User login
Dermatofibrosarcoma Protuberans
To the Editor:
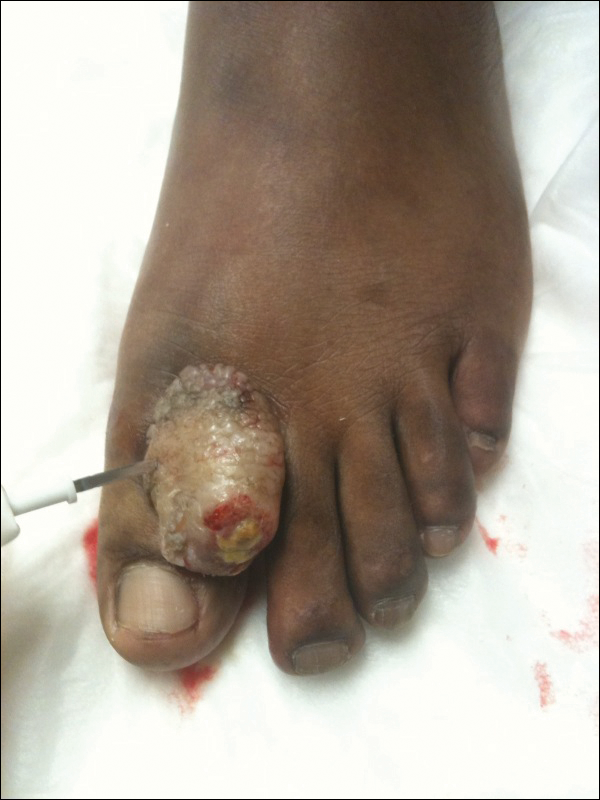
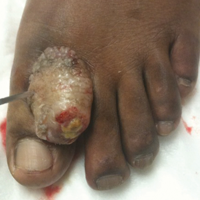
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
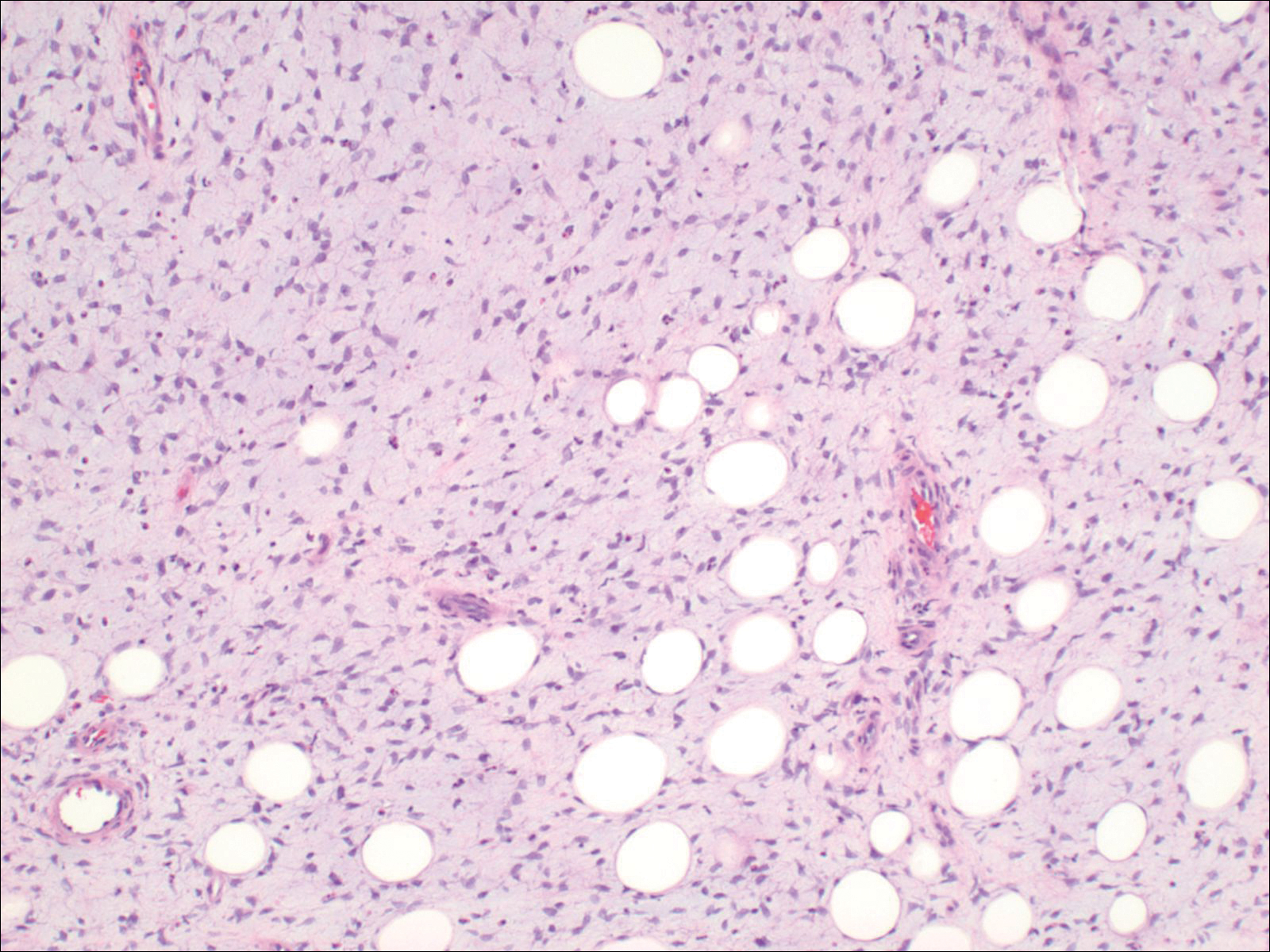
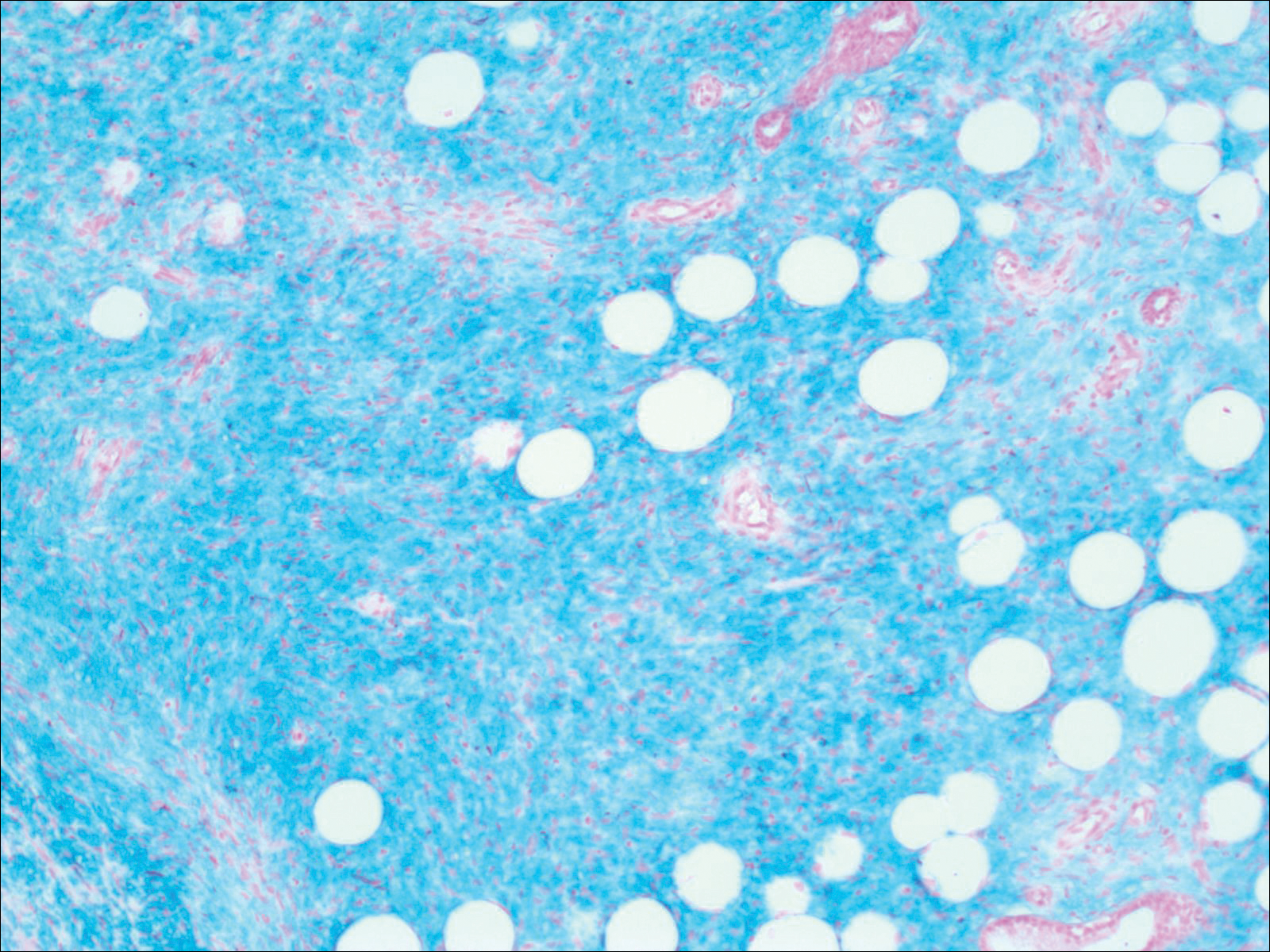
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
To the Editor:
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
To the Editor:
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
Practice Points
- Consider dermatofibrosarcoma protuberans for a keloidlike enlarging lesion when there is no history of trauma or prior keloid formation.
- Treatments such as Mohs micrographic surgery or oral imatinib mesylate can provide lower recurrence rates in appropriate patients as stand-alone or adjuvant therapy.
Temporal Triangular Alopecia Acquired in Adulthood
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
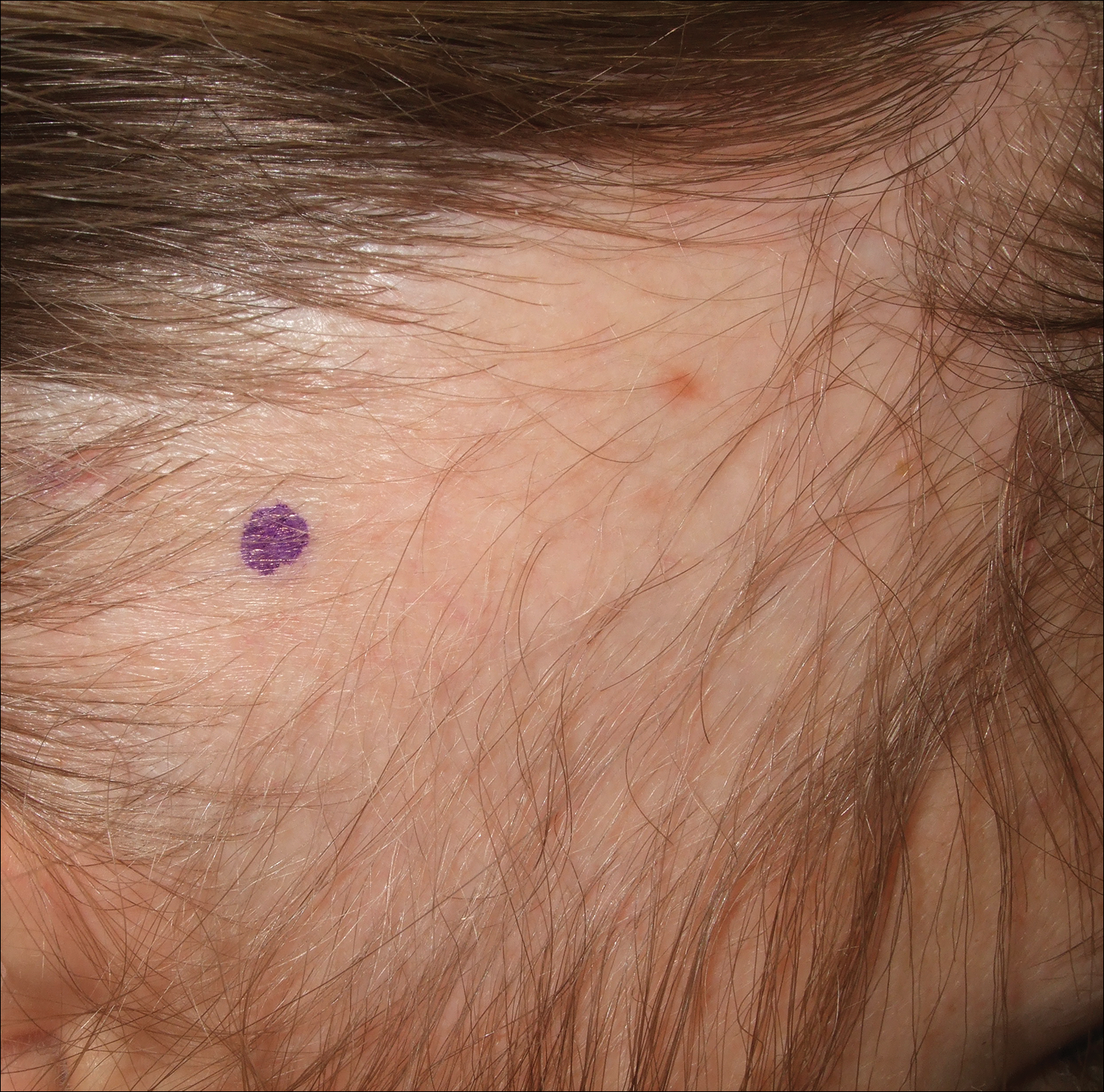
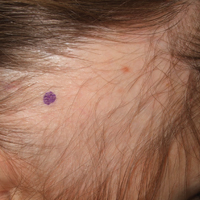
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
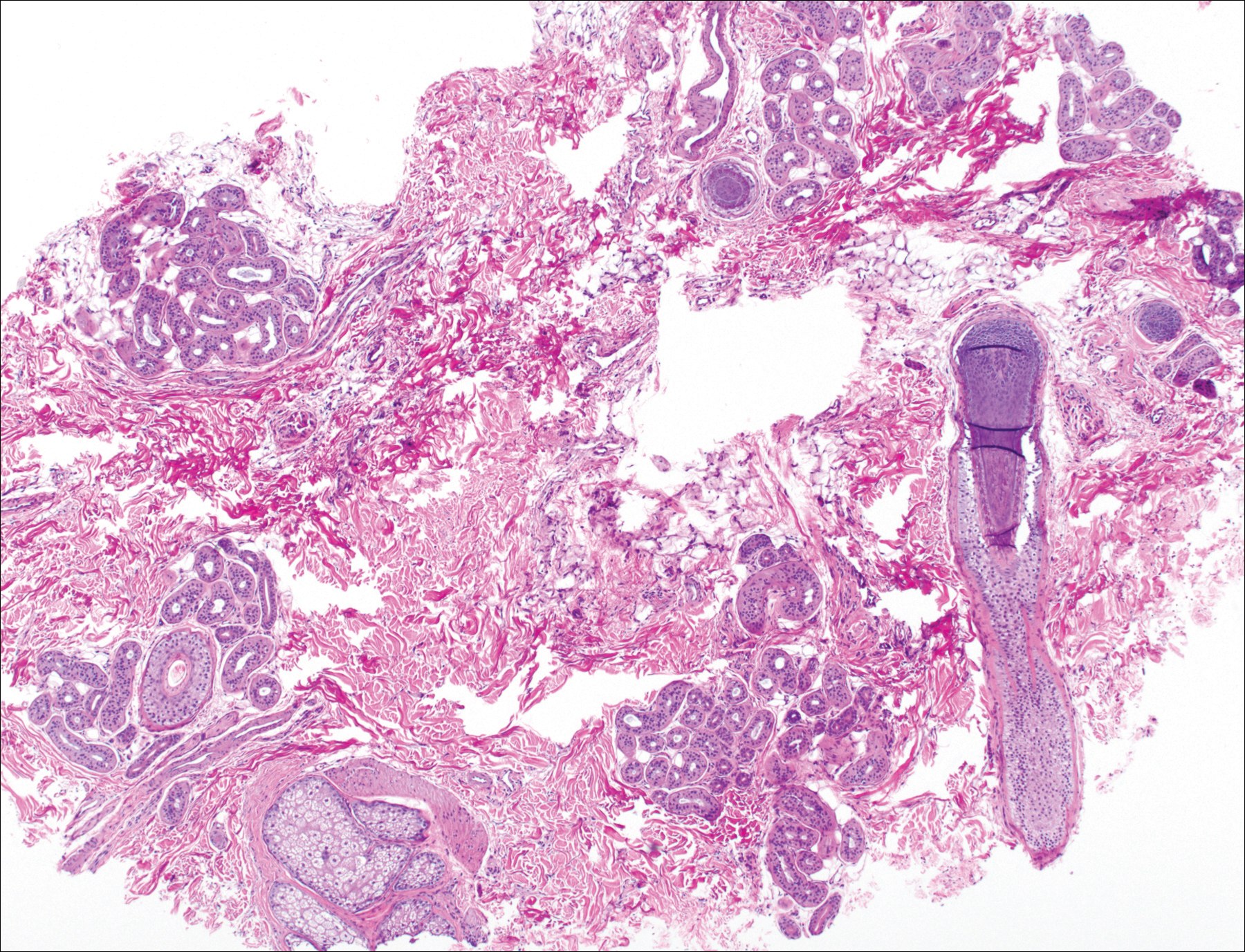
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
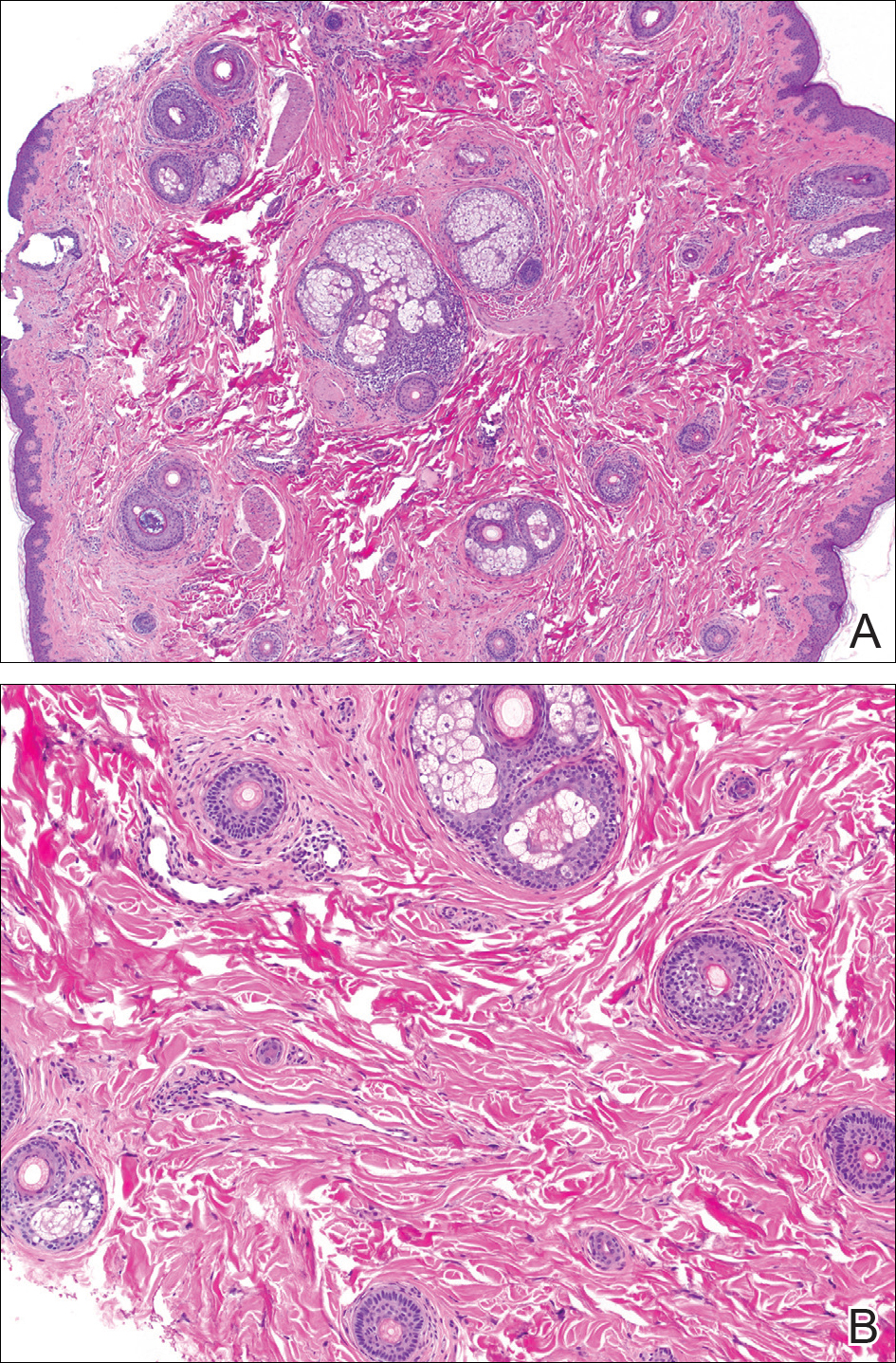
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
Practice Points
- Temporal triangular alopecia (TTA) in adults often is confused with alopecia areata.
- An acquired, persistent, unchanging, circumscribed hairless spot in an adult that does not respond to intralesional corticosteroids may represent TTA.
- Hair miniaturization without peribulbar inflammation is consistent with a diagnosis of TTA.
Netherton Syndrome in Association With Vitamin D Deficiency
To the Editor:
Netherton syndrome (NS) is a rare genodermatosis that presents with erythroderma accompanied with failure to thrive in the neonatal period. Ichthyosis linearis circumflexa, or double-edged scale, is a typical skin finding. Chronic severe atopic dermatitis with diffuse generalized xerosis usually develops and often is associated with elevated IgE levels; however, a feature most associated with and crucial for the diagnosis of NS is trichorrhexis invaginata, or bamboo hair, that causes patchy hair thinning. The triad of ichthyosis linearis circumflexa, atopic dermatitis, and trichorrhexis invaginata is diagnostic of NS. Several other clinical features, including delayed growth, skeletal age delay, and short stature also can develop during its clinical course.1
Netherton syndrome is an autosomal-recessive disorder resulting from a mutation in the SPINK5 gene, which encodes a serine protease inhibitor important in skin barrier formation and immunity.2 Thus, frequent infections are common in these patients. Current treatment options include emollients and topical anti-inflammatory agents to minimize and control the classic manifestations of NS.
A 10-year-old girl with a history of allergic rhinitis and multiple food allergies presented to the dermatology clinic with a long history of diffuse generalized xerosis and erythema with areas of lichenification and scaly patches on the face, trunk, and extremities. She was born prematurely at 34 weeks and developed scaling and erythema involving most of the body shortly after birth. She exhibited severe failure to thrive that necessitated placement of a gastrostomy feeding tube at 8 months of age, resulting in satisfactory weight gain and the tube was later removed. A liver biopsy obtained at that time revealed early intrahepatic duct obstruction and early cirrhosis. She continued to have severe atopic dermatitis, poor growth, milk intolerance, and frequent infections. She had a history of dysfunctional voiding, necessitating the use of oxybutynin. The patient also was taking desmopressin to help with insensible water losses. She had no family history of dermatologic disorders.
At presentation she had diffuse scaling and erythema around the nasal vestibule and bilateral oral commissures. She also was noted to have coarse, brittle, and sparse scalp hair and eyebrows. Her current medications included hydrocortisone cream 2.5%, loratadine 10 mg daily, desmopressin 0.1 mg twice daily, and oxybutynin. Laboratory DNA analysis revealed 2 deletion mutations involving the SPINK5 gene that combined with physical findings led to the diagnosis of NS. Due to her severe growth retardation (approximately 6 SDs below the mean), she was referred to the pediatric endocrinology department. Our patient’s skeletal age was markedly delayed (6.5 years), and she was vitamin D deficient with a total vitamin D level of 16 ng/mL (reference range, 30–80 ng/mL). She is now under the care of a dietitian and taking a vitamin D supplement of 2000 IU of vitamin D3 daily. Growth hormone therapy trials have not been helpful.
An important feature of NS is growth retardation, which is multifactorial, resulting from increased caloric requirements, percutaneous fluid loss, and food allergies. Komatsu et al3 proposed that the SPINK5 inhibitory domain in addition to its role in skin barrier function is involved in regulating proteolytic processing of growth hormone in the pituitary gland. Its dysfunction may lead to a decrease in human growth hormone levels, resulting in short stature.3 This association suggested that our patient would be a good candidate for growth hormone therapy.
Furthermore, our patient was found to be vitamin D deficient, which was not surprising, as cholecalciferol (vitamin D3) is synthesized in the epidermis with UV exposure. This finding suggests that vitamin D deficiency should be suspected in patients with an impaired skin barrier. In addition to calcium regulation and bone mineralization, vitamin D plays a preventative role in cardiovascular disease, autoimmune diseases such as Crohn disease and multiple sclerosis, type 2 diabetes mellitus, infectious diseases such as tuberculosis and influenza, and many cancers.4
Vitamin D has 2 primary derivatives: (1) vitamin D3 from the skin and dietary animal sources, and (2) ergocalciferol (vitamin D2), which is obtained primarily from dietary plant sources and fortified foods. The most common test for vitamin D sufficiency is an assay for serum 25-hydroxyvitamin D (25[OH]D) concentration; 25(OH)D is derived primarily from vitamin D3, which is 3 times more potent than vitamin D2 in the production of 25(OH)D.5 The American Academy of Pediatrics recommends vitamin D replacement therapy for children with 25(OH)D levels less than 20 ng/mL (50 nmol/L) or in children who are clinically symptomatic.6 The Endocrine Society Clinical Practice Guidelines suggest screening for vitamin D deficiency only in individuals at risk.7 We suggest that serum vitamin D testing should be routine in children with NS and other atopic dermatitis conditions in which UV absorption may be impaired.
