User login
When a fetus survives methotrexate exposure
CASE A 28-year-old woman, gravida 4, para 3, requested medical termination of her pregnancy at approximately 7 weeks’ gestation. She was given intramuscular methotrexate 50 mg/m2 and oral misoprostol 400 mcg. Nine weeks later, she presented at our primary care clinic complaining of mild pelvic pain. Given her history, we ordered transvaginal ultrasound, which showed a viable pregnancy with an average ultrasound age of 16 weeks’ gestation and grossly normal fetal anatomy. We counseled the patient regarding the risks associated with maintaining the pregnancy after exposure to methotrexate, and she and her husband elected to proceed with the pregnancy.
Throughout the pregnancy, a perinatologist conducted serial 2-dimensional ultrasounds. At 30 weeks’ gestation, ultrasound revealed mild hydrocephalus but yielded poor visualization of the kidneys, heart, and spine. A repeat ultrasound at 34 weeks demonstrated unchanged hydrocephalus and normal fetal anatomy with appropriate interval growth. A fetal echocardiogram in utero showed no cardiac anomalies. At 39 weeks, the patient underwent induction of labor due to severe oligohydramnios. A viable female infant weighing 2456 g was delivered by spontaneous vaginal delivery, with Apgar scores of 8 and 9 at 1 and 5 minutes, respectively.
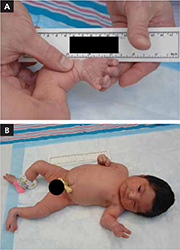
The infant was small for her gestational age. Her hands had 4 digits each, including the thumb (FIGURE 1A). The upper extremities were shorter than expected in relation to the infant’s torso and were locked in 90 degrees of flexion at the elbow (FIGURE 1B). Her lower extremities were both normal in length, but her left ankle joint was everted with considerable laxity at the tibiotalar joint. The infant’s mandible deviated to the right. An ultrasound of her head showed dilation of the lateral ventricle, consistent with nonobstructive hydrocephalus. A skeletal survey revealed bilateral radial-humeral synostosis at approximately 90 degrees of flexion. The second day after birth, the infant was transferred to a tertiary medical facility for further evaluation by pediatric subspecialists.
A pediatric orthopedic surgeon noted bilateral hip dysplasia and fitted the infant for a Pavlik harness. Ultrasound of the spine identified a dysmorphic sacrum with a thickened conus medullaris ending at the level of L2-L3, with increased risk for a tethered spinal cord. To correct the hydrocephalus, a neurosurgeon recommended intervention in the neonatal period. The parents were counseled to expect some degree of developmental disability, and karyotyping was performed to rule out potential genetically linked syndromes.
FIGURE 1
Methotrexate exposure led to these congenital anomalies
Methotrexate exposure at 7 weeks’ gestation resulted in this child having 4 digits on each hand (A), shortened arms locked in 90 degrees of flexion at the elbow, and an everted left ankle joint (B).
Risks associated with methotrexate
In the 1960s, methotrexate was commonly used as an abortifacient.1 Its use for that purpose became rare, however, after multiple reports from the 1950s to the 1970s of congenital malformations in infants exposed to the drug in utero, either inadvertently or after attempted abortion.1 In the 1990s, its use increased again in conjunction with misoprostol, primarily for medical management of suspected ectopic pregnancies and less often for elective terminations. Use of the combination resulted in fewer reports of congenital anomalies.1
The failure rate of medical abortion using methotrexate varies. In 1999, one study reported an 8% failure rate when methotrexate was used with misoprostol.2 In 2004, methotrexate alone led to a failure rate of 31% in medical termination of early pregnancy.3 In 2005, another study of methotrexate and misoprostol used in combination for elective termination reported a failure rate of 2% to 10%.4
Risk is not always foreseen
Methotrexate is widely used to treat such conditions as neoplastic disease and autoimmune disorders. Unintended exposure of a fetus to methotrexate is a very real possibility when the drug is used to treat a mother’s rheumatoid arthritis, psoriasis, or systemic lupus erythematosus. Methotrexate is a folic acid antagonist that produces its most teratogenic effects between 6 and 8 weeks postconception.4 Anomalies associated with methotrexate exposure include skull defects, central nervous system abnormalities, limb defects, gastrointestinal and cardiopulmonary defects, developmental delay, and cognitive impairment.4 Even at low doses and with short-term exposure, methotrexate can cause substantial fetal anomalies. In 2002, a woman who was unknowingly 3.5 weeks pregnant used oral methotrexate 7.5 mg/d for 2 days to treat her psoriasis.5 During a fetal anatomy sonogram at 18 weeks’ gestation, multiple anomalies were noted and later confirmed at fetopsy.
A meta-analysis on the safety of methotrexate in treating rheumatoid arthritis concluded that, for doses typical in this setting, data were lacking regarding the safety and risks of the drug during conception, pregnancy, and lactation.6 The review said that rheumatologists should discourage patients from continuing methotrexate if they wish to become pregnant, and that any continuing pregnancy should be closely monitored.6
The exact malformation rate after in utero exposure to methotrexate is unknown.7 Kozlowski et al reported 10 pregnancies in which the fetus was exposed to low-dose methotrexate (5 mg orally every week) for the treatment of rheumatoid arthritis.8 Five of the pregnancies were carried to term and the newborns exhibited no abnormalities, thus illustrating the drug’s variability for teratogenicity. The risk is real, however, and methotrexate can remain in human tissue for up to 8 months, thereby putting a fetus at risk for exposure even after a mother has discontinued the drug.2
The importance of primary care counsel
Incidental exposure. Inform any pregnant patient who has used methotrexate of the potential for congenital anomalies. The capacity to make educated decisions about elective termination of pregnancy requires a full disclosure of risks. In particular, ultrasound may not identify teratogenic effects from methotrexate exposure, and antenatal diagnosis of congenital anomalies is uncommon.9 Diagnosis is usually made at delivery. Thorough counseling on this point is imperative to prevent a false reassurance of having a normal fetus.
Did medical termination fail? Despite methotrexate’s widespread use for pregnancy termination, insufficient published data exist to guide the counseling of patients who have experienced a failed termination. Nevertheless, primary care physicians are often called on to counsel such patients.
Only about half of women who undergo a medical termination procedure attend follow-up visits with the abortion provider.10 One reason is the distance some patients travel and the associated costs. A 2000 report showed that 87% of counties in the United States lack even a single abortion provider, and that approximately 25% of women travel 50 miles or more for their abortions.11
Financial hardship leads some women to opt for continuing a pregnancy after a failed elective termination.1 That was the case with our patient. When she began experiencing pelvic pain after the termination procedure, she did not return to the abortion clinic, but instead sought guidance from her primary care physician at our medical center. After learning that she was 16 weeks pregnant, she opted to proceed with the pregnancy because she couldn’t afford a second elective termination.
Primary care involvement makes sense for other reasons as well. Protocols requiring in-person follow-up appointments after elective termination may not make the best use of the medical system.10 The high proportion of “no shows” can lead to scheduling difficulties and reduce a provider’s availability to perform abortions. This in turn would lead to a loss of income for the provider and could possibly increase the total cost of medical care.
One proposed solution has been to teach women how to recognize the signs and symptoms of a successful abortion or possible complications. However, a study of methotrexate-misoprostol abortion in the United States showed that women were often unable to assess whether they had successfully aborted.10 Of 50 women, 28 thought they had aborted by day 9, and 13 of those (46%) were still pregnant.10 A patient’s overestimation of her ability to make such judgments is thought to be another reason for the low follow-up rates post termination.
When termination is performed—regardless of the modality used—it is imperative to confirm that it was successful. Primary care providers, who are usually accessible and offer cost-effective care, can provide such confirmation. In addition, primary care physicians may need to address the psychological stress caused by elective termination.
1. Wheeler M, O’Meara P, Stanford M. Fetal methotrexate and misoprostol exposure: the past revisited. Teratology. 2002;66:73-76.
2. Carbonell Esteve JL, Varela L, Velazco A, et al. 25 mg or 50 mg of oral methotrexate followed by vaginal misoprostol 7 days after for early abortion: a randomized trial. Gynecol Obstet Invest. 1999;47:182-187.
3. Addar MH. Methotrexate embryopathy in a surviving intrauterine fetus after presumed diagnosis of ectopic pregnancy: case report. J Obstet Gynecol Can. 2004;26:1001-1003.
4. Yedlinsky NT, Morgan FC, Whitecar PW. Anomalies associated with failed methotrexate and misoprostol termination. Obstet Gynecol. 2005;105:1203-1205.
5. Nguyen C, Duhl AJ, Escallon CS, et al. Multiple anomalies in a fetus exposed to low-dose methotrexate in the first trimester. Obstet Gynecol. 2002;99:599-602.
6. Martinez Lopez JA, Loza E, Carmona L. Systemic review of the safety of methotrexate in rheumatoid arthritis regarding the reproductive system (fertility, pregnancy and breastfeeding). Clin Exp Rheumatol. 2009;27:678-684.
7. Goffman D, Cole DS, Bobby P, et al. Failed methotrexate termination of pregnancy: a case report. J Perinatol. 2006;26:645-647.
8. Kozlowski RD, Steinbrunner JV, MacKenzie AH, et al. Outcome of first trimester exposure to low dose methotrexate in eight patients with rheumatic disease. Am J Med. 1990;88:589-592.
9. Chapa JB, Hibbard JU, Weber EM, et al. Prenatal diagnosis of methotrexate embryopathy. Obstet Gynecol. 2003;101:1104-1107.
10. Grossman D, Ellertson C, Grimes DA, et al. Routine follow-up visits after first trimester induced abortion. Obstet Gynecol. 2004;103:738-745.
11. Finer LB, Henshaw SK. Abortion incidence and services in the United States in 2000. Perspect Sex Reprod Health. 2003;35:6-15.
CORRESPONDENCE Tammy Donoway, DO, Family Medicine, Womack Army Medical Center, 4-2817 Reilly Road, Fort Bragg, NC 28310; [email protected]
CASE A 28-year-old woman, gravida 4, para 3, requested medical termination of her pregnancy at approximately 7 weeks’ gestation. She was given intramuscular methotrexate 50 mg/m2 and oral misoprostol 400 mcg. Nine weeks later, she presented at our primary care clinic complaining of mild pelvic pain. Given her history, we ordered transvaginal ultrasound, which showed a viable pregnancy with an average ultrasound age of 16 weeks’ gestation and grossly normal fetal anatomy. We counseled the patient regarding the risks associated with maintaining the pregnancy after exposure to methotrexate, and she and her husband elected to proceed with the pregnancy.
Throughout the pregnancy, a perinatologist conducted serial 2-dimensional ultrasounds. At 30 weeks’ gestation, ultrasound revealed mild hydrocephalus but yielded poor visualization of the kidneys, heart, and spine. A repeat ultrasound at 34 weeks demonstrated unchanged hydrocephalus and normal fetal anatomy with appropriate interval growth. A fetal echocardiogram in utero showed no cardiac anomalies. At 39 weeks, the patient underwent induction of labor due to severe oligohydramnios. A viable female infant weighing 2456 g was delivered by spontaneous vaginal delivery, with Apgar scores of 8 and 9 at 1 and 5 minutes, respectively.
The infant was small for her gestational age. Her hands had 4 digits each, including the thumb (FIGURE 1A). The upper extremities were shorter than expected in relation to the infant’s torso and were locked in 90 degrees of flexion at the elbow (FIGURE 1B). Her lower extremities were both normal in length, but her left ankle joint was everted with considerable laxity at the tibiotalar joint. The infant’s mandible deviated to the right. An ultrasound of her head showed dilation of the lateral ventricle, consistent with nonobstructive hydrocephalus. A skeletal survey revealed bilateral radial-humeral synostosis at approximately 90 degrees of flexion. The second day after birth, the infant was transferred to a tertiary medical facility for further evaluation by pediatric subspecialists.
A pediatric orthopedic surgeon noted bilateral hip dysplasia and fitted the infant for a Pavlik harness. Ultrasound of the spine identified a dysmorphic sacrum with a thickened conus medullaris ending at the level of L2-L3, with increased risk for a tethered spinal cord. To correct the hydrocephalus, a neurosurgeon recommended intervention in the neonatal period. The parents were counseled to expect some degree of developmental disability, and karyotyping was performed to rule out potential genetically linked syndromes.
FIGURE 1
Methotrexate exposure led to these congenital anomalies
Methotrexate exposure at 7 weeks’ gestation resulted in this child having 4 digits on each hand (A), shortened arms locked in 90 degrees of flexion at the elbow, and an everted left ankle joint (B).
Risks associated with methotrexate
In the 1960s, methotrexate was commonly used as an abortifacient.1 Its use for that purpose became rare, however, after multiple reports from the 1950s to the 1970s of congenital malformations in infants exposed to the drug in utero, either inadvertently or after attempted abortion.1 In the 1990s, its use increased again in conjunction with misoprostol, primarily for medical management of suspected ectopic pregnancies and less often for elective terminations. Use of the combination resulted in fewer reports of congenital anomalies.1
The failure rate of medical abortion using methotrexate varies. In 1999, one study reported an 8% failure rate when methotrexate was used with misoprostol.2 In 2004, methotrexate alone led to a failure rate of 31% in medical termination of early pregnancy.3 In 2005, another study of methotrexate and misoprostol used in combination for elective termination reported a failure rate of 2% to 10%.4
Risk is not always foreseen
Methotrexate is widely used to treat such conditions as neoplastic disease and autoimmune disorders. Unintended exposure of a fetus to methotrexate is a very real possibility when the drug is used to treat a mother’s rheumatoid arthritis, psoriasis, or systemic lupus erythematosus. Methotrexate is a folic acid antagonist that produces its most teratogenic effects between 6 and 8 weeks postconception.4 Anomalies associated with methotrexate exposure include skull defects, central nervous system abnormalities, limb defects, gastrointestinal and cardiopulmonary defects, developmental delay, and cognitive impairment.4 Even at low doses and with short-term exposure, methotrexate can cause substantial fetal anomalies. In 2002, a woman who was unknowingly 3.5 weeks pregnant used oral methotrexate 7.5 mg/d for 2 days to treat her psoriasis.5 During a fetal anatomy sonogram at 18 weeks’ gestation, multiple anomalies were noted and later confirmed at fetopsy.
A meta-analysis on the safety of methotrexate in treating rheumatoid arthritis concluded that, for doses typical in this setting, data were lacking regarding the safety and risks of the drug during conception, pregnancy, and lactation.6 The review said that rheumatologists should discourage patients from continuing methotrexate if they wish to become pregnant, and that any continuing pregnancy should be closely monitored.6
The exact malformation rate after in utero exposure to methotrexate is unknown.7 Kozlowski et al reported 10 pregnancies in which the fetus was exposed to low-dose methotrexate (5 mg orally every week) for the treatment of rheumatoid arthritis.8 Five of the pregnancies were carried to term and the newborns exhibited no abnormalities, thus illustrating the drug’s variability for teratogenicity. The risk is real, however, and methotrexate can remain in human tissue for up to 8 months, thereby putting a fetus at risk for exposure even after a mother has discontinued the drug.2
The importance of primary care counsel
Incidental exposure. Inform any pregnant patient who has used methotrexate of the potential for congenital anomalies. The capacity to make educated decisions about elective termination of pregnancy requires a full disclosure of risks. In particular, ultrasound may not identify teratogenic effects from methotrexate exposure, and antenatal diagnosis of congenital anomalies is uncommon.9 Diagnosis is usually made at delivery. Thorough counseling on this point is imperative to prevent a false reassurance of having a normal fetus.
Did medical termination fail? Despite methotrexate’s widespread use for pregnancy termination, insufficient published data exist to guide the counseling of patients who have experienced a failed termination. Nevertheless, primary care physicians are often called on to counsel such patients.
Only about half of women who undergo a medical termination procedure attend follow-up visits with the abortion provider.10 One reason is the distance some patients travel and the associated costs. A 2000 report showed that 87% of counties in the United States lack even a single abortion provider, and that approximately 25% of women travel 50 miles or more for their abortions.11
Financial hardship leads some women to opt for continuing a pregnancy after a failed elective termination.1 That was the case with our patient. When she began experiencing pelvic pain after the termination procedure, she did not return to the abortion clinic, but instead sought guidance from her primary care physician at our medical center. After learning that she was 16 weeks pregnant, she opted to proceed with the pregnancy because she couldn’t afford a second elective termination.
Primary care involvement makes sense for other reasons as well. Protocols requiring in-person follow-up appointments after elective termination may not make the best use of the medical system.10 The high proportion of “no shows” can lead to scheduling difficulties and reduce a provider’s availability to perform abortions. This in turn would lead to a loss of income for the provider and could possibly increase the total cost of medical care.
One proposed solution has been to teach women how to recognize the signs and symptoms of a successful abortion or possible complications. However, a study of methotrexate-misoprostol abortion in the United States showed that women were often unable to assess whether they had successfully aborted.10 Of 50 women, 28 thought they had aborted by day 9, and 13 of those (46%) were still pregnant.10 A patient’s overestimation of her ability to make such judgments is thought to be another reason for the low follow-up rates post termination.
When termination is performed—regardless of the modality used—it is imperative to confirm that it was successful. Primary care providers, who are usually accessible and offer cost-effective care, can provide such confirmation. In addition, primary care physicians may need to address the psychological stress caused by elective termination.
CASE A 28-year-old woman, gravida 4, para 3, requested medical termination of her pregnancy at approximately 7 weeks’ gestation. She was given intramuscular methotrexate 50 mg/m2 and oral misoprostol 400 mcg. Nine weeks later, she presented at our primary care clinic complaining of mild pelvic pain. Given her history, we ordered transvaginal ultrasound, which showed a viable pregnancy with an average ultrasound age of 16 weeks’ gestation and grossly normal fetal anatomy. We counseled the patient regarding the risks associated with maintaining the pregnancy after exposure to methotrexate, and she and her husband elected to proceed with the pregnancy.
Throughout the pregnancy, a perinatologist conducted serial 2-dimensional ultrasounds. At 30 weeks’ gestation, ultrasound revealed mild hydrocephalus but yielded poor visualization of the kidneys, heart, and spine. A repeat ultrasound at 34 weeks demonstrated unchanged hydrocephalus and normal fetal anatomy with appropriate interval growth. A fetal echocardiogram in utero showed no cardiac anomalies. At 39 weeks, the patient underwent induction of labor due to severe oligohydramnios. A viable female infant weighing 2456 g was delivered by spontaneous vaginal delivery, with Apgar scores of 8 and 9 at 1 and 5 minutes, respectively.
The infant was small for her gestational age. Her hands had 4 digits each, including the thumb (FIGURE 1A). The upper extremities were shorter than expected in relation to the infant’s torso and were locked in 90 degrees of flexion at the elbow (FIGURE 1B). Her lower extremities were both normal in length, but her left ankle joint was everted with considerable laxity at the tibiotalar joint. The infant’s mandible deviated to the right. An ultrasound of her head showed dilation of the lateral ventricle, consistent with nonobstructive hydrocephalus. A skeletal survey revealed bilateral radial-humeral synostosis at approximately 90 degrees of flexion. The second day after birth, the infant was transferred to a tertiary medical facility for further evaluation by pediatric subspecialists.
A pediatric orthopedic surgeon noted bilateral hip dysplasia and fitted the infant for a Pavlik harness. Ultrasound of the spine identified a dysmorphic sacrum with a thickened conus medullaris ending at the level of L2-L3, with increased risk for a tethered spinal cord. To correct the hydrocephalus, a neurosurgeon recommended intervention in the neonatal period. The parents were counseled to expect some degree of developmental disability, and karyotyping was performed to rule out potential genetically linked syndromes.
FIGURE 1
Methotrexate exposure led to these congenital anomalies
Methotrexate exposure at 7 weeks’ gestation resulted in this child having 4 digits on each hand (A), shortened arms locked in 90 degrees of flexion at the elbow, and an everted left ankle joint (B).
Risks associated with methotrexate
In the 1960s, methotrexate was commonly used as an abortifacient.1 Its use for that purpose became rare, however, after multiple reports from the 1950s to the 1970s of congenital malformations in infants exposed to the drug in utero, either inadvertently or after attempted abortion.1 In the 1990s, its use increased again in conjunction with misoprostol, primarily for medical management of suspected ectopic pregnancies and less often for elective terminations. Use of the combination resulted in fewer reports of congenital anomalies.1
The failure rate of medical abortion using methotrexate varies. In 1999, one study reported an 8% failure rate when methotrexate was used with misoprostol.2 In 2004, methotrexate alone led to a failure rate of 31% in medical termination of early pregnancy.3 In 2005, another study of methotrexate and misoprostol used in combination for elective termination reported a failure rate of 2% to 10%.4
Risk is not always foreseen
Methotrexate is widely used to treat such conditions as neoplastic disease and autoimmune disorders. Unintended exposure of a fetus to methotrexate is a very real possibility when the drug is used to treat a mother’s rheumatoid arthritis, psoriasis, or systemic lupus erythematosus. Methotrexate is a folic acid antagonist that produces its most teratogenic effects between 6 and 8 weeks postconception.4 Anomalies associated with methotrexate exposure include skull defects, central nervous system abnormalities, limb defects, gastrointestinal and cardiopulmonary defects, developmental delay, and cognitive impairment.4 Even at low doses and with short-term exposure, methotrexate can cause substantial fetal anomalies. In 2002, a woman who was unknowingly 3.5 weeks pregnant used oral methotrexate 7.5 mg/d for 2 days to treat her psoriasis.5 During a fetal anatomy sonogram at 18 weeks’ gestation, multiple anomalies were noted and later confirmed at fetopsy.
A meta-analysis on the safety of methotrexate in treating rheumatoid arthritis concluded that, for doses typical in this setting, data were lacking regarding the safety and risks of the drug during conception, pregnancy, and lactation.6 The review said that rheumatologists should discourage patients from continuing methotrexate if they wish to become pregnant, and that any continuing pregnancy should be closely monitored.6
The exact malformation rate after in utero exposure to methotrexate is unknown.7 Kozlowski et al reported 10 pregnancies in which the fetus was exposed to low-dose methotrexate (5 mg orally every week) for the treatment of rheumatoid arthritis.8 Five of the pregnancies were carried to term and the newborns exhibited no abnormalities, thus illustrating the drug’s variability for teratogenicity. The risk is real, however, and methotrexate can remain in human tissue for up to 8 months, thereby putting a fetus at risk for exposure even after a mother has discontinued the drug.2
The importance of primary care counsel
Incidental exposure. Inform any pregnant patient who has used methotrexate of the potential for congenital anomalies. The capacity to make educated decisions about elective termination of pregnancy requires a full disclosure of risks. In particular, ultrasound may not identify teratogenic effects from methotrexate exposure, and antenatal diagnosis of congenital anomalies is uncommon.9 Diagnosis is usually made at delivery. Thorough counseling on this point is imperative to prevent a false reassurance of having a normal fetus.
Did medical termination fail? Despite methotrexate’s widespread use for pregnancy termination, insufficient published data exist to guide the counseling of patients who have experienced a failed termination. Nevertheless, primary care physicians are often called on to counsel such patients.
Only about half of women who undergo a medical termination procedure attend follow-up visits with the abortion provider.10 One reason is the distance some patients travel and the associated costs. A 2000 report showed that 87% of counties in the United States lack even a single abortion provider, and that approximately 25% of women travel 50 miles or more for their abortions.11
Financial hardship leads some women to opt for continuing a pregnancy after a failed elective termination.1 That was the case with our patient. When she began experiencing pelvic pain after the termination procedure, she did not return to the abortion clinic, but instead sought guidance from her primary care physician at our medical center. After learning that she was 16 weeks pregnant, she opted to proceed with the pregnancy because she couldn’t afford a second elective termination.
Primary care involvement makes sense for other reasons as well. Protocols requiring in-person follow-up appointments after elective termination may not make the best use of the medical system.10 The high proportion of “no shows” can lead to scheduling difficulties and reduce a provider’s availability to perform abortions. This in turn would lead to a loss of income for the provider and could possibly increase the total cost of medical care.
One proposed solution has been to teach women how to recognize the signs and symptoms of a successful abortion or possible complications. However, a study of methotrexate-misoprostol abortion in the United States showed that women were often unable to assess whether they had successfully aborted.10 Of 50 women, 28 thought they had aborted by day 9, and 13 of those (46%) were still pregnant.10 A patient’s overestimation of her ability to make such judgments is thought to be another reason for the low follow-up rates post termination.
When termination is performed—regardless of the modality used—it is imperative to confirm that it was successful. Primary care providers, who are usually accessible and offer cost-effective care, can provide such confirmation. In addition, primary care physicians may need to address the psychological stress caused by elective termination.
1. Wheeler M, O’Meara P, Stanford M. Fetal methotrexate and misoprostol exposure: the past revisited. Teratology. 2002;66:73-76.
2. Carbonell Esteve JL, Varela L, Velazco A, et al. 25 mg or 50 mg of oral methotrexate followed by vaginal misoprostol 7 days after for early abortion: a randomized trial. Gynecol Obstet Invest. 1999;47:182-187.
3. Addar MH. Methotrexate embryopathy in a surviving intrauterine fetus after presumed diagnosis of ectopic pregnancy: case report. J Obstet Gynecol Can. 2004;26:1001-1003.
4. Yedlinsky NT, Morgan FC, Whitecar PW. Anomalies associated with failed methotrexate and misoprostol termination. Obstet Gynecol. 2005;105:1203-1205.
5. Nguyen C, Duhl AJ, Escallon CS, et al. Multiple anomalies in a fetus exposed to low-dose methotrexate in the first trimester. Obstet Gynecol. 2002;99:599-602.
6. Martinez Lopez JA, Loza E, Carmona L. Systemic review of the safety of methotrexate in rheumatoid arthritis regarding the reproductive system (fertility, pregnancy and breastfeeding). Clin Exp Rheumatol. 2009;27:678-684.
7. Goffman D, Cole DS, Bobby P, et al. Failed methotrexate termination of pregnancy: a case report. J Perinatol. 2006;26:645-647.
8. Kozlowski RD, Steinbrunner JV, MacKenzie AH, et al. Outcome of first trimester exposure to low dose methotrexate in eight patients with rheumatic disease. Am J Med. 1990;88:589-592.
9. Chapa JB, Hibbard JU, Weber EM, et al. Prenatal diagnosis of methotrexate embryopathy. Obstet Gynecol. 2003;101:1104-1107.
10. Grossman D, Ellertson C, Grimes DA, et al. Routine follow-up visits after first trimester induced abortion. Obstet Gynecol. 2004;103:738-745.
11. Finer LB, Henshaw SK. Abortion incidence and services in the United States in 2000. Perspect Sex Reprod Health. 2003;35:6-15.
CORRESPONDENCE Tammy Donoway, DO, Family Medicine, Womack Army Medical Center, 4-2817 Reilly Road, Fort Bragg, NC 28310; [email protected]
1. Wheeler M, O’Meara P, Stanford M. Fetal methotrexate and misoprostol exposure: the past revisited. Teratology. 2002;66:73-76.
2. Carbonell Esteve JL, Varela L, Velazco A, et al. 25 mg or 50 mg of oral methotrexate followed by vaginal misoprostol 7 days after for early abortion: a randomized trial. Gynecol Obstet Invest. 1999;47:182-187.
3. Addar MH. Methotrexate embryopathy in a surviving intrauterine fetus after presumed diagnosis of ectopic pregnancy: case report. J Obstet Gynecol Can. 2004;26:1001-1003.
4. Yedlinsky NT, Morgan FC, Whitecar PW. Anomalies associated with failed methotrexate and misoprostol termination. Obstet Gynecol. 2005;105:1203-1205.
5. Nguyen C, Duhl AJ, Escallon CS, et al. Multiple anomalies in a fetus exposed to low-dose methotrexate in the first trimester. Obstet Gynecol. 2002;99:599-602.
6. Martinez Lopez JA, Loza E, Carmona L. Systemic review of the safety of methotrexate in rheumatoid arthritis regarding the reproductive system (fertility, pregnancy and breastfeeding). Clin Exp Rheumatol. 2009;27:678-684.
7. Goffman D, Cole DS, Bobby P, et al. Failed methotrexate termination of pregnancy: a case report. J Perinatol. 2006;26:645-647.
8. Kozlowski RD, Steinbrunner JV, MacKenzie AH, et al. Outcome of first trimester exposure to low dose methotrexate in eight patients with rheumatic disease. Am J Med. 1990;88:589-592.
9. Chapa JB, Hibbard JU, Weber EM, et al. Prenatal diagnosis of methotrexate embryopathy. Obstet Gynecol. 2003;101:1104-1107.
10. Grossman D, Ellertson C, Grimes DA, et al. Routine follow-up visits after first trimester induced abortion. Obstet Gynecol. 2004;103:738-745.
11. Finer LB, Henshaw SK. Abortion incidence and services in the United States in 2000. Perspect Sex Reprod Health. 2003;35:6-15.
CORRESPONDENCE Tammy Donoway, DO, Family Medicine, Womack Army Medical Center, 4-2817 Reilly Road, Fort Bragg, NC 28310; [email protected]
The work-up for mixed hyperlipidemia: A case study
A 42-year-old man with type 2 diabetes mellitus and hypertension was referred to our clinic for assessment of mixed hyperlipidemia found on routine investigation. Results of our physical examination were unremarkable. The patient had no xanthomatous deposits. His family history was strongly positive for type 2 diabetes. His medications included ramipril, glyburide, and hydrochlorothiazide.
In our further laboratory testing, a fasting blood sample revealed a grossly lipemic serum, with a total cholesterol level of 536.34 mg/dL (normal range=146.94-201.08 mg/dL), total triglyceride level of 5927.4 mg/dL (normal=31.15-151.3 mg/dL), and high-density cholesterol (HDL-C) level of 23.4 mg/dL (normal=35.1-93.6 mg/dL). His thyroid-stimulating hormone (TSH) level was 0.94 mIU/L (normal=0.49-4.67 mIU/L).
Results were in the normal range for urea, creatinine, electrolytes, bilirubin, alkaline phosphatase, alanine aminotransferase, and albumin. Hemoglobin A1c (HbA1c) was 9.5%.
Following clues to an accurate diagnosis
When the lipid phenotype is a mixed hyperlipidemia—a common disorder that becomes more prevalent with increasing age—investigate potential underlying disorders such as diabetes mellitus, renal failure, hypothyroidism, and chronic liver disease (TABLE 1). Ask about alcohol intake and use of medications including glucocorticoids and oral contraceptives. And explore the family history, particularly for premature heart disease, pancreatitis, and known lipid disorders. Epidemiologic studies have shown that higher-than-normal triglyceride levels increase the risk of coronary artery disease (CAD), and triglyceride levels greater than 500.44 mg/dL are associated with pancreatitis.1
What to look for in the physical examination. Measure body mass index (BMI), check blood pressure and carotid and peripheral pulses, and palpate the liver and thyroid. Inspect palms, soles, extensor surfaces of the arms, buttocks, and tendinous attachments for xanthomatous deposits.
Lab work. Order a fasting glucose test, renal panel, thyroid function tests, and a liver panel to detect or rule out diabetes, hypothyroidism, and renal and liver disease. Typically, in dyslipidemia due to excessive alcohol intake or estrogen use, HDL cholesterol is disproportionately elevated (TABLE 1). Patients with hypertriglyceridemia may also present with acute pancreatitis and relatively low amylase levels, due to interference by triglyceride-rich lipoproteins that can show falsely low amylase levels. Removal of chylomicrons from plasma by centrifugation before laboratory testing can eliminate such artifacts.2 In addition, hypertriglyceridemia can interfere with biochemical measurement of glucose, leading to falsely normal levels in these patients.3
To further refine the diagnosis, order lipoprotein electrophoresis, which identifies mixed hyperlipidemias according to the Fredrickson classification (types I–V).4
TABLE 1
Secondary causes of hyperlipidemia
| Underlying cause | Chylomicrons | VLDL | LDL | HDL | IDL | Lp(a) |
|---|---|---|---|---|---|---|
| Acromegaly | + | + | ||||
| Acute intermittent porphyria | + | |||||
| Alcohol | + | + | ||||
| Anorexia nervosa | + | |||||
| Autoimmune disease | + | + | ||||
| Cushing’s disease | + | |||||
| Diabetes mellitus (type 2) | + | + | – | |||
| Glucocorticoids | + | |||||
| Hepatitis | + | |||||
| Hypothyroidism | + | + | + | |||
| Liver disease (severe) | – | |||||
| Monoclonal gammopathies | + | |||||
| Multiple myeloma | + | |||||
| Nephrotic syndrome | + | + | ||||
| Obesity | + | – | ||||
| Oral contraceptives | + | + | ||||
| Renal failure | + | + | ||||
| +, elevated; –, reduced. HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; Lp(a), lipoprotein a; VLDL, very low-density lipoprotein. Adapted from: Rader DJ, Hobbs HH. Harrison’s Principles of Internal Medicine. 2012.10 | ||||||
Making sense of findings
Although patients with type 2 diabetes and hyperlipidemia most often have the type IV variety, they can also have other types, including type V. In uncontrolled diabetes, increased lipid metabolism mobilizes fat stores and increases VLDL and chylomicrons in plasma. Lipoprotein lipase activity is insulin dependent and is transiently reduced in insulin-deficient states, further increasing triglyceride levels.5
Hypothyroidism is classically associated with elevated plasma LDL cholesterol, but is also sometimes linked with high plasma triglycerides. The elevated plasma LDL cholesterol in hypothyroidism is due to reduced expression of LDL receptors resulting in impaired clearance of LDL.6 Elevated triglycerides in hypothyroidism are due to decreased lipoprotein lipase activity.7
Suspect primary (familial) hyperlipidemia (TABLE 2) if blood test results exclude such disorders as diabetes or hypothyroidism, and excessive alcohol intake and medication use have been ruled out. Some genetic causes of hyperchylomicronemia are rare and include familial lipoprotein lipase deficiency and apoprotein C-II deficiency. The differential diagnosis of mixed hyperlipidemia also includes familial combined hyperlipidemia (FCHL), familial dysbetalipoproteinemia, and familial hypertriglyceridemia.
FCHL can be difficult to differentiate from dyslipidemia of metabolic syndrome. A dominant inheritance pattern favors a diagnosis of FCHL, while environmental factors are more important in dyslipidemia of metabolic syndrome.8
TABLE 2
Primary hyperlipidemia
| Genetic disorder (Frederickson type) | Typical clinical findings |
|---|---|
| Familial lipoprotein lipase deficiency (type I) | Eruptive xanthomas, hepatosplenomegaly, pancreatitis |
| Familial apoprotein C-II deficiency (type I) | Eruptive xanthomas, hepatosplenomegaly, pancreatitis |
| Familial combined hyperlipidemia (type IIb) | Coronary or peripheral atherosclerosis |
| Familial dysbetalipoproteinemia (type III) | Palmar and tuberous xanthomas, coronary or peripheral atherosclerosis |
| Familial hypertriglyceridemia (type IV or V) | Eruptive xanthomas (type V) |
| Adapted from: Rader DJ, Hobbs HH. Harrison’s Principles of Internal Medicine. 2012.10 | |
How my patient’s case resolved
My patient’s case was consistent with secondary dyslipidemia due to diabetes and metabolic syndrome. But patients with triglyceride levels above 2001.77 mg/dL almost always have both a secondary and a genetic form of hyperlipidemia.9 My colleagues and I suspected Fredrickson’s type V hyperlipoproteinemia because of the high triglycerides. This was confirmed when the lipoprotein electrophoresis showed decreased alpha, increased prebeta, and normal beta fractions and chylomicronemia.
Treatment. Therapy choices differ depending on the type of mixed hyperlipidemia a patient has. However, fibrates are usually needed in addition to statins. (Of note: Statin-induced myopathy is more likely in patients who are also taking fibrates, so careful monitoring is important.)
I added fenofibrate, metformin, and rosuvastatin to the patient’s regimen, which included ramipril, glyburide, and hydrochlorothiazide. I also recommended lifestyle modifications and arranged a consultation with a dietician.
Four weeks later, his fasting lipid profile had improved: Total serum cholesterol level was 213.45 mg/dL, triglyceride level was 825.5 mg/dL, and HDL-C level was 37.05 mg/dL. Apolipoprotein B100 was 2.54 g/L (normal=0.59-1.46 g/L). At follow-up 3 months later, the patient’s total cholesterol level was 145.9 mg/dL, triglyceride level was 330.4 mg/dL, and HDL-C level was 27.84 mg/dL.
CORRESPONDENCE H.U. Rehman, MB, Clinical Associate Professor, Department of Medicine, Regina Qu’Appelle Health Region, Regina General Hospital, 1440 14th Avenue, Regina, SK, S4P 0W5, Canada; [email protected]
1. Sarwar N, Danesh J, Eiriksdottir G, et al. Triglycerides and the risk of coronary heart disease. 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450-458.
2. Chait A, Brunzell JD. Chylomicronemia syndrome. Adv Intern Med. 1992;37:249-273.
3. Rumbak MJ, Hughes TA, Kitabchi AE. Pseudonormoglycemia in diabetic ketoacidosis with elevated triglycerides. Am J Emerg Med. 1991;9:61-63.
4. Jialal I. A practical approach to the laboratory diagnosis of dyslipidemia. Am J Clin Pathol. 1996;106:128-138.
5. McLean AG, Petersons CJ, Hooper AJ, et al. Extreme diabetic lipaemia associated with a novel lipoprotein gene mutation. Clin Chem Acta. 2009;406:167-169.
6. Heimberg M, Olubadew JO, Wilcox HG. Plasma lipoproteins and regulation of hepatic metabolism of fatty acids in altered thyroid states. Endocr Rev. 1985;6:590-607.
7. Valdemarsson S, Hansson P, Hedner P, et al. Relations between thyroid function, hepatic and lipoprotein lipase activities, and plasma lipoprotein concentrations. Acta Endocrinol. 1983;104:50-56.
8. Cabezas MC, Rabelink TJ. Familial combined hyperlipidemia: The case of triglycerides. In: Betteridge DJ, ed. Case Studies in Lipid Management. London, England: Informa UK; 2007:85–93.
9. Martin D, McCann E, Glynn P. Rheologic reflection in hypertriglyceridemia-induced pancreatitis. South Med J. 2009;102:1049-1051.
10. Rader DJ, Hobbs HH. Disorders of lipoprotein metabolism. In: Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill Companies, Inc; 2012:3145-3161.
A 42-year-old man with type 2 diabetes mellitus and hypertension was referred to our clinic for assessment of mixed hyperlipidemia found on routine investigation. Results of our physical examination were unremarkable. The patient had no xanthomatous deposits. His family history was strongly positive for type 2 diabetes. His medications included ramipril, glyburide, and hydrochlorothiazide.
In our further laboratory testing, a fasting blood sample revealed a grossly lipemic serum, with a total cholesterol level of 536.34 mg/dL (normal range=146.94-201.08 mg/dL), total triglyceride level of 5927.4 mg/dL (normal=31.15-151.3 mg/dL), and high-density cholesterol (HDL-C) level of 23.4 mg/dL (normal=35.1-93.6 mg/dL). His thyroid-stimulating hormone (TSH) level was 0.94 mIU/L (normal=0.49-4.67 mIU/L).
Results were in the normal range for urea, creatinine, electrolytes, bilirubin, alkaline phosphatase, alanine aminotransferase, and albumin. Hemoglobin A1c (HbA1c) was 9.5%.
Following clues to an accurate diagnosis
When the lipid phenotype is a mixed hyperlipidemia—a common disorder that becomes more prevalent with increasing age—investigate potential underlying disorders such as diabetes mellitus, renal failure, hypothyroidism, and chronic liver disease (TABLE 1). Ask about alcohol intake and use of medications including glucocorticoids and oral contraceptives. And explore the family history, particularly for premature heart disease, pancreatitis, and known lipid disorders. Epidemiologic studies have shown that higher-than-normal triglyceride levels increase the risk of coronary artery disease (CAD), and triglyceride levels greater than 500.44 mg/dL are associated with pancreatitis.1
What to look for in the physical examination. Measure body mass index (BMI), check blood pressure and carotid and peripheral pulses, and palpate the liver and thyroid. Inspect palms, soles, extensor surfaces of the arms, buttocks, and tendinous attachments for xanthomatous deposits.
Lab work. Order a fasting glucose test, renal panel, thyroid function tests, and a liver panel to detect or rule out diabetes, hypothyroidism, and renal and liver disease. Typically, in dyslipidemia due to excessive alcohol intake or estrogen use, HDL cholesterol is disproportionately elevated (TABLE 1). Patients with hypertriglyceridemia may also present with acute pancreatitis and relatively low amylase levels, due to interference by triglyceride-rich lipoproteins that can show falsely low amylase levels. Removal of chylomicrons from plasma by centrifugation before laboratory testing can eliminate such artifacts.2 In addition, hypertriglyceridemia can interfere with biochemical measurement of glucose, leading to falsely normal levels in these patients.3
To further refine the diagnosis, order lipoprotein electrophoresis, which identifies mixed hyperlipidemias according to the Fredrickson classification (types I–V).4
TABLE 1
Secondary causes of hyperlipidemia
| Underlying cause | Chylomicrons | VLDL | LDL | HDL | IDL | Lp(a) |
|---|---|---|---|---|---|---|
| Acromegaly | + | + | ||||
| Acute intermittent porphyria | + | |||||
| Alcohol | + | + | ||||
| Anorexia nervosa | + | |||||
| Autoimmune disease | + | + | ||||
| Cushing’s disease | + | |||||
| Diabetes mellitus (type 2) | + | + | – | |||
| Glucocorticoids | + | |||||
| Hepatitis | + | |||||
| Hypothyroidism | + | + | + | |||
| Liver disease (severe) | – | |||||
| Monoclonal gammopathies | + | |||||
| Multiple myeloma | + | |||||
| Nephrotic syndrome | + | + | ||||
| Obesity | + | – | ||||
| Oral contraceptives | + | + | ||||
| Renal failure | + | + | ||||
| +, elevated; –, reduced. HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; Lp(a), lipoprotein a; VLDL, very low-density lipoprotein. Adapted from: Rader DJ, Hobbs HH. Harrison’s Principles of Internal Medicine. 2012.10 | ||||||
Making sense of findings
Although patients with type 2 diabetes and hyperlipidemia most often have the type IV variety, they can also have other types, including type V. In uncontrolled diabetes, increased lipid metabolism mobilizes fat stores and increases VLDL and chylomicrons in plasma. Lipoprotein lipase activity is insulin dependent and is transiently reduced in insulin-deficient states, further increasing triglyceride levels.5
Hypothyroidism is classically associated with elevated plasma LDL cholesterol, but is also sometimes linked with high plasma triglycerides. The elevated plasma LDL cholesterol in hypothyroidism is due to reduced expression of LDL receptors resulting in impaired clearance of LDL.6 Elevated triglycerides in hypothyroidism are due to decreased lipoprotein lipase activity.7
Suspect primary (familial) hyperlipidemia (TABLE 2) if blood test results exclude such disorders as diabetes or hypothyroidism, and excessive alcohol intake and medication use have been ruled out. Some genetic causes of hyperchylomicronemia are rare and include familial lipoprotein lipase deficiency and apoprotein C-II deficiency. The differential diagnosis of mixed hyperlipidemia also includes familial combined hyperlipidemia (FCHL), familial dysbetalipoproteinemia, and familial hypertriglyceridemia.
FCHL can be difficult to differentiate from dyslipidemia of metabolic syndrome. A dominant inheritance pattern favors a diagnosis of FCHL, while environmental factors are more important in dyslipidemia of metabolic syndrome.8
TABLE 2
Primary hyperlipidemia
| Genetic disorder (Frederickson type) | Typical clinical findings |
|---|---|
| Familial lipoprotein lipase deficiency (type I) | Eruptive xanthomas, hepatosplenomegaly, pancreatitis |
| Familial apoprotein C-II deficiency (type I) | Eruptive xanthomas, hepatosplenomegaly, pancreatitis |
| Familial combined hyperlipidemia (type IIb) | Coronary or peripheral atherosclerosis |
| Familial dysbetalipoproteinemia (type III) | Palmar and tuberous xanthomas, coronary or peripheral atherosclerosis |
| Familial hypertriglyceridemia (type IV or V) | Eruptive xanthomas (type V) |
| Adapted from: Rader DJ, Hobbs HH. Harrison’s Principles of Internal Medicine. 2012.10 | |
How my patient’s case resolved
My patient’s case was consistent with secondary dyslipidemia due to diabetes and metabolic syndrome. But patients with triglyceride levels above 2001.77 mg/dL almost always have both a secondary and a genetic form of hyperlipidemia.9 My colleagues and I suspected Fredrickson’s type V hyperlipoproteinemia because of the high triglycerides. This was confirmed when the lipoprotein electrophoresis showed decreased alpha, increased prebeta, and normal beta fractions and chylomicronemia.
Treatment. Therapy choices differ depending on the type of mixed hyperlipidemia a patient has. However, fibrates are usually needed in addition to statins. (Of note: Statin-induced myopathy is more likely in patients who are also taking fibrates, so careful monitoring is important.)
I added fenofibrate, metformin, and rosuvastatin to the patient’s regimen, which included ramipril, glyburide, and hydrochlorothiazide. I also recommended lifestyle modifications and arranged a consultation with a dietician.
Four weeks later, his fasting lipid profile had improved: Total serum cholesterol level was 213.45 mg/dL, triglyceride level was 825.5 mg/dL, and HDL-C level was 37.05 mg/dL. Apolipoprotein B100 was 2.54 g/L (normal=0.59-1.46 g/L). At follow-up 3 months later, the patient’s total cholesterol level was 145.9 mg/dL, triglyceride level was 330.4 mg/dL, and HDL-C level was 27.84 mg/dL.
CORRESPONDENCE H.U. Rehman, MB, Clinical Associate Professor, Department of Medicine, Regina Qu’Appelle Health Region, Regina General Hospital, 1440 14th Avenue, Regina, SK, S4P 0W5, Canada; [email protected]
A 42-year-old man with type 2 diabetes mellitus and hypertension was referred to our clinic for assessment of mixed hyperlipidemia found on routine investigation. Results of our physical examination were unremarkable. The patient had no xanthomatous deposits. His family history was strongly positive for type 2 diabetes. His medications included ramipril, glyburide, and hydrochlorothiazide.
In our further laboratory testing, a fasting blood sample revealed a grossly lipemic serum, with a total cholesterol level of 536.34 mg/dL (normal range=146.94-201.08 mg/dL), total triglyceride level of 5927.4 mg/dL (normal=31.15-151.3 mg/dL), and high-density cholesterol (HDL-C) level of 23.4 mg/dL (normal=35.1-93.6 mg/dL). His thyroid-stimulating hormone (TSH) level was 0.94 mIU/L (normal=0.49-4.67 mIU/L).
Results were in the normal range for urea, creatinine, electrolytes, bilirubin, alkaline phosphatase, alanine aminotransferase, and albumin. Hemoglobin A1c (HbA1c) was 9.5%.
Following clues to an accurate diagnosis
When the lipid phenotype is a mixed hyperlipidemia—a common disorder that becomes more prevalent with increasing age—investigate potential underlying disorders such as diabetes mellitus, renal failure, hypothyroidism, and chronic liver disease (TABLE 1). Ask about alcohol intake and use of medications including glucocorticoids and oral contraceptives. And explore the family history, particularly for premature heart disease, pancreatitis, and known lipid disorders. Epidemiologic studies have shown that higher-than-normal triglyceride levels increase the risk of coronary artery disease (CAD), and triglyceride levels greater than 500.44 mg/dL are associated with pancreatitis.1
What to look for in the physical examination. Measure body mass index (BMI), check blood pressure and carotid and peripheral pulses, and palpate the liver and thyroid. Inspect palms, soles, extensor surfaces of the arms, buttocks, and tendinous attachments for xanthomatous deposits.
Lab work. Order a fasting glucose test, renal panel, thyroid function tests, and a liver panel to detect or rule out diabetes, hypothyroidism, and renal and liver disease. Typically, in dyslipidemia due to excessive alcohol intake or estrogen use, HDL cholesterol is disproportionately elevated (TABLE 1). Patients with hypertriglyceridemia may also present with acute pancreatitis and relatively low amylase levels, due to interference by triglyceride-rich lipoproteins that can show falsely low amylase levels. Removal of chylomicrons from plasma by centrifugation before laboratory testing can eliminate such artifacts.2 In addition, hypertriglyceridemia can interfere with biochemical measurement of glucose, leading to falsely normal levels in these patients.3
To further refine the diagnosis, order lipoprotein electrophoresis, which identifies mixed hyperlipidemias according to the Fredrickson classification (types I–V).4
TABLE 1
Secondary causes of hyperlipidemia
| Underlying cause | Chylomicrons | VLDL | LDL | HDL | IDL | Lp(a) |
|---|---|---|---|---|---|---|
| Acromegaly | + | + | ||||
| Acute intermittent porphyria | + | |||||
| Alcohol | + | + | ||||
| Anorexia nervosa | + | |||||
| Autoimmune disease | + | + | ||||
| Cushing’s disease | + | |||||
| Diabetes mellitus (type 2) | + | + | – | |||
| Glucocorticoids | + | |||||
| Hepatitis | + | |||||
| Hypothyroidism | + | + | + | |||
| Liver disease (severe) | – | |||||
| Monoclonal gammopathies | + | |||||
| Multiple myeloma | + | |||||
| Nephrotic syndrome | + | + | ||||
| Obesity | + | – | ||||
| Oral contraceptives | + | + | ||||
| Renal failure | + | + | ||||
| +, elevated; –, reduced. HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; Lp(a), lipoprotein a; VLDL, very low-density lipoprotein. Adapted from: Rader DJ, Hobbs HH. Harrison’s Principles of Internal Medicine. 2012.10 | ||||||
Making sense of findings
Although patients with type 2 diabetes and hyperlipidemia most often have the type IV variety, they can also have other types, including type V. In uncontrolled diabetes, increased lipid metabolism mobilizes fat stores and increases VLDL and chylomicrons in plasma. Lipoprotein lipase activity is insulin dependent and is transiently reduced in insulin-deficient states, further increasing triglyceride levels.5
Hypothyroidism is classically associated with elevated plasma LDL cholesterol, but is also sometimes linked with high plasma triglycerides. The elevated plasma LDL cholesterol in hypothyroidism is due to reduced expression of LDL receptors resulting in impaired clearance of LDL.6 Elevated triglycerides in hypothyroidism are due to decreased lipoprotein lipase activity.7
Suspect primary (familial) hyperlipidemia (TABLE 2) if blood test results exclude such disorders as diabetes or hypothyroidism, and excessive alcohol intake and medication use have been ruled out. Some genetic causes of hyperchylomicronemia are rare and include familial lipoprotein lipase deficiency and apoprotein C-II deficiency. The differential diagnosis of mixed hyperlipidemia also includes familial combined hyperlipidemia (FCHL), familial dysbetalipoproteinemia, and familial hypertriglyceridemia.
FCHL can be difficult to differentiate from dyslipidemia of metabolic syndrome. A dominant inheritance pattern favors a diagnosis of FCHL, while environmental factors are more important in dyslipidemia of metabolic syndrome.8
TABLE 2
Primary hyperlipidemia
| Genetic disorder (Frederickson type) | Typical clinical findings |
|---|---|
| Familial lipoprotein lipase deficiency (type I) | Eruptive xanthomas, hepatosplenomegaly, pancreatitis |
| Familial apoprotein C-II deficiency (type I) | Eruptive xanthomas, hepatosplenomegaly, pancreatitis |
| Familial combined hyperlipidemia (type IIb) | Coronary or peripheral atherosclerosis |
| Familial dysbetalipoproteinemia (type III) | Palmar and tuberous xanthomas, coronary or peripheral atherosclerosis |
| Familial hypertriglyceridemia (type IV or V) | Eruptive xanthomas (type V) |
| Adapted from: Rader DJ, Hobbs HH. Harrison’s Principles of Internal Medicine. 2012.10 | |
How my patient’s case resolved
My patient’s case was consistent with secondary dyslipidemia due to diabetes and metabolic syndrome. But patients with triglyceride levels above 2001.77 mg/dL almost always have both a secondary and a genetic form of hyperlipidemia.9 My colleagues and I suspected Fredrickson’s type V hyperlipoproteinemia because of the high triglycerides. This was confirmed when the lipoprotein electrophoresis showed decreased alpha, increased prebeta, and normal beta fractions and chylomicronemia.
Treatment. Therapy choices differ depending on the type of mixed hyperlipidemia a patient has. However, fibrates are usually needed in addition to statins. (Of note: Statin-induced myopathy is more likely in patients who are also taking fibrates, so careful monitoring is important.)
I added fenofibrate, metformin, and rosuvastatin to the patient’s regimen, which included ramipril, glyburide, and hydrochlorothiazide. I also recommended lifestyle modifications and arranged a consultation with a dietician.
Four weeks later, his fasting lipid profile had improved: Total serum cholesterol level was 213.45 mg/dL, triglyceride level was 825.5 mg/dL, and HDL-C level was 37.05 mg/dL. Apolipoprotein B100 was 2.54 g/L (normal=0.59-1.46 g/L). At follow-up 3 months later, the patient’s total cholesterol level was 145.9 mg/dL, triglyceride level was 330.4 mg/dL, and HDL-C level was 27.84 mg/dL.
CORRESPONDENCE H.U. Rehman, MB, Clinical Associate Professor, Department of Medicine, Regina Qu’Appelle Health Region, Regina General Hospital, 1440 14th Avenue, Regina, SK, S4P 0W5, Canada; [email protected]
1. Sarwar N, Danesh J, Eiriksdottir G, et al. Triglycerides and the risk of coronary heart disease. 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450-458.
2. Chait A, Brunzell JD. Chylomicronemia syndrome. Adv Intern Med. 1992;37:249-273.
3. Rumbak MJ, Hughes TA, Kitabchi AE. Pseudonormoglycemia in diabetic ketoacidosis with elevated triglycerides. Am J Emerg Med. 1991;9:61-63.
4. Jialal I. A practical approach to the laboratory diagnosis of dyslipidemia. Am J Clin Pathol. 1996;106:128-138.
5. McLean AG, Petersons CJ, Hooper AJ, et al. Extreme diabetic lipaemia associated with a novel lipoprotein gene mutation. Clin Chem Acta. 2009;406:167-169.
6. Heimberg M, Olubadew JO, Wilcox HG. Plasma lipoproteins and regulation of hepatic metabolism of fatty acids in altered thyroid states. Endocr Rev. 1985;6:590-607.
7. Valdemarsson S, Hansson P, Hedner P, et al. Relations between thyroid function, hepatic and lipoprotein lipase activities, and plasma lipoprotein concentrations. Acta Endocrinol. 1983;104:50-56.
8. Cabezas MC, Rabelink TJ. Familial combined hyperlipidemia: The case of triglycerides. In: Betteridge DJ, ed. Case Studies in Lipid Management. London, England: Informa UK; 2007:85–93.
9. Martin D, McCann E, Glynn P. Rheologic reflection in hypertriglyceridemia-induced pancreatitis. South Med J. 2009;102:1049-1051.
10. Rader DJ, Hobbs HH. Disorders of lipoprotein metabolism. In: Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill Companies, Inc; 2012:3145-3161.
1. Sarwar N, Danesh J, Eiriksdottir G, et al. Triglycerides and the risk of coronary heart disease. 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450-458.
2. Chait A, Brunzell JD. Chylomicronemia syndrome. Adv Intern Med. 1992;37:249-273.
3. Rumbak MJ, Hughes TA, Kitabchi AE. Pseudonormoglycemia in diabetic ketoacidosis with elevated triglycerides. Am J Emerg Med. 1991;9:61-63.
4. Jialal I. A practical approach to the laboratory diagnosis of dyslipidemia. Am J Clin Pathol. 1996;106:128-138.
5. McLean AG, Petersons CJ, Hooper AJ, et al. Extreme diabetic lipaemia associated with a novel lipoprotein gene mutation. Clin Chem Acta. 2009;406:167-169.
6. Heimberg M, Olubadew JO, Wilcox HG. Plasma lipoproteins and regulation of hepatic metabolism of fatty acids in altered thyroid states. Endocr Rev. 1985;6:590-607.
7. Valdemarsson S, Hansson P, Hedner P, et al. Relations between thyroid function, hepatic and lipoprotein lipase activities, and plasma lipoprotein concentrations. Acta Endocrinol. 1983;104:50-56.
8. Cabezas MC, Rabelink TJ. Familial combined hyperlipidemia: The case of triglycerides. In: Betteridge DJ, ed. Case Studies in Lipid Management. London, England: Informa UK; 2007:85–93.
9. Martin D, McCann E, Glynn P. Rheologic reflection in hypertriglyceridemia-induced pancreatitis. South Med J. 2009;102:1049-1051.
10. Rader DJ, Hobbs HH. Disorders of lipoprotein metabolism. In: Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill Companies, Inc; 2012:3145-3161.
Combatting the cough that won’t quit
• Always include postnasal drip, asthma, and gastroesophageal reflux disease in the differential diagnosis for persistent cough, regardless of clinical signs and symptoms. B
• Do not rely on a patient’s description of the character and timing of the cough or the absence (or presence) of sputum to narrow down the differential diagnosis. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Margaret M, a 52-year-old nonsmoker, came to our clinic because of a persistent cough that had started about 4 weeks earlier. She had tried multiple over-the-counter cough suppressants, including dextromethorphan and guaifenesin, as well as cough drops, but none had been effective.
Margaret denied having had a cold or respiratory infection in the past few months or being in close contact with anyone with a chronic cough, and she had never had an asthma diagnosis. In response to a question about previous coughing episodes, the patient recalled having had several bouts of chronic cough in the past, including one about a year ago.
While Margaret had no known allergies, she did have occasional heartburn, which an antacid—or, at times, a drink of water—always relieved. Thyroid medication and calcium were the only things she took on a regular basis, separated by several hours to avoid problems with absorption.
Patients like Margaret, who seek help from their primary care physician only after attempting to combat a persistent cough on their own, may be quite frustrated by the time they arrive in your office. They’re counting on you to provide a cure. Fortunately, you’re likely to find it, as the differential diagnosis for subacute cough (a cough of 3-8 weeks’ duration) is limited.
Nonetheless, finding the cause of a subacute or chronic cough (lasting >8 weeks) is sometimes a matter of trial and error. Postnasal drip (also known as upper airway cough syndrome, or UACS), asthma, and gastroesophageal reflux disease (GERD) are the most common causes,1,2 followed by postinfectious cough, nonasthmatic eosinophilic bronchitis (NAEB), and pertussis.3 Although these conditions are all relatively well known, they are not always easy to detect: Some disorders, including UACS, asthma, and GERD, may be “silent,” with persistent cough the only presenting sign or symptom.4 In other cases, more than one condition may be contributing to the cough.

Starting with trials of empiric therapy for the most common causes of persistent cough—with sequential therapy and diagnostic tests, as needed—is far more effective than searching for relatively uncommon or obscure conditions. Following such a protocol, as detailed in the algorithm (FIGURE)4-7 we’ve developed and in the text that follows, can help you combat subacute and chronic cough in a cost-effective, timely way.
FIGURE
Dx and treatment when persistent cough is the only symptom4-7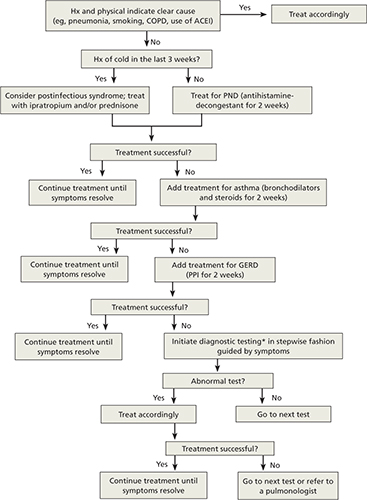
*May include CXR, PPD, B pertussis IgG or IgA, spirometry with methacholine inhalation challenge, barium swallow, prolonged pH monitoring, sinus CT, and sputum eosinophil count, excluding any tests that have already been performed.
ACEI, angiotensin-converting enzyme inhibitor; CT, computed tomography; COPD, chronic obstructive pulmonary disease; CXR, chest x-ray; GERD, gastroesophageal reflux disease; IgA, immunoglobulin A; IgG, immunoglobulin G; PND, postnasal drip; PPD, purified protein derivative; PPI, proton pump inhibitor.
Treat all patients for upper airway cough syndrome
Postnasal drip—renamed UACS by the guideline committee of the American Association of Chest Physicians because it isn’t clear whether the cough is caused by irritation from direct contact with postnasal drip or by inflammation of cough receptors in the upper airway—is the most common cause of chronic cough.6
The differential diagnosis for UACS, which is implicated in about 34% of cases of persistent cough, includes allergic, postinfectious, and occupational rhinitis; rhinitis due to anatomic abnormalities or physical or chemical irritants, rhinitis medicamentosa, and rhinitis of pregnancy; bacterial sinusitis; and allergic fungal sinusitis.8
The signs and symptoms of UACS are nonspecific, and a definitive diagnosis typically cannot be made from the medical history and physical examination alone. What’s more, the absence of any of the usual clinical findings—eg, rhinorrhea and excess sputum production—should not preclude an empiric trial with a first-generation antihistamine-decongestant combination such as brompheniramine/sustained-release pseudoephedrine. Second-and third-generation combination products, such as fexofenadine/pseudoephedrine, should not be used, as they are not effective in treating UACS.4
CASE Margaret’s physical exam was unremarkable. Her vital signs were stable, she had no cervical lymphadenopathy, and her chest was clear on auscultation. She had a dry cough that occurred twice during the exam, but not on inspiration.
The patient’s work-up included office spirometry, which was normal; a nasopharyngeal culture for Bordetella pertussis was negative. We prescribed a 2-week course of therapy with brompheniramine/sustained-release pseudoephedrine and scheduled a return visit shortly after it was completed.
There is no gold standard diagnostic test to confirm or rule out postnasal drip as the cause of cough. CT scanning of sinuses has a poor positive predictive value and is no longer recommended as part of an initial work-up,9 but may be useful for patients whose symptoms persist longer than 3 weeks.
Consider bronchodilator Tx when asthma is suspected
Cough-variant asthma is the second most common cause of persistent cough, and is responsible for an estimated 28% of cases.6 Asthma is the easiest of the conditions included in the differential diagnosis for persistent cough to establish in an office setting. The challenge is to remember to consider it in patients who present with cough but no sign of the classic expiratory wheezing. When you suspect that a patient has asthma, consider empiric bronchodilator therapy—or conduct spirometry testing.
Spirometric values of forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) <70% and a positive bronchodilator response (≥12%) are consistent with an asthma diagnosis. Management of asthma depends on severity, and patients should be evaluated based on the National Heart, Lung, and Blood Institute’s National Asthma Education and Prevention Program Guidelines for the Diagnosis and Management of Asthma.10
It is crucial to ask patients with asthma (and, indeed, to ask all patients with a persistent cough) about exposure to secondhand smoke, and to stress the importance of avoiding smoking and secondary exposure. Individuals who are regularly exposed to secondhand smoke report more nasal symptoms and greater use of nasal decongestants compared with people with no exposure to smoke;11 they also have poor control of asthma.12-14
Cough unresolved? Add therapy for GERD
Although GERD is primarily associated with heartburn and gastrointestinal distress, it is not unusual for cough to be its only sign or symptom.15 In fact, GERD is the third most common cause of subacute cough—affecting about 21% of patients who seek help for cough at primary care practices.3
CASE Margaret returned to the clinic shortly after completion of a 2-week course of brompheniramine/sustained-release pseudoephedrine, and reported that she was still coughing frequently—and that the medication had brought little improvement. Because of her history of heartburn, we added a 2-week trial with a proton pump inhibitor (PPI)—omeprazole 20 mg/d.
While there are diagnostic tests for GERD, including a pH probe of the esophagus, a barium esophagogram, and manometry testing, empiric therapy with a PPI—starting with a trial of at least 2 weeks—often eliminates the troublesome cough.16 If the patient responds to treatment, the medication can be continued. Risks associated with long-term PPI therapy include osteoporosis and interference with calcium and magnesium absorption,17 so it is important to monitor patients taking them and to discontinue treatment as soon as the cough symptoms resolve.
Have you ruled out postinfectious cough?
If a patient has a cough that has lingered for 3 to 8 weeks after his or her recovery from an acute upper respiratory infection (URI), postinfectious cough may be the reason.18,19 Such a cough is subacute and self-limiting. (If the cough lasts >8 weeks after an acute illness, other diagnoses, such as chronic infection, are more likely.)
The pathogenesis for postinfectious cough may be related to postviral airway inflammation or bronchial hyperresponsiveness, and antibiotics are not indicated.4 Patients may be treated with a bronchodilator such as ipratropium rather than a beta-agonist or inhaled corticosteroids; oral tapered prednisone can be prescribed, if needed, for severe paroxysms, although there is limited evidence of its efficacy.20 Central antitussive agents such as codeine and dextromethorphan can be used when other measures fail to bring relief.
