additional tests for false-positive results, or cost-effectiveness. However, it is important for hospitalists to recognize undiagnosed cancer is common in unexplained VTE and warrants at least a limited-screening approach with more extensive screening.
Bottom line: Although the prevalence of undiagnosed cancer is common in VTE, extensive screening did not offer a cancer-related mortality benefit. CT of the abdomen and pelvis did, however, lead to a greater number of cancer diagnoses in patients with unexplained VTE.
Citation: Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149: 323-333.
Does the use of preadmission statins decrease the risk of death, bacteremia, and pulmonary complications in patients admitted with pneumonia?
Background: Both experimental and clinical studies have suggested statins improve outcomes in severe infections, such as sepsis. This is thought to be due to the antithrombotic, anti-inflammatory, and immunomodulatory effects of statins. However, previous studies on the effect of statins on pneumonia have conflicting outcomes.
Study design: Population-based cohort study of 29,900 patients.
Setting: Danish Health Registry.
Synopsis: Researchers studied patients ages 15 years and older hospitalized with pneumonia for the first time between January 1997 and December 2004. While statin users had more co-morbidities than nonusers, the 30-day mortality was 10.3% in users, compared with 15.7% in nonusers, corresponding to an adjusted 30-day mortality rate ratio of 0.69 (95% CI of 0.58-0.82). The 90-day mortality ratio was 16.8% in users, compared with 22.4% in nonusers, corresponding to an adjusted 90-day mortality ratio of 0.75 (95% CI of 0.65-0.86). Former use of statins was not associated with a decreased risk of death. The adjusted risk for bacteremia and pulmonary complications was not significantly different between nonusers and users.
Because this was an observational study, a causal relationship cannot be determined. Hospitalists should be alerted to the possibility statins might, in the future, prove to be a standard treatment modality in pneumonia. A randomized, double-blind trial might help further determine the effect of the acute use of statins on pneumonia outcomes.
Bottom line: Preadmission statin use is associated with a decrease in 30- and 90-day mortality in pneumonia.
Citation: Thomsen RW, Riis A, Kornum JB, Christensen S, Johnsen SP, Sorensen HT. Preadmission use of statins and outcomes after hospitalization with pneumonia. Arch Intern Med. 2008;168(19):2081-2087.
Do outcomes differ when patients with acute myocardial infarction (MI) undergo PCI with drug-eluting stents (DES) compared with bare-metal stents?
Background: Randomized trials comparing drug-eluting stents with bare-metal stents in acute MI have been limited in size and duration. Concern exists regarding higher mortality among patients with ST-elevation MI treated with DES.
Study design: Observational, cohort study.
Setting: Patients were identified from a state-mandated database, in which all PCI performed in Massachusetts are reported.
Synopsis: Between April 2003 and September 2004, 7,217 eligible patients underwent stenting for acute MI. They were assigned to either the DES group or the bare-metal stent (BMS) group using propensity score matching. Patients in the DES group had lower mortality at two years, compared to a matched cohort of patients in the BMS group with MI (10.7% vs. 12.8%; absolute risk difference, -2.1%, CI, -3.8% to -0.4%). A statistically significant difference was noted among patients with or without ST-elevation MI.
The rates of target vessel revascularization at two years with MI were significantly lower among patients receiving DES than among those receiving BMS (9.6% vs. 14.5%; risk difference, -4.9%; CI, -6.1% to -3.1%).
The study is limited by its observational nature and residual confounding bias after matching. Importantly, this study was performed to determine if DESs were harmful, and the finding of reduced mortality was unanticipated.
Bottom line: Although patients with acute MI treated with drug-eluting stents had lower mortality and repeat revascularization rates compared with bare-metal stents, this outcome merits confirmation in randomized trials.
Citation: Mauri L, Silbaugh TS, Garg P, et al. Drug-eluting or bare-metal stents for acute myocardial infarction. N Engl J Med. 2008;359 (13):1330-1342.
In This Edition
- Perioperative smoking cessation reduces postoperative complications.
- Quality of life is not diminished in heart failure patients receiving defibrillator therapy.
- Simplification of the revised Geneva score may be useful in assessing for pulmonary embolism.
- Non-cardiac surgery after drug eluting stents not associated with major cardiac risk.
- Major cardiac risk is lowest 90 days after bare metal stent percutaneous coronary intervention.
- Extensive cancer screening in unexplained VTE detects more malignancies, but does not affect cancer related mortality.
- Preadmission use of statins decreases after hospitalization mortality in pneumonia.
- Drug-eluting stents are better than bare-metal stents for PCI in acute MI, but additional randomized study is needed.
Does a short duration of perioperative smoking cessation lead to a reduction in postoperative complications?
Background: Prior studies have demonstrated a reduction in postoperative complications when patients stop smoking in the perioperative period. However, they have not clearly shown what effect a fairly short duration of cessation, such as a period of only four weeks, has on the frequency of complications.
Study design: Randomized controlled trial.
Setting: Four university-affiliated hospitals in Sweden.
Synopsis: Using 117 patients who were daily smokers for less than one year between the ages of 18-79 who were scheduled for elective general or orthopedic surgery, this study showed that a smoking-cessation intervention initiated as little as four weeks prior to surgery resulted in fewer postoperative complications. The complication rate was reduced from 41% in the control group to 21% in the intervention group, which received cessation counseling and nicotine-replacement therapy. The relative risk reduction was 49% (95% confidence interval, 3-40) with a number needed to treat of five.
Because this was a randomized controlled trial with a large observed benefit, it appears to be reasonable to endorse perioperative smoking cessation as late as four weeks before an elective surgery. The study was limited in its ability to detect a difference in wound infections by the small sample size and the possibility patients might have unblinded themselves to outcome assessors, causing an overestimation of the effect of the intervention on the primary outcome of all complications.
Bottom line: Perioperative smoking cessation reduces postoperative complications even when started just four weeks prior to surgery.
Citation: Lindstrom D, Azodi OS, Wladis A, et al. Effects of a perioperative smoking cessation intervention on postoperative complications. Ann Surg. 2008;248(5):739-745.
Does implantable-defibrillator therapy cause deterioration in quality of life for patients with heart failure?
Background: Patients with depressed left-ventricular function are known to have improved survival after receiving implantable cardioverter defibrillators (ICDs). However, there is concern ICD therapy can prolong survival at the expense of a diminished quality of life.
Study design: Randomized placebo-controlled trial.
Setting: Multiple centers in the U.S., Canada, and New Zealand.
Synopsis: Using 2,479 patients from the Sudden Cardiac Death in Heart Failure trial who were 18 and older and had stable heart failure and depressed left-ventricular function, this study demonstrated no significant quality-of-life difference at 30 months when compared with patients who received ICD, amiodarone, and state-of-the-art medical therapy or an amiodarone placebo and state-of-the-art medical therapy. While functional status did not differ at any time between the three groups, psychological well-being was improved in the ICD group at three months (p=0.01) and 12 months (p=0.03) when compared with the placebo group, but at 30 months there was no difference between the groups.
While the trial was randomized and placebo-controlled, the investigators were unable to blind patients or outcome assessors. Nevertheless, the lack of deterioration of quality of life in ICD patients is reassuring.
Bottom line: Placement of ICDs in heart failure patients with a high risk of sudden cardiac death does not appear to decrease quality of life.
Citation: Mark DB, Anstrom KJ, Sun JL, et al. Quality of life with defibrillator therapy or amiodarone in heart failure. N Engl J Med. 2008;359:999-1008.
Can a simplified, revised Geneva score retain diagnostic accuracy and clinical utility?
Background: The revised Geneva score is a validated and objective clinical decision rule, but has multiple variables with different weights. This can make the tool cumbersome and difficult to remember, and could lead to inaccurate calculations and misjudgments in patient care.
Study design: Retrospective cohort study.
Setting: Four university-affiliated European hospitals.
Synopsis: Using data from two prior prospective trials involving patients with suspected pulmonary embolism (PE), this study showed re-analysis of these patients with a simplified, revised Geneva score, which gives only one point to each clinical factor, resulted in the same level of diagnostic accuracy. Specifically, data from 1,049 patients was used to construct a receiver-operating characteristic curve analysis comparing the standardized and simplified Geneva score, which showed areas under the curve of 0.75 (95% confidence interval 0.71-0.78) and 0.74 (0.70-0.77), respectively. Additionally, the safety of using this clinical tool to rule out PE was demonstrated when using both a three-level (low-intermediate probability) and a dichotomized scheme (PE unlikely) in combination with a negative D-dimer test.
The retrospective nature of the study was its major limitation. The authors suggest a prospective study to complete validation of the simplified, revised Geneva score.
Bottom line: With prospective analysis, it might be possible to further validate a simplified, revised Geneva score.
Citation: Klok FA, Mos ICM, Nijkeuter M, et al. Simplification of the revised Geneva score for assessing clinical probability of pulmonary embolism. Arch Intern Med. 2008;168(19):2131-2136.
Is the rate of postoperative major adverse cardiac events (MACEs) inversely related to time after percutaneous coronary intervention (PCI) with a drug-eluting stent (DES)?
Background: The American College of Cardiology and the American Heart Association recently released an advisory that included a recommendation to delay elective noncardiac surgery (NCS) for one year after DES placement. However, no large study addresses the timing of NCS after PCI with DES.
Study design: Retrospective observational study.
Setting: Mayo Clinic, Rochester, Minn.
Synopsis: Looking at 520 patients who had NCS after DES at the Mayo Clinic, 5.4% experienced MACEs, but the rate of MACEs was not significantly associated with the time after stent placement to surgery (p=0.337). However, observed rates of MACEs were lower after one year. Elderly patients and those going for emergent surgery are at the highest risk for MACE. Bleeding complications were not associated with antiplatelet use.
Although this study does not provide a clear cutoff time for when it is safe to proceed to NCS after DES, it is somewhat reassuring to see the relatively small number of MACEs and the lack of bleeding complications associated with antiplatelet use. However, careful coordination between hospitalists, cardiologists, anesthesiologists, and surgeons is still needed when coordinating NCS after DES, especially in the elderly or during emergent situations.
Bottom line: While time to noncardiac surgery after drug-eluting stent placement is not associated with major adverse cardiac events, observed rates of events are lower after one year.
Citation: Rabbitts JA, Nuttall GA, Brown MJ, et al. Cardiac risk of non-cardiac surgery after percutaneous coronary intervention with drug-eluting stents. Anesthesiology.2008;109: 596-604.
Is the risk of MACEs and bleeding events for patients undergoing NCS related to the time interval between PCI with bare-metal stent?
Background: In order to prevent thrombosis of bare-metal stents (BMS) placed during percutaneous coronary intervention (PCI), antiplatelet therapy is used. This poses a risk of bleeding, if surgery is needed during the antiplatelet therapy. The American College of Cardiology and the American Heart Association recommends delaying NCS for at least six weeks after PCI with BMS.
Study design: Retrospective observational study.
Setting: Mayo Clinic, Rochester, Minn.
Synopsis: Looking at 899 patients who had NCS within one year of PCI with BMS at the Mayo Clinic between Jan. 1, 1990, and Jan. 1, 2005, this study found that when NCS was done 30 days or less after PCI with BMS, the MACEs rate was 10.5%, compared with 2.8% when NCS was done 91 or more days after PCI with BMS. After a multivariable analysis, it also was shown bleeding events were not associated with time between PCI with BMS and NCS.
While the American College of Cardiology and the American Heart Association recommends delaying NCS for at least six weeks after PCI with BMS, waiting at least 90 days would permit completion of antiplatelet therapy and re-endothelialization of the stent.
Bottom line: The risk of MACEs with noncardiac surgery is lowest when performed at least 90 days after PCI with bare-metal stent.
Citation: Nuttall GA, Brown MJ, Stombaugh JW, et al. Time and cardiac risk of surgery after bare-metal stent, percutaneous coronary intervention. Anesthesiology. 2008;109: 588-595.
Should we screen extensively for cancer in patients with newly diagnosed venous thromboembolism (VTE)?
Background: It is well known VTE can be the first manifestation of previously undiagnosed cancer. Retrospective studies have suggested “limited” cancer screening, including a history and physical examination, along with basic blood work, adequately identifies malignancy in patients with unexplained VTE. However, more recent prospective studies have suggested more extensive screening, which includes imaging studies or tumor-marker measurement, can increase the rate of cancer detection.
Study design: Systematic review.
Setting: Literature search using MEDLINE, EMBASE, the Cochrane Register of Controlled Trials, and evidence-based medicine reviews.
Synopsis: Thirty-six studies of 9,516 patients with VTE reported the period prevalence of previously undiagnosed cancer from baseline to 12 months was 6.3% (95% confidence interval (CI) of 5.6% to 6.9%) in all patients with VTE, and was even higher in patients with unprovoked VTE, 10% (95% CI 8.6% to 11.3%). Of the 34 articles used for prevalence assessment, an extensive screening strategy using CT scans of the abdomen and pelvis increased the proportion of previously undiagnosed cancer detection from 49.4% (CI, 40.2% to 58.5%; limited screening) to 69.7% (CI, 61.1% to 77.8%) in patients with unprovoked VTE. Ultrasonography of the abdomen and pelvis and tumor-marker screening did not result in a clinically significant increase in the frequency of cancer detection.
Four studies compared the rate of detection of early-stage, previously undiagnosed cancer between the limited and extensive screening strategies. Extensive screening led to an absolute decrease in cancer-related mortality of 1.9%, but this difference was not statistically significant.
In this systematic review, there is a great deal of heterogeneity in the studies. Most of the studies did not look at whether an increase in detection of new malignant conditions resulted in a change in the detection rate of early-stage cancer, or a decrease in cancer-related morbidity, cancer-related mortality, or overall mortality. Furthermore, the studies did not assess the consequences of extensive screening, such as patient anxiety and discomfort, testing complications, burden of additional tests for false-positive results, or cost-effectiveness. However, it is important for hospitalists to recognize undiagnosed cancer is common in unexplained VTE and warrants at least a limited-screening approach with more extensive screening.
Bottom line: Although the prevalence of undiagnosed cancer is common in VTE, extensive screening did not offer a cancer-related mortality benefit. CT of the abdomen and pelvis did, however, lead to a greater number of cancer diagnoses in patients with unexplained VTE.
Citation: Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149: 323-333.
Does the use of preadmission statins decrease the risk of death, bacteremia, and pulmonary complications in patients admitted with pneumonia?
Background: Both experimental and clinical studies have suggested statins improve outcomes in severe infections, such as sepsis. This is thought to be due to the antithrombotic, anti-inflammatory, and immunomodulatory effects of statins. However, previous studies on the effect of statins on pneumonia have conflicting outcomes.
Study design: Population-based cohort study of 29,900 patients.
Setting: Danish Health Registry.
Synopsis: Researchers studied patients ages 15 years and older hospitalized with pneumonia for the first time between January 1997 and December 2004. While statin users had more co-morbidities than nonusers, the 30-day mortality was 10.3% in users, compared with 15.7% in nonusers, corresponding to an adjusted 30-day mortality rate ratio of 0.69 (95% CI of 0.58-0.82). The 90-day mortality ratio was 16.8% in users, compared with 22.4% in nonusers, corresponding to an adjusted 90-day mortality ratio of 0.75 (95% CI of 0.65-0.86). Former use of statins was not associated with a decreased risk of death. The adjusted risk for bacteremia and pulmonary complications was not significantly different between nonusers and users.