- Sun J, Linden K. Netherton syndrome: a case report and review of the literature. Int J Dermatol. 2006;45:693-697.
- Bitoun E, Chavanas S, Irvine AD, et al. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol. 2002;118:352-361.
- Komatsu N, Saijoh K, Otsuki N, et al. Proteolytic processing of human growth hormone by multiple tissue kallikreins and regulation by the serine protease inhibitor Kazal-Type5 (SPINK5) protein. Clin Chim Acta. 2007;377:228-236.
- Wacker M, Holick MF. Vitamin D—effects on skeletal and extraskeletal health and the need for supplementation. Nutrients. 2013;5:111-148.
- Armas LA, Hollis BW, Heaney RP. Vitamin D2 is much less effective than vitamin D3 in humans. J Clin Endocrinol Metab. 2004;89:5387-5391.
- Madhusmita M, Pacaud D, Collett-Solberg PF, et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. 2008;122:398-417.
- Holick MF, Binkley NC, Bisckoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
To the Editor:
Netherton syndrome (NS) is a rare genodermatosis that presents with erythroderma accompanied with failure to thrive in the neonatal period. Ichthyosis linearis circumflexa, or double-edged scale, is a typical skin finding. Chronic severe atopic dermatitis with diffuse generalized xerosis usually develops and often is associated with elevated IgE levels; however, a feature most associated with and crucial for the diagnosis of NS is trichorrhexis invaginata, or bamboo hair, that causes patchy hair thinning. The triad of ichthyosis linearis circumflexa, atopic dermatitis, and trichorrhexis invaginata is diagnostic of NS. Several other clinical features, including delayed growth, skeletal age delay, and short stature also can develop during its clinical course.1
Netherton syndrome is an autosomal-recessive disorder resulting from a mutation in the SPINK5 gene, which encodes a serine protease inhibitor important in skin barrier formation and immunity.2 Thus, frequent infections are common in these patients. Current treatment options include emollients and topical anti-inflammatory agents to minimize and control the classic manifestations of NS.
A 10-year-old girl with a history of allergic rhinitis and multiple food allergies presented to the dermatology clinic with a long history of diffuse generalized xerosis and erythema with areas of lichenification and scaly patches on the face, trunk, and extremities. She was born prematurely at 34 weeks and developed scaling and erythema involving most of the body shortly after birth. She exhibited severe failure to thrive that necessitated placement of a gastrostomy feeding tube at 8 months of age, resulting in satisfactory weight gain and the tube was later removed. A liver biopsy obtained at that time revealed early intrahepatic duct obstruction and early cirrhosis. She continued to have severe atopic dermatitis, poor growth, milk intolerance, and frequent infections. She had a history of dysfunctional voiding, necessitating the use of oxybutynin. The patient also was taking desmopressin to help with insensible water losses. She had no family history of dermatologic disorders.
At presentation she had diffuse scaling and erythema around the nasal vestibule and bilateral oral commissures. She also was noted to have coarse, brittle, and sparse scalp hair and eyebrows. Her current medications included hydrocortisone cream 2.5%, loratadine 10 mg daily, desmopressin 0.1 mg twice daily, and oxybutynin. Laboratory DNA analysis revealed 2 deletion mutations involving the SPINK5 gene that combined with physical findings led to the diagnosis of NS. Due to her severe growth retardation (approximately 6 SDs below the mean), she was referred to the pediatric endocrinology department. Our patient’s skeletal age was markedly delayed (6.5 years), and she was vitamin D deficient with a total vitamin D level of 16 ng/mL (reference range, 30–80 ng/mL). She is now under the care of a dietitian and taking a vitamin D supplement of 2000 IU of vitamin D3 daily. Growth hormone therapy trials have not been helpful.
An important feature of NS is growth retardation, which is multifactorial, resulting from increased caloric requirements, percutaneous fluid loss, and food allergies. Komatsu et al3 proposed that the SPINK5 inhibitory domain in addition to its role in skin barrier function is involved in regulating proteolytic processing of growth hormone in the pituitary gland. Its dysfunction may lead to a decrease in human growth hormone levels, resulting in short stature.3 This association suggested that our patient would be a good candidate for growth hormone therapy.
Furthermore, our patient was found to be vitamin D deficient, which was not surprising, as cholecalciferol (vitamin D3) is synthesized in the epidermis with UV exposure. This finding suggests that vitamin D deficiency should be suspected in patients with an impaired skin barrier. In addition to calcium regulation and bone mineralization, vitamin D plays a preventative role in cardiovascular disease, autoimmune diseases such as Crohn disease and multiple sclerosis, type 2 diabetes mellitus, infectious diseases such as tuberculosis and influenza, and many cancers.4
Vitamin D has 2 primary derivatives: (1) vitamin D3 from the skin and dietary animal sources, and (2) ergocalciferol (vitamin D2), which is obtained primarily from dietary plant sources and fortified foods. The most common test for vitamin D sufficiency is an assay for serum 25-hydroxyvitamin D (25[OH]D) concentration; 25(OH)D is derived primarily from vitamin D3, which is 3 times more potent than vitamin D2 in the production of 25(OH)D.5 The American Academy of Pediatrics recommends vitamin D replacement therapy for children with 25(OH)D levels less than 20 ng/mL (50 nmol/L) or in children who are clinically symptomatic.6 The Endocrine Society Clinical Practice Guidelines suggest screening for vitamin D deficiency only in individuals at risk.7 We suggest that serum vitamin D testing should be routine in children with NS and other atopic dermatitis conditions in which UV absorption may be impaired.
To the Editor:
Netherton syndrome (NS) is a rare genodermatosis that presents with erythroderma accompanied with failure to thrive in the neonatal period. Ichthyosis linearis circumflexa, or double-edged scale, is a typical skin finding. Chronic severe atopic dermatitis with diffuse generalized xerosis usually develops and often is associated with elevated IgE levels; however, a feature most associated with and crucial for the diagnosis of NS is trichorrhexis invaginata, or bamboo hair, that causes patchy hair thinning. The triad of ichthyosis linearis circumflexa, atopic dermatitis, and trichorrhexis invaginata is diagnostic of NS. Several other clinical features, including delayed growth, skeletal age delay, and short stature also can develop during its clinical course.1
Netherton syndrome is an autosomal-recessive disorder resulting from a mutation in the SPINK5 gene, which encodes a serine protease inhibitor important in skin barrier formation and immunity.2 Thus, frequent infections are common in these patients. Current treatment options include emollients and topical anti-inflammatory agents to minimize and control the classic manifestations of NS.
A 10-year-old girl with a history of allergic rhinitis and multiple food allergies presented to the dermatology clinic with a long history of diffuse generalized xerosis and erythema with areas of lichenification and scaly patches on the face, trunk, and extremities. She was born prematurely at 34 weeks and developed scaling and erythema involving most of the body shortly after birth. She exhibited severe failure to thrive that necessitated placement of a gastrostomy feeding tube at 8 months of age, resulting in satisfactory weight gain and the tube was later removed. A liver biopsy obtained at that time revealed early intrahepatic duct obstruction and early cirrhosis. She continued to have severe atopic dermatitis, poor growth, milk intolerance, and frequent infections. She had a history of dysfunctional voiding, necessitating the use of oxybutynin. The patient also was taking desmopressin to help with insensible water losses. She had no family history of dermatologic disorders.
At presentation she had diffuse scaling and erythema around the nasal vestibule and bilateral oral commissures. She also was noted to have coarse, brittle, and sparse scalp hair and eyebrows. Her current medications included hydrocortisone cream 2.5%, loratadine 10 mg daily, desmopressin 0.1 mg twice daily, and oxybutynin. Laboratory DNA analysis revealed 2 deletion mutations involving the SPINK5 gene that combined with physical findings led to the diagnosis of NS. Due to her severe growth retardation (approximately 6 SDs below the mean), she was referred to the pediatric endocrinology department. Our patient’s skeletal age was markedly delayed (6.5 years), and she was vitamin D deficient with a total vitamin D level of 16 ng/mL (reference range, 30–80 ng/mL). She is now under the care of a dietitian and taking a vitamin D supplement of 2000 IU of vitamin D3 daily. Growth hormone therapy trials have not been helpful.
An important feature of NS is growth retardation, which is multifactorial, resulting from increased caloric requirements, percutaneous fluid loss, and food allergies. Komatsu et al3 proposed that the SPINK5 inhibitory domain in addition to its role in skin barrier function is involved in regulating proteolytic processing of growth hormone in the pituitary gland. Its dysfunction may lead to a decrease in human growth hormone levels, resulting in short stature.3 This association suggested that our patient would be a good candidate for growth hormone therapy.
Furthermore, our patient was found to be vitamin D deficient, which was not surprising, as cholecalciferol (vitamin D3) is synthesized in the epidermis with UV exposure. This finding suggests that vitamin D deficiency should be suspected in patients with an impaired skin barrier. In addition to calcium regulation and bone mineralization, vitamin D plays a preventative role in cardiovascular disease, autoimmune diseases such as Crohn disease and multiple sclerosis, type 2 diabetes mellitus, infectious diseases such as tuberculosis and influenza, and many cancers.4
Vitamin D has 2 primary derivatives: (1) vitamin D3 from the skin and dietary animal sources, and (2) ergocalciferol (vitamin D2), which is obtained primarily from dietary plant sources and fortified foods. The most common test for vitamin D sufficiency is an assay for serum 25-hydroxyvitamin D (25[OH]D) concentration; 25(OH)D is derived primarily from vitamin D3, which is 3 times more potent than vitamin D2 in the production of 25(OH)D.5 The American Academy of Pediatrics recommends vitamin D replacement therapy for children with 25(OH)D levels less than 20 ng/mL (50 nmol/L) or in children who are clinically symptomatic.6 The Endocrine Society Clinical Practice Guidelines suggest screening for vitamin D deficiency only in individuals at risk.7 We suggest that serum vitamin D testing should be routine in children with NS and other atopic dermatitis conditions in which UV absorption may be impaired.
- Sun J, Linden K. Netherton syndrome: a case report and review of the literature. Int J Dermatol. 2006;45:693-697.
- Bitoun E, Chavanas S, Irvine AD, et al. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol. 2002;118:352-361.
- Komatsu N, Saijoh K, Otsuki N, et al. Proteolytic processing of human growth hormone by multiple tissue kallikreins and regulation by the serine protease inhibitor Kazal-Type5 (SPINK5) protein. Clin Chim Acta. 2007;377:228-236.
- Wacker M, Holick MF. Vitamin D—effects on skeletal and extraskeletal health and the need for supplementation. Nutrients. 2013;5:111-148.
- Armas LA, Hollis BW, Heaney RP. Vitamin D2 is much less effective than vitamin D3 in humans. J Clin Endocrinol Metab. 2004;89:5387-5391.
- Madhusmita M, Pacaud D, Collett-Solberg PF, et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. 2008;122:398-417.
- Holick MF, Binkley NC, Bisckoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
- Sun J, Linden K. Netherton syndrome: a case report and review of the literature. Int J Dermatol. 2006;45:693-697.
- Bitoun E, Chavanas S, Irvine AD, et al. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol. 2002;118:352-361.
- Komatsu N, Saijoh K, Otsuki N, et al. Proteolytic processing of human growth hormone by multiple tissue kallikreins and regulation by the serine protease inhibitor Kazal-Type5 (SPINK5) protein. Clin Chim Acta. 2007;377:228-236.
- Wacker M, Holick MF. Vitamin D—effects on skeletal and extraskeletal health and the need for supplementation. Nutrients. 2013;5:111-148.
- Armas LA, Hollis BW, Heaney RP. Vitamin D2 is much less effective than vitamin D3 in humans. J Clin Endocrinol Metab. 2004;89:5387-5391.
- Madhusmita M, Pacaud D, Collett-Solberg PF, et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. 2008;122:398-417.
- Holick MF, Binkley NC, Bisckoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
Practice Points
- Netherton syndrome (NS) is characterized by severe atopic dermatitis, ichthyosis linearis circumflexa, and trichorrhexis invaginata.
- Children with NS are at increased risk for vitamin D deficiency.
- Consider screening patients with chronic severe dermatitis for vitamin D deficiency.

Segmental Vitiligo–like Hypopigmentation Associated With Metastatic Melanoma
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
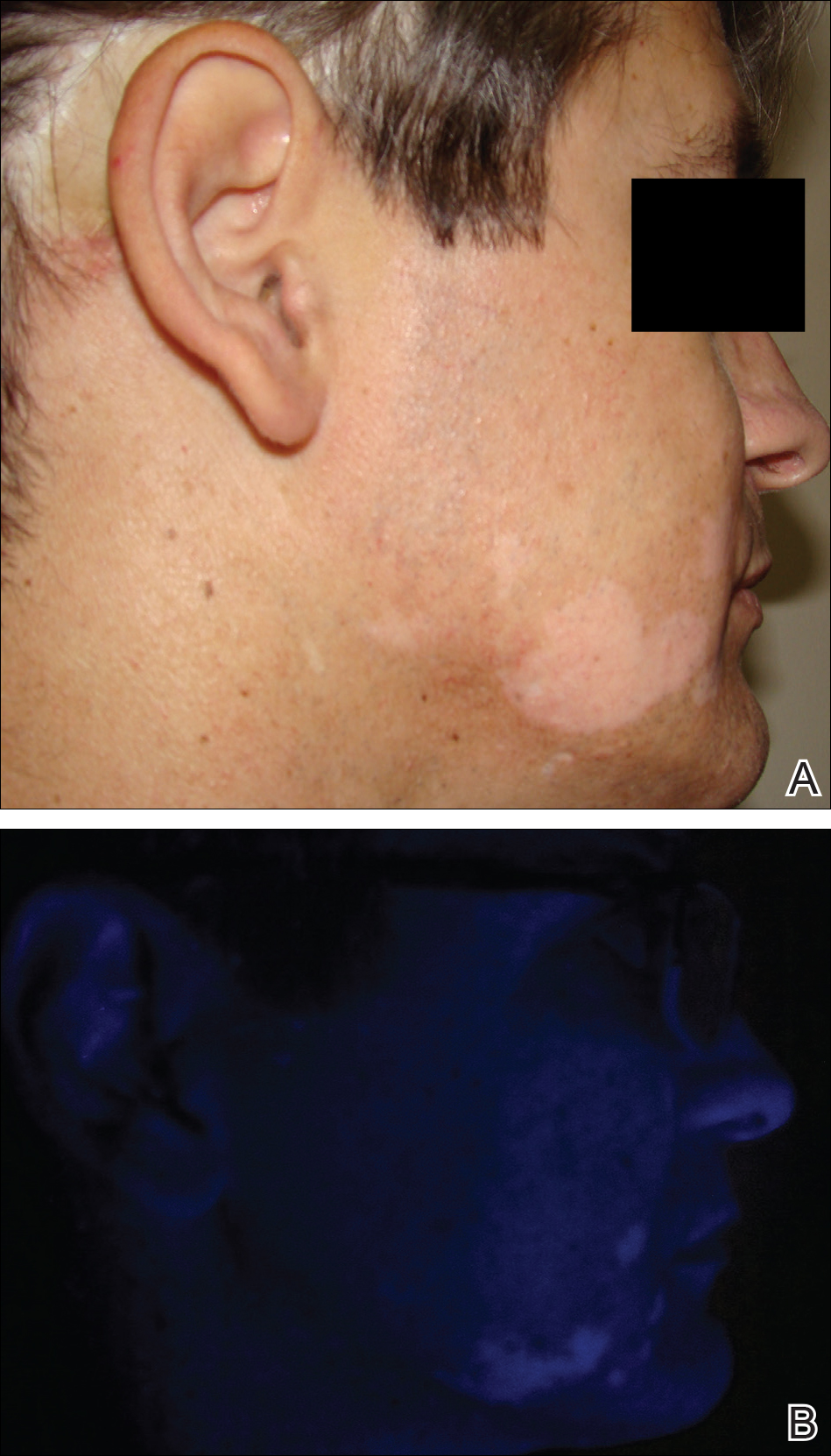
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
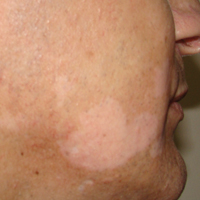
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
Prac
- Melanoma-associated hypopigmentation usually manifests as common vitiligo; however, little is known about the pathophysiology of segmental vitiligo–like hypopigmentation associated with melanoma.
- This case of segmental vitiligo–like hypopigmentation associated with melanoma sheds light on possible autoimmune and mosaic disease etiology.
Granulomatous Cheilitis Mimicking Angioedema
To the Editor:
Granulomatous cheilitis (GC), also known as Miescher cheilitis, belongs to a larger class of diseases known as orofacial granulomatoses (OFGs), a set of diseases distinguished by their clinical and pathologic features of facial edema and granulomatous inflammation.1-3 Granulomatous cheilitis, a monosymptomatic variant of a more extensive disease known as Melkersson-Rosenthal syndrome (MRS), presents with labial swelling mimicking angioedema. Timely diagnosis of GC and MRS reduces the number of unnecessary tests, health care costs, and unnecessary patient burden. We present a case of idiopathic persistent swelling of the upper lip that was originally misdiagnosed as angioedema.
A 13-year-old white adolescent boy was referred to the allergy-immunology clinic for an alternate opinion regarding a presumed diagnosis of angioedema. He presented with prominent persistent swelling of the upper lip of 1 year’s duration associated with fissuring and discomfort while eating, which led to weight loss of more than 4.5 kg. The patient denied any history of facial asymmetry, paralysis, dental infections, or gastrointestinal tract symptoms. Additionally, he was not on any medications. His parents reported variable symptomatic worsening associated with egg ingestion, but avoiding egg did not provide any symptomatic relief. The swelling was unresponsive to multiple and prolonged courses of antihistamines and oral glucocorticoids. The patient’s medical history revealed no similar episodes of unexplained swelling, and family history was negative for angioedema. On examination, the upper lip was tender with a firm rubbery consistency. No other areas of swelling were noted. Angular cheilosis and minor labial mucosal ulcerations also were observed (Figure).
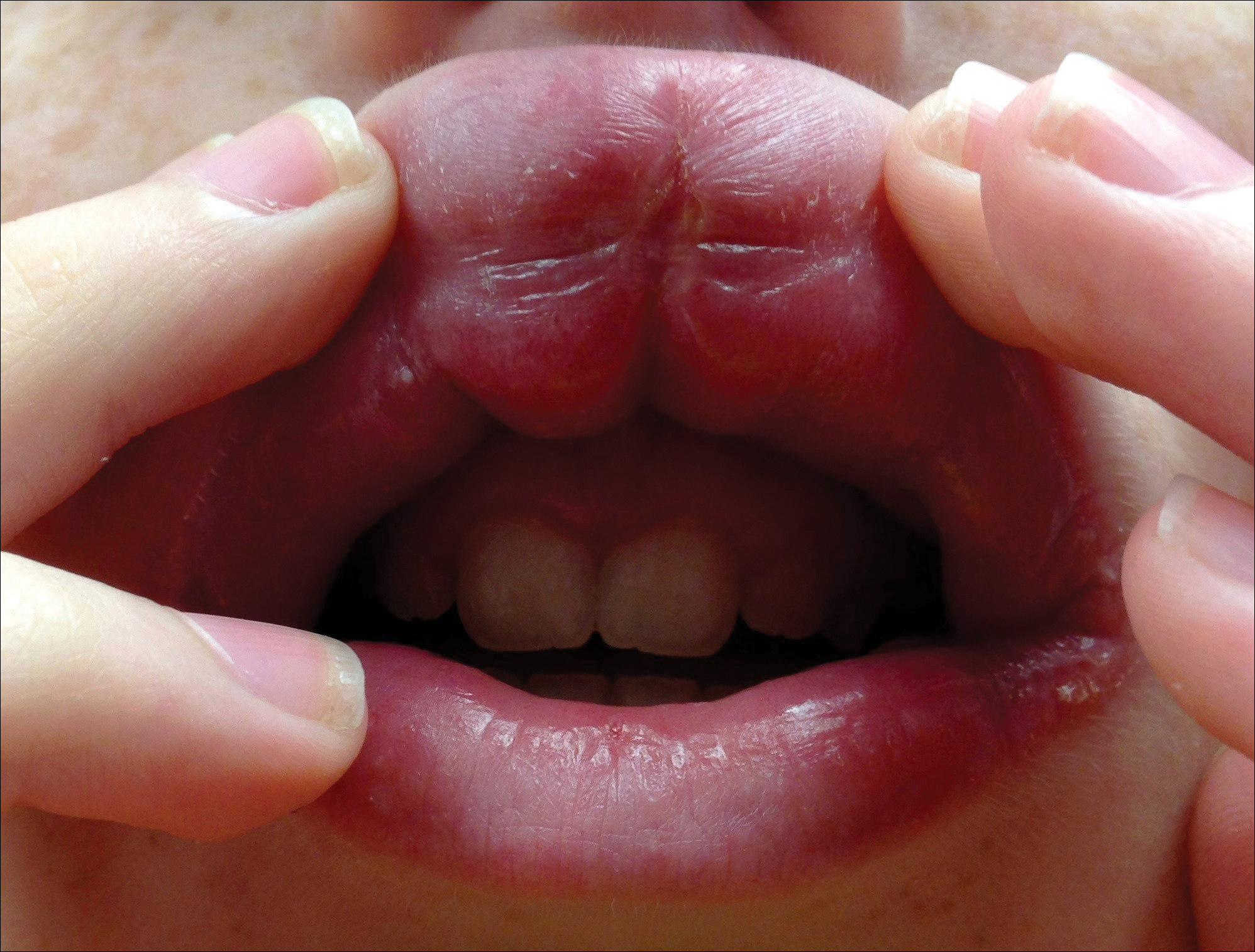
The persistent nature of the lip swelling and findings of fissures were not consistent with angioedema. Furthermore, prior laboratory studies did not reveal evidence of hereditary or acquired angioedema, and a complete blood cell count with differential was within reference range. Although the clinical suspicion for egg allergy was low, a blood test for serum-specific IgE showed a mild reactivity to egg allergen. The patient was referred to an oral surgeon for biopsy, which revealed dermal foci of noncaseating granulomas consistent with the preliminary diagnosis of GC.
Intralesional triamcinolone injections were initiated with marked improvement. Shortly after the initial improvement, however, the symptoms recurred, which necessitated several additional intralesional triamcinolone injections, again with remarkable improvement. Approximately 1.5 years later, the patient presented with recurrence of the lip swelling and admitted to having episodic diarrhea and abdominal cramps. He was referred to a pediatric gastroenterologist and a colonoscopy with biopsy confirmed Crohn disease. He was started on azathioprine followed by infliximab. A few months after this treatment was initiated, both his lip swelling and gastrointestinal tract symptoms remarkably improved. He has been maintained on this regimen and in the most recent follow-up had no recurrence of GC. He is scheduled to have another colonoscopy.
Granulomatous cheilitis is a rare chronic inflammatory condition characterized clinically by persistent lip swelling and histologically by granulomatous inflammation in the absence of systemic granulomatous disorders.4 Granulomatous cheilitis falls under the umbrella of OFGs. When it is paired with facial paralysis and fissuring of the tongue, it is specifically referred to as MRS. The prevalence of GC has historically been difficult to ascertain. In a review, an estimated incidence of 0.08% in the general population was reported with no predilection for race, sex, or age.4,5 Initially, the swelling of GC can be misdiagnosed as angioedema; therefore, it is imperative to include OFG and GC in the differential diagnosis of facial angioedema.3 Other possible diagnoses to consider include contact dermatitis, foreign-body reactions, infection, and reactions to medications such as angiotensin-converting enzyme inhibitors and nonsteroidal anti-inflammatory drugs.5 Chronic lymphedema and other granulomatous diseases also should be considered in the differential diagnosis. Isolated lymphedema of the head and neck, though rare, typically is seen following surgical or radiological interventions for cancer. Lymphatic fibrosis also can occur in the setting of chronic inflammatory skin conditions but is not typically the first presenting symptom, as was seen in our patient.6 Although granulomatous diseases such as sarcoidosis may be difficult to clinically and histologically differentiate from GC, isolated orofacial swelling in sarcoidosis is rare. If clinical suspicion for sarcoidosis does exist, however, a negative chest radiograph as well as serum calcium and angiotensin-converting enzyme levels within reference range may help differentiate GC from sarcoidosis. In our patient, the clinical suspicion for sarcoidosis was low given his clinical history, young age, and race.
The etiology of MRS and GC currently is unknown. Genetic factors, food allergies, infectious processes, and aberrant immunologic functions all have been proposed as possible mechanisms.1-3,7,8 Genetic factors, such as HLA antigen subtypes, have been investigated but have not shown a definitive correlation.8 Numerous food allergens have been suggested as causative factors in OFG via a type of delayed hypersensitivity reaction,7 with cinnamon and benzoate reported as 2 of the most cited entities.9,10 Currently, it is believed that both of these mechanisms may play an exacerbating role to an otherwise unknown disease process.7,8 The infectious process most often associated with GC is Mycobacterium tuberculosis; however, similar to genetics and food allergens, causality has not been determined.4,7 At the present time, the best evidence points to an immunologic basis of GC with the inciting event being a random influx of inflammatory cells.7,11
There is a known association between GC and Crohn disease, especially when oral lesions are present.1,9 Granulomatous cheilitis can be considered an extraintestinal manifestation of Crohn disease.Up to 20% of OFG patients eventually go on to develop Crohn disease, with some reports being even higher when OFG presents in childhood.1,9 One study proposed that both GC and Crohn disease patients shared similar histopathologic and immunopathologic features including a helper T cell (TH1)–predominant inflammatory reaction.11
The treatment of GC is challenging, with most evidence coming from sporadic case reports. Given the relatively high rate of cinnamon and benzoate hypersensitivity seen in GC patients, it has been postulated that a diet lacking in them will improve the disease. At least one study has reported positive clinical outcomes from diets lacking in cinnamon and benzoate and in fact recommended it as a potential first-line treatment.10 The mainstay of treatment, however, is corticosteroids, but continued use is discouraged due to their large side-effect profile.12 Currently, the most agreed upon treatment for patients with isolated GC is intralesional triamcinolone injections.12 Despite the robust initial response often seen with triamcinolone injections, it is not uncommon for the benefit to be short-lived, requiring additional treatments.1,5,12 Newer medical therapies that have shown promise largely are centered on anti–tumor necrosis factor α medications such as infliximab and adalimumab.13,14 It is postulated that due to the potential overlapping pathophysiology between Crohn disease and GC, there may be utility in using the same treatments.13 In situations where medical therapy fails or in extremely disfiguring cases of GC and MRS, surgical cheiloplasty is performed to reduce lip size and improve cosmetic appearance.12 In a small study, reduction cheiloplasty gave satisfactory functional and cosmetic outcomes in all 7 patients reviewed at a median follow-up of 6.5 years.15
This case emphasizes the importance of paying close attention to history and physical examination features in developing any differential diagnosis. In this patient, persistent orofacial swelling with associated mucosal ulcerations were sufficient to exclude drug-induced, idiopathic, hereditary, and acquired angioedema. The clinical history coupled with the biopsy results yielded a confident diagnosis of GC. Furthermore, similar presentations should raise concern for a subclinical inflammatory bowel disease such as Crohn disease.