Nonasthmatic eosinophilic bronchitis does not impede airflow
NAEB is less well known than the conditions discussed thus far, but it is a relatively common cause of persistent cough.21-23 In some studies, up to 13% of patients with subacute cough were diagnosed with NAEB.6
Unlike asthma, NAEB is not associated with abnormalities in airway function; patients have no dyspnea and no wheezing, and no obstruction of airflow.24 Patients will have FEV1 >80% and FEV1/FVC >75% on spirometric examination, a negative response to bronchoprovocation, and, typically, an elevated sputum eosinophil count of >3%. Because induced sputum or bronchoscopic washings are difficult, exhaled nitric oxide testing is another option. If these tests are not available, a trial of inhaled steroids is indicated, even if neither spirometry nor bronchoprovocation testing was abnormal.9
Patients with NAEB respond well to inhaled corticosteroids, and budesonide 400 mcg twice a day or prednisolone 30 mg daily may be prescribed. It is also important to remove airway irritants. Long-term follow-up studies of patients with NAEB have had conflicting results. One study found that most cases resolve completely;23 another showed a need for long-term treatment, and suggested that patients with NAEB may be at increased risk for asthma and chronic obstructive pulmonary disease. 25
Paroxysmal cough, whoops point to pertussis
When a patient has paroxysms of cough, posttussive vomiting, and/or an inspiratory whooping sound, B pertussis infection is the likely culprit.26-28 A definitive diagnosis of pertussis, or whooping cough, may be based on a positive culture from a nasopharyngeal aspirate swab.29 Suspected cases can be confirmed with a polymerase chain reaction test, and a presumptive diagnosis may be made as a result of a 4-fold increase in immunoglobulin G or immunoglobulin A antibodies for B pertussis.4
A macrolide antibiotic, usually azithromycin, is the standard treatment for pertussis.30-32 Patients should be isolated for 5 days from the start of treatment. Antibiotic therapy will reduce the risk of transmission, but will not affect the duration of the cough, which may be 6 to 8 weeks. Long-acting beta-agonists, antihistamines, and corticosteroids should not be used to treat pertussis.4
CASE After a 2-week course of omeprazole 20 mg daily, Margaret was coughing much less. We extended the prescription, and by the end of the next 4 weeks, she was no longer coughing. After 2 months, both the PPI and the antihistamine/decongestant were discontinued. We advised her to institute antireflux measures, such as elevating her head at night and not eating after 6 pm, and she has not had a relapse.
CORRESPONDENCE Rebecca H. Gladu, MD, FAAFP, San Jacinto Methodist Hospital, 4401 Garth Road, Baytown, TX 77521; [email protected]
1. Corrao WM. Chronic persistent cough: diagnosis and treatment update. Pediatr Ann. 1996;25:162-168.
2. Holmes RL, Fadden CT. Evaluation of the patient with chronic cough. Am Fam Physician. 2004;69:2159-2166.
3. Irwin RS, Curley FJ, French CL. Chronic cough. The spectrum and frequency of causes, key components of the diagnostic evaluation, and outcome of specific therapy. Am Rev Respir Dis. 1990;141:640-647.
4. Irwin RS, Baumann MH, Bolser DC, et al. Diagnosis and management of cough. Executive summary: ACCP evidence-based practice guideline. Chest. 2006;129(1 suppl):1S-23S.
5. Pratter MR, Bartter T, Akers S, et al. An algorithmic approach to chronic cough. Ann Intern Med. 1993;119:977-983.
6. Pratter MR, Brightling CE, Boulet LP, et al. An empiric integrative approach to the management of cough: ACCP evidence-based clinical practice guidelines. Chest. 2006;129 (1 suppl):222S-231S.
7. Irwin RS, Madison JM. Anatomical diagnostic protocol in evaluating chronic cough with specific reference to gastroesophageal reflux disease. Am J Med. 2000;108(suppl 4a):126S-130S.
8. Irwin RS, Corrao WM, Pratter MR. Chronic persistent cough in the adult: the spectrum and frequency of causes and successful outcome of specific therapy. Am Rev Respir Dis. 1981;123 (4 Pt 1):413-417.
9. Birring SS. Controversies in the evaluation and management of chronic cough. Am J Respir Crit Care Med. 2011;183:708-715.
10. National Asthma Education and Prevention Program. Expert Panel Report 3 (EPR-3): guidelines for the diagnosis and management of asthma-summary report 2007. J Allergy Clin Immunol. 2007;120(5 suppl):S94-S138.
11. Reh DD, Lin SY, Clipp SL, et al. Secondhand tobacco smoke exposure and chronic rhinosinusitis: a population-based case-control study. Am J Rhinol Allergy. 2009;23:562-567.
12. Stapleton M, Howard-Thompson A, George C, et al. Smoking and asthma. J Am Board Fam Med. 2011;24:313-322.
13. Hersoug LG, Husemoen LL, Sigsgaard T, et al. Indoor exposure to environmental cigarette smoke, but not other inhaled particulates associates with respiratory symptoms and diminished lung function in adults. Respirology. 2010;15:993-1000.
14. Self TH, Wallace JL, Gray LA, et al. Are we failing to document adequate smoking histories? A brief review 1999-2009. Curr Med Res Opin. 2010;26:1691-1696.
15. Sontag SJ. The spectrum of pulmonary symptoms due to gastroesophageal reflux. Thorac Surg Clin. 2005;15:353-368.
16. Irwin RS. Chronic cough due to gastroesophageal reflux. ACCP evidence-based clinical practice guidelines. Chest. 2006;129(suppl 1):80S-94S.
17. Chen J, Yuan YC, Leontiadis GI, et al. Recent safety concerns with proton pump inhibitors. J Clin Gastroenterol. 2012;46:93-114.
18. Braman SS. Postinfectious cough: ACCP evidence-based practice guidelines. Chest. 2006;129(suppl 1):138S-146S.
19. Pratter MR. Cough and the common cold: ACCP evidence-based practice guidelines. Chest. 2006;129(suppl 1):72S-74S.
20. Chang AB, McKean M, Morris P. Inhaled anticholinergics for prolonged non-specific cough in children. Cochrane Database Syst Rev. 2004;(1):CD004358.-
21. Brightling CE, Ward R, Goh KL, et al. Eosinophilic bronchitis is an important cause of chronic cough. Am J Respir Crit Care Med. 1999;160:406-410.
22. Gonlugur U, Gonlugur TE. Eosinophilic bronchitis without asthma. Int Arch Allergy Immunol. 2008;147:1-5.
23. Brightling CE. Cough due to asthma and nonasthmatic eosinophilic bronchitis. Lung. 2010;188 (suppl 1):S13-S17.
24. Gibson PG, Hargreave FE, Girgis-Gabardo, et al. Chronic cough with eosinophilic bronchitis: examination for variable airflow obstruction and response to corticosteroid. Clin Exp Allergy. 1995;25:127-132.
25. Berry MA, Hargadon B, McKenna S, et al. Observational study of the natural history of eosinophilic bronchitis. Clin Exp Allergy. 2005;35:598-601.
26. Antico A, Fabozzi F, Scipiotti C. Pertussis in adults. A study in an Italian population with chronic cough. Monaldi Arch Chest Dis. 2002;57:247-252.
27. Birkebaek NH, Kristiansen M, Seefeldt T, et al. Bordetella pertussis and chronic cough in adults. Clin Infect Dis. 1999;29:1239-1242.
28. Kapaskelis AM, Vouloumanou EK, Rafailidis PI, et al. High prevalence of antibody titers against Bordetella pertussis in an adult population with prolonged cough. Respir Med. 2008;102:1586-1591.
29. Cornia PB, Hersh AL, Lipsky BA, et al. Does this coughing adolescent or adult patient have pertussis? JAMA. 2010;304:890-896.
30. Devasia RA, Jones TF, Collier B, et al. Compliance with azithromycin versus erythromycin in the setting of a pertussis outbreak. Am J Med Sci. 2009;337:176-178.
31. Poe RH, Harder RV, Israel RH, et al. Chronic persistent cough. Experience in diagnosis and outcome using an anatomic diagnostic protocol. Chest. 1989;95:723-728.
32. Altunaji SM, Kukuruzovic RH, Curtis NC, et al. Antibiotics for whooping cough (pertussis). Cochrane Database Syst Rev. 2007;(3):CD004404.-
• Always include postnasal drip, asthma, and gastroesophageal reflux disease in the differential diagnosis for persistent cough, regardless of clinical signs and symptoms. B
• Do not rely on a patient’s description of the character and timing of the cough or the absence (or presence) of sputum to narrow down the differential diagnosis. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Margaret M, a 52-year-old nonsmoker, came to our clinic because of a persistent cough that had started about 4 weeks earlier. She had tried multiple over-the-counter cough suppressants, including dextromethorphan and guaifenesin, as well as cough drops, but none had been effective.
Margaret denied having had a cold or respiratory infection in the past few months or being in close contact with anyone with a chronic cough, and she had never had an asthma diagnosis. In response to a question about previous coughing episodes, the patient recalled having had several bouts of chronic cough in the past, including one about a year ago.
While Margaret had no known allergies, she did have occasional heartburn, which an antacid—or, at times, a drink of water—always relieved. Thyroid medication and calcium were the only things she took on a regular basis, separated by several hours to avoid problems with absorption.
Patients like Margaret, who seek help from their primary care physician only after attempting to combat a persistent cough on their own, may be quite frustrated by the time they arrive in your office. They’re counting on you to provide a cure. Fortunately, you’re likely to find it, as the differential diagnosis for subacute cough (a cough of 3-8 weeks’ duration) is limited.
Nonetheless, finding the cause of a subacute or chronic cough (lasting >8 weeks) is sometimes a matter of trial and error. Postnasal drip (also known as upper airway cough syndrome, or UACS), asthma, and gastroesophageal reflux disease (GERD) are the most common causes,1,2 followed by postinfectious cough, nonasthmatic eosinophilic bronchitis (NAEB), and pertussis.3 Although these conditions are all relatively well known, they are not always easy to detect: Some disorders, including UACS, asthma, and GERD, may be “silent,” with persistent cough the only presenting sign or symptom.4 In other cases, more than one condition may be contributing to the cough.
Starting with trials of empiric therapy for the most common causes of persistent cough—with sequential therapy and diagnostic tests, as needed—is far more effective than searching for relatively uncommon or obscure conditions. Following such a protocol, as detailed in the algorithm (FIGURE)4-7 we’ve developed and in the text that follows, can help you combat subacute and chronic cough in a cost-effective, timely way.
FIGURE
Dx and treatment when persistent cough is the only symptom4-7
*May include CXR, PPD, B pertussis IgG or IgA, spirometry with methacholine inhalation challenge, barium swallow, prolonged pH monitoring, sinus CT, and sputum eosinophil count, excluding any tests that have already been performed.
ACEI, angiotensin-converting enzyme inhibitor; CT, computed tomography; COPD, chronic obstructive pulmonary disease; CXR, chest x-ray; GERD, gastroesophageal reflux disease; IgA, immunoglobulin A; IgG, immunoglobulin G; PND, postnasal drip; PPD, purified protein derivative; PPI, proton pump inhibitor.
Treat all patients for upper airway cough syndrome
Postnasal drip—renamed UACS by the guideline committee of the American Association of Chest Physicians because it isn’t clear whether the cough is caused by irritation from direct contact with postnasal drip or by inflammation of cough receptors in the upper airway—is the most common cause of chronic cough.6
The differential diagnosis for UACS, which is implicated in about 34% of cases of persistent cough, includes allergic, postinfectious, and occupational rhinitis; rhinitis due to anatomic abnormalities or physical or chemical irritants, rhinitis medicamentosa, and rhinitis of pregnancy; bacterial sinusitis; and allergic fungal sinusitis.8
The signs and symptoms of UACS are nonspecific, and a definitive diagnosis typically cannot be made from the medical history and physical examination alone. What’s more, the absence of any of the usual clinical findings—eg, rhinorrhea and excess sputum production—should not preclude an empiric trial with a first-generation antihistamine-decongestant combination such as brompheniramine/sustained-release pseudoephedrine. Second-and third-generation combination products, such as fexofenadine/pseudoephedrine, should not be used, as they are not effective in treating UACS.4
CASE Margaret’s physical exam was unremarkable. Her vital signs were stable, she had no cervical lymphadenopathy, and her chest was clear on auscultation. She had a dry cough that occurred twice during the exam, but not on inspiration.
The patient’s work-up included office spirometry, which was normal; a nasopharyngeal culture for Bordetella pertussis was negative. We prescribed a 2-week course of therapy with brompheniramine/sustained-release pseudoephedrine and scheduled a return visit shortly after it was completed.
There is no gold standard diagnostic test to confirm or rule out postnasal drip as the cause of cough. CT scanning of sinuses has a poor positive predictive value and is no longer recommended as part of an initial work-up,9 but may be useful for patients whose symptoms persist longer than 3 weeks.
Consider bronchodilator Tx when asthma is suspected
Cough-variant asthma is the second most common cause of persistent cough, and is responsible for an estimated 28% of cases.6 Asthma is the easiest of the conditions included in the differential diagnosis for persistent cough to establish in an office setting. The challenge is to remember to consider it in patients who present with cough but no sign of the classic expiratory wheezing. When you suspect that a patient has asthma, consider empiric bronchodilator therapy—or conduct spirometry testing.
Spirometric values of forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) <70% and a positive bronchodilator response (≥12%) are consistent with an asthma diagnosis. Management of asthma depends on severity, and patients should be evaluated based on the National Heart, Lung, and Blood Institute’s National Asthma Education and Prevention Program Guidelines for the Diagnosis and Management of Asthma.10
It is crucial to ask patients with asthma (and, indeed, to ask all patients with a persistent cough) about exposure to secondhand smoke, and to stress the importance of avoiding smoking and secondary exposure. Individuals who are regularly exposed to secondhand smoke report more nasal symptoms and greater use of nasal decongestants compared with people with no exposure to smoke;11 they also have poor control of asthma.12-14
Cough unresolved? Add therapy for GERD
Although GERD is primarily associated with heartburn and gastrointestinal distress, it is not unusual for cough to be its only sign or symptom.15 In fact, GERD is the third most common cause of subacute cough—affecting about 21% of patients who seek help for cough at primary care practices.3
CASE Margaret returned to the clinic shortly after completion of a 2-week course of brompheniramine/sustained-release pseudoephedrine, and reported that she was still coughing frequently—and that the medication had brought little improvement. Because of her history of heartburn, we added a 2-week trial with a proton pump inhibitor (PPI)—omeprazole 20 mg/d.
While there are diagnostic tests for GERD, including a pH probe of the esophagus, a barium esophagogram, and manometry testing, empiric therapy with a PPI—starting with a trial of at least 2 weeks—often eliminates the troublesome cough.16 If the patient responds to treatment, the medication can be continued. Risks associated with long-term PPI therapy include osteoporosis and interference with calcium and magnesium absorption,17 so it is important to monitor patients taking them and to discontinue treatment as soon as the cough symptoms resolve.
Have you ruled out postinfectious cough?
If a patient has a cough that has lingered for 3 to 8 weeks after his or her recovery from an acute upper respiratory infection (URI), postinfectious cough may be the reason.18,19 Such a cough is subacute and self-limiting. (If the cough lasts >8 weeks after an acute illness, other diagnoses, such as chronic infection, are more likely.)
The pathogenesis for postinfectious cough may be related to postviral airway inflammation or bronchial hyperresponsiveness, and antibiotics are not indicated.4 Patients may be treated with a bronchodilator such as ipratropium rather than a beta-agonist or inhaled corticosteroids; oral tapered prednisone can be prescribed, if needed, for severe paroxysms, although there is limited evidence of its efficacy.20 Central antitussive agents such as codeine and dextromethorphan can be used when other measures fail to bring relief.
Nonasthmatic eosinophilic bronchitis does not impede airflow
NAEB is less well known than the conditions discussed thus far, but it is a relatively common cause of persistent cough.21-23 In some studies, up to 13% of patients with subacute cough were diagnosed with NAEB.6
Unlike asthma, NAEB is not associated with abnormalities in airway function; patients have no dyspnea and no wheezing, and no obstruction of airflow.24 Patients will have FEV1 >80% and FEV1/FVC >75% on spirometric examination, a negative response to bronchoprovocation, and, typically, an elevated sputum eosinophil count of >3%. Because induced sputum or bronchoscopic washings are difficult, exhaled nitric oxide testing is another option. If these tests are not available, a trial of inhaled steroids is indicated, even if neither spirometry nor bronchoprovocation testing was abnormal.9
Patients with NAEB respond well to inhaled corticosteroids, and budesonide 400 mcg twice a day or prednisolone 30 mg daily may be prescribed. It is also important to remove airway irritants. Long-term follow-up studies of patients with NAEB have had conflicting results. One study found that most cases resolve completely;23 another showed a need for long-term treatment, and suggested that patients with NAEB may be at increased risk for asthma and chronic obstructive pulmonary disease. 25
Paroxysmal cough, whoops point to pertussis
When a patient has paroxysms of cough, posttussive vomiting, and/or an inspiratory whooping sound, B pertussis infection is the likely culprit.26-28 A definitive diagnosis of pertussis, or whooping cough, may be based on a positive culture from a nasopharyngeal aspirate swab.29 Suspected cases can be confirmed with a polymerase chain reaction test, and a presumptive diagnosis may be made as a result of a 4-fold increase in immunoglobulin G or immunoglobulin A antibodies for B pertussis.4
A macrolide antibiotic, usually azithromycin, is the standard treatment for pertussis.30-32 Patients should be isolated for 5 days from the start of treatment. Antibiotic therapy will reduce the risk of transmission, but will not affect the duration of the cough, which may be 6 to 8 weeks. Long-acting beta-agonists, antihistamines, and corticosteroids should not be used to treat pertussis.4
CASE After a 2-week course of omeprazole 20 mg daily, Margaret was coughing much less. We extended the prescription, and by the end of the next 4 weeks, she was no longer coughing. After 2 months, both the PPI and the antihistamine/decongestant were discontinued. We advised her to institute antireflux measures, such as elevating her head at night and not eating after 6 pm, and she has not had a relapse.
CORRESPONDENCE Rebecca H. Gladu, MD, FAAFP, San Jacinto Methodist Hospital, 4401 Garth Road, Baytown, TX 77521; [email protected]
• Always include postnasal drip, asthma, and gastroesophageal reflux disease in the differential diagnosis for persistent cough, regardless of clinical signs and symptoms. B
• Do not rely on a patient’s description of the character and timing of the cough or the absence (or presence) of sputum to narrow down the differential diagnosis. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Margaret M, a 52-year-old nonsmoker, came to our clinic because of a persistent cough that had started about 4 weeks earlier. She had tried multiple over-the-counter cough suppressants, including dextromethorphan and guaifenesin, as well as cough drops, but none had been effective.
Margaret denied having had a cold or respiratory infection in the past few months or being in close contact with anyone with a chronic cough, and she had never had an asthma diagnosis. In response to a question about previous coughing episodes, the patient recalled having had several bouts of chronic cough in the past, including one about a year ago.
While Margaret had no known allergies, she did have occasional heartburn, which an antacid—or, at times, a drink of water—always relieved. Thyroid medication and calcium were the only things she took on a regular basis, separated by several hours to avoid problems with absorption.
Patients like Margaret, who seek help from their primary care physician only after attempting to combat a persistent cough on their own, may be quite frustrated by the time they arrive in your office. They’re counting on you to provide a cure. Fortunately, you’re likely to find it, as the differential diagnosis for subacute cough (a cough of 3-8 weeks’ duration) is limited.
Nonetheless, finding the cause of a subacute or chronic cough (lasting >8 weeks) is sometimes a matter of trial and error. Postnasal drip (also known as upper airway cough syndrome, or UACS), asthma, and gastroesophageal reflux disease (GERD) are the most common causes,1,2 followed by postinfectious cough, nonasthmatic eosinophilic bronchitis (NAEB), and pertussis.3 Although these conditions are all relatively well known, they are not always easy to detect: Some disorders, including UACS, asthma, and GERD, may be “silent,” with persistent cough the only presenting sign or symptom.4 In other cases, more than one condition may be contributing to the cough.
Starting with trials of empiric therapy for the most common causes of persistent cough—with sequential therapy and diagnostic tests, as needed—is far more effective than searching for relatively uncommon or obscure conditions. Following such a protocol, as detailed in the algorithm (FIGURE)4-7 we’ve developed and in the text that follows, can help you combat subacute and chronic cough in a cost-effective, timely way.
FIGURE
Dx and treatment when persistent cough is the only symptom4-7
*May include CXR, PPD, B pertussis IgG or IgA, spirometry with methacholine inhalation challenge, barium swallow, prolonged pH monitoring, sinus CT, and sputum eosinophil count, excluding any tests that have already been performed.
ACEI, angiotensin-converting enzyme inhibitor; CT, computed tomography; COPD, chronic obstructive pulmonary disease; CXR, chest x-ray; GERD, gastroesophageal reflux disease; IgA, immunoglobulin A; IgG, immunoglobulin G; PND, postnasal drip; PPD, purified protein derivative; PPI, proton pump inhibitor.
Treat all patients for upper airway cough syndrome
Postnasal drip—renamed UACS by the guideline committee of the American Association of Chest Physicians because it isn’t clear whether the cough is caused by irritation from direct contact with postnasal drip or by inflammation of cough receptors in the upper airway—is the most common cause of chronic cough.6
The differential diagnosis for UACS, which is implicated in about 34% of cases of persistent cough, includes allergic, postinfectious, and occupational rhinitis; rhinitis due to anatomic abnormalities or physical or chemical irritants, rhinitis medicamentosa, and rhinitis of pregnancy; bacterial sinusitis; and allergic fungal sinusitis.8
The signs and symptoms of UACS are nonspecific, and a definitive diagnosis typically cannot be made from the medical history and physical examination alone. What’s more, the absence of any of the usual clinical findings—eg, rhinorrhea and excess sputum production—should not preclude an empiric trial with a first-generation antihistamine-decongestant combination such as brompheniramine/sustained-release pseudoephedrine. Second-and third-generation combination products, such as fexofenadine/pseudoephedrine, should not be used, as they are not effective in treating UACS.4
CASE Margaret’s physical exam was unremarkable. Her vital signs were stable, she had no cervical lymphadenopathy, and her chest was clear on auscultation. She had a dry cough that occurred twice during the exam, but not on inspiration.
The patient’s work-up included office spirometry, which was normal; a nasopharyngeal culture for Bordetella pertussis was negative. We prescribed a 2-week course of therapy with brompheniramine/sustained-release pseudoephedrine and scheduled a return visit shortly after it was completed.
There is no gold standard diagnostic test to confirm or rule out postnasal drip as the cause of cough. CT scanning of sinuses has a poor positive predictive value and is no longer recommended as part of an initial work-up,9 but may be useful for patients whose symptoms persist longer than 3 weeks.
Consider bronchodilator Tx when asthma is suspected
Cough-variant asthma is the second most common cause of persistent cough, and is responsible for an estimated 28% of cases.6 Asthma is the easiest of the conditions included in the differential diagnosis for persistent cough to establish in an office setting. The challenge is to remember to consider it in patients who present with cough but no sign of the classic expiratory wheezing. When you suspect that a patient has asthma, consider empiric bronchodilator therapy—or conduct spirometry testing.
Spirometric values of forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) <70% and a positive bronchodilator response (≥12%) are consistent with an asthma diagnosis. Management of asthma depends on severity, and patients should be evaluated based on the National Heart, Lung, and Blood Institute’s National Asthma Education and Prevention Program Guidelines for the Diagnosis and Management of Asthma.10
It is crucial to ask patients with asthma (and, indeed, to ask all patients with a persistent cough) about exposure to secondhand smoke, and to stress the importance of avoiding smoking and secondary exposure. Individuals who are regularly exposed to secondhand smoke report more nasal symptoms and greater use of nasal decongestants compared with people with no exposure to smoke;11 they also have poor control of asthma.12-14
Cough unresolved? Add therapy for GERD
Although GERD is primarily associated with heartburn and gastrointestinal distress, it is not unusual for cough to be its only sign or symptom.15 In fact, GERD is the third most common cause of subacute cough—affecting about 21% of patients who seek help for cough at primary care practices.3
CASE Margaret returned to the clinic shortly after completion of a 2-week course of brompheniramine/sustained-release pseudoephedrine, and reported that she was still coughing frequently—and that the medication had brought little improvement. Because of her history of heartburn, we added a 2-week trial with a proton pump inhibitor (PPI)—omeprazole 20 mg/d.
While there are diagnostic tests for GERD, including a pH probe of the esophagus, a barium esophagogram, and manometry testing, empiric therapy with a PPI—starting with a trial of at least 2 weeks—often eliminates the troublesome cough.16 If the patient responds to treatment, the medication can be continued. Risks associated with long-term PPI therapy include osteoporosis and interference with calcium and magnesium absorption,17 so it is important to monitor patients taking them and to discontinue treatment as soon as the cough symptoms resolve.
Have you ruled out postinfectious cough?
If a patient has a cough that has lingered for 3 to 8 weeks after his or her recovery from an acute upper respiratory infection (URI), postinfectious cough may be the reason.18,19 Such a cough is subacute and self-limiting. (If the cough lasts >8 weeks after an acute illness, other diagnoses, such as chronic infection, are more likely.)
The pathogenesis for postinfectious cough may be related to postviral airway inflammation or bronchial hyperresponsiveness, and antibiotics are not indicated.4 Patients may be treated with a bronchodilator such as ipratropium rather than a beta-agonist or inhaled corticosteroids; oral tapered prednisone can be prescribed, if needed, for severe paroxysms, although there is limited evidence of its efficacy.20 Central antitussive agents such as codeine and dextromethorphan can be used when other measures fail to bring relief.
Nonasthmatic eosinophilic bronchitis does not impede airflow
NAEB is less well known than the conditions discussed thus far, but it is a relatively common cause of persistent cough.21-23 In some studies, up to 13% of patients with subacute cough were diagnosed with NAEB.6
Unlike asthma, NAEB is not associated with abnormalities in airway function; patients have no dyspnea and no wheezing, and no obstruction of airflow.24 Patients will have FEV1 >80% and FEV1/FVC >75% on spirometric examination, a negative response to bronchoprovocation, and, typically, an elevated sputum eosinophil count of >3%. Because induced sputum or bronchoscopic washings are difficult, exhaled nitric oxide testing is another option. If these tests are not available, a trial of inhaled steroids is indicated, even if neither spirometry nor bronchoprovocation testing was abnormal.9
Patients with NAEB respond well to inhaled corticosteroids, and budesonide 400 mcg twice a day or prednisolone 30 mg daily may be prescribed. It is also important to remove airway irritants. Long-term follow-up studies of patients with NAEB have had conflicting results. One study found that most cases resolve completely;23 another showed a need for long-term treatment, and suggested that patients with NAEB may be at increased risk for asthma and chronic obstructive pulmonary disease. 25
Paroxysmal cough, whoops point to pertussis
When a patient has paroxysms of cough, posttussive vomiting, and/or an inspiratory whooping sound, B pertussis infection is the likely culprit.26-28 A definitive diagnosis of pertussis, or whooping cough, may be based on a positive culture from a nasopharyngeal aspirate swab.29 Suspected cases can be confirmed with a polymerase chain reaction test, and a presumptive diagnosis may be made as a result of a 4-fold increase in immunoglobulin G or immunoglobulin A antibodies for B pertussis.4
A macrolide antibiotic, usually azithromycin, is the standard treatment for pertussis.30-32 Patients should be isolated for 5 days from the start of treatment. Antibiotic therapy will reduce the risk of transmission, but will not affect the duration of the cough, which may be 6 to 8 weeks. Long-acting beta-agonists, antihistamines, and corticosteroids should not be used to treat pertussis.4
CASE After a 2-week course of omeprazole 20 mg daily, Margaret was coughing much less. We extended the prescription, and by the end of the next 4 weeks, she was no longer coughing. After 2 months, both the PPI and the antihistamine/decongestant were discontinued. We advised her to institute antireflux measures, such as elevating her head at night and not eating after 6 pm, and she has not had a relapse.
CORRESPONDENCE Rebecca H. Gladu, MD, FAAFP, San Jacinto Methodist Hospital, 4401 Garth Road, Baytown, TX 77521; [email protected]
1. Corrao WM. Chronic persistent cough: diagnosis and treatment update. Pediatr Ann. 1996;25:162-168.
2. Holmes RL, Fadden CT. Evaluation of the patient with chronic cough. Am Fam Physician. 2004;69:2159-2166.
3. Irwin RS, Curley FJ, French CL. Chronic cough. The spectrum and frequency of causes, key components of the diagnostic evaluation, and outcome of specific therapy. Am Rev Respir Dis. 1990;141:640-647.
4. Irwin RS, Baumann MH, Bolser DC, et al. Diagnosis and management of cough. Executive summary: ACCP evidence-based practice guideline. Chest. 2006;129(1 suppl):1S-23S.
5. Pratter MR, Bartter T, Akers S, et al. An algorithmic approach to chronic cough. Ann Intern Med. 1993;119:977-983.
6. Pratter MR, Brightling CE, Boulet LP, et al. An empiric integrative approach to the management of cough: ACCP evidence-based clinical practice guidelines. Chest. 2006;129 (1 suppl):222S-231S.
7. Irwin RS, Madison JM. Anatomical diagnostic protocol in evaluating chronic cough with specific reference to gastroesophageal reflux disease. Am J Med. 2000;108(suppl 4a):126S-130S.
8. Irwin RS, Corrao WM, Pratter MR. Chronic persistent cough in the adult: the spectrum and frequency of causes and successful outcome of specific therapy. Am Rev Respir Dis. 1981;123 (4 Pt 1):413-417.
9. Birring SS. Controversies in the evaluation and management of chronic cough. Am J Respir Crit Care Med. 2011;183:708-715.
10. National Asthma Education and Prevention Program. Expert Panel Report 3 (EPR-3): guidelines for the diagnosis and management of asthma-summary report 2007. J Allergy Clin Immunol. 2007;120(5 suppl):S94-S138.
11. Reh DD, Lin SY, Clipp SL, et al. Secondhand tobacco smoke exposure and chronic rhinosinusitis: a population-based case-control study. Am J Rhinol Allergy. 2009;23:562-567.
12. Stapleton M, Howard-Thompson A, George C, et al. Smoking and asthma. J Am Board Fam Med. 2011;24:313-322.
13. Hersoug LG, Husemoen LL, Sigsgaard T, et al. Indoor exposure to environmental cigarette smoke, but not other inhaled particulates associates with respiratory symptoms and diminished lung function in adults. Respirology. 2010;15:993-1000.
14. Self TH, Wallace JL, Gray LA, et al. Are we failing to document adequate smoking histories? A brief review 1999-2009. Curr Med Res Opin. 2010;26:1691-1696.
15. Sontag SJ. The spectrum of pulmonary symptoms due to gastroesophageal reflux. Thorac Surg Clin. 2005;15:353-368.
16. Irwin RS. Chronic cough due to gastroesophageal reflux. ACCP evidence-based clinical practice guidelines. Chest. 2006;129(suppl 1):80S-94S.
17. Chen J, Yuan YC, Leontiadis GI, et al. Recent safety concerns with proton pump inhibitors. J Clin Gastroenterol. 2012;46:93-114.
18. Braman SS. Postinfectious cough: ACCP evidence-based practice guidelines. Chest. 2006;129(suppl 1):138S-146S.
19. Pratter MR. Cough and the common cold: ACCP evidence-based practice guidelines. Chest. 2006;129(suppl 1):72S-74S.
20. Chang AB, McKean M, Morris P. Inhaled anticholinergics for prolonged non-specific cough in children. Cochrane Database Syst Rev. 2004;(1):CD004358.-
21. Brightling CE, Ward R, Goh KL, et al. Eosinophilic bronchitis is an important cause of chronic cough. Am J Respir Crit Care Med. 1999;160:406-410.
22. Gonlugur U, Gonlugur TE. Eosinophilic bronchitis without asthma. Int Arch Allergy Immunol. 2008;147:1-5.
23. Brightling CE. Cough due to asthma and nonasthmatic eosinophilic bronchitis. Lung. 2010;188 (suppl 1):S13-S17.
24. Gibson PG, Hargreave FE, Girgis-Gabardo, et al. Chronic cough with eosinophilic bronchitis: examination for variable airflow obstruction and response to corticosteroid. Clin Exp Allergy. 1995;25:127-132.
25. Berry MA, Hargadon B, McKenna S, et al. Observational study of the natural history of eosinophilic bronchitis. Clin Exp Allergy. 2005;35:598-601.
26. Antico A, Fabozzi F, Scipiotti C. Pertussis in adults. A study in an Italian population with chronic cough. Monaldi Arch Chest Dis. 2002;57:247-252.
27. Birkebaek NH, Kristiansen M, Seefeldt T, et al. Bordetella pertussis and chronic cough in adults. Clin Infect Dis. 1999;29:1239-1242.
28. Kapaskelis AM, Vouloumanou EK, Rafailidis PI, et al. High prevalence of antibody titers against Bordetella pertussis in an adult population with prolonged cough. Respir Med. 2008;102:1586-1591.
29. Cornia PB, Hersh AL, Lipsky BA, et al. Does this coughing adolescent or adult patient have pertussis? JAMA. 2010;304:890-896.
30. Devasia RA, Jones TF, Collier B, et al. Compliance with azithromycin versus erythromycin in the setting of a pertussis outbreak. Am J Med Sci. 2009;337:176-178.
31. Poe RH, Harder RV, Israel RH, et al. Chronic persistent cough. Experience in diagnosis and outcome using an anatomic diagnostic protocol. Chest. 1989;95:723-728.
32. Altunaji SM, Kukuruzovic RH, Curtis NC, et al. Antibiotics for whooping cough (pertussis). Cochrane Database Syst Rev. 2007;(3):CD004404.-
1. Corrao WM. Chronic persistent cough: diagnosis and treatment update. Pediatr Ann. 1996;25:162-168.
2. Holmes RL, Fadden CT. Evaluation of the patient with chronic cough. Am Fam Physician. 2004;69:2159-2166.
3. Irwin RS, Curley FJ, French CL. Chronic cough. The spectrum and frequency of causes, key components of the diagnostic evaluation, and outcome of specific therapy. Am Rev Respir Dis. 1990;141:640-647.
4. Irwin RS, Baumann MH, Bolser DC, et al. Diagnosis and management of cough. Executive summary: ACCP evidence-based practice guideline. Chest. 2006;129(1 suppl):1S-23S.
5. Pratter MR, Bartter T, Akers S, et al. An algorithmic approach to chronic cough. Ann Intern Med. 1993;119:977-983.
6. Pratter MR, Brightling CE, Boulet LP, et al. An empiric integrative approach to the management of cough: ACCP evidence-based clinical practice guidelines. Chest. 2006;129 (1 suppl):222S-231S.
7. Irwin RS, Madison JM. Anatomical diagnostic protocol in evaluating chronic cough with specific reference to gastroesophageal reflux disease. Am J Med. 2000;108(suppl 4a):126S-130S.
8. Irwin RS, Corrao WM, Pratter MR. Chronic persistent cough in the adult: the spectrum and frequency of causes and successful outcome of specific therapy. Am Rev Respir Dis. 1981;123 (4 Pt 1):413-417.
9. Birring SS. Controversies in the evaluation and management of chronic cough. Am J Respir Crit Care Med. 2011;183:708-715.
10. National Asthma Education and Prevention Program. Expert Panel Report 3 (EPR-3): guidelines for the diagnosis and management of asthma-summary report 2007. J Allergy Clin Immunol. 2007;120(5 suppl):S94-S138.
11. Reh DD, Lin SY, Clipp SL, et al. Secondhand tobacco smoke exposure and chronic rhinosinusitis: a population-based case-control study. Am J Rhinol Allergy. 2009;23:562-567.
12. Stapleton M, Howard-Thompson A, George C, et al. Smoking and asthma. J Am Board Fam Med. 2011;24:313-322.
13. Hersoug LG, Husemoen LL, Sigsgaard T, et al. Indoor exposure to environmental cigarette smoke, but not other inhaled particulates associates with respiratory symptoms and diminished lung function in adults. Respirology. 2010;15:993-1000.
14. Self TH, Wallace JL, Gray LA, et al. Are we failing to document adequate smoking histories? A brief review 1999-2009. Curr Med Res Opin. 2010;26:1691-1696.
15. Sontag SJ. The spectrum of pulmonary symptoms due to gastroesophageal reflux. Thorac Surg Clin. 2005;15:353-368.
16. Irwin RS. Chronic cough due to gastroesophageal reflux. ACCP evidence-based clinical practice guidelines. Chest. 2006;129(suppl 1):80S-94S.
17. Chen J, Yuan YC, Leontiadis GI, et al. Recent safety concerns with proton pump inhibitors. J Clin Gastroenterol. 2012;46:93-114.
18. Braman SS. Postinfectious cough: ACCP evidence-based practice guidelines. Chest. 2006;129(suppl 1):138S-146S.
19. Pratter MR. Cough and the common cold: ACCP evidence-based practice guidelines. Chest. 2006;129(suppl 1):72S-74S.
20. Chang AB, McKean M, Morris P. Inhaled anticholinergics for prolonged non-specific cough in children. Cochrane Database Syst Rev. 2004;(1):CD004358.-
21. Brightling CE, Ward R, Goh KL, et al. Eosinophilic bronchitis is an important cause of chronic cough. Am J Respir Crit Care Med. 1999;160:406-410.
22. Gonlugur U, Gonlugur TE. Eosinophilic bronchitis without asthma. Int Arch Allergy Immunol. 2008;147:1-5.
23. Brightling CE. Cough due to asthma and nonasthmatic eosinophilic bronchitis. Lung. 2010;188 (suppl 1):S13-S17.
24. Gibson PG, Hargreave FE, Girgis-Gabardo, et al. Chronic cough with eosinophilic bronchitis: examination for variable airflow obstruction and response to corticosteroid. Clin Exp Allergy. 1995;25:127-132.
25. Berry MA, Hargadon B, McKenna S, et al. Observational study of the natural history of eosinophilic bronchitis. Clin Exp Allergy. 2005;35:598-601.
26. Antico A, Fabozzi F, Scipiotti C. Pertussis in adults. A study in an Italian population with chronic cough. Monaldi Arch Chest Dis. 2002;57:247-252.
27. Birkebaek NH, Kristiansen M, Seefeldt T, et al. Bordetella pertussis and chronic cough in adults. Clin Infect Dis. 1999;29:1239-1242.
28. Kapaskelis AM, Vouloumanou EK, Rafailidis PI, et al. High prevalence of antibody titers against Bordetella pertussis in an adult population with prolonged cough. Respir Med. 2008;102:1586-1591.
29. Cornia PB, Hersh AL, Lipsky BA, et al. Does this coughing adolescent or adult patient have pertussis? JAMA. 2010;304:890-896.
30. Devasia RA, Jones TF, Collier B, et al. Compliance with azithromycin versus erythromycin in the setting of a pertussis outbreak. Am J Med Sci. 2009;337:176-178.
31. Poe RH, Harder RV, Israel RH, et al. Chronic persistent cough. Experience in diagnosis and outcome using an anatomic diagnostic protocol. Chest. 1989;95:723-728.
32. Altunaji SM, Kukuruzovic RH, Curtis NC, et al. Antibiotics for whooping cough (pertussis). Cochrane Database Syst Rev. 2007;(3):CD004404.-
Infertility: Help for couples starts with you
• Evaluate the fallopian tubes and their patency when menstruation is normal. Also, consider arranging for a hysterosalpingogram. B
• Suspect polycystic ovarian syndrome when adiposity, acne, and hirsutism with menstrual irregularity are factors. B
• Suspect androgen deficiency when a man’s arm span is >2 cm longer than his height, or when he has experienced a loss of pubic, axillary, or facial hair. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
During an annual visit, a patient confides in you that she and her husband have been trying to get pregnant for a year, but haven’t had any success. She tells you that she’s starting to get worried.
How do you advise her? What are your next steps?
The approach to evaluating infertility complaints is usually straightforward and can lead to a positive outcome. Not surprisingly, the dialogue often begins with you, the family physician. Your attention to clues in each partner’s history can do much to get to the heart of the problem. And even if a couple requires a specialty referral, it’s best to be familiar with the more extensive evaluation and management options they’ll encounter to help them anticipate likely discussions in their consultations. In this article, we review the best evidence for the care of your patients who want to conceive.
Who’s affected?
Infertility difficulties may be attributable to one or both partners, may be multifactorial, or may be unexplained (TABLE).
In women, infertility is the inability to conceive after 1 year of unprotected regular intercourse in those younger than 35 years, and after 6 months in those 35 years and older.1 Fecundability is the probability of achieving pregnancy in 1 menstrual cycle. Normal fecundability with a single menstrual cycle is ~20%, peaking between the ages of 20 and 24 years.2 Fecundability decreases slightly at age 32 and declines progressively and more rapidly after age 40. Spontaneous miscarriage is a factor; its rate in younger women is ~10% and in women >40 years is ~40%.2 Overall, approximately 13% of women between the ages of 15 and 44 have fecundity impairment, with more than 6 million women in the United States affected.2
About 24% of all cases of infertility are due to male factors—seminiferous dysfunction, including problems with motility, morphology, and volume of sperm; primary hypogonadism; posttesticular defects; and hypothalamic pituitary disease.3 Recent observational trends show declines in fertility among men older than 40, and among men from different areas in the country, thus raising the issue of the role that environmental pollutants or toxins may play. Supposed increases in urogenital abnormalities and testicular cancers may also contribute to declining fertility rates.4,5
TABLE
Consider these factors in cases of suspected infertility3,6,21,23,40,41
| Major causes of infertility | Infertility risk factors | |
|---|---|---|
| Female | ||
| Cause | Contribution | |
| Endocrine factors | 45%-55% |
|
| PCOS Thyroid Diabetes mellitus Prolactinemia | 21%-28% 10%-20% 10%-20% 7% | |
| Tubal and peritoneal pathology | 30%-40% | |
| Ovulatory dysfunction* | 15% | |
| Cervical and uterine factors | <5% | |
| Male | ||
| Cause | Contribution | |
| Seminiferous tubule dysfunction | 60%-80% |
|
| Posttesticular defects | 10%-20% | |
| Primary hypogonadism | 10%-15% | |
| Hypothalamic pituitary disease | 1%-2% | |
| *Assuming appropriate ovarian reserve, indicated by follicle-stimulating hormone (FSH) level <10 mIU/mL, FSH-to-luteinizing hormone ratio <2, and estradiol level <50 pg/mL. GU, genitourinary; PCOS, polycystic ovarian syndrome; STIs, sexually transmitted infections | ||
Zero in on these areas of the history
As with any diagnostic work-up, the most important aspect of an infertility evaluation is the history. Document menstrual cycle length and regularity and the timing of intercourse. (Ideally, this would be done when a couple first decides to conceive.) It’s important to know how long the couple has been trying to become pregnant. More time may be all they need to achieve pregnancy. Educate them on reproductive cycles and optimal timing to achieve pregnancy. Some women experience lower abdominal pain (mittelschmerz) signifying release of an egg from the ovary, which can help identify the time of ovulation.
Remember, too, the role that a couple’s psychological state can play; worries over suspected infertility may cause anxiety, anger, depression, and marital troubles.
Is it her?
Regular menstrual cycles—menses occurring every 21 to 35 days—carry an ovulation probability of 95% with each cycle.6 With normal menses, ovulatory dysfunction is an unlikely cause of infertility. If menstrual cycles are irregular, ovulatory function is not normal and cyclical. Explore the woman’s medical, surgical, and gynecologic histories, looking particularly for thyroid disease, galactorrhea, hirsutism, pelvic or abdominal pain, dysmenorrhea, dyspareunia, pelvic inflammatory disease (PID), and abdominal or pelvic surgery.
The fallopian tubes. When menstruation is normal, evaluate the fallopian tubes and their patency; 30% to 40% of infertility cases can be related to peritoneal pathology.3,7 Inability to conceive in a previous relationship, history of PID, or prior tubal surgery all correlate to infertility. Ten percent of patients with a history of one PID episode and 54% to 75% of patients with 3 episodes will have patency issues.7
Consider arranging for a hysterosalpingogram (HSG) in all patients as part of an initial work-up for infertility.8 HSG is useful in evaluating tubal patency and the uterine cavity, and it can be therapeutic. HSG is not useful in detecting peritubal adhesions or endometriosis; patients in whom you suspect these conditions should undergo diagnostic laparoscopy. If abnormalities are found on HSG, refer patients to a reproductive endocrinologist to evaluate treatment options.
Chlamydia trachomatis IgG antibody testing can predict the presence of tubal disease. For women with low risk of tubal disease, it may be more cost effective to test for the Chlamydia antibody and proceed with HSG if the result is positive. Antibody testing is also useful for women with an allergy to contrast dye who cannot undergo an HSG. If the antibody test result is positive, consider arranging for a sonohysterogram to evaluate for the presence of fluid in the cul-de-sac, or an intrauterine infusion of saline to evaluate the patency of at least one tube.9
Ovulatory function. To assess ovulatory function, measure a midluteal-phase serum progesterone level, drawn 1 week before the expected day of menses (Day 21 of a 28-day cycle). A level >3 ng/mL is evidence of ovulation. Over-the-counter ovulation kits detect the luteinizing hormone (LH) surge but have false-positive and false-negative rates of 5% to 10%, respectively.10 Recording basal body temperature is a noninvasive and inexpensive means of evaluating ovulation. The patient must record temperatures at exactly the same time each day. Have her log the temperatures and watch for a spike that occurs 1 to 2 days after the LH surge. The average woman’s temperature rises above 98ºF in progressing from the follicular to the luteal phase. Since the spike occurs 1 to 2 days after ovulation, this method is best used for many months so the woman can predict her cycle.11
Once timing of ovulation has been established, you can check lab results at Day 3 of the woman’s cycle for follicle-stimulating hormone (FSH), LH, estradiol, thyroid-stimulating hormone (TSH), prolactin, and 2-hour fasting glucose tolerance. In addition to polycystic ovarian syndrome (PCOS), patients may have ovulatory dysfunction secondary to glucose intolerance.
A clomiphene (Clomid) challenge can help in assessing ovarian reserve. Administer 100 mg clomiphene on Days 5 through 9 of the patient’s cycle, and check FSH and estradiol levels on Day 10. With diminished ovarian reserve, FSH will increase to >12 mIU/mL and estradiol to >300 pg/mL.12 If this occurs, consider referring for an ultrasound measurement of antral follicle count. The presence of 4 to 10 follicles measuring 2 to 10 mm in diameter suggests adequate reserve.13
Although not widely available in the United States, the test for antimüllerian hormone (AMH) levels may be useful in reflecting the size of the primordial follicle pool. At menopause, the level is undetectable. A level above 0.5 ng/mL correlates with good ovarian reserve; levels <0.15 ng/mL suggest poor response to in vitro fertilization (IVF).14
Endocrine factors account for 45% to 55% of female infertility and include thyroid disease, PCOS, diabetes mellitus, prolactinemia, and luteal phase defects. Subclinical hypothyroidism, often evidenced only by high levels of TSH, decreases the chance of a successful pregnancy. This can occur even if the dysfunction is not severe enough to affect cycle regularity.15 Clinical hypo- or hyperthyroidism can affect ovulation by interfering with normal hormonal feedback loops, and correcting thyroid disease can improve fertility.
Galactorrhea discovered during the history and physical may be caused by elevated prolactin levels, which also inhibit normal ovulatory function. Chronically elevated prolactin levels in patients with PCOS can be attributed to elevated estrogen levels. Adiposity, acne, and hirsutism with menstrual irregularity can indicate PCOS as the primary cause, and your work-up should focus on a hyperandrogenic state.16 Low or normal FSH levels are common in patients with PCOS. Also test for 17a-hydroxyprogestrone and serum testosterone levels.17
Endometriosis. How endometriosis affects fertility is controversial. One hypothesis is that it is associated with overproduction of prostaglandins, metalloproteinases, cytokines, and chemokines. The inflammatory process impairs ovarian, peritoneal, tubal, and endometrial function.18
Discourage the use of the postcoital test. Patients may inquire about this test, in which the cervical mucus is obtained after intercourse to assess stretch ability and sperm motility. This test has been used for more than a century, but has poor predictive value and is not recommended.19
Or is it him?
Inquire about sexual development and medical history, including mumps orchitis or other infections, sinopulmonary symptoms suggesting cystic fibrosis, sexually transmitted infections (STIs) and genitourinary infections, and surgical procedures of the inguinal and scrotal areas. Also ask about prescription and illicit drug use, environmental exposures, and sexual history.
Physical exam. Look for signs of androgen deficiency, such as an arm span >2 cm longer than height (eunuchoidal proportions), or loss of pubic, axillary, or facial hair.20 Examine the external genitalia to evaluate for complete sexual development (Tanner stage of 5). The scrotum can provide clues to disorders that can affect sperm maturation and transport. Examination may reveal absence of the vas deferens, epididymal thickening, varicocele, or hernia.21 Testicular volume, if <15 mL with testicular length <3.6 cm, can point to a decreased number of seminiferous tubules.21
Semen analysis. If the physical examination is normal, analyze semen for volume and pH; microscopic debris and agglutination; sperm concentration, motility, and morphology; leukocyte count; and immature germ cells. Have the man abstain from sex for 2 to 7 days before semen collection. If collection is not possible to do in the office, the patient can drop it off at a lab within an hour of collection. Analyze 2 samples at least 2 weeks apart.22
More detailed semen analysis can be done, especially if evaluation of the female partner does not reveal a cause of infertility. Tests include sperm autoantibodies, sperm biochemistry, semen culture, sperm function tests, and sperm-cervical mucus interaction. Typically, these tests and further evaluation of the male partner after an abnormal semen analysis are best done by a urologist specializing in reproduction.
Oligospermia or azoospermia point toward hypogonadism. Elevated morning FSH and low total testosterone correlate with primary hypogonadism, whereas low levels of both hormones correlate with secondary hypogonadism. Hyperprolactinemia is a cause of secondary hypogonadism.3 Low volume of semen can be further evaluated by testing a postejaculatory urinalysis and transrectal ultrasonography to rule out retrograde ejaculation and ejaculatory duct obstruction.23
Fixing the problem
Focus initial counseling for couples on lifestyle modifications. Advise patients to quit smoking, reduce excessive caffeine and alcohol consumption, and engage in intercourse every day or every other day around ovulation. Patients should also avoid lubricants and douching as they can interfere with sperm deposition.
Managing female infertility
Tubal, pelvic, and uterine infertility. Patients with bilateral tubal obstruction may wish to undergo tubal reconstruction, especially if IVF treatments are not readily available to them. Counsel them that surgery for proximal tubal occlusion is not effective and the risk of ectopic pregnancy in the future is high, at approximately 20%.24 Because of the low efficacy of surgery and high ectopic rate, most patients with tubal disease favor IVF. Patients with endometriosis sometimes benefit from laser ablation or surgical resection, but often do well with intrauterine insemination (IUI) or IVF in conjunction with ovulation induction.25 Uterine abnormalities including submucous fibroid, endometrial polyp, septate uterus, or uterine synechiae frequently benefit from surgical correction.26 Patients with irreparable defects may want to consider a surrogate.
Ovulatory dysfunction. Anovulation can be hypogonadotropic hypogonadal (secondary to functional factors such as exercise and weight), normogonadotropic normoestrogenic with PCOS, or hypergonadotropic hypoestrogenic infertility (premature ovarian failure).
A body mass index >17 and <27 kg/m2 is optimal to achieve fertility and to sustain a healthy pregnancy.27 Individuals who are obese or very thin or who overexercise and do not respond to behavioral modification are known to benefit from pulsatile gonadotropin-releasing hormone therapy. This treatment, however, is not available in the United States.28
Dopaminergic agents can restore normal ovulation in patients with hyperprolactinemia,29 but they should receive ovulation induction first. Patients who have glucose intolerance may benefit from an insulin-sensitizing agent such as metformin. It is particularly useful if patients also have PCOS; however, it is not an FDA-approved indication for the medication. Clomiphene has recently been shown to result in a higher rate of ovulation, but not pregnancy, than metformin.30
Most patients with ovulatory dysfunction are best treated with clomiphene.31 Give 50 mg of the drug on cycle Days 3 through 7; ovulation occurs between Days 10 through 15.12 If, after the first cycle, pregnancy has not occurred, increase the dose by 50 mg with each cycle, to a maximum of 150 mg daily.32 Higher doses are not FDA approved, nor are they more effective. Clomiphene is most effective in the first 6 cycles, and the American Congress of Obstetricians and Gynecologists recommends limiting its use to fewer than 12 cycles due to the risk of ovarian neoplasm.33 Clomiphene yields an ovulation rate of 73% and a pregnancy rate of 36% per cycle. Multiple births, primarily twinning, occur at a rate of 8% to 13%.33 If clomiphene is unsuccessful, refer patients to a reproductive endocrinologist for evaluation for IVF and injectable ovulation-inducing agents.
Managing male infertility
Men who have hyperprolactinemic infertility can often be treated with dopaminergic agents such as bromocriptine. Inform them that normal spermatogenesis can take 3 to 6 months. Gonadotropin therapy may be effective for patients with hypothalamic or pituitary diseases. Surgery may correct obstruction, but may not actually increase pregnancy rates. Repairing a varicocele, for instance, increases sperm counts but not conception rates.34 Other obstructive problems may need sperm extraction followed by IUI or IVF, with or without intracytoplasmic sperm injection, where the sperm is injected into the ovum in the lab before implantation.34
Managing unexplained infertility
Fifteen percent of infertility is unexplained.35 Assisting these patients is challenging. Performing IUI with or without clomiphene, or giving clomiphene alone is often attempted. Pregnancy rates are 2% for expectant management, 5% for IUI alone, 9.5% for clomiphene alone, and 19% for combined IUI with clomiphene.36 Gonadotropins are no more effective in achieving conception than clomiphene, but gonadotropin injection and IUI together are more effective than no treatment.37 IVF, if successful, leads to pregnancy in the shortest amount of time. But it is the most costly intervention and the most likely to result in multiple births. In randomized controlled trials, however, IVF has not proved beneficial for unexplained infertility.38
Trends likely to affect fertility treatment
Currently in the United States, there is little regulation to guide reproductive technologies. But there is a trend, varying by state, toward legislation similar to child protection laws and adoption services, under which couples are evaluated for suitability as parents for the potential child’s safety.39 Other countries have acts regulating reproductive technologies and infertility services. England focuses on the child’s welfare; Australia restricts access by eligibility requirements.39 In the United States we may see similar policies, especially as controversy grows regarding multiple births. Cost is a factor in the treatment of infertility. Education and household income correlate with the amount of money spent on fertility care.
CORRESPONDENCE Heather Bell, MD, Center for Family Medicine, 1115 East 20th Street, Sioux Falls, SD 57105; [email protected]
1. Practice Committee of the American Society for Reproductive Medicine. Definitions of infertility and recurrent pregnancy loss. Fertil Steril. 2008;90(suppl):S60.-
2. Boivin J, Bunting L, Collins JA, et al. International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Hum Reprod. 2007;22:1506-1512.
3. Hull MGR, Glazener CMJ, Kelly NJ, et al. Population studies of causes, treatment, and outcome of infertility. BMJ. 1985;291:1693-1697.
4. Carlsen E, Giwercman A, Keiding N, et al. Evidence for decreasing quality of semen during past 50 years. BMJ. 1992;305:609-613.
5. de La Rochebrochard E, Thonneau P. Paternal age >or=40 years: an important risk factor for infertility. Am J Obstet Gynecol. 2003;189:901-905.
6. Behre HM, Kuhlage J, Gassner C, et al. Prediction of ovulation by urinary hormone measurements with the home use ClearPlan Fertility Monitor: comparison with transvaginal ultrasound scans and serum hormone measurements. Hum Reprod. 2000;15:2478-2482.
7. Brassard M, AinMelk Y, Baillargeon J. Basic infertility including polycystic ovary syndrome. Med Clin North Am. 2008;92:1163-1192.
8. Papaioannou S, Bourdrez P, Varma R, et al. Tubal evaluation in the investigation of subfertility: a structured comparison of tests. BJOG. 2004;111:1313-1321.
9. Mol BW, Collins JA, Van Der Veen F, et al. Cost-effectiveness of hysterosalpingography, laparoscopy, and Chlamydia antibody testing in subfertile couples. Fertil Steril. 2001;75:571-580.
10. Corson SL. Self-prediction of ovulation using a urinary luteinizing hormone test. J Reprod Med. 1986;31(8 suppl):760-763.
11. Kambic R, Gray RH. Interobserver variation in estimation of day of conception intercourse using selected natural family planning charts. Fertil Steril. 1989;51:430-434.
12. Wu CH, Winkel CA. The effect of therapy initiation day on clomiphene citrate therapy. Fertil Steril. 1989;52:564-568.
13. Chang MY, Chiang CH, Hsieh TT, et al. Use of the antral follicle count to predict the outcome of assisted reproductive technologies. Fertil Steril. 1998;69:505-510.
14. de Vet A, Laven JS, de Jong FH, et al. Antimüllerian hormone serum levels: a putative marker for ovarian aging. Fertil Steril. 2002;77:357-362.
15. De Sutter P. Rational diagnosis and treatment in infertility. Best Pract Res Clin Obstet Gynaecol. 2006;20:647-664.
16. Ehrmann DA. Polycystic ovary syndrome. N Engl J Med. 2005;352:1223-1236.
17. Azziz R, Zacur HA. 21-Hydroxylase deficiency in female hyperandrogenism, screening and diagnosis. J Clin Endocrinol Metab. 1989;69:577-584.
18. Bulun SE. Endometriosis. N Engl J Med. 2009;360:268-279.
19. van der Steeg JW, Steures P, Eijkemans MJ, et al. Should the post-coital test (PCT) be part of the routine fertility work-up? Hum Reprod. 2004;19:1373-1379.
20. Themmen APN, Huhtaniemi IT. Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev. 2000;21:551-583.
21. Rowe PJ. WHO Manual for the Standardization Investigation and Diagnosis of the Infertile Couple. New York, NY: Cambridge University Press; 1993.
22. World Health Organization Department of Reproductive Health and Research World Health Organization Laboratory Manual for the Examination and Processing of Human Semen. 5th ed. Geneva, Switzerland: World Health Organization; 2010.
23. Male Infertility Best Practice Policy Committee of the American Urological Association; Practical Committee of the American Society for Reproductive Medicine. Report on optimal evaluation of the infertile male. Fertil Steril. 2004;82(suppl 1):S123-S130.
24. Honoré GM, Holden AE, Schenken RS. Pathophysiology and management of proximal tubal blockage. Fertil Steril. 1999;71:785-795.
25. Tummon IS, Asher LF, Martin JS, et al. Randomized controlled trial of superovulation and insemination for infertility associated with minimal or mild endometriosis. Fertil Steril. 1997;68:8-12.
26. Heinonen PK, Saarikoski S, Pystynen P. Reproductive performance of women with uterine anomalies. An evaluation of 182 cases. Acta Obstet Gynecol Scand. 1982;61:157-162.
27. Frisch RE. The right weight: body fat, menarche and ovulation. Baillieres Clin Obstet Gynaecol. 1990;4:419-439.
28. Abraham S, Mira M, Llewellyn-Jones D. Should ovulation be induced in women recovering from an eating disorder or who are compulsive exercisers? Fertil Steril. 1990;53:566-568.
29. Crosignan PG. Management of hyperprolactinemia in infertility. J Reprod Med. 1999;44(12 suppl):1116-1120.
30. Baran S, Api M, Godsedef BP, et al. Comparison of metformin and clomiphene citrate therapy for induction in the polycystic ovary syndrome. Arch Gynecol Obstet. 2010;282:439-443.
31. Kerin JF, Liu JH, Phillipou G, et al. Evidence for a hypothalamic site of action of clomiphene citrate in women. J Clin Endocrinol Metab. 1985;61:265-268.
32. Huang KE. The primary treatment of luteal phase inadequacy: progesterone versus clomiphene citrate. Am J Obstet Gynecol. 1986;155:824-828.
33. Homburg R. Clomiphene citrate—end of an era? A mini-review. Hum Reprod. 2005;20:2043-2051.
34. Hirsh A. Male subfertility. BMJ. 2003;327:669-672.
35. Collins JA, Crosignani PG. Unexplained infertility: a review of diagnosis, prognosis, treatment efficacy and management. Int J Gynaecol Obstet. 1992;39:267-275.
36. Fisch P, Casper RF, Brown SE, et al. Unexplained infertility: evaluation of treatment with clomiphene citrate and human chorionic gonadotropin. Fertil Steril. 1989;51:828-833.
37. Verhulst SM, Cohlen BJ, Hughes E, et al. Intra-uterine insemination for unexplained subfertility. Cochrane Database Syst Rev. 2006;(4):CD001838.-
38. Pandian Z, Bhattacharya S, Vale L, et al. In vitro fertilization for unexplained subfertility. Cochrane Database Syst Rev. 2005;(2):CD003357.-
39. Liu C. Restricting access to infertility services: what is a justified limitation on reproductive freedom? Minn J Law Sci Technol. 2009;10:291. Available at: http://mjlst.umn.edu/uploads/QJ/WD/QJWDUZ1D-pV28sHvArqp6A/101_liu.pdf. Accessed May 4, 2011.
40. Practice Committee of the American Society for Reproductive Medicine. Optimal evaluation of the infertile female. Fertil Steril. 2006;86(suppl 1):S264-S267.
41. Practice Committee of the American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil Steril. 2004;82(suppl 1):S33-S39.
• Evaluate the fallopian tubes and their patency when menstruation is normal. Also, consider arranging for a hysterosalpingogram. B
• Suspect polycystic ovarian syndrome when adiposity, acne, and hirsutism with menstrual irregularity are factors. B
• Suspect androgen deficiency when a man’s arm span is >2 cm longer than his height, or when he has experienced a loss of pubic, axillary, or facial hair. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
During an annual visit, a patient confides in you that she and her husband have been trying to get pregnant for a year, but haven’t had any success. She tells you that she’s starting to get worried.
How do you advise her? What are your next steps?
The approach to evaluating infertility complaints is usually straightforward and can lead to a positive outcome. Not surprisingly, the dialogue often begins with you, the family physician. Your attention to clues in each partner’s history can do much to get to the heart of the problem. And even if a couple requires a specialty referral, it’s best to be familiar with the more extensive evaluation and management options they’ll encounter to help them anticipate likely discussions in their consultations. In this article, we review the best evidence for the care of your patients who want to conceive.
Who’s affected?
Infertility difficulties may be attributable to one or both partners, may be multifactorial, or may be unexplained (TABLE).
In women, infertility is the inability to conceive after 1 year of unprotected regular intercourse in those younger than 35 years, and after 6 months in those 35 years and older.1 Fecundability is the probability of achieving pregnancy in 1 menstrual cycle. Normal fecundability with a single menstrual cycle is ~20%, peaking between the ages of 20 and 24 years.2 Fecundability decreases slightly at age 32 and declines progressively and more rapidly after age 40. Spontaneous miscarriage is a factor; its rate in younger women is ~10% and in women >40 years is ~40%.2 Overall, approximately 13% of women between the ages of 15 and 44 have fecundity impairment, with more than 6 million women in the United States affected.2
About 24% of all cases of infertility are due to male factors—seminiferous dysfunction, including problems with motility, morphology, and volume of sperm; primary hypogonadism; posttesticular defects; and hypothalamic pituitary disease.3 Recent observational trends show declines in fertility among men older than 40, and among men from different areas in the country, thus raising the issue of the role that environmental pollutants or toxins may play. Supposed increases in urogenital abnormalities and testicular cancers may also contribute to declining fertility rates.4,5
TABLE
Consider these factors in cases of suspected infertility3,6,21,23,40,41
| Major causes of infertility | Infertility risk factors | |
|---|---|---|
| Female | ||
| Cause | Contribution | |
| Endocrine factors | 45%-55% |
|
| PCOS Thyroid Diabetes mellitus Prolactinemia | 21%-28% 10%-20% 10%-20% 7% | |
| Tubal and peritoneal pathology | 30%-40% | |
| Ovulatory dysfunction* | 15% | |
| Cervical and uterine factors | <5% | |
| Male | ||
| Cause | Contribution | |
| Seminiferous tubule dysfunction | 60%-80% |
|
| Posttesticular defects | 10%-20% | |
| Primary hypogonadism | 10%-15% | |
| Hypothalamic pituitary disease | 1%-2% | |
| *Assuming appropriate ovarian reserve, indicated by follicle-stimulating hormone (FSH) level <10 mIU/mL, FSH-to-luteinizing hormone ratio <2, and estradiol level <50 pg/mL. GU, genitourinary; PCOS, polycystic ovarian syndrome; STIs, sexually transmitted infections | ||
Zero in on these areas of the history
As with any diagnostic work-up, the most important aspect of an infertility evaluation is the history. Document menstrual cycle length and regularity and the timing of intercourse. (Ideally, this would be done when a couple first decides to conceive.) It’s important to know how long the couple has been trying to become pregnant. More time may be all they need to achieve pregnancy. Educate them on reproductive cycles and optimal timing to achieve pregnancy. Some women experience lower abdominal pain (mittelschmerz) signifying release of an egg from the ovary, which can help identify the time of ovulation.
Remember, too, the role that a couple’s psychological state can play; worries over suspected infertility may cause anxiety, anger, depression, and marital troubles.
Is it her?
Regular menstrual cycles—menses occurring every 21 to 35 days—carry an ovulation probability of 95% with each cycle.6 With normal menses, ovulatory dysfunction is an unlikely cause of infertility. If menstrual cycles are irregular, ovulatory function is not normal and cyclical. Explore the woman’s medical, surgical, and gynecologic histories, looking particularly for thyroid disease, galactorrhea, hirsutism, pelvic or abdominal pain, dysmenorrhea, dyspareunia, pelvic inflammatory disease (PID), and abdominal or pelvic surgery.
The fallopian tubes. When menstruation is normal, evaluate the fallopian tubes and their patency; 30% to 40% of infertility cases can be related to peritoneal pathology.3,7 Inability to conceive in a previous relationship, history of PID, or prior tubal surgery all correlate to infertility. Ten percent of patients with a history of one PID episode and 54% to 75% of patients with 3 episodes will have patency issues.7
Consider arranging for a hysterosalpingogram (HSG) in all patients as part of an initial work-up for infertility.8 HSG is useful in evaluating tubal patency and the uterine cavity, and it can be therapeutic. HSG is not useful in detecting peritubal adhesions or endometriosis; patients in whom you suspect these conditions should undergo diagnostic laparoscopy. If abnormalities are found on HSG, refer patients to a reproductive endocrinologist to evaluate treatment options.
