User login
Osteoporosis: What about men?
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4

Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.

The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20

Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23

Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4
Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.
The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20
Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23
Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4
Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.
The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20
Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23
Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.

Hepatitis C: How to fine-tune your approach
› Screen at-risk patients and all those born between 1945 and 1965 for hepatitis C virus (HCV) infection. B
› Screen HCV-positive patients for level of fibrosis and for conditions that may accelerate liver disease, including alcohol use, hepatitis B virus, and human immunodeficiency virus. B
› Continuously monitor patients with chronic HCV for the development of cirrhosis and hepatocellular carcinoma. A
› Refer patients to specialty care for HCV treatment and, if they have cirrhosis, for potential transplant evaluation. C
› Counsel HCV-positive patients about how to avoid transmission to others. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hepatitis C virus (HCV) infection is a leading cause of chronic liver disease. Over the next few decades, the number of deaths per year due to complications of HCV such as liver failure and hepatocellular carcinoma (HCC) is predicted to more than triple to 36,000 by 2032.1
Fortunately, major advances in drug therapy have made it possible to cure patients of HCV, and treatment is now less complex, of shorter duration, and better tolerated than it once was. To help family physicians maximize the care they provide to these patients, we’ve summarized screening recommendations from the Centers for Disease Control and Prevention (CDC), innovative alternatives to biopsy for staging liver disease, and counseling points to cover with patients.
A common, usually silent infection with potentially fatal complications
According to the National Health and Nutrition Examination Survey (NHANES), an estimated 2.7 to 3.9 million people in the United States are chronically infected with HCV, about threefourths of whom were born between 1945 and 1965 (the “baby boomer” generation).2 However, by adding “unaccounted groups” (eg, incarcerated, homeless, and active duty military) to these estimates, the number of people with HCV is likely more than 5.2 million.3
HCV is a ribonucleic acid (RNA) virus capable of mutating at a high rate to escape detection and clearance by the host’s immune system.4 Most patients with HCV are asymptomatic during the acute and chronic phases of infection, and may have a silent infection for decades. In fact, 65% to 75% of patients with HCV are unaware of their infection.5
Approximately 20% of chronically infected patients develop cirrhosis after 20 years and, once they do, the annual rate of HCC and liver decompensation is about 5%.6-8 Risk factors for advancement to cirrhosis includes male sex, alcohol consumption, co-infection with human immunodeficiency virus (HIV) or hepatitis B virus (HBV), immunosuppression, having had HCV infection for a long time, becoming infected with HCV after age 40, and not having responded to previous treatment.9
Chronic HCV infection can lead to extrahepatic manifestations such as essential mixed cryoglobulinemia, porphyria cutanea tarda, membranoproliferative glomerulonephritis, lymphoma, and glucose intolerance.10 There is also growing evidence that HCV infection affects cognitive function in the absence of fibrosis and hepatic encephalopathy. Several studies show that HCV-infected patients score poorly on neuropsychological testing for verbal learning, attention, memory, and executive function.11 This may be related to the expression of receptors for HCV by the brain’s microvascular endothelial cells.12
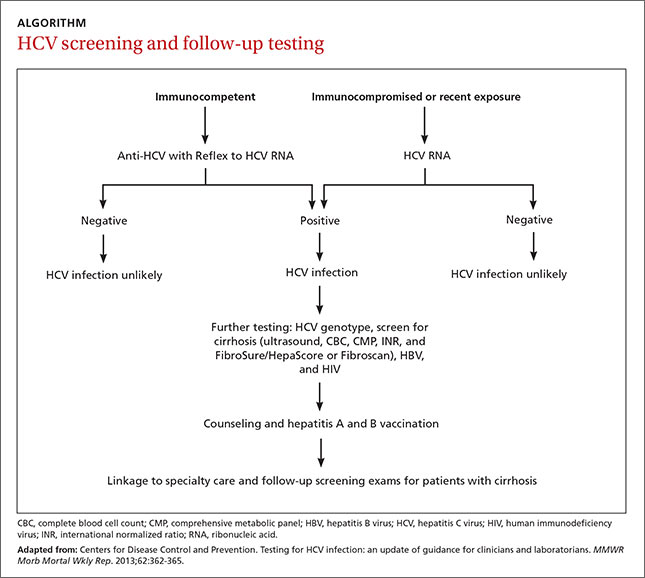
Screening recommendations. Given the high prevalence of HCV infection among baby boomers, the CDC decided in 2012 to recommend one-time HCV screening for all patients born between 1945 and 1965.13 This is in addition to risk-based screening for all patients who have a history of injection drug use, those on long-term hemodialysis or with tattoos obtained in unregulated settings, offspring of HCV-infected mothers, and those with health-care associated exposures (TABLE13). In 2013, the US Preventive Services Task Force upgraded its recommendation to match those of the CDC.14

Despite these recommendations, which are expected to increase detection of HCV among asymptomatic persons who do not know they are infected, there remain significant barriers to HCV testing. These include poor access to primary care and preventive services, lack of knowledge and awareness of the disease among patients and providers, and a lack of studies that support a universal screening approach for HCV.5,15,16 One tool that might help overcome some of these barriers and aid family physicians in the screening process is automatic reminders or standing lab orders for HCV testing in electronic medical records systems.
Screening for HCV can be done using any of the US Food and Drug Administration (FDA)-approved tests for the anti-HCV antibody, which have sensitivities and specificities greater than 99%.17 A positive screening result should be confirmed with an HCV RNA test. However, for practical purposes, ordering the anti-HCV test with reflex to the HCV RNA test decreases the number of blood draws and office visits required of the patient. The reflex confirmation allows the physician to deliver the patient’s full diagnosis and reduces the psychological distress associated with waiting for confirmatory results. The HCV RNA test (alone) should be used, however, in immunocompromised patients, those who may have had exposure to HCV in the past 6 months, and those suspected of having an HCV re-infection after having cleared the virus.18
Look for the evidence of liver disease
Family physicians should order several additional tests for patients found to have chronic HCV infection before referring such patients to a specialist (ALGORITHM). Work-up should include the complete blood count, HCV genotype (which will help guide treatment), liver function tests, international normalized ratio test, and ultrasound of the liver.18 In addition, all HCV-positive patients should be tested for HIV and HBV, because these co-infections may accelerate liver fibrosis.19,20
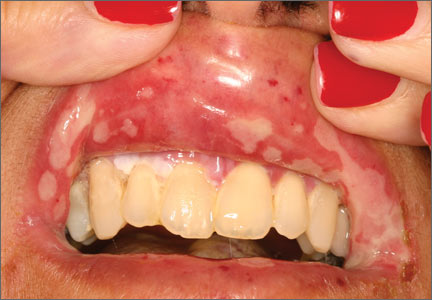
All patients with chronic HCV infection should also be screened for the presence of fibrosis and cirrhosis, as this will influence treatment choice and duration. Signs of cirrhosis that may be evident on physical exam include jaundice, spider angiomata, palmar erythema, encephalopathy with asterixis, and fluid overload, especially ascites. Cirrhosis can be classified clinically as compensated (stage 1 with no varices present and stage 2 with varices present) and decompensated (stages 3 and 4), which is defined as cirrhosis with signs of severe portal hypertension (bleeding varices, ascites, hepatic encephalopathy) or liver insufficiency (jaundice).21 Patients with decompensated cirrhosis should be managed by a liver transplant center. For more on cirrhosis, see “Cirrhosis complications: Keeping them under control” (J Fam Pract. 2015;64:338-342).
Several noninvasive alternatives to liver biopsy
Historically, liver biopsy has been the gold standard for staging liver disease. The Metavir scoring system is a histological assessment of the degree of inflammatory activity and the stage of fibrosis.22 The degree of inflammation activity, which is a precursor of fibrosis, is scored from A0 (no activity) to A3 (severe activity). The staging of fibrosis involves a 5-stage scoring system: F0 (chronic hepatitis without fibrosis); F1 (portal fibrosis without septae); F2 (portal fibrosis with rare septae); F3 (many septae without cirrhosis); or F4 (cirrhosis).
That said, noninvasive tests have largely supplanted liver biopsy for fibrosis screening.
For example, the FibroSure test uses the patient’s age, gender, and a combination of 6 serum markers of liver function in a computational algorithm to generate a quantitative indicator of liver fibrosis, with a score of 0.0 to 1.0 that corresponds to the Metavir fibrosis score (F0-F4), and an inflammatory activity score (A0-A3).23 Similarly, HepaScore uses several noninvasive markers to calculate a score from 0.00 to 1.00. A score ≤0.2 accurately excludes significant fibrosis. However, a score of ≥0.55 or higher corresponds to a Metavir score of at least F2, and in such cases further testing would be needed to evaluate for cirrhosis.24
FDA-approved in 2013, transient elastography (FibroScan) is another noninvasive alternative to liver biopsy for determining the stage of liver disease. This bedside test uses ultrasound technology to measure liver stiffness and provides a score ranging from 0 to 75 kPA that correlates with the Metavir score. Although not yet widely available in the United States, FibroScan is becoming increasingly popular as a rapid and noninvasive screening tool for cirrhosis.25
Identifying cirrhosis in patients who have HCV is crucial because such patients need prompt care from a specialist. In addition to receiving HCV treatment, patients with cirrhosis also need regular liver ultrasound exams to screen for HCC (every 6 months) and esophagogastroduodenoscopy to screen for esophageal and gastric varices.26
Advise patients to avoid alcohol, lose weight
Counsel patients who test positive for HCV infection about making lifestyle changes to avoid further liver damage and transmission of HCV to others. Infectious diseases and hepatology society guidelines recommend vaccination against hepatitis A and B for all HCV-infected patients who are not immune to these viruses because acute co-infection could lead to severe acute liver injury.18,27 Urge all HCV-infected patients to completely abstain from alcohol and, if necessary, refer them to an addiction specialist, because excess alcohol consumption is strongly associated with the development of cirrhosis and HCC.28,29
Comorbid conditions such as metabolic syndrome, obesity, and hyperlipidemia can worsen the prognosis for HCV-infected patients; therefore, intense counseling on weight loss is recommended.30 Statins are safe and beneficial for HCV patients with hypercholesterolemia and compensated cirrhosis.31
Teach patients that the primary mode of transmission of HCV is through infected blood. Sexual transmission of HCV has been well documented in HIV-positive men who have sex with men.32 Although the risk of transmission of HCV among heterosexual couples is extremely low, it is possible, and patients should be counseled accordingly.33 Transmission of HCV from mother to the baby occurs in up to 6% of births and most commonly occurs during delivery.34
Newer treatments are highly effective and well tolderated
HCV treatment has changed dramatically over the past few years. Previous treatments for HCV, particularly those containing interferon, were known for their poor tolerability due to adverse effects and low cure rates. Compared to previous therapies, the new interferon-free direct-acting antiviral (DAA) regimens are not only less complex but also shorter in duration, ranging from 8 to 24 weeks depending on the patient’s viral load, stage of liver disease, and previous treatment experience.18 The specific agents and dosages used in DAA regimens aren’t described here because these regimens are rapidly changing. However, continuously updated treatment recommendations from the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America are available at http://www.hcvguidelines.org.
The goal of HCV treatment is cure as evidenced by a sustained virologic response (SVR), which is defined as the absence of HCV RNA 12 weeks or more after completing treatment.35,36 In general, for the most common genotypes of HCV, treatment with a DAA regimen results in a SVR in ≥95% of patients.18 Achieving SVR is associated with a 50% reduction in all-cause mortality, a 90% reduction in liver-associated mortality, and a >70% reduction in the risk of developing HCC.27,37,38 SVR also has been shown to have a significant effect on reducing extrahepatic manifestations of HCV infection, such as cryoglobulinemia and lymphoma.39-41
Current barriers to the newer, highly effective hepatitis C virus (HCV) infection treatments are largely financial. Although insurance companies have been able to negotiate substantial discounts from the high wholesale price of treatment, many insurance programs require prior authorizations and will approve treatment only for patients with advanced liver fibrosis. In our experience, many patients are left to wait for their liver disease to progress before their insurance company will agree to cover treatment.
In addition, many insurance companies have mandated that only subspecialists prescribe these medications. However, infectious diseases and hepatology specialists and their support staffs are often overburdened with paperwork and phone calls related to prior authorizations and justification of treatment, which can add to delays in treatment.
There is already evidence that treatment of all patients with HCV is cost-effective and leads to better healthcare outcomes42 and there are indications that these barriers will decrease over time, with prices already dropping significantly due to increasing competition between drug companies.
The DAAs are well tolerated and have good safety profiles. In phase III clinical trials of today’s most commonly used DAA regimens, the discontinuation rate was <1% in non-cirrhotic patients and 2% in those with cirrhosis.18 The most commonly reported adverse effects were nausea, fatigue, and headache. DAAs may have drug-drug interactions; therefore, careful medication reconciliation should be performed before initiating treatment.18
Prioritizing treatment. Current evidence supports treatment for all patients with HCV except those with a life expectancy of <12 months.18 Evidence indicates that treatment becomes less effective as a patient’s liver injury progresses to cirrhosis. Due to the high cost of available treatments, however, many insurers have imposed strict criteria for coverage. (See “Barriers to HCV Treatment,” above.42)
The highest priority for treatment has been given to patients with advanced liver fibrosis, compensated cirrhosis, those who have received a liver transplant, and those with severe extrahepatic manifestations (eg, mixed cryoglobulinemia and end-organ disease such as nephropathy). Treatment is also prioritized for high-risk populations (eg, patients with HBV and HIV co-infection, diabetes mellitus) and patients who are at high risk of transmitting the virus (eg, individuals who inject drugs or are incarcerated, men who have sex with men, women of childbearing age, hemodialysis patients, and health care professionals who perform exposure-prone procedures).18
While it may eventually become feasible for family physicians to treat HCV-infected patients, the rapid evolution and significant cost of treatment, as well as the challenges in obtaining insurance coverage, have kept HCV treatment largely in the domain of specialists, at least for now. In the interim, family physicians play a crucial role by screening, diagnosing, and counseling patients with this infection, referring them to specialty care, and providing ongoing monitoring for signs of HCC and esophageal and gastric varices.
CORRESPONDENCE
Laura Wangensteen, MD, Department of Family Medicine, Drexel University, 3401 South Market Street #105 A, Philadelphia, PA 19104; [email protected]
1. Rein DB, Wittenborn JS, Weinbaum CM, et al. Forecasting the morbidity and mortality associated with prevalent cases of precirrhotic chronic hepatitis C in the United States. Dig Liver Dis. 2011;43:66-72.
2. Armstrong GL, Wasley A, Simard EP, et al. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705-714.
3. Chak E, Talal AH, Sherman KE, et al. Hepatitis C virus infection in USA: an estimate of true prevalence. Liver Int. 2011;31:1090-1101.
4. Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103-107.
5. Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology. 2010;51:729-733.
6. Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis. 2000;20:17-35.
7. El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36:S74-S83.
8. Westbrook RH, Dusheiko G. Natural history of hepatitis C. J Hepatol. 2014;61:S58-S68.
9. McCaughan GW, George J. Fibrosis progression in chronic hepatitis C virus infection. Gut. 2004;53:318-321.
10. El-Serag HB, Hampel H, Yeh C, et al. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology. 2002;36:1439-1445.
11. Solinas A, Piras MR, Deplano A. Cognitive dysfunction and hepatitis C virus infection. World J Hepatol. 2015;7:922-925.
12. Fletcher NF, Wilson GK, Murray J, et al. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology. 2012;142:634-643.e6.
13. Smith BD, Morgan RL, Beckett GA, et al; Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945-1965. MMWR Recomm Rep. 2012;61:1-32.
14. US Preventive Services Task Force. Final recommendation statement on hepatitis C screening, June 2013. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/hepatitis-c-screening. Accessed on December 28, 2014.
15. Arora S, Thornton K, Murata G, et al. Outcomes of treatment for hepatitis C virus infection by primary care providers. N Engl J Med. 2011;364:2199-2207.
16. Morrill JA, Shrestha M, Grant RW. Barriers to the treatment of hepatitis C. Patient, provider, and system factors. J Gen Intern Med. 2005;20:754-758.
17. Shivkumar S, Peeling R, Jafari Y, et al. Accuracy of rapid and pointof- care screening tests for hepatitis C: a systematic review and meta-analysis. Ann Intern Med. 2012;157:558-566.
18. American Association for the Study of Liver Diseases; Infectious Diseases Society of America; International Antiviral Society—USA. HCV guidance: Recommendations for testing, managing, and treating hepatitis C. HCV guidelines Web site. Available at: http://www.hcvguidelines.org. Accessed May 25, 2015.
19. Zarski JP, Bohn B, Bastie A, et al. Characteristics of patients with dual infection by hepatitis B and C viruses. J Hepatol. 1998;28:27-33.
20. Graham CS, Baden LR, Yu E, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis. 2001;33:562-569.
21. Garcia-Tsao G, Friedman S, Iredale J, et al. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology. 2010;51:1445-1449.
22. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 1996;24:289-293.
23. Ngo Y, Munteanu M, Messous D, et al. A prospective analysis of the prognostic value of biomarkers (FibroTest) in patients with chronic hepatitis C. Clin Chem. 2006;52:1887-1896.
24. Becker L, Salameh W, Sferruzza A, et al. Validation of hepascore, compared with simple indices of fibrosis, in patients with chronic hepatitis C virus infection in United States. Clin Gastroenterol Hepatol. 2009;7:696-701.
25. Bonder A, Afdhal N. Utilization of FibroScan in clinical practice. Curr Gastroenterol Rep. 2014;16:372.
26. Garcia-Tsao G, Sanyal AJ, Grace ND, et al; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46:922-938.
27. Ghany MG, Strader DB, Thomas DL, et al; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335-1374.
28. Pessione F, Degos F, Marcellin P, et al. Effect of alcohol consumption on serum hepatitis C virus RNA and histological lesions in chronic hepatitis C. Hepatology. 1998;27:1717-1722.
29. Mueller S, Millonig G, Seitz HK. Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J Gastroenterol. 2009;15:3462-3471.
30. Ortiz V, Berenguer M, Rayón JM, et al. Contribution of obesity to hepatitis C-related fibrosis progression. Am J Gastroenterol. 2002;97:2408-2414.
31. Lewis JH, Mortensen ME, Zweig S, et al; Pravastatin in Chronic Liver Disease Study Investigators. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well-compensated chronic liver disease: Results of a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Hepatology. 2007;46:1453-1463.
32. Gamage DG, Read TR, Bradshaw CS, et al. Incidence of hepatitis-C among HIV infected men who have sex with men (MSM) attending a sexual health service: a cohort study. BMC Infect Dis. 2011;11:39.
33. Terrault NA, Dodge JL, Murphy EL, et al. Sexual transmission of hepatitis C virus among monogamous heterosexual couples: the HCV partners study. Hepatology. 2013;57:881-889.
34. Yeung LT, King SM, Roberts EA. Mother-to-infant transmission of hepatitis C virus. Hepatology. 2001;34:223-229.
35. Swain MG, Lai MY, Shiffman ML, et al. A sustained virologic response is durable in patients with chronic hepatitis C treated with peginterferon alfa-2a and ribavirin. Gastroenterology. 2010;139:1593-1601.
36. Thomas AM, Kattakuzhy S, Jones S, et al. SVR durability: HCV patients treated with IFN-free DAA regimens. Presented at: Conference on Retroviruses and Opportunistic Infections (CROI); February, 2015; Seattle, Washington. Abstract 653.
37. Backus LI, Boothroyd DB, Phillips BR, et al. A sustained virologic response reduces risk of all-cause mortality in patients with hepatitis C. Clin Gastroenterol Hepatol. 2011;9:509-516.e1.
38. Russo MW. Antiviral therapy for hepatitis C is associated with improved clinical outcomes in patients with advanced fibrosis. Expert Rev Gastroenterol Hepatol. 2010;4:535-539.
39. Fabrizi F, Dixit V, Messa P. Antiviral therapy of symptomatic HCVassociated mixed cryoglobulinemia: meta-analysis of clinical studies. J Med Virol. 2013;85:1019-1027.
40. Takahashi K, Nishida N, Kawabata H, et al. Regression of Hodgkin lymphoma in response to antiviral therapy for hepatitis C virus infection. Intern Med. 2012;51:2745-2747.
41. Gisbert JP, García-Buey L, Pajares JM, et al. Systematic review: regression of lymphoproliferative disorders after treatment for hepatitis C infection. Aliment Pharmacol Ther. 2005;21:653-662.
42. Najafzadeh M, Andersson K, Shrank WH, et al. Cost-effectiveness of novel regimens for the treatment of hepatitis C virus. Ann Intern Med. 2015;162:407-419.
› Screen at-risk patients and all those born between 1945 and 1965 for hepatitis C virus (HCV) infection. B
› Screen HCV-positive patients for level of fibrosis and for conditions that may accelerate liver disease, including alcohol use, hepatitis B virus, and human immunodeficiency virus. B
› Continuously monitor patients with chronic HCV for the development of cirrhosis and hepatocellular carcinoma. A
› Refer patients to specialty care for HCV treatment and, if they have cirrhosis, for potential transplant evaluation. C
› Counsel HCV-positive patients about how to avoid transmission to others. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hepatitis C virus (HCV) infection is a leading cause of chronic liver disease. Over the next few decades, the number of deaths per year due to complications of HCV such as liver failure and hepatocellular carcinoma (HCC) is predicted to more than triple to 36,000 by 2032.1
Fortunately, major advances in drug therapy have made it possible to cure patients of HCV, and treatment is now less complex, of shorter duration, and better tolerated than it once was. To help family physicians maximize the care they provide to these patients, we’ve summarized screening recommendations from the Centers for Disease Control and Prevention (CDC), innovative alternatives to biopsy for staging liver disease, and counseling points to cover with patients.
A common, usually silent infection with potentially fatal complications
According to the National Health and Nutrition Examination Survey (NHANES), an estimated 2.7 to 3.9 million people in the United States are chronically infected with HCV, about threefourths of whom were born between 1945 and 1965 (the “baby boomer” generation).2 However, by adding “unaccounted groups” (eg, incarcerated, homeless, and active duty military) to these estimates, the number of people with HCV is likely more than 5.2 million.3
HCV is a ribonucleic acid (RNA) virus capable of mutating at a high rate to escape detection and clearance by the host’s immune system.4 Most patients with HCV are asymptomatic during the acute and chronic phases of infection, and may have a silent infection for decades. In fact, 65% to 75% of patients with HCV are unaware of their infection.5
Approximately 20% of chronically infected patients develop cirrhosis after 20 years and, once they do, the annual rate of HCC and liver decompensation is about 5%.6-8 Risk factors for advancement to cirrhosis includes male sex, alcohol consumption, co-infection with human immunodeficiency virus (HIV) or hepatitis B virus (HBV), immunosuppression, having had HCV infection for a long time, becoming infected with HCV after age 40, and not having responded to previous treatment.9
Chronic HCV infection can lead to extrahepatic manifestations such as essential mixed cryoglobulinemia, porphyria cutanea tarda, membranoproliferative glomerulonephritis, lymphoma, and glucose intolerance.10 There is also growing evidence that HCV infection affects cognitive function in the absence of fibrosis and hepatic encephalopathy. Several studies show that HCV-infected patients score poorly on neuropsychological testing for verbal learning, attention, memory, and executive function.11 This may be related to the expression of receptors for HCV by the brain’s microvascular endothelial cells.12
Screening recommendations. Given the high prevalence of HCV infection among baby boomers, the CDC decided in 2012 to recommend one-time HCV screening for all patients born between 1945 and 1965.13 This is in addition to risk-based screening for all patients who have a history of injection drug use, those on long-term hemodialysis or with tattoos obtained in unregulated settings, offspring of HCV-infected mothers, and those with health-care associated exposures (TABLE13). In 2013, the US Preventive Services Task Force upgraded its recommendation to match those of the CDC.14
Despite these recommendations, which are expected to increase detection of HCV among asymptomatic persons who do not know they are infected, there remain significant barriers to HCV testing. These include poor access to primary care and preventive services, lack of knowledge and awareness of the disease among patients and providers, and a lack of studies that support a universal screening approach for HCV.5,15,16 One tool that might help overcome some of these barriers and aid family physicians in the screening process is automatic reminders or standing lab orders for HCV testing in electronic medical records systems.
Screening for HCV can be done using any of the US Food and Drug Administration (FDA)-approved tests for the anti-HCV antibody, which have sensitivities and specificities greater than 99%.17 A positive screening result should be confirmed with an HCV RNA test. However, for practical purposes, ordering the anti-HCV test with reflex to the HCV RNA test decreases the number of blood draws and office visits required of the patient. The reflex confirmation allows the physician to deliver the patient’s full diagnosis and reduces the psychological distress associated with waiting for confirmatory results. The HCV RNA test (alone) should be used, however, in immunocompromised patients, those who may have had exposure to HCV in the past 6 months, and those suspected of having an HCV re-infection after having cleared the virus.18
Look for the evidence of liver disease
Family physicians should order several additional tests for patients found to have chronic HCV infection before referring such patients to a specialist (ALGORITHM). Work-up should include the complete blood count, HCV genotype (which will help guide treatment), liver function tests, international normalized ratio test, and ultrasound of the liver.18 In addition, all HCV-positive patients should be tested for HIV and HBV, because these co-infections may accelerate liver fibrosis.19,20
All patients with chronic HCV infection should also be screened for the presence of fibrosis and cirrhosis, as this will influence treatment choice and duration. Signs of cirrhosis that may be evident on physical exam include jaundice, spider angiomata, palmar erythema, encephalopathy with asterixis, and fluid overload, especially ascites. Cirrhosis can be classified clinically as compensated (stage 1 with no varices present and stage 2 with varices present) and decompensated (stages 3 and 4), which is defined as cirrhosis with signs of severe portal hypertension (bleeding varices, ascites, hepatic encephalopathy) or liver insufficiency (jaundice).21 Patients with decompensated cirrhosis should be managed by a liver transplant center. For more on cirrhosis, see “Cirrhosis complications: Keeping them under control” (J Fam Pract. 2015;64:338-342).
Several noninvasive alternatives to liver biopsy
Historically, liver biopsy has been the gold standard for staging liver disease. The Metavir scoring system is a histological assessment of the degree of inflammatory activity and the stage of fibrosis.22 The degree of inflammation activity, which is a precursor of fibrosis, is scored from A0 (no activity) to A3 (severe activity). The staging of fibrosis involves a 5-stage scoring system: F0 (chronic hepatitis without fibrosis); F1 (portal fibrosis without septae); F2 (portal fibrosis with rare septae); F3 (many septae without cirrhosis); or F4 (cirrhosis).
That said, noninvasive tests have largely supplanted liver biopsy for fibrosis screening.
For example, the FibroSure test uses the patient’s age, gender, and a combination of 6 serum markers of liver function in a computational algorithm to generate a quantitative indicator of liver fibrosis, with a score of 0.0 to 1.0 that corresponds to the Metavir fibrosis score (F0-F4), and an inflammatory activity score (A0-A3).23 Similarly, HepaScore uses several noninvasive markers to calculate a score from 0.00 to 1.00. A score ≤0.2 accurately excludes significant fibrosis. However, a score of ≥0.55 or higher corresponds to a Metavir score of at least F2, and in such cases further testing would be needed to evaluate for cirrhosis.24
FDA-approved in 2013, transient elastography (FibroScan) is another noninvasive alternative to liver biopsy for determining the stage of liver disease. This bedside test uses ultrasound technology to measure liver stiffness and provides a score ranging from 0 to 75 kPA that correlates with the Metavir score. Although not yet widely available in the United States, FibroScan is becoming increasingly popular as a rapid and noninvasive screening tool for cirrhosis.25
Identifying cirrhosis in patients who have HCV is crucial because such patients need prompt care from a specialist. In addition to receiving HCV treatment, patients with cirrhosis also need regular liver ultrasound exams to screen for HCC (every 6 months) and esophagogastroduodenoscopy to screen for esophageal and gastric varices.26
Advise patients to avoid alcohol, lose weight
Counsel patients who test positive for HCV infection about making lifestyle changes to avoid further liver damage and transmission of HCV to others. Infectious diseases and hepatology society guidelines recommend vaccination against hepatitis A and B for all HCV-infected patients who are not immune to these viruses because acute co-infection could lead to severe acute liver injury.18,27 Urge all HCV-infected patients to completely abstain from alcohol and, if necessary, refer them to an addiction specialist, because excess alcohol consumption is strongly associated with the development of cirrhosis and HCC.28,29
Comorbid conditions such as metabolic syndrome, obesity, and hyperlipidemia can worsen the prognosis for HCV-infected patients; therefore, intense counseling on weight loss is recommended.30 Statins are safe and beneficial for HCV patients with hypercholesterolemia and compensated cirrhosis.31
Teach patients that the primary mode of transmission of HCV is through infected blood. Sexual transmission of HCV has been well documented in HIV-positive men who have sex with men.32 Although the risk of transmission of HCV among heterosexual couples is extremely low, it is possible, and patients should be counseled accordingly.33 Transmission of HCV from mother to the baby occurs in up to 6% of births and most commonly occurs during delivery.34
Newer treatments are highly effective and well tolderated
HCV treatment has changed dramatically over the past few years. Previous treatments for HCV, particularly those containing interferon, were known for their poor tolerability due to adverse effects and low cure rates. Compared to previous therapies, the new interferon-free direct-acting antiviral (DAA) regimens are not only less complex but also shorter in duration, ranging from 8 to 24 weeks depending on the patient’s viral load, stage of liver disease, and previous treatment experience.18 The specific agents and dosages used in DAA regimens aren’t described here because these regimens are rapidly changing. However, continuously updated treatment recommendations from the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America are available at http://www.hcvguidelines.org.
The goal of HCV treatment is cure as evidenced by a sustained virologic response (SVR), which is defined as the absence of HCV RNA 12 weeks or more after completing treatment.35,36 In general, for the most common genotypes of HCV, treatment with a DAA regimen results in a SVR in ≥95% of patients.18 Achieving SVR is associated with a 50% reduction in all-cause mortality, a 90% reduction in liver-associated mortality, and a >70% reduction in the risk of developing HCC.27,37,38 SVR also has been shown to have a significant effect on reducing extrahepatic manifestations of HCV infection, such as cryoglobulinemia and lymphoma.39-41
Current barriers to the newer, highly effective hepatitis C virus (HCV) infection treatments are largely financial. Although insurance companies have been able to negotiate substantial discounts from the high wholesale price of treatment, many insurance programs require prior authorizations and will approve treatment only for patients with advanced liver fibrosis. In our experience, many patients are left to wait for their liver disease to progress before their insurance company will agree to cover treatment.
In addition, many insurance companies have mandated that only subspecialists prescribe these medications. However, infectious diseases and hepatology specialists and their support staffs are often overburdened with paperwork and phone calls related to prior authorizations and justification of treatment, which can add to delays in treatment.
There is already evidence that treatment of all patients with HCV is cost-effective and leads to better healthcare outcomes42 and there are indications that these barriers will decrease over time, with prices already dropping significantly due to increasing competition between drug companies.
The DAAs are well tolerated and have good safety profiles. In phase III clinical trials of today’s most commonly used DAA regimens, the discontinuation rate was <1% in non-cirrhotic patients and 2% in those with cirrhosis.18 The most commonly reported adverse effects were nausea, fatigue, and headache. DAAs may have drug-drug interactions; therefore, careful medication reconciliation should be performed before initiating treatment.18
Prioritizing treatment. Current evidence supports treatment for all patients with HCV except those with a life expectancy of <12 months.18 Evidence indicates that treatment becomes less effective as a patient’s liver injury progresses to cirrhosis. Due to the high cost of available treatments, however, many insurers have imposed strict criteria for coverage. (See “Barriers to HCV Treatment,” above.42)
The highest priority for treatment has been given to patients with advanced liver fibrosis, compensated cirrhosis, those who have received a liver transplant, and those with severe extrahepatic manifestations (eg, mixed cryoglobulinemia and end-organ disease such as nephropathy). Treatment is also prioritized for high-risk populations (eg, patients with HBV and HIV co-infection, diabetes mellitus) and patients who are at high risk of transmitting the virus (eg, individuals who inject drugs or are incarcerated, men who have sex with men, women of childbearing age, hemodialysis patients, and health care professionals who perform exposure-prone procedures).18
While it may eventually become feasible for family physicians to treat HCV-infected patients, the rapid evolution and significant cost of treatment, as well as the challenges in obtaining insurance coverage, have kept HCV treatment largely in the domain of specialists, at least for now. In the interim, family physicians play a crucial role by screening, diagnosing, and counseling patients with this infection, referring them to specialty care, and providing ongoing monitoring for signs of HCC and esophageal and gastric varices.
CORRESPONDENCE
Laura Wangensteen, MD, Department of Family Medicine, Drexel University, 3401 South Market Street #105 A, Philadelphia, PA 19104; [email protected]
› Screen at-risk patients and all those born between 1945 and 1965 for hepatitis C virus (HCV) infection. B
› Screen HCV-positive patients for level of fibrosis and for conditions that may accelerate liver disease, including alcohol use, hepatitis B virus, and human immunodeficiency virus. B
› Continuously monitor patients with chronic HCV for the development of cirrhosis and hepatocellular carcinoma. A
› Refer patients to specialty care for HCV treatment and, if they have cirrhosis, for potential transplant evaluation. C
› Counsel HCV-positive patients about how to avoid transmission to others. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hepatitis C virus (HCV) infection is a leading cause of chronic liver disease. Over the next few decades, the number of deaths per year due to complications of HCV such as liver failure and hepatocellular carcinoma (HCC) is predicted to more than triple to 36,000 by 2032.1
Fortunately, major advances in drug therapy have made it possible to cure patients of HCV, and treatment is now less complex, of shorter duration, and better tolerated than it once was. To help family physicians maximize the care they provide to these patients, we’ve summarized screening recommendations from the Centers for Disease Control and Prevention (CDC), innovative alternatives to biopsy for staging liver disease, and counseling points to cover with patients.
A common, usually silent infection with potentially fatal complications
According to the National Health and Nutrition Examination Survey (NHANES), an estimated 2.7 to 3.9 million people in the United States are chronically infected with HCV, about threefourths of whom were born between 1945 and 1965 (the “baby boomer” generation).2 However, by adding “unaccounted groups” (eg, incarcerated, homeless, and active duty military) to these estimates, the number of people with HCV is likely more than 5.2 million.3
HCV is a ribonucleic acid (RNA) virus capable of mutating at a high rate to escape detection and clearance by the host’s immune system.4 Most patients with HCV are asymptomatic during the acute and chronic phases of infection, and may have a silent infection for decades. In fact, 65% to 75% of patients with HCV are unaware of their infection.5
Approximately 20% of chronically infected patients develop cirrhosis after 20 years and, once they do, the annual rate of HCC and liver decompensation is about 5%.6-8 Risk factors for advancement to cirrhosis includes male sex, alcohol consumption, co-infection with human immunodeficiency virus (HIV) or hepatitis B virus (HBV), immunosuppression, having had HCV infection for a long time, becoming infected with HCV after age 40, and not having responded to previous treatment.9
Chronic HCV infection can lead to extrahepatic manifestations such as essential mixed cryoglobulinemia, porphyria cutanea tarda, membranoproliferative glomerulonephritis, lymphoma, and glucose intolerance.10 There is also growing evidence that HCV infection affects cognitive function in the absence of fibrosis and hepatic encephalopathy. Several studies show that HCV-infected patients score poorly on neuropsychological testing for verbal learning, attention, memory, and executive function.11 This may be related to the expression of receptors for HCV by the brain’s microvascular endothelial cells.12
Screening recommendations. Given the high prevalence of HCV infection among baby boomers, the CDC decided in 2012 to recommend one-time HCV screening for all patients born between 1945 and 1965.13 This is in addition to risk-based screening for all patients who have a history of injection drug use, those on long-term hemodialysis or with tattoos obtained in unregulated settings, offspring of HCV-infected mothers, and those with health-care associated exposures (TABLE13). In 2013, the US Preventive Services Task Force upgraded its recommendation to match those of the CDC.14
Despite these recommendations, which are expected to increase detection of HCV among asymptomatic persons who do not know they are infected, there remain significant barriers to HCV testing. These include poor access to primary care and preventive services, lack of knowledge and awareness of the disease among patients and providers, and a lack of studies that support a universal screening approach for HCV.5,15,16 One tool that might help overcome some of these barriers and aid family physicians in the screening process is automatic reminders or standing lab orders for HCV testing in electronic medical records systems.
Screening for HCV can be done using any of the US Food and Drug Administration (FDA)-approved tests for the anti-HCV antibody, which have sensitivities and specificities greater than 99%.17 A positive screening result should be confirmed with an HCV RNA test. However, for practical purposes, ordering the anti-HCV test with reflex to the HCV RNA test decreases the number of blood draws and office visits required of the patient. The reflex confirmation allows the physician to deliver the patient’s full diagnosis and reduces the psychological distress associated with waiting for confirmatory results. The HCV RNA test (alone) should be used, however, in immunocompromised patients, those who may have had exposure to HCV in the past 6 months, and those suspected of having an HCV re-infection after having cleared the virus.18
Look for the evidence of liver disease
Family physicians should order several additional tests for patients found to have chronic HCV infection before referring such patients to a specialist (ALGORITHM). Work-up should include the complete blood count, HCV genotype (which will help guide treatment), liver function tests, international normalized ratio test, and ultrasound of the liver.18 In addition, all HCV-positive patients should be tested for HIV and HBV, because these co-infections may accelerate liver fibrosis.19,20
All patients with chronic HCV infection should also be screened for the presence of fibrosis and cirrhosis, as this will influence treatment choice and duration. Signs of cirrhosis that may be evident on physical exam include jaundice, spider angiomata, palmar erythema, encephalopathy with asterixis, and fluid overload, especially ascites. Cirrhosis can be classified clinically as compensated (stage 1 with no varices present and stage 2 with varices present) and decompensated (stages 3 and 4), which is defined as cirrhosis with signs of severe portal hypertension (bleeding varices, ascites, hepatic encephalopathy) or liver insufficiency (jaundice).21 Patients with decompensated cirrhosis should be managed by a liver transplant center. For more on cirrhosis, see “Cirrhosis complications: Keeping them under control” (J Fam Pract. 2015;64:338-342).
Several noninvasive alternatives to liver biopsy
Historically, liver biopsy has been the gold standard for staging liver disease. The Metavir scoring system is a histological assessment of the degree of inflammatory activity and the stage of fibrosis.22 The degree of inflammation activity, which is a precursor of fibrosis, is scored from A0 (no activity) to A3 (severe activity). The staging of fibrosis involves a 5-stage scoring system: F0 (chronic hepatitis without fibrosis); F1 (portal fibrosis without septae); F2 (portal fibrosis with rare septae); F3 (many septae without cirrhosis); or F4 (cirrhosis).
That said, noninvasive tests have largely supplanted liver biopsy for fibrosis screening.
For example, the FibroSure test uses the patient’s age, gender, and a combination of 6 serum markers of liver function in a computational algorithm to generate a quantitative indicator of liver fibrosis, with a score of 0.0 to 1.0 that corresponds to the Metavir fibrosis score (F0-F4), and an inflammatory activity score (A0-A3).23 Similarly, HepaScore uses several noninvasive markers to calculate a score from 0.00 to 1.00. A score ≤0.2 accurately excludes significant fibrosis. However, a score of ≥0.55 or higher corresponds to a Metavir score of at least F2, and in such cases further testing would be needed to evaluate for cirrhosis.24
FDA-approved in 2013, transient elastography (FibroScan) is another noninvasive alternative to liver biopsy for determining the stage of liver disease. This bedside test uses ultrasound technology to measure liver stiffness and provides a score ranging from 0 to 75 kPA that correlates with the Metavir score. Although not yet widely available in the United States, FibroScan is becoming increasingly popular as a rapid and noninvasive screening tool for cirrhosis.25
Identifying cirrhosis in patients who have HCV is crucial because such patients need prompt care from a specialist. In addition to receiving HCV treatment, patients with cirrhosis also need regular liver ultrasound exams to screen for HCC (every 6 months) and esophagogastroduodenoscopy to screen for esophageal and gastric varices.26
Advise patients to avoid alcohol, lose weight
Counsel patients who test positive for HCV infection about making lifestyle changes to avoid further liver damage and transmission of HCV to others. Infectious diseases and hepatology society guidelines recommend vaccination against hepatitis A and B for all HCV-infected patients who are not immune to these viruses because acute co-infection could lead to severe acute liver injury.18,27 Urge all HCV-infected patients to completely abstain from alcohol and, if necessary, refer them to an addiction specialist, because excess alcohol consumption is strongly associated with the development of cirrhosis and HCC.28,29
Comorbid conditions such as metabolic syndrome, obesity, and hyperlipidemia can worsen the prognosis for HCV-infected patients; therefore, intense counseling on weight loss is recommended.30 Statins are safe and beneficial for HCV patients with hypercholesterolemia and compensated cirrhosis.31
Teach patients that the primary mode of transmission of HCV is through infected blood. Sexual transmission of HCV has been well documented in HIV-positive men who have sex with men.32 Although the risk of transmission of HCV among heterosexual couples is extremely low, it is possible, and patients should be counseled accordingly.33 Transmission of HCV from mother to the baby occurs in up to 6% of births and most commonly occurs during delivery.34
Newer treatments are highly effective and well tolderated
HCV treatment has changed dramatically over the past few years. Previous treatments for HCV, particularly those containing interferon, were known for their poor tolerability due to adverse effects and low cure rates. Compared to previous therapies, the new interferon-free direct-acting antiviral (DAA) regimens are not only less complex but also shorter in duration, ranging from 8 to 24 weeks depending on the patient’s viral load, stage of liver disease, and previous treatment experience.18 The specific agents and dosages used in DAA regimens aren’t described here because these regimens are rapidly changing. However, continuously updated treatment recommendations from the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America are available at http://www.hcvguidelines.org.
The goal of HCV treatment is cure as evidenced by a sustained virologic response (SVR), which is defined as the absence of HCV RNA 12 weeks or more after completing treatment.35,36 In general, for the most common genotypes of HCV, treatment with a DAA regimen results in a SVR in ≥95% of patients.18 Achieving SVR is associated with a 50% reduction in all-cause mortality, a 90% reduction in liver-associated mortality, and a >70% reduction in the risk of developing HCC.27,37,38 SVR also has been shown to have a significant effect on reducing extrahepatic manifestations of HCV infection, such as cryoglobulinemia and lymphoma.39-41
Current barriers to the newer, highly effective hepatitis C virus (HCV) infection treatments are largely financial. Although insurance companies have been able to negotiate substantial discounts from the high wholesale price of treatment, many insurance programs require prior authorizations and will approve treatment only for patients with advanced liver fibrosis. In our experience, many patients are left to wait for their liver disease to progress before their insurance company will agree to cover treatment.
In addition, many insurance companies have mandated that only subspecialists prescribe these medications. However, infectious diseases and hepatology specialists and their support staffs are often overburdened with paperwork and phone calls related to prior authorizations and justification of treatment, which can add to delays in treatment.
There is already evidence that treatment of all patients with HCV is cost-effective and leads to better healthcare outcomes42 and there are indications that these barriers will decrease over time, with prices already dropping significantly due to increasing competition between drug companies.
The DAAs are well tolerated and have good safety profiles. In phase III clinical trials of today’s most commonly used DAA regimens, the discontinuation rate was <1% in non-cirrhotic patients and 2% in those with cirrhosis.18 The most commonly reported adverse effects were nausea, fatigue, and headache. DAAs may have drug-drug interactions; therefore, careful medication reconciliation should be performed before initiating treatment.18
Prioritizing treatment. Current evidence supports treatment for all patients with HCV except those with a life expectancy of <12 months.18 Evidence indicates that treatment becomes less effective as a patient’s liver injury progresses to cirrhosis. Due to the high cost of available treatments, however, many insurers have imposed strict criteria for coverage. (See “Barriers to HCV Treatment,” above.42)
The highest priority for treatment has been given to patients with advanced liver fibrosis, compensated cirrhosis, those who have received a liver transplant, and those with severe extrahepatic manifestations (eg, mixed cryoglobulinemia and end-organ disease such as nephropathy). Treatment is also prioritized for high-risk populations (eg, patients with HBV and HIV co-infection, diabetes mellitus) and patients who are at high risk of transmitting the virus (eg, individuals who inject drugs or are incarcerated, men who have sex with men, women of childbearing age, hemodialysis patients, and health care professionals who perform exposure-prone procedures).18
While it may eventually become feasible for family physicians to treat HCV-infected patients, the rapid evolution and significant cost of treatment, as well as the challenges in obtaining insurance coverage, have kept HCV treatment largely in the domain of specialists, at least for now. In the interim, family physicians play a crucial role by screening, diagnosing, and counseling patients with this infection, referring them to specialty care, and providing ongoing monitoring for signs of HCC and esophageal and gastric varices.
CORRESPONDENCE
Laura Wangensteen, MD, Department of Family Medicine, Drexel University, 3401 South Market Street #105 A, Philadelphia, PA 19104; [email protected]
1. Rein DB, Wittenborn JS, Weinbaum CM, et al. Forecasting the morbidity and mortality associated with prevalent cases of precirrhotic chronic hepatitis C in the United States. Dig Liver Dis. 2011;43:66-72.
2. Armstrong GL, Wasley A, Simard EP, et al. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705-714.
3. Chak E, Talal AH, Sherman KE, et al. Hepatitis C virus infection in USA: an estimate of true prevalence. Liver Int. 2011;31:1090-1101.
4. Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103-107.
5. Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology. 2010;51:729-733.
6. Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis. 2000;20:17-35.
7. El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36:S74-S83.
8. Westbrook RH, Dusheiko G. Natural history of hepatitis C. J Hepatol. 2014;61:S58-S68.
9. McCaughan GW, George J. Fibrosis progression in chronic hepatitis C virus infection. Gut. 2004;53:318-321.
10. El-Serag HB, Hampel H, Yeh C, et al. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology. 2002;36:1439-1445.
11. Solinas A, Piras MR, Deplano A. Cognitive dysfunction and hepatitis C virus infection. World J Hepatol. 2015;7:922-925.
12. Fletcher NF, Wilson GK, Murray J, et al. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology. 2012;142:634-643.e6.
13. Smith BD, Morgan RL, Beckett GA, et al; Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945-1965. MMWR Recomm Rep. 2012;61:1-32.
14. US Preventive Services Task Force. Final recommendation statement on hepatitis C screening, June 2013. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/hepatitis-c-screening. Accessed on December 28, 2014.
15. Arora S, Thornton K, Murata G, et al. Outcomes of treatment for hepatitis C virus infection by primary care providers. N Engl J Med. 2011;364:2199-2207.
16. Morrill JA, Shrestha M, Grant RW. Barriers to the treatment of hepatitis C. Patient, provider, and system factors. J Gen Intern Med. 2005;20:754-758.
17. Shivkumar S, Peeling R, Jafari Y, et al. Accuracy of rapid and pointof- care screening tests for hepatitis C: a systematic review and meta-analysis. Ann Intern Med. 2012;157:558-566.
18. American Association for the Study of Liver Diseases; Infectious Diseases Society of America; International Antiviral Society—USA. HCV guidance: Recommendations for testing, managing, and treating hepatitis C. HCV guidelines Web site. Available at: http://www.hcvguidelines.org. Accessed May 25, 2015.
19. Zarski JP, Bohn B, Bastie A, et al. Characteristics of patients with dual infection by hepatitis B and C viruses. J Hepatol. 1998;28:27-33.
20. Graham CS, Baden LR, Yu E, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis. 2001;33:562-569.
21. Garcia-Tsao G, Friedman S, Iredale J, et al. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology. 2010;51:1445-1449.
22. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 1996;24:289-293.
23. Ngo Y, Munteanu M, Messous D, et al. A prospective analysis of the prognostic value of biomarkers (FibroTest) in patients with chronic hepatitis C. Clin Chem. 2006;52:1887-1896.
24. Becker L, Salameh W, Sferruzza A, et al. Validation of hepascore, compared with simple indices of fibrosis, in patients with chronic hepatitis C virus infection in United States. Clin Gastroenterol Hepatol. 2009;7:696-701.
25. Bonder A, Afdhal N. Utilization of FibroScan in clinical practice. Curr Gastroenterol Rep. 2014;16:372.
26. Garcia-Tsao G, Sanyal AJ, Grace ND, et al; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46:922-938.
27. Ghany MG, Strader DB, Thomas DL, et al; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335-1374.
28. Pessione F, Degos F, Marcellin P, et al. Effect of alcohol consumption on serum hepatitis C virus RNA and histological lesions in chronic hepatitis C. Hepatology. 1998;27:1717-1722.
29. Mueller S, Millonig G, Seitz HK. Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J Gastroenterol. 2009;15:3462-3471.
30. Ortiz V, Berenguer M, Rayón JM, et al. Contribution of obesity to hepatitis C-related fibrosis progression. Am J Gastroenterol. 2002;97:2408-2414.
31. Lewis JH, Mortensen ME, Zweig S, et al; Pravastatin in Chronic Liver Disease Study Investigators. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well-compensated chronic liver disease: Results of a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Hepatology. 2007;46:1453-1463.
32. Gamage DG, Read TR, Bradshaw CS, et al. Incidence of hepatitis-C among HIV infected men who have sex with men (MSM) attending a sexual health service: a cohort study. BMC Infect Dis. 2011;11:39.
33. Terrault NA, Dodge JL, Murphy EL, et al. Sexual transmission of hepatitis C virus among monogamous heterosexual couples: the HCV partners study. Hepatology. 2013;57:881-889.
34. Yeung LT, King SM, Roberts EA. Mother-to-infant transmission of hepatitis C virus. Hepatology. 2001;34:223-229.
35. Swain MG, Lai MY, Shiffman ML, et al. A sustained virologic response is durable in patients with chronic hepatitis C treated with peginterferon alfa-2a and ribavirin. Gastroenterology. 2010;139:1593-1601.
36. Thomas AM, Kattakuzhy S, Jones S, et al. SVR durability: HCV patients treated with IFN-free DAA regimens. Presented at: Conference on Retroviruses and Opportunistic Infections (CROI); February, 2015; Seattle, Washington. Abstract 653.
37. Backus LI, Boothroyd DB, Phillips BR, et al. A sustained virologic response reduces risk of all-cause mortality in patients with hepatitis C. Clin Gastroenterol Hepatol. 2011;9:509-516.e1.
38. Russo MW. Antiviral therapy for hepatitis C is associated with improved clinical outcomes in patients with advanced fibrosis. Expert Rev Gastroenterol Hepatol. 2010;4:535-539.
39. Fabrizi F, Dixit V, Messa P. Antiviral therapy of symptomatic HCVassociated mixed cryoglobulinemia: meta-analysis of clinical studies. J Med Virol. 2013;85:1019-1027.
40. Takahashi K, Nishida N, Kawabata H, et al. Regression of Hodgkin lymphoma in response to antiviral therapy for hepatitis C virus infection. Intern Med. 2012;51:2745-2747.
41. Gisbert JP, García-Buey L, Pajares JM, et al. Systematic review: regression of lymphoproliferative disorders after treatment for hepatitis C infection. Aliment Pharmacol Ther. 2005;21:653-662.
42. Najafzadeh M, Andersson K, Shrank WH, et al. Cost-effectiveness of novel regimens for the treatment of hepatitis C virus. Ann Intern Med. 2015;162:407-419.
1. Rein DB, Wittenborn JS, Weinbaum CM, et al. Forecasting the morbidity and mortality associated with prevalent cases of precirrhotic chronic hepatitis C in the United States. Dig Liver Dis. 2011;43:66-72.
2. Armstrong GL, Wasley A, Simard EP, et al. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705-714.
3. Chak E, Talal AH, Sherman KE, et al. Hepatitis C virus infection in USA: an estimate of true prevalence. Liver Int. 2011;31:1090-1101.
4. Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103-107.
5. Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology. 2010;51:729-733.
6. Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis. 2000;20:17-35.
7. El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36:S74-S83.
8. Westbrook RH, Dusheiko G. Natural history of hepatitis C. J Hepatol. 2014;61:S58-S68.
9. McCaughan GW, George J. Fibrosis progression in chronic hepatitis C virus infection. Gut. 2004;53:318-321.
10. El-Serag HB, Hampel H, Yeh C, et al. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology. 2002;36:1439-1445.
11. Solinas A, Piras MR, Deplano A. Cognitive dysfunction and hepatitis C virus infection. World J Hepatol. 2015;7:922-925.
12. Fletcher NF, Wilson GK, Murray J, et al. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology. 2012;142:634-643.e6.
13. Smith BD, Morgan RL, Beckett GA, et al; Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945-1965. MMWR Recomm Rep. 2012;61:1-32.
14. US Preventive Services Task Force. Final recommendation statement on hepatitis C screening, June 2013. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/hepatitis-c-screening. Accessed on December 28, 2014.
15. Arora S, Thornton K, Murata G, et al. Outcomes of treatment for hepatitis C virus infection by primary care providers. N Engl J Med. 2011;364:2199-2207.
16. Morrill JA, Shrestha M, Grant RW. Barriers to the treatment of hepatitis C. Patient, provider, and system factors. J Gen Intern Med. 2005;20:754-758.
17. Shivkumar S, Peeling R, Jafari Y, et al. Accuracy of rapid and pointof- care screening tests for hepatitis C: a systematic review and meta-analysis. Ann Intern Med. 2012;157:558-566.
18. American Association for the Study of Liver Diseases; Infectious Diseases Society of America; International Antiviral Society—USA. HCV guidance: Recommendations for testing, managing, and treating hepatitis C. HCV guidelines Web site. Available at: http://www.hcvguidelines.org. Accessed May 25, 2015.
19. Zarski JP, Bohn B, Bastie A, et al. Characteristics of patients with dual infection by hepatitis B and C viruses. J Hepatol. 1998;28:27-33.
20. Graham CS, Baden LR, Yu E, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis. 2001;33:562-569.
21. Garcia-Tsao G, Friedman S, Iredale J, et al. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology. 2010;51:1445-1449.
22. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 1996;24:289-293.
23. Ngo Y, Munteanu M, Messous D, et al. A prospective analysis of the prognostic value of biomarkers (FibroTest) in patients with chronic hepatitis C. Clin Chem. 2006;52:1887-1896.
24. Becker L, Salameh W, Sferruzza A, et al. Validation of hepascore, compared with simple indices of fibrosis, in patients with chronic hepatitis C virus infection in United States. Clin Gastroenterol Hepatol. 2009;7:696-701.
25. Bonder A, Afdhal N. Utilization of FibroScan in clinical practice. Curr Gastroenterol Rep. 2014;16:372.
26. Garcia-Tsao G, Sanyal AJ, Grace ND, et al; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46:922-938.
27. Ghany MG, Strader DB, Thomas DL, et al; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335-1374.
28. Pessione F, Degos F, Marcellin P, et al. Effect of alcohol consumption on serum hepatitis C virus RNA and histological lesions in chronic hepatitis C. Hepatology. 1998;27:1717-1722.
29. Mueller S, Millonig G, Seitz HK. Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J Gastroenterol. 2009;15:3462-3471.
30. Ortiz V, Berenguer M, Rayón JM, et al. Contribution of obesity to hepatitis C-related fibrosis progression. Am J Gastroenterol. 2002;97:2408-2414.
31. Lewis JH, Mortensen ME, Zweig S, et al; Pravastatin in Chronic Liver Disease Study Investigators. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well-compensated chronic liver disease: Results of a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Hepatology. 2007;46:1453-1463.
32. Gamage DG, Read TR, Bradshaw CS, et al. Incidence of hepatitis-C among HIV infected men who have sex with men (MSM) attending a sexual health service: a cohort study. BMC Infect Dis. 2011;11:39.
33. Terrault NA, Dodge JL, Murphy EL, et al. Sexual transmission of hepatitis C virus among monogamous heterosexual couples: the HCV partners study. Hepatology. 2013;57:881-889.
34. Yeung LT, King SM, Roberts EA. Mother-to-infant transmission of hepatitis C virus. Hepatology. 2001;34:223-229.
35. Swain MG, Lai MY, Shiffman ML, et al. A sustained virologic response is durable in patients with chronic hepatitis C treated with peginterferon alfa-2a and ribavirin. Gastroenterology. 2010;139:1593-1601.
36. Thomas AM, Kattakuzhy S, Jones S, et al. SVR durability: HCV patients treated with IFN-free DAA regimens. Presented at: Conference on Retroviruses and Opportunistic Infections (CROI); February, 2015; Seattle, Washington. Abstract 653.
37. Backus LI, Boothroyd DB, Phillips BR, et al. A sustained virologic response reduces risk of all-cause mortality in patients with hepatitis C. Clin Gastroenterol Hepatol. 2011;9:509-516.e1.
38. Russo MW. Antiviral therapy for hepatitis C is associated with improved clinical outcomes in patients with advanced fibrosis. Expert Rev Gastroenterol Hepatol. 2010;4:535-539.
39. Fabrizi F, Dixit V, Messa P. Antiviral therapy of symptomatic HCVassociated mixed cryoglobulinemia: meta-analysis of clinical studies. J Med Virol. 2013;85:1019-1027.
40. Takahashi K, Nishida N, Kawabata H, et al. Regression of Hodgkin lymphoma in response to antiviral therapy for hepatitis C virus infection. Intern Med. 2012;51:2745-2747.
41. Gisbert JP, García-Buey L, Pajares JM, et al. Systematic review: regression of lymphoproliferative disorders after treatment for hepatitis C infection. Aliment Pharmacol Ther. 2005;21:653-662.
42. Najafzadeh M, Andersson K, Shrank WH, et al. Cost-effectiveness of novel regimens for the treatment of hepatitis C virus. Ann Intern Med. 2015;162:407-419.