- Rose AE, Leger M, Chu J, et al. Cheilitis granulomatosa. Dermatol Online J. 2011;17:15.
- Vibhute NA, Vibhute AH, Nilima DR. Cheilitis granulomatosa: a case report with review of literature. Indian J Dermatol. 2013;58:242.
- Kakimoto C, Sparks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189.
- McCartan BE, Healy CM, McCreary CE, et al. Characteristics of patients with orofacial granulomatosis. Oral Dis. 2011;17:696-704.
- Critchlow WA, Chang D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213.
- Withey S, Pracy P, Vaz F, et al. Sensory deprivation as a consequence of severe head and neck lymphoedema. J Laryngol Otol. 2001;115:62-64.
- Grave B, McCullough M, Wiesenfeld D. Orofacial granulomatosis—a 20-year review. Oral Dis. 2009;15:46-51.
- Gibson J, Wray D. Human leucocyte antigen typing in orofacial. Br J Dermatol. 2000;143:1119-1121.
- Campbell H, Escudier M, Patel P, et al. Distinguishing orofacial granulomatosis from Crohn’s disease: two separate disease entities? Inflamm Bowel Dis. 2011;17:2109-2115.
- White A, Nunes C, Escudier M, et al. Improvement in orofacial granulomatosis on a cinnamon- and benzoate-free diet. Inflamm Bowel Dis. 2006;12:508-514.
- Freysdottir J, Zhang S, Tilakaratne WM, et al. Oral biopsies from patients with orofacial granulomatosis with histology resembling Crohn’s disease have a prominent Th1 environment. Inflamm Bowel Dis. 2007;13:439-445.
- Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
- Peitsch WK, Kemmler N, Goerdt S, et al. Infliximab: a novel treatment option for refractory orofacial granulomatosis. Acta Derm Venereol. 2007;87:265-266.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
- Kruse-Lösler B, Presser D, Metze D, et al. Surgical treatment of persistent macrocheilia in patients with Melkersson-Rosenthal syndrome and cheilitis granulomatosa. Arch Dermatol. 2005;141:1085-1091.
To the Editor:
Granulomatous cheilitis (GC), also known as Miescher cheilitis, belongs to a larger class of diseases known as orofacial granulomatoses (OFGs), a set of diseases distinguished by their clinical and pathologic features of facial edema and granulomatous inflammation.1-3 Granulomatous cheilitis, a monosymptomatic variant of a more extensive disease known as Melkersson-Rosenthal syndrome (MRS), presents with labial swelling mimicking angioedema. Timely diagnosis of GC and MRS reduces the number of unnecessary tests, health care costs, and unnecessary patient burden. We present a case of idiopathic persistent swelling of the upper lip that was originally misdiagnosed as angioedema.
A 13-year-old white adolescent boy was referred to the allergy-immunology clinic for an alternate opinion regarding a presumed diagnosis of angioedema. He presented with prominent persistent swelling of the upper lip of 1 year’s duration associated with fissuring and discomfort while eating, which led to weight loss of more than 4.5 kg. The patient denied any history of facial asymmetry, paralysis, dental infections, or gastrointestinal tract symptoms. Additionally, he was not on any medications. His parents reported variable symptomatic worsening associated with egg ingestion, but avoiding egg did not provide any symptomatic relief. The swelling was unresponsive to multiple and prolonged courses of antihistamines and oral glucocorticoids. The patient’s medical history revealed no similar episodes of unexplained swelling, and family history was negative for angioedema. On examination, the upper lip was tender with a firm rubbery consistency. No other areas of swelling were noted. Angular cheilosis and minor labial mucosal ulcerations also were observed (Figure).
The persistent nature of the lip swelling and findings of fissures were not consistent with angioedema. Furthermore, prior laboratory studies did not reveal evidence of hereditary or acquired angioedema, and a complete blood cell count with differential was within reference range. Although the clinical suspicion for egg allergy was low, a blood test for serum-specific IgE showed a mild reactivity to egg allergen. The patient was referred to an oral surgeon for biopsy, which revealed dermal foci of noncaseating granulomas consistent with the preliminary diagnosis of GC.
Intralesional triamcinolone injections were initiated with marked improvement. Shortly after the initial improvement, however, the symptoms recurred, which necessitated several additional intralesional triamcinolone injections, again with remarkable improvement. Approximately 1.5 years later, the patient presented with recurrence of the lip swelling and admitted to having episodic diarrhea and abdominal cramps. He was referred to a pediatric gastroenterologist and a colonoscopy with biopsy confirmed Crohn disease. He was started on azathioprine followed by infliximab. A few months after this treatment was initiated, both his lip swelling and gastrointestinal tract symptoms remarkably improved. He has been maintained on this regimen and in the most recent follow-up had no recurrence of GC. He is scheduled to have another colonoscopy.
Granulomatous cheilitis is a rare chronic inflammatory condition characterized clinically by persistent lip swelling and histologically by granulomatous inflammation in the absence of systemic granulomatous disorders.4 Granulomatous cheilitis falls under the umbrella of OFGs. When it is paired with facial paralysis and fissuring of the tongue, it is specifically referred to as MRS. The prevalence of GC has historically been difficult to ascertain. In a review, an estimated incidence of 0.08% in the general population was reported with no predilection for race, sex, or age.4,5 Initially, the swelling of GC can be misdiagnosed as angioedema; therefore, it is imperative to include OFG and GC in the differential diagnosis of facial angioedema.3 Other possible diagnoses to consider include contact dermatitis, foreign-body reactions, infection, and reactions to medications such as angiotensin-converting enzyme inhibitors and nonsteroidal anti-inflammatory drugs.5 Chronic lymphedema and other granulomatous diseases also should be considered in the differential diagnosis. Isolated lymphedema of the head and neck, though rare, typically is seen following surgical or radiological interventions for cancer. Lymphatic fibrosis also can occur in the setting of chronic inflammatory skin conditions but is not typically the first presenting symptom, as was seen in our patient.6 Although granulomatous diseases such as sarcoidosis may be difficult to clinically and histologically differentiate from GC, isolated orofacial swelling in sarcoidosis is rare. If clinical suspicion for sarcoidosis does exist, however, a negative chest radiograph as well as serum calcium and angiotensin-converting enzyme levels within reference range may help differentiate GC from sarcoidosis. In our patient, the clinical suspicion for sarcoidosis was low given his clinical history, young age, and race.
The etiology of MRS and GC currently is unknown. Genetic factors, food allergies, infectious processes, and aberrant immunologic functions all have been proposed as possible mechanisms.1-3,7,8 Genetic factors, such as HLA antigen subtypes, have been investigated but have not shown a definitive correlation.8 Numerous food allergens have been suggested as causative factors in OFG via a type of delayed hypersensitivity reaction,7 with cinnamon and benzoate reported as 2 of the most cited entities.9,10 Currently, it is believed that both of these mechanisms may play an exacerbating role to an otherwise unknown disease process.7,8 The infectious process most often associated with GC is Mycobacterium tuberculosis; however, similar to genetics and food allergens, causality has not been determined.4,7 At the present time, the best evidence points to an immunologic basis of GC with the inciting event being a random influx of inflammatory cells.7,11
There is a known association between GC and Crohn disease, especially when oral lesions are present.1,9 Granulomatous cheilitis can be considered an extraintestinal manifestation of Crohn disease.Up to 20% of OFG patients eventually go on to develop Crohn disease, with some reports being even higher when OFG presents in childhood.1,9 One study proposed that both GC and Crohn disease patients shared similar histopathologic and immunopathologic features including a helper T cell (TH1)–predominant inflammatory reaction.11
The treatment of GC is challenging, with most evidence coming from sporadic case reports. Given the relatively high rate of cinnamon and benzoate hypersensitivity seen in GC patients, it has been postulated that a diet lacking in them will improve the disease. At least one study has reported positive clinical outcomes from diets lacking in cinnamon and benzoate and in fact recommended it as a potential first-line treatment.10 The mainstay of treatment, however, is corticosteroids, but continued use is discouraged due to their large side-effect profile.12 Currently, the most agreed upon treatment for patients with isolated GC is intralesional triamcinolone injections.12 Despite the robust initial response often seen with triamcinolone injections, it is not uncommon for the benefit to be short-lived, requiring additional treatments.1,5,12 Newer medical therapies that have shown promise largely are centered on anti–tumor necrosis factor α medications such as infliximab and adalimumab.13,14 It is postulated that due to the potential overlapping pathophysiology between Crohn disease and GC, there may be utility in using the same treatments.13 In situations where medical therapy fails or in extremely disfiguring cases of GC and MRS, surgical cheiloplasty is performed to reduce lip size and improve cosmetic appearance.12 In a small study, reduction cheiloplasty gave satisfactory functional and cosmetic outcomes in all 7 patients reviewed at a median follow-up of 6.5 years.15
This case emphasizes the importance of paying close attention to history and physical examination features in developing any differential diagnosis. In this patient, persistent orofacial swelling with associated mucosal ulcerations were sufficient to exclude drug-induced, idiopathic, hereditary, and acquired angioedema. The clinical history coupled with the biopsy results yielded a confident diagnosis of GC. Furthermore, similar presentations should raise concern for a subclinical inflammatory bowel disease such as Crohn disease.
To the Editor:
Granulomatous cheilitis (GC), also known as Miescher cheilitis, belongs to a larger class of diseases known as orofacial granulomatoses (OFGs), a set of diseases distinguished by their clinical and pathologic features of facial edema and granulomatous inflammation.1-3 Granulomatous cheilitis, a monosymptomatic variant of a more extensive disease known as Melkersson-Rosenthal syndrome (MRS), presents with labial swelling mimicking angioedema. Timely diagnosis of GC and MRS reduces the number of unnecessary tests, health care costs, and unnecessary patient burden. We present a case of idiopathic persistent swelling of the upper lip that was originally misdiagnosed as angioedema.
A 13-year-old white adolescent boy was referred to the allergy-immunology clinic for an alternate opinion regarding a presumed diagnosis of angioedema. He presented with prominent persistent swelling of the upper lip of 1 year’s duration associated with fissuring and discomfort while eating, which led to weight loss of more than 4.5 kg. The patient denied any history of facial asymmetry, paralysis, dental infections, or gastrointestinal tract symptoms. Additionally, he was not on any medications. His parents reported variable symptomatic worsening associated with egg ingestion, but avoiding egg did not provide any symptomatic relief. The swelling was unresponsive to multiple and prolonged courses of antihistamines and oral glucocorticoids. The patient’s medical history revealed no similar episodes of unexplained swelling, and family history was negative for angioedema. On examination, the upper lip was tender with a firm rubbery consistency. No other areas of swelling were noted. Angular cheilosis and minor labial mucosal ulcerations also were observed (Figure).
The persistent nature of the lip swelling and findings of fissures were not consistent with angioedema. Furthermore, prior laboratory studies did not reveal evidence of hereditary or acquired angioedema, and a complete blood cell count with differential was within reference range. Although the clinical suspicion for egg allergy was low, a blood test for serum-specific IgE showed a mild reactivity to egg allergen. The patient was referred to an oral surgeon for biopsy, which revealed dermal foci of noncaseating granulomas consistent with the preliminary diagnosis of GC.
Intralesional triamcinolone injections were initiated with marked improvement. Shortly after the initial improvement, however, the symptoms recurred, which necessitated several additional intralesional triamcinolone injections, again with remarkable improvement. Approximately 1.5 years later, the patient presented with recurrence of the lip swelling and admitted to having episodic diarrhea and abdominal cramps. He was referred to a pediatric gastroenterologist and a colonoscopy with biopsy confirmed Crohn disease. He was started on azathioprine followed by infliximab. A few months after this treatment was initiated, both his lip swelling and gastrointestinal tract symptoms remarkably improved. He has been maintained on this regimen and in the most recent follow-up had no recurrence of GC. He is scheduled to have another colonoscopy.
Granulomatous cheilitis is a rare chronic inflammatory condition characterized clinically by persistent lip swelling and histologically by granulomatous inflammation in the absence of systemic granulomatous disorders.4 Granulomatous cheilitis falls under the umbrella of OFGs. When it is paired with facial paralysis and fissuring of the tongue, it is specifically referred to as MRS. The prevalence of GC has historically been difficult to ascertain. In a review, an estimated incidence of 0.08% in the general population was reported with no predilection for race, sex, or age.4,5 Initially, the swelling of GC can be misdiagnosed as angioedema; therefore, it is imperative to include OFG and GC in the differential diagnosis of facial angioedema.3 Other possible diagnoses to consider include contact dermatitis, foreign-body reactions, infection, and reactions to medications such as angiotensin-converting enzyme inhibitors and nonsteroidal anti-inflammatory drugs.5 Chronic lymphedema and other granulomatous diseases also should be considered in the differential diagnosis. Isolated lymphedema of the head and neck, though rare, typically is seen following surgical or radiological interventions for cancer. Lymphatic fibrosis also can occur in the setting of chronic inflammatory skin conditions but is not typically the first presenting symptom, as was seen in our patient.6 Although granulomatous diseases such as sarcoidosis may be difficult to clinically and histologically differentiate from GC, isolated orofacial swelling in sarcoidosis is rare. If clinical suspicion for sarcoidosis does exist, however, a negative chest radiograph as well as serum calcium and angiotensin-converting enzyme levels within reference range may help differentiate GC from sarcoidosis. In our patient, the clinical suspicion for sarcoidosis was low given his clinical history, young age, and race.
The etiology of MRS and GC currently is unknown. Genetic factors, food allergies, infectious processes, and aberrant immunologic functions all have been proposed as possible mechanisms.1-3,7,8 Genetic factors, such as HLA antigen subtypes, have been investigated but have not shown a definitive correlation.8 Numerous food allergens have been suggested as causative factors in OFG via a type of delayed hypersensitivity reaction,7 with cinnamon and benzoate reported as 2 of the most cited entities.9,10 Currently, it is believed that both of these mechanisms may play an exacerbating role to an otherwise unknown disease process.7,8 The infectious process most often associated with GC is Mycobacterium tuberculosis; however, similar to genetics and food allergens, causality has not been determined.4,7 At the present time, the best evidence points to an immunologic basis of GC with the inciting event being a random influx of inflammatory cells.7,11
There is a known association between GC and Crohn disease, especially when oral lesions are present.1,9 Granulomatous cheilitis can be considered an extraintestinal manifestation of Crohn disease.Up to 20% of OFG patients eventually go on to develop Crohn disease, with some reports being even higher when OFG presents in childhood.1,9 One study proposed that both GC and Crohn disease patients shared similar histopathologic and immunopathologic features including a helper T cell (TH1)–predominant inflammatory reaction.11
The treatment of GC is challenging, with most evidence coming from sporadic case reports. Given the relatively high rate of cinnamon and benzoate hypersensitivity seen in GC patients, it has been postulated that a diet lacking in them will improve the disease. At least one study has reported positive clinical outcomes from diets lacking in cinnamon and benzoate and in fact recommended it as a potential first-line treatment.10 The mainstay of treatment, however, is corticosteroids, but continued use is discouraged due to their large side-effect profile.12 Currently, the most agreed upon treatment for patients with isolated GC is intralesional triamcinolone injections.12 Despite the robust initial response often seen with triamcinolone injections, it is not uncommon for the benefit to be short-lived, requiring additional treatments.1,5,12 Newer medical therapies that have shown promise largely are centered on anti–tumor necrosis factor α medications such as infliximab and adalimumab.13,14 It is postulated that due to the potential overlapping pathophysiology between Crohn disease and GC, there may be utility in using the same treatments.13 In situations where medical therapy fails or in extremely disfiguring cases of GC and MRS, surgical cheiloplasty is performed to reduce lip size and improve cosmetic appearance.12 In a small study, reduction cheiloplasty gave satisfactory functional and cosmetic outcomes in all 7 patients reviewed at a median follow-up of 6.5 years.15
This case emphasizes the importance of paying close attention to history and physical examination features in developing any differential diagnosis. In this patient, persistent orofacial swelling with associated mucosal ulcerations were sufficient to exclude drug-induced, idiopathic, hereditary, and acquired angioedema. The clinical history coupled with the biopsy results yielded a confident diagnosis of GC. Furthermore, similar presentations should raise concern for a subclinical inflammatory bowel disease such as Crohn disease.
- Rose AE, Leger M, Chu J, et al. Cheilitis granulomatosa. Dermatol Online J. 2011;17:15.
- Vibhute NA, Vibhute AH, Nilima DR. Cheilitis granulomatosa: a case report with review of literature. Indian J Dermatol. 2013;58:242.
- Kakimoto C, Sparks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189.
- McCartan BE, Healy CM, McCreary CE, et al. Characteristics of patients with orofacial granulomatosis. Oral Dis. 2011;17:696-704.
- Critchlow WA, Chang D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213.
- Withey S, Pracy P, Vaz F, et al. Sensory deprivation as a consequence of severe head and neck lymphoedema. J Laryngol Otol. 2001;115:62-64.
- Grave B, McCullough M, Wiesenfeld D. Orofacial granulomatosis—a 20-year review. Oral Dis. 2009;15:46-51.
- Gibson J, Wray D. Human leucocyte antigen typing in orofacial. Br J Dermatol. 2000;143:1119-1121.
- Campbell H, Escudier M, Patel P, et al. Distinguishing orofacial granulomatosis from Crohn’s disease: two separate disease entities? Inflamm Bowel Dis. 2011;17:2109-2115.
- White A, Nunes C, Escudier M, et al. Improvement in orofacial granulomatosis on a cinnamon- and benzoate-free diet. Inflamm Bowel Dis. 2006;12:508-514.
- Freysdottir J, Zhang S, Tilakaratne WM, et al. Oral biopsies from patients with orofacial granulomatosis with histology resembling Crohn’s disease have a prominent Th1 environment. Inflamm Bowel Dis. 2007;13:439-445.
- Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
- Peitsch WK, Kemmler N, Goerdt S, et al. Infliximab: a novel treatment option for refractory orofacial granulomatosis. Acta Derm Venereol. 2007;87:265-266.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
- Kruse-Lösler B, Presser D, Metze D, et al. Surgical treatment of persistent macrocheilia in patients with Melkersson-Rosenthal syndrome and cheilitis granulomatosa. Arch Dermatol. 2005;141:1085-1091.
- Rose AE, Leger M, Chu J, et al. Cheilitis granulomatosa. Dermatol Online J. 2011;17:15.
- Vibhute NA, Vibhute AH, Nilima DR. Cheilitis granulomatosa: a case report with review of literature. Indian J Dermatol. 2013;58:242.
- Kakimoto C, Sparks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189.
- McCartan BE, Healy CM, McCreary CE, et al. Characteristics of patients with orofacial granulomatosis. Oral Dis. 2011;17:696-704.
- Critchlow WA, Chang D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213.
- Withey S, Pracy P, Vaz F, et al. Sensory deprivation as a consequence of severe head and neck lymphoedema. J Laryngol Otol. 2001;115:62-64.
- Grave B, McCullough M, Wiesenfeld D. Orofacial granulomatosis—a 20-year review. Oral Dis. 2009;15:46-51.
- Gibson J, Wray D. Human leucocyte antigen typing in orofacial. Br J Dermatol. 2000;143:1119-1121.
- Campbell H, Escudier M, Patel P, et al. Distinguishing orofacial granulomatosis from Crohn’s disease: two separate disease entities? Inflamm Bowel Dis. 2011;17:2109-2115.
- White A, Nunes C, Escudier M, et al. Improvement in orofacial granulomatosis on a cinnamon- and benzoate-free diet. Inflamm Bowel Dis. 2006;12:508-514.
- Freysdottir J, Zhang S, Tilakaratne WM, et al. Oral biopsies from patients with orofacial granulomatosis with histology resembling Crohn’s disease have a prominent Th1 environment. Inflamm Bowel Dis. 2007;13:439-445.
- Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
- Peitsch WK, Kemmler N, Goerdt S, et al. Infliximab: a novel treatment option for refractory orofacial granulomatosis. Acta Derm Venereol. 2007;87:265-266.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
- Kruse-Lösler B, Presser D, Metze D, et al. Surgical treatment of persistent macrocheilia in patients with Melkersson-Rosenthal syndrome and cheilitis granulomatosa. Arch Dermatol. 2005;141:1085-1091.
Practice Points
- Granulomatous cheilitis (GC) is a rare diagnosis that can present as an isolated disease or in association with another disease, most commonly an inflammatory bowel disease (ie, Crohn disease).
- Often misdiagnosed as angioedema, GC can be differentiated primarily based on history and clinical examination.
- Intervention such as intralesional steroid injection is effective in the primary form; however, treatment of the underlying condition, such as Crohn disease, is needed when the 2 conditions are associated.
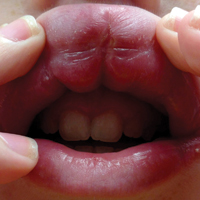
Unilateral Verrucous Porokeratosis of the Gluteal Cleft
To the Editor:
Verrucous porokeratosis of the gluteal cleft is a rare skin condition that has distinct clinical and histologic features. A review of 5 cases described a characteristic clinical presentation of a butterfly-shaped bilateral gluteal cleft lesion on most patients.1 We present an unusual case of verrucous porokeratosis presenting as a unilateral single lesion in the gluteal area that emulated seborrheic keratosis with histology consistent with verrucous porokeratosis. This case adds to the variable presentation of this unusual disease.
A 40-year-old man who presented to the dermatology clinic for a follow-up on a basal cell carcinoma of the temple region was concerned about a lesion on the left buttock of 1 year’s duration. Physical examination revealed a unilateral hyperkeratotic plaque that clinically resembled seborrheic keratosis (Figure 1). Biopsy revealed hyperkeratosis with numerous columns of parakeratosis, psoriasiform epidermal hyperplasia (Figures 2A and 2B), dyskeratotic keratinocytes (Figure 2C), pigment incontinence, and mild superficial chronic inflammation consistent with verrucous porokeratosis. The patient was treated with urea lotion but ultimately was lost to follow-up.
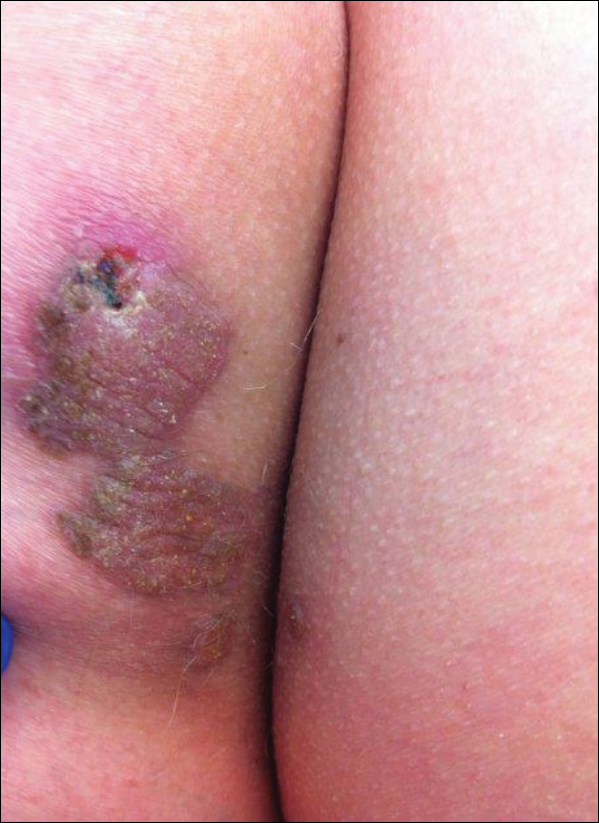
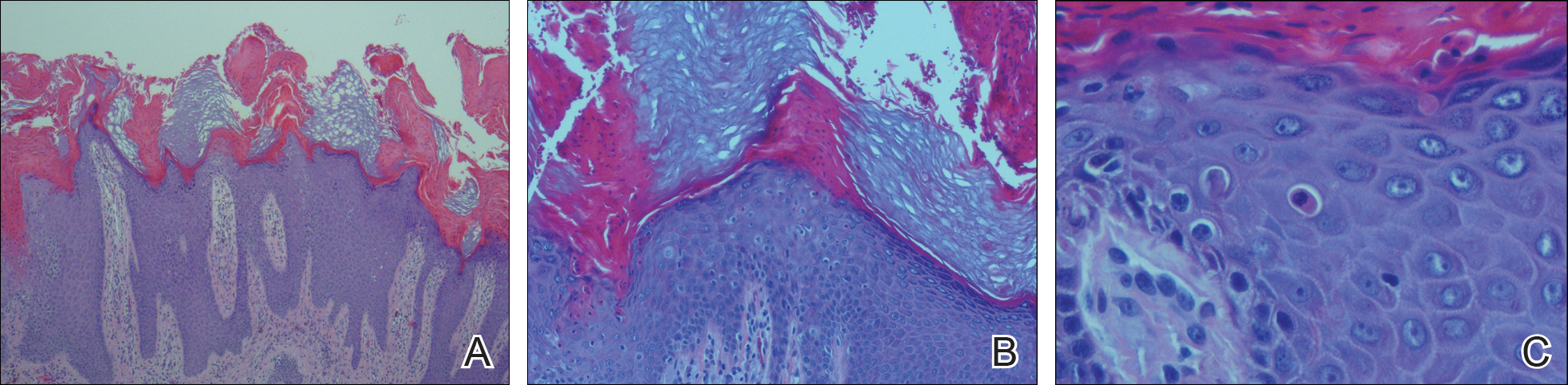
We present a unique case of unilateral verrucous porokeratosis of the gluteal cleft. The clinical differential diagnosis included seborrheic keratosis, condyloma acuminata, and inflammatory linear verrucous epidermal nevus. Histopathology was consistent with verrucous porokeratosis. Porokeratosis is a heterogeneous group of keratinization disorders containing several described variants such as classici porokeratosis of Mibelli, disseminated superficial porokeratosis, porokeratosis palmaris et plantaris disseminata, linear porokeratosis, and punctuate porokeratosis.1,2 Most patients present clinically with plaquelike bilateral (butterfly) lesions with threadlike (ridge) borders, though some patients initially have a unilateral lesion that subsequently develops into a bilateral lesion.1 The clinical course is slow growing, but it can potentially give rise to malignancies such as squamous cell carcinoma.3 Histologically, numerous columns of parakeratosis overlying epidermal cells with attenuated granular layer are observed with the concentric cornoid lamellae considered unique to the verrucous variant.1 Although our patient had only a single unilateral lesion on the gluteal cleft, the histology was consistent with verrucous porokeratosis. Our case adds to the growing clinical presentations of this unusual disease.