Chlamydia trachomatis IgG antibody testing can predict the presence of tubal disease. For women with low risk of tubal disease, it may be more cost effective to test for the Chlamydia antibody and proceed with HSG if the result is positive. Antibody testing is also useful for women with an allergy to contrast dye who cannot undergo an HSG. If the antibody test result is positive, consider arranging for a sonohysterogram to evaluate for the presence of fluid in the cul-de-sac, or an intrauterine infusion of saline to evaluate the patency of at least one tube.9
Ovulatory function. To assess ovulatory function, measure a midluteal-phase serum progesterone level, drawn 1 week before the expected day of menses (Day 21 of a 28-day cycle). A level >3 ng/mL is evidence of ovulation. Over-the-counter ovulation kits detect the luteinizing hormone (LH) surge but have false-positive and false-negative rates of 5% to 10%, respectively.10 Recording basal body temperature is a noninvasive and inexpensive means of evaluating ovulation. The patient must record temperatures at exactly the same time each day. Have her log the temperatures and watch for a spike that occurs 1 to 2 days after the LH surge. The average woman’s temperature rises above 98ºF in progressing from the follicular to the luteal phase. Since the spike occurs 1 to 2 days after ovulation, this method is best used for many months so the woman can predict her cycle.11
Once timing of ovulation has been established, you can check lab results at Day 3 of the woman’s cycle for follicle-stimulating hormone (FSH), LH, estradiol, thyroid-stimulating hormone (TSH), prolactin, and 2-hour fasting glucose tolerance. In addition to polycystic ovarian syndrome (PCOS), patients may have ovulatory dysfunction secondary to glucose intolerance.
A clomiphene (Clomid) challenge can help in assessing ovarian reserve. Administer 100 mg clomiphene on Days 5 through 9 of the patient’s cycle, and check FSH and estradiol levels on Day 10. With diminished ovarian reserve, FSH will increase to >12 mIU/mL and estradiol to >300 pg/mL.12 If this occurs, consider referring for an ultrasound measurement of antral follicle count. The presence of 4 to 10 follicles measuring 2 to 10 mm in diameter suggests adequate reserve.13
Although not widely available in the United States, the test for antimüllerian hormone (AMH) levels may be useful in reflecting the size of the primordial follicle pool. At menopause, the level is undetectable. A level above 0.5 ng/mL correlates with good ovarian reserve; levels <0.15 ng/mL suggest poor response to in vitro fertilization (IVF).14
Endocrine factors account for 45% to 55% of female infertility and include thyroid disease, PCOS, diabetes mellitus, prolactinemia, and luteal phase defects. Subclinical hypothyroidism, often evidenced only by high levels of TSH, decreases the chance of a successful pregnancy. This can occur even if the dysfunction is not severe enough to affect cycle regularity.15 Clinical hypo- or hyperthyroidism can affect ovulation by interfering with normal hormonal feedback loops, and correcting thyroid disease can improve fertility.
Galactorrhea discovered during the history and physical may be caused by elevated prolactin levels, which also inhibit normal ovulatory function. Chronically elevated prolactin levels in patients with PCOS can be attributed to elevated estrogen levels. Adiposity, acne, and hirsutism with menstrual irregularity can indicate PCOS as the primary cause, and your work-up should focus on a hyperandrogenic state.16 Low or normal FSH levels are common in patients with PCOS. Also test for 17a-hydroxyprogestrone and serum testosterone levels.17
Endometriosis. How endometriosis affects fertility is controversial. One hypothesis is that it is associated with overproduction of prostaglandins, metalloproteinases, cytokines, and chemokines. The inflammatory process impairs ovarian, peritoneal, tubal, and endometrial function.18
Discourage the use of the postcoital test. Patients may inquire about this test, in which the cervical mucus is obtained after intercourse to assess stretch ability and sperm motility. This test has been used for more than a century, but has poor predictive value and is not recommended.19
Or is it him?
Inquire about sexual development and medical history, including mumps orchitis or other infections, sinopulmonary symptoms suggesting cystic fibrosis, sexually transmitted infections (STIs) and genitourinary infections, and surgical procedures of the inguinal and scrotal areas. Also ask about prescription and illicit drug use, environmental exposures, and sexual history.
Physical exam. Look for signs of androgen deficiency, such as an arm span >2 cm longer than height (eunuchoidal proportions), or loss of pubic, axillary, or facial hair.20 Examine the external genitalia to evaluate for complete sexual development (Tanner stage of 5). The scrotum can provide clues to disorders that can affect sperm maturation and transport. Examination may reveal absence of the vas deferens, epididymal thickening, varicocele, or hernia.21 Testicular volume, if <15 mL with testicular length <3.6 cm, can point to a decreased number of seminiferous tubules.21
Semen analysis. If the physical examination is normal, analyze semen for volume and pH; microscopic debris and agglutination; sperm concentration, motility, and morphology; leukocyte count; and immature germ cells. Have the man abstain from sex for 2 to 7 days before semen collection. If collection is not possible to do in the office, the patient can drop it off at a lab within an hour of collection. Analyze 2 samples at least 2 weeks apart.22
More detailed semen analysis can be done, especially if evaluation of the female partner does not reveal a cause of infertility. Tests include sperm autoantibodies, sperm biochemistry, semen culture, sperm function tests, and sperm-cervical mucus interaction. Typically, these tests and further evaluation of the male partner after an abnormal semen analysis are best done by a urologist specializing in reproduction.
Oligospermia or azoospermia point toward hypogonadism. Elevated morning FSH and low total testosterone correlate with primary hypogonadism, whereas low levels of both hormones correlate with secondary hypogonadism. Hyperprolactinemia is a cause of secondary hypogonadism.3 Low volume of semen can be further evaluated by testing a postejaculatory urinalysis and transrectal ultrasonography to rule out retrograde ejaculation and ejaculatory duct obstruction.23
Fixing the problem
Focus initial counseling for couples on lifestyle modifications. Advise patients to quit smoking, reduce excessive caffeine and alcohol consumption, and engage in intercourse every day or every other day around ovulation. Patients should also avoid lubricants and douching as they can interfere with sperm deposition.
Managing female infertility
Tubal, pelvic, and uterine infertility. Patients with bilateral tubal obstruction may wish to undergo tubal reconstruction, especially if IVF treatments are not readily available to them. Counsel them that surgery for proximal tubal occlusion is not effective and the risk of ectopic pregnancy in the future is high, at approximately 20%.24 Because of the low efficacy of surgery and high ectopic rate, most patients with tubal disease favor IVF. Patients with endometriosis sometimes benefit from laser ablation or surgical resection, but often do well with intrauterine insemination (IUI) or IVF in conjunction with ovulation induction.25 Uterine abnormalities including submucous fibroid, endometrial polyp, septate uterus, or uterine synechiae frequently benefit from surgical correction.26 Patients with irreparable defects may want to consider a surrogate.
Ovulatory dysfunction. Anovulation can be hypogonadotropic hypogonadal (secondary to functional factors such as exercise and weight), normogonadotropic normoestrogenic with PCOS, or hypergonadotropic hypoestrogenic infertility (premature ovarian failure).
A body mass index >17 and <27 kg/m2 is optimal to achieve fertility and to sustain a healthy pregnancy.27 Individuals who are obese or very thin or who overexercise and do not respond to behavioral modification are known to benefit from pulsatile gonadotropin-releasing hormone therapy. This treatment, however, is not available in the United States.28
Dopaminergic agents can restore normal ovulation in patients with hyperprolactinemia,29 but they should receive ovulation induction first. Patients who have glucose intolerance may benefit from an insulin-sensitizing agent such as metformin. It is particularly useful if patients also have PCOS; however, it is not an FDA-approved indication for the medication. Clomiphene has recently been shown to result in a higher rate of ovulation, but not pregnancy, than metformin.30
Most patients with ovulatory dysfunction are best treated with clomiphene.31 Give 50 mg of the drug on cycle Days 3 through 7; ovulation occurs between Days 10 through 15.12 If, after the first cycle, pregnancy has not occurred, increase the dose by 50 mg with each cycle, to a maximum of 150 mg daily.32 Higher doses are not FDA approved, nor are they more effective. Clomiphene is most effective in the first 6 cycles, and the American Congress of Obstetricians and Gynecologists recommends limiting its use to fewer than 12 cycles due to the risk of ovarian neoplasm.33 Clomiphene yields an ovulation rate of 73% and a pregnancy rate of 36% per cycle. Multiple births, primarily twinning, occur at a rate of 8% to 13%.33 If clomiphene is unsuccessful, refer patients to a reproductive endocrinologist for evaluation for IVF and injectable ovulation-inducing agents.
Managing male infertility
Men who have hyperprolactinemic infertility can often be treated with dopaminergic agents such as bromocriptine. Inform them that normal spermatogenesis can take 3 to 6 months. Gonadotropin therapy may be effective for patients with hypothalamic or pituitary diseases. Surgery may correct obstruction, but may not actually increase pregnancy rates. Repairing a varicocele, for instance, increases sperm counts but not conception rates.34 Other obstructive problems may need sperm extraction followed by IUI or IVF, with or without intracytoplasmic sperm injection, where the sperm is injected into the ovum in the lab before implantation.34
Managing unexplained infertility
Fifteen percent of infertility is unexplained.35 Assisting these patients is challenging. Performing IUI with or without clomiphene, or giving clomiphene alone is often attempted. Pregnancy rates are 2% for expectant management, 5% for IUI alone, 9.5% for clomiphene alone, and 19% for combined IUI with clomiphene.36 Gonadotropins are no more effective in achieving conception than clomiphene, but gonadotropin injection and IUI together are more effective than no treatment.37 IVF, if successful, leads to pregnancy in the shortest amount of time. But it is the most costly intervention and the most likely to result in multiple births. In randomized controlled trials, however, IVF has not proved beneficial for unexplained infertility.38
Trends likely to affect fertility treatment
Currently in the United States, there is little regulation to guide reproductive technologies. But there is a trend, varying by state, toward legislation similar to child protection laws and adoption services, under which couples are evaluated for suitability as parents for the potential child’s safety.39 Other countries have acts regulating reproductive technologies and infertility services. England focuses on the child’s welfare; Australia restricts access by eligibility requirements.39 In the United States we may see similar policies, especially as controversy grows regarding multiple births. Cost is a factor in the treatment of infertility. Education and household income correlate with the amount of money spent on fertility care.
CORRESPONDENCE Heather Bell, MD, Center for Family Medicine, 1115 East 20th Street, Sioux Falls, SD 57105; [email protected]
• Evaluate the fallopian tubes and their patency when menstruation is normal. Also, consider arranging for a hysterosalpingogram. B
• Suspect polycystic ovarian syndrome when adiposity, acne, and hirsutism with menstrual irregularity are factors. B
• Suspect androgen deficiency when a man’s arm span is >2 cm longer than his height, or when he has experienced a loss of pubic, axillary, or facial hair. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
During an annual visit, a patient confides in you that she and her husband have been trying to get pregnant for a year, but haven’t had any success. She tells you that she’s starting to get worried.
How do you advise her? What are your next steps?
The approach to evaluating infertility complaints is usually straightforward and can lead to a positive outcome. Not surprisingly, the dialogue often begins with you, the family physician. Your attention to clues in each partner’s history can do much to get to the heart of the problem. And even if a couple requires a specialty referral, it’s best to be familiar with the more extensive evaluation and management options they’ll encounter to help them anticipate likely discussions in their consultations. In this article, we review the best evidence for the care of your patients who want to conceive.
Who’s affected?
Infertility difficulties may be attributable to one or both partners, may be multifactorial, or may be unexplained (TABLE).
In women, infertility is the inability to conceive after 1 year of unprotected regular intercourse in those younger than 35 years, and after 6 months in those 35 years and older.1 Fecundability is the probability of achieving pregnancy in 1 menstrual cycle. Normal fecundability with a single menstrual cycle is ~20%, peaking between the ages of 20 and 24 years.2 Fecundability decreases slightly at age 32 and declines progressively and more rapidly after age 40. Spontaneous miscarriage is a factor; its rate in younger women is ~10% and in women >40 years is ~40%.2 Overall, approximately 13% of women between the ages of 15 and 44 have fecundity impairment, with more than 6 million women in the United States affected.2
About 24% of all cases of infertility are due to male factors—seminiferous dysfunction, including problems with motility, morphology, and volume of sperm; primary hypogonadism; posttesticular defects; and hypothalamic pituitary disease.3 Recent observational trends show declines in fertility among men older than 40, and among men from different areas in the country, thus raising the issue of the role that environmental pollutants or toxins may play. Supposed increases in urogenital abnormalities and testicular cancers may also contribute to declining fertility rates.4,5
TABLE
Consider these factors in cases of suspected infertility3,6,21,23,40,41
| Major causes of infertility | Infertility risk factors | |
|---|---|---|
| Female | ||
| Cause | Contribution | |
| Endocrine factors | 45%-55% |
|
| PCOS Thyroid Diabetes mellitus Prolactinemia | 21%-28% 10%-20% 10%-20% 7% | |
| Tubal and peritoneal pathology | 30%-40% | |
| Ovulatory dysfunction* | 15% | |
| Cervical and uterine factors | <5% | |
| Male | ||
| Cause | Contribution | |
| Seminiferous tubule dysfunction | 60%-80% |
|
| Posttesticular defects | 10%-20% | |
| Primary hypogonadism | 10%-15% | |
| Hypothalamic pituitary disease | 1%-2% | |
| *Assuming appropriate ovarian reserve, indicated by follicle-stimulating hormone (FSH) level <10 mIU/mL, FSH-to-luteinizing hormone ratio <2, and estradiol level <50 pg/mL. GU, genitourinary; PCOS, polycystic ovarian syndrome; STIs, sexually transmitted infections | ||
Zero in on these areas of the history
As with any diagnostic work-up, the most important aspect of an infertility evaluation is the history. Document menstrual cycle length and regularity and the timing of intercourse. (Ideally, this would be done when a couple first decides to conceive.) It’s important to know how long the couple has been trying to become pregnant. More time may be all they need to achieve pregnancy. Educate them on reproductive cycles and optimal timing to achieve pregnancy. Some women experience lower abdominal pain (mittelschmerz) signifying release of an egg from the ovary, which can help identify the time of ovulation.
Remember, too, the role that a couple’s psychological state can play; worries over suspected infertility may cause anxiety, anger, depression, and marital troubles.
Is it her?
Regular menstrual cycles—menses occurring every 21 to 35 days—carry an ovulation probability of 95% with each cycle.6 With normal menses, ovulatory dysfunction is an unlikely cause of infertility. If menstrual cycles are irregular, ovulatory function is not normal and cyclical. Explore the woman’s medical, surgical, and gynecologic histories, looking particularly for thyroid disease, galactorrhea, hirsutism, pelvic or abdominal pain, dysmenorrhea, dyspareunia, pelvic inflammatory disease (PID), and abdominal or pelvic surgery.
The fallopian tubes. When menstruation is normal, evaluate the fallopian tubes and their patency; 30% to 40% of infertility cases can be related to peritoneal pathology.3,7 Inability to conceive in a previous relationship, history of PID, or prior tubal surgery all correlate to infertility. Ten percent of patients with a history of one PID episode and 54% to 75% of patients with 3 episodes will have patency issues.7
Consider arranging for a hysterosalpingogram (HSG) in all patients as part of an initial work-up for infertility.8 HSG is useful in evaluating tubal patency and the uterine cavity, and it can be therapeutic. HSG is not useful in detecting peritubal adhesions or endometriosis; patients in whom you suspect these conditions should undergo diagnostic laparoscopy. If abnormalities are found on HSG, refer patients to a reproductive endocrinologist to evaluate treatment options.
Chlamydia trachomatis IgG antibody testing can predict the presence of tubal disease. For women with low risk of tubal disease, it may be more cost effective to test for the Chlamydia antibody and proceed with HSG if the result is positive. Antibody testing is also useful for women with an allergy to contrast dye who cannot undergo an HSG. If the antibody test result is positive, consider arranging for a sonohysterogram to evaluate for the presence of fluid in the cul-de-sac, or an intrauterine infusion of saline to evaluate the patency of at least one tube.9
Ovulatory function. To assess ovulatory function, measure a midluteal-phase serum progesterone level, drawn 1 week before the expected day of menses (Day 21 of a 28-day cycle). A level >3 ng/mL is evidence of ovulation. Over-the-counter ovulation kits detect the luteinizing hormone (LH) surge but have false-positive and false-negative rates of 5% to 10%, respectively.10 Recording basal body temperature is a noninvasive and inexpensive means of evaluating ovulation. The patient must record temperatures at exactly the same time each day. Have her log the temperatures and watch for a spike that occurs 1 to 2 days after the LH surge. The average woman’s temperature rises above 98ºF in progressing from the follicular to the luteal phase. Since the spike occurs 1 to 2 days after ovulation, this method is best used for many months so the woman can predict her cycle.11
Once timing of ovulation has been established, you can check lab results at Day 3 of the woman’s cycle for follicle-stimulating hormone (FSH), LH, estradiol, thyroid-stimulating hormone (TSH), prolactin, and 2-hour fasting glucose tolerance. In addition to polycystic ovarian syndrome (PCOS), patients may have ovulatory dysfunction secondary to glucose intolerance.
A clomiphene (Clomid) challenge can help in assessing ovarian reserve. Administer 100 mg clomiphene on Days 5 through 9 of the patient’s cycle, and check FSH and estradiol levels on Day 10. With diminished ovarian reserve, FSH will increase to >12 mIU/mL and estradiol to >300 pg/mL.12 If this occurs, consider referring for an ultrasound measurement of antral follicle count. The presence of 4 to 10 follicles measuring 2 to 10 mm in diameter suggests adequate reserve.13
Although not widely available in the United States, the test for antimüllerian hormone (AMH) levels may be useful in reflecting the size of the primordial follicle pool. At menopause, the level is undetectable. A level above 0.5 ng/mL correlates with good ovarian reserve; levels <0.15 ng/mL suggest poor response to in vitro fertilization (IVF).14
Endocrine factors account for 45% to 55% of female infertility and include thyroid disease, PCOS, diabetes mellitus, prolactinemia, and luteal phase defects. Subclinical hypothyroidism, often evidenced only by high levels of TSH, decreases the chance of a successful pregnancy. This can occur even if the dysfunction is not severe enough to affect cycle regularity.15 Clinical hypo- or hyperthyroidism can affect ovulation by interfering with normal hormonal feedback loops, and correcting thyroid disease can improve fertility.
Galactorrhea discovered during the history and physical may be caused by elevated prolactin levels, which also inhibit normal ovulatory function. Chronically elevated prolactin levels in patients with PCOS can be attributed to elevated estrogen levels. Adiposity, acne, and hirsutism with menstrual irregularity can indicate PCOS as the primary cause, and your work-up should focus on a hyperandrogenic state.16 Low or normal FSH levels are common in patients with PCOS. Also test for 17a-hydroxyprogestrone and serum testosterone levels.17
Endometriosis. How endometriosis affects fertility is controversial. One hypothesis is that it is associated with overproduction of prostaglandins, metalloproteinases, cytokines, and chemokines. The inflammatory process impairs ovarian, peritoneal, tubal, and endometrial function.18
Discourage the use of the postcoital test. Patients may inquire about this test, in which the cervical mucus is obtained after intercourse to assess stretch ability and sperm motility. This test has been used for more than a century, but has poor predictive value and is not recommended.19
Or is it him?
Inquire about sexual development and medical history, including mumps orchitis or other infections, sinopulmonary symptoms suggesting cystic fibrosis, sexually transmitted infections (STIs) and genitourinary infections, and surgical procedures of the inguinal and scrotal areas. Also ask about prescription and illicit drug use, environmental exposures, and sexual history.
Physical exam. Look for signs of androgen deficiency, such as an arm span >2 cm longer than height (eunuchoidal proportions), or loss of pubic, axillary, or facial hair.20 Examine the external genitalia to evaluate for complete sexual development (Tanner stage of 5). The scrotum can provide clues to disorders that can affect sperm maturation and transport. Examination may reveal absence of the vas deferens, epididymal thickening, varicocele, or hernia.21 Testicular volume, if <15 mL with testicular length <3.6 cm, can point to a decreased number of seminiferous tubules.21
Semen analysis. If the physical examination is normal, analyze semen for volume and pH; microscopic debris and agglutination; sperm concentration, motility, and morphology; leukocyte count; and immature germ cells. Have the man abstain from sex for 2 to 7 days before semen collection. If collection is not possible to do in the office, the patient can drop it off at a lab within an hour of collection. Analyze 2 samples at least 2 weeks apart.22
More detailed semen analysis can be done, especially if evaluation of the female partner does not reveal a cause of infertility. Tests include sperm autoantibodies, sperm biochemistry, semen culture, sperm function tests, and sperm-cervical mucus interaction. Typically, these tests and further evaluation of the male partner after an abnormal semen analysis are best done by a urologist specializing in reproduction.
Oligospermia or azoospermia point toward hypogonadism. Elevated morning FSH and low total testosterone correlate with primary hypogonadism, whereas low levels of both hormones correlate with secondary hypogonadism. Hyperprolactinemia is a cause of secondary hypogonadism.3 Low volume of semen can be further evaluated by testing a postejaculatory urinalysis and transrectal ultrasonography to rule out retrograde ejaculation and ejaculatory duct obstruction.23
Fixing the problem
Focus initial counseling for couples on lifestyle modifications. Advise patients to quit smoking, reduce excessive caffeine and alcohol consumption, and engage in intercourse every day or every other day around ovulation. Patients should also avoid lubricants and douching as they can interfere with sperm deposition.
Managing female infertility
Tubal, pelvic, and uterine infertility. Patients with bilateral tubal obstruction may wish to undergo tubal reconstruction, especially if IVF treatments are not readily available to them. Counsel them that surgery for proximal tubal occlusion is not effective and the risk of ectopic pregnancy in the future is high, at approximately 20%.24 Because of the low efficacy of surgery and high ectopic rate, most patients with tubal disease favor IVF. Patients with endometriosis sometimes benefit from laser ablation or surgical resection, but often do well with intrauterine insemination (IUI) or IVF in conjunction with ovulation induction.25 Uterine abnormalities including submucous fibroid, endometrial polyp, septate uterus, or uterine synechiae frequently benefit from surgical correction.26 Patients with irreparable defects may want to consider a surrogate.
Ovulatory dysfunction. Anovulation can be hypogonadotropic hypogonadal (secondary to functional factors such as exercise and weight), normogonadotropic normoestrogenic with PCOS, or hypergonadotropic hypoestrogenic infertility (premature ovarian failure).
A body mass index >17 and <27 kg/m2 is optimal to achieve fertility and to sustain a healthy pregnancy.27 Individuals who are obese or very thin or who overexercise and do not respond to behavioral modification are known to benefit from pulsatile gonadotropin-releasing hormone therapy. This treatment, however, is not available in the United States.28
Dopaminergic agents can restore normal ovulation in patients with hyperprolactinemia,29 but they should receive ovulation induction first. Patients who have glucose intolerance may benefit from an insulin-sensitizing agent such as metformin. It is particularly useful if patients also have PCOS; however, it is not an FDA-approved indication for the medication. Clomiphene has recently been shown to result in a higher rate of ovulation, but not pregnancy, than metformin.30
Most patients with ovulatory dysfunction are best treated with clomiphene.31 Give 50 mg of the drug on cycle Days 3 through 7; ovulation occurs between Days 10 through 15.12 If, after the first cycle, pregnancy has not occurred, increase the dose by 50 mg with each cycle, to a maximum of 150 mg daily.32 Higher doses are not FDA approved, nor are they more effective. Clomiphene is most effective in the first 6 cycles, and the American Congress of Obstetricians and Gynecologists recommends limiting its use to fewer than 12 cycles due to the risk of ovarian neoplasm.33 Clomiphene yields an ovulation rate of 73% and a pregnancy rate of 36% per cycle. Multiple births, primarily twinning, occur at a rate of 8% to 13%.33 If clomiphene is unsuccessful, refer patients to a reproductive endocrinologist for evaluation for IVF and injectable ovulation-inducing agents.
Managing male infertility
Men who have hyperprolactinemic infertility can often be treated with dopaminergic agents such as bromocriptine. Inform them that normal spermatogenesis can take 3 to 6 months. Gonadotropin therapy may be effective for patients with hypothalamic or pituitary diseases. Surgery may correct obstruction, but may not actually increase pregnancy rates. Repairing a varicocele, for instance, increases sperm counts but not conception rates.34 Other obstructive problems may need sperm extraction followed by IUI or IVF, with or without intracytoplasmic sperm injection, where the sperm is injected into the ovum in the lab before implantation.34
Managing unexplained infertility
Fifteen percent of infertility is unexplained.35 Assisting these patients is challenging. Performing IUI with or without clomiphene, or giving clomiphene alone is often attempted. Pregnancy rates are 2% for expectant management, 5% for IUI alone, 9.5% for clomiphene alone, and 19% for combined IUI with clomiphene.36 Gonadotropins are no more effective in achieving conception than clomiphene, but gonadotropin injection and IUI together are more effective than no treatment.37 IVF, if successful, leads to pregnancy in the shortest amount of time. But it is the most costly intervention and the most likely to result in multiple births. In randomized controlled trials, however, IVF has not proved beneficial for unexplained infertility.38
Trends likely to affect fertility treatment
Currently in the United States, there is little regulation to guide reproductive technologies. But there is a trend, varying by state, toward legislation similar to child protection laws and adoption services, under which couples are evaluated for suitability as parents for the potential child’s safety.39 Other countries have acts regulating reproductive technologies and infertility services. England focuses on the child’s welfare; Australia restricts access by eligibility requirements.39 In the United States we may see similar policies, especially as controversy grows regarding multiple births. Cost is a factor in the treatment of infertility. Education and household income correlate with the amount of money spent on fertility care.
CORRESPONDENCE Heather Bell, MD, Center for Family Medicine, 1115 East 20th Street, Sioux Falls, SD 57105; [email protected]
1. Practice Committee of the American Society for Reproductive Medicine. Definitions of infertility and recurrent pregnancy loss. Fertil Steril. 2008;90(suppl):S60.-
2. Boivin J, Bunting L, Collins JA, et al. International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Hum Reprod. 2007;22:1506-1512.
3. Hull MGR, Glazener CMJ, Kelly NJ, et al. Population studies of causes, treatment, and outcome of infertility. BMJ. 1985;291:1693-1697.
4. Carlsen E, Giwercman A, Keiding N, et al. Evidence for decreasing quality of semen during past 50 years. BMJ. 1992;305:609-613.
5. de La Rochebrochard E, Thonneau P. Paternal age >or=40 years: an important risk factor for infertility. Am J Obstet Gynecol. 2003;189:901-905.
6. Behre HM, Kuhlage J, Gassner C, et al. Prediction of ovulation by urinary hormone measurements with the home use ClearPlan Fertility Monitor: comparison with transvaginal ultrasound scans and serum hormone measurements. Hum Reprod. 2000;15:2478-2482.
7. Brassard M, AinMelk Y, Baillargeon J. Basic infertility including polycystic ovary syndrome. Med Clin North Am. 2008;92:1163-1192.
8. Papaioannou S, Bourdrez P, Varma R, et al. Tubal evaluation in the investigation of subfertility: a structured comparison of tests. BJOG. 2004;111:1313-1321.
9. Mol BW, Collins JA, Van Der Veen F, et al. Cost-effectiveness of hysterosalpingography, laparoscopy, and Chlamydia antibody testing in subfertile couples. Fertil Steril. 2001;75:571-580.
10. Corson SL. Self-prediction of ovulation using a urinary luteinizing hormone test. J Reprod Med. 1986;31(8 suppl):760-763.
11. Kambic R, Gray RH. Interobserver variation in estimation of day of conception intercourse using selected natural family planning charts. Fertil Steril. 1989;51:430-434.
12. Wu CH, Winkel CA. The effect of therapy initiation day on clomiphene citrate therapy. Fertil Steril. 1989;52:564-568.
13. Chang MY, Chiang CH, Hsieh TT, et al. Use of the antral follicle count to predict the outcome of assisted reproductive technologies. Fertil Steril. 1998;69:505-510.
14. de Vet A, Laven JS, de Jong FH, et al. Antimüllerian hormone serum levels: a putative marker for ovarian aging. Fertil Steril. 2002;77:357-362.
15. De Sutter P. Rational diagnosis and treatment in infertility. Best Pract Res Clin Obstet Gynaecol. 2006;20:647-664.
16. Ehrmann DA. Polycystic ovary syndrome. N Engl J Med. 2005;352:1223-1236.
17. Azziz R, Zacur HA. 21-Hydroxylase deficiency in female hyperandrogenism, screening and diagnosis. J Clin Endocrinol Metab. 1989;69:577-584.
18. Bulun SE. Endometriosis. N Engl J Med. 2009;360:268-279.
19. van der Steeg JW, Steures P, Eijkemans MJ, et al. Should the post-coital test (PCT) be part of the routine fertility work-up? Hum Reprod. 2004;19:1373-1379.
20. Themmen APN, Huhtaniemi IT. Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev. 2000;21:551-583.
21. Rowe PJ. WHO Manual for the Standardization Investigation and Diagnosis of the Infertile Couple. New York, NY: Cambridge University Press; 1993.
22. World Health Organization Department of Reproductive Health and Research World Health Organization Laboratory Manual for the Examination and Processing of Human Semen. 5th ed. Geneva, Switzerland: World Health Organization; 2010.
23. Male Infertility Best Practice Policy Committee of the American Urological Association; Practical Committee of the American Society for Reproductive Medicine. Report on optimal evaluation of the infertile male. Fertil Steril. 2004;82(suppl 1):S123-S130.
24. Honoré GM, Holden AE, Schenken RS. Pathophysiology and management of proximal tubal blockage. Fertil Steril. 1999;71:785-795.
25. Tummon IS, Asher LF, Martin JS, et al. Randomized controlled trial of superovulation and insemination for infertility associated with minimal or mild endometriosis. Fertil Steril. 1997;68:8-12.
26. Heinonen PK, Saarikoski S, Pystynen P. Reproductive performance of women with uterine anomalies. An evaluation of 182 cases. Acta Obstet Gynecol Scand. 1982;61:157-162.
27. Frisch RE. The right weight: body fat, menarche and ovulation. Baillieres Clin Obstet Gynaecol. 1990;4:419-439.
28. Abraham S, Mira M, Llewellyn-Jones D. Should ovulation be induced in women recovering from an eating disorder or who are compulsive exercisers? Fertil Steril. 1990;53:566-568.
29. Crosignan PG. Management of hyperprolactinemia in infertility. J Reprod Med. 1999;44(12 suppl):1116-1120.
30. Baran S, Api M, Godsedef BP, et al. Comparison of metformin and clomiphene citrate therapy for induction in the polycystic ovary syndrome. Arch Gynecol Obstet. 2010;282:439-443.
31. Kerin JF, Liu JH, Phillipou G, et al. Evidence for a hypothalamic site of action of clomiphene citrate in women. J Clin Endocrinol Metab. 1985;61:265-268.
32. Huang KE. The primary treatment of luteal phase inadequacy: progesterone versus clomiphene citrate. Am J Obstet Gynecol. 1986;155:824-828.
33. Homburg R. Clomiphene citrate—end of an era? A mini-review. Hum Reprod. 2005;20:2043-2051.
34. Hirsh A. Male subfertility. BMJ. 2003;327:669-672.
35. Collins JA, Crosignani PG. Unexplained infertility: a review of diagnosis, prognosis, treatment efficacy and management. Int J Gynaecol Obstet. 1992;39:267-275.
36. Fisch P, Casper RF, Brown SE, et al. Unexplained infertility: evaluation of treatment with clomiphene citrate and human chorionic gonadotropin. Fertil Steril. 1989;51:828-833.
37. Verhulst SM, Cohlen BJ, Hughes E, et al. Intra-uterine insemination for unexplained subfertility. Cochrane Database Syst Rev. 2006;(4):CD001838.-
38. Pandian Z, Bhattacharya S, Vale L, et al. In vitro fertilization for unexplained subfertility. Cochrane Database Syst Rev. 2005;(2):CD003357.-
39. Liu C. Restricting access to infertility services: what is a justified limitation on reproductive freedom? Minn J Law Sci Technol. 2009;10:291. Available at: http://mjlst.umn.edu/uploads/QJ/WD/QJWDUZ1D-pV28sHvArqp6A/101_liu.pdf. Accessed May 4, 2011.
40. Practice Committee of the American Society for Reproductive Medicine. Optimal evaluation of the infertile female. Fertil Steril. 2006;86(suppl 1):S264-S267.
41. Practice Committee of the American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil Steril. 2004;82(suppl 1):S33-S39.
1. Practice Committee of the American Society for Reproductive Medicine. Definitions of infertility and recurrent pregnancy loss. Fertil Steril. 2008;90(suppl):S60.-
2. Boivin J, Bunting L, Collins JA, et al. International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Hum Reprod. 2007;22:1506-1512.
3. Hull MGR, Glazener CMJ, Kelly NJ, et al. Population studies of causes, treatment, and outcome of infertility. BMJ. 1985;291:1693-1697.
4. Carlsen E, Giwercman A, Keiding N, et al. Evidence for decreasing quality of semen during past 50 years. BMJ. 1992;305:609-613.
5. de La Rochebrochard E, Thonneau P. Paternal age >or=40 years: an important risk factor for infertility. Am J Obstet Gynecol. 2003;189:901-905.
6. Behre HM, Kuhlage J, Gassner C, et al. Prediction of ovulation by urinary hormone measurements with the home use ClearPlan Fertility Monitor: comparison with transvaginal ultrasound scans and serum hormone measurements. Hum Reprod. 2000;15:2478-2482.
7. Brassard M, AinMelk Y, Baillargeon J. Basic infertility including polycystic ovary syndrome. Med Clin North Am. 2008;92:1163-1192.
8. Papaioannou S, Bourdrez P, Varma R, et al. Tubal evaluation in the investigation of subfertility: a structured comparison of tests. BJOG. 2004;111:1313-1321.
9. Mol BW, Collins JA, Van Der Veen F, et al. Cost-effectiveness of hysterosalpingography, laparoscopy, and Chlamydia antibody testing in subfertile couples. Fertil Steril. 2001;75:571-580.
10. Corson SL. Self-prediction of ovulation using a urinary luteinizing hormone test. J Reprod Med. 1986;31(8 suppl):760-763.
11. Kambic R, Gray RH. Interobserver variation in estimation of day of conception intercourse using selected natural family planning charts. Fertil Steril. 1989;51:430-434.
12. Wu CH, Winkel CA. The effect of therapy initiation day on clomiphene citrate therapy. Fertil Steril. 1989;52:564-568.
13. Chang MY, Chiang CH, Hsieh TT, et al. Use of the antral follicle count to predict the outcome of assisted reproductive technologies. Fertil Steril. 1998;69:505-510.
14. de Vet A, Laven JS, de Jong FH, et al. Antimüllerian hormone serum levels: a putative marker for ovarian aging. Fertil Steril. 2002;77:357-362.
15. De Sutter P. Rational diagnosis and treatment in infertility. Best Pract Res Clin Obstet Gynaecol. 2006;20:647-664.
16. Ehrmann DA. Polycystic ovary syndrome. N Engl J Med. 2005;352:1223-1236.
17. Azziz R, Zacur HA. 21-Hydroxylase deficiency in female hyperandrogenism, screening and diagnosis. J Clin Endocrinol Metab. 1989;69:577-584.
18. Bulun SE. Endometriosis. N Engl J Med. 2009;360:268-279.
19. van der Steeg JW, Steures P, Eijkemans MJ, et al. Should the post-coital test (PCT) be part of the routine fertility work-up? Hum Reprod. 2004;19:1373-1379.
20. Themmen APN, Huhtaniemi IT. Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev. 2000;21:551-583.
21. Rowe PJ. WHO Manual for the Standardization Investigation and Diagnosis of the Infertile Couple. New York, NY: Cambridge University Press; 1993.
22. World Health Organization Department of Reproductive Health and Research World Health Organization Laboratory Manual for the Examination and Processing of Human Semen. 5th ed. Geneva, Switzerland: World Health Organization; 2010.
23. Male Infertility Best Practice Policy Committee of the American Urological Association; Practical Committee of the American Society for Reproductive Medicine. Report on optimal evaluation of the infertile male. Fertil Steril. 2004;82(suppl 1):S123-S130.
24. Honoré GM, Holden AE, Schenken RS. Pathophysiology and management of proximal tubal blockage. Fertil Steril. 1999;71:785-795.
25. Tummon IS, Asher LF, Martin JS, et al. Randomized controlled trial of superovulation and insemination for infertility associated with minimal or mild endometriosis. Fertil Steril. 1997;68:8-12.
26. Heinonen PK, Saarikoski S, Pystynen P. Reproductive performance of women with uterine anomalies. An evaluation of 182 cases. Acta Obstet Gynecol Scand. 1982;61:157-162.
27. Frisch RE. The right weight: body fat, menarche and ovulation. Baillieres Clin Obstet Gynaecol. 1990;4:419-439.
28. Abraham S, Mira M, Llewellyn-Jones D. Should ovulation be induced in women recovering from an eating disorder or who are compulsive exercisers? Fertil Steril. 1990;53:566-568.
29. Crosignan PG. Management of hyperprolactinemia in infertility. J Reprod Med. 1999;44(12 suppl):1116-1120.
30. Baran S, Api M, Godsedef BP, et al. Comparison of metformin and clomiphene citrate therapy for induction in the polycystic ovary syndrome. Arch Gynecol Obstet. 2010;282:439-443.
31. Kerin JF, Liu JH, Phillipou G, et al. Evidence for a hypothalamic site of action of clomiphene citrate in women. J Clin Endocrinol Metab. 1985;61:265-268.
32. Huang KE. The primary treatment of luteal phase inadequacy: progesterone versus clomiphene citrate. Am J Obstet Gynecol. 1986;155:824-828.
33. Homburg R. Clomiphene citrate—end of an era? A mini-review. Hum Reprod. 2005;20:2043-2051.
34. Hirsh A. Male subfertility. BMJ. 2003;327:669-672.
35. Collins JA, Crosignani PG. Unexplained infertility: a review of diagnosis, prognosis, treatment efficacy and management. Int J Gynaecol Obstet. 1992;39:267-275.
36. Fisch P, Casper RF, Brown SE, et al. Unexplained infertility: evaluation of treatment with clomiphene citrate and human chorionic gonadotropin. Fertil Steril. 1989;51:828-833.
37. Verhulst SM, Cohlen BJ, Hughes E, et al. Intra-uterine insemination for unexplained subfertility. Cochrane Database Syst Rev. 2006;(4):CD001838.-
38. Pandian Z, Bhattacharya S, Vale L, et al. In vitro fertilization for unexplained subfertility. Cochrane Database Syst Rev. 2005;(2):CD003357.-
39. Liu C. Restricting access to infertility services: what is a justified limitation on reproductive freedom? Minn J Law Sci Technol. 2009;10:291. Available at: http://mjlst.umn.edu/uploads/QJ/WD/QJWDUZ1D-pV28sHvArqp6A/101_liu.pdf. Accessed May 4, 2011.
40. Practice Committee of the American Society for Reproductive Medicine. Optimal evaluation of the infertile female. Fertil Steril. 2006;86(suppl 1):S264-S267.
41. Practice Committee of the American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil Steril. 2004;82(suppl 1):S33-S39.
Derm emergencies— detecting early signs of trouble
• Consider starting a course of systemic corticosteroids for a patient with erythroderma, fever, and multiorgan involvement when you strongly suspect a drug reaction is the cause—and have ruled out infection. C
• Suspect Stevens– Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) in a patient with widespread and rapidly progressive desquamation, fever, hypotension, and end-organ involvement. C
• In assessing the severity of skin pain, consider the location; involvement of the eyes, perineum, and hands are associated with greater morbidity. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The usual approach to dermatologic conditions—honed pattern recognition, a deliberate differential diagnosis, and empiric treatment with longer follow-up—runs counter to the response that dermatologic red flags require. Because patients with signs and symptoms associated with dermatologic emergencies have the potential for rapid clinical deterioration, urgent action is paramount.
With this in mind, we’ve focused on 4 red flags—erythroderma, desquamation, skin pain, and petechiae/purpura—as a starting point, rather than presenting a list of dermatologic emergencies and discussing each diagnosis in turn. The text, tables, and images on the pages that follow will increase your awareness of dermatologic presentations that require an immediate response and help you differentiate between signs and symptoms of serious skin disorders and benign findings that might be described as red flag mimics (TABLE 1).
TABLE 1
Conditions that mimic dermatologic emergencies
| Red skin | |
| Diagnosis | Key discriminating features |
| Allergic contact dermatitis | Itchy, rather than painful |
| Red man syndrome | History of vancomycin infusion |
| Stasis dermatitis | Stasis dermatitis location (lower extremities), pruritus |
| Sunburn | History, sun-exposed areas |
| Desquamation | |
| Diagnosis | Key discriminating features |
| Bullous impetigo | Localized; no systemic manifestations |
| Postinfectious desquamation | Subungual location common; occurs during convalescent phase of illness |
| Petechiae and purpura | |
| Diagnosis | Key discriminating features |
| Local trauma | History and location |
| Pigmented purpuric dermatosis | History and healthy appearance |
| Viral exanthema | Healthy appearance |
Erythroderma: Red skin that’s life-threatening
From an etymological perspective, “erythroderma” simply means red skin. Clinically, however, it is defined as extensive erythema, typically covering ≥90% of the skin surface (FIGURE 1). True erythroderma can be life-threatening and must always be considered a dermatologic emergency.1,2
Diligent monitoring of the speed of progression and the presence of fever, systemic symptoms, and multiorgan dysfunction is essential. In a case review of 56 children who presented to an emergency department with fever and erythroderma, 45% progressed to shock.3 Some common causes of erythroderma are psoriasis; contact, atopic, and seborrheic dermatitis; pityriasis rubra pilaris; cutaneous T-cell lymphoma; drug reaction; and toxic shock syndrome (TSS).4
Is it a drug reaction? Erythroderma, fever, and evidence of multiorgan involvement in a patient taking any medication—not just a new one—prompts consideration of a drug reaction. Antiepileptics, dapsone, and sulfonamides are the most frequent offenders.5
DRESS syndrome (drug reaction with eosinophilia and systemic symptoms) is characterized by fever, lymphadenopathy, elevated liver enzymes, and leukocytosis with eosinophilia, as well as erythroderma. The rash may be urticarial or morbilliform in appearance; petechiae, purpura, and blisters may be present, as well.
Because fever, leukocytosis, and transaminitis are also suggestive of an infectious etiology, DRESS syndrome is frequently overlooked. As a result, its true incidence is unknown. Estimates range from about one in 1000 to one in 10,000 drug exposures.6
In addition to discontinuing the medication, treatment for DRESS calls for systemic corticosteroids—which may actually be harmful when infection, rather than a drug reaction, is the cause. Thus, it is necessary to maintain a high index of suspicion and to thoroughly review the medication history of
any patient who presents with erythroderma and systemic symptoms.
When to suspect toxic shock syndrome. Consider TSS in any patient with erythroderma and hypotension, as well as laboratory evidence of end-organ involvement (including transaminitis, elevated creatinine, anemia, thrombocytopenia, and elevated creatinine kinase). Diagnostic criteria are detailed in FIGURE 2.7 Group A Streptococcus and Staphylococcus aureus are the classic infectious causes, but other bacteria have been implicated, as well. In most cases, the responsible bacterium is not known initially.
Because the toxins produced by these streptococcal and staphylococcal strains act as superantigens that fuel the immune response and worsen the shock, patients with a presumptive diagnosis of TSS should begin empiric treatment with an antimicrobial agent that inhibits toxin synthesis, such as clindamycin, immediately.8,9
FIGURE 1
Erythema covering the chest and arms
This patient was given a diagnosis of erythrodermic psoriasis.
FIGURE 2
Diagnostic criteria for toxic shock syndrome
ARDS, acute respiratory distress syndrome; BUN, blood urea nitrogen; CNS, central nervous system; CPK, creatinine phosphokinase; DIC, disseminated intravascular coagulation; GAS, group A Streptococcus; GI, gastrointestinal; RMSF, Rocky Mountain spotted fever.
Adapted from: Pickering LK, et al, eds. Red Book: 2009 Report of the Committee on Infectious Diseases. 2009.7
Desquamation/blistering: Act quickly when it’s widespread
Although desquamation can be seen in benign skin conditions, widespread desquamation with or without bullae requires careful evaluation and a rapid response. Separation, either at the dermal-epidermal junction or intraepidermally, raises the specter of 2 emergent conditions: the Stevens–Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) spectrum and staphylococcal scalded skin syndrome (SSSS). Mucosal involvement is another red flag, if the patient appears ill and the desquamation is progressing rapidly. Conjunctival involvement, in particular, is associated with greater morbidity, and a consult with an ophthalmologist is prude
Rapid progression is a hallmark of SJS/TEN. Desquamation that’s widespread and rapidly progressive in a patient with fever, hypotension, and end-organ involvement is suggestive of SJS/TEN (FIGURE 3). Medications, including allopurinol, antimicrobials, and antiepileptics, are frequent culprits.10,11
Nonsteroidal anti-inflammatory drugs (NSAIDs) have also been linked to SJS/TEN.10 Given their widespread use (1-2 per million users per week), however, the likelihood of NSAIDs leading to SJS/TEN is exceedingly low.12
Signs and symptoms of SJS/TEN may include target lesions with dusky centers, erythroderma, or significant pain without any visible skin abnormality, typically accompanied by fever and malaise. Widespread sloughing of the skin may be seen within several hours.11
Admission to an intensive care unit—preferably a burn unit—is suggested for aggressive fluid resuscitation and management of shock and end-organ dysfunction. Intravenous immunoglobulin G (IVIG) and steroids are often used, although there is little consensus as to the most effective treatment.13 Mortality from TEN approaches 50%.13
SSSS can present at any age. Newborns often present with SSSS during their first week of life: Widespread erythema is quickly followed by fragile blisters, which may have already ruptured by the time the infant receives medical attention. Mucosal surfaces are not typically involved. Nikolsky’s sign (separation of the upper epidermis with gentle pressure) is a classic finding.
Infants with SSSS are frequently irritable, suggesting that the skin may be painful. Cultures from unruptured bullae will be negative as the blisters represent a cutaneous reaction to an infection, rather than a skin infection, but blood, urine, and nasopharynx cultures may be positive. Systemic treatment with nafcillin or oxacillin should be initiated, and supportive skin care provided.14,15 Clindamycin or vancomycin should be used in parts of the country in which methicillin-resistant Staphylococcus aureus is prevalent. In very young infants, the outcome of SSSS is generally favorable. Not so with adults.
Because mature kidneys have a greater ability to excrete exfoliative toxins, SSSS primarily affects adults with significant comorbidities—and has a much poorer prognosis.16 You may also see chronic autoimmune bullous diseases, such as bullous pemphigoid and pemphigus vulgaris, with widespread desquamation and blistering, in the adult population. Untreated, the secondary infection and electrolyte disturbances from fluid loss associated with pemphigus vulgaris can be fatal.
Desquamation is a late finding in Kawasaki disease. Desquamation is often cited as a potential skin finding in children with Kawasaki disease (KD) (FIGURE 4), but usually not until the convalescent stage.17 (Desquamation may also appear during the recovery period of several other infections, including scarlet fever and TSS.) IVIG can prevent coronary aneurysm, the major complication of KD, but only if it is administered during the acute phase of the illness. Therefore, early diagnosis of KD (TABLE 2)18—before desquamation occurs—is critical.19
FIGURE 3
Desquamation, full-thickness epidermal necrosis on the upper back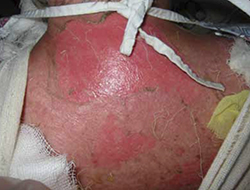
Erythroderma and widespread denudation on the upper back of a patient who was given a diagnosis of toxic epidermal necrolysis.
FIGURE 4
Desquamation on a young patient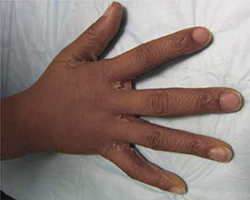
Desquamation associated with Kawasaki disease (shown on the hand of a child) usually occurs during the convalescent stage.
TABLE 2
Diagnostic criteria for Kawasaki disease
| Fever for ≥5 days, and 4 out of 5 criteria (required): |
|---|
|
| Supporting findings: |
|
| CRP, C-reactive protein; ESR, erythrocyte sedimentation rate. Source: Kawasaki Disease Research Committee. Pediatr Int. 2005.18 |
Skin pain is always a red flag
Widespread skin pain should always be taken seriously, as it is rarely associated with minor dermatoses.20 Infectious cellulitis is the most likely diagnosis of a painful erythematous skin lesion. Patients with cellulitis do not usually have erythroderma, as the affected area tends to be very localized.
Cellulitis may be over- or undertreated. Once cellulitis has been diagnosed, the next thing to consider is severity. A recent retrospective study found that misclassification of skin and soft-tissue infections may result in both significant overtreatment of mild soft-tissue infections and dangerous undertreatment of severe infections, with consequent morbidity.21
Location is a key consideration, as cellulitis in certain locations—including the eye, perineum, and hand—carries an increased risk of morbidity. Orbital cellulitis—which may be characterized by proptosis, ophthalmoplegia, and pain with extraocular movements—most often results from initial sinusitis, and can lead to vision loss, intracranial infection, and significant invasive disease. Prompt antimicrobial therapy and urgent ophthalmologic consultation are essential, as operative drainage may be required.22,23
When the perineum is involved, a careful exam must be performed to determine the limits of the affected area. Although Fournier’s gangrene is uncommon, it is a life-threatening infection. In one small retrospective study, more than half of the patients presented with a perianal abscess.24
Similarly, the hand is vulnerable to significant infection, particularly if it is inoculated with bacteria from human mouth flora (the well-described “fight bite”). In a review of 100 cases of human fight bites, 18 patients ultimately required amputation.25
Early necrotizing fasciitis is often missed. Clinicians generally expect painful lesions to also have erythema, swelling, and increased warmth—the cardinal signs of inflammation. As a result, early necrotizing fasciitis, which initially presents with pain out of proportion to other dermatologic findings, may be overlooked. In fact, pain can precede significant skin findings by 24 to 48 hours; prior to that, only mild erythema or swelling (with minimal pain, in some cases) may be evident.26,27
The general pattern, however, is for a site with exquisite tenderness to evolve into a smooth, swollen area, then to develop dusky plaques and late-stage full thickness necrosis with hemorrhagic bullae.26 At that point, necrosis can render the skin insensate. Case reviews have found necrotizing fasciitis to be protean, with only 3 findings—erythema, edema, and tenderness beyond the expected lesion borders—present in most patients.27 Assiduous attention to skin pain in the presence of any other skin manifestation is the key to early diagnosis and rapid treatment.
Petechiae/purpura may be severe or benign
Petechiae are flat, pinpoint, nonblanching spots caused by intradermal hemorrhage associated with a wide variety of conditions, ranging from benign (local trauma) to severe (eg, disseminated intravascular coagulation [DIC] and sepsis). Similarly, purpura—larger lesions that may be palpable—can accompany less severe diseases, such as Henoch-Schönlein purpura (HSP), or life-threatening conditions like sepsis and DIC (FIGURE 5). Here, as in many other dermatologic conditions, the key differentiating features are location (local vs diffuse), speed of progression, and signs and symptoms of systemic illness.
FIGURE 5
Signs of a life-threatening condition
Hemorrhagic bullae with surrounding erythema on the lateral thigh of a patient with purpura fulminans from bacterial sepsis.
Localized petechiae are common with direct trauma, as well as barotrauma associated with coughing, vomiting, or even asphyxiation. Location is an important clue. Periorbital petechiae and petechiae on the chest above the nipple line suggest that the lesions were caused by the force of the barotrauma, rather than systemic disease.28 A careful history and physical exam are needed to rule out serious underlying conditions, such as pneumonia, dehydration, and abdominal obstruction.
Petechiae out of proportion to the force applied may be an indication of an underlying bleeding diathesis, including thrombocytopenia, coagulation defects, and some fulminant infections. Idiopathic thrombocytopenic purpura (ITP) and HSP may present with more widespread petechiae/purpura, but without fever or systemic symptoms. ITP can develop spontaneously, after a viral infection or after a child’s inoculation with the measles-mumps-rubella vaccine.29 ITP typically presents as easy bruising and petechiae out of proportion to the condition that caused it. These patients, as a rule, will have a healthy appearance.
Treatment (with steroids, IVIG, or anti-D immunoglobulin) is generally not required for children with ITP unless they have bleeding that is mucosal or substantial, as spontaneous remission is expected. Adults, who are more likely to develop chronic ITP, may benefit from treatment.30
HSP occurs most commonly in children, who may have palpable purpura, typically in the lower extremities, as well as arthritis or arthralgia, abdominal pain, and renal involvement that can progress from microscopic hematuria or proteinuria to renal insufficiency.31 Typically, children whose disease is in the acute phase do not appear to be sick, with an important exception: Those who develop hemorrhage and edema in the bowel wall, resulting in intussusception, have significant abdominal pain and are more likely to need surgical reduction.32
Diffuse petechiae in the absence of any trauma, accompanied by significant signs of systemic illness, may be an indication of fulminant infection, including meningococcemia, DIC, and Rocky Mountain spotted fever (RMSF). (Fever and diffuse petechiae can also be seen in viral exanthema, but patients usually look well and the rash often has both blanching and petechial components.33)
When a returning traveler presents with a rash and systemic symptoms, it is important to take a thorough history and to consider infections endemic to the area visited. RMSF may initially be localized to the wrists and progress to widespread petechiae over hours to days. Because the cutaneous findings may not be as fulminant—and up to 10% of patients with RMSF have no rash at all34—attention to the noncutaneous features is important. Fever, headache, neurologic symptoms, joint complaints, and abdominal pain (or only a few of these manifestations) in the context of potential tick bite exposure should prompt consideration of RMSF.35
Keep in mind, too, that in cases of fulminant infections such as meningococcemia and DIC, the hallmark purpura fulminans may not be present initially.36 Although the initial cutaneous findings may be subtle, however, such patients will appear quite ill, and their condition will deteriorate rapidly. Because prompt antibiotic therapy can save life and limb, a high index of suspicion should be maintained for any patient who presents with a rash in the setting of fever and hypotension or other evidence of shock.
1. Botella-Estrada R, Sanmartin O, Oliver V, et al. Erythroderma. A clinicopathological study of 56 cases. Arch Dermatol. 1994;130:1503-1507.
2. King LE, Jr, Dufresne RG, Jr, Lovett GL, et al. Erythroderma: review of 82 cases. South Med J. 1986;79:1210-1215.
3. Byer RL, Bachur RG. Clinical deterioration among patients with fever and erythroderma. Pediatrics. 2006;118:2450-2460.
4. Yuan XY, Guo JY, Dang YP, et al. Erythroderma: a clinical-etiological study of 82 cases. Eur J Dermatol. 2010;20:373-377.
5. Walsh SA, Creamer D. Drug reaction with eosinophilia and systemic symptoms (DRESS): a clinical update and review of current thinking. Clin Exp Dermatol. 2010;36:6-11.
6. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
7. Pickering LK, Baker CJ, Kimberlin DW, et al. eds Red Book: 2009 Report of the Committee on Infectious Diseases. 28th ed. Elk Grove Village, Ill: American Academy of Pediatrics; 2009.
8. Silversides JA, Lappin E, Ferguson AJ. Staphylococcal toxic shock syndrome: mechanisms and management. Curr Infect Dis Rep. 2011;12:392-400.
9. Lappin E, Ferguson AJ. Gram-positive toxic shock syndromes. Lancet Infect Dis. 2009;9:281-290.
10. Sanmarkan AD, Sori T, Thappa DM, et al. Retrospective analysis of Stevens-Johnson syndrome and toxic epidermal necrolysis over a period of 10 years. Indian J Dermatol. 2011;56:25-29.
11. Fritsch PO, Sidoroff A. Drug-induced Stevens-Johnson syndrome/toxic epidermal necrolysis. Am J Clin Dermatol. 2000;1:349-360.
12. Ward KE, Archambault R, Mersfelder TL. Severe adverse skin reactions to nonsteroidal antiinflammatory drugs: a review of the literature. Am J Health Syst Pharm. 2010;67:206-213.
13. Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
14. Patel GK, Finlay AY. Staphylococcal scalded skin syndrome: diagnosis and management. Am J Clin Dermatol. 2003;4:165-175.
15. Berk DR, Bayliss SJ. MRSA, staphylococcal scalded skin syndrome, and other cutaneous bacterial emergencies. Pediatr Ann. 2010;39:627-633.
16. Dobson CM, King CM. Adult staphylococcal scalded skin syndrome: histological pitfalls and new diagnostic perspectives. Br J Dermatol. 2003;148:1068-1069.
17. Wang S, Best BM, Burns JC. Periungual desquamation in patients with Kawasaki disease. Pediatr Infect Dis J. 2009;28:538-539.
18. Kawasaki Disease Research Committee. Revision of diagnostic guidelines for Kawasaki disease (the 5th rev ed). Pediatr Int. 2005;47:232-234.
19. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771.
20. Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed. 2009;94:50-54.
21. Koerner R, Johnson AP. Changes in the classification and management of skin and soft tissue infections. J Antimicrob Chemother. 2010;66:232-234.
22. Botting AM, McIntosh D, Mahadevan M. Paediatric pre- and post-septal periorbital infections are different diseases. A retrospective review of 262 cases. Int J Pediatr Otorhinolaryngol. 2008;72:377-383.
23. Liu IT, Kao SC, Wang AG, et al. Preseptal and orbital cellulitis: a 10-year review of hospitalized patients. J Chin Med Assoc. 2006;69:415-422.
24. Koukouras D, Kallidonis P, Panagoloulos C, et al. Fournier’s gangrene, a urologic and surgical emergency: presentation of a multi-institutional experience with 45 cases. Urol Int. 2011;86:167-172.
25. Mennen U, Howells CJ. Human fight-bite injuries of the hand. A study of 100 cases within 18 months. J Hand Surg Br. 1991;16:431-435.
26. Morgan MS. Diagnosis and management of necrotising fasciitis: a multiparametric approach. J Hosp Infect. 2010;75:249-257.
27. Sarani B, Strong M, Pascual J, et al. Necrotizing fasciitis: current concepts and review of the literature. J Am Coll Surg. 2009;208:279-288.
28. Baker RC, Seguin JH, Leslie N, Gilchrist MJ, Myers MG. Fever and petechiae in children. Pediatrics. 1989;84:1051-1055.
29. Mantadakis E, Farmaki E, Buchanan GR. Thrombocytopenic purpura after measles-mumps-rubella vaccination: a systematic review of the literature and guidance for management. J Pediatr. 2010;156:623-628.
30. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190-4207.
31. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
32. Choong CK, Beasley SW. Intra-abdominal manifestations of Henoch-Schönlein purpura. J Paediatr Child Health. 1998;34:405-409.
33. Klinkhammer MD, Colletti JE. Pediatric myth: fever and petechiae. CJEM. 2008;10:479-482.
34. Sexton DJ, Kaye KS. Rocky Mountain spotted fever. Med Clin North Am. 2002;86:351-360, vii-viii.
35. Elston DM. Tick bites and skin rashes. Curr Opin Infect Dis. 2010;23:132-138.
36. Milonovich LM. Meningococcemia: epidemiology, pathophysiology, and management. J Pediatr Health Care. 2007;21:75-80.
CORRESPONDENCE Stephen A. Martin, MD, EdM, Barre Family Health Center, 151 Worcester Road, Barre, MA 01005; [email protected]
| Derm emergency or red flag mimic? You decide Peter A. Lio, MD |
Alisa McQueen, MD
Pediatric Emergency Medicine, The University of Chicago Pritzker School of Medicine
Stephen A. Martin, MD, EdM
Department of Family Medicine and Community Health, University of Massachusetts Medical School, Worcester
[email protected]
Peter A. Lio, MD
Dermatology and Pediatrics, Northwestern University Feinberg School of Medicine, Chicago
The authors reported no potential conflict of interest relevant to this article.
| Derm emergency or red flag mimic? You decide Peter A. Lio, MD |
Alisa McQueen, MD
Pediatric Emergency Medicine, The University of Chicago Pritzker School of Medicine
Stephen A. Martin, MD, EdM
Department of Family Medicine and Community Health, University of Massachusetts Medical School, Worcester
[email protected]
Peter A. Lio, MD
Dermatology and Pediatrics, Northwestern University Feinberg School of Medicine, Chicago
The authors reported no potential conflict of interest relevant to this article.
| Derm emergency or red flag mimic? You decide Peter A. Lio, MD |
Alisa McQueen, MD
Pediatric Emergency Medicine, The University of Chicago Pritzker School of Medicine
Stephen A. Martin, MD, EdM
Department of Family Medicine and Community Health, University of Massachusetts Medical School, Worcester
[email protected]
Peter A. Lio, MD
Dermatology and Pediatrics, Northwestern University Feinberg School of Medicine, Chicago
The authors reported no potential conflict of interest relevant to this article.
• Consider starting a course of systemic corticosteroids for a patient with erythroderma, fever, and multiorgan involvement when you strongly suspect a drug reaction is the cause—and have ruled out infection. C
• Suspect Stevens– Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) in a patient with widespread and rapidly progressive desquamation, fever, hypotension, and end-organ involvement. C
• In assessing the severity of skin pain, consider the location; involvement of the eyes, perineum, and hands are associated with greater morbidity. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The usual approach to dermatologic conditions—honed pattern recognition, a deliberate differential diagnosis, and empiric treatment with longer follow-up—runs counter to the response that dermatologic red flags require. Because patients with signs and symptoms associated with dermatologic emergencies have the potential for rapid clinical deterioration, urgent action is paramount.
With this in mind, we’ve focused on 4 red flags—erythroderma, desquamation, skin pain, and petechiae/purpura—as a starting point, rather than presenting a list of dermatologic emergencies and discussing each diagnosis in turn. The text, tables, and images on the pages that follow will increase your awareness of dermatologic presentations that require an immediate response and help you differentiate between signs and symptoms of serious skin disorders and benign findings that might be described as red flag mimics (TABLE 1).
TABLE 1
Conditions that mimic dermatologic emergencies
| Red skin | |
| Diagnosis | Key discriminating features |
| Allergic contact dermatitis | Itchy, rather than painful |
| Red man syndrome | History of vancomycin infusion |
| Stasis dermatitis | Stasis dermatitis location (lower extremities), pruritus |
| Sunburn | History, sun-exposed areas |
| Desquamation | |
| Diagnosis | Key discriminating features |
| Bullous impetigo | Localized; no systemic manifestations |
| Postinfectious desquamation | Subungual location common; occurs during convalescent phase of illness |
| Petechiae and purpura | |
| Diagnosis | Key discriminating features |
| Local trauma | History and location |
| Pigmented purpuric dermatosis | History and healthy appearance |
| Viral exanthema | Healthy appearance |
Erythroderma: Red skin that’s life-threatening
From an etymological perspective, “erythroderma” simply means red skin. Clinically, however, it is defined as extensive erythema, typically covering ≥90% of the skin surface (FIGURE 1). True erythroderma can be life-threatening and must always be considered a dermatologic emergency.1,2
Diligent monitoring of the speed of progression and the presence of fever, systemic symptoms, and multiorgan dysfunction is essential. In a case review of 56 children who presented to an emergency department with fever and erythroderma, 45% progressed to shock.3 Some common causes of erythroderma are psoriasis; contact, atopic, and seborrheic dermatitis; pityriasis rubra pilaris; cutaneous T-cell lymphoma; drug reaction; and toxic shock syndrome (TSS).4
Is it a drug reaction? Erythroderma, fever, and evidence of multiorgan involvement in a patient taking any medication—not just a new one—prompts consideration of a drug reaction. Antiepileptics, dapsone, and sulfonamides are the most frequent offenders.5
DRESS syndrome (drug reaction with eosinophilia and systemic symptoms) is characterized by fever, lymphadenopathy, elevated liver enzymes, and leukocytosis with eosinophilia, as well as erythroderma. The rash may be urticarial or morbilliform in appearance; petechiae, purpura, and blisters may be present, as well.
Because fever, leukocytosis, and transaminitis are also suggestive of an infectious etiology, DRESS syndrome is frequently overlooked. As a result, its true incidence is unknown. Estimates range from about one in 1000 to one in 10,000 drug exposures.6
In addition to discontinuing the medication, treatment for DRESS calls for systemic corticosteroids—which may actually be harmful when infection, rather than a drug reaction, is the cause. Thus, it is necessary to maintain a high index of suspicion and to thoroughly review the medication history of
any patient who presents with erythroderma and systemic symptoms.
When to suspect toxic shock syndrome. Consider TSS in any patient with erythroderma and hypotension, as well as laboratory evidence of end-organ involvement (including transaminitis, elevated creatinine, anemia, thrombocytopenia, and elevated creatinine kinase). Diagnostic criteria are detailed in FIGURE 2.7 Group A Streptococcus and Staphylococcus aureus are the classic infectious causes, but other bacteria have been implicated, as well. In most cases, the responsible bacterium is not known initially.
Because the toxins produced by these streptococcal and staphylococcal strains act as superantigens that fuel the immune response and worsen the shock, patients with a presumptive diagnosis of TSS should begin empiric treatment with an antimicrobial agent that inhibits toxin synthesis, such as clindamycin, immediately.8,9
FIGURE 1
Erythema covering the chest and arms
This patient was given a diagnosis of erythrodermic psoriasis.
FIGURE 2
Diagnostic criteria for toxic shock syndrome
ARDS, acute respiratory distress syndrome; BUN, blood urea nitrogen; CNS, central nervous system; CPK, creatinine phosphokinase; DIC, disseminated intravascular coagulation; GAS, group A Streptococcus; GI, gastrointestinal; RMSF, Rocky Mountain spotted fever.