Because this was an observational study, a causal relationship cannot be determined. Hospitalists should be alerted to the possibility statins might, in the future, prove to be a standard treatment modality in pneumonia. A randomized, double-blind trial might help further determine the effect of the acute use of statins on pneumonia outcomes.
Bottom line: Preadmission statin use is associated with a decrease in 30- and 90-day mortality in pneumonia.
Citation: Thomsen RW, Riis A, Kornum JB, Christensen S, Johnsen SP, Sorensen HT. Preadmission use of statins and outcomes after hospitalization with pneumonia. Arch Intern Med. 2008;168(19):2081-2087.
Do outcomes differ when patients with acute myocardial infarction (MI) undergo PCI with drug-eluting stents (DES) compared with bare-metal stents?
Background: Randomized trials comparing drug-eluting stents with bare-metal stents in acute MI have been limited in size and duration. Concern exists regarding higher mortality among patients with ST-elevation MI treated with DES.
Study design: Observational, cohort study.
Setting: Patients were identified from a state-mandated database, in which all PCI performed in Massachusetts are reported.
Synopsis: Between April 2003 and September 2004, 7,217 eligible patients underwent stenting for acute MI. They were assigned to either the DES group or the bare-metal stent (BMS) group using propensity score matching. Patients in the DES group had lower mortality at two years, compared to a matched cohort of patients in the BMS group with MI (10.7% vs. 12.8%; absolute risk difference, -2.1%, CI, -3.8% to -0.4%). A statistically significant difference was noted among patients with or without ST-elevation MI.
The rates of target vessel revascularization at two years with MI were significantly lower among patients receiving DES than among those receiving BMS (9.6% vs. 14.5%; risk difference, -4.9%; CI, -6.1% to -3.1%).
The study is limited by its observational nature and residual confounding bias after matching. Importantly, this study was performed to determine if DESs were harmful, and the finding of reduced mortality was unanticipated.
Bottom line: Although patients with acute MI treated with drug-eluting stents had lower mortality and repeat revascularization rates compared with bare-metal stents, this outcome merits confirmation in randomized trials.
Citation: Mauri L, Silbaugh TS, Garg P, et al. Drug-eluting or bare-metal stents for acute myocardial infarction. N Engl J Med. 2008;359 (13):1330-1342.
SHM Welcomes New Board Member

It has been a good year for Eric Howell, MD, FHM. Not only is he the 2009 recipient of SHM’s Award for Excellence in Teaching, but he also is the society’s newest board member.
Dr. Howell, who studied engineering before becoming a hospitalist, is director of the hospitalist division at Johns Hopkins Bayview Medical Center in Baltimore. An SHM member since 2000, he mentors 120 medical students as an assistant professor at the Johns Hopkins School of Medicine.
Dr. Howell, who is chair of SHM’s Leadership Committee and a member of the Public Policy Committee, was added to the 12-seat board this spring. Dr. Howell recently spoke to TH eWire about his award and his new position.
Question: What do you hope to accomplish during your SHM board tenure?
Answer:I am passionate about hospital medicine and enjoy advocating for hospitalists. I thought being a board member would be a mix of these two passions. My goal is to help hospitalists improve the care of their patients.
Q: How did you develop this passion for HM?
A: I love fixing broken things—improving on the hospital processes. I am an electrical engineer, and there is no specialty more based on systems improvement than hospital medicine. When I started, hospitalists were the red-headed stepchildren. Now they are implementers of quality improvement. I love patients, but I really love looking at systems and improving them.
Q: What do you see as the future of HM?
A: I hope it becomes a national leader for patients in hospital and quality improvement. I hope we set the stage nationally for changes in healthcare, such as [President] Obama is proposing. Hospitalists as individuals have been instrumental in advocating for patients. I’d like to see hospital medicine become the doctor-patient advocate.
It has been a good year for Eric Howell, MD, FHM. Not only is he the 2009 recipient of SHM’s Award for Excellence in Teaching, but he also is the society’s newest board member.
Dr. Howell, who studied engineering before becoming a hospitalist, is director of the hospitalist division at Johns Hopkins Bayview Medical Center in Baltimore. An SHM member since 2000, he mentors 120 medical students as an assistant professor at the Johns Hopkins School of Medicine.
Dr. Howell, who is chair of SHM’s Leadership Committee and a member of the Public Policy Committee, was added to the 12-seat board this spring. Dr. Howell recently spoke to TH eWire about his award and his new position.
Question: What do you hope to accomplish during your SHM board tenure?
Answer:I am passionate about hospital medicine and enjoy advocating for hospitalists. I thought being a board member would be a mix of these two passions. My goal is to help hospitalists improve the care of their patients.
Q: How did you develop this passion for HM?
A: I love fixing broken things—improving on the hospital processes. I am an electrical engineer, and there is no specialty more based on systems improvement than hospital medicine. When I started, hospitalists were the red-headed stepchildren. Now they are implementers of quality improvement. I love patients, but I really love looking at systems and improving them.
Q: What do you see as the future of HM?
A: I hope it becomes a national leader for patients in hospital and quality improvement. I hope we set the stage nationally for changes in healthcare, such as [President] Obama is proposing. Hospitalists as individuals have been instrumental in advocating for patients. I’d like to see hospital medicine become the doctor-patient advocate.
It has been a good year for Eric Howell, MD, FHM. Not only is he the 2009 recipient of SHM’s Award for Excellence in Teaching, but he also is the society’s newest board member.
Dr. Howell, who studied engineering before becoming a hospitalist, is director of the hospitalist division at Johns Hopkins Bayview Medical Center in Baltimore. An SHM member since 2000, he mentors 120 medical students as an assistant professor at the Johns Hopkins School of Medicine.
Dr. Howell, who is chair of SHM’s Leadership Committee and a member of the Public Policy Committee, was added to the 12-seat board this spring. Dr. Howell recently spoke to TH eWire about his award and his new position.
Question: What do you hope to accomplish during your SHM board tenure?
Answer:I am passionate about hospital medicine and enjoy advocating for hospitalists. I thought being a board member would be a mix of these two passions. My goal is to help hospitalists improve the care of their patients.
Q: How did you develop this passion for HM?
A: I love fixing broken things—improving on the hospital processes. I am an electrical engineer, and there is no specialty more based on systems improvement than hospital medicine. When I started, hospitalists were the red-headed stepchildren. Now they are implementers of quality improvement. I love patients, but I really love looking at systems and improving them.
Q: What do you see as the future of HM?
A: I hope it becomes a national leader for patients in hospital and quality improvement. I hope we set the stage nationally for changes in healthcare, such as [President] Obama is proposing. Hospitalists as individuals have been instrumental in advocating for patients. I’d like to see hospital medicine become the doctor-patient advocate.
More Money, More Problems
Hospitalists should be mindful that President Obama's half-billion-dollar commitment to new funding for community health centers (CHCs) could translate into unexpected compensation and burnout issues in the coming years, according to an SHM Public Policy Committee member.
Felix Aguirre, MD, FHM, vice president of medical affairs for IPC: The Hospitalist Company, says that until long-term healthcare reform is implemented, Obama’s $493 million in CHC grants "should be a wash."
Dr. Aguirre cautions that American Recovery and Reinvestment Act funding could have unintended consequences. For example, some clinics might raise compensation standards to retain or recruit hospitalists in order to deal with increased patient census. That increase could force local bidding wars for hospitalists at a time when supply is short.
“They will have a bit more money to attract those hospitalists,” Dr. Aguirre says.
In San Antonio, where Dr. Aguirre works, the effect could be even more pronounced, as one in four Texans are uninsured and more likely to take advantage of federally qualified health centers. In fact, 2007 federal data show roughly 40% of CHC patients were uninsured.
U.S. Health and Human Services officials say the stimulus money will provide care to nearly 3 million additional patients in the next two years, including roughly 1 million people without insurance. The added workload could cause burnout in hospitalists serving those institutions but who specialize in HM in part for the quality of life and scheduling perks that it affords.
"The ones that work with the CHCs would have more volume, but less chance of collecting on it, unless they have some arrangement to collect on that," Dr. Aguirre says. "There will be stress with increased volume."
Hospitalists should be mindful that President Obama's half-billion-dollar commitment to new funding for community health centers (CHCs) could translate into unexpected compensation and burnout issues in the coming years, according to an SHM Public Policy Committee member.
Felix Aguirre, MD, FHM, vice president of medical affairs for IPC: The Hospitalist Company, says that until long-term healthcare reform is implemented, Obama’s $493 million in CHC grants "should be a wash."
Dr. Aguirre cautions that American Recovery and Reinvestment Act funding could have unintended consequences. For example, some clinics might raise compensation standards to retain or recruit hospitalists in order to deal with increased patient census. That increase could force local bidding wars for hospitalists at a time when supply is short.
“They will have a bit more money to attract those hospitalists,” Dr. Aguirre says.
In San Antonio, where Dr. Aguirre works, the effect could be even more pronounced, as one in four Texans are uninsured and more likely to take advantage of federally qualified health centers. In fact, 2007 federal data show roughly 40% of CHC patients were uninsured.
U.S. Health and Human Services officials say the stimulus money will provide care to nearly 3 million additional patients in the next two years, including roughly 1 million people without insurance. The added workload could cause burnout in hospitalists serving those institutions but who specialize in HM in part for the quality of life and scheduling perks that it affords.
"The ones that work with the CHCs would have more volume, but less chance of collecting on it, unless they have some arrangement to collect on that," Dr. Aguirre says. "There will be stress with increased volume."
Hospitalists should be mindful that President Obama's half-billion-dollar commitment to new funding for community health centers (CHCs) could translate into unexpected compensation and burnout issues in the coming years, according to an SHM Public Policy Committee member.
Felix Aguirre, MD, FHM, vice president of medical affairs for IPC: The Hospitalist Company, says that until long-term healthcare reform is implemented, Obama’s $493 million in CHC grants "should be a wash."
Dr. Aguirre cautions that American Recovery and Reinvestment Act funding could have unintended consequences. For example, some clinics might raise compensation standards to retain or recruit hospitalists in order to deal with increased patient census. That increase could force local bidding wars for hospitalists at a time when supply is short.
“They will have a bit more money to attract those hospitalists,” Dr. Aguirre says.
In San Antonio, where Dr. Aguirre works, the effect could be even more pronounced, as one in four Texans are uninsured and more likely to take advantage of federally qualified health centers. In fact, 2007 federal data show roughly 40% of CHC patients were uninsured.
U.S. Health and Human Services officials say the stimulus money will provide care to nearly 3 million additional patients in the next two years, including roughly 1 million people without insurance. The added workload could cause burnout in hospitalists serving those institutions but who specialize in HM in part for the quality of life and scheduling perks that it affords.
"The ones that work with the CHCs would have more volume, but less chance of collecting on it, unless they have some arrangement to collect on that," Dr. Aguirre says. "There will be stress with increased volume."
Night-Shift Solutions
Karim Godamunne, MD, watched the moving images on the computer screen as he maneuvered the joystick with his hand. Using the computer screen as a guide, he traversed hallways, entered rooms, and zoomed the camera lens in on patients and equipment—all with a slight flick of the controller.
Sounds like a doc playing video games in the back office, right? But entertainment wasn’t what Dr. Godamunne, a hospitalist medical director with Eagle Hospital Physicians in Atlanta, was after. He was busy overseeing a study on admitting ED patients to St. Joseph’s Hospital in Atlanta, but he and the other participating physicians weren’t physically in the ED: With the help of a robot, a computer, and a secure high-speed Internet connection, the physicians obtained patients’ medical histories, performed physical exams, and admitted them in about the same time it normally takes on-site doctors.
“It’s like a video game, but much more. That’s how I describe it to people,” Dr. Godamunne says of the technology used in the study. “You have to be able to visualize what you’re doing.”
About 10 Eagle hospitalists participated in a pilot program last year that aimed to determine whether ED patients could be admitted by remote hospitalists using the RP-7 robot, which was developed by Santa Barbara, Calif.-based InTouch Health. Eagle was so pleased with the small study’s results that it began offering its remote-robot program to hospitals last October and anticipates deploying the first robot for HM work this spring. Eagle CEO Robert Young, MD, MPH, conceived the study and considers his company’s fledgling telemedicine program a solution to the hospitalist shortage, particularly for covering night shifts.
The Future of Round-the-Clock Rounds

Is telemedicine the right fit for your HM group? Consider the following pros and cons before you make a decision:
BENEFITS
- Less burnout for hospitalists, leading to higher productivity and longevity;
- Easier hospitalist recruitment, because applicants know they are less likely to work nights;
- Ability to cover multiple hospitals at one time, mitigating the hospitalist shortage that many areas face;
- Enhanced access for hospitalists and their patients to medical specialists;
- Higher technology comfort level among many patients; and
- Ability to expand hospital market area by employing telemedicine technology at outlying medical clinics and offices.
BARRIERS
- Reimbursement obstacles for Medicare and some private health insurers;
- Medical licensing can be time-consuming for telemedicine doctors who serve patients in multiple states;
- Difficulty getting buy-in from on-site doctors and primary-care physicians who are worried about liability and telemedicine doctors’ competence;
- Fear of lawsuits by patients treated via telemedicine;
- Startup technology and training costs; and
- Potential patient resistance to telemedicine.
“Eagle’s experience is that many hospitalists will be skeptical at first, but once they see it in action, not only does much of the resistance go away, but some become champions for its use,” Dr. Young says. “It is largely a matter of exposure to and experience in using the technology.”
While increasingly common in hospital ICUs and radiology departments, telemedicine is catching on more slowly in HM. Experts and practicing hospitalists cite reimbursement hiccups, a laborious medical licensing process, technology costs, physician and patient resistance, and risk aversion as the main reasons telemedicine isn’t embraced throughout HM. Some think it will take a concerted government effort to nudge hospitals and HM groups to buy into the technology.
Nevertheless, a growing number of physicians and administrators think telemedicine is inevitable, especially as the demand for HM services outpaces the supply. As in within the Eagle system, some hospitalists are positioning themselves to capitalize on the advancing technology.
The Future Isn’t Far

“I think it’s going to explode,” says Yomi Olusanya, MD, a hospitalist in rural Rolla, Mo., and founder of The Night Hospitalist Co., LLC, a startup that is busy developing a business model to provide nighttime hospital coverage via telemedicine. “I think with increased costs and the shortage of physicians, hospitalist groups are not going to have any choice but to find alternative ways of doing business. I really believe that.”
Dr. Olusanya envisions establishing a team of about 10 telehospitalists who would handle cross-coverage calls at multiple hospitals in multiple states. The hospitalists would use a mobile cart fashioned with a high-resolution, dual-focus video screen; a video camera; and diagnostic equipment, such as a digital stethoscope, to aid in physical exams. Hospital clients would be given a toll-free number to call to connect with a telehospitalist between
7 p.m. and 7 a.m., and on-site nurses would simply wheel the mobile cart into a patient’s room to begin the care. All overnight changes in medical management would be transmitted to the correct hospital floors for insertion into patients’ medical records. The Night Hospitalist plans to cover malpractice insurance for its physicians and charge a nightly rate, which would vary depending on the length of the contract.
The mobile cart costs between $20,000 and $30,000, and Dr. Olusanya is contemplating absorbing that expense just to get groups interested. At this point, he’s not promising prospective clients cost savings. Instead, he’s offering them a way to lighten the physician workload in order to increase productivity, job satisfaction and career longevity.