- Takiguchi R, White K, Clifton W, et al. Verrucous porokeratosis of the gluteal cleft (porokeratosis stychotropica): a rare disorder easily misdiagnosed. J Cutan Pathol. 2010;37:802-807.
- McGuigan K, Shurman D, Campanelli C, et al. Porokeratosis ptychotropica: a clinically distinct variant of porokeratosis. J Am Acad Dermatol. 2009;60:501-503.
- Malek J, Chedraoui A, Kibbi AG, et al. Genitogluteal porokeratosis: 10 years to make the diagnosis! Am J Dermatopathol. 2009;31:604-606.
To the Editor:
Verrucous porokeratosis of the gluteal cleft is a rare skin condition that has distinct clinical and histologic features. A review of 5 cases described a characteristic clinical presentation of a butterfly-shaped bilateral gluteal cleft lesion on most patients.1 We present an unusual case of verrucous porokeratosis presenting as a unilateral single lesion in the gluteal area that emulated seborrheic keratosis with histology consistent with verrucous porokeratosis. This case adds to the variable presentation of this unusual disease.
A 40-year-old man who presented to the dermatology clinic for a follow-up on a basal cell carcinoma of the temple region was concerned about a lesion on the left buttock of 1 year’s duration. Physical examination revealed a unilateral hyperkeratotic plaque that clinically resembled seborrheic keratosis (Figure 1). Biopsy revealed hyperkeratosis with numerous columns of parakeratosis, psoriasiform epidermal hyperplasia (Figures 2A and 2B), dyskeratotic keratinocytes (Figure 2C), pigment incontinence, and mild superficial chronic inflammation consistent with verrucous porokeratosis. The patient was treated with urea lotion but ultimately was lost to follow-up.
We present a unique case of unilateral verrucous porokeratosis of the gluteal cleft. The clinical differential diagnosis included seborrheic keratosis, condyloma acuminata, and inflammatory linear verrucous epidermal nevus. Histopathology was consistent with verrucous porokeratosis. Porokeratosis is a heterogeneous group of keratinization disorders containing several described variants such as classici porokeratosis of Mibelli, disseminated superficial porokeratosis, porokeratosis palmaris et plantaris disseminata, linear porokeratosis, and punctuate porokeratosis.1,2 Most patients present clinically with plaquelike bilateral (butterfly) lesions with threadlike (ridge) borders, though some patients initially have a unilateral lesion that subsequently develops into a bilateral lesion.1 The clinical course is slow growing, but it can potentially give rise to malignancies such as squamous cell carcinoma.3 Histologically, numerous columns of parakeratosis overlying epidermal cells with attenuated granular layer are observed with the concentric cornoid lamellae considered unique to the verrucous variant.1 Although our patient had only a single unilateral lesion on the gluteal cleft, the histology was consistent with verrucous porokeratosis. Our case adds to the growing clinical presentations of this unusual disease.
To the Editor:
Verrucous porokeratosis of the gluteal cleft is a rare skin condition that has distinct clinical and histologic features. A review of 5 cases described a characteristic clinical presentation of a butterfly-shaped bilateral gluteal cleft lesion on most patients.1 We present an unusual case of verrucous porokeratosis presenting as a unilateral single lesion in the gluteal area that emulated seborrheic keratosis with histology consistent with verrucous porokeratosis. This case adds to the variable presentation of this unusual disease.
A 40-year-old man who presented to the dermatology clinic for a follow-up on a basal cell carcinoma of the temple region was concerned about a lesion on the left buttock of 1 year’s duration. Physical examination revealed a unilateral hyperkeratotic plaque that clinically resembled seborrheic keratosis (Figure 1). Biopsy revealed hyperkeratosis with numerous columns of parakeratosis, psoriasiform epidermal hyperplasia (Figures 2A and 2B), dyskeratotic keratinocytes (Figure 2C), pigment incontinence, and mild superficial chronic inflammation consistent with verrucous porokeratosis. The patient was treated with urea lotion but ultimately was lost to follow-up.
We present a unique case of unilateral verrucous porokeratosis of the gluteal cleft. The clinical differential diagnosis included seborrheic keratosis, condyloma acuminata, and inflammatory linear verrucous epidermal nevus. Histopathology was consistent with verrucous porokeratosis. Porokeratosis is a heterogeneous group of keratinization disorders containing several described variants such as classici porokeratosis of Mibelli, disseminated superficial porokeratosis, porokeratosis palmaris et plantaris disseminata, linear porokeratosis, and punctuate porokeratosis.1,2 Most patients present clinically with plaquelike bilateral (butterfly) lesions with threadlike (ridge) borders, though some patients initially have a unilateral lesion that subsequently develops into a bilateral lesion.1 The clinical course is slow growing, but it can potentially give rise to malignancies such as squamous cell carcinoma.3 Histologically, numerous columns of parakeratosis overlying epidermal cells with attenuated granular layer are observed with the concentric cornoid lamellae considered unique to the verrucous variant.1 Although our patient had only a single unilateral lesion on the gluteal cleft, the histology was consistent with verrucous porokeratosis. Our case adds to the growing clinical presentations of this unusual disease.
- Takiguchi R, White K, Clifton W, et al. Verrucous porokeratosis of the gluteal cleft (porokeratosis stychotropica): a rare disorder easily misdiagnosed. J Cutan Pathol. 2010;37:802-807.
- McGuigan K, Shurman D, Campanelli C, et al. Porokeratosis ptychotropica: a clinically distinct variant of porokeratosis. J Am Acad Dermatol. 2009;60:501-503.
- Malek J, Chedraoui A, Kibbi AG, et al. Genitogluteal porokeratosis: 10 years to make the diagnosis! Am J Dermatopathol. 2009;31:604-606.
- Takiguchi R, White K, Clifton W, et al. Verrucous porokeratosis of the gluteal cleft (porokeratosis stychotropica): a rare disorder easily misdiagnosed. J Cutan Pathol. 2010;37:802-807.
- McGuigan K, Shurman D, Campanelli C, et al. Porokeratosis ptychotropica: a clinically distinct variant of porokeratosis. J Am Acad Dermatol. 2009;60:501-503.
- Malek J, Chedraoui A, Kibbi AG, et al. Genitogluteal porokeratosis: 10 years to make the diagnosis! Am J Dermatopathol. 2009;31:604-606.
Practice Point
- Porokeratosis of the gluteal cleft typically is bilateral but may be unilateral.
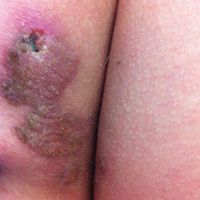
Transverse Melanonychia and Palmar Hyperpigmentation Secondary to Hydroxyurea Therapy
To the Editor:
An 85-year-old woman with a history of hypertension, hyperlipidemia, stroke, hypothyroidism, chronic obstructive pulmonary disease, and chronic myeloproliferative disorder presented to our clinic for evaluation of brown lesions on the hands and discoloration of the fingernails and toenails of 4 months’ duration. Six months prior to visiting our clinic she was admitted to the hospital for a pulmonary embolism. On admission she was noted to have a platelet count of more than 2 million/μL (reference range, 150,000–350,000/μL). She received urgent plasmapheresis and started hydroxyurea 500 mg twice daily, which she continued as an outpatient.
On physical examination at our clinic she had diffusely scattered red and brown macules on the bilateral palms and transverse hyperpigmented bands of various intensities on all fingernails and toenails (Figure). Her platelet count was 372,000/μL, white blood cell count was 5200/μL (reference range, 4500–11,000/μL), hemoglobin was 12.6 g/dL (reference range, 14.0–17.5 g/dL), hematocrit was 39.0% (reference range, 41%–50%), and mean corpuscular volume was 87.5 fL per red cell (reference range, 80–96 fL per red cell).
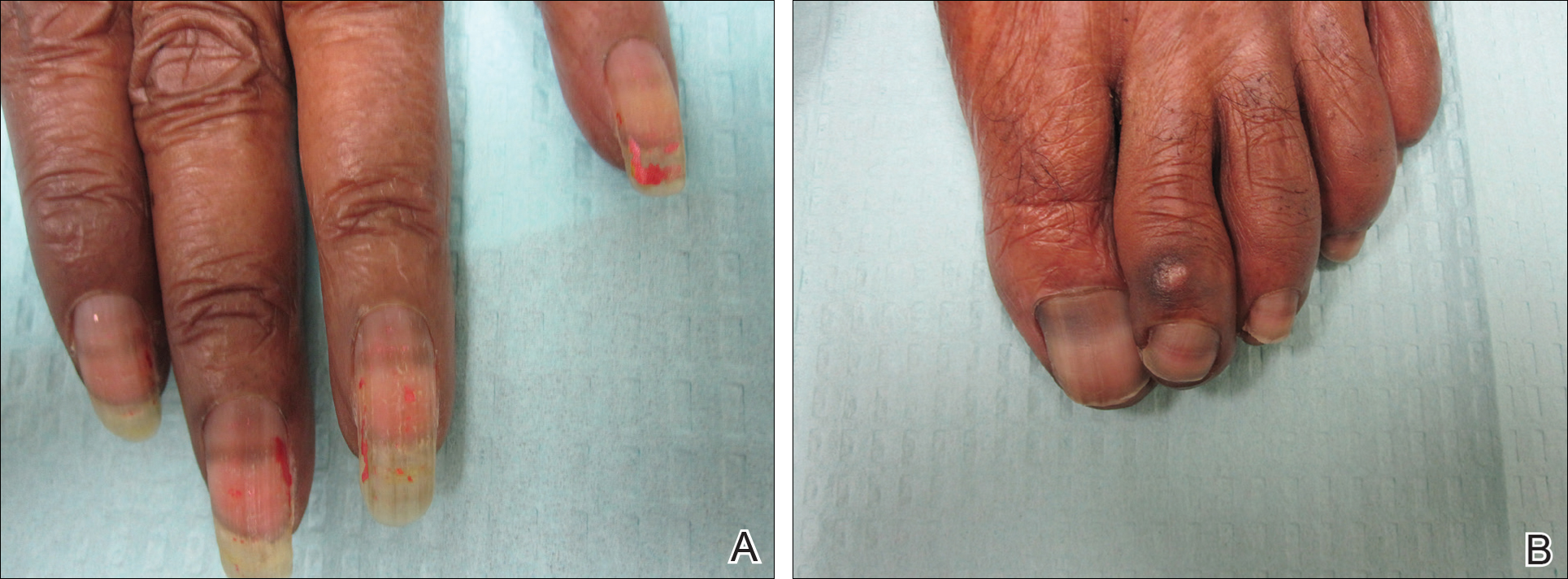
The patient was diagnosed with hydroxyurea-induced nail hyperpigmentation and was counseled on the benign nature of the condition. Three months later her platelet count decreased to below 100,000/μL, and hydroxyurea was discontinued. She noticed considerable improvement in the lesions on the hands and nails with the cessation of hydroxyurea.
Hydroxyurea is a cytostatic agent that has been used for more than 40 years in the treatment of myeloproliferative disorders including chronic myelogenous leukemia, polycythemia vera, essential thrombocythemia, and sickle cell anemia.1 It inhibits ribonucleoside diphosphate reductase and promotes cell death in the S phase of the cell cycle.1-3
Several adverse cutaneous reactions have been associated with hydroxyurea including increased pigmentation, hyperkeratosis, skin atrophy, xerosis, lichenoid eruptions, palmoplantar keratoderma, cutaneous vasculitis, alopecia, chronic leg ulcers, cutaneous carcinomas, and melanonychia.3,4
Hydroxyurea-induced melanonychia most often occurs after several months of therapy but has been reported to occur as early as 4 months and as late as 5 years after initiating the drug.1,4-6 The prevalence of melanonychia in the general population has been estimated at 1% and is thought to increase to approximately 4% in patients treated with hydroxyurea.1,2,6,7 The prevalence of affected individuals increases with age; it is more common in females as well as black and Hispanic patients.2
Multiple patterns of hydroxyurea-induced melanonychia have been described, including longitudinal bands, transverse bands, and diffuse hyperpigmentation.1-3,6 By far the most common pattern described in the literature is longitudinal banding1-3,8; transverse bands are more rare. Although there are sporadic case reports linking the transverse bands with hydroxyurea, these bands occur more frequently with systemic chemotherapy such as doxorubicin and cyclosphosphamide.1,6
The exact pathogenesis of hydroxyurea-induced melanonychia remains unclear, though it is thought to result from focal melanogenesis in the nail bed or matrix followed by deposition of melanin granules on the nail plate.5,8 When these melanocytes are activated, melanosomes filled with melanin are transferred to differentiating matrix cells, which migrate distally as they become nail plate oncocytes, resulting in a visible band of pigmentation in the nail plate.2 There also may be a genetic and photosensitivity component.1,2
Prior case series have described spontaneous remission of nail hyperpigmentation following discontinuation of hydroxyurea therapy.1 In many patients, however, the chronic nature of the myeloproliferative disorder and lack of alternative treatments make a therapeutic change difficult. Although the melanonychia itself is benign, it may precede the appearance of more serious mucocutaneous side effects, such as skin ulceration or development of cutaneous carcinomas, so careful monitoring should be performed.2
Our patient presented with melanonychia that was transverse, polydactylic, monochromic, stable in size and shape, and associated with palmar hyperpigmentation. Of note, the pigmentation remitted over time along with discontinuation of the drug. Although this presentation did not warrant a nail matrix biopsy, it should be noted that patients with single nail melanonychia suspicious for melanoma should have a biopsy, even with concomitant use of hydroxyurea.2 Although transverse melanonychia most commonly is associated with other systemic chemotherapeutics, in the absence of such medications hydroxyurea was the likely culprit in our patient. The palmar hyperpigmentation, which has previously been reported with hydroxyurea use, further solidifies the diagnosis.
- Aste N, Futmo G, Contu F, et al. Nail pigmentation caused by hydroxyurea: report of 9 cases. J Am Acad Dermatol. 2002;47:146-147.
- Murray N, Tapia P, Porcell J, et al. Acquired melanonychia in Chilean patients with essential thrombocythemia treated with hydroxyurea: a report of 7 clinical cases and review of the literature [published online February 7, 2013]. ISRN Dermatol. 2013;2013:325246.
- Utas S. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:469-470.
- Saraceno R, Teoli M, Chimenti S. Hydroxyurea associated with concomitant occurrence of diffuse longitudinal melanonychia and multiple squamous cell carcinomas in an elderly subject. Clin Ther. 2008;30:1324-1329.
- Cohen AD, Hallel-Halevy D, Hatskelzon L, et al. Longitudinal melanonychia associated with hydroxyurea therapy in a patient with essential thrombocytosis. J Eur Acad Dermatol. 1999;13:137-139.
- Hernández-Martín A, Ros-Forteza S, de Unamuno P. Longitudinal, transverse, and diffuse nail hyperpigmentation induced by hydroxyurea. J Am Acad Dermatol. 1999;41(2, pt 2):333-334.
- Kwong Y. Hydroxyurea-induced nail pigmentation. J Am Acad Dermatol. 1996;35:275-276.
- O’Branski E, Ware R, Prose N, et al. Skin and nail changes in children with sickle cell anemia receiving hydroxyurea therapy. J Am Acad Dermatol. 2001;44:859-861.
To the Editor:
An 85-year-old woman with a history of hypertension, hyperlipidemia, stroke, hypothyroidism, chronic obstructive pulmonary disease, and chronic myeloproliferative disorder presented to our clinic for evaluation of brown lesions on the hands and discoloration of the fingernails and toenails of 4 months’ duration. Six months prior to visiting our clinic she was admitted to the hospital for a pulmonary embolism. On admission she was noted to have a platelet count of more than 2 million/μL (reference range, 150,000–350,000/μL). She received urgent plasmapheresis and started hydroxyurea 500 mg twice daily, which she continued as an outpatient.
On physical examination at our clinic she had diffusely scattered red and brown macules on the bilateral palms and transverse hyperpigmented bands of various intensities on all fingernails and toenails (Figure). Her platelet count was 372,000/μL, white blood cell count was 5200/μL (reference range, 4500–11,000/μL), hemoglobin was 12.6 g/dL (reference range, 14.0–17.5 g/dL), hematocrit was 39.0% (reference range, 41%–50%), and mean corpuscular volume was 87.5 fL per red cell (reference range, 80–96 fL per red cell).
The patient was diagnosed with hydroxyurea-induced nail hyperpigmentation and was counseled on the benign nature of the condition. Three months later her platelet count decreased to below 100,000/μL, and hydroxyurea was discontinued. She noticed considerable improvement in the lesions on the hands and nails with the cessation of hydroxyurea.
Hydroxyurea is a cytostatic agent that has been used for more than 40 years in the treatment of myeloproliferative disorders including chronic myelogenous leukemia, polycythemia vera, essential thrombocythemia, and sickle cell anemia.1 It inhibits ribonucleoside diphosphate reductase and promotes cell death in the S phase of the cell cycle.1-3
Several adverse cutaneous reactions have been associated with hydroxyurea including increased pigmentation, hyperkeratosis, skin atrophy, xerosis, lichenoid eruptions, palmoplantar keratoderma, cutaneous vasculitis, alopecia, chronic leg ulcers, cutaneous carcinomas, and melanonychia.3,4
Hydroxyurea-induced melanonychia most often occurs after several months of therapy but has been reported to occur as early as 4 months and as late as 5 years after initiating the drug.1,4-6 The prevalence of melanonychia in the general population has been estimated at 1% and is thought to increase to approximately 4% in patients treated with hydroxyurea.1,2,6,7 The prevalence of affected individuals increases with age; it is more common in females as well as black and Hispanic patients.2
Multiple patterns of hydroxyurea-induced melanonychia have been described, including longitudinal bands, transverse bands, and diffuse hyperpigmentation.1-3,6 By far the most common pattern described in the literature is longitudinal banding1-3,8; transverse bands are more rare. Although there are sporadic case reports linking the transverse bands with hydroxyurea, these bands occur more frequently with systemic chemotherapy such as doxorubicin and cyclosphosphamide.1,6
The exact pathogenesis of hydroxyurea-induced melanonychia remains unclear, though it is thought to result from focal melanogenesis in the nail bed or matrix followed by deposition of melanin granules on the nail plate.5,8 When these melanocytes are activated, melanosomes filled with melanin are transferred to differentiating matrix cells, which migrate distally as they become nail plate oncocytes, resulting in a visible band of pigmentation in the nail plate.2 There also may be a genetic and photosensitivity component.1,2
Prior case series have described spontaneous remission of nail hyperpigmentation following discontinuation of hydroxyurea therapy.1 In many patients, however, the chronic nature of the myeloproliferative disorder and lack of alternative treatments make a therapeutic change difficult. Although the melanonychia itself is benign, it may precede the appearance of more serious mucocutaneous side effects, such as skin ulceration or development of cutaneous carcinomas, so careful monitoring should be performed.2
Our patient presented with melanonychia that was transverse, polydactylic, monochromic, stable in size and shape, and associated with palmar hyperpigmentation. Of note, the pigmentation remitted over time along with discontinuation of the drug. Although this presentation did not warrant a nail matrix biopsy, it should be noted that patients with single nail melanonychia suspicious for melanoma should have a biopsy, even with concomitant use of hydroxyurea.2 Although transverse melanonychia most commonly is associated with other systemic chemotherapeutics, in the absence of such medications hydroxyurea was the likely culprit in our patient. The palmar hyperpigmentation, which has previously been reported with hydroxyurea use, further solidifies the diagnosis.
To the Editor:
An 85-year-old woman with a history of hypertension, hyperlipidemia, stroke, hypothyroidism, chronic obstructive pulmonary disease, and chronic myeloproliferative disorder presented to our clinic for evaluation of brown lesions on the hands and discoloration of the fingernails and toenails of 4 months’ duration. Six months prior to visiting our clinic she was admitted to the hospital for a pulmonary embolism. On admission she was noted to have a platelet count of more than 2 million/μL (reference range, 150,000–350,000/μL). She received urgent plasmapheresis and started hydroxyurea 500 mg twice daily, which she continued as an outpatient.
On physical examination at our clinic she had diffusely scattered red and brown macules on the bilateral palms and transverse hyperpigmented bands of various intensities on all fingernails and toenails (Figure). Her platelet count was 372,000/μL, white blood cell count was 5200/μL (reference range, 4500–11,000/μL), hemoglobin was 12.6 g/dL (reference range, 14.0–17.5 g/dL), hematocrit was 39.0% (reference range, 41%–50%), and mean corpuscular volume was 87.5 fL per red cell (reference range, 80–96 fL per red cell).
The patient was diagnosed with hydroxyurea-induced nail hyperpigmentation and was counseled on the benign nature of the condition. Three months later her platelet count decreased to below 100,000/μL, and hydroxyurea was discontinued. She noticed considerable improvement in the lesions on the hands and nails with the cessation of hydroxyurea.
Hydroxyurea is a cytostatic agent that has been used for more than 40 years in the treatment of myeloproliferative disorders including chronic myelogenous leukemia, polycythemia vera, essential thrombocythemia, and sickle cell anemia.1 It inhibits ribonucleoside diphosphate reductase and promotes cell death in the S phase of the cell cycle.1-3
Several adverse cutaneous reactions have been associated with hydroxyurea including increased pigmentation, hyperkeratosis, skin atrophy, xerosis, lichenoid eruptions, palmoplantar keratoderma, cutaneous vasculitis, alopecia, chronic leg ulcers, cutaneous carcinomas, and melanonychia.3,4
Hydroxyurea-induced melanonychia most often occurs after several months of therapy but has been reported to occur as early as 4 months and as late as 5 years after initiating the drug.1,4-6 The prevalence of melanonychia in the general population has been estimated at 1% and is thought to increase to approximately 4% in patients treated with hydroxyurea.1,2,6,7 The prevalence of affected individuals increases with age; it is more common in females as well as black and Hispanic patients.2
Multiple patterns of hydroxyurea-induced melanonychia have been described, including longitudinal bands, transverse bands, and diffuse hyperpigmentation.1-3,6 By far the most common pattern described in the literature is longitudinal banding1-3,8; transverse bands are more rare. Although there are sporadic case reports linking the transverse bands with hydroxyurea, these bands occur more frequently with systemic chemotherapy such as doxorubicin and cyclosphosphamide.1,6
The exact pathogenesis of hydroxyurea-induced melanonychia remains unclear, though it is thought to result from focal melanogenesis in the nail bed or matrix followed by deposition of melanin granules on the nail plate.5,8 When these melanocytes are activated, melanosomes filled with melanin are transferred to differentiating matrix cells, which migrate distally as they become nail plate oncocytes, resulting in a visible band of pigmentation in the nail plate.2 There also may be a genetic and photosensitivity component.1,2
Prior case series have described spontaneous remission of nail hyperpigmentation following discontinuation of hydroxyurea therapy.1 In many patients, however, the chronic nature of the myeloproliferative disorder and lack of alternative treatments make a therapeutic change difficult. Although the melanonychia itself is benign, it may precede the appearance of more serious mucocutaneous side effects, such as skin ulceration or development of cutaneous carcinomas, so careful monitoring should be performed.2
Our patient presented with melanonychia that was transverse, polydactylic, monochromic, stable in size and shape, and associated with palmar hyperpigmentation. Of note, the pigmentation remitted over time along with discontinuation of the drug. Although this presentation did not warrant a nail matrix biopsy, it should be noted that patients with single nail melanonychia suspicious for melanoma should have a biopsy, even with concomitant use of hydroxyurea.2 Although transverse melanonychia most commonly is associated with other systemic chemotherapeutics, in the absence of such medications hydroxyurea was the likely culprit in our patient. The palmar hyperpigmentation, which has previously been reported with hydroxyurea use, further solidifies the diagnosis.
- Aste N, Futmo G, Contu F, et al. Nail pigmentation caused by hydroxyurea: report of 9 cases. J Am Acad Dermatol. 2002;47:146-147.
- Murray N, Tapia P, Porcell J, et al. Acquired melanonychia in Chilean patients with essential thrombocythemia treated with hydroxyurea: a report of 7 clinical cases and review of the literature [published online February 7, 2013]. ISRN Dermatol. 2013;2013:325246.
- Utas S. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:469-470.
- Saraceno R, Teoli M, Chimenti S. Hydroxyurea associated with concomitant occurrence of diffuse longitudinal melanonychia and multiple squamous cell carcinomas in an elderly subject. Clin Ther. 2008;30:1324-1329.
- Cohen AD, Hallel-Halevy D, Hatskelzon L, et al. Longitudinal melanonychia associated with hydroxyurea therapy in a patient with essential thrombocytosis. J Eur Acad Dermatol. 1999;13:137-139.
- Hernández-Martín A, Ros-Forteza S, de Unamuno P. Longitudinal, transverse, and diffuse nail hyperpigmentation induced by hydroxyurea. J Am Acad Dermatol. 1999;41(2, pt 2):333-334.
- Kwong Y. Hydroxyurea-induced nail pigmentation. J Am Acad Dermatol. 1996;35:275-276.
- O’Branski E, Ware R, Prose N, et al. Skin and nail changes in children with sickle cell anemia receiving hydroxyurea therapy. J Am Acad Dermatol. 2001;44:859-861.
- Aste N, Futmo G, Contu F, et al. Nail pigmentation caused by hydroxyurea: report of 9 cases. J Am Acad Dermatol. 2002;47:146-147.
- Murray N, Tapia P, Porcell J, et al. Acquired melanonychia in Chilean patients with essential thrombocythemia treated with hydroxyurea: a report of 7 clinical cases and review of the literature [published online February 7, 2013]. ISRN Dermatol. 2013;2013:325246.
- Utas S. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:469-470.
- Saraceno R, Teoli M, Chimenti S. Hydroxyurea associated with concomitant occurrence of diffuse longitudinal melanonychia and multiple squamous cell carcinomas in an elderly subject. Clin Ther. 2008;30:1324-1329.
- Cohen AD, Hallel-Halevy D, Hatskelzon L, et al. Longitudinal melanonychia associated with hydroxyurea therapy in a patient with essential thrombocytosis. J Eur Acad Dermatol. 1999;13:137-139.
- Hernández-Martín A, Ros-Forteza S, de Unamuno P. Longitudinal, transverse, and diffuse nail hyperpigmentation induced by hydroxyurea. J Am Acad Dermatol. 1999;41(2, pt 2):333-334.
- Kwong Y. Hydroxyurea-induced nail pigmentation. J Am Acad Dermatol. 1996;35:275-276.
- O’Branski E, Ware R, Prose N, et al. Skin and nail changes in children with sickle cell anemia receiving hydroxyurea therapy. J Am Acad Dermatol. 2001;44:859-861.
Practice Points
- Transverse melanonychia may result as a side effect of hydroxyurea.
- Discontinuation of hydroxyurea typically results in a resolution of symptoms. If the medication cannot be stopped, however, pigmentary changes may precede the development of severe mucocutaneous side effects and close monitoring is warranted.
- Patients with single nail melanonychia suspicious for melanoma should have a biopsy, even with concomitant use of hydroxyurea.
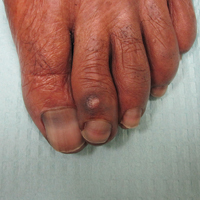
Serpentine Supravenous Hyperpigmentation Following Cisplatin and Pemetrexed Chemotherapy
To the Editor:
Serpentine supravenous hyperpigmentation (SSH) is a rare phenomenon characterized by linear hyperpigmentation of the skin overlying veins secondary to intravenous antineoplastic therapy. The term was first suggested by Hrushesky1 in 1976 as an uncommon side effect of administering intravenous 5-fluorouracil (5-FU). Although 5-FU is the most frequent offending agent, cases involving treatment with actinomycin, cyclophosphamide, docetaxel, fotemustine, nitrogen mustard, nitrosoureas, taxanes, and triazinate, as well as various combinations of chemotherapeutic agents, also have been observed.2,3 We present the case of SSH following a cisplatin and pemetrexed chemotherapy regimen.
A 52-year-old man with newly diagnosed inoperable adenocarcinoma in the left upper lung lobe received 2 cycles of treatment with cisplatin 138 mg and pemetrexed 920 mg 21 days apart. The first cycle of chemotherapy was delivered intravenously through the left forearm and the second cycle through the right forearm. Each infusion was followed by a 20-cc 0.9% saline flush. The patient developed nausea, vomiting, diarrhea, and hyperpigmentation tracing the path of infusion on the right arm as well as a slight darkness on the left arm that were noted by medical staff. At that time, cisplatin was discontinued from the chemotherapeutic regimen.
A port-a-cath was inserted into the patient’s right upper chest 4 weeks later and was used for subsequent infusions. Carboplatin 450 mg was initiated with pemetrexed thereafter. The patient was seen in the dermatology clinic 3 weeks after the insertion of the port-a-cath for evaluation of diffuse tinea versicolor of the trunk. Further examination of the arms revealed asymptomatic serpiginous hyperpigmentation overlying the superficial venous network tracing from the prior intravenous access points in the bilateral forearms to the upper arms (Figure). There was no evidence of extravasation or phlebitis prior to the hyperpigmentation. The patient was continued on pemetrexed and was subsequently lost to follow-up.
Cisplatin was the first member of the platinum-based chemotherapeutic agent class and is now one of the most potent and widely used in the treatment of solid malignancies. The cytotoxic mode of action is primarily mediated through interaction with DNA to form intrastrand cross-link adducts leading to aberrant mitosis and culminating in the activation of apoptosis. A variety of dermatologic complications have been reported with cisplatin chemotherapy including melanonychia, oral mucosal hyperpigmentation, hypersensitivity reactions, extravasation,4 Raynaud phenomenon, and flushing.5
Two cases of SSH have been reported following combination chemotherapy with cisplatin included in the regimen. A 61-year-old man with inoperable esophageal squamous cell carcinoma received cisplatin and 5-FU in addition to concurrent radiotherapy.6 After worsening renal function, cisplatin promptly was replaced with leucovorin. The patient developed SSH after the eighth infusion of 5-FU–leucovorin delivered through a peripheral catheter over a 24-hour period. The cutaneous side effect was attributed to the use of intravenous 5-FU.6 The second case involved a 48-year-old woman diagnosed with Paget disease of the breast who received adjuvant therapy with 12 courses of once-daily 5-FU and docetaxel for 5 years as well as 2 courses of vinorelbine and 1 course of cisplatin and etoposide for lung metastases.7 Serpentine supravenous hyperpigmentation lesions slowly developed over approximately 6 months. Based on the literature, the authors speculated that 5-FU and vinorelbine were most likely to be responsible. They noted, however, the inability to clarify the relationship between the onset of skin lesions and the time course of the chemotherapy.7 Although these cases do not directly implicate cisplatin as the cause of SSH, the possibility of a delayed reaction or augmentation of another drug’s effect cannot be excluded.
Pemetrexed, on the other hand, has not been associated with SSH. Several cutaneous adverse reactions have been reported, including acute generalized exanthematous pustulosis, alopecia, pityriasis lichenoides, radiation recall dermatitis, toxic epidermal necrolysis, and urticarial vasculitis.8 Three cases of pemetrexed-induced skin hyperpigmentation including the palms of the hands and soles of the feet as well as diffuse hyperpigmentation sparing only the palms and soles have been reported.8-10
Similar cases of SSH have demonstrated histopathologic findings with increased basal melanin synthesis and occasional melanophages in the papillary dermis without inflammatory changes.7,11 Although the unique serpentine pattern of hyperpigmentation is instantly recognizable, clinical differential diagnosis may include thrombophlebitis, cutis marmorata, erythema ab igne, livedo reticularis, and lichen planus.2,12
The exact mechanism of SSH has not been conclusively elucidated. Several studies postulate that direct cytotoxic damage causes loss of endothelial integrity permitting the extravasation of the agent to the overlying epidermis and interfering with melanogenesis.2,6,11 Other hypotheses include direct stimulation of melanocytes, depletion of reduced thioredoxin leading to tyrosinase stimulation, hyperthermia-related changes including reduced cytokine production and/or increased expression of melanocyte-stimulating hormone receptor, subclinical phlebitis leading to postinflammatory hyperpigmentation, or hyperpigmentation secondary to increased blood flow in certain areas and therefore increased drug deposition.12,13
Currently, there is no specific therapy recommended for SSH and the pigment may persist anywhere from a few months to more than a year after completing chemotherapy.2,7 Although discontinuing the offending agent would certainly prevent further development, due to the benign nature of the reaction, modifying therapy based on cutaneous findings alone is not recommended.12 Several authors have suggested avoiding peripheral infusions of chemotherapeutic agents known to cause SSH or have recommended using a permanent central venous catheter.6,7 Another option, which needs further investigation, is the administration of an abundant flush following chemotherapy. This technique was described in a case report of a 47-year-old man who developed persistent SSH in the right forearm following docetaxel injection.13 Copious venous washing with 1000 mL of isotonic saline solution following the second infusion in the unaffected arm prevented discoloration. The lack of subsequent reaction may support the theory that direct toxic effect on the vascular endothelium results in hyperpigmentation of the supravenous skin.13
Serpentine supravenous hyperpigmentation is an uncommon cutaneous reaction secondary to antineoplastic therapies. Given the widespread use of chemotherapeutic regimens, dermatologists should be aware of the reaction. Additional studies are warranted to better elucidate the pathogenesis and investigate how infusion techniques might aid in the prevention of skin discoloration. Although this side effect originally was described in relation to 5-FU, subsequent observations have included other chemotherapeutic agents. In light of the findings presented in this report, cisplatin and pemetrexed should be considered on the list of offending agents. Ultimately, patients should be reassured that the lesions are benign, self-limiting, and gradually resolve on their own in most cases.12
- Hrushesky WJ. Letter: serpentine supravenous fluorouracil hyperpigmentation. JAMA. 1976;236:138.
- Ghosh SK, Bandyopadhyay D, Ghoshal L, et al. Letter: docetaxel-induced supravenous serpentine dermatitis. Dermatol Online J. 2011;17:16.
- Pujol RM, Rocamora V, Lopez-Pousa A, et al. Persistent supravenous erythematous eruption: a rare local complication of intravenous 5-fluorouracil therapy. J Am Acad Dermatol. 1998;39:839-842.
- Kufe DW, Pollock RE, Weichsebaum RR, et al, eds. Holland-Frei Cancer Medicine. 6th ed. Hamilton, Ontario, Canada: BC Decker Inc; 2000.
- Mann MW, Berk DR, Popkin DL, et al. Handbook of Dermatology: A Practical Manual. Hoboken, NJ: Wiley-Blackwell; 2009.
- Chan CC, Lin SJ. Serpentine supravenous hyperpigmentation. N Engl J Med. 2010;29:363.
- Ouyang Y-H, Chu C-Y, Hu S-L. Linear hyperpigmentation of the left hand following chemotherapy. Dermatol Sinica. 2004;22:262-263.
- Piérard-Franchimont C, Quatresooz P, Reginster MA, et al. Revisiting cutaneous adverse reactions to pemetrexed. Oncol Lett. 2011;2:769-772.
- Buchinger K, Stahel R, Niggemeier V, et al. Pemetrexed-induced neutropenic enteritis and severe cutaneous hyperpigmentation in a patient with malignant pleural mesothelioma. Lung Cancer. 2013;80:347-349.
- Schallier D, Decoster L, De Greve J. Pemetrexed-induced hyperpigmentation of the skin. Anticancer Res. 2011;31:1753-1755.
- Rao R, Balachandran C. Serpentine supravenous pigmentation. a rare vasculocutaneous effect induced by systemic 5-fluoruracil. Indian J Dermatol Venereol Leprol. 2010;76:714-715.
- Geddes ER, Cohen PR. Antineoplastic agent-associated serpentine supravenous hyperpigmentation: superficial venous system hyperpigmentation following intravenous chemotherapy. South Med J. 2010;103:231-235.
- Ayodogan I, Kavak A, Parlak AH, et al. Persistent serpentine supravenous hyperpigmented eruption associated with docetaxel. J Eur Acad Dermatol Venereol. 2005;19:345-347.
To the Editor:
Serpentine supravenous hyperpigmentation (SSH) is a rare phenomenon characterized by linear hyperpigmentation of the skin overlying veins secondary to intravenous antineoplastic therapy. The term was first suggested by Hrushesky1 in 1976 as an uncommon side effect of administering intravenous 5-fluorouracil (5-FU). Although 5-FU is the most frequent offending agent, cases involving treatment with actinomycin, cyclophosphamide, docetaxel, fotemustine, nitrogen mustard, nitrosoureas, taxanes, and triazinate, as well as various combinations of chemotherapeutic agents, also have been observed.2,3 We present the case of SSH following a cisplatin and pemetrexed chemotherapy regimen.
A 52-year-old man with newly diagnosed inoperable adenocarcinoma in the left upper lung lobe received 2 cycles of treatment with cisplatin 138 mg and pemetrexed 920 mg 21 days apart. The first cycle of chemotherapy was delivered intravenously through the left forearm and the second cycle through the right forearm. Each infusion was followed by a 20-cc 0.9% saline flush. The patient developed nausea, vomiting, diarrhea, and hyperpigmentation tracing the path of infusion on the right arm as well as a slight darkness on the left arm that were noted by medical staff. At that time, cisplatin was discontinued from the chemotherapeutic regimen.
A port-a-cath was inserted into the patient’s right upper chest 4 weeks later and was used for subsequent infusions. Carboplatin 450 mg was initiated with pemetrexed thereafter. The patient was seen in the dermatology clinic 3 weeks after the insertion of the port-a-cath for evaluation of diffuse tinea versicolor of the trunk. Further examination of the arms revealed asymptomatic serpiginous hyperpigmentation overlying the superficial venous network tracing from the prior intravenous access points in the bilateral forearms to the upper arms (Figure). There was no evidence of extravasation or phlebitis prior to the hyperpigmentation. The patient was continued on pemetrexed and was subsequently lost to follow-up.
Cisplatin was the first member of the platinum-based chemotherapeutic agent class and is now one of the most potent and widely used in the treatment of solid malignancies. The cytotoxic mode of action is primarily mediated through interaction with DNA to form intrastrand cross-link adducts leading to aberrant mitosis and culminating in the activation of apoptosis. A variety of dermatologic complications have been reported with cisplatin chemotherapy including melanonychia, oral mucosal hyperpigmentation, hypersensitivity reactions, extravasation,4 Raynaud phenomenon, and flushing.5
Two cases of SSH have been reported following combination chemotherapy with cisplatin included in the regimen. A 61-year-old man with inoperable esophageal squamous cell carcinoma received cisplatin and 5-FU in addition to concurrent radiotherapy.6 After worsening renal function, cisplatin promptly was replaced with leucovorin. The patient developed SSH after the eighth infusion of 5-FU–leucovorin delivered through a peripheral catheter over a 24-hour period. The cutaneous side effect was attributed to the use of intravenous 5-FU.6 The second case involved a 48-year-old woman diagnosed with Paget disease of the breast who received adjuvant therapy with 12 courses of once-daily 5-FU and docetaxel for 5 years as well as 2 courses of vinorelbine and 1 course of cisplatin and etoposide for lung metastases.7 Serpentine supravenous hyperpigmentation lesions slowly developed over approximately 6 months. Based on the literature, the authors speculated that 5-FU and vinorelbine were most likely to be responsible. They noted, however, the inability to clarify the relationship between the onset of skin lesions and the time course of the chemotherapy.7 Although these cases do not directly implicate cisplatin as the cause of SSH, the possibility of a delayed reaction or augmentation of another drug’s effect cannot be excluded.
Pemetrexed, on the other hand, has not been associated with SSH. Several cutaneous adverse reactions have been reported, including acute generalized exanthematous pustulosis, alopecia, pityriasis lichenoides, radiation recall dermatitis, toxic epidermal necrolysis, and urticarial vasculitis.8 Three cases of pemetrexed-induced skin hyperpigmentation including the palms of the hands and soles of the feet as well as diffuse hyperpigmentation sparing only the palms and soles have been reported.8-10
Similar cases of SSH have demonstrated histopathologic findings with increased basal melanin synthesis and occasional melanophages in the papillary dermis without inflammatory changes.7,11 Although the unique serpentine pattern of hyperpigmentation is instantly recognizable, clinical differential diagnosis may include thrombophlebitis, cutis marmorata, erythema ab igne, livedo reticularis, and lichen planus.2,12
The exact mechanism of SSH has not been conclusively elucidated. Several studies postulate that direct cytotoxic damage causes loss of endothelial integrity permitting the extravasation of the agent to the overlying epidermis and interfering with melanogenesis.2,6,11 Other hypotheses include direct stimulation of melanocytes, depletion of reduced thioredoxin leading to tyrosinase stimulation, hyperthermia-related changes including reduced cytokine production and/or increased expression of melanocyte-stimulating hormone receptor, subclinical phlebitis leading to postinflammatory hyperpigmentation, or hyperpigmentation secondary to increased blood flow in certain areas and therefore increased drug deposition.12,13
Currently, there is no specific therapy recommended for SSH and the pigment may persist anywhere from a few months to more than a year after completing chemotherapy.2,7 Although discontinuing the offending agent would certainly prevent further development, due to the benign nature of the reaction, modifying therapy based on cutaneous findings alone is not recommended.12 Several authors have suggested avoiding peripheral infusions of chemotherapeutic agents known to cause SSH or have recommended using a permanent central venous catheter.6,7 Another option, which needs further investigation, is the administration of an abundant flush following chemotherapy. This technique was described in a case report of a 47-year-old man who developed persistent SSH in the right forearm following docetaxel injection.13 Copious venous washing with 1000 mL of isotonic saline solution following the second infusion in the unaffected arm prevented discoloration. The lack of subsequent reaction may support the theory that direct toxic effect on the vascular endothelium results in hyperpigmentation of the supravenous skin.13
Serpentine supravenous hyperpigmentation is an uncommon cutaneous reaction secondary to antineoplastic therapies. Given the widespread use of chemotherapeutic regimens, dermatologists should be aware of the reaction. Additional studies are warranted to better elucidate the pathogenesis and investigate how infusion techniques might aid in the prevention of skin discoloration. Although this side effect originally was described in relation to 5-FU, subsequent observations have included other chemotherapeutic agents. In light of the findings presented in this report, cisplatin and pemetrexed should be considered on the list of offending agents. Ultimately, patients should be reassured that the lesions are benign, self-limiting, and gradually resolve on their own in most cases.12
To the Editor:
Serpentine supravenous hyperpigmentation (SSH) is a rare phenomenon characterized by linear hyperpigmentation of the skin overlying veins secondary to intravenous antineoplastic therapy. The term was first suggested by Hrushesky1 in 1976 as an uncommon side effect of administering intravenous 5-fluorouracil (5-FU). Although 5-FU is the most frequent offending agent, cases involving treatment with actinomycin, cyclophosphamide, docetaxel, fotemustine, nitrogen mustard, nitrosoureas, taxanes, and triazinate, as well as various combinations of chemotherapeutic agents, also have been observed.2,3 We present the case of SSH following a cisplatin and pemetrexed chemotherapy regimen.
A 52-year-old man with newly diagnosed inoperable adenocarcinoma in the left upper lung lobe received 2 cycles of treatment with cisplatin 138 mg and pemetrexed 920 mg 21 days apart. The first cycle of chemotherapy was delivered intravenously through the left forearm and the second cycle through the right forearm. Each infusion was followed by a 20-cc 0.9% saline flush. The patient developed nausea, vomiting, diarrhea, and hyperpigmentation tracing the path of infusion on the right arm as well as a slight darkness on the left arm that were noted by medical staff. At that time, cisplatin was discontinued from the chemotherapeutic regimen.
A port-a-cath was inserted into the patient’s right upper chest 4 weeks later and was used for subsequent infusions. Carboplatin 450 mg was initiated with pemetrexed thereafter. The patient was seen in the dermatology clinic 3 weeks after the insertion of the port-a-cath for evaluation of diffuse tinea versicolor of the trunk. Further examination of the arms revealed asymptomatic serpiginous hyperpigmentation overlying the superficial venous network tracing from the prior intravenous access points in the bilateral forearms to the upper arms (Figure). There was no evidence of extravasation or phlebitis prior to the hyperpigmentation. The patient was continued on pemetrexed and was subsequently lost to follow-up.
Cisplatin was the first member of the platinum-based chemotherapeutic agent class and is now one of the most potent and widely used in the treatment of solid malignancies. The cytotoxic mode of action is primarily mediated through interaction with DNA to form intrastrand cross-link adducts leading to aberrant mitosis and culminating in the activation of apoptosis. A variety of dermatologic complications have been reported with cisplatin chemotherapy including melanonychia, oral mucosal hyperpigmentation, hypersensitivity reactions, extravasation,4 Raynaud phenomenon, and flushing.5
Two cases of SSH have been reported following combination chemotherapy with cisplatin included in the regimen. A 61-year-old man with inoperable esophageal squamous cell carcinoma received cisplatin and 5-FU in addition to concurrent radiotherapy.6 After worsening renal function, cisplatin promptly was replaced with leucovorin. The patient developed SSH after the eighth infusion of 5-FU–leucovorin delivered through a peripheral catheter over a 24-hour period. The cutaneous side effect was attributed to the use of intravenous 5-FU.6 The second case involved a 48-year-old woman diagnosed with Paget disease of the breast who received adjuvant therapy with 12 courses of once-daily 5-FU and docetaxel for 5 years as well as 2 courses of vinorelbine and 1 course of cisplatin and etoposide for lung metastases.7 Serpentine supravenous hyperpigmentation lesions slowly developed over approximately 6 months. Based on the literature, the authors speculated that 5-FU and vinorelbine were most likely to be responsible. They noted, however, the inability to clarify the relationship between the onset of skin lesions and the time course of the chemotherapy.7 Although these cases do not directly implicate cisplatin as the cause of SSH, the possibility of a delayed reaction or augmentation of another drug’s effect cannot be excluded.
Pemetrexed, on the other hand, has not been associated with SSH. Several cutaneous adverse reactions have been reported, including acute generalized exanthematous pustulosis, alopecia, pityriasis lichenoides, radiation recall dermatitis, toxic epidermal necrolysis, and urticarial vasculitis.8 Three cases of pemetrexed-induced skin hyperpigmentation including the palms of the hands and soles of the feet as well as diffuse hyperpigmentation sparing only the palms and soles have been reported.8-10
Similar cases of SSH have demonstrated histopathologic findings with increased basal melanin synthesis and occasional melanophages in the papillary dermis without inflammatory changes.7,11 Although the unique serpentine pattern of hyperpigmentation is instantly recognizable, clinical differential diagnosis may include thrombophlebitis, cutis marmorata, erythema ab igne, livedo reticularis, and lichen planus.2,12
The exact mechanism of SSH has not been conclusively elucidated. Several studies postulate that direct cytotoxic damage causes loss of endothelial integrity permitting the extravasation of the agent to the overlying epidermis and interfering with melanogenesis.2,6,11 Other hypotheses include direct stimulation of melanocytes, depletion of reduced thioredoxin leading to tyrosinase stimulation, hyperthermia-related changes including reduced cytokine production and/or increased expression of melanocyte-stimulating hormone receptor, subclinical phlebitis leading to postinflammatory hyperpigmentation, or hyperpigmentation secondary to increased blood flow in certain areas and therefore increased drug deposition.12,13
Currently, there is no specific therapy recommended for SSH and the pigment may persist anywhere from a few months to more than a year after completing chemotherapy.2,7 Although discontinuing the offending agent would certainly prevent further development, due to the benign nature of the reaction, modifying therapy based on cutaneous findings alone is not recommended.12 Several authors have suggested avoiding peripheral infusions of chemotherapeutic agents known to cause SSH or have recommended using a permanent central venous catheter.6,7 Another option, which needs further investigation, is the administration of an abundant flush following chemotherapy. This technique was described in a case report of a 47-year-old man who developed persistent SSH in the right forearm following docetaxel injection.13 Copious venous washing with 1000 mL of isotonic saline solution following the second infusion in the unaffected arm prevented discoloration. The lack of subsequent reaction may support the theory that direct toxic effect on the vascular endothelium results in hyperpigmentation of the supravenous skin.13
Serpentine supravenous hyperpigmentation is an uncommon cutaneous reaction secondary to antineoplastic therapies. Given the widespread use of chemotherapeutic regimens, dermatologists should be aware of the reaction. Additional studies are warranted to better elucidate the pathogenesis and investigate how infusion techniques might aid in the prevention of skin discoloration. Although this side effect originally was described in relation to 5-FU, subsequent observations have included other chemotherapeutic agents. In light of the findings presented in this report, cisplatin and pemetrexed should be considered on the list of offending agents. Ultimately, patients should be reassured that the lesions are benign, self-limiting, and gradually resolve on their own in most cases.12
- Hrushesky WJ. Letter: serpentine supravenous fluorouracil hyperpigmentation. JAMA. 1976;236:138.
- Ghosh SK, Bandyopadhyay D, Ghoshal L, et al. Letter: docetaxel-induced supravenous serpentine dermatitis. Dermatol Online J. 2011;17:16.
- Pujol RM, Rocamora V, Lopez-Pousa A, et al. Persistent supravenous erythematous eruption: a rare local complication of intravenous 5-fluorouracil therapy. J Am Acad Dermatol. 1998;39:839-842.
- Kufe DW, Pollock RE, Weichsebaum RR, et al, eds. Holland-Frei Cancer Medicine. 6th ed. Hamilton, Ontario, Canada: BC Decker Inc; 2000.
- Mann MW, Berk DR, Popkin DL, et al. Handbook of Dermatology: A Practical Manual. Hoboken, NJ: Wiley-Blackwell; 2009.
- Chan CC, Lin SJ. Serpentine supravenous hyperpigmentation. N Engl J Med. 2010;29:363.
- Ouyang Y-H, Chu C-Y, Hu S-L. Linear hyperpigmentation of the left hand following chemotherapy. Dermatol Sinica. 2004;22:262-263.
- Piérard-Franchimont C, Quatresooz P, Reginster MA, et al. Revisiting cutaneous adverse reactions to pemetrexed. Oncol Lett. 2011;2:769-772.
- Buchinger K, Stahel R, Niggemeier V, et al. Pemetrexed-induced neutropenic enteritis and severe cutaneous hyperpigmentation in a patient with malignant pleural mesothelioma. Lung Cancer. 2013;80:347-349.
- Schallier D, Decoster L, De Greve J. Pemetrexed-induced hyperpigmentation of the skin. Anticancer Res. 2011;31:1753-1755.
- Rao R, Balachandran C. Serpentine supravenous pigmentation. a rare vasculocutaneous effect induced by systemic 5-fluoruracil. Indian J Dermatol Venereol Leprol. 2010;76:714-715.
- Geddes ER, Cohen PR. Antineoplastic agent-associated serpentine supravenous hyperpigmentation: superficial venous system hyperpigmentation following intravenous chemotherapy. South Med J. 2010;103:231-235.
