User login
Glucosuria Is Not Always Due to Diabetes
Familial renal glucosuria is an uncommon, rarely documented condition wherein the absence of other renal or endocrine conditions and with a normal serum glucose level, glucosuria persists due to an isolated defect in the nephron’s proximal tubule. Seemingly, in these patients, the body’s physiologic function mimics that of sodiumglucose cotransporter-2 (SGLT2)-inhibiting medications with the glucose cotransporter being selectively targeted for promoting renal excretion of glucose. This has implications for the patient’s prospective development of hyperglycemic diseases, urinary tract infections (UTIs), and potentially even cardiovascular disease. Though it is a generally asymptomatic condition, it is one that seasoned clinicians should investigate given the future impacts and considerations required for their patients.
Case Presentation
Mr. A was a 28-year-old male with no medical history nor prescription medication use who presented to the nephrology clinic at Eglin Air Force Base, Florida, in June 2019 for a workup of asymptomatic glucosuria. The condition was discovered on a routine urinalysis in October 2015 at the initial presentation at Eglin Air Force Base, when the patient was being evaluated by his primary care physician for acute, benign headache with fever and chills. Urinalysis testing was performed in October 2015 and resulted in a urine glucose of 500 mg/dL (2+). He was directed to the emergency department for further evaluation, reciprocating the results.
On further laboratory testing in October 2015, his blood glucose was normal at 75 mg/dL; hemoglobin A1c was 5.5%. On repeat urinalysis 2 weeks later, his urinary glucose was found to be 500 mg/dL (2+). Each time, the elevated urinary glucose was the only abnormal finding: There was no concurrent hematuria, proteinuria, or ketonuria. The patient reported he had no associated symptoms, including nausea, vomiting, abdominal pain, dysuria, polyuria, and increased thirst. He was not taking any prescription medications, including SGLT2 inhibitors. His presenting headache and fever resolved with supportive care and was considered unrelated to his additional workup.
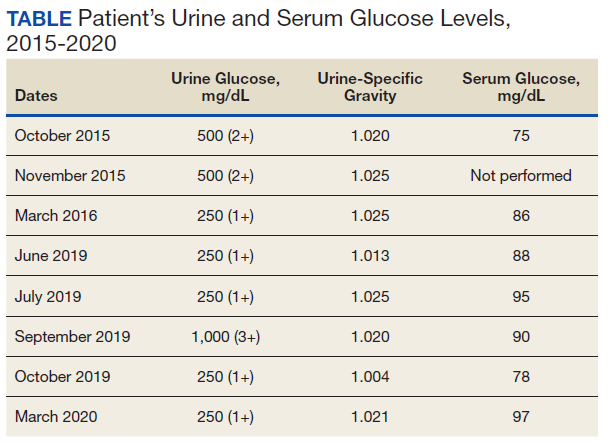
A diagnostic evaluation ensued from 2015 to 2020, including follow-up urinalyses, metabolic panels, complete blood counts, urine protein electrophoresis (UPEP), urine creatinine, urine electrolytes, 25-OH vitamin D level, κ/λ light chain panel, and serum protein electrophoresis (SPEP). The results of all diagnostic workup throughout the entirety of his evaluation were found to be normal. In 2020, his 25-OH vitamin D level was borderline low at 29.4 ng/mL. His κ/λ ratio was normal at 1.65, and his serum albumin protein electrophoresis was 4.74 g/dL, marginally elevated, but his SPEP and UPEP were normal, as were urine protein levels, total gamma globulin, and no monoclonal gamma spike noted on pathology review. Serum uric acid, and urine phosphorous were both normal. His serum creatinine and electrolytes were all within normal limits. Over the 5 years of intermittent monitoring, the maximum amount of glucosuria was 1,000 mg/dL (3+) and the minimum was 250 mg/dL (1+). There was a gap of monitoring from March 2016 until June 2019 due to the patient receiving care from offsite health care providers without shared documentation of specific laboratory values, but notes documenting persistent glucosuria (Table).
Analysis
Building the initial differential diagnosis for this patient began with confirming that he had isolated glucosuria, and not glucosuria secondary to elevated serum glucose. Additionally, conditions related to generalized proximal tubule dysfunction, acute or chronic impaired renal function, and neoplasms, including multiple myeloma (MM), were eliminated because this patient did not have the other specific findings associated with these conditions.
Proximal tubulopathies, including proximal renal tubular acidosis (type 2) and Fanconi syndrome, was initially a leading diagnosis in this patient. Isolated proximal renal tubular acidosis (RTA) (type 2) is uncommon and pathophysiologically involves reduced proximal tubular reabsorption of bicarbonate, resulting in low serum bicarbonate and metabolic acidosis. Patients with isolated proximal RTA (type 2) typically present in infancy with failure to thrive, tachypnea, recurrent vomiting, and feeding difficulties. These symptoms do not meet our patient’s clinical presentation. Fanconi syndrome involves a specific disruption in the proximal tubular apical sodium uptake mechanism affecting the transmembrane sodium gradient and the sodium-potassium- ATPase pump. Fanconi syndrome, therefore, would not only present with glucosuria, but also classically with proteinuria, hypophosphatemia, hypokalemia, and a hyperchloremic metabolic acidosis.
Chronic or acute renal disease may present with glucosuria, but one would expect additional findings including elevated serum creatinine, elevated urinary creatinine, 25-OH vitamin D deficiency, or anemia of chronic disease. Other potential diagnoses included MM and similar neoplasms. MM also would present with glucosuria with proteinuria, an elevated κ/λ light chain ratio, and an elevated SPEP and concern for bone lytic lesions, which were not present. A related disorder, monoclonal gammopathy of renal significance (MGRS), akin to monoclonal gammopathy of unknown significance (MGUS), presents with proteinuria with evidence of renal injury. While this patient had a marginally elevated κ/λ light chain ratio, the remainder of his SPEP and UPEP were normal, and evaluation by a hematologist/ oncologist and pathology review of laboratory findings confirmed no additional evidence for MM, including no monoclonal γ spike. With no evidence of renal injury with a normal serum creatinine and glomerular filtration rate, MGRS was eliminated from the differential as it did not meet the International Myeloma Working Group diagnostic criteria.1 The elevated κ/λ ratio with normal renal function is attributed to polyclonal immunoglobulin elevation, which may occur more commonly with uncomplicated acute viral illnesses.
Diagnosis
The differential homed in on a targeted defect in the proximal tubular SGLT2 gene as the final diagnosis causing isolated glucosuria. Familial renal glucosuria (FRG), a condition caused by a mutation in the SLC5A2 gene that codes for the SGLT2 has been identified in the literature as causing cases with nearly identical presentations to this patient.2,3 This condition is often found in otherwise healthy, asymptomatic patients in whom isolated glucosuria was identified on routine urinalysis testing.
Due to isolated case reports sharing this finding and the asymptomatic nature of the condition, specific data pertaining to its prevalence are not available. Case studies of other affected individuals have not noted adverse effects (AEs), such as UTIs or hypotension specifically.2,3 The patient was referred for genetic testing for this gene mutation; however, he was unable to obtain the test due to lack of insurance coverage. Mr. A has no other family members that have been evaluated for or identified as having this condition. Despite the name, FRG has an unknown inheritance pattern and is attributed to a variety of missense mutations in the SLC5A2 gene.4,5
Discussion
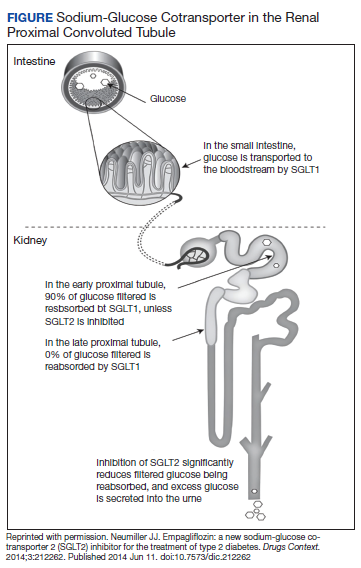
The SGLT2 gene believed to be mutated in this patient has recently become wellknown. The inhibition of the SGLT2 transport protein has become an important tool in the management of type 2 diabetes mellitus (T2DM) independent of the insulin pathway. The SGLT2 in the proximal convoluted tubule of the kidney reabsorbs the majority, 98%, of the renal glucose for reabsorption, and the remaining glucose is reabsorbed by the SGLT2 gene in the more distal portion of the proximal tubule in healthy individuals.4,6 The normal renal threshold for glucose reabsorption in a patient with a normal glomerular filtration rate is equivalent to a serum glucose concentration of 180 mg/dL, even higher in patients with T2DM due to upregulation of the SGLT2 inhibitors. SGLT2 inhibitors, such as canagliflozin, dapagliflozin, and empagliflozin, selectively inhibit this cotransporter, reducing the threshold from 40 to 120 mg/dL, thereby significantly increasing the renal excretion of glucose.4 The patient’s mutation in question and clinical presentation aligned with a naturally occurring mimicry of this drug’s mechanism of action (Figure).
Arguably, one of the more significant benefits to using this new class of oral antihyperglycemics, aside from the noninferior glycemic control compared with that of other first-line agents, is the added metabolic benefit. To date, SGLT2 inhibitors have been found to decrease blood pressure in all studies of the medications and promote moderate weight loss.7 SGLT2 inhibitors have not only demonstrated significant cardiovascular (CV) benefits, linked with the aforementioned metabolic benefits, but also have reduced hospitalizations for heart failure in patients with T2DM and those without.7 The EMPA-REG OUTCOME trial showed a 38% relative risk reduction in CV events in empagliflozin vs placebo.4,8 However, it is unknown whether patients with the SLC5A2 mutation also benefit from these CV benefits akin to the SGLT2 inhibiting medications, and it is and worthy of studying via longterm follow-up with patients similar to this.
This SLC5A2 mutation causing FRG selectively inhibiting SGLT2 function effectively causes this patient’s natural physiology to mimic that of these new oral antihyperglycemic medications. Patients with FRG should be counseled regarding this condition and the implications it has on their overall health. At this time, there is no formal recommendation for short-term or longterm management of patients with FRG; observation and routine preventive care monitoring based on US Preventive Services Task Force screening recommendations apply to this population in line with the general population.
This condition is not known to be associated with hypotension or hypoglycemia, and to some extent, it can be theorized that patients with this condition may have inherent protection of development of hyperglycemia. 4 Akin to patients on SGLT2 inhibitors, these patients may be at an increased risk of UTIs and genital infections, including mycotic infections due to glycemic-related imbalance in the normal flora of the urinary tract.9 Other serious AEs of SGLT2 inhibitors, such as diabetic ketoacidosis, osteoporosis and related fractures, and acute pancreatitis, should be shared with FRG patients, though they are unlikely to be at increased risk for this condition in the setting of normal serum glucose and electrolyte levels. Notably, the osteoporosis risk is small, and specific other risk factors pertinent to individual patient’s medical history, and canagliflozin exclusively. If a patient with FRG develops T2DM after diagnosis, it is imperative that they inform physicians of their condition, because SGLT2-inhibiting drugs will be ineffective in this subset of patients, necessitating increased clinical judgment in selecting an appropriate antihyperglycemic agent in this population.
Conclusions
FRG is an uncommon diagnosis of exclusion that presents with isolated glucosuria in the setting of normal serum glucose. The patient generally presents asymptomatically with a urinalysis completed for other reasons, and the patient may or may not have a family history of similar findings. The condition is of particular interest given that its SGLT2 mutation mimics the effect of SGLT2 inhibitors used for T2DM. More monitoring of patients with this condition will be required for documentation regarding long-term implications, including development of further renal disease, T2DM, or CV disease.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12). doi:10.1016/s1470-2045(14)70442-5
2. Calado J, Sznajer Y, Metzger D, et al. Twenty-one additional cases of familial renal glucosuria: absence of genetic heterogeneity, high prevalence of private mutations and further evidence of volume depletion. Nephrol Dial Transplant. 2008;23(12):3874-3879. doi.org/10.1093/ndt/gfn386
3. Kim KM, Kwon SK, Kim HY. A case of isolated glycosuria mediated by an SLC5A2 gene mutation and characterized by postprandial heavy glycosuria without salt wasting. Electrolyte Blood Press. 2016;14(2):35-37. doi:10.5049/EBP.2016.14.2.35
4. Hsia DS, Grove O, Cefalu WT. An update on sodiumglucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr Opin Endocrinol Diabetes Obes. 2017;24(1):73-79. doi:10.1097/MED.0000000000000311
5. Kleta R. Renal glucosuria due to SGLT2 mutations. Mol Genet Metab. 2004;82(1):56-58. doi:10.1016/j.ymgme.2004.01.018
6. Neumiller JJ. Empagliflozin: a new sodium-glucose co-transporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. Drugs Context. 2014;3:212262. doi:10.7573/dic.212262
7. Raz I, Cernea S, Cahn A. SGLT2 inhibitors for primary prevention of cardiovascular events. J Diabetes. 2020;12(1):5- 7. doi:10.1111/1753-0407.13004
8. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/nejmoa1504720
9. Mcgill JB, Subramanian S. Safety of sodium-glucose cotransporter 2 inhibitors. Am J Cardiol. 2019;124(suppl 1):S45-S52. doi:10.1016/j.amjcard.2019.10.029
Familial renal glucosuria is an uncommon, rarely documented condition wherein the absence of other renal or endocrine conditions and with a normal serum glucose level, glucosuria persists due to an isolated defect in the nephron’s proximal tubule. Seemingly, in these patients, the body’s physiologic function mimics that of sodiumglucose cotransporter-2 (SGLT2)-inhibiting medications with the glucose cotransporter being selectively targeted for promoting renal excretion of glucose. This has implications for the patient’s prospective development of hyperglycemic diseases, urinary tract infections (UTIs), and potentially even cardiovascular disease. Though it is a generally asymptomatic condition, it is one that seasoned clinicians should investigate given the future impacts and considerations required for their patients.
Case Presentation
Mr. A was a 28-year-old male with no medical history nor prescription medication use who presented to the nephrology clinic at Eglin Air Force Base, Florida, in June 2019 for a workup of asymptomatic glucosuria. The condition was discovered on a routine urinalysis in October 2015 at the initial presentation at Eglin Air Force Base, when the patient was being evaluated by his primary care physician for acute, benign headache with fever and chills. Urinalysis testing was performed in October 2015 and resulted in a urine glucose of 500 mg/dL (2+). He was directed to the emergency department for further evaluation, reciprocating the results.
On further laboratory testing in October 2015, his blood glucose was normal at 75 mg/dL; hemoglobin A1c was 5.5%. On repeat urinalysis 2 weeks later, his urinary glucose was found to be 500 mg/dL (2+). Each time, the elevated urinary glucose was the only abnormal finding: There was no concurrent hematuria, proteinuria, or ketonuria. The patient reported he had no associated symptoms, including nausea, vomiting, abdominal pain, dysuria, polyuria, and increased thirst. He was not taking any prescription medications, including SGLT2 inhibitors. His presenting headache and fever resolved with supportive care and was considered unrelated to his additional workup.
A diagnostic evaluation ensued from 2015 to 2020, including follow-up urinalyses, metabolic panels, complete blood counts, urine protein electrophoresis (UPEP), urine creatinine, urine electrolytes, 25-OH vitamin D level, κ/λ light chain panel, and serum protein electrophoresis (SPEP). The results of all diagnostic workup throughout the entirety of his evaluation were found to be normal. In 2020, his 25-OH vitamin D level was borderline low at 29.4 ng/mL. His κ/λ ratio was normal at 1.65, and his serum albumin protein electrophoresis was 4.74 g/dL, marginally elevated, but his SPEP and UPEP were normal, as were urine protein levels, total gamma globulin, and no monoclonal gamma spike noted on pathology review. Serum uric acid, and urine phosphorous were both normal. His serum creatinine and electrolytes were all within normal limits. Over the 5 years of intermittent monitoring, the maximum amount of glucosuria was 1,000 mg/dL (3+) and the minimum was 250 mg/dL (1+). There was a gap of monitoring from March 2016 until June 2019 due to the patient receiving care from offsite health care providers without shared documentation of specific laboratory values, but notes documenting persistent glucosuria (Table).
Analysis
Building the initial differential diagnosis for this patient began with confirming that he had isolated glucosuria, and not glucosuria secondary to elevated serum glucose. Additionally, conditions related to generalized proximal tubule dysfunction, acute or chronic impaired renal function, and neoplasms, including multiple myeloma (MM), were eliminated because this patient did not have the other specific findings associated with these conditions.
Proximal tubulopathies, including proximal renal tubular acidosis (type 2) and Fanconi syndrome, was initially a leading diagnosis in this patient. Isolated proximal renal tubular acidosis (RTA) (type 2) is uncommon and pathophysiologically involves reduced proximal tubular reabsorption of bicarbonate, resulting in low serum bicarbonate and metabolic acidosis. Patients with isolated proximal RTA (type 2) typically present in infancy with failure to thrive, tachypnea, recurrent vomiting, and feeding difficulties. These symptoms do not meet our patient’s clinical presentation. Fanconi syndrome involves a specific disruption in the proximal tubular apical sodium uptake mechanism affecting the transmembrane sodium gradient and the sodium-potassium- ATPase pump. Fanconi syndrome, therefore, would not only present with glucosuria, but also classically with proteinuria, hypophosphatemia, hypokalemia, and a hyperchloremic metabolic acidosis.
Chronic or acute renal disease may present with glucosuria, but one would expect additional findings including elevated serum creatinine, elevated urinary creatinine, 25-OH vitamin D deficiency, or anemia of chronic disease. Other potential diagnoses included MM and similar neoplasms. MM also would present with glucosuria with proteinuria, an elevated κ/λ light chain ratio, and an elevated SPEP and concern for bone lytic lesions, which were not present. A related disorder, monoclonal gammopathy of renal significance (MGRS), akin to monoclonal gammopathy of unknown significance (MGUS), presents with proteinuria with evidence of renal injury. While this patient had a marginally elevated κ/λ light chain ratio, the remainder of his SPEP and UPEP were normal, and evaluation by a hematologist/ oncologist and pathology review of laboratory findings confirmed no additional evidence for MM, including no monoclonal γ spike. With no evidence of renal injury with a normal serum creatinine and glomerular filtration rate, MGRS was eliminated from the differential as it did not meet the International Myeloma Working Group diagnostic criteria.1 The elevated κ/λ ratio with normal renal function is attributed to polyclonal immunoglobulin elevation, which may occur more commonly with uncomplicated acute viral illnesses.
Diagnosis
The differential homed in on a targeted defect in the proximal tubular SGLT2 gene as the final diagnosis causing isolated glucosuria. Familial renal glucosuria (FRG), a condition caused by a mutation in the SLC5A2 gene that codes for the SGLT2 has been identified in the literature as causing cases with nearly identical presentations to this patient.2,3 This condition is often found in otherwise healthy, asymptomatic patients in whom isolated glucosuria was identified on routine urinalysis testing.
Due to isolated case reports sharing this finding and the asymptomatic nature of the condition, specific data pertaining to its prevalence are not available. Case studies of other affected individuals have not noted adverse effects (AEs), such as UTIs or hypotension specifically.2,3 The patient was referred for genetic testing for this gene mutation; however, he was unable to obtain the test due to lack of insurance coverage. Mr. A has no other family members that have been evaluated for or identified as having this condition. Despite the name, FRG has an unknown inheritance pattern and is attributed to a variety of missense mutations in the SLC5A2 gene.4,5
Discussion
The SGLT2 gene believed to be mutated in this patient has recently become wellknown. The inhibition of the SGLT2 transport protein has become an important tool in the management of type 2 diabetes mellitus (T2DM) independent of the insulin pathway. The SGLT2 in the proximal convoluted tubule of the kidney reabsorbs the majority, 98%, of the renal glucose for reabsorption, and the remaining glucose is reabsorbed by the SGLT2 gene in the more distal portion of the proximal tubule in healthy individuals.4,6 The normal renal threshold for glucose reabsorption in a patient with a normal glomerular filtration rate is equivalent to a serum glucose concentration of 180 mg/dL, even higher in patients with T2DM due to upregulation of the SGLT2 inhibitors. SGLT2 inhibitors, such as canagliflozin, dapagliflozin, and empagliflozin, selectively inhibit this cotransporter, reducing the threshold from 40 to 120 mg/dL, thereby significantly increasing the renal excretion of glucose.4 The patient’s mutation in question and clinical presentation aligned with a naturally occurring mimicry of this drug’s mechanism of action (Figure).
Arguably, one of the more significant benefits to using this new class of oral antihyperglycemics, aside from the noninferior glycemic control compared with that of other first-line agents, is the added metabolic benefit. To date, SGLT2 inhibitors have been found to decrease blood pressure in all studies of the medications and promote moderate weight loss.7 SGLT2 inhibitors have not only demonstrated significant cardiovascular (CV) benefits, linked with the aforementioned metabolic benefits, but also have reduced hospitalizations for heart failure in patients with T2DM and those without.7 The EMPA-REG OUTCOME trial showed a 38% relative risk reduction in CV events in empagliflozin vs placebo.4,8 However, it is unknown whether patients with the SLC5A2 mutation also benefit from these CV benefits akin to the SGLT2 inhibiting medications, and it is and worthy of studying via longterm follow-up with patients similar to this.
This SLC5A2 mutation causing FRG selectively inhibiting SGLT2 function effectively causes this patient’s natural physiology to mimic that of these new oral antihyperglycemic medications. Patients with FRG should be counseled regarding this condition and the implications it has on their overall health. At this time, there is no formal recommendation for short-term or longterm management of patients with FRG; observation and routine preventive care monitoring based on US Preventive Services Task Force screening recommendations apply to this population in line with the general population.
This condition is not known to be associated with hypotension or hypoglycemia, and to some extent, it can be theorized that patients with this condition may have inherent protection of development of hyperglycemia. 4 Akin to patients on SGLT2 inhibitors, these patients may be at an increased risk of UTIs and genital infections, including mycotic infections due to glycemic-related imbalance in the normal flora of the urinary tract.9 Other serious AEs of SGLT2 inhibitors, such as diabetic ketoacidosis, osteoporosis and related fractures, and acute pancreatitis, should be shared with FRG patients, though they are unlikely to be at increased risk for this condition in the setting of normal serum glucose and electrolyte levels. Notably, the osteoporosis risk is small, and specific other risk factors pertinent to individual patient’s medical history, and canagliflozin exclusively. If a patient with FRG develops T2DM after diagnosis, it is imperative that they inform physicians of their condition, because SGLT2-inhibiting drugs will be ineffective in this subset of patients, necessitating increased clinical judgment in selecting an appropriate antihyperglycemic agent in this population.
Conclusions
FRG is an uncommon diagnosis of exclusion that presents with isolated glucosuria in the setting of normal serum glucose. The patient generally presents asymptomatically with a urinalysis completed for other reasons, and the patient may or may not have a family history of similar findings. The condition is of particular interest given that its SGLT2 mutation mimics the effect of SGLT2 inhibitors used for T2DM. More monitoring of patients with this condition will be required for documentation regarding long-term implications, including development of further renal disease, T2DM, or CV disease.
Familial renal glucosuria is an uncommon, rarely documented condition wherein the absence of other renal or endocrine conditions and with a normal serum glucose level, glucosuria persists due to an isolated defect in the nephron’s proximal tubule. Seemingly, in these patients, the body’s physiologic function mimics that of sodiumglucose cotransporter-2 (SGLT2)-inhibiting medications with the glucose cotransporter being selectively targeted for promoting renal excretion of glucose. This has implications for the patient’s prospective development of hyperglycemic diseases, urinary tract infections (UTIs), and potentially even cardiovascular disease. Though it is a generally asymptomatic condition, it is one that seasoned clinicians should investigate given the future impacts and considerations required for their patients.
Case Presentation
Mr. A was a 28-year-old male with no medical history nor prescription medication use who presented to the nephrology clinic at Eglin Air Force Base, Florida, in June 2019 for a workup of asymptomatic glucosuria. The condition was discovered on a routine urinalysis in October 2015 at the initial presentation at Eglin Air Force Base, when the patient was being evaluated by his primary care physician for acute, benign headache with fever and chills. Urinalysis testing was performed in October 2015 and resulted in a urine glucose of 500 mg/dL (2+). He was directed to the emergency department for further evaluation, reciprocating the results.
On further laboratory testing in October 2015, his blood glucose was normal at 75 mg/dL; hemoglobin A1c was 5.5%. On repeat urinalysis 2 weeks later, his urinary glucose was found to be 500 mg/dL (2+). Each time, the elevated urinary glucose was the only abnormal finding: There was no concurrent hematuria, proteinuria, or ketonuria. The patient reported he had no associated symptoms, including nausea, vomiting, abdominal pain, dysuria, polyuria, and increased thirst. He was not taking any prescription medications, including SGLT2 inhibitors. His presenting headache and fever resolved with supportive care and was considered unrelated to his additional workup.
A diagnostic evaluation ensued from 2015 to 2020, including follow-up urinalyses, metabolic panels, complete blood counts, urine protein electrophoresis (UPEP), urine creatinine, urine electrolytes, 25-OH vitamin D level, κ/λ light chain panel, and serum protein electrophoresis (SPEP). The results of all diagnostic workup throughout the entirety of his evaluation were found to be normal. In 2020, his 25-OH vitamin D level was borderline low at 29.4 ng/mL. His κ/λ ratio was normal at 1.65, and his serum albumin protein electrophoresis was 4.74 g/dL, marginally elevated, but his SPEP and UPEP were normal, as were urine protein levels, total gamma globulin, and no monoclonal gamma spike noted on pathology review. Serum uric acid, and urine phosphorous were both normal. His serum creatinine and electrolytes were all within normal limits. Over the 5 years of intermittent monitoring, the maximum amount of glucosuria was 1,000 mg/dL (3+) and the minimum was 250 mg/dL (1+). There was a gap of monitoring from March 2016 until June 2019 due to the patient receiving care from offsite health care providers without shared documentation of specific laboratory values, but notes documenting persistent glucosuria (Table).
Analysis
Building the initial differential diagnosis for this patient began with confirming that he had isolated glucosuria, and not glucosuria secondary to elevated serum glucose. Additionally, conditions related to generalized proximal tubule dysfunction, acute or chronic impaired renal function, and neoplasms, including multiple myeloma (MM), were eliminated because this patient did not have the other specific findings associated with these conditions.
Proximal tubulopathies, including proximal renal tubular acidosis (type 2) and Fanconi syndrome, was initially a leading diagnosis in this patient. Isolated proximal renal tubular acidosis (RTA) (type 2) is uncommon and pathophysiologically involves reduced proximal tubular reabsorption of bicarbonate, resulting in low serum bicarbonate and metabolic acidosis. Patients with isolated proximal RTA (type 2) typically present in infancy with failure to thrive, tachypnea, recurrent vomiting, and feeding difficulties. These symptoms do not meet our patient’s clinical presentation. Fanconi syndrome involves a specific disruption in the proximal tubular apical sodium uptake mechanism affecting the transmembrane sodium gradient and the sodium-potassium- ATPase pump. Fanconi syndrome, therefore, would not only present with glucosuria, but also classically with proteinuria, hypophosphatemia, hypokalemia, and a hyperchloremic metabolic acidosis.
Chronic or acute renal disease may present with glucosuria, but one would expect additional findings including elevated serum creatinine, elevated urinary creatinine, 25-OH vitamin D deficiency, or anemia of chronic disease. Other potential diagnoses included MM and similar neoplasms. MM also would present with glucosuria with proteinuria, an elevated κ/λ light chain ratio, and an elevated SPEP and concern for bone lytic lesions, which were not present. A related disorder, monoclonal gammopathy of renal significance (MGRS), akin to monoclonal gammopathy of unknown significance (MGUS), presents with proteinuria with evidence of renal injury. While this patient had a marginally elevated κ/λ light chain ratio, the remainder of his SPEP and UPEP were normal, and evaluation by a hematologist/ oncologist and pathology review of laboratory findings confirmed no additional evidence for MM, including no monoclonal γ spike. With no evidence of renal injury with a normal serum creatinine and glomerular filtration rate, MGRS was eliminated from the differential as it did not meet the International Myeloma Working Group diagnostic criteria.1 The elevated κ/λ ratio with normal renal function is attributed to polyclonal immunoglobulin elevation, which may occur more commonly with uncomplicated acute viral illnesses.
Diagnosis
The differential homed in on a targeted defect in the proximal tubular SGLT2 gene as the final diagnosis causing isolated glucosuria. Familial renal glucosuria (FRG), a condition caused by a mutation in the SLC5A2 gene that codes for the SGLT2 has been identified in the literature as causing cases with nearly identical presentations to this patient.2,3 This condition is often found in otherwise healthy, asymptomatic patients in whom isolated glucosuria was identified on routine urinalysis testing.
Due to isolated case reports sharing this finding and the asymptomatic nature of the condition, specific data pertaining to its prevalence are not available. Case studies of other affected individuals have not noted adverse effects (AEs), such as UTIs or hypotension specifically.2,3 The patient was referred for genetic testing for this gene mutation; however, he was unable to obtain the test due to lack of insurance coverage. Mr. A has no other family members that have been evaluated for or identified as having this condition. Despite the name, FRG has an unknown inheritance pattern and is attributed to a variety of missense mutations in the SLC5A2 gene.4,5
Discussion
The SGLT2 gene believed to be mutated in this patient has recently become wellknown. The inhibition of the SGLT2 transport protein has become an important tool in the management of type 2 diabetes mellitus (T2DM) independent of the insulin pathway. The SGLT2 in the proximal convoluted tubule of the kidney reabsorbs the majority, 98%, of the renal glucose for reabsorption, and the remaining glucose is reabsorbed by the SGLT2 gene in the more distal portion of the proximal tubule in healthy individuals.4,6 The normal renal threshold for glucose reabsorption in a patient with a normal glomerular filtration rate is equivalent to a serum glucose concentration of 180 mg/dL, even higher in patients with T2DM due to upregulation of the SGLT2 inhibitors. SGLT2 inhibitors, such as canagliflozin, dapagliflozin, and empagliflozin, selectively inhibit this cotransporter, reducing the threshold from 40 to 120 mg/dL, thereby significantly increasing the renal excretion of glucose.4 The patient’s mutation in question and clinical presentation aligned with a naturally occurring mimicry of this drug’s mechanism of action (Figure).
Arguably, one of the more significant benefits to using this new class of oral antihyperglycemics, aside from the noninferior glycemic control compared with that of other first-line agents, is the added metabolic benefit. To date, SGLT2 inhibitors have been found to decrease blood pressure in all studies of the medications and promote moderate weight loss.7 SGLT2 inhibitors have not only demonstrated significant cardiovascular (CV) benefits, linked with the aforementioned metabolic benefits, but also have reduced hospitalizations for heart failure in patients with T2DM and those without.7 The EMPA-REG OUTCOME trial showed a 38% relative risk reduction in CV events in empagliflozin vs placebo.4,8 However, it is unknown whether patients with the SLC5A2 mutation also benefit from these CV benefits akin to the SGLT2 inhibiting medications, and it is and worthy of studying via longterm follow-up with patients similar to this.
This SLC5A2 mutation causing FRG selectively inhibiting SGLT2 function effectively causes this patient’s natural physiology to mimic that of these new oral antihyperglycemic medications. Patients with FRG should be counseled regarding this condition and the implications it has on their overall health. At this time, there is no formal recommendation for short-term or longterm management of patients with FRG; observation and routine preventive care monitoring based on US Preventive Services Task Force screening recommendations apply to this population in line with the general population.
This condition is not known to be associated with hypotension or hypoglycemia, and to some extent, it can be theorized that patients with this condition may have inherent protection of development of hyperglycemia. 4 Akin to patients on SGLT2 inhibitors, these patients may be at an increased risk of UTIs and genital infections, including mycotic infections due to glycemic-related imbalance in the normal flora of the urinary tract.9 Other serious AEs of SGLT2 inhibitors, such as diabetic ketoacidosis, osteoporosis and related fractures, and acute pancreatitis, should be shared with FRG patients, though they are unlikely to be at increased risk for this condition in the setting of normal serum glucose and electrolyte levels. Notably, the osteoporosis risk is small, and specific other risk factors pertinent to individual patient’s medical history, and canagliflozin exclusively. If a patient with FRG develops T2DM after diagnosis, it is imperative that they inform physicians of their condition, because SGLT2-inhibiting drugs will be ineffective in this subset of patients, necessitating increased clinical judgment in selecting an appropriate antihyperglycemic agent in this population.
Conclusions
FRG is an uncommon diagnosis of exclusion that presents with isolated glucosuria in the setting of normal serum glucose. The patient generally presents asymptomatically with a urinalysis completed for other reasons, and the patient may or may not have a family history of similar findings. The condition is of particular interest given that its SGLT2 mutation mimics the effect of SGLT2 inhibitors used for T2DM. More monitoring of patients with this condition will be required for documentation regarding long-term implications, including development of further renal disease, T2DM, or CV disease.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12). doi:10.1016/s1470-2045(14)70442-5
2. Calado J, Sznajer Y, Metzger D, et al. Twenty-one additional cases of familial renal glucosuria: absence of genetic heterogeneity, high prevalence of private mutations and further evidence of volume depletion. Nephrol Dial Transplant. 2008;23(12):3874-3879. doi.org/10.1093/ndt/gfn386
3. Kim KM, Kwon SK, Kim HY. A case of isolated glycosuria mediated by an SLC5A2 gene mutation and characterized by postprandial heavy glycosuria without salt wasting. Electrolyte Blood Press. 2016;14(2):35-37. doi:10.5049/EBP.2016.14.2.35
4. Hsia DS, Grove O, Cefalu WT. An update on sodiumglucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr Opin Endocrinol Diabetes Obes. 2017;24(1):73-79. doi:10.1097/MED.0000000000000311
5. Kleta R. Renal glucosuria due to SGLT2 mutations. Mol Genet Metab. 2004;82(1):56-58. doi:10.1016/j.ymgme.2004.01.018
6. Neumiller JJ. Empagliflozin: a new sodium-glucose co-transporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. Drugs Context. 2014;3:212262. doi:10.7573/dic.212262
7. Raz I, Cernea S, Cahn A. SGLT2 inhibitors for primary prevention of cardiovascular events. J Diabetes. 2020;12(1):5- 7. doi:10.1111/1753-0407.13004
8. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/nejmoa1504720
9. Mcgill JB, Subramanian S. Safety of sodium-glucose cotransporter 2 inhibitors. Am J Cardiol. 2019;124(suppl 1):S45-S52. doi:10.1016/j.amjcard.2019.10.029
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12). doi:10.1016/s1470-2045(14)70442-5
2. Calado J, Sznajer Y, Metzger D, et al. Twenty-one additional cases of familial renal glucosuria: absence of genetic heterogeneity, high prevalence of private mutations and further evidence of volume depletion. Nephrol Dial Transplant. 2008;23(12):3874-3879. doi.org/10.1093/ndt/gfn386
3. Kim KM, Kwon SK, Kim HY. A case of isolated glycosuria mediated by an SLC5A2 gene mutation and characterized by postprandial heavy glycosuria without salt wasting. Electrolyte Blood Press. 2016;14(2):35-37. doi:10.5049/EBP.2016.14.2.35
4. Hsia DS, Grove O, Cefalu WT. An update on sodiumglucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr Opin Endocrinol Diabetes Obes. 2017;24(1):73-79. doi:10.1097/MED.0000000000000311
5. Kleta R. Renal glucosuria due to SGLT2 mutations. Mol Genet Metab. 2004;82(1):56-58. doi:10.1016/j.ymgme.2004.01.018
6. Neumiller JJ. Empagliflozin: a new sodium-glucose co-transporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. Drugs Context. 2014;3:212262. doi:10.7573/dic.212262
7. Raz I, Cernea S, Cahn A. SGLT2 inhibitors for primary prevention of cardiovascular events. J Diabetes. 2020;12(1):5- 7. doi:10.1111/1753-0407.13004
8. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128. doi:10.1056/nejmoa1504720
9. Mcgill JB, Subramanian S. Safety of sodium-glucose cotransporter 2 inhibitors. Am J Cardiol. 2019;124(suppl 1):S45-S52. doi:10.1016/j.amjcard.2019.10.029
Truncus Bicaroticus With Arteria Lusoria: A Rare Combination of Aortic Root Anatomy Complicating Cardiac Catheterization
While most patients with arteria lusoria and common carotid trunk conditions are asymptomatic, discovery of such anomalies periprocedurally may affect the cardiac catheterization access site, catheter selection, and additional imaging.
Branching of the great vessels from the aorta normally progresses with the brachiocephalic trunk as the first takeoff followed by the left common carotid and left subclavian artery in approximately 85% of cases.1 Variants of great vessel branching patterns include the so-called bovine arch, arteria lusoria or aberrant right subclavian artery (ARSA), aberrant origin of the vertebral arteries, and truncus bicaroticus, or common origin of the carotid arteries (COCA). These aberrancies are quite rare, some with an incidence of < 1%.1,2
These vascular anomalies become clinically relevant when they pose difficulty for operators in surgical and interventional specialties, necessitating unique approaches, catheters, and techniques to overcome. We present a case of concomitant aortic arch abnormalities during a diagnostic workup for transcatheter aortic valve replacement (TAVR) in a patient with previous coronary artery bypass grafting (CABG).
Case Presentation
A 66-year-old woman with coronary artery disease (CAD) status post-CABG and stage D1 aortic stenosis (AS) presented with exertional dyspnea. She was referred for coronary angiography as part of a workup for TAVR. Echocardiography confirmed severe AS with a peak velocity of 4.1 m/s, mean pressure gradient of 50 mm Hg, and an aortic valve area of 0.7 cm2. The patient was scheduled for cardiac catheterization with anticipated left radial artery approach for intubation and opacification of the left internal mammary artery (LIMA). However, this approach was abandoned during the procedure due to discovery of aberrant left radial artery anatomy, and the procedure was completed via femoral access.
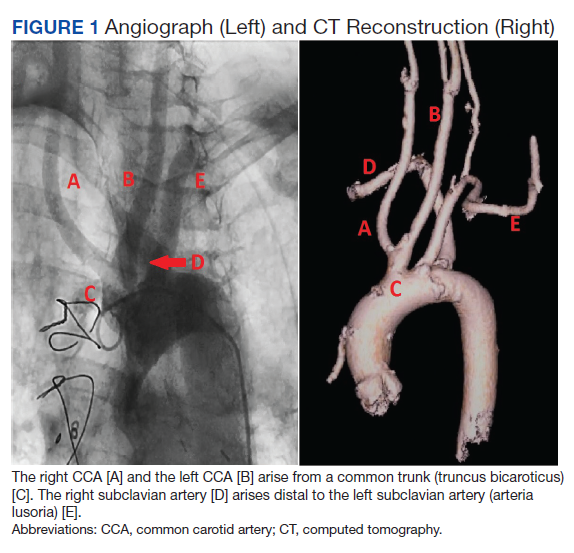
Subsequent coronary angiography revealed 3-vessel CAD, patent saphenous vein grafts (SVG) to the right coronary artery (RCA) and a diagonal branch vessel with an occluded SVG to the left circumflex. Difficulty was encountered when engaging the left subclavian artery using a JR 4.0 diagnostic catheter for LIMA angiography. Nonselective angiography of the aortic arch was performed and demonstrated an uncommon anatomical variant (Figure 1, left). The right common carotid artery (CCA) [A] and the left CCA [B] arose from a single trunk, consistent with truncus bicaroticus or COCA [C]. The right subclavian artery [D] originated distal to the left subclavian artery otherwise known as arteria lusoria or ARSA forming an incomplete vascular ring [E]. Selective engagement of the left subclavian artery remained problematic even with the use of specialty arch catheters (Headhunter and LIMA catheters). The procedure concluded without confirming patency of the LIMA graft. A total of 145 mL of Omnipaque (iohexol injection) contrast was used for the procedure, and no adverse events occurred.
Same-day access of the ipsilateral ulnar artery was not pursued because of the risk of hand ischemia. The patient underwent repeat catheterization utilizing left ulnar artery access after adequate recovery time from the initial left radial approach. Selective LIMA angiography was achieved and demonstrated a patent LIMA to LAD graft. A computed tomography (CT) aorta for purposes of TAVR planning was able to reconstruct the aortic arch vasculature (Figure 1, right) confirming the presence of both ARSA and COCA. The patient went on to undergo successful TAVR with subsequent improvement of clinical symptoms.
Discussion
Arteria lusoria is defined as an anomalous right subclavian artery arising distal to the origin of the left subclavian artery on the aortic arch. It has an estimated incidence of 0.5 to 2% and occurs as a consequence of abnormal embryologic involution of the right fourth aortic arch and right proximal dorsal aorta. This causes the origin of the right subclavian artery to shift onto the descending aorta and cross the mediastinum from left to right, passing behind the esophagus and the trachea.1,3-5
ARSA is often associated with other anatomic abnormalities, including COCA, right-sided aortic arch, interrupted aortic arch, aortic coarctation, tetralogy of Fallot, truncus arteriosus, transposition of the great arteries, atrial septal defects, and ventricular septal defects.Underlying genetic disorders, such as Edwards, Down, DiGeorge syndromes, aneurysms, and arterioesophageal fistulae can accompany these vascular malformations.6
COCA, such as we encountered, is the presence of a single branch from the aorta giving off both right and left common carotid arteries. It has an incidence of < 0.1% in isolation and is discovered most often in cadaveric dissections or incidentally on imaging.1 Its embryologic origin results from the third pair of cervical aortic arches persisting as a common bicarotid trunk.1,4,5 The combination of ARSA and COCA is rare. Of the 0.5 to 2% of ARSA cases discovered, only 20% of those cases present with associated COCA for a combined prevalence estimated at < 0.05%.7
The majority of patients with either anatomic abnormality are asymptomatic. However, a few classic clinical manifestations have been described. ARSA can rarely present with dysphagia lusoria, a condition resulting from an incomplete vascular ring formed by the abnormal course of the right subclavian compressing the esophagus. Although not seen in our patient, it should be considered in the differential diagnosis for dysphagia.1,2,7 Ortner syndrome can result from right laryngeal nerve compression and palsy resultant from the aberrant course of the right subclavian artery.8 Another clinically relevant feature of ARSA is the presence of a diverticulum of Kommerell or dilatation at the origin of the right subclavian artery. It is a type of retroesophageal diverticulum resulting from persistence of a segment of the right sixth aortic arch.9 Finally, the spatial arrangement of ARSA increases risk for injury during head and neck surgical procedures, such as thyroidectomy, tracheotomy, and lymph node dissection of the right paratracheal fossa.6 Although the incidence is not well described, COCA has been described in several case reports as causing tracheal compression with dyspnea and in some cases, ischemic stroke.4,5,10
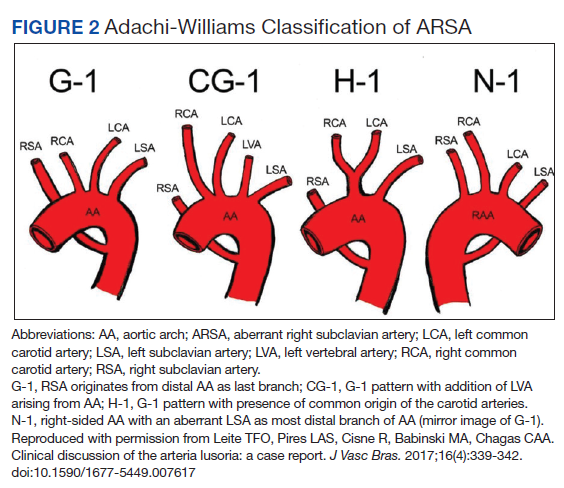
Diagnosis
The diagnosis of ARSA and COCA is often made incidentally on diagnostic imaging studies such as endovascular imaging, CT angiography, magnetic resonance (MR) angiography, postmortem cadaveric dissections, or, as in our case, during cardiac catheterization.11,12 A classification system for aortic arch branching patterns exists published by Adachi and Williams.6 The classification includes ARSA and differentiates it into 4 subtypes (Figure 2). Our patient exhibited type H-1, indicating ARSA as the distal most branch of the aortic arch with coexistence of COCA.6 The primary clinical implication of ARSA and COCA in our case was increased difficulty and complexity when performing coronary angiography. Available literature has well characterized the challenges operators encounter when cannulating aberrant great vessel anatomy, often electing to perform nonselective aortography to define a patient’s anatomy.7,9,13 A comparison of diagnostic imaging techniques for vascular rings such as ARSA have shown MR, CT, and endovascular angiography to be the most reliable modalities to delineate vascular anatomy.14
Methods
Due to the presence of CABG in our patient, left radial and ulnar artery approaches were used rather than a right radial artery approach. Engagement of the LIMA is performed most commonly with left radial or femoral artery access using an internal mammary catheter that has a more steeply angled tip (80º-85º) compared with the standard JR catheter. An accessory left radial artery anatomic variant was encountered in our case precluding left radial approach. In addition, abnormal takeoffs of the great vessels thwarted multiple attempts at intubation of the LSA (Figure 1, right). Some data suggest CT imaging can be of assistance in establishing patency of bypass grafts in CABG patients.15 This can be considered an option if branch-vessel anatomy remains unclear. Our patient exhibited several risk factors for stroke, including female gender, hypertension, and prior CABG. These and other risk factors may influence clinical decisions such as continued catheter manipulation, choice of catheter type, and further contrast studies.16
Nonselective angiography in these cases often can require excessive iodinated contrast, exposing the patient to increased risk of contrast-induced nephropathy (CIN).7,17 Although the amount of contrast used in our case was average for diagnostic catheterization,the patient went on to undergo a second catheterization and CT angiography to establish LIMA graft patency.17 CT imaging reconstruction elucidated her aberrant branch-vessel anatomy. Patients are at increased risk of CIN with contrast loads < 200 mL per study, and this effect is compounded when the patient is elderly, has diabetes mellitus, and/or antecedent renal disease.18 Attention to the patient’s preoperative glomerular filtration rate, avoidance of nephrotoxic agents, and intraoperative left ventricular end-diastolic pressure during cardiac catheterization with postcontrast administration of IV isotonic fluids have been shown to prevent CIN.19,20 In the POSEIDON trial, fluid administration on a sliding scale based on the left ventricular end-diastolic pressure resulted in lower absolute risk of CIN postcatheterization vs standard postprocedure hydration in cardiac catheterization.21 Further, the now widespread use of low and iso-osmolar contrast agents further reduces the risk of CIN.22
For cardiac catheter laboratory operators, it is important to note that ARSA is more frequently encountered due to increased use of the transradial approach to coronary angiography.11 It should be suspected when accessing the ascending aorta proves exceptionally challenging and the catheter has a predilection for entering the descending aorta.11 While more technically demanding, 2 cases described by Allen and colleagues exhibited safe and successful entry into the ascending aorta with catheter rotation and hydrophilic support wires indicating the right radial approach is feasible despite presence of ARSA.12 Several patient-initiated maneuvers can be utilized to aid in accessing the ascending aorta. For example, deep inspiration to reduce the angulation between the aortic arch and ARSA. The use of curved catheters, such as Amplatz left, internal mammary catheter, or Simmons catheter may be considered to cannulate the ascending aorta if ARSA is encountered. Complications associated with a transradial approach include dissection and intramural hematoma. Minor bleeds and vasospasm also can occur secondary to increased procedural duration.6,8
Treatment
ARSA and COCA are considered normal anatomic variants and no treatment is indicated if the patient is asymptomatic. If symptoms are present, they often arise from aneurysmal or occlusive complications of the vascular anatomy. In patients with isolated ARSA and mild dysphasia or reflux symptoms, the use of prokinetics and antireflux medications may provide relief. It is important to note the coexistence of ARSA and COCA is more likely to produce esophageal compression compared to ARSA alone due to formation of a more complete vascular ring. Surgical management has been described in severe cases of ARSA involving risk of aneurysm rupture, right upper limb ischemia, or compression of the esophagus or trachea.
Several surgical approaches have been described, including simple ligation and division of ARSA and reimplantation of the RSA into the right CCA or ascending aorta.5 A recent review of 180 cases of ARSA diagnosed on CT angiography with concomitant common carotid trunk in half of studied individuals focused on a hybrid open and intravascular procedure. This procedure involved a double transposition or bypass (LSA to left common carotid artery and ARSA to the right CCA) followed by implantation of a thoracic stent graft. Few cases are eligible for these procedures or require them for definitive treatment.23
Conclusions
Recognition of aortic arch anatomical variants such as our case of ARSA with concomitant COCA may influence clinician decisions in various specialties, such as interventional cardiology, interventional neurology, cardiothoracic surgery, and gastroenterology. While most patients with these conditions are asymptomatic, some may present with dysphagia, dyspnea, and/or stroke symptoms. In our practice, discovery of such anomalies periprocedurally may affect cardiac catheterization access site, catheter selection, and additional imaging. The presence of arteria lusoria can be of critical importance when encountering a patient with myocardial infarction as switching from transradial to transfemoral approach may be required to gain access to the ascending aorta. Overall, transradial coronary angiography and percutaneous coronary intervention is not contraindicated in the setting of ARSA/COCA and can be safely performed by an experienced operator.
It is important for surgical specialists to be aware of the coexistence of anomalies where the discovery of one aberrancy can signal coexistent variant anatomy. If aortic arch anatomy is unclear, it is useful to perform nonselective angiography and/or further imaging with CT angiography. Knowledge of abnormal aortic arch anatomy can decrease fluoroscopy time and contrast load administered, thereby reducing potential periprocedural adverse events.
1. Kurt MA, An I, Ikiz I. A case with coincidence of aberrant right subclavian artery and common origin of the carotid arteries. Ann Anat. 1997;179(2):175-176. doi:10.1016/s0940-9602(97)80100-8
2. Klinkhamer AC. Aberrant right subclavian artery. Clinical and roentgenologic aspects. Am J Roentgenol Radium Ther Nucl Med. 1966;97(2):438-446. doi:10.2214/ajr.97.2.438
3. Türkvatan A, Büyükbayraktar FG, Olçer T, Cumhur T. Congenital anomalies of the aortic arch: evaluation with the use of multidetector computed tomography. Korean J Radiol. 2009;10(2):176-184. doi:10.3348/kjr.2009.10.2.176
4. Ozateş M, Nazaroglu H, Uyar A. MR angiography in diagnosis of aberrant right subclavian artery associated with common carotid trunk. Eur Radiol. 2000;10(9):1503. doi:10.1007/s003300000335
5. Poultsides GA, Lolis ED, Vasquez J, Drezner AD, Venieratos D. Common origins of carotid and subclavian arterial systems: report of a rare aortic arch variant. Ann Vasc Surg. 2004;18(5):597-600. doi:10.1007/s10016-004-0060-3
6. Leite TFO, Pires LAS, Cisne R, Babinski MA, Chagas CAA. Clinical discussion of the arteria lusoria: a case report. J Vasc Bras. 2017;16(4):339-342. doi:10.1590/1677-5449.007617
7. Tsai IC, Tzeng WS, Lee T, et al. Vertebral and carotid artery anomalies in patients with aberrant right subclavian arteries. Pediatr Radiol. 2007;37(10):1007-1012. doi:10.1007/s00247-007-0574-2
8. Rafiq A, Chutani S, Krim NR. Incidental finding of arteria lusoria during transradial coronary catheterization: significance in interventional cardiology. Catheter Cardiovasc Interv. 2018;91(7):1283-1286. doi:10.1002/ccd.27439
9. Priya S, Thomas R, Nagpal P, Sharma A, Steigner M. Congenital anomalies of the aortic arch. Cardiovasc Diagn Ther. 2018;8(suppl 1):S26-S44. doi:10.21037/cdt.2017.10.15
10. Khatri R, Maud A, Rodriguez GJ. Aberrant right subclavian artery and common carotid trunk. J Vasc Interv Neurol. 2010;3(1):33-34.
11. Valsecchi O, Vassileva A, Musumeci G, et al. Failure of transradial approach during coronary interventions: anatomic considerations. Catheter Cardiovasc Interv. 2006;67(6):870-878. doi:10.1002/ccd.20732
12. Allen D, Bews H, Vo M, Kass M, Jassal DS, Ravandi A. Arteria lusoria: an anomalous finding during right transradial coronary intervention. Case Rep Cardiol. 2016;2016:8079856. doi:10.1155/2016/8079856
13. Fineschi M, Iadanza A, Sinicropi G, Pierli C. Images in cardiology: angiographic evidence of aberrant right subclavian artery associated with common carotid trunk. Heart. 2002;88(2):158. doi:10.1136/heart.88.2.158
14. van Son JA, Julsrud PR, Hagler DJ, et al. Imaging strategies for vascular rings. Ann Thorac Surg. 1994;57(3):604-610. doi:10.1016/0003-4975(94)90552-5
15. Lee R, Lim J, Kaw G, Wan G, Ng K, Ho KT. Comprehensive noninvasive evaluation of bypass grafts and native coronary arteries in patients after coronary bypass surgery: accuracy of 64-slice multidetector computed tomography compared to invasive coronary angiography. J Cardiovasc Med (Hagerstown). 2010;11(2):81-90. doi:10.2459/JCM.0b013e32832f3e2e
16. Hamon M, Baron JC, Viader F, Hamon M. Periprocedural stroke and cardiac catheterization. Circulation. 2008;118(6): 678-683. doi:10.1161/CIRCULATIONAHA.108.784504
17. Hwang JR, D’Alfonso S, Kostuk WJ, et al. Contrast volume use in manual vs automated contrast injection systems for diagnostic coronary angiography and percutaneous coronary interventions. Can J Cardiol. 2013;29(3):372-376. doi:10.1016/j.cjca.2012.11.023
18. Rich MW, Crecelius CA. Incidence, risk factors, and clinical course of acute renal insufficiency after cardiac catheterization in patients 70 years of age or older. A prospective study. Arch Intern Med. 1990;150(6):1237-1242.
19. Davenport MS, Khalatbari S, Cohan RH, Dillman JR, Myles JD, Ellis JH. Contrast material-induced nephrotoxicity and intravenous low-osmolality iodinated contrast material: risk stratification by using estimated glomerular filtration rate. Radiology. 2013;268(3):719-728. doi:10.1148/radiol.13122276
20. American College of Radiology. ACR Manual on Contrast Media 2020. American College of Radiology; 2020:33-34. Accessed January 15, 2021. https://www.acr.org/-/media/ACR/Files/Clinical-Resources/Contrast_Media.pdf
21. Brar SS, Aharonian V, Mansukhani P, et al. Haemodynamic-guided fluid administration for the prevention of contrast-induced acute kidney injury: the POSEIDON randomised controlled trial. Lancet. 2014;383(9931):1814-1823. doi:10.1016/S0140-6736(14)60689-9
22. Aoun J, Nicolas D, Brown JR, Jaber BL. Maximum allowable contrast dose and prevention of acute kidney injury following cardiovascular procedures. Curr Opin Nephrol Hypertens. 2018;27(2):121-129. doi:10.1097/MNH.0000000000000389
23. Settembre N, Saba C, Bouziane Z, Jeannon F, Mandry D, Malikov S. Hybrid treatment of the aberrant right subclavian artery (arteria lusoria): feasibility study on 180 angio-CTs. Ann Vasc Surg. 2017;44:229-233. doi:10.1016/j.avsg.2017.03.172
While most patients with arteria lusoria and common carotid trunk conditions are asymptomatic, discovery of such anomalies periprocedurally may affect the cardiac catheterization access site, catheter selection, and additional imaging.
While most patients with arteria lusoria and common carotid trunk conditions are asymptomatic, discovery of such anomalies periprocedurally may affect the cardiac catheterization access site, catheter selection, and additional imaging.
Branching of the great vessels from the aorta normally progresses with the brachiocephalic trunk as the first takeoff followed by the left common carotid and left subclavian artery in approximately 85% of cases.1 Variants of great vessel branching patterns include the so-called bovine arch, arteria lusoria or aberrant right subclavian artery (ARSA), aberrant origin of the vertebral arteries, and truncus bicaroticus, or common origin of the carotid arteries (COCA). These aberrancies are quite rare, some with an incidence of < 1%.1,2
These vascular anomalies become clinically relevant when they pose difficulty for operators in surgical and interventional specialties, necessitating unique approaches, catheters, and techniques to overcome. We present a case of concomitant aortic arch abnormalities during a diagnostic workup for transcatheter aortic valve replacement (TAVR) in a patient with previous coronary artery bypass grafting (CABG).
Case Presentation
A 66-year-old woman with coronary artery disease (CAD) status post-CABG and stage D1 aortic stenosis (AS) presented with exertional dyspnea. She was referred for coronary angiography as part of a workup for TAVR. Echocardiography confirmed severe AS with a peak velocity of 4.1 m/s, mean pressure gradient of 50 mm Hg, and an aortic valve area of 0.7 cm2. The patient was scheduled for cardiac catheterization with anticipated left radial artery approach for intubation and opacification of the left internal mammary artery (LIMA). However, this approach was abandoned during the procedure due to discovery of aberrant left radial artery anatomy, and the procedure was completed via femoral access.
Subsequent coronary angiography revealed 3-vessel CAD, patent saphenous vein grafts (SVG) to the right coronary artery (RCA) and a diagonal branch vessel with an occluded SVG to the left circumflex. Difficulty was encountered when engaging the left subclavian artery using a JR 4.0 diagnostic catheter for LIMA angiography. Nonselective angiography of the aortic arch was performed and demonstrated an uncommon anatomical variant (Figure 1, left). The right common carotid artery (CCA) [A] and the left CCA [B] arose from a single trunk, consistent with truncus bicaroticus or COCA [C]. The right subclavian artery [D] originated distal to the left subclavian artery otherwise known as arteria lusoria or ARSA forming an incomplete vascular ring [E]. Selective engagement of the left subclavian artery remained problematic even with the use of specialty arch catheters (Headhunter and LIMA catheters). The procedure concluded without confirming patency of the LIMA graft. A total of 145 mL of Omnipaque (iohexol injection) contrast was used for the procedure, and no adverse events occurred.
Same-day access of the ipsilateral ulnar artery was not pursued because of the risk of hand ischemia. The patient underwent repeat catheterization utilizing left ulnar artery access after adequate recovery time from the initial left radial approach. Selective LIMA angiography was achieved and demonstrated a patent LIMA to LAD graft. A computed tomography (CT) aorta for purposes of TAVR planning was able to reconstruct the aortic arch vasculature (Figure 1, right) confirming the presence of both ARSA and COCA. The patient went on to undergo successful TAVR with subsequent improvement of clinical symptoms.
Discussion
Arteria lusoria is defined as an anomalous right subclavian artery arising distal to the origin of the left subclavian artery on the aortic arch. It has an estimated incidence of 0.5 to 2% and occurs as a consequence of abnormal embryologic involution of the right fourth aortic arch and right proximal dorsal aorta. This causes the origin of the right subclavian artery to shift onto the descending aorta and cross the mediastinum from left to right, passing behind the esophagus and the trachea.1,3-5
ARSA is often associated with other anatomic abnormalities, including COCA, right-sided aortic arch, interrupted aortic arch, aortic coarctation, tetralogy of Fallot, truncus arteriosus, transposition of the great arteries, atrial septal defects, and ventricular septal defects.Underlying genetic disorders, such as Edwards, Down, DiGeorge syndromes, aneurysms, and arterioesophageal fistulae can accompany these vascular malformations.6
COCA, such as we encountered, is the presence of a single branch from the aorta giving off both right and left common carotid arteries. It has an incidence of < 0.1% in isolation and is discovered most often in cadaveric dissections or incidentally on imaging.1 Its embryologic origin results from the third pair of cervical aortic arches persisting as a common bicarotid trunk.1,4,5 The combination of ARSA and COCA is rare. Of the 0.5 to 2% of ARSA cases discovered, only 20% of those cases present with associated COCA for a combined prevalence estimated at < 0.05%.7
The majority of patients with either anatomic abnormality are asymptomatic. However, a few classic clinical manifestations have been described. ARSA can rarely present with dysphagia lusoria, a condition resulting from an incomplete vascular ring formed by the abnormal course of the right subclavian compressing the esophagus. Although not seen in our patient, it should be considered in the differential diagnosis for dysphagia.1,2,7 Ortner syndrome can result from right laryngeal nerve compression and palsy resultant from the aberrant course of the right subclavian artery.8 Another clinically relevant feature of ARSA is the presence of a diverticulum of Kommerell or dilatation at the origin of the right subclavian artery. It is a type of retroesophageal diverticulum resulting from persistence of a segment of the right sixth aortic arch.9 Finally, the spatial arrangement of ARSA increases risk for injury during head and neck surgical procedures, such as thyroidectomy, tracheotomy, and lymph node dissection of the right paratracheal fossa.6 Although the incidence is not well described, COCA has been described in several case reports as causing tracheal compression with dyspnea and in some cases, ischemic stroke.4,5,10
Diagnosis
The diagnosis of ARSA and COCA is often made incidentally on diagnostic imaging studies such as endovascular imaging, CT angiography, magnetic resonance (MR) angiography, postmortem cadaveric dissections, or, as in our case, during cardiac catheterization.11,12 A classification system for aortic arch branching patterns exists published by Adachi and Williams.6 The classification includes ARSA and differentiates it into 4 subtypes (Figure 2). Our patient exhibited type H-1, indicating ARSA as the distal most branch of the aortic arch with coexistence of COCA.6 The primary clinical implication of ARSA and COCA in our case was increased difficulty and complexity when performing coronary angiography. Available literature has well characterized the challenges operators encounter when cannulating aberrant great vessel anatomy, often electing to perform nonselective aortography to define a patient’s anatomy.7,9,13 A comparison of diagnostic imaging techniques for vascular rings such as ARSA have shown MR, CT, and endovascular angiography to be the most reliable modalities to delineate vascular anatomy.14
Methods
Due to the presence of CABG in our patient, left radial and ulnar artery approaches were used rather than a right radial artery approach. Engagement of the LIMA is performed most commonly with left radial or femoral artery access using an internal mammary catheter that has a more steeply angled tip (80º-85º) compared with the standard JR catheter. An accessory left radial artery anatomic variant was encountered in our case precluding left radial approach. In addition, abnormal takeoffs of the great vessels thwarted multiple attempts at intubation of the LSA (Figure 1, right). Some data suggest CT imaging can be of assistance in establishing patency of bypass grafts in CABG patients.15 This can be considered an option if branch-vessel anatomy remains unclear. Our patient exhibited several risk factors for stroke, including female gender, hypertension, and prior CABG. These and other risk factors may influence clinical decisions such as continued catheter manipulation, choice of catheter type, and further contrast studies.16
Nonselective angiography in these cases often can require excessive iodinated contrast, exposing the patient to increased risk of contrast-induced nephropathy (CIN).7,17 Although the amount of contrast used in our case was average for diagnostic catheterization,the patient went on to undergo a second catheterization and CT angiography to establish LIMA graft patency.17 CT imaging reconstruction elucidated her aberrant branch-vessel anatomy. Patients are at increased risk of CIN with contrast loads < 200 mL per study, and this effect is compounded when the patient is elderly, has diabetes mellitus, and/or antecedent renal disease.18 Attention to the patient’s preoperative glomerular filtration rate, avoidance of nephrotoxic agents, and intraoperative left ventricular end-diastolic pressure during cardiac catheterization with postcontrast administration of IV isotonic fluids have been shown to prevent CIN.19,20 In the POSEIDON trial, fluid administration on a sliding scale based on the left ventricular end-diastolic pressure resulted in lower absolute risk of CIN postcatheterization vs standard postprocedure hydration in cardiac catheterization.21 Further, the now widespread use of low and iso-osmolar contrast agents further reduces the risk of CIN.22
For cardiac catheter laboratory operators, it is important to note that ARSA is more frequently encountered due to increased use of the transradial approach to coronary angiography.11 It should be suspected when accessing the ascending aorta proves exceptionally challenging and the catheter has a predilection for entering the descending aorta.11 While more technically demanding, 2 cases described by Allen and colleagues exhibited safe and successful entry into the ascending aorta with catheter rotation and hydrophilic support wires indicating the right radial approach is feasible despite presence of ARSA.12 Several patient-initiated maneuvers can be utilized to aid in accessing the ascending aorta. For example, deep inspiration to reduce the angulation between the aortic arch and ARSA. The use of curved catheters, such as Amplatz left, internal mammary catheter, or Simmons catheter may be considered to cannulate the ascending aorta if ARSA is encountered. Complications associated with a transradial approach include dissection and intramural hematoma. Minor bleeds and vasospasm also can occur secondary to increased procedural duration.6,8
Treatment
ARSA and COCA are considered normal anatomic variants and no treatment is indicated if the patient is asymptomatic. If symptoms are present, they often arise from aneurysmal or occlusive complications of the vascular anatomy. In patients with isolated ARSA and mild dysphasia or reflux symptoms, the use of prokinetics and antireflux medications may provide relief. It is important to note the coexistence of ARSA and COCA is more likely to produce esophageal compression compared to ARSA alone due to formation of a more complete vascular ring. Surgical management has been described in severe cases of ARSA involving risk of aneurysm rupture, right upper limb ischemia, or compression of the esophagus or trachea.
Several surgical approaches have been described, including simple ligation and division of ARSA and reimplantation of the RSA into the right CCA or ascending aorta.5 A recent review of 180 cases of ARSA diagnosed on CT angiography with concomitant common carotid trunk in half of studied individuals focused on a hybrid open and intravascular procedure. This procedure involved a double transposition or bypass (LSA to left common carotid artery and ARSA to the right CCA) followed by implantation of a thoracic stent graft. Few cases are eligible for these procedures or require them for definitive treatment.23
Conclusions
Recognition of aortic arch anatomical variants such as our case of ARSA with concomitant COCA may influence clinician decisions in various specialties, such as interventional cardiology, interventional neurology, cardiothoracic surgery, and gastroenterology. While most patients with these conditions are asymptomatic, some may present with dysphagia, dyspnea, and/or stroke symptoms. In our practice, discovery of such anomalies periprocedurally may affect cardiac catheterization access site, catheter selection, and additional imaging. The presence of arteria lusoria can be of critical importance when encountering a patient with myocardial infarction as switching from transradial to transfemoral approach may be required to gain access to the ascending aorta. Overall, transradial coronary angiography and percutaneous coronary intervention is not contraindicated in the setting of ARSA/COCA and can be safely performed by an experienced operator.
It is important for surgical specialists to be aware of the coexistence of anomalies where the discovery of one aberrancy can signal coexistent variant anatomy. If aortic arch anatomy is unclear, it is useful to perform nonselective angiography and/or further imaging with CT angiography. Knowledge of abnormal aortic arch anatomy can decrease fluoroscopy time and contrast load administered, thereby reducing potential periprocedural adverse events.
Branching of the great vessels from the aorta normally progresses with the brachiocephalic trunk as the first takeoff followed by the left common carotid and left subclavian artery in approximately 85% of cases.1 Variants of great vessel branching patterns include the so-called bovine arch, arteria lusoria or aberrant right subclavian artery (ARSA), aberrant origin of the vertebral arteries, and truncus bicaroticus, or common origin of the carotid arteries (COCA). These aberrancies are quite rare, some with an incidence of < 1%.1,2
These vascular anomalies become clinically relevant when they pose difficulty for operators in surgical and interventional specialties, necessitating unique approaches, catheters, and techniques to overcome. We present a case of concomitant aortic arch abnormalities during a diagnostic workup for transcatheter aortic valve replacement (TAVR) in a patient with previous coronary artery bypass grafting (CABG).
Case Presentation
A 66-year-old woman with coronary artery disease (CAD) status post-CABG and stage D1 aortic stenosis (AS) presented with exertional dyspnea. She was referred for coronary angiography as part of a workup for TAVR. Echocardiography confirmed severe AS with a peak velocity of 4.1 m/s, mean pressure gradient of 50 mm Hg, and an aortic valve area of 0.7 cm2. The patient was scheduled for cardiac catheterization with anticipated left radial artery approach for intubation and opacification of the left internal mammary artery (LIMA). However, this approach was abandoned during the procedure due to discovery of aberrant left radial artery anatomy, and the procedure was completed via femoral access.
Subsequent coronary angiography revealed 3-vessel CAD, patent saphenous vein grafts (SVG) to the right coronary artery (RCA) and a diagonal branch vessel with an occluded SVG to the left circumflex. Difficulty was encountered when engaging the left subclavian artery using a JR 4.0 diagnostic catheter for LIMA angiography. Nonselective angiography of the aortic arch was performed and demonstrated an uncommon anatomical variant (Figure 1, left). The right common carotid artery (CCA) [A] and the left CCA [B] arose from a single trunk, consistent with truncus bicaroticus or COCA [C]. The right subclavian artery [D] originated distal to the left subclavian artery otherwise known as arteria lusoria or ARSA forming an incomplete vascular ring [E]. Selective engagement of the left subclavian artery remained problematic even with the use of specialty arch catheters (Headhunter and LIMA catheters). The procedure concluded without confirming patency of the LIMA graft. A total of 145 mL of Omnipaque (iohexol injection) contrast was used for the procedure, and no adverse events occurred.
Same-day access of the ipsilateral ulnar artery was not pursued because of the risk of hand ischemia. The patient underwent repeat catheterization utilizing left ulnar artery access after adequate recovery time from the initial left radial approach. Selective LIMA angiography was achieved and demonstrated a patent LIMA to LAD graft. A computed tomography (CT) aorta for purposes of TAVR planning was able to reconstruct the aortic arch vasculature (Figure 1, right) confirming the presence of both ARSA and COCA. The patient went on to undergo successful TAVR with subsequent improvement of clinical symptoms.
Discussion
Arteria lusoria is defined as an anomalous right subclavian artery arising distal to the origin of the left subclavian artery on the aortic arch. It has an estimated incidence of 0.5 to 2% and occurs as a consequence of abnormal embryologic involution of the right fourth aortic arch and right proximal dorsal aorta. This causes the origin of the right subclavian artery to shift onto the descending aorta and cross the mediastinum from left to right, passing behind the esophagus and the trachea.1,3-5
ARSA is often associated with other anatomic abnormalities, including COCA, right-sided aortic arch, interrupted aortic arch, aortic coarctation, tetralogy of Fallot, truncus arteriosus, transposition of the great arteries, atrial septal defects, and ventricular septal defects.Underlying genetic disorders, such as Edwards, Down, DiGeorge syndromes, aneurysms, and arterioesophageal fistulae can accompany these vascular malformations.6
COCA, such as we encountered, is the presence of a single branch from the aorta giving off both right and left common carotid arteries. It has an incidence of < 0.1% in isolation and is discovered most often in cadaveric dissections or incidentally on imaging.1 Its embryologic origin results from the third pair of cervical aortic arches persisting as a common bicarotid trunk.1,4,5 The combination of ARSA and COCA is rare. Of the 0.5 to 2% of ARSA cases discovered, only 20% of those cases present with associated COCA for a combined prevalence estimated at < 0.05%.7
The majority of patients with either anatomic abnormality are asymptomatic. However, a few classic clinical manifestations have been described. ARSA can rarely present with dysphagia lusoria, a condition resulting from an incomplete vascular ring formed by the abnormal course of the right subclavian compressing the esophagus. Although not seen in our patient, it should be considered in the differential diagnosis for dysphagia.1,2,7 Ortner syndrome can result from right laryngeal nerve compression and palsy resultant from the aberrant course of the right subclavian artery.8 Another clinically relevant feature of ARSA is the presence of a diverticulum of Kommerell or dilatation at the origin of the right subclavian artery. It is a type of retroesophageal diverticulum resulting from persistence of a segment of the right sixth aortic arch.9 Finally, the spatial arrangement of ARSA increases risk for injury during head and neck surgical procedures, such as thyroidectomy, tracheotomy, and lymph node dissection of the right paratracheal fossa.6 Although the incidence is not well described, COCA has been described in several case reports as causing tracheal compression with dyspnea and in some cases, ischemic stroke.4,5,10
Diagnosis
The diagnosis of ARSA and COCA is often made incidentally on diagnostic imaging studies such as endovascular imaging, CT angiography, magnetic resonance (MR) angiography, postmortem cadaveric dissections, or, as in our case, during cardiac catheterization.11,12 A classification system for aortic arch branching patterns exists published by Adachi and Williams.6 The classification includes ARSA and differentiates it into 4 subtypes (Figure 2). Our patient exhibited type H-1, indicating ARSA as the distal most branch of the aortic arch with coexistence of COCA.6 The primary clinical implication of ARSA and COCA in our case was increased difficulty and complexity when performing coronary angiography. Available literature has well characterized the challenges operators encounter when cannulating aberrant great vessel anatomy, often electing to perform nonselective aortography to define a patient’s anatomy.7,9,13 A comparison of diagnostic imaging techniques for vascular rings such as ARSA have shown MR, CT, and endovascular angiography to be the most reliable modalities to delineate vascular anatomy.14
Methods
Due to the presence of CABG in our patient, left radial and ulnar artery approaches were used rather than a right radial artery approach. Engagement of the LIMA is performed most commonly with left radial or femoral artery access using an internal mammary catheter that has a more steeply angled tip (80º-85º) compared with the standard JR catheter. An accessory left radial artery anatomic variant was encountered in our case precluding left radial approach. In addition, abnormal takeoffs of the great vessels thwarted multiple attempts at intubation of the LSA (Figure 1, right). Some data suggest CT imaging can be of assistance in establishing patency of bypass grafts in CABG patients.15 This can be considered an option if branch-vessel anatomy remains unclear. Our patient exhibited several risk factors for stroke, including female gender, hypertension, and prior CABG. These and other risk factors may influence clinical decisions such as continued catheter manipulation, choice of catheter type, and further contrast studies.16
Nonselective angiography in these cases often can require excessive iodinated contrast, exposing the patient to increased risk of contrast-induced nephropathy (CIN).7,17 Although the amount of contrast used in our case was average for diagnostic catheterization,the patient went on to undergo a second catheterization and CT angiography to establish LIMA graft patency.17 CT imaging reconstruction elucidated her aberrant branch-vessel anatomy. Patients are at increased risk of CIN with contrast loads < 200 mL per study, and this effect is compounded when the patient is elderly, has diabetes mellitus, and/or antecedent renal disease.18 Attention to the patient’s preoperative glomerular filtration rate, avoidance of nephrotoxic agents, and intraoperative left ventricular end-diastolic pressure during cardiac catheterization with postcontrast administration of IV isotonic fluids have been shown to prevent CIN.19,20 In the POSEIDON trial, fluid administration on a sliding scale based on the left ventricular end-diastolic pressure resulted in lower absolute risk of CIN postcatheterization vs standard postprocedure hydration in cardiac catheterization.21 Further, the now widespread use of low and iso-osmolar contrast agents further reduces the risk of CIN.22
For cardiac catheter laboratory operators, it is important to note that ARSA is more frequently encountered due to increased use of the transradial approach to coronary angiography.11 It should be suspected when accessing the ascending aorta proves exceptionally challenging and the catheter has a predilection for entering the descending aorta.11 While more technically demanding, 2 cases described by Allen and colleagues exhibited safe and successful entry into the ascending aorta with catheter rotation and hydrophilic support wires indicating the right radial approach is feasible despite presence of ARSA.12 Several patient-initiated maneuvers can be utilized to aid in accessing the ascending aorta. For example, deep inspiration to reduce the angulation between the aortic arch and ARSA. The use of curved catheters, such as Amplatz left, internal mammary catheter, or Simmons catheter may be considered to cannulate the ascending aorta if ARSA is encountered. Complications associated with a transradial approach include dissection and intramural hematoma. Minor bleeds and vasospasm also can occur secondary to increased procedural duration.6,8
Treatment
ARSA and COCA are considered normal anatomic variants and no treatment is indicated if the patient is asymptomatic. If symptoms are present, they often arise from aneurysmal or occlusive complications of the vascular anatomy. In patients with isolated ARSA and mild dysphasia or reflux symptoms, the use of prokinetics and antireflux medications may provide relief. It is important to note the coexistence of ARSA and COCA is more likely to produce esophageal compression compared to ARSA alone due to formation of a more complete vascular ring. Surgical management has been described in severe cases of ARSA involving risk of aneurysm rupture, right upper limb ischemia, or compression of the esophagus or trachea.
Several surgical approaches have been described, including simple ligation and division of ARSA and reimplantation of the RSA into the right CCA or ascending aorta.5 A recent review of 180 cases of ARSA diagnosed on CT angiography with concomitant common carotid trunk in half of studied individuals focused on a hybrid open and intravascular procedure. This procedure involved a double transposition or bypass (LSA to left common carotid artery and ARSA to the right CCA) followed by implantation of a thoracic stent graft. Few cases are eligible for these procedures or require them for definitive treatment.23
Conclusions
Recognition of aortic arch anatomical variants such as our case of ARSA with concomitant COCA may influence clinician decisions in various specialties, such as interventional cardiology, interventional neurology, cardiothoracic surgery, and gastroenterology. While most patients with these conditions are asymptomatic, some may present with dysphagia, dyspnea, and/or stroke symptoms. In our practice, discovery of such anomalies periprocedurally may affect cardiac catheterization access site, catheter selection, and additional imaging. The presence of arteria lusoria can be of critical importance when encountering a patient with myocardial infarction as switching from transradial to transfemoral approach may be required to gain access to the ascending aorta. Overall, transradial coronary angiography and percutaneous coronary intervention is not contraindicated in the setting of ARSA/COCA and can be safely performed by an experienced operator.
It is important for surgical specialists to be aware of the coexistence of anomalies where the discovery of one aberrancy can signal coexistent variant anatomy. If aortic arch anatomy is unclear, it is useful to perform nonselective angiography and/or further imaging with CT angiography. Knowledge of abnormal aortic arch anatomy can decrease fluoroscopy time and contrast load administered, thereby reducing potential periprocedural adverse events.
1. Kurt MA, An I, Ikiz I. A case with coincidence of aberrant right subclavian artery and common origin of the carotid arteries. Ann Anat. 1997;179(2):175-176. doi:10.1016/s0940-9602(97)80100-8
2. Klinkhamer AC. Aberrant right subclavian artery. Clinical and roentgenologic aspects. Am J Roentgenol Radium Ther Nucl Med. 1966;97(2):438-446. doi:10.2214/ajr.97.2.438
3. Türkvatan A, Büyükbayraktar FG, Olçer T, Cumhur T. Congenital anomalies of the aortic arch: evaluation with the use of multidetector computed tomography. Korean J Radiol. 2009;10(2):176-184. doi:10.3348/kjr.2009.10.2.176
4. Ozateş M, Nazaroglu H, Uyar A. MR angiography in diagnosis of aberrant right subclavian artery associated with common carotid trunk. Eur Radiol. 2000;10(9):1503. doi:10.1007/s003300000335
5. Poultsides GA, Lolis ED, Vasquez J, Drezner AD, Venieratos D. Common origins of carotid and subclavian arterial systems: report of a rare aortic arch variant. Ann Vasc Surg. 2004;18(5):597-600. doi:10.1007/s10016-004-0060-3
6. Leite TFO, Pires LAS, Cisne R, Babinski MA, Chagas CAA. Clinical discussion of the arteria lusoria: a case report. J Vasc Bras. 2017;16(4):339-342. doi:10.1590/1677-5449.007617
7. Tsai IC, Tzeng WS, Lee T, et al. Vertebral and carotid artery anomalies in patients with aberrant right subclavian arteries. Pediatr Radiol. 2007;37(10):1007-1012. doi:10.1007/s00247-007-0574-2
8. Rafiq A, Chutani S, Krim NR. Incidental finding of arteria lusoria during transradial coronary catheterization: significance in interventional cardiology. Catheter Cardiovasc Interv. 2018;91(7):1283-1286. doi:10.1002/ccd.27439
9. Priya S, Thomas R, Nagpal P, Sharma A, Steigner M. Congenital anomalies of the aortic arch. Cardiovasc Diagn Ther. 2018;8(suppl 1):S26-S44. doi:10.21037/cdt.2017.10.15
10. Khatri R, Maud A, Rodriguez GJ. Aberrant right subclavian artery and common carotid trunk. J Vasc Interv Neurol. 2010;3(1):33-34.
11. Valsecchi O, Vassileva A, Musumeci G, et al. Failure of transradial approach during coronary interventions: anatomic considerations. Catheter Cardiovasc Interv. 2006;67(6):870-878. doi:10.1002/ccd.20732
12. Allen D, Bews H, Vo M, Kass M, Jassal DS, Ravandi A. Arteria lusoria: an anomalous finding during right transradial coronary intervention. Case Rep Cardiol. 2016;2016:8079856. doi:10.1155/2016/8079856
13. Fineschi M, Iadanza A, Sinicropi G, Pierli C. Images in cardiology: angiographic evidence of aberrant right subclavian artery associated with common carotid trunk. Heart. 2002;88(2):158. doi:10.1136/heart.88.2.158
14. van Son JA, Julsrud PR, Hagler DJ, et al. Imaging strategies for vascular rings. Ann Thorac Surg. 1994;57(3):604-610. doi:10.1016/0003-4975(94)90552-5
15. Lee R, Lim J, Kaw G, Wan G, Ng K, Ho KT. Comprehensive noninvasive evaluation of bypass grafts and native coronary arteries in patients after coronary bypass surgery: accuracy of 64-slice multidetector computed tomography compared to invasive coronary angiography. J Cardiovasc Med (Hagerstown). 2010;11(2):81-90. doi:10.2459/JCM.0b013e32832f3e2e
16. Hamon M, Baron JC, Viader F, Hamon M. Periprocedural stroke and cardiac catheterization. Circulation. 2008;118(6): 678-683. doi:10.1161/CIRCULATIONAHA.108.784504
17. Hwang JR, D’Alfonso S, Kostuk WJ, et al. Contrast volume use in manual vs automated contrast injection systems for diagnostic coronary angiography and percutaneous coronary interventions. Can J Cardiol. 2013;29(3):372-376. doi:10.1016/j.cjca.2012.11.023
18. Rich MW, Crecelius CA. Incidence, risk factors, and clinical course of acute renal insufficiency after cardiac catheterization in patients 70 years of age or older. A prospective study. Arch Intern Med. 1990;150(6):1237-1242.
19. Davenport MS, Khalatbari S, Cohan RH, Dillman JR, Myles JD, Ellis JH. Contrast material-induced nephrotoxicity and intravenous low-osmolality iodinated contrast material: risk stratification by using estimated glomerular filtration rate. Radiology. 2013;268(3):719-728. doi:10.1148/radiol.13122276
20. American College of Radiology. ACR Manual on Contrast Media 2020. American College of Radiology; 2020:33-34. Accessed January 15, 2021. https://www.acr.org/-/media/ACR/Files/Clinical-Resources/Contrast_Media.pdf
21. Brar SS, Aharonian V, Mansukhani P, et al. Haemodynamic-guided fluid administration for the prevention of contrast-induced acute kidney injury: the POSEIDON randomised controlled trial. Lancet. 2014;383(9931):1814-1823. doi:10.1016/S0140-6736(14)60689-9
22. Aoun J, Nicolas D, Brown JR, Jaber BL. Maximum allowable contrast dose and prevention of acute kidney injury following cardiovascular procedures. Curr Opin Nephrol Hypertens. 2018;27(2):121-129. doi:10.1097/MNH.0000000000000389
23. Settembre N, Saba C, Bouziane Z, Jeannon F, Mandry D, Malikov S. Hybrid treatment of the aberrant right subclavian artery (arteria lusoria): feasibility study on 180 angio-CTs. Ann Vasc Surg. 2017;44:229-233. doi:10.1016/j.avsg.2017.03.172
1. Kurt MA, An I, Ikiz I. A case with coincidence of aberrant right subclavian artery and common origin of the carotid arteries. Ann Anat. 1997;179(2):175-176. doi:10.1016/s0940-9602(97)80100-8
2. Klinkhamer AC. Aberrant right subclavian artery. Clinical and roentgenologic aspects. Am J Roentgenol Radium Ther Nucl Med. 1966;97(2):438-446. doi:10.2214/ajr.97.2.438
3. Türkvatan A, Büyükbayraktar FG, Olçer T, Cumhur T. Congenital anomalies of the aortic arch: evaluation with the use of multidetector computed tomography. Korean J Radiol. 2009;10(2):176-184. doi:10.3348/kjr.2009.10.2.176
4. Ozateş M, Nazaroglu H, Uyar A. MR angiography in diagnosis of aberrant right subclavian artery associated with common carotid trunk. Eur Radiol. 2000;10(9):1503. doi:10.1007/s003300000335
5. Poultsides GA, Lolis ED, Vasquez J, Drezner AD, Venieratos D. Common origins of carotid and subclavian arterial systems: report of a rare aortic arch variant. Ann Vasc Surg. 2004;18(5):597-600. doi:10.1007/s10016-004-0060-3
6. Leite TFO, Pires LAS, Cisne R, Babinski MA, Chagas CAA. Clinical discussion of the arteria lusoria: a case report. J Vasc Bras. 2017;16(4):339-342. doi:10.1590/1677-5449.007617
7. Tsai IC, Tzeng WS, Lee T, et al. Vertebral and carotid artery anomalies in patients with aberrant right subclavian arteries. Pediatr Radiol. 2007;37(10):1007-1012. doi:10.1007/s00247-007-0574-2
8. Rafiq A, Chutani S, Krim NR. Incidental finding of arteria lusoria during transradial coronary catheterization: significance in interventional cardiology. Catheter Cardiovasc Interv. 2018;91(7):1283-1286. doi:10.1002/ccd.27439
9. Priya S, Thomas R, Nagpal P, Sharma A, Steigner M. Congenital anomalies of the aortic arch. Cardiovasc Diagn Ther. 2018;8(suppl 1):S26-S44. doi:10.21037/cdt.2017.10.15
10. Khatri R, Maud A, Rodriguez GJ. Aberrant right subclavian artery and common carotid trunk. J Vasc Interv Neurol. 2010;3(1):33-34.
11. Valsecchi O, Vassileva A, Musumeci G, et al. Failure of transradial approach during coronary interventions: anatomic considerations. Catheter Cardiovasc Interv. 2006;67(6):870-878. doi:10.1002/ccd.20732
12. Allen D, Bews H, Vo M, Kass M, Jassal DS, Ravandi A. Arteria lusoria: an anomalous finding during right transradial coronary intervention. Case Rep Cardiol. 2016;2016:8079856. doi:10.1155/2016/8079856
13. Fineschi M, Iadanza A, Sinicropi G, Pierli C. Images in cardiology: angiographic evidence of aberrant right subclavian artery associated with common carotid trunk. Heart. 2002;88(2):158. doi:10.1136/heart.88.2.158
14. van Son JA, Julsrud PR, Hagler DJ, et al. Imaging strategies for vascular rings. Ann Thorac Surg. 1994;57(3):604-610. doi:10.1016/0003-4975(94)90552-5
15. Lee R, Lim J, Kaw G, Wan G, Ng K, Ho KT. Comprehensive noninvasive evaluation of bypass grafts and native coronary arteries in patients after coronary bypass surgery: accuracy of 64-slice multidetector computed tomography compared to invasive coronary angiography. J Cardiovasc Med (Hagerstown). 2010;11(2):81-90. doi:10.2459/JCM.0b013e32832f3e2e
16. Hamon M, Baron JC, Viader F, Hamon M. Periprocedural stroke and cardiac catheterization. Circulation. 2008;118(6): 678-683. doi:10.1161/CIRCULATIONAHA.108.784504
17. Hwang JR, D’Alfonso S, Kostuk WJ, et al. Contrast volume use in manual vs automated contrast injection systems for diagnostic coronary angiography and percutaneous coronary interventions. Can J Cardiol. 2013;29(3):372-376. doi:10.1016/j.cjca.2012.11.023
18. Rich MW, Crecelius CA. Incidence, risk factors, and clinical course of acute renal insufficiency after cardiac catheterization in patients 70 years of age or older. A prospective study. Arch Intern Med. 1990;150(6):1237-1242.
19. Davenport MS, Khalatbari S, Cohan RH, Dillman JR, Myles JD, Ellis JH. Contrast material-induced nephrotoxicity and intravenous low-osmolality iodinated contrast material: risk stratification by using estimated glomerular filtration rate. Radiology. 2013;268(3):719-728. doi:10.1148/radiol.13122276
20. American College of Radiology. ACR Manual on Contrast Media 2020. American College of Radiology; 2020:33-34. Accessed January 15, 2021. https://www.acr.org/-/media/ACR/Files/Clinical-Resources/Contrast_Media.pdf
21. Brar SS, Aharonian V, Mansukhani P, et al. Haemodynamic-guided fluid administration for the prevention of contrast-induced acute kidney injury: the POSEIDON randomised controlled trial. Lancet. 2014;383(9931):1814-1823. doi:10.1016/S0140-6736(14)60689-9
22. Aoun J, Nicolas D, Brown JR, Jaber BL. Maximum allowable contrast dose and prevention of acute kidney injury following cardiovascular procedures. Curr Opin Nephrol Hypertens. 2018;27(2):121-129. doi:10.1097/MNH.0000000000000389
23. Settembre N, Saba C, Bouziane Z, Jeannon F, Mandry D, Malikov S. Hybrid treatment of the aberrant right subclavian artery (arteria lusoria): feasibility study on 180 angio-CTs. Ann Vasc Surg. 2017;44:229-233. doi:10.1016/j.avsg.2017.03.172
A Case Series of Catheter-Directed Thrombolysis With Mechanical Thrombectomy for Treating Severe Deep Vein Thrombosis
Two cases of extensive symptomatic deep vein thrombosis without phlegmasia cerulea dolens were successfully treated with an endovascular technique that combines catheter-directed thrombolysis and mechanical thrombectomy.
Deep vein thrombosis (DVT) is a frequently encountered medical condition with about 1 in 1,000 adults diagnosed annually.1,2 Up to one-half of patients who receive a diagnosis will experience long-term complications in the affected limb.1 Anticoagulation is the treatment of choice for DVT in the absence of any contraindications.3 Thrombolytic therapies (eg, systemic thrombolysis, catheter-directed thrombolysis with or without thrombectomy) historically have been reserved for patients who present with phlegmasia cerulea dolens (PCD), a severe condition involving venous obstruction within the extremities that causes impaired arterial blood supply and cyanosis that can lead to limb loss and death.4
The role of thrombolytic therapy is less clear in patients without PCD who present with extensive or symptomatic lower extremity DVT that causes significant pain, edema, and functional disability. Proximal lower extremity DVT (thrombus above the knee and above the popliteal vein) and particularly those involving the iliac or common femoral vein (ie, iliofemoral DVT) carry a significant risk of recurrent thromboembolism as well as postthrombotic syndrome (PTS), a complication of DVT resulting in chronic leg pain, edema, skin discoloration, and venous ulcers.5
The goal of thrombolytic therapy is to prevent thrombus propagation, recurrent thromboembolism, and PTS, in addition to providing more rapid pain relief and improvement in limb function.
Catheter-directed thrombolysis can be combined with catheter-directed thrombectomy using the same endovascular technique. This combination is called a pharmacomechanical thrombectomy or a pharmacomechanical thromobolysis and can offer more rapid removal of thrombus and decreased infusion times of thrombolytic drug.8 Pharmacomechanical thrombolysis is a relatively new technique, so the choice of thrombolytic therapy will depend on procedural expertise and resource availability. Early interventional radiology consultation (or vascular surgery in some centers) can assist in determining appropriate candidates for thrombolytic therapies. Here we present 2 cases of extensive symptomatic DVT successfully treated with catheter-directed pharmacomechanical thrombolysis.
Case 1
A 61-year-old male current smoker with a history of obesity and hypertension presented to the West Los Angeles Veterans Affairs Medical Center emergency department (ED) with 2 days of progressive pain and swelling in the right lower extremity (RLE) after sustaining a calf injury the preceding week. The patient rated pain as 9 on a 10-point scale and reported no other symptoms. He reported no prior history of venous thromboembolism (VTE) or family history of thrombophilia.
A physical examination was notable for stable vital signs and normal cardiopulmonary examination. There was extensive RLE edema below the knee with tenderness to palpation and shiny taut skin. The neurovascular examination of the RLE was normal. Laboratory studies were notable only for a mild leukocytosis. Compression ultrasound with Doppler of the RLE demonstrated an acute thrombus of the right femoral vein extending to the popliteal vein.
The patient was prescribed enoxaparin 90 mg every 12 hours for anticoagulation. After 36 hours of anticoagulation, he continued to experience severe RLE pain and swelling limiting ambulation. Interventional radiology was consulted, and catheter-directed pharmacomechanical thrombolysis of the RLE was pursued given the persistence of significant symptoms. Intraprocedure venogram demonstrated thrombi filling the entirety of the right femoral and popliteal veins (Figure 1A). This was treated with catheter-directed pulse-spray thrombolysis with 12 mg of tissue plasminogen activator (tPA).
After a 20-minute incubation period, a thrombectomy was performed several times along the femoral vein and popliteal vein, using an AngioJet device. A follow-up venogram revealed a small amount of residual thrombi in the right suprageniculate popliteal vein and right femoral vein. This entire segment was further treated with angioplasty, and a postintervention venogram demonstrated patency of the right suprageniculate popliteal vein and right femoral vein with minimal residual thrombi and with brisk venous flow (Figure 1B). Immediately after the procedure, the patient’s RLE pain significantly improved. On day 2 postprocedure, the patient’s RLE edema resolved, and the patient was able to resume normal ambulation. There were no bleeding complications. The patient was discharged with oral anticoagulation therapy.
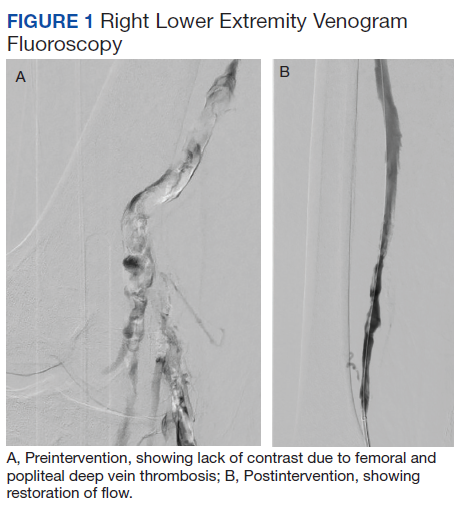
Case 2
A male aged 78 years with a history of hypertension, hyperlipidemia, and benign prostatic hypertrophy presented to the ED with 10 days of progressive pain and swelling in the left lower extremity (LLE). The patient noted decreased mobility over recent months and was using a front wheel walker while recovering from surgical repair of a hamstring tendon injury. He reported taking a transcontinental flight around the same time that his LLE pain began. The patient reported no prior history of VTE or family history of thrombophilia.
A physical examination was notable for stable vital signs with a normal cardiopulmonary examination. There was extensive LLE edema up to the proximal thigh without erythema or cyanosis, and his skin was taut and tender. Neurovascular examination of the LLE was normal. Laboratory studies were unremarkable. Compression ultrasonography with Doppler of the LLE demonstrated an extensive acute occlusive thrombus within the left common femoral, entire left femoral, and left popliteal veins.
After evaluating the patient, the Vascular Surgery service did not feel there was evidence of compartment syndrome nor PCD. The patient received unfractionated heparin anticoagulation therapy and the LLE was elevated continuously. After 24 hours of anticoagulation therapy, the patient continued to have significant pain and was unable to ambulate. The case was presented in a joint Interventional Radiology/Vascular Surgery conference and the decision was made to pursue pharmacomechanic thrombolysis given the significant extent of thrombotic burden.
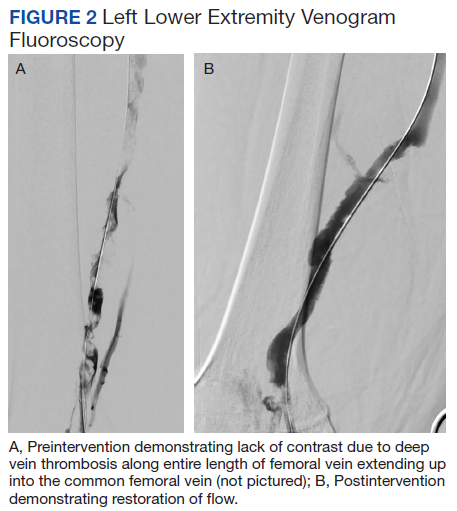
The patient underwent successful catheter-directed pharmacomechanic thrombolysis via pulse-spray thrombolysis of 15 mg of tPA using the Boston Scientific AngioJet Thrombectomy System, and angioplasty with no immediate complications (Figure 2). The patient noted dramatic improvement in LLE pain and swelling 1 day postprocedure and was able to ambulate. He developed mild asymptomatic hematuria, which resolved within 12 hours and without an associated drop in hemoglobin. The patient was transitioned to oral anticoagulation and discharged to an acute rehabilitation unit on postprocedure day 2.
Discussion
Anticoagulation is the preferred therapy for most patients with acute uncomplicated lower extremity DVT. PCD is the only widely accepted indication for thrombolytic therapy in patients with acute lower extremity DVT. However, in the absence of PCD, management of complicated DVT where there are either significant symptoms, extensive clot burden, or proximal location is less clear due to the paucity of clinical data. For example, in the case of iliofemoral DVT, thrombosis of the iliofemoral region is associated with an increased risk of pulmonary embolism, limb malperfusion, and PTS when compared with other types of DVT.5,6
Earlier retrospective observational studies in patients with acute DVT found that the addition of either systemic thrombolysis or catheter-directed thrombolysis to anticoagulation increased rates of clot lysis but did not lead to a reduction in clinical outcomes such as recurrent thromboembolism, mortality, or the rate of PTS.10-12 Additionally, both systemic thrombolytic therapy and catheter-directed thrombolytic therapy were associated with higher rates of major bleeding. However, these studies included all patients with acute DVT without selecting for criteria, such as proximal location of DVT, severe symptoms, or extensive clot burden. Because thrombolytic therapy is proven to provide more rapid and immediate clot lysis (whereas conventional anticoagulation prevents thrombus extension and recurrence but does not dissolve the clot), it is reasonable to suggest that a subpopulation of patients with extensive or symptomatic DVT may benefit from immediate clot lysis, thereby restoring limb perfusion and avoiding limb gangrene while preserving venous function and preventing PTS.
Mixed Study Results
The 2012 CaVenT study is one of the few randomized controlled trials to assess outcomes comparing conventional anticoagulation alone to anticoagulation with catheter-directed thrombolysis in patients with acute lower extremity DVT.13 Study patients did not undergo catheter-directed mechanical thrombectomy. Patients in this study consisted solely of those with first-time iliofemoral DVT. Long-term outcomes at 24-month follow-up showed that additional catheter-directed thrombolysis reduced the risk of PTS when compared with those who were treated with anticoagulation alone (41.1% vs 55.6%, P = .047). The difference in PTS corresponded to an absolute risk reduction of 14.4% (95% CI, 0.2-27.9), and the number needed to treat was 7 (95% CI, 4-502). There was a clinically relevant bleeding complication rate of 8.9% in the thrombolysis group with none leading to a permanently impaired outcome.
These results could not be confirmed by a more recent randomized control trial in 2017 conducted by Vedantham and colleagues.14 In this trial, patients with acute proximal DVT (femoral and iliofemoral DVT) were randomized to receive either anticoagulation alone or anticoagulation plus pharmacomechanical thrombolysis. In the pharmacomechanic thrombolysis group, the overall incidence of PTS and recurrent VTE was not reduced over the 24-month follow-up period. Those who developed PTS in the pharmacomechanical thrombolysis group had lower severity scores, as there was a significant reduction in moderate-to-severe PTS in this group. There also were more early major bleeds in the pharmacomechanic thrombolysis group (1.7%, with no fatal or intracranial bleeds) when compared with the control group; however, this bleeding complication rate was much less than what was noted in the CaVenT study. Additionally, there was a significant decrease in both lower extremity pain and edema in the pharmacomechanical thrombolysis group at 10 days and 30 days postintervention.
Given the mixed results of these 2 randomized controlled trials, further studies are warranted to clarify the role of thrombolytic therapies in preventing major events such as recurrent VTE and PTS, especially given the increased risk of bleeding observed with thrombolytic therapies. The 2016 American College of Chest Physicians guidelines recommend anticoagulation as monotherapy vs thrombolytics, systemic or catheter-directed thrombolysis as designated treatment modalities.3 These guidelines are rated “Grade 2C”, which reflect a weak recommendation based on low-quality evidence. While these recommendations do not comment on additional considerations, such as DVT clot burden, location, or severity of symptoms, the guidelines do state that patients who attach a high value to the prevention of PTS and a lower value to the risk of bleeding with catheter-directed therapy are likely to choose catheter-directed therapy over anticoagulation alone.
Case Studies Analyses
In our first case presentation, pharma-comechanic thrombolysis was pursued because the patient presented with severesymptoms and did not experience any symptomatic improvement after 36 hours of anticoagulation. It is unclear whether a longer duration of anticoagulation might have improved the severity of his symptoms. When considering the level of pain, edema, and inability to ambulate, thrombolytic therapy was considered the most appropriate choice for treatment. Pharmacomechanic thrombolysis was successful, resulting in complete clot lysis, significant decrease in pain and edema with total recovery of ambulatory abilities, no bleeding complications, and prevention of any potential clinical deterioration, such as phlegmasia cerulea dolens. The patient is now 12 months postprocedure without symptoms of PTS or recurrent thromboembolic events. Continued follow-up that monitors the development of PTS will be necessary for at least 2 years postprocedure.
In the second case, our patient experienced some improvement in pain after 24 hours of anticoagulation alone. However, considering the extensive proximal clot burden involving the entire femoral and common femoral veins, the treatment teams believed it was likely that this patient would experience a prolonged recovery time and increased morbidity on anticoagulant therapy alone. Pharmacomechanic thrombolysis was again successful with almost immediate resolution of pain and edema, and recovery of ambulatory abilities on postprocedure day 1. The patient is now 6 months postprocedure without any symptoms of PTS or recurrent thromboembolic events.
In both case presentations, the presenting symptoms, methods of treatment, and immediate symptomatic improvement postintervention were similar. The patient in Case 2 had more extensive clot burden, a more proximal location of clot, and was classified as having an iliofemoral DVT because the thrombus included the common femoral vein; the decision for intervention in this case was more weighted on clot burden and location rather than on the significant symptoms of severe pain and difficulty with ambulation seen in Case 1. However, it is noteworthy that in Case 2 our patient also experienced significant improvement in pain, swelling, and ambulation postintervention. Complications were minimal and limited to Case 2 where our patient experienced mild asymptomatic hematuria likely related to the catheter-directed tPA that resolved spontaneously within hours and did not cause further complications. Additionally, it is likely that the length of hospital stay was decreased significantly in both cases given the rapid improvement in symptoms and recovery of ambulatory abilities.
High-Risk Patients
Given the successful treatment results in these 2 cases, we believe that there is a subset of higher-risk patients with severe symptomatic proximal DVT but without PCD that may benefit from the addition of thrombolytic therapies to anticoagulation. These patients may present with significant pain, difficulty ambulating, and will likely have extensive proximal clot burden. Immediate thrombolytic intervention can achieve rapid symptom relief, which, in turn, can decrease morbidity by decreasing length of hospitalization, improving ambulation, and possibly decreasing the incidence or severity of future PTS. Positive outcomes may be easier to predict for those with obvious features of pain, edema, and difficulty ambulating, which may be more readily reversed by rapid clot reversal/removal.
These patients should be considered on a case-by-case basis. For example, the severity of pain can be balanced against the patient’s risk factors for bleeding because rapid thrombus lysis or immediate thrombus removal will likely reduce the pain. Patients who attach a high value to functional quality (eg, both patients in this case study experienced significant difficulty ambulating), quicker recovery, and decreased hospitalization duration may be more likely to choose the addition of thrombolytic therapies over anticoagulation alone and accept the higher risk of bleed.
Finally, additional studies involving variations in methodology should be examined, including whether pharmacomechanic thrombolysis may be safer in terms of bleeding than catheter-directed thrombolysis alone, as suggested by the lower bleeding rates seen in the pharmacomechanic study by Vedantham and colleagues when compared with the CaVenT study.13,14 Patients in the CaVenT study received an infusion of 20 mg of alteplase over a maximum of 96 hours. Patients in the pharmacomechanic study by Vedanthem and colleagues received either a rapid pulsed delivery of alteplase over a single procedural session (
Conclusions
There is a relative lack of high-quality data examining thrombolytic therapies in the setting of acute lower extremity DVT. Recent studies have prioritized evaluation of the posttreatment incidence of PTS, recurrent thromboembolism, and risk of bleeding caused by thrombolytic therapies. Results are mixed thus far, and further studies are necessary to clarify a more definitive role for thrombolytic therapies, particularly in established higher-risk populations with proximal DVT. In this case series, we highlighted 2 patients with extensive proximal DVT burden with significant symptoms who experienced almost complete resolution of symptoms immediately following thrombolytic therapies. We postulate that even in the absence of PCD, there is a subset of patients with severe symptoms in the setting of acute proximal lower extremity DVT that clearly benefit from thrombolytic therapies.
1. Centers for Disease Control and Prevention. Venous Thromboembolism (Blood Clots). Updated February 7, 2020. Accessed January 11, 2021. https://www.cdc.gov/ncbddd/dvt/data.html
2. White RH. The epidemiology of venous thromboembolism. Circulation. 2003;107(23 Suppl 1):I4-I8. doi:10.1161/01.CIR.0000078468.11849.66
3. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report [published correction appears in Chest. 2016 Oct;150(4):988]. Chest. 2016;149(2):315-352. doi:10.1016/j.chest.2015.11.026
4. Sarwar S, Narra S, Munir A. Phlegmasia cerulea dolens. Tex Heart Inst J. 2009;36(1):76-77.
5. Nyamekye I, Merker L. Management of proximal deep vein thrombosis. Phlebology. 2012;27 Suppl 2:61-72. doi:10.1258/phleb.2012.012s37
6. Abhishek M, Sukriti K, Purav S, et al. Comparison of catheter-directed thrombolysis vs systemic thrombolysis in pulmonary embolism: a propensity match analysis. Chest. 2017;152(4): A1047. doi:10.1016/j.chest.2017.08.1080
7. Sista AK, Kearon C. Catheter-directed thrombolysis for pulmonary embolism: where do we stand? JACC Cardiovasc Interv. 2015;8(10):1393-1395. doi:10.1016/j.jcin.2015.06.009
8. Robertson L, McBride O, Burdess A. Pharmacomechanical thrombectomy for iliofemoral deep vein thrombosis. Cochrane Database Syst Rev. 2016;11(11):CD011536. Published 2016 Nov 4. doi:10.1002/14651858.CD011536.pub2
9. Kahn SR, Shbaklo H, Lamping DL, et al. Determinants of health-related quality of life during the 2 years following deep vein thrombosis. J Thromb Haemost. 2008;6(7):1105-1112. doi:10.1111/j.1538-7836.2008.03002.x
10. Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines [published correction appears in Chest. 2012 Dec;142(6):1698-1704]. Chest. 2012;141(2 Suppl):e419S-e496S. doi:10.1378/chest.11-2301
11. Bashir R, Zack CJ, Zhao H, Comerota AJ, Bove AA. Comparative outcomes of catheter-directed thrombolysis plus anticoagulation vs anticoagulation alone to treat lower-extremity proximal deep vein thrombosis. JAMA Intern Med. 2014;174(9):1494-1501. doi:10.1001/jamainternmed.2014.3415
12. Watson L, Broderick C, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev. 2016;11(11):CD002783. Published 2016 Nov 10. doi:10.1002/14651858.CD002783.pub4
13. Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet. 2012;379(9810):31-38. doi:10.1016/S0140-6736(11)61753-4
14. Vedantham S, Goldhaber SZ, Julian JA, et al; ATTRACT Trial Investigators. Pharmacomechanical catheter-directed thrombolysis for deep-vein thrombosis. N Engl J Med. 2017;377(23):2240-2252. doi:10.1056/NEJMoa1615066
Two cases of extensive symptomatic deep vein thrombosis without phlegmasia cerulea dolens were successfully treated with an endovascular technique that combines catheter-directed thrombolysis and mechanical thrombectomy.
Two cases of extensive symptomatic deep vein thrombosis without phlegmasia cerulea dolens were successfully treated with an endovascular technique that combines catheter-directed thrombolysis and mechanical thrombectomy.
Deep vein thrombosis (DVT) is a frequently encountered medical condition with about 1 in 1,000 adults diagnosed annually.1,2 Up to one-half of patients who receive a diagnosis will experience long-term complications in the affected limb.1 Anticoagulation is the treatment of choice for DVT in the absence of any contraindications.3 Thrombolytic therapies (eg, systemic thrombolysis, catheter-directed thrombolysis with or without thrombectomy) historically have been reserved for patients who present with phlegmasia cerulea dolens (PCD), a severe condition involving venous obstruction within the extremities that causes impaired arterial blood supply and cyanosis that can lead to limb loss and death.4
The role of thrombolytic therapy is less clear in patients without PCD who present with extensive or symptomatic lower extremity DVT that causes significant pain, edema, and functional disability. Proximal lower extremity DVT (thrombus above the knee and above the popliteal vein) and particularly those involving the iliac or common femoral vein (ie, iliofemoral DVT) carry a significant risk of recurrent thromboembolism as well as postthrombotic syndrome (PTS), a complication of DVT resulting in chronic leg pain, edema, skin discoloration, and venous ulcers.5
The goal of thrombolytic therapy is to prevent thrombus propagation, recurrent thromboembolism, and PTS, in addition to providing more rapid pain relief and improvement in limb function.
Catheter-directed thrombolysis can be combined with catheter-directed thrombectomy using the same endovascular technique. This combination is called a pharmacomechanical thrombectomy or a pharmacomechanical thromobolysis and can offer more rapid removal of thrombus and decreased infusion times of thrombolytic drug.8 Pharmacomechanical thrombolysis is a relatively new technique, so the choice of thrombolytic therapy will depend on procedural expertise and resource availability. Early interventional radiology consultation (or vascular surgery in some centers) can assist in determining appropriate candidates for thrombolytic therapies. Here we present 2 cases of extensive symptomatic DVT successfully treated with catheter-directed pharmacomechanical thrombolysis.
Case 1
A 61-year-old male current smoker with a history of obesity and hypertension presented to the West Los Angeles Veterans Affairs Medical Center emergency department (ED) with 2 days of progressive pain and swelling in the right lower extremity (RLE) after sustaining a calf injury the preceding week. The patient rated pain as 9 on a 10-point scale and reported no other symptoms. He reported no prior history of venous thromboembolism (VTE) or family history of thrombophilia.
A physical examination was notable for stable vital signs and normal cardiopulmonary examination. There was extensive RLE edema below the knee with tenderness to palpation and shiny taut skin. The neurovascular examination of the RLE was normal. Laboratory studies were notable only for a mild leukocytosis. Compression ultrasound with Doppler of the RLE demonstrated an acute thrombus of the right femoral vein extending to the popliteal vein.
The patient was prescribed enoxaparin 90 mg every 12 hours for anticoagulation. After 36 hours of anticoagulation, he continued to experience severe RLE pain and swelling limiting ambulation. Interventional radiology was consulted, and catheter-directed pharmacomechanical thrombolysis of the RLE was pursued given the persistence of significant symptoms. Intraprocedure venogram demonstrated thrombi filling the entirety of the right femoral and popliteal veins (Figure 1A). This was treated with catheter-directed pulse-spray thrombolysis with 12 mg of tissue plasminogen activator (tPA).
After a 20-minute incubation period, a thrombectomy was performed several times along the femoral vein and popliteal vein, using an AngioJet device. A follow-up venogram revealed a small amount of residual thrombi in the right suprageniculate popliteal vein and right femoral vein. This entire segment was further treated with angioplasty, and a postintervention venogram demonstrated patency of the right suprageniculate popliteal vein and right femoral vein with minimal residual thrombi and with brisk venous flow (Figure 1B). Immediately after the procedure, the patient’s RLE pain significantly improved. On day 2 postprocedure, the patient’s RLE edema resolved, and the patient was able to resume normal ambulation. There were no bleeding complications. The patient was discharged with oral anticoagulation therapy.
Case 2
A male aged 78 years with a history of hypertension, hyperlipidemia, and benign prostatic hypertrophy presented to the ED with 10 days of progressive pain and swelling in the left lower extremity (LLE). The patient noted decreased mobility over recent months and was using a front wheel walker while recovering from surgical repair of a hamstring tendon injury. He reported taking a transcontinental flight around the same time that his LLE pain began. The patient reported no prior history of VTE or family history of thrombophilia.
A physical examination was notable for stable vital signs with a normal cardiopulmonary examination. There was extensive LLE edema up to the proximal thigh without erythema or cyanosis, and his skin was taut and tender. Neurovascular examination of the LLE was normal. Laboratory studies were unremarkable. Compression ultrasonography with Doppler of the LLE demonstrated an extensive acute occlusive thrombus within the left common femoral, entire left femoral, and left popliteal veins.
After evaluating the patient, the Vascular Surgery service did not feel there was evidence of compartment syndrome nor PCD. The patient received unfractionated heparin anticoagulation therapy and the LLE was elevated continuously. After 24 hours of anticoagulation therapy, the patient continued to have significant pain and was unable to ambulate. The case was presented in a joint Interventional Radiology/Vascular Surgery conference and the decision was made to pursue pharmacomechanic thrombolysis given the significant extent of thrombotic burden.
The patient underwent successful catheter-directed pharmacomechanic thrombolysis via pulse-spray thrombolysis of 15 mg of tPA using the Boston Scientific AngioJet Thrombectomy System, and angioplasty with no immediate complications (Figure 2). The patient noted dramatic improvement in LLE pain and swelling 1 day postprocedure and was able to ambulate. He developed mild asymptomatic hematuria, which resolved within 12 hours and without an associated drop in hemoglobin. The patient was transitioned to oral anticoagulation and discharged to an acute rehabilitation unit on postprocedure day 2.
Discussion
Anticoagulation is the preferred therapy for most patients with acute uncomplicated lower extremity DVT. PCD is the only widely accepted indication for thrombolytic therapy in patients with acute lower extremity DVT. However, in the absence of PCD, management of complicated DVT where there are either significant symptoms, extensive clot burden, or proximal location is less clear due to the paucity of clinical data. For example, in the case of iliofemoral DVT, thrombosis of the iliofemoral region is associated with an increased risk of pulmonary embolism, limb malperfusion, and PTS when compared with other types of DVT.5,6
Earlier retrospective observational studies in patients with acute DVT found that the addition of either systemic thrombolysis or catheter-directed thrombolysis to anticoagulation increased rates of clot lysis but did not lead to a reduction in clinical outcomes such as recurrent thromboembolism, mortality, or the rate of PTS.10-12 Additionally, both systemic thrombolytic therapy and catheter-directed thrombolytic therapy were associated with higher rates of major bleeding. However, these studies included all patients with acute DVT without selecting for criteria, such as proximal location of DVT, severe symptoms, or extensive clot burden. Because thrombolytic therapy is proven to provide more rapid and immediate clot lysis (whereas conventional anticoagulation prevents thrombus extension and recurrence but does not dissolve the clot), it is reasonable to suggest that a subpopulation of patients with extensive or symptomatic DVT may benefit from immediate clot lysis, thereby restoring limb perfusion and avoiding limb gangrene while preserving venous function and preventing PTS.
Mixed Study Results
The 2012 CaVenT study is one of the few randomized controlled trials to assess outcomes comparing conventional anticoagulation alone to anticoagulation with catheter-directed thrombolysis in patients with acute lower extremity DVT.13 Study patients did not undergo catheter-directed mechanical thrombectomy. Patients in this study consisted solely of those with first-time iliofemoral DVT. Long-term outcomes at 24-month follow-up showed that additional catheter-directed thrombolysis reduced the risk of PTS when compared with those who were treated with anticoagulation alone (41.1% vs 55.6%, P = .047). The difference in PTS corresponded to an absolute risk reduction of 14.4% (95% CI, 0.2-27.9), and the number needed to treat was 7 (95% CI, 4-502). There was a clinically relevant bleeding complication rate of 8.9% in the thrombolysis group with none leading to a permanently impaired outcome.
These results could not be confirmed by a more recent randomized control trial in 2017 conducted by Vedantham and colleagues.14 In this trial, patients with acute proximal DVT (femoral and iliofemoral DVT) were randomized to receive either anticoagulation alone or anticoagulation plus pharmacomechanical thrombolysis. In the pharmacomechanic thrombolysis group, the overall incidence of PTS and recurrent VTE was not reduced over the 24-month follow-up period. Those who developed PTS in the pharmacomechanical thrombolysis group had lower severity scores, as there was a significant reduction in moderate-to-severe PTS in this group. There also were more early major bleeds in the pharmacomechanic thrombolysis group (1.7%, with no fatal or intracranial bleeds) when compared with the control group; however, this bleeding complication rate was much less than what was noted in the CaVenT study. Additionally, there was a significant decrease in both lower extremity pain and edema in the pharmacomechanical thrombolysis group at 10 days and 30 days postintervention.
Given the mixed results of these 2 randomized controlled trials, further studies are warranted to clarify the role of thrombolytic therapies in preventing major events such as recurrent VTE and PTS, especially given the increased risk of bleeding observed with thrombolytic therapies. The 2016 American College of Chest Physicians guidelines recommend anticoagulation as monotherapy vs thrombolytics, systemic or catheter-directed thrombolysis as designated treatment modalities.3 These guidelines are rated “Grade 2C”, which reflect a weak recommendation based on low-quality evidence. While these recommendations do not comment on additional considerations, such as DVT clot burden, location, or severity of symptoms, the guidelines do state that patients who attach a high value to the prevention of PTS and a lower value to the risk of bleeding with catheter-directed therapy are likely to choose catheter-directed therapy over anticoagulation alone.
Case Studies Analyses
In our first case presentation, pharma-comechanic thrombolysis was pursued because the patient presented with severesymptoms and did not experience any symptomatic improvement after 36 hours of anticoagulation. It is unclear whether a longer duration of anticoagulation might have improved the severity of his symptoms. When considering the level of pain, edema, and inability to ambulate, thrombolytic therapy was considered the most appropriate choice for treatment. Pharmacomechanic thrombolysis was successful, resulting in complete clot lysis, significant decrease in pain and edema with total recovery of ambulatory abilities, no bleeding complications, and prevention of any potential clinical deterioration, such as phlegmasia cerulea dolens. The patient is now 12 months postprocedure without symptoms of PTS or recurrent thromboembolic events. Continued follow-up that monitors the development of PTS will be necessary for at least 2 years postprocedure.
In the second case, our patient experienced some improvement in pain after 24 hours of anticoagulation alone. However, considering the extensive proximal clot burden involving the entire femoral and common femoral veins, the treatment teams believed it was likely that this patient would experience a prolonged recovery time and increased morbidity on anticoagulant therapy alone. Pharmacomechanic thrombolysis was again successful with almost immediate resolution of pain and edema, and recovery of ambulatory abilities on postprocedure day 1. The patient is now 6 months postprocedure without any symptoms of PTS or recurrent thromboembolic events.
In both case presentations, the presenting symptoms, methods of treatment, and immediate symptomatic improvement postintervention were similar. The patient in Case 2 had more extensive clot burden, a more proximal location of clot, and was classified as having an iliofemoral DVT because the thrombus included the common femoral vein; the decision for intervention in this case was more weighted on clot burden and location rather than on the significant symptoms of severe pain and difficulty with ambulation seen in Case 1. However, it is noteworthy that in Case 2 our patient also experienced significant improvement in pain, swelling, and ambulation postintervention. Complications were minimal and limited to Case 2 where our patient experienced mild asymptomatic hematuria likely related to the catheter-directed tPA that resolved spontaneously within hours and did not cause further complications. Additionally, it is likely that the length of hospital stay was decreased significantly in both cases given the rapid improvement in symptoms and recovery of ambulatory abilities.
High-Risk Patients
Given the successful treatment results in these 2 cases, we believe that there is a subset of higher-risk patients with severe symptomatic proximal DVT but without PCD that may benefit from the addition of thrombolytic therapies to anticoagulation. These patients may present with significant pain, difficulty ambulating, and will likely have extensive proximal clot burden. Immediate thrombolytic intervention can achieve rapid symptom relief, which, in turn, can decrease morbidity by decreasing length of hospitalization, improving ambulation, and possibly decreasing the incidence or severity of future PTS. Positive outcomes may be easier to predict for those with obvious features of pain, edema, and difficulty ambulating, which may be more readily reversed by rapid clot reversal/removal.
These patients should be considered on a case-by-case basis. For example, the severity of pain can be balanced against the patient’s risk factors for bleeding because rapid thrombus lysis or immediate thrombus removal will likely reduce the pain. Patients who attach a high value to functional quality (eg, both patients in this case study experienced significant difficulty ambulating), quicker recovery, and decreased hospitalization duration may be more likely to choose the addition of thrombolytic therapies over anticoagulation alone and accept the higher risk of bleed.
Finally, additional studies involving variations in methodology should be examined, including whether pharmacomechanic thrombolysis may be safer in terms of bleeding than catheter-directed thrombolysis alone, as suggested by the lower bleeding rates seen in the pharmacomechanic study by Vedantham and colleagues when compared with the CaVenT study.13,14 Patients in the CaVenT study received an infusion of 20 mg of alteplase over a maximum of 96 hours. Patients in the pharmacomechanic study by Vedanthem and colleagues received either a rapid pulsed delivery of alteplase over a single procedural session (
Conclusions
There is a relative lack of high-quality data examining thrombolytic therapies in the setting of acute lower extremity DVT. Recent studies have prioritized evaluation of the posttreatment incidence of PTS, recurrent thromboembolism, and risk of bleeding caused by thrombolytic therapies. Results are mixed thus far, and further studies are necessary to clarify a more definitive role for thrombolytic therapies, particularly in established higher-risk populations with proximal DVT. In this case series, we highlighted 2 patients with extensive proximal DVT burden with significant symptoms who experienced almost complete resolution of symptoms immediately following thrombolytic therapies. We postulate that even in the absence of PCD, there is a subset of patients with severe symptoms in the setting of acute proximal lower extremity DVT that clearly benefit from thrombolytic therapies.
Deep vein thrombosis (DVT) is a frequently encountered medical condition with about 1 in 1,000 adults diagnosed annually.1,2 Up to one-half of patients who receive a diagnosis will experience long-term complications in the affected limb.1 Anticoagulation is the treatment of choice for DVT in the absence of any contraindications.3 Thrombolytic therapies (eg, systemic thrombolysis, catheter-directed thrombolysis with or without thrombectomy) historically have been reserved for patients who present with phlegmasia cerulea dolens (PCD), a severe condition involving venous obstruction within the extremities that causes impaired arterial blood supply and cyanosis that can lead to limb loss and death.4
The role of thrombolytic therapy is less clear in patients without PCD who present with extensive or symptomatic lower extremity DVT that causes significant pain, edema, and functional disability. Proximal lower extremity DVT (thrombus above the knee and above the popliteal vein) and particularly those involving the iliac or common femoral vein (ie, iliofemoral DVT) carry a significant risk of recurrent thromboembolism as well as postthrombotic syndrome (PTS), a complication of DVT resulting in chronic leg pain, edema, skin discoloration, and venous ulcers.5
The goal of thrombolytic therapy is to prevent thrombus propagation, recurrent thromboembolism, and PTS, in addition to providing more rapid pain relief and improvement in limb function.
Catheter-directed thrombolysis can be combined with catheter-directed thrombectomy using the same endovascular technique. This combination is called a pharmacomechanical thrombectomy or a pharmacomechanical thromobolysis and can offer more rapid removal of thrombus and decreased infusion times of thrombolytic drug.8 Pharmacomechanical thrombolysis is a relatively new technique, so the choice of thrombolytic therapy will depend on procedural expertise and resource availability. Early interventional radiology consultation (or vascular surgery in some centers) can assist in determining appropriate candidates for thrombolytic therapies. Here we present 2 cases of extensive symptomatic DVT successfully treated with catheter-directed pharmacomechanical thrombolysis.
Case 1
A 61-year-old male current smoker with a history of obesity and hypertension presented to the West Los Angeles Veterans Affairs Medical Center emergency department (ED) with 2 days of progressive pain and swelling in the right lower extremity (RLE) after sustaining a calf injury the preceding week. The patient rated pain as 9 on a 10-point scale and reported no other symptoms. He reported no prior history of venous thromboembolism (VTE) or family history of thrombophilia.
A physical examination was notable for stable vital signs and normal cardiopulmonary examination. There was extensive RLE edema below the knee with tenderness to palpation and shiny taut skin. The neurovascular examination of the RLE was normal. Laboratory studies were notable only for a mild leukocytosis. Compression ultrasound with Doppler of the RLE demonstrated an acute thrombus of the right femoral vein extending to the popliteal vein.
The patient was prescribed enoxaparin 90 mg every 12 hours for anticoagulation. After 36 hours of anticoagulation, he continued to experience severe RLE pain and swelling limiting ambulation. Interventional radiology was consulted, and catheter-directed pharmacomechanical thrombolysis of the RLE was pursued given the persistence of significant symptoms. Intraprocedure venogram demonstrated thrombi filling the entirety of the right femoral and popliteal veins (Figure 1A). This was treated with catheter-directed pulse-spray thrombolysis with 12 mg of tissue plasminogen activator (tPA).
After a 20-minute incubation period, a thrombectomy was performed several times along the femoral vein and popliteal vein, using an AngioJet device. A follow-up venogram revealed a small amount of residual thrombi in the right suprageniculate popliteal vein and right femoral vein. This entire segment was further treated with angioplasty, and a postintervention venogram demonstrated patency of the right suprageniculate popliteal vein and right femoral vein with minimal residual thrombi and with brisk venous flow (Figure 1B). Immediately after the procedure, the patient’s RLE pain significantly improved. On day 2 postprocedure, the patient’s RLE edema resolved, and the patient was able to resume normal ambulation. There were no bleeding complications. The patient was discharged with oral anticoagulation therapy.
Case 2
A male aged 78 years with a history of hypertension, hyperlipidemia, and benign prostatic hypertrophy presented to the ED with 10 days of progressive pain and swelling in the left lower extremity (LLE). The patient noted decreased mobility over recent months and was using a front wheel walker while recovering from surgical repair of a hamstring tendon injury. He reported taking a transcontinental flight around the same time that his LLE pain began. The patient reported no prior history of VTE or family history of thrombophilia.
A physical examination was notable for stable vital signs with a normal cardiopulmonary examination. There was extensive LLE edema up to the proximal thigh without erythema or cyanosis, and his skin was taut and tender. Neurovascular examination of the LLE was normal. Laboratory studies were unremarkable. Compression ultrasonography with Doppler of the LLE demonstrated an extensive acute occlusive thrombus within the left common femoral, entire left femoral, and left popliteal veins.
After evaluating the patient, the Vascular Surgery service did not feel there was evidence of compartment syndrome nor PCD. The patient received unfractionated heparin anticoagulation therapy and the LLE was elevated continuously. After 24 hours of anticoagulation therapy, the patient continued to have significant pain and was unable to ambulate. The case was presented in a joint Interventional Radiology/Vascular Surgery conference and the decision was made to pursue pharmacomechanic thrombolysis given the significant extent of thrombotic burden.
The patient underwent successful catheter-directed pharmacomechanic thrombolysis via pulse-spray thrombolysis of 15 mg of tPA using the Boston Scientific AngioJet Thrombectomy System, and angioplasty with no immediate complications (Figure 2). The patient noted dramatic improvement in LLE pain and swelling 1 day postprocedure and was able to ambulate. He developed mild asymptomatic hematuria, which resolved within 12 hours and without an associated drop in hemoglobin. The patient was transitioned to oral anticoagulation and discharged to an acute rehabilitation unit on postprocedure day 2.
Discussion
Anticoagulation is the preferred therapy for most patients with acute uncomplicated lower extremity DVT. PCD is the only widely accepted indication for thrombolytic therapy in patients with acute lower extremity DVT. However, in the absence of PCD, management of complicated DVT where there are either significant symptoms, extensive clot burden, or proximal location is less clear due to the paucity of clinical data. For example, in the case of iliofemoral DVT, thrombosis of the iliofemoral region is associated with an increased risk of pulmonary embolism, limb malperfusion, and PTS when compared with other types of DVT.5,6
Earlier retrospective observational studies in patients with acute DVT found that the addition of either systemic thrombolysis or catheter-directed thrombolysis to anticoagulation increased rates of clot lysis but did not lead to a reduction in clinical outcomes such as recurrent thromboembolism, mortality, or the rate of PTS.10-12 Additionally, both systemic thrombolytic therapy and catheter-directed thrombolytic therapy were associated with higher rates of major bleeding. However, these studies included all patients with acute DVT without selecting for criteria, such as proximal location of DVT, severe symptoms, or extensive clot burden. Because thrombolytic therapy is proven to provide more rapid and immediate clot lysis (whereas conventional anticoagulation prevents thrombus extension and recurrence but does not dissolve the clot), it is reasonable to suggest that a subpopulation of patients with extensive or symptomatic DVT may benefit from immediate clot lysis, thereby restoring limb perfusion and avoiding limb gangrene while preserving venous function and preventing PTS.
Mixed Study Results
The 2012 CaVenT study is one of the few randomized controlled trials to assess outcomes comparing conventional anticoagulation alone to anticoagulation with catheter-directed thrombolysis in patients with acute lower extremity DVT.13 Study patients did not undergo catheter-directed mechanical thrombectomy. Patients in this study consisted solely of those with first-time iliofemoral DVT. Long-term outcomes at 24-month follow-up showed that additional catheter-directed thrombolysis reduced the risk of PTS when compared with those who were treated with anticoagulation alone (41.1% vs 55.6%, P = .047). The difference in PTS corresponded to an absolute risk reduction of 14.4% (95% CI, 0.2-27.9), and the number needed to treat was 7 (95% CI, 4-502). There was a clinically relevant bleeding complication rate of 8.9% in the thrombolysis group with none leading to a permanently impaired outcome.
These results could not be confirmed by a more recent randomized control trial in 2017 conducted by Vedantham and colleagues.14 In this trial, patients with acute proximal DVT (femoral and iliofemoral DVT) were randomized to receive either anticoagulation alone or anticoagulation plus pharmacomechanical thrombolysis. In the pharmacomechanic thrombolysis group, the overall incidence of PTS and recurrent VTE was not reduced over the 24-month follow-up period. Those who developed PTS in the pharmacomechanical thrombolysis group had lower severity scores, as there was a significant reduction in moderate-to-severe PTS in this group. There also were more early major bleeds in the pharmacomechanic thrombolysis group (1.7%, with no fatal or intracranial bleeds) when compared with the control group; however, this bleeding complication rate was much less than what was noted in the CaVenT study. Additionally, there was a significant decrease in both lower extremity pain and edema in the pharmacomechanical thrombolysis group at 10 days and 30 days postintervention.
Given the mixed results of these 2 randomized controlled trials, further studies are warranted to clarify the role of thrombolytic therapies in preventing major events such as recurrent VTE and PTS, especially given the increased risk of bleeding observed with thrombolytic therapies. The 2016 American College of Chest Physicians guidelines recommend anticoagulation as monotherapy vs thrombolytics, systemic or catheter-directed thrombolysis as designated treatment modalities.3 These guidelines are rated “Grade 2C”, which reflect a weak recommendation based on low-quality evidence. While these recommendations do not comment on additional considerations, such as DVT clot burden, location, or severity of symptoms, the guidelines do state that patients who attach a high value to the prevention of PTS and a lower value to the risk of bleeding with catheter-directed therapy are likely to choose catheter-directed therapy over anticoagulation alone.
Case Studies Analyses
In our first case presentation, pharma-comechanic thrombolysis was pursued because the patient presented with severesymptoms and did not experience any symptomatic improvement after 36 hours of anticoagulation. It is unclear whether a longer duration of anticoagulation might have improved the severity of his symptoms. When considering the level of pain, edema, and inability to ambulate, thrombolytic therapy was considered the most appropriate choice for treatment. Pharmacomechanic thrombolysis was successful, resulting in complete clot lysis, significant decrease in pain and edema with total recovery of ambulatory abilities, no bleeding complications, and prevention of any potential clinical deterioration, such as phlegmasia cerulea dolens. The patient is now 12 months postprocedure without symptoms of PTS or recurrent thromboembolic events. Continued follow-up that monitors the development of PTS will be necessary for at least 2 years postprocedure.
In the second case, our patient experienced some improvement in pain after 24 hours of anticoagulation alone. However, considering the extensive proximal clot burden involving the entire femoral and common femoral veins, the treatment teams believed it was likely that this patient would experience a prolonged recovery time and increased morbidity on anticoagulant therapy alone. Pharmacomechanic thrombolysis was again successful with almost immediate resolution of pain and edema, and recovery of ambulatory abilities on postprocedure day 1. The patient is now 6 months postprocedure without any symptoms of PTS or recurrent thromboembolic events.
In both case presentations, the presenting symptoms, methods of treatment, and immediate symptomatic improvement postintervention were similar. The patient in Case 2 had more extensive clot burden, a more proximal location of clot, and was classified as having an iliofemoral DVT because the thrombus included the common femoral vein; the decision for intervention in this case was more weighted on clot burden and location rather than on the significant symptoms of severe pain and difficulty with ambulation seen in Case 1. However, it is noteworthy that in Case 2 our patient also experienced significant improvement in pain, swelling, and ambulation postintervention. Complications were minimal and limited to Case 2 where our patient experienced mild asymptomatic hematuria likely related to the catheter-directed tPA that resolved spontaneously within hours and did not cause further complications. Additionally, it is likely that the length of hospital stay was decreased significantly in both cases given the rapid improvement in symptoms and recovery of ambulatory abilities.
High-Risk Patients
Given the successful treatment results in these 2 cases, we believe that there is a subset of higher-risk patients with severe symptomatic proximal DVT but without PCD that may benefit from the addition of thrombolytic therapies to anticoagulation. These patients may present with significant pain, difficulty ambulating, and will likely have extensive proximal clot burden. Immediate thrombolytic intervention can achieve rapid symptom relief, which, in turn, can decrease morbidity by decreasing length of hospitalization, improving ambulation, and possibly decreasing the incidence or severity of future PTS. Positive outcomes may be easier to predict for those with obvious features of pain, edema, and difficulty ambulating, which may be more readily reversed by rapid clot reversal/removal.
These patients should be considered on a case-by-case basis. For example, the severity of pain can be balanced against the patient’s risk factors for bleeding because rapid thrombus lysis or immediate thrombus removal will likely reduce the pain. Patients who attach a high value to functional quality (eg, both patients in this case study experienced significant difficulty ambulating), quicker recovery, and decreased hospitalization duration may be more likely to choose the addition of thrombolytic therapies over anticoagulation alone and accept the higher risk of bleed.
Finally, additional studies involving variations in methodology should be examined, including whether pharmacomechanic thrombolysis may be safer in terms of bleeding than catheter-directed thrombolysis alone, as suggested by the lower bleeding rates seen in the pharmacomechanic study by Vedantham and colleagues when compared with the CaVenT study.13,14 Patients in the CaVenT study received an infusion of 20 mg of alteplase over a maximum of 96 hours. Patients in the pharmacomechanic study by Vedanthem and colleagues received either a rapid pulsed delivery of alteplase over a single procedural session (
Conclusions
There is a relative lack of high-quality data examining thrombolytic therapies in the setting of acute lower extremity DVT. Recent studies have prioritized evaluation of the posttreatment incidence of PTS, recurrent thromboembolism, and risk of bleeding caused by thrombolytic therapies. Results are mixed thus far, and further studies are necessary to clarify a more definitive role for thrombolytic therapies, particularly in established higher-risk populations with proximal DVT. In this case series, we highlighted 2 patients with extensive proximal DVT burden with significant symptoms who experienced almost complete resolution of symptoms immediately following thrombolytic therapies. We postulate that even in the absence of PCD, there is a subset of patients with severe symptoms in the setting of acute proximal lower extremity DVT that clearly benefit from thrombolytic therapies.
1. Centers for Disease Control and Prevention. Venous Thromboembolism (Blood Clots). Updated February 7, 2020. Accessed January 11, 2021. https://www.cdc.gov/ncbddd/dvt/data.html
2. White RH. The epidemiology of venous thromboembolism. Circulation. 2003;107(23 Suppl 1):I4-I8. doi:10.1161/01.CIR.0000078468.11849.66
3. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report [published correction appears in Chest. 2016 Oct;150(4):988]. Chest. 2016;149(2):315-352. doi:10.1016/j.chest.2015.11.026
4. Sarwar S, Narra S, Munir A. Phlegmasia cerulea dolens. Tex Heart Inst J. 2009;36(1):76-77.
5. Nyamekye I, Merker L. Management of proximal deep vein thrombosis. Phlebology. 2012;27 Suppl 2:61-72. doi:10.1258/phleb.2012.012s37
6. Abhishek M, Sukriti K, Purav S, et al. Comparison of catheter-directed thrombolysis vs systemic thrombolysis in pulmonary embolism: a propensity match analysis. Chest. 2017;152(4): A1047. doi:10.1016/j.chest.2017.08.1080
7. Sista AK, Kearon C. Catheter-directed thrombolysis for pulmonary embolism: where do we stand? JACC Cardiovasc Interv. 2015;8(10):1393-1395. doi:10.1016/j.jcin.2015.06.009
8. Robertson L, McBride O, Burdess A. Pharmacomechanical thrombectomy for iliofemoral deep vein thrombosis. Cochrane Database Syst Rev. 2016;11(11):CD011536. Published 2016 Nov 4. doi:10.1002/14651858.CD011536.pub2
9. Kahn SR, Shbaklo H, Lamping DL, et al. Determinants of health-related quality of life during the 2 years following deep vein thrombosis. J Thromb Haemost. 2008;6(7):1105-1112. doi:10.1111/j.1538-7836.2008.03002.x
10. Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines [published correction appears in Chest. 2012 Dec;142(6):1698-1704]. Chest. 2012;141(2 Suppl):e419S-e496S. doi:10.1378/chest.11-2301
11. Bashir R, Zack CJ, Zhao H, Comerota AJ, Bove AA. Comparative outcomes of catheter-directed thrombolysis plus anticoagulation vs anticoagulation alone to treat lower-extremity proximal deep vein thrombosis. JAMA Intern Med. 2014;174(9):1494-1501. doi:10.1001/jamainternmed.2014.3415
12. Watson L, Broderick C, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev. 2016;11(11):CD002783. Published 2016 Nov 10. doi:10.1002/14651858.CD002783.pub4
13. Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet. 2012;379(9810):31-38. doi:10.1016/S0140-6736(11)61753-4
14. Vedantham S, Goldhaber SZ, Julian JA, et al; ATTRACT Trial Investigators. Pharmacomechanical catheter-directed thrombolysis for deep-vein thrombosis. N Engl J Med. 2017;377(23):2240-2252. doi:10.1056/NEJMoa1615066
1. Centers for Disease Control and Prevention. Venous Thromboembolism (Blood Clots). Updated February 7, 2020. Accessed January 11, 2021. https://www.cdc.gov/ncbddd/dvt/data.html
2. White RH. The epidemiology of venous thromboembolism. Circulation. 2003;107(23 Suppl 1):I4-I8. doi:10.1161/01.CIR.0000078468.11849.66
3. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report [published correction appears in Chest. 2016 Oct;150(4):988]. Chest. 2016;149(2):315-352. doi:10.1016/j.chest.2015.11.026
4. Sarwar S, Narra S, Munir A. Phlegmasia cerulea dolens. Tex Heart Inst J. 2009;36(1):76-77.
5. Nyamekye I, Merker L. Management of proximal deep vein thrombosis. Phlebology. 2012;27 Suppl 2:61-72. doi:10.1258/phleb.2012.012s37
6. Abhishek M, Sukriti K, Purav S, et al. Comparison of catheter-directed thrombolysis vs systemic thrombolysis in pulmonary embolism: a propensity match analysis. Chest. 2017;152(4): A1047. doi:10.1016/j.chest.2017.08.1080
7. Sista AK, Kearon C. Catheter-directed thrombolysis for pulmonary embolism: where do we stand? JACC Cardiovasc Interv. 2015;8(10):1393-1395. doi:10.1016/j.jcin.2015.06.009
8. Robertson L, McBride O, Burdess A. Pharmacomechanical thrombectomy for iliofemoral deep vein thrombosis. Cochrane Database Syst Rev. 2016;11(11):CD011536. Published 2016 Nov 4. doi:10.1002/14651858.CD011536.pub2
9. Kahn SR, Shbaklo H, Lamping DL, et al. Determinants of health-related quality of life during the 2 years following deep vein thrombosis. J Thromb Haemost. 2008;6(7):1105-1112. doi:10.1111/j.1538-7836.2008.03002.x
10. Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines [published correction appears in Chest. 2012 Dec;142(6):1698-1704]. Chest. 2012;141(2 Suppl):e419S-e496S. doi:10.1378/chest.11-2301
11. Bashir R, Zack CJ, Zhao H, Comerota AJ, Bove AA. Comparative outcomes of catheter-directed thrombolysis plus anticoagulation vs anticoagulation alone to treat lower-extremity proximal deep vein thrombosis. JAMA Intern Med. 2014;174(9):1494-1501. doi:10.1001/jamainternmed.2014.3415
12. Watson L, Broderick C, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev. 2016;11(11):CD002783. Published 2016 Nov 10. doi:10.1002/14651858.CD002783.pub4
13. Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet. 2012;379(9810):31-38. doi:10.1016/S0140-6736(11)61753-4
14. Vedantham S, Goldhaber SZ, Julian JA, et al; ATTRACT Trial Investigators. Pharmacomechanical catheter-directed thrombolysis for deep-vein thrombosis. N Engl J Med. 2017;377(23):2240-2252. doi:10.1056/NEJMoa1615066
Brodalumab in an Organ Transplant Recipient With Psoriasis
The treatment landscape for psoriasis has evolved rapidly over the last decade. Biologic therapies have demonstrated robust efficacy and acceptable safety profiles among many patients with moderate to severe plaque psoriasis. However, the use of biologics among immunocompromised patients with psoriasis rarely is discussed in the literature. As new biologics for psoriasis are being developed, a critical gap exists in the literature regarding the safety and efficacy of these medications in immunocompromised patients. Per American Academy of Dermatology–National Psoriasis Foundation guidelines, caution should be exercised when using biologics in patients with immunocompromising conditions.1 In organ transplant recipients, the potential risks of combining systemic medications used for organ transplantation and biologic treatments for psoriasis are unknown.2
In the posttransplant period, the immunosuppressive regimens for transplantation likely will improve psoriasis. However, patients with organ transplant and psoriasis still experience flares that can be challenging to treat.3 Prior treatment modalities to prevent psoriasis flares in organ transplant recipients have relied largely on topical therapies, posttransplant immunosuppressive medications (eg, cyclosporine, tacrolimus, mycophenolate mofetil) that prevent graft rejection, and systemic corticosteroids. We report a case of a 50-year-old man with a recent history of liver transplantation who presented with severe plaque psoriasis and psoriatic arthritis.
Case Report
A 50-year-old man presented to the dermatology clinic with moderate to severe plaque psoriasis and psoriatic arthritis that had been present for 15 years. His plaque psoriasis covered approximately 40% of the body surface area, including the scalp, trunk, arms, and legs. In addition, he had diffuse joint pain in the hands and feet; a radiograph revealed active psoriatic arthritis involving the joints of the fingers and toes.
One year prior to presentation to our dermatology clinic, the patient underwent an an orthotopic liver transplant for history of Child-Pugh class C liver cirrhosis secondary to untreated hepatitis C virus (HCV) and alcohol use that was complicated by hepatocellular carcinoma. He acquired a high-risk donor liver that was HCV positive with HCV genotype 1a. Starting 2 months after the transplant, he underwent 12 weeks of treatment for HCV with glecaprevir-pibrentasvir. Once his HCV treatment course was completed, he achieved a sustained virologic response with an undetectable viral load. To prevent transplant rejection, he was on chronic immunosuppression with tacrolimus, a calcineurin inhibitor, and mycophenolate mofetil, an inhibitor of inosine monophosphate dehydrogenase whose action leads to decreased proliferation of T cells and B cells.
The patient’s psoriasis initially was treated with triamcinolone acetonide ointment 0.1% applied twice daily to the psoriasis lesions for 1 year by another dermatologist. However, his psoriasis progressed to involve 40% of the body surface area. Following our evaluation 1 year posttransplant, the patient was started on subcutaneous brodalumab 210 mg at weeks 0, 1, and 2, then every 2 weeks thereafter. Approximately 10 weeks after initiation of brodalumab, the patient’s psoriasis was completely clear, and he was asymptomatic from psoriatic arthritis. The patient’s improvement persisted at 6 months, and his liver enzymes, including alkaline phosphatase, total bilirubin, alanine transaminase, and aspartate transaminase, continued to be within reference range. To date, there has been no evidence of posttransplant complications such as graft-vs-host disease, serious infections, or skin cancers.
Comment
Increased Risk for Infection and Malignancies in Transplant Patients
Transplant patients are on immunosuppressive regimens that increase their risk for infection and malignancies. For example, high doses of immunosuppresants predispose these patients to reactivation of viral infections, including BK and JC viruses.4 In addition, the incidence of squamous cell carcinoma is 65- to 250-fold higher in transplant patients compared to the general population.5 The risk for Merkel cell carcinoma is increased after solid organ transplantation compared to the general population.6 Importantly, transplant patients have a higher mortality from skin cancers than other types of cancers, including breast and colon cancer.7
Psoriasis in Organ Transplant Recipients
Psoriasis is a chronic, immune-mediated, inflammatory disease with a prevalence of approximately 3% in the United States.8 Approximately one-third of patients with psoriasis develop psoriatic arthritis.9 Organ transplant recipients with psoriasis and psoriatic arthritis represent a unique patient population whereby their use of chronic immunosuppressive medications to prevent graft rejection may put them at risk for developing infections and malignancies.
Special Considerations for Brodalumab
Brodalumab is an immunomodulatory biologic that binds to and inhibits IL-17RA, thereby inhibiting the actions of IL-17A, F, E, and C.2 The blockade of IL-17RA by brodalumab has been shown to result in reversal of psoriatic phenotype and gene expression patterns.10 Brodalumab was chosen as the treatment in our patient because it has a rapid onset of action, sustained efficacy, and an acceptable safety profile.11 Brodalumab is well tolerated, with approximately 60% of patients achieving clearance long-term.12 Candidal infections can occur in patients with brodalumab, but the rates are low and they are reversible with antifungal treatment.13 The increased mucocutaneous candidal infections are consistent with medications whose mechanism of action is IL-17 inhibition.14,15 The most common adverse reactions found were nasopharyngitis and headache.16 The causal link between brodalumab and suicidality has not been established.17
The use of brodalumab for psoriasis in organ transplant recipients has not been previously reported in the literature. A few case reports have been published on the successful use of etanercept and ixekizumab as biologic treatment options for psoriasis in transplant patients.18-23 In addition to choosing an appropriate biologic for psoriasis in transplant patients, transplant providers may evaluate the choice of immunosuppression regimen for the organ transplant in the context of psoriasis. In a retrospective analysis of liver transplant patients with psoriasis, Foroncewicz et al3 found cyclosporine, which was used as an antirejection immunosuppressive agent in the posttransplant period, to be more effective than tacrolimus in treating recurrent psoriasis in liver transplant recipients.
Our case illustrates one example of the successful use of brodalumab in a patient with a solid organ transplant. Our patient’s psoriasis and symptoms of psoriatic arthritis greatly improved after initiation of brodalumab. In the posttransplant period, the patient did not develop graft-vs-host disease, infections, malignancies, depression, or suicidal ideation while taking brodalumab.
Conclusion
It is important that the patient, dermatology team, and transplant team work together to navigate the challenges and relatively unknown landscape of psoriasis treatment in organ transplant recipients. As the number of organ transplant recipients continues to increase, this issue will become more clinically relevant. Case reports and future prospective studies will continue to inform us regarding the role of biologics in psoriasis treatment posttransplantation.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80:1029-1072.
- Prussick R, Wu JJ, Armstrong AW, et al. Psoriasis in solid organ transplant patients: best practice recommendations from The Medical Board of the National Psoriasis Foundation. J Dermatol Treat. 2018;29:329-333.
- Foroncewicz B, Mucha K, Lerut J, et al. Cyclosporine is superior to tacrolimus in liver transplant recipients with recurrent psoriasis. Ann Transplant. 2014;19:427-433.
- Boukoum H, Nahdi I, Sahtout W, et al. BK and JC virus infections in healthy patients compared to kidney transplant recipients in Tunisia. Microbial Pathogenesis. 2016;97:204-208.
- Bouwes Bavinck JN, Euvrard S, Naldi L, et al. Keratotic skin lesions and other risk factors are associated with skin cancer in organ-transplant recipients: a case-control study in The Netherlands, United Kingdom, Germany, France, and Italy. J Invest Dermatol. 2007;127:1647-1656.
- Clark CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after transplant. Clin Oncol. 2019;31:779-788.
- Lakhani NA, Saraiya M, Thompson TD, et al. Total body skin examination for skin cancer screening among U.S. adults from 2000 to 2010. Prev Med. 2014;61:75-80.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Alinaghi F, Calov M, Kristensen LE, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80:251-265.
- Russell CB, Rand H, Bigler J, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol. 2014;192:3828-3836.
- Foulkes AC, Warren RB. Brodalumab in psoriasis: evidence to date and clinical potential. Drugs Context. 2019;8:212570. doi:10.7573/dic.212570
- Puig L, Lebwohl M, Bachelez H, et al. Long-term efficacy and safety of brodalumab in the treatment of psoriasis: 120-week results from the randomized, double-blind, placebo- and active comparator-controlled phase 3 AMAGINE-2 trial. J Am Acad Dermatol. 2020;82:352-359.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab and ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299-311.
- Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65-68.
- Farahnik B, Beroukhim B, Abrouk M, et al. Brodalumab for the treatment of psoriasis: a review of Phase III trials. Dermatol Ther. 2016;6:111-124.
- Lebwohl MG, Papp KA, Marangell LB, et al. Psychiatric adverse events during treatment with brodalumab: analysis of psoriasis clinical trials. J Am Acad Dermatol. 2018;78:81-89.
- DeSimone C, Perino F, Caldarola G, et al. Treatment of psoriasis with etanercept in immunocompromised patients: two case reports. J Int Med Res. 2016;44:67-71.
- Madankumar R, Teperman LW, Stein JA. Use of etanercept for psoriasis in a liver transplant recipient. JAAD Case Rep. 2015;1:S36-S37.
- Collazo MH, González JR, Torres EA. Etanercept therapy for psoriasis in a patient with concomitant hepatitis C and liver transplant. P R Health Sci J. 2008;27:346-347.
- Hoover WD. Etanercept therapy for severe plaque psoriasis in a patient who underwent a liver transplant. Cutis. 2007;80:211-214.
- Brokalaki EI, Voshege N, Witzke O, et al. Treatment of severe psoriasis with etanercept in a pancreas-kidney transplant recipient. Transplant Proc. 2012;44:2776-2777.
- Lora V, Graceffa D, De Felice C, et al. Treatment of severe psoriasis with ixekizumab in a liver transplant recipient with concomitant hepatitis B virus infection. Dermatol Ther. 2019;32:E12909.
The treatment landscape for psoriasis has evolved rapidly over the last decade. Biologic therapies have demonstrated robust efficacy and acceptable safety profiles among many patients with moderate to severe plaque psoriasis. However, the use of biologics among immunocompromised patients with psoriasis rarely is discussed in the literature. As new biologics for psoriasis are being developed, a critical gap exists in the literature regarding the safety and efficacy of these medications in immunocompromised patients. Per American Academy of Dermatology–National Psoriasis Foundation guidelines, caution should be exercised when using biologics in patients with immunocompromising conditions.1 In organ transplant recipients, the potential risks of combining systemic medications used for organ transplantation and biologic treatments for psoriasis are unknown.2
In the posttransplant period, the immunosuppressive regimens for transplantation likely will improve psoriasis. However, patients with organ transplant and psoriasis still experience flares that can be challenging to treat.3 Prior treatment modalities to prevent psoriasis flares in organ transplant recipients have relied largely on topical therapies, posttransplant immunosuppressive medications (eg, cyclosporine, tacrolimus, mycophenolate mofetil) that prevent graft rejection, and systemic corticosteroids. We report a case of a 50-year-old man with a recent history of liver transplantation who presented with severe plaque psoriasis and psoriatic arthritis.
Case Report
A 50-year-old man presented to the dermatology clinic with moderate to severe plaque psoriasis and psoriatic arthritis that had been present for 15 years. His plaque psoriasis covered approximately 40% of the body surface area, including the scalp, trunk, arms, and legs. In addition, he had diffuse joint pain in the hands and feet; a radiograph revealed active psoriatic arthritis involving the joints of the fingers and toes.
One year prior to presentation to our dermatology clinic, the patient underwent an an orthotopic liver transplant for history of Child-Pugh class C liver cirrhosis secondary to untreated hepatitis C virus (HCV) and alcohol use that was complicated by hepatocellular carcinoma. He acquired a high-risk donor liver that was HCV positive with HCV genotype 1a. Starting 2 months after the transplant, he underwent 12 weeks of treatment for HCV with glecaprevir-pibrentasvir. Once his HCV treatment course was completed, he achieved a sustained virologic response with an undetectable viral load. To prevent transplant rejection, he was on chronic immunosuppression with tacrolimus, a calcineurin inhibitor, and mycophenolate mofetil, an inhibitor of inosine monophosphate dehydrogenase whose action leads to decreased proliferation of T cells and B cells.
The patient’s psoriasis initially was treated with triamcinolone acetonide ointment 0.1% applied twice daily to the psoriasis lesions for 1 year by another dermatologist. However, his psoriasis progressed to involve 40% of the body surface area. Following our evaluation 1 year posttransplant, the patient was started on subcutaneous brodalumab 210 mg at weeks 0, 1, and 2, then every 2 weeks thereafter. Approximately 10 weeks after initiation of brodalumab, the patient’s psoriasis was completely clear, and he was asymptomatic from psoriatic arthritis. The patient’s improvement persisted at 6 months, and his liver enzymes, including alkaline phosphatase, total bilirubin, alanine transaminase, and aspartate transaminase, continued to be within reference range. To date, there has been no evidence of posttransplant complications such as graft-vs-host disease, serious infections, or skin cancers.
Comment
Increased Risk for Infection and Malignancies in Transplant Patients
Transplant patients are on immunosuppressive regimens that increase their risk for infection and malignancies. For example, high doses of immunosuppresants predispose these patients to reactivation of viral infections, including BK and JC viruses.4 In addition, the incidence of squamous cell carcinoma is 65- to 250-fold higher in transplant patients compared to the general population.5 The risk for Merkel cell carcinoma is increased after solid organ transplantation compared to the general population.6 Importantly, transplant patients have a higher mortality from skin cancers than other types of cancers, including breast and colon cancer.7
Psoriasis in Organ Transplant Recipients
Psoriasis is a chronic, immune-mediated, inflammatory disease with a prevalence of approximately 3% in the United States.8 Approximately one-third of patients with psoriasis develop psoriatic arthritis.9 Organ transplant recipients with psoriasis and psoriatic arthritis represent a unique patient population whereby their use of chronic immunosuppressive medications to prevent graft rejection may put them at risk for developing infections and malignancies.
Special Considerations for Brodalumab
Brodalumab is an immunomodulatory biologic that binds to and inhibits IL-17RA, thereby inhibiting the actions of IL-17A, F, E, and C.2 The blockade of IL-17RA by brodalumab has been shown to result in reversal of psoriatic phenotype and gene expression patterns.10 Brodalumab was chosen as the treatment in our patient because it has a rapid onset of action, sustained efficacy, and an acceptable safety profile.11 Brodalumab is well tolerated, with approximately 60% of patients achieving clearance long-term.12 Candidal infections can occur in patients with brodalumab, but the rates are low and they are reversible with antifungal treatment.13 The increased mucocutaneous candidal infections are consistent with medications whose mechanism of action is IL-17 inhibition.14,15 The most common adverse reactions found were nasopharyngitis and headache.16 The causal link between brodalumab and suicidality has not been established.17
The use of brodalumab for psoriasis in organ transplant recipients has not been previously reported in the literature. A few case reports have been published on the successful use of etanercept and ixekizumab as biologic treatment options for psoriasis in transplant patients.18-23 In addition to choosing an appropriate biologic for psoriasis in transplant patients, transplant providers may evaluate the choice of immunosuppression regimen for the organ transplant in the context of psoriasis. In a retrospective analysis of liver transplant patients with psoriasis, Foroncewicz et al3 found cyclosporine, which was used as an antirejection immunosuppressive agent in the posttransplant period, to be more effective than tacrolimus in treating recurrent psoriasis in liver transplant recipients.
Our case illustrates one example of the successful use of brodalumab in a patient with a solid organ transplant. Our patient’s psoriasis and symptoms of psoriatic arthritis greatly improved after initiation of brodalumab. In the posttransplant period, the patient did not develop graft-vs-host disease, infections, malignancies, depression, or suicidal ideation while taking brodalumab.
Conclusion
It is important that the patient, dermatology team, and transplant team work together to navigate the challenges and relatively unknown landscape of psoriasis treatment in organ transplant recipients. As the number of organ transplant recipients continues to increase, this issue will become more clinically relevant. Case reports and future prospective studies will continue to inform us regarding the role of biologics in psoriasis treatment posttransplantation.
The treatment landscape for psoriasis has evolved rapidly over the last decade. Biologic therapies have demonstrated robust efficacy and acceptable safety profiles among many patients with moderate to severe plaque psoriasis. However, the use of biologics among immunocompromised patients with psoriasis rarely is discussed in the literature. As new biologics for psoriasis are being developed, a critical gap exists in the literature regarding the safety and efficacy of these medications in immunocompromised patients. Per American Academy of Dermatology–National Psoriasis Foundation guidelines, caution should be exercised when using biologics in patients with immunocompromising conditions.1 In organ transplant recipients, the potential risks of combining systemic medications used for organ transplantation and biologic treatments for psoriasis are unknown.2
In the posttransplant period, the immunosuppressive regimens for transplantation likely will improve psoriasis. However, patients with organ transplant and psoriasis still experience flares that can be challenging to treat.3 Prior treatment modalities to prevent psoriasis flares in organ transplant recipients have relied largely on topical therapies, posttransplant immunosuppressive medications (eg, cyclosporine, tacrolimus, mycophenolate mofetil) that prevent graft rejection, and systemic corticosteroids. We report a case of a 50-year-old man with a recent history of liver transplantation who presented with severe plaque psoriasis and psoriatic arthritis.
Case Report
A 50-year-old man presented to the dermatology clinic with moderate to severe plaque psoriasis and psoriatic arthritis that had been present for 15 years. His plaque psoriasis covered approximately 40% of the body surface area, including the scalp, trunk, arms, and legs. In addition, he had diffuse joint pain in the hands and feet; a radiograph revealed active psoriatic arthritis involving the joints of the fingers and toes.
One year prior to presentation to our dermatology clinic, the patient underwent an an orthotopic liver transplant for history of Child-Pugh class C liver cirrhosis secondary to untreated hepatitis C virus (HCV) and alcohol use that was complicated by hepatocellular carcinoma. He acquired a high-risk donor liver that was HCV positive with HCV genotype 1a. Starting 2 months after the transplant, he underwent 12 weeks of treatment for HCV with glecaprevir-pibrentasvir. Once his HCV treatment course was completed, he achieved a sustained virologic response with an undetectable viral load. To prevent transplant rejection, he was on chronic immunosuppression with tacrolimus, a calcineurin inhibitor, and mycophenolate mofetil, an inhibitor of inosine monophosphate dehydrogenase whose action leads to decreased proliferation of T cells and B cells.
The patient’s psoriasis initially was treated with triamcinolone acetonide ointment 0.1% applied twice daily to the psoriasis lesions for 1 year by another dermatologist. However, his psoriasis progressed to involve 40% of the body surface area. Following our evaluation 1 year posttransplant, the patient was started on subcutaneous brodalumab 210 mg at weeks 0, 1, and 2, then every 2 weeks thereafter. Approximately 10 weeks after initiation of brodalumab, the patient’s psoriasis was completely clear, and he was asymptomatic from psoriatic arthritis. The patient’s improvement persisted at 6 months, and his liver enzymes, including alkaline phosphatase, total bilirubin, alanine transaminase, and aspartate transaminase, continued to be within reference range. To date, there has been no evidence of posttransplant complications such as graft-vs-host disease, serious infections, or skin cancers.
Comment
Increased Risk for Infection and Malignancies in Transplant Patients
Transplant patients are on immunosuppressive regimens that increase their risk for infection and malignancies. For example, high doses of immunosuppresants predispose these patients to reactivation of viral infections, including BK and JC viruses.4 In addition, the incidence of squamous cell carcinoma is 65- to 250-fold higher in transplant patients compared to the general population.5 The risk for Merkel cell carcinoma is increased after solid organ transplantation compared to the general population.6 Importantly, transplant patients have a higher mortality from skin cancers than other types of cancers, including breast and colon cancer.7
Psoriasis in Organ Transplant Recipients
Psoriasis is a chronic, immune-mediated, inflammatory disease with a prevalence of approximately 3% in the United States.8 Approximately one-third of patients with psoriasis develop psoriatic arthritis.9 Organ transplant recipients with psoriasis and psoriatic arthritis represent a unique patient population whereby their use of chronic immunosuppressive medications to prevent graft rejection may put them at risk for developing infections and malignancies.
Special Considerations for Brodalumab
Brodalumab is an immunomodulatory biologic that binds to and inhibits IL-17RA, thereby inhibiting the actions of IL-17A, F, E, and C.2 The blockade of IL-17RA by brodalumab has been shown to result in reversal of psoriatic phenotype and gene expression patterns.10 Brodalumab was chosen as the treatment in our patient because it has a rapid onset of action, sustained efficacy, and an acceptable safety profile.11 Brodalumab is well tolerated, with approximately 60% of patients achieving clearance long-term.12 Candidal infections can occur in patients with brodalumab, but the rates are low and they are reversible with antifungal treatment.13 The increased mucocutaneous candidal infections are consistent with medications whose mechanism of action is IL-17 inhibition.14,15 The most common adverse reactions found were nasopharyngitis and headache.16 The causal link between brodalumab and suicidality has not been established.17
The use of brodalumab for psoriasis in organ transplant recipients has not been previously reported in the literature. A few case reports have been published on the successful use of etanercept and ixekizumab as biologic treatment options for psoriasis in transplant patients.18-23 In addition to choosing an appropriate biologic for psoriasis in transplant patients, transplant providers may evaluate the choice of immunosuppression regimen for the organ transplant in the context of psoriasis. In a retrospective analysis of liver transplant patients with psoriasis, Foroncewicz et al3 found cyclosporine, which was used as an antirejection immunosuppressive agent in the posttransplant period, to be more effective than tacrolimus in treating recurrent psoriasis in liver transplant recipients.
Our case illustrates one example of the successful use of brodalumab in a patient with a solid organ transplant. Our patient’s psoriasis and symptoms of psoriatic arthritis greatly improved after initiation of brodalumab. In the posttransplant period, the patient did not develop graft-vs-host disease, infections, malignancies, depression, or suicidal ideation while taking brodalumab.
Conclusion
It is important that the patient, dermatology team, and transplant team work together to navigate the challenges and relatively unknown landscape of psoriasis treatment in organ transplant recipients. As the number of organ transplant recipients continues to increase, this issue will become more clinically relevant. Case reports and future prospective studies will continue to inform us regarding the role of biologics in psoriasis treatment posttransplantation.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80:1029-1072.
- Prussick R, Wu JJ, Armstrong AW, et al. Psoriasis in solid organ transplant patients: best practice recommendations from The Medical Board of the National Psoriasis Foundation. J Dermatol Treat. 2018;29:329-333.
- Foroncewicz B, Mucha K, Lerut J, et al. Cyclosporine is superior to tacrolimus in liver transplant recipients with recurrent psoriasis. Ann Transplant. 2014;19:427-433.
- Boukoum H, Nahdi I, Sahtout W, et al. BK and JC virus infections in healthy patients compared to kidney transplant recipients in Tunisia. Microbial Pathogenesis. 2016;97:204-208.
- Bouwes Bavinck JN, Euvrard S, Naldi L, et al. Keratotic skin lesions and other risk factors are associated with skin cancer in organ-transplant recipients: a case-control study in The Netherlands, United Kingdom, Germany, France, and Italy. J Invest Dermatol. 2007;127:1647-1656.
- Clark CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after transplant. Clin Oncol. 2019;31:779-788.
- Lakhani NA, Saraiya M, Thompson TD, et al. Total body skin examination for skin cancer screening among U.S. adults from 2000 to 2010. Prev Med. 2014;61:75-80.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Alinaghi F, Calov M, Kristensen LE, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80:251-265.
- Russell CB, Rand H, Bigler J, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol. 2014;192:3828-3836.
- Foulkes AC, Warren RB. Brodalumab in psoriasis: evidence to date and clinical potential. Drugs Context. 2019;8:212570. doi:10.7573/dic.212570
- Puig L, Lebwohl M, Bachelez H, et al. Long-term efficacy and safety of brodalumab in the treatment of psoriasis: 120-week results from the randomized, double-blind, placebo- and active comparator-controlled phase 3 AMAGINE-2 trial. J Am Acad Dermatol. 2020;82:352-359.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab and ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299-311.
- Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65-68.
- Farahnik B, Beroukhim B, Abrouk M, et al. Brodalumab for the treatment of psoriasis: a review of Phase III trials. Dermatol Ther. 2016;6:111-124.
- Lebwohl MG, Papp KA, Marangell LB, et al. Psychiatric adverse events during treatment with brodalumab: analysis of psoriasis clinical trials. J Am Acad Dermatol. 2018;78:81-89.
- DeSimone C, Perino F, Caldarola G, et al. Treatment of psoriasis with etanercept in immunocompromised patients: two case reports. J Int Med Res. 2016;44:67-71.
- Madankumar R, Teperman LW, Stein JA. Use of etanercept for psoriasis in a liver transplant recipient. JAAD Case Rep. 2015;1:S36-S37.
- Collazo MH, González JR, Torres EA. Etanercept therapy for psoriasis in a patient with concomitant hepatitis C and liver transplant. P R Health Sci J. 2008;27:346-347.
- Hoover WD. Etanercept therapy for severe plaque psoriasis in a patient who underwent a liver transplant. Cutis. 2007;80:211-214.
- Brokalaki EI, Voshege N, Witzke O, et al. Treatment of severe psoriasis with etanercept in a pancreas-kidney transplant recipient. Transplant Proc. 2012;44:2776-2777.
- Lora V, Graceffa D, De Felice C, et al. Treatment of severe psoriasis with ixekizumab in a liver transplant recipient with concomitant hepatitis B virus infection. Dermatol Ther. 2019;32:E12909.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80:1029-1072.
- Prussick R, Wu JJ, Armstrong AW, et al. Psoriasis in solid organ transplant patients: best practice recommendations from The Medical Board of the National Psoriasis Foundation. J Dermatol Treat. 2018;29:329-333.
- Foroncewicz B, Mucha K, Lerut J, et al. Cyclosporine is superior to tacrolimus in liver transplant recipients with recurrent psoriasis. Ann Transplant. 2014;19:427-433.
- Boukoum H, Nahdi I, Sahtout W, et al. BK and JC virus infections in healthy patients compared to kidney transplant recipients in Tunisia. Microbial Pathogenesis. 2016;97:204-208.
- Bouwes Bavinck JN, Euvrard S, Naldi L, et al. Keratotic skin lesions and other risk factors are associated with skin cancer in organ-transplant recipients: a case-control study in The Netherlands, United Kingdom, Germany, France, and Italy. J Invest Dermatol. 2007;127:1647-1656.
- Clark CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after transplant. Clin Oncol. 2019;31:779-788.
- Lakhani NA, Saraiya M, Thompson TD, et al. Total body skin examination for skin cancer screening among U.S. adults from 2000 to 2010. Prev Med. 2014;61:75-80.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Alinaghi F, Calov M, Kristensen LE, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80:251-265.
- Russell CB, Rand H, Bigler J, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol. 2014;192:3828-3836.
- Foulkes AC, Warren RB. Brodalumab in psoriasis: evidence to date and clinical potential. Drugs Context. 2019;8:212570. doi:10.7573/dic.212570
- Puig L, Lebwohl M, Bachelez H, et al. Long-term efficacy and safety of brodalumab in the treatment of psoriasis: 120-week results from the randomized, double-blind, placebo- and active comparator-controlled phase 3 AMAGINE-2 trial. J Am Acad Dermatol. 2020;82:352-359.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab and ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299-311.
- Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65-68.
- Farahnik B, Beroukhim B, Abrouk M, et al. Brodalumab for the treatment of psoriasis: a review of Phase III trials. Dermatol Ther. 2016;6:111-124.
- Lebwohl MG, Papp KA, Marangell LB, et al. Psychiatric adverse events during treatment with brodalumab: analysis of psoriasis clinical trials. J Am Acad Dermatol. 2018;78:81-89.
- DeSimone C, Perino F, Caldarola G, et al. Treatment of psoriasis with etanercept in immunocompromised patients: two case reports. J Int Med Res. 2016;44:67-71.
- Madankumar R, Teperman LW, Stein JA. Use of etanercept for psoriasis in a liver transplant recipient. JAAD Case Rep. 2015;1:S36-S37.
- Collazo MH, González JR, Torres EA. Etanercept therapy for psoriasis in a patient with concomitant hepatitis C and liver transplant. P R Health Sci J. 2008;27:346-347.
- Hoover WD. Etanercept therapy for severe plaque psoriasis in a patient who underwent a liver transplant. Cutis. 2007;80:211-214.
- Brokalaki EI, Voshege N, Witzke O, et al. Treatment of severe psoriasis with etanercept in a pancreas-kidney transplant recipient. Transplant Proc. 2012;44:2776-2777.
- Lora V, Graceffa D, De Felice C, et al. Treatment of severe psoriasis with ixekizumab in a liver transplant recipient with concomitant hepatitis B virus infection. Dermatol Ther. 2019;32:E12909.
Practice Points
- Immunocompromised patients, such as organ transplant recipients, require careful benefit-risk consideration when selecting a systemic agent for psoriasis.
- Brodalumab, an IL-17RA antagonist, was used to treat a patient with psoriasis who had undergone solid organ transplant with excellent response and good tolerability.
- Further studies are needed to evaluate the benefits and risks of using biologic treatments in patients with psoriasis who are organ transplant recipients.
Unilateral Verrucous Psoriasis
Case Report
An 80-year-old man with a history of hypertension and coronary artery disease presented to the dermatology clinic with a rash characterized by multiple asymptomatic plaques with overlying verrucous nodules on the left side of the abdomen, back, and leg (Figure 1). He reported that these “growths” appeared 20 years prior to presentation, shortly after coronary artery bypass surgery with a saphenous vein graft. The patient initially was given a diagnosis of verruca vulgaris and then biopsy-proven psoriasis later that year. At that time, he refused systemic treatment and was treated instead with triamcinolone acetonide ointment, with periodic surgical removal of bothersome lesions.
At the current presentation, physical examination revealed many hyperkeratotic, yellow-gray, verrucous nodules overlying scaly, erythematous, sharply demarcated plaques, exclusively on the left side of the body, including the left side of the abdomen, back, and leg. The differential diagnosis included linear psoriasis and inflammatory linear verrucous epidermal nevus (ILVEN).
Skin biopsy showed irregular psoriasiform epidermal hyperplasia with acanthosis, hyperkeratosis, and papillomatosis, with convergence of the rete ridges, known as buttressing (Figure 2A). There were tortuous dilated blood vessels in the dermal papillae, epidermal neutrophils at the tip of the suprapapillary plates, and Munro microabscesses in the stratum corneum (Figure 2B). Koilocytes were absent, and periodic acid–Schiff staining was negative. Taken together, clinical and histologic features led to a diagnosis of unilateral verrucous psoriasis.
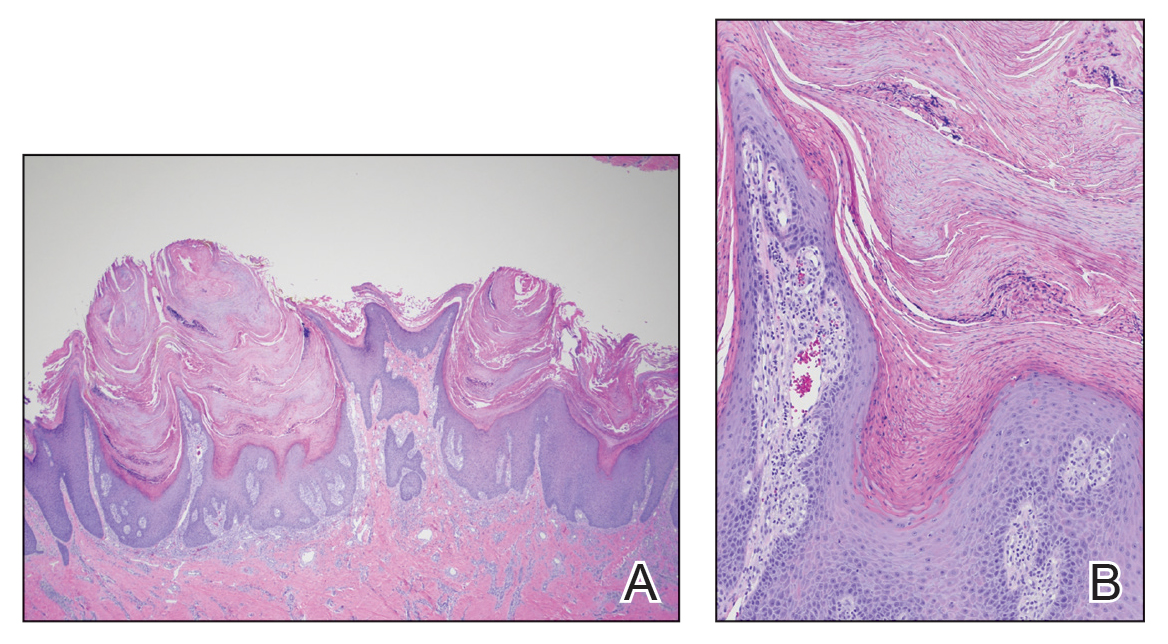
Comment
Presentation and Histology
Verrucous psoriasis is a variant of psoriasis that presents with wartlike clinical features and overlapping histologic features of verruca and psoriasis. It typically arises in patients with established psoriasis but can occur de novo.
Histologic features of verrucous psoriasis include epidermal hyperplasia with acanthosis, papillomatosis, and epidermal buttressing.1 It has been hypothesized that notable hyperkeratosis observed in these lesions is induced by repeat trauma to the extremities in patients with established psoriasis or by anoxia from conditions that predispose to poor circulation, such as diabetes mellitus and pulmonary disease.1,2
Pathogenesis
Most reported cases of verrucous psoriasis arose atop pre-existing psoriasis lesions.3,4 The relevance of our patient’s verrucous psoriasis to his prior coronary artery bypass surgery with saphenous vein graft is unknown; however, the distribution of lesions, timing of psoriasis onset in relation to the surgical procedure, and recent data proposing a role for neuropeptide responses to nerve injury in the development of psoriasis, taken together, provide an argument for a role for surgical trauma in the development of our patient’s condition.
Treatment
Although verrucous psoriasis presents both diagnostic and therapeutic challenges, there are some reports of improvement with topical or intralesional corticosteroids in combination with keratolytics,3 coal tar,5 and oral methotrexate.6 In addition, there are rare reports of successful treatment with biologics. A case report showed successful resolution with adalimumab,4 and a case of erythrodermic verrucous psoriasis showed moderate improvement with ustekinumab after other failed treatments.7
Differential Diagnosis
Psoriasis typically presents in a symmetric distribution, with rare reported cases of unilateral distribution. Two cases of unilateral psoriasis arising after a surgical procedure have been reported, one after mastectomy and the other after neurosurgery.8,9 Other cases of unilateral psoriasis are reported to have arisen in adolescents and young adults idiopathically.
A case of linear psoriasis arising in the distribution of the sciatic nerve in a patient with radiculopathy implicated tumor necrosis factor α, neuropeptides, and nerve growth factor released in response to compression as possible etiologic agents.10 However, none of the reported cases of linear psoriasis, or reported cases of unilateral psoriasis, exhibited verrucous features clinically or histologically. In our patient, distribution of the lesions appeared less typically blaschkoid than in linear psoriasis, and the presence of exophytic wartlike growths throughout the lesions was not characteristic of linear psoriasis.
Late-adulthood onset in this patient in addition to the absence of typical histologic features of ILVEN, including alternating orthokeratosis and parakeratosis,11 make a diagnosis of ILVEN less likely; ILVEN can be distinguished from linear psoriasis based on later age of onset and responsiveness to antipsoriatic therapy of linear psoriasis.12
Conclusion
We describe a unique presentation of an already rare variant of psoriasis that can be difficult to diagnose clinically. The unilateral distribution of lesions in this patient can create further diagnostic confusion with other entities, such as ILVEN and linear psoriasis, though it can be distinguished from those diseases based on histologic features. Our aim is that this report improves recognition of this unusual presentation of verrucous psoriasis in clinical settings and decreases delays in diagnosis and treatment.
- Khalil FK, Keehn CA, Saeed S, et al. Verrucous psoriasis: a distinctive clinicopathologic variant of psoriasis. Am J Dermatopathol. 2005;27:204-207.
- Wakamatsu K, Naniwa K, Hagiya Y, et al. Psoriasis verrucosa. J Dermatol. 2010;37:1060-1062.
- Monroe HR, Hillman JD, Chiu MW. A case of verrucous psoriasis. Dermatol Online J. 2011;17:10.
- Maejima H, Katayama C, Watarai A, et al. A case of psoriasis verrucosa successfully treated with adalimumab. J Drugs Dermatol. 2012;11:E74-E75.
- Erkek E, Bozdog˘an O. Annular verrucous psoriasis with exaggerated papillomatosis. Am J Dermatopathol. 2001;23:133-135.
- Hall L, Marks V, Tyler W. Verrucous psoriasis: a clinical and histopathologic mimicker of verruca vulgaris. J Am Acad Dermatol. 2013;68(4 suppl 1):AB218.
- Curtis AR, Yosipovitch G. Erythrodermic verrucous psoriasis. J Dermatolog Treat. 2012;23:215-218.
- Kim M, Jung JY, Na SY, et al. Unilateral psoriasis in a woman with ipsilateral post-mastectomy lymphedema. Ann Dermatol. 2011;23(suppl 3):S303-S305.
- Reyter I, Woodley D. Widespread unilateral plaques in a 68-year-old woman after neurosurgery. Arch Dermatol. 2004;140:1531-1536.
- Galluzzo M, Talamonti M, Di Stefani A, et al. Linear psoriasis following the typical distribution of the sciatic nerve. J Dermatol Case Rep. 2015;9:6-11.
- Sengupta S, Das JK, Gangopadhyay A. Naevoid psoriasis and ILVEN: same coin, two faces? Indian J Dermatol. 2012;57:489-491.
- Morag C, Metzker A. Inflammatory linear verrucous epidermal nevus: report of seven new cases and review of the literature. Pediatr Dermatol. 1985;3:15-18.
Case Report
An 80-year-old man with a history of hypertension and coronary artery disease presented to the dermatology clinic with a rash characterized by multiple asymptomatic plaques with overlying verrucous nodules on the left side of the abdomen, back, and leg (Figure 1). He reported that these “growths” appeared 20 years prior to presentation, shortly after coronary artery bypass surgery with a saphenous vein graft. The patient initially was given a diagnosis of verruca vulgaris and then biopsy-proven psoriasis later that year. At that time, he refused systemic treatment and was treated instead with triamcinolone acetonide ointment, with periodic surgical removal of bothersome lesions.
At the current presentation, physical examination revealed many hyperkeratotic, yellow-gray, verrucous nodules overlying scaly, erythematous, sharply demarcated plaques, exclusively on the left side of the body, including the left side of the abdomen, back, and leg. The differential diagnosis included linear psoriasis and inflammatory linear verrucous epidermal nevus (ILVEN).
Skin biopsy showed irregular psoriasiform epidermal hyperplasia with acanthosis, hyperkeratosis, and papillomatosis, with convergence of the rete ridges, known as buttressing (Figure 2A). There were tortuous dilated blood vessels in the dermal papillae, epidermal neutrophils at the tip of the suprapapillary plates, and Munro microabscesses in the stratum corneum (Figure 2B). Koilocytes were absent, and periodic acid–Schiff staining was negative. Taken together, clinical and histologic features led to a diagnosis of unilateral verrucous psoriasis.
Comment
Presentation and Histology
Verrucous psoriasis is a variant of psoriasis that presents with wartlike clinical features and overlapping histologic features of verruca and psoriasis. It typically arises in patients with established psoriasis but can occur de novo.
Histologic features of verrucous psoriasis include epidermal hyperplasia with acanthosis, papillomatosis, and epidermal buttressing.1 It has been hypothesized that notable hyperkeratosis observed in these lesions is induced by repeat trauma to the extremities in patients with established psoriasis or by anoxia from conditions that predispose to poor circulation, such as diabetes mellitus and pulmonary disease.1,2
Pathogenesis
Most reported cases of verrucous psoriasis arose atop pre-existing psoriasis lesions.3,4 The relevance of our patient’s verrucous psoriasis to his prior coronary artery bypass surgery with saphenous vein graft is unknown; however, the distribution of lesions, timing of psoriasis onset in relation to the surgical procedure, and recent data proposing a role for neuropeptide responses to nerve injury in the development of psoriasis, taken together, provide an argument for a role for surgical trauma in the development of our patient’s condition.
Treatment
Although verrucous psoriasis presents both diagnostic and therapeutic challenges, there are some reports of improvement with topical or intralesional corticosteroids in combination with keratolytics,3 coal tar,5 and oral methotrexate.6 In addition, there are rare reports of successful treatment with biologics. A case report showed successful resolution with adalimumab,4 and a case of erythrodermic verrucous psoriasis showed moderate improvement with ustekinumab after other failed treatments.7
Differential Diagnosis
Psoriasis typically presents in a symmetric distribution, with rare reported cases of unilateral distribution. Two cases of unilateral psoriasis arising after a surgical procedure have been reported, one after mastectomy and the other after neurosurgery.8,9 Other cases of unilateral psoriasis are reported to have arisen in adolescents and young adults idiopathically.
A case of linear psoriasis arising in the distribution of the sciatic nerve in a patient with radiculopathy implicated tumor necrosis factor α, neuropeptides, and nerve growth factor released in response to compression as possible etiologic agents.10 However, none of the reported cases of linear psoriasis, or reported cases of unilateral psoriasis, exhibited verrucous features clinically or histologically. In our patient, distribution of the lesions appeared less typically blaschkoid than in linear psoriasis, and the presence of exophytic wartlike growths throughout the lesions was not characteristic of linear psoriasis.
Late-adulthood onset in this patient in addition to the absence of typical histologic features of ILVEN, including alternating orthokeratosis and parakeratosis,11 make a diagnosis of ILVEN less likely; ILVEN can be distinguished from linear psoriasis based on later age of onset and responsiveness to antipsoriatic therapy of linear psoriasis.12
Conclusion
We describe a unique presentation of an already rare variant of psoriasis that can be difficult to diagnose clinically. The unilateral distribution of lesions in this patient can create further diagnostic confusion with other entities, such as ILVEN and linear psoriasis, though it can be distinguished from those diseases based on histologic features. Our aim is that this report improves recognition of this unusual presentation of verrucous psoriasis in clinical settings and decreases delays in diagnosis and treatment.
Case Report
An 80-year-old man with a history of hypertension and coronary artery disease presented to the dermatology clinic with a rash characterized by multiple asymptomatic plaques with overlying verrucous nodules on the left side of the abdomen, back, and leg (Figure 1). He reported that these “growths” appeared 20 years prior to presentation, shortly after coronary artery bypass surgery with a saphenous vein graft. The patient initially was given a diagnosis of verruca vulgaris and then biopsy-proven psoriasis later that year. At that time, he refused systemic treatment and was treated instead with triamcinolone acetonide ointment, with periodic surgical removal of bothersome lesions.
At the current presentation, physical examination revealed many hyperkeratotic, yellow-gray, verrucous nodules overlying scaly, erythematous, sharply demarcated plaques, exclusively on the left side of the body, including the left side of the abdomen, back, and leg. The differential diagnosis included linear psoriasis and inflammatory linear verrucous epidermal nevus (ILVEN).
Skin biopsy showed irregular psoriasiform epidermal hyperplasia with acanthosis, hyperkeratosis, and papillomatosis, with convergence of the rete ridges, known as buttressing (Figure 2A). There were tortuous dilated blood vessels in the dermal papillae, epidermal neutrophils at the tip of the suprapapillary plates, and Munro microabscesses in the stratum corneum (Figure 2B). Koilocytes were absent, and periodic acid–Schiff staining was negative. Taken together, clinical and histologic features led to a diagnosis of unilateral verrucous psoriasis.
Comment
Presentation and Histology
Verrucous psoriasis is a variant of psoriasis that presents with wartlike clinical features and overlapping histologic features of verruca and psoriasis. It typically arises in patients with established psoriasis but can occur de novo.
Histologic features of verrucous psoriasis include epidermal hyperplasia with acanthosis, papillomatosis, and epidermal buttressing.1 It has been hypothesized that notable hyperkeratosis observed in these lesions is induced by repeat trauma to the extremities in patients with established psoriasis or by anoxia from conditions that predispose to poor circulation, such as diabetes mellitus and pulmonary disease.1,2
Pathogenesis
Most reported cases of verrucous psoriasis arose atop pre-existing psoriasis lesions.3,4 The relevance of our patient’s verrucous psoriasis to his prior coronary artery bypass surgery with saphenous vein graft is unknown; however, the distribution of lesions, timing of psoriasis onset in relation to the surgical procedure, and recent data proposing a role for neuropeptide responses to nerve injury in the development of psoriasis, taken together, provide an argument for a role for surgical trauma in the development of our patient’s condition.
Treatment
Although verrucous psoriasis presents both diagnostic and therapeutic challenges, there are some reports of improvement with topical or intralesional corticosteroids in combination with keratolytics,3 coal tar,5 and oral methotrexate.6 In addition, there are rare reports of successful treatment with biologics. A case report showed successful resolution with adalimumab,4 and a case of erythrodermic verrucous psoriasis showed moderate improvement with ustekinumab after other failed treatments.7
Differential Diagnosis
Psoriasis typically presents in a symmetric distribution, with rare reported cases of unilateral distribution. Two cases of unilateral psoriasis arising after a surgical procedure have been reported, one after mastectomy and the other after neurosurgery.8,9 Other cases of unilateral psoriasis are reported to have arisen in adolescents and young adults idiopathically.
A case of linear psoriasis arising in the distribution of the sciatic nerve in a patient with radiculopathy implicated tumor necrosis factor α, neuropeptides, and nerve growth factor released in response to compression as possible etiologic agents.10 However, none of the reported cases of linear psoriasis, or reported cases of unilateral psoriasis, exhibited verrucous features clinically or histologically. In our patient, distribution of the lesions appeared less typically blaschkoid than in linear psoriasis, and the presence of exophytic wartlike growths throughout the lesions was not characteristic of linear psoriasis.
Late-adulthood onset in this patient in addition to the absence of typical histologic features of ILVEN, including alternating orthokeratosis and parakeratosis,11 make a diagnosis of ILVEN less likely; ILVEN can be distinguished from linear psoriasis based on later age of onset and responsiveness to antipsoriatic therapy of linear psoriasis.12
Conclusion
We describe a unique presentation of an already rare variant of psoriasis that can be difficult to diagnose clinically. The unilateral distribution of lesions in this patient can create further diagnostic confusion with other entities, such as ILVEN and linear psoriasis, though it can be distinguished from those diseases based on histologic features. Our aim is that this report improves recognition of this unusual presentation of verrucous psoriasis in clinical settings and decreases delays in diagnosis and treatment.
- Khalil FK, Keehn CA, Saeed S, et al. Verrucous psoriasis: a distinctive clinicopathologic variant of psoriasis. Am J Dermatopathol. 2005;27:204-207.
- Wakamatsu K, Naniwa K, Hagiya Y, et al. Psoriasis verrucosa. J Dermatol. 2010;37:1060-1062.
- Monroe HR, Hillman JD, Chiu MW. A case of verrucous psoriasis. Dermatol Online J. 2011;17:10.
- Maejima H, Katayama C, Watarai A, et al. A case of psoriasis verrucosa successfully treated with adalimumab. J Drugs Dermatol. 2012;11:E74-E75.
- Erkek E, Bozdog˘an O. Annular verrucous psoriasis with exaggerated papillomatosis. Am J Dermatopathol. 2001;23:133-135.
- Hall L, Marks V, Tyler W. Verrucous psoriasis: a clinical and histopathologic mimicker of verruca vulgaris. J Am Acad Dermatol. 2013;68(4 suppl 1):AB218.
- Curtis AR, Yosipovitch G. Erythrodermic verrucous psoriasis. J Dermatolog Treat. 2012;23:215-218.
- Kim M, Jung JY, Na SY, et al. Unilateral psoriasis in a woman with ipsilateral post-mastectomy lymphedema. Ann Dermatol. 2011;23(suppl 3):S303-S305.
- Reyter I, Woodley D. Widespread unilateral plaques in a 68-year-old woman after neurosurgery. Arch Dermatol. 2004;140:1531-1536.
- Galluzzo M, Talamonti M, Di Stefani A, et al. Linear psoriasis following the typical distribution of the sciatic nerve. J Dermatol Case Rep. 2015;9:6-11.
- Sengupta S, Das JK, Gangopadhyay A. Naevoid psoriasis and ILVEN: same coin, two faces? Indian J Dermatol. 2012;57:489-491.
- Morag C, Metzker A. Inflammatory linear verrucous epidermal nevus: report of seven new cases and review of the literature. Pediatr Dermatol. 1985;3:15-18.
- Khalil FK, Keehn CA, Saeed S, et al. Verrucous psoriasis: a distinctive clinicopathologic variant of psoriasis. Am J Dermatopathol. 2005;27:204-207.
- Wakamatsu K, Naniwa K, Hagiya Y, et al. Psoriasis verrucosa. J Dermatol. 2010;37:1060-1062.
- Monroe HR, Hillman JD, Chiu MW. A case of verrucous psoriasis. Dermatol Online J. 2011;17:10.
- Maejima H, Katayama C, Watarai A, et al. A case of psoriasis verrucosa successfully treated with adalimumab. J Drugs Dermatol. 2012;11:E74-E75.
- Erkek E, Bozdog˘an O. Annular verrucous psoriasis with exaggerated papillomatosis. Am J Dermatopathol. 2001;23:133-135.
- Hall L, Marks V, Tyler W. Verrucous psoriasis: a clinical and histopathologic mimicker of verruca vulgaris. J Am Acad Dermatol. 2013;68(4 suppl 1):AB218.
- Curtis AR, Yosipovitch G. Erythrodermic verrucous psoriasis. J Dermatolog Treat. 2012;23:215-218.
- Kim M, Jung JY, Na SY, et al. Unilateral psoriasis in a woman with ipsilateral post-mastectomy lymphedema. Ann Dermatol. 2011;23(suppl 3):S303-S305.
- Reyter I, Woodley D. Widespread unilateral plaques in a 68-year-old woman after neurosurgery. Arch Dermatol. 2004;140:1531-1536.
- Galluzzo M, Talamonti M, Di Stefani A, et al. Linear psoriasis following the typical distribution of the sciatic nerve. J Dermatol Case Rep. 2015;9:6-11.
- Sengupta S, Das JK, Gangopadhyay A. Naevoid psoriasis and ILVEN: same coin, two faces? Indian J Dermatol. 2012;57:489-491.
- Morag C, Metzker A. Inflammatory linear verrucous epidermal nevus: report of seven new cases and review of the literature. Pediatr Dermatol. 1985;3:15-18.
Practice Points
- Verrucous psoriasis is a rare variant of psoriasis characterized by hypertrophic verrucous papules and plaques on an erythematous base.
- Histologically, verrucous psoriasis presents with overlapping features of verruca and psoriasis.
- Although psoriasis typically presents in a symmetric distribution, unilateral psoriasis can occur either de novo in younger patients or after surgical trauma in older patients.
An Unusual Presentation of Cutaneous Metastatic Lobular Breast Carcinoma
In women, breast cancer is the leading cancer diagnosis and the second leading cause of cancer-related death,1 as well as the most common malignancy to metastasize to the skin.2 Cutaneous breast carcinoma may present as cutaneous metastasis or can occur secondary to direct tumor extension. Five percent to 10% of women with breast cancer will present clinically with metastatic cutaneous disease, most commonly as a recurrence of early-stage breast carcinoma.2
In a published meta-analysis that investigated the incidence of tumors most commonly found to metastasize to the skin, Krathen et al3 found that cutaneous metastases occurred in 24% of patients with breast cancer (N=1903). In 2 large retrospective studies from tumor registry data, breast cancer was found to be the most common tumor involving metastasis to the skin, and 3.5% of the breast cancer cases identified in the registry had cutaneous metastasis as the presenting sign (n=35) at time of diagnosis.4
We report an unusual presentation of cutaneous metastatic lobular breast carcinoma that involved diffuse cutaneous lesions and rapid progression from onset of the breast mass to development of clinically apparent metastatic skin lesions.
Case Report
A 59-year-old woman with an unremarkable medical history presented to our dermatology clinic for evaluation of new widespread lesions that developed over a period of months. The eruption was asymptomatic and consisted of numerous bumpy lesions that reportedly started on the patient’s neck and progressively spread to involve the trunk. Physical examination revealed multiple flesh-colored, firm nodules scattered across the upper back, neck, and chest (Figure 1). Bilateral cervical and axillary lymphadenopathy also was noted. Upon questioning regarding family history of malignancy, the patient reported that her brother had been diagnosed with colon cancer. Although she was not up to date on age-appropriate malignancy screenings, she did report having a diagnostic mammogram 1 year prior that revealed a suspicious lesion on the left breast. A repeat mammogram of the left breast 6 months later was read as unremarkable.
Two 3-mm representative punch biopsies were performed. Hematoxylin and eosin staining revealed a basket-weave stratum corneum with underlying epidermal atrophy. A relatively monomorphic epithelioid cell infiltrate extending from the superficial reticular dermis into the deep dermis and displaying an open chromatin pattern and pink cytoplasm was observed, as well as dermal collagen thickening. Linear, single-filing cells along with focal irregular nests and scattered cells were observed (Figure 2). Immunohistochemical staining was positive for cytokeratin 7 (Figure 3A), epithelial membrane antigen, and estrogen receptor (Figure 3B) along with gross cystic disease fluid protein 15; focal progesterone receptor positivity also was present. Cytokeratin 20, cytokeratin 5/6, carcinoembryonic antigen, p63, CDX2, paired box gene 8, thyroid transcription factor 1, and human epidermal growth factor receptor 2/neu stains were negative. Findings identified in both biopsies were consistent with metastatic cutaneous lobular breast carcinoma.
A complete blood cell count and complete metabolic panels were within normal limits, aside from a mildly elevated alkaline phosphatase level. Breast ultrasonography was unremarkable. Stereotactic breast magnetic resonance imaging (MRI) revealed a 9.4-cm mass in the upper outer quadrant of the right breast as well as enlarged lymph nodes 2.2 cm from the left axilla. A subsequent bone scan demonstrated focal activity in the left lateral fourth rib, left costochondral junction, and right anterolateral fifth rib—it was unclear whether these lesions were metastatic or secondary to trauma from a fall the patient reportedly had sustained 2 weeks prior. Lumbar MRI without gadolinium contrast revealed extensive abnormal heterogeneous signal intensity of osseous structures consistent with osseous metastasis.
Subsequent diagnostic bilateral breast ultrasonography and percutaneous left lymph node biopsy revealed pathology consistent with metastatic lobular breast carcinoma with near total effacement of the lymph node and extracapsular extension concordant with previous MRI findings. The mass in the upper outer quadrant of the right breast that previously was observed on MRI was not identifiable on this ultrasound. It was recommended that the patient pursue MRI-guided breast biopsy to have the breast lesion further characterized. She was referred to surgical oncology at a tertiary center for management; however, the patient was lost to follow-up, and there are no records available indicating the patient pursued any treatment. Although we were unable to confirm the patient’s breast lesion that previously was seen on MRI was the cause of the metastatic disease, the overall clinical picture supported metastatic lobular breast carcinoma.
Comment
Tumor metastasis to the skin accounts for approximately 2% of all skin cancers5 and typically is observed in advanced stages of cancer. In women, breast carcinoma is the most common type of cancer to exhibit this behavior.2 Invasive ductal carcinoma represents the most common histologic subtype of breast cancer overall,6,7 and breast adenocarcinomas, including lobular and ductal breast carcinomas, are the most common histologic subtypes to exhibit metastatic cutaneous lesions.8
Invasive lobular breast carcinoma represents approximately 10% of invasive breast cancer cases. Compared to invasive ductal carcinoma, there tends to be a delay in diagnosis often leading to larger tumor sizes relative to the former upon detection and with lymph node invasion. These findings may be explained by the greater difficulty of detecting invasive lobular carcinomas by mammography and clinical breast examination compared to invasive ductal carcinomas.9-11 Additionally, invasive lobular carcinomas are more likely to be positive for estrogen and progesterone receptors compared to invasive ductal carcinomas,12 which also was consistent in our case.
Cutaneous metastases of breast cancer most commonly are found on the anterior chest wall and can present as a wide spectrum of lesions, with nodules as the most common primary dermatologic manifestation.13 Cutaneous metastatic lesions commonly have been described as firm, mobile, round or oval, solitary or grouped nodules. The color of the nodules varies and may be flesh-colored, brown, blue, black, pink, and/or red-brown. The lesions often are asymptomatic but may ulcerate.2
In our case, the distribution of lesions was a unique aspect that is not typical of most cases of metastatic cutaneous breast carcinoma. The nodules appeared more scattered and involved multiple body regions, including the back, neck, and chest. Although cutaneous breast cancer metastases have been documented to extend to these body regions, a review of PubMed articles indexed for MEDLINE using the terms cutaneous metastatic lobular breast carcinoma, breast carcinoma, and metastatic breast cancer suggested that it is uncommon for these multiple areas to be simultaneously affected.4,14 Rather, the more common clinical presentation of cutaneous metastatic breast carcinoma is as a solitary nodule or group of nodules localized to a single anatomic region.14
Another notable feature of our case was the rapid development of the cutaneous lesions relative to the primary tumor. This patient developed diffuse lesions over a period of several months, and given that her mammogram performed the previous year was negative for any abnormalities, one could suggest that the metastatic lesions developed less than a year from onset of the primary tumor. A previous study involving 41 patients with a known clinical primary visceral malignancy (ie, breast, lung, colon, esophageal, gastric, pancreatic, kidney, thyroid, prostate, or ovarian origin) found that it takes approximately 3 years on average for cutaneous metastases to develop from the onset of cancer diagnosis (range, 1–177 months).14 In the aforementioned study, 94% of patients had stage III or IV disease at time of skin metastasis, with the majority of those demonstrating stage IV disease. However, it also is possible that these breast tumors evaded detection or were too small to be identified on prior imaging.14 A review of our patient’s medical records did not indicate documentation of any visual or palpable breast changes prior to the onset of the clinically detected metastatic nodules.
Conclusion
Biopsy with immunohistochemical staining ultimately yielded the diagnosis of metastatic lobular breast carcinoma in our patient. Providers should be aware of the varying clinical presentations that may arise in the setting of cutaneous metastasis. When faced with lesions suspicious for cutaneous metastasis, biopsy is warranted to determine the correct diagnosis and ensure appropriate management. Upon diagnosis of cutaneous metastasis, prompt coordination with the primary care provider and appropriate referral to multidisciplinary teams is necessary. Clinical providers also should maintain a high index of suspicion when evaluating patients with cutaneous metastasis who have a history of normal malignancy screenings.
- American Cancer Society. Cancer facts & figures 2015. Accessed January 7, 2021. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2015/cancer-facts-and-figures-2015.pdf
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Krathen RA, Orengo IF, Rosen T. Cutaneous metastasis: a meta-analysis of data. South Med J. 2003;96:164-167.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. a retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Li CI, Anderson BO, Daling JR, et al. Trends in incidence rates of invasive lobular and ductal breast carcinoma. JAMA. 2003;289:1421-1424.
- Li CI, Daling JR. Changes in breast cancer incidence rates in the United States by histologic subtype and race/ethnicity, 1995 to 2004. Cancer Epidemiol Biomarkers Prev. 2007;16:2773-2780.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Dixon J, Anderson R, Page D, et al. Infiltrating lobular carcioma of the breast. Histopathology. 1982;6:149-161.
- Yeatman T, Cantor AB, Smith TJ, et al. Tumor biology of infiltrating lobular carcinoma: implications for management. Ann Surg. 1995;222:549-559.
- Silverstein M, Lewinski BS, Waisman JR, et al. Infiltrating lobular carcinoma: is it different from infiltrating duct carcinoma? Cancer. 1994;73:1673-1677.
- Li CI, Uribe DJ, Daling JR. Clinical characteristics of different histologic types of breast cancer. Br J Cancer. 2005;93:1046-1052.
- Mordenti C, Peris K, Fargnoli M, et al. Cutaneous metastatic breast carcinoma. Acta Dermatovenerol. 2000;9:143-148.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
In women, breast cancer is the leading cancer diagnosis and the second leading cause of cancer-related death,1 as well as the most common malignancy to metastasize to the skin.2 Cutaneous breast carcinoma may present as cutaneous metastasis or can occur secondary to direct tumor extension. Five percent to 10% of women with breast cancer will present clinically with metastatic cutaneous disease, most commonly as a recurrence of early-stage breast carcinoma.2
In a published meta-analysis that investigated the incidence of tumors most commonly found to metastasize to the skin, Krathen et al3 found that cutaneous metastases occurred in 24% of patients with breast cancer (N=1903). In 2 large retrospective studies from tumor registry data, breast cancer was found to be the most common tumor involving metastasis to the skin, and 3.5% of the breast cancer cases identified in the registry had cutaneous metastasis as the presenting sign (n=35) at time of diagnosis.4
We report an unusual presentation of cutaneous metastatic lobular breast carcinoma that involved diffuse cutaneous lesions and rapid progression from onset of the breast mass to development of clinically apparent metastatic skin lesions.
Case Report
A 59-year-old woman with an unremarkable medical history presented to our dermatology clinic for evaluation of new widespread lesions that developed over a period of months. The eruption was asymptomatic and consisted of numerous bumpy lesions that reportedly started on the patient’s neck and progressively spread to involve the trunk. Physical examination revealed multiple flesh-colored, firm nodules scattered across the upper back, neck, and chest (Figure 1). Bilateral cervical and axillary lymphadenopathy also was noted. Upon questioning regarding family history of malignancy, the patient reported that her brother had been diagnosed with colon cancer. Although she was not up to date on age-appropriate malignancy screenings, she did report having a diagnostic mammogram 1 year prior that revealed a suspicious lesion on the left breast. A repeat mammogram of the left breast 6 months later was read as unremarkable.
Two 3-mm representative punch biopsies were performed. Hematoxylin and eosin staining revealed a basket-weave stratum corneum with underlying epidermal atrophy. A relatively monomorphic epithelioid cell infiltrate extending from the superficial reticular dermis into the deep dermis and displaying an open chromatin pattern and pink cytoplasm was observed, as well as dermal collagen thickening. Linear, single-filing cells along with focal irregular nests and scattered cells were observed (Figure 2). Immunohistochemical staining was positive for cytokeratin 7 (Figure 3A), epithelial membrane antigen, and estrogen receptor (Figure 3B) along with gross cystic disease fluid protein 15; focal progesterone receptor positivity also was present. Cytokeratin 20, cytokeratin 5/6, carcinoembryonic antigen, p63, CDX2, paired box gene 8, thyroid transcription factor 1, and human epidermal growth factor receptor 2/neu stains were negative. Findings identified in both biopsies were consistent with metastatic cutaneous lobular breast carcinoma.
A complete blood cell count and complete metabolic panels were within normal limits, aside from a mildly elevated alkaline phosphatase level. Breast ultrasonography was unremarkable. Stereotactic breast magnetic resonance imaging (MRI) revealed a 9.4-cm mass in the upper outer quadrant of the right breast as well as enlarged lymph nodes 2.2 cm from the left axilla. A subsequent bone scan demonstrated focal activity in the left lateral fourth rib, left costochondral junction, and right anterolateral fifth rib—it was unclear whether these lesions were metastatic or secondary to trauma from a fall the patient reportedly had sustained 2 weeks prior. Lumbar MRI without gadolinium contrast revealed extensive abnormal heterogeneous signal intensity of osseous structures consistent with osseous metastasis.
Subsequent diagnostic bilateral breast ultrasonography and percutaneous left lymph node biopsy revealed pathology consistent with metastatic lobular breast carcinoma with near total effacement of the lymph node and extracapsular extension concordant with previous MRI findings. The mass in the upper outer quadrant of the right breast that previously was observed on MRI was not identifiable on this ultrasound. It was recommended that the patient pursue MRI-guided breast biopsy to have the breast lesion further characterized. She was referred to surgical oncology at a tertiary center for management; however, the patient was lost to follow-up, and there are no records available indicating the patient pursued any treatment. Although we were unable to confirm the patient’s breast lesion that previously was seen on MRI was the cause of the metastatic disease, the overall clinical picture supported metastatic lobular breast carcinoma.
Comment
Tumor metastasis to the skin accounts for approximately 2% of all skin cancers5 and typically is observed in advanced stages of cancer. In women, breast carcinoma is the most common type of cancer to exhibit this behavior.2 Invasive ductal carcinoma represents the most common histologic subtype of breast cancer overall,6,7 and breast adenocarcinomas, including lobular and ductal breast carcinomas, are the most common histologic subtypes to exhibit metastatic cutaneous lesions.8
Invasive lobular breast carcinoma represents approximately 10% of invasive breast cancer cases. Compared to invasive ductal carcinoma, there tends to be a delay in diagnosis often leading to larger tumor sizes relative to the former upon detection and with lymph node invasion. These findings may be explained by the greater difficulty of detecting invasive lobular carcinomas by mammography and clinical breast examination compared to invasive ductal carcinomas.9-11 Additionally, invasive lobular carcinomas are more likely to be positive for estrogen and progesterone receptors compared to invasive ductal carcinomas,12 which also was consistent in our case.
Cutaneous metastases of breast cancer most commonly are found on the anterior chest wall and can present as a wide spectrum of lesions, with nodules as the most common primary dermatologic manifestation.13 Cutaneous metastatic lesions commonly have been described as firm, mobile, round or oval, solitary or grouped nodules. The color of the nodules varies and may be flesh-colored, brown, blue, black, pink, and/or red-brown. The lesions often are asymptomatic but may ulcerate.2
In our case, the distribution of lesions was a unique aspect that is not typical of most cases of metastatic cutaneous breast carcinoma. The nodules appeared more scattered and involved multiple body regions, including the back, neck, and chest. Although cutaneous breast cancer metastases have been documented to extend to these body regions, a review of PubMed articles indexed for MEDLINE using the terms cutaneous metastatic lobular breast carcinoma, breast carcinoma, and metastatic breast cancer suggested that it is uncommon for these multiple areas to be simultaneously affected.4,14 Rather, the more common clinical presentation of cutaneous metastatic breast carcinoma is as a solitary nodule or group of nodules localized to a single anatomic region.14
Another notable feature of our case was the rapid development of the cutaneous lesions relative to the primary tumor. This patient developed diffuse lesions over a period of several months, and given that her mammogram performed the previous year was negative for any abnormalities, one could suggest that the metastatic lesions developed less than a year from onset of the primary tumor. A previous study involving 41 patients with a known clinical primary visceral malignancy (ie, breast, lung, colon, esophageal, gastric, pancreatic, kidney, thyroid, prostate, or ovarian origin) found that it takes approximately 3 years on average for cutaneous metastases to develop from the onset of cancer diagnosis (range, 1–177 months).14 In the aforementioned study, 94% of patients had stage III or IV disease at time of skin metastasis, with the majority of those demonstrating stage IV disease. However, it also is possible that these breast tumors evaded detection or were too small to be identified on prior imaging.14 A review of our patient’s medical records did not indicate documentation of any visual or palpable breast changes prior to the onset of the clinically detected metastatic nodules.
Conclusion
Biopsy with immunohistochemical staining ultimately yielded the diagnosis of metastatic lobular breast carcinoma in our patient. Providers should be aware of the varying clinical presentations that may arise in the setting of cutaneous metastasis. When faced with lesions suspicious for cutaneous metastasis, biopsy is warranted to determine the correct diagnosis and ensure appropriate management. Upon diagnosis of cutaneous metastasis, prompt coordination with the primary care provider and appropriate referral to multidisciplinary teams is necessary. Clinical providers also should maintain a high index of suspicion when evaluating patients with cutaneous metastasis who have a history of normal malignancy screenings.
In women, breast cancer is the leading cancer diagnosis and the second leading cause of cancer-related death,1 as well as the most common malignancy to metastasize to the skin.2 Cutaneous breast carcinoma may present as cutaneous metastasis or can occur secondary to direct tumor extension. Five percent to 10% of women with breast cancer will present clinically with metastatic cutaneous disease, most commonly as a recurrence of early-stage breast carcinoma.2
In a published meta-analysis that investigated the incidence of tumors most commonly found to metastasize to the skin, Krathen et al3 found that cutaneous metastases occurred in 24% of patients with breast cancer (N=1903). In 2 large retrospective studies from tumor registry data, breast cancer was found to be the most common tumor involving metastasis to the skin, and 3.5% of the breast cancer cases identified in the registry had cutaneous metastasis as the presenting sign (n=35) at time of diagnosis.4
We report an unusual presentation of cutaneous metastatic lobular breast carcinoma that involved diffuse cutaneous lesions and rapid progression from onset of the breast mass to development of clinically apparent metastatic skin lesions.
Case Report
A 59-year-old woman with an unremarkable medical history presented to our dermatology clinic for evaluation of new widespread lesions that developed over a period of months. The eruption was asymptomatic and consisted of numerous bumpy lesions that reportedly started on the patient’s neck and progressively spread to involve the trunk. Physical examination revealed multiple flesh-colored, firm nodules scattered across the upper back, neck, and chest (Figure 1). Bilateral cervical and axillary lymphadenopathy also was noted. Upon questioning regarding family history of malignancy, the patient reported that her brother had been diagnosed with colon cancer. Although she was not up to date on age-appropriate malignancy screenings, she did report having a diagnostic mammogram 1 year prior that revealed a suspicious lesion on the left breast. A repeat mammogram of the left breast 6 months later was read as unremarkable.
Two 3-mm representative punch biopsies were performed. Hematoxylin and eosin staining revealed a basket-weave stratum corneum with underlying epidermal atrophy. A relatively monomorphic epithelioid cell infiltrate extending from the superficial reticular dermis into the deep dermis and displaying an open chromatin pattern and pink cytoplasm was observed, as well as dermal collagen thickening. Linear, single-filing cells along with focal irregular nests and scattered cells were observed (Figure 2). Immunohistochemical staining was positive for cytokeratin 7 (Figure 3A), epithelial membrane antigen, and estrogen receptor (Figure 3B) along with gross cystic disease fluid protein 15; focal progesterone receptor positivity also was present. Cytokeratin 20, cytokeratin 5/6, carcinoembryonic antigen, p63, CDX2, paired box gene 8, thyroid transcription factor 1, and human epidermal growth factor receptor 2/neu stains were negative. Findings identified in both biopsies were consistent with metastatic cutaneous lobular breast carcinoma.
A complete blood cell count and complete metabolic panels were within normal limits, aside from a mildly elevated alkaline phosphatase level. Breast ultrasonography was unremarkable. Stereotactic breast magnetic resonance imaging (MRI) revealed a 9.4-cm mass in the upper outer quadrant of the right breast as well as enlarged lymph nodes 2.2 cm from the left axilla. A subsequent bone scan demonstrated focal activity in the left lateral fourth rib, left costochondral junction, and right anterolateral fifth rib—it was unclear whether these lesions were metastatic or secondary to trauma from a fall the patient reportedly had sustained 2 weeks prior. Lumbar MRI without gadolinium contrast revealed extensive abnormal heterogeneous signal intensity of osseous structures consistent with osseous metastasis.
Subsequent diagnostic bilateral breast ultrasonography and percutaneous left lymph node biopsy revealed pathology consistent with metastatic lobular breast carcinoma with near total effacement of the lymph node and extracapsular extension concordant with previous MRI findings. The mass in the upper outer quadrant of the right breast that previously was observed on MRI was not identifiable on this ultrasound. It was recommended that the patient pursue MRI-guided breast biopsy to have the breast lesion further characterized. She was referred to surgical oncology at a tertiary center for management; however, the patient was lost to follow-up, and there are no records available indicating the patient pursued any treatment. Although we were unable to confirm the patient’s breast lesion that previously was seen on MRI was the cause of the metastatic disease, the overall clinical picture supported metastatic lobular breast carcinoma.
Comment
Tumor metastasis to the skin accounts for approximately 2% of all skin cancers5 and typically is observed in advanced stages of cancer. In women, breast carcinoma is the most common type of cancer to exhibit this behavior.2 Invasive ductal carcinoma represents the most common histologic subtype of breast cancer overall,6,7 and breast adenocarcinomas, including lobular and ductal breast carcinomas, are the most common histologic subtypes to exhibit metastatic cutaneous lesions.8
Invasive lobular breast carcinoma represents approximately 10% of invasive breast cancer cases. Compared to invasive ductal carcinoma, there tends to be a delay in diagnosis often leading to larger tumor sizes relative to the former upon detection and with lymph node invasion. These findings may be explained by the greater difficulty of detecting invasive lobular carcinomas by mammography and clinical breast examination compared to invasive ductal carcinomas.9-11 Additionally, invasive lobular carcinomas are more likely to be positive for estrogen and progesterone receptors compared to invasive ductal carcinomas,12 which also was consistent in our case.
Cutaneous metastases of breast cancer most commonly are found on the anterior chest wall and can present as a wide spectrum of lesions, with nodules as the most common primary dermatologic manifestation.13 Cutaneous metastatic lesions commonly have been described as firm, mobile, round or oval, solitary or grouped nodules. The color of the nodules varies and may be flesh-colored, brown, blue, black, pink, and/or red-brown. The lesions often are asymptomatic but may ulcerate.2
In our case, the distribution of lesions was a unique aspect that is not typical of most cases of metastatic cutaneous breast carcinoma. The nodules appeared more scattered and involved multiple body regions, including the back, neck, and chest. Although cutaneous breast cancer metastases have been documented to extend to these body regions, a review of PubMed articles indexed for MEDLINE using the terms cutaneous metastatic lobular breast carcinoma, breast carcinoma, and metastatic breast cancer suggested that it is uncommon for these multiple areas to be simultaneously affected.4,14 Rather, the more common clinical presentation of cutaneous metastatic breast carcinoma is as a solitary nodule or group of nodules localized to a single anatomic region.14
Another notable feature of our case was the rapid development of the cutaneous lesions relative to the primary tumor. This patient developed diffuse lesions over a period of several months, and given that her mammogram performed the previous year was negative for any abnormalities, one could suggest that the metastatic lesions developed less than a year from onset of the primary tumor. A previous study involving 41 patients with a known clinical primary visceral malignancy (ie, breast, lung, colon, esophageal, gastric, pancreatic, kidney, thyroid, prostate, or ovarian origin) found that it takes approximately 3 years on average for cutaneous metastases to develop from the onset of cancer diagnosis (range, 1–177 months).14 In the aforementioned study, 94% of patients had stage III or IV disease at time of skin metastasis, with the majority of those demonstrating stage IV disease. However, it also is possible that these breast tumors evaded detection or were too small to be identified on prior imaging.14 A review of our patient’s medical records did not indicate documentation of any visual or palpable breast changes prior to the onset of the clinically detected metastatic nodules.
Conclusion
Biopsy with immunohistochemical staining ultimately yielded the diagnosis of metastatic lobular breast carcinoma in our patient. Providers should be aware of the varying clinical presentations that may arise in the setting of cutaneous metastasis. When faced with lesions suspicious for cutaneous metastasis, biopsy is warranted to determine the correct diagnosis and ensure appropriate management. Upon diagnosis of cutaneous metastasis, prompt coordination with the primary care provider and appropriate referral to multidisciplinary teams is necessary. Clinical providers also should maintain a high index of suspicion when evaluating patients with cutaneous metastasis who have a history of normal malignancy screenings.
- American Cancer Society. Cancer facts & figures 2015. Accessed January 7, 2021. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2015/cancer-facts-and-figures-2015.pdf
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Krathen RA, Orengo IF, Rosen T. Cutaneous metastasis: a meta-analysis of data. South Med J. 2003;96:164-167.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. a retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Li CI, Anderson BO, Daling JR, et al. Trends in incidence rates of invasive lobular and ductal breast carcinoma. JAMA. 2003;289:1421-1424.
- Li CI, Daling JR. Changes in breast cancer incidence rates in the United States by histologic subtype and race/ethnicity, 1995 to 2004. Cancer Epidemiol Biomarkers Prev. 2007;16:2773-2780.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Dixon J, Anderson R, Page D, et al. Infiltrating lobular carcioma of the breast. Histopathology. 1982;6:149-161.
- Yeatman T, Cantor AB, Smith TJ, et al. Tumor biology of infiltrating lobular carcinoma: implications for management. Ann Surg. 1995;222:549-559.
- Silverstein M, Lewinski BS, Waisman JR, et al. Infiltrating lobular carcinoma: is it different from infiltrating duct carcinoma? Cancer. 1994;73:1673-1677.
- Li CI, Uribe DJ, Daling JR. Clinical characteristics of different histologic types of breast cancer. Br J Cancer. 2005;93:1046-1052.
- Mordenti C, Peris K, Fargnoli M, et al. Cutaneous metastatic breast carcinoma. Acta Dermatovenerol. 2000;9:143-148.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- American Cancer Society. Cancer facts & figures 2015. Accessed January 7, 2021. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2015/cancer-facts-and-figures-2015.pdf
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Krathen RA, Orengo IF, Rosen T. Cutaneous metastasis: a meta-analysis of data. South Med J. 2003;96:164-167.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. a retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Li CI, Anderson BO, Daling JR, et al. Trends in incidence rates of invasive lobular and ductal breast carcinoma. JAMA. 2003;289:1421-1424.
- Li CI, Daling JR. Changes in breast cancer incidence rates in the United States by histologic subtype and race/ethnicity, 1995 to 2004. Cancer Epidemiol Biomarkers Prev. 2007;16:2773-2780.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Dixon J, Anderson R, Page D, et al. Infiltrating lobular carcioma of the breast. Histopathology. 1982;6:149-161.
- Yeatman T, Cantor AB, Smith TJ, et al. Tumor biology of infiltrating lobular carcinoma: implications for management. Ann Surg. 1995;222:549-559.
- Silverstein M, Lewinski BS, Waisman JR, et al. Infiltrating lobular carcinoma: is it different from infiltrating duct carcinoma? Cancer. 1994;73:1673-1677.
- Li CI, Uribe DJ, Daling JR. Clinical characteristics of different histologic types of breast cancer. Br J Cancer. 2005;93:1046-1052.
- Mordenti C, Peris K, Fargnoli M, et al. Cutaneous metastatic breast carcinoma. Acta Dermatovenerol. 2000;9:143-148.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
Practice Points
- Clinical providers should be aware of the varying presentations of metastatic cutaneous breast carcinomas.
- Clinicians should remain alert to the possibility of breast cancer as a cause of cutaneous metastases, even in patients with recent negative breast cancer screening.
60-year-old man • chronic cough • history of GERD & dyslipidemia • throat tickle • Dx?
THE CASE
A 60-year-old man with a past medical history of gastroesophageal reflux disease (GERD) and dyslipidemia presented to his family physician for evaluation of chronic cough. Five years prior, the patient had developed a high fever and respiratory symptoms, including a cough, and was believed to have had severe otitis media. He was treated with multiple courses of antibiotics and corticosteroids for persistent otitis media. Although the condition eventually resolved, his cough continued.
The persistent cough prompted the patient to consult a succession of specialists. First, he saw a gastroenterologist; following an esophagogastroduodenoscopy, he was prescribed pantoprazole. Despite the proton-pump inhibitor (PPI) therapy, the cough remained. Next, he had multiple visits with an otolaryngologist but that yielded no specific diagnosis for the cough. He also saw an allergist-immunologist, who identified a ragweed allergy, gave him a diagnosis of cough-variant asthma, and prescribed antihistamines and mometasone furoate and formoterol fumarate dihydrate. Neither was helpful.
After 5 years of frustration, the patient complained to his family physician that he still had a cough and “a tickle” in his throat that was worsened by speaking and drinking cold beverages. He denied fever, shortness of breath, nausea, vomiting, or any other associated symptoms.
THE DIAGNOSIS
The failed treatment attempts with antihistamines, corticosteroids, bronchodilators, and PPI therapy excluded multiple etiologies for the cough. The throat discomfort and feeling of a “tickle” prompted us to consider a nerve-related disorder on the differential. The diagnosis of laryngeal sensory neuropathy (LSN) was considered.
DISCUSSION
LSN is a relatively uncommon cause of chronic refractory cough that can also manifest with throat discomfort, dysphagia, and dysphonia.1 It is thought to result from some type of insult to the recurrent laryngeal nerve or superior laryngeal nerve via viral infections, metabolic changes, or mechanical trauma, leading to a change in the firing threshold.2 The hypothesis of nerve damage is supported by the increased incidence of LSN in patients with goiters and those with type 2 diabetes.3,4 When there is a decrease in the laryngeal sensory threshold, dysfunctional laryngeal behavior results, leading to symptoms such as persistent cough and throat clearing.
Diagnosis. LSN is often diagnosed clinically, after GERD, allergies, asthma, angiotensin-converting enzyme inhibitor intake, and psychogenic disorders have been ruled out.1 Our patient had a prior diagnosis or investigation of nearly all of these conditions. Other clues pointing to an LSN diagnosis include a cough lasting 8 weeks or more, recurrent sensory disturbances (such as a tickle) of instantaneous onset before each cough episode, triggers that can include talking or a change in air temperature, daily coughing episodes numbering in the 10s to 100s, and a nonproductive cough.5,6
Beyond clinical clues, laryngeal electromyography, which evaluates the neuromuscular system in the larynx by recording action potentials generated in the laryngeal muscles during contraction, can be used for diagnosis.4 Videostroboscopy, which allows for an enlarged and slow motion view of the vocal cords, can also be used.
Continue to: Treatment
Treatment. To both confirm the diagnosis and treat the patient in a rapid, practical fashion, a trial of a neuromodulating agent such as pregabalin or gabapentin can be employed.6-9 A study identifying 28 LSN patients found symptomatic relief in 68% of patients taking gabapentin 100 to 900 mg/d.2 In another study, 12 LSN patients given pregabalin found relief after a 1-month regimen.1 Another study of 12 patients showed amitriptyline hydrochloride and gabapentin provided a positive response in 2 months, and the addition of reflux precautions and acid-suppression therapy was helpful.9 Finally, a group of 32 patients trialed on 3 different medications (amitriptyline, desipramine, and gabapentin) found similar efficacy among the 3.6
Another option. Aside from medications, botulinum toxin type A has been shown in a case series to directly decrease laryngeal hypertonicity and possibly reduce neurogenic inflammation and neuropeptide-mediated cough.10 Another study found that 18 patients with neurogenic cough who received superior laryngeal nerve blocks had cough severity index scores decrease from an average of 26.8 pretreatment to 14.6 posttreatment (P < .0001).11
Our patient agreed to a trial of gabapentin 300 mg once a day, with titration up to a maximum of 900 mg tid. When the patient returned to the clinic 4 months later, he reported that when he reached 300 mg bid, the cough completely resolved.
THE TAKEAWAY
A persistent cough with minimal identifiable triggers is a huge disruption to a patient’s life; having to visit multiple specialists before receiving a diagnosis compounds that. In our patient’s case, the process took 5 years, which underscores how important it is that LSN be considered in the differential diagnosis. Since this is generally a diagnosis of exclusion, it is important to take a careful history of a patient with a chronic cough. If LSN seems likely, trialing a patient on neuromodulating medication is the next best step, with dose titration if necessary.
Selena R. Pasadyn, 675 West 130th Street, Hinckley, OH, 44233; [email protected]
1. Halum SL, Sycamore DL, McRae BR. A new treatment option for laryngeal sensory neuropathy. Laryngoscope. 2009;119:1844-1847.
2. Lee B, Woo P. Chronic cough as a sign of laryngeal sensory neuropathy: diagnosis and treatment. Ann Otol Rhinol Laryngol. 2005;114:253-257.
3. Hamdan AL, Jabour J, Azar ST. Goiter and laryngeal sensory neuropathy. Int J Otolaryngol. 2013;2013:765265.
4. Hamdan AL, Dowli A, Barazi R, et al. Laryngeal sensory neuropathy in patients with diabetes mellitus. J Laryngol Otol. 2014;128:725-729.
5. Bastian RW, Vaidya AM, Delsupehe KG. Sensory neuropathic cough: a common and treatable cause of chronic cough. Otolaryngol Head Neck Surg. 2006;135:17-21.
6. Bastian ZJ, Bastian RW. The use of neuralgia medications to treat sensory neuropathic cough: our experience in a retrospective cohort of thirty-two patients. PeerJ. 2015;3:e816.
7. Van de Kerkhove C, Goeminne PC, Van Bleyenbergh P, et al. A cohort description and analysis of the effect of gabapentin on idiopathic cough. Cough. 2012;8:9.
8. Mishriki YY. Laryngeal neuropathy as a cause of chronic intractable cough. Am J Med. 2007;120:e5.
9. Norris BK, Schweinfurth JM. Management of recurrent laryngeal sensory neuropathic symptoms. Ann Otol Rhinol Laryngol. 2010;119:188-191.
10. Chu MW, Lieser JD, Sinacori JT. Use of botulinum toxin type a for chronic cough: a neuropathic model. Arch Otolaryngol Head Neck Surg. 2010;136:447.
11. Simpson CB, Tibbetts KM, Loochtan MJ, et al. Treatment of chronic neurogenic cough with in-office superior laryngeal nerve block. Laryngoscope. 2018;128:1898-1903.
THE CASE
A 60-year-old man with a past medical history of gastroesophageal reflux disease (GERD) and dyslipidemia presented to his family physician for evaluation of chronic cough. Five years prior, the patient had developed a high fever and respiratory symptoms, including a cough, and was believed to have had severe otitis media. He was treated with multiple courses of antibiotics and corticosteroids for persistent otitis media. Although the condition eventually resolved, his cough continued.
The persistent cough prompted the patient to consult a succession of specialists. First, he saw a gastroenterologist; following an esophagogastroduodenoscopy, he was prescribed pantoprazole. Despite the proton-pump inhibitor (PPI) therapy, the cough remained. Next, he had multiple visits with an otolaryngologist but that yielded no specific diagnosis for the cough. He also saw an allergist-immunologist, who identified a ragweed allergy, gave him a diagnosis of cough-variant asthma, and prescribed antihistamines and mometasone furoate and formoterol fumarate dihydrate. Neither was helpful.
After 5 years of frustration, the patient complained to his family physician that he still had a cough and “a tickle” in his throat that was worsened by speaking and drinking cold beverages. He denied fever, shortness of breath, nausea, vomiting, or any other associated symptoms.
THE DIAGNOSIS
The failed treatment attempts with antihistamines, corticosteroids, bronchodilators, and PPI therapy excluded multiple etiologies for the cough. The throat discomfort and feeling of a “tickle” prompted us to consider a nerve-related disorder on the differential. The diagnosis of laryngeal sensory neuropathy (LSN) was considered.
DISCUSSION
LSN is a relatively uncommon cause of chronic refractory cough that can also manifest with throat discomfort, dysphagia, and dysphonia.1 It is thought to result from some type of insult to the recurrent laryngeal nerve or superior laryngeal nerve via viral infections, metabolic changes, or mechanical trauma, leading to a change in the firing threshold.2 The hypothesis of nerve damage is supported by the increased incidence of LSN in patients with goiters and those with type 2 diabetes.3,4 When there is a decrease in the laryngeal sensory threshold, dysfunctional laryngeal behavior results, leading to symptoms such as persistent cough and throat clearing.
Diagnosis. LSN is often diagnosed clinically, after GERD, allergies, asthma, angiotensin-converting enzyme inhibitor intake, and psychogenic disorders have been ruled out.1 Our patient had a prior diagnosis or investigation of nearly all of these conditions. Other clues pointing to an LSN diagnosis include a cough lasting 8 weeks or more, recurrent sensory disturbances (such as a tickle) of instantaneous onset before each cough episode, triggers that can include talking or a change in air temperature, daily coughing episodes numbering in the 10s to 100s, and a nonproductive cough.5,6
Beyond clinical clues, laryngeal electromyography, which evaluates the neuromuscular system in the larynx by recording action potentials generated in the laryngeal muscles during contraction, can be used for diagnosis.4 Videostroboscopy, which allows for an enlarged and slow motion view of the vocal cords, can also be used.
Continue to: Treatment
Treatment. To both confirm the diagnosis and treat the patient in a rapid, practical fashion, a trial of a neuromodulating agent such as pregabalin or gabapentin can be employed.6-9 A study identifying 28 LSN patients found symptomatic relief in 68% of patients taking gabapentin 100 to 900 mg/d.2 In another study, 12 LSN patients given pregabalin found relief after a 1-month regimen.1 Another study of 12 patients showed amitriptyline hydrochloride and gabapentin provided a positive response in 2 months, and the addition of reflux precautions and acid-suppression therapy was helpful.9 Finally, a group of 32 patients trialed on 3 different medications (amitriptyline, desipramine, and gabapentin) found similar efficacy among the 3.6
Another option. Aside from medications, botulinum toxin type A has been shown in a case series to directly decrease laryngeal hypertonicity and possibly reduce neurogenic inflammation and neuropeptide-mediated cough.10 Another study found that 18 patients with neurogenic cough who received superior laryngeal nerve blocks had cough severity index scores decrease from an average of 26.8 pretreatment to 14.6 posttreatment (P < .0001).11
Our patient agreed to a trial of gabapentin 300 mg once a day, with titration up to a maximum of 900 mg tid. When the patient returned to the clinic 4 months later, he reported that when he reached 300 mg bid, the cough completely resolved.
THE TAKEAWAY
A persistent cough with minimal identifiable triggers is a huge disruption to a patient’s life; having to visit multiple specialists before receiving a diagnosis compounds that. In our patient’s case, the process took 5 years, which underscores how important it is that LSN be considered in the differential diagnosis. Since this is generally a diagnosis of exclusion, it is important to take a careful history of a patient with a chronic cough. If LSN seems likely, trialing a patient on neuromodulating medication is the next best step, with dose titration if necessary.
Selena R. Pasadyn, 675 West 130th Street, Hinckley, OH, 44233; [email protected]
THE CASE
A 60-year-old man with a past medical history of gastroesophageal reflux disease (GERD) and dyslipidemia presented to his family physician for evaluation of chronic cough. Five years prior, the patient had developed a high fever and respiratory symptoms, including a cough, and was believed to have had severe otitis media. He was treated with multiple courses of antibiotics and corticosteroids for persistent otitis media. Although the condition eventually resolved, his cough continued.
The persistent cough prompted the patient to consult a succession of specialists. First, he saw a gastroenterologist; following an esophagogastroduodenoscopy, he was prescribed pantoprazole. Despite the proton-pump inhibitor (PPI) therapy, the cough remained. Next, he had multiple visits with an otolaryngologist but that yielded no specific diagnosis for the cough. He also saw an allergist-immunologist, who identified a ragweed allergy, gave him a diagnosis of cough-variant asthma, and prescribed antihistamines and mometasone furoate and formoterol fumarate dihydrate. Neither was helpful.
After 5 years of frustration, the patient complained to his family physician that he still had a cough and “a tickle” in his throat that was worsened by speaking and drinking cold beverages. He denied fever, shortness of breath, nausea, vomiting, or any other associated symptoms.
THE DIAGNOSIS
The failed treatment attempts with antihistamines, corticosteroids, bronchodilators, and PPI therapy excluded multiple etiologies for the cough. The throat discomfort and feeling of a “tickle” prompted us to consider a nerve-related disorder on the differential. The diagnosis of laryngeal sensory neuropathy (LSN) was considered.
DISCUSSION
LSN is a relatively uncommon cause of chronic refractory cough that can also manifest with throat discomfort, dysphagia, and dysphonia.1 It is thought to result from some type of insult to the recurrent laryngeal nerve or superior laryngeal nerve via viral infections, metabolic changes, or mechanical trauma, leading to a change in the firing threshold.2 The hypothesis of nerve damage is supported by the increased incidence of LSN in patients with goiters and those with type 2 diabetes.3,4 When there is a decrease in the laryngeal sensory threshold, dysfunctional laryngeal behavior results, leading to symptoms such as persistent cough and throat clearing.
Diagnosis. LSN is often diagnosed clinically, after GERD, allergies, asthma, angiotensin-converting enzyme inhibitor intake, and psychogenic disorders have been ruled out.1 Our patient had a prior diagnosis or investigation of nearly all of these conditions. Other clues pointing to an LSN diagnosis include a cough lasting 8 weeks or more, recurrent sensory disturbances (such as a tickle) of instantaneous onset before each cough episode, triggers that can include talking or a change in air temperature, daily coughing episodes numbering in the 10s to 100s, and a nonproductive cough.5,6
Beyond clinical clues, laryngeal electromyography, which evaluates the neuromuscular system in the larynx by recording action potentials generated in the laryngeal muscles during contraction, can be used for diagnosis.4 Videostroboscopy, which allows for an enlarged and slow motion view of the vocal cords, can also be used.
Continue to: Treatment
Treatment. To both confirm the diagnosis and treat the patient in a rapid, practical fashion, a trial of a neuromodulating agent such as pregabalin or gabapentin can be employed.6-9 A study identifying 28 LSN patients found symptomatic relief in 68% of patients taking gabapentin 100 to 900 mg/d.2 In another study, 12 LSN patients given pregabalin found relief after a 1-month regimen.1 Another study of 12 patients showed amitriptyline hydrochloride and gabapentin provided a positive response in 2 months, and the addition of reflux precautions and acid-suppression therapy was helpful.9 Finally, a group of 32 patients trialed on 3 different medications (amitriptyline, desipramine, and gabapentin) found similar efficacy among the 3.6
Another option. Aside from medications, botulinum toxin type A has been shown in a case series to directly decrease laryngeal hypertonicity and possibly reduce neurogenic inflammation and neuropeptide-mediated cough.10 Another study found that 18 patients with neurogenic cough who received superior laryngeal nerve blocks had cough severity index scores decrease from an average of 26.8 pretreatment to 14.6 posttreatment (P < .0001).11
Our patient agreed to a trial of gabapentin 300 mg once a day, with titration up to a maximum of 900 mg tid. When the patient returned to the clinic 4 months later, he reported that when he reached 300 mg bid, the cough completely resolved.
THE TAKEAWAY
A persistent cough with minimal identifiable triggers is a huge disruption to a patient’s life; having to visit multiple specialists before receiving a diagnosis compounds that. In our patient’s case, the process took 5 years, which underscores how important it is that LSN be considered in the differential diagnosis. Since this is generally a diagnosis of exclusion, it is important to take a careful history of a patient with a chronic cough. If LSN seems likely, trialing a patient on neuromodulating medication is the next best step, with dose titration if necessary.
Selena R. Pasadyn, 675 West 130th Street, Hinckley, OH, 44233; [email protected]
1. Halum SL, Sycamore DL, McRae BR. A new treatment option for laryngeal sensory neuropathy. Laryngoscope. 2009;119:1844-1847.
2. Lee B, Woo P. Chronic cough as a sign of laryngeal sensory neuropathy: diagnosis and treatment. Ann Otol Rhinol Laryngol. 2005;114:253-257.
3. Hamdan AL, Jabour J, Azar ST. Goiter and laryngeal sensory neuropathy. Int J Otolaryngol. 2013;2013:765265.
4. Hamdan AL, Dowli A, Barazi R, et al. Laryngeal sensory neuropathy in patients with diabetes mellitus. J Laryngol Otol. 2014;128:725-729.
5. Bastian RW, Vaidya AM, Delsupehe KG. Sensory neuropathic cough: a common and treatable cause of chronic cough. Otolaryngol Head Neck Surg. 2006;135:17-21.
6. Bastian ZJ, Bastian RW. The use of neuralgia medications to treat sensory neuropathic cough: our experience in a retrospective cohort of thirty-two patients. PeerJ. 2015;3:e816.
7. Van de Kerkhove C, Goeminne PC, Van Bleyenbergh P, et al. A cohort description and analysis of the effect of gabapentin on idiopathic cough. Cough. 2012;8:9.
8. Mishriki YY. Laryngeal neuropathy as a cause of chronic intractable cough. Am J Med. 2007;120:e5.
9. Norris BK, Schweinfurth JM. Management of recurrent laryngeal sensory neuropathic symptoms. Ann Otol Rhinol Laryngol. 2010;119:188-191.
10. Chu MW, Lieser JD, Sinacori JT. Use of botulinum toxin type a for chronic cough: a neuropathic model. Arch Otolaryngol Head Neck Surg. 2010;136:447.
11. Simpson CB, Tibbetts KM, Loochtan MJ, et al. Treatment of chronic neurogenic cough with in-office superior laryngeal nerve block. Laryngoscope. 2018;128:1898-1903.
1. Halum SL, Sycamore DL, McRae BR. A new treatment option for laryngeal sensory neuropathy. Laryngoscope. 2009;119:1844-1847.
2. Lee B, Woo P. Chronic cough as a sign of laryngeal sensory neuropathy: diagnosis and treatment. Ann Otol Rhinol Laryngol. 2005;114:253-257.
3. Hamdan AL, Jabour J, Azar ST. Goiter and laryngeal sensory neuropathy. Int J Otolaryngol. 2013;2013:765265.
4. Hamdan AL, Dowli A, Barazi R, et al. Laryngeal sensory neuropathy in patients with diabetes mellitus. J Laryngol Otol. 2014;128:725-729.
5. Bastian RW, Vaidya AM, Delsupehe KG. Sensory neuropathic cough: a common and treatable cause of chronic cough. Otolaryngol Head Neck Surg. 2006;135:17-21.
6. Bastian ZJ, Bastian RW. The use of neuralgia medications to treat sensory neuropathic cough: our experience in a retrospective cohort of thirty-two patients. PeerJ. 2015;3:e816.
7. Van de Kerkhove C, Goeminne PC, Van Bleyenbergh P, et al. A cohort description and analysis of the effect of gabapentin on idiopathic cough. Cough. 2012;8:9.
8. Mishriki YY. Laryngeal neuropathy as a cause of chronic intractable cough. Am J Med. 2007;120:e5.
9. Norris BK, Schweinfurth JM. Management of recurrent laryngeal sensory neuropathic symptoms. Ann Otol Rhinol Laryngol. 2010;119:188-191.
10. Chu MW, Lieser JD, Sinacori JT. Use of botulinum toxin type a for chronic cough: a neuropathic model. Arch Otolaryngol Head Neck Surg. 2010;136:447.
11. Simpson CB, Tibbetts KM, Loochtan MJ, et al. Treatment of chronic neurogenic cough with in-office superior laryngeal nerve block. Laryngoscope. 2018;128:1898-1903.

20-year-old man • sudden-onset chest pain • worsening pain with cough and exertion • Dx?
THE CASE
A 20-year-old man presented to our clinic with a 3-day history of nonradiating chest pain located at the center of his chest. Past medical history included idiopathic neonatal giant-cell hepatitis and subsequent liver transplant at 1 month of age; he had been followed by the transplant team without rejection or infection and was in otherwise good health prior to the chest pain.
On the day of symptom onset, he was walking inside his house and fell to his knees with a chest pain described as “a punch” to the center of the chest that lasted for a few seconds. He was able to continue his daily activities without limitation despite a constant, squeezing, centrally located chest pain. The pain worsened with cough and exertion.
A few hours later, he went to an urgent care center for evaluation. There, he reported, his chest radiograph and electrocardiogram (EKG) results were normal and he was given a diagnosis of musculoskeletal chest pain. Over the next 3 days, his chest pain persisted but did not worsen. He was taking 500 mg of naproxen every 8 hours with no improvement. No other acute or chronic medications were being taken. He had no significant family history. A review of systems was otherwise negative.
On physical exam, his vital statistics included a height of 6’4”; weight, 261 lb; body mass index, 31.8; temperature, 98.7 °F; blood pressure, 134/77 mm Hg; heart rate, 92 beats/min; respiratory rate, 18 breaths/min; and oxygen saturation, 96%. Throughout the exam, he demonstrated no acute distress, appeared well, and was talkative; however, he reported having a “constant, squeezing” chest pain that did not worsen with palpation of the chest. The rest of his physical exam was unremarkable.
Although he reported that his EKG and chest radiograph were normal 3 days prior, repeat chest radiograph and EKG were ordered due to his unexplained, active chest pain and the lack of immediate access to the prior results.
THE DIAGNOSIS
The chest radiograph (FIGURE 1A) showed a “mildly ectatic ascending thoracic aorta” that had increased since a chest radiograph from 6 years prior (FIGURE 1B) and “was concerning for an aneurysm.” Computed tomography (CT) angiography (FIGURE 2) then confirmed a 7-cm aneurysm of the ascending aorta, with findings suggestive of a retrograde ascending aortic dissection.

DISCUSSION
The average age of a patient with acute aortic dissection (AAD) is 63 years; only 7% occur in people younger than 40.1 AAD is often accompanied by a predisposing risk factor such as a connective tissue disease, bicuspid aortic valve, longstanding hypertension, trauma, or larger aortic dimensions.2,3 Younger patients are more likely to have predisposing risk factors of Marfan syndrome, prior aortic surgery, or a bicuspid aortic valve.3

Continue to: A literature review did not reveal...
A literature review did not reveal any known correlation between the patient’s history of giant-cell hepatitis or antirejection therapy with thoracic aortic dissection. Furthermore, liver transplant is not known to be a specific risk factor for AAD in pediatric patients or outside the immediate postoperative period. Therefore, there were no known predisposing risk factors for AAD in our patient.
The most common clinical feature of AAD is chest pain, which occurs in 75% of patients.1 Other clinical symptoms include hypertension and diaphoresis.2,4 However, classic clinical findings are not always displayed, making the diagnosis difficult.2,4 The classical description of “tearing pain” is seen in only 51% of patients, and 5% to 15% of patients present without any pain.1
Commonly missed or misdiagnosed. The diagnosis of AAD has been missed during the initial exam in 38% of patients.4 As seen in our case, symptoms may be initially diagnosed as musculoskeletal chest pain. Based on symptoms, AAD can be incorrectly diagnosed as an acute myocardial infarction or vascular embolization.2,4
Every hour after symptom onset, the mortality rate of untreated AAD increases 1% to 2%,with no difference based on age.3,4 Different reports have shown mortality rates between 7% and 30%.4
Effective imaging is crucial to the diagnosis and treatment of AAD, given the occurrence of atypical presentation, missed diagnosis, and high mortality rate.4 A chest radiograph will show a widened mediastinum, but the preferred diagnostic tests are a CT or transthoracic echocardiogram.2,4 Once the diagnosis of AAD is confirmed, an aortic angiogram is the preferred test to determine the extent of the dissection prior to surgical treatment.2
Continue to: Classification dictates treatment
Classification dictates treatment. AAD is classified based on where the dissection of the aorta occurs. If the dissection involves the ascending aorta, it is classified as a type A AAD and should immediately be treated with emergent surgery in order to prevent complications including myocardial infarction, cardiac tamponade, and aortic rupture.2,4,5 If the dissection is limited to the descending aorta, it is classified as a type B AAD and can be medically managed by controlling pain and lowering blood pressure; if symptoms persist, surgical management may be required.2 After hospital discharge, AAD patients are followed closely with medical therapy, serial imaging, and reoperation if necessary.4
Our patient underwent emergent surgery for aortic root/ascending aortic replacement with a mechanical valve. He tolerated the procedure well. Surgical tissue pathology of the aortic segment showed a wall of elastic vessel with medial degeneration and dissection, and the tissue pathology of the aorta leaflets showed valvular tissue with myxoid degeneration.
THE TAKEAWAY
It is critical to keep AAD in the differential diagnosis of a patient presenting with acute onset of chest pain, as AAD often has an atypical presentation and can easily be misdiagnosed. Effective imaging is crucial to diagnosis, and immediate treatment is essential to patient survival.
CORRESPONDENCE
Rachel A. Reedy, PA, University of Florida, Department of General Pediatrics, 7046 SW Archer Road, Gainesville, FL 32608; [email protected]
1. Pineault J, Ouimet D, Pichette V, Vallée M. A case of aortic dissection in a young adult: a refresher of the literature of this “great masquerader.” Int J Gen Med. 2011;4:889-893.
2. Agabegi SS, Agabegi ElD, Ring AC. Diseases of the cardiovascular system. In: Jackson A, ed. Step-up to Medicine. 3rd ed. Lippincott Williams & Wilkins; 2012:54-55.
3. Januzzi JL, Isselbacher EM, Fattori R, et al. Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol. 2004;43:665-669.
4. Tsai TT, Trimarchi S, Nienaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg. 2009;37:149-159.
5. Trimarchi S, Eagle KA, Nienaber CA, et al. Role of age in acute type A aortic dissection outcome: Report from the International Registry of Acute Aortic Dissection (IRAD). J Thorac Cardiovasc Surg. 2010;140:784-789.
THE CASE
A 20-year-old man presented to our clinic with a 3-day history of nonradiating chest pain located at the center of his chest. Past medical history included idiopathic neonatal giant-cell hepatitis and subsequent liver transplant at 1 month of age; he had been followed by the transplant team without rejection or infection and was in otherwise good health prior to the chest pain.
On the day of symptom onset, he was walking inside his house and fell to his knees with a chest pain described as “a punch” to the center of the chest that lasted for a few seconds. He was able to continue his daily activities without limitation despite a constant, squeezing, centrally located chest pain. The pain worsened with cough and exertion.
A few hours later, he went to an urgent care center for evaluation. There, he reported, his chest radiograph and electrocardiogram (EKG) results were normal and he was given a diagnosis of musculoskeletal chest pain. Over the next 3 days, his chest pain persisted but did not worsen. He was taking 500 mg of naproxen every 8 hours with no improvement. No other acute or chronic medications were being taken. He had no significant family history. A review of systems was otherwise negative.
On physical exam, his vital statistics included a height of 6’4”; weight, 261 lb; body mass index, 31.8; temperature, 98.7 °F; blood pressure, 134/77 mm Hg; heart rate, 92 beats/min; respiratory rate, 18 breaths/min; and oxygen saturation, 96%. Throughout the exam, he demonstrated no acute distress, appeared well, and was talkative; however, he reported having a “constant, squeezing” chest pain that did not worsen with palpation of the chest. The rest of his physical exam was unremarkable.
Although he reported that his EKG and chest radiograph were normal 3 days prior, repeat chest radiograph and EKG were ordered due to his unexplained, active chest pain and the lack of immediate access to the prior results.
THE DIAGNOSIS
The chest radiograph (FIGURE 1A) showed a “mildly ectatic ascending thoracic aorta” that had increased since a chest radiograph from 6 years prior (FIGURE 1B) and “was concerning for an aneurysm.” Computed tomography (CT) angiography (FIGURE 2) then confirmed a 7-cm aneurysm of the ascending aorta, with findings suggestive of a retrograde ascending aortic dissection.
DISCUSSION
The average age of a patient with acute aortic dissection (AAD) is 63 years; only 7% occur in people younger than 40.1 AAD is often accompanied by a predisposing risk factor such as a connective tissue disease, bicuspid aortic valve, longstanding hypertension, trauma, or larger aortic dimensions.2,3 Younger patients are more likely to have predisposing risk factors of Marfan syndrome, prior aortic surgery, or a bicuspid aortic valve.3
Continue to: A literature review did not reveal...
A literature review did not reveal any known correlation between the patient’s history of giant-cell hepatitis or antirejection therapy with thoracic aortic dissection. Furthermore, liver transplant is not known to be a specific risk factor for AAD in pediatric patients or outside the immediate postoperative period. Therefore, there were no known predisposing risk factors for AAD in our patient.
The most common clinical feature of AAD is chest pain, which occurs in 75% of patients.1 Other clinical symptoms include hypertension and diaphoresis.2,4 However, classic clinical findings are not always displayed, making the diagnosis difficult.2,4 The classical description of “tearing pain” is seen in only 51% of patients, and 5% to 15% of patients present without any pain.1
Commonly missed or misdiagnosed. The diagnosis of AAD has been missed during the initial exam in 38% of patients.4 As seen in our case, symptoms may be initially diagnosed as musculoskeletal chest pain. Based on symptoms, AAD can be incorrectly diagnosed as an acute myocardial infarction or vascular embolization.2,4
Every hour after symptom onset, the mortality rate of untreated AAD increases 1% to 2%,with no difference based on age.3,4 Different reports have shown mortality rates between 7% and 30%.4
Effective imaging is crucial to the diagnosis and treatment of AAD, given the occurrence of atypical presentation, missed diagnosis, and high mortality rate.4 A chest radiograph will show a widened mediastinum, but the preferred diagnostic tests are a CT or transthoracic echocardiogram.2,4 Once the diagnosis of AAD is confirmed, an aortic angiogram is the preferred test to determine the extent of the dissection prior to surgical treatment.2
Continue to: Classification dictates treatment
Classification dictates treatment. AAD is classified based on where the dissection of the aorta occurs. If the dissection involves the ascending aorta, it is classified as a type A AAD and should immediately be treated with emergent surgery in order to prevent complications including myocardial infarction, cardiac tamponade, and aortic rupture.2,4,5 If the dissection is limited to the descending aorta, it is classified as a type B AAD and can be medically managed by controlling pain and lowering blood pressure; if symptoms persist, surgical management may be required.2 After hospital discharge, AAD patients are followed closely with medical therapy, serial imaging, and reoperation if necessary.4
Our patient underwent emergent surgery for aortic root/ascending aortic replacement with a mechanical valve. He tolerated the procedure well. Surgical tissue pathology of the aortic segment showed a wall of elastic vessel with medial degeneration and dissection, and the tissue pathology of the aorta leaflets showed valvular tissue with myxoid degeneration.
THE TAKEAWAY
It is critical to keep AAD in the differential diagnosis of a patient presenting with acute onset of chest pain, as AAD often has an atypical presentation and can easily be misdiagnosed. Effective imaging is crucial to diagnosis, and immediate treatment is essential to patient survival.
CORRESPONDENCE
Rachel A. Reedy, PA, University of Florida, Department of General Pediatrics, 7046 SW Archer Road, Gainesville, FL 32608; [email protected]
THE CASE
A 20-year-old man presented to our clinic with a 3-day history of nonradiating chest pain located at the center of his chest. Past medical history included idiopathic neonatal giant-cell hepatitis and subsequent liver transplant at 1 month of age; he had been followed by the transplant team without rejection or infection and was in otherwise good health prior to the chest pain.
On the day of symptom onset, he was walking inside his house and fell to his knees with a chest pain described as “a punch” to the center of the chest that lasted for a few seconds. He was able to continue his daily activities without limitation despite a constant, squeezing, centrally located chest pain. The pain worsened with cough and exertion.
A few hours later, he went to an urgent care center for evaluation. There, he reported, his chest radiograph and electrocardiogram (EKG) results were normal and he was given a diagnosis of musculoskeletal chest pain. Over the next 3 days, his chest pain persisted but did not worsen. He was taking 500 mg of naproxen every 8 hours with no improvement. No other acute or chronic medications were being taken. He had no significant family history. A review of systems was otherwise negative.
On physical exam, his vital statistics included a height of 6’4”; weight, 261 lb; body mass index, 31.8; temperature, 98.7 °F; blood pressure, 134/77 mm Hg; heart rate, 92 beats/min; respiratory rate, 18 breaths/min; and oxygen saturation, 96%. Throughout the exam, he demonstrated no acute distress, appeared well, and was talkative; however, he reported having a “constant, squeezing” chest pain that did not worsen with palpation of the chest. The rest of his physical exam was unremarkable.
Although he reported that his EKG and chest radiograph were normal 3 days prior, repeat chest radiograph and EKG were ordered due to his unexplained, active chest pain and the lack of immediate access to the prior results.
THE DIAGNOSIS
The chest radiograph (FIGURE 1A) showed a “mildly ectatic ascending thoracic aorta” that had increased since a chest radiograph from 6 years prior (FIGURE 1B) and “was concerning for an aneurysm.” Computed tomography (CT) angiography (FIGURE 2) then confirmed a 7-cm aneurysm of the ascending aorta, with findings suggestive of a retrograde ascending aortic dissection.
DISCUSSION
The average age of a patient with acute aortic dissection (AAD) is 63 years; only 7% occur in people younger than 40.1 AAD is often accompanied by a predisposing risk factor such as a connective tissue disease, bicuspid aortic valve, longstanding hypertension, trauma, or larger aortic dimensions.2,3 Younger patients are more likely to have predisposing risk factors of Marfan syndrome, prior aortic surgery, or a bicuspid aortic valve.3
Continue to: A literature review did not reveal...
A literature review did not reveal any known correlation between the patient’s history of giant-cell hepatitis or antirejection therapy with thoracic aortic dissection. Furthermore, liver transplant is not known to be a specific risk factor for AAD in pediatric patients or outside the immediate postoperative period. Therefore, there were no known predisposing risk factors for AAD in our patient.
The most common clinical feature of AAD is chest pain, which occurs in 75% of patients.1 Other clinical symptoms include hypertension and diaphoresis.2,4 However, classic clinical findings are not always displayed, making the diagnosis difficult.2,4 The classical description of “tearing pain” is seen in only 51% of patients, and 5% to 15% of patients present without any pain.1
Commonly missed or misdiagnosed. The diagnosis of AAD has been missed during the initial exam in 38% of patients.4 As seen in our case, symptoms may be initially diagnosed as musculoskeletal chest pain. Based on symptoms, AAD can be incorrectly diagnosed as an acute myocardial infarction or vascular embolization.2,4
Every hour after symptom onset, the mortality rate of untreated AAD increases 1% to 2%,with no difference based on age.3,4 Different reports have shown mortality rates between 7% and 30%.4
Effective imaging is crucial to the diagnosis and treatment of AAD, given the occurrence of atypical presentation, missed diagnosis, and high mortality rate.4 A chest radiograph will show a widened mediastinum, but the preferred diagnostic tests are a CT or transthoracic echocardiogram.2,4 Once the diagnosis of AAD is confirmed, an aortic angiogram is the preferred test to determine the extent of the dissection prior to surgical treatment.2
Continue to: Classification dictates treatment
Classification dictates treatment. AAD is classified based on where the dissection of the aorta occurs. If the dissection involves the ascending aorta, it is classified as a type A AAD and should immediately be treated with emergent surgery in order to prevent complications including myocardial infarction, cardiac tamponade, and aortic rupture.2,4,5 If the dissection is limited to the descending aorta, it is classified as a type B AAD and can be medically managed by controlling pain and lowering blood pressure; if symptoms persist, surgical management may be required.2 After hospital discharge, AAD patients are followed closely with medical therapy, serial imaging, and reoperation if necessary.4
Our patient underwent emergent surgery for aortic root/ascending aortic replacement with a mechanical valve. He tolerated the procedure well. Surgical tissue pathology of the aortic segment showed a wall of elastic vessel with medial degeneration and dissection, and the tissue pathology of the aorta leaflets showed valvular tissue with myxoid degeneration.
THE TAKEAWAY
It is critical to keep AAD in the differential diagnosis of a patient presenting with acute onset of chest pain, as AAD often has an atypical presentation and can easily be misdiagnosed. Effective imaging is crucial to diagnosis, and immediate treatment is essential to patient survival.
CORRESPONDENCE
Rachel A. Reedy, PA, University of Florida, Department of General Pediatrics, 7046 SW Archer Road, Gainesville, FL 32608; [email protected]
1. Pineault J, Ouimet D, Pichette V, Vallée M. A case of aortic dissection in a young adult: a refresher of the literature of this “great masquerader.” Int J Gen Med. 2011;4:889-893.
2. Agabegi SS, Agabegi ElD, Ring AC. Diseases of the cardiovascular system. In: Jackson A, ed. Step-up to Medicine. 3rd ed. Lippincott Williams & Wilkins; 2012:54-55.
3. Januzzi JL, Isselbacher EM, Fattori R, et al. Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol. 2004;43:665-669.
4. Tsai TT, Trimarchi S, Nienaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg. 2009;37:149-159.
5. Trimarchi S, Eagle KA, Nienaber CA, et al. Role of age in acute type A aortic dissection outcome: Report from the International Registry of Acute Aortic Dissection (IRAD). J Thorac Cardiovasc Surg. 2010;140:784-789.
1. Pineault J, Ouimet D, Pichette V, Vallée M. A case of aortic dissection in a young adult: a refresher of the literature of this “great masquerader.” Int J Gen Med. 2011;4:889-893.
2. Agabegi SS, Agabegi ElD, Ring AC. Diseases of the cardiovascular system. In: Jackson A, ed. Step-up to Medicine. 3rd ed. Lippincott Williams & Wilkins; 2012:54-55.
3. Januzzi JL, Isselbacher EM, Fattori R, et al. Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol. 2004;43:665-669.
4. Tsai TT, Trimarchi S, Nienaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg. 2009;37:149-159.
5. Trimarchi S, Eagle KA, Nienaber CA, et al. Role of age in acute type A aortic dissection outcome: Report from the International Registry of Acute Aortic Dissection (IRAD). J Thorac Cardiovasc Surg. 2010;140:784-789.

Cutaneous Insulin-Derived Amyloidosis Presenting as Hyperkeratotic Nodules
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Practice Points
- Deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.
- Patients with insulin-derived (AIns) amyloidosis may initially present after noticing nodular deposits.
- Insulin injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis.