User login
Management of high-grade pleomorphic sarcoma with colon metastasis
Soft tissue sarcomas (STS) are a heterogeneous group of tumors of mesenchymal origin that represent a rare form of adult malignancy. According to the World Health Organization classification system, there are more than 100 histologic subtypes of sarcoma based on tissue of origin. Staging criteria most commonly use the American Joint Committee on Cancer’s TNM Classification of Malignant Tumours. About 50% of soft tissue sarcomas originate in the lower extremity.1 Advancements in the use of multimodal therapy have reduced the need for amputation and allowed for equally effective treatment strategies that use limb-sparing surgical resections.
Although most sarcoma metastases spread in a hematogenous fashion, nodal spread is underestimated. Certain histologic subtypes carry a higher predilection for nodal involvement: these include rhabdomyosarcoma, synovial sarcoma, epithelioid sarcoma, vascular sarcoma, and clear-cell sarcoma.2-4 Fong and colleagues have reported that 2.6% of sarcomas have lymphatic spread.3 In the current report, we describe a rare observation of locoregional pelvic nodal metastases from a large undifferentiated pleomorphic sarcoma of the right thigh.
Case presentation and summary
A 63-year-old white woman had a 1-year history of a right thigh mass and an unintentional weight loss of 40 lb. After a year of chiropractic care, she was referred to a physician because of palpable inguinal adenopathy and a 20-cm mass in the medial compartment of the right thigh, with heterogeneous appearance on a magnetic-resonance imaging scan (Figure 1). The patient was referred to the sarcoma transdisciplinary team for evaluation. She was diagnosed by core needle biopsy with a high-grade malignant epithelioid and spindle-cell neoplasm, favoring pleomorphic sarcoma. The metastatic work-up confirmed locoregional right inguinal and retroperitoneal lymph node disease, with 2 lung nodules that were too small to characterize. She also had a paraneoplastic leukocytosis with a white blood cell count of 45,500 cells/ml (normal 10,500 cells/ml).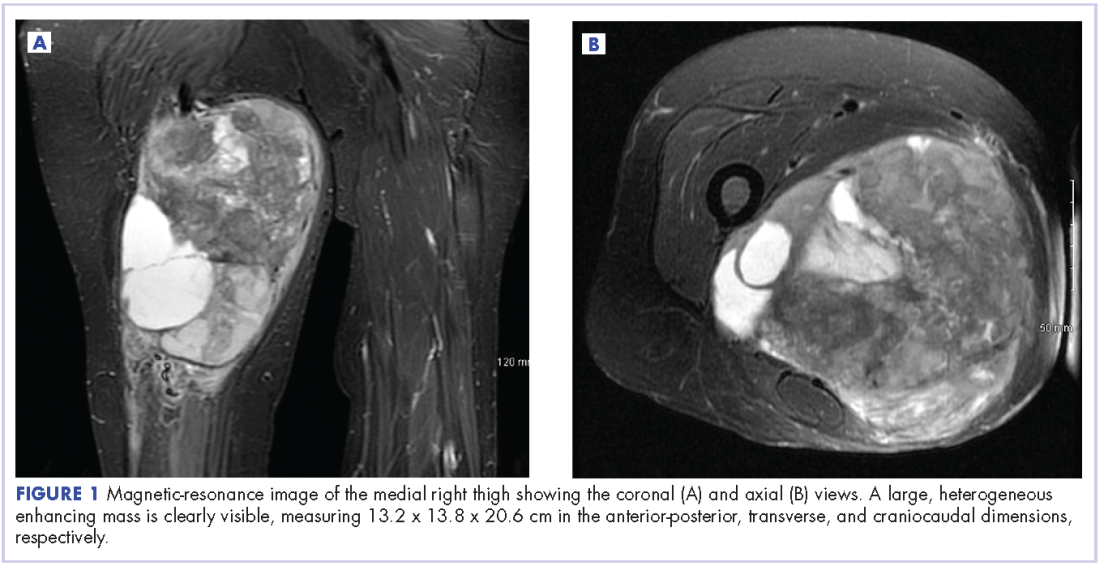
About 8 months after surgery, she presented to the emergency room with a 3-day history of blood per rectum, anemia, and fatigue. She also reported a weight loss of 10 lb in the previous month. She was admitted for hydration and monitoring. Although computed-tomography (CT) scans of the abdomen and chest done during the previous month and had been interpreted as having no evidence of recurrence or any lymph node disease, the results of an inpatient colonoscopy revealed 2 colonic masses, 1 in the ascending colon and another in the transverse colon. The biopsy findings were consistent with undifferentiated pleomorphic sarcoma, favoring epithelioid histology. The CT scans were re-evaluated in light of these colonoscopic findings. These masses were visible retrospectively on imaging but had been interpreted as stool given the lack of abnormality on imaging 3 months before.
Adequate re-staging was complete, and without other evidence of disease, an extended right hemicolectomy was performed. The postoperative pathology report described geographically 2 distinct masses: a 7-cm mass in the ascending colon, about 3 cm from the ileocecal valve; and a 4-cm mass in the transverse colon, about 7.5 cm from the distal margin of resection. Both masses were identified as high-grade pleomorphic sarcoma. Again, all nodes recovered were negative for malignancy (0/5). Of note is that the background colonic mucosa showed active multifocal colitis with deep inflammatory activity likely consistent with a paraneoplastic syndrome.
Discussion
Surgery remains the primary treatment modality for localized soft tissue sarcoma. Obtaining a margin-free resection, while maintaining optimal function, is the objective with extremity sarcoma. In addition to surgical resection, doxorubicin-based adjuvant chemotherapy remains the standard of care with modest improvement in overall survival and disease-free survival, especially in sarcomas of the extremities.5,6 Gemcitabine also has activity in soft tissue sarcomas7 and might synergize with docetaxel. High response rates of 53% with fixed dose infusion rates of these agents in uterine leiomyosarcoma led the Sarcoma Alliance for Research through Collaboration investigators to consider this regimen for other STS.8 An overall response rate of 16% was noted across all sarcoma subtypes, and undifferentiated pleomorphic sarcoma had a response rate of 32%.
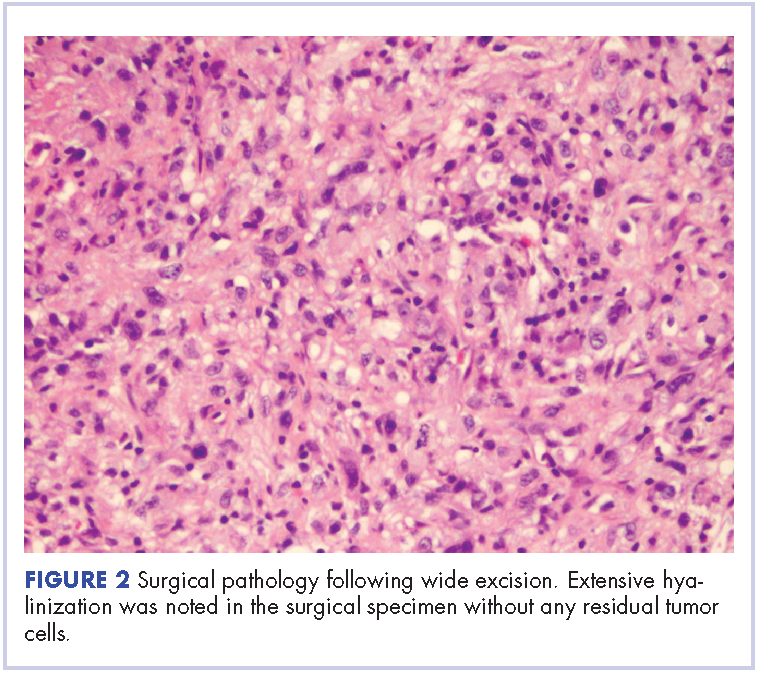
In our experience, a modified schedule of gemcitabine and docetaxel is better tolerated than the standard every 3 weeks regimen or doxorubicin-based chemotherapies. As previously described, gemcitabine and docetaxel were administered to this patient every 2 weeks.9 This regimen, in our experience, is less toxic and preserves the dose intensity of the regimen. A complete pathologic necrosis of the primary tumor and regional nodal basin was observed, as well as pulmonary nodule regression, enabling an R0-wide excision and regional lymphadenectomy. The colonic recurrence was surprising, but easily managed with surgical resection.
Pathologic complete response after neoadjuvant chemotherapy is a rare event, observed in about 10% of patients. However, when observed, complete pathologic necrosis (>95%) provided a distant recurrence-free survival of 100% at 3 years.10-12
Our patient, despite achieving a complete pathologic response and excellent initial local control, ultimately experienced an isolated metastatic recurrence in the colon within 1 year of therapy. Data supports performing metastatectomy for stage IV extremity sarcoma for isolated pulmonary or hepatic burden in selected patients with improved survival rates.13,14 As far as we know, there is no published literature describing isolated colon metastases in the absence of liver burden from lower extremity soft tissue sarcomas, or the outcome of surgical resection and adjuvant therapy in these cases.
In conclusion, high-grade pleomorphic sarcoma often follows an aggressive clinical course with ultimately local and distant recurrence. Use of multimodal therapy may have a role in improving local control. Complete pathologic necrosis is a rare event that is predictive of improved outcome. Our case represents an unusual pattern of recurrence among patients with a complete pathologic response to neoadjuvant therapy with isolated colon metastases. Timely, comprehensive management together with vigilant surveillance remain key priorities in the long-term management of high-risk sarcoma.
1. Lawrence W Jr, Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas. A pattern of care survey of the American College of Surgeons. Ann Surg. 1987;205:349-359.
2. Riad S, Griffin AM, Liberman B, et al. Lymph node metastasis in soft tissue sarcoma in an extremity. Clin Orthop Relat Res. 2004:129-134.
3. Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg. 1993;217:72-77.
4. Mazeron JJ, Suit HD. Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer. 1987;60:1800-1808.
5. Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft tissue sarcoma of adults: meta-analysis of individual data. Lancet. 1997;350:1647-1654.
6. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft tissue sarcoma. Cancer. 2008;113:573-581.
7. Patel SR, Gandhi V, Jenkins J, et al. Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol. 2001;19:3483-3489.
8. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25:2755-2763.
9. Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Ann Oncol. 2012;23:785-790.
10. MacDermed DM, Miller LL, Peabody TD, et al. Primary tumor necrosis predicts distant control in locally advanced soft tissue sarcomas after preoperative concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2010;76:1147-1153.
11. Eilber FC, Rosen G, Eckardt J, et al. Treatment-induced pathologic necrosis: a predictor of local recurrence and survival in patients receiving neoadjuvant therapy for high-grade extremity soft tissue sarcomas. J Clin Oncol. 2001;19:3203-3209.
12. Shah D, Borys D, Martinez SR, et al. Complete pathologic response to neoadjuvant radiotherapy is predictive of oncological outcome in patients with soft tissue sarcoma. Anticancer Res. 2012;32:3911-3915.
13. Rehders A, Peiper M, Stoecklein NH, et al. Hepatic metastasectomy for soft tissue sarcomas: is it justified? World J Surg. 2009;33:111-117.
14. Pfannschmidt J, Hoffmann H, Schneider T, Dienemann H. Pulmonary metastasectomy for soft tissue sarcomas: is it justified? Recent Results Cancer Res. 2009;179:321-336.
Soft tissue sarcomas (STS) are a heterogeneous group of tumors of mesenchymal origin that represent a rare form of adult malignancy. According to the World Health Organization classification system, there are more than 100 histologic subtypes of sarcoma based on tissue of origin. Staging criteria most commonly use the American Joint Committee on Cancer’s TNM Classification of Malignant Tumours. About 50% of soft tissue sarcomas originate in the lower extremity.1 Advancements in the use of multimodal therapy have reduced the need for amputation and allowed for equally effective treatment strategies that use limb-sparing surgical resections.
Although most sarcoma metastases spread in a hematogenous fashion, nodal spread is underestimated. Certain histologic subtypes carry a higher predilection for nodal involvement: these include rhabdomyosarcoma, synovial sarcoma, epithelioid sarcoma, vascular sarcoma, and clear-cell sarcoma.2-4 Fong and colleagues have reported that 2.6% of sarcomas have lymphatic spread.3 In the current report, we describe a rare observation of locoregional pelvic nodal metastases from a large undifferentiated pleomorphic sarcoma of the right thigh.
Case presentation and summary
A 63-year-old white woman had a 1-year history of a right thigh mass and an unintentional weight loss of 40 lb. After a year of chiropractic care, she was referred to a physician because of palpable inguinal adenopathy and a 20-cm mass in the medial compartment of the right thigh, with heterogeneous appearance on a magnetic-resonance imaging scan (Figure 1). The patient was referred to the sarcoma transdisciplinary team for evaluation. She was diagnosed by core needle biopsy with a high-grade malignant epithelioid and spindle-cell neoplasm, favoring pleomorphic sarcoma. The metastatic work-up confirmed locoregional right inguinal and retroperitoneal lymph node disease, with 2 lung nodules that were too small to characterize. She also had a paraneoplastic leukocytosis with a white blood cell count of 45,500 cells/ml (normal 10,500 cells/ml).
About 8 months after surgery, she presented to the emergency room with a 3-day history of blood per rectum, anemia, and fatigue. She also reported a weight loss of 10 lb in the previous month. She was admitted for hydration and monitoring. Although computed-tomography (CT) scans of the abdomen and chest done during the previous month and had been interpreted as having no evidence of recurrence or any lymph node disease, the results of an inpatient colonoscopy revealed 2 colonic masses, 1 in the ascending colon and another in the transverse colon. The biopsy findings were consistent with undifferentiated pleomorphic sarcoma, favoring epithelioid histology. The CT scans were re-evaluated in light of these colonoscopic findings. These masses were visible retrospectively on imaging but had been interpreted as stool given the lack of abnormality on imaging 3 months before.
Adequate re-staging was complete, and without other evidence of disease, an extended right hemicolectomy was performed. The postoperative pathology report described geographically 2 distinct masses: a 7-cm mass in the ascending colon, about 3 cm from the ileocecal valve; and a 4-cm mass in the transverse colon, about 7.5 cm from the distal margin of resection. Both masses were identified as high-grade pleomorphic sarcoma. Again, all nodes recovered were negative for malignancy (0/5). Of note is that the background colonic mucosa showed active multifocal colitis with deep inflammatory activity likely consistent with a paraneoplastic syndrome.
Discussion
Surgery remains the primary treatment modality for localized soft tissue sarcoma. Obtaining a margin-free resection, while maintaining optimal function, is the objective with extremity sarcoma. In addition to surgical resection, doxorubicin-based adjuvant chemotherapy remains the standard of care with modest improvement in overall survival and disease-free survival, especially in sarcomas of the extremities.5,6 Gemcitabine also has activity in soft tissue sarcomas7 and might synergize with docetaxel. High response rates of 53% with fixed dose infusion rates of these agents in uterine leiomyosarcoma led the Sarcoma Alliance for Research through Collaboration investigators to consider this regimen for other STS.8 An overall response rate of 16% was noted across all sarcoma subtypes, and undifferentiated pleomorphic sarcoma had a response rate of 32%.
In our experience, a modified schedule of gemcitabine and docetaxel is better tolerated than the standard every 3 weeks regimen or doxorubicin-based chemotherapies. As previously described, gemcitabine and docetaxel were administered to this patient every 2 weeks.9 This regimen, in our experience, is less toxic and preserves the dose intensity of the regimen. A complete pathologic necrosis of the primary tumor and regional nodal basin was observed, as well as pulmonary nodule regression, enabling an R0-wide excision and regional lymphadenectomy. The colonic recurrence was surprising, but easily managed with surgical resection.
Pathologic complete response after neoadjuvant chemotherapy is a rare event, observed in about 10% of patients. However, when observed, complete pathologic necrosis (>95%) provided a distant recurrence-free survival of 100% at 3 years.10-12
Our patient, despite achieving a complete pathologic response and excellent initial local control, ultimately experienced an isolated metastatic recurrence in the colon within 1 year of therapy. Data supports performing metastatectomy for stage IV extremity sarcoma for isolated pulmonary or hepatic burden in selected patients with improved survival rates.13,14 As far as we know, there is no published literature describing isolated colon metastases in the absence of liver burden from lower extremity soft tissue sarcomas, or the outcome of surgical resection and adjuvant therapy in these cases.
In conclusion, high-grade pleomorphic sarcoma often follows an aggressive clinical course with ultimately local and distant recurrence. Use of multimodal therapy may have a role in improving local control. Complete pathologic necrosis is a rare event that is predictive of improved outcome. Our case represents an unusual pattern of recurrence among patients with a complete pathologic response to neoadjuvant therapy with isolated colon metastases. Timely, comprehensive management together with vigilant surveillance remain key priorities in the long-term management of high-risk sarcoma.
Soft tissue sarcomas (STS) are a heterogeneous group of tumors of mesenchymal origin that represent a rare form of adult malignancy. According to the World Health Organization classification system, there are more than 100 histologic subtypes of sarcoma based on tissue of origin. Staging criteria most commonly use the American Joint Committee on Cancer’s TNM Classification of Malignant Tumours. About 50% of soft tissue sarcomas originate in the lower extremity.1 Advancements in the use of multimodal therapy have reduced the need for amputation and allowed for equally effective treatment strategies that use limb-sparing surgical resections.
Although most sarcoma metastases spread in a hematogenous fashion, nodal spread is underestimated. Certain histologic subtypes carry a higher predilection for nodal involvement: these include rhabdomyosarcoma, synovial sarcoma, epithelioid sarcoma, vascular sarcoma, and clear-cell sarcoma.2-4 Fong and colleagues have reported that 2.6% of sarcomas have lymphatic spread.3 In the current report, we describe a rare observation of locoregional pelvic nodal metastases from a large undifferentiated pleomorphic sarcoma of the right thigh.
Case presentation and summary
A 63-year-old white woman had a 1-year history of a right thigh mass and an unintentional weight loss of 40 lb. After a year of chiropractic care, she was referred to a physician because of palpable inguinal adenopathy and a 20-cm mass in the medial compartment of the right thigh, with heterogeneous appearance on a magnetic-resonance imaging scan (Figure 1). The patient was referred to the sarcoma transdisciplinary team for evaluation. She was diagnosed by core needle biopsy with a high-grade malignant epithelioid and spindle-cell neoplasm, favoring pleomorphic sarcoma. The metastatic work-up confirmed locoregional right inguinal and retroperitoneal lymph node disease, with 2 lung nodules that were too small to characterize. She also had a paraneoplastic leukocytosis with a white blood cell count of 45,500 cells/ml (normal 10,500 cells/ml).
About 8 months after surgery, she presented to the emergency room with a 3-day history of blood per rectum, anemia, and fatigue. She also reported a weight loss of 10 lb in the previous month. She was admitted for hydration and monitoring. Although computed-tomography (CT) scans of the abdomen and chest done during the previous month and had been interpreted as having no evidence of recurrence or any lymph node disease, the results of an inpatient colonoscopy revealed 2 colonic masses, 1 in the ascending colon and another in the transverse colon. The biopsy findings were consistent with undifferentiated pleomorphic sarcoma, favoring epithelioid histology. The CT scans were re-evaluated in light of these colonoscopic findings. These masses were visible retrospectively on imaging but had been interpreted as stool given the lack of abnormality on imaging 3 months before.
Adequate re-staging was complete, and without other evidence of disease, an extended right hemicolectomy was performed. The postoperative pathology report described geographically 2 distinct masses: a 7-cm mass in the ascending colon, about 3 cm from the ileocecal valve; and a 4-cm mass in the transverse colon, about 7.5 cm from the distal margin of resection. Both masses were identified as high-grade pleomorphic sarcoma. Again, all nodes recovered were negative for malignancy (0/5). Of note is that the background colonic mucosa showed active multifocal colitis with deep inflammatory activity likely consistent with a paraneoplastic syndrome.
Discussion
Surgery remains the primary treatment modality for localized soft tissue sarcoma. Obtaining a margin-free resection, while maintaining optimal function, is the objective with extremity sarcoma. In addition to surgical resection, doxorubicin-based adjuvant chemotherapy remains the standard of care with modest improvement in overall survival and disease-free survival, especially in sarcomas of the extremities.5,6 Gemcitabine also has activity in soft tissue sarcomas7 and might synergize with docetaxel. High response rates of 53% with fixed dose infusion rates of these agents in uterine leiomyosarcoma led the Sarcoma Alliance for Research through Collaboration investigators to consider this regimen for other STS.8 An overall response rate of 16% was noted across all sarcoma subtypes, and undifferentiated pleomorphic sarcoma had a response rate of 32%.
In our experience, a modified schedule of gemcitabine and docetaxel is better tolerated than the standard every 3 weeks regimen or doxorubicin-based chemotherapies. As previously described, gemcitabine and docetaxel were administered to this patient every 2 weeks.9 This regimen, in our experience, is less toxic and preserves the dose intensity of the regimen. A complete pathologic necrosis of the primary tumor and regional nodal basin was observed, as well as pulmonary nodule regression, enabling an R0-wide excision and regional lymphadenectomy. The colonic recurrence was surprising, but easily managed with surgical resection.
Pathologic complete response after neoadjuvant chemotherapy is a rare event, observed in about 10% of patients. However, when observed, complete pathologic necrosis (>95%) provided a distant recurrence-free survival of 100% at 3 years.10-12
Our patient, despite achieving a complete pathologic response and excellent initial local control, ultimately experienced an isolated metastatic recurrence in the colon within 1 year of therapy. Data supports performing metastatectomy for stage IV extremity sarcoma for isolated pulmonary or hepatic burden in selected patients with improved survival rates.13,14 As far as we know, there is no published literature describing isolated colon metastases in the absence of liver burden from lower extremity soft tissue sarcomas, or the outcome of surgical resection and adjuvant therapy in these cases.
In conclusion, high-grade pleomorphic sarcoma often follows an aggressive clinical course with ultimately local and distant recurrence. Use of multimodal therapy may have a role in improving local control. Complete pathologic necrosis is a rare event that is predictive of improved outcome. Our case represents an unusual pattern of recurrence among patients with a complete pathologic response to neoadjuvant therapy with isolated colon metastases. Timely, comprehensive management together with vigilant surveillance remain key priorities in the long-term management of high-risk sarcoma.
1. Lawrence W Jr, Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas. A pattern of care survey of the American College of Surgeons. Ann Surg. 1987;205:349-359.
2. Riad S, Griffin AM, Liberman B, et al. Lymph node metastasis in soft tissue sarcoma in an extremity. Clin Orthop Relat Res. 2004:129-134.
3. Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg. 1993;217:72-77.
4. Mazeron JJ, Suit HD. Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer. 1987;60:1800-1808.
5. Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft tissue sarcoma of adults: meta-analysis of individual data. Lancet. 1997;350:1647-1654.
6. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft tissue sarcoma. Cancer. 2008;113:573-581.
7. Patel SR, Gandhi V, Jenkins J, et al. Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol. 2001;19:3483-3489.
8. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25:2755-2763.
9. Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Ann Oncol. 2012;23:785-790.
10. MacDermed DM, Miller LL, Peabody TD, et al. Primary tumor necrosis predicts distant control in locally advanced soft tissue sarcomas after preoperative concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2010;76:1147-1153.
11. Eilber FC, Rosen G, Eckardt J, et al. Treatment-induced pathologic necrosis: a predictor of local recurrence and survival in patients receiving neoadjuvant therapy for high-grade extremity soft tissue sarcomas. J Clin Oncol. 2001;19:3203-3209.
12. Shah D, Borys D, Martinez SR, et al. Complete pathologic response to neoadjuvant radiotherapy is predictive of oncological outcome in patients with soft tissue sarcoma. Anticancer Res. 2012;32:3911-3915.
13. Rehders A, Peiper M, Stoecklein NH, et al. Hepatic metastasectomy for soft tissue sarcomas: is it justified? World J Surg. 2009;33:111-117.
14. Pfannschmidt J, Hoffmann H, Schneider T, Dienemann H. Pulmonary metastasectomy for soft tissue sarcomas: is it justified? Recent Results Cancer Res. 2009;179:321-336.
1. Lawrence W Jr, Donegan WL, Natarajan N, Mettlin C, Beart R, Winchester D. Adult soft tissue sarcomas. A pattern of care survey of the American College of Surgeons. Ann Surg. 1987;205:349-359.
2. Riad S, Griffin AM, Liberman B, et al. Lymph node metastasis in soft tissue sarcoma in an extremity. Clin Orthop Relat Res. 2004:129-134.
3. Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg. 1993;217:72-77.
4. Mazeron JJ, Suit HD. Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer. 1987;60:1800-1808.
5. Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft tissue sarcoma of adults: meta-analysis of individual data. Lancet. 1997;350:1647-1654.
6. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft tissue sarcoma. Cancer. 2008;113:573-581.
7. Patel SR, Gandhi V, Jenkins J, et al. Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. J Clin Oncol. 2001;19:3483-3489.
8. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25:2755-2763.
9. Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Ann Oncol. 2012;23:785-790.
10. MacDermed DM, Miller LL, Peabody TD, et al. Primary tumor necrosis predicts distant control in locally advanced soft tissue sarcomas after preoperative concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2010;76:1147-1153.
11. Eilber FC, Rosen G, Eckardt J, et al. Treatment-induced pathologic necrosis: a predictor of local recurrence and survival in patients receiving neoadjuvant therapy for high-grade extremity soft tissue sarcomas. J Clin Oncol. 2001;19:3203-3209.
12. Shah D, Borys D, Martinez SR, et al. Complete pathologic response to neoadjuvant radiotherapy is predictive of oncological outcome in patients with soft tissue sarcoma. Anticancer Res. 2012;32:3911-3915.
13. Rehders A, Peiper M, Stoecklein NH, et al. Hepatic metastasectomy for soft tissue sarcomas: is it justified? World J Surg. 2009;33:111-117.
14. Pfannschmidt J, Hoffmann H, Schneider T, Dienemann H. Pulmonary metastasectomy for soft tissue sarcomas: is it justified? Recent Results Cancer Res. 2009;179:321-336.
Intramedullary spinal cord and leptomeningeal metastases presenting as cauda equina syndrome in a patient with melanoma
The incidence of malignant melanoma has been rising in the United States, especially among non-Hispanic white men and women. Death rates have increased for those aged 65 years or older, and incidence rates have increased for all age groups.1 It is a serious public health issue.
Given the unique biology of melanoma, metastatic disease can present in a variety of ways. In most cases, the lymph nodes and lungs are involved.2 The incidence of brain metastases is 10%-40%, however the percentage may be even higher based on reported incidence of autopsy reports.3 The most common forms of metastatic melanoma to the spine are vertebral and intramedullary.4 Specifically, leptomeningeal involvement can be found in 20% of patients in clinical studies and 44%-70% in autopsy series of patients with central nervous system (CNS) metastatic disease.5 Despite its incidence, leptomeningeal disease (LMD) from melanoma is rarely discussed in the literature and the diagnosis may be difficult. Even rarer is the documented presentation of intramedullary spinal cord metastases, or “drop metastases.”6 In our review of the literature, we found no published case reports to date of drop metastases from melanoma causing cauda equina syndrome.
The prognosis of patients with metastatic melanoma with brain metastases is very poor, with a median overall survival of about 4 months reported in several studies.7-9 Prognosis is even worse for patients with leptomeningeal involvement, and median survival without therapy is about 4-6 weeks.10 A combination of intrathecal and systemic chemotherapy can be used to treat LMD.11
Case presentation and summary
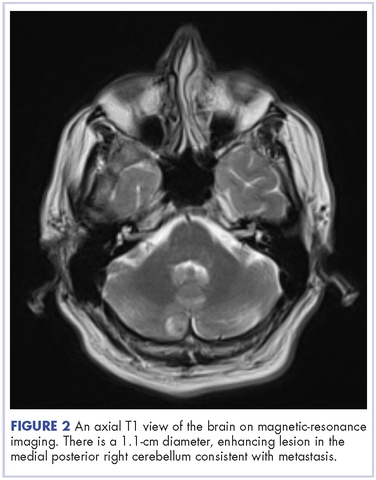
This is the case of a 56-year-old man with history of metastatic melanoma that had been initially diagnosed about 4 years before the current case presentation. Original sites of disease were a supraclavicular lymph node and solitary liver metastasis, both of which were resected. The patient then developed biopsy-proven lung involvement that required left and right wedge resections. Mutation testing for BRAF V600E and BRAF V600K was sent and not detected. Therefore the patient did not receive any BRAF-targeted therapies. Subsequently, recurrent metastatic disease to the brain with 2 dominant lesions in the cerebellum and the occiput as well as numerous small lesions at the gray-white matter junction was identified (Figure 1 and Figure 2).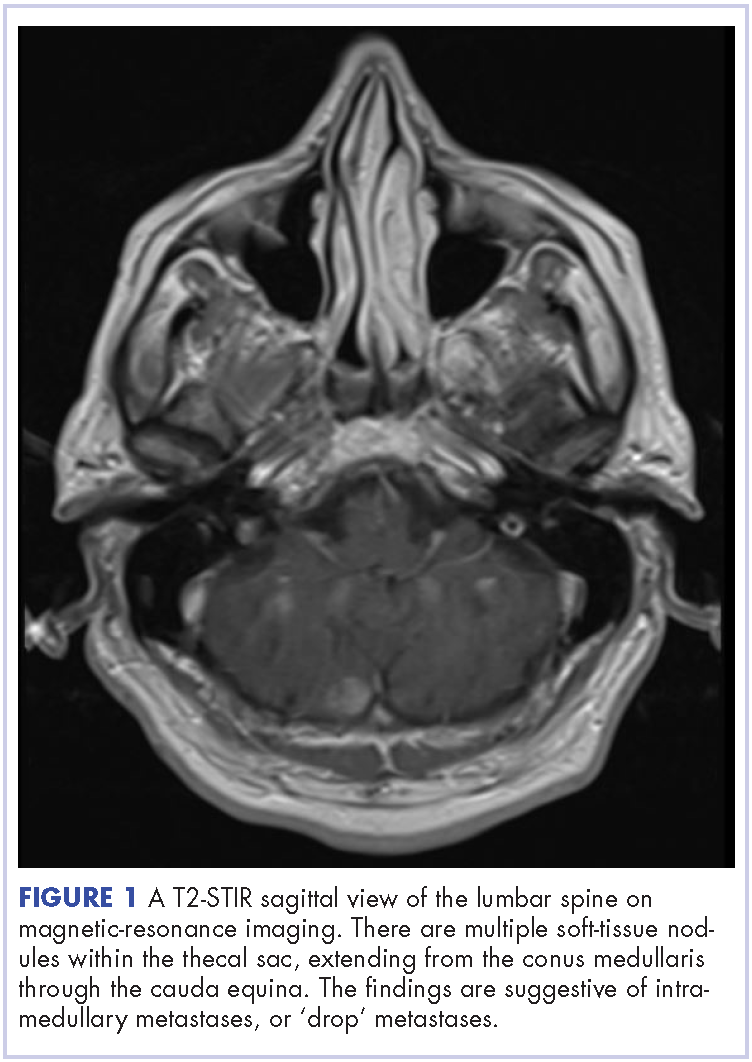

The patient received whole-brain radiation (30 Gy in 10 fractions of 3 Gy each). There was no evidence of disease in his spine at that time. About 2 weeks after completing whole-brain radiation, the patient presented to the hospital with left lower extremity weakness, urinary retention, bowel incontinence, saddle anesthesia, and malaise. The symptoms had begun after he had finished whole-brain radiation and weakness progressed to the point at which he need a cane to be able to walk. A physical examination was significant for hyporreflexia, decreased strength and sensitivity on left lower extremity, saddle anesthesia, and lumbar spinal tenderness to palpation. The results of magnetic-resonance imaging (MRI) of the spine revealed multiple soft-tissue nodules extending from the conus medullaris throughout the cauda equina, consistent with intramedullary metastases, as well as concomitant leptomeningeal involvement (Figure 3).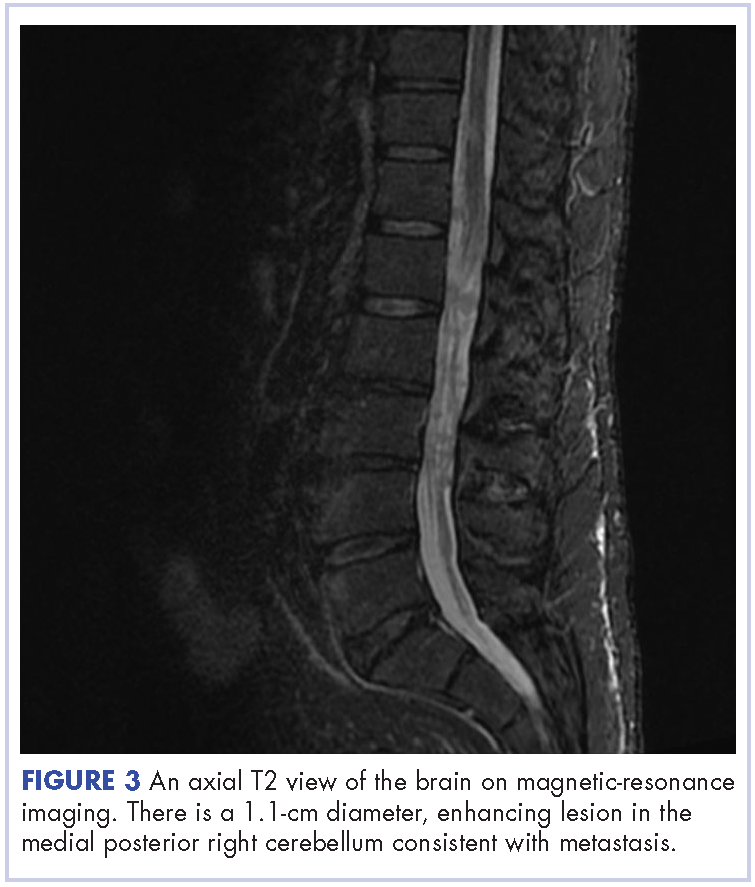
The patient was started on steroids with minimal improvement in neurologic function. We consulted with our neurosurgery colleagues, but learned that no direct surgical intervention could be performed because of widespread involvement. We then proceeded with radiation, 30 Gy in 10 fractions to the lumbar spine. Intrathecal chemotherapy with methotrexate (12 mg twice a week) was also started, with a plan to complete 4 weeks. Shortly after starting radiation therapy and methotrexate, we observed clinical improvement in the patient, with mildly increased left lower extremity strength and increased ambulation with a physical therapist.
Cerebrospinal fluid studies (CSF) showed clearance of malignant cells after 2 treatments of intrathecal methotrexate as well as improvement in CSF chemistry parameters: the patient’s protein level decreased from 1,095 mg/dL to 42 mg/dL (15-45 mg/dL) and his glucose level increased from 3 mg/dL to 73 mg/dL (40-85 mg/dL) However, after completing 3 weeks of intrathecal chemotherapy, the hospital course was complicated by leukopenia, thrombocytopenia, and spontaneous intracranial hemorrhage. The cytopenias were thought to be secondary to systemic effect of intrathecal methotrexate in conjunction with the radiation treatments to the spine. Intrathecal chemotherapy was held.
The patient was not a candidate for systemic immunotherapy because of his decline in performance status. He continued to deteriorate neurologically, and the family decided to pursue inpatient hospice. He died a week after transfer to hospice and 5 weeks after the initial diagnosis of leptomeningeal and intramedullary metastases.
Conclusions
Although metastatic melanoma to the brain is not uncommon, leptomeningeal and intramedullary drop metastases are an infrequent presentation. Even more rare are intramedullary drop metastasis that are significant enough to cause cauda equina syndrome, as with our patient. The incidence of LMD has increased over the years and may continue to increase, likely because of the improved overall survival and a prolonged control of extracranial disease with newly approved systemic therapeutic drugs, such as molecularly targeted therapy and immunotherapy.12 Intramedullary metastases are extremely rare, but reported incidence has seemed to be increasing due to detection with MRI. Currently there are fewer than 100 case reports of intramedullary spinal cord metastasis.6 In one retrospective study, 40 patients with intramedullary metastatic disease secondary to systemic cancer were identified during 1980-1993.6 About half of those cases were from lung cancer, the second most common was breast cancer.
CNS involvement by melanoma can have debilitating complications and confers a poor prognosis. In another retrospective study, several patient characteristics were found to be associated with significantly shorter survival in patients with known brain metastases, including presence of neurologic symptoms and leptomeningeal involvement.3
Malignant cells can reach the CSF by several routes: direct extension, hematogenous, venous access, venous drainage from bone marrow and cranial and peripheral nerves. Once the tumor has reached the CSF, it can seed any portion of the nervous system that has contact with the CSF and become entangled among the cauda equine.13
Given the rarity of leptomeningeal and intramedullary involvement of melanoma, there are no standard treatment guidelines. Treatment for LMD usually consists of intrathecal and systemic chemotherapy. Commonly used intrathecal agents are methotrexate, liposomal cytarabine, and thiopeta.11 The goals of treatment are to improve or stabilize neurologic status of the patient and ideally prolong survival. The choice of agent for intrathecal chemotherapy is guided by the primary tumor, however, there is no strong evidence to choose one agent over the other.12,14 Methotrexate or cytarabine are generally recommended in the National Comprehensive Cancer Network (NCCN) guidelines. Targeted therapy toward the primary tumor is occasionally used for treatment of LMD, for example rituximab can be given intrathecally for lymphoma,15 and trastuzumab has been given intrathecally for breast cancer.16 No intrathecal targeted agents are currently available for melanoma. Administration of intrathecal chemotherapy is given via lumbar puncture or Ommaya reservoir. Induction intrathecal chemotherapy is recommended by NCCN to be given for 4-6 weeks. The schedule of administration varies based on the agent used. Most systemic chemotherapy has poor CSF penetration, which is the basis behind using chemotherapy intrathecally in these patients.14 However, novel therapies for melanoma, such as ipilimumab, have shown activity in the CNS, and it is not known if intrathecal chemotherapy will continue to play role in the management of LMD.17
Systemic therapy for metastatic melanoma has changed with the development of novel agents, which have shown better efficacy than traditional chemotherapy. The recommendation for first-line systemic therapy of metastatic unresectable melanoma is based on several factors: BRAF mutation status, tempo of disease, and presence or absence of cancer-related symptoms. Immunotherapy for metastatic melanoma that is unresectable includes anti-programmed cell death protein-1 (PD-1) monotherapy (nivolumab or pembrolizumab) or combination therapy with nivolumab plus ipilimumab. Targeted therapy is preferred in cases with an identified BRAF mutation. Combination therapy with dabrafenib plus trametinib or with vemurafenib plus cobimetinib is recommended. Single-agent therapy may also be used with dabrafenib or vemurafenib.18
Ipilimumab is a monoclonal antibody that blocks cytotoxic T-lymphocyte antigen-4 to potentiate an anti-tumor T-cell response that was approved in 2011 by the US Food and Drug Administration for the treatment of melanoma. A randomized, phase 3 clinical trial showed an increase in overall survival in patients with unresectable metastatic disease who had received previous treatment.19 Before that, no therapy had been shown to improve overall survival in patients with metastatic melanoma. Patients with CNS metastases were included in this study.19
The activity of ipilimumab specifically in patients with brain metastasis was further studied in a phase 2 trial that enrolled 72 patients, 1 cohort with symptomatic brain metastases and the other cohort with asymptomatic brain metastases.20 After 12 weeks of therapy, response was assessed by modified World Health Organization criteria for disease control (complete response plus partial response plus stable disease). In all, 18% of patients with asymptomatic brain metastasis achieved disease control, compared with 10% of patients with symptomatic brain metastases. When the brain alone was assessed, 24% of asymptomatic patients and 10% of symptomatic patients achieved disease control. No unexpected toxic effects occurred during the study. Anti-PD1 therapy such as nivolumab, which has shown durable responses in metastatic melanoma, has no published results specifically in patients with active brain metastases.
Of the BRAF-targeted therapy, dabrafenib and vemurafenib have also been studied in patients with brain metastases. For darafenib, 172 patients with BRAF-mutated metastatic melanoma were included in a phase 2 clinical trial that showed an intracranial response of 39% in previously untreated patients and 31% in patients whose brain metastases had progressed after previous local treatment.21 Vemurafenib has also shown intracranial response in a phase 2 clinical trial.22
The role of the aforementioned therapies in patients with metastatic melanoma with CNS disease should not be overlooked because these patients are typically excluded from clinical trials. As already noted, agents such as ipilimumab and the dabrafenib–vemurafenib combination have been studied in patients with brain metastases and have shown disease control, but more studies are needed to truly assess whether there is an improvement in overall survival and whether that will change treatment guidelines. Although patients with parenchymal brain metastases were included in these studies, it is not clear how patients with LMD and intramedullary spinal cord metastases, such as our patient, would be affected. It is also not clear whether intrathecal chemotherapy will continue to play a role in management of metastatic melanoma with LMD, especially if these newer agents have CNS activity in addition to controlling extracranial disease. Although rarely documented, leptomeningeal and intramedullary metastatic disease will likely become increasingly recognized as patients with cancer live longer and diagnostic studies improve. These initial studies showing intracranial disease control show compelling evidence to continue enrolling patients with active CNS disease in clinical trials.
1. Jemal A, Saraiya M, Patel P, et al. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992-2006. J Am Acad Dermatol. 2011;65(5 Suppl 1):S17.e1-S17.e11.
2. Patel JK, Didolkar MS, Pickren JW, Moore RH. Metastatic pattern of malignant melanoma: a study of 216 autopsy cases. Am J Surg. 1978;135(6):807-810.
3. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
4. Sun L, Song Y, Gong Q. Easily misdiagnosed delayed metastatic intraspinal extradural melanoma of the lumbar spine: a case report and review of the literature. Oncol Lett. 2013;5(6):1799-1802.
5. Moseley R, Davies A, Bourne S, et al. Neoplastic meningitis in malignant melanoma: diagnosis with monoclonal antibiodies. J Neurol Neurosurg Psychiatry. 1989;52:991-886.
6. Schiff D, O’Neill B. Intramedullary spinal cord metastases clinical features and treatment outcome. Neurology. 1996;47(4):906-912.
7. Fife KM, Colman MH, Stevens G, et al. Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol. 2004;22(7):1293-1300.
8. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
9. Sampson JH, Carter JH Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
10. Abernethy AP. Central nervous system tumors. In: Loprinzi C, ed. ASCO-SEP: Medical Oncology Self-evaluation Program. 4th ed. Alexandria, VA: American Society of Clinical Oncology, 2015. Page 396. Print.
11. Pape E, Desmedt E, Zairi , et al. Leptomeningeal metastasis in melanoma: a prospective clinical study of nine patients. In Vivo. 2012;26(6):1079-1086.
12. Pavlidis N. The diagnostic and therapeutic management of leptomeningeal carcinomatosis. Ann Oncol. 2004;15(Suppl 4):iv285-291.
13. DeAngelis L, Posner JB. Neurologic complications of cancer. 2nd ed. New York, NY: Oxford University Press; 2008.
14. Chamberlain, M. Leptomeningeal metastasis. Curr Opin Oncol. 2010;22:627-635.
15. Chamberlain M, Johnston S, Van Horn A, Glantz MJ. Recurrent lymphomatous meningitis treated with intra-CSF rituximab and liposomal ara-C. J Neurooncol. 2009;91(3):271-277.
16. Zagouri F, Sergentanis T, Bartsch R, et al. Intrathecal administration of trastuzumab for the treatment of meningeal carcinomatosis in HER2-positive metastatic breast cancer: a systematic review and pooled analysis. Breast Cancer Res Treat. 2013;139(1):13-22.
17. Silk A, Bassetti M, West BT, Tsien C, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2(6):899-906.
18. [Behind paywall.] National Comprehensive Cancer Network. Melanoma (version 2.2016). http://www.nccn.org/professionals/physician_gls/pdf/melanoma.pdf. November 10, 2016. Accessed February 28, 2016
19. Hodi F, O’Day S, McDermott D, et al. Improved survival with ipilimumab in patients with metastatic melanoma. NEJM. 2010;363(8):711-723.
20. Margolin K, Ernstoff M, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459-465.
21. Long G, Trefzer U, Davies M, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
22. McArthur GA, Maio M, Arance A, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicenter study. Ann Oncol. 2017;
The incidence of malignant melanoma has been rising in the United States, especially among non-Hispanic white men and women. Death rates have increased for those aged 65 years or older, and incidence rates have increased for all age groups.1 It is a serious public health issue.
Given the unique biology of melanoma, metastatic disease can present in a variety of ways. In most cases, the lymph nodes and lungs are involved.2 The incidence of brain metastases is 10%-40%, however the percentage may be even higher based on reported incidence of autopsy reports.3 The most common forms of metastatic melanoma to the spine are vertebral and intramedullary.4 Specifically, leptomeningeal involvement can be found in 20% of patients in clinical studies and 44%-70% in autopsy series of patients with central nervous system (CNS) metastatic disease.5 Despite its incidence, leptomeningeal disease (LMD) from melanoma is rarely discussed in the literature and the diagnosis may be difficult. Even rarer is the documented presentation of intramedullary spinal cord metastases, or “drop metastases.”6 In our review of the literature, we found no published case reports to date of drop metastases from melanoma causing cauda equina syndrome.
The prognosis of patients with metastatic melanoma with brain metastases is very poor, with a median overall survival of about 4 months reported in several studies.7-9 Prognosis is even worse for patients with leptomeningeal involvement, and median survival without therapy is about 4-6 weeks.10 A combination of intrathecal and systemic chemotherapy can be used to treat LMD.11
Case presentation and summary
This is the case of a 56-year-old man with history of metastatic melanoma that had been initially diagnosed about 4 years before the current case presentation. Original sites of disease were a supraclavicular lymph node and solitary liver metastasis, both of which were resected. The patient then developed biopsy-proven lung involvement that required left and right wedge resections. Mutation testing for BRAF V600E and BRAF V600K was sent and not detected. Therefore the patient did not receive any BRAF-targeted therapies. Subsequently, recurrent metastatic disease to the brain with 2 dominant lesions in the cerebellum and the occiput as well as numerous small lesions at the gray-white matter junction was identified (Figure 1 and Figure 2).
The patient received whole-brain radiation (30 Gy in 10 fractions of 3 Gy each). There was no evidence of disease in his spine at that time. About 2 weeks after completing whole-brain radiation, the patient presented to the hospital with left lower extremity weakness, urinary retention, bowel incontinence, saddle anesthesia, and malaise. The symptoms had begun after he had finished whole-brain radiation and weakness progressed to the point at which he need a cane to be able to walk. A physical examination was significant for hyporreflexia, decreased strength and sensitivity on left lower extremity, saddle anesthesia, and lumbar spinal tenderness to palpation. The results of magnetic-resonance imaging (MRI) of the spine revealed multiple soft-tissue nodules extending from the conus medullaris throughout the cauda equina, consistent with intramedullary metastases, as well as concomitant leptomeningeal involvement (Figure 3).
The patient was started on steroids with minimal improvement in neurologic function. We consulted with our neurosurgery colleagues, but learned that no direct surgical intervention could be performed because of widespread involvement. We then proceeded with radiation, 30 Gy in 10 fractions to the lumbar spine. Intrathecal chemotherapy with methotrexate (12 mg twice a week) was also started, with a plan to complete 4 weeks. Shortly after starting radiation therapy and methotrexate, we observed clinical improvement in the patient, with mildly increased left lower extremity strength and increased ambulation with a physical therapist.
Cerebrospinal fluid studies (CSF) showed clearance of malignant cells after 2 treatments of intrathecal methotrexate as well as improvement in CSF chemistry parameters: the patient’s protein level decreased from 1,095 mg/dL to 42 mg/dL (15-45 mg/dL) and his glucose level increased from 3 mg/dL to 73 mg/dL (40-85 mg/dL) However, after completing 3 weeks of intrathecal chemotherapy, the hospital course was complicated by leukopenia, thrombocytopenia, and spontaneous intracranial hemorrhage. The cytopenias were thought to be secondary to systemic effect of intrathecal methotrexate in conjunction with the radiation treatments to the spine. Intrathecal chemotherapy was held.
The patient was not a candidate for systemic immunotherapy because of his decline in performance status. He continued to deteriorate neurologically, and the family decided to pursue inpatient hospice. He died a week after transfer to hospice and 5 weeks after the initial diagnosis of leptomeningeal and intramedullary metastases.
Conclusions
Although metastatic melanoma to the brain is not uncommon, leptomeningeal and intramedullary drop metastases are an infrequent presentation. Even more rare are intramedullary drop metastasis that are significant enough to cause cauda equina syndrome, as with our patient. The incidence of LMD has increased over the years and may continue to increase, likely because of the improved overall survival and a prolonged control of extracranial disease with newly approved systemic therapeutic drugs, such as molecularly targeted therapy and immunotherapy.12 Intramedullary metastases are extremely rare, but reported incidence has seemed to be increasing due to detection with MRI. Currently there are fewer than 100 case reports of intramedullary spinal cord metastasis.6 In one retrospective study, 40 patients with intramedullary metastatic disease secondary to systemic cancer were identified during 1980-1993.6 About half of those cases were from lung cancer, the second most common was breast cancer.
CNS involvement by melanoma can have debilitating complications and confers a poor prognosis. In another retrospective study, several patient characteristics were found to be associated with significantly shorter survival in patients with known brain metastases, including presence of neurologic symptoms and leptomeningeal involvement.3
Malignant cells can reach the CSF by several routes: direct extension, hematogenous, venous access, venous drainage from bone marrow and cranial and peripheral nerves. Once the tumor has reached the CSF, it can seed any portion of the nervous system that has contact with the CSF and become entangled among the cauda equine.13
Given the rarity of leptomeningeal and intramedullary involvement of melanoma, there are no standard treatment guidelines. Treatment for LMD usually consists of intrathecal and systemic chemotherapy. Commonly used intrathecal agents are methotrexate, liposomal cytarabine, and thiopeta.11 The goals of treatment are to improve or stabilize neurologic status of the patient and ideally prolong survival. The choice of agent for intrathecal chemotherapy is guided by the primary tumor, however, there is no strong evidence to choose one agent over the other.12,14 Methotrexate or cytarabine are generally recommended in the National Comprehensive Cancer Network (NCCN) guidelines. Targeted therapy toward the primary tumor is occasionally used for treatment of LMD, for example rituximab can be given intrathecally for lymphoma,15 and trastuzumab has been given intrathecally for breast cancer.16 No intrathecal targeted agents are currently available for melanoma. Administration of intrathecal chemotherapy is given via lumbar puncture or Ommaya reservoir. Induction intrathecal chemotherapy is recommended by NCCN to be given for 4-6 weeks. The schedule of administration varies based on the agent used. Most systemic chemotherapy has poor CSF penetration, which is the basis behind using chemotherapy intrathecally in these patients.14 However, novel therapies for melanoma, such as ipilimumab, have shown activity in the CNS, and it is not known if intrathecal chemotherapy will continue to play role in the management of LMD.17
Systemic therapy for metastatic melanoma has changed with the development of novel agents, which have shown better efficacy than traditional chemotherapy. The recommendation for first-line systemic therapy of metastatic unresectable melanoma is based on several factors: BRAF mutation status, tempo of disease, and presence or absence of cancer-related symptoms. Immunotherapy for metastatic melanoma that is unresectable includes anti-programmed cell death protein-1 (PD-1) monotherapy (nivolumab or pembrolizumab) or combination therapy with nivolumab plus ipilimumab. Targeted therapy is preferred in cases with an identified BRAF mutation. Combination therapy with dabrafenib plus trametinib or with vemurafenib plus cobimetinib is recommended. Single-agent therapy may also be used with dabrafenib or vemurafenib.18
Ipilimumab is a monoclonal antibody that blocks cytotoxic T-lymphocyte antigen-4 to potentiate an anti-tumor T-cell response that was approved in 2011 by the US Food and Drug Administration for the treatment of melanoma. A randomized, phase 3 clinical trial showed an increase in overall survival in patients with unresectable metastatic disease who had received previous treatment.19 Before that, no therapy had been shown to improve overall survival in patients with metastatic melanoma. Patients with CNS metastases were included in this study.19
The activity of ipilimumab specifically in patients with brain metastasis was further studied in a phase 2 trial that enrolled 72 patients, 1 cohort with symptomatic brain metastases and the other cohort with asymptomatic brain metastases.20 After 12 weeks of therapy, response was assessed by modified World Health Organization criteria for disease control (complete response plus partial response plus stable disease). In all, 18% of patients with asymptomatic brain metastasis achieved disease control, compared with 10% of patients with symptomatic brain metastases. When the brain alone was assessed, 24% of asymptomatic patients and 10% of symptomatic patients achieved disease control. No unexpected toxic effects occurred during the study. Anti-PD1 therapy such as nivolumab, which has shown durable responses in metastatic melanoma, has no published results specifically in patients with active brain metastases.
Of the BRAF-targeted therapy, dabrafenib and vemurafenib have also been studied in patients with brain metastases. For darafenib, 172 patients with BRAF-mutated metastatic melanoma were included in a phase 2 clinical trial that showed an intracranial response of 39% in previously untreated patients and 31% in patients whose brain metastases had progressed after previous local treatment.21 Vemurafenib has also shown intracranial response in a phase 2 clinical trial.22
The role of the aforementioned therapies in patients with metastatic melanoma with CNS disease should not be overlooked because these patients are typically excluded from clinical trials. As already noted, agents such as ipilimumab and the dabrafenib–vemurafenib combination have been studied in patients with brain metastases and have shown disease control, but more studies are needed to truly assess whether there is an improvement in overall survival and whether that will change treatment guidelines. Although patients with parenchymal brain metastases were included in these studies, it is not clear how patients with LMD and intramedullary spinal cord metastases, such as our patient, would be affected. It is also not clear whether intrathecal chemotherapy will continue to play a role in management of metastatic melanoma with LMD, especially if these newer agents have CNS activity in addition to controlling extracranial disease. Although rarely documented, leptomeningeal and intramedullary metastatic disease will likely become increasingly recognized as patients with cancer live longer and diagnostic studies improve. These initial studies showing intracranial disease control show compelling evidence to continue enrolling patients with active CNS disease in clinical trials.
The incidence of malignant melanoma has been rising in the United States, especially among non-Hispanic white men and women. Death rates have increased for those aged 65 years or older, and incidence rates have increased for all age groups.1 It is a serious public health issue.
Given the unique biology of melanoma, metastatic disease can present in a variety of ways. In most cases, the lymph nodes and lungs are involved.2 The incidence of brain metastases is 10%-40%, however the percentage may be even higher based on reported incidence of autopsy reports.3 The most common forms of metastatic melanoma to the spine are vertebral and intramedullary.4 Specifically, leptomeningeal involvement can be found in 20% of patients in clinical studies and 44%-70% in autopsy series of patients with central nervous system (CNS) metastatic disease.5 Despite its incidence, leptomeningeal disease (LMD) from melanoma is rarely discussed in the literature and the diagnosis may be difficult. Even rarer is the documented presentation of intramedullary spinal cord metastases, or “drop metastases.”6 In our review of the literature, we found no published case reports to date of drop metastases from melanoma causing cauda equina syndrome.
The prognosis of patients with metastatic melanoma with brain metastases is very poor, with a median overall survival of about 4 months reported in several studies.7-9 Prognosis is even worse for patients with leptomeningeal involvement, and median survival without therapy is about 4-6 weeks.10 A combination of intrathecal and systemic chemotherapy can be used to treat LMD.11
Case presentation and summary
This is the case of a 56-year-old man with history of metastatic melanoma that had been initially diagnosed about 4 years before the current case presentation. Original sites of disease were a supraclavicular lymph node and solitary liver metastasis, both of which were resected. The patient then developed biopsy-proven lung involvement that required left and right wedge resections. Mutation testing for BRAF V600E and BRAF V600K was sent and not detected. Therefore the patient did not receive any BRAF-targeted therapies. Subsequently, recurrent metastatic disease to the brain with 2 dominant lesions in the cerebellum and the occiput as well as numerous small lesions at the gray-white matter junction was identified (Figure 1 and Figure 2).
The patient received whole-brain radiation (30 Gy in 10 fractions of 3 Gy each). There was no evidence of disease in his spine at that time. About 2 weeks after completing whole-brain radiation, the patient presented to the hospital with left lower extremity weakness, urinary retention, bowel incontinence, saddle anesthesia, and malaise. The symptoms had begun after he had finished whole-brain radiation and weakness progressed to the point at which he need a cane to be able to walk. A physical examination was significant for hyporreflexia, decreased strength and sensitivity on left lower extremity, saddle anesthesia, and lumbar spinal tenderness to palpation. The results of magnetic-resonance imaging (MRI) of the spine revealed multiple soft-tissue nodules extending from the conus medullaris throughout the cauda equina, consistent with intramedullary metastases, as well as concomitant leptomeningeal involvement (Figure 3).
The patient was started on steroids with minimal improvement in neurologic function. We consulted with our neurosurgery colleagues, but learned that no direct surgical intervention could be performed because of widespread involvement. We then proceeded with radiation, 30 Gy in 10 fractions to the lumbar spine. Intrathecal chemotherapy with methotrexate (12 mg twice a week) was also started, with a plan to complete 4 weeks. Shortly after starting radiation therapy and methotrexate, we observed clinical improvement in the patient, with mildly increased left lower extremity strength and increased ambulation with a physical therapist.
Cerebrospinal fluid studies (CSF) showed clearance of malignant cells after 2 treatments of intrathecal methotrexate as well as improvement in CSF chemistry parameters: the patient’s protein level decreased from 1,095 mg/dL to 42 mg/dL (15-45 mg/dL) and his glucose level increased from 3 mg/dL to 73 mg/dL (40-85 mg/dL) However, after completing 3 weeks of intrathecal chemotherapy, the hospital course was complicated by leukopenia, thrombocytopenia, and spontaneous intracranial hemorrhage. The cytopenias were thought to be secondary to systemic effect of intrathecal methotrexate in conjunction with the radiation treatments to the spine. Intrathecal chemotherapy was held.
The patient was not a candidate for systemic immunotherapy because of his decline in performance status. He continued to deteriorate neurologically, and the family decided to pursue inpatient hospice. He died a week after transfer to hospice and 5 weeks after the initial diagnosis of leptomeningeal and intramedullary metastases.
Conclusions
Although metastatic melanoma to the brain is not uncommon, leptomeningeal and intramedullary drop metastases are an infrequent presentation. Even more rare are intramedullary drop metastasis that are significant enough to cause cauda equina syndrome, as with our patient. The incidence of LMD has increased over the years and may continue to increase, likely because of the improved overall survival and a prolonged control of extracranial disease with newly approved systemic therapeutic drugs, such as molecularly targeted therapy and immunotherapy.12 Intramedullary metastases are extremely rare, but reported incidence has seemed to be increasing due to detection with MRI. Currently there are fewer than 100 case reports of intramedullary spinal cord metastasis.6 In one retrospective study, 40 patients with intramedullary metastatic disease secondary to systemic cancer were identified during 1980-1993.6 About half of those cases were from lung cancer, the second most common was breast cancer.
CNS involvement by melanoma can have debilitating complications and confers a poor prognosis. In another retrospective study, several patient characteristics were found to be associated with significantly shorter survival in patients with known brain metastases, including presence of neurologic symptoms and leptomeningeal involvement.3
Malignant cells can reach the CSF by several routes: direct extension, hematogenous, venous access, venous drainage from bone marrow and cranial and peripheral nerves. Once the tumor has reached the CSF, it can seed any portion of the nervous system that has contact with the CSF and become entangled among the cauda equine.13
Given the rarity of leptomeningeal and intramedullary involvement of melanoma, there are no standard treatment guidelines. Treatment for LMD usually consists of intrathecal and systemic chemotherapy. Commonly used intrathecal agents are methotrexate, liposomal cytarabine, and thiopeta.11 The goals of treatment are to improve or stabilize neurologic status of the patient and ideally prolong survival. The choice of agent for intrathecal chemotherapy is guided by the primary tumor, however, there is no strong evidence to choose one agent over the other.12,14 Methotrexate or cytarabine are generally recommended in the National Comprehensive Cancer Network (NCCN) guidelines. Targeted therapy toward the primary tumor is occasionally used for treatment of LMD, for example rituximab can be given intrathecally for lymphoma,15 and trastuzumab has been given intrathecally for breast cancer.16 No intrathecal targeted agents are currently available for melanoma. Administration of intrathecal chemotherapy is given via lumbar puncture or Ommaya reservoir. Induction intrathecal chemotherapy is recommended by NCCN to be given for 4-6 weeks. The schedule of administration varies based on the agent used. Most systemic chemotherapy has poor CSF penetration, which is the basis behind using chemotherapy intrathecally in these patients.14 However, novel therapies for melanoma, such as ipilimumab, have shown activity in the CNS, and it is not known if intrathecal chemotherapy will continue to play role in the management of LMD.17
Systemic therapy for metastatic melanoma has changed with the development of novel agents, which have shown better efficacy than traditional chemotherapy. The recommendation for first-line systemic therapy of metastatic unresectable melanoma is based on several factors: BRAF mutation status, tempo of disease, and presence or absence of cancer-related symptoms. Immunotherapy for metastatic melanoma that is unresectable includes anti-programmed cell death protein-1 (PD-1) monotherapy (nivolumab or pembrolizumab) or combination therapy with nivolumab plus ipilimumab. Targeted therapy is preferred in cases with an identified BRAF mutation. Combination therapy with dabrafenib plus trametinib or with vemurafenib plus cobimetinib is recommended. Single-agent therapy may also be used with dabrafenib or vemurafenib.18
Ipilimumab is a monoclonal antibody that blocks cytotoxic T-lymphocyte antigen-4 to potentiate an anti-tumor T-cell response that was approved in 2011 by the US Food and Drug Administration for the treatment of melanoma. A randomized, phase 3 clinical trial showed an increase in overall survival in patients with unresectable metastatic disease who had received previous treatment.19 Before that, no therapy had been shown to improve overall survival in patients with metastatic melanoma. Patients with CNS metastases were included in this study.19
The activity of ipilimumab specifically in patients with brain metastasis was further studied in a phase 2 trial that enrolled 72 patients, 1 cohort with symptomatic brain metastases and the other cohort with asymptomatic brain metastases.20 After 12 weeks of therapy, response was assessed by modified World Health Organization criteria for disease control (complete response plus partial response plus stable disease). In all, 18% of patients with asymptomatic brain metastasis achieved disease control, compared with 10% of patients with symptomatic brain metastases. When the brain alone was assessed, 24% of asymptomatic patients and 10% of symptomatic patients achieved disease control. No unexpected toxic effects occurred during the study. Anti-PD1 therapy such as nivolumab, which has shown durable responses in metastatic melanoma, has no published results specifically in patients with active brain metastases.
Of the BRAF-targeted therapy, dabrafenib and vemurafenib have also been studied in patients with brain metastases. For darafenib, 172 patients with BRAF-mutated metastatic melanoma were included in a phase 2 clinical trial that showed an intracranial response of 39% in previously untreated patients and 31% in patients whose brain metastases had progressed after previous local treatment.21 Vemurafenib has also shown intracranial response in a phase 2 clinical trial.22
The role of the aforementioned therapies in patients with metastatic melanoma with CNS disease should not be overlooked because these patients are typically excluded from clinical trials. As already noted, agents such as ipilimumab and the dabrafenib–vemurafenib combination have been studied in patients with brain metastases and have shown disease control, but more studies are needed to truly assess whether there is an improvement in overall survival and whether that will change treatment guidelines. Although patients with parenchymal brain metastases were included in these studies, it is not clear how patients with LMD and intramedullary spinal cord metastases, such as our patient, would be affected. It is also not clear whether intrathecal chemotherapy will continue to play a role in management of metastatic melanoma with LMD, especially if these newer agents have CNS activity in addition to controlling extracranial disease. Although rarely documented, leptomeningeal and intramedullary metastatic disease will likely become increasingly recognized as patients with cancer live longer and diagnostic studies improve. These initial studies showing intracranial disease control show compelling evidence to continue enrolling patients with active CNS disease in clinical trials.
1. Jemal A, Saraiya M, Patel P, et al. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992-2006. J Am Acad Dermatol. 2011;65(5 Suppl 1):S17.e1-S17.e11.
2. Patel JK, Didolkar MS, Pickren JW, Moore RH. Metastatic pattern of malignant melanoma: a study of 216 autopsy cases. Am J Surg. 1978;135(6):807-810.
3. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
4. Sun L, Song Y, Gong Q. Easily misdiagnosed delayed metastatic intraspinal extradural melanoma of the lumbar spine: a case report and review of the literature. Oncol Lett. 2013;5(6):1799-1802.
5. Moseley R, Davies A, Bourne S, et al. Neoplastic meningitis in malignant melanoma: diagnosis with monoclonal antibiodies. J Neurol Neurosurg Psychiatry. 1989;52:991-886.
6. Schiff D, O’Neill B. Intramedullary spinal cord metastases clinical features and treatment outcome. Neurology. 1996;47(4):906-912.
7. Fife KM, Colman MH, Stevens G, et al. Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol. 2004;22(7):1293-1300.
8. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
9. Sampson JH, Carter JH Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
10. Abernethy AP. Central nervous system tumors. In: Loprinzi C, ed. ASCO-SEP: Medical Oncology Self-evaluation Program. 4th ed. Alexandria, VA: American Society of Clinical Oncology, 2015. Page 396. Print.
11. Pape E, Desmedt E, Zairi , et al. Leptomeningeal metastasis in melanoma: a prospective clinical study of nine patients. In Vivo. 2012;26(6):1079-1086.
12. Pavlidis N. The diagnostic and therapeutic management of leptomeningeal carcinomatosis. Ann Oncol. 2004;15(Suppl 4):iv285-291.
13. DeAngelis L, Posner JB. Neurologic complications of cancer. 2nd ed. New York, NY: Oxford University Press; 2008.
14. Chamberlain, M. Leptomeningeal metastasis. Curr Opin Oncol. 2010;22:627-635.
15. Chamberlain M, Johnston S, Van Horn A, Glantz MJ. Recurrent lymphomatous meningitis treated with intra-CSF rituximab and liposomal ara-C. J Neurooncol. 2009;91(3):271-277.
16. Zagouri F, Sergentanis T, Bartsch R, et al. Intrathecal administration of trastuzumab for the treatment of meningeal carcinomatosis in HER2-positive metastatic breast cancer: a systematic review and pooled analysis. Breast Cancer Res Treat. 2013;139(1):13-22.
17. Silk A, Bassetti M, West BT, Tsien C, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2(6):899-906.
18. [Behind paywall.] National Comprehensive Cancer Network. Melanoma (version 2.2016). http://www.nccn.org/professionals/physician_gls/pdf/melanoma.pdf. November 10, 2016. Accessed February 28, 2016
19. Hodi F, O’Day S, McDermott D, et al. Improved survival with ipilimumab in patients with metastatic melanoma. NEJM. 2010;363(8):711-723.
20. Margolin K, Ernstoff M, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459-465.
21. Long G, Trefzer U, Davies M, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
22. McArthur GA, Maio M, Arance A, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicenter study. Ann Oncol. 2017;
1. Jemal A, Saraiya M, Patel P, et al. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992-2006. J Am Acad Dermatol. 2011;65(5 Suppl 1):S17.e1-S17.e11.
2. Patel JK, Didolkar MS, Pickren JW, Moore RH. Metastatic pattern of malignant melanoma: a study of 216 autopsy cases. Am J Surg. 1978;135(6):807-810.
3. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
4. Sun L, Song Y, Gong Q. Easily misdiagnosed delayed metastatic intraspinal extradural melanoma of the lumbar spine: a case report and review of the literature. Oncol Lett. 2013;5(6):1799-1802.
5. Moseley R, Davies A, Bourne S, et al. Neoplastic meningitis in malignant melanoma: diagnosis with monoclonal antibiodies. J Neurol Neurosurg Psychiatry. 1989;52:991-886.
6. Schiff D, O’Neill B. Intramedullary spinal cord metastases clinical features and treatment outcome. Neurology. 1996;47(4):906-912.
7. Fife KM, Colman MH, Stevens G, et al. Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol. 2004;22(7):1293-1300.
8. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
9. Sampson JH, Carter JH Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
10. Abernethy AP. Central nervous system tumors. In: Loprinzi C, ed. ASCO-SEP: Medical Oncology Self-evaluation Program. 4th ed. Alexandria, VA: American Society of Clinical Oncology, 2015. Page 396. Print.
11. Pape E, Desmedt E, Zairi , et al. Leptomeningeal metastasis in melanoma: a prospective clinical study of nine patients. In Vivo. 2012;26(6):1079-1086.
12. Pavlidis N. The diagnostic and therapeutic management of leptomeningeal carcinomatosis. Ann Oncol. 2004;15(Suppl 4):iv285-291.
13. DeAngelis L, Posner JB. Neurologic complications of cancer. 2nd ed. New York, NY: Oxford University Press; 2008.
14. Chamberlain, M. Leptomeningeal metastasis. Curr Opin Oncol. 2010;22:627-635.
15. Chamberlain M, Johnston S, Van Horn A, Glantz MJ. Recurrent lymphomatous meningitis treated with intra-CSF rituximab and liposomal ara-C. J Neurooncol. 2009;91(3):271-277.
16. Zagouri F, Sergentanis T, Bartsch R, et al. Intrathecal administration of trastuzumab for the treatment of meningeal carcinomatosis in HER2-positive metastatic breast cancer: a systematic review and pooled analysis. Breast Cancer Res Treat. 2013;139(1):13-22.
17. Silk A, Bassetti M, West BT, Tsien C, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2(6):899-906.
18. [Behind paywall.] National Comprehensive Cancer Network. Melanoma (version 2.2016). http://www.nccn.org/professionals/physician_gls/pdf/melanoma.pdf. November 10, 2016. Accessed February 28, 2016
19. Hodi F, O’Day S, McDermott D, et al. Improved survival with ipilimumab in patients with metastatic melanoma. NEJM. 2010;363(8):711-723.
20. Margolin K, Ernstoff M, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459-465.
21. Long G, Trefzer U, Davies M, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
22. McArthur GA, Maio M, Arance A, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicenter study. Ann Oncol. 2017;
Presumed Serum Sickness Following Thymoglobulin Treatment of Acute Cellular Rejection of a Cardiac Allograft
Serum sickness was first described by von Pirquet and Schick1 as a constellation of signs and symptoms displayed in patients receiving equine serum as an antitoxin for the treatment of scarlet fever and diphtheria. Serum sickness is an immune complex–mediated hypersensitivity reaction that can be clinically diagnosed in patients who present with fever, rash, and polyarthralgia or polyarthritis following exposure to heterologous serum proteins.2,3 Symptom onset typically occurs within 1 to 2 weeks of first exposure to the serum, and resolution frequently occurs with discontinuation of the offending agent. Other symptoms may include malaise, gastrointestinal tract concerns, headache, blurred vision, or lymphadenopathy.4 Proteinuria, hematuria, and a transient decrease in creatinine clearance also have been reported in serum sickness.4
Serum sickness is caused by a type III immune complex–mediated hypersensitivity reaction to heterologous rabbit or equine serum proteins. Nonhuman proteins present in antithymocyte globulin (ATG) stimulate the production of IgG, IgM, IgA, and IgE antibodies.2-4 If the resultant immune complexes overwhelm the mononuclear phagocyte system, these complexes are deposited in blood vessels and tissues, which leads to complement activation and the production of complement fragments such as C3a and C5a.5 C3a is an anaphylatoxin that causes mast cell degranulation and the consequent formation of urticarial lesions. C5a is a neutrophil chemoattractant that promotes inflammation at the site of complement deposition.
Serum sickness–like reactions may occur days to weeks following administration of certain drugs, such as cefaclor or penicillin. Although the symptoms and timing of serum sickness–like reactions are similar to serum sickness, they are not caused by an immune complex–mediated mechanism and are believed to be secondary to an idiosyncratic delayed drug reaction.6
Thymoglobulin, a type of ATG, is a polyclonal antibody generated in rabbits that targets numerous human epitopes, including cell surface markers on T cells (CD2, CD3, CD4, CD8), B cells (CD21, CD19, CD40), and adhesion molecules (CD6, CD25, CD44, CD45, and the integrin LFA-1 [lymphocyte function-associated antigen-1]).7,8 Thymoglobulin has proven efficacy in the setting of cardiac transplantation.9-11 Although calcineurin inhibitors form the foundation in the armamentarium of immunosuppressive agents in cardiac transplantation, their nephrotoxicity has limited their unrestrained use in patients.9 By delaying the need for calcineurin inhibitors, thymoglobulin preserves greater renal function without increasing the risk for acute rejection.9,10 Akin to its use in the patient presented in this case report, thymoglobulin also is used in the treatment of acute cellular rejection in heart transplant recipients with signs of heart failure.11
Case Report
A 35-year-old man with a history of familial cardiomyopathy who underwent orthotopic heart transplantation presented with grade 3R acute cellular rejection. The patient’s immunosuppressive regimen consisted of thymoglobulin 150 mg once daily, tacrolimus 2.5 mg twice daily, hydrocortisone 100 mg once daily, and mycophenolate mofetil 1000 mg twice daily. On day 7 of thymoglobulin treatment, the dermatology department was consulted to evaluate a pruritic eruption. The patient reported that he noticed redness of the palms and soles, as well as redness accentuated in the axilla, groin, and other skin creases 2 days prior. The patient also reported symmetric bilateral hand pain that had started 1 day following rash onset. He denied fever and remained afebrile throughout his hospitalization.
On physical examination, the patient displayed a blanching, erythematous, edematous, evanescent macular rash with some areas of wheal formation symmetrically distributed in the bilateral axillae, inframammary folds, and groin (Figure, A and B). The palms and soles were tender with diffuse blanching erythema. The eruption was accentuated at the lateral and medial borders of both feet (Figure, C). There was concern that the patient may have a form of serum sickness with a blunted incomplete response due to his concomitant use of immunosuppressive agents. Shortly after evaluation, the patient left the hospital against medical advice before the recommended evaluation and systemic workup could be implemented.
The patient returned for an outpatient appointment approximately 1 week later. Medical records indicated that the patient’s skin eruption had resolved. Tests for antithymoglobulin antibodies at this visit were negative. The antithymoglobulin antibody enzyme-linked immunosorbent assay has a diagnostic sensitivity of 86%12 and large interlaboratory variability.13 Given the presence of other features of serum sickness, a false-negative result was considered by dermatology. Nonetheless, one must consider other differential diagnoses, including a simple cutaneous adverse drug eruption or viral exanthem that might have in fact been causative.
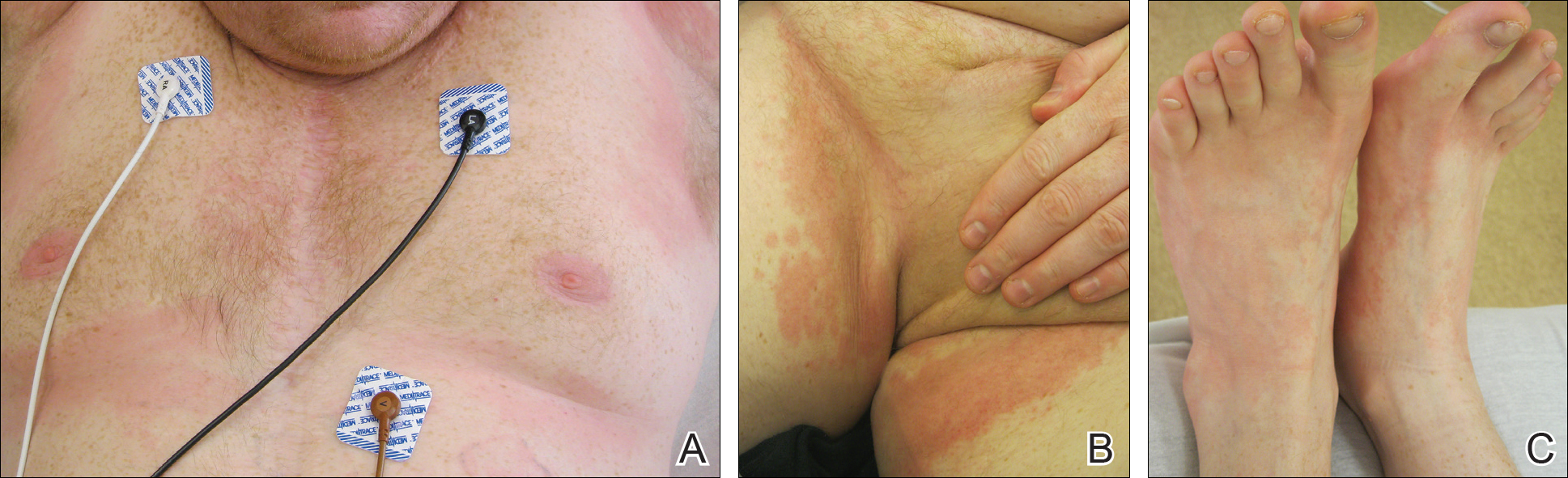
Comment
We present an atypical case of possible serum sickness in a heart transplant recipient following thymoglobulin treatment of acute cellular rejection of the cardiac allograft. Serum sickness is a clinical diagnosis supported by laboratory data. Some authors have suggested major and minor diagnostic criteria to aid with the diagnosis.7 Major diagnostic criteria include onset more than 7 days after the initial thymoglobulin administration, persistent high fevers (temperature, >38.4°C), persistent arthritis/arthralgia, and positive heterologous antibodies on enzyme-linked immunosorbent assay. Minor diagnostic criteria include rash, acute renal failure, trismus, and low serum complement (C3 and C4).
The variable cutaneous presentations of serum sickness are important to recognize in the process of making the correct diagnosis. Rash is frequently reported in serum sickness, with some studies displaying rates of up to 93%.4,14 The skin findings are most frequently described as urticarial or serpiginous macular lesions.3 Other variations of the eruption exist, and morbilliform eruptions or a combination of morbilliform and urticarial eruptions have been reported.3 It is important to judge cutaneous eruptions of serum sickness within the context of the potential cytopenia in a patient being treated with ATG. As such, purpuric eruptions have been attributed to serum sickness in thrombocytopenic patients receiving ATG for bone marrow failure.14
Usually, cutaneous eruptions of serum sickness initially are identified in the groin, axilla, and periumbilical region, and then they proceed to include the trunk and extremities. Erythema of the palms and soles frequently is described as well as a linear accentuation of the rash along the lateral and medial borders of the feet and hands at the margin of the plantar or palmar skin, respectively.14 The mucous membranes frequently are spared in serum sickness.
Despite the lack of evidence-based guidelines, case series and literature reviews have suggested a treatment regimen for serum sickness,7,15-18 calling for immediate withdrawal of the offending agent. Antihistamines may be added to control pruritus and rash. Patients with high fever, a progressive rash, or severe arthralgia have benefited from short courses of oral16,18 or intravenous7,17 glucocorticoids. The extent of the eruption in our patient was concerning, particularly because he was already receiving systemic corticosteroids in conjunction with other immunosuppressives, which may have explained his lack of fever.
Because our patient satisfied some diagnostic criteria for serum sickness and failed to satisfy others, our team was faced with the challenge of balancing the risks of possible serum sickness with the risks of the potential for progressive cardiac rejection from the withdrawal of thymoglobulin.7 There is some evidence in the literature for the use of therapeutic plasma exchange (TPE) for the treatment of serum sickness if the offending agent could not be discontinued. Tanriover et al19 presented a case series of 5 renal transplant recipients treated with thymoglobulin who developed serum sickness. The diagnosis of serum sickness was made clinically and augmented by the presence of antiheterologous antibodies. All 5 patients had persistent symptoms of serum sickness despite 2 days of glucocorticoid treatment. Interestingly, 3 patients had complete resolution of all symptoms after a single TPE treatment, and 2 patients achieved resolution of fever and arthritis after 2 consecutive days of TPE treatments.19 Because plasmapheresis is used to treat cardiac allograft rejection in patients showing signs of heart failure,11 the employment of TPE in these patients may have dual beneficial effects of concurrently treating serum sickness and allograft rejection.
Given the patient’s noncompliance and leaving the hospital against medical advice, a full workup was not able to be pursued in this case, though fortunately the eruption and his other symptoms had resolved by the time he was seen for outpatient follow-up 1 week later. Noncompliance with immunosuppressive therapy is a considerable risk factor for morbidity and mortality following heart transplantation. These patients have more transplant coronary artery disease and substantially shorter clinical event-free time.20 Our patient demonstrates the need for proactive compliance-enhancing interventions in heart transplant patients who experience allograft rejection.
- von Pirquet C, Schick B. Serum Sickness. Schick B, trans-ed. Baltimore, MD; Williams & Wilkins; 1951.
- Vincent C, Revillard JP. Antibody response to horse gamma-globulin in recipients of renal allografts: relationship with transplant crises and transplant survival. Transplantation. 1977;24:141-147.
- Lawley TJ, Bielory L, Gascon P, et al. A prospective clinical and immunologic analysis of patients with serum sickness. N Engl J Med. 1984;311:1407-1413.
- Bielory L, Gascon P, Lawley TJ, et al. Human serum sickness: a prospective analysis of 35 patients treated with equine anti-thymocyte globulin for bone marrow failure. Medicine (Baltimore). 1988;67:40-57.
- Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276-J286.
- Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587-1591.
- Lundquist AL, Chari RS, Wood JH, et al. Serum sickness following rabbit antithymocyte-globulin induction in a liver transplant recipient: case report and literature review. Liver Transpl. 2007;13:647-650.
- Bourdage JS, Hamlin DM. Comparative polyclonal antithymocyte globulin and antilymphocyte/antilymphoblast globulin anti-CD antigen analysis by flow cytometry. Transplantation. 1995;59:1194-1200.
- Zuckermann AO, Aliabadi AZ. Calcineurin-inhibitor minimization protocols in heart transplantation. Transpl Int. 2009;22:78-89.
- Cantarovich M, Giannetti N, Barkun J, et al. Antithymocyte globulin induction allows a prolonged delay in the initiation of cyclosporine in heart transplant patients with postoperative renal dysfunction. Transplantation. 2004;78:779-781.
- Patel JK, Kittleson M, Kobashigawa JA. Cardiac allograft rejection. Surgeon. 2010;9:160-167.
- Tatum AH, Bollinger RR, Sanfilippo F. Rapid serologic diagnosis of serum sickness from antithymocyte globulin therapy using enzyme immunoassay. Transplantation. 1984;38:582-586.
- Kimball JA, Pescovitz MD, Book BK, et al. Reduced human IgG anti-ATGAM antibody formation in renal transplant recipients receiving mycophenolate mofetil. Transplantation. 1995;60:1379-1383.
- Bielory L, Yancey KB, Young NS, et al. Cutaneous manifestations of serum sickness in patients receiving antithymocyte globulin. J Am Acad Dermatol. 1985;13:411-417.
- Joubert GI, Hadad K, Matsui D, et al. Selection of treatment of cefaclor-associated urticarial, serum sickness-like reactions and erythema multiforme by emergency pediatricians: lack of a uniform standard of care. Can J Clin Pharmacol. 1999;6:197-201.
- Clark BM, Kotti GH, Shah AD, et al. Severe serum sickness reaction to oral and intramuscular penicillin. Pharmacotherapy. 2006;26:705-708.
- Finger E, Scheinberg M. Development of serum sickness-like symptoms after rituximab infusion in two patients with severe hypergammaglobulinemia. J Clin Rheumatol. 2007;13:94-95.
- Tatum AJ, Ditto AM, Patterson R. Severe serum sickness-like reaction to oral penicillin drugs: three case reports. Ann Allergy Asthma Immunol. 2001;86:330-334.
- Tanriover B, Chuang P, Fishbach B, et al. Polyclonal antibody-induced serum sickness in renal transplant recipients: treatment with therapeutic plasma exchange. Transplantation. 2005;80:279-281.
- Dobbels F, De Geest S, van Cleemput J, et al. Effect of late medication non-compliance on outcome after heart transplantation: a 5-year follow-up. J Heart Lung Transplant. 2004;23:1245-1251.
Serum sickness was first described by von Pirquet and Schick1 as a constellation of signs and symptoms displayed in patients receiving equine serum as an antitoxin for the treatment of scarlet fever and diphtheria. Serum sickness is an immune complex–mediated hypersensitivity reaction that can be clinically diagnosed in patients who present with fever, rash, and polyarthralgia or polyarthritis following exposure to heterologous serum proteins.2,3 Symptom onset typically occurs within 1 to 2 weeks of first exposure to the serum, and resolution frequently occurs with discontinuation of the offending agent. Other symptoms may include malaise, gastrointestinal tract concerns, headache, blurred vision, or lymphadenopathy.4 Proteinuria, hematuria, and a transient decrease in creatinine clearance also have been reported in serum sickness.4
Serum sickness is caused by a type III immune complex–mediated hypersensitivity reaction to heterologous rabbit or equine serum proteins. Nonhuman proteins present in antithymocyte globulin (ATG) stimulate the production of IgG, IgM, IgA, and IgE antibodies.2-4 If the resultant immune complexes overwhelm the mononuclear phagocyte system, these complexes are deposited in blood vessels and tissues, which leads to complement activation and the production of complement fragments such as C3a and C5a.5 C3a is an anaphylatoxin that causes mast cell degranulation and the consequent formation of urticarial lesions. C5a is a neutrophil chemoattractant that promotes inflammation at the site of complement deposition.
Serum sickness–like reactions may occur days to weeks following administration of certain drugs, such as cefaclor or penicillin. Although the symptoms and timing of serum sickness–like reactions are similar to serum sickness, they are not caused by an immune complex–mediated mechanism and are believed to be secondary to an idiosyncratic delayed drug reaction.6
Thymoglobulin, a type of ATG, is a polyclonal antibody generated in rabbits that targets numerous human epitopes, including cell surface markers on T cells (CD2, CD3, CD4, CD8), B cells (CD21, CD19, CD40), and adhesion molecules (CD6, CD25, CD44, CD45, and the integrin LFA-1 [lymphocyte function-associated antigen-1]).7,8 Thymoglobulin has proven efficacy in the setting of cardiac transplantation.9-11 Although calcineurin inhibitors form the foundation in the armamentarium of immunosuppressive agents in cardiac transplantation, their nephrotoxicity has limited their unrestrained use in patients.9 By delaying the need for calcineurin inhibitors, thymoglobulin preserves greater renal function without increasing the risk for acute rejection.9,10 Akin to its use in the patient presented in this case report, thymoglobulin also is used in the treatment of acute cellular rejection in heart transplant recipients with signs of heart failure.11
Case Report
A 35-year-old man with a history of familial cardiomyopathy who underwent orthotopic heart transplantation presented with grade 3R acute cellular rejection. The patient’s immunosuppressive regimen consisted of thymoglobulin 150 mg once daily, tacrolimus 2.5 mg twice daily, hydrocortisone 100 mg once daily, and mycophenolate mofetil 1000 mg twice daily. On day 7 of thymoglobulin treatment, the dermatology department was consulted to evaluate a pruritic eruption. The patient reported that he noticed redness of the palms and soles, as well as redness accentuated in the axilla, groin, and other skin creases 2 days prior. The patient also reported symmetric bilateral hand pain that had started 1 day following rash onset. He denied fever and remained afebrile throughout his hospitalization.
On physical examination, the patient displayed a blanching, erythematous, edematous, evanescent macular rash with some areas of wheal formation symmetrically distributed in the bilateral axillae, inframammary folds, and groin (Figure, A and B). The palms and soles were tender with diffuse blanching erythema. The eruption was accentuated at the lateral and medial borders of both feet (Figure, C). There was concern that the patient may have a form of serum sickness with a blunted incomplete response due to his concomitant use of immunosuppressive agents. Shortly after evaluation, the patient left the hospital against medical advice before the recommended evaluation and systemic workup could be implemented.
The patient returned for an outpatient appointment approximately 1 week later. Medical records indicated that the patient’s skin eruption had resolved. Tests for antithymoglobulin antibodies at this visit were negative. The antithymoglobulin antibody enzyme-linked immunosorbent assay has a diagnostic sensitivity of 86%12 and large interlaboratory variability.13 Given the presence of other features of serum sickness, a false-negative result was considered by dermatology. Nonetheless, one must consider other differential diagnoses, including a simple cutaneous adverse drug eruption or viral exanthem that might have in fact been causative.
Comment
We present an atypical case of possible serum sickness in a heart transplant recipient following thymoglobulin treatment of acute cellular rejection of the cardiac allograft. Serum sickness is a clinical diagnosis supported by laboratory data. Some authors have suggested major and minor diagnostic criteria to aid with the diagnosis.7 Major diagnostic criteria include onset more than 7 days after the initial thymoglobulin administration, persistent high fevers (temperature, >38.4°C), persistent arthritis/arthralgia, and positive heterologous antibodies on enzyme-linked immunosorbent assay. Minor diagnostic criteria include rash, acute renal failure, trismus, and low serum complement (C3 and C4).
The variable cutaneous presentations of serum sickness are important to recognize in the process of making the correct diagnosis. Rash is frequently reported in serum sickness, with some studies displaying rates of up to 93%.4,14 The skin findings are most frequently described as urticarial or serpiginous macular lesions.3 Other variations of the eruption exist, and morbilliform eruptions or a combination of morbilliform and urticarial eruptions have been reported.3 It is important to judge cutaneous eruptions of serum sickness within the context of the potential cytopenia in a patient being treated with ATG. As such, purpuric eruptions have been attributed to serum sickness in thrombocytopenic patients receiving ATG for bone marrow failure.14
Usually, cutaneous eruptions of serum sickness initially are identified in the groin, axilla, and periumbilical region, and then they proceed to include the trunk and extremities. Erythema of the palms and soles frequently is described as well as a linear accentuation of the rash along the lateral and medial borders of the feet and hands at the margin of the plantar or palmar skin, respectively.14 The mucous membranes frequently are spared in serum sickness.
Despite the lack of evidence-based guidelines, case series and literature reviews have suggested a treatment regimen for serum sickness,7,15-18 calling for immediate withdrawal of the offending agent. Antihistamines may be added to control pruritus and rash. Patients with high fever, a progressive rash, or severe arthralgia have benefited from short courses of oral16,18 or intravenous7,17 glucocorticoids. The extent of the eruption in our patient was concerning, particularly because he was already receiving systemic corticosteroids in conjunction with other immunosuppressives, which may have explained his lack of fever.
Because our patient satisfied some diagnostic criteria for serum sickness and failed to satisfy others, our team was faced with the challenge of balancing the risks of possible serum sickness with the risks of the potential for progressive cardiac rejection from the withdrawal of thymoglobulin.7 There is some evidence in the literature for the use of therapeutic plasma exchange (TPE) for the treatment of serum sickness if the offending agent could not be discontinued. Tanriover et al19 presented a case series of 5 renal transplant recipients treated with thymoglobulin who developed serum sickness. The diagnosis of serum sickness was made clinically and augmented by the presence of antiheterologous antibodies. All 5 patients had persistent symptoms of serum sickness despite 2 days of glucocorticoid treatment. Interestingly, 3 patients had complete resolution of all symptoms after a single TPE treatment, and 2 patients achieved resolution of fever and arthritis after 2 consecutive days of TPE treatments.19 Because plasmapheresis is used to treat cardiac allograft rejection in patients showing signs of heart failure,11 the employment of TPE in these patients may have dual beneficial effects of concurrently treating serum sickness and allograft rejection.
Given the patient’s noncompliance and leaving the hospital against medical advice, a full workup was not able to be pursued in this case, though fortunately the eruption and his other symptoms had resolved by the time he was seen for outpatient follow-up 1 week later. Noncompliance with immunosuppressive therapy is a considerable risk factor for morbidity and mortality following heart transplantation. These patients have more transplant coronary artery disease and substantially shorter clinical event-free time.20 Our patient demonstrates the need for proactive compliance-enhancing interventions in heart transplant patients who experience allograft rejection.
Serum sickness was first described by von Pirquet and Schick1 as a constellation of signs and symptoms displayed in patients receiving equine serum as an antitoxin for the treatment of scarlet fever and diphtheria. Serum sickness is an immune complex–mediated hypersensitivity reaction that can be clinically diagnosed in patients who present with fever, rash, and polyarthralgia or polyarthritis following exposure to heterologous serum proteins.2,3 Symptom onset typically occurs within 1 to 2 weeks of first exposure to the serum, and resolution frequently occurs with discontinuation of the offending agent. Other symptoms may include malaise, gastrointestinal tract concerns, headache, blurred vision, or lymphadenopathy.4 Proteinuria, hematuria, and a transient decrease in creatinine clearance also have been reported in serum sickness.4
Serum sickness is caused by a type III immune complex–mediated hypersensitivity reaction to heterologous rabbit or equine serum proteins. Nonhuman proteins present in antithymocyte globulin (ATG) stimulate the production of IgG, IgM, IgA, and IgE antibodies.2-4 If the resultant immune complexes overwhelm the mononuclear phagocyte system, these complexes are deposited in blood vessels and tissues, which leads to complement activation and the production of complement fragments such as C3a and C5a.5 C3a is an anaphylatoxin that causes mast cell degranulation and the consequent formation of urticarial lesions. C5a is a neutrophil chemoattractant that promotes inflammation at the site of complement deposition.
Serum sickness–like reactions may occur days to weeks following administration of certain drugs, such as cefaclor or penicillin. Although the symptoms and timing of serum sickness–like reactions are similar to serum sickness, they are not caused by an immune complex–mediated mechanism and are believed to be secondary to an idiosyncratic delayed drug reaction.6
Thymoglobulin, a type of ATG, is a polyclonal antibody generated in rabbits that targets numerous human epitopes, including cell surface markers on T cells (CD2, CD3, CD4, CD8), B cells (CD21, CD19, CD40), and adhesion molecules (CD6, CD25, CD44, CD45, and the integrin LFA-1 [lymphocyte function-associated antigen-1]).7,8 Thymoglobulin has proven efficacy in the setting of cardiac transplantation.9-11 Although calcineurin inhibitors form the foundation in the armamentarium of immunosuppressive agents in cardiac transplantation, their nephrotoxicity has limited their unrestrained use in patients.9 By delaying the need for calcineurin inhibitors, thymoglobulin preserves greater renal function without increasing the risk for acute rejection.9,10 Akin to its use in the patient presented in this case report, thymoglobulin also is used in the treatment of acute cellular rejection in heart transplant recipients with signs of heart failure.11
Case Report
A 35-year-old man with a history of familial cardiomyopathy who underwent orthotopic heart transplantation presented with grade 3R acute cellular rejection. The patient’s immunosuppressive regimen consisted of thymoglobulin 150 mg once daily, tacrolimus 2.5 mg twice daily, hydrocortisone 100 mg once daily, and mycophenolate mofetil 1000 mg twice daily. On day 7 of thymoglobulin treatment, the dermatology department was consulted to evaluate a pruritic eruption. The patient reported that he noticed redness of the palms and soles, as well as redness accentuated in the axilla, groin, and other skin creases 2 days prior. The patient also reported symmetric bilateral hand pain that had started 1 day following rash onset. He denied fever and remained afebrile throughout his hospitalization.
On physical examination, the patient displayed a blanching, erythematous, edematous, evanescent macular rash with some areas of wheal formation symmetrically distributed in the bilateral axillae, inframammary folds, and groin (Figure, A and B). The palms and soles were tender with diffuse blanching erythema. The eruption was accentuated at the lateral and medial borders of both feet (Figure, C). There was concern that the patient may have a form of serum sickness with a blunted incomplete response due to his concomitant use of immunosuppressive agents. Shortly after evaluation, the patient left the hospital against medical advice before the recommended evaluation and systemic workup could be implemented.
The patient returned for an outpatient appointment approximately 1 week later. Medical records indicated that the patient’s skin eruption had resolved. Tests for antithymoglobulin antibodies at this visit were negative. The antithymoglobulin antibody enzyme-linked immunosorbent assay has a diagnostic sensitivity of 86%12 and large interlaboratory variability.13 Given the presence of other features of serum sickness, a false-negative result was considered by dermatology. Nonetheless, one must consider other differential diagnoses, including a simple cutaneous adverse drug eruption or viral exanthem that might have in fact been causative.
Comment
We present an atypical case of possible serum sickness in a heart transplant recipient following thymoglobulin treatment of acute cellular rejection of the cardiac allograft. Serum sickness is a clinical diagnosis supported by laboratory data. Some authors have suggested major and minor diagnostic criteria to aid with the diagnosis.7 Major diagnostic criteria include onset more than 7 days after the initial thymoglobulin administration, persistent high fevers (temperature, >38.4°C), persistent arthritis/arthralgia, and positive heterologous antibodies on enzyme-linked immunosorbent assay. Minor diagnostic criteria include rash, acute renal failure, trismus, and low serum complement (C3 and C4).
The variable cutaneous presentations of serum sickness are important to recognize in the process of making the correct diagnosis. Rash is frequently reported in serum sickness, with some studies displaying rates of up to 93%.4,14 The skin findings are most frequently described as urticarial or serpiginous macular lesions.3 Other variations of the eruption exist, and morbilliform eruptions or a combination of morbilliform and urticarial eruptions have been reported.3 It is important to judge cutaneous eruptions of serum sickness within the context of the potential cytopenia in a patient being treated with ATG. As such, purpuric eruptions have been attributed to serum sickness in thrombocytopenic patients receiving ATG for bone marrow failure.14
Usually, cutaneous eruptions of serum sickness initially are identified in the groin, axilla, and periumbilical region, and then they proceed to include the trunk and extremities. Erythema of the palms and soles frequently is described as well as a linear accentuation of the rash along the lateral and medial borders of the feet and hands at the margin of the plantar or palmar skin, respectively.14 The mucous membranes frequently are spared in serum sickness.
Despite the lack of evidence-based guidelines, case series and literature reviews have suggested a treatment regimen for serum sickness,7,15-18 calling for immediate withdrawal of the offending agent. Antihistamines may be added to control pruritus and rash. Patients with high fever, a progressive rash, or severe arthralgia have benefited from short courses of oral16,18 or intravenous7,17 glucocorticoids. The extent of the eruption in our patient was concerning, particularly because he was already receiving systemic corticosteroids in conjunction with other immunosuppressives, which may have explained his lack of fever.
Because our patient satisfied some diagnostic criteria for serum sickness and failed to satisfy others, our team was faced with the challenge of balancing the risks of possible serum sickness with the risks of the potential for progressive cardiac rejection from the withdrawal of thymoglobulin.7 There is some evidence in the literature for the use of therapeutic plasma exchange (TPE) for the treatment of serum sickness if the offending agent could not be discontinued. Tanriover et al19 presented a case series of 5 renal transplant recipients treated with thymoglobulin who developed serum sickness. The diagnosis of serum sickness was made clinically and augmented by the presence of antiheterologous antibodies. All 5 patients had persistent symptoms of serum sickness despite 2 days of glucocorticoid treatment. Interestingly, 3 patients had complete resolution of all symptoms after a single TPE treatment, and 2 patients achieved resolution of fever and arthritis after 2 consecutive days of TPE treatments.19 Because plasmapheresis is used to treat cardiac allograft rejection in patients showing signs of heart failure,11 the employment of TPE in these patients may have dual beneficial effects of concurrently treating serum sickness and allograft rejection.
Given the patient’s noncompliance and leaving the hospital against medical advice, a full workup was not able to be pursued in this case, though fortunately the eruption and his other symptoms had resolved by the time he was seen for outpatient follow-up 1 week later. Noncompliance with immunosuppressive therapy is a considerable risk factor for morbidity and mortality following heart transplantation. These patients have more transplant coronary artery disease and substantially shorter clinical event-free time.20 Our patient demonstrates the need for proactive compliance-enhancing interventions in heart transplant patients who experience allograft rejection.
- von Pirquet C, Schick B. Serum Sickness. Schick B, trans-ed. Baltimore, MD; Williams & Wilkins; 1951.
- Vincent C, Revillard JP. Antibody response to horse gamma-globulin in recipients of renal allografts: relationship with transplant crises and transplant survival. Transplantation. 1977;24:141-147.
- Lawley TJ, Bielory L, Gascon P, et al. A prospective clinical and immunologic analysis of patients with serum sickness. N Engl J Med. 1984;311:1407-1413.
- Bielory L, Gascon P, Lawley TJ, et al. Human serum sickness: a prospective analysis of 35 patients treated with equine anti-thymocyte globulin for bone marrow failure. Medicine (Baltimore). 1988;67:40-57.
- Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276-J286.
- Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587-1591.
- Lundquist AL, Chari RS, Wood JH, et al. Serum sickness following rabbit antithymocyte-globulin induction in a liver transplant recipient: case report and literature review. Liver Transpl. 2007;13:647-650.
- Bourdage JS, Hamlin DM. Comparative polyclonal antithymocyte globulin and antilymphocyte/antilymphoblast globulin anti-CD antigen analysis by flow cytometry. Transplantation. 1995;59:1194-1200.
- Zuckermann AO, Aliabadi AZ. Calcineurin-inhibitor minimization protocols in heart transplantation. Transpl Int. 2009;22:78-89.
- Cantarovich M, Giannetti N, Barkun J, et al. Antithymocyte globulin induction allows a prolonged delay in the initiation of cyclosporine in heart transplant patients with postoperative renal dysfunction. Transplantation. 2004;78:779-781.
- Patel JK, Kittleson M, Kobashigawa JA. Cardiac allograft rejection. Surgeon. 2010;9:160-167.
- Tatum AH, Bollinger RR, Sanfilippo F. Rapid serologic diagnosis of serum sickness from antithymocyte globulin therapy using enzyme immunoassay. Transplantation. 1984;38:582-586.
- Kimball JA, Pescovitz MD, Book BK, et al. Reduced human IgG anti-ATGAM antibody formation in renal transplant recipients receiving mycophenolate mofetil. Transplantation. 1995;60:1379-1383.
- Bielory L, Yancey KB, Young NS, et al. Cutaneous manifestations of serum sickness in patients receiving antithymocyte globulin. J Am Acad Dermatol. 1985;13:411-417.
- Joubert GI, Hadad K, Matsui D, et al. Selection of treatment of cefaclor-associated urticarial, serum sickness-like reactions and erythema multiforme by emergency pediatricians: lack of a uniform standard of care. Can J Clin Pharmacol. 1999;6:197-201.
- Clark BM, Kotti GH, Shah AD, et al. Severe serum sickness reaction to oral and intramuscular penicillin. Pharmacotherapy. 2006;26:705-708.
- Finger E, Scheinberg M. Development of serum sickness-like symptoms after rituximab infusion in two patients with severe hypergammaglobulinemia. J Clin Rheumatol. 2007;13:94-95.
- Tatum AJ, Ditto AM, Patterson R. Severe serum sickness-like reaction to oral penicillin drugs: three case reports. Ann Allergy Asthma Immunol. 2001;86:330-334.
- Tanriover B, Chuang P, Fishbach B, et al. Polyclonal antibody-induced serum sickness in renal transplant recipients: treatment with therapeutic plasma exchange. Transplantation. 2005;80:279-281.
- Dobbels F, De Geest S, van Cleemput J, et al. Effect of late medication non-compliance on outcome after heart transplantation: a 5-year follow-up. J Heart Lung Transplant. 2004;23:1245-1251.
- von Pirquet C, Schick B. Serum Sickness. Schick B, trans-ed. Baltimore, MD; Williams & Wilkins; 1951.
- Vincent C, Revillard JP. Antibody response to horse gamma-globulin in recipients of renal allografts: relationship with transplant crises and transplant survival. Transplantation. 1977;24:141-147.
- Lawley TJ, Bielory L, Gascon P, et al. A prospective clinical and immunologic analysis of patients with serum sickness. N Engl J Med. 1984;311:1407-1413.
- Bielory L, Gascon P, Lawley TJ, et al. Human serum sickness: a prospective analysis of 35 patients treated with equine anti-thymocyte globulin for bone marrow failure. Medicine (Baltimore). 1988;67:40-57.
- Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276-J286.
- Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587-1591.
- Lundquist AL, Chari RS, Wood JH, et al. Serum sickness following rabbit antithymocyte-globulin induction in a liver transplant recipient: case report and literature review. Liver Transpl. 2007;13:647-650.
- Bourdage JS, Hamlin DM. Comparative polyclonal antithymocyte globulin and antilymphocyte/antilymphoblast globulin anti-CD antigen analysis by flow cytometry. Transplantation. 1995;59:1194-1200.
- Zuckermann AO, Aliabadi AZ. Calcineurin-inhibitor minimization protocols in heart transplantation. Transpl Int. 2009;22:78-89.
- Cantarovich M, Giannetti N, Barkun J, et al. Antithymocyte globulin induction allows a prolonged delay in the initiation of cyclosporine in heart transplant patients with postoperative renal dysfunction. Transplantation. 2004;78:779-781.
- Patel JK, Kittleson M, Kobashigawa JA. Cardiac allograft rejection. Surgeon. 2010;9:160-167.
- Tatum AH, Bollinger RR, Sanfilippo F. Rapid serologic diagnosis of serum sickness from antithymocyte globulin therapy using enzyme immunoassay. Transplantation. 1984;38:582-586.
- Kimball JA, Pescovitz MD, Book BK, et al. Reduced human IgG anti-ATGAM antibody formation in renal transplant recipients receiving mycophenolate mofetil. Transplantation. 1995;60:1379-1383.
- Bielory L, Yancey KB, Young NS, et al. Cutaneous manifestations of serum sickness in patients receiving antithymocyte globulin. J Am Acad Dermatol. 1985;13:411-417.
- Joubert GI, Hadad K, Matsui D, et al. Selection of treatment of cefaclor-associated urticarial, serum sickness-like reactions and erythema multiforme by emergency pediatricians: lack of a uniform standard of care. Can J Clin Pharmacol. 1999;6:197-201.
- Clark BM, Kotti GH, Shah AD, et al. Severe serum sickness reaction to oral and intramuscular penicillin. Pharmacotherapy. 2006;26:705-708.
- Finger E, Scheinberg M. Development of serum sickness-like symptoms after rituximab infusion in two patients with severe hypergammaglobulinemia. J Clin Rheumatol. 2007;13:94-95.
- Tatum AJ, Ditto AM, Patterson R. Severe serum sickness-like reaction to oral penicillin drugs: three case reports. Ann Allergy Asthma Immunol. 2001;86:330-334.
- Tanriover B, Chuang P, Fishbach B, et al. Polyclonal antibody-induced serum sickness in renal transplant recipients: treatment with therapeutic plasma exchange. Transplantation. 2005;80:279-281.
- Dobbels F, De Geest S, van Cleemput J, et al. Effect of late medication non-compliance on outcome after heart transplantation: a 5-year follow-up. J Heart Lung Transplant. 2004;23:1245-1251.
Practice Points
- Serum sickness can be seen in patients treated with thymoglobulin to prevent transplant rejection.
- Serum sickness can display multiple cutaneous manifestation, thus making it an important entity for dermatologists.
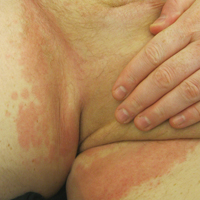
Photosensitive Atopic Dermatitis Exacerbated by UVB Exposure
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
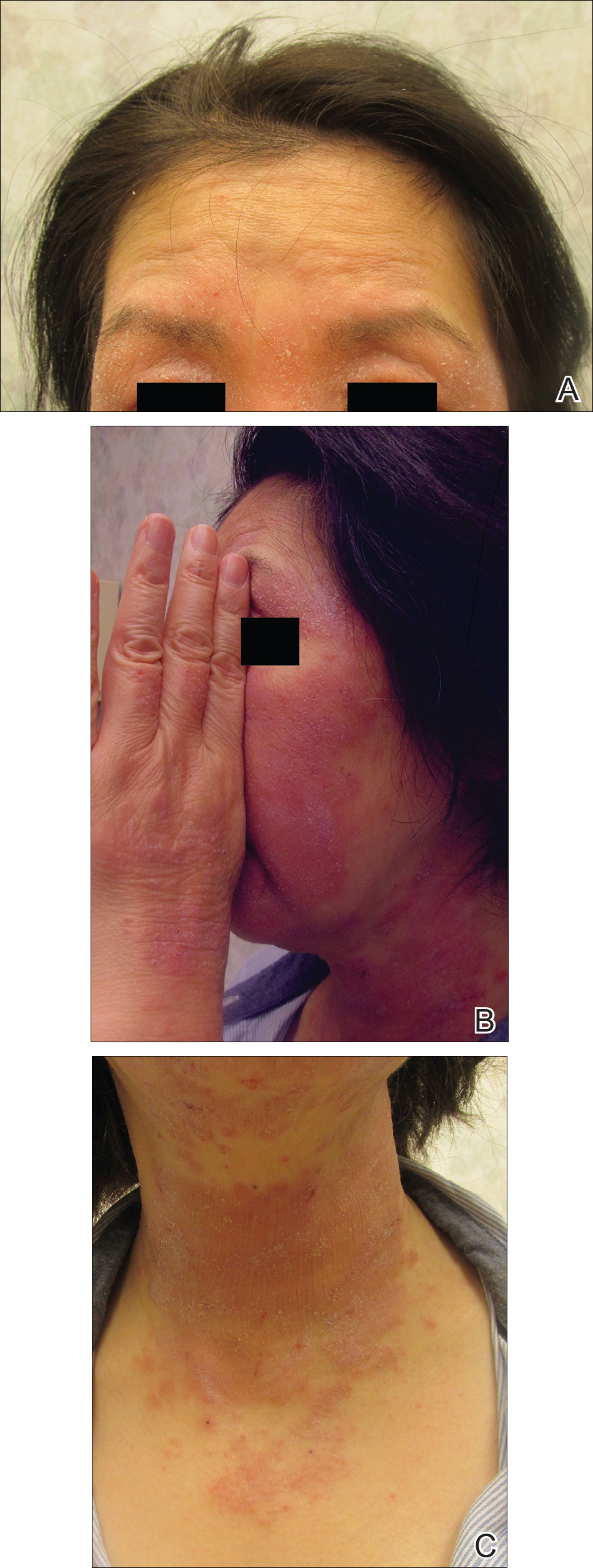
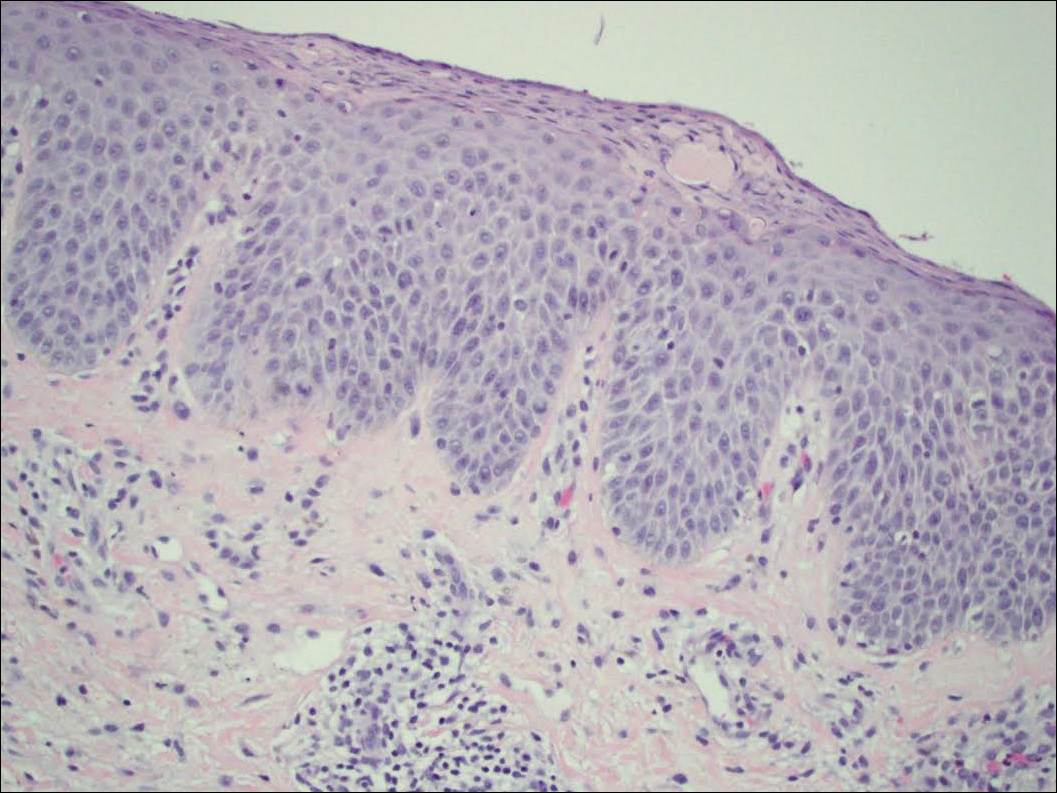
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
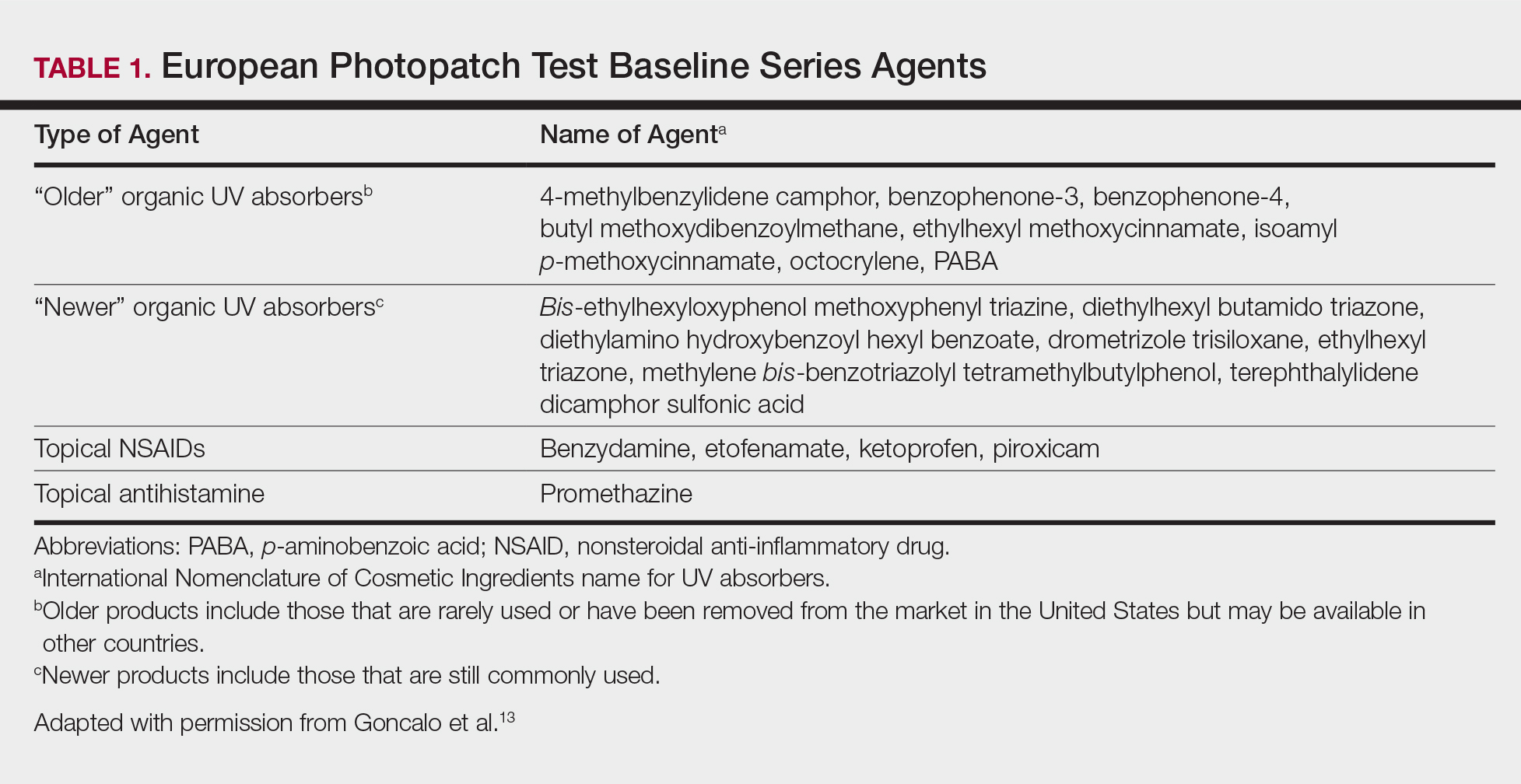
Differential Diagnosis
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
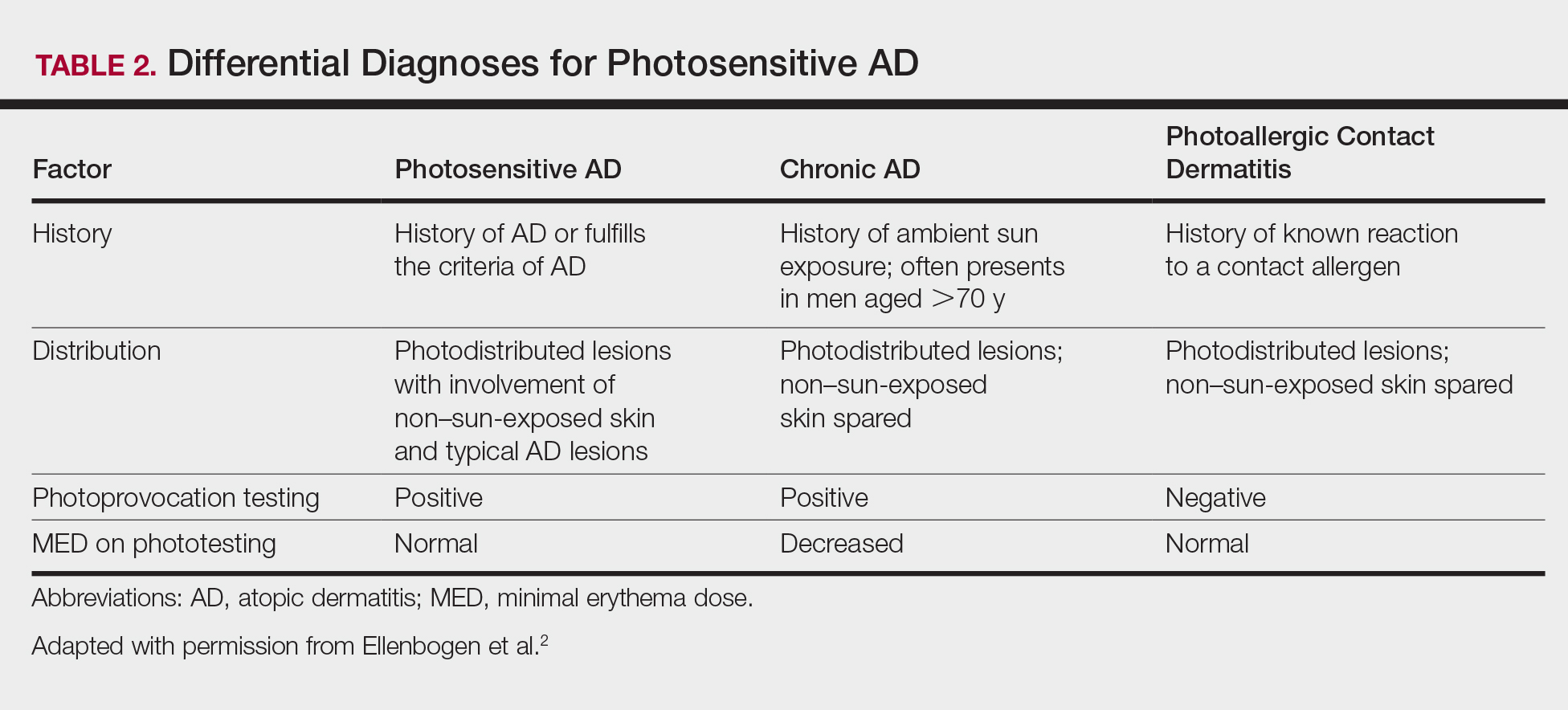
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
Differential Diagnosis
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
Differential Diagnosis
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
Practice Points
- Photosensitive atopic dermatitis (AD) is rare but should be considered in patients with uncontrolled AD with a rash on sun-exposed skin.
- A thorough history and physical examination of these patients can provide the necessary clues for further workup.
- Phototesting should be performed to confirm the diagnosis and evaluate the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response.
- Photoprovocation and photopatch testing also can be useful to confirm the diagnosis.
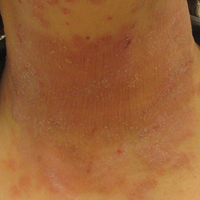
Immigrant with stomach pain, distension, nausea, and fever • Dx?
THE CASE
A 34-year-old Eritrean man presented to the emergency department with complaints of diffuse abdominal pain and distention. He had emigrated to the United States 3 months earlier, following 5 years in a refugee camp in Ethiopia. Two weeks earlier, the patient sought care at his primary care clinic and was diagnosed with post-operative urinary retention and constipation following a recent hemorrhoidectomy. A Foley catheter was inserted and provided a short period of relief.
Following the visit, however, his abdominal pain worsened. He also experienced increasing abdominal distention, a declining appetite, and persistent nausea. The patient said that he was unable to urinate and had not had a bowel movement in 6 days. He also described fevers, drenching night sweats, chills, and a 4-kg weight loss over 2 months.
On physical examination, the patient had a wasted appearance. He was afebrile, alert, and oriented, but anxious and writhing in pain. An abdominal examination revealed some distention, generalized guarding, and tenderness. There was dullness to percussion in all regions without rebound, and no caput medusa was noted. The remainder of the physical examination was unremarkable.
Pertinent laboratory values included negative screens for human immunodeficiency virus (HIV) 1 and 2, and a purified protein derivative test that produced 10 mm of induration at 48 hours. An interferon-gamma release assay was not performed following these results. A computerized tomography (CT) scan of the abdomen and pelvis with intravenous and oral contrast revealed thickening of the peritoneal lining with infiltration of the mesenteric fat and large loculated fluid collections in the abdominal cavity (FIGURE). A CT scan of the patient’s lungs showed some mild atelectasis with left-sided effusion.
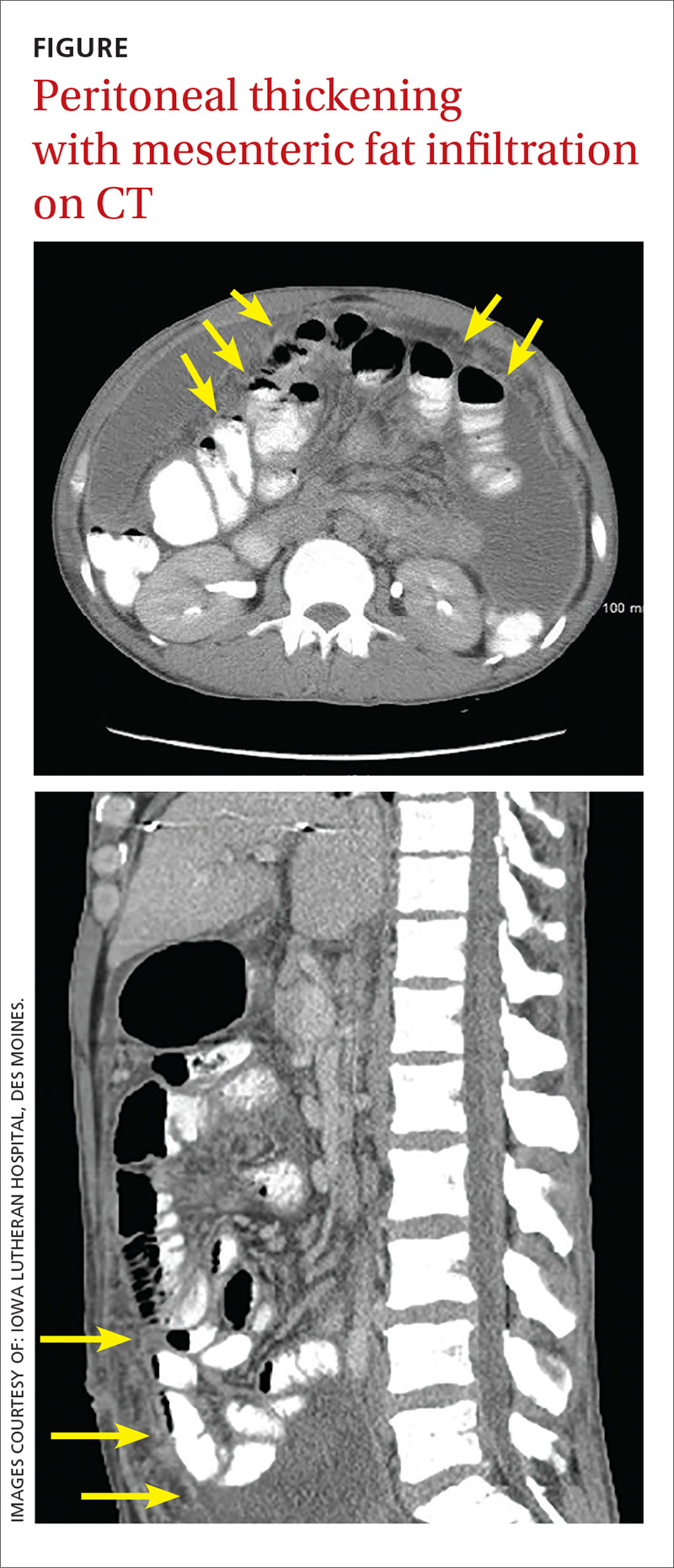
After hospital admission, the patient spiked fevers as high as 103.3° F and developed progressively worsening ascites. An ultrasound-guided paracentesis was performed, during which almost 2 liters of yellow, hazy fluid was removed. Fluid and blood cultures were negative.
THE DIAGNOSIS
With a high clinical suspicion for tuberculosis (TB) peritonitis, we requested a surgical consultation and a peritoneal biopsy was performed. The patient was started on ethambutol, isoniazid, pyrazinamide, pyridoxine, and rifampin while the biopsy results were pending.
Pathology subsequently confirmed a diagnosis of TB peritonitis, reporting dense fibroconnective tissue with areas of chronic inflammation and occasional accumulations of histiocytes with multinucleated giant cells showing granulomatous inflammation. An acid-fast (AF) bacilli stain for Mycobacteria showed a single curved bacillus compatible with Mycobacterium tuberculosis.
The patient was discharged following a 3-week hospital stay. At his follow-up visit several weeks later, the patient reported marked improvement and increasing exercise tolerance. He had gained weight, and the abdominal distention and tenderness had resolved.
DISCUSSION
Worldwide, TB is one of the top 10 causes of death. The World Health Organization estimates that there were 1.4 million TB deaths globally in 2015.1 And while rates of TB are decreasing in the United States, there was a resurgence from 1985 to 1992.2 This was attributable to the HIV/acquired immunodeficiency syndrome epidemic, increased immigration from countries endemic for TB, and deterioration of the TB public health infrastructure.3
Transmission. M tuberculosis is a rod-shaped, nonspore-forming AF bacillus that typically infects the lungs, but may infect other areas of the body. Transmission typically occurs via airborne spread of droplets from an infected individual. Possible other methods of disease dissemination include ingestion of infected sputum, hematogenous spread from active pulmonary TB, or ingestion of contaminated milk or food.
M tuberculosis elicits a proinflammatory phase, which facilitates the formation of a granuloma within the host tissues. The host’s immune response to M tuberculosis plays a role in the risk of developing this type of TB.3
TB presentation is classified as pulmonary, extrapulmonary, or both. Clinicians are generally attentive to the classic symptoms of pulmonary TB: cough, weight loss, night sweats, and fever. Presentation of extrapulmonary TB, however, may vary.4
According to one study, the most common presenting symptoms for peritoneal TB are weight loss, abdominal pain, and/or fever, all of which our patient experienced.5 In addition, our patient was an immigrant from Africa, and black patients have been shown to have a significantly higher incidence of extrapulmonary TB than their nonblack counterparts.6 Although our patient was HIV-negative, a recent meta-analysis confirmed the strong association between extrapulmonary TB and HIV, emphasizing the importance of including HIV screens in the standard work-up for TB.7
Other symptoms may include microcytosis, anemia, thrombocytosis, and an elevated erythrocyte sedimentation rate. Although a chest x-ray is often negative, advanced imaging, such as CT or magnetic resonance imaging, is often abnormal and may point to the diagnosis.5
Treatment of extrapulmonary TB is generally the same as that for pulmonary TB and, interestingly, the incidence of multi-drug resistant extrapulmonary TB is not necessarily higher than it is for pulmonary TB (<1% vs 1.6%).3,7 In light of this, a standard regimen—like the one our patient received—is generally utilized for 6 to 9 months. Nonetheless, resistance testing should still be performed.3,4
THE TAKEAWAY
While considered uncommon, more than 20% of TB cases in the United States are extrapulmonary (the most common form is TB lymphadenitis).7,8 It is imperative to identify appropriate risk factors, including associated comorbidities, patient characteristics, and population/endemic differences in immigrant populations.
In this case, although the symptom combination of persistent abdominal pain, fever, and weight loss may not trigger suspicion of a TB diagnosis in isolation, combining the symptoms with knowledge of the patient’s immigration status should at least raise an eyebrow. Given their nonpulmonary symptoms, many of these patients will not present to pulmonologists, making diagnosis particularly relevant to primary care.
1. World Health Organization. Global tuberculosis report 2016. Available at: http://www.who.int/tb/publications/global_report/gtbr2016_executive_summary.pdf?ua=1. Accessed August 22, 2017.
2. Peto HM, Pratt RH, Harrington TA, et al. Epidemiology of extrapulmonary tuberculosis in the United States, 1993-2006. Clin Infect Dis. 2009;49:1350-1357.
3. Centers for Disease Control and Prevention. Reported Tuberculosis in the United States, 2006. Available at: http://digitallibrary.utah.gov/awweb/awarchive?type=file&item=56908. Accessed August 3, 2017.
4. World Health Organization. Global tuberculosis report 2012. Available at: http://apps.who.int/medicinedocs/documents/s19908en/s19908en.pdf. Accessed July 27, 2017.
5. Ramesh J, Banait GS, Ormerod LP. Abdominal tuberculosis in a district general hospital: a retrospective review of 86 cases. QJM. 2008;101:189-195.
6. Fiske CT, Griffin MR, Erin H, et al. Black race, sex and extrapulmonary tuberculosis risk: an observational study. BMC Infect Dis. 2010;10:16.
7. Naing C, Mak JW, Maung M, et al. Meta-analysis: the association between HIV infection and extrapulmonary tuberculosis. Lung. 2013;191:27-34.
8. Neelakantan S, Nair PP, Emmanuel RV, et al. Diversities in presentations of extrapulmonary tuberculosis. BMJ Case Rep. 2013.
THE CASE
A 34-year-old Eritrean man presented to the emergency department with complaints of diffuse abdominal pain and distention. He had emigrated to the United States 3 months earlier, following 5 years in a refugee camp in Ethiopia. Two weeks earlier, the patient sought care at his primary care clinic and was diagnosed with post-operative urinary retention and constipation following a recent hemorrhoidectomy. A Foley catheter was inserted and provided a short period of relief.
Following the visit, however, his abdominal pain worsened. He also experienced increasing abdominal distention, a declining appetite, and persistent nausea. The patient said that he was unable to urinate and had not had a bowel movement in 6 days. He also described fevers, drenching night sweats, chills, and a 4-kg weight loss over 2 months.
On physical examination, the patient had a wasted appearance. He was afebrile, alert, and oriented, but anxious and writhing in pain. An abdominal examination revealed some distention, generalized guarding, and tenderness. There was dullness to percussion in all regions without rebound, and no caput medusa was noted. The remainder of the physical examination was unremarkable.
Pertinent laboratory values included negative screens for human immunodeficiency virus (HIV) 1 and 2, and a purified protein derivative test that produced 10 mm of induration at 48 hours. An interferon-gamma release assay was not performed following these results. A computerized tomography (CT) scan of the abdomen and pelvis with intravenous and oral contrast revealed thickening of the peritoneal lining with infiltration of the mesenteric fat and large loculated fluid collections in the abdominal cavity (FIGURE). A CT scan of the patient’s lungs showed some mild atelectasis with left-sided effusion.
After hospital admission, the patient spiked fevers as high as 103.3° F and developed progressively worsening ascites. An ultrasound-guided paracentesis was performed, during which almost 2 liters of yellow, hazy fluid was removed. Fluid and blood cultures were negative.
THE DIAGNOSIS
With a high clinical suspicion for tuberculosis (TB) peritonitis, we requested a surgical consultation and a peritoneal biopsy was performed. The patient was started on ethambutol, isoniazid, pyrazinamide, pyridoxine, and rifampin while the biopsy results were pending.
Pathology subsequently confirmed a diagnosis of TB peritonitis, reporting dense fibroconnective tissue with areas of chronic inflammation and occasional accumulations of histiocytes with multinucleated giant cells showing granulomatous inflammation. An acid-fast (AF) bacilli stain for Mycobacteria showed a single curved bacillus compatible with Mycobacterium tuberculosis.
The patient was discharged following a 3-week hospital stay. At his follow-up visit several weeks later, the patient reported marked improvement and increasing exercise tolerance. He had gained weight, and the abdominal distention and tenderness had resolved.
DISCUSSION
Worldwide, TB is one of the top 10 causes of death. The World Health Organization estimates that there were 1.4 million TB deaths globally in 2015.1 And while rates of TB are decreasing in the United States, there was a resurgence from 1985 to 1992.2 This was attributable to the HIV/acquired immunodeficiency syndrome epidemic, increased immigration from countries endemic for TB, and deterioration of the TB public health infrastructure.3
Transmission. M tuberculosis is a rod-shaped, nonspore-forming AF bacillus that typically infects the lungs, but may infect other areas of the body. Transmission typically occurs via airborne spread of droplets from an infected individual. Possible other methods of disease dissemination include ingestion of infected sputum, hematogenous spread from active pulmonary TB, or ingestion of contaminated milk or food.
M tuberculosis elicits a proinflammatory phase, which facilitates the formation of a granuloma within the host tissues. The host’s immune response to M tuberculosis plays a role in the risk of developing this type of TB.3
TB presentation is classified as pulmonary, extrapulmonary, or both. Clinicians are generally attentive to the classic symptoms of pulmonary TB: cough, weight loss, night sweats, and fever. Presentation of extrapulmonary TB, however, may vary.4
According to one study, the most common presenting symptoms for peritoneal TB are weight loss, abdominal pain, and/or fever, all of which our patient experienced.5 In addition, our patient was an immigrant from Africa, and black patients have been shown to have a significantly higher incidence of extrapulmonary TB than their nonblack counterparts.6 Although our patient was HIV-negative, a recent meta-analysis confirmed the strong association between extrapulmonary TB and HIV, emphasizing the importance of including HIV screens in the standard work-up for TB.7
Other symptoms may include microcytosis, anemia, thrombocytosis, and an elevated erythrocyte sedimentation rate. Although a chest x-ray is often negative, advanced imaging, such as CT or magnetic resonance imaging, is often abnormal and may point to the diagnosis.5
Treatment of extrapulmonary TB is generally the same as that for pulmonary TB and, interestingly, the incidence of multi-drug resistant extrapulmonary TB is not necessarily higher than it is for pulmonary TB (<1% vs 1.6%).3,7 In light of this, a standard regimen—like the one our patient received—is generally utilized for 6 to 9 months. Nonetheless, resistance testing should still be performed.3,4
THE TAKEAWAY
While considered uncommon, more than 20% of TB cases in the United States are extrapulmonary (the most common form is TB lymphadenitis).7,8 It is imperative to identify appropriate risk factors, including associated comorbidities, patient characteristics, and population/endemic differences in immigrant populations.
In this case, although the symptom combination of persistent abdominal pain, fever, and weight loss may not trigger suspicion of a TB diagnosis in isolation, combining the symptoms with knowledge of the patient’s immigration status should at least raise an eyebrow. Given their nonpulmonary symptoms, many of these patients will not present to pulmonologists, making diagnosis particularly relevant to primary care.
THE CASE
A 34-year-old Eritrean man presented to the emergency department with complaints of diffuse abdominal pain and distention. He had emigrated to the United States 3 months earlier, following 5 years in a refugee camp in Ethiopia. Two weeks earlier, the patient sought care at his primary care clinic and was diagnosed with post-operative urinary retention and constipation following a recent hemorrhoidectomy. A Foley catheter was inserted and provided a short period of relief.
Following the visit, however, his abdominal pain worsened. He also experienced increasing abdominal distention, a declining appetite, and persistent nausea. The patient said that he was unable to urinate and had not had a bowel movement in 6 days. He also described fevers, drenching night sweats, chills, and a 4-kg weight loss over 2 months.
On physical examination, the patient had a wasted appearance. He was afebrile, alert, and oriented, but anxious and writhing in pain. An abdominal examination revealed some distention, generalized guarding, and tenderness. There was dullness to percussion in all regions without rebound, and no caput medusa was noted. The remainder of the physical examination was unremarkable.
Pertinent laboratory values included negative screens for human immunodeficiency virus (HIV) 1 and 2, and a purified protein derivative test that produced 10 mm of induration at 48 hours. An interferon-gamma release assay was not performed following these results. A computerized tomography (CT) scan of the abdomen and pelvis with intravenous and oral contrast revealed thickening of the peritoneal lining with infiltration of the mesenteric fat and large loculated fluid collections in the abdominal cavity (FIGURE). A CT scan of the patient’s lungs showed some mild atelectasis with left-sided effusion.
After hospital admission, the patient spiked fevers as high as 103.3° F and developed progressively worsening ascites. An ultrasound-guided paracentesis was performed, during which almost 2 liters of yellow, hazy fluid was removed. Fluid and blood cultures were negative.
THE DIAGNOSIS
With a high clinical suspicion for tuberculosis (TB) peritonitis, we requested a surgical consultation and a peritoneal biopsy was performed. The patient was started on ethambutol, isoniazid, pyrazinamide, pyridoxine, and rifampin while the biopsy results were pending.
Pathology subsequently confirmed a diagnosis of TB peritonitis, reporting dense fibroconnective tissue with areas of chronic inflammation and occasional accumulations of histiocytes with multinucleated giant cells showing granulomatous inflammation. An acid-fast (AF) bacilli stain for Mycobacteria showed a single curved bacillus compatible with Mycobacterium tuberculosis.
The patient was discharged following a 3-week hospital stay. At his follow-up visit several weeks later, the patient reported marked improvement and increasing exercise tolerance. He had gained weight, and the abdominal distention and tenderness had resolved.
DISCUSSION
Worldwide, TB is one of the top 10 causes of death. The World Health Organization estimates that there were 1.4 million TB deaths globally in 2015.1 And while rates of TB are decreasing in the United States, there was a resurgence from 1985 to 1992.2 This was attributable to the HIV/acquired immunodeficiency syndrome epidemic, increased immigration from countries endemic for TB, and deterioration of the TB public health infrastructure.3
Transmission. M tuberculosis is a rod-shaped, nonspore-forming AF bacillus that typically infects the lungs, but may infect other areas of the body. Transmission typically occurs via airborne spread of droplets from an infected individual. Possible other methods of disease dissemination include ingestion of infected sputum, hematogenous spread from active pulmonary TB, or ingestion of contaminated milk or food.
M tuberculosis elicits a proinflammatory phase, which facilitates the formation of a granuloma within the host tissues. The host’s immune response to M tuberculosis plays a role in the risk of developing this type of TB.3
TB presentation is classified as pulmonary, extrapulmonary, or both. Clinicians are generally attentive to the classic symptoms of pulmonary TB: cough, weight loss, night sweats, and fever. Presentation of extrapulmonary TB, however, may vary.4
According to one study, the most common presenting symptoms for peritoneal TB are weight loss, abdominal pain, and/or fever, all of which our patient experienced.5 In addition, our patient was an immigrant from Africa, and black patients have been shown to have a significantly higher incidence of extrapulmonary TB than their nonblack counterparts.6 Although our patient was HIV-negative, a recent meta-analysis confirmed the strong association between extrapulmonary TB and HIV, emphasizing the importance of including HIV screens in the standard work-up for TB.7
Other symptoms may include microcytosis, anemia, thrombocytosis, and an elevated erythrocyte sedimentation rate. Although a chest x-ray is often negative, advanced imaging, such as CT or magnetic resonance imaging, is often abnormal and may point to the diagnosis.5
Treatment of extrapulmonary TB is generally the same as that for pulmonary TB and, interestingly, the incidence of multi-drug resistant extrapulmonary TB is not necessarily higher than it is for pulmonary TB (<1% vs 1.6%).3,7 In light of this, a standard regimen—like the one our patient received—is generally utilized for 6 to 9 months. Nonetheless, resistance testing should still be performed.3,4
THE TAKEAWAY
While considered uncommon, more than 20% of TB cases in the United States are extrapulmonary (the most common form is TB lymphadenitis).7,8 It is imperative to identify appropriate risk factors, including associated comorbidities, patient characteristics, and population/endemic differences in immigrant populations.
In this case, although the symptom combination of persistent abdominal pain, fever, and weight loss may not trigger suspicion of a TB diagnosis in isolation, combining the symptoms with knowledge of the patient’s immigration status should at least raise an eyebrow. Given their nonpulmonary symptoms, many of these patients will not present to pulmonologists, making diagnosis particularly relevant to primary care.
1. World Health Organization. Global tuberculosis report 2016. Available at: http://www.who.int/tb/publications/global_report/gtbr2016_executive_summary.pdf?ua=1. Accessed August 22, 2017.
2. Peto HM, Pratt RH, Harrington TA, et al. Epidemiology of extrapulmonary tuberculosis in the United States, 1993-2006. Clin Infect Dis. 2009;49:1350-1357.
3. Centers for Disease Control and Prevention. Reported Tuberculosis in the United States, 2006. Available at: http://digitallibrary.utah.gov/awweb/awarchive?type=file&item=56908. Accessed August 3, 2017.
4. World Health Organization. Global tuberculosis report 2012. Available at: http://apps.who.int/medicinedocs/documents/s19908en/s19908en.pdf. Accessed July 27, 2017.
5. Ramesh J, Banait GS, Ormerod LP. Abdominal tuberculosis in a district general hospital: a retrospective review of 86 cases. QJM. 2008;101:189-195.
6. Fiske CT, Griffin MR, Erin H, et al. Black race, sex and extrapulmonary tuberculosis risk: an observational study. BMC Infect Dis. 2010;10:16.
7. Naing C, Mak JW, Maung M, et al. Meta-analysis: the association between HIV infection and extrapulmonary tuberculosis. Lung. 2013;191:27-34.
8. Neelakantan S, Nair PP, Emmanuel RV, et al. Diversities in presentations of extrapulmonary tuberculosis. BMJ Case Rep. 2013.
1. World Health Organization. Global tuberculosis report 2016. Available at: http://www.who.int/tb/publications/global_report/gtbr2016_executive_summary.pdf?ua=1. Accessed August 22, 2017.
2. Peto HM, Pratt RH, Harrington TA, et al. Epidemiology of extrapulmonary tuberculosis in the United States, 1993-2006. Clin Infect Dis. 2009;49:1350-1357.
3. Centers for Disease Control and Prevention. Reported Tuberculosis in the United States, 2006. Available at: http://digitallibrary.utah.gov/awweb/awarchive?type=file&item=56908. Accessed August 3, 2017.
4. World Health Organization. Global tuberculosis report 2012. Available at: http://apps.who.int/medicinedocs/documents/s19908en/s19908en.pdf. Accessed July 27, 2017.
5. Ramesh J, Banait GS, Ormerod LP. Abdominal tuberculosis in a district general hospital: a retrospective review of 86 cases. QJM. 2008;101:189-195.
6. Fiske CT, Griffin MR, Erin H, et al. Black race, sex and extrapulmonary tuberculosis risk: an observational study. BMC Infect Dis. 2010;10:16.
7. Naing C, Mak JW, Maung M, et al. Meta-analysis: the association between HIV infection and extrapulmonary tuberculosis. Lung. 2013;191:27-34.
8. Neelakantan S, Nair PP, Emmanuel RV, et al. Diversities in presentations of extrapulmonary tuberculosis. BMJ Case Rep. 2013.
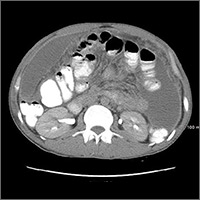
Postpartum Treatment of Metastatic Recurrent Giant Cell Tumor of Capitate Bone of Wrist
Take-Home Points
- GCT of bones of the wrist is rare. This article is the only report of a wrist GCT during pregnancy that we could identify.
- Routine treatment usually consists of surgical excision with local adjuvant, and in the wrist, often results in reduced wrist motion.
- GCT of the wrist is more aggressive than the more common locations in long bones, with higher local recurrence rates if treated with surgery alone.
- Diagnosis is often delayed for GCT of the wrist, due to insufficient imaging, which should include CT or MRI.
- For pregnant women with GCT, local adjuvant treatments can be used in addition to surgery. Following pregnancy, denosumab can be used systemically, and can be effective with metastatic or unresectable disease.
Giant cell tumor (GCT) of bone accounts for about 5% of primary bone tumors.1-3 Only 3% to 5% of GCTs occur in the hand.4,5 Wrist involvement, which is rare, most often involves the hamate bone.5-7 Capitate bone involvement is exceedingly rare.8-11 Although histologically benign, GCT can recur locally after treatment with curettage alone, and lung metastases are found in 2% to 5% of cases.2,12-14 Therefore, en bloc tumor excision is preferred in the setting of cortical erosion or soft-tissue involvement.1,4,8 Wrist joint motion is inevitably reduced, and bone graft donor-site morbidity is significant.6-8
In the unusual case reported here, GCT presented in the capitate bone and, after the patient became pregnant, recurred in the hamate and trapezoid bones with soft-tissue extension and lung metastases. The capitate was excised en bloc and reconstructed with an interposition of polymethylmethacrylate bone cement. Pulmonary metastases developed, and the GCT expanded to involve multiple carpal bones and the bases of the second through fourth metacarpals. A 10-month course of systemic chemotherapy with the RANK ligand (RANKL) inhibitor denosumab was started after the pregnancy. After this treatment, the patient underwent both tumor resection and reconstruction with autogenous bicortical iliac crest bone graft (ICBG) carefully designed to preserve range of motion and maintain the fingers in anatomical position. Treatment with denosumab was continued after surgery. Although this case offers no endpoint for postoperative chemotherapy with denosumab, preoperative treatment dramatically reduced the GCT and permitted limb-sparing reconstruction. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old right-handed woman with atraumatic swelling of the left wrist presented to an orthopedic surgeon at an outside facility. Physical examination revealed tender fullness on the dorsum of the wrist, slightly reduced range of motion and grip strength, and a neurovascularly intact wrist. The diagnosis was periarticular cyst, and the patient underwent physical therapy. Two years later, the swelling returned, tenderness was increasing, and symptoms did not resolve with cast immobilization. A radiograph showed a lytic lesion in the capitate bone (Figure 1).[[{"fid":"202332","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"1"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 1.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"1":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 1.","field_file_image_credit[und][0][value]":""}}}]]
GCT was diagnosed with percutaneous needle biopsy. A preoperative chest radiograph was reported normal. For initial treatment, the capitate and trapezoid bones were resected en bloc through a dorsal approach. Reconstruction consisted of limited arthrodesis using bone cement without additional fixation.
At 6-month follow-up, the patient was pregnant, and there was a recurrence of the wrist lesion. During the first 2 months of pregnancy, swelling and pain rapidly progressed, and a palpable mass formed. Radiographs showed a lytic lesion extending into the hamate bone (Figure 2), and magnetic resonance imaging (MRI) showed articular extension of the lesion with involvement of the base of the fourth metacarpal. [[{"fid":"202334","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"2"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 2.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"2":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 2.","field_file_image_credit[und][0][value]":""}}}]]Targeted anti-RANKL therapy was not recommended (and was not available at the patient’s home hospital). The patient deferred surgical treatment because of the pregnancy, which proved otherwise uneventful and ended with a full-term delivery.
After the pregnancy, radiographs of the wrist showed complete destruction of the hamate and trapezium bones, with erosion of the bases of the second through fourth metacarpals (Figure 3A). [[{"fid":"202335","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"3"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 3.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"3":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 3.","field_file_image_credit[und][0][value]":""}}}]]The patient presented at our institution 4 years after initial diagnosis. Computed tomography (CT) of the chest showed numerous bilateral pulmonary nodular opacities. Wrist imaging showed soft-tissue extension (Figure 3B). The diagnosis of recurrent metastatic GCT was confirmed with needle biopsies of the wrist mass and the right lung nodule.
Systemic chemotherapy was initiated with 120 mg of denosumab, given subcutaneously on days 1, 8, and 15 and then monthly during the 10 months leading up to surgery. Serum calcium was monitored during treatment and remained within the normal range the entire time, except for once at the start of therapy, when it dropped to 6.8 mg/dL. After 8 months, the soft-tissue mass, originally 8 cm × 8 cm × 6 cm, shrunk and stabilized at 5 cm × 4 cm × 4 cm (Figure 3B), and a bony shell reformed around it. Nodules in both lung fields showed response to denosumab.
Histologic examination revealed scattered osteoclast-like, multinucleated giant cells, consistent with a recurrent lesion (Figure 4). [[{"fid":"202336","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"4"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 4.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"4":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 4.","field_file_image_credit[und][0][value]":""}}}]]After 10 months of treatment with denosumab, the patient underwent resection (dorsal approach) of the residual cement, the soft-tissue mass, the affected carpal bones, half of the third metacarpal, and the second and fourth metacarpal bases. The proximal carpal row was preserved after no intra-articular involvement was verified. The closet margin was marginal; the tumor mass abutted without encompassing the flexor tendons and median nerve. The tumor was meticulously elevated from the neurovascular and tendinous structures, which were not sacrificed. Hydrogen peroxide was used for local adjuvant treatment. Bicortical autogenous ICBG was placed between the remaining scaphoid, lunate, and metacarpal bones. The second, third, and fourth metacarpal bases were stabilized on the overlapping outer table of ICBG with 2.0-mm plates and miniscrews (Figure 5A). Kirschner wires were used to stabilize the proximal bone graft and the scapholunate fossa. Cancellous bone graft was packed between the structural bone graft and neighboring unaffected carpal bones (Figure 5A). Immobilization with a short-arm thumb spica cast was maintained for 6 weeks after surgery and was followed by a 12-week rehabilitation program. The patient returned to normal activities when plain radiographs showed solid bony union (Figure 5B). Fourteen months after initial surgery, tenolysis was performed to free the extensor tendons (index, middle, and ring fingers on dorsum of left hand) from adhesions to the bone graft. At 37-month follow-up (Figure 5C), there was no clinical or radiographic evidence of progression in the wrist.[[{"fid":"202337","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"5"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 5.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"5":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 5.","field_file_image_credit[und][0][value]":""}}}]]
The patient had bilateral pulmonary metastases (Figures 6A, 6B). Treatment with denosumab produced an initial response (smaller pulmonary lesions) and subsequent stability. After 12 months of treatment with denosumab, the patient underwent left thoracotomy and wedge resection of pulmonary metastases on the left. Pathologic evaluation revealed pulmonary parenchyma with calcification and ossification and limited viable tumor. Given the dramatic effects on the left pulmonary metastases, denosumab was continued, and surgical intervention on the right was not attempted. Pulmonary metastases were stable afterward (Figure 6C).[[{"fid":"202338","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"6"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 6.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"6":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 6.","field_file_image_credit[und][0][value]":""}}}]]
At 54-month follow-up, systemic treatment with denosumab was continued. The patient had no pain in the wrist or hand and was able to use the left hand normally. There was some fissuring of the third and fourth digits over each other. However, the patient had good grip strength and was using eating utensils, picking up water bottles, and engaging in other activities without difficulty.
Discussion
GCT isolated to the carpus is rare. However, compared with GCT in the more common locations in long bones, it is also more aggressive, and its local recurrence rates are higher, probably 60% or more if treated with curettage alone.15 Therefore, excision augmented with adjuvant treatment is recommended.2,7 Use of bone cement in the hand is relatively uncommon.4,5,7-10
The diagnosis of GCT in the carpus is difficult and often delayed. The initial complaint is usually mild wrist pain after relatively mild trauma.5 The first reported case of GCT in the lunate bone was mistakenly thought to be Kienbock disease.5 Similarly, our patient was initially given a nononcologic diagnosis, which prompted conservative management.
Whether the biological behavior of GCT in the carpus differs from that of GCT in other sites is unclear. The high recurrence rates might be attributable in part to suboptimal curettage.5,6 En bloc resections of involved bone inevitably result in carpal instability or loss of wrist motion if arthrodesis is performed.4-7,11 In the present case, resection was followed by limited arthrodesis to mitigate motion losses.
Multifocal GCT in the carpal bones often affects younger patients and has a high rate of recurrence.7,16 In the present case, the patient’s pregnancy delayed treatment and allowed tumor extension into soft tissues and metacarpal bones. Given her young age, en bloc tumor resection was performed, with the proximal carpal row spared to preserve wrist motion. ICBG was carefully shaped to match the defect that remained after tumor resection.7 Supporting wrist height to prevent carpal collapse provided a stable base for remaining distal segments of the second through fourth metacarpals. After short-arm thumb spica casting and early rehabilitation, the patient recovered wrist motion and use of the involved fingers distal to the carpometacarpal joints.
In pregnant women, GCTs have been found primarily in the long bones and spine but are rare.17-21 A review of the literature (1950-present) revealed that the present article is the first report of GCT in the hand or wrist bones of a pregnant woman.18,20,21 There is no consensus as to whether surgical excision should be performed during pregnancy.18,20,21 In 1 unusual case, at 18 weeks’ gestation GCT in the distal femur was resected with curettage and bone grafting, and there were no complications.21 Therefore, pregnancy termination is not indicated for GCT.
The relationship between tumorigenesis and pregnancy is unclear.18,20,21 Empirically, pregnancy is thought to promote tumor growth.18,20 Estrogen and progesterone levels are elevated during pregnancy, potentially influencing tumor cells that are hormonally sensitive.18,20 An early report in which reverse transcription–polymerase chain reaction showed estrogen receptor expression in GCT osteoclast-like cells was followed by several studies that failed to find estrogen receptors at the protein level.19 In contrast, progesterone receptors were found in 50% of GCTs in a study.22 However, the etiopathogenic significance of this is unclear. In pregnant women, vascular endothelial growth factor, placental growth factor, and other growth factors induce osteoclast formation.23 ß-Human chorionic gonadotropin expression (ß-hCG) has been found in 58% of cases, with some showing ß-hCG elevation in the serum.24 Other studies have focused on an immunologic explanation for occurrence of GCT during pregnancy.18 Oncofetal antigens, which are similar to fetal antigens, have been found in fibrosarcoma and in an osteosarcoma cell line but not in GCT.18-20 Thus, though occurrence during pregnancy may be coincidental given the frequency of GCT in women of childbearing age, it is plausible that tumor growth may be enhanced by pregnancy. More studies are needed to understand the relationship between giant cell proliferation and pregnancy-related growth factors and hormones.
With GCT, the rate of pulmonary metastases ranges from 0% to 4%; these metastases are usually diagnosed at time of local recurrence, or 2 years to 3 years after initial GCT diagnosis.2,3,12,14,25 Lung metastases secondary to GCT in the hand or foot bones are rare; our literature review identified only 4 cases.12,14 Risk factors for lung metastasis include local recurrence, aggressive appearance (Enneking grade 3) on radiograph, Ki-67 antigen expression, and distal radius location.14 The mechanism of metastasis is unknown.12,14
Lung metastases are usually excised, but they may spontaneously evolve toward necrosis and ossification.12 In cases in which surgery is unfeasible, chemotherapy (eg, with doxorubicin) has been used to control progression.12,14 Radiation can cause sarcomatous transformation and is contraindicated. Interferon26-28 and other antiangiogenic strategies have been successfully used in systemic therapy for GCT of bone. More recently, bisphosphonates29-32 and denosumab33 have been investigated.29,32-36 The limited toxicity of denosumab makes the drug a very attractive treatment option for recurrent or unresectable GCT of bone.33 Reported rates of mortality from lung metastases have ranged from 0% to 40%.14 There is evidence that control of lung metastases during the first 3 years after diagnosis is important for favorable outcomes.2,3
Malignant stromal cells of GCT of bone have been known to secrete RANKL, which recruits osteoclasts and osteoclast precursor cells, which in turn generate aggressive osteolytic activity.33,37 Denosumab, a monoclonal antibody that inhibits RANKL, is effective in stopping osteoclastic activity. In a phase 2 trial of denosumab in the treatment of GCT of bone, 96% of treated patients with unresectable disease showed no progression at 13 months.38 In addition, 74% of treated patients who had resectable disease but were likely to have morbid surgery did not require surgery, and 62% of treated patients who underwent surgery were able to have a less morbid procedure. Forty-one percent to 58% of treated patients had a reduction in tumor size.
Denosumab is very well tolerated. The phase 2 trial found serious adverse events in 9% of patients, and in 5% of cases the drug was discontinued because of toxicity.38 Serious adverse events include osteonecrosis of jaw, hypocalcemia, and hypophosphatemia.37 Electrolyte changes with denosumab are easy to monitor and manage. Although the favorable toxicity profile of denosumab allows for long-term therapy, the data on therapy duration in patients with unresectable disease are unclear. Patients who discontinue therapy should be closely monitored, as disease can progress in this setting.37
In contrast to GCT of larger bones, GCT of the wrist is rare and typically more aggressive, and has higher local recurrence rates. In many cases, diagnosis is delayed by insufficient imaging, which optimally should include either CT or MRI (Table). [[{"fid":"202341","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"7"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Table.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"7":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Table.","field_file_image_credit[und][0][value]":""}}}]]For pregnant women with GCT, options include surgical resection with curettage and local adjuvant treatment. After pregnancy, denosumab can be used systemically, and can be effective with metastatic or unresectable disease. Surgical treatment in the wrist can be challenging when partial or complete resections of carpal bones are required. Occupational therapy is recommended for optimization of hand function after surgery.
1. Balke M, Ahrens H, Streitbuerger A, et al. Treatment options for recurrent giant cell tumors of bone. J Cancer Res Clin Oncol. 2009;135(1):149-158.
2. Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Giant cell tumor of bone risk factors for recurrence. Clin Orthop Relat Res. 2011;469(2):591-599.
3. Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Recurrent giant cell tumor of long bones: analysis of surgical management. Clin Orthop Relat Res. 2011;469(4):1181-1187.
4. Averill RM, Smith RJ, Campbell CJ. Giant-cell tumors of the bones of the hand. J Hand Surg Am. 1980;5(1):39-50.
5. Shigematsu K, Kobata Y, Yajima H, Kawamura K, Maegawa N, Takakura Y. Giant-cell tumors of the carpus. J Hand Surg Am. 2006;31(7):1214-1219.
6. Gupta GG, Lucas GL, Pirela-Cruz M. Multifocal giant cell tumor of the capitate, hamate, and triquetrum: a case report. J Hand Surg Am. 1995;20(6):1003-1006.
7. Tarng YW, Yang SW, Hsu CJ. Surgical treatment of multifocal giant cell tumor of carpal bones with preservation of wrist function: case report. J Hand Surg Am. 2009;34(2):262-265.
8. Angelini A, Mavrogenis AF, Ruggieri P. Giant cell tumor of the capitate. Musculoskelet Surg. 2011;95(1):45-48.
9. Howard FM, Lassen K. Giant cell tumor of the capitate. J Hand Surg Am. 1984;9(2):272-274.
10. McDonald DJ, Schajowicz F. Giant cell tumor of the capitate. A case report. Clin Orthop Relat Res. 1992(279):264-268.
11. Wilson SC, Cascio BM, Plauche HR. Giant-cell tumor of the capitate. Orthopedics. 2001;24(11):1085-1086.
12. Combalia-Aleu A, Sastre S, Fernández-de-Retana P, Tomás X, Palacin A. Giant cell tumor of the talus with pulmonary metastasis: seven years follow up. Foot. 2006;16(2):107-111.
13. Donthineni R, Boriani L, Ofluoglu O, Bandiera S. Metastatic behaviour of giant cell tumour of the spine. Int Orthop. 2009;33(2):497-501.
14. Jacopin S, Viehweger E, Glard Y, et al. Fatal lung metastasis secondary to index finger giant cell tumor in an 8-year-old child. Orthop Traumatol Surg Res. 2010;96(3):310-313.
15. Plate AM, Lee SJ, Steiner G, Posner MA. Tumor-like lesions and benign tumors of the hand and wrist. J Am Acad Orthop Surg. 2003;11(2):129-141.
16. Moreel P, Le Viet D. Failure of initial surgical treatment of a giant cell tumor of the capitate and its salvage: a case report [in French]. Chir Main. 2006;25(6):315-318.
17. Caillouette JC, Mattar N. Massive peripheral giant-cell reparative granuloma of the jaw: a pregnancy dependent tumor. Trans Pac Coast Obstet Gynecol Soc. 1978;45:78-81.
18. Kathiresan AS, Johnson JN, Hood BJ, Montoya SP, Vanni S, Gonzalez-Quintero VH. Giant cell bone tumor of the thoracic spine presenting in late pregnancy. Obstet Gynecol. 2011;118(2 pt 2):428-431.
19. Komiya S, Zenmyo M, Inoue A. Bone tumors in the pelvis presenting growth during pregnancy. Arch Orthop Trauma Surg. 1999;119(1-2):22-29.
20. Ross AE, Bojescul JA, Kuklo TR. Giant cell tumor: a case report of recurrence during pregnancy. Spine. 2005;30(12):E332-3E35.
21. Sharma JB, Chanana C, Rastogi, et al. Successful pregnancy outcome with elective caesarean section following two attempts of surgical excision of large giant cell tumor of the lower limb during pregnancy. Arch Gynecol Obstet. 2006;274(5):313-315.
22. Demertzis N, Kotsiandri F, Giotis I, Apostolikas N. Giant-cell tumors of bone and progesterone receptors. Orthopedics. 2003;26(12):1209-1212.
23. Taylor RM, Kashima TG, Knowles HJ, Athanasou NA. VEGF, FLT3 ligand, PlGF and HGF can substitute for M-CSF to induce human osteoclast formation: implications for giant cell tumour pathobiology. Lab Invest. 2012;92(10):1398-1406.
24. Lawless ME, Jour G, Hoch BL, Rendi MH. Beta-human chorionic gonadotropin expression in recurrent and metastatic giant cell tumors of bone: a potential mimicker of germ cell tumor. Int J Surg Pathol. 2014;22(7):617-622.
25. Viswanathan S, Jambhekar NA. Metastatic giant cell tumor of bone: are there associated factors and best treatment modalities? Clin Orthop Relat Res. 2010;468(3):827-833.
26. Kaban LB, Troulis MJ, Ebb D, August M, Hornicek FJ, Dodson TB. Antiangiogenic therapy with interferon alpha for giant cell lesions of the jaws. J Oral Maxillofac Surg. 2002;60(10):1103-1111.
27. Kaiser U, Neumann K, Havemann K. Generalised giant-cell tumour of bone: successful treatment of pulmonary metastases with interferon alpha, a case report. J Cancer Res Clin Oncol. 1993;119(5):301-303.
28. Dickerman JD. Interferon and giant cell tumors. Pediatrics. 1999;103(6 pt 1):1282-1283.
29. Balke M, Campanacci L, Gebert C, et al. Bisphosphonate treatment of aggressive primary, recurrent and metastatic giant cell tumour of bone. BMC Cancer. 2010;10:462.
30. Gille O, Oliveira Bde A, Guerin P, Lepreux S, Richez C, Vital JM. Regression of giant cell tumor of the cervical spine with bisphosphonate as single therapy. Spine. 2012;37(6):E396-E399.
31. Moriceau G, Ory B, Gobin B, et al. Therapeutic approach of primary bone tumours by bisphosphonates. Curr Pharm Des. 2010;16(27):2981-2987.
32. Tse LF, Wong KC, Kumta SM, Huang L, Chow TC, Griffith JF. Bisphosphonates reduce local recurrence in extremity giant cell tumor of bone: a case–control study. Bone. 2008;42(1):68-73.
33. Thomas D, Henshaw R, Skubitz K, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11(3):275-280.
34. Balke M, Hardes J. Denosumab: a breakthrough in treatment of giant-cell tumour of bone? Lancet Oncol. 2010;11(3):218-219.
35. Kyrgidis A, Toulis K. Safety and efficacy of denosumab in giant-cell tumour of bone. Lancet Oncol. 2010;11(6):513-514.
36. Thomas D, Carriere P, Jacobs I. Safety of denosumab in giant-cell tumour of bone. Lancet Oncol. 2010;11(9):815.
37. Skubitz KM. Giant cell tumor of bone: current treatment options. Curr Treat Options Oncol. 2014;15(3):507-518.
38. Chawla S, Henshaw R, Seeger L, et al. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: interim analysis of an open-label, parallel-group, phase 2 study. Lancet Oncol. 2013;14(9):901-908.
Take-Home Points
- GCT of bones of the wrist is rare. This article is the only report of a wrist GCT during pregnancy that we could identify.
- Routine treatment usually consists of surgical excision with local adjuvant, and in the wrist, often results in reduced wrist motion.
- GCT of the wrist is more aggressive than the more common locations in long bones, with higher local recurrence rates if treated with surgery alone.
- Diagnosis is often delayed for GCT of the wrist, due to insufficient imaging, which should include CT or MRI.
- For pregnant women with GCT, local adjuvant treatments can be used in addition to surgery. Following pregnancy, denosumab can be used systemically, and can be effective with metastatic or unresectable disease.
Giant cell tumor (GCT) of bone accounts for about 5% of primary bone tumors.1-3 Only 3% to 5% of GCTs occur in the hand.4,5 Wrist involvement, which is rare, most often involves the hamate bone.5-7 Capitate bone involvement is exceedingly rare.8-11 Although histologically benign, GCT can recur locally after treatment with curettage alone, and lung metastases are found in 2% to 5% of cases.2,12-14 Therefore, en bloc tumor excision is preferred in the setting of cortical erosion or soft-tissue involvement.1,4,8 Wrist joint motion is inevitably reduced, and bone graft donor-site morbidity is significant.6-8
In the unusual case reported here, GCT presented in the capitate bone and, after the patient became pregnant, recurred in the hamate and trapezoid bones with soft-tissue extension and lung metastases. The capitate was excised en bloc and reconstructed with an interposition of polymethylmethacrylate bone cement. Pulmonary metastases developed, and the GCT expanded to involve multiple carpal bones and the bases of the second through fourth metacarpals. A 10-month course of systemic chemotherapy with the RANK ligand (RANKL) inhibitor denosumab was started after the pregnancy. After this treatment, the patient underwent both tumor resection and reconstruction with autogenous bicortical iliac crest bone graft (ICBG) carefully designed to preserve range of motion and maintain the fingers in anatomical position. Treatment with denosumab was continued after surgery. Although this case offers no endpoint for postoperative chemotherapy with denosumab, preoperative treatment dramatically reduced the GCT and permitted limb-sparing reconstruction. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old right-handed woman with atraumatic swelling of the left wrist presented to an orthopedic surgeon at an outside facility. Physical examination revealed tender fullness on the dorsum of the wrist, slightly reduced range of motion and grip strength, and a neurovascularly intact wrist. The diagnosis was periarticular cyst, and the patient underwent physical therapy. Two years later, the swelling returned, tenderness was increasing, and symptoms did not resolve with cast immobilization. A radiograph showed a lytic lesion in the capitate bone (Figure 1).[[{"fid":"202332","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"1"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 1.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"1":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 1.","field_file_image_credit[und][0][value]":""}}}]]
GCT was diagnosed with percutaneous needle biopsy. A preoperative chest radiograph was reported normal. For initial treatment, the capitate and trapezoid bones were resected en bloc through a dorsal approach. Reconstruction consisted of limited arthrodesis using bone cement without additional fixation.
At 6-month follow-up, the patient was pregnant, and there was a recurrence of the wrist lesion. During the first 2 months of pregnancy, swelling and pain rapidly progressed, and a palpable mass formed. Radiographs showed a lytic lesion extending into the hamate bone (Figure 2), and magnetic resonance imaging (MRI) showed articular extension of the lesion with involvement of the base of the fourth metacarpal. [[{"fid":"202334","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"2"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 2.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"2":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 2.","field_file_image_credit[und][0][value]":""}}}]]Targeted anti-RANKL therapy was not recommended (and was not available at the patient’s home hospital). The patient deferred surgical treatment because of the pregnancy, which proved otherwise uneventful and ended with a full-term delivery.
After the pregnancy, radiographs of the wrist showed complete destruction of the hamate and trapezium bones, with erosion of the bases of the second through fourth metacarpals (Figure 3A). [[{"fid":"202335","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"3"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 3.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"3":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 3.","field_file_image_credit[und][0][value]":""}}}]]The patient presented at our institution 4 years after initial diagnosis. Computed tomography (CT) of the chest showed numerous bilateral pulmonary nodular opacities. Wrist imaging showed soft-tissue extension (Figure 3B). The diagnosis of recurrent metastatic GCT was confirmed with needle biopsies of the wrist mass and the right lung nodule.
Systemic chemotherapy was initiated with 120 mg of denosumab, given subcutaneously on days 1, 8, and 15 and then monthly during the 10 months leading up to surgery. Serum calcium was monitored during treatment and remained within the normal range the entire time, except for once at the start of therapy, when it dropped to 6.8 mg/dL. After 8 months, the soft-tissue mass, originally 8 cm × 8 cm × 6 cm, shrunk and stabilized at 5 cm × 4 cm × 4 cm (Figure 3B), and a bony shell reformed around it. Nodules in both lung fields showed response to denosumab.
Histologic examination revealed scattered osteoclast-like, multinucleated giant cells, consistent with a recurrent lesion (Figure 4). [[{"fid":"202336","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"4"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 4.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"4":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 4.","field_file_image_credit[und][0][value]":""}}}]]After 10 months of treatment with denosumab, the patient underwent resection (dorsal approach) of the residual cement, the soft-tissue mass, the affected carpal bones, half of the third metacarpal, and the second and fourth metacarpal bases. The proximal carpal row was preserved after no intra-articular involvement was verified. The closet margin was marginal; the tumor mass abutted without encompassing the flexor tendons and median nerve. The tumor was meticulously elevated from the neurovascular and tendinous structures, which were not sacrificed. Hydrogen peroxide was used for local adjuvant treatment. Bicortical autogenous ICBG was placed between the remaining scaphoid, lunate, and metacarpal bones. The second, third, and fourth metacarpal bases were stabilized on the overlapping outer table of ICBG with 2.0-mm plates and miniscrews (Figure 5A). Kirschner wires were used to stabilize the proximal bone graft and the scapholunate fossa. Cancellous bone graft was packed between the structural bone graft and neighboring unaffected carpal bones (Figure 5A). Immobilization with a short-arm thumb spica cast was maintained for 6 weeks after surgery and was followed by a 12-week rehabilitation program. The patient returned to normal activities when plain radiographs showed solid bony union (Figure 5B). Fourteen months after initial surgery, tenolysis was performed to free the extensor tendons (index, middle, and ring fingers on dorsum of left hand) from adhesions to the bone graft. At 37-month follow-up (Figure 5C), there was no clinical or radiographic evidence of progression in the wrist.[[{"fid":"202337","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"5"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 5.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"5":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 5.","field_file_image_credit[und][0][value]":""}}}]]
The patient had bilateral pulmonary metastases (Figures 6A, 6B). Treatment with denosumab produced an initial response (smaller pulmonary lesions) and subsequent stability. After 12 months of treatment with denosumab, the patient underwent left thoracotomy and wedge resection of pulmonary metastases on the left. Pathologic evaluation revealed pulmonary parenchyma with calcification and ossification and limited viable tumor. Given the dramatic effects on the left pulmonary metastases, denosumab was continued, and surgical intervention on the right was not attempted. Pulmonary metastases were stable afterward (Figure 6C).[[{"fid":"202338","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"6"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 6.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"6":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 6.","field_file_image_credit[und][0][value]":""}}}]]
At 54-month follow-up, systemic treatment with denosumab was continued. The patient had no pain in the wrist or hand and was able to use the left hand normally. There was some fissuring of the third and fourth digits over each other. However, the patient had good grip strength and was using eating utensils, picking up water bottles, and engaging in other activities without difficulty.
Discussion
GCT isolated to the carpus is rare. However, compared with GCT in the more common locations in long bones, it is also more aggressive, and its local recurrence rates are higher, probably 60% or more if treated with curettage alone.15 Therefore, excision augmented with adjuvant treatment is recommended.2,7 Use of bone cement in the hand is relatively uncommon.4,5,7-10
The diagnosis of GCT in the carpus is difficult and often delayed. The initial complaint is usually mild wrist pain after relatively mild trauma.5 The first reported case of GCT in the lunate bone was mistakenly thought to be Kienbock disease.5 Similarly, our patient was initially given a nononcologic diagnosis, which prompted conservative management.
Whether the biological behavior of GCT in the carpus differs from that of GCT in other sites is unclear. The high recurrence rates might be attributable in part to suboptimal curettage.5,6 En bloc resections of involved bone inevitably result in carpal instability or loss of wrist motion if arthrodesis is performed.4-7,11 In the present case, resection was followed by limited arthrodesis to mitigate motion losses.
Multifocal GCT in the carpal bones often affects younger patients and has a high rate of recurrence.7,16 In the present case, the patient’s pregnancy delayed treatment and allowed tumor extension into soft tissues and metacarpal bones. Given her young age, en bloc tumor resection was performed, with the proximal carpal row spared to preserve wrist motion. ICBG was carefully shaped to match the defect that remained after tumor resection.7 Supporting wrist height to prevent carpal collapse provided a stable base for remaining distal segments of the second through fourth metacarpals. After short-arm thumb spica casting and early rehabilitation, the patient recovered wrist motion and use of the involved fingers distal to the carpometacarpal joints.
In pregnant women, GCTs have been found primarily in the long bones and spine but are rare.17-21 A review of the literature (1950-present) revealed that the present article is the first report of GCT in the hand or wrist bones of a pregnant woman.18,20,21 There is no consensus as to whether surgical excision should be performed during pregnancy.18,20,21 In 1 unusual case, at 18 weeks’ gestation GCT in the distal femur was resected with curettage and bone grafting, and there were no complications.21 Therefore, pregnancy termination is not indicated for GCT.
The relationship between tumorigenesis and pregnancy is unclear.18,20,21 Empirically, pregnancy is thought to promote tumor growth.18,20 Estrogen and progesterone levels are elevated during pregnancy, potentially influencing tumor cells that are hormonally sensitive.18,20 An early report in which reverse transcription–polymerase chain reaction showed estrogen receptor expression in GCT osteoclast-like cells was followed by several studies that failed to find estrogen receptors at the protein level.19 In contrast, progesterone receptors were found in 50% of GCTs in a study.22 However, the etiopathogenic significance of this is unclear. In pregnant women, vascular endothelial growth factor, placental growth factor, and other growth factors induce osteoclast formation.23 ß-Human chorionic gonadotropin expression (ß-hCG) has been found in 58% of cases, with some showing ß-hCG elevation in the serum.24 Other studies have focused on an immunologic explanation for occurrence of GCT during pregnancy.18 Oncofetal antigens, which are similar to fetal antigens, have been found in fibrosarcoma and in an osteosarcoma cell line but not in GCT.18-20 Thus, though occurrence during pregnancy may be coincidental given the frequency of GCT in women of childbearing age, it is plausible that tumor growth may be enhanced by pregnancy. More studies are needed to understand the relationship between giant cell proliferation and pregnancy-related growth factors and hormones.
With GCT, the rate of pulmonary metastases ranges from 0% to 4%; these metastases are usually diagnosed at time of local recurrence, or 2 years to 3 years after initial GCT diagnosis.2,3,12,14,25 Lung metastases secondary to GCT in the hand or foot bones are rare; our literature review identified only 4 cases.12,14 Risk factors for lung metastasis include local recurrence, aggressive appearance (Enneking grade 3) on radiograph, Ki-67 antigen expression, and distal radius location.14 The mechanism of metastasis is unknown.12,14
Lung metastases are usually excised, but they may spontaneously evolve toward necrosis and ossification.12 In cases in which surgery is unfeasible, chemotherapy (eg, with doxorubicin) has been used to control progression.12,14 Radiation can cause sarcomatous transformation and is contraindicated. Interferon26-28 and other antiangiogenic strategies have been successfully used in systemic therapy for GCT of bone. More recently, bisphosphonates29-32 and denosumab33 have been investigated.29,32-36 The limited toxicity of denosumab makes the drug a very attractive treatment option for recurrent or unresectable GCT of bone.33 Reported rates of mortality from lung metastases have ranged from 0% to 40%.14 There is evidence that control of lung metastases during the first 3 years after diagnosis is important for favorable outcomes.2,3
Malignant stromal cells of GCT of bone have been known to secrete RANKL, which recruits osteoclasts and osteoclast precursor cells, which in turn generate aggressive osteolytic activity.33,37 Denosumab, a monoclonal antibody that inhibits RANKL, is effective in stopping osteoclastic activity. In a phase 2 trial of denosumab in the treatment of GCT of bone, 96% of treated patients with unresectable disease showed no progression at 13 months.38 In addition, 74% of treated patients who had resectable disease but were likely to have morbid surgery did not require surgery, and 62% of treated patients who underwent surgery were able to have a less morbid procedure. Forty-one percent to 58% of treated patients had a reduction in tumor size.
Denosumab is very well tolerated. The phase 2 trial found serious adverse events in 9% of patients, and in 5% of cases the drug was discontinued because of toxicity.38 Serious adverse events include osteonecrosis of jaw, hypocalcemia, and hypophosphatemia.37 Electrolyte changes with denosumab are easy to monitor and manage. Although the favorable toxicity profile of denosumab allows for long-term therapy, the data on therapy duration in patients with unresectable disease are unclear. Patients who discontinue therapy should be closely monitored, as disease can progress in this setting.37
In contrast to GCT of larger bones, GCT of the wrist is rare and typically more aggressive, and has higher local recurrence rates. In many cases, diagnosis is delayed by insufficient imaging, which optimally should include either CT or MRI (Table). [[{"fid":"202341","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"7"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Table.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"7":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Table.","field_file_image_credit[und][0][value]":""}}}]]For pregnant women with GCT, options include surgical resection with curettage and local adjuvant treatment. After pregnancy, denosumab can be used systemically, and can be effective with metastatic or unresectable disease. Surgical treatment in the wrist can be challenging when partial or complete resections of carpal bones are required. Occupational therapy is recommended for optimization of hand function after surgery.
Take-Home Points
- GCT of bones of the wrist is rare. This article is the only report of a wrist GCT during pregnancy that we could identify.
- Routine treatment usually consists of surgical excision with local adjuvant, and in the wrist, often results in reduced wrist motion.
- GCT of the wrist is more aggressive than the more common locations in long bones, with higher local recurrence rates if treated with surgery alone.
- Diagnosis is often delayed for GCT of the wrist, due to insufficient imaging, which should include CT or MRI.
- For pregnant women with GCT, local adjuvant treatments can be used in addition to surgery. Following pregnancy, denosumab can be used systemically, and can be effective with metastatic or unresectable disease.
Giant cell tumor (GCT) of bone accounts for about 5% of primary bone tumors.1-3 Only 3% to 5% of GCTs occur in the hand.4,5 Wrist involvement, which is rare, most often involves the hamate bone.5-7 Capitate bone involvement is exceedingly rare.8-11 Although histologically benign, GCT can recur locally after treatment with curettage alone, and lung metastases are found in 2% to 5% of cases.2,12-14 Therefore, en bloc tumor excision is preferred in the setting of cortical erosion or soft-tissue involvement.1,4,8 Wrist joint motion is inevitably reduced, and bone graft donor-site morbidity is significant.6-8
In the unusual case reported here, GCT presented in the capitate bone and, after the patient became pregnant, recurred in the hamate and trapezoid bones with soft-tissue extension and lung metastases. The capitate was excised en bloc and reconstructed with an interposition of polymethylmethacrylate bone cement. Pulmonary metastases developed, and the GCT expanded to involve multiple carpal bones and the bases of the second through fourth metacarpals. A 10-month course of systemic chemotherapy with the RANK ligand (RANKL) inhibitor denosumab was started after the pregnancy. After this treatment, the patient underwent both tumor resection and reconstruction with autogenous bicortical iliac crest bone graft (ICBG) carefully designed to preserve range of motion and maintain the fingers in anatomical position. Treatment with denosumab was continued after surgery. Although this case offers no endpoint for postoperative chemotherapy with denosumab, preoperative treatment dramatically reduced the GCT and permitted limb-sparing reconstruction. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old right-handed woman with atraumatic swelling of the left wrist presented to an orthopedic surgeon at an outside facility. Physical examination revealed tender fullness on the dorsum of the wrist, slightly reduced range of motion and grip strength, and a neurovascularly intact wrist. The diagnosis was periarticular cyst, and the patient underwent physical therapy. Two years later, the swelling returned, tenderness was increasing, and symptoms did not resolve with cast immobilization. A radiograph showed a lytic lesion in the capitate bone (Figure 1).[[{"fid":"202332","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"1"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 1.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"1":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 1.","field_file_image_credit[und][0][value]":""}}}]]
GCT was diagnosed with percutaneous needle biopsy. A preoperative chest radiograph was reported normal. For initial treatment, the capitate and trapezoid bones were resected en bloc through a dorsal approach. Reconstruction consisted of limited arthrodesis using bone cement without additional fixation.
At 6-month follow-up, the patient was pregnant, and there was a recurrence of the wrist lesion. During the first 2 months of pregnancy, swelling and pain rapidly progressed, and a palpable mass formed. Radiographs showed a lytic lesion extending into the hamate bone (Figure 2), and magnetic resonance imaging (MRI) showed articular extension of the lesion with involvement of the base of the fourth metacarpal. [[{"fid":"202334","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"2"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 2.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"2":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 2.","field_file_image_credit[und][0][value]":""}}}]]Targeted anti-RANKL therapy was not recommended (and was not available at the patient’s home hospital). The patient deferred surgical treatment because of the pregnancy, which proved otherwise uneventful and ended with a full-term delivery.
After the pregnancy, radiographs of the wrist showed complete destruction of the hamate and trapezium bones, with erosion of the bases of the second through fourth metacarpals (Figure 3A). [[{"fid":"202335","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"3"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 3.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"3":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 3.","field_file_image_credit[und][0][value]":""}}}]]The patient presented at our institution 4 years after initial diagnosis. Computed tomography (CT) of the chest showed numerous bilateral pulmonary nodular opacities. Wrist imaging showed soft-tissue extension (Figure 3B). The diagnosis of recurrent metastatic GCT was confirmed with needle biopsies of the wrist mass and the right lung nodule.
Systemic chemotherapy was initiated with 120 mg of denosumab, given subcutaneously on days 1, 8, and 15 and then monthly during the 10 months leading up to surgery. Serum calcium was monitored during treatment and remained within the normal range the entire time, except for once at the start of therapy, when it dropped to 6.8 mg/dL. After 8 months, the soft-tissue mass, originally 8 cm × 8 cm × 6 cm, shrunk and stabilized at 5 cm × 4 cm × 4 cm (Figure 3B), and a bony shell reformed around it. Nodules in both lung fields showed response to denosumab.
Histologic examination revealed scattered osteoclast-like, multinucleated giant cells, consistent with a recurrent lesion (Figure 4). [[{"fid":"202336","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"4"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 4.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"4":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 4.","field_file_image_credit[und][0][value]":""}}}]]After 10 months of treatment with denosumab, the patient underwent resection (dorsal approach) of the residual cement, the soft-tissue mass, the affected carpal bones, half of the third metacarpal, and the second and fourth metacarpal bases. The proximal carpal row was preserved after no intra-articular involvement was verified. The closet margin was marginal; the tumor mass abutted without encompassing the flexor tendons and median nerve. The tumor was meticulously elevated from the neurovascular and tendinous structures, which were not sacrificed. Hydrogen peroxide was used for local adjuvant treatment. Bicortical autogenous ICBG was placed between the remaining scaphoid, lunate, and metacarpal bones. The second, third, and fourth metacarpal bases were stabilized on the overlapping outer table of ICBG with 2.0-mm plates and miniscrews (Figure 5A). Kirschner wires were used to stabilize the proximal bone graft and the scapholunate fossa. Cancellous bone graft was packed between the structural bone graft and neighboring unaffected carpal bones (Figure 5A). Immobilization with a short-arm thumb spica cast was maintained for 6 weeks after surgery and was followed by a 12-week rehabilitation program. The patient returned to normal activities when plain radiographs showed solid bony union (Figure 5B). Fourteen months after initial surgery, tenolysis was performed to free the extensor tendons (index, middle, and ring fingers on dorsum of left hand) from adhesions to the bone graft. At 37-month follow-up (Figure 5C), there was no clinical or radiographic evidence of progression in the wrist.[[{"fid":"202337","view_mode":"medstat_image_flush_left","attributes":{"class":"media-element file-medstat-image-flush-left","data-delta":"5"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 5.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"5":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Figure 5.","field_file_image_credit[und][0][value]":""}}}]]
The patient had bilateral pulmonary metastases (Figures 6A, 6B). Treatment with denosumab produced an initial response (smaller pulmonary lesions) and subsequent stability. After 12 months of treatment with denosumab, the patient underwent left thoracotomy and wedge resection of pulmonary metastases on the left. Pathologic evaluation revealed pulmonary parenchyma with calcification and ossification and limited viable tumor. Given the dramatic effects on the left pulmonary metastases, denosumab was continued, and surgical intervention on the right was not attempted. Pulmonary metastases were stable afterward (Figure 6C).[[{"fid":"202338","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"6"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 6.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"6":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Figure 6.","field_file_image_credit[und][0][value]":""}}}]]
At 54-month follow-up, systemic treatment with denosumab was continued. The patient had no pain in the wrist or hand and was able to use the left hand normally. There was some fissuring of the third and fourth digits over each other. However, the patient had good grip strength and was using eating utensils, picking up water bottles, and engaging in other activities without difficulty.
Discussion
GCT isolated to the carpus is rare. However, compared with GCT in the more common locations in long bones, it is also more aggressive, and its local recurrence rates are higher, probably 60% or more if treated with curettage alone.15 Therefore, excision augmented with adjuvant treatment is recommended.2,7 Use of bone cement in the hand is relatively uncommon.4,5,7-10
The diagnosis of GCT in the carpus is difficult and often delayed. The initial complaint is usually mild wrist pain after relatively mild trauma.5 The first reported case of GCT in the lunate bone was mistakenly thought to be Kienbock disease.5 Similarly, our patient was initially given a nononcologic diagnosis, which prompted conservative management.
Whether the biological behavior of GCT in the carpus differs from that of GCT in other sites is unclear. The high recurrence rates might be attributable in part to suboptimal curettage.5,6 En bloc resections of involved bone inevitably result in carpal instability or loss of wrist motion if arthrodesis is performed.4-7,11 In the present case, resection was followed by limited arthrodesis to mitigate motion losses.
Multifocal GCT in the carpal bones often affects younger patients and has a high rate of recurrence.7,16 In the present case, the patient’s pregnancy delayed treatment and allowed tumor extension into soft tissues and metacarpal bones. Given her young age, en bloc tumor resection was performed, with the proximal carpal row spared to preserve wrist motion. ICBG was carefully shaped to match the defect that remained after tumor resection.7 Supporting wrist height to prevent carpal collapse provided a stable base for remaining distal segments of the second through fourth metacarpals. After short-arm thumb spica casting and early rehabilitation, the patient recovered wrist motion and use of the involved fingers distal to the carpometacarpal joints.
In pregnant women, GCTs have been found primarily in the long bones and spine but are rare.17-21 A review of the literature (1950-present) revealed that the present article is the first report of GCT in the hand or wrist bones of a pregnant woman.18,20,21 There is no consensus as to whether surgical excision should be performed during pregnancy.18,20,21 In 1 unusual case, at 18 weeks’ gestation GCT in the distal femur was resected with curettage and bone grafting, and there were no complications.21 Therefore, pregnancy termination is not indicated for GCT.
The relationship between tumorigenesis and pregnancy is unclear.18,20,21 Empirically, pregnancy is thought to promote tumor growth.18,20 Estrogen and progesterone levels are elevated during pregnancy, potentially influencing tumor cells that are hormonally sensitive.18,20 An early report in which reverse transcription–polymerase chain reaction showed estrogen receptor expression in GCT osteoclast-like cells was followed by several studies that failed to find estrogen receptors at the protein level.19 In contrast, progesterone receptors were found in 50% of GCTs in a study.22 However, the etiopathogenic significance of this is unclear. In pregnant women, vascular endothelial growth factor, placental growth factor, and other growth factors induce osteoclast formation.23 ß-Human chorionic gonadotropin expression (ß-hCG) has been found in 58% of cases, with some showing ß-hCG elevation in the serum.24 Other studies have focused on an immunologic explanation for occurrence of GCT during pregnancy.18 Oncofetal antigens, which are similar to fetal antigens, have been found in fibrosarcoma and in an osteosarcoma cell line but not in GCT.18-20 Thus, though occurrence during pregnancy may be coincidental given the frequency of GCT in women of childbearing age, it is plausible that tumor growth may be enhanced by pregnancy. More studies are needed to understand the relationship between giant cell proliferation and pregnancy-related growth factors and hormones.
With GCT, the rate of pulmonary metastases ranges from 0% to 4%; these metastases are usually diagnosed at time of local recurrence, or 2 years to 3 years after initial GCT diagnosis.2,3,12,14,25 Lung metastases secondary to GCT in the hand or foot bones are rare; our literature review identified only 4 cases.12,14 Risk factors for lung metastasis include local recurrence, aggressive appearance (Enneking grade 3) on radiograph, Ki-67 antigen expression, and distal radius location.14 The mechanism of metastasis is unknown.12,14
Lung metastases are usually excised, but they may spontaneously evolve toward necrosis and ossification.12 In cases in which surgery is unfeasible, chemotherapy (eg, with doxorubicin) has been used to control progression.12,14 Radiation can cause sarcomatous transformation and is contraindicated. Interferon26-28 and other antiangiogenic strategies have been successfully used in systemic therapy for GCT of bone. More recently, bisphosphonates29-32 and denosumab33 have been investigated.29,32-36 The limited toxicity of denosumab makes the drug a very attractive treatment option for recurrent or unresectable GCT of bone.33 Reported rates of mortality from lung metastases have ranged from 0% to 40%.14 There is evidence that control of lung metastases during the first 3 years after diagnosis is important for favorable outcomes.2,3
Malignant stromal cells of GCT of bone have been known to secrete RANKL, which recruits osteoclasts and osteoclast precursor cells, which in turn generate aggressive osteolytic activity.33,37 Denosumab, a monoclonal antibody that inhibits RANKL, is effective in stopping osteoclastic activity. In a phase 2 trial of denosumab in the treatment of GCT of bone, 96% of treated patients with unresectable disease showed no progression at 13 months.38 In addition, 74% of treated patients who had resectable disease but were likely to have morbid surgery did not require surgery, and 62% of treated patients who underwent surgery were able to have a less morbid procedure. Forty-one percent to 58% of treated patients had a reduction in tumor size.
Denosumab is very well tolerated. The phase 2 trial found serious adverse events in 9% of patients, and in 5% of cases the drug was discontinued because of toxicity.38 Serious adverse events include osteonecrosis of jaw, hypocalcemia, and hypophosphatemia.37 Electrolyte changes with denosumab are easy to monitor and manage. Although the favorable toxicity profile of denosumab allows for long-term therapy, the data on therapy duration in patients with unresectable disease are unclear. Patients who discontinue therapy should be closely monitored, as disease can progress in this setting.37
In contrast to GCT of larger bones, GCT of the wrist is rare and typically more aggressive, and has higher local recurrence rates. In many cases, diagnosis is delayed by insufficient imaging, which optimally should include either CT or MRI (Table). [[{"fid":"202341","view_mode":"medstat_image_flush_right","attributes":{"class":"media-element file-medstat-image-flush-right","data-delta":"7"},"fields":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Table.","field_file_image_credit[und][0][value]":"","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"7":{"format":"medstat_image_flush_right","field_file_image_caption[und][0][value]":"Table.","field_file_image_credit[und][0][value]":""}}}]]For pregnant women with GCT, options include surgical resection with curettage and local adjuvant treatment. After pregnancy, denosumab can be used systemically, and can be effective with metastatic or unresectable disease. Surgical treatment in the wrist can be challenging when partial or complete resections of carpal bones are required. Occupational therapy is recommended for optimization of hand function after surgery.
1. Balke M, Ahrens H, Streitbuerger A, et al. Treatment options for recurrent giant cell tumors of bone. J Cancer Res Clin Oncol. 2009;135(1):149-158.
2. Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Giant cell tumor of bone risk factors for recurrence. Clin Orthop Relat Res. 2011;469(2):591-599.
3. Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Recurrent giant cell tumor of long bones: analysis of surgical management. Clin Orthop Relat Res. 2011;469(4):1181-1187.
4. Averill RM, Smith RJ, Campbell CJ. Giant-cell tumors of the bones of the hand. J Hand Surg Am. 1980;5(1):39-50.
5. Shigematsu K, Kobata Y, Yajima H, Kawamura K, Maegawa N, Takakura Y. Giant-cell tumors of the carpus. J Hand Surg Am. 2006;31(7):1214-1219.
6. Gupta GG, Lucas GL, Pirela-Cruz M. Multifocal giant cell tumor of the capitate, hamate, and triquetrum: a case report. J Hand Surg Am. 1995;20(6):1003-1006.
7. Tarng YW, Yang SW, Hsu CJ. Surgical treatment of multifocal giant cell tumor of carpal bones with preservation of wrist function: case report. J Hand Surg Am. 2009;34(2):262-265.
8. Angelini A, Mavrogenis AF, Ruggieri P. Giant cell tumor of the capitate. Musculoskelet Surg. 2011;95(1):45-48.
9. Howard FM, Lassen K. Giant cell tumor of the capitate. J Hand Surg Am. 1984;9(2):272-274.
10. McDonald DJ, Schajowicz F. Giant cell tumor of the capitate. A case report. Clin Orthop Relat Res. 1992(279):264-268.
11. Wilson SC, Cascio BM, Plauche HR. Giant-cell tumor of the capitate. Orthopedics. 2001;24(11):1085-1086.
12. Combalia-Aleu A, Sastre S, Fernández-de-Retana P, Tomás X, Palacin A. Giant cell tumor of the talus with pulmonary metastasis: seven years follow up. Foot. 2006;16(2):107-111.
13. Donthineni R, Boriani L, Ofluoglu O, Bandiera S. Metastatic behaviour of giant cell tumour of the spine. Int Orthop. 2009;33(2):497-501.
14. Jacopin S, Viehweger E, Glard Y, et al. Fatal lung metastasis secondary to index finger giant cell tumor in an 8-year-old child. Orthop Traumatol Surg Res. 2010;96(3):310-313.
15. Plate AM, Lee SJ, Steiner G, Posner MA. Tumor-like lesions and benign tumors of the hand and wrist. J Am Acad Orthop Surg. 2003;11(2):129-141.
16. Moreel P, Le Viet D. Failure of initial surgical treatment of a giant cell tumor of the capitate and its salvage: a case report [in French]. Chir Main. 2006;25(6):315-318.
17. Caillouette JC, Mattar N. Massive peripheral giant-cell reparative granuloma of the jaw: a pregnancy dependent tumor. Trans Pac Coast Obstet Gynecol Soc. 1978;45:78-81.
18. Kathiresan AS, Johnson JN, Hood BJ, Montoya SP, Vanni S, Gonzalez-Quintero VH. Giant cell bone tumor of the thoracic spine presenting in late pregnancy. Obstet Gynecol. 2011;118(2 pt 2):428-431.
19. Komiya S, Zenmyo M, Inoue A. Bone tumors in the pelvis presenting growth during pregnancy. Arch Orthop Trauma Surg. 1999;119(1-2):22-29.
20. Ross AE, Bojescul JA, Kuklo TR. Giant cell tumor: a case report of recurrence during pregnancy. Spine. 2005;30(12):E332-3E35.
21. Sharma JB, Chanana C, Rastogi, et al. Successful pregnancy outcome with elective caesarean section following two attempts of surgical excision of large giant cell tumor of the lower limb during pregnancy. Arch Gynecol Obstet. 2006;274(5):313-315.
22. Demertzis N, Kotsiandri F, Giotis I, Apostolikas N. Giant-cell tumors of bone and progesterone receptors. Orthopedics. 2003;26(12):1209-1212.
23. Taylor RM, Kashima TG, Knowles HJ, Athanasou NA. VEGF, FLT3 ligand, PlGF and HGF can substitute for M-CSF to induce human osteoclast formation: implications for giant cell tumour pathobiology. Lab Invest. 2012;92(10):1398-1406.
24. Lawless ME, Jour G, Hoch BL, Rendi MH. Beta-human chorionic gonadotropin expression in recurrent and metastatic giant cell tumors of bone: a potential mimicker of germ cell tumor. Int J Surg Pathol. 2014;22(7):617-622.
25. Viswanathan S, Jambhekar NA. Metastatic giant cell tumor of bone: are there associated factors and best treatment modalities? Clin Orthop Relat Res. 2010;468(3):827-833.
26. Kaban LB, Troulis MJ, Ebb D, August M, Hornicek FJ, Dodson TB. Antiangiogenic therapy with interferon alpha for giant cell lesions of the jaws. J Oral Maxillofac Surg. 2002;60(10):1103-1111.
27. Kaiser U, Neumann K, Havemann K. Generalised giant-cell tumour of bone: successful treatment of pulmonary metastases with interferon alpha, a case report. J Cancer Res Clin Oncol. 1993;119(5):301-303.
28. Dickerman JD. Interferon and giant cell tumors. Pediatrics. 1999;103(6 pt 1):1282-1283.
29. Balke M, Campanacci L, Gebert C, et al. Bisphosphonate treatment of aggressive primary, recurrent and metastatic giant cell tumour of bone. BMC Cancer. 2010;10:462.
30. Gille O, Oliveira Bde A, Guerin P, Lepreux S, Richez C, Vital JM. Regression of giant cell tumor of the cervical spine with bisphosphonate as single therapy. Spine. 2012;37(6):E396-E399.
31. Moriceau G, Ory B, Gobin B, et al. Therapeutic approach of primary bone tumours by bisphosphonates. Curr Pharm Des. 2010;16(27):2981-2987.
32. Tse LF, Wong KC, Kumta SM, Huang L, Chow TC, Griffith JF. Bisphosphonates reduce local recurrence in extremity giant cell tumor of bone: a case–control study. Bone. 2008;42(1):68-73.
33. Thomas D, Henshaw R, Skubitz K, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11(3):275-280.
34. Balke M, Hardes J. Denosumab: a breakthrough in treatment of giant-cell tumour of bone? Lancet Oncol. 2010;11(3):218-219.
35. Kyrgidis A, Toulis K. Safety and efficacy of denosumab in giant-cell tumour of bone. Lancet Oncol. 2010;11(6):513-514.
36. Thomas D, Carriere P, Jacobs I. Safety of denosumab in giant-cell tumour of bone. Lancet Oncol. 2010;11(9):815.
37. Skubitz KM. Giant cell tumor of bone: current treatment options. Curr Treat Options Oncol. 2014;15(3):507-518.
38. Chawla S, Henshaw R, Seeger L, et al. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: interim analysis of an open-label, parallel-group, phase 2 study. Lancet Oncol. 2013;14(9):901-908.
1. Balke M, Ahrens H, Streitbuerger A, et al. Treatment options for recurrent giant cell tumors of bone. J Cancer Res Clin Oncol. 2009;135(1):149-158.
2. Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Giant cell tumor of bone risk factors for recurrence. Clin Orthop Relat Res. 2011;469(2):591-599.
3. Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Recurrent giant cell tumor of long bones: analysis of surgical management. Clin Orthop Relat Res. 2011;469(4):1181-1187.
4. Averill RM, Smith RJ, Campbell CJ. Giant-cell tumors of the bones of the hand. J Hand Surg Am. 1980;5(1):39-50.
5. Shigematsu K, Kobata Y, Yajima H, Kawamura K, Maegawa N, Takakura Y. Giant-cell tumors of the carpus. J Hand Surg Am. 2006;31(7):1214-1219.
6. Gupta GG, Lucas GL, Pirela-Cruz M. Multifocal giant cell tumor of the capitate, hamate, and triquetrum: a case report. J Hand Surg Am. 1995;20(6):1003-1006.
7. Tarng YW, Yang SW, Hsu CJ. Surgical treatment of multifocal giant cell tumor of carpal bones with preservation of wrist function: case report. J Hand Surg Am. 2009;34(2):262-265.
8. Angelini A, Mavrogenis AF, Ruggieri P. Giant cell tumor of the capitate. Musculoskelet Surg. 2011;95(1):45-48.
9. Howard FM, Lassen K. Giant cell tumor of the capitate. J Hand Surg Am. 1984;9(2):272-274.
10. McDonald DJ, Schajowicz F. Giant cell tumor of the capitate. A case report. Clin Orthop Relat Res. 1992(279):264-268.
11. Wilson SC, Cascio BM, Plauche HR. Giant-cell tumor of the capitate. Orthopedics. 2001;24(11):1085-1086.
12. Combalia-Aleu A, Sastre S, Fernández-de-Retana P, Tomás X, Palacin A. Giant cell tumor of the talus with pulmonary metastasis: seven years follow up. Foot. 2006;16(2):107-111.
13. Donthineni R, Boriani L, Ofluoglu O, Bandiera S. Metastatic behaviour of giant cell tumour of the spine. Int Orthop. 2009;33(2):497-501.
14. Jacopin S, Viehweger E, Glard Y, et al. Fatal lung metastasis secondary to index finger giant cell tumor in an 8-year-old child. Orthop Traumatol Surg Res. 2010;96(3):310-313.
15. Plate AM, Lee SJ, Steiner G, Posner MA. Tumor-like lesions and benign tumors of the hand and wrist. J Am Acad Orthop Surg. 2003;11(2):129-141.
16. Moreel P, Le Viet D. Failure of initial surgical treatment of a giant cell tumor of the capitate and its salvage: a case report [in French]. Chir Main. 2006;25(6):315-318.
17. Caillouette JC, Mattar N. Massive peripheral giant-cell reparative granuloma of the jaw: a pregnancy dependent tumor. Trans Pac Coast Obstet Gynecol Soc. 1978;45:78-81.
18. Kathiresan AS, Johnson JN, Hood BJ, Montoya SP, Vanni S, Gonzalez-Quintero VH. Giant cell bone tumor of the thoracic spine presenting in late pregnancy. Obstet Gynecol. 2011;118(2 pt 2):428-431.
19. Komiya S, Zenmyo M, Inoue A. Bone tumors in the pelvis presenting growth during pregnancy. Arch Orthop Trauma Surg. 1999;119(1-2):22-29.
20. Ross AE, Bojescul JA, Kuklo TR. Giant cell tumor: a case report of recurrence during pregnancy. Spine. 2005;30(12):E332-3E35.
21. Sharma JB, Chanana C, Rastogi, et al. Successful pregnancy outcome with elective caesarean section following two attempts of surgical excision of large giant cell tumor of the lower limb during pregnancy. Arch Gynecol Obstet. 2006;274(5):313-315.
22. Demertzis N, Kotsiandri F, Giotis I, Apostolikas N. Giant-cell tumors of bone and progesterone receptors. Orthopedics. 2003;26(12):1209-1212.
23. Taylor RM, Kashima TG, Knowles HJ, Athanasou NA. VEGF, FLT3 ligand, PlGF and HGF can substitute for M-CSF to induce human osteoclast formation: implications for giant cell tumour pathobiology. Lab Invest. 2012;92(10):1398-1406.
24. Lawless ME, Jour G, Hoch BL, Rendi MH. Beta-human chorionic gonadotropin expression in recurrent and metastatic giant cell tumors of bone: a potential mimicker of germ cell tumor. Int J Surg Pathol. 2014;22(7):617-622.
25. Viswanathan S, Jambhekar NA. Metastatic giant cell tumor of bone: are there associated factors and best treatment modalities? Clin Orthop Relat Res. 2010;468(3):827-833.
26. Kaban LB, Troulis MJ, Ebb D, August M, Hornicek FJ, Dodson TB. Antiangiogenic therapy with interferon alpha for giant cell lesions of the jaws. J Oral Maxillofac Surg. 2002;60(10):1103-1111.
27. Kaiser U, Neumann K, Havemann K. Generalised giant-cell tumour of bone: successful treatment of pulmonary metastases with interferon alpha, a case report. J Cancer Res Clin Oncol. 1993;119(5):301-303.
28. Dickerman JD. Interferon and giant cell tumors. Pediatrics. 1999;103(6 pt 1):1282-1283.
29. Balke M, Campanacci L, Gebert C, et al. Bisphosphonate treatment of aggressive primary, recurrent and metastatic giant cell tumour of bone. BMC Cancer. 2010;10:462.
30. Gille O, Oliveira Bde A, Guerin P, Lepreux S, Richez C, Vital JM. Regression of giant cell tumor of the cervical spine with bisphosphonate as single therapy. Spine. 2012;37(6):E396-E399.
31. Moriceau G, Ory B, Gobin B, et al. Therapeutic approach of primary bone tumours by bisphosphonates. Curr Pharm Des. 2010;16(27):2981-2987.
32. Tse LF, Wong KC, Kumta SM, Huang L, Chow TC, Griffith JF. Bisphosphonates reduce local recurrence in extremity giant cell tumor of bone: a case–control study. Bone. 2008;42(1):68-73.
33. Thomas D, Henshaw R, Skubitz K, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11(3):275-280.
34. Balke M, Hardes J. Denosumab: a breakthrough in treatment of giant-cell tumour of bone? Lancet Oncol. 2010;11(3):218-219.
35. Kyrgidis A, Toulis K. Safety and efficacy of denosumab in giant-cell tumour of bone. Lancet Oncol. 2010;11(6):513-514.
36. Thomas D, Carriere P, Jacobs I. Safety of denosumab in giant-cell tumour of bone. Lancet Oncol. 2010;11(9):815.
37. Skubitz KM. Giant cell tumor of bone: current treatment options. Curr Treat Options Oncol. 2014;15(3):507-518.
38. Chawla S, Henshaw R, Seeger L, et al. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: interim analysis of an open-label, parallel-group, phase 2 study. Lancet Oncol. 2013;14(9):901-908.
Amyopathic Dermatomyositis With Plantar Keratoderma Responding to Methotrexate Therapy
Case Report
A 54-year-old woman presented with a painful pruritic rash on the hands and feet of 7 years’ duration. She reported intermittent joint pain but denied muscle weakness. Physical examination revealed fissured fingertips and heavy scaling of the palms and lateral fingers (Figure 1). Violaceous scaly papules were seen on the distal and proximal interphalangeal joints (Figure 2). A severe plantar keratoderma also was noted (Figure 3). Pink scaly plaques were present on the bilateral elbows and postauricular skin. Diffuse mat telangiectases covered the malar skin. Extensive poikilodermatous skin changes covered approximately 20% of the total body surface area. Salt-and-pepper patches and papules were noted over the bilateral thighs. She reported an uncertain history of recent radiographs of one or both hands, which showed no joint degeneration characteristic of psoriatic arthritis. She previously had been given a diagnosis of psoriasis by an outside dermatologist but was not responding to topical therapy.

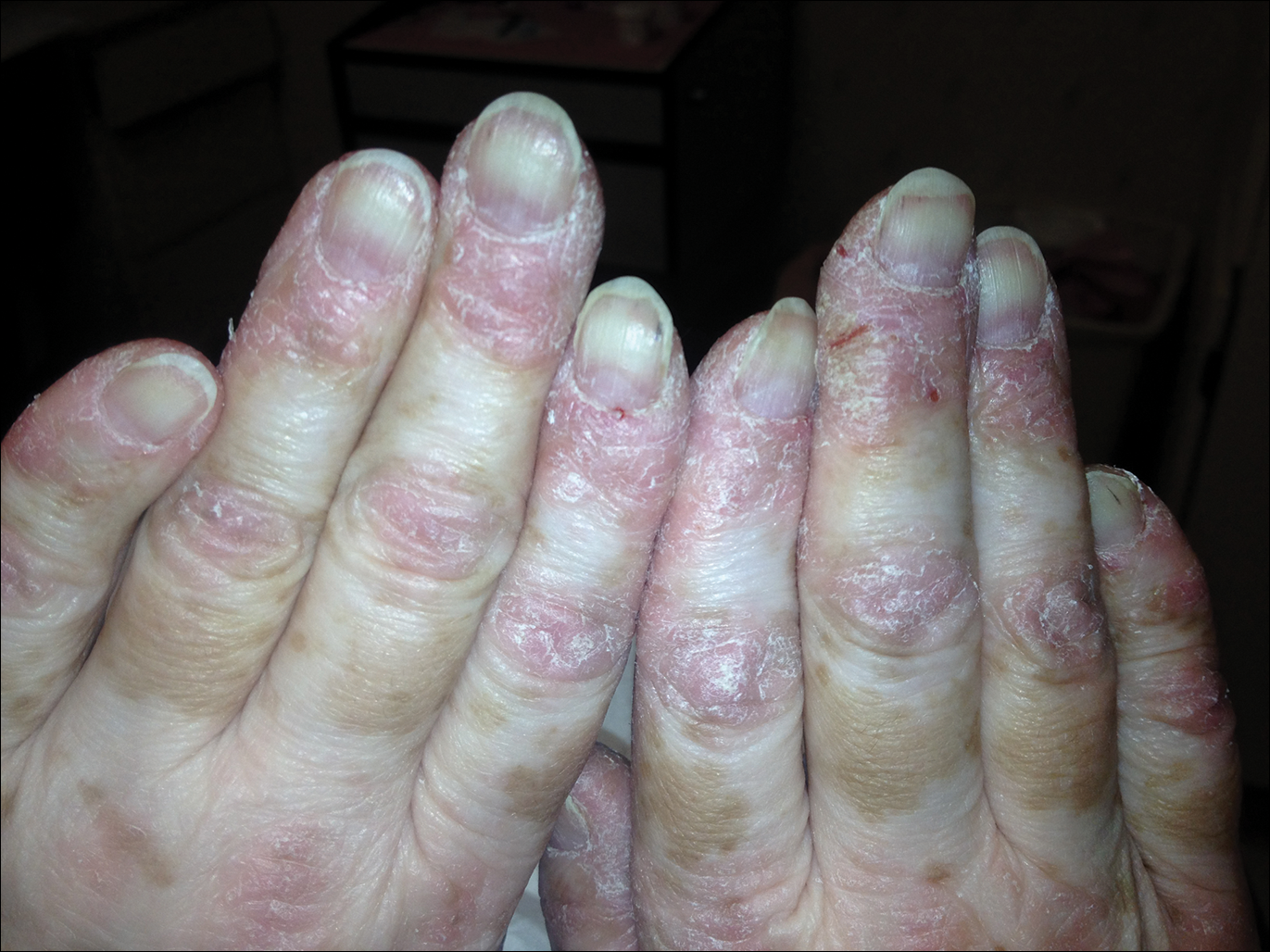
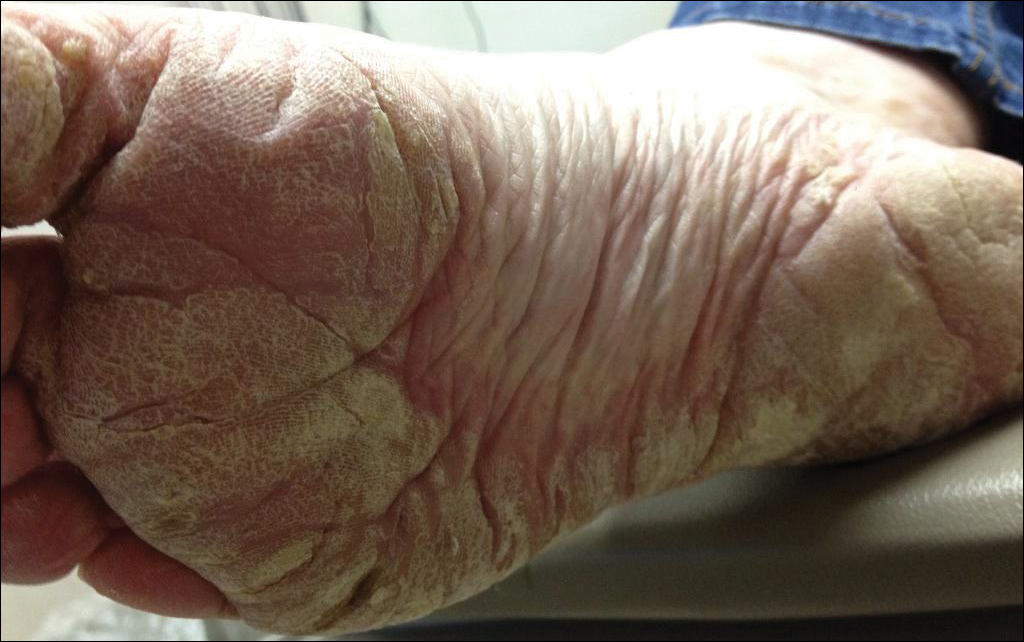
Several skin biopsies showed histologic evidence of dermatomyositis (DM)(Figure 4). Prominent basement thickening also was seen on periodic acid–Schiff staining (not shown). Laboratory workup showed negative antinuclear antibodies and anti–Jo-1, anti-Ku, and anti-Mi2 antibodies. Muscle enzymes including creatinine kinase and aldolase were within reference range. Pelvic ultrasonography and mammography were negative. Pulmonary function tests were unremarkable. High-resolution chest computed tomography (CT) was ordered because of a history of chronic cough; however, no evidence of malignancy or interstitial lung disease was seen. The patient was diagnosed with amyopathic dermatomyositis (ADM). Rheumatology was consulted and initiated oral hydroxychloroquine therapy. After 3 months, the patient’s cutaneous disease did not respond and she reported having headaches associated with this medication; therefore, methotrexate was started. Within 2 months of treatment, full resolution of the plantar keratoderma (Figure 5) and clearance of the scaling/fissuring of the hands as well as the psoriatic-appearing plaques on the elbows was noted.
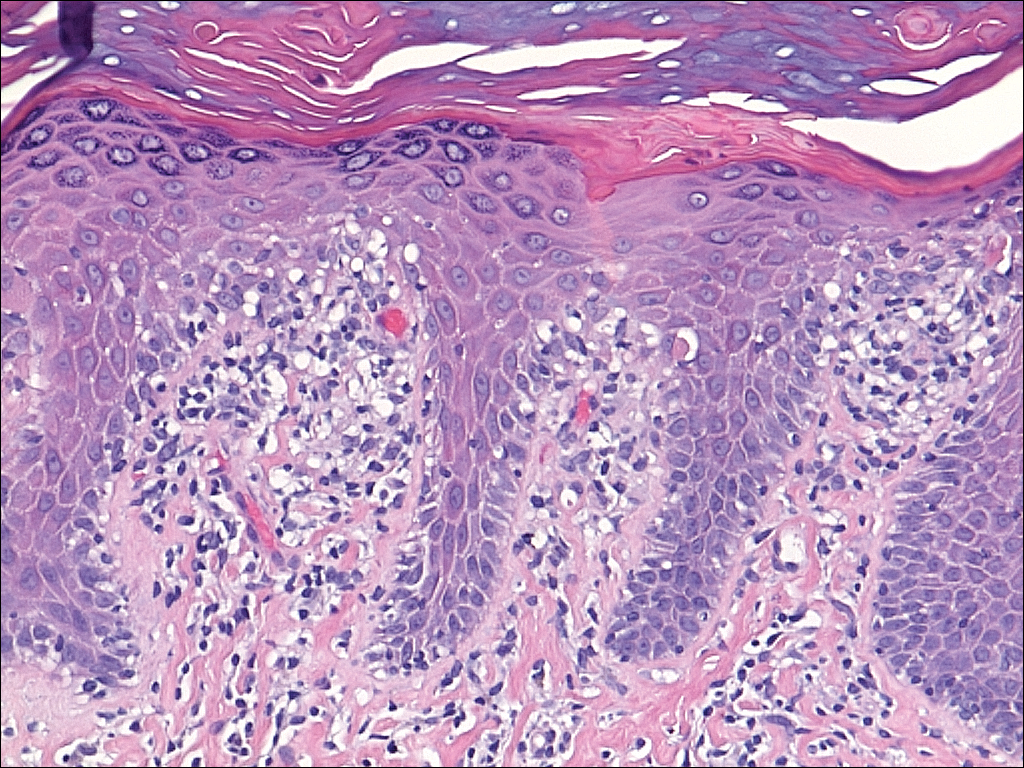
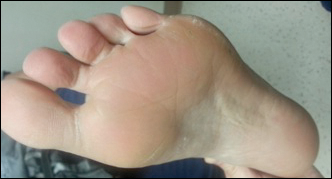
Comment
Amyopathic DM is a subset of DM that accounts for 10% to 20% of DM cases.1,2 Sontheimer’s3 diagnostic criteria for ADM require histopathologic confirmation of the hallmark skin findings of classic DM and lack of muscle weakness or muscle enzyme (creatine kinase/aldolase) elevation for at least 2 years.
Similar to classic DM, ADM typically presents in the fifth decade of life and has a female predilection.1,4 The term hypomyopathic DM is used to describe patients who exhibit classic skin findings and evidence of muscle involvement on magnetic resonance imaging, electromyography, biopsy, or serum enzymes but have no clinical evidence of muscle weakness for at least 6 months. Together, hypomyopathic DM and ADM are referred to as clinically ADM (CADM). Patients who have met the criteria for hypomyopathic DM or ADM may later develop frank myopathy, progressing to a diagnosis of CADM, which may occur in as many as 10% to 13% of cases of CADM.1,2 Clinical evidence of muscle weakness typically is heralded by elevation of creatine kinase and aldolase; therefore, patients with ADM should have muscle enzymes periodically checked.
Cutaneous findings of ADM are the same as the hallmark skin findings in CADM.3 Poikiloderma appears as thin telangiectatic skin in a background of mottled hyperpigmentation and hypopigmentation. It represents chronic inflammation and often occurs in sun-exposed areas. Poikiloderma located on the posterior neck and shoulders is known as the shawl sign and on the lateral thighs as the holster sign.5 The term mechanic’s hands is used to describe the clinical finding of palmar erythema with scaling and fissuring of the fingertips.6 Scalp findings include erythematous, atrophic, scaly plaques resembling psoriasis and nonscarring alopecia.7 Gottron papules are nearly pathognomonic for DM. These violaceous papules often are pruritic and found over the finger joints, in contrast to the hand rash of lupus erythematosus that involves the skin between finger joints.8 Psoriatic-appearing plaques overlying the elbows and knees are known as Gottron sign and can contribute to misdiagnosis as psoriasis.8 The classic heliotrope rash presents as a violaceous hue in the periorbital area and may be associated with periorbital edema.9 Calcinosis cutis is common in CADM but rarely is reported in ADM.10 Nail findings include periungual hyperemia, cuticular overgrowth, and nail bed changes due to avascular areas and dilated capillaries. The cutaneous histopathologic findings in ADM are the same as with CADM: a smudged dermoepidermal interface, vacuolar alterations of the basal layer, and dermal mucin deposits.
Palmoplantar keratoderma rarely is reported as a cutaneous finding in DM. The finding of keratoderma has mainly been reported in association with Wong-type DM, a rare subtype of DM with features of pityriasis rubra pilaris.11-13 Palmoplantar keratoderma also has been reported in a case of an ADM-like hydroxyurea-induced eruption14 and as an early presenting feature in one patient with CADM and one with juvenile DM.15,16
The autoantibody profile in patients with ADM varies from that of CADM and can be helpful in both diagnosis and prognosis. Similar to CADM, the majority of patients with ADM have positive antinuclear antibodies.2,17 Anti–Jo-1 (an anti–aminoacyl-transfer RNA synthetase) antibody frequently is found in CADM but rarely in ADM.2 Anti–Jo-1 is predictive of interstitial lung disease (ILD) in CADM. Positive anti–Jo-1 in combination with Raynaud phenomenon and mechanic’s hands is referred to as antisynthetase syndrome in patients with CADM.18,19 An antibody uniquely linked with CADM is the anti–CADM-140/MDA5 antibody and can be a marker of rapidly progressing ILD in these patients.20 Anti–Mi-2 is another myositis-specific antibody not commonly found in ADM but is present in 15% to 30% of DM cases.2,21 In CADM, the anti–Mi-2 antibody is associated with the shawl sign, ragged cuticles, and carpal tunnel syndrome and has a favorable prognosis.17,21 Myositis-associated autoantibodies (eg, anti-Ku) are found in patients with symptoms overlapping both DM and scleroderma or other connective tissue diseases.22 More recently described, the anti-p155/140 antibody is highly specific (up to 89%) for occult malignancy in DM.23
Lung disease is an important association in ADM. When it develops, it may be more aggressive compared to lung disease associated with CADM.24-26 In a systematic review of 197 cases of ADM by Gerami et al,2 10% of patients had ILD, and it was fatal in 42% of cases. Most cases of ILD associated with CADM were diagnosed as interstitial pneumonitis or diffuse alveolar disease; bronchiolitis obliterans organizing pneumonia and basilar fibrosis also were recorded.2 Anti–Jo-1 antibodies often accompany lung disease in CADM but are not typically found in lung disease associated with ADM. The anti–CADM-140/MDA5 antibody is associated with an increased risk for rapidly progressing ILD in patients with CADM.20 Recommended baseline screening for lung disease in DM includes chest radiography, pulmonary function tests with diffusion capacity,8 and in some instances high-resolution chest CT.27 Follow-up visits should include screening for symptoms of ILD such as cough, shortness of breath, or dyspnea. Treatment of myopathy-associated ILD is systemic steroids combined with various immunosuppressants including cyclophosphamide, azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus, and intravenous immunoglobulin.28,29
The risk of malignancy in ADM is thought to be similar to the rate of 20% to 25% found in CADM.1,30-32 The most commonly reported malignancies associated with ADM are nasopharyngeal, breast, lung, ovarian, colorectal, pancreatic, and stomach cancers and lymphoma/leukemia.2,33 Patients with ADM should be screened for malignancy at diagnosis, then yearly for 3 years.8,31,33 In addition to history, physical examination, and age/sex-appropriate screening, a complete blood cell count, chemistry panel, urinalysis, stool guaiac, CA 125, CA 19-9, chest radiograph, and abdominal ultrasound should be performed. For women, mammography and pelvic ultrasonography should be completed.31 Some experts also recommend a full-body CT scan. Because Asian patients have a higher risk for nasopharyngeal carcinoma, referral to an ear, nose, and throat surgeon for direct visualization also can be considered.33 The risk of cancer in patients with DM compared to the general population is increased for at least the first 5 years after diagnosis, but most associated cancers are found within the first 3 years.34
Several therapies have been found useful in ADM. Because lesions often are photoexacerbated, sun protection is essential. Antimalarials such as hydroxychloroquine are considered first-line therapy. Clinicians must be aware of 2 possible hydroxychloroquine side effects that can uniquely confuse the clinical picture in ADM. The first is a rash, most often morbilliform and pruritic, that occurs in DM more frequently than in other diseases.35 The second is a myopathy found in as many as 6.7% of patients using antimalarials for rheumatic disease,36 which can clinically mimic the progression of ADM to CADM.37 Two small retrospective case series found that methotrexate was beneficial in ADM.38,39 Methotrexate also has been reported as an efficacious treatment of ILD in patients with connective tissue diseases.40,41 Intravenous immunoglobulin and other immunosuppressants are additional agents to be considered.42
In summary, ADM is an important subset of DM and is more likely to present to dermatology practices than to other specialists. Amyopathic DM shares cutaneous findings with DM, and both overlap and differ with respect to other key disease characteristics including autoantibody profile, associated lung disease, and malignancy risk. Palmoplantar keratoderma is a rarely reported skin finding in DM. We report a case of ADM with the unique finding of severe plantar keratoderma. The fact that our patient’s keratoderma and other skin findings resolved concomitantly during methotrexate therapy leads us to believe that the keratoderma was a unique skin manifestation of the ADM itself.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30.
- Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54:597-613.
- Sontheimer RD. Cutaneous features of classic dermatomyositis and amyopathic dermatomyositis. Curr Opin Rheumatol. 1999;11:475-482.
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Stahl NI, Klippel JH, Decker JL. A cutaneous lesion associated with myositis. Ann Intern Med. 1979;91:577-579.
- Kasteler JS, Callen JP. Scalp involvement in dermatomyositis. often overlooked or misdiagnosed. JAMA. 1994;272:1939-1941.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Russo T, Piccolo V, Ruocco E, et al. The heliotrope sign of dermatomyositis: the correct meaning of the term heliotrope. Arch Dermatol. 2012;148:1178.
- Peñate Y, Guillermo N, Melwani P, et al. Calcinosis cutis associated with amyopathic dermatomyositis: response to intravenous immunoglobulin. J Am Acad Dermatol. 2009;60:1076-1077.
- Requena L, Grilli R, Soriano L, et al. Dermatomyositis with a pityriasis rubra pilaris-like eruption: a little-known distinctive cutaneous manifestation of dermatomyositis. Br J Dermatol. 1997;136:768-771.
- Lupton JR, Figueroa P, Berberian BJ, et al. An unusual presentation of dermatomyositis: the type Wong variant revisited. J Am Acad Dermatol. 2000;43(5 part 2):908-912.
- Caporali R, Cavagna L, Bellosta M, et al. Inflammatory myopathy in a patient with cutaneous findings of pityriasis rubra pilaris: a case of Wong’s dermatomyositis. Clin Rheumatol. 2004;23:63-65.
- Nofal A, El-Din ES. Hydroxyurea-induced dermatomyositis: true amyopathic dermatomyositis or dermatomyositis-like eruption? Int J Dermatol. 2012;51:535-541.
- See Y, Rooney M, Woo P. Palmar plantar hyperkeratosis—a previously undescribed skin manifestation of juvenile dermatomyositis. Br J Rheumatol. 1997;36(8):917-919.
- Chang LY, Yang LJ, Wu YJJ. Keratoderma plantaris and mechanic’s hands as the initial presentation in a case of dermatomyositis. Dermatol Sinica. 2002;20:329-334.
- Love L, Leff R, Fraser D, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70:360-374.
- Marguerie C, Bunn CC, Beynon HL, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med. 1990;77:1019-1038.
- Marie I, Hatron PY, Hachulla E, et al. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol. 1998;25:1336-1343.
- Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52:1571-1576.
- Dimachkie MM. Idiopathic inflammatory myopathies. J Neuroimmunol. 2011;231:32-42.
- Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther. 2011;13:209.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, et al. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol. 2010;22:627-632.
- Kang EH, Lee EB, Shin KC, et al. Interstitial lung disease in patients with polymyositis, dermatomyositis, and amyopathic dermatomyositis. Rheumatology (Oxford). 2005;44:1282-1286.
- Ye S, Chen XX, Lu XY, et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: a retrospective cohort study. Clin Rheumatol. 2007;26:1647-1654.
- Mukae H, Ishimoto H, Sakamoto N, et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest. 2009;136:1341-1347.
- Fathi M, Dastmalchi, M, Rasmussen E, et al. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis. 2004;63:297-301.
- Kalluri M, Oddis CV. Pulmonary manifestations of the idiopathic inflammatory myopathies. Clin Chest Med. 2010;31:501-512.
- Mira-Avendano IC, Parambil JG, Yadav R, et al. A retrospective review of clinical features and treatment outcomes in steroid-resistant interstitial lung disease from polymyositis/dermatomyositis. Respir Med. 2013;107:890-896.
- Klein RQ, Teal V, Taylor L, et al. Number, characteristics, and classification of patients with dermatomyositis seen by dermatology and rheumatology departments at a large tertiary medical center. J Am Acad Dermatol. 2007;57:937-943.
- Sontheimer RD. Clinically amyopathic dermatomyositis: what can we now tell our patients? Arch Dermatol. 2010;146:76-80.
- Azuma K, Yamada H, Ohkubo M, et al. Incidence and predictive factors for malignancies in 136 Japanese patients with dermatomyositis, polymyositis and clinically amyopathic dermatomyositis. Mod Rheumatol. 2011;21:178-183.
- Femia AN, Vleugels RA, Callen JP. Cutaneous dermatomyositis: an updated review of treatment options and internal associations. Am J Clin Dermatol. 2013;14:291-313.
- Buchbinder R, Forbes A, Hall S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based cohort study. Ann Intern Med. 2001;134:1087-1095.
- Pelle MT, Callen JP. Adverse cutaneous reactions to hydroxychloroquine are more common in patients with dermatomyositis than in patients with cutaneous lupus erythematosus. Arch Dermatol. 2002;138:1231-1233.
- Casado E, Gratacós J, Tolosa C, et al. Antimalarial myopathy: an underdiagnosed complication? prospective longitudinal study of 119 patients. Ann Rheum Dis. 2006;65:385-390.
- Zieglschmid-Adams ME, Pandya AG, Cohen SB, et al. Treatment of dermatomyositis with methotrexate. J Am Acad Dermatol. 1995;32(5, pt 1):754-757.
- Foulke G, Baccon J, Marks JG, et al. Antimalarial myopathy in amyopathic dermatomyositis. Arch Dermatol. 2012;148:1100-1101.
- Kasteler JS, Callen JP. Low-dose methotrexate administered weekly is an effective corticosteroid-sparing agent for the treatment of the cutaneous manifestations of dermatomyositis. J Am Acad Dermatol. 1997;36:67-71.
- Scott DG, Bacon PA. Response to methotrexate in fibrosing alveolitis associated with connective tissue disease. Thorax. 1980;35:725-731.
- Fink SD, Kremer JM. Successful treatment of interstitial lung disease in systemic lupus erythematosus with methotrexate. J Rheumatol. 1995;22:967-969.
- Ernste FC, Reed AM. Idiopathic inflammatory myopathies: current trends in pathogenesis, clinical features, and up-to-date treatment recommendations. Mayo Clin Proc. 2013;88:83-105.
Case Report
A 54-year-old woman presented with a painful pruritic rash on the hands and feet of 7 years’ duration. She reported intermittent joint pain but denied muscle weakness. Physical examination revealed fissured fingertips and heavy scaling of the palms and lateral fingers (Figure 1). Violaceous scaly papules were seen on the distal and proximal interphalangeal joints (Figure 2). A severe plantar keratoderma also was noted (Figure 3). Pink scaly plaques were present on the bilateral elbows and postauricular skin. Diffuse mat telangiectases covered the malar skin. Extensive poikilodermatous skin changes covered approximately 20% of the total body surface area. Salt-and-pepper patches and papules were noted over the bilateral thighs. She reported an uncertain history of recent radiographs of one or both hands, which showed no joint degeneration characteristic of psoriatic arthritis. She previously had been given a diagnosis of psoriasis by an outside dermatologist but was not responding to topical therapy.
Several skin biopsies showed histologic evidence of dermatomyositis (DM)(Figure 4). Prominent basement thickening also was seen on periodic acid–Schiff staining (not shown). Laboratory workup showed negative antinuclear antibodies and anti–Jo-1, anti-Ku, and anti-Mi2 antibodies. Muscle enzymes including creatinine kinase and aldolase were within reference range. Pelvic ultrasonography and mammography were negative. Pulmonary function tests were unremarkable. High-resolution chest computed tomography (CT) was ordered because of a history of chronic cough; however, no evidence of malignancy or interstitial lung disease was seen. The patient was diagnosed with amyopathic dermatomyositis (ADM). Rheumatology was consulted and initiated oral hydroxychloroquine therapy. After 3 months, the patient’s cutaneous disease did not respond and she reported having headaches associated with this medication; therefore, methotrexate was started. Within 2 months of treatment, full resolution of the plantar keratoderma (Figure 5) and clearance of the scaling/fissuring of the hands as well as the psoriatic-appearing plaques on the elbows was noted.
Comment
Amyopathic DM is a subset of DM that accounts for 10% to 20% of DM cases.1,2 Sontheimer’s3 diagnostic criteria for ADM require histopathologic confirmation of the hallmark skin findings of classic DM and lack of muscle weakness or muscle enzyme (creatine kinase/aldolase) elevation for at least 2 years.
Similar to classic DM, ADM typically presents in the fifth decade of life and has a female predilection.1,4 The term hypomyopathic DM is used to describe patients who exhibit classic skin findings and evidence of muscle involvement on magnetic resonance imaging, electromyography, biopsy, or serum enzymes but have no clinical evidence of muscle weakness for at least 6 months. Together, hypomyopathic DM and ADM are referred to as clinically ADM (CADM). Patients who have met the criteria for hypomyopathic DM or ADM may later develop frank myopathy, progressing to a diagnosis of CADM, which may occur in as many as 10% to 13% of cases of CADM.1,2 Clinical evidence of muscle weakness typically is heralded by elevation of creatine kinase and aldolase; therefore, patients with ADM should have muscle enzymes periodically checked.
Cutaneous findings of ADM are the same as the hallmark skin findings in CADM.3 Poikiloderma appears as thin telangiectatic skin in a background of mottled hyperpigmentation and hypopigmentation. It represents chronic inflammation and often occurs in sun-exposed areas. Poikiloderma located on the posterior neck and shoulders is known as the shawl sign and on the lateral thighs as the holster sign.5 The term mechanic’s hands is used to describe the clinical finding of palmar erythema with scaling and fissuring of the fingertips.6 Scalp findings include erythematous, atrophic, scaly plaques resembling psoriasis and nonscarring alopecia.7 Gottron papules are nearly pathognomonic for DM. These violaceous papules often are pruritic and found over the finger joints, in contrast to the hand rash of lupus erythematosus that involves the skin between finger joints.8 Psoriatic-appearing plaques overlying the elbows and knees are known as Gottron sign and can contribute to misdiagnosis as psoriasis.8 The classic heliotrope rash presents as a violaceous hue in the periorbital area and may be associated with periorbital edema.9 Calcinosis cutis is common in CADM but rarely is reported in ADM.10 Nail findings include periungual hyperemia, cuticular overgrowth, and nail bed changes due to avascular areas and dilated capillaries. The cutaneous histopathologic findings in ADM are the same as with CADM: a smudged dermoepidermal interface, vacuolar alterations of the basal layer, and dermal mucin deposits.
Palmoplantar keratoderma rarely is reported as a cutaneous finding in DM. The finding of keratoderma has mainly been reported in association with Wong-type DM, a rare subtype of DM with features of pityriasis rubra pilaris.11-13 Palmoplantar keratoderma also has been reported in a case of an ADM-like hydroxyurea-induced eruption14 and as an early presenting feature in one patient with CADM and one with juvenile DM.15,16
The autoantibody profile in patients with ADM varies from that of CADM and can be helpful in both diagnosis and prognosis. Similar to CADM, the majority of patients with ADM have positive antinuclear antibodies.2,17 Anti–Jo-1 (an anti–aminoacyl-transfer RNA synthetase) antibody frequently is found in CADM but rarely in ADM.2 Anti–Jo-1 is predictive of interstitial lung disease (ILD) in CADM. Positive anti–Jo-1 in combination with Raynaud phenomenon and mechanic’s hands is referred to as antisynthetase syndrome in patients with CADM.18,19 An antibody uniquely linked with CADM is the anti–CADM-140/MDA5 antibody and can be a marker of rapidly progressing ILD in these patients.20 Anti–Mi-2 is another myositis-specific antibody not commonly found in ADM but is present in 15% to 30% of DM cases.2,21 In CADM, the anti–Mi-2 antibody is associated with the shawl sign, ragged cuticles, and carpal tunnel syndrome and has a favorable prognosis.17,21 Myositis-associated autoantibodies (eg, anti-Ku) are found in patients with symptoms overlapping both DM and scleroderma or other connective tissue diseases.22 More recently described, the anti-p155/140 antibody is highly specific (up to 89%) for occult malignancy in DM.23
Lung disease is an important association in ADM. When it develops, it may be more aggressive compared to lung disease associated with CADM.24-26 In a systematic review of 197 cases of ADM by Gerami et al,2 10% of patients had ILD, and it was fatal in 42% of cases. Most cases of ILD associated with CADM were diagnosed as interstitial pneumonitis or diffuse alveolar disease; bronchiolitis obliterans organizing pneumonia and basilar fibrosis also were recorded.2 Anti–Jo-1 antibodies often accompany lung disease in CADM but are not typically found in lung disease associated with ADM. The anti–CADM-140/MDA5 antibody is associated with an increased risk for rapidly progressing ILD in patients with CADM.20 Recommended baseline screening for lung disease in DM includes chest radiography, pulmonary function tests with diffusion capacity,8 and in some instances high-resolution chest CT.27 Follow-up visits should include screening for symptoms of ILD such as cough, shortness of breath, or dyspnea. Treatment of myopathy-associated ILD is systemic steroids combined with various immunosuppressants including cyclophosphamide, azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus, and intravenous immunoglobulin.28,29
The risk of malignancy in ADM is thought to be similar to the rate of 20% to 25% found in CADM.1,30-32 The most commonly reported malignancies associated with ADM are nasopharyngeal, breast, lung, ovarian, colorectal, pancreatic, and stomach cancers and lymphoma/leukemia.2,33 Patients with ADM should be screened for malignancy at diagnosis, then yearly for 3 years.8,31,33 In addition to history, physical examination, and age/sex-appropriate screening, a complete blood cell count, chemistry panel, urinalysis, stool guaiac, CA 125, CA 19-9, chest radiograph, and abdominal ultrasound should be performed. For women, mammography and pelvic ultrasonography should be completed.31 Some experts also recommend a full-body CT scan. Because Asian patients have a higher risk for nasopharyngeal carcinoma, referral to an ear, nose, and throat surgeon for direct visualization also can be considered.33 The risk of cancer in patients with DM compared to the general population is increased for at least the first 5 years after diagnosis, but most associated cancers are found within the first 3 years.34
Several therapies have been found useful in ADM. Because lesions often are photoexacerbated, sun protection is essential. Antimalarials such as hydroxychloroquine are considered first-line therapy. Clinicians must be aware of 2 possible hydroxychloroquine side effects that can uniquely confuse the clinical picture in ADM. The first is a rash, most often morbilliform and pruritic, that occurs in DM more frequently than in other diseases.35 The second is a myopathy found in as many as 6.7% of patients using antimalarials for rheumatic disease,36 which can clinically mimic the progression of ADM to CADM.37 Two small retrospective case series found that methotrexate was beneficial in ADM.38,39 Methotrexate also has been reported as an efficacious treatment of ILD in patients with connective tissue diseases.40,41 Intravenous immunoglobulin and other immunosuppressants are additional agents to be considered.42
In summary, ADM is an important subset of DM and is more likely to present to dermatology practices than to other specialists. Amyopathic DM shares cutaneous findings with DM, and both overlap and differ with respect to other key disease characteristics including autoantibody profile, associated lung disease, and malignancy risk. Palmoplantar keratoderma is a rarely reported skin finding in DM. We report a case of ADM with the unique finding of severe plantar keratoderma. The fact that our patient’s keratoderma and other skin findings resolved concomitantly during methotrexate therapy leads us to believe that the keratoderma was a unique skin manifestation of the ADM itself.
Case Report
A 54-year-old woman presented with a painful pruritic rash on the hands and feet of 7 years’ duration. She reported intermittent joint pain but denied muscle weakness. Physical examination revealed fissured fingertips and heavy scaling of the palms and lateral fingers (Figure 1). Violaceous scaly papules were seen on the distal and proximal interphalangeal joints (Figure 2). A severe plantar keratoderma also was noted (Figure 3). Pink scaly plaques were present on the bilateral elbows and postauricular skin. Diffuse mat telangiectases covered the malar skin. Extensive poikilodermatous skin changes covered approximately 20% of the total body surface area. Salt-and-pepper patches and papules were noted over the bilateral thighs. She reported an uncertain history of recent radiographs of one or both hands, which showed no joint degeneration characteristic of psoriatic arthritis. She previously had been given a diagnosis of psoriasis by an outside dermatologist but was not responding to topical therapy.
Several skin biopsies showed histologic evidence of dermatomyositis (DM)(Figure 4). Prominent basement thickening also was seen on periodic acid–Schiff staining (not shown). Laboratory workup showed negative antinuclear antibodies and anti–Jo-1, anti-Ku, and anti-Mi2 antibodies. Muscle enzymes including creatinine kinase and aldolase were within reference range. Pelvic ultrasonography and mammography were negative. Pulmonary function tests were unremarkable. High-resolution chest computed tomography (CT) was ordered because of a history of chronic cough; however, no evidence of malignancy or interstitial lung disease was seen. The patient was diagnosed with amyopathic dermatomyositis (ADM). Rheumatology was consulted and initiated oral hydroxychloroquine therapy. After 3 months, the patient’s cutaneous disease did not respond and she reported having headaches associated with this medication; therefore, methotrexate was started. Within 2 months of treatment, full resolution of the plantar keratoderma (Figure 5) and clearance of the scaling/fissuring of the hands as well as the psoriatic-appearing plaques on the elbows was noted.
Comment
Amyopathic DM is a subset of DM that accounts for 10% to 20% of DM cases.1,2 Sontheimer’s3 diagnostic criteria for ADM require histopathologic confirmation of the hallmark skin findings of classic DM and lack of muscle weakness or muscle enzyme (creatine kinase/aldolase) elevation for at least 2 years.
Similar to classic DM, ADM typically presents in the fifth decade of life and has a female predilection.1,4 The term hypomyopathic DM is used to describe patients who exhibit classic skin findings and evidence of muscle involvement on magnetic resonance imaging, electromyography, biopsy, or serum enzymes but have no clinical evidence of muscle weakness for at least 6 months. Together, hypomyopathic DM and ADM are referred to as clinically ADM (CADM). Patients who have met the criteria for hypomyopathic DM or ADM may later develop frank myopathy, progressing to a diagnosis of CADM, which may occur in as many as 10% to 13% of cases of CADM.1,2 Clinical evidence of muscle weakness typically is heralded by elevation of creatine kinase and aldolase; therefore, patients with ADM should have muscle enzymes periodically checked.
Cutaneous findings of ADM are the same as the hallmark skin findings in CADM.3 Poikiloderma appears as thin telangiectatic skin in a background of mottled hyperpigmentation and hypopigmentation. It represents chronic inflammation and often occurs in sun-exposed areas. Poikiloderma located on the posterior neck and shoulders is known as the shawl sign and on the lateral thighs as the holster sign.5 The term mechanic’s hands is used to describe the clinical finding of palmar erythema with scaling and fissuring of the fingertips.6 Scalp findings include erythematous, atrophic, scaly plaques resembling psoriasis and nonscarring alopecia.7 Gottron papules are nearly pathognomonic for DM. These violaceous papules often are pruritic and found over the finger joints, in contrast to the hand rash of lupus erythematosus that involves the skin between finger joints.8 Psoriatic-appearing plaques overlying the elbows and knees are known as Gottron sign and can contribute to misdiagnosis as psoriasis.8 The classic heliotrope rash presents as a violaceous hue in the periorbital area and may be associated with periorbital edema.9 Calcinosis cutis is common in CADM but rarely is reported in ADM.10 Nail findings include periungual hyperemia, cuticular overgrowth, and nail bed changes due to avascular areas and dilated capillaries. The cutaneous histopathologic findings in ADM are the same as with CADM: a smudged dermoepidermal interface, vacuolar alterations of the basal layer, and dermal mucin deposits.
Palmoplantar keratoderma rarely is reported as a cutaneous finding in DM. The finding of keratoderma has mainly been reported in association with Wong-type DM, a rare subtype of DM with features of pityriasis rubra pilaris.11-13 Palmoplantar keratoderma also has been reported in a case of an ADM-like hydroxyurea-induced eruption14 and as an early presenting feature in one patient with CADM and one with juvenile DM.15,16
The autoantibody profile in patients with ADM varies from that of CADM and can be helpful in both diagnosis and prognosis. Similar to CADM, the majority of patients with ADM have positive antinuclear antibodies.2,17 Anti–Jo-1 (an anti–aminoacyl-transfer RNA synthetase) antibody frequently is found in CADM but rarely in ADM.2 Anti–Jo-1 is predictive of interstitial lung disease (ILD) in CADM. Positive anti–Jo-1 in combination with Raynaud phenomenon and mechanic’s hands is referred to as antisynthetase syndrome in patients with CADM.18,19 An antibody uniquely linked with CADM is the anti–CADM-140/MDA5 antibody and can be a marker of rapidly progressing ILD in these patients.20 Anti–Mi-2 is another myositis-specific antibody not commonly found in ADM but is present in 15% to 30% of DM cases.2,21 In CADM, the anti–Mi-2 antibody is associated with the shawl sign, ragged cuticles, and carpal tunnel syndrome and has a favorable prognosis.17,21 Myositis-associated autoantibodies (eg, anti-Ku) are found in patients with symptoms overlapping both DM and scleroderma or other connective tissue diseases.22 More recently described, the anti-p155/140 antibody is highly specific (up to 89%) for occult malignancy in DM.23
Lung disease is an important association in ADM. When it develops, it may be more aggressive compared to lung disease associated with CADM.24-26 In a systematic review of 197 cases of ADM by Gerami et al,2 10% of patients had ILD, and it was fatal in 42% of cases. Most cases of ILD associated with CADM were diagnosed as interstitial pneumonitis or diffuse alveolar disease; bronchiolitis obliterans organizing pneumonia and basilar fibrosis also were recorded.2 Anti–Jo-1 antibodies often accompany lung disease in CADM but are not typically found in lung disease associated with ADM. The anti–CADM-140/MDA5 antibody is associated with an increased risk for rapidly progressing ILD in patients with CADM.20 Recommended baseline screening for lung disease in DM includes chest radiography, pulmonary function tests with diffusion capacity,8 and in some instances high-resolution chest CT.27 Follow-up visits should include screening for symptoms of ILD such as cough, shortness of breath, or dyspnea. Treatment of myopathy-associated ILD is systemic steroids combined with various immunosuppressants including cyclophosphamide, azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus, and intravenous immunoglobulin.28,29
The risk of malignancy in ADM is thought to be similar to the rate of 20% to 25% found in CADM.1,30-32 The most commonly reported malignancies associated with ADM are nasopharyngeal, breast, lung, ovarian, colorectal, pancreatic, and stomach cancers and lymphoma/leukemia.2,33 Patients with ADM should be screened for malignancy at diagnosis, then yearly for 3 years.8,31,33 In addition to history, physical examination, and age/sex-appropriate screening, a complete blood cell count, chemistry panel, urinalysis, stool guaiac, CA 125, CA 19-9, chest radiograph, and abdominal ultrasound should be performed. For women, mammography and pelvic ultrasonography should be completed.31 Some experts also recommend a full-body CT scan. Because Asian patients have a higher risk for nasopharyngeal carcinoma, referral to an ear, nose, and throat surgeon for direct visualization also can be considered.33 The risk of cancer in patients with DM compared to the general population is increased for at least the first 5 years after diagnosis, but most associated cancers are found within the first 3 years.34
Several therapies have been found useful in ADM. Because lesions often are photoexacerbated, sun protection is essential. Antimalarials such as hydroxychloroquine are considered first-line therapy. Clinicians must be aware of 2 possible hydroxychloroquine side effects that can uniquely confuse the clinical picture in ADM. The first is a rash, most often morbilliform and pruritic, that occurs in DM more frequently than in other diseases.35 The second is a myopathy found in as many as 6.7% of patients using antimalarials for rheumatic disease,36 which can clinically mimic the progression of ADM to CADM.37 Two small retrospective case series found that methotrexate was beneficial in ADM.38,39 Methotrexate also has been reported as an efficacious treatment of ILD in patients with connective tissue diseases.40,41 Intravenous immunoglobulin and other immunosuppressants are additional agents to be considered.42
In summary, ADM is an important subset of DM and is more likely to present to dermatology practices than to other specialists. Amyopathic DM shares cutaneous findings with DM, and both overlap and differ with respect to other key disease characteristics including autoantibody profile, associated lung disease, and malignancy risk. Palmoplantar keratoderma is a rarely reported skin finding in DM. We report a case of ADM with the unique finding of severe plantar keratoderma. The fact that our patient’s keratoderma and other skin findings resolved concomitantly during methotrexate therapy leads us to believe that the keratoderma was a unique skin manifestation of the ADM itself.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30.
- Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54:597-613.
- Sontheimer RD. Cutaneous features of classic dermatomyositis and amyopathic dermatomyositis. Curr Opin Rheumatol. 1999;11:475-482.
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Stahl NI, Klippel JH, Decker JL. A cutaneous lesion associated with myositis. Ann Intern Med. 1979;91:577-579.
- Kasteler JS, Callen JP. Scalp involvement in dermatomyositis. often overlooked or misdiagnosed. JAMA. 1994;272:1939-1941.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Russo T, Piccolo V, Ruocco E, et al. The heliotrope sign of dermatomyositis: the correct meaning of the term heliotrope. Arch Dermatol. 2012;148:1178.
- Peñate Y, Guillermo N, Melwani P, et al. Calcinosis cutis associated with amyopathic dermatomyositis: response to intravenous immunoglobulin. J Am Acad Dermatol. 2009;60:1076-1077.
- Requena L, Grilli R, Soriano L, et al. Dermatomyositis with a pityriasis rubra pilaris-like eruption: a little-known distinctive cutaneous manifestation of dermatomyositis. Br J Dermatol. 1997;136:768-771.
- Lupton JR, Figueroa P, Berberian BJ, et al. An unusual presentation of dermatomyositis: the type Wong variant revisited. J Am Acad Dermatol. 2000;43(5 part 2):908-912.
- Caporali R, Cavagna L, Bellosta M, et al. Inflammatory myopathy in a patient with cutaneous findings of pityriasis rubra pilaris: a case of Wong’s dermatomyositis. Clin Rheumatol. 2004;23:63-65.
- Nofal A, El-Din ES. Hydroxyurea-induced dermatomyositis: true amyopathic dermatomyositis or dermatomyositis-like eruption? Int J Dermatol. 2012;51:535-541.
- See Y, Rooney M, Woo P. Palmar plantar hyperkeratosis—a previously undescribed skin manifestation of juvenile dermatomyositis. Br J Rheumatol. 1997;36(8):917-919.
- Chang LY, Yang LJ, Wu YJJ. Keratoderma plantaris and mechanic’s hands as the initial presentation in a case of dermatomyositis. Dermatol Sinica. 2002;20:329-334.
- Love L, Leff R, Fraser D, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70:360-374.
- Marguerie C, Bunn CC, Beynon HL, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med. 1990;77:1019-1038.
- Marie I, Hatron PY, Hachulla E, et al. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol. 1998;25:1336-1343.
- Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52:1571-1576.
- Dimachkie MM. Idiopathic inflammatory myopathies. J Neuroimmunol. 2011;231:32-42.
- Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther. 2011;13:209.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, et al. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol. 2010;22:627-632.
- Kang EH, Lee EB, Shin KC, et al. Interstitial lung disease in patients with polymyositis, dermatomyositis, and amyopathic dermatomyositis. Rheumatology (Oxford). 2005;44:1282-1286.
- Ye S, Chen XX, Lu XY, et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: a retrospective cohort study. Clin Rheumatol. 2007;26:1647-1654.
- Mukae H, Ishimoto H, Sakamoto N, et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest. 2009;136:1341-1347.
- Fathi M, Dastmalchi, M, Rasmussen E, et al. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis. 2004;63:297-301.
- Kalluri M, Oddis CV. Pulmonary manifestations of the idiopathic inflammatory myopathies. Clin Chest Med. 2010;31:501-512.
- Mira-Avendano IC, Parambil JG, Yadav R, et al. A retrospective review of clinical features and treatment outcomes in steroid-resistant interstitial lung disease from polymyositis/dermatomyositis. Respir Med. 2013;107:890-896.
- Klein RQ, Teal V, Taylor L, et al. Number, characteristics, and classification of patients with dermatomyositis seen by dermatology and rheumatology departments at a large tertiary medical center. J Am Acad Dermatol. 2007;57:937-943.
- Sontheimer RD. Clinically amyopathic dermatomyositis: what can we now tell our patients? Arch Dermatol. 2010;146:76-80.
- Azuma K, Yamada H, Ohkubo M, et al. Incidence and predictive factors for malignancies in 136 Japanese patients with dermatomyositis, polymyositis and clinically amyopathic dermatomyositis. Mod Rheumatol. 2011;21:178-183.
- Femia AN, Vleugels RA, Callen JP. Cutaneous dermatomyositis: an updated review of treatment options and internal associations. Am J Clin Dermatol. 2013;14:291-313.
- Buchbinder R, Forbes A, Hall S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based cohort study. Ann Intern Med. 2001;134:1087-1095.
- Pelle MT, Callen JP. Adverse cutaneous reactions to hydroxychloroquine are more common in patients with dermatomyositis than in patients with cutaneous lupus erythematosus. Arch Dermatol. 2002;138:1231-1233.
- Casado E, Gratacós J, Tolosa C, et al. Antimalarial myopathy: an underdiagnosed complication? prospective longitudinal study of 119 patients. Ann Rheum Dis. 2006;65:385-390.
- Zieglschmid-Adams ME, Pandya AG, Cohen SB, et al. Treatment of dermatomyositis with methotrexate. J Am Acad Dermatol. 1995;32(5, pt 1):754-757.
- Foulke G, Baccon J, Marks JG, et al. Antimalarial myopathy in amyopathic dermatomyositis. Arch Dermatol. 2012;148:1100-1101.
- Kasteler JS, Callen JP. Low-dose methotrexate administered weekly is an effective corticosteroid-sparing agent for the treatment of the cutaneous manifestations of dermatomyositis. J Am Acad Dermatol. 1997;36:67-71.
- Scott DG, Bacon PA. Response to methotrexate in fibrosing alveolitis associated with connective tissue disease. Thorax. 1980;35:725-731.
- Fink SD, Kremer JM. Successful treatment of interstitial lung disease in systemic lupus erythematosus with methotrexate. J Rheumatol. 1995;22:967-969.
- Ernste FC, Reed AM. Idiopathic inflammatory myopathies: current trends in pathogenesis, clinical features, and up-to-date treatment recommendations. Mayo Clin Proc. 2013;88:83-105.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30.
- Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54:597-613.
- Sontheimer RD. Cutaneous features of classic dermatomyositis and amyopathic dermatomyositis. Curr Opin Rheumatol. 1999;11:475-482.
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Stahl NI, Klippel JH, Decker JL. A cutaneous lesion associated with myositis. Ann Intern Med. 1979;91:577-579.
- Kasteler JS, Callen JP. Scalp involvement in dermatomyositis. often overlooked or misdiagnosed. JAMA. 1994;272:1939-1941.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Russo T, Piccolo V, Ruocco E, et al. The heliotrope sign of dermatomyositis: the correct meaning of the term heliotrope. Arch Dermatol. 2012;148:1178.
- Peñate Y, Guillermo N, Melwani P, et al. Calcinosis cutis associated with amyopathic dermatomyositis: response to intravenous immunoglobulin. J Am Acad Dermatol. 2009;60:1076-1077.
- Requena L, Grilli R, Soriano L, et al. Dermatomyositis with a pityriasis rubra pilaris-like eruption: a little-known distinctive cutaneous manifestation of dermatomyositis. Br J Dermatol. 1997;136:768-771.
- Lupton JR, Figueroa P, Berberian BJ, et al. An unusual presentation of dermatomyositis: the type Wong variant revisited. J Am Acad Dermatol. 2000;43(5 part 2):908-912.
- Caporali R, Cavagna L, Bellosta M, et al. Inflammatory myopathy in a patient with cutaneous findings of pityriasis rubra pilaris: a case of Wong’s dermatomyositis. Clin Rheumatol. 2004;23:63-65.
- Nofal A, El-Din ES. Hydroxyurea-induced dermatomyositis: true amyopathic dermatomyositis or dermatomyositis-like eruption? Int J Dermatol. 2012;51:535-541.
- See Y, Rooney M, Woo P. Palmar plantar hyperkeratosis—a previously undescribed skin manifestation of juvenile dermatomyositis. Br J Rheumatol. 1997;36(8):917-919.
- Chang LY, Yang LJ, Wu YJJ. Keratoderma plantaris and mechanic’s hands as the initial presentation in a case of dermatomyositis. Dermatol Sinica. 2002;20:329-334.
- Love L, Leff R, Fraser D, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70:360-374.
- Marguerie C, Bunn CC, Beynon HL, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med. 1990;77:1019-1038.
- Marie I, Hatron PY, Hachulla E, et al. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol. 1998;25:1336-1343.
- Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52:1571-1576.
- Dimachkie MM. Idiopathic inflammatory myopathies. J Neuroimmunol. 2011;231:32-42.
- Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther. 2011;13:209.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, et al. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol. 2010;22:627-632.
- Kang EH, Lee EB, Shin KC, et al. Interstitial lung disease in patients with polymyositis, dermatomyositis, and amyopathic dermatomyositis. Rheumatology (Oxford). 2005;44:1282-1286.
- Ye S, Chen XX, Lu XY, et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: a retrospective cohort study. Clin Rheumatol. 2007;26:1647-1654.
- Mukae H, Ishimoto H, Sakamoto N, et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest. 2009;136:1341-1347.
- Fathi M, Dastmalchi, M, Rasmussen E, et al. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis. 2004;63:297-301.
- Kalluri M, Oddis CV. Pulmonary manifestations of the idiopathic inflammatory myopathies. Clin Chest Med. 2010;31:501-512.
- Mira-Avendano IC, Parambil JG, Yadav R, et al. A retrospective review of clinical features and treatment outcomes in steroid-resistant interstitial lung disease from polymyositis/dermatomyositis. Respir Med. 2013;107:890-896.
- Klein RQ, Teal V, Taylor L, et al. Number, characteristics, and classification of patients with dermatomyositis seen by dermatology and rheumatology departments at a large tertiary medical center. J Am Acad Dermatol. 2007;57:937-943.
- Sontheimer RD. Clinically amyopathic dermatomyositis: what can we now tell our patients? Arch Dermatol. 2010;146:76-80.
- Azuma K, Yamada H, Ohkubo M, et al. Incidence and predictive factors for malignancies in 136 Japanese patients with dermatomyositis, polymyositis and clinically amyopathic dermatomyositis. Mod Rheumatol. 2011;21:178-183.
- Femia AN, Vleugels RA, Callen JP. Cutaneous dermatomyositis: an updated review of treatment options and internal associations. Am J Clin Dermatol. 2013;14:291-313.
- Buchbinder R, Forbes A, Hall S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based cohort study. Ann Intern Med. 2001;134:1087-1095.
- Pelle MT, Callen JP. Adverse cutaneous reactions to hydroxychloroquine are more common in patients with dermatomyositis than in patients with cutaneous lupus erythematosus. Arch Dermatol. 2002;138:1231-1233.
- Casado E, Gratacós J, Tolosa C, et al. Antimalarial myopathy: an underdiagnosed complication? prospective longitudinal study of 119 patients. Ann Rheum Dis. 2006;65:385-390.
- Zieglschmid-Adams ME, Pandya AG, Cohen SB, et al. Treatment of dermatomyositis with methotrexate. J Am Acad Dermatol. 1995;32(5, pt 1):754-757.
- Foulke G, Baccon J, Marks JG, et al. Antimalarial myopathy in amyopathic dermatomyositis. Arch Dermatol. 2012;148:1100-1101.
- Kasteler JS, Callen JP. Low-dose methotrexate administered weekly is an effective corticosteroid-sparing agent for the treatment of the cutaneous manifestations of dermatomyositis. J Am Acad Dermatol. 1997;36:67-71.
- Scott DG, Bacon PA. Response to methotrexate in fibrosing alveolitis associated with connective tissue disease. Thorax. 1980;35:725-731.
- Fink SD, Kremer JM. Successful treatment of interstitial lung disease in systemic lupus erythematosus with methotrexate. J Rheumatol. 1995;22:967-969.
- Ernste FC, Reed AM. Idiopathic inflammatory myopathies: current trends in pathogenesis, clinical features, and up-to-date treatment recommendations. Mayo Clin Proc. 2013;88:83-105.
Practice Points
- Dermatomyositis (DM) can present without muscular weakness as clinically amyopathic dermatomyositis (CADM).
- Clinically amyopathic dermatomyositis has cutaneous findings that can mimic other diseases including psoriasis.
- Clinically amyopathic dermatomyositis may have similar systemic associations as DM in general, such as an increased risk for malignancies.
- Treatments to consider for CADM should include systemic methotrexate.
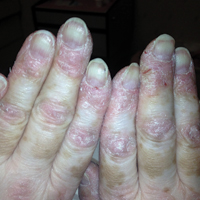
Traumatic Ulcerative Granuloma With Stromal Eosinophilia: A Malignant-Appearing Benign Lesion
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.

The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.


Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.
The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.
Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.
The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.
Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
Practice Points
- Immunohistochemical staining of traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) may suggest an underlying lymphoproliferative disorder.
- Early recognition of TUGSE, which often is malignant appearing, is key, with watchful waiting as the mainstay therapy.
- Adjunctive therapy for TUGSE includes prednisolone and oral analgesics.
Practitioner Cognitive Reframing: Working More Effectively in Addictions
I am a behavioral health-licensed clinical social worker, an approved motivational enhancement therapy provider, and the point of contact for substance use disorders at the Berks CBOC of the Lebanon VAMC in Wyomissing, Pennsylvania. Recently, an exasperated primary care provider at the Berks CBOC approached me about working with patients with substance use disorders and asked “How do you work with this challenging population?”
The question was cause for some introspection. When I started working in addictions, I had to modify my approach to work effectively with this population. I experienced a paradigm shift in which I no longer assigned myself credit or blame for a veteran’s continued sobriety or relapse: Each patient is responsible for his or her progress in the recovery journey. When a patient has a relapse, I remind myself that statistically relapse is a probability for the majority of those in recovery; even multiple relapses are common. Therefore, another way of viewing relapse is that the relapse itself may bring the veteran a step closer to permanent abstinence.
Cognitive Reframing
Consider a toddler learning to walk. Parents and caregivers of the child expect the child to fall quite a few times before he or she has mastered walking. The parents and caregivers don’t get angry, take the situation personally, or feel manipulated by the child’s “failure.” Instead, the parents offer the child emotional support and encouragement.
Arguably, the practitioner should take a similar stance—emotionally supportive and encouraging—in combination with dialogue guided by motivational interviewing to support change and help the veteran get back on track. Although not inevitable, relapse is a normal part of recovery.
Practitioners should avoid either scolding or praising patients. Scolding doesn’t help in the recovery journey, and if the veteran learns that the practitioner reacts by scolding after learning of the relapse, then it is less likely that he or she will be open about future missteps. In fact, the veteran may not return at all. The goal is to have the patient come back when relapse occurs to help him or her progress to recovery.
Too much praise can cause similar problems. A veteran accustomed to praise when he or she is doing well, may be too embarrassed or ashamed to return to ask for help after a relapse. Instead the practitioner should use affirmations, which are widely discussed in motivational interviewing (MI) literature.
“I want to be clean and sober” or “I want to stop drinking” are vague statements that should raise a red flag for experienced practitioners. Is this patient just telling me what he or she thinks I want to hear to respond? That may be the case but also may be an assumption. Instead these assertions could be regarded as global treatment goals, and the task of the practitioner is to help the veteran develop objectives and interventions in relation to this goal. These broad statements can be a starting point in MI. These words can sound just as foreign to a patient who isn’t sure whether becoming clean and sober is a possibility. The veteran may not have the confidence to reach the goal of being clean and sober, and these statements may seem awkward and out of sync with his or her facial expression and body language.
Showing disbelief in a veteran also can have negative consequences. The veteran might feel that “even my therapist/health care provider doesn’t believe I can become clean and sober.” Instead, I remind myself that we all must manipulate our environment for survival. I find it more valuable to think of the veteran as being resourceful rather than manipulative.
This point may seem self-evident, but it took me a while to catch on: Most of the change in the recovery journey transpires outside the practitioner’s office. I had to embrace this truism and be prepared when the veteran returned for the next session. The task, then, is to determine at this moment where the veteran is in his or her recovery journey instead of continuing the conversation from the previous session. The previous session may be irrelevant. Thinking this way was an adjustment for me. One of my favorite therapy approaches was to consciously continue a conversation from the previous session to demonstrate that I remembered what the patient had said, thereby showing that I care.
In addition, get to know the patient underneath, behind, and before the substance use disorder. Knowing and liking the veteran helps me avoid burnout, bringing me back to my values and the reason I became a therapist. I make efforts in my thinking process to convert “alcoholic” or “drug abuser” into a more helpful “client addicted to alcohol” or “client with a substance use disorder.” The veteran should not be labeled.
Motivational Interviewing
Oftentimes what propels veterans forward in their recovery is the cognitive dissonance created between who they were and how they acted before substance addiction and how they act now. Conversation steered in this direction, fueled by MI, can enhance a veteran’s motivation to change and ready the veteran to change behaviors. Listen to the veteran’s account of loved ones and remember the names of these important family members and friends. Weave into the conversation the names of these loved ones when the veteran makes statements about becoming a better son/father/grandfather or daughter/mother/grandmother. These references make the goal concrete . A loved one can even be a pet; and for some, the desire to be a more competent and reliable pet owner can be a strong motivation. Give the veteran an opportunity to describe his or her strengths and bask in a self-description. In recovery, it is critical to identify strengths that can be built on to sustain recovery.
Veterans who are confronting a substance use disorder may approach the practitioner with a “fix it for me” attitude as a mental inventory is being taken of all of the negative consequences (eg, homelessness, legal issues, or unemployment). Getting the veteran to take ownership of the problem and the solution is key.
I don’t promise to fix things, instead I engage the veteran in problem-solving. I offer to team up with the veteran in this process, and I promise my best efforts but not outcomes. I avoid giving advice and work to empower the veteran to make sound decisions. Veterans who make their own decisions feel as though they have more control over their lives.
Working with the substance use disorder population is challenging but rewarding when a practitioner can embrace some of the paradigms described in this article. Practitioners may need to do some cognitive reframing within their own thinking, as I described in this article, to become more effective in the field and to help avoid burnout.
I am a behavioral health-licensed clinical social worker, an approved motivational enhancement therapy provider, and the point of contact for substance use disorders at the Berks CBOC of the Lebanon VAMC in Wyomissing, Pennsylvania. Recently, an exasperated primary care provider at the Berks CBOC approached me about working with patients with substance use disorders and asked “How do you work with this challenging population?”
The question was cause for some introspection. When I started working in addictions, I had to modify my approach to work effectively with this population. I experienced a paradigm shift in which I no longer assigned myself credit or blame for a veteran’s continued sobriety or relapse: Each patient is responsible for his or her progress in the recovery journey. When a patient has a relapse, I remind myself that statistically relapse is a probability for the majority of those in recovery; even multiple relapses are common. Therefore, another way of viewing relapse is that the relapse itself may bring the veteran a step closer to permanent abstinence.
Cognitive Reframing
Consider a toddler learning to walk. Parents and caregivers of the child expect the child to fall quite a few times before he or she has mastered walking. The parents and caregivers don’t get angry, take the situation personally, or feel manipulated by the child’s “failure.” Instead, the parents offer the child emotional support and encouragement.
Arguably, the practitioner should take a similar stance—emotionally supportive and encouraging—in combination with dialogue guided by motivational interviewing to support change and help the veteran get back on track. Although not inevitable, relapse is a normal part of recovery.
Practitioners should avoid either scolding or praising patients. Scolding doesn’t help in the recovery journey, and if the veteran learns that the practitioner reacts by scolding after learning of the relapse, then it is less likely that he or she will be open about future missteps. In fact, the veteran may not return at all. The goal is to have the patient come back when relapse occurs to help him or her progress to recovery.
Too much praise can cause similar problems. A veteran accustomed to praise when he or she is doing well, may be too embarrassed or ashamed to return to ask for help after a relapse. Instead the practitioner should use affirmations, which are widely discussed in motivational interviewing (MI) literature.
“I want to be clean and sober” or “I want to stop drinking” are vague statements that should raise a red flag for experienced practitioners. Is this patient just telling me what he or she thinks I want to hear to respond? That may be the case but also may be an assumption. Instead these assertions could be regarded as global treatment goals, and the task of the practitioner is to help the veteran develop objectives and interventions in relation to this goal. These broad statements can be a starting point in MI. These words can sound just as foreign to a patient who isn’t sure whether becoming clean and sober is a possibility. The veteran may not have the confidence to reach the goal of being clean and sober, and these statements may seem awkward and out of sync with his or her facial expression and body language.
Showing disbelief in a veteran also can have negative consequences. The veteran might feel that “even my therapist/health care provider doesn’t believe I can become clean and sober.” Instead, I remind myself that we all must manipulate our environment for survival. I find it more valuable to think of the veteran as being resourceful rather than manipulative.
This point may seem self-evident, but it took me a while to catch on: Most of the change in the recovery journey transpires outside the practitioner’s office. I had to embrace this truism and be prepared when the veteran returned for the next session. The task, then, is to determine at this moment where the veteran is in his or her recovery journey instead of continuing the conversation from the previous session. The previous session may be irrelevant. Thinking this way was an adjustment for me. One of my favorite therapy approaches was to consciously continue a conversation from the previous session to demonstrate that I remembered what the patient had said, thereby showing that I care.
In addition, get to know the patient underneath, behind, and before the substance use disorder. Knowing and liking the veteran helps me avoid burnout, bringing me back to my values and the reason I became a therapist. I make efforts in my thinking process to convert “alcoholic” or “drug abuser” into a more helpful “client addicted to alcohol” or “client with a substance use disorder.” The veteran should not be labeled.
Motivational Interviewing
Oftentimes what propels veterans forward in their recovery is the cognitive dissonance created between who they were and how they acted before substance addiction and how they act now. Conversation steered in this direction, fueled by MI, can enhance a veteran’s motivation to change and ready the veteran to change behaviors. Listen to the veteran’s account of loved ones and remember the names of these important family members and friends. Weave into the conversation the names of these loved ones when the veteran makes statements about becoming a better son/father/grandfather or daughter/mother/grandmother. These references make the goal concrete . A loved one can even be a pet; and for some, the desire to be a more competent and reliable pet owner can be a strong motivation. Give the veteran an opportunity to describe his or her strengths and bask in a self-description. In recovery, it is critical to identify strengths that can be built on to sustain recovery.
Veterans who are confronting a substance use disorder may approach the practitioner with a “fix it for me” attitude as a mental inventory is being taken of all of the negative consequences (eg, homelessness, legal issues, or unemployment). Getting the veteran to take ownership of the problem and the solution is key.
I don’t promise to fix things, instead I engage the veteran in problem-solving. I offer to team up with the veteran in this process, and I promise my best efforts but not outcomes. I avoid giving advice and work to empower the veteran to make sound decisions. Veterans who make their own decisions feel as though they have more control over their lives.
Working with the substance use disorder population is challenging but rewarding when a practitioner can embrace some of the paradigms described in this article. Practitioners may need to do some cognitive reframing within their own thinking, as I described in this article, to become more effective in the field and to help avoid burnout.
I am a behavioral health-licensed clinical social worker, an approved motivational enhancement therapy provider, and the point of contact for substance use disorders at the Berks CBOC of the Lebanon VAMC in Wyomissing, Pennsylvania. Recently, an exasperated primary care provider at the Berks CBOC approached me about working with patients with substance use disorders and asked “How do you work with this challenging population?”
The question was cause for some introspection. When I started working in addictions, I had to modify my approach to work effectively with this population. I experienced a paradigm shift in which I no longer assigned myself credit or blame for a veteran’s continued sobriety or relapse: Each patient is responsible for his or her progress in the recovery journey. When a patient has a relapse, I remind myself that statistically relapse is a probability for the majority of those in recovery; even multiple relapses are common. Therefore, another way of viewing relapse is that the relapse itself may bring the veteran a step closer to permanent abstinence.
Cognitive Reframing
Consider a toddler learning to walk. Parents and caregivers of the child expect the child to fall quite a few times before he or she has mastered walking. The parents and caregivers don’t get angry, take the situation personally, or feel manipulated by the child’s “failure.” Instead, the parents offer the child emotional support and encouragement.
Arguably, the practitioner should take a similar stance—emotionally supportive and encouraging—in combination with dialogue guided by motivational interviewing to support change and help the veteran get back on track. Although not inevitable, relapse is a normal part of recovery.
Practitioners should avoid either scolding or praising patients. Scolding doesn’t help in the recovery journey, and if the veteran learns that the practitioner reacts by scolding after learning of the relapse, then it is less likely that he or she will be open about future missteps. In fact, the veteran may not return at all. The goal is to have the patient come back when relapse occurs to help him or her progress to recovery.
Too much praise can cause similar problems. A veteran accustomed to praise when he or she is doing well, may be too embarrassed or ashamed to return to ask for help after a relapse. Instead the practitioner should use affirmations, which are widely discussed in motivational interviewing (MI) literature.
“I want to be clean and sober” or “I want to stop drinking” are vague statements that should raise a red flag for experienced practitioners. Is this patient just telling me what he or she thinks I want to hear to respond? That may be the case but also may be an assumption. Instead these assertions could be regarded as global treatment goals, and the task of the practitioner is to help the veteran develop objectives and interventions in relation to this goal. These broad statements can be a starting point in MI. These words can sound just as foreign to a patient who isn’t sure whether becoming clean and sober is a possibility. The veteran may not have the confidence to reach the goal of being clean and sober, and these statements may seem awkward and out of sync with his or her facial expression and body language.
Showing disbelief in a veteran also can have negative consequences. The veteran might feel that “even my therapist/health care provider doesn’t believe I can become clean and sober.” Instead, I remind myself that we all must manipulate our environment for survival. I find it more valuable to think of the veteran as being resourceful rather than manipulative.
This point may seem self-evident, but it took me a while to catch on: Most of the change in the recovery journey transpires outside the practitioner’s office. I had to embrace this truism and be prepared when the veteran returned for the next session. The task, then, is to determine at this moment where the veteran is in his or her recovery journey instead of continuing the conversation from the previous session. The previous session may be irrelevant. Thinking this way was an adjustment for me. One of my favorite therapy approaches was to consciously continue a conversation from the previous session to demonstrate that I remembered what the patient had said, thereby showing that I care.
In addition, get to know the patient underneath, behind, and before the substance use disorder. Knowing and liking the veteran helps me avoid burnout, bringing me back to my values and the reason I became a therapist. I make efforts in my thinking process to convert “alcoholic” or “drug abuser” into a more helpful “client addicted to alcohol” or “client with a substance use disorder.” The veteran should not be labeled.
Motivational Interviewing
Oftentimes what propels veterans forward in their recovery is the cognitive dissonance created between who they were and how they acted before substance addiction and how they act now. Conversation steered in this direction, fueled by MI, can enhance a veteran’s motivation to change and ready the veteran to change behaviors. Listen to the veteran’s account of loved ones and remember the names of these important family members and friends. Weave into the conversation the names of these loved ones when the veteran makes statements about becoming a better son/father/grandfather or daughter/mother/grandmother. These references make the goal concrete . A loved one can even be a pet; and for some, the desire to be a more competent and reliable pet owner can be a strong motivation. Give the veteran an opportunity to describe his or her strengths and bask in a self-description. In recovery, it is critical to identify strengths that can be built on to sustain recovery.
Veterans who are confronting a substance use disorder may approach the practitioner with a “fix it for me” attitude as a mental inventory is being taken of all of the negative consequences (eg, homelessness, legal issues, or unemployment). Getting the veteran to take ownership of the problem and the solution is key.
I don’t promise to fix things, instead I engage the veteran in problem-solving. I offer to team up with the veteran in this process, and I promise my best efforts but not outcomes. I avoid giving advice and work to empower the veteran to make sound decisions. Veterans who make their own decisions feel as though they have more control over their lives.
Working with the substance use disorder population is challenging but rewarding when a practitioner can embrace some of the paradigms described in this article. Practitioners may need to do some cognitive reframing within their own thinking, as I described in this article, to become more effective in the field and to help avoid burnout.