- Ayodogan I, Kavak A, Parlak AH, et al. Persistent serpentine supravenous hyperpigmented eruption associated with docetaxel. J Eur Acad Dermatol Venereol. 2005;19:345-347.
- Hrushesky WJ. Letter: serpentine supravenous fluorouracil hyperpigmentation. JAMA. 1976;236:138.
- Ghosh SK, Bandyopadhyay D, Ghoshal L, et al. Letter: docetaxel-induced supravenous serpentine dermatitis. Dermatol Online J. 2011;17:16.
- Pujol RM, Rocamora V, Lopez-Pousa A, et al. Persistent supravenous erythematous eruption: a rare local complication of intravenous 5-fluorouracil therapy. J Am Acad Dermatol. 1998;39:839-842.
- Kufe DW, Pollock RE, Weichsebaum RR, et al, eds. Holland-Frei Cancer Medicine. 6th ed. Hamilton, Ontario, Canada: BC Decker Inc; 2000.
- Mann MW, Berk DR, Popkin DL, et al. Handbook of Dermatology: A Practical Manual. Hoboken, NJ: Wiley-Blackwell; 2009.
- Chan CC, Lin SJ. Serpentine supravenous hyperpigmentation. N Engl J Med. 2010;29:363.
- Ouyang Y-H, Chu C-Y, Hu S-L. Linear hyperpigmentation of the left hand following chemotherapy. Dermatol Sinica. 2004;22:262-263.
- Piérard-Franchimont C, Quatresooz P, Reginster MA, et al. Revisiting cutaneous adverse reactions to pemetrexed. Oncol Lett. 2011;2:769-772.
- Buchinger K, Stahel R, Niggemeier V, et al. Pemetrexed-induced neutropenic enteritis and severe cutaneous hyperpigmentation in a patient with malignant pleural mesothelioma. Lung Cancer. 2013;80:347-349.
- Schallier D, Decoster L, De Greve J. Pemetrexed-induced hyperpigmentation of the skin. Anticancer Res. 2011;31:1753-1755.
- Rao R, Balachandran C. Serpentine supravenous pigmentation. a rare vasculocutaneous effect induced by systemic 5-fluoruracil. Indian J Dermatol Venereol Leprol. 2010;76:714-715.
- Geddes ER, Cohen PR. Antineoplastic agent-associated serpentine supravenous hyperpigmentation: superficial venous system hyperpigmentation following intravenous chemotherapy. South Med J. 2010;103:231-235.
- Ayodogan I, Kavak A, Parlak AH, et al. Persistent serpentine supravenous hyperpigmented eruption associated with docetaxel. J Eur Acad Dermatol Venereol. 2005;19:345-347.
Practice Points
- A variety of dermatologic complications have been reported with cisplatin chemotherapy, including serpentine supravenous hyperpigmentation (SSH); however, pemetrexed has not been associated with SSH.
- Although discontinuing the offending agent would certainly prevent further development, due to the benign nature of the reaction, modifying therapy based on cutaneous findings alone is not recommended.
Recovery of Hair in the Psoriatic Plaques of a Patient With Coexistent Alopecia Universalis
To the Editor:
Both alopecia areata (AA) and psoriasis vulgaris are chronic relapsing autoimmune diseases, with AA causing nonscarring hair loss in approximately 0.1% to 0.2%1 of the population with a lifetime risk of 1.7%,2 and psoriasis more broadly impacting 1.5% to 2% of the population.3 The helper T cell (TH1) cytokine milieu is pathogenic in both conditions.4-6 IFN-γ knockout mice, unlike their wild-type counterparts, do not exhibit AA.7 Psoriasis is notably improved by IL-10 injections, which dampen the TH1 response.8 Distinct from AA, TH17 and TH22 cells have been implicated as key players in psoriasis pathogenesis, along with the associated IL-17 and IL-22 cytokines.9-12
Few cases of patients with concurrent AA and psoriasis have been described. Interestingly, these cases document normal hair regrowth in the areas of psoriasis.13-16 These cases may offer unique insight into the immune factors driving each disease. We describe a case of a man with both alopecia universalis (AU) and psoriasis who developed hair regrowth in some of the psoriatic plaques.
A 34-year-old man with concurrent AU and psoriasis who had not used any systemic or topical medication for either condition in the last year presented to our clinic seeking treatment. The patient had a history of alopecia totalis as a toddler that completely resolved by 4 years of age with the use of squaric acid dibutylester (SADBE). At 31 years of age, the alopecia recurred and was localized to the scalp. It was partially responsive to intralesional triamcinolone acetonide. The patient’s alopecia worsened over the 2 years following recurrence, ultimately progressing to AU. Two months after the alopecia recurrence, he developed the first psoriatic plaques. As the plaque psoriasis progressed, systemic therapy was initiated, first methotrexate and then etanercept. Shortly after developing AU, he lost his health insurance and discontinued all therapy. The patient’s psoriasis began to recur approximately 3 months after stopping etanercept. He was not using any other psoriasis medications. At that time, he noted terminal hair regrowth within some of the psoriatic plaques. No terminal hairs grew outside of the psoriatic plaques, and all regions with growth had previously been without hair for an extended period of time. The patient presented to our clinic approximately 1 year later. He had no other medical conditions and no relevant family history.
On initial physical examination, he had nonscarring hair loss involving nearly 100% of the body with psoriatic plaques on approximately 30% of the body surface area. Regions of terminal hair growth were confined to some but not all of the psoriatic plaques (Figure). Interestingly, the terminal hairs were primarily localized to the thickest central regions of the plaques. The patient’s psoriasis was treated with a combination of topical clobetasol and calcipotriene. In addition, he was started on tacrolimus ointment to the face and eyebrows for the AA. Maintenance of terminal hair within a region of topically treated psoriasis on the forearm persisted at the 2-month follow-up despite complete clearance of the corresponding psoriatic plaque. A small psoriatic plaque on the scalp cleared early with topical therapy without noticeable hair regrowth. The patient subsequently was started on contact immunotherapy with SADBE and intralesional triamcinolone acetonide for the scalp alopecia without satisfactory response. He decided to discontinue further attempts at treating the alopecia and requested to be restarted on etanercept therapy for recalcitrant psoriatic plaques. His psoriasis responded well to this therapy and he continues to be followed in our psoriasis clinic. One year after clearance of the treated psoriatic plaques, the corresponding terminal hairs persist.
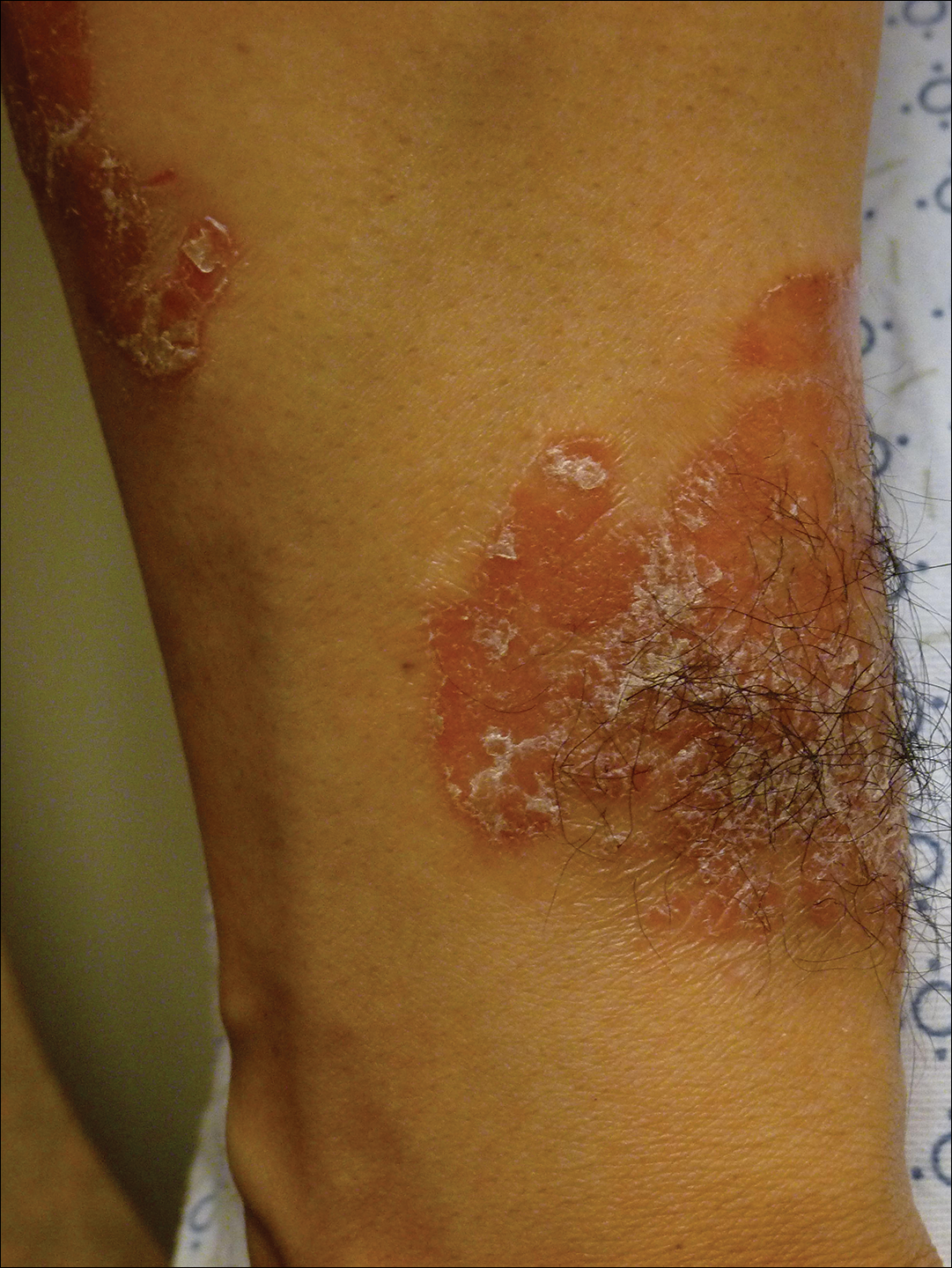
Contact immunotherapy, most commonly with diphenylcyclopropenone or SADBE, is reported to have a 50% to 60% success rate in extensive AA, with a broad range of 9% to 87%17; however, randomized controlled trials testing the efficacy of contact immunotherapy are lacking. Although the mechanism of action of these topical sensitizers is not clearly delineated, it has been postulated that by inducing a new type of inflammatory response in the region, the immunologic milieu is changed, allowing the hair to grow. Some proposed mechanisms include promoting perifollicular lymphocyte apoptosis, preventing new recruitment of autoreactive lymphocytes, and allowing for the correction of aberrant major histocompatibility complex expression on the hair matrix epithelium to regain follicle immune privilege.18-20
Iatrogenic immunotherapy may work analogously to the natural immune system deviation demonstrated in our patient. Psoriasis and AA are believed to form competing immune cells and cytokine milieus, thus explaining how an individual with AA could regain normal hair growth in areas of psoriasis.15,16 The Renbök phenomenon, or reverse Köbner phenomenon, coined by Happle et al13 can be used to describe both the iatrogenic and natural cases of dermatologic disease improvement in response to secondary insults.14
A complex cascade of immune cells and cytokines coordinate AA pathogenesis. In the acute stage of AA, an inflammatory infiltrate of CD4+ T cells, CD8+ T cells, and antigen-presenting cells target anagen phase follicles, with a higher CD4+:CD8+ ratio in clinically active disease.21-23 Subcutaneous injections of either CD4+ or CD8+ lymphocyte subsets from mice with AA into normal-haired mice induces disease. However, CD8+ T cell injections rapidly produce apparent hair loss, whereas CD4+ T cells cause hair loss after several weeks, suggesting that CD8+ T cells directly modulate AA hair loss and CD4+ T cells act as an aide.24 The growth, differentiation, and survival of CD8+ T cells are stimulated by IL-2 and IFN-γ. Alopecia areata biopsies demonstrate a prevalence of TH1 cytokines, and patients with localized AA, alopecia totalis, and AU have notably higher serum IFN-γ levels compared to controls.25 In murine models, IL-1α and IL-1β increase during the catagen phase of the hair cycle and peak during the telogen phase.26 Excessive IL-1β expression is detected in the early stages of human disease, and certain IL-1β polymorphisms are associated with severe forms of AA.26 The role of tumor necrosis factor (TNF) α in AA is not well understood. In vitro studies show it inhibits hair growth, suggesting the cytokine may play a role in AA.27 However, anti–TNF-α therapy is not effective in AA, and case reports propose these therapies rarely induce AA.28-31
The TH1 response is likewise critical to psoriatic plaque development. IFN-γ and TNF-α are overexpressed in psoriatic plaques.32 IFN-γ has an antiproliferative and differentiation-inducing effect on normal keratinocytes, but psoriatic epithelial cells in vitro respond differently to the cytokine with a notably diminished growth inhibition.33,34 One explanation for the role of IFN-γ is that it stimulates dendritic cells to produce IL-1 and IL-23.35 IL-23 activates TH17 cells36; TH1 and TH17 conditions produce IL-22 whose serum level correlates with disease severity.37-39 IL-22 induces keratinocyte proliferation and migration and inhibits keratinocyte differentiation, helping account for hallmarks of the disease.40 Patients with psoriasis have increased levels of TH1, TH17, and TH22 cells, as well as their associated cytokines, in the skin and blood compared to controls.4,11,32,39,41
Alopecia areata and psoriasis are regulated by complex and still not entirely understood immune interactions. The fact that many of the same therapies are used to treat both diseases emphasizes both their overlapping characteristics and the lack of targeted therapy. It is unclear if and how the topical or systemic therapies used in our patient to treat one disease affected the natural history of the other condition. It is important to highlight, however, that the patient had not been treated for months when he developed the psoriatic plaques with hair regrowth. Other case reports also document hair regrowth in untreated plaques,13,16 making it unlikely to be a side effect of the medication regimen. For both psoriasis and AA, the immune cell composition and cytokine levels in the skin or serum vary throughout a patient’s disease course depending on severity of disease or response to treatment.6,39,42,43 Therefore, we hypothesize that the 2 conditions interact in a similarly distinct manner based on each disease’s stage and intensity in the patient. Both our patient’s course thus far and the various presentations described by other groups support this hypothesis. Our patient had a small region of psoriasis on the scalp that cleared without any terminal hair growth. He also had larger plaques on the forearms that developed hair growth most predominantly within the thicker regions of the plaques. His unique presentation highlights the fluidity of the immune factors driving psoriasis vulgaris and AA.
- Safavi K. Prevalence of alopecia areata in the First National Health and Nutrition Examination Survey. Arch Dermatol. 1992;128:702.
- Safavi KH, Muller SA, Suman VJ, et al. Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70:628-633.
- Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York, NY: McGraw-Hill; 2009.
- Austin LM, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Ghoreishi M, Martinka M, Dutz JP. Type 1 interferon signature in the scalp lesions of alopecia areata. Br J Dermatol. 2010;163:57-62.
- Rossi A, Cantisani C, Carlesimo M, et al. Serum concentrations of IL-2, IL-6, IL-12 and TNF-α in patients with alopecia areata. Int J Immunopathol Pharmacol. 2012;25:781-788.
- Freyschmidt-Paul P, McElwee KJ, Hoffmann R, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515-521.
- Reich K, Garbe C, Blaschke V, et al. Response of psoriasis to interleukin-10 is associated with suppression of cutaneous type 1 inflammation, downregulation of the epidermal interleukin-8/CXCR2 pathway and normalization of keratinocyte maturation. J Invest Dermatol. 2001;116:319-329.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648-651.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Zaba LC, Suárez-Fariñas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022-1030.e395.
- Happle R, van der Steen PHM, Perret CM. The Renbök phenomenon: an inverse Köebner reaction observed in alopecia areata. Eur J Dermatol. 1991;2:39-40.
- Ito T, Hashizume H, Takigawa M. Contact immunotherapy-induced Renbök phenomenon in a patient with alopecia areata and psoriasis vulgaris. Eur J Dermatol. 2010;20:126-127.
- Criado PR, Valente NY, Michalany NS, et al. An unusual association between scalp psoriasis and ophiasic alopecia areata: the Renbök phenomenon. Clin Exp Dermatol. 2007;32:320-321.
- Harris JE, Seykora JT, Lee RA. Renbök phenomenon and contact sensitization in a patient with alopecia universalis. Arch Dermatol. 2010;146:422-425.
- Alkhalifah A. Topical and intralesional therapies for alopecia areata. Dermatol Ther. 2011;24:355-363.
- Herbst V, Zöller M, Kissling S, et al. Diphenylcyclopropenone treatment of alopecia areata induces apoptosis of perifollicular lymphocytes. Eur J Dermatol. 2006;16:537-542.
- Zöller M, Freyschmidt-Paul P, Vitacolonna M, et al. Chronic delayed-type hypersensitivity reaction as a means to treat alopecia areata. Clin Exp Immunol. 2004;135:398-408.
- Bröcker EB, Echternacht-Happle K, Hamm H, et al. Abnormal expression of class I and class II major histocompatibility antigens in alopecia areata: modulation by topical immunotherapy. J Invest Dermatol. 1987;88:564-568.
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11:216-223.
- Perret C, Wiesner-Menzel L, Happle R. Immunohistochemical analysis of T-cell subsets in the peribulbar and intrabulbar infiltrates of alopecia areata. Acta Derm Venereol. 1984;64:26-30.
- Wiesner-Menzel L, Happle R. Intrabulbar and peribulbar accumulation of dendritic OKT 6-positive cells in alopecia areata. Arch Dermatol Res. 1984;276:333-334.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8+ cells induces localized hair loss whereas CD4+/CD25– cells promote systemic alopecia areata and CD4+/CD25+ cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- Arca E, Muşabak U, Akar A, et al. Interferon-gamma in alopecia areata. Eur J Dermatol. 2004;14:33-36.
- Hoffmann R. The potential role of cytokines and T cells in alopecia areata. J Investig Dermatol Symp Proc. 1999;4:235-238.
- Philpott MP, Sanders DA, Bowen J, et al. Effects of interleukins, colony-stimulating factor and tumour necrosis factor on human hair follicle growth in vitro: a possible role for interleukin-1 and tumour necrosis factor-alpha in alopecia areata. Br J Dermatol. 1996;135:942-948.
- Le Bidre E, Chaby G, Martin L, et al. Alopecia areata during anti-TNF alpha therapy: nine cases. Ann Dermatol Venereol. 2011;138:285-293.
- Ferran M, Calvet J, Almirall M, et al. Alopecia areata as another immune-mediated disease developed in patients treated with tumour necrosis factor-α blocker agents: report of five cases and review of the literature. J Eur Acad Dermatol Venereol. 2011;25:479-484.
- Pan Y, Rao NA. Alopecia areata during etanercept therapy. Ocul Immunol Inflamm. 2009;17:127-129.
- Pelivani N, Hassan AS, Braathen LR, et al. Alopecia areata universalis elicited during treatment with adalimumab. Dermatology. 2008;216:320-323.
- Uyemura K, Yamamura M, Fivenson DF, et al. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701-705.
- Baker BS, Powles AV, Valdimarsson H, et al. An altered response by psoriatic keratinocytes to gamma interferon. Scan J Immunol. 1988;28:735-740.
- Jackson M, Howie SE, Weller R, et al. Psoriatic keratinocytes show reduced IRF-1 and STAT-1alpha activation in response to gamma-IFN. FASEB J. 1999;13:495-502.
- Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385-422.
- McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314-324.
- Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650-657.
- Boniface K, Blumenschein WM, Brovont-Porth K, et al. Human Th17 cells comprise heterogeneous subsets including IFN-gamma-producing cells with distinct properties from the Th1 lineage. J Immunol. 2010;185:679-687.
- Kagami S, Rizzo HL, Lee JJ, et al. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130:1373-1383.
- Boniface K, Bernard FX, Garcia M, et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695-3702.
- Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129:2175-2183.
- Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5:699-711.
- Hoffmann R, Wenzel E, Huth A, et al. Cytokine mRNA levels in alopecia areata before and after treatment with the contact allergen diphenylcyclopropenone. J Invest Dermatol. 1994;103:530-533.
To the Editor:
Both alopecia areata (AA) and psoriasis vulgaris are chronic relapsing autoimmune diseases, with AA causing nonscarring hair loss in approximately 0.1% to 0.2%1 of the population with a lifetime risk of 1.7%,2 and psoriasis more broadly impacting 1.5% to 2% of the population.3 The helper T cell (TH1) cytokine milieu is pathogenic in both conditions.4-6 IFN-γ knockout mice, unlike their wild-type counterparts, do not exhibit AA.7 Psoriasis is notably improved by IL-10 injections, which dampen the TH1 response.8 Distinct from AA, TH17 and TH22 cells have been implicated as key players in psoriasis pathogenesis, along with the associated IL-17 and IL-22 cytokines.9-12
Few cases of patients with concurrent AA and psoriasis have been described. Interestingly, these cases document normal hair regrowth in the areas of psoriasis.13-16 These cases may offer unique insight into the immune factors driving each disease. We describe a case of a man with both alopecia universalis (AU) and psoriasis who developed hair regrowth in some of the psoriatic plaques.
A 34-year-old man with concurrent AU and psoriasis who had not used any systemic or topical medication for either condition in the last year presented to our clinic seeking treatment. The patient had a history of alopecia totalis as a toddler that completely resolved by 4 years of age with the use of squaric acid dibutylester (SADBE). At 31 years of age, the alopecia recurred and was localized to the scalp. It was partially responsive to intralesional triamcinolone acetonide. The patient’s alopecia worsened over the 2 years following recurrence, ultimately progressing to AU. Two months after the alopecia recurrence, he developed the first psoriatic plaques. As the plaque psoriasis progressed, systemic therapy was initiated, first methotrexate and then etanercept. Shortly after developing AU, he lost his health insurance and discontinued all therapy. The patient’s psoriasis began to recur approximately 3 months after stopping etanercept. He was not using any other psoriasis medications. At that time, he noted terminal hair regrowth within some of the psoriatic plaques. No terminal hairs grew outside of the psoriatic plaques, and all regions with growth had previously been without hair for an extended period of time. The patient presented to our clinic approximately 1 year later. He had no other medical conditions and no relevant family history.
On initial physical examination, he had nonscarring hair loss involving nearly 100% of the body with psoriatic plaques on approximately 30% of the body surface area. Regions of terminal hair growth were confined to some but not all of the psoriatic plaques (Figure). Interestingly, the terminal hairs were primarily localized to the thickest central regions of the plaques. The patient’s psoriasis was treated with a combination of topical clobetasol and calcipotriene. In addition, he was started on tacrolimus ointment to the face and eyebrows for the AA. Maintenance of terminal hair within a region of topically treated psoriasis on the forearm persisted at the 2-month follow-up despite complete clearance of the corresponding psoriatic plaque. A small psoriatic plaque on the scalp cleared early with topical therapy without noticeable hair regrowth. The patient subsequently was started on contact immunotherapy with SADBE and intralesional triamcinolone acetonide for the scalp alopecia without satisfactory response. He decided to discontinue further attempts at treating the alopecia and requested to be restarted on etanercept therapy for recalcitrant psoriatic plaques. His psoriasis responded well to this therapy and he continues to be followed in our psoriasis clinic. One year after clearance of the treated psoriatic plaques, the corresponding terminal hairs persist.
Contact immunotherapy, most commonly with diphenylcyclopropenone or SADBE, is reported to have a 50% to 60% success rate in extensive AA, with a broad range of 9% to 87%17; however, randomized controlled trials testing the efficacy of contact immunotherapy are lacking. Although the mechanism of action of these topical sensitizers is not clearly delineated, it has been postulated that by inducing a new type of inflammatory response in the region, the immunologic milieu is changed, allowing the hair to grow. Some proposed mechanisms include promoting perifollicular lymphocyte apoptosis, preventing new recruitment of autoreactive lymphocytes, and allowing for the correction of aberrant major histocompatibility complex expression on the hair matrix epithelium to regain follicle immune privilege.18-20
Iatrogenic immunotherapy may work analogously to the natural immune system deviation demonstrated in our patient. Psoriasis and AA are believed to form competing immune cells and cytokine milieus, thus explaining how an individual with AA could regain normal hair growth in areas of psoriasis.15,16 The Renbök phenomenon, or reverse Köbner phenomenon, coined by Happle et al13 can be used to describe both the iatrogenic and natural cases of dermatologic disease improvement in response to secondary insults.14
A complex cascade of immune cells and cytokines coordinate AA pathogenesis. In the acute stage of AA, an inflammatory infiltrate of CD4+ T cells, CD8+ T cells, and antigen-presenting cells target anagen phase follicles, with a higher CD4+:CD8+ ratio in clinically active disease.21-23 Subcutaneous injections of either CD4+ or CD8+ lymphocyte subsets from mice with AA into normal-haired mice induces disease. However, CD8+ T cell injections rapidly produce apparent hair loss, whereas CD4+ T cells cause hair loss after several weeks, suggesting that CD8+ T cells directly modulate AA hair loss and CD4+ T cells act as an aide.24 The growth, differentiation, and survival of CD8+ T cells are stimulated by IL-2 and IFN-γ. Alopecia areata biopsies demonstrate a prevalence of TH1 cytokines, and patients with localized AA, alopecia totalis, and AU have notably higher serum IFN-γ levels compared to controls.25 In murine models, IL-1α and IL-1β increase during the catagen phase of the hair cycle and peak during the telogen phase.26 Excessive IL-1β expression is detected in the early stages of human disease, and certain IL-1β polymorphisms are associated with severe forms of AA.26 The role of tumor necrosis factor (TNF) α in AA is not well understood. In vitro studies show it inhibits hair growth, suggesting the cytokine may play a role in AA.27 However, anti–TNF-α therapy is not effective in AA, and case reports propose these therapies rarely induce AA.28-31
The TH1 response is likewise critical to psoriatic plaque development. IFN-γ and TNF-α are overexpressed in psoriatic plaques.32 IFN-γ has an antiproliferative and differentiation-inducing effect on normal keratinocytes, but psoriatic epithelial cells in vitro respond differently to the cytokine with a notably diminished growth inhibition.33,34 One explanation for the role of IFN-γ is that it stimulates dendritic cells to produce IL-1 and IL-23.35 IL-23 activates TH17 cells36; TH1 and TH17 conditions produce IL-22 whose serum level correlates with disease severity.37-39 IL-22 induces keratinocyte proliferation and migration and inhibits keratinocyte differentiation, helping account for hallmarks of the disease.40 Patients with psoriasis have increased levels of TH1, TH17, and TH22 cells, as well as their associated cytokines, in the skin and blood compared to controls.4,11,32,39,41
Alopecia areata and psoriasis are regulated by complex and still not entirely understood immune interactions. The fact that many of the same therapies are used to treat both diseases emphasizes both their overlapping characteristics and the lack of targeted therapy. It is unclear if and how the topical or systemic therapies used in our patient to treat one disease affected the natural history of the other condition. It is important to highlight, however, that the patient had not been treated for months when he developed the psoriatic plaques with hair regrowth. Other case reports also document hair regrowth in untreated plaques,13,16 making it unlikely to be a side effect of the medication regimen. For both psoriasis and AA, the immune cell composition and cytokine levels in the skin or serum vary throughout a patient’s disease course depending on severity of disease or response to treatment.6,39,42,43 Therefore, we hypothesize that the 2 conditions interact in a similarly distinct manner based on each disease’s stage and intensity in the patient. Both our patient’s course thus far and the various presentations described by other groups support this hypothesis. Our patient had a small region of psoriasis on the scalp that cleared without any terminal hair growth. He also had larger plaques on the forearms that developed hair growth most predominantly within the thicker regions of the plaques. His unique presentation highlights the fluidity of the immune factors driving psoriasis vulgaris and AA.