Zeroing in on the cause of your patient's facial pain
› Advise patients who have a temporomandibular disorder that in addition to taking their medication as prescribed, they should limit activities that require moving their jaw, modify their diet, and minimize stress; they may require physical therapy and therapeutic exercises. C
› Consider prescribing a tricyclic antidepressant for patients with persistent idiopathic facial pain. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Facial pain is a common complaint: Up to 22% of adults in the United States experience orofacial pain during any 6-month period.1 Yet this type of pain can be difficult to diagnose due to the many structures of the face and mouth, pain referral patterns, and insufficient diagnostic tools.
Specifically, extraoral facial pain can be the result of temporomandibular disorders, neuropathic disorders, vascular disorders, or atypical causes, whereas facial pain stemming from inside the mouth can have a dental or nondental cause (FIGURE). Overlapping characteristics can make it difficult to distinguish these disorders. To help you to better diagnose and manage facial pain, we describe the most common causes and underlying pathological processes.
Extraoral facial pain
Extraoral pain refers to the pain that occurs on the face outside of the oral cavity. The TABLE2-15 summarizes the site, timing and severity, aggravating factors, history and exam findings, and management of several common causes of extraoral facial pain.
Musculoskeletal pain
Temporomandibular disorders (TMD) are a broad group of problems that affect the temporomandibular joint (TMJ), muscles of mastication, and/or associated bony and soft tissue structures.6 They may occur secondary to malocclusion, traumatic injuries, oral parafunctional habits (eg, bruxism), hormonal influences, or psychogenic factors.6 TMD is more prevalent in women, with a peak occurrence between ages 20 and 40 years.6,8
TMD can be articular (intracapsular) or nonarticular (extracapsular). Nonarticular disorders (>50% of TMD) usually affect the muscles of mastication and include chronic conditions such as fibromyalgia, muscle strain, and myopathies.8 Muscle-related pain and dysfunction are believed to arise from parafunctional habits such as bruxism or clenching. Articular disorders include synovitis/capsulitis, joint effusion, trauma/fracture, internal derangement (disturbance in the normal anatomic relationship between the disc and condyle), arthritis, and neoplasm.16
What you’ll see. Orofacial pain (usually dull and located in the preauricular region), joint noise, and restricted jaw function are key signs and symptoms of TMD. Exacerbation of pain with mandibular functions (eg, chewing, yawning, or swallowing) is a pathognomonic sign. Joint sounds such as clicking or crepitus are common. In most cases, crepitus correlates with osteoarthritis.6 Nonspecific TMD symptoms include headache, earache, insomnia, tinnitus, and neck and shoulder pain.6
The gold standard of diagnosis of TMD consists of taking a detailed history, evaluating the patient’s head and neck, and conducting a general physical examination and behavioral/psychological assessment.17 Imaging of the TMJ and associated structures is essential.17
Treatment. Nonsteroidal anti-inflammatory drugs, opioids, muscle relaxants, antidepressants, anticonvulsants, anxiolytics, and corticosteroids are options for treating TMD.6,8 Isometric jaw exercises, maxillomandibular appliances, and physical therapy are valuable adjuncts for pain relief. Advise patients to establish a self-care routine to reduce TMJ pain that might include changing their head posture or sleeping position, and limiting activities that require using their jaw, such as clenching, bruxism, and excessive gum chewing. Some patients may need to adopt a non-chewing diet that consists of liquid or pureed food. Massage and moist heat can help relax muscles of mastication and improve range of motion.
Approximately 5% of patients with TMD undergo surgery, typically simple arthrocentesis, arthroscopy, arthrotomy, or modified condylotomy.6 Total joint replacement is indicated only for patients with severely damaged joints with end-stage disease when all other conservative treatments have failed. Joint replacement primarily restores form and function; pain relief is a secondary benefit.8
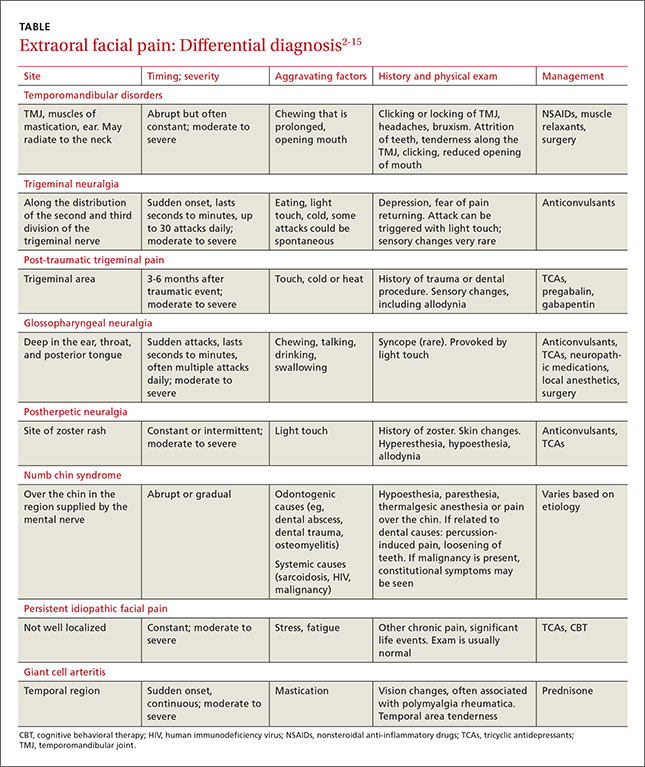
Neuropathic pain
Trigeminal neuralgia (TN) is sudden, usually unilateral, severe, brief, stabbing, recurrent episodes of pain in the distribution of one or more branches of the trigeminal nerve.9 It most commonly presents in the lower 2 branches of the trigeminal nerve and usually is caused by compression of the trigeminal nerve root by vascular or nonvascular causes.4 The pain is severe and can profoundly impact a patient’s quality of life.
TN attacks typically last from a few seconds to up to 2 minutes. As many as 30 attacks can occur daily, with refractory periods between attacks. After the initial attack, individuals are left with a residual dull or burning pain. TN can be triggered by face washing, teeth brushing, speaking, eating, shaving, or cold wind.4
Diagnosis can be tricky because more than half of patients with TN experience less severe pain after the main sharp attack; this presentation is called TN type II.7 A detailed patient history and careful evaluation can help identify patients with TN type II. TN can be misdiagnosed as TMD, especially if it presents unilaterally.15
Treatment. Anticonvulsants are the primary medications used to treat TN.
Post-traumatic trigeminal pain is usually the result of an injury or dental procedure, such as facial trauma, tooth extraction, root canal, or dental implants.12,18,19 Nerve injury is assumed to be the cause. This type of pain can start within 3 to 6 months of a trauma. It is located in the trigeminal area and patients describe it as burning, tingling and, at times, sharp.15 Patients who have sustained injury to the lingual or inferior alveolar nerves have reported feeling “pins and needles.”12
Common triggers include temperature changes or simple touch. Not all injuries result in pain; some patients may have only sensory impairment15 or sensory deficits such as allodynia or hypoesthesia.
Treatment. The first line of treatment for post-traumatic trigeminal pain is tricyclic antidepressants (TCAs) followed by pregabalin or gabapentin.14
Glossopharyngeal neuralgia (GN) is similar in presentation to TN but is much rarer.15 GN pain occurs deep in the throat, ear, or posterior tongue.15 When the pain occurs in the inner ear, GN can be misdiagnosed as TMD. In most cases, no cause of GN can be determined.
Patients describe GN pain as shooting, sharp, and electrical shock-like, lasting from seconds to minutes, with recurrent attacks throughout the day. Like TN, GN can present as episodes of attacks that last weeks to months. Triggers include chewing, drinking, swallowing, and talking, as well as light touch.13,15 Some patients with GN experience syncope due to the anatomical proximity of the vagus nerve.14
Treatment. Anticonvulsants are the first-line treatment for GN. Local anesthetics or surgery can be considered for patients who don’t improve after medical therapy.15
Postherpetic neuralgia (PHN) can cause facial pain when the characteristic vesicular rash of the varicella zoster virus (shingles) occurs on the face. PHN usually affects the first division of trigeminal nerve, but the second and third divisions can be affected as well.13
What you’ll see. The acute phase of PHN begins a few days before the initial rash has resolved and can last up to a month after. A new pain may begin one to 6 months after the initial rash has healed.20 This pain, which patients often describe as sharp, stabbing, or burning, can be constant or intermittent. Dysesthesia, hypoesthesia, and allodynia may also occur within the affected dermatome.
PHN is usually diagnosed based on the patient’s history and clinical presentation. However, direct fluorescent antibody stain, viral culture, or polymerase chain reaction performed on vesicular fluid from a herpetic lesion during the initial rash are the laboratory tests of choice if confirmation is needed.
Treatment. PHN is managed with anticonvulsants and TCAs.
Numb chin syndrome (NCS) is characterized by hypoesthesia, paresthesia, thermalgesic anesthesia, or pain over the chin in the region supplied by the mental nerve, a terminal branch of the mandibular division of the trigeminal nerve.5,21,22
NCS can be caused by odontogenic conditions, such as dental abscess, dental anesthesia, dental trauma, or osteomyelitis; systemic conditions such as amyloidosis, sickle cell disease, sarcoidosis, multiple sclerosis, human immunodeficiency virus, or diabetes; or malignancies such as lymphoma, leukemia, breast cancer, lung cancer, prostate cancer, or head and neck cancers.21 In one study of patients with NCS, cancer was the cause of the condition in 89% of patients.22
What you’ll see. NCS is characterized by numbness of the skin in the lower lip, chin and mucous membrane inside the lip that extends to the midline.5 Depending upon the etiology, patients may present with percussion-induced pain, loosening of teeth, sequestra, and mobility of fractured segments. Patients with metastatic malignancy may develop constitutional symptoms.
Making the diagnosis. Panoramic radiography is a useful starting point. If possible, a computerized tomography scan of the head and neck should also be done. Nuclear bone scintigraphy (bone scanning) may help identify bone disease such as osteomyelitis. A biopsy may be needed if a mass lesion is present.
Treatment. In NCS that is the result of a dental etiology, the prognosis usually is good. For example, NCS that is the result of an abscess usually resolves after the abscess is drained. However, if NCS is caused by metastasis, the prognosis is grim; the average length of survival after diagnosis is approximately 5 months if NCS is caused by mandibular metastasis and 12 months if leptomeningeal metastasis is present. Treatment does little to affect the outcome in these cases.21,22
Atypical pain
Persistent idiopathic facial pain (PIFP), previously known as atypical facial pain, is a persistent facial pain that does not have the classical characteristics of cranial neuralgias and for which there is no obvious cause.2,10,23 PIFP is not triggered by any of the factors that typically precipitate neuralgias.2 The onset may be spontaneous or associated with dental intervention or facial injury, but it usually does not have a demonstrable local cause.24,25
Neuropathic mechanisms that might be at work in PIFP include nociceptor sensitization, phenotypic changes and ectopic activity from the nociceptors, central sensitization possibly maintained by ongoing activity from initially damaged peripheral tissues, sympathetic abnormal activity, alteration of segmental inhibitory control, or hyperactivity or hypoactivity of descending controls.2
PIFP is most frequently reported in women in their 40s and 50s.25 The history of a patient with PIFP often include mood disorders, chronic pain, or poor coping skills.14 Patients complain of a steady, unilateral, poorly localized pain that is deep, constant, aching, pulling, or crushing. It is usually present all day, every day. The constancy of the pain is its distinguishing feature. In the beginning, this pain may be in a limited area on one side of the face, usually the nasolabial folds or the angle of the mandible. Later, it may affect both sides of the face and extend to the neck and upper limbs.23,24 Most patients with PIFP report other symptoms, including headache, neck and backache, dermatitis, pruritus, irritable bowel, and dysfunctional uterine bleeding.26
Making the diagnosis. A targeted history and accurate clinical examination are essential.2,10 Although there are no formal diagnostic criteria, a patient can be assumed to have PIFP if:2,10
• There is pain in the face for most of the day or all day, every day.
• Initially, the pain may be confined to a portion of the face, but it is poorly localized and deep.
• The pain is not associated with other physical signs or loss of sensation.
• Imaging does not reveal an obvious anatomic or structural cause.
Treatment. Treatment of PIFP can be difficult and unsatisfactory.23 Counseling to educate patients about the chronic and nonmalignant nature of the illness is the mainstay of treatment, followed by pharmacotherapy.23 TCAs have shown a moderate effect in several trials. Gabapentin, topiramate, carbamazepine, and pregabalin also have shown limited to modest benefit in some patients. Surgical therapies appear to be of little or no use.23 Experimental treatments such as pulsed radiofrequency, low-energy level diode laser have shown success in small studies.10,23
Vascular pain
Giant cell arteritis (GCA) is a systemic, chronic vasculitis involving the large and medium-sized vessels, mainly the extracranial branches of the carotid artery.6,11 It predominantly affects people older than age 50 and is more common among women and those of Scandinavian ethnicity.27
The cause of GCA is unclear. Genetic predisposition linked to humoral and cellmediated immunity is believed to play a role.28 Familial aggregation and predominance of the HLA-DR4 allele has been reported in patients with GCA.6
What you’ll see. The most common signs and symptoms of GCA are temporal headache (seen in two-thirds of patients), jaw claudication and tenderness, and swelling of the temporal artery.6,11 The headache of GCA usually is unilateral, severe, boring or lancinating, and localized to the temporal or occipital regions of the scalp.6 Other orofacial manifestations include trismus, throat pain that develops while chewing, changes in tongue sensation and tongue claudication, tooth pain, dysphagia, dysarthria, submandibular mass, lip and chin numbness, macroglossia, glossitis, lip and tongue necrosis, and facial swelling.11
Visual symptoms include diplopia, ptosis, and possibly blindness if treatment is not instituted at first suspicion. Ocular symptoms result from anterior ischemic optic neuropathy, posterior ischemic optic neuropathy, or central retinal or cilioretinal artery occlusion.6,28 Patients have also reported low-grade fever, asthenia, anorexia, weight loss, and generalized aches.11,28
Making the diagnosis. Arterial biopsy is the gold standard for diagnosis of GCA. It is usually performed on the temporal artery and is positive in 80% to 95% of people with the condition.28 Other useful lab tests include erythrocyte sedimentation rate (ESR; elevated), white blood cell count (mildly elevated), and C-reactive protein (elevated).
Treatment. Prednisone is used to treat GCA, in initial doses ranging from 30 to 80 mg. A maintenance dose may be required for up to 2 years, with close follow-up and periodic ESR measurements.28
Malignancy is a rare cause of facial pain. The pain may be due to metastasis of extracranial bony or soft tissue as it compresses cervical and cranial nerves.3 Lung cancer can cause referred pain in the periauricular region by compressing the vagus nerve, and this pain can be misdiagnosed as dental pain, atypical facial pain, TMD, or TN.3,29 The facial pain of lung cancer is unilateral and on the same side as the lung neoplasm, and commonly is referred to the jaw, ear, or temporal region. While many patients have continuous pain, some report intermittent pain or pain that lasts for hours.3 Facial pain caused by a malignancy is differentiated from other sources of facial pain by the presence of associated symptoms such as weight loss, cough, and hemoptysis.
Treatment. Treatment can include radiation or chemotherapy.29
The mouth is often the source of lower facial pain
Pain in the oral cavity is the most common cause of pain in the lower face.15 Intraoral pain usually is caused by disease in the following structures:
1. Dentition (eg, caries, dentin sensitivity, pulpal disease)
2. Periodontium (eg, gingivitis, acute or chronic periodontal disease, sensitivity related to gum recession, alveolar bone pathology)
3. Other soft and hard tissues, such as the palate, floor of mouth, buccal mucosa, non-tooth supporting bone, and tongue (eg, mucosal diseases, neoplasms, pain related to parafunction or trauma).
Rarely, intraoral pain may be referred. For example, myofascial pain might cause diffuse tooth pain.30
See TABLE W131-35 at the end of this article for a summary of the etiology, signs/symptoms, diagnosis, and management of these and other dental causes of oral facial pain.
Nondental causes of oral facial pain can be associated with oral mucosal disorders, malignant disease and its therapy, salivary gland disorders, maxillary sinusitis, burning mouth syndrome, or atypical odontalgia. See TABLE W236-41 for a more detailed description of these conditions.
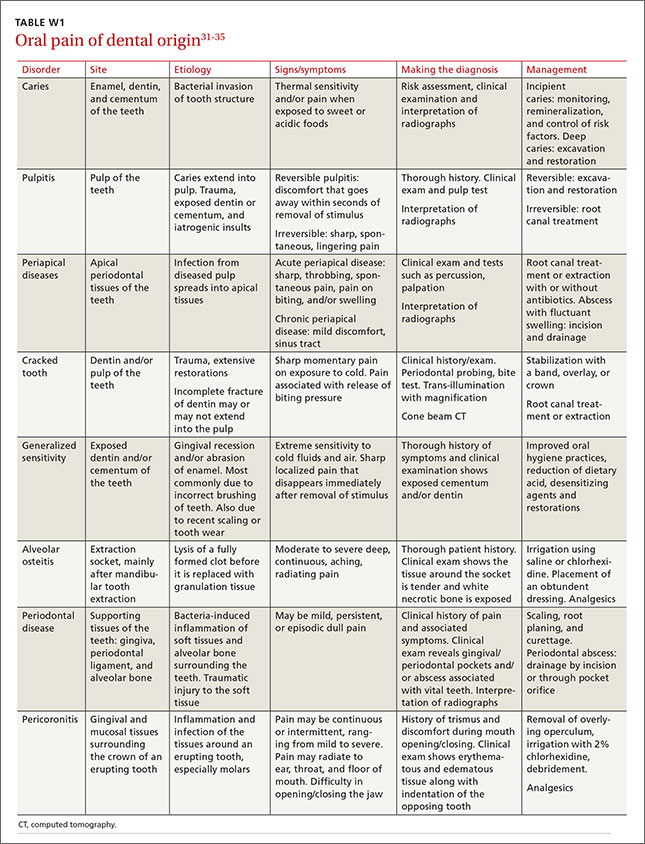
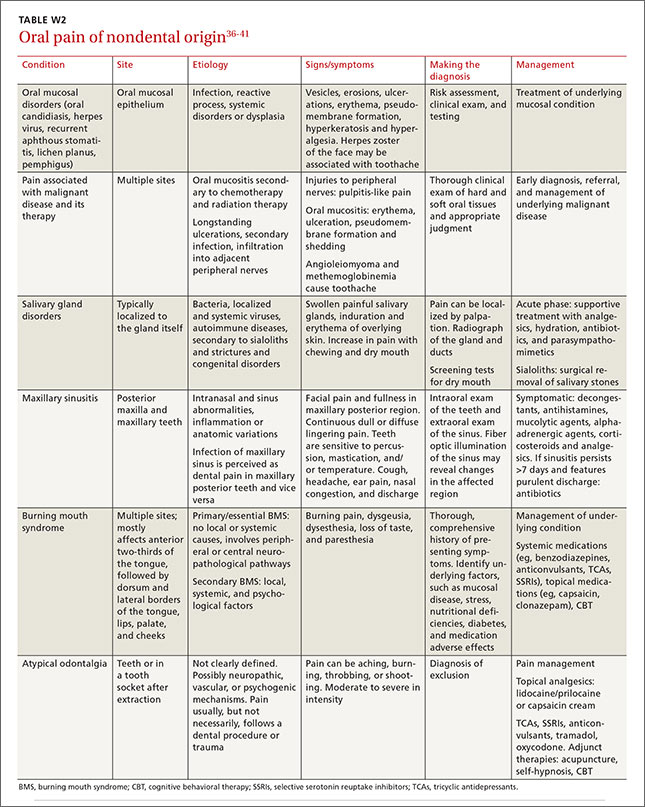
CORRESPONDENCE
Tamer H. Said, MD, MetroHealth Medical Center, 2500 MetroHealth Drive, Cleveland, Ohio 44109; [email protected]
1. Lipton JA, Ship JA, Larach-Robinson D. Estimated prevalence and distribution of reported orofacial pain in the United States. J Am Dent Assoc. 1993;124:115-1121.
2. Agostoni E, Frigerio R, Santoro P. Atypical facial pain: clinical considerations and differential diagnosis. Neurol Sci. 2005;26:S71-S74.
3. Bajwa Z, Ho C, Khan S, et al. Overview of craniofacial pain. UpTo-Date Web site. Available at: http://www.uptodate.com/contents/overview-of-craniofacial-pain. Accessed January 28, 2015.
4. Bendtsen L, Birk S, Kasch H, et al. Reference programme: Diagnosis and treatment of headache disorders and facial pain. Danish Headache Society, 2nd Edition, 2012. J Headache Pain. 2012;13:S1-S29.
5. Divya KS, Moran NA, Atkin PA. Numb chin syndrome: a case series and discussion. Br Dent J. 2010;208:157-160.
6. Kapur N, Kamel IR, Herlich A. Oral and craniofacial pain: diagnosis, pathophysiology, and treatment. Int Anesthesiol Clin. 2003;41:115-150.
7. Limonadi FM, McCartney S, Burchiel KJ. Design of an artificial neural network for diagnosis of facial pain syndromes. Stereotact Funct Neurosurg. 2006;84:212-220.
8. Liu F, Steinkeler A. Epidemiology, diagnosis, and treatment of temporomandibular disorders. Dent Clin North Am. 2013;57:465-479.
9. Merskey H, Bogduk N (eds). Classification of Chronic Pain. Descriptors of Chronic Pain Syndromes and Definition of Pain Terms, 2nd ed. Seattle, WA: International Association for the Study of Pain Press; 1994.
10. Nguyen CT, Wang MB. Complementary and integrative treatments: atypical facial pain. Otolaryngol Clin North Am. 2013;46:367-382.
11. Reiter S, Winocur E, Goldsmith C, et al. Giant cell arteritis misdiagnosed as temporomandibular disorder: a case report and review of the literature. J Orofac Pain. 2009;23:360-365.
12. Renton T, Adey-Viscuso D, Meechan JG, et al. Trigeminal nerve injuries in relation to local anaesthesia in mandibular injections. Br Dent J. 2010;209:E15.
13. Shephard MK, Macgregor EA, Zakrzewska JM. Orofacial pain: a guide for the headache physician. Headache. 2014;54:22-39.
14. Zakrzewska JM. Differential diagnosis of facial pain and guidelines for management. Br J Anaesth. 2013;111:95-104.
15. Zakrzewska JM. Multi-dimensionality of chronic pain of the oral cavity and face. J Headache Pain. 2013;14:37.
16. Herb K, Cho S, Stiles MA. Temporomandibular joint pain and dysfunction. Curr Pain Headache Rep. 2006;10:408-414.
17. American Society of Temporomandibular Joint Surgeons. Guidelines for diagnosis and management of disorders involving the temporomandibular joint and related musculoskeletal structures. Cranio. 2003;21:68-76.
18. Benoliel R, Zadik Y, Eliav E, et al. Peripheral painful traumatic trigeminal neuropathy: clinical features in 91 cases and proposal of novel diagnostic criteria. J Orofac Pain. 2012;26:49-58.
19. Brooke RI. Atypical odontalgia. A report of twenty-two cases. Oral Surg Oral Med Oral Pathol. 1980;49:196-199.
20. Bouhassira D, Chassany O, Gaillat J, et al. Patient perspective on herpes zoster and its complications: an observational prospective study in patients aged over 50 years in general practice. Pain. 2012;153:342-349.
21. Baskaran RK, Krishnamoorthy, Smith M. Numb chin syndrome—a reflection of systemic malignancy. World J Surg Oncol. 2006;4:52.
22. Lata J, Kumar P. Numb chin syndrome: a case report and review of the literature. Indian J Dent Res. 2010;21:135-137.
23. Cornelissen P, van Kleef M, Mekhail N, et al. Evidence-based interventional pain medicine according to clinical diagnoses. 3. Persistent idiopathic facial pain. Pain Pract. 2009;9:443-448.
24. Didier H, Marchetti C, Borromeo G, et al. Persistent idiopathic facial pain: multidisciplinary approach and assumption of comorbidity. Neurol Sci. 2010;31:S189-S195.
25. Klasser G. Management of persistent idiopathic facial pain. J Can Dent Assoc. 2013;79:d71.
26. Abiko Y, Matsuoka H, Chiba I, et al. Current evidence on atypical odontalgia: diagnosis and clinical management. Int J Dent. 2012;2012:518548.
27. Sheldon CA, White VA, Holland SP. Giant cell arteritis presenting with bilateral loss of vision and jaw pain: reminder of a potentially devastating condition. J Can Dent Assoc. 2011;77:b55.
28. Rockey JG, Anand R. Tongue necrosis secondary to temporal arteritis: a case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:471-473.
29. Sarlani E, Schwartz AH, Greenspan JD, et al. Facial pain as first manifestation of lung cancer: a case of lung cancer-related cluster headache and a review of the literature. J Orofac Pain. 2003;17:262-267.
30. Kumar A, Brennan MT. Differential diagnosis of orofacial pain and temporomandibular disorder. Dent Clin North Am. 2013;57:419-428.
31. Laudenbach JM, Simon Z. Common dental and periodontal diseases: evaluation and management. Med Clin North Am. 2014;98:1239-1260.
32. Napeñas JJ. Intraoral pain disorders. Dent Clin North Am. 2013;57:429-447.
33. Vickers ER, Zakrzewska JM. Dental causes of orofacial pain. In: Orofacial Pain. Zakrzewska JM, ed. Oxford, UK: Oxford University Press; 2009:69-81.
34. Pierse JE, Dym H, Clarkson E. Diagnosis and management of common postextraction complications. Dent Clin North Am. 2012;56:75-93.
35. Renton T. Dental (odontogenic) pain. Br J Pain. 2011;5:2-7.
36. Yatani H, Komiyama O, Matsuka Y, et al. Systematic review and recommendations for nonodontogenic toothache. J Oral Rehabil. 2014;41:843-852.
37. Klasser GD, Fischer DJ, Epstein JB. Burning mouth syndrome: recognition, understanding, and management. Oral Maxillofac Surg Clin North Am. 2008;20:255-271.
38. Balasubramaniam R, Turner LN, Fischer D, et al. Non-odontogenic toothache revisited. Open Journal of Stomatology. 2011;1:92-102.
39. Patton LL, Siegel MA, Benoliel R, et al. Management of burning mouth syndrome: systematic review and management recommendations. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:S39.e1-e13.
40. Cascarini L, McGurk M. Epidemiology of salivary gland infections. Oral Maxillofac Surg Clin North Am. 2009;21:353-357.
41. Hegarty AM, Zakrzewska JM. Differential diagnosis for orofacial pain, including sinusitis, TMD, trigeminal neuralgia. Dent Update. 2011;38:396-400,402-403,405-406.
› Advise patients who have a temporomandibular disorder that in addition to taking their medication as prescribed, they should limit activities that require moving their jaw, modify their diet, and minimize stress; they may require physical therapy and therapeutic exercises. C
› Consider prescribing a tricyclic antidepressant for patients with persistent idiopathic facial pain. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Facial pain is a common complaint: Up to 22% of adults in the United States experience orofacial pain during any 6-month period.1 Yet this type of pain can be difficult to diagnose due to the many structures of the face and mouth, pain referral patterns, and insufficient diagnostic tools.
Specifically, extraoral facial pain can be the result of temporomandibular disorders, neuropathic disorders, vascular disorders, or atypical causes, whereas facial pain stemming from inside the mouth can have a dental or nondental cause (FIGURE). Overlapping characteristics can make it difficult to distinguish these disorders. To help you to better diagnose and manage facial pain, we describe the most common causes and underlying pathological processes.
Extraoral facial pain
Extraoral pain refers to the pain that occurs on the face outside of the oral cavity. The TABLE2-15 summarizes the site, timing and severity, aggravating factors, history and exam findings, and management of several common causes of extraoral facial pain.
Musculoskeletal pain
Temporomandibular disorders (TMD) are a broad group of problems that affect the temporomandibular joint (TMJ), muscles of mastication, and/or associated bony and soft tissue structures.6 They may occur secondary to malocclusion, traumatic injuries, oral parafunctional habits (eg, bruxism), hormonal influences, or psychogenic factors.6 TMD is more prevalent in women, with a peak occurrence between ages 20 and 40 years.6,8
TMD can be articular (intracapsular) or nonarticular (extracapsular). Nonarticular disorders (>50% of TMD) usually affect the muscles of mastication and include chronic conditions such as fibromyalgia, muscle strain, and myopathies.8 Muscle-related pain and dysfunction are believed to arise from parafunctional habits such as bruxism or clenching. Articular disorders include synovitis/capsulitis, joint effusion, trauma/fracture, internal derangement (disturbance in the normal anatomic relationship between the disc and condyle), arthritis, and neoplasm.16
What you’ll see. Orofacial pain (usually dull and located in the preauricular region), joint noise, and restricted jaw function are key signs and symptoms of TMD. Exacerbation of pain with mandibular functions (eg, chewing, yawning, or swallowing) is a pathognomonic sign. Joint sounds such as clicking or crepitus are common. In most cases, crepitus correlates with osteoarthritis.6 Nonspecific TMD symptoms include headache, earache, insomnia, tinnitus, and neck and shoulder pain.6
The gold standard of diagnosis of TMD consists of taking a detailed history, evaluating the patient’s head and neck, and conducting a general physical examination and behavioral/psychological assessment.17 Imaging of the TMJ and associated structures is essential.17
Treatment. Nonsteroidal anti-inflammatory drugs, opioids, muscle relaxants, antidepressants, anticonvulsants, anxiolytics, and corticosteroids are options for treating TMD.6,8 Isometric jaw exercises, maxillomandibular appliances, and physical therapy are valuable adjuncts for pain relief. Advise patients to establish a self-care routine to reduce TMJ pain that might include changing their head posture or sleeping position, and limiting activities that require using their jaw, such as clenching, bruxism, and excessive gum chewing. Some patients may need to adopt a non-chewing diet that consists of liquid or pureed food. Massage and moist heat can help relax muscles of mastication and improve range of motion.
Approximately 5% of patients with TMD undergo surgery, typically simple arthrocentesis, arthroscopy, arthrotomy, or modified condylotomy.6 Total joint replacement is indicated only for patients with severely damaged joints with end-stage disease when all other conservative treatments have failed. Joint replacement primarily restores form and function; pain relief is a secondary benefit.8
Neuropathic pain
Trigeminal neuralgia (TN) is sudden, usually unilateral, severe, brief, stabbing, recurrent episodes of pain in the distribution of one or more branches of the trigeminal nerve.9 It most commonly presents in the lower 2 branches of the trigeminal nerve and usually is caused by compression of the trigeminal nerve root by vascular or nonvascular causes.4 The pain is severe and can profoundly impact a patient’s quality of life.
TN attacks typically last from a few seconds to up to 2 minutes. As many as 30 attacks can occur daily, with refractory periods between attacks. After the initial attack, individuals are left with a residual dull or burning pain. TN can be triggered by face washing, teeth brushing, speaking, eating, shaving, or cold wind.4
Diagnosis can be tricky because more than half of patients with TN experience less severe pain after the main sharp attack; this presentation is called TN type II.7 A detailed patient history and careful evaluation can help identify patients with TN type II. TN can be misdiagnosed as TMD, especially if it presents unilaterally.15
Treatment. Anticonvulsants are the primary medications used to treat TN.
Post-traumatic trigeminal pain is usually the result of an injury or dental procedure, such as facial trauma, tooth extraction, root canal, or dental implants.12,18,19 Nerve injury is assumed to be the cause. This type of pain can start within 3 to 6 months of a trauma. It is located in the trigeminal area and patients describe it as burning, tingling and, at times, sharp.15 Patients who have sustained injury to the lingual or inferior alveolar nerves have reported feeling “pins and needles.”12
Common triggers include temperature changes or simple touch. Not all injuries result in pain; some patients may have only sensory impairment15 or sensory deficits such as allodynia or hypoesthesia.
Treatment. The first line of treatment for post-traumatic trigeminal pain is tricyclic antidepressants (TCAs) followed by pregabalin or gabapentin.14
Glossopharyngeal neuralgia (GN) is similar in presentation to TN but is much rarer.15 GN pain occurs deep in the throat, ear, or posterior tongue.15 When the pain occurs in the inner ear, GN can be misdiagnosed as TMD. In most cases, no cause of GN can be determined.
Patients describe GN pain as shooting, sharp, and electrical shock-like, lasting from seconds to minutes, with recurrent attacks throughout the day. Like TN, GN can present as episodes of attacks that last weeks to months. Triggers include chewing, drinking, swallowing, and talking, as well as light touch.13,15 Some patients with GN experience syncope due to the anatomical proximity of the vagus nerve.14
Treatment. Anticonvulsants are the first-line treatment for GN. Local anesthetics or surgery can be considered for patients who don’t improve after medical therapy.15
Postherpetic neuralgia (PHN) can cause facial pain when the characteristic vesicular rash of the varicella zoster virus (shingles) occurs on the face. PHN usually affects the first division of trigeminal nerve, but the second and third divisions can be affected as well.13
What you’ll see. The acute phase of PHN begins a few days before the initial rash has resolved and can last up to a month after. A new pain may begin one to 6 months after the initial rash has healed.20 This pain, which patients often describe as sharp, stabbing, or burning, can be constant or intermittent. Dysesthesia, hypoesthesia, and allodynia may also occur within the affected dermatome.
PHN is usually diagnosed based on the patient’s history and clinical presentation. However, direct fluorescent antibody stain, viral culture, or polymerase chain reaction performed on vesicular fluid from a herpetic lesion during the initial rash are the laboratory tests of choice if confirmation is needed.
Treatment. PHN is managed with anticonvulsants and TCAs.
Numb chin syndrome (NCS) is characterized by hypoesthesia, paresthesia, thermalgesic anesthesia, or pain over the chin in the region supplied by the mental nerve, a terminal branch of the mandibular division of the trigeminal nerve.5,21,22
NCS can be caused by odontogenic conditions, such as dental abscess, dental anesthesia, dental trauma, or osteomyelitis; systemic conditions such as amyloidosis, sickle cell disease, sarcoidosis, multiple sclerosis, human immunodeficiency virus, or diabetes; or malignancies such as lymphoma, leukemia, breast cancer, lung cancer, prostate cancer, or head and neck cancers.21 In one study of patients with NCS, cancer was the cause of the condition in 89% of patients.22
What you’ll see. NCS is characterized by numbness of the skin in the lower lip, chin and mucous membrane inside the lip that extends to the midline.5 Depending upon the etiology, patients may present with percussion-induced pain, loosening of teeth, sequestra, and mobility of fractured segments. Patients with metastatic malignancy may develop constitutional symptoms.
Making the diagnosis. Panoramic radiography is a useful starting point. If possible, a computerized tomography scan of the head and neck should also be done. Nuclear bone scintigraphy (bone scanning) may help identify bone disease such as osteomyelitis. A biopsy may be needed if a mass lesion is present.
Treatment. In NCS that is the result of a dental etiology, the prognosis usually is good. For example, NCS that is the result of an abscess usually resolves after the abscess is drained. However, if NCS is caused by metastasis, the prognosis is grim; the average length of survival after diagnosis is approximately 5 months if NCS is caused by mandibular metastasis and 12 months if leptomeningeal metastasis is present. Treatment does little to affect the outcome in these cases.21,22
Atypical pain
Persistent idiopathic facial pain (PIFP), previously known as atypical facial pain, is a persistent facial pain that does not have the classical characteristics of cranial neuralgias and for which there is no obvious cause.2,10,23 PIFP is not triggered by any of the factors that typically precipitate neuralgias.2 The onset may be spontaneous or associated with dental intervention or facial injury, but it usually does not have a demonstrable local cause.24,25
Neuropathic mechanisms that might be at work in PIFP include nociceptor sensitization, phenotypic changes and ectopic activity from the nociceptors, central sensitization possibly maintained by ongoing activity from initially damaged peripheral tissues, sympathetic abnormal activity, alteration of segmental inhibitory control, or hyperactivity or hypoactivity of descending controls.2
PIFP is most frequently reported in women in their 40s and 50s.25 The history of a patient with PIFP often include mood disorders, chronic pain, or poor coping skills.14 Patients complain of a steady, unilateral, poorly localized pain that is deep, constant, aching, pulling, or crushing. It is usually present all day, every day. The constancy of the pain is its distinguishing feature. In the beginning, this pain may be in a limited area on one side of the face, usually the nasolabial folds or the angle of the mandible. Later, it may affect both sides of the face and extend to the neck and upper limbs.23,24 Most patients with PIFP report other symptoms, including headache, neck and backache, dermatitis, pruritus, irritable bowel, and dysfunctional uterine bleeding.26
Making the diagnosis. A targeted history and accurate clinical examination are essential.2,10 Although there are no formal diagnostic criteria, a patient can be assumed to have PIFP if:2,10
• There is pain in the face for most of the day or all day, every day.
• Initially, the pain may be confined to a portion of the face, but it is poorly localized and deep.
• The pain is not associated with other physical signs or loss of sensation.
• Imaging does not reveal an obvious anatomic or structural cause.
Treatment. Treatment of PIFP can be difficult and unsatisfactory.23 Counseling to educate patients about the chronic and nonmalignant nature of the illness is the mainstay of treatment, followed by pharmacotherapy.23 TCAs have shown a moderate effect in several trials. Gabapentin, topiramate, carbamazepine, and pregabalin also have shown limited to modest benefit in some patients. Surgical therapies appear to be of little or no use.23 Experimental treatments such as pulsed radiofrequency, low-energy level diode laser have shown success in small studies.10,23
Vascular pain
Giant cell arteritis (GCA) is a systemic, chronic vasculitis involving the large and medium-sized vessels, mainly the extracranial branches of the carotid artery.6,11 It predominantly affects people older than age 50 and is more common among women and those of Scandinavian ethnicity.27
The cause of GCA is unclear. Genetic predisposition linked to humoral and cellmediated immunity is believed to play a role.28 Familial aggregation and predominance of the HLA-DR4 allele has been reported in patients with GCA.6
What you’ll see. The most common signs and symptoms of GCA are temporal headache (seen in two-thirds of patients), jaw claudication and tenderness, and swelling of the temporal artery.6,11 The headache of GCA usually is unilateral, severe, boring or lancinating, and localized to the temporal or occipital regions of the scalp.6 Other orofacial manifestations include trismus, throat pain that develops while chewing, changes in tongue sensation and tongue claudication, tooth pain, dysphagia, dysarthria, submandibular mass, lip and chin numbness, macroglossia, glossitis, lip and tongue necrosis, and facial swelling.11
Visual symptoms include diplopia, ptosis, and possibly blindness if treatment is not instituted at first suspicion. Ocular symptoms result from anterior ischemic optic neuropathy, posterior ischemic optic neuropathy, or central retinal or cilioretinal artery occlusion.6,28 Patients have also reported low-grade fever, asthenia, anorexia, weight loss, and generalized aches.11,28
Making the diagnosis. Arterial biopsy is the gold standard for diagnosis of GCA. It is usually performed on the temporal artery and is positive in 80% to 95% of people with the condition.28 Other useful lab tests include erythrocyte sedimentation rate (ESR; elevated), white blood cell count (mildly elevated), and C-reactive protein (elevated).
Treatment. Prednisone is used to treat GCA, in initial doses ranging from 30 to 80 mg. A maintenance dose may be required for up to 2 years, with close follow-up and periodic ESR measurements.28
Malignancy is a rare cause of facial pain. The pain may be due to metastasis of extracranial bony or soft tissue as it compresses cervical and cranial nerves.3 Lung cancer can cause referred pain in the periauricular region by compressing the vagus nerve, and this pain can be misdiagnosed as dental pain, atypical facial pain, TMD, or TN.3,29 The facial pain of lung cancer is unilateral and on the same side as the lung neoplasm, and commonly is referred to the jaw, ear, or temporal region. While many patients have continuous pain, some report intermittent pain or pain that lasts for hours.3 Facial pain caused by a malignancy is differentiated from other sources of facial pain by the presence of associated symptoms such as weight loss, cough, and hemoptysis.
Treatment. Treatment can include radiation or chemotherapy.29
The mouth is often the source of lower facial pain
Pain in the oral cavity is the most common cause of pain in the lower face.15 Intraoral pain usually is caused by disease in the following structures:
1. Dentition (eg, caries, dentin sensitivity, pulpal disease)
2. Periodontium (eg, gingivitis, acute or chronic periodontal disease, sensitivity related to gum recession, alveolar bone pathology)
3. Other soft and hard tissues, such as the palate, floor of mouth, buccal mucosa, non-tooth supporting bone, and tongue (eg, mucosal diseases, neoplasms, pain related to parafunction or trauma).
Rarely, intraoral pain may be referred. For example, myofascial pain might cause diffuse tooth pain.30
See TABLE W131-35 at the end of this article for a summary of the etiology, signs/symptoms, diagnosis, and management of these and other dental causes of oral facial pain.
Nondental causes of oral facial pain can be associated with oral mucosal disorders, malignant disease and its therapy, salivary gland disorders, maxillary sinusitis, burning mouth syndrome, or atypical odontalgia. See TABLE W236-41 for a more detailed description of these conditions.
CORRESPONDENCE
Tamer H. Said, MD, MetroHealth Medical Center, 2500 MetroHealth Drive, Cleveland, Ohio 44109; [email protected]
› Advise patients who have a temporomandibular disorder that in addition to taking their medication as prescribed, they should limit activities that require moving their jaw, modify their diet, and minimize stress; they may require physical therapy and therapeutic exercises. C
› Consider prescribing a tricyclic antidepressant for patients with persistent idiopathic facial pain. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Facial pain is a common complaint: Up to 22% of adults in the United States experience orofacial pain during any 6-month period.1 Yet this type of pain can be difficult to diagnose due to the many structures of the face and mouth, pain referral patterns, and insufficient diagnostic tools.
Specifically, extraoral facial pain can be the result of temporomandibular disorders, neuropathic disorders, vascular disorders, or atypical causes, whereas facial pain stemming from inside the mouth can have a dental or nondental cause (FIGURE). Overlapping characteristics can make it difficult to distinguish these disorders. To help you to better diagnose and manage facial pain, we describe the most common causes and underlying pathological processes.
Extraoral facial pain
Extraoral pain refers to the pain that occurs on the face outside of the oral cavity. The TABLE2-15 summarizes the site, timing and severity, aggravating factors, history and exam findings, and management of several common causes of extraoral facial pain.
Musculoskeletal pain
Temporomandibular disorders (TMD) are a broad group of problems that affect the temporomandibular joint (TMJ), muscles of mastication, and/or associated bony and soft tissue structures.6 They may occur secondary to malocclusion, traumatic injuries, oral parafunctional habits (eg, bruxism), hormonal influences, or psychogenic factors.6 TMD is more prevalent in women, with a peak occurrence between ages 20 and 40 years.6,8
TMD can be articular (intracapsular) or nonarticular (extracapsular). Nonarticular disorders (>50% of TMD) usually affect the muscles of mastication and include chronic conditions such as fibromyalgia, muscle strain, and myopathies.8 Muscle-related pain and dysfunction are believed to arise from parafunctional habits such as bruxism or clenching. Articular disorders include synovitis/capsulitis, joint effusion, trauma/fracture, internal derangement (disturbance in the normal anatomic relationship between the disc and condyle), arthritis, and neoplasm.16
What you’ll see. Orofacial pain (usually dull and located in the preauricular region), joint noise, and restricted jaw function are key signs and symptoms of TMD. Exacerbation of pain with mandibular functions (eg, chewing, yawning, or swallowing) is a pathognomonic sign. Joint sounds such as clicking or crepitus are common. In most cases, crepitus correlates with osteoarthritis.6 Nonspecific TMD symptoms include headache, earache, insomnia, tinnitus, and neck and shoulder pain.6
The gold standard of diagnosis of TMD consists of taking a detailed history, evaluating the patient’s head and neck, and conducting a general physical examination and behavioral/psychological assessment.17 Imaging of the TMJ and associated structures is essential.17
Treatment. Nonsteroidal anti-inflammatory drugs, opioids, muscle relaxants, antidepressants, anticonvulsants, anxiolytics, and corticosteroids are options for treating TMD.6,8 Isometric jaw exercises, maxillomandibular appliances, and physical therapy are valuable adjuncts for pain relief. Advise patients to establish a self-care routine to reduce TMJ pain that might include changing their head posture or sleeping position, and limiting activities that require using their jaw, such as clenching, bruxism, and excessive gum chewing. Some patients may need to adopt a non-chewing diet that consists of liquid or pureed food. Massage and moist heat can help relax muscles of mastication and improve range of motion.
Approximately 5% of patients with TMD undergo surgery, typically simple arthrocentesis, arthroscopy, arthrotomy, or modified condylotomy.6 Total joint replacement is indicated only for patients with severely damaged joints with end-stage disease when all other conservative treatments have failed. Joint replacement primarily restores form and function; pain relief is a secondary benefit.8
Neuropathic pain
Trigeminal neuralgia (TN) is sudden, usually unilateral, severe, brief, stabbing, recurrent episodes of pain in the distribution of one or more branches of the trigeminal nerve.9 It most commonly presents in the lower 2 branches of the trigeminal nerve and usually is caused by compression of the trigeminal nerve root by vascular or nonvascular causes.4 The pain is severe and can profoundly impact a patient’s quality of life.
TN attacks typically last from a few seconds to up to 2 minutes. As many as 30 attacks can occur daily, with refractory periods between attacks. After the initial attack, individuals are left with a residual dull or burning pain. TN can be triggered by face washing, teeth brushing, speaking, eating, shaving, or cold wind.4
Diagnosis can be tricky because more than half of patients with TN experience less severe pain after the main sharp attack; this presentation is called TN type II.7 A detailed patient history and careful evaluation can help identify patients with TN type II. TN can be misdiagnosed as TMD, especially if it presents unilaterally.15
Treatment. Anticonvulsants are the primary medications used to treat TN.
Post-traumatic trigeminal pain is usually the result of an injury or dental procedure, such as facial trauma, tooth extraction, root canal, or dental implants.12,18,19 Nerve injury is assumed to be the cause. This type of pain can start within 3 to 6 months of a trauma. It is located in the trigeminal area and patients describe it as burning, tingling and, at times, sharp.15 Patients who have sustained injury to the lingual or inferior alveolar nerves have reported feeling “pins and needles.”12
Common triggers include temperature changes or simple touch. Not all injuries result in pain; some patients may have only sensory impairment15 or sensory deficits such as allodynia or hypoesthesia.
Treatment. The first line of treatment for post-traumatic trigeminal pain is tricyclic antidepressants (TCAs) followed by pregabalin or gabapentin.14
Glossopharyngeal neuralgia (GN) is similar in presentation to TN but is much rarer.15 GN pain occurs deep in the throat, ear, or posterior tongue.15 When the pain occurs in the inner ear, GN can be misdiagnosed as TMD. In most cases, no cause of GN can be determined.
Patients describe GN pain as shooting, sharp, and electrical shock-like, lasting from seconds to minutes, with recurrent attacks throughout the day. Like TN, GN can present as episodes of attacks that last weeks to months. Triggers include chewing, drinking, swallowing, and talking, as well as light touch.13,15 Some patients with GN experience syncope due to the anatomical proximity of the vagus nerve.14
Treatment. Anticonvulsants are the first-line treatment for GN. Local anesthetics or surgery can be considered for patients who don’t improve after medical therapy.15
Postherpetic neuralgia (PHN) can cause facial pain when the characteristic vesicular rash of the varicella zoster virus (shingles) occurs on the face. PHN usually affects the first division of trigeminal nerve, but the second and third divisions can be affected as well.13
What you’ll see. The acute phase of PHN begins a few days before the initial rash has resolved and can last up to a month after. A new pain may begin one to 6 months after the initial rash has healed.20 This pain, which patients often describe as sharp, stabbing, or burning, can be constant or intermittent. Dysesthesia, hypoesthesia, and allodynia may also occur within the affected dermatome.
PHN is usually diagnosed based on the patient’s history and clinical presentation. However, direct fluorescent antibody stain, viral culture, or polymerase chain reaction performed on vesicular fluid from a herpetic lesion during the initial rash are the laboratory tests of choice if confirmation is needed.
Treatment. PHN is managed with anticonvulsants and TCAs.
Numb chin syndrome (NCS) is characterized by hypoesthesia, paresthesia, thermalgesic anesthesia, or pain over the chin in the region supplied by the mental nerve, a terminal branch of the mandibular division of the trigeminal nerve.5,21,22
NCS can be caused by odontogenic conditions, such as dental abscess, dental anesthesia, dental trauma, or osteomyelitis; systemic conditions such as amyloidosis, sickle cell disease, sarcoidosis, multiple sclerosis, human immunodeficiency virus, or diabetes; or malignancies such as lymphoma, leukemia, breast cancer, lung cancer, prostate cancer, or head and neck cancers.21 In one study of patients with NCS, cancer was the cause of the condition in 89% of patients.22
What you’ll see. NCS is characterized by numbness of the skin in the lower lip, chin and mucous membrane inside the lip that extends to the midline.5 Depending upon the etiology, patients may present with percussion-induced pain, loosening of teeth, sequestra, and mobility of fractured segments. Patients with metastatic malignancy may develop constitutional symptoms.
Making the diagnosis. Panoramic radiography is a useful starting point. If possible, a computerized tomography scan of the head and neck should also be done. Nuclear bone scintigraphy (bone scanning) may help identify bone disease such as osteomyelitis. A biopsy may be needed if a mass lesion is present.
Treatment. In NCS that is the result of a dental etiology, the prognosis usually is good. For example, NCS that is the result of an abscess usually resolves after the abscess is drained. However, if NCS is caused by metastasis, the prognosis is grim; the average length of survival after diagnosis is approximately 5 months if NCS is caused by mandibular metastasis and 12 months if leptomeningeal metastasis is present. Treatment does little to affect the outcome in these cases.21,22
Atypical pain
Persistent idiopathic facial pain (PIFP), previously known as atypical facial pain, is a persistent facial pain that does not have the classical characteristics of cranial neuralgias and for which there is no obvious cause.2,10,23 PIFP is not triggered by any of the factors that typically precipitate neuralgias.2 The onset may be spontaneous or associated with dental intervention or facial injury, but it usually does not have a demonstrable local cause.24,25
Neuropathic mechanisms that might be at work in PIFP include nociceptor sensitization, phenotypic changes and ectopic activity from the nociceptors, central sensitization possibly maintained by ongoing activity from initially damaged peripheral tissues, sympathetic abnormal activity, alteration of segmental inhibitory control, or hyperactivity or hypoactivity of descending controls.2
PIFP is most frequently reported in women in their 40s and 50s.25 The history of a patient with PIFP often include mood disorders, chronic pain, or poor coping skills.14 Patients complain of a steady, unilateral, poorly localized pain that is deep, constant, aching, pulling, or crushing. It is usually present all day, every day. The constancy of the pain is its distinguishing feature. In the beginning, this pain may be in a limited area on one side of the face, usually the nasolabial folds or the angle of the mandible. Later, it may affect both sides of the face and extend to the neck and upper limbs.23,24 Most patients with PIFP report other symptoms, including headache, neck and backache, dermatitis, pruritus, irritable bowel, and dysfunctional uterine bleeding.26
Making the diagnosis. A targeted history and accurate clinical examination are essential.2,10 Although there are no formal diagnostic criteria, a patient can be assumed to have PIFP if:2,10
• There is pain in the face for most of the day or all day, every day.
• Initially, the pain may be confined to a portion of the face, but it is poorly localized and deep.
• The pain is not associated with other physical signs or loss of sensation.
• Imaging does not reveal an obvious anatomic or structural cause.
Treatment. Treatment of PIFP can be difficult and unsatisfactory.23 Counseling to educate patients about the chronic and nonmalignant nature of the illness is the mainstay of treatment, followed by pharmacotherapy.23 TCAs have shown a moderate effect in several trials. Gabapentin, topiramate, carbamazepine, and pregabalin also have shown limited to modest benefit in some patients. Surgical therapies appear to be of little or no use.23 Experimental treatments such as pulsed radiofrequency, low-energy level diode laser have shown success in small studies.10,23
Vascular pain
Giant cell arteritis (GCA) is a systemic, chronic vasculitis involving the large and medium-sized vessels, mainly the extracranial branches of the carotid artery.6,11 It predominantly affects people older than age 50 and is more common among women and those of Scandinavian ethnicity.27
The cause of GCA is unclear. Genetic predisposition linked to humoral and cellmediated immunity is believed to play a role.28 Familial aggregation and predominance of the HLA-DR4 allele has been reported in patients with GCA.6
What you’ll see. The most common signs and symptoms of GCA are temporal headache (seen in two-thirds of patients), jaw claudication and tenderness, and swelling of the temporal artery.6,11 The headache of GCA usually is unilateral, severe, boring or lancinating, and localized to the temporal or occipital regions of the scalp.6 Other orofacial manifestations include trismus, throat pain that develops while chewing, changes in tongue sensation and tongue claudication, tooth pain, dysphagia, dysarthria, submandibular mass, lip and chin numbness, macroglossia, glossitis, lip and tongue necrosis, and facial swelling.11
Visual symptoms include diplopia, ptosis, and possibly blindness if treatment is not instituted at first suspicion. Ocular symptoms result from anterior ischemic optic neuropathy, posterior ischemic optic neuropathy, or central retinal or cilioretinal artery occlusion.6,28 Patients have also reported low-grade fever, asthenia, anorexia, weight loss, and generalized aches.11,28
Making the diagnosis. Arterial biopsy is the gold standard for diagnosis of GCA. It is usually performed on the temporal artery and is positive in 80% to 95% of people with the condition.28 Other useful lab tests include erythrocyte sedimentation rate (ESR; elevated), white blood cell count (mildly elevated), and C-reactive protein (elevated).
Treatment. Prednisone is used to treat GCA, in initial doses ranging from 30 to 80 mg. A maintenance dose may be required for up to 2 years, with close follow-up and periodic ESR measurements.28
Malignancy is a rare cause of facial pain. The pain may be due to metastasis of extracranial bony or soft tissue as it compresses cervical and cranial nerves.3 Lung cancer can cause referred pain in the periauricular region by compressing the vagus nerve, and this pain can be misdiagnosed as dental pain, atypical facial pain, TMD, or TN.3,29 The facial pain of lung cancer is unilateral and on the same side as the lung neoplasm, and commonly is referred to the jaw, ear, or temporal region. While many patients have continuous pain, some report intermittent pain or pain that lasts for hours.3 Facial pain caused by a malignancy is differentiated from other sources of facial pain by the presence of associated symptoms such as weight loss, cough, and hemoptysis.
Treatment. Treatment can include radiation or chemotherapy.29
The mouth is often the source of lower facial pain
Pain in the oral cavity is the most common cause of pain in the lower face.15 Intraoral pain usually is caused by disease in the following structures:
1. Dentition (eg, caries, dentin sensitivity, pulpal disease)
2. Periodontium (eg, gingivitis, acute or chronic periodontal disease, sensitivity related to gum recession, alveolar bone pathology)
3. Other soft and hard tissues, such as the palate, floor of mouth, buccal mucosa, non-tooth supporting bone, and tongue (eg, mucosal diseases, neoplasms, pain related to parafunction or trauma).
Rarely, intraoral pain may be referred. For example, myofascial pain might cause diffuse tooth pain.30
See TABLE W131-35 at the end of this article for a summary of the etiology, signs/symptoms, diagnosis, and management of these and other dental causes of oral facial pain.
Nondental causes of oral facial pain can be associated with oral mucosal disorders, malignant disease and its therapy, salivary gland disorders, maxillary sinusitis, burning mouth syndrome, or atypical odontalgia. See TABLE W236-41 for a more detailed description of these conditions.
CORRESPONDENCE
Tamer H. Said, MD, MetroHealth Medical Center, 2500 MetroHealth Drive, Cleveland, Ohio 44109; [email protected]
1. Lipton JA, Ship JA, Larach-Robinson D. Estimated prevalence and distribution of reported orofacial pain in the United States. J Am Dent Assoc. 1993;124:115-1121.
2. Agostoni E, Frigerio R, Santoro P. Atypical facial pain: clinical considerations and differential diagnosis. Neurol Sci. 2005;26:S71-S74.
3. Bajwa Z, Ho C, Khan S, et al. Overview of craniofacial pain. UpTo-Date Web site. Available at: http://www.uptodate.com/contents/overview-of-craniofacial-pain. Accessed January 28, 2015.
4. Bendtsen L, Birk S, Kasch H, et al. Reference programme: Diagnosis and treatment of headache disorders and facial pain. Danish Headache Society, 2nd Edition, 2012. J Headache Pain. 2012;13:S1-S29.
5. Divya KS, Moran NA, Atkin PA. Numb chin syndrome: a case series and discussion. Br Dent J. 2010;208:157-160.
6. Kapur N, Kamel IR, Herlich A. Oral and craniofacial pain: diagnosis, pathophysiology, and treatment. Int Anesthesiol Clin. 2003;41:115-150.
7. Limonadi FM, McCartney S, Burchiel KJ. Design of an artificial neural network for diagnosis of facial pain syndromes. Stereotact Funct Neurosurg. 2006;84:212-220.
8. Liu F, Steinkeler A. Epidemiology, diagnosis, and treatment of temporomandibular disorders. Dent Clin North Am. 2013;57:465-479.
9. Merskey H, Bogduk N (eds). Classification of Chronic Pain. Descriptors of Chronic Pain Syndromes and Definition of Pain Terms, 2nd ed. Seattle, WA: International Association for the Study of Pain Press; 1994.
10. Nguyen CT, Wang MB. Complementary and integrative treatments: atypical facial pain. Otolaryngol Clin North Am. 2013;46:367-382.
11. Reiter S, Winocur E, Goldsmith C, et al. Giant cell arteritis misdiagnosed as temporomandibular disorder: a case report and review of the literature. J Orofac Pain. 2009;23:360-365.
12. Renton T, Adey-Viscuso D, Meechan JG, et al. Trigeminal nerve injuries in relation to local anaesthesia in mandibular injections. Br Dent J. 2010;209:E15.
13. Shephard MK, Macgregor EA, Zakrzewska JM. Orofacial pain: a guide for the headache physician. Headache. 2014;54:22-39.
14. Zakrzewska JM. Differential diagnosis of facial pain and guidelines for management. Br J Anaesth. 2013;111:95-104.
15. Zakrzewska JM. Multi-dimensionality of chronic pain of the oral cavity and face. J Headache Pain. 2013;14:37.
16. Herb K, Cho S, Stiles MA. Temporomandibular joint pain and dysfunction. Curr Pain Headache Rep. 2006;10:408-414.
17. American Society of Temporomandibular Joint Surgeons. Guidelines for diagnosis and management of disorders involving the temporomandibular joint and related musculoskeletal structures. Cranio. 2003;21:68-76.
18. Benoliel R, Zadik Y, Eliav E, et al. Peripheral painful traumatic trigeminal neuropathy: clinical features in 91 cases and proposal of novel diagnostic criteria. J Orofac Pain. 2012;26:49-58.
19. Brooke RI. Atypical odontalgia. A report of twenty-two cases. Oral Surg Oral Med Oral Pathol. 1980;49:196-199.
20. Bouhassira D, Chassany O, Gaillat J, et al. Patient perspective on herpes zoster and its complications: an observational prospective study in patients aged over 50 years in general practice. Pain. 2012;153:342-349.
21. Baskaran RK, Krishnamoorthy, Smith M. Numb chin syndrome—a reflection of systemic malignancy. World J Surg Oncol. 2006;4:52.
22. Lata J, Kumar P. Numb chin syndrome: a case report and review of the literature. Indian J Dent Res. 2010;21:135-137.
23. Cornelissen P, van Kleef M, Mekhail N, et al. Evidence-based interventional pain medicine according to clinical diagnoses. 3. Persistent idiopathic facial pain. Pain Pract. 2009;9:443-448.
24. Didier H, Marchetti C, Borromeo G, et al. Persistent idiopathic facial pain: multidisciplinary approach and assumption of comorbidity. Neurol Sci. 2010;31:S189-S195.
25. Klasser G. Management of persistent idiopathic facial pain. J Can Dent Assoc. 2013;79:d71.
26. Abiko Y, Matsuoka H, Chiba I, et al. Current evidence on atypical odontalgia: diagnosis and clinical management. Int J Dent. 2012;2012:518548.
27. Sheldon CA, White VA, Holland SP. Giant cell arteritis presenting with bilateral loss of vision and jaw pain: reminder of a potentially devastating condition. J Can Dent Assoc. 2011;77:b55.
28. Rockey JG, Anand R. Tongue necrosis secondary to temporal arteritis: a case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:471-473.
29. Sarlani E, Schwartz AH, Greenspan JD, et al. Facial pain as first manifestation of lung cancer: a case of lung cancer-related cluster headache and a review of the literature. J Orofac Pain. 2003;17:262-267.
30. Kumar A, Brennan MT. Differential diagnosis of orofacial pain and temporomandibular disorder. Dent Clin North Am. 2013;57:419-428.
31. Laudenbach JM, Simon Z. Common dental and periodontal diseases: evaluation and management. Med Clin North Am. 2014;98:1239-1260.
32. Napeñas JJ. Intraoral pain disorders. Dent Clin North Am. 2013;57:429-447.
33. Vickers ER, Zakrzewska JM. Dental causes of orofacial pain. In: Orofacial Pain. Zakrzewska JM, ed. Oxford, UK: Oxford University Press; 2009:69-81.
34. Pierse JE, Dym H, Clarkson E. Diagnosis and management of common postextraction complications. Dent Clin North Am. 2012;56:75-93.
35. Renton T. Dental (odontogenic) pain. Br J Pain. 2011;5:2-7.
36. Yatani H, Komiyama O, Matsuka Y, et al. Systematic review and recommendations for nonodontogenic toothache. J Oral Rehabil. 2014;41:843-852.
37. Klasser GD, Fischer DJ, Epstein JB. Burning mouth syndrome: recognition, understanding, and management. Oral Maxillofac Surg Clin North Am. 2008;20:255-271.
38. Balasubramaniam R, Turner LN, Fischer D, et al. Non-odontogenic toothache revisited. Open Journal of Stomatology. 2011;1:92-102.
39. Patton LL, Siegel MA, Benoliel R, et al. Management of burning mouth syndrome: systematic review and management recommendations. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:S39.e1-e13.
40. Cascarini L, McGurk M. Epidemiology of salivary gland infections. Oral Maxillofac Surg Clin North Am. 2009;21:353-357.
41. Hegarty AM, Zakrzewska JM. Differential diagnosis for orofacial pain, including sinusitis, TMD, trigeminal neuralgia. Dent Update. 2011;38:396-400,402-403,405-406.
1. Lipton JA, Ship JA, Larach-Robinson D. Estimated prevalence and distribution of reported orofacial pain in the United States. J Am Dent Assoc. 1993;124:115-1121.
2. Agostoni E, Frigerio R, Santoro P. Atypical facial pain: clinical considerations and differential diagnosis. Neurol Sci. 2005;26:S71-S74.
3. Bajwa Z, Ho C, Khan S, et al. Overview of craniofacial pain. UpTo-Date Web site. Available at: http://www.uptodate.com/contents/overview-of-craniofacial-pain. Accessed January 28, 2015.
4. Bendtsen L, Birk S, Kasch H, et al. Reference programme: Diagnosis and treatment of headache disorders and facial pain. Danish Headache Society, 2nd Edition, 2012. J Headache Pain. 2012;13:S1-S29.
5. Divya KS, Moran NA, Atkin PA. Numb chin syndrome: a case series and discussion. Br Dent J. 2010;208:157-160.
6. Kapur N, Kamel IR, Herlich A. Oral and craniofacial pain: diagnosis, pathophysiology, and treatment. Int Anesthesiol Clin. 2003;41:115-150.
7. Limonadi FM, McCartney S, Burchiel KJ. Design of an artificial neural network for diagnosis of facial pain syndromes. Stereotact Funct Neurosurg. 2006;84:212-220.
8. Liu F, Steinkeler A. Epidemiology, diagnosis, and treatment of temporomandibular disorders. Dent Clin North Am. 2013;57:465-479.
9. Merskey H, Bogduk N (eds). Classification of Chronic Pain. Descriptors of Chronic Pain Syndromes and Definition of Pain Terms, 2nd ed. Seattle, WA: International Association for the Study of Pain Press; 1994.
10. Nguyen CT, Wang MB. Complementary and integrative treatments: atypical facial pain. Otolaryngol Clin North Am. 2013;46:367-382.
11. Reiter S, Winocur E, Goldsmith C, et al. Giant cell arteritis misdiagnosed as temporomandibular disorder: a case report and review of the literature. J Orofac Pain. 2009;23:360-365.
12. Renton T, Adey-Viscuso D, Meechan JG, et al. Trigeminal nerve injuries in relation to local anaesthesia in mandibular injections. Br Dent J. 2010;209:E15.
13. Shephard MK, Macgregor EA, Zakrzewska JM. Orofacial pain: a guide for the headache physician. Headache. 2014;54:22-39.
14. Zakrzewska JM. Differential diagnosis of facial pain and guidelines for management. Br J Anaesth. 2013;111:95-104.
15. Zakrzewska JM. Multi-dimensionality of chronic pain of the oral cavity and face. J Headache Pain. 2013;14:37.
16. Herb K, Cho S, Stiles MA. Temporomandibular joint pain and dysfunction. Curr Pain Headache Rep. 2006;10:408-414.
17. American Society of Temporomandibular Joint Surgeons. Guidelines for diagnosis and management of disorders involving the temporomandibular joint and related musculoskeletal structures. Cranio. 2003;21:68-76.
18. Benoliel R, Zadik Y, Eliav E, et al. Peripheral painful traumatic trigeminal neuropathy: clinical features in 91 cases and proposal of novel diagnostic criteria. J Orofac Pain. 2012;26:49-58.
19. Brooke RI. Atypical odontalgia. A report of twenty-two cases. Oral Surg Oral Med Oral Pathol. 1980;49:196-199.
20. Bouhassira D, Chassany O, Gaillat J, et al. Patient perspective on herpes zoster and its complications: an observational prospective study in patients aged over 50 years in general practice. Pain. 2012;153:342-349.
21. Baskaran RK, Krishnamoorthy, Smith M. Numb chin syndrome—a reflection of systemic malignancy. World J Surg Oncol. 2006;4:52.
22. Lata J, Kumar P. Numb chin syndrome: a case report and review of the literature. Indian J Dent Res. 2010;21:135-137.
23. Cornelissen P, van Kleef M, Mekhail N, et al. Evidence-based interventional pain medicine according to clinical diagnoses. 3. Persistent idiopathic facial pain. Pain Pract. 2009;9:443-448.
24. Didier H, Marchetti C, Borromeo G, et al. Persistent idiopathic facial pain: multidisciplinary approach and assumption of comorbidity. Neurol Sci. 2010;31:S189-S195.
25. Klasser G. Management of persistent idiopathic facial pain. J Can Dent Assoc. 2013;79:d71.
26. Abiko Y, Matsuoka H, Chiba I, et al. Current evidence on atypical odontalgia: diagnosis and clinical management. Int J Dent. 2012;2012:518548.
27. Sheldon CA, White VA, Holland SP. Giant cell arteritis presenting with bilateral loss of vision and jaw pain: reminder of a potentially devastating condition. J Can Dent Assoc. 2011;77:b55.
28. Rockey JG, Anand R. Tongue necrosis secondary to temporal arteritis: a case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:471-473.
29. Sarlani E, Schwartz AH, Greenspan JD, et al. Facial pain as first manifestation of lung cancer: a case of lung cancer-related cluster headache and a review of the literature. J Orofac Pain. 2003;17:262-267.
30. Kumar A, Brennan MT. Differential diagnosis of orofacial pain and temporomandibular disorder. Dent Clin North Am. 2013;57:419-428.
31. Laudenbach JM, Simon Z. Common dental and periodontal diseases: evaluation and management. Med Clin North Am. 2014;98:1239-1260.
32. Napeñas JJ. Intraoral pain disorders. Dent Clin North Am. 2013;57:429-447.
33. Vickers ER, Zakrzewska JM. Dental causes of orofacial pain. In: Orofacial Pain. Zakrzewska JM, ed. Oxford, UK: Oxford University Press; 2009:69-81.
34. Pierse JE, Dym H, Clarkson E. Diagnosis and management of common postextraction complications. Dent Clin North Am. 2012;56:75-93.
35. Renton T. Dental (odontogenic) pain. Br J Pain. 2011;5:2-7.
36. Yatani H, Komiyama O, Matsuka Y, et al. Systematic review and recommendations for nonodontogenic toothache. J Oral Rehabil. 2014;41:843-852.
37. Klasser GD, Fischer DJ, Epstein JB. Burning mouth syndrome: recognition, understanding, and management. Oral Maxillofac Surg Clin North Am. 2008;20:255-271.
38. Balasubramaniam R, Turner LN, Fischer D, et al. Non-odontogenic toothache revisited. Open Journal of Stomatology. 2011;1:92-102.
39. Patton LL, Siegel MA, Benoliel R, et al. Management of burning mouth syndrome: systematic review and management recommendations. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:S39.e1-e13.
40. Cascarini L, McGurk M. Epidemiology of salivary gland infections. Oral Maxillofac Surg Clin North Am. 2009;21:353-357.
41. Hegarty AM, Zakrzewska JM. Differential diagnosis for orofacial pain, including sinusitis, TMD, trigeminal neuralgia. Dent Update. 2011;38:396-400,402-403,405-406.

