User login
5-year-old boy • calf pain • fever • cough & rhinitis • Dx?
THE CASE
A 5-year-old previously healthy white boy presented to clinic with bilateral calf pain and refusal to bear weight since awakening that morning. Associated symptoms included a 3-day history of generalized fatigue, subjective fevers, cough, congestion, and rhinitis. The night prior to presentation, he showed no symptoms of gait abnormalities, muscle pain, or weakness. There was no history of similar symptoms, trauma, overexertion, foreign travel, or family history of musculoskeletal disease. He was fully immunized, except for the annual influenza vaccine. He was not taking any medications. This case occurred before the onset of the COVID-19 pandemic.
Objective findings included fever of 101 °F, refusal to bear weight, and symmetrical bilateral tenderness to palpation of the gastrocnemius-soleus complex. Pain was elicited with passive dorsiflexion. There was no erythema, edema, or sensory deficits, and the distal leg compartments were soft. There was normal range of motion of the hips, knees, and ankles. Dorsalis pedis pulses were 2+, and patella reflexes were 2/4 bilaterally.
Lab results included a white blood cell count of 2500/μL (normal range, 4500 to 11,000/μL);absolute neutrophil count, 900/μL (1500 to 8000/μL); platelet count, 131,000/μL (150,000 to 450,000/μL); creatine kinase level, 869 IU/L (22 to 198 U/L); and aspartate aminotransferase level, 116 U/L (8 to 33 U/L). A rapid influenza swab was positive for influenza B. Plain films of the bilateral hips and lower extremities were unremarkable. C-reactive protein (CRP) level, urinalysis, and renal function tests were within normal limits. Creatine kinase (CK) level peaked (1935 U/L; normal range, 22 to 198 U/L) within the first 24 hours of presentation and then trended down.
The Diagnosis
The patient’s sudden onset of symmetrical bilateral calf pain in the setting of an upper respiratory tract infection was extremely suspicious for benign acute childhood myositis (BACM). Lab work and radiologic evaluation were performed to rule out more ominous causes of refusal to bear weight. The suspicion of BACM was further validated by influenza B serology, an elevated CK, and a normal CRP.
Discussion
BACM was first described by Lundberg in 1957.1 The overall incidence and prevalence are unclear.2 A viral prodrome involving rhinorrhea, low-grade fever, sore throat, cough, and malaise typically precedes bilateral calf pain by 3 days.2-4 Myositis symptoms typically last for 4 days.3 While several infectious etiologies have been linked to this condition, influenza B has the greatest association.5,6
❚ Patient population. BACM occurs predominately in school-aged children (6-8 years old) and has a male-to-female ratio of 2:1.3,5,6 In a retrospective study of 219 children, BACM was strongly associated with male gender and ages 6 to 9 years.3 In another retrospective study of 54 children,80% of patients were male, and the mean age was 7.3 years.5
❚ Key symptoms and differential. The distinguishing feature of BACM is bilateral symmetric gastrocnemius-soleus tenderness.2,4 Additionally, the lack of neurologic symptoms is an important differentiator, as long as refusal to bear weight is not mistaken for weakness.6 These features help to distinguish BACM from other items in the differential, including trauma, Guillain-Barre syndrome, osteomyelitis, malignancy, deep vein thrombosis, and inherited musculoskeletal disorders.2
Continue to: Labratory evaluation...
❚ Laboratory evaluation will often show mild neutropenia, thrombocytopenia, and mild elevation in CK.7,8 CRP is typically normal.4,7,9 In a retrospective study of 28 admissions for BACM from 2001 to 2012, common findings included leukopenia (35%), neutropenia (25%), and thrombocytopenia (21%). The median CK value was 4181 U/L.4 In another analysis of BACM cases, mean CK was 1872 U/L.5
❚ Biopsy is unnecessary; however, calf muscle samples from 11 of 12 children with suspected BACM due to influenza B infection were consistent with patchy necrosis without significant myositis.10
❚ Complications. Rhabdomyolysis, although rare, has been reported with BACM. In 1 analysis, 10 of 316 patients with influenza-associated myositis developed rhabdomyolysis; 8 experienced renal failure. Rhabdomyolysis was 4 times more likely to occur in girls, and 86% of cases were associated with influenza A.6 Common manifestations of rhabdomyolysis associated with influenza include diffuse myopathy, gross hematuria, and myoglobinuria.6
❚ Treatment is mainly supportive.4,8,9 Antivirals typically are not indicated, as the bilateral calf pain manifests during the recovery phase of the illness.4,9,11 BACM is self-limited and should resolve within 3 days of myositis manifestation.2 Patients should follow up in 2 to 3 weeks to verify symptom resolution.2
If muscle pain, swelling, and tenderness worsen, further work-up is indicated. In more severe cases, including those involving renal failure, intensive care management and even dialysis may be necessary.4,6
❚ Our patient was hospitalized due to fever in the setting of neutropenia. Ultimately, he was treated with acetaminophen and intravenous fluids for mild dehydration and elevated CK levels. He was discharged home after 3 days, at which time he had complete resolution of pain and was able to resume normal activities.
The Takeaway
Benign acute childhood myositis is a self-limited disorder with an excellent prognosis. It has a typical presentation and therefore should be a clinical diagnosis; however, investigative studies may be warranted to rule out more ominous causes. Reassurance to family that the condition should self-resolve in a few days is important. Close follow-up should be scheduled to ensure resolution of symptoms.
CORRESPONDENCE
Nicholas A. Rathjen, DO, William Beaumont Army Medical Center, Department of Soldier and Family Care, 11335 SSG Sims Street, Fort Bliss, TX 79918; nicholas.a.rathjen@gmail. com
- Lundberg A. Myalgia cruris epidemica. Acta Paediatr. 1957;46:18-31. doi: 10.1111/j.1651-2227.1957.tb08627.x
- Magee H, Goldman RD. Viral myositis in children. Can Fam Physician. 2017;63:365-368.
- Mall S, Buchholz U, Tibussek D, et al. A large outbreak of influenza B-associated benign acute childhood myositis in Germany, 2007/2008. Pediatr Infect Dis J. 2011;30:e142-e146. doi: 10.1097/INF.0b013e318217e356
- Santos JA, Albuquerque C, Lito D, et al. Benign acute childhood myositis: an alarming condition with an excellent prognosis! Am J Emerg Med. 2014;32:1418-1419. doi: 10.1016/j.ajem.2014.08.022
- Rosenberg T, Heitner S, Scolnik D, et al. Outcome of benign acute childhood myositis: the experience of 2 large tertiary care pediatric hospitals. Pediatr Emerg Care. 2018;34:400-402. doi: 10.1097/PEC.0000000000000830
- Agyeman P, Duppenthaler A, Heininger U, et al. Influenza-associated myositis in children. Infection. 2004;32:199-203. doi: 10.1007/s15010-004-4003-2
- Mackay MT, Kornberg AJ, Shield LK, et al. Benign acute childhood myositis: laboratory and clinical features. Neurology. 1999;53:2127-2131. doi: 10.1212/wnl.53.9.2127
- Neocleous C, Spanou C, Mpampalis E, et al. Unnecessary diagnostic investigations in benign acute childhood myositis: a case series report. Scott Med J. 2012;57:182. doi: 10.1258/smj.2012.012023
- Felipe Cavagnaro SM, Alejandra Aird G, Ingrid Harwardt R, et al. Benign acute childhood myositis: clinical series and literature review. Rev Chil Pediatr. 2017;88:268-274. doi: 10.1016/j.rchipe.2016.07.002
- Bove KE, Hilton PK, Partin J, et al. Morphology of acute myopathy associated with influenza B infection. Pediatric Pathology. 1983;1:51-66. https://doi.org/10.3109/15513818309048284
- Koliou M, Hadjiloizou S, Ourani S, et al. A case of benign acute childhood myositis associated with influenza A (HINI) virus infection. Clin Microbiol Infect. 2010;16:193-195. doi: 10.1111/j.1469-0691.2009.03064.x
THE CASE
A 5-year-old previously healthy white boy presented to clinic with bilateral calf pain and refusal to bear weight since awakening that morning. Associated symptoms included a 3-day history of generalized fatigue, subjective fevers, cough, congestion, and rhinitis. The night prior to presentation, he showed no symptoms of gait abnormalities, muscle pain, or weakness. There was no history of similar symptoms, trauma, overexertion, foreign travel, or family history of musculoskeletal disease. He was fully immunized, except for the annual influenza vaccine. He was not taking any medications. This case occurred before the onset of the COVID-19 pandemic.
Objective findings included fever of 101 °F, refusal to bear weight, and symmetrical bilateral tenderness to palpation of the gastrocnemius-soleus complex. Pain was elicited with passive dorsiflexion. There was no erythema, edema, or sensory deficits, and the distal leg compartments were soft. There was normal range of motion of the hips, knees, and ankles. Dorsalis pedis pulses were 2+, and patella reflexes were 2/4 bilaterally.
Lab results included a white blood cell count of 2500/μL (normal range, 4500 to 11,000/μL);absolute neutrophil count, 900/μL (1500 to 8000/μL); platelet count, 131,000/μL (150,000 to 450,000/μL); creatine kinase level, 869 IU/L (22 to 198 U/L); and aspartate aminotransferase level, 116 U/L (8 to 33 U/L). A rapid influenza swab was positive for influenza B. Plain films of the bilateral hips and lower extremities were unremarkable. C-reactive protein (CRP) level, urinalysis, and renal function tests were within normal limits. Creatine kinase (CK) level peaked (1935 U/L; normal range, 22 to 198 U/L) within the first 24 hours of presentation and then trended down.
The Diagnosis
The patient’s sudden onset of symmetrical bilateral calf pain in the setting of an upper respiratory tract infection was extremely suspicious for benign acute childhood myositis (BACM). Lab work and radiologic evaluation were performed to rule out more ominous causes of refusal to bear weight. The suspicion of BACM was further validated by influenza B serology, an elevated CK, and a normal CRP.
Discussion
BACM was first described by Lundberg in 1957.1 The overall incidence and prevalence are unclear.2 A viral prodrome involving rhinorrhea, low-grade fever, sore throat, cough, and malaise typically precedes bilateral calf pain by 3 days.2-4 Myositis symptoms typically last for 4 days.3 While several infectious etiologies have been linked to this condition, influenza B has the greatest association.5,6
❚ Patient population. BACM occurs predominately in school-aged children (6-8 years old) and has a male-to-female ratio of 2:1.3,5,6 In a retrospective study of 219 children, BACM was strongly associated with male gender and ages 6 to 9 years.3 In another retrospective study of 54 children,80% of patients were male, and the mean age was 7.3 years.5
❚ Key symptoms and differential. The distinguishing feature of BACM is bilateral symmetric gastrocnemius-soleus tenderness.2,4 Additionally, the lack of neurologic symptoms is an important differentiator, as long as refusal to bear weight is not mistaken for weakness.6 These features help to distinguish BACM from other items in the differential, including trauma, Guillain-Barre syndrome, osteomyelitis, malignancy, deep vein thrombosis, and inherited musculoskeletal disorders.2
Continue to: Labratory evaluation...
❚ Laboratory evaluation will often show mild neutropenia, thrombocytopenia, and mild elevation in CK.7,8 CRP is typically normal.4,7,9 In a retrospective study of 28 admissions for BACM from 2001 to 2012, common findings included leukopenia (35%), neutropenia (25%), and thrombocytopenia (21%). The median CK value was 4181 U/L.4 In another analysis of BACM cases, mean CK was 1872 U/L.5
❚ Biopsy is unnecessary; however, calf muscle samples from 11 of 12 children with suspected BACM due to influenza B infection were consistent with patchy necrosis without significant myositis.10
❚ Complications. Rhabdomyolysis, although rare, has been reported with BACM. In 1 analysis, 10 of 316 patients with influenza-associated myositis developed rhabdomyolysis; 8 experienced renal failure. Rhabdomyolysis was 4 times more likely to occur in girls, and 86% of cases were associated with influenza A.6 Common manifestations of rhabdomyolysis associated with influenza include diffuse myopathy, gross hematuria, and myoglobinuria.6
❚ Treatment is mainly supportive.4,8,9 Antivirals typically are not indicated, as the bilateral calf pain manifests during the recovery phase of the illness.4,9,11 BACM is self-limited and should resolve within 3 days of myositis manifestation.2 Patients should follow up in 2 to 3 weeks to verify symptom resolution.2
If muscle pain, swelling, and tenderness worsen, further work-up is indicated. In more severe cases, including those involving renal failure, intensive care management and even dialysis may be necessary.4,6
❚ Our patient was hospitalized due to fever in the setting of neutropenia. Ultimately, he was treated with acetaminophen and intravenous fluids for mild dehydration and elevated CK levels. He was discharged home after 3 days, at which time he had complete resolution of pain and was able to resume normal activities.
The Takeaway
Benign acute childhood myositis is a self-limited disorder with an excellent prognosis. It has a typical presentation and therefore should be a clinical diagnosis; however, investigative studies may be warranted to rule out more ominous causes. Reassurance to family that the condition should self-resolve in a few days is important. Close follow-up should be scheduled to ensure resolution of symptoms.
CORRESPONDENCE
Nicholas A. Rathjen, DO, William Beaumont Army Medical Center, Department of Soldier and Family Care, 11335 SSG Sims Street, Fort Bliss, TX 79918; nicholas.a.rathjen@gmail. com
THE CASE
A 5-year-old previously healthy white boy presented to clinic with bilateral calf pain and refusal to bear weight since awakening that morning. Associated symptoms included a 3-day history of generalized fatigue, subjective fevers, cough, congestion, and rhinitis. The night prior to presentation, he showed no symptoms of gait abnormalities, muscle pain, or weakness. There was no history of similar symptoms, trauma, overexertion, foreign travel, or family history of musculoskeletal disease. He was fully immunized, except for the annual influenza vaccine. He was not taking any medications. This case occurred before the onset of the COVID-19 pandemic.
Objective findings included fever of 101 °F, refusal to bear weight, and symmetrical bilateral tenderness to palpation of the gastrocnemius-soleus complex. Pain was elicited with passive dorsiflexion. There was no erythema, edema, or sensory deficits, and the distal leg compartments were soft. There was normal range of motion of the hips, knees, and ankles. Dorsalis pedis pulses were 2+, and patella reflexes were 2/4 bilaterally.
Lab results included a white blood cell count of 2500/μL (normal range, 4500 to 11,000/μL);absolute neutrophil count, 900/μL (1500 to 8000/μL); platelet count, 131,000/μL (150,000 to 450,000/μL); creatine kinase level, 869 IU/L (22 to 198 U/L); and aspartate aminotransferase level, 116 U/L (8 to 33 U/L). A rapid influenza swab was positive for influenza B. Plain films of the bilateral hips and lower extremities were unremarkable. C-reactive protein (CRP) level, urinalysis, and renal function tests were within normal limits. Creatine kinase (CK) level peaked (1935 U/L; normal range, 22 to 198 U/L) within the first 24 hours of presentation and then trended down.
The Diagnosis
The patient’s sudden onset of symmetrical bilateral calf pain in the setting of an upper respiratory tract infection was extremely suspicious for benign acute childhood myositis (BACM). Lab work and radiologic evaluation were performed to rule out more ominous causes of refusal to bear weight. The suspicion of BACM was further validated by influenza B serology, an elevated CK, and a normal CRP.
Discussion
BACM was first described by Lundberg in 1957.1 The overall incidence and prevalence are unclear.2 A viral prodrome involving rhinorrhea, low-grade fever, sore throat, cough, and malaise typically precedes bilateral calf pain by 3 days.2-4 Myositis symptoms typically last for 4 days.3 While several infectious etiologies have been linked to this condition, influenza B has the greatest association.5,6
❚ Patient population. BACM occurs predominately in school-aged children (6-8 years old) and has a male-to-female ratio of 2:1.3,5,6 In a retrospective study of 219 children, BACM was strongly associated with male gender and ages 6 to 9 years.3 In another retrospective study of 54 children,80% of patients were male, and the mean age was 7.3 years.5
❚ Key symptoms and differential. The distinguishing feature of BACM is bilateral symmetric gastrocnemius-soleus tenderness.2,4 Additionally, the lack of neurologic symptoms is an important differentiator, as long as refusal to bear weight is not mistaken for weakness.6 These features help to distinguish BACM from other items in the differential, including trauma, Guillain-Barre syndrome, osteomyelitis, malignancy, deep vein thrombosis, and inherited musculoskeletal disorders.2
Continue to: Labratory evaluation...
❚ Laboratory evaluation will often show mild neutropenia, thrombocytopenia, and mild elevation in CK.7,8 CRP is typically normal.4,7,9 In a retrospective study of 28 admissions for BACM from 2001 to 2012, common findings included leukopenia (35%), neutropenia (25%), and thrombocytopenia (21%). The median CK value was 4181 U/L.4 In another analysis of BACM cases, mean CK was 1872 U/L.5
❚ Biopsy is unnecessary; however, calf muscle samples from 11 of 12 children with suspected BACM due to influenza B infection were consistent with patchy necrosis without significant myositis.10
❚ Complications. Rhabdomyolysis, although rare, has been reported with BACM. In 1 analysis, 10 of 316 patients with influenza-associated myositis developed rhabdomyolysis; 8 experienced renal failure. Rhabdomyolysis was 4 times more likely to occur in girls, and 86% of cases were associated with influenza A.6 Common manifestations of rhabdomyolysis associated with influenza include diffuse myopathy, gross hematuria, and myoglobinuria.6
❚ Treatment is mainly supportive.4,8,9 Antivirals typically are not indicated, as the bilateral calf pain manifests during the recovery phase of the illness.4,9,11 BACM is self-limited and should resolve within 3 days of myositis manifestation.2 Patients should follow up in 2 to 3 weeks to verify symptom resolution.2
If muscle pain, swelling, and tenderness worsen, further work-up is indicated. In more severe cases, including those involving renal failure, intensive care management and even dialysis may be necessary.4,6
❚ Our patient was hospitalized due to fever in the setting of neutropenia. Ultimately, he was treated with acetaminophen and intravenous fluids for mild dehydration and elevated CK levels. He was discharged home after 3 days, at which time he had complete resolution of pain and was able to resume normal activities.
The Takeaway
Benign acute childhood myositis is a self-limited disorder with an excellent prognosis. It has a typical presentation and therefore should be a clinical diagnosis; however, investigative studies may be warranted to rule out more ominous causes. Reassurance to family that the condition should self-resolve in a few days is important. Close follow-up should be scheduled to ensure resolution of symptoms.
CORRESPONDENCE
Nicholas A. Rathjen, DO, William Beaumont Army Medical Center, Department of Soldier and Family Care, 11335 SSG Sims Street, Fort Bliss, TX 79918; nicholas.a.rathjen@gmail. com
- Lundberg A. Myalgia cruris epidemica. Acta Paediatr. 1957;46:18-31. doi: 10.1111/j.1651-2227.1957.tb08627.x
- Magee H, Goldman RD. Viral myositis in children. Can Fam Physician. 2017;63:365-368.
- Mall S, Buchholz U, Tibussek D, et al. A large outbreak of influenza B-associated benign acute childhood myositis in Germany, 2007/2008. Pediatr Infect Dis J. 2011;30:e142-e146. doi: 10.1097/INF.0b013e318217e356
- Santos JA, Albuquerque C, Lito D, et al. Benign acute childhood myositis: an alarming condition with an excellent prognosis! Am J Emerg Med. 2014;32:1418-1419. doi: 10.1016/j.ajem.2014.08.022
- Rosenberg T, Heitner S, Scolnik D, et al. Outcome of benign acute childhood myositis: the experience of 2 large tertiary care pediatric hospitals. Pediatr Emerg Care. 2018;34:400-402. doi: 10.1097/PEC.0000000000000830
- Agyeman P, Duppenthaler A, Heininger U, et al. Influenza-associated myositis in children. Infection. 2004;32:199-203. doi: 10.1007/s15010-004-4003-2
- Mackay MT, Kornberg AJ, Shield LK, et al. Benign acute childhood myositis: laboratory and clinical features. Neurology. 1999;53:2127-2131. doi: 10.1212/wnl.53.9.2127
- Neocleous C, Spanou C, Mpampalis E, et al. Unnecessary diagnostic investigations in benign acute childhood myositis: a case series report. Scott Med J. 2012;57:182. doi: 10.1258/smj.2012.012023
- Felipe Cavagnaro SM, Alejandra Aird G, Ingrid Harwardt R, et al. Benign acute childhood myositis: clinical series and literature review. Rev Chil Pediatr. 2017;88:268-274. doi: 10.1016/j.rchipe.2016.07.002
- Bove KE, Hilton PK, Partin J, et al. Morphology of acute myopathy associated with influenza B infection. Pediatric Pathology. 1983;1:51-66. https://doi.org/10.3109/15513818309048284
- Koliou M, Hadjiloizou S, Ourani S, et al. A case of benign acute childhood myositis associated with influenza A (HINI) virus infection. Clin Microbiol Infect. 2010;16:193-195. doi: 10.1111/j.1469-0691.2009.03064.x
- Lundberg A. Myalgia cruris epidemica. Acta Paediatr. 1957;46:18-31. doi: 10.1111/j.1651-2227.1957.tb08627.x
- Magee H, Goldman RD. Viral myositis in children. Can Fam Physician. 2017;63:365-368.
- Mall S, Buchholz U, Tibussek D, et al. A large outbreak of influenza B-associated benign acute childhood myositis in Germany, 2007/2008. Pediatr Infect Dis J. 2011;30:e142-e146. doi: 10.1097/INF.0b013e318217e356
- Santos JA, Albuquerque C, Lito D, et al. Benign acute childhood myositis: an alarming condition with an excellent prognosis! Am J Emerg Med. 2014;32:1418-1419. doi: 10.1016/j.ajem.2014.08.022
- Rosenberg T, Heitner S, Scolnik D, et al. Outcome of benign acute childhood myositis: the experience of 2 large tertiary care pediatric hospitals. Pediatr Emerg Care. 2018;34:400-402. doi: 10.1097/PEC.0000000000000830
- Agyeman P, Duppenthaler A, Heininger U, et al. Influenza-associated myositis in children. Infection. 2004;32:199-203. doi: 10.1007/s15010-004-4003-2
- Mackay MT, Kornberg AJ, Shield LK, et al. Benign acute childhood myositis: laboratory and clinical features. Neurology. 1999;53:2127-2131. doi: 10.1212/wnl.53.9.2127
- Neocleous C, Spanou C, Mpampalis E, et al. Unnecessary diagnostic investigations in benign acute childhood myositis: a case series report. Scott Med J. 2012;57:182. doi: 10.1258/smj.2012.012023
- Felipe Cavagnaro SM, Alejandra Aird G, Ingrid Harwardt R, et al. Benign acute childhood myositis: clinical series and literature review. Rev Chil Pediatr. 2017;88:268-274. doi: 10.1016/j.rchipe.2016.07.002
- Bove KE, Hilton PK, Partin J, et al. Morphology of acute myopathy associated with influenza B infection. Pediatric Pathology. 1983;1:51-66. https://doi.org/10.3109/15513818309048284
- Koliou M, Hadjiloizou S, Ourani S, et al. A case of benign acute childhood myositis associated with influenza A (HINI) virus infection. Clin Microbiol Infect. 2010;16:193-195. doi: 10.1111/j.1469-0691.2009.03064.x
Stopping Empagliflozin Unmasks Heart Failure
SGLT2 inhibitors have been shown to have a role in the management of heart failure in patients with type 2 diabetes mellitus, but there is a risk of exacerbation when discontinued.
About 40% of patients with heart failure (HF) also have type 2 diabetes mellitus (T2DM).1 Certain sodium-glucose cotransporter-2 (SGLT2) inhibitors have benefited patients with HF.2 We report a case of a patient with T2DM who had signs and symptoms of hypervolemia after discontinuing the SGLT2 inhibitor empagliflozin. The patient was found to have previously undiagnosed HF.
Case Presentation
A 58-year-old male presented for care at Malcolm Randall Veterans Affairs Medical Center in Gainseville, Florida, diabetes clinic. The patient was diagnosed with T2DM at age 32 years. At 36 years, he was started on subcutaneous insulin injections, and was switched to insulin pump therapy in his early 40s. At the time of evaluation, the T2DM was managed using an insulin pump, metformin, and acarbose. He had been prescribed empagliflozin 10 mg several months before presentation, but the medication ran out about 1 month prior to evaluation, and additional refills were unavailable.
The patient reported a 1-month history of worsening exertional shortness of breath, decreased exercise tolerance, and lower extremity swelling. Vitals signs, including respiratory rate and oxygen saturation were within normal limits. Bibasilar crackles and bilateral 2+ pitting pedal edema were noted. The remaining examination was unrevealing. His most recent glycated hemoglobin A1c level from 1 month prior to the presentation was 6.4%.
Given the patient’s shortness of breath and evidence of fluid overload on examination, brain natriuretic peptide was obtained and was significantly elevated at 5,895 pg/mL. A transthoracic echocardiogram revealed left ventricular ejection fraction < 20%. The patient was started on furosemide 40 mg, pending receipt of empagliflozin. A cardiology evaluation also was recommended.
Cardiac catheterization identified significant obstructions to the left anterior descending and left circumflex arteries. The patient underwent successful percutaneous coronary intervention to these areas. Following initiation of medications and coronary revascularization, the patient reported significant symptom improvement. At the follow-up evaluation 8 weeks later, he was symptom free, and his physical examination was consistent with euvolemia.
Discussion
T2DM has been associated with adverse cardiovascular outcomes, including atherosclerotic heart disease and HF. There are several theories about the relationship between T2DM and HF, though the exact pathophysiology of this relationship is unknown.3,4 One theory suggests diabetic cardiomyopathy as the cause. In patients with diabetic cardiomyopathy, there is early development of diastolic dysfunction, which eventually progresses to ventricular dysfunction. There is continued stimulation of the renin-angiotensin-aldosterone system that leads to death of cardiomyocytes, fibrosis, and remodeling, which worsens pump failure.5
SGLT2 inhibitors decrease hyperglycemia and hyperinsulinemia, potentially reducing HF risk. SGLT2 inhibitors decrease blood glucose levels by inhibiting SGLT2 in the proximal tubule, leading to a decrease of glucose reabsorption and an increase in excretion.6,7 The EMPA-REG OUTCOME trial looked at cardiovascular outcomes in patients with T2DM at high risk for adverse cardiac events. There was a significant risk reduction in deaths and hospitalizations for HF in patients treated with empagliflozin.8
The EMPRISE study specifically examined empagliflozin and its effects on hospitalization for HF.2 When compared with patients treated with sitagliptin, there was a statistically significant decrease in hospitalization for HF in patients with T2DM, both with and without preexisting cardiovascular disease.
This case highlights the relationship between T2DM and HF. We also show how the use of empagliflozin may have helped manage the patient’s undiagnosed HF and how its discontinuation luckily unmasked it. Routine evaluation for HF in patients with T2DM is not done, but likely there are patients who would benefit, especially given the strong, albeit less known, association between these 2 conditions.
Further studies are needed to determine the type of patients who would benefit most from HF screening. For now, the best practice is to obtain a complete medical history that includes current and recently discontinued medications as well a thorough physical examination for signs of fluid overload and cardiovascular compromise. Patients who may have signs concerning for HF can have appropriate testing and intervention.
Conclusions
SGLT2 inhibitors have been shown to have a role in the management of HF in patients with T2DM. There is a risk of exacerbation or unmasking of HF when discontinuing SGLT2 inhibitors. To our knowledge, this is the first paper describing the discovery of HF following interruption of SGLT2 inhibitor treatment. The clinician and patient should monitor for signs and symptoms of fluid overload when stopping therapy. Further research into the benefits of a more comprehensive evaluation is needed.