To the Editor:
Both alopecia areata (AA) and psoriasis vulgaris are chronic relapsing autoimmune diseases, with AA causing nonscarring hair loss in approximately 0.1% to 0.2%1 of the population with a lifetime risk of 1.7%,2 and psoriasis more broadly impacting 1.5% to 2% of the population.3 The helper T cell (TH1) cytokine milieu is pathogenic in both conditions.4-6 IFN-γ knockout mice, unlike their wild-type counterparts, do not exhibit AA.7 Psoriasis is notably improved by IL-10 injections, which dampen the TH1 response.8 Distinct from AA, TH17 and TH22 cells have been implicated as key players in psoriasis pathogenesis, along with the associated IL-17 and IL-22 cytokines.9-12
Few cases of patients with concurrent AA and psoriasis have been described. Interestingly, these cases document normal hair regrowth in the areas of psoriasis.13-16 These cases may offer unique insight into the immune factors driving each disease. We describe a case of a man with both alopecia universalis (AU) and psoriasis who developed hair regrowth in some of the psoriatic plaques.
A 34-year-old man with concurrent AU and psoriasis who had not used any systemic or topical medication for either condition in the last year presented to our clinic seeking treatment. The patient had a history of alopecia totalis as a toddler that completely resolved by 4 years of age with the use of squaric acid dibutylester (SADBE). At 31 years of age, the alopecia recurred and was localized to the scalp. It was partially responsive to intralesional triamcinolone acetonide. The patient’s alopecia worsened over the 2 years following recurrence, ultimately progressing to AU. Two months after the alopecia recurrence, he developed the first psoriatic plaques. As the plaque psoriasis progressed, systemic therapy was initiated, first methotrexate and then etanercept. Shortly after developing AU, he lost his health insurance and discontinued all therapy. The patient’s psoriasis began to recur approximately 3 months after stopping etanercept. He was not using any other psoriasis medications. At that time, he noted terminal hair regrowth within some of the psoriatic plaques. No terminal hairs grew outside of the psoriatic plaques, and all regions with growth had previously been without hair for an extended period of time. The patient presented to our clinic approximately 1 year later. He had no other medical conditions and no relevant family history.
On initial physical examination, he had nonscarring hair loss involving nearly 100% of the body with psoriatic plaques on approximately 30% of the body surface area. Regions of terminal hair growth were confined to some but not all of the psoriatic plaques (Figure). Interestingly, the terminal hairs were primarily localized to the thickest central regions of the plaques. The patient’s psoriasis was treated with a combination of topical clobetasol and calcipotriene. In addition, he was started on tacrolimus ointment to the face and eyebrows for the AA. Maintenance of terminal hair within a region of topically treated psoriasis on the forearm persisted at the 2-month follow-up despite complete clearance of the corresponding psoriatic plaque. A small psoriatic plaque on the scalp cleared early with topical therapy without noticeable hair regrowth. The patient subsequently was started on contact immunotherapy with SADBE and intralesional triamcinolone acetonide for the scalp alopecia without satisfactory response. He decided to discontinue further attempts at treating the alopecia and requested to be restarted on etanercept therapy for recalcitrant psoriatic plaques. His psoriasis responded well to this therapy and he continues to be followed in our psoriasis clinic. One year after clearance of the treated psoriatic plaques, the corresponding terminal hairs persist.
Contact immunotherapy, most commonly with diphenylcyclopropenone or SADBE, is reported to have a 50% to 60% success rate in extensive AA, with a broad range of 9% to 87%17; however, randomized controlled trials testing the efficacy of contact immunotherapy are lacking. Although the mechanism of action of these topical sensitizers is not clearly delineated, it has been postulated that by inducing a new type of inflammatory response in the region, the immunologic milieu is changed, allowing the hair to grow. Some proposed mechanisms include promoting perifollicular lymphocyte apoptosis, preventing new recruitment of autoreactive lymphocytes, and allowing for the correction of aberrant major histocompatibility complex expression on the hair matrix epithelium to regain follicle immune privilege.18-20
Iatrogenic immunotherapy may work analogously to the natural immune system deviation demonstrated in our patient. Psoriasis and AA are believed to form competing immune cells and cytokine milieus, thus explaining how an individual with AA could regain normal hair growth in areas of psoriasis.15,16 The Renbök phenomenon, or reverse Köbner phenomenon, coined by Happle et al13 can be used to describe both the iatrogenic and natural cases of dermatologic disease improvement in response to secondary insults.14
A complex cascade of immune cells and cytokines coordinate AA pathogenesis. In the acute stage of AA, an inflammatory infiltrate of CD4+ T cells, CD8+ T cells, and antigen-presenting cells target anagen phase follicles, with a higher CD4+:CD8+ ratio in clinically active disease.21-23 Subcutaneous injections of either CD4+ or CD8+ lymphocyte subsets from mice with AA into normal-haired mice induces disease. However, CD8+ T cell injections rapidly produce apparent hair loss, whereas CD4+ T cells cause hair loss after several weeks, suggesting that CD8+ T cells directly modulate AA hair loss and CD4+ T cells act as an aide.24 The growth, differentiation, and survival of CD8+ T cells are stimulated by IL-2 and IFN-γ. Alopecia areata biopsies demonstrate a prevalence of TH1 cytokines, and patients with localized AA, alopecia totalis, and AU have notably higher serum IFN-γ levels compared to controls.25 In murine models, IL-1α and IL-1β increase during the catagen phase of the hair cycle and peak during the telogen phase.26 Excessive IL-1β expression is detected in the early stages of human disease, and certain IL-1β polymorphisms are associated with severe forms of AA.26 The role of tumor necrosis factor (TNF) α in AA is not well understood. In vitro studies show it inhibits hair growth, suggesting the cytokine may play a role in AA.27 However, anti–TNF-α therapy is not effective in AA, and case reports propose these therapies rarely induce AA.28-31
The TH1 response is likewise critical to psoriatic plaque development. IFN-γ and TNF-α are overexpressed in psoriatic plaques.32 IFN-γ has an antiproliferative and differentiation-inducing effect on normal keratinocytes, but psoriatic epithelial cells in vitro respond differently to the cytokine with a notably diminished growth inhibition.33,34 One explanation for the role of IFN-γ is that it stimulates dendritic cells to produce IL-1 and IL-23.35 IL-23 activates TH17 cells36; TH1 and TH17 conditions produce IL-22 whose serum level correlates with disease severity.37-39 IL-22 induces keratinocyte proliferation and migration and inhibits keratinocyte differentiation, helping account for hallmarks of the disease.40 Patients with psoriasis have increased levels of TH1, TH17, and TH22 cells, as well as their associated cytokines, in the skin and blood compared to controls.4,11,32,39,41
Alopecia areata and psoriasis are regulated by complex and still not entirely understood immune interactions. The fact that many of the same therapies are used to treat both diseases emphasizes both their overlapping characteristics and the lack of targeted therapy. It is unclear if and how the topical or systemic therapies used in our patient to treat one disease affected the natural history of the other condition. It is important to highlight, however, that the patient had not been treated for months when he developed the psoriatic plaques with hair regrowth. Other case reports also document hair regrowth in untreated plaques,13,16 making it unlikely to be a side effect of the medication regimen. For both psoriasis and AA, the immune cell composition and cytokine levels in the skin or serum vary throughout a patient’s disease course depending on severity of disease or response to treatment.6,39,42,43 Therefore, we hypothesize that the 2 conditions interact in a similarly distinct manner based on each disease’s stage and intensity in the patient. Both our patient’s course thus far and the various presentations described by other groups support this hypothesis. Our patient had a small region of psoriasis on the scalp that cleared without any terminal hair growth. He also had larger plaques on the forearms that developed hair growth most predominantly within the thicker regions of the plaques. His unique presentation highlights the fluidity of the immune factors driving psoriasis vulgaris and AA.
- Safavi K. Prevalence of alopecia areata in the First National Health and Nutrition Examination Survey. Arch Dermatol. 1992;128:702.
- Safavi KH, Muller SA, Suman VJ, et al. Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70:628-633.
- Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York, NY: McGraw-Hill; 2009.
- Austin LM, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Ghoreishi M, Martinka M, Dutz JP. Type 1 interferon signature in the scalp lesions of alopecia areata. Br J Dermatol. 2010;163:57-62.
- Rossi A, Cantisani C, Carlesimo M, et al. Serum concentrations of IL-2, IL-6, IL-12 and TNF-α in patients with alopecia areata. Int J Immunopathol Pharmacol. 2012;25:781-788.
- Freyschmidt-Paul P, McElwee KJ, Hoffmann R, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515-521.
- Reich K, Garbe C, Blaschke V, et al. Response of psoriasis to interleukin-10 is associated with suppression of cutaneous type 1 inflammation, downregulation of the epidermal interleukin-8/CXCR2 pathway and normalization of keratinocyte maturation. J Invest Dermatol. 2001;116:319-329.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648-651.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Zaba LC, Suárez-Fariñas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022-1030.e395.
- Happle R, van der Steen PHM, Perret CM. The Renbök phenomenon: an inverse Köebner reaction observed in alopecia areata. Eur J Dermatol. 1991;2:39-40.
- Ito T, Hashizume H, Takigawa M. Contact immunotherapy-induced Renbök phenomenon in a patient with alopecia areata and psoriasis vulgaris. Eur J Dermatol. 2010;20:126-127.
- Criado PR, Valente NY, Michalany NS, et al. An unusual association between scalp psoriasis and ophiasic alopecia areata: the Renbök phenomenon. Clin Exp Dermatol. 2007;32:320-321.
- Harris JE, Seykora JT, Lee RA. Renbök phenomenon and contact sensitization in a patient with alopecia universalis. Arch Dermatol. 2010;146:422-425.
- Alkhalifah A. Topical and intralesional therapies for alopecia areata. Dermatol Ther. 2011;24:355-363.
- Herbst V, Zöller M, Kissling S, et al. Diphenylcyclopropenone treatment of alopecia areata induces apoptosis of perifollicular lymphocytes. Eur J Dermatol. 2006;16:537-542.
- Zöller M, Freyschmidt-Paul P, Vitacolonna M, et al. Chronic delayed-type hypersensitivity reaction as a means to treat alopecia areata. Clin Exp Immunol. 2004;135:398-408.
- Bröcker EB, Echternacht-Happle K, Hamm H, et al. Abnormal expression of class I and class II major histocompatibility antigens in alopecia areata: modulation by topical immunotherapy. J Invest Dermatol. 1987;88:564-568.
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11:216-223.
- Perret C, Wiesner-Menzel L, Happle R. Immunohistochemical analysis of T-cell subsets in the peribulbar and intrabulbar infiltrates of alopecia areata. Acta Derm Venereol. 1984;64:26-30.
- Wiesner-Menzel L, Happle R. Intrabulbar and peribulbar accumulation of dendritic OKT 6-positive cells in alopecia areata. Arch Dermatol Res. 1984;276:333-334.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8+ cells induces localized hair loss whereas CD4+/CD25– cells promote systemic alopecia areata and CD4+/CD25+ cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- Arca E, Muşabak U, Akar A, et al. Interferon-gamma in alopecia areata. Eur J Dermatol. 2004;14:33-36.
- Hoffmann R. The potential role of cytokines and T cells in alopecia areata. J Investig Dermatol Symp Proc. 1999;4:235-238.
- Philpott MP, Sanders DA, Bowen J, et al. Effects of interleukins, colony-stimulating factor and tumour necrosis factor on human hair follicle growth in vitro: a possible role for interleukin-1 and tumour necrosis factor-alpha in alopecia areata. Br J Dermatol. 1996;135:942-948.
- Le Bidre E, Chaby G, Martin L, et al. Alopecia areata during anti-TNF alpha therapy: nine cases. Ann Dermatol Venereol. 2011;138:285-293.
- Ferran M, Calvet J, Almirall M, et al. Alopecia areata as another immune-mediated disease developed in patients treated with tumour necrosis factor-α blocker agents: report of five cases and review of the literature. J Eur Acad Dermatol Venereol. 2011;25:479-484.
- Pan Y, Rao NA. Alopecia areata during etanercept therapy. Ocul Immunol Inflamm. 2009;17:127-129.
- Pelivani N, Hassan AS, Braathen LR, et al. Alopecia areata universalis elicited during treatment with adalimumab. Dermatology. 2008;216:320-323.
- Uyemura K, Yamamura M, Fivenson DF, et al. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701-705.
- Baker BS, Powles AV, Valdimarsson H, et al. An altered response by psoriatic keratinocytes to gamma interferon. Scan J Immunol. 1988;28:735-740.
- Jackson M, Howie SE, Weller R, et al. Psoriatic keratinocytes show reduced IRF-1 and STAT-1alpha activation in response to gamma-IFN. FASEB J. 1999;13:495-502.
- Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385-422.
- McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314-324.
- Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650-657.
- Boniface K, Blumenschein WM, Brovont-Porth K, et al. Human Th17 cells comprise heterogeneous subsets including IFN-gamma-producing cells with distinct properties from the Th1 lineage. J Immunol. 2010;185:679-687.
- Kagami S, Rizzo HL, Lee JJ, et al. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130:1373-1383.
- Boniface K, Bernard FX, Garcia M, et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695-3702.
- Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129:2175-2183.
- Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5:699-711.
- Hoffmann R, Wenzel E, Huth A, et al. Cytokine mRNA levels in alopecia areata before and after treatment with the contact allergen diphenylcyclopropenone. J Invest Dermatol. 1994;103:530-533.
- Safavi K. Prevalence of alopecia areata in the First National Health and Nutrition Examination Survey. Arch Dermatol. 1992;128:702.
- Safavi KH, Muller SA, Suman VJ, et al. Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70:628-633.
- Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York, NY: McGraw-Hill; 2009.
- Austin LM, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Ghoreishi M, Martinka M, Dutz JP. Type 1 interferon signature in the scalp lesions of alopecia areata. Br J Dermatol. 2010;163:57-62.
- Rossi A, Cantisani C, Carlesimo M, et al. Serum concentrations of IL-2, IL-6, IL-12 and TNF-α in patients with alopecia areata. Int J Immunopathol Pharmacol. 2012;25:781-788.
- Freyschmidt-Paul P, McElwee KJ, Hoffmann R, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515-521.
- Reich K, Garbe C, Blaschke V, et al. Response of psoriasis to interleukin-10 is associated with suppression of cutaneous type 1 inflammation, downregulation of the epidermal interleukin-8/CXCR2 pathway and normalization of keratinocyte maturation. J Invest Dermatol. 2001;116:319-329.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648-651.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Zaba LC, Suárez-Fariñas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022-1030.e395.
- Happle R, van der Steen PHM, Perret CM. The Renbök phenomenon: an inverse Köebner reaction observed in alopecia areata. Eur J Dermatol. 1991;2:39-40.
- Ito T, Hashizume H, Takigawa M. Contact immunotherapy-induced Renbök phenomenon in a patient with alopecia areata and psoriasis vulgaris. Eur J Dermatol. 2010;20:126-127.
- Criado PR, Valente NY, Michalany NS, et al. An unusual association between scalp psoriasis and ophiasic alopecia areata: the Renbök phenomenon. Clin Exp Dermatol. 2007;32:320-321.
- Harris JE, Seykora JT, Lee RA. Renbök phenomenon and contact sensitization in a patient with alopecia universalis. Arch Dermatol. 2010;146:422-425.
- Alkhalifah A. Topical and intralesional therapies for alopecia areata. Dermatol Ther. 2011;24:355-363.
- Herbst V, Zöller M, Kissling S, et al. Diphenylcyclopropenone treatment of alopecia areata induces apoptosis of perifollicular lymphocytes. Eur J Dermatol. 2006;16:537-542.
- Zöller M, Freyschmidt-Paul P, Vitacolonna M, et al. Chronic delayed-type hypersensitivity reaction as a means to treat alopecia areata. Clin Exp Immunol. 2004;135:398-408.
- Bröcker EB, Echternacht-Happle K, Hamm H, et al. Abnormal expression of class I and class II major histocompatibility antigens in alopecia areata: modulation by topical immunotherapy. J Invest Dermatol. 1987;88:564-568.
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11:216-223.
- Perret C, Wiesner-Menzel L, Happle R. Immunohistochemical analysis of T-cell subsets in the peribulbar and intrabulbar infiltrates of alopecia areata. Acta Derm Venereol. 1984;64:26-30.
- Wiesner-Menzel L, Happle R. Intrabulbar and peribulbar accumulation of dendritic OKT 6-positive cells in alopecia areata. Arch Dermatol Res. 1984;276:333-334.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8+ cells induces localized hair loss whereas CD4+/CD25– cells promote systemic alopecia areata and CD4+/CD25+ cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- Arca E, Muşabak U, Akar A, et al. Interferon-gamma in alopecia areata. Eur J Dermatol. 2004;14:33-36.
- Hoffmann R. The potential role of cytokines and T cells in alopecia areata. J Investig Dermatol Symp Proc. 1999;4:235-238.
- Philpott MP, Sanders DA, Bowen J, et al. Effects of interleukins, colony-stimulating factor and tumour necrosis factor on human hair follicle growth in vitro: a possible role for interleukin-1 and tumour necrosis factor-alpha in alopecia areata. Br J Dermatol. 1996;135:942-948.
- Le Bidre E, Chaby G, Martin L, et al. Alopecia areata during anti-TNF alpha therapy: nine cases. Ann Dermatol Venereol. 2011;138:285-293.
- Ferran M, Calvet J, Almirall M, et al. Alopecia areata as another immune-mediated disease developed in patients treated with tumour necrosis factor-α blocker agents: report of five cases and review of the literature. J Eur Acad Dermatol Venereol. 2011;25:479-484.
- Pan Y, Rao NA. Alopecia areata during etanercept therapy. Ocul Immunol Inflamm. 2009;17:127-129.
- Pelivani N, Hassan AS, Braathen LR, et al. Alopecia areata universalis elicited during treatment with adalimumab. Dermatology. 2008;216:320-323.
- Uyemura K, Yamamura M, Fivenson DF, et al. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701-705.
- Baker BS, Powles AV, Valdimarsson H, et al. An altered response by psoriatic keratinocytes to gamma interferon. Scan J Immunol. 1988;28:735-740.
- Jackson M, Howie SE, Weller R, et al. Psoriatic keratinocytes show reduced IRF-1 and STAT-1alpha activation in response to gamma-IFN. FASEB J. 1999;13:495-502.
- Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385-422.
- McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314-324.
- Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650-657.
- Boniface K, Blumenschein WM, Brovont-Porth K, et al. Human Th17 cells comprise heterogeneous subsets including IFN-gamma-producing cells with distinct properties from the Th1 lineage. J Immunol. 2010;185:679-687.
- Kagami S, Rizzo HL, Lee JJ, et al. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130:1373-1383.
- Boniface K, Bernard FX, Garcia M, et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695-3702.
- Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129:2175-2183.
- Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5:699-711.
- Hoffmann R, Wenzel E, Huth A, et al. Cytokine mRNA levels in alopecia areata before and after treatment with the contact allergen diphenylcyclopropenone. J Invest Dermatol. 1994;103:530-533.
Practice Points
- The Renbök phenomenon, or reverse Köbner phenomenon, describes cases where secondary insults improve dermatologic disease.
- Current evidence suggests that alopecia areata (AA) is driven by a helper T cell (TH1) response whereas psoriasis vulgaris is driven by TH1, TH17, and TH22.
- Patients with concurrent AA and psoriasis can develop normal hair regrowth confined to the psoriatic plaques. Developing methods to artificially alter the cytokine milieu in affected skin may lead to new therapeutic options for each condition.
Severe Refractory Atopic Dermatitis With Elevated Serum IgE Treated With Omalizumab
To the Editor:
Atopic dermatitis (AD) is a common skin condition with an increasing prevalence, affecting up to 20% of children and 3% of adults.1,2 More than 80% of patients with AD have elevated IgE levels.3,4 IgE modulates the inflammatory response in AD in several ways including “a biphasic immediate/late phase reaction, allergen presentation by IgE-bearing Langerhans cells, allergen-induced activation of IgE-bearing macrophages, and IgE autoreactivity to human proteins.”5 Historically, most therapies have focused on mitigating the allergic symptoms caused by degranulated effector cells, such as antihistamines. However, a new class of biologically engineered medications (eg, anti-IgE [omalizumab]) aim to prevent the initiation of the allergic response.6 Variable success has been reported using omalizumab in the treatment of AD, though the majority of studies have shown improvement, especially when used in combination with conventional therapies.4,5,7-22 Omalizumab dosage is determined by body weight and pretreatment serum total IgE levels and is administered via subcutaneous injections every 2 to 4 weeks.6,7,23-26 However, the dosing tables are based on asthma therapy, in which serum IgE levels may be much lower than chronic AD,7,24 and the appropriate dosage in AD patients with markedly elevated IgE is unclear. We report an interesting case of a 57-year-old man with erythroderma from long-standing severe chronic AD that was unresponsive to conventional therapy as well as an associated serum IgE level of 17,183 IU/mL who dramatically improved when omalizumab was added to his treatment regimen.
A 57-year-old man presented with essentially 100% body surface area involvement of AD with erythroderma and pruritus. Severe AD developed at infancy and cleared at 5 years of age; childhood onset of asthma was responsive to theophylline and oral inhalers. He developed recurrent AD and asthma at 38 years of age, which was progressive and developed into severe recalcitrant erythroderma by 50 years of age. His AD was unresponsive to multiple therapies, including topical steroids, antibiotics, tacrolimus, bleach baths, antihistamines, methotrexate (15 mg weekly for 1 year, then 12.5 mg weekly for 6 months), UVB phototherapy, and psoralen plus UVA photochemotherapy. He had minimal improvement with cyclosporine (200 mg daily for 4 weeks) and mycophenolate mofetil (3 g daily), and required systemic steroids for relief. The skin was violaceous and lichenified (Figure, A). Laboratory studies were normal, except for a serum IgE level of 17,183 IU/mL (reference range, <150 IU/mL) and peripheral blood eosinophilia up to 29.8% (reference range, 1%–5%) of the differential. Skin biopsies showed AD progressing to lichen simplex chronicus. Omalizumab was added to the therapeutic regimen at a dose of 375 mg every 2 weeks, with noticeable improvement after 3 months. The patient had approximately 80% to 90% clearing with resolution of erythroderma and pruritus, and only limited residual lichenification (Figure, B). The mycophenolate was tapered slowly, and the patient experienced a mild flare at 1 g daily. He is presently on 1 g of mycophenolate daily and omalizumab (375 mg every 2 weeks) and remains remarkably improved. His IgE level decreased to 11,983 IU/mL.
Omalizumab is a monoclonal IgG1 antibody that specifically binds to the FcεRI domain of serum IgE. It blocks binding to high-affinity receptors on effector cells, primarily mast cells, basophils, macrophages, and dendritic cells; it also decreases free IgE serum levels and downregulates the IgE receptor.4,6-10,23-25,27,28 Currently, omalizumab is US Food and Drug Administration approved for moderate to severe persistent asthma in patients 6 years or older with a positive aeroallergen skin test and IgE levels up to 700 IU/mL.6,7,23-25,27,28
However, scattered case reports and small case series have described variable success in the treatment of severe AD that is unresponsive to conventional therapy in patients with markedly elevated serum IgE levels.4,5,7-22 The majority of patients (approximately 80% of published cases yielded by a PubMed search of articles indexed for MEDLINE using the search terms omalizumab and atopic dermatitis) showed improvement when measured by clinical severity scores and quality of life improvement, especially when used in conjunction with conventional therapy. Possible reasons for reported treatment failure include insufficient dosage, lack of established treatment guidelines for markedly elevated serum IgE levels, severity of disease, or variable response with failure to lower IgE level below a required threshold.7,9,23,24,27
Krathen and Hsu9 reported treatment failure with omalizumab for AD in 3 patients with serum IgE levels ranging from 5440 and 24,400 IU/mL, and one review indicated omalizumab may work best in patients with only moderately elevated serum IgE levels.21 However, Toledo et al18 reported efficacy of low-dose omalizumab for pretreatment IgE levels up to 30,000 IU/mL in 6 of 11 reported cases. The pretreatment serum IgE level is not predictive of response, and lowering the serum IgE level without normalization can be efficacious,12,23 as in the current case. Serum IgE levels are not used for monitoring therapeutic response or calculating future dosing, given potential increases in serum IgE levels during and after therapy (for up to 12 months) secondary to the formation of anti-IgE:IgE complexes.6,28 Omalizumab appears most effective when used in combination with conventional therapies. Hopefully ongoing studies will further elucidate the role of omalizumab in recalcitrant AD with elevated serum IgE levels.