Treating depression: What works besides meds?
› Recommend cognitive behavioral therapy, interpersonal therapy, or problem-solving therapy for the treatment of depression in patients of all ages. A
› Consider prescribing exercise as a stand-alone or adjunctive treatment for patients with depression. B
› Advise patients who ask about omega-3 fatty acid supplements that formulations with a high eicosapentaenoic acid (EPA) to docosahexaenoic acid (DHA) ratio (2:1) may be a useful “add-on” to their current regimen. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Steve J, age 43, comes to your clinic looking uncharacteristically glum. He was recently downsized from his job and misses his former colleagues. His job loss has caused a financial strain for his family, and he admits to crying in the shower when he thinks about how his life has turned out. Mr. J tells you that he’s gotten a part-time job, but he’s already called in sick several times. On those sick days he “stayed in bed all day and slept.” He says that when he does go to work, he rarely interacts with his coworkers and his concentration is poor. He tells you he wakes up early in the morning on most days and cannot return to sleep, despite being “tired all the time.” He denies suicidal ideation. Mr. J has never felt this way before, which is what prompted his visit today, but he thinks it is “weak to take a pill to feel better.”
What nonpharmacologic options can you offer him?
CASE 2 › Kerri S is a 27-year-old mother of 2 who comes to your clinic to establish care. She tells you about a recent recurrence of depressed mood, which she feels is due to the stress of moving to the area. She is experiencing sleep-onset insomnia and concentration lapses. Her appetite is poor (self-reported 8-lb weight loss in 2 months) and she lacks the motivation to engage in her daily activities, saying, “I wouldn’t even get out of bed if my kids didn’t need me.” She notes that she is constantly irritable and has completely lost her sex drive. Unlike her prior depressive episode, she has not had any suicidal thoughts. Mrs. S was previously successfully treated with paroxetine, 20 mg/d, but she is not interested in restarting her medication because she is still breastfeeding her toddler.
Are there evidence-based options for her care that do not include medication?
Major depressive disorder (MDD) is widespread and often disabling, affecting nearly 8% of people ages 12 and older at any given time.1 Thus, it’s crucial to be familiar with the diverse array of evidence-based treatment options from which patients can choose. Although medications are an essential treatment option for patients with severe depression, their value for patients with mild to moderate depression is often limited.2 In addition, when antidepressants aren’t combined with psychosocial interventions, discontinuing them is associated with relapse.3
Fortunately, research has found that certain nonpharmacologic interventions—including psychotherapies, somatic therapies, and dietary supplements—can have either therapeutic or adjunctive benefits for treating depression, and can be provided in ways that are time- and cost-effective. This article reviews the evidence supporting several options in each of these treatment categories.
Evidence backs several types of psychotherapy
Several recent meta-analyses suggest that a variety of psychotherapeutic treatments may hold promise for your patients with depression.4,5 When analyses were limited to larger studies in order to decrease the risk of bias, cognitive behavioral therapy (CBT), interpersonal therapy (IPT), and problem-solving therapy (PST) all resulted in moderate to large improvement in depressive symptoms when compared to wait-list controls.4 These findings were echoed in a recent systematic review/meta-analysis that focused on depressed primary care patients. Linde et al5 found that the number needed to treat (NNT) to achieve one response (≥50% reduction in score on a depression scale) using any type of psychotherapy was 10, and the NNT to achieve one remission (scoring below a predefined score on a depression scale) was 15.
Psychotherapy can be effective when provided in individual and group settings,6 as well as via telephone, the Internet, or software programs.7 (For a list of self-help, computerized, and Internet-based resources, see TABLE W1 below.)
CBT has been studied for several decades and there’s strong evidence for its efficacy.6 Recent investigations have suggested that CBT delivered in less resource-intensive modes (such as via computer program, Internet, telephone, or videoconferencing) can be as effective as face-to-face CBT.6,8 CBT has been shown to be helpful for a wide range of patients,6 improves outcomes over standard primary care treatment,9 and provides a useful adjunct to medication in treatment-resistant severe depression.10
Behavioral activation (BA), which generally is included as a component of CBT, has received support as an independent treatment, and may produce therapeutic results similar to CBT11 and PST (which we’ll discuss in a bit).12 The core components of BA are scheduling pleasant activities and increasing the patient’s positive interactions with his or her environment by decreasing avoidance, withdrawal, and inactivity.11 Compared to CBT, BA is easier for clinicians to learn and incorporate into primary care visits, and it may be especially useful as an adjunctive or first-step intervention in outpatient clinics.11 Like CBT, BA can be effective in diverse patient groups13,14 and can be provided using novel delivery modes, such as via the Internet.15
IPT is a supportive, structured, brief therapy (12-16 visits) that focuses on helping patients identify and solve current situation- and relationship-based problems that stem from or contribute to their depression.16 Enhancing the patient’s interpersonal communication—including improving social skills, assertiveness, and appropriate expression of anger—is typically a component of IPT. Like CBT, IPT has been found to be effective for treating depression when administered in person, in group therapy, or via the phone or Internet, and across a broad age range.17-19
PST involves teaching patients a structured problem-solving process to decrease interpersonal strain and improve positive life experiences.20 Patients are taught to define their problem, generate and evaluate multiple solutions for it, implement a plan for the solution, and evaluate the results. In addition to being used to successfully treat adults,4,5 PST has been adapted effectively to treat adolescents16 and older adults.18
Somatic therapies are also an option
Exercise has long been considered a possible depression treatment due to its activity on endorphin, monoamine, and cortisol levels and via increased social and general activity. A 2013 Cochrane review of 39 randomized control trials (RCTs; N=2326) assessed whether exercise was effective for treating depression in adults.21 Thirty-five trials found a moderate effect size when specifically comparing exercise to no treatment or control interventions. The effect size was reduced, however, when analyses were restricted to trials with the highest methodological quality. There was no statistically significant difference when exercise was compared to pharmacologic treatment or psychotherapy.
Although the amount of research is meager, small but statistically significant improvements have also been found for older adults22 and children/adolescents.23 There is no consensus on the type, frequency, or intensity of exercise needed to achieve benefit. However, because nearly all studies for all age groups have found that exercise has no adverse psychological effects and substantial positive physical effects, exercise should be recommended to all patients with depression unless contraindicated.
Yoga (both exercise-based and meditation-based) has been evaluated both as a sole treatment and as an adjunctive treatment for depression. Several studies have supported the impact of yoga, particularly in pregnant women,24 although the evidence for its efficacy is inconsistent, with yoga frequently failing to improve upon the outcome of waitlist control.25 The evidence for meditation and mindfulness is more consistently positive, with these interventions equaling or exceeding “treatment as usual,” other psychotherapies, and antidepressants in numerous RCTs.25
Electroconvulsive therapy (ECT) has a substantial evidence base supporting its efficacy.26 ECT has been used for decades, although stigma, cardiac and memory risks, and risks of anesthesia often limit its use. Benefits of ECT include a rapid response relative to pharmacotherapy (>50% of patients respond by the end of the first week of ECT)27 and a strong response in older patients.28
In repetitive transcranial magnetic stimulation (rTMS), electromagnetic coils are placed on a patient’s head to deliver electromagnetic pulses that stimulate areas of the brain that regulate mood. Although rTMS is not widely available, a growing body of evidence supports its use for treating depression, including a meta-analysis of 34 RCTs that included 1383 patients.29 A multisite RCT (N=190) that was not industry-funded reported a 15% response rate and 60% maintenance of remission at 3 months (NNT=12).30 Although ECT is more effective than rTMS, rTMS appears useful for treatment-resistant depression, and can be used as an adjunctive treatment.29,31
Dietary supplements may be best used as adjuncts
St. John’s wort (Hypericum perforatum), which contains 2 bioactive ingredients (hyperforin and hypericin), has been effectively used to treat depression.32 A 2008 Cochrane review that was limited to high-quality trials involving patients meeting Diagnostic and Statistical Manual of Mental Disorders, 4th Edition criteria for depression identified 29 trials (N=5489), of which 18 involved comparisons with placebo and 17 with standard antidepressants.33 Patients’ depression was rated mild to moderate in 19 studies and moderate to severe in 9 studies. Trials examined 4 to 12 weeks of treatment with Hypericum extracts. This study (and several published since) provides strong clinical evidence supporting the efficacy of St. John’s wort for mild to moderate depression. There is insufficient evidence for its use for severe major depression.33TABLE 1 contains dosing information for St. John’s wort and other supplements used to treat depression.34-36
S-adenosyl-L-methionine (SAMe). In a 2003 systematic review,37 1600 mg/d of oral SAMe was found to significantly benefit patients with depression in 4 of 5 studies, as did parenteral SAMe (7 of 7 trials). Another review of 48 studies found SAMe was safe and effective for depression.38 SAMe has been proposed for use alone or in combination with an antidepressant.
Folate and folic acid. Low folate levels have been associated with a less robust response to antidepressants in patients with MDD,39 and higher folate levels appear to be associated with better antidepressant response.40 A 2003 Cochrane review suggested folate might have a role in treating depression.39 A 2009 study found folate supplementation could reduce depressive symptoms for patients with normal baseline folate levels as well as those with low levels.41 Although the evidence is equivocal, folate augmentation may enhance antidepressant efficacy or improve response/remission rates.41,42
It seems reasonable to check folate levels in depressed patients, and address deficiencies by instructing patients to increase their dietary intake of folate or to take supplements. Augmenting antidepressants with folate appears to be low-risk and possibly helpful in maintaining remission.
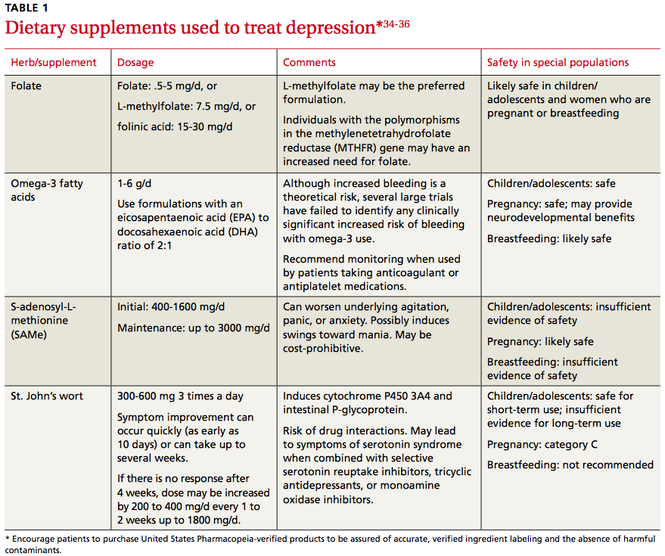
Omega-3 fatty acids. There is substantial evidence that omega-3 fatty acids can prevent and treat depression.43,44 Recent meta-analyses support the use of omega-3 fatty acids as monotherapy and augmentation, but only formulations that contain a high eicosapentaenoic acid (EPA) to docosahexaenoic acid (DHA) ratio (EPA/DHA 2:1).45,46 Omega-3 supplementation has been used with positive results in older adults, children,47 pregnant women,48 and women with postpartum depression.49 Although initial research into omega-3 treatment of depression appears promising, augmentation of standard antidepressant therapy may be a good conservative option.
Use a validated tool to monitor response to treatment
You can enhance outcomes for your patients with depression if you schedule routine follow-up visits with them to gauge adherence to recommendations, monitor response to treatment, and increase the intensity of care when response is inadequate.50 The most important aspect of monitoring response is to use a standardized instrument that quantifies symptoms at every visit.
The Patient Health Questionnaire 9-item depression assessment (PHQ-9)—which is free—has been validated for depression screening and monitoring of treatment response in primary care patients.51 A decrease of 5 points on the PHQ-9 is the minimum considered to be clinically significant.52 Other well-validated, although lengthier, self-report depression assessment and monitoring instruments include the Beck Depression Inventory-revised and the Zung Depression Scale.
CASE 1 › Mr. J is not enjoying his new job or engaging with new coworkers to replace the positive social experiences he had at his previous job. Together, you set a goal of increasing social involvement by having him make plans to see at least one friend per weekend. Because he indicates that he is unlikely to follow through with a therapy referral, you encourage him to try an online CBT program, start an exercise regimen, or take a SAMe supplement. Mr. Jackson agrees to try the CBT and exercise (moderate intensity, 30 minutes 3-4 times per week), but does not want to take SAMe. He agrees to an assessment of his folate levels, which are normal.
Mr. J starts the online CBT program, which reinforces the exercise and social activity prescription you provided. He establishes a regular exercise routine with a good friend. After one month, his mood has started to improve and he has added regular participation in a hobby (woodworking), as well as volunteer work, which he finds fulfilling. You plan to continue monitoring his depression and his adherence to the treatment plan.
CASE 2 › The recent move has decreased Mrs. S’s interactions with family and long-time friends. Because she had previously expressed interest in exercise, you encourage her to join a local “Mommy and Me” exercise and support group for mothers of toddlers. She is willing to participate in psychotherapy, so you provide a referral to a local therapist with expertise in IPT. You also discuss with Mrs. S the possible benefits of omega-3 fatty acid supplementation, which appears to be safe during breastfeeding.34
Mrs. S begins therapy and exercise classes, but can’t motivate herself to continue either of these activities. She becomes discouraged because she’s unable to easily find an omega-3 fatty acid supplement with the ratio you specified (EPA/DHA 2:1). When you see her 2 weeks later, her depression has worsened.
Because you are concerned her suicidality will return, you revisit the pros and cons of taking an antidepressant. Although small amounts of antidepressants can be passed from mother to infant via breastmilk, the amount varies by specific medication, as do the potential risks. Mrs. S decides to resume taking paroxetine 20 mg/d and eventually, once her motivation improves, she’s able to add psychotherapy and exercise to her maintenance/relapse prevention regimen. After you discuss with her the possibility that B vitamin supplementation may assist in maintenance of remission, she adds L-methylfolate 7.5 mg/day to her regimen.
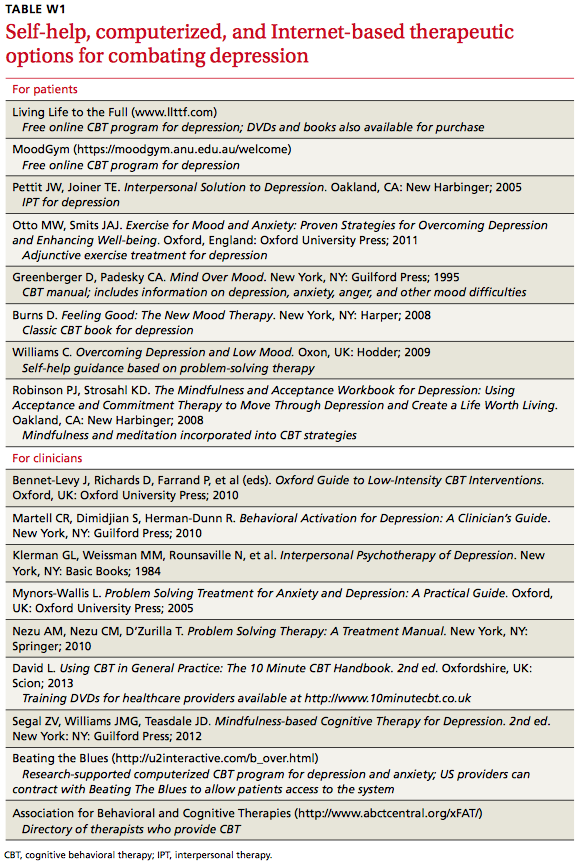
CORRESPONDENCE
Michele M. Larzelere, PhD; LSUHSC Department of Family Medicine; 200 W. Esplanade Avenue, Suite 409; Kenner, LA 70065; [email protected]
1. Centers for Disease Control and Prevention (CDC). QuickStats: Prevalence of Current Depression Among Persons Aged ≥12 Years, by Age Group and Sex — United States, National Health and Nutrition Examination Survey, 2007–2010. CDC Morbidity and Mortality Weekly Report Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6051a7.htm. Accessed June 11, 2015.
2. Fournier J, DeRubeis RJ, Hollon SD, et al. Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA. 2010;303:47-53.
3. Dobson KS, Hollon SD, Dimidjian S, et al. Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the prevention of relapse and recurrence in major depression. J Consult Clin Psychol. 2008;76:468-477.
4. Barth J, Munder T, Gerger H, et al. Comparative efficacy of seven psychotherapeutic interventions for patients with depression: A network meta-analysis. PLoS Med. 2013;10:e1001454.
5. Linde K, Sigterman K, Kriston L, et al. Effectiveness of psychological treatments for depressive disorders in primary care: systematic review and meta-analysis. Ann Fam Med. 2015;13:56-68.
6. DeRubeis RJ, Webb CA, Tang TZ, et al. Cognitive therapy. In: Dobson KS, ed. Handbook of Cognitive Behavioral Therapies, 3rd ed. New York, NY: Guilford; 2009:277-316.
7. Andersson G, Cuijpers P. Internet-based and other computerized psychological treatments for adult depression: a meta-analysis. Cogn Behav Ther. 2008;38:196-205.
8. Andersson G, Cuijpers P, Carlbring P, et al. Guided internet-based vs. face-to-face cognitive behavior therapy for psychiatric and somatic disorders: a systematic review and meta-analysis. World Psychiatry. 2014;13:288-295.
9. Twomey C, O’Reilly G, Byrne M. Effectiveness of cognitive behavioral therapy for anxiety and depression in primary care: a meta-analysis. Fam Pract. 2015;32:3-15.
10. Zhou X, Michael K, Liu Y, et al. Systematic review of management for treatment-resistant depression in adolescents. BMC Psychiatry. 2014;14:340.
11. Ekers D, Webster L, Van Straten A, et al. Behavioural activation for depression: An update of meta-analysis of effectiveness and sub group analysis. PLoS One. 2014;9:e100100.
12. Alexopoulos GS, Raue PJ, Kiosses DN, et al. Comparing engage with PST in late-life major depression: A preliminary report. Am J Geriatr Psychiatry. 2015;23:506-513.
13. Soucy Chartier I, Provencher MD. Behavioral activation for depression: Efficacy, effectiveness, and dissemination. J Affect Disord. 2013;145:292-299.
14. McCauley E, Gudmundson G, Schloredt K, et al. The Adolescent Behavior Activation Program: Adapting behavioral activation as a treatment for depression in adolescence. J Clin Child Adolesc Psychol. 2015;1-14. [Epub ahead of print].
15. Carlbring P, Hägglund M, Luthström A, et al. Internet-based behavioral activation and acceptance-based treatment for depression: a randomized controlled trial. J Affect Disord. 2013;148:331-337.
16. Markowitz JC, Weissman MM. Interpersonal psychotherapy: principles and applications. World Psychiatry. 2004; 3:136-139.
17. Kersting A, Kroker K, Schlicht S, et al. Efficacy of a cognitive-behavioral internet-based therapy in parents after the loss of a child during pregnancy: pilot data from a randomized controlled trial. Arch Womens Mental Health. 2011;14:465-477.
18. Francis J, Kumar A. Psychological treatment of late-life depression. Psychiatr Clin North Am. 2013;36:561-575.
19. Picardi A, Gaetano P. Psychotherapy of mood disorders. Clin Pract Epidemiol Ment Health. 2014;10:140-158.
20. Bell AC, D’Zurilla TJ. Problem-solving therapy for depression: a meta-analysis. Clin Psychol Review. 2009;29:348-353.
21. Cooney GM, Dwan K, Greig CA, et al. Exercise for depression. Cochrane Database Syst Rev. 2013;9:CD004366
22. Brindle C, Spanjers K, Patel S, et al. Effect of exercise on depression severity in older people: systematic review and meta-analysis of randomized controlled trials. B J Psychiatry. 2012;201:180-185.
23. Brown HE, Pearson N, Braithwaite RE, et al. Physical activity interventions and depression in children and adolescents: a systematic review and meta-analysis. Sports Med. 2013;43:195-206.
24. Gong H, Ni C, Shen X, et al. Yoga for prenatal depression: a systematic review and meta-analysis. BMC Psychiatry. 2015;15:14.
25. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
26. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357:1939-1945.
27. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65:485-491.
28. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23:274-282.
29. Slotema CW, Blom JD, Hoek HW, et al. Should we expand the toolbox of psychiatric treatment methods to include Repetitive Transcranial Magnetic Stimulation (rTMS)? A metaanalysis of the efficacy of rTMS in psychiatric disorders. J Clin Psychiatry. 2010;71:873-884.
30. George MS, Lisanby SH, Avery D, et al. Daily left prefrontal transcranial magnetic stimulation therapy for major depressive disorder: a sham-controlled randomized trial. Arch Gen Psychiatry. 2010;67:507-516
31. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham controlled studies. BMC Psychiatry. 2014;14:342.
32. Brown RP, Gerberg PL, Muskin PR. Mood disorders. In: Brown RP, Gerbarg PL, Muskin P. How to Use Herbs, Nutrients and Yoga in Mental Health. New York, NY: WW Norton & Company; 2009.
33. Linde K, Berner MM, Kriston L. St John’s wort for major depression. Cochrane Database Syst Rev. 2008;(4):CD000448.
34. Natural Medicines Comprehensive Database. Natural Medicines Comprehensive Database Web site. Available at: http://naturaldatabase.therapeuticresearch.com/home.aspx. Accessed March 1, 2015.
35. Harris WS. Expert opinion: omega-3 fatty acids and bleeding-cause for concern? Am J Cardiol. 2007;99:44C-6C.
36. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force Report. J Clin Psychiatry. 2010;71:669-681.
37. Papakostas GI, Alpert JE, Fava M. S-adenosyl-methionine in depression: a comprehensive review of the literature. Curr Psychiatry Reports. 2003;5:460-466.
38. Brown RP, Gerbarg PL, Bottiglieri T. S-Adenosylmethionine (SAMe) for depression: biochemical and clinical evidence. Psychiatr Ann. 2002;32:29-44.
39. Taylor MJ, Carney S, Geddes J, et al. Folate for depressive disorders. Cochrane Database Syst Rev. 2003;(2):CD003390.
40. Alpert M, Silva RR, Pouget ER. Prediction of treatment response in geriatric depression from baseline folate level: interaction with an SSRI or a tricyclic antidepressant. J Clin Psychopharmacol. 2003;23:309-313.
41. Fava M, Mischoulon D. Folate in depression: efficacy, safety, differences in formulations, and clinical issues. J Clin Psychiatry. 2009;70(suppl 5):12-17.
42. Almeida OP, Ford AH, Hirani V, et al. B vitamins to enhance treatment response to antidepressants in middle-aged and older adults: results from the B-VITAGE randomised, double-blind, placebo-controlled trial. Br J Psychiatry. 2014;205:450-457.
43. Grosso G, Galvano F, Marventano S, et al. Omega-3 fatty acids and depression: scientific evidence and biological mechanisms. Oxid Med Cell Longev. 2014;2014:313570.
44. Appleton KM, Rogers PJ, Ness AR. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91:757-770.
45. Grosso G, Pajak A, Marventano S, et al. Role of omega-3 fatty acids in the treatment of depressive disorders: a comprehensive metaanalysis of randomized clinical trials. PLoS One. 2014;9:e96905.
46. Martins JG, Bentsen H, Puri BK. Eicosapentaenoic acid appears to be the key omega-3 fatty acid component associated with efficacy in major depressive disorder: a critique of Bloch and Hannestad and updated meta-analysis. Mol Psychiatry. 2012;17:1144-1149.
47. Nemets H, Nemets B, Apter A, et al. Omega-3 treatment of childhood depression: a controlled, double-blind pilot study. Am J Psychiatry. 2006;163:1098-1100.
48. Su KP, Huang SY, Chiu TH. Omega-3 fatty acids for major depressive disorder during pregnancy: Results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2008;69:644-651.
49. Freeman MP, Davis M, Sinha P, et al. Omega-3 fatty acids and supportive psychotherapy for perinatal depression: a randomized placebo-controlled study. J Affect Disord. 2008;110:142-148.
50. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement (ICSI). Health Care Guideline: Adult depression in primary care. 16th ed. September 2013. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Accessed June 9, 2015.
51. Kroenke K, Spitzer RL, Williams JBW, et al. The Patient Health Questionnaire Somatic, Anxiety, and Depressive Symptom Scales: a systematic review. Gen Hosp Psychiatry. 2010;32:345-359.
52. Trivedi MH. Tools and strategies for ongoing assessment of depression:
a measurement-based approach to remission. J Clin Psychiatry. 2009;70:26-31.
› Recommend cognitive behavioral therapy, interpersonal therapy, or problem-solving therapy for the treatment of depression in patients of all ages. A
› Consider prescribing exercise as a stand-alone or adjunctive treatment for patients with depression. B
› Advise patients who ask about omega-3 fatty acid supplements that formulations with a high eicosapentaenoic acid (EPA) to docosahexaenoic acid (DHA) ratio (2:1) may be a useful “add-on” to their current regimen. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Steve J, age 43, comes to your clinic looking uncharacteristically glum. He was recently downsized from his job and misses his former colleagues. His job loss has caused a financial strain for his family, and he admits to crying in the shower when he thinks about how his life has turned out. Mr. J tells you that he’s gotten a part-time job, but he’s already called in sick several times. On those sick days he “stayed in bed all day and slept.” He says that when he does go to work, he rarely interacts with his coworkers and his concentration is poor. He tells you he wakes up early in the morning on most days and cannot return to sleep, despite being “tired all the time.” He denies suicidal ideation. Mr. J has never felt this way before, which is what prompted his visit today, but he thinks it is “weak to take a pill to feel better.”
What nonpharmacologic options can you offer him?
CASE 2 › Kerri S is a 27-year-old mother of 2 who comes to your clinic to establish care. She tells you about a recent recurrence of depressed mood, which she feels is due to the stress of moving to the area. She is experiencing sleep-onset insomnia and concentration lapses. Her appetite is poor (self-reported 8-lb weight loss in 2 months) and she lacks the motivation to engage in her daily activities, saying, “I wouldn’t even get out of bed if my kids didn’t need me.” She notes that she is constantly irritable and has completely lost her sex drive. Unlike her prior depressive episode, she has not had any suicidal thoughts. Mrs. S was previously successfully treated with paroxetine, 20 mg/d, but she is not interested in restarting her medication because she is still breastfeeding her toddler.
Are there evidence-based options for her care that do not include medication?
Major depressive disorder (MDD) is widespread and often disabling, affecting nearly 8% of people ages 12 and older at any given time.1 Thus, it’s crucial to be familiar with the diverse array of evidence-based treatment options from which patients can choose. Although medications are an essential treatment option for patients with severe depression, their value for patients with mild to moderate depression is often limited.2 In addition, when antidepressants aren’t combined with psychosocial interventions, discontinuing them is associated with relapse.3
Fortunately, research has found that certain nonpharmacologic interventions—including psychotherapies, somatic therapies, and dietary supplements—can have either therapeutic or adjunctive benefits for treating depression, and can be provided in ways that are time- and cost-effective. This article reviews the evidence supporting several options in each of these treatment categories.
Evidence backs several types of psychotherapy
Several recent meta-analyses suggest that a variety of psychotherapeutic treatments may hold promise for your patients with depression.4,5 When analyses were limited to larger studies in order to decrease the risk of bias, cognitive behavioral therapy (CBT), interpersonal therapy (IPT), and problem-solving therapy (PST) all resulted in moderate to large improvement in depressive symptoms when compared to wait-list controls.4 These findings were echoed in a recent systematic review/meta-analysis that focused on depressed primary care patients. Linde et al5 found that the number needed to treat (NNT) to achieve one response (≥50% reduction in score on a depression scale) using any type of psychotherapy was 10, and the NNT to achieve one remission (scoring below a predefined score on a depression scale) was 15.
Psychotherapy can be effective when provided in individual and group settings,6 as well as via telephone, the Internet, or software programs.7 (For a list of self-help, computerized, and Internet-based resources, see TABLE W1 below.)
CBT has been studied for several decades and there’s strong evidence for its efficacy.6 Recent investigations have suggested that CBT delivered in less resource-intensive modes (such as via computer program, Internet, telephone, or videoconferencing) can be as effective as face-to-face CBT.6,8 CBT has been shown to be helpful for a wide range of patients,6 improves outcomes over standard primary care treatment,9 and provides a useful adjunct to medication in treatment-resistant severe depression.10
Behavioral activation (BA), which generally is included as a component of CBT, has received support as an independent treatment, and may produce therapeutic results similar to CBT11 and PST (which we’ll discuss in a bit).12 The core components of BA are scheduling pleasant activities and increasing the patient’s positive interactions with his or her environment by decreasing avoidance, withdrawal, and inactivity.11 Compared to CBT, BA is easier for clinicians to learn and incorporate into primary care visits, and it may be especially useful as an adjunctive or first-step intervention in outpatient clinics.11 Like CBT, BA can be effective in diverse patient groups13,14 and can be provided using novel delivery modes, such as via the Internet.15
IPT is a supportive, structured, brief therapy (12-16 visits) that focuses on helping patients identify and solve current situation- and relationship-based problems that stem from or contribute to their depression.16 Enhancing the patient’s interpersonal communication—including improving social skills, assertiveness, and appropriate expression of anger—is typically a component of IPT. Like CBT, IPT has been found to be effective for treating depression when administered in person, in group therapy, or via the phone or Internet, and across a broad age range.17-19
PST involves teaching patients a structured problem-solving process to decrease interpersonal strain and improve positive life experiences.20 Patients are taught to define their problem, generate and evaluate multiple solutions for it, implement a plan for the solution, and evaluate the results. In addition to being used to successfully treat adults,4,5 PST has been adapted effectively to treat adolescents16 and older adults.18
Somatic therapies are also an option
Exercise has long been considered a possible depression treatment due to its activity on endorphin, monoamine, and cortisol levels and via increased social and general activity. A 2013 Cochrane review of 39 randomized control trials (RCTs; N=2326) assessed whether exercise was effective for treating depression in adults.21 Thirty-five trials found a moderate effect size when specifically comparing exercise to no treatment or control interventions. The effect size was reduced, however, when analyses were restricted to trials with the highest methodological quality. There was no statistically significant difference when exercise was compared to pharmacologic treatment or psychotherapy.
Although the amount of research is meager, small but statistically significant improvements have also been found for older adults22 and children/adolescents.23 There is no consensus on the type, frequency, or intensity of exercise needed to achieve benefit. However, because nearly all studies for all age groups have found that exercise has no adverse psychological effects and substantial positive physical effects, exercise should be recommended to all patients with depression unless contraindicated.
Yoga (both exercise-based and meditation-based) has been evaluated both as a sole treatment and as an adjunctive treatment for depression. Several studies have supported the impact of yoga, particularly in pregnant women,24 although the evidence for its efficacy is inconsistent, with yoga frequently failing to improve upon the outcome of waitlist control.25 The evidence for meditation and mindfulness is more consistently positive, with these interventions equaling or exceeding “treatment as usual,” other psychotherapies, and antidepressants in numerous RCTs.25
Electroconvulsive therapy (ECT) has a substantial evidence base supporting its efficacy.26 ECT has been used for decades, although stigma, cardiac and memory risks, and risks of anesthesia often limit its use. Benefits of ECT include a rapid response relative to pharmacotherapy (>50% of patients respond by the end of the first week of ECT)27 and a strong response in older patients.28
In repetitive transcranial magnetic stimulation (rTMS), electromagnetic coils are placed on a patient’s head to deliver electromagnetic pulses that stimulate areas of the brain that regulate mood. Although rTMS is not widely available, a growing body of evidence supports its use for treating depression, including a meta-analysis of 34 RCTs that included 1383 patients.29 A multisite RCT (N=190) that was not industry-funded reported a 15% response rate and 60% maintenance of remission at 3 months (NNT=12).30 Although ECT is more effective than rTMS, rTMS appears useful for treatment-resistant depression, and can be used as an adjunctive treatment.29,31
Dietary supplements may be best used as adjuncts
St. John’s wort (Hypericum perforatum), which contains 2 bioactive ingredients (hyperforin and hypericin), has been effectively used to treat depression.32 A 2008 Cochrane review that was limited to high-quality trials involving patients meeting Diagnostic and Statistical Manual of Mental Disorders, 4th Edition criteria for depression identified 29 trials (N=5489), of which 18 involved comparisons with placebo and 17 with standard antidepressants.33 Patients’ depression was rated mild to moderate in 19 studies and moderate to severe in 9 studies. Trials examined 4 to 12 weeks of treatment with Hypericum extracts. This study (and several published since) provides strong clinical evidence supporting the efficacy of St. John’s wort for mild to moderate depression. There is insufficient evidence for its use for severe major depression.33TABLE 1 contains dosing information for St. John’s wort and other supplements used to treat depression.34-36
S-adenosyl-L-methionine (SAMe). In a 2003 systematic review,37 1600 mg/d of oral SAMe was found to significantly benefit patients with depression in 4 of 5 studies, as did parenteral SAMe (7 of 7 trials). Another review of 48 studies found SAMe was safe and effective for depression.38 SAMe has been proposed for use alone or in combination with an antidepressant.
Folate and folic acid. Low folate levels have been associated with a less robust response to antidepressants in patients with MDD,39 and higher folate levels appear to be associated with better antidepressant response.40 A 2003 Cochrane review suggested folate might have a role in treating depression.39 A 2009 study found folate supplementation could reduce depressive symptoms for patients with normal baseline folate levels as well as those with low levels.41 Although the evidence is equivocal, folate augmentation may enhance antidepressant efficacy or improve response/remission rates.41,42
It seems reasonable to check folate levels in depressed patients, and address deficiencies by instructing patients to increase their dietary intake of folate or to take supplements. Augmenting antidepressants with folate appears to be low-risk and possibly helpful in maintaining remission.
Omega-3 fatty acids. There is substantial evidence that omega-3 fatty acids can prevent and treat depression.43,44 Recent meta-analyses support the use of omega-3 fatty acids as monotherapy and augmentation, but only formulations that contain a high eicosapentaenoic acid (EPA) to docosahexaenoic acid (DHA) ratio (EPA/DHA 2:1).45,46 Omega-3 supplementation has been used with positive results in older adults, children,47 pregnant women,48 and women with postpartum depression.49 Although initial research into omega-3 treatment of depression appears promising, augmentation of standard antidepressant therapy may be a good conservative option.
Use a validated tool to monitor response to treatment
You can enhance outcomes for your patients with depression if you schedule routine follow-up visits with them to gauge adherence to recommendations, monitor response to treatment, and increase the intensity of care when response is inadequate.50 The most important aspect of monitoring response is to use a standardized instrument that quantifies symptoms at every visit.
The Patient Health Questionnaire 9-item depression assessment (PHQ-9)—which is free—has been validated for depression screening and monitoring of treatment response in primary care patients.51 A decrease of 5 points on the PHQ-9 is the minimum considered to be clinically significant.52 Other well-validated, although lengthier, self-report depression assessment and monitoring instruments include the Beck Depression Inventory-revised and the Zung Depression Scale.
CASE 1 › Mr. J is not enjoying his new job or engaging with new coworkers to replace the positive social experiences he had at his previous job. Together, you set a goal of increasing social involvement by having him make plans to see at least one friend per weekend. Because he indicates that he is unlikely to follow through with a therapy referral, you encourage him to try an online CBT program, start an exercise regimen, or take a SAMe supplement. Mr. Jackson agrees to try the CBT and exercise (moderate intensity, 30 minutes 3-4 times per week), but does not want to take SAMe. He agrees to an assessment of his folate levels, which are normal.
Mr. J starts the online CBT program, which reinforces the exercise and social activity prescription you provided. He establishes a regular exercise routine with a good friend. After one month, his mood has started to improve and he has added regular participation in a hobby (woodworking), as well as volunteer work, which he finds fulfilling. You plan to continue monitoring his depression and his adherence to the treatment plan.
CASE 2 › The recent move has decreased Mrs. S’s interactions with family and long-time friends. Because she had previously expressed interest in exercise, you encourage her to join a local “Mommy and Me” exercise and support group for mothers of toddlers. She is willing to participate in psychotherapy, so you provide a referral to a local therapist with expertise in IPT. You also discuss with Mrs. S the possible benefits of omega-3 fatty acid supplementation, which appears to be safe during breastfeeding.34
Mrs. S begins therapy and exercise classes, but can’t motivate herself to continue either of these activities. She becomes discouraged because she’s unable to easily find an omega-3 fatty acid supplement with the ratio you specified (EPA/DHA 2:1). When you see her 2 weeks later, her depression has worsened.
Because you are concerned her suicidality will return, you revisit the pros and cons of taking an antidepressant. Although small amounts of antidepressants can be passed from mother to infant via breastmilk, the amount varies by specific medication, as do the potential risks. Mrs. S decides to resume taking paroxetine 20 mg/d and eventually, once her motivation improves, she’s able to add psychotherapy and exercise to her maintenance/relapse prevention regimen. After you discuss with her the possibility that B vitamin supplementation may assist in maintenance of remission, she adds L-methylfolate 7.5 mg/day to her regimen.
CORRESPONDENCE
Michele M. Larzelere, PhD; LSUHSC Department of Family Medicine; 200 W. Esplanade Avenue, Suite 409; Kenner, LA 70065; [email protected]
› Recommend cognitive behavioral therapy, interpersonal therapy, or problem-solving therapy for the treatment of depression in patients of all ages. A
› Consider prescribing exercise as a stand-alone or adjunctive treatment for patients with depression. B
› Advise patients who ask about omega-3 fatty acid supplements that formulations with a high eicosapentaenoic acid (EPA) to docosahexaenoic acid (DHA) ratio (2:1) may be a useful “add-on” to their current regimen. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 › Steve J, age 43, comes to your clinic looking uncharacteristically glum. He was recently downsized from his job and misses his former colleagues. His job loss has caused a financial strain for his family, and he admits to crying in the shower when he thinks about how his life has turned out. Mr. J tells you that he’s gotten a part-time job, but he’s already called in sick several times. On those sick days he “stayed in bed all day and slept.” He says that when he does go to work, he rarely interacts with his coworkers and his concentration is poor. He tells you he wakes up early in the morning on most days and cannot return to sleep, despite being “tired all the time.” He denies suicidal ideation. Mr. J has never felt this way before, which is what prompted his visit today, but he thinks it is “weak to take a pill to feel better.”
What nonpharmacologic options can you offer him?
CASE 2 › Kerri S is a 27-year-old mother of 2 who comes to your clinic to establish care. She tells you about a recent recurrence of depressed mood, which she feels is due to the stress of moving to the area. She is experiencing sleep-onset insomnia and concentration lapses. Her appetite is poor (self-reported 8-lb weight loss in 2 months) and she lacks the motivation to engage in her daily activities, saying, “I wouldn’t even get out of bed if my kids didn’t need me.” She notes that she is constantly irritable and has completely lost her sex drive. Unlike her prior depressive episode, she has not had any suicidal thoughts. Mrs. S was previously successfully treated with paroxetine, 20 mg/d, but she is not interested in restarting her medication because she is still breastfeeding her toddler.
Are there evidence-based options for her care that do not include medication?
Major depressive disorder (MDD) is widespread and often disabling, affecting nearly 8% of people ages 12 and older at any given time.1 Thus, it’s crucial to be familiar with the diverse array of evidence-based treatment options from which patients can choose. Although medications are an essential treatment option for patients with severe depression, their value for patients with mild to moderate depression is often limited.2 In addition, when antidepressants aren’t combined with psychosocial interventions, discontinuing them is associated with relapse.3
Fortunately, research has found that certain nonpharmacologic interventions—including psychotherapies, somatic therapies, and dietary supplements—can have either therapeutic or adjunctive benefits for treating depression, and can be provided in ways that are time- and cost-effective. This article reviews the evidence supporting several options in each of these treatment categories.
Evidence backs several types of psychotherapy
Several recent meta-analyses suggest that a variety of psychotherapeutic treatments may hold promise for your patients with depression.4,5 When analyses were limited to larger studies in order to decrease the risk of bias, cognitive behavioral therapy (CBT), interpersonal therapy (IPT), and problem-solving therapy (PST) all resulted in moderate to large improvement in depressive symptoms when compared to wait-list controls.4 These findings were echoed in a recent systematic review/meta-analysis that focused on depressed primary care patients. Linde et al5 found that the number needed to treat (NNT) to achieve one response (≥50% reduction in score on a depression scale) using any type of psychotherapy was 10, and the NNT to achieve one remission (scoring below a predefined score on a depression scale) was 15.
Psychotherapy can be effective when provided in individual and group settings,6 as well as via telephone, the Internet, or software programs.7 (For a list of self-help, computerized, and Internet-based resources, see TABLE W1 below.)
CBT has been studied for several decades and there’s strong evidence for its efficacy.6 Recent investigations have suggested that CBT delivered in less resource-intensive modes (such as via computer program, Internet, telephone, or videoconferencing) can be as effective as face-to-face CBT.6,8 CBT has been shown to be helpful for a wide range of patients,6 improves outcomes over standard primary care treatment,9 and provides a useful adjunct to medication in treatment-resistant severe depression.10
Behavioral activation (BA), which generally is included as a component of CBT, has received support as an independent treatment, and may produce therapeutic results similar to CBT11 and PST (which we’ll discuss in a bit).12 The core components of BA are scheduling pleasant activities and increasing the patient’s positive interactions with his or her environment by decreasing avoidance, withdrawal, and inactivity.11 Compared to CBT, BA is easier for clinicians to learn and incorporate into primary care visits, and it may be especially useful as an adjunctive or first-step intervention in outpatient clinics.11 Like CBT, BA can be effective in diverse patient groups13,14 and can be provided using novel delivery modes, such as via the Internet.15
IPT is a supportive, structured, brief therapy (12-16 visits) that focuses on helping patients identify and solve current situation- and relationship-based problems that stem from or contribute to their depression.16 Enhancing the patient’s interpersonal communication—including improving social skills, assertiveness, and appropriate expression of anger—is typically a component of IPT. Like CBT, IPT has been found to be effective for treating depression when administered in person, in group therapy, or via the phone or Internet, and across a broad age range.17-19
PST involves teaching patients a structured problem-solving process to decrease interpersonal strain and improve positive life experiences.20 Patients are taught to define their problem, generate and evaluate multiple solutions for it, implement a plan for the solution, and evaluate the results. In addition to being used to successfully treat adults,4,5 PST has been adapted effectively to treat adolescents16 and older adults.18
Somatic therapies are also an option
Exercise has long been considered a possible depression treatment due to its activity on endorphin, monoamine, and cortisol levels and via increased social and general activity. A 2013 Cochrane review of 39 randomized control trials (RCTs; N=2326) assessed whether exercise was effective for treating depression in adults.21 Thirty-five trials found a moderate effect size when specifically comparing exercise to no treatment or control interventions. The effect size was reduced, however, when analyses were restricted to trials with the highest methodological quality. There was no statistically significant difference when exercise was compared to pharmacologic treatment or psychotherapy.
Although the amount of research is meager, small but statistically significant improvements have also been found for older adults22 and children/adolescents.23 There is no consensus on the type, frequency, or intensity of exercise needed to achieve benefit. However, because nearly all studies for all age groups have found that exercise has no adverse psychological effects and substantial positive physical effects, exercise should be recommended to all patients with depression unless contraindicated.
Yoga (both exercise-based and meditation-based) has been evaluated both as a sole treatment and as an adjunctive treatment for depression. Several studies have supported the impact of yoga, particularly in pregnant women,24 although the evidence for its efficacy is inconsistent, with yoga frequently failing to improve upon the outcome of waitlist control.25 The evidence for meditation and mindfulness is more consistently positive, with these interventions equaling or exceeding “treatment as usual,” other psychotherapies, and antidepressants in numerous RCTs.25
Electroconvulsive therapy (ECT) has a substantial evidence base supporting its efficacy.26 ECT has been used for decades, although stigma, cardiac and memory risks, and risks of anesthesia often limit its use. Benefits of ECT include a rapid response relative to pharmacotherapy (>50% of patients respond by the end of the first week of ECT)27 and a strong response in older patients.28
In repetitive transcranial magnetic stimulation (rTMS), electromagnetic coils are placed on a patient’s head to deliver electromagnetic pulses that stimulate areas of the brain that regulate mood. Although rTMS is not widely available, a growing body of evidence supports its use for treating depression, including a meta-analysis of 34 RCTs that included 1383 patients.29 A multisite RCT (N=190) that was not industry-funded reported a 15% response rate and 60% maintenance of remission at 3 months (NNT=12).30 Although ECT is more effective than rTMS, rTMS appears useful for treatment-resistant depression, and can be used as an adjunctive treatment.29,31
Dietary supplements may be best used as adjuncts
St. John’s wort (Hypericum perforatum), which contains 2 bioactive ingredients (hyperforin and hypericin), has been effectively used to treat depression.32 A 2008 Cochrane review that was limited to high-quality trials involving patients meeting Diagnostic and Statistical Manual of Mental Disorders, 4th Edition criteria for depression identified 29 trials (N=5489), of which 18 involved comparisons with placebo and 17 with standard antidepressants.33 Patients’ depression was rated mild to moderate in 19 studies and moderate to severe in 9 studies. Trials examined 4 to 12 weeks of treatment with Hypericum extracts. This study (and several published since) provides strong clinical evidence supporting the efficacy of St. John’s wort for mild to moderate depression. There is insufficient evidence for its use for severe major depression.33TABLE 1 contains dosing information for St. John’s wort and other supplements used to treat depression.34-36
S-adenosyl-L-methionine (SAMe). In a 2003 systematic review,37 1600 mg/d of oral SAMe was found to significantly benefit patients with depression in 4 of 5 studies, as did parenteral SAMe (7 of 7 trials). Another review of 48 studies found SAMe was safe and effective for depression.38 SAMe has been proposed for use alone or in combination with an antidepressant.
Folate and folic acid. Low folate levels have been associated with a less robust response to antidepressants in patients with MDD,39 and higher folate levels appear to be associated with better antidepressant response.40 A 2003 Cochrane review suggested folate might have a role in treating depression.39 A 2009 study found folate supplementation could reduce depressive symptoms for patients with normal baseline folate levels as well as those with low levels.41 Although the evidence is equivocal, folate augmentation may enhance antidepressant efficacy or improve response/remission rates.41,42
It seems reasonable to check folate levels in depressed patients, and address deficiencies by instructing patients to increase their dietary intake of folate or to take supplements. Augmenting antidepressants with folate appears to be low-risk and possibly helpful in maintaining remission.
Omega-3 fatty acids. There is substantial evidence that omega-3 fatty acids can prevent and treat depression.43,44 Recent meta-analyses support the use of omega-3 fatty acids as monotherapy and augmentation, but only formulations that contain a high eicosapentaenoic acid (EPA) to docosahexaenoic acid (DHA) ratio (EPA/DHA 2:1).45,46 Omega-3 supplementation has been used with positive results in older adults, children,47 pregnant women,48 and women with postpartum depression.49 Although initial research into omega-3 treatment of depression appears promising, augmentation of standard antidepressant therapy may be a good conservative option.
Use a validated tool to monitor response to treatment
You can enhance outcomes for your patients with depression if you schedule routine follow-up visits with them to gauge adherence to recommendations, monitor response to treatment, and increase the intensity of care when response is inadequate.50 The most important aspect of monitoring response is to use a standardized instrument that quantifies symptoms at every visit.
The Patient Health Questionnaire 9-item depression assessment (PHQ-9)—which is free—has been validated for depression screening and monitoring of treatment response in primary care patients.51 A decrease of 5 points on the PHQ-9 is the minimum considered to be clinically significant.52 Other well-validated, although lengthier, self-report depression assessment and monitoring instruments include the Beck Depression Inventory-revised and the Zung Depression Scale.
CASE 1 › Mr. J is not enjoying his new job or engaging with new coworkers to replace the positive social experiences he had at his previous job. Together, you set a goal of increasing social involvement by having him make plans to see at least one friend per weekend. Because he indicates that he is unlikely to follow through with a therapy referral, you encourage him to try an online CBT program, start an exercise regimen, or take a SAMe supplement. Mr. Jackson agrees to try the CBT and exercise (moderate intensity, 30 minutes 3-4 times per week), but does not want to take SAMe. He agrees to an assessment of his folate levels, which are normal.
Mr. J starts the online CBT program, which reinforces the exercise and social activity prescription you provided. He establishes a regular exercise routine with a good friend. After one month, his mood has started to improve and he has added regular participation in a hobby (woodworking), as well as volunteer work, which he finds fulfilling. You plan to continue monitoring his depression and his adherence to the treatment plan.
CASE 2 › The recent move has decreased Mrs. S’s interactions with family and long-time friends. Because she had previously expressed interest in exercise, you encourage her to join a local “Mommy and Me” exercise and support group for mothers of toddlers. She is willing to participate in psychotherapy, so you provide a referral to a local therapist with expertise in IPT. You also discuss with Mrs. S the possible benefits of omega-3 fatty acid supplementation, which appears to be safe during breastfeeding.34
Mrs. S begins therapy and exercise classes, but can’t motivate herself to continue either of these activities. She becomes discouraged because she’s unable to easily find an omega-3 fatty acid supplement with the ratio you specified (EPA/DHA 2:1). When you see her 2 weeks later, her depression has worsened.
Because you are concerned her suicidality will return, you revisit the pros and cons of taking an antidepressant. Although small amounts of antidepressants can be passed from mother to infant via breastmilk, the amount varies by specific medication, as do the potential risks. Mrs. S decides to resume taking paroxetine 20 mg/d and eventually, once her motivation improves, she’s able to add psychotherapy and exercise to her maintenance/relapse prevention regimen. After you discuss with her the possibility that B vitamin supplementation may assist in maintenance of remission, she adds L-methylfolate 7.5 mg/day to her regimen.
CORRESPONDENCE
Michele M. Larzelere, PhD; LSUHSC Department of Family Medicine; 200 W. Esplanade Avenue, Suite 409; Kenner, LA 70065; [email protected]
1. Centers for Disease Control and Prevention (CDC). QuickStats: Prevalence of Current Depression Among Persons Aged ≥12 Years, by Age Group and Sex — United States, National Health and Nutrition Examination Survey, 2007–2010. CDC Morbidity and Mortality Weekly Report Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6051a7.htm. Accessed June 11, 2015.
2. Fournier J, DeRubeis RJ, Hollon SD, et al. Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA. 2010;303:47-53.
3. Dobson KS, Hollon SD, Dimidjian S, et al. Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the prevention of relapse and recurrence in major depression. J Consult Clin Psychol. 2008;76:468-477.
4. Barth J, Munder T, Gerger H, et al. Comparative efficacy of seven psychotherapeutic interventions for patients with depression: A network meta-analysis. PLoS Med. 2013;10:e1001454.
5. Linde K, Sigterman K, Kriston L, et al. Effectiveness of psychological treatments for depressive disorders in primary care: systematic review and meta-analysis. Ann Fam Med. 2015;13:56-68.
6. DeRubeis RJ, Webb CA, Tang TZ, et al. Cognitive therapy. In: Dobson KS, ed. Handbook of Cognitive Behavioral Therapies, 3rd ed. New York, NY: Guilford; 2009:277-316.
7. Andersson G, Cuijpers P. Internet-based and other computerized psychological treatments for adult depression: a meta-analysis. Cogn Behav Ther. 2008;38:196-205.
8. Andersson G, Cuijpers P, Carlbring P, et al. Guided internet-based vs. face-to-face cognitive behavior therapy for psychiatric and somatic disorders: a systematic review and meta-analysis. World Psychiatry. 2014;13:288-295.
9. Twomey C, O’Reilly G, Byrne M. Effectiveness of cognitive behavioral therapy for anxiety and depression in primary care: a meta-analysis. Fam Pract. 2015;32:3-15.
10. Zhou X, Michael K, Liu Y, et al. Systematic review of management for treatment-resistant depression in adolescents. BMC Psychiatry. 2014;14:340.
11. Ekers D, Webster L, Van Straten A, et al. Behavioural activation for depression: An update of meta-analysis of effectiveness and sub group analysis. PLoS One. 2014;9:e100100.
12. Alexopoulos GS, Raue PJ, Kiosses DN, et al. Comparing engage with PST in late-life major depression: A preliminary report. Am J Geriatr Psychiatry. 2015;23:506-513.
13. Soucy Chartier I, Provencher MD. Behavioral activation for depression: Efficacy, effectiveness, and dissemination. J Affect Disord. 2013;145:292-299.
14. McCauley E, Gudmundson G, Schloredt K, et al. The Adolescent Behavior Activation Program: Adapting behavioral activation as a treatment for depression in adolescence. J Clin Child Adolesc Psychol. 2015;1-14. [Epub ahead of print].
15. Carlbring P, Hägglund M, Luthström A, et al. Internet-based behavioral activation and acceptance-based treatment for depression: a randomized controlled trial. J Affect Disord. 2013;148:331-337.
16. Markowitz JC, Weissman MM. Interpersonal psychotherapy: principles and applications. World Psychiatry. 2004; 3:136-139.
17. Kersting A, Kroker K, Schlicht S, et al. Efficacy of a cognitive-behavioral internet-based therapy in parents after the loss of a child during pregnancy: pilot data from a randomized controlled trial. Arch Womens Mental Health. 2011;14:465-477.
18. Francis J, Kumar A. Psychological treatment of late-life depression. Psychiatr Clin North Am. 2013;36:561-575.
19. Picardi A, Gaetano P. Psychotherapy of mood disorders. Clin Pract Epidemiol Ment Health. 2014;10:140-158.
20. Bell AC, D’Zurilla TJ. Problem-solving therapy for depression: a meta-analysis. Clin Psychol Review. 2009;29:348-353.
21. Cooney GM, Dwan K, Greig CA, et al. Exercise for depression. Cochrane Database Syst Rev. 2013;9:CD004366
22. Brindle C, Spanjers K, Patel S, et al. Effect of exercise on depression severity in older people: systematic review and meta-analysis of randomized controlled trials. B J Psychiatry. 2012;201:180-185.
23. Brown HE, Pearson N, Braithwaite RE, et al. Physical activity interventions and depression in children and adolescents: a systematic review and meta-analysis. Sports Med. 2013;43:195-206.
24. Gong H, Ni C, Shen X, et al. Yoga for prenatal depression: a systematic review and meta-analysis. BMC Psychiatry. 2015;15:14.
25. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
26. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357:1939-1945.
27. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65:485-491.
28. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23:274-282.
29. Slotema CW, Blom JD, Hoek HW, et al. Should we expand the toolbox of psychiatric treatment methods to include Repetitive Transcranial Magnetic Stimulation (rTMS)? A metaanalysis of the efficacy of rTMS in psychiatric disorders. J Clin Psychiatry. 2010;71:873-884.
30. George MS, Lisanby SH, Avery D, et al. Daily left prefrontal transcranial magnetic stimulation therapy for major depressive disorder: a sham-controlled randomized trial. Arch Gen Psychiatry. 2010;67:507-516
31. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham controlled studies. BMC Psychiatry. 2014;14:342.
32. Brown RP, Gerberg PL, Muskin PR. Mood disorders. In: Brown RP, Gerbarg PL, Muskin P. How to Use Herbs, Nutrients and Yoga in Mental Health. New York, NY: WW Norton & Company; 2009.
33. Linde K, Berner MM, Kriston L. St John’s wort for major depression. Cochrane Database Syst Rev. 2008;(4):CD000448.
34. Natural Medicines Comprehensive Database. Natural Medicines Comprehensive Database Web site. Available at: http://naturaldatabase.therapeuticresearch.com/home.aspx. Accessed March 1, 2015.
35. Harris WS. Expert opinion: omega-3 fatty acids and bleeding-cause for concern? Am J Cardiol. 2007;99:44C-6C.
36. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force Report. J Clin Psychiatry. 2010;71:669-681.
37. Papakostas GI, Alpert JE, Fava M. S-adenosyl-methionine in depression: a comprehensive review of the literature. Curr Psychiatry Reports. 2003;5:460-466.
38. Brown RP, Gerbarg PL, Bottiglieri T. S-Adenosylmethionine (SAMe) for depression: biochemical and clinical evidence. Psychiatr Ann. 2002;32:29-44.
39. Taylor MJ, Carney S, Geddes J, et al. Folate for depressive disorders. Cochrane Database Syst Rev. 2003;(2):CD003390.
40. Alpert M, Silva RR, Pouget ER. Prediction of treatment response in geriatric depression from baseline folate level: interaction with an SSRI or a tricyclic antidepressant. J Clin Psychopharmacol. 2003;23:309-313.
41. Fava M, Mischoulon D. Folate in depression: efficacy, safety, differences in formulations, and clinical issues. J Clin Psychiatry. 2009;70(suppl 5):12-17.
42. Almeida OP, Ford AH, Hirani V, et al. B vitamins to enhance treatment response to antidepressants in middle-aged and older adults: results from the B-VITAGE randomised, double-blind, placebo-controlled trial. Br J Psychiatry. 2014;205:450-457.
43. Grosso G, Galvano F, Marventano S, et al. Omega-3 fatty acids and depression: scientific evidence and biological mechanisms. Oxid Med Cell Longev. 2014;2014:313570.
44. Appleton KM, Rogers PJ, Ness AR. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91:757-770.
45. Grosso G, Pajak A, Marventano S, et al. Role of omega-3 fatty acids in the treatment of depressive disorders: a comprehensive metaanalysis of randomized clinical trials. PLoS One. 2014;9:e96905.
46. Martins JG, Bentsen H, Puri BK. Eicosapentaenoic acid appears to be the key omega-3 fatty acid component associated with efficacy in major depressive disorder: a critique of Bloch and Hannestad and updated meta-analysis. Mol Psychiatry. 2012;17:1144-1149.
47. Nemets H, Nemets B, Apter A, et al. Omega-3 treatment of childhood depression: a controlled, double-blind pilot study. Am J Psychiatry. 2006;163:1098-1100.
48. Su KP, Huang SY, Chiu TH. Omega-3 fatty acids for major depressive disorder during pregnancy: Results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2008;69:644-651.
49. Freeman MP, Davis M, Sinha P, et al. Omega-3 fatty acids and supportive psychotherapy for perinatal depression: a randomized placebo-controlled study. J Affect Disord. 2008;110:142-148.
50. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement (ICSI). Health Care Guideline: Adult depression in primary care. 16th ed. September 2013. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Accessed June 9, 2015.
51. Kroenke K, Spitzer RL, Williams JBW, et al. The Patient Health Questionnaire Somatic, Anxiety, and Depressive Symptom Scales: a systematic review. Gen Hosp Psychiatry. 2010;32:345-359.
52. Trivedi MH. Tools and strategies for ongoing assessment of depression:
a measurement-based approach to remission. J Clin Psychiatry. 2009;70:26-31.
1. Centers for Disease Control and Prevention (CDC). QuickStats: Prevalence of Current Depression Among Persons Aged ≥12 Years, by Age Group and Sex — United States, National Health and Nutrition Examination Survey, 2007–2010. CDC Morbidity and Mortality Weekly Report Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6051a7.htm. Accessed June 11, 2015.
2. Fournier J, DeRubeis RJ, Hollon SD, et al. Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA. 2010;303:47-53.
3. Dobson KS, Hollon SD, Dimidjian S, et al. Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the prevention of relapse and recurrence in major depression. J Consult Clin Psychol. 2008;76:468-477.
4. Barth J, Munder T, Gerger H, et al. Comparative efficacy of seven psychotherapeutic interventions for patients with depression: A network meta-analysis. PLoS Med. 2013;10:e1001454.
5. Linde K, Sigterman K, Kriston L, et al. Effectiveness of psychological treatments for depressive disorders in primary care: systematic review and meta-analysis. Ann Fam Med. 2015;13:56-68.
6. DeRubeis RJ, Webb CA, Tang TZ, et al. Cognitive therapy. In: Dobson KS, ed. Handbook of Cognitive Behavioral Therapies, 3rd ed. New York, NY: Guilford; 2009:277-316.
7. Andersson G, Cuijpers P. Internet-based and other computerized psychological treatments for adult depression: a meta-analysis. Cogn Behav Ther. 2008;38:196-205.
8. Andersson G, Cuijpers P, Carlbring P, et al. Guided internet-based vs. face-to-face cognitive behavior therapy for psychiatric and somatic disorders: a systematic review and meta-analysis. World Psychiatry. 2014;13:288-295.
9. Twomey C, O’Reilly G, Byrne M. Effectiveness of cognitive behavioral therapy for anxiety and depression in primary care: a meta-analysis. Fam Pract. 2015;32:3-15.
10. Zhou X, Michael K, Liu Y, et al. Systematic review of management for treatment-resistant depression in adolescents. BMC Psychiatry. 2014;14:340.
11. Ekers D, Webster L, Van Straten A, et al. Behavioural activation for depression: An update of meta-analysis of effectiveness and sub group analysis. PLoS One. 2014;9:e100100.
12. Alexopoulos GS, Raue PJ, Kiosses DN, et al. Comparing engage with PST in late-life major depression: A preliminary report. Am J Geriatr Psychiatry. 2015;23:506-513.
13. Soucy Chartier I, Provencher MD. Behavioral activation for depression: Efficacy, effectiveness, and dissemination. J Affect Disord. 2013;145:292-299.
14. McCauley E, Gudmundson G, Schloredt K, et al. The Adolescent Behavior Activation Program: Adapting behavioral activation as a treatment for depression in adolescence. J Clin Child Adolesc Psychol. 2015;1-14. [Epub ahead of print].
15. Carlbring P, Hägglund M, Luthström A, et al. Internet-based behavioral activation and acceptance-based treatment for depression: a randomized controlled trial. J Affect Disord. 2013;148:331-337.
16. Markowitz JC, Weissman MM. Interpersonal psychotherapy: principles and applications. World Psychiatry. 2004; 3:136-139.
17. Kersting A, Kroker K, Schlicht S, et al. Efficacy of a cognitive-behavioral internet-based therapy in parents after the loss of a child during pregnancy: pilot data from a randomized controlled trial. Arch Womens Mental Health. 2011;14:465-477.
18. Francis J, Kumar A. Psychological treatment of late-life depression. Psychiatr Clin North Am. 2013;36:561-575.
19. Picardi A, Gaetano P. Psychotherapy of mood disorders. Clin Pract Epidemiol Ment Health. 2014;10:140-158.
20. Bell AC, D’Zurilla TJ. Problem-solving therapy for depression: a meta-analysis. Clin Psychol Review. 2009;29:348-353.
21. Cooney GM, Dwan K, Greig CA, et al. Exercise for depression. Cochrane Database Syst Rev. 2013;9:CD004366
22. Brindle C, Spanjers K, Patel S, et al. Effect of exercise on depression severity in older people: systematic review and meta-analysis of randomized controlled trials. B J Psychiatry. 2012;201:180-185.
23. Brown HE, Pearson N, Braithwaite RE, et al. Physical activity interventions and depression in children and adolescents: a systematic review and meta-analysis. Sports Med. 2013;43:195-206.
24. Gong H, Ni C, Shen X, et al. Yoga for prenatal depression: a systematic review and meta-analysis. BMC Psychiatry. 2015;15:14.
25. D’Silva S, Poscablo C, Habousha R, et al. Mind-body medicine therapies for a range of depression severity: a systematic review. Psychosomatics. 2012;53:407-423.
26. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357:1939-1945.
27. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65:485-491.
28. Rhebergen D, Huisman A, Bouckaert F, et al. Older age is associated with rapid remission of depression after electroconvulsive therapy: a latent class growth analysis. Am J Geriatr Psychiatry. 2015;23:274-282.
29. Slotema CW, Blom JD, Hoek HW, et al. Should we expand the toolbox of psychiatric treatment methods to include Repetitive Transcranial Magnetic Stimulation (rTMS)? A metaanalysis of the efficacy of rTMS in psychiatric disorders. J Clin Psychiatry. 2010;71:873-884.
30. George MS, Lisanby SH, Avery D, et al. Daily left prefrontal transcranial magnetic stimulation therapy for major depressive disorder: a sham-controlled randomized trial. Arch Gen Psychiatry. 2010;67:507-516
31. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham controlled studies. BMC Psychiatry. 2014;14:342.
32. Brown RP, Gerberg PL, Muskin PR. Mood disorders. In: Brown RP, Gerbarg PL, Muskin P. How to Use Herbs, Nutrients and Yoga in Mental Health. New York, NY: WW Norton & Company; 2009.
33. Linde K, Berner MM, Kriston L. St John’s wort for major depression. Cochrane Database Syst Rev. 2008;(4):CD000448.
34. Natural Medicines Comprehensive Database. Natural Medicines Comprehensive Database Web site. Available at: http://naturaldatabase.therapeuticresearch.com/home.aspx. Accessed March 1, 2015.
35. Harris WS. Expert opinion: omega-3 fatty acids and bleeding-cause for concern? Am J Cardiol. 2007;99:44C-6C.
36. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force Report. J Clin Psychiatry. 2010;71:669-681.
37. Papakostas GI, Alpert JE, Fava M. S-adenosyl-methionine in depression: a comprehensive review of the literature. Curr Psychiatry Reports. 2003;5:460-466.
38. Brown RP, Gerbarg PL, Bottiglieri T. S-Adenosylmethionine (SAMe) for depression: biochemical and clinical evidence. Psychiatr Ann. 2002;32:29-44.
39. Taylor MJ, Carney S, Geddes J, et al. Folate for depressive disorders. Cochrane Database Syst Rev. 2003;(2):CD003390.
40. Alpert M, Silva RR, Pouget ER. Prediction of treatment response in geriatric depression from baseline folate level: interaction with an SSRI or a tricyclic antidepressant. J Clin Psychopharmacol. 2003;23:309-313.
41. Fava M, Mischoulon D. Folate in depression: efficacy, safety, differences in formulations, and clinical issues. J Clin Psychiatry. 2009;70(suppl 5):12-17.
42. Almeida OP, Ford AH, Hirani V, et al. B vitamins to enhance treatment response to antidepressants in middle-aged and older adults: results from the B-VITAGE randomised, double-blind, placebo-controlled trial. Br J Psychiatry. 2014;205:450-457.
43. Grosso G, Galvano F, Marventano S, et al. Omega-3 fatty acids and depression: scientific evidence and biological mechanisms. Oxid Med Cell Longev. 2014;2014:313570.
44. Appleton KM, Rogers PJ, Ness AR. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91:757-770.
45. Grosso G, Pajak A, Marventano S, et al. Role of omega-3 fatty acids in the treatment of depressive disorders: a comprehensive metaanalysis of randomized clinical trials. PLoS One. 2014;9:e96905.
46. Martins JG, Bentsen H, Puri BK. Eicosapentaenoic acid appears to be the key omega-3 fatty acid component associated with efficacy in major depressive disorder: a critique of Bloch and Hannestad and updated meta-analysis. Mol Psychiatry. 2012;17:1144-1149.
47. Nemets H, Nemets B, Apter A, et al. Omega-3 treatment of childhood depression: a controlled, double-blind pilot study. Am J Psychiatry. 2006;163:1098-1100.
48. Su KP, Huang SY, Chiu TH. Omega-3 fatty acids for major depressive disorder during pregnancy: Results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2008;69:644-651.
49. Freeman MP, Davis M, Sinha P, et al. Omega-3 fatty acids and supportive psychotherapy for perinatal depression: a randomized placebo-controlled study. J Affect Disord. 2008;110:142-148.
50. Mitchell J, Trangle M, Degnan B, et al. Institute for Clinical Systems Improvement (ICSI). Health Care Guideline: Adult depression in primary care. 16th ed. September 2013. Available at: https://www.icsi.org/_asset/fnhdm3/Depr-Interactive0512b.pdf. Accessed June 9, 2015.
51. Kroenke K, Spitzer RL, Williams JBW, et al. The Patient Health Questionnaire Somatic, Anxiety, and Depressive Symptom Scales: a systematic review. Gen Hosp Psychiatry. 2010;32:345-359.
52. Trivedi MH. Tools and strategies for ongoing assessment of depression:
a measurement-based approach to remission. J Clin Psychiatry. 2009;70:26-31.