“We’re trying to sell the idea to hospitalists,” he says. “This is so new that I’m trying to figure out the best model.” After originally including hospital admissions in his business model, he ultimately decided to focus exclusively on cross-coverage calls and leave the admissions to an on-site physician. “At this point, I don’t see the telemed machine in the ED doing an admission of a new patient, because it becomes less efficient,” he explains.
Conversely, Eagle Hospital Physicians’ remote-robot program is designed to do hospital admissions. The RP-7 robot is mobile enough to aid in cross-coverage, but hospitals must be careful not to overburden the machine with floor calls because it takes the robot longer to travel around the hospital than it does for an on-site physician, says Betty Abbott, Eagle’s chief operating officer.
—Betty Abbott, COO, Eagle Hospital Physicians, Atlanta
Through the robot, which stands 5 feet 6 inches tall, a remote hospitalist can interact with a patient, the patient’s family, and the physician or nurse through a live, two-way audio and video system. The remote hospitalist can move the robot’s head to view charts and vital signs on monitors, zoom in to look at a patient’s pupils, and use several diagnostic tools with the help of an on-site health provider to conduct a patient exam, Abbott says. The remote hospitalist also can split the robot’s screen to show a patient X-rays, test results, videos, or other multimedia imaging.
“Certainly, using a robot to interact with patients takes some thought,” Abbott says. “Doctors have to be good at using the robot to act like a human being rather than simply a stationary screen in a room.”
The robot received high marks from patients, hospitalists, ED staff, and healthcare providers who participated in the pilot program at St. Joseph’s Hospital, according to the results of Eagle’s unpublished study. The technology is user-friendly enough that all types of healthcare providers can be trained to use it, says Dr. Godamunne. He designed and helped implement the study, and he found patients quickly adapted to the robot once they focused on the physicians’ faces on the screen.
Financial, Philosophical Hurdles
Hospitalists like Suman Narumanchi, MD, who leads the HM team at Resurrection Medical Center in Chicago, surmise most patients and their primary-care physicians expect doctors—not a robot or telemed cart—to physically be at the bedside in the hospital. As a result, if something goes wrong, the patient and their primary-care physician might respond with lawsuits. For that reason, “there has to be consistency in telemedicine,” Dr. Narumanchi says. “I just think at this point, it is probably a different level of care based on pure luck, because you don’t know who is going to be working that particular night.”
The concept raises interesting questions, says Eric Samson, DO, HM director for IN Compass Health Inc. in Greensboro, N.C. “Such as that of accountability and ownership of outcomes. On the other hand, it seems enticing to limit the multitude of distractions that occur through nighttime floor calls by implementing a cross-cover specialist fielding floor calls from a more-humane time zone—‘Hey, I’m working night call, but during bankers’ hours.’ ”

Protocols vary from hospital to hospital, and it will be difficult for telehospitalists who cover multiple facilities to learn the differences, says John Nelson, MD, FACP, co-founder and past president of SHM, and principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm in La Quinta, Calif. The job becomes even harder if one or more of the hospitals does not have electronic medical records (EMRs) and instead has to fax patient records to the telehospitalist, he says. Before hospitals invest in this expensive technology, a better solution might be to invent another way to address night coverage, such as allowing nonphysician providers (e.g., nurses) sign off on routine items that now require a doctor’s signature, he says.
Robert Cimasi, president of Health Capital Consultants, a St. Louis-based healthcare financial and economic consulting firm, says telemedicine’s ability to connect patients with distant specialists and allow hospitals to share doctors is tremendous, but agrees the technology is expensive and shouldn’t be entered into without a solid game plan, staff buy-in, and a long-term outlook. Although telemedicine proponents insist EMRs aren’t necessary, Cimasi advises hospitals serious about telemedicine to invest in EMRs, along with electronic order entry for their pharmacies and a secure computer network.
“A lot of hospitals aren’t going to have the capital capacity to do this without government help,” Cimasi explains. “The question is whether the political will is there to have a sustained period of investment.”
Eagle’s remote-robot program is less expensive than hiring a nocturnist or using a locum tenens physician, Dr. Young says. He predicts rural hospitals will benefit the most from his company’s program and other telemedicine services in the market because rural hospitals are most affected by the shortage of inpatient physicians. That might be the case, but if telemedicine is to ever make inroads among hospitalists, it will happen at urban hospitals first because they have the patient populations to support it, Dr. Nelson says.
“At larger hospitals where hospitalists are very busy admitting patients and busy checking patients already admitted, I could see using telemedicine to do the cross-coverage,” he says. “But in a small hospital, that wouldn’t make much sense, because there’s not enough patient volume.” TH
Lisa Ryan is a freelance writer based in New Jersey.
Karim Godamunne, MD, watched the moving images on the computer screen as he maneuvered the joystick with his hand. Using the computer screen as a guide, he traversed hallways, entered rooms, and zoomed the camera lens in on patients and equipment—all with a slight flick of the controller.
Sounds like a doc playing video games in the back office, right? But entertainment wasn’t what Dr. Godamunne, a hospitalist medical director with Eagle Hospital Physicians in Atlanta, was after. He was busy overseeing a study on admitting ED patients to St. Joseph’s Hospital in Atlanta, but he and the other participating physicians weren’t physically in the ED: With the help of a robot, a computer, and a secure high-speed Internet connection, the physicians obtained patients’ medical histories, performed physical exams, and admitted them in about the same time it normally takes on-site doctors.
“It’s like a video game, but much more. That’s how I describe it to people,” Dr. Godamunne says of the technology used in the study. “You have to be able to visualize what you’re doing.”
About 10 Eagle hospitalists participated in a pilot program last year that aimed to determine whether ED patients could be admitted by remote hospitalists using the RP-7 robot, which was developed by Santa Barbara, Calif.-based InTouch Health. Eagle was so pleased with the small study’s results that it began offering its remote-robot program to hospitals last October and anticipates deploying the first robot for HM work this spring. Eagle CEO Robert Young, MD, MPH, conceived the study and considers his company’s fledgling telemedicine program a solution to the hospitalist shortage, particularly for covering night shifts.
The Future of Round-the-Clock Rounds
Is telemedicine the right fit for your HM group? Consider the following pros and cons before you make a decision:
BENEFITS
- Less burnout for hospitalists, leading to higher productivity and longevity;
- Easier hospitalist recruitment, because applicants know they are less likely to work nights;
- Ability to cover multiple hospitals at one time, mitigating the hospitalist shortage that many areas face;
- Enhanced access for hospitalists and their patients to medical specialists;
- Higher technology comfort level among many patients; and
- Ability to expand hospital market area by employing telemedicine technology at outlying medical clinics and offices.
BARRIERS
- Reimbursement obstacles for Medicare and some private health insurers;
- Medical licensing can be time-consuming for telemedicine doctors who serve patients in multiple states;
- Difficulty getting buy-in from on-site doctors and primary-care physicians who are worried about liability and telemedicine doctors’ competence;
- Fear of lawsuits by patients treated via telemedicine;
- Startup technology and training costs; and
- Potential patient resistance to telemedicine.
“Eagle’s experience is that many hospitalists will be skeptical at first, but once they see it in action, not only does much of the resistance go away, but some become champions for its use,” Dr. Young says. “It is largely a matter of exposure to and experience in using the technology.”
While increasingly common in hospital ICUs and radiology departments, telemedicine is catching on more slowly in HM. Experts and practicing hospitalists cite reimbursement hiccups, a laborious medical licensing process, technology costs, physician and patient resistance, and risk aversion as the main reasons telemedicine isn’t embraced throughout HM. Some think it will take a concerted government effort to nudge hospitals and HM groups to buy into the technology.
Nevertheless, a growing number of physicians and administrators think telemedicine is inevitable, especially as the demand for HM services outpaces the supply. As in within the Eagle system, some hospitalists are positioning themselves to capitalize on the advancing technology.
The Future Isn’t Far
“I think it’s going to explode,” says Yomi Olusanya, MD, a hospitalist in rural Rolla, Mo., and founder of The Night Hospitalist Co., LLC, a startup that is busy developing a business model to provide nighttime hospital coverage via telemedicine. “I think with increased costs and the shortage of physicians, hospitalist groups are not going to have any choice but to find alternative ways of doing business. I really believe that.”
Dr. Olusanya envisions establishing a team of about 10 telehospitalists who would handle cross-coverage calls at multiple hospitals in multiple states. The hospitalists would use a mobile cart fashioned with a high-resolution, dual-focus video screen; a video camera; and diagnostic equipment, such as a digital stethoscope, to aid in physical exams. Hospital clients would be given a toll-free number to call to connect with a telehospitalist between
7 p.m. and 7 a.m., and on-site nurses would simply wheel the mobile cart into a patient’s room to begin the care. All overnight changes in medical management would be transmitted to the correct hospital floors for insertion into patients’ medical records. The Night Hospitalist plans to cover malpractice insurance for its physicians and charge a nightly rate, which would vary depending on the length of the contract.
The mobile cart costs between $20,000 and $30,000, and Dr. Olusanya is contemplating absorbing that expense just to get groups interested. At this point, he’s not promising prospective clients cost savings. Instead, he’s offering them a way to lighten the physician workload in order to increase productivity, job satisfaction and career longevity.
“We’re trying to sell the idea to hospitalists,” he says. “This is so new that I’m trying to figure out the best model.” After originally including hospital admissions in his business model, he ultimately decided to focus exclusively on cross-coverage calls and leave the admissions to an on-site physician. “At this point, I don’t see the telemed machine in the ED doing an admission of a new patient, because it becomes less efficient,” he explains.
Conversely, Eagle Hospital Physicians’ remote-robot program is designed to do hospital admissions. The RP-7 robot is mobile enough to aid in cross-coverage, but hospitals must be careful not to overburden the machine with floor calls because it takes the robot longer to travel around the hospital than it does for an on-site physician, says Betty Abbott, Eagle’s chief operating officer.
—Betty Abbott, COO, Eagle Hospital Physicians, Atlanta
Through the robot, which stands 5 feet 6 inches tall, a remote hospitalist can interact with a patient, the patient’s family, and the physician or nurse through a live, two-way audio and video system. The remote hospitalist can move the robot’s head to view charts and vital signs on monitors, zoom in to look at a patient’s pupils, and use several diagnostic tools with the help of an on-site health provider to conduct a patient exam, Abbott says. The remote hospitalist also can split the robot’s screen to show a patient X-rays, test results, videos, or other multimedia imaging.
“Certainly, using a robot to interact with patients takes some thought,” Abbott says. “Doctors have to be good at using the robot to act like a human being rather than simply a stationary screen in a room.”
The robot received high marks from patients, hospitalists, ED staff, and healthcare providers who participated in the pilot program at St. Joseph’s Hospital, according to the results of Eagle’s unpublished study. The technology is user-friendly enough that all types of healthcare providers can be trained to use it, says Dr. Godamunne. He designed and helped implement the study, and he found patients quickly adapted to the robot once they focused on the physicians’ faces on the screen.
Financial, Philosophical Hurdles
Hospitalists like Suman Narumanchi, MD, who leads the HM team at Resurrection Medical Center in Chicago, surmise most patients and their primary-care physicians expect doctors—not a robot or telemed cart—to physically be at the bedside in the hospital. As a result, if something goes wrong, the patient and their primary-care physician might respond with lawsuits. For that reason, “there has to be consistency in telemedicine,” Dr. Narumanchi says. “I just think at this point, it is probably a different level of care based on pure luck, because you don’t know who is going to be working that particular night.”
The concept raises interesting questions, says Eric Samson, DO, HM director for IN Compass Health Inc. in Greensboro, N.C. “Such as that of accountability and ownership of outcomes. On the other hand, it seems enticing to limit the multitude of distractions that occur through nighttime floor calls by implementing a cross-cover specialist fielding floor calls from a more-humane time zone—‘Hey, I’m working night call, but during bankers’ hours.’ ”
Protocols vary from hospital to hospital, and it will be difficult for telehospitalists who cover multiple facilities to learn the differences, says John Nelson, MD, FACP, co-founder and past president of SHM, and principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm in La Quinta, Calif. The job becomes even harder if one or more of the hospitals does not have electronic medical records (EMRs) and instead has to fax patient records to the telehospitalist, he says. Before hospitals invest in this expensive technology, a better solution might be to invent another way to address night coverage, such as allowing nonphysician providers (e.g., nurses) sign off on routine items that now require a doctor’s signature, he says.
Robert Cimasi, president of Health Capital Consultants, a St. Louis-based healthcare financial and economic consulting firm, says telemedicine’s ability to connect patients with distant specialists and allow hospitals to share doctors is tremendous, but agrees the technology is expensive and shouldn’t be entered into without a solid game plan, staff buy-in, and a long-term outlook. Although telemedicine proponents insist EMRs aren’t necessary, Cimasi advises hospitals serious about telemedicine to invest in EMRs, along with electronic order entry for their pharmacies and a secure computer network.
“A lot of hospitals aren’t going to have the capital capacity to do this without government help,” Cimasi explains. “The question is whether the political will is there to have a sustained period of investment.”
Eagle’s remote-robot program is less expensive than hiring a nocturnist or using a locum tenens physician, Dr. Young says. He predicts rural hospitals will benefit the most from his company’s program and other telemedicine services in the market because rural hospitals are most affected by the shortage of inpatient physicians. That might be the case, but if telemedicine is to ever make inroads among hospitalists, it will happen at urban hospitals first because they have the patient populations to support it, Dr. Nelson says.
“At larger hospitals where hospitalists are very busy admitting patients and busy checking patients already admitted, I could see using telemedicine to do the cross-coverage,” he says. “But in a small hospital, that wouldn’t make much sense, because there’s not enough patient volume.” TH
Lisa Ryan is a freelance writer based in New Jersey.
Karim Godamunne, MD, watched the moving images on the computer screen as he maneuvered the joystick with his hand. Using the computer screen as a guide, he traversed hallways, entered rooms, and zoomed the camera lens in on patients and equipment—all with a slight flick of the controller.
Sounds like a doc playing video games in the back office, right? But entertainment wasn’t what Dr. Godamunne, a hospitalist medical director with Eagle Hospital Physicians in Atlanta, was after. He was busy overseeing a study on admitting ED patients to St. Joseph’s Hospital in Atlanta, but he and the other participating physicians weren’t physically in the ED: With the help of a robot, a computer, and a secure high-speed Internet connection, the physicians obtained patients’ medical histories, performed physical exams, and admitted them in about the same time it normally takes on-site doctors.
“It’s like a video game, but much more. That’s how I describe it to people,” Dr. Godamunne says of the technology used in the study. “You have to be able to visualize what you’re doing.”
About 10 Eagle hospitalists participated in a pilot program last year that aimed to determine whether ED patients could be admitted by remote hospitalists using the RP-7 robot, which was developed by Santa Barbara, Calif.-based InTouch Health. Eagle was so pleased with the small study’s results that it began offering its remote-robot program to hospitals last October and anticipates deploying the first robot for HM work this spring. Eagle CEO Robert Young, MD, MPH, conceived the study and considers his company’s fledgling telemedicine program a solution to the hospitalist shortage, particularly for covering night shifts.