Adapted from: Pickering LK, et al, eds. Red Book: 2009 Report of the Committee on Infectious Diseases. 2009.7
Desquamation/blistering: Act quickly when it’s widespread
Although desquamation can be seen in benign skin conditions, widespread desquamation with or without bullae requires careful evaluation and a rapid response. Separation, either at the dermal-epidermal junction or intraepidermally, raises the specter of 2 emergent conditions: the Stevens–Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) spectrum and staphylococcal scalded skin syndrome (SSSS). Mucosal involvement is another red flag, if the patient appears ill and the desquamation is progressing rapidly. Conjunctival involvement, in particular, is associated with greater morbidity, and a consult with an ophthalmologist is prude
Rapid progression is a hallmark of SJS/TEN. Desquamation that’s widespread and rapidly progressive in a patient with fever, hypotension, and end-organ involvement is suggestive of SJS/TEN (FIGURE 3). Medications, including allopurinol, antimicrobials, and antiepileptics, are frequent culprits.10,11
Nonsteroidal anti-inflammatory drugs (NSAIDs) have also been linked to SJS/TEN.10 Given their widespread use (1-2 per million users per week), however, the likelihood of NSAIDs leading to SJS/TEN is exceedingly low.12
Signs and symptoms of SJS/TEN may include target lesions with dusky centers, erythroderma, or significant pain without any visible skin abnormality, typically accompanied by fever and malaise. Widespread sloughing of the skin may be seen within several hours.11
Admission to an intensive care unit—preferably a burn unit—is suggested for aggressive fluid resuscitation and management of shock and end-organ dysfunction. Intravenous immunoglobulin G (IVIG) and steroids are often used, although there is little consensus as to the most effective treatment.13 Mortality from TEN approaches 50%.13
SSSS can present at any age. Newborns often present with SSSS during their first week of life: Widespread erythema is quickly followed by fragile blisters, which may have already ruptured by the time the infant receives medical attention. Mucosal surfaces are not typically involved. Nikolsky’s sign (separation of the upper epidermis with gentle pressure) is a classic finding.
Infants with SSSS are frequently irritable, suggesting that the skin may be painful. Cultures from unruptured bullae will be negative as the blisters represent a cutaneous reaction to an infection, rather than a skin infection, but blood, urine, and nasopharynx cultures may be positive. Systemic treatment with nafcillin or oxacillin should be initiated, and supportive skin care provided.14,15 Clindamycin or vancomycin should be used in parts of the country in which methicillin-resistant Staphylococcus aureus is prevalent. In very young infants, the outcome of SSSS is generally favorable. Not so with adults.
Because mature kidneys have a greater ability to excrete exfoliative toxins, SSSS primarily affects adults with significant comorbidities—and has a much poorer prognosis.16 You may also see chronic autoimmune bullous diseases, such as bullous pemphigoid and pemphigus vulgaris, with widespread desquamation and blistering, in the adult population. Untreated, the secondary infection and electrolyte disturbances from fluid loss associated with pemphigus vulgaris can be fatal.
Desquamation is a late finding in Kawasaki disease. Desquamation is often cited as a potential skin finding in children with Kawasaki disease (KD) (FIGURE 4), but usually not until the convalescent stage.17 (Desquamation may also appear during the recovery period of several other infections, including scarlet fever and TSS.) IVIG can prevent coronary aneurysm, the major complication of KD, but only if it is administered during the acute phase of the illness. Therefore, early diagnosis of KD (TABLE 2)18—before desquamation occurs—is critical.19
FIGURE 3
Desquamation, full-thickness epidermal necrosis on the upper back
Erythroderma and widespread denudation on the upper back of a patient who was given a diagnosis of toxic epidermal necrolysis.
FIGURE 4
Desquamation on a young patient
Desquamation associated with Kawasaki disease (shown on the hand of a child) usually occurs during the convalescent stage.
TABLE 2
Diagnostic criteria for Kawasaki disease
| Fever for ≥5 days, and 4 out of 5 criteria (required): |
|---|
|
| Supporting findings: |
|
| CRP, C-reactive protein; ESR, erythrocyte sedimentation rate. Source: Kawasaki Disease Research Committee. Pediatr Int. 2005.18 |
Skin pain is always a red flag
Widespread skin pain should always be taken seriously, as it is rarely associated with minor dermatoses.20 Infectious cellulitis is the most likely diagnosis of a painful erythematous skin lesion. Patients with cellulitis do not usually have erythroderma, as the affected area tends to be very localized.
Cellulitis may be over- or undertreated. Once cellulitis has been diagnosed, the next thing to consider is severity. A recent retrospective study found that misclassification of skin and soft-tissue infections may result in both significant overtreatment of mild soft-tissue infections and dangerous undertreatment of severe infections, with consequent morbidity.21
Location is a key consideration, as cellulitis in certain locations—including the eye, perineum, and hand—carries an increased risk of morbidity. Orbital cellulitis—which may be characterized by proptosis, ophthalmoplegia, and pain with extraocular movements—most often results from initial sinusitis, and can lead to vision loss, intracranial infection, and significant invasive disease. Prompt antimicrobial therapy and urgent ophthalmologic consultation are essential, as operative drainage may be required.22,23
When the perineum is involved, a careful exam must be performed to determine the limits of the affected area. Although Fournier’s gangrene is uncommon, it is a life-threatening infection. In one small retrospective study, more than half of the patients presented with a perianal abscess.24
Similarly, the hand is vulnerable to significant infection, particularly if it is inoculated with bacteria from human mouth flora (the well-described “fight bite”). In a review of 100 cases of human fight bites, 18 patients ultimately required amputation.25
Early necrotizing fasciitis is often missed. Clinicians generally expect painful lesions to also have erythema, swelling, and increased warmth—the cardinal signs of inflammation. As a result, early necrotizing fasciitis, which initially presents with pain out of proportion to other dermatologic findings, may be overlooked. In fact, pain can precede significant skin findings by 24 to 48 hours; prior to that, only mild erythema or swelling (with minimal pain, in some cases) may be evident.26,27
The general pattern, however, is for a site with exquisite tenderness to evolve into a smooth, swollen area, then to develop dusky plaques and late-stage full thickness necrosis with hemorrhagic bullae.26 At that point, necrosis can render the skin insensate. Case reviews have found necrotizing fasciitis to be protean, with only 3 findings—erythema, edema, and tenderness beyond the expected lesion borders—present in most patients.27 Assiduous attention to skin pain in the presence of any other skin manifestation is the key to early diagnosis and rapid treatment.
Petechiae/purpura may be severe or benign
Petechiae are flat, pinpoint, nonblanching spots caused by intradermal hemorrhage associated with a wide variety of conditions, ranging from benign (local trauma) to severe (eg, disseminated intravascular coagulation [DIC] and sepsis). Similarly, purpura—larger lesions that may be palpable—can accompany less severe diseases, such as Henoch-Schönlein purpura (HSP), or life-threatening conditions like sepsis and DIC (FIGURE 5). Here, as in many other dermatologic conditions, the key differentiating features are location (local vs diffuse), speed of progression, and signs and symptoms of systemic illness.
FIGURE 5
Signs of a life-threatening condition
Hemorrhagic bullae with surrounding erythema on the lateral thigh of a patient with purpura fulminans from bacterial sepsis.
Localized petechiae are common with direct trauma, as well as barotrauma associated with coughing, vomiting, or even asphyxiation. Location is an important clue. Periorbital petechiae and petechiae on the chest above the nipple line suggest that the lesions were caused by the force of the barotrauma, rather than systemic disease.28 A careful history and physical exam are needed to rule out serious underlying conditions, such as pneumonia, dehydration, and abdominal obstruction.
Petechiae out of proportion to the force applied may be an indication of an underlying bleeding diathesis, including thrombocytopenia, coagulation defects, and some fulminant infections. Idiopathic thrombocytopenic purpura (ITP) and HSP may present with more widespread petechiae/purpura, but without fever or systemic symptoms. ITP can develop spontaneously, after a viral infection or after a child’s inoculation with the measles-mumps-rubella vaccine.29 ITP typically presents as easy bruising and petechiae out of proportion to the condition that caused it. These patients, as a rule, will have a healthy appearance.
Treatment (with steroids, IVIG, or anti-D immunoglobulin) is generally not required for children with ITP unless they have bleeding that is mucosal or substantial, as spontaneous remission is expected. Adults, who are more likely to develop chronic ITP, may benefit from treatment.30
HSP occurs most commonly in children, who may have palpable purpura, typically in the lower extremities, as well as arthritis or arthralgia, abdominal pain, and renal involvement that can progress from microscopic hematuria or proteinuria to renal insufficiency.31 Typically, children whose disease is in the acute phase do not appear to be sick, with an important exception: Those who develop hemorrhage and edema in the bowel wall, resulting in intussusception, have significant abdominal pain and are more likely to need surgical reduction.32
Diffuse petechiae in the absence of any trauma, accompanied by significant signs of systemic illness, may be an indication of fulminant infection, including meningococcemia, DIC, and Rocky Mountain spotted fever (RMSF). (Fever and diffuse petechiae can also be seen in viral exanthema, but patients usually look well and the rash often has both blanching and petechial components.33)
When a returning traveler presents with a rash and systemic symptoms, it is important to take a thorough history and to consider infections endemic to the area visited. RMSF may initially be localized to the wrists and progress to widespread petechiae over hours to days. Because the cutaneous findings may not be as fulminant—and up to 10% of patients with RMSF have no rash at all34—attention to the noncutaneous features is important. Fever, headache, neurologic symptoms, joint complaints, and abdominal pain (or only a few of these manifestations) in the context of potential tick bite exposure should prompt consideration of RMSF.35
Keep in mind, too, that in cases of fulminant infections such as meningococcemia and DIC, the hallmark purpura fulminans may not be present initially.36 Although the initial cutaneous findings may be subtle, however, such patients will appear quite ill, and their condition will deteriorate rapidly. Because prompt antibiotic therapy can save life and limb, a high index of suspicion should be maintained for any patient who presents with a rash in the setting of fever and hypotension or other evidence of shock.
• Consider starting a course of systemic corticosteroids for a patient with erythroderma, fever, and multiorgan involvement when you strongly suspect a drug reaction is the cause—and have ruled out infection. C
• Suspect Stevens– Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) in a patient with widespread and rapidly progressive desquamation, fever, hypotension, and end-organ involvement. C
• In assessing the severity of skin pain, consider the location; involvement of the eyes, perineum, and hands are associated with greater morbidity. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The usual approach to dermatologic conditions—honed pattern recognition, a deliberate differential diagnosis, and empiric treatment with longer follow-up—runs counter to the response that dermatologic red flags require. Because patients with signs and symptoms associated with dermatologic emergencies have the potential for rapid clinical deterioration, urgent action is paramount.
With this in mind, we’ve focused on 4 red flags—erythroderma, desquamation, skin pain, and petechiae/purpura—as a starting point, rather than presenting a list of dermatologic emergencies and discussing each diagnosis in turn. The text, tables, and images on the pages that follow will increase your awareness of dermatologic presentations that require an immediate response and help you differentiate between signs and symptoms of serious skin disorders and benign findings that might be described as red flag mimics (TABLE 1).
TABLE 1
Conditions that mimic dermatologic emergencies
| Red skin | |
| Diagnosis | Key discriminating features |
| Allergic contact dermatitis | Itchy, rather than painful |
| Red man syndrome | History of vancomycin infusion |
| Stasis dermatitis | Stasis dermatitis location (lower extremities), pruritus |
| Sunburn | History, sun-exposed areas |
| Desquamation | |
| Diagnosis | Key discriminating features |
| Bullous impetigo | Localized; no systemic manifestations |
| Postinfectious desquamation | Subungual location common; occurs during convalescent phase of illness |
| Petechiae and purpura | |
| Diagnosis | Key discriminating features |
| Local trauma | History and location |
| Pigmented purpuric dermatosis | History and healthy appearance |
| Viral exanthema | Healthy appearance |
Erythroderma: Red skin that’s life-threatening
From an etymological perspective, “erythroderma” simply means red skin. Clinically, however, it is defined as extensive erythema, typically covering ≥90% of the skin surface (FIGURE 1). True erythroderma can be life-threatening and must always be considered a dermatologic emergency.1,2
Diligent monitoring of the speed of progression and the presence of fever, systemic symptoms, and multiorgan dysfunction is essential. In a case review of 56 children who presented to an emergency department with fever and erythroderma, 45% progressed to shock.3 Some common causes of erythroderma are psoriasis; contact, atopic, and seborrheic dermatitis; pityriasis rubra pilaris; cutaneous T-cell lymphoma; drug reaction; and toxic shock syndrome (TSS).4
Is it a drug reaction? Erythroderma, fever, and evidence of multiorgan involvement in a patient taking any medication—not just a new one—prompts consideration of a drug reaction. Antiepileptics, dapsone, and sulfonamides are the most frequent offenders.5
DRESS syndrome (drug reaction with eosinophilia and systemic symptoms) is characterized by fever, lymphadenopathy, elevated liver enzymes, and leukocytosis with eosinophilia, as well as erythroderma. The rash may be urticarial or morbilliform in appearance; petechiae, purpura, and blisters may be present, as well.
Because fever, leukocytosis, and transaminitis are also suggestive of an infectious etiology, DRESS syndrome is frequently overlooked. As a result, its true incidence is unknown. Estimates range from about one in 1000 to one in 10,000 drug exposures.6
In addition to discontinuing the medication, treatment for DRESS calls for systemic corticosteroids—which may actually be harmful when infection, rather than a drug reaction, is the cause. Thus, it is necessary to maintain a high index of suspicion and to thoroughly review the medication history of
any patient who presents with erythroderma and systemic symptoms.
When to suspect toxic shock syndrome. Consider TSS in any patient with erythroderma and hypotension, as well as laboratory evidence of end-organ involvement (including transaminitis, elevated creatinine, anemia, thrombocytopenia, and elevated creatinine kinase). Diagnostic criteria are detailed in FIGURE 2.7 Group A Streptococcus and Staphylococcus aureus are the classic infectious causes, but other bacteria have been implicated, as well. In most cases, the responsible bacterium is not known initially.
Because the toxins produced by these streptococcal and staphylococcal strains act as superantigens that fuel the immune response and worsen the shock, patients with a presumptive diagnosis of TSS should begin empiric treatment with an antimicrobial agent that inhibits toxin synthesis, such as clindamycin, immediately.8,9
FIGURE 1
Erythema covering the chest and arms
This patient was given a diagnosis of erythrodermic psoriasis.
FIGURE 2
Diagnostic criteria for toxic shock syndrome
ARDS, acute respiratory distress syndrome; BUN, blood urea nitrogen; CNS, central nervous system; CPK, creatinine phosphokinase; DIC, disseminated intravascular coagulation; GAS, group A Streptococcus; GI, gastrointestinal; RMSF, Rocky Mountain spotted fever.
Adapted from: Pickering LK, et al, eds. Red Book: 2009 Report of the Committee on Infectious Diseases. 2009.7
Desquamation/blistering: Act quickly when it’s widespread
Although desquamation can be seen in benign skin conditions, widespread desquamation with or without bullae requires careful evaluation and a rapid response. Separation, either at the dermal-epidermal junction or intraepidermally, raises the specter of 2 emergent conditions: the Stevens–Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) spectrum and staphylococcal scalded skin syndrome (SSSS). Mucosal involvement is another red flag, if the patient appears ill and the desquamation is progressing rapidly. Conjunctival involvement, in particular, is associated with greater morbidity, and a consult with an ophthalmologist is prude
Rapid progression is a hallmark of SJS/TEN. Desquamation that’s widespread and rapidly progressive in a patient with fever, hypotension, and end-organ involvement is suggestive of SJS/TEN (FIGURE 3). Medications, including allopurinol, antimicrobials, and antiepileptics, are frequent culprits.10,11
Nonsteroidal anti-inflammatory drugs (NSAIDs) have also been linked to SJS/TEN.10 Given their widespread use (1-2 per million users per week), however, the likelihood of NSAIDs leading to SJS/TEN is exceedingly low.12
Signs and symptoms of SJS/TEN may include target lesions with dusky centers, erythroderma, or significant pain without any visible skin abnormality, typically accompanied by fever and malaise. Widespread sloughing of the skin may be seen within several hours.11
Admission to an intensive care unit—preferably a burn unit—is suggested for aggressive fluid resuscitation and management of shock and end-organ dysfunction. Intravenous immunoglobulin G (IVIG) and steroids are often used, although there is little consensus as to the most effective treatment.13 Mortality from TEN approaches 50%.13
SSSS can present at any age. Newborns often present with SSSS during their first week of life: Widespread erythema is quickly followed by fragile blisters, which may have already ruptured by the time the infant receives medical attention. Mucosal surfaces are not typically involved. Nikolsky’s sign (separation of the upper epidermis with gentle pressure) is a classic finding.
Infants with SSSS are frequently irritable, suggesting that the skin may be painful. Cultures from unruptured bullae will be negative as the blisters represent a cutaneous reaction to an infection, rather than a skin infection, but blood, urine, and nasopharynx cultures may be positive. Systemic treatment with nafcillin or oxacillin should be initiated, and supportive skin care provided.14,15 Clindamycin or vancomycin should be used in parts of the country in which methicillin-resistant Staphylococcus aureus is prevalent. In very young infants, the outcome of SSSS is generally favorable. Not so with adults.
Because mature kidneys have a greater ability to excrete exfoliative toxins, SSSS primarily affects adults with significant comorbidities—and has a much poorer prognosis.16 You may also see chronic autoimmune bullous diseases, such as bullous pemphigoid and pemphigus vulgaris, with widespread desquamation and blistering, in the adult population. Untreated, the secondary infection and electrolyte disturbances from fluid loss associated with pemphigus vulgaris can be fatal.
Desquamation is a late finding in Kawasaki disease. Desquamation is often cited as a potential skin finding in children with Kawasaki disease (KD) (FIGURE 4), but usually not until the convalescent stage.17 (Desquamation may also appear during the recovery period of several other infections, including scarlet fever and TSS.) IVIG can prevent coronary aneurysm, the major complication of KD, but only if it is administered during the acute phase of the illness. Therefore, early diagnosis of KD (TABLE 2)18—before desquamation occurs—is critical.19
FIGURE 3
Desquamation, full-thickness epidermal necrosis on the upper back
Erythroderma and widespread denudation on the upper back of a patient who was given a diagnosis of toxic epidermal necrolysis.
FIGURE 4
Desquamation on a young patient
Desquamation associated with Kawasaki disease (shown on the hand of a child) usually occurs during the convalescent stage.
TABLE 2
Diagnostic criteria for Kawasaki disease
| Fever for ≥5 days, and 4 out of 5 criteria (required): |
|---|
|
| Supporting findings: |
|
| CRP, C-reactive protein; ESR, erythrocyte sedimentation rate. Source: Kawasaki Disease Research Committee. Pediatr Int. 2005.18 |
Skin pain is always a red flag
Widespread skin pain should always be taken seriously, as it is rarely associated with minor dermatoses.20 Infectious cellulitis is the most likely diagnosis of a painful erythematous skin lesion. Patients with cellulitis do not usually have erythroderma, as the affected area tends to be very localized.
Cellulitis may be over- or undertreated. Once cellulitis has been diagnosed, the next thing to consider is severity. A recent retrospective study found that misclassification of skin and soft-tissue infections may result in both significant overtreatment of mild soft-tissue infections and dangerous undertreatment of severe infections, with consequent morbidity.21
Location is a key consideration, as cellulitis in certain locations—including the eye, perineum, and hand—carries an increased risk of morbidity. Orbital cellulitis—which may be characterized by proptosis, ophthalmoplegia, and pain with extraocular movements—most often results from initial sinusitis, and can lead to vision loss, intracranial infection, and significant invasive disease. Prompt antimicrobial therapy and urgent ophthalmologic consultation are essential, as operative drainage may be required.22,23
When the perineum is involved, a careful exam must be performed to determine the limits of the affected area. Although Fournier’s gangrene is uncommon, it is a life-threatening infection. In one small retrospective study, more than half of the patients presented with a perianal abscess.24
Similarly, the hand is vulnerable to significant infection, particularly if it is inoculated with bacteria from human mouth flora (the well-described “fight bite”). In a review of 100 cases of human fight bites, 18 patients ultimately required amputation.25
Early necrotizing fasciitis is often missed. Clinicians generally expect painful lesions to also have erythema, swelling, and increased warmth—the cardinal signs of inflammation. As a result, early necrotizing fasciitis, which initially presents with pain out of proportion to other dermatologic findings, may be overlooked. In fact, pain can precede significant skin findings by 24 to 48 hours; prior to that, only mild erythema or swelling (with minimal pain, in some cases) may be evident.26,27
The general pattern, however, is for a site with exquisite tenderness to evolve into a smooth, swollen area, then to develop dusky plaques and late-stage full thickness necrosis with hemorrhagic bullae.26 At that point, necrosis can render the skin insensate. Case reviews have found necrotizing fasciitis to be protean, with only 3 findings—erythema, edema, and tenderness beyond the expected lesion borders—present in most patients.27 Assiduous attention to skin pain in the presence of any other skin manifestation is the key to early diagnosis and rapid treatment.
Petechiae/purpura may be severe or benign
Petechiae are flat, pinpoint, nonblanching spots caused by intradermal hemorrhage associated with a wide variety of conditions, ranging from benign (local trauma) to severe (eg, disseminated intravascular coagulation [DIC] and sepsis). Similarly, purpura—larger lesions that may be palpable—can accompany less severe diseases, such as Henoch-Schönlein purpura (HSP), or life-threatening conditions like sepsis and DIC (FIGURE 5). Here, as in many other dermatologic conditions, the key differentiating features are location (local vs diffuse), speed of progression, and signs and symptoms of systemic illness.
FIGURE 5
Signs of a life-threatening condition
Hemorrhagic bullae with surrounding erythema on the lateral thigh of a patient with purpura fulminans from bacterial sepsis.
Localized petechiae are common with direct trauma, as well as barotrauma associated with coughing, vomiting, or even asphyxiation. Location is an important clue. Periorbital petechiae and petechiae on the chest above the nipple line suggest that the lesions were caused by the force of the barotrauma, rather than systemic disease.28 A careful history and physical exam are needed to rule out serious underlying conditions, such as pneumonia, dehydration, and abdominal obstruction.
Petechiae out of proportion to the force applied may be an indication of an underlying bleeding diathesis, including thrombocytopenia, coagulation defects, and some fulminant infections. Idiopathic thrombocytopenic purpura (ITP) and HSP may present with more widespread petechiae/purpura, but without fever or systemic symptoms. ITP can develop spontaneously, after a viral infection or after a child’s inoculation with the measles-mumps-rubella vaccine.29 ITP typically presents as easy bruising and petechiae out of proportion to the condition that caused it. These patients, as a rule, will have a healthy appearance.
Treatment (with steroids, IVIG, or anti-D immunoglobulin) is generally not required for children with ITP unless they have bleeding that is mucosal or substantial, as spontaneous remission is expected. Adults, who are more likely to develop chronic ITP, may benefit from treatment.30
HSP occurs most commonly in children, who may have palpable purpura, typically in the lower extremities, as well as arthritis or arthralgia, abdominal pain, and renal involvement that can progress from microscopic hematuria or proteinuria to renal insufficiency.31 Typically, children whose disease is in the acute phase do not appear to be sick, with an important exception: Those who develop hemorrhage and edema in the bowel wall, resulting in intussusception, have significant abdominal pain and are more likely to need surgical reduction.32
Diffuse petechiae in the absence of any trauma, accompanied by significant signs of systemic illness, may be an indication of fulminant infection, including meningococcemia, DIC, and Rocky Mountain spotted fever (RMSF). (Fever and diffuse petechiae can also be seen in viral exanthema, but patients usually look well and the rash often has both blanching and petechial components.33)
When a returning traveler presents with a rash and systemic symptoms, it is important to take a thorough history and to consider infections endemic to the area visited. RMSF may initially be localized to the wrists and progress to widespread petechiae over hours to days. Because the cutaneous findings may not be as fulminant—and up to 10% of patients with RMSF have no rash at all34—attention to the noncutaneous features is important. Fever, headache, neurologic symptoms, joint complaints, and abdominal pain (or only a few of these manifestations) in the context of potential tick bite exposure should prompt consideration of RMSF.35
Keep in mind, too, that in cases of fulminant infections such as meningococcemia and DIC, the hallmark purpura fulminans may not be present initially.36 Although the initial cutaneous findings may be subtle, however, such patients will appear quite ill, and their condition will deteriorate rapidly. Because prompt antibiotic therapy can save life and limb, a high index of suspicion should be maintained for any patient who presents with a rash in the setting of fever and hypotension or other evidence of shock.
1. Botella-Estrada R, Sanmartin O, Oliver V, et al. Erythroderma. A clinicopathological study of 56 cases. Arch Dermatol. 1994;130:1503-1507.
2. King LE, Jr, Dufresne RG, Jr, Lovett GL, et al. Erythroderma: review of 82 cases. South Med J. 1986;79:1210-1215.
3. Byer RL, Bachur RG. Clinical deterioration among patients with fever and erythroderma. Pediatrics. 2006;118:2450-2460.
4. Yuan XY, Guo JY, Dang YP, et al. Erythroderma: a clinical-etiological study of 82 cases. Eur J Dermatol. 2010;20:373-377.
5. Walsh SA, Creamer D. Drug reaction with eosinophilia and systemic symptoms (DRESS): a clinical update and review of current thinking. Clin Exp Dermatol. 2010;36:6-11.
6. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
7. Pickering LK, Baker CJ, Kimberlin DW, et al. eds Red Book: 2009 Report of the Committee on Infectious Diseases. 28th ed. Elk Grove Village, Ill: American Academy of Pediatrics; 2009.
8. Silversides JA, Lappin E, Ferguson AJ. Staphylococcal toxic shock syndrome: mechanisms and management. Curr Infect Dis Rep. 2011;12:392-400.
9. Lappin E, Ferguson AJ. Gram-positive toxic shock syndromes. Lancet Infect Dis. 2009;9:281-290.
10. Sanmarkan AD, Sori T, Thappa DM, et al. Retrospective analysis of Stevens-Johnson syndrome and toxic epidermal necrolysis over a period of 10 years. Indian J Dermatol. 2011;56:25-29.
11. Fritsch PO, Sidoroff A. Drug-induced Stevens-Johnson syndrome/toxic epidermal necrolysis. Am J Clin Dermatol. 2000;1:349-360.
12. Ward KE, Archambault R, Mersfelder TL. Severe adverse skin reactions to nonsteroidal antiinflammatory drugs: a review of the literature. Am J Health Syst Pharm. 2010;67:206-213.
13. Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
14. Patel GK, Finlay AY. Staphylococcal scalded skin syndrome: diagnosis and management. Am J Clin Dermatol. 2003;4:165-175.
15. Berk DR, Bayliss SJ. MRSA, staphylococcal scalded skin syndrome, and other cutaneous bacterial emergencies. Pediatr Ann. 2010;39:627-633.
16. Dobson CM, King CM. Adult staphylococcal scalded skin syndrome: histological pitfalls and new diagnostic perspectives. Br J Dermatol. 2003;148:1068-1069.
17. Wang S, Best BM, Burns JC. Periungual desquamation in patients with Kawasaki disease. Pediatr Infect Dis J. 2009;28:538-539.
18. Kawasaki Disease Research Committee. Revision of diagnostic guidelines for Kawasaki disease (the 5th rev ed). Pediatr Int. 2005;47:232-234.
19. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771.
20. Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed. 2009;94:50-54.
21. Koerner R, Johnson AP. Changes in the classification and management of skin and soft tissue infections. J Antimicrob Chemother. 2010;66:232-234.
22. Botting AM, McIntosh D, Mahadevan M. Paediatric pre- and post-septal periorbital infections are different diseases. A retrospective review of 262 cases. Int J Pediatr Otorhinolaryngol. 2008;72:377-383.
23. Liu IT, Kao SC, Wang AG, et al. Preseptal and orbital cellulitis: a 10-year review of hospitalized patients. J Chin Med Assoc. 2006;69:415-422.
24. Koukouras D, Kallidonis P, Panagoloulos C, et al. Fournier’s gangrene, a urologic and surgical emergency: presentation of a multi-institutional experience with 45 cases. Urol Int. 2011;86:167-172.
25. Mennen U, Howells CJ. Human fight-bite injuries of the hand. A study of 100 cases within 18 months. J Hand Surg Br. 1991;16:431-435.
26. Morgan MS. Diagnosis and management of necrotising fasciitis: a multiparametric approach. J Hosp Infect. 2010;75:249-257.
27. Sarani B, Strong M, Pascual J, et al. Necrotizing fasciitis: current concepts and review of the literature. J Am Coll Surg. 2009;208:279-288.
28. Baker RC, Seguin JH, Leslie N, Gilchrist MJ, Myers MG. Fever and petechiae in children. Pediatrics. 1989;84:1051-1055.
29. Mantadakis E, Farmaki E, Buchanan GR. Thrombocytopenic purpura after measles-mumps-rubella vaccination: a systematic review of the literature and guidance for management. J Pediatr. 2010;156:623-628.
30. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190-4207.
31. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
32. Choong CK, Beasley SW. Intra-abdominal manifestations of Henoch-Schönlein purpura. J Paediatr Child Health. 1998;34:405-409.
33. Klinkhammer MD, Colletti JE. Pediatric myth: fever and petechiae. CJEM. 2008;10:479-482.
34. Sexton DJ, Kaye KS. Rocky Mountain spotted fever. Med Clin North Am. 2002;86:351-360, vii-viii.
35. Elston DM. Tick bites and skin rashes. Curr Opin Infect Dis. 2010;23:132-138.
36. Milonovich LM. Meningococcemia: epidemiology, pathophysiology, and management. J Pediatr Health Care. 2007;21:75-80.
CORRESPONDENCE Stephen A. Martin, MD, EdM, Barre Family Health Center, 151 Worcester Road, Barre, MA 01005; [email protected]
1. Botella-Estrada R, Sanmartin O, Oliver V, et al. Erythroderma. A clinicopathological study of 56 cases. Arch Dermatol. 1994;130:1503-1507.
2. King LE, Jr, Dufresne RG, Jr, Lovett GL, et al. Erythroderma: review of 82 cases. South Med J. 1986;79:1210-1215.
3. Byer RL, Bachur RG. Clinical deterioration among patients with fever and erythroderma. Pediatrics. 2006;118:2450-2460.
4. Yuan XY, Guo JY, Dang YP, et al. Erythroderma: a clinical-etiological study of 82 cases. Eur J Dermatol. 2010;20:373-377.
5. Walsh SA, Creamer D. Drug reaction with eosinophilia and systemic symptoms (DRESS): a clinical update and review of current thinking. Clin Exp Dermatol. 2010;36:6-11.
6. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
7. Pickering LK, Baker CJ, Kimberlin DW, et al. eds Red Book: 2009 Report of the Committee on Infectious Diseases. 28th ed. Elk Grove Village, Ill: American Academy of Pediatrics; 2009.
8. Silversides JA, Lappin E, Ferguson AJ. Staphylococcal toxic shock syndrome: mechanisms and management. Curr Infect Dis Rep. 2011;12:392-400.
9. Lappin E, Ferguson AJ. Gram-positive toxic shock syndromes. Lancet Infect Dis. 2009;9:281-290.
10. Sanmarkan AD, Sori T, Thappa DM, et al. Retrospective analysis of Stevens-Johnson syndrome and toxic epidermal necrolysis over a period of 10 years. Indian J Dermatol. 2011;56:25-29.
11. Fritsch PO, Sidoroff A. Drug-induced Stevens-Johnson syndrome/toxic epidermal necrolysis. Am J Clin Dermatol. 2000;1:349-360.
12. Ward KE, Archambault R, Mersfelder TL. Severe adverse skin reactions to nonsteroidal antiinflammatory drugs: a review of the literature. Am J Health Syst Pharm. 2010;67:206-213.
13. Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
14. Patel GK, Finlay AY. Staphylococcal scalded skin syndrome: diagnosis and management. Am J Clin Dermatol. 2003;4:165-175.
15. Berk DR, Bayliss SJ. MRSA, staphylococcal scalded skin syndrome, and other cutaneous bacterial emergencies. Pediatr Ann. 2010;39:627-633.
16. Dobson CM, King CM. Adult staphylococcal scalded skin syndrome: histological pitfalls and new diagnostic perspectives. Br J Dermatol. 2003;148:1068-1069.
17. Wang S, Best BM, Burns JC. Periungual desquamation in patients with Kawasaki disease. Pediatr Infect Dis J. 2009;28:538-539.
18. Kawasaki Disease Research Committee. Revision of diagnostic guidelines for Kawasaki disease (the 5th rev ed). Pediatr Int. 2005;47:232-234.
19. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747-2771.
20. Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed. 2009;94:50-54.
21. Koerner R, Johnson AP. Changes in the classification and management of skin and soft tissue infections. J Antimicrob Chemother. 2010;66:232-234.
22. Botting AM, McIntosh D, Mahadevan M. Paediatric pre- and post-septal periorbital infections are different diseases. A retrospective review of 262 cases. Int J Pediatr Otorhinolaryngol. 2008;72:377-383.
23. Liu IT, Kao SC, Wang AG, et al. Preseptal and orbital cellulitis: a 10-year review of hospitalized patients. J Chin Med Assoc. 2006;69:415-422.
24. Koukouras D, Kallidonis P, Panagoloulos C, et al. Fournier’s gangrene, a urologic and surgical emergency: presentation of a multi-institutional experience with 45 cases. Urol Int. 2011;86:167-172.
25. Mennen U, Howells CJ. Human fight-bite injuries of the hand. A study of 100 cases within 18 months. J Hand Surg Br. 1991;16:431-435.
26. Morgan MS. Diagnosis and management of necrotising fasciitis: a multiparametric approach. J Hosp Infect. 2010;75:249-257.
27. Sarani B, Strong M, Pascual J, et al. Necrotizing fasciitis: current concepts and review of the literature. J Am Coll Surg. 2009;208:279-288.
28. Baker RC, Seguin JH, Leslie N, Gilchrist MJ, Myers MG. Fever and petechiae in children. Pediatrics. 1989;84:1051-1055.
29. Mantadakis E, Farmaki E, Buchanan GR. Thrombocytopenic purpura after measles-mumps-rubella vaccination: a systematic review of the literature and guidance for management. J Pediatr. 2010;156:623-628.
30. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190-4207.
31. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
32. Choong CK, Beasley SW. Intra-abdominal manifestations of Henoch-Schönlein purpura. J Paediatr Child Health. 1998;34:405-409.
33. Klinkhammer MD, Colletti JE. Pediatric myth: fever and petechiae. CJEM. 2008;10:479-482.
34. Sexton DJ, Kaye KS. Rocky Mountain spotted fever. Med Clin North Am. 2002;86:351-360, vii-viii.
35. Elston DM. Tick bites and skin rashes. Curr Opin Infect Dis. 2010;23:132-138.
36. Milonovich LM. Meningococcemia: epidemiology, pathophysiology, and management. J Pediatr Health Care. 2007;21:75-80.
CORRESPONDENCE Stephen A. Martin, MD, EdM, Barre Family Health Center, 151 Worcester Road, Barre, MA 01005; [email protected]
Genetic blood disorders: Questions you need to ask
• Advise patients at risk for genetic disorders to be tested before starting a family, as carrier status identified at birth is often lost to follow up. C
• Keep genetic blood disorders in mind for patients of all ages; the most common form of porphyria, as well as hereditary hemochromatosis, often remains hidden until well into adulthood. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Some inherited diseases are found as a result of routine neonatal screening. Others remain hidden for years—until a pregnant woman is tested, an older patient receives a particular laboratory test, or signs and symptoms of a previously undetected disorder emerge.
To recognize the signs and respond appropriately, you need to know which patient populations are at heightened risk for which inherited diseases. It is crucial, too, to routinely obtain a comprehensive medical history.
General questions about the health status of family members may be sufficient when there is no reason to suspect a genetic disease. When you have evidence or a reasonable suspicion of a particular disorder, however, a targeted family history (TABLE) is essential. It is crucial, too, to thoroughly document any evidence of an inherited condition found at birth, as well as to advise patients of reproductive age who may be carriers to seek genetic testing or counseling when they begin thinking about starting a family. This review focuses on inherited blood disorders, but these principles apply to other genetic diseases, as well.
TABLE
Family history as a guide to genetic disease
| General (ask all patients) |
|---|
Are your parents alive?
Do you have any siblings?
Are there any diseases that run in your family (eg, breast or colon cancer, heart disease, type 2 diabetes, or genetic disorders)? |
| Targeted family history (tailored to diagnosis or suspected disorder) |
| Has anyone else in your family had this disease? Has anyone in the family:
|
| Family/social history |
| Who is living in the home with you (or your child)? Are there any children who may be affected or may be silent carriers? Would you be willing to see a genetic counselor? |
Sickle cell disease
About one in 500 African Americans are born with sickle cell disease (SCD)—perhaps the best-known inherited blood disorder. Far more (approximately one in 12) are carriers. SCD also affects people of Hispanic origin, although the incidence (about one in 36,000) is much lower than that of blacks. Overall, an estimated 70,000 to 100,000 US residents have SCD.1,2
Inheritance
The genetics of SCD are fairly straightforward: A mutation in the hemoglobin beta chain results in a single amino acid substitution of the normal glutamic acid.3 The mode of inheritance is autosomal recessive. Simply put, patients with SCD have 2 abnormal genes (one from each parent) that cause their red blood cells to change shape. People with sickle cell trait have only one abnormal gene. They cannot develop SCD themselves, but they are carriers of the disease.
Testing
SCD is typically diagnosed as a result of routine neonatal screening (screening newborns for SCD is mandatory in every state).4 The gold standard test is hemoglobin electrophoresis. Although gene sequencing can be used to distinguish SCD from other hemoglobinopathies in equivocal cases, this is rarely necessary.5
Clinical presentation
Infants with SCD are typically asymptomatic for the first few months of life. This is because of the persistence of fetal hemoglobin, which is not predisposed to sickling.6
In many cases, hand/mouth syndrome—a painful swelling of the hands and feet—is the first physical manifestation of SCD and is treated with pain medication and an increase in fluids. The clinical course of SCD, although somewhat variable, is characterized by sickle cell and pain crises.
Sickle cell crisis occurs when a precipitating factor, such as a temperature change, dehydration, or infection, induces the abnormal genes to polymerize and deform the red blood cells, which then hemolyze. Microinfarctions can occur throughout the body, causing intense pain. The hematocrit may fall precipitously. If the bone marrow is unable to replace the hemolyzed red blood cells, an aplastic crisis may result.
A pain crisis is similar, with one key difference: Hematocrit levels do not drop precipitously. It is not always possible to distinguish between a sickle cell crisis and a pain crisis within the first few hours of presentation. However, prompt recognition and rapid treatment of an acute flare-up (with analgesia, oxygen, hydration, and a search for the precipitating cause) can sometimes prevent the drop in hematocrit associated with a sickle cell crisis.7
Treatment and follow-up
A rapid response to flare-ups is a key reason for primary care physicians and patients with SCD (or their parents) to develop a close relationship. Physicians caring for such patients must ensure that they receive adequate pain management, control of sickling events, and, when necessary, access to emergency services without delay. Blood transfusions are needed to treat severe anemia, which may develop as a result of an infection or enlargement of the spleen.
Beyond the crisis. Management of SCD involves more than responding to crises. Magnesium, taken regularly, can stabilize red cell membranes, and hydroxyurea, which increases fetal hemoglobin, may reduce the frequency of attacks.8 Screening for folate and B12 deficiency should be considered prior to supplementation.9 Bone marrow transplantation and gene therapy are being explored, as well.10
Growing awareness of SCD and improvements in disease management have extended the life expectancy of patients with the disease in recent years to the mid-40s, on average.11 Advances in our understanding of SCD, however, have done little to reduce the incidence of the disease.6
Part of the problem is that sickle cell trait—like carrier status of other genetic diseases identified at birth—is often forgotten or overlooked by the time the patient reaches reproductive age. Family physicians can help by educating parents of a newborn with an SCD diagnosis—or with sickle cell trait—about the risks of passing on the disorder. It is equally important to advise teens and young adults to find out whether they or their partners are carriers, stressing the need for both parties to be tested before they marry or start a family.
Thalassemia
Originally described by Thomas Cooley in 1925,12 thalassemia is named for the Greek word for sea (thalassa)—reflecting the fact that for many years, those most affected lived in the vicinity of the Mediterranean Sea.13 Unlike SCD, which is caused by a qualitative problem involving the beta chain of hemoglobin, thalassemia is characterized by quantitative defects in the synthesis of either the alpha or beta hemoglobin chain.
Thus, thalassemia is a spectrum disorder designated in part by the chain of hemoglobin (alpha or beta) that is affected. The HbVar Database of Human Hemoglobin Variants and Thalassemia Mutations (http://globin.bx.psu.edu/hbvar/) lists hundreds of mutations associated with various subtypes, with particular forms of the disease found in certain ethnic groups.
The number of Americans affected by thalassemia is not known. It is estimated, however, that 15% of African Americans (the US population with the highest incidence of any form of thalassemia) are affected by alpha thalassemia.14 The incidence of beta thalassemia among Mediterranean, African, and Middle Eastern populations ranges from about 5% to 25%.15
Inheritance
Most people have 4 genes for the alpha chain of hemoglobin (2 from each parent) and 2 genes for the beta chain of hemoglobin (one maternal and one paternal). The severity of the disorder typically depends on the number of genes affected.
Individuals in which one alpha gene is missing or damaged are silent carriers, while those with 2 affected genes may have a subclinical anemia, often mistaken for iron deficiency anemia. Those in which 3 alpha genes are missing or defective have hemoglobin H disease, a serious condition that causes an enlarged liver and spleen and hemolytic anemia. When all 4 alpha genes are affected, the result is hydrops fetalis, a condition that leads to the death of the fetus or newborn.16
Beta thalassemia—the group of disorders caused by reduced or absent synthesis of the beta chains of hemoglobin—is further classified as minor, intermediate, or major (also known as Cooley’s anemia). People with minor beta thalassemia are typically asymptomatic; those with major thalassemia are severely affected, and often die by the age of 20 years.17
Testing
Routine neonatal screening can detect both alpha and beta thalassemia and identify asymptomatic carriers. Confirmation testing is widely available by hemoglobin electrophoresis. Antenatal testing can be performed via amniocentesis or chorionic villus sampling, and is indicated in countries in which thalassemia is relatively prevalent. In the United States, we typically take a family history and may screen the parents for carrier status.18
Clinical presentation
Mild forms of thalassemia are often mistaken for iron deficiency anemia, and it is likely that the prevalence of this inherited disease is underestimated.19 Patients at the intermediate level often need transfusions under circumstances in which they’re not typically required, such as childbirth. Those with thalassemia major develop splenomegaly and bone malformations. Although patients with thalassemia major have not been expected to live much beyond adulthood, the disorder has been treated successfully with bone marrow transplantation in recent years.20
Treatment and follow-up
Patients who are severely affected by thalassemia need frequent transfusions; ensuring that they receive emergency services, as needed, is key. When caring for such patients—or for individuals who have symptoms suggestive of thalassemia or whose children are found to be carriers—targeted questions about family medical history are necessary, as well. Asking whether anyone in the family has required blood transfusions or had “problems with their blood” may help you detect patterns suggestive of a family history of thalassemia.
As is the case with sickle cell trait, however, the significance (or evidence) of carrier status may be lost by the time a child reaches reproductive age. Thus, the medical records of patients found to be carriers of any genetic disorder should be flagged, and individuals from affected racial/ethnic groups should be routinely advised to review their own and their partner’s genetic status as part of the family planning process. Parents who fail to consider carrier status may have children who inherit SCD, thalassemia, or a combination of these diseases.21,22
Porphyria
Porphyria may encompass an even wider spectrum of disorders than thalassemia. The various clinical entities have little in common other than their pathophysiology—disordered heme synthesis. As heme is synthesized, it transitions through several highly reactive states, and deficiencies in different areas result in different syndromes. When there is a deficiency in one of the enzymes in the heme degradation pathway, reactive metabolites upstream of the defect may occur.
However, all porphyrias have one common feature: the accumulation of porphyrins or their precursors, which sometimes gives the urine a reddish color. A number of porphyrias are extremely rare—only 5 cases of dehydrogenase deficiency porphyria have been reported, for example, and the incidence of congenital erythropoietic porphyria is <1 in a million.23,24
Others occur a bit more frequently in particular ethnic groups. Variegate porphyria, a hepatic form of the disorder associated with acute attacks and photosensitivity, is particularly common among the white South African population, for example.25 Among Eastern Europeans, the incidence of porphyria cutanea tarda (PCT)—the most common porphyria—may be as high as 1 in 5000.25 The “tarda” in the name reflects the fact that, unlike other porphyrias, onset of PCT occurs later in life.26
Inheritance
Inheritance varies by condition. PCT is autosomal dominant, as are most porphyrias; acute intermittent porphyria, however, is autosomal recessive. Some cases of PCT are acquired, occurring as a result of exposure to environmental or infectious agents.27
Testing
A random urine porphobilinogen (PBG) is a useful screening test for porphyria, although checking urine for fluorescence may be the most readily available clinical examination. Further delineation using urine and fecal porphyrins is usually not readily available, and may require sending specimens to a specialized laboratory. “GeneReviews” (http://www.ncbi.nlm.nih.gov/sites/GeneTests/review?db=GeneTests) is a useful resource for locating such labs; the site also provides educational material about genetic diseases as well as information about specimen collection and billing.
Clinical presentation
Characterized by irritable and erratic behavior provoked by sunlight (with varying degrees of hirsutism) and ameliorated by ingesting fresh blood, porphyria could have been the inspiration for the vampire legends.28 Although symptoms vary from one type of porphyria to another, most affect either the nervous system or the skin. Some porphyrias, including PCT, are associated with a blistering, photosensitive rash.29
The acute porphyrias—a grouping of several variants, including acute intermittent porphyria and variegate porphyria—typically cause severe abdominal pain and neurologic symptoms, while erythropoietic porphyria patients may present with anemia, hypo- and hyperpigmentation of the skin, red urine, and reddish coloration of the teeth (FIGURE).29 Porphyrias are an often-overlooked cause of neuropathy, as well.30
FIGURE
Erythropoietic porphyria: Distinguishing characteristics
Patients with erythropoietic porphyria, like the one shown here, often present with reddish coloration of the teeth and red urine.
Treatment and follow-up
The abdominal pain associated with porphyria can be treated with dextrose infusions, analgesics, and hematin. Long-term management includes monitoring for cirrhosis, iron overload, and possibly, hepatocellular carcinoma.27
The treatment for anemia resulting from ineffective erythropoiesis—which ironically, results in iron overload—is phlebotomy, with 400 mL of blood removed every 2 weeks until the iron overload is relieved.31 Erythropoietin may be used to treat anemia resulting from phlebotomy, and is thought to mobilize iron stores.32
For patients with PCT (and any other porphyria associated with photosensitivity), avoidance of sunlight—and any known precipitating factors—is essential. Hydroxychloroquine 200 mg can be given by mouth twice a week.33 Fresh blisters should be kept clean and free from infection.
In addition to symptom management, it is important to learn as much as possible about the family history of patients who have, or whom you suspect of having, any variant of this little-known genetic disease. Start with these 2 questions:
- Do you have family members who are unusually sensitive to the sun?
- Do you have any family members who always seem to be in the hospital but no one knows what’s wrong with them?
Hemochromatosis
Hereditary hemochromatosis (HH), also referred to as iron overload disease, is a little-known genetic disease that primarily affects Caucasians of Northern European descent, although other ethnic groups may also be affected.34,35
Iron is such a precious commodity that no mechanism has evolved for excreting it. During times of systemic infection, there is a tendency for the body to “hide” iron from invading bacteria, resulting in an increased serum and tissue ferritin—and causing chronic anemia if this continues for long periods of time. The balance of iron homeostasis depends on the regulation of iron absorption, which HH interferes with. In patients with HH, the excess iron builds up in the body, particularly in the liver, heart, pancreas, joints, and pituitary gland, and can cause tissue and organ damage.
Inheritance
Although our understanding of the mechanism by which HH occurs may not be complete, it appears that most cases involve a dysregulation of hepcidin, the major regulator of iron transfer. About 10% of white Americans are carriers of the disorder, which is autosomal recessive, and approximately 0.3% to 0.5% have the double mutation and are therefore at high risk for developing HH.34,35 In many cases, the disease does not develop until middle age.
Testing
HH often goes undetected for years. In some cases, routine lab tests that reveal a polycythemia with a high serum iron and a high ferritin and elevated liver enzymes are the first indication of a problem.
While iron studies can raise the suspicion of HH, however, pinning down the genetic mutation can be a little more complicated. The most common allele in Caucasians results from a C282Y mutation thought to be traced to a Celtic or Viking who lived several centuries ago. For this reason, patients with signs of iron overload who are of northern European ancestry are sometimes tested immediately for the gene associated with HH. Patients who present with cirrhosis may have had a liver biopsy before HH was suspected, and may be offered genetic testing, as well.36
Clinical presentation
Iron overload results in iron deposits in many tissues, with varying results: Infiltration of the liver can cause cirrhosis, infiltration of the pancreas can lead to diabetes, and infiltration of the skin results in a bronzed appearance. Diabetes is a primary complication when HH—sometimes referred to as “bronze diabetes”—goes untreated.37
Treatment
With the exception of a small amount of iron that is sloughed off in dead skin cells each day, bleeding is the only way to rid the body of excess iron. Most patients with HH can be treated with phlebotomy (See “A case for phlebotomized blood)”. One unit of whole blood is removed at approximately 2-week intervals until the serum ferritin is <20 ng/mL. This process takes about 13 months.38
Unlike other genetic disorders, HH can be completely controlled—provided it is detected before major organ damage occurs. Thus, it is particularly important that all family members of an individual diagnosed with HH—or found to be a carrier of the disorder—undergo genetic testing. They should also be asked whether they have (or have had) any relatives who frequently donate blood or have been told to give blood frequently.
A referral for genetic counseling may be indicated, not only for families affected by HH, but for those suspected of having (or carrying) other genetic disorders, as well.
The problem is straightforward: The United States has a shortage of donor blood, and phlebotomized blood from patients with hereditary hemochromatosis (HH) is available in large quantities—and is an excellent source of blood for patients with severe anemia. Yet blood banks often discard it.39
Sweden has used phlebotomies as a source of donor blood since 1984, with no ill effects.40 Until 1999, US blood banks were permitted to use blood from patients with HH, provided they indicated on the label that it came from a patient with the disorder. Since then, the US Food and Drug Administration (FDA) has permitted blood banks to apply for a variance permitting them to use this blood without labeling it as such. The standard safety measures apply to the phlebotomized blood, of course—and the change in this provision reflects the fact that blood from patients with HH is not harmful to recipients in any way.
The FDA maintains a list of establishments that have received such a variance (http://www.fda.gov/BiologicsBloodVaccines/BloodBloodProducts/RegulationoftheBloodSupply/Variances/ucm164649.htm) As of November 2011, there were more than 100 blood banks on that list.
CORRESPONDENCE S. Paul Starr, MD, LSU Family Medicine, 200 West Esplanade #412, Kenner, LA 70065; [email protected]
1. Centers for Disease Control and Prevention. Sickle dell disease. Available at: http://www.cdc.gov/ncbddd/sicklecell/data.html. Accessed. Accessed July 5, 2011.
2. Brousseau DC, Panepinto JA, Nimmer M, et al. The number of people with sickle-cell disease in the United States: national and state estimates. Am J Hematol. 2009;85:77-78.
3. Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326-328.
4. National Heart, Lung, and Blood Institute. What is sickle cell anemia? Available at: http://www.nhlbi.nih.gov/health/health-topics/topics/sca/. Accessed December 14, 2011.
5. National Center for Biotechnology Information. Sickle cell disease. Gene Reviews. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1377/. Accessed December 14, 2011.
6. Serjeant GR. One hundred years of sickle cell disease. Br J Haematol. 2010;151:425-429.
7. National Heart, Lung, and Blood Institute. What are the signs and symptoms of sickle cell disease. Available at: http://www.nhlbi.nih.gov/health/health-topics/topics/sca/signs.html. Accessed December 14, 2011.
8. Candrilli SD, O’Brien SH, Ware RE, et al. Hydroxyurea adherence and associated outcomes among Medicaid enrollees with sickle cell disease. Am J Hematol. 2011;86:273-277.
9. Hoffer LJ. Folate supplementation in sickle cell anemia. N Engl J Med. 2003;349:813.-
10. ClinicalTrials.gov. Evaluating the safety and effectiveness of bone marrow transplants in children with sickle cell disease (BMT CTN #0601) (The SCURT study). Available at: http://clinicaltrials.gov/ct2/show/NCT00745420?term=sickle+cell+bone+marrow+transplant&rank=1. Accessed December 15, 2011.
11. McKerrell TD, Cohen HW, Billett HH. The older sickle cell patient. Am J Hematol. 2004;76:101-106.
12. Cooley TB, Lee P. Series of cases of splenomegaly in children with anemia and peculiar bon changes. Trans Am Pediatr Soc. 1925;37:29.-
13. Thalassemia. Stedman’s Medical Dictionary. 24th ed. Baltimore, Md: Williams & Wilkins; 1982:1435.
14. Johnson CS, Tegos C, Beutler E. Alpha thalassemia; prevalence and hematologic findings in American blacks. Arch Intern Med. 1982;142:1280-1282.
15. Al-Awamy BH. Thalassemia syndromes in Saudi Arabia; meta-analysis of local studies. Saudi Med J. 2000;21:8-17.
16. Lie-Injo L. Alpha-chain thalassemia and hydrops fetalis in Malaya: report of five cases. Blood. 1962;20:581-590.
17. Chouliaris G, Berdoukas V, Ladis V, et al. Impact of magnetic resonance imaging on cardiac mortality in thalassemia major. J Magn Reson Imaging. 2011;34:56-59.
18. Mendilcioglu I, Yakut S, Keser I, et al. Prenatal diagnosis of beta-thalassemia and other hemoglobinopathies in southwestern Turkey. Hemoglobin. 2011;35:47-55.
19. Vichinsky EP. Changing patterns of thalassemia worldwide. Ann NY Acad Sci. 2005;1054:18-24.
20. Cao A, Moi P, Galanello R. Recent advances in beta-thalassemias. Pediatr Rep. 2011;3:e17.-
21. Texas Department of State Health Services. Sickle cell + thalassemia. Available at: http://www.dshs.state.tx.us/newborn/thala.shtm. Accessed December 14, 2011.
22. Weatherall DJ. History of genetic disease: thalassaemia: the long road from bedside to genome. Nat Rev Genet. 2004;5:625-631.
23. Jaffe EK, Stith L. ALAD porphyria is a conformational disease. Am J Hum Genet. 2007;80:329-337.
24. Thunell S, Floderus Y, Henrichson A, et al. Porphyria in Sweden. Physiol Res. 2006;55(suppl 2):S109-S118.
25. James WD, Berger T, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Saunders; 2006.
26. Gross U, Hoffman GF, Doss MO. Erythropoietic and hepatic porphyries. J Inherit Metab Dis. 2000;23:641-661.
27. American Porphyria Foundation. About porphyria. Available at: http://www.porphyriafoundation.com/about_porphyria. Accessed December 19, 2011.
28. Boffey PM. Rare disease proposed as cause for “vampires.” New York Times. May 31, 1985. Available at: http://www.nytimes.com/1985/05/31/us/rare-disease-proposed-as-cause-for-vampires.html. Accessed December 15, 2011.
29. Khachemoune A, Blyumin M. Red urine and photosensitive skin rash. J Fam Pract. 2009;58:200-202.
30. Lin CS, Lee NJ, Park SB, et al. Purple pigments: the pathophysiology of acute porphyric neuropathy. Clin Neurophysiol. 2011;122:2336-2344.
31. Seubert S, Seubert A, Stella AM, et al. Results of treatment of porphyria cutanea tarda with bloodletting and chloroquine. Z Hautkr. 1990;65:223-225.
32. Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135:281-292.
33. Cainelli T, Di Padova C, Marchesi L, et al. Hydroxychloroquine versus phlebotomy in the treatment of porphyria cutanea tarda. Br J Dermatol. 1983;108:593-600.
34. McKusick-Nathans Institute of Genetic Medicine. Mendelian Inheritance in Man: A Catalog of Human Genes and Genetic Disorders. Vol .3. Baltimore, Md: Johns Hopkins University; 1998:2305.
35. National Digestive Diseases National Clearinghouse. Hemochromatosis. Available at: http://digestive.niddk.nih.gov/diseases/pubs/hemochromatosis. Accessed December 14, 2011.
36. Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393-408.
37. American Diabetes Association. Hemochromatosis. Available at: http://www.diabetes.org/living-with-diabetes/complications/related-conditions/hemochromatosis.html. Accessed December 15, 2011.
38. Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol. 1986;114:621-629.
39. American Hemochromatosis Society. Approved blood banks. Available at: http://www.americanhs.org/approvedbanks.htm. Accessed December 14, 2011.
40. Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010;116:317-325.
• Advise patients at risk for genetic disorders to be tested before starting a family, as carrier status identified at birth is often lost to follow up. C
• Keep genetic blood disorders in mind for patients of all ages; the most common form of porphyria, as well as hereditary hemochromatosis, often remains hidden until well into adulthood. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Some inherited diseases are found as a result of routine neonatal screening. Others remain hidden for years—until a pregnant woman is tested, an older patient receives a particular laboratory test, or signs and symptoms of a previously undetected disorder emerge.
To recognize the signs and respond appropriately, you need to know which patient populations are at heightened risk for which inherited diseases. It is crucial, too, to routinely obtain a comprehensive medical history.
General questions about the health status of family members may be sufficient when there is no reason to suspect a genetic disease. When you have evidence or a reasonable suspicion of a particular disorder, however, a targeted family history (TABLE) is essential. It is crucial, too, to thoroughly document any evidence of an inherited condition found at birth, as well as to advise patients of reproductive age who may be carriers to seek genetic testing or counseling when they begin thinking about starting a family. This review focuses on inherited blood disorders, but these principles apply to other genetic diseases, as well.
TABLE
Family history as a guide to genetic disease
| General (ask all patients) |
|---|
Are your parents alive?
Do you have any siblings?
Are there any diseases that run in your family (eg, breast or colon cancer, heart disease, type 2 diabetes, or genetic disorders)? |
| Targeted family history (tailored to diagnosis or suspected disorder) |
| Has anyone else in your family had this disease? Has anyone in the family:
|
| Family/social history |
| Who is living in the home with you (or your child)? Are there any children who may be affected or may be silent carriers? Would you be willing to see a genetic counselor? |
Sickle cell disease
About one in 500 African Americans are born with sickle cell disease (SCD)—perhaps the best-known inherited blood disorder. Far more (approximately one in 12) are carriers. SCD also affects people of Hispanic origin, although the incidence (about one in 36,000) is much lower than that of blacks. Overall, an estimated 70,000 to 100,000 US residents have SCD.1,2
Inheritance
The genetics of SCD are fairly straightforward: A mutation in the hemoglobin beta chain results in a single amino acid substitution of the normal glutamic acid.3 The mode of inheritance is autosomal recessive. Simply put, patients with SCD have 2 abnormal genes (one from each parent) that cause their red blood cells to change shape. People with sickle cell trait have only one abnormal gene. They cannot develop SCD themselves, but they are carriers of the disease.
Testing
SCD is typically diagnosed as a result of routine neonatal screening (screening newborns for SCD is mandatory in every state).4 The gold standard test is hemoglobin electrophoresis. Although gene sequencing can be used to distinguish SCD from other hemoglobinopathies in equivocal cases, this is rarely necessary.5
Clinical presentation
Infants with SCD are typically asymptomatic for the first few months of life. This is because of the persistence of fetal hemoglobin, which is not predisposed to sickling.6
In many cases, hand/mouth syndrome—a painful swelling of the hands and feet—is the first physical manifestation of SCD and is treated with pain medication and an increase in fluids. The clinical course of SCD, although somewhat variable, is characterized by sickle cell and pain crises.
Sickle cell crisis occurs when a precipitating factor, such as a temperature change, dehydration, or infection, induces the abnormal genes to polymerize and deform the red blood cells, which then hemolyze. Microinfarctions can occur throughout the body, causing intense pain. The hematocrit may fall precipitously. If the bone marrow is unable to replace the hemolyzed red blood cells, an aplastic crisis may result.
A pain crisis is similar, with one key difference: Hematocrit levels do not drop precipitously. It is not always possible to distinguish between a sickle cell crisis and a pain crisis within the first few hours of presentation. However, prompt recognition and rapid treatment of an acute flare-up (with analgesia, oxygen, hydration, and a search for the precipitating cause) can sometimes prevent the drop in hematocrit associated with a sickle cell crisis.7
Treatment and follow-up
A rapid response to flare-ups is a key reason for primary care physicians and patients with SCD (or their parents) to develop a close relationship. Physicians caring for such patients must ensure that they receive adequate pain management, control of sickling events, and, when necessary, access to emergency services without delay. Blood transfusions are needed to treat severe anemia, which may develop as a result of an infection or enlargement of the spleen.
Beyond the crisis. Management of SCD involves more than responding to crises. Magnesium, taken regularly, can stabilize red cell membranes, and hydroxyurea, which increases fetal hemoglobin, may reduce the frequency of attacks.8 Screening for folate and B12 deficiency should be considered prior to supplementation.9 Bone marrow transplantation and gene therapy are being explored, as well.10
Growing awareness of SCD and improvements in disease management have extended the life expectancy of patients with the disease in recent years to the mid-40s, on average.11 Advances in our understanding of SCD, however, have done little to reduce the incidence of the disease.6
Part of the problem is that sickle cell trait—like carrier status of other genetic diseases identified at birth—is often forgotten or overlooked by the time the patient reaches reproductive age. Family physicians can help by educating parents of a newborn with an SCD diagnosis—or with sickle cell trait—about the risks of passing on the disorder. It is equally important to advise teens and young adults to find out whether they or their partners are carriers, stressing the need for both parties to be tested before they marry or start a family.
Thalassemia
Originally described by Thomas Cooley in 1925,12 thalassemia is named for the Greek word for sea (thalassa)—reflecting the fact that for many years, those most affected lived in the vicinity of the Mediterranean Sea.13 Unlike SCD, which is caused by a qualitative problem involving the beta chain of hemoglobin, thalassemia is characterized by quantitative defects in the synthesis of either the alpha or beta hemoglobin chain.
Thus, thalassemia is a spectrum disorder designated in part by the chain of hemoglobin (alpha or beta) that is affected. The HbVar Database of Human Hemoglobin Variants and Thalassemia Mutations (http://globin.bx.psu.edu/hbvar/) lists hundreds of mutations associated with various subtypes, with particular forms of the disease found in certain ethnic groups.
The number of Americans affected by thalassemia is not known. It is estimated, however, that 15% of African Americans (the US population with the highest incidence of any form of thalassemia) are affected by alpha thalassemia.14 The incidence of beta thalassemia among Mediterranean, African, and Middle Eastern populations ranges from about 5% to 25%.15
Inheritance
Most people have 4 genes for the alpha chain of hemoglobin (2 from each parent) and 2 genes for the beta chain of hemoglobin (one maternal and one paternal). The severity of the disorder typically depends on the number of genes affected.
Individuals in which one alpha gene is missing or damaged are silent carriers, while those with 2 affected genes may have a subclinical anemia, often mistaken for iron deficiency anemia. Those in which 3 alpha genes are missing or defective have hemoglobin H disease, a serious condition that causes an enlarged liver and spleen and hemolytic anemia. When all 4 alpha genes are affected, the result is hydrops fetalis, a condition that leads to the death of the fetus or newborn.16
Beta thalassemia—the group of disorders caused by reduced or absent synthesis of the beta chains of hemoglobin—is further classified as minor, intermediate, or major (also known as Cooley’s anemia). People with minor beta thalassemia are typically asymptomatic; those with major thalassemia are severely affected, and often die by the age of 20 years.17
Testing
Routine neonatal screening can detect both alpha and beta thalassemia and identify asymptomatic carriers. Confirmation testing is widely available by hemoglobin electrophoresis. Antenatal testing can be performed via amniocentesis or chorionic villus sampling, and is indicated in countries in which thalassemia is relatively prevalent. In the United States, we typically take a family history and may screen the parents for carrier status.18
Clinical presentation
Mild forms of thalassemia are often mistaken for iron deficiency anemia, and it is likely that the prevalence of this inherited disease is underestimated.19 Patients at the intermediate level often need transfusions under circumstances in which they’re not typically required, such as childbirth. Those with thalassemia major develop splenomegaly and bone malformations. Although patients with thalassemia major have not been expected to live much beyond adulthood, the disorder has been treated successfully with bone marrow transplantation in recent years.20
Treatment and follow-up
Patients who are severely affected by thalassemia need frequent transfusions; ensuring that they receive emergency services, as needed, is key. When caring for such patients—or for individuals who have symptoms suggestive of thalassemia or whose children are found to be carriers—targeted questions about family medical history are necessary, as well. Asking whether anyone in the family has required blood transfusions or had “problems with their blood” may help you detect patterns suggestive of a family history of thalassemia.
As is the case with sickle cell trait, however, the significance (or evidence) of carrier status may be lost by the time a child reaches reproductive age. Thus, the medical records of patients found to be carriers of any genetic disorder should be flagged, and individuals from affected racial/ethnic groups should be routinely advised to review their own and their partner’s genetic status as part of the family planning process. Parents who fail to consider carrier status may have children who inherit SCD, thalassemia, or a combination of these diseases.21,22
Porphyria
Porphyria may encompass an even wider spectrum of disorders than thalassemia. The various clinical entities have little in common other than their pathophysiology—disordered heme synthesis. As heme is synthesized, it transitions through several highly reactive states, and deficiencies in different areas result in different syndromes. When there is a deficiency in one of the enzymes in the heme degradation pathway, reactive metabolites upstream of the defect may occur.