Long-acting reversible contraception: Who, what, when, and how
› Suggest long-acting reversible contraception (LARC), including intrauterine devices (IUDs), as a first-line method of contraception to most women, including adolescents and nulliparous women. A
› Offer immediate post-placental insertion of LARC when counseling women who have barriers to seeking contraception at a postpartum visit or are unlikely to return for a postpartum visit. B
› Treat sexually transmitted infections in most cases without removing an IUD that is already in place. Consider removing the IUD, however, if there is no clinical improvement after 2 to 3 days of antibiotics. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The number of women using long-acting reversible contraception (LARC) in the United States has been increasing, with current use accounting for approximately 18% of reversible contraception, according to the National Survey of Family Growth.1,2 LARC includes any method of contraception that lasts ≥3 years, is easily reversed, and does not rely on the user to maintain efficacy. Five LARC devices are available in the United States: 4 intrauterine devices (IUDs) and one subdermal implant.
The number of women using LARC is surprisingly low, given that it is considered a first-line contraceptive method for most women and adolescents,3 and when compared with other forms of reversible contraception, is more efficacious,4-6 has higher satisfaction rates,7-9 and higher rates of continuation.9 In fact, the Contraceptive CHOICE Project—a St. Louis community-based research program promoting and enabling access to reversible contraceptive methods—has shown that when appropriate counseling is provided and cost barriers are removed, up to 79% of women choose LARC as their preferred method of contraception.10
CASES › Jenny, who is 16 years old, comes to your office with her mother to discuss contraceptive options. She is nulliparous, has regular menses, and, aside from a body mass index (BMI) of 28, has no medical problems. Her mother is concerned about Jenny becoming pregnant while she is still in high school.
Maria D, a 32-year-old G2P1, comes in for a prenatal visit with her husband. She tells you that after delivery she is interested in a long-acting contraceptive, but is planning on breastfeeding and does not want anything to interfere with that.
What LARC options do these and other patients have?
The 4 IUDs and one implant approved for use are all viable options depending on a patient’s preference and comorbidities (TABLE 1).3-9,11-15 The copper IUD is the oldest method of LARC available and the only one that is nonhormonal. It is approved by the Food and Drug Administration (FDA) for use up to 10 years,11 but studies support its effectiveness for up to 12 years.16
The remaining IUDs (Skyla, Liletta, Mirena) contain varying amounts of the progestin levonorgestrel (LNG), released by each device at a slightly different rate that declines over time. Skyla releases a significantly lower dose of hormone than Liletta or Mirena.12-14 Skyla and Liletta are FDA-approved for up to 3 years of use,12,13 and Liletta is currently undergoing trials to gain approval for use up to 5 years. Mirena is FDA-approved for use up to 5 years,14 but studies have shown that it can be effective for 7 years.4,16
The only implant available in the US is Nexplanon, a plastic rod containing 68 mg of etonorgestrel. It is inserted subdermally and is FDA-approved for use up to 3 years.15
Through systemic hormonal effects, the primary mechanism of action of the implant is prevention of ovulation. Additionally, the implant has been shown to inhibit endometrial proliferation and cervical mucus thickening, both of which may contribute to the implant’s overall effectiveness.17 In contrast, both the copper IUD and the LNG-IUDs work primarily by preventing fertilization. The LNG-IUDs also exhibit local hormonal effects (endometrial atrophy and thickened cervical mucus) that contribute to their effectiveness.17
Who is eligible for LARC?
LARC is suitable for the vast majority of women of reproductive age. For most multiparous women ≥20 years, all LARC devices are classified as category 1 (use without restriction) in the Centers for Disease Control and Prevention’s (CDC) US Medical Eligibility Criteria (US MEC).3 For women <20 years, the implant is also considered category 1, but IUDs in this age group are classified as category 2 (recommended with the caution that advantages usually outweigh risks) because of concerns about an increased risk of IUD expulsion and the increased prevalence of sexually transmitted infections (STIs) in adolescents.3 Contraindications to use of LARC vary depending on the method chosen (TABLE 1).3
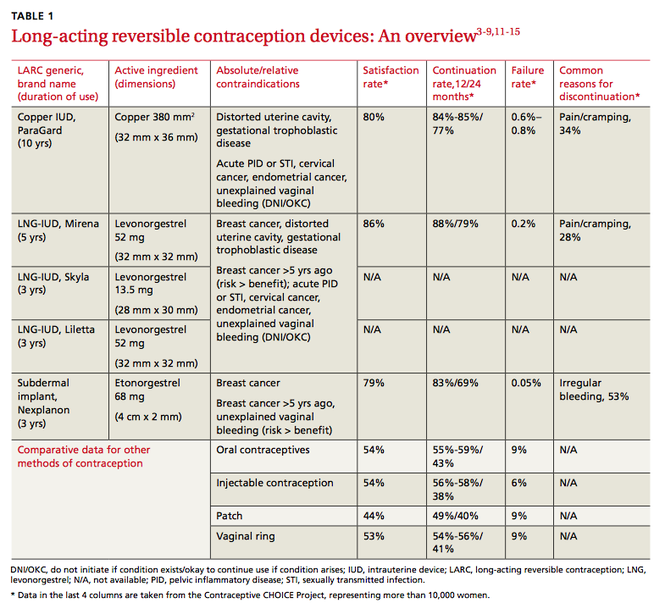
There has been concern about the efficacy of implants in overweight women because the original trials of subdermal implants excluded women >130% of ideal body weight. However, according to the Contraceptive CHOICE Project, overweight and obese women enrolled in its program did not experience reduced contraceptive efficacy when using the implant when compared with normal-weight women.18
When can LARC devices be inserted?
LARC device insertion is possible at any time during the menstrual cycle. An algorithm to guide initiation of LARC is available through the Reproductive Health Access Project’s Web site at http://www.reproductiveaccess.org/wp-content/uploads/2014/12/quickstart_algorithm.pdf.
Rule out pregnancy before placing any LARC device. The copper IUD can be inserted at any time during the menstrual cycle without the need for back-up contraception.11,19 In contrast, for LNG-IUDs, back-up contraception is recommended for 7 days unless the insertion is done during the first 7 days of the menstrual cycle.12-14,19
For the implant, recommendations about when to insert are based on a woman’s previous method of contraception (TABLE 2).15 If insertion is done at a time other than when recommended, advise patients to use barrier protection for 7 days after insertion.4,15,19
Other issues often arise and cause concern about whether and when a LARC device can be inserted, including the possibility of undiagnosed STI, time elapsed since delivery, and advisability of use when breastfeeding.
Sexually transmitted infections and IUDs
Whether or not a woman chooses to receive an IUD, follow routine CDC guidelines in determining if a patient is a candidate for STI screening.20 If a woman wants an IUD and routine screening is recommended, you can perform screening on the day of IUD insertion.4,19 For women with an IUD already in place who are diagnosed with an STI, treat the infection while leaving the IUD in place.19 For women with a known or suspected STI who do not have an IUD already, treat the STI before inserting the IUD. The American Congress of Obstetricians and Gynecologists (ACOG) advises postponing insertion of an IUD until a negative STI test result is obtained 3 to 4 weeks after treatment completion.4
Breastfeeding concerns and timing of insertion postpartum
The US MEC classifies insertion of the copper IUD as category 1 for all postpartum women, regardless of breastfeeding status, if placed >4 weeks postpartum or immediately postpartum (defined as within 10 minutes of the delivery of the placenta). IUD placement is category 2 (recommended with the caution that advantages usually outweigh risks) if placed ≥10 minutes after placental delivery (until 4 weeks postpartum) because of an increased risk of expulsion.3
The US MEC also considers use of the implant and LNG-IUDs in breastfeeding women as category 1 if the device is placed at ≥4 weeks postpartum. Insertion at <4 weeks postpartum is considered category 2 because of concerns for decreased breast milk supply.3 However, studies on whether progestin-containing LARC devices affect breastfeeding have yielded varying results. In one randomized controlled trial (RCT) of 69 breastfeeding women using the implant, breastfeeding duration and milk production were not dependent on the timing of insertion after delivery.21 Another RCT of 96 women using LNG-IUDs showed fewer women continued to breastfeed at 6 months when their LNG-IUD was inserted immediately postpartum, compared with waiting 6 weeks.22
In addition to a concern about breast milk supply, breastfeeding women have a higher risk for uterine perforation from IUDs, especially during the first 36 weeks after delivery.23
Several studies have shown that there is a lower repeat pregnancy rate among women who receive immediate postpartum LARC placement.24 However, even if IUD insertion is performed immediately postpartum, there is a higher expulsion rate than when the IUD is inserted ≥4 weeks postpartum. The expulsion rates for insertion <10 minutes after vaginal delivery range from 9.5% to 15% for the copper IUD to as high as 24% for the LNG-IUDs. Expulsion rates for all IUDs are slightly lower for cesarean delivery.4,25,26 ACOG supports immediate post-placental placement for women with barriers to postpartum care or limited access to contraception.4
How can I help my patients make an informed choice?
Provide counseling on efficacy, common adverse effects, risks, and complications.
Efficacy is high
The failure rate of LARC is equal to, or lower than, that of female sterilization and is significantly lower than that of oral contraceptives (TABLE 1).4-6 Not only are LARC devices extremely effective, they have a higher rate of satisfaction than any other reversible contraceptive (TABLE 1).7,8
Common adverse effects
The most common adverse effect seen with all LARC devices is an alteration in menstrual bleeding, and a frequent adverse effect with IUDs is pain. Vaginitis is less common and can be seen with any of the devices. The progestin-containing LARC devices are associated with hormonal effects: vaginitis, headache, weight gain, acne, breast pain, hair loss, and emotional lability.12-15
Copper IUD. Many women using the copper IUD experience either a transient increase in menstrual bleeding lasting for a few months or inter-menstrual bleeding that tends to continue for the duration of use.4,17 However, according to data from the Contraceptive CHOICE Project, the most common reason cited for early discontinuation of the copper IUD is pain and cramping.9
LNG-IUDs. Like the copper IUD, many users of LNG-IUDs experience an initial increase in menstrual bleeding. However, unlike the other LARC devices, 20% to 33% of Mirena users are likely to experience amenorrhea after one year of use and 70% at 2 years.4,14 According to package inserts, amenorrhea after 3 years is less common with both Skyla (12%) and Liletta (38%).12,13 As with the copper IUD, based on data from the Contraceptive CHOICE Project, the most common reason cited for early discontinuation of LNG-IUDs is pain and cramping.9
Subdermal implant. Changes in menses in women using the subdermal implant range from amenorrhea (22%) to prolonged bleeding (18%).15,17 Although it is difficult to predict which pattern a particular woman will experience, heavier women are more likely to have heavier bleeding patterns, and initial bleeding patterns are predictive of future ones.4 The most common reason women choose to discontinue use of the implant is abnormal bleeding.4,9,27,28
Newer IUDs do not increase risk of STIs
Many patients and clinicians erroneously believe that IUDs increase the risk of STIs and therefore assume that patients with a history of STI are not appropriate candidates for an IUD.29 There is a slightly increased risk of pelvic inflammatory disease (PID) in the first 21 days after insertion of an IUD. However, in contrast to older IUDs, currently available IUDs do not increase the general risk for STIs.17,30
Risk of infertility is nil
There is no risk of infertility from use of currently available LARCs. For those who want to become pregnant, fertility typically returns immediately after removal of the device, regardless of which method of LARC is used.11-15,30
Complications of IUD insertion
Uterine perforation. Uterine perforation occurs in 0.8 to 2.1 per 1000 women, usually at the time of IUD placement. If IUD strings are not visible during a speculum examination, locate the IUD with ultrasound.4,17,30 If the IUD is in the abdomen, refer to a gynecologist for laparoscopic removal and select another form of contraception for use in the interim.30
Expulsion. Rates of expulsion are low, occurring in less than 10% of women4,17 and are not affected by parity or BMI.31 Expulsion rates are higher when the IUD is inserted immediately postpartum.4,25,26 Adolescents also have a 2-fold higher risk of uterine expulsion than older women.31
Ectopic pregnancy. Although a woman’s overall risk of ectopic pregnancy is not increased by using an IUD,4 it is true that if a woman becomes pregnant with an IUD in place, the pregnancy is more likely to be ectopic. Thus, if pregnancy is confirmed in a woman with an IUD in place, rule out ectopic pregnancy.
The FDA and the World Health Organization recommend that if an intrauterine pregnancy is confirmed with an IUD in place and the strings are visible, the IUD should be removed.4 Although removing the IUD increases the risk of spontaneous abortion (SAB) as compared with pregnancies without an IUD in place, the risk of SAB is still lower than if the IUD is left in place.4 Additional risks of continuing a pregnancy with an IUD in place include increased risks of preterm labor, chorioamnionitis, and septic abortion.4,30
Complications of subdermal implant insertion
After insertion of the implant, women usually experience temporary bruising and soreness at the insertion site. Less than 1% of women develop an infection or hematoma.17 There is a low risk of nerve damage if the implant is inserted too deeply.15 Removal of the subdermal implant is recommended if pregnancy occurs.15
CASE DECISIONS › Jenny has been using oral contraceptive pills, but not regularly. You suggest that LARC may be a better option and counsel her that if she does choose an IUD or the implant, it is likely that her menses will change. You provide information and reassurance that LARC is safe to use in adolescents. Jenny says she would like to try an implant. Six months later, Jenny returns and says the implant is working well. She has some irregular bleeding, but it is not bothersome.
You review with Ms. D the types of LARC devices available and reassure her that all are safe to use once breastfeeding is established. Ms. D says she would like to use an IUD and elects to wait until her postpartum visit to have an IUD inserted. Ms. D returns 6 months after IUD insertion; breastfeeding is going well, and she has not had any menstrual bleeding since delivery.
CORRESPONDENCE
Karyn Kolman, MD, 2800 East Ajo Way, Room 3006, Tucson, AZ 85713; [email protected]
1. Daniels K, Daugherty J, Jones J. Current contraceptive status among women aged 15-44: United States 2011-2013. NCHS data brief, no. 173. Hyattsville, MD: National Center for Health Statistics, 2014.
2. Branum AM, Jones J. Trends in long-acting reversible contraception use among US women aged 15-44. NCHS data brief, no. 188. Hyattsville, MD: National Center for Health Statistics, 2015.
3. Centers for Disease Control and Prevention (CDC). US medical eligibility criteria for contraceptive use, 2010. MMWR Recomm Rep. 2010;59:1-86.
4. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No 121: Long-acting reversible contraception: Implants and intrauterine devices. Obstet Gynecol. 2011;118:184-196.
5. Pickle S, Wu J, Burbank-Schmitt E. Prevention of unintended pregnancy: a focus on long-acting reversible contraception. Prim Care. 2014;41:239-260.
6. Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med. 2012;366:1998-2007.
7. Peipert JF, Zhao Q, Allsworth JE, et al. Continuation and satisfaction of reversible contraception. Obstet Gynecol. 2011;117:1105-1113.
8. O’Neil-Callahan M, Peipert JF, Zhao Q, et al. Twenty-four-month continuation of reversible contraception. Obstet Gynecol. 2013;122:1083-1091.
9. Grunloh DS, Casner T, Secura GM, et al. Characteristics associated with discontinuation of long-acting reversible contraception within the first 6 months of use. Obstet Gynecol. 2011;117:705-719.
10. Birgisson NE, Zhao Q, Secura GM, et al. Preventing unintended pregnancy: the contraceptive CHOICE project in review. J Womens Health (Larchmt). 2015;24:349-353.
11. ParaGard T 380A. (intrauterine copper contraceptive) [package insert]. Sellersville, PA : Teva Pharmaceuticals USA, Inc., 2013.
12. Skyla (levonorgestrel-releasing intrauterine system) [package insert]. Wayne, NJ : Bayer HealthCare Pharmaceuticals, Inc., 2013.
13. Liletta (levonorgestrel-releasing intrauterine system) [package insert]. Parsippany, NJ : Actavis Pharma, Inc., 2015.
14. Mirena (levonorgestrel-releasing intrauterine system) [package insert]. Whippany, NJ : Bayer HealthCare Pharmaceuticals, Inc., 2014.
15. Nexplanon (etongestrel implant) [package insert]. Whitehouse Station, NJ: Merck & Co Inc.; 2014.
16. Wu JP, Pickle S. Extended use of the intrauterine device: a literature review and recommendations for clinical practice. Contraception. 2014;89:495-503.
17. Stoddard A, McNicholas C, Peipert JF. Efficacy and safety of long-acting reversible contraception. Drugs. 2011;71:969-980.
18. Xu H, Wade JA, Peipert JF, et al. Contraceptive failure rates of etonogestrel subdermal implants in overweight and obese women. Obstet Gynecol. 2012;120:21-26.
19. Centers for Disease Control and Prevention (CDC). US selected practice recommendations for contraceptive use. MMWR Recomm Rep. 2013;62:1-60.
20. Centers for Disease Control and Prevention (CDC). Sexually transmitted disease treatment guidelines. MMWR Recomm Rep. 2010;59:1-110.
21. Gurtcheff SE, Turok DK, Stoddard G, et al. Lactogenesis after early postpartum use of the contraceptive implant: a randomized controlled trial. Obstet Gynecol. 2011;117:1114-1121.
22. Chen BA, Reeves MF, Creinin MD, et al. Postplacental or delayed levonorgestrel intrauterine device insertion and breast-feeding duration. Contraception. 2011;84:499-504.
23. Heinemann K, Reed S, Moehner S, et al. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91:274-279.
24. Tocce K, Sheeder J, Python J, et al. Long acting reversible contraception in postpartum adolescents: early initiation of etonogestrel implant is superior to IUDs in the outpatient setting. J Pediatr Adolesc Gynecol. 2012;25:59-63.
25. Mwalwanda CS, Black KI. Immediate post-partum initiation of intrauterine contraception and implants: a review of the safety and guidelines for use. Aust N Z J Obstet Gynaecol. 2013;53:331-337.
26. Sober, S, Schreiber CA. Postpartum contraception. Clin Obstet Gynecol. 2014;57:763-776.
27. Dickerson LM, Diaz VA, Jordon J, et al. Satisfaction, early removal, and side effects associated with long-acting reversible contraception. Fam Med. 2013;45:701-707.
28. Berenson AB, Tan A, Hirth JM. Complications and continuation rates associated with 2 types of long-acting contraception. Am J Obstet Gynecol. 2015;212:e1-e8.
29. Kavanaugh ML, Frowirth L, Jerman J, et al. Long-acting reversible contraception for adolescents and young adults: patient and provider perspectives. J Pediatr Adolesc Gynecol. 2013;86:86-95.
30. Espey E, Ogburn T. Long-acting reversible contraceptives: intrauterine devices and the contraceptive implant. Obstet Gynecol. 2011;117:705-719.
31. Madden T, McNicholas C, Zhao Q, et al. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014;124:718-726.
› Suggest long-acting reversible contraception (LARC), including intrauterine devices (IUDs), as a first-line method of contraception to most women, including adolescents and nulliparous women. A
› Offer immediate post-placental insertion of LARC when counseling women who have barriers to seeking contraception at a postpartum visit or are unlikely to return for a postpartum visit. B
› Treat sexually transmitted infections in most cases without removing an IUD that is already in place. Consider removing the IUD, however, if there is no clinical improvement after 2 to 3 days of antibiotics. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The number of women using long-acting reversible contraception (LARC) in the United States has been increasing, with current use accounting for approximately 18% of reversible contraception, according to the National Survey of Family Growth.1,2 LARC includes any method of contraception that lasts ≥3 years, is easily reversed, and does not rely on the user to maintain efficacy. Five LARC devices are available in the United States: 4 intrauterine devices (IUDs) and one subdermal implant.
The number of women using LARC is surprisingly low, given that it is considered a first-line contraceptive method for most women and adolescents,3 and when compared with other forms of reversible contraception, is more efficacious,4-6 has higher satisfaction rates,7-9 and higher rates of continuation.9 In fact, the Contraceptive CHOICE Project—a St. Louis community-based research program promoting and enabling access to reversible contraceptive methods—has shown that when appropriate counseling is provided and cost barriers are removed, up to 79% of women choose LARC as their preferred method of contraception.10
CASES › Jenny, who is 16 years old, comes to your office with her mother to discuss contraceptive options. She is nulliparous, has regular menses, and, aside from a body mass index (BMI) of 28, has no medical problems. Her mother is concerned about Jenny becoming pregnant while she is still in high school.
Maria D, a 32-year-old G2P1, comes in for a prenatal visit with her husband. She tells you that after delivery she is interested in a long-acting contraceptive, but is planning on breastfeeding and does not want anything to interfere with that.
What LARC options do these and other patients have?
The 4 IUDs and one implant approved for use are all viable options depending on a patient’s preference and comorbidities (TABLE 1).3-9,11-15 The copper IUD is the oldest method of LARC available and the only one that is nonhormonal. It is approved by the Food and Drug Administration (FDA) for use up to 10 years,11 but studies support its effectiveness for up to 12 years.16
The remaining IUDs (Skyla, Liletta, Mirena) contain varying amounts of the progestin levonorgestrel (LNG), released by each device at a slightly different rate that declines over time. Skyla releases a significantly lower dose of hormone than Liletta or Mirena.12-14 Skyla and Liletta are FDA-approved for up to 3 years of use,12,13 and Liletta is currently undergoing trials to gain approval for use up to 5 years. Mirena is FDA-approved for use up to 5 years,14 but studies have shown that it can be effective for 7 years.4,16
The only implant available in the US is Nexplanon, a plastic rod containing 68 mg of etonorgestrel. It is inserted subdermally and is FDA-approved for use up to 3 years.15
Through systemic hormonal effects, the primary mechanism of action of the implant is prevention of ovulation. Additionally, the implant has been shown to inhibit endometrial proliferation and cervical mucus thickening, both of which may contribute to the implant’s overall effectiveness.17 In contrast, both the copper IUD and the LNG-IUDs work primarily by preventing fertilization. The LNG-IUDs also exhibit local hormonal effects (endometrial atrophy and thickened cervical mucus) that contribute to their effectiveness.17
Who is eligible for LARC?
LARC is suitable for the vast majority of women of reproductive age. For most multiparous women ≥20 years, all LARC devices are classified as category 1 (use without restriction) in the Centers for Disease Control and Prevention’s (CDC) US Medical Eligibility Criteria (US MEC).3 For women <20 years, the implant is also considered category 1, but IUDs in this age group are classified as category 2 (recommended with the caution that advantages usually outweigh risks) because of concerns about an increased risk of IUD expulsion and the increased prevalence of sexually transmitted infections (STIs) in adolescents.3 Contraindications to use of LARC vary depending on the method chosen (TABLE 1).3
There has been concern about the efficacy of implants in overweight women because the original trials of subdermal implants excluded women >130% of ideal body weight. However, according to the Contraceptive CHOICE Project, overweight and obese women enrolled in its program did not experience reduced contraceptive efficacy when using the implant when compared with normal-weight women.18
When can LARC devices be inserted?
LARC device insertion is possible at any time during the menstrual cycle. An algorithm to guide initiation of LARC is available through the Reproductive Health Access Project’s Web site at http://www.reproductiveaccess.org/wp-content/uploads/2014/12/quickstart_algorithm.pdf.
Rule out pregnancy before placing any LARC device. The copper IUD can be inserted at any time during the menstrual cycle without the need for back-up contraception.11,19 In contrast, for LNG-IUDs, back-up contraception is recommended for 7 days unless the insertion is done during the first 7 days of the menstrual cycle.12-14,19
For the implant, recommendations about when to insert are based on a woman’s previous method of contraception (TABLE 2).15 If insertion is done at a time other than when recommended, advise patients to use barrier protection for 7 days after insertion.4,15,19
Other issues often arise and cause concern about whether and when a LARC device can be inserted, including the possibility of undiagnosed STI, time elapsed since delivery, and advisability of use when breastfeeding.
Sexually transmitted infections and IUDs
Whether or not a woman chooses to receive an IUD, follow routine CDC guidelines in determining if a patient is a candidate for STI screening.20 If a woman wants an IUD and routine screening is recommended, you can perform screening on the day of IUD insertion.4,19 For women with an IUD already in place who are diagnosed with an STI, treat the infection while leaving the IUD in place.19 For women with a known or suspected STI who do not have an IUD already, treat the STI before inserting the IUD. The American Congress of Obstetricians and Gynecologists (ACOG) advises postponing insertion of an IUD until a negative STI test result is obtained 3 to 4 weeks after treatment completion.4
Breastfeeding concerns and timing of insertion postpartum
The US MEC classifies insertion of the copper IUD as category 1 for all postpartum women, regardless of breastfeeding status, if placed >4 weeks postpartum or immediately postpartum (defined as within 10 minutes of the delivery of the placenta). IUD placement is category 2 (recommended with the caution that advantages usually outweigh risks) if placed ≥10 minutes after placental delivery (until 4 weeks postpartum) because of an increased risk of expulsion.3
The US MEC also considers use of the implant and LNG-IUDs in breastfeeding women as category 1 if the device is placed at ≥4 weeks postpartum. Insertion at <4 weeks postpartum is considered category 2 because of concerns for decreased breast milk supply.3 However, studies on whether progestin-containing LARC devices affect breastfeeding have yielded varying results. In one randomized controlled trial (RCT) of 69 breastfeeding women using the implant, breastfeeding duration and milk production were not dependent on the timing of insertion after delivery.21 Another RCT of 96 women using LNG-IUDs showed fewer women continued to breastfeed at 6 months when their LNG-IUD was inserted immediately postpartum, compared with waiting 6 weeks.22
In addition to a concern about breast milk supply, breastfeeding women have a higher risk for uterine perforation from IUDs, especially during the first 36 weeks after delivery.23
Several studies have shown that there is a lower repeat pregnancy rate among women who receive immediate postpartum LARC placement.24 However, even if IUD insertion is performed immediately postpartum, there is a higher expulsion rate than when the IUD is inserted ≥4 weeks postpartum. The expulsion rates for insertion <10 minutes after vaginal delivery range from 9.5% to 15% for the copper IUD to as high as 24% for the LNG-IUDs. Expulsion rates for all IUDs are slightly lower for cesarean delivery.4,25,26 ACOG supports immediate post-placental placement for women with barriers to postpartum care or limited access to contraception.4
How can I help my patients make an informed choice?
Provide counseling on efficacy, common adverse effects, risks, and complications.
Efficacy is high
The failure rate of LARC is equal to, or lower than, that of female sterilization and is significantly lower than that of oral contraceptives (TABLE 1).4-6 Not only are LARC devices extremely effective, they have a higher rate of satisfaction than any other reversible contraceptive (TABLE 1).7,8
Common adverse effects
The most common adverse effect seen with all LARC devices is an alteration in menstrual bleeding, and a frequent adverse effect with IUDs is pain. Vaginitis is less common and can be seen with any of the devices. The progestin-containing LARC devices are associated with hormonal effects: vaginitis, headache, weight gain, acne, breast pain, hair loss, and emotional lability.12-15
Copper IUD. Many women using the copper IUD experience either a transient increase in menstrual bleeding lasting for a few months or inter-menstrual bleeding that tends to continue for the duration of use.4,17 However, according to data from the Contraceptive CHOICE Project, the most common reason cited for early discontinuation of the copper IUD is pain and cramping.9
LNG-IUDs. Like the copper IUD, many users of LNG-IUDs experience an initial increase in menstrual bleeding. However, unlike the other LARC devices, 20% to 33% of Mirena users are likely to experience amenorrhea after one year of use and 70% at 2 years.4,14 According to package inserts, amenorrhea after 3 years is less common with both Skyla (12%) and Liletta (38%).12,13 As with the copper IUD, based on data from the Contraceptive CHOICE Project, the most common reason cited for early discontinuation of LNG-IUDs is pain and cramping.9
Subdermal implant. Changes in menses in women using the subdermal implant range from amenorrhea (22%) to prolonged bleeding (18%).15,17 Although it is difficult to predict which pattern a particular woman will experience, heavier women are more likely to have heavier bleeding patterns, and initial bleeding patterns are predictive of future ones.4 The most common reason women choose to discontinue use of the implant is abnormal bleeding.4,9,27,28
Newer IUDs do not increase risk of STIs
Many patients and clinicians erroneously believe that IUDs increase the risk of STIs and therefore assume that patients with a history of STI are not appropriate candidates for an IUD.29 There is a slightly increased risk of pelvic inflammatory disease (PID) in the first 21 days after insertion of an IUD. However, in contrast to older IUDs, currently available IUDs do not increase the general risk for STIs.17,30
Risk of infertility is nil
There is no risk of infertility from use of currently available LARCs. For those who want to become pregnant, fertility typically returns immediately after removal of the device, regardless of which method of LARC is used.11-15,30
Complications of IUD insertion
Uterine perforation. Uterine perforation occurs in 0.8 to 2.1 per 1000 women, usually at the time of IUD placement. If IUD strings are not visible during a speculum examination, locate the IUD with ultrasound.4,17,30 If the IUD is in the abdomen, refer to a gynecologist for laparoscopic removal and select another form of contraception for use in the interim.30
Expulsion. Rates of expulsion are low, occurring in less than 10% of women4,17 and are not affected by parity or BMI.31 Expulsion rates are higher when the IUD is inserted immediately postpartum.4,25,26 Adolescents also have a 2-fold higher risk of uterine expulsion than older women.31
Ectopic pregnancy. Although a woman’s overall risk of ectopic pregnancy is not increased by using an IUD,4 it is true that if a woman becomes pregnant with an IUD in place, the pregnancy is more likely to be ectopic. Thus, if pregnancy is confirmed in a woman with an IUD in place, rule out ectopic pregnancy.
The FDA and the World Health Organization recommend that if an intrauterine pregnancy is confirmed with an IUD in place and the strings are visible, the IUD should be removed.4 Although removing the IUD increases the risk of spontaneous abortion (SAB) as compared with pregnancies without an IUD in place, the risk of SAB is still lower than if the IUD is left in place.4 Additional risks of continuing a pregnancy with an IUD in place include increased risks of preterm labor, chorioamnionitis, and septic abortion.4,30
Complications of subdermal implant insertion
After insertion of the implant, women usually experience temporary bruising and soreness at the insertion site. Less than 1% of women develop an infection or hematoma.17 There is a low risk of nerve damage if the implant is inserted too deeply.15 Removal of the subdermal implant is recommended if pregnancy occurs.15
CASE DECISIONS › Jenny has been using oral contraceptive pills, but not regularly. You suggest that LARC may be a better option and counsel her that if she does choose an IUD or the implant, it is likely that her menses will change. You provide information and reassurance that LARC is safe to use in adolescents. Jenny says she would like to try an implant. Six months later, Jenny returns and says the implant is working well. She has some irregular bleeding, but it is not bothersome.
You review with Ms. D the types of LARC devices available and reassure her that all are safe to use once breastfeeding is established. Ms. D says she would like to use an IUD and elects to wait until her postpartum visit to have an IUD inserted. Ms. D returns 6 months after IUD insertion; breastfeeding is going well, and she has not had any menstrual bleeding since delivery.
CORRESPONDENCE
Karyn Kolman, MD, 2800 East Ajo Way, Room 3006, Tucson, AZ 85713; [email protected]
› Suggest long-acting reversible contraception (LARC), including intrauterine devices (IUDs), as a first-line method of contraception to most women, including adolescents and nulliparous women. A
› Offer immediate post-placental insertion of LARC when counseling women who have barriers to seeking contraception at a postpartum visit or are unlikely to return for a postpartum visit. B
› Treat sexually transmitted infections in most cases without removing an IUD that is already in place. Consider removing the IUD, however, if there is no clinical improvement after 2 to 3 days of antibiotics. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The number of women using long-acting reversible contraception (LARC) in the United States has been increasing, with current use accounting for approximately 18% of reversible contraception, according to the National Survey of Family Growth.1,2 LARC includes any method of contraception that lasts ≥3 years, is easily reversed, and does not rely on the user to maintain efficacy. Five LARC devices are available in the United States: 4 intrauterine devices (IUDs) and one subdermal implant.
The number of women using LARC is surprisingly low, given that it is considered a first-line contraceptive method for most women and adolescents,3 and when compared with other forms of reversible contraception, is more efficacious,4-6 has higher satisfaction rates,7-9 and higher rates of continuation.9 In fact, the Contraceptive CHOICE Project—a St. Louis community-based research program promoting and enabling access to reversible contraceptive methods—has shown that when appropriate counseling is provided and cost barriers are removed, up to 79% of women choose LARC as their preferred method of contraception.10
CASES › Jenny, who is 16 years old, comes to your office with her mother to discuss contraceptive options. She is nulliparous, has regular menses, and, aside from a body mass index (BMI) of 28, has no medical problems. Her mother is concerned about Jenny becoming pregnant while she is still in high school.
Maria D, a 32-year-old G2P1, comes in for a prenatal visit with her husband. She tells you that after delivery she is interested in a long-acting contraceptive, but is planning on breastfeeding and does not want anything to interfere with that.
What LARC options do these and other patients have?
The 4 IUDs and one implant approved for use are all viable options depending on a patient’s preference and comorbidities (TABLE 1).3-9,11-15 The copper IUD is the oldest method of LARC available and the only one that is nonhormonal. It is approved by the Food and Drug Administration (FDA) for use up to 10 years,11 but studies support its effectiveness for up to 12 years.16
The remaining IUDs (Skyla, Liletta, Mirena) contain varying amounts of the progestin levonorgestrel (LNG), released by each device at a slightly different rate that declines over time. Skyla releases a significantly lower dose of hormone than Liletta or Mirena.12-14 Skyla and Liletta are FDA-approved for up to 3 years of use,12,13 and Liletta is currently undergoing trials to gain approval for use up to 5 years. Mirena is FDA-approved for use up to 5 years,14 but studies have shown that it can be effective for 7 years.4,16
The only implant available in the US is Nexplanon, a plastic rod containing 68 mg of etonorgestrel. It is inserted subdermally and is FDA-approved for use up to 3 years.15
Through systemic hormonal effects, the primary mechanism of action of the implant is prevention of ovulation. Additionally, the implant has been shown to inhibit endometrial proliferation and cervical mucus thickening, both of which may contribute to the implant’s overall effectiveness.17 In contrast, both the copper IUD and the LNG-IUDs work primarily by preventing fertilization. The LNG-IUDs also exhibit local hormonal effects (endometrial atrophy and thickened cervical mucus) that contribute to their effectiveness.17
Who is eligible for LARC?
LARC is suitable for the vast majority of women of reproductive age. For most multiparous women ≥20 years, all LARC devices are classified as category 1 (use without restriction) in the Centers for Disease Control and Prevention’s (CDC) US Medical Eligibility Criteria (US MEC).3 For women <20 years, the implant is also considered category 1, but IUDs in this age group are classified as category 2 (recommended with the caution that advantages usually outweigh risks) because of concerns about an increased risk of IUD expulsion and the increased prevalence of sexually transmitted infections (STIs) in adolescents.3 Contraindications to use of LARC vary depending on the method chosen (TABLE 1).3
There has been concern about the efficacy of implants in overweight women because the original trials of subdermal implants excluded women >130% of ideal body weight. However, according to the Contraceptive CHOICE Project, overweight and obese women enrolled in its program did not experience reduced contraceptive efficacy when using the implant when compared with normal-weight women.18
When can LARC devices be inserted?
LARC device insertion is possible at any time during the menstrual cycle. An algorithm to guide initiation of LARC is available through the Reproductive Health Access Project’s Web site at http://www.reproductiveaccess.org/wp-content/uploads/2014/12/quickstart_algorithm.pdf.
Rule out pregnancy before placing any LARC device. The copper IUD can be inserted at any time during the menstrual cycle without the need for back-up contraception.11,19 In contrast, for LNG-IUDs, back-up contraception is recommended for 7 days unless the insertion is done during the first 7 days of the menstrual cycle.12-14,19
For the implant, recommendations about when to insert are based on a woman’s previous method of contraception (TABLE 2).15 If insertion is done at a time other than when recommended, advise patients to use barrier protection for 7 days after insertion.4,15,19
Other issues often arise and cause concern about whether and when a LARC device can be inserted, including the possibility of undiagnosed STI, time elapsed since delivery, and advisability of use when breastfeeding.
Sexually transmitted infections and IUDs
Whether or not a woman chooses to receive an IUD, follow routine CDC guidelines in determining if a patient is a candidate for STI screening.20 If a woman wants an IUD and routine screening is recommended, you can perform screening on the day of IUD insertion.4,19 For women with an IUD already in place who are diagnosed with an STI, treat the infection while leaving the IUD in place.19 For women with a known or suspected STI who do not have an IUD already, treat the STI before inserting the IUD. The American Congress of Obstetricians and Gynecologists (ACOG) advises postponing insertion of an IUD until a negative STI test result is obtained 3 to 4 weeks after treatment completion.4
Breastfeeding concerns and timing of insertion postpartum
The US MEC classifies insertion of the copper IUD as category 1 for all postpartum women, regardless of breastfeeding status, if placed >4 weeks postpartum or immediately postpartum (defined as within 10 minutes of the delivery of the placenta). IUD placement is category 2 (recommended with the caution that advantages usually outweigh risks) if placed ≥10 minutes after placental delivery (until 4 weeks postpartum) because of an increased risk of expulsion.3
The US MEC also considers use of the implant and LNG-IUDs in breastfeeding women as category 1 if the device is placed at ≥4 weeks postpartum. Insertion at <4 weeks postpartum is considered category 2 because of concerns for decreased breast milk supply.3 However, studies on whether progestin-containing LARC devices affect breastfeeding have yielded varying results. In one randomized controlled trial (RCT) of 69 breastfeeding women using the implant, breastfeeding duration and milk production were not dependent on the timing of insertion after delivery.21 Another RCT of 96 women using LNG-IUDs showed fewer women continued to breastfeed at 6 months when their LNG-IUD was inserted immediately postpartum, compared with waiting 6 weeks.22
In addition to a concern about breast milk supply, breastfeeding women have a higher risk for uterine perforation from IUDs, especially during the first 36 weeks after delivery.23
Several studies have shown that there is a lower repeat pregnancy rate among women who receive immediate postpartum LARC placement.24 However, even if IUD insertion is performed immediately postpartum, there is a higher expulsion rate than when the IUD is inserted ≥4 weeks postpartum. The expulsion rates for insertion <10 minutes after vaginal delivery range from 9.5% to 15% for the copper IUD to as high as 24% for the LNG-IUDs. Expulsion rates for all IUDs are slightly lower for cesarean delivery.4,25,26 ACOG supports immediate post-placental placement for women with barriers to postpartum care or limited access to contraception.4
How can I help my patients make an informed choice?
Provide counseling on efficacy, common adverse effects, risks, and complications.
Efficacy is high
The failure rate of LARC is equal to, or lower than, that of female sterilization and is significantly lower than that of oral contraceptives (TABLE 1).4-6 Not only are LARC devices extremely effective, they have a higher rate of satisfaction than any other reversible contraceptive (TABLE 1).7,8
Common adverse effects
The most common adverse effect seen with all LARC devices is an alteration in menstrual bleeding, and a frequent adverse effect with IUDs is pain. Vaginitis is less common and can be seen with any of the devices. The progestin-containing LARC devices are associated with hormonal effects: vaginitis, headache, weight gain, acne, breast pain, hair loss, and emotional lability.12-15
Copper IUD. Many women using the copper IUD experience either a transient increase in menstrual bleeding lasting for a few months or inter-menstrual bleeding that tends to continue for the duration of use.4,17 However, according to data from the Contraceptive CHOICE Project, the most common reason cited for early discontinuation of the copper IUD is pain and cramping.9
LNG-IUDs. Like the copper IUD, many users of LNG-IUDs experience an initial increase in menstrual bleeding. However, unlike the other LARC devices, 20% to 33% of Mirena users are likely to experience amenorrhea after one year of use and 70% at 2 years.4,14 According to package inserts, amenorrhea after 3 years is less common with both Skyla (12%) and Liletta (38%).12,13 As with the copper IUD, based on data from the Contraceptive CHOICE Project, the most common reason cited for early discontinuation of LNG-IUDs is pain and cramping.9
Subdermal implant. Changes in menses in women using the subdermal implant range from amenorrhea (22%) to prolonged bleeding (18%).15,17 Although it is difficult to predict which pattern a particular woman will experience, heavier women are more likely to have heavier bleeding patterns, and initial bleeding patterns are predictive of future ones.4 The most common reason women choose to discontinue use of the implant is abnormal bleeding.4,9,27,28
Newer IUDs do not increase risk of STIs
Many patients and clinicians erroneously believe that IUDs increase the risk of STIs and therefore assume that patients with a history of STI are not appropriate candidates for an IUD.29 There is a slightly increased risk of pelvic inflammatory disease (PID) in the first 21 days after insertion of an IUD. However, in contrast to older IUDs, currently available IUDs do not increase the general risk for STIs.17,30
Risk of infertility is nil
There is no risk of infertility from use of currently available LARCs. For those who want to become pregnant, fertility typically returns immediately after removal of the device, regardless of which method of LARC is used.11-15,30
Complications of IUD insertion
Uterine perforation. Uterine perforation occurs in 0.8 to 2.1 per 1000 women, usually at the time of IUD placement. If IUD strings are not visible during a speculum examination, locate the IUD with ultrasound.4,17,30 If the IUD is in the abdomen, refer to a gynecologist for laparoscopic removal and select another form of contraception for use in the interim.30
Expulsion. Rates of expulsion are low, occurring in less than 10% of women4,17 and are not affected by parity or BMI.31 Expulsion rates are higher when the IUD is inserted immediately postpartum.4,25,26 Adolescents also have a 2-fold higher risk of uterine expulsion than older women.31
Ectopic pregnancy. Although a woman’s overall risk of ectopic pregnancy is not increased by using an IUD,4 it is true that if a woman becomes pregnant with an IUD in place, the pregnancy is more likely to be ectopic. Thus, if pregnancy is confirmed in a woman with an IUD in place, rule out ectopic pregnancy.
The FDA and the World Health Organization recommend that if an intrauterine pregnancy is confirmed with an IUD in place and the strings are visible, the IUD should be removed.4 Although removing the IUD increases the risk of spontaneous abortion (SAB) as compared with pregnancies without an IUD in place, the risk of SAB is still lower than if the IUD is left in place.4 Additional risks of continuing a pregnancy with an IUD in place include increased risks of preterm labor, chorioamnionitis, and septic abortion.4,30
Complications of subdermal implant insertion
After insertion of the implant, women usually experience temporary bruising and soreness at the insertion site. Less than 1% of women develop an infection or hematoma.17 There is a low risk of nerve damage if the implant is inserted too deeply.15 Removal of the subdermal implant is recommended if pregnancy occurs.15
CASE DECISIONS › Jenny has been using oral contraceptive pills, but not regularly. You suggest that LARC may be a better option and counsel her that if she does choose an IUD or the implant, it is likely that her menses will change. You provide information and reassurance that LARC is safe to use in adolescents. Jenny says she would like to try an implant. Six months later, Jenny returns and says the implant is working well. She has some irregular bleeding, but it is not bothersome.
You review with Ms. D the types of LARC devices available and reassure her that all are safe to use once breastfeeding is established. Ms. D says she would like to use an IUD and elects to wait until her postpartum visit to have an IUD inserted. Ms. D returns 6 months after IUD insertion; breastfeeding is going well, and she has not had any menstrual bleeding since delivery.
CORRESPONDENCE
Karyn Kolman, MD, 2800 East Ajo Way, Room 3006, Tucson, AZ 85713; [email protected]
1. Daniels K, Daugherty J, Jones J. Current contraceptive status among women aged 15-44: United States 2011-2013. NCHS data brief, no. 173. Hyattsville, MD: National Center for Health Statistics, 2014.
2. Branum AM, Jones J. Trends in long-acting reversible contraception use among US women aged 15-44. NCHS data brief, no. 188. Hyattsville, MD: National Center for Health Statistics, 2015.
3. Centers for Disease Control and Prevention (CDC). US medical eligibility criteria for contraceptive use, 2010. MMWR Recomm Rep. 2010;59:1-86.
4. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No 121: Long-acting reversible contraception: Implants and intrauterine devices. Obstet Gynecol. 2011;118:184-196.
5. Pickle S, Wu J, Burbank-Schmitt E. Prevention of unintended pregnancy: a focus on long-acting reversible contraception. Prim Care. 2014;41:239-260.
6. Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med. 2012;366:1998-2007.
7. Peipert JF, Zhao Q, Allsworth JE, et al. Continuation and satisfaction of reversible contraception. Obstet Gynecol. 2011;117:1105-1113.
8. O’Neil-Callahan M, Peipert JF, Zhao Q, et al. Twenty-four-month continuation of reversible contraception. Obstet Gynecol. 2013;122:1083-1091.
9. Grunloh DS, Casner T, Secura GM, et al. Characteristics associated with discontinuation of long-acting reversible contraception within the first 6 months of use. Obstet Gynecol. 2011;117:705-719.
10. Birgisson NE, Zhao Q, Secura GM, et al. Preventing unintended pregnancy: the contraceptive CHOICE project in review. J Womens Health (Larchmt). 2015;24:349-353.
11. ParaGard T 380A. (intrauterine copper contraceptive) [package insert]. Sellersville, PA : Teva Pharmaceuticals USA, Inc., 2013.
12. Skyla (levonorgestrel-releasing intrauterine system) [package insert]. Wayne, NJ : Bayer HealthCare Pharmaceuticals, Inc., 2013.
13. Liletta (levonorgestrel-releasing intrauterine system) [package insert]. Parsippany, NJ : Actavis Pharma, Inc., 2015.
14. Mirena (levonorgestrel-releasing intrauterine system) [package insert]. Whippany, NJ : Bayer HealthCare Pharmaceuticals, Inc., 2014.
15. Nexplanon (etongestrel implant) [package insert]. Whitehouse Station, NJ: Merck & Co Inc.; 2014.
16. Wu JP, Pickle S. Extended use of the intrauterine device: a literature review and recommendations for clinical practice. Contraception. 2014;89:495-503.
17. Stoddard A, McNicholas C, Peipert JF. Efficacy and safety of long-acting reversible contraception. Drugs. 2011;71:969-980.
18. Xu H, Wade JA, Peipert JF, et al. Contraceptive failure rates of etonogestrel subdermal implants in overweight and obese women. Obstet Gynecol. 2012;120:21-26.
19. Centers for Disease Control and Prevention (CDC). US selected practice recommendations for contraceptive use. MMWR Recomm Rep. 2013;62:1-60.
20. Centers for Disease Control and Prevention (CDC). Sexually transmitted disease treatment guidelines. MMWR Recomm Rep. 2010;59:1-110.
21. Gurtcheff SE, Turok DK, Stoddard G, et al. Lactogenesis after early postpartum use of the contraceptive implant: a randomized controlled trial. Obstet Gynecol. 2011;117:1114-1121.
22. Chen BA, Reeves MF, Creinin MD, et al. Postplacental or delayed levonorgestrel intrauterine device insertion and breast-feeding duration. Contraception. 2011;84:499-504.
23. Heinemann K, Reed S, Moehner S, et al. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91:274-279.
24. Tocce K, Sheeder J, Python J, et al. Long acting reversible contraception in postpartum adolescents: early initiation of etonogestrel implant is superior to IUDs in the outpatient setting. J Pediatr Adolesc Gynecol. 2012;25:59-63.
25. Mwalwanda CS, Black KI. Immediate post-partum initiation of intrauterine contraception and implants: a review of the safety and guidelines for use. Aust N Z J Obstet Gynaecol. 2013;53:331-337.
26. Sober, S, Schreiber CA. Postpartum contraception. Clin Obstet Gynecol. 2014;57:763-776.
27. Dickerson LM, Diaz VA, Jordon J, et al. Satisfaction, early removal, and side effects associated with long-acting reversible contraception. Fam Med. 2013;45:701-707.
28. Berenson AB, Tan A, Hirth JM. Complications and continuation rates associated with 2 types of long-acting contraception. Am J Obstet Gynecol. 2015;212:e1-e8.
29. Kavanaugh ML, Frowirth L, Jerman J, et al. Long-acting reversible contraception for adolescents and young adults: patient and provider perspectives. J Pediatr Adolesc Gynecol. 2013;86:86-95.
30. Espey E, Ogburn T. Long-acting reversible contraceptives: intrauterine devices and the contraceptive implant. Obstet Gynecol. 2011;117:705-719.
31. Madden T, McNicholas C, Zhao Q, et al. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014;124:718-726.
1. Daniels K, Daugherty J, Jones J. Current contraceptive status among women aged 15-44: United States 2011-2013. NCHS data brief, no. 173. Hyattsville, MD: National Center for Health Statistics, 2014.
2. Branum AM, Jones J. Trends in long-acting reversible contraception use among US women aged 15-44. NCHS data brief, no. 188. Hyattsville, MD: National Center for Health Statistics, 2015.
3. Centers for Disease Control and Prevention (CDC). US medical eligibility criteria for contraceptive use, 2010. MMWR Recomm Rep. 2010;59:1-86.
4. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No 121: Long-acting reversible contraception: Implants and intrauterine devices. Obstet Gynecol. 2011;118:184-196.
5. Pickle S, Wu J, Burbank-Schmitt E. Prevention of unintended pregnancy: a focus on long-acting reversible contraception. Prim Care. 2014;41:239-260.
6. Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med. 2012;366:1998-2007.
7. Peipert JF, Zhao Q, Allsworth JE, et al. Continuation and satisfaction of reversible contraception. Obstet Gynecol. 2011;117:1105-1113.
8. O’Neil-Callahan M, Peipert JF, Zhao Q, et al. Twenty-four-month continuation of reversible contraception. Obstet Gynecol. 2013;122:1083-1091.
9. Grunloh DS, Casner T, Secura GM, et al. Characteristics associated with discontinuation of long-acting reversible contraception within the first 6 months of use. Obstet Gynecol. 2011;117:705-719.
10. Birgisson NE, Zhao Q, Secura GM, et al. Preventing unintended pregnancy: the contraceptive CHOICE project in review. J Womens Health (Larchmt). 2015;24:349-353.
11. ParaGard T 380A. (intrauterine copper contraceptive) [package insert]. Sellersville, PA : Teva Pharmaceuticals USA, Inc., 2013.
12. Skyla (levonorgestrel-releasing intrauterine system) [package insert]. Wayne, NJ : Bayer HealthCare Pharmaceuticals, Inc., 2013.
13. Liletta (levonorgestrel-releasing intrauterine system) [package insert]. Parsippany, NJ : Actavis Pharma, Inc., 2015.
14. Mirena (levonorgestrel-releasing intrauterine system) [package insert]. Whippany, NJ : Bayer HealthCare Pharmaceuticals, Inc., 2014.
15. Nexplanon (etongestrel implant) [package insert]. Whitehouse Station, NJ: Merck & Co Inc.; 2014.
16. Wu JP, Pickle S. Extended use of the intrauterine device: a literature review and recommendations for clinical practice. Contraception. 2014;89:495-503.
17. Stoddard A, McNicholas C, Peipert JF. Efficacy and safety of long-acting reversible contraception. Drugs. 2011;71:969-980.
18. Xu H, Wade JA, Peipert JF, et al. Contraceptive failure rates of etonogestrel subdermal implants in overweight and obese women. Obstet Gynecol. 2012;120:21-26.
19. Centers for Disease Control and Prevention (CDC). US selected practice recommendations for contraceptive use. MMWR Recomm Rep. 2013;62:1-60.
20. Centers for Disease Control and Prevention (CDC). Sexually transmitted disease treatment guidelines. MMWR Recomm Rep. 2010;59:1-110.
21. Gurtcheff SE, Turok DK, Stoddard G, et al. Lactogenesis after early postpartum use of the contraceptive implant: a randomized controlled trial. Obstet Gynecol. 2011;117:1114-1121.
22. Chen BA, Reeves MF, Creinin MD, et al. Postplacental or delayed levonorgestrel intrauterine device insertion and breast-feeding duration. Contraception. 2011;84:499-504.
23. Heinemann K, Reed S, Moehner S, et al. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91:274-279.
24. Tocce K, Sheeder J, Python J, et al. Long acting reversible contraception in postpartum adolescents: early initiation of etonogestrel implant is superior to IUDs in the outpatient setting. J Pediatr Adolesc Gynecol. 2012;25:59-63.
25. Mwalwanda CS, Black KI. Immediate post-partum initiation of intrauterine contraception and implants: a review of the safety and guidelines for use. Aust N Z J Obstet Gynaecol. 2013;53:331-337.
26. Sober, S, Schreiber CA. Postpartum contraception. Clin Obstet Gynecol. 2014;57:763-776.
27. Dickerson LM, Diaz VA, Jordon J, et al. Satisfaction, early removal, and side effects associated with long-acting reversible contraception. Fam Med. 2013;45:701-707.
28. Berenson AB, Tan A, Hirth JM. Complications and continuation rates associated with 2 types of long-acting contraception. Am J Obstet Gynecol. 2015;212:e1-e8.
29. Kavanaugh ML, Frowirth L, Jerman J, et al. Long-acting reversible contraception for adolescents and young adults: patient and provider perspectives. J Pediatr Adolesc Gynecol. 2013;86:86-95.
30. Espey E, Ogburn T. Long-acting reversible contraceptives: intrauterine devices and the contraceptive implant. Obstet Gynecol. 2011;117:705-719.
31. Madden T, McNicholas C, Zhao Q, et al. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014;124:718-726.
Familial hypercholesterolemia: Clues to catching it early
› When one member of a family has early heart disease, screen the entire family for familial hypercholesterolemia (FH). A
› Consider all patients with FH as being at high risk for coronary heart disease, regardless of their Framingham Risk Score. C
› Treat FH patients with statins early to avoid cardiovascular events. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Familial hypercholesterolemia (FH) poses a “silent” threat to patients with the condition, putting them at great risk of a coronary event. This genetic disorder, in which one or more mutations cause extremely high low-density lipoprotein (LDL) cholesterol levels, goes undiagnosed in approximately 80% of patients who have it.1 As a result, men with FH have a >50% risk of coronary heart disease (CHD) by age 50 and women with FH have a 30% risk of CHD by age 60.2 Patients with FH face a much higher risk of dying from a coronary event than those in the general population.3 For example, women between the ages of 20 and 39 who have this disorder are 125 times more likely to die of a coronary event than those who don’t.3
Unfortunately, FH can be difficult to diagnose. Some patients have physical findings, but these features can be subtle and easily missed. Typically, however, FH is diagnosed based on a patient’s cholesterol level and family history. By implementing screening and early treatment for FH, you may be able to initiate treatment that can temper the development of atherosclerosis and possibly extend a patient’s life.4
Two forms of the disorder, although one is more common
There are 2 types of FH:
Heterozygous FH (HeFH) occurs in about 1 in 300 to 500 people, which makes it more common than Down syndrome.5 More than a half a million people in the United States have HeFH.6
Homozygous FH (HoFH) is more serious than HeFH, and less common, affecting one in 1 million people. Homozygous carriers suffer from CHD much earlier than those with HeFH; some die within the first few years of life.7
Regardless of whether an affected individual inherited FH from one or both parents, more than one thousand mutations are known to cause inadequate clearance of LDL from the bloodstream.8 One of the most common mutations is a defective LDL receptor gene. Other abnormalities are known to occur with the proprotein convertase subtilisin/kexin type 9 (PCSK9) and apolipoprotein B genes.9