The Future of Round-the-Clock Rounds
Is telemedicine the right fit for your HM group? Consider the following pros and cons before you make a decision:
BENEFITS
- Less burnout for hospitalists, leading to higher productivity and longevity;
- Easier hospitalist recruitment, because applicants know they are less likely to work nights;
- Ability to cover multiple hospitals at one time, mitigating the hospitalist shortage that many areas face;
- Enhanced access for hospitalists and their patients to medical specialists;
- Higher technology comfort level among many patients; and
- Ability to expand hospital market area by employing telemedicine technology at outlying medical clinics and offices.
BARRIERS
- Reimbursement obstacles for Medicare and some private health insurers;
- Medical licensing can be time-consuming for telemedicine doctors who serve patients in multiple states;
- Difficulty getting buy-in from on-site doctors and primary-care physicians who are worried about liability and telemedicine doctors’ competence;
- Fear of lawsuits by patients treated via telemedicine;
- Startup technology and training costs; and
- Potential patient resistance to telemedicine.
“Eagle’s experience is that many hospitalists will be skeptical at first, but once they see it in action, not only does much of the resistance go away, but some become champions for its use,” Dr. Young says. “It is largely a matter of exposure to and experience in using the technology.”
While increasingly common in hospital ICUs and radiology departments, telemedicine is catching on more slowly in HM. Experts and practicing hospitalists cite reimbursement hiccups, a laborious medical licensing process, technology costs, physician and patient resistance, and risk aversion as the main reasons telemedicine isn’t embraced throughout HM. Some think it will take a concerted government effort to nudge hospitals and HM groups to buy into the technology.
Nevertheless, a growing number of physicians and administrators think telemedicine is inevitable, especially as the demand for HM services outpaces the supply. As in within the Eagle system, some hospitalists are positioning themselves to capitalize on the advancing technology.
The Future Isn’t Far
“I think it’s going to explode,” says Yomi Olusanya, MD, a hospitalist in rural Rolla, Mo., and founder of The Night Hospitalist Co., LLC, a startup that is busy developing a business model to provide nighttime hospital coverage via telemedicine. “I think with increased costs and the shortage of physicians, hospitalist groups are not going to have any choice but to find alternative ways of doing business. I really believe that.”
Dr. Olusanya envisions establishing a team of about 10 telehospitalists who would handle cross-coverage calls at multiple hospitals in multiple states. The hospitalists would use a mobile cart fashioned with a high-resolution, dual-focus video screen; a video camera; and diagnostic equipment, such as a digital stethoscope, to aid in physical exams. Hospital clients would be given a toll-free number to call to connect with a telehospitalist between
7 p.m. and 7 a.m., and on-site nurses would simply wheel the mobile cart into a patient’s room to begin the care. All overnight changes in medical management would be transmitted to the correct hospital floors for insertion into patients’ medical records. The Night Hospitalist plans to cover malpractice insurance for its physicians and charge a nightly rate, which would vary depending on the length of the contract.
The mobile cart costs between $20,000 and $30,000, and Dr. Olusanya is contemplating absorbing that expense just to get groups interested. At this point, he’s not promising prospective clients cost savings. Instead, he’s offering them a way to lighten the physician workload in order to increase productivity, job satisfaction and career longevity.
“We’re trying to sell the idea to hospitalists,” he says. “This is so new that I’m trying to figure out the best model.” After originally including hospital admissions in his business model, he ultimately decided to focus exclusively on cross-coverage calls and leave the admissions to an on-site physician. “At this point, I don’t see the telemed machine in the ED doing an admission of a new patient, because it becomes less efficient,” he explains.
Conversely, Eagle Hospital Physicians’ remote-robot program is designed to do hospital admissions. The RP-7 robot is mobile enough to aid in cross-coverage, but hospitals must be careful not to overburden the machine with floor calls because it takes the robot longer to travel around the hospital than it does for an on-site physician, says Betty Abbott, Eagle’s chief operating officer.
—Betty Abbott, COO, Eagle Hospital Physicians, Atlanta
Through the robot, which stands 5 feet 6 inches tall, a remote hospitalist can interact with a patient, the patient’s family, and the physician or nurse through a live, two-way audio and video system. The remote hospitalist can move the robot’s head to view charts and vital signs on monitors, zoom in to look at a patient’s pupils, and use several diagnostic tools with the help of an on-site health provider to conduct a patient exam, Abbott says. The remote hospitalist also can split the robot’s screen to show a patient X-rays, test results, videos, or other multimedia imaging.
“Certainly, using a robot to interact with patients takes some thought,” Abbott says. “Doctors have to be good at using the robot to act like a human being rather than simply a stationary screen in a room.”
The robot received high marks from patients, hospitalists, ED staff, and healthcare providers who participated in the pilot program at St. Joseph’s Hospital, according to the results of Eagle’s unpublished study. The technology is user-friendly enough that all types of healthcare providers can be trained to use it, says Dr. Godamunne. He designed and helped implement the study, and he found patients quickly adapted to the robot once they focused on the physicians’ faces on the screen.
Financial, Philosophical Hurdles
Hospitalists like Suman Narumanchi, MD, who leads the HM team at Resurrection Medical Center in Chicago, surmise most patients and their primary-care physicians expect doctors—not a robot or telemed cart—to physically be at the bedside in the hospital. As a result, if something goes wrong, the patient and their primary-care physician might respond with lawsuits. For that reason, “there has to be consistency in telemedicine,” Dr. Narumanchi says. “I just think at this point, it is probably a different level of care based on pure luck, because you don’t know who is going to be working that particular night.”
The concept raises interesting questions, says Eric Samson, DO, HM director for IN Compass Health Inc. in Greensboro, N.C. “Such as that of accountability and ownership of outcomes. On the other hand, it seems enticing to limit the multitude of distractions that occur through nighttime floor calls by implementing a cross-cover specialist fielding floor calls from a more-humane time zone—‘Hey, I’m working night call, but during bankers’ hours.’ ”
Protocols vary from hospital to hospital, and it will be difficult for telehospitalists who cover multiple facilities to learn the differences, says John Nelson, MD, FACP, co-founder and past president of SHM, and principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm in La Quinta, Calif. The job becomes even harder if one or more of the hospitals does not have electronic medical records (EMRs) and instead has to fax patient records to the telehospitalist, he says. Before hospitals invest in this expensive technology, a better solution might be to invent another way to address night coverage, such as allowing nonphysician providers (e.g., nurses) sign off on routine items that now require a doctor’s signature, he says.
Robert Cimasi, president of Health Capital Consultants, a St. Louis-based healthcare financial and economic consulting firm, says telemedicine’s ability to connect patients with distant specialists and allow hospitals to share doctors is tremendous, but agrees the technology is expensive and shouldn’t be entered into without a solid game plan, staff buy-in, and a long-term outlook. Although telemedicine proponents insist EMRs aren’t necessary, Cimasi advises hospitals serious about telemedicine to invest in EMRs, along with electronic order entry for their pharmacies and a secure computer network.
“A lot of hospitals aren’t going to have the capital capacity to do this without government help,” Cimasi explains. “The question is whether the political will is there to have a sustained period of investment.”
Eagle’s remote-robot program is less expensive than hiring a nocturnist or using a locum tenens physician, Dr. Young says. He predicts rural hospitals will benefit the most from his company’s program and other telemedicine services in the market because rural hospitals are most affected by the shortage of inpatient physicians. That might be the case, but if telemedicine is to ever make inroads among hospitalists, it will happen at urban hospitals first because they have the patient populations to support it, Dr. Nelson says.
“At larger hospitals where hospitalists are very busy admitting patients and busy checking patients already admitted, I could see using telemedicine to do the cross-coverage,” he says. “But in a small hospital, that wouldn’t make much sense, because there’s not enough patient volume.” TH
Lisa Ryan is a freelance writer based in New Jersey.
Stage 2 Sarcoidosis
A 50‐year‐old man presented to the emergency department with progressive shortness of breath for 6 months. He described a dry cough, left‐sided chest pain, malaise, night sweats, and a 15‐pound weight loss. The patient had never smoked cigarettes, but he had been exposed to asbestos and wood dust when working at a sawmill. His physical examination was remarkable for decreased breath sounds at the left lung base. The admission blood tests were within normal limits. Chest radiography and a computed tomography (CT) scan of the chest were performed (the CT scan is shown in Figures 1 and 2). The CT scan showed a left pleural effusion with subpleural and peribronchovascular nodules. Also demonstrated on the CT scan were bilateral hilar and mediastinal lymphadenopathies with faint central calcification. As the left‐sided pleural effusion was initially suspected to be malignant, a thoracentesis was performed, and it revealed an exudative effusion. The total white cell count in fluid was 2100/L (lymphocytes, 76%), and cultures for aerobic and anaerobic bacteria, acid fasting bacilli, and fungi were negative. Cytology was negative for malignant cells. On the basis of the findings in the lung parenchyma and the presence of mediastinal lymphadenopathies, fiber‐optic bronchoscopy with bronchoalveolar lavage, protected specimen brushing, transbronchial needle aspiration, and transbronchial biopsies were performed. Mediastinal lymph node cytology was negative for malignant cells, whereas transbronchial biopsies showed noncaseating granulomas (Figure 3). At that time, our differential diagnoses of noncaseating granulomas included mycobacterium infection (although this usually presents caseating granulomas), berylliosis, histoplasmosis, and sarcoidosis. The tuberculin skin test (purified protein derivative) and serology for human immunodeficiency virus were negative. Bronchoalveolar lavage and cultures of lung tissue biopsies as well as needle aspiration from mediastinal lymph nodes were negative for mycobacterial, fungal, and bacterial organisms. The beryllium lymphocyte proliferation test was normal. Serologic antibodies for Aspergillus, Blastomyces, Coccidioides, and Histoplasma were negative. The urinary Histoplasma antigen was negative as well. The Department of Infectious Diseases was consulted, and an empirical treatment for histoplasmosis with itraconazole was started on the basis of the residence of the patient and the presence of noncaseating granulomas. After 1 month of antifungal treatment, there was no significant improvement. Video‐assisted thoracoscopic surgery with pleural biopsy was performed because of persistent pleural effusion and concern about an underlying infectious or malignant process. Pleural biopsies showed noncaseating granulomas (Figure 4). Pleural fluid was sent for adenosine deaminase (17 U/L) and flow cytometry (CD4/CD8 2.71). Cultures and cytology remained negative. A diagnosis of stage 2 sarcoidosis with pleural involvement was made, and treatment with prednisone was started.
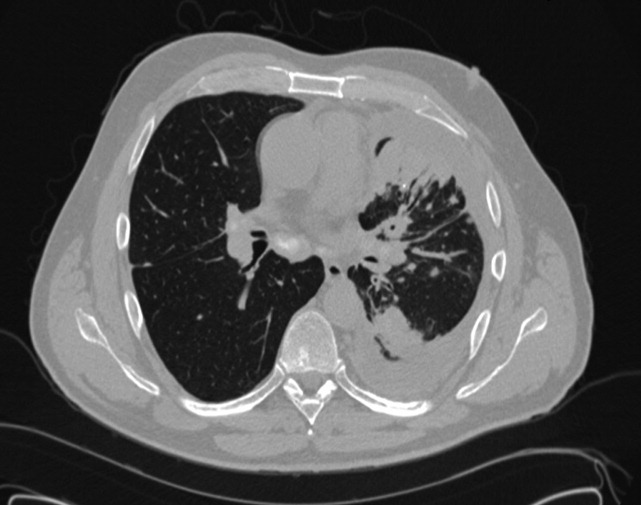
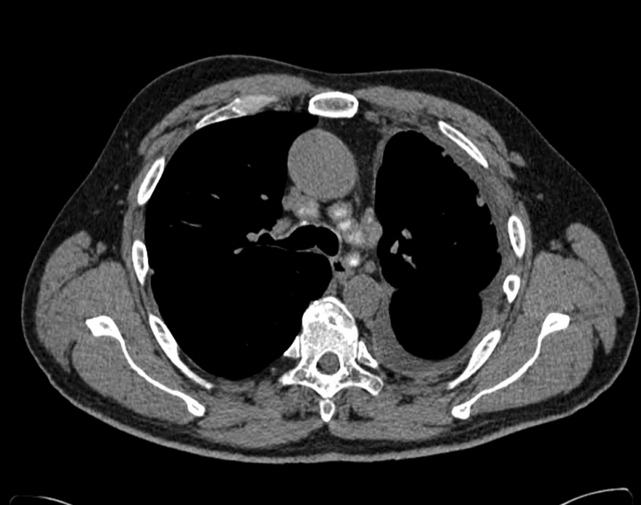
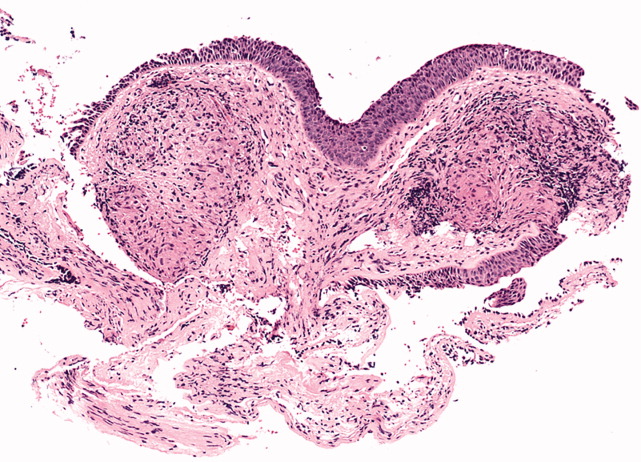

Discussion
The overall prevalence of pleural involvement in sarcoidosis is about 3%. Patients with pleural sarcoidosis tend to be between 30 and 50 years of age, in contrast to the usual presentation of sarcoidosis between 20 and 30 years of age. The most common forms of pleural involvement are pleural effusions, pneumothorax, pleural thickening, and pleural nodules.1 Most effusions are usually small or modest in size, with few reports describing massive effusions.2 Recurrent pleural and pericardial effusions due to sarcoidosis have been reported as well.3 The fluid is typically a lymphocytic exudate, and almost all cases describe a CD4 predominant lymphocytic effusion with CD4/CD8 ratios ranging from 2.35 to 8.6.1 The presence of bloody pleural effusions in sarcoidosis most likely represents the rupture of small vessels that are compressed or infiltrated by granulomas.4
The majority of patients with reported sarcoid pleural effusions have stage 2 disease. With the progression of parenchymal disease, the prevalence of pleural effusions decreases, whereas pleural thickening and pneumothorax increase.5 It is important to emphasize that 40% of pleural effusions in sarcoidosis may be due to other causes, such as tuberculosis and mesothelioma. Our patient was initially treated with itraconazole as histoplasmosis is most prevalent in the Central and Eastern United States, especially in Ohio River valleys, where this patient lived.