However, all porphyrias have one common feature: the accumulation of porphyrins or their precursors, which sometimes gives the urine a reddish color. A number of porphyrias are extremely rare—only 5 cases of dehydrogenase deficiency porphyria have been reported, for example, and the incidence of congenital erythropoietic porphyria is <1 in a million.23,24
Others occur a bit more frequently in particular ethnic groups. Variegate porphyria, a hepatic form of the disorder associated with acute attacks and photosensitivity, is particularly common among the white South African population, for example.25 Among Eastern Europeans, the incidence of porphyria cutanea tarda (PCT)—the most common porphyria—may be as high as 1 in 5000.25 The “tarda” in the name reflects the fact that, unlike other porphyrias, onset of PCT occurs later in life.26
Inheritance
Inheritance varies by condition. PCT is autosomal dominant, as are most porphyrias; acute intermittent porphyria, however, is autosomal recessive. Some cases of PCT are acquired, occurring as a result of exposure to environmental or infectious agents.27
Testing
A random urine porphobilinogen (PBG) is a useful screening test for porphyria, although checking urine for fluorescence may be the most readily available clinical examination. Further delineation using urine and fecal porphyrins is usually not readily available, and may require sending specimens to a specialized laboratory. “GeneReviews” (http://www.ncbi.nlm.nih.gov/sites/GeneTests/review?db=GeneTests) is a useful resource for locating such labs; the site also provides educational material about genetic diseases as well as information about specimen collection and billing.
Clinical presentation
Characterized by irritable and erratic behavior provoked by sunlight (with varying degrees of hirsutism) and ameliorated by ingesting fresh blood, porphyria could have been the inspiration for the vampire legends.28 Although symptoms vary from one type of porphyria to another, most affect either the nervous system or the skin. Some porphyrias, including PCT, are associated with a blistering, photosensitive rash.29
The acute porphyrias—a grouping of several variants, including acute intermittent porphyria and variegate porphyria—typically cause severe abdominal pain and neurologic symptoms, while erythropoietic porphyria patients may present with anemia, hypo- and hyperpigmentation of the skin, red urine, and reddish coloration of the teeth (FIGURE).29 Porphyrias are an often-overlooked cause of neuropathy, as well.30
FIGURE
Erythropoietic porphyria: Distinguishing characteristics
Patients with erythropoietic porphyria, like the one shown here, often present with reddish coloration of the teeth and red urine.
Treatment and follow-up
The abdominal pain associated with porphyria can be treated with dextrose infusions, analgesics, and hematin. Long-term management includes monitoring for cirrhosis, iron overload, and possibly, hepatocellular carcinoma.27
The treatment for anemia resulting from ineffective erythropoiesis—which ironically, results in iron overload—is phlebotomy, with 400 mL of blood removed every 2 weeks until the iron overload is relieved.31 Erythropoietin may be used to treat anemia resulting from phlebotomy, and is thought to mobilize iron stores.32
For patients with PCT (and any other porphyria associated with photosensitivity), avoidance of sunlight—and any known precipitating factors—is essential. Hydroxychloroquine 200 mg can be given by mouth twice a week.33 Fresh blisters should be kept clean and free from infection.
In addition to symptom management, it is important to learn as much as possible about the family history of patients who have, or whom you suspect of having, any variant of this little-known genetic disease. Start with these 2 questions:
- Do you have family members who are unusually sensitive to the sun?
- Do you have any family members who always seem to be in the hospital but no one knows what’s wrong with them?
Hemochromatosis
Hereditary hemochromatosis (HH), also referred to as iron overload disease, is a little-known genetic disease that primarily affects Caucasians of Northern European descent, although other ethnic groups may also be affected.34,35
Iron is such a precious commodity that no mechanism has evolved for excreting it. During times of systemic infection, there is a tendency for the body to “hide” iron from invading bacteria, resulting in an increased serum and tissue ferritin—and causing chronic anemia if this continues for long periods of time. The balance of iron homeostasis depends on the regulation of iron absorption, which HH interferes with. In patients with HH, the excess iron builds up in the body, particularly in the liver, heart, pancreas, joints, and pituitary gland, and can cause tissue and organ damage.
Inheritance
Although our understanding of the mechanism by which HH occurs may not be complete, it appears that most cases involve a dysregulation of hepcidin, the major regulator of iron transfer. About 10% of white Americans are carriers of the disorder, which is autosomal recessive, and approximately 0.3% to 0.5% have the double mutation and are therefore at high risk for developing HH.34,35 In many cases, the disease does not develop until middle age.
Testing
HH often goes undetected for years. In some cases, routine lab tests that reveal a polycythemia with a high serum iron and a high ferritin and elevated liver enzymes are the first indication of a problem.
While iron studies can raise the suspicion of HH, however, pinning down the genetic mutation can be a little more complicated. The most common allele in Caucasians results from a C282Y mutation thought to be traced to a Celtic or Viking who lived several centuries ago. For this reason, patients with signs of iron overload who are of northern European ancestry are sometimes tested immediately for the gene associated with HH. Patients who present with cirrhosis may have had a liver biopsy before HH was suspected, and may be offered genetic testing, as well.36
Clinical presentation
Iron overload results in iron deposits in many tissues, with varying results: Infiltration of the liver can cause cirrhosis, infiltration of the pancreas can lead to diabetes, and infiltration of the skin results in a bronzed appearance. Diabetes is a primary complication when HH—sometimes referred to as “bronze diabetes”—goes untreated.37
Treatment
With the exception of a small amount of iron that is sloughed off in dead skin cells each day, bleeding is the only way to rid the body of excess iron. Most patients with HH can be treated with phlebotomy (See “A case for phlebotomized blood)”. One unit of whole blood is removed at approximately 2-week intervals until the serum ferritin is <20 ng/mL. This process takes about 13 months.38
Unlike other genetic disorders, HH can be completely controlled—provided it is detected before major organ damage occurs. Thus, it is particularly important that all family members of an individual diagnosed with HH—or found to be a carrier of the disorder—undergo genetic testing. They should also be asked whether they have (or have had) any relatives who frequently donate blood or have been told to give blood frequently.
A referral for genetic counseling may be indicated, not only for families affected by HH, but for those suspected of having (or carrying) other genetic disorders, as well.
The problem is straightforward: The United States has a shortage of donor blood, and phlebotomized blood from patients with hereditary hemochromatosis (HH) is available in large quantities—and is an excellent source of blood for patients with severe anemia. Yet blood banks often discard it.39
Sweden has used phlebotomies as a source of donor blood since 1984, with no ill effects.40 Until 1999, US blood banks were permitted to use blood from patients with HH, provided they indicated on the label that it came from a patient with the disorder. Since then, the US Food and Drug Administration (FDA) has permitted blood banks to apply for a variance permitting them to use this blood without labeling it as such. The standard safety measures apply to the phlebotomized blood, of course—and the change in this provision reflects the fact that blood from patients with HH is not harmful to recipients in any way.
The FDA maintains a list of establishments that have received such a variance (http://www.fda.gov/BiologicsBloodVaccines/BloodBloodProducts/RegulationoftheBloodSupply/Variances/ucm164649.htm) As of November 2011, there were more than 100 blood banks on that list.
CORRESPONDENCE S. Paul Starr, MD, LSU Family Medicine, 200 West Esplanade #412, Kenner, LA 70065; [email protected]
• Advise patients at risk for genetic disorders to be tested before starting a family, as carrier status identified at birth is often lost to follow up. C
• Keep genetic blood disorders in mind for patients of all ages; the most common form of porphyria, as well as hereditary hemochromatosis, often remains hidden until well into adulthood. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Some inherited diseases are found as a result of routine neonatal screening. Others remain hidden for years—until a pregnant woman is tested, an older patient receives a particular laboratory test, or signs and symptoms of a previously undetected disorder emerge.
To recognize the signs and respond appropriately, you need to know which patient populations are at heightened risk for which inherited diseases. It is crucial, too, to routinely obtain a comprehensive medical history.
General questions about the health status of family members may be sufficient when there is no reason to suspect a genetic disease. When you have evidence or a reasonable suspicion of a particular disorder, however, a targeted family history (TABLE) is essential. It is crucial, too, to thoroughly document any evidence of an inherited condition found at birth, as well as to advise patients of reproductive age who may be carriers to seek genetic testing or counseling when they begin thinking about starting a family. This review focuses on inherited blood disorders, but these principles apply to other genetic diseases, as well.
TABLE
Family history as a guide to genetic disease
| General (ask all patients) |
|---|
Are your parents alive?
Do you have any siblings?
Are there any diseases that run in your family (eg, breast or colon cancer, heart disease, type 2 diabetes, or genetic disorders)? |
| Targeted family history (tailored to diagnosis or suspected disorder) |
| Has anyone else in your family had this disease? Has anyone in the family:
|
| Family/social history |
| Who is living in the home with you (or your child)? Are there any children who may be affected or may be silent carriers? Would you be willing to see a genetic counselor? |
Sickle cell disease
About one in 500 African Americans are born with sickle cell disease (SCD)—perhaps the best-known inherited blood disorder. Far more (approximately one in 12) are carriers. SCD also affects people of Hispanic origin, although the incidence (about one in 36,000) is much lower than that of blacks. Overall, an estimated 70,000 to 100,000 US residents have SCD.1,2
Inheritance
The genetics of SCD are fairly straightforward: A mutation in the hemoglobin beta chain results in a single amino acid substitution of the normal glutamic acid.3 The mode of inheritance is autosomal recessive. Simply put, patients with SCD have 2 abnormal genes (one from each parent) that cause their red blood cells to change shape. People with sickle cell trait have only one abnormal gene. They cannot develop SCD themselves, but they are carriers of the disease.
Testing
SCD is typically diagnosed as a result of routine neonatal screening (screening newborns for SCD is mandatory in every state).4 The gold standard test is hemoglobin electrophoresis. Although gene sequencing can be used to distinguish SCD from other hemoglobinopathies in equivocal cases, this is rarely necessary.5
Clinical presentation
Infants with SCD are typically asymptomatic for the first few months of life. This is because of the persistence of fetal hemoglobin, which is not predisposed to sickling.6
In many cases, hand/mouth syndrome—a painful swelling of the hands and feet—is the first physical manifestation of SCD and is treated with pain medication and an increase in fluids. The clinical course of SCD, although somewhat variable, is characterized by sickle cell and pain crises.
Sickle cell crisis occurs when a precipitating factor, such as a temperature change, dehydration, or infection, induces the abnormal genes to polymerize and deform the red blood cells, which then hemolyze. Microinfarctions can occur throughout the body, causing intense pain. The hematocrit may fall precipitously. If the bone marrow is unable to replace the hemolyzed red blood cells, an aplastic crisis may result.
A pain crisis is similar, with one key difference: Hematocrit levels do not drop precipitously. It is not always possible to distinguish between a sickle cell crisis and a pain crisis within the first few hours of presentation. However, prompt recognition and rapid treatment of an acute flare-up (with analgesia, oxygen, hydration, and a search for the precipitating cause) can sometimes prevent the drop in hematocrit associated with a sickle cell crisis.7
Treatment and follow-up
A rapid response to flare-ups is a key reason for primary care physicians and patients with SCD (or their parents) to develop a close relationship. Physicians caring for such patients must ensure that they receive adequate pain management, control of sickling events, and, when necessary, access to emergency services without delay. Blood transfusions are needed to treat severe anemia, which may develop as a result of an infection or enlargement of the spleen.
Beyond the crisis. Management of SCD involves more than responding to crises. Magnesium, taken regularly, can stabilize red cell membranes, and hydroxyurea, which increases fetal hemoglobin, may reduce the frequency of attacks.8 Screening for folate and B12 deficiency should be considered prior to supplementation.9 Bone marrow transplantation and gene therapy are being explored, as well.10
Growing awareness of SCD and improvements in disease management have extended the life expectancy of patients with the disease in recent years to the mid-40s, on average.11 Advances in our understanding of SCD, however, have done little to reduce the incidence of the disease.6
Part of the problem is that sickle cell trait—like carrier status of other genetic diseases identified at birth—is often forgotten or overlooked by the time the patient reaches reproductive age. Family physicians can help by educating parents of a newborn with an SCD diagnosis—or with sickle cell trait—about the risks of passing on the disorder. It is equally important to advise teens and young adults to find out whether they or their partners are carriers, stressing the need for both parties to be tested before they marry or start a family.
Thalassemia
Originally described by Thomas Cooley in 1925,12 thalassemia is named for the Greek word for sea (thalassa)—reflecting the fact that for many years, those most affected lived in the vicinity of the Mediterranean Sea.13 Unlike SCD, which is caused by a qualitative problem involving the beta chain of hemoglobin, thalassemia is characterized by quantitative defects in the synthesis of either the alpha or beta hemoglobin chain.
Thus, thalassemia is a spectrum disorder designated in part by the chain of hemoglobin (alpha or beta) that is affected. The HbVar Database of Human Hemoglobin Variants and Thalassemia Mutations (http://globin.bx.psu.edu/hbvar/) lists hundreds of mutations associated with various subtypes, with particular forms of the disease found in certain ethnic groups.
The number of Americans affected by thalassemia is not known. It is estimated, however, that 15% of African Americans (the US population with the highest incidence of any form of thalassemia) are affected by alpha thalassemia.14 The incidence of beta thalassemia among Mediterranean, African, and Middle Eastern populations ranges from about 5% to 25%.15
Inheritance
Most people have 4 genes for the alpha chain of hemoglobin (2 from each parent) and 2 genes for the beta chain of hemoglobin (one maternal and one paternal). The severity of the disorder typically depends on the number of genes affected.
Individuals in which one alpha gene is missing or damaged are silent carriers, while those with 2 affected genes may have a subclinical anemia, often mistaken for iron deficiency anemia. Those in which 3 alpha genes are missing or defective have hemoglobin H disease, a serious condition that causes an enlarged liver and spleen and hemolytic anemia. When all 4 alpha genes are affected, the result is hydrops fetalis, a condition that leads to the death of the fetus or newborn.16
Beta thalassemia—the group of disorders caused by reduced or absent synthesis of the beta chains of hemoglobin—is further classified as minor, intermediate, or major (also known as Cooley’s anemia). People with minor beta thalassemia are typically asymptomatic; those with major thalassemia are severely affected, and often die by the age of 20 years.17
Testing
Routine neonatal screening can detect both alpha and beta thalassemia and identify asymptomatic carriers. Confirmation testing is widely available by hemoglobin electrophoresis. Antenatal testing can be performed via amniocentesis or chorionic villus sampling, and is indicated in countries in which thalassemia is relatively prevalent. In the United States, we typically take a family history and may screen the parents for carrier status.18
Clinical presentation
Mild forms of thalassemia are often mistaken for iron deficiency anemia, and it is likely that the prevalence of this inherited disease is underestimated.19 Patients at the intermediate level often need transfusions under circumstances in which they’re not typically required, such as childbirth. Those with thalassemia major develop splenomegaly and bone malformations. Although patients with thalassemia major have not been expected to live much beyond adulthood, the disorder has been treated successfully with bone marrow transplantation in recent years.20
Treatment and follow-up
Patients who are severely affected by thalassemia need frequent transfusions; ensuring that they receive emergency services, as needed, is key. When caring for such patients—or for individuals who have symptoms suggestive of thalassemia or whose children are found to be carriers—targeted questions about family medical history are necessary, as well. Asking whether anyone in the family has required blood transfusions or had “problems with their blood” may help you detect patterns suggestive of a family history of thalassemia.
As is the case with sickle cell trait, however, the significance (or evidence) of carrier status may be lost by the time a child reaches reproductive age. Thus, the medical records of patients found to be carriers of any genetic disorder should be flagged, and individuals from affected racial/ethnic groups should be routinely advised to review their own and their partner’s genetic status as part of the family planning process. Parents who fail to consider carrier status may have children who inherit SCD, thalassemia, or a combination of these diseases.21,22
Porphyria
Porphyria may encompass an even wider spectrum of disorders than thalassemia. The various clinical entities have little in common other than their pathophysiology—disordered heme synthesis. As heme is synthesized, it transitions through several highly reactive states, and deficiencies in different areas result in different syndromes. When there is a deficiency in one of the enzymes in the heme degradation pathway, reactive metabolites upstream of the defect may occur.
However, all porphyrias have one common feature: the accumulation of porphyrins or their precursors, which sometimes gives the urine a reddish color. A number of porphyrias are extremely rare—only 5 cases of dehydrogenase deficiency porphyria have been reported, for example, and the incidence of congenital erythropoietic porphyria is <1 in a million.23,24
Others occur a bit more frequently in particular ethnic groups. Variegate porphyria, a hepatic form of the disorder associated with acute attacks and photosensitivity, is particularly common among the white South African population, for example.25 Among Eastern Europeans, the incidence of porphyria cutanea tarda (PCT)—the most common porphyria—may be as high as 1 in 5000.25 The “tarda” in the name reflects the fact that, unlike other porphyrias, onset of PCT occurs later in life.26
Inheritance
Inheritance varies by condition. PCT is autosomal dominant, as are most porphyrias; acute intermittent porphyria, however, is autosomal recessive. Some cases of PCT are acquired, occurring as a result of exposure to environmental or infectious agents.27
Testing
A random urine porphobilinogen (PBG) is a useful screening test for porphyria, although checking urine for fluorescence may be the most readily available clinical examination. Further delineation using urine and fecal porphyrins is usually not readily available, and may require sending specimens to a specialized laboratory. “GeneReviews” (http://www.ncbi.nlm.nih.gov/sites/GeneTests/review?db=GeneTests) is a useful resource for locating such labs; the site also provides educational material about genetic diseases as well as information about specimen collection and billing.
Clinical presentation
Characterized by irritable and erratic behavior provoked by sunlight (with varying degrees of hirsutism) and ameliorated by ingesting fresh blood, porphyria could have been the inspiration for the vampire legends.28 Although symptoms vary from one type of porphyria to another, most affect either the nervous system or the skin. Some porphyrias, including PCT, are associated with a blistering, photosensitive rash.29
The acute porphyrias—a grouping of several variants, including acute intermittent porphyria and variegate porphyria—typically cause severe abdominal pain and neurologic symptoms, while erythropoietic porphyria patients may present with anemia, hypo- and hyperpigmentation of the skin, red urine, and reddish coloration of the teeth (FIGURE).29 Porphyrias are an often-overlooked cause of neuropathy, as well.30
FIGURE
Erythropoietic porphyria: Distinguishing characteristics
Patients with erythropoietic porphyria, like the one shown here, often present with reddish coloration of the teeth and red urine.
Treatment and follow-up
The abdominal pain associated with porphyria can be treated with dextrose infusions, analgesics, and hematin. Long-term management includes monitoring for cirrhosis, iron overload, and possibly, hepatocellular carcinoma.27
The treatment for anemia resulting from ineffective erythropoiesis—which ironically, results in iron overload—is phlebotomy, with 400 mL of blood removed every 2 weeks until the iron overload is relieved.31 Erythropoietin may be used to treat anemia resulting from phlebotomy, and is thought to mobilize iron stores.32
For patients with PCT (and any other porphyria associated with photosensitivity), avoidance of sunlight—and any known precipitating factors—is essential. Hydroxychloroquine 200 mg can be given by mouth twice a week.33 Fresh blisters should be kept clean and free from infection.
In addition to symptom management, it is important to learn as much as possible about the family history of patients who have, or whom you suspect of having, any variant of this little-known genetic disease. Start with these 2 questions:
- Do you have family members who are unusually sensitive to the sun?
- Do you have any family members who always seem to be in the hospital but no one knows what’s wrong with them?
Hemochromatosis
Hereditary hemochromatosis (HH), also referred to as iron overload disease, is a little-known genetic disease that primarily affects Caucasians of Northern European descent, although other ethnic groups may also be affected.34,35
Iron is such a precious commodity that no mechanism has evolved for excreting it. During times of systemic infection, there is a tendency for the body to “hide” iron from invading bacteria, resulting in an increased serum and tissue ferritin—and causing chronic anemia if this continues for long periods of time. The balance of iron homeostasis depends on the regulation of iron absorption, which HH interferes with. In patients with HH, the excess iron builds up in the body, particularly in the liver, heart, pancreas, joints, and pituitary gland, and can cause tissue and organ damage.
Inheritance
Although our understanding of the mechanism by which HH occurs may not be complete, it appears that most cases involve a dysregulation of hepcidin, the major regulator of iron transfer. About 10% of white Americans are carriers of the disorder, which is autosomal recessive, and approximately 0.3% to 0.5% have the double mutation and are therefore at high risk for developing HH.34,35 In many cases, the disease does not develop until middle age.
Testing
HH often goes undetected for years. In some cases, routine lab tests that reveal a polycythemia with a high serum iron and a high ferritin and elevated liver enzymes are the first indication of a problem.
While iron studies can raise the suspicion of HH, however, pinning down the genetic mutation can be a little more complicated. The most common allele in Caucasians results from a C282Y mutation thought to be traced to a Celtic or Viking who lived several centuries ago. For this reason, patients with signs of iron overload who are of northern European ancestry are sometimes tested immediately for the gene associated with HH. Patients who present with cirrhosis may have had a liver biopsy before HH was suspected, and may be offered genetic testing, as well.36
Clinical presentation
Iron overload results in iron deposits in many tissues, with varying results: Infiltration of the liver can cause cirrhosis, infiltration of the pancreas can lead to diabetes, and infiltration of the skin results in a bronzed appearance. Diabetes is a primary complication when HH—sometimes referred to as “bronze diabetes”—goes untreated.37
Treatment
With the exception of a small amount of iron that is sloughed off in dead skin cells each day, bleeding is the only way to rid the body of excess iron. Most patients with HH can be treated with phlebotomy (See “A case for phlebotomized blood)”. One unit of whole blood is removed at approximately 2-week intervals until the serum ferritin is <20 ng/mL. This process takes about 13 months.38
Unlike other genetic disorders, HH can be completely controlled—provided it is detected before major organ damage occurs. Thus, it is particularly important that all family members of an individual diagnosed with HH—or found to be a carrier of the disorder—undergo genetic testing. They should also be asked whether they have (or have had) any relatives who frequently donate blood or have been told to give blood frequently.
A referral for genetic counseling may be indicated, not only for families affected by HH, but for those suspected of having (or carrying) other genetic disorders, as well.
The problem is straightforward: The United States has a shortage of donor blood, and phlebotomized blood from patients with hereditary hemochromatosis (HH) is available in large quantities—and is an excellent source of blood for patients with severe anemia. Yet blood banks often discard it.39
Sweden has used phlebotomies as a source of donor blood since 1984, with no ill effects.40 Until 1999, US blood banks were permitted to use blood from patients with HH, provided they indicated on the label that it came from a patient with the disorder. Since then, the US Food and Drug Administration (FDA) has permitted blood banks to apply for a variance permitting them to use this blood without labeling it as such. The standard safety measures apply to the phlebotomized blood, of course—and the change in this provision reflects the fact that blood from patients with HH is not harmful to recipients in any way.
The FDA maintains a list of establishments that have received such a variance (http://www.fda.gov/BiologicsBloodVaccines/BloodBloodProducts/RegulationoftheBloodSupply/Variances/ucm164649.htm) As of November 2011, there were more than 100 blood banks on that list.
CORRESPONDENCE S. Paul Starr, MD, LSU Family Medicine, 200 West Esplanade #412, Kenner, LA 70065; [email protected]
1. Centers for Disease Control and Prevention. Sickle dell disease. Available at: http://www.cdc.gov/ncbddd/sicklecell/data.html. Accessed. Accessed July 5, 2011.
2. Brousseau DC, Panepinto JA, Nimmer M, et al. The number of people with sickle-cell disease in the United States: national and state estimates. Am J Hematol. 2009;85:77-78.
3. Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326-328.
4. National Heart, Lung, and Blood Institute. What is sickle cell anemia? Available at: http://www.nhlbi.nih.gov/health/health-topics/topics/sca/. Accessed December 14, 2011.
5. National Center for Biotechnology Information. Sickle cell disease. Gene Reviews. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1377/. Accessed December 14, 2011.
6. Serjeant GR. One hundred years of sickle cell disease. Br J Haematol. 2010;151:425-429.
7. National Heart, Lung, and Blood Institute. What are the signs and symptoms of sickle cell disease. Available at: http://www.nhlbi.nih.gov/health/health-topics/topics/sca/signs.html. Accessed December 14, 2011.
8. Candrilli SD, O’Brien SH, Ware RE, et al. Hydroxyurea adherence and associated outcomes among Medicaid enrollees with sickle cell disease. Am J Hematol. 2011;86:273-277.
9. Hoffer LJ. Folate supplementation in sickle cell anemia. N Engl J Med. 2003;349:813.-
10. ClinicalTrials.gov. Evaluating the safety and effectiveness of bone marrow transplants in children with sickle cell disease (BMT CTN #0601) (The SCURT study). Available at: http://clinicaltrials.gov/ct2/show/NCT00745420?term=sickle+cell+bone+marrow+transplant&rank=1. Accessed December 15, 2011.
11. McKerrell TD, Cohen HW, Billett HH. The older sickle cell patient. Am J Hematol. 2004;76:101-106.
12. Cooley TB, Lee P. Series of cases of splenomegaly in children with anemia and peculiar bon changes. Trans Am Pediatr Soc. 1925;37:29.-
13. Thalassemia. Stedman’s Medical Dictionary. 24th ed. Baltimore, Md: Williams & Wilkins; 1982:1435.
14. Johnson CS, Tegos C, Beutler E. Alpha thalassemia; prevalence and hematologic findings in American blacks. Arch Intern Med. 1982;142:1280-1282.
15. Al-Awamy BH. Thalassemia syndromes in Saudi Arabia; meta-analysis of local studies. Saudi Med J. 2000;21:8-17.
16. Lie-Injo L. Alpha-chain thalassemia and hydrops fetalis in Malaya: report of five cases. Blood. 1962;20:581-590.
17. Chouliaris G, Berdoukas V, Ladis V, et al. Impact of magnetic resonance imaging on cardiac mortality in thalassemia major. J Magn Reson Imaging. 2011;34:56-59.
18. Mendilcioglu I, Yakut S, Keser I, et al. Prenatal diagnosis of beta-thalassemia and other hemoglobinopathies in southwestern Turkey. Hemoglobin. 2011;35:47-55.
19. Vichinsky EP. Changing patterns of thalassemia worldwide. Ann NY Acad Sci. 2005;1054:18-24.
20. Cao A, Moi P, Galanello R. Recent advances in beta-thalassemias. Pediatr Rep. 2011;3:e17.-
21. Texas Department of State Health Services. Sickle cell + thalassemia. Available at: http://www.dshs.state.tx.us/newborn/thala.shtm. Accessed December 14, 2011.
22. Weatherall DJ. History of genetic disease: thalassaemia: the long road from bedside to genome. Nat Rev Genet. 2004;5:625-631.
23. Jaffe EK, Stith L. ALAD porphyria is a conformational disease. Am J Hum Genet. 2007;80:329-337.
24. Thunell S, Floderus Y, Henrichson A, et al. Porphyria in Sweden. Physiol Res. 2006;55(suppl 2):S109-S118.
25. James WD, Berger T, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Saunders; 2006.
26. Gross U, Hoffman GF, Doss MO. Erythropoietic and hepatic porphyries. J Inherit Metab Dis. 2000;23:641-661.
27. American Porphyria Foundation. About porphyria. Available at: http://www.porphyriafoundation.com/about_porphyria. Accessed December 19, 2011.
28. Boffey PM. Rare disease proposed as cause for “vampires.” New York Times. May 31, 1985. Available at: http://www.nytimes.com/1985/05/31/us/rare-disease-proposed-as-cause-for-vampires.html. Accessed December 15, 2011.
29. Khachemoune A, Blyumin M. Red urine and photosensitive skin rash. J Fam Pract. 2009;58:200-202.
30. Lin CS, Lee NJ, Park SB, et al. Purple pigments: the pathophysiology of acute porphyric neuropathy. Clin Neurophysiol. 2011;122:2336-2344.
31. Seubert S, Seubert A, Stella AM, et al. Results of treatment of porphyria cutanea tarda with bloodletting and chloroquine. Z Hautkr. 1990;65:223-225.
32. Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135:281-292.
33. Cainelli T, Di Padova C, Marchesi L, et al. Hydroxychloroquine versus phlebotomy in the treatment of porphyria cutanea tarda. Br J Dermatol. 1983;108:593-600.
34. McKusick-Nathans Institute of Genetic Medicine. Mendelian Inheritance in Man: A Catalog of Human Genes and Genetic Disorders. Vol .3. Baltimore, Md: Johns Hopkins University; 1998:2305.
35. National Digestive Diseases National Clearinghouse. Hemochromatosis. Available at: http://digestive.niddk.nih.gov/diseases/pubs/hemochromatosis. Accessed December 14, 2011.
36. Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393-408.
37. American Diabetes Association. Hemochromatosis. Available at: http://www.diabetes.org/living-with-diabetes/complications/related-conditions/hemochromatosis.html. Accessed December 15, 2011.
38. Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol. 1986;114:621-629.
39. American Hemochromatosis Society. Approved blood banks. Available at: http://www.americanhs.org/approvedbanks.htm. Accessed December 14, 2011.
40. Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010;116:317-325.
1. Centers for Disease Control and Prevention. Sickle dell disease. Available at: http://www.cdc.gov/ncbddd/sicklecell/data.html. Accessed. Accessed July 5, 2011.
2. Brousseau DC, Panepinto JA, Nimmer M, et al. The number of people with sickle-cell disease in the United States: national and state estimates. Am J Hematol. 2009;85:77-78.
3. Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326-328.
4. National Heart, Lung, and Blood Institute. What is sickle cell anemia? Available at: http://www.nhlbi.nih.gov/health/health-topics/topics/sca/. Accessed December 14, 2011.
5. National Center for Biotechnology Information. Sickle cell disease. Gene Reviews. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1377/. Accessed December 14, 2011.
6. Serjeant GR. One hundred years of sickle cell disease. Br J Haematol. 2010;151:425-429.
7. National Heart, Lung, and Blood Institute. What are the signs and symptoms of sickle cell disease. Available at: http://www.nhlbi.nih.gov/health/health-topics/topics/sca/signs.html. Accessed December 14, 2011.
8. Candrilli SD, O’Brien SH, Ware RE, et al. Hydroxyurea adherence and associated outcomes among Medicaid enrollees with sickle cell disease. Am J Hematol. 2011;86:273-277.
9. Hoffer LJ. Folate supplementation in sickle cell anemia. N Engl J Med. 2003;349:813.-
10. ClinicalTrials.gov. Evaluating the safety and effectiveness of bone marrow transplants in children with sickle cell disease (BMT CTN #0601) (The SCURT study). Available at: http://clinicaltrials.gov/ct2/show/NCT00745420?term=sickle+cell+bone+marrow+transplant&rank=1. Accessed December 15, 2011.
11. McKerrell TD, Cohen HW, Billett HH. The older sickle cell patient. Am J Hematol. 2004;76:101-106.
12. Cooley TB, Lee P. Series of cases of splenomegaly in children with anemia and peculiar bon changes. Trans Am Pediatr Soc. 1925;37:29.-
13. Thalassemia. Stedman’s Medical Dictionary. 24th ed. Baltimore, Md: Williams & Wilkins; 1982:1435.
14. Johnson CS, Tegos C, Beutler E. Alpha thalassemia; prevalence and hematologic findings in American blacks. Arch Intern Med. 1982;142:1280-1282.
15. Al-Awamy BH. Thalassemia syndromes in Saudi Arabia; meta-analysis of local studies. Saudi Med J. 2000;21:8-17.
16. Lie-Injo L. Alpha-chain thalassemia and hydrops fetalis in Malaya: report of five cases. Blood. 1962;20:581-590.
17. Chouliaris G, Berdoukas V, Ladis V, et al. Impact of magnetic resonance imaging on cardiac mortality in thalassemia major. J Magn Reson Imaging. 2011;34:56-59.
18. Mendilcioglu I, Yakut S, Keser I, et al. Prenatal diagnosis of beta-thalassemia and other hemoglobinopathies in southwestern Turkey. Hemoglobin. 2011;35:47-55.
19. Vichinsky EP. Changing patterns of thalassemia worldwide. Ann NY Acad Sci. 2005;1054:18-24.
20. Cao A, Moi P, Galanello R. Recent advances in beta-thalassemias. Pediatr Rep. 2011;3:e17.-
21. Texas Department of State Health Services. Sickle cell + thalassemia. Available at: http://www.dshs.state.tx.us/newborn/thala.shtm. Accessed December 14, 2011.
22. Weatherall DJ. History of genetic disease: thalassaemia: the long road from bedside to genome. Nat Rev Genet. 2004;5:625-631.
23. Jaffe EK, Stith L. ALAD porphyria is a conformational disease. Am J Hum Genet. 2007;80:329-337.
24. Thunell S, Floderus Y, Henrichson A, et al. Porphyria in Sweden. Physiol Res. 2006;55(suppl 2):S109-S118.
25. James WD, Berger T, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Saunders; 2006.
26. Gross U, Hoffman GF, Doss MO. Erythropoietic and hepatic porphyries. J Inherit Metab Dis. 2000;23:641-661.
27. American Porphyria Foundation. About porphyria. Available at: http://www.porphyriafoundation.com/about_porphyria. Accessed December 19, 2011.
28. Boffey PM. Rare disease proposed as cause for “vampires.” New York Times. May 31, 1985. Available at: http://www.nytimes.com/1985/05/31/us/rare-disease-proposed-as-cause-for-vampires.html. Accessed December 15, 2011.
29. Khachemoune A, Blyumin M. Red urine and photosensitive skin rash. J Fam Pract. 2009;58:200-202.
30. Lin CS, Lee NJ, Park SB, et al. Purple pigments: the pathophysiology of acute porphyric neuropathy. Clin Neurophysiol. 2011;122:2336-2344.
31. Seubert S, Seubert A, Stella AM, et al. Results of treatment of porphyria cutanea tarda with bloodletting and chloroquine. Z Hautkr. 1990;65:223-225.
32. Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135:281-292.
33. Cainelli T, Di Padova C, Marchesi L, et al. Hydroxychloroquine versus phlebotomy in the treatment of porphyria cutanea tarda. Br J Dermatol. 1983;108:593-600.
34. McKusick-Nathans Institute of Genetic Medicine. Mendelian Inheritance in Man: A Catalog of Human Genes and Genetic Disorders. Vol .3. Baltimore, Md: Johns Hopkins University; 1998:2305.
35. National Digestive Diseases National Clearinghouse. Hemochromatosis. Available at: http://digestive.niddk.nih.gov/diseases/pubs/hemochromatosis. Accessed December 14, 2011.
36. Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393-408.
37. American Diabetes Association. Hemochromatosis. Available at: http://www.diabetes.org/living-with-diabetes/complications/related-conditions/hemochromatosis.html. Accessed December 15, 2011.
38. Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol. 1986;114:621-629.
39. American Hemochromatosis Society. Approved blood banks. Available at: http://www.americanhs.org/approvedbanks.htm. Accessed December 14, 2011.
40. Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010;116:317-325.
Exercise-induced proteinuria?
• Rely on a spot urine microalbumin-to-creatinine or protein-to-creatinine ratio to accurately assess proteinuria. B
• Repeat testing if routine urinalysis detects proteinuria—especially if the patient reports having exercised in the previous 24 hours. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE As part of a routine physical examination, urinalysis reveals that a patient new to your practice is excreting an excessive level of protein. The patient is physically fit and shared during the history taking that he is an avid runner. The physical examination and other laboratory values were unremarkable. How concerned should you be about the finding of proteinuria?
Exercise-induced proteinuria is generally benign and a function of the intensity—rather than the duration—of exercise.1 It occurs most often among athletes participating in such sports as running, swimming, rowing, football, or boxing.2 It’s also transient—lasting 24 to 48 hours.1 Recognizing exercise-induced proteinuria is fairly straightforward—once you know what to look for.
But first, a word about the processes at work.
Diverse processes that work alone—or together
The normal range of protein excretion in healthy individuals is 150 to 200 mg of protein per day, of which albumin constitutes 10 to 20 mg.3 Individuals with proteinuria persistently higher than this level need further evaluation.
Diverse processes leading to proteinuria—working alone or concomitantly—occur at the level of the nephron.3
Glomerular proteinuria results from increased filtration of macromolecules such as albumin across the glomerular capillary barrier. This type of proteinuria can occur with different glomerulopathies, upright posture, or exercise.4
Researchers have not identified the mechanisms leading to postexercise proteinuria, but there are several theories. (For more on this, see “Why does exercise increase protein excretion?”.)
Tubular proteinuria is due to a deranged tubular apparatus with an intact glomerulus. This results in the escape of β2-microglobulin and immunoglobulin light chains from proximal tubular reabsorption. It is often missed on dipstick testing, which detects only albumin. This type of proteinuria is usually seen in tubulointerstitial diseases or in patients with idiopathic nephrotic syndrome.5
Overflow proteinuria occurs when small molecular light chains escape the glomerular filtration barrier and overwhelm the tubular reabsorptive capacity. This type of proteinuria can be seen in multiple myeloma, and is detectable by protein-to-creatinine ratio or urine protein electrophoresis.
The root cause of exercise-induced proteinuria is unclear, but the renin-angiotensin system (RAS) and prostaglandins are thought to play a major role. The plasma concentration of angiotensin II increases during exercise, leading to filtration of protein through the glomerular membrane.30 And angiotensin-converting enzyme (ACE) inhibitors have been shown to significantly diminish exercise-induced proteinuria, thus supporting this theory.31,32
Also, strenuous exercise increases sympathetic nervous system activity as well as blood levels of catecholamines, thereby increasing permeability of the glomerular capillary membrane.33 Furthermore, lactate increases with strenuous exercise and causes conformational changes in serum proteins that, when coupled with glomerular barrier changes, can lead to increased permeability and protein excretion.
The surest means of detecting proteinuria
Albumin excretion >300 mg/d is called macroalbuminuria, overt proteinuria, or dipstick-positive proteinuria. Albumin persistently excreted in the urine between 30 and 300 mg/d is referred to as microalbuminuria.
Because microalbuminuria is not detectable by a standard urine dipstick test, some providers routinely screen for protein with the microalbumin-to-creatinine ratio. A first-voided morning urine specimen is recommended, but random urine samples are an acceptable alternative.6 The microalbumin-to-creatinine ratio is recommended as a screen for early diabetic nephropathy and other kidney diseases. And a positive test result may also suggest increased risk of cardiovascular disease.6 Microalbuminuria is defined as persistent albumin excretion between 30 and 300 mg/d.7
When exercise is a factor, here’s what to look for
As noted earlier, exercise-induced proteinuria is a function of the intensity of the exercise. Moderate and strenuous (vigorous) exercise are the 2 types of exercise that come into play when discussing proteinuria. Differentiating them is not precise, but is often defined by maximal oxygen consumption (vigorous=60% of VO2max; moderate <60% VO2max); metabolic equivalents (vigorous=6 METS; moderate <6 METS); walking/running speeds (various); and heart rate reserve (vigorous=60% HRR; moderate <60% HRR).8
Moderate exercise produces glomerular proteinuria, with an increase in macromolecular (albumin) filtration across the glomerular barrier. Strenuous exercise increases glomerular filtration of low-molecular-weight proteins (β2-microglobulin), which overwhelm the reabsorbing capacity of the tubular apparatus, causing temporary dysfunction and tubular proteinuria.9 Thus, the pathophysiology is mixed, with a major contribution from glomerular proteinuria.10
Strenuous exercise can cause protein excretion to exceed 1.5 mg/min.11 However, it seldom rises beyond 1 to 2 g/d,4 and this increase usually reverts to normal physiologic levels within 24 to 48 hours after exercise.12
Exercise-induced proteinuria is biphasic.13 Increased protein excretion occurs 30 minutes after exercise and is related to changes in intraglomerular hemodynamics and the resulting saturation of the renal tubules. Around 24 hours after exercise, oxidative stress on the glomeruli causes another slight elevation in albumin excretion without changes in β2-microglobulin, thereby indicating glomerular proteinuria exclusively.
Even the pros aren’t exempt. Exercise-induced proteinuria does not decrease with regular physical training. This was demonstrated in a study of 10 well-trained professional cyclists for whom strenuous exercise increased overnight protein excretion of both tubular and glomerular origin despite ongoing regular physical training.14
Creatine supplements do not increase proteinuria. A study of creatine supplementation in animal models noted no changes in 24-hour proteinuria or albumin excretion in both normal and two-thirds-nephrectomized animals.15 Another study compared creatine use with nonuse in athletes who had been training regularly and strenuously (12- 18 h/wk) for 5 to 10 years. They were evaluated for 10 months to 5 years. The groups exhibited equivalent urine excretion rates for albumin and creatinine, with no deleterious effect on kidney function.16
What happens when chronic disease is factored into the exercise equation?
Patients with a 2- to 20-year history of insulin-dependent diabetes without chronic kidney disease (CKD) who exhibited normal albumin excretion at baseline were more likely to develop proteinuria after exercise than healthy controls.17,18 The postulated cause was undetected glomerular changes due to diabetes. An exercise-provocation test may one day be useful in predicting future development of nephropathy, but further studies are needed.19-21
Exercise increases proteinuria immediately in individuals with metabolic disorders like obesity, through a mechanism different from diabetes mellitus. Proteinuria in the obese population is thought to be glomerular in origin, as opposed to both tubular and glomerular proteinuria in diabetic nephropathy.22,23
In CKD, low-intensity exercise long term does not promote proteinuria or lead to rapid progression of CKD. In one study, obese patients (body mass index >30 kg/m2) with diabetes and CKD stage II to IV who exercised 3 times weekly (aerobic training for 6 weeks, followed by 18 weeks of supervised home exercise) increased their stamina and exhibited slight, statistically insignificant decreases in resting systolic blood pressure and 24-hour proteinuria.24 A 12-week low-intensity aquatic exercise program for 26 patients with mild to moderate CKD decreased blood pressure and proteinuria and slightly improved glomerular filtration rate (GFR).25 These results for proteinuria and GFR were shown previously in rats with subtotal nephrectomy.26
Elevated urinary albumin excretion with exercise is significantly higher in patients with acromegaly when compared with normal healthy subjects. The underlying pathology is thought to occur at the glomerular filtration barrier with intact tubular function. Somatostatin analog treatment for acromegaly leads to reductions in postexercise albuminuria.27,28
How to manage suspected exercise-induced proteinuria
When interpreting the meaning of proteinuria detected on routine urinalysis, keep in mind the temporal relevance between exercise and urine collection. If urine is found to have been collected within 24 hours of intense exercise, repeat testing in the absence of prior exercise on at least one other occasion to differentiate between transient and persistent proteinuria. In confirming transient proteinuria after exercise, reassure the patient that it is a benign condition. This holds true as well for routine microalbumin-to-creatinine urine testing in patients with diabetes who exercise. If the result of a repeat test is high, turn your attention to another possible cause of proteinuria, such as diabetic nephropathy.
Screening for proteinuria during sports preparticipation examinations is not recommended because the diagnostic utility is low.29 Researchers performed urine dipstick testing for protein, blood, and glucose in preparticipation assessments of 701 students.29 They detected proteinuria in 40 students and glucosuria in one. Follow-up testing with first-voided morning urine specimens and glucose tolerance testing was normal in all students.
CORRESPONDENCE Fahad Saeed, MD, 313 Brook Hollow, Hanover, NH 03755; [email protected]
1. Poortmans JR. Exercise and renal function. Sports Med. 1984;1:125-153.
2. Gebke KB. Genitourinary system. In: McKeag DB, Moeller JL, eds. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2007;234.-
3. Venkat KK. Proteinuria and microalbuminuria in adults: significance, evaluation, and treatment. South Med J. 2004;97:969-979.
4. Rose BD. Pathophysiology of Renal Disease. 2nd ed. New York, NY: McGraw-Hill; 1987;11-16.
5. Sesso R, Santos AP, Nishida SK, et al. Prediction of steroid responsiveness in the idiopathic nephrotic syndrome using urinary retinol-binding protein and beta-2-microglobulin. Ann Intern Med. 1992;116:905-909.
6. Levey AS, Coresh J, Balk E, et al. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139:137-147.
7. Family Practice Notebook Urine protein to creatinine ratio. Available at: http://www.fpnotebook.com/urology/lab/urnprtntcrtnrt.htm. Accessed August 9, 2011.
8. Swain DP, Franklin BA. Comparison of cardioprotective benefits of vigorous versus moderate intensity aerobic exercise. Am J Cardiol. 2006;97:141-147.
9. Poortmans JR, Labilloy D. The influence of work intensity on postexercise proteinuria. Eur J Appl Physiol Occup Physiol. 1988;57:260-263.
10. Estivi P, Urbino R, Tetta C, et al. Urinary protein excretion induced by exercise: effect of a mountain agonistic footrace in healthy subjects. Renal function and mountain footrace. J Sports Med Phys Fitness. 1992;32:196-200.
11. Poortmans JR, Brauman H, Staroukine M, et al. Indirect evidence of glomerular/tubular mixed-type postexercise proteinuria in healthy humans. Am J Physiol. 1988;254:F277-F283.
12. Heathcote KL, Wilson MP, Quest DW, et al. Prevalence and duration of exercise induced albuminuria in healthy people. Clin Invest Med. 2009;32:E261-E265.
13. Sentürk UK, Kuru O, Koçer G, et al. Biphasic pattern of exercise-induced proteinuria in sedentary and trained men. Nephron Physiol. 2007;105:22-32.
14. Clerico A, Giammattei C, Cecchini L, et al. Exercise-induced proteinuria in well-trained athletes. Clin Chem. 1990;36:562-564.
15. Taes YE, Delanghe JR, Wuyts B, et al. Creatine supplementation does not affect kidney function in an animal model with pre-existing renal failure. Nephrol Dial Transplant. 2003;18:258-264.
16. Poortmans JR, Francaux M. Long-term oral creatine supplementation does not impair renal function in healthy athletes. Med Sci Sports Exerc. 1999;31:1108-1110.
17. Mogensen CE, Vittinghus E, Sølling K. Abnormal albumin excretion after two provocative renal tests in diabetes: physical exercise and lysine injection. Kidney Int. 1979;16:385-393.
18. Vittinghus E, Mogensen CE. Albumin excretion during physical exercise in diabetes. Studies on the effect of insulin treatment and of the renal haemodynamic response. Acta Endocrinol Suppl (Copenh). 1981;242:61-62.
19. Watts GF, Williams I, Morris RW, et al. An acceptable exercise test to study microalbuminuria in type 1 diabetes. Diabet Med. 1989;6:787-792.
20. Pan X, Wang P, Hu N, et al. A physiologically based pharmacokinetic model characterizing mechanism-based inhibition of CYP1A2 for predicting theophylline/antofloxacin interaction in both rats and humans. Drug Metab Pharmacokinet. 2011;26:387-398.
21. O’Brien SF, Watts GF, Powrie JK, et al. Exercise testing as a long-term predictor of the development of microalbuminuria in normoalbuminuric IDDM patients. Diabetes Care. 1995;18:1602-1605.
22. Hidaka S, Kaneko O, Shirai M, et al. Do obesity and non-insulin dependent diabetes mellitus aggravate exercise-induced microproteinuria? Clin Chim Acta. 1998;275:115-126.
23. Hidaka S, Kakuta S, Okada H, et al. Exercise-induced proteinuria in diseases with metabolic disorders. Contrib Nephrol. 1990;83:136-143.
24. Leehey DJ, Moinuddin I, Bast JP, et al. Aerobic exercise in obese diabetic patients with chronic kidney disease: a randomized and controlled pilot study. Cardiovasc Diabetol. 2009;8:62.-
25. Pechter U, Ots M, Mesikepp S, et al. Beneficial effects of water-based exercise in patients with chronic kidney disease. Int J Rehabil Res. 2003;26:153-156.
26. Heifets M, Davis TA, Tegtmeyer E, et al. Exercise training ameliorates progressive renal disease in rats with subtotal nephrectomy. Kidney Int. 1987;32:815-820.
27. Manelli F, Bossoni S, Burattin A, et al. Exercise-induced microalbuminuria in patients with active acromegaly: acute effects of slow-release lanreotide, a long-acting somatostatin analog. Metabolism. 2000;49:634-639.
28. Hoogenberg K, Sluiter WJ, Dullaart RP. Effect of growth hormone and insulin-like growth factor I on urinary albumin excretion: studies in acromegaly and growth hormone deficiency. Acta Endocrinol (Copenh). 1993;129:151-157.
29. Goldberg B, Saraniti A, Witman P, et al. Pre-participation sports assessment—an objective evaluation. Pediatrics. 1980;66:736-745.
30. Garrett WE, Kirkendall DT, Squire DL. eds. Principles and Practice of Primary Care Sports Medicine. Philadelphia, Pa: Lippincott Williams & Wilkins; 2001;299-310.
31. Cosenzi A, Carraro M, Sacerdote A, et al. Involvement of the renin angiotensin system in the pathogenesis of postexercise proteinuria. Scand J Urol Nephrol. 1993;27:301-304.
32. Székács B, Vajo Z, Dachman W. Effect of ACE inhibition by benazepril, enalapril and captopril on chronic and post exercise proteinuria. Acta Physiol Hung. 1996;84:361-367.
33. Poortmans JR, Haggenmacher C, Vanderstraeten J. Postexercise proteinuria in humans and its adrenergic component. J Sports Med Phys Fitness. 2001;41:95-100.
• Rely on a spot urine microalbumin-to-creatinine or protein-to-creatinine ratio to accurately assess proteinuria. B
• Repeat testing if routine urinalysis detects proteinuria—especially if the patient reports having exercised in the previous 24 hours. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE As part of a routine physical examination, urinalysis reveals that a patient new to your practice is excreting an excessive level of protein. The patient is physically fit and shared during the history taking that he is an avid runner. The physical examination and other laboratory values were unremarkable. How concerned should you be about the finding of proteinuria?
Exercise-induced proteinuria is generally benign and a function of the intensity—rather than the duration—of exercise.1 It occurs most often among athletes participating in such sports as running, swimming, rowing, football, or boxing.2 It’s also transient—lasting 24 to 48 hours.1 Recognizing exercise-induced proteinuria is fairly straightforward—once you know what to look for.
But first, a word about the processes at work.
Diverse processes that work alone—or together
The normal range of protein excretion in healthy individuals is 150 to 200 mg of protein per day, of which albumin constitutes 10 to 20 mg.3 Individuals with proteinuria persistently higher than this level need further evaluation.
Diverse processes leading to proteinuria—working alone or concomitantly—occur at the level of the nephron.3
Glomerular proteinuria results from increased filtration of macromolecules such as albumin across the glomerular capillary barrier. This type of proteinuria can occur with different glomerulopathies, upright posture, or exercise.4
Researchers have not identified the mechanisms leading to postexercise proteinuria, but there are several theories. (For more on this, see “Why does exercise increase protein excretion?”.)
Tubular proteinuria is due to a deranged tubular apparatus with an intact glomerulus. This results in the escape of β2-microglobulin and immunoglobulin light chains from proximal tubular reabsorption. It is often missed on dipstick testing, which detects only albumin. This type of proteinuria is usually seen in tubulointerstitial diseases or in patients with idiopathic nephrotic syndrome.5
Overflow proteinuria occurs when small molecular light chains escape the glomerular filtration barrier and overwhelm the tubular reabsorptive capacity. This type of proteinuria can be seen in multiple myeloma, and is detectable by protein-to-creatinine ratio or urine protein electrophoresis.
The root cause of exercise-induced proteinuria is unclear, but the renin-angiotensin system (RAS) and prostaglandins are thought to play a major role. The plasma concentration of angiotensin II increases during exercise, leading to filtration of protein through the glomerular membrane.30 And angiotensin-converting enzyme (ACE) inhibitors have been shown to significantly diminish exercise-induced proteinuria, thus supporting this theory.31,32
Also, strenuous exercise increases sympathetic nervous system activity as well as blood levels of catecholamines, thereby increasing permeability of the glomerular capillary membrane.33 Furthermore, lactate increases with strenuous exercise and causes conformational changes in serum proteins that, when coupled with glomerular barrier changes, can lead to increased permeability and protein excretion.
The surest means of detecting proteinuria
Albumin excretion >300 mg/d is called macroalbuminuria, overt proteinuria, or dipstick-positive proteinuria. Albumin persistently excreted in the urine between 30 and 300 mg/d is referred to as microalbuminuria.
Because microalbuminuria is not detectable by a standard urine dipstick test, some providers routinely screen for protein with the microalbumin-to-creatinine ratio. A first-voided morning urine specimen is recommended, but random urine samples are an acceptable alternative.6 The microalbumin-to-creatinine ratio is recommended as a screen for early diabetic nephropathy and other kidney diseases. And a positive test result may also suggest increased risk of cardiovascular disease.6 Microalbuminuria is defined as persistent albumin excretion between 30 and 300 mg/d.7
When exercise is a factor, here’s what to look for
As noted earlier, exercise-induced proteinuria is a function of the intensity of the exercise. Moderate and strenuous (vigorous) exercise are the 2 types of exercise that come into play when discussing proteinuria. Differentiating them is not precise, but is often defined by maximal oxygen consumption (vigorous=60% of VO2max; moderate <60% VO2max); metabolic equivalents (vigorous=6 METS; moderate <6 METS); walking/running speeds (various); and heart rate reserve (vigorous=60% HRR; moderate <60% HRR).8
Moderate exercise produces glomerular proteinuria, with an increase in macromolecular (albumin) filtration across the glomerular barrier. Strenuous exercise increases glomerular filtration of low-molecular-weight proteins (β2-microglobulin), which overwhelm the reabsorbing capacity of the tubular apparatus, causing temporary dysfunction and tubular proteinuria.9 Thus, the pathophysiology is mixed, with a major contribution from glomerular proteinuria.10
Strenuous exercise can cause protein excretion to exceed 1.5 mg/min.11 However, it seldom rises beyond 1 to 2 g/d,4 and this increase usually reverts to normal physiologic levels within 24 to 48 hours after exercise.12
Exercise-induced proteinuria is biphasic.13 Increased protein excretion occurs 30 minutes after exercise and is related to changes in intraglomerular hemodynamics and the resulting saturation of the renal tubules. Around 24 hours after exercise, oxidative stress on the glomeruli causes another slight elevation in albumin excretion without changes in β2-microglobulin, thereby indicating glomerular proteinuria exclusively.
Even the pros aren’t exempt. Exercise-induced proteinuria does not decrease with regular physical training. This was demonstrated in a study of 10 well-trained professional cyclists for whom strenuous exercise increased overnight protein excretion of both tubular and glomerular origin despite ongoing regular physical training.14
Creatine supplements do not increase proteinuria. A study of creatine supplementation in animal models noted no changes in 24-hour proteinuria or albumin excretion in both normal and two-thirds-nephrectomized animals.15 Another study compared creatine use with nonuse in athletes who had been training regularly and strenuously (12- 18 h/wk) for 5 to 10 years. They were evaluated for 10 months to 5 years. The groups exhibited equivalent urine excretion rates for albumin and creatinine, with no deleterious effect on kidney function.16
What happens when chronic disease is factored into the exercise equation?
Patients with a 2- to 20-year history of insulin-dependent diabetes without chronic kidney disease (CKD) who exhibited normal albumin excretion at baseline were more likely to develop proteinuria after exercise than healthy controls.17,18 The postulated cause was undetected glomerular changes due to diabetes. An exercise-provocation test may one day be useful in predicting future development of nephropathy, but further studies are needed.19-21
Exercise increases proteinuria immediately in individuals with metabolic disorders like obesity, through a mechanism different from diabetes mellitus. Proteinuria in the obese population is thought to be glomerular in origin, as opposed to both tubular and glomerular proteinuria in diabetic nephropathy.22,23
In CKD, low-intensity exercise long term does not promote proteinuria or lead to rapid progression of CKD. In one study, obese patients (body mass index >30 kg/m2) with diabetes and CKD stage II to IV who exercised 3 times weekly (aerobic training for 6 weeks, followed by 18 weeks of supervised home exercise) increased their stamina and exhibited slight, statistically insignificant decreases in resting systolic blood pressure and 24-hour proteinuria.24 A 12-week low-intensity aquatic exercise program for 26 patients with mild to moderate CKD decreased blood pressure and proteinuria and slightly improved glomerular filtration rate (GFR).25 These results for proteinuria and GFR were shown previously in rats with subtotal nephrectomy.26
Elevated urinary albumin excretion with exercise is significantly higher in patients with acromegaly when compared with normal healthy subjects. The underlying pathology is thought to occur at the glomerular filtration barrier with intact tubular function. Somatostatin analog treatment for acromegaly leads to reductions in postexercise albuminuria.27,28
How to manage suspected exercise-induced proteinuria
When interpreting the meaning of proteinuria detected on routine urinalysis, keep in mind the temporal relevance between exercise and urine collection. If urine is found to have been collected within 24 hours of intense exercise, repeat testing in the absence of prior exercise on at least one other occasion to differentiate between transient and persistent proteinuria. In confirming transient proteinuria after exercise, reassure the patient that it is a benign condition. This holds true as well for routine microalbumin-to-creatinine urine testing in patients with diabetes who exercise. If the result of a repeat test is high, turn your attention to another possible cause of proteinuria, such as diabetic nephropathy.
Screening for proteinuria during sports preparticipation examinations is not recommended because the diagnostic utility is low.29 Researchers performed urine dipstick testing for protein, blood, and glucose in preparticipation assessments of 701 students.29 They detected proteinuria in 40 students and glucosuria in one. Follow-up testing with first-voided morning urine specimens and glucose tolerance testing was normal in all students.
CORRESPONDENCE Fahad Saeed, MD, 313 Brook Hollow, Hanover, NH 03755; [email protected]
• Rely on a spot urine microalbumin-to-creatinine or protein-to-creatinine ratio to accurately assess proteinuria. B
• Repeat testing if routine urinalysis detects proteinuria—especially if the patient reports having exercised in the previous 24 hours. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE As part of a routine physical examination, urinalysis reveals that a patient new to your practice is excreting an excessive level of protein. The patient is physically fit and shared during the history taking that he is an avid runner. The physical examination and other laboratory values were unremarkable. How concerned should you be about the finding of proteinuria?
Exercise-induced proteinuria is generally benign and a function of the intensity—rather than the duration—of exercise.1 It occurs most often among athletes participating in such sports as running, swimming, rowing, football, or boxing.2 It’s also transient—lasting 24 to 48 hours.1 Recognizing exercise-induced proteinuria is fairly straightforward—once you know what to look for.
But first, a word about the processes at work.
Diverse processes that work alone—or together
The normal range of protein excretion in healthy individuals is 150 to 200 mg of protein per day, of which albumin constitutes 10 to 20 mg.3 Individuals with proteinuria persistently higher than this level need further evaluation.
Diverse processes leading to proteinuria—working alone or concomitantly—occur at the level of the nephron.3
Glomerular proteinuria results from increased filtration of macromolecules such as albumin across the glomerular capillary barrier. This type of proteinuria can occur with different glomerulopathies, upright posture, or exercise.4
Researchers have not identified the mechanisms leading to postexercise proteinuria, but there are several theories. (For more on this, see “Why does exercise increase protein excretion?”.)
Tubular proteinuria is due to a deranged tubular apparatus with an intact glomerulus. This results in the escape of β2-microglobulin and immunoglobulin light chains from proximal tubular reabsorption. It is often missed on dipstick testing, which detects only albumin. This type of proteinuria is usually seen in tubulointerstitial diseases or in patients with idiopathic nephrotic syndrome.5
Overflow proteinuria occurs when small molecular light chains escape the glomerular filtration barrier and overwhelm the tubular reabsorptive capacity. This type of proteinuria can be seen in multiple myeloma, and is detectable by protein-to-creatinine ratio or urine protein electrophoresis.
The root cause of exercise-induced proteinuria is unclear, but the renin-angiotensin system (RAS) and prostaglandins are thought to play a major role. The plasma concentration of angiotensin II increases during exercise, leading to filtration of protein through the glomerular membrane.30 And angiotensin-converting enzyme (ACE) inhibitors have been shown to significantly diminish exercise-induced proteinuria, thus supporting this theory.31,32
Also, strenuous exercise increases sympathetic nervous system activity as well as blood levels of catecholamines, thereby increasing permeability of the glomerular capillary membrane.33 Furthermore, lactate increases with strenuous exercise and causes conformational changes in serum proteins that, when coupled with glomerular barrier changes, can lead to increased permeability and protein excretion.
The surest means of detecting proteinuria
Albumin excretion >300 mg/d is called macroalbuminuria, overt proteinuria, or dipstick-positive proteinuria. Albumin persistently excreted in the urine between 30 and 300 mg/d is referred to as microalbuminuria.
Because microalbuminuria is not detectable by a standard urine dipstick test, some providers routinely screen for protein with the microalbumin-to-creatinine ratio. A first-voided morning urine specimen is recommended, but random urine samples are an acceptable alternative.6 The microalbumin-to-creatinine ratio is recommended as a screen for early diabetic nephropathy and other kidney diseases. And a positive test result may also suggest increased risk of cardiovascular disease.6 Microalbuminuria is defined as persistent albumin excretion between 30 and 300 mg/d.7
When exercise is a factor, here’s what to look for
As noted earlier, exercise-induced proteinuria is a function of the intensity of the exercise. Moderate and strenuous (vigorous) exercise are the 2 types of exercise that come into play when discussing proteinuria. Differentiating them is not precise, but is often defined by maximal oxygen consumption (vigorous=60% of VO2max; moderate <60% VO2max); metabolic equivalents (vigorous=6 METS; moderate <6 METS); walking/running speeds (various); and heart rate reserve (vigorous=60% HRR; moderate <60% HRR).8
Moderate exercise produces glomerular proteinuria, with an increase in macromolecular (albumin) filtration across the glomerular barrier. Strenuous exercise increases glomerular filtration of low-molecular-weight proteins (β2-microglobulin), which overwhelm the reabsorbing capacity of the tubular apparatus, causing temporary dysfunction and tubular proteinuria.9 Thus, the pathophysiology is mixed, with a major contribution from glomerular proteinuria.10
Strenuous exercise can cause protein excretion to exceed 1.5 mg/min.11 However, it seldom rises beyond 1 to 2 g/d,4 and this increase usually reverts to normal physiologic levels within 24 to 48 hours after exercise.12
Exercise-induced proteinuria is biphasic.13 Increased protein excretion occurs 30 minutes after exercise and is related to changes in intraglomerular hemodynamics and the resulting saturation of the renal tubules. Around 24 hours after exercise, oxidative stress on the glomeruli causes another slight elevation in albumin excretion without changes in β2-microglobulin, thereby indicating glomerular proteinuria exclusively.
Even the pros aren’t exempt. Exercise-induced proteinuria does not decrease with regular physical training. This was demonstrated in a study of 10 well-trained professional cyclists for whom strenuous exercise increased overnight protein excretion of both tubular and glomerular origin despite ongoing regular physical training.14
Creatine supplements do not increase proteinuria. A study of creatine supplementation in animal models noted no changes in 24-hour proteinuria or albumin excretion in both normal and two-thirds-nephrectomized animals.15 Another study compared creatine use with nonuse in athletes who had been training regularly and strenuously (12- 18 h/wk) for 5 to 10 years. They were evaluated for 10 months to 5 years. The groups exhibited equivalent urine excretion rates for albumin and creatinine, with no deleterious effect on kidney function.16
What happens when chronic disease is factored into the exercise equation?
Patients with a 2- to 20-year history of insulin-dependent diabetes without chronic kidney disease (CKD) who exhibited normal albumin excretion at baseline were more likely to develop proteinuria after exercise than healthy controls.17,18 The postulated cause was undetected glomerular changes due to diabetes. An exercise-provocation test may one day be useful in predicting future development of nephropathy, but further studies are needed.19-21
Exercise increases proteinuria immediately in individuals with metabolic disorders like obesity, through a mechanism different from diabetes mellitus. Proteinuria in the obese population is thought to be glomerular in origin, as opposed to both tubular and glomerular proteinuria in diabetic nephropathy.22,23
In CKD, low-intensity exercise long term does not promote proteinuria or lead to rapid progression of CKD. In one study, obese patients (body mass index >30 kg/m2) with diabetes and CKD stage II to IV who exercised 3 times weekly (aerobic training for 6 weeks, followed by 18 weeks of supervised home exercise) increased their stamina and exhibited slight, statistically insignificant decreases in resting systolic blood pressure and 24-hour proteinuria.24 A 12-week low-intensity aquatic exercise program for 26 patients with mild to moderate CKD decreased blood pressure and proteinuria and slightly improved glomerular filtration rate (GFR).25 These results for proteinuria and GFR were shown previously in rats with subtotal nephrectomy.26
Elevated urinary albumin excretion with exercise is significantly higher in patients with acromegaly when compared with normal healthy subjects. The underlying pathology is thought to occur at the glomerular filtration barrier with intact tubular function. Somatostatin analog treatment for acromegaly leads to reductions in postexercise albuminuria.27,28
How to manage suspected exercise-induced proteinuria
When interpreting the meaning of proteinuria detected on routine urinalysis, keep in mind the temporal relevance between exercise and urine collection. If urine is found to have been collected within 24 hours of intense exercise, repeat testing in the absence of prior exercise on at least one other occasion to differentiate between transient and persistent proteinuria. In confirming transient proteinuria after exercise, reassure the patient that it is a benign condition. This holds true as well for routine microalbumin-to-creatinine urine testing in patients with diabetes who exercise. If the result of a repeat test is high, turn your attention to another possible cause of proteinuria, such as diabetic nephropathy.
Screening for proteinuria during sports preparticipation examinations is not recommended because the diagnostic utility is low.29 Researchers performed urine dipstick testing for protein, blood, and glucose in preparticipation assessments of 701 students.29 They detected proteinuria in 40 students and glucosuria in one. Follow-up testing with first-voided morning urine specimens and glucose tolerance testing was normal in all students.
CORRESPONDENCE Fahad Saeed, MD, 313 Brook Hollow, Hanover, NH 03755; [email protected]
1. Poortmans JR. Exercise and renal function. Sports Med. 1984;1:125-153.
2. Gebke KB. Genitourinary system. In: McKeag DB, Moeller JL, eds. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2007;234.-
3. Venkat KK. Proteinuria and microalbuminuria in adults: significance, evaluation, and treatment. South Med J. 2004;97:969-979.
4. Rose BD. Pathophysiology of Renal Disease. 2nd ed. New York, NY: McGraw-Hill; 1987;11-16.
5. Sesso R, Santos AP, Nishida SK, et al. Prediction of steroid responsiveness in the idiopathic nephrotic syndrome using urinary retinol-binding protein and beta-2-microglobulin. Ann Intern Med. 1992;116:905-909.