Start with screening
Suspect FH in patients who have a family history of premature heart disease. Also consider the patient’s ethnic background. The prevalence of FH is as high as one in 100 among certain groups, including French Canadians, Christian Lebanese, and 3 populations in South Africa (Ashkenazi Jews, Dutch Afrikaners, and Asian Indians).10
When there is high suspicion of FH based on a patient’s family history or ethnicity, additional screening is warranted for any patient older than age 2.11 If a patient’s family history is incomplete (eg, adoption, single-parent family), a lower threshold for screening is appropriate.
Lipid screening includes measuring serum total, LDL, and high-density lipoprotein cholesterol in either fasting or non-fasting samples. The United States Preventive Services Task Force (USPSTF) offers gender-specific recommendations for lipid disorder screening in the general population. For men, universal screening is recommended starting at age 35, and screening for those at increased risk of CHD should start at age 20.12
For women, the USPSTF recommends lipid screening only for those over age 20 who are at increased risk for CHD; such screening is strongly recommended for high-risk women ages 45 and older. In light of the serious consequences associated with FH, the National Lipid Association recommends lipid screening for all adults starting at age 20 (TABLE 1).13
What about kids? The recommendations for lipid screening in children and adolescents are mixed. Both the USPSTF and the American Academy of Family Physicians indicate that there is insufficient evidence to screen for lipid disorders in asymptomatic children and adolescents.14,15 However, in a set of recommendations based on expert opinion, the National Heart, Lung, and Blood Institute (NHLBI) suggests universal screening for younger patients with a non-fasting lipid profile once between ages 9 to 11 and again between ages 17 to 21.16 The American Academy of Pediatrics has adopted the NHLBI recommendations.17
Physical exam findings that suggest familial hypercholesterolemia
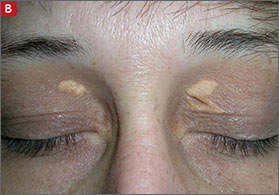
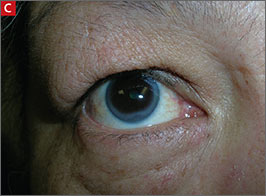
Tendon xanthomas (A), a thickening of the soft tissue as a result of infiltration by lipid-rich cells, most commonly occur at the Achilles and metacarpal tendons, but also can be seen at the patellar and triceps tendons.
Tuberous xanthomas or xanthelasmas (B) are waxy-appearing growths that appear to be pasted on the skin in areas around the face, commonly the eyelids.
Arcus corneae (C) is an opaque ring around the outer edge of the cornea.
Use validated criteria to make the diagnosis
Include FH in your differential diagnosis when evaluating patients with very high LDL levels. However, rule out possible secondary causes of elevated LDL before rendering a conclusion. Hypothyroidism, nephrotic syndrome, diabetes, and liver disease are among the most common secondary causes of high LDL cholesterol.13
Several validated criteria sets can be used to establish an FH diagnosis. No single criteria set is more valid or more widely adopted around the world. All 3 of the most commonly used criteria sets take into account family history and a patient’s LDL level, and 2 of the 3 factor in physical findings (TABLE 2).9
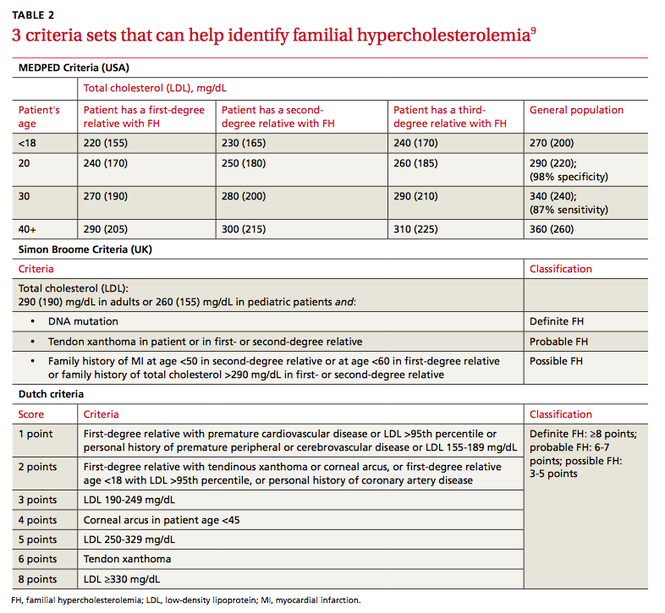
Physical exam findings that suggest FH can be subtle (FIGURE). Tendon xanthomas are a thickening of the soft tissue as a result of infiltration by lipid-rich cells. They most commonly occur at the Achilles and metacarpal tendons, but can also be seen at the patellar and triceps tendons. Xanthomas may not be readily visible, so it’s important to run your fingers over these areas to detect nodularity or thickening. While the presence of a tendon xanthoma makes FH highly likely, they are present in less than half of patients with FH.17
Tuberous xanthomas or xanthelasmas are waxy-appearing growths that may look yellow or orange and appear to be pasted on the skin in areas around the face, commonly the eyelids. The presence of xanthelasmas in a patient younger than age 25 suggests FH.
Finally, arcus corneae is an opaque ring around the outer edge of the cornea. When this is seen in patients younger than age 45, it’s suggestive of FH.13 If you note tendon xanthomas, xanthelasmas, or arcus corneae while examining any of your patients, be sure to order an LDL level if it hasn’t already been done.
Is genetic testing necessary?
The only way to make a definitive diagnosis of FH is to find a mutation in a gene known to affect LDL metabolism. However, because genetic testing is expensive—and because more than one thousand different genetic defects can contribute to FH—it’s not practical to test every patient. Furthermore, since an estimated 20% of the mutations that contribute to FH have not yet been clearly delineated, a “normal” result on a genetic test might be misleading.5 Therefore, the diagnosis of FH usually is a clinical one. After clinically diagnosing a patient with FH, it’s imperative to screen first-degree family members by measuring their LDL cholesterol levels.
Lifestyle changes, statins can ward off CHD
Lifestyle modifications (ie, improved diet and exercise) and statins are the treatments of choice for patients with FH. Before starting pharmacotherapy, patients should undergo 3 months of lifestyle modification to assess how well this approach improves their lipid levels, assuming the patient doesn’t have additional risk factors such as hypertension or tobacco use, in which case he or she might require immediate pharmacotherapy. Statins can be initiated simultaneously with lifestyle choices in patients with an LDL >190 mg/dL.18
Lifestyle modification. Although FH is a genetic problem, patients should be encouraged to make healthy choices regarding diet and exercise. While the best choices may not get FH patients to their LDL goal, better choices may mean that patients can take fewer medications, or lower doses of them. Healthy lifestyle choices can also have other positive effects on cardiovascular risk (eg, lowering blood pressure).
Patients can’t be expected to navigate their food choices alone, and several visits with a dietician will likely be needed. It’s important to emphasize the family influence on diet and get the entire family involved with making healthy food choices.
In addition to addressing diet and exercise, be sure to encourage patients to abstain from tobacco and manage stress as part of their overall effort to reduce the likelihood of a cardiovascular event.
Statins. Early treatment of FH with statins can delay initial coronary events and prolong life.19 In a 12.5-year study of 2146 patients with FH, approximately 80% of patients treated with statins survived without experiencing CHD, compared to slightly less than 40% of those who were not treated with statins.19 Patients treated with statins had a 76% reduction in risk of CHD compared to those who didn’t receive statins.19 Even low doses of statins started early have been shown to help avoid myocardial infarction in adults with FH.20
The goal of treatment for FH is to reduced LDL levels by 50%.21 In pediatric patients, treating to an LDL level of 130 mg/dL is an alternative goal.21 Because it’s challenging to achieve this goal with improved diet and exercise alone, treatment with a statin is often necessary.22
Statins can be used in children as young as age 8, or even earlier in homozygous FH.6 While a physician might be hesitant to start a chronic medication in a young patient, research shows that earlier intervention results in additional years of life.23 To date, no significant adverse effects of statins in pediatric patients have been identified, and statins have not been shown to impair growth.24,25 Young female patients should be counseled about the adverse effects statins can have on a fetus if the patient becomes pregnant while taking the medication.
Navigating the waters of statin treatment
Musculoskeletal symptoms are the most common adverse effect reported by patients taking statins. A thorough assessment of a patient’s muscle complaints is necessary to avoid prematurely concluding that he or she cannot tolerate statins.
A study in which “statin-intolerant” patients were re-challenged found that more than 90% of patients could tolerate statins through the course of the one-year study and that it was likely that the patients’ initial muscle complaints were not due to statin use.23 (To read more about potential adverse events of statins, see “Statin adverse effects: Sorting out the evidence,” J Fam Pract. 2014;63:497-506.).
If LDL levels in a patient with HeFH remain at or above 160 mg/dL, intensifying treatment by adding another lipid-lowering medication might be warranted.22 For patients with HoFH, in whom the condition is more quickly life-threatening, there are additional choices, including LDL apheresis and medications such as mipomersen and lomitapide. Both of these medications can cause hepatotoxicity, and are available only through a Risk Evaluation and Mitigation Strategy program, which means they can only be prescribed by certified physicians. PCSK9 inhibitors are in the pipeline and may one day help patients with HoFH by addressing one of the genetic causes of this disorder.
CORRESPONDENCE
Richard Safeer, MD, 6704 Curtis Court, Glen Burnie, MD 21060; [email protected]
1. Datta BN, McDowell IF, Rees A. Integrating provision of specialist lipid services with cascade testing for familial hypercholesterolaemia. Curr Opin Lipidol. 2010;21:366-371.
2. DeMott K, Nherera L, Shaw EJ, et al. Clinical Guidelines and Evidence Review for Familial Hypercholesterolaemia: The Identification and Management of Adults and Children with Familial Hypercholesterolaemia. 2008. London, UK: National Collaborating Centre for Primary Care and Royal College of General Practitioners.
3. Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis. 1999;142:105-112.
4. Kavey RE, Allada V, Daniels SR, et al. American Heart Association Expert Panel on Population and Prevention Science; American Heart Association Council on Cardiovascular Disease in the Young; American Heart Association Council on Epidemiology and Prevention. Cardiovascular risk reduction in high-risk pediatric patients. Circulation. 2006;114:2710-2738.
5. Rees A. Familial hypercholesterolaemia: underdiagnosed and undertreated. Eur Heart J. 2008;29:2583-2584.
6. Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients. Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S1-8.
7. Moriarty PM. LDL-apheresis therapy. Curr Treat Options Cardiovasc Med. 2006;8:282-288.
8. Goldstein JL, Brown MS. The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979;13:259-289.
9. Fahed AC, Nemer GM. Familial hypercholesterolemia: the lipids or the genes? Nutr Metab (Lond). 2011;8:23.
10. Goldstein J, Hobbs H, Brown M. Familial hypercholesterolemia. In: Scriver C, Baudet A, Sly W, et al, eds. The Metabolic Basis of Inherited Disease. New York, NY: McGraw-Hill; 2001: 2863-2913.
11. Kwiterovich, PO. Clinical and laboratory assessment of cardiovascular risk in children: Guidelines for screening, evaluation and treatment. J Clin Lipidol. 2008;2:248-266.
12. US Preventive Services Task Force. Lipid disorders in adults (cholesterol, dyslipidemia): Screening. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/lipid-disorders-in-adults-cholesterol-dyslipidemia-screening. Accessed July 6, 2015.
13. Hopkins PN, Toth PP, Ballantyne CM, et al; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S9-17.
14. US Preventive Services Task Force. Screening for lipid disorders in children: US Preventive Services Task Force recommendation statement. Pediatrics. 2007;120;e215-219.
15. American Academy of Family Physicians. Summary of recommendations for clinical preventive services. American Academy of Family Physicians Web site. Available at: http://www.aafp.org/dam/AAFP/documents/patient_care/clinical_recommendations/cps-recommendations.pdf. Accessed May 2, 2015.
16. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128:S213-56.
17. American Academy of Pediatrics. 2014 Recommendations for Pediatric Preventive Health Care. American Academy of Pediatrics Web site. Available at: http://pediatrics.aappublications.org/content/133/3/568.full.pdf+html. Accessed May 2, 2015.
18. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889-2934.
19. Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478-3490a.
20. Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423.
21. Daniels SR, Gidding SS, de Ferranti SD; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Pediatric aspects of familial hypercholesterolemias: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S30-37.
22. Robinson JG, Goldberg AC; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S18-29.
23. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158:526-534.
24. Eiland LS, Luttrell PK. Use of statins for dyslipidemia in the pediatric population. J Pediatr Pharmacol Ther. 2010;15:160-172.
25. O’Gorman CS, Higgins MF, O’Neill MB. Systematic review and metaanalysis of statins for heterozygous familial hypercholesterolemia in children: evaluation of cholesterol changes and side effects. Pediatr Cardiol. 2009;30:482-489.
› When one member of a family has early heart disease, screen the entire family for familial hypercholesterolemia (FH). A
› Consider all patients with FH as being at high risk for coronary heart disease, regardless of their Framingham Risk Score. C
› Treat FH patients with statins early to avoid cardiovascular events. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Familial hypercholesterolemia (FH) poses a “silent” threat to patients with the condition, putting them at great risk of a coronary event. This genetic disorder, in which one or more mutations cause extremely high low-density lipoprotein (LDL) cholesterol levels, goes undiagnosed in approximately 80% of patients who have it.1 As a result, men with FH have a >50% risk of coronary heart disease (CHD) by age 50 and women with FH have a 30% risk of CHD by age 60.2 Patients with FH face a much higher risk of dying from a coronary event than those in the general population.3 For example, women between the ages of 20 and 39 who have this disorder are 125 times more likely to die of a coronary event than those who don’t.3
Unfortunately, FH can be difficult to diagnose. Some patients have physical findings, but these features can be subtle and easily missed. Typically, however, FH is diagnosed based on a patient’s cholesterol level and family history. By implementing screening and early treatment for FH, you may be able to initiate treatment that can temper the development of atherosclerosis and possibly extend a patient’s life.4
Two forms of the disorder, although one is more common
There are 2 types of FH:
Heterozygous FH (HeFH) occurs in about 1 in 300 to 500 people, which makes it more common than Down syndrome.5 More than a half a million people in the United States have HeFH.6
Homozygous FH (HoFH) is more serious than HeFH, and less common, affecting one in 1 million people. Homozygous carriers suffer from CHD much earlier than those with HeFH; some die within the first few years of life.7
Regardless of whether an affected individual inherited FH from one or both parents, more than one thousand mutations are known to cause inadequate clearance of LDL from the bloodstream.8 One of the most common mutations is a defective LDL receptor gene. Other abnormalities are known to occur with the proprotein convertase subtilisin/kexin type 9 (PCSK9) and apolipoprotein B genes.9
Start with screening
Suspect FH in patients who have a family history of premature heart disease. Also consider the patient’s ethnic background. The prevalence of FH is as high as one in 100 among certain groups, including French Canadians, Christian Lebanese, and 3 populations in South Africa (Ashkenazi Jews, Dutch Afrikaners, and Asian Indians).10
When there is high suspicion of FH based on a patient’s family history or ethnicity, additional screening is warranted for any patient older than age 2.11 If a patient’s family history is incomplete (eg, adoption, single-parent family), a lower threshold for screening is appropriate.
Lipid screening includes measuring serum total, LDL, and high-density lipoprotein cholesterol in either fasting or non-fasting samples. The United States Preventive Services Task Force (USPSTF) offers gender-specific recommendations for lipid disorder screening in the general population. For men, universal screening is recommended starting at age 35, and screening for those at increased risk of CHD should start at age 20.12
For women, the USPSTF recommends lipid screening only for those over age 20 who are at increased risk for CHD; such screening is strongly recommended for high-risk women ages 45 and older. In light of the serious consequences associated with FH, the National Lipid Association recommends lipid screening for all adults starting at age 20 (TABLE 1).13
What about kids? The recommendations for lipid screening in children and adolescents are mixed. Both the USPSTF and the American Academy of Family Physicians indicate that there is insufficient evidence to screen for lipid disorders in asymptomatic children and adolescents.14,15 However, in a set of recommendations based on expert opinion, the National Heart, Lung, and Blood Institute (NHLBI) suggests universal screening for younger patients with a non-fasting lipid profile once between ages 9 to 11 and again between ages 17 to 21.16 The American Academy of Pediatrics has adopted the NHLBI recommendations.17
Physical exam findings that suggest familial hypercholesterolemia
Tendon xanthomas (A), a thickening of the soft tissue as a result of infiltration by lipid-rich cells, most commonly occur at the Achilles and metacarpal tendons, but also can be seen at the patellar and triceps tendons.
Tuberous xanthomas or xanthelasmas (B) are waxy-appearing growths that appear to be pasted on the skin in areas around the face, commonly the eyelids.
Arcus corneae (C) is an opaque ring around the outer edge of the cornea.
Use validated criteria to make the diagnosis
Include FH in your differential diagnosis when evaluating patients with very high LDL levels. However, rule out possible secondary causes of elevated LDL before rendering a conclusion. Hypothyroidism, nephrotic syndrome, diabetes, and liver disease are among the most common secondary causes of high LDL cholesterol.13
Several validated criteria sets can be used to establish an FH diagnosis. No single criteria set is more valid or more widely adopted around the world. All 3 of the most commonly used criteria sets take into account family history and a patient’s LDL level, and 2 of the 3 factor in physical findings (TABLE 2).9
Physical exam findings that suggest FH can be subtle (FIGURE). Tendon xanthomas are a thickening of the soft tissue as a result of infiltration by lipid-rich cells. They most commonly occur at the Achilles and metacarpal tendons, but can also be seen at the patellar and triceps tendons. Xanthomas may not be readily visible, so it’s important to run your fingers over these areas to detect nodularity or thickening. While the presence of a tendon xanthoma makes FH highly likely, they are present in less than half of patients with FH.17
Tuberous xanthomas or xanthelasmas are waxy-appearing growths that may look yellow or orange and appear to be pasted on the skin in areas around the face, commonly the eyelids. The presence of xanthelasmas in a patient younger than age 25 suggests FH.
Finally, arcus corneae is an opaque ring around the outer edge of the cornea. When this is seen in patients younger than age 45, it’s suggestive of FH.13 If you note tendon xanthomas, xanthelasmas, or arcus corneae while examining any of your patients, be sure to order an LDL level if it hasn’t already been done.
Is genetic testing necessary?
The only way to make a definitive diagnosis of FH is to find a mutation in a gene known to affect LDL metabolism. However, because genetic testing is expensive—and because more than one thousand different genetic defects can contribute to FH—it’s not practical to test every patient. Furthermore, since an estimated 20% of the mutations that contribute to FH have not yet been clearly delineated, a “normal” result on a genetic test might be misleading.5 Therefore, the diagnosis of FH usually is a clinical one. After clinically diagnosing a patient with FH, it’s imperative to screen first-degree family members by measuring their LDL cholesterol levels.
Lifestyle changes, statins can ward off CHD
Lifestyle modifications (ie, improved diet and exercise) and statins are the treatments of choice for patients with FH. Before starting pharmacotherapy, patients should undergo 3 months of lifestyle modification to assess how well this approach improves their lipid levels, assuming the patient doesn’t have additional risk factors such as hypertension or tobacco use, in which case he or she might require immediate pharmacotherapy. Statins can be initiated simultaneously with lifestyle choices in patients with an LDL >190 mg/dL.18
Lifestyle modification. Although FH is a genetic problem, patients should be encouraged to make healthy choices regarding diet and exercise. While the best choices may not get FH patients to their LDL goal, better choices may mean that patients can take fewer medications, or lower doses of them. Healthy lifestyle choices can also have other positive effects on cardiovascular risk (eg, lowering blood pressure).
Patients can’t be expected to navigate their food choices alone, and several visits with a dietician will likely be needed. It’s important to emphasize the family influence on diet and get the entire family involved with making healthy food choices.
In addition to addressing diet and exercise, be sure to encourage patients to abstain from tobacco and manage stress as part of their overall effort to reduce the likelihood of a cardiovascular event.
Statins. Early treatment of FH with statins can delay initial coronary events and prolong life.19 In a 12.5-year study of 2146 patients with FH, approximately 80% of patients treated with statins survived without experiencing CHD, compared to slightly less than 40% of those who were not treated with statins.19 Patients treated with statins had a 76% reduction in risk of CHD compared to those who didn’t receive statins.19 Even low doses of statins started early have been shown to help avoid myocardial infarction in adults with FH.20
The goal of treatment for FH is to reduced LDL levels by 50%.21 In pediatric patients, treating to an LDL level of 130 mg/dL is an alternative goal.21 Because it’s challenging to achieve this goal with improved diet and exercise alone, treatment with a statin is often necessary.22
Statins can be used in children as young as age 8, or even earlier in homozygous FH.6 While a physician might be hesitant to start a chronic medication in a young patient, research shows that earlier intervention results in additional years of life.23 To date, no significant adverse effects of statins in pediatric patients have been identified, and statins have not been shown to impair growth.24,25 Young female patients should be counseled about the adverse effects statins can have on a fetus if the patient becomes pregnant while taking the medication.
Navigating the waters of statin treatment
Musculoskeletal symptoms are the most common adverse effect reported by patients taking statins. A thorough assessment of a patient’s muscle complaints is necessary to avoid prematurely concluding that he or she cannot tolerate statins.
A study in which “statin-intolerant” patients were re-challenged found that more than 90% of patients could tolerate statins through the course of the one-year study and that it was likely that the patients’ initial muscle complaints were not due to statin use.23 (To read more about potential adverse events of statins, see “Statin adverse effects: Sorting out the evidence,” J Fam Pract. 2014;63:497-506.).
If LDL levels in a patient with HeFH remain at or above 160 mg/dL, intensifying treatment by adding another lipid-lowering medication might be warranted.22 For patients with HoFH, in whom the condition is more quickly life-threatening, there are additional choices, including LDL apheresis and medications such as mipomersen and lomitapide. Both of these medications can cause hepatotoxicity, and are available only through a Risk Evaluation and Mitigation Strategy program, which means they can only be prescribed by certified physicians. PCSK9 inhibitors are in the pipeline and may one day help patients with HoFH by addressing one of the genetic causes of this disorder.
CORRESPONDENCE
Richard Safeer, MD, 6704 Curtis Court, Glen Burnie, MD 21060; [email protected]
› When one member of a family has early heart disease, screen the entire family for familial hypercholesterolemia (FH). A
› Consider all patients with FH as being at high risk for coronary heart disease, regardless of their Framingham Risk Score. C
› Treat FH patients with statins early to avoid cardiovascular events. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Familial hypercholesterolemia (FH) poses a “silent” threat to patients with the condition, putting them at great risk of a coronary event. This genetic disorder, in which one or more mutations cause extremely high low-density lipoprotein (LDL) cholesterol levels, goes undiagnosed in approximately 80% of patients who have it.1 As a result, men with FH have a >50% risk of coronary heart disease (CHD) by age 50 and women with FH have a 30% risk of CHD by age 60.2 Patients with FH face a much higher risk of dying from a coronary event than those in the general population.3 For example, women between the ages of 20 and 39 who have this disorder are 125 times more likely to die of a coronary event than those who don’t.3
Unfortunately, FH can be difficult to diagnose. Some patients have physical findings, but these features can be subtle and easily missed. Typically, however, FH is diagnosed based on a patient’s cholesterol level and family history. By implementing screening and early treatment for FH, you may be able to initiate treatment that can temper the development of atherosclerosis and possibly extend a patient’s life.4
Two forms of the disorder, although one is more common
There are 2 types of FH:
Heterozygous FH (HeFH) occurs in about 1 in 300 to 500 people, which makes it more common than Down syndrome.5 More than a half a million people in the United States have HeFH.6
Homozygous FH (HoFH) is more serious than HeFH, and less common, affecting one in 1 million people. Homozygous carriers suffer from CHD much earlier than those with HeFH; some die within the first few years of life.7
Regardless of whether an affected individual inherited FH from one or both parents, more than one thousand mutations are known to cause inadequate clearance of LDL from the bloodstream.8 One of the most common mutations is a defective LDL receptor gene. Other abnormalities are known to occur with the proprotein convertase subtilisin/kexin type 9 (PCSK9) and apolipoprotein B genes.9
Start with screening
Suspect FH in patients who have a family history of premature heart disease. Also consider the patient’s ethnic background. The prevalence of FH is as high as one in 100 among certain groups, including French Canadians, Christian Lebanese, and 3 populations in South Africa (Ashkenazi Jews, Dutch Afrikaners, and Asian Indians).10
When there is high suspicion of FH based on a patient’s family history or ethnicity, additional screening is warranted for any patient older than age 2.11 If a patient’s family history is incomplete (eg, adoption, single-parent family), a lower threshold for screening is appropriate.
Lipid screening includes measuring serum total, LDL, and high-density lipoprotein cholesterol in either fasting or non-fasting samples. The United States Preventive Services Task Force (USPSTF) offers gender-specific recommendations for lipid disorder screening in the general population. For men, universal screening is recommended starting at age 35, and screening for those at increased risk of CHD should start at age 20.12
For women, the USPSTF recommends lipid screening only for those over age 20 who are at increased risk for CHD; such screening is strongly recommended for high-risk women ages 45 and older. In light of the serious consequences associated with FH, the National Lipid Association recommends lipid screening for all adults starting at age 20 (TABLE 1).13
What about kids? The recommendations for lipid screening in children and adolescents are mixed. Both the USPSTF and the American Academy of Family Physicians indicate that there is insufficient evidence to screen for lipid disorders in asymptomatic children and adolescents.14,15 However, in a set of recommendations based on expert opinion, the National Heart, Lung, and Blood Institute (NHLBI) suggests universal screening for younger patients with a non-fasting lipid profile once between ages 9 to 11 and again between ages 17 to 21.16 The American Academy of Pediatrics has adopted the NHLBI recommendations.17
Physical exam findings that suggest familial hypercholesterolemia
Tendon xanthomas (A), a thickening of the soft tissue as a result of infiltration by lipid-rich cells, most commonly occur at the Achilles and metacarpal tendons, but also can be seen at the patellar and triceps tendons.
Tuberous xanthomas or xanthelasmas (B) are waxy-appearing growths that appear to be pasted on the skin in areas around the face, commonly the eyelids.
Arcus corneae (C) is an opaque ring around the outer edge of the cornea.
Use validated criteria to make the diagnosis
Include FH in your differential diagnosis when evaluating patients with very high LDL levels. However, rule out possible secondary causes of elevated LDL before rendering a conclusion. Hypothyroidism, nephrotic syndrome, diabetes, and liver disease are among the most common secondary causes of high LDL cholesterol.13
Several validated criteria sets can be used to establish an FH diagnosis. No single criteria set is more valid or more widely adopted around the world. All 3 of the most commonly used criteria sets take into account family history and a patient’s LDL level, and 2 of the 3 factor in physical findings (TABLE 2).9
Physical exam findings that suggest FH can be subtle (FIGURE). Tendon xanthomas are a thickening of the soft tissue as a result of infiltration by lipid-rich cells. They most commonly occur at the Achilles and metacarpal tendons, but can also be seen at the patellar and triceps tendons. Xanthomas may not be readily visible, so it’s important to run your fingers over these areas to detect nodularity or thickening. While the presence of a tendon xanthoma makes FH highly likely, they are present in less than half of patients with FH.17
Tuberous xanthomas or xanthelasmas are waxy-appearing growths that may look yellow or orange and appear to be pasted on the skin in areas around the face, commonly the eyelids. The presence of xanthelasmas in a patient younger than age 25 suggests FH.
Finally, arcus corneae is an opaque ring around the outer edge of the cornea. When this is seen in patients younger than age 45, it’s suggestive of FH.13 If you note tendon xanthomas, xanthelasmas, or arcus corneae while examining any of your patients, be sure to order an LDL level if it hasn’t already been done.
Is genetic testing necessary?
The only way to make a definitive diagnosis of FH is to find a mutation in a gene known to affect LDL metabolism. However, because genetic testing is expensive—and because more than one thousand different genetic defects can contribute to FH—it’s not practical to test every patient. Furthermore, since an estimated 20% of the mutations that contribute to FH have not yet been clearly delineated, a “normal” result on a genetic test might be misleading.5 Therefore, the diagnosis of FH usually is a clinical one. After clinically diagnosing a patient with FH, it’s imperative to screen first-degree family members by measuring their LDL cholesterol levels.
Lifestyle changes, statins can ward off CHD
Lifestyle modifications (ie, improved diet and exercise) and statins are the treatments of choice for patients with FH. Before starting pharmacotherapy, patients should undergo 3 months of lifestyle modification to assess how well this approach improves their lipid levels, assuming the patient doesn’t have additional risk factors such as hypertension or tobacco use, in which case he or she might require immediate pharmacotherapy. Statins can be initiated simultaneously with lifestyle choices in patients with an LDL >190 mg/dL.18
Lifestyle modification. Although FH is a genetic problem, patients should be encouraged to make healthy choices regarding diet and exercise. While the best choices may not get FH patients to their LDL goal, better choices may mean that patients can take fewer medications, or lower doses of them. Healthy lifestyle choices can also have other positive effects on cardiovascular risk (eg, lowering blood pressure).
Patients can’t be expected to navigate their food choices alone, and several visits with a dietician will likely be needed. It’s important to emphasize the family influence on diet and get the entire family involved with making healthy food choices.
In addition to addressing diet and exercise, be sure to encourage patients to abstain from tobacco and manage stress as part of their overall effort to reduce the likelihood of a cardiovascular event.
Statins. Early treatment of FH with statins can delay initial coronary events and prolong life.19 In a 12.5-year study of 2146 patients with FH, approximately 80% of patients treated with statins survived without experiencing CHD, compared to slightly less than 40% of those who were not treated with statins.19 Patients treated with statins had a 76% reduction in risk of CHD compared to those who didn’t receive statins.19 Even low doses of statins started early have been shown to help avoid myocardial infarction in adults with FH.20
The goal of treatment for FH is to reduced LDL levels by 50%.21 In pediatric patients, treating to an LDL level of 130 mg/dL is an alternative goal.21 Because it’s challenging to achieve this goal with improved diet and exercise alone, treatment with a statin is often necessary.22
Statins can be used in children as young as age 8, or even earlier in homozygous FH.6 While a physician might be hesitant to start a chronic medication in a young patient, research shows that earlier intervention results in additional years of life.23 To date, no significant adverse effects of statins in pediatric patients have been identified, and statins have not been shown to impair growth.24,25 Young female patients should be counseled about the adverse effects statins can have on a fetus if the patient becomes pregnant while taking the medication.
Navigating the waters of statin treatment
Musculoskeletal symptoms are the most common adverse effect reported by patients taking statins. A thorough assessment of a patient’s muscle complaints is necessary to avoid prematurely concluding that he or she cannot tolerate statins.
A study in which “statin-intolerant” patients were re-challenged found that more than 90% of patients could tolerate statins through the course of the one-year study and that it was likely that the patients’ initial muscle complaints were not due to statin use.23 (To read more about potential adverse events of statins, see “Statin adverse effects: Sorting out the evidence,” J Fam Pract. 2014;63:497-506.).
If LDL levels in a patient with HeFH remain at or above 160 mg/dL, intensifying treatment by adding another lipid-lowering medication might be warranted.22 For patients with HoFH, in whom the condition is more quickly life-threatening, there are additional choices, including LDL apheresis and medications such as mipomersen and lomitapide. Both of these medications can cause hepatotoxicity, and are available only through a Risk Evaluation and Mitigation Strategy program, which means they can only be prescribed by certified physicians. PCSK9 inhibitors are in the pipeline and may one day help patients with HoFH by addressing one of the genetic causes of this disorder.
CORRESPONDENCE
Richard Safeer, MD, 6704 Curtis Court, Glen Burnie, MD 21060; [email protected]
1. Datta BN, McDowell IF, Rees A. Integrating provision of specialist lipid services with cascade testing for familial hypercholesterolaemia. Curr Opin Lipidol. 2010;21:366-371.
2. DeMott K, Nherera L, Shaw EJ, et al. Clinical Guidelines and Evidence Review for Familial Hypercholesterolaemia: The Identification and Management of Adults and Children with Familial Hypercholesterolaemia. 2008. London, UK: National Collaborating Centre for Primary Care and Royal College of General Practitioners.
3. Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis. 1999;142:105-112.
4. Kavey RE, Allada V, Daniels SR, et al. American Heart Association Expert Panel on Population and Prevention Science; American Heart Association Council on Cardiovascular Disease in the Young; American Heart Association Council on Epidemiology and Prevention. Cardiovascular risk reduction in high-risk pediatric patients. Circulation. 2006;114:2710-2738.
5. Rees A. Familial hypercholesterolaemia: underdiagnosed and undertreated. Eur Heart J. 2008;29:2583-2584.
6. Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients. Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S1-8.
7. Moriarty PM. LDL-apheresis therapy. Curr Treat Options Cardiovasc Med. 2006;8:282-288.
8. Goldstein JL, Brown MS. The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979;13:259-289.
9. Fahed AC, Nemer GM. Familial hypercholesterolemia: the lipids or the genes? Nutr Metab (Lond). 2011;8:23.
10. Goldstein J, Hobbs H, Brown M. Familial hypercholesterolemia. In: Scriver C, Baudet A, Sly W, et al, eds. The Metabolic Basis of Inherited Disease. New York, NY: McGraw-Hill; 2001: 2863-2913.
11. Kwiterovich, PO. Clinical and laboratory assessment of cardiovascular risk in children: Guidelines for screening, evaluation and treatment. J Clin Lipidol. 2008;2:248-266.
12. US Preventive Services Task Force. Lipid disorders in adults (cholesterol, dyslipidemia): Screening. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/lipid-disorders-in-adults-cholesterol-dyslipidemia-screening. Accessed July 6, 2015.
13. Hopkins PN, Toth PP, Ballantyne CM, et al; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S9-17.
14. US Preventive Services Task Force. Screening for lipid disorders in children: US Preventive Services Task Force recommendation statement. Pediatrics. 2007;120;e215-219.
15. American Academy of Family Physicians. Summary of recommendations for clinical preventive services. American Academy of Family Physicians Web site. Available at: http://www.aafp.org/dam/AAFP/documents/patient_care/clinical_recommendations/cps-recommendations.pdf. Accessed May 2, 2015.
16. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128:S213-56.
17. American Academy of Pediatrics. 2014 Recommendations for Pediatric Preventive Health Care. American Academy of Pediatrics Web site. Available at: http://pediatrics.aappublications.org/content/133/3/568.full.pdf+html. Accessed May 2, 2015.
18. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889-2934.
19. Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478-3490a.
20. Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423.
21. Daniels SR, Gidding SS, de Ferranti SD; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Pediatric aspects of familial hypercholesterolemias: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S30-37.
22. Robinson JG, Goldberg AC; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S18-29.
23. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158:526-534.
24. Eiland LS, Luttrell PK. Use of statins for dyslipidemia in the pediatric population. J Pediatr Pharmacol Ther. 2010;15:160-172.
25. O’Gorman CS, Higgins MF, O’Neill MB. Systematic review and metaanalysis of statins for heterozygous familial hypercholesterolemia in children: evaluation of cholesterol changes and side effects. Pediatr Cardiol. 2009;30:482-489.
1. Datta BN, McDowell IF, Rees A. Integrating provision of specialist lipid services with cascade testing for familial hypercholesterolaemia. Curr Opin Lipidol. 2010;21:366-371.
2. DeMott K, Nherera L, Shaw EJ, et al. Clinical Guidelines and Evidence Review for Familial Hypercholesterolaemia: The Identification and Management of Adults and Children with Familial Hypercholesterolaemia. 2008. London, UK: National Collaborating Centre for Primary Care and Royal College of General Practitioners.
3. Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis. 1999;142:105-112.
4. Kavey RE, Allada V, Daniels SR, et al. American Heart Association Expert Panel on Population and Prevention Science; American Heart Association Council on Cardiovascular Disease in the Young; American Heart Association Council on Epidemiology and Prevention. Cardiovascular risk reduction in high-risk pediatric patients. Circulation. 2006;114:2710-2738.
5. Rees A. Familial hypercholesterolaemia: underdiagnosed and undertreated. Eur Heart J. 2008;29:2583-2584.
6. Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients. Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S1-8.
7. Moriarty PM. LDL-apheresis therapy. Curr Treat Options Cardiovasc Med. 2006;8:282-288.
8. Goldstein JL, Brown MS. The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979;13:259-289.
9. Fahed AC, Nemer GM. Familial hypercholesterolemia: the lipids or the genes? Nutr Metab (Lond). 2011;8:23.
10. Goldstein J, Hobbs H, Brown M. Familial hypercholesterolemia. In: Scriver C, Baudet A, Sly W, et al, eds. The Metabolic Basis of Inherited Disease. New York, NY: McGraw-Hill; 2001: 2863-2913.
11. Kwiterovich, PO. Clinical and laboratory assessment of cardiovascular risk in children: Guidelines for screening, evaluation and treatment. J Clin Lipidol. 2008;2:248-266.
12. US Preventive Services Task Force. Lipid disorders in adults (cholesterol, dyslipidemia): Screening. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/lipid-disorders-in-adults-cholesterol-dyslipidemia-screening. Accessed July 6, 2015.
13. Hopkins PN, Toth PP, Ballantyne CM, et al; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S9-17.
14. US Preventive Services Task Force. Screening for lipid disorders in children: US Preventive Services Task Force recommendation statement. Pediatrics. 2007;120;e215-219.
15. American Academy of Family Physicians. Summary of recommendations for clinical preventive services. American Academy of Family Physicians Web site. Available at: http://www.aafp.org/dam/AAFP/documents/patient_care/clinical_recommendations/cps-recommendations.pdf. Accessed May 2, 2015.
16. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128:S213-56.
17. American Academy of Pediatrics. 2014 Recommendations for Pediatric Preventive Health Care. American Academy of Pediatrics Web site. Available at: http://pediatrics.aappublications.org/content/133/3/568.full.pdf+html. Accessed May 2, 2015.
18. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889-2934.
19. Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478-3490a.
20. Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423.
21. Daniels SR, Gidding SS, de Ferranti SD; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Pediatric aspects of familial hypercholesterolemias: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S30-37.
22. Robinson JG, Goldberg AC; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S18-29.
23. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158:526-534.
24. Eiland LS, Luttrell PK. Use of statins for dyslipidemia in the pediatric population. J Pediatr Pharmacol Ther. 2010;15:160-172.
25. O’Gorman CS, Higgins MF, O’Neill MB. Systematic review and metaanalysis of statins for heterozygous familial hypercholesterolemia in children: evaluation of cholesterol changes and side effects. Pediatr Cardiol. 2009;30:482-489.
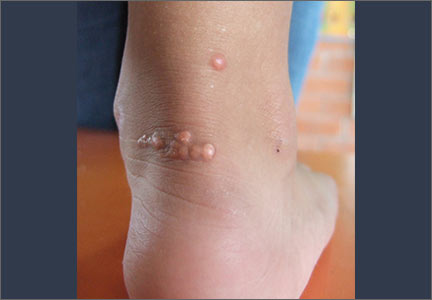
Targeting neuropathic pain: Consider these alternatives
Anticonvulsants, antidepressants, and opioids are the most frequently prescribed medications for neuropathic pain.1 But some patients are unable to tolerate the adverse effects of these drugs, and others achieve only partial pain relief. What can you offer them?
Combinations of prescription medications are generally considered more effective than monotherapy for painful peripheral neuropathy,1 but it is unclear which combinations are best. Alternative therapies—several of which have some evidence of safety and efficacy in treating peripheral neuropathy—are another option. Yet trials with alternative therapies, alone or in combination with prescription drugs, are rarely considered.
In fact, physicians are often unfamiliar with these therapies. Many are concerned about the absence of US Food and Drug Administration approval for alternative therapies and the variability in quality control associated with the lack of oversight, as well. Making recommendations about the duration of therapy also presents a challenge because most studies of supplements are relatively short. What’s more, alternative treatments are rarely covered by third-party payers.
Nonetheless, the therapies detailed in the text and TABLE2-12 that follow are generally well tolerated and appear to be safe. Adding them to your arsenal of therapeutic choices for patients with painful peripheral neuropathy may increase your ability to provide successful treatment.
Acetyl-L-carnitine (ALC)
ALC occurs naturally in the body as L-carnitine and acetyl-carnitine esters, which are converted to carnitines by intracellular enzymes and cell membrane transporters.2 ALC has been studied in patients with neuropathy associated with human immunodeficiency virus (HIV), cancer, and diabetes. Potential mechanisms of action include the correction of a deficiency that may be causing the neuropathy (which sometimes occurs in HIV-positive patients13 or those taking anticonvulsants14), a direct antioxidant effect, or an enhanced response to nerve growth factor.13
ALC can be given intramuscularly (IM) or orally in doses of 2000 to 3000 mg/d. In one randomized placebo-controlled trial (N=333), patients with diabetic neuropathy received 1000 mg IM followed by an oral dose of 2000 mg every day for a year.6 Mean pain scores decreased by 39%, with 67% of those receiving ALC vs 23% of those on placebo showing moderate to marked improvement.
In a pooled analysis (N=1257) of 2 randomized controlled trials (RCTs), patients with diabetes took 1000 mg ALC 3 times daily or placebo for a year.7 Cohort pain scores improved by 40% from baseline in the ALC group compared with a 24% improvement for those in the placebo group.
THE BOTTOM LINE ALC is well tolerated, with minor adverse effects such as headache and nausea reported.6,7 It should not be given to patients taking acenocoumarol or warfarin, however. A major interaction causing an elevated international normalized ratio has been found to occur when either agent is combined with L-carnitine2 and could theoretically occur with ALC, as well. No other drug-drug interactions have been documented.2
Alpha lipoic acid (ALA)
Both a fat- and a water-soluble vitamin that is usually obtained from the diet, ALA regenerates endogenous antioxidants like vitamins C and E and glutathione. It is this regenerative mechanism that it is believed to alleviate diabetic neuropathy.2 ALA 600 mg/d appears to be effective, although studies suggest that intravenous (IV) use is more effective than oral administration.
A meta-analysis of 4 RCTs (N=653), 2 with ALA taken orally and 2 involving IV administration, is a case in point.3 The pooled standardized mean difference estimated from all trials showed a reduction in total symptom scores of −2.26 (95% confidence interval [CI], −3.12 to −1.41; P=.00001), with 0 indicating no symptoms, 3 indicating severe symptoms, and a maximum score of 14.64 if all symptoms were severe and continuous. Subgroup analyses revealed a reduction of −1.78 (95% CI, −2.45 to −1.10; P=.00001) for oral ALA and −2.81 (95% CI, −4.16 to −1.46; P=.0001) for IV administration. Doses >600 mg/d did not improve efficacy, but did increase adverse effects such as nausea, vomiting, and dizziness.
In a multicenter RCT (N=460) of ALA 600 mg/d for 4 years, however, no improvement in the primary endpoint (a composite of neuropathy impairment scores and 7 neurophysiologic tests) was found.15 Although there was a statistically significant improvement in symptoms of neuropathy (−0.68 with ALA compared with +0.61 with placebo), the change was too small to be considered clinically significant.
ALA did slow the progression of neuropathy, however, with 29% of patients in the treatment group experiencing worsening symptoms compared with 38% of those on placebo. There was no difference in tolerability or discontinuation of treatment between the 2 groups.
A recent observational study (N=101) compared the efficacy of pregabalin, carbamazepine, and ALA over a 21-month period.4 Although those taking pregabalin had the best response rate, all 3 treatments led to significant improvement in the burning associated with neuropathic pain.

ALA 100 mg bid has been investigated as part of a 3-drug combination (with pregabalin 75 mg bid and methylcobalamin 750 mcg bid) compared with monotherapy (pregabalin 75 mg bid) in an open randomized study (N=30) for 12 weeks.16 While there was a trend toward improvement in pain relief, sleep interference, and nerve function in the combination therapy group, no statistically significant difference between the 2 groups was found. Nonetheless, more than a third (36%) had a global assessment rating of “excellent” vs one in 5 (20%) of those on pregabalin alone.
THE BOTTOM LINE Overall, ALA is well tolerated; the most common adverse effects are nausea and skin rash. IV administration is more effective than oral administration, but may cause nausea, headache, and an allergic reaction at the injection site.2 ALA does have the potential for an interaction with chemotherapy and thyroid hormone and may decrease the effectiveness of these therapies.2
B vitamins
Deficiencies of vitamin B1 (thiamine), B6 (pyridoxine), B12 (cyanocobalamin), and folate are known causes of neuropathy, and correcting them often improves or eliminates the symptoms.13 Vitamin B12 deficiency is commonly seen in patients taking metformin;14 these patients may benefit from supplementation with B12 1000 mcg/d.
Many of the B vitamins have been studied for treatment of neuropathy, but benfotiamine (a lipid-soluble form of thiamine) is thought to be the best option because it is better absorbed across cell membranes than other B vitamins.9 A Cochrane review found that benfotiamine alone may be effective for both diabetic and alcoholic neuropathy and that short-term use of higher doses of vitamin B complex (25 mg B1 or 320 mg benfotiamine + 50-720 mg B6 + 1000 mcg B12 daily) may reduce neuropathic pain.9
A randomized multicenter trial (N=214) found that adding a supplement containing L-methylfolate 3 mg, pyridoxal 5-phosphate 35 mg, and methylcobalamin 2 mg twice daily to other medications (eg, pregabalin, gabapentin, or duloxetine) improved symptoms of diabetic neuropathy.10 At 24 weeks, those receiving the combination therapy had a 26% decrease in pain symptoms compared with a 15% decrease for those on medication alone, with no significant adverse effects.
THE BOTTOM LINE Overall, vitamin B supplementation is well tolerated and appears to be more effective in relieving neuropathic pain than medication alone.9,14 But larger studies are needed before its efficacy in treating patients who do not have a deficiency can be established.
Capsaicin
Capsaicin, an ingredient found in peppers, works by binding to nociceptors to selectively stimulate afferent C fibers. This causes the release of substance P, a neurotransmitter that mediates pain, leading to its depletion and resulting in desensitization.2 Several meta-analyses and systematic reviews have found that topical capsaicin can be very effective, both as an adjunctive treatment and as monotherapy for neuropathic pain.11,17,18 The concentration used in the studies was 0.075% capsaicin cream, applied 3 to 4 times a day for 6 to 12 weeks, compared with placebo creams. In all categories studied, capsaicin was either statistically significant or trending in its favor, with the exception of adverse effects.
Capsaicin led to an improvement in daily activities and ability to sleep and a reduction in pain as measured with a visual analog scale and physician global evaluation.11,17,18
The most notable adverse effects were a burning sensation on the skin and coughing and sneezing caused by inhalation of dried cream. Although the adverse effects were expected to improve after 2 to 7 days of use, a significant number of participants withdrew from the study.
A 7-study meta-analysis showed the effectiveness of an 8% capsaicin patch for treatment of post-herpetic neuralgia and HIV-associated neuropathy.12 The patch, available only by prescription, was worn every day for 4 weeks (60 minutes daily for post-herpetic neuralgia and 30 minutes a day for HIV-associated neuropathy). The pooled results were statistically significant, but the patch was less effective for patients ages 18 to 40 years and for those of Asian descent. It can be used with other analgesics or as monotherapy, with few adverse reactions.12,19
THE BOTTOM LINE Since capsaicin is a topical medication, there are no relevant drug-drug interactions. Patients should be cautioned to wash their hands after application, however, and to avoid contact with eyes and open wounds.