The prevalence of a pneumothorax in sarcoidosis is up to 4%.1 Pleural thickening can be demonstrated in 11% to 71% of patients with pleural sarcoidosis, and 10% to 20% of these cases have thickening without effusion. Detection of subpleural nodules and cysts has been possible since the introduction of high‐resolution CT scans. Their prevalence in sarcoidosis ranges from 22% to 76%, and they are often described as masses that correspond to nodules seen in both parietal and visceral surfaces. Hilar or mediastinal lymphadenopathy is present on CT in 47% to 94% of patients with sarcoidosis. Lymph node enlargement is usually bilateral, most commonly with right‐sided predominance. The most involved stations are the right lower paratracheal, right hilar, subcarinal, aortopulmonary window, and right interlobar stations. Nodal calcification is noted in 53% with eggshell calcification present in 9%. The enlargement of internal mammary and pericardial lymph nodes requires the exclusion of lymphoma.6
The management of pleural sarcoidosis should be individualized because a majority of these effusions resolve spontaneously in 1 to 3 months.5 There have been reports of resolution in 2 weeks with steroid therapy. Incomplete resolution of the pleural effusions with progression to chronic pleural thickening or a trapped lung has been reported. There is agreement that oral corticosteroid treatment should be considered in patients with severe persistent or progressively worsening respiratory symptoms or declining lung function. Severe symptoms can be considered as those that interfere with essential aspects of the patient's daily life.7 The initial dosage of oral prednisone recommended by the American Thoracic Society, the European Respiratory Society, and the World Association of Sarcoidosis and Other Granulomatous Disorders guidelines is 20 to 40 mg/day.8 Further evaluation is recommended after 1 to 3 months. If the patient responds, the dose should be reduced gradually to a maintenance dose, such as 5 to 15 mg/day of prednisolone. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and Other Granulomatous Disorders guidelines advise treatment for at least 1 year. Immunosuppressive, cytotoxic, and immunomodulatory agents have been used to treat patients failing or experiencing adverse effects of steroids. Favorable responses have been reported with methotrexate, leflunomide, azathioprine, cyclophosphamide, chlorambucil, cyclosporine A, antimalarials, tumor necrosis factor inhibitors, and thalidomide. Because of potential serious toxicities associated with cyclophosphamide and chlorambucil, these agents are not recommended.9
Our patient presented with pleural sarcoidosis with a pleural effusion and nodules. Treatment with 20 mg of prednisone daily was started initially. Four weeks after discharge, he was still dyspneic and had persistent left pleural effusion. He also had gained a significant amount of weight and developed bilateral lower extremity edema; these were thought to be secondary to prednisone treatment. Steroids were subsequently tapered, and leflunomide was started. His symptoms improved dramatically after 1 month of treatment with leflunomide and steroids, and 3 months later, his pleural effusion had completely resolved.
- ,.Pleural involvement in sarcoidosis.Curr Opin Pulm Med.2000;6(5):455–468.
- ,.Pleural sarcoidosis with massive effusion and lung entrapment.Kans Med.1990;91(4):103–105.
- ,,,,.Recurrent pleural and pericardial effusions due to sarcoidosis.PLoS Med.2005;2(3):e63.
- ,,,,.Pulmonary sarcoidosis with associated bloody pleurisy.Intern Med.2002;41(11):1021–1023.
- ,,,,.Pleural effusions in a series of 181 outpatients with sarcoidosis.Chest.2006;129(6):1599–1604.
- ,,,,.Imaging in sarcoidosis.Semin Respir Crit Care Med.2007;28(1):102–120.
- .Guidelines for the use of corticosteroids in the treatment of pulmonary sarcoidosis.Drugs.2007;67(8):1139–1147.
- ,,, et al.ATS/ERS/WASOG statement on sarcoidosis.Sarcoidosis Vasc Diffuse Lung Dis.1999;16(2):149–173.
- ,,,.Pulmonary sarcoidosis.Semin Respir Crit Care Med.2007;28(1):53–74.
A 50‐year‐old man presented to the emergency department with progressive shortness of breath for 6 months. He described a dry cough, left‐sided chest pain, malaise, night sweats, and a 15‐pound weight loss. The patient had never smoked cigarettes, but he had been exposed to asbestos and wood dust when working at a sawmill. His physical examination was remarkable for decreased breath sounds at the left lung base. The admission blood tests were within normal limits. Chest radiography and a computed tomography (CT) scan of the chest were performed (the CT scan is shown in Figures 1 and 2). The CT scan showed a left pleural effusion with subpleural and peribronchovascular nodules. Also demonstrated on the CT scan were bilateral hilar and mediastinal lymphadenopathies with faint central calcification. As the left‐sided pleural effusion was initially suspected to be malignant, a thoracentesis was performed, and it revealed an exudative effusion. The total white cell count in fluid was 2100/L (lymphocytes, 76%), and cultures for aerobic and anaerobic bacteria, acid fasting bacilli, and fungi were negative. Cytology was negative for malignant cells. On the basis of the findings in the lung parenchyma and the presence of mediastinal lymphadenopathies, fiber‐optic bronchoscopy with bronchoalveolar lavage, protected specimen brushing, transbronchial needle aspiration, and transbronchial biopsies were performed. Mediastinal lymph node cytology was negative for malignant cells, whereas transbronchial biopsies showed noncaseating granulomas (Figure 3). At that time, our differential diagnoses of noncaseating granulomas included mycobacterium infection (although this usually presents caseating granulomas), berylliosis, histoplasmosis, and sarcoidosis. The tuberculin skin test (purified protein derivative) and serology for human immunodeficiency virus were negative. Bronchoalveolar lavage and cultures of lung tissue biopsies as well as needle aspiration from mediastinal lymph nodes were negative for mycobacterial, fungal, and bacterial organisms. The beryllium lymphocyte proliferation test was normal. Serologic antibodies for Aspergillus, Blastomyces, Coccidioides, and Histoplasma were negative. The urinary Histoplasma antigen was negative as well. The Department of Infectious Diseases was consulted, and an empirical treatment for histoplasmosis with itraconazole was started on the basis of the residence of the patient and the presence of noncaseating granulomas. After 1 month of antifungal treatment, there was no significant improvement. Video‐assisted thoracoscopic surgery with pleural biopsy was performed because of persistent pleural effusion and concern about an underlying infectious or malignant process. Pleural biopsies showed noncaseating granulomas (Figure 4). Pleural fluid was sent for adenosine deaminase (17 U/L) and flow cytometry (CD4/CD8 2.71). Cultures and cytology remained negative. A diagnosis of stage 2 sarcoidosis with pleural involvement was made, and treatment with prednisone was started.
Discussion
The overall prevalence of pleural involvement in sarcoidosis is about 3%. Patients with pleural sarcoidosis tend to be between 30 and 50 years of age, in contrast to the usual presentation of sarcoidosis between 20 and 30 years of age. The most common forms of pleural involvement are pleural effusions, pneumothorax, pleural thickening, and pleural nodules.1 Most effusions are usually small or modest in size, with few reports describing massive effusions.2 Recurrent pleural and pericardial effusions due to sarcoidosis have been reported as well.3 The fluid is typically a lymphocytic exudate, and almost all cases describe a CD4 predominant lymphocytic effusion with CD4/CD8 ratios ranging from 2.35 to 8.6.1 The presence of bloody pleural effusions in sarcoidosis most likely represents the rupture of small vessels that are compressed or infiltrated by granulomas.4
The majority of patients with reported sarcoid pleural effusions have stage 2 disease. With the progression of parenchymal disease, the prevalence of pleural effusions decreases, whereas pleural thickening and pneumothorax increase.5 It is important to emphasize that 40% of pleural effusions in sarcoidosis may be due to other causes, such as tuberculosis and mesothelioma. Our patient was initially treated with itraconazole as histoplasmosis is most prevalent in the Central and Eastern United States, especially in Ohio River valleys, where this patient lived.
The prevalence of a pneumothorax in sarcoidosis is up to 4%.1 Pleural thickening can be demonstrated in 11% to 71% of patients with pleural sarcoidosis, and 10% to 20% of these cases have thickening without effusion. Detection of subpleural nodules and cysts has been possible since the introduction of high‐resolution CT scans. Their prevalence in sarcoidosis ranges from 22% to 76%, and they are often described as masses that correspond to nodules seen in both parietal and visceral surfaces. Hilar or mediastinal lymphadenopathy is present on CT in 47% to 94% of patients with sarcoidosis. Lymph node enlargement is usually bilateral, most commonly with right‐sided predominance. The most involved stations are the right lower paratracheal, right hilar, subcarinal, aortopulmonary window, and right interlobar stations. Nodal calcification is noted in 53% with eggshell calcification present in 9%. The enlargement of internal mammary and pericardial lymph nodes requires the exclusion of lymphoma.6
The management of pleural sarcoidosis should be individualized because a majority of these effusions resolve spontaneously in 1 to 3 months.5 There have been reports of resolution in 2 weeks with steroid therapy. Incomplete resolution of the pleural effusions with progression to chronic pleural thickening or a trapped lung has been reported. There is agreement that oral corticosteroid treatment should be considered in patients with severe persistent or progressively worsening respiratory symptoms or declining lung function. Severe symptoms can be considered as those that interfere with essential aspects of the patient's daily life.7 The initial dosage of oral prednisone recommended by the American Thoracic Society, the European Respiratory Society, and the World Association of Sarcoidosis and Other Granulomatous Disorders guidelines is 20 to 40 mg/day.8 Further evaluation is recommended after 1 to 3 months. If the patient responds, the dose should be reduced gradually to a maintenance dose, such as 5 to 15 mg/day of prednisolone. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and Other Granulomatous Disorders guidelines advise treatment for at least 1 year. Immunosuppressive, cytotoxic, and immunomodulatory agents have been used to treat patients failing or experiencing adverse effects of steroids. Favorable responses have been reported with methotrexate, leflunomide, azathioprine, cyclophosphamide, chlorambucil, cyclosporine A, antimalarials, tumor necrosis factor inhibitors, and thalidomide. Because of potential serious toxicities associated with cyclophosphamide and chlorambucil, these agents are not recommended.9
Our patient presented with pleural sarcoidosis with a pleural effusion and nodules. Treatment with 20 mg of prednisone daily was started initially. Four weeks after discharge, he was still dyspneic and had persistent left pleural effusion. He also had gained a significant amount of weight and developed bilateral lower extremity edema; these were thought to be secondary to prednisone treatment. Steroids were subsequently tapered, and leflunomide was started. His symptoms improved dramatically after 1 month of treatment with leflunomide and steroids, and 3 months later, his pleural effusion had completely resolved.
A 50‐year‐old man presented to the emergency department with progressive shortness of breath for 6 months. He described a dry cough, left‐sided chest pain, malaise, night sweats, and a 15‐pound weight loss. The patient had never smoked cigarettes, but he had been exposed to asbestos and wood dust when working at a sawmill. His physical examination was remarkable for decreased breath sounds at the left lung base. The admission blood tests were within normal limits. Chest radiography and a computed tomography (CT) scan of the chest were performed (the CT scan is shown in Figures 1 and 2). The CT scan showed a left pleural effusion with subpleural and peribronchovascular nodules. Also demonstrated on the CT scan were bilateral hilar and mediastinal lymphadenopathies with faint central calcification. As the left‐sided pleural effusion was initially suspected to be malignant, a thoracentesis was performed, and it revealed an exudative effusion. The total white cell count in fluid was 2100/L (lymphocytes, 76%), and cultures for aerobic and anaerobic bacteria, acid fasting bacilli, and fungi were negative. Cytology was negative for malignant cells. On the basis of the findings in the lung parenchyma and the presence of mediastinal lymphadenopathies, fiber‐optic bronchoscopy with bronchoalveolar lavage, protected specimen brushing, transbronchial needle aspiration, and transbronchial biopsies were performed. Mediastinal lymph node cytology was negative for malignant cells, whereas transbronchial biopsies showed noncaseating granulomas (Figure 3). At that time, our differential diagnoses of noncaseating granulomas included mycobacterium infection (although this usually presents caseating granulomas), berylliosis, histoplasmosis, and sarcoidosis. The tuberculin skin test (purified protein derivative) and serology for human immunodeficiency virus were negative. Bronchoalveolar lavage and cultures of lung tissue biopsies as well as needle aspiration from mediastinal lymph nodes were negative for mycobacterial, fungal, and bacterial organisms. The beryllium lymphocyte proliferation test was normal. Serologic antibodies for Aspergillus, Blastomyces, Coccidioides, and Histoplasma were negative. The urinary Histoplasma antigen was negative as well. The Department of Infectious Diseases was consulted, and an empirical treatment for histoplasmosis with itraconazole was started on the basis of the residence of the patient and the presence of noncaseating granulomas. After 1 month of antifungal treatment, there was no significant improvement. Video‐assisted thoracoscopic surgery with pleural biopsy was performed because of persistent pleural effusion and concern about an underlying infectious or malignant process. Pleural biopsies showed noncaseating granulomas (Figure 4). Pleural fluid was sent for adenosine deaminase (17 U/L) and flow cytometry (CD4/CD8 2.71). Cultures and cytology remained negative. A diagnosis of stage 2 sarcoidosis with pleural involvement was made, and treatment with prednisone was started.
Discussion
The overall prevalence of pleural involvement in sarcoidosis is about 3%. Patients with pleural sarcoidosis tend to be between 30 and 50 years of age, in contrast to the usual presentation of sarcoidosis between 20 and 30 years of age. The most common forms of pleural involvement are pleural effusions, pneumothorax, pleural thickening, and pleural nodules.1 Most effusions are usually small or modest in size, with few reports describing massive effusions.2 Recurrent pleural and pericardial effusions due to sarcoidosis have been reported as well.3 The fluid is typically a lymphocytic exudate, and almost all cases describe a CD4 predominant lymphocytic effusion with CD4/CD8 ratios ranging from 2.35 to 8.6.1 The presence of bloody pleural effusions in sarcoidosis most likely represents the rupture of small vessels that are compressed or infiltrated by granulomas.4
The majority of patients with reported sarcoid pleural effusions have stage 2 disease. With the progression of parenchymal disease, the prevalence of pleural effusions decreases, whereas pleural thickening and pneumothorax increase.5 It is important to emphasize that 40% of pleural effusions in sarcoidosis may be due to other causes, such as tuberculosis and mesothelioma. Our patient was initially treated with itraconazole as histoplasmosis is most prevalent in the Central and Eastern United States, especially in Ohio River valleys, where this patient lived.
The prevalence of a pneumothorax in sarcoidosis is up to 4%.1 Pleural thickening can be demonstrated in 11% to 71% of patients with pleural sarcoidosis, and 10% to 20% of these cases have thickening without effusion. Detection of subpleural nodules and cysts has been possible since the introduction of high‐resolution CT scans. Their prevalence in sarcoidosis ranges from 22% to 76%, and they are often described as masses that correspond to nodules seen in both parietal and visceral surfaces. Hilar or mediastinal lymphadenopathy is present on CT in 47% to 94% of patients with sarcoidosis. Lymph node enlargement is usually bilateral, most commonly with right‐sided predominance. The most involved stations are the right lower paratracheal, right hilar, subcarinal, aortopulmonary window, and right interlobar stations. Nodal calcification is noted in 53% with eggshell calcification present in 9%. The enlargement of internal mammary and pericardial lymph nodes requires the exclusion of lymphoma.6
The management of pleural sarcoidosis should be individualized because a majority of these effusions resolve spontaneously in 1 to 3 months.5 There have been reports of resolution in 2 weeks with steroid therapy. Incomplete resolution of the pleural effusions with progression to chronic pleural thickening or a trapped lung has been reported. There is agreement that oral corticosteroid treatment should be considered in patients with severe persistent or progressively worsening respiratory symptoms or declining lung function. Severe symptoms can be considered as those that interfere with essential aspects of the patient's daily life.7 The initial dosage of oral prednisone recommended by the American Thoracic Society, the European Respiratory Society, and the World Association of Sarcoidosis and Other Granulomatous Disorders guidelines is 20 to 40 mg/day.8 Further evaluation is recommended after 1 to 3 months. If the patient responds, the dose should be reduced gradually to a maintenance dose, such as 5 to 15 mg/day of prednisolone. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and Other Granulomatous Disorders guidelines advise treatment for at least 1 year. Immunosuppressive, cytotoxic, and immunomodulatory agents have been used to treat patients failing or experiencing adverse effects of steroids. Favorable responses have been reported with methotrexate, leflunomide, azathioprine, cyclophosphamide, chlorambucil, cyclosporine A, antimalarials, tumor necrosis factor inhibitors, and thalidomide. Because of potential serious toxicities associated with cyclophosphamide and chlorambucil, these agents are not recommended.9
Our patient presented with pleural sarcoidosis with a pleural effusion and nodules. Treatment with 20 mg of prednisone daily was started initially. Four weeks after discharge, he was still dyspneic and had persistent left pleural effusion. He also had gained a significant amount of weight and developed bilateral lower extremity edema; these were thought to be secondary to prednisone treatment. Steroids were subsequently tapered, and leflunomide was started. His symptoms improved dramatically after 1 month of treatment with leflunomide and steroids, and 3 months later, his pleural effusion had completely resolved.