6. Levey AS, Coresh J, Balk E, et al. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139:137-147.
7. Family Practice Notebook Urine protein to creatinine ratio. Available at: http://www.fpnotebook.com/urology/lab/urnprtntcrtnrt.htm. Accessed August 9, 2011.
8. Swain DP, Franklin BA. Comparison of cardioprotective benefits of vigorous versus moderate intensity aerobic exercise. Am J Cardiol. 2006;97:141-147.
9. Poortmans JR, Labilloy D. The influence of work intensity on postexercise proteinuria. Eur J Appl Physiol Occup Physiol. 1988;57:260-263.
10. Estivi P, Urbino R, Tetta C, et al. Urinary protein excretion induced by exercise: effect of a mountain agonistic footrace in healthy subjects. Renal function and mountain footrace. J Sports Med Phys Fitness. 1992;32:196-200.
11. Poortmans JR, Brauman H, Staroukine M, et al. Indirect evidence of glomerular/tubular mixed-type postexercise proteinuria in healthy humans. Am J Physiol. 1988;254:F277-F283.
12. Heathcote KL, Wilson MP, Quest DW, et al. Prevalence and duration of exercise induced albuminuria in healthy people. Clin Invest Med. 2009;32:E261-E265.
13. Sentürk UK, Kuru O, Koçer G, et al. Biphasic pattern of exercise-induced proteinuria in sedentary and trained men. Nephron Physiol. 2007;105:22-32.
14. Clerico A, Giammattei C, Cecchini L, et al. Exercise-induced proteinuria in well-trained athletes. Clin Chem. 1990;36:562-564.
15. Taes YE, Delanghe JR, Wuyts B, et al. Creatine supplementation does not affect kidney function in an animal model with pre-existing renal failure. Nephrol Dial Transplant. 2003;18:258-264.
16. Poortmans JR, Francaux M. Long-term oral creatine supplementation does not impair renal function in healthy athletes. Med Sci Sports Exerc. 1999;31:1108-1110.
17. Mogensen CE, Vittinghus E, Sølling K. Abnormal albumin excretion after two provocative renal tests in diabetes: physical exercise and lysine injection. Kidney Int. 1979;16:385-393.
18. Vittinghus E, Mogensen CE. Albumin excretion during physical exercise in diabetes. Studies on the effect of insulin treatment and of the renal haemodynamic response. Acta Endocrinol Suppl (Copenh). 1981;242:61-62.
19. Watts GF, Williams I, Morris RW, et al. An acceptable exercise test to study microalbuminuria in type 1 diabetes. Diabet Med. 1989;6:787-792.
20. Pan X, Wang P, Hu N, et al. A physiologically based pharmacokinetic model characterizing mechanism-based inhibition of CYP1A2 for predicting theophylline/antofloxacin interaction in both rats and humans. Drug Metab Pharmacokinet. 2011;26:387-398.
21. O’Brien SF, Watts GF, Powrie JK, et al. Exercise testing as a long-term predictor of the development of microalbuminuria in normoalbuminuric IDDM patients. Diabetes Care. 1995;18:1602-1605.
22. Hidaka S, Kaneko O, Shirai M, et al. Do obesity and non-insulin dependent diabetes mellitus aggravate exercise-induced microproteinuria? Clin Chim Acta. 1998;275:115-126.
23. Hidaka S, Kakuta S, Okada H, et al. Exercise-induced proteinuria in diseases with metabolic disorders. Contrib Nephrol. 1990;83:136-143.
24. Leehey DJ, Moinuddin I, Bast JP, et al. Aerobic exercise in obese diabetic patients with chronic kidney disease: a randomized and controlled pilot study. Cardiovasc Diabetol. 2009;8:62.-
25. Pechter U, Ots M, Mesikepp S, et al. Beneficial effects of water-based exercise in patients with chronic kidney disease. Int J Rehabil Res. 2003;26:153-156.
26. Heifets M, Davis TA, Tegtmeyer E, et al. Exercise training ameliorates progressive renal disease in rats with subtotal nephrectomy. Kidney Int. 1987;32:815-820.
27. Manelli F, Bossoni S, Burattin A, et al. Exercise-induced microalbuminuria in patients with active acromegaly: acute effects of slow-release lanreotide, a long-acting somatostatin analog. Metabolism. 2000;49:634-639.
28. Hoogenberg K, Sluiter WJ, Dullaart RP. Effect of growth hormone and insulin-like growth factor I on urinary albumin excretion: studies in acromegaly and growth hormone deficiency. Acta Endocrinol (Copenh). 1993;129:151-157.
29. Goldberg B, Saraniti A, Witman P, et al. Pre-participation sports assessment—an objective evaluation. Pediatrics. 1980;66:736-745.
30. Garrett WE, Kirkendall DT, Squire DL. eds. Principles and Practice of Primary Care Sports Medicine. Philadelphia, Pa: Lippincott Williams & Wilkins; 2001;299-310.
31. Cosenzi A, Carraro M, Sacerdote A, et al. Involvement of the renin angiotensin system in the pathogenesis of postexercise proteinuria. Scand J Urol Nephrol. 1993;27:301-304.
32. Székács B, Vajo Z, Dachman W. Effect of ACE inhibition by benazepril, enalapril and captopril on chronic and post exercise proteinuria. Acta Physiol Hung. 1996;84:361-367.
33. Poortmans JR, Haggenmacher C, Vanderstraeten J. Postexercise proteinuria in humans and its adrenergic component. J Sports Med Phys Fitness. 2001;41:95-100.
1. Poortmans JR. Exercise and renal function. Sports Med. 1984;1:125-153.
2. Gebke KB. Genitourinary system. In: McKeag DB, Moeller JL, eds. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2007;234.-
3. Venkat KK. Proteinuria and microalbuminuria in adults: significance, evaluation, and treatment. South Med J. 2004;97:969-979.
4. Rose BD. Pathophysiology of Renal Disease. 2nd ed. New York, NY: McGraw-Hill; 1987;11-16.
5. Sesso R, Santos AP, Nishida SK, et al. Prediction of steroid responsiveness in the idiopathic nephrotic syndrome using urinary retinol-binding protein and beta-2-microglobulin. Ann Intern Med. 1992;116:905-909.
6. Levey AS, Coresh J, Balk E, et al. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139:137-147.
7. Family Practice Notebook Urine protein to creatinine ratio. Available at: http://www.fpnotebook.com/urology/lab/urnprtntcrtnrt.htm. Accessed August 9, 2011.
8. Swain DP, Franklin BA. Comparison of cardioprotective benefits of vigorous versus moderate intensity aerobic exercise. Am J Cardiol. 2006;97:141-147.
9. Poortmans JR, Labilloy D. The influence of work intensity on postexercise proteinuria. Eur J Appl Physiol Occup Physiol. 1988;57:260-263.
10. Estivi P, Urbino R, Tetta C, et al. Urinary protein excretion induced by exercise: effect of a mountain agonistic footrace in healthy subjects. Renal function and mountain footrace. J Sports Med Phys Fitness. 1992;32:196-200.
11. Poortmans JR, Brauman H, Staroukine M, et al. Indirect evidence of glomerular/tubular mixed-type postexercise proteinuria in healthy humans. Am J Physiol. 1988;254:F277-F283.
12. Heathcote KL, Wilson MP, Quest DW, et al. Prevalence and duration of exercise induced albuminuria in healthy people. Clin Invest Med. 2009;32:E261-E265.
13. Sentürk UK, Kuru O, Koçer G, et al. Biphasic pattern of exercise-induced proteinuria in sedentary and trained men. Nephron Physiol. 2007;105:22-32.
14. Clerico A, Giammattei C, Cecchini L, et al. Exercise-induced proteinuria in well-trained athletes. Clin Chem. 1990;36:562-564.
15. Taes YE, Delanghe JR, Wuyts B, et al. Creatine supplementation does not affect kidney function in an animal model with pre-existing renal failure. Nephrol Dial Transplant. 2003;18:258-264.
16. Poortmans JR, Francaux M. Long-term oral creatine supplementation does not impair renal function in healthy athletes. Med Sci Sports Exerc. 1999;31:1108-1110.
17. Mogensen CE, Vittinghus E, Sølling K. Abnormal albumin excretion after two provocative renal tests in diabetes: physical exercise and lysine injection. Kidney Int. 1979;16:385-393.
18. Vittinghus E, Mogensen CE. Albumin excretion during physical exercise in diabetes. Studies on the effect of insulin treatment and of the renal haemodynamic response. Acta Endocrinol Suppl (Copenh). 1981;242:61-62.
19. Watts GF, Williams I, Morris RW, et al. An acceptable exercise test to study microalbuminuria in type 1 diabetes. Diabet Med. 1989;6:787-792.
20. Pan X, Wang P, Hu N, et al. A physiologically based pharmacokinetic model characterizing mechanism-based inhibition of CYP1A2 for predicting theophylline/antofloxacin interaction in both rats and humans. Drug Metab Pharmacokinet. 2011;26:387-398.
21. O’Brien SF, Watts GF, Powrie JK, et al. Exercise testing as a long-term predictor of the development of microalbuminuria in normoalbuminuric IDDM patients. Diabetes Care. 1995;18:1602-1605.
22. Hidaka S, Kaneko O, Shirai M, et al. Do obesity and non-insulin dependent diabetes mellitus aggravate exercise-induced microproteinuria? Clin Chim Acta. 1998;275:115-126.
23. Hidaka S, Kakuta S, Okada H, et al. Exercise-induced proteinuria in diseases with metabolic disorders. Contrib Nephrol. 1990;83:136-143.
24. Leehey DJ, Moinuddin I, Bast JP, et al. Aerobic exercise in obese diabetic patients with chronic kidney disease: a randomized and controlled pilot study. Cardiovasc Diabetol. 2009;8:62.-
25. Pechter U, Ots M, Mesikepp S, et al. Beneficial effects of water-based exercise in patients with chronic kidney disease. Int J Rehabil Res. 2003;26:153-156.
26. Heifets M, Davis TA, Tegtmeyer E, et al. Exercise training ameliorates progressive renal disease in rats with subtotal nephrectomy. Kidney Int. 1987;32:815-820.
27. Manelli F, Bossoni S, Burattin A, et al. Exercise-induced microalbuminuria in patients with active acromegaly: acute effects of slow-release lanreotide, a long-acting somatostatin analog. Metabolism. 2000;49:634-639.
28. Hoogenberg K, Sluiter WJ, Dullaart RP. Effect of growth hormone and insulin-like growth factor I on urinary albumin excretion: studies in acromegaly and growth hormone deficiency. Acta Endocrinol (Copenh). 1993;129:151-157.
29. Goldberg B, Saraniti A, Witman P, et al. Pre-participation sports assessment—an objective evaluation. Pediatrics. 1980;66:736-745.
30. Garrett WE, Kirkendall DT, Squire DL. eds. Principles and Practice of Primary Care Sports Medicine. Philadelphia, Pa: Lippincott Williams & Wilkins; 2001;299-310.
31. Cosenzi A, Carraro M, Sacerdote A, et al. Involvement of the renin angiotensin system in the pathogenesis of postexercise proteinuria. Scand J Urol Nephrol. 1993;27:301-304.
32. Székács B, Vajo Z, Dachman W. Effect of ACE inhibition by benazepril, enalapril and captopril on chronic and post exercise proteinuria. Acta Physiol Hung. 1996;84:361-367.
33. Poortmans JR, Haggenmacher C, Vanderstraeten J. Postexercise proteinuria in humans and its adrenergic component. J Sports Med Phys Fitness. 2001;41:95-100.
Shoulder pain: 3 cases to test your diagnostic skills
Shoulder pain is a common reason for visits to primary care physicians, who are most likely to diagnose it as rotator cuff tendinitis1,2—often erroneously. The complexity of the joint and the overlapping pathologies that may present as shoulder pain highlight the need to take a closer look when dealing with this diagnostic challenge.
Often, a targeted medical history—including the mechanism of injury and pain-provoking and pain-relieving factors—and a problem-based physical examination (incorporating many of the maneuvers highlighted in the text and tables that follow) will lead to an accurate diagnosis without the need for imaging studies. We recommend that imaging be reserved for patients who don’t respond to conventional treatments, cases in which the diagnosis is in doubt, and instances in which surgical intervention is being considered.
The 3 cases* that follow, and the take-away message incorporated in each, will give you an opportunity to test—and to hone—your shoulder pain diagnostic skill.
CASE 1 The history: Jesse, a 17-year-old student who’s active in football and track, came in during track season complaining of severe left shoulder pain. He denied any traumatic event or previous injury to the shoulder, but reported that any motion involving the shoulder caused pain. It hurt at night, the patient said, when he lay on his left side.
The physical: No muscle atrophy, redness, or swelling was evident, nor was there any indication of asymmetry or ecchymosis in the affected area. Jesse’s neck range of motion was normal; he had a very hard time with any active motion of the shoulder, however, because of the pain.
Evaluation of scapular motion demonstrated scapular dyskinesis3,4 without winging. Passive motion of the glenohumeral joint was much better than active motion. Strength testing appeared to be grossly intact but was limited by the pain. Shoulder impingement testing was positive. Sensation and deep tendon reflexes were intact.
Patients' names have been changed to protect their privacy
What’s the diagnosis?
Subacromial bursitis, suggested by the patient’s pain and altered scapular motion, was our working diagnosis, and we administered a subacromial injection of corticosteroid with lidocaine, for diagnostic as well as therapeutic purposes. Reexamination after the injection revealed immediate partial improvement in resting pain, range of motion, and strength. We referred Jesse to physical therapy with a focus on scapular stabilization and rotator cuff strength.
Three months later, Jesse returned to our office, complaining of weakness in his left shoulder. The pain had subsided a week after his first appointment, so he’d never gone to physical therapy. The weakness, which he had first noticed about 2 months after starting a lifting program in preparation for football season, was limited to resistance exercises, especially overhead shoulder presses and bench press. There were no other changes in his history, and he reported no reinjuries.
Physical examination revealed atrophy of the supraspinatus and infraspinatus muscles (FIGURE 1) and external rotation and shoulder abduction (in the scapular plane) resistance tests revealed weakness of these muscles. There was no scapular winging. The cervical spine exam was normal, and neurovascular status was intact in both upper extremities.
FIGURE 1
Severe shoulder pain, followed by weakness
Physical examination reveals atrophy of the patient’s supraspinatus (^) and infraspinatus (+) muscles.
New evidence points to nerve injury. Based on Jesse’s current history and physical, nerve injury was our new working diagnosis. (We considered the possibility of a rotator cuff tear, but this was not corroborated by the history.)
We ordered an electromyogram/nerve conduction velocity study to localize the lesion. The test revealed a brachial plexitis/neuritis (also known as Parsonage-Turner syndrome or brachial amyotrophy). The etiology of most atraumatic brachial plexopathies is unknown, and most are thought to be viral or autoimmune in nature.5,6
A classic case of Parsonage-Turner syndrome. The typical presentation of Parsonage-Turner syndrome (like Jesse’s) is one of acute, intense shoulder pain for no known reason. After 1 to 3 weeks, the pain resolves and the patient is left with weakness, usually of the supraspinatus and infraspinatus muscles. The weakness typically resolves with time, but full resolution may take 6 to 9 months.5,6 (In Jesse’s case, it took about 6 months.)
The take-away message: Look beyond the shoulder
As this case illustrates, not all shoulder pain originates in the shoulder. When evaluating shoulder pain, it is essential to consider other causes. The differential diagnosis for shoulder pain includes cervical spine disorders, cholecystitis (right shoulder), diaphragmatic irritation (eg, in the case of splenic rupture, usually involving the left shoulder), cardiac disease, and thoracic outlet syndrome.7
Evaluation of the cervical spine should be part of a complete shoulder examination. It is vital to follow a systematic approach that carefully assesses the cervical region for the possibility of nerve root impingement and radicular dysfunction masquerading as a primary shoulder disorder. (TABLE 18,9 details pain and sensory distribution patterns, reflex involvement, and potential motor impairments associated with various spinal nerve root levels.)
TABLE 1
Assessing the cervical spine
| Nerve root | Pain distribution | Sensory distribution | Reflex changes | Motor involvement |
|---|---|---|---|---|
| C5 | Lateral neck/upper trapezius | Lateral arm | Biceps | Deltoid, biceps |
| C6 | Base of neck/upper trapezius to superior glenohumeral joint | Radial aspect of distal forearm, thenar eminence, and index finger | Brachioradialis | Biceps, extensor carpi radialis longus and brevis (wrist extension) |
| C7 | Base of neck, almost entire upper quadrant of the back | Third finger | Triceps | Triceps, wrist flexion, finger extension |
| C8 | No shoulder pain | 4th and 5th fingers, distal half of forearm (ulna side) | None | Finger flexion (grip strength) |
| Adapted from: Miller JD, et al. Am Fam Physician. 20008; Eubanks JD. Am Fam Physician. 2010.9 | ||||
Practitioners should develop their own approach to “clearing the neck.” A logical order is to note posture of the head/neck/shoulders, observe active motion, perform palpation and provocative tests, and then assess neurologic function with sensation/reflex/strength testing. Provocative tests that can help to identify cervical involvement relating to shoulder pain include Spurling’s maneuver, axial compression test, abduction relief sign, and Lhermitte’s sign.10,11
CASE 2 The history: Mark, a 17-year-old, right-handed volleyball player, presented with right shoulder pain, which he felt whenever he spiked or served the ball. The pain started last season, Mark said, diminished during the months when he wasn’t playing, then got progressively worse as his activity level increased. The pain was in the posterior aspect of the shoulder.
The physical: Physical examination revealed a well-developed, but thin (6’4”, 170 pounds) young man who was not in distress. The general examination was benign, and a joint-specific exam showed no asymmetry or atrophy on inspection and no tenderness to palpation over the posterior and anterior soft tissues of the right shoulder. Rotator cuff testing yielded intact strength for all 4 muscles, but external rotation and supraspinatus testing elicited pain. The crank test, drawer sign, load and shift test, relocation test, and sulcus sign, detailed in TABLE 2,12-14 were all positive for shoulder instability; the Clunk and O’Brien tests were negative, and the contralateral shoulder exam was within normal limits. General joint laxity was observed, with the ability to oppose the thumb to the volar forearm and hyperextension noted in both elbows and knees. There were no outward signs of connective tissue disease.
Because of the chronicity of Mark’s pain and the progressive nature of his symptoms, we ordered radiographs, including anterior-posterior, lateral axillary, and scapular Y views. These films showed a nearly skeletally mature male without bony abnormalities; the humeral head was well located in the glenoid.
TABLE 2
Testing for shoulder instability12-14
| Test | Procedure | Positive result/implication |
|---|---|---|
| Apprehension | Patient supine, arm abducted 90º, externally rotated with anteriorly directed force applied to humeral head | Pain/apprehension with force suggests anterior instability |
| Relocation* | Patient supine, posteriorly directed force applied to humeral head | Relief with force suggests anterior instability |
| Crank | Patient sitting, arm abducted 90º, elbow flexed to 90º, humerus supported with forced external rotation | Pain/apprehension with forced external rotation suggests anterior instability |
| Load and shift | Patient supine, arm held by examiner and abducted 90º, force applied along axis of humerus to "seat" the humerus within the glenoid, followed by anterior force directed to humeral head | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Drawer | Patient sitting, arm at side, proximal humeral shaft grasped by examiner, seating the humeral head within the glenoid then applying anterior translational force | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Sulcus | Patient sitting, arm at side, forearm grasped by examiner with an inferior/caudally directed force applied | Sulcus or depression seen inferior to acromion as humeral head subluxes posteriorly, pathognomonic for multidirectional instability |
| Clunk | Patient supine, examiner grasps at forearm and humeral shaft, with humeral head seated within the fossa, taking the arm through passive ROM from extension through forward flexion | Clunk sound or clicking sensation suggests labral tear |
| O’Brien | Patient sitting, arm is forward flexed to 90º and fully adducted and internally rotated; patient resists downward motion. If pain is elicited, the maneuver is repeated in external rotation | Pain elicited with resisted downward motion in internal rotation but relieved with external rotation suggests labral pathology |
| *Perform only if apprehension test is positive. ROM, range of motion | ||
What’s the diagnosis?
Multidirectional instability with recurrent subluxations and probable acute rotator cuff tendinitis was our provisional diagnosis. Treatment focused on physical therapy, with a concentration on scapular stabilization and rotator cuff strengthening.
Shoulder instability is relatively common and represents a spectrum of disorders ranging from dislocation to subluxation to simple laxity.12,13 A complete loss of humeral articulation within the glenoid fossa is evidence of dislocation, whereas subluxation includes approximation of the humeral head to the limits of the glenoid rim. Dislocation typically results from trauma, whereas subluxation can be the result of microtrauma and repetitive overuse injury. Anterior instability is the most common type and is reported in as many as 95% of all dislocations.13
The take-away message: Rule out instability
The shoulder is one of the most complex joints in the body. The rotator cuff structures, the glenoid labrum, and the collective capsular ligaments provide structural stability to the glenohumeral joint.12,13 The shoulder is vulnerable to instability because the shallow glenoid fossa offers little bony support for the humeral head. Thus, instability should always be included in an assessment of shoulder pain.
Key factors to consider in identifying shoulder instability include the location of the pain, the direction of traumatic force applied, the presence of a known complete dislocation vs apprehension with specific movement, the position of the arm in which pain is elicited, a previous occurrence of instability (subluxation or dislocation), and the presence of tingling or numbness.12-14 The maneuvers detailed in TABLE 212-14 can help identify instability, as they did in this case. Patients with hypermobility are at increased risk for shoulder instability, so a targeted exam and patient history aimed at identifying signs and symptoms of hypermobility is needed, as well.
Ask the patient to attempt to:
- bend the thumb to the volar forearm
- place hands to the floor with hyperextended knees
- perform maximal hyperextension of the fifth metacarpophalangeal joint (>90° is a positive result).
Findings from the medical history that indicate a predisposition to instability include generalized joint laxity, Ehlers-Danlos syndrome, Marfan syndrome, osteogenesis imperfecta, hyperhomocysteinuria, hyperlysinemia, benign joint hypermobility syndrome, juvenile rheumatoid arthritis, and previous shoulder or patellar dislocations.
Imaging tips: Scapular Y and/or axillary lateral views should always be included when ordering imaging studies for suspected instability/dislocations, as 50% of posterior dislocations are missed on standard shoulder x-rays that do not include them.12 In reviewing the x-rays, it is important to look for signs of a compression fracture of the posterior humeral head (known as a Hill-Sachs lesion) for anterior shoulder dislocations, and fractures to the anterior glenoid rim (known as a Bankart lesion).12-14
CASE 3 The history: Robert, a right-handed, 50-year-old motorcycle instructor, came to our office because of chronic right shoulder pain. The pain, located over the anterior portion of the glenohumeral joint, developed insidiously about 3 or 4 years ago, the patient reported. He had finally decided to seek help because he’d recently experienced an acute exacerbation of pain brought on by shoveling snow, after which he also noticed associated weakness, a clicking/popping on active motion, and mild loss of motion.
The physical: Robert’s cervical spine exam was unremarkable. He demonstrated full active range of motion (ROM) without exacerbation of right shoulder symptoms, and special tests for disc pathology at the neck were negative. Active ROM testing of the right shoulder revealed full abduction, with only minimal pain; full flexion, with moderate pain noted initially at 49°; full extension, with a painful arc noted at 50°; and full horizontal adduction, with a painful arc noted at the halfway point. The testing also revealed that his right thumb was 3 inches lower than the left on reaching for the opposite scapula. At the superior aspect of the acromioclavicular (AC) joint, 2+ tenderness was noted; 3+ tenderness was noted at the greater tubercle of the humerus.
After inspecting the shoulder region for alterations in bony landmarks, muscle bulk, carrying position, and movement characteristics, palpation of the region was performed.
When assessing shoulder strength, there are a variety of tests for each functioning component of the rotator cuff structures (TABLE 3).15-17 Manual muscle tests revealed: 4-/5 on external rotation (French horn test), 3+/5 on the lift-off test, and 5-/5 on all other tests for right shoulder function. Impingement testing was slightly positive, or pain producing, on Hawkins and Neer tests.18,19 For the Hawkins test, the examiner flexes the arm to 90° of shoulder flexion with the elbow flexed at 90°, then internally rotates the shoulder. For the Neer test, the arm is fully elevated in the scapular plane and internally rotated by the examiner.
The subscapularis muscle, which functions primarily in internal rotation, is tested by the French horn and lift-off tests. The teres minor muscle, which performs external rotation, is tested by the French horn test of external rotation. And the supraspinatus muscle, which performs abduction and external rotation, is tested by the empty can (also known as the Jobe) and full can tests. Some researchers suggest that the empty can test is better for diagnosing impingement, based on evidence showing that the full can test is better at diagnosing supraspinatus tears because it causes less pain during testing.20
TABLE 3
Suspect rotator cuff involvement?7,15-17
What’s the diagnosis?
Rotator cuff tear was suspected because Robert had positive elements of the “rotator cuff triad”—supraspinatus weakness (as indicated by a positive empty can test), external rotation weakness (revealed by the French horn test), and a positive Hawkins impingement test. We ordered diagnostic studies, including plain radiographs, which revealed degenerative changes at the acromioclavicular joint, decreased acromiohumeral interval, and no significant changes at the glenohumeral joint (FIGURE 2), and magnetic resonance imaging (MRI) of the right shoulder. The MRI revealed a large, full-thickness rotator cuff tear of the supraspinatus tendon with retraction. A torn and retracted biceps tendon and AC joint osteoarthritis were also shown, likely causing a mass effect on the supraspinatus. The patient underwent surgery to repair the torn rotator cuff, with excellent results.
FIGURE 2
Chronic right shoulder pain
An AP view of the patient’s right shoulder shows acromioclavicular joint narrowing and degeneration and subtle narrowing of the acromiohumeral interval.
The take-away message: Keep the rotator-cuff triad in mind
Because none of the tests that comprise the triad is specific enough alone to diagnose a rotator cuff tear,15,20,21 Murrell and Walton16 suggested that the 3 tests be considered together for diagnostic purposes. If all 3 are positive, there is a 98% chance of a rotator cuff tear; if 2 tests are positive and the patient is older than 60 years, the findings are suggestive of a tear; and if all 3 tests (plus the drop arm test) are negative, there is less than a 5% chance of a major rotator cuff tear.16
CORRESPONDENCE Nilesh Shah, MD, Summa Center for Sports Health, 20 Olive Street, Suite 201, Akron, OH 44310; [email protected]
1. Van der Windt DA, Koes BW, De Jong BA, et al. Shoulder disorders in general practice: incidence, patient characteristics, and management. Ann Rheum Dis. 1995;54:959-964.
2. Johansson K, Adolfsson L, Foldevi M. Attitudes toward management of patients with subacromial pain in Swedish primary care. Fam Pract. 1999;16:233-237.
3. Kibler WB, McMullen J. Scapular dyskinesis and its relation to shoulder pain. J Am Acad Orthop Surg. 2003;11:142-151.
4. Kibler WB. The role of the scapula in athletic shoulder function. Am J Sports Med. 1998;26:325-337.
5. Vanermen B, Aertgeerts M, Hoogmartens M, et al. The syndrome of Parsonage and Turner. Discussion of clinical features with a review of 8 cases. Acta Orthop Belg. 1991;57:414-419.
6. Misamore GW, Lehman DE. Parsonage-Turner syndrome (acute brachial neuritis). J Bone Joint Surg. 1996;78:1405-1408.
7. Stevenson J, Trojian T. Evaluation of shoulder pain. J Fam Pract. 2002;51:605-611.
8. Miller JD, Pruitt RN, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
9. Eubanks JD. Cervical radiculopathy: nonoperative management of neck pain and radicular symptoms. Am Fam Physician. 2010;81:33-40.
10. Malanga GA, Landes P, Nadler SF. Provocative tests in cervical spine examination: historical basis and scientific analyses. Pain Physician. 2003;6:199-205.
11. Huston M, Ellis R. eds. Textbook of Musculoskeletal Medicine. Oxford, UK: Oxford University Press; 2005.
12. Mahaffey BL, Smith PA. Shoulder instability in young athletes. Am Fam Physician. 1999;59:2773-2782, 2787.
13. Petron DJ, Khan U. The shoulder and upper extremity. In: McKeag DB, Moeller JL. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Wolters Kluwer, Lippincott Williams & Wilkins; 2007:359–373.
14. Woodward TW, Best TM. The painful shoulder: part I. clinical evaluation. Am Fam Physician. 2000;61:3079-3088.
15. Kelly BT, Kadrmas WR, Speer KP. The manual muscle examination for rotator cuff strength: an electromyographic investigation. Am J Sports Med. 1996;24:581-588.
16. Murrell GA, Walton JR. Diagnosis of rotator cuff tears. Lancet. 2001;357:769-770.
17. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3:347-352.
18. Neer CS, Welsh RP. The shoulder in sports. Orthop Clin North Am. 1977;8:583-591.
19. Hawkins RJ, Kennedy JC. Impingement syndrome in athletics. Am J Sports Med. 1980;8:151-163.
20. Itoi E, Kido T, Sano A, et al. Which is more useful, the “full can test” or the “empty can test,” in detecting the torn supraspinatus tendon? Am J Sports Med. 1999;27:65-68.
21. Boettcher CE, Ginn KA, Cathers I. The ‘empty can’ and ‘full can’ tests do not selectively activate supraspinatus. J Sci Med Sport. 2009;12:435-439.
Shoulder pain is a common reason for visits to primary care physicians, who are most likely to diagnose it as rotator cuff tendinitis1,2—often erroneously. The complexity of the joint and the overlapping pathologies that may present as shoulder pain highlight the need to take a closer look when dealing with this diagnostic challenge.
Often, a targeted medical history—including the mechanism of injury and pain-provoking and pain-relieving factors—and a problem-based physical examination (incorporating many of the maneuvers highlighted in the text and tables that follow) will lead to an accurate diagnosis without the need for imaging studies. We recommend that imaging be reserved for patients who don’t respond to conventional treatments, cases in which the diagnosis is in doubt, and instances in which surgical intervention is being considered.
The 3 cases* that follow, and the take-away message incorporated in each, will give you an opportunity to test—and to hone—your shoulder pain diagnostic skill.
CASE 1 The history: Jesse, a 17-year-old student who’s active in football and track, came in during track season complaining of severe left shoulder pain. He denied any traumatic event or previous injury to the shoulder, but reported that any motion involving the shoulder caused pain. It hurt at night, the patient said, when he lay on his left side.
The physical: No muscle atrophy, redness, or swelling was evident, nor was there any indication of asymmetry or ecchymosis in the affected area. Jesse’s neck range of motion was normal; he had a very hard time with any active motion of the shoulder, however, because of the pain.
Evaluation of scapular motion demonstrated scapular dyskinesis3,4 without winging. Passive motion of the glenohumeral joint was much better than active motion. Strength testing appeared to be grossly intact but was limited by the pain. Shoulder impingement testing was positive. Sensation and deep tendon reflexes were intact.
Patients' names have been changed to protect their privacy
What’s the diagnosis?
Subacromial bursitis, suggested by the patient’s pain and altered scapular motion, was our working diagnosis, and we administered a subacromial injection of corticosteroid with lidocaine, for diagnostic as well as therapeutic purposes. Reexamination after the injection revealed immediate partial improvement in resting pain, range of motion, and strength. We referred Jesse to physical therapy with a focus on scapular stabilization and rotator cuff strength.
Three months later, Jesse returned to our office, complaining of weakness in his left shoulder. The pain had subsided a week after his first appointment, so he’d never gone to physical therapy. The weakness, which he had first noticed about 2 months after starting a lifting program in preparation for football season, was limited to resistance exercises, especially overhead shoulder presses and bench press. There were no other changes in his history, and he reported no reinjuries.
Physical examination revealed atrophy of the supraspinatus and infraspinatus muscles (FIGURE 1) and external rotation and shoulder abduction (in the scapular plane) resistance tests revealed weakness of these muscles. There was no scapular winging. The cervical spine exam was normal, and neurovascular status was intact in both upper extremities.
FIGURE 1
Severe shoulder pain, followed by weakness
Physical examination reveals atrophy of the patient’s supraspinatus (^) and infraspinatus (+) muscles.
New evidence points to nerve injury. Based on Jesse’s current history and physical, nerve injury was our new working diagnosis. (We considered the possibility of a rotator cuff tear, but this was not corroborated by the history.)
We ordered an electromyogram/nerve conduction velocity study to localize the lesion. The test revealed a brachial plexitis/neuritis (also known as Parsonage-Turner syndrome or brachial amyotrophy). The etiology of most atraumatic brachial plexopathies is unknown, and most are thought to be viral or autoimmune in nature.5,6
A classic case of Parsonage-Turner syndrome. The typical presentation of Parsonage-Turner syndrome (like Jesse’s) is one of acute, intense shoulder pain for no known reason. After 1 to 3 weeks, the pain resolves and the patient is left with weakness, usually of the supraspinatus and infraspinatus muscles. The weakness typically resolves with time, but full resolution may take 6 to 9 months.5,6 (In Jesse’s case, it took about 6 months.)
The take-away message: Look beyond the shoulder
As this case illustrates, not all shoulder pain originates in the shoulder. When evaluating shoulder pain, it is essential to consider other causes. The differential diagnosis for shoulder pain includes cervical spine disorders, cholecystitis (right shoulder), diaphragmatic irritation (eg, in the case of splenic rupture, usually involving the left shoulder), cardiac disease, and thoracic outlet syndrome.7
Evaluation of the cervical spine should be part of a complete shoulder examination. It is vital to follow a systematic approach that carefully assesses the cervical region for the possibility of nerve root impingement and radicular dysfunction masquerading as a primary shoulder disorder. (TABLE 18,9 details pain and sensory distribution patterns, reflex involvement, and potential motor impairments associated with various spinal nerve root levels.)
TABLE 1
Assessing the cervical spine
| Nerve root | Pain distribution | Sensory distribution | Reflex changes | Motor involvement |
|---|---|---|---|---|
| C5 | Lateral neck/upper trapezius | Lateral arm | Biceps | Deltoid, biceps |
| C6 | Base of neck/upper trapezius to superior glenohumeral joint | Radial aspect of distal forearm, thenar eminence, and index finger | Brachioradialis | Biceps, extensor carpi radialis longus and brevis (wrist extension) |
| C7 | Base of neck, almost entire upper quadrant of the back | Third finger | Triceps | Triceps, wrist flexion, finger extension |
| C8 | No shoulder pain | 4th and 5th fingers, distal half of forearm (ulna side) | None | Finger flexion (grip strength) |
| Adapted from: Miller JD, et al. Am Fam Physician. 20008; Eubanks JD. Am Fam Physician. 2010.9 | ||||
Practitioners should develop their own approach to “clearing the neck.” A logical order is to note posture of the head/neck/shoulders, observe active motion, perform palpation and provocative tests, and then assess neurologic function with sensation/reflex/strength testing. Provocative tests that can help to identify cervical involvement relating to shoulder pain include Spurling’s maneuver, axial compression test, abduction relief sign, and Lhermitte’s sign.10,11
CASE 2 The history: Mark, a 17-year-old, right-handed volleyball player, presented with right shoulder pain, which he felt whenever he spiked or served the ball. The pain started last season, Mark said, diminished during the months when he wasn’t playing, then got progressively worse as his activity level increased. The pain was in the posterior aspect of the shoulder.
The physical: Physical examination revealed a well-developed, but thin (6’4”, 170 pounds) young man who was not in distress. The general examination was benign, and a joint-specific exam showed no asymmetry or atrophy on inspection and no tenderness to palpation over the posterior and anterior soft tissues of the right shoulder. Rotator cuff testing yielded intact strength for all 4 muscles, but external rotation and supraspinatus testing elicited pain. The crank test, drawer sign, load and shift test, relocation test, and sulcus sign, detailed in TABLE 2,12-14 were all positive for shoulder instability; the Clunk and O’Brien tests were negative, and the contralateral shoulder exam was within normal limits. General joint laxity was observed, with the ability to oppose the thumb to the volar forearm and hyperextension noted in both elbows and knees. There were no outward signs of connective tissue disease.
Because of the chronicity of Mark’s pain and the progressive nature of his symptoms, we ordered radiographs, including anterior-posterior, lateral axillary, and scapular Y views. These films showed a nearly skeletally mature male without bony abnormalities; the humeral head was well located in the glenoid.
TABLE 2
Testing for shoulder instability12-14
| Test | Procedure | Positive result/implication |
|---|---|---|
| Apprehension | Patient supine, arm abducted 90º, externally rotated with anteriorly directed force applied to humeral head | Pain/apprehension with force suggests anterior instability |
| Relocation* | Patient supine, posteriorly directed force applied to humeral head | Relief with force suggests anterior instability |
| Crank | Patient sitting, arm abducted 90º, elbow flexed to 90º, humerus supported with forced external rotation | Pain/apprehension with forced external rotation suggests anterior instability |
| Load and shift | Patient supine, arm held by examiner and abducted 90º, force applied along axis of humerus to "seat" the humerus within the glenoid, followed by anterior force directed to humeral head | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Drawer | Patient sitting, arm at side, proximal humeral shaft grasped by examiner, seating the humeral head within the glenoid then applying anterior translational force | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Sulcus | Patient sitting, arm at side, forearm grasped by examiner with an inferior/caudally directed force applied | Sulcus or depression seen inferior to acromion as humeral head subluxes posteriorly, pathognomonic for multidirectional instability |
| Clunk | Patient supine, examiner grasps at forearm and humeral shaft, with humeral head seated within the fossa, taking the arm through passive ROM from extension through forward flexion | Clunk sound or clicking sensation suggests labral tear |
| O’Brien | Patient sitting, arm is forward flexed to 90º and fully adducted and internally rotated; patient resists downward motion. If pain is elicited, the maneuver is repeated in external rotation | Pain elicited with resisted downward motion in internal rotation but relieved with external rotation suggests labral pathology |
| *Perform only if apprehension test is positive. ROM, range of motion | ||
What’s the diagnosis?
Multidirectional instability with recurrent subluxations and probable acute rotator cuff tendinitis was our provisional diagnosis. Treatment focused on physical therapy, with a concentration on scapular stabilization and rotator cuff strengthening.
Shoulder instability is relatively common and represents a spectrum of disorders ranging from dislocation to subluxation to simple laxity.12,13 A complete loss of humeral articulation within the glenoid fossa is evidence of dislocation, whereas subluxation includes approximation of the humeral head to the limits of the glenoid rim. Dislocation typically results from trauma, whereas subluxation can be the result of microtrauma and repetitive overuse injury. Anterior instability is the most common type and is reported in as many as 95% of all dislocations.13
The take-away message: Rule out instability
The shoulder is one of the most complex joints in the body. The rotator cuff structures, the glenoid labrum, and the collective capsular ligaments provide structural stability to the glenohumeral joint.12,13 The shoulder is vulnerable to instability because the shallow glenoid fossa offers little bony support for the humeral head. Thus, instability should always be included in an assessment of shoulder pain.
Key factors to consider in identifying shoulder instability include the location of the pain, the direction of traumatic force applied, the presence of a known complete dislocation vs apprehension with specific movement, the position of the arm in which pain is elicited, a previous occurrence of instability (subluxation or dislocation), and the presence of tingling or numbness.12-14 The maneuvers detailed in TABLE 212-14 can help identify instability, as they did in this case. Patients with hypermobility are at increased risk for shoulder instability, so a targeted exam and patient history aimed at identifying signs and symptoms of hypermobility is needed, as well.
Ask the patient to attempt to:
- bend the thumb to the volar forearm
- place hands to the floor with hyperextended knees
- perform maximal hyperextension of the fifth metacarpophalangeal joint (>90° is a positive result).
Findings from the medical history that indicate a predisposition to instability include generalized joint laxity, Ehlers-Danlos syndrome, Marfan syndrome, osteogenesis imperfecta, hyperhomocysteinuria, hyperlysinemia, benign joint hypermobility syndrome, juvenile rheumatoid arthritis, and previous shoulder or patellar dislocations.
Imaging tips: Scapular Y and/or axillary lateral views should always be included when ordering imaging studies for suspected instability/dislocations, as 50% of posterior dislocations are missed on standard shoulder x-rays that do not include them.12 In reviewing the x-rays, it is important to look for signs of a compression fracture of the posterior humeral head (known as a Hill-Sachs lesion) for anterior shoulder dislocations, and fractures to the anterior glenoid rim (known as a Bankart lesion).12-14
CASE 3 The history: Robert, a right-handed, 50-year-old motorcycle instructor, came to our office because of chronic right shoulder pain. The pain, located over the anterior portion of the glenohumeral joint, developed insidiously about 3 or 4 years ago, the patient reported. He had finally decided to seek help because he’d recently experienced an acute exacerbation of pain brought on by shoveling snow, after which he also noticed associated weakness, a clicking/popping on active motion, and mild loss of motion.
The physical: Robert’s cervical spine exam was unremarkable. He demonstrated full active range of motion (ROM) without exacerbation of right shoulder symptoms, and special tests for disc pathology at the neck were negative. Active ROM testing of the right shoulder revealed full abduction, with only minimal pain; full flexion, with moderate pain noted initially at 49°; full extension, with a painful arc noted at 50°; and full horizontal adduction, with a painful arc noted at the halfway point. The testing also revealed that his right thumb was 3 inches lower than the left on reaching for the opposite scapula. At the superior aspect of the acromioclavicular (AC) joint, 2+ tenderness was noted; 3+ tenderness was noted at the greater tubercle of the humerus.
After inspecting the shoulder region for alterations in bony landmarks, muscle bulk, carrying position, and movement characteristics, palpation of the region was performed.
When assessing shoulder strength, there are a variety of tests for each functioning component of the rotator cuff structures (TABLE 3).15-17 Manual muscle tests revealed: 4-/5 on external rotation (French horn test), 3+/5 on the lift-off test, and 5-/5 on all other tests for right shoulder function. Impingement testing was slightly positive, or pain producing, on Hawkins and Neer tests.18,19 For the Hawkins test, the examiner flexes the arm to 90° of shoulder flexion with the elbow flexed at 90°, then internally rotates the shoulder. For the Neer test, the arm is fully elevated in the scapular plane and internally rotated by the examiner.
The subscapularis muscle, which functions primarily in internal rotation, is tested by the French horn and lift-off tests. The teres minor muscle, which performs external rotation, is tested by the French horn test of external rotation. And the supraspinatus muscle, which performs abduction and external rotation, is tested by the empty can (also known as the Jobe) and full can tests. Some researchers suggest that the empty can test is better for diagnosing impingement, based on evidence showing that the full can test is better at diagnosing supraspinatus tears because it causes less pain during testing.20
TABLE 3
Suspect rotator cuff involvement?7,15-17
What’s the diagnosis?
Rotator cuff tear was suspected because Robert had positive elements of the “rotator cuff triad”—supraspinatus weakness (as indicated by a positive empty can test), external rotation weakness (revealed by the French horn test), and a positive Hawkins impingement test. We ordered diagnostic studies, including plain radiographs, which revealed degenerative changes at the acromioclavicular joint, decreased acromiohumeral interval, and no significant changes at the glenohumeral joint (FIGURE 2), and magnetic resonance imaging (MRI) of the right shoulder. The MRI revealed a large, full-thickness rotator cuff tear of the supraspinatus tendon with retraction. A torn and retracted biceps tendon and AC joint osteoarthritis were also shown, likely causing a mass effect on the supraspinatus. The patient underwent surgery to repair the torn rotator cuff, with excellent results.
FIGURE 2
Chronic right shoulder pain
An AP view of the patient’s right shoulder shows acromioclavicular joint narrowing and degeneration and subtle narrowing of the acromiohumeral interval.
The take-away message: Keep the rotator-cuff triad in mind
Because none of the tests that comprise the triad is specific enough alone to diagnose a rotator cuff tear,15,20,21 Murrell and Walton16 suggested that the 3 tests be considered together for diagnostic purposes. If all 3 are positive, there is a 98% chance of a rotator cuff tear; if 2 tests are positive and the patient is older than 60 years, the findings are suggestive of a tear; and if all 3 tests (plus the drop arm test) are negative, there is less than a 5% chance of a major rotator cuff tear.16
CORRESPONDENCE Nilesh Shah, MD, Summa Center for Sports Health, 20 Olive Street, Suite 201, Akron, OH 44310; [email protected]
Shoulder pain is a common reason for visits to primary care physicians, who are most likely to diagnose it as rotator cuff tendinitis1,2—often erroneously. The complexity of the joint and the overlapping pathologies that may present as shoulder pain highlight the need to take a closer look when dealing with this diagnostic challenge.
Often, a targeted medical history—including the mechanism of injury and pain-provoking and pain-relieving factors—and a problem-based physical examination (incorporating many of the maneuvers highlighted in the text and tables that follow) will lead to an accurate diagnosis without the need for imaging studies. We recommend that imaging be reserved for patients who don’t respond to conventional treatments, cases in which the diagnosis is in doubt, and instances in which surgical intervention is being considered.
The 3 cases* that follow, and the take-away message incorporated in each, will give you an opportunity to test—and to hone—your shoulder pain diagnostic skill.
CASE 1 The history: Jesse, a 17-year-old student who’s active in football and track, came in during track season complaining of severe left shoulder pain. He denied any traumatic event or previous injury to the shoulder, but reported that any motion involving the shoulder caused pain. It hurt at night, the patient said, when he lay on his left side.
The physical: No muscle atrophy, redness, or swelling was evident, nor was there any indication of asymmetry or ecchymosis in the affected area. Jesse’s neck range of motion was normal; he had a very hard time with any active motion of the shoulder, however, because of the pain.
Evaluation of scapular motion demonstrated scapular dyskinesis3,4 without winging. Passive motion of the glenohumeral joint was much better than active motion. Strength testing appeared to be grossly intact but was limited by the pain. Shoulder impingement testing was positive. Sensation and deep tendon reflexes were intact.
Patients' names have been changed to protect their privacy
What’s the diagnosis?
Subacromial bursitis, suggested by the patient’s pain and altered scapular motion, was our working diagnosis, and we administered a subacromial injection of corticosteroid with lidocaine, for diagnostic as well as therapeutic purposes. Reexamination after the injection revealed immediate partial improvement in resting pain, range of motion, and strength. We referred Jesse to physical therapy with a focus on scapular stabilization and rotator cuff strength.
Three months later, Jesse returned to our office, complaining of weakness in his left shoulder. The pain had subsided a week after his first appointment, so he’d never gone to physical therapy. The weakness, which he had first noticed about 2 months after starting a lifting program in preparation for football season, was limited to resistance exercises, especially overhead shoulder presses and bench press. There were no other changes in his history, and he reported no reinjuries.
Physical examination revealed atrophy of the supraspinatus and infraspinatus muscles (FIGURE 1) and external rotation and shoulder abduction (in the scapular plane) resistance tests revealed weakness of these muscles. There was no scapular winging. The cervical spine exam was normal, and neurovascular status was intact in both upper extremities.
FIGURE 1
Severe shoulder pain, followed by weakness
Physical examination reveals atrophy of the patient’s supraspinatus (^) and infraspinatus (+) muscles.
New evidence points to nerve injury. Based on Jesse’s current history and physical, nerve injury was our new working diagnosis. (We considered the possibility of a rotator cuff tear, but this was not corroborated by the history.)
We ordered an electromyogram/nerve conduction velocity study to localize the lesion. The test revealed a brachial plexitis/neuritis (also known as Parsonage-Turner syndrome or brachial amyotrophy). The etiology of most atraumatic brachial plexopathies is unknown, and most are thought to be viral or autoimmune in nature.5,6
A classic case of Parsonage-Turner syndrome. The typical presentation of Parsonage-Turner syndrome (like Jesse’s) is one of acute, intense shoulder pain for no known reason. After 1 to 3 weeks, the pain resolves and the patient is left with weakness, usually of the supraspinatus and infraspinatus muscles. The weakness typically resolves with time, but full resolution may take 6 to 9 months.5,6 (In Jesse’s case, it took about 6 months.)
The take-away message: Look beyond the shoulder
As this case illustrates, not all shoulder pain originates in the shoulder. When evaluating shoulder pain, it is essential to consider other causes. The differential diagnosis for shoulder pain includes cervical spine disorders, cholecystitis (right shoulder), diaphragmatic irritation (eg, in the case of splenic rupture, usually involving the left shoulder), cardiac disease, and thoracic outlet syndrome.7
Evaluation of the cervical spine should be part of a complete shoulder examination. It is vital to follow a systematic approach that carefully assesses the cervical region for the possibility of nerve root impingement and radicular dysfunction masquerading as a primary shoulder disorder. (TABLE 18,9 details pain and sensory distribution patterns, reflex involvement, and potential motor impairments associated with various spinal nerve root levels.)
TABLE 1
Assessing the cervical spine
| Nerve root | Pain distribution | Sensory distribution | Reflex changes | Motor involvement |
|---|---|---|---|---|
| C5 | Lateral neck/upper trapezius | Lateral arm | Biceps | Deltoid, biceps |
| C6 | Base of neck/upper trapezius to superior glenohumeral joint | Radial aspect of distal forearm, thenar eminence, and index finger | Brachioradialis | Biceps, extensor carpi radialis longus and brevis (wrist extension) |
| C7 | Base of neck, almost entire upper quadrant of the back | Third finger | Triceps | Triceps, wrist flexion, finger extension |
| C8 | No shoulder pain | 4th and 5th fingers, distal half of forearm (ulna side) | None | Finger flexion (grip strength) |
| Adapted from: Miller JD, et al. Am Fam Physician. 20008; Eubanks JD. Am Fam Physician. 2010.9 | ||||
Practitioners should develop their own approach to “clearing the neck.” A logical order is to note posture of the head/neck/shoulders, observe active motion, perform palpation and provocative tests, and then assess neurologic function with sensation/reflex/strength testing. Provocative tests that can help to identify cervical involvement relating to shoulder pain include Spurling’s maneuver, axial compression test, abduction relief sign, and Lhermitte’s sign.10,11
CASE 2 The history: Mark, a 17-year-old, right-handed volleyball player, presented with right shoulder pain, which he felt whenever he spiked or served the ball. The pain started last season, Mark said, diminished during the months when he wasn’t playing, then got progressively worse as his activity level increased. The pain was in the posterior aspect of the shoulder.
The physical: Physical examination revealed a well-developed, but thin (6’4”, 170 pounds) young man who was not in distress. The general examination was benign, and a joint-specific exam showed no asymmetry or atrophy on inspection and no tenderness to palpation over the posterior and anterior soft tissues of the right shoulder. Rotator cuff testing yielded intact strength for all 4 muscles, but external rotation and supraspinatus testing elicited pain. The crank test, drawer sign, load and shift test, relocation test, and sulcus sign, detailed in TABLE 2,12-14 were all positive for shoulder instability; the Clunk and O’Brien tests were negative, and the contralateral shoulder exam was within normal limits. General joint laxity was observed, with the ability to oppose the thumb to the volar forearm and hyperextension noted in both elbows and knees. There were no outward signs of connective tissue disease.
Because of the chronicity of Mark’s pain and the progressive nature of his symptoms, we ordered radiographs, including anterior-posterior, lateral axillary, and scapular Y views. These films showed a nearly skeletally mature male without bony abnormalities; the humeral head was well located in the glenoid.
TABLE 2
Testing for shoulder instability12-14
| Test | Procedure | Positive result/implication |
|---|---|---|
| Apprehension | Patient supine, arm abducted 90º, externally rotated with anteriorly directed force applied to humeral head | Pain/apprehension with force suggests anterior instability |
| Relocation* | Patient supine, posteriorly directed force applied to humeral head | Relief with force suggests anterior instability |
| Crank | Patient sitting, arm abducted 90º, elbow flexed to 90º, humerus supported with forced external rotation | Pain/apprehension with forced external rotation suggests anterior instability |
| Load and shift | Patient supine, arm held by examiner and abducted 90º, force applied along axis of humerus to "seat" the humerus within the glenoid, followed by anterior force directed to humeral head | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Drawer | Patient sitting, arm at side, proximal humeral shaft grasped by examiner, seating the humeral head within the glenoid then applying anterior translational force | Pain and appreciable translation felt with anterior force suggest anterior instability |
| Sulcus | Patient sitting, arm at side, forearm grasped by examiner with an inferior/caudally directed force applied | Sulcus or depression seen inferior to acromion as humeral head subluxes posteriorly, pathognomonic for multidirectional instability |
| Clunk | Patient supine, examiner grasps at forearm and humeral shaft, with humeral head seated within the fossa, taking the arm through passive ROM from extension through forward flexion | Clunk sound or clicking sensation suggests labral tear |
| O’Brien | Patient sitting, arm is forward flexed to 90º and fully adducted and internally rotated; patient resists downward motion. If pain is elicited, the maneuver is repeated in external rotation | Pain elicited with resisted downward motion in internal rotation but relieved with external rotation suggests labral pathology |
| *Perform only if apprehension test is positive. ROM, range of motion | ||
What’s the diagnosis?
Multidirectional instability with recurrent subluxations and probable acute rotator cuff tendinitis was our provisional diagnosis. Treatment focused on physical therapy, with a concentration on scapular stabilization and rotator cuff strengthening.
Shoulder instability is relatively common and represents a spectrum of disorders ranging from dislocation to subluxation to simple laxity.12,13 A complete loss of humeral articulation within the glenoid fossa is evidence of dislocation, whereas subluxation includes approximation of the humeral head to the limits of the glenoid rim. Dislocation typically results from trauma, whereas subluxation can be the result of microtrauma and repetitive overuse injury. Anterior instability is the most common type and is reported in as many as 95% of all dislocations.13
The take-away message: Rule out instability
The shoulder is one of the most complex joints in the body. The rotator cuff structures, the glenoid labrum, and the collective capsular ligaments provide structural stability to the glenohumeral joint.12,13 The shoulder is vulnerable to instability because the shallow glenoid fossa offers little bony support for the humeral head. Thus, instability should always be included in an assessment of shoulder pain.
Key factors to consider in identifying shoulder instability include the location of the pain, the direction of traumatic force applied, the presence of a known complete dislocation vs apprehension with specific movement, the position of the arm in which pain is elicited, a previous occurrence of instability (subluxation or dislocation), and the presence of tingling or numbness.12-14 The maneuvers detailed in TABLE 212-14 can help identify instability, as they did in this case. Patients with hypermobility are at increased risk for shoulder instability, so a targeted exam and patient history aimed at identifying signs and symptoms of hypermobility is needed, as well.
Ask the patient to attempt to:
- bend the thumb to the volar forearm
- place hands to the floor with hyperextended knees
- perform maximal hyperextension of the fifth metacarpophalangeal joint (>90° is a positive result).
Findings from the medical history that indicate a predisposition to instability include generalized joint laxity, Ehlers-Danlos syndrome, Marfan syndrome, osteogenesis imperfecta, hyperhomocysteinuria, hyperlysinemia, benign joint hypermobility syndrome, juvenile rheumatoid arthritis, and previous shoulder or patellar dislocations.
Imaging tips: Scapular Y and/or axillary lateral views should always be included when ordering imaging studies for suspected instability/dislocations, as 50% of posterior dislocations are missed on standard shoulder x-rays that do not include them.12 In reviewing the x-rays, it is important to look for signs of a compression fracture of the posterior humeral head (known as a Hill-Sachs lesion) for anterior shoulder dislocations, and fractures to the anterior glenoid rim (known as a Bankart lesion).12-14
CASE 3 The history: Robert, a right-handed, 50-year-old motorcycle instructor, came to our office because of chronic right shoulder pain. The pain, located over the anterior portion of the glenohumeral joint, developed insidiously about 3 or 4 years ago, the patient reported. He had finally decided to seek help because he’d recently experienced an acute exacerbation of pain brought on by shoveling snow, after which he also noticed associated weakness, a clicking/popping on active motion, and mild loss of motion.
The physical: Robert’s cervical spine exam was unremarkable. He demonstrated full active range of motion (ROM) without exacerbation of right shoulder symptoms, and special tests for disc pathology at the neck were negative. Active ROM testing of the right shoulder revealed full abduction, with only minimal pain; full flexion, with moderate pain noted initially at 49°; full extension, with a painful arc noted at 50°; and full horizontal adduction, with a painful arc noted at the halfway point. The testing also revealed that his right thumb was 3 inches lower than the left on reaching for the opposite scapula. At the superior aspect of the acromioclavicular (AC) joint, 2+ tenderness was noted; 3+ tenderness was noted at the greater tubercle of the humerus.
After inspecting the shoulder region for alterations in bony landmarks, muscle bulk, carrying position, and movement characteristics, palpation of the region was performed.
When assessing shoulder strength, there are a variety of tests for each functioning component of the rotator cuff structures (TABLE 3).15-17 Manual muscle tests revealed: 4-/5 on external rotation (French horn test), 3+/5 on the lift-off test, and 5-/5 on all other tests for right shoulder function. Impingement testing was slightly positive, or pain producing, on Hawkins and Neer tests.18,19 For the Hawkins test, the examiner flexes the arm to 90° of shoulder flexion with the elbow flexed at 90°, then internally rotates the shoulder. For the Neer test, the arm is fully elevated in the scapular plane and internally rotated by the examiner.
The subscapularis muscle, which functions primarily in internal rotation, is tested by the French horn and lift-off tests. The teres minor muscle, which performs external rotation, is tested by the French horn test of external rotation. And the supraspinatus muscle, which performs abduction and external rotation, is tested by the empty can (also known as the Jobe) and full can tests. Some researchers suggest that the empty can test is better for diagnosing impingement, based on evidence showing that the full can test is better at diagnosing supraspinatus tears because it causes less pain during testing.20
TABLE 3
Suspect rotator cuff involvement?7,15-17
What’s the diagnosis?
Rotator cuff tear was suspected because Robert had positive elements of the “rotator cuff triad”—supraspinatus weakness (as indicated by a positive empty can test), external rotation weakness (revealed by the French horn test), and a positive Hawkins impingement test. We ordered diagnostic studies, including plain radiographs, which revealed degenerative changes at the acromioclavicular joint, decreased acromiohumeral interval, and no significant changes at the glenohumeral joint (FIGURE 2), and magnetic resonance imaging (MRI) of the right shoulder. The MRI revealed a large, full-thickness rotator cuff tear of the supraspinatus tendon with retraction. A torn and retracted biceps tendon and AC joint osteoarthritis were also shown, likely causing a mass effect on the supraspinatus. The patient underwent surgery to repair the torn rotator cuff, with excellent results.
FIGURE 2
Chronic right shoulder pain
An AP view of the patient’s right shoulder shows acromioclavicular joint narrowing and degeneration and subtle narrowing of the acromiohumeral interval.
The take-away message: Keep the rotator-cuff triad in mind
Because none of the tests that comprise the triad is specific enough alone to diagnose a rotator cuff tear,15,20,21 Murrell and Walton16 suggested that the 3 tests be considered together for diagnostic purposes. If all 3 are positive, there is a 98% chance of a rotator cuff tear; if 2 tests are positive and the patient is older than 60 years, the findings are suggestive of a tear; and if all 3 tests (plus the drop arm test) are negative, there is less than a 5% chance of a major rotator cuff tear.16
CORRESPONDENCE Nilesh Shah, MD, Summa Center for Sports Health, 20 Olive Street, Suite 201, Akron, OH 44310; [email protected]
1. Van der Windt DA, Koes BW, De Jong BA, et al. Shoulder disorders in general practice: incidence, patient characteristics, and management. Ann Rheum Dis. 1995;54:959-964.
2. Johansson K, Adolfsson L, Foldevi M. Attitudes toward management of patients with subacromial pain in Swedish primary care. Fam Pract. 1999;16:233-237.
3. Kibler WB, McMullen J. Scapular dyskinesis and its relation to shoulder pain. J Am Acad Orthop Surg. 2003;11:142-151.
4. Kibler WB. The role of the scapula in athletic shoulder function. Am J Sports Med. 1998;26:325-337.
5. Vanermen B, Aertgeerts M, Hoogmartens M, et al. The syndrome of Parsonage and Turner. Discussion of clinical features with a review of 8 cases. Acta Orthop Belg. 1991;57:414-419.
6. Misamore GW, Lehman DE. Parsonage-Turner syndrome (acute brachial neuritis). J Bone Joint Surg. 1996;78:1405-1408.
7. Stevenson J, Trojian T. Evaluation of shoulder pain. J Fam Pract. 2002;51:605-611.
8. Miller JD, Pruitt RN, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
9. Eubanks JD. Cervical radiculopathy: nonoperative management of neck pain and radicular symptoms. Am Fam Physician. 2010;81:33-40.
10. Malanga GA, Landes P, Nadler SF. Provocative tests in cervical spine examination: historical basis and scientific analyses. Pain Physician. 2003;6:199-205.
11. Huston M, Ellis R. eds. Textbook of Musculoskeletal Medicine. Oxford, UK: Oxford University Press; 2005.
12. Mahaffey BL, Smith PA. Shoulder instability in young athletes. Am Fam Physician. 1999;59:2773-2782, 2787.
13. Petron DJ, Khan U. The shoulder and upper extremity. In: McKeag DB, Moeller JL. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Wolters Kluwer, Lippincott Williams & Wilkins; 2007:359–373.
14. Woodward TW, Best TM. The painful shoulder: part I. clinical evaluation. Am Fam Physician. 2000;61:3079-3088.
15. Kelly BT, Kadrmas WR, Speer KP. The manual muscle examination for rotator cuff strength: an electromyographic investigation. Am J Sports Med. 1996;24:581-588.
16. Murrell GA, Walton JR. Diagnosis of rotator cuff tears. Lancet. 2001;357:769-770.
17. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3:347-352.
18. Neer CS, Welsh RP. The shoulder in sports. Orthop Clin North Am. 1977;8:583-591.
19. Hawkins RJ, Kennedy JC. Impingement syndrome in athletics. Am J Sports Med. 1980;8:151-163.
20. Itoi E, Kido T, Sano A, et al. Which is more useful, the “full can test” or the “empty can test,” in detecting the torn supraspinatus tendon? Am J Sports Med. 1999;27:65-68.
21. Boettcher CE, Ginn KA, Cathers I. The ‘empty can’ and ‘full can’ tests do not selectively activate supraspinatus. J Sci Med Sport. 2009;12:435-439.
1. Van der Windt DA, Koes BW, De Jong BA, et al. Shoulder disorders in general practice: incidence, patient characteristics, and management. Ann Rheum Dis. 1995;54:959-964.
2. Johansson K, Adolfsson L, Foldevi M. Attitudes toward management of patients with subacromial pain in Swedish primary care. Fam Pract. 1999;16:233-237.
3. Kibler WB, McMullen J. Scapular dyskinesis and its relation to shoulder pain. J Am Acad Orthop Surg. 2003;11:142-151.
4. Kibler WB. The role of the scapula in athletic shoulder function. Am J Sports Med. 1998;26:325-337.
5. Vanermen B, Aertgeerts M, Hoogmartens M, et al. The syndrome of Parsonage and Turner. Discussion of clinical features with a review of 8 cases. Acta Orthop Belg. 1991;57:414-419.
6. Misamore GW, Lehman DE. Parsonage-Turner syndrome (acute brachial neuritis). J Bone Joint Surg. 1996;78:1405-1408.
7. Stevenson J, Trojian T. Evaluation of shoulder pain. J Fam Pract. 2002;51:605-611.
8. Miller JD, Pruitt RN, McDonald TJ. Acute brachial plexus neuritis: an uncommon cause of shoulder pain. Am Fam Physician. 2000;62:2067-2072.
9. Eubanks JD. Cervical radiculopathy: nonoperative management of neck pain and radicular symptoms. Am Fam Physician. 2010;81:33-40.
10. Malanga GA, Landes P, Nadler SF. Provocative tests in cervical spine examination: historical basis and scientific analyses. Pain Physician. 2003;6:199-205.
11. Huston M, Ellis R. eds. Textbook of Musculoskeletal Medicine. Oxford, UK: Oxford University Press; 2005.
12. Mahaffey BL, Smith PA. Shoulder instability in young athletes. Am Fam Physician. 1999;59:2773-2782, 2787.
13. Petron DJ, Khan U. The shoulder and upper extremity. In: McKeag DB, Moeller JL. ACSM’s Primary Care Sports Medicine. 2nd ed. Philadelphia, Pa: Wolters Kluwer, Lippincott Williams & Wilkins; 2007:359–373.
14. Woodward TW, Best TM. The painful shoulder: part I. clinical evaluation. Am Fam Physician. 2000;61:3079-3088.
15. Kelly BT, Kadrmas WR, Speer KP. The manual muscle examination for rotator cuff strength: an electromyographic investigation. Am J Sports Med. 1996;24:581-588.
16. Murrell GA, Walton JR. Diagnosis of rotator cuff tears. Lancet. 2001;357:769-770.
17. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3:347-352.
18. Neer CS, Welsh RP. The shoulder in sports. Orthop Clin North Am. 1977;8:583-591.
19. Hawkins RJ, Kennedy JC. Impingement syndrome in athletics. Am J Sports Med. 1980;8:151-163.
20. Itoi E, Kido T, Sano A, et al. Which is more useful, the “full can test” or the “empty can test,” in detecting the torn supraspinatus tendon? Am J Sports Med. 1999;27:65-68.
21. Boettcher CE, Ginn KA, Cathers I. The ‘empty can’ and ‘full can’ tests do not selectively activate supraspinatus. J Sci Med Sport. 2009;12:435-439.
Diabetes and alcohol use: Detecting at-risk drinking
• Ask a question such as “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” to ascertain a patient’s quantity of alcohol use. A
• Apply elements of the FRAMES approach to help patients curtail at-risk drinking—eg, use elevated HbA1c levels as evidence of a need to change behavior. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
There are enough challenges in controlling diabetes mellitus without the hindrance of undetected problematic alcohol use. The good news is that asking a single nonthreatening question can help you detect at-risk drinking—defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) as 5 or more drinks on one occasion or more than 14 drinks per week for men; and 4 or more drinks on one occasion or more than 7 drinks per week for women.1,2 And, for patients who may be compromising their diabetes care and overall health through problem drinking, brief intervention techniques used in the office can enable them to reduce alcohol consumption significantly.