Gamma linolenic acid (GLA)
Also known as evening primrose oil, GLA is an omega-6 fatty acid that’s an important constituent of neuronal cell membranes—and believed to decrease neuropathic pain by having some anti-inflammatory effects.2 This suggests that therapy with GLA has the potential to improve neuronal phospholipid structure and microcirculation.2
Two placebo-controlled trials (N=22,111) showed improvement in pain scores and multiple neurophysiologic assessments in patients with diabetes treated with GLA (360-480 mg/d).20,21 The treatment was well tolerated, but the beneficial effect was more pronounced in those with less severe diabetes.
THE BOTTOM LINE The dose of GLA studied (8 to 12 capsules daily) could lead to problems with patient adherence. In addition, GLA should be used with caution in patients who are taking antiplatelet medication or have seizure disorders.2
Magnesium (Mg)
Mg is highly involved in multiple enzyme systems throughout the body. Although it is very well absorbed from dietary sources,2 patients with diabetes, liver disease, and hormonal imbalances, as well as the elderly, are often deficient in Mg. It is unclear how this affects peripheral neuropathy.13
Mg may have an antinociceptive effect by decreasing intracellular calcium influx and antagonizing N-methyl-D-aspartate receptors and associated nerve signaling.22 A small RCT (N=80) showed Mg to decrease the severity of neuropathic back pain.22 Patients received Mg sulfate 1 g IV, given over 4 hours, every day for 2 weeks. The infusion was then replaced with Mg oxide 400 mg plus Mg gluconate 100 mg, taken orally twice daily for 4 weeks. An improvement in mean pain score was seen as early as 2 weeks, and scores had decreased by 2.8 points (on a 0-10-point scale) at 6 months.
Another small RCT (N=45) gave patients with neuropathy of postherpetic, traumatic, or surgical (but not diabetic) origin Mg chloride 838 mg orally 3 times a day for 4 weeks.23 The supplement was taken with meals. Mean pain scores in the treatment group decreased by 3 points, but this was not significantly different from the improvement seen in those on placebo.
In a similar study, patients (N=110) with type 1 diabetes and a normal serum Mg but an insufficiency as measured by erythrocyte Mg were given Mg gluconate 300 mg or placebo daily for 5 years.8 The supplement slowed the progression of peripheral neuropathy (only 12% of those receiving Mg gluconate experienced a significant worsening of symptoms over the course of the study, compared with 61% of those in the placebo group), but in most cases, it did not lead to an improvement.
No consistent approach to Mg supplementation has been studied, which makes recommending a particular route, dose, or formulation challenging. There is evidence that oral Mg, particularly in the form of Mg oxide, can cause diarrhea, especially in doses >350 mg/d. Mg gluconate and Mg chloride are better tolerated; Mg carbonate should be avoided due to poor oral absorption.2
BOTTOM LINE Mg supplementation appears to slow the progression of diabetic peripheral neuropathy, but is unsafe for patients with renal dysfunction, cardiac conduction abnormalities, or elevated Mg levels.2 Caution is required, too, when considering Mg supplementation for patients taking anticoagulants, bisphosphonates, digoxin, potassium-sparing diuretics, or tetracycline antibiotics.2
CORRESPONDENCE
Mary Onysko, PharmD, BCPS, University of Wyoming, School of Pharmacy Health Sciences Center, Room 292, 1000 E. University Avenue, Laramie, WY 82071; [email protected]
1. Chaparro LE, Wiffen PJ, Moore RA, et al. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst Rev. 2012:(7):CD008943.
2. Natural Medicines Comprehensive Database. Natural Medicines Comprehensive Database Web site. Available at: http://naturaldatabase.therapeuticresearch.com. Accessed January 4, 2015.
3. Mijnhout GS, Kollen BJ, Alkhalaf A, et al. Alpha lipoic acid for symptomatic peripheral neuropathy in patients with diabetes: a meta-analysis of randomized controlled trials. Int J Endocrinol. 2012;2012:456279.
4. Patel N, Mishra V, Patel P, et al. A study of the use of carbamazepine, pregabalin and alpha lipoic acid in patients of diabetic neuropathy. J Diabetes Metab Disord. 2014;13:62.
5. Bertolotto F, Massone A. Combination of alpha lipoic acid and superoxide dismutase leads to physiological and symptomatic improvements in diabetic neuropathy. Drugs R D. 2012;12:29-34.
6. De Grandis D, Minardi C. Acetyl-L-carnitine (levacecarnine) in the treatment of diabetic neuropathy. A long-term, randomised, double-blind, placebo-controlled study. Drugs R D. 2002;3:223-231.
7. Sima AA, Calvani M, Mehra M, et al; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care. 2005;28:89-94.
8. De Leeuw, Engelen W, De Block C, et al. Long term magnesium supplementation influences favourably the natural evolution of neuropathy in Mg-depleted type 1 diabetic patients (T1dm). Magnes Res. 2004;17:109-114.
9. Ang CD, Alviar MJM, Dans AL, et al. Vitamin B for treating peripheral neuropathy. Cochrane Database Syst Rev. 2008;(3):CD004573.
10. Fonseca VA, Lavery LA, Thethi TK, et al. Metanx in type 2 diabetes
with peripheral neuropathy: a randomized trial. Am J Med. 2013;126:141-149.
11. Mason L, Moore RA, Derry S, et al. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991.
12. Mou J, Paillard F, Turnbull B, et al. Efficacy of Qutenza® (capsaicin) 8% patch for neuropathic pain: a meta-analysis of the Qutenza Clinical Trials Database. Pain. 2013;154:1632-1639.
13. Head KA. Peripheral neuropathy: pathogenic mechanisms and alternative therapies. Altern Med Rev. 2006; 11:294-329.
14. Miranda-Massari JR, Gonzalez MJ, Jimenez FJ, et al. Metabolic correction in the management of diabetic peripheral neuropathy: improving clinical results beyond symptom control. Curr Clin Pharmacol. 2011; 6:260-273.
15. Ziegler D, Low PA, Litchy WJ, et al. Efficacy and safety of antioxidant treatment with a-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care. 2011;34:2054-2060.
16. Vasudevan D, Naik MM, Mukaddam QI. Efficacy and safety of methylcobalamin, alpha lipoic acid and pregabalin combination versus pregabalin monotherapy in improving pain and nerve conduction velocity in type 2 diabetes associated impaired peripheral neuropathic condition. [MAINTAIN]: Results of a pilot study. Ann Indian Acad Neurol. 2014;17:19-24.
17. Halat KM, Dennehy CE. Botanicals and dietary supplements in diabetic peripheral neuropathy. J Am Board Fam Pract. 2003;16:47-57.
18. Donofrio P, Walker F, Hunt V, et al. Treatment of painful diabetic neuropathy with topical capsaicin: A multicenter, double-blind, vehicle-controlled study. Arch Int Med. 1991;151:2225-2229.
19. Derry S, Rice ASC, Cole P, et al. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2013;(2):CD007393.
20. Keen H, Payan J, Allawi J, et al. Treatment of diabetic neuropathy with gamma-linolenic acid. The gamma-Linolenic Acid Multicenter Trial Group. Diabetes Care. 1993;16:8-15.
21. Jamal GA, Carmichael H. The effect of gamma linolenic acid on human diabetic peripheral neuropathy: a double blind placebo controlled trial. Diabetic Med. 1990;7:319-323.
22. Yousef AA, Al-deeb AE. A double-blinded randomised controlled study of the value of sequential intravenous and oral magnesium therapy in patients with chronic low back pain with a neuropathic component. Anaesthesia. 2013;68:260-266.
23. Pickering G, Morel V, Simen E. Oral magnesium treatment in patients with neuropathic pain: a randomized clinical trial. Magnes Res. 2011;24:28-35.
Anticonvulsants, antidepressants, and opioids are the most frequently prescribed medications for neuropathic pain.1 But some patients are unable to tolerate the adverse effects of these drugs, and others achieve only partial pain relief. What can you offer them?
Combinations of prescription medications are generally considered more effective than monotherapy for painful peripheral neuropathy,1 but it is unclear which combinations are best. Alternative therapies—several of which have some evidence of safety and efficacy in treating peripheral neuropathy—are another option. Yet trials with alternative therapies, alone or in combination with prescription drugs, are rarely considered.
In fact, physicians are often unfamiliar with these therapies. Many are concerned about the absence of US Food and Drug Administration approval for alternative therapies and the variability in quality control associated with the lack of oversight, as well. Making recommendations about the duration of therapy also presents a challenge because most studies of supplements are relatively short. What’s more, alternative treatments are rarely covered by third-party payers.
Nonetheless, the therapies detailed in the text and TABLE2-12 that follow are generally well tolerated and appear to be safe. Adding them to your arsenal of therapeutic choices for patients with painful peripheral neuropathy may increase your ability to provide successful treatment.
Acetyl-L-carnitine (ALC)
ALC occurs naturally in the body as L-carnitine and acetyl-carnitine esters, which are converted to carnitines by intracellular enzymes and cell membrane transporters.2 ALC has been studied in patients with neuropathy associated with human immunodeficiency virus (HIV), cancer, and diabetes. Potential mechanisms of action include the correction of a deficiency that may be causing the neuropathy (which sometimes occurs in HIV-positive patients13 or those taking anticonvulsants14), a direct antioxidant effect, or an enhanced response to nerve growth factor.13
ALC can be given intramuscularly (IM) or orally in doses of 2000 to 3000 mg/d. In one randomized placebo-controlled trial (N=333), patients with diabetic neuropathy received 1000 mg IM followed by an oral dose of 2000 mg every day for a year.6 Mean pain scores decreased by 39%, with 67% of those receiving ALC vs 23% of those on placebo showing moderate to marked improvement.
In a pooled analysis (N=1257) of 2 randomized controlled trials (RCTs), patients with diabetes took 1000 mg ALC 3 times daily or placebo for a year.7 Cohort pain scores improved by 40% from baseline in the ALC group compared with a 24% improvement for those in the placebo group.
THE BOTTOM LINE ALC is well tolerated, with minor adverse effects such as headache and nausea reported.6,7 It should not be given to patients taking acenocoumarol or warfarin, however. A major interaction causing an elevated international normalized ratio has been found to occur when either agent is combined with L-carnitine2 and could theoretically occur with ALC, as well. No other drug-drug interactions have been documented.2
Alpha lipoic acid (ALA)
Both a fat- and a water-soluble vitamin that is usually obtained from the diet, ALA regenerates endogenous antioxidants like vitamins C and E and glutathione. It is this regenerative mechanism that it is believed to alleviate diabetic neuropathy.2 ALA 600 mg/d appears to be effective, although studies suggest that intravenous (IV) use is more effective than oral administration.
A meta-analysis of 4 RCTs (N=653), 2 with ALA taken orally and 2 involving IV administration, is a case in point.3 The pooled standardized mean difference estimated from all trials showed a reduction in total symptom scores of −2.26 (95% confidence interval [CI], −3.12 to −1.41; P=.00001), with 0 indicating no symptoms, 3 indicating severe symptoms, and a maximum score of 14.64 if all symptoms were severe and continuous. Subgroup analyses revealed a reduction of −1.78 (95% CI, −2.45 to −1.10; P=.00001) for oral ALA and −2.81 (95% CI, −4.16 to −1.46; P=.0001) for IV administration. Doses >600 mg/d did not improve efficacy, but did increase adverse effects such as nausea, vomiting, and dizziness.
In a multicenter RCT (N=460) of ALA 600 mg/d for 4 years, however, no improvement in the primary endpoint (a composite of neuropathy impairment scores and 7 neurophysiologic tests) was found.15 Although there was a statistically significant improvement in symptoms of neuropathy (−0.68 with ALA compared with +0.61 with placebo), the change was too small to be considered clinically significant.
ALA did slow the progression of neuropathy, however, with 29% of patients in the treatment group experiencing worsening symptoms compared with 38% of those on placebo. There was no difference in tolerability or discontinuation of treatment between the 2 groups.
A recent observational study (N=101) compared the efficacy of pregabalin, carbamazepine, and ALA over a 21-month period.4 Although those taking pregabalin had the best response rate, all 3 treatments led to significant improvement in the burning associated with neuropathic pain.
ALA 100 mg bid has been investigated as part of a 3-drug combination (with pregabalin 75 mg bid and methylcobalamin 750 mcg bid) compared with monotherapy (pregabalin 75 mg bid) in an open randomized study (N=30) for 12 weeks.16 While there was a trend toward improvement in pain relief, sleep interference, and nerve function in the combination therapy group, no statistically significant difference between the 2 groups was found. Nonetheless, more than a third (36%) had a global assessment rating of “excellent” vs one in 5 (20%) of those on pregabalin alone.
THE BOTTOM LINE Overall, ALA is well tolerated; the most common adverse effects are nausea and skin rash. IV administration is more effective than oral administration, but may cause nausea, headache, and an allergic reaction at the injection site.2 ALA does have the potential for an interaction with chemotherapy and thyroid hormone and may decrease the effectiveness of these therapies.2
B vitamins
Deficiencies of vitamin B1 (thiamine), B6 (pyridoxine), B12 (cyanocobalamin), and folate are known causes of neuropathy, and correcting them often improves or eliminates the symptoms.13 Vitamin B12 deficiency is commonly seen in patients taking metformin;14 these patients may benefit from supplementation with B12 1000 mcg/d.
Many of the B vitamins have been studied for treatment of neuropathy, but benfotiamine (a lipid-soluble form of thiamine) is thought to be the best option because it is better absorbed across cell membranes than other B vitamins.9 A Cochrane review found that benfotiamine alone may be effective for both diabetic and alcoholic neuropathy and that short-term use of higher doses of vitamin B complex (25 mg B1 or 320 mg benfotiamine + 50-720 mg B6 + 1000 mcg B12 daily) may reduce neuropathic pain.9
A randomized multicenter trial (N=214) found that adding a supplement containing L-methylfolate 3 mg, pyridoxal 5-phosphate 35 mg, and methylcobalamin 2 mg twice daily to other medications (eg, pregabalin, gabapentin, or duloxetine) improved symptoms of diabetic neuropathy.10 At 24 weeks, those receiving the combination therapy had a 26% decrease in pain symptoms compared with a 15% decrease for those on medication alone, with no significant adverse effects.
THE BOTTOM LINE Overall, vitamin B supplementation is well tolerated and appears to be more effective in relieving neuropathic pain than medication alone.9,14 But larger studies are needed before its efficacy in treating patients who do not have a deficiency can be established.
Capsaicin
Capsaicin, an ingredient found in peppers, works by binding to nociceptors to selectively stimulate afferent C fibers. This causes the release of substance P, a neurotransmitter that mediates pain, leading to its depletion and resulting in desensitization.2 Several meta-analyses and systematic reviews have found that topical capsaicin can be very effective, both as an adjunctive treatment and as monotherapy for neuropathic pain.11,17,18 The concentration used in the studies was 0.075% capsaicin cream, applied 3 to 4 times a day for 6 to 12 weeks, compared with placebo creams. In all categories studied, capsaicin was either statistically significant or trending in its favor, with the exception of adverse effects.
Capsaicin led to an improvement in daily activities and ability to sleep and a reduction in pain as measured with a visual analog scale and physician global evaluation.11,17,18
The most notable adverse effects were a burning sensation on the skin and coughing and sneezing caused by inhalation of dried cream. Although the adverse effects were expected to improve after 2 to 7 days of use, a significant number of participants withdrew from the study.
A 7-study meta-analysis showed the effectiveness of an 8% capsaicin patch for treatment of post-herpetic neuralgia and HIV-associated neuropathy.12 The patch, available only by prescription, was worn every day for 4 weeks (60 minutes daily for post-herpetic neuralgia and 30 minutes a day for HIV-associated neuropathy). The pooled results were statistically significant, but the patch was less effective for patients ages 18 to 40 years and for those of Asian descent. It can be used with other analgesics or as monotherapy, with few adverse reactions.12,19
THE BOTTOM LINE Since capsaicin is a topical medication, there are no relevant drug-drug interactions. Patients should be cautioned to wash their hands after application, however, and to avoid contact with eyes and open wounds.
Gamma linolenic acid (GLA)
Also known as evening primrose oil, GLA is an omega-6 fatty acid that’s an important constituent of neuronal cell membranes—and believed to decrease neuropathic pain by having some anti-inflammatory effects.2 This suggests that therapy with GLA has the potential to improve neuronal phospholipid structure and microcirculation.2
Two placebo-controlled trials (N=22,111) showed improvement in pain scores and multiple neurophysiologic assessments in patients with diabetes treated with GLA (360-480 mg/d).20,21 The treatment was well tolerated, but the beneficial effect was more pronounced in those with less severe diabetes.
THE BOTTOM LINE The dose of GLA studied (8 to 12 capsules daily) could lead to problems with patient adherence. In addition, GLA should be used with caution in patients who are taking antiplatelet medication or have seizure disorders.2
Magnesium (Mg)
Mg is highly involved in multiple enzyme systems throughout the body. Although it is very well absorbed from dietary sources,2 patients with diabetes, liver disease, and hormonal imbalances, as well as the elderly, are often deficient in Mg. It is unclear how this affects peripheral neuropathy.13
Mg may have an antinociceptive effect by decreasing intracellular calcium influx and antagonizing N-methyl-D-aspartate receptors and associated nerve signaling.22 A small RCT (N=80) showed Mg to decrease the severity of neuropathic back pain.22 Patients received Mg sulfate 1 g IV, given over 4 hours, every day for 2 weeks. The infusion was then replaced with Mg oxide 400 mg plus Mg gluconate 100 mg, taken orally twice daily for 4 weeks. An improvement in mean pain score was seen as early as 2 weeks, and scores had decreased by 2.8 points (on a 0-10-point scale) at 6 months.
Another small RCT (N=45) gave patients with neuropathy of postherpetic, traumatic, or surgical (but not diabetic) origin Mg chloride 838 mg orally 3 times a day for 4 weeks.23 The supplement was taken with meals. Mean pain scores in the treatment group decreased by 3 points, but this was not significantly different from the improvement seen in those on placebo.
In a similar study, patients (N=110) with type 1 diabetes and a normal serum Mg but an insufficiency as measured by erythrocyte Mg were given Mg gluconate 300 mg or placebo daily for 5 years.8 The supplement slowed the progression of peripheral neuropathy (only 12% of those receiving Mg gluconate experienced a significant worsening of symptoms over the course of the study, compared with 61% of those in the placebo group), but in most cases, it did not lead to an improvement.
No consistent approach to Mg supplementation has been studied, which makes recommending a particular route, dose, or formulation challenging. There is evidence that oral Mg, particularly in the form of Mg oxide, can cause diarrhea, especially in doses >350 mg/d. Mg gluconate and Mg chloride are better tolerated; Mg carbonate should be avoided due to poor oral absorption.2
BOTTOM LINE Mg supplementation appears to slow the progression of diabetic peripheral neuropathy, but is unsafe for patients with renal dysfunction, cardiac conduction abnormalities, or elevated Mg levels.2 Caution is required, too, when considering Mg supplementation for patients taking anticoagulants, bisphosphonates, digoxin, potassium-sparing diuretics, or tetracycline antibiotics.2
CORRESPONDENCE
Mary Onysko, PharmD, BCPS, University of Wyoming, School of Pharmacy Health Sciences Center, Room 292, 1000 E. University Avenue, Laramie, WY 82071; [email protected]
Anticonvulsants, antidepressants, and opioids are the most frequently prescribed medications for neuropathic pain.1 But some patients are unable to tolerate the adverse effects of these drugs, and others achieve only partial pain relief. What can you offer them?
Combinations of prescription medications are generally considered more effective than monotherapy for painful peripheral neuropathy,1 but it is unclear which combinations are best. Alternative therapies—several of which have some evidence of safety and efficacy in treating peripheral neuropathy—are another option. Yet trials with alternative therapies, alone or in combination with prescription drugs, are rarely considered.
In fact, physicians are often unfamiliar with these therapies. Many are concerned about the absence of US Food and Drug Administration approval for alternative therapies and the variability in quality control associated with the lack of oversight, as well. Making recommendations about the duration of therapy also presents a challenge because most studies of supplements are relatively short. What’s more, alternative treatments are rarely covered by third-party payers.
Nonetheless, the therapies detailed in the text and TABLE2-12 that follow are generally well tolerated and appear to be safe. Adding them to your arsenal of therapeutic choices for patients with painful peripheral neuropathy may increase your ability to provide successful treatment.
Acetyl-L-carnitine (ALC)
ALC occurs naturally in the body as L-carnitine and acetyl-carnitine esters, which are converted to carnitines by intracellular enzymes and cell membrane transporters.2 ALC has been studied in patients with neuropathy associated with human immunodeficiency virus (HIV), cancer, and diabetes. Potential mechanisms of action include the correction of a deficiency that may be causing the neuropathy (which sometimes occurs in HIV-positive patients13 or those taking anticonvulsants14), a direct antioxidant effect, or an enhanced response to nerve growth factor.13
ALC can be given intramuscularly (IM) or orally in doses of 2000 to 3000 mg/d. In one randomized placebo-controlled trial (N=333), patients with diabetic neuropathy received 1000 mg IM followed by an oral dose of 2000 mg every day for a year.6 Mean pain scores decreased by 39%, with 67% of those receiving ALC vs 23% of those on placebo showing moderate to marked improvement.
In a pooled analysis (N=1257) of 2 randomized controlled trials (RCTs), patients with diabetes took 1000 mg ALC 3 times daily or placebo for a year.7 Cohort pain scores improved by 40% from baseline in the ALC group compared with a 24% improvement for those in the placebo group.
THE BOTTOM LINE ALC is well tolerated, with minor adverse effects such as headache and nausea reported.6,7 It should not be given to patients taking acenocoumarol or warfarin, however. A major interaction causing an elevated international normalized ratio has been found to occur when either agent is combined with L-carnitine2 and could theoretically occur with ALC, as well. No other drug-drug interactions have been documented.2
Alpha lipoic acid (ALA)
Both a fat- and a water-soluble vitamin that is usually obtained from the diet, ALA regenerates endogenous antioxidants like vitamins C and E and glutathione. It is this regenerative mechanism that it is believed to alleviate diabetic neuropathy.2 ALA 600 mg/d appears to be effective, although studies suggest that intravenous (IV) use is more effective than oral administration.
A meta-analysis of 4 RCTs (N=653), 2 with ALA taken orally and 2 involving IV administration, is a case in point.3 The pooled standardized mean difference estimated from all trials showed a reduction in total symptom scores of −2.26 (95% confidence interval [CI], −3.12 to −1.41; P=.00001), with 0 indicating no symptoms, 3 indicating severe symptoms, and a maximum score of 14.64 if all symptoms were severe and continuous. Subgroup analyses revealed a reduction of −1.78 (95% CI, −2.45 to −1.10; P=.00001) for oral ALA and −2.81 (95% CI, −4.16 to −1.46; P=.0001) for IV administration. Doses >600 mg/d did not improve efficacy, but did increase adverse effects such as nausea, vomiting, and dizziness.
In a multicenter RCT (N=460) of ALA 600 mg/d for 4 years, however, no improvement in the primary endpoint (a composite of neuropathy impairment scores and 7 neurophysiologic tests) was found.15 Although there was a statistically significant improvement in symptoms of neuropathy (−0.68 with ALA compared with +0.61 with placebo), the change was too small to be considered clinically significant.
ALA did slow the progression of neuropathy, however, with 29% of patients in the treatment group experiencing worsening symptoms compared with 38% of those on placebo. There was no difference in tolerability or discontinuation of treatment between the 2 groups.
A recent observational study (N=101) compared the efficacy of pregabalin, carbamazepine, and ALA over a 21-month period.4 Although those taking pregabalin had the best response rate, all 3 treatments led to significant improvement in the burning associated with neuropathic pain.
ALA 100 mg bid has been investigated as part of a 3-drug combination (with pregabalin 75 mg bid and methylcobalamin 750 mcg bid) compared with monotherapy (pregabalin 75 mg bid) in an open randomized study (N=30) for 12 weeks.16 While there was a trend toward improvement in pain relief, sleep interference, and nerve function in the combination therapy group, no statistically significant difference between the 2 groups was found. Nonetheless, more than a third (36%) had a global assessment rating of “excellent” vs one in 5 (20%) of those on pregabalin alone.
THE BOTTOM LINE Overall, ALA is well tolerated; the most common adverse effects are nausea and skin rash. IV administration is more effective than oral administration, but may cause nausea, headache, and an allergic reaction at the injection site.2 ALA does have the potential for an interaction with chemotherapy and thyroid hormone and may decrease the effectiveness of these therapies.2
B vitamins
Deficiencies of vitamin B1 (thiamine), B6 (pyridoxine), B12 (cyanocobalamin), and folate are known causes of neuropathy, and correcting them often improves or eliminates the symptoms.13 Vitamin B12 deficiency is commonly seen in patients taking metformin;14 these patients may benefit from supplementation with B12 1000 mcg/d.
Many of the B vitamins have been studied for treatment of neuropathy, but benfotiamine (a lipid-soluble form of thiamine) is thought to be the best option because it is better absorbed across cell membranes than other B vitamins.9 A Cochrane review found that benfotiamine alone may be effective for both diabetic and alcoholic neuropathy and that short-term use of higher doses of vitamin B complex (25 mg B1 or 320 mg benfotiamine + 50-720 mg B6 + 1000 mcg B12 daily) may reduce neuropathic pain.9
A randomized multicenter trial (N=214) found that adding a supplement containing L-methylfolate 3 mg, pyridoxal 5-phosphate 35 mg, and methylcobalamin 2 mg twice daily to other medications (eg, pregabalin, gabapentin, or duloxetine) improved symptoms of diabetic neuropathy.10 At 24 weeks, those receiving the combination therapy had a 26% decrease in pain symptoms compared with a 15% decrease for those on medication alone, with no significant adverse effects.
THE BOTTOM LINE Overall, vitamin B supplementation is well tolerated and appears to be more effective in relieving neuropathic pain than medication alone.9,14 But larger studies are needed before its efficacy in treating patients who do not have a deficiency can be established.
Capsaicin
Capsaicin, an ingredient found in peppers, works by binding to nociceptors to selectively stimulate afferent C fibers. This causes the release of substance P, a neurotransmitter that mediates pain, leading to its depletion and resulting in desensitization.2 Several meta-analyses and systematic reviews have found that topical capsaicin can be very effective, both as an adjunctive treatment and as monotherapy for neuropathic pain.11,17,18 The concentration used in the studies was 0.075% capsaicin cream, applied 3 to 4 times a day for 6 to 12 weeks, compared with placebo creams. In all categories studied, capsaicin was either statistically significant or trending in its favor, with the exception of adverse effects.
Capsaicin led to an improvement in daily activities and ability to sleep and a reduction in pain as measured with a visual analog scale and physician global evaluation.11,17,18
The most notable adverse effects were a burning sensation on the skin and coughing and sneezing caused by inhalation of dried cream. Although the adverse effects were expected to improve after 2 to 7 days of use, a significant number of participants withdrew from the study.
A 7-study meta-analysis showed the effectiveness of an 8% capsaicin patch for treatment of post-herpetic neuralgia and HIV-associated neuropathy.12 The patch, available only by prescription, was worn every day for 4 weeks (60 minutes daily for post-herpetic neuralgia and 30 minutes a day for HIV-associated neuropathy). The pooled results were statistically significant, but the patch was less effective for patients ages 18 to 40 years and for those of Asian descent. It can be used with other analgesics or as monotherapy, with few adverse reactions.12,19
THE BOTTOM LINE Since capsaicin is a topical medication, there are no relevant drug-drug interactions. Patients should be cautioned to wash their hands after application, however, and to avoid contact with eyes and open wounds.
Gamma linolenic acid (GLA)
Also known as evening primrose oil, GLA is an omega-6 fatty acid that’s an important constituent of neuronal cell membranes—and believed to decrease neuropathic pain by having some anti-inflammatory effects.2 This suggests that therapy with GLA has the potential to improve neuronal phospholipid structure and microcirculation.2
Two placebo-controlled trials (N=22,111) showed improvement in pain scores and multiple neurophysiologic assessments in patients with diabetes treated with GLA (360-480 mg/d).20,21 The treatment was well tolerated, but the beneficial effect was more pronounced in those with less severe diabetes.
THE BOTTOM LINE The dose of GLA studied (8 to 12 capsules daily) could lead to problems with patient adherence. In addition, GLA should be used with caution in patients who are taking antiplatelet medication or have seizure disorders.2
Magnesium (Mg)
Mg is highly involved in multiple enzyme systems throughout the body. Although it is very well absorbed from dietary sources,2 patients with diabetes, liver disease, and hormonal imbalances, as well as the elderly, are often deficient in Mg. It is unclear how this affects peripheral neuropathy.13
Mg may have an antinociceptive effect by decreasing intracellular calcium influx and antagonizing N-methyl-D-aspartate receptors and associated nerve signaling.22 A small RCT (N=80) showed Mg to decrease the severity of neuropathic back pain.22 Patients received Mg sulfate 1 g IV, given over 4 hours, every day for 2 weeks. The infusion was then replaced with Mg oxide 400 mg plus Mg gluconate 100 mg, taken orally twice daily for 4 weeks. An improvement in mean pain score was seen as early as 2 weeks, and scores had decreased by 2.8 points (on a 0-10-point scale) at 6 months.
Another small RCT (N=45) gave patients with neuropathy of postherpetic, traumatic, or surgical (but not diabetic) origin Mg chloride 838 mg orally 3 times a day for 4 weeks.23 The supplement was taken with meals. Mean pain scores in the treatment group decreased by 3 points, but this was not significantly different from the improvement seen in those on placebo.
In a similar study, patients (N=110) with type 1 diabetes and a normal serum Mg but an insufficiency as measured by erythrocyte Mg were given Mg gluconate 300 mg or placebo daily for 5 years.8 The supplement slowed the progression of peripheral neuropathy (only 12% of those receiving Mg gluconate experienced a significant worsening of symptoms over the course of the study, compared with 61% of those in the placebo group), but in most cases, it did not lead to an improvement.
No consistent approach to Mg supplementation has been studied, which makes recommending a particular route, dose, or formulation challenging. There is evidence that oral Mg, particularly in the form of Mg oxide, can cause diarrhea, especially in doses >350 mg/d. Mg gluconate and Mg chloride are better tolerated; Mg carbonate should be avoided due to poor oral absorption.2
BOTTOM LINE Mg supplementation appears to slow the progression of diabetic peripheral neuropathy, but is unsafe for patients with renal dysfunction, cardiac conduction abnormalities, or elevated Mg levels.2 Caution is required, too, when considering Mg supplementation for patients taking anticoagulants, bisphosphonates, digoxin, potassium-sparing diuretics, or tetracycline antibiotics.2
CORRESPONDENCE
Mary Onysko, PharmD, BCPS, University of Wyoming, School of Pharmacy Health Sciences Center, Room 292, 1000 E. University Avenue, Laramie, WY 82071; [email protected]
1. Chaparro LE, Wiffen PJ, Moore RA, et al. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst Rev. 2012:(7):CD008943.
2. Natural Medicines Comprehensive Database. Natural Medicines Comprehensive Database Web site. Available at: http://naturaldatabase.therapeuticresearch.com. Accessed January 4, 2015.
3. Mijnhout GS, Kollen BJ, Alkhalaf A, et al. Alpha lipoic acid for symptomatic peripheral neuropathy in patients with diabetes: a meta-analysis of randomized controlled trials. Int J Endocrinol. 2012;2012:456279.
4. Patel N, Mishra V, Patel P, et al. A study of the use of carbamazepine, pregabalin and alpha lipoic acid in patients of diabetic neuropathy. J Diabetes Metab Disord. 2014;13:62.
5. Bertolotto F, Massone A. Combination of alpha lipoic acid and superoxide dismutase leads to physiological and symptomatic improvements in diabetic neuropathy. Drugs R D. 2012;12:29-34.
6. De Grandis D, Minardi C. Acetyl-L-carnitine (levacecarnine) in the treatment of diabetic neuropathy. A long-term, randomised, double-blind, placebo-controlled study. Drugs R D. 2002;3:223-231.
7. Sima AA, Calvani M, Mehra M, et al; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care. 2005;28:89-94.
8. De Leeuw, Engelen W, De Block C, et al. Long term magnesium supplementation influences favourably the natural evolution of neuropathy in Mg-depleted type 1 diabetic patients (T1dm). Magnes Res. 2004;17:109-114.
9. Ang CD, Alviar MJM, Dans AL, et al. Vitamin B for treating peripheral neuropathy. Cochrane Database Syst Rev. 2008;(3):CD004573.
10. Fonseca VA, Lavery LA, Thethi TK, et al. Metanx in type 2 diabetes
with peripheral neuropathy: a randomized trial. Am J Med. 2013;126:141-149.
11. Mason L, Moore RA, Derry S, et al. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991.
12. Mou J, Paillard F, Turnbull B, et al. Efficacy of Qutenza® (capsaicin) 8% patch for neuropathic pain: a meta-analysis of the Qutenza Clinical Trials Database. Pain. 2013;154:1632-1639.
13. Head KA. Peripheral neuropathy: pathogenic mechanisms and alternative therapies. Altern Med Rev. 2006; 11:294-329.
14. Miranda-Massari JR, Gonzalez MJ, Jimenez FJ, et al. Metabolic correction in the management of diabetic peripheral neuropathy: improving clinical results beyond symptom control. Curr Clin Pharmacol. 2011; 6:260-273.
15. Ziegler D, Low PA, Litchy WJ, et al. Efficacy and safety of antioxidant treatment with a-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care. 2011;34:2054-2060.
16. Vasudevan D, Naik MM, Mukaddam QI. Efficacy and safety of methylcobalamin, alpha lipoic acid and pregabalin combination versus pregabalin monotherapy in improving pain and nerve conduction velocity in type 2 diabetes associated impaired peripheral neuropathic condition. [MAINTAIN]: Results of a pilot study. Ann Indian Acad Neurol. 2014;17:19-24.
17. Halat KM, Dennehy CE. Botanicals and dietary supplements in diabetic peripheral neuropathy. J Am Board Fam Pract. 2003;16:47-57.
18. Donofrio P, Walker F, Hunt V, et al. Treatment of painful diabetic neuropathy with topical capsaicin: A multicenter, double-blind, vehicle-controlled study. Arch Int Med. 1991;151:2225-2229.
19. Derry S, Rice ASC, Cole P, et al. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2013;(2):CD007393.
20. Keen H, Payan J, Allawi J, et al. Treatment of diabetic neuropathy with gamma-linolenic acid. The gamma-Linolenic Acid Multicenter Trial Group. Diabetes Care. 1993;16:8-15.
21. Jamal GA, Carmichael H. The effect of gamma linolenic acid on human diabetic peripheral neuropathy: a double blind placebo controlled trial. Diabetic Med. 1990;7:319-323.
22. Yousef AA, Al-deeb AE. A double-blinded randomised controlled study of the value of sequential intravenous and oral magnesium therapy in patients with chronic low back pain with a neuropathic component. Anaesthesia. 2013;68:260-266.
23. Pickering G, Morel V, Simen E. Oral magnesium treatment in patients with neuropathic pain: a randomized clinical trial. Magnes Res. 2011;24:28-35.
1. Chaparro LE, Wiffen PJ, Moore RA, et al. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst Rev. 2012:(7):CD008943.
2. Natural Medicines Comprehensive Database. Natural Medicines Comprehensive Database Web site. Available at: http://naturaldatabase.therapeuticresearch.com. Accessed January 4, 2015.
3. Mijnhout GS, Kollen BJ, Alkhalaf A, et al. Alpha lipoic acid for symptomatic peripheral neuropathy in patients with diabetes: a meta-analysis of randomized controlled trials. Int J Endocrinol. 2012;2012:456279.
4. Patel N, Mishra V, Patel P, et al. A study of the use of carbamazepine, pregabalin and alpha lipoic acid in patients of diabetic neuropathy. J Diabetes Metab Disord. 2014;13:62.
5. Bertolotto F, Massone A. Combination of alpha lipoic acid and superoxide dismutase leads to physiological and symptomatic improvements in diabetic neuropathy. Drugs R D. 2012;12:29-34.
6. De Grandis D, Minardi C. Acetyl-L-carnitine (levacecarnine) in the treatment of diabetic neuropathy. A long-term, randomised, double-blind, placebo-controlled study. Drugs R D. 2002;3:223-231.
7. Sima AA, Calvani M, Mehra M, et al; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care. 2005;28:89-94.
8. De Leeuw, Engelen W, De Block C, et al. Long term magnesium supplementation influences favourably the natural evolution of neuropathy in Mg-depleted type 1 diabetic patients (T1dm). Magnes Res. 2004;17:109-114.
9. Ang CD, Alviar MJM, Dans AL, et al. Vitamin B for treating peripheral neuropathy. Cochrane Database Syst Rev. 2008;(3):CD004573.
10. Fonseca VA, Lavery LA, Thethi TK, et al. Metanx in type 2 diabetes
with peripheral neuropathy: a randomized trial. Am J Med. 2013;126:141-149.
11. Mason L, Moore RA, Derry S, et al. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991.
12. Mou J, Paillard F, Turnbull B, et al. Efficacy of Qutenza® (capsaicin) 8% patch for neuropathic pain: a meta-analysis of the Qutenza Clinical Trials Database. Pain. 2013;154:1632-1639.
13. Head KA. Peripheral neuropathy: pathogenic mechanisms and alternative therapies. Altern Med Rev. 2006; 11:294-329.
14. Miranda-Massari JR, Gonzalez MJ, Jimenez FJ, et al. Metabolic correction in the management of diabetic peripheral neuropathy: improving clinical results beyond symptom control. Curr Clin Pharmacol. 2011; 6:260-273.
15. Ziegler D, Low PA, Litchy WJ, et al. Efficacy and safety of antioxidant treatment with a-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care. 2011;34:2054-2060.
16. Vasudevan D, Naik MM, Mukaddam QI. Efficacy and safety of methylcobalamin, alpha lipoic acid and pregabalin combination versus pregabalin monotherapy in improving pain and nerve conduction velocity in type 2 diabetes associated impaired peripheral neuropathic condition. [MAINTAIN]: Results of a pilot study. Ann Indian Acad Neurol. 2014;17:19-24.
17. Halat KM, Dennehy CE. Botanicals and dietary supplements in diabetic peripheral neuropathy. J Am Board Fam Pract. 2003;16:47-57.
18. Donofrio P, Walker F, Hunt V, et al. Treatment of painful diabetic neuropathy with topical capsaicin: A multicenter, double-blind, vehicle-controlled study. Arch Int Med. 1991;151:2225-2229.
19. Derry S, Rice ASC, Cole P, et al. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2013;(2):CD007393.
20. Keen H, Payan J, Allawi J, et al. Treatment of diabetic neuropathy with gamma-linolenic acid. The gamma-Linolenic Acid Multicenter Trial Group. Diabetes Care. 1993;16:8-15.
21. Jamal GA, Carmichael H. The effect of gamma linolenic acid on human diabetic peripheral neuropathy: a double blind placebo controlled trial. Diabetic Med. 1990;7:319-323.
22. Yousef AA, Al-deeb AE. A double-blinded randomised controlled study of the value of sequential intravenous and oral magnesium therapy in patients with chronic low back pain with a neuropathic component. Anaesthesia. 2013;68:260-266.
23. Pickering G, Morel V, Simen E. Oral magnesium treatment in patients with neuropathic pain: a randomized clinical trial. Magnes Res. 2011;24:28-35.

Monoclonal gammopathy of undetermined significance: Using risk stratification to guide follow-up
› For monoclonal gammopathy of undetermined significance (MGUS) patients at low risk, repeat serum protein electrophoresis (SPE) in 6 months. If no significant elevation of M-protein is found, repeat SPE every 2 to 3 years. A
› For patients with smoldering multiple myeloma, order SPE every 2 to 3 months in the first year following diagnosis; repeat every 4 to 6 months in the following year and every 6 to 12 months thereafter. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › A 54-year-old man’s lab results following a routine annual examination reveal a level of IgM M-protein just under 1.5 g/dL. All other lab values, including free light chain (FLC) ratio and bone marrow exam, are normal. No clinical evidence of a related disorder is found. What is the risk that this patient’s condition could progress toward multiple myeloma, and how would you follow up?
The patient with a monoclonal gammopathy has an abnormal proliferation of monoclonal plasma cells that secrete an immunoglobulin, M-protein. This proliferation occurs most often in the bone marrow but can also be found in extra-medullary body tissue. The condition can begin insidiously, remain stable, or progress to frank malignancy causing bone and end-organ destruction. The major challenge is to separate stable, asymptomatic patients who require no treatment from patients with progressive, symptomatic myeloma who require immediate treatment.
An increased, measurable level of serum monoclonal immunoglobulins or FLCs is called monoclonal gammopathy of undetermined significance (MGUS) when there is <3 g/dL monoclonal protein in the serum, <10% monoclonal plasma cells in the bone marrow, and an absence of beta-cell proliferative disorders, lytic bone lesions, anemia, hypercalcemia, or renal insufficiency (TABLE 1).1,2 Serum and marrow measurements exceeding these values indicate progression of disease to a premalignancy stage. Continued proliferation of plasma cells in the bone marrow results in anemia and bone destruction, while the increase in M-protein leads to end-organ destruction. This final malignant state is multiple myeloma (MM).

Detailed classification of MGUS: A roadmap for monitoring patients
Extensive epidemiologic and clinical studies have refined the classification of MGUS3-5 and related disorders (TABLES 2-4),3 providing physicians with guidance on how to monitor patients. There are 3 kinds of monoclonal gammopathies, each reflecting a particular type of immunoglobulin involvement—non-IgM, IgM, or light chain. Additionally, within each type of gammopathy, patient-specific characteristics determine 3 categories of clinical significance: premalignancy with low risk of progression (1%-2% per year3); premalignancy with high risk of progression (10% per year3); and malignancy.
Non-IgM MGUS with a high risk of progression is designated smoldering multiple myeloma (SMM) (TABLE 2).3 IgM MGUS with a high risk of progression is defined as smoldering Waldenström macroglobulinemia (SWM), with a predisposition to progress to Waldenström macroglobulinemia (WM) and, rarely, to IgM MM (TABLE 3).3
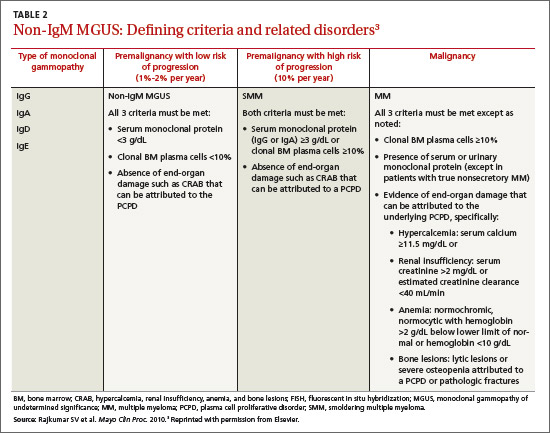
More recently, it has been reported that approximately 20% of the cases of MM belong to a new entity called light-chain MM that features an absence of heavy chain (IgG, IgA, IgM, IgD, or IgE) secretion in serum.6 The premalignant precursor is light-chain MGUS (LC-MGUS). The criteria for LC-MGUS and idiopathic Bence Jones proteinuria are found in TABLE 4.3 Idiopathic Bence Jones proteinuria is equivalent to SMM and SWM due to its higher risk of progression (10%/year)3 to light-chain MM.
Prevalence of MGUS
In general, the prevalence of all types of MGUS increases with age and is affected by race, sex, family history, immunosuppression, and pesticide exposure. The Caucasian American population >50 years exhibits a prevalence of MGUS of approximately 3.2%;7 the African American population exhibits a significantly higher prevalence of 5.9% to 8.4%.7 Native Asians have a lower rate of MM, and, as expected, a lower MGUS prevalence than is seen in the Western population (Thailand ≈2.3%;8 Korea ≈3.3%;9 Japan ≈2.1%;10 China ≈0.8%11). The overall prevalence of the 3 types of MGUS is 4.2% in Caucasians.6
Distinguishing stable from progressive disease
The Mayo Clinic’s risk stratification model12 further specifies risk of disease progression based on 3 indicators: serum M-protein concentration, Ig isotype of M-protein, and serum FLC ratio.
MGUS. A marked increase in risk for disease progression is associated with a serum M-protein concentration ≥1.5 g/dL, a non-IgG isotype, or an abnormal serum FLC ratio (<0.26 or >1.65, reflecting an increase in either the kappa or lambda light chain).12 An MGUS patient exhibiting all 3 of these features has a 58% absolute risk of developing MM after 20 years of follow-up. A patient with 2 of the 3 abnormalities has a 37% risk of progressing to MM, and one who has just one abnormality has a 21% risk. In contrast, an MGUS patient who has an M-protein level <1.5 g/dL, an IgG isotype, and normal FLC range has only a 5% risk of progression to MM in the same 20 years.12
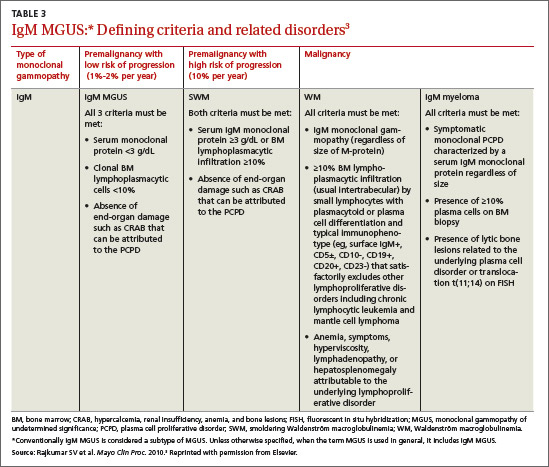
The Spanish Group risk stratification model13 is based on 2 risk factors: a high proportion of abnormal plasma cells (aPC) within the bone marrow plasma cell (BMPC) compartment (ie, ≥95% CD56+/CD19-); and an evolving subtype of the disease (defined as an increase in the level of serum M-protein by at least 10% during the first 6 months of follow-up, or a progressive and constant increase of the M-protein until overt MM develops). The 7-year cumulative probability of progression of MGUS to MM: 2% for patients with neither risk factor, 16% with one risk factor, and 72% with both risk factors.13
SMM. Classification of this progressive state is defined by a serum level of monoclonal protein (IgG, IgA, IgD, or IgE) ≥3 g/dL or a concentration of clonal bone marrow plasma cells ≥10%; and by an absence of end-organ damage such as hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB) that can be attributed to a plasma cell proliferative disorder (TABLE 2).3 Both laboratory and clinical criteria must be met.
According to the Mayo Clinic risk stratification model, likelihood of progression reflects combinations of 3 factors: bone marrow plasmacytosis ≥10%, a serum M-protein level ≥3 g/dL, and a serum FLC ratio ≤0.125 or ≥8.14 Using this stratification scheme, the risk over 10 years of progressing from SMM to MM is 84% for those with all 3 risk factors, 65% with 2 factors, and 52% with one factor.14 As SMM is defined, there is no upper limit of bone marrow involvement. However, Rajkumar et al15 found that progression time was significantly shorter (P<.001) among patients with ≥60% bone marrow involvement, compared with those having <60% involvement.
The Spanish Group risk stratification model13 uses the same model applied to MGUS: a proportion of abnormal plasma cells in the BMPC compartment ≥95% CD56+/CD19-; and an evolving subtype of disease. The 3-year cumulative probability of progression of SMM to MM is 46% for those with both risk factors, 12% for those with one factor, and <1% for those with no risk factors.13

LC-MGUS. The classification of LC-MGUS (TABLE 4)3 is primarily from a Mayo Clinic study6 and research on risk stratification is underway at 2 other institutions. False-positive results are possible in patients with renal16 and inflammatory17 disorders.
Applying risk stratification to patient management
The current approach to a patient with clearly defined MGUS is a prudent “watch and wait” strategy that specifies monitoring details based on risk category (ALGORITHM).1,18
MGUS. In the low-risk MGUS group (IgG subtype, M-protein <1.5 g/dL, and normal FLC ratio)3 there is no need for bone marrow examination or skeletal radiography. Repeat the serum protein electrophoresis (SPE) in 6 months, and if there is no significant elevation of M-protein, repeat the SPE every 2 to 3 years.1,19,20 However, if other findings are suggestive of plasma cell malignancy (anemia, renal insufficiency, hypercalcemia, or bone lesions), bone marrow examination and computed tomographic (CT) scan are advised. Further evaluation of an incidental detection of MGUS is also important since it is occasionally associated with bone diseases,21 arterial and venous thrombosis,22 and an increased risk (P<.05) of developing bacterial (pneumonia, osteomyelitis, septicemia, pyelonephritis, cellulitis, endocarditis, and meningitis) and viral (influenza and herpes zoster) infections.23
Patients in the intermediate- and high-risk MGUS groups with serum monoclonal protein ≥1.5 g/dL, IgA or IgM subtype or an abnormal FLC ratio should undergo tests for CRAB and have bone marrow aspirate and biopsy with cytogenetics, flow cytometry, and fluorescence in situ hybridization (FISH). Patients with IgM MGUS should also undergo a CT scan of the abdomen to rule out the presence of asymptomatic retroperitoneal lymph nodes.1,19 If the BM examination and CT scan yield negative results, repeat SPE and complete blood count (CBC) after 6 months and annually thereafter for life. IgD or IgE MGUS is rare, and patients exhibit a progression similar to the 20-year risk seen with MGUS generally.
SMM. Given the increased risk of progression from SMM to MM compared with MGUS (all risk groups), the 2010 International Myeloma Working Group (IMWG) has suggested monitoring SMM patients more frequently—ie, SPE every 2 to 3 months in the first year following diagnosis.1 Repeat SPE in the second year every 4 to 6 months, and, if results are clinically stable, every 6 to 12 months thereafter. In addition to a baseline bone marrow examination (including cytogenetics, flow cytometry, and FISH studies), consider ordering magnetic resonance imaging of the spine and pelvis to detect occult lesions, as their presence predicts a more rapid progression to MM.24 During the course of the follow-up, evaluate any unexplained anemia or renal function impairment for its origin. A report of MGUS progression over more than a decade to SMM and then to MM illustrates prudent monitoring of a patient.25
LC-MGUS. Once LC-MGUS is detected, first rule out AL-amyloidosis, light-chain deposition disease, or cast nephropathy. If no malignant state is present, repeat the FLC serum assay every 6 months with renal function tests. Idiopathic Bence Jones proteinuria and LC-MGUS have some overlap and both entities put patients at risk for developing MM or amyloidosis. It is not uncommon for MGUS to be accompanied by Bence Jones proteinuria.
In addition to a thorough history and physical examination, recommended followup for both of these entities includes CBC, creatinine, serum FLC, and 24-hour urine protein electrophoresis.6 With idiopathic Bence Jones proteinuria, a monoclonal protein evident on urine protein electrophoresis at >500 mg/24 hr must be followed up with tests for other signs of malignancy (CRAB) and BM examination to exclude the possibility of MM.6
Treatment of MGUS to prevent progression
Multiple myeloma is still an incurable disease. Since MGUS is a precursor of MM, attempts have been made to either slow its progression or eradicate it. Several independent intervention studies26 for the precursor diseases MGUS and SMM have been conducted or are ongoing. Thus far, no conclusive preventive treatment has been found and the 2010 IMWG guidelines do not recommend preventive therapy for MGUS and SMM patients by means of any drug, unless it is a part of a clinical trial.1
CASE › The patient profiled at the start of this article has one abnormal risk factor (IgM isotype) and has a low risk of progression to MM. Management should follow the steps outlined in the ALGORITHM1,18 for low-risk IgM MGUS: repeat SPE, CBC, and CT scan in 6 months and annually thereafter. If any abnormality is observed, rule out the possibilities of IgM SWM, IgM WM, or rapid progression to MM, and consider referral to an oncologist.
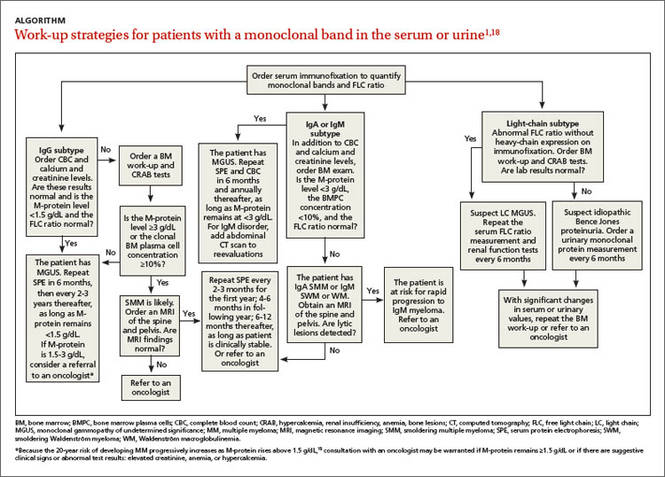
CORRESPONDENCE
John M. Boltri, MD, Department of Family and Community Medicine, Northeast Ohio Medical University, College of Medicine, 4209 St. Rt. 44, PO Box 95, Rootstown, Ohio 44272; [email protected].
ACKNOWLEDGEMENTS
The authors thank Kenneth F. Tucker, MD (Webber Cancer Center, St John Macomb-Oakland Hospital, Warren, Mich) and Elizabeth Sykes, MD (Professor, Oakland University, William Beaumont School of Medicine, Rochester, Mich) for their review of this article.
1. Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24:1121-1127.
2. Swerdlow SH, Campro E, Harris NL, et al. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IRAC Press; 2008.
3. Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc. 2010;85:945-948.
4. Korde N, Kristinsson SY, Landgren O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood. 2011;117:5573-5581.
5. Landgren O, Kyle RA, Rajkumar SV. From myeloma precursor disease to multiple myeloma: new diagnostic concepts and opportunities for early intervention. Clin Cancer Res. 2011;17:1243-1252.
6. Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet. 2010;375:1721-1728.
7. Wadhera RK, Rajkumar SV. Prevalence of monoclonal gammopathy of undetermined significance: a systematic review. Mayo Clin Proc. 2010;85:933-942.
8. Watanaboonyongcharoen P, Nakorn TN, Rojnuckarin P. Prevalence of monoclonal gammopathy of undetermined significance in Thailand. Int J Hematol. 2012;95:176-181.
9. Park HK, Lee KR, Kim YJ, et al. Prevalence of monoclonal gammopathy of undetermined significance in an elderly urban Korean population. Am J Hematol. 2011;86:752-755.
10. Iwanaga M, Tagawa M, Tsukasaki K, et al. Prevalence of monoclonal gammopathy of undetermined significance: study of 52,802 persons in Nagasaki City, Japan. Mayo Clin Proc. 2007;82:1474-1479.
11. Wu SP, Minter A, Costello R, et al. MGUS prevalence in an ethnically Chinese population in Hong Kong. Blood. 2013;121:2363-2364.
12. Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106:812-817.
13. Pérez-Persona E, Mateo G, García-Sanz R, et al. Risk of progression in smouldering myeloma and monoclonal gammopathies of unknown significance: comparative analysis of the evolution of monoclonal component and multiparameter flow cytometry of bone marrow plasma cells. Br J Haematol. 2010;148:110-114.
14. Dispenzieri A, Kyle RA, Katzmann JA, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111:785-789.
15. Rajkumar SV, Larson D, Kyle RA. Diagnosis of smoldering multiple myeloma. N Engl J Med. 2011;365:474-475.
16. Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol. 2008;3:1684-1690.
17. Gottenberg JE, Aucouturier F, Goetz J, et al. Serum immunoglobulin free light chain assessment in rheumatoid arthritis and primary Sjögren’s syndrome. Ann Rheum Dis. 2007;66:23-27.
18. Kyle RA, Buadi F, Rajkumar SV. Management of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Oncology. 2011;25:578-586.
19. Landgren O, Waxman AJ. Multiple myeloma precursor disease. JAMA. 2010;304:2397-2404.
20. Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood. 2010;116:2019-2025.
21. Minter AR, Simpson H, Weiss BM, et al. Bone disease from monoclonal gammopathy of undetermined significance to multiple myeloma: pathogenesis, interventions, and future opportunities. Semin Hematol. 2011;48:55-65.
22. Za T, De Stefano V, Rossi E, et al; Multiple Myeloma GIMEMALatium Region Working Group. Arterial and venous thrombosis in patients with monoclonal gammopathy of undetermined significance: incidence and risk factors in a cohort of 1491 patients. Br J Haematol. 2013;160:673-679.
23. Kristinsson SY, Tang M, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance and risk of infections: a population based study. Haematologica. 2012;97:854-858.
24. Hillengass J, Fechtner K, Weber MA, et al. Prognostic significance of focal lesions in whole-body magnetic resonance imaging in patients with asymptomatic multiple myeloma. J Clin Oncol. 2010;28:1606-1610.
25. Yancey MA, Waxman AJ, Landgren O. A case study progression to multiple myeloma. Clin J Oncol Nurs. 2010;14:419-422.
26. ClinicalTrials.gov. Available at: http://www.clinicaltrials.gov/ct2/results?term=MGUS and http://www.clinicaltrials.gov/ct2/results?term=SMM. Accessed June 23, 2015.
› For monoclonal gammopathy of undetermined significance (MGUS) patients at low risk, repeat serum protein electrophoresis (SPE) in 6 months. If no significant elevation of M-protein is found, repeat SPE every 2 to 3 years. A
› For patients with smoldering multiple myeloma, order SPE every 2 to 3 months in the first year following diagnosis; repeat every 4 to 6 months in the following year and every 6 to 12 months thereafter. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › A 54-year-old man’s lab results following a routine annual examination reveal a level of IgM M-protein just under 1.5 g/dL. All other lab values, including free light chain (FLC) ratio and bone marrow exam, are normal. No clinical evidence of a related disorder is found. What is the risk that this patient’s condition could progress toward multiple myeloma, and how would you follow up?
The patient with a monoclonal gammopathy has an abnormal proliferation of monoclonal plasma cells that secrete an immunoglobulin, M-protein. This proliferation occurs most often in the bone marrow but can also be found in extra-medullary body tissue. The condition can begin insidiously, remain stable, or progress to frank malignancy causing bone and end-organ destruction. The major challenge is to separate stable, asymptomatic patients who require no treatment from patients with progressive, symptomatic myeloma who require immediate treatment.
An increased, measurable level of serum monoclonal immunoglobulins or FLCs is called monoclonal gammopathy of undetermined significance (MGUS) when there is <3 g/dL monoclonal protein in the serum, <10% monoclonal plasma cells in the bone marrow, and an absence of beta-cell proliferative disorders, lytic bone lesions, anemia, hypercalcemia, or renal insufficiency (TABLE 1).1,2 Serum and marrow measurements exceeding these values indicate progression of disease to a premalignancy stage. Continued proliferation of plasma cells in the bone marrow results in anemia and bone destruction, while the increase in M-protein leads to end-organ destruction. This final malignant state is multiple myeloma (MM).
Detailed classification of MGUS: A roadmap for monitoring patients
Extensive epidemiologic and clinical studies have refined the classification of MGUS3-5 and related disorders (TABLES 2-4),3 providing physicians with guidance on how to monitor patients. There are 3 kinds of monoclonal gammopathies, each reflecting a particular type of immunoglobulin involvement—non-IgM, IgM, or light chain. Additionally, within each type of gammopathy, patient-specific characteristics determine 3 categories of clinical significance: premalignancy with low risk of progression (1%-2% per year3); premalignancy with high risk of progression (10% per year3); and malignancy.
Non-IgM MGUS with a high risk of progression is designated smoldering multiple myeloma (SMM) (TABLE 2).3 IgM MGUS with a high risk of progression is defined as smoldering Waldenström macroglobulinemia (SWM), with a predisposition to progress to Waldenström macroglobulinemia (WM) and, rarely, to IgM MM (TABLE 3).3
More recently, it has been reported that approximately 20% of the cases of MM belong to a new entity called light-chain MM that features an absence of heavy chain (IgG, IgA, IgM, IgD, or IgE) secretion in serum.6 The premalignant precursor is light-chain MGUS (LC-MGUS). The criteria for LC-MGUS and idiopathic Bence Jones proteinuria are found in TABLE 4.3 Idiopathic Bence Jones proteinuria is equivalent to SMM and SWM due to its higher risk of progression (10%/year)3 to light-chain MM.
Prevalence of MGUS
In general, the prevalence of all types of MGUS increases with age and is affected by race, sex, family history, immunosuppression, and pesticide exposure. The Caucasian American population >50 years exhibits a prevalence of MGUS of approximately 3.2%;7 the African American population exhibits a significantly higher prevalence of 5.9% to 8.4%.7 Native Asians have a lower rate of MM, and, as expected, a lower MGUS prevalence than is seen in the Western population (Thailand ≈2.3%;8 Korea ≈3.3%;9 Japan ≈2.1%;10 China ≈0.8%11). The overall prevalence of the 3 types of MGUS is 4.2% in Caucasians.6
Distinguishing stable from progressive disease
The Mayo Clinic’s risk stratification model12 further specifies risk of disease progression based on 3 indicators: serum M-protein concentration, Ig isotype of M-protein, and serum FLC ratio.
MGUS. A marked increase in risk for disease progression is associated with a serum M-protein concentration ≥1.5 g/dL, a non-IgG isotype, or an abnormal serum FLC ratio (<0.26 or >1.65, reflecting an increase in either the kappa or lambda light chain).12 An MGUS patient exhibiting all 3 of these features has a 58% absolute risk of developing MM after 20 years of follow-up. A patient with 2 of the 3 abnormalities has a 37% risk of progressing to MM, and one who has just one abnormality has a 21% risk. In contrast, an MGUS patient who has an M-protein level <1.5 g/dL, an IgG isotype, and normal FLC range has only a 5% risk of progression to MM in the same 20 years.12
The Spanish Group risk stratification model13 is based on 2 risk factors: a high proportion of abnormal plasma cells (aPC) within the bone marrow plasma cell (BMPC) compartment (ie, ≥95% CD56+/CD19-); and an evolving subtype of the disease (defined as an increase in the level of serum M-protein by at least 10% during the first 6 months of follow-up, or a progressive and constant increase of the M-protein until overt MM develops). The 7-year cumulative probability of progression of MGUS to MM: 2% for patients with neither risk factor, 16% with one risk factor, and 72% with both risk factors.13
SMM. Classification of this progressive state is defined by a serum level of monoclonal protein (IgG, IgA, IgD, or IgE) ≥3 g/dL or a concentration of clonal bone marrow plasma cells ≥10%; and by an absence of end-organ damage such as hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB) that can be attributed to a plasma cell proliferative disorder (TABLE 2).3 Both laboratory and clinical criteria must be met.
According to the Mayo Clinic risk stratification model, likelihood of progression reflects combinations of 3 factors: bone marrow plasmacytosis ≥10%, a serum M-protein level ≥3 g/dL, and a serum FLC ratio ≤0.125 or ≥8.14 Using this stratification scheme, the risk over 10 years of progressing from SMM to MM is 84% for those with all 3 risk factors, 65% with 2 factors, and 52% with one factor.14 As SMM is defined, there is no upper limit of bone marrow involvement. However, Rajkumar et al15 found that progression time was significantly shorter (P<.001) among patients with ≥60% bone marrow involvement, compared with those having <60% involvement.
The Spanish Group risk stratification model13 uses the same model applied to MGUS: a proportion of abnormal plasma cells in the BMPC compartment ≥95% CD56+/CD19-; and an evolving subtype of disease. The 3-year cumulative probability of progression of SMM to MM is 46% for those with both risk factors, 12% for those with one factor, and <1% for those with no risk factors.13
LC-MGUS. The classification of LC-MGUS (TABLE 4)3 is primarily from a Mayo Clinic study6 and research on risk stratification is underway at 2 other institutions. False-positive results are possible in patients with renal16 and inflammatory17 disorders.
Applying risk stratification to patient management
The current approach to a patient with clearly defined MGUS is a prudent “watch and wait” strategy that specifies monitoring details based on risk category (ALGORITHM).1,18
MGUS. In the low-risk MGUS group (IgG subtype, M-protein <1.5 g/dL, and normal FLC ratio)3 there is no need for bone marrow examination or skeletal radiography. Repeat the serum protein electrophoresis (SPE) in 6 months, and if there is no significant elevation of M-protein, repeat the SPE every 2 to 3 years.1,19,20 However, if other findings are suggestive of plasma cell malignancy (anemia, renal insufficiency, hypercalcemia, or bone lesions), bone marrow examination and computed tomographic (CT) scan are advised. Further evaluation of an incidental detection of MGUS is also important since it is occasionally associated with bone diseases,21 arterial and venous thrombosis,22 and an increased risk (P<.05) of developing bacterial (pneumonia, osteomyelitis, septicemia, pyelonephritis, cellulitis, endocarditis, and meningitis) and viral (influenza and herpes zoster) infections.23
Patients in the intermediate- and high-risk MGUS groups with serum monoclonal protein ≥1.5 g/dL, IgA or IgM subtype or an abnormal FLC ratio should undergo tests for CRAB and have bone marrow aspirate and biopsy with cytogenetics, flow cytometry, and fluorescence in situ hybridization (FISH). Patients with IgM MGUS should also undergo a CT scan of the abdomen to rule out the presence of asymptomatic retroperitoneal lymph nodes.1,19 If the BM examination and CT scan yield negative results, repeat SPE and complete blood count (CBC) after 6 months and annually thereafter for life. IgD or IgE MGUS is rare, and patients exhibit a progression similar to the 20-year risk seen with MGUS generally.
SMM. Given the increased risk of progression from SMM to MM compared with MGUS (all risk groups), the 2010 International Myeloma Working Group (IMWG) has suggested monitoring SMM patients more frequently—ie, SPE every 2 to 3 months in the first year following diagnosis.1 Repeat SPE in the second year every 4 to 6 months, and, if results are clinically stable, every 6 to 12 months thereafter. In addition to a baseline bone marrow examination (including cytogenetics, flow cytometry, and FISH studies), consider ordering magnetic resonance imaging of the spine and pelvis to detect occult lesions, as their presence predicts a more rapid progression to MM.24 During the course of the follow-up, evaluate any unexplained anemia or renal function impairment for its origin. A report of MGUS progression over more than a decade to SMM and then to MM illustrates prudent monitoring of a patient.25
LC-MGUS. Once LC-MGUS is detected, first rule out AL-amyloidosis, light-chain deposition disease, or cast nephropathy. If no malignant state is present, repeat the FLC serum assay every 6 months with renal function tests. Idiopathic Bence Jones proteinuria and LC-MGUS have some overlap and both entities put patients at risk for developing MM or amyloidosis. It is not uncommon for MGUS to be accompanied by Bence Jones proteinuria.
In addition to a thorough history and physical examination, recommended followup for both of these entities includes CBC, creatinine, serum FLC, and 24-hour urine protein electrophoresis.6 With idiopathic Bence Jones proteinuria, a monoclonal protein evident on urine protein electrophoresis at >500 mg/24 hr must be followed up with tests for other signs of malignancy (CRAB) and BM examination to exclude the possibility of MM.6
Treatment of MGUS to prevent progression
Multiple myeloma is still an incurable disease. Since MGUS is a precursor of MM, attempts have been made to either slow its progression or eradicate it. Several independent intervention studies26 for the precursor diseases MGUS and SMM have been conducted or are ongoing. Thus far, no conclusive preventive treatment has been found and the 2010 IMWG guidelines do not recommend preventive therapy for MGUS and SMM patients by means of any drug, unless it is a part of a clinical trial.1
CASE › The patient profiled at the start of this article has one abnormal risk factor (IgM isotype) and has a low risk of progression to MM. Management should follow the steps outlined in the ALGORITHM1,18 for low-risk IgM MGUS: repeat SPE, CBC, and CT scan in 6 months and annually thereafter. If any abnormality is observed, rule out the possibilities of IgM SWM, IgM WM, or rapid progression to MM, and consider referral to an oncologist.
CORRESPONDENCE
John M. Boltri, MD, Department of Family and Community Medicine, Northeast Ohio Medical University, College of Medicine, 4209 St. Rt. 44, PO Box 95, Rootstown, Ohio 44272; [email protected].
ACKNOWLEDGEMENTS
The authors thank Kenneth F. Tucker, MD (Webber Cancer Center, St John Macomb-Oakland Hospital, Warren, Mich) and Elizabeth Sykes, MD (Professor, Oakland University, William Beaumont School of Medicine, Rochester, Mich) for their review of this article.
› For monoclonal gammopathy of undetermined significance (MGUS) patients at low risk, repeat serum protein electrophoresis (SPE) in 6 months. If no significant elevation of M-protein is found, repeat SPE every 2 to 3 years. A
› For patients with smoldering multiple myeloma, order SPE every 2 to 3 months in the first year following diagnosis; repeat every 4 to 6 months in the following year and every 6 to 12 months thereafter. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › A 54-year-old man’s lab results following a routine annual examination reveal a level of IgM M-protein just under 1.5 g/dL. All other lab values, including free light chain (FLC) ratio and bone marrow exam, are normal. No clinical evidence of a related disorder is found. What is the risk that this patient’s condition could progress toward multiple myeloma, and how would you follow up?
The patient with a monoclonal gammopathy has an abnormal proliferation of monoclonal plasma cells that secrete an immunoglobulin, M-protein. This proliferation occurs most often in the bone marrow but can also be found in extra-medullary body tissue. The condition can begin insidiously, remain stable, or progress to frank malignancy causing bone and end-organ destruction. The major challenge is to separate stable, asymptomatic patients who require no treatment from patients with progressive, symptomatic myeloma who require immediate treatment.
An increased, measurable level of serum monoclonal immunoglobulins or FLCs is called monoclonal gammopathy of undetermined significance (MGUS) when there is <3 g/dL monoclonal protein in the serum, <10% monoclonal plasma cells in the bone marrow, and an absence of beta-cell proliferative disorders, lytic bone lesions, anemia, hypercalcemia, or renal insufficiency (TABLE 1).1,2 Serum and marrow measurements exceeding these values indicate progression of disease to a premalignancy stage. Continued proliferation of plasma cells in the bone marrow results in anemia and bone destruction, while the increase in M-protein leads to end-organ destruction. This final malignant state is multiple myeloma (MM).
Detailed classification of MGUS: A roadmap for monitoring patients
Extensive epidemiologic and clinical studies have refined the classification of MGUS3-5 and related disorders (TABLES 2-4),3 providing physicians with guidance on how to monitor patients. There are 3 kinds of monoclonal gammopathies, each reflecting a particular type of immunoglobulin involvement—non-IgM, IgM, or light chain. Additionally, within each type of gammopathy, patient-specific characteristics determine 3 categories of clinical significance: premalignancy with low risk of progression (1%-2% per year3); premalignancy with high risk of progression (10% per year3); and malignancy.
Non-IgM MGUS with a high risk of progression is designated smoldering multiple myeloma (SMM) (TABLE 2).3 IgM MGUS with a high risk of progression is defined as smoldering Waldenström macroglobulinemia (SWM), with a predisposition to progress to Waldenström macroglobulinemia (WM) and, rarely, to IgM MM (TABLE 3).3
More recently, it has been reported that approximately 20% of the cases of MM belong to a new entity called light-chain MM that features an absence of heavy chain (IgG, IgA, IgM, IgD, or IgE) secretion in serum.6 The premalignant precursor is light-chain MGUS (LC-MGUS). The criteria for LC-MGUS and idiopathic Bence Jones proteinuria are found in TABLE 4.3 Idiopathic Bence Jones proteinuria is equivalent to SMM and SWM due to its higher risk of progression (10%/year)3 to light-chain MM.
Prevalence of MGUS
In general, the prevalence of all types of MGUS increases with age and is affected by race, sex, family history, immunosuppression, and pesticide exposure. The Caucasian American population >50 years exhibits a prevalence of MGUS of approximately 3.2%;7 the African American population exhibits a significantly higher prevalence of 5.9% to 8.4%.7 Native Asians have a lower rate of MM, and, as expected, a lower MGUS prevalence than is seen in the Western population (Thailand ≈2.3%;8 Korea ≈3.3%;9 Japan ≈2.1%;10 China ≈0.8%11). The overall prevalence of the 3 types of MGUS is 4.2% in Caucasians.6
Distinguishing stable from progressive disease
The Mayo Clinic’s risk stratification model12 further specifies risk of disease progression based on 3 indicators: serum M-protein concentration, Ig isotype of M-protein, and serum FLC ratio.
MGUS. A marked increase in risk for disease progression is associated with a serum M-protein concentration ≥1.5 g/dL, a non-IgG isotype, or an abnormal serum FLC ratio (<0.26 or >1.65, reflecting an increase in either the kappa or lambda light chain).12 An MGUS patient exhibiting all 3 of these features has a 58% absolute risk of developing MM after 20 years of follow-up. A patient with 2 of the 3 abnormalities has a 37% risk of progressing to MM, and one who has just one abnormality has a 21% risk. In contrast, an MGUS patient who has an M-protein level <1.5 g/dL, an IgG isotype, and normal FLC range has only a 5% risk of progression to MM in the same 20 years.12
The Spanish Group risk stratification model13 is based on 2 risk factors: a high proportion of abnormal plasma cells (aPC) within the bone marrow plasma cell (BMPC) compartment (ie, ≥95% CD56+/CD19-); and an evolving subtype of the disease (defined as an increase in the level of serum M-protein by at least 10% during the first 6 months of follow-up, or a progressive and constant increase of the M-protein until overt MM develops). The 7-year cumulative probability of progression of MGUS to MM: 2% for patients with neither risk factor, 16% with one risk factor, and 72% with both risk factors.13
SMM. Classification of this progressive state is defined by a serum level of monoclonal protein (IgG, IgA, IgD, or IgE) ≥3 g/dL or a concentration of clonal bone marrow plasma cells ≥10%; and by an absence of end-organ damage such as hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB) that can be attributed to a plasma cell proliferative disorder (TABLE 2).3 Both laboratory and clinical criteria must be met.
According to the Mayo Clinic risk stratification model, likelihood of progression reflects combinations of 3 factors: bone marrow plasmacytosis ≥10%, a serum M-protein level ≥3 g/dL, and a serum FLC ratio ≤0.125 or ≥8.14 Using this stratification scheme, the risk over 10 years of progressing from SMM to MM is 84% for those with all 3 risk factors, 65% with 2 factors, and 52% with one factor.14 As SMM is defined, there is no upper limit of bone marrow involvement. However, Rajkumar et al15 found that progression time was significantly shorter (P<.001) among patients with ≥60% bone marrow involvement, compared with those having <60% involvement.
The Spanish Group risk stratification model13 uses the same model applied to MGUS: a proportion of abnormal plasma cells in the BMPC compartment ≥95% CD56+/CD19-; and an evolving subtype of disease. The 3-year cumulative probability of progression of SMM to MM is 46% for those with both risk factors, 12% for those with one factor, and <1% for those with no risk factors.13
LC-MGUS. The classification of LC-MGUS (TABLE 4)3 is primarily from a Mayo Clinic study6 and research on risk stratification is underway at 2 other institutions. False-positive results are possible in patients with renal16 and inflammatory17 disorders.
Applying risk stratification to patient management
The current approach to a patient with clearly defined MGUS is a prudent “watch and wait” strategy that specifies monitoring details based on risk category (ALGORITHM).1,18
MGUS. In the low-risk MGUS group (IgG subtype, M-protein <1.5 g/dL, and normal FLC ratio)3 there is no need for bone marrow examination or skeletal radiography. Repeat the serum protein electrophoresis (SPE) in 6 months, and if there is no significant elevation of M-protein, repeat the SPE every 2 to 3 years.1,19,20 However, if other findings are suggestive of plasma cell malignancy (anemia, renal insufficiency, hypercalcemia, or bone lesions), bone marrow examination and computed tomographic (CT) scan are advised. Further evaluation of an incidental detection of MGUS is also important since it is occasionally associated with bone diseases,21 arterial and venous thrombosis,22 and an increased risk (P<.05) of developing bacterial (pneumonia, osteomyelitis, septicemia, pyelonephritis, cellulitis, endocarditis, and meningitis) and viral (influenza and herpes zoster) infections.23
Patients in the intermediate- and high-risk MGUS groups with serum monoclonal protein ≥1.5 g/dL, IgA or IgM subtype or an abnormal FLC ratio should undergo tests for CRAB and have bone marrow aspirate and biopsy with cytogenetics, flow cytometry, and fluorescence in situ hybridization (FISH). Patients with IgM MGUS should also undergo a CT scan of the abdomen to rule out the presence of asymptomatic retroperitoneal lymph nodes.1,19 If the BM examination and CT scan yield negative results, repeat SPE and complete blood count (CBC) after 6 months and annually thereafter for life. IgD or IgE MGUS is rare, and patients exhibit a progression similar to the 20-year risk seen with MGUS generally.
SMM. Given the increased risk of progression from SMM to MM compared with MGUS (all risk groups), the 2010 International Myeloma Working Group (IMWG) has suggested monitoring SMM patients more frequently—ie, SPE every 2 to 3 months in the first year following diagnosis.1 Repeat SPE in the second year every 4 to 6 months, and, if results are clinically stable, every 6 to 12 months thereafter. In addition to a baseline bone marrow examination (including cytogenetics, flow cytometry, and FISH studies), consider ordering magnetic resonance imaging of the spine and pelvis to detect occult lesions, as their presence predicts a more rapid progression to MM.24 During the course of the follow-up, evaluate any unexplained anemia or renal function impairment for its origin. A report of MGUS progression over more than a decade to SMM and then to MM illustrates prudent monitoring of a patient.25
LC-MGUS. Once LC-MGUS is detected, first rule out AL-amyloidosis, light-chain deposition disease, or cast nephropathy. If no malignant state is present, repeat the FLC serum assay every 6 months with renal function tests. Idiopathic Bence Jones proteinuria and LC-MGUS have some overlap and both entities put patients at risk for developing MM or amyloidosis. It is not uncommon for MGUS to be accompanied by Bence Jones proteinuria.
In addition to a thorough history and physical examination, recommended followup for both of these entities includes CBC, creatinine, serum FLC, and 24-hour urine protein electrophoresis.6 With idiopathic Bence Jones proteinuria, a monoclonal protein evident on urine protein electrophoresis at >500 mg/24 hr must be followed up with tests for other signs of malignancy (CRAB) and BM examination to exclude the possibility of MM.6
Treatment of MGUS to prevent progression
Multiple myeloma is still an incurable disease. Since MGUS is a precursor of MM, attempts have been made to either slow its progression or eradicate it. Several independent intervention studies26 for the precursor diseases MGUS and SMM have been conducted or are ongoing. Thus far, no conclusive preventive treatment has been found and the 2010 IMWG guidelines do not recommend preventive therapy for MGUS and SMM patients by means of any drug, unless it is a part of a clinical trial.1
CASE › The patient profiled at the start of this article has one abnormal risk factor (IgM isotype) and has a low risk of progression to MM. Management should follow the steps outlined in the ALGORITHM1,18 for low-risk IgM MGUS: repeat SPE, CBC, and CT scan in 6 months and annually thereafter. If any abnormality is observed, rule out the possibilities of IgM SWM, IgM WM, or rapid progression to MM, and consider referral to an oncologist.
CORRESPONDENCE
John M. Boltri, MD, Department of Family and Community Medicine, Northeast Ohio Medical University, College of Medicine, 4209 St. Rt. 44, PO Box 95, Rootstown, Ohio 44272; [email protected].
ACKNOWLEDGEMENTS
The authors thank Kenneth F. Tucker, MD (Webber Cancer Center, St John Macomb-Oakland Hospital, Warren, Mich) and Elizabeth Sykes, MD (Professor, Oakland University, William Beaumont School of Medicine, Rochester, Mich) for their review of this article.
1. Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24:1121-1127.
2. Swerdlow SH, Campro E, Harris NL, et al. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IRAC Press; 2008.
3. Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc. 2010;85:945-948.
4. Korde N, Kristinsson SY, Landgren O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood. 2011;117:5573-5581.
5. Landgren O, Kyle RA, Rajkumar SV. From myeloma precursor disease to multiple myeloma: new diagnostic concepts and opportunities for early intervention. Clin Cancer Res. 2011;17:1243-1252.
6. Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet. 2010;375:1721-1728.
7. Wadhera RK, Rajkumar SV. Prevalence of monoclonal gammopathy of undetermined significance: a systematic review. Mayo Clin Proc. 2010;85:933-942.
8. Watanaboonyongcharoen P, Nakorn TN, Rojnuckarin P. Prevalence of monoclonal gammopathy of undetermined significance in Thailand. Int J Hematol. 2012;95:176-181.
9. Park HK, Lee KR, Kim YJ, et al. Prevalence of monoclonal gammopathy of undetermined significance in an elderly urban Korean population. Am J Hematol. 2011;86:752-755.
10. Iwanaga M, Tagawa M, Tsukasaki K, et al. Prevalence of monoclonal gammopathy of undetermined significance: study of 52,802 persons in Nagasaki City, Japan. Mayo Clin Proc. 2007;82:1474-1479.
11. Wu SP, Minter A, Costello R, et al. MGUS prevalence in an ethnically Chinese population in Hong Kong. Blood. 2013;121:2363-2364.
12. Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106:812-817.
13. Pérez-Persona E, Mateo G, García-Sanz R, et al. Risk of progression in smouldering myeloma and monoclonal gammopathies of unknown significance: comparative analysis of the evolution of monoclonal component and multiparameter flow cytometry of bone marrow plasma cells. Br J Haematol. 2010;148:110-114.
14. Dispenzieri A, Kyle RA, Katzmann JA, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111:785-789.
15. Rajkumar SV, Larson D, Kyle RA. Diagnosis of smoldering multiple myeloma. N Engl J Med. 2011;365:474-475.
16. Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol. 2008;3:1684-1690.
17. Gottenberg JE, Aucouturier F, Goetz J, et al. Serum immunoglobulin free light chain assessment in rheumatoid arthritis and primary Sjögren’s syndrome. Ann Rheum Dis. 2007;66:23-27.
18. Kyle RA, Buadi F, Rajkumar SV. Management of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Oncology. 2011;25:578-586.
19. Landgren O, Waxman AJ. Multiple myeloma precursor disease. JAMA. 2010;304:2397-2404.
20. Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood. 2010;116:2019-2025.
21. Minter AR, Simpson H, Weiss BM, et al. Bone disease from monoclonal gammopathy of undetermined significance to multiple myeloma: pathogenesis, interventions, and future opportunities. Semin Hematol. 2011;48:55-65.
22. Za T, De Stefano V, Rossi E, et al; Multiple Myeloma GIMEMALatium Region Working Group. Arterial and venous thrombosis in patients with monoclonal gammopathy of undetermined significance: incidence and risk factors in a cohort of 1491 patients. Br J Haematol. 2013;160:673-679.
23. Kristinsson SY, Tang M, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance and risk of infections: a population based study. Haematologica. 2012;97:854-858.
24. Hillengass J, Fechtner K, Weber MA, et al. Prognostic significance of focal lesions in whole-body magnetic resonance imaging in patients with asymptomatic multiple myeloma. J Clin Oncol. 2010;28:1606-1610.
25. Yancey MA, Waxman AJ, Landgren O. A case study progression to multiple myeloma. Clin J Oncol Nurs. 2010;14:419-422.
26. ClinicalTrials.gov. Available at: http://www.clinicaltrials.gov/ct2/results?term=MGUS and http://www.clinicaltrials.gov/ct2/results?term=SMM. Accessed June 23, 2015.
1. Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24:1121-1127.
2. Swerdlow SH, Campro E, Harris NL, et al. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IRAC Press; 2008.
3. Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc. 2010;85:945-948.
4. Korde N, Kristinsson SY, Landgren O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood. 2011;117:5573-5581.
5. Landgren O, Kyle RA, Rajkumar SV. From myeloma precursor disease to multiple myeloma: new diagnostic concepts and opportunities for early intervention. Clin Cancer Res. 2011;17:1243-1252.
6. Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet. 2010;375:1721-1728.
7. Wadhera RK, Rajkumar SV. Prevalence of monoclonal gammopathy of undetermined significance: a systematic review. Mayo Clin Proc. 2010;85:933-942.
8. Watanaboonyongcharoen P, Nakorn TN, Rojnuckarin P. Prevalence of monoclonal gammopathy of undetermined significance in Thailand. Int J Hematol. 2012;95:176-181.
9. Park HK, Lee KR, Kim YJ, et al. Prevalence of monoclonal gammopathy of undetermined significance in an elderly urban Korean population. Am J Hematol. 2011;86:752-755.
10. Iwanaga M, Tagawa M, Tsukasaki K, et al. Prevalence of monoclonal gammopathy of undetermined significance: study of 52,802 persons in Nagasaki City, Japan. Mayo Clin Proc. 2007;82:1474-1479.
11. Wu SP, Minter A, Costello R, et al. MGUS prevalence in an ethnically Chinese population in Hong Kong. Blood. 2013;121:2363-2364.
12. Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106:812-817.
13. Pérez-Persona E, Mateo G, García-Sanz R, et al. Risk of progression in smouldering myeloma and monoclonal gammopathies of unknown significance: comparative analysis of the evolution of monoclonal component and multiparameter flow cytometry of bone marrow plasma cells. Br J Haematol. 2010;148:110-114.
14. Dispenzieri A, Kyle RA, Katzmann JA, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111:785-789.
15. Rajkumar SV, Larson D, Kyle RA. Diagnosis of smoldering multiple myeloma. N Engl J Med. 2011;365:474-475.
16. Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol. 2008;3:1684-1690.
17. Gottenberg JE, Aucouturier F, Goetz J, et al. Serum immunoglobulin free light chain assessment in rheumatoid arthritis and primary Sjögren’s syndrome. Ann Rheum Dis. 2007;66:23-27.
18. Kyle RA, Buadi F, Rajkumar SV. Management of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Oncology. 2011;25:578-586.
19. Landgren O, Waxman AJ. Multiple myeloma precursor disease. JAMA. 2010;304:2397-2404.
20. Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood. 2010;116:2019-2025.
21. Minter AR, Simpson H, Weiss BM, et al. Bone disease from monoclonal gammopathy of undetermined significance to multiple myeloma: pathogenesis, interventions, and future opportunities. Semin Hematol. 2011;48:55-65.
22. Za T, De Stefano V, Rossi E, et al; Multiple Myeloma GIMEMALatium Region Working Group. Arterial and venous thrombosis in patients with monoclonal gammopathy of undetermined significance: incidence and risk factors in a cohort of 1491 patients. Br J Haematol. 2013;160:673-679.
23. Kristinsson SY, Tang M, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance and risk of infections: a population based study. Haematologica. 2012;97:854-858.
24. Hillengass J, Fechtner K, Weber MA, et al. Prognostic significance of focal lesions in whole-body magnetic resonance imaging in patients with asymptomatic multiple myeloma. J Clin Oncol. 2010;28:1606-1610.
25. Yancey MA, Waxman AJ, Landgren O. A case study progression to multiple myeloma. Clin J Oncol Nurs. 2010;14:419-422.
26. ClinicalTrials.gov. Available at: http://www.clinicaltrials.gov/ct2/results?term=MGUS and http://www.clinicaltrials.gov/ct2/results?term=SMM. Accessed June 23, 2015.
The art & science of prescribing
› To increase adherence, give patients treatment options, ensure that they participate in discussions of treatment, and empower them to reach "informed collaboration" as opposed to informed consent. A
› Ask patients to tell you in their own words what they understand about the treatment they have chosen. A
› At each follow-up visit, anticipate nonadherence, ask nonjudgmental questions about missed medication doses and sexual adverse effects, and offer simple solutions. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Medication nonadherence is a major—and remediable—contributor to poor outcomes, leading to approximately 125,000 preventable deaths,1 worsening of acute and chronic conditions, and billions of dollars in avoidable costs related to increased hospitalizations and emergency visits each year.2,3 Nonadherence rates are 20% to 30% among patients being treated for cancer and acute illness3 and 50% to 60% for chronic conditions, with an average of 50% of all patients taking their medication incorrectly—or not at all.2,4,5
What’s more, nonadherence disrupts the physician-patient relationship6—a serious problem, given that feeling understood is often the most critical component of recovery.7-9
With that in mind, the words used to describe the problem have changed. Compliance and noncompliance, the older labels, were based on the assumptions that patients are passive recipients of medical advice that they should follow without question and that they are to blame for not doing so. Adherence and nonadherence, on the other hand, emphasize mutual agreement and the patient’s freedom to follow the doctor’s recommendations or not, without blame if he or she decides not to do so.10
Many systemic approaches have been tried to maximize adherence, including disease management (eg, Web-based assessment tools, clinical guidelines, and call center-based triage), smart phone apps11 (for reminders and monitoring), and paying for or subsidizing the cost of drugs for those who can’t afford them. All have met with limited success.12 Based on a thorough review of the literature, we suggest a different approach.