- ,.Pleural involvement in sarcoidosis.Curr Opin Pulm Med.2000;6(5):455–468.
- ,.Pleural sarcoidosis with massive effusion and lung entrapment.Kans Med.1990;91(4):103–105.
- ,,,,.Recurrent pleural and pericardial effusions due to sarcoidosis.PLoS Med.2005;2(3):e63.
- ,,,,.Pulmonary sarcoidosis with associated bloody pleurisy.Intern Med.2002;41(11):1021–1023.
- ,,,,.Pleural effusions in a series of 181 outpatients with sarcoidosis.Chest.2006;129(6):1599–1604.
- ,,,,.Imaging in sarcoidosis.Semin Respir Crit Care Med.2007;28(1):102–120.
- .Guidelines for the use of corticosteroids in the treatment of pulmonary sarcoidosis.Drugs.2007;67(8):1139–1147.
- ,,, et al.ATS/ERS/WASOG statement on sarcoidosis.Sarcoidosis Vasc Diffuse Lung Dis.1999;16(2):149–173.
- ,,,.Pulmonary sarcoidosis.Semin Respir Crit Care Med.2007;28(1):53–74.
- ,.Pleural involvement in sarcoidosis.Curr Opin Pulm Med.2000;6(5):455–468.
- ,.Pleural sarcoidosis with massive effusion and lung entrapment.Kans Med.1990;91(4):103–105.
- ,,,,.Recurrent pleural and pericardial effusions due to sarcoidosis.PLoS Med.2005;2(3):e63.
- ,,,,.Pulmonary sarcoidosis with associated bloody pleurisy.Intern Med.2002;41(11):1021–1023.
- ,,,,.Pleural effusions in a series of 181 outpatients with sarcoidosis.Chest.2006;129(6):1599–1604.
- ,,,,.Imaging in sarcoidosis.Semin Respir Crit Care Med.2007;28(1):102–120.
- .Guidelines for the use of corticosteroids in the treatment of pulmonary sarcoidosis.Drugs.2007;67(8):1139–1147.
- ,,, et al.ATS/ERS/WASOG statement on sarcoidosis.Sarcoidosis Vasc Diffuse Lung Dis.1999;16(2):149–173.
- ,,,.Pulmonary sarcoidosis.Semin Respir Crit Care Med.2007;28(1):53–74.
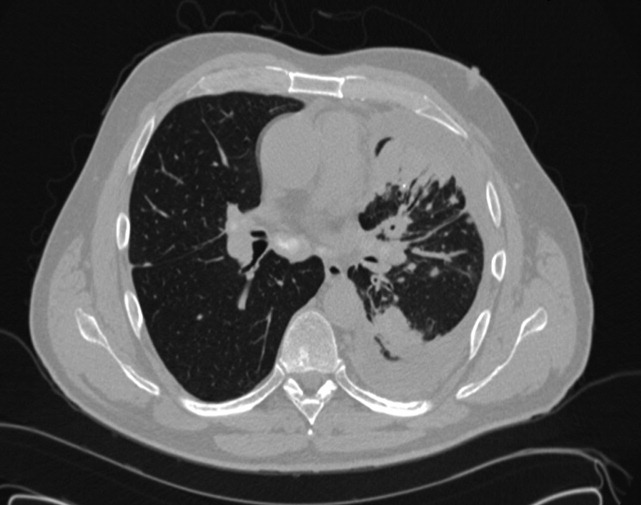
Rube Goldberg Coordinated Care
I was 12 years old before I knew her actual name, Le Thi Canh, because we always called her Ba ngoai. She was my grandmother, and this is the story of how she died.
To see my grandmother for the warrior woman that she was to me, you have to know that her farmer father sent her to the city for schooling because he didn't know what else to do with a daughter who was so smart. In early 20th‐century Vietnam, this was an unusual thing to do with a girl. She met my grandfather there, when he was a campus activist, helping him hand out nationalist leaflets. He introduced her to his Communist friends. After the French jailed my grandfather, my grandmother courted him by sending him long letters and care packages while he was in prison. When he was finally released, they married and started a family while he struggled financially as a newspaper publisher and at other odd jobs. But in 1947 his own Communist comrades killed him as part of a party purge. He had been forewarned, and opted to go quietly rather than try to escape because he was promised that this would guarantee her safety. Before they killed him, somewhere in the mountains, my grandfather gave a soldier friend a poem he wrote for his wife. When she told me this story 8 years ago, more than half a century later, she recited his love missive from memory.
At the time of her husband's death, my grandmother had 6 children, the last born just a few weeks before. After a few years of scraping by (she ran her own one‐room school for a while), she decided to leave Hanoi, and migrated south to Saigon with her brood. She was a famously strict parent, to hear my aunts and uncles tell it. She watched them like a hawk, worked full‐time, put them all through school, and eventually rose to a leadership position in the Ministry of Social Work in South Vietnam. My memories of Saigon life are punctuated by scenes of siblings and cousins running around at her regal house, yellow stucco with porticos and black iron gates, at a corner turn in the road, past a cemetery.
On this side of the world, to see her, you would never have thought that my grandmother had led such an epic life. She never worked again after immigrating with us in 1975. She lived on Social Security checks, gardened, said Buddhist prayers, and was nanny to her grandchildren. She watched soap operas religiously, and could report their full plot lines while sitting and knitting. She bundled her many sadnesses in a contented, 4‐foot 9‐inch frame.
Having no home of her own, she would move from one child's house to another every few months so as not to wear out her welcome. But her children lived in Pennsylvania, New Jersey, Florida, Texas, and Maryland. And in most of these cities, she had a different primary care physician. She has 21 grandchildren; 8 of us are physicians. Yet the aunts and uncles told us very little about her medical care. She preferred older‐generation Vietnamese physicians and I'm not sure that they were all competent, but her children did not want to argue with an octogenarian war survivor, and we deferred to their judgment. So we would find out only incidentally, for example, that a doctor prescribed her tuberculosis drugs for a visit to Vietnam.
For many years, Ba ngoai had no major medical problems. She was hypothyroid and hypertensive but on medication and generally high functioning. She had a lumpectomy for early‐stage breast cancer. Then, a year or so after she told me the story of my grandfather's death, Alzheimer's set in. It became harder for her to report symptoms reliably, and she became mildly depressed. Her grandchildren were now birthing our own babies, and we offered these as a distraction, trying to surround her with celebrations of these new fruits of her life labors.
Ba ngoai's decline worsened 3 years ago. She became more easily fatigued, depressed, and confused. A few months before she died, she started to get dyspneic, and couldn't go for short walks any more. In retrospect, I think that her prescription for thyroid replacement somehow fell through the cracks, probably in the transfer of care from one city to another, although there remains a great deal of confusion in the family about exactly what happened. Her thyroxine levels dwindled. One evening in October of that year, at my uncle's house in Maryland, she became severely short of breath and nearly unconscious. They called her Maryland PCP, who sent her to an emergency room at a local hospital. She was admitted in severe congestive heart failure. When the hospitalist spoke with my mother and uncle, he explained that he could take some fluid off her lungs, but that she might need to be intubated and admitted to intensive care. Looking back, I guessed that she probably needed pressors and invasive monitoring. He asked them, Is this what you want? My uncle said, No, it's not. And the hospitalist and the huddle of relatives decided she should come home.
The question was, into whose care would she be discharged? My elders were wary of contacting her PCP, partly because some blamed him for not catching and addressing her symptoms sooner, partly because to even confront him with this perception would cause him, and hence them, to lose face. This seemed too excruciating a scenario to them.
So at last, my uncle called my brother, the oldest grandchild and a very talented clinician. My brother is a pulmonologist, the kind of physician who once did a history and physical on a patient complaining only of Really feeling bad, Doc, and confidently started a steroid infusion before returning the next day with test results confirming his suspicion of Wegener's granulomatosis. He took my grandmother's physical by phone, and told my uncle to increase her furosemide dose. Then he said, I'm on call, but I'll be down there tomorrow. Call everyone together. Most of my relatives were already in town; they had come at news of her decline. She became alert enough for a couple of days to see and recognize most of the faces around her, like so many markers on a long journey. And then she died, slipped away.
I find it hard to define good coordination of care. My instinct as a researcher is to list measurable elements, but the tools we currently have generate metrics that are either reductionistsuch as how rapidly a physician returns a patient's callor so global that they no longer seem actionablesuch as patient satisfaction. But if such metrics set the goal in the distance, it seems useful to also define its counterpartdiscoordinationas a marker of the reality we would like to leave behind us as far as possible. Discoordination includes elements of discontinuity (lost patient history), fragmentation (actions by multiple players), overuse and/or inappropriate use of services, and ultimately, ineffective care (that is, the patient's needs go unmet).
My experience of discoordination was that of a Rube Goldberg contraption. It's composed of innumerable subtasks, each cleverly designed as the easiest solution to a seemingly short‐term problem, as quick fixes, but that in aggregate generate such chaos that the ultimate purpose is lost. They include acts of denial, lies to avoid embarrassment or conflict, and choices of convenience. My mother and her siblings accommodated my grandmother's choice of physicians by (secretly) not always adhering to care recommendations they didn't agree with, instead of challenging her. They took her to different physicians in different cities rather than risk embarrassing (due to an exaggerated sense of the smallness of the Vietnamese community) any one physician by dropping him. Her grandchildren, despite our medical training, found it culturally easier to defer to our elders than to intervene in substandard care. And none of her physicians aggressively followed up to ensure that a frail Alzheimer's patient was getting the care she needed. This is not to suggest that coordination is a simple task because Rube Goldberg machines make simple tasks complicated. Rather, it is a depiction of how indirectly we tend to address the problem.
I imagine a different course of events for my grandmother in the absence of discoordination. What if her children and physicians had understood and acknowledged to one another that her care was fragmented and therefore suboptimal? What if we grandchildren had confronted both Ba Ngoai and our parents sooner about their choice of physicians and offered to take on more of the burden of helping with her care decisions? Would we, as physicians, have been better able to ensure that her providers made rational clinical decisions? And what if she and her family had consistently recognized a single physician as her medical home? Snowbirding is hardly a rare phenomenon among Medicare patients; we could have designated one physician as primarily responsible for coordinating her care even without limiting her travel.
Care coordination is an inherently human activity. Supportive elements such as efficient transfer of medical information, resources for patient education and self‐care, and adequate reimbursement can take us to the brink of, but not actually bridge, the chasm that we want to cross. Traversing that divide sometimes requires settling turf issues over undesirable responsibilities between different physicians and between physicians and other providers; clarifying who has primary responsibility for different types of decisions (I lead on cardiac issues and her son leads on health maintenance); and the violation of cultural norms of patients, families, and/or providers. These can be uncomfortable, unpleasant conversations that at times seem beside the point. But in aggregate, they are the work of coordination, because they force us to align our expectations of one another. No level of information technology could have dismantled the Rube Goldberg machine that trapped my grandmother. Her last of many lessons for me was that emotional courage, honesty, and perseverance offer a much more direct path through the muck.
I was 12 years old before I knew her actual name, Le Thi Canh, because we always called her Ba ngoai. She was my grandmother, and this is the story of how she died.
To see my grandmother for the warrior woman that she was to me, you have to know that her farmer father sent her to the city for schooling because he didn't know what else to do with a daughter who was so smart. In early 20th‐century Vietnam, this was an unusual thing to do with a girl. She met my grandfather there, when he was a campus activist, helping him hand out nationalist leaflets. He introduced her to his Communist friends. After the French jailed my grandfather, my grandmother courted him by sending him long letters and care packages while he was in prison. When he was finally released, they married and started a family while he struggled financially as a newspaper publisher and at other odd jobs. But in 1947 his own Communist comrades killed him as part of a party purge. He had been forewarned, and opted to go quietly rather than try to escape because he was promised that this would guarantee her safety. Before they killed him, somewhere in the mountains, my grandfather gave a soldier friend a poem he wrote for his wife. When she told me this story 8 years ago, more than half a century later, she recited his love missive from memory.
At the time of her husband's death, my grandmother had 6 children, the last born just a few weeks before. After a few years of scraping by (she ran her own one‐room school for a while), she decided to leave Hanoi, and migrated south to Saigon with her brood. She was a famously strict parent, to hear my aunts and uncles tell it. She watched them like a hawk, worked full‐time, put them all through school, and eventually rose to a leadership position in the Ministry of Social Work in South Vietnam. My memories of Saigon life are punctuated by scenes of siblings and cousins running around at her regal house, yellow stucco with porticos and black iron gates, at a corner turn in the road, past a cemetery.
On this side of the world, to see her, you would never have thought that my grandmother had led such an epic life. She never worked again after immigrating with us in 1975. She lived on Social Security checks, gardened, said Buddhist prayers, and was nanny to her grandchildren. She watched soap operas religiously, and could report their full plot lines while sitting and knitting. She bundled her many sadnesses in a contented, 4‐foot 9‐inch frame.
Having no home of her own, she would move from one child's house to another every few months so as not to wear out her welcome. But her children lived in Pennsylvania, New Jersey, Florida, Texas, and Maryland. And in most of these cities, she had a different primary care physician. She has 21 grandchildren; 8 of us are physicians. Yet the aunts and uncles told us very little about her medical care. She preferred older‐generation Vietnamese physicians and I'm not sure that they were all competent, but her children did not want to argue with an octogenarian war survivor, and we deferred to their judgment. So we would find out only incidentally, for example, that a doctor prescribed her tuberculosis drugs for a visit to Vietnam.
For many years, Ba ngoai had no major medical problems. She was hypothyroid and hypertensive but on medication and generally high functioning. She had a lumpectomy for early‐stage breast cancer. Then, a year or so after she told me the story of my grandfather's death, Alzheimer's set in. It became harder for her to report symptoms reliably, and she became mildly depressed. Her grandchildren were now birthing our own babies, and we offered these as a distraction, trying to surround her with celebrations of these new fruits of her life labors.