When alcohol becomes a problem in diabetes care
Several studies have explored the long-term benefits of moderate alcohol use on glycemic control—with mixed results. A 2007 study found that diabetes patients who drink 1 glass of wine per day exhibited a lower fasting glucose level than abstainers after 3 months.3 There was no difference, however, on postprandial glucose levels. A 2008 study found that individuals who drank one to 2 glasses of wine per day for a month had lower fasting serum insulin levels relative to when they have abstained for a month,4 although levels of fasting plasma cholesterol, HDL cholesterol, glucose, and hemoglobin A1c (HbA1c) remained unchanged relative to periods of abstinence.4
Furthermore, rates of coronary heart disease and CHD mortality in a meta-analysis were significantly lower in 3 categories of alcohol consumption (<6 g/d, 6 to <18 g/d, and ≥18 g/d) compared with abstinence.5 Nondrinkers also had a greater risk of total mortality compared with the lightest drinking group. Notably, however, the lower limit of the highest drinking category was only 1.5 drinks per day.
How big is the problem? In a study of insulin-treated patients seen for severe hypoglycemia, 17% had been drinking before the episode.6 In a primary care sample, 28% of randomly selected patients with type 2 diabetes met Diagnostic and Statistical Manual of Mental Disorders-IV criteria for a lifetime incidence of alcohol abuse and 13% met either current or lifetime criteria for alcohol dependence.7 Another study of primary care patients with diabetes8 found that 13.4% met NIAAA criteria for at-risk drinking; 11.1% of these at-risk drinkers met criteria for current alcohol dependence. (According to the NIAAA, the rate of at-risk/heavy drinking among US adults is 30%, and about one in 4 heavy drinkers meets the criteria for alcohol abuse or dependence.1)
Detrimental effects with immoderate drinking. Individuals who engage in at-risk drinking, as defined by the NIAAA, are at increased risk for alcohol dependence9 and associated complications such as diabetic neuropathy and retinopathy,10 atherosclerosis,11 and total and CHD mortality.3,12 Heavy drinking also interferes with neuroendocrine, gastrointestinal, and sexual function,13 and its interaction with diabetes increases the risk for hepatocellular carcinoma after controlling for hepatitis B and C serology.14
Interference with diabetes control. Research examining the short-term effect of alcohol use has produced contradictory results, partly due to differences among studies, such as whether alcohol is administered with a meal and whether a fasting glucose level is measured.15 However, alcohol affects glycemic control and, when used excessively, can impair glucose production.16,17 Alcohol may induce hypoglycemia,10,18 and even small amounts may jeopardize diabetes control.13 In a study of patients with insulin-treated diabetes, alcohol use in the presence of mild hypoglycemia increased diastolic blood pressure or exacerbated hypoglycemia-related cognitive deficits.19 Another concern—in both the short and long term—is that alcohol interacts negatively with certain diabetes medications. It is more likely to induce hypoglycemia in the presence of sulfonylurea medications.10 Chlorpropamide decreases the rate of ethanol elimination from the blood.20 And, in those taking metformin, excessive alcohol use elevates risk for lactic acidosis. 21
Diminished self-care. Alcohol use can interfere with self-care,22,23 which is a crucial component of diabetes treatment.24 It may lead to reduced eating16 or to decreased willingness to adhere to prescribed dietary regimens.13 It also impairs other self-care behaviors13,15,25 such as self-monitoring blood glucose and showing up for medical appointments.26 In a large, diverse sample of patients with diabetes,24 heavy drinkers had the highest rates of morbidity. Importantly, alcohol and diabetes self-care behavior were significantly negatively associated. Studies of ethnic minority samples have yielded comparable results.27
Assessing alcohol use: Obstacles and solutions
Although alcohol use can be readily evaluated during routine primary care appointments, it is often neglected, perhaps due to a lack of awareness about its impact on diabetes.15 Those who are most often assessed tend to have a psychiatric diagnosis or other condition raising a red flag for physicians.28 When internists, general practitioners, and psychiatrists were questioned in a study regarding patients’ alcohol and drug use, all 3 groups were misinformed about which substance-use treatments were empirically supported29 and did not believe that treatment for alcohol abuse held much promise. Another study showed that physicians can be reluctant to screen for alcohol use because of the difficulty in recognizing a problem, the perceived unimportance of alcohol use as a health risk, a supposed lack of adequate intervention tools, and a fear of stigmatizing patients.30 Physicians are more likely to discuss alcohol use under certain extreme conditions such as when a patient smells of alcohol.
Multiple opportunities to ask in the VA system. In the Veterans Health Administration, primary care VA providers have reported that prompts for alcohol screening embedded in computerized progress notes, clinical reminder lists, and annual health evaluation forms encourage them to assess alcohol use. Other useful materials include manual checklists and reference cards.31 These providers also report that education, feedback on rates of alcohol screening, and increased supervision facilitate assessment. Finally, providers indicate that asking nurses or clerical staff to administer the screen improves completion rates.
Ask a simple question
“How often have you had a drink containing alcohol in the last year?” or “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” are questions that can help you compare a patient’s alcohol use to the at-risk drinking cutoffs established by the NIAAA.1,2
Recent research has also validated the use of a single question in identifying NIAAA-defined at-risk drinking.32 Simply ask patients, “How many times in the past year have you had X or more drinks in a day?” (X=5 for men or 4 for women). The screen is positive when a patient acknowledges having done so at least once in the past year. This question was 81.8% sensitive and 79.3% specific for unhealthy alcohol use, and 87.9% sensitive and 66.8% specific for current alcohol abuse or dependence.32 Advantages of this method are its brevity, ease of scoring, validity in the primary care setting,32 and ease of recollection for treatment providers (TABLE 1).1,2
TABLE 1
Ask these simple questions to assess alcohol use1,2
| To assess… | Ask… |
|---|---|
| Frequency of alcohol use | “How often do you drink alcohol (including beer or wine)?” or “How often have you had a drink containing alcohol in the last year?” |
| Quantity of alcohol use | “When you do drink alcohol, how many standard* drinks do you have?” or “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” |
| Binge drinking | For men: “How often do you have 5 or more standard* drinks on one occasion?” For women: “How often do you have 4 or more standard* drinks on one occasion?” |
| Alcohol use with the NIAAA single-question screen | “How many times in the past year have you had X or more drinks in a day?” (X = 5 for men and X = 4 for women; positive response ≥1) |
| *A standard drink is equal to 12 oz. of regular beer, 8 to 9 oz. of malt liquor, 5 oz. of wine, or 1.5 oz. of 80-proof spirits.1 NIAAA, National Institute on Alcohol Abuse and Alcoholism. | |
Brief intervention works in primary care
Brief interventions for drinking have strong empirical support. In a review of treatments for alcohol abuse and dependence,33 brief intervention was one of only 2 “efficacious” treatments.
Although some individual studies of brief alcohol interventions in primary care have not shown favorable results, several systematic reviews have demonstrated the efficacy of such interventions in this setting. General practitioner–delivered brief interventions led to significantly better patient outcomes compared with standard care, and “very brief advice” resulted in reductions in alcohol consumption overall and in the percentage of “excessive drinkers.”34 In a review of health behavior interventions,35 brief interventions reduced risky or harmful drinking. In one of 2 meta-analyses that support this finding, brief interventions with primary care patients not seeking treatment for alcohol abuse yielded small-to-medium effect sizes relative to control conditions.36 In the other study, brief interventions significantly reduced longer term alcohol use in primary care patients.37
The US Preventive Services Task Force conducted a systematic review of behavioral counseling interventions and recommends screening and brief interventions for unhealthy drinking in primary care.38,39 Its findings indicate that alcohol use declines significantly after brief interventions containing at least 2 of the following elements: feedback regarding drinking, advice to reduce drinking, or goal setting.
Brief advice is a form of intervention that shows considerable promise in primary care.40-42 Two 10- to 15-minute sessions have led to significant reductions in the mean number of drinks and frequency of excessive drinking in the 7 days before a follow-up interview, as well as a reduction in binge drinking episodes in the previous 30 days.41,42
One study produced positive results with just a 5- to 10-minute counseling session involving advice for drinking goals delivered by primary care providers as part of a routine medical visit.43 This intervention led to significant decreases in alcohol use at a 6-month follow-up for high-risk drinkers compared with controls.43 Brief interventions additionally work within the time constraints of a busy primary care practice and are cost effective.
Some primary care providers think a specialist should conduct interventions and suggest that having a specialist immediately available would enable intervention.31 In fact, some research has supported the idea of special training. In a European study, primary care providers reported that more practical training, information about brief intervention studies, personal training, and lectures would facilitate interventions.44
Applying brief alcohol interventions to diabetes patients
Newer research has tested the efficacy of alcohol interventions with diabetes patients in the primary care setting. In one study,45 brief advice was given in 2 15-minute sessions and 2 5-minute follow-up telephone calls. Compared with controls, significantly more participants who received the intervention reduced heavy drinking from baseline to follow-up. One caveat is that patients with hypertension were included in the sample, making it difficult to determine the impact of the intervention on diabetes patients specifically.
In a small study of patients with diabetes exhibiting at-risk drinking,8 a single-session intervention based on motivational interviewing (MI) principles46 gave participants personalized feedback in relation to sex-based norms of drinking rates and HbA1c and triglyceride laboratory results. Patients were given information on the physiologic effects of alcohol on diabetes, the potential interactions between alcohol and diabetes medications, and the effect of alcohol on diabetes self-care behavior. They were asked to identify pros and cons of their drinking and to develop personal change goals. One of 2 PhD-level clinical psychologists trained in MI administered the single 50-minute intervention. By 1 month and continuing through to the 6-month follow-up, participants had reduced the proportion of drinking days, mean number of daily drinks, and proportion of heavy drinking days.
Ramsey and colleagues47 extended this work by comparatively examining a group of patients exhibiting at-risk drinking who received no intervention. The results favored the intervention group, with a medium-to-large effect size for the proportion of drinking days, a medium effect size for the reduction of mean number of daily drinks, and a small-to-medium effect size in the reduction of heavy drinking days. Furthermore, in the intervention group there was a trend toward better diabetes adherence behavior.
Implementing brief intervention in practice. Despite differences among interventions, the elements of brief interventions tend to be similar.48 Incorporating these elements in the primary care setting provides a useful framework that will likely prove beneficial. Specifically, brief interventions typically contain elements of the FRAMES (TABLE 2)46 acronym:
- Feedback about one’s drinking relative to others
- Responsibility for deciding to change
- Advice to change drinking
- Menu of options for implementing a change strategy
- Empathic listening
- Self-efficacy enhancement.
Decision-making models indicate that expectations about the effects of behavior change play a significant role in determining whether a decision to change is made.49 The perceived costs and benefits of changing drinking50,51 and positive52 and negative alcohol expectancies53,54 predict future alcohol use. For patients with diabetes who are at-risk drinkers, primary care appointments may provide “teachable moments” in which brief advice can have a significant impact—particularly when patients are told laboratory test results; advised about the sugar and carbohydrate content of alcohol; or given information regarding the effect of alcohol on diabetes, medications, and self-care behavior. Finally, primary care providers will also likely have knowledge of a patient’s comorbid conditions (eg, depression) that may relate to diabetes or alcohol use.
TABLE 2
How to implement the FRAMES approach in brief interventions46
| FRAMES elements | Sample statements |
|---|---|
| Feedback about one’s drinking relative to others | “Based on what you told me, you are drinking an average of 21 drinks per week, which puts you above the cutoff for at-risk drinking” or “According to what you told me, it looks like you are drinking more than 88% of men (or women) in the United States.” |
| Responsibility for deciding to change | “You’re in the best position to decide how you would like to make a change” or “Although reducing your drinking would be good for your health, it’s up to you to decide when you’re ready to make a change.” |
| Advice to change drinking | “Your test results indicate your HbA1c, an important measure of blood sugar, is elevated; making a change in your drinking will likely improve your blood sugar” or “I am concerned about the effect on your health of drinking alcohol while taking your diabetes medications. Making a change in your drinking is likely to protect you from complications.” |
| Menu of options for implementing a change strategy | “If it’s okay with you, I can share what has worked for others whose drinking is similar to yours. Some people alternate a drink containing alcohol with water or diet soda. Others will bring only a certain amount of money with them when they go to a bar.” |
| Empathic listening | “It sounds like this has been a concern" or "I know that change can be difficult.” |
| Self-efficacy enhancement | “I wonder if you could use some of the same strategies you used to lose 10 pounds last year?” or “It sounds like you have some ideas for how to make this happen.” |
CORRESPONDENCE Patricia A. Engler, PhD, DGIM, 111 Plain Street Building, Providence, RI 02903; [email protected]
1. National Institute on Alcohol Abuse and Alcoholism. Helping patients who drink too much: a clinician’s guide. 2005. Available at: http://pubs.niaaa.nih.gov/publications/Practitioner/CliniciansGuide2005/guide.pdf. Accessed November 8, 2011.
2. Bradley KA, Kivlahan DR, Williams E. Brief approaches to alcohol screening: practical alternatives for primary care. J Gen Intern Med 2009;24:881-883.
3. Shai I, Fraser D, Wainstein J, et al. Glycemic effects of moderate alcohol intake among patients with type 2 diabetes. Diabetes Care 2007;30:3011-3016.
4. Bantle AE, Tomas W, Bantle JP. Metabolic effects of alcohol in the form of wine in persons with type 2 diabetes mellitus. Metabolism 2008;57:241-245.
5. Koppes LLJ, Dekker JM, Hendriks HFJ, et al. Meta-analysis of the relationship between alcohol consumption and coronary heart disease and mortality in type 2 diabetic patients. Diabetologia 2006;49:648-652.
6. Pedersen-Bjergaard U, Reubsaet JLE, Nielsen SL, et al. Psychoactive drugs, alcohol, and severe hypoglycemia in insulin-treated diabetes: analysis of 141 cases. Am J Med 2005;118:307-310.
7. Fleming M, Mundt M. Carbohydrate-deficient transferrin: validity of a new alcohol biomarker in a sample of patients with diabetes and hypertension. J Am Board Fam Pract 2004;17:247-255.
8. Engler PA, Ramsey SE, Stein MD. Brief alcohol intervention among diabetic patients: a pilot study. Presented at: Annual Meeting of the Society for Behavioral Medicine; March 26–29, 2008; San Diego, CA.
9. Saha TD, Stinson FS, Grant BF. The role of alcohol consumption in future classifications of alcohol use disorders. Drug Alcohol Depend 2007;89:82-92.
10. Shai I, Rimm EB, Schulze MB, et al. Moderate alcohol intake and markers of inflammation and endothelial dysfunction among diabetic men. Diabetologia 2004;14:1760-1767.
11. Wakabayashi I, Kobaba-Wakabayashi R, Masuda H. Relation of drinking alcohol to atherosclerotic risk in type 2 diabetes. Diabetes Care 2002;25:1223-1228.
12. Diem M, Deplazes M, Fajfr R, et al. Effects of alcohol consumption on mortality in patients with type 2 diabetes mellitus. Diabetologia 2003;46:1581-1585.
13. Cox WM, Blount JP, Crowe PA, et al. Diabetic patients’ alcohol use and quality of life: relationships with prescribed treatment compliance among older males. Alcohol Clin Exp Res 1996;20:327-331.
14. Yuan JM, Govindarajan S, Arakawa K, et al. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer 2004;101:1009-1017.
15. Howard AA, Arnsten JH, Gourevitch MN. Effect of alcohol consumption on diabetes mellitus. Ann Intern Med 2004;140:211-219.
16. Glasgow AM, Tynan D, Schwartz R, et al. Alcohol and drug use in teenagers with diabetes mellitus. J Adolesc Health 1991;12:11-14.
17. Turner BC, Jenkins E, Kerr D, et al. The effect of evening alcohol consumption on next-morning glucose control in type 1 diabetes. Diabetes Care 2001;24:1888-1893.
18. Richardson T, Weiss M, Thomas P, et al. Day after the night before. Influence of evening alcohol on risk of hypoglycemia in patients with type 1 diabetes. Diabetes Care. 2005;28:1801-1802.
19. Cheyne EH, Sherwin RS, Lunt MJ, et al. Influence of alcohol on cognitive performance during mild hypoglycaemia: implications for type 1 diabetes. Diabet Med 2004;21:230-237.
20. Lao B, Czyzyk A, Szutowski M, et al. Alcohol tolerance in patients with non-insulin-dependent (type 2) diabetes treated with sulphonylurea derivatives. Arzneimittelforschung 1994;44:727-734.
21. PDR Staff Physicians’ Desk Reference 2003. 57th ed. Montvale, NJ: Medical Economics Company; 2003.
22. Ramchandani N, Cantey-Kiser JM, Alter CA, et al. Self-reported factors that affect glycemic control in college students with type 1 diabetes. Diabetes Educ 2000;26:656-666.
23. Kyngas H. Compliance of adolescents with chronic disease. J Clin Nurs 2000;9:549-556.
24. Ahmed AT, Karter AJ, Liu J. Alcohol consumption is inversely associated with adherence to diabetes self-care behaviours. Diabet Med 2006;23:795-802.
25. Karter AJ, Ferrara A, Darbinian JA, et al. Self-monitoring of blood glucose. Diabetes Care 2004;23:477-483.
26. Chew LD, Nelson KM, Young BA, et al. Association between alcohol consumption and diabetes preventative practices. Fam Med 2005;37:589-594.
27. Johnson KH, Bazargan M, Bing E. Alcohol consumption and compliance among inner-city minority patients with type 2 diabetes mellitus. Arch Fam Med 2000;9:964-970.
28. D’Amico EJ, Paddock SM, Burnam A, et al. Identification of and guidance for problem drinking by general medical providers. Med Care 2005;43:229-236.
29. Roche AM, Parle MD, Stubbs JM, et al. Management and treatment efficacy of drug and alcohol problems: what do doctors believe? Addiction. 1995;90:1357-1366.
30. Aira M, Kauhanen J, Larivaara P, et al. Differences in brief interventions on excessive drinking and smoking by primary care physicians: qualitative study. Prev Med 2004;38:473-478.
31. Barry KL, Blow FC, Willenbring M, et al. Use of alcohol screening and brief interventions in primary care settings: implementation and barriers. Subst Abus 2004;25:27-36.
32. Smith PC, Schmidt SM, Allensworth-Davies D, et al. Primary care validation of a single-question alcohol screening test. J Gen Intern Med 2009;24:783-788.
33. McCrady BS. Alcohol use disorders and the Division 12 Task Force of the American Psychological Association. Psychol Addict Behav 2000;14:267-276.
34. Richmond RL, Anderson P. Research in general practice for smokers and excessive drinkers in Australia and the UK. I. Interpretation of results. Addiction 1994;89:35-40.
35. Goldstein MG, Whitlock EP, DePue J. Multiple behavioral risk factor interventions in primary care. Am J Prev Med 2004;27:61-79.
36. Moyer A, Finney JW, Swearingen CE, et al. Brief interventions for alcohol problems: a meta-analytic review of controlled investigations in treatment-seeking and non-treatment-seeking populations. Addiction 2002;97:279-292.
37. Bertholet N, Daeppen JB, Wietlisbach V, et al. Reduction of alcohol consumption by brief alcohol intervention in primary care: a systematic review and meta-analysis. Arch Intern Med 2005;165:986-995.
38. Whitlock EP, Polen MR, Green CA, et al. Behavioral counseling interventions in primary care to reduce risky/harmful alcohol use by adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2004;140:557-568.
39. U. S. Preventive Services Task Force. Screening for problem drinking. In: DiGuiseppi C, Atkins D, Woolf SH, Kamerow DB, eds. Guide to Clinical Preventive Services. 2nd ed. Alexandria, VA: International Medical Services; 1996;567-582.
40. Anderson P, Scott E. The effect of general practitioners’ advice to heavy drinking men. Br J Addict 1992;87:891-900.
41. Fleming MF, Mundt MP, French MT, et al. Brief physician advice for problem drinkers: long-term efficacy and benefit-cost analysis. Alcohol Clin Exp Res 2002;26:36-43.
42. Fleming MF, Barry KL, Manwell LB, et al. Brief physician advice for problem alcohol drinkers. JAMA 1997;277:1039-1045.
43. Ockene JK, Adams A, Hurley TG, et al. Brief physician- and nurse practitioner-delivered counseling for high-risk drinkers. Arch Intern Med 1999;159:2198-2205.
44. Aalto M, Pekuri P, Seppa K. Primary health care nurses’ and physicians’ attitudes, knowledge and beliefs regarding brief intervention for heavy drinkers. Addiction 2001;96:305-311.
45. Fleming M, Brown R, Brown D. The efficacy of a brief alcohol intervention combined with %CDT feedback in patients being treated for type 2 diabetes and/or hypertension. J Stud Alcohol 2004;65:631-637.
46. Miller WR, Rollnick S. Motivational Interviewing: Preparing People for Change. New York: Guilford Press; 2002.
47. Ramsey SE, Engler PA, Harrington M, et al. A brief alcohol intervention with at-risk drinking diabetics. Subst Abus 2010;4:1-8.
48. Bien TH, Miller WR, Tonigan JS. Brief interventions for alcohol problems: a review. Addiction 1993;88:315-335.
49. Sutton S. Social-psychological approaches to understanding addictive behaviours: attitude-behaviour and decision-making models. Br J Addict 1987;82:355-370.
50. Cunningham JA, Sobell LC, Gavin DR, et al. Assessing motivation for change: preliminary development and evaluation of a scale measuring the costs and benefits of changing alcohol or drug use. Psychol Addict Behav 1997;11:107-114.
51. Rollnick S, Morgan M, Heather N. The development of a brief scale to measure outcome expectations of reduced consumption among excessive drinkers. Addict Behav 1996;21:377-387.
52. Brown SA. Reinforcement expectancies and alcoholism treatment outcome after a one-year follow-up. J Stud Alcohol 1985;46:304-308.
53. Jones BT, McMahon J. Negative alcohol expectancy predicts post-treatment abstinence survivorship: the whether, when and why of relapse to a first drink. Addiction 1994;89:1653-1665.
54. Jones BT, McMahon J. Negative and positive alcohol expectancies as predictors of abstinence after discharge from a residential treatment program: a one-month and three-month follow-up study in men. J Stud Alcohol 1994;55:543-548.
• Ask a question such as “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” to ascertain a patient’s quantity of alcohol use. A
• Apply elements of the FRAMES approach to help patients curtail at-risk drinking—eg, use elevated HbA1c levels as evidence of a need to change behavior. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
There are enough challenges in controlling diabetes mellitus without the hindrance of undetected problematic alcohol use. The good news is that asking a single nonthreatening question can help you detect at-risk drinking—defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) as 5 or more drinks on one occasion or more than 14 drinks per week for men; and 4 or more drinks on one occasion or more than 7 drinks per week for women.1,2 And, for patients who may be compromising their diabetes care and overall health through problem drinking, brief intervention techniques used in the office can enable them to reduce alcohol consumption significantly.
When alcohol becomes a problem in diabetes care
Several studies have explored the long-term benefits of moderate alcohol use on glycemic control—with mixed results. A 2007 study found that diabetes patients who drink 1 glass of wine per day exhibited a lower fasting glucose level than abstainers after 3 months.3 There was no difference, however, on postprandial glucose levels. A 2008 study found that individuals who drank one to 2 glasses of wine per day for a month had lower fasting serum insulin levels relative to when they have abstained for a month,4 although levels of fasting plasma cholesterol, HDL cholesterol, glucose, and hemoglobin A1c (HbA1c) remained unchanged relative to periods of abstinence.4
Furthermore, rates of coronary heart disease and CHD mortality in a meta-analysis were significantly lower in 3 categories of alcohol consumption (<6 g/d, 6 to <18 g/d, and ≥18 g/d) compared with abstinence.5 Nondrinkers also had a greater risk of total mortality compared with the lightest drinking group. Notably, however, the lower limit of the highest drinking category was only 1.5 drinks per day.
How big is the problem? In a study of insulin-treated patients seen for severe hypoglycemia, 17% had been drinking before the episode.6 In a primary care sample, 28% of randomly selected patients with type 2 diabetes met Diagnostic and Statistical Manual of Mental Disorders-IV criteria for a lifetime incidence of alcohol abuse and 13% met either current or lifetime criteria for alcohol dependence.7 Another study of primary care patients with diabetes8 found that 13.4% met NIAAA criteria for at-risk drinking; 11.1% of these at-risk drinkers met criteria for current alcohol dependence. (According to the NIAAA, the rate of at-risk/heavy drinking among US adults is 30%, and about one in 4 heavy drinkers meets the criteria for alcohol abuse or dependence.1)
Detrimental effects with immoderate drinking. Individuals who engage in at-risk drinking, as defined by the NIAAA, are at increased risk for alcohol dependence9 and associated complications such as diabetic neuropathy and retinopathy,10 atherosclerosis,11 and total and CHD mortality.3,12 Heavy drinking also interferes with neuroendocrine, gastrointestinal, and sexual function,13 and its interaction with diabetes increases the risk for hepatocellular carcinoma after controlling for hepatitis B and C serology.14
Interference with diabetes control. Research examining the short-term effect of alcohol use has produced contradictory results, partly due to differences among studies, such as whether alcohol is administered with a meal and whether a fasting glucose level is measured.15 However, alcohol affects glycemic control and, when used excessively, can impair glucose production.16,17 Alcohol may induce hypoglycemia,10,18 and even small amounts may jeopardize diabetes control.13 In a study of patients with insulin-treated diabetes, alcohol use in the presence of mild hypoglycemia increased diastolic blood pressure or exacerbated hypoglycemia-related cognitive deficits.19 Another concern—in both the short and long term—is that alcohol interacts negatively with certain diabetes medications. It is more likely to induce hypoglycemia in the presence of sulfonylurea medications.10 Chlorpropamide decreases the rate of ethanol elimination from the blood.20 And, in those taking metformin, excessive alcohol use elevates risk for lactic acidosis. 21
Diminished self-care. Alcohol use can interfere with self-care,22,23 which is a crucial component of diabetes treatment.24 It may lead to reduced eating16 or to decreased willingness to adhere to prescribed dietary regimens.13 It also impairs other self-care behaviors13,15,25 such as self-monitoring blood glucose and showing up for medical appointments.26 In a large, diverse sample of patients with diabetes,24 heavy drinkers had the highest rates of morbidity. Importantly, alcohol and diabetes self-care behavior were significantly negatively associated. Studies of ethnic minority samples have yielded comparable results.27
Assessing alcohol use: Obstacles and solutions
Although alcohol use can be readily evaluated during routine primary care appointments, it is often neglected, perhaps due to a lack of awareness about its impact on diabetes.15 Those who are most often assessed tend to have a psychiatric diagnosis or other condition raising a red flag for physicians.28 When internists, general practitioners, and psychiatrists were questioned in a study regarding patients’ alcohol and drug use, all 3 groups were misinformed about which substance-use treatments were empirically supported29 and did not believe that treatment for alcohol abuse held much promise. Another study showed that physicians can be reluctant to screen for alcohol use because of the difficulty in recognizing a problem, the perceived unimportance of alcohol use as a health risk, a supposed lack of adequate intervention tools, and a fear of stigmatizing patients.30 Physicians are more likely to discuss alcohol use under certain extreme conditions such as when a patient smells of alcohol.
Multiple opportunities to ask in the VA system. In the Veterans Health Administration, primary care VA providers have reported that prompts for alcohol screening embedded in computerized progress notes, clinical reminder lists, and annual health evaluation forms encourage them to assess alcohol use. Other useful materials include manual checklists and reference cards.31 These providers also report that education, feedback on rates of alcohol screening, and increased supervision facilitate assessment. Finally, providers indicate that asking nurses or clerical staff to administer the screen improves completion rates.
Ask a simple question
“How often have you had a drink containing alcohol in the last year?” or “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” are questions that can help you compare a patient’s alcohol use to the at-risk drinking cutoffs established by the NIAAA.1,2
Recent research has also validated the use of a single question in identifying NIAAA-defined at-risk drinking.32 Simply ask patients, “How many times in the past year have you had X or more drinks in a day?” (X=5 for men or 4 for women). The screen is positive when a patient acknowledges having done so at least once in the past year. This question was 81.8% sensitive and 79.3% specific for unhealthy alcohol use, and 87.9% sensitive and 66.8% specific for current alcohol abuse or dependence.32 Advantages of this method are its brevity, ease of scoring, validity in the primary care setting,32 and ease of recollection for treatment providers (TABLE 1).1,2
TABLE 1
Ask these simple questions to assess alcohol use1,2
| To assess… | Ask… |
|---|---|
| Frequency of alcohol use | “How often do you drink alcohol (including beer or wine)?” or “How often have you had a drink containing alcohol in the last year?” |
| Quantity of alcohol use | “When you do drink alcohol, how many standard* drinks do you have?” or “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” |
| Binge drinking | For men: “How often do you have 5 or more standard* drinks on one occasion?” For women: “How often do you have 4 or more standard* drinks on one occasion?” |
| Alcohol use with the NIAAA single-question screen | “How many times in the past year have you had X or more drinks in a day?” (X = 5 for men and X = 4 for women; positive response ≥1) |
| *A standard drink is equal to 12 oz. of regular beer, 8 to 9 oz. of malt liquor, 5 oz. of wine, or 1.5 oz. of 80-proof spirits.1 NIAAA, National Institute on Alcohol Abuse and Alcoholism. | |
Brief intervention works in primary care
Brief interventions for drinking have strong empirical support. In a review of treatments for alcohol abuse and dependence,33 brief intervention was one of only 2 “efficacious” treatments.
Although some individual studies of brief alcohol interventions in primary care have not shown favorable results, several systematic reviews have demonstrated the efficacy of such interventions in this setting. General practitioner–delivered brief interventions led to significantly better patient outcomes compared with standard care, and “very brief advice” resulted in reductions in alcohol consumption overall and in the percentage of “excessive drinkers.”34 In a review of health behavior interventions,35 brief interventions reduced risky or harmful drinking. In one of 2 meta-analyses that support this finding, brief interventions with primary care patients not seeking treatment for alcohol abuse yielded small-to-medium effect sizes relative to control conditions.36 In the other study, brief interventions significantly reduced longer term alcohol use in primary care patients.37
The US Preventive Services Task Force conducted a systematic review of behavioral counseling interventions and recommends screening and brief interventions for unhealthy drinking in primary care.38,39 Its findings indicate that alcohol use declines significantly after brief interventions containing at least 2 of the following elements: feedback regarding drinking, advice to reduce drinking, or goal setting.
Brief advice is a form of intervention that shows considerable promise in primary care.40-42 Two 10- to 15-minute sessions have led to significant reductions in the mean number of drinks and frequency of excessive drinking in the 7 days before a follow-up interview, as well as a reduction in binge drinking episodes in the previous 30 days.41,42
One study produced positive results with just a 5- to 10-minute counseling session involving advice for drinking goals delivered by primary care providers as part of a routine medical visit.43 This intervention led to significant decreases in alcohol use at a 6-month follow-up for high-risk drinkers compared with controls.43 Brief interventions additionally work within the time constraints of a busy primary care practice and are cost effective.
Some primary care providers think a specialist should conduct interventions and suggest that having a specialist immediately available would enable intervention.31 In fact, some research has supported the idea of special training. In a European study, primary care providers reported that more practical training, information about brief intervention studies, personal training, and lectures would facilitate interventions.44
Applying brief alcohol interventions to diabetes patients
Newer research has tested the efficacy of alcohol interventions with diabetes patients in the primary care setting. In one study,45 brief advice was given in 2 15-minute sessions and 2 5-minute follow-up telephone calls. Compared with controls, significantly more participants who received the intervention reduced heavy drinking from baseline to follow-up. One caveat is that patients with hypertension were included in the sample, making it difficult to determine the impact of the intervention on diabetes patients specifically.
In a small study of patients with diabetes exhibiting at-risk drinking,8 a single-session intervention based on motivational interviewing (MI) principles46 gave participants personalized feedback in relation to sex-based norms of drinking rates and HbA1c and triglyceride laboratory results. Patients were given information on the physiologic effects of alcohol on diabetes, the potential interactions between alcohol and diabetes medications, and the effect of alcohol on diabetes self-care behavior. They were asked to identify pros and cons of their drinking and to develop personal change goals. One of 2 PhD-level clinical psychologists trained in MI administered the single 50-minute intervention. By 1 month and continuing through to the 6-month follow-up, participants had reduced the proportion of drinking days, mean number of daily drinks, and proportion of heavy drinking days.
Ramsey and colleagues47 extended this work by comparatively examining a group of patients exhibiting at-risk drinking who received no intervention. The results favored the intervention group, with a medium-to-large effect size for the proportion of drinking days, a medium effect size for the reduction of mean number of daily drinks, and a small-to-medium effect size in the reduction of heavy drinking days. Furthermore, in the intervention group there was a trend toward better diabetes adherence behavior.
Implementing brief intervention in practice. Despite differences among interventions, the elements of brief interventions tend to be similar.48 Incorporating these elements in the primary care setting provides a useful framework that will likely prove beneficial. Specifically, brief interventions typically contain elements of the FRAMES (TABLE 2)46 acronym:
- Feedback about one’s drinking relative to others
- Responsibility for deciding to change
- Advice to change drinking
- Menu of options for implementing a change strategy
- Empathic listening
- Self-efficacy enhancement.
Decision-making models indicate that expectations about the effects of behavior change play a significant role in determining whether a decision to change is made.49 The perceived costs and benefits of changing drinking50,51 and positive52 and negative alcohol expectancies53,54 predict future alcohol use. For patients with diabetes who are at-risk drinkers, primary care appointments may provide “teachable moments” in which brief advice can have a significant impact—particularly when patients are told laboratory test results; advised about the sugar and carbohydrate content of alcohol; or given information regarding the effect of alcohol on diabetes, medications, and self-care behavior. Finally, primary care providers will also likely have knowledge of a patient’s comorbid conditions (eg, depression) that may relate to diabetes or alcohol use.
TABLE 2
How to implement the FRAMES approach in brief interventions46
| FRAMES elements | Sample statements |
|---|---|
| Feedback about one’s drinking relative to others | “Based on what you told me, you are drinking an average of 21 drinks per week, which puts you above the cutoff for at-risk drinking” or “According to what you told me, it looks like you are drinking more than 88% of men (or women) in the United States.” |
| Responsibility for deciding to change | “You’re in the best position to decide how you would like to make a change” or “Although reducing your drinking would be good for your health, it’s up to you to decide when you’re ready to make a change.” |
| Advice to change drinking | “Your test results indicate your HbA1c, an important measure of blood sugar, is elevated; making a change in your drinking will likely improve your blood sugar” or “I am concerned about the effect on your health of drinking alcohol while taking your diabetes medications. Making a change in your drinking is likely to protect you from complications.” |
| Menu of options for implementing a change strategy | “If it’s okay with you, I can share what has worked for others whose drinking is similar to yours. Some people alternate a drink containing alcohol with water or diet soda. Others will bring only a certain amount of money with them when they go to a bar.” |
| Empathic listening | “It sounds like this has been a concern" or "I know that change can be difficult.” |
| Self-efficacy enhancement | “I wonder if you could use some of the same strategies you used to lose 10 pounds last year?” or “It sounds like you have some ideas for how to make this happen.” |
CORRESPONDENCE Patricia A. Engler, PhD, DGIM, 111 Plain Street Building, Providence, RI 02903; [email protected]
• Ask a question such as “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” to ascertain a patient’s quantity of alcohol use. A
• Apply elements of the FRAMES approach to help patients curtail at-risk drinking—eg, use elevated HbA1c levels as evidence of a need to change behavior. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
There are enough challenges in controlling diabetes mellitus without the hindrance of undetected problematic alcohol use. The good news is that asking a single nonthreatening question can help you detect at-risk drinking—defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) as 5 or more drinks on one occasion or more than 14 drinks per week for men; and 4 or more drinks on one occasion or more than 7 drinks per week for women.1,2 And, for patients who may be compromising their diabetes care and overall health through problem drinking, brief intervention techniques used in the office can enable them to reduce alcohol consumption significantly.
When alcohol becomes a problem in diabetes care
Several studies have explored the long-term benefits of moderate alcohol use on glycemic control—with mixed results. A 2007 study found that diabetes patients who drink 1 glass of wine per day exhibited a lower fasting glucose level than abstainers after 3 months.3 There was no difference, however, on postprandial glucose levels. A 2008 study found that individuals who drank one to 2 glasses of wine per day for a month had lower fasting serum insulin levels relative to when they have abstained for a month,4 although levels of fasting plasma cholesterol, HDL cholesterol, glucose, and hemoglobin A1c (HbA1c) remained unchanged relative to periods of abstinence.4
Furthermore, rates of coronary heart disease and CHD mortality in a meta-analysis were significantly lower in 3 categories of alcohol consumption (<6 g/d, 6 to <18 g/d, and ≥18 g/d) compared with abstinence.5 Nondrinkers also had a greater risk of total mortality compared with the lightest drinking group. Notably, however, the lower limit of the highest drinking category was only 1.5 drinks per day.
How big is the problem? In a study of insulin-treated patients seen for severe hypoglycemia, 17% had been drinking before the episode.6 In a primary care sample, 28% of randomly selected patients with type 2 diabetes met Diagnostic and Statistical Manual of Mental Disorders-IV criteria for a lifetime incidence of alcohol abuse and 13% met either current or lifetime criteria for alcohol dependence.7 Another study of primary care patients with diabetes8 found that 13.4% met NIAAA criteria for at-risk drinking; 11.1% of these at-risk drinkers met criteria for current alcohol dependence. (According to the NIAAA, the rate of at-risk/heavy drinking among US adults is 30%, and about one in 4 heavy drinkers meets the criteria for alcohol abuse or dependence.1)
Detrimental effects with immoderate drinking. Individuals who engage in at-risk drinking, as defined by the NIAAA, are at increased risk for alcohol dependence9 and associated complications such as diabetic neuropathy and retinopathy,10 atherosclerosis,11 and total and CHD mortality.3,12 Heavy drinking also interferes with neuroendocrine, gastrointestinal, and sexual function,13 and its interaction with diabetes increases the risk for hepatocellular carcinoma after controlling for hepatitis B and C serology.14
Interference with diabetes control. Research examining the short-term effect of alcohol use has produced contradictory results, partly due to differences among studies, such as whether alcohol is administered with a meal and whether a fasting glucose level is measured.15 However, alcohol affects glycemic control and, when used excessively, can impair glucose production.16,17 Alcohol may induce hypoglycemia,10,18 and even small amounts may jeopardize diabetes control.13 In a study of patients with insulin-treated diabetes, alcohol use in the presence of mild hypoglycemia increased diastolic blood pressure or exacerbated hypoglycemia-related cognitive deficits.19 Another concern—in both the short and long term—is that alcohol interacts negatively with certain diabetes medications. It is more likely to induce hypoglycemia in the presence of sulfonylurea medications.10 Chlorpropamide decreases the rate of ethanol elimination from the blood.20 And, in those taking metformin, excessive alcohol use elevates risk for lactic acidosis. 21
Diminished self-care. Alcohol use can interfere with self-care,22,23 which is a crucial component of diabetes treatment.24 It may lead to reduced eating16 or to decreased willingness to adhere to prescribed dietary regimens.13 It also impairs other self-care behaviors13,15,25 such as self-monitoring blood glucose and showing up for medical appointments.26 In a large, diverse sample of patients with diabetes,24 heavy drinkers had the highest rates of morbidity. Importantly, alcohol and diabetes self-care behavior were significantly negatively associated. Studies of ethnic minority samples have yielded comparable results.27
Assessing alcohol use: Obstacles and solutions
Although alcohol use can be readily evaluated during routine primary care appointments, it is often neglected, perhaps due to a lack of awareness about its impact on diabetes.15 Those who are most often assessed tend to have a psychiatric diagnosis or other condition raising a red flag for physicians.28 When internists, general practitioners, and psychiatrists were questioned in a study regarding patients’ alcohol and drug use, all 3 groups were misinformed about which substance-use treatments were empirically supported29 and did not believe that treatment for alcohol abuse held much promise. Another study showed that physicians can be reluctant to screen for alcohol use because of the difficulty in recognizing a problem, the perceived unimportance of alcohol use as a health risk, a supposed lack of adequate intervention tools, and a fear of stigmatizing patients.30 Physicians are more likely to discuss alcohol use under certain extreme conditions such as when a patient smells of alcohol.
Multiple opportunities to ask in the VA system. In the Veterans Health Administration, primary care VA providers have reported that prompts for alcohol screening embedded in computerized progress notes, clinical reminder lists, and annual health evaluation forms encourage them to assess alcohol use. Other useful materials include manual checklists and reference cards.31 These providers also report that education, feedback on rates of alcohol screening, and increased supervision facilitate assessment. Finally, providers indicate that asking nurses or clerical staff to administer the screen improves completion rates.
Ask a simple question
“How often have you had a drink containing alcohol in the last year?” or “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” are questions that can help you compare a patient’s alcohol use to the at-risk drinking cutoffs established by the NIAAA.1,2
Recent research has also validated the use of a single question in identifying NIAAA-defined at-risk drinking.32 Simply ask patients, “How many times in the past year have you had X or more drinks in a day?” (X=5 for men or 4 for women). The screen is positive when a patient acknowledges having done so at least once in the past year. This question was 81.8% sensitive and 79.3% specific for unhealthy alcohol use, and 87.9% sensitive and 66.8% specific for current alcohol abuse or dependence.32 Advantages of this method are its brevity, ease of scoring, validity in the primary care setting,32 and ease of recollection for treatment providers (TABLE 1).1,2
TABLE 1
Ask these simple questions to assess alcohol use1,2
| To assess… | Ask… |
|---|---|
| Frequency of alcohol use | “How often do you drink alcohol (including beer or wine)?” or “How often have you had a drink containing alcohol in the last year?” |
| Quantity of alcohol use | “When you do drink alcohol, how many standard* drinks do you have?” or “How many drinks containing alcohol did you have on a typical day when you were drinking in the last year?” |
| Binge drinking | For men: “How often do you have 5 or more standard* drinks on one occasion?” For women: “How often do you have 4 or more standard* drinks on one occasion?” |
| Alcohol use with the NIAAA single-question screen | “How many times in the past year have you had X or more drinks in a day?” (X = 5 for men and X = 4 for women; positive response ≥1) |
| *A standard drink is equal to 12 oz. of regular beer, 8 to 9 oz. of malt liquor, 5 oz. of wine, or 1.5 oz. of 80-proof spirits.1 NIAAA, National Institute on Alcohol Abuse and Alcoholism. | |
Brief intervention works in primary care
Brief interventions for drinking have strong empirical support. In a review of treatments for alcohol abuse and dependence,33 brief intervention was one of only 2 “efficacious” treatments.
Although some individual studies of brief alcohol interventions in primary care have not shown favorable results, several systematic reviews have demonstrated the efficacy of such interventions in this setting. General practitioner–delivered brief interventions led to significantly better patient outcomes compared with standard care, and “very brief advice” resulted in reductions in alcohol consumption overall and in the percentage of “excessive drinkers.”34 In a review of health behavior interventions,35 brief interventions reduced risky or harmful drinking. In one of 2 meta-analyses that support this finding, brief interventions with primary care patients not seeking treatment for alcohol abuse yielded small-to-medium effect sizes relative to control conditions.36 In the other study, brief interventions significantly reduced longer term alcohol use in primary care patients.37
The US Preventive Services Task Force conducted a systematic review of behavioral counseling interventions and recommends screening and brief interventions for unhealthy drinking in primary care.38,39 Its findings indicate that alcohol use declines significantly after brief interventions containing at least 2 of the following elements: feedback regarding drinking, advice to reduce drinking, or goal setting.
Brief advice is a form of intervention that shows considerable promise in primary care.40-42 Two 10- to 15-minute sessions have led to significant reductions in the mean number of drinks and frequency of excessive drinking in the 7 days before a follow-up interview, as well as a reduction in binge drinking episodes in the previous 30 days.41,42
One study produced positive results with just a 5- to 10-minute counseling session involving advice for drinking goals delivered by primary care providers as part of a routine medical visit.43 This intervention led to significant decreases in alcohol use at a 6-month follow-up for high-risk drinkers compared with controls.43 Brief interventions additionally work within the time constraints of a busy primary care practice and are cost effective.
Some primary care providers think a specialist should conduct interventions and suggest that having a specialist immediately available would enable intervention.31 In fact, some research has supported the idea of special training. In a European study, primary care providers reported that more practical training, information about brief intervention studies, personal training, and lectures would facilitate interventions.44
Applying brief alcohol interventions to diabetes patients
Newer research has tested the efficacy of alcohol interventions with diabetes patients in the primary care setting. In one study,45 brief advice was given in 2 15-minute sessions and 2 5-minute follow-up telephone calls. Compared with controls, significantly more participants who received the intervention reduced heavy drinking from baseline to follow-up. One caveat is that patients with hypertension were included in the sample, making it difficult to determine the impact of the intervention on diabetes patients specifically.
In a small study of patients with diabetes exhibiting at-risk drinking,8 a single-session intervention based on motivational interviewing (MI) principles46 gave participants personalized feedback in relation to sex-based norms of drinking rates and HbA1c and triglyceride laboratory results. Patients were given information on the physiologic effects of alcohol on diabetes, the potential interactions between alcohol and diabetes medications, and the effect of alcohol on diabetes self-care behavior. They were asked to identify pros and cons of their drinking and to develop personal change goals. One of 2 PhD-level clinical psychologists trained in MI administered the single 50-minute intervention. By 1 month and continuing through to the 6-month follow-up, participants had reduced the proportion of drinking days, mean number of daily drinks, and proportion of heavy drinking days.
Ramsey and colleagues47 extended this work by comparatively examining a group of patients exhibiting at-risk drinking who received no intervention. The results favored the intervention group, with a medium-to-large effect size for the proportion of drinking days, a medium effect size for the reduction of mean number of daily drinks, and a small-to-medium effect size in the reduction of heavy drinking days. Furthermore, in the intervention group there was a trend toward better diabetes adherence behavior.
Implementing brief intervention in practice. Despite differences among interventions, the elements of brief interventions tend to be similar.48 Incorporating these elements in the primary care setting provides a useful framework that will likely prove beneficial. Specifically, brief interventions typically contain elements of the FRAMES (TABLE 2)46 acronym:
- Feedback about one’s drinking relative to others
- Responsibility for deciding to change
- Advice to change drinking
- Menu of options for implementing a change strategy
- Empathic listening
- Self-efficacy enhancement.
Decision-making models indicate that expectations about the effects of behavior change play a significant role in determining whether a decision to change is made.49 The perceived costs and benefits of changing drinking50,51 and positive52 and negative alcohol expectancies53,54 predict future alcohol use. For patients with diabetes who are at-risk drinkers, primary care appointments may provide “teachable moments” in which brief advice can have a significant impact—particularly when patients are told laboratory test results; advised about the sugar and carbohydrate content of alcohol; or given information regarding the effect of alcohol on diabetes, medications, and self-care behavior. Finally, primary care providers will also likely have knowledge of a patient’s comorbid conditions (eg, depression) that may relate to diabetes or alcohol use.
TABLE 2
How to implement the FRAMES approach in brief interventions46
| FRAMES elements | Sample statements |
|---|---|
| Feedback about one’s drinking relative to others | “Based on what you told me, you are drinking an average of 21 drinks per week, which puts you above the cutoff for at-risk drinking” or “According to what you told me, it looks like you are drinking more than 88% of men (or women) in the United States.” |
| Responsibility for deciding to change | “You’re in the best position to decide how you would like to make a change” or “Although reducing your drinking would be good for your health, it’s up to you to decide when you’re ready to make a change.” |
| Advice to change drinking | “Your test results indicate your HbA1c, an important measure of blood sugar, is elevated; making a change in your drinking will likely improve your blood sugar” or “I am concerned about the effect on your health of drinking alcohol while taking your diabetes medications. Making a change in your drinking is likely to protect you from complications.” |
| Menu of options for implementing a change strategy | “If it’s okay with you, I can share what has worked for others whose drinking is similar to yours. Some people alternate a drink containing alcohol with water or diet soda. Others will bring only a certain amount of money with them when they go to a bar.” |
| Empathic listening | “It sounds like this has been a concern" or "I know that change can be difficult.” |
| Self-efficacy enhancement | “I wonder if you could use some of the same strategies you used to lose 10 pounds last year?” or “It sounds like you have some ideas for how to make this happen.” |
CORRESPONDENCE Patricia A. Engler, PhD, DGIM, 111 Plain Street Building, Providence, RI 02903; [email protected]
1. National Institute on Alcohol Abuse and Alcoholism. Helping patients who drink too much: a clinician’s guide. 2005. Available at: http://pubs.niaaa.nih.gov/publications/Practitioner/CliniciansGuide2005/guide.pdf. Accessed November 8, 2011.
2. Bradley KA, Kivlahan DR, Williams E. Brief approaches to alcohol screening: practical alternatives for primary care. J Gen Intern Med 2009;24:881-883.
3. Shai I, Fraser D, Wainstein J, et al. Glycemic effects of moderate alcohol intake among patients with type 2 diabetes. Diabetes Care 2007;30:3011-3016.
4. Bantle AE, Tomas W, Bantle JP. Metabolic effects of alcohol in the form of wine in persons with type 2 diabetes mellitus. Metabolism 2008;57:241-245.
5. Koppes LLJ, Dekker JM, Hendriks HFJ, et al. Meta-analysis of the relationship between alcohol consumption and coronary heart disease and mortality in type 2 diabetic patients. Diabetologia 2006;49:648-652.
6. Pedersen-Bjergaard U, Reubsaet JLE, Nielsen SL, et al. Psychoactive drugs, alcohol, and severe hypoglycemia in insulin-treated diabetes: analysis of 141 cases. Am J Med 2005;118:307-310.
7. Fleming M, Mundt M. Carbohydrate-deficient transferrin: validity of a new alcohol biomarker in a sample of patients with diabetes and hypertension. J Am Board Fam Pract 2004;17:247-255.
8. Engler PA, Ramsey SE, Stein MD. Brief alcohol intervention among diabetic patients: a pilot study. Presented at: Annual Meeting of the Society for Behavioral Medicine; March 26–29, 2008; San Diego, CA.
9. Saha TD, Stinson FS, Grant BF. The role of alcohol consumption in future classifications of alcohol use disorders. Drug Alcohol Depend 2007;89:82-92.
10. Shai I, Rimm EB, Schulze MB, et al. Moderate alcohol intake and markers of inflammation and endothelial dysfunction among diabetic men. Diabetologia 2004;14:1760-1767.
11. Wakabayashi I, Kobaba-Wakabayashi R, Masuda H. Relation of drinking alcohol to atherosclerotic risk in type 2 diabetes. Diabetes Care 2002;25:1223-1228.
12. Diem M, Deplazes M, Fajfr R, et al. Effects of alcohol consumption on mortality in patients with type 2 diabetes mellitus. Diabetologia 2003;46:1581-1585.
13. Cox WM, Blount JP, Crowe PA, et al. Diabetic patients’ alcohol use and quality of life: relationships with prescribed treatment compliance among older males. Alcohol Clin Exp Res 1996;20:327-331.
14. Yuan JM, Govindarajan S, Arakawa K, et al. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer 2004;101:1009-1017.
15. Howard AA, Arnsten JH, Gourevitch MN. Effect of alcohol consumption on diabetes mellitus. Ann Intern Med 2004;140:211-219.
16. Glasgow AM, Tynan D, Schwartz R, et al. Alcohol and drug use in teenagers with diabetes mellitus. J Adolesc Health 1991;12:11-14.
17. Turner BC, Jenkins E, Kerr D, et al. The effect of evening alcohol consumption on next-morning glucose control in type 1 diabetes. Diabetes Care 2001;24:1888-1893.
18. Richardson T, Weiss M, Thomas P, et al. Day after the night before. Influence of evening alcohol on risk of hypoglycemia in patients with type 1 diabetes. Diabetes Care. 2005;28:1801-1802.
19. Cheyne EH, Sherwin RS, Lunt MJ, et al. Influence of alcohol on cognitive performance during mild hypoglycaemia: implications for type 1 diabetes. Diabet Med 2004;21:230-237.
20. Lao B, Czyzyk A, Szutowski M, et al. Alcohol tolerance in patients with non-insulin-dependent (type 2) diabetes treated with sulphonylurea derivatives. Arzneimittelforschung 1994;44:727-734.
21. PDR Staff Physicians’ Desk Reference 2003. 57th ed. Montvale, NJ: Medical Economics Company; 2003.
22. Ramchandani N, Cantey-Kiser JM, Alter CA, et al. Self-reported factors that affect glycemic control in college students with type 1 diabetes. Diabetes Educ 2000;26:656-666.
23. Kyngas H. Compliance of adolescents with chronic disease. J Clin Nurs 2000;9:549-556.
24. Ahmed AT, Karter AJ, Liu J. Alcohol consumption is inversely associated with adherence to diabetes self-care behaviours. Diabet Med 2006;23:795-802.
25. Karter AJ, Ferrara A, Darbinian JA, et al. Self-monitoring of blood glucose. Diabetes Care 2004;23:477-483.
26. Chew LD, Nelson KM, Young BA, et al. Association between alcohol consumption and diabetes preventative practices. Fam Med 2005;37:589-594.
27. Johnson KH, Bazargan M, Bing E. Alcohol consumption and compliance among inner-city minority patients with type 2 diabetes mellitus. Arch Fam Med 2000;9:964-970.
28. D’Amico EJ, Paddock SM, Burnam A, et al. Identification of and guidance for problem drinking by general medical providers. Med Care 2005;43:229-236.
29. Roche AM, Parle MD, Stubbs JM, et al. Management and treatment efficacy of drug and alcohol problems: what do doctors believe? Addiction. 1995;90:1357-1366.
30. Aira M, Kauhanen J, Larivaara P, et al. Differences in brief interventions on excessive drinking and smoking by primary care physicians: qualitative study. Prev Med 2004;38:473-478.
31. Barry KL, Blow FC, Willenbring M, et al. Use of alcohol screening and brief interventions in primary care settings: implementation and barriers. Subst Abus 2004;25:27-36.
32. Smith PC, Schmidt SM, Allensworth-Davies D, et al. Primary care validation of a single-question alcohol screening test. J Gen Intern Med 2009;24:783-788.
33. McCrady BS. Alcohol use disorders and the Division 12 Task Force of the American Psychological Association. Psychol Addict Behav 2000;14:267-276.
34. Richmond RL, Anderson P. Research in general practice for smokers and excessive drinkers in Australia and the UK. I. Interpretation of results. Addiction 1994;89:35-40.
35. Goldstein MG, Whitlock EP, DePue J. Multiple behavioral risk factor interventions in primary care. Am J Prev Med 2004;27:61-79.
36. Moyer A, Finney JW, Swearingen CE, et al. Brief interventions for alcohol problems: a meta-analytic review of controlled investigations in treatment-seeking and non-treatment-seeking populations. Addiction 2002;97:279-292.
37. Bertholet N, Daeppen JB, Wietlisbach V, et al. Reduction of alcohol consumption by brief alcohol intervention in primary care: a systematic review and meta-analysis. Arch Intern Med 2005;165:986-995.
38. Whitlock EP, Polen MR, Green CA, et al. Behavioral counseling interventions in primary care to reduce risky/harmful alcohol use by adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2004;140:557-568.
39. U. S. Preventive Services Task Force. Screening for problem drinking. In: DiGuiseppi C, Atkins D, Woolf SH, Kamerow DB, eds. Guide to Clinical Preventive Services. 2nd ed. Alexandria, VA: International Medical Services; 1996;567-582.
40. Anderson P, Scott E. The effect of general practitioners’ advice to heavy drinking men. Br J Addict 1992;87:891-900.
41. Fleming MF, Mundt MP, French MT, et al. Brief physician advice for problem drinkers: long-term efficacy and benefit-cost analysis. Alcohol Clin Exp Res 2002;26:36-43.
42. Fleming MF, Barry KL, Manwell LB, et al. Brief physician advice for problem alcohol drinkers. JAMA 1997;277:1039-1045.
43. Ockene JK, Adams A, Hurley TG, et al. Brief physician- and nurse practitioner-delivered counseling for high-risk drinkers. Arch Intern Med 1999;159:2198-2205.
44. Aalto M, Pekuri P, Seppa K. Primary health care nurses’ and physicians’ attitudes, knowledge and beliefs regarding brief intervention for heavy drinkers. Addiction 2001;96:305-311.
45. Fleming M, Brown R, Brown D. The efficacy of a brief alcohol intervention combined with %CDT feedback in patients being treated for type 2 diabetes and/or hypertension. J Stud Alcohol 2004;65:631-637.
46. Miller WR, Rollnick S. Motivational Interviewing: Preparing People for Change. New York: Guilford Press; 2002.
47. Ramsey SE, Engler PA, Harrington M, et al. A brief alcohol intervention with at-risk drinking diabetics. Subst Abus 2010;4:1-8.
48. Bien TH, Miller WR, Tonigan JS. Brief interventions for alcohol problems: a review. Addiction 1993;88:315-335.
49. Sutton S. Social-psychological approaches to understanding addictive behaviours: attitude-behaviour and decision-making models. Br J Addict 1987;82:355-370.
50. Cunningham JA, Sobell LC, Gavin DR, et al. Assessing motivation for change: preliminary development and evaluation of a scale measuring the costs and benefits of changing alcohol or drug use. Psychol Addict Behav 1997;11:107-114.
51. Rollnick S, Morgan M, Heather N. The development of a brief scale to measure outcome expectations of reduced consumption among excessive drinkers. Addict Behav 1996;21:377-387.
52. Brown SA. Reinforcement expectancies and alcoholism treatment outcome after a one-year follow-up. J Stud Alcohol 1985;46:304-308.
53. Jones BT, McMahon J. Negative alcohol expectancy predicts post-treatment abstinence survivorship: the whether, when and why of relapse to a first drink. Addiction 1994;89:1653-1665.
54. Jones BT, McMahon J. Negative and positive alcohol expectancies as predictors of abstinence after discharge from a residential treatment program: a one-month and three-month follow-up study in men. J Stud Alcohol 1994;55:543-548.
1. National Institute on Alcohol Abuse and Alcoholism. Helping patients who drink too much: a clinician’s guide. 2005. Available at: http://pubs.niaaa.nih.gov/publications/Practitioner/CliniciansGuide2005/guide.pdf. Accessed November 8, 2011.
2. Bradley KA, Kivlahan DR, Williams E. Brief approaches to alcohol screening: practical alternatives for primary care. J Gen Intern Med 2009;24:881-883.
3. Shai I, Fraser D, Wainstein J, et al. Glycemic effects of moderate alcohol intake among patients with type 2 diabetes. Diabetes Care 2007;30:3011-3016.
4. Bantle AE, Tomas W, Bantle JP. Metabolic effects of alcohol in the form of wine in persons with type 2 diabetes mellitus. Metabolism 2008;57:241-245.
5. Koppes LLJ, Dekker JM, Hendriks HFJ, et al. Meta-analysis of the relationship between alcohol consumption and coronary heart disease and mortality in type 2 diabetic patients. Diabetologia 2006;49:648-652.
6. Pedersen-Bjergaard U, Reubsaet JLE, Nielsen SL, et al. Psychoactive drugs, alcohol, and severe hypoglycemia in insulin-treated diabetes: analysis of 141 cases. Am J Med 2005;118:307-310.
7. Fleming M, Mundt M. Carbohydrate-deficient transferrin: validity of a new alcohol biomarker in a sample of patients with diabetes and hypertension. J Am Board Fam Pract 2004;17:247-255.
8. Engler PA, Ramsey SE, Stein MD. Brief alcohol intervention among diabetic patients: a pilot study. Presented at: Annual Meeting of the Society for Behavioral Medicine; March 26–29, 2008; San Diego, CA.
9. Saha TD, Stinson FS, Grant BF. The role of alcohol consumption in future classifications of alcohol use disorders. Drug Alcohol Depend 2007;89:82-92.
10. Shai I, Rimm EB, Schulze MB, et al. Moderate alcohol intake and markers of inflammation and endothelial dysfunction among diabetic men. Diabetologia 2004;14:1760-1767.
11. Wakabayashi I, Kobaba-Wakabayashi R, Masuda H. Relation of drinking alcohol to atherosclerotic risk in type 2 diabetes. Diabetes Care 2002;25:1223-1228.
12. Diem M, Deplazes M, Fajfr R, et al. Effects of alcohol consumption on mortality in patients with type 2 diabetes mellitus. Diabetologia 2003;46:1581-1585.
13. Cox WM, Blount JP, Crowe PA, et al. Diabetic patients’ alcohol use and quality of life: relationships with prescribed treatment compliance among older males. Alcohol Clin Exp Res 1996;20:327-331.
14. Yuan JM, Govindarajan S, Arakawa K, et al. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer 2004;101:1009-1017.
15. Howard AA, Arnsten JH, Gourevitch MN. Effect of alcohol consumption on diabetes mellitus. Ann Intern Med 2004;140:211-219.
16. Glasgow AM, Tynan D, Schwartz R, et al. Alcohol and drug use in teenagers with diabetes mellitus. J Adolesc Health 1991;12:11-14.
17. Turner BC, Jenkins E, Kerr D, et al. The effect of evening alcohol consumption on next-morning glucose control in type 1 diabetes. Diabetes Care 2001;24:1888-1893.
18. Richardson T, Weiss M, Thomas P, et al. Day after the night before. Influence of evening alcohol on risk of hypoglycemia in patients with type 1 diabetes. Diabetes Care. 2005;28:1801-1802.
19. Cheyne EH, Sherwin RS, Lunt MJ, et al. Influence of alcohol on cognitive performance during mild hypoglycaemia: implications for type 1 diabetes. Diabet Med 2004;21:230-237.
20. Lao B, Czyzyk A, Szutowski M, et al. Alcohol tolerance in patients with non-insulin-dependent (type 2) diabetes treated with sulphonylurea derivatives. Arzneimittelforschung 1994;44:727-734.
21. PDR Staff Physicians’ Desk Reference 2003. 57th ed. Montvale, NJ: Medical Economics Company; 2003.
22. Ramchandani N, Cantey-Kiser JM, Alter CA, et al. Self-reported factors that affect glycemic control in college students with type 1 diabetes. Diabetes Educ 2000;26:656-666.
23. Kyngas H. Compliance of adolescents with chronic disease. J Clin Nurs 2000;9:549-556.
24. Ahmed AT, Karter AJ, Liu J. Alcohol consumption is inversely associated with adherence to diabetes self-care behaviours. Diabet Med 2006;23:795-802.
25. Karter AJ, Ferrara A, Darbinian JA, et al. Self-monitoring of blood glucose. Diabetes Care 2004;23:477-483.
26. Chew LD, Nelson KM, Young BA, et al. Association between alcohol consumption and diabetes preventative practices. Fam Med 2005;37:589-594.
27. Johnson KH, Bazargan M, Bing E. Alcohol consumption and compliance among inner-city minority patients with type 2 diabetes mellitus. Arch Fam Med 2000;9:964-970.
28. D’Amico EJ, Paddock SM, Burnam A, et al. Identification of and guidance for problem drinking by general medical providers. Med Care 2005;43:229-236.
29. Roche AM, Parle MD, Stubbs JM, et al. Management and treatment efficacy of drug and alcohol problems: what do doctors believe? Addiction. 1995;90:1357-1366.
30. Aira M, Kauhanen J, Larivaara P, et al. Differences in brief interventions on excessive drinking and smoking by primary care physicians: qualitative study. Prev Med 2004;38:473-478.
31. Barry KL, Blow FC, Willenbring M, et al. Use of alcohol screening and brief interventions in primary care settings: implementation and barriers. Subst Abus 2004;25:27-36.
32. Smith PC, Schmidt SM, Allensworth-Davies D, et al. Primary care validation of a single-question alcohol screening test. J Gen Intern Med 2009;24:783-788.
33. McCrady BS. Alcohol use disorders and the Division 12 Task Force of the American Psychological Association. Psychol Addict Behav 2000;14:267-276.
34. Richmond RL, Anderson P. Research in general practice for smokers and excessive drinkers in Australia and the UK. I. Interpretation of results. Addiction 1994;89:35-40.
35. Goldstein MG, Whitlock EP, DePue J. Multiple behavioral risk factor interventions in primary care. Am J Prev Med 2004;27:61-79.
36. Moyer A, Finney JW, Swearingen CE, et al. Brief interventions for alcohol problems: a meta-analytic review of controlled investigations in treatment-seeking and non-treatment-seeking populations. Addiction 2002;97:279-292.
37. Bertholet N, Daeppen JB, Wietlisbach V, et al. Reduction of alcohol consumption by brief alcohol intervention in primary care: a systematic review and meta-analysis. Arch Intern Med 2005;165:986-995.
38. Whitlock EP, Polen MR, Green CA, et al. Behavioral counseling interventions in primary care to reduce risky/harmful alcohol use by adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2004;140:557-568.
39. U. S. Preventive Services Task Force. Screening for problem drinking. In: DiGuiseppi C, Atkins D, Woolf SH, Kamerow DB, eds. Guide to Clinical Preventive Services. 2nd ed. Alexandria, VA: International Medical Services; 1996;567-582.
40. Anderson P, Scott E. The effect of general practitioners’ advice to heavy drinking men. Br J Addict 1992;87:891-900.
41. Fleming MF, Mundt MP, French MT, et al. Brief physician advice for problem drinkers: long-term efficacy and benefit-cost analysis. Alcohol Clin Exp Res 2002;26:36-43.
42. Fleming MF, Barry KL, Manwell LB, et al. Brief physician advice for problem alcohol drinkers. JAMA 1997;277:1039-1045.
43. Ockene JK, Adams A, Hurley TG, et al. Brief physician- and nurse practitioner-delivered counseling for high-risk drinkers. Arch Intern Med 1999;159:2198-2205.
44. Aalto M, Pekuri P, Seppa K. Primary health care nurses’ and physicians’ attitudes, knowledge and beliefs regarding brief intervention for heavy drinkers. Addiction 2001;96:305-311.