1. Thomas MC. Type 2 diabetes and heart failure: challenges and solutions. Curr Cardiol Rev. 2016;12(3):249-255. doi:10.2174/1573403X12666160606120254
2. Patorno E, Pawar A, Franklin J, et al. Empagliflozin and the risk of heart failure hospitalization in routine clinical care: a first analysis from the EMPRISE study. Circulation. 2019;139(25):2822-2830. doi:10.1161/CIRCULATIONAHA.118.039177
3. Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41(1):11-13. doi:10.2337/dci17-0052
4. Thrainsdottir I, Aspelund T, Thorheirsson G, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavík study. Diabetes Care. 2005;28(3):612-616. doi:10.2337/diacare.28.3.612
5. Bell D, Goncalves E. Heart failure in the patient with diabetes: epidemiology, aetiology, prognosis, therapy and the effect of glucose-lowering medications. Diabetes Obes Metab. 2019;21(6):1277-1290. doi:10.1111/dom.13652
6. Nair S, Wilding JPH. Sodium glucose cotransporter 2 Inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95(1):34-42. doi:10.1210/jc.2009-0473
7. Ali A, Bain S, Hicks D, et al; Improving Diabetes Steering Committee. SGLT2 inhibitors: cardiovascular benefits beyond HbA1c- translating evidence into practice. Diabetes Ther. 2019;10(5):1595-1622. doi:10.1007/s13300-019-0657-8
8. Zinman B, Wanner C, Lachin J, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/NEJMoa1504720
SGLT2 inhibitors have been shown to have a role in the management of heart failure in patients with type 2 diabetes mellitus, but there is a risk of exacerbation when discontinued.
SGLT2 inhibitors have been shown to have a role in the management of heart failure in patients with type 2 diabetes mellitus, but there is a risk of exacerbation when discontinued.
About 40% of patients with heart failure (HF) also have type 2 diabetes mellitus (T2DM).1 Certain sodium-glucose cotransporter-2 (SGLT2) inhibitors have benefited patients with HF.2 We report a case of a patient with T2DM who had signs and symptoms of hypervolemia after discontinuing the SGLT2 inhibitor empagliflozin. The patient was found to have previously undiagnosed HF.
Case Presentation
A 58-year-old male presented for care at Malcolm Randall Veterans Affairs Medical Center in Gainseville, Florida, diabetes clinic. The patient was diagnosed with T2DM at age 32 years. At 36 years, he was started on subcutaneous insulin injections, and was switched to insulin pump therapy in his early 40s. At the time of evaluation, the T2DM was managed using an insulin pump, metformin, and acarbose. He had been prescribed empagliflozin 10 mg several months before presentation, but the medication ran out about 1 month prior to evaluation, and additional refills were unavailable.
The patient reported a 1-month history of worsening exertional shortness of breath, decreased exercise tolerance, and lower extremity swelling. Vitals signs, including respiratory rate and oxygen saturation were within normal limits. Bibasilar crackles and bilateral 2+ pitting pedal edema were noted. The remaining examination was unrevealing. His most recent glycated hemoglobin A1c level from 1 month prior to the presentation was 6.4%.
Given the patient’s shortness of breath and evidence of fluid overload on examination, brain natriuretic peptide was obtained and was significantly elevated at 5,895 pg/mL. A transthoracic echocardiogram revealed left ventricular ejection fraction < 20%. The patient was started on furosemide 40 mg, pending receipt of empagliflozin. A cardiology evaluation also was recommended.
Cardiac catheterization identified significant obstructions to the left anterior descending and left circumflex arteries. The patient underwent successful percutaneous coronary intervention to these areas. Following initiation of medications and coronary revascularization, the patient reported significant symptom improvement. At the follow-up evaluation 8 weeks later, he was symptom free, and his physical examination was consistent with euvolemia.
Discussion
T2DM has been associated with adverse cardiovascular outcomes, including atherosclerotic heart disease and HF. There are several theories about the relationship between T2DM and HF, though the exact pathophysiology of this relationship is unknown.3,4 One theory suggests diabetic cardiomyopathy as the cause. In patients with diabetic cardiomyopathy, there is early development of diastolic dysfunction, which eventually progresses to ventricular dysfunction. There is continued stimulation of the renin-angiotensin-aldosterone system that leads to death of cardiomyocytes, fibrosis, and remodeling, which worsens pump failure.5
SGLT2 inhibitors decrease hyperglycemia and hyperinsulinemia, potentially reducing HF risk. SGLT2 inhibitors decrease blood glucose levels by inhibiting SGLT2 in the proximal tubule, leading to a decrease of glucose reabsorption and an increase in excretion.6,7 The EMPA-REG OUTCOME trial looked at cardiovascular outcomes in patients with T2DM at high risk for adverse cardiac events. There was a significant risk reduction in deaths and hospitalizations for HF in patients treated with empagliflozin.8
The EMPRISE study specifically examined empagliflozin and its effects on hospitalization for HF.2 When compared with patients treated with sitagliptin, there was a statistically significant decrease in hospitalization for HF in patients with T2DM, both with and without preexisting cardiovascular disease.
This case highlights the relationship between T2DM and HF. We also show how the use of empagliflozin may have helped manage the patient’s undiagnosed HF and how its discontinuation luckily unmasked it. Routine evaluation for HF in patients with T2DM is not done, but likely there are patients who would benefit, especially given the strong, albeit less known, association between these 2 conditions.
Further studies are needed to determine the type of patients who would benefit most from HF screening. For now, the best practice is to obtain a complete medical history that includes current and recently discontinued medications as well a thorough physical examination for signs of fluid overload and cardiovascular compromise. Patients who may have signs concerning for HF can have appropriate testing and intervention.
Conclusions
SGLT2 inhibitors have been shown to have a role in the management of HF in patients with T2DM. There is a risk of exacerbation or unmasking of HF when discontinuing SGLT2 inhibitors. To our knowledge, this is the first paper describing the discovery of HF following interruption of SGLT2 inhibitor treatment. The clinician and patient should monitor for signs and symptoms of fluid overload when stopping therapy. Further research into the benefits of a more comprehensive evaluation is needed.
About 40% of patients with heart failure (HF) also have type 2 diabetes mellitus (T2DM).1 Certain sodium-glucose cotransporter-2 (SGLT2) inhibitors have benefited patients with HF.2 We report a case of a patient with T2DM who had signs and symptoms of hypervolemia after discontinuing the SGLT2 inhibitor empagliflozin. The patient was found to have previously undiagnosed HF.
Case Presentation
A 58-year-old male presented for care at Malcolm Randall Veterans Affairs Medical Center in Gainseville, Florida, diabetes clinic. The patient was diagnosed with T2DM at age 32 years. At 36 years, he was started on subcutaneous insulin injections, and was switched to insulin pump therapy in his early 40s. At the time of evaluation, the T2DM was managed using an insulin pump, metformin, and acarbose. He had been prescribed empagliflozin 10 mg several months before presentation, but the medication ran out about 1 month prior to evaluation, and additional refills were unavailable.
The patient reported a 1-month history of worsening exertional shortness of breath, decreased exercise tolerance, and lower extremity swelling. Vitals signs, including respiratory rate and oxygen saturation were within normal limits. Bibasilar crackles and bilateral 2+ pitting pedal edema were noted. The remaining examination was unrevealing. His most recent glycated hemoglobin A1c level from 1 month prior to the presentation was 6.4%.
Given the patient’s shortness of breath and evidence of fluid overload on examination, brain natriuretic peptide was obtained and was significantly elevated at 5,895 pg/mL. A transthoracic echocardiogram revealed left ventricular ejection fraction < 20%. The patient was started on furosemide 40 mg, pending receipt of empagliflozin. A cardiology evaluation also was recommended.
Cardiac catheterization identified significant obstructions to the left anterior descending and left circumflex arteries. The patient underwent successful percutaneous coronary intervention to these areas. Following initiation of medications and coronary revascularization, the patient reported significant symptom improvement. At the follow-up evaluation 8 weeks later, he was symptom free, and his physical examination was consistent with euvolemia.
Discussion
T2DM has been associated with adverse cardiovascular outcomes, including atherosclerotic heart disease and HF. There are several theories about the relationship between T2DM and HF, though the exact pathophysiology of this relationship is unknown.3,4 One theory suggests diabetic cardiomyopathy as the cause. In patients with diabetic cardiomyopathy, there is early development of diastolic dysfunction, which eventually progresses to ventricular dysfunction. There is continued stimulation of the renin-angiotensin-aldosterone system that leads to death of cardiomyocytes, fibrosis, and remodeling, which worsens pump failure.5
SGLT2 inhibitors decrease hyperglycemia and hyperinsulinemia, potentially reducing HF risk. SGLT2 inhibitors decrease blood glucose levels by inhibiting SGLT2 in the proximal tubule, leading to a decrease of glucose reabsorption and an increase in excretion.6,7 The EMPA-REG OUTCOME trial looked at cardiovascular outcomes in patients with T2DM at high risk for adverse cardiac events. There was a significant risk reduction in deaths and hospitalizations for HF in patients treated with empagliflozin.8
The EMPRISE study specifically examined empagliflozin and its effects on hospitalization for HF.2 When compared with patients treated with sitagliptin, there was a statistically significant decrease in hospitalization for HF in patients with T2DM, both with and without preexisting cardiovascular disease.
This case highlights the relationship between T2DM and HF. We also show how the use of empagliflozin may have helped manage the patient’s undiagnosed HF and how its discontinuation luckily unmasked it. Routine evaluation for HF in patients with T2DM is not done, but likely there are patients who would benefit, especially given the strong, albeit less known, association between these 2 conditions.
Further studies are needed to determine the type of patients who would benefit most from HF screening. For now, the best practice is to obtain a complete medical history that includes current and recently discontinued medications as well a thorough physical examination for signs of fluid overload and cardiovascular compromise. Patients who may have signs concerning for HF can have appropriate testing and intervention.
Conclusions
SGLT2 inhibitors have been shown to have a role in the management of HF in patients with T2DM. There is a risk of exacerbation or unmasking of HF when discontinuing SGLT2 inhibitors. To our knowledge, this is the first paper describing the discovery of HF following interruption of SGLT2 inhibitor treatment. The clinician and patient should monitor for signs and symptoms of fluid overload when stopping therapy. Further research into the benefits of a more comprehensive evaluation is needed.
1. Thomas MC. Type 2 diabetes and heart failure: challenges and solutions. Curr Cardiol Rev. 2016;12(3):249-255. doi:10.2174/1573403X12666160606120254
2. Patorno E, Pawar A, Franklin J, et al. Empagliflozin and the risk of heart failure hospitalization in routine clinical care: a first analysis from the EMPRISE study. Circulation. 2019;139(25):2822-2830. doi:10.1161/CIRCULATIONAHA.118.039177
3. Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41(1):11-13. doi:10.2337/dci17-0052
4. Thrainsdottir I, Aspelund T, Thorheirsson G, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavík study. Diabetes Care. 2005;28(3):612-616. doi:10.2337/diacare.28.3.612
5. Bell D, Goncalves E. Heart failure in the patient with diabetes: epidemiology, aetiology, prognosis, therapy and the effect of glucose-lowering medications. Diabetes Obes Metab. 2019;21(6):1277-1290. doi:10.1111/dom.13652
6. Nair S, Wilding JPH. Sodium glucose cotransporter 2 Inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95(1):34-42. doi:10.1210/jc.2009-0473
7. Ali A, Bain S, Hicks D, et al; Improving Diabetes Steering Committee. SGLT2 inhibitors: cardiovascular benefits beyond HbA1c- translating evidence into practice. Diabetes Ther. 2019;10(5):1595-1622. doi:10.1007/s13300-019-0657-8
8. Zinman B, Wanner C, Lachin J, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/NEJMoa1504720
1. Thomas MC. Type 2 diabetes and heart failure: challenges and solutions. Curr Cardiol Rev. 2016;12(3):249-255. doi:10.2174/1573403X12666160606120254
2. Patorno E, Pawar A, Franklin J, et al. Empagliflozin and the risk of heart failure hospitalization in routine clinical care: a first analysis from the EMPRISE study. Circulation. 2019;139(25):2822-2830. doi:10.1161/CIRCULATIONAHA.118.039177
3. Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41(1):11-13. doi:10.2337/dci17-0052
4. Thrainsdottir I, Aspelund T, Thorheirsson G, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavík study. Diabetes Care. 2005;28(3):612-616. doi:10.2337/diacare.28.3.612
5. Bell D, Goncalves E. Heart failure in the patient with diabetes: epidemiology, aetiology, prognosis, therapy and the effect of glucose-lowering medications. Diabetes Obes Metab. 2019;21(6):1277-1290. doi:10.1111/dom.13652
6. Nair S, Wilding JPH. Sodium glucose cotransporter 2 Inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95(1):34-42. doi:10.1210/jc.2009-0473
7. Ali A, Bain S, Hicks D, et al; Improving Diabetes Steering Committee. SGLT2 inhibitors: cardiovascular benefits beyond HbA1c- translating evidence into practice. Diabetes Ther. 2019;10(5):1595-1622. doi:10.1007/s13300-019-0657-8
8. Zinman B, Wanner C, Lachin J, et al; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/NEJMoa1504720
Nivolumab-Induced Granuloma Annulare
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
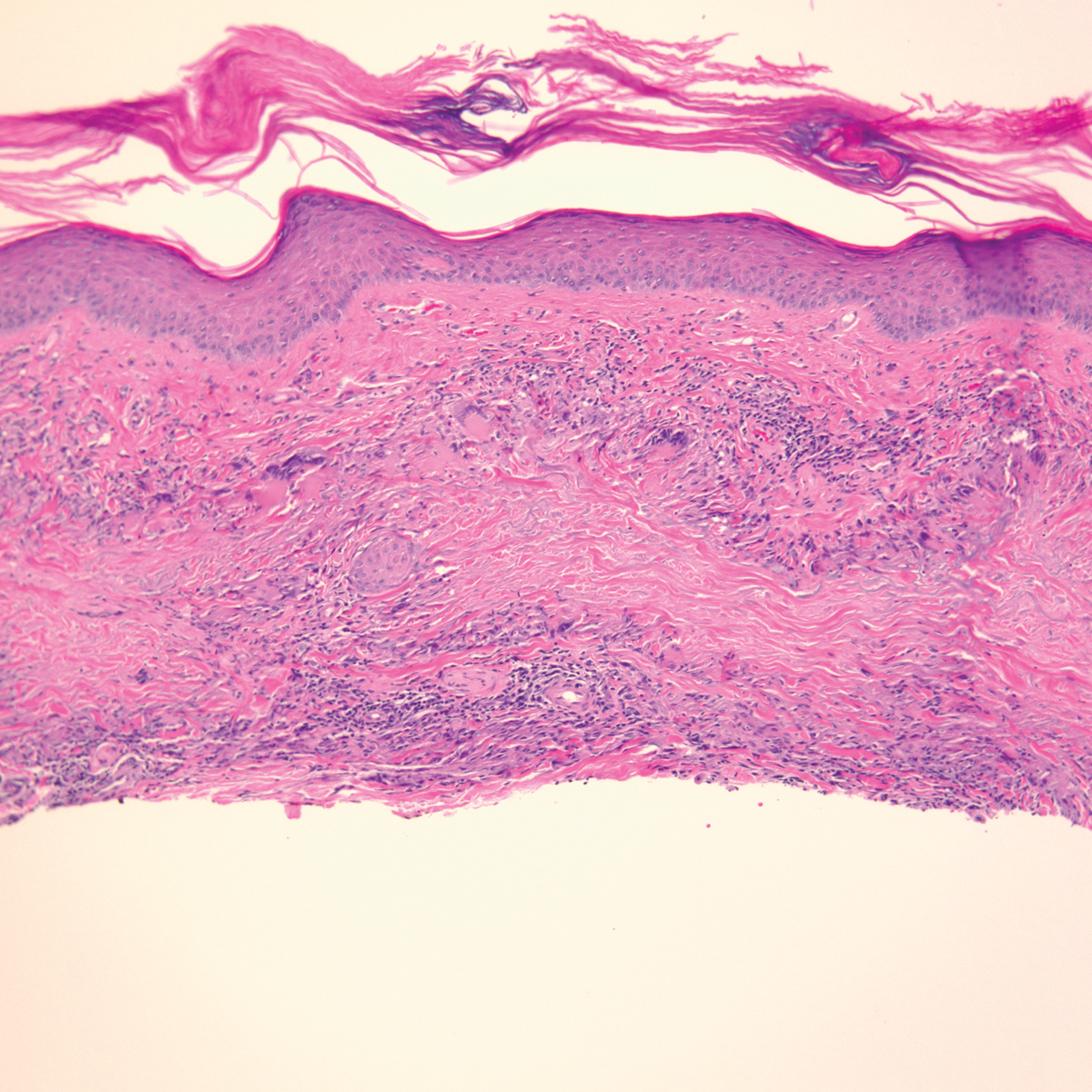
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
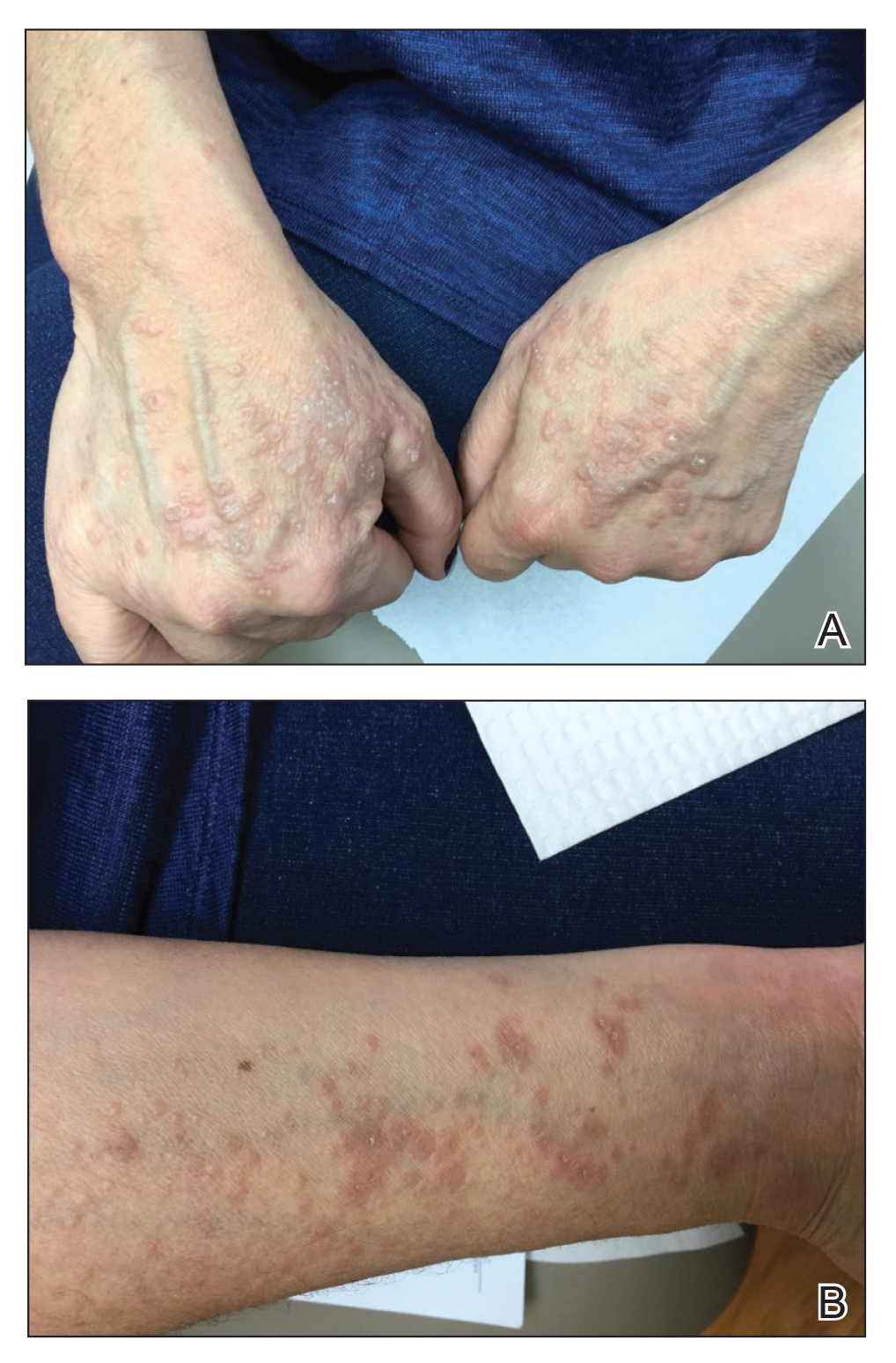
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
Granuloma annulare (GA) is a benign, cutaneous, granulomatous disease of unclear etiology. Typically, GA presents in young adults as asymptomatic, annular, flesh-colored to pink papules and plaques, commonly on the upper and lower extremities. Histologically, GA is characterized by mucin deposition, palisading or an interstitial granulomatous pattern, and collagen and elastic fiber degeneration.1
Granuloma annulare has been associated with various medications and medical conditions, including diabetes mellitus, hyperlipidemia, thyroid disease, and HIV.1 More recently, immune-checkpoint inhibitors (ICIs) have been reported to trigger GA.2 We report a case of nivolumab-induced GA in a 54-year-old woman.
Case Report
A 54-year-old woman presented with an itchy rash on the upper extremities, face, and chest of 4 months’ duration. The patient noted that the rash started on the hands and progressed to include the arms, face, and chest. She also reported associated mild tenderness. She had a history of stage IV non–small-cell lung carcinoma with metastases to the ribs and adrenal glands. She had been started on biweekly intravenous infusions of the ICI nivolumab by her oncologist approximately 1 year prior to the current presentation after failing a course of conventional chemotherapy. The most recent positron emission tomography–computed tomography scan 1 month prior to presentation showed a stable lung mass with radiologic disappearance of metastases, indicating a favorable response to nivolumab. The patient also had a history of hypothyroidism and depression, which were treated with oral levothyroxine 75 μg once daily and oral sertraline 50 mg once daily, respectively, both for longer than 5 years.
Physical examination revealed annular, erythematous, flat-topped papules, some with surmounting fine scale, coalescing into larger plaques along the dorsal surface of the hands and arms (Figure 1) as well as the forehead and chest. A biopsy of a papule on the dorsal aspect of the left hand revealed nodules of histiocytes admixed with Langerhans giant cells within the dermis; mucin was noted centrally within some nodules (Figure 2). Periodic acid–Schiff staining was negative for fungal elements compared to control. Polarization of the specimen was negative for foreign bodies. The biopsy findings therefore were consistent with a diagnosis of GA.
A 3-month treatment course of betamethasone dipropionate 0.05% cream twice daily failed. Narrowband UVB phototherapy was then initiated at 3 sessions weekly. The eruption of GA improved after 3 months of phototherapy. Subsequently, the patient was lost to follow-up.
Comment
Discovery of specific immune checkpoints in tumor-induced immunosuppression revolutionized oncologic therapy. An example is the programmed cell-death protein 1 (PD-1) receptor that is expressed on activated immune cells, including T cells and macrophages.3,4 Upon binding to the PD-1 ligand (PD-L1), T-cell proliferation is inhibited, resulting in downregulation of the immune response. As a result, tumor cells have evolved to overexpress PD-L1 to evade immunologic detection.3 Nivolumab, a fully human IgG4 antibody to PD-1, has emerged along with other ICIs as effective treatments for numerous cancers, including melanoma and non–small-cell lung cancer. By disrupting downregulation of T cells, ICIs improve immune-mediated antitumor activity.3
However, the resulting immunologic disturbance by ICIs has been reported to induce various cutaneous and systemic immune-mediated adverse reactions, including granulomatous reactions such as sarcoidosis, GA, and a cutaneous sarcoidlike granulomatous reaction.1,2,5,6 Our patient represents a rare case of nivolumab-induced GA.
Recent evidence suggests that GA might be caused in part by a cell-mediated hypersensitivity reaction that is regulated by a helper T cell subset 1 inflammatory reaction. Through release of cytokines by activated CD4+ T cells, macrophages are recruited, forming the granulomatous pattern and secreting enzymes that can degrade connective tissue. Nivolumab and other ICIs can thus trigger this reaction because their blockade of PD-1 enhances T cell–mediated immune reactions.2 In addition, because macrophages themselves also express PD-1, ICIs can directly enhance macrophage recruitment and proliferation, further increasing the risk of a granulomatous reaction.4
Interestingly, cutaneous adverse reactions to nivolumab have been associated with improved survival in melanoma patients.7 The nature of this association with granulomatous reactions in general and with GA specifically remains to be determined.
Conclusion
Since the approval of the first PD-1 inhibitors, pembrolizumab and nivolumab, in 2014, other ICIs targeting the immune checkpoint pathway have been developed. Newer agents targeting PD-L1 (avelumab, atezolizumab, and durvalumab) were recently approved. Additionally, cemiplimab, another PD-1 inhibitor, was approved by the US Food and Drug Administration in 2018 for the treatment of advanced cutaneous squamous cell carcinoma.8 Indications for all ICIs also have expanded considerably.3 Therefore, the incidence of immune-mediated adverse reactions, including GA, is bound to increase. Physicians should be cognizant of this association to accurately diagnose and effectively treat adverse reactions in patients who are taking ICIs.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015.03.055
- Wu J, Kwong BY, Martires KJ, et al. Granuloma annulare associated with immune checkpoint inhibitors. J Eur Acad Dermatol. 2018;32:E124-E126. doi:10.1111/jdv.14617
- Gong J, Chehrazi-Raffle A, Reddi S, et al. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. doi:10.1186/s40425-018-0316-z
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499. doi:10.1038/nature22396
- Birnbaum MR, Ma MW, Fleisig S, et al. Nivolumab-related cutaneous sarcoidosis in a patient with lung adenocarcinoma. JAAD Case Rep. 2017;3:208-211. doi:10.1016/j.jdcr.2017.02.015
- Danlos F-X, Pagès C, Baroudjian B, et al. Nivolumab-induced sarcoid-like granulomatous reaction in a patient with advanced melanoma. Chest. 2016;149:E133-E136. doi:10.1016/j.chest.2015.10.082
- Freeman-Keller M, Kim Y, Cronin H, et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. 2016;22:886-894. doi:10.1158/1078-0432.CCR-15-1136
- Migden MR, Rischin D, Schmults CD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-351. doi:10.1056/NEJMoa1805131
Practice Points
- Immune-related adverse events (irAEs) frequently occur in patients on immunotherapy, with the skin representing the most common site of involvement.
- Although rare, granulomatous reactions such as granuloma annulare increasingly are recognized as potential irAEs.
- Clinicians should be aware of this novel association to accurately diagnose and effectively treat adverse reactions in patients receiving immunotherapy.
20-year-old woman • 2 syncopal episodes • nausea • dizziness • Dx?
THE CASE
A 20-year-old woman presented to clinic with a chief complaint of 2 syncopal episodes within 10 minutes of each other. She reported that in both cases, she felt nauseated and dizzy before losing consciousness. She lost consciousness for a few seconds during the first episode and a few minutes during the second episode. Both episodes were unwitnessed.
The patient denied any fasting, vomiting, diarrhea, palpitations, chest pain, incontinence, oral trauma, headaches, fevers, chills, or tremors. Her last menstrual period started 3 days prior to presentation. The patient was taking sertraline 25 mg once daily for anxiety and depression and norethindrone acetate–ethinyl estradiol tablets 20 µg daily for birth control. She also was finishing a 7-day course of metronidazole for bacterial vaginosis. She reported having started the sertraline about 10 days prior to the syncopal episodes. She denied any personal history of drug or alcohol use, syncope, seizures, or any other medical conditions. Family history was negative for any cardiac or neurologic conditions.
The patient appeared euvolemic on exam. Overall, the review of the respiratory, cardiac, and neurologic systems was unremarkable. An electrocardiogram, obtained in clinic, showed a normal sinus rhythm and QT interval. Orthostatic blood pressure and heart rate measurements were as follows: supine, 122/83 mm Hg and 67 beats/min; seated, 118/87 mm Hg and 60 beats/min; and standing, 123/83 mm Hg and 95 beats/min. In addition to the increase in pulse between sitting and standing, the patient reported feeling nauseated when transitioning to a standing position.
Laboratory work-up included a comprehensive metabolic panel, complete blood count, and thyroid-stimulating hormone test. The results showed mild erythrocytosis with a hematocrit and hemoglobin of 46.1% and 15.6 g/dL respectively, as well as mild hypercalcemia (10.4 mg/dL).
THE DIAGNOSIS
An increase in heart rate of more than 30 beats/min when the patient went from a sitting to a standing position pointed to a diagnosis of postural orthostatic tachycardia syndrome (POTS). This prompted us to stop the sertraline.