Ba ngoai's decline worsened 3 years ago. She became more easily fatigued, depressed, and confused. A few months before she died, she started to get dyspneic, and couldn't go for short walks any more. In retrospect, I think that her prescription for thyroid replacement somehow fell through the cracks, probably in the transfer of care from one city to another, although there remains a great deal of confusion in the family about exactly what happened. Her thyroxine levels dwindled. One evening in October of that year, at my uncle's house in Maryland, she became severely short of breath and nearly unconscious. They called her Maryland PCP, who sent her to an emergency room at a local hospital. She was admitted in severe congestive heart failure. When the hospitalist spoke with my mother and uncle, he explained that he could take some fluid off her lungs, but that she might need to be intubated and admitted to intensive care. Looking back, I guessed that she probably needed pressors and invasive monitoring. He asked them, Is this what you want? My uncle said, No, it's not. And the hospitalist and the huddle of relatives decided she should come home.
The question was, into whose care would she be discharged? My elders were wary of contacting her PCP, partly because some blamed him for not catching and addressing her symptoms sooner, partly because to even confront him with this perception would cause him, and hence them, to lose face. This seemed too excruciating a scenario to them.
So at last, my uncle called my brother, the oldest grandchild and a very talented clinician. My brother is a pulmonologist, the kind of physician who once did a history and physical on a patient complaining only of Really feeling bad, Doc, and confidently started a steroid infusion before returning the next day with test results confirming his suspicion of Wegener's granulomatosis. He took my grandmother's physical by phone, and told my uncle to increase her furosemide dose. Then he said, I'm on call, but I'll be down there tomorrow. Call everyone together. Most of my relatives were already in town; they had come at news of her decline. She became alert enough for a couple of days to see and recognize most of the faces around her, like so many markers on a long journey. And then she died, slipped away.
I find it hard to define good coordination of care. My instinct as a researcher is to list measurable elements, but the tools we currently have generate metrics that are either reductionistsuch as how rapidly a physician returns a patient's callor so global that they no longer seem actionablesuch as patient satisfaction. But if such metrics set the goal in the distance, it seems useful to also define its counterpartdiscoordinationas a marker of the reality we would like to leave behind us as far as possible. Discoordination includes elements of discontinuity (lost patient history), fragmentation (actions by multiple players), overuse and/or inappropriate use of services, and ultimately, ineffective care (that is, the patient's needs go unmet).
My experience of discoordination was that of a Rube Goldberg contraption. It's composed of innumerable subtasks, each cleverly designed as the easiest solution to a seemingly short‐term problem, as quick fixes, but that in aggregate generate such chaos that the ultimate purpose is lost. They include acts of denial, lies to avoid embarrassment or conflict, and choices of convenience. My mother and her siblings accommodated my grandmother's choice of physicians by (secretly) not always adhering to care recommendations they didn't agree with, instead of challenging her. They took her to different physicians in different cities rather than risk embarrassing (due to an exaggerated sense of the smallness of the Vietnamese community) any one physician by dropping him. Her grandchildren, despite our medical training, found it culturally easier to defer to our elders than to intervene in substandard care. And none of her physicians aggressively followed up to ensure that a frail Alzheimer's patient was getting the care she needed. This is not to suggest that coordination is a simple task because Rube Goldberg machines make simple tasks complicated. Rather, it is a depiction of how indirectly we tend to address the problem.
I imagine a different course of events for my grandmother in the absence of discoordination. What if her children and physicians had understood and acknowledged to one another that her care was fragmented and therefore suboptimal? What if we grandchildren had confronted both Ba Ngoai and our parents sooner about their choice of physicians and offered to take on more of the burden of helping with her care decisions? Would we, as physicians, have been better able to ensure that her providers made rational clinical decisions? And what if she and her family had consistently recognized a single physician as her medical home? Snowbirding is hardly a rare phenomenon among Medicare patients; we could have designated one physician as primarily responsible for coordinating her care even without limiting her travel.
Care coordination is an inherently human activity. Supportive elements such as efficient transfer of medical information, resources for patient education and self‐care, and adequate reimbursement can take us to the brink of, but not actually bridge, the chasm that we want to cross. Traversing that divide sometimes requires settling turf issues over undesirable responsibilities between different physicians and between physicians and other providers; clarifying who has primary responsibility for different types of decisions (I lead on cardiac issues and her son leads on health maintenance); and the violation of cultural norms of patients, families, and/or providers. These can be uncomfortable, unpleasant conversations that at times seem beside the point. But in aggregate, they are the work of coordination, because they force us to align our expectations of one another. No level of information technology could have dismantled the Rube Goldberg machine that trapped my grandmother. Her last of many lessons for me was that emotional courage, honesty, and perseverance offer a much more direct path through the muck.
I was 12 years old before I knew her actual name, Le Thi Canh, because we always called her Ba ngoai. She was my grandmother, and this is the story of how she died.
To see my grandmother for the warrior woman that she was to me, you have to know that her farmer father sent her to the city for schooling because he didn't know what else to do with a daughter who was so smart. In early 20th‐century Vietnam, this was an unusual thing to do with a girl. She met my grandfather there, when he was a campus activist, helping him hand out nationalist leaflets. He introduced her to his Communist friends. After the French jailed my grandfather, my grandmother courted him by sending him long letters and care packages while he was in prison. When he was finally released, they married and started a family while he struggled financially as a newspaper publisher and at other odd jobs. But in 1947 his own Communist comrades killed him as part of a party purge. He had been forewarned, and opted to go quietly rather than try to escape because he was promised that this would guarantee her safety. Before they killed him, somewhere in the mountains, my grandfather gave a soldier friend a poem he wrote for his wife. When she told me this story 8 years ago, more than half a century later, she recited his love missive from memory.
At the time of her husband's death, my grandmother had 6 children, the last born just a few weeks before. After a few years of scraping by (she ran her own one‐room school for a while), she decided to leave Hanoi, and migrated south to Saigon with her brood. She was a famously strict parent, to hear my aunts and uncles tell it. She watched them like a hawk, worked full‐time, put them all through school, and eventually rose to a leadership position in the Ministry of Social Work in South Vietnam. My memories of Saigon life are punctuated by scenes of siblings and cousins running around at her regal house, yellow stucco with porticos and black iron gates, at a corner turn in the road, past a cemetery.
On this side of the world, to see her, you would never have thought that my grandmother had led such an epic life. She never worked again after immigrating with us in 1975. She lived on Social Security checks, gardened, said Buddhist prayers, and was nanny to her grandchildren. She watched soap operas religiously, and could report their full plot lines while sitting and knitting. She bundled her many sadnesses in a contented, 4‐foot 9‐inch frame.
Having no home of her own, she would move from one child's house to another every few months so as not to wear out her welcome. But her children lived in Pennsylvania, New Jersey, Florida, Texas, and Maryland. And in most of these cities, she had a different primary care physician. She has 21 grandchildren; 8 of us are physicians. Yet the aunts and uncles told us very little about her medical care. She preferred older‐generation Vietnamese physicians and I'm not sure that they were all competent, but her children did not want to argue with an octogenarian war survivor, and we deferred to their judgment. So we would find out only incidentally, for example, that a doctor prescribed her tuberculosis drugs for a visit to Vietnam.
For many years, Ba ngoai had no major medical problems. She was hypothyroid and hypertensive but on medication and generally high functioning. She had a lumpectomy for early‐stage breast cancer. Then, a year or so after she told me the story of my grandfather's death, Alzheimer's set in. It became harder for her to report symptoms reliably, and she became mildly depressed. Her grandchildren were now birthing our own babies, and we offered these as a distraction, trying to surround her with celebrations of these new fruits of her life labors.
Ba ngoai's decline worsened 3 years ago. She became more easily fatigued, depressed, and confused. A few months before she died, she started to get dyspneic, and couldn't go for short walks any more. In retrospect, I think that her prescription for thyroid replacement somehow fell through the cracks, probably in the transfer of care from one city to another, although there remains a great deal of confusion in the family about exactly what happened. Her thyroxine levels dwindled. One evening in October of that year, at my uncle's house in Maryland, she became severely short of breath and nearly unconscious. They called her Maryland PCP, who sent her to an emergency room at a local hospital. She was admitted in severe congestive heart failure. When the hospitalist spoke with my mother and uncle, he explained that he could take some fluid off her lungs, but that she might need to be intubated and admitted to intensive care. Looking back, I guessed that she probably needed pressors and invasive monitoring. He asked them, Is this what you want? My uncle said, No, it's not. And the hospitalist and the huddle of relatives decided she should come home.
The question was, into whose care would she be discharged? My elders were wary of contacting her PCP, partly because some blamed him for not catching and addressing her symptoms sooner, partly because to even confront him with this perception would cause him, and hence them, to lose face. This seemed too excruciating a scenario to them.
So at last, my uncle called my brother, the oldest grandchild and a very talented clinician. My brother is a pulmonologist, the kind of physician who once did a history and physical on a patient complaining only of Really feeling bad, Doc, and confidently started a steroid infusion before returning the next day with test results confirming his suspicion of Wegener's granulomatosis. He took my grandmother's physical by phone, and told my uncle to increase her furosemide dose. Then he said, I'm on call, but I'll be down there tomorrow. Call everyone together. Most of my relatives were already in town; they had come at news of her decline. She became alert enough for a couple of days to see and recognize most of the faces around her, like so many markers on a long journey. And then she died, slipped away.
I find it hard to define good coordination of care. My instinct as a researcher is to list measurable elements, but the tools we currently have generate metrics that are either reductionistsuch as how rapidly a physician returns a patient's callor so global that they no longer seem actionablesuch as patient satisfaction. But if such metrics set the goal in the distance, it seems useful to also define its counterpartdiscoordinationas a marker of the reality we would like to leave behind us as far as possible. Discoordination includes elements of discontinuity (lost patient history), fragmentation (actions by multiple players), overuse and/or inappropriate use of services, and ultimately, ineffective care (that is, the patient's needs go unmet).
My experience of discoordination was that of a Rube Goldberg contraption. It's composed of innumerable subtasks, each cleverly designed as the easiest solution to a seemingly short‐term problem, as quick fixes, but that in aggregate generate such chaos that the ultimate purpose is lost. They include acts of denial, lies to avoid embarrassment or conflict, and choices of convenience. My mother and her siblings accommodated my grandmother's choice of physicians by (secretly) not always adhering to care recommendations they didn't agree with, instead of challenging her. They took her to different physicians in different cities rather than risk embarrassing (due to an exaggerated sense of the smallness of the Vietnamese community) any one physician by dropping him. Her grandchildren, despite our medical training, found it culturally easier to defer to our elders than to intervene in substandard care. And none of her physicians aggressively followed up to ensure that a frail Alzheimer's patient was getting the care she needed. This is not to suggest that coordination is a simple task because Rube Goldberg machines make simple tasks complicated. Rather, it is a depiction of how indirectly we tend to address the problem.
I imagine a different course of events for my grandmother in the absence of discoordination. What if her children and physicians had understood and acknowledged to one another that her care was fragmented and therefore suboptimal? What if we grandchildren had confronted both Ba Ngoai and our parents sooner about their choice of physicians and offered to take on more of the burden of helping with her care decisions? Would we, as physicians, have been better able to ensure that her providers made rational clinical decisions? And what if she and her family had consistently recognized a single physician as her medical home? Snowbirding is hardly a rare phenomenon among Medicare patients; we could have designated one physician as primarily responsible for coordinating her care even without limiting her travel.
Care coordination is an inherently human activity. Supportive elements such as efficient transfer of medical information, resources for patient education and self‐care, and adequate reimbursement can take us to the brink of, but not actually bridge, the chasm that we want to cross. Traversing that divide sometimes requires settling turf issues over undesirable responsibilities between different physicians and between physicians and other providers; clarifying who has primary responsibility for different types of decisions (I lead on cardiac issues and her son leads on health maintenance); and the violation of cultural norms of patients, families, and/or providers. These can be uncomfortable, unpleasant conversations that at times seem beside the point. But in aggregate, they are the work of coordination, because they force us to align our expectations of one another. No level of information technology could have dismantled the Rube Goldberg machine that trapped my grandmother. Her last of many lessons for me was that emotional courage, honesty, and perseverance offer a much more direct path through the muck.
BOOSTing the Hospital Discharge
Hospitalists recognize the importance of the care transition from the inpatient setting to the outpatient setting, despite being described as causing a divorce between inpatient and outpatient care.1 If you do not believe this, just glance at the table of contents for this issue of the Journal of Hospital Medicine, which has 5 reports on research about various aspects of the hospital discharge transition complemented by an eloquent story of how a hospitalist facilitated the care coordination of one family's matriarch.2 An accompanying editorial proposes that hospitalists embrace the need of patients and their caregivers for care coordination.3 Thankfully, a growing number of academic hospitalists are focusing their efforts on identifying problems in the process and evaluating potential interventions to optimize it.
The hospital discharge process commonly has been an afterthought, concluding a typically intense experience for patients, some of whom may have begun the episode of hospitalization near death. After diagnostic evaluations and treatments, a patient has achieved stable enough status to be discharged home, and the inpatient physician has signed off with a simple may go in the written orders. The physician may feel absolved of responsibility as he expects the nurses to take care of instructions and to find transportation home for the patient. Unfortunately, this experience often is consistent with Webster's definition of discharge: to relieve of a charge, load, or burden unload release from an obligation. Some patients may feel like a Nolan Ryan fastball flying out of the hospital, but with no one to catch them.
Recognizing how the hospital discharge transition to home can be a perilous process fraught with failure,4 we laid out a research agenda for transitions of care. We are gratified to see the robust response from researchers published in this issue of the Journal of Hospital Medicine. The studies range from the description of a new tool to assess patients' mobility before discharge5 to evidence that the length of stay is prolonged (ie, delayed discharge) when the discharge diagnosis differs from that made on admission.6 Chen and colleagues analyzed the timing of discharge during the day and found that the duration of the discharge process was influenced by the need for consultation or a procedure prior to discharge; this finding is not surprising to practicing hospitalists. We agree with their conclusion that broad institutional efforts will be needed to facilitate the process. Hospitalists are part of a system and must engage the entire team to improve efficiency.
O'Leary and fellow hospitalists7 at Northwestern Memorial Hospital focused on creating a better discharge summary within their electronic health record with the aim of improved overall quality of the summaries and, just as important, timely completion. Despite some research indicating that absence of adequate communication between primary care providers and inpatient medical teams is not associated with adverse clinical outcomes,8 other research has demonstrated that it does affect outcomes and probably affects rehospitalization rates.9, 10 Moreover, another article in this issue describes a project undertaken at Baylor Health Care System (Dallas, TX) that demonstrated a reduction in emergency department visits and readmissions within 30 days post‐discharge among high‐risk elderly medical patients when a targeted care bundle was used.11 The results from this intervention, which consisted of medication counseling/reconciliation by a clinical pharmacist, condition‐specific enhanced discharge planning by a care coordinator, and phone follow‐up, confirm recent results from 2 similar studies.12, 13 These studies provide support for the idea that straightforward changes in the discharge process can improve patient outcomes.
Today in the United States, hospitalists likely care for the majority of hospitalized older patients.14 We strongly encourage them to use evidence‐based approaches to optimize the discharge process in their hospitals, and fortunately, clear guidance is available. Because of generous funding from the John A. Hartford Foundation, Project BOOST (Better Outcomes for Older Adults Through Safe Transitions) is mentoring 30 hospitals in an effort to implement the BOOST toolkit and improve their discharge transition processes.15 Another cost‐effective method involves the use of transition coaches to help the most vulnerable older patients with complex care needs.16 This approach is now being implemented by more than 100 healthcare organizations worldwide.17
Heartened by these exciting initiatives, we applaud the Society of Hospital Medicine's collaboration with the American College of Physicians, the Society of General Internal Medicine, the American Geriatrics Society, and the Society of Academic Emergency Medicine to produce a consensus policy statement on transitions of care that provides guiding principles for transitions both into and out of the hospital.18 Soon, all hospitalized patients and their caregivers may receive robust education prior to discharge, confirmation of their understanding with the teach‐back approach, medication reconciliation, and clear instructions for follow‐up, and the patient's primary care provider will be aware of all that has happened. Patients should expect nothing less than hospitalists ensuring their seamless transition from hospital to home.