- Schultz-Larsen F, Diepgen T, Svennson A. The occurrence of atopic dermatitis in north Europe: an international questionnaire study. J Am Acad Dermatol. 1996;34:760-764.
- Laughter D, Istvan JA, Tofte SJ, et al. The presence of atopic dermatitis in Oregon schoolchildren. J Am Acad Dermatol. 2000;43:649-655.
- Jones HE, Inouye JC, McGerity JL, et al. Atopic disease and serum immunoglobulin-E. Br J Dermatol. 1975;92:17-25.
- Abramovits W. A clinician’s paradigm in the treatment of atopic dermatitis. J Am Acad Dermatol. 2005;53(1, suppl 1):570-577.
- Leung D, Soter N. Cellular and immunologic mechanisms in atopic dermatitis. J Am Acad Dermatol. 2001;44(suppl):S1-S12.
- US Food and Drug Administration. Briefing document on safety. Omalizumab (Xolair) (recombinant humanized monoclonal antibody to IgE) for treatment of allergic asthma. http://www.fda.gov/ohrms/dockets/ac/03/briefing/3952B1_02_FDA-Xolair-Safety.pdf. Published April 18, 2003. Accessed June 23, 2014.
- Lane JE, Cheyney JM, Lane TN, et al. Treatment of recalcitrant atopic dermatitis with omalizumab. J Am Acad Dermatol. 2006;54:68-72.
- Caruso C, Gaeta F, Valluzzi RL, et al. Omalizumab efficacy in a girl with atopic eczema. Allergy. 2010;65:278-279.
- Krathen RA, Hsu S. Failure of omalizumab for the treatment of severe atopic dermatitis. J Am Dermatol. 2005;53:338-340.
- Fernández-Antón Martínez MC, Leis-Dosil V, Alfageme-Roldán F, et al. Omalizumab for the treatment of Atopic Dermatitis. Actas Dermosifiliogr. 2012;103:624-628.
- Pelaia G, Gallelli L, Romeo P, et al. Omalizumab decreases exacerbation frequency, oral intake of corticosteroids and peripheral blood eosinophils in atopic patients with uncontrolled asthma. Int J Clin Pharmacol Ther. 2011;49:713-721.
- Belloni B, Ziai M, Lim A, et al. Low-dose anti-IgE therapy in patients with atopic eczema with high serum IgE levels. J Allergy Clin Immunol. 2007;120:1223-1225.
- Park SY, Choi MR, Na JI, et al. Recalcitrant atopic dermatitis treated with omalizumab. Ann Dermatol. 2010;22:349-352.
- Velling P, Skowasch D, Pabst S. Improvement of quality of life in patients with concomitant allergic asthma and atopic dermatitis: one year follow-up of omalizumab therapy. Eur J Med Res. 2011;15:407-410.
- Amrol D. Anti-immunoglobulin E in the treatment of refractory atopic dermatitis. South Med J. 2010;103:554-558.
- Heil PM, Maurer D, Klein B, et al. Omalizumab therapy in atopic dermatitis: depletion of IgE does not improve the clinical course-a randomized, placebo-controlled and double blind study. J Dtsch Dermatol Ges. 2010;8:990-998.
- Ramírez del Pozo ME, Contreras Contreras E, López Tiro J, et al. Omalizumab (anti-IgE antibody) in the treatment of severe atopic dermatitis. J Investig Allergol Clin Immunol. 2011;21:416-417.
- Toledo F, Silvestre JF, Muñoz C. Combined therapy with low-dose omalizumab and intravenous immunoglobulin for severe atopic dermatitis: report of four cases. J Eur Acad Dermatol Venereol. 2012;26:1325-1327.
- Sheinkopf LE, Rafi AW, Katz RM. Efficacy of omalizumab in the treatment of atopic dermatitis: a pilot study. Allergy Asthma Proc. 2008;29:530-537.
- Incorvia C, Pravettoni C, Mauro M, et al. Effectiveness of omalizumab in a patient with severe asthma and atopic dermatitis. Monaldi Arch Chest Dis. 2008:69:78-80.
- Schmitt J, Schäkel K. Omalizumab as a therapeutic option in atopic eczema. Current evidence and potential benefit [in German]. Hautarzt. 2007;58:130-132.
- Thaiwat S, Sangasapaviliya A. Omalizumab treatment in severe atopic dermatitis. Asian Pac J Allergy Immunol. 2011;29:357-360.
- Kopp MV. Omalizumab: anti-IgE therapy in allergy. Curr Allergy Asthma Rep. 2011;11:101-106.
- Vichyanond P. Omalizumab in allergic diseases, a recent review. Asian Pc J Allergy Immunol. 2011;29:209-219.
- Scheinfeld N. Omalizumab. A recombinant humanized monoclonal IgE-blocking antibody. Dermatol Online J. 2005;11:2.
- Lowe PJ, Georgiou P, Canvin J. Revision of omalizumab dosing table for dosing every 4 instead of 2 weeks for specific ranges of bodyweight and baseline IgE [published online ahead of print December 8, 2014]. Regul Toxicol Pharmacol. 2015;71:68-77.
- Vigo PG, Girgis KR, Pfuetze BL, et al. Efficacy of anti-IgE therapy in patients with atopic dermatitis. J Am Acad Dermatol. 2006;55:168-170.
- Hanifin J, Chan S. Biochemical and immunologic mechanisms in atopic dermatitis: new targets for emerging therapies. J Am Acad Dermatol. 1999;41:72-77.
To the Editor:
Atopic dermatitis (AD) is a common skin condition with an increasing prevalence, affecting up to 20% of children and 3% of adults.1,2 More than 80% of patients with AD have elevated IgE levels.3,4 IgE modulates the inflammatory response in AD in several ways including “a biphasic immediate/late phase reaction, allergen presentation by IgE-bearing Langerhans cells, allergen-induced activation of IgE-bearing macrophages, and IgE autoreactivity to human proteins.”5 Historically, most therapies have focused on mitigating the allergic symptoms caused by degranulated effector cells, such as antihistamines. However, a new class of biologically engineered medications (eg, anti-IgE [omalizumab]) aim to prevent the initiation of the allergic response.6 Variable success has been reported using omalizumab in the treatment of AD, though the majority of studies have shown improvement, especially when used in combination with conventional therapies.4,5,7-22 Omalizumab dosage is determined by body weight and pretreatment serum total IgE levels and is administered via subcutaneous injections every 2 to 4 weeks.6,7,23-26 However, the dosing tables are based on asthma therapy, in which serum IgE levels may be much lower than chronic AD,7,24 and the appropriate dosage in AD patients with markedly elevated IgE is unclear. We report an interesting case of a 57-year-old man with erythroderma from long-standing severe chronic AD that was unresponsive to conventional therapy as well as an associated serum IgE level of 17,183 IU/mL who dramatically improved when omalizumab was added to his treatment regimen.
A 57-year-old man presented with essentially 100% body surface area involvement of AD with erythroderma and pruritus. Severe AD developed at infancy and cleared at 5 years of age; childhood onset of asthma was responsive to theophylline and oral inhalers. He developed recurrent AD and asthma at 38 years of age, which was progressive and developed into severe recalcitrant erythroderma by 50 years of age. His AD was unresponsive to multiple therapies, including topical steroids, antibiotics, tacrolimus, bleach baths, antihistamines, methotrexate (15 mg weekly for 1 year, then 12.5 mg weekly for 6 months), UVB phototherapy, and psoralen plus UVA photochemotherapy. He had minimal improvement with cyclosporine (200 mg daily for 4 weeks) and mycophenolate mofetil (3 g daily), and required systemic steroids for relief. The skin was violaceous and lichenified (Figure, A). Laboratory studies were normal, except for a serum IgE level of 17,183 IU/mL (reference range, <150 IU/mL) and peripheral blood eosinophilia up to 29.8% (reference range, 1%–5%) of the differential. Skin biopsies showed AD progressing to lichen simplex chronicus. Omalizumab was added to the therapeutic regimen at a dose of 375 mg every 2 weeks, with noticeable improvement after 3 months. The patient had approximately 80% to 90% clearing with resolution of erythroderma and pruritus, and only limited residual lichenification (Figure, B). The mycophenolate was tapered slowly, and the patient experienced a mild flare at 1 g daily. He is presently on 1 g of mycophenolate daily and omalizumab (375 mg every 2 weeks) and remains remarkably improved. His IgE level decreased to 11,983 IU/mL.
Omalizumab is a monoclonal IgG1 antibody that specifically binds to the FcεRI domain of serum IgE. It blocks binding to high-affinity receptors on effector cells, primarily mast cells, basophils, macrophages, and dendritic cells; it also decreases free IgE serum levels and downregulates the IgE receptor.4,6-10,23-25,27,28 Currently, omalizumab is US Food and Drug Administration approved for moderate to severe persistent asthma in patients 6 years or older with a positive aeroallergen skin test and IgE levels up to 700 IU/mL.6,7,23-25,27,28
However, scattered case reports and small case series have described variable success in the treatment of severe AD that is unresponsive to conventional therapy in patients with markedly elevated serum IgE levels.4,5,7-22 The majority of patients (approximately 80% of published cases yielded by a PubMed search of articles indexed for MEDLINE using the search terms omalizumab and atopic dermatitis) showed improvement when measured by clinical severity scores and quality of life improvement, especially when used in conjunction with conventional therapy. Possible reasons for reported treatment failure include insufficient dosage, lack of established treatment guidelines for markedly elevated serum IgE levels, severity of disease, or variable response with failure to lower IgE level below a required threshold.7,9,23,24,27
Krathen and Hsu9 reported treatment failure with omalizumab for AD in 3 patients with serum IgE levels ranging from 5440 and 24,400 IU/mL, and one review indicated omalizumab may work best in patients with only moderately elevated serum IgE levels.21 However, Toledo et al18 reported efficacy of low-dose omalizumab for pretreatment IgE levels up to 30,000 IU/mL in 6 of 11 reported cases. The pretreatment serum IgE level is not predictive of response, and lowering the serum IgE level without normalization can be efficacious,12,23 as in the current case. Serum IgE levels are not used for monitoring therapeutic response or calculating future dosing, given potential increases in serum IgE levels during and after therapy (for up to 12 months) secondary to the formation of anti-IgE:IgE complexes.6,28 Omalizumab appears most effective when used in combination with conventional therapies. Hopefully ongoing studies will further elucidate the role of omalizumab in recalcitrant AD with elevated serum IgE levels.
To the Editor:
Atopic dermatitis (AD) is a common skin condition with an increasing prevalence, affecting up to 20% of children and 3% of adults.1,2 More than 80% of patients with AD have elevated IgE levels.3,4 IgE modulates the inflammatory response in AD in several ways including “a biphasic immediate/late phase reaction, allergen presentation by IgE-bearing Langerhans cells, allergen-induced activation of IgE-bearing macrophages, and IgE autoreactivity to human proteins.”5 Historically, most therapies have focused on mitigating the allergic symptoms caused by degranulated effector cells, such as antihistamines. However, a new class of biologically engineered medications (eg, anti-IgE [omalizumab]) aim to prevent the initiation of the allergic response.6 Variable success has been reported using omalizumab in the treatment of AD, though the majority of studies have shown improvement, especially when used in combination with conventional therapies.4,5,7-22 Omalizumab dosage is determined by body weight and pretreatment serum total IgE levels and is administered via subcutaneous injections every 2 to 4 weeks.6,7,23-26 However, the dosing tables are based on asthma therapy, in which serum IgE levels may be much lower than chronic AD,7,24 and the appropriate dosage in AD patients with markedly elevated IgE is unclear. We report an interesting case of a 57-year-old man with erythroderma from long-standing severe chronic AD that was unresponsive to conventional therapy as well as an associated serum IgE level of 17,183 IU/mL who dramatically improved when omalizumab was added to his treatment regimen.
A 57-year-old man presented with essentially 100% body surface area involvement of AD with erythroderma and pruritus. Severe AD developed at infancy and cleared at 5 years of age; childhood onset of asthma was responsive to theophylline and oral inhalers. He developed recurrent AD and asthma at 38 years of age, which was progressive and developed into severe recalcitrant erythroderma by 50 years of age. His AD was unresponsive to multiple therapies, including topical steroids, antibiotics, tacrolimus, bleach baths, antihistamines, methotrexate (15 mg weekly for 1 year, then 12.5 mg weekly for 6 months), UVB phototherapy, and psoralen plus UVA photochemotherapy. He had minimal improvement with cyclosporine (200 mg daily for 4 weeks) and mycophenolate mofetil (3 g daily), and required systemic steroids for relief. The skin was violaceous and lichenified (Figure, A). Laboratory studies were normal, except for a serum IgE level of 17,183 IU/mL (reference range, <150 IU/mL) and peripheral blood eosinophilia up to 29.8% (reference range, 1%–5%) of the differential. Skin biopsies showed AD progressing to lichen simplex chronicus. Omalizumab was added to the therapeutic regimen at a dose of 375 mg every 2 weeks, with noticeable improvement after 3 months. The patient had approximately 80% to 90% clearing with resolution of erythroderma and pruritus, and only limited residual lichenification (Figure, B). The mycophenolate was tapered slowly, and the patient experienced a mild flare at 1 g daily. He is presently on 1 g of mycophenolate daily and omalizumab (375 mg every 2 weeks) and remains remarkably improved. His IgE level decreased to 11,983 IU/mL.
Omalizumab is a monoclonal IgG1 antibody that specifically binds to the FcεRI domain of serum IgE. It blocks binding to high-affinity receptors on effector cells, primarily mast cells, basophils, macrophages, and dendritic cells; it also decreases free IgE serum levels and downregulates the IgE receptor.4,6-10,23-25,27,28 Currently, omalizumab is US Food and Drug Administration approved for moderate to severe persistent asthma in patients 6 years or older with a positive aeroallergen skin test and IgE levels up to 700 IU/mL.6,7,23-25,27,28
However, scattered case reports and small case series have described variable success in the treatment of severe AD that is unresponsive to conventional therapy in patients with markedly elevated serum IgE levels.4,5,7-22 The majority of patients (approximately 80% of published cases yielded by a PubMed search of articles indexed for MEDLINE using the search terms omalizumab and atopic dermatitis) showed improvement when measured by clinical severity scores and quality of life improvement, especially when used in conjunction with conventional therapy. Possible reasons for reported treatment failure include insufficient dosage, lack of established treatment guidelines for markedly elevated serum IgE levels, severity of disease, or variable response with failure to lower IgE level below a required threshold.7,9,23,24,27
Krathen and Hsu9 reported treatment failure with omalizumab for AD in 3 patients with serum IgE levels ranging from 5440 and 24,400 IU/mL, and one review indicated omalizumab may work best in patients with only moderately elevated serum IgE levels.21 However, Toledo et al18 reported efficacy of low-dose omalizumab for pretreatment IgE levels up to 30,000 IU/mL in 6 of 11 reported cases. The pretreatment serum IgE level is not predictive of response, and lowering the serum IgE level without normalization can be efficacious,12,23 as in the current case. Serum IgE levels are not used for monitoring therapeutic response or calculating future dosing, given potential increases in serum IgE levels during and after therapy (for up to 12 months) secondary to the formation of anti-IgE:IgE complexes.6,28 Omalizumab appears most effective when used in combination with conventional therapies. Hopefully ongoing studies will further elucidate the role of omalizumab in recalcitrant AD with elevated serum IgE levels.
- Schultz-Larsen F, Diepgen T, Svennson A. The occurrence of atopic dermatitis in north Europe: an international questionnaire study. J Am Acad Dermatol. 1996;34:760-764.
- Laughter D, Istvan JA, Tofte SJ, et al. The presence of atopic dermatitis in Oregon schoolchildren. J Am Acad Dermatol. 2000;43:649-655.
- Jones HE, Inouye JC, McGerity JL, et al. Atopic disease and serum immunoglobulin-E. Br J Dermatol. 1975;92:17-25.
- Abramovits W. A clinician’s paradigm in the treatment of atopic dermatitis. J Am Acad Dermatol. 2005;53(1, suppl 1):570-577.
- Leung D, Soter N. Cellular and immunologic mechanisms in atopic dermatitis. J Am Acad Dermatol. 2001;44(suppl):S1-S12.
- US Food and Drug Administration. Briefing document on safety. Omalizumab (Xolair) (recombinant humanized monoclonal antibody to IgE) for treatment of allergic asthma. http://www.fda.gov/ohrms/dockets/ac/03/briefing/3952B1_02_FDA-Xolair-Safety.pdf. Published April 18, 2003. Accessed June 23, 2014.
- Lane JE, Cheyney JM, Lane TN, et al. Treatment of recalcitrant atopic dermatitis with omalizumab. J Am Acad Dermatol. 2006;54:68-72.
- Caruso C, Gaeta F, Valluzzi RL, et al. Omalizumab efficacy in a girl with atopic eczema. Allergy. 2010;65:278-279.
- Krathen RA, Hsu S. Failure of omalizumab for the treatment of severe atopic dermatitis. J Am Dermatol. 2005;53:338-340.
- Fernández-Antón Martínez MC, Leis-Dosil V, Alfageme-Roldán F, et al. Omalizumab for the treatment of Atopic Dermatitis. Actas Dermosifiliogr. 2012;103:624-628.
- Pelaia G, Gallelli L, Romeo P, et al. Omalizumab decreases exacerbation frequency, oral intake of corticosteroids and peripheral blood eosinophils in atopic patients with uncontrolled asthma. Int J Clin Pharmacol Ther. 2011;49:713-721.
- Belloni B, Ziai M, Lim A, et al. Low-dose anti-IgE therapy in patients with atopic eczema with high serum IgE levels. J Allergy Clin Immunol. 2007;120:1223-1225.
- Park SY, Choi MR, Na JI, et al. Recalcitrant atopic dermatitis treated with omalizumab. Ann Dermatol. 2010;22:349-352.
- Velling P, Skowasch D, Pabst S. Improvement of quality of life in patients with concomitant allergic asthma and atopic dermatitis: one year follow-up of omalizumab therapy. Eur J Med Res. 2011;15:407-410.
- Amrol D. Anti-immunoglobulin E in the treatment of refractory atopic dermatitis. South Med J. 2010;103:554-558.
- Heil PM, Maurer D, Klein B, et al. Omalizumab therapy in atopic dermatitis: depletion of IgE does not improve the clinical course-a randomized, placebo-controlled and double blind study. J Dtsch Dermatol Ges. 2010;8:990-998.
- Ramírez del Pozo ME, Contreras Contreras E, López Tiro J, et al. Omalizumab (anti-IgE antibody) in the treatment of severe atopic dermatitis. J Investig Allergol Clin Immunol. 2011;21:416-417.
- Toledo F, Silvestre JF, Muñoz C. Combined therapy with low-dose omalizumab and intravenous immunoglobulin for severe atopic dermatitis: report of four cases. J Eur Acad Dermatol Venereol. 2012;26:1325-1327.
- Sheinkopf LE, Rafi AW, Katz RM. Efficacy of omalizumab in the treatment of atopic dermatitis: a pilot study. Allergy Asthma Proc. 2008;29:530-537.
- Incorvia C, Pravettoni C, Mauro M, et al. Effectiveness of omalizumab in a patient with severe asthma and atopic dermatitis. Monaldi Arch Chest Dis. 2008:69:78-80.
- Schmitt J, Schäkel K. Omalizumab as a therapeutic option in atopic eczema. Current evidence and potential benefit [in German]. Hautarzt. 2007;58:130-132.
- Thaiwat S, Sangasapaviliya A. Omalizumab treatment in severe atopic dermatitis. Asian Pac J Allergy Immunol. 2011;29:357-360.
- Kopp MV. Omalizumab: anti-IgE therapy in allergy. Curr Allergy Asthma Rep. 2011;11:101-106.
- Vichyanond P. Omalizumab in allergic diseases, a recent review. Asian Pc J Allergy Immunol. 2011;29:209-219.
- Scheinfeld N. Omalizumab. A recombinant humanized monoclonal IgE-blocking antibody. Dermatol Online J. 2005;11:2.
- Lowe PJ, Georgiou P, Canvin J. Revision of omalizumab dosing table for dosing every 4 instead of 2 weeks for specific ranges of bodyweight and baseline IgE [published online ahead of print December 8, 2014]. Regul Toxicol Pharmacol. 2015;71:68-77.
- Vigo PG, Girgis KR, Pfuetze BL, et al. Efficacy of anti-IgE therapy in patients with atopic dermatitis. J Am Acad Dermatol. 2006;55:168-170.
- Hanifin J, Chan S. Biochemical and immunologic mechanisms in atopic dermatitis: new targets for emerging therapies. J Am Acad Dermatol. 1999;41:72-77.
- Schultz-Larsen F, Diepgen T, Svennson A. The occurrence of atopic dermatitis in north Europe: an international questionnaire study. J Am Acad Dermatol. 1996;34:760-764.
- Laughter D, Istvan JA, Tofte SJ, et al. The presence of atopic dermatitis in Oregon schoolchildren. J Am Acad Dermatol. 2000;43:649-655.
- Jones HE, Inouye JC, McGerity JL, et al. Atopic disease and serum immunoglobulin-E. Br J Dermatol. 1975;92:17-25.
- Abramovits W. A clinician’s paradigm in the treatment of atopic dermatitis. J Am Acad Dermatol. 2005;53(1, suppl 1):570-577.
- Leung D, Soter N. Cellular and immunologic mechanisms in atopic dermatitis. J Am Acad Dermatol. 2001;44(suppl):S1-S12.
- US Food and Drug Administration. Briefing document on safety. Omalizumab (Xolair) (recombinant humanized monoclonal antibody to IgE) for treatment of allergic asthma. http://www.fda.gov/ohrms/dockets/ac/03/briefing/3952B1_02_FDA-Xolair-Safety.pdf. Published April 18, 2003. Accessed June 23, 2014.
- Lane JE, Cheyney JM, Lane TN, et al. Treatment of recalcitrant atopic dermatitis with omalizumab. J Am Acad Dermatol. 2006;54:68-72.
- Caruso C, Gaeta F, Valluzzi RL, et al. Omalizumab efficacy in a girl with atopic eczema. Allergy. 2010;65:278-279.
- Krathen RA, Hsu S. Failure of omalizumab for the treatment of severe atopic dermatitis. J Am Dermatol. 2005;53:338-340.
- Fernández-Antón Martínez MC, Leis-Dosil V, Alfageme-Roldán F, et al. Omalizumab for the treatment of Atopic Dermatitis. Actas Dermosifiliogr. 2012;103:624-628.
- Pelaia G, Gallelli L, Romeo P, et al. Omalizumab decreases exacerbation frequency, oral intake of corticosteroids and peripheral blood eosinophils in atopic patients with uncontrolled asthma. Int J Clin Pharmacol Ther. 2011;49:713-721.
- Belloni B, Ziai M, Lim A, et al. Low-dose anti-IgE therapy in patients with atopic eczema with high serum IgE levels. J Allergy Clin Immunol. 2007;120:1223-1225.
- Park SY, Choi MR, Na JI, et al. Recalcitrant atopic dermatitis treated with omalizumab. Ann Dermatol. 2010;22:349-352.
- Velling P, Skowasch D, Pabst S. Improvement of quality of life in patients with concomitant allergic asthma and atopic dermatitis: one year follow-up of omalizumab therapy. Eur J Med Res. 2011;15:407-410.
- Amrol D. Anti-immunoglobulin E in the treatment of refractory atopic dermatitis. South Med J. 2010;103:554-558.
- Heil PM, Maurer D, Klein B, et al. Omalizumab therapy in atopic dermatitis: depletion of IgE does not improve the clinical course-a randomized, placebo-controlled and double blind study. J Dtsch Dermatol Ges. 2010;8:990-998.
- Ramírez del Pozo ME, Contreras Contreras E, López Tiro J, et al. Omalizumab (anti-IgE antibody) in the treatment of severe atopic dermatitis. J Investig Allergol Clin Immunol. 2011;21:416-417.
- Toledo F, Silvestre JF, Muñoz C. Combined therapy with low-dose omalizumab and intravenous immunoglobulin for severe atopic dermatitis: report of four cases. J Eur Acad Dermatol Venereol. 2012;26:1325-1327.
- Sheinkopf LE, Rafi AW, Katz RM. Efficacy of omalizumab in the treatment of atopic dermatitis: a pilot study. Allergy Asthma Proc. 2008;29:530-537.
- Incorvia C, Pravettoni C, Mauro M, et al. Effectiveness of omalizumab in a patient with severe asthma and atopic dermatitis. Monaldi Arch Chest Dis. 2008:69:78-80.
- Schmitt J, Schäkel K. Omalizumab as a therapeutic option in atopic eczema. Current evidence and potential benefit [in German]. Hautarzt. 2007;58:130-132.
- Thaiwat S, Sangasapaviliya A. Omalizumab treatment in severe atopic dermatitis. Asian Pac J Allergy Immunol. 2011;29:357-360.
- Kopp MV. Omalizumab: anti-IgE therapy in allergy. Curr Allergy Asthma Rep. 2011;11:101-106.
- Vichyanond P. Omalizumab in allergic diseases, a recent review. Asian Pc J Allergy Immunol. 2011;29:209-219.
- Scheinfeld N. Omalizumab. A recombinant humanized monoclonal IgE-blocking antibody. Dermatol Online J. 2005;11:2.
- Lowe PJ, Georgiou P, Canvin J. Revision of omalizumab dosing table for dosing every 4 instead of 2 weeks for specific ranges of bodyweight and baseline IgE [published online ahead of print December 8, 2014]. Regul Toxicol Pharmacol. 2015;71:68-77.
- Vigo PG, Girgis KR, Pfuetze BL, et al. Efficacy of anti-IgE therapy in patients with atopic dermatitis. J Am Acad Dermatol. 2006;55:168-170.
- Hanifin J, Chan S. Biochemical and immunologic mechanisms in atopic dermatitis: new targets for emerging therapies. J Am Acad Dermatol. 1999;41:72-77.
Practice Points
- Omalizumab can be effective in treating patients with severe recalcitrant atopic dermatitis with markedly elevated serum IgE.
- Omalizumab appears most effective when used in combination with conventional therapies.