Evidence-based efforts by clinicians are the key to effective prescribing and maximal adherence. In the text and table that follow, we summarize physician and patient factors that influence adherence and present optimal prescribing guidelines.
Listen carefully, then respond
Whether patients are seeing a primary care physician or a specialist, they want their doctors to spend more time with them and to give them more comprehensive information about their condition.13-15 The interaction should begin with the physician listening carefully to the patient before responding, but all too often this is not the case.
Family physicians have been found to interrupt patients 23 seconds after asking a question.16 To improve communication, listen quietly until the patient finishes presenting his or her complaints and agenda for the visit. Then ask, “Is there anything else that’s important for me to know?”17
Be more forthcoming
It is equally important for physicians to respond fully, but this is often not the case. A study involving internists found that in patient encounters lasting 20 minutes, physicians devoted little more than one minute, on average, to explaining the patient’s medical condition. The research showed that many physicians greatly overestimated the time they spent doing so.13
Studies have also shown that clinicians tell patients the name of the drug they’re prescribing 74% of the time and state its purpose 87% of the time, but discuss potential adverse effects and duration of treatment a mere 34% of the time. More than 4 in 10 patients are not told the frequency or timing of doses or the number of tablets to take.18
To improve communication, take the following steps when it’s your turn to talk:
Avoid medical jargon. Technical language (eg, edema) and medical shorthand (eg, history) is a significant barrier to patient understanding. In one study of more than 800 pediatrician visits, such speech was found to be detrimental more than half of the time. Although many mothers were confused by the terms, they rarely asked for clarification.19
It has been suggested that doctors and patients have engaged in a “communication conspiracy.”20 In one study, even after obstetricians and gynecologists had identified terms that they knew their patients did not understand, they continued to use them, and in only 15% of visits where unfamiliar terms were used did the patients admit that they did not understand them.21 Part of the problem may be that patients believe they must be seen as undemanding and compliant if they are to receive optimal attention from their physicians.22
Compounding the problem is the fact that clinicians’ use of highly technical language doubles when they are pressured for time,20 suggesting that this behavior could become more widespread as the demand for greater efficiency on the part of physicians increases.
Simplify the treatment regimen. It also helps to keep treatment regimens as straightforward as possible. Prescribing multiple medications simultaneously or giving patients a more complicated regimen decreases adherence. In one study, adherence rates of 84% were achieved when the regimen called for once-a-day dosing, but dropped to 59% when patients were instructed to take their medication 3 times a day.23
Ask the patient to summarize. Using simple terms and clear, succinct explanations promotes understanding, but asking the patient to summarize what you’ve just said is an ideal way to find out just how much he or she grasped. “What will you tell your family about your diagnosis and treatment?” you might ask, or “Tell me what you plan to do to ensure that you follow the prescribed regimen.”
This is particularly important when patients are not native English speakers or when the news is bad. Patients find it particularly tough to understand difficult messages, such as a poor prognosis,24 and are often unaware of their poor comprehension. This was underscored by a study of emergency department (ED) patients, in which 78% demonstrated deficient comprehension in at least one domain (eg, post-ED care, diagnosis, cause) but only 20% recognized their lack of understanding.25
Asking patients if they have any other questions is a crucial step in ensuring complete understanding.21,26
Take steps to maximize patient recall
Even when patients understand what they’ve heard, research suggests they may not retain it. Overall, 40% to 80% of medical information is forgotten immediately, and almost half of what is retained is incorrect.27,28 This is a serious problem, as understanding and accurate recall increase patient satisfaction and the likelihood of adherence to treatment (FIGURE W1).28,29
There are 3 basic explanations for poor recall: factors related to the clinician, such as the use of difficult medical terminology; the mode of communication (eg, spoken vs written); and factors related to the patient, such as a low level of education or learning disability.29-32
Being as specific as possible and spending more time explaining the diagnosis and treatment has been shown to enhance patient recall. In an experiment in which patients read advice on how to develop self-control over their eating, the use of simple language and specific instructions, rather than general rules, increased recall.33 Providing generic information by whatever means does little to improve recall and might even inhibit it.
Linking advice to the patient’s chief complaint, thereby creating a “teachable moment,” is also helpful.34 For example, you might tell a patient with a kidney infection that “Your backache is also because of the kidney infection. Both the backache and the burning during urination should be better about 3 days after you start these pills.”
Watch your affect. How relaxed or worried you appear also influences patient recall. In a recent study, 40 women at risk for breast cancer viewed videotapes of an oncologist presenting mammogram results. Compared to women whose results were conveyed by a physician who appeared relaxed, those who had the same findings presented by a physician who seemed worried perceived their clinical situation to be more severe, developed higher anxiety, and recalled significantly less of what they were told.35
Use multiple means of communication. In a comparison study, patients who received verbal lists of actions for managing fever and sore mouth accompanied by pictographs—images that represented the information presented—had a correct recall rate of 85%; those who received the verbal information alone had a recall rate of only 14%.36,37
A review of recall in cancer patients also found that tailoring communication to the individual—providing an audiotape of the consultation, for instance, or having the patient bring a list of questions and addressing them one by one—is most effective.36 Another study assessed the retention of pediatric patients and their parents when they received either a verbal report alone or a verbal report plus written information or visuals. The researchers concluded that children and their parents should receive verbal reports only when such reports are supplemented with written information or visuals.37
The large body of research on learning and memory has proven useful in designing educational materials for those with poor reading skills. When images were used to convey meaning to 21 adults in a job training program—all with less than fifth grade reading skills—they had on average 85% correct recall immediately after the training and 71% recall 4 weeks later. Although the impact on symptom management and patient quality of life has yet to be studied, these findings suggest that pictures can help people with low literacy recall and retain complex information.38
Overall, while written or recorded instructions appear to improve recall in most situations,39 images have been shown to have the greatest impact.36,37,40
Is the patient ready to adhere to treatment?
No matter how well or by what means you communicate, some patients are not ready for change. Patients in the “precontemplation” stage of change—who may not even recognize the need for change, let alone consider it—can benefit from supportive education and motivational interviewing, while those in the “contemplation” stage need support and convincing to reach the “preparation” stage. It is only in the “action” stage, however, that a patient is ready to collaborate with his or her physician in agreeing on and adhering to treatment.40
Comorbid depression is a common condition, particularly in those with chronic illness, and one of the strongest predictors of nonadherence.1,41 Thus, depression screening for all patients who are chronically or severely ill or nonadherent is strongly recommended, followed by treatment when appropriate.41
“Informed collaboration” is critical
Research shows that if both physician and patient agree on the individual’s medical problem, it will be improved or resolved at follow-up in about half of all cases. In contrast, when the physician alone sees the patient’s condition as a problem, just over a quarter of cases improve, regardless of the severity.42 Compounding this difficulty is the finding that patients fail to report up to two-thirds of their most important health problems.43 When physicians identify them, discord and denial typically result.42
Thus, concordance (we prefer the term “informed collaboration”)—an overt agreement reached after a discussion in which the physician shares expert knowledge, then listens to and respects the feelings and beliefs of the patient with regard to how, when, or whether he or she will take the recommended treatment44—is crucial.42,43,45,46
One way to reach informed collaboration is to give patients problem lists or letters summarizing their health problems in simple and specific terms after each visit, in hopes that the written communication will encourage discussion and a physician-patient partnership in addressing them.43 In a recent study of 967 psychiatric outpatients, adherence was significantly higher among those who cited concordance between their preferences and their treatment and felt that they had participated in decision making.47
Problems can arise at any time
Even after a patient starts out fully adhering to his medication regimen, several issues can derail treatment. Inability to afford the medication is one potential problem.48 Adverse effects are another major reason for discontinuation. Sexual dysfunction, caused by a number of drugs, is embarrassing to many patients and frequently goes unaddressed.49 Thus, a patient may stop taking the medication without saying why—seemingly for no apparent reason. The best approach is to ask specifically why it was discontinued, including direct questions about sexual adverse effects.
Prescribing recommendations
We believe that the outcome of treatment is being determined from the moment a patient steps into your office. Thus, we’ve compiled an evidence-based checklist (TABLE)24,33,40,41,47,49,50 with broad areas for discussion that constitute the art and science of prescribing. These fall into 3 main areas: 1) what to say before you write a prescription; 2) how to get patient buy-in (informed collaboration, rather than informed consent) when you’re ready to write the prescription; and 3) what to address to boost the likelihood of continued adherence at follow-up visits.
It is clear that allowing adequate patient participation and arriving at concordance and overt agreement lead to better clinical outcomes.51 The sequential steps we recommend may take a few extra minutes up front, but without them, nonadherence is highly likely. While physicians are supportive of shared decision making in theory, they are often less confident that this is achieved in practice.52,53
It may help to keep in mind that every step need not be carried out by the physician. Using other members of the health care team, such as a nurse, medical assistant, or health coach, to provide patient education and support and take the patient through a number of the steps that are included in a physician visit has become increasingly necessary—and is easily accommodated in this case.
As the physician, you bear the final responsibility to ensure that the critical elements—particularly the overt agreement—are addressed. Ultimately supporting your patient's decision and reinforcing it will ensure continued adherence.
CORRESPONDENCE
Swati Shivale, MBBS, Department of Psychiatry, SUNY Upstate Medical University, 750 Adams Street, Syracuse, NY 13210; [email protected]
1. Martin LR, Summer LW, Haskard KB, et al. The challenge of patient adherence. Ther Clin Risk Manage. 2005;1:189-199.
2. Jha AK, Aubert R, Yao J, et al. Greater adherence to diabetes drugs is linked to less hospital use and could save nearly $5 billion annually. Health Aff. 2012;8:1836-1846.
3. DiMatteo MR. Variations in patients’ adherence to medical recommendations: a quantitative review of 50 years of research. Med Care. 2004;42:200–209.
4. Brown MT, Bussell JK. Medication adherence: WHO cares? Mayo Clin Proc. 2011;86:304-314.
5. Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manage Healthcare Policy. 2014;7:35-44.
6. Ansell B. Not getting to goal: the clinical costs of noncompliance. J Managed Care Pharm. 2008;14(Suppl):6-b.
7. Van Kleef GA, van den Berg H, Heerdink MW. The persuasive power of emotions: effects of emotional expressions on attitude formation and change. J Appl Psychol. 2014. Nov 17 [Epub ahead of print].
8. Wright JM, Lee C, Chambers GK. Real-world effectiveness of antihypertensive drugs. Can Med Assoc J. 2000;162:190–191.
9. Dunbar J, Agras W. Compliance with medical instructions. In: Ferguson J, Taylor C, eds. The Comprehensive Handbook of Behavioral Medicine. New York, NY: Springer; 1980:115–145.
10. Horne R, Weinman J, Barber N, et al. Concordance, adherence and compliance in medicine taking. Report for the National Coordinating Centre for NHS Service Delivery and Organisation R & D (NCCSDO). December 2005. University of Leeds, School of Healthcare. Available at: http://www.netscc.ac.uk/hsdr/files/project/SDO_FR_08-1412-076_V01.pdf. Accessed June 18, 2015.
11. Dayer L, Heldenbrand S, Anderson P, et al. Smartphone medication adherence apps: potential benefits to patients and providers. J Am Pharm Assoc. 2013; 53:172-181.
12. Jaarsma T, van der Wal ML, Lesman-Leegte I, et al. Effect of moderate or intensive disease management program on outcome in patients with heart failure: coordinating study evaluating outcomes of advising and counseling in heart failure (COACH). Arch Intern Med. 2008;168:316-324.
13. Waitzkin H. Doctor-patient communication. Clinical implications of social scientific research. JAMA. 1984;252:2441–2446.
14. Freeman GK, Horder JP, Howie JGR, et al. Evolving general practice consultation in Britain: issues of length and context. BMJ. 2002;324:880-882.
15. Beisecker AE, Beisecker TD. Patient information-seeking behaviors when communicating with doctors. Med Care. 1990;28:19-28.
16. Marvel M, Epstein R, Flowers K, et al. Soliciting the patient’s agenda: have we improved? JAMA. 1999; 281:283-287.
17. Barrier P, Li T, Jensen N. Two words to improve physician-patient communication: what else? Mayo Clin Proc. 2003;78:211-214.
18. Tarn D, Heritage J, Paterniti D, et al. Physician communications when prescribing new medications. Arch Internal Med. 2006;166:1855-1862.
19. Korsch BM, Gozzi EK, Francis V. Gaps in doctor-patient communication, doctor-patient interaction and patient satisfaction. Pediatrics. 1968;42:855-871.
20. Lipton HL, Svarstad BL. Parental expectations of a multi-disciplinary clinic for children with developmental disabilities. J Health Soc Behav. 1974;15:157-166.
21. McKinlay JB. Who is really ignorant--physician or patient? J Health Soc Behav. 1975;16:3-11.
22. Nehring V, Geach B. Patients’ evaluation of their care: why they don’t complain. Nurs Outlook. 1973; 21:317-321.
23. De las Cuevas C, Peñate W, de Rivera L. To what extent is treatment adherence of psychiatric patients influenced by their participation in shared decision making? Patient Preference Adherence. 2014;8:1547–1553.
24. Tuckett D, Boulton M, Olson C, et al. Meetings Between Experts–An Approach to Sharing Ideas in Medical Consultations. London, UK: Tavistock Publications; 1985.
25. Engel K, Heisler M, Smith D, et al. Patient comprehension of emergency department care and instructions: are patients aware of when they do not understand? Ann Emerg Med. 2009;53:454-461.
26. Viswanathan M, Golin CE, Jones CD, et al. Interventions to improve adherence to self-administered medications for chronic diseases in the United States: a systematic review. Ann Intern Med. 2012;157:785-795.
27. McGuire LC. Remembering what the doctor said: organization and older adults’ memory for medical information. Exp Aging Res. 1996;22:403-428.
28. Anderson JL, Dodman S, Kopelman M, et al. Patient information recall in a rheumatology clinic. Rheumatol Rehab. 1979;18:18-22.
29. Ley P. Communicating with Patients. New York, NY: Croom Helm; 1988.
30. Ley P. Primacy, rated importance, and the recall of medical statements. J Health Soc Beh. 1972;13:311-317.
31. Ley P, Bradshaw PW, Eaves D, et al. A method for increasing patients’ recall of information presented by doctors. Psychol Med. 1973;3:217-220.
32. Kessels R. Patients’ memory for medical information. J Royal Soc Med. 2003;96:219-222.
33. Bradshaw PW, Ley P, Kincey JA. Recall of medical advice: comprehensibility and specificity. Br J Clin Psychol. 1975;14:55-82.
34. Flocke S, Stange K. Direct observation and patient recall of health behavior advice. Prev Med. 2004;38:34-349.
35. Shapiro DE, Boggs SR, Melamed BG, et al. The effect of varied physician affect on recall, anxiety, and perceptions in women at risk for breast cancer: an analogue study. Health Psychol. 1992;11:61-66.
36. van der Meulen N, Jansen J, van Dulmen S, et al. Interventions to improve recall of medical information in cancer patients: a systematic review of the literature. Psychooncology. 2008;17:857-868.
37. Houts PS, Bachrach R, Witmer JT, et al. Using pictographs to enhance recall of spoken medical instructions. Patient Educ Couns. 1998;35:83-88.
38. Watson P, McKinstry B. A systematic review of interventions to improve recall of medical advice in healthcare consultations. J Royal Soc Med. 2009;102:235-243.
39. Houts PS, Witmer JT, Egeth HE, et al. Using pictographs to enhance recall of spoken medical instructions II. Patient Educ Couns. 2001;43:231-242.
40. Prochaska J, Norcross J, DiClemente C. Changing for Good. New York, NY: Avon; 1995.
41. DiMatteo M, Lepper H, Croghan T. Depression is a risk factor for non-compliance in medical treatment: a meta-analysis of the effects of anxiety and depression in patient adherence. Arch Int Med. 2000;160: 2101-2107.
42. Starfield B, Wray C, Hess K, et al. The influence of patientpractitioner agreement on outcome of care. Am J Pub Health. 1981;71:127–131.
43. Scheitel SM, Boland BJ, Wollan PC, et al. Patient-physician agreement about medical diagnoses and cardiovascular risk factors in the ambulatory general medical examination. Mayo Clin Proc. 1996;71: 1131-1137.
44. Bell JS, Airaksinen MS, Lyles A, et al. Concordance is not synonymous with compliance or adherence. Br J Clin Pharmacol. 2007;64:710-711.
45. Staiger T, Jarvik J, Deyo R, et al. Patient-physician agreement as a predictor of outcomes in patients with back pain. J Gen Int Med. 2005;20:935-937.
46. Stewart M, Brown J, Donner A, et al. The impact of patient-centered care on outcomes. J Fam Pract. 2000;49:796-804.
47. Saini SD, Schoenfeld P, Kaulback K, et al. Effect of medication dosing frequency on adherence in chronic diseases. Am J Manage Care. 2009;15:e22–e33.
48. Kedenge SV, Kangwana BP, Waweru EW, et al. Understanding the impact of subsidizing artemisinin-based combination therapies (ACTs) in the retail sector–results from focus group discussions in rural Kenya. PLoS One. 2013;8:e54371.
49. Santini I, De Lauretis I, Roncone R, et al. Psychotropic-associated sexual dysfunctions: a survey of clinical pharmacology and medication-associated practice. Clin Ter. 2014;165:e243-e252.
50. Ibrahim S, Hossam M, Belal D. Study of non-compliance among chronic hemodialysis patients and its impact on patients’ outcomes. Saudi J Kidney Dis Transpl. 2015;26:243-249.
51. Légaré F, Stacey D, Turcotte S, et al. Interventions for improving the adoption of shared decision making by healthcare professionals. Cochrane Database Syst Rev. 2014;(9):CD006732.
52. Cox K, Stevenson F, Britten N, et al. A Systematic Review of Communication between Patients and Healthcare Professionals about Medicine Taking and Prescribing. London, UK: GKT Concordance Unit Kings College; 2004.
53. Edwards A, Elwyn G. Involving patients in decision making and communicating risk: a longitudinal evaluation of doctors’ attitudes and confidence during a randomized trial. J Eval Clin Pract. 2004;10:431-437.
› To increase adherence, give patients treatment options, ensure that they participate in discussions of treatment, and empower them to reach "informed collaboration" as opposed to informed consent. A
› Ask patients to tell you in their own words what they understand about the treatment they have chosen. A
› At each follow-up visit, anticipate nonadherence, ask nonjudgmental questions about missed medication doses and sexual adverse effects, and offer simple solutions. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Medication nonadherence is a major—and remediable—contributor to poor outcomes, leading to approximately 125,000 preventable deaths,1 worsening of acute and chronic conditions, and billions of dollars in avoidable costs related to increased hospitalizations and emergency visits each year.2,3 Nonadherence rates are 20% to 30% among patients being treated for cancer and acute illness3 and 50% to 60% for chronic conditions, with an average of 50% of all patients taking their medication incorrectly—or not at all.2,4,5
What’s more, nonadherence disrupts the physician-patient relationship6—a serious problem, given that feeling understood is often the most critical component of recovery.7-9
With that in mind, the words used to describe the problem have changed. Compliance and noncompliance, the older labels, were based on the assumptions that patients are passive recipients of medical advice that they should follow without question and that they are to blame for not doing so. Adherence and nonadherence, on the other hand, emphasize mutual agreement and the patient’s freedom to follow the doctor’s recommendations or not, without blame if he or she decides not to do so.10
Many systemic approaches have been tried to maximize adherence, including disease management (eg, Web-based assessment tools, clinical guidelines, and call center-based triage), smart phone apps11 (for reminders and monitoring), and paying for or subsidizing the cost of drugs for those who can’t afford them. All have met with limited success.12 Based on a thorough review of the literature, we suggest a different approach.
Evidence-based efforts by clinicians are the key to effective prescribing and maximal adherence. In the text and table that follow, we summarize physician and patient factors that influence adherence and present optimal prescribing guidelines.
Listen carefully, then respond
Whether patients are seeing a primary care physician or a specialist, they want their doctors to spend more time with them and to give them more comprehensive information about their condition.13-15 The interaction should begin with the physician listening carefully to the patient before responding, but all too often this is not the case.
Family physicians have been found to interrupt patients 23 seconds after asking a question.16 To improve communication, listen quietly until the patient finishes presenting his or her complaints and agenda for the visit. Then ask, “Is there anything else that’s important for me to know?”17
Be more forthcoming
It is equally important for physicians to respond fully, but this is often not the case. A study involving internists found that in patient encounters lasting 20 minutes, physicians devoted little more than one minute, on average, to explaining the patient’s medical condition. The research showed that many physicians greatly overestimated the time they spent doing so.13
Studies have also shown that clinicians tell patients the name of the drug they’re prescribing 74% of the time and state its purpose 87% of the time, but discuss potential adverse effects and duration of treatment a mere 34% of the time. More than 4 in 10 patients are not told the frequency or timing of doses or the number of tablets to take.18
To improve communication, take the following steps when it’s your turn to talk:
Avoid medical jargon. Technical language (eg, edema) and medical shorthand (eg, history) is a significant barrier to patient understanding. In one study of more than 800 pediatrician visits, such speech was found to be detrimental more than half of the time. Although many mothers were confused by the terms, they rarely asked for clarification.19
It has been suggested that doctors and patients have engaged in a “communication conspiracy.”20 In one study, even after obstetricians and gynecologists had identified terms that they knew their patients did not understand, they continued to use them, and in only 15% of visits where unfamiliar terms were used did the patients admit that they did not understand them.21 Part of the problem may be that patients believe they must be seen as undemanding and compliant if they are to receive optimal attention from their physicians.22
Compounding the problem is the fact that clinicians’ use of highly technical language doubles when they are pressured for time,20 suggesting that this behavior could become more widespread as the demand for greater efficiency on the part of physicians increases.
Simplify the treatment regimen. It also helps to keep treatment regimens as straightforward as possible. Prescribing multiple medications simultaneously or giving patients a more complicated regimen decreases adherence. In one study, adherence rates of 84% were achieved when the regimen called for once-a-day dosing, but dropped to 59% when patients were instructed to take their medication 3 times a day.23
Ask the patient to summarize. Using simple terms and clear, succinct explanations promotes understanding, but asking the patient to summarize what you’ve just said is an ideal way to find out just how much he or she grasped. “What will you tell your family about your diagnosis and treatment?” you might ask, or “Tell me what you plan to do to ensure that you follow the prescribed regimen.”
This is particularly important when patients are not native English speakers or when the news is bad. Patients find it particularly tough to understand difficult messages, such as a poor prognosis,24 and are often unaware of their poor comprehension. This was underscored by a study of emergency department (ED) patients, in which 78% demonstrated deficient comprehension in at least one domain (eg, post-ED care, diagnosis, cause) but only 20% recognized their lack of understanding.25
Asking patients if they have any other questions is a crucial step in ensuring complete understanding.21,26
Take steps to maximize patient recall
Even when patients understand what they’ve heard, research suggests they may not retain it. Overall, 40% to 80% of medical information is forgotten immediately, and almost half of what is retained is incorrect.27,28 This is a serious problem, as understanding and accurate recall increase patient satisfaction and the likelihood of adherence to treatment (FIGURE W1).28,29
There are 3 basic explanations for poor recall: factors related to the clinician, such as the use of difficult medical terminology; the mode of communication (eg, spoken vs written); and factors related to the patient, such as a low level of education or learning disability.29-32
Being as specific as possible and spending more time explaining the diagnosis and treatment has been shown to enhance patient recall. In an experiment in which patients read advice on how to develop self-control over their eating, the use of simple language and specific instructions, rather than general rules, increased recall.33 Providing generic information by whatever means does little to improve recall and might even inhibit it.
Linking advice to the patient’s chief complaint, thereby creating a “teachable moment,” is also helpful.34 For example, you might tell a patient with a kidney infection that “Your backache is also because of the kidney infection. Both the backache and the burning during urination should be better about 3 days after you start these pills.”
Watch your affect. How relaxed or worried you appear also influences patient recall. In a recent study, 40 women at risk for breast cancer viewed videotapes of an oncologist presenting mammogram results. Compared to women whose results were conveyed by a physician who appeared relaxed, those who had the same findings presented by a physician who seemed worried perceived their clinical situation to be more severe, developed higher anxiety, and recalled significantly less of what they were told.35
Use multiple means of communication. In a comparison study, patients who received verbal lists of actions for managing fever and sore mouth accompanied by pictographs—images that represented the information presented—had a correct recall rate of 85%; those who received the verbal information alone had a recall rate of only 14%.36,37
A review of recall in cancer patients also found that tailoring communication to the individual—providing an audiotape of the consultation, for instance, or having the patient bring a list of questions and addressing them one by one—is most effective.36 Another study assessed the retention of pediatric patients and their parents when they received either a verbal report alone or a verbal report plus written information or visuals. The researchers concluded that children and their parents should receive verbal reports only when such reports are supplemented with written information or visuals.37
The large body of research on learning and memory has proven useful in designing educational materials for those with poor reading skills. When images were used to convey meaning to 21 adults in a job training program—all with less than fifth grade reading skills—they had on average 85% correct recall immediately after the training and 71% recall 4 weeks later. Although the impact on symptom management and patient quality of life has yet to be studied, these findings suggest that pictures can help people with low literacy recall and retain complex information.38
Overall, while written or recorded instructions appear to improve recall in most situations,39 images have been shown to have the greatest impact.36,37,40
Is the patient ready to adhere to treatment?
No matter how well or by what means you communicate, some patients are not ready for change. Patients in the “precontemplation” stage of change—who may not even recognize the need for change, let alone consider it—can benefit from supportive education and motivational interviewing, while those in the “contemplation” stage need support and convincing to reach the “preparation” stage. It is only in the “action” stage, however, that a patient is ready to collaborate with his or her physician in agreeing on and adhering to treatment.40
Comorbid depression is a common condition, particularly in those with chronic illness, and one of the strongest predictors of nonadherence.1,41 Thus, depression screening for all patients who are chronically or severely ill or nonadherent is strongly recommended, followed by treatment when appropriate.41
“Informed collaboration” is critical
Research shows that if both physician and patient agree on the individual’s medical problem, it will be improved or resolved at follow-up in about half of all cases. In contrast, when the physician alone sees the patient’s condition as a problem, just over a quarter of cases improve, regardless of the severity.42 Compounding this difficulty is the finding that patients fail to report up to two-thirds of their most important health problems.43 When physicians identify them, discord and denial typically result.42
Thus, concordance (we prefer the term “informed collaboration”)—an overt agreement reached after a discussion in which the physician shares expert knowledge, then listens to and respects the feelings and beliefs of the patient with regard to how, when, or whether he or she will take the recommended treatment44—is crucial.42,43,45,46
One way to reach informed collaboration is to give patients problem lists or letters summarizing their health problems in simple and specific terms after each visit, in hopes that the written communication will encourage discussion and a physician-patient partnership in addressing them.43 In a recent study of 967 psychiatric outpatients, adherence was significantly higher among those who cited concordance between their preferences and their treatment and felt that they had participated in decision making.47
Problems can arise at any time
Even after a patient starts out fully adhering to his medication regimen, several issues can derail treatment. Inability to afford the medication is one potential problem.48 Adverse effects are another major reason for discontinuation. Sexual dysfunction, caused by a number of drugs, is embarrassing to many patients and frequently goes unaddressed.49 Thus, a patient may stop taking the medication without saying why—seemingly for no apparent reason. The best approach is to ask specifically why it was discontinued, including direct questions about sexual adverse effects.
Prescribing recommendations
We believe that the outcome of treatment is being determined from the moment a patient steps into your office. Thus, we’ve compiled an evidence-based checklist (TABLE)24,33,40,41,47,49,50 with broad areas for discussion that constitute the art and science of prescribing. These fall into 3 main areas: 1) what to say before you write a prescription; 2) how to get patient buy-in (informed collaboration, rather than informed consent) when you’re ready to write the prescription; and 3) what to address to boost the likelihood of continued adherence at follow-up visits.
It is clear that allowing adequate patient participation and arriving at concordance and overt agreement lead to better clinical outcomes.51 The sequential steps we recommend may take a few extra minutes up front, but without them, nonadherence is highly likely. While physicians are supportive of shared decision making in theory, they are often less confident that this is achieved in practice.52,53
It may help to keep in mind that every step need not be carried out by the physician. Using other members of the health care team, such as a nurse, medical assistant, or health coach, to provide patient education and support and take the patient through a number of the steps that are included in a physician visit has become increasingly necessary—and is easily accommodated in this case.
As the physician, you bear the final responsibility to ensure that the critical elements—particularly the overt agreement—are addressed. Ultimately supporting your patient's decision and reinforcing it will ensure continued adherence.
CORRESPONDENCE
Swati Shivale, MBBS, Department of Psychiatry, SUNY Upstate Medical University, 750 Adams Street, Syracuse, NY 13210; [email protected]
› To increase adherence, give patients treatment options, ensure that they participate in discussions of treatment, and empower them to reach "informed collaboration" as opposed to informed consent. A
› Ask patients to tell you in their own words what they understand about the treatment they have chosen. A
› At each follow-up visit, anticipate nonadherence, ask nonjudgmental questions about missed medication doses and sexual adverse effects, and offer simple solutions. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Medication nonadherence is a major—and remediable—contributor to poor outcomes, leading to approximately 125,000 preventable deaths,1 worsening of acute and chronic conditions, and billions of dollars in avoidable costs related to increased hospitalizations and emergency visits each year.2,3 Nonadherence rates are 20% to 30% among patients being treated for cancer and acute illness3 and 50% to 60% for chronic conditions, with an average of 50% of all patients taking their medication incorrectly—or not at all.2,4,5
What’s more, nonadherence disrupts the physician-patient relationship6—a serious problem, given that feeling understood is often the most critical component of recovery.7-9
With that in mind, the words used to describe the problem have changed. Compliance and noncompliance, the older labels, were based on the assumptions that patients are passive recipients of medical advice that they should follow without question and that they are to blame for not doing so. Adherence and nonadherence, on the other hand, emphasize mutual agreement and the patient’s freedom to follow the doctor’s recommendations or not, without blame if he or she decides not to do so.10
Many systemic approaches have been tried to maximize adherence, including disease management (eg, Web-based assessment tools, clinical guidelines, and call center-based triage), smart phone apps11 (for reminders and monitoring), and paying for or subsidizing the cost of drugs for those who can’t afford them. All have met with limited success.12 Based on a thorough review of the literature, we suggest a different approach.
Evidence-based efforts by clinicians are the key to effective prescribing and maximal adherence. In the text and table that follow, we summarize physician and patient factors that influence adherence and present optimal prescribing guidelines.
Listen carefully, then respond
Whether patients are seeing a primary care physician or a specialist, they want their doctors to spend more time with them and to give them more comprehensive information about their condition.13-15 The interaction should begin with the physician listening carefully to the patient before responding, but all too often this is not the case.
Family physicians have been found to interrupt patients 23 seconds after asking a question.16 To improve communication, listen quietly until the patient finishes presenting his or her complaints and agenda for the visit. Then ask, “Is there anything else that’s important for me to know?”17
Be more forthcoming
It is equally important for physicians to respond fully, but this is often not the case. A study involving internists found that in patient encounters lasting 20 minutes, physicians devoted little more than one minute, on average, to explaining the patient’s medical condition. The research showed that many physicians greatly overestimated the time they spent doing so.13
Studies have also shown that clinicians tell patients the name of the drug they’re prescribing 74% of the time and state its purpose 87% of the time, but discuss potential adverse effects and duration of treatment a mere 34% of the time. More than 4 in 10 patients are not told the frequency or timing of doses or the number of tablets to take.18
To improve communication, take the following steps when it’s your turn to talk:
Avoid medical jargon. Technical language (eg, edema) and medical shorthand (eg, history) is a significant barrier to patient understanding. In one study of more than 800 pediatrician visits, such speech was found to be detrimental more than half of the time. Although many mothers were confused by the terms, they rarely asked for clarification.19
It has been suggested that doctors and patients have engaged in a “communication conspiracy.”20 In one study, even after obstetricians and gynecologists had identified terms that they knew their patients did not understand, they continued to use them, and in only 15% of visits where unfamiliar terms were used did the patients admit that they did not understand them.21 Part of the problem may be that patients believe they must be seen as undemanding and compliant if they are to receive optimal attention from their physicians.22
Compounding the problem is the fact that clinicians’ use of highly technical language doubles when they are pressured for time,20 suggesting that this behavior could become more widespread as the demand for greater efficiency on the part of physicians increases.
Simplify the treatment regimen. It also helps to keep treatment regimens as straightforward as possible. Prescribing multiple medications simultaneously or giving patients a more complicated regimen decreases adherence. In one study, adherence rates of 84% were achieved when the regimen called for once-a-day dosing, but dropped to 59% when patients were instructed to take their medication 3 times a day.23
Ask the patient to summarize. Using simple terms and clear, succinct explanations promotes understanding, but asking the patient to summarize what you’ve just said is an ideal way to find out just how much he or she grasped. “What will you tell your family about your diagnosis and treatment?” you might ask, or “Tell me what you plan to do to ensure that you follow the prescribed regimen.”
This is particularly important when patients are not native English speakers or when the news is bad. Patients find it particularly tough to understand difficult messages, such as a poor prognosis,24 and are often unaware of their poor comprehension. This was underscored by a study of emergency department (ED) patients, in which 78% demonstrated deficient comprehension in at least one domain (eg, post-ED care, diagnosis, cause) but only 20% recognized their lack of understanding.25
Asking patients if they have any other questions is a crucial step in ensuring complete understanding.21,26
Take steps to maximize patient recall
Even when patients understand what they’ve heard, research suggests they may not retain it. Overall, 40% to 80% of medical information is forgotten immediately, and almost half of what is retained is incorrect.27,28 This is a serious problem, as understanding and accurate recall increase patient satisfaction and the likelihood of adherence to treatment (FIGURE W1).28,29
There are 3 basic explanations for poor recall: factors related to the clinician, such as the use of difficult medical terminology; the mode of communication (eg, spoken vs written); and factors related to the patient, such as a low level of education or learning disability.29-32
Being as specific as possible and spending more time explaining the diagnosis and treatment has been shown to enhance patient recall. In an experiment in which patients read advice on how to develop self-control over their eating, the use of simple language and specific instructions, rather than general rules, increased recall.33 Providing generic information by whatever means does little to improve recall and might even inhibit it.
Linking advice to the patient’s chief complaint, thereby creating a “teachable moment,” is also helpful.34 For example, you might tell a patient with a kidney infection that “Your backache is also because of the kidney infection. Both the backache and the burning during urination should be better about 3 days after you start these pills.”
Watch your affect. How relaxed or worried you appear also influences patient recall. In a recent study, 40 women at risk for breast cancer viewed videotapes of an oncologist presenting mammogram results. Compared to women whose results were conveyed by a physician who appeared relaxed, those who had the same findings presented by a physician who seemed worried perceived their clinical situation to be more severe, developed higher anxiety, and recalled significantly less of what they were told.35
Use multiple means of communication. In a comparison study, patients who received verbal lists of actions for managing fever and sore mouth accompanied by pictographs—images that represented the information presented—had a correct recall rate of 85%; those who received the verbal information alone had a recall rate of only 14%.36,37
A review of recall in cancer patients also found that tailoring communication to the individual—providing an audiotape of the consultation, for instance, or having the patient bring a list of questions and addressing them one by one—is most effective.36 Another study assessed the retention of pediatric patients and their parents when they received either a verbal report alone or a verbal report plus written information or visuals. The researchers concluded that children and their parents should receive verbal reports only when such reports are supplemented with written information or visuals.37
The large body of research on learning and memory has proven useful in designing educational materials for those with poor reading skills. When images were used to convey meaning to 21 adults in a job training program—all with less than fifth grade reading skills—they had on average 85% correct recall immediately after the training and 71% recall 4 weeks later. Although the impact on symptom management and patient quality of life has yet to be studied, these findings suggest that pictures can help people with low literacy recall and retain complex information.38
Overall, while written or recorded instructions appear to improve recall in most situations,39 images have been shown to have the greatest impact.36,37,40
Is the patient ready to adhere to treatment?
No matter how well or by what means you communicate, some patients are not ready for change. Patients in the “precontemplation” stage of change—who may not even recognize the need for change, let alone consider it—can benefit from supportive education and motivational interviewing, while those in the “contemplation” stage need support and convincing to reach the “preparation” stage. It is only in the “action” stage, however, that a patient is ready to collaborate with his or her physician in agreeing on and adhering to treatment.40
Comorbid depression is a common condition, particularly in those with chronic illness, and one of the strongest predictors of nonadherence.1,41 Thus, depression screening for all patients who are chronically or severely ill or nonadherent is strongly recommended, followed by treatment when appropriate.41
“Informed collaboration” is critical
Research shows that if both physician and patient agree on the individual’s medical problem, it will be improved or resolved at follow-up in about half of all cases. In contrast, when the physician alone sees the patient’s condition as a problem, just over a quarter of cases improve, regardless of the severity.42 Compounding this difficulty is the finding that patients fail to report up to two-thirds of their most important health problems.43 When physicians identify them, discord and denial typically result.42
Thus, concordance (we prefer the term “informed collaboration”)—an overt agreement reached after a discussion in which the physician shares expert knowledge, then listens to and respects the feelings and beliefs of the patient with regard to how, when, or whether he or she will take the recommended treatment44—is crucial.42,43,45,46
One way to reach informed collaboration is to give patients problem lists or letters summarizing their health problems in simple and specific terms after each visit, in hopes that the written communication will encourage discussion and a physician-patient partnership in addressing them.43 In a recent study of 967 psychiatric outpatients, adherence was significantly higher among those who cited concordance between their preferences and their treatment and felt that they had participated in decision making.47
Problems can arise at any time
Even after a patient starts out fully adhering to his medication regimen, several issues can derail treatment. Inability to afford the medication is one potential problem.48 Adverse effects are another major reason for discontinuation. Sexual dysfunction, caused by a number of drugs, is embarrassing to many patients and frequently goes unaddressed.49 Thus, a patient may stop taking the medication without saying why—seemingly for no apparent reason. The best approach is to ask specifically why it was discontinued, including direct questions about sexual adverse effects.
Prescribing recommendations
We believe that the outcome of treatment is being determined from the moment a patient steps into your office. Thus, we’ve compiled an evidence-based checklist (TABLE)24,33,40,41,47,49,50 with broad areas for discussion that constitute the art and science of prescribing. These fall into 3 main areas: 1) what to say before you write a prescription; 2) how to get patient buy-in (informed collaboration, rather than informed consent) when you’re ready to write the prescription; and 3) what to address to boost the likelihood of continued adherence at follow-up visits.
It is clear that allowing adequate patient participation and arriving at concordance and overt agreement lead to better clinical outcomes.51 The sequential steps we recommend may take a few extra minutes up front, but without them, nonadherence is highly likely. While physicians are supportive of shared decision making in theory, they are often less confident that this is achieved in practice.52,53
It may help to keep in mind that every step need not be carried out by the physician. Using other members of the health care team, such as a nurse, medical assistant, or health coach, to provide patient education and support and take the patient through a number of the steps that are included in a physician visit has become increasingly necessary—and is easily accommodated in this case.
As the physician, you bear the final responsibility to ensure that the critical elements—particularly the overt agreement—are addressed. Ultimately supporting your patient's decision and reinforcing it will ensure continued adherence.
CORRESPONDENCE
Swati Shivale, MBBS, Department of Psychiatry, SUNY Upstate Medical University, 750 Adams Street, Syracuse, NY 13210; [email protected]
1. Martin LR, Summer LW, Haskard KB, et al. The challenge of patient adherence. Ther Clin Risk Manage. 2005;1:189-199.
2. Jha AK, Aubert R, Yao J, et al. Greater adherence to diabetes drugs is linked to less hospital use and could save nearly $5 billion annually. Health Aff. 2012;8:1836-1846.
3. DiMatteo MR. Variations in patients’ adherence to medical recommendations: a quantitative review of 50 years of research. Med Care. 2004;42:200–209.
4. Brown MT, Bussell JK. Medication adherence: WHO cares? Mayo Clin Proc. 2011;86:304-314.
5. Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manage Healthcare Policy. 2014;7:35-44.
6. Ansell B. Not getting to goal: the clinical costs of noncompliance. J Managed Care Pharm. 2008;14(Suppl):6-b.
7. Van Kleef GA, van den Berg H, Heerdink MW. The persuasive power of emotions: effects of emotional expressions on attitude formation and change. J Appl Psychol. 2014. Nov 17 [Epub ahead of print].
8. Wright JM, Lee C, Chambers GK. Real-world effectiveness of antihypertensive drugs. Can Med Assoc J. 2000;162:190–191.
9. Dunbar J, Agras W. Compliance with medical instructions. In: Ferguson J, Taylor C, eds. The Comprehensive Handbook of Behavioral Medicine. New York, NY: Springer; 1980:115–145.
10. Horne R, Weinman J, Barber N, et al. Concordance, adherence and compliance in medicine taking. Report for the National Coordinating Centre for NHS Service Delivery and Organisation R & D (NCCSDO). December 2005. University of Leeds, School of Healthcare. Available at: http://www.netscc.ac.uk/hsdr/files/project/SDO_FR_08-1412-076_V01.pdf. Accessed June 18, 2015.
11. Dayer L, Heldenbrand S, Anderson P, et al. Smartphone medication adherence apps: potential benefits to patients and providers. J Am Pharm Assoc. 2013; 53:172-181.
12. Jaarsma T, van der Wal ML, Lesman-Leegte I, et al. Effect of moderate or intensive disease management program on outcome in patients with heart failure: coordinating study evaluating outcomes of advising and counseling in heart failure (COACH). Arch Intern Med. 2008;168:316-324.
13. Waitzkin H. Doctor-patient communication. Clinical implications of social scientific research. JAMA. 1984;252:2441–2446.
14. Freeman GK, Horder JP, Howie JGR, et al. Evolving general practice consultation in Britain: issues of length and context. BMJ. 2002;324:880-882.
15. Beisecker AE, Beisecker TD. Patient information-seeking behaviors when communicating with doctors. Med Care. 1990;28:19-28.
16. Marvel M, Epstein R, Flowers K, et al. Soliciting the patient’s agenda: have we improved? JAMA. 1999; 281:283-287.
17. Barrier P, Li T, Jensen N. Two words to improve physician-patient communication: what else? Mayo Clin Proc. 2003;78:211-214.
18. Tarn D, Heritage J, Paterniti D, et al. Physician communications when prescribing new medications. Arch Internal Med. 2006;166:1855-1862.
19. Korsch BM, Gozzi EK, Francis V. Gaps in doctor-patient communication, doctor-patient interaction and patient satisfaction. Pediatrics. 1968;42:855-871.
20. Lipton HL, Svarstad BL. Parental expectations of a multi-disciplinary clinic for children with developmental disabilities. J Health Soc Behav. 1974;15:157-166.
21. McKinlay JB. Who is really ignorant--physician or patient? J Health Soc Behav. 1975;16:3-11.
22. Nehring V, Geach B. Patients’ evaluation of their care: why they don’t complain. Nurs Outlook. 1973; 21:317-321.
23. De las Cuevas C, Peñate W, de Rivera L. To what extent is treatment adherence of psychiatric patients influenced by their participation in shared decision making? Patient Preference Adherence. 2014;8:1547–1553.
24. Tuckett D, Boulton M, Olson C, et al. Meetings Between Experts–An Approach to Sharing Ideas in Medical Consultations. London, UK: Tavistock Publications; 1985.
25. Engel K, Heisler M, Smith D, et al. Patient comprehension of emergency department care and instructions: are patients aware of when they do not understand? Ann Emerg Med. 2009;53:454-461.
26. Viswanathan M, Golin CE, Jones CD, et al. Interventions to improve adherence to self-administered medications for chronic diseases in the United States: a systematic review. Ann Intern Med. 2012;157:785-795.
27. McGuire LC. Remembering what the doctor said: organization and older adults’ memory for medical information. Exp Aging Res. 1996;22:403-428.
28. Anderson JL, Dodman S, Kopelman M, et al. Patient information recall in a rheumatology clinic. Rheumatol Rehab. 1979;18:18-22.
29. Ley P. Communicating with Patients. New York, NY: Croom Helm; 1988.
30. Ley P. Primacy, rated importance, and the recall of medical statements. J Health Soc Beh. 1972;13:311-317.
31. Ley P, Bradshaw PW, Eaves D, et al. A method for increasing patients’ recall of information presented by doctors. Psychol Med. 1973;3:217-220.
32. Kessels R. Patients’ memory for medical information. J Royal Soc Med. 2003;96:219-222.
33. Bradshaw PW, Ley P, Kincey JA. Recall of medical advice: comprehensibility and specificity. Br J Clin Psychol. 1975;14:55-82.
34. Flocke S, Stange K. Direct observation and patient recall of health behavior advice. Prev Med. 2004;38:34-349.
35. Shapiro DE, Boggs SR, Melamed BG, et al. The effect of varied physician affect on recall, anxiety, and perceptions in women at risk for breast cancer: an analogue study. Health Psychol. 1992;11:61-66.
36. van der Meulen N, Jansen J, van Dulmen S, et al. Interventions to improve recall of medical information in cancer patients: a systematic review of the literature. Psychooncology. 2008;17:857-868.
37. Houts PS, Bachrach R, Witmer JT, et al. Using pictographs to enhance recall of spoken medical instructions. Patient Educ Couns. 1998;35:83-88.
38. Watson P, McKinstry B. A systematic review of interventions to improve recall of medical advice in healthcare consultations. J Royal Soc Med. 2009;102:235-243.
39. Houts PS, Witmer JT, Egeth HE, et al. Using pictographs to enhance recall of spoken medical instructions II. Patient Educ Couns. 2001;43:231-242.
40. Prochaska J, Norcross J, DiClemente C. Changing for Good. New York, NY: Avon; 1995.
41. DiMatteo M, Lepper H, Croghan T. Depression is a risk factor for non-compliance in medical treatment: a meta-analysis of the effects of anxiety and depression in patient adherence. Arch Int Med. 2000;160: 2101-2107.
42. Starfield B, Wray C, Hess K, et al. The influence of patientpractitioner agreement on outcome of care. Am J Pub Health. 1981;71:127–131.
43. Scheitel SM, Boland BJ, Wollan PC, et al. Patient-physician agreement about medical diagnoses and cardiovascular risk factors in the ambulatory general medical examination. Mayo Clin Proc. 1996;71: 1131-1137.
44. Bell JS, Airaksinen MS, Lyles A, et al. Concordance is not synonymous with compliance or adherence. Br J Clin Pharmacol. 2007;64:710-711.
45. Staiger T, Jarvik J, Deyo R, et al. Patient-physician agreement as a predictor of outcomes in patients with back pain. J Gen Int Med. 2005;20:935-937.
46. Stewart M, Brown J, Donner A, et al. The impact of patient-centered care on outcomes. J Fam Pract. 2000;49:796-804.
47. Saini SD, Schoenfeld P, Kaulback K, et al. Effect of medication dosing frequency on adherence in chronic diseases. Am J Manage Care. 2009;15:e22–e33.
48. Kedenge SV, Kangwana BP, Waweru EW, et al. Understanding the impact of subsidizing artemisinin-based combination therapies (ACTs) in the retail sector–results from focus group discussions in rural Kenya. PLoS One. 2013;8:e54371.
49. Santini I, De Lauretis I, Roncone R, et al. Psychotropic-associated sexual dysfunctions: a survey of clinical pharmacology and medication-associated practice. Clin Ter. 2014;165:e243-e252.
50. Ibrahim S, Hossam M, Belal D. Study of non-compliance among chronic hemodialysis patients and its impact on patients’ outcomes. Saudi J Kidney Dis Transpl. 2015;26:243-249.
51. Légaré F, Stacey D, Turcotte S, et al. Interventions for improving the adoption of shared decision making by healthcare professionals. Cochrane Database Syst Rev. 2014;(9):CD006732.
52. Cox K, Stevenson F, Britten N, et al. A Systematic Review of Communication between Patients and Healthcare Professionals about Medicine Taking and Prescribing. London, UK: GKT Concordance Unit Kings College; 2004.
53. Edwards A, Elwyn G. Involving patients in decision making and communicating risk: a longitudinal evaluation of doctors’ attitudes and confidence during a randomized trial. J Eval Clin Pract. 2004;10:431-437.
1. Martin LR, Summer LW, Haskard KB, et al. The challenge of patient adherence. Ther Clin Risk Manage. 2005;1:189-199.
2. Jha AK, Aubert R, Yao J, et al. Greater adherence to diabetes drugs is linked to less hospital use and could save nearly $5 billion annually. Health Aff. 2012;8:1836-1846.
3. DiMatteo MR. Variations in patients’ adherence to medical recommendations: a quantitative review of 50 years of research. Med Care. 2004;42:200–209.
4. Brown MT, Bussell JK. Medication adherence: WHO cares? Mayo Clin Proc. 2011;86:304-314.
5. Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manage Healthcare Policy. 2014;7:35-44.
6. Ansell B. Not getting to goal: the clinical costs of noncompliance. J Managed Care Pharm. 2008;14(Suppl):6-b.
7. Van Kleef GA, van den Berg H, Heerdink MW. The persuasive power of emotions: effects of emotional expressions on attitude formation and change. J Appl Psychol. 2014. Nov 17 [Epub ahead of print].
8. Wright JM, Lee C, Chambers GK. Real-world effectiveness of antihypertensive drugs. Can Med Assoc J. 2000;162:190–191.
9. Dunbar J, Agras W. Compliance with medical instructions. In: Ferguson J, Taylor C, eds. The Comprehensive Handbook of Behavioral Medicine. New York, NY: Springer; 1980:115–145.
10. Horne R, Weinman J, Barber N, et al. Concordance, adherence and compliance in medicine taking. Report for the National Coordinating Centre for NHS Service Delivery and Organisation R & D (NCCSDO). December 2005. University of Leeds, School of Healthcare. Available at: http://www.netscc.ac.uk/hsdr/files/project/SDO_FR_08-1412-076_V01.pdf. Accessed June 18, 2015.
11. Dayer L, Heldenbrand S, Anderson P, et al. Smartphone medication adherence apps: potential benefits to patients and providers. J Am Pharm Assoc. 2013; 53:172-181.
12. Jaarsma T, van der Wal ML, Lesman-Leegte I, et al. Effect of moderate or intensive disease management program on outcome in patients with heart failure: coordinating study evaluating outcomes of advising and counseling in heart failure (COACH). Arch Intern Med. 2008;168:316-324.
13. Waitzkin H. Doctor-patient communication. Clinical implications of social scientific research. JAMA. 1984;252:2441–2446.
14. Freeman GK, Horder JP, Howie JGR, et al. Evolving general practice consultation in Britain: issues of length and context. BMJ. 2002;324:880-882.
15. Beisecker AE, Beisecker TD. Patient information-seeking behaviors when communicating with doctors. Med Care. 1990;28:19-28.
16. Marvel M, Epstein R, Flowers K, et al. Soliciting the patient’s agenda: have we improved? JAMA. 1999; 281:283-287.
17. Barrier P, Li T, Jensen N. Two words to improve physician-patient communication: what else? Mayo Clin Proc. 2003;78:211-214.
18. Tarn D, Heritage J, Paterniti D, et al. Physician communications when prescribing new medications. Arch Internal Med. 2006;166:1855-1862.
19. Korsch BM, Gozzi EK, Francis V. Gaps in doctor-patient communication, doctor-patient interaction and patient satisfaction. Pediatrics. 1968;42:855-871.
20. Lipton HL, Svarstad BL. Parental expectations of a multi-disciplinary clinic for children with developmental disabilities. J Health Soc Behav. 1974;15:157-166.
21. McKinlay JB. Who is really ignorant--physician or patient? J Health Soc Behav. 1975;16:3-11.
22. Nehring V, Geach B. Patients’ evaluation of their care: why they don’t complain. Nurs Outlook. 1973; 21:317-321.
23. De las Cuevas C, Peñate W, de Rivera L. To what extent is treatment adherence of psychiatric patients influenced by their participation in shared decision making? Patient Preference Adherence. 2014;8:1547–1553.
24. Tuckett D, Boulton M, Olson C, et al. Meetings Between Experts–An Approach to Sharing Ideas in Medical Consultations. London, UK: Tavistock Publications; 1985.
25. Engel K, Heisler M, Smith D, et al. Patient comprehension of emergency department care and instructions: are patients aware of when they do not understand? Ann Emerg Med. 2009;53:454-461.
26. Viswanathan M, Golin CE, Jones CD, et al. Interventions to improve adherence to self-administered medications for chronic diseases in the United States: a systematic review. Ann Intern Med. 2012;157:785-795.
27. McGuire LC. Remembering what the doctor said: organization and older adults’ memory for medical information. Exp Aging Res. 1996;22:403-428.
28. Anderson JL, Dodman S, Kopelman M, et al. Patient information recall in a rheumatology clinic. Rheumatol Rehab. 1979;18:18-22.
29. Ley P. Communicating with Patients. New York, NY: Croom Helm; 1988.
30. Ley P. Primacy, rated importance, and the recall of medical statements. J Health Soc Beh. 1972;13:311-317.
31. Ley P, Bradshaw PW, Eaves D, et al. A method for increasing patients’ recall of information presented by doctors. Psychol Med. 1973;3:217-220.
32. Kessels R. Patients’ memory for medical information. J Royal Soc Med. 2003;96:219-222.
33. Bradshaw PW, Ley P, Kincey JA. Recall of medical advice: comprehensibility and specificity. Br J Clin Psychol. 1975;14:55-82.
34. Flocke S, Stange K. Direct observation and patient recall of health behavior advice. Prev Med. 2004;38:34-349.
35. Shapiro DE, Boggs SR, Melamed BG, et al. The effect of varied physician affect on recall, anxiety, and perceptions in women at risk for breast cancer: an analogue study. Health Psychol. 1992;11:61-66.
36. van der Meulen N, Jansen J, van Dulmen S, et al. Interventions to improve recall of medical information in cancer patients: a systematic review of the literature. Psychooncology. 2008;17:857-868.
37. Houts PS, Bachrach R, Witmer JT, et al. Using pictographs to enhance recall of spoken medical instructions. Patient Educ Couns. 1998;35:83-88.
38. Watson P, McKinstry B. A systematic review of interventions to improve recall of medical advice in healthcare consultations. J Royal Soc Med. 2009;102:235-243.
39. Houts PS, Witmer JT, Egeth HE, et al. Using pictographs to enhance recall of spoken medical instructions II. Patient Educ Couns. 2001;43:231-242.
40. Prochaska J, Norcross J, DiClemente C. Changing for Good. New York, NY: Avon; 1995.
41. DiMatteo M, Lepper H, Croghan T. Depression is a risk factor for non-compliance in medical treatment: a meta-analysis of the effects of anxiety and depression in patient adherence. Arch Int Med. 2000;160: 2101-2107.
42. Starfield B, Wray C, Hess K, et al. The influence of patientpractitioner agreement on outcome of care. Am J Pub Health. 1981;71:127–131.
43. Scheitel SM, Boland BJ, Wollan PC, et al. Patient-physician agreement about medical diagnoses and cardiovascular risk factors in the ambulatory general medical examination. Mayo Clin Proc. 1996;71: 1131-1137.
44. Bell JS, Airaksinen MS, Lyles A, et al. Concordance is not synonymous with compliance or adherence. Br J Clin Pharmacol. 2007;64:710-711.
45. Staiger T, Jarvik J, Deyo R, et al. Patient-physician agreement as a predictor of outcomes in patients with back pain. J Gen Int Med. 2005;20:935-937.
46. Stewart M, Brown J, Donner A, et al. The impact of patient-centered care on outcomes. J Fam Pract. 2000;49:796-804.
47. Saini SD, Schoenfeld P, Kaulback K, et al. Effect of medication dosing frequency on adherence in chronic diseases. Am J Manage Care. 2009;15:e22–e33.
48. Kedenge SV, Kangwana BP, Waweru EW, et al. Understanding the impact of subsidizing artemisinin-based combination therapies (ACTs) in the retail sector–results from focus group discussions in rural Kenya. PLoS One. 2013;8:e54371.
49. Santini I, De Lauretis I, Roncone R, et al. Psychotropic-associated sexual dysfunctions: a survey of clinical pharmacology and medication-associated practice. Clin Ter. 2014;165:e243-e252.
50. Ibrahim S, Hossam M, Belal D. Study of non-compliance among chronic hemodialysis patients and its impact on patients’ outcomes. Saudi J Kidney Dis Transpl. 2015;26:243-249.
51. Légaré F, Stacey D, Turcotte S, et al. Interventions for improving the adoption of shared decision making by healthcare professionals. Cochrane Database Syst Rev. 2014;(9):CD006732.
52. Cox K, Stevenson F, Britten N, et al. A Systematic Review of Communication between Patients and Healthcare Professionals about Medicine Taking and Prescribing. London, UK: GKT Concordance Unit Kings College; 2004.
53. Edwards A, Elwyn G. Involving patients in decision making and communicating risk: a longitudinal evaluation of doctors’ attitudes and confidence during a randomized trial. J Eval Clin Pract. 2004;10:431-437.

Oral lesions you can’t afford to miss
› Perform a biopsy and carefully monitor all potentially malignant oral lesions, including leukoplakia and erythroplakia. A
› Consider evaluation for human immunodeficiency virus infection for any patient who has acute necrotizing ulcerative gingivitis/acute necrotizing ulcerative periodontitis, recurrent candidiasis, or oral hairy leukoplakia. A
› Include melanoma in the differential diagnosis of oral pigmented lesions that have any features of cutaneous melanoma (eg, asymmetry, irregular borders, or variable or changing color). A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Family physicians can play an essential role in managing their patients’ oral health by promptly recognizing and diagnosing conditions that demand further medical attention, including non-odontogenic and odontogenic infections, primary oral mucosal diseases, oral manifestations of systemic disease, and malignancy. Many conditions are amenable to treatment by the family physician, while others will require referral to a specialist.
This article and accompanying photo guide describe the types of lesions you may encounter during examinations of the oral cavity, and the corresponding diagnoses.
Be vigilant for non-odontogenic conditions that may require urgent treatment
There are several uncommon, acute non-odontogenic conditions that affect the oral cavity that, when severe, can require urgent medical attention and possible hospitalization.
Primary herpes simplex virus 1 (HSV-1) infection is generally subclinical, but some patients develop significant oral disease—called primary herpetic gingivostomatitis—that’s characterized by painful diffuse, irregular, crop-like ulcerations throughout the oral cavity and lips1 (FIGURE 1). The gingiva is nearly universally affected, which distinguishes this condition from erythema multiforme and aphthous stomatitis, which are described below. The incidence is highest in children, followed by adolescents and young adults.2
Erythema multiforme. This mucocutaneous hypersensitivity reaction can be limited to the oral cavity and lips, without accompanying skin lesions. Flu-like symptoms, including fever and chills, are followed by acute onset of diffuse oral ulcerations that are generally limited to non-keratinized mucosa, and spare the gingiva (FIGURE 2).3 Ulceration and crusting of the lips is common.
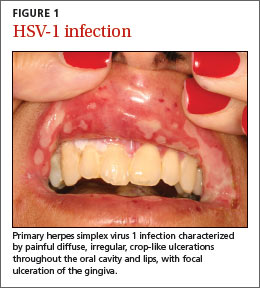
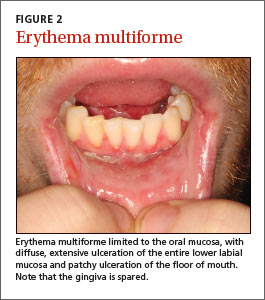
Aphthous stomatitis. Recurrent aphthous stomatitis (RAS) is a common immune-mediated inflammatory condition characterized by “canker sores,” or small round/ovoid ulcers with a well-defined erythematous halo (FIGURE 3). Lesions almost exclusively affect non-keratinized mucosa (and never the lip vermilion) and heal within 7 to 10 days, although “major” (>0.5 cm) lesions may persist much longer (FIGURE 4). A herpetiform pattern with multiple coalescing ulcers closely mimics HSV.
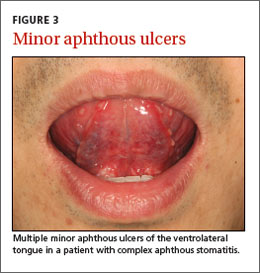

Small subsets of patients develop “complex” RAS, which is characterized by continuous and often multiple ulcerations that may extend into the esophagus, with associated chronic pain and compromised intake.2 RAS associated with systemic conditions is reviewed below.
How to spot signs of common dental diseases
In 2010 in the United States, close to 1.4 million emergency department visits and about $1 billion in hospital charges were due to dental problems.4 Approximately 40% of these visits were made by individuals without insurance.4 Due to a lack of dental insurance, patients may present to a medical professional rather than a dental professional. Additionally, uninsured individuals may neglect their dental problem until it becomes a medical emergency. Family physicians need to recognize dental disease and be able to provide basic management of emergencies.5
Dental abscess. A dental abscess can arise from pulpal infection (due to progression of caries) or periodontal infection (due to progression of periodontal disease). Pain symptoms are variable; however, intense, spontaneous cyclical pain is generally characteristic of a dental abscess of pulpal etiology, whereas a periodontal abscess can have less obvious symptoms. Swelling intraorally or extraorally indicates the spread of a localized infection (FIGURE 5).
Severe infection and swelling can limit mouth opening and function, and in extreme cases may obstruct swallowing and even breathing (eg, Ludwig’s angina). Affected teeth may or may not demonstrate obvious findings of advanced dental disease, such as gross caries, fracture, heavy calculus deposits, or marked periodontal attachment loss. Oral examination may reveal a parulis (focal erythematous swelling of the adjacent gingiva with a central draining sinus tract) (FIGURE 6) and percussion of the affected tooth is generally painful.
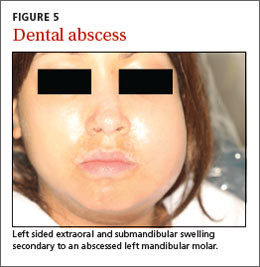
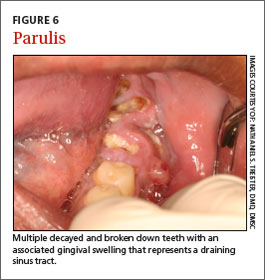
Pericoronitis is infection and swelling of the gingival tissues that surround a tooth, typically in association with a partially erupted third molar. Signs and symptoms include pain, discomfort with eating and swallowing, and limited mouth opening. Examination demonstrates gingival inflammation around a tooth, with or without purulence (FIGURE 7).
Acute necrotizing ulcerative gingivitis (ANUG) and periodontitis (ANUP) are severe conditions that are typically associated with psychological stress, severe malnutrition, and immunosuppression in patients with preexisting gingivitis or periodontitis.6 ANUG is associated with intense gingival pain, halitosis, generalized erythema, and destruction of the gingival papilla, often with bleeding.7 ANUP is a more advanced condition associated with damage and loss of the periodontium (including bone), often with loose teeth (FIGURE 8).8
Trauma. Dental trauma can be limited to the teeth and soft tissues, while more severe injuries can also affect the jaw bone.9 Accidental falls, assault, and motor vehicle traffic accidents are the most common causes of facial fractures in the United States, and are often associated with dentoalveolar trauma (FIGURE 9). The most commonly fractured facial bone is the mandible, characterized by painful opening and closing and an incomplete or altered bite.10
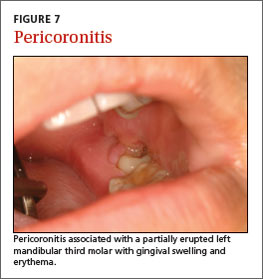
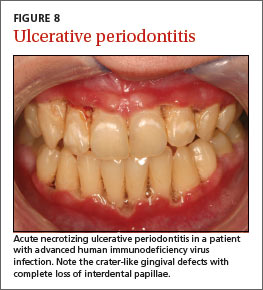
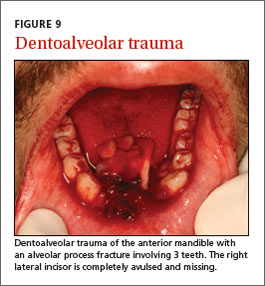
Oral symptoms may be the first sign of systemic disease
Inflammatory bowel disease. Crohn’s disease may affect the gastrointestinal tract anywhere from the mouth to the anus and may initially present with oral findings that may not correlate with abdominal symptoms. Oral Crohn’s disease may present as mucosal cobblestoning, mucosal tags, deep linear ulcerations, gingival hyperplasia, lip fissuring, aphthous ulcers, and angular cheilitis (FIGURES 10 AND 11). Other features may include diffuse, painless swelling of the lips and mucosal erythema.
Pyostomatitis vegetans is an uncommon condition typically associated with ulcerative colitis that is characterized by serpentine pustules that coalesce in a “snail track” pattern (FIGURE 12).11
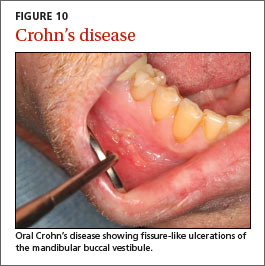
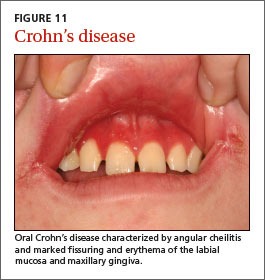
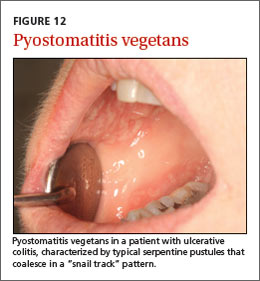
Dermatologic/vesiculo-bullous diseases. Vesiculo-bullous lesions in the mouth may be seen in pemphigus vulgaris or bullous pemphigoid. Pemphigus vulgaris is an autoimmune intraepithelial blistering disease that often first presents in the oral cavity as flaccid bullae or painful ulcerations, prior to the onset of skin lesions (FIGURE 13).
Mucous membrane pemphigoid is an autoimmune subepithelial disease that affects mucous membranes and the skin. Characteristic oral mucosal blisters quickly rupture and form ulcerations, which may be present in the absence of other mucosal involvement (eg, anus, genitalia, nose or throat) (FIGURE 14).12 Painful aphthous ulcers are common. When oral ulcerations are diffuse and recurrent, they may be the first sign of Behçet’s disease, a multisystem autoimmune vasculitis.13
Oral lichen planus is a chronic immunemediated mucocutaneous disease that is often limited to the oral cavity. It presents with characteristic radiating white striations of the buccal mucosa and tongue, often with associated erythema and ulcerations (FIGURE 15).14
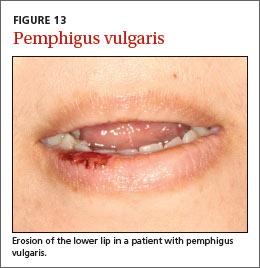
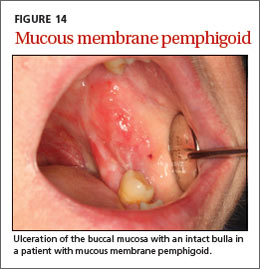
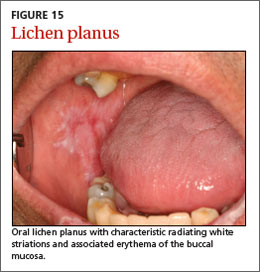
Rheumatologic conditions. Systemic or discoid lupus erythematosus may present with oral findings that largely resemble those of oral lichen planus (FIGURE 16).15 Sjögren’s syndrome is an autoimmune disease with characteristic xerostomia, which can lead to oral discomfort, dysphagia, recurrent candidiasis, and rampant dental caries.
Other conditions to watch for. Erosion of the enamel on the lingual surface of the teeth may be a sign of gastroesophageal reflux disease or bulimia (FIGURE 17). Examination of the oral mucosa can reveal typical white plaques of oral candidiasis (FIGURE 18), which may be associated with systemic immune suppression as well as salivary gland dysfunction.
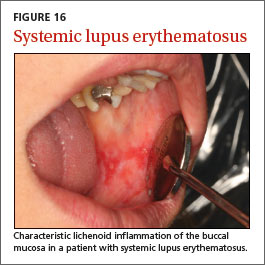
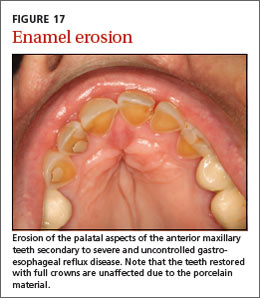
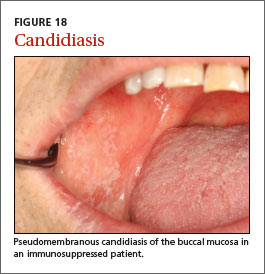
Oral conditions that have been associated with human immunodeficiency virus infection include ANUG/ANUP, recurrent candidiasis, and oral hairy leukoplakia (FIGURE 19). In the absence of known HIV infection, patients who present with any of these oral conditions should be evaluated for HIV infection.13 (For more on recognizing the signs of HIV infection in patients without classic risk factors, see “HIV: 3 cases that hid in plain site” [J Fam Pract. 2015;64:20-26]).
Atrophic glossitis may indicate a vitamin B deficiency. Thrombocytopenia and leukemia may present with oral petechiae, purpura, oral hematomas, or hemorrhagic bullae (FIGURE 20).13 Painless pseudomembranous mucosal erosions may be a presentation for secondary syphilis.16
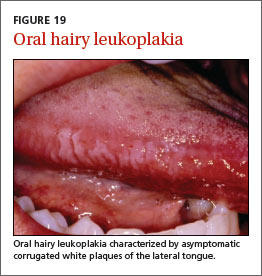
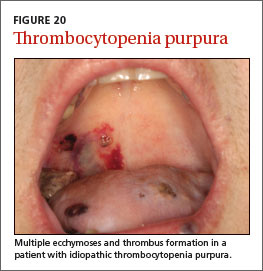
Look for signs that suggest malignancy
In the United States, oral and pharyngeal cancers account for approximately 40,000 cases of cancer and 8000 deaths each year.17 More than 90% of these are squamous cell carcinomas (SCCs); the remainder are mainly salivary gland tumors, lymphoma, and other infrequent cancers.18
SCC of the oral cavity most commonly occurs on the tongue but can develop in any site, presenting as mucosal ulcers, plaques, or masses that do not heal (FIGURE 21). Tobacco and alcohol use are associated with up to 80% of cases of SCC of the head and neck.18 Some oropharyngeal SCCs are associated with human papillomavirus infection type 16.19
Potentially malignant oral lesions include leukoplakia and erythroplakia. Leukoplakia is a white patch or plaque of the oral mucosa that can’t be explained by any other clinical diagnosis (FIGURE 22). These lesions are at risk for malignant transformation and may demonstrate dysplasia or frank SCC on biopsy.20 Proliferative verrucous leukoplakia is a unique form of leukoplakia that’s characterized by a wrinkled appearance that is often multifocal; the condition is associated with a higher risk of malignant transformation.
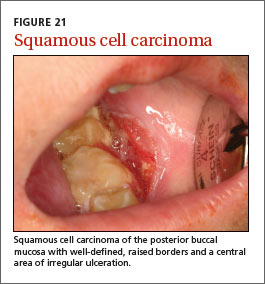
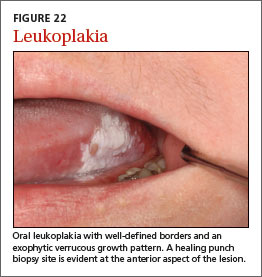
Erythroplakia is a red patch that similarly can’t be explained by another diagnosis. It has a very high risk of malignant transformation over time. All potentially malignant oral lesions, including leukoplakia and erythroplakia, require biopsy and careful monitoring.
Non-SCC cancers. Salivary gland tumors are rare and most commonly occur in patients ages 55 to 65 years. Most neoplasms (70%-85%) occur in the parotid gland, while 8% to 15% develop in the submandibular salivary gland and less than 1% involve the sublingual gland.21 Minor salivary gland tissue, especially in the lips and palate, may also be affected (FIGURE 23). Patients present with circumscribed, fixed or movable, painless, soft or firm masses in a salivary gland.
Melanoma should be included in the differential diagnosis of oral pigmented lesions that have any features of cutaneous melanoma, such as asymmetry, irregular borders, or variable or changing color.22
Hematologic malignancies may initially present (or demonstrate evidence of relapse) in the oral cavity. Leukemia typically presents with sheet-like overgrowth and swelling of the gingiva with associated erythema and bleeding (FIGURE 24), whereas lymphoma typically presents as a solitary mass or ulceration. Solid tumors that metastasize to the oral cavity may present with localized unexplained soft or hard tissue growths, with or without associated neurologic symptoms (eg, paresthesia).
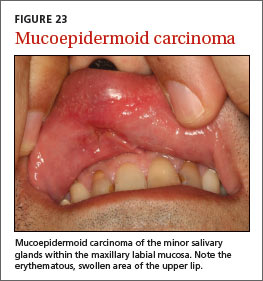
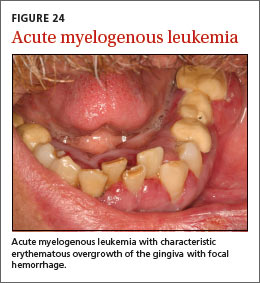
CORRESPONDENCE
William D. Anderson, III, MD, DABFM, FAAFP, University of South Carolina School of Medicine, 15 Medical Park, Suite 300, Columbia, SC 29203; [email protected]
1. Spruance SL. The natural history of recurrent oral-facial herpes simplex virus infection. Semin Dermatol. 1992;11:200-206.
2. Balasubramaniam R, Kuperstein AS, Stoopler ET. Update on oral herpes virus infections. Dent Clin North Am. 2014;58:265-280.
3. Lozada-Nur F, Gorsky M, Silverman S Jr. Oral erythema multiforme: clinical observations and treatment of 95 patients. Oral Surg Oral Med Oral Pathol. 1989;67:36-40.
4. Allareddy V, Rampa S, Lee MK, et al. Hospital-based emergency department visits involving dental conditions: profile and predictors of poor outcomes and resource utilization. J Am Dent Assoc. 2014;145:331-337.
5. Allareddy V, Lin CY, Shah A, et al. Outcomes in patients hospitalized for periapical abscess in the United States: an analysis involving the use of a nationwide inpatient sample. J Am Dent Assoc. 2010;141:1107-1116.
6. Folayan MO. The epidemiology, etiology, and pathophysiology of acute necrotizing ulcerative gingivitis associated with malnutrition. J Contemp Dent Pract. 2004;5:28-41.
7. Atout RN, Todescan S. Managing patients with necrotizing ulcerative gingivitis. J Can Dent Assoc. 2013;79:d46.
8. Todescan S, Nizar R. Managing patients with necrotizing ulcerative periodontitis. J Can Dent Assoc. 2013;79:d44.
9. Allareddy V, Allareddy V, Nalliah RP. Epidemiology of facial fracture injuries. J Oral Maxillofac Surg. 2011;69:2613-2618.
10. Nalliah RP, Allareddy V, Kim MK, et al. Economics of facial fracture reductions in the United States over 12 months. Dent Traumatol. 2013;29:115-120.
11. Padmavathi B, Sharma S, Astekar M, et al. Oral Crohn’s disease. J Oral Maxillofac Pathol. 2014;18(suppl 1):S139-S142.
12. Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
13. Chi AC, Neville BW, Krayer JW, et al. Oral manifestations of systemic disease. Am Fam Physician. 2010;82:1381-1388.
14. Lavanya N, Jayanthi P, Rao UK, et al. Oral lichen planus: An update on pathogenesis and treatment. J Oral Maxillofac Pathol. 2011;15:127-132.
15. Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemic lupus erythematosus. Autoimmune Dis. 2012;2012:834291.
16. Ficarra G, Carlos R. Syphilis: The renaissance of an old disease with oral implications. Head Neck Pathol. 2009;3:195-206.
17. Siegel R, Ward E, Brawley O, et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212-236.
18. Licitra L, Locati LD, Bossi P, et al. Head and neck tumors other than squamous cell carcinoma. Curr Opin Oncol. 2004;16:236-241.
19. Gillison ML, Broutian T, Pickard RK, et al. Prevalence of oral HPV infection in the United States, 2009-2010. JAMA. 2012;307:693-703.
20. Silverman S Jr., Gorsky M, Lozada F. Oral leukoplakia and malignant transformation: A follow‐up study of 257 patients. Cancer. 1984;53:563-568.
21. Spiro RH. Salivary neoplasms: overview of a 35-year experience with 2807 patients. Head Neck Surg. 1986;8:177-184.
22. DeMatos P, Tyler DS, Seigler HF. Malignant melanoma of the mucous membranes: a review of 119 cases. Ann Surg Oncol. 1998;5:733-742.
› Perform a biopsy and carefully monitor all potentially malignant oral lesions, including leukoplakia and erythroplakia. A
› Consider evaluation for human immunodeficiency virus infection for any patient who has acute necrotizing ulcerative gingivitis/acute necrotizing ulcerative periodontitis, recurrent candidiasis, or oral hairy leukoplakia. A
› Include melanoma in the differential diagnosis of oral pigmented lesions that have any features of cutaneous melanoma (eg, asymmetry, irregular borders, or variable or changing color). A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Family physicians can play an essential role in managing their patients’ oral health by promptly recognizing and diagnosing conditions that demand further medical attention, including non-odontogenic and odontogenic infections, primary oral mucosal diseases, oral manifestations of systemic disease, and malignancy. Many conditions are amenable to treatment by the family physician, while others will require referral to a specialist.
This article and accompanying photo guide describe the types of lesions you may encounter during examinations of the oral cavity, and the corresponding diagnoses.
Be vigilant for non-odontogenic conditions that may require urgent treatment
There are several uncommon, acute non-odontogenic conditions that affect the oral cavity that, when severe, can require urgent medical attention and possible hospitalization.
Primary herpes simplex virus 1 (HSV-1) infection is generally subclinical, but some patients develop significant oral disease—called primary herpetic gingivostomatitis—that’s characterized by painful diffuse, irregular, crop-like ulcerations throughout the oral cavity and lips1 (FIGURE 1). The gingiva is nearly universally affected, which distinguishes this condition from erythema multiforme and aphthous stomatitis, which are described below. The incidence is highest in children, followed by adolescents and young adults.2
Erythema multiforme. This mucocutaneous hypersensitivity reaction can be limited to the oral cavity and lips, without accompanying skin lesions. Flu-like symptoms, including fever and chills, are followed by acute onset of diffuse oral ulcerations that are generally limited to non-keratinized mucosa, and spare the gingiva (FIGURE 2).3 Ulceration and crusting of the lips is common.
Aphthous stomatitis. Recurrent aphthous stomatitis (RAS) is a common immune-mediated inflammatory condition characterized by “canker sores,” or small round/ovoid ulcers with a well-defined erythematous halo (FIGURE 3). Lesions almost exclusively affect non-keratinized mucosa (and never the lip vermilion) and heal within 7 to 10 days, although “major” (>0.5 cm) lesions may persist much longer (FIGURE 4). A herpetiform pattern with multiple coalescing ulcers closely mimics HSV.
Small subsets of patients develop “complex” RAS, which is characterized by continuous and often multiple ulcerations that may extend into the esophagus, with associated chronic pain and compromised intake.2 RAS associated with systemic conditions is reviewed below.
How to spot signs of common dental diseases
In 2010 in the United States, close to 1.4 million emergency department visits and about $1 billion in hospital charges were due to dental problems.4 Approximately 40% of these visits were made by individuals without insurance.4 Due to a lack of dental insurance, patients may present to a medical professional rather than a dental professional. Additionally, uninsured individuals may neglect their dental problem until it becomes a medical emergency. Family physicians need to recognize dental disease and be able to provide basic management of emergencies.5
Dental abscess. A dental abscess can arise from pulpal infection (due to progression of caries) or periodontal infection (due to progression of periodontal disease). Pain symptoms are variable; however, intense, spontaneous cyclical pain is generally characteristic of a dental abscess of pulpal etiology, whereas a periodontal abscess can have less obvious symptoms. Swelling intraorally or extraorally indicates the spread of a localized infection (FIGURE 5).
Severe infection and swelling can limit mouth opening and function, and in extreme cases may obstruct swallowing and even breathing (eg, Ludwig’s angina). Affected teeth may or may not demonstrate obvious findings of advanced dental disease, such as gross caries, fracture, heavy calculus deposits, or marked periodontal attachment loss. Oral examination may reveal a parulis (focal erythematous swelling of the adjacent gingiva with a central draining sinus tract) (FIGURE 6) and percussion of the affected tooth is generally painful.
Pericoronitis is infection and swelling of the gingival tissues that surround a tooth, typically in association with a partially erupted third molar. Signs and symptoms include pain, discomfort with eating and swallowing, and limited mouth opening. Examination demonstrates gingival inflammation around a tooth, with or without purulence (FIGURE 7).
Acute necrotizing ulcerative gingivitis (ANUG) and periodontitis (ANUP) are severe conditions that are typically associated with psychological stress, severe malnutrition, and immunosuppression in patients with preexisting gingivitis or periodontitis.6 ANUG is associated with intense gingival pain, halitosis, generalized erythema, and destruction of the gingival papilla, often with bleeding.7 ANUP is a more advanced condition associated with damage and loss of the periodontium (including bone), often with loose teeth (FIGURE 8).8
Trauma. Dental trauma can be limited to the teeth and soft tissues, while more severe injuries can also affect the jaw bone.9 Accidental falls, assault, and motor vehicle traffic accidents are the most common causes of facial fractures in the United States, and are often associated with dentoalveolar trauma (FIGURE 9). The most commonly fractured facial bone is the mandible, characterized by painful opening and closing and an incomplete or altered bite.10
Oral symptoms may be the first sign of systemic disease
Inflammatory bowel disease. Crohn’s disease may affect the gastrointestinal tract anywhere from the mouth to the anus and may initially present with oral findings that may not correlate with abdominal symptoms. Oral Crohn’s disease may present as mucosal cobblestoning, mucosal tags, deep linear ulcerations, gingival hyperplasia, lip fissuring, aphthous ulcers, and angular cheilitis (FIGURES 10 AND 11). Other features may include diffuse, painless swelling of the lips and mucosal erythema.
Pyostomatitis vegetans is an uncommon condition typically associated with ulcerative colitis that is characterized by serpentine pustules that coalesce in a “snail track” pattern (FIGURE 12).11
Dermatologic/vesiculo-bullous diseases. Vesiculo-bullous lesions in the mouth may be seen in pemphigus vulgaris or bullous pemphigoid. Pemphigus vulgaris is an autoimmune intraepithelial blistering disease that often first presents in the oral cavity as flaccid bullae or painful ulcerations, prior to the onset of skin lesions (FIGURE 13).
Mucous membrane pemphigoid is an autoimmune subepithelial disease that affects mucous membranes and the skin. Characteristic oral mucosal blisters quickly rupture and form ulcerations, which may be present in the absence of other mucosal involvement (eg, anus, genitalia, nose or throat) (FIGURE 14).12 Painful aphthous ulcers are common. When oral ulcerations are diffuse and recurrent, they may be the first sign of Behçet’s disease, a multisystem autoimmune vasculitis.13
Oral lichen planus is a chronic immunemediated mucocutaneous disease that is often limited to the oral cavity. It presents with characteristic radiating white striations of the buccal mucosa and tongue, often with associated erythema and ulcerations (FIGURE 15).14
Rheumatologic conditions. Systemic or discoid lupus erythematosus may present with oral findings that largely resemble those of oral lichen planus (FIGURE 16).15 Sjögren’s syndrome is an autoimmune disease with characteristic xerostomia, which can lead to oral discomfort, dysphagia, recurrent candidiasis, and rampant dental caries.
Other conditions to watch for. Erosion of the enamel on the lingual surface of the teeth may be a sign of gastroesophageal reflux disease or bulimia (FIGURE 17). Examination of the oral mucosa can reveal typical white plaques of oral candidiasis (FIGURE 18), which may be associated with systemic immune suppression as well as salivary gland dysfunction.
Oral conditions that have been associated with human immunodeficiency virus infection include ANUG/ANUP, recurrent candidiasis, and oral hairy leukoplakia (FIGURE 19). In the absence of known HIV infection, patients who present with any of these oral conditions should be evaluated for HIV infection.13 (For more on recognizing the signs of HIV infection in patients without classic risk factors, see “HIV: 3 cases that hid in plain site” [J Fam Pract. 2015;64:20-26]).
Atrophic glossitis may indicate a vitamin B deficiency. Thrombocytopenia and leukemia may present with oral petechiae, purpura, oral hematomas, or hemorrhagic bullae (FIGURE 20).13 Painless pseudomembranous mucosal erosions may be a presentation for secondary syphilis.16
Look for signs that suggest malignancy
In the United States, oral and pharyngeal cancers account for approximately 40,000 cases of cancer and 8000 deaths each year.17 More than 90% of these are squamous cell carcinomas (SCCs); the remainder are mainly salivary gland tumors, lymphoma, and other infrequent cancers.18
SCC of the oral cavity most commonly occurs on the tongue but can develop in any site, presenting as mucosal ulcers, plaques, or masses that do not heal (FIGURE 21). Tobacco and alcohol use are associated with up to 80% of cases of SCC of the head and neck.18 Some oropharyngeal SCCs are associated with human papillomavirus infection type 16.19
Potentially malignant oral lesions include leukoplakia and erythroplakia. Leukoplakia is a white patch or plaque of the oral mucosa that can’t be explained by any other clinical diagnosis (FIGURE 22). These lesions are at risk for malignant transformation and may demonstrate dysplasia or frank SCC on biopsy.20 Proliferative verrucous leukoplakia is a unique form of leukoplakia that’s characterized by a wrinkled appearance that is often multifocal; the condition is associated with a higher risk of malignant transformation.
Erythroplakia is a red patch that similarly can’t be explained by another diagnosis. It has a very high risk of malignant transformation over time. All potentially malignant oral lesions, including leukoplakia and erythroplakia, require biopsy and careful monitoring.
Non-SCC cancers. Salivary gland tumors are rare and most commonly occur in patients ages 55 to 65 years. Most neoplasms (70%-85%) occur in the parotid gland, while 8% to 15% develop in the submandibular salivary gland and less than 1% involve the sublingual gland.21 Minor salivary gland tissue, especially in the lips and palate, may also be affected (FIGURE 23). Patients present with circumscribed, fixed or movable, painless, soft or firm masses in a salivary gland.
Melanoma should be included in the differential diagnosis of oral pigmented lesions that have any features of cutaneous melanoma, such as asymmetry, irregular borders, or variable or changing color.22
Hematologic malignancies may initially present (or demonstrate evidence of relapse) in the oral cavity. Leukemia typically presents with sheet-like overgrowth and swelling of the gingiva with associated erythema and bleeding (FIGURE 24), whereas lymphoma typically presents as a solitary mass or ulceration. Solid tumors that metastasize to the oral cavity may present with localized unexplained soft or hard tissue growths, with or without associated neurologic symptoms (eg, paresthesia).
CORRESPONDENCE
William D. Anderson, III, MD, DABFM, FAAFP, University of South Carolina School of Medicine, 15 Medical Park, Suite 300, Columbia, SC 29203; [email protected]
› Perform a biopsy and carefully monitor all potentially malignant oral lesions, including leukoplakia and erythroplakia. A
› Consider evaluation for human immunodeficiency virus infection for any patient who has acute necrotizing ulcerative gingivitis/acute necrotizing ulcerative periodontitis, recurrent candidiasis, or oral hairy leukoplakia. A
› Include melanoma in the differential diagnosis of oral pigmented lesions that have any features of cutaneous melanoma (eg, asymmetry, irregular borders, or variable or changing color). A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Family physicians can play an essential role in managing their patients’ oral health by promptly recognizing and diagnosing conditions that demand further medical attention, including non-odontogenic and odontogenic infections, primary oral mucosal diseases, oral manifestations of systemic disease, and malignancy. Many conditions are amenable to treatment by the family physician, while others will require referral to a specialist.
This article and accompanying photo guide describe the types of lesions you may encounter during examinations of the oral cavity, and the corresponding diagnoses.
Be vigilant for non-odontogenic conditions that may require urgent treatment
There are several uncommon, acute non-odontogenic conditions that affect the oral cavity that, when severe, can require urgent medical attention and possible hospitalization.
Primary herpes simplex virus 1 (HSV-1) infection is generally subclinical, but some patients develop significant oral disease—called primary herpetic gingivostomatitis—that’s characterized by painful diffuse, irregular, crop-like ulcerations throughout the oral cavity and lips1 (FIGURE 1). The gingiva is nearly universally affected, which distinguishes this condition from erythema multiforme and aphthous stomatitis, which are described below. The incidence is highest in children, followed by adolescents and young adults.2
Erythema multiforme. This mucocutaneous hypersensitivity reaction can be limited to the oral cavity and lips, without accompanying skin lesions. Flu-like symptoms, including fever and chills, are followed by acute onset of diffuse oral ulcerations that are generally limited to non-keratinized mucosa, and spare the gingiva (FIGURE 2).3 Ulceration and crusting of the lips is common.
Aphthous stomatitis. Recurrent aphthous stomatitis (RAS) is a common immune-mediated inflammatory condition characterized by “canker sores,” or small round/ovoid ulcers with a well-defined erythematous halo (FIGURE 3). Lesions almost exclusively affect non-keratinized mucosa (and never the lip vermilion) and heal within 7 to 10 days, although “major” (>0.5 cm) lesions may persist much longer (FIGURE 4). A herpetiform pattern with multiple coalescing ulcers closely mimics HSV.
Small subsets of patients develop “complex” RAS, which is characterized by continuous and often multiple ulcerations that may extend into the esophagus, with associated chronic pain and compromised intake.2 RAS associated with systemic conditions is reviewed below.
How to spot signs of common dental diseases
In 2010 in the United States, close to 1.4 million emergency department visits and about $1 billion in hospital charges were due to dental problems.4 Approximately 40% of these visits were made by individuals without insurance.4 Due to a lack of dental insurance, patients may present to a medical professional rather than a dental professional. Additionally, uninsured individuals may neglect their dental problem until it becomes a medical emergency. Family physicians need to recognize dental disease and be able to provide basic management of emergencies.5
Dental abscess. A dental abscess can arise from pulpal infection (due to progression of caries) or periodontal infection (due to progression of periodontal disease). Pain symptoms are variable; however, intense, spontaneous cyclical pain is generally characteristic of a dental abscess of pulpal etiology, whereas a periodontal abscess can have less obvious symptoms. Swelling intraorally or extraorally indicates the spread of a localized infection (FIGURE 5).
Severe infection and swelling can limit mouth opening and function, and in extreme cases may obstruct swallowing and even breathing (eg, Ludwig’s angina). Affected teeth may or may not demonstrate obvious findings of advanced dental disease, such as gross caries, fracture, heavy calculus deposits, or marked periodontal attachment loss. Oral examination may reveal a parulis (focal erythematous swelling of the adjacent gingiva with a central draining sinus tract) (FIGURE 6) and percussion of the affected tooth is generally painful.
Pericoronitis is infection and swelling of the gingival tissues that surround a tooth, typically in association with a partially erupted third molar. Signs and symptoms include pain, discomfort with eating and swallowing, and limited mouth opening. Examination demonstrates gingival inflammation around a tooth, with or without purulence (FIGURE 7).
Acute necrotizing ulcerative gingivitis (ANUG) and periodontitis (ANUP) are severe conditions that are typically associated with psychological stress, severe malnutrition, and immunosuppression in patients with preexisting gingivitis or periodontitis.6 ANUG is associated with intense gingival pain, halitosis, generalized erythema, and destruction of the gingival papilla, often with bleeding.7 ANUP is a more advanced condition associated with damage and loss of the periodontium (including bone), often with loose teeth (FIGURE 8).8
Trauma. Dental trauma can be limited to the teeth and soft tissues, while more severe injuries can also affect the jaw bone.9 Accidental falls, assault, and motor vehicle traffic accidents are the most common causes of facial fractures in the United States, and are often associated with dentoalveolar trauma (FIGURE 9). The most commonly fractured facial bone is the mandible, characterized by painful opening and closing and an incomplete or altered bite.10
Oral symptoms may be the first sign of systemic disease
Inflammatory bowel disease. Crohn’s disease may affect the gastrointestinal tract anywhere from the mouth to the anus and may initially present with oral findings that may not correlate with abdominal symptoms. Oral Crohn’s disease may present as mucosal cobblestoning, mucosal tags, deep linear ulcerations, gingival hyperplasia, lip fissuring, aphthous ulcers, and angular cheilitis (FIGURES 10 AND 11). Other features may include diffuse, painless swelling of the lips and mucosal erythema.
Pyostomatitis vegetans is an uncommon condition typically associated with ulcerative colitis that is characterized by serpentine pustules that coalesce in a “snail track” pattern (FIGURE 12).11
Dermatologic/vesiculo-bullous diseases. Vesiculo-bullous lesions in the mouth may be seen in pemphigus vulgaris or bullous pemphigoid. Pemphigus vulgaris is an autoimmune intraepithelial blistering disease that often first presents in the oral cavity as flaccid bullae or painful ulcerations, prior to the onset of skin lesions (FIGURE 13).
Mucous membrane pemphigoid is an autoimmune subepithelial disease that affects mucous membranes and the skin. Characteristic oral mucosal blisters quickly rupture and form ulcerations, which may be present in the absence of other mucosal involvement (eg, anus, genitalia, nose or throat) (FIGURE 14).12 Painful aphthous ulcers are common. When oral ulcerations are diffuse and recurrent, they may be the first sign of Behçet’s disease, a multisystem autoimmune vasculitis.13
Oral lichen planus is a chronic immunemediated mucocutaneous disease that is often limited to the oral cavity. It presents with characteristic radiating white striations of the buccal mucosa and tongue, often with associated erythema and ulcerations (FIGURE 15).14
Rheumatologic conditions. Systemic or discoid lupus erythematosus may present with oral findings that largely resemble those of oral lichen planus (FIGURE 16).15 Sjögren’s syndrome is an autoimmune disease with characteristic xerostomia, which can lead to oral discomfort, dysphagia, recurrent candidiasis, and rampant dental caries.
Other conditions to watch for. Erosion of the enamel on the lingual surface of the teeth may be a sign of gastroesophageal reflux disease or bulimia (FIGURE 17). Examination of the oral mucosa can reveal typical white plaques of oral candidiasis (FIGURE 18), which may be associated with systemic immune suppression as well as salivary gland dysfunction.
Oral conditions that have been associated with human immunodeficiency virus infection include ANUG/ANUP, recurrent candidiasis, and oral hairy leukoplakia (FIGURE 19). In the absence of known HIV infection, patients who present with any of these oral conditions should be evaluated for HIV infection.13 (For more on recognizing the signs of HIV infection in patients without classic risk factors, see “HIV: 3 cases that hid in plain site” [J Fam Pract. 2015;64:20-26]).
Atrophic glossitis may indicate a vitamin B deficiency. Thrombocytopenia and leukemia may present with oral petechiae, purpura, oral hematomas, or hemorrhagic bullae (FIGURE 20).13 Painless pseudomembranous mucosal erosions may be a presentation for secondary syphilis.16
Look for signs that suggest malignancy
In the United States, oral and pharyngeal cancers account for approximately 40,000 cases of cancer and 8000 deaths each year.17 More than 90% of these are squamous cell carcinomas (SCCs); the remainder are mainly salivary gland tumors, lymphoma, and other infrequent cancers.18
SCC of the oral cavity most commonly occurs on the tongue but can develop in any site, presenting as mucosal ulcers, plaques, or masses that do not heal (FIGURE 21). Tobacco and alcohol use are associated with up to 80% of cases of SCC of the head and neck.18 Some oropharyngeal SCCs are associated with human papillomavirus infection type 16.19
Potentially malignant oral lesions include leukoplakia and erythroplakia. Leukoplakia is a white patch or plaque of the oral mucosa that can’t be explained by any other clinical diagnosis (FIGURE 22). These lesions are at risk for malignant transformation and may demonstrate dysplasia or frank SCC on biopsy.20 Proliferative verrucous leukoplakia is a unique form of leukoplakia that’s characterized by a wrinkled appearance that is often multifocal; the condition is associated with a higher risk of malignant transformation.
Erythroplakia is a red patch that similarly can’t be explained by another diagnosis. It has a very high risk of malignant transformation over time. All potentially malignant oral lesions, including leukoplakia and erythroplakia, require biopsy and careful monitoring.
Non-SCC cancers. Salivary gland tumors are rare and most commonly occur in patients ages 55 to 65 years. Most neoplasms (70%-85%) occur in the parotid gland, while 8% to 15% develop in the submandibular salivary gland and less than 1% involve the sublingual gland.21 Minor salivary gland tissue, especially in the lips and palate, may also be affected (FIGURE 23). Patients present with circumscribed, fixed or movable, painless, soft or firm masses in a salivary gland.
Melanoma should be included in the differential diagnosis of oral pigmented lesions that have any features of cutaneous melanoma, such as asymmetry, irregular borders, or variable or changing color.22
Hematologic malignancies may initially present (or demonstrate evidence of relapse) in the oral cavity. Leukemia typically presents with sheet-like overgrowth and swelling of the gingiva with associated erythema and bleeding (FIGURE 24), whereas lymphoma typically presents as a solitary mass or ulceration. Solid tumors that metastasize to the oral cavity may present with localized unexplained soft or hard tissue growths, with or without associated neurologic symptoms (eg, paresthesia).
CORRESPONDENCE
William D. Anderson, III, MD, DABFM, FAAFP, University of South Carolina School of Medicine, 15 Medical Park, Suite 300, Columbia, SC 29203; [email protected]
1. Spruance SL. The natural history of recurrent oral-facial herpes simplex virus infection. Semin Dermatol. 1992;11:200-206.
2. Balasubramaniam R, Kuperstein AS, Stoopler ET. Update on oral herpes virus infections. Dent Clin North Am. 2014;58:265-280.
3. Lozada-Nur F, Gorsky M, Silverman S Jr. Oral erythema multiforme: clinical observations and treatment of 95 patients. Oral Surg Oral Med Oral Pathol. 1989;67:36-40.
4. Allareddy V, Rampa S, Lee MK, et al. Hospital-based emergency department visits involving dental conditions: profile and predictors of poor outcomes and resource utilization. J Am Dent Assoc. 2014;145:331-337.
5. Allareddy V, Lin CY, Shah A, et al. Outcomes in patients hospitalized for periapical abscess in the United States: an analysis involving the use of a nationwide inpatient sample. J Am Dent Assoc. 2010;141:1107-1116.
6. Folayan MO. The epidemiology, etiology, and pathophysiology of acute necrotizing ulcerative gingivitis associated with malnutrition. J Contemp Dent Pract. 2004;5:28-41.
7. Atout RN, Todescan S. Managing patients with necrotizing ulcerative gingivitis. J Can Dent Assoc. 2013;79:d46.
8. Todescan S, Nizar R. Managing patients with necrotizing ulcerative periodontitis. J Can Dent Assoc. 2013;79:d44.
9. Allareddy V, Allareddy V, Nalliah RP. Epidemiology of facial fracture injuries. J Oral Maxillofac Surg. 2011;69:2613-2618.
10. Nalliah RP, Allareddy V, Kim MK, et al. Economics of facial fracture reductions in the United States over 12 months. Dent Traumatol. 2013;29:115-120.
11. Padmavathi B, Sharma S, Astekar M, et al. Oral Crohn’s disease. J Oral Maxillofac Pathol. 2014;18(suppl 1):S139-S142.
12. Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
13. Chi AC, Neville BW, Krayer JW, et al. Oral manifestations of systemic disease. Am Fam Physician. 2010;82:1381-1388.
14. Lavanya N, Jayanthi P, Rao UK, et al. Oral lichen planus: An update on pathogenesis and treatment. J Oral Maxillofac Pathol. 2011;15:127-132.
15. Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemic lupus erythematosus. Autoimmune Dis. 2012;2012:834291.
16. Ficarra G, Carlos R. Syphilis: The renaissance of an old disease with oral implications. Head Neck Pathol. 2009;3:195-206.
17. Siegel R, Ward E, Brawley O, et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212-236.
18. Licitra L, Locati LD, Bossi P, et al. Head and neck tumors other than squamous cell carcinoma. Curr Opin Oncol. 2004;16:236-241.
19. Gillison ML, Broutian T, Pickard RK, et al. Prevalence of oral HPV infection in the United States, 2009-2010. JAMA. 2012;307:693-703.
20. Silverman S Jr., Gorsky M, Lozada F. Oral leukoplakia and malignant transformation: A follow‐up study of 257 patients. Cancer. 1984;53:563-568.
21. Spiro RH. Salivary neoplasms: overview of a 35-year experience with 2807 patients. Head Neck Surg. 1986;8:177-184.
22. DeMatos P, Tyler DS, Seigler HF. Malignant melanoma of the mucous membranes: a review of 119 cases. Ann Surg Oncol. 1998;5:733-742.
1. Spruance SL. The natural history of recurrent oral-facial herpes simplex virus infection. Semin Dermatol. 1992;11:200-206.
2. Balasubramaniam R, Kuperstein AS, Stoopler ET. Update on oral herpes virus infections. Dent Clin North Am. 2014;58:265-280.
3. Lozada-Nur F, Gorsky M, Silverman S Jr. Oral erythema multiforme: clinical observations and treatment of 95 patients. Oral Surg Oral Med Oral Pathol. 1989;67:36-40.
4. Allareddy V, Rampa S, Lee MK, et al. Hospital-based emergency department visits involving dental conditions: profile and predictors of poor outcomes and resource utilization. J Am Dent Assoc. 2014;145:331-337.
5. Allareddy V, Lin CY, Shah A, et al. Outcomes in patients hospitalized for periapical abscess in the United States: an analysis involving the use of a nationwide inpatient sample. J Am Dent Assoc. 2010;141:1107-1116.
6. Folayan MO. The epidemiology, etiology, and pathophysiology of acute necrotizing ulcerative gingivitis associated with malnutrition. J Contemp Dent Pract. 2004;5:28-41.
7. Atout RN, Todescan S. Managing patients with necrotizing ulcerative gingivitis. J Can Dent Assoc. 2013;79:d46.
8. Todescan S, Nizar R. Managing patients with necrotizing ulcerative periodontitis. J Can Dent Assoc. 2013;79:d44.
9. Allareddy V, Allareddy V, Nalliah RP. Epidemiology of facial fracture injuries. J Oral Maxillofac Surg. 2011;69:2613-2618.
10. Nalliah RP, Allareddy V, Kim MK, et al. Economics of facial fracture reductions in the United States over 12 months. Dent Traumatol. 2013;29:115-120.
11. Padmavathi B, Sharma S, Astekar M, et al. Oral Crohn’s disease. J Oral Maxillofac Pathol. 2014;18(suppl 1):S139-S142.
12. Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
13. Chi AC, Neville BW, Krayer JW, et al. Oral manifestations of systemic disease. Am Fam Physician. 2010;82:1381-1388.
14. Lavanya N, Jayanthi P, Rao UK, et al. Oral lichen planus: An update on pathogenesis and treatment. J Oral Maxillofac Pathol. 2011;15:127-132.
15. Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemic lupus erythematosus. Autoimmune Dis. 2012;2012:834291.
16. Ficarra G, Carlos R. Syphilis: The renaissance of an old disease with oral implications. Head Neck Pathol. 2009;3:195-206.
17. Siegel R, Ward E, Brawley O, et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212-236.
18. Licitra L, Locati LD, Bossi P, et al. Head and neck tumors other than squamous cell carcinoma. Curr Opin Oncol. 2004;16:236-241.
19. Gillison ML, Broutian T, Pickard RK, et al. Prevalence of oral HPV infection in the United States, 2009-2010. JAMA. 2012;307:693-703.
20. Silverman S Jr., Gorsky M, Lozada F. Oral leukoplakia and malignant transformation: A follow‐up study of 257 patients. Cancer. 1984;53:563-568.
21. Spiro RH. Salivary neoplasms: overview of a 35-year experience with 2807 patients. Head Neck Surg. 1986;8:177-184.
22. DeMatos P, Tyler DS, Seigler HF. Malignant melanoma of the mucous membranes: a review of 119 cases. Ann Surg Oncol. 1998;5:733-742.