- ,,,.Hospitalists and care transitions: the divorce of inpatient and outpatient care.Health Aff.2008;27:1315–1327.
- .Dismantling Rube Goldberg: cutting through chaos to achieve coordinated care.J Hosp Med.2009;4:259–260.
- ,,.A new narrative for hospitalists.J Hosp Med.2009;4:207–208.
- ,.Executing high‐quality care transitions: a call to do it right.J Hosp Med.2007;2:287–290.
- ,,.Home alone: mobility independence before discharge.J Hosp Med.2009;4:252–254.
- ,,,,.Discrepancy between admission and discharge diagnoses as a predictor of hospital length of stay.J Hosp Med.2009;4:234–239.
- ,,, et al.,Creating a better discharge summary: improvement in quality and timeliness using an electronic discharge summary.J Hosp Med.2009;4:219–225.
- ,,, et al.Association of communication between hospital‐based physicians and primary care providers with patient outcomes.J Gen Intern Med.2009;24:381–386.
- ,,,,,.Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care.JAMA.2007;297:831–841.
- ,,.Rehospitalizations among patients in the Medicare fee‐for‐service program.N Engl J Med. In press.
- ,,, et al.Reduction of 30‐day post‐discharge hospital readmission or ED visit rates in high‐risk elderly medical patients through delivery of a targeted care bundle.J Hosp Med.2009;4:211–218.
- ,,, et al.A reengineered hospital discharge program to decrease rehospitalization: a randomized trial.Ann Intern Med.2009;150:178–187.
- ,,,.Redefining and redesigning hospital discharge to enhance patient care: a randomized controlled study.J Gen Intern Med.2008;23:1228–1233.
- ,,,.Growth in the care of older patients by hospitalists in the United States.N Engl J Med.2009;360:1102–1112.
- Society of Hospital Medicine. BOOSTing Care Transitions Resource Room. Available at: http://www.hospitalmedicine.org. Accessed March2009.
- ,,,.The care transitions intervention: results of a randomized controlled trial.Arch Intern Med.2006;166:1822–1828.
- Care Transitions Program. Available at: http://www.caretransitions.org. Accessed March2009.
- ,,, et al.Transitions of care consensus policy statement. American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College of Emergency Physicians, and Society of Academic Emergency Medicine.J Hosp Med. In press.
Hospitalists recognize the importance of the care transition from the inpatient setting to the outpatient setting, despite being described as causing a divorce between inpatient and outpatient care.1 If you do not believe this, just glance at the table of contents for this issue of the Journal of Hospital Medicine, which has 5 reports on research about various aspects of the hospital discharge transition complemented by an eloquent story of how a hospitalist facilitated the care coordination of one family's matriarch.2 An accompanying editorial proposes that hospitalists embrace the need of patients and their caregivers for care coordination.3 Thankfully, a growing number of academic hospitalists are focusing their efforts on identifying problems in the process and evaluating potential interventions to optimize it.
The hospital discharge process commonly has been an afterthought, concluding a typically intense experience for patients, some of whom may have begun the episode of hospitalization near death. After diagnostic evaluations and treatments, a patient has achieved stable enough status to be discharged home, and the inpatient physician has signed off with a simple may go in the written orders. The physician may feel absolved of responsibility as he expects the nurses to take care of instructions and to find transportation home for the patient. Unfortunately, this experience often is consistent with Webster's definition of discharge: to relieve of a charge, load, or burden unload release from an obligation. Some patients may feel like a Nolan Ryan fastball flying out of the hospital, but with no one to catch them.
Recognizing how the hospital discharge transition to home can be a perilous process fraught with failure,4 we laid out a research agenda for transitions of care. We are gratified to see the robust response from researchers published in this issue of the Journal of Hospital Medicine. The studies range from the description of a new tool to assess patients' mobility before discharge5 to evidence that the length of stay is prolonged (ie, delayed discharge) when the discharge diagnosis differs from that made on admission.6 Chen and colleagues analyzed the timing of discharge during the day and found that the duration of the discharge process was influenced by the need for consultation or a procedure prior to discharge; this finding is not surprising to practicing hospitalists. We agree with their conclusion that broad institutional efforts will be needed to facilitate the process. Hospitalists are part of a system and must engage the entire team to improve efficiency.
O'Leary and fellow hospitalists7 at Northwestern Memorial Hospital focused on creating a better discharge summary within their electronic health record with the aim of improved overall quality of the summaries and, just as important, timely completion. Despite some research indicating that absence of adequate communication between primary care providers and inpatient medical teams is not associated with adverse clinical outcomes,8 other research has demonstrated that it does affect outcomes and probably affects rehospitalization rates.9, 10 Moreover, another article in this issue describes a project undertaken at Baylor Health Care System (Dallas, TX) that demonstrated a reduction in emergency department visits and readmissions within 30 days post‐discharge among high‐risk elderly medical patients when a targeted care bundle was used.11 The results from this intervention, which consisted of medication counseling/reconciliation by a clinical pharmacist, condition‐specific enhanced discharge planning by a care coordinator, and phone follow‐up, confirm recent results from 2 similar studies.12, 13 These studies provide support for the idea that straightforward changes in the discharge process can improve patient outcomes.
Today in the United States, hospitalists likely care for the majority of hospitalized older patients.14 We strongly encourage them to use evidence‐based approaches to optimize the discharge process in their hospitals, and fortunately, clear guidance is available. Because of generous funding from the John A. Hartford Foundation, Project BOOST (Better Outcomes for Older Adults Through Safe Transitions) is mentoring 30 hospitals in an effort to implement the BOOST toolkit and improve their discharge transition processes.15 Another cost‐effective method involves the use of transition coaches to help the most vulnerable older patients with complex care needs.16 This approach is now being implemented by more than 100 healthcare organizations worldwide.17
Heartened by these exciting initiatives, we applaud the Society of Hospital Medicine's collaboration with the American College of Physicians, the Society of General Internal Medicine, the American Geriatrics Society, and the Society of Academic Emergency Medicine to produce a consensus policy statement on transitions of care that provides guiding principles for transitions both into and out of the hospital.18 Soon, all hospitalized patients and their caregivers may receive robust education prior to discharge, confirmation of their understanding with the teach‐back approach, medication reconciliation, and clear instructions for follow‐up, and the patient's primary care provider will be aware of all that has happened. Patients should expect nothing less than hospitalists ensuring their seamless transition from hospital to home.
Hospitalists recognize the importance of the care transition from the inpatient setting to the outpatient setting, despite being described as causing a divorce between inpatient and outpatient care.1 If you do not believe this, just glance at the table of contents for this issue of the Journal of Hospital Medicine, which has 5 reports on research about various aspects of the hospital discharge transition complemented by an eloquent story of how a hospitalist facilitated the care coordination of one family's matriarch.2 An accompanying editorial proposes that hospitalists embrace the need of patients and their caregivers for care coordination.3 Thankfully, a growing number of academic hospitalists are focusing their efforts on identifying problems in the process and evaluating potential interventions to optimize it.
The hospital discharge process commonly has been an afterthought, concluding a typically intense experience for patients, some of whom may have begun the episode of hospitalization near death. After diagnostic evaluations and treatments, a patient has achieved stable enough status to be discharged home, and the inpatient physician has signed off with a simple may go in the written orders. The physician may feel absolved of responsibility as he expects the nurses to take care of instructions and to find transportation home for the patient. Unfortunately, this experience often is consistent with Webster's definition of discharge: to relieve of a charge, load, or burden unload release from an obligation. Some patients may feel like a Nolan Ryan fastball flying out of the hospital, but with no one to catch them.
Recognizing how the hospital discharge transition to home can be a perilous process fraught with failure,4 we laid out a research agenda for transitions of care. We are gratified to see the robust response from researchers published in this issue of the Journal of Hospital Medicine. The studies range from the description of a new tool to assess patients' mobility before discharge5 to evidence that the length of stay is prolonged (ie, delayed discharge) when the discharge diagnosis differs from that made on admission.6 Chen and colleagues analyzed the timing of discharge during the day and found that the duration of the discharge process was influenced by the need for consultation or a procedure prior to discharge; this finding is not surprising to practicing hospitalists. We agree with their conclusion that broad institutional efforts will be needed to facilitate the process. Hospitalists are part of a system and must engage the entire team to improve efficiency.
O'Leary and fellow hospitalists7 at Northwestern Memorial Hospital focused on creating a better discharge summary within their electronic health record with the aim of improved overall quality of the summaries and, just as important, timely completion. Despite some research indicating that absence of adequate communication between primary care providers and inpatient medical teams is not associated with adverse clinical outcomes,8 other research has demonstrated that it does affect outcomes and probably affects rehospitalization rates.9, 10 Moreover, another article in this issue describes a project undertaken at Baylor Health Care System (Dallas, TX) that demonstrated a reduction in emergency department visits and readmissions within 30 days post‐discharge among high‐risk elderly medical patients when a targeted care bundle was used.11 The results from this intervention, which consisted of medication counseling/reconciliation by a clinical pharmacist, condition‐specific enhanced discharge planning by a care coordinator, and phone follow‐up, confirm recent results from 2 similar studies.12, 13 These studies provide support for the idea that straightforward changes in the discharge process can improve patient outcomes.
Today in the United States, hospitalists likely care for the majority of hospitalized older patients.14 We strongly encourage them to use evidence‐based approaches to optimize the discharge process in their hospitals, and fortunately, clear guidance is available. Because of generous funding from the John A. Hartford Foundation, Project BOOST (Better Outcomes for Older Adults Through Safe Transitions) is mentoring 30 hospitals in an effort to implement the BOOST toolkit and improve their discharge transition processes.15 Another cost‐effective method involves the use of transition coaches to help the most vulnerable older patients with complex care needs.16 This approach is now being implemented by more than 100 healthcare organizations worldwide.17
Heartened by these exciting initiatives, we applaud the Society of Hospital Medicine's collaboration with the American College of Physicians, the Society of General Internal Medicine, the American Geriatrics Society, and the Society of Academic Emergency Medicine to produce a consensus policy statement on transitions of care that provides guiding principles for transitions both into and out of the hospital.18 Soon, all hospitalized patients and their caregivers may receive robust education prior to discharge, confirmation of their understanding with the teach‐back approach, medication reconciliation, and clear instructions for follow‐up, and the patient's primary care provider will be aware of all that has happened. Patients should expect nothing less than hospitalists ensuring their seamless transition from hospital to home.
- ,,,.Hospitalists and care transitions: the divorce of inpatient and outpatient care.Health Aff.2008;27:1315–1327.
- .Dismantling Rube Goldberg: cutting through chaos to achieve coordinated care.J Hosp Med.2009;4:259–260.
- ,,.A new narrative for hospitalists.J Hosp Med.2009;4:207–208.
- ,.Executing high‐quality care transitions: a call to do it right.J Hosp Med.2007;2:287–290.
- ,,.Home alone: mobility independence before discharge.J Hosp Med.2009;4:252–254.
- ,,,,.Discrepancy between admission and discharge diagnoses as a predictor of hospital length of stay.J Hosp Med.2009;4:234–239.
- ,,, et al.,Creating a better discharge summary: improvement in quality and timeliness using an electronic discharge summary.J Hosp Med.2009;4:219–225.
- ,,, et al.Association of communication between hospital‐based physicians and primary care providers with patient outcomes.J Gen Intern Med.2009;24:381–386.
- ,,,,,.Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care.JAMA.2007;297:831–841.
- ,,.Rehospitalizations among patients in the Medicare fee‐for‐service program.N Engl J Med. In press.
- ,,, et al.Reduction of 30‐day post‐discharge hospital readmission or ED visit rates in high‐risk elderly medical patients through delivery of a targeted care bundle.J Hosp Med.2009;4:211–218.
- ,,, et al.A reengineered hospital discharge program to decrease rehospitalization: a randomized trial.Ann Intern Med.2009;150:178–187.
- ,,,.Redefining and redesigning hospital discharge to enhance patient care: a randomized controlled study.J Gen Intern Med.2008;23:1228–1233.
- ,,,.Growth in the care of older patients by hospitalists in the United States.N Engl J Med.2009;360:1102–1112.
- Society of Hospital Medicine. BOOSTing Care Transitions Resource Room. Available at: http://www.hospitalmedicine.org. Accessed March2009.
- ,,,.The care transitions intervention: results of a randomized controlled trial.Arch Intern Med.2006;166:1822–1828.
- Care Transitions Program. Available at: http://www.caretransitions.org. Accessed March2009.
- ,,, et al.Transitions of care consensus policy statement. American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College of Emergency Physicians, and Society of Academic Emergency Medicine.J Hosp Med. In press.
- ,,,.Hospitalists and care transitions: the divorce of inpatient and outpatient care.Health Aff.2008;27:1315–1327.
- .Dismantling Rube Goldberg: cutting through chaos to achieve coordinated care.J Hosp Med.2009;4:259–260.
- ,,.A new narrative for hospitalists.J Hosp Med.2009;4:207–208.
- ,.Executing high‐quality care transitions: a call to do it right.J Hosp Med.2007;2:287–290.
- ,,.Home alone: mobility independence before discharge.J Hosp Med.2009;4:252–254.
- ,,,,.Discrepancy between admission and discharge diagnoses as a predictor of hospital length of stay.J Hosp Med.2009;4:234–239.
- ,,, et al.,Creating a better discharge summary: improvement in quality and timeliness using an electronic discharge summary.J Hosp Med.2009;4:219–225.
- ,,, et al.Association of communication between hospital‐based physicians and primary care providers with patient outcomes.J Gen Intern Med.2009;24:381–386.
- ,,,,,.Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care.JAMA.2007;297:831–841.
- ,,.Rehospitalizations among patients in the Medicare fee‐for‐service program.N Engl J Med. In press.
- ,,, et al.Reduction of 30‐day post‐discharge hospital readmission or ED visit rates in high‐risk elderly medical patients through delivery of a targeted care bundle.J Hosp Med.2009;4:211–218.
- ,,, et al.A reengineered hospital discharge program to decrease rehospitalization: a randomized trial.Ann Intern Med.2009;150:178–187.
- ,,,.Redefining and redesigning hospital discharge to enhance patient care: a randomized controlled study.J Gen Intern Med.2008;23:1228–1233.
- ,,,.Growth in the care of older patients by hospitalists in the United States.N Engl J Med.2009;360:1102–1112.
- Society of Hospital Medicine. BOOSTing Care Transitions Resource Room. Available at: http://www.hospitalmedicine.org. Accessed March2009.
- ,,,.The care transitions intervention: results of a randomized controlled trial.Arch Intern Med.2006;166:1822–1828.
- Care Transitions Program. Available at: http://www.caretransitions.org. Accessed March2009.
- ,,, et al.Transitions of care consensus policy statement. American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College of Emergency Physicians, and Society of Academic Emergency Medicine.J Hosp Med. In press.