45. Fleming M, Brown R, Brown D. The efficacy of a brief alcohol intervention combined with %CDT feedback in patients being treated for type 2 diabetes and/or hypertension. J Stud Alcohol 2004;65:631-637.
46. Miller WR, Rollnick S. Motivational Interviewing: Preparing People for Change. New York: Guilford Press; 2002.
47. Ramsey SE, Engler PA, Harrington M, et al. A brief alcohol intervention with at-risk drinking diabetics. Subst Abus 2010;4:1-8.
48. Bien TH, Miller WR, Tonigan JS. Brief interventions for alcohol problems: a review. Addiction 1993;88:315-335.
49. Sutton S. Social-psychological approaches to understanding addictive behaviours: attitude-behaviour and decision-making models. Br J Addict 1987;82:355-370.
50. Cunningham JA, Sobell LC, Gavin DR, et al. Assessing motivation for change: preliminary development and evaluation of a scale measuring the costs and benefits of changing alcohol or drug use. Psychol Addict Behav 1997;11:107-114.
51. Rollnick S, Morgan M, Heather N. The development of a brief scale to measure outcome expectations of reduced consumption among excessive drinkers. Addict Behav 1996;21:377-387.
52. Brown SA. Reinforcement expectancies and alcoholism treatment outcome after a one-year follow-up. J Stud Alcohol 1985;46:304-308.
53. Jones BT, McMahon J. Negative alcohol expectancy predicts post-treatment abstinence survivorship: the whether, when and why of relapse to a first drink. Addiction 1994;89:1653-1665.
54. Jones BT, McMahon J. Negative and positive alcohol expectancies as predictors of abstinence after discharge from a residential treatment program: a one-month and three-month follow-up study in men. J Stud Alcohol 1994;55:543-548.
Detecting and treating delirium—key interventions you may be missing
• Nonpharmacologic interventions are the mainstay of treatment for delirium. B
• When medication is needed, atypical antipsychotics are as effective as typical antipsychotics for treating delirium in elderly patients, and have fewer side effects. B
• Benzodiazepines should be avoided in elderly patients with delirium that is not associated with alcohol withdrawal. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mr. D, a 75-year-old patient with a history of hypertension and congestive heart failure, sustained a femoral neck fracture and was admitted to the hospital for surgery. He underwent open reduction and internal fixation and was doing well postoperatively, until Day 2—when his primary care physician made morning rounds and noted that Mr. D was somnolent. The nurse on duty assured the physician that Mr. D was fine and “was awake and alert earlier,” and attributed his somnolence to the oxycodone (10 mg) the patient was taking for pain. The physician ordered a reduction in dosage.
If Mr. D had been your patient, would you have considered other possible causes of his somnolence? Or do you think the physician’s action was sufficient?
Derived from Latin, the word delirium literally means “off the [ploughed] track.”1 Dozens of terms have been used to describe delirium, with acute confusion state, organic brain syndrome, acute brain syndrome, and toxic psychosis among them.
Delirium has been reported to occur in 15% to 30% of patients on general medical units,2 about 40% of postoperative patients, and up to 70% of terminally ill patients.3 The true prevalence is hard to determine, as up to 66% of cases may be missed.4
Delirium is being diagnosed more frequently, however—a likely result of a growing geriatric population, increased longevity, and greater awareness of the condition. Each year, an estimated 2.3 million US residents are affected, leading to prolonged hospitalization; poor functional outcomes; the development or worsening of dementia; increased nursing home placement; and a significant burden for families and the US health care system.5
Delirium is also associated with an increase in mortality.6,7 The mortality rate among hospitalized patients who develop delirium is reported to be 18%, rising to an estimated 47% within the first 3 months after discharge.6 Greater awareness of risk factors, rapid recognition of signs and symptoms of delirium, and early intervention—detailed in the text and tables that follow—will lead to better outcomes.
Assessing risk, evaluating mental status
In addition to advanced age, risk factors for delirium (TABLE 1)8-14 include alcohol use, brain dysfunction, comorbidities, hypertension, malignancy, anticholinergic medications, anemia, metabolic abnormalities, and male sex. In patients who, like Mr. D, have numerous risk factors, early—and frequent—evaluation of mental status is needed. One way to do this is to treat mental status as a vital sign, to be included in the assessment of every elderly patient.15
The Confusion Assessment Method, a quick and easy-to-use delirium screening tool (TABLE 2), has a sensitivity of 94% to 100% and a specificity of 90% to 95%.16,17 There are a number of other screening tools, including the widely used Mini-Mental State Exam (MMSE), as well as the Delirium Rating Scale, Delirium Symptom Interview, and Delirium Severity Scale.
TABLE 1
Risk factors for delirium8-14
Advanced age Alcohol use Brain dysfunction (dementia, epilepsy) Hypertension Male sex Malignancy Medications (mainly anticholinergic) Metabolic abnormalities:
Old age Preoperative anemia Preoperative metabolic abnormalities |
| BUN, blood urea nitrogen; Cr, creatinine; Na, sodium. |
TABLE 2
Screening for delirium: The Confusion Assessment Method*16,17
| Criteria | Evidence Yes to questions 1, 2, and 3 plus 4 or 5 (or both) suggests a delirium diagnosis |
|---|---|
| 1. Acute onset | Is there evidence of an acute change in mental status from the patient’s baseline? |
| 2. Fluctuating course | Did the abnormal behavior fluctuate during the day—ie, tend to come and go or increase and decrease in severity? |
| 3. Inattention | Did the patient have difficulty focusing attention, eg, being easily distractible or having difficulty keeping track of what was being said? |
| PLUS | |
| 4. Disorganized thinking | Was the patient’s thinking disorganized or incoherent, such as rambling or irrelevant conversation, unclear or illogical flow of ideas, or unpredictable switching from subject to subject? |
| 5. Altered level of consciousness | Would you rate the patient’s level of consciousness as (any of the following): – Vigilant (hyperalert) – Lethargic (drowsy, easily aroused) – Stupor (difficult to arouse) – Coma (unarousable) |
| *CAM shortened version worksheet. Adapted from: Inouye SK et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941-948; Inouye SK. Confusion Assessment Method (CAM): Training Manual and Coding Guide. Copyright 2003, Hospital Elder Life Program, LLC. | |
Arriving at a delirium diagnosis
The clinical presentation of delirium is characterized by acute—and reversible—impairment of cognition, attention, orientation, and memory, and disruption of the normal sleep/wake cycle. The Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR) criteria for a delirium diagnosis include all of the following:
- disturbance of consciousness, with a reduced ability to focus, sustain, or shift attention
- change in cognition, or a perceptual disturbance, that is not accounted for by a preexisting or developing dementia
- rapid onset of cognitive impairment, with fluctuation likely during the course of the day
- evidence from the history, physical exam, or laboratory findings that the disturbed consciousness is a direct physiological consequence of a general medical condition.17
There are 3 basic types of delirium, each associated with a different psychomotor disturbance.
- Hyperactive delirium—the least common—is characterized by restlessness and agitation, and is therefore the easiest to diagnose.
- Hypoactive delirium is characterized by psychomotor retardation and hypoalertness. It is often misdiagnosed as depression, and has the poorest prognosis.
- Mixed delirium—the most common—is characterized by symptoms that fluctuate between hyper- and hypoactivity.18
CASE By lunchtime, Mr. D had awakened; however, he needed help with his meal. After eating, he slept for the rest of the day. At night, a nurse paged the resident to report that the patient’s blood pressure was 82/60 mm Hg and his heart rate was 115. The physician ordered an intravenous fluid bolus, which corrected the patient’s hypotension, but only temporarily.
The fluctuating nature of delirium—most notably, in patients’ level of alertness—is helpful in establishing a diagnosis. The history and physical exam are the gold standard tools, both for diagnosing delirium and identifying the underlying cause (TABLE 3).19,20 A review of the patient’s medications should be a key component of the medical history, as drugs—particularly those with anticholinergic properties—are often associated with delirium. Environmental shifts, including hospitalization and a disruption of the normal sleep/wake cycle, endocrine disorders, infection, and nutritional deficiencies are also potential causes of delirium, among others.
If history and physical exam fail to identify the underlying cause, laboratory testing, including complete blood count, complete metabolic profile, and urinalysis, should be done. Brain imaging is usually not needed for individuals with symptoms of delirium, but computed tomography (CT) may be indicated if a patient’s condition continues to deteriorate while the underlying cause remains unidentified.21 Electroencephalography (EEG) may be used to confirm a delirium diagnosis that’s uncertain, in a patient with underlying dementia, for instance. (In more than 16% of cases of delirium, the cause is unknown.22)
The most common structural abnormalities found in patients with delirium are brain atrophy and increased white matter lesions, as well as basal ganglia lesions.23 Single-photon emission CT (SPECT) shows a reduction of regional cerebral perfusion by 50%,24 while EEG shows slowing of the posterior dominant rhythm and increased generalized slow-wave activity.25
TABLE 3
A DELIRIUM mnemonic to get to the heart of the problem19,20
| Cause | Comment |
|---|---|
| Drugs | Drug classes: Anesthesia, anticholinergics, anticonvulsants, antiemetics, antihistamines, antihypertensives, antimicrobials, antipsychotics, benzodiazepines, corticosteroids, hypnotics, H2 blockers, muscle relaxants, NSAIDs, opioids, SSRIs, tricyclic antidepressants Drugs: digoxin, levodopa, lithium, theophylline OTCs: henbane, Jimson weed, mandrake, Atropa belladonna extract |
| Environmental | Change of environment, sensory deprivation, sleep deprivation |
| Endocrine | Hyperparathyroidism, hyper-/hypothyroidism |
| Low perfusion | MI, pulmonary embolism, CVA |
| Infection | Pneumonia, sepsis, systemic infection, UTI |
| Retention | Fecal impaction, urinary retention |
| Intoxication | Alcohol, illegal drugs/drug overdose |
| Undernutrition | Malnutrition, thiamin deficiency, vitamin B12 deficiency |
| Metabolic | Acid-base disturbances, fluid and electrolyte abnormalities, hepatic or uremic encephalopathy, hypercarbia, hyper-/hypoglycemia, hyperosmolality, hypoxia |
| Subdural | History of falls |
| CVA, cerebrovascular accident; MI, myocardial infarction; NSAIDs, nonsteroidal anti-inflammatory drugs; OTCs, over-the-counter agents; SSRIs, selective serotonin reuptake inhibitors; UTI, urinary tract infection. | |
Treating (or preventing) delirium: Start with these steps
Nonpharmacologic interventions are the mainstay of treatment for patients with delirium, and may also help to prevent the development of delirium in patients at risk. One key measure is to correct, or avoid, disruptions in the patient’s normal sleep/wake cycle—eg, restoring circadian rhythm by avoiding,
to the extent possible, awakening the patient at night for medication or vital signs. Preventing sensory deprivation, by ensuring that the patient’s eyeglasses and hearing aid are nearby and that there is a clock and calendar nearby and adequate light, is also helpful. Other key interventions (TABLE 4)26-28 include:
- limiting medications associated with delirium (and eliminating any nonessential medication)
- improving nutrition and ambulation
- correcting electrolyte and fluid disturbances
- treating infection
- involving family members in patient care
- ensuring that patients receive adequate pain management
- avoiding transfers (if the patient is hospitalized) and trying to secure a single room.
Several studies have evaluated the effectiveness of nonpharmacologic interventions in preventing or lowering the incidence of delirium. A large multicomponent delirium prevention study of patients >70 years on general medical units focused on managing risk factors. The interventions studied included (1) avoidance of sensory deprivation, (2) early mobilization, (3) treating dehydration, (4) implementing noise reduction strategies and sleep enhancement programs, and (5) avoiding the use of sleep medications. These interventions proved to be effective not only in lowering the incidence of delirium, but in shortening the duration of delirium in affected patients (NNT=20).27
One study found that proactively using a geriatric consultation model (ie, implementing standardized protocols for the management of 6 risk factors) for elderly hospitalized patients led to a reduction in the incidence of delirium by more than a third.26 Admission to a specialized geriatric unit is associated with a lower incidence of delirium compared with being hospitalized on a general medical unit.29
Reducing the incidence of postoperative delirium. Bright light therapy (a light intensity of 5000 lux with a distance from the light source of 100 cm), implemented postoperatively, may play a role in reducing the incidence of delirium, research suggests.30 Music may be helpful, as well. An RCT involving patients (>65 years) undergoing elective knee or hip surgery found that those who listened to classical music postoperatively had a lower incidence of delirium.31 Similarly, playing music in nursing homes has been shown to decrease aggressive behavior and agitation.32
TABLE 4
Helpful interventions in the hospital or at home26-28
|
When medication is needed, proceed with caution
None of the medications currently used to treat delirium are approved by the US Food and Drug Administration for this indication, and many of them have substantial side effects. Nonetheless, palliative or symptomatic treatment requires some form of sedation for agitated patients with delirium. Thus, it is necessary to strike a balance in order to manage the symptoms of delirium and avoid potential side effects (primarily, sedation). Overly sedating patients can confuse the clinical picture of delirium and make it difficult to differentiate between ongoing delirium and medication side effects. Medication should be started at a low, but frequent, dose to achieve an effective therapeutic level, after which a lower maintenance dose can be used until the cause of delirium is resolved.
Antipsychotics are the cornerstone of drug treatment
Haloperidol has traditionally been used to treat delirium33 and has proven effectiveness. However, it is associated with increased risk of extrapyramidal manifestations compared with atypical antipsychotics.
Atypical antipsychotics (olanzapine, risperidone, quetiapine) are increasingly being used to treat delirium because they have fewer extrapyramidal side effects.34 With the exception of olanzapine (available in intramuscular and oral disintegrating form), atypical antipsychotics are available only in oral form, which may limit their usefulness as a treatment for agitated, delirious patients.
Risperidone (at a dose ranging from 0.25 to 1 mg/d) and olanzapine (1.25 to 2.5 mg/d) have shown similar efficacy to haloperidol (0.75 to 1.5 mg/d) in both the prevention and treatment of delirium, but with fewer extrapyramidal side effects.35-39 Quetiapine, a second-generation antipsychotic, is widely used to treat inpatient delirium, although there are no large RCTs comparing it with placebo. One pilot study and another open-label trial found the drug to be beneficial for patients with delirium, with fewer extrapyramidal side effects than haloperidol.40,41
Do a risk-benefit analysis. The use of antipsychotics in elderly patients with delirium has been associated with increased morbidity and mortality. The incidence of stroke and death were higher for community-dwelling patients (NNH=100) and patients in long-term care (N=67) who received typical or atypical antipsychotics for 6 months compared with that of patients who did not receive any antipsychotics.42,43 Thus, a risk-benefit analysis should be done before prescribing antipsychotics for elderly patients. Both typical and atypical antipsychotics carry black box warnings of increased mortality rates in the elderly.
Other drugs for delirium? More research is needed
Cholinesterase inhibitors. Procholinergic agents would be expected to be helpful in treating delirium, as cholinergic deficiency has been implicated as a predisposing factor for delirium and medications with anticholinergic effects have been shown to induce delirium. However, several studies of cholinesterase inhibitors have not found this to be the case.44-47
Benzodiazepines. There is no evidence to support the use of benzodiazepines in the treatment of delirium, except when the delirium is related to alcohol withdrawal.48 When indicated, the use of a short-acting benzodiazepine such as lorazepam is preferred for elderly patients (vs long-acting agents like diazepam) because of its shorter half-life and better side effect profile.2 Drowsiness, ataxia, and disinhibition are common side effects of benzodiazepines.
Gabapentin. A pilot study conducted to assess the efficacy of gabapentin (900 mg/d) for the prevention of postoperative delirium found a significantly lower incidence of delirium among patients who received gabapentin compared with placebo. This may be associated with gabapentin’s opioid-sparing effect.49 Larger studies are needed to recommend for or against the use of gabapentin in patients receiving opiates.
Further study of the pathophysiology of delirium is needed, as well, to increase our ability to prevent and treat it.
CASE After receiving the IV fluid bolus, Mr. D became increasingly short of breath and required more oxygen to keep his oxygen saturation in the 90s. Labs were ordered during morning rounds, and the patient was found to have urosepsis. He was admitted to the ICU in septic shock, and was intubated and died several days later.
In retrospect, it was determined that Mr. D had developed hypoactive delirium brought on by the infection—and that his somnolence on the second postoperative day was not a sign of overmedication. Had this been recognized early on through the use of an appropriate screening tool, the outcome would likely have been more favorable.
CORRESPONDENCE Abdulraouf Ghandour, MD, Green Meadows Clinic University Physicians, 3217 Providence Road, Columbia, MO 65203; [email protected]
1. Casselman WG. Dictionary of Medical Derivations. The Real Meaning of Medical Terms. New York, NY: Informa Healthcare; 1998.
2. Kiely DK, Bergmann MA, Murphy KM, et al. Delirium among newly admitted postacute facility patients, prevalence, symptoms, and severity. J Gerontol Biol Sci Med Sci. 2003;58:M441-M445.
3. Inouye SK, Charpentier PA. Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275:852-857.
4. Inouye SK. The dilemma of delirium: clinical and research controversies regarding diagnosis and evaluation of delirium in hospitalized elderly medical patients. Am J Med. 1994;97:278-288.
5. Pompei P, Foreman M, Rudberg M, et al. Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42:809-815.
6. Kolbeinsson H, Jonsson A. Delirium and dementia in acute medical admissions of elderly patients in Iceland. Acta Psychiatr Scand. 1993;87:123-127.
7. Cole MG, Primeau FJ. Prognosis of delirium in elderly hospital patients. CMAJ. 1993;149:41-46.
8. Rahkonen T, Eloniemi-Sulkava U, Halonen P, et al. Delirium in the non-demented oldest old in the general population: risk factors and prognosis. Int J Geriatr Psychiatry. 2001;16:415-421.
9. Edlund A, Lundstrom M, Brannstrom B, et al. Delirium before and after operation for femoral neck fracture. J Am Geriatr Soc. 2001;49:1335-1340.
10. Andersson EM, Gustafson L, Hallberg IR. Acute confusional state in elderly orthopaedic patients: factors of importance for detection in nursing care. Int J Geriatr Psychiatry. 2001;16:7-17.
11. Inouye SK, Viscoli CM, Horwitz RI, et al. A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119:474-481.
12. Marcantonio ER, Juarez G, Goldman L, et al. The relationship of postoperative delirium with psychoactive medications. JAMA. 1994;272:1518-1522.
13. Marcantonio ER, Goldman L, Orav EJ, et al. The association of intraoperative factors with the development of postoperative delirium. Am J Med. 1998;105:380-384.
14. Tune L, Carr S, Hoag E, et al. Anticholinergic effects of drugs commonly prescribed for the elderly: potential means for assessing risk of delirium. Am J Psychiatry. 1992;149:1393-1394.
15. Flaherty JH, Shay K, Weir C, et al. The development of a mental status vital sign for use across the spectrum of care . J Am Med Dir Assoc. 2009;10:379-380.
16. Inouye SK, Van Dyck CH, Alessi CA, et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941-948.
17. Inouye SK. Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, Conn: Yale University School of Medicine; 2003.
18. Halter J, Ouslander J, Tinetti M, et al. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York, NY: McGraw-Hill; 2009;648-658.
19. Eriksson S. Social and environmental contributants to delirium in the elderly. Dement Geriatr Cogn Disord. 1999;10:350-352.
20. Francis J, Martin D, Kapoor WN. A prospective study of delirium in hospitalized elderly. JAMA. 1990;263:1097-1101.
21. Francis J, Hilko EM, Kapoor WN. Acute mental change: when are head scans needed? Clin Res. 1991;39:103.-
22. Rudberg MA, Pompei P, Foreman MD, et al. The natural history of delirium in older hospitalized patients: a syndrome of heterogeneity. Age Ageing. 1997;26:169-174.
23. Soiza RL, Sharma V, Ferguson K, et al. Neuroimaging studies of delirium: a systematic review. J Psychosom Res. 2008;65:239-248.
24. Fong TG, Bogardus ST Jr, Daftary A, et al. Cerebral perfusion changes in older delirious patients using 99mTc HMPAO SPECT. J Gerontol A Biol Sci Med Sci. 2006;61:1294-1299.
25. Jacobson SA, Leuchter AF, Walter DO. Conventional and quantitative EEG in the diagnosis of delirium among the elderly. J Neurol Neurosurg Psychiatry. 1993;56:153-158.
26. Marcantonio ER, Flacker JM, Wright RJ, et al. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc. 2001;49:516-522.
27. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340:669-676.
28. Weber JB, Coverdale JH, Kunik ME. Delirium: current trends in prevention and treatment. Intern Med J. 2004;34:115-121.
29. Bo M, Martini B, Ruatta C, et al. Geriatric ward hospitalization reduced incidence delirium among older medical inpatients. Am J Geriatr Psychiatry. 2009;17:760-768.
30. Taguchi T, Yano M, Kido Y. Influence of bright light therapy on postoperative patients: a pilot study. Intensive Crit Care Nurs. 2007;23:289-297.
31. McCaffrey R, Locsin R. The effect of music listening on acute confusion and delirium in elders undergoing elective hip and knee surgery. J Clin Nurs. 2004;13:91-96.
32. Remington R. Calming music and hand massage with agitated elderly. Nurs Res. 2004;51:317-323.
33. Seitz DP, Gill SS, van Zyl LT. Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11-21.
34. Schwartz T, Masand PS. The role of atypical antipsychotics in the treatment of delirium. Psychosomatics. 2002;43:171-174.
35. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.-
36. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium. Chongqing Med J. 2004;8:1234-1237.
37. Han CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45:297-301.
38. Kim SW, Yoo JA, Lee SY, et al. Risperidone versus olanzapine for the treatment of delirium. Hum Psychopharmacol. 2010;25:298-302.
39. Prakanrattana U, Prapaitrakool S. Efficacy of risperidone for prevention of postoperative delirium in cardiac surgery. Anaesth Intensive Care. 2007;35:714-719.
40. Maneeton B, Maneeton N, Srisurapanont M. An open-label study of quetiapine for delirium. J Med Assoc Thai. 2007;90:2158-2163.
41. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38:419-427.
42. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775-786.
43. Wang PS, Schneeweiss S, Avorn J, et al. Death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335-2341.
44. Liptzin B, Laki A, Garb JL, et al. Donepezil in the prevention and treatment of post-surgical delirium. Am J Geriatr Psychiatry. 2005;13:1100-1106.
45. Sampson EL, Raven PR, Ndhlovu PN, et al. A randomized, double-blind, placebo-controlled trial of donepezil hydrochloride (Aricept) for reducing the incidence of postoperative delirium after elective total hip replacement. Int J Geriatr Psychiatry. 2007;22:343-349.
46. Gamberini M, Bolliger D, Lurati Buse GA, et al. Rivastigmine for the prevention of postoperative delirium in elderly patients undergoing elective cardiac surgery—a randomized controlled trial. Crit Care Med. 2009;37:1762-1768.
47. Overshott R, Vernon M, Morris J, et al. Rivastigmine in the treatment of delirium in older people: a pilot study. Int Psychogeriatr. 2010;22:812-818.
48. Lonergan E, Luxenberg J, Areosa Sastre A. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(4):CD006379.-
49. Leung JM, Sands LP, Rico M, et al. Pilot clinical trial of gabapentin to decrease postoperative delirium in older patients. Neurology. 2006;67:1251-1253.
• Nonpharmacologic interventions are the mainstay of treatment for delirium. B
• When medication is needed, atypical antipsychotics are as effective as typical antipsychotics for treating delirium in elderly patients, and have fewer side effects. B
• Benzodiazepines should be avoided in elderly patients with delirium that is not associated with alcohol withdrawal. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mr. D, a 75-year-old patient with a history of hypertension and congestive heart failure, sustained a femoral neck fracture and was admitted to the hospital for surgery. He underwent open reduction and internal fixation and was doing well postoperatively, until Day 2—when his primary care physician made morning rounds and noted that Mr. D was somnolent. The nurse on duty assured the physician that Mr. D was fine and “was awake and alert earlier,” and attributed his somnolence to the oxycodone (10 mg) the patient was taking for pain. The physician ordered a reduction in dosage.
If Mr. D had been your patient, would you have considered other possible causes of his somnolence? Or do you think the physician’s action was sufficient?
Derived from Latin, the word delirium literally means “off the [ploughed] track.”1 Dozens of terms have been used to describe delirium, with acute confusion state, organic brain syndrome, acute brain syndrome, and toxic psychosis among them.
Delirium has been reported to occur in 15% to 30% of patients on general medical units,2 about 40% of postoperative patients, and up to 70% of terminally ill patients.3 The true prevalence is hard to determine, as up to 66% of cases may be missed.4
Delirium is being diagnosed more frequently, however—a likely result of a growing geriatric population, increased longevity, and greater awareness of the condition. Each year, an estimated 2.3 million US residents are affected, leading to prolonged hospitalization; poor functional outcomes; the development or worsening of dementia; increased nursing home placement; and a significant burden for families and the US health care system.5
Delirium is also associated with an increase in mortality.6,7 The mortality rate among hospitalized patients who develop delirium is reported to be 18%, rising to an estimated 47% within the first 3 months after discharge.6 Greater awareness of risk factors, rapid recognition of signs and symptoms of delirium, and early intervention—detailed in the text and tables that follow—will lead to better outcomes.
Assessing risk, evaluating mental status
In addition to advanced age, risk factors for delirium (TABLE 1)8-14 include alcohol use, brain dysfunction, comorbidities, hypertension, malignancy, anticholinergic medications, anemia, metabolic abnormalities, and male sex. In patients who, like Mr. D, have numerous risk factors, early—and frequent—evaluation of mental status is needed. One way to do this is to treat mental status as a vital sign, to be included in the assessment of every elderly patient.15
The Confusion Assessment Method, a quick and easy-to-use delirium screening tool (TABLE 2), has a sensitivity of 94% to 100% and a specificity of 90% to 95%.16,17 There are a number of other screening tools, including the widely used Mini-Mental State Exam (MMSE), as well as the Delirium Rating Scale, Delirium Symptom Interview, and Delirium Severity Scale.
TABLE 1
Risk factors for delirium8-14
Advanced age Alcohol use Brain dysfunction (dementia, epilepsy) Hypertension Male sex Malignancy Medications (mainly anticholinergic) Metabolic abnormalities:
Old age Preoperative anemia Preoperative metabolic abnormalities |
| BUN, blood urea nitrogen; Cr, creatinine; Na, sodium. |
TABLE 2
Screening for delirium: The Confusion Assessment Method*16,17
| Criteria | Evidence Yes to questions 1, 2, and 3 plus 4 or 5 (or both) suggests a delirium diagnosis |
|---|---|
| 1. Acute onset | Is there evidence of an acute change in mental status from the patient’s baseline? |
| 2. Fluctuating course | Did the abnormal behavior fluctuate during the day—ie, tend to come and go or increase and decrease in severity? |
| 3. Inattention | Did the patient have difficulty focusing attention, eg, being easily distractible or having difficulty keeping track of what was being said? |
| PLUS | |
| 4. Disorganized thinking | Was the patient’s thinking disorganized or incoherent, such as rambling or irrelevant conversation, unclear or illogical flow of ideas, or unpredictable switching from subject to subject? |
| 5. Altered level of consciousness | Would you rate the patient’s level of consciousness as (any of the following): – Vigilant (hyperalert) – Lethargic (drowsy, easily aroused) – Stupor (difficult to arouse) – Coma (unarousable) |
| *CAM shortened version worksheet. Adapted from: Inouye SK et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941-948; Inouye SK. Confusion Assessment Method (CAM): Training Manual and Coding Guide. Copyright 2003, Hospital Elder Life Program, LLC. | |
Arriving at a delirium diagnosis
The clinical presentation of delirium is characterized by acute—and reversible—impairment of cognition, attention, orientation, and memory, and disruption of the normal sleep/wake cycle. The Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR) criteria for a delirium diagnosis include all of the following:
- disturbance of consciousness, with a reduced ability to focus, sustain, or shift attention
- change in cognition, or a perceptual disturbance, that is not accounted for by a preexisting or developing dementia
- rapid onset of cognitive impairment, with fluctuation likely during the course of the day
- evidence from the history, physical exam, or laboratory findings that the disturbed consciousness is a direct physiological consequence of a general medical condition.17
There are 3 basic types of delirium, each associated with a different psychomotor disturbance.
- Hyperactive delirium—the least common—is characterized by restlessness and agitation, and is therefore the easiest to diagnose.
- Hypoactive delirium is characterized by psychomotor retardation and hypoalertness. It is often misdiagnosed as depression, and has the poorest prognosis.
- Mixed delirium—the most common—is characterized by symptoms that fluctuate between hyper- and hypoactivity.18
CASE By lunchtime, Mr. D had awakened; however, he needed help with his meal. After eating, he slept for the rest of the day. At night, a nurse paged the resident to report that the patient’s blood pressure was 82/60 mm Hg and his heart rate was 115. The physician ordered an intravenous fluid bolus, which corrected the patient’s hypotension, but only temporarily.
The fluctuating nature of delirium—most notably, in patients’ level of alertness—is helpful in establishing a diagnosis. The history and physical exam are the gold standard tools, both for diagnosing delirium and identifying the underlying cause (TABLE 3).19,20 A review of the patient’s medications should be a key component of the medical history, as drugs—particularly those with anticholinergic properties—are often associated with delirium. Environmental shifts, including hospitalization and a disruption of the normal sleep/wake cycle, endocrine disorders, infection, and nutritional deficiencies are also potential causes of delirium, among others.
If history and physical exam fail to identify the underlying cause, laboratory testing, including complete blood count, complete metabolic profile, and urinalysis, should be done. Brain imaging is usually not needed for individuals with symptoms of delirium, but computed tomography (CT) may be indicated if a patient’s condition continues to deteriorate while the underlying cause remains unidentified.21 Electroencephalography (EEG) may be used to confirm a delirium diagnosis that’s uncertain, in a patient with underlying dementia, for instance. (In more than 16% of cases of delirium, the cause is unknown.22)
The most common structural abnormalities found in patients with delirium are brain atrophy and increased white matter lesions, as well as basal ganglia lesions.23 Single-photon emission CT (SPECT) shows a reduction of regional cerebral perfusion by 50%,24 while EEG shows slowing of the posterior dominant rhythm and increased generalized slow-wave activity.25
TABLE 3
A DELIRIUM mnemonic to get to the heart of the problem19,20
| Cause | Comment |
|---|---|
| Drugs | Drug classes: Anesthesia, anticholinergics, anticonvulsants, antiemetics, antihistamines, antihypertensives, antimicrobials, antipsychotics, benzodiazepines, corticosteroids, hypnotics, H2 blockers, muscle relaxants, NSAIDs, opioids, SSRIs, tricyclic antidepressants Drugs: digoxin, levodopa, lithium, theophylline OTCs: henbane, Jimson weed, mandrake, Atropa belladonna extract |
| Environmental | Change of environment, sensory deprivation, sleep deprivation |
| Endocrine | Hyperparathyroidism, hyper-/hypothyroidism |
| Low perfusion | MI, pulmonary embolism, CVA |
| Infection | Pneumonia, sepsis, systemic infection, UTI |
| Retention | Fecal impaction, urinary retention |
| Intoxication | Alcohol, illegal drugs/drug overdose |
| Undernutrition | Malnutrition, thiamin deficiency, vitamin B12 deficiency |
| Metabolic | Acid-base disturbances, fluid and electrolyte abnormalities, hepatic or uremic encephalopathy, hypercarbia, hyper-/hypoglycemia, hyperosmolality, hypoxia |
| Subdural | History of falls |
| CVA, cerebrovascular accident; MI, myocardial infarction; NSAIDs, nonsteroidal anti-inflammatory drugs; OTCs, over-the-counter agents; SSRIs, selective serotonin reuptake inhibitors; UTI, urinary tract infection. | |
Treating (or preventing) delirium: Start with these steps
Nonpharmacologic interventions are the mainstay of treatment for patients with delirium, and may also help to prevent the development of delirium in patients at risk. One key measure is to correct, or avoid, disruptions in the patient’s normal sleep/wake cycle—eg, restoring circadian rhythm by avoiding,
to the extent possible, awakening the patient at night for medication or vital signs. Preventing sensory deprivation, by ensuring that the patient’s eyeglasses and hearing aid are nearby and that there is a clock and calendar nearby and adequate light, is also helpful. Other key interventions (TABLE 4)26-28 include:
- limiting medications associated with delirium (and eliminating any nonessential medication)
- improving nutrition and ambulation
- correcting electrolyte and fluid disturbances
- treating infection
- involving family members in patient care
- ensuring that patients receive adequate pain management
- avoiding transfers (if the patient is hospitalized) and trying to secure a single room.
Several studies have evaluated the effectiveness of nonpharmacologic interventions in preventing or lowering the incidence of delirium. A large multicomponent delirium prevention study of patients >70 years on general medical units focused on managing risk factors. The interventions studied included (1) avoidance of sensory deprivation, (2) early mobilization, (3) treating dehydration, (4) implementing noise reduction strategies and sleep enhancement programs, and (5) avoiding the use of sleep medications. These interventions proved to be effective not only in lowering the incidence of delirium, but in shortening the duration of delirium in affected patients (NNT=20).27
One study found that proactively using a geriatric consultation model (ie, implementing standardized protocols for the management of 6 risk factors) for elderly hospitalized patients led to a reduction in the incidence of delirium by more than a third.26 Admission to a specialized geriatric unit is associated with a lower incidence of delirium compared with being hospitalized on a general medical unit.29
Reducing the incidence of postoperative delirium. Bright light therapy (a light intensity of 5000 lux with a distance from the light source of 100 cm), implemented postoperatively, may play a role in reducing the incidence of delirium, research suggests.30 Music may be helpful, as well. An RCT involving patients (>65 years) undergoing elective knee or hip surgery found that those who listened to classical music postoperatively had a lower incidence of delirium.31 Similarly, playing music in nursing homes has been shown to decrease aggressive behavior and agitation.32
TABLE 4
Helpful interventions in the hospital or at home26-28
|
When medication is needed, proceed with caution
None of the medications currently used to treat delirium are approved by the US Food and Drug Administration for this indication, and many of them have substantial side effects. Nonetheless, palliative or symptomatic treatment requires some form of sedation for agitated patients with delirium. Thus, it is necessary to strike a balance in order to manage the symptoms of delirium and avoid potential side effects (primarily, sedation). Overly sedating patients can confuse the clinical picture of delirium and make it difficult to differentiate between ongoing delirium and medication side effects. Medication should be started at a low, but frequent, dose to achieve an effective therapeutic level, after which a lower maintenance dose can be used until the cause of delirium is resolved.
Antipsychotics are the cornerstone of drug treatment
Haloperidol has traditionally been used to treat delirium33 and has proven effectiveness. However, it is associated with increased risk of extrapyramidal manifestations compared with atypical antipsychotics.
Atypical antipsychotics (olanzapine, risperidone, quetiapine) are increasingly being used to treat delirium because they have fewer extrapyramidal side effects.34 With the exception of olanzapine (available in intramuscular and oral disintegrating form), atypical antipsychotics are available only in oral form, which may limit their usefulness as a treatment for agitated, delirious patients.
Risperidone (at a dose ranging from 0.25 to 1 mg/d) and olanzapine (1.25 to 2.5 mg/d) have shown similar efficacy to haloperidol (0.75 to 1.5 mg/d) in both the prevention and treatment of delirium, but with fewer extrapyramidal side effects.35-39 Quetiapine, a second-generation antipsychotic, is widely used to treat inpatient delirium, although there are no large RCTs comparing it with placebo. One pilot study and another open-label trial found the drug to be beneficial for patients with delirium, with fewer extrapyramidal side effects than haloperidol.40,41
Do a risk-benefit analysis. The use of antipsychotics in elderly patients with delirium has been associated with increased morbidity and mortality. The incidence of stroke and death were higher for community-dwelling patients (NNH=100) and patients in long-term care (N=67) who received typical or atypical antipsychotics for 6 months compared with that of patients who did not receive any antipsychotics.42,43 Thus, a risk-benefit analysis should be done before prescribing antipsychotics for elderly patients. Both typical and atypical antipsychotics carry black box warnings of increased mortality rates in the elderly.
Other drugs for delirium? More research is needed
Cholinesterase inhibitors. Procholinergic agents would be expected to be helpful in treating delirium, as cholinergic deficiency has been implicated as a predisposing factor for delirium and medications with anticholinergic effects have been shown to induce delirium. However, several studies of cholinesterase inhibitors have not found this to be the case.44-47
Benzodiazepines. There is no evidence to support the use of benzodiazepines in the treatment of delirium, except when the delirium is related to alcohol withdrawal.48 When indicated, the use of a short-acting benzodiazepine such as lorazepam is preferred for elderly patients (vs long-acting agents like diazepam) because of its shorter half-life and better side effect profile.2 Drowsiness, ataxia, and disinhibition are common side effects of benzodiazepines.
Gabapentin. A pilot study conducted to assess the efficacy of gabapentin (900 mg/d) for the prevention of postoperative delirium found a significantly lower incidence of delirium among patients who received gabapentin compared with placebo. This may be associated with gabapentin’s opioid-sparing effect.49 Larger studies are needed to recommend for or against the use of gabapentin in patients receiving opiates.
Further study of the pathophysiology of delirium is needed, as well, to increase our ability to prevent and treat it.
CASE After receiving the IV fluid bolus, Mr. D became increasingly short of breath and required more oxygen to keep his oxygen saturation in the 90s. Labs were ordered during morning rounds, and the patient was found to have urosepsis. He was admitted to the ICU in septic shock, and was intubated and died several days later.
In retrospect, it was determined that Mr. D had developed hypoactive delirium brought on by the infection—and that his somnolence on the second postoperative day was not a sign of overmedication. Had this been recognized early on through the use of an appropriate screening tool, the outcome would likely have been more favorable.
CORRESPONDENCE Abdulraouf Ghandour, MD, Green Meadows Clinic University Physicians, 3217 Providence Road, Columbia, MO 65203; [email protected]
• Nonpharmacologic interventions are the mainstay of treatment for delirium. B
• When medication is needed, atypical antipsychotics are as effective as typical antipsychotics for treating delirium in elderly patients, and have fewer side effects. B
• Benzodiazepines should be avoided in elderly patients with delirium that is not associated with alcohol withdrawal. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mr. D, a 75-year-old patient with a history of hypertension and congestive heart failure, sustained a femoral neck fracture and was admitted to the hospital for surgery. He underwent open reduction and internal fixation and was doing well postoperatively, until Day 2—when his primary care physician made morning rounds and noted that Mr. D was somnolent. The nurse on duty assured the physician that Mr. D was fine and “was awake and alert earlier,” and attributed his somnolence to the oxycodone (10 mg) the patient was taking for pain. The physician ordered a reduction in dosage.
If Mr. D had been your patient, would you have considered other possible causes of his somnolence? Or do you think the physician’s action was sufficient?
Derived from Latin, the word delirium literally means “off the [ploughed] track.”1 Dozens of terms have been used to describe delirium, with acute confusion state, organic brain syndrome, acute brain syndrome, and toxic psychosis among them.
Delirium has been reported to occur in 15% to 30% of patients on general medical units,2 about 40% of postoperative patients, and up to 70% of terminally ill patients.3 The true prevalence is hard to determine, as up to 66% of cases may be missed.4
Delirium is being diagnosed more frequently, however—a likely result of a growing geriatric population, increased longevity, and greater awareness of the condition. Each year, an estimated 2.3 million US residents are affected, leading to prolonged hospitalization; poor functional outcomes; the development or worsening of dementia; increased nursing home placement; and a significant burden for families and the US health care system.5
Delirium is also associated with an increase in mortality.6,7 The mortality rate among hospitalized patients who develop delirium is reported to be 18%, rising to an estimated 47% within the first 3 months after discharge.6 Greater awareness of risk factors, rapid recognition of signs and symptoms of delirium, and early intervention—detailed in the text and tables that follow—will lead to better outcomes.
Assessing risk, evaluating mental status
In addition to advanced age, risk factors for delirium (TABLE 1)8-14 include alcohol use, brain dysfunction, comorbidities, hypertension, malignancy, anticholinergic medications, anemia, metabolic abnormalities, and male sex. In patients who, like Mr. D, have numerous risk factors, early—and frequent—evaluation of mental status is needed. One way to do this is to treat mental status as a vital sign, to be included in the assessment of every elderly patient.15
The Confusion Assessment Method, a quick and easy-to-use delirium screening tool (TABLE 2), has a sensitivity of 94% to 100% and a specificity of 90% to 95%.16,17 There are a number of other screening tools, including the widely used Mini-Mental State Exam (MMSE), as well as the Delirium Rating Scale, Delirium Symptom Interview, and Delirium Severity Scale.
TABLE 1
Risk factors for delirium8-14
Advanced age Alcohol use Brain dysfunction (dementia, epilepsy) Hypertension Male sex Malignancy Medications (mainly anticholinergic) Metabolic abnormalities:
Old age Preoperative anemia Preoperative metabolic abnormalities |
| BUN, blood urea nitrogen; Cr, creatinine; Na, sodium. |
TABLE 2
Screening for delirium: The Confusion Assessment Method*16,17
| Criteria | Evidence Yes to questions 1, 2, and 3 plus 4 or 5 (or both) suggests a delirium diagnosis |
|---|---|
| 1. Acute onset | Is there evidence of an acute change in mental status from the patient’s baseline? |
| 2. Fluctuating course | Did the abnormal behavior fluctuate during the day—ie, tend to come and go or increase and decrease in severity? |
| 3. Inattention | Did the patient have difficulty focusing attention, eg, being easily distractible or having difficulty keeping track of what was being said? |
| PLUS | |
| 4. Disorganized thinking | Was the patient’s thinking disorganized or incoherent, such as rambling or irrelevant conversation, unclear or illogical flow of ideas, or unpredictable switching from subject to subject? |
| 5. Altered level of consciousness | Would you rate the patient’s level of consciousness as (any of the following): – Vigilant (hyperalert) – Lethargic (drowsy, easily aroused) – Stupor (difficult to arouse) – Coma (unarousable) |
| *CAM shortened version worksheet. Adapted from: Inouye SK et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941-948; Inouye SK. Confusion Assessment Method (CAM): Training Manual and Coding Guide. Copyright 2003, Hospital Elder Life Program, LLC. | |
Arriving at a delirium diagnosis
The clinical presentation of delirium is characterized by acute—and reversible—impairment of cognition, attention, orientation, and memory, and disruption of the normal sleep/wake cycle. The Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR) criteria for a delirium diagnosis include all of the following:
- disturbance of consciousness, with a reduced ability to focus, sustain, or shift attention
- change in cognition, or a perceptual disturbance, that is not accounted for by a preexisting or developing dementia
- rapid onset of cognitive impairment, with fluctuation likely during the course of the day
- evidence from the history, physical exam, or laboratory findings that the disturbed consciousness is a direct physiological consequence of a general medical condition.17
There are 3 basic types of delirium, each associated with a different psychomotor disturbance.
- Hyperactive delirium—the least common—is characterized by restlessness and agitation, and is therefore the easiest to diagnose.
- Hypoactive delirium is characterized by psychomotor retardation and hypoalertness. It is often misdiagnosed as depression, and has the poorest prognosis.
- Mixed delirium—the most common—is characterized by symptoms that fluctuate between hyper- and hypoactivity.18
CASE By lunchtime, Mr. D had awakened; however, he needed help with his meal. After eating, he slept for the rest of the day. At night, a nurse paged the resident to report that the patient’s blood pressure was 82/60 mm Hg and his heart rate was 115. The physician ordered an intravenous fluid bolus, which corrected the patient’s hypotension, but only temporarily.
The fluctuating nature of delirium—most notably, in patients’ level of alertness—is helpful in establishing a diagnosis. The history and physical exam are the gold standard tools, both for diagnosing delirium and identifying the underlying cause (TABLE 3).19,20 A review of the patient’s medications should be a key component of the medical history, as drugs—particularly those with anticholinergic properties—are often associated with delirium. Environmental shifts, including hospitalization and a disruption of the normal sleep/wake cycle, endocrine disorders, infection, and nutritional deficiencies are also potential causes of delirium, among others.
If history and physical exam fail to identify the underlying cause, laboratory testing, including complete blood count, complete metabolic profile, and urinalysis, should be done. Brain imaging is usually not needed for individuals with symptoms of delirium, but computed tomography (CT) may be indicated if a patient’s condition continues to deteriorate while the underlying cause remains unidentified.21 Electroencephalography (EEG) may be used to confirm a delirium diagnosis that’s uncertain, in a patient with underlying dementia, for instance. (In more than 16% of cases of delirium, the cause is unknown.22)
The most common structural abnormalities found in patients with delirium are brain atrophy and increased white matter lesions, as well as basal ganglia lesions.23 Single-photon emission CT (SPECT) shows a reduction of regional cerebral perfusion by 50%,24 while EEG shows slowing of the posterior dominant rhythm and increased generalized slow-wave activity.25
TABLE 3
A DELIRIUM mnemonic to get to the heart of the problem19,20
| Cause | Comment |
|---|---|
| Drugs | Drug classes: Anesthesia, anticholinergics, anticonvulsants, antiemetics, antihistamines, antihypertensives, antimicrobials, antipsychotics, benzodiazepines, corticosteroids, hypnotics, H2 blockers, muscle relaxants, NSAIDs, opioids, SSRIs, tricyclic antidepressants Drugs: digoxin, levodopa, lithium, theophylline OTCs: henbane, Jimson weed, mandrake, Atropa belladonna extract |
| Environmental | Change of environment, sensory deprivation, sleep deprivation |
| Endocrine | Hyperparathyroidism, hyper-/hypothyroidism |
| Low perfusion | MI, pulmonary embolism, CVA |
| Infection | Pneumonia, sepsis, systemic infection, UTI |
| Retention | Fecal impaction, urinary retention |
| Intoxication | Alcohol, illegal drugs/drug overdose |
| Undernutrition | Malnutrition, thiamin deficiency, vitamin B12 deficiency |
| Metabolic | Acid-base disturbances, fluid and electrolyte abnormalities, hepatic or uremic encephalopathy, hypercarbia, hyper-/hypoglycemia, hyperosmolality, hypoxia |
| Subdural | History of falls |
| CVA, cerebrovascular accident; MI, myocardial infarction; NSAIDs, nonsteroidal anti-inflammatory drugs; OTCs, over-the-counter agents; SSRIs, selective serotonin reuptake inhibitors; UTI, urinary tract infection. | |
Treating (or preventing) delirium: Start with these steps
Nonpharmacologic interventions are the mainstay of treatment for patients with delirium, and may also help to prevent the development of delirium in patients at risk. One key measure is to correct, or avoid, disruptions in the patient’s normal sleep/wake cycle—eg, restoring circadian rhythm by avoiding,
to the extent possible, awakening the patient at night for medication or vital signs. Preventing sensory deprivation, by ensuring that the patient’s eyeglasses and hearing aid are nearby and that there is a clock and calendar nearby and adequate light, is also helpful. Other key interventions (TABLE 4)26-28 include:
- limiting medications associated with delirium (and eliminating any nonessential medication)
- improving nutrition and ambulation
- correcting electrolyte and fluid disturbances
- treating infection
- involving family members in patient care
- ensuring that patients receive adequate pain management
- avoiding transfers (if the patient is hospitalized) and trying to secure a single room.
Several studies have evaluated the effectiveness of nonpharmacologic interventions in preventing or lowering the incidence of delirium. A large multicomponent delirium prevention study of patients >70 years on general medical units focused on managing risk factors. The interventions studied included (1) avoidance of sensory deprivation, (2) early mobilization, (3) treating dehydration, (4) implementing noise reduction strategies and sleep enhancement programs, and (5) avoiding the use of sleep medications. These interventions proved to be effective not only in lowering the incidence of delirium, but in shortening the duration of delirium in affected patients (NNT=20).27
One study found that proactively using a geriatric consultation model (ie, implementing standardized protocols for the management of 6 risk factors) for elderly hospitalized patients led to a reduction in the incidence of delirium by more than a third.26 Admission to a specialized geriatric unit is associated with a lower incidence of delirium compared with being hospitalized on a general medical unit.29
Reducing the incidence of postoperative delirium. Bright light therapy (a light intensity of 5000 lux with a distance from the light source of 100 cm), implemented postoperatively, may play a role in reducing the incidence of delirium, research suggests.30 Music may be helpful, as well. An RCT involving patients (>65 years) undergoing elective knee or hip surgery found that those who listened to classical music postoperatively had a lower incidence of delirium.31 Similarly, playing music in nursing homes has been shown to decrease aggressive behavior and agitation.32
TABLE 4
Helpful interventions in the hospital or at home26-28
|
When medication is needed, proceed with caution
None of the medications currently used to treat delirium are approved by the US Food and Drug Administration for this indication, and many of them have substantial side effects. Nonetheless, palliative or symptomatic treatment requires some form of sedation for agitated patients with delirium. Thus, it is necessary to strike a balance in order to manage the symptoms of delirium and avoid potential side effects (primarily, sedation). Overly sedating patients can confuse the clinical picture of delirium and make it difficult to differentiate between ongoing delirium and medication side effects. Medication should be started at a low, but frequent, dose to achieve an effective therapeutic level, after which a lower maintenance dose can be used until the cause of delirium is resolved.
Antipsychotics are the cornerstone of drug treatment
Haloperidol has traditionally been used to treat delirium33 and has proven effectiveness. However, it is associated with increased risk of extrapyramidal manifestations compared with atypical antipsychotics.
Atypical antipsychotics (olanzapine, risperidone, quetiapine) are increasingly being used to treat delirium because they have fewer extrapyramidal side effects.34 With the exception of olanzapine (available in intramuscular and oral disintegrating form), atypical antipsychotics are available only in oral form, which may limit their usefulness as a treatment for agitated, delirious patients.
Risperidone (at a dose ranging from 0.25 to 1 mg/d) and olanzapine (1.25 to 2.5 mg/d) have shown similar efficacy to haloperidol (0.75 to 1.5 mg/d) in both the prevention and treatment of delirium, but with fewer extrapyramidal side effects.35-39 Quetiapine, a second-generation antipsychotic, is widely used to treat inpatient delirium, although there are no large RCTs comparing it with placebo. One pilot study and another open-label trial found the drug to be beneficial for patients with delirium, with fewer extrapyramidal side effects than haloperidol.40,41
Do a risk-benefit analysis. The use of antipsychotics in elderly patients with delirium has been associated with increased morbidity and mortality. The incidence of stroke and death were higher for community-dwelling patients (NNH=100) and patients in long-term care (N=67) who received typical or atypical antipsychotics for 6 months compared with that of patients who did not receive any antipsychotics.42,43 Thus, a risk-benefit analysis should be done before prescribing antipsychotics for elderly patients. Both typical and atypical antipsychotics carry black box warnings of increased mortality rates in the elderly.
Other drugs for delirium? More research is needed
Cholinesterase inhibitors. Procholinergic agents would be expected to be helpful in treating delirium, as cholinergic deficiency has been implicated as a predisposing factor for delirium and medications with anticholinergic effects have been shown to induce delirium. However, several studies of cholinesterase inhibitors have not found this to be the case.44-47
Benzodiazepines. There is no evidence to support the use of benzodiazepines in the treatment of delirium, except when the delirium is related to alcohol withdrawal.48 When indicated, the use of a short-acting benzodiazepine such as lorazepam is preferred for elderly patients (vs long-acting agents like diazepam) because of its shorter half-life and better side effect profile.2 Drowsiness, ataxia, and disinhibition are common side effects of benzodiazepines.
Gabapentin. A pilot study conducted to assess the efficacy of gabapentin (900 mg/d) for the prevention of postoperative delirium found a significantly lower incidence of delirium among patients who received gabapentin compared with placebo. This may be associated with gabapentin’s opioid-sparing effect.49 Larger studies are needed to recommend for or against the use of gabapentin in patients receiving opiates.
Further study of the pathophysiology of delirium is needed, as well, to increase our ability to prevent and treat it.
CASE After receiving the IV fluid bolus, Mr. D became increasingly short of breath and required more oxygen to keep his oxygen saturation in the 90s. Labs were ordered during morning rounds, and the patient was found to have urosepsis. He was admitted to the ICU in septic shock, and was intubated and died several days later.
In retrospect, it was determined that Mr. D had developed hypoactive delirium brought on by the infection—and that his somnolence on the second postoperative day was not a sign of overmedication. Had this been recognized early on through the use of an appropriate screening tool, the outcome would likely have been more favorable.
CORRESPONDENCE Abdulraouf Ghandour, MD, Green Meadows Clinic University Physicians, 3217 Providence Road, Columbia, MO 65203; [email protected]
1. Casselman WG. Dictionary of Medical Derivations. The Real Meaning of Medical Terms. New York, NY: Informa Healthcare; 1998.
2. Kiely DK, Bergmann MA, Murphy KM, et al. Delirium among newly admitted postacute facility patients, prevalence, symptoms, and severity. J Gerontol Biol Sci Med Sci. 2003;58:M441-M445.
3. Inouye SK, Charpentier PA. Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275:852-857.
4. Inouye SK. The dilemma of delirium: clinical and research controversies regarding diagnosis and evaluation of delirium in hospitalized elderly medical patients. Am J Med. 1994;97:278-288.
5. Pompei P, Foreman M, Rudberg M, et al. Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42:809-815.
6. Kolbeinsson H, Jonsson A. Delirium and dementia in acute medical admissions of elderly patients in Iceland. Acta Psychiatr Scand. 1993;87:123-127.
7. Cole MG, Primeau FJ. Prognosis of delirium in elderly hospital patients. CMAJ. 1993;149:41-46.
8. Rahkonen T, Eloniemi-Sulkava U, Halonen P, et al. Delirium in the non-demented oldest old in the general population: risk factors and prognosis. Int J Geriatr Psychiatry. 2001;16:415-421.
9. Edlund A, Lundstrom M, Brannstrom B, et al. Delirium before and after operation for femoral neck fracture. J Am Geriatr Soc. 2001;49:1335-1340.
10. Andersson EM, Gustafson L, Hallberg IR. Acute confusional state in elderly orthopaedic patients: factors of importance for detection in nursing care. Int J Geriatr Psychiatry. 2001;16:7-17.
11. Inouye SK, Viscoli CM, Horwitz RI, et al. A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119:474-481.
12. Marcantonio ER, Juarez G, Goldman L, et al. The relationship of postoperative delirium with psychoactive medications. JAMA. 1994;272:1518-1522.
13. Marcantonio ER, Goldman L, Orav EJ, et al. The association of intraoperative factors with the development of postoperative delirium. Am J Med. 1998;105:380-384.
14. Tune L, Carr S, Hoag E, et al. Anticholinergic effects of drugs commonly prescribed for the elderly: potential means for assessing risk of delirium. Am J Psychiatry. 1992;149:1393-1394.
15. Flaherty JH, Shay K, Weir C, et al. The development of a mental status vital sign for use across the spectrum of care . J Am Med Dir Assoc. 2009;10:379-380.
16. Inouye SK, Van Dyck CH, Alessi CA, et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941-948.
17. Inouye SK. Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, Conn: Yale University School of Medicine; 2003.
18. Halter J, Ouslander J, Tinetti M, et al. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York, NY: McGraw-Hill; 2009;648-658.
19. Eriksson S. Social and environmental contributants to delirium in the elderly. Dement Geriatr Cogn Disord. 1999;10:350-352.
20. Francis J, Martin D, Kapoor WN. A prospective study of delirium in hospitalized elderly. JAMA. 1990;263:1097-1101.
21. Francis J, Hilko EM, Kapoor WN. Acute mental change: when are head scans needed? Clin Res. 1991;39:103.-
22. Rudberg MA, Pompei P, Foreman MD, et al. The natural history of delirium in older hospitalized patients: a syndrome of heterogeneity. Age Ageing. 1997;26:169-174.
23. Soiza RL, Sharma V, Ferguson K, et al. Neuroimaging studies of delirium: a systematic review. J Psychosom Res. 2008;65:239-248.
24. Fong TG, Bogardus ST Jr, Daftary A, et al. Cerebral perfusion changes in older delirious patients using 99mTc HMPAO SPECT. J Gerontol A Biol Sci Med Sci. 2006;61:1294-1299.
25. Jacobson SA, Leuchter AF, Walter DO. Conventional and quantitative EEG in the diagnosis of delirium among the elderly. J Neurol Neurosurg Psychiatry. 1993;56:153-158.
26. Marcantonio ER, Flacker JM, Wright RJ, et al. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc. 2001;49:516-522.
27. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340:669-676.
28. Weber JB, Coverdale JH, Kunik ME. Delirium: current trends in prevention and treatment. Intern Med J. 2004;34:115-121.
29. Bo M, Martini B, Ruatta C, et al. Geriatric ward hospitalization reduced incidence delirium among older medical inpatients. Am J Geriatr Psychiatry. 2009;17:760-768.
30. Taguchi T, Yano M, Kido Y. Influence of bright light therapy on postoperative patients: a pilot study. Intensive Crit Care Nurs. 2007;23:289-297.
31. McCaffrey R, Locsin R. The effect of music listening on acute confusion and delirium in elders undergoing elective hip and knee surgery. J Clin Nurs. 2004;13:91-96.
32. Remington R. Calming music and hand massage with agitated elderly. Nurs Res. 2004;51:317-323.
33. Seitz DP, Gill SS, van Zyl LT. Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11-21.
34. Schwartz T, Masand PS. The role of atypical antipsychotics in the treatment of delirium. Psychosomatics. 2002;43:171-174.
35. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.-
36. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium. Chongqing Med J. 2004;8:1234-1237.
37. Han CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45:297-301.
38. Kim SW, Yoo JA, Lee SY, et al. Risperidone versus olanzapine for the treatment of delirium. Hum Psychopharmacol. 2010;25:298-302.
39. Prakanrattana U, Prapaitrakool S. Efficacy of risperidone for prevention of postoperative delirium in cardiac surgery. Anaesth Intensive Care. 2007;35:714-719.
40. Maneeton B, Maneeton N, Srisurapanont M. An open-label study of quetiapine for delirium. J Med Assoc Thai. 2007;90:2158-2163.
41. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38:419-427.
42. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775-786.
43. Wang PS, Schneeweiss S, Avorn J, et al. Death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335-2341.
44. Liptzin B, Laki A, Garb JL, et al. Donepezil in the prevention and treatment of post-surgical delirium. Am J Geriatr Psychiatry. 2005;13:1100-1106.
45. Sampson EL, Raven PR, Ndhlovu PN, et al. A randomized, double-blind, placebo-controlled trial of donepezil hydrochloride (Aricept) for reducing the incidence of postoperative delirium after elective total hip replacement. Int J Geriatr Psychiatry. 2007;22:343-349.
46. Gamberini M, Bolliger D, Lurati Buse GA, et al. Rivastigmine for the prevention of postoperative delirium in elderly patients undergoing elective cardiac surgery—a randomized controlled trial. Crit Care Med. 2009;37:1762-1768.
47. Overshott R, Vernon M, Morris J, et al. Rivastigmine in the treatment of delirium in older people: a pilot study. Int Psychogeriatr. 2010;22:812-818.
48. Lonergan E, Luxenberg J, Areosa Sastre A. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(4):CD006379.-
49. Leung JM, Sands LP, Rico M, et al. Pilot clinical trial of gabapentin to decrease postoperative delirium in older patients. Neurology. 2006;67:1251-1253.
1. Casselman WG. Dictionary of Medical Derivations. The Real Meaning of Medical Terms. New York, NY: Informa Healthcare; 1998.
2. Kiely DK, Bergmann MA, Murphy KM, et al. Delirium among newly admitted postacute facility patients, prevalence, symptoms, and severity. J Gerontol Biol Sci Med Sci. 2003;58:M441-M445.
3. Inouye SK, Charpentier PA. Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275:852-857.
4. Inouye SK. The dilemma of delirium: clinical and research controversies regarding diagnosis and evaluation of delirium in hospitalized elderly medical patients. Am J Med. 1994;97:278-288.
5. Pompei P, Foreman M, Rudberg M, et al. Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42:809-815.
6. Kolbeinsson H, Jonsson A. Delirium and dementia in acute medical admissions of elderly patients in Iceland. Acta Psychiatr Scand. 1993;87:123-127.
7. Cole MG, Primeau FJ. Prognosis of delirium in elderly hospital patients. CMAJ. 1993;149:41-46.
8. Rahkonen T, Eloniemi-Sulkava U, Halonen P, et al. Delirium in the non-demented oldest old in the general population: risk factors and prognosis. Int J Geriatr Psychiatry. 2001;16:415-421.
9. Edlund A, Lundstrom M, Brannstrom B, et al. Delirium before and after operation for femoral neck fracture. J Am Geriatr Soc. 2001;49:1335-1340.
10. Andersson EM, Gustafson L, Hallberg IR. Acute confusional state in elderly orthopaedic patients: factors of importance for detection in nursing care. Int J Geriatr Psychiatry. 2001;16:7-17.
11. Inouye SK, Viscoli CM, Horwitz RI, et al. A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119:474-481.
12. Marcantonio ER, Juarez G, Goldman L, et al. The relationship of postoperative delirium with psychoactive medications. JAMA. 1994;272:1518-1522.
13. Marcantonio ER, Goldman L, Orav EJ, et al. The association of intraoperative factors with the development of postoperative delirium. Am J Med. 1998;105:380-384.
14. Tune L, Carr S, Hoag E, et al. Anticholinergic effects of drugs commonly prescribed for the elderly: potential means for assessing risk of delirium. Am J Psychiatry. 1992;149:1393-1394.
15. Flaherty JH, Shay K, Weir C, et al. The development of a mental status vital sign for use across the spectrum of care . J Am Med Dir Assoc. 2009;10:379-380.
16. Inouye SK, Van Dyck CH, Alessi CA, et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941-948.
17. Inouye SK. Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, Conn: Yale University School of Medicine; 2003.
18. Halter J, Ouslander J, Tinetti M, et al. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York, NY: McGraw-Hill; 2009;648-658.
19. Eriksson S. Social and environmental contributants to delirium in the elderly. Dement Geriatr Cogn Disord. 1999;10:350-352.
20. Francis J, Martin D, Kapoor WN. A prospective study of delirium in hospitalized elderly. JAMA. 1990;263:1097-1101.
21. Francis J, Hilko EM, Kapoor WN. Acute mental change: when are head scans needed? Clin Res. 1991;39:103.-
22. Rudberg MA, Pompei P, Foreman MD, et al. The natural history of delirium in older hospitalized patients: a syndrome of heterogeneity. Age Ageing. 1997;26:169-174.
23. Soiza RL, Sharma V, Ferguson K, et al. Neuroimaging studies of delirium: a systematic review. J Psychosom Res. 2008;65:239-248.
24. Fong TG, Bogardus ST Jr, Daftary A, et al. Cerebral perfusion changes in older delirious patients using 99mTc HMPAO SPECT. J Gerontol A Biol Sci Med Sci. 2006;61:1294-1299.
25. Jacobson SA, Leuchter AF, Walter DO. Conventional and quantitative EEG in the diagnosis of delirium among the elderly. J Neurol Neurosurg Psychiatry. 1993;56:153-158.
26. Marcantonio ER, Flacker JM, Wright RJ, et al. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc. 2001;49:516-522.
27. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340:669-676.
28. Weber JB, Coverdale JH, Kunik ME. Delirium: current trends in prevention and treatment. Intern Med J. 2004;34:115-121.
29. Bo M, Martini B, Ruatta C, et al. Geriatric ward hospitalization reduced incidence delirium among older medical inpatients. Am J Geriatr Psychiatry. 2009;17:760-768.
30. Taguchi T, Yano M, Kido Y. Influence of bright light therapy on postoperative patients: a pilot study. Intensive Crit Care Nurs. 2007;23:289-297.
31. McCaffrey R, Locsin R. The effect of music listening on acute confusion and delirium in elders undergoing elective hip and knee surgery. J Clin Nurs. 2004;13:91-96.
32. Remington R. Calming music and hand massage with agitated elderly. Nurs Res. 2004;51:317-323.
33. Seitz DP, Gill SS, van Zyl LT. Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11-21.
34. Schwartz T, Masand PS. The role of atypical antipsychotics in the treatment of delirium. Psychosomatics. 2002;43:171-174.
35. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.-
36. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium. Chongqing Med J. 2004;8:1234-1237.
37. Han CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45:297-301.
38. Kim SW, Yoo JA, Lee SY, et al. Risperidone versus olanzapine for the treatment of delirium. Hum Psychopharmacol. 2010;25:298-302.
39. Prakanrattana U, Prapaitrakool S. Efficacy of risperidone for prevention of postoperative delirium in cardiac surgery. Anaesth Intensive Care. 2007;35:714-719.
40. Maneeton B, Maneeton N, Srisurapanont M. An open-label study of quetiapine for delirium. J Med Assoc Thai. 2007;90:2158-2163.
41. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38:419-427.
42. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775-786.
43. Wang PS, Schneeweiss S, Avorn J, et al. Death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335-2341.
44. Liptzin B, Laki A, Garb JL, et al. Donepezil in the prevention and treatment of post-surgical delirium. Am J Geriatr Psychiatry. 2005;13:1100-1106.
45. Sampson EL, Raven PR, Ndhlovu PN, et al. A randomized, double-blind, placebo-controlled trial of donepezil hydrochloride (Aricept) for reducing the incidence of postoperative delirium after elective total hip replacement. Int J Geriatr Psychiatry. 2007;22:343-349.
46. Gamberini M, Bolliger D, Lurati Buse GA, et al. Rivastigmine for the prevention of postoperative delirium in elderly patients undergoing elective cardiac surgery—a randomized controlled trial. Crit Care Med. 2009;37:1762-1768.
47. Overshott R, Vernon M, Morris J, et al. Rivastigmine in the treatment of delirium in older people: a pilot study. Int Psychogeriatr. 2010;22:812-818.
48. Lonergan E, Luxenberg J, Areosa Sastre A. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(4):CD006379.-
49. Leung JM, Sands LP, Rico M, et al. Pilot clinical trial of gabapentin to decrease postoperative delirium in older patients. Neurology. 2006;67:1251-1253.