DISCUSSION
POTS is a type of intolerance to orthostasis related to a significant increase in pulse without resulting hypotension upon standing. Other symptoms that accompany this change in position include dizziness, lightheadedness, blurry vision, and fatigue. Syncope occurs in about 40% of patients with POTS, which may be more frequent than for patients with orthostatic hypotension.1
The overall prevalence of POTS is 0.2% to 1%; however, it is generally seen in a 5:1 female-to-male ratio.2,3 POTS is often idiopathic. That said, it can also be caused by medication adverse effects, hypovolemia, and stressors, including vaccinations, viral infections, trauma, and emotional triggers. On physical exam, this patient did not appear to be hypovolemic, and she reported normal oral intake prior to this visit. Since the patient had started taking sertraline about 10 days prior to her syncopal episodes, we suspected POTS secondary to sertraline use was the likely etiology in this otherwise healthy young woman.
Continue to: Syncope could indicate a larger cardiovascular problem
Syncope could indicate a larger cardiovascular problem
The differential diagnosis of dizziness with loss of consciousness includes anemia, vasovagal syncope, orthostatic hypotension, dehydration, electrolyte imbalance, arrhythmia, prolonged QT syndrome, cardiac valve or structure abnormality, and seizure. Most of these differentials can be ruled out from basic laboratory tests or cardiac imaging. In POTS, the diagnostic work-up is essentially normal compared to other causes of syncope. Orthostatic hypotension, for example, is similar; however, there is an additional change in the arterial blood pressure.
Unintended adverse effects
Selective serotonin reuptake inhibitors (SSRIs), such as sertraline, are known to have fewer cardiovascular adverse effects compared to older antidepressants such as tricyclic antidepressants and monoamine oxidase inhibitors.4 However, case reports have shown an association between SSRIs and syncope.4-6 SSRIs have also been tied to increased heart rate variability.7
Nearly 2 weeks after stopping sertraline, our patient presented to clinic and was given a diagnosis of streptococcal pharyngitis. She said she’d had no additional syncopal episodes. Twenty days after sertraline cessation, the patient returned for follow-up. Her blood pressure and heart rate were as follows: supine, 112/68 mm Hg and 61 beats/min; seated, 113/74 mm Hg and 87 beats/min; and standing, 108/74 mm Hg and 78 beats/min.
Thus, after cessation of sertraline, her orthostatic heart rate changes were smaller than when she was first examined. Her vital signs showed an increase in pulse of 26 beats/min between lying and sitting, without any reports of nausea. She had no further complaints of dizziness or syncopal episodes.
THE TAKEAWAY
We don’t always know how a patient will respond to a newly prescribed medication or lifestyle change. A proper review of a patient’s history and medication use is a pivotal first step in making any diagnosis.
CORRESPONDENCE
Courtney Lynn Dominguez, MD, 4220 North Roxboro Street, Durham, NC 27704; [email protected]
1. Ojha A, McNeeley K, Heller E, et al. Orthostatic syndromes differ in syncope frequency. Am J Med. 2010;123:245-249. doi: 10.1016/j.amjmed.2009.09.018
2. Arnold AC, Ng J, Raj SR. Postural tachycardia syndrome—diagnosis, physiology, and prognosis. Auton Neurosci. 2018;215:3-11. doi: 10.1016/j.autneu.2018.02.005
3. Fedorowski A. Postural orthostatic tachycardia syndrome: clinical presentation, aetiology and management. J Intern Med. 2018;285:352-366. doi:10.1111/joim.12852
4. Pacher P, Ungvari Z, Kecskemeti V, et al. Review of cardiovascular effects of fluoxetine, a selective serotonin reuptake inhibitor, compared to tricyclic antidepressants. Curr Med Chem. 1998;5:381-390.
5. Feder R. Bradycardia and syncope induced by fluoxetine. J Clin Psychiatry. 1991;52:139.
6. Ellison JM, Milofsky JE, Ely E. Fluoxetine-induced bradycardia and syncope in two patients. J Clin Psychiatry. 1990;51:385-386.
7. Tucker P, Adamson P, Miranda R Jr, et al. Paroxetine increases heart rate variability in panic disorder. J Clin Psychopharmacol. 1997;17:370-376. doi: 10.1097/00004714-199710000-00006
THE CASE
A 20-year-old woman presented to clinic with a chief complaint of 2 syncopal episodes within 10 minutes of each other. She reported that in both cases, she felt nauseated and dizzy before losing consciousness. She lost consciousness for a few seconds during the first episode and a few minutes during the second episode. Both episodes were unwitnessed.
The patient denied any fasting, vomiting, diarrhea, palpitations, chest pain, incontinence, oral trauma, headaches, fevers, chills, or tremors. Her last menstrual period started 3 days prior to presentation. The patient was taking sertraline 25 mg once daily for anxiety and depression and norethindrone acetate–ethinyl estradiol tablets 20 µg daily for birth control. She also was finishing a 7-day course of metronidazole for bacterial vaginosis. She reported having started the sertraline about 10 days prior to the syncopal episodes. She denied any personal history of drug or alcohol use, syncope, seizures, or any other medical conditions. Family history was negative for any cardiac or neurologic conditions.
The patient appeared euvolemic on exam. Overall, the review of the respiratory, cardiac, and neurologic systems was unremarkable. An electrocardiogram, obtained in clinic, showed a normal sinus rhythm and QT interval. Orthostatic blood pressure and heart rate measurements were as follows: supine, 122/83 mm Hg and 67 beats/min; seated, 118/87 mm Hg and 60 beats/min; and standing, 123/83 mm Hg and 95 beats/min. In addition to the increase in pulse between sitting and standing, the patient reported feeling nauseated when transitioning to a standing position.
Laboratory work-up included a comprehensive metabolic panel, complete blood count, and thyroid-stimulating hormone test. The results showed mild erythrocytosis with a hematocrit and hemoglobin of 46.1% and 15.6 g/dL respectively, as well as mild hypercalcemia (10.4 mg/dL).
THE DIAGNOSIS
An increase in heart rate of more than 30 beats/min when the patient went from a sitting to a standing position pointed to a diagnosis of postural orthostatic tachycardia syndrome (POTS). This prompted us to stop the sertraline.
DISCUSSION
POTS is a type of intolerance to orthostasis related to a significant increase in pulse without resulting hypotension upon standing. Other symptoms that accompany this change in position include dizziness, lightheadedness, blurry vision, and fatigue. Syncope occurs in about 40% of patients with POTS, which may be more frequent than for patients with orthostatic hypotension.1
The overall prevalence of POTS is 0.2% to 1%; however, it is generally seen in a 5:1 female-to-male ratio.2,3 POTS is often idiopathic. That said, it can also be caused by medication adverse effects, hypovolemia, and stressors, including vaccinations, viral infections, trauma, and emotional triggers. On physical exam, this patient did not appear to be hypovolemic, and she reported normal oral intake prior to this visit. Since the patient had started taking sertraline about 10 days prior to her syncopal episodes, we suspected POTS secondary to sertraline use was the likely etiology in this otherwise healthy young woman.
Continue to: Syncope could indicate a larger cardiovascular problem
Syncope could indicate a larger cardiovascular problem
The differential diagnosis of dizziness with loss of consciousness includes anemia, vasovagal syncope, orthostatic hypotension, dehydration, electrolyte imbalance, arrhythmia, prolonged QT syndrome, cardiac valve or structure abnormality, and seizure. Most of these differentials can be ruled out from basic laboratory tests or cardiac imaging. In POTS, the diagnostic work-up is essentially normal compared to other causes of syncope. Orthostatic hypotension, for example, is similar; however, there is an additional change in the arterial blood pressure.
Unintended adverse effects
Selective serotonin reuptake inhibitors (SSRIs), such as sertraline, are known to have fewer cardiovascular adverse effects compared to older antidepressants such as tricyclic antidepressants and monoamine oxidase inhibitors.4 However, case reports have shown an association between SSRIs and syncope.4-6 SSRIs have also been tied to increased heart rate variability.7
Nearly 2 weeks after stopping sertraline, our patient presented to clinic and was given a diagnosis of streptococcal pharyngitis. She said she’d had no additional syncopal episodes. Twenty days after sertraline cessation, the patient returned for follow-up. Her blood pressure and heart rate were as follows: supine, 112/68 mm Hg and 61 beats/min; seated, 113/74 mm Hg and 87 beats/min; and standing, 108/74 mm Hg and 78 beats/min.
Thus, after cessation of sertraline, her orthostatic heart rate changes were smaller than when she was first examined. Her vital signs showed an increase in pulse of 26 beats/min between lying and sitting, without any reports of nausea. She had no further complaints of dizziness or syncopal episodes.
THE TAKEAWAY
We don’t always know how a patient will respond to a newly prescribed medication or lifestyle change. A proper review of a patient’s history and medication use is a pivotal first step in making any diagnosis.
CORRESPONDENCE
Courtney Lynn Dominguez, MD, 4220 North Roxboro Street, Durham, NC 27704; [email protected]
THE CASE
A 20-year-old woman presented to clinic with a chief complaint of 2 syncopal episodes within 10 minutes of each other. She reported that in both cases, she felt nauseated and dizzy before losing consciousness. She lost consciousness for a few seconds during the first episode and a few minutes during the second episode. Both episodes were unwitnessed.
The patient denied any fasting, vomiting, diarrhea, palpitations, chest pain, incontinence, oral trauma, headaches, fevers, chills, or tremors. Her last menstrual period started 3 days prior to presentation. The patient was taking sertraline 25 mg once daily for anxiety and depression and norethindrone acetate–ethinyl estradiol tablets 20 µg daily for birth control. She also was finishing a 7-day course of metronidazole for bacterial vaginosis. She reported having started the sertraline about 10 days prior to the syncopal episodes. She denied any personal history of drug or alcohol use, syncope, seizures, or any other medical conditions. Family history was negative for any cardiac or neurologic conditions.
The patient appeared euvolemic on exam. Overall, the review of the respiratory, cardiac, and neurologic systems was unremarkable. An electrocardiogram, obtained in clinic, showed a normal sinus rhythm and QT interval. Orthostatic blood pressure and heart rate measurements were as follows: supine, 122/83 mm Hg and 67 beats/min; seated, 118/87 mm Hg and 60 beats/min; and standing, 123/83 mm Hg and 95 beats/min. In addition to the increase in pulse between sitting and standing, the patient reported feeling nauseated when transitioning to a standing position.
Laboratory work-up included a comprehensive metabolic panel, complete blood count, and thyroid-stimulating hormone test. The results showed mild erythrocytosis with a hematocrit and hemoglobin of 46.1% and 15.6 g/dL respectively, as well as mild hypercalcemia (10.4 mg/dL).
THE DIAGNOSIS
An increase in heart rate of more than 30 beats/min when the patient went from a sitting to a standing position pointed to a diagnosis of postural orthostatic tachycardia syndrome (POTS). This prompted us to stop the sertraline.
DISCUSSION
POTS is a type of intolerance to orthostasis related to a significant increase in pulse without resulting hypotension upon standing. Other symptoms that accompany this change in position include dizziness, lightheadedness, blurry vision, and fatigue. Syncope occurs in about 40% of patients with POTS, which may be more frequent than for patients with orthostatic hypotension.1
The overall prevalence of POTS is 0.2% to 1%; however, it is generally seen in a 5:1 female-to-male ratio.2,3 POTS is often idiopathic. That said, it can also be caused by medication adverse effects, hypovolemia, and stressors, including vaccinations, viral infections, trauma, and emotional triggers. On physical exam, this patient did not appear to be hypovolemic, and she reported normal oral intake prior to this visit. Since the patient had started taking sertraline about 10 days prior to her syncopal episodes, we suspected POTS secondary to sertraline use was the likely etiology in this otherwise healthy young woman.
Continue to: Syncope could indicate a larger cardiovascular problem
Syncope could indicate a larger cardiovascular problem
The differential diagnosis of dizziness with loss of consciousness includes anemia, vasovagal syncope, orthostatic hypotension, dehydration, electrolyte imbalance, arrhythmia, prolonged QT syndrome, cardiac valve or structure abnormality, and seizure. Most of these differentials can be ruled out from basic laboratory tests or cardiac imaging. In POTS, the diagnostic work-up is essentially normal compared to other causes of syncope. Orthostatic hypotension, for example, is similar; however, there is an additional change in the arterial blood pressure.
Unintended adverse effects
Selective serotonin reuptake inhibitors (SSRIs), such as sertraline, are known to have fewer cardiovascular adverse effects compared to older antidepressants such as tricyclic antidepressants and monoamine oxidase inhibitors.4 However, case reports have shown an association between SSRIs and syncope.4-6 SSRIs have also been tied to increased heart rate variability.7
Nearly 2 weeks after stopping sertraline, our patient presented to clinic and was given a diagnosis of streptococcal pharyngitis. She said she’d had no additional syncopal episodes. Twenty days after sertraline cessation, the patient returned for follow-up. Her blood pressure and heart rate were as follows: supine, 112/68 mm Hg and 61 beats/min; seated, 113/74 mm Hg and 87 beats/min; and standing, 108/74 mm Hg and 78 beats/min.
Thus, after cessation of sertraline, her orthostatic heart rate changes were smaller than when she was first examined. Her vital signs showed an increase in pulse of 26 beats/min between lying and sitting, without any reports of nausea. She had no further complaints of dizziness or syncopal episodes.
THE TAKEAWAY
We don’t always know how a patient will respond to a newly prescribed medication or lifestyle change. A proper review of a patient’s history and medication use is a pivotal first step in making any diagnosis.
CORRESPONDENCE
Courtney Lynn Dominguez, MD, 4220 North Roxboro Street, Durham, NC 27704; [email protected]
1. Ojha A, McNeeley K, Heller E, et al. Orthostatic syndromes differ in syncope frequency. Am J Med. 2010;123:245-249. doi: 10.1016/j.amjmed.2009.09.018
2. Arnold AC, Ng J, Raj SR. Postural tachycardia syndrome—diagnosis, physiology, and prognosis. Auton Neurosci. 2018;215:3-11. doi: 10.1016/j.autneu.2018.02.005
3. Fedorowski A. Postural orthostatic tachycardia syndrome: clinical presentation, aetiology and management. J Intern Med. 2018;285:352-366. doi:10.1111/joim.12852
4. Pacher P, Ungvari Z, Kecskemeti V, et al. Review of cardiovascular effects of fluoxetine, a selective serotonin reuptake inhibitor, compared to tricyclic antidepressants. Curr Med Chem. 1998;5:381-390.
5. Feder R. Bradycardia and syncope induced by fluoxetine. J Clin Psychiatry. 1991;52:139.
6. Ellison JM, Milofsky JE, Ely E. Fluoxetine-induced bradycardia and syncope in two patients. J Clin Psychiatry. 1990;51:385-386.
7. Tucker P, Adamson P, Miranda R Jr, et al. Paroxetine increases heart rate variability in panic disorder. J Clin Psychopharmacol. 1997;17:370-376. doi: 10.1097/00004714-199710000-00006
1. Ojha A, McNeeley K, Heller E, et al. Orthostatic syndromes differ in syncope frequency. Am J Med. 2010;123:245-249. doi: 10.1016/j.amjmed.2009.09.018
2. Arnold AC, Ng J, Raj SR. Postural tachycardia syndrome—diagnosis, physiology, and prognosis. Auton Neurosci. 2018;215:3-11. doi: 10.1016/j.autneu.2018.02.005
3. Fedorowski A. Postural orthostatic tachycardia syndrome: clinical presentation, aetiology and management. J Intern Med. 2018;285:352-366. doi:10.1111/joim.12852
4. Pacher P, Ungvari Z, Kecskemeti V, et al. Review of cardiovascular effects of fluoxetine, a selective serotonin reuptake inhibitor, compared to tricyclic antidepressants. Curr Med Chem. 1998;5:381-390.
5. Feder R. Bradycardia and syncope induced by fluoxetine. J Clin Psychiatry. 1991;52:139.
6. Ellison JM, Milofsky JE, Ely E. Fluoxetine-induced bradycardia and syncope in two patients. J Clin Psychiatry. 1990;51:385-386.
7. Tucker P, Adamson P, Miranda R Jr, et al. Paroxetine increases heart rate variability in panic disorder. J Clin Psychopharmacol. 1997;17:370-376. doi: 10.1097/00004714-199710000-00006

Treating Hepatitis C Virus Reinfection With 8 Weeks of Ledipasvir/Sofosbuvir Achieves Sustained Virologic Response
Three patients reinfected with hepatitis C virus after a sustained virologic response were considered treatment naïve and treated with a short-course direct acting antiviral regimen.
To decrease the incidence and prevalence of hepatitis C virus (HCV) in the United States, hepatology experts, public health officials, and patient advocates agree that linkage to care is essential for treatment of people who inject drugs (PWID). The most recent surveillance report from the Centers for Disease Control and Prevention (CDC) estimates that injection drug use accounts for the transmission of approximately 72% of new HCV infections.1,2
Although recent studies of direct-acting antiviral (DAA) agents have not been designed to investigate the long-term rates of reinfection in this population, various population-based studies in multiple countries have attempted to describe the rate of reinfection for this cohort.3-7 This rate varies widely based on the defined population of PWID, definition of reinfection, and the prevalence of HCV in a given PWID population. However, studies have consistently shown a relatively low historic rate of reinfection, which varies from 1 to 5 per 100 person-years in patients who have ever injected drugs, to 3 to 33 per 100 person-years in patients who continue injection drug use (IDU). Higher rates are found in those who engage in high-risk behaviors such as needle sharing.3-7 Yet, the US opioid crisis is attributable to a recent rise in both overall incidence and reinfections, highlighting the importance of determining the best treatment strategy for those who become reinfected.1
Current HCV guidelines from the American Association for the Study of Liver Diseases AASLD) and Infectious Diseases Society of America (IDSA) encourage access to retreatment for PWID who become reinfected, stating that new reinfections should follow treatment-naïve therapy recommendations.8 However, to date this recommendation has not been validated by published clinical trials or patient case reports. This is likely due in part both to the small number of reinfections among PWID requiring retreatment and barriers to payment for treatment, particularly for individuals with substance use disorders.9 While this recommendation can be found under the key population section for the “Identification and Management of HCV in People Who Inject Drugs,” health care providers (HCPs) may easily miss this statement if they alternatively refer to the “Treatment-Experienced” section that recommends escalation to either sofosbuvir/velpatasvir/voxilaprevir or glecaprevir/pibrentasvir in patients who are NS5A inhibitor DAA-experienced.8 Anecdotally, the first instinct for many HCPs when considering a treatment regimen for a reinfected patient is to refer to treatment-experienced regimen recommendations rather than appreciating the reinfected virus to be treatment naïve.
A treatment-escalation approach could have the consequence of limiting the number of times a patient could undergo treatment on successive reinfections. Additionally, these retreatment regimens often are more expensive, resulting in further cost barriers for payors approving retreatment for individuals with HCV reinfection. In contrast, demonstrating efficacy of a less costly short-course regimen would support increased access to initial and retreatment courses for PWID. The implications of enabling improved access to care is essential in the setting of the ongoing opioid epidemic in the United States.
Given the perspective that the virus should be considered treatment naïve for patients who become reinfected, we describe here 3 cases of patients previously achieving sustained virologic response (SVR) being retreated with the cost-effective 8-week regimen of ledipasvir/sofosbuvir following reinfection.
Case Reports
Case 1
A 59-year-old male presented for his third treatment course for HCV genotype 1a. The patient initially underwent 76 weeks of interferon-based HCV treatment in 2007 and 2008, from which he was determined to have achieved SVR in 24 weeks (SVR24) in April 2009. His viral load remained undetected through February 2010 but subsequently had detectable virus again in 2011 following relapsed use of alcohol, cocaine, and injection drugs. The patient elected to await approval of DAAs and eventually completed an 8-week regimen of ledipasvir/sofosbuvir from May to July 2016, achieving SVR24 in December 2016. The patient’s viral load was rechecked in October 2018 and he was again viremic following recent IDU, suggesting a second reinfection.
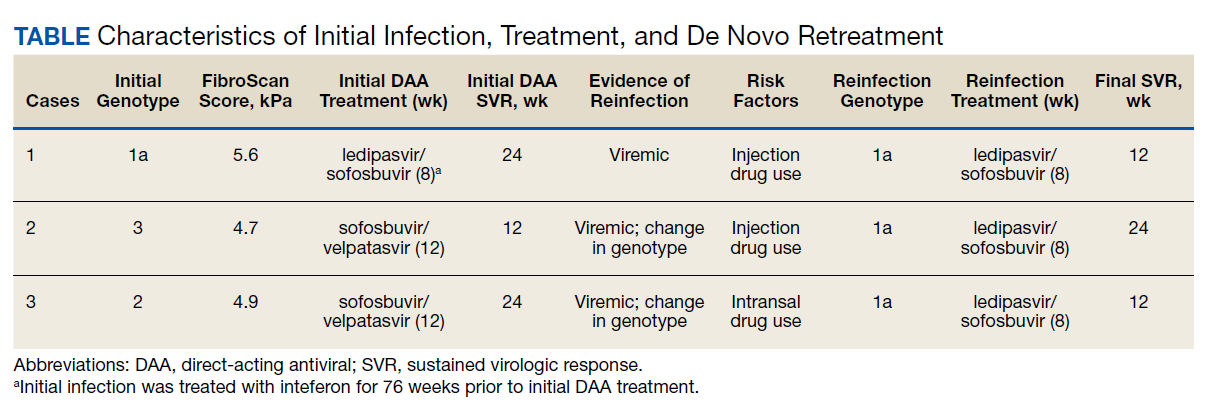
In preparation for his third HCV treatment, the patient was included in shared decision making to consider retreating his de novo infection as treatment naïve to provide a briefer (ie, 8 weeks) and more cost-effective treatment given his low likelihood of advanced fibrotic liver disease—his FibroScan score was 6.5 kPa, whereas scores ≥ 12.5 kPa in patients with chronic HCV suggest a higher likelihood of cirrhosis.10 At week 4, the patient’s viral load was undetected, he completed his 8-week regimen of ledipasvir/sofosbuvir as planned and achieved SVR12 (Table). He had reported excellent adherence throughout treatment with assistance of a pill box and validated by a reported pill count.
Case 2
A 32-year-old male presented with HCV genotype 1a. Like case 1, this patient had a low FibroScan score of 4.7 kPa. He was previously infected with genotype 3 and completed a 12-week course of sofosbuvir/velpatasvir in November 2016. He achieved SVR12 as evidenced by an undetected viral load in February 2017 despite questionable adherence throughout and relapsed use of heroin by the end of his regimen. He continued intermittent IDU and presented in October 2018 with a detectable viral load, now with genotype 1a. The patient similarly agreed to undergo an 8-week regimen of ledipasvir/sofosbuvir, considering his de novo infection to be treatment naïve. His viral load at treatment week 3 was quantitatively negative while qualitatively detectable at < 15 U/mL. He completed his treatment course in March 2019 and was determined to have achieved SVR24 in September 2019.
Case 3
A 51-year-old male presented with a history of HCV genotype 1a and a low FibroScan score (4.9 kPa ). The patient was previously infected with genotype 2 and had achieved SVR24 following a 12-week regimen of sofosbuvir/velpatasvir in 2017. The patient subsequently was reinfected with genotype 1a and completed an 8-week course of ledipasvir/sofosbuvir in May 2019. The patient had his SVR12 lab drawn 9 days early and was undetectable at that time. He reported 0 missed doses during treatment and achieved an undetected viral load by treatment week 4.
Discussion
We demonstrate that HCV reinfection after treatment with previous interferon and/or DAA-based regimens can be treated with less costly 8-week treatment regimens. Current guidelines include a statement allowing for reinfected patients to follow initial treatment guidelines, but this statement has previously lacked published evidence and may be overlooked by HCPs who refer to recommendations for treatment-experienced patients. Given the increasing likelihood of HCPs encountering patients who have become reinfected with HCV after achieving SVR from a DAA regimen, further delineation may be needed in the recommendations for treatment-experienced patients to highlight the important nuance of recognizing that reinfections should follow initial treatment guidance.
While all 3 of these cases met criteria for the least costly and simplest 1 pill once daily 8-week regimen of ledipasvir/sofosbuvir, patients requiring retreatment with alternative genotypes or evidence of advanced fibrotic liver disease could benefit from a similar approach of using the least expensive and/or shortest duration regimen for which they meet eligibility. With this approach, coverage could be further expanded to the PWID population to help limit HCV transmission amid the opioid crisis.1
Studies have established that PWID are able to achieve similar SVR efficacy rates similar to that of the general population when treated in the setting of an interdisciplinary treatment team that offers collaborative management of complex psychosocial comorbidities and harm reduction strategies.11,12 These integrative patient-centric strategies may include personalized behavioral health pretreatment evaluations, access to substance use treatment, harm reduction counseling, needle exchange programs, and close follow-up by a case manager.2,13 Current DAA regimens combined with 1 or more of these strategies have demonstrated SVR12 rates of 90 to 95% for initial treatment regimens.11 These high SVR12 rates were even achieved in a recent study in which 74% (76/103) of participants had self-reported IDU within 30 days of HCV treatment start and similar IDU rates throughout treatment.12 A meta-analysis, including real-world studies of DAA treatment outcomes yielded a pooled SVR of 88% (95% CI, 83‐92%) for recent PWID and 91% (95% CI, 88‐95%) for individuals using opiate substitution therapy (OST).14 Additionally, linking PWID with OST also reduces risk for reinfection.14,15
For any patient with detectable HCV after completing the initial DAA regimen, it is important to distinguish between relapse and reinfection. SVR12 is generally synonymous with a clinical cure. Patients with ongoing risk factors posttreatment should continue to have their HCV viral load monitored for evidence of reinfection. Patients without known risk factors may benefit from repeat viral load only if there is clinical concern for reinfection, for example, a rise in liver enzymes.
We have shown that patients with ongoing risk factors who are reinfected can be treated successfully with cost-effective 8-week regimens. For comparison this 8-week regimen of ledipasvir/sofosbuvir has an average wholesale price (AWP) of $28,800, while alternative regimens approved for treatment-naïve patients vary in AWP from $31,680 to $43,200, and regimens approved for retreatment of DAA failures have an AWP as high as $89,712.
An 8-week treatment regimen for both initial and reinfection regimens affords many advantages in medication adherence and both medication and provider resource cost-effectiveness. First, new HCV reinfections are disproportionally younger individuals often with complex psychosocial issues that impact retention in treatment. An 8-week course of treatment can be initiated concurrently with substance abuse treatment programs, including intensive outpatient programs and residential treatment programs that are usually at least 28 days. Many of these programs provide aftercare options that would extend the entire course of treatment. These opportunities afford individuals to receive HCV treatment in a setting that supports medication adherence, sobriety efforts, and education on harm reduction to reduce risk for reinfection.
Finally, statistical models indicate eradication of HCV will require scaling up the treatment of PWID in conjunction with harm reduction strategies such as OST and needle exchange programs.16 In contrast, there are low risks associated with retreatment given these medications are well-tolerated, treatment of PWID lowers the risk of further HCV transmission, and the understanding of these reinfections being treatment naïve disavows concerns of these patients having resistance to regimens that cleared their prior infections. The opportunity to provide retreatment without escalating regimen complexity or cost increases access to care for a vulnerable population while aiding in the eradication of HCV.
1. Centers for Disease Control and Prevention. Viral Hepatitis Surveillance - United States, 2018. Updated August 28, 2020. Accessed May 18, 2021. https://www.cdc.gov/hepatitis/statistics/2018surveillance/HepC.htm 2. Grebely J, Robaeys G, Bruggmann P, et al; International Network for Hepatitis in Substance Users. Recommendations for the management of hepatitis C virus infection among people who inject drugs. Int J Drug Policy. 2015;26(10):1028-1038. doi:10.1016/j.drugpo.2015.07.005
3. Marco A, Esteban JI, Solé C, et al. Hepatitis C virus reinfection among prisoners with sustained virological response after treatment for chronic hepatitis C. J Hepatol. 2013;59(1):45-51. doi:10.1016/j.jhep.2013.03.008
4. Midgard H, Bjøro B, Mæland A, et al. Hepatitis C reinfection after sustained virological response. J Hepatol. 2016;64(5):1020-1026. doi:10.1016/j.jhep.2016.01.001
5. Currie SL, Ryan JC, Tracy D, et al. A prospective study to examine persistent HCV reinfection in injection drug users who have previously cleared the virus [published correction appears in Drug Alcohol Depend. 2008 Jul;96(1-2):192]. Drug Alcohol Depend. 2008;93(1-2):148-154. doi:10.1016/j.drugalcdep.2007.09.011
6. Grady BP, Vanhommerig JW, Schinkel J, et al. Low incidence of reinfection with the hepatitis C virus following treatment in active drug users in Amsterdam. Eur J Gastroenterol Hepatol. 2012;24(11):1302-1307. doi:10.1097/MEG.0b013e32835702a8
7. Grebely J, Pham ST, Matthews GV, et al; ATAHC Study Group. Hepatitis C virus reinfection and superinfection among treated and untreated participants with recent infection. Hepatology. 2012;55(4):1058-1069. doi:10.1002/hep.24754
8. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. HCV Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C. Accessed May 26, 2021. https://www.hcvguidelines.org
9. National Viral Hepatitis Roundtable, Center for Health Law and Policy Innovation, Harvard Law School. Hepatitis C: The State of Medicaid Access. 2017 National Summary Report. Updated October 23, 2017. Accessed May 26, 2021. https://hepcstage.wpengine.com/wp-content/uploads/2017/10/State-of-HepC_2017_FINAL.pdf
10. Singh S, Muir AJ, Dieterich DT, Falck-Ytter YT. American Gastroenterological Association Institute technical review on the role of elastography in chronic liver diseases. Gastroenterology. 2017;152(6):1544-1577. doi:10.1053/j.gastro.2017.03.016
11. Dore GJ, Altice F, Litwin AH, et al; C-EDGE CO-STAR Study Group. Elbasvir-grazoprevir to treat hepatitis C virus infection in persons receiving opioid agonist therapy: a randomized trial. Ann Intern Med. 2016;165(9):625-634. doi:10.7326/M16-0816
12. Grebely J, Dalgard O, Conway B, et al; SIMPLIFY Study Group. Sofosbuvir and velpatasvir for hepatitis C virus infection in people with recent injection drug use (SIMPLIFY): an open-label, single-arm, phase 4, multicentre trial. Lancet Gastroenterol Hepatol. 2018;3(3):153-161. doi:10.1016/S2468-1253(17)30404-1
13. Cos TA, Bartholomew TS, Huynh, KJ. Role of behavioral health providers in treating hepatitis C. Professional Psychol Res Pract. 2019;50(4):246–254. doi:10.1037/pro0000243
14. Latham NH, Doyle JS, Palmer AY, et al. Staying hepatitis C negative: a systematic review and meta-analysis of cure and reinfection in people who inject drugs. Liver Int. 2019;39(12):2244-2260. doi:10.1111/liv.14152
15. Platt L, Minozzi S, Reed J, et al. Needle syringe programmes and opioid substitution therapy for preventing hepatitis C transmission in people who inject drugs. Cochrane Database Syst Rev. 2017;9(9):CD012021. Published 2017 Sep 18. doi:10.1002/14651858.CD012021.pub2
16. Fraser H, Martin NK, Brummer-Korvenkontio H, et al. Model projections on the impact of HCV treatment in the prevention of HCV transmission among people who inject drugs in Europe. J Hepatol. 2018;68(3):402-411. doi:10.1016/j.jhep.2017.10.010
Three patients reinfected with hepatitis C virus after a sustained virologic response were considered treatment naïve and treated with a short-course direct acting antiviral regimen.
Three patients reinfected with hepatitis C virus after a sustained virologic response were considered treatment naïve and treated with a short-course direct acting antiviral regimen.
To decrease the incidence and prevalence of hepatitis C virus (HCV) in the United States, hepatology experts, public health officials, and patient advocates agree that linkage to care is essential for treatment of people who inject drugs (PWID). The most recent surveillance report from the Centers for Disease Control and Prevention (CDC) estimates that injection drug use accounts for the transmission of approximately 72% of new HCV infections.1,2
Although recent studies of direct-acting antiviral (DAA) agents have not been designed to investigate the long-term rates of reinfection in this population, various population-based studies in multiple countries have attempted to describe the rate of reinfection for this cohort.3-7 This rate varies widely based on the defined population of PWID, definition of reinfection, and the prevalence of HCV in a given PWID population. However, studies have consistently shown a relatively low historic rate of reinfection, which varies from 1 to 5 per 100 person-years in patients who have ever injected drugs, to 3 to 33 per 100 person-years in patients who continue injection drug use (IDU). Higher rates are found in those who engage in high-risk behaviors such as needle sharing.3-7 Yet, the US opioid crisis is attributable to a recent rise in both overall incidence and reinfections, highlighting the importance of determining the best treatment strategy for those who become reinfected.1
Current HCV guidelines from the American Association for the Study of Liver Diseases AASLD) and Infectious Diseases Society of America (IDSA) encourage access to retreatment for PWID who become reinfected, stating that new reinfections should follow treatment-naïve therapy recommendations.8 However, to date this recommendation has not been validated by published clinical trials or patient case reports. This is likely due in part both to the small number of reinfections among PWID requiring retreatment and barriers to payment for treatment, particularly for individuals with substance use disorders.9 While this recommendation can be found under the key population section for the “Identification and Management of HCV in People Who Inject Drugs,” health care providers (HCPs) may easily miss this statement if they alternatively refer to the “Treatment-Experienced” section that recommends escalation to either sofosbuvir/velpatasvir/voxilaprevir or glecaprevir/pibrentasvir in patients who are NS5A inhibitor DAA-experienced.8 Anecdotally, the first instinct for many HCPs when considering a treatment regimen for a reinfected patient is to refer to treatment-experienced regimen recommendations rather than appreciating the reinfected virus to be treatment naïve.
A treatment-escalation approach could have the consequence of limiting the number of times a patient could undergo treatment on successive reinfections. Additionally, these retreatment regimens often are more expensive, resulting in further cost barriers for payors approving retreatment for individuals with HCV reinfection. In contrast, demonstrating efficacy of a less costly short-course regimen would support increased access to initial and retreatment courses for PWID. The implications of enabling improved access to care is essential in the setting of the ongoing opioid epidemic in the United States.
Given the perspective that the virus should be considered treatment naïve for patients who become reinfected, we describe here 3 cases of patients previously achieving sustained virologic response (SVR) being retreated with the cost-effective 8-week regimen of ledipasvir/sofosbuvir following reinfection.
Case Reports
Case 1
A 59-year-old male presented for his third treatment course for HCV genotype 1a. The patient initially underwent 76 weeks of interferon-based HCV treatment in 2007 and 2008, from which he was determined to have achieved SVR in 24 weeks (SVR24) in April 2009. His viral load remained undetected through February 2010 but subsequently had detectable virus again in 2011 following relapsed use of alcohol, cocaine, and injection drugs. The patient elected to await approval of DAAs and eventually completed an 8-week regimen of ledipasvir/sofosbuvir from May to July 2016, achieving SVR24 in December 2016. The patient’s viral load was rechecked in October 2018 and he was again viremic following recent IDU, suggesting a second reinfection.
In preparation for his third HCV treatment, the patient was included in shared decision making to consider retreating his de novo infection as treatment naïve to provide a briefer (ie, 8 weeks) and more cost-effective treatment given his low likelihood of advanced fibrotic liver disease—his FibroScan score was 6.5 kPa, whereas scores ≥ 12.5 kPa in patients with chronic HCV suggest a higher likelihood of cirrhosis.10 At week 4, the patient’s viral load was undetected, he completed his 8-week regimen of ledipasvir/sofosbuvir as planned and achieved SVR12 (Table). He had reported excellent adherence throughout treatment with assistance of a pill box and validated by a reported pill count.
Case 2
A 32-year-old male presented with HCV genotype 1a. Like case 1, this patient had a low FibroScan score of 4.7 kPa. He was previously infected with genotype 3 and completed a 12-week course of sofosbuvir/velpatasvir in November 2016. He achieved SVR12 as evidenced by an undetected viral load in February 2017 despite questionable adherence throughout and relapsed use of heroin by the end of his regimen. He continued intermittent IDU and presented in October 2018 with a detectable viral load, now with genotype 1a. The patient similarly agreed to undergo an 8-week regimen of ledipasvir/sofosbuvir, considering his de novo infection to be treatment naïve. His viral load at treatment week 3 was quantitatively negative while qualitatively detectable at < 15 U/mL. He completed his treatment course in March 2019 and was determined to have achieved SVR24 in September 2019.
Case 3
A 51-year-old male presented with a history of HCV genotype 1a and a low FibroScan score (4.9 kPa ). The patient was previously infected with genotype 2 and had achieved SVR24 following a 12-week regimen of sofosbuvir/velpatasvir in 2017. The patient subsequently was reinfected with genotype 1a and completed an 8-week course of ledipasvir/sofosbuvir in May 2019. The patient had his SVR12 lab drawn 9 days early and was undetectable at that time. He reported 0 missed doses during treatment and achieved an undetected viral load by treatment week 4.
Discussion
We demonstrate that HCV reinfection after treatment with previous interferon and/or DAA-based regimens can be treated with less costly 8-week treatment regimens. Current guidelines include a statement allowing for reinfected patients to follow initial treatment guidelines, but this statement has previously lacked published evidence and may be overlooked by HCPs who refer to recommendations for treatment-experienced patients. Given the increasing likelihood of HCPs encountering patients who have become reinfected with HCV after achieving SVR from a DAA regimen, further delineation may be needed in the recommendations for treatment-experienced patients to highlight the important nuance of recognizing that reinfections should follow initial treatment guidance.
While all 3 of these cases met criteria for the least costly and simplest 1 pill once daily 8-week regimen of ledipasvir/sofosbuvir, patients requiring retreatment with alternative genotypes or evidence of advanced fibrotic liver disease could benefit from a similar approach of using the least expensive and/or shortest duration regimen for which they meet eligibility. With this approach, coverage could be further expanded to the PWID population to help limit HCV transmission amid the opioid crisis.1
Studies have established that PWID are able to achieve similar SVR efficacy rates similar to that of the general population when treated in the setting of an interdisciplinary treatment team that offers collaborative management of complex psychosocial comorbidities and harm reduction strategies.11,12 These integrative patient-centric strategies may include personalized behavioral health pretreatment evaluations, access to substance use treatment, harm reduction counseling, needle exchange programs, and close follow-up by a case manager.2,13 Current DAA regimens combined with 1 or more of these strategies have demonstrated SVR12 rates of 90 to 95% for initial treatment regimens.11 These high SVR12 rates were even achieved in a recent study in which 74% (76/103) of participants had self-reported IDU within 30 days of HCV treatment start and similar IDU rates throughout treatment.12 A meta-analysis, including real-world studies of DAA treatment outcomes yielded a pooled SVR of 88% (95% CI, 83‐92%) for recent PWID and 91% (95% CI, 88‐95%) for individuals using opiate substitution therapy (OST).14 Additionally, linking PWID with OST also reduces risk for reinfection.14,15
For any patient with detectable HCV after completing the initial DAA regimen, it is important to distinguish between relapse and reinfection. SVR12 is generally synonymous with a clinical cure. Patients with ongoing risk factors posttreatment should continue to have their HCV viral load monitored for evidence of reinfection. Patients without known risk factors may benefit from repeat viral load only if there is clinical concern for reinfection, for example, a rise in liver enzymes.
We have shown that patients with ongoing risk factors who are reinfected can be treated successfully with cost-effective 8-week regimens. For comparison this 8-week regimen of ledipasvir/sofosbuvir has an average wholesale price (AWP) of $28,800, while alternative regimens approved for treatment-naïve patients vary in AWP from $31,680 to $43,200, and regimens approved for retreatment of DAA failures have an AWP as high as $89,712.
An 8-week treatment regimen for both initial and reinfection regimens affords many advantages in medication adherence and both medication and provider resource cost-effectiveness. First, new HCV reinfections are disproportionally younger individuals often with complex psychosocial issues that impact retention in treatment. An 8-week course of treatment can be initiated concurrently with substance abuse treatment programs, including intensive outpatient programs and residential treatment programs that are usually at least 28 days. Many of these programs provide aftercare options that would extend the entire course of treatment. These opportunities afford individuals to receive HCV treatment in a setting that supports medication adherence, sobriety efforts, and education on harm reduction to reduce risk for reinfection.
Finally, statistical models indicate eradication of HCV will require scaling up the treatment of PWID in conjunction with harm reduction strategies such as OST and needle exchange programs.16 In contrast, there are low risks associated with retreatment given these medications are well-tolerated, treatment of PWID lowers the risk of further HCV transmission, and the understanding of these reinfections being treatment naïve disavows concerns of these patients having resistance to regimens that cleared their prior infections. The opportunity to provide retreatment without escalating regimen complexity or cost increases access to care for a vulnerable population while aiding in the eradication of HCV.
To decrease the incidence and prevalence of hepatitis C virus (HCV) in the United States, hepatology experts, public health officials, and patient advocates agree that linkage to care is essential for treatment of people who inject drugs (PWID). The most recent surveillance report from the Centers for Disease Control and Prevention (CDC) estimates that injection drug use accounts for the transmission of approximately 72% of new HCV infections.1,2
Although recent studies of direct-acting antiviral (DAA) agents have not been designed to investigate the long-term rates of reinfection in this population, various population-based studies in multiple countries have attempted to describe the rate of reinfection for this cohort.3-7 This rate varies widely based on the defined population of PWID, definition of reinfection, and the prevalence of HCV in a given PWID population. However, studies have consistently shown a relatively low historic rate of reinfection, which varies from 1 to 5 per 100 person-years in patients who have ever injected drugs, to 3 to 33 per 100 person-years in patients who continue injection drug use (IDU). Higher rates are found in those who engage in high-risk behaviors such as needle sharing.3-7 Yet, the US opioid crisis is attributable to a recent rise in both overall incidence and reinfections, highlighting the importance of determining the best treatment strategy for those who become reinfected.1
Current HCV guidelines from the American Association for the Study of Liver Diseases AASLD) and Infectious Diseases Society of America (IDSA) encourage access to retreatment for PWID who become reinfected, stating that new reinfections should follow treatment-naïve therapy recommendations.8 However, to date this recommendation has not been validated by published clinical trials or patient case reports. This is likely due in part both to the small number of reinfections among PWID requiring retreatment and barriers to payment for treatment, particularly for individuals with substance use disorders.9 While this recommendation can be found under the key population section for the “Identification and Management of HCV in People Who Inject Drugs,” health care providers (HCPs) may easily miss this statement if they alternatively refer to the “Treatment-Experienced” section that recommends escalation to either sofosbuvir/velpatasvir/voxilaprevir or glecaprevir/pibrentasvir in patients who are NS5A inhibitor DAA-experienced.8 Anecdotally, the first instinct for many HCPs when considering a treatment regimen for a reinfected patient is to refer to treatment-experienced regimen recommendations rather than appreciating the reinfected virus to be treatment naïve.
A treatment-escalation approach could have the consequence of limiting the number of times a patient could undergo treatment on successive reinfections. Additionally, these retreatment regimens often are more expensive, resulting in further cost barriers for payors approving retreatment for individuals with HCV reinfection. In contrast, demonstrating efficacy of a less costly short-course regimen would support increased access to initial and retreatment courses for PWID. The implications of enabling improved access to care is essential in the setting of the ongoing opioid epidemic in the United States.
Given the perspective that the virus should be considered treatment naïve for patients who become reinfected, we describe here 3 cases of patients previously achieving sustained virologic response (SVR) being retreated with the cost-effective 8-week regimen of ledipasvir/sofosbuvir following reinfection.
Case Reports
Case 1
A 59-year-old male presented for his third treatment course for HCV genotype 1a. The patient initially underwent 76 weeks of interferon-based HCV treatment in 2007 and 2008, from which he was determined to have achieved SVR in 24 weeks (SVR24) in April 2009. His viral load remained undetected through February 2010 but subsequently had detectable virus again in 2011 following relapsed use of alcohol, cocaine, and injection drugs. The patient elected to await approval of DAAs and eventually completed an 8-week regimen of ledipasvir/sofosbuvir from May to July 2016, achieving SVR24 in December 2016. The patient’s viral load was rechecked in October 2018 and he was again viremic following recent IDU, suggesting a second reinfection.
In preparation for his third HCV treatment, the patient was included in shared decision making to consider retreating his de novo infection as treatment naïve to provide a briefer (ie, 8 weeks) and more cost-effective treatment given his low likelihood of advanced fibrotic liver disease—his FibroScan score was 6.5 kPa, whereas scores ≥ 12.5 kPa in patients with chronic HCV suggest a higher likelihood of cirrhosis.10 At week 4, the patient’s viral load was undetected, he completed his 8-week regimen of ledipasvir/sofosbuvir as planned and achieved SVR12 (Table). He had reported excellent adherence throughout treatment with assistance of a pill box and validated by a reported pill count.
Case 2
A 32-year-old male presented with HCV genotype 1a. Like case 1, this patient had a low FibroScan score of 4.7 kPa. He was previously infected with genotype 3 and completed a 12-week course of sofosbuvir/velpatasvir in November 2016. He achieved SVR12 as evidenced by an undetected viral load in February 2017 despite questionable adherence throughout and relapsed use of heroin by the end of his regimen. He continued intermittent IDU and presented in October 2018 with a detectable viral load, now with genotype 1a. The patient similarly agreed to undergo an 8-week regimen of ledipasvir/sofosbuvir, considering his de novo infection to be treatment naïve. His viral load at treatment week 3 was quantitatively negative while qualitatively detectable at < 15 U/mL. He completed his treatment course in March 2019 and was determined to have achieved SVR24 in September 2019.
Case 3
A 51-year-old male presented with a history of HCV genotype 1a and a low FibroScan score (4.9 kPa ). The patient was previously infected with genotype 2 and had achieved SVR24 following a 12-week regimen of sofosbuvir/velpatasvir in 2017. The patient subsequently was reinfected with genotype 1a and completed an 8-week course of ledipasvir/sofosbuvir in May 2019. The patient had his SVR12 lab drawn 9 days early and was undetectable at that time. He reported 0 missed doses during treatment and achieved an undetected viral load by treatment week 4.
Discussion
We demonstrate that HCV reinfection after treatment with previous interferon and/or DAA-based regimens can be treated with less costly 8-week treatment regimens. Current guidelines include a statement allowing for reinfected patients to follow initial treatment guidelines, but this statement has previously lacked published evidence and may be overlooked by HCPs who refer to recommendations for treatment-experienced patients. Given the increasing likelihood of HCPs encountering patients who have become reinfected with HCV after achieving SVR from a DAA regimen, further delineation may be needed in the recommendations for treatment-experienced patients to highlight the important nuance of recognizing that reinfections should follow initial treatment guidance.
While all 3 of these cases met criteria for the least costly and simplest 1 pill once daily 8-week regimen of ledipasvir/sofosbuvir, patients requiring retreatment with alternative genotypes or evidence of advanced fibrotic liver disease could benefit from a similar approach of using the least expensive and/or shortest duration regimen for which they meet eligibility. With this approach, coverage could be further expanded to the PWID population to help limit HCV transmission amid the opioid crisis.1
Studies have established that PWID are able to achieve similar SVR efficacy rates similar to that of the general population when treated in the setting of an interdisciplinary treatment team that offers collaborative management of complex psychosocial comorbidities and harm reduction strategies.11,12 These integrative patient-centric strategies may include personalized behavioral health pretreatment evaluations, access to substance use treatment, harm reduction counseling, needle exchange programs, and close follow-up by a case manager.2,13 Current DAA regimens combined with 1 or more of these strategies have demonstrated SVR12 rates of 90 to 95% for initial treatment regimens.11 These high SVR12 rates were even achieved in a recent study in which 74% (76/103) of participants had self-reported IDU within 30 days of HCV treatment start and similar IDU rates throughout treatment.12 A meta-analysis, including real-world studies of DAA treatment outcomes yielded a pooled SVR of 88% (95% CI, 83‐92%) for recent PWID and 91% (95% CI, 88‐95%) for individuals using opiate substitution therapy (OST).14 Additionally, linking PWID with OST also reduces risk for reinfection.14,15
For any patient with detectable HCV after completing the initial DAA regimen, it is important to distinguish between relapse and reinfection. SVR12 is generally synonymous with a clinical cure. Patients with ongoing risk factors posttreatment should continue to have their HCV viral load monitored for evidence of reinfection. Patients without known risk factors may benefit from repeat viral load only if there is clinical concern for reinfection, for example, a rise in liver enzymes.
We have shown that patients with ongoing risk factors who are reinfected can be treated successfully with cost-effective 8-week regimens. For comparison this 8-week regimen of ledipasvir/sofosbuvir has an average wholesale price (AWP) of $28,800, while alternative regimens approved for treatment-naïve patients vary in AWP from $31,680 to $43,200, and regimens approved for retreatment of DAA failures have an AWP as high as $89,712.
An 8-week treatment regimen for both initial and reinfection regimens affords many advantages in medication adherence and both medication and provider resource cost-effectiveness. First, new HCV reinfections are disproportionally younger individuals often with complex psychosocial issues that impact retention in treatment. An 8-week course of treatment can be initiated concurrently with substance abuse treatment programs, including intensive outpatient programs and residential treatment programs that are usually at least 28 days. Many of these programs provide aftercare options that would extend the entire course of treatment. These opportunities afford individuals to receive HCV treatment in a setting that supports medication adherence, sobriety efforts, and education on harm reduction to reduce risk for reinfection.
Finally, statistical models indicate eradication of HCV will require scaling up the treatment of PWID in conjunction with harm reduction strategies such as OST and needle exchange programs.16 In contrast, there are low risks associated with retreatment given these medications are well-tolerated, treatment of PWID lowers the risk of further HCV transmission, and the understanding of these reinfections being treatment naïve disavows concerns of these patients having resistance to regimens that cleared their prior infections. The opportunity to provide retreatment without escalating regimen complexity or cost increases access to care for a vulnerable population while aiding in the eradication of HCV.
1. Centers for Disease Control and Prevention. Viral Hepatitis Surveillance - United States, 2018. Updated August 28, 2020. Accessed May 18, 2021. https://www.cdc.gov/hepatitis/statistics/2018surveillance/HepC.htm 2. Grebely J, Robaeys G, Bruggmann P, et al; International Network for Hepatitis in Substance Users. Recommendations for the management of hepatitis C virus infection among people who inject drugs. Int J Drug Policy. 2015;26(10):1028-1038. doi:10.1016/j.drugpo.2015.07.005
3. Marco A, Esteban JI, Solé C, et al. Hepatitis C virus reinfection among prisoners with sustained virological response after treatment for chronic hepatitis C. J Hepatol. 2013;59(1):45-51. doi:10.1016/j.jhep.2013.03.008
4. Midgard H, Bjøro B, Mæland A, et al. Hepatitis C reinfection after sustained virological response. J Hepatol. 2016;64(5):1020-1026. doi:10.1016/j.jhep.2016.01.001
5. Currie SL, Ryan JC, Tracy D, et al. A prospective study to examine persistent HCV reinfection in injection drug users who have previously cleared the virus [published correction appears in Drug Alcohol Depend. 2008 Jul;96(1-2):192]. Drug Alcohol Depend. 2008;93(1-2):148-154. doi:10.1016/j.drugalcdep.2007.09.011
6. Grady BP, Vanhommerig JW, Schinkel J, et al. Low incidence of reinfection with the hepatitis C virus following treatment in active drug users in Amsterdam. Eur J Gastroenterol Hepatol. 2012;24(11):1302-1307. doi:10.1097/MEG.0b013e32835702a8
7. Grebely J, Pham ST, Matthews GV, et al; ATAHC Study Group. Hepatitis C virus reinfection and superinfection among treated and untreated participants with recent infection. Hepatology. 2012;55(4):1058-1069. doi:10.1002/hep.24754
8. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. HCV Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C. Accessed May 26, 2021. https://www.hcvguidelines.org
9. National Viral Hepatitis Roundtable, Center for Health Law and Policy Innovation, Harvard Law School. Hepatitis C: The State of Medicaid Access. 2017 National Summary Report. Updated October 23, 2017. Accessed May 26, 2021. https://hepcstage.wpengine.com/wp-content/uploads/2017/10/State-of-HepC_2017_FINAL.pdf
10. Singh S, Muir AJ, Dieterich DT, Falck-Ytter YT. American Gastroenterological Association Institute technical review on the role of elastography in chronic liver diseases. Gastroenterology. 2017;152(6):1544-1577. doi:10.1053/j.gastro.2017.03.016
11. Dore GJ, Altice F, Litwin AH, et al; C-EDGE CO-STAR Study Group. Elbasvir-grazoprevir to treat hepatitis C virus infection in persons receiving opioid agonist therapy: a randomized trial. Ann Intern Med. 2016;165(9):625-634. doi:10.7326/M16-0816
12. Grebely J, Dalgard O, Conway B, et al; SIMPLIFY Study Group. Sofosbuvir and velpatasvir for hepatitis C virus infection in people with recent injection drug use (SIMPLIFY): an open-label, single-arm, phase 4, multicentre trial. Lancet Gastroenterol Hepatol. 2018;3(3):153-161. doi:10.1016/S2468-1253(17)30404-1
13. Cos TA, Bartholomew TS, Huynh, KJ. Role of behavioral health providers in treating hepatitis C. Professional Psychol Res Pract. 2019;50(4):246–254. doi:10.1037/pro0000243
14. Latham NH, Doyle JS, Palmer AY, et al. Staying hepatitis C negative: a systematic review and meta-analysis of cure and reinfection in people who inject drugs. Liver Int. 2019;39(12):2244-2260. doi:10.1111/liv.14152
15. Platt L, Minozzi S, Reed J, et al. Needle syringe programmes and opioid substitution therapy for preventing hepatitis C transmission in people who inject drugs. Cochrane Database Syst Rev. 2017;9(9):CD012021. Published 2017 Sep 18. doi:10.1002/14651858.CD012021.pub2
16. Fraser H, Martin NK, Brummer-Korvenkontio H, et al. Model projections on the impact of HCV treatment in the prevention of HCV transmission among people who inject drugs in Europe. J Hepatol. 2018;68(3):402-411. doi:10.1016/j.jhep.2017.10.010
1. Centers for Disease Control and Prevention. Viral Hepatitis Surveillance - United States, 2018. Updated August 28, 2020. Accessed May 18, 2021. https://www.cdc.gov/hepatitis/statistics/2018surveillance/HepC.htm 2. Grebely J, Robaeys G, Bruggmann P, et al; International Network for Hepatitis in Substance Users. Recommendations for the management of hepatitis C virus infection among people who inject drugs. Int J Drug Policy. 2015;26(10):1028-1038. doi:10.1016/j.drugpo.2015.07.005
3. Marco A, Esteban JI, Solé C, et al. Hepatitis C virus reinfection among prisoners with sustained virological response after treatment for chronic hepatitis C. J Hepatol. 2013;59(1):45-51. doi:10.1016/j.jhep.2013.03.008
4. Midgard H, Bjøro B, Mæland A, et al. Hepatitis C reinfection after sustained virological response. J Hepatol. 2016;64(5):1020-1026. doi:10.1016/j.jhep.2016.01.001
5. Currie SL, Ryan JC, Tracy D, et al. A prospective study to examine persistent HCV reinfection in injection drug users who have previously cleared the virus [published correction appears in Drug Alcohol Depend. 2008 Jul;96(1-2):192]. Drug Alcohol Depend. 2008;93(1-2):148-154. doi:10.1016/j.drugalcdep.2007.09.011
6. Grady BP, Vanhommerig JW, Schinkel J, et al. Low incidence of reinfection with the hepatitis C virus following treatment in active drug users in Amsterdam. Eur J Gastroenterol Hepatol. 2012;24(11):1302-1307. doi:10.1097/MEG.0b013e32835702a8
7. Grebely J, Pham ST, Matthews GV, et al; ATAHC Study Group. Hepatitis C virus reinfection and superinfection among treated and untreated participants with recent infection. Hepatology. 2012;55(4):1058-1069. doi:10.1002/hep.24754
8. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. HCV Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C. Accessed May 26, 2021. https://www.hcvguidelines.org
9. National Viral Hepatitis Roundtable, Center for Health Law and Policy Innovation, Harvard Law School. Hepatitis C: The State of Medicaid Access. 2017 National Summary Report. Updated October 23, 2017. Accessed May 26, 2021. https://hepcstage.wpengine.com/wp-content/uploads/2017/10/State-of-HepC_2017_FINAL.pdf
10. Singh S, Muir AJ, Dieterich DT, Falck-Ytter YT. American Gastroenterological Association Institute technical review on the role of elastography in chronic liver diseases. Gastroenterology. 2017;152(6):1544-1577. doi:10.1053/j.gastro.2017.03.016
11. Dore GJ, Altice F, Litwin AH, et al; C-EDGE CO-STAR Study Group. Elbasvir-grazoprevir to treat hepatitis C virus infection in persons receiving opioid agonist therapy: a randomized trial. Ann Intern Med. 2016;165(9):625-634. doi:10.7326/M16-0816
12. Grebely J, Dalgard O, Conway B, et al; SIMPLIFY Study Group. Sofosbuvir and velpatasvir for hepatitis C virus infection in people with recent injection drug use (SIMPLIFY): an open-label, single-arm, phase 4, multicentre trial. Lancet Gastroenterol Hepatol. 2018;3(3):153-161. doi:10.1016/S2468-1253(17)30404-1
13. Cos TA, Bartholomew TS, Huynh, KJ. Role of behavioral health providers in treating hepatitis C. Professional Psychol Res Pract. 2019;50(4):246–254. doi:10.1037/pro0000243
14. Latham NH, Doyle JS, Palmer AY, et al. Staying hepatitis C negative: a systematic review and meta-analysis of cure and reinfection in people who inject drugs. Liver Int. 2019;39(12):2244-2260. doi:10.1111/liv.14152
15. Platt L, Minozzi S, Reed J, et al. Needle syringe programmes and opioid substitution therapy for preventing hepatitis C transmission in people who inject drugs. Cochrane Database Syst Rev. 2017;9(9):CD012021. Published 2017 Sep 18. doi:10.1002/14651858.CD012021.pub2
16. Fraser H, Martin NK, Brummer-Korvenkontio H, et al. Model projections on the impact of HCV treatment in the prevention of HCV transmission among people who inject drugs in Europe. J Hepatol. 2018;68(3):402-411. doi:10.1016/j.jhep.2017.10.010
Thinking Outside the ‘Cage’
A 74-year-old male veteran presented at an urgent care clinic in Aguadilla, Puerto Rico, with a sharp, nonradiating, left-sided precordial chest pain that started while cleaning his house and gardening. The patient described the pain as 9 on the 10-point Wong-Baker FACES Pain Rating Scale, lasting about 5 to 10 minutes and was alleviated with rest. The patient’s medical history consisted of multiple comorbidities, including a mitral valve replacement with a Star-Edwards valve (ball in cage) in 1987. The electrocardiogram performed at the clinic showed no acute ischemic changes. Due to the persistent pain, the patient was transferred to Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, for further evaluation and management. On arrival, the patient had an international normalized ratio (INR) of 2.22; elevated high-sensitive troponin enzyme readings of 56 ng/L at 6:38 PM (0h); 61 ng/L at 7:38 PM (1h); and 83 ng/L at 9:47 PM (3h), reference range, 0-22 ng/L, and changes that prompted admission to the cardiac critical care unit. Two days later, a follow-up enzyme level was 52 ng/L. Cardiac catheterization revealed an acute filling defect at mid-left anterior descending artery and remaining coronary arteries with < 25% atherosclerosis (Figure). A myocardial perfusion study was performed for myocardial viability. The results showed a small, reversible perfusion defect involving the apical-septal wall with the remaining left ventricular myocardium appearing viable. Aspirin was added to the patient’s anticoagulation regimen of warfarin. Once target INR was reached, the patient was discharged home without recurrence of angina.
- What is your diagnosis?
- How would you treat this patient?
Acute coronary syndrome (ACS) consists of clinical suspicion of myocardial ischemia or laboratory confirmation of myocardial infarction (MI). ACS includes 3 major entities: non-ST elevation MI (NSTEMI), unstable angina, and ST-elevation MI (STEMI). ACS usually occurs as a result of a reduced supply of oxygenated blood to the myocardium, which is caused by restriction or occlusion of at least 1 of the coronary arteries. This alteration in blood flow is commonly secondary to a rupture of an atherosclerotic plaque or spontaneous dissection of a coronary artery. In rare cases, this reduction in blood flow is caused by a coronary embolism (CE) arising from a prosthetic heart valve.1,2
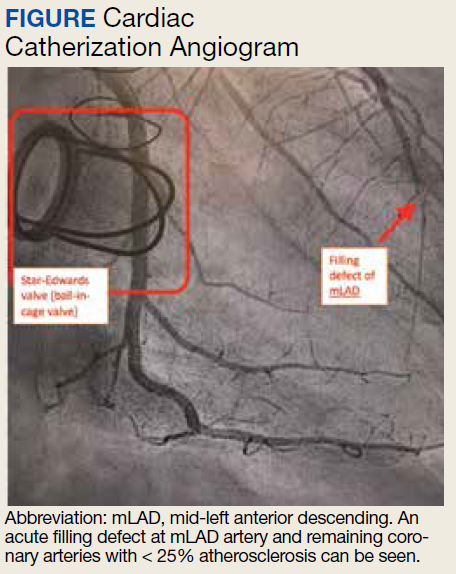
One of the first descriptions of CE was provided by Rudolf Virchow in the 1850s from postmortem autopsy findings.3 At that time, these coronary findings were associated with intracardiac mural thrombus or infective endocarditis. During the 1940s, CE was described in living patients who had survived a MI, and outcomes were not as catastrophic as originally believed. In the 1960s, a higher than usual association between prosthetic valves and CE was suspected and later confirmed by the invention and implementation of coronary angiography. Multiple studies have been published that confirm the association between prosthetic valves (especially in the mitral position), atrial fibrillation (AF), and a higher than usual rate of CEs.4,5
Discussion
The prevalence of this disease has varied during the years. Data from autopsies of patients with ACS and evidence of thromboembolic material in coronary arteries originally estimated a prevalence as high as 13%.6,7 After the invention of diagnostic angiography, consensus studies have established the prevalence to be approximately 3% in patient with ACS.1 The prevalence may be higher in patient with significant risk factors that may increase the probability of CEs, like prosthetic heart valves and AF.2
In 2015 Shibata and colleagues proposed a scoring system for the diagnosis of CE. The scoring system consisted of major and minor criteria.6 Diagnosis of CE is established by ≥ 2 major criteria; 1 major and 2 minor; or ≥ 3 minor criteria. This scoring system increases the diagnostic probability of the disease.1,6
The major criteria are angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components (met by this patient); concomitant coronary emboli in multiple coronary vascular territories; concomitant systemic embolization without left ventricular thrombus attributable to acute MI; histological evidence of venous origin of coronary embolic material; and evidence of an embolic source based on transthoracic echocardiography, transesophageal echocardiography, computed tomography, or magnetic resonance imaging.1,6 The minor criteria are 25% stenosis on coronary angiography except for the culprit lesion (met by this patient); presence of emboli risk factors, such as prosthetic heart valve (met by this patient); and AF.1,6
Management of CE remains controversial; aspiration of thrombus may be considered in the acute setting and with evidence of a heavy thrombus formation. This may allow for restoration of flow and retrieval of thrombus formation for histopathologic evaluation. However, it is important to mention that in the setting of STEMI, aspiration has been shown to increase risk of stroke and lead to increased morbidity. If aspiration of thrombus provides good restoration of flow, there is no need for further percutaneous intervention. Benefits of aspiration in low thrombus burden are not well established and do not provide any additional benefit compared with those of anticoagulation.6-11
Anticoagulation should be initiated in patients with AF and low bleeding risk, even when CHA2DS2-VASc (congestive heart failure, hypertension, aged ≥ 75 years, diabetes mellitus, stroke or transient ischemic attack, vascular disease, aged 65 to 74 years, sex category) score is low. In patients with prolonged immobilization, recent surgery, pregnancy, use of oral contraceptives/tamoxifen, or other reversible risks, 3 months of anticoagulation has been shown to be sufficient. In the setting of active cancer or known thrombophilia, prolonged anticoagulation is recommended. Thrombophilia testing is not recommended in the setting of CE.1
The America College of Cardiology/American Heart Association guidelines for valvular heart disease recommend that patients with mechanical prosthetic aortic valves should be started on a vitamin K antagonist with a target INR of 2 to 3. (Class 1A). Prosthetic mitral and high thromboembolic valves require a higher INR target above 3.0. The addition of antiplatelet agents, such as aspirin in doses of 75 to 100 mg, should be started to decrease risk of thromboembolic disease in all patients with prosthetic heart valves.12
CE is not a common cause of ACS. Nevertheless, it was considered in the differential diagnosis of this patient, and diagnostic criteria were reviewed. This patient met the diagnostic criteria for a definitive diagnosis of CE. These included 1 major and 2 minor criteria: angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components; < 25% stenosis on coronary angiography except for the culprit lesion; and presence of emboli risk factors (prosthetic heart valve).
CE is rare, and review of the literature reveals that it accounts for < 3% of all ACS cases. Despite its rarity, it is important to recognize its risk factors, which include prosthetic heart valves, valvuloplasty, vasculitis, AF, left ventricular aneurysm, and endocarditis. The difference in treatment between CE and the most frequently encountered etiologies of ACS reveals the importance in recognizing this syndrome. Management of CE remains controversial. Nevertheless, when the culprit lesion is located in a distal portion of the vessel involved, as was seen in our patient, and in cases where there is a low thrombi burden, anticoagulation instead of thrombectomy is usually preferred. Patients with prosthetic mechanical valves have a high incidence of thromboembolism. This sometimes leads to thrombi formation in uncommon locations. Guidelines of therapy in these patients recommend that all prosthetic mechanical valves should be treated with both antiplatelet and anticoagulation therapies to reduce the risk of thrombi formation.
Conclusion
Physicians involved in diagnosing ACS should be aware of the risk factors for CE and always consider it while evaluating patients and developing the differential diagnosis.
1. Raphael CE, Heit JA, Reeder GS, et al. Coronary embolus: an underappreciated cause of acute coronary syndromes. JACC Cardiovasc Interv. 2018;11(2):172-180. doi:10.1016/j.jcin.2017.08.057
2. Popovic B, Agrinier N, Bouchahda N, et al. Coronary embolism among ST-segment-elevation myocardial infarction patients: mechanisms and management. Circ Cardiovasc Interv. 2018;11(1):e005587. doi:10.1161/CIRCINTERVENTIONS.117.005587
3. Oakley C, Yusuf R, Hollman A. Coronary embolism and angina in mitral stenosis. Br Heart J. 1961;23(4):357-369. doi:10.1136/hrt.23.4.357
4. Charles RG, Epstein EJ. Diagnosis of coronary embolism: a review. J R Soc Med. 1983;76(10):863-869.
5. Bawell MB, Moragues V, Shrader EL. Coronary embolism. Circulation. 1956;14(6):1159-1163. doi:10.1161/01.cir.14.6.1159
6. Shibata T, Kawakami S, Noguchi T, et al. Prevalence, clinical features, and prognosis of acute myocardial infarction attributable to coronary artery embolism. Circulation. 2015;132(4):241-250. doi:10.1161/CIRCULATIONAHA.114.015134
7. Prizel KR, Hutchins GM, Bulkley BH. Coronary artery embolism and myocardial infarction. Ann Intern Med. 1978;88(2):155-161. doi:10.7326/0003-4819-88-2-155
8. Lacunza-Ruiz FJ, Muñoz-Esparza C, García-de-Lara J. Coronary embolism and thrombosis of prosthetic mitral valve. JACC Cardiovasc Interv. 2014;7(10):e127-e128. doi:10.1016/j.jcin.2014.02.025
9. Jolly SS, Cairns JA, Yusuf S, et al. Outcomes after thrombus aspiration for ST elevation myocardial infarction: 1-year follow-up of the prospective randomised TOTAL trial. Lancet. 2016;387(10014):127-135. doi:10.1016/S0140-6736(15)00448-1
10. Fröbert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction [published correction appears in N Engl J Med. 2014 Aug 21;371(8):786]. N Engl J Med. 2013;369(17):1587-1597. doi:10.1056/NEJMoa1308789
11. Kalçık M, Yesin M, Gürsoy MO, Karakoyun S, Özkan M. Treatment strategies for prosthetic valve thrombosis-derived coronary embolism. JACC Cardiovasc Interv. 2015;8(5):756-757. doi:10.1016/j.jcin.2014.11.019
12. Nishimura RA, Otto CM, Bonow RO, et al. 2017 AHA/ACC focused update of the 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017;135(25):e1159-e1195. doi:10.1161/CIR.0000000000000503
A 74-year-old male veteran presented at an urgent care clinic in Aguadilla, Puerto Rico, with a sharp, nonradiating, left-sided precordial chest pain that started while cleaning his house and gardening. The patient described the pain as 9 on the 10-point Wong-Baker FACES Pain Rating Scale, lasting about 5 to 10 minutes and was alleviated with rest. The patient’s medical history consisted of multiple comorbidities, including a mitral valve replacement with a Star-Edwards valve (ball in cage) in 1987. The electrocardiogram performed at the clinic showed no acute ischemic changes. Due to the persistent pain, the patient was transferred to Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, for further evaluation and management. On arrival, the patient had an international normalized ratio (INR) of 2.22; elevated high-sensitive troponin enzyme readings of 56 ng/L at 6:38 PM (0h); 61 ng/L at 7:38 PM (1h); and 83 ng/L at 9:47 PM (3h), reference range, 0-22 ng/L, and changes that prompted admission to the cardiac critical care unit. Two days later, a follow-up enzyme level was 52 ng/L. Cardiac catheterization revealed an acute filling defect at mid-left anterior descending artery and remaining coronary arteries with < 25% atherosclerosis (Figure). A myocardial perfusion study was performed for myocardial viability. The results showed a small, reversible perfusion defect involving the apical-septal wall with the remaining left ventricular myocardium appearing viable. Aspirin was added to the patient’s anticoagulation regimen of warfarin. Once target INR was reached, the patient was discharged home without recurrence of angina.
- What is your diagnosis?
- How would you treat this patient?
Acute coronary syndrome (ACS) consists of clinical suspicion of myocardial ischemia or laboratory confirmation of myocardial infarction (MI). ACS includes 3 major entities: non-ST elevation MI (NSTEMI), unstable angina, and ST-elevation MI (STEMI). ACS usually occurs as a result of a reduced supply of oxygenated blood to the myocardium, which is caused by restriction or occlusion of at least 1 of the coronary arteries. This alteration in blood flow is commonly secondary to a rupture of an atherosclerotic plaque or spontaneous dissection of a coronary artery. In rare cases, this reduction in blood flow is caused by a coronary embolism (CE) arising from a prosthetic heart valve.1,2
One of the first descriptions of CE was provided by Rudolf Virchow in the 1850s from postmortem autopsy findings.3 At that time, these coronary findings were associated with intracardiac mural thrombus or infective endocarditis. During the 1940s, CE was described in living patients who had survived a MI, and outcomes were not as catastrophic as originally believed. In the 1960s, a higher than usual association between prosthetic valves and CE was suspected and later confirmed by the invention and implementation of coronary angiography. Multiple studies have been published that confirm the association between prosthetic valves (especially in the mitral position), atrial fibrillation (AF), and a higher than usual rate of CEs.4,5
Discussion
The prevalence of this disease has varied during the years. Data from autopsies of patients with ACS and evidence of thromboembolic material in coronary arteries originally estimated a prevalence as high as 13%.6,7 After the invention of diagnostic angiography, consensus studies have established the prevalence to be approximately 3% in patient with ACS.1 The prevalence may be higher in patient with significant risk factors that may increase the probability of CEs, like prosthetic heart valves and AF.2
In 2015 Shibata and colleagues proposed a scoring system for the diagnosis of CE. The scoring system consisted of major and minor criteria.6 Diagnosis of CE is established by ≥ 2 major criteria; 1 major and 2 minor; or ≥ 3 minor criteria. This scoring system increases the diagnostic probability of the disease.1,6
The major criteria are angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components (met by this patient); concomitant coronary emboli in multiple coronary vascular territories; concomitant systemic embolization without left ventricular thrombus attributable to acute MI; histological evidence of venous origin of coronary embolic material; and evidence of an embolic source based on transthoracic echocardiography, transesophageal echocardiography, computed tomography, or magnetic resonance imaging.1,6 The minor criteria are 25% stenosis on coronary angiography except for the culprit lesion (met by this patient); presence of emboli risk factors, such as prosthetic heart valve (met by this patient); and AF.1,6
Management of CE remains controversial; aspiration of thrombus may be considered in the acute setting and with evidence of a heavy thrombus formation. This may allow for restoration of flow and retrieval of thrombus formation for histopathologic evaluation. However, it is important to mention that in the setting of STEMI, aspiration has been shown to increase risk of stroke and lead to increased morbidity. If aspiration of thrombus provides good restoration of flow, there is no need for further percutaneous intervention. Benefits of aspiration in low thrombus burden are not well established and do not provide any additional benefit compared with those of anticoagulation.6-11
Anticoagulation should be initiated in patients with AF and low bleeding risk, even when CHA2DS2-VASc (congestive heart failure, hypertension, aged ≥ 75 years, diabetes mellitus, stroke or transient ischemic attack, vascular disease, aged 65 to 74 years, sex category) score is low. In patients with prolonged immobilization, recent surgery, pregnancy, use of oral contraceptives/tamoxifen, or other reversible risks, 3 months of anticoagulation has been shown to be sufficient. In the setting of active cancer or known thrombophilia, prolonged anticoagulation is recommended. Thrombophilia testing is not recommended in the setting of CE.1
The America College of Cardiology/American Heart Association guidelines for valvular heart disease recommend that patients with mechanical prosthetic aortic valves should be started on a vitamin K antagonist with a target INR of 2 to 3. (Class 1A). Prosthetic mitral and high thromboembolic valves require a higher INR target above 3.0. The addition of antiplatelet agents, such as aspirin in doses of 75 to 100 mg, should be started to decrease risk of thromboembolic disease in all patients with prosthetic heart valves.12
CE is not a common cause of ACS. Nevertheless, it was considered in the differential diagnosis of this patient, and diagnostic criteria were reviewed. This patient met the diagnostic criteria for a definitive diagnosis of CE. These included 1 major and 2 minor criteria: angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components; < 25% stenosis on coronary angiography except for the culprit lesion; and presence of emboli risk factors (prosthetic heart valve).
CE is rare, and review of the literature reveals that it accounts for < 3% of all ACS cases. Despite its rarity, it is important to recognize its risk factors, which include prosthetic heart valves, valvuloplasty, vasculitis, AF, left ventricular aneurysm, and endocarditis. The difference in treatment between CE and the most frequently encountered etiologies of ACS reveals the importance in recognizing this syndrome. Management of CE remains controversial. Nevertheless, when the culprit lesion is located in a distal portion of the vessel involved, as was seen in our patient, and in cases where there is a low thrombi burden, anticoagulation instead of thrombectomy is usually preferred. Patients with prosthetic mechanical valves have a high incidence of thromboembolism. This sometimes leads to thrombi formation in uncommon locations. Guidelines of therapy in these patients recommend that all prosthetic mechanical valves should be treated with both antiplatelet and anticoagulation therapies to reduce the risk of thrombi formation.
Conclusion
Physicians involved in diagnosing ACS should be aware of the risk factors for CE and always consider it while evaluating patients and developing the differential diagnosis.
A 74-year-old male veteran presented at an urgent care clinic in Aguadilla, Puerto Rico, with a sharp, nonradiating, left-sided precordial chest pain that started while cleaning his house and gardening. The patient described the pain as 9 on the 10-point Wong-Baker FACES Pain Rating Scale, lasting about 5 to 10 minutes and was alleviated with rest. The patient’s medical history consisted of multiple comorbidities, including a mitral valve replacement with a Star-Edwards valve (ball in cage) in 1987. The electrocardiogram performed at the clinic showed no acute ischemic changes. Due to the persistent pain, the patient was transferred to Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, for further evaluation and management. On arrival, the patient had an international normalized ratio (INR) of 2.22; elevated high-sensitive troponin enzyme readings of 56 ng/L at 6:38 PM (0h); 61 ng/L at 7:38 PM (1h); and 83 ng/L at 9:47 PM (3h), reference range, 0-22 ng/L, and changes that prompted admission to the cardiac critical care unit. Two days later, a follow-up enzyme level was 52 ng/L. Cardiac catheterization revealed an acute filling defect at mid-left anterior descending artery and remaining coronary arteries with < 25% atherosclerosis (Figure). A myocardial perfusion study was performed for myocardial viability. The results showed a small, reversible perfusion defect involving the apical-septal wall with the remaining left ventricular myocardium appearing viable. Aspirin was added to the patient’s anticoagulation regimen of warfarin. Once target INR was reached, the patient was discharged home without recurrence of angina.
- What is your diagnosis?
- How would you treat this patient?
Acute coronary syndrome (ACS) consists of clinical suspicion of myocardial ischemia or laboratory confirmation of myocardial infarction (MI). ACS includes 3 major entities: non-ST elevation MI (NSTEMI), unstable angina, and ST-elevation MI (STEMI). ACS usually occurs as a result of a reduced supply of oxygenated blood to the myocardium, which is caused by restriction or occlusion of at least 1 of the coronary arteries. This alteration in blood flow is commonly secondary to a rupture of an atherosclerotic plaque or spontaneous dissection of a coronary artery. In rare cases, this reduction in blood flow is caused by a coronary embolism (CE) arising from a prosthetic heart valve.1,2
One of the first descriptions of CE was provided by Rudolf Virchow in the 1850s from postmortem autopsy findings.3 At that time, these coronary findings were associated with intracardiac mural thrombus or infective endocarditis. During the 1940s, CE was described in living patients who had survived a MI, and outcomes were not as catastrophic as originally believed. In the 1960s, a higher than usual association between prosthetic valves and CE was suspected and later confirmed by the invention and implementation of coronary angiography. Multiple studies have been published that confirm the association between prosthetic valves (especially in the mitral position), atrial fibrillation (AF), and a higher than usual rate of CEs.4,5
Discussion
The prevalence of this disease has varied during the years. Data from autopsies of patients with ACS and evidence of thromboembolic material in coronary arteries originally estimated a prevalence as high as 13%.6,7 After the invention of diagnostic angiography, consensus studies have established the prevalence to be approximately 3% in patient with ACS.1 The prevalence may be higher in patient with significant risk factors that may increase the probability of CEs, like prosthetic heart valves and AF.2
In 2015 Shibata and colleagues proposed a scoring system for the diagnosis of CE. The scoring system consisted of major and minor criteria.6 Diagnosis of CE is established by ≥ 2 major criteria; 1 major and 2 minor; or ≥ 3 minor criteria. This scoring system increases the diagnostic probability of the disease.1,6
The major criteria are angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components (met by this patient); concomitant coronary emboli in multiple coronary vascular territories; concomitant systemic embolization without left ventricular thrombus attributable to acute MI; histological evidence of venous origin of coronary embolic material; and evidence of an embolic source based on transthoracic echocardiography, transesophageal echocardiography, computed tomography, or magnetic resonance imaging.1,6 The minor criteria are 25% stenosis on coronary angiography except for the culprit lesion (met by this patient); presence of emboli risk factors, such as prosthetic heart valve (met by this patient); and AF.1,6
Management of CE remains controversial; aspiration of thrombus may be considered in the acute setting and with evidence of a heavy thrombus formation. This may allow for restoration of flow and retrieval of thrombus formation for histopathologic evaluation. However, it is important to mention that in the setting of STEMI, aspiration has been shown to increase risk of stroke and lead to increased morbidity. If aspiration of thrombus provides good restoration of flow, there is no need for further percutaneous intervention. Benefits of aspiration in low thrombus burden are not well established and do not provide any additional benefit compared with those of anticoagulation.6-11
Anticoagulation should be initiated in patients with AF and low bleeding risk, even when CHA2DS2-VASc (congestive heart failure, hypertension, aged ≥ 75 years, diabetes mellitus, stroke or transient ischemic attack, vascular disease, aged 65 to 74 years, sex category) score is low. In patients with prolonged immobilization, recent surgery, pregnancy, use of oral contraceptives/tamoxifen, or other reversible risks, 3 months of anticoagulation has been shown to be sufficient. In the setting of active cancer or known thrombophilia, prolonged anticoagulation is recommended. Thrombophilia testing is not recommended in the setting of CE.1
The America College of Cardiology/American Heart Association guidelines for valvular heart disease recommend that patients with mechanical prosthetic aortic valves should be started on a vitamin K antagonist with a target INR of 2 to 3. (Class 1A). Prosthetic mitral and high thromboembolic valves require a higher INR target above 3.0. The addition of antiplatelet agents, such as aspirin in doses of 75 to 100 mg, should be started to decrease risk of thromboembolic disease in all patients with prosthetic heart valves.12
CE is not a common cause of ACS. Nevertheless, it was considered in the differential diagnosis of this patient, and diagnostic criteria were reviewed. This patient met the diagnostic criteria for a definitive diagnosis of CE. These included 1 major and 2 minor criteria: angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components; < 25% stenosis on coronary angiography except for the culprit lesion; and presence of emboli risk factors (prosthetic heart valve).
CE is rare, and review of the literature reveals that it accounts for < 3% of all ACS cases. Despite its rarity, it is important to recognize its risk factors, which include prosthetic heart valves, valvuloplasty, vasculitis, AF, left ventricular aneurysm, and endocarditis. The difference in treatment between CE and the most frequently encountered etiologies of ACS reveals the importance in recognizing this syndrome. Management of CE remains controversial. Nevertheless, when the culprit lesion is located in a distal portion of the vessel involved, as was seen in our patient, and in cases where there is a low thrombi burden, anticoagulation instead of thrombectomy is usually preferred. Patients with prosthetic mechanical valves have a high incidence of thromboembolism. This sometimes leads to thrombi formation in uncommon locations. Guidelines of therapy in these patients recommend that all prosthetic mechanical valves should be treated with both antiplatelet and anticoagulation therapies to reduce the risk of thrombi formation.
Conclusion
Physicians involved in diagnosing ACS should be aware of the risk factors for CE and always consider it while evaluating patients and developing the differential diagnosis.
1. Raphael CE, Heit JA, Reeder GS, et al. Coronary embolus: an underappreciated cause of acute coronary syndromes. JACC Cardiovasc Interv. 2018;11(2):172-180. doi:10.1016/j.jcin.2017.08.057
2. Popovic B, Agrinier N, Bouchahda N, et al. Coronary embolism among ST-segment-elevation myocardial infarction patients: mechanisms and management. Circ Cardiovasc Interv. 2018;11(1):e005587. doi:10.1161/CIRCINTERVENTIONS.117.005587
3. Oakley C, Yusuf R, Hollman A. Coronary embolism and angina in mitral stenosis. Br Heart J. 1961;23(4):357-369. doi:10.1136/hrt.23.4.357
4. Charles RG, Epstein EJ. Diagnosis of coronary embolism: a review. J R Soc Med. 1983;76(10):863-869.
5. Bawell MB, Moragues V, Shrader EL. Coronary embolism. Circulation. 1956;14(6):1159-1163. doi:10.1161/01.cir.14.6.1159
6. Shibata T, Kawakami S, Noguchi T, et al. Prevalence, clinical features, and prognosis of acute myocardial infarction attributable to coronary artery embolism. Circulation. 2015;132(4):241-250. doi:10.1161/CIRCULATIONAHA.114.015134
7. Prizel KR, Hutchins GM, Bulkley BH. Coronary artery embolism and myocardial infarction. Ann Intern Med. 1978;88(2):155-161. doi:10.7326/0003-4819-88-2-155
8. Lacunza-Ruiz FJ, Muñoz-Esparza C, García-de-Lara J. Coronary embolism and thrombosis of prosthetic mitral valve. JACC Cardiovasc Interv. 2014;7(10):e127-e128. doi:10.1016/j.jcin.2014.02.025
9. Jolly SS, Cairns JA, Yusuf S, et al. Outcomes after thrombus aspiration for ST elevation myocardial infarction: 1-year follow-up of the prospective randomised TOTAL trial. Lancet. 2016;387(10014):127-135. doi:10.1016/S0140-6736(15)00448-1
10. Fröbert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction [published correction appears in N Engl J Med. 2014 Aug 21;371(8):786]. N Engl J Med. 2013;369(17):1587-1597. doi:10.1056/NEJMoa1308789
11. Kalçık M, Yesin M, Gürsoy MO, Karakoyun S, Özkan M. Treatment strategies for prosthetic valve thrombosis-derived coronary embolism. JACC Cardiovasc Interv. 2015;8(5):756-757. doi:10.1016/j.jcin.2014.11.019
12. Nishimura RA, Otto CM, Bonow RO, et al. 2017 AHA/ACC focused update of the 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017;135(25):e1159-e1195. doi:10.1161/CIR.0000000000000503
1. Raphael CE, Heit JA, Reeder GS, et al. Coronary embolus: an underappreciated cause of acute coronary syndromes. JACC Cardiovasc Interv. 2018;11(2):172-180. doi:10.1016/j.jcin.2017.08.057
2. Popovic B, Agrinier N, Bouchahda N, et al. Coronary embolism among ST-segment-elevation myocardial infarction patients: mechanisms and management. Circ Cardiovasc Interv. 2018;11(1):e005587. doi:10.1161/CIRCINTERVENTIONS.117.005587
3. Oakley C, Yusuf R, Hollman A. Coronary embolism and angina in mitral stenosis. Br Heart J. 1961;23(4):357-369. doi:10.1136/hrt.23.4.357
4. Charles RG, Epstein EJ. Diagnosis of coronary embolism: a review. J R Soc Med. 1983;76(10):863-869.
5. Bawell MB, Moragues V, Shrader EL. Coronary embolism. Circulation. 1956;14(6):1159-1163. doi:10.1161/01.cir.14.6.1159
6. Shibata T, Kawakami S, Noguchi T, et al. Prevalence, clinical features, and prognosis of acute myocardial infarction attributable to coronary artery embolism. Circulation. 2015;132(4):241-250. doi:10.1161/CIRCULATIONAHA.114.015134
7. Prizel KR, Hutchins GM, Bulkley BH. Coronary artery embolism and myocardial infarction. Ann Intern Med. 1978;88(2):155-161. doi:10.7326/0003-4819-88-2-155
8. Lacunza-Ruiz FJ, Muñoz-Esparza C, García-de-Lara J. Coronary embolism and thrombosis of prosthetic mitral valve. JACC Cardiovasc Interv. 2014;7(10):e127-e128. doi:10.1016/j.jcin.2014.02.025
9. Jolly SS, Cairns JA, Yusuf S, et al. Outcomes after thrombus aspiration for ST elevation myocardial infarction: 1-year follow-up of the prospective randomised TOTAL trial. Lancet. 2016;387(10014):127-135. doi:10.1016/S0140-6736(15)00448-1
10. Fröbert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction [published correction appears in N Engl J Med. 2014 Aug 21;371(8):786]. N Engl J Med. 2013;369(17):1587-1597. doi:10.1056/NEJMoa1308789
11. Kalçık M, Yesin M, Gürsoy MO, Karakoyun S, Özkan M. Treatment strategies for prosthetic valve thrombosis-derived coronary embolism. JACC Cardiovasc Interv. 2015;8(5):756-757. doi:10.1016/j.jcin.2014.11.019
12. Nishimura RA, Otto CM, Bonow RO, et al. 2017 AHA/ACC focused update of the 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017;135(25):e1159-e1195. doi:10.1161/CIR.0000000000000503
Left-Sided Amyand Hernia: Case Report and Review of the Literature
Left-sided Amyand hernia is a rare condition that requires a high degree of clinical suspicion to correctly diagnose.
The presence of the vermiform appendix within an inguinal hernia sac is termed an Amyand hernia. While the incidence of Amyand hernia in the general population is thought to be exceedingly rare, the presence of a left-sided Amyand hernia is even more rare due to the normal anatomical position of the appendix on the right side. Left-sided Amyand hernia presents a novel diagnosis that necessitates a high degree of clinical suspicion and special consideration during patient workup and operative treatment. We describe such a case and provide a review of all reports in the literature of this rare finding.
Case Presentation
A male aged 62 years presented to the emergency department of the Michael E. DeBakey Veterans Affairs Medical Center in Houston, Texas, in acute distress after experiencing 5 days of nausea and pain in his lower abdomen. The patient’s history was significant for cocaine abuse and a left-sided inguinal hernia that was repaired about 15 years prior to this visit. He reported having no bowel movements for the past 5 days and no other symptoms, including vomiting, hematemesis, and trauma to the abdomen. The patient’s abdominal pain was located in the suprapubic and periumbilical regions. Upon palpation of the lower abdomen, a firm, protruding mass was identified in the left lower quadrant and suspected to be a left-sided inguinal hernia.
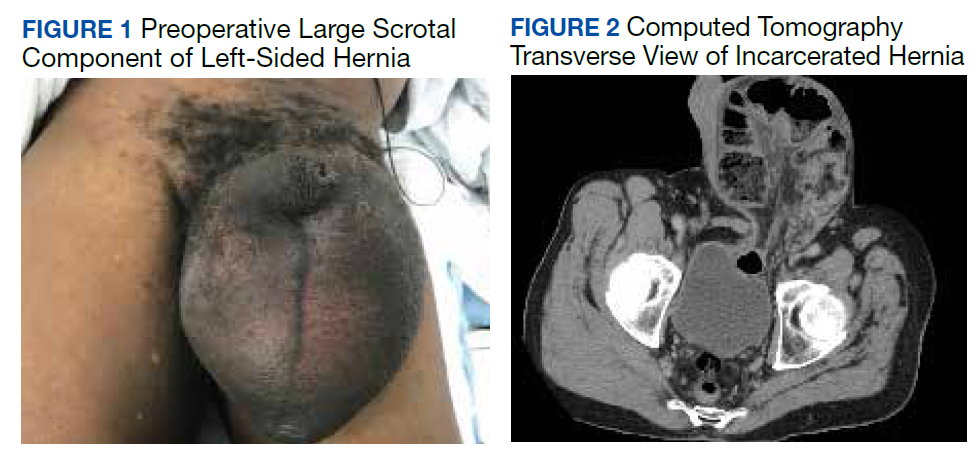
A scout film and computed tomography (CT) scan of the abdomen taken on the same day that the patient presented to the emergency department confirmed the presence of a large left-sided inguinal hernia with possible bowel strangulation involving the colon (Figures 1, 2, and 3). The patient was diagnosed with an incarcerated recurrent left inguinal hernia and was taken emergently to the operating suite. General anesthesia and an ilioinguinal nerve block were performed. An inguinal incision was made on the left side, and the large hernia sac was identified and separated from the scrotum and spermatic cord structures.
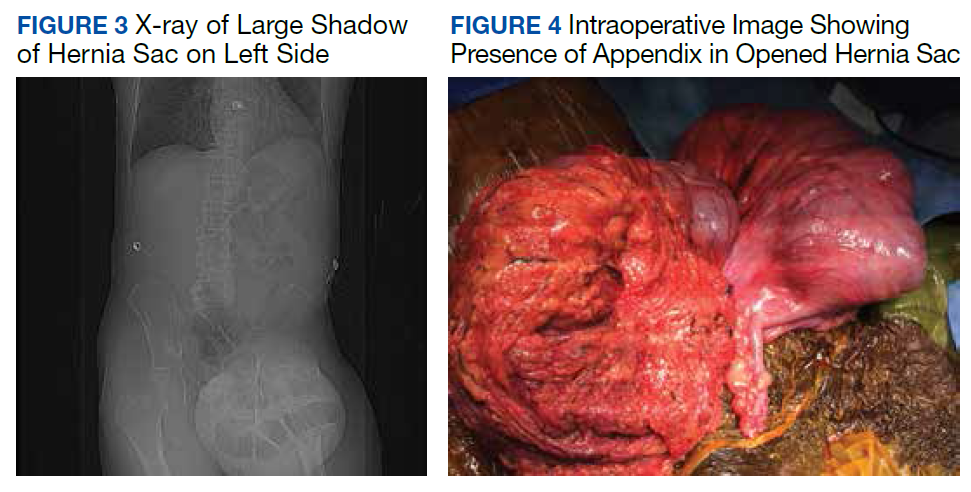
On visual inspection, the hernia was identified as both a direct and an indirect inguinal hernia, making it a pantaloon hernia. The hernia sac was opened, and contents of the herniated sac were found to include the omentum, a loop of transverse colon, as well as the entire cecum and appendix, confirming the diagnosis of an Amyand hernia (Figure 4). Though the bowel was initially dusky, all the bowel became pink and appeared to be viable after detorsion of the bowel. Diagnostic laparoscopy through a 5-mm port was performed to assess the remainder of the bowel located intra-abdominally. The remaining intra-abdominal bowel appeared healthy and without obvious signs of ischemia, twisting, or malrotation. The large hernia defect was repaired with a polypropylene mesh.
Discussion
An Amyand hernia is an inguinal hernia in which the vermiform appendix is located within the hernial sac. Named after the French surgeon Claudius Amyand who first documented such a case during an appendectomy in 1735, the Amyand hernia is rare and is thought to occur in < 1% of inguinal hernias.1 Given the normal anatomical position of the appendix on the right side of the body, most Amyand hernias occur in a right-sided inguinal hernia.
A literature review yielded 25 reported instances of a left-sided Amyand hernia (Table 1) including this report. The true age of incidence of Amyand hernia for each patient is difficult to determine, as many patients do not present until pain or discomfort reaches high levels, often many years after hernia formation. Additionally, some cases of left-sided Amyand hernia described herein, including our case, are recurrent cases of a previous hernia that have been surgically repaired.2-20
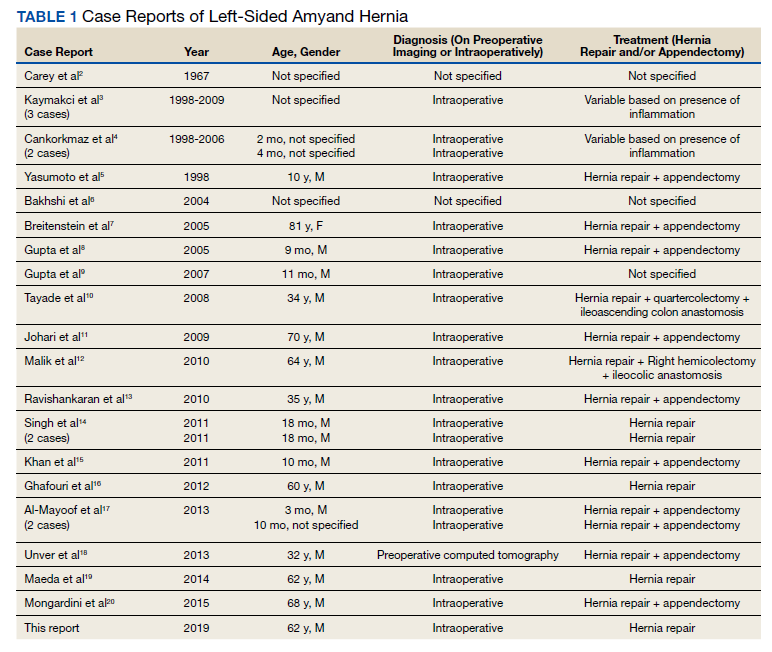
Presentation of Amyand hernia often resembles that of a complicated inguinal hernia, acute appendicitis, or both. Hence, clinicians should consider this a possibility when patients present with signs and symptoms that could otherwise be thought to be originating from an incarcerated, strangulated, or recurrent hernia. Specifically, these signs and symptoms include a tender, nonreducible mass in the inguinal region, acute lower abdominal pain, nausea, or signs of intestinal obstruction such as failure to produce bowel movements.4,17 Because of the unusual anatomy in patients presenting with left-sided Amyand hernia, tenderness at the McBurney point usually is absent and not a useful diagnostic tool to rule out acute appendicitis.
A literature review indicates that an Amyand hernia on either side tends to occur in males more often than it does in females. The rate of diagnosis of Amyand hernia also has been reported to be 3 times higher in children than it is in adults due to failure of the processus vaginalis to obliterate during development.21 Our literature review supports this finding, as 16 of the documented 25 cases of left-sided Amyand hernia were reported in males. Additionally, information regarding gender was not found in 6 cases, suggesting a potential for an even higher prevalence in males.
Explanations as to why the appendix is on the left side in these patients include developmental anomalies, such as situs inversus, intestinal rotation, mobile cecum, or an abnormally long appendix.3,8 In our case, the likely causative culprit was a mobile cecum, as there was neither indication of intestinal malformation, rotation, nor of an abnormally long appendix during surgery. Additionally, pre-operative radiologic studies, clinical evaluation, and electrocardiogram did not suggest the presence of situs inversus.
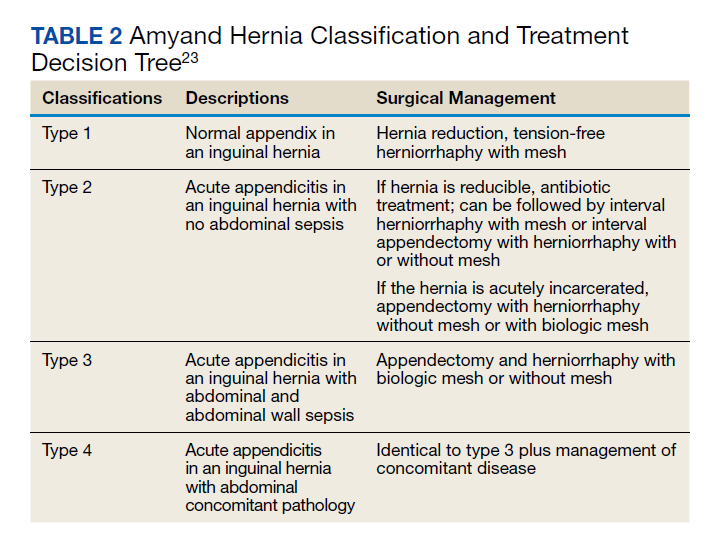
Treatment of Amyand hernia usually follows the landmark classification algorithm set forth in 2007 by Losanoff and Basson (Table 2).22 This system stratifies treatment based on intraoperative findings of the appendix and surrounding structures, ranging from type 1, which involves a normal appendix within the hernia, to type 4, which includes acute appendicitis with additional abdominal pathology. Our patient presented with a type 1 Amyand hernia and appendectomy was foregone as per the guidelines; however, there have been numerous reported cases of surgeons opting for prophylactic appendectomy in the case of a normal appearing appendix and surrounding structures. The decision to act independent of the Losanoff and Basson classification underscores the lack of true standardization, namely, when it comes to a treatment approach for type 1 Amyand hernias. Nonetheless, many contend that indiscriminately performing appendectomies in all cases of left-sided Amyand hernia is useful as a prophylactic measure, as cases of future appendicitis in these patients will have atypical presentations based on the contralateral location of the appendix.6,11,17
Others disagree, citing that prophylactic appendectomy in the case of a normal looking appendix is unnecessary and complicates an otherwise sterile surgery (clean wound classification) with the removal of an appendix containing fecal matter and gut microbiota (converted into a clean contaminated or a contaminated wound classification).17 Additionally, it is thought that in the cases of middle-aged or geriatric patients where the chances of appendicitis are far less, the risks of detriment from prophylactic appendectomy may outweigh the benefits. In these cases, a macroscopic view of the appendix based on visual examination during the operation should guide decision making.4
While the decision to remove a healthy-appearing appendix remains contentious, the decision for or against placement of a heterogenous hernia mesh has proven to be binary, with near universally accepted criteria. If signs of perforation or infection are present in the hernia sac, then surgeons will forego hernioplasty with mesh for simple herniorrhaphy. This contraindication for mesh placement is due to the increased risk of mesh infection, wound infection, and fistulae associated with the introduction of a foreign structure to an active infection site.2
While most cases of Amyand hernia are diagnosed intraoperatively, there have been documented cases of preoperative diagnosis using ultrasonography and CT imaging modalities.19,23,24 In all cases, the presence of the vermiform appendix within the hernia sac can complicate diagnosis and treatment, and preoperative knowledge of this condition may help to guide physician decision making. Identifying Amyand hernia via CT scan is not only useful for alerting physicians of a potentially inflamed appendix within the hernia sac, but also may create opportunities for the use of other treatment modalities. For example, laparoscopic Amyand hernia reduction, an approach that was performed successfully and documented for the first time by Vermillion and colleagues, was made possible by preoperative diagnosis and can potentially result in improved patient outcomes.25
Regardless, while standardization of treatment for Amyand hernia has not yet occurred, it is clear that improved preoperative diagnosis, especially in the case of an unanticipated left-sided Amyand hernia, can allow for better planning and use of a wider variety of treatment modalities. The main impediment to this approach is that suspected cases of appendicitis and inguinal hernias (the most common preoperative diagnoses of Amyand hernia) usually are diagnosed clinically without the need of additional imaging studies like CT or ultrasound. In accordance with the guiding principle of radiation safety of exposing patients to “as low as reasonably achievable” (ALARA) radiation and with consideration of expediency of care and cost efficiency, we recommend physicians continue to screen for and treat cases of potentially emergent appendicitis and/or inguinal hernia as per the conventional methodology. The best approach may involve increasing preoperative diagnoses of left-sided Amyand hernias via physician awareness of this rare finding, as well as evaluating imaging studies that have previously been obtained in order to narrow a broad differential diagnosis.
Conclusions
Left-sided Amyand hernia is an exceptionally rare condition whose preoperative diagnosis remains difficult to establish but whose treatment decision tree is significantly impacted by the condition.
1. Franko J, Raftopoulos I, Sulkowski R. A rare variation of Amyand’s hernia. Am J Gastroenterol. 2002;97(10):2684-2685. doi:10.1111/j.1572-0241.2002.06060.x
2. Carey LC. Acute appendicitis occurring in hernias: a report of 10 cases. Surgery. 1967;61(2):236-238.
3. Kaymakci A, Akillioglu I, Akkoyun I, Guven S, Ozdemir A, Gulen S. Amyand’s hernia: a series of 30 cases in children. Hernia. 2009;13(6):609-612. doi:10.1007/s10029-009-0528-8
4. Cankorkmaz L, Ozer H, Guney C, Atalar MH, Arslan MS, Koyluoglu G. Amyand’s hernia in the children: a single center experience. Surgery. 2010;147(1):140-143. doi:10.1016/j.surg.2009.09.038
5. Yasumoto R, Kawano M, Kawanishi H, et al. Left acute scrotum associated with appendicitis. Int J Urol. 1998;5(1):108-110. doi:10.1111/j.1442-2042.1998.tb00254.x
6. Bakhshi GD, Bhandarwar AH, Govila AA. Acute appendicitis in left scrotum. Indian J Gastroenterol. 2004;23(5):195.
7. Breitenstein S, Eisenbach C, Wille G, Decurtins M. Incarcerated vermiform appendix in a left-sided inguinal hernia. Hernia. 2005;9(1):100-102. doi:10.1007/s10029-004-0263-0
8. Gupta S, Sharma R, Kaushik R. Left-sided Amyand’s hernia. Singapore Med J. 2005;46(8):424-425.
9. Gupta N, Wilkinson TV, Wilkinson A, Akhtar M. Left-sided incarcerated Amyand’s hernia. Indian J Surg. 2007;69(1):17-18.
10. Tayade, MB, Bakhshi GD, Borisa AD, Deshpande G, Joshi N. A rare combination of left sided Amyand’s and Richter’s hernia. Bombay Hosp J. 2008;50(4): 644-645
11. Johari HG, Paydar S, Davani SZ, Eskandari S, Johari MG. Left-sided Amyand hernia. Ann Saudi Med. 2009;29(4):321-322. doi:10.4103/0256-4947.55305
12. Ali SM, Malik KA, Al-Qadhi H. Amyand’s Hernia: Study of four cases and literature review. Sultan Qaboos Univ Med J. 2012;12(2):232-236. doi:10.12816/0003119
13. Ravishankaran P, Mohan G, Srinivasan A, Ravindran G, Ramalingam A. Left sided amyand’s hernia, a rare occurrence: A Case Report. Indian J Surg. 2013;75(3):247-248. doi:10.1007/s12262-010-0223-0
14. Singh K, Singh RR, Kaur S. Amyand’s hernia. J Indian Assoc Pediatr Surg. 2011;16(4):171-172. doi:10.4103/0971-9261.86890
15. Khan TS, Wani ML, Bijli AH, et al. Amyand’s hernia: a rare occurrence. Ann Nigerian Med. 2011;5(2):62-64.doi:10.4103/0331-3131.92955
16. Ghafouri A, Anbara T, Foroutankia R. A rare case report of appendix and cecum in the sac of left inguinal hernia (left Amyand’s hernia). Med J Islam Repub Iran. 2012;26(2):94-95.
17. Al-Mayoof AF, Al-Ani BH. Left-sided amyand hernia: report of two cases with review of literature. European J Pediatr Surg Rep. 2014;2(1):63-66. doi:10.1055/s-0033-1347131
18. Unver M, Ozturk S, Karaman K, Turgut E. Left sided Amyand’s hernia. World J Gastrointest Surg. 2013;5(10):285-286. doi:10.4240/wjgs.v5.i10.285
19. Maeda K, Kunieda K, Kawai M, et al. Giant left-sided inguinoscrotal hernia containing the cecum and appendix (giant left-sided Amyand’s hernia). Clin Case Rep. 2014;2(6):254-257. doi:10.1002/ccr3.104
20. Mongardini M, Maturo A, De Anna L, et al. Appendiceal abscess in a giant left-sided inguinoscrotal hernia: a rare case of Amyand hernia. Springerplus. 2015;4:378. Published 2015 Jul 26. doi:10.1186/s40064-015-1162-9
21. Ivanschuk G, Cesmebasi A, Sorenson EP, Blaak C, Loukas M, Tubbs SR. Amyand’s hernia: a review. Med Sci Monit. 2014;20:140-146. Published 2014 Jan 28. doi:10.12659/MSM.889873
22. Losanoff JE, Basson MD. Amyand hernia: what lies beneath--a proposed classification scheme to determine management. Am Surg. 2007;73(12):1288-1290.
23. Coulier B, Pacary J, Broze B. Sonographic diagnosis of appendicitis within a right inguinal hernia (Amyand’s hernia). J Clin Ultrasound. 2006;34(9):454-457. doi:10.1002/jcu.20266
24. Vehbi H, Agirgun C, Agirgun F, Dogan Y. Preoperative diagnosis of Amyand’s hernia by ultrasound and computed tomography. Turk J Emerg Med. 2016;16(2):72-74. Published 2016 May 8. doi:10.1016/j.tjem.2015.11.014
25. Vermillion JM, Abernathy SW, Snyder SK. Laparoscopic reduction of Amyand’s hernia. Hernia. 1999;3:159-160. doi:10.1007/BF01195318
Left-sided Amyand hernia is a rare condition that requires a high degree of clinical suspicion to correctly diagnose.
Left-sided Amyand hernia is a rare condition that requires a high degree of clinical suspicion to correctly diagnose.
The presence of the vermiform appendix within an inguinal hernia sac is termed an Amyand hernia. While the incidence of Amyand hernia in the general population is thought to be exceedingly rare, the presence of a left-sided Amyand hernia is even more rare due to the normal anatomical position of the appendix on the right side. Left-sided Amyand hernia presents a novel diagnosis that necessitates a high degree of clinical suspicion and special consideration during patient workup and operative treatment. We describe such a case and provide a review of all reports in the literature of this rare finding.
Case Presentation
A male aged 62 years presented to the emergency department of the Michael E. DeBakey Veterans Affairs Medical Center in Houston, Texas, in acute distress after experiencing 5 days of nausea and pain in his lower abdomen. The patient’s history was significant for cocaine abuse and a left-sided inguinal hernia that was repaired about 15 years prior to this visit. He reported having no bowel movements for the past 5 days and no other symptoms, including vomiting, hematemesis, and trauma to the abdomen. The patient’s abdominal pain was located in the suprapubic and periumbilical regions. Upon palpation of the lower abdomen, a firm, protruding mass was identified in the left lower quadrant and suspected to be a left-sided inguinal hernia.
A scout film and computed tomography (CT) scan of the abdomen taken on the same day that the patient presented to the emergency department confirmed the presence of a large left-sided inguinal hernia with possible bowel strangulation involving the colon (Figures 1, 2, and 3). The patient was diagnosed with an incarcerated recurrent left inguinal hernia and was taken emergently to the operating suite. General anesthesia and an ilioinguinal nerve block were performed. An inguinal incision was made on the left side, and the large hernia sac was identified and separated from the scrotum and spermatic cord structures.
On visual inspection, the hernia was identified as both a direct and an indirect inguinal hernia, making it a pantaloon hernia. The hernia sac was opened, and contents of the herniated sac were found to include the omentum, a loop of transverse colon, as well as the entire cecum and appendix, confirming the diagnosis of an Amyand hernia (Figure 4). Though the bowel was initially dusky, all the bowel became pink and appeared to be viable after detorsion of the bowel. Diagnostic laparoscopy through a 5-mm port was performed to assess the remainder of the bowel located intra-abdominally. The remaining intra-abdominal bowel appeared healthy and without obvious signs of ischemia, twisting, or malrotation. The large hernia defect was repaired with a polypropylene mesh.
Discussion
An Amyand hernia is an inguinal hernia in which the vermiform appendix is located within the hernial sac. Named after the French surgeon Claudius Amyand who first documented such a case during an appendectomy in 1735, the Amyand hernia is rare and is thought to occur in < 1% of inguinal hernias.1 Given the normal anatomical position of the appendix on the right side of the body, most Amyand hernias occur in a right-sided inguinal hernia.
A literature review yielded 25 reported instances of a left-sided Amyand hernia (Table 1) including this report. The true age of incidence of Amyand hernia for each patient is difficult to determine, as many patients do not present until pain or discomfort reaches high levels, often many years after hernia formation. Additionally, some cases of left-sided Amyand hernia described herein, including our case, are recurrent cases of a previous hernia that have been surgically repaired.2-20
Presentation of Amyand hernia often resembles that of a complicated inguinal hernia, acute appendicitis, or both. Hence, clinicians should consider this a possibility when patients present with signs and symptoms that could otherwise be thought to be originating from an incarcerated, strangulated, or recurrent hernia. Specifically, these signs and symptoms include a tender, nonreducible mass in the inguinal region, acute lower abdominal pain, nausea, or signs of intestinal obstruction such as failure to produce bowel movements.4,17 Because of the unusual anatomy in patients presenting with left-sided Amyand hernia, tenderness at the McBurney point usually is absent and not a useful diagnostic tool to rule out acute appendicitis.
A literature review indicates that an Amyand hernia on either side tends to occur in males more often than it does in females. The rate of diagnosis of Amyand hernia also has been reported to be 3 times higher in children than it is in adults due to failure of the processus vaginalis to obliterate during development.21 Our literature review supports this finding, as 16 of the documented 25 cases of left-sided Amyand hernia were reported in males. Additionally, information regarding gender was not found in 6 cases, suggesting a potential for an even higher prevalence in males.
Explanations as to why the appendix is on the left side in these patients include developmental anomalies, such as situs inversus, intestinal rotation, mobile cecum, or an abnormally long appendix.3,8 In our case, the likely causative culprit was a mobile cecum, as there was neither indication of intestinal malformation, rotation, nor of an abnormally long appendix during surgery. Additionally, pre-operative radiologic studies, clinical evaluation, and electrocardiogram did not suggest the presence of situs inversus.
Treatment of Amyand hernia usually follows the landmark classification algorithm set forth in 2007 by Losanoff and Basson (Table 2).22 This system stratifies treatment based on intraoperative findings of the appendix and surrounding structures, ranging from type 1, which involves a normal appendix within the hernia, to type 4, which includes acute appendicitis with additional abdominal pathology. Our patient presented with a type 1 Amyand hernia and appendectomy was foregone as per the guidelines; however, there have been numerous reported cases of surgeons opting for prophylactic appendectomy in the case of a normal appearing appendix and surrounding structures. The decision to act independent of the Losanoff and Basson classification underscores the lack of true standardization, namely, when it comes to a treatment approach for type 1 Amyand hernias. Nonetheless, many contend that indiscriminately performing appendectomies in all cases of left-sided Amyand hernia is useful as a prophylactic measure, as cases of future appendicitis in these patients will have atypical presentations based on the contralateral location of the appendix.6,11,17
Others disagree, citing that prophylactic appendectomy in the case of a normal looking appendix is unnecessary and complicates an otherwise sterile surgery (clean wound classification) with the removal of an appendix containing fecal matter and gut microbiota (converted into a clean contaminated or a contaminated wound classification).17 Additionally, it is thought that in the cases of middle-aged or geriatric patients where the chances of appendicitis are far less, the risks of detriment from prophylactic appendectomy may outweigh the benefits. In these cases, a macroscopic view of the appendix based on visual examination during the operation should guide decision making.4
While the decision to remove a healthy-appearing appendix remains contentious, the decision for or against placement of a heterogenous hernia mesh has proven to be binary, with near universally accepted criteria. If signs of perforation or infection are present in the hernia sac, then surgeons will forego hernioplasty with mesh for simple herniorrhaphy. This contraindication for mesh placement is due to the increased risk of mesh infection, wound infection, and fistulae associated with the introduction of a foreign structure to an active infection site.2
While most cases of Amyand hernia are diagnosed intraoperatively, there have been documented cases of preoperative diagnosis using ultrasonography and CT imaging modalities.19,23,24 In all cases, the presence of the vermiform appendix within the hernia sac can complicate diagnosis and treatment, and preoperative knowledge of this condition may help to guide physician decision making. Identifying Amyand hernia via CT scan is not only useful for alerting physicians of a potentially inflamed appendix within the hernia sac, but also may create opportunities for the use of other treatment modalities. For example, laparoscopic Amyand hernia reduction, an approach that was performed successfully and documented for the first time by Vermillion and colleagues, was made possible by preoperative diagnosis and can potentially result in improved patient outcomes.25
Regardless, while standardization of treatment for Amyand hernia has not yet occurred, it is clear that improved preoperative diagnosis, especially in the case of an unanticipated left-sided Amyand hernia, can allow for better planning and use of a wider variety of treatment modalities. The main impediment to this approach is that suspected cases of appendicitis and inguinal hernias (the most common preoperative diagnoses of Amyand hernia) usually are diagnosed clinically without the need of additional imaging studies like CT or ultrasound. In accordance with the guiding principle of radiation safety of exposing patients to “as low as reasonably achievable” (ALARA) radiation and with consideration of expediency of care and cost efficiency, we recommend physicians continue to screen for and treat cases of potentially emergent appendicitis and/or inguinal hernia as per the conventional methodology. The best approach may involve increasing preoperative diagnoses of left-sided Amyand hernias via physician awareness of this rare finding, as well as evaluating imaging studies that have previously been obtained in order to narrow a broad differential diagnosis.
Conclusions
Left-sided Amyand hernia is an exceptionally rare condition whose preoperative diagnosis remains difficult to establish but whose treatment decision tree is significantly impacted by the condition.
The presence of the vermiform appendix within an inguinal hernia sac is termed an Amyand hernia. While the incidence of Amyand hernia in the general population is thought to be exceedingly rare, the presence of a left-sided Amyand hernia is even more rare due to the normal anatomical position of the appendix on the right side. Left-sided Amyand hernia presents a novel diagnosis that necessitates a high degree of clinical suspicion and special consideration during patient workup and operative treatment. We describe such a case and provide a review of all reports in the literature of this rare finding.
Case Presentation
A male aged 62 years presented to the emergency department of the Michael E. DeBakey Veterans Affairs Medical Center in Houston, Texas, in acute distress after experiencing 5 days of nausea and pain in his lower abdomen. The patient’s history was significant for cocaine abuse and a left-sided inguinal hernia that was repaired about 15 years prior to this visit. He reported having no bowel movements for the past 5 days and no other symptoms, including vomiting, hematemesis, and trauma to the abdomen. The patient’s abdominal pain was located in the suprapubic and periumbilical regions. Upon palpation of the lower abdomen, a firm, protruding mass was identified in the left lower quadrant and suspected to be a left-sided inguinal hernia.
A scout film and computed tomography (CT) scan of the abdomen taken on the same day that the patient presented to the emergency department confirmed the presence of a large left-sided inguinal hernia with possible bowel strangulation involving the colon (Figures 1, 2, and 3). The patient was diagnosed with an incarcerated recurrent left inguinal hernia and was taken emergently to the operating suite. General anesthesia and an ilioinguinal nerve block were performed. An inguinal incision was made on the left side, and the large hernia sac was identified and separated from the scrotum and spermatic cord structures.
On visual inspection, the hernia was identified as both a direct and an indirect inguinal hernia, making it a pantaloon hernia. The hernia sac was opened, and contents of the herniated sac were found to include the omentum, a loop of transverse colon, as well as the entire cecum and appendix, confirming the diagnosis of an Amyand hernia (Figure 4). Though the bowel was initially dusky, all the bowel became pink and appeared to be viable after detorsion of the bowel. Diagnostic laparoscopy through a 5-mm port was performed to assess the remainder of the bowel located intra-abdominally. The remaining intra-abdominal bowel appeared healthy and without obvious signs of ischemia, twisting, or malrotation. The large hernia defect was repaired with a polypropylene mesh.
Discussion
An Amyand hernia is an inguinal hernia in which the vermiform appendix is located within the hernial sac. Named after the French surgeon Claudius Amyand who first documented such a case during an appendectomy in 1735, the Amyand hernia is rare and is thought to occur in < 1% of inguinal hernias.1 Given the normal anatomical position of the appendix on the right side of the body, most Amyand hernias occur in a right-sided inguinal hernia.
A literature review yielded 25 reported instances of a left-sided Amyand hernia (Table 1) including this report. The true age of incidence of Amyand hernia for each patient is difficult to determine, as many patients do not present until pain or discomfort reaches high levels, often many years after hernia formation. Additionally, some cases of left-sided Amyand hernia described herein, including our case, are recurrent cases of a previous hernia that have been surgically repaired.2-20
Presentation of Amyand hernia often resembles that of a complicated inguinal hernia, acute appendicitis, or both. Hence, clinicians should consider this a possibility when patients present with signs and symptoms that could otherwise be thought to be originating from an incarcerated, strangulated, or recurrent hernia. Specifically, these signs and symptoms include a tender, nonreducible mass in the inguinal region, acute lower abdominal pain, nausea, or signs of intestinal obstruction such as failure to produce bowel movements.4,17 Because of the unusual anatomy in patients presenting with left-sided Amyand hernia, tenderness at the McBurney point usually is absent and not a useful diagnostic tool to rule out acute appendicitis.
A literature review indicates that an Amyand hernia on either side tends to occur in males more often than it does in females. The rate of diagnosis of Amyand hernia also has been reported to be 3 times higher in children than it is in adults due to failure of the processus vaginalis to obliterate during development.21 Our literature review supports this finding, as 16 of the documented 25 cases of left-sided Amyand hernia were reported in males. Additionally, information regarding gender was not found in 6 cases, suggesting a potential for an even higher prevalence in males.
Explanations as to why the appendix is on the left side in these patients include developmental anomalies, such as situs inversus, intestinal rotation, mobile cecum, or an abnormally long appendix.3,8 In our case, the likely causative culprit was a mobile cecum, as there was neither indication of intestinal malformation, rotation, nor of an abnormally long appendix during surgery. Additionally, pre-operative radiologic studies, clinical evaluation, and electrocardiogram did not suggest the presence of situs inversus.
Treatment of Amyand hernia usually follows the landmark classification algorithm set forth in 2007 by Losanoff and Basson (Table 2).22 This system stratifies treatment based on intraoperative findings of the appendix and surrounding structures, ranging from type 1, which involves a normal appendix within the hernia, to type 4, which includes acute appendicitis with additional abdominal pathology. Our patient presented with a type 1 Amyand hernia and appendectomy was foregone as per the guidelines; however, there have been numerous reported cases of surgeons opting for prophylactic appendectomy in the case of a normal appearing appendix and surrounding structures. The decision to act independent of the Losanoff and Basson classification underscores the lack of true standardization, namely, when it comes to a treatment approach for type 1 Amyand hernias. Nonetheless, many contend that indiscriminately performing appendectomies in all cases of left-sided Amyand hernia is useful as a prophylactic measure, as cases of future appendicitis in these patients will have atypical presentations based on the contralateral location of the appendix.6,11,17
Others disagree, citing that prophylactic appendectomy in the case of a normal looking appendix is unnecessary and complicates an otherwise sterile surgery (clean wound classification) with the removal of an appendix containing fecal matter and gut microbiota (converted into a clean contaminated or a contaminated wound classification).17 Additionally, it is thought that in the cases of middle-aged or geriatric patients where the chances of appendicitis are far less, the risks of detriment from prophylactic appendectomy may outweigh the benefits. In these cases, a macroscopic view of the appendix based on visual examination during the operation should guide decision making.4
While the decision to remove a healthy-appearing appendix remains contentious, the decision for or against placement of a heterogenous hernia mesh has proven to be binary, with near universally accepted criteria. If signs of perforation or infection are present in the hernia sac, then surgeons will forego hernioplasty with mesh for simple herniorrhaphy. This contraindication for mesh placement is due to the increased risk of mesh infection, wound infection, and fistulae associated with the introduction of a foreign structure to an active infection site.2
While most cases of Amyand hernia are diagnosed intraoperatively, there have been documented cases of preoperative diagnosis using ultrasonography and CT imaging modalities.19,23,24 In all cases, the presence of the vermiform appendix within the hernia sac can complicate diagnosis and treatment, and preoperative knowledge of this condition may help to guide physician decision making. Identifying Amyand hernia via CT scan is not only useful for alerting physicians of a potentially inflamed appendix within the hernia sac, but also may create opportunities for the use of other treatment modalities. For example, laparoscopic Amyand hernia reduction, an approach that was performed successfully and documented for the first time by Vermillion and colleagues, was made possible by preoperative diagnosis and can potentially result in improved patient outcomes.25
Regardless, while standardization of treatment for Amyand hernia has not yet occurred, it is clear that improved preoperative diagnosis, especially in the case of an unanticipated left-sided Amyand hernia, can allow for better planning and use of a wider variety of treatment modalities. The main impediment to this approach is that suspected cases of appendicitis and inguinal hernias (the most common preoperative diagnoses of Amyand hernia) usually are diagnosed clinically without the need of additional imaging studies like CT or ultrasound. In accordance with the guiding principle of radiation safety of exposing patients to “as low as reasonably achievable” (ALARA) radiation and with consideration of expediency of care and cost efficiency, we recommend physicians continue to screen for and treat cases of potentially emergent appendicitis and/or inguinal hernia as per the conventional methodology. The best approach may involve increasing preoperative diagnoses of left-sided Amyand hernias via physician awareness of this rare finding, as well as evaluating imaging studies that have previously been obtained in order to narrow a broad differential diagnosis.
Conclusions
Left-sided Amyand hernia is an exceptionally rare condition whose preoperative diagnosis remains difficult to establish but whose treatment decision tree is significantly impacted by the condition.
1. Franko J, Raftopoulos I, Sulkowski R. A rare variation of Amyand’s hernia. Am J Gastroenterol. 2002;97(10):2684-2685. doi:10.1111/j.1572-0241.2002.06060.x
2. Carey LC. Acute appendicitis occurring in hernias: a report of 10 cases. Surgery. 1967;61(2):236-238.
3. Kaymakci A, Akillioglu I, Akkoyun I, Guven S, Ozdemir A, Gulen S. Amyand’s hernia: a series of 30 cases in children. Hernia. 2009;13(6):609-612. doi:10.1007/s10029-009-0528-8
4. Cankorkmaz L, Ozer H, Guney C, Atalar MH, Arslan MS, Koyluoglu G. Amyand’s hernia in the children: a single center experience. Surgery. 2010;147(1):140-143. doi:10.1016/j.surg.2009.09.038
5. Yasumoto R, Kawano M, Kawanishi H, et al. Left acute scrotum associated with appendicitis. Int J Urol. 1998;5(1):108-110. doi:10.1111/j.1442-2042.1998.tb00254.x
6. Bakhshi GD, Bhandarwar AH, Govila AA. Acute appendicitis in left scrotum. Indian J Gastroenterol. 2004;23(5):195.
7. Breitenstein S, Eisenbach C, Wille G, Decurtins M. Incarcerated vermiform appendix in a left-sided inguinal hernia. Hernia. 2005;9(1):100-102. doi:10.1007/s10029-004-0263-0
8. Gupta S, Sharma R, Kaushik R. Left-sided Amyand’s hernia. Singapore Med J. 2005;46(8):424-425.
9. Gupta N, Wilkinson TV, Wilkinson A, Akhtar M. Left-sided incarcerated Amyand’s hernia. Indian J Surg. 2007;69(1):17-18.
10. Tayade, MB, Bakhshi GD, Borisa AD, Deshpande G, Joshi N. A rare combination of left sided Amyand’s and Richter’s hernia. Bombay Hosp J. 2008;50(4): 644-645
11. Johari HG, Paydar S, Davani SZ, Eskandari S, Johari MG. Left-sided Amyand hernia. Ann Saudi Med. 2009;29(4):321-322. doi:10.4103/0256-4947.55305
12. Ali SM, Malik KA, Al-Qadhi H. Amyand’s Hernia: Study of four cases and literature review. Sultan Qaboos Univ Med J. 2012;12(2):232-236. doi:10.12816/0003119
13. Ravishankaran P, Mohan G, Srinivasan A, Ravindran G, Ramalingam A. Left sided amyand’s hernia, a rare occurrence: A Case Report. Indian J Surg. 2013;75(3):247-248. doi:10.1007/s12262-010-0223-0
14. Singh K, Singh RR, Kaur S. Amyand’s hernia. J Indian Assoc Pediatr Surg. 2011;16(4):171-172. doi:10.4103/0971-9261.86890
15. Khan TS, Wani ML, Bijli AH, et al. Amyand’s hernia: a rare occurrence. Ann Nigerian Med. 2011;5(2):62-64.doi:10.4103/0331-3131.92955
16. Ghafouri A, Anbara T, Foroutankia R. A rare case report of appendix and cecum in the sac of left inguinal hernia (left Amyand’s hernia). Med J Islam Repub Iran. 2012;26(2):94-95.
17. Al-Mayoof AF, Al-Ani BH. Left-sided amyand hernia: report of two cases with review of literature. European J Pediatr Surg Rep. 2014;2(1):63-66. doi:10.1055/s-0033-1347131
18. Unver M, Ozturk S, Karaman K, Turgut E. Left sided Amyand’s hernia. World J Gastrointest Surg. 2013;5(10):285-286. doi:10.4240/wjgs.v5.i10.285
19. Maeda K, Kunieda K, Kawai M, et al. Giant left-sided inguinoscrotal hernia containing the cecum and appendix (giant left-sided Amyand’s hernia). Clin Case Rep. 2014;2(6):254-257. doi:10.1002/ccr3.104
20. Mongardini M, Maturo A, De Anna L, et al. Appendiceal abscess in a giant left-sided inguinoscrotal hernia: a rare case of Amyand hernia. Springerplus. 2015;4:378. Published 2015 Jul 26. doi:10.1186/s40064-015-1162-9
21. Ivanschuk G, Cesmebasi A, Sorenson EP, Blaak C, Loukas M, Tubbs SR. Amyand’s hernia: a review. Med Sci Monit. 2014;20:140-146. Published 2014 Jan 28. doi:10.12659/MSM.889873
22. Losanoff JE, Basson MD. Amyand hernia: what lies beneath--a proposed classification scheme to determine management. Am Surg. 2007;73(12):1288-1290.
23. Coulier B, Pacary J, Broze B. Sonographic diagnosis of appendicitis within a right inguinal hernia (Amyand’s hernia). J Clin Ultrasound. 2006;34(9):454-457. doi:10.1002/jcu.20266
24. Vehbi H, Agirgun C, Agirgun F, Dogan Y. Preoperative diagnosis of Amyand’s hernia by ultrasound and computed tomography. Turk J Emerg Med. 2016;16(2):72-74. Published 2016 May 8. doi:10.1016/j.tjem.2015.11.014
25. Vermillion JM, Abernathy SW, Snyder SK. Laparoscopic reduction of Amyand’s hernia. Hernia. 1999;3:159-160. doi:10.1007/BF01195318
1. Franko J, Raftopoulos I, Sulkowski R. A rare variation of Amyand’s hernia. Am J Gastroenterol. 2002;97(10):2684-2685. doi:10.1111/j.1572-0241.2002.06060.x
2. Carey LC. Acute appendicitis occurring in hernias: a report of 10 cases. Surgery. 1967;61(2):236-238.
3. Kaymakci A, Akillioglu I, Akkoyun I, Guven S, Ozdemir A, Gulen S. Amyand’s hernia: a series of 30 cases in children. Hernia. 2009;13(6):609-612. doi:10.1007/s10029-009-0528-8
4. Cankorkmaz L, Ozer H, Guney C, Atalar MH, Arslan MS, Koyluoglu G. Amyand’s hernia in the children: a single center experience. Surgery. 2010;147(1):140-143. doi:10.1016/j.surg.2009.09.038
5. Yasumoto R, Kawano M, Kawanishi H, et al. Left acute scrotum associated with appendicitis. Int J Urol. 1998;5(1):108-110. doi:10.1111/j.1442-2042.1998.tb00254.x
6. Bakhshi GD, Bhandarwar AH, Govila AA. Acute appendicitis in left scrotum. Indian J Gastroenterol. 2004;23(5):195.
7. Breitenstein S, Eisenbach C, Wille G, Decurtins M. Incarcerated vermiform appendix in a left-sided inguinal hernia. Hernia. 2005;9(1):100-102. doi:10.1007/s10029-004-0263-0
8. Gupta S, Sharma R, Kaushik R. Left-sided Amyand’s hernia. Singapore Med J. 2005;46(8):424-425.
9. Gupta N, Wilkinson TV, Wilkinson A, Akhtar M. Left-sided incarcerated Amyand’s hernia. Indian J Surg. 2007;69(1):17-18.
10. Tayade, MB, Bakhshi GD, Borisa AD, Deshpande G, Joshi N. A rare combination of left sided Amyand’s and Richter’s hernia. Bombay Hosp J. 2008;50(4): 644-645
11. Johari HG, Paydar S, Davani SZ, Eskandari S, Johari MG. Left-sided Amyand hernia. Ann Saudi Med. 2009;29(4):321-322. doi:10.4103/0256-4947.55305
12. Ali SM, Malik KA, Al-Qadhi H. Amyand’s Hernia: Study of four cases and literature review. Sultan Qaboos Univ Med J. 2012;12(2):232-236. doi:10.12816/0003119
13. Ravishankaran P, Mohan G, Srinivasan A, Ravindran G, Ramalingam A. Left sided amyand’s hernia, a rare occurrence: A Case Report. Indian J Surg. 2013;75(3):247-248. doi:10.1007/s12262-010-0223-0
14. Singh K, Singh RR, Kaur S. Amyand’s hernia. J Indian Assoc Pediatr Surg. 2011;16(4):171-172. doi:10.4103/0971-9261.86890
15. Khan TS, Wani ML, Bijli AH, et al. Amyand’s hernia: a rare occurrence. Ann Nigerian Med. 2011;5(2):62-64.doi:10.4103/0331-3131.92955
16. Ghafouri A, Anbara T, Foroutankia R. A rare case report of appendix and cecum in the sac of left inguinal hernia (left Amyand’s hernia). Med J Islam Repub Iran. 2012;26(2):94-95.
17. Al-Mayoof AF, Al-Ani BH. Left-sided amyand hernia: report of two cases with review of literature. European J Pediatr Surg Rep. 2014;2(1):63-66. doi:10.1055/s-0033-1347131
18. Unver M, Ozturk S, Karaman K, Turgut E. Left sided Amyand’s hernia. World J Gastrointest Surg. 2013;5(10):285-286. doi:10.4240/wjgs.v5.i10.285
19. Maeda K, Kunieda K, Kawai M, et al. Giant left-sided inguinoscrotal hernia containing the cecum and appendix (giant left-sided Amyand’s hernia). Clin Case Rep. 2014;2(6):254-257. doi:10.1002/ccr3.104
20. Mongardini M, Maturo A, De Anna L, et al. Appendiceal abscess in a giant left-sided inguinoscrotal hernia: a rare case of Amyand hernia. Springerplus. 2015;4:378. Published 2015 Jul 26. doi:10.1186/s40064-015-1162-9
21. Ivanschuk G, Cesmebasi A, Sorenson EP, Blaak C, Loukas M, Tubbs SR. Amyand’s hernia: a review. Med Sci Monit. 2014;20:140-146. Published 2014 Jan 28. doi:10.12659/MSM.889873
22. Losanoff JE, Basson MD. Amyand hernia: what lies beneath--a proposed classification scheme to determine management. Am Surg. 2007;73(12):1288-1290.
23. Coulier B, Pacary J, Broze B. Sonographic diagnosis of appendicitis within a right inguinal hernia (Amyand’s hernia). J Clin Ultrasound. 2006;34(9):454-457. doi:10.1002/jcu.20266
24. Vehbi H, Agirgun C, Agirgun F, Dogan Y. Preoperative diagnosis of Amyand’s hernia by ultrasound and computed tomography. Turk J Emerg Med. 2016;16(2):72-74. Published 2016 May 8. doi:10.1016/j.tjem.2015.11.014
25. Vermillion JM, Abernathy SW, Snyder SK. Laparoscopic reduction of Amyand’s hernia. Hernia. 1999;3:159-160. doi:10.1007/BF01195318
How to Save a Limb: Identification of Pyoderma Gangrenosum
Case Report
A 67-year-old woman presented with a painful expanding ulcer on the left leg and a new nearby ulcer of 2 months’ duration. She initially was seen 2 months prior for a wound on the left knee due to a fall as well as cellulitis, which was treated with intravenous vancomycin and ceftriaxone. Wound cultures were negative for bacteria, and she was discharged without antibiotics. She presented to the emergency department 1 month later for malodorous discharge of the first ulcer with zero systemic inflammatory response syndrome criteria; no fever; and no abnormal heart rate, respiratory rate, or leukocyte count. She was discharged with wound care. After 3 weeks, she returned with a second ulcer and worsening drainage but zero systemic inflammatory response syndrome criteria. She had a medical history of Crohn disease with 9-year remission, atrial fibrillation, pacemaker, mitral valve replacement, chronic obstructive pulmonary disease, and a 51 pack-year smoking history.
Physical examination of the left leg revealed a 3×3-cm deep lesion (ulcer A) on the distal left thigh located superomedial to the knee (Figure 1) as well as a 2×1-cm deep lesion (ulcer B) on the anteromedial knee with undermining and tunneling (Figure 2). A large amount of malodorous tan bloody discharge was present on both ulcers. There were no signs of induration or crepitus.Due to concerns of skin and soft tissue infection (SSTI) or osteomyelitis, a bone scan and wound and blood cultures were ordered. The patient was started on vancomycin and piperacillin-tazobactam in the emergency department, which later was augmented with cefepime. Trauma surgery scheduled debridement for the following morning with suspicion of necrotizing fasciitis. Additional consultations were requested, including infectious disease, wound care, and dermatology. Dermatology evaluated the wound, performed a punch biopsy, and canceled debridement due to unclear diagnosis. The clinical differential at that time included pyoderma gangrenosum (PG), atypical vasculitis, or infection. Additional workup revealed positive antineutrophil cytoplasmic antibodies but negative proteinase 3 and myeloperoxidase, disfavoring vasculitis. Wound cultures grew Staphylococcus aureus and Pseudomonas aeruginosa.
Histologic evaluation revealed deep dermal necrosis with a mixed inflammatory infiltrate (Figure 3) and no organisms or vasculitis. Antibiotics were discontinued, and she was discharged on a 14-day course of prednisone 60 mg daily for empirical treatment of PG with dermatology follow-up. Medical management included a 6-month course of dapsone that was extended to 7 months because of an intensive care unit stay for a cerebrovascular accident. Daily dosing was as follows: 100 mg for 5 months, 50 mg for 1 month, and 25 mg for 1 month, then stopped. She was followed with serial complete blood cell count every 1 to 2 months and home-health wound care. One month after dapsone initiation, the ulcers decreased in size. Ulcer B was fully healed after 4 months, and ulcer A was nearly closed at 6 months without any new flares.
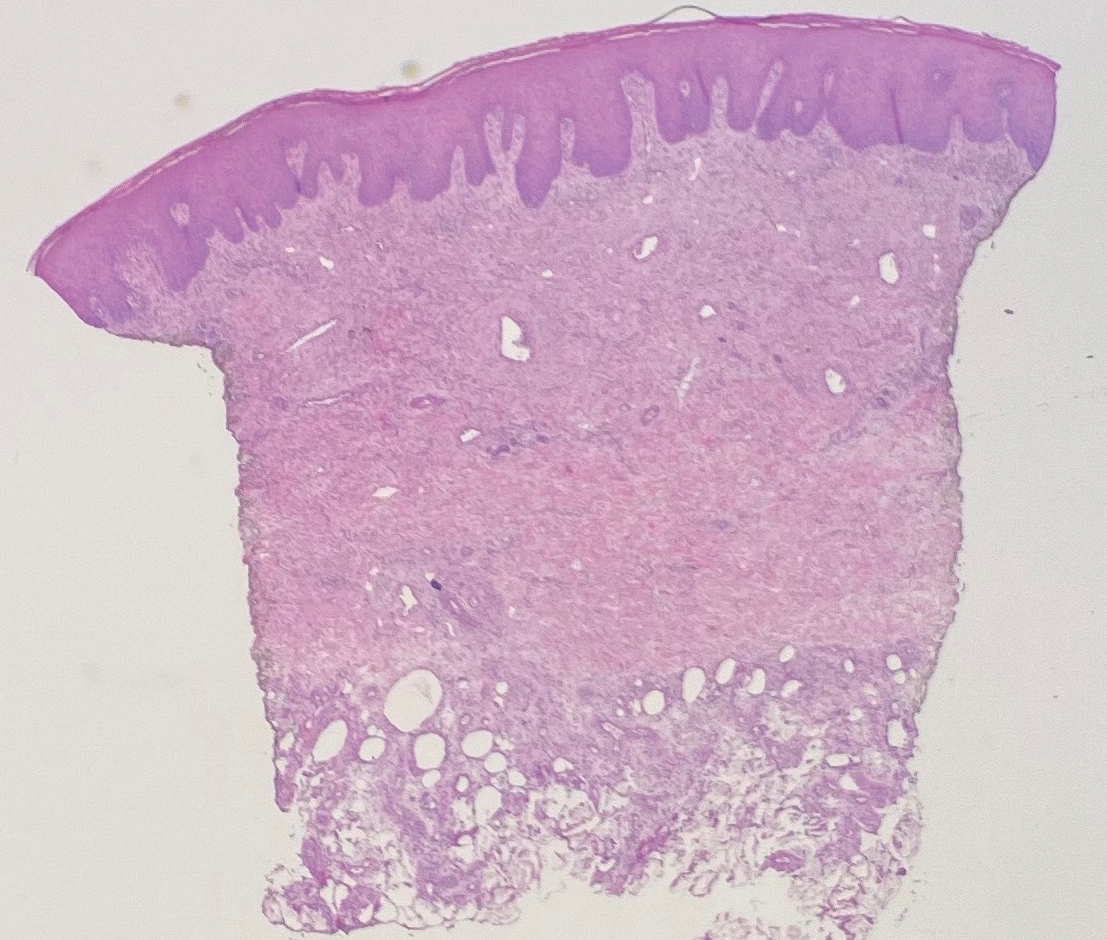
Comment
Pyoderma gangrenosum is a rare inflammatory skin condition that classically presents as tender papules or pustules evolving into painful ulcers, most commonly on the lower extremities. Pyoderma gangrenosum has a propensity to exhibit pathergy, the hyperreactivity of the skin in response to minor trauma. This phenomenon in PG manifests as the rapid evolution from pustule to ulceration with violaceous undermining borders.
Diagnosis of PG
Pyoderma gangrenosum has been described as a diagnosis of exclusion, as its findings frequently mimic SSTIs. Important findings to obtain are histology, history, ulcer morphology, and response to treatment.
In 2018, Maverakis et al1 proposed diagnostic criteria for classic ulcerative PG (Table 1). A diagnosis of PG can be made if the patient meets 1 major criterion and 4 minor criteria. Our case met 0 major criteria and 5 minor criteria: history of inflammatory bowel disease (IBD); history of pustule ulcerating within 4 days of appearing; peripheral erythema, undermining border, and tenderness at ulceration site; multiple ulcerations, with at least 1 on an anterior lower leg; and decreased ulcer size within 1 month of initiating immunosuppressive medication(s). Although our patient’s biopsy demonstrated a mixed infiltrate, PG was not excluded due to spontaneous resolution at the time of biopsy, emphasizing the need to biopsy subsequent new lesions if neutrophils are not initially seen.1 Pyoderma gangrenosum frequently is associated with IBD, most often Crohn disease, as seen in our patient.2-4 Although IBD classically is associated with smoking, studies have yet to conclude if smoking is a predictive factor of PG.5 Our patient presented with an initial ulcer that evolved into 2 ulcers, similar to a case of bilateral ulcers.6
Differential Diagnosis of PG
Other possible diagnoses to consider are SSTI and vasculitis, the latter being disfavored by no evidence of vasculitis on biopsy and negative titers for proteinase 3 and myeloperoxidase antibodies. However, the presence of either, similar to a mixed infiltrate, does not exclude a diagnosis of PG, as they can occur simultaneously. Consequently, superinfection of a chronically open wound can occur due to underlying PG.7 The differences between PG and SSTI are listed in Table 2.
Although we know PG involves neutrophilic dysfunction, the pathophysiology remains poorly understood, contributing to the lack of clinical guidelines.8 Therefore, the diagnosis of PG often is delayed and is associated with severe consequences such as necrotizing fasciitis, osteomyelitis, cosmetic morbidity, and limb amputation.9,10 Dermatologic consultation can aid in early diagnosis and avoid amputation.7,10 Amputation has been used as a last resort to preserve optimal outcomes in patients with severe PG.11
Management of PG
A gold standard of treatment of PG does not exist, but the goal is to promote wound healing. Patients with limited disease typically can be managed with wound care and topical steroids or calcineurin inhibitors, though data on efficacy are limited. However, our patient had more extensive disease and needed to be treated with systemic therapy. First-line therapy for extensive disease includes oral prednisone or cyclosporine for patients who cannot tolerate systemic corticosteroids.12 Second-line and adjunctive therapy options include dapsone, minocycline, methotrexate, and infliximab. Our patient was prescribed a 7-month course of dapsone with outpatient dermatology and demonstrated resolution of both ulcers. Dapsone was tapered from a daily dose of 100 mg to 50 mg to 25 mg to none over the course of 2 to 3 months. Close monitoring with wound care is recommended, and petroleum jelly can be used for dry skin around the lesion for comfort.
Conclusion
The diagnosis of PG is challenging because it relies heavily on clinical signs and often mimics SSTI. Gathering a detailed medical history is critical to make the diagnosis of PG. In a patient with associated features of PG, dermatologic consultation and biopsy of skin lesions should be considered. Physicians should evaluate for suspected PG prior to proceeding with surgical intervention to avoid unnecessary amputation. The diagnostic criteria for classic ulcerative PG are gaining wider acceptance and are a useful tool for clinicians.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- Bisarya K, Azzopardi S, Lye G, et al. Necrotizing fasciitis versus pyoderma gangrenosum: securing the correct diagnosis! a case report and literature review. Eplasty. 2011;11:E24.
- Perricone G, Vangeli M. Pyoderma gangrenosum in ulcerative colitis. N Engl J Med. 2018;379:E7.
- Ashchyan HJ, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Ampuero J, Rojas-Feria M, Castro-Fernández M, et al. Predictive factors for erythema nodosum and pyoderma gangrenosum in inflammatory bowel disease. J Gastroenterol Hepatol. 2014;29:291-295.
- Ebner DW, Hu M, Poterucha TH. 29-year-old woman with fever and bilateral lower extremity lesions. Mayo Clin Proc. 2018;93:1659-1663.
- Marzak H, Von Hunolstein JJ, Lipsker D, et al. Management of a superinfected pyoderma gangrenosum after pacemaker implant. HeartRhythm Case Rep. 2018;5:63-65.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Saffie MG, Shroff A. A case of pyoderma gangrenosum misdiagnosed as necrotizing infection: a potential diagnostic catastrophe. Case Rep Infect Dis. 2018;2018:8907542.
- Haag CK, Nutan F, Cyrus JW, et al. Pyoderma gangrenosum misdiagnosis resulting in amputation: a review. J Trauma Acute Care Surg. 2019;86:307-313.
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol. 2019;155:79-84.
- Alavi A, French LE, Davis MD, et al. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18:355-372.
Case Report
A 67-year-old woman presented with a painful expanding ulcer on the left leg and a new nearby ulcer of 2 months’ duration. She initially was seen 2 months prior for a wound on the left knee due to a fall as well as cellulitis, which was treated with intravenous vancomycin and ceftriaxone. Wound cultures were negative for bacteria, and she was discharged without antibiotics. She presented to the emergency department 1 month later for malodorous discharge of the first ulcer with zero systemic inflammatory response syndrome criteria; no fever; and no abnormal heart rate, respiratory rate, or leukocyte count. She was discharged with wound care. After 3 weeks, she returned with a second ulcer and worsening drainage but zero systemic inflammatory response syndrome criteria. She had a medical history of Crohn disease with 9-year remission, atrial fibrillation, pacemaker, mitral valve replacement, chronic obstructive pulmonary disease, and a 51 pack-year smoking history.
Physical examination of the left leg revealed a 3×3-cm deep lesion (ulcer A) on the distal left thigh located superomedial to the knee (Figure 1) as well as a 2×1-cm deep lesion (ulcer B) on the anteromedial knee with undermining and tunneling (Figure 2). A large amount of malodorous tan bloody discharge was present on both ulcers. There were no signs of induration or crepitus.Due to concerns of skin and soft tissue infection (SSTI) or osteomyelitis, a bone scan and wound and blood cultures were ordered. The patient was started on vancomycin and piperacillin-tazobactam in the emergency department, which later was augmented with cefepime. Trauma surgery scheduled debridement for the following morning with suspicion of necrotizing fasciitis. Additional consultations were requested, including infectious disease, wound care, and dermatology. Dermatology evaluated the wound, performed a punch biopsy, and canceled debridement due to unclear diagnosis. The clinical differential at that time included pyoderma gangrenosum (PG), atypical vasculitis, or infection. Additional workup revealed positive antineutrophil cytoplasmic antibodies but negative proteinase 3 and myeloperoxidase, disfavoring vasculitis. Wound cultures grew Staphylococcus aureus and Pseudomonas aeruginosa.
Histologic evaluation revealed deep dermal necrosis with a mixed inflammatory infiltrate (Figure 3) and no organisms or vasculitis. Antibiotics were discontinued, and she was discharged on a 14-day course of prednisone 60 mg daily for empirical treatment of PG with dermatology follow-up. Medical management included a 6-month course of dapsone that was extended to 7 months because of an intensive care unit stay for a cerebrovascular accident. Daily dosing was as follows: 100 mg for 5 months, 50 mg for 1 month, and 25 mg for 1 month, then stopped. She was followed with serial complete blood cell count every 1 to 2 months and home-health wound care. One month after dapsone initiation, the ulcers decreased in size. Ulcer B was fully healed after 4 months, and ulcer A was nearly closed at 6 months without any new flares.
Comment
Pyoderma gangrenosum is a rare inflammatory skin condition that classically presents as tender papules or pustules evolving into painful ulcers, most commonly on the lower extremities. Pyoderma gangrenosum has a propensity to exhibit pathergy, the hyperreactivity of the skin in response to minor trauma. This phenomenon in PG manifests as the rapid evolution from pustule to ulceration with violaceous undermining borders.
Diagnosis of PG
Pyoderma gangrenosum has been described as a diagnosis of exclusion, as its findings frequently mimic SSTIs. Important findings to obtain are histology, history, ulcer morphology, and response to treatment.
In 2018, Maverakis et al1 proposed diagnostic criteria for classic ulcerative PG (Table 1). A diagnosis of PG can be made if the patient meets 1 major criterion and 4 minor criteria. Our case met 0 major criteria and 5 minor criteria: history of inflammatory bowel disease (IBD); history of pustule ulcerating within 4 days of appearing; peripheral erythema, undermining border, and tenderness at ulceration site; multiple ulcerations, with at least 1 on an anterior lower leg; and decreased ulcer size within 1 month of initiating immunosuppressive medication(s). Although our patient’s biopsy demonstrated a mixed infiltrate, PG was not excluded due to spontaneous resolution at the time of biopsy, emphasizing the need to biopsy subsequent new lesions if neutrophils are not initially seen.1 Pyoderma gangrenosum frequently is associated with IBD, most often Crohn disease, as seen in our patient.2-4 Although IBD classically is associated with smoking, studies have yet to conclude if smoking is a predictive factor of PG.5 Our patient presented with an initial ulcer that evolved into 2 ulcers, similar to a case of bilateral ulcers.6
Differential Diagnosis of PG
Other possible diagnoses to consider are SSTI and vasculitis, the latter being disfavored by no evidence of vasculitis on biopsy and negative titers for proteinase 3 and myeloperoxidase antibodies. However, the presence of either, similar to a mixed infiltrate, does not exclude a diagnosis of PG, as they can occur simultaneously. Consequently, superinfection of a chronically open wound can occur due to underlying PG.7 The differences between PG and SSTI are listed in Table 2.
Although we know PG involves neutrophilic dysfunction, the pathophysiology remains poorly understood, contributing to the lack of clinical guidelines.8 Therefore, the diagnosis of PG often is delayed and is associated with severe consequences such as necrotizing fasciitis, osteomyelitis, cosmetic morbidity, and limb amputation.9,10 Dermatologic consultation can aid in early diagnosis and avoid amputation.7,10 Amputation has been used as a last resort to preserve optimal outcomes in patients with severe PG.11
Management of PG
A gold standard of treatment of PG does not exist, but the goal is to promote wound healing. Patients with limited disease typically can be managed with wound care and topical steroids or calcineurin inhibitors, though data on efficacy are limited. However, our patient had more extensive disease and needed to be treated with systemic therapy. First-line therapy for extensive disease includes oral prednisone or cyclosporine for patients who cannot tolerate systemic corticosteroids.12 Second-line and adjunctive therapy options include dapsone, minocycline, methotrexate, and infliximab. Our patient was prescribed a 7-month course of dapsone with outpatient dermatology and demonstrated resolution of both ulcers. Dapsone was tapered from a daily dose of 100 mg to 50 mg to 25 mg to none over the course of 2 to 3 months. Close monitoring with wound care is recommended, and petroleum jelly can be used for dry skin around the lesion for comfort.
Conclusion
The diagnosis of PG is challenging because it relies heavily on clinical signs and often mimics SSTI. Gathering a detailed medical history is critical to make the diagnosis of PG. In a patient with associated features of PG, dermatologic consultation and biopsy of skin lesions should be considered. Physicians should evaluate for suspected PG prior to proceeding with surgical intervention to avoid unnecessary amputation. The diagnostic criteria for classic ulcerative PG are gaining wider acceptance and are a useful tool for clinicians.
Case Report
A 67-year-old woman presented with a painful expanding ulcer on the left leg and a new nearby ulcer of 2 months’ duration. She initially was seen 2 months prior for a wound on the left knee due to a fall as well as cellulitis, which was treated with intravenous vancomycin and ceftriaxone. Wound cultures were negative for bacteria, and she was discharged without antibiotics. She presented to the emergency department 1 month later for malodorous discharge of the first ulcer with zero systemic inflammatory response syndrome criteria; no fever; and no abnormal heart rate, respiratory rate, or leukocyte count. She was discharged with wound care. After 3 weeks, she returned with a second ulcer and worsening drainage but zero systemic inflammatory response syndrome criteria. She had a medical history of Crohn disease with 9-year remission, atrial fibrillation, pacemaker, mitral valve replacement, chronic obstructive pulmonary disease, and a 51 pack-year smoking history.
Physical examination of the left leg revealed a 3×3-cm deep lesion (ulcer A) on the distal left thigh located superomedial to the knee (Figure 1) as well as a 2×1-cm deep lesion (ulcer B) on the anteromedial knee with undermining and tunneling (Figure 2). A large amount of malodorous tan bloody discharge was present on both ulcers. There were no signs of induration or crepitus.Due to concerns of skin and soft tissue infection (SSTI) or osteomyelitis, a bone scan and wound and blood cultures were ordered. The patient was started on vancomycin and piperacillin-tazobactam in the emergency department, which later was augmented with cefepime. Trauma surgery scheduled debridement for the following morning with suspicion of necrotizing fasciitis. Additional consultations were requested, including infectious disease, wound care, and dermatology. Dermatology evaluated the wound, performed a punch biopsy, and canceled debridement due to unclear diagnosis. The clinical differential at that time included pyoderma gangrenosum (PG), atypical vasculitis, or infection. Additional workup revealed positive antineutrophil cytoplasmic antibodies but negative proteinase 3 and myeloperoxidase, disfavoring vasculitis. Wound cultures grew Staphylococcus aureus and Pseudomonas aeruginosa.
Histologic evaluation revealed deep dermal necrosis with a mixed inflammatory infiltrate (Figure 3) and no organisms or vasculitis. Antibiotics were discontinued, and she was discharged on a 14-day course of prednisone 60 mg daily for empirical treatment of PG with dermatology follow-up. Medical management included a 6-month course of dapsone that was extended to 7 months because of an intensive care unit stay for a cerebrovascular accident. Daily dosing was as follows: 100 mg for 5 months, 50 mg for 1 month, and 25 mg for 1 month, then stopped. She was followed with serial complete blood cell count every 1 to 2 months and home-health wound care. One month after dapsone initiation, the ulcers decreased in size. Ulcer B was fully healed after 4 months, and ulcer A was nearly closed at 6 months without any new flares.
Comment
Pyoderma gangrenosum is a rare inflammatory skin condition that classically presents as tender papules or pustules evolving into painful ulcers, most commonly on the lower extremities. Pyoderma gangrenosum has a propensity to exhibit pathergy, the hyperreactivity of the skin in response to minor trauma. This phenomenon in PG manifests as the rapid evolution from pustule to ulceration with violaceous undermining borders.
Diagnosis of PG
Pyoderma gangrenosum has been described as a diagnosis of exclusion, as its findings frequently mimic SSTIs. Important findings to obtain are histology, history, ulcer morphology, and response to treatment.
In 2018, Maverakis et al1 proposed diagnostic criteria for classic ulcerative PG (Table 1). A diagnosis of PG can be made if the patient meets 1 major criterion and 4 minor criteria. Our case met 0 major criteria and 5 minor criteria: history of inflammatory bowel disease (IBD); history of pustule ulcerating within 4 days of appearing; peripheral erythema, undermining border, and tenderness at ulceration site; multiple ulcerations, with at least 1 on an anterior lower leg; and decreased ulcer size within 1 month of initiating immunosuppressive medication(s). Although our patient’s biopsy demonstrated a mixed infiltrate, PG was not excluded due to spontaneous resolution at the time of biopsy, emphasizing the need to biopsy subsequent new lesions if neutrophils are not initially seen.1 Pyoderma gangrenosum frequently is associated with IBD, most often Crohn disease, as seen in our patient.2-4 Although IBD classically is associated with smoking, studies have yet to conclude if smoking is a predictive factor of PG.5 Our patient presented with an initial ulcer that evolved into 2 ulcers, similar to a case of bilateral ulcers.6
Differential Diagnosis of PG
Other possible diagnoses to consider are SSTI and vasculitis, the latter being disfavored by no evidence of vasculitis on biopsy and negative titers for proteinase 3 and myeloperoxidase antibodies. However, the presence of either, similar to a mixed infiltrate, does not exclude a diagnosis of PG, as they can occur simultaneously. Consequently, superinfection of a chronically open wound can occur due to underlying PG.7 The differences between PG and SSTI are listed in Table 2.
Although we know PG involves neutrophilic dysfunction, the pathophysiology remains poorly understood, contributing to the lack of clinical guidelines.8 Therefore, the diagnosis of PG often is delayed and is associated with severe consequences such as necrotizing fasciitis, osteomyelitis, cosmetic morbidity, and limb amputation.9,10 Dermatologic consultation can aid in early diagnosis and avoid amputation.7,10 Amputation has been used as a last resort to preserve optimal outcomes in patients with severe PG.11
Management of PG
A gold standard of treatment of PG does not exist, but the goal is to promote wound healing. Patients with limited disease typically can be managed with wound care and topical steroids or calcineurin inhibitors, though data on efficacy are limited. However, our patient had more extensive disease and needed to be treated with systemic therapy. First-line therapy for extensive disease includes oral prednisone or cyclosporine for patients who cannot tolerate systemic corticosteroids.12 Second-line and adjunctive therapy options include dapsone, minocycline, methotrexate, and infliximab. Our patient was prescribed a 7-month course of dapsone with outpatient dermatology and demonstrated resolution of both ulcers. Dapsone was tapered from a daily dose of 100 mg to 50 mg to 25 mg to none over the course of 2 to 3 months. Close monitoring with wound care is recommended, and petroleum jelly can be used for dry skin around the lesion for comfort.
Conclusion
The diagnosis of PG is challenging because it relies heavily on clinical signs and often mimics SSTI. Gathering a detailed medical history is critical to make the diagnosis of PG. In a patient with associated features of PG, dermatologic consultation and biopsy of skin lesions should be considered. Physicians should evaluate for suspected PG prior to proceeding with surgical intervention to avoid unnecessary amputation. The diagnostic criteria for classic ulcerative PG are gaining wider acceptance and are a useful tool for clinicians.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- Bisarya K, Azzopardi S, Lye G, et al. Necrotizing fasciitis versus pyoderma gangrenosum: securing the correct diagnosis! a case report and literature review. Eplasty. 2011;11:E24.
- Perricone G, Vangeli M. Pyoderma gangrenosum in ulcerative colitis. N Engl J Med. 2018;379:E7.
- Ashchyan HJ, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Ampuero J, Rojas-Feria M, Castro-Fernández M, et al. Predictive factors for erythema nodosum and pyoderma gangrenosum in inflammatory bowel disease. J Gastroenterol Hepatol. 2014;29:291-295.
- Ebner DW, Hu M, Poterucha TH. 29-year-old woman with fever and bilateral lower extremity lesions. Mayo Clin Proc. 2018;93:1659-1663.
- Marzak H, Von Hunolstein JJ, Lipsker D, et al. Management of a superinfected pyoderma gangrenosum after pacemaker implant. HeartRhythm Case Rep. 2018;5:63-65.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Saffie MG, Shroff A. A case of pyoderma gangrenosum misdiagnosed as necrotizing infection: a potential diagnostic catastrophe. Case Rep Infect Dis. 2018;2018:8907542.
- Haag CK, Nutan F, Cyrus JW, et al. Pyoderma gangrenosum misdiagnosis resulting in amputation: a review. J Trauma Acute Care Surg. 2019;86:307-313.
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol. 2019;155:79-84.
- Alavi A, French LE, Davis MD, et al. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18:355-372.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- Bisarya K, Azzopardi S, Lye G, et al. Necrotizing fasciitis versus pyoderma gangrenosum: securing the correct diagnosis! a case report and literature review. Eplasty. 2011;11:E24.
- Perricone G, Vangeli M. Pyoderma gangrenosum in ulcerative colitis. N Engl J Med. 2018;379:E7.
- Ashchyan HJ, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Ampuero J, Rojas-Feria M, Castro-Fernández M, et al. Predictive factors for erythema nodosum and pyoderma gangrenosum in inflammatory bowel disease. J Gastroenterol Hepatol. 2014;29:291-295.
- Ebner DW, Hu M, Poterucha TH. 29-year-old woman with fever and bilateral lower extremity lesions. Mayo Clin Proc. 2018;93:1659-1663.
- Marzak H, Von Hunolstein JJ, Lipsker D, et al. Management of a superinfected pyoderma gangrenosum after pacemaker implant. HeartRhythm Case Rep. 2018;5:63-65.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Saffie MG, Shroff A. A case of pyoderma gangrenosum misdiagnosed as necrotizing infection: a potential diagnostic catastrophe. Case Rep Infect Dis. 2018;2018:8907542.
- Haag CK, Nutan F, Cyrus JW, et al. Pyoderma gangrenosum misdiagnosis resulting in amputation: a review. J Trauma Acute Care Surg. 2019;86:307-313.
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol. 2019;155:79-84.
- Alavi A, French LE, Davis MD, et al. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18:355-372.
Practice Points
- Pyoderma gangrenosum (PG) frequently is misdiagnosed due to its similar presentation to other skin and soft tissue infections (SSTIs). Patients with known risk factors for PG should be evaluated with a high index of suspicion to ensure early diagnosis and avoid serious complications. Common associations include inflammatory bowel disease (IBD), hematologic malignancies, and rheumatologic disorders.
- Response to treatment may be used to guide management when the diagnosis of SSTIs vs PG cannot be distinguished with clinical and histologic findings alone. In a worsening ulcer that has failed antibiotic therapy, clinicians should consider the diagnosis of PG and the risk of pathergy prior to surgical intervention such as debridement.
- Although typically a diagnosis of exclusion, clinicians can consider the use of diagnostic criteria for PG in patients of high clinical suspicion. A trial of immunosuppressants can be considered after infection has been ruled out.
Ulcerative Heliotrope Rash in Antimelanoma Differentiation–Associated Gene 5 Dermatomyositis
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
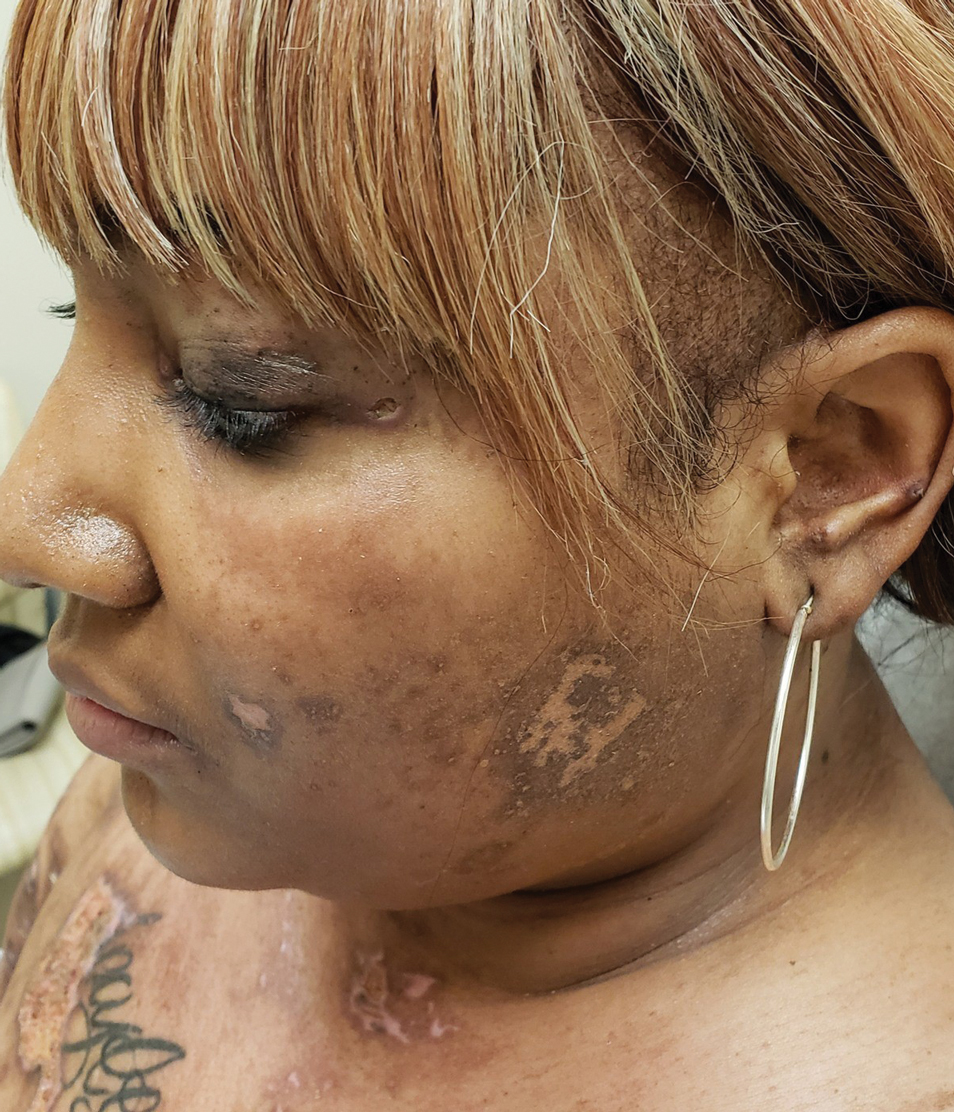
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
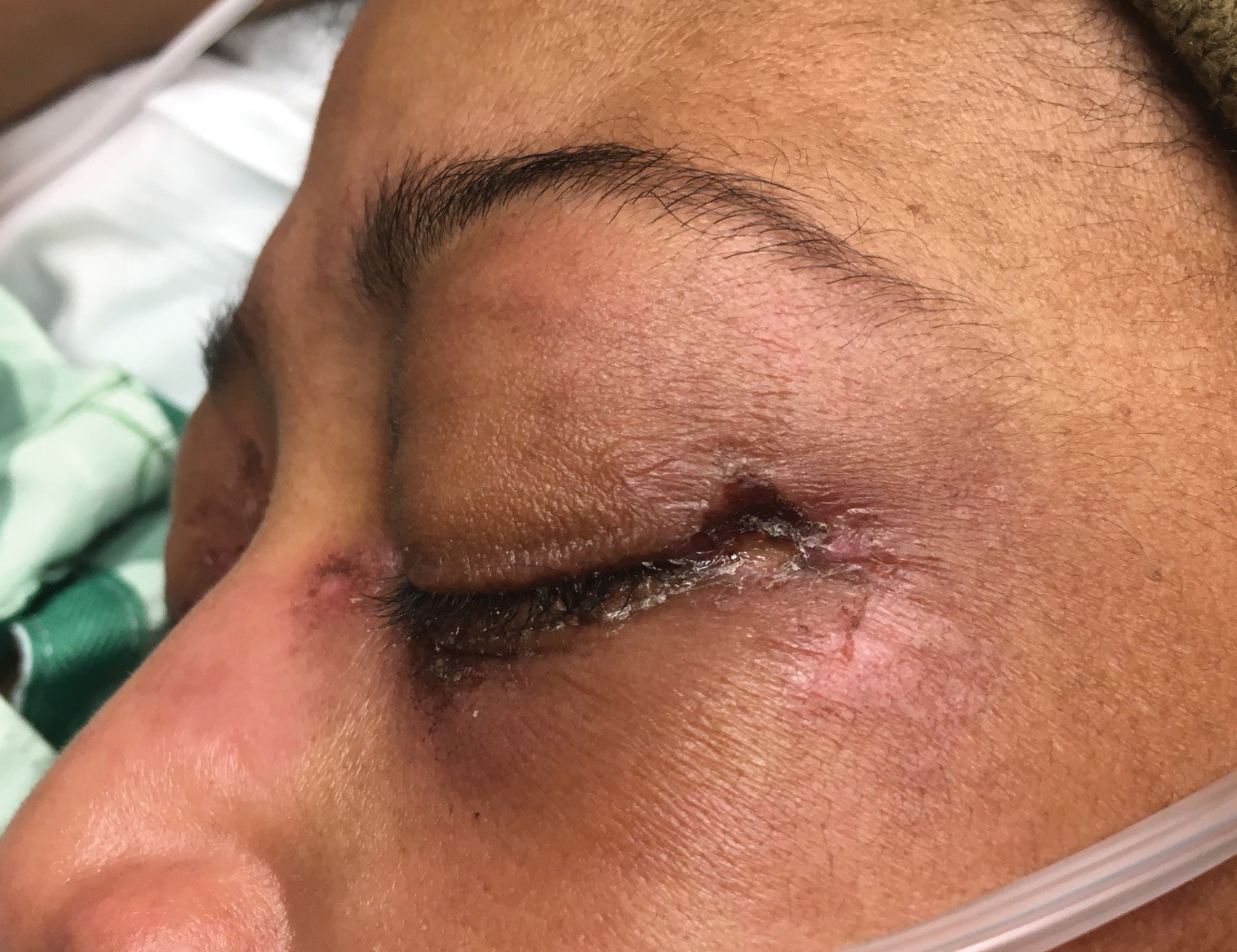
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
Practice Points
- Antimelanoma differentiation–associated gene 5 dermatomyositis (anti-MDA5 DM) can present with an ulcerative heliotrope rash.
- Ulceration of the heliotrope rash in anti-MDA5 DM may indicate disease progression.
- Rapidly progressive interstitial lung disease is highly associated with anti-MDA